User login
Comorbidities may cut effectiveness of psoriasis biologics
PARIS – in response to biologic therapy, according to the results of the prospective observational PSO-BIO-REAL study.

The clinical importance of this finding lies in the fact that comorbidities are highly prevalent among patients with moderate to severe psoriasis. Indeed, fully 64% of the 846 participants in PSO-BIO-REAL had at least one major comorbid condition at baseline, Finn Ziegler said at the annual congress of the European Academy of Dermatology and Venereology.
“I think this reflects a picture that has been seen in other studies,” noted Mr. Ziegler, director of global patient access at Leo Pharma in Ballerup, Denmark.
The purpose of the 12-month PSO-BIO-REAL (PSOriasis treated with BIOlogics in REAL life) study was to assess the effectiveness of a variety of biologic agents in a real-world population typical of patients encountered in routine clinical practice, as opposed to more restrictive format of often-cited randomized trials, which generally feature a lengthy list of exclusions. One-third of participants were from the United States, with the rest drawn from four Western European countries. Their mean age was 47 years, with an 18.4-year history of psoriasis and a baseline Psoriasis Area and Severity Index (PASI) score of 14.3.
Sixty percent of participants were starting treatment with a biologic agent for the first time. The other 40% had prior biologic experience. At physician discretion, 61% of enrollees were put on a tumor necrosis factor inhibitor, either etanercept (Enbrel), adalimumab (Humira), or infliximab (Remicade); 30% initiated treatment with the interleukin-12/23 inhibitor ustekinumab (Stelara); and 9% received secukinumab (Cosentyx), an interleukin-17 inhibitor.
The five most common comorbid conditions present at baseline were hypertension, present in 33.5% of participants; psoriatic arthritis (PsA), present in 28.1%; hyperlipidemia, 20.9%; diabetes, 13.9%, and depression, present in 13.7% of the psoriasis patients.
Baseline comorbidities were significantly more common among the biologic-experienced patients. For example, their prevalence of hypertension was 42%, compared with 28% in the biologic-naive group. PsA was present in 35% of the biologic-experienced and 23% of the biologic-naive patients. Nineteen percent of biologic-experienced patients had diabetes at baseline, as did 11% of the biologic-naive group.
During the 12-month study, 3.7% of patients developed a new comorbidity, the most common being anxiety, hypertension, PsA, depression, and hyperlipidemia.
The primary outcome in the study was the complete clearance rate – a PASI 100 response – at 6 months. It ranged from a high of 31% in patients with no baseline comorbid conditions to a low of 16.5% in those with three or more. The results were similar at 12 months.
Conversely, an inadequate therapeutic response as defined by a PASI 50 or less at 6 months occurred in 15% of psoriasis patients with no baseline comorbidities, 27% with one, 35% with two comorbid conditions, and 28% with three or more.
The major caveat regarding this study is that the observed association between comorbid conditions and complete clearance rates doesn’t prove causality, Mr. Ziegler noted.
The PSO-BIO-REAL study was sponsored by Amgen, AstraZeneca, and Leo Pharma. Mr. Ziegler is a Leo executive.
SOURCE: Ziegler F. EADV Congress, Abstract FC04.01.
PARIS – in response to biologic therapy, according to the results of the prospective observational PSO-BIO-REAL study.
The clinical importance of this finding lies in the fact that comorbidities are highly prevalent among patients with moderate to severe psoriasis. Indeed, fully 64% of the 846 participants in PSO-BIO-REAL had at least one major comorbid condition at baseline, Finn Ziegler said at the annual congress of the European Academy of Dermatology and Venereology.
“I think this reflects a picture that has been seen in other studies,” noted Mr. Ziegler, director of global patient access at Leo Pharma in Ballerup, Denmark.
The purpose of the 12-month PSO-BIO-REAL (PSOriasis treated with BIOlogics in REAL life) study was to assess the effectiveness of a variety of biologic agents in a real-world population typical of patients encountered in routine clinical practice, as opposed to more restrictive format of often-cited randomized trials, which generally feature a lengthy list of exclusions. One-third of participants were from the United States, with the rest drawn from four Western European countries. Their mean age was 47 years, with an 18.4-year history of psoriasis and a baseline Psoriasis Area and Severity Index (PASI) score of 14.3.
Sixty percent of participants were starting treatment with a biologic agent for the first time. The other 40% had prior biologic experience. At physician discretion, 61% of enrollees were put on a tumor necrosis factor inhibitor, either etanercept (Enbrel), adalimumab (Humira), or infliximab (Remicade); 30% initiated treatment with the interleukin-12/23 inhibitor ustekinumab (Stelara); and 9% received secukinumab (Cosentyx), an interleukin-17 inhibitor.
The five most common comorbid conditions present at baseline were hypertension, present in 33.5% of participants; psoriatic arthritis (PsA), present in 28.1%; hyperlipidemia, 20.9%; diabetes, 13.9%, and depression, present in 13.7% of the psoriasis patients.
Baseline comorbidities were significantly more common among the biologic-experienced patients. For example, their prevalence of hypertension was 42%, compared with 28% in the biologic-naive group. PsA was present in 35% of the biologic-experienced and 23% of the biologic-naive patients. Nineteen percent of biologic-experienced patients had diabetes at baseline, as did 11% of the biologic-naive group.
During the 12-month study, 3.7% of patients developed a new comorbidity, the most common being anxiety, hypertension, PsA, depression, and hyperlipidemia.
The primary outcome in the study was the complete clearance rate – a PASI 100 response – at 6 months. It ranged from a high of 31% in patients with no baseline comorbid conditions to a low of 16.5% in those with three or more. The results were similar at 12 months.
Conversely, an inadequate therapeutic response as defined by a PASI 50 or less at 6 months occurred in 15% of psoriasis patients with no baseline comorbidities, 27% with one, 35% with two comorbid conditions, and 28% with three or more.
The major caveat regarding this study is that the observed association between comorbid conditions and complete clearance rates doesn’t prove causality, Mr. Ziegler noted.
The PSO-BIO-REAL study was sponsored by Amgen, AstraZeneca, and Leo Pharma. Mr. Ziegler is a Leo executive.
SOURCE: Ziegler F. EADV Congress, Abstract FC04.01.
PARIS – in response to biologic therapy, according to the results of the prospective observational PSO-BIO-REAL study.
The clinical importance of this finding lies in the fact that comorbidities are highly prevalent among patients with moderate to severe psoriasis. Indeed, fully 64% of the 846 participants in PSO-BIO-REAL had at least one major comorbid condition at baseline, Finn Ziegler said at the annual congress of the European Academy of Dermatology and Venereology.
“I think this reflects a picture that has been seen in other studies,” noted Mr. Ziegler, director of global patient access at Leo Pharma in Ballerup, Denmark.
The purpose of the 12-month PSO-BIO-REAL (PSOriasis treated with BIOlogics in REAL life) study was to assess the effectiveness of a variety of biologic agents in a real-world population typical of patients encountered in routine clinical practice, as opposed to more restrictive format of often-cited randomized trials, which generally feature a lengthy list of exclusions. One-third of participants were from the United States, with the rest drawn from four Western European countries. Their mean age was 47 years, with an 18.4-year history of psoriasis and a baseline Psoriasis Area and Severity Index (PASI) score of 14.3.
Sixty percent of participants were starting treatment with a biologic agent for the first time. The other 40% had prior biologic experience. At physician discretion, 61% of enrollees were put on a tumor necrosis factor inhibitor, either etanercept (Enbrel), adalimumab (Humira), or infliximab (Remicade); 30% initiated treatment with the interleukin-12/23 inhibitor ustekinumab (Stelara); and 9% received secukinumab (Cosentyx), an interleukin-17 inhibitor.
The five most common comorbid conditions present at baseline were hypertension, present in 33.5% of participants; psoriatic arthritis (PsA), present in 28.1%; hyperlipidemia, 20.9%; diabetes, 13.9%, and depression, present in 13.7% of the psoriasis patients.
Baseline comorbidities were significantly more common among the biologic-experienced patients. For example, their prevalence of hypertension was 42%, compared with 28% in the biologic-naive group. PsA was present in 35% of the biologic-experienced and 23% of the biologic-naive patients. Nineteen percent of biologic-experienced patients had diabetes at baseline, as did 11% of the biologic-naive group.
During the 12-month study, 3.7% of patients developed a new comorbidity, the most common being anxiety, hypertension, PsA, depression, and hyperlipidemia.
The primary outcome in the study was the complete clearance rate – a PASI 100 response – at 6 months. It ranged from a high of 31% in patients with no baseline comorbid conditions to a low of 16.5% in those with three or more. The results were similar at 12 months.
Conversely, an inadequate therapeutic response as defined by a PASI 50 or less at 6 months occurred in 15% of psoriasis patients with no baseline comorbidities, 27% with one, 35% with two comorbid conditions, and 28% with three or more.
The major caveat regarding this study is that the observed association between comorbid conditions and complete clearance rates doesn’t prove causality, Mr. Ziegler noted.
The PSO-BIO-REAL study was sponsored by Amgen, AstraZeneca, and Leo Pharma. Mr. Ziegler is a Leo executive.
SOURCE: Ziegler F. EADV Congress, Abstract FC04.01.
REPORTING FROM THE EADV CONGRESS
Key clinical point: As the number of baseline comorbid conditions increases, the complete clearance rate in response to biologic agents for psoriasis falls.
Major finding: The complete clearance rate after 6 months of biologic therapy ranged from a high of 31% in patients with no baseline comorbid conditions to a low of 16.5% in those with three or more.
Study details: This multinational, prospective, observational, 12-month study included 846 patients initiating biologic therapy for moderate to severe psoriasis.
Disclosures: The PSO-BIO-REAL study was sponsored by Amgen, AstraZeneca, and Leo Pharma and was presented by a Leo executive.
Source: Ziegler F. EADV Congress, Abstract FC04.01.
Brodalumab raced past ustekinumab to PASI 100
PARIS – The interleukin-17 receptor inhibitor

That’s according to a post hoc pooled analysis of the phase 3 randomized AMAGINE-1 and -3 trials that Kristian Reich, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
Other interleukin-17 inhibitors have also outperformed ustekinumab (Stelara) in head-to-head, randomized trials. What’s unique about this new secondary analysis of the AMAGINE trials is the demonstration that the complete clearance rate – that is, 100% improvement in Psoriasis Area and Severity Index (PASI) – with brodalumab (Siliq) was consistent, regardless of a psoriasis patient’s prior treatment history, according to Dr. Reich, professor of dermatology at Georg-August-University in Göttingen, Germany, and a partner at the Dermatologikum Hamburg.
“I don’t want to niche brodalumab as a rescue drug; but if you need a response in a patient who has failed a biologic, then obviously, this is a pretty good choice,” he said.
Typically, psoriasis patients who have previously failed to respond favorably to a biologic agent have a substantially lower complete clearance rate when placed on another biologic than do those who are biologic naive or haven’t been on a nonbiologic systemic therapy.
“I think it’s interesting that there is very little impact of previous treatment response with regard to this analysis when it comes to brodalumab,” the dermatologist observed. “It goes down a little bit, but if you compare it to ustekinumab, you see a very good robustness despite previous therapy.”
His presentation focused on the 339 AMAGINE-2 or AMAGINE-3 participants randomized to brodalumab at the approved dose of 210 mg by subcutaneous injection every 2 weeks, or to subcutaneous ustekinumab at the approved dose of 45 mg or 90 mg, depending upon body weight, on day 1, week 4, and then every 12 weeks in the 52-week trials.
It took 14 weeks for 50% of patients assigned to brodalumab to achieve a PASI 100 response, and 44 weeks to accomplish the same in the ustekinumab group. At 52 weeks, the PASI 100 response rate was 76% for brodalumab and 52% for ustekinumab.
This was a competing-risk analysis – a methodology relatively new to dermatology – in which the coprimary endpoint was inadequate response to treatment, as defined by a static Physician’s Global Assessment score of 3 or more or two consecutive sPGAs of at least 2 over a 4-week interval at any point from week 16 on. The inadequate response rate was 20% in the brodalumab group and 40% with ustekinumab.
Looking in the brodalumab group at PASI 100 response rates in relation to prior treatments, the complete clearance rate at week 52 was 76% in those with no prior systemic treatment at study entry, 78% in those with a history of nonbiologic systemic treatment, 75% in patients who hadn’t experienced treatment failure when previously on another biologic agent, and 70% in those with a baseline history of failure on a different biologic.
The corresponding PASI 100 rates in the ustekinumab group were strikingly lower, at 58%, 55%, 41%, and 30%.
Leo Pharma funded Dr. Reich’s post hoc analysis; Leo markets brodalumab in Europe. Dr. Reich reported receiving research funding from and serving as a consultant to that pharmaceutical company and numerous others involved in developing new drugs for psoriasis and atopic dermatitis.
SOURCE: Reich K. EADV Congress, Abstract #FC03.06.
PARIS – The interleukin-17 receptor inhibitor
That’s according to a post hoc pooled analysis of the phase 3 randomized AMAGINE-1 and -3 trials that Kristian Reich, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
Other interleukin-17 inhibitors have also outperformed ustekinumab (Stelara) in head-to-head, randomized trials. What’s unique about this new secondary analysis of the AMAGINE trials is the demonstration that the complete clearance rate – that is, 100% improvement in Psoriasis Area and Severity Index (PASI) – with brodalumab (Siliq) was consistent, regardless of a psoriasis patient’s prior treatment history, according to Dr. Reich, professor of dermatology at Georg-August-University in Göttingen, Germany, and a partner at the Dermatologikum Hamburg.
“I don’t want to niche brodalumab as a rescue drug; but if you need a response in a patient who has failed a biologic, then obviously, this is a pretty good choice,” he said.
Typically, psoriasis patients who have previously failed to respond favorably to a biologic agent have a substantially lower complete clearance rate when placed on another biologic than do those who are biologic naive or haven’t been on a nonbiologic systemic therapy.
“I think it’s interesting that there is very little impact of previous treatment response with regard to this analysis when it comes to brodalumab,” the dermatologist observed. “It goes down a little bit, but if you compare it to ustekinumab, you see a very good robustness despite previous therapy.”
His presentation focused on the 339 AMAGINE-2 or AMAGINE-3 participants randomized to brodalumab at the approved dose of 210 mg by subcutaneous injection every 2 weeks, or to subcutaneous ustekinumab at the approved dose of 45 mg or 90 mg, depending upon body weight, on day 1, week 4, and then every 12 weeks in the 52-week trials.
It took 14 weeks for 50% of patients assigned to brodalumab to achieve a PASI 100 response, and 44 weeks to accomplish the same in the ustekinumab group. At 52 weeks, the PASI 100 response rate was 76% for brodalumab and 52% for ustekinumab.
This was a competing-risk analysis – a methodology relatively new to dermatology – in which the coprimary endpoint was inadequate response to treatment, as defined by a static Physician’s Global Assessment score of 3 or more or two consecutive sPGAs of at least 2 over a 4-week interval at any point from week 16 on. The inadequate response rate was 20% in the brodalumab group and 40% with ustekinumab.
Looking in the brodalumab group at PASI 100 response rates in relation to prior treatments, the complete clearance rate at week 52 was 76% in those with no prior systemic treatment at study entry, 78% in those with a history of nonbiologic systemic treatment, 75% in patients who hadn’t experienced treatment failure when previously on another biologic agent, and 70% in those with a baseline history of failure on a different biologic.
The corresponding PASI 100 rates in the ustekinumab group were strikingly lower, at 58%, 55%, 41%, and 30%.
Leo Pharma funded Dr. Reich’s post hoc analysis; Leo markets brodalumab in Europe. Dr. Reich reported receiving research funding from and serving as a consultant to that pharmaceutical company and numerous others involved in developing new drugs for psoriasis and atopic dermatitis.
SOURCE: Reich K. EADV Congress, Abstract #FC03.06.
PARIS – The interleukin-17 receptor inhibitor
That’s according to a post hoc pooled analysis of the phase 3 randomized AMAGINE-1 and -3 trials that Kristian Reich, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
Other interleukin-17 inhibitors have also outperformed ustekinumab (Stelara) in head-to-head, randomized trials. What’s unique about this new secondary analysis of the AMAGINE trials is the demonstration that the complete clearance rate – that is, 100% improvement in Psoriasis Area and Severity Index (PASI) – with brodalumab (Siliq) was consistent, regardless of a psoriasis patient’s prior treatment history, according to Dr. Reich, professor of dermatology at Georg-August-University in Göttingen, Germany, and a partner at the Dermatologikum Hamburg.
“I don’t want to niche brodalumab as a rescue drug; but if you need a response in a patient who has failed a biologic, then obviously, this is a pretty good choice,” he said.
Typically, psoriasis patients who have previously failed to respond favorably to a biologic agent have a substantially lower complete clearance rate when placed on another biologic than do those who are biologic naive or haven’t been on a nonbiologic systemic therapy.
“I think it’s interesting that there is very little impact of previous treatment response with regard to this analysis when it comes to brodalumab,” the dermatologist observed. “It goes down a little bit, but if you compare it to ustekinumab, you see a very good robustness despite previous therapy.”
His presentation focused on the 339 AMAGINE-2 or AMAGINE-3 participants randomized to brodalumab at the approved dose of 210 mg by subcutaneous injection every 2 weeks, or to subcutaneous ustekinumab at the approved dose of 45 mg or 90 mg, depending upon body weight, on day 1, week 4, and then every 12 weeks in the 52-week trials.
It took 14 weeks for 50% of patients assigned to brodalumab to achieve a PASI 100 response, and 44 weeks to accomplish the same in the ustekinumab group. At 52 weeks, the PASI 100 response rate was 76% for brodalumab and 52% for ustekinumab.
This was a competing-risk analysis – a methodology relatively new to dermatology – in which the coprimary endpoint was inadequate response to treatment, as defined by a static Physician’s Global Assessment score of 3 or more or two consecutive sPGAs of at least 2 over a 4-week interval at any point from week 16 on. The inadequate response rate was 20% in the brodalumab group and 40% with ustekinumab.
Looking in the brodalumab group at PASI 100 response rates in relation to prior treatments, the complete clearance rate at week 52 was 76% in those with no prior systemic treatment at study entry, 78% in those with a history of nonbiologic systemic treatment, 75% in patients who hadn’t experienced treatment failure when previously on another biologic agent, and 70% in those with a baseline history of failure on a different biologic.
The corresponding PASI 100 rates in the ustekinumab group were strikingly lower, at 58%, 55%, 41%, and 30%.
Leo Pharma funded Dr. Reich’s post hoc analysis; Leo markets brodalumab in Europe. Dr. Reich reported receiving research funding from and serving as a consultant to that pharmaceutical company and numerous others involved in developing new drugs for psoriasis and atopic dermatitis.
SOURCE: Reich K. EADV Congress, Abstract #FC03.06.
REPORTING FROM THE EADV CONGRESS
Key clinical point: Complete clearance rates in psoriasis patients on brodalumab were similar regardless of treatment history.
Major finding: Half of brodalumab-treated patients with moderate-to-severe psoriasis experienced complete clearance at 14 weeks; it took 44 weeks in patients assigned to ustekinumab.
Study details: This was a post hoc analysis of 52-week outcomes in more than 900 participants in the phase 3 AMAGINE-2 and AMAGINE-3 randomized head-to-head comparisons of brodalumab and ustekinumab.
Disclosures: Leo Pharma funded the post hoc analysis. The presenter reported receiving research funding from and serving as a consultant to that pharmaceutical company and numerous others involved in developing new drugs for psoriasis and atopic dermatitis.
Source: Reich K. EADV Congress, Abstract #FC03.06.
Adult atopic dermatitis is fraught with dermatologic comorbidities
PARIS – : that is, from alopecia areata to vitiligo, and points in between, according to a large German national study.

These dermatologic comorbidities are in addition to already well established strong associations between AD and allergic rhinitis and bronchial asthma, as well as the emerging evidence of increased risk for depression, sleep impairment, suicidality, and other nondermatologic conditions, Nicole Zander noted at the annual congress of the European Academy of Dermatology and Venereology.
“Atopic dermatitis patients seem to be at substantial risk for a variety of comorbidities. It’s important to recognize the whole spectrum of comorbidities as a prerequisite for provision of patient-centered care,” said Ms. Zander, a research associate at the University of Hamburg Institute for Health Services Research in Dermatology and Nursing.
This is a priority in Germany, where the prevalence of AD has doubled or even tripled in the last 3 decades, she added.
She presented a snapshot of the comorbid conditions associated with AD based upon two German large national datasets: Structured skin screening examinations conducted by dermatologists in 162,269 adults working in more than 500 German companies and national medical claims data for 1,349,671 adults aged 18 years and up.
The annual point prevalence for adult AD was 1.4% in the occupational screening, while in the claims dataset it was 3.7%. The true prevalence, as well as that of the dermatologic comorbidities, probably lies somewhere in between, since both of these enormous datasets have their limitations. The claims dataset may contain coding errors, plus many of the skin disorders entombed in that database were diagnosed by nondermatologists. And the occupational dataset is vulnerable to what epidemiologists call the healthy worker effect: some people with severe AD might be absent from work or on disability, with a resultant lower point prevalence of the disorder in the workplace, she explained.
Both datasets documented a clear decline in the prevalence of AD with advancing age. In the medical claims database, for example, the prevalence was 6.6% among 18- and 19-year-olds, 5.4% for patients in their 20s, 3.9% for those in their 30s, 3.4% for those in their 40s, 3.0% for patients in their 50s, 2.7% for those in their 70s, and 2.4% for patients in their 80s.
This is the most likely explanation for the significantly reduced risks of stroke, hypertension, diabetes, hyperlipidemia, and ischemic heart disease seen in AD patients in the claims dataset. These are all conditions that become much more common with advancing age, whereas AD is skewed toward a younger population. And for purposes of this study the data weren’t age adjusted, Ms. Zander observed.
Ms. Zander reported having no financial conflicts regarding her study, conducted without commercial support.
PARIS – : that is, from alopecia areata to vitiligo, and points in between, according to a large German national study.
These dermatologic comorbidities are in addition to already well established strong associations between AD and allergic rhinitis and bronchial asthma, as well as the emerging evidence of increased risk for depression, sleep impairment, suicidality, and other nondermatologic conditions, Nicole Zander noted at the annual congress of the European Academy of Dermatology and Venereology.
“Atopic dermatitis patients seem to be at substantial risk for a variety of comorbidities. It’s important to recognize the whole spectrum of comorbidities as a prerequisite for provision of patient-centered care,” said Ms. Zander, a research associate at the University of Hamburg Institute for Health Services Research in Dermatology and Nursing.
This is a priority in Germany, where the prevalence of AD has doubled or even tripled in the last 3 decades, she added.
She presented a snapshot of the comorbid conditions associated with AD based upon two German large national datasets: Structured skin screening examinations conducted by dermatologists in 162,269 adults working in more than 500 German companies and national medical claims data for 1,349,671 adults aged 18 years and up.
The annual point prevalence for adult AD was 1.4% in the occupational screening, while in the claims dataset it was 3.7%. The true prevalence, as well as that of the dermatologic comorbidities, probably lies somewhere in between, since both of these enormous datasets have their limitations. The claims dataset may contain coding errors, plus many of the skin disorders entombed in that database were diagnosed by nondermatologists. And the occupational dataset is vulnerable to what epidemiologists call the healthy worker effect: some people with severe AD might be absent from work or on disability, with a resultant lower point prevalence of the disorder in the workplace, she explained.
Both datasets documented a clear decline in the prevalence of AD with advancing age. In the medical claims database, for example, the prevalence was 6.6% among 18- and 19-year-olds, 5.4% for patients in their 20s, 3.9% for those in their 30s, 3.4% for those in their 40s, 3.0% for patients in their 50s, 2.7% for those in their 70s, and 2.4% for patients in their 80s.
This is the most likely explanation for the significantly reduced risks of stroke, hypertension, diabetes, hyperlipidemia, and ischemic heart disease seen in AD patients in the claims dataset. These are all conditions that become much more common with advancing age, whereas AD is skewed toward a younger population. And for purposes of this study the data weren’t age adjusted, Ms. Zander observed.
Ms. Zander reported having no financial conflicts regarding her study, conducted without commercial support.
PARIS – : that is, from alopecia areata to vitiligo, and points in between, according to a large German national study.
These dermatologic comorbidities are in addition to already well established strong associations between AD and allergic rhinitis and bronchial asthma, as well as the emerging evidence of increased risk for depression, sleep impairment, suicidality, and other nondermatologic conditions, Nicole Zander noted at the annual congress of the European Academy of Dermatology and Venereology.
“Atopic dermatitis patients seem to be at substantial risk for a variety of comorbidities. It’s important to recognize the whole spectrum of comorbidities as a prerequisite for provision of patient-centered care,” said Ms. Zander, a research associate at the University of Hamburg Institute for Health Services Research in Dermatology and Nursing.
This is a priority in Germany, where the prevalence of AD has doubled or even tripled in the last 3 decades, she added.
She presented a snapshot of the comorbid conditions associated with AD based upon two German large national datasets: Structured skin screening examinations conducted by dermatologists in 162,269 adults working in more than 500 German companies and national medical claims data for 1,349,671 adults aged 18 years and up.
The annual point prevalence for adult AD was 1.4% in the occupational screening, while in the claims dataset it was 3.7%. The true prevalence, as well as that of the dermatologic comorbidities, probably lies somewhere in between, since both of these enormous datasets have their limitations. The claims dataset may contain coding errors, plus many of the skin disorders entombed in that database were diagnosed by nondermatologists. And the occupational dataset is vulnerable to what epidemiologists call the healthy worker effect: some people with severe AD might be absent from work or on disability, with a resultant lower point prevalence of the disorder in the workplace, she explained.
Both datasets documented a clear decline in the prevalence of AD with advancing age. In the medical claims database, for example, the prevalence was 6.6% among 18- and 19-year-olds, 5.4% for patients in their 20s, 3.9% for those in their 30s, 3.4% for those in their 40s, 3.0% for patients in their 50s, 2.7% for those in their 70s, and 2.4% for patients in their 80s.
This is the most likely explanation for the significantly reduced risks of stroke, hypertension, diabetes, hyperlipidemia, and ischemic heart disease seen in AD patients in the claims dataset. These are all conditions that become much more common with advancing age, whereas AD is skewed toward a younger population. And for purposes of this study the data weren’t age adjusted, Ms. Zander observed.
Ms. Zander reported having no financial conflicts regarding her study, conducted without commercial support.
REPORTING FROM THE EADV CONGRESS
Key clinical point: Adult atopic dermatitis patients have higher prevalence of psoriasis, alopecia areata, vitiligo, and other dermatologic disorders.
Major finding: The risk of contact dermatitis is increased 3.4- to 5.6-fold.
Study details: This German population-based study used both a medical claims database including more than 1.3 million adults and dermatologist-conducted occupational skin screening in more than 162,000 workers.
Disclosures: The study presenter reported having no financial conflicts regarding her study, conducted without commercial support.
Danish study finds reassuring data on pregnancy outcomes in atopic dermatitis patients
PARIS – over an 18-year period, which showed no increased risk of pregnancy and birth problems other than modestly increased risks of premature rupture of membranes and neonatal staphylococcal septicemia, according to Jacob P. Thyssen, MD, PhD.

At a session of the European Task Force of Atopic Dermatitis held in conjunction with the annual congress of the European Academy of Dermatology and Venereology, he presented a case control study of 10,668 births to Danish women with atopic dermatitis (AD) during 1997-2014. They were matched 1:10 by age, parity, and birth year to mothers without AD.
The risk of premature rupture of membranes was 15% higher in mothers with AD. And while the increased relative risk of neonatal staphylococcal septicemia was more substantial – a 145% increase – this was in fact a rare complication, observed Dr. Thyssen, a dermatologist at the University of Copenhagen.
There was no significant difference between women with or without AD in rates of preeclampsia, prematurity, pregnancy-induced hypertension, placenta previa, placental abruption, neonatal nonstaphylococcal septicemia, or other complications. The two groups had a similar number of visits to physicians and midwives during pregnancy.
Moreover, although the body mass index was similar in women with or without AD, the risk of gestational diabetes in women with the disease was significantly reduced by 21%; their risk of having a large-for-gestational-age baby with a birth weight of 4,500 g or more was also significantly lower than in controls.
Women received less treatment for AD during their pregnancy than they did beforehand. While pregnant, their disease was managed predominantly with topical corticosteroids and UV therapy. There was very little use of superpotent topical steroids, topical calcineurin inhibitors, or immunosuppressants, although 10% of pregnant women received systemic corticosteroids for their AD.
Dr. Thyssen reported serving as a scientific adviser and paid speaker for Leo Pharma, Roche, Eli Lilly, and Sanofi-Genzyme, although this study was conducted without commercial support.
PARIS – over an 18-year period, which showed no increased risk of pregnancy and birth problems other than modestly increased risks of premature rupture of membranes and neonatal staphylococcal septicemia, according to Jacob P. Thyssen, MD, PhD.
At a session of the European Task Force of Atopic Dermatitis held in conjunction with the annual congress of the European Academy of Dermatology and Venereology, he presented a case control study of 10,668 births to Danish women with atopic dermatitis (AD) during 1997-2014. They were matched 1:10 by age, parity, and birth year to mothers without AD.
The risk of premature rupture of membranes was 15% higher in mothers with AD. And while the increased relative risk of neonatal staphylococcal septicemia was more substantial – a 145% increase – this was in fact a rare complication, observed Dr. Thyssen, a dermatologist at the University of Copenhagen.
There was no significant difference between women with or without AD in rates of preeclampsia, prematurity, pregnancy-induced hypertension, placenta previa, placental abruption, neonatal nonstaphylococcal septicemia, or other complications. The two groups had a similar number of visits to physicians and midwives during pregnancy.
Moreover, although the body mass index was similar in women with or without AD, the risk of gestational diabetes in women with the disease was significantly reduced by 21%; their risk of having a large-for-gestational-age baby with a birth weight of 4,500 g or more was also significantly lower than in controls.
Women received less treatment for AD during their pregnancy than they did beforehand. While pregnant, their disease was managed predominantly with topical corticosteroids and UV therapy. There was very little use of superpotent topical steroids, topical calcineurin inhibitors, or immunosuppressants, although 10% of pregnant women received systemic corticosteroids for their AD.
Dr. Thyssen reported serving as a scientific adviser and paid speaker for Leo Pharma, Roche, Eli Lilly, and Sanofi-Genzyme, although this study was conducted without commercial support.
PARIS – over an 18-year period, which showed no increased risk of pregnancy and birth problems other than modestly increased risks of premature rupture of membranes and neonatal staphylococcal septicemia, according to Jacob P. Thyssen, MD, PhD.
At a session of the European Task Force of Atopic Dermatitis held in conjunction with the annual congress of the European Academy of Dermatology and Venereology, he presented a case control study of 10,668 births to Danish women with atopic dermatitis (AD) during 1997-2014. They were matched 1:10 by age, parity, and birth year to mothers without AD.
The risk of premature rupture of membranes was 15% higher in mothers with AD. And while the increased relative risk of neonatal staphylococcal septicemia was more substantial – a 145% increase – this was in fact a rare complication, observed Dr. Thyssen, a dermatologist at the University of Copenhagen.
There was no significant difference between women with or without AD in rates of preeclampsia, prematurity, pregnancy-induced hypertension, placenta previa, placental abruption, neonatal nonstaphylococcal septicemia, or other complications. The two groups had a similar number of visits to physicians and midwives during pregnancy.
Moreover, although the body mass index was similar in women with or without AD, the risk of gestational diabetes in women with the disease was significantly reduced by 21%; their risk of having a large-for-gestational-age baby with a birth weight of 4,500 g or more was also significantly lower than in controls.
Women received less treatment for AD during their pregnancy than they did beforehand. While pregnant, their disease was managed predominantly with topical corticosteroids and UV therapy. There was very little use of superpotent topical steroids, topical calcineurin inhibitors, or immunosuppressants, although 10% of pregnant women received systemic corticosteroids for their AD.
Dr. Thyssen reported serving as a scientific adviser and paid speaker for Leo Pharma, Roche, Eli Lilly, and Sanofi-Genzyme, although this study was conducted without commercial support.
REPORTING FROM THE EADV CONGRESS
Key clinical point: Birth complications are uncommon for women with atopic dermatitis in pregnancy.
Major finding: The risk of premature rupture of membranes was increased by 15% in women with atopic dermatitis in pregnancy, but their risk of gestational diabetes was reduced by 21%.
Study details: This case control study included 10,668 births to Danish women with atopic dermatitis and 10 times as many matched controls without the disease.
Disclosures: The study presenter reported serving as a scientific adviser and paid speaker for Leo Pharma, Roche, Eli Lilly, and Sanofi-Genzyme, although this study was conducted without commercial support.
Difelikefalin shows promise for hemodialysis-associated itch
PARIS – while achieving across-the-board clinically meaningful improvements in quality of life measures in patients with hemodialysis-associated pruritus in a phase 2 study, Frédérique Menzaghi, PhD, reported at the annual congress of the European Academy of Dermatology and Venereology.

At present there is no approved medication in the United States or Europe for the often intense itching associated with chronic kidney disease. Off-label treatments have limited efficacy.
Dr. Menzaghi is senior vice president for research and development at Cara Therapeutics, which is developing difelikefalin.
More than half – 60% to 70% – of patients on hemodialysis for end-stage renal disease experience chronic pruritus, as do a smaller proportion of individuals with chronic kidney disease (CKD) not requiring dialysis. CKD-associated pruritus is a day-and-night itch that makes life miserable for affected patients. Not only must they endure the predictable complications of skin excoriation, including impetigo, ulcerations, papules, and prurigo nodularis, but they also experience sleep disruption, depressed mood, and a 10%-20% increased mortality risk compared with CKD patients without pruritus.
Difelikefalin is a potent and selective peripheral kappa opioid receptor agonist that doesn’t activate mu or delta opioid receptors. It’s a synthetic drug that mimics endogenous dynorphin. Its key attribute is that it doesn’t cross the blood/brain barrier, so it doesn’t pose a risk for adverse events caused by activation of central opioid receptors. Difelikefalin has two mechanisms of action in CKD-associated pruritus: an antipruritic effect due to inhibition of ion channels responsible for afferent peripheral nerve activity; and an anti-inflammatory effect mediated by activation of kappa opioid receptors expressed by immune system cells, according to Dr. Menzaghi.
She reported on 174 hemodialysis patients with moderate to severe CKD-associated pruritus who were randomized to a double-blind, phase 2, dose-ranging study featuring an intravenous bolus of difelikefalin at 0.5, 1.0, or 1.5 mcg/kg or placebo given immediately after each of the thrice-weekly hemodialysis sessions for 8 weeks.
An oral formulation of difelikefalin is also under investigation for treatment of CKD-associated pruritus. The IV version is being developed for hemodialysis patients because difelikefalin is renally excreted.
“We’re taking advantage of the fact that their kidneys aren’t working. The drug stays in the system until the next dialysis because it can’t be eliminated. It’s quite convenient for these patients,” she explained.
The primary endpoint in the phase 2 study was change from baseline through week 8 in the weekly average of a patient’s daily self-rated 0-10 worst itching intensity numeric rating scale (NRS) scores. All participants had to have a baseline NRS score of at least 4, considered the lower threshold for moderate itch. In fact, the mean baseline score was 6.7-7.1 in the four study arms.
The results
Sixty-four percent of patients on difelikefalin 0.5 mcg/kg – the most effective dose – experienced at least a 3-point reduction, compared with 29% of placebo-treated controls. And a 4-point or greater reduction in NRS from baseline was documented in 51% of patients on difelikefalin at 0.5 mcg/kg, compared with 24% of controls.
Although a 4-point difference is widely considered to represent clinically meaningful improvement in atopic dermatitis studies, Dr. Menzaghi said psychometric analyses of the difelikefalin trial data indicated that a 3-point or greater improvement in NRS score was associated with clinically meaningful change.
“Our data suggest that a 4-point change may not be generalizable to all conditions,” she said.
Hemodialysis patients with severe baseline itch typically improved to moderate itch on difelikefalin, while those with baseline moderate itch – that is, an NRS of 4-6 – dropped down to mild or no itch while on the drug.
“But that’s just a number. The question is, is that really clinically meaningful?” Dr. Menzaghi noted.
The answer, she continued, is yes. A high correlation was seen between reduction in itch intensity and improvement in quality of life. Scores on the 5-D Itch Scale and Skindex-10 improved two- to threefold more in the difelikefalin 0.5-mcg group than in controls. So did scores on the 12-item Medical Outcomes Study Sleep Scale assessing sleep restlessness, awakening during sleep, and trouble falling asleep.
“We think these results suggest that peripheral kappa opioid receptors play an integral role in the modulation of itch signals and represent a primary target for the development of antipruritic agents,” said Dr. Menzaghi.
Indeed, a phase 3 randomized trial of difelikefalin 0.5 mcg/kg versus placebo in 350 hemodialysis patients with CKD-associated itch is ongoing in the United States, Europe, Australia, and Korea. Also ongoing is a phase 2 U.S. study of oral difelikefalin in patients with CKD-associated pruritus, many of whom are not on hemodialysis. In January, the company announced that enrollment in a phase 3 U.S. study of difelikefalin injection (0.5 mcg/kg) in hemodialysis patients with moderate to severe CKD-associated pruritus had been completed. The trials are funded by Cara Therapeutics.
SOURCE: Menzaghi F. EADV Congress, Abstract FC0.4.7.
PARIS – while achieving across-the-board clinically meaningful improvements in quality of life measures in patients with hemodialysis-associated pruritus in a phase 2 study, Frédérique Menzaghi, PhD, reported at the annual congress of the European Academy of Dermatology and Venereology.
At present there is no approved medication in the United States or Europe for the often intense itching associated with chronic kidney disease. Off-label treatments have limited efficacy.
Dr. Menzaghi is senior vice president for research and development at Cara Therapeutics, which is developing difelikefalin.
More than half – 60% to 70% – of patients on hemodialysis for end-stage renal disease experience chronic pruritus, as do a smaller proportion of individuals with chronic kidney disease (CKD) not requiring dialysis. CKD-associated pruritus is a day-and-night itch that makes life miserable for affected patients. Not only must they endure the predictable complications of skin excoriation, including impetigo, ulcerations, papules, and prurigo nodularis, but they also experience sleep disruption, depressed mood, and a 10%-20% increased mortality risk compared with CKD patients without pruritus.
Difelikefalin is a potent and selective peripheral kappa opioid receptor agonist that doesn’t activate mu or delta opioid receptors. It’s a synthetic drug that mimics endogenous dynorphin. Its key attribute is that it doesn’t cross the blood/brain barrier, so it doesn’t pose a risk for adverse events caused by activation of central opioid receptors. Difelikefalin has two mechanisms of action in CKD-associated pruritus: an antipruritic effect due to inhibition of ion channels responsible for afferent peripheral nerve activity; and an anti-inflammatory effect mediated by activation of kappa opioid receptors expressed by immune system cells, according to Dr. Menzaghi.
She reported on 174 hemodialysis patients with moderate to severe CKD-associated pruritus who were randomized to a double-blind, phase 2, dose-ranging study featuring an intravenous bolus of difelikefalin at 0.5, 1.0, or 1.5 mcg/kg or placebo given immediately after each of the thrice-weekly hemodialysis sessions for 8 weeks.
An oral formulation of difelikefalin is also under investigation for treatment of CKD-associated pruritus. The IV version is being developed for hemodialysis patients because difelikefalin is renally excreted.
“We’re taking advantage of the fact that their kidneys aren’t working. The drug stays in the system until the next dialysis because it can’t be eliminated. It’s quite convenient for these patients,” she explained.
The primary endpoint in the phase 2 study was change from baseline through week 8 in the weekly average of a patient’s daily self-rated 0-10 worst itching intensity numeric rating scale (NRS) scores. All participants had to have a baseline NRS score of at least 4, considered the lower threshold for moderate itch. In fact, the mean baseline score was 6.7-7.1 in the four study arms.
The results
Sixty-four percent of patients on difelikefalin 0.5 mcg/kg – the most effective dose – experienced at least a 3-point reduction, compared with 29% of placebo-treated controls. And a 4-point or greater reduction in NRS from baseline was documented in 51% of patients on difelikefalin at 0.5 mcg/kg, compared with 24% of controls.
Although a 4-point difference is widely considered to represent clinically meaningful improvement in atopic dermatitis studies, Dr. Menzaghi said psychometric analyses of the difelikefalin trial data indicated that a 3-point or greater improvement in NRS score was associated with clinically meaningful change.
“Our data suggest that a 4-point change may not be generalizable to all conditions,” she said.
Hemodialysis patients with severe baseline itch typically improved to moderate itch on difelikefalin, while those with baseline moderate itch – that is, an NRS of 4-6 – dropped down to mild or no itch while on the drug.
“But that’s just a number. The question is, is that really clinically meaningful?” Dr. Menzaghi noted.
The answer, she continued, is yes. A high correlation was seen between reduction in itch intensity and improvement in quality of life. Scores on the 5-D Itch Scale and Skindex-10 improved two- to threefold more in the difelikefalin 0.5-mcg group than in controls. So did scores on the 12-item Medical Outcomes Study Sleep Scale assessing sleep restlessness, awakening during sleep, and trouble falling asleep.
“We think these results suggest that peripheral kappa opioid receptors play an integral role in the modulation of itch signals and represent a primary target for the development of antipruritic agents,” said Dr. Menzaghi.
Indeed, a phase 3 randomized trial of difelikefalin 0.5 mcg/kg versus placebo in 350 hemodialysis patients with CKD-associated itch is ongoing in the United States, Europe, Australia, and Korea. Also ongoing is a phase 2 U.S. study of oral difelikefalin in patients with CKD-associated pruritus, many of whom are not on hemodialysis. In January, the company announced that enrollment in a phase 3 U.S. study of difelikefalin injection (0.5 mcg/kg) in hemodialysis patients with moderate to severe CKD-associated pruritus had been completed. The trials are funded by Cara Therapeutics.
SOURCE: Menzaghi F. EADV Congress, Abstract FC0.4.7.
PARIS – while achieving across-the-board clinically meaningful improvements in quality of life measures in patients with hemodialysis-associated pruritus in a phase 2 study, Frédérique Menzaghi, PhD, reported at the annual congress of the European Academy of Dermatology and Venereology.
At present there is no approved medication in the United States or Europe for the often intense itching associated with chronic kidney disease. Off-label treatments have limited efficacy.
Dr. Menzaghi is senior vice president for research and development at Cara Therapeutics, which is developing difelikefalin.
More than half – 60% to 70% – of patients on hemodialysis for end-stage renal disease experience chronic pruritus, as do a smaller proportion of individuals with chronic kidney disease (CKD) not requiring dialysis. CKD-associated pruritus is a day-and-night itch that makes life miserable for affected patients. Not only must they endure the predictable complications of skin excoriation, including impetigo, ulcerations, papules, and prurigo nodularis, but they also experience sleep disruption, depressed mood, and a 10%-20% increased mortality risk compared with CKD patients without pruritus.
Difelikefalin is a potent and selective peripheral kappa opioid receptor agonist that doesn’t activate mu or delta opioid receptors. It’s a synthetic drug that mimics endogenous dynorphin. Its key attribute is that it doesn’t cross the blood/brain barrier, so it doesn’t pose a risk for adverse events caused by activation of central opioid receptors. Difelikefalin has two mechanisms of action in CKD-associated pruritus: an antipruritic effect due to inhibition of ion channels responsible for afferent peripheral nerve activity; and an anti-inflammatory effect mediated by activation of kappa opioid receptors expressed by immune system cells, according to Dr. Menzaghi.
She reported on 174 hemodialysis patients with moderate to severe CKD-associated pruritus who were randomized to a double-blind, phase 2, dose-ranging study featuring an intravenous bolus of difelikefalin at 0.5, 1.0, or 1.5 mcg/kg or placebo given immediately after each of the thrice-weekly hemodialysis sessions for 8 weeks.
An oral formulation of difelikefalin is also under investigation for treatment of CKD-associated pruritus. The IV version is being developed for hemodialysis patients because difelikefalin is renally excreted.
“We’re taking advantage of the fact that their kidneys aren’t working. The drug stays in the system until the next dialysis because it can’t be eliminated. It’s quite convenient for these patients,” she explained.
The primary endpoint in the phase 2 study was change from baseline through week 8 in the weekly average of a patient’s daily self-rated 0-10 worst itching intensity numeric rating scale (NRS) scores. All participants had to have a baseline NRS score of at least 4, considered the lower threshold for moderate itch. In fact, the mean baseline score was 6.7-7.1 in the four study arms.
The results
Sixty-four percent of patients on difelikefalin 0.5 mcg/kg – the most effective dose – experienced at least a 3-point reduction, compared with 29% of placebo-treated controls. And a 4-point or greater reduction in NRS from baseline was documented in 51% of patients on difelikefalin at 0.5 mcg/kg, compared with 24% of controls.
Although a 4-point difference is widely considered to represent clinically meaningful improvement in atopic dermatitis studies, Dr. Menzaghi said psychometric analyses of the difelikefalin trial data indicated that a 3-point or greater improvement in NRS score was associated with clinically meaningful change.
“Our data suggest that a 4-point change may not be generalizable to all conditions,” she said.
Hemodialysis patients with severe baseline itch typically improved to moderate itch on difelikefalin, while those with baseline moderate itch – that is, an NRS of 4-6 – dropped down to mild or no itch while on the drug.
“But that’s just a number. The question is, is that really clinically meaningful?” Dr. Menzaghi noted.
The answer, she continued, is yes. A high correlation was seen between reduction in itch intensity and improvement in quality of life. Scores on the 5-D Itch Scale and Skindex-10 improved two- to threefold more in the difelikefalin 0.5-mcg group than in controls. So did scores on the 12-item Medical Outcomes Study Sleep Scale assessing sleep restlessness, awakening during sleep, and trouble falling asleep.
“We think these results suggest that peripheral kappa opioid receptors play an integral role in the modulation of itch signals and represent a primary target for the development of antipruritic agents,” said Dr. Menzaghi.
Indeed, a phase 3 randomized trial of difelikefalin 0.5 mcg/kg versus placebo in 350 hemodialysis patients with CKD-associated itch is ongoing in the United States, Europe, Australia, and Korea. Also ongoing is a phase 2 U.S. study of oral difelikefalin in patients with CKD-associated pruritus, many of whom are not on hemodialysis. In January, the company announced that enrollment in a phase 3 U.S. study of difelikefalin injection (0.5 mcg/kg) in hemodialysis patients with moderate to severe CKD-associated pruritus had been completed. The trials are funded by Cara Therapeutics.
SOURCE: Menzaghi F. EADV Congress, Abstract FC0.4.7.
REPORTING FROM THE EADV CONGRESS
Key clinical point: Moderate to severe chronic itching associated with chronic kidney disease is a common and underrecognized problem with a huge quality of life impact.
Major finding: Sixty-four percent of hemodialysis patients on difelikefalin 0.5 mcg/kg experienced at least a 3-point reduction on a 0-10 worst daily itch numeric rating scale, compared with 29% of placebo-treated controls.
Study details: This phase 2, multicenter, 8-week, double-blind study comprised 174 patients with moderate to severe hemodialysis-related itching.
Disclosures: The study was sponsored by Cara Therapeutics and presented by a company officer.
Source: Menzaghi F. EADV Congress, Abstract FC0.4.7.
Longterm maintenance of PASI 75 responses observed with tildrakizumab
PARIS – Dermatologists are likely to do a double-take when they see the long-term efficacy and safety data for tildrakizumab (Ilumya), a high-affinity humanized monoclonal antibody targeting interleukin-23 p19, relative to the performance of older and more familiar biologic agents with other targets in psoriasis, Diamant Thaçi, MD, predicted at the annual congress of the European Academy of Dermatology and Venereology.

“The time to relapse [off tildrakizumab] is very different from what we are used to with other biologics; for example, the tumor necrosis factor inhibitors,” observed Dr. Thaçi, professor and chairman of the department of dermatology at the University of Lübeck (Germany).
He presented the 148-week, follow-up results of a pooled analysis of the open-label extension studies of reSURFACE 1 and reSURFACE 2, two pivotal phase 3 randomized double-blind international trials of 1,862 patients with moderate to severe chronic plaque psoriasis. The primary outcomes through week 12, which were instrumental in gaining marketing approval for tildrakizumab for treating psoriasis in 2018 from the Food and Drug Administration and the European Medicines Agency, have been published in the Lancet (2017 Jul 15;390[10091]:276-88).
Dr. Thaçi’s analysis of the 148-week outcomes was restricted to the patients who had at least a 75% improvement from baseline in Psoriasis Area and Severity Index scores (PASI 75) at week 28. Nearly 80% of patients on tildrakizumab reached that threshold at week 28 in reSURFACE 1, as did 73% in reSURFACE 2.
The question asked in the extension study was, How do responders to tildrakizumab at 28 weeks fare after nearly 3 years on the drug? And the answer was: very well. Maintenance of at least a PASI 75 response was observed at 148 weeks in 91% of patients on tildrakizumab at the approved 100-mg dose and 92% of those on the 200-mg dose. The FDA-approved regimen is 100 mg by subcutaneous injection at weeks 0 and 4, and then every 12 weeks after that.
An intriguing feature of reSURFACE 1 was that a subset of PASI 75 responders at week 28 got taken off tildrakizumab at that point and switched to double-blind placebo, then restarted on their earlier dose of tildrakizumab upon relapse, which was defined as loss of at least 50% of the achieved on-drug PASI improvement.
At week 64, fully 48 weeks after their last dose of tildrakizumab, the relapse rate was 54% in the group formerly on 100 mg of tildrakizumab and slightly better at 47% in those formerly on 200 mg. The median time to relapse was 226 days in the 100-mg group and 258 days in the higher-dose arm. Those are exceptionally long times to relapse, and it’s useful information to file away in the event a psoriasis patient needs to discontinue biologic therapy for a period of time, Dr. Thaçi observed.
At week 64 – again, off active treatment since week 16 – 63% of the tildrakizumab 100-mg group had lost their previous PASI 75 response, as had 52% who were formerly on tildrakizumab at 200 mg.
The long-term safety profile of tildrakizumab paralleled that of placebo. For example, the exposure-adjusted adverse event rates of serious infections and major adverse cardiovascular events were closely similar in the placebo, tildrakizumab 100 mg, and tildrakizumab 200 mg groups.
There were two notable between-group differences in adverse events of interest: injection site reactions occurred at a rate of 5.36 per 100 person-years with placebo, compared with 1.94 and 2.3 per 100 person-years with tildrakizumab at 100 and 200 mg, respectively; and the incidence of nonmelanoma skin cancer was 0.97 cases per 100 person-years in the placebo arm, versus 0.5 and 0.49 cases per 100 person-years in the two tildrakizumab arms.
Dr. Thaçi did not present PASI 90 response outcomes because, at the time the reSURFACE trials were planned, PASI 75 was considered state of the art. The PASI 90 data are still being crunched but will be available soon. The 4- and 5-year follow-up data from the long-term extension studies are also on their way.
The reSURFACE 1 and reSURFACE 2 trials and their extension studies were funded by Sun Pharma and Merck. Dr. Thaçi reported receiving research grants from and serving as a consultant and paid scientific advisor to those pharmaceutical companies and more than a dozen others.
PARIS – Dermatologists are likely to do a double-take when they see the long-term efficacy and safety data for tildrakizumab (Ilumya), a high-affinity humanized monoclonal antibody targeting interleukin-23 p19, relative to the performance of older and more familiar biologic agents with other targets in psoriasis, Diamant Thaçi, MD, predicted at the annual congress of the European Academy of Dermatology and Venereology.
“The time to relapse [off tildrakizumab] is very different from what we are used to with other biologics; for example, the tumor necrosis factor inhibitors,” observed Dr. Thaçi, professor and chairman of the department of dermatology at the University of Lübeck (Germany).
He presented the 148-week, follow-up results of a pooled analysis of the open-label extension studies of reSURFACE 1 and reSURFACE 2, two pivotal phase 3 randomized double-blind international trials of 1,862 patients with moderate to severe chronic plaque psoriasis. The primary outcomes through week 12, which were instrumental in gaining marketing approval for tildrakizumab for treating psoriasis in 2018 from the Food and Drug Administration and the European Medicines Agency, have been published in the Lancet (2017 Jul 15;390[10091]:276-88).
Dr. Thaçi’s analysis of the 148-week outcomes was restricted to the patients who had at least a 75% improvement from baseline in Psoriasis Area and Severity Index scores (PASI 75) at week 28. Nearly 80% of patients on tildrakizumab reached that threshold at week 28 in reSURFACE 1, as did 73% in reSURFACE 2.
The question asked in the extension study was, How do responders to tildrakizumab at 28 weeks fare after nearly 3 years on the drug? And the answer was: very well. Maintenance of at least a PASI 75 response was observed at 148 weeks in 91% of patients on tildrakizumab at the approved 100-mg dose and 92% of those on the 200-mg dose. The FDA-approved regimen is 100 mg by subcutaneous injection at weeks 0 and 4, and then every 12 weeks after that.
An intriguing feature of reSURFACE 1 was that a subset of PASI 75 responders at week 28 got taken off tildrakizumab at that point and switched to double-blind placebo, then restarted on their earlier dose of tildrakizumab upon relapse, which was defined as loss of at least 50% of the achieved on-drug PASI improvement.
At week 64, fully 48 weeks after their last dose of tildrakizumab, the relapse rate was 54% in the group formerly on 100 mg of tildrakizumab and slightly better at 47% in those formerly on 200 mg. The median time to relapse was 226 days in the 100-mg group and 258 days in the higher-dose arm. Those are exceptionally long times to relapse, and it’s useful information to file away in the event a psoriasis patient needs to discontinue biologic therapy for a period of time, Dr. Thaçi observed.
At week 64 – again, off active treatment since week 16 – 63% of the tildrakizumab 100-mg group had lost their previous PASI 75 response, as had 52% who were formerly on tildrakizumab at 200 mg.
The long-term safety profile of tildrakizumab paralleled that of placebo. For example, the exposure-adjusted adverse event rates of serious infections and major adverse cardiovascular events were closely similar in the placebo, tildrakizumab 100 mg, and tildrakizumab 200 mg groups.
There were two notable between-group differences in adverse events of interest: injection site reactions occurred at a rate of 5.36 per 100 person-years with placebo, compared with 1.94 and 2.3 per 100 person-years with tildrakizumab at 100 and 200 mg, respectively; and the incidence of nonmelanoma skin cancer was 0.97 cases per 100 person-years in the placebo arm, versus 0.5 and 0.49 cases per 100 person-years in the two tildrakizumab arms.
Dr. Thaçi did not present PASI 90 response outcomes because, at the time the reSURFACE trials were planned, PASI 75 was considered state of the art. The PASI 90 data are still being crunched but will be available soon. The 4- and 5-year follow-up data from the long-term extension studies are also on their way.
The reSURFACE 1 and reSURFACE 2 trials and their extension studies were funded by Sun Pharma and Merck. Dr. Thaçi reported receiving research grants from and serving as a consultant and paid scientific advisor to those pharmaceutical companies and more than a dozen others.
PARIS – Dermatologists are likely to do a double-take when they see the long-term efficacy and safety data for tildrakizumab (Ilumya), a high-affinity humanized monoclonal antibody targeting interleukin-23 p19, relative to the performance of older and more familiar biologic agents with other targets in psoriasis, Diamant Thaçi, MD, predicted at the annual congress of the European Academy of Dermatology and Venereology.
“The time to relapse [off tildrakizumab] is very different from what we are used to with other biologics; for example, the tumor necrosis factor inhibitors,” observed Dr. Thaçi, professor and chairman of the department of dermatology at the University of Lübeck (Germany).
He presented the 148-week, follow-up results of a pooled analysis of the open-label extension studies of reSURFACE 1 and reSURFACE 2, two pivotal phase 3 randomized double-blind international trials of 1,862 patients with moderate to severe chronic plaque psoriasis. The primary outcomes through week 12, which were instrumental in gaining marketing approval for tildrakizumab for treating psoriasis in 2018 from the Food and Drug Administration and the European Medicines Agency, have been published in the Lancet (2017 Jul 15;390[10091]:276-88).
Dr. Thaçi’s analysis of the 148-week outcomes was restricted to the patients who had at least a 75% improvement from baseline in Psoriasis Area and Severity Index scores (PASI 75) at week 28. Nearly 80% of patients on tildrakizumab reached that threshold at week 28 in reSURFACE 1, as did 73% in reSURFACE 2.
The question asked in the extension study was, How do responders to tildrakizumab at 28 weeks fare after nearly 3 years on the drug? And the answer was: very well. Maintenance of at least a PASI 75 response was observed at 148 weeks in 91% of patients on tildrakizumab at the approved 100-mg dose and 92% of those on the 200-mg dose. The FDA-approved regimen is 100 mg by subcutaneous injection at weeks 0 and 4, and then every 12 weeks after that.
An intriguing feature of reSURFACE 1 was that a subset of PASI 75 responders at week 28 got taken off tildrakizumab at that point and switched to double-blind placebo, then restarted on their earlier dose of tildrakizumab upon relapse, which was defined as loss of at least 50% of the achieved on-drug PASI improvement.
At week 64, fully 48 weeks after their last dose of tildrakizumab, the relapse rate was 54% in the group formerly on 100 mg of tildrakizumab and slightly better at 47% in those formerly on 200 mg. The median time to relapse was 226 days in the 100-mg group and 258 days in the higher-dose arm. Those are exceptionally long times to relapse, and it’s useful information to file away in the event a psoriasis patient needs to discontinue biologic therapy for a period of time, Dr. Thaçi observed.
At week 64 – again, off active treatment since week 16 – 63% of the tildrakizumab 100-mg group had lost their previous PASI 75 response, as had 52% who were formerly on tildrakizumab at 200 mg.
The long-term safety profile of tildrakizumab paralleled that of placebo. For example, the exposure-adjusted adverse event rates of serious infections and major adverse cardiovascular events were closely similar in the placebo, tildrakizumab 100 mg, and tildrakizumab 200 mg groups.
There were two notable between-group differences in adverse events of interest: injection site reactions occurred at a rate of 5.36 per 100 person-years with placebo, compared with 1.94 and 2.3 per 100 person-years with tildrakizumab at 100 and 200 mg, respectively; and the incidence of nonmelanoma skin cancer was 0.97 cases per 100 person-years in the placebo arm, versus 0.5 and 0.49 cases per 100 person-years in the two tildrakizumab arms.
Dr. Thaçi did not present PASI 90 response outcomes because, at the time the reSURFACE trials were planned, PASI 75 was considered state of the art. The PASI 90 data are still being crunched but will be available soon. The 4- and 5-year follow-up data from the long-term extension studies are also on their way.
The reSURFACE 1 and reSURFACE 2 trials and their extension studies were funded by Sun Pharma and Merck. Dr. Thaçi reported receiving research grants from and serving as a consultant and paid scientific advisor to those pharmaceutical companies and more than a dozen others.
REPORTING FROM THE EADV CONGRESS
Key clinical point: Inhibition of interleukin-23 p19 via tildrakizumab pays major long-term dividends.
Major finding: Of patients with a PASI 75 response to tildrakizumab 100 mg at 6 months, 91% maintained that level of response through 148 weeks.
Study details: This was a long-term, prospective, open-label extension study of the phase 3 reSURFACE 1 and 2 trials of 1,862 psoriasis patients.
Disclosures: The reSURFACE 1 and reSURFACE 2 trials and their extension study were funded by Sun Pharma and Merck. The presenter reported receiving research grants from and serving as a consultant to those pharmaceutical companies and more than a dozen others.
CONDOR trial: Most psoriasis patients can be downshifted to reduced-dose biologics
PARIS – with long-term maintenance of disease control and no adverse consequences, Juul van den Reek, MD, PhD, reported at the annual congress of the European Academy of Dermatology and Venereology.

She presented the results of the CONDOR trial, the first-ever formal, randomized, controlled trial of tightly regulated dose reduction of biologics, compared with usual care standard-dose therapy. “Our current advice is we think you can try to reduce the dose because there are a lot of patients who benefit from this,” declared Dr. van den Reek, a dermatologist at Radboud University, Nijmegen, the Netherlands.
The advantages of this strategy are twofold: lower expenditures for this costly collection of medications and less exposure to any long-term, drug-related health risks, she noted.
CONDOR was a Dutch six-center, 12-month, open-label, unblinded, noninferiority, randomized trial including 111 patients. Participants had to have stable low disease activity as defined by both Psoriasis Area and Severity Index (PASI) and Dermatology Life Quality Index (DLQI) scores of 5 or less for at least 6 months while on standard-dose etanercept (Enbrel), adalimumab (Humira), or ustekinumab (Stelara) prior to enrollment. In fact, the average baseline PASI score was less than 2, with a DLQI of 0.
Participants were randomized to usual care – the customary approved dose of biologic therapy – or a drop down to 67% of that dose, achieved through prolongation of the dosing interval. If the reduced-dose patients kept their PASI and DLQI scores at 5 or less for 3 months straight, they dropped further to 50% of their original dose. However, patients who exceeded those thresholds were immediately returned to their previously effective dose.
The primary endpoint in this noninferiority trial was the difference in mean PASI scores between the dose-reduction and usual-care groups at 12 months. The prespecified margin for noninferiority was a difference of 0.5 PASI points. And that’s where the results get dicey: The mean difference turned out to be 1.1 PASI points in favor of usual care, meaning that, according to the study ground rules, dose reduction was not statistically noninferior. In hindsight, however, that 0.5-point margin was ill-considered and too narrowly defined.
“Within the chosen margins, the dose-reduction strategy seemed inferior. But what is the clinical relevance of a mean difference of 1.1 PASI points, when the accepted minimal clinically important difference is 3.2 points?” Dr. van den Reek observed.
There was no significant between-group difference in DLQI scores at 12 months. Nor did the two study arms differ in terms of the prespecified secondary endpoint of persistent disease flares as defined by a PASI or DLQI greater than 5 for 3 consecutive months: five patients in the reduced-dose group and three in the usual-care arm experienced such flares. There were no serious adverse events or other safety signals related to the intervention.
At 12 months, 50% of patients in the dose-reduction group were well maintained on 50% of their original approved-dose biologic and another 17% were doing well on 67% of their former dose.
Session chair Dedee Murrell, MD, professor of dermatology at the University of New South Wales, Sydney, noted that neither patients nor dermatologists were blinded as to treatment status in CONDOR. She then asked the question on everybody’s minds: Was there any loss of treatment efficacy when patients in the dose-reduction arm needed to resume higher-dose therapy?
No, Dr. van den Reek replied. She added that planned future CONDOR analyses include a cost-effectiveness determination as well as measurement of serum drug levels and identification of antidrug antibodies, information that might prove helpful in identifying an enriched population of patients most likely to respond favorably to biologic dose reduction. In addition, CONDOR-X, a long-term extension study, is ongoing in order to learn how patients on reduced-dose biologics fare after the 12-month mark.
The CONDOR trial was funded by the Netherlands Organization for Health Research and Development; Dr. van den Reek reported having no financial conflicts of interest.
PARIS – with long-term maintenance of disease control and no adverse consequences, Juul van den Reek, MD, PhD, reported at the annual congress of the European Academy of Dermatology and Venereology.
She presented the results of the CONDOR trial, the first-ever formal, randomized, controlled trial of tightly regulated dose reduction of biologics, compared with usual care standard-dose therapy. “Our current advice is we think you can try to reduce the dose because there are a lot of patients who benefit from this,” declared Dr. van den Reek, a dermatologist at Radboud University, Nijmegen, the Netherlands.
The advantages of this strategy are twofold: lower expenditures for this costly collection of medications and less exposure to any long-term, drug-related health risks, she noted.
CONDOR was a Dutch six-center, 12-month, open-label, unblinded, noninferiority, randomized trial including 111 patients. Participants had to have stable low disease activity as defined by both Psoriasis Area and Severity Index (PASI) and Dermatology Life Quality Index (DLQI) scores of 5 or less for at least 6 months while on standard-dose etanercept (Enbrel), adalimumab (Humira), or ustekinumab (Stelara) prior to enrollment. In fact, the average baseline PASI score was less than 2, with a DLQI of 0.
Participants were randomized to usual care – the customary approved dose of biologic therapy – or a drop down to 67% of that dose, achieved through prolongation of the dosing interval. If the reduced-dose patients kept their PASI and DLQI scores at 5 or less for 3 months straight, they dropped further to 50% of their original dose. However, patients who exceeded those thresholds were immediately returned to their previously effective dose.
The primary endpoint in this noninferiority trial was the difference in mean PASI scores between the dose-reduction and usual-care groups at 12 months. The prespecified margin for noninferiority was a difference of 0.5 PASI points. And that’s where the results get dicey: The mean difference turned out to be 1.1 PASI points in favor of usual care, meaning that, according to the study ground rules, dose reduction was not statistically noninferior. In hindsight, however, that 0.5-point margin was ill-considered and too narrowly defined.
“Within the chosen margins, the dose-reduction strategy seemed inferior. But what is the clinical relevance of a mean difference of 1.1 PASI points, when the accepted minimal clinically important difference is 3.2 points?” Dr. van den Reek observed.
There was no significant between-group difference in DLQI scores at 12 months. Nor did the two study arms differ in terms of the prespecified secondary endpoint of persistent disease flares as defined by a PASI or DLQI greater than 5 for 3 consecutive months: five patients in the reduced-dose group and three in the usual-care arm experienced such flares. There were no serious adverse events or other safety signals related to the intervention.
At 12 months, 50% of patients in the dose-reduction group were well maintained on 50% of their original approved-dose biologic and another 17% were doing well on 67% of their former dose.
Session chair Dedee Murrell, MD, professor of dermatology at the University of New South Wales, Sydney, noted that neither patients nor dermatologists were blinded as to treatment status in CONDOR. She then asked the question on everybody’s minds: Was there any loss of treatment efficacy when patients in the dose-reduction arm needed to resume higher-dose therapy?
No, Dr. van den Reek replied. She added that planned future CONDOR analyses include a cost-effectiveness determination as well as measurement of serum drug levels and identification of antidrug antibodies, information that might prove helpful in identifying an enriched population of patients most likely to respond favorably to biologic dose reduction. In addition, CONDOR-X, a long-term extension study, is ongoing in order to learn how patients on reduced-dose biologics fare after the 12-month mark.
The CONDOR trial was funded by the Netherlands Organization for Health Research and Development; Dr. van den Reek reported having no financial conflicts of interest.
PARIS – with long-term maintenance of disease control and no adverse consequences, Juul van den Reek, MD, PhD, reported at the annual congress of the European Academy of Dermatology and Venereology.
She presented the results of the CONDOR trial, the first-ever formal, randomized, controlled trial of tightly regulated dose reduction of biologics, compared with usual care standard-dose therapy. “Our current advice is we think you can try to reduce the dose because there are a lot of patients who benefit from this,” declared Dr. van den Reek, a dermatologist at Radboud University, Nijmegen, the Netherlands.
The advantages of this strategy are twofold: lower expenditures for this costly collection of medications and less exposure to any long-term, drug-related health risks, she noted.
CONDOR was a Dutch six-center, 12-month, open-label, unblinded, noninferiority, randomized trial including 111 patients. Participants had to have stable low disease activity as defined by both Psoriasis Area and Severity Index (PASI) and Dermatology Life Quality Index (DLQI) scores of 5 or less for at least 6 months while on standard-dose etanercept (Enbrel), adalimumab (Humira), or ustekinumab (Stelara) prior to enrollment. In fact, the average baseline PASI score was less than 2, with a DLQI of 0.
Participants were randomized to usual care – the customary approved dose of biologic therapy – or a drop down to 67% of that dose, achieved through prolongation of the dosing interval. If the reduced-dose patients kept their PASI and DLQI scores at 5 or less for 3 months straight, they dropped further to 50% of their original dose. However, patients who exceeded those thresholds were immediately returned to their previously effective dose.
The primary endpoint in this noninferiority trial was the difference in mean PASI scores between the dose-reduction and usual-care groups at 12 months. The prespecified margin for noninferiority was a difference of 0.5 PASI points. And that’s where the results get dicey: The mean difference turned out to be 1.1 PASI points in favor of usual care, meaning that, according to the study ground rules, dose reduction was not statistically noninferior. In hindsight, however, that 0.5-point margin was ill-considered and too narrowly defined.
“Within the chosen margins, the dose-reduction strategy seemed inferior. But what is the clinical relevance of a mean difference of 1.1 PASI points, when the accepted minimal clinically important difference is 3.2 points?” Dr. van den Reek observed.
There was no significant between-group difference in DLQI scores at 12 months. Nor did the two study arms differ in terms of the prespecified secondary endpoint of persistent disease flares as defined by a PASI or DLQI greater than 5 for 3 consecutive months: five patients in the reduced-dose group and three in the usual-care arm experienced such flares. There were no serious adverse events or other safety signals related to the intervention.
At 12 months, 50% of patients in the dose-reduction group were well maintained on 50% of their original approved-dose biologic and another 17% were doing well on 67% of their former dose.
Session chair Dedee Murrell, MD, professor of dermatology at the University of New South Wales, Sydney, noted that neither patients nor dermatologists were blinded as to treatment status in CONDOR. She then asked the question on everybody’s minds: Was there any loss of treatment efficacy when patients in the dose-reduction arm needed to resume higher-dose therapy?
No, Dr. van den Reek replied. She added that planned future CONDOR analyses include a cost-effectiveness determination as well as measurement of serum drug levels and identification of antidrug antibodies, information that might prove helpful in identifying an enriched population of patients most likely to respond favorably to biologic dose reduction. In addition, CONDOR-X, a long-term extension study, is ongoing in order to learn how patients on reduced-dose biologics fare after the 12-month mark.
The CONDOR trial was funded by the Netherlands Organization for Health Research and Development; Dr. van den Reek reported having no financial conflicts of interest.
REPORTING FROM THE EADV CONGRESS
Key clinical point: An attempt at dose reduction is worthwhile in psoriasis patients well controlled on full-dose biologic therapy.
Major finding: Two-thirds of psoriasis patients maintained disease control after 12 months on reduced-dose biologic therapy.
Study details: This was a Dutch six-center, 12-month, open-label, unblinded, noninferiority, randomized trial of 111 psoriasis patients with stable low disease activity on standard-dose biologics at enrollment.
Disclosures: The CONDOR trial was funded by the Netherlands Organization for Health Research and Development; the presenter reported having no financial conflicts of interest.
New worldwide atopic dermatitis survey brings big surprises
PARIS – A major worldwide survey of the 12-month prevalence of atopic dermatitis (AD) across the course of life provides new insights into global disease trends, Jonathan I. Silverberg, MD, PhD, reported at the annual congress of the European Academy of Dermatology and Venereology.

Among the most important takeaways from this Internet-based survey of more than 273,645 infants, children, and adults in 18 countries across five continents conducted in 2017 was that “global atopic dermatitis prevalence appears to be higher in adults, at 10%, than in younger cohorts, where it’s 4%-8%, which I think is quite provocative and requires further study and confirmation,” said Dr. Silverberg, a dermatologist at Northwestern University in Chicago.
“Let’s keep in mind that there’s this accepted dogma in the literature than atopic dermatitis is somehow only a childhood disorder – it doesn’t affect adults. Well, these data tell a very different story because we’re actually seeing overall highest prevalences throughout the world occurring in adulthood,” based on the U.K. Working Party’s Diagnostic Criteria for Atopic Dermatitis (Br J Dermatol. 1994 Sep;131[3]:383-96).
This is the biggest epidemiologic survey ever to examine the 12-month prevalence and severity of AD around the world for both adults and children. Survey respondents included 172,627 adults aged 18 years and older, 34,212 adolescents aged 12-17 years, 54,806 children aged 2-11 years, and more than 12,000 infants.
Key findings from the study include the following:
- AD prevalence rates varied widely from country to country around the world, as well as by age groups (see graphic).
- The highest rate in adults was observed in China. South Korea had the highest rates in both children and adolescents. The top AD rates in infancy occurred in France and the United Kingdom.
- Rates across the age spectrum were consistently lowest in Israel and Switzerland.

“These kinds of patterns raise fascinating questions about the potential risk factors or protective factors that happen in different countries. There are some startling differences in terms of the different regions,” Dr. Silverberg observed. “Certain regions of the world really stand out as having much higher prevalences, particularly China and South Korea, and then as you get into the adult years, Brazil and Mexico, which I think are areas that, at least in the global atopic dermatitis epidemiology community, are not quite as well recognized as being hot spots for atopic dermatitis.”
Indeed, the 12-month prevalence rate of AD among adults was 14% in Mexico and 12% in Brazil, as compared with 13% in Saudi Arabia, 11% in Australia and Spain, 10% in Canada and the United Kingdom, and 9% in the United States.
The prevalence was generally lowest in infants, then jumped substantially within countries during the childhood years, declined slightly in adolescents, and then peaked in adulthood.
AD severity was assessed using PO-SCORAD, the Patient-Oriented Scoring AD measure. Most affected individuals had moderate AD as defined by a PO-SCORAD score of 25-50. Across the age spectrum, the highest proportion of infants with AD who had moderate disease was in China, with 72%. In Taiwan, 63% of children with AD had moderate disease, as did 68% of adolescents and an equal proportion of adults.
In the United Kingdom, 49% percent of infants with AD had severe disease, making that country the world leader in the youngest age group. Severe AD was most common among Turkish children, where 30% of kids with the skin disease had a PO-SCORAD score greater than 50. In Brazil, 31% of adolescents with AD had severe disease, the world’s highest rate in that age group. Among adults with AD, the world’s highest rate of severe disease was 25%, which was seen in the United States, Brazil, and Saudi Arabia.
Across the age spectrum, Japan had a consistently lower-end, overall, 12-month AD prevalence rate of 5%. Germany, Italy, and France had overall rates of 6%, 7%, and 8%, respectively. The rate was 9% in the United States and Canada, and it was 10% in Australia.
Dr. Silverberg performed validation analyses using the Patient-Oriented Eczema Measure (POEM) and diagnostic criteria similar to the earlier landmark International Study of Asthma and Allergies in Childhood, or ISAAC (Lancet. 1998 Apr 25;351[9111]:1225-32). This was a huge study that excluded the United States, leaving a hole in the epidemiologic picture of the disease that the new survey fills. The validation analyses were supportive of the main findings based on the U.K. Working Party criteria.
Dr. Silverberg reported serving as a consultant to Pfizer, which sponsored the global epidemiologic survey, as well as to roughly a dozen other pharmaceutical companies.
SOURCE: Silverberg JI. EADV Congress, Abstract FC01.01.
PARIS – A major worldwide survey of the 12-month prevalence of atopic dermatitis (AD) across the course of life provides new insights into global disease trends, Jonathan I. Silverberg, MD, PhD, reported at the annual congress of the European Academy of Dermatology and Venereology.
Among the most important takeaways from this Internet-based survey of more than 273,645 infants, children, and adults in 18 countries across five continents conducted in 2017 was that “global atopic dermatitis prevalence appears to be higher in adults, at 10%, than in younger cohorts, where it’s 4%-8%, which I think is quite provocative and requires further study and confirmation,” said Dr. Silverberg, a dermatologist at Northwestern University in Chicago.
“Let’s keep in mind that there’s this accepted dogma in the literature than atopic dermatitis is somehow only a childhood disorder – it doesn’t affect adults. Well, these data tell a very different story because we’re actually seeing overall highest prevalences throughout the world occurring in adulthood,” based on the U.K. Working Party’s Diagnostic Criteria for Atopic Dermatitis (Br J Dermatol. 1994 Sep;131[3]:383-96).
This is the biggest epidemiologic survey ever to examine the 12-month prevalence and severity of AD around the world for both adults and children. Survey respondents included 172,627 adults aged 18 years and older, 34,212 adolescents aged 12-17 years, 54,806 children aged 2-11 years, and more than 12,000 infants.
Key findings from the study include the following:
- AD prevalence rates varied widely from country to country around the world, as well as by age groups (see graphic).
- The highest rate in adults was observed in China. South Korea had the highest rates in both children and adolescents. The top AD rates in infancy occurred in France and the United Kingdom.
- Rates across the age spectrum were consistently lowest in Israel and Switzerland.
“These kinds of patterns raise fascinating questions about the potential risk factors or protective factors that happen in different countries. There are some startling differences in terms of the different regions,” Dr. Silverberg observed. “Certain regions of the world really stand out as having much higher prevalences, particularly China and South Korea, and then as you get into the adult years, Brazil and Mexico, which I think are areas that, at least in the global atopic dermatitis epidemiology community, are not quite as well recognized as being hot spots for atopic dermatitis.”
Indeed, the 12-month prevalence rate of AD among adults was 14% in Mexico and 12% in Brazil, as compared with 13% in Saudi Arabia, 11% in Australia and Spain, 10% in Canada and the United Kingdom, and 9% in the United States.
The prevalence was generally lowest in infants, then jumped substantially within countries during the childhood years, declined slightly in adolescents, and then peaked in adulthood.
AD severity was assessed using PO-SCORAD, the Patient-Oriented Scoring AD measure. Most affected individuals had moderate AD as defined by a PO-SCORAD score of 25-50. Across the age spectrum, the highest proportion of infants with AD who had moderate disease was in China, with 72%. In Taiwan, 63% of children with AD had moderate disease, as did 68% of adolescents and an equal proportion of adults.
In the United Kingdom, 49% percent of infants with AD had severe disease, making that country the world leader in the youngest age group. Severe AD was most common among Turkish children, where 30% of kids with the skin disease had a PO-SCORAD score greater than 50. In Brazil, 31% of adolescents with AD had severe disease, the world’s highest rate in that age group. Among adults with AD, the world’s highest rate of severe disease was 25%, which was seen in the United States, Brazil, and Saudi Arabia.
Across the age spectrum, Japan had a consistently lower-end, overall, 12-month AD prevalence rate of 5%. Germany, Italy, and France had overall rates of 6%, 7%, and 8%, respectively. The rate was 9% in the United States and Canada, and it was 10% in Australia.
Dr. Silverberg performed validation analyses using the Patient-Oriented Eczema Measure (POEM) and diagnostic criteria similar to the earlier landmark International Study of Asthma and Allergies in Childhood, or ISAAC (Lancet. 1998 Apr 25;351[9111]:1225-32). This was a huge study that excluded the United States, leaving a hole in the epidemiologic picture of the disease that the new survey fills. The validation analyses were supportive of the main findings based on the U.K. Working Party criteria.
Dr. Silverberg reported serving as a consultant to Pfizer, which sponsored the global epidemiologic survey, as well as to roughly a dozen other pharmaceutical companies.
SOURCE: Silverberg JI. EADV Congress, Abstract FC01.01.
PARIS – A major worldwide survey of the 12-month prevalence of atopic dermatitis (AD) across the course of life provides new insights into global disease trends, Jonathan I. Silverberg, MD, PhD, reported at the annual congress of the European Academy of Dermatology and Venereology.
Among the most important takeaways from this Internet-based survey of more than 273,645 infants, children, and adults in 18 countries across five continents conducted in 2017 was that “global atopic dermatitis prevalence appears to be higher in adults, at 10%, than in younger cohorts, where it’s 4%-8%, which I think is quite provocative and requires further study and confirmation,” said Dr. Silverberg, a dermatologist at Northwestern University in Chicago.
“Let’s keep in mind that there’s this accepted dogma in the literature than atopic dermatitis is somehow only a childhood disorder – it doesn’t affect adults. Well, these data tell a very different story because we’re actually seeing overall highest prevalences throughout the world occurring in adulthood,” based on the U.K. Working Party’s Diagnostic Criteria for Atopic Dermatitis (Br J Dermatol. 1994 Sep;131[3]:383-96).
This is the biggest epidemiologic survey ever to examine the 12-month prevalence and severity of AD around the world for both adults and children. Survey respondents included 172,627 adults aged 18 years and older, 34,212 adolescents aged 12-17 years, 54,806 children aged 2-11 years, and more than 12,000 infants.
Key findings from the study include the following:
- AD prevalence rates varied widely from country to country around the world, as well as by age groups (see graphic).
- The highest rate in adults was observed in China. South Korea had the highest rates in both children and adolescents. The top AD rates in infancy occurred in France and the United Kingdom.
- Rates across the age spectrum were consistently lowest in Israel and Switzerland.
“These kinds of patterns raise fascinating questions about the potential risk factors or protective factors that happen in different countries. There are some startling differences in terms of the different regions,” Dr. Silverberg observed. “Certain regions of the world really stand out as having much higher prevalences, particularly China and South Korea, and then as you get into the adult years, Brazil and Mexico, which I think are areas that, at least in the global atopic dermatitis epidemiology community, are not quite as well recognized as being hot spots for atopic dermatitis.”
Indeed, the 12-month prevalence rate of AD among adults was 14% in Mexico and 12% in Brazil, as compared with 13% in Saudi Arabia, 11% in Australia and Spain, 10% in Canada and the United Kingdom, and 9% in the United States.
The prevalence was generally lowest in infants, then jumped substantially within countries during the childhood years, declined slightly in adolescents, and then peaked in adulthood.
AD severity was assessed using PO-SCORAD, the Patient-Oriented Scoring AD measure. Most affected individuals had moderate AD as defined by a PO-SCORAD score of 25-50. Across the age spectrum, the highest proportion of infants with AD who had moderate disease was in China, with 72%. In Taiwan, 63% of children with AD had moderate disease, as did 68% of adolescents and an equal proportion of adults.
In the United Kingdom, 49% percent of infants with AD had severe disease, making that country the world leader in the youngest age group. Severe AD was most common among Turkish children, where 30% of kids with the skin disease had a PO-SCORAD score greater than 50. In Brazil, 31% of adolescents with AD had severe disease, the world’s highest rate in that age group. Among adults with AD, the world’s highest rate of severe disease was 25%, which was seen in the United States, Brazil, and Saudi Arabia.
Across the age spectrum, Japan had a consistently lower-end, overall, 12-month AD prevalence rate of 5%. Germany, Italy, and France had overall rates of 6%, 7%, and 8%, respectively. The rate was 9% in the United States and Canada, and it was 10% in Australia.
Dr. Silverberg performed validation analyses using the Patient-Oriented Eczema Measure (POEM) and diagnostic criteria similar to the earlier landmark International Study of Asthma and Allergies in Childhood, or ISAAC (Lancet. 1998 Apr 25;351[9111]:1225-32). This was a huge study that excluded the United States, leaving a hole in the epidemiologic picture of the disease that the new survey fills. The validation analyses were supportive of the main findings based on the U.K. Working Party criteria.
Dr. Silverberg reported serving as a consultant to Pfizer, which sponsored the global epidemiologic survey, as well as to roughly a dozen other pharmaceutical companies.
SOURCE: Silverberg JI. EADV Congress, Abstract FC01.01.
REPORTING FROM THE EADV CONGRESS
Key clinical point: Worldwide, the 12-month prevalence of atopic dermatitis (AD) varies substantially but is unexpectedly highest in adults.
Major finding: The global 12-month prevalence of AD in adults is 10%, substantially higher than in infants, children, or adolescents.
Study details: This was an Internet survey of 273,654 subjects conducted in 2017 in 18 countries on five continents.
Disclosures: The presenter reported serving as a consultant to Pfizer, the study sponsor, as well as to roughly a dozen other pharmaceutical companies.
Source: Silverberg JI. EADV Congress, Abstract FC01.01.
Novel topical JAK inhibitor shows promise for atopic dermatitis
PARIS – A cream formulation of ruxolitinib, a selective inhibitor of Janus kinase (JAK) 1 and 2, outperformed triamcinolone cream 0.1% and vehicle control in a large, phase 2, dose-ranging, randomized trial in patients with atopic dermatitis (AD), Brian S. Kim, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.

This novel topical JAK inhibitor not only modulates inflammatory cytokines involved in the pathogenesis of AD, including interleukin-4, -5, -13, and -31, but Dr. Kim and his coinvestigators also demonstrated that ruxolitinib has antipruritic effects achieved by acting directly on sensory nerve fibers.
“Ultimately, said Dr. Kim, a dermatologist and codirector of the Center for the Study of Itch at Washington University, St. Louis.
The trial included 307 adults, mean age 35 years, with a median 21-year disease history and a mean of 7.3 flares within the past 12 months. Dr. Kim characterized the study population as having AD of “high-moderate” severity, with a mean involved body surface area of 9.7%, half of patients having a baseline Eczema Area and Severity Index (EASI) score greater than 7, and having a mean itch numeric rating scale of 7. Two-thirds of patients had an Investigator’s Global Assessment (IGA) score of 3 and the rest had scores of 2.
Patients were randomized to one of six study arms entailing 8 weeks of double-blind therapy: ruxolitinib cream 1.5% once daily, 1.5% twice daily; 0.5% once daily; 0.15% once daily; twice-daily vehicle; or triamcinolone cream 0.1% twice a day for 4 weeks followed by 4 weeks of vehicle.
All the ruxolitinib regimens provided dose- and time-dependent efficacy, compared with vehicle. The best results were seen with ruxolitinib 1.5% twice daily, which outperformed triamcinolone cream.
The primary study endpoint was change in EASI score from baseline to week 4, but the week 2 and week 8 data were also informative. Key secondary endpoints included the proportion of subjects achieving an EASI-75 response and/or an IGA response, which required improvement to an IGA score of 0 or 1 with at least a 2-point reduction from baseline.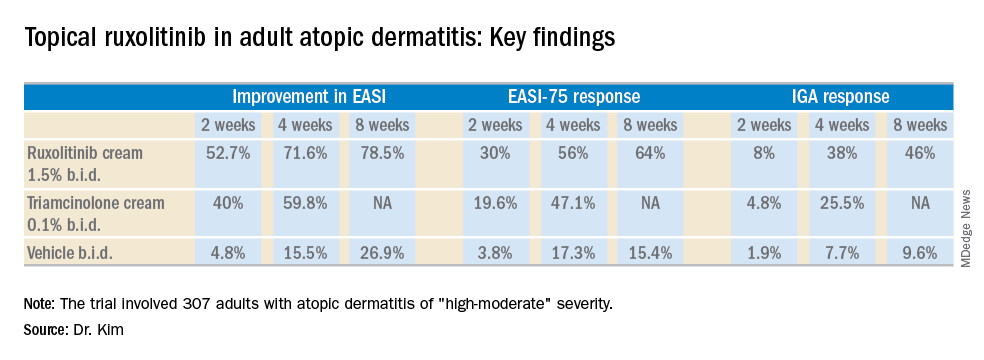
As for itch, ruxolitinib cream provided rapid and sustained improvement, said Dr. Kim. Indeed, within the first 2 days of the study, the ruxolitinib 1.5% twice-daily group had a mean 1.8-point reduction on the numeric rating scale, compared with a 0.2-point drop with vehicle and a 1-point drop with triamcinolone cream twice a day. By week 4, the twice-daily ruxolitinib 1.5% group had about a 4-point drop from baseline, the once-daily ruxolitinib 1.5% group had a 3.5-point drop, and the triamcinolone-treated patients had a 2.5-point drop.
Topical ruxolitinib was not associated with any significant safety or tolerability issues, and there were no clinically significant application site reactions, according to the dermatologist.
Session cochair Konstantine Buxtorf Friedli, MD, a Swiss dermatologist, commented that she could easily imagine this topical JAK inhibitor also being useful in other diseases with itch.
Dr. Kim reported serving as a consultant to and recipient of research funding from Incyte, which sponsored the study.
PARIS – A cream formulation of ruxolitinib, a selective inhibitor of Janus kinase (JAK) 1 and 2, outperformed triamcinolone cream 0.1% and vehicle control in a large, phase 2, dose-ranging, randomized trial in patients with atopic dermatitis (AD), Brian S. Kim, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
This novel topical JAK inhibitor not only modulates inflammatory cytokines involved in the pathogenesis of AD, including interleukin-4, -5, -13, and -31, but Dr. Kim and his coinvestigators also demonstrated that ruxolitinib has antipruritic effects achieved by acting directly on sensory nerve fibers.
“Ultimately, said Dr. Kim, a dermatologist and codirector of the Center for the Study of Itch at Washington University, St. Louis.
The trial included 307 adults, mean age 35 years, with a median 21-year disease history and a mean of 7.3 flares within the past 12 months. Dr. Kim characterized the study population as having AD of “high-moderate” severity, with a mean involved body surface area of 9.7%, half of patients having a baseline Eczema Area and Severity Index (EASI) score greater than 7, and having a mean itch numeric rating scale of 7. Two-thirds of patients had an Investigator’s Global Assessment (IGA) score of 3 and the rest had scores of 2.
Patients were randomized to one of six study arms entailing 8 weeks of double-blind therapy: ruxolitinib cream 1.5% once daily, 1.5% twice daily; 0.5% once daily; 0.15% once daily; twice-daily vehicle; or triamcinolone cream 0.1% twice a day for 4 weeks followed by 4 weeks of vehicle.
All the ruxolitinib regimens provided dose- and time-dependent efficacy, compared with vehicle. The best results were seen with ruxolitinib 1.5% twice daily, which outperformed triamcinolone cream.
The primary study endpoint was change in EASI score from baseline to week 4, but the week 2 and week 8 data were also informative. Key secondary endpoints included the proportion of subjects achieving an EASI-75 response and/or an IGA response, which required improvement to an IGA score of 0 or 1 with at least a 2-point reduction from baseline.
As for itch, ruxolitinib cream provided rapid and sustained improvement, said Dr. Kim. Indeed, within the first 2 days of the study, the ruxolitinib 1.5% twice-daily group had a mean 1.8-point reduction on the numeric rating scale, compared with a 0.2-point drop with vehicle and a 1-point drop with triamcinolone cream twice a day. By week 4, the twice-daily ruxolitinib 1.5% group had about a 4-point drop from baseline, the once-daily ruxolitinib 1.5% group had a 3.5-point drop, and the triamcinolone-treated patients had a 2.5-point drop.
Topical ruxolitinib was not associated with any significant safety or tolerability issues, and there were no clinically significant application site reactions, according to the dermatologist.
Session cochair Konstantine Buxtorf Friedli, MD, a Swiss dermatologist, commented that she could easily imagine this topical JAK inhibitor also being useful in other diseases with itch.
Dr. Kim reported serving as a consultant to and recipient of research funding from Incyte, which sponsored the study.
PARIS – A cream formulation of ruxolitinib, a selective inhibitor of Janus kinase (JAK) 1 and 2, outperformed triamcinolone cream 0.1% and vehicle control in a large, phase 2, dose-ranging, randomized trial in patients with atopic dermatitis (AD), Brian S. Kim, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
This novel topical JAK inhibitor not only modulates inflammatory cytokines involved in the pathogenesis of AD, including interleukin-4, -5, -13, and -31, but Dr. Kim and his coinvestigators also demonstrated that ruxolitinib has antipruritic effects achieved by acting directly on sensory nerve fibers.
“Ultimately, said Dr. Kim, a dermatologist and codirector of the Center for the Study of Itch at Washington University, St. Louis.
The trial included 307 adults, mean age 35 years, with a median 21-year disease history and a mean of 7.3 flares within the past 12 months. Dr. Kim characterized the study population as having AD of “high-moderate” severity, with a mean involved body surface area of 9.7%, half of patients having a baseline Eczema Area and Severity Index (EASI) score greater than 7, and having a mean itch numeric rating scale of 7. Two-thirds of patients had an Investigator’s Global Assessment (IGA) score of 3 and the rest had scores of 2.
Patients were randomized to one of six study arms entailing 8 weeks of double-blind therapy: ruxolitinib cream 1.5% once daily, 1.5% twice daily; 0.5% once daily; 0.15% once daily; twice-daily vehicle; or triamcinolone cream 0.1% twice a day for 4 weeks followed by 4 weeks of vehicle.
All the ruxolitinib regimens provided dose- and time-dependent efficacy, compared with vehicle. The best results were seen with ruxolitinib 1.5% twice daily, which outperformed triamcinolone cream.
The primary study endpoint was change in EASI score from baseline to week 4, but the week 2 and week 8 data were also informative. Key secondary endpoints included the proportion of subjects achieving an EASI-75 response and/or an IGA response, which required improvement to an IGA score of 0 or 1 with at least a 2-point reduction from baseline.
As for itch, ruxolitinib cream provided rapid and sustained improvement, said Dr. Kim. Indeed, within the first 2 days of the study, the ruxolitinib 1.5% twice-daily group had a mean 1.8-point reduction on the numeric rating scale, compared with a 0.2-point drop with vehicle and a 1-point drop with triamcinolone cream twice a day. By week 4, the twice-daily ruxolitinib 1.5% group had about a 4-point drop from baseline, the once-daily ruxolitinib 1.5% group had a 3.5-point drop, and the triamcinolone-treated patients had a 2.5-point drop.
Topical ruxolitinib was not associated with any significant safety or tolerability issues, and there were no clinically significant application site reactions, according to the dermatologist.
Session cochair Konstantine Buxtorf Friedli, MD, a Swiss dermatologist, commented that she could easily imagine this topical JAK inhibitor also being useful in other diseases with itch.
Dr. Kim reported serving as a consultant to and recipient of research funding from Incyte, which sponsored the study.
REPORTING FROM THE EADV CONGRESS
Key clinical point: A novel topical Janus kinase inhibitor may provide a valuable alternative to potent topical steroids in atopic dermatitis.
Major finding: At week 4, the mean improvement in Eczema Area and Severity Index score was 72% with ruxolitinib cream 1.5% twice a day, compared with 60% with triamcinolone cream 0.1% twice a day.
Study details: This 8-week, phase 2 clinical trial included 307 adult atopic dermatitis patients randomized to ruxolitinib cream, triamcinolone cream, or vehicle.
Disclosures: The study was sponsored by Incyte. The presenter reported serving as a consultant to and recipient of research funding from the company.
Ligelizumab outperformed omalizumab for refractory chronic spontaneous urticaria
.

“For sure, ligelizumab is the highlight of this year in urticariology,” Marcus Maurer, MD, declared at the annual congress of the European Academy of Dermatology and Venereology. An ongoing phase 3 trial will now compare more than 1,000 patients with CSU who will be randomized to ligelizumab, omalizumab, or placebo.
Like omalizumab (Xolair), which is approved in the United States and Europe for treatment of CSU, ligelizumab is a humanized anti-IgE monoclonal antibody. But the investigational agent binds to IgE with greater affinity than omalizumab, and this translated into greater therapeutic efficacy in the multicenter, double-blind, placebo-controlled clinical trial, explained Dr. Maurer, professor of dermatology and allergy at Charité University in Berlin.
Study participants, all refractory to histamine1 antihistamines and in many cases to leukotriene receptor antagonists as well, were randomized to omalizumab at 300 mg, placebo, or to ligelizumab at 24 mg, 72 mg, or 240 mg administered by subcutaneous injection every 4 weeks for 20 weeks. The study showed that the effective dose of ligelizumab lies somewhere between 72 and 240 mg; the 24-mg dose won’t be pursued in further studies.
“Three things are important in the comparison between ligelizumab and omalizumab: First, ligelizumab works faster – and omalizumab is a fast-working drug in urticaria. As early as week 4 after initiation of treatment, ligelizumab resulted in a significantly higher response rate,” he said.
Second, a complete response rate as defined by an Urticaria Activity Score over the past 7 days (UAS7) of 0 was achieved by more than 50% of patients on ligelizumab at 240 mg, a rate twice that seen in the omalizumab group. Indeed, more patients were symptom-free on ligelizumab at 72 mg or 240 mg than on omalizumab throughout the 20-week study.
And third, time to relapse after treatment discontinuation was markedly longer with ligelizumab.
“Once you stop the treatment, we expect patients to come back because we didn’t cure the disease, we blocked the signs and symptoms by blocking mast cell degranulation. Relapse after the last injection occurred at about 4 weeks with omalizumab versus 10 weeks for ligelizumab on average. That’s amazing,” Dr. Maurer said.
At week 20, the mean reductions from baseline in UAS7 scores were 13.6 points with placebo, 15.2 points with the lowest dose of ligelizumab, 18.2 points with omalizumab, 23.1 points with ligelizumab at 72 mg, and 22.5 points for ligelizumab at 240 mg.
The side effect profiles for both biologics were essentially the same as for placebo with the exception of a 5.9% rate of mild injection site reactions with ligelizumab at the 240-mg dose versus 2.3% with placebo.
Many clinicians have noticed a significant limitation of omalizumab: It is less effective in patients with more complex CSU having an autoimmune overlay, type 2b angioedema, and/or long disease duration.
“This does not seem to be the case with ligelizumab. Even for the difficult-to-treat subpopulations of CSU, ligelizumab appears to be a drug that can protect against mast cell degranulation. We see a reduction in angioedema activity; we see a reduction in wheal size and number; we see a reduction in the itch – so across all the symptoms in the difficult subpopulations, this is the better drug. Now we have to make it work in the phase 3 trials to bring it to clinical practice,” he said.
Dr. Maurer reported receiving research funding from and serving as an advisor to and paid speaker for Novartis, which markets omalizumab and is developing ligelizumab.
.
“For sure, ligelizumab is the highlight of this year in urticariology,” Marcus Maurer, MD, declared at the annual congress of the European Academy of Dermatology and Venereology. An ongoing phase 3 trial will now compare more than 1,000 patients with CSU who will be randomized to ligelizumab, omalizumab, or placebo.
Like omalizumab (Xolair), which is approved in the United States and Europe for treatment of CSU, ligelizumab is a humanized anti-IgE monoclonal antibody. But the investigational agent binds to IgE with greater affinity than omalizumab, and this translated into greater therapeutic efficacy in the multicenter, double-blind, placebo-controlled clinical trial, explained Dr. Maurer, professor of dermatology and allergy at Charité University in Berlin.
Study participants, all refractory to histamine1 antihistamines and in many cases to leukotriene receptor antagonists as well, were randomized to omalizumab at 300 mg, placebo, or to ligelizumab at 24 mg, 72 mg, or 240 mg administered by subcutaneous injection every 4 weeks for 20 weeks. The study showed that the effective dose of ligelizumab lies somewhere between 72 and 240 mg; the 24-mg dose won’t be pursued in further studies.
“Three things are important in the comparison between ligelizumab and omalizumab: First, ligelizumab works faster – and omalizumab is a fast-working drug in urticaria. As early as week 4 after initiation of treatment, ligelizumab resulted in a significantly higher response rate,” he said.
Second, a complete response rate as defined by an Urticaria Activity Score over the past 7 days (UAS7) of 0 was achieved by more than 50% of patients on ligelizumab at 240 mg, a rate twice that seen in the omalizumab group. Indeed, more patients were symptom-free on ligelizumab at 72 mg or 240 mg than on omalizumab throughout the 20-week study.
And third, time to relapse after treatment discontinuation was markedly longer with ligelizumab.
“Once you stop the treatment, we expect patients to come back because we didn’t cure the disease, we blocked the signs and symptoms by blocking mast cell degranulation. Relapse after the last injection occurred at about 4 weeks with omalizumab versus 10 weeks for ligelizumab on average. That’s amazing,” Dr. Maurer said.
At week 20, the mean reductions from baseline in UAS7 scores were 13.6 points with placebo, 15.2 points with the lowest dose of ligelizumab, 18.2 points with omalizumab, 23.1 points with ligelizumab at 72 mg, and 22.5 points for ligelizumab at 240 mg.
The side effect profiles for both biologics were essentially the same as for placebo with the exception of a 5.9% rate of mild injection site reactions with ligelizumab at the 240-mg dose versus 2.3% with placebo.
Many clinicians have noticed a significant limitation of omalizumab: It is less effective in patients with more complex CSU having an autoimmune overlay, type 2b angioedema, and/or long disease duration.
“This does not seem to be the case with ligelizumab. Even for the difficult-to-treat subpopulations of CSU, ligelizumab appears to be a drug that can protect against mast cell degranulation. We see a reduction in angioedema activity; we see a reduction in wheal size and number; we see a reduction in the itch – so across all the symptoms in the difficult subpopulations, this is the better drug. Now we have to make it work in the phase 3 trials to bring it to clinical practice,” he said.
Dr. Maurer reported receiving research funding from and serving as an advisor to and paid speaker for Novartis, which markets omalizumab and is developing ligelizumab.
.
“For sure, ligelizumab is the highlight of this year in urticariology,” Marcus Maurer, MD, declared at the annual congress of the European Academy of Dermatology and Venereology. An ongoing phase 3 trial will now compare more than 1,000 patients with CSU who will be randomized to ligelizumab, omalizumab, or placebo.
Like omalizumab (Xolair), which is approved in the United States and Europe for treatment of CSU, ligelizumab is a humanized anti-IgE monoclonal antibody. But the investigational agent binds to IgE with greater affinity than omalizumab, and this translated into greater therapeutic efficacy in the multicenter, double-blind, placebo-controlled clinical trial, explained Dr. Maurer, professor of dermatology and allergy at Charité University in Berlin.
Study participants, all refractory to histamine1 antihistamines and in many cases to leukotriene receptor antagonists as well, were randomized to omalizumab at 300 mg, placebo, or to ligelizumab at 24 mg, 72 mg, or 240 mg administered by subcutaneous injection every 4 weeks for 20 weeks. The study showed that the effective dose of ligelizumab lies somewhere between 72 and 240 mg; the 24-mg dose won’t be pursued in further studies.
“Three things are important in the comparison between ligelizumab and omalizumab: First, ligelizumab works faster – and omalizumab is a fast-working drug in urticaria. As early as week 4 after initiation of treatment, ligelizumab resulted in a significantly higher response rate,” he said.
Second, a complete response rate as defined by an Urticaria Activity Score over the past 7 days (UAS7) of 0 was achieved by more than 50% of patients on ligelizumab at 240 mg, a rate twice that seen in the omalizumab group. Indeed, more patients were symptom-free on ligelizumab at 72 mg or 240 mg than on omalizumab throughout the 20-week study.
And third, time to relapse after treatment discontinuation was markedly longer with ligelizumab.
“Once you stop the treatment, we expect patients to come back because we didn’t cure the disease, we blocked the signs and symptoms by blocking mast cell degranulation. Relapse after the last injection occurred at about 4 weeks with omalizumab versus 10 weeks for ligelizumab on average. That’s amazing,” Dr. Maurer said.
At week 20, the mean reductions from baseline in UAS7 scores were 13.6 points with placebo, 15.2 points with the lowest dose of ligelizumab, 18.2 points with omalizumab, 23.1 points with ligelizumab at 72 mg, and 22.5 points for ligelizumab at 240 mg.
The side effect profiles for both biologics were essentially the same as for placebo with the exception of a 5.9% rate of mild injection site reactions with ligelizumab at the 240-mg dose versus 2.3% with placebo.
Many clinicians have noticed a significant limitation of omalizumab: It is less effective in patients with more complex CSU having an autoimmune overlay, type 2b angioedema, and/or long disease duration.
“This does not seem to be the case with ligelizumab. Even for the difficult-to-treat subpopulations of CSU, ligelizumab appears to be a drug that can protect against mast cell degranulation. We see a reduction in angioedema activity; we see a reduction in wheal size and number; we see a reduction in the itch – so across all the symptoms in the difficult subpopulations, this is the better drug. Now we have to make it work in the phase 3 trials to bring it to clinical practice,” he said.
Dr. Maurer reported receiving research funding from and serving as an advisor to and paid speaker for Novartis, which markets omalizumab and is developing ligelizumab.
REPORTING FROM THE EADV CONGRESS
Key clinical point: Ligelizumab shows considerable promise for chronic spontaneous urticaria.
Major finding: The complete response rate was two times higher with ligelizumab than it was with omalizumab.
Study details: This phase 2b randomized, double-blind, multicenter, active- and placebo-controlled, 20-week study included 382 patients with chronic spontaneous urticaria.
Disclosures: The presenter reported receiving research funding from and serving as an advisor to and paid speaker for Novartis, which is developing ligelizumab.