User login
ICYMI: Rivaroxaban reduces VTE incidence in ambulatory cancer patients
While treatment with rivaroxaban did not significantly reduce venous thromboembolism incidence in high-risk ambulatory patients with cancer over the entire course of a 180-day intervention period (6.0% vs. 8.8% in controls; hazard ratio, 0.66; 95% confidence interval, 0.40-1.09), it did reduce major bleeding incidence while patients were on treatment (2.0% vs. 6.4%; HR, 0.40; 95% CI, 0.20 0.80), according to results from the multicenter, randomized, double-blind, placebo-controlled, parallel-group, phase 3b CASSINI trial published in the New England Journal of Medicine (2019 Feb 20. doi: 10.1056/NEJMoa1814630).
We reported this story at the annual meeting of the American Society of Hematology before it was published in the journal. Find our coverage at the link below.
While treatment with rivaroxaban did not significantly reduce venous thromboembolism incidence in high-risk ambulatory patients with cancer over the entire course of a 180-day intervention period (6.0% vs. 8.8% in controls; hazard ratio, 0.66; 95% confidence interval, 0.40-1.09), it did reduce major bleeding incidence while patients were on treatment (2.0% vs. 6.4%; HR, 0.40; 95% CI, 0.20 0.80), according to results from the multicenter, randomized, double-blind, placebo-controlled, parallel-group, phase 3b CASSINI trial published in the New England Journal of Medicine (2019 Feb 20. doi: 10.1056/NEJMoa1814630).
We reported this story at the annual meeting of the American Society of Hematology before it was published in the journal. Find our coverage at the link below.
While treatment with rivaroxaban did not significantly reduce venous thromboembolism incidence in high-risk ambulatory patients with cancer over the entire course of a 180-day intervention period (6.0% vs. 8.8% in controls; hazard ratio, 0.66; 95% confidence interval, 0.40-1.09), it did reduce major bleeding incidence while patients were on treatment (2.0% vs. 6.4%; HR, 0.40; 95% CI, 0.20 0.80), according to results from the multicenter, randomized, double-blind, placebo-controlled, parallel-group, phase 3b CASSINI trial published in the New England Journal of Medicine (2019 Feb 20. doi: 10.1056/NEJMoa1814630).
We reported this story at the annual meeting of the American Society of Hematology before it was published in the journal. Find our coverage at the link below.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
FDA grants priority review to polatuzumab vedotin for DLBCL

With this BLA, Genentech is seeking approval for polatuzumab vedotin in combination with bendamustine and rituximab (BR) to treat patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL).
The FDA grants priority review to applications for products that are expected to provide significant improvements in the treatment, diagnosis, or prevention of serious conditions. The agency aims to take action on a priority review application within 6 months, rather than the standard 10 months.
The FDA is expected to make a decision on this BLA by Aug. 19, 2019.
The BLA is supported by a phase 1b/2 trial (NCT02257567) of patients with relapsed or refractory follicular lymphoma or DLBCL who received polatuzumab vedotin in combination with BR or obinutuzumab.
The trial’s phase 2 stage included 80 DLBCL patients who were randomized to receive BR or BR plus polatuzumab vedotin, according to Genentech.
The complete response rate was 40% in the polatuzumab vedotin arm and 18% in the BR arm. The median duration of response was 10.3 months and 4.1 months, respectively (hazard ratio [HR] = 0.44).
The median progression-free survival was 7.6 months in the polatuzumab vedotin arm and 2.0 months in the BR arm (HR = 0.34).
Among patients who were ineligible for a transplant, the median overall survival (an exploratory endpoint) was 12.4 months in the polatuzumab vedotin arm and 4.7 months in the BR arm (HR = 0.42).
Patients who received polatuzumab vedotin had higher rates of grade 3-4 cytopenias, compared with patients who received BR alone. Rates of infection and transfusion were similar between the arms.
With this BLA, Genentech is seeking approval for polatuzumab vedotin in combination with bendamustine and rituximab (BR) to treat patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL).
The FDA grants priority review to applications for products that are expected to provide significant improvements in the treatment, diagnosis, or prevention of serious conditions. The agency aims to take action on a priority review application within 6 months, rather than the standard 10 months.
The FDA is expected to make a decision on this BLA by Aug. 19, 2019.
The BLA is supported by a phase 1b/2 trial (NCT02257567) of patients with relapsed or refractory follicular lymphoma or DLBCL who received polatuzumab vedotin in combination with BR or obinutuzumab.
The trial’s phase 2 stage included 80 DLBCL patients who were randomized to receive BR or BR plus polatuzumab vedotin, according to Genentech.
The complete response rate was 40% in the polatuzumab vedotin arm and 18% in the BR arm. The median duration of response was 10.3 months and 4.1 months, respectively (hazard ratio [HR] = 0.44).
The median progression-free survival was 7.6 months in the polatuzumab vedotin arm and 2.0 months in the BR arm (HR = 0.34).
Among patients who were ineligible for a transplant, the median overall survival (an exploratory endpoint) was 12.4 months in the polatuzumab vedotin arm and 4.7 months in the BR arm (HR = 0.42).
Patients who received polatuzumab vedotin had higher rates of grade 3-4 cytopenias, compared with patients who received BR alone. Rates of infection and transfusion were similar between the arms.
With this BLA, Genentech is seeking approval for polatuzumab vedotin in combination with bendamustine and rituximab (BR) to treat patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL).
The FDA grants priority review to applications for products that are expected to provide significant improvements in the treatment, diagnosis, or prevention of serious conditions. The agency aims to take action on a priority review application within 6 months, rather than the standard 10 months.
The FDA is expected to make a decision on this BLA by Aug. 19, 2019.
The BLA is supported by a phase 1b/2 trial (NCT02257567) of patients with relapsed or refractory follicular lymphoma or DLBCL who received polatuzumab vedotin in combination with BR or obinutuzumab.
The trial’s phase 2 stage included 80 DLBCL patients who were randomized to receive BR or BR plus polatuzumab vedotin, according to Genentech.
The complete response rate was 40% in the polatuzumab vedotin arm and 18% in the BR arm. The median duration of response was 10.3 months and 4.1 months, respectively (hazard ratio [HR] = 0.44).
The median progression-free survival was 7.6 months in the polatuzumab vedotin arm and 2.0 months in the BR arm (HR = 0.34).
Among patients who were ineligible for a transplant, the median overall survival (an exploratory endpoint) was 12.4 months in the polatuzumab vedotin arm and 4.7 months in the BR arm (HR = 0.42).
Patients who received polatuzumab vedotin had higher rates of grade 3-4 cytopenias, compared with patients who received BR alone. Rates of infection and transfusion were similar between the arms.
Supplementary compression doesn’t improve DVT odds in critically ill
SAN DIEGO – In critically ill patients receiving pharmacologic thromboprophylaxis, (DVT), according to a new trial.

“I was surprised. My hypothesis was that it would work,” said lead author Yaseen M. Arabi, MD, chairman of the intensive care department at King Saud bin Abdulaziz University for Health Sciences, Riyadh, Saudi Arabia.
Many physicians routinely carry out the practice on the assumption that IPC should lead to better blood flow and further cut DVT risk. The procedure carries few risks, aside from patient discomfort. “The main issue is that it’s not needed. It might be useful in patients who are not receiving heparin or low-molecular-weight heparin,” said Dr. Arabi, who presented the results of the study at the Critical Care Congress sponsored by the Society of Critical Care Medicine. The study was simultaneously published online in the New England Journal of Medicine.
Unfractionated or low-molecular-weight heparin reduces the risk of DVT by about 50%, but about 5%-20% of critically ill patients will develop DVT in spite of treatment, and mechanical thromboprophylaxis reduces DVT risk, compared with no prophylaxis. Some researchers have attempted to address whether adjunct intermittent pneumatic compression could further reduce DVT risk, but their studies were marked by a lack of controls, unoptimized pharmacologic regimens, and other limitations.
The trial included 2,003 adults from 20 sites in Saudi Arabia, Canada, Australia, and India, who were expected to have an intensive care unit stay of at least 72 hours. They were randomized to receive IPC combined with pharmacologic thromboprophylaxis (pneumatic compression group) or pharmacologic thromboprophylaxis alone (control).
The proportion of patients receiving unfractionated heparin versus low-molecular-weight heparin was similar between the two groups, with about 58% treated with unfractionated heparin.
A total of 3.9% of patients in the pneumatic compression group experienced incident proximal DVT, compared with 4.2% of controls (relative risk, 0.93; P =.74). A total of 3.4% experienced prevalent proximal DVT, compared with 2.7% of controls (RR, 1.29; 95% confidence interval, 0.78-2.12). There was no significant difference in the incidence of any lower-limb DVT (9.6% vs. 8.4%; RR, 1.14; 95% CI, 0.86-1.51).
There was no difference between the two groups in a composite outcome that included pulmonary embolism or all prevalent and incident lower-limb DVT (RR, 1.11; 95% CI, 0.85-1.44), and there were no between-group differences with respect to lower-limb skin injury or ischemia.
The results should change practice among those who still provide adjunct intermittent pneumatic compression, however surprising physicians may find these new results to be, according to Dr. Arabi: “People believed strongly that (adjunct IPC) should work, but you need to be evidence based, and here it showed no difference. But that’s why we do studies, right?”
The study was funded by King Abdulaziz City for Science and Technology and King Abdullah International Medical Research Center. Dr. Arabi has no relevant financial conflicts.
SOURCE: Arabi Y et al. CCC48, Abstract 142. N Engl J Med Feb 18. doi: 10.1056/NEJMoa1816150.
SAN DIEGO – In critically ill patients receiving pharmacologic thromboprophylaxis, (DVT), according to a new trial.
“I was surprised. My hypothesis was that it would work,” said lead author Yaseen M. Arabi, MD, chairman of the intensive care department at King Saud bin Abdulaziz University for Health Sciences, Riyadh, Saudi Arabia.
Many physicians routinely carry out the practice on the assumption that IPC should lead to better blood flow and further cut DVT risk. The procedure carries few risks, aside from patient discomfort. “The main issue is that it’s not needed. It might be useful in patients who are not receiving heparin or low-molecular-weight heparin,” said Dr. Arabi, who presented the results of the study at the Critical Care Congress sponsored by the Society of Critical Care Medicine. The study was simultaneously published online in the New England Journal of Medicine.
Unfractionated or low-molecular-weight heparin reduces the risk of DVT by about 50%, but about 5%-20% of critically ill patients will develop DVT in spite of treatment, and mechanical thromboprophylaxis reduces DVT risk, compared with no prophylaxis. Some researchers have attempted to address whether adjunct intermittent pneumatic compression could further reduce DVT risk, but their studies were marked by a lack of controls, unoptimized pharmacologic regimens, and other limitations.
The trial included 2,003 adults from 20 sites in Saudi Arabia, Canada, Australia, and India, who were expected to have an intensive care unit stay of at least 72 hours. They were randomized to receive IPC combined with pharmacologic thromboprophylaxis (pneumatic compression group) or pharmacologic thromboprophylaxis alone (control).
The proportion of patients receiving unfractionated heparin versus low-molecular-weight heparin was similar between the two groups, with about 58% treated with unfractionated heparin.
A total of 3.9% of patients in the pneumatic compression group experienced incident proximal DVT, compared with 4.2% of controls (relative risk, 0.93; P =.74). A total of 3.4% experienced prevalent proximal DVT, compared with 2.7% of controls (RR, 1.29; 95% confidence interval, 0.78-2.12). There was no significant difference in the incidence of any lower-limb DVT (9.6% vs. 8.4%; RR, 1.14; 95% CI, 0.86-1.51).
There was no difference between the two groups in a composite outcome that included pulmonary embolism or all prevalent and incident lower-limb DVT (RR, 1.11; 95% CI, 0.85-1.44), and there were no between-group differences with respect to lower-limb skin injury or ischemia.
The results should change practice among those who still provide adjunct intermittent pneumatic compression, however surprising physicians may find these new results to be, according to Dr. Arabi: “People believed strongly that (adjunct IPC) should work, but you need to be evidence based, and here it showed no difference. But that’s why we do studies, right?”
The study was funded by King Abdulaziz City for Science and Technology and King Abdullah International Medical Research Center. Dr. Arabi has no relevant financial conflicts.
SOURCE: Arabi Y et al. CCC48, Abstract 142. N Engl J Med Feb 18. doi: 10.1056/NEJMoa1816150.
SAN DIEGO – In critically ill patients receiving pharmacologic thromboprophylaxis, (DVT), according to a new trial.
“I was surprised. My hypothesis was that it would work,” said lead author Yaseen M. Arabi, MD, chairman of the intensive care department at King Saud bin Abdulaziz University for Health Sciences, Riyadh, Saudi Arabia.
Many physicians routinely carry out the practice on the assumption that IPC should lead to better blood flow and further cut DVT risk. The procedure carries few risks, aside from patient discomfort. “The main issue is that it’s not needed. It might be useful in patients who are not receiving heparin or low-molecular-weight heparin,” said Dr. Arabi, who presented the results of the study at the Critical Care Congress sponsored by the Society of Critical Care Medicine. The study was simultaneously published online in the New England Journal of Medicine.
Unfractionated or low-molecular-weight heparin reduces the risk of DVT by about 50%, but about 5%-20% of critically ill patients will develop DVT in spite of treatment, and mechanical thromboprophylaxis reduces DVT risk, compared with no prophylaxis. Some researchers have attempted to address whether adjunct intermittent pneumatic compression could further reduce DVT risk, but their studies were marked by a lack of controls, unoptimized pharmacologic regimens, and other limitations.
The trial included 2,003 adults from 20 sites in Saudi Arabia, Canada, Australia, and India, who were expected to have an intensive care unit stay of at least 72 hours. They were randomized to receive IPC combined with pharmacologic thromboprophylaxis (pneumatic compression group) or pharmacologic thromboprophylaxis alone (control).
The proportion of patients receiving unfractionated heparin versus low-molecular-weight heparin was similar between the two groups, with about 58% treated with unfractionated heparin.
A total of 3.9% of patients in the pneumatic compression group experienced incident proximal DVT, compared with 4.2% of controls (relative risk, 0.93; P =.74). A total of 3.4% experienced prevalent proximal DVT, compared with 2.7% of controls (RR, 1.29; 95% confidence interval, 0.78-2.12). There was no significant difference in the incidence of any lower-limb DVT (9.6% vs. 8.4%; RR, 1.14; 95% CI, 0.86-1.51).
There was no difference between the two groups in a composite outcome that included pulmonary embolism or all prevalent and incident lower-limb DVT (RR, 1.11; 95% CI, 0.85-1.44), and there were no between-group differences with respect to lower-limb skin injury or ischemia.
The results should change practice among those who still provide adjunct intermittent pneumatic compression, however surprising physicians may find these new results to be, according to Dr. Arabi: “People believed strongly that (adjunct IPC) should work, but you need to be evidence based, and here it showed no difference. But that’s why we do studies, right?”
The study was funded by King Abdulaziz City for Science and Technology and King Abdullah International Medical Research Center. Dr. Arabi has no relevant financial conflicts.
SOURCE: Arabi Y et al. CCC48, Abstract 142. N Engl J Med Feb 18. doi: 10.1056/NEJMoa1816150.
REPORTING FROM CCC48
FDA approves turoctocog alfa pegol for hemophilia A
The agency approved turoctocog alfa pegol for use as routine prophylaxis to reduce the frequency of bleeding episodes, for on-demand treatment and control of bleeding episodes, and for perioperative management of bleeding in adults and children with hemophilia A.
Turoctocog alfa pegol will not be available in the United States before 2020, according to Novo Nordisk. The company cannot yet launch the product because of third-party intellectual property agreements.
The FDA’s approval of turoctocog alfa pegol was supported by results from the pathfinder 2 (NCT01480180), pathfinder 3 (NCT01489111), and pathfinder 5 (NCT01731600) trials.
The trials included children, adolescents, and adults with previously treated, severe hemophilia A and no history of inhibitors. Turoctocog alfa pegol was considered effective and well tolerated in these trials.
Pooled results from pathfinder 2 and pathfinder 5 were presented at the 2018 annual meeting of the American Society of Hematology (Blood. 2018:132:1177).
Results from pathfinder 3 were previously published in Haemophilia (2017. Sep;23[5]:689-96).
The agency approved turoctocog alfa pegol for use as routine prophylaxis to reduce the frequency of bleeding episodes, for on-demand treatment and control of bleeding episodes, and for perioperative management of bleeding in adults and children with hemophilia A.
Turoctocog alfa pegol will not be available in the United States before 2020, according to Novo Nordisk. The company cannot yet launch the product because of third-party intellectual property agreements.
The FDA’s approval of turoctocog alfa pegol was supported by results from the pathfinder 2 (NCT01480180), pathfinder 3 (NCT01489111), and pathfinder 5 (NCT01731600) trials.
The trials included children, adolescents, and adults with previously treated, severe hemophilia A and no history of inhibitors. Turoctocog alfa pegol was considered effective and well tolerated in these trials.
Pooled results from pathfinder 2 and pathfinder 5 were presented at the 2018 annual meeting of the American Society of Hematology (Blood. 2018:132:1177).
Results from pathfinder 3 were previously published in Haemophilia (2017. Sep;23[5]:689-96).
The agency approved turoctocog alfa pegol for use as routine prophylaxis to reduce the frequency of bleeding episodes, for on-demand treatment and control of bleeding episodes, and for perioperative management of bleeding in adults and children with hemophilia A.
Turoctocog alfa pegol will not be available in the United States before 2020, according to Novo Nordisk. The company cannot yet launch the product because of third-party intellectual property agreements.
The FDA’s approval of turoctocog alfa pegol was supported by results from the pathfinder 2 (NCT01480180), pathfinder 3 (NCT01489111), and pathfinder 5 (NCT01731600) trials.
The trials included children, adolescents, and adults with previously treated, severe hemophilia A and no history of inhibitors. Turoctocog alfa pegol was considered effective and well tolerated in these trials.
Pooled results from pathfinder 2 and pathfinder 5 were presented at the 2018 annual meeting of the American Society of Hematology (Blood. 2018:132:1177).
Results from pathfinder 3 were previously published in Haemophilia (2017. Sep;23[5]:689-96).
Researchers characterize new subtype of high-grade DLBCL
(DLBCL)
Patients with this subtype, dubbed “molecular high-grade” (MHG) DLBCL, were more likely to have germinal center B-cell-like (GCB) DLBCL, MYC rearrangements, and double-hit lymphoma.
When compared to other DLBCL patients, those with MHG DLBCL had inferior progression-free and overall survival.
Chulin Sha, PhD, of the University of Leeds (England), and colleagues reported these findings in the Journal of Clinical Oncology. The findings were published alongside a related editorial and a similar study from another group.
Dr. Sha and colleagues began their study by applying a previously developed gene expression classifier (Genome Med. 2015 Jul 1;7[1]:64) to 928 DLBCL patients enrolled in the REMoDL-B trial. REMoDL-B was designed to compare rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisolone (R-CHOP) to R-CHOP plus bortezomib (Hematol Oncol. 2017;35:130-1).
Dr. Sha and colleagues looked for somatic mutations in 400 REMoDL-B patient samples that were sequenced for a 70-gene panel.
The team also tested 360 samples for MYC, BCL2, and BCL6 chromosomal rearrangements using fluorescent in situ hybridization, and they tested 355 samples for MYC and BCL2 protein expression with immunohistochemistry.
Characteristics of MHG DLBCL
The researchers identified 83 REMoDL-B patients as having MHG DLBCL (9%). Most of the MHG patients had GCB DLBCL (90%), 48.6% had MYC rearrangements, and 36.1% had double-hit lymphoma.
Patients with MHG DLBCL had higher International Prognostic Index scores (P = .004), greater tumor bulk (P = .007), higher disease stage (P = .06), and higher lactate dehydrogenase levels (P less than .001) than patients with non-MHG DLBCL.
Although most MHG patients had GCB DLBCL, the researchers found key differences between patients with MHG DLBCL and non-MHG GCB DLBCL. MHG patients were significantly more likely than patients with non-MHG GCB DLBCL to have mutations in KMT2D, BCL2, MYC, and DDX3X. Additionally, some genes frequently mutated in GCB DLBCL — such as B2M, SGK1, and NFKBIA — were rare in MHG DLBCL.
Dr. Sha and colleagues also compared the MHG patients to 70 patients with Burkitt lymphoma (BL) who had been analyzed in a previous study (Genome Med. 2015 Jul 1;7[1]:64).
The researchers found that BL has more upregulated genes than GCB (2,483 genes) and MHG DLBCL (1,784 genes), and MHG DLBCL has more upregulated genes than GCB DLBCL (382 genes). The team observed a similar pattern with downregulated genes and said this suggests “MHG is an intermediate group but closer to GCB than to BL.”
The researchers also found, however, that “MHG and BL share high expression of signatures that contain cell-cycle genes, ribosome biogenesis, MYC overexpression, and TCF3 targets, which suggests a shared proliferative phenotype.”
The team determined that MHG has “a highly proliferative phenotype and shares features with centroblasts of the germinal center dark zone.”
Another discovery was that MHG patients in the REMoDL-B trial had worse progression-free survival (PFS) than their peers.
Among patients who received R-CHOP, the estimated 3-year PFS was:
- 37% for MHG patients
- 78% for patients with GCB DLBCL
- 64% for patients with activated B-cell like (ABC) DLBCL
- 65% for patients with unclassified DLBCL.
Among patients who received bortezomib plus R-CHOP, there was a trend toward improved PFS for patients with MHG DLBCL (58%; P = .08).
Validation cohort
Dr. Sha and colleagues validated their initial findings using RNA sequencing data from another group of DLBCL patients (Cell. 2017 Oct 5;171[2]:481-94.e15). This data set included 624 patients who received rituximab-based therapy.
Seventy-two patients in this group had MHG DLBCL (11.5%), and most MHG patients had GCB DLBCL (82%).
The researchers said the MHG group in this cohort “showed similar associations with clinical variables” and a “similar mutation spectrum” as the MHG group in the REMoDL-B cohort. Additionally, MHG patients in the validation cohort had inferior overall survival (P less than .001) compared to patients with non-MHG GCB DLBCL.
Dr. Sha and colleagues said the poor prognosis in MHG patients in both cohorts suggests a need for different treatment approaches in this group.
In the related editorial, Wing C. Chan, MD, of City of Hope Medical Center in Duarte, Calif., echoed that sentiment and said it will be important to include patients with high-risk DLBCL in clinical trials.
“Their tumors should be comprehensively characterize[d] for correlative analysis to determine the molecular lesions that underlie their biology and response to treatment,” Dr. Chan wrote.
Dr. Chan disclosed a patent for a diagnostic algorithm on GCB/ABC-type DLBCL and a patent on a diagnostic algorithm for peripheral T-cell lymphoma.
Dr. Sha and colleagues disclosed relationships with a range of pharmaceutical companies. The team’s research was supported by a grant from Bloodwise.
The REMoDL-B trial was endorsed by Cancer Research UK and was funded by Janssen-Cillag.
SOURCES: Sha C et al. J Clin Oncol. 2019 Jan 20;37(3):202-12. doi: 10.1200/JCO.18.01314; Chan WC. J Clin Oncol. 2019 Jan 20;37(3):175-7. doi: 10.1200/JCO.18.01910; Ennishi D et al. J Clin Oncol. 2019 Jan 20;37(3):190-201. doi: 10.1200/JCO.18.01583
(DLBCL)
Patients with this subtype, dubbed “molecular high-grade” (MHG) DLBCL, were more likely to have germinal center B-cell-like (GCB) DLBCL, MYC rearrangements, and double-hit lymphoma.
When compared to other DLBCL patients, those with MHG DLBCL had inferior progression-free and overall survival.
Chulin Sha, PhD, of the University of Leeds (England), and colleagues reported these findings in the Journal of Clinical Oncology. The findings were published alongside a related editorial and a similar study from another group.
Dr. Sha and colleagues began their study by applying a previously developed gene expression classifier (Genome Med. 2015 Jul 1;7[1]:64) to 928 DLBCL patients enrolled in the REMoDL-B trial. REMoDL-B was designed to compare rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisolone (R-CHOP) to R-CHOP plus bortezomib (Hematol Oncol. 2017;35:130-1).
Dr. Sha and colleagues looked for somatic mutations in 400 REMoDL-B patient samples that were sequenced for a 70-gene panel.
The team also tested 360 samples for MYC, BCL2, and BCL6 chromosomal rearrangements using fluorescent in situ hybridization, and they tested 355 samples for MYC and BCL2 protein expression with immunohistochemistry.
Characteristics of MHG DLBCL
The researchers identified 83 REMoDL-B patients as having MHG DLBCL (9%). Most of the MHG patients had GCB DLBCL (90%), 48.6% had MYC rearrangements, and 36.1% had double-hit lymphoma.
Patients with MHG DLBCL had higher International Prognostic Index scores (P = .004), greater tumor bulk (P = .007), higher disease stage (P = .06), and higher lactate dehydrogenase levels (P less than .001) than patients with non-MHG DLBCL.
Although most MHG patients had GCB DLBCL, the researchers found key differences between patients with MHG DLBCL and non-MHG GCB DLBCL. MHG patients were significantly more likely than patients with non-MHG GCB DLBCL to have mutations in KMT2D, BCL2, MYC, and DDX3X. Additionally, some genes frequently mutated in GCB DLBCL — such as B2M, SGK1, and NFKBIA — were rare in MHG DLBCL.
Dr. Sha and colleagues also compared the MHG patients to 70 patients with Burkitt lymphoma (BL) who had been analyzed in a previous study (Genome Med. 2015 Jul 1;7[1]:64).
The researchers found that BL has more upregulated genes than GCB (2,483 genes) and MHG DLBCL (1,784 genes), and MHG DLBCL has more upregulated genes than GCB DLBCL (382 genes). The team observed a similar pattern with downregulated genes and said this suggests “MHG is an intermediate group but closer to GCB than to BL.”
The researchers also found, however, that “MHG and BL share high expression of signatures that contain cell-cycle genes, ribosome biogenesis, MYC overexpression, and TCF3 targets, which suggests a shared proliferative phenotype.”
The team determined that MHG has “a highly proliferative phenotype and shares features with centroblasts of the germinal center dark zone.”
Another discovery was that MHG patients in the REMoDL-B trial had worse progression-free survival (PFS) than their peers.
Among patients who received R-CHOP, the estimated 3-year PFS was:
- 37% for MHG patients
- 78% for patients with GCB DLBCL
- 64% for patients with activated B-cell like (ABC) DLBCL
- 65% for patients with unclassified DLBCL.
Among patients who received bortezomib plus R-CHOP, there was a trend toward improved PFS for patients with MHG DLBCL (58%; P = .08).
Validation cohort
Dr. Sha and colleagues validated their initial findings using RNA sequencing data from another group of DLBCL patients (Cell. 2017 Oct 5;171[2]:481-94.e15). This data set included 624 patients who received rituximab-based therapy.
Seventy-two patients in this group had MHG DLBCL (11.5%), and most MHG patients had GCB DLBCL (82%).
The researchers said the MHG group in this cohort “showed similar associations with clinical variables” and a “similar mutation spectrum” as the MHG group in the REMoDL-B cohort. Additionally, MHG patients in the validation cohort had inferior overall survival (P less than .001) compared to patients with non-MHG GCB DLBCL.
Dr. Sha and colleagues said the poor prognosis in MHG patients in both cohorts suggests a need for different treatment approaches in this group.
In the related editorial, Wing C. Chan, MD, of City of Hope Medical Center in Duarte, Calif., echoed that sentiment and said it will be important to include patients with high-risk DLBCL in clinical trials.
“Their tumors should be comprehensively characterize[d] for correlative analysis to determine the molecular lesions that underlie their biology and response to treatment,” Dr. Chan wrote.
Dr. Chan disclosed a patent for a diagnostic algorithm on GCB/ABC-type DLBCL and a patent on a diagnostic algorithm for peripheral T-cell lymphoma.
Dr. Sha and colleagues disclosed relationships with a range of pharmaceutical companies. The team’s research was supported by a grant from Bloodwise.
The REMoDL-B trial was endorsed by Cancer Research UK and was funded by Janssen-Cillag.
SOURCES: Sha C et al. J Clin Oncol. 2019 Jan 20;37(3):202-12. doi: 10.1200/JCO.18.01314; Chan WC. J Clin Oncol. 2019 Jan 20;37(3):175-7. doi: 10.1200/JCO.18.01910; Ennishi D et al. J Clin Oncol. 2019 Jan 20;37(3):190-201. doi: 10.1200/JCO.18.01583
(DLBCL)
Patients with this subtype, dubbed “molecular high-grade” (MHG) DLBCL, were more likely to have germinal center B-cell-like (GCB) DLBCL, MYC rearrangements, and double-hit lymphoma.
When compared to other DLBCL patients, those with MHG DLBCL had inferior progression-free and overall survival.
Chulin Sha, PhD, of the University of Leeds (England), and colleagues reported these findings in the Journal of Clinical Oncology. The findings were published alongside a related editorial and a similar study from another group.
Dr. Sha and colleagues began their study by applying a previously developed gene expression classifier (Genome Med. 2015 Jul 1;7[1]:64) to 928 DLBCL patients enrolled in the REMoDL-B trial. REMoDL-B was designed to compare rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisolone (R-CHOP) to R-CHOP plus bortezomib (Hematol Oncol. 2017;35:130-1).
Dr. Sha and colleagues looked for somatic mutations in 400 REMoDL-B patient samples that were sequenced for a 70-gene panel.
The team also tested 360 samples for MYC, BCL2, and BCL6 chromosomal rearrangements using fluorescent in situ hybridization, and they tested 355 samples for MYC and BCL2 protein expression with immunohistochemistry.
Characteristics of MHG DLBCL
The researchers identified 83 REMoDL-B patients as having MHG DLBCL (9%). Most of the MHG patients had GCB DLBCL (90%), 48.6% had MYC rearrangements, and 36.1% had double-hit lymphoma.
Patients with MHG DLBCL had higher International Prognostic Index scores (P = .004), greater tumor bulk (P = .007), higher disease stage (P = .06), and higher lactate dehydrogenase levels (P less than .001) than patients with non-MHG DLBCL.
Although most MHG patients had GCB DLBCL, the researchers found key differences between patients with MHG DLBCL and non-MHG GCB DLBCL. MHG patients were significantly more likely than patients with non-MHG GCB DLBCL to have mutations in KMT2D, BCL2, MYC, and DDX3X. Additionally, some genes frequently mutated in GCB DLBCL — such as B2M, SGK1, and NFKBIA — were rare in MHG DLBCL.
Dr. Sha and colleagues also compared the MHG patients to 70 patients with Burkitt lymphoma (BL) who had been analyzed in a previous study (Genome Med. 2015 Jul 1;7[1]:64).
The researchers found that BL has more upregulated genes than GCB (2,483 genes) and MHG DLBCL (1,784 genes), and MHG DLBCL has more upregulated genes than GCB DLBCL (382 genes). The team observed a similar pattern with downregulated genes and said this suggests “MHG is an intermediate group but closer to GCB than to BL.”
The researchers also found, however, that “MHG and BL share high expression of signatures that contain cell-cycle genes, ribosome biogenesis, MYC overexpression, and TCF3 targets, which suggests a shared proliferative phenotype.”
The team determined that MHG has “a highly proliferative phenotype and shares features with centroblasts of the germinal center dark zone.”
Another discovery was that MHG patients in the REMoDL-B trial had worse progression-free survival (PFS) than their peers.
Among patients who received R-CHOP, the estimated 3-year PFS was:
- 37% for MHG patients
- 78% for patients with GCB DLBCL
- 64% for patients with activated B-cell like (ABC) DLBCL
- 65% for patients with unclassified DLBCL.
Among patients who received bortezomib plus R-CHOP, there was a trend toward improved PFS for patients with MHG DLBCL (58%; P = .08).
Validation cohort
Dr. Sha and colleagues validated their initial findings using RNA sequencing data from another group of DLBCL patients (Cell. 2017 Oct 5;171[2]:481-94.e15). This data set included 624 patients who received rituximab-based therapy.
Seventy-two patients in this group had MHG DLBCL (11.5%), and most MHG patients had GCB DLBCL (82%).
The researchers said the MHG group in this cohort “showed similar associations with clinical variables” and a “similar mutation spectrum” as the MHG group in the REMoDL-B cohort. Additionally, MHG patients in the validation cohort had inferior overall survival (P less than .001) compared to patients with non-MHG GCB DLBCL.
Dr. Sha and colleagues said the poor prognosis in MHG patients in both cohorts suggests a need for different treatment approaches in this group.
In the related editorial, Wing C. Chan, MD, of City of Hope Medical Center in Duarte, Calif., echoed that sentiment and said it will be important to include patients with high-risk DLBCL in clinical trials.
“Their tumors should be comprehensively characterize[d] for correlative analysis to determine the molecular lesions that underlie their biology and response to treatment,” Dr. Chan wrote.
Dr. Chan disclosed a patent for a diagnostic algorithm on GCB/ABC-type DLBCL and a patent on a diagnostic algorithm for peripheral T-cell lymphoma.
Dr. Sha and colleagues disclosed relationships with a range of pharmaceutical companies. The team’s research was supported by a grant from Bloodwise.
The REMoDL-B trial was endorsed by Cancer Research UK and was funded by Janssen-Cillag.
SOURCES: Sha C et al. J Clin Oncol. 2019 Jan 20;37(3):202-12. doi: 10.1200/JCO.18.01314; Chan WC. J Clin Oncol. 2019 Jan 20;37(3):175-7. doi: 10.1200/JCO.18.01910; Ennishi D et al. J Clin Oncol. 2019 Jan 20;37(3):190-201. doi: 10.1200/JCO.18.01583
FROM THE JOURNAL OF CLINICAL ONCOLOGY
CAR T-cell therapies difficult to compare
One chimeric antigen receptor (CAR) T-cell therapy may appear better than another, but confounding factors make it difficult to compare these therapies effectively, according to a review published in the Journal of Clinical Oncology.
Caron A. Jacobson, MD, of the Dana-Farber Cancer Institute in Boston, reviewed results from three trials of CAR T-cell therapies in patients with B-cell non-Hodgkin lymphoma (B-NHL).
She noted that cross-trial comparisons are always limited, but such comparisons of CAR T-cell therapies are hindered by several confounding factors.
Dr. Jacobson said differences in manufacturing procedures and turnaround time, differences in patient eligibility and management, and the complexity of CAR T-cell therapies make it difficult to compare results from three CAR-T trials in B-NHL:
• The ZUMA-1 trial (NCT02348216) of axicabtagene ciloleucel (axi-cel, Yescarta)
• The JULIET trial (NCT02445248) of tisagenlecleucel (t-cel, Kymriah)
• The TRANSCEND-NHL-001 trial (NCT02631044) of lisocabtagene maraleucel (liso-cel, JCAR017).
Looking at response rates alone, axi-cel appears the most promising. The overall response rate (ORR) was 82% with axi-cel, 75% with liso-cel, and 52% with t-cel.
When considering cytokine release syndrome (CRS), liso-cel appears the safest. The rate of CRS was 93% with axi-cel (13% grade 3 or higher), 58% with t-cel (22% grade 3 or higher), and 39% with liso-cel (1% grade 3 or higher).
However, as Dr. Jacobson pointed out, it’s impossible to know if these differences in efficacy and toxicity are “statistically meaningful.”
Dr. Jacobson also noted that bridging therapy may have affected these results, as it might reduce tumor burden and increase toxicity, but bridging therapy was not used uniformly across these trials.
Most patients received bridging therapy before t-cel, none received it before axi-cel, and the use of bridging therapy was not reported in the trial of liso-cel.
“It is not possible to know whether patients treated on the ZUMA-1 trial, who were more likely to receive their CAR T cells, were healthier and more fit than patients on other studies or, because they were not allowed to receive bridging therapy, were actually sicker with a higher tumor burden and were therefore at risk for greater toxicity,” Dr. Jacobson wrote.
The fact that ZUMA-1 patients were more likely to receive CAR T cells brings up another issue—the difference between the reported results and the intent-to-treat (ITT) results in these trials. Since most patients on ZUMA-1 received the study treatment, there isn't much difference between the reported results and ITT results. However, about a third of patients who underwent apheresis on the JULIET trial did not ultimately receive CAR T cells, which means a bigger difference between the reported results and ITT results.
In ZUMA-1, 111 patients underwent leukapheresis, and 101 received treatment with axi-cel and were evaluable for efficacy. So the ORR was 75% (83/111) in the ITT population, compared to 82% in the population evaluable for efficacy.
In JULIET, 165 patients underwent leukapheresis, 111 received t-cel, and 93 were evaluable. The ORR was 30% (48/161) in the ITT population, compared to 52% in the evaluable population.
In TRANSCEND-NHL-001, 134 patients underwent leukapheresis, 114 patients received liso-cel, and 102 were evaluable. The ORR was 63% (77/122) in the ITT population, compared to 75% in the evaluable population.
Dr. Jacobson said these differences can be explained, in part, by differences in manufacturing. The time to manufacture cells was longer on the JULIET trial than on ZUMA-1, which may have been due to differences in transfection and manufacturing procedures as well as manufacturing ability.
In addition, differences in patient eligibility may have played a role, as healthier patients might be able to tolerate a longer manufacturing period than sicker patients.
Unfortunately, these differences cannot be accounted for without a randomized trial, but Dr. Jacobson said a randomized trial of these therapies is unlikely to occur.
“[S]o perhaps the best answers will come from institutions that have experience with all three products,” she wrote. “And in these cases, physicians and institutions will have to decide to what extent they would sacrifice efficacy for improved safety or sacrifice safety for improved reliability and consistency of treatment delivery.”
Dr. Jacobson disclosed relationships with Kite Pharma/Gilead Sciences, Bayer AG, Pfizer, Precision BioSciences, Novartis, Celgene, and Cowen.
SOURCE: Jacobson CA. J Clin Oncol. 2019 Feb 1;37(4):328-35. doi: 10.1200/JCO.18.01457
One chimeric antigen receptor (CAR) T-cell therapy may appear better than another, but confounding factors make it difficult to compare these therapies effectively, according to a review published in the Journal of Clinical Oncology.
Caron A. Jacobson, MD, of the Dana-Farber Cancer Institute in Boston, reviewed results from three trials of CAR T-cell therapies in patients with B-cell non-Hodgkin lymphoma (B-NHL).
She noted that cross-trial comparisons are always limited, but such comparisons of CAR T-cell therapies are hindered by several confounding factors.
Dr. Jacobson said differences in manufacturing procedures and turnaround time, differences in patient eligibility and management, and the complexity of CAR T-cell therapies make it difficult to compare results from three CAR-T trials in B-NHL:
• The ZUMA-1 trial (NCT02348216) of axicabtagene ciloleucel (axi-cel, Yescarta)
• The JULIET trial (NCT02445248) of tisagenlecleucel (t-cel, Kymriah)
• The TRANSCEND-NHL-001 trial (NCT02631044) of lisocabtagene maraleucel (liso-cel, JCAR017).
Looking at response rates alone, axi-cel appears the most promising. The overall response rate (ORR) was 82% with axi-cel, 75% with liso-cel, and 52% with t-cel.
When considering cytokine release syndrome (CRS), liso-cel appears the safest. The rate of CRS was 93% with axi-cel (13% grade 3 or higher), 58% with t-cel (22% grade 3 or higher), and 39% with liso-cel (1% grade 3 or higher).
However, as Dr. Jacobson pointed out, it’s impossible to know if these differences in efficacy and toxicity are “statistically meaningful.”
Dr. Jacobson also noted that bridging therapy may have affected these results, as it might reduce tumor burden and increase toxicity, but bridging therapy was not used uniformly across these trials.
Most patients received bridging therapy before t-cel, none received it before axi-cel, and the use of bridging therapy was not reported in the trial of liso-cel.
“It is not possible to know whether patients treated on the ZUMA-1 trial, who were more likely to receive their CAR T cells, were healthier and more fit than patients on other studies or, because they were not allowed to receive bridging therapy, were actually sicker with a higher tumor burden and were therefore at risk for greater toxicity,” Dr. Jacobson wrote.
The fact that ZUMA-1 patients were more likely to receive CAR T cells brings up another issue—the difference between the reported results and the intent-to-treat (ITT) results in these trials. Since most patients on ZUMA-1 received the study treatment, there isn't much difference between the reported results and ITT results. However, about a third of patients who underwent apheresis on the JULIET trial did not ultimately receive CAR T cells, which means a bigger difference between the reported results and ITT results.
In ZUMA-1, 111 patients underwent leukapheresis, and 101 received treatment with axi-cel and were evaluable for efficacy. So the ORR was 75% (83/111) in the ITT population, compared to 82% in the population evaluable for efficacy.
In JULIET, 165 patients underwent leukapheresis, 111 received t-cel, and 93 were evaluable. The ORR was 30% (48/161) in the ITT population, compared to 52% in the evaluable population.
In TRANSCEND-NHL-001, 134 patients underwent leukapheresis, 114 patients received liso-cel, and 102 were evaluable. The ORR was 63% (77/122) in the ITT population, compared to 75% in the evaluable population.
Dr. Jacobson said these differences can be explained, in part, by differences in manufacturing. The time to manufacture cells was longer on the JULIET trial than on ZUMA-1, which may have been due to differences in transfection and manufacturing procedures as well as manufacturing ability.
In addition, differences in patient eligibility may have played a role, as healthier patients might be able to tolerate a longer manufacturing period than sicker patients.
Unfortunately, these differences cannot be accounted for without a randomized trial, but Dr. Jacobson said a randomized trial of these therapies is unlikely to occur.
“[S]o perhaps the best answers will come from institutions that have experience with all three products,” she wrote. “And in these cases, physicians and institutions will have to decide to what extent they would sacrifice efficacy for improved safety or sacrifice safety for improved reliability and consistency of treatment delivery.”
Dr. Jacobson disclosed relationships with Kite Pharma/Gilead Sciences, Bayer AG, Pfizer, Precision BioSciences, Novartis, Celgene, and Cowen.
SOURCE: Jacobson CA. J Clin Oncol. 2019 Feb 1;37(4):328-35. doi: 10.1200/JCO.18.01457
One chimeric antigen receptor (CAR) T-cell therapy may appear better than another, but confounding factors make it difficult to compare these therapies effectively, according to a review published in the Journal of Clinical Oncology.
Caron A. Jacobson, MD, of the Dana-Farber Cancer Institute in Boston, reviewed results from three trials of CAR T-cell therapies in patients with B-cell non-Hodgkin lymphoma (B-NHL).
She noted that cross-trial comparisons are always limited, but such comparisons of CAR T-cell therapies are hindered by several confounding factors.
Dr. Jacobson said differences in manufacturing procedures and turnaround time, differences in patient eligibility and management, and the complexity of CAR T-cell therapies make it difficult to compare results from three CAR-T trials in B-NHL:
• The ZUMA-1 trial (NCT02348216) of axicabtagene ciloleucel (axi-cel, Yescarta)
• The JULIET trial (NCT02445248) of tisagenlecleucel (t-cel, Kymriah)
• The TRANSCEND-NHL-001 trial (NCT02631044) of lisocabtagene maraleucel (liso-cel, JCAR017).
Looking at response rates alone, axi-cel appears the most promising. The overall response rate (ORR) was 82% with axi-cel, 75% with liso-cel, and 52% with t-cel.
When considering cytokine release syndrome (CRS), liso-cel appears the safest. The rate of CRS was 93% with axi-cel (13% grade 3 or higher), 58% with t-cel (22% grade 3 or higher), and 39% with liso-cel (1% grade 3 or higher).
However, as Dr. Jacobson pointed out, it’s impossible to know if these differences in efficacy and toxicity are “statistically meaningful.”
Dr. Jacobson also noted that bridging therapy may have affected these results, as it might reduce tumor burden and increase toxicity, but bridging therapy was not used uniformly across these trials.
Most patients received bridging therapy before t-cel, none received it before axi-cel, and the use of bridging therapy was not reported in the trial of liso-cel.
“It is not possible to know whether patients treated on the ZUMA-1 trial, who were more likely to receive their CAR T cells, were healthier and more fit than patients on other studies or, because they were not allowed to receive bridging therapy, were actually sicker with a higher tumor burden and were therefore at risk for greater toxicity,” Dr. Jacobson wrote.
The fact that ZUMA-1 patients were more likely to receive CAR T cells brings up another issue—the difference between the reported results and the intent-to-treat (ITT) results in these trials. Since most patients on ZUMA-1 received the study treatment, there isn't much difference between the reported results and ITT results. However, about a third of patients who underwent apheresis on the JULIET trial did not ultimately receive CAR T cells, which means a bigger difference between the reported results and ITT results.
In ZUMA-1, 111 patients underwent leukapheresis, and 101 received treatment with axi-cel and were evaluable for efficacy. So the ORR was 75% (83/111) in the ITT population, compared to 82% in the population evaluable for efficacy.
In JULIET, 165 patients underwent leukapheresis, 111 received t-cel, and 93 were evaluable. The ORR was 30% (48/161) in the ITT population, compared to 52% in the evaluable population.
In TRANSCEND-NHL-001, 134 patients underwent leukapheresis, 114 patients received liso-cel, and 102 were evaluable. The ORR was 63% (77/122) in the ITT population, compared to 75% in the evaluable population.
Dr. Jacobson said these differences can be explained, in part, by differences in manufacturing. The time to manufacture cells was longer on the JULIET trial than on ZUMA-1, which may have been due to differences in transfection and manufacturing procedures as well as manufacturing ability.
In addition, differences in patient eligibility may have played a role, as healthier patients might be able to tolerate a longer manufacturing period than sicker patients.
Unfortunately, these differences cannot be accounted for without a randomized trial, but Dr. Jacobson said a randomized trial of these therapies is unlikely to occur.
“[S]o perhaps the best answers will come from institutions that have experience with all three products,” she wrote. “And in these cases, physicians and institutions will have to decide to what extent they would sacrifice efficacy for improved safety or sacrifice safety for improved reliability and consistency of treatment delivery.”
Dr. Jacobson disclosed relationships with Kite Pharma/Gilead Sciences, Bayer AG, Pfizer, Precision BioSciences, Novartis, Celgene, and Cowen.
SOURCE: Jacobson CA. J Clin Oncol. 2019 Feb 1;37(4):328-35. doi: 10.1200/JCO.18.01457
FROM JOURNAL OF CLINICAL ONCOLOGY
CMS proposes coverage of CAR T-cell therapy in trials
The Centers for Medicare & Medicaid Services has proposed to cover chimeric antigen receptor (CAR) T-cell therapy for cancer patients participating in clinical trials that study the treatment’s effectiveness, according to a Feb. 15 announcement.
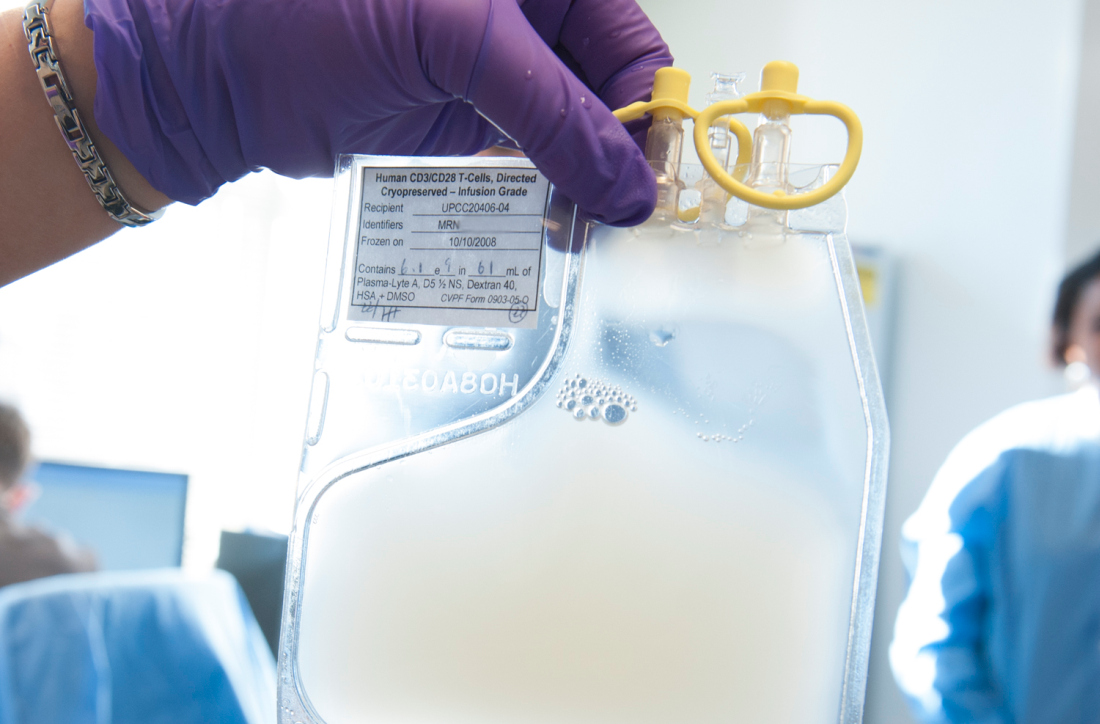
The proposed national coverage determination would require CMS to cover CAR T-cell therapies nationwide when the treatment is offered in CMS-approved registries or clinical studies in which patients are monitored for 2 or more years following treatment.
Results from the studies would help CMS identify which patients benefit most from CAR T-cell therapies and inform future coverage decisions, CMS Administrator Seema Verma said.
“CAR T-cell therapy was the first FDA-approved gene therapy, marking the beginning of an entirely new approach to treating serious and even life-threatening diseases,” Ms. Verma said in a statement. “Today’s proposed coverage decision would improve access to this therapy while deepening CMS’s understanding of how patients in Medicare respond to it, so the agency can ensure that it is paying for CAR T-cell therapy for cases in which the benefits outweigh the risks.”
As part of the proposal, CMS would cover autologous treatment with T cells expressing at least one chimeric antigen receptor (CAR) through coverage with evidence development when prescribed by a treating oncologist and performed in a hospital, according to a summary of the proposal.
The patient and hospital must meet specific criteria to be eligible for coverage, including that patients have relapsed or refractory cancer and do not have a comorbidity that would otherwise preclude patient benefit.
Hospitals, meanwhile, must have a cellular therapy program consisting of an integrated medical team that includes a clinical program director, a quality manager, and at least one physician experienced in cellular therapy, among other requirements.
CMS also would require that treatment is an FDA-approved biologic, providing targeted therapy for a known antigen expressed in the patient’s cancer according to an FDA indication. Repeat treatment would be covered only when a new primary cancer diagnosis is made by the treating oncologist and certain patient conditions are met.
Both inpatient and outpatient settings for the CAR T-cell therapy treatment are acceptable under the proposal. In either case, the patient and the hospital must be participating in a prospective, national, audited registry that consecutively enrolls patients, accepts all manufactured products, follows the patient for at least 2 years, and addresses a set of approved evidence-development questions. Additionally, all registries must be reviewed and approved by CMS.
The proposed national coverage determination was the result of an Aug. 22, 2018 meeting of the Medicare Evidence Development & Coverage Advisory Committee. The committee provides CMS with an external assessment of the appropriateness of therapies under review.
Public comments about the CAR T-cell therapy proposal will be accepted online here until March 15. A final decision on the proposal is expected by May 2019.
The agency’s proposal follows an Aug. 17 final rule by CMS that sets a new payment scheme for inpatient administration of two CAR T-cell therapies. The rule categorizes CAR T-cell therapies under the umbrella of the renamed Medicare Severity–Diagnosis Related Groups 016 – Autologous Bone Marrow Transplant with CC/MCC or T-cell Immunotherapy – and assigns ICD-10 PCS procedure codes XW033C3 and XW043C3 to the use of axicabtagene ciloleucel (Yescarta) and tisagenlecleucel (Kymriah) in the inpatient setting for fiscal year 2019, which began in October 2018. CMS also approved a temporary New Technology Add-On Payment for use of the therapies with a maximum threshold of $186,500.
In April 2018, CMS announced payment rates for outpatient administration of the two drugs, settling on $395,380 for axicabtagene ciloleucel and $500,839 for tisagenlecleucel. The two medications have list prices of $373,000 and $475,000, respectively.
The Centers for Medicare & Medicaid Services has proposed to cover chimeric antigen receptor (CAR) T-cell therapy for cancer patients participating in clinical trials that study the treatment’s effectiveness, according to a Feb. 15 announcement.
The proposed national coverage determination would require CMS to cover CAR T-cell therapies nationwide when the treatment is offered in CMS-approved registries or clinical studies in which patients are monitored for 2 or more years following treatment.
Results from the studies would help CMS identify which patients benefit most from CAR T-cell therapies and inform future coverage decisions, CMS Administrator Seema Verma said.
“CAR T-cell therapy was the first FDA-approved gene therapy, marking the beginning of an entirely new approach to treating serious and even life-threatening diseases,” Ms. Verma said in a statement. “Today’s proposed coverage decision would improve access to this therapy while deepening CMS’s understanding of how patients in Medicare respond to it, so the agency can ensure that it is paying for CAR T-cell therapy for cases in which the benefits outweigh the risks.”
As part of the proposal, CMS would cover autologous treatment with T cells expressing at least one chimeric antigen receptor (CAR) through coverage with evidence development when prescribed by a treating oncologist and performed in a hospital, according to a summary of the proposal.
The patient and hospital must meet specific criteria to be eligible for coverage, including that patients have relapsed or refractory cancer and do not have a comorbidity that would otherwise preclude patient benefit.
Hospitals, meanwhile, must have a cellular therapy program consisting of an integrated medical team that includes a clinical program director, a quality manager, and at least one physician experienced in cellular therapy, among other requirements.
CMS also would require that treatment is an FDA-approved biologic, providing targeted therapy for a known antigen expressed in the patient’s cancer according to an FDA indication. Repeat treatment would be covered only when a new primary cancer diagnosis is made by the treating oncologist and certain patient conditions are met.
Both inpatient and outpatient settings for the CAR T-cell therapy treatment are acceptable under the proposal. In either case, the patient and the hospital must be participating in a prospective, national, audited registry that consecutively enrolls patients, accepts all manufactured products, follows the patient for at least 2 years, and addresses a set of approved evidence-development questions. Additionally, all registries must be reviewed and approved by CMS.
The proposed national coverage determination was the result of an Aug. 22, 2018 meeting of the Medicare Evidence Development & Coverage Advisory Committee. The committee provides CMS with an external assessment of the appropriateness of therapies under review.
Public comments about the CAR T-cell therapy proposal will be accepted online here until March 15. A final decision on the proposal is expected by May 2019.
The agency’s proposal follows an Aug. 17 final rule by CMS that sets a new payment scheme for inpatient administration of two CAR T-cell therapies. The rule categorizes CAR T-cell therapies under the umbrella of the renamed Medicare Severity–Diagnosis Related Groups 016 – Autologous Bone Marrow Transplant with CC/MCC or T-cell Immunotherapy – and assigns ICD-10 PCS procedure codes XW033C3 and XW043C3 to the use of axicabtagene ciloleucel (Yescarta) and tisagenlecleucel (Kymriah) in the inpatient setting for fiscal year 2019, which began in October 2018. CMS also approved a temporary New Technology Add-On Payment for use of the therapies with a maximum threshold of $186,500.
In April 2018, CMS announced payment rates for outpatient administration of the two drugs, settling on $395,380 for axicabtagene ciloleucel and $500,839 for tisagenlecleucel. The two medications have list prices of $373,000 and $475,000, respectively.
The Centers for Medicare & Medicaid Services has proposed to cover chimeric antigen receptor (CAR) T-cell therapy for cancer patients participating in clinical trials that study the treatment’s effectiveness, according to a Feb. 15 announcement.
The proposed national coverage determination would require CMS to cover CAR T-cell therapies nationwide when the treatment is offered in CMS-approved registries or clinical studies in which patients are monitored for 2 or more years following treatment.
Results from the studies would help CMS identify which patients benefit most from CAR T-cell therapies and inform future coverage decisions, CMS Administrator Seema Verma said.
“CAR T-cell therapy was the first FDA-approved gene therapy, marking the beginning of an entirely new approach to treating serious and even life-threatening diseases,” Ms. Verma said in a statement. “Today’s proposed coverage decision would improve access to this therapy while deepening CMS’s understanding of how patients in Medicare respond to it, so the agency can ensure that it is paying for CAR T-cell therapy for cases in which the benefits outweigh the risks.”
As part of the proposal, CMS would cover autologous treatment with T cells expressing at least one chimeric antigen receptor (CAR) through coverage with evidence development when prescribed by a treating oncologist and performed in a hospital, according to a summary of the proposal.
The patient and hospital must meet specific criteria to be eligible for coverage, including that patients have relapsed or refractory cancer and do not have a comorbidity that would otherwise preclude patient benefit.
Hospitals, meanwhile, must have a cellular therapy program consisting of an integrated medical team that includes a clinical program director, a quality manager, and at least one physician experienced in cellular therapy, among other requirements.
CMS also would require that treatment is an FDA-approved biologic, providing targeted therapy for a known antigen expressed in the patient’s cancer according to an FDA indication. Repeat treatment would be covered only when a new primary cancer diagnosis is made by the treating oncologist and certain patient conditions are met.
Both inpatient and outpatient settings for the CAR T-cell therapy treatment are acceptable under the proposal. In either case, the patient and the hospital must be participating in a prospective, national, audited registry that consecutively enrolls patients, accepts all manufactured products, follows the patient for at least 2 years, and addresses a set of approved evidence-development questions. Additionally, all registries must be reviewed and approved by CMS.
The proposed national coverage determination was the result of an Aug. 22, 2018 meeting of the Medicare Evidence Development & Coverage Advisory Committee. The committee provides CMS with an external assessment of the appropriateness of therapies under review.
Public comments about the CAR T-cell therapy proposal will be accepted online here until March 15. A final decision on the proposal is expected by May 2019.
The agency’s proposal follows an Aug. 17 final rule by CMS that sets a new payment scheme for inpatient administration of two CAR T-cell therapies. The rule categorizes CAR T-cell therapies under the umbrella of the renamed Medicare Severity–Diagnosis Related Groups 016 – Autologous Bone Marrow Transplant with CC/MCC or T-cell Immunotherapy – and assigns ICD-10 PCS procedure codes XW033C3 and XW043C3 to the use of axicabtagene ciloleucel (Yescarta) and tisagenlecleucel (Kymriah) in the inpatient setting for fiscal year 2019, which began in October 2018. CMS also approved a temporary New Technology Add-On Payment for use of the therapies with a maximum threshold of $186,500.
In April 2018, CMS announced payment rates for outpatient administration of the two drugs, settling on $395,380 for axicabtagene ciloleucel and $500,839 for tisagenlecleucel. The two medications have list prices of $373,000 and $475,000, respectively.
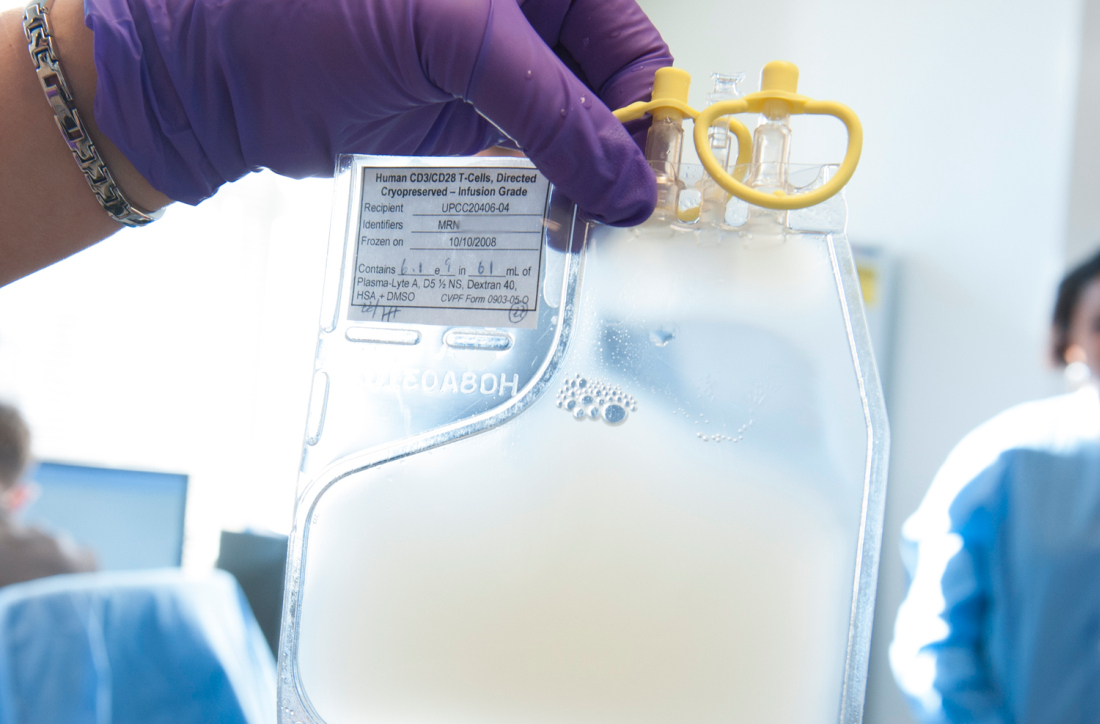
Adding palbociclib upped responses in previously treated MCL
An early study adding palbociclib to ibrutinib in previously treated patients with mantle cell lymphoma (MCL) showed a higher complete response rate than what has previously been reported for single-agent ibrutinib, according to investigators.
Results from the phase 1 trial (NCT02159755) support preclinical models, suggesting that the CDK4/6 inhibitor palbociclib may be able to help overcome resistance to ibrutinib, an inhibitor of Bruton’s tyrosine kinase (BTK).
These findings set the stage for an ongoing phase 2 multicenter study, reported lead author Peter Martin, MD, of Weill Cornell Medicine in New York and his colleagues.
The present study involved 27 patients with previously treated MCL, the investigators wrote in Blood. Of these, 21 were men and 6 were women, all of whom had adequate organ and bone marrow function, good performance status, and no previous treatment with CDK4/6 or BTK inhibitors.
Patients were randomly grouped into five dose levels of each drug: Ibrutinib doses ranged from 280-560 mg, and palbociclib from 75-125 mg. Ibrutinib was given daily and palbociclib was administered for 21 out of 28 days per cycle. Therapy continued until withdrawal, unacceptable toxicity, or disease progression.
The primary objective was to determine phase 2 dose. Secondarily, the investigators sought to determine activity and toxicity profiles. The maximum tolerated doses were ibrutinib 560 mg daily plus palbociclib 100 mg on days 1-21 of each 28-day cycle.
Across all patients, the complete response rate was 37%, compared with 21% for ibrutinib monotherapy in a previous trial. About two-thirds of patients had a response of any kind, which aligns closely with the overall response rate previously reported for ibrutinib alone (67% vs. 68%). After a median follow-up of 25.6 months in survivors, the 2-year progression free survival was 59.4%. The two-year overall survival rate was 60.6%.
The dose-limiting toxicity was grade 3 rash, which occurred in two out of five patients treated at the highest doses. The most common grade 3 or higher toxicities were neutropenia (41%) and thrombocytopenia (30%), followed by hypertension (15%), febrile neutropenia (15%), lung infection (11%), fatigue (7%), upper respiratory tract infection (7%), hyperglycemia (7%), rash (7%), myalgia (7%), and increased alanine transaminase/aspartate aminotransferase (7%).
“Although BTK-inhibitor-based combinations appear promising, the degree to which they improve upon single-agent ibrutinib is unclear,” the investigators wrote, noting that a phase 2 trial (NCT03478514) is currently underway and uses the maximum tolerated doses.
The phase 1 trial was sponsored by the National Cancer Institute. Study funding was provided by the Sarah Cannon Fund at the HCA Foundation. The investigators reported financial relationships with Janssen, Gilead, AstraZeneca, Celgene, Karyopharm, and others.
SOURCE: Martin P et al. Blood. 2019 Jan 28. doi: 10.1182/blood-2018-11-886457.
An early study adding palbociclib to ibrutinib in previously treated patients with mantle cell lymphoma (MCL) showed a higher complete response rate than what has previously been reported for single-agent ibrutinib, according to investigators.
Results from the phase 1 trial (NCT02159755) support preclinical models, suggesting that the CDK4/6 inhibitor palbociclib may be able to help overcome resistance to ibrutinib, an inhibitor of Bruton’s tyrosine kinase (BTK).
These findings set the stage for an ongoing phase 2 multicenter study, reported lead author Peter Martin, MD, of Weill Cornell Medicine in New York and his colleagues.
The present study involved 27 patients with previously treated MCL, the investigators wrote in Blood. Of these, 21 were men and 6 were women, all of whom had adequate organ and bone marrow function, good performance status, and no previous treatment with CDK4/6 or BTK inhibitors.
Patients were randomly grouped into five dose levels of each drug: Ibrutinib doses ranged from 280-560 mg, and palbociclib from 75-125 mg. Ibrutinib was given daily and palbociclib was administered for 21 out of 28 days per cycle. Therapy continued until withdrawal, unacceptable toxicity, or disease progression.
The primary objective was to determine phase 2 dose. Secondarily, the investigators sought to determine activity and toxicity profiles. The maximum tolerated doses were ibrutinib 560 mg daily plus palbociclib 100 mg on days 1-21 of each 28-day cycle.
Across all patients, the complete response rate was 37%, compared with 21% for ibrutinib monotherapy in a previous trial. About two-thirds of patients had a response of any kind, which aligns closely with the overall response rate previously reported for ibrutinib alone (67% vs. 68%). After a median follow-up of 25.6 months in survivors, the 2-year progression free survival was 59.4%. The two-year overall survival rate was 60.6%.
The dose-limiting toxicity was grade 3 rash, which occurred in two out of five patients treated at the highest doses. The most common grade 3 or higher toxicities were neutropenia (41%) and thrombocytopenia (30%), followed by hypertension (15%), febrile neutropenia (15%), lung infection (11%), fatigue (7%), upper respiratory tract infection (7%), hyperglycemia (7%), rash (7%), myalgia (7%), and increased alanine transaminase/aspartate aminotransferase (7%).
“Although BTK-inhibitor-based combinations appear promising, the degree to which they improve upon single-agent ibrutinib is unclear,” the investigators wrote, noting that a phase 2 trial (NCT03478514) is currently underway and uses the maximum tolerated doses.
The phase 1 trial was sponsored by the National Cancer Institute. Study funding was provided by the Sarah Cannon Fund at the HCA Foundation. The investigators reported financial relationships with Janssen, Gilead, AstraZeneca, Celgene, Karyopharm, and others.
SOURCE: Martin P et al. Blood. 2019 Jan 28. doi: 10.1182/blood-2018-11-886457.
An early study adding palbociclib to ibrutinib in previously treated patients with mantle cell lymphoma (MCL) showed a higher complete response rate than what has previously been reported for single-agent ibrutinib, according to investigators.
Results from the phase 1 trial (NCT02159755) support preclinical models, suggesting that the CDK4/6 inhibitor palbociclib may be able to help overcome resistance to ibrutinib, an inhibitor of Bruton’s tyrosine kinase (BTK).
These findings set the stage for an ongoing phase 2 multicenter study, reported lead author Peter Martin, MD, of Weill Cornell Medicine in New York and his colleagues.
The present study involved 27 patients with previously treated MCL, the investigators wrote in Blood. Of these, 21 were men and 6 were women, all of whom had adequate organ and bone marrow function, good performance status, and no previous treatment with CDK4/6 or BTK inhibitors.
Patients were randomly grouped into five dose levels of each drug: Ibrutinib doses ranged from 280-560 mg, and palbociclib from 75-125 mg. Ibrutinib was given daily and palbociclib was administered for 21 out of 28 days per cycle. Therapy continued until withdrawal, unacceptable toxicity, or disease progression.
The primary objective was to determine phase 2 dose. Secondarily, the investigators sought to determine activity and toxicity profiles. The maximum tolerated doses were ibrutinib 560 mg daily plus palbociclib 100 mg on days 1-21 of each 28-day cycle.
Across all patients, the complete response rate was 37%, compared with 21% for ibrutinib monotherapy in a previous trial. About two-thirds of patients had a response of any kind, which aligns closely with the overall response rate previously reported for ibrutinib alone (67% vs. 68%). After a median follow-up of 25.6 months in survivors, the 2-year progression free survival was 59.4%. The two-year overall survival rate was 60.6%.
The dose-limiting toxicity was grade 3 rash, which occurred in two out of five patients treated at the highest doses. The most common grade 3 or higher toxicities were neutropenia (41%) and thrombocytopenia (30%), followed by hypertension (15%), febrile neutropenia (15%), lung infection (11%), fatigue (7%), upper respiratory tract infection (7%), hyperglycemia (7%), rash (7%), myalgia (7%), and increased alanine transaminase/aspartate aminotransferase (7%).
“Although BTK-inhibitor-based combinations appear promising, the degree to which they improve upon single-agent ibrutinib is unclear,” the investigators wrote, noting that a phase 2 trial (NCT03478514) is currently underway and uses the maximum tolerated doses.
The phase 1 trial was sponsored by the National Cancer Institute. Study funding was provided by the Sarah Cannon Fund at the HCA Foundation. The investigators reported financial relationships with Janssen, Gilead, AstraZeneca, Celgene, Karyopharm, and others.
SOURCE: Martin P et al. Blood. 2019 Jan 28. doi: 10.1182/blood-2018-11-886457.
FROM BLOOD
Key clinical point:
Major finding: The complete response rate for the combination treatment was 37%.
Study details: A prospective, phase 1 trial of 27 patients with previously treated MCL.
Disclosures: The trial was sponsored by the National Cancer Institute. Funding was provided by the Sarah Cannon Fund at the HCA Foundation. The investigators reported financial relationships with Janssen, Gilead, AstraZeneca, Celgene, Karyopharm, and others.
Source: Martin P et al. Blood. 2019 Jan 28. doi: 10.1182/blood-2018-11-886457.
EC approves BV plus AVD for Hodgkin lymphoma
The to treat adults with previously untreated, CD30+, stage IV Hodgkin lymphoma (HL).
This is the fifth approved indication for BV (adults with CD30+ HL at increased risk of relapse or progression after autologous stem cell transplant (ASCT); relapsed or refractory, CD30+ HL after ASCT or at least two prior therapies when ASCT or multi-agent chemotherapy is not an option; relapsed or refractory systemic anaplastic large-cell lymphoma; and CD30+ cutaneous T-cell lymphoma after at least one prior systemic therapy.
The EC’s approval of BV plus AVD is supported by the phase 3 ECHELON-1 trial (N Engl J Med. 2018;378:331-44).
ECHELON-1 included 1,334 patients with advanced HL who received BV plus AVD (n = 664) or doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD, n = 670) as frontline treatment.
The study's primary endpoint was modified progression-free survival (PFS), which was defined as time to progression, death, or evidence of non-complete response after completion of frontline therapy followed by subsequent anticancer therapy.
According to an independent review committee, BV plus AVD provided a significant improvement in modified PFS. The 2-year modified PFS rate was 82% in the BV-AVD arm and 77% in the ABVD arm (hazard ratio = 0.77; P = .04).
There was no significant difference between the treatment arms in response rates or overall survival.
The overall incidence of adverse events (AEs) was 99% in the BV-AVD arm and 98% in the ABVD arm. The incidence of grade 3 or higher AEs was 83% and 66%, respectively. The incidence of serious AEs was 43% and 27%, respectively.
Neutropenia, febrile neutropenia, and peripheral neuropathy were more common with BV-AVD, while pulmonary toxicity was more common with ABVD.
The ECHELON-1 trial was sponsored by Millennium Pharmaceuticals (a Takeda company) in collaboration with Seattle Genetics.
The to treat adults with previously untreated, CD30+, stage IV Hodgkin lymphoma (HL).
This is the fifth approved indication for BV (adults with CD30+ HL at increased risk of relapse or progression after autologous stem cell transplant (ASCT); relapsed or refractory, CD30+ HL after ASCT or at least two prior therapies when ASCT or multi-agent chemotherapy is not an option; relapsed or refractory systemic anaplastic large-cell lymphoma; and CD30+ cutaneous T-cell lymphoma after at least one prior systemic therapy.
The EC’s approval of BV plus AVD is supported by the phase 3 ECHELON-1 trial (N Engl J Med. 2018;378:331-44).
ECHELON-1 included 1,334 patients with advanced HL who received BV plus AVD (n = 664) or doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD, n = 670) as frontline treatment.
The study's primary endpoint was modified progression-free survival (PFS), which was defined as time to progression, death, or evidence of non-complete response after completion of frontline therapy followed by subsequent anticancer therapy.
According to an independent review committee, BV plus AVD provided a significant improvement in modified PFS. The 2-year modified PFS rate was 82% in the BV-AVD arm and 77% in the ABVD arm (hazard ratio = 0.77; P = .04).
There was no significant difference between the treatment arms in response rates or overall survival.
The overall incidence of adverse events (AEs) was 99% in the BV-AVD arm and 98% in the ABVD arm. The incidence of grade 3 or higher AEs was 83% and 66%, respectively. The incidence of serious AEs was 43% and 27%, respectively.
Neutropenia, febrile neutropenia, and peripheral neuropathy were more common with BV-AVD, while pulmonary toxicity was more common with ABVD.
The ECHELON-1 trial was sponsored by Millennium Pharmaceuticals (a Takeda company) in collaboration with Seattle Genetics.
The to treat adults with previously untreated, CD30+, stage IV Hodgkin lymphoma (HL).
This is the fifth approved indication for BV (adults with CD30+ HL at increased risk of relapse or progression after autologous stem cell transplant (ASCT); relapsed or refractory, CD30+ HL after ASCT or at least two prior therapies when ASCT or multi-agent chemotherapy is not an option; relapsed or refractory systemic anaplastic large-cell lymphoma; and CD30+ cutaneous T-cell lymphoma after at least one prior systemic therapy.
The EC’s approval of BV plus AVD is supported by the phase 3 ECHELON-1 trial (N Engl J Med. 2018;378:331-44).
ECHELON-1 included 1,334 patients with advanced HL who received BV plus AVD (n = 664) or doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD, n = 670) as frontline treatment.
The study's primary endpoint was modified progression-free survival (PFS), which was defined as time to progression, death, or evidence of non-complete response after completion of frontline therapy followed by subsequent anticancer therapy.
According to an independent review committee, BV plus AVD provided a significant improvement in modified PFS. The 2-year modified PFS rate was 82% in the BV-AVD arm and 77% in the ABVD arm (hazard ratio = 0.77; P = .04).
There was no significant difference between the treatment arms in response rates or overall survival.
The overall incidence of adverse events (AEs) was 99% in the BV-AVD arm and 98% in the ABVD arm. The incidence of grade 3 or higher AEs was 83% and 66%, respectively. The incidence of serious AEs was 43% and 27%, respectively.
Neutropenia, febrile neutropenia, and peripheral neuropathy were more common with BV-AVD, while pulmonary toxicity was more common with ABVD.
The ECHELON-1 trial was sponsored by Millennium Pharmaceuticals (a Takeda company) in collaboration with Seattle Genetics.
Fitusiran is reversible during dosing suspension
PRAGUE – Fitusiran, a novel small interfering RNA therapeutic that decreases antithrombin (AT) synthesis and bleeding in patients with hemophilia A or B, with or without inhibitors, is reversible upon dosing cessation and becomes effective again with resumption of dosing. That finding is based on data obtained before, during, and after a phase 2 study dosing suspension.

In September 2017, the fitusiran open-label extension study was suspended to investigate a fatal thrombotic event. The suspension was lifted 3 months later, and the trial continued with reduced doses of replacement factors or bypassing agents for breakthrough bleeds.
The pause in the study was used to gather data about the effects of dosing cessation and resumption, which were reported by coauthor Craig Benson, MD, of Sanofi Genzyme in Cambridge, Mass. Dr. Benson presented findings at the annual congress of the European Association for Haemophilia and Allied Disorders on behalf of lead author John Pasi, MD, PhD, of the Haemophilia Centre at the Royal London Hospital and colleagues.
Prior to the suspension, 28 patients with hemophilia A or B, with or without inhibitors, were given 50-80 mg of fitusiran for up to 20 months with a median dose duration of 11 months. During dosing suspension, the investigators measured AT levels, thrombin generation, and annualized bleeding rates (ABR).
“We can see that the antithrombin knockdown effect of fitusiran is reversible with dosing hold,” Dr. Benson said, referring to an upward trend of median AT level.
Within 4 months of stopping fitusiran, median AT level was 60% of normal. This level continued to rise to about 90% by month 7, before returning to normal at month 11.
“This is an AT recovery we had previously seen by a few subjects that had discontinued [fitusiran] prior to the clinical hold,” Dr. Benson said, “but here, with a much larger sample size, we see a fairly consistent AT recovery.”
As expected, while AT levels rose, thrombin generation showed an inverse trend. “We see a similar temporal pattern,” Dr. Benson said. “The bulk of thrombin generation decrease is seen by the 4th month off fitusiran.”
When patients restarted fitusiran after the suspension was lifted, AT levels dropped to about 30% of normal by month 1 and about 20% of normal from month 2 onward. Again, in an inverse manner, thrombin generation increased.
Clinically, changes in AT and thrombin generation before, during, and after study suspension were reflected in median ABR, which rose from 1.43 events per year prior to cessation to 6.47 events per year during treatment hold before falling to 1.25 events per year after restarting fitusiran.
Overall, the results support the efficacy and reversibility of fitusiran. The agent is continuing to be studied in the phase 3 ATLAS trials.
The study was funded by Sanofi Genzyme and Alnylam. Dr. Benson is an employee of Sanofi Genzyme. Other investigators reported financial ties to Sanofi Genzyme, Alnylam, Baxalta, Octapharma, Pfizer, Shire, and others.
SOURCE: Pasi J et al. EAHAD 2019, Abstract OR16.
PRAGUE – Fitusiran, a novel small interfering RNA therapeutic that decreases antithrombin (AT) synthesis and bleeding in patients with hemophilia A or B, with or without inhibitors, is reversible upon dosing cessation and becomes effective again with resumption of dosing. That finding is based on data obtained before, during, and after a phase 2 study dosing suspension.
In September 2017, the fitusiran open-label extension study was suspended to investigate a fatal thrombotic event. The suspension was lifted 3 months later, and the trial continued with reduced doses of replacement factors or bypassing agents for breakthrough bleeds.
The pause in the study was used to gather data about the effects of dosing cessation and resumption, which were reported by coauthor Craig Benson, MD, of Sanofi Genzyme in Cambridge, Mass. Dr. Benson presented findings at the annual congress of the European Association for Haemophilia and Allied Disorders on behalf of lead author John Pasi, MD, PhD, of the Haemophilia Centre at the Royal London Hospital and colleagues.
Prior to the suspension, 28 patients with hemophilia A or B, with or without inhibitors, were given 50-80 mg of fitusiran for up to 20 months with a median dose duration of 11 months. During dosing suspension, the investigators measured AT levels, thrombin generation, and annualized bleeding rates (ABR).
“We can see that the antithrombin knockdown effect of fitusiran is reversible with dosing hold,” Dr. Benson said, referring to an upward trend of median AT level.
Within 4 months of stopping fitusiran, median AT level was 60% of normal. This level continued to rise to about 90% by month 7, before returning to normal at month 11.
“This is an AT recovery we had previously seen by a few subjects that had discontinued [fitusiran] prior to the clinical hold,” Dr. Benson said, “but here, with a much larger sample size, we see a fairly consistent AT recovery.”
As expected, while AT levels rose, thrombin generation showed an inverse trend. “We see a similar temporal pattern,” Dr. Benson said. “The bulk of thrombin generation decrease is seen by the 4th month off fitusiran.”
When patients restarted fitusiran after the suspension was lifted, AT levels dropped to about 30% of normal by month 1 and about 20% of normal from month 2 onward. Again, in an inverse manner, thrombin generation increased.
Clinically, changes in AT and thrombin generation before, during, and after study suspension were reflected in median ABR, which rose from 1.43 events per year prior to cessation to 6.47 events per year during treatment hold before falling to 1.25 events per year after restarting fitusiran.
Overall, the results support the efficacy and reversibility of fitusiran. The agent is continuing to be studied in the phase 3 ATLAS trials.
The study was funded by Sanofi Genzyme and Alnylam. Dr. Benson is an employee of Sanofi Genzyme. Other investigators reported financial ties to Sanofi Genzyme, Alnylam, Baxalta, Octapharma, Pfizer, Shire, and others.
SOURCE: Pasi J et al. EAHAD 2019, Abstract OR16.
PRAGUE – Fitusiran, a novel small interfering RNA therapeutic that decreases antithrombin (AT) synthesis and bleeding in patients with hemophilia A or B, with or without inhibitors, is reversible upon dosing cessation and becomes effective again with resumption of dosing. That finding is based on data obtained before, during, and after a phase 2 study dosing suspension.
In September 2017, the fitusiran open-label extension study was suspended to investigate a fatal thrombotic event. The suspension was lifted 3 months later, and the trial continued with reduced doses of replacement factors or bypassing agents for breakthrough bleeds.
The pause in the study was used to gather data about the effects of dosing cessation and resumption, which were reported by coauthor Craig Benson, MD, of Sanofi Genzyme in Cambridge, Mass. Dr. Benson presented findings at the annual congress of the European Association for Haemophilia and Allied Disorders on behalf of lead author John Pasi, MD, PhD, of the Haemophilia Centre at the Royal London Hospital and colleagues.
Prior to the suspension, 28 patients with hemophilia A or B, with or without inhibitors, were given 50-80 mg of fitusiran for up to 20 months with a median dose duration of 11 months. During dosing suspension, the investigators measured AT levels, thrombin generation, and annualized bleeding rates (ABR).
“We can see that the antithrombin knockdown effect of fitusiran is reversible with dosing hold,” Dr. Benson said, referring to an upward trend of median AT level.
Within 4 months of stopping fitusiran, median AT level was 60% of normal. This level continued to rise to about 90% by month 7, before returning to normal at month 11.
“This is an AT recovery we had previously seen by a few subjects that had discontinued [fitusiran] prior to the clinical hold,” Dr. Benson said, “but here, with a much larger sample size, we see a fairly consistent AT recovery.”
As expected, while AT levels rose, thrombin generation showed an inverse trend. “We see a similar temporal pattern,” Dr. Benson said. “The bulk of thrombin generation decrease is seen by the 4th month off fitusiran.”
When patients restarted fitusiran after the suspension was lifted, AT levels dropped to about 30% of normal by month 1 and about 20% of normal from month 2 onward. Again, in an inverse manner, thrombin generation increased.
Clinically, changes in AT and thrombin generation before, during, and after study suspension were reflected in median ABR, which rose from 1.43 events per year prior to cessation to 6.47 events per year during treatment hold before falling to 1.25 events per year after restarting fitusiran.
Overall, the results support the efficacy and reversibility of fitusiran. The agent is continuing to be studied in the phase 3 ATLAS trials.
The study was funded by Sanofi Genzyme and Alnylam. Dr. Benson is an employee of Sanofi Genzyme. Other investigators reported financial ties to Sanofi Genzyme, Alnylam, Baxalta, Octapharma, Pfizer, Shire, and others.
SOURCE: Pasi J et al. EAHAD 2019, Abstract OR16.
REPORTING FROM EAHAD 2019
Key clinical point: Major finding: Median antithrombin returned to levels greater than 60% of normal 4 months after stopping fitusiran.
Study details: A phase 2, open-label study involving 28 patients with hemophilia A or B.
Disclosures: The study was funded by Sanofi Genzyme and Alnylam. Dr. Benson is an employee of Sanofi Genzyme. Other investigators reported financial affiliations with Sanofi Genzyme, Alnylam, Baxalta, Octapharma, Pfizer, Shire, and others.
Source: Pasi J et al. EAHAD 2019, Abstract OR16.