User login
Secukinumab shows promise in hidradenitis suppurativa
PARIS – David Rosmarin, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.

“It was especially notable that secukinumab was effective in five of the six patients who had previously failed anti-TNF [tumor necrosis factor] therapy,” said Dr. Rosmarin, a dermatologist at Tufts University, Boston.
At present, the sole medication approved for treatment of hidradenitis suppurativa (HS) is the TNF inhibitor adalimumab (Humira). The rationale for investigating secukinumab (Cosentyx), a biologic that blocks the interleukin-17A receptor and is approved for treatment of psoriasis, psoriatic arthritis, and ankylosing spondylitis, lies in the observation that HS lesions exhibit an increased ratio of Th17- to T-regulatory cells, compared with normal skin. Lesional skin also features elevated IL-17 levels. These abnormalities can be reversed by anti-TNF therapy, he explained.
Dr. Rosmarin presented a 28-week, open-label study in which 18 patients with moderate to severe HS received an induction regimen consisting of 300 mg secukinumab given subcutaneously once weekly for 5 weeks and were then randomized to the same dose of the biologic given either every 2 or 4 weeks until week 24.
The primary endpoint was achievement of a Hidradenitis Suppurativa Clinical Response (HiSCR) at 24 weeks. This requires no increase in the number of draining fistulae or abscesses, compared with baseline, plus at least a 50% reduction in total inflammatory nodules. Secondary endpoints were the mean change from baseline in the Sartorius Scale as well as on the Dermatology Life Quality Index (DLQI).
Participants were an average of 33 years and had a disease duration of 12 years; 14 patients were Hurley Stage II, the rest Stage III. Their mean baseline DLQI was 12.
“Patients with hidradenitis suppurativa have a horrible quality of life,” Dr. Rosmarin commented. “When we compare it to patients who have atopic dermatitis, psoriasis, or chronic idiopathic urticaria, oftentimes patients with hidradenitis suppurativa suffer the worst and have the most quality of life impairment.”
Of the 18 patients, 14 – 7 of 9 in each group – achieved HiSCR. This included five of the six patients who were previous nonresponders to adalimumab or another anti-TNF biologic.
“The other thing we noticed was the rapidity of response, which is important to a lot of our patients. It took an average of 7 weeks to achieve HiSCR, and eight patients achieved HiSCR during the induction phase of treatment,” the dermatologist said.
Mean scores on the Sartorius Scale dropped by 28%. Similarly, scores on the DLQI improved by a mean of 3.6 points, or 26%. Nine patients experienced a reduction of 5 points or more on the DLQI. “This happened largely in the first 1-2 months of therapy,” Dr. Rosmarin continued.
Secukinumab was well tolerated. There were no treatment discontinuations because of adverse events. Four patients, all in the biweekly dosing arm, developed Candida infections, all easily cured using topical ketoconazole.
The next step will be to conduct a large, placebo-controlled, randomized trial to firmly establish the efficacy of secukinumab for HS. Also, the optimal dosing of the biologic for induction and long-term maintenance therapy have yet to be determined. Over the long term, it will be important to see whether marked improvement in HS is accompanied by a reduction in the elevated cardiovascular risk associated with this inflammatory disease, he added.
In 2019, a trial will get underway to compare two doses of secukinumab for patients with HS. Based on a search of clinical trials at ClinicalTrials.gov, a wide range of monoclonal antibody therapies are being investigated for the treatment of HS.
The results of this preliminary study of secukinumab emphasize the importance of the Th17 pathway in HS and open the door to alternative strategies targeting this pathway. Dr. Rosmarin noted that he and his coinvestigators have collected a case series of positive responses to guselkumab (Tremfya), which targets the IL-23 p19 subunit, which also lies along the Th17 pathway.
The secukinumab study was sponsored by Novartis. Dr. Rosmarin reported serving as a consultant to or on speakers’ bureaus for that company and more than half a dozen other pharmaceutical companies.
PARIS – David Rosmarin, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“It was especially notable that secukinumab was effective in five of the six patients who had previously failed anti-TNF [tumor necrosis factor] therapy,” said Dr. Rosmarin, a dermatologist at Tufts University, Boston.
At present, the sole medication approved for treatment of hidradenitis suppurativa (HS) is the TNF inhibitor adalimumab (Humira). The rationale for investigating secukinumab (Cosentyx), a biologic that blocks the interleukin-17A receptor and is approved for treatment of psoriasis, psoriatic arthritis, and ankylosing spondylitis, lies in the observation that HS lesions exhibit an increased ratio of Th17- to T-regulatory cells, compared with normal skin. Lesional skin also features elevated IL-17 levels. These abnormalities can be reversed by anti-TNF therapy, he explained.
Dr. Rosmarin presented a 28-week, open-label study in which 18 patients with moderate to severe HS received an induction regimen consisting of 300 mg secukinumab given subcutaneously once weekly for 5 weeks and were then randomized to the same dose of the biologic given either every 2 or 4 weeks until week 24.
The primary endpoint was achievement of a Hidradenitis Suppurativa Clinical Response (HiSCR) at 24 weeks. This requires no increase in the number of draining fistulae or abscesses, compared with baseline, plus at least a 50% reduction in total inflammatory nodules. Secondary endpoints were the mean change from baseline in the Sartorius Scale as well as on the Dermatology Life Quality Index (DLQI).
Participants were an average of 33 years and had a disease duration of 12 years; 14 patients were Hurley Stage II, the rest Stage III. Their mean baseline DLQI was 12.
“Patients with hidradenitis suppurativa have a horrible quality of life,” Dr. Rosmarin commented. “When we compare it to patients who have atopic dermatitis, psoriasis, or chronic idiopathic urticaria, oftentimes patients with hidradenitis suppurativa suffer the worst and have the most quality of life impairment.”
Of the 18 patients, 14 – 7 of 9 in each group – achieved HiSCR. This included five of the six patients who were previous nonresponders to adalimumab or another anti-TNF biologic.
“The other thing we noticed was the rapidity of response, which is important to a lot of our patients. It took an average of 7 weeks to achieve HiSCR, and eight patients achieved HiSCR during the induction phase of treatment,” the dermatologist said.
Mean scores on the Sartorius Scale dropped by 28%. Similarly, scores on the DLQI improved by a mean of 3.6 points, or 26%. Nine patients experienced a reduction of 5 points or more on the DLQI. “This happened largely in the first 1-2 months of therapy,” Dr. Rosmarin continued.
Secukinumab was well tolerated. There were no treatment discontinuations because of adverse events. Four patients, all in the biweekly dosing arm, developed Candida infections, all easily cured using topical ketoconazole.
The next step will be to conduct a large, placebo-controlled, randomized trial to firmly establish the efficacy of secukinumab for HS. Also, the optimal dosing of the biologic for induction and long-term maintenance therapy have yet to be determined. Over the long term, it will be important to see whether marked improvement in HS is accompanied by a reduction in the elevated cardiovascular risk associated with this inflammatory disease, he added.
In 2019, a trial will get underway to compare two doses of secukinumab for patients with HS. Based on a search of clinical trials at ClinicalTrials.gov, a wide range of monoclonal antibody therapies are being investigated for the treatment of HS.
The results of this preliminary study of secukinumab emphasize the importance of the Th17 pathway in HS and open the door to alternative strategies targeting this pathway. Dr. Rosmarin noted that he and his coinvestigators have collected a case series of positive responses to guselkumab (Tremfya), which targets the IL-23 p19 subunit, which also lies along the Th17 pathway.
The secukinumab study was sponsored by Novartis. Dr. Rosmarin reported serving as a consultant to or on speakers’ bureaus for that company and more than half a dozen other pharmaceutical companies.
PARIS – David Rosmarin, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“It was especially notable that secukinumab was effective in five of the six patients who had previously failed anti-TNF [tumor necrosis factor] therapy,” said Dr. Rosmarin, a dermatologist at Tufts University, Boston.
At present, the sole medication approved for treatment of hidradenitis suppurativa (HS) is the TNF inhibitor adalimumab (Humira). The rationale for investigating secukinumab (Cosentyx), a biologic that blocks the interleukin-17A receptor and is approved for treatment of psoriasis, psoriatic arthritis, and ankylosing spondylitis, lies in the observation that HS lesions exhibit an increased ratio of Th17- to T-regulatory cells, compared with normal skin. Lesional skin also features elevated IL-17 levels. These abnormalities can be reversed by anti-TNF therapy, he explained.
Dr. Rosmarin presented a 28-week, open-label study in which 18 patients with moderate to severe HS received an induction regimen consisting of 300 mg secukinumab given subcutaneously once weekly for 5 weeks and were then randomized to the same dose of the biologic given either every 2 or 4 weeks until week 24.
The primary endpoint was achievement of a Hidradenitis Suppurativa Clinical Response (HiSCR) at 24 weeks. This requires no increase in the number of draining fistulae or abscesses, compared with baseline, plus at least a 50% reduction in total inflammatory nodules. Secondary endpoints were the mean change from baseline in the Sartorius Scale as well as on the Dermatology Life Quality Index (DLQI).
Participants were an average of 33 years and had a disease duration of 12 years; 14 patients were Hurley Stage II, the rest Stage III. Their mean baseline DLQI was 12.
“Patients with hidradenitis suppurativa have a horrible quality of life,” Dr. Rosmarin commented. “When we compare it to patients who have atopic dermatitis, psoriasis, or chronic idiopathic urticaria, oftentimes patients with hidradenitis suppurativa suffer the worst and have the most quality of life impairment.”
Of the 18 patients, 14 – 7 of 9 in each group – achieved HiSCR. This included five of the six patients who were previous nonresponders to adalimumab or another anti-TNF biologic.
“The other thing we noticed was the rapidity of response, which is important to a lot of our patients. It took an average of 7 weeks to achieve HiSCR, and eight patients achieved HiSCR during the induction phase of treatment,” the dermatologist said.
Mean scores on the Sartorius Scale dropped by 28%. Similarly, scores on the DLQI improved by a mean of 3.6 points, or 26%. Nine patients experienced a reduction of 5 points or more on the DLQI. “This happened largely in the first 1-2 months of therapy,” Dr. Rosmarin continued.
Secukinumab was well tolerated. There were no treatment discontinuations because of adverse events. Four patients, all in the biweekly dosing arm, developed Candida infections, all easily cured using topical ketoconazole.
The next step will be to conduct a large, placebo-controlled, randomized trial to firmly establish the efficacy of secukinumab for HS. Also, the optimal dosing of the biologic for induction and long-term maintenance therapy have yet to be determined. Over the long term, it will be important to see whether marked improvement in HS is accompanied by a reduction in the elevated cardiovascular risk associated with this inflammatory disease, he added.
In 2019, a trial will get underway to compare two doses of secukinumab for patients with HS. Based on a search of clinical trials at ClinicalTrials.gov, a wide range of monoclonal antibody therapies are being investigated for the treatment of HS.
The results of this preliminary study of secukinumab emphasize the importance of the Th17 pathway in HS and open the door to alternative strategies targeting this pathway. Dr. Rosmarin noted that he and his coinvestigators have collected a case series of positive responses to guselkumab (Tremfya), which targets the IL-23 p19 subunit, which also lies along the Th17 pathway.
The secukinumab study was sponsored by Novartis. Dr. Rosmarin reported serving as a consultant to or on speakers’ bureaus for that company and more than half a dozen other pharmaceutical companies.
REPORTING FROM THE EADV CONGRESS
Key clinical point: Secukinumab shows considerable promise for treatment of hidradenitis suppurativa.
Major finding: Hidradenitis suppurativa improved markedly in response to secukinumab in 14 of 18 patients.
Study details: This prospective, open-label, 28-week study included 18 patients with hidradenitis suppurativa who were randomized to one of two secukinumab dosing regimens.
Disclosures: The study was sponsored by Novartis. The presenter reported serving as a consultant to or on speakers’ bureaus for that company and more than half a dozen other pharmaceutical companies.
Delusional infestation: not so rare
PARIS – Ever wonder, when encountering an occasional patient afflicted with delusional infestation, just how common this mental disorder is?
John J. Kohorst, MD, and his coinvestigators at the Mayo Clinic in Rochester, Minn., have the evidence-based answer.
The age- and sex-adjusted point prevalence of delusional infestation among Olmsted County, Minn., residents on the final day of 2010 was 27.3 cases per 100,000 person-years, he reported at the annual congress of the European Academy of Dermatology and Venereology.
“This is the than previously suspected,” according to the dermatologist.
He and his coinvestigators retrospectively analyzed data from the Rochester Epidemiology Project. They identified 22 female and 13 male county residents with a firm diagnosis of delusional infestation, also known as delusional parasitosis. This disorder is marked by a patient’s fixed false belief that they are infested with insects, worms, or other pathogens.
The prevalence was similar in men and women. The most striking study finding was how heavily age-dependent delusional infestation was. Before age 40, the prevalence was a mere 1.2 cases per 100,000 person-years. Among 40- to 59-year-old Olmsted County residents, it was 35/100,000, jumping to 64.5/100,000 in the 60- to 79-year-old age bracket, then doubling to 130.1 cases per 100,000 person-years in individuals aged 80 or older.
Dr. Kohorst reported having no financial conflicts regarding his study, conducted free of commercial support.
PARIS – Ever wonder, when encountering an occasional patient afflicted with delusional infestation, just how common this mental disorder is?
John J. Kohorst, MD, and his coinvestigators at the Mayo Clinic in Rochester, Minn., have the evidence-based answer.
The age- and sex-adjusted point prevalence of delusional infestation among Olmsted County, Minn., residents on the final day of 2010 was 27.3 cases per 100,000 person-years, he reported at the annual congress of the European Academy of Dermatology and Venereology.
“This is the than previously suspected,” according to the dermatologist.
He and his coinvestigators retrospectively analyzed data from the Rochester Epidemiology Project. They identified 22 female and 13 male county residents with a firm diagnosis of delusional infestation, also known as delusional parasitosis. This disorder is marked by a patient’s fixed false belief that they are infested with insects, worms, or other pathogens.
The prevalence was similar in men and women. The most striking study finding was how heavily age-dependent delusional infestation was. Before age 40, the prevalence was a mere 1.2 cases per 100,000 person-years. Among 40- to 59-year-old Olmsted County residents, it was 35/100,000, jumping to 64.5/100,000 in the 60- to 79-year-old age bracket, then doubling to 130.1 cases per 100,000 person-years in individuals aged 80 or older.
Dr. Kohorst reported having no financial conflicts regarding his study, conducted free of commercial support.
PARIS – Ever wonder, when encountering an occasional patient afflicted with delusional infestation, just how common this mental disorder is?
John J. Kohorst, MD, and his coinvestigators at the Mayo Clinic in Rochester, Minn., have the evidence-based answer.
The age- and sex-adjusted point prevalence of delusional infestation among Olmsted County, Minn., residents on the final day of 2010 was 27.3 cases per 100,000 person-years, he reported at the annual congress of the European Academy of Dermatology and Venereology.
“This is the than previously suspected,” according to the dermatologist.
He and his coinvestigators retrospectively analyzed data from the Rochester Epidemiology Project. They identified 22 female and 13 male county residents with a firm diagnosis of delusional infestation, also known as delusional parasitosis. This disorder is marked by a patient’s fixed false belief that they are infested with insects, worms, or other pathogens.
The prevalence was similar in men and women. The most striking study finding was how heavily age-dependent delusional infestation was. Before age 40, the prevalence was a mere 1.2 cases per 100,000 person-years. Among 40- to 59-year-old Olmsted County residents, it was 35/100,000, jumping to 64.5/100,000 in the 60- to 79-year-old age bracket, then doubling to 130.1 cases per 100,000 person-years in individuals aged 80 or older.
Dr. Kohorst reported having no financial conflicts regarding his study, conducted free of commercial support.
REPORTING FROM THE EADV CONGRESS
Key clinical point: Delusional infestation may be more common than previously suspected, particularly among older age groups.
Major finding: The age- and sex-adjusted point prevalence of delusional infestation among residents of one county in southeastern Minnesota is 27.3 cases per 100,000 person-years.
Study details: This was a retrospective analysis of data from the Rochester (Minn.) Epidemiology Project.
Disclosures: The presenter reported having no financial conflicts regarding his study, conducted free of commercial support.
Topical treatment with retinoid/benzoyl peroxide combination reduced acne scars
PARIS – Treatment with the fixed combination in a multicenter, randomized trial, Brigitte Dreno, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.

“To my knowledge, this is the first time that we have seen a topical therapy showing a reduction in atrophic acne scars,” said Dr. Dreno, professor and chair of the department of dermatology at Nantes (France) University Hospital.
She reported on 67 adolescents and adults with mainly moderate facial acne randomized to treat half their face with adapalene 0.3%/benzoyl peroxide 2.5% gel (Epiduo Forte) and the other half with the product’s vehicle daily for 6 months. Investigators were blinded as to which side was which. At baseline, patients averaged 40 acne lesions and 12 scars per half face.
The primary efficacy endpoint was the atrophic acne scar count per half face at week 24. At that point, the mean total was 9.5 scars on the active treatment side, compared with 13.3 on the control side. This translated to a statistically significant and clinically meaningful 15.5% decrease in scars with active treatment versus a 14.4% increase with vehicle. The between-side difference achieved statistical significance at week 1 and remained so at all follow-up visits through week 24.
By Scar Global Assessment at week 24, 32.9% of half faces treated with the combination product were rated clear or almost clear, compared with 16.4% with vehicle.
At 24 weeks, 24.1% of participants reported having moderately or very visible holes or indents on the active treatment side of their face, compared with 51.8% on the control side. The number of inflammatory acne lesions fell by 86.7% with the active treatment and 57.9% with vehicle over the course of 24 weeks. Again, the difference became statistically significant starting at week 1. By the Investigator’s Global Assessment at week 24, 64.2% of adapalene/benzoyl peroxide gel–treated faces were rated clear or almost clear, as were 19.4% with vehicle. In addition, 32% of patients reported a marked improvement in skin texture on their active treatment side at 24 weeks, as did 14% on the control side.
The salutary effect on acne scars documented with a topical therapy in this study represents a real advance in clinical care.
“Facial acne scars are a very important and difficult problem for our patients and also for dermatologists,” Dr. Dreno observed, adding that the evidence base for procedural interventions for acne scars, such as dermabrasion and laser resurfacing, is not top quality.
Not surprisingly with a topical retinoid, skin irritation was the most common treatment-emergent adverse event, reported by 14.9% of patients on their active treatment side and 6% with vehicle. This side effect was typically mild and resolved within the first 2-3 weeks.
The improvement in preexisting acne scars documented in this trial was probably caused by drug-induced remodeling of the dermal matrix, according to Dr. Dreno.
The study was funded by Galderma. Dr. Dreno reported receiving research grants from and/or serving as a consultant to Galderma, Bioderma, Pierre Fabre, and La Roche–Posay.
PARIS – Treatment with the fixed combination in a multicenter, randomized trial, Brigitte Dreno, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“To my knowledge, this is the first time that we have seen a topical therapy showing a reduction in atrophic acne scars,” said Dr. Dreno, professor and chair of the department of dermatology at Nantes (France) University Hospital.
She reported on 67 adolescents and adults with mainly moderate facial acne randomized to treat half their face with adapalene 0.3%/benzoyl peroxide 2.5% gel (Epiduo Forte) and the other half with the product’s vehicle daily for 6 months. Investigators were blinded as to which side was which. At baseline, patients averaged 40 acne lesions and 12 scars per half face.
The primary efficacy endpoint was the atrophic acne scar count per half face at week 24. At that point, the mean total was 9.5 scars on the active treatment side, compared with 13.3 on the control side. This translated to a statistically significant and clinically meaningful 15.5% decrease in scars with active treatment versus a 14.4% increase with vehicle. The between-side difference achieved statistical significance at week 1 and remained so at all follow-up visits through week 24.
By Scar Global Assessment at week 24, 32.9% of half faces treated with the combination product were rated clear or almost clear, compared with 16.4% with vehicle.
At 24 weeks, 24.1% of participants reported having moderately or very visible holes or indents on the active treatment side of their face, compared with 51.8% on the control side. The number of inflammatory acne lesions fell by 86.7% with the active treatment and 57.9% with vehicle over the course of 24 weeks. Again, the difference became statistically significant starting at week 1. By the Investigator’s Global Assessment at week 24, 64.2% of adapalene/benzoyl peroxide gel–treated faces were rated clear or almost clear, as were 19.4% with vehicle. In addition, 32% of patients reported a marked improvement in skin texture on their active treatment side at 24 weeks, as did 14% on the control side.
The salutary effect on acne scars documented with a topical therapy in this study represents a real advance in clinical care.
“Facial acne scars are a very important and difficult problem for our patients and also for dermatologists,” Dr. Dreno observed, adding that the evidence base for procedural interventions for acne scars, such as dermabrasion and laser resurfacing, is not top quality.
Not surprisingly with a topical retinoid, skin irritation was the most common treatment-emergent adverse event, reported by 14.9% of patients on their active treatment side and 6% with vehicle. This side effect was typically mild and resolved within the first 2-3 weeks.
The improvement in preexisting acne scars documented in this trial was probably caused by drug-induced remodeling of the dermal matrix, according to Dr. Dreno.
The study was funded by Galderma. Dr. Dreno reported receiving research grants from and/or serving as a consultant to Galderma, Bioderma, Pierre Fabre, and La Roche–Posay.
PARIS – Treatment with the fixed combination in a multicenter, randomized trial, Brigitte Dreno, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“To my knowledge, this is the first time that we have seen a topical therapy showing a reduction in atrophic acne scars,” said Dr. Dreno, professor and chair of the department of dermatology at Nantes (France) University Hospital.
She reported on 67 adolescents and adults with mainly moderate facial acne randomized to treat half their face with adapalene 0.3%/benzoyl peroxide 2.5% gel (Epiduo Forte) and the other half with the product’s vehicle daily for 6 months. Investigators were blinded as to which side was which. At baseline, patients averaged 40 acne lesions and 12 scars per half face.
The primary efficacy endpoint was the atrophic acne scar count per half face at week 24. At that point, the mean total was 9.5 scars on the active treatment side, compared with 13.3 on the control side. This translated to a statistically significant and clinically meaningful 15.5% decrease in scars with active treatment versus a 14.4% increase with vehicle. The between-side difference achieved statistical significance at week 1 and remained so at all follow-up visits through week 24.
By Scar Global Assessment at week 24, 32.9% of half faces treated with the combination product were rated clear or almost clear, compared with 16.4% with vehicle.
At 24 weeks, 24.1% of participants reported having moderately or very visible holes or indents on the active treatment side of their face, compared with 51.8% on the control side. The number of inflammatory acne lesions fell by 86.7% with the active treatment and 57.9% with vehicle over the course of 24 weeks. Again, the difference became statistically significant starting at week 1. By the Investigator’s Global Assessment at week 24, 64.2% of adapalene/benzoyl peroxide gel–treated faces were rated clear or almost clear, as were 19.4% with vehicle. In addition, 32% of patients reported a marked improvement in skin texture on their active treatment side at 24 weeks, as did 14% on the control side.
The salutary effect on acne scars documented with a topical therapy in this study represents a real advance in clinical care.
“Facial acne scars are a very important and difficult problem for our patients and also for dermatologists,” Dr. Dreno observed, adding that the evidence base for procedural interventions for acne scars, such as dermabrasion and laser resurfacing, is not top quality.
Not surprisingly with a topical retinoid, skin irritation was the most common treatment-emergent adverse event, reported by 14.9% of patients on their active treatment side and 6% with vehicle. This side effect was typically mild and resolved within the first 2-3 weeks.
The improvement in preexisting acne scars documented in this trial was probably caused by drug-induced remodeling of the dermal matrix, according to Dr. Dreno.
The study was funded by Galderma. Dr. Dreno reported receiving research grants from and/or serving as a consultant to Galderma, Bioderma, Pierre Fabre, and La Roche–Posay.
REPORTING FROM THE EADV CONGRESS
Key clinical point: A fixed combination adapalene 0.3%/benzoyl peroxide 2.5% gel reduced the number of preexisting facial atrophic acne scars and prevented new scar formation.
Major finding: Facial atrophic acne scar count dropped by 15.5% with 6 months of treatment with adapalene 0.3%/benzoyl peroxide 2.5% gel while increasing by 14.4% with vehicle.
Study details: This was a 6-month, prospective, multicenter, randomized, vehicle-controlled, split-face study involving 67 acne patients.
Disclosures: The study was funded by Galderma. The presenter reported receiving research grants from and/or serving as a consultant to Galderma, Bioderma, Pierre Fabre, and La Roche–Posay.
Serlopitant quells prurigo nodularis in phase 2 study
PARIS – The in a 127-patient, randomized, placebo-controlled trial, Sonja Stander, MD, said at the annual congress of the European Academy of Dermatology and Venereology.

She used her speaker’s platform not only to present key outcomes of the phase 2 study, but also to explain some of the underappreciated aspects of prurigo nodularis and how the results shed new light on the pathogenesis of the disease.
“There is a knowledge gap regarding this disease,” observed Dr. Stander, professor of dermatology at the University of Munster (Germany).
Prurigo nodularis is a chronic skin condition characterized by numerous intensely pruritic, symmetrically distributed, erosive nodular lesions. As Dr. Stander and her fellow members of the EADV Task Force Pruritus recently reported, prurigo nodularis should be considered as one of the clinical manifestations of chronic prurigo, hallmarks of which include neuronal sensitization to itch and development of an itch-scratch cycle (J Eur Acad Dermatol Venereol. 2018 Jul;32[7]:1059-65). At present, there is no approved treatment for prurigo nodularis in the United States or Europe.
Despite its name, prurigo nodularis is not just about the itch. Additional symptoms were frequently reported by participants in the serlopitant study. Indeed, while 97% of participants reported experiencing devilish pruritus, 52% reported a burning sensation on involved skin, 41% said the lesions were painful, and 36% indicated they were plagued by a stinging sensation. All of these disturbing sensations were significantly reduced with 8 weeks of serlopitant in the trial.

The clinical implications of these baseline findings are clear: “We have to ask not only about itch, but also about pain and burning and stinging, which are really an issue with these patients,” the dermatologist said.
Dr. Stander noted that prurigo nodularis has a large negative impact on quality of life as reflected in study participants’ mean baseline Dermatology Life Quality Index score of 61. And it’s a disease that sticks around: At enrollment, 38% of subjects had a 1- to 5-year history of prurigo nodularis, 20% had had the disease for 5-10 years, and fully 29% had had prurigo nodularis for longer than 10 years.
“This is not a chronic relapsing disease. These patients had prurigo nodularis all the time,” Dr. Stander said.
Consistent with the findings of other studies, half of patients with prurigo nodularis had an atopic predisposition as defined by an Erlangen Atopy Questionnaire score of 10 or higher.
Patients in the phase 2 study were randomized to either a 15-mg loading dose of oral serlopitant followed by 5 mg/day for 8 weeks or to placebo. All four of the most common manifestations of prurigo nodularis – itching, burning, pain, and stinging – were reduced to a significantly greater extent in the serlopitant group than with placebo. For example, the proportion of serlopitant-treated patients who rated their itching as none or mild went from zero at baseline to 54% at week 8, as compared with 26% among placebo-treated controls. After 8 weeks, 46% of serlopitant-treated patients rated their itching as moderate, severe, or very severe, as did 71% of controls.
Similarly, the proportion of patients in the serlopitant group who reported no or only a mild burning sensation climbed from 41% at baseline to 73% after 8 weeks. In the control group, the proportion improved from 36% to 47% over the course of 8 weeks. At week 8, 27% of patients on serlopitant characterized themselves as experiencing a moderate, severe, or very severe burning sensation; the rate was twice as high in controls.
Although the stinging sensation associated with prurigo nodularis was significantly reduced by serlopitant, the effect was less robust than with the other common sensory expressions of the disease. The proportion of patients with no or mild stinging increased by an absolute 23% after 8 weeks on serlopitant and by 6% with placebo. At week 8, 22% of patients treated with the neurokinin-1 receptor antagonist complained of a moderate to very severe stinging sensation, as did 40% of controls.
As a neurokinin-1 receptor antagonist, serlopitant inhibits substance P. The drug’s efficacy suggests that substance P plays a role in inducing the itch and other sensory symptoms of prurigo nodularis, according to Dr. Stander.
Based upon the favorable phase 2 results, multiple phase 3 clinical trials of serlopitant for prurigo nodularis are underway in the United States and elsewhere, including a year-long safety study.
Serlopitant is being developed by Menlo Therapeutics. Dr. Stander reported receiving research funding from and serving as a consultant to the company.
PARIS – The in a 127-patient, randomized, placebo-controlled trial, Sonja Stander, MD, said at the annual congress of the European Academy of Dermatology and Venereology.
She used her speaker’s platform not only to present key outcomes of the phase 2 study, but also to explain some of the underappreciated aspects of prurigo nodularis and how the results shed new light on the pathogenesis of the disease.
“There is a knowledge gap regarding this disease,” observed Dr. Stander, professor of dermatology at the University of Munster (Germany).
Prurigo nodularis is a chronic skin condition characterized by numerous intensely pruritic, symmetrically distributed, erosive nodular lesions. As Dr. Stander and her fellow members of the EADV Task Force Pruritus recently reported, prurigo nodularis should be considered as one of the clinical manifestations of chronic prurigo, hallmarks of which include neuronal sensitization to itch and development of an itch-scratch cycle (J Eur Acad Dermatol Venereol. 2018 Jul;32[7]:1059-65). At present, there is no approved treatment for prurigo nodularis in the United States or Europe.
Despite its name, prurigo nodularis is not just about the itch. Additional symptoms were frequently reported by participants in the serlopitant study. Indeed, while 97% of participants reported experiencing devilish pruritus, 52% reported a burning sensation on involved skin, 41% said the lesions were painful, and 36% indicated they were plagued by a stinging sensation. All of these disturbing sensations were significantly reduced with 8 weeks of serlopitant in the trial.
The clinical implications of these baseline findings are clear: “We have to ask not only about itch, but also about pain and burning and stinging, which are really an issue with these patients,” the dermatologist said.
Dr. Stander noted that prurigo nodularis has a large negative impact on quality of life as reflected in study participants’ mean baseline Dermatology Life Quality Index score of 61. And it’s a disease that sticks around: At enrollment, 38% of subjects had a 1- to 5-year history of prurigo nodularis, 20% had had the disease for 5-10 years, and fully 29% had had prurigo nodularis for longer than 10 years.
“This is not a chronic relapsing disease. These patients had prurigo nodularis all the time,” Dr. Stander said.
Consistent with the findings of other studies, half of patients with prurigo nodularis had an atopic predisposition as defined by an Erlangen Atopy Questionnaire score of 10 or higher.
Patients in the phase 2 study were randomized to either a 15-mg loading dose of oral serlopitant followed by 5 mg/day for 8 weeks or to placebo. All four of the most common manifestations of prurigo nodularis – itching, burning, pain, and stinging – were reduced to a significantly greater extent in the serlopitant group than with placebo. For example, the proportion of serlopitant-treated patients who rated their itching as none or mild went from zero at baseline to 54% at week 8, as compared with 26% among placebo-treated controls. After 8 weeks, 46% of serlopitant-treated patients rated their itching as moderate, severe, or very severe, as did 71% of controls.
Similarly, the proportion of patients in the serlopitant group who reported no or only a mild burning sensation climbed from 41% at baseline to 73% after 8 weeks. In the control group, the proportion improved from 36% to 47% over the course of 8 weeks. At week 8, 27% of patients on serlopitant characterized themselves as experiencing a moderate, severe, or very severe burning sensation; the rate was twice as high in controls.
Although the stinging sensation associated with prurigo nodularis was significantly reduced by serlopitant, the effect was less robust than with the other common sensory expressions of the disease. The proportion of patients with no or mild stinging increased by an absolute 23% after 8 weeks on serlopitant and by 6% with placebo. At week 8, 22% of patients treated with the neurokinin-1 receptor antagonist complained of a moderate to very severe stinging sensation, as did 40% of controls.
As a neurokinin-1 receptor antagonist, serlopitant inhibits substance P. The drug’s efficacy suggests that substance P plays a role in inducing the itch and other sensory symptoms of prurigo nodularis, according to Dr. Stander.
Based upon the favorable phase 2 results, multiple phase 3 clinical trials of serlopitant for prurigo nodularis are underway in the United States and elsewhere, including a year-long safety study.
Serlopitant is being developed by Menlo Therapeutics. Dr. Stander reported receiving research funding from and serving as a consultant to the company.
PARIS – The in a 127-patient, randomized, placebo-controlled trial, Sonja Stander, MD, said at the annual congress of the European Academy of Dermatology and Venereology.
She used her speaker’s platform not only to present key outcomes of the phase 2 study, but also to explain some of the underappreciated aspects of prurigo nodularis and how the results shed new light on the pathogenesis of the disease.
“There is a knowledge gap regarding this disease,” observed Dr. Stander, professor of dermatology at the University of Munster (Germany).
Prurigo nodularis is a chronic skin condition characterized by numerous intensely pruritic, symmetrically distributed, erosive nodular lesions. As Dr. Stander and her fellow members of the EADV Task Force Pruritus recently reported, prurigo nodularis should be considered as one of the clinical manifestations of chronic prurigo, hallmarks of which include neuronal sensitization to itch and development of an itch-scratch cycle (J Eur Acad Dermatol Venereol. 2018 Jul;32[7]:1059-65). At present, there is no approved treatment for prurigo nodularis in the United States or Europe.
Despite its name, prurigo nodularis is not just about the itch. Additional symptoms were frequently reported by participants in the serlopitant study. Indeed, while 97% of participants reported experiencing devilish pruritus, 52% reported a burning sensation on involved skin, 41% said the lesions were painful, and 36% indicated they were plagued by a stinging sensation. All of these disturbing sensations were significantly reduced with 8 weeks of serlopitant in the trial.
The clinical implications of these baseline findings are clear: “We have to ask not only about itch, but also about pain and burning and stinging, which are really an issue with these patients,” the dermatologist said.
Dr. Stander noted that prurigo nodularis has a large negative impact on quality of life as reflected in study participants’ mean baseline Dermatology Life Quality Index score of 61. And it’s a disease that sticks around: At enrollment, 38% of subjects had a 1- to 5-year history of prurigo nodularis, 20% had had the disease for 5-10 years, and fully 29% had had prurigo nodularis for longer than 10 years.
“This is not a chronic relapsing disease. These patients had prurigo nodularis all the time,” Dr. Stander said.
Consistent with the findings of other studies, half of patients with prurigo nodularis had an atopic predisposition as defined by an Erlangen Atopy Questionnaire score of 10 or higher.
Patients in the phase 2 study were randomized to either a 15-mg loading dose of oral serlopitant followed by 5 mg/day for 8 weeks or to placebo. All four of the most common manifestations of prurigo nodularis – itching, burning, pain, and stinging – were reduced to a significantly greater extent in the serlopitant group than with placebo. For example, the proportion of serlopitant-treated patients who rated their itching as none or mild went from zero at baseline to 54% at week 8, as compared with 26% among placebo-treated controls. After 8 weeks, 46% of serlopitant-treated patients rated their itching as moderate, severe, or very severe, as did 71% of controls.
Similarly, the proportion of patients in the serlopitant group who reported no or only a mild burning sensation climbed from 41% at baseline to 73% after 8 weeks. In the control group, the proportion improved from 36% to 47% over the course of 8 weeks. At week 8, 27% of patients on serlopitant characterized themselves as experiencing a moderate, severe, or very severe burning sensation; the rate was twice as high in controls.
Although the stinging sensation associated with prurigo nodularis was significantly reduced by serlopitant, the effect was less robust than with the other common sensory expressions of the disease. The proportion of patients with no or mild stinging increased by an absolute 23% after 8 weeks on serlopitant and by 6% with placebo. At week 8, 22% of patients treated with the neurokinin-1 receptor antagonist complained of a moderate to very severe stinging sensation, as did 40% of controls.
As a neurokinin-1 receptor antagonist, serlopitant inhibits substance P. The drug’s efficacy suggests that substance P plays a role in inducing the itch and other sensory symptoms of prurigo nodularis, according to Dr. Stander.
Based upon the favorable phase 2 results, multiple phase 3 clinical trials of serlopitant for prurigo nodularis are underway in the United States and elsewhere, including a year-long safety study.
Serlopitant is being developed by Menlo Therapeutics. Dr. Stander reported receiving research funding from and serving as a consultant to the company.
REPORTING FROM THE EADV CONGRESS
Key clinical point: Serlopitant decreases the disruptive itch, burning, pain, and stinging of prurigo nodularis.
Major finding: The proportion of prurigo nodularis patients who rated their itching as none or mild went from zero at baseline to 54% after 8 weeks on oral serlopitant.
Study details: This 8-week, randomized, double-blind phase 2 study included 127 patients with prurigo nodularis who were randomized to oral serlopitant or placebo.
Disclosures: Serlopitant is being developed by Menlo Therapeutics. The presenter reported receiving research funding from and serving as a consultant to the company.
A new era arrives in alopecia areata therapy
PARIS – Each of two oral Janus kinase inhibitors individually showed impressive efficacy and acceptable safety and tolerability for treatment of chronic moderate to severe alopecia areata in a phase 2 study that promises to alter the therapeutic landscape in this challenging disease, Rodney Sinclair, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.

“My personal view is that these results represent a paradigm shift in the treatment of alopecia areata. Perhaps a line in the sand. On the one side, clinical observation, case reports, and small case series, but no reliably effective treatment. And on the other side of the line, investigational new drugs specifically targeting the pathogenesis, tested in prospective, multicenter, double-blind, placebo-controlled studies – and perhaps the first instance of evidence-based medicine arriving for alopecia areata. And I think that our patients with alopecia areata now have reason to be optimistic,” the dermatologist said.
“No reliably effective therapies now exist for alopecia areata, especially for patients with chronic extensive disease. There is an unmet need for a reliably effective therapy with a benefit-to-risk ratio that is appropriate for long-term use,” he added.
The phase 2 multicenter, placebo-controlled, double-blind study randomized 142 patients with chronic alopecia of at least 6 months duration and 50% or greater hair loss at baseline to an , or placebo for 24 weeks. The first 4 weeks of the double-blind trial involved induction dosing of the JAK3 inhibitor at 200 mg per day and the TYK2/JAK 1 inhibitor at 60 mg per day. Thereafter, participants switched to maintenance dosing at 50 mg and 30 mg per day, respectively.
Participants had a median 2.3 years of disease duration; 62 patients had alopecia totalis, and 42 had alopecia universalis.
The primary efficacy endpoint was the mean change from baseline to week 24 in the Severity of Alopecia Tool (SALT) score, a well-validated metric widely utilized in hair loss studies. The mean baseline score was 88.1. At week 24, the score had dropped by a mean of 33.6% in the JAK 3 inhibitor group, 49.5% in the TYK2/JAK1 inhibitor group, and zero in placebo-treated controls.
A key secondary endpoint known as the SALT 30, reflecting at least 30% improvement in SALT score, was achieved in 47.9% of patients on the oral JAK 3 inhibitor, 59.6% of those on the TYK2/JAK1 inhibitor, and by no one in the control group. The utter unresponsiveness of placebo-treated controls confirms the investigators’ success in recruiting a study population with truly chronic alopecia areata, Dr. Sinclair noted.
Of note, 25% of patients on the JAK 3 inhibitor and 31.9% on the TYK2/JAK1 inhibitor achieved a SALT 90 response, and 12.5% and 12.8% reached SALT 100.
Significant improvement on the Eyelash Assessment and Eyebrow Assessment Scales occurred in 45.2% and 36.2% of the JAK 3 inhibitor group and in 60.7% and 51.7% of the TYK2/JAK 1 inhibitor group.
The study is ongoing. It’s Dr. Sinclair’s anecdotal impression that further improvement is occurring in the active treatment arms with continued therapy after 24 weeks, but that remains to be seen.
Adverse events were similar in nature and frequency in all three study arms with one notable exception: two cases of rhabdomyolysis in the TYK2/JAK 1 inhibitor group.Session chair DeDee Murrell, MD, homed in on that piece of information, pressing for further details.
“Are you concerned? Were there predictors?” asked Dr. Murrell, professor of dermatology at the University of New South Wales in Sydney.
The two affected patients had muscle pain and elevated creatinine kinase levels. “They didn’t feel particularly unwell but were withdrawn as a precaution, after which the condition quickly resolved,” Dr. Sinclair said.
The investigators were unable to identify any risk factors for rhabdomyolysis in the study population, he added.
Dr. Sinclair reported receiving research grants from and serving as a consultant to Pfizer, which sponsored the phase 2 study, as well as more than a dozen other pharmaceutical companies.
PARIS – Each of two oral Janus kinase inhibitors individually showed impressive efficacy and acceptable safety and tolerability for treatment of chronic moderate to severe alopecia areata in a phase 2 study that promises to alter the therapeutic landscape in this challenging disease, Rodney Sinclair, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“My personal view is that these results represent a paradigm shift in the treatment of alopecia areata. Perhaps a line in the sand. On the one side, clinical observation, case reports, and small case series, but no reliably effective treatment. And on the other side of the line, investigational new drugs specifically targeting the pathogenesis, tested in prospective, multicenter, double-blind, placebo-controlled studies – and perhaps the first instance of evidence-based medicine arriving for alopecia areata. And I think that our patients with alopecia areata now have reason to be optimistic,” the dermatologist said.
“No reliably effective therapies now exist for alopecia areata, especially for patients with chronic extensive disease. There is an unmet need for a reliably effective therapy with a benefit-to-risk ratio that is appropriate for long-term use,” he added.
The phase 2 multicenter, placebo-controlled, double-blind study randomized 142 patients with chronic alopecia of at least 6 months duration and 50% or greater hair loss at baseline to an , or placebo for 24 weeks. The first 4 weeks of the double-blind trial involved induction dosing of the JAK3 inhibitor at 200 mg per day and the TYK2/JAK 1 inhibitor at 60 mg per day. Thereafter, participants switched to maintenance dosing at 50 mg and 30 mg per day, respectively.
Participants had a median 2.3 years of disease duration; 62 patients had alopecia totalis, and 42 had alopecia universalis.
The primary efficacy endpoint was the mean change from baseline to week 24 in the Severity of Alopecia Tool (SALT) score, a well-validated metric widely utilized in hair loss studies. The mean baseline score was 88.1. At week 24, the score had dropped by a mean of 33.6% in the JAK 3 inhibitor group, 49.5% in the TYK2/JAK1 inhibitor group, and zero in placebo-treated controls.
A key secondary endpoint known as the SALT 30, reflecting at least 30% improvement in SALT score, was achieved in 47.9% of patients on the oral JAK 3 inhibitor, 59.6% of those on the TYK2/JAK1 inhibitor, and by no one in the control group. The utter unresponsiveness of placebo-treated controls confirms the investigators’ success in recruiting a study population with truly chronic alopecia areata, Dr. Sinclair noted.
Of note, 25% of patients on the JAK 3 inhibitor and 31.9% on the TYK2/JAK1 inhibitor achieved a SALT 90 response, and 12.5% and 12.8% reached SALT 100.
Significant improvement on the Eyelash Assessment and Eyebrow Assessment Scales occurred in 45.2% and 36.2% of the JAK 3 inhibitor group and in 60.7% and 51.7% of the TYK2/JAK 1 inhibitor group.
The study is ongoing. It’s Dr. Sinclair’s anecdotal impression that further improvement is occurring in the active treatment arms with continued therapy after 24 weeks, but that remains to be seen.
Adverse events were similar in nature and frequency in all three study arms with one notable exception: two cases of rhabdomyolysis in the TYK2/JAK 1 inhibitor group.Session chair DeDee Murrell, MD, homed in on that piece of information, pressing for further details.
“Are you concerned? Were there predictors?” asked Dr. Murrell, professor of dermatology at the University of New South Wales in Sydney.
The two affected patients had muscle pain and elevated creatinine kinase levels. “They didn’t feel particularly unwell but were withdrawn as a precaution, after which the condition quickly resolved,” Dr. Sinclair said.
The investigators were unable to identify any risk factors for rhabdomyolysis in the study population, he added.
Dr. Sinclair reported receiving research grants from and serving as a consultant to Pfizer, which sponsored the phase 2 study, as well as more than a dozen other pharmaceutical companies.
PARIS – Each of two oral Janus kinase inhibitors individually showed impressive efficacy and acceptable safety and tolerability for treatment of chronic moderate to severe alopecia areata in a phase 2 study that promises to alter the therapeutic landscape in this challenging disease, Rodney Sinclair, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“My personal view is that these results represent a paradigm shift in the treatment of alopecia areata. Perhaps a line in the sand. On the one side, clinical observation, case reports, and small case series, but no reliably effective treatment. And on the other side of the line, investigational new drugs specifically targeting the pathogenesis, tested in prospective, multicenter, double-blind, placebo-controlled studies – and perhaps the first instance of evidence-based medicine arriving for alopecia areata. And I think that our patients with alopecia areata now have reason to be optimistic,” the dermatologist said.
“No reliably effective therapies now exist for alopecia areata, especially for patients with chronic extensive disease. There is an unmet need for a reliably effective therapy with a benefit-to-risk ratio that is appropriate for long-term use,” he added.
The phase 2 multicenter, placebo-controlled, double-blind study randomized 142 patients with chronic alopecia of at least 6 months duration and 50% or greater hair loss at baseline to an , or placebo for 24 weeks. The first 4 weeks of the double-blind trial involved induction dosing of the JAK3 inhibitor at 200 mg per day and the TYK2/JAK 1 inhibitor at 60 mg per day. Thereafter, participants switched to maintenance dosing at 50 mg and 30 mg per day, respectively.
Participants had a median 2.3 years of disease duration; 62 patients had alopecia totalis, and 42 had alopecia universalis.
The primary efficacy endpoint was the mean change from baseline to week 24 in the Severity of Alopecia Tool (SALT) score, a well-validated metric widely utilized in hair loss studies. The mean baseline score was 88.1. At week 24, the score had dropped by a mean of 33.6% in the JAK 3 inhibitor group, 49.5% in the TYK2/JAK1 inhibitor group, and zero in placebo-treated controls.
A key secondary endpoint known as the SALT 30, reflecting at least 30% improvement in SALT score, was achieved in 47.9% of patients on the oral JAK 3 inhibitor, 59.6% of those on the TYK2/JAK1 inhibitor, and by no one in the control group. The utter unresponsiveness of placebo-treated controls confirms the investigators’ success in recruiting a study population with truly chronic alopecia areata, Dr. Sinclair noted.
Of note, 25% of patients on the JAK 3 inhibitor and 31.9% on the TYK2/JAK1 inhibitor achieved a SALT 90 response, and 12.5% and 12.8% reached SALT 100.
Significant improvement on the Eyelash Assessment and Eyebrow Assessment Scales occurred in 45.2% and 36.2% of the JAK 3 inhibitor group and in 60.7% and 51.7% of the TYK2/JAK 1 inhibitor group.
The study is ongoing. It’s Dr. Sinclair’s anecdotal impression that further improvement is occurring in the active treatment arms with continued therapy after 24 weeks, but that remains to be seen.
Adverse events were similar in nature and frequency in all three study arms with one notable exception: two cases of rhabdomyolysis in the TYK2/JAK 1 inhibitor group.Session chair DeDee Murrell, MD, homed in on that piece of information, pressing for further details.
“Are you concerned? Were there predictors?” asked Dr. Murrell, professor of dermatology at the University of New South Wales in Sydney.
The two affected patients had muscle pain and elevated creatinine kinase levels. “They didn’t feel particularly unwell but were withdrawn as a precaution, after which the condition quickly resolved,” Dr. Sinclair said.
The investigators were unable to identify any risk factors for rhabdomyolysis in the study population, he added.
Dr. Sinclair reported receiving research grants from and serving as a consultant to Pfizer, which sponsored the phase 2 study, as well as more than a dozen other pharmaceutical companies.
REPORTING FROM THE EADV CONGRESS
Key clinical point: Targeted therapy with Janus kinase inhibitors shows great promise in chronic alopecia areata.
Major finding: Mean placebo-subtracted improvement in Severity of Alopecia Tool scores was 50% after 24 weeks of treatment with an oral TYK2/JAK 1 inhibitor.
Study details: This was a prospective, multicenter, double-blind, placebo-controlled, 24-week phase 2 study of 142 patients with chronic moderate to severe alopecia areata.
Disclosures: The presenter reported receiving research grants from and serving as a consultant to Pfizer, which sponsored the phase 2 study, as well as to more than a dozen other pharmaceutical companies.
Dupilumab positive in phase 3 study for treating adolescent atopic dermatitis
PARIS – Dupilumab scorched its way through a landmark pivotal phase 3 clinical trial in adolescents with moderate to severe atopic dermatitis (AD), achieving unprecedented clinically meaningful improvements in signs and symptoms of the disease along with important quality of life benefits, Eric L. Simpson, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.

Indeed, akin to that previously shown in adults with moderate to severe AD in phase 3 trials that earned the biologic U.S. and European regulatory approval in the adult population, noted Dr. Simpson, professor of dermatology at Oregon Health & Science University, Portland.
This positive phase 3 study represents a major development in pediatric dermatology because of the pressing unmet need for better treatments for teens with moderate to severe AD whose disease can’t be controlled with topical therapies. The adolescent years are, after all, a critical period in growth and development, and a debilitating, uncontrolled disease can reshape that experience in unwelcome ways.
“Atopic dermatitis profoundly affects quality of life in adolescents and their family: The itching affects mood and sleep, these patients commonly have anxiety and depression, and the chronic and relapsing nature of the disease adversely affects the family,” the dermatologist observed.
Currently, no systemic agent is approved for pediatric patients with AD because evidence demonstrating a favorable benefit-to-risk profile has been lacking. The dupilumab study was the first-ever phase 3 trial of a biologic in such a population.
The phase 3 adolescent trial was a 16-week, randomized, double-blind, multicenter, placebo-controlled study of 251 patients aged 12-17 years with moderate to severe AD, which could not be adequately controlled with topical therapies. Participants averaged 14 years of age, with a 12-year history of AD. “These patients had the disease basically their whole life,” Dr. Simpson noted.
They were more severely affected than participants in the adult clinical trials, with mean Eczema Area And Severity Index (EASI) scores in the mid-30s and an average 56% involved body surface area. The adolescent AD patients had a heavy burden of comorbid allergic type 2 immune comorbidity: Fully 92% of them had documented asthma, food allergy, allergic rhinitis, and/or some other form of allergic comorbidity. The majority of the teens were categorized as having severe AD, whereas most participants in the adult phase 3 trials of dupilumab had moderate disease. That distinction becomes relevant in comparing the trial results.
Participants were randomized to once-monthly subcutaneous injections of dupilumab at 300 mg, following a 600-mg loading dose, or to an injection of 200 mg or 300 mg every 2 weeks with an initial dose of 400 mg or 600 mg based upon a body weight cutoff of 60 kg, or to biweekly placebo injections.
The coprimary endpoints were the proportion of patients who achieved an EASI 75 response at week 16 and achievement of an Investigator’s Global Assessment (IGA) score of 0 or 1, meaning clear or almost clear, on a 5-point scale at week 16.
This trial introduced an important new design feature that physicians can expect to see more of in the future: regulatory agencies now want to see the effects of monotherapy in pivotal studies in AD. Previously, participants in AD studies of systemic agents could also utilize topical steroids as needed. No longer. In the adolescent dupilumab study, resort to rescue topical steroids led to exclusion from inclusion in the primary outcome results. Not surprisingly, this lack of access to rescue medication resulted in a 60% dropout rate by 16 weeks in placebo-treated controls, a 30% dropout rate in teens on dupilumab every 4 weeks, and a 20% dropout rate with biweekly dupilumab.
The EASI 75 rate at week 16 was 8.2% with placebo, 38.1% with monthly dupilumab, and 41.5% with biweekly dosing. The other coprimary endpoint – an IGA of 0 or 1 at 16 weeks – was achieved in 2.4% of controls, 17.9% with dupilumab at 300 mg every 4 weeks, and 41.5% with biweekly dosing.
Turning to secondary endpoints, Dr. Simpson reported that baseline peak pruritus Numeric Rating Scale scores dropped by 19% with placebo at 16 weeks, compared with reductions of 45.5% and 47.9% with monthly and biweekly dupilumab, respectively.
An EASI 50 response,while not as impressive as an EASI 75, is nonetheless considered clinically meaningful improvement. It was attained in 12.9% on placebo and 54.8% and 61% on monthly and biweekly dupilumab.
From a mean baseline score of 13.6 on the Children’s Dermatology Life Quality Index, scores improved over the course of 16 weeks by a mean of 5.1 points with placebo, 8.5 points with monthly dupilumab, and 8.8 points with biweekly biologic therapy. The same pattern was noted with regard to the Patient-Oriented Eczema Measure or POEM.
Adverse events mirrored those documented in the pivotal trials in adults: increased rates of mild to moderate conjunctivitis: 4.7% with placebo, 10.8% with monthly dupilumab, and 9.8% with biweekly dupilumab, along with single-digit rates of injection-site reactions. As in adult AD patients, however, these side effects were counterbalanced by significantly reduced rates of skin infections in the adolescent group: 20% during 16 weeks with placebo, and 13.3% and 11% with monthly and biweekly biologic therapy, respectively.
Study participants had a mean baseline score of 12.5 on the Hospital Anxiety and Depression Scale, which is categorized as clinically significant psychiatric disease. The impact of dupilumab therapy on those scores will be the topic of a future presentation, Dr. Simpson said.
Across the board, specific outcomes were consistently numerically better in patients who received dupilumab biweekly than monthly, albeit not statistically significantly so. Dr. Simpson thinks he knows why: Lab studies showed that the mean serum concentration of functional dupilumab in patients who got the biologic biweekly was nearly twice that for the group with monthly dosing.
At first glance, the IGA “clear” or “almost clear” response rates seen with dupilumab in the adolescent study appeared to be less robust than in the adult pivotal phase 3 trials, such as the SOLO 1 and SOLO 2 trials (N Engl J Med. 2016 Dec 15;375[24]:2335-48), also led by Dr. Simpson.
“I think that’s because of the greater severity of that baseline adolescent population,” he commented. “It made for a much lower placebo response rate. But when you correct for the placebo-subtracted difference, the rates are actually pretty similar, and a lot of the other endpoints are the same or even better than in SOLO.”
After his presentation, Dr. Simpson commented, “This is huge. This is the study we’ve been waiting for.”
Elsewhere at the EADV congress, Emma Guttman-Yassky, MD, PhD, deemed the pivotal phase 3 trial in adolescent AD patients one of the meeting’s highlights. And it’s a harbinger of more good things to come, because the investigational drug pipeline for AD is full of promising candidates addressing the disease from a variety of novel directions. The long therapeutic drought in AD appears to have finally ended, observed Dr. Guttman-Yassky, professor and vice-chair of the department of dermatology at Icahn School of Medicine at Mount Sinai, New York.

That’s welcome news because AD is the most common inflammatory skin disease, both in adults, where the latest data puts the prevalence at 7%-10%, and in children, where the global rate is 15%-25%.
And while at the EADV congress only the 16-week data were reported in the new adolescent study, there is reason to be optimistic that the benefits will remain durable over time. Dr. Guttman-Yassky cited the published 52-week data from the large phase 3 CHRONOS trial in adults with moderate to severe AD. In that trial, in which patients could use concomitant topical steroids, the EASI 75 rates at week 16 were 69% in patients on dupilumab at 300 mg every 2 weeks, 64% with 300 mg weekly, and 23% with placebo. Reassuringly, at 52 weeks the EASI 75 response rates were essentially unchanged: 65%, 64%, and 22% (Lancet. 2017 Jun 10;389[10086]:2287-303).
“This is what we are seeking: not just treatment that is able to quickly modify disease, but we want the treatment to be sustained, and of course we want it to be safe for our patients because we know we cannot use cyclosporine for a long time,” Dr. Guttman-Yassky said.
Dr. Simpson reported serving as a consultant to and recipient of research grants from Sanofi and Regeneron, which sponsored the adolescent study, as well as more than a dozen other pharmaceutical companies. Dr. Guttman-Yassky reported serving as an advisor and consultant, and has received grants/research funding from Regeneron, and multiple other companies.
PARIS – Dupilumab scorched its way through a landmark pivotal phase 3 clinical trial in adolescents with moderate to severe atopic dermatitis (AD), achieving unprecedented clinically meaningful improvements in signs and symptoms of the disease along with important quality of life benefits, Eric L. Simpson, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
Indeed, akin to that previously shown in adults with moderate to severe AD in phase 3 trials that earned the biologic U.S. and European regulatory approval in the adult population, noted Dr. Simpson, professor of dermatology at Oregon Health & Science University, Portland.
This positive phase 3 study represents a major development in pediatric dermatology because of the pressing unmet need for better treatments for teens with moderate to severe AD whose disease can’t be controlled with topical therapies. The adolescent years are, after all, a critical period in growth and development, and a debilitating, uncontrolled disease can reshape that experience in unwelcome ways.
“Atopic dermatitis profoundly affects quality of life in adolescents and their family: The itching affects mood and sleep, these patients commonly have anxiety and depression, and the chronic and relapsing nature of the disease adversely affects the family,” the dermatologist observed.
Currently, no systemic agent is approved for pediatric patients with AD because evidence demonstrating a favorable benefit-to-risk profile has been lacking. The dupilumab study was the first-ever phase 3 trial of a biologic in such a population.
The phase 3 adolescent trial was a 16-week, randomized, double-blind, multicenter, placebo-controlled study of 251 patients aged 12-17 years with moderate to severe AD, which could not be adequately controlled with topical therapies. Participants averaged 14 years of age, with a 12-year history of AD. “These patients had the disease basically their whole life,” Dr. Simpson noted.
They were more severely affected than participants in the adult clinical trials, with mean Eczema Area And Severity Index (EASI) scores in the mid-30s and an average 56% involved body surface area. The adolescent AD patients had a heavy burden of comorbid allergic type 2 immune comorbidity: Fully 92% of them had documented asthma, food allergy, allergic rhinitis, and/or some other form of allergic comorbidity. The majority of the teens were categorized as having severe AD, whereas most participants in the adult phase 3 trials of dupilumab had moderate disease. That distinction becomes relevant in comparing the trial results.
Participants were randomized to once-monthly subcutaneous injections of dupilumab at 300 mg, following a 600-mg loading dose, or to an injection of 200 mg or 300 mg every 2 weeks with an initial dose of 400 mg or 600 mg based upon a body weight cutoff of 60 kg, or to biweekly placebo injections.
The coprimary endpoints were the proportion of patients who achieved an EASI 75 response at week 16 and achievement of an Investigator’s Global Assessment (IGA) score of 0 or 1, meaning clear or almost clear, on a 5-point scale at week 16.
This trial introduced an important new design feature that physicians can expect to see more of in the future: regulatory agencies now want to see the effects of monotherapy in pivotal studies in AD. Previously, participants in AD studies of systemic agents could also utilize topical steroids as needed. No longer. In the adolescent dupilumab study, resort to rescue topical steroids led to exclusion from inclusion in the primary outcome results. Not surprisingly, this lack of access to rescue medication resulted in a 60% dropout rate by 16 weeks in placebo-treated controls, a 30% dropout rate in teens on dupilumab every 4 weeks, and a 20% dropout rate with biweekly dupilumab.
The EASI 75 rate at week 16 was 8.2% with placebo, 38.1% with monthly dupilumab, and 41.5% with biweekly dosing. The other coprimary endpoint – an IGA of 0 or 1 at 16 weeks – was achieved in 2.4% of controls, 17.9% with dupilumab at 300 mg every 4 weeks, and 41.5% with biweekly dosing.
Turning to secondary endpoints, Dr. Simpson reported that baseline peak pruritus Numeric Rating Scale scores dropped by 19% with placebo at 16 weeks, compared with reductions of 45.5% and 47.9% with monthly and biweekly dupilumab, respectively.
An EASI 50 response,while not as impressive as an EASI 75, is nonetheless considered clinically meaningful improvement. It was attained in 12.9% on placebo and 54.8% and 61% on monthly and biweekly dupilumab.
From a mean baseline score of 13.6 on the Children’s Dermatology Life Quality Index, scores improved over the course of 16 weeks by a mean of 5.1 points with placebo, 8.5 points with monthly dupilumab, and 8.8 points with biweekly biologic therapy. The same pattern was noted with regard to the Patient-Oriented Eczema Measure or POEM.
Adverse events mirrored those documented in the pivotal trials in adults: increased rates of mild to moderate conjunctivitis: 4.7% with placebo, 10.8% with monthly dupilumab, and 9.8% with biweekly dupilumab, along with single-digit rates of injection-site reactions. As in adult AD patients, however, these side effects were counterbalanced by significantly reduced rates of skin infections in the adolescent group: 20% during 16 weeks with placebo, and 13.3% and 11% with monthly and biweekly biologic therapy, respectively.
Study participants had a mean baseline score of 12.5 on the Hospital Anxiety and Depression Scale, which is categorized as clinically significant psychiatric disease. The impact of dupilumab therapy on those scores will be the topic of a future presentation, Dr. Simpson said.
Across the board, specific outcomes were consistently numerically better in patients who received dupilumab biweekly than monthly, albeit not statistically significantly so. Dr. Simpson thinks he knows why: Lab studies showed that the mean serum concentration of functional dupilumab in patients who got the biologic biweekly was nearly twice that for the group with monthly dosing.
At first glance, the IGA “clear” or “almost clear” response rates seen with dupilumab in the adolescent study appeared to be less robust than in the adult pivotal phase 3 trials, such as the SOLO 1 and SOLO 2 trials (N Engl J Med. 2016 Dec 15;375[24]:2335-48), also led by Dr. Simpson.
“I think that’s because of the greater severity of that baseline adolescent population,” he commented. “It made for a much lower placebo response rate. But when you correct for the placebo-subtracted difference, the rates are actually pretty similar, and a lot of the other endpoints are the same or even better than in SOLO.”
After his presentation, Dr. Simpson commented, “This is huge. This is the study we’ve been waiting for.”
Elsewhere at the EADV congress, Emma Guttman-Yassky, MD, PhD, deemed the pivotal phase 3 trial in adolescent AD patients one of the meeting’s highlights. And it’s a harbinger of more good things to come, because the investigational drug pipeline for AD is full of promising candidates addressing the disease from a variety of novel directions. The long therapeutic drought in AD appears to have finally ended, observed Dr. Guttman-Yassky, professor and vice-chair of the department of dermatology at Icahn School of Medicine at Mount Sinai, New York.
That’s welcome news because AD is the most common inflammatory skin disease, both in adults, where the latest data puts the prevalence at 7%-10%, and in children, where the global rate is 15%-25%.
And while at the EADV congress only the 16-week data were reported in the new adolescent study, there is reason to be optimistic that the benefits will remain durable over time. Dr. Guttman-Yassky cited the published 52-week data from the large phase 3 CHRONOS trial in adults with moderate to severe AD. In that trial, in which patients could use concomitant topical steroids, the EASI 75 rates at week 16 were 69% in patients on dupilumab at 300 mg every 2 weeks, 64% with 300 mg weekly, and 23% with placebo. Reassuringly, at 52 weeks the EASI 75 response rates were essentially unchanged: 65%, 64%, and 22% (Lancet. 2017 Jun 10;389[10086]:2287-303).
“This is what we are seeking: not just treatment that is able to quickly modify disease, but we want the treatment to be sustained, and of course we want it to be safe for our patients because we know we cannot use cyclosporine for a long time,” Dr. Guttman-Yassky said.
Dr. Simpson reported serving as a consultant to and recipient of research grants from Sanofi and Regeneron, which sponsored the adolescent study, as well as more than a dozen other pharmaceutical companies. Dr. Guttman-Yassky reported serving as an advisor and consultant, and has received grants/research funding from Regeneron, and multiple other companies.
PARIS – Dupilumab scorched its way through a landmark pivotal phase 3 clinical trial in adolescents with moderate to severe atopic dermatitis (AD), achieving unprecedented clinically meaningful improvements in signs and symptoms of the disease along with important quality of life benefits, Eric L. Simpson, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
Indeed, akin to that previously shown in adults with moderate to severe AD in phase 3 trials that earned the biologic U.S. and European regulatory approval in the adult population, noted Dr. Simpson, professor of dermatology at Oregon Health & Science University, Portland.
This positive phase 3 study represents a major development in pediatric dermatology because of the pressing unmet need for better treatments for teens with moderate to severe AD whose disease can’t be controlled with topical therapies. The adolescent years are, after all, a critical period in growth and development, and a debilitating, uncontrolled disease can reshape that experience in unwelcome ways.
“Atopic dermatitis profoundly affects quality of life in adolescents and their family: The itching affects mood and sleep, these patients commonly have anxiety and depression, and the chronic and relapsing nature of the disease adversely affects the family,” the dermatologist observed.
Currently, no systemic agent is approved for pediatric patients with AD because evidence demonstrating a favorable benefit-to-risk profile has been lacking. The dupilumab study was the first-ever phase 3 trial of a biologic in such a population.
The phase 3 adolescent trial was a 16-week, randomized, double-blind, multicenter, placebo-controlled study of 251 patients aged 12-17 years with moderate to severe AD, which could not be adequately controlled with topical therapies. Participants averaged 14 years of age, with a 12-year history of AD. “These patients had the disease basically their whole life,” Dr. Simpson noted.
They were more severely affected than participants in the adult clinical trials, with mean Eczema Area And Severity Index (EASI) scores in the mid-30s and an average 56% involved body surface area. The adolescent AD patients had a heavy burden of comorbid allergic type 2 immune comorbidity: Fully 92% of them had documented asthma, food allergy, allergic rhinitis, and/or some other form of allergic comorbidity. The majority of the teens were categorized as having severe AD, whereas most participants in the adult phase 3 trials of dupilumab had moderate disease. That distinction becomes relevant in comparing the trial results.
Participants were randomized to once-monthly subcutaneous injections of dupilumab at 300 mg, following a 600-mg loading dose, or to an injection of 200 mg or 300 mg every 2 weeks with an initial dose of 400 mg or 600 mg based upon a body weight cutoff of 60 kg, or to biweekly placebo injections.
The coprimary endpoints were the proportion of patients who achieved an EASI 75 response at week 16 and achievement of an Investigator’s Global Assessment (IGA) score of 0 or 1, meaning clear or almost clear, on a 5-point scale at week 16.
This trial introduced an important new design feature that physicians can expect to see more of in the future: regulatory agencies now want to see the effects of monotherapy in pivotal studies in AD. Previously, participants in AD studies of systemic agents could also utilize topical steroids as needed. No longer. In the adolescent dupilumab study, resort to rescue topical steroids led to exclusion from inclusion in the primary outcome results. Not surprisingly, this lack of access to rescue medication resulted in a 60% dropout rate by 16 weeks in placebo-treated controls, a 30% dropout rate in teens on dupilumab every 4 weeks, and a 20% dropout rate with biweekly dupilumab.
The EASI 75 rate at week 16 was 8.2% with placebo, 38.1% with monthly dupilumab, and 41.5% with biweekly dosing. The other coprimary endpoint – an IGA of 0 or 1 at 16 weeks – was achieved in 2.4% of controls, 17.9% with dupilumab at 300 mg every 4 weeks, and 41.5% with biweekly dosing.
Turning to secondary endpoints, Dr. Simpson reported that baseline peak pruritus Numeric Rating Scale scores dropped by 19% with placebo at 16 weeks, compared with reductions of 45.5% and 47.9% with monthly and biweekly dupilumab, respectively.
An EASI 50 response,while not as impressive as an EASI 75, is nonetheless considered clinically meaningful improvement. It was attained in 12.9% on placebo and 54.8% and 61% on monthly and biweekly dupilumab.
From a mean baseline score of 13.6 on the Children’s Dermatology Life Quality Index, scores improved over the course of 16 weeks by a mean of 5.1 points with placebo, 8.5 points with monthly dupilumab, and 8.8 points with biweekly biologic therapy. The same pattern was noted with regard to the Patient-Oriented Eczema Measure or POEM.
Adverse events mirrored those documented in the pivotal trials in adults: increased rates of mild to moderate conjunctivitis: 4.7% with placebo, 10.8% with monthly dupilumab, and 9.8% with biweekly dupilumab, along with single-digit rates of injection-site reactions. As in adult AD patients, however, these side effects were counterbalanced by significantly reduced rates of skin infections in the adolescent group: 20% during 16 weeks with placebo, and 13.3% and 11% with monthly and biweekly biologic therapy, respectively.
Study participants had a mean baseline score of 12.5 on the Hospital Anxiety and Depression Scale, which is categorized as clinically significant psychiatric disease. The impact of dupilumab therapy on those scores will be the topic of a future presentation, Dr. Simpson said.
Across the board, specific outcomes were consistently numerically better in patients who received dupilumab biweekly than monthly, albeit not statistically significantly so. Dr. Simpson thinks he knows why: Lab studies showed that the mean serum concentration of functional dupilumab in patients who got the biologic biweekly was nearly twice that for the group with monthly dosing.
At first glance, the IGA “clear” or “almost clear” response rates seen with dupilumab in the adolescent study appeared to be less robust than in the adult pivotal phase 3 trials, such as the SOLO 1 and SOLO 2 trials (N Engl J Med. 2016 Dec 15;375[24]:2335-48), also led by Dr. Simpson.
“I think that’s because of the greater severity of that baseline adolescent population,” he commented. “It made for a much lower placebo response rate. But when you correct for the placebo-subtracted difference, the rates are actually pretty similar, and a lot of the other endpoints are the same or even better than in SOLO.”
After his presentation, Dr. Simpson commented, “This is huge. This is the study we’ve been waiting for.”
Elsewhere at the EADV congress, Emma Guttman-Yassky, MD, PhD, deemed the pivotal phase 3 trial in adolescent AD patients one of the meeting’s highlights. And it’s a harbinger of more good things to come, because the investigational drug pipeline for AD is full of promising candidates addressing the disease from a variety of novel directions. The long therapeutic drought in AD appears to have finally ended, observed Dr. Guttman-Yassky, professor and vice-chair of the department of dermatology at Icahn School of Medicine at Mount Sinai, New York.
That’s welcome news because AD is the most common inflammatory skin disease, both in adults, where the latest data puts the prevalence at 7%-10%, and in children, where the global rate is 15%-25%.
And while at the EADV congress only the 16-week data were reported in the new adolescent study, there is reason to be optimistic that the benefits will remain durable over time. Dr. Guttman-Yassky cited the published 52-week data from the large phase 3 CHRONOS trial in adults with moderate to severe AD. In that trial, in which patients could use concomitant topical steroids, the EASI 75 rates at week 16 were 69% in patients on dupilumab at 300 mg every 2 weeks, 64% with 300 mg weekly, and 23% with placebo. Reassuringly, at 52 weeks the EASI 75 response rates were essentially unchanged: 65%, 64%, and 22% (Lancet. 2017 Jun 10;389[10086]:2287-303).
“This is what we are seeking: not just treatment that is able to quickly modify disease, but we want the treatment to be sustained, and of course we want it to be safe for our patients because we know we cannot use cyclosporine for a long time,” Dr. Guttman-Yassky said.
Dr. Simpson reported serving as a consultant to and recipient of research grants from Sanofi and Regeneron, which sponsored the adolescent study, as well as more than a dozen other pharmaceutical companies. Dr. Guttman-Yassky reported serving as an advisor and consultant, and has received grants/research funding from Regeneron, and multiple other companies.
REPORTING FROM THE EADV CONGRESS
Key clinical point: Dupilumab gets solid green-light evidence for use in teens with atopic dermatitis (AD).
Major finding: Dupilumab was as safe and effective in adolescents with moderate to severe AD as previously established in adult patients.
Study details: This prospective, randomized, double-blind, placebo-controlled, 16-week pivotal phase 3 trial included 251 adolescents with moderate to severe AD.
Disclosures: The presenter reported serving as a consultant to and recipient of research grants from Sanofi and Regeneron, which sponsored the adolescent study, as well as more than a dozen other pharmaceutical companies.
Topical cyclosporine safely tamed atopic dermatitis in 4-week study
PARIS – A first-of-its-kind Ana M. Giménez-Arnau, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.

The 5% cyclosporine topical spray, known as Cyclatop, showed significantly better results across the board than its vehicle, even during the first week of treatment in the 4-week, multicenter, Spanish, double-blind, randomized trial, which included 44 patients with mild or moderate atopic dermatitis (AD), according to Dr. Giménez-Arnau, a dermatologist at Hospital del Mar and the Autonomous University of Barcelona.
“Besides the clinical efficacy, the study also demonstrated that, when cyclosporine was detectable in the blood, the highest blood level was at least 200-fold less than after systemic administration of cyclosporine at therapeutic doses,” she noted.
The motivation to develop a topical formulation of cyclosporine stemmed from the need to find substitutes for topical corticosteroids, especially in the pediatric population, where steroid phobia is rampant among parents. And while systemic cyclosporine is approved by European regulators for treatment of difficult cases of AD and is widely utilized off label for this purpose in the United States, the fact is that it is an immunosuppressant that paints with a broad brush and is best utilized for a matter of weeks as induction therapy.
But developing a topical formulation of cyclosporine suitable for long-term use posed many challenges. Lack of stability in cream and ointment formulations was a recurring issue. “Cyclosporine is a very big molecule, which is not easy to work with topically,” she explained. “The challenge was to find a stable formulation with good skin penetration, but without systemic absorption.”
Indeed, researchers at Barcelona-based Spherium Biomed evaluated more than 100 prototype compounds in animal models before settling on a proprietary oil emulsion formulation of 5% cyclosporine delivered via a spray without propellant gas.
Key study findings
The 44 study participants had a mean baseline of 8.3% body surface area involvement. As a condition of participation, they needed to have similar lesional areas bilaterally. They treated involved areas on one side of the body twice daily with Cyclatop, while they sprayed those on the opposite side with its vehicle.
From a mean baseline Eczema Area and Severity Index (EASI) score of 5.5, EASI scores improved by an average of 3.2 points after 28 days of cyclosporine spray, compared with 1.7 points with vehicle. Atopic Dermatitis Area and Severity Index (ADSI) scores improved from a mean baseline of 6.5 by 3.6 points with topical cyclosporine versus 2.4 points with vehicle.
At week 3, an EASI 75 response – that is, at least a 75% reduction from baseline EASI scores – was achieved at 44.4% of actively treated sites, compared with 25.9% of control sites. ADSI 75 rates at 3 weeks were 33.3% and 11.1%, respectively. An Investigator’s Global Assessment of clear or almost clear was reached at week 4 at 61.5% of active treatment sites, compared with 42.3% of vehicle-treated sites.
Itching responded dramatically to topical cyclosporine. From a mean baseline score of 3.3 on a standard 10-point pruritus visual analog scale, cyclosporine spray–treated areas showed a mean 1.2-point decrease at week 4, compared with a 0.4-point reduction at vehicle-treated areas. About 50% of the reduction in pruritus scores was achieved within the first week of active treatment. Moreover, among patients with moderate as opposed to mild itching scores at baseline, who had a mean pruritus score of 5.6, topical cyclosporine spray resulted in a mean 3.3-point reduction at week 4, Dr. Giménez-Arnau continued.
No safety signals emerged in this initial study. Side effects associated with the cyclosporine spray were the same as with its vehicle, and in exit interviews, more than 85% of patients indicated they were satisfied with the comfort and practicality of topical cyclosporine.
Session chair DeDee Murrell, MD, professor of dermatology at the University of New South Wales, Sydney, noted that the study was restricted to patients with less than 10% body surface area of involvement.
“Are you concerned that if you use this product over widespread areas, as is quite common in eczema, that you might get positive blood levels?” she asked.
“We don’t know. We should check that,” Dr. Giménez-Arnau replied. She added that more studies need to be done before cyclosporine spray is ready for the market. These studies will need to address the optimal dosing schedule and duration, the spectrum of disease severity where the topical spray works best, and other key issues.
Cyclatop is being developed by Spherium Biomed, which sponsored the study. Dr. Giménez-Arnau reported receiving research grants from and/or serving as a consultant to roughly half a dozen pharmaceutical companies.
PARIS – A first-of-its-kind Ana M. Giménez-Arnau, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
The 5% cyclosporine topical spray, known as Cyclatop, showed significantly better results across the board than its vehicle, even during the first week of treatment in the 4-week, multicenter, Spanish, double-blind, randomized trial, which included 44 patients with mild or moderate atopic dermatitis (AD), according to Dr. Giménez-Arnau, a dermatologist at Hospital del Mar and the Autonomous University of Barcelona.
“Besides the clinical efficacy, the study also demonstrated that, when cyclosporine was detectable in the blood, the highest blood level was at least 200-fold less than after systemic administration of cyclosporine at therapeutic doses,” she noted.
The motivation to develop a topical formulation of cyclosporine stemmed from the need to find substitutes for topical corticosteroids, especially in the pediatric population, where steroid phobia is rampant among parents. And while systemic cyclosporine is approved by European regulators for treatment of difficult cases of AD and is widely utilized off label for this purpose in the United States, the fact is that it is an immunosuppressant that paints with a broad brush and is best utilized for a matter of weeks as induction therapy.
But developing a topical formulation of cyclosporine suitable for long-term use posed many challenges. Lack of stability in cream and ointment formulations was a recurring issue. “Cyclosporine is a very big molecule, which is not easy to work with topically,” she explained. “The challenge was to find a stable formulation with good skin penetration, but without systemic absorption.”
Indeed, researchers at Barcelona-based Spherium Biomed evaluated more than 100 prototype compounds in animal models before settling on a proprietary oil emulsion formulation of 5% cyclosporine delivered via a spray without propellant gas.
Key study findings
The 44 study participants had a mean baseline of 8.3% body surface area involvement. As a condition of participation, they needed to have similar lesional areas bilaterally. They treated involved areas on one side of the body twice daily with Cyclatop, while they sprayed those on the opposite side with its vehicle.
From a mean baseline Eczema Area and Severity Index (EASI) score of 5.5, EASI scores improved by an average of 3.2 points after 28 days of cyclosporine spray, compared with 1.7 points with vehicle. Atopic Dermatitis Area and Severity Index (ADSI) scores improved from a mean baseline of 6.5 by 3.6 points with topical cyclosporine versus 2.4 points with vehicle.
At week 3, an EASI 75 response – that is, at least a 75% reduction from baseline EASI scores – was achieved at 44.4% of actively treated sites, compared with 25.9% of control sites. ADSI 75 rates at 3 weeks were 33.3% and 11.1%, respectively. An Investigator’s Global Assessment of clear or almost clear was reached at week 4 at 61.5% of active treatment sites, compared with 42.3% of vehicle-treated sites.
Itching responded dramatically to topical cyclosporine. From a mean baseline score of 3.3 on a standard 10-point pruritus visual analog scale, cyclosporine spray–treated areas showed a mean 1.2-point decrease at week 4, compared with a 0.4-point reduction at vehicle-treated areas. About 50% of the reduction in pruritus scores was achieved within the first week of active treatment. Moreover, among patients with moderate as opposed to mild itching scores at baseline, who had a mean pruritus score of 5.6, topical cyclosporine spray resulted in a mean 3.3-point reduction at week 4, Dr. Giménez-Arnau continued.
No safety signals emerged in this initial study. Side effects associated with the cyclosporine spray were the same as with its vehicle, and in exit interviews, more than 85% of patients indicated they were satisfied with the comfort and practicality of topical cyclosporine.
Session chair DeDee Murrell, MD, professor of dermatology at the University of New South Wales, Sydney, noted that the study was restricted to patients with less than 10% body surface area of involvement.
“Are you concerned that if you use this product over widespread areas, as is quite common in eczema, that you might get positive blood levels?” she asked.
“We don’t know. We should check that,” Dr. Giménez-Arnau replied. She added that more studies need to be done before cyclosporine spray is ready for the market. These studies will need to address the optimal dosing schedule and duration, the spectrum of disease severity where the topical spray works best, and other key issues.
Cyclatop is being developed by Spherium Biomed, which sponsored the study. Dr. Giménez-Arnau reported receiving research grants from and/or serving as a consultant to roughly half a dozen pharmaceutical companies.
PARIS – A first-of-its-kind Ana M. Giménez-Arnau, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
The 5% cyclosporine topical spray, known as Cyclatop, showed significantly better results across the board than its vehicle, even during the first week of treatment in the 4-week, multicenter, Spanish, double-blind, randomized trial, which included 44 patients with mild or moderate atopic dermatitis (AD), according to Dr. Giménez-Arnau, a dermatologist at Hospital del Mar and the Autonomous University of Barcelona.
“Besides the clinical efficacy, the study also demonstrated that, when cyclosporine was detectable in the blood, the highest blood level was at least 200-fold less than after systemic administration of cyclosporine at therapeutic doses,” she noted.
The motivation to develop a topical formulation of cyclosporine stemmed from the need to find substitutes for topical corticosteroids, especially in the pediatric population, where steroid phobia is rampant among parents. And while systemic cyclosporine is approved by European regulators for treatment of difficult cases of AD and is widely utilized off label for this purpose in the United States, the fact is that it is an immunosuppressant that paints with a broad brush and is best utilized for a matter of weeks as induction therapy.
But developing a topical formulation of cyclosporine suitable for long-term use posed many challenges. Lack of stability in cream and ointment formulations was a recurring issue. “Cyclosporine is a very big molecule, which is not easy to work with topically,” she explained. “The challenge was to find a stable formulation with good skin penetration, but without systemic absorption.”
Indeed, researchers at Barcelona-based Spherium Biomed evaluated more than 100 prototype compounds in animal models before settling on a proprietary oil emulsion formulation of 5% cyclosporine delivered via a spray without propellant gas.
Key study findings
The 44 study participants had a mean baseline of 8.3% body surface area involvement. As a condition of participation, they needed to have similar lesional areas bilaterally. They treated involved areas on one side of the body twice daily with Cyclatop, while they sprayed those on the opposite side with its vehicle.
From a mean baseline Eczema Area and Severity Index (EASI) score of 5.5, EASI scores improved by an average of 3.2 points after 28 days of cyclosporine spray, compared with 1.7 points with vehicle. Atopic Dermatitis Area and Severity Index (ADSI) scores improved from a mean baseline of 6.5 by 3.6 points with topical cyclosporine versus 2.4 points with vehicle.
At week 3, an EASI 75 response – that is, at least a 75% reduction from baseline EASI scores – was achieved at 44.4% of actively treated sites, compared with 25.9% of control sites. ADSI 75 rates at 3 weeks were 33.3% and 11.1%, respectively. An Investigator’s Global Assessment of clear or almost clear was reached at week 4 at 61.5% of active treatment sites, compared with 42.3% of vehicle-treated sites.
Itching responded dramatically to topical cyclosporine. From a mean baseline score of 3.3 on a standard 10-point pruritus visual analog scale, cyclosporine spray–treated areas showed a mean 1.2-point decrease at week 4, compared with a 0.4-point reduction at vehicle-treated areas. About 50% of the reduction in pruritus scores was achieved within the first week of active treatment. Moreover, among patients with moderate as opposed to mild itching scores at baseline, who had a mean pruritus score of 5.6, topical cyclosporine spray resulted in a mean 3.3-point reduction at week 4, Dr. Giménez-Arnau continued.
No safety signals emerged in this initial study. Side effects associated with the cyclosporine spray were the same as with its vehicle, and in exit interviews, more than 85% of patients indicated they were satisfied with the comfort and practicality of topical cyclosporine.
Session chair DeDee Murrell, MD, professor of dermatology at the University of New South Wales, Sydney, noted that the study was restricted to patients with less than 10% body surface area of involvement.
“Are you concerned that if you use this product over widespread areas, as is quite common in eczema, that you might get positive blood levels?” she asked.
“We don’t know. We should check that,” Dr. Giménez-Arnau replied. She added that more studies need to be done before cyclosporine spray is ready for the market. These studies will need to address the optimal dosing schedule and duration, the spectrum of disease severity where the topical spray works best, and other key issues.
Cyclatop is being developed by Spherium Biomed, which sponsored the study. Dr. Giménez-Arnau reported receiving research grants from and/or serving as a consultant to roughly half a dozen pharmaceutical companies.
REPORTING FROM THE EADV CONGRESS
Key clinical point: Cyclosporine 5% topical spray shows promise for atopic dermatitis.
Major finding: About 62% of patients with mild to moderate atopic dermatitis were clear or almost clear after 4 weeks of twice-daily cyclosporine 5% topical spray.
Study details: This prospective, multicenter, double-blind, vehicle-controlled study included 44 children and adults with mild or moderate atopic dermatitis.
Disclosures: The study was sponsored by Spherium Biomed. The presenter reported receiving research grants from and/or serving as a consultant to that and roughly half a dozen other pharmaceutical companies.
Pediatric data on novel axillary hyperhidrosis treatment reported
PARIS – Two compelling reasons exist to take excessive sweating in children and adolescents more seriously, Lawrence J. Green, MD, asserted at the annual congress of the European Academy of Dermatology and Venereology.

One is that this is a surprisingly common and embarrassing medical condition that can have a profound adverse developmental impact in young people at a time when they are engaged in forming their self-image.
The other reason to get serious about addressing primary axillary hyperhidrosis in pediatric patients is the recent approval of glycopyrronium tosylate as a topical therapy, Dr. Green, a dermatologist at George Washington University, Washington. The treatment, glycopyrronium pads (Qbrexza), was approved by the Food and Drug Administration for the topical treatment of primary axillary hyperhidrosis in patients aged 9 years and older in June 2018, and will be available in October 2018.
He presented new data from a 44-week, open-label extension of two pivotal 4-week, phase 3, randomized, double-blind, placebo-controlled trials known as ATMOS-1 and ATMOS-2. The new post hoc analysis from the extension study, known as the ARIDO study, provides reassurance that the product remains both safe and durably effective with longterm use.
Dr. Green’s analysis focused on the 44 pediatric participants aged 9-16 years. That’s because even though primary axillary hyperhidrosis affects people of all ages, with an estimated 4.8% prevalence in the U.S. population – 5.3 million people – it is more common in children and adolescents than adults. And it hits them particularly hard.
“Hyperhidrosis is largely underdiagnosed and undertreated, particularly among pediatric patients,” he said. “The impact on quality of life is comparable to or greater than acne, psoriasis, or eczema.”
The glycopyrronium pad is self-applied as a once-daily wipe. Glycopyrronium is an anticholinergic agent, which blocks sweat production by inhibiting the receptors that activate sweat glands.
Dr. Green noted several key findings from the 44-week ARIDO analysis, presented for the first time at the EADV congress.
The median absolute decrease in sweat production in pediatric patients at 44 weeks as measured gravimetrically was 50.3 mg per 5 minutes from a baseline of 150 mg per 5 minutes, comparable with the mean 75 mg reduction from a baseline of 175 mg in the 507-patient older cohort. However, Dr. Green advised not to make too much of this endpoint, as sweat production is notoriously difficult to measure accurately. In addition, an individual’s sweat rate can vary widely depending upon a multitude of factors, including ambient temperature and even what a patient is thinking about. The FDA recognizes this and therefore elevated several validated patient-reported outcomes to the status of coprimary endpoints in the clinical trials.
A positive result on one such patient-reported outcome, the Hyperhidrosis Severity Scale, was achieved in 57% of pediatric patients and 64% of adults at week 44 of open-label therapy. This required at least a 2-grade improvement from baseline, when roughly 60% of youths had a score of 3 and the remainder scored 4 on the 1-4 point scale.
From a mean baseline score of 9.2 on the Children’s Dermatology Life Quality Index, the pediatric group averaged a mean 6.2-point improvement at week 44, while adults experienced a mean 8.7-point improvement on the Dermatology Life Quality Index from a baseline of 11.25.
There was no diminution in treatment efficacy through 44 weeks, Dr. Green noted. Treatment-emergent adverse events consisted largely of transient mild to moderate anticholinergic effects, which seldom led to study discontinuation.
Dilated pupils and blurred vision were more common in children than adults (7.9% and 10.5% vs. 5.1% and 6.4%, respectively). “Why that is I can only speculate. Kids do tend to touch their eyes more often than adults. Pretty much everything else was the same. The adverse events can be worked around by educating people to use the pads appropriately. We saw the anticholinergic side effects more often in the first 4 weeks of the double-blind trials than in the longterm extension because once patients learned how to use the pad and not touch themselves afterwards, the adverse events came down,” he said.
The studies were sponsored by Dermira. Dr. Green has received research funding from and been a consultant to the company.
PARIS – Two compelling reasons exist to take excessive sweating in children and adolescents more seriously, Lawrence J. Green, MD, asserted at the annual congress of the European Academy of Dermatology and Venereology.
One is that this is a surprisingly common and embarrassing medical condition that can have a profound adverse developmental impact in young people at a time when they are engaged in forming their self-image.
The other reason to get serious about addressing primary axillary hyperhidrosis in pediatric patients is the recent approval of glycopyrronium tosylate as a topical therapy, Dr. Green, a dermatologist at George Washington University, Washington. The treatment, glycopyrronium pads (Qbrexza), was approved by the Food and Drug Administration for the topical treatment of primary axillary hyperhidrosis in patients aged 9 years and older in June 2018, and will be available in October 2018.
He presented new data from a 44-week, open-label extension of two pivotal 4-week, phase 3, randomized, double-blind, placebo-controlled trials known as ATMOS-1 and ATMOS-2. The new post hoc analysis from the extension study, known as the ARIDO study, provides reassurance that the product remains both safe and durably effective with longterm use.
Dr. Green’s analysis focused on the 44 pediatric participants aged 9-16 years. That’s because even though primary axillary hyperhidrosis affects people of all ages, with an estimated 4.8% prevalence in the U.S. population – 5.3 million people – it is more common in children and adolescents than adults. And it hits them particularly hard.
“Hyperhidrosis is largely underdiagnosed and undertreated, particularly among pediatric patients,” he said. “The impact on quality of life is comparable to or greater than acne, psoriasis, or eczema.”
The glycopyrronium pad is self-applied as a once-daily wipe. Glycopyrronium is an anticholinergic agent, which blocks sweat production by inhibiting the receptors that activate sweat glands.
Dr. Green noted several key findings from the 44-week ARIDO analysis, presented for the first time at the EADV congress.
The median absolute decrease in sweat production in pediatric patients at 44 weeks as measured gravimetrically was 50.3 mg per 5 minutes from a baseline of 150 mg per 5 minutes, comparable with the mean 75 mg reduction from a baseline of 175 mg in the 507-patient older cohort. However, Dr. Green advised not to make too much of this endpoint, as sweat production is notoriously difficult to measure accurately. In addition, an individual’s sweat rate can vary widely depending upon a multitude of factors, including ambient temperature and even what a patient is thinking about. The FDA recognizes this and therefore elevated several validated patient-reported outcomes to the status of coprimary endpoints in the clinical trials.
A positive result on one such patient-reported outcome, the Hyperhidrosis Severity Scale, was achieved in 57% of pediatric patients and 64% of adults at week 44 of open-label therapy. This required at least a 2-grade improvement from baseline, when roughly 60% of youths had a score of 3 and the remainder scored 4 on the 1-4 point scale.
From a mean baseline score of 9.2 on the Children’s Dermatology Life Quality Index, the pediatric group averaged a mean 6.2-point improvement at week 44, while adults experienced a mean 8.7-point improvement on the Dermatology Life Quality Index from a baseline of 11.25.
There was no diminution in treatment efficacy through 44 weeks, Dr. Green noted. Treatment-emergent adverse events consisted largely of transient mild to moderate anticholinergic effects, which seldom led to study discontinuation.
Dilated pupils and blurred vision were more common in children than adults (7.9% and 10.5% vs. 5.1% and 6.4%, respectively). “Why that is I can only speculate. Kids do tend to touch their eyes more often than adults. Pretty much everything else was the same. The adverse events can be worked around by educating people to use the pads appropriately. We saw the anticholinergic side effects more often in the first 4 weeks of the double-blind trials than in the longterm extension because once patients learned how to use the pad and not touch themselves afterwards, the adverse events came down,” he said.
The studies were sponsored by Dermira. Dr. Green has received research funding from and been a consultant to the company.
PARIS – Two compelling reasons exist to take excessive sweating in children and adolescents more seriously, Lawrence J. Green, MD, asserted at the annual congress of the European Academy of Dermatology and Venereology.
One is that this is a surprisingly common and embarrassing medical condition that can have a profound adverse developmental impact in young people at a time when they are engaged in forming their self-image.
The other reason to get serious about addressing primary axillary hyperhidrosis in pediatric patients is the recent approval of glycopyrronium tosylate as a topical therapy, Dr. Green, a dermatologist at George Washington University, Washington. The treatment, glycopyrronium pads (Qbrexza), was approved by the Food and Drug Administration for the topical treatment of primary axillary hyperhidrosis in patients aged 9 years and older in June 2018, and will be available in October 2018.
He presented new data from a 44-week, open-label extension of two pivotal 4-week, phase 3, randomized, double-blind, placebo-controlled trials known as ATMOS-1 and ATMOS-2. The new post hoc analysis from the extension study, known as the ARIDO study, provides reassurance that the product remains both safe and durably effective with longterm use.
Dr. Green’s analysis focused on the 44 pediatric participants aged 9-16 years. That’s because even though primary axillary hyperhidrosis affects people of all ages, with an estimated 4.8% prevalence in the U.S. population – 5.3 million people – it is more common in children and adolescents than adults. And it hits them particularly hard.
“Hyperhidrosis is largely underdiagnosed and undertreated, particularly among pediatric patients,” he said. “The impact on quality of life is comparable to or greater than acne, psoriasis, or eczema.”
The glycopyrronium pad is self-applied as a once-daily wipe. Glycopyrronium is an anticholinergic agent, which blocks sweat production by inhibiting the receptors that activate sweat glands.
Dr. Green noted several key findings from the 44-week ARIDO analysis, presented for the first time at the EADV congress.
The median absolute decrease in sweat production in pediatric patients at 44 weeks as measured gravimetrically was 50.3 mg per 5 minutes from a baseline of 150 mg per 5 minutes, comparable with the mean 75 mg reduction from a baseline of 175 mg in the 507-patient older cohort. However, Dr. Green advised not to make too much of this endpoint, as sweat production is notoriously difficult to measure accurately. In addition, an individual’s sweat rate can vary widely depending upon a multitude of factors, including ambient temperature and even what a patient is thinking about. The FDA recognizes this and therefore elevated several validated patient-reported outcomes to the status of coprimary endpoints in the clinical trials.
A positive result on one such patient-reported outcome, the Hyperhidrosis Severity Scale, was achieved in 57% of pediatric patients and 64% of adults at week 44 of open-label therapy. This required at least a 2-grade improvement from baseline, when roughly 60% of youths had a score of 3 and the remainder scored 4 on the 1-4 point scale.
From a mean baseline score of 9.2 on the Children’s Dermatology Life Quality Index, the pediatric group averaged a mean 6.2-point improvement at week 44, while adults experienced a mean 8.7-point improvement on the Dermatology Life Quality Index from a baseline of 11.25.
There was no diminution in treatment efficacy through 44 weeks, Dr. Green noted. Treatment-emergent adverse events consisted largely of transient mild to moderate anticholinergic effects, which seldom led to study discontinuation.
Dilated pupils and blurred vision were more common in children than adults (7.9% and 10.5% vs. 5.1% and 6.4%, respectively). “Why that is I can only speculate. Kids do tend to touch their eyes more often than adults. Pretty much everything else was the same. The adverse events can be worked around by educating people to use the pads appropriately. We saw the anticholinergic side effects more often in the first 4 weeks of the double-blind trials than in the longterm extension because once patients learned how to use the pad and not touch themselves afterwards, the adverse events came down,” he said.
The studies were sponsored by Dermira. Dr. Green has received research funding from and been a consultant to the company.
REPORTING FROM THE EADV CONGRESS
Key clinical point: Glycopyrronium tosylate pads address a common and undertreated medical condition in children: primary axillary hyperhidrosis.
Major finding: Mean scores on the Children’s Dermatology Life Quality Index improved by an average of 6.2 points from a baseline of 9.2 in children aged 9-16 years with primary axillary hyperhidrosis treated with once-daily glycopyrronium tosylate pads during an open-label, 44-week study.
Study details: This was a post hoc analysis of 44 patients aged 9-16 years and 507 patients aged 17 years and older who participated in a 44-week, open-label extension study of once-daily glycopyrronium tosylate pads for treatment of primary axillary hyperhidrosis.
Disclosures: The study was sponsored by Dermira. The presenter has received research funding from and been a consultant to the company.
Trifarotene cream for acne meets all endpoints in twin phase 3 trials
PARIS – A novel kinder, gentler topical retinoid met all of its primary and secondary endpoints in two identical phase 3 randomized trials totaling 2,420 patients with moderate acne vulgaris on both the face and trunk.

Trifarotene cream 50 mcg/g selectively targets the gamma retinoic acid receptor. This unique selectivity for just one of the three retinoic acid receptors results in less of the classic retinoid side effects – redness, scaling, dryness, stinging, burning – that can limit the clinical utility of existing retinoids. This was borne out by the high completion rates in trifarotene-treated participants in the two 12-week trials: 88.2% in the PERFECT 1 trial and 92.7% in PERFECT 2, compared with rates of 89.8% and 93.9% in vehicle-treated controls, Jerry K.L. Tan, MD, said at the annual congress of the European Academy of Dermatology and Venereology.
“Most adverse events involved local intolerance occurring at application sites and were mild and transient,” Dr. Tan, a dermatologist at the University of Western Ontario, Windsor, and head of Windsor Clinical Research, reported.
PERFECT 1 and 2 were multicenter, double-blind, randomized, vehicle-controlled, 12-week phase 3 trials. Of note, these were the first-ever large-scale randomized trials to simultaneously evaluate a topical therapy for treatment of both facial and truncal acne. This ambitious goal created some unique challenges, which Dr. Tan described.
Participants, all of whom had moderate acne vulgaris on the face and trunk, ranged in age from 9 to 58 years, with a mean age of 19 years. They were randomized to once-daily application of trifarotene cream 50 mcg/g or its vehicle in the evening.
Primary and secondary outcomes
One major efficacy endpoint on the face was achievement of Investigator Global Assessment success as defined by a score of 0 or 1, meaning clear or almost clear, coupled with at least a 2-grade improvement from baseline to week 12. In PERFECT 1 and 2 this was achieved by 29.7% and 42.8% of trifarotene cream-treated patients, response rates significantly better than the 20% and 25.8% in vehicle-treated controls.
Another endpoint for facial therapy were the absolute reductions from baseline in facial inflammatory and noninflammatory acne lesions. The mean reduction in inflammatory lesion count in trifarotene-treated patients was 19.6% in PERFECT 1 and 24.6% in PERFECT 2, both significantly better than the mean 15.8% and 19.6% decreases in controls. Noninflammatory facial lesion counts dropped by 26.7% and 30.4% with trifarotene, versus 18.9% and 22.3% with vehicle.
The efficacy yardsticks utilized on the trunk were the same as on the face except that Physician Global Assessment was the terminology utilized in lieu of Investigator Global Assessment. Physician Global Assessment success on the trunk was achieved by 35.8% and 41.1% of trifarotene-treated patients in the two trials, as compared with 25.7% and 30.1% of controls.
The mean reductions in truncal inflammatory lesion count obtained with trifarotene cream were 22% and 26.1%, both significantly better than the 18.8% and 20.3% rates with vehicle.
Treatment-emergent adverse events leading to study discontinuation occurred in 1.9% of trifarotene-treated patients in one trial and 1% in the other.
One audience member commented that the vehicle response rates looked too strong for that compound to be inert. Dr. Tan agreed. “The vehicle looks really good. One of the issues with vehicles is that many of them have to contain products to prevent decay, fermentation, and proliferation of yeast and bacteria. So I quite agree: I think many of our vehicles do have active ingredients,” he replied. “If you look at the topical dapsone trials, the vehicles look amazing.”
“The other possibility is that there’s what we call ‘investigator creep,’” the dermatologist continued. “It’s the notion that you have no idea what the patients are getting, but they look like maybe they’re getting better, so you grade it as better.”
Dr. Tan reported serving as an advisor and consultant to, speaker for, and recipient of research grants from Galderma, which sponsored the two phase 3 trials. The company is also developing trifarotene for the treatment of lamellar ichthyosis.
PARIS – A novel kinder, gentler topical retinoid met all of its primary and secondary endpoints in two identical phase 3 randomized trials totaling 2,420 patients with moderate acne vulgaris on both the face and trunk.
Trifarotene cream 50 mcg/g selectively targets the gamma retinoic acid receptor. This unique selectivity for just one of the three retinoic acid receptors results in less of the classic retinoid side effects – redness, scaling, dryness, stinging, burning – that can limit the clinical utility of existing retinoids. This was borne out by the high completion rates in trifarotene-treated participants in the two 12-week trials: 88.2% in the PERFECT 1 trial and 92.7% in PERFECT 2, compared with rates of 89.8% and 93.9% in vehicle-treated controls, Jerry K.L. Tan, MD, said at the annual congress of the European Academy of Dermatology and Venereology.
“Most adverse events involved local intolerance occurring at application sites and were mild and transient,” Dr. Tan, a dermatologist at the University of Western Ontario, Windsor, and head of Windsor Clinical Research, reported.
PERFECT 1 and 2 were multicenter, double-blind, randomized, vehicle-controlled, 12-week phase 3 trials. Of note, these were the first-ever large-scale randomized trials to simultaneously evaluate a topical therapy for treatment of both facial and truncal acne. This ambitious goal created some unique challenges, which Dr. Tan described.
Participants, all of whom had moderate acne vulgaris on the face and trunk, ranged in age from 9 to 58 years, with a mean age of 19 years. They were randomized to once-daily application of trifarotene cream 50 mcg/g or its vehicle in the evening.
Primary and secondary outcomes
One major efficacy endpoint on the face was achievement of Investigator Global Assessment success as defined by a score of 0 or 1, meaning clear or almost clear, coupled with at least a 2-grade improvement from baseline to week 12. In PERFECT 1 and 2 this was achieved by 29.7% and 42.8% of trifarotene cream-treated patients, response rates significantly better than the 20% and 25.8% in vehicle-treated controls.
Another endpoint for facial therapy were the absolute reductions from baseline in facial inflammatory and noninflammatory acne lesions. The mean reduction in inflammatory lesion count in trifarotene-treated patients was 19.6% in PERFECT 1 and 24.6% in PERFECT 2, both significantly better than the mean 15.8% and 19.6% decreases in controls. Noninflammatory facial lesion counts dropped by 26.7% and 30.4% with trifarotene, versus 18.9% and 22.3% with vehicle.
The efficacy yardsticks utilized on the trunk were the same as on the face except that Physician Global Assessment was the terminology utilized in lieu of Investigator Global Assessment. Physician Global Assessment success on the trunk was achieved by 35.8% and 41.1% of trifarotene-treated patients in the two trials, as compared with 25.7% and 30.1% of controls.
The mean reductions in truncal inflammatory lesion count obtained with trifarotene cream were 22% and 26.1%, both significantly better than the 18.8% and 20.3% rates with vehicle.
Treatment-emergent adverse events leading to study discontinuation occurred in 1.9% of trifarotene-treated patients in one trial and 1% in the other.
One audience member commented that the vehicle response rates looked too strong for that compound to be inert. Dr. Tan agreed. “The vehicle looks really good. One of the issues with vehicles is that many of them have to contain products to prevent decay, fermentation, and proliferation of yeast and bacteria. So I quite agree: I think many of our vehicles do have active ingredients,” he replied. “If you look at the topical dapsone trials, the vehicles look amazing.”
“The other possibility is that there’s what we call ‘investigator creep,’” the dermatologist continued. “It’s the notion that you have no idea what the patients are getting, but they look like maybe they’re getting better, so you grade it as better.”
Dr. Tan reported serving as an advisor and consultant to, speaker for, and recipient of research grants from Galderma, which sponsored the two phase 3 trials. The company is also developing trifarotene for the treatment of lamellar ichthyosis.
PARIS – A novel kinder, gentler topical retinoid met all of its primary and secondary endpoints in two identical phase 3 randomized trials totaling 2,420 patients with moderate acne vulgaris on both the face and trunk.
Trifarotene cream 50 mcg/g selectively targets the gamma retinoic acid receptor. This unique selectivity for just one of the three retinoic acid receptors results in less of the classic retinoid side effects – redness, scaling, dryness, stinging, burning – that can limit the clinical utility of existing retinoids. This was borne out by the high completion rates in trifarotene-treated participants in the two 12-week trials: 88.2% in the PERFECT 1 trial and 92.7% in PERFECT 2, compared with rates of 89.8% and 93.9% in vehicle-treated controls, Jerry K.L. Tan, MD, said at the annual congress of the European Academy of Dermatology and Venereology.
“Most adverse events involved local intolerance occurring at application sites and were mild and transient,” Dr. Tan, a dermatologist at the University of Western Ontario, Windsor, and head of Windsor Clinical Research, reported.
PERFECT 1 and 2 were multicenter, double-blind, randomized, vehicle-controlled, 12-week phase 3 trials. Of note, these were the first-ever large-scale randomized trials to simultaneously evaluate a topical therapy for treatment of both facial and truncal acne. This ambitious goal created some unique challenges, which Dr. Tan described.
Participants, all of whom had moderate acne vulgaris on the face and trunk, ranged in age from 9 to 58 years, with a mean age of 19 years. They were randomized to once-daily application of trifarotene cream 50 mcg/g or its vehicle in the evening.
Primary and secondary outcomes
One major efficacy endpoint on the face was achievement of Investigator Global Assessment success as defined by a score of 0 or 1, meaning clear or almost clear, coupled with at least a 2-grade improvement from baseline to week 12. In PERFECT 1 and 2 this was achieved by 29.7% and 42.8% of trifarotene cream-treated patients, response rates significantly better than the 20% and 25.8% in vehicle-treated controls.
Another endpoint for facial therapy were the absolute reductions from baseline in facial inflammatory and noninflammatory acne lesions. The mean reduction in inflammatory lesion count in trifarotene-treated patients was 19.6% in PERFECT 1 and 24.6% in PERFECT 2, both significantly better than the mean 15.8% and 19.6% decreases in controls. Noninflammatory facial lesion counts dropped by 26.7% and 30.4% with trifarotene, versus 18.9% and 22.3% with vehicle.
The efficacy yardsticks utilized on the trunk were the same as on the face except that Physician Global Assessment was the terminology utilized in lieu of Investigator Global Assessment. Physician Global Assessment success on the trunk was achieved by 35.8% and 41.1% of trifarotene-treated patients in the two trials, as compared with 25.7% and 30.1% of controls.
The mean reductions in truncal inflammatory lesion count obtained with trifarotene cream were 22% and 26.1%, both significantly better than the 18.8% and 20.3% rates with vehicle.
Treatment-emergent adverse events leading to study discontinuation occurred in 1.9% of trifarotene-treated patients in one trial and 1% in the other.
One audience member commented that the vehicle response rates looked too strong for that compound to be inert. Dr. Tan agreed. “The vehicle looks really good. One of the issues with vehicles is that many of them have to contain products to prevent decay, fermentation, and proliferation of yeast and bacteria. So I quite agree: I think many of our vehicles do have active ingredients,” he replied. “If you look at the topical dapsone trials, the vehicles look amazing.”
“The other possibility is that there’s what we call ‘investigator creep,’” the dermatologist continued. “It’s the notion that you have no idea what the patients are getting, but they look like maybe they’re getting better, so you grade it as better.”
Dr. Tan reported serving as an advisor and consultant to, speaker for, and recipient of research grants from Galderma, which sponsored the two phase 3 trials. The company is also developing trifarotene for the treatment of lamellar ichthyosis.
REPORTING FROM THE EADV CONGRESS
Key clinical point:
Major finding: Treatment-emergent adverse events leading to study discontinuation occurred in 1.9% of trifarotene cream–treated patients in one trial and 1% in the other.
Study details: PERFECT 1 and PERFECT 2 were identically designed 12-week phase 3 randomized trials including 2,420 patients with moderate facial and truncal acne.
Disclosures: The study presenter reported serving as an advisor, consultant to, speaker for, and recipient of research grants from Galderma, which sponsored the two phase 3 trials.
Novel oral agent shows unprecedented efficacy in psoriasis
PARIS – A novel in a phase 2 clinical trial including 267 adults with moderate to severe disease, James G. Krueger, MD, PhD, reported at the annual congress of the European Academy of Dermatology and Venereology.

“I would say the clinical response here is almost dead-on as a copy for ustekinumab, which is an [injectable interleukin] IL-23/IL-12 blocker. And we’re only at 12 weeks here; some of the curves look like they’re on a trajectory to go up further in terms of improvement. So I’m getting a performance with an oral drug that is just so much better than the approved alternatives that we have,” said Dr. Krueger, head of the laboratory of investigative dermatology and professor in clinical investigation at Rockefeller University in New York.
Oral apremilast (Otezla), for example, can’t touch those PASI 75 response rates in patients with moderate to severe psoriasis. Indeed, many psoriasis experts favor reserving apremilast for patients with moderate disease.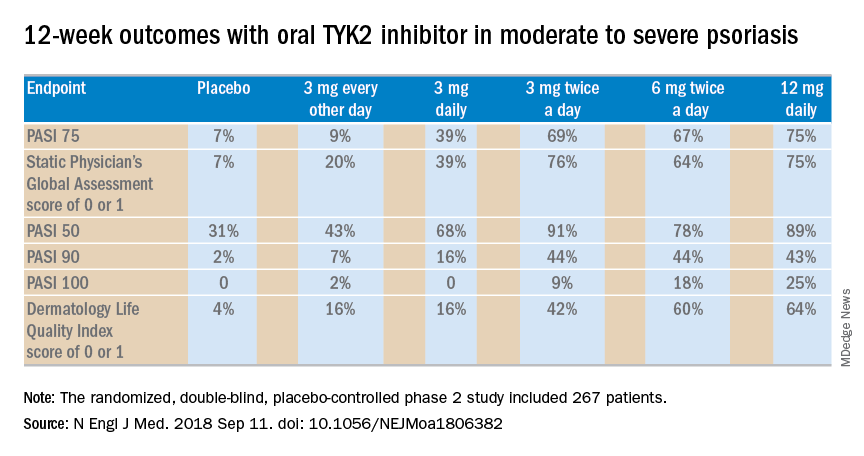
The 12-week, double-blind, placebo-controlled study was conducted at 82 sites in the United States and seven other countries. In this dose-ranging study, participants were randomized to the oral selective tyrosine kinase 2 (TYK2) inhibitor, known for the time being as BMS-986165, at 3 mg every other day, 3 mg daily, 3 mg twice a day, 6 mg twice a day, 12 mg daily, or to placebo.
The primary outcome was a 75% or greater reduction from baseline in Psoriasis Area and Severity Index score (PASI 75) at week 12. The TYK2 inhibitor outperformed placebo in dose-dependent fashion starting at the 3 mg/day dose. The PASI 75 rate was 7% with placebo, 9% with 3 mg of BMS-986165 every other day, 39% with 3 mg daily, 69% with 3 mg BID, 67% with 6 mg BID, and 75% with 12 mg/day. All secondary endpoints followed suit.
A striking finding in the phase 2 study was that when the drug was stopped for a month at the end of the 12-week treatment period, for the most part, the PASI 75 response and other clinical benefits were retained.
“I would contrast this to experiments that I have personally done with cyclosporine, where I have cleared people with cyclosporine, stopped it, and a month later every single patient has rip-roaring disease back. So I think this TYK2 inhibitor has some different performance features than just blocking a downstream T-cell transduction molecule,” observed the dermatologist, who is credited as the discoverer of the importance of the T cell in psoriasis pathogenesis.
The strong multidimensional evidence of clinical efficacy in the phase 2 study was supported mechanistically by analysis of skin biopsies obtained on study days 1, 15, and 85. The laboratory studies showed that the oral drug improved molecular, cellular, and clinical biomarkers associated with treatment efficacy. For example, at doses of 3 mg twice a day or higher, the TYK2 inhibitor reduced expression of IL-19 and IL-36A, which are key drivers of keratinocyte activation and epidermal hyperplasia. The drug also markedly decreased expression of genes in the Th17 pathway and essentially normalized expression of the proinflammatory genes beta defensin and S100A9.
In contrast to the Janus kinase (JAK) 1/3 and JAK 2 inhibitors in development for treatment of psoriasis, which paint with a much broader brush, the TYK2 inhibitor is highly selective for IL-23, IL-12, and interferon alpha.
“Previous studies have shown pan-JAK inhibition can be very effective in remitting psoriasis. The problem is that if one inhibits JAK1 and JAK3, one blocks the transduction of effector cytokines that are essentially there for protective immunity. That could lead to undesirable levels of immunosuppression,” Dr. Krueger explained.
The most important cytokine in the pathogenesis of psoriasis is clearly IL-23, he continued. In cell-based assays, the TYK2 inhibitor has been shown to be 100 times more selective in inhibiting IL-23 , IL-12, and interferon-alpha than JAK 1/3 inhibitors and 3,000 times more selective than JAK 2 inhibitors. This high degree of selectivity makes for fewer off-target effects and for a favorable safety profile.
“There were no major safety signals that would lead you to be concerned,” Dr. Krueger said. Indeed, based upon the encouraging safety and efficacy demonstrated this phase 2 study, a phase 3 program known as POETYK-PSO is underway (POETYK-PSO-1 and POETYK-PSO-2).
The phase 2 clinical trial results were published online in conjunction with the EADV congress.
The TYK2 inhibitor is being developed by Bristol-Myers Squibb. Dr. Krueger reported receiving personal fees as well as research grants paid directly to Rockefeller University from that pharmaceutical company and numerous others.
Source: Papp K et al. N Engl J Med. 2018 Sep 11. doi: 10.1056/NEJMoa1806382.
PARIS – A novel in a phase 2 clinical trial including 267 adults with moderate to severe disease, James G. Krueger, MD, PhD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“I would say the clinical response here is almost dead-on as a copy for ustekinumab, which is an [injectable interleukin] IL-23/IL-12 blocker. And we’re only at 12 weeks here; some of the curves look like they’re on a trajectory to go up further in terms of improvement. So I’m getting a performance with an oral drug that is just so much better than the approved alternatives that we have,” said Dr. Krueger, head of the laboratory of investigative dermatology and professor in clinical investigation at Rockefeller University in New York.
Oral apremilast (Otezla), for example, can’t touch those PASI 75 response rates in patients with moderate to severe psoriasis. Indeed, many psoriasis experts favor reserving apremilast for patients with moderate disease.
The 12-week, double-blind, placebo-controlled study was conducted at 82 sites in the United States and seven other countries. In this dose-ranging study, participants were randomized to the oral selective tyrosine kinase 2 (TYK2) inhibitor, known for the time being as BMS-986165, at 3 mg every other day, 3 mg daily, 3 mg twice a day, 6 mg twice a day, 12 mg daily, or to placebo.
The primary outcome was a 75% or greater reduction from baseline in Psoriasis Area and Severity Index score (PASI 75) at week 12. The TYK2 inhibitor outperformed placebo in dose-dependent fashion starting at the 3 mg/day dose. The PASI 75 rate was 7% with placebo, 9% with 3 mg of BMS-986165 every other day, 39% with 3 mg daily, 69% with 3 mg BID, 67% with 6 mg BID, and 75% with 12 mg/day. All secondary endpoints followed suit.
A striking finding in the phase 2 study was that when the drug was stopped for a month at the end of the 12-week treatment period, for the most part, the PASI 75 response and other clinical benefits were retained.
“I would contrast this to experiments that I have personally done with cyclosporine, where I have cleared people with cyclosporine, stopped it, and a month later every single patient has rip-roaring disease back. So I think this TYK2 inhibitor has some different performance features than just blocking a downstream T-cell transduction molecule,” observed the dermatologist, who is credited as the discoverer of the importance of the T cell in psoriasis pathogenesis.
The strong multidimensional evidence of clinical efficacy in the phase 2 study was supported mechanistically by analysis of skin biopsies obtained on study days 1, 15, and 85. The laboratory studies showed that the oral drug improved molecular, cellular, and clinical biomarkers associated with treatment efficacy. For example, at doses of 3 mg twice a day or higher, the TYK2 inhibitor reduced expression of IL-19 and IL-36A, which are key drivers of keratinocyte activation and epidermal hyperplasia. The drug also markedly decreased expression of genes in the Th17 pathway and essentially normalized expression of the proinflammatory genes beta defensin and S100A9.
In contrast to the Janus kinase (JAK) 1/3 and JAK 2 inhibitors in development for treatment of psoriasis, which paint with a much broader brush, the TYK2 inhibitor is highly selective for IL-23, IL-12, and interferon alpha.
“Previous studies have shown pan-JAK inhibition can be very effective in remitting psoriasis. The problem is that if one inhibits JAK1 and JAK3, one blocks the transduction of effector cytokines that are essentially there for protective immunity. That could lead to undesirable levels of immunosuppression,” Dr. Krueger explained.
The most important cytokine in the pathogenesis of psoriasis is clearly IL-23, he continued. In cell-based assays, the TYK2 inhibitor has been shown to be 100 times more selective in inhibiting IL-23 , IL-12, and interferon-alpha than JAK 1/3 inhibitors and 3,000 times more selective than JAK 2 inhibitors. This high degree of selectivity makes for fewer off-target effects and for a favorable safety profile.
“There were no major safety signals that would lead you to be concerned,” Dr. Krueger said. Indeed, based upon the encouraging safety and efficacy demonstrated this phase 2 study, a phase 3 program known as POETYK-PSO is underway (POETYK-PSO-1 and POETYK-PSO-2).
The phase 2 clinical trial results were published online in conjunction with the EADV congress.
The TYK2 inhibitor is being developed by Bristol-Myers Squibb. Dr. Krueger reported receiving personal fees as well as research grants paid directly to Rockefeller University from that pharmaceutical company and numerous others.
Source: Papp K et al. N Engl J Med. 2018 Sep 11. doi: 10.1056/NEJMoa1806382.
PARIS – A novel in a phase 2 clinical trial including 267 adults with moderate to severe disease, James G. Krueger, MD, PhD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“I would say the clinical response here is almost dead-on as a copy for ustekinumab, which is an [injectable interleukin] IL-23/IL-12 blocker. And we’re only at 12 weeks here; some of the curves look like they’re on a trajectory to go up further in terms of improvement. So I’m getting a performance with an oral drug that is just so much better than the approved alternatives that we have,” said Dr. Krueger, head of the laboratory of investigative dermatology and professor in clinical investigation at Rockefeller University in New York.
Oral apremilast (Otezla), for example, can’t touch those PASI 75 response rates in patients with moderate to severe psoriasis. Indeed, many psoriasis experts favor reserving apremilast for patients with moderate disease.
The 12-week, double-blind, placebo-controlled study was conducted at 82 sites in the United States and seven other countries. In this dose-ranging study, participants were randomized to the oral selective tyrosine kinase 2 (TYK2) inhibitor, known for the time being as BMS-986165, at 3 mg every other day, 3 mg daily, 3 mg twice a day, 6 mg twice a day, 12 mg daily, or to placebo.
The primary outcome was a 75% or greater reduction from baseline in Psoriasis Area and Severity Index score (PASI 75) at week 12. The TYK2 inhibitor outperformed placebo in dose-dependent fashion starting at the 3 mg/day dose. The PASI 75 rate was 7% with placebo, 9% with 3 mg of BMS-986165 every other day, 39% with 3 mg daily, 69% with 3 mg BID, 67% with 6 mg BID, and 75% with 12 mg/day. All secondary endpoints followed suit.
A striking finding in the phase 2 study was that when the drug was stopped for a month at the end of the 12-week treatment period, for the most part, the PASI 75 response and other clinical benefits were retained.
“I would contrast this to experiments that I have personally done with cyclosporine, where I have cleared people with cyclosporine, stopped it, and a month later every single patient has rip-roaring disease back. So I think this TYK2 inhibitor has some different performance features than just blocking a downstream T-cell transduction molecule,” observed the dermatologist, who is credited as the discoverer of the importance of the T cell in psoriasis pathogenesis.
The strong multidimensional evidence of clinical efficacy in the phase 2 study was supported mechanistically by analysis of skin biopsies obtained on study days 1, 15, and 85. The laboratory studies showed that the oral drug improved molecular, cellular, and clinical biomarkers associated with treatment efficacy. For example, at doses of 3 mg twice a day or higher, the TYK2 inhibitor reduced expression of IL-19 and IL-36A, which are key drivers of keratinocyte activation and epidermal hyperplasia. The drug also markedly decreased expression of genes in the Th17 pathway and essentially normalized expression of the proinflammatory genes beta defensin and S100A9.
In contrast to the Janus kinase (JAK) 1/3 and JAK 2 inhibitors in development for treatment of psoriasis, which paint with a much broader brush, the TYK2 inhibitor is highly selective for IL-23, IL-12, and interferon alpha.
“Previous studies have shown pan-JAK inhibition can be very effective in remitting psoriasis. The problem is that if one inhibits JAK1 and JAK3, one blocks the transduction of effector cytokines that are essentially there for protective immunity. That could lead to undesirable levels of immunosuppression,” Dr. Krueger explained.
The most important cytokine in the pathogenesis of psoriasis is clearly IL-23, he continued. In cell-based assays, the TYK2 inhibitor has been shown to be 100 times more selective in inhibiting IL-23 , IL-12, and interferon-alpha than JAK 1/3 inhibitors and 3,000 times more selective than JAK 2 inhibitors. This high degree of selectivity makes for fewer off-target effects and for a favorable safety profile.
“There were no major safety signals that would lead you to be concerned,” Dr. Krueger said. Indeed, based upon the encouraging safety and efficacy demonstrated this phase 2 study, a phase 3 program known as POETYK-PSO is underway (POETYK-PSO-1 and POETYK-PSO-2).
The phase 2 clinical trial results were published online in conjunction with the EADV congress.
The TYK2 inhibitor is being developed by Bristol-Myers Squibb. Dr. Krueger reported receiving personal fees as well as research grants paid directly to Rockefeller University from that pharmaceutical company and numerous others.
Source: Papp K et al. N Engl J Med. 2018 Sep 11. doi: 10.1056/NEJMoa1806382.
REPORTING FROM THE EADV CONGRESS
Key clinical point: A novel selective tyrosine kinase 2 inhibitor achieves response rates previously unheard of in oral therapy for moderate to severe psoriasis.
Major finding: At the top dose of oral BMS-986165 studied to date, the PASI 75 rate at 12 weeks was 75%.
Study details: This eight-country, randomized, double-blind, placebo-controlled phase 2 study included 267 patients with moderate to severe psoriasis.
Disclosures: The study was sponsored by Bristol-Myers Squibb. The presenter reported receiving personal fees and institutional research grants from that pharmaceutical company and numerous others.
Source: Papp K et al. N Engl J Med. 2018 Sep 11. doi: 10.1056/NEJMoa1806382.