User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
div[contains(@class, 'main-prefix')]
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
Children and COVID: New cases, vaccinations both decline
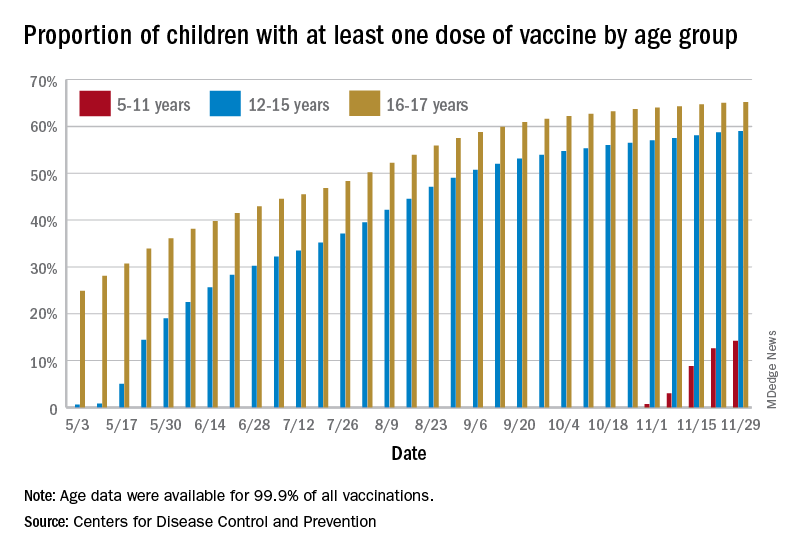
States reported 131,828 new pediatric cases for the week of Nov. 19-25, a decline of 7.1% over the previous week but still enough to surpass 100,000 for the 16th consecutive week. The weekly count had risen for 3 straight weeks since the last decrease in late October, the American Academy of Pediatrics and the Children’s Hospital Association said Nov. 30 in their weekly COVID report.
The AAP/CHA analysis, based on data from state and territorial health departments, puts the total number of cases in children at 6.9 million since the pandemic began, representing 17.0% of cases in Americans of all ages. The Centers for Disease Control and Prevention, which uses an age limit of 18 years to define a child, unlike some states, reports numbers of 6.1 million and 15.5%.
New vaccinations among the youngest eligible children, those aged 5-11 years, were down for the second week in a row after reaching almost 1.7 million during the first full week after approval on Nov. 2. Since then, the vaccination counts have been 1.2 million (Nov. 16-22) and 333,000 (Nov. 23-29), the CDC said on its COVID Data Tracker. A similar drop in the last week – from 127,000 to just 50,000 – also was seen for those aged 12-17 years.
Altogether, 14.2% of children aged 5-11, almost 4.1 million individuals, have received at least one dose of the vaccine, compared with 59.0% (10 million) of the 12- to 15-year-olds and 65.2% (5.5 million) of those aged 16-17. Just under 1% of the youngest group has been fully vaccinated, versus 49.0% and 55.8% for the older children, the CDC said.
It has been reported that Pfizer and BioNTech, which produce the only COVID vaccine approved for children, are planning to apply to the Food and Drug Administration during the first week of December for authorization for a booster dose for 16- and 17-year-olds.
States reported 131,828 new pediatric cases for the week of Nov. 19-25, a decline of 7.1% over the previous week but still enough to surpass 100,000 for the 16th consecutive week. The weekly count had risen for 3 straight weeks since the last decrease in late October, the American Academy of Pediatrics and the Children’s Hospital Association said Nov. 30 in their weekly COVID report.
The AAP/CHA analysis, based on data from state and territorial health departments, puts the total number of cases in children at 6.9 million since the pandemic began, representing 17.0% of cases in Americans of all ages. The Centers for Disease Control and Prevention, which uses an age limit of 18 years to define a child, unlike some states, reports numbers of 6.1 million and 15.5%.
New vaccinations among the youngest eligible children, those aged 5-11 years, were down for the second week in a row after reaching almost 1.7 million during the first full week after approval on Nov. 2. Since then, the vaccination counts have been 1.2 million (Nov. 16-22) and 333,000 (Nov. 23-29), the CDC said on its COVID Data Tracker. A similar drop in the last week – from 127,000 to just 50,000 – also was seen for those aged 12-17 years.
Altogether, 14.2% of children aged 5-11, almost 4.1 million individuals, have received at least one dose of the vaccine, compared with 59.0% (10 million) of the 12- to 15-year-olds and 65.2% (5.5 million) of those aged 16-17. Just under 1% of the youngest group has been fully vaccinated, versus 49.0% and 55.8% for the older children, the CDC said.
It has been reported that Pfizer and BioNTech, which produce the only COVID vaccine approved for children, are planning to apply to the Food and Drug Administration during the first week of December for authorization for a booster dose for 16- and 17-year-olds.
States reported 131,828 new pediatric cases for the week of Nov. 19-25, a decline of 7.1% over the previous week but still enough to surpass 100,000 for the 16th consecutive week. The weekly count had risen for 3 straight weeks since the last decrease in late October, the American Academy of Pediatrics and the Children’s Hospital Association said Nov. 30 in their weekly COVID report.
The AAP/CHA analysis, based on data from state and territorial health departments, puts the total number of cases in children at 6.9 million since the pandemic began, representing 17.0% of cases in Americans of all ages. The Centers for Disease Control and Prevention, which uses an age limit of 18 years to define a child, unlike some states, reports numbers of 6.1 million and 15.5%.
New vaccinations among the youngest eligible children, those aged 5-11 years, were down for the second week in a row after reaching almost 1.7 million during the first full week after approval on Nov. 2. Since then, the vaccination counts have been 1.2 million (Nov. 16-22) and 333,000 (Nov. 23-29), the CDC said on its COVID Data Tracker. A similar drop in the last week – from 127,000 to just 50,000 – also was seen for those aged 12-17 years.
Altogether, 14.2% of children aged 5-11, almost 4.1 million individuals, have received at least one dose of the vaccine, compared with 59.0% (10 million) of the 12- to 15-year-olds and 65.2% (5.5 million) of those aged 16-17. Just under 1% of the youngest group has been fully vaccinated, versus 49.0% and 55.8% for the older children, the CDC said.
It has been reported that Pfizer and BioNTech, which produce the only COVID vaccine approved for children, are planning to apply to the Food and Drug Administration during the first week of December for authorization for a booster dose for 16- and 17-year-olds.
Fauci: Omicron ‘very different from other variants’
The newly detected Omicron COVID-19 variant may be highly infectious and less responsive to available vaccines than other variants, but it is too early to know how it compares to the Delta variant, top infectious disease official Anthony S. Fauci, MD, said Nov. 30.
Dr. Fauci, speaking at a White House COVID-19 briefing, said there’s a “very unusual constellation of changes” across the COVID-19 genome that indicates it is unlike any variant we have seen so far.
“This mutational profile is very different from other variants of interest and concern, and although some mutations are also found in Delta, this is not Delta,” Dr. Fauci said. “These mutations have been associated with increased transmissibility and immune evasion.”
Omicron is the fifth designated COVID-19 variant of concern.
Detected first in South Africa, Omicron has been found in 20 countries so far. There are no known cases yet in the United States, but it has been detected in Canada.
Omicron has more than 30 mutations to the spike protein, the part of the virus that binds to human cells, Dr. Fauci said.
Cross-protection from boosters
Though the mutations suggest there is increased transmission of this variant, he said it is too soon to know how this compares to the Delta variant. And although the vaccines may not be as effective against Omicron, Dr. Fauci said there will likely be some protection.
“Remember, as with other variants, although partial immune escape may occur, vaccines, particularly boosters, give a level of antibodies that even with variants like Delta give you a degree of cross-protection, particularly against severe disease,” he said.
“When we say that although these mutations suggest a diminution of protection and a degree of immune evasion, we still, from experience with Delta, can make a reasonable conclusion that you would not eliminate all protection against this particular variant,” Dr. Fauci said.
So far, there is no reason to believe Omicron will cause more severe illness than other variants of concern.
“Although some preliminary information from South Africa suggests no unusual symptoms associated with variant, we do not know, and it is too early to tell,” Dr. Fauci said.
He recommended that people continue to wear masks, wash hands, and avoid crowded indoor venues. Most importantly, he recommended that everyone get their vaccines and boosters.
“One thing has become clear over the last 20 months: We can’t predict the future, but we can be prepared for it,” CDC Director Rochelle P. Walensky, MD, said at the briefing. “We have far more tools to fight the variant today than we did at this time last year.”
A version of this story first appeared on Medscape.com.
The newly detected Omicron COVID-19 variant may be highly infectious and less responsive to available vaccines than other variants, but it is too early to know how it compares to the Delta variant, top infectious disease official Anthony S. Fauci, MD, said Nov. 30.
Dr. Fauci, speaking at a White House COVID-19 briefing, said there’s a “very unusual constellation of changes” across the COVID-19 genome that indicates it is unlike any variant we have seen so far.
“This mutational profile is very different from other variants of interest and concern, and although some mutations are also found in Delta, this is not Delta,” Dr. Fauci said. “These mutations have been associated with increased transmissibility and immune evasion.”
Omicron is the fifth designated COVID-19 variant of concern.
Detected first in South Africa, Omicron has been found in 20 countries so far. There are no known cases yet in the United States, but it has been detected in Canada.
Omicron has more than 30 mutations to the spike protein, the part of the virus that binds to human cells, Dr. Fauci said.
Cross-protection from boosters
Though the mutations suggest there is increased transmission of this variant, he said it is too soon to know how this compares to the Delta variant. And although the vaccines may not be as effective against Omicron, Dr. Fauci said there will likely be some protection.
“Remember, as with other variants, although partial immune escape may occur, vaccines, particularly boosters, give a level of antibodies that even with variants like Delta give you a degree of cross-protection, particularly against severe disease,” he said.
“When we say that although these mutations suggest a diminution of protection and a degree of immune evasion, we still, from experience with Delta, can make a reasonable conclusion that you would not eliminate all protection against this particular variant,” Dr. Fauci said.
So far, there is no reason to believe Omicron will cause more severe illness than other variants of concern.
“Although some preliminary information from South Africa suggests no unusual symptoms associated with variant, we do not know, and it is too early to tell,” Dr. Fauci said.
He recommended that people continue to wear masks, wash hands, and avoid crowded indoor venues. Most importantly, he recommended that everyone get their vaccines and boosters.
“One thing has become clear over the last 20 months: We can’t predict the future, but we can be prepared for it,” CDC Director Rochelle P. Walensky, MD, said at the briefing. “We have far more tools to fight the variant today than we did at this time last year.”
A version of this story first appeared on Medscape.com.
The newly detected Omicron COVID-19 variant may be highly infectious and less responsive to available vaccines than other variants, but it is too early to know how it compares to the Delta variant, top infectious disease official Anthony S. Fauci, MD, said Nov. 30.
Dr. Fauci, speaking at a White House COVID-19 briefing, said there’s a “very unusual constellation of changes” across the COVID-19 genome that indicates it is unlike any variant we have seen so far.
“This mutational profile is very different from other variants of interest and concern, and although some mutations are also found in Delta, this is not Delta,” Dr. Fauci said. “These mutations have been associated with increased transmissibility and immune evasion.”
Omicron is the fifth designated COVID-19 variant of concern.
Detected first in South Africa, Omicron has been found in 20 countries so far. There are no known cases yet in the United States, but it has been detected in Canada.
Omicron has more than 30 mutations to the spike protein, the part of the virus that binds to human cells, Dr. Fauci said.
Cross-protection from boosters
Though the mutations suggest there is increased transmission of this variant, he said it is too soon to know how this compares to the Delta variant. And although the vaccines may not be as effective against Omicron, Dr. Fauci said there will likely be some protection.
“Remember, as with other variants, although partial immune escape may occur, vaccines, particularly boosters, give a level of antibodies that even with variants like Delta give you a degree of cross-protection, particularly against severe disease,” he said.
“When we say that although these mutations suggest a diminution of protection and a degree of immune evasion, we still, from experience with Delta, can make a reasonable conclusion that you would not eliminate all protection against this particular variant,” Dr. Fauci said.
So far, there is no reason to believe Omicron will cause more severe illness than other variants of concern.
“Although some preliminary information from South Africa suggests no unusual symptoms associated with variant, we do not know, and it is too early to tell,” Dr. Fauci said.
He recommended that people continue to wear masks, wash hands, and avoid crowded indoor venues. Most importantly, he recommended that everyone get their vaccines and boosters.
“One thing has become clear over the last 20 months: We can’t predict the future, but we can be prepared for it,” CDC Director Rochelle P. Walensky, MD, said at the briefing. “We have far more tools to fight the variant today than we did at this time last year.”
A version of this story first appeared on Medscape.com.
FDA panel backs first pill for COVID-19 by a small margin
, according to a panel of experts that advises the Food and Drug Administration on its regulatory decisions for these types of drugs.

The FDA’s Antimicrobial Drugs Advisory Committee narrowly voted to authorize the drug molnupiravir, voting 13 to 10 to support emergency use, which requires a medication to meet a lower standard of evidence than does full approval.
The FDA is not bound by the committee’s vote but typically follows its advice.
If authorized by the agency, molnupiravir would be the first antiviral agent available as a pill to treat COVID-19. Other therapies to treat the infection are available — monoclonal antibodies and the drug remdesivir — but they are given by infusion.
The United Kingdom has already authorized the use of Merck’s drug.
“This was clearly a difficult decision,” said committee member Michael Green, MD, a pediatric infectious disease expert at the University of Pittsburg School of Medicine.
Green said he voted yes, and that the drug’s ability to prevent deaths in the study weighed heavily on his decision. He said given uncertainties around the drug both the company and FDA should keep a close eye on patients taking the drug going forward.
“Should an alternative oral agent become available that had a better safety profile and equal or better efficacy profile, the agency might reconsider its authorization,” he said.
Others didn’t agree that the drug should be allowed onto the market.
“I voted no,” said Jennifer Le, PharmD, a professor of clinical pharmacy at the University of California. Dr. Le said the modest benefit of the medication didn’t outweigh all the potential safety issues. “I think I just need more efficacy and safety data,” she said.
Initial results from the first half of people enrolled in the clinical trial found the pill cut the risk of hospitalization or death by 50% in patients at higher risk of severe outcomes from COVID-19.
But later results, released just days before the meeting, showed that the drug’s effectiveness had dropped to about 30%.
In the updated analysis, 48 patients out of the 709 who were taking the drug were hospitalized or died within 29 days compared to 68 out of 699 who randomly got the placebo. There was one death in the group that got molnupiravir compared to nine in the placebo group. Nearly all those deaths occurred during the first phase of the study.
On Nov. 30 Merck explained that the drug’s efficacy appeared to fall, in part, because the placebo group had experienced fewer hospitalizations and deaths than expected during the second half of the study, making the drug look less beneficial by comparison.
The company said it wasn’t sure why patients in the placebo group had fared so much better in later trial enrollments.
“The efficacy of this product is not overwhelmingly good,” said committee member David Hardy, MD, an infectious disease expert at Charles Drew University School of Medicine in Los Angeles. “And I think that makes all of us a little uncomfortable about whether this is an advanced therapeutic because it’s an oral medication rather than an intravenous medication,” he said during the panel’s deliberations.
“I think we have to be very careful about how we’re going to allow people to use this,” Dr. Hardy said.
Many who voted for authorization thought use of the drug should be restricted to unvaccinated people who were at high risk of severe COVID-19 outcomes, the same population enrolled in the clinical trial. People in the trial were considered at higher risk if they were over age 60, had cancer, chronic kidney disease, chronic obstructive pulmonary disease, were obese, or had heart disease or diabetes.
There are some significant limitations of the study that may affect how the drug is used. Vaccinated people couldn’t enroll in the study, so it’s not known if the medication would have any benefit for them. Nearly two-thirds of the U.S. population is fully vaccinated. The study found no additional benefit of the medication compared to the placebo in people who had detectable antibodies, presumably from a prior infection.
Animal studies found that the drug — which kills the virus by forcing it to make errors as it copies its genetic material inside cells — could disrupt bone formation. For that reason, the manufacturer and the FDA agreed that it should not be used in anyone younger than age 18.
Animal studies also indicated that the drug could cause birth defects. For that reason, the company said the drug shouldn’t be given to women who are pregnant or breastfeeding and said doctors should make sure women of childbearing age aren’t pregnant before taking the medication.
Some members of the panel felt that pregnant women and their doctors should be given the choice of whether or not to use the drug, given that pregnant women are at high risk for severe COVID-19 outcomes and infused therapies may not be available in all settings.
Other members of the committee said they were uncomfortable authorizing the drug given its potential to mutate the virus.
The drug, which forces the virus to mutate as it copies its RNA, eventually causes the virus to make so many errors in its genetic material that it can no longer make more of itself and the immune system clears it out of the body.
But it takes a few days to work — the drug is designed to be taken for 5 consecutive days -- and studies of the viral loads of patients taking the drug show that through the first 2 days, viral loads remain detectable as these mutations occur.
Studies by the FDA show some of those mutations in the spike protein are the same ones that have helped the virus become more transmissible and escape the protection of vaccines.
So the question is whether someone taking the medication could develop a dangerous mutation and then infect someone else, sparking the spread of a new variant.
Nicholas Kartsonis, MD, a vice president at Merck, said that the company was still analyzing data.
“Even if the probability is very low — 1 in 10,000 or 1 in 100,000 -- that this drug would induce an escape mutant for which the vaccines we have would not cover, that would be catastrophic for the whole world, actually,” said committee member James Hildreth, MD, an immunologist and president of Meharry Medical College, Nashville. “Do you have sufficient data on the likelihood of that happening?” he asked Dr. Kartsonis of Merck.
“So we don’t,” Dr. Kartsonis said.
He said, in theory, the risk of mutation with molnupiravir is the same as seen with the use of vaccines or monoclonal antibody therapies. Dr. Hildreth wasn’t satisfied with that answer.
“With all respect, the mechanism of your drug is to drive [genetic mutations], so it’s not the same as the vaccine. It’s not the same as monoclonal antibodies,” he said.
Dr. Hildreth later said he didn’t feel comfortable voting for authorization given the uncertainties around escape mutants. He voted no.
“It was an easy vote for me,” he said.
A version of this article first appeared on Medscape.com.
, according to a panel of experts that advises the Food and Drug Administration on its regulatory decisions for these types of drugs.
The FDA’s Antimicrobial Drugs Advisory Committee narrowly voted to authorize the drug molnupiravir, voting 13 to 10 to support emergency use, which requires a medication to meet a lower standard of evidence than does full approval.
The FDA is not bound by the committee’s vote but typically follows its advice.
If authorized by the agency, molnupiravir would be the first antiviral agent available as a pill to treat COVID-19. Other therapies to treat the infection are available — monoclonal antibodies and the drug remdesivir — but they are given by infusion.
The United Kingdom has already authorized the use of Merck’s drug.
“This was clearly a difficult decision,” said committee member Michael Green, MD, a pediatric infectious disease expert at the University of Pittsburg School of Medicine.
Green said he voted yes, and that the drug’s ability to prevent deaths in the study weighed heavily on his decision. He said given uncertainties around the drug both the company and FDA should keep a close eye on patients taking the drug going forward.
“Should an alternative oral agent become available that had a better safety profile and equal or better efficacy profile, the agency might reconsider its authorization,” he said.
Others didn’t agree that the drug should be allowed onto the market.
“I voted no,” said Jennifer Le, PharmD, a professor of clinical pharmacy at the University of California. Dr. Le said the modest benefit of the medication didn’t outweigh all the potential safety issues. “I think I just need more efficacy and safety data,” she said.
Initial results from the first half of people enrolled in the clinical trial found the pill cut the risk of hospitalization or death by 50% in patients at higher risk of severe outcomes from COVID-19.
But later results, released just days before the meeting, showed that the drug’s effectiveness had dropped to about 30%.
In the updated analysis, 48 patients out of the 709 who were taking the drug were hospitalized or died within 29 days compared to 68 out of 699 who randomly got the placebo. There was one death in the group that got molnupiravir compared to nine in the placebo group. Nearly all those deaths occurred during the first phase of the study.
On Nov. 30 Merck explained that the drug’s efficacy appeared to fall, in part, because the placebo group had experienced fewer hospitalizations and deaths than expected during the second half of the study, making the drug look less beneficial by comparison.
The company said it wasn’t sure why patients in the placebo group had fared so much better in later trial enrollments.
“The efficacy of this product is not overwhelmingly good,” said committee member David Hardy, MD, an infectious disease expert at Charles Drew University School of Medicine in Los Angeles. “And I think that makes all of us a little uncomfortable about whether this is an advanced therapeutic because it’s an oral medication rather than an intravenous medication,” he said during the panel’s deliberations.
“I think we have to be very careful about how we’re going to allow people to use this,” Dr. Hardy said.
Many who voted for authorization thought use of the drug should be restricted to unvaccinated people who were at high risk of severe COVID-19 outcomes, the same population enrolled in the clinical trial. People in the trial were considered at higher risk if they were over age 60, had cancer, chronic kidney disease, chronic obstructive pulmonary disease, were obese, or had heart disease or diabetes.
There are some significant limitations of the study that may affect how the drug is used. Vaccinated people couldn’t enroll in the study, so it’s not known if the medication would have any benefit for them. Nearly two-thirds of the U.S. population is fully vaccinated. The study found no additional benefit of the medication compared to the placebo in people who had detectable antibodies, presumably from a prior infection.
Animal studies found that the drug — which kills the virus by forcing it to make errors as it copies its genetic material inside cells — could disrupt bone formation. For that reason, the manufacturer and the FDA agreed that it should not be used in anyone younger than age 18.
Animal studies also indicated that the drug could cause birth defects. For that reason, the company said the drug shouldn’t be given to women who are pregnant or breastfeeding and said doctors should make sure women of childbearing age aren’t pregnant before taking the medication.
Some members of the panel felt that pregnant women and their doctors should be given the choice of whether or not to use the drug, given that pregnant women are at high risk for severe COVID-19 outcomes and infused therapies may not be available in all settings.
Other members of the committee said they were uncomfortable authorizing the drug given its potential to mutate the virus.
The drug, which forces the virus to mutate as it copies its RNA, eventually causes the virus to make so many errors in its genetic material that it can no longer make more of itself and the immune system clears it out of the body.
But it takes a few days to work — the drug is designed to be taken for 5 consecutive days -- and studies of the viral loads of patients taking the drug show that through the first 2 days, viral loads remain detectable as these mutations occur.
Studies by the FDA show some of those mutations in the spike protein are the same ones that have helped the virus become more transmissible and escape the protection of vaccines.
So the question is whether someone taking the medication could develop a dangerous mutation and then infect someone else, sparking the spread of a new variant.
Nicholas Kartsonis, MD, a vice president at Merck, said that the company was still analyzing data.
“Even if the probability is very low — 1 in 10,000 or 1 in 100,000 -- that this drug would induce an escape mutant for which the vaccines we have would not cover, that would be catastrophic for the whole world, actually,” said committee member James Hildreth, MD, an immunologist and president of Meharry Medical College, Nashville. “Do you have sufficient data on the likelihood of that happening?” he asked Dr. Kartsonis of Merck.
“So we don’t,” Dr. Kartsonis said.
He said, in theory, the risk of mutation with molnupiravir is the same as seen with the use of vaccines or monoclonal antibody therapies. Dr. Hildreth wasn’t satisfied with that answer.
“With all respect, the mechanism of your drug is to drive [genetic mutations], so it’s not the same as the vaccine. It’s not the same as monoclonal antibodies,” he said.
Dr. Hildreth later said he didn’t feel comfortable voting for authorization given the uncertainties around escape mutants. He voted no.
“It was an easy vote for me,” he said.
A version of this article first appeared on Medscape.com.
, according to a panel of experts that advises the Food and Drug Administration on its regulatory decisions for these types of drugs.
The FDA’s Antimicrobial Drugs Advisory Committee narrowly voted to authorize the drug molnupiravir, voting 13 to 10 to support emergency use, which requires a medication to meet a lower standard of evidence than does full approval.
The FDA is not bound by the committee’s vote but typically follows its advice.
If authorized by the agency, molnupiravir would be the first antiviral agent available as a pill to treat COVID-19. Other therapies to treat the infection are available — monoclonal antibodies and the drug remdesivir — but they are given by infusion.
The United Kingdom has already authorized the use of Merck’s drug.
“This was clearly a difficult decision,” said committee member Michael Green, MD, a pediatric infectious disease expert at the University of Pittsburg School of Medicine.
Green said he voted yes, and that the drug’s ability to prevent deaths in the study weighed heavily on his decision. He said given uncertainties around the drug both the company and FDA should keep a close eye on patients taking the drug going forward.
“Should an alternative oral agent become available that had a better safety profile and equal or better efficacy profile, the agency might reconsider its authorization,” he said.
Others didn’t agree that the drug should be allowed onto the market.
“I voted no,” said Jennifer Le, PharmD, a professor of clinical pharmacy at the University of California. Dr. Le said the modest benefit of the medication didn’t outweigh all the potential safety issues. “I think I just need more efficacy and safety data,” she said.
Initial results from the first half of people enrolled in the clinical trial found the pill cut the risk of hospitalization or death by 50% in patients at higher risk of severe outcomes from COVID-19.
But later results, released just days before the meeting, showed that the drug’s effectiveness had dropped to about 30%.
In the updated analysis, 48 patients out of the 709 who were taking the drug were hospitalized or died within 29 days compared to 68 out of 699 who randomly got the placebo. There was one death in the group that got molnupiravir compared to nine in the placebo group. Nearly all those deaths occurred during the first phase of the study.
On Nov. 30 Merck explained that the drug’s efficacy appeared to fall, in part, because the placebo group had experienced fewer hospitalizations and deaths than expected during the second half of the study, making the drug look less beneficial by comparison.
The company said it wasn’t sure why patients in the placebo group had fared so much better in later trial enrollments.
“The efficacy of this product is not overwhelmingly good,” said committee member David Hardy, MD, an infectious disease expert at Charles Drew University School of Medicine in Los Angeles. “And I think that makes all of us a little uncomfortable about whether this is an advanced therapeutic because it’s an oral medication rather than an intravenous medication,” he said during the panel’s deliberations.
“I think we have to be very careful about how we’re going to allow people to use this,” Dr. Hardy said.
Many who voted for authorization thought use of the drug should be restricted to unvaccinated people who were at high risk of severe COVID-19 outcomes, the same population enrolled in the clinical trial. People in the trial were considered at higher risk if they were over age 60, had cancer, chronic kidney disease, chronic obstructive pulmonary disease, were obese, or had heart disease or diabetes.
There are some significant limitations of the study that may affect how the drug is used. Vaccinated people couldn’t enroll in the study, so it’s not known if the medication would have any benefit for them. Nearly two-thirds of the U.S. population is fully vaccinated. The study found no additional benefit of the medication compared to the placebo in people who had detectable antibodies, presumably from a prior infection.
Animal studies found that the drug — which kills the virus by forcing it to make errors as it copies its genetic material inside cells — could disrupt bone formation. For that reason, the manufacturer and the FDA agreed that it should not be used in anyone younger than age 18.
Animal studies also indicated that the drug could cause birth defects. For that reason, the company said the drug shouldn’t be given to women who are pregnant or breastfeeding and said doctors should make sure women of childbearing age aren’t pregnant before taking the medication.
Some members of the panel felt that pregnant women and their doctors should be given the choice of whether or not to use the drug, given that pregnant women are at high risk for severe COVID-19 outcomes and infused therapies may not be available in all settings.
Other members of the committee said they were uncomfortable authorizing the drug given its potential to mutate the virus.
The drug, which forces the virus to mutate as it copies its RNA, eventually causes the virus to make so many errors in its genetic material that it can no longer make more of itself and the immune system clears it out of the body.
But it takes a few days to work — the drug is designed to be taken for 5 consecutive days -- and studies of the viral loads of patients taking the drug show that through the first 2 days, viral loads remain detectable as these mutations occur.
Studies by the FDA show some of those mutations in the spike protein are the same ones that have helped the virus become more transmissible and escape the protection of vaccines.
So the question is whether someone taking the medication could develop a dangerous mutation and then infect someone else, sparking the spread of a new variant.
Nicholas Kartsonis, MD, a vice president at Merck, said that the company was still analyzing data.
“Even if the probability is very low — 1 in 10,000 or 1 in 100,000 -- that this drug would induce an escape mutant for which the vaccines we have would not cover, that would be catastrophic for the whole world, actually,” said committee member James Hildreth, MD, an immunologist and president of Meharry Medical College, Nashville. “Do you have sufficient data on the likelihood of that happening?” he asked Dr. Kartsonis of Merck.
“So we don’t,” Dr. Kartsonis said.
He said, in theory, the risk of mutation with molnupiravir is the same as seen with the use of vaccines or monoclonal antibody therapies. Dr. Hildreth wasn’t satisfied with that answer.
“With all respect, the mechanism of your drug is to drive [genetic mutations], so it’s not the same as the vaccine. It’s not the same as monoclonal antibodies,” he said.
Dr. Hildreth later said he didn’t feel comfortable voting for authorization given the uncertainties around escape mutants. He voted no.
“It was an easy vote for me,” he said.
A version of this article first appeared on Medscape.com.
Geospatial maps show areas of Africa that need HIV services
Can geospatial mapping fill in the gaps in areas lagging behind in global efforts to end the HIV epidemic?
That’s what Diego Cuadros, PhD, assistant professor of health geography and disease modeling at the University of Cincinnati, set out to learn using geospatial data combined with prevalence data to identify the most underserved areas in Sub-Saharan Africa (SSA) for HIV services.
Study findings, which were published Nov. 24 in PLOS Global Public Health, highlight that as many as 1.5 million people living with HIV (PLHIV) in SSA have more than an hour’s motorized travel time both ways to access care, while roughly 3 million must set aside, at minimum, 30 minutes. When the only mode of transportation is walking, as much as 95.3% of underserved areas are faced with at least 30 minutes travel time.
This is simply the tip of the overall problem, Dr. Cuadros told this news organization.
“We are able to estimate how many people [whose] quality of life is being affected by HIV because they are not on treatment and most probably, HIV incidence is high in those areas. But [it’s not as simple as just] increasing the number of health care facilities,” he said. “We need to find strategies to be able to cover this population.”
Dr. Cuadros also noted that the problem goes both ways. “It’s hard for them to move, and it’s [also] hard to reach them,” he explained.
Mapping care, or lack thereof
Dr. Cuadros and team used two primary sources of data to generate high-resolution maps of underserved SSA areas: estimated number of PLHIV between the ages of 15 and 49 years in 47 SSA countries paired with population density and global map of travel time to the nearest health facility by motorized and nonmotorized (that is, walking) transportation. Combining these data allowed them to then detail the distance from access to care for every 5 km².
The mapping exercise showed that 90.5% of the total territory, in which about 7 million PLHIV resided, had more than 10 minutes motorized travel time to the nearest health care facility, while 74.6% were within 30 minutes, and 58.9% were within 60 minutes. Increases in threshold travel times (from 10 to 60 minutes) corresponded directly to declines in the average proportion of underserved areas (from 80.9% to 42.6%). However, in certain countries like Sudan and Mauritania, 99.4% of the areas were underserved at the 10 minute threshold, while more than 90% were underserved at the 60 minute threshold.
Corresponding rates for nonmotorized access to health services were similar: 88.7% (~17.6 million) PLHIV had 10 minutes walking time to health care services, while 57.8% (~11.5 million) had at least 30 minutes, and 33.0% (~6.6 million), at least 60 minutes. Likewise, as threshold times increased from 10 to 60 minutes, the percentage of affected PLHIV declined (to roughly 50% in two-thirds of the countries). But more than 70% of PLHIV resided in underserved areas in countries like Equatorial Guinea, Eritrea, South Sudan, and Sudan.
Geographical allocation of health service facilities underscores treatment gaps
“We think that most PLHIV live in urban areas or close to urban areas, and most of the health care facilities in Africa are concentrated in those areas. But [roughly 8 million people with HIV] are living in rural areas, and for most, movement is very difficult,” explained Dr. Cuadros, meaning that the majority are not on treatment despite the high incidence of HIV.
Inarguably, the pandemic has interrupted HIV services and treatment substantially on the African continent, further challenging any efforts to translate these study findings into actionable strategies.
“We’ve known for quite a while that distance and travel times and travel expenses are known risks for nonadherence, for lack of access to diagnostics, for people at risk for exposure,” Chris Beyrer, MD, MPH, Desmond M. Tutu Professor of Public Health and Human Rights at the Johns Hopkins Bloomberg School of Public Health, Baltimore, said in an interview. (Dr. Beyrer was not involved in the study.)
“What’s new is the ability to really look at this across geographies and really home in on how many people face very long times and distances for travel. That’s a really important contribution,” he said.
Dr. Cuadros pointed out that these hard-to-reach populations are key to achieving the UNAID’s HIV elimination targets. “We’re going to have these pockets of transmission that are going to be really important for epidemic control,” he explained.
Toward that end, the onus appears to extend well beyond solutions that emphasize difficulty in reaching people from the provider perspective. “There’s quite a lot of what you might want to think of as blaming the victim for when people miss appointments, don’t appear to be adherent, [or] can’t stay reliably suppressed,” said Dr. Beyrer.
“It’s really important for providers in general to include in history and intake how far people have come, what their challenges are with travel, to really pay attention to those issues. Having this elegant analysis, this level of detail, is an important first step,” he added.
Dr. Cuadros has disclosed no relevant financial relationships. Dr. Beyrer reports a consulting agreement with Merck.
A version of this article first appeared on Medscape.com.
Can geospatial mapping fill in the gaps in areas lagging behind in global efforts to end the HIV epidemic?
That’s what Diego Cuadros, PhD, assistant professor of health geography and disease modeling at the University of Cincinnati, set out to learn using geospatial data combined with prevalence data to identify the most underserved areas in Sub-Saharan Africa (SSA) for HIV services.
Study findings, which were published Nov. 24 in PLOS Global Public Health, highlight that as many as 1.5 million people living with HIV (PLHIV) in SSA have more than an hour’s motorized travel time both ways to access care, while roughly 3 million must set aside, at minimum, 30 minutes. When the only mode of transportation is walking, as much as 95.3% of underserved areas are faced with at least 30 minutes travel time.
This is simply the tip of the overall problem, Dr. Cuadros told this news organization.
“We are able to estimate how many people [whose] quality of life is being affected by HIV because they are not on treatment and most probably, HIV incidence is high in those areas. But [it’s not as simple as just] increasing the number of health care facilities,” he said. “We need to find strategies to be able to cover this population.”
Dr. Cuadros also noted that the problem goes both ways. “It’s hard for them to move, and it’s [also] hard to reach them,” he explained.
Mapping care, or lack thereof
Dr. Cuadros and team used two primary sources of data to generate high-resolution maps of underserved SSA areas: estimated number of PLHIV between the ages of 15 and 49 years in 47 SSA countries paired with population density and global map of travel time to the nearest health facility by motorized and nonmotorized (that is, walking) transportation. Combining these data allowed them to then detail the distance from access to care for every 5 km².
The mapping exercise showed that 90.5% of the total territory, in which about 7 million PLHIV resided, had more than 10 minutes motorized travel time to the nearest health care facility, while 74.6% were within 30 minutes, and 58.9% were within 60 minutes. Increases in threshold travel times (from 10 to 60 minutes) corresponded directly to declines in the average proportion of underserved areas (from 80.9% to 42.6%). However, in certain countries like Sudan and Mauritania, 99.4% of the areas were underserved at the 10 minute threshold, while more than 90% were underserved at the 60 minute threshold.
Corresponding rates for nonmotorized access to health services were similar: 88.7% (~17.6 million) PLHIV had 10 minutes walking time to health care services, while 57.8% (~11.5 million) had at least 30 minutes, and 33.0% (~6.6 million), at least 60 minutes. Likewise, as threshold times increased from 10 to 60 minutes, the percentage of affected PLHIV declined (to roughly 50% in two-thirds of the countries). But more than 70% of PLHIV resided in underserved areas in countries like Equatorial Guinea, Eritrea, South Sudan, and Sudan.
Geographical allocation of health service facilities underscores treatment gaps
“We think that most PLHIV live in urban areas or close to urban areas, and most of the health care facilities in Africa are concentrated in those areas. But [roughly 8 million people with HIV] are living in rural areas, and for most, movement is very difficult,” explained Dr. Cuadros, meaning that the majority are not on treatment despite the high incidence of HIV.
Inarguably, the pandemic has interrupted HIV services and treatment substantially on the African continent, further challenging any efforts to translate these study findings into actionable strategies.
“We’ve known for quite a while that distance and travel times and travel expenses are known risks for nonadherence, for lack of access to diagnostics, for people at risk for exposure,” Chris Beyrer, MD, MPH, Desmond M. Tutu Professor of Public Health and Human Rights at the Johns Hopkins Bloomberg School of Public Health, Baltimore, said in an interview. (Dr. Beyrer was not involved in the study.)
“What’s new is the ability to really look at this across geographies and really home in on how many people face very long times and distances for travel. That’s a really important contribution,” he said.
Dr. Cuadros pointed out that these hard-to-reach populations are key to achieving the UNAID’s HIV elimination targets. “We’re going to have these pockets of transmission that are going to be really important for epidemic control,” he explained.
Toward that end, the onus appears to extend well beyond solutions that emphasize difficulty in reaching people from the provider perspective. “There’s quite a lot of what you might want to think of as blaming the victim for when people miss appointments, don’t appear to be adherent, [or] can’t stay reliably suppressed,” said Dr. Beyrer.
“It’s really important for providers in general to include in history and intake how far people have come, what their challenges are with travel, to really pay attention to those issues. Having this elegant analysis, this level of detail, is an important first step,” he added.
Dr. Cuadros has disclosed no relevant financial relationships. Dr. Beyrer reports a consulting agreement with Merck.
A version of this article first appeared on Medscape.com.
Can geospatial mapping fill in the gaps in areas lagging behind in global efforts to end the HIV epidemic?
That’s what Diego Cuadros, PhD, assistant professor of health geography and disease modeling at the University of Cincinnati, set out to learn using geospatial data combined with prevalence data to identify the most underserved areas in Sub-Saharan Africa (SSA) for HIV services.
Study findings, which were published Nov. 24 in PLOS Global Public Health, highlight that as many as 1.5 million people living with HIV (PLHIV) in SSA have more than an hour’s motorized travel time both ways to access care, while roughly 3 million must set aside, at minimum, 30 minutes. When the only mode of transportation is walking, as much as 95.3% of underserved areas are faced with at least 30 minutes travel time.
This is simply the tip of the overall problem, Dr. Cuadros told this news organization.
“We are able to estimate how many people [whose] quality of life is being affected by HIV because they are not on treatment and most probably, HIV incidence is high in those areas. But [it’s not as simple as just] increasing the number of health care facilities,” he said. “We need to find strategies to be able to cover this population.”
Dr. Cuadros also noted that the problem goes both ways. “It’s hard for them to move, and it’s [also] hard to reach them,” he explained.
Mapping care, or lack thereof
Dr. Cuadros and team used two primary sources of data to generate high-resolution maps of underserved SSA areas: estimated number of PLHIV between the ages of 15 and 49 years in 47 SSA countries paired with population density and global map of travel time to the nearest health facility by motorized and nonmotorized (that is, walking) transportation. Combining these data allowed them to then detail the distance from access to care for every 5 km².
The mapping exercise showed that 90.5% of the total territory, in which about 7 million PLHIV resided, had more than 10 minutes motorized travel time to the nearest health care facility, while 74.6% were within 30 minutes, and 58.9% were within 60 minutes. Increases in threshold travel times (from 10 to 60 minutes) corresponded directly to declines in the average proportion of underserved areas (from 80.9% to 42.6%). However, in certain countries like Sudan and Mauritania, 99.4% of the areas were underserved at the 10 minute threshold, while more than 90% were underserved at the 60 minute threshold.
Corresponding rates for nonmotorized access to health services were similar: 88.7% (~17.6 million) PLHIV had 10 minutes walking time to health care services, while 57.8% (~11.5 million) had at least 30 minutes, and 33.0% (~6.6 million), at least 60 minutes. Likewise, as threshold times increased from 10 to 60 minutes, the percentage of affected PLHIV declined (to roughly 50% in two-thirds of the countries). But more than 70% of PLHIV resided in underserved areas in countries like Equatorial Guinea, Eritrea, South Sudan, and Sudan.
Geographical allocation of health service facilities underscores treatment gaps
“We think that most PLHIV live in urban areas or close to urban areas, and most of the health care facilities in Africa are concentrated in those areas. But [roughly 8 million people with HIV] are living in rural areas, and for most, movement is very difficult,” explained Dr. Cuadros, meaning that the majority are not on treatment despite the high incidence of HIV.
Inarguably, the pandemic has interrupted HIV services and treatment substantially on the African continent, further challenging any efforts to translate these study findings into actionable strategies.
“We’ve known for quite a while that distance and travel times and travel expenses are known risks for nonadherence, for lack of access to diagnostics, for people at risk for exposure,” Chris Beyrer, MD, MPH, Desmond M. Tutu Professor of Public Health and Human Rights at the Johns Hopkins Bloomberg School of Public Health, Baltimore, said in an interview. (Dr. Beyrer was not involved in the study.)
“What’s new is the ability to really look at this across geographies and really home in on how many people face very long times and distances for travel. That’s a really important contribution,” he said.
Dr. Cuadros pointed out that these hard-to-reach populations are key to achieving the UNAID’s HIV elimination targets. “We’re going to have these pockets of transmission that are going to be really important for epidemic control,” he explained.
Toward that end, the onus appears to extend well beyond solutions that emphasize difficulty in reaching people from the provider perspective. “There’s quite a lot of what you might want to think of as blaming the victim for when people miss appointments, don’t appear to be adherent, [or] can’t stay reliably suppressed,” said Dr. Beyrer.
“It’s really important for providers in general to include in history and intake how far people have come, what their challenges are with travel, to really pay attention to those issues. Having this elegant analysis, this level of detail, is an important first step,” he added.
Dr. Cuadros has disclosed no relevant financial relationships. Dr. Beyrer reports a consulting agreement with Merck.
A version of this article first appeared on Medscape.com.
Merck’s COVID-19 pill may be less effective than first hoped
According to an analysis by scientists at the Food and Drug Administration, the experimental pill cut the risk of hospitalization or death from COVID-19 by about 30%, compared to a placebo, and the pill showed no benefit for people with antibodies against COVID-19 from prior infection.
The updated analysis showed 48 hospitalizations or deaths among study participants who were randomly assigned to take the antiviral drug, compared to 68 among those who took a placebo.
Those results come from the full set of 1,433 patients who were randomized in the clinical trial, which just became available last week.
Initial results from the first 775 patients enrolled in the clinical trial, which were issued in a company news release in October, had said the drug cut the risk of hospitalization or death for patients at high risk of severe disease by about 50%.
Merck has been producing millions of doses of molnupiravir, which is the first antiviral pill to treat COVID-19 infections. The United Kingdom’s drug regulator authorized use of the medication in early November. The company said it expected to distribute the medication globally by the end of 2021.
In October, two Indian drug companies halted late-stage clinical trials of a generic version of molnupiravir after the studies failed to find any benefit to patients with moderate COVID-19. Trials in patients with milder symptoms are still ongoing.
On Nov. 27, the New England Journal of Medicine postponed its planned early release of the molnupiravir study results, citing “new information.”
The medication is designed to be given as four pills taken every 12 hours for 5 days. It’s most effective when taken within the first few days of new symptoms, something that requires convenient and affordable testing.
The new results seem to put molnupiravir far below the effectiveness of existing treatments.
The infused monoclonal antibody cocktail REGEN-COV, which the FDA has already authorized for emergency use, is about 85% effective at preventing hospitalization or death in patients who are at risk for severe COVID-19 outcomes, and it appears to be just as effective in people who already have antibodies against COVID-19, which is why it is being given to both vaccinated and unvaccinated patients, the FDA said.
In early November, Pfizer said its experimental antiviral pill Paxlovid cut the risk of hospitalization or death by 89%.
In briefing documents posted ahead of an advisory committee meeting Nov. 30, the FDA highlights other potential safety issues with the Merck drug, which works by causing the virus to make mistakes as it copies itself, eventually causing the virus to mutate itself to death.
The agency has asked the advisory committee to weigh in on the right patient population for the drug: Should pregnant women get it? Could the drug harm a developing fetus?
Should vaccinated people with breakthrough infections get it? Would it work for them? People with reduced immune function are more likely to get a breakthrough infection. They’re also more likely to shed virus for a longer period of time, making them perfect incubators for variants. What could happen if we give this type of patient a drug that increases mutations?
And what about mutations caused by the medication? Could they increase the potential for more variants? The agency concluded the risk of this happening was low.
In animal studies, the drug impacted bone formation. For this reason, the agency has agreed with the drug company that molnupiravir should not be given to anyone under the age of 18.
Aside from these concerns, the FDA says there were no major safety issues among people who took part in the clinical trial, though they acknowledge that number is small.
A version of this article first appeared on WebMD.com.
According to an analysis by scientists at the Food and Drug Administration, the experimental pill cut the risk of hospitalization or death from COVID-19 by about 30%, compared to a placebo, and the pill showed no benefit for people with antibodies against COVID-19 from prior infection.
The updated analysis showed 48 hospitalizations or deaths among study participants who were randomly assigned to take the antiviral drug, compared to 68 among those who took a placebo.
Those results come from the full set of 1,433 patients who were randomized in the clinical trial, which just became available last week.
Initial results from the first 775 patients enrolled in the clinical trial, which were issued in a company news release in October, had said the drug cut the risk of hospitalization or death for patients at high risk of severe disease by about 50%.
Merck has been producing millions of doses of molnupiravir, which is the first antiviral pill to treat COVID-19 infections. The United Kingdom’s drug regulator authorized use of the medication in early November. The company said it expected to distribute the medication globally by the end of 2021.
In October, two Indian drug companies halted late-stage clinical trials of a generic version of molnupiravir after the studies failed to find any benefit to patients with moderate COVID-19. Trials in patients with milder symptoms are still ongoing.
On Nov. 27, the New England Journal of Medicine postponed its planned early release of the molnupiravir study results, citing “new information.”
The medication is designed to be given as four pills taken every 12 hours for 5 days. It’s most effective when taken within the first few days of new symptoms, something that requires convenient and affordable testing.
The new results seem to put molnupiravir far below the effectiveness of existing treatments.
The infused monoclonal antibody cocktail REGEN-COV, which the FDA has already authorized for emergency use, is about 85% effective at preventing hospitalization or death in patients who are at risk for severe COVID-19 outcomes, and it appears to be just as effective in people who already have antibodies against COVID-19, which is why it is being given to both vaccinated and unvaccinated patients, the FDA said.
In early November, Pfizer said its experimental antiviral pill Paxlovid cut the risk of hospitalization or death by 89%.
In briefing documents posted ahead of an advisory committee meeting Nov. 30, the FDA highlights other potential safety issues with the Merck drug, which works by causing the virus to make mistakes as it copies itself, eventually causing the virus to mutate itself to death.
The agency has asked the advisory committee to weigh in on the right patient population for the drug: Should pregnant women get it? Could the drug harm a developing fetus?
Should vaccinated people with breakthrough infections get it? Would it work for them? People with reduced immune function are more likely to get a breakthrough infection. They’re also more likely to shed virus for a longer period of time, making them perfect incubators for variants. What could happen if we give this type of patient a drug that increases mutations?
And what about mutations caused by the medication? Could they increase the potential for more variants? The agency concluded the risk of this happening was low.
In animal studies, the drug impacted bone formation. For this reason, the agency has agreed with the drug company that molnupiravir should not be given to anyone under the age of 18.
Aside from these concerns, the FDA says there were no major safety issues among people who took part in the clinical trial, though they acknowledge that number is small.
A version of this article first appeared on WebMD.com.
According to an analysis by scientists at the Food and Drug Administration, the experimental pill cut the risk of hospitalization or death from COVID-19 by about 30%, compared to a placebo, and the pill showed no benefit for people with antibodies against COVID-19 from prior infection.
The updated analysis showed 48 hospitalizations or deaths among study participants who were randomly assigned to take the antiviral drug, compared to 68 among those who took a placebo.
Those results come from the full set of 1,433 patients who were randomized in the clinical trial, which just became available last week.
Initial results from the first 775 patients enrolled in the clinical trial, which were issued in a company news release in October, had said the drug cut the risk of hospitalization or death for patients at high risk of severe disease by about 50%.
Merck has been producing millions of doses of molnupiravir, which is the first antiviral pill to treat COVID-19 infections. The United Kingdom’s drug regulator authorized use of the medication in early November. The company said it expected to distribute the medication globally by the end of 2021.
In October, two Indian drug companies halted late-stage clinical trials of a generic version of molnupiravir after the studies failed to find any benefit to patients with moderate COVID-19. Trials in patients with milder symptoms are still ongoing.
On Nov. 27, the New England Journal of Medicine postponed its planned early release of the molnupiravir study results, citing “new information.”
The medication is designed to be given as four pills taken every 12 hours for 5 days. It’s most effective when taken within the first few days of new symptoms, something that requires convenient and affordable testing.
The new results seem to put molnupiravir far below the effectiveness of existing treatments.
The infused monoclonal antibody cocktail REGEN-COV, which the FDA has already authorized for emergency use, is about 85% effective at preventing hospitalization or death in patients who are at risk for severe COVID-19 outcomes, and it appears to be just as effective in people who already have antibodies against COVID-19, which is why it is being given to both vaccinated and unvaccinated patients, the FDA said.
In early November, Pfizer said its experimental antiviral pill Paxlovid cut the risk of hospitalization or death by 89%.
In briefing documents posted ahead of an advisory committee meeting Nov. 30, the FDA highlights other potential safety issues with the Merck drug, which works by causing the virus to make mistakes as it copies itself, eventually causing the virus to mutate itself to death.
The agency has asked the advisory committee to weigh in on the right patient population for the drug: Should pregnant women get it? Could the drug harm a developing fetus?
Should vaccinated people with breakthrough infections get it? Would it work for them? People with reduced immune function are more likely to get a breakthrough infection. They’re also more likely to shed virus for a longer period of time, making them perfect incubators for variants. What could happen if we give this type of patient a drug that increases mutations?
And what about mutations caused by the medication? Could they increase the potential for more variants? The agency concluded the risk of this happening was low.
In animal studies, the drug impacted bone formation. For this reason, the agency has agreed with the drug company that molnupiravir should not be given to anyone under the age of 18.
Aside from these concerns, the FDA says there were no major safety issues among people who took part in the clinical trial, though they acknowledge that number is small.
A version of this article first appeared on WebMD.com.
Did prior authorization refusals lead to this patient’s death?
Ramy Sedhom, MD, a medical oncologist and a palliative care physician at Penn Medicine Princeton Health in Plainsboro, N.J., will always wonder if prior authorization refusals led to his patient’s death.
The patient had advanced gastric cancer and the insurer initially denied a PET scan to rule out metastatic disease. When the scan was eventually allowed, it revealed that the cancer had spread.
Standard treatment would have been difficult for the patient, an older individual with comorbidities. But Dr. Sedhom knew that a European study had reported equal efficacy and fewer side effects with a reduced chemotherapy regimen, and he thought that was the best approach in this situation.
The insurer disagreed with Dr. Sedhom’s decision and, while the two argued, the patient’s symptoms worsened. He was admitted to the hospital, where he experienced a decline in function, common for older patients. “Long story short, he was never able to seek treatment and then transitioned to hospice,” Dr. Sedhom said. “It was one of those situations where there was a 3- to 4-week delay in what should have been standard care.”
. Nearly 4 years after major organizations — American Hospital Association, America’s Health Insurance Plans, American Medical Association, Blue Cross Blue Shield Association, and others — signed a consensus statement agreeing to improve the prior authorization process, physicians say little progress has been made.
Indeed, 83% of physicians say that the number of prior authorizations required for prescription medications and medical services has increased over the last 5 years, according to survey results released earlier this year.
“It’s decidedly worse — there’s no question about it,” said Andrew R. Spector, MD, a neurologist and sleep medicine specialist at Duke Health in Durham, N.C. “Drugs that I used to get without prior authorizations now require them.”
When Vignesh I. Doraiswamy, MD, an internal medicine hospitalist at the Ohio State University Wexner Medical Center in Columbus, discharged a patient with Clostridioides difficile infection, he followed clinical guidelines to prescribe vancomycin for 10 to 14 days. “And the insurance company said, ‘Well, yeah, we only authorize about 5 days,’ which just makes no sense,” Dr. Doraiswamy said. “There’s nowhere in any literature that says 5 days is sufficient. What worries me is that is the standard of care we are supposed to give and yet we are unable to.”
Yash B. Jobanputra, MD, a cardiology fellow at Saint Vincent Hospital in Worcester, Mass., laments that prior authorization is used in situations that simply do not make common sense. During his residency, a woman who had tested positive for the BRCA gene mutation with a strong family history of breast cancer needed a breast ultrasound and an MRI scan every 6 months to 1 year. Despite the documentation that she was at extremely high risk for developing breast cancer, he had to go through prior authorization every time she was due for new images.
“I had to call the insurance company, they would put me on hold, I would wait to speak to a physician — and the end response would be, ‘Yeah, this is what needs to be done,’” he said. “But having established her positive status once should be enough really. I shouldn’t have to go through the circus all over again.”
Prior authorization is also being used for routine diagnostics, such as a Holter monitor for patients complaining of heart palpitations. “Depending on the insurance, for some patients we can give it to them in the clinic right away,” Dr. Jobanputra said. “Whereas some others we have to wait until we get prior authorization from the insurance company and the patient has to come back again to the hospital to get the monitor. That is a delay in patient care.”
The delays also extend to emergency care, Dr. Doraiswamy said. He cites the example of a heart attack patient who needed an emergency heart catheterization but ran into a prior authorization delay. “I just said, ‘Try your best not to get stressed’ which is not easy for a patient finding out their stay wasn’t covered when they had just been through a heart attack,” he said. “Then I spent 20 to 30 minutes — most of it on hold — to answer the question ‘Why did this patient need to get admitted?’ “
Physicians feel disrespected because that type of prior authorization hassle is just busywork. “Rarely is a valid stay that was initially denied, not eventually accepted,” Dr. Doraiswamy said. “But why couldn’t they have just seen that the guy had a heart attack and he obviously needed to be in the hospital?”
For Dr. Spector, the Duke Health sleep medicine specialist, prior authorization is not just a speed bump, it’s a full stop. Insurers have started mandating a multiple sleep latency test (MSLT) to confirm narcolepsy before covering medication to treat the condition. “We know that the MSLT is very often wrong,” he said. “There are a lot of times we’re dealing with patients with narcolepsy who simply don’t meet the testing criteria that the insurance requires, and payers will not accept our clinical judgment.”
In his view, the prior authorization landscape is worsening — and not only because a “faulty test” is being used to deny treatment. “The appeal process is worse,” Dr. Spector said. “I used to be able to get on the phone and do a peer-to-peer review with a physician who I could reason with… but that doesn’t happen anymore. There is virtually no way to bypass these blanket rules.”
Other survey findings also stand in direct contradiction of the 2018 consensus agreement:
A large majority (87%) of physicians report that prior authorization interferes with continuity of care, even though the industry groups agreed that patients should be protected from treatment disruption when there is a formulary or treatment-coverage change.
Despite a consensus to encourage transparency and easy accessibility of prior authorization requirements, 68% of physicians reported that it is difficult to determine whether a prescription medication requires prior authorization, and 58% report that it’s difficult for medical services.
Phone and fax are the most commonly used methods for completing prior authorizations, despite agreement that electronic prior authorization, using existing national standard transactions, should be accelerated. Fewer than one quarter of physicians said that their electronic health record system supports electronic prior authorization for prescription medications.
Dr. Spector wants to see legislation that forces insurers to live up to some of the tenets of the 2018 consensus statement. In September, a new Texas law went into effect, exempting physicians from prior authorization if, during the previous six months, 90% of their treatments met an insurer›s medical necessity criteria. In January, the recently approved Prior Authorization Reform Act in Illinois will reduce the number of services subject to prior authorization, mandate a prior authorization decision within 5 days, and set disciplinary measures for health plans that do not comply, among other things.
“What gives me hope is that at least somewhere in the country, somebody is doing something,” Dr. Spector said. “And if it goes well, maybe other insurers will adopt it. I’m really hoping they demonstrate that the money they can save on the administration of all the appeals and prior authorization paperwork can actually go into caring for patients.”
In addition to state-level action, reform may also be advancing at the federal level. In October, a bill was introduced in the U.S. Senate that mirrors a prior authorization reform bill introduced in the House of Representatives last May. Both bills have broad bipartisan support; the House bill has more than 235 co-sponsors.
In an interview with this news organization, Rep. Ami Bera, MD, (D-CA) said it is “very realistic” that the bill will become law during this session of Congress. “We do think this bill will get marked up in committee and hopefully we can get it to the floor either as a stand-alone bill where we know we have the votes to pass it or as part of a larger legislative package,” he said.
If approved, the Improving Seniors’ Timely Access to Care Act of 2021 would require that Medicare Advantage plans minimize the use of prior authorization for routinely approved services; require real-time decisions for certain requests; report the extent of their use of prior authorization and their rate of approvals or denials, among other things; and establish an electronic prior authorization system.
Medicare Advantage plans are private insurers that are regulated by the Centers for Medicare & Medicaid Services (CMS), which will create the specific rules and penalties associated with the reforms, if they become law. “One would presume that a condition of being a Medicare Advantage plan is that you’re going to have to comply with these new regulations,” said Katie Orrico, senior vice president of health policy and advocacy for the American Association of Neurological Surgeons and Congress of Neurological Surgeons (AANS/CNS). “So they will have some amount of teeth in the form of a mandate.”
The AANS and CNS are part of the Regulatory Relief Coalition, a group of 14 national physician specialty organizations. Winning prior authorization reform in the Medicare Advantage plans is part of its bigger strategy. “If those commercial plans have to follow a set of rules and processes for Medicare, then why not just expand those same processes to all other parts of their business?” Ms. Orrico said.
Despite his frustration with their prior authorization processes, Dr. Doraiswamy, the Ohio State hospitalist, agrees that working to improve insurers’ practices is the best way forward. “It’s so easy to make them look like these evil, giant conglomerations that exist solely to suck money and not care about anyone’s health, but I don’t know if that’s necessarily the case,” he said. “We really have to figure out how best to work with insurance companies to make sure that, while they are profit-generating institutions, that [profit] shouldn’t come at the cost of patient care.”
A version of this article first appeared on Medscape.com.
Ramy Sedhom, MD, a medical oncologist and a palliative care physician at Penn Medicine Princeton Health in Plainsboro, N.J., will always wonder if prior authorization refusals led to his patient’s death.
The patient had advanced gastric cancer and the insurer initially denied a PET scan to rule out metastatic disease. When the scan was eventually allowed, it revealed that the cancer had spread.
Standard treatment would have been difficult for the patient, an older individual with comorbidities. But Dr. Sedhom knew that a European study had reported equal efficacy and fewer side effects with a reduced chemotherapy regimen, and he thought that was the best approach in this situation.
The insurer disagreed with Dr. Sedhom’s decision and, while the two argued, the patient’s symptoms worsened. He was admitted to the hospital, where he experienced a decline in function, common for older patients. “Long story short, he was never able to seek treatment and then transitioned to hospice,” Dr. Sedhom said. “It was one of those situations where there was a 3- to 4-week delay in what should have been standard care.”
. Nearly 4 years after major organizations — American Hospital Association, America’s Health Insurance Plans, American Medical Association, Blue Cross Blue Shield Association, and others — signed a consensus statement agreeing to improve the prior authorization process, physicians say little progress has been made.
Indeed, 83% of physicians say that the number of prior authorizations required for prescription medications and medical services has increased over the last 5 years, according to survey results released earlier this year.
“It’s decidedly worse — there’s no question about it,” said Andrew R. Spector, MD, a neurologist and sleep medicine specialist at Duke Health in Durham, N.C. “Drugs that I used to get without prior authorizations now require them.”
When Vignesh I. Doraiswamy, MD, an internal medicine hospitalist at the Ohio State University Wexner Medical Center in Columbus, discharged a patient with Clostridioides difficile infection, he followed clinical guidelines to prescribe vancomycin for 10 to 14 days. “And the insurance company said, ‘Well, yeah, we only authorize about 5 days,’ which just makes no sense,” Dr. Doraiswamy said. “There’s nowhere in any literature that says 5 days is sufficient. What worries me is that is the standard of care we are supposed to give and yet we are unable to.”
Yash B. Jobanputra, MD, a cardiology fellow at Saint Vincent Hospital in Worcester, Mass., laments that prior authorization is used in situations that simply do not make common sense. During his residency, a woman who had tested positive for the BRCA gene mutation with a strong family history of breast cancer needed a breast ultrasound and an MRI scan every 6 months to 1 year. Despite the documentation that she was at extremely high risk for developing breast cancer, he had to go through prior authorization every time she was due for new images.
“I had to call the insurance company, they would put me on hold, I would wait to speak to a physician — and the end response would be, ‘Yeah, this is what needs to be done,’” he said. “But having established her positive status once should be enough really. I shouldn’t have to go through the circus all over again.”
Prior authorization is also being used for routine diagnostics, such as a Holter monitor for patients complaining of heart palpitations. “Depending on the insurance, for some patients we can give it to them in the clinic right away,” Dr. Jobanputra said. “Whereas some others we have to wait until we get prior authorization from the insurance company and the patient has to come back again to the hospital to get the monitor. That is a delay in patient care.”
The delays also extend to emergency care, Dr. Doraiswamy said. He cites the example of a heart attack patient who needed an emergency heart catheterization but ran into a prior authorization delay. “I just said, ‘Try your best not to get stressed’ which is not easy for a patient finding out their stay wasn’t covered when they had just been through a heart attack,” he said. “Then I spent 20 to 30 minutes — most of it on hold — to answer the question ‘Why did this patient need to get admitted?’ “
Physicians feel disrespected because that type of prior authorization hassle is just busywork. “Rarely is a valid stay that was initially denied, not eventually accepted,” Dr. Doraiswamy said. “But why couldn’t they have just seen that the guy had a heart attack and he obviously needed to be in the hospital?”
For Dr. Spector, the Duke Health sleep medicine specialist, prior authorization is not just a speed bump, it’s a full stop. Insurers have started mandating a multiple sleep latency test (MSLT) to confirm narcolepsy before covering medication to treat the condition. “We know that the MSLT is very often wrong,” he said. “There are a lot of times we’re dealing with patients with narcolepsy who simply don’t meet the testing criteria that the insurance requires, and payers will not accept our clinical judgment.”
In his view, the prior authorization landscape is worsening — and not only because a “faulty test” is being used to deny treatment. “The appeal process is worse,” Dr. Spector said. “I used to be able to get on the phone and do a peer-to-peer review with a physician who I could reason with… but that doesn’t happen anymore. There is virtually no way to bypass these blanket rules.”
Other survey findings also stand in direct contradiction of the 2018 consensus agreement:
A large majority (87%) of physicians report that prior authorization interferes with continuity of care, even though the industry groups agreed that patients should be protected from treatment disruption when there is a formulary or treatment-coverage change.
Despite a consensus to encourage transparency and easy accessibility of prior authorization requirements, 68% of physicians reported that it is difficult to determine whether a prescription medication requires prior authorization, and 58% report that it’s difficult for medical services.
Phone and fax are the most commonly used methods for completing prior authorizations, despite agreement that electronic prior authorization, using existing national standard transactions, should be accelerated. Fewer than one quarter of physicians said that their electronic health record system supports electronic prior authorization for prescription medications.
Dr. Spector wants to see legislation that forces insurers to live up to some of the tenets of the 2018 consensus statement. In September, a new Texas law went into effect, exempting physicians from prior authorization if, during the previous six months, 90% of their treatments met an insurer›s medical necessity criteria. In January, the recently approved Prior Authorization Reform Act in Illinois will reduce the number of services subject to prior authorization, mandate a prior authorization decision within 5 days, and set disciplinary measures for health plans that do not comply, among other things.
“What gives me hope is that at least somewhere in the country, somebody is doing something,” Dr. Spector said. “And if it goes well, maybe other insurers will adopt it. I’m really hoping they demonstrate that the money they can save on the administration of all the appeals and prior authorization paperwork can actually go into caring for patients.”
In addition to state-level action, reform may also be advancing at the federal level. In October, a bill was introduced in the U.S. Senate that mirrors a prior authorization reform bill introduced in the House of Representatives last May. Both bills have broad bipartisan support; the House bill has more than 235 co-sponsors.
In an interview with this news organization, Rep. Ami Bera, MD, (D-CA) said it is “very realistic” that the bill will become law during this session of Congress. “We do think this bill will get marked up in committee and hopefully we can get it to the floor either as a stand-alone bill where we know we have the votes to pass it or as part of a larger legislative package,” he said.
If approved, the Improving Seniors’ Timely Access to Care Act of 2021 would require that Medicare Advantage plans minimize the use of prior authorization for routinely approved services; require real-time decisions for certain requests; report the extent of their use of prior authorization and their rate of approvals or denials, among other things; and establish an electronic prior authorization system.
Medicare Advantage plans are private insurers that are regulated by the Centers for Medicare & Medicaid Services (CMS), which will create the specific rules and penalties associated with the reforms, if they become law. “One would presume that a condition of being a Medicare Advantage plan is that you’re going to have to comply with these new regulations,” said Katie Orrico, senior vice president of health policy and advocacy for the American Association of Neurological Surgeons and Congress of Neurological Surgeons (AANS/CNS). “So they will have some amount of teeth in the form of a mandate.”
The AANS and CNS are part of the Regulatory Relief Coalition, a group of 14 national physician specialty organizations. Winning prior authorization reform in the Medicare Advantage plans is part of its bigger strategy. “If those commercial plans have to follow a set of rules and processes for Medicare, then why not just expand those same processes to all other parts of their business?” Ms. Orrico said.
Despite his frustration with their prior authorization processes, Dr. Doraiswamy, the Ohio State hospitalist, agrees that working to improve insurers’ practices is the best way forward. “It’s so easy to make them look like these evil, giant conglomerations that exist solely to suck money and not care about anyone’s health, but I don’t know if that’s necessarily the case,” he said. “We really have to figure out how best to work with insurance companies to make sure that, while they are profit-generating institutions, that [profit] shouldn’t come at the cost of patient care.”
A version of this article first appeared on Medscape.com.
Ramy Sedhom, MD, a medical oncologist and a palliative care physician at Penn Medicine Princeton Health in Plainsboro, N.J., will always wonder if prior authorization refusals led to his patient’s death.
The patient had advanced gastric cancer and the insurer initially denied a PET scan to rule out metastatic disease. When the scan was eventually allowed, it revealed that the cancer had spread.
Standard treatment would have been difficult for the patient, an older individual with comorbidities. But Dr. Sedhom knew that a European study had reported equal efficacy and fewer side effects with a reduced chemotherapy regimen, and he thought that was the best approach in this situation.
The insurer disagreed with Dr. Sedhom’s decision and, while the two argued, the patient’s symptoms worsened. He was admitted to the hospital, where he experienced a decline in function, common for older patients. “Long story short, he was never able to seek treatment and then transitioned to hospice,” Dr. Sedhom said. “It was one of those situations where there was a 3- to 4-week delay in what should have been standard care.”
. Nearly 4 years after major organizations — American Hospital Association, America’s Health Insurance Plans, American Medical Association, Blue Cross Blue Shield Association, and others — signed a consensus statement agreeing to improve the prior authorization process, physicians say little progress has been made.
Indeed, 83% of physicians say that the number of prior authorizations required for prescription medications and medical services has increased over the last 5 years, according to survey results released earlier this year.
“It’s decidedly worse — there’s no question about it,” said Andrew R. Spector, MD, a neurologist and sleep medicine specialist at Duke Health in Durham, N.C. “Drugs that I used to get without prior authorizations now require them.”
When Vignesh I. Doraiswamy, MD, an internal medicine hospitalist at the Ohio State University Wexner Medical Center in Columbus, discharged a patient with Clostridioides difficile infection, he followed clinical guidelines to prescribe vancomycin for 10 to 14 days. “And the insurance company said, ‘Well, yeah, we only authorize about 5 days,’ which just makes no sense,” Dr. Doraiswamy said. “There’s nowhere in any literature that says 5 days is sufficient. What worries me is that is the standard of care we are supposed to give and yet we are unable to.”
Yash B. Jobanputra, MD, a cardiology fellow at Saint Vincent Hospital in Worcester, Mass., laments that prior authorization is used in situations that simply do not make common sense. During his residency, a woman who had tested positive for the BRCA gene mutation with a strong family history of breast cancer needed a breast ultrasound and an MRI scan every 6 months to 1 year. Despite the documentation that she was at extremely high risk for developing breast cancer, he had to go through prior authorization every time she was due for new images.
“I had to call the insurance company, they would put me on hold, I would wait to speak to a physician — and the end response would be, ‘Yeah, this is what needs to be done,’” he said. “But having established her positive status once should be enough really. I shouldn’t have to go through the circus all over again.”
Prior authorization is also being used for routine diagnostics, such as a Holter monitor for patients complaining of heart palpitations. “Depending on the insurance, for some patients we can give it to them in the clinic right away,” Dr. Jobanputra said. “Whereas some others we have to wait until we get prior authorization from the insurance company and the patient has to come back again to the hospital to get the monitor. That is a delay in patient care.”
The delays also extend to emergency care, Dr. Doraiswamy said. He cites the example of a heart attack patient who needed an emergency heart catheterization but ran into a prior authorization delay. “I just said, ‘Try your best not to get stressed’ which is not easy for a patient finding out their stay wasn’t covered when they had just been through a heart attack,” he said. “Then I spent 20 to 30 minutes — most of it on hold — to answer the question ‘Why did this patient need to get admitted?’ “
Physicians feel disrespected because that type of prior authorization hassle is just busywork. “Rarely is a valid stay that was initially denied, not eventually accepted,” Dr. Doraiswamy said. “But why couldn’t they have just seen that the guy had a heart attack and he obviously needed to be in the hospital?”
For Dr. Spector, the Duke Health sleep medicine specialist, prior authorization is not just a speed bump, it’s a full stop. Insurers have started mandating a multiple sleep latency test (MSLT) to confirm narcolepsy before covering medication to treat the condition. “We know that the MSLT is very often wrong,” he said. “There are a lot of times we’re dealing with patients with narcolepsy who simply don’t meet the testing criteria that the insurance requires, and payers will not accept our clinical judgment.”
In his view, the prior authorization landscape is worsening — and not only because a “faulty test” is being used to deny treatment. “The appeal process is worse,” Dr. Spector said. “I used to be able to get on the phone and do a peer-to-peer review with a physician who I could reason with… but that doesn’t happen anymore. There is virtually no way to bypass these blanket rules.”
Other survey findings also stand in direct contradiction of the 2018 consensus agreement:
A large majority (87%) of physicians report that prior authorization interferes with continuity of care, even though the industry groups agreed that patients should be protected from treatment disruption when there is a formulary or treatment-coverage change.
Despite a consensus to encourage transparency and easy accessibility of prior authorization requirements, 68% of physicians reported that it is difficult to determine whether a prescription medication requires prior authorization, and 58% report that it’s difficult for medical services.
Phone and fax are the most commonly used methods for completing prior authorizations, despite agreement that electronic prior authorization, using existing national standard transactions, should be accelerated. Fewer than one quarter of physicians said that their electronic health record system supports electronic prior authorization for prescription medications.
Dr. Spector wants to see legislation that forces insurers to live up to some of the tenets of the 2018 consensus statement. In September, a new Texas law went into effect, exempting physicians from prior authorization if, during the previous six months, 90% of their treatments met an insurer›s medical necessity criteria. In January, the recently approved Prior Authorization Reform Act in Illinois will reduce the number of services subject to prior authorization, mandate a prior authorization decision within 5 days, and set disciplinary measures for health plans that do not comply, among other things.
“What gives me hope is that at least somewhere in the country, somebody is doing something,” Dr. Spector said. “And if it goes well, maybe other insurers will adopt it. I’m really hoping they demonstrate that the money they can save on the administration of all the appeals and prior authorization paperwork can actually go into caring for patients.”
In addition to state-level action, reform may also be advancing at the federal level. In October, a bill was introduced in the U.S. Senate that mirrors a prior authorization reform bill introduced in the House of Representatives last May. Both bills have broad bipartisan support; the House bill has more than 235 co-sponsors.
In an interview with this news organization, Rep. Ami Bera, MD, (D-CA) said it is “very realistic” that the bill will become law during this session of Congress. “We do think this bill will get marked up in committee and hopefully we can get it to the floor either as a stand-alone bill where we know we have the votes to pass it or as part of a larger legislative package,” he said.
If approved, the Improving Seniors’ Timely Access to Care Act of 2021 would require that Medicare Advantage plans minimize the use of prior authorization for routinely approved services; require real-time decisions for certain requests; report the extent of their use of prior authorization and their rate of approvals or denials, among other things; and establish an electronic prior authorization system.
Medicare Advantage plans are private insurers that are regulated by the Centers for Medicare & Medicaid Services (CMS), which will create the specific rules and penalties associated with the reforms, if they become law. “One would presume that a condition of being a Medicare Advantage plan is that you’re going to have to comply with these new regulations,” said Katie Orrico, senior vice president of health policy and advocacy for the American Association of Neurological Surgeons and Congress of Neurological Surgeons (AANS/CNS). “So they will have some amount of teeth in the form of a mandate.”
The AANS and CNS are part of the Regulatory Relief Coalition, a group of 14 national physician specialty organizations. Winning prior authorization reform in the Medicare Advantage plans is part of its bigger strategy. “If those commercial plans have to follow a set of rules and processes for Medicare, then why not just expand those same processes to all other parts of their business?” Ms. Orrico said.
Despite his frustration with their prior authorization processes, Dr. Doraiswamy, the Ohio State hospitalist, agrees that working to improve insurers’ practices is the best way forward. “It’s so easy to make them look like these evil, giant conglomerations that exist solely to suck money and not care about anyone’s health, but I don’t know if that’s necessarily the case,” he said. “We really have to figure out how best to work with insurance companies to make sure that, while they are profit-generating institutions, that [profit] shouldn’t come at the cost of patient care.”
A version of this article first appeared on Medscape.com.
FDA approves first drug for treatment of resistant cytomegalovirus infection
The Food and Drug Administration has approved the first treatment for posttransplant cytomegalovirus (CMV) that is resistant to other drugs.
There are an estimated 200,000 adult transplants every year globally. CMV, a type of herpes virus, is one of the most common infections in transplant patients, occurring in 16%-56% of solid organ transplant recipients and 30%-70% of hematopoietic stem cell transplant recipients, according to Takeda Pharmaceutical Company Limited, the company that manufactures Livtencity. For immunosuppressed transplant patients, CMV infection can lead to complications that include loss of the transplanted or organ or even death.
“Cytomegalovirus infections that are resistant or do not respond to available drugs are of even greater concern,” John Farley, MD, MPH, the director of the Office of Infectious Diseases in the FDA’s Center for Drug Evaluation and Research, said in a statement. “Today’s approval helps meet a significant unmet medical need by providing a treatment option for this patient population.”
Livtencity, which is taken orally, works by preventing the activity of the enzyme responsible for virus replication. The approval, announced Nov. 23, was based on a phase 3 clinical trial that compared Livtencity with conventional antiviral treatments in the achievement of CMV DNA concentration levels below what is measurable in transplant patients with CMV infection that is refractory or treatment-resistant. After 8 weeks, of the 235 patients who received Livtencity, 56% achieved this primary endpoint, compared with 24% of the 117 patients who received conventional antiviral treatments, the press release says.
The most reported adverse reactions of Livtencity were taste disturbance, nausea, diarrhea, vomiting, and fatigue.
“We are grateful for the contributions of the patients and clinicians who participated in our clinical trials, as well as the dedication of our scientists and researchers,” Ramona Sequeira, president of the Takeda’s U.S. Business Unit and Global Portfolio Commercialization, said in a statement. “People undergoing transplants have a lengthy and complex health care journey; with the approval of this treatment, we’re proud to offer these individuals a new oral antiviral to fight CMV infection and disease.”
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has approved the first treatment for posttransplant cytomegalovirus (CMV) that is resistant to other drugs.
There are an estimated 200,000 adult transplants every year globally. CMV, a type of herpes virus, is one of the most common infections in transplant patients, occurring in 16%-56% of solid organ transplant recipients and 30%-70% of hematopoietic stem cell transplant recipients, according to Takeda Pharmaceutical Company Limited, the company that manufactures Livtencity. For immunosuppressed transplant patients, CMV infection can lead to complications that include loss of the transplanted or organ or even death.
“Cytomegalovirus infections that are resistant or do not respond to available drugs are of even greater concern,” John Farley, MD, MPH, the director of the Office of Infectious Diseases in the FDA’s Center for Drug Evaluation and Research, said in a statement. “Today’s approval helps meet a significant unmet medical need by providing a treatment option for this patient population.”
Livtencity, which is taken orally, works by preventing the activity of the enzyme responsible for virus replication. The approval, announced Nov. 23, was based on a phase 3 clinical trial that compared Livtencity with conventional antiviral treatments in the achievement of CMV DNA concentration levels below what is measurable in transplant patients with CMV infection that is refractory or treatment-resistant. After 8 weeks, of the 235 patients who received Livtencity, 56% achieved this primary endpoint, compared with 24% of the 117 patients who received conventional antiviral treatments, the press release says.
The most reported adverse reactions of Livtencity were taste disturbance, nausea, diarrhea, vomiting, and fatigue.
“We are grateful for the contributions of the patients and clinicians who participated in our clinical trials, as well as the dedication of our scientists and researchers,” Ramona Sequeira, president of the Takeda’s U.S. Business Unit and Global Portfolio Commercialization, said in a statement. “People undergoing transplants have a lengthy and complex health care journey; with the approval of this treatment, we’re proud to offer these individuals a new oral antiviral to fight CMV infection and disease.”
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has approved the first treatment for posttransplant cytomegalovirus (CMV) that is resistant to other drugs.
There are an estimated 200,000 adult transplants every year globally. CMV, a type of herpes virus, is one of the most common infections in transplant patients, occurring in 16%-56% of solid organ transplant recipients and 30%-70% of hematopoietic stem cell transplant recipients, according to Takeda Pharmaceutical Company Limited, the company that manufactures Livtencity. For immunosuppressed transplant patients, CMV infection can lead to complications that include loss of the transplanted or organ or even death.
“Cytomegalovirus infections that are resistant or do not respond to available drugs are of even greater concern,” John Farley, MD, MPH, the director of the Office of Infectious Diseases in the FDA’s Center for Drug Evaluation and Research, said in a statement. “Today’s approval helps meet a significant unmet medical need by providing a treatment option for this patient population.”
Livtencity, which is taken orally, works by preventing the activity of the enzyme responsible for virus replication. The approval, announced Nov. 23, was based on a phase 3 clinical trial that compared Livtencity with conventional antiviral treatments in the achievement of CMV DNA concentration levels below what is measurable in transplant patients with CMV infection that is refractory or treatment-resistant. After 8 weeks, of the 235 patients who received Livtencity, 56% achieved this primary endpoint, compared with 24% of the 117 patients who received conventional antiviral treatments, the press release says.
The most reported adverse reactions of Livtencity were taste disturbance, nausea, diarrhea, vomiting, and fatigue.
“We are grateful for the contributions of the patients and clinicians who participated in our clinical trials, as well as the dedication of our scientists and researchers,” Ramona Sequeira, president of the Takeda’s U.S. Business Unit and Global Portfolio Commercialization, said in a statement. “People undergoing transplants have a lengthy and complex health care journey; with the approval of this treatment, we’re proud to offer these individuals a new oral antiviral to fight CMV infection and disease.”
A version of this article first appeared on Medscape.com.
Big drop in U.S. cervical cancer rates, mortality in younger women
The analysis adds to a growing body of evidence demonstrating vaccine-associated changes in cervical cancer incidence and mortality.
Previous data from the United Kingdom, published earlier in November, showed that cervical cancer rates were 87% lower among girls who received the HPV vaccine compared to previously unvaccinated generations. Based on the analysis, the authors concluded that the UK’s HPV immunization program “almost eliminated cervical cancer” in women born since September 1995.
The latest study, published Nov. 29 in JAMA Pediatrics , reports a 38% drop in cervical cancer incidence and a 43% decline in mortality among young women and girls after HPV vaccination was introduced in the United States.
“These results are encouraging,” Peter Sasieni, MD, of King’s College London, and senior author on the U.K. study, told this news organization in an email.
The difference in incidence rates between the U.K. and U.S. studies, Dr. Sasieni explained, is likely due to HPV vaccine coverage not expanding as significantly in the United States as it has in the United Kingdom, and “thus one would anticipate a lower impact on the population in the U.S.”
In the U.S. analysis, Justin Barnes, MD, a radiation oncology resident at Washington University, St. Louis, and colleagues examined cervical cancer incidence between January 2001 and December 2017 using Surveillance, Epidemiology, and End Results and National Program of Cancer Registries data as well as mortality data from the National Center for Health Statistics.
Dr. Barnes and colleagues then compared changes in cervical cancer incidence and mortality between prevaccination years (January 2001 to December 2005) and postvaccination years (January 2010 to December 2017) among three age cohorts – 15-24 years, 25-29 years, and 30-39 years.
“The older 2 groups were included as comparison, given their low vaccination rates,” Dr. Barnes and colleagues explained.
Results show that between the prevaccination and postvaccination periods, the incidence of cervical cancer dropped by 38% in the youngest cohort and by only 16% in the middle-aged group and 8% in the oldest cohort.
Women and girls in the youngest group saw a striking drop in mortality: a 43% decline, which translated to a mortality rate of 0.6 per 100,000.
On the other hand, the authors report a 4.7% decline in mortality in the oldest group and a 4.3% increase in mortality in the middle-aged group – translating to a mortality rate of 1.89 per 100,000 and 0.57 per 100,000, respectively.
Overall, “these nationwide data showed decreased cervical cancer incidence and mortality among women and girls aged 15-24 years after HPV vaccine introduction,” Dr. Barnes and colleagues wrote. The changes in cervical cancer incidence and mortality observed in the youngest age group “were greater than changes in those aged 25 to 29 years and 30 to 39 years, suggesting possible associations with HPV vaccination.”
This analysis lines up with previous evidence from U.S. epidemiologic data, which “have shown decreased cervical cancer incidence after vaccine implementation in women and girls aged 15 to 24 years but not older women.”
Although “the number of deaths and hence the number of potentially averted deaths in young women and girls was small,” the study adds to the current literature by “providing suggestive evidence for vaccine-associated decreases in cervical cancer mortality,” investigators concluded.
The authors have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The analysis adds to a growing body of evidence demonstrating vaccine-associated changes in cervical cancer incidence and mortality.
Previous data from the United Kingdom, published earlier in November, showed that cervical cancer rates were 87% lower among girls who received the HPV vaccine compared to previously unvaccinated generations. Based on the analysis, the authors concluded that the UK’s HPV immunization program “almost eliminated cervical cancer” in women born since September 1995.
The latest study, published Nov. 29 in JAMA Pediatrics , reports a 38% drop in cervical cancer incidence and a 43% decline in mortality among young women and girls after HPV vaccination was introduced in the United States.
“These results are encouraging,” Peter Sasieni, MD, of King’s College London, and senior author on the U.K. study, told this news organization in an email.
The difference in incidence rates between the U.K. and U.S. studies, Dr. Sasieni explained, is likely due to HPV vaccine coverage not expanding as significantly in the United States as it has in the United Kingdom, and “thus one would anticipate a lower impact on the population in the U.S.”
In the U.S. analysis, Justin Barnes, MD, a radiation oncology resident at Washington University, St. Louis, and colleagues examined cervical cancer incidence between January 2001 and December 2017 using Surveillance, Epidemiology, and End Results and National Program of Cancer Registries data as well as mortality data from the National Center for Health Statistics.
Dr. Barnes and colleagues then compared changes in cervical cancer incidence and mortality between prevaccination years (January 2001 to December 2005) and postvaccination years (January 2010 to December 2017) among three age cohorts – 15-24 years, 25-29 years, and 30-39 years.
“The older 2 groups were included as comparison, given their low vaccination rates,” Dr. Barnes and colleagues explained.
Results show that between the prevaccination and postvaccination periods, the incidence of cervical cancer dropped by 38% in the youngest cohort and by only 16% in the middle-aged group and 8% in the oldest cohort.
Women and girls in the youngest group saw a striking drop in mortality: a 43% decline, which translated to a mortality rate of 0.6 per 100,000.
On the other hand, the authors report a 4.7% decline in mortality in the oldest group and a 4.3% increase in mortality in the middle-aged group – translating to a mortality rate of 1.89 per 100,000 and 0.57 per 100,000, respectively.
Overall, “these nationwide data showed decreased cervical cancer incidence and mortality among women and girls aged 15-24 years after HPV vaccine introduction,” Dr. Barnes and colleagues wrote. The changes in cervical cancer incidence and mortality observed in the youngest age group “were greater than changes in those aged 25 to 29 years and 30 to 39 years, suggesting possible associations with HPV vaccination.”
This analysis lines up with previous evidence from U.S. epidemiologic data, which “have shown decreased cervical cancer incidence after vaccine implementation in women and girls aged 15 to 24 years but not older women.”
Although “the number of deaths and hence the number of potentially averted deaths in young women and girls was small,” the study adds to the current literature by “providing suggestive evidence for vaccine-associated decreases in cervical cancer mortality,” investigators concluded.
The authors have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The analysis adds to a growing body of evidence demonstrating vaccine-associated changes in cervical cancer incidence and mortality.
Previous data from the United Kingdom, published earlier in November, showed that cervical cancer rates were 87% lower among girls who received the HPV vaccine compared to previously unvaccinated generations. Based on the analysis, the authors concluded that the UK’s HPV immunization program “almost eliminated cervical cancer” in women born since September 1995.
The latest study, published Nov. 29 in JAMA Pediatrics , reports a 38% drop in cervical cancer incidence and a 43% decline in mortality among young women and girls after HPV vaccination was introduced in the United States.
“These results are encouraging,” Peter Sasieni, MD, of King’s College London, and senior author on the U.K. study, told this news organization in an email.
The difference in incidence rates between the U.K. and U.S. studies, Dr. Sasieni explained, is likely due to HPV vaccine coverage not expanding as significantly in the United States as it has in the United Kingdom, and “thus one would anticipate a lower impact on the population in the U.S.”
In the U.S. analysis, Justin Barnes, MD, a radiation oncology resident at Washington University, St. Louis, and colleagues examined cervical cancer incidence between January 2001 and December 2017 using Surveillance, Epidemiology, and End Results and National Program of Cancer Registries data as well as mortality data from the National Center for Health Statistics.
Dr. Barnes and colleagues then compared changes in cervical cancer incidence and mortality between prevaccination years (January 2001 to December 2005) and postvaccination years (January 2010 to December 2017) among three age cohorts – 15-24 years, 25-29 years, and 30-39 years.
“The older 2 groups were included as comparison, given their low vaccination rates,” Dr. Barnes and colleagues explained.
Results show that between the prevaccination and postvaccination periods, the incidence of cervical cancer dropped by 38% in the youngest cohort and by only 16% in the middle-aged group and 8% in the oldest cohort.
Women and girls in the youngest group saw a striking drop in mortality: a 43% decline, which translated to a mortality rate of 0.6 per 100,000.
On the other hand, the authors report a 4.7% decline in mortality in the oldest group and a 4.3% increase in mortality in the middle-aged group – translating to a mortality rate of 1.89 per 100,000 and 0.57 per 100,000, respectively.
Overall, “these nationwide data showed decreased cervical cancer incidence and mortality among women and girls aged 15-24 years after HPV vaccine introduction,” Dr. Barnes and colleagues wrote. The changes in cervical cancer incidence and mortality observed in the youngest age group “were greater than changes in those aged 25 to 29 years and 30 to 39 years, suggesting possible associations with HPV vaccination.”
This analysis lines up with previous evidence from U.S. epidemiologic data, which “have shown decreased cervical cancer incidence after vaccine implementation in women and girls aged 15 to 24 years but not older women.”
Although “the number of deaths and hence the number of potentially averted deaths in young women and girls was small,” the study adds to the current literature by “providing suggestive evidence for vaccine-associated decreases in cervical cancer mortality,” investigators concluded.
The authors have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM JAMA PEDIATRICS
Pfizer COVID vaccine is 100% effective in adolescents: Study
Pfizer announced on Nov. 22 that its COVID-19 vaccine provided long-term protection against the virus in a late-stage clinical trial among adolescents ages 12-15.
A two-dose series was 100% effective against COVID-19, which was measured between 7 days and 4 months after the second dose.
“As the global health community works to increase the number of vaccinated people around the world, these additional data provide further confidence in our vaccine safety and effectiveness profile in adolescents,” Albert Bourla, PhD, chairman and CEO of Pfizer, said in a statement.
The clinical trial researchers found no serious safety concerns while following patients for 6 months. The adverse events were consistent with other clinical safety data for the vaccine, the company said.
Pfizer will incorporate the data into its submissions for full regulatory approval of the vaccine for ages 12-15 in the United States and worldwide.
The company will request clearance for a 30-mcg dose of the vaccines for ages 12 and older. The shot received FDA emergency use authorization for ages 12-15 in May and full approval for ages 16 and older in August.
The study included 2,228 clinical trial participants who were monitored between November 2020 and September 2021. There were 30 confirmed symptomatic cases of COVID-19 in the placebo group that didn’t receive the vaccine and 0 COVID-19 cases among the vaccinated group.
The efficacy was consistently high across gender, race, ethnicity, and health conditions, the company said.
“This is especially important as we see rates of COVID-19 climbing in this age group in some regions, while vaccine uptake has slowed,” Mr. Bourla said. “We look forward to sharing these data with the FDA and other regulators.”
A version of this article first appeared on WebMD.com.
Pfizer announced on Nov. 22 that its COVID-19 vaccine provided long-term protection against the virus in a late-stage clinical trial among adolescents ages 12-15.
A two-dose series was 100% effective against COVID-19, which was measured between 7 days and 4 months after the second dose.
“As the global health community works to increase the number of vaccinated people around the world, these additional data provide further confidence in our vaccine safety and effectiveness profile in adolescents,” Albert Bourla, PhD, chairman and CEO of Pfizer, said in a statement.
The clinical trial researchers found no serious safety concerns while following patients for 6 months. The adverse events were consistent with other clinical safety data for the vaccine, the company said.
Pfizer will incorporate the data into its submissions for full regulatory approval of the vaccine for ages 12-15 in the United States and worldwide.
The company will request clearance for a 30-mcg dose of the vaccines for ages 12 and older. The shot received FDA emergency use authorization for ages 12-15 in May and full approval for ages 16 and older in August.
The study included 2,228 clinical trial participants who were monitored between November 2020 and September 2021. There were 30 confirmed symptomatic cases of COVID-19 in the placebo group that didn’t receive the vaccine and 0 COVID-19 cases among the vaccinated group.
The efficacy was consistently high across gender, race, ethnicity, and health conditions, the company said.
“This is especially important as we see rates of COVID-19 climbing in this age group in some regions, while vaccine uptake has slowed,” Mr. Bourla said. “We look forward to sharing these data with the FDA and other regulators.”
A version of this article first appeared on WebMD.com.
Pfizer announced on Nov. 22 that its COVID-19 vaccine provided long-term protection against the virus in a late-stage clinical trial among adolescents ages 12-15.
A two-dose series was 100% effective against COVID-19, which was measured between 7 days and 4 months after the second dose.
“As the global health community works to increase the number of vaccinated people around the world, these additional data provide further confidence in our vaccine safety and effectiveness profile in adolescents,” Albert Bourla, PhD, chairman and CEO of Pfizer, said in a statement.
The clinical trial researchers found no serious safety concerns while following patients for 6 months. The adverse events were consistent with other clinical safety data for the vaccine, the company said.
Pfizer will incorporate the data into its submissions for full regulatory approval of the vaccine for ages 12-15 in the United States and worldwide.
The company will request clearance for a 30-mcg dose of the vaccines for ages 12 and older. The shot received FDA emergency use authorization for ages 12-15 in May and full approval for ages 16 and older in August.
The study included 2,228 clinical trial participants who were monitored between November 2020 and September 2021. There were 30 confirmed symptomatic cases of COVID-19 in the placebo group that didn’t receive the vaccine and 0 COVID-19 cases among the vaccinated group.
The efficacy was consistently high across gender, race, ethnicity, and health conditions, the company said.
“This is especially important as we see rates of COVID-19 climbing in this age group in some regions, while vaccine uptake has slowed,” Mr. Bourla said. “We look forward to sharing these data with the FDA and other regulators.”
A version of this article first appeared on WebMD.com.
Surveillance for measles is a victim of the COVID pandemic
Although the estimated annual number of measles deaths decreased 94% from 2000 to 2020, the COVID-19 pandemic took a toll on both measles vaccination and surveillance, according to a recent report in Morbidity and Mortality Weekly Report.
The number of World Health Organization member states that achieved more than 90% coverage with the first dose of the measles vaccine (MCV1) declined 37% from 2019 to 2020. In 2020, 23 million infants did not receive MCV1 through routine immunization services, and another 93 million were affected by the postponement of mass immunizations or supplementary immunization activities because of the pandemic. Also, endemic transmission was reestablished in nine countries that had previously eliminated measles.
But perhaps the most overlooked aspect of COVID-19 is its effect on surveillance.
“The entire COVID pandemic really put a lot of strain on the surveillance systems, not only for measles but for all vaccine-preventable disease, because there’s a lot of overlap in the staff who work for surveillance,” said Katrina Kretsinger, MD, a medical epidemiologist at the Centers for Disease Control and Prevention, who contributed to the MMWR report.
Because of the stress on the systems, a lot fewer specimens were tested, she said in an interview. And it’s not just measles that is at risk. This has had an impact on the Global Polio Eradication Initiative, which lost staff.
In addition, many vaccination campaigns “were postponed and curtailed throughout 2020,” Dr. Kretsinger said. The strengthening of surveillance systems – and immunization systems, more broadly – needs to be a priority.
“It’s not clear that the children who were missed during that year were subsequently caught up,” she explained. Having a “cohort of children who have missed measles vaccine creates the reservoir of susceptibility that will provide the nidus for the next big outbreak.”
Measles is the indicator disease. That could mean a resurgence of other vaccine-preventable diseases as well.
This report “was written by some of the world’s experts in measles, and it raises concerns about potential resurgence of measles,” said Walter Orenstein, MD, professor of medicine, epidemiology, global health, and pediatrics at Emory University, Atlanta. “Measles is sort of a canary in the coal mine. If you look at vaccine-preventable diseases, measles is probably the most contagious, so the herd-immunity threshold is highest. Usually on the order of 92%-94% immunity is needed to stop transmission.”
“Measles is the indicator disease,” he said in an interview. “That could mean a resurgence of other vaccine-preventable diseases as well.” Outbreaks don’t just affect the countries where infections are occurring, they “also affect our own domestic health security.”
“Some sort of periodic intensified routine immunization” would be helpful, said Dr. Kretsinger, who recommends “going through and selectively doing some sort of intensified efforts to catch children up early for the entire range of vaccines that they may have missed.”
“Some of these capture campaigns in areas that are thought to have the major problem would be very, very important,” agreed Dr. Orenstein. “A school entry check is one way of trying to look at kids, let’s say at 4-6 years of age, in schools around the world,” offering doses if they’re unvaccinated or inadequately vaccinated. “Another is to try to improve surveillance and try to understand if the cases are vaccine failure or failure to vaccinate.”
“Where the health systems are the most fragile is where those gaps will be the last to be filled, if they are at all, and where we have the basic concerns,” Dr. Kretsinger explained.
“Years ago, WHO recognized that vaccine hesitancy is a top global health threat,” said Dr. Orenstein. “People may not see these diseases so they don’t mean much to them. Since vaccines, we’re victims of our own success.” There’s also a lot of incorrect information circulating.
“We need to realize – and it’s been shown with COVID – that a decision not to vaccinate is not just a decision for your own child. It’s a community decision,” he pointed out. “It’s not my freedom to drive drunk, because not only do I put myself at risk, but others can’t control the car. We have speed limits and other examples where we restrict personal choice because it can adversely affect individuals.”
“My favorite line is vaccines don’t save lives, vaccinations save lives,” Dr. Orenstein said. “The vaccine dose that remains in the vial is 0% effective, no matter what the clinical trials show. And the issue, I think, is that we need to determine how to convince the hesitant to get confident enough to accept vaccination. For that, there is behavioral research; there’s a whole bunch of things that need to be supported. Just purchasing the vaccine doesn’t get it into the bodies.”
Dr. Kretsinger and Dr. Orenstein disclosed no relevant financial relationships .
A version of this article first appeared on Medscape.com.
Although the estimated annual number of measles deaths decreased 94% from 2000 to 2020, the COVID-19 pandemic took a toll on both measles vaccination and surveillance, according to a recent report in Morbidity and Mortality Weekly Report.
The number of World Health Organization member states that achieved more than 90% coverage with the first dose of the measles vaccine (MCV1) declined 37% from 2019 to 2020. In 2020, 23 million infants did not receive MCV1 through routine immunization services, and another 93 million were affected by the postponement of mass immunizations or supplementary immunization activities because of the pandemic. Also, endemic transmission was reestablished in nine countries that had previously eliminated measles.
But perhaps the most overlooked aspect of COVID-19 is its effect on surveillance.
“The entire COVID pandemic really put a lot of strain on the surveillance systems, not only for measles but for all vaccine-preventable disease, because there’s a lot of overlap in the staff who work for surveillance,” said Katrina Kretsinger, MD, a medical epidemiologist at the Centers for Disease Control and Prevention, who contributed to the MMWR report.
Because of the stress on the systems, a lot fewer specimens were tested, she said in an interview. And it’s not just measles that is at risk. This has had an impact on the Global Polio Eradication Initiative, which lost staff.
In addition, many vaccination campaigns “were postponed and curtailed throughout 2020,” Dr. Kretsinger said. The strengthening of surveillance systems – and immunization systems, more broadly – needs to be a priority.
“It’s not clear that the children who were missed during that year were subsequently caught up,” she explained. Having a “cohort of children who have missed measles vaccine creates the reservoir of susceptibility that will provide the nidus for the next big outbreak.”
Measles is the indicator disease. That could mean a resurgence of other vaccine-preventable diseases as well.
This report “was written by some of the world’s experts in measles, and it raises concerns about potential resurgence of measles,” said Walter Orenstein, MD, professor of medicine, epidemiology, global health, and pediatrics at Emory University, Atlanta. “Measles is sort of a canary in the coal mine. If you look at vaccine-preventable diseases, measles is probably the most contagious, so the herd-immunity threshold is highest. Usually on the order of 92%-94% immunity is needed to stop transmission.”
“Measles is the indicator disease,” he said in an interview. “That could mean a resurgence of other vaccine-preventable diseases as well.” Outbreaks don’t just affect the countries where infections are occurring, they “also affect our own domestic health security.”
“Some sort of periodic intensified routine immunization” would be helpful, said Dr. Kretsinger, who recommends “going through and selectively doing some sort of intensified efforts to catch children up early for the entire range of vaccines that they may have missed.”
“Some of these capture campaigns in areas that are thought to have the major problem would be very, very important,” agreed Dr. Orenstein. “A school entry check is one way of trying to look at kids, let’s say at 4-6 years of age, in schools around the world,” offering doses if they’re unvaccinated or inadequately vaccinated. “Another is to try to improve surveillance and try to understand if the cases are vaccine failure or failure to vaccinate.”
“Where the health systems are the most fragile is where those gaps will be the last to be filled, if they are at all, and where we have the basic concerns,” Dr. Kretsinger explained.
“Years ago, WHO recognized that vaccine hesitancy is a top global health threat,” said Dr. Orenstein. “People may not see these diseases so they don’t mean much to them. Since vaccines, we’re victims of our own success.” There’s also a lot of incorrect information circulating.
“We need to realize – and it’s been shown with COVID – that a decision not to vaccinate is not just a decision for your own child. It’s a community decision,” he pointed out. “It’s not my freedom to drive drunk, because not only do I put myself at risk, but others can’t control the car. We have speed limits and other examples where we restrict personal choice because it can adversely affect individuals.”
“My favorite line is vaccines don’t save lives, vaccinations save lives,” Dr. Orenstein said. “The vaccine dose that remains in the vial is 0% effective, no matter what the clinical trials show. And the issue, I think, is that we need to determine how to convince the hesitant to get confident enough to accept vaccination. For that, there is behavioral research; there’s a whole bunch of things that need to be supported. Just purchasing the vaccine doesn’t get it into the bodies.”
Dr. Kretsinger and Dr. Orenstein disclosed no relevant financial relationships .
A version of this article first appeared on Medscape.com.
Although the estimated annual number of measles deaths decreased 94% from 2000 to 2020, the COVID-19 pandemic took a toll on both measles vaccination and surveillance, according to a recent report in Morbidity and Mortality Weekly Report.
The number of World Health Organization member states that achieved more than 90% coverage with the first dose of the measles vaccine (MCV1) declined 37% from 2019 to 2020. In 2020, 23 million infants did not receive MCV1 through routine immunization services, and another 93 million were affected by the postponement of mass immunizations or supplementary immunization activities because of the pandemic. Also, endemic transmission was reestablished in nine countries that had previously eliminated measles.
But perhaps the most overlooked aspect of COVID-19 is its effect on surveillance.
“The entire COVID pandemic really put a lot of strain on the surveillance systems, not only for measles but for all vaccine-preventable disease, because there’s a lot of overlap in the staff who work for surveillance,” said Katrina Kretsinger, MD, a medical epidemiologist at the Centers for Disease Control and Prevention, who contributed to the MMWR report.
Because of the stress on the systems, a lot fewer specimens were tested, she said in an interview. And it’s not just measles that is at risk. This has had an impact on the Global Polio Eradication Initiative, which lost staff.
In addition, many vaccination campaigns “were postponed and curtailed throughout 2020,” Dr. Kretsinger said. The strengthening of surveillance systems – and immunization systems, more broadly – needs to be a priority.
“It’s not clear that the children who were missed during that year were subsequently caught up,” she explained. Having a “cohort of children who have missed measles vaccine creates the reservoir of susceptibility that will provide the nidus for the next big outbreak.”
Measles is the indicator disease. That could mean a resurgence of other vaccine-preventable diseases as well.
This report “was written by some of the world’s experts in measles, and it raises concerns about potential resurgence of measles,” said Walter Orenstein, MD, professor of medicine, epidemiology, global health, and pediatrics at Emory University, Atlanta. “Measles is sort of a canary in the coal mine. If you look at vaccine-preventable diseases, measles is probably the most contagious, so the herd-immunity threshold is highest. Usually on the order of 92%-94% immunity is needed to stop transmission.”
“Measles is the indicator disease,” he said in an interview. “That could mean a resurgence of other vaccine-preventable diseases as well.” Outbreaks don’t just affect the countries where infections are occurring, they “also affect our own domestic health security.”
“Some sort of periodic intensified routine immunization” would be helpful, said Dr. Kretsinger, who recommends “going through and selectively doing some sort of intensified efforts to catch children up early for the entire range of vaccines that they may have missed.”
“Some of these capture campaigns in areas that are thought to have the major problem would be very, very important,” agreed Dr. Orenstein. “A school entry check is one way of trying to look at kids, let’s say at 4-6 years of age, in schools around the world,” offering doses if they’re unvaccinated or inadequately vaccinated. “Another is to try to improve surveillance and try to understand if the cases are vaccine failure or failure to vaccinate.”
“Where the health systems are the most fragile is where those gaps will be the last to be filled, if they are at all, and where we have the basic concerns,” Dr. Kretsinger explained.
“Years ago, WHO recognized that vaccine hesitancy is a top global health threat,” said Dr. Orenstein. “People may not see these diseases so they don’t mean much to them. Since vaccines, we’re victims of our own success.” There’s also a lot of incorrect information circulating.
“We need to realize – and it’s been shown with COVID – that a decision not to vaccinate is not just a decision for your own child. It’s a community decision,” he pointed out. “It’s not my freedom to drive drunk, because not only do I put myself at risk, but others can’t control the car. We have speed limits and other examples where we restrict personal choice because it can adversely affect individuals.”
“My favorite line is vaccines don’t save lives, vaccinations save lives,” Dr. Orenstein said. “The vaccine dose that remains in the vial is 0% effective, no matter what the clinical trials show. And the issue, I think, is that we need to determine how to convince the hesitant to get confident enough to accept vaccination. For that, there is behavioral research; there’s a whole bunch of things that need to be supported. Just purchasing the vaccine doesn’t get it into the bodies.”
Dr. Kretsinger and Dr. Orenstein disclosed no relevant financial relationships .
A version of this article first appeared on Medscape.com.