User login
Anastrozole-Induced Subacute Cutaneous Lupus Erythematosus
Drug-induced subacute cutaneous lupus erythematosus (DI-SCLE) was first described in 1985 in 5 patients who had been taking hydrochlorothiazide.1 The skin lesions in these patients were identical to those seen in idiopathic subacute cutaneous lupus erythematosus (SCLE) and were accompanied by the same autoantibodies (anti-Ro/Sjögren syndrome antigen A [SS-A] and anti-La/Sjögren syndrome antigen B [SS-B]) and HLA type (HLA-DR2/DR3) that are known to be associated with idiopathic SCLE. The skin lesions of SCLE in these 5 patients resolved spontaneously after discontinuing hydrochlorothiazide; however, anti-Ro/SS-A antibodies persisted in all except 1 patient.1 Over the last decade, an increasing number of drugs from different classes have been implicated to be associated with DI-SCLE. Since the concept of DI-SCLE was introduced, it has been reported to look identical to idiopathic SCLE, both clinically and histopathologically; however, one report suggested that the 2 entities can be distinguished based on clinical variations.2 In general, patients with DI-SCLE develop the same anti-Ro antibodies as seen in idiopathic SCLE. In addition, although the rash in DI-SCLE typically resolves with withdrawal of the offending drug, the antibodies tend to persist. Herein, we report a case of a patient being treated with an aromatase inhibitor who presented with clinical, serologic, and histopathologic evidence of DI-SCLE.
Case Report
A 69-year-old woman diagnosed with breast cancer 4 years prior to her presentation to dermatology initially underwent a lumpectomy and radiation treatment. She was subsequently started on anastrozole 2 years later. After 16 months of treatment with anastrozole, she developed an erythematous scaly rash on sun-exposed areas of the skin. The patient was seen by an outside dermatologist who treated her for a patient-perceived drug rash based on biopsy results that simply demonstrated interface dermatitis. She was treated with both topical and oral steroids with little improvement and therefore presented to our office approximately 6 months after starting treatment seeking a second opinion.
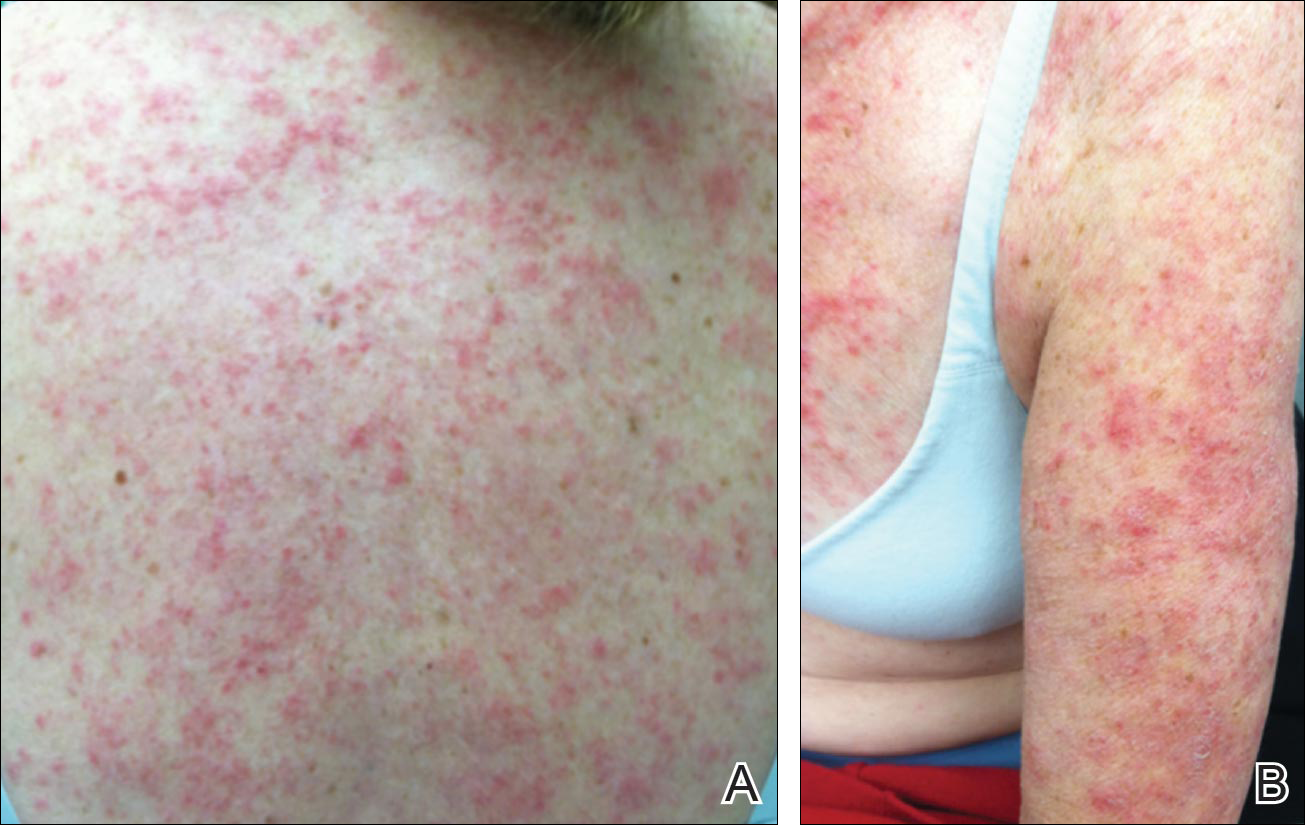
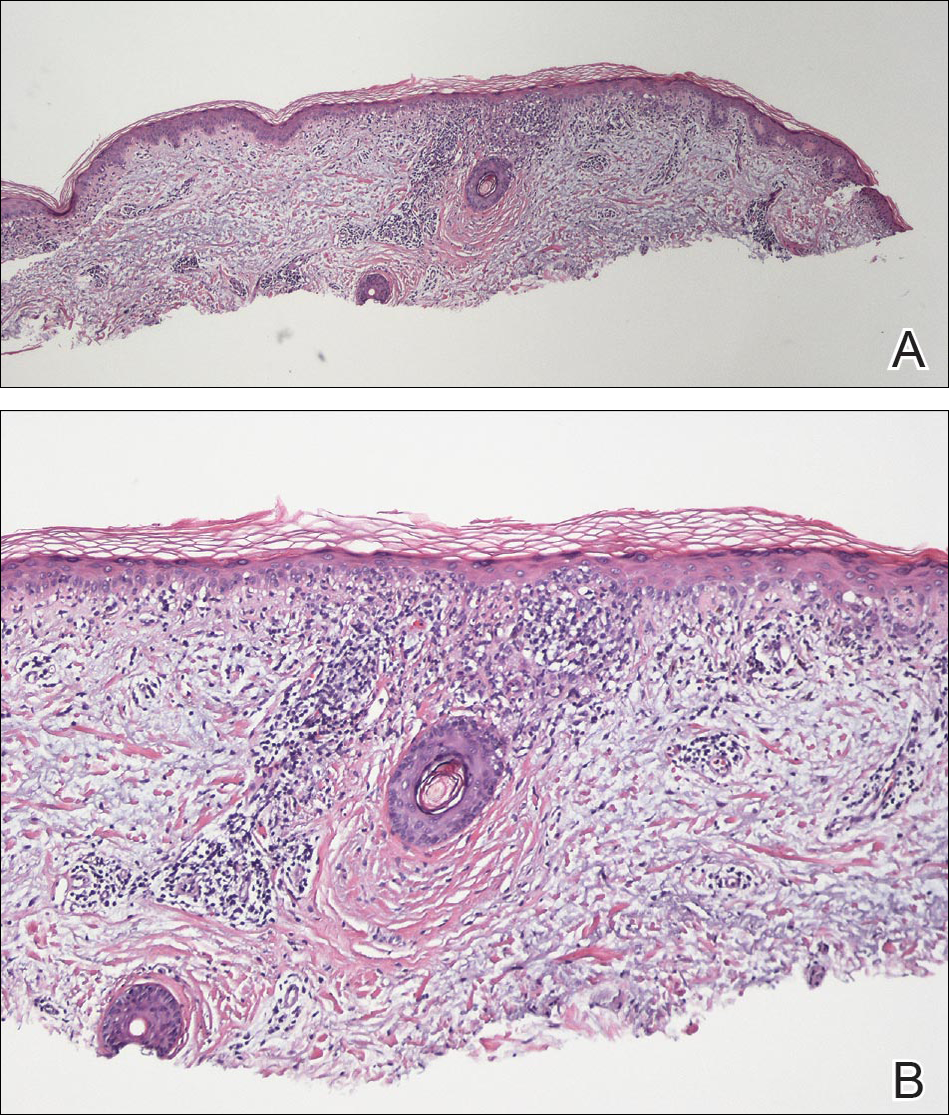
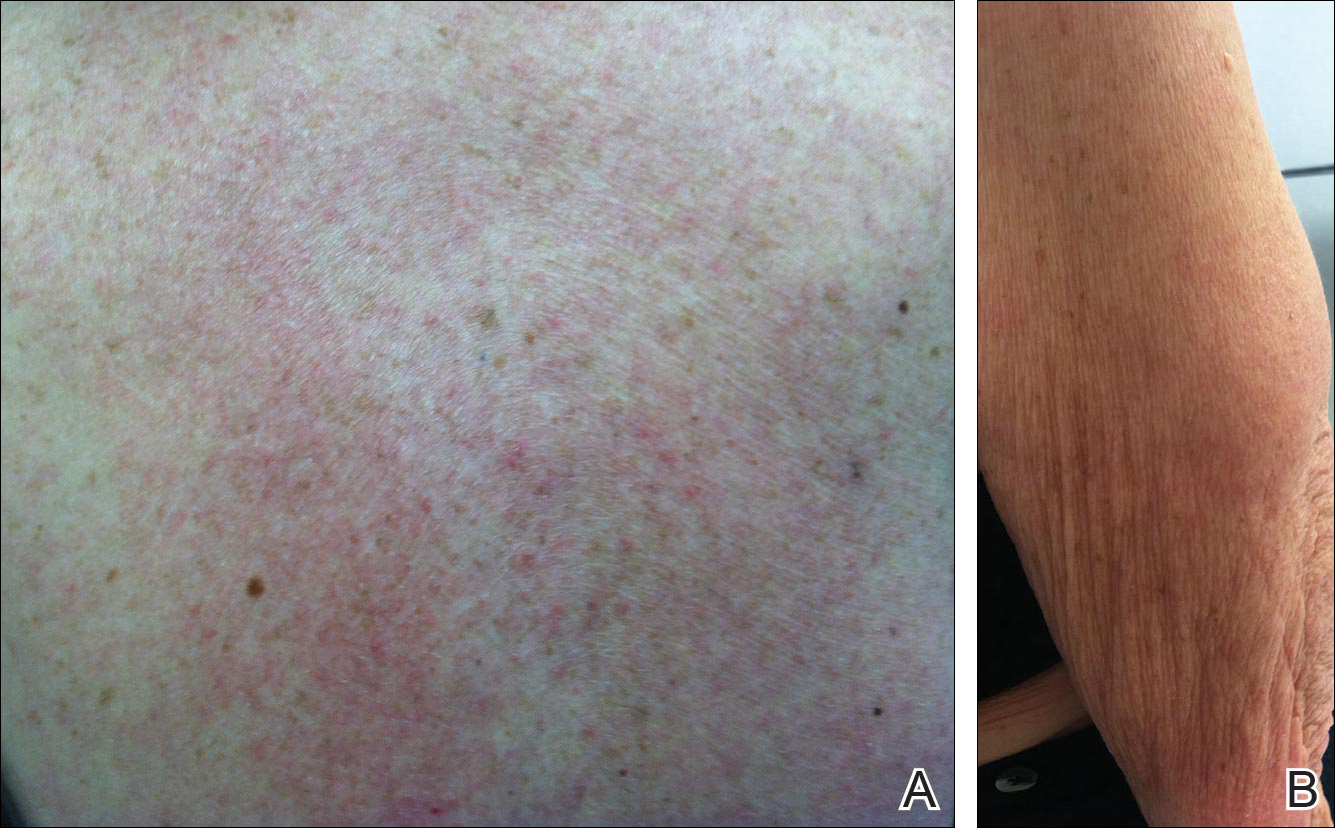
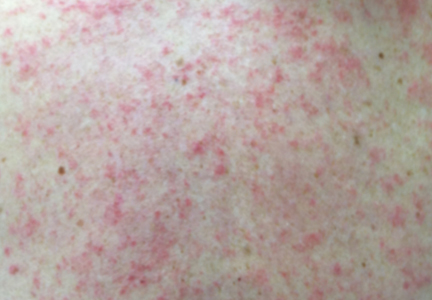
Physical examination revealed numerous erythematous scaly papules and plaques in a photodistributed pattern on the chest, back, legs, and arms (Figure 1). On further questioning, the patient noted that the rash became worse when she was at the beach or playing tennis outside as well as under indoor lights. A repeat biopsy was performed, revealing interface and perivascular dermatitis with an infiltrate composed of lymphocytes, histiocytes, and scattered pigment-laden macrophages (Figure 2). Given the appearance and distribution of the rash as well as the clinical scenario, drug-induced lupus was suspected. Anastrozole was the only medication being taken. Laboratory evaluation was performed and was negative for antinuclear antibodies, antihistone antibodies, and anti-La/SS-B antibodies but was positive for anti-Ro/SS-A antibodies (>8.0 U [reference range, <1.0 U]). Based on these findings, anastrozole-induced SCLE was the most likely explanation for this presentation. The patient was started on a sun-protective regimen (ie, wide-brimmed hat, daily sunscreen) and anastrozole was discontinued by her oncologist; the combination led to moderate improvement in symptoms. One week later, oral hydroxychloroquine 200 mg twice daily was started, which led to notable improvement (Figure 3). The patient was seen for 2 additional follow-up visits, each time with sustained resolution of the rash. The hydroxychloroquine was then stopped at her last visit 3 months after diagnosis. The patient was subsequently lost to follow-up.
Comment
Presentation of SCLE
Subacute cutaneous lupus erythematosus is a form of lupus erythematosus characterized by nonscarring, annular, scaly, erythematous plaques that occur on sun-exposed skin. The lesions are classically distributed on the upper back, chest, dorsal arms, and lateral neck but also can be found in other locations.3,4 Subacute cutaneous lupus erythematosus may be idiopathic; may occur in patients with systemic lupus erythematosus, Sjögren syndrome, or deficiency of the second component of complement (C2d); or may be drug induced.5 On histology SCLE presents as a lichenoid tissue reaction with focal vacuolization of the epidermal basal layer and perivascular lymphocytic infiltrate. On direct immunofluorescence, both idiopathic and drug-induced SCLE present with granular deposition of IgM, IgG, and C3 in a bandlike array at the dermoepidermal junction and circulating anti-Ro/SS-A antibodies. Therefore, histopathologically and immunologically, DI-SCLE is indistinguishable from idiopathic cases.6
Differential Diagnosis
It was previously thought that the clinical presentation of DI-SCLE and idiopathic SCLE were indistinguishable; however, Marzano et al2 described remarkable differences in the cutaneous manifestations of the 2 diseases. Drug-induced SCLE lesions are more widespread, occur more frequently on the legs, and may be bullous or erythema multiforme–like versus the idiopathic lesions, which tend to be more concentrated on the upper body and classically present as scaly erythematous plaques. Additionally, malar rash and vasculitic lesions, such as purpura and necrotic-ulcerative lesions, are seen more often in DI-SCLE.
Drug-induced systemic lupus erythematosus (DI-SLE) is a lupuslike syndrome that can be differentiated from DI-SCLE by virtue of its clinical and serological presentation. It differs from DI-SCLE in that DI-SLE typically does not present with skin symptoms; rather, systemic symptoms such as fever, weight loss, arthralgia, polyarthritis, pericarditis, and pleuritis are more commonly seen. Additionally, it has been associated with antihistone antibodies.4 More than 80 drugs have been reported to cause DI-SLE, including procainamide, hydralazine, and quinidine.7
To be classified as either DI-SCLE or DI-SLE, symptoms need to present after administration of the triggering drug and must resolve after the drug is discontinued.7 The drugs most commonly associated with DI-SCLE are thiazides, calcium channel blockers, tumor necrosis factor α inhibitors, angiotensin-converting enzyme inhibitors, and terbinafine, with few cases citing anastrozole as the inciting agent.4,6,8,9 The incubation period for DI-SCLE varies substantially. Thiazide diuretics and calcium channel blockers typically have the longest incubation period, ranging from 6 months to 5 years for thiazides,1,6,10,11 while calcium channel blockers have an average incubation period of 3 years.12 Drug-induced SCLE associated with antifungals, however, usually is much more rapid in onset; the incubation period on average is 5 weeks for terbinafine and 2 weeks for griseofulvin.13-15
Antiestrogen Drugs and SCLE
Anastrozole, the inciting agent in our case, is a third-generation, selective, nonsteroidal, aromatase inhibitor with no progestogenic, androgenic, or estrogenic activity. Anastrozole, when taken at its recommended dosage of 1 mg daily, will suppress estradiol. It is used as an adjuvant treatment of estrogen-sensitive breast cancer in postmenopausal women. In contrast to a prior case of DI-SCLE secondary to anastrozole in which the incubation period was approximately 1 month,8 our patient had an incubation period of approximately 16 months. Tamoxifen, another antiestrogen drug, also has been associated with DI-SCLE.9 In cases of tamoxifen-induced SCLE, the incubation period was several years, which is more similar to our patient. Although these drugs do not have the same mechanism of action, they both have antiestrogen properties.9 A systemic review of DI-SCLE reported that incubation periods between drug exposure and appearance of DI-SCLE varied greatly and were drug class dependent. It is possible that reactions associated with antiestrogen medications have a delayed presentation; however, given there are limited cases of anastrozole-induced DI-SCLE, we cannot make a clear statement on incubation periods.6
Reports of DI-SCLE caused by antiestrogen drugs are particularly interesting because sex hormones in relation to lupus disease activity have been the subject of debate for decades. Women are considerably more likely to develop autoimmune diseases than men, suggesting that steroid hormones, especially estrogen and progesterone, influence the immune system.16 Estrogen actions are proinflammatory, while the actions of progesterone, androgens, and glucocorticoids are anti-inflammatory.17 Studies in women with lupus revealed an increased rate of mild- to moderate-intensity disease flares associated with estrogen-containing hormone replace-ment therapy.18-20
Over the years, several antiestrogen therapies have been used in murine models, which showed remarkable clinical improvement in the course of SLE. The precise mechanisms involved in disease immunomodulation by these therapies have not been elucidated.21-23 It is thought that estrogen plays a role in the synthesis and expression of Ro antigens on the surface of keratinocytes, increasing the fixation of anti-Ro antibodies in keratinocytes and provoking the appearance of a cutaneous eruption in patients with a susceptible HLA profile.6
Conclusion
We report a rare case of SCLE induced by anastrozole use. Cases such as ours and others that implicate antiestrogen drugs in association with DI-SCLE are particularly noteworthy, considering many studies are looking at the potential usefulness of antiestrogen therapy in the treatment of SLE. Further research on this relationship is warranted.
- Reed B, Huff J, Jones S, et al. Subacute cutaneous lupus erythematosus associated with hydrochlorothiazide therapy. Ann Intern Med. 1985;103:49-51.
- Marzano A, Lazzari R, Polloni I, et al. Drug-induced subacute cutaneous lupus erythematosus: evidence for differences from its idiopathic counterpart. Br J Dermatol. 2011;165:335-341.
- Bonsmann G, Schiller M, Luger T, et al. Terbinafine-induced subacute cutaneous lupus erythematosus. J Am Acad Dermatol. 2001;44:925-931.
- Callen J. Review: drug induced subacute cutaneous lupus erythematosus. Lupus. 2010;19:1107-1111.
- Lin J, Callen JP. Subacute cutaneous lupus erythematosus (SCLE). Medscape website. http://emedicine.medscape.com/article/1065657-overview. Updated March 7, 2016. Accessed April 29, 2016.
- Lowe GC, Henderson CL, Grau RH, et al. A systematic review of drug-induced subacute cutaneous lupus erythematosus. Br J Dermatol. 2011;164:465-472.
- Vedove C, Giglio M, Schena D, et al. Drug-induced lupus erythematosus. Arch Dermatol Res. 2009;301:99-105.
- Trancart M, Cavailhes A, Balme B, et al. Anastrozole-induced subacute cutaneous lupus erythematosus [published online December 6, 2007]. Br J Dermatol. 2008;158:628-629.
- Fumal I, Danchin A, Cosserat F, et al. Subacute cutaneous lupus erythematosus associated with tamoxifen therapy: two cases. Dermatology. 2005;210:251-252.
- Brown C, Deng J. Thiazide diuretics induce cutaneous lupus-like adverse reaction. J Toxicol Clin Toxicol. 1995;33:729-733.
- Sontheimer R. Subacute cutaneous lupus erythematosus: 25-year evolution of a prototypic subset (subphenotype) of lupus erythematosus defined by characteristic cutaneous, pathological, immunological, and genetic findings. Autoimmun Rev. 2005;4:253-263.
- Crowson A, Magro C. Subacute cutaneous lupus erythematosus arising in the setting of calcium channel blocker therapy. Hum Pathol. 1997;28:67-73.
- Lorentz K, Booken N, Goerdt S, et al. Subacute cutaneous lupus erythematosus induced by terbinafine: case report and review of literature. J Dtsch Dermatol Ges. 2008;6:823-837.
- Kasperkiewicz M, Anemüller W, Angelova-Fischer I, et al. Subacute cutaneous lupus erythematosus associated with terbinafine. Clin Exp Dermatol. 2009;34:403-404.
- Miyagawa S, Okuchi T, Shiomi Y, et al. Subacute cutaneous lupus erythematosus lesions precipitated by griseofulvin. J Am Acad Dermatol. 1989;21:343-346.
- Inman RD. Immunologic sex differences and the female predominance in systemic lupus erythematosus. Arthritis Rheum. 1978;21:849-854.
- Cutolo M, Wilder RL. Different roles of androgens and estrogens in the susceptibility to autoimmune rheumatic diseases. Rheum Dis Clin North Am. 2000;26:825-839.
- Petri M. Sex hormones and systemic lupus erythematosus. Lupus. 2008;17:412-415.
- Lateef A, Petri M. Hormone replacement and contraceptive therapy in autoimmune diseases [published online January 18, 2012]. J Autoimmun. 2012;38:J170-J176.
- Buyon JP, Petri M, Kim MY, et al. The effect of combined estrogen and progesterone hormone replacement therapy on disease activity in systemic lupus erythematosus: a randomized trial. Ann Intern Med. 2005;142:954-962.
- Wu W, Suen J, Lin B, et al. Tamoxifen alleviates disease severity and decreases double negative T cells in autoimmune MRL-lpr/lpr mice. Immunology. 2000;100:110-118.
- Dayan M, Zinger H, Kalush F, et al. The beneficial effects of treatment with tamoxifen and anti-oestradiol antibody on experimental systemic lupus erythematosus are associated with cytokine modulations. Immunology. 1997;90:101-108.
- Sthoeger Z, Zinger H, Mozes E. Beneficial effects of the anti-oestrogen tamoxifen on systemic lupus erythematosus of (NZBxNZW)F1 female mice are associated with specific reduction of IgG3 autoantibodies. Ann Rheum Dis. 2003;62:341-346.
Drug-induced subacute cutaneous lupus erythematosus (DI-SCLE) was first described in 1985 in 5 patients who had been taking hydrochlorothiazide.1 The skin lesions in these patients were identical to those seen in idiopathic subacute cutaneous lupus erythematosus (SCLE) and were accompanied by the same autoantibodies (anti-Ro/Sjögren syndrome antigen A [SS-A] and anti-La/Sjögren syndrome antigen B [SS-B]) and HLA type (HLA-DR2/DR3) that are known to be associated with idiopathic SCLE. The skin lesions of SCLE in these 5 patients resolved spontaneously after discontinuing hydrochlorothiazide; however, anti-Ro/SS-A antibodies persisted in all except 1 patient.1 Over the last decade, an increasing number of drugs from different classes have been implicated to be associated with DI-SCLE. Since the concept of DI-SCLE was introduced, it has been reported to look identical to idiopathic SCLE, both clinically and histopathologically; however, one report suggested that the 2 entities can be distinguished based on clinical variations.2 In general, patients with DI-SCLE develop the same anti-Ro antibodies as seen in idiopathic SCLE. In addition, although the rash in DI-SCLE typically resolves with withdrawal of the offending drug, the antibodies tend to persist. Herein, we report a case of a patient being treated with an aromatase inhibitor who presented with clinical, serologic, and histopathologic evidence of DI-SCLE.
Case Report
A 69-year-old woman diagnosed with breast cancer 4 years prior to her presentation to dermatology initially underwent a lumpectomy and radiation treatment. She was subsequently started on anastrozole 2 years later. After 16 months of treatment with anastrozole, she developed an erythematous scaly rash on sun-exposed areas of the skin. The patient was seen by an outside dermatologist who treated her for a patient-perceived drug rash based on biopsy results that simply demonstrated interface dermatitis. She was treated with both topical and oral steroids with little improvement and therefore presented to our office approximately 6 months after starting treatment seeking a second opinion.
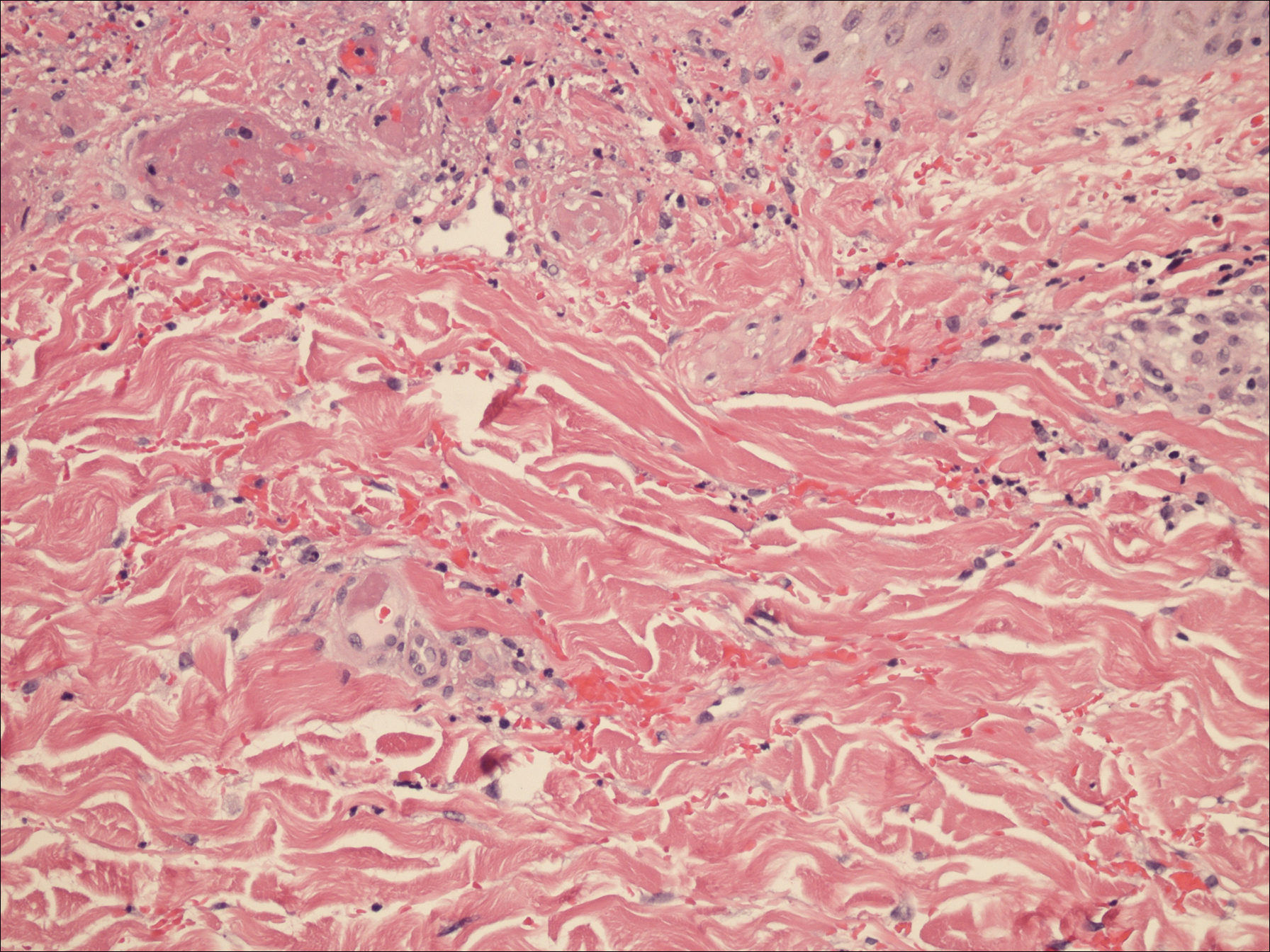
Physical examination revealed numerous erythematous scaly papules and plaques in a photodistributed pattern on the chest, back, legs, and arms (Figure 1). On further questioning, the patient noted that the rash became worse when she was at the beach or playing tennis outside as well as under indoor lights. A repeat biopsy was performed, revealing interface and perivascular dermatitis with an infiltrate composed of lymphocytes, histiocytes, and scattered pigment-laden macrophages (Figure 2). Given the appearance and distribution of the rash as well as the clinical scenario, drug-induced lupus was suspected. Anastrozole was the only medication being taken. Laboratory evaluation was performed and was negative for antinuclear antibodies, antihistone antibodies, and anti-La/SS-B antibodies but was positive for anti-Ro/SS-A antibodies (>8.0 U [reference range, <1.0 U]). Based on these findings, anastrozole-induced SCLE was the most likely explanation for this presentation. The patient was started on a sun-protective regimen (ie, wide-brimmed hat, daily sunscreen) and anastrozole was discontinued by her oncologist; the combination led to moderate improvement in symptoms. One week later, oral hydroxychloroquine 200 mg twice daily was started, which led to notable improvement (Figure 3). The patient was seen for 2 additional follow-up visits, each time with sustained resolution of the rash. The hydroxychloroquine was then stopped at her last visit 3 months after diagnosis. The patient was subsequently lost to follow-up.
Comment
Presentation of SCLE
Subacute cutaneous lupus erythematosus is a form of lupus erythematosus characterized by nonscarring, annular, scaly, erythematous plaques that occur on sun-exposed skin. The lesions are classically distributed on the upper back, chest, dorsal arms, and lateral neck but also can be found in other locations.3,4 Subacute cutaneous lupus erythematosus may be idiopathic; may occur in patients with systemic lupus erythematosus, Sjögren syndrome, or deficiency of the second component of complement (C2d); or may be drug induced.5 On histology SCLE presents as a lichenoid tissue reaction with focal vacuolization of the epidermal basal layer and perivascular lymphocytic infiltrate. On direct immunofluorescence, both idiopathic and drug-induced SCLE present with granular deposition of IgM, IgG, and C3 in a bandlike array at the dermoepidermal junction and circulating anti-Ro/SS-A antibodies. Therefore, histopathologically and immunologically, DI-SCLE is indistinguishable from idiopathic cases.6
Differential Diagnosis
It was previously thought that the clinical presentation of DI-SCLE and idiopathic SCLE were indistinguishable; however, Marzano et al2 described remarkable differences in the cutaneous manifestations of the 2 diseases. Drug-induced SCLE lesions are more widespread, occur more frequently on the legs, and may be bullous or erythema multiforme–like versus the idiopathic lesions, which tend to be more concentrated on the upper body and classically present as scaly erythematous plaques. Additionally, malar rash and vasculitic lesions, such as purpura and necrotic-ulcerative lesions, are seen more often in DI-SCLE.
Drug-induced systemic lupus erythematosus (DI-SLE) is a lupuslike syndrome that can be differentiated from DI-SCLE by virtue of its clinical and serological presentation. It differs from DI-SCLE in that DI-SLE typically does not present with skin symptoms; rather, systemic symptoms such as fever, weight loss, arthralgia, polyarthritis, pericarditis, and pleuritis are more commonly seen. Additionally, it has been associated with antihistone antibodies.4 More than 80 drugs have been reported to cause DI-SLE, including procainamide, hydralazine, and quinidine.7
To be classified as either DI-SCLE or DI-SLE, symptoms need to present after administration of the triggering drug and must resolve after the drug is discontinued.7 The drugs most commonly associated with DI-SCLE are thiazides, calcium channel blockers, tumor necrosis factor α inhibitors, angiotensin-converting enzyme inhibitors, and terbinafine, with few cases citing anastrozole as the inciting agent.4,6,8,9 The incubation period for DI-SCLE varies substantially. Thiazide diuretics and calcium channel blockers typically have the longest incubation period, ranging from 6 months to 5 years for thiazides,1,6,10,11 while calcium channel blockers have an average incubation period of 3 years.12 Drug-induced SCLE associated with antifungals, however, usually is much more rapid in onset; the incubation period on average is 5 weeks for terbinafine and 2 weeks for griseofulvin.13-15
Antiestrogen Drugs and SCLE
Anastrozole, the inciting agent in our case, is a third-generation, selective, nonsteroidal, aromatase inhibitor with no progestogenic, androgenic, or estrogenic activity. Anastrozole, when taken at its recommended dosage of 1 mg daily, will suppress estradiol. It is used as an adjuvant treatment of estrogen-sensitive breast cancer in postmenopausal women. In contrast to a prior case of DI-SCLE secondary to anastrozole in which the incubation period was approximately 1 month,8 our patient had an incubation period of approximately 16 months. Tamoxifen, another antiestrogen drug, also has been associated with DI-SCLE.9 In cases of tamoxifen-induced SCLE, the incubation period was several years, which is more similar to our patient. Although these drugs do not have the same mechanism of action, they both have antiestrogen properties.9 A systemic review of DI-SCLE reported that incubation periods between drug exposure and appearance of DI-SCLE varied greatly and were drug class dependent. It is possible that reactions associated with antiestrogen medications have a delayed presentation; however, given there are limited cases of anastrozole-induced DI-SCLE, we cannot make a clear statement on incubation periods.6
Reports of DI-SCLE caused by antiestrogen drugs are particularly interesting because sex hormones in relation to lupus disease activity have been the subject of debate for decades. Women are considerably more likely to develop autoimmune diseases than men, suggesting that steroid hormones, especially estrogen and progesterone, influence the immune system.16 Estrogen actions are proinflammatory, while the actions of progesterone, androgens, and glucocorticoids are anti-inflammatory.17 Studies in women with lupus revealed an increased rate of mild- to moderate-intensity disease flares associated with estrogen-containing hormone replace-ment therapy.18-20
Over the years, several antiestrogen therapies have been used in murine models, which showed remarkable clinical improvement in the course of SLE. The precise mechanisms involved in disease immunomodulation by these therapies have not been elucidated.21-23 It is thought that estrogen plays a role in the synthesis and expression of Ro antigens on the surface of keratinocytes, increasing the fixation of anti-Ro antibodies in keratinocytes and provoking the appearance of a cutaneous eruption in patients with a susceptible HLA profile.6
Conclusion
We report a rare case of SCLE induced by anastrozole use. Cases such as ours and others that implicate antiestrogen drugs in association with DI-SCLE are particularly noteworthy, considering many studies are looking at the potential usefulness of antiestrogen therapy in the treatment of SLE. Further research on this relationship is warranted.
Drug-induced subacute cutaneous lupus erythematosus (DI-SCLE) was first described in 1985 in 5 patients who had been taking hydrochlorothiazide.1 The skin lesions in these patients were identical to those seen in idiopathic subacute cutaneous lupus erythematosus (SCLE) and were accompanied by the same autoantibodies (anti-Ro/Sjögren syndrome antigen A [SS-A] and anti-La/Sjögren syndrome antigen B [SS-B]) and HLA type (HLA-DR2/DR3) that are known to be associated with idiopathic SCLE. The skin lesions of SCLE in these 5 patients resolved spontaneously after discontinuing hydrochlorothiazide; however, anti-Ro/SS-A antibodies persisted in all except 1 patient.1 Over the last decade, an increasing number of drugs from different classes have been implicated to be associated with DI-SCLE. Since the concept of DI-SCLE was introduced, it has been reported to look identical to idiopathic SCLE, both clinically and histopathologically; however, one report suggested that the 2 entities can be distinguished based on clinical variations.2 In general, patients with DI-SCLE develop the same anti-Ro antibodies as seen in idiopathic SCLE. In addition, although the rash in DI-SCLE typically resolves with withdrawal of the offending drug, the antibodies tend to persist. Herein, we report a case of a patient being treated with an aromatase inhibitor who presented with clinical, serologic, and histopathologic evidence of DI-SCLE.
Case Report
A 69-year-old woman diagnosed with breast cancer 4 years prior to her presentation to dermatology initially underwent a lumpectomy and radiation treatment. She was subsequently started on anastrozole 2 years later. After 16 months of treatment with anastrozole, she developed an erythematous scaly rash on sun-exposed areas of the skin. The patient was seen by an outside dermatologist who treated her for a patient-perceived drug rash based on biopsy results that simply demonstrated interface dermatitis. She was treated with both topical and oral steroids with little improvement and therefore presented to our office approximately 6 months after starting treatment seeking a second opinion.
Physical examination revealed numerous erythematous scaly papules and plaques in a photodistributed pattern on the chest, back, legs, and arms (Figure 1). On further questioning, the patient noted that the rash became worse when she was at the beach or playing tennis outside as well as under indoor lights. A repeat biopsy was performed, revealing interface and perivascular dermatitis with an infiltrate composed of lymphocytes, histiocytes, and scattered pigment-laden macrophages (Figure 2). Given the appearance and distribution of the rash as well as the clinical scenario, drug-induced lupus was suspected. Anastrozole was the only medication being taken. Laboratory evaluation was performed and was negative for antinuclear antibodies, antihistone antibodies, and anti-La/SS-B antibodies but was positive for anti-Ro/SS-A antibodies (>8.0 U [reference range, <1.0 U]). Based on these findings, anastrozole-induced SCLE was the most likely explanation for this presentation. The patient was started on a sun-protective regimen (ie, wide-brimmed hat, daily sunscreen) and anastrozole was discontinued by her oncologist; the combination led to moderate improvement in symptoms. One week later, oral hydroxychloroquine 200 mg twice daily was started, which led to notable improvement (Figure 3). The patient was seen for 2 additional follow-up visits, each time with sustained resolution of the rash. The hydroxychloroquine was then stopped at her last visit 3 months after diagnosis. The patient was subsequently lost to follow-up.
Comment
Presentation of SCLE
Subacute cutaneous lupus erythematosus is a form of lupus erythematosus characterized by nonscarring, annular, scaly, erythematous plaques that occur on sun-exposed skin. The lesions are classically distributed on the upper back, chest, dorsal arms, and lateral neck but also can be found in other locations.3,4 Subacute cutaneous lupus erythematosus may be idiopathic; may occur in patients with systemic lupus erythematosus, Sjögren syndrome, or deficiency of the second component of complement (C2d); or may be drug induced.5 On histology SCLE presents as a lichenoid tissue reaction with focal vacuolization of the epidermal basal layer and perivascular lymphocytic infiltrate. On direct immunofluorescence, both idiopathic and drug-induced SCLE present with granular deposition of IgM, IgG, and C3 in a bandlike array at the dermoepidermal junction and circulating anti-Ro/SS-A antibodies. Therefore, histopathologically and immunologically, DI-SCLE is indistinguishable from idiopathic cases.6
Differential Diagnosis
It was previously thought that the clinical presentation of DI-SCLE and idiopathic SCLE were indistinguishable; however, Marzano et al2 described remarkable differences in the cutaneous manifestations of the 2 diseases. Drug-induced SCLE lesions are more widespread, occur more frequently on the legs, and may be bullous or erythema multiforme–like versus the idiopathic lesions, which tend to be more concentrated on the upper body and classically present as scaly erythematous plaques. Additionally, malar rash and vasculitic lesions, such as purpura and necrotic-ulcerative lesions, are seen more often in DI-SCLE.
Drug-induced systemic lupus erythematosus (DI-SLE) is a lupuslike syndrome that can be differentiated from DI-SCLE by virtue of its clinical and serological presentation. It differs from DI-SCLE in that DI-SLE typically does not present with skin symptoms; rather, systemic symptoms such as fever, weight loss, arthralgia, polyarthritis, pericarditis, and pleuritis are more commonly seen. Additionally, it has been associated with antihistone antibodies.4 More than 80 drugs have been reported to cause DI-SLE, including procainamide, hydralazine, and quinidine.7
To be classified as either DI-SCLE or DI-SLE, symptoms need to present after administration of the triggering drug and must resolve after the drug is discontinued.7 The drugs most commonly associated with DI-SCLE are thiazides, calcium channel blockers, tumor necrosis factor α inhibitors, angiotensin-converting enzyme inhibitors, and terbinafine, with few cases citing anastrozole as the inciting agent.4,6,8,9 The incubation period for DI-SCLE varies substantially. Thiazide diuretics and calcium channel blockers typically have the longest incubation period, ranging from 6 months to 5 years for thiazides,1,6,10,11 while calcium channel blockers have an average incubation period of 3 years.12 Drug-induced SCLE associated with antifungals, however, usually is much more rapid in onset; the incubation period on average is 5 weeks for terbinafine and 2 weeks for griseofulvin.13-15
Antiestrogen Drugs and SCLE
Anastrozole, the inciting agent in our case, is a third-generation, selective, nonsteroidal, aromatase inhibitor with no progestogenic, androgenic, or estrogenic activity. Anastrozole, when taken at its recommended dosage of 1 mg daily, will suppress estradiol. It is used as an adjuvant treatment of estrogen-sensitive breast cancer in postmenopausal women. In contrast to a prior case of DI-SCLE secondary to anastrozole in which the incubation period was approximately 1 month,8 our patient had an incubation period of approximately 16 months. Tamoxifen, another antiestrogen drug, also has been associated with DI-SCLE.9 In cases of tamoxifen-induced SCLE, the incubation period was several years, which is more similar to our patient. Although these drugs do not have the same mechanism of action, they both have antiestrogen properties.9 A systemic review of DI-SCLE reported that incubation periods between drug exposure and appearance of DI-SCLE varied greatly and were drug class dependent. It is possible that reactions associated with antiestrogen medications have a delayed presentation; however, given there are limited cases of anastrozole-induced DI-SCLE, we cannot make a clear statement on incubation periods.6
Reports of DI-SCLE caused by antiestrogen drugs are particularly interesting because sex hormones in relation to lupus disease activity have been the subject of debate for decades. Women are considerably more likely to develop autoimmune diseases than men, suggesting that steroid hormones, especially estrogen and progesterone, influence the immune system.16 Estrogen actions are proinflammatory, while the actions of progesterone, androgens, and glucocorticoids are anti-inflammatory.17 Studies in women with lupus revealed an increased rate of mild- to moderate-intensity disease flares associated with estrogen-containing hormone replace-ment therapy.18-20
Over the years, several antiestrogen therapies have been used in murine models, which showed remarkable clinical improvement in the course of SLE. The precise mechanisms involved in disease immunomodulation by these therapies have not been elucidated.21-23 It is thought that estrogen plays a role in the synthesis and expression of Ro antigens on the surface of keratinocytes, increasing the fixation of anti-Ro antibodies in keratinocytes and provoking the appearance of a cutaneous eruption in patients with a susceptible HLA profile.6
Conclusion
We report a rare case of SCLE induced by anastrozole use. Cases such as ours and others that implicate antiestrogen drugs in association with DI-SCLE are particularly noteworthy, considering many studies are looking at the potential usefulness of antiestrogen therapy in the treatment of SLE. Further research on this relationship is warranted.
- Reed B, Huff J, Jones S, et al. Subacute cutaneous lupus erythematosus associated with hydrochlorothiazide therapy. Ann Intern Med. 1985;103:49-51.
- Marzano A, Lazzari R, Polloni I, et al. Drug-induced subacute cutaneous lupus erythematosus: evidence for differences from its idiopathic counterpart. Br J Dermatol. 2011;165:335-341.
- Bonsmann G, Schiller M, Luger T, et al. Terbinafine-induced subacute cutaneous lupus erythematosus. J Am Acad Dermatol. 2001;44:925-931.
- Callen J. Review: drug induced subacute cutaneous lupus erythematosus. Lupus. 2010;19:1107-1111.
- Lin J, Callen JP. Subacute cutaneous lupus erythematosus (SCLE). Medscape website. http://emedicine.medscape.com/article/1065657-overview. Updated March 7, 2016. Accessed April 29, 2016.
- Lowe GC, Henderson CL, Grau RH, et al. A systematic review of drug-induced subacute cutaneous lupus erythematosus. Br J Dermatol. 2011;164:465-472.
- Vedove C, Giglio M, Schena D, et al. Drug-induced lupus erythematosus. Arch Dermatol Res. 2009;301:99-105.
- Trancart M, Cavailhes A, Balme B, et al. Anastrozole-induced subacute cutaneous lupus erythematosus [published online December 6, 2007]. Br J Dermatol. 2008;158:628-629.
- Fumal I, Danchin A, Cosserat F, et al. Subacute cutaneous lupus erythematosus associated with tamoxifen therapy: two cases. Dermatology. 2005;210:251-252.
- Brown C, Deng J. Thiazide diuretics induce cutaneous lupus-like adverse reaction. J Toxicol Clin Toxicol. 1995;33:729-733.
- Sontheimer R. Subacute cutaneous lupus erythematosus: 25-year evolution of a prototypic subset (subphenotype) of lupus erythematosus defined by characteristic cutaneous, pathological, immunological, and genetic findings. Autoimmun Rev. 2005;4:253-263.
- Crowson A, Magro C. Subacute cutaneous lupus erythematosus arising in the setting of calcium channel blocker therapy. Hum Pathol. 1997;28:67-73.
- Lorentz K, Booken N, Goerdt S, et al. Subacute cutaneous lupus erythematosus induced by terbinafine: case report and review of literature. J Dtsch Dermatol Ges. 2008;6:823-837.
- Kasperkiewicz M, Anemüller W, Angelova-Fischer I, et al. Subacute cutaneous lupus erythematosus associated with terbinafine. Clin Exp Dermatol. 2009;34:403-404.
- Miyagawa S, Okuchi T, Shiomi Y, et al. Subacute cutaneous lupus erythematosus lesions precipitated by griseofulvin. J Am Acad Dermatol. 1989;21:343-346.
- Inman RD. Immunologic sex differences and the female predominance in systemic lupus erythematosus. Arthritis Rheum. 1978;21:849-854.
- Cutolo M, Wilder RL. Different roles of androgens and estrogens in the susceptibility to autoimmune rheumatic diseases. Rheum Dis Clin North Am. 2000;26:825-839.
- Petri M. Sex hormones and systemic lupus erythematosus. Lupus. 2008;17:412-415.
- Lateef A, Petri M. Hormone replacement and contraceptive therapy in autoimmune diseases [published online January 18, 2012]. J Autoimmun. 2012;38:J170-J176.
- Buyon JP, Petri M, Kim MY, et al. The effect of combined estrogen and progesterone hormone replacement therapy on disease activity in systemic lupus erythematosus: a randomized trial. Ann Intern Med. 2005;142:954-962.
- Wu W, Suen J, Lin B, et al. Tamoxifen alleviates disease severity and decreases double negative T cells in autoimmune MRL-lpr/lpr mice. Immunology. 2000;100:110-118.
- Dayan M, Zinger H, Kalush F, et al. The beneficial effects of treatment with tamoxifen and anti-oestradiol antibody on experimental systemic lupus erythematosus are associated with cytokine modulations. Immunology. 1997;90:101-108.
- Sthoeger Z, Zinger H, Mozes E. Beneficial effects of the anti-oestrogen tamoxifen on systemic lupus erythematosus of (NZBxNZW)F1 female mice are associated with specific reduction of IgG3 autoantibodies. Ann Rheum Dis. 2003;62:341-346.
- Reed B, Huff J, Jones S, et al. Subacute cutaneous lupus erythematosus associated with hydrochlorothiazide therapy. Ann Intern Med. 1985;103:49-51.
- Marzano A, Lazzari R, Polloni I, et al. Drug-induced subacute cutaneous lupus erythematosus: evidence for differences from its idiopathic counterpart. Br J Dermatol. 2011;165:335-341.
- Bonsmann G, Schiller M, Luger T, et al. Terbinafine-induced subacute cutaneous lupus erythematosus. J Am Acad Dermatol. 2001;44:925-931.
- Callen J. Review: drug induced subacute cutaneous lupus erythematosus. Lupus. 2010;19:1107-1111.
- Lin J, Callen JP. Subacute cutaneous lupus erythematosus (SCLE). Medscape website. http://emedicine.medscape.com/article/1065657-overview. Updated March 7, 2016. Accessed April 29, 2016.
- Lowe GC, Henderson CL, Grau RH, et al. A systematic review of drug-induced subacute cutaneous lupus erythematosus. Br J Dermatol. 2011;164:465-472.
- Vedove C, Giglio M, Schena D, et al. Drug-induced lupus erythematosus. Arch Dermatol Res. 2009;301:99-105.
- Trancart M, Cavailhes A, Balme B, et al. Anastrozole-induced subacute cutaneous lupus erythematosus [published online December 6, 2007]. Br J Dermatol. 2008;158:628-629.
- Fumal I, Danchin A, Cosserat F, et al. Subacute cutaneous lupus erythematosus associated with tamoxifen therapy: two cases. Dermatology. 2005;210:251-252.
- Brown C, Deng J. Thiazide diuretics induce cutaneous lupus-like adverse reaction. J Toxicol Clin Toxicol. 1995;33:729-733.
- Sontheimer R. Subacute cutaneous lupus erythematosus: 25-year evolution of a prototypic subset (subphenotype) of lupus erythematosus defined by characteristic cutaneous, pathological, immunological, and genetic findings. Autoimmun Rev. 2005;4:253-263.
- Crowson A, Magro C. Subacute cutaneous lupus erythematosus arising in the setting of calcium channel blocker therapy. Hum Pathol. 1997;28:67-73.
- Lorentz K, Booken N, Goerdt S, et al. Subacute cutaneous lupus erythematosus induced by terbinafine: case report and review of literature. J Dtsch Dermatol Ges. 2008;6:823-837.
- Kasperkiewicz M, Anemüller W, Angelova-Fischer I, et al. Subacute cutaneous lupus erythematosus associated with terbinafine. Clin Exp Dermatol. 2009;34:403-404.
- Miyagawa S, Okuchi T, Shiomi Y, et al. Subacute cutaneous lupus erythematosus lesions precipitated by griseofulvin. J Am Acad Dermatol. 1989;21:343-346.
- Inman RD. Immunologic sex differences and the female predominance in systemic lupus erythematosus. Arthritis Rheum. 1978;21:849-854.
- Cutolo M, Wilder RL. Different roles of androgens and estrogens in the susceptibility to autoimmune rheumatic diseases. Rheum Dis Clin North Am. 2000;26:825-839.
- Petri M. Sex hormones and systemic lupus erythematosus. Lupus. 2008;17:412-415.
- Lateef A, Petri M. Hormone replacement and contraceptive therapy in autoimmune diseases [published online January 18, 2012]. J Autoimmun. 2012;38:J170-J176.
- Buyon JP, Petri M, Kim MY, et al. The effect of combined estrogen and progesterone hormone replacement therapy on disease activity in systemic lupus erythematosus: a randomized trial. Ann Intern Med. 2005;142:954-962.
- Wu W, Suen J, Lin B, et al. Tamoxifen alleviates disease severity and decreases double negative T cells in autoimmune MRL-lpr/lpr mice. Immunology. 2000;100:110-118.
- Dayan M, Zinger H, Kalush F, et al. The beneficial effects of treatment with tamoxifen and anti-oestradiol antibody on experimental systemic lupus erythematosus are associated with cytokine modulations. Immunology. 1997;90:101-108.
- Sthoeger Z, Zinger H, Mozes E. Beneficial effects of the anti-oestrogen tamoxifen on systemic lupus erythematosus of (NZBxNZW)F1 female mice are associated with specific reduction of IgG3 autoantibodies. Ann Rheum Dis. 2003;62:341-346.
Practice Points
- There are numerous cases of drug-induced subacute cutaneous lupus erythematosus (DI-SCLE) published in the literature; however, there are limited reports with anastrozole implicated as the causative agent.
- Cases of DI-SCLE are clinically and histologically indistinguishable from idiopathic cases. It is important to recognize and withdraw the offending agent.
Prednisone and Vardenafil Hydrochloride for Refractory Levamisole-Induced Vasculitis
Levamisole is an immunomodulatory drug that had been used to treat various medical conditions, including parasitic infections, nephrotic syndrome, and colorectal cancer,1 before being withdrawn from the US market in 2000. The most common reasons for levamisole discontinuation were leukopenia and rashes (1%–2%),1 many of which included leg ulcers and necrotizing purpura of the ears.1,2 The drug is currently available only as a deworming agent in veterinary medicine.
Since 2007, increasing amounts of levamisole have been used as an adulterant in cocaine. In 2007, less than 10% of cocaine was contaminated with levamisole, with an increase to 77% by 2010.3 In addition, 78% of 249 urine toxicology screens that were positive for cocaine in an inner city hospital also tested positive for levamisole.4 Levamisole-cut cocaine has become a concern because it is associated with a life-threatening syndrome involving a necrotizing purpuric rash, autoantibody production, and leukopenia.5
Levamisole-induced vasculitis is an independent entity from cocaine-induced vasculitis, which is associated with skin findings ranging from palpable purpura and chronic ulcers to digital infarction secondary to its vasospastic activity.6-8 Cocaine-induced vasculopathy has been related to cytoplasmic antineutrophil cytoplasmic antibody positivity and often resembles Wegener granulomatosis.6 Although both cocaine and levamisole have reportedly caused acrally distributed purpura and vasculopathy, levamisole is specifically associated with retiform purpura, ear involvement, and leukopenia.6,9 In addition, levamisole-induced skin reactions have been linked to specific antibodies, including antinuclear, antiphospholipid, and perinuclear antineutrophil cytoplasmic antibody (p-ANCA).2,5-7,9-14
We present a case of refractory levamisole-induced vasculitis and review its clinical presentation, diagnostic approach, laboratory findings, histology, and management. Furthermore, we discuss the possibility of a new treatment option for levamisole-induced vasculitis for patients with refractory disease or for patients who continue to use levamisole.
Case Report
A 49-year-old man with a history of polysubstance abuse presented with intermittent fevers and painful swollen ears as well as joint pain of 3 weeks’ duration. One week after the lesions developed on the ears, similar lesions were seen on the legs, arms, and trunk. He admitted to cocaine use 3 weeks prior to presentation when the symptoms began.
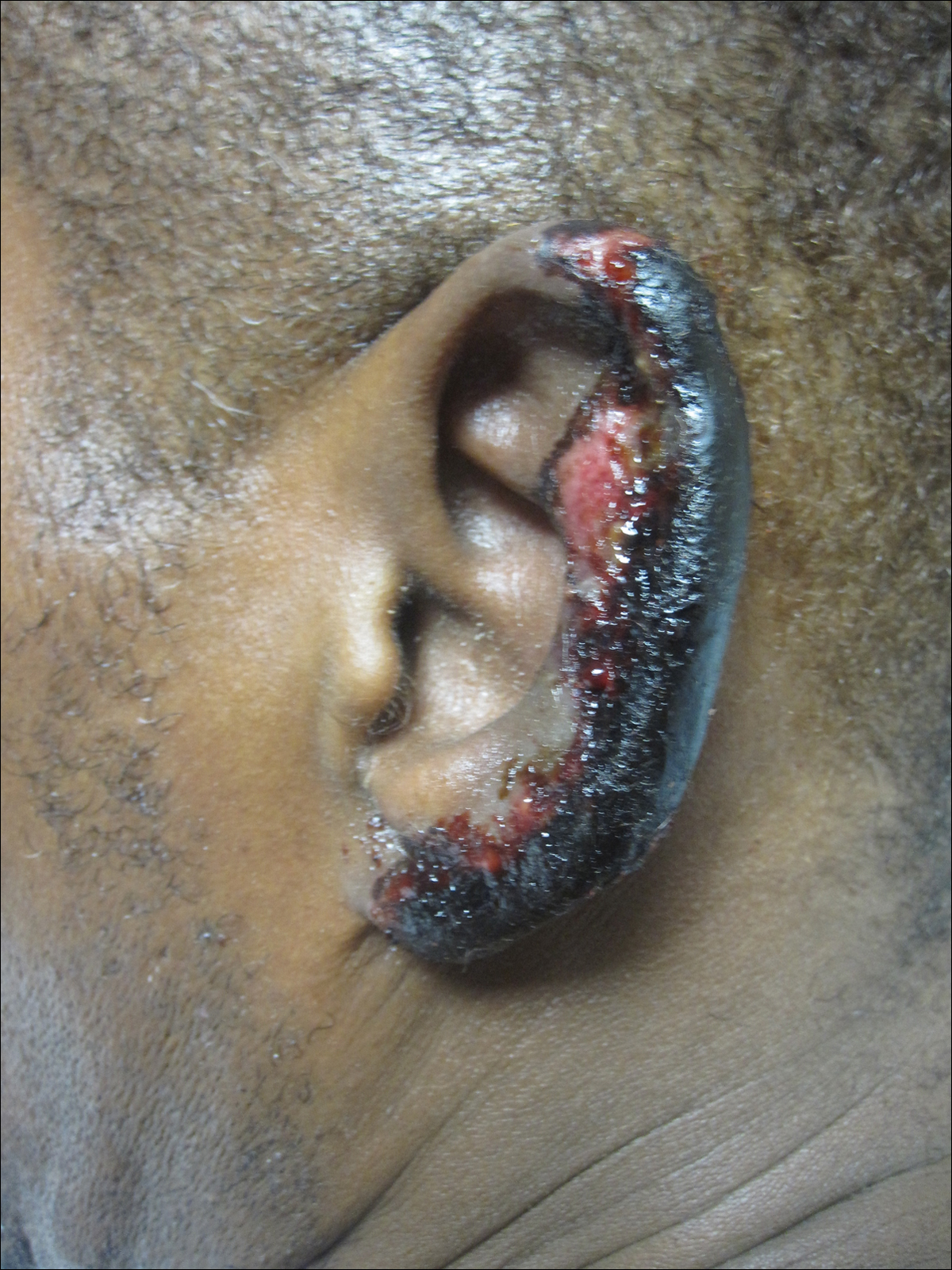
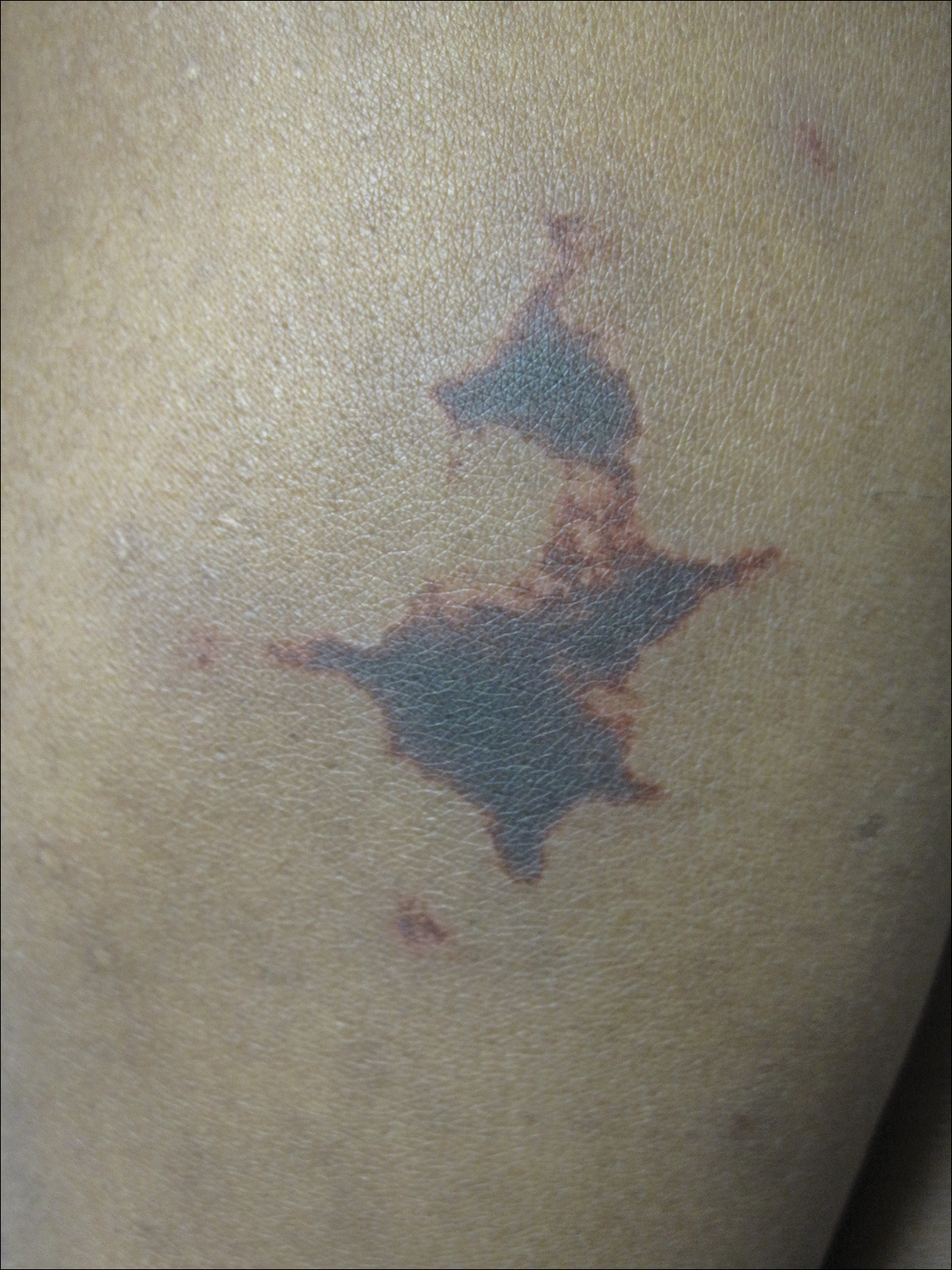
On physical examination, violaceous patches with necrotic bleeding edges and overlying black eschars were noted on the helices, antihelices, and ear lobules bilaterally (Figure 1). Retiform, purpuric to dark brown patches, some with signs of epidermal necrosis, were scattered on the arms, legs, and chest (Figure 2).
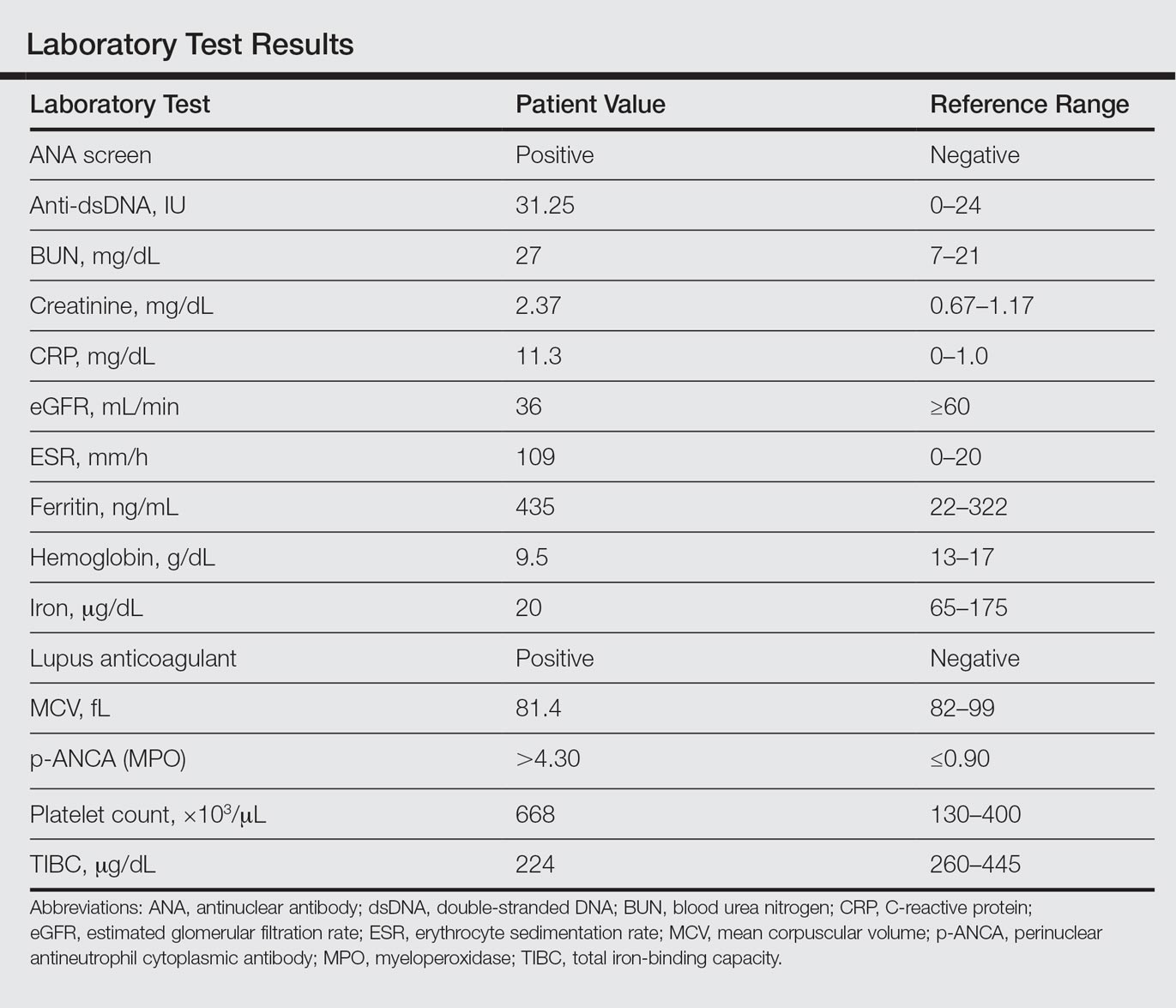
Laboratory examination revealed renal failure, anemia of chronic disease, and thrombocytosis (Table). The patient also screened positive for lupus anticoagulant and antinuclear antibodies and had elevated p-ANCA and anti–double-stranded DNA (Table). He also had an elevated sedimentation rate (109 mm/h [reference range, 0–20 mm/h]) and C-reactive protein level (11.3 mg/dL [reference range, 0–1.0 mg/dL])(Table). Urine toxicology was positive for cocaine.
A punch biopsy of the left thigh was performed on the edge of a retiform purpuric patch. Histopathologic examination revealed epidermal necrosis with subjacent intraluminal vascular thrombi along with extravasated red blood cells and neutrophilic debris (leukocytoclasis) and fibrin in and around vessel walls, consistent with vasculitis (Figure 3).
The patient was admitted to the hospital for pain management and wound care. Despite cocaine cessation and oral prednisone taper, the lesions on the legs worsened over the next several weeks. His condition was further complicated by wound infections, nonhealing ulcers, and subjective fevers and chills requiring frequent hospitalization. The patient was managed by the dermatology department as an outpatient and in clinic between hospital visits. He was treated with antibiotics, ulcer debridement, compression wraps, and aspirin (81 mg once daily) with moderate improvement.
Ten weeks after the first visit, the patient returned with worsening and recurrent leg and ear lesions. He denied any cocaine use since the initial hospital admission; however, a toxicology screen was never obtained. It was decided that the patient would need additional treatment along with traditional trigger (cocaine) avoidance and wound care. Combined treatment with aspirin (81 mg once daily), oral prednisone (40 mg once daily), and vardenafil hydrochloride (20 mg twice weekly) was initiated. At the end of week 1, the patient began to exhibit signs of improvement, which continued over the next 4 weeks. He was then lost to follow-up.
Comment
Our patient presented with severe necrotizing cutaneous vasculitis, likely secondary to levamisole exposure. Some of our patient’s cutaneous findings may be explained exclusively on the basis of cocaine exposure, but the characteristic lesion distribution and histopathologic findings along with the evidence of autoantibody positivity and concurrent arthralgias make the combination of levamisole and cocaine a more likely cause. Similar skin lesions were first described in children treated with levamisole for nephrotic syndrome.2 The most common site of clinical involvement in these children was the ears, as seen in our patient. Our patient tested positive for p-ANCA, which is the most commonly reported autoantibody associated with this patient population. Sixty-one percent (20/33) of patients with levamisole-induced vasculitis from 2 separate reviews showed p-ANCA positivity.7,10
On histopathology, our patient’s skin biopsy findings were consistent with those of prior reports of levamisole-induced vasculitis, which describe patterns of thrombotic vasculitis, leukocytoclasis, and fibrin deposition or occlusive disease.2,6,7,9-14 Mixed histologic findings of vasculitis and thrombosis, usually with varying ages of thrombi, are characteristic of levamisole-induced purpura. In addition, the disease can present nonspecifically with pure microvascular thrombosis without vasculitis, especially later in the course.9
The recommended management of levamisole-induced vasculitis currently involves the withdrawal of the culprit adulterated cocaine along with supportive treatment. Spontaneous and complete clinical resolution of lesions has been reported within 2 to 3 weeks and serology normalization within 2 to 14 months of levamisole cessation.2,6 A 2011 review of patients with levamisole-induced vasculitis reported 66% (19/29) of cases with either full cutaneous resolution after levamisole withdrawal or recurrence with resumed use, supporting a causal relationship.7 Walsh et al9 described 2 patients with recurrent and exacerbated retiform purpura following cocaine binges. Both of these patients had urine samples that tested positive for levamisole.9 In more severe cases, medications shown to be effective include colchicine, polidocanol, antibiotics, methotrexate, anticoagulants, and most commonly systemic corticosteroids.7,10,11,15 Nonsteroidal anti-inflammatory drugs were successful in treating lesions in 2 patients with concurrent arthralgia.7 Rarely, patients have required surgical debridement or skin grafting due to advanced disease at initial presentation.9,12-14 One of the most severe cases of levamisole-induced vasculitis reported in the literature involved 52% of the patient’s total body surface area with skin, soft tissue, and bony necrosis requiring nasal amputation, upper lip excision, skin grafting, and extremity amputation.14 Another severe case with widespread skin involvement was recently reported.16
For unclear reasons, our patient continued to develop cutaneous lesions despite self-reported cocaine cessation. Complete resolution required the combination of vardenafil, prednisone, and aspirin, along with debridement and wound care. Vardenafil, a selective phosphodiesterase 5 inhibitor, enhances the effect of nitrous oxide by increasing levels of cyclic guanosine monophosphate,17 which results in smooth muscle relaxation and vasodilatation. The primary indication for vardenafil is the treatment of erectile dysfunction, but it often is used off label in diseases that may benefit from vasodilatation. Because of its mechanism of action, it is understandable that a vasodilator such as vardenafil could be therapeutic in a condition associated with thrombosis. Moreover, the autoinflammatory nature of levamisole-induced vasculitis makes corticosteroid treatment effective. Given the 10-week delay in improvement, we suspect that it was the combination of treatment or an individual agent that led to our patient’s eventual recovery.
There are few reports in the literature focusing on optimal treatment of levamisole-induced vasculitis and none that mention alternative management for patients who continue to develop new lesions despite cocaine avoidance. Although the discontinuation of levamisole seems to be imperative for resolution of cutaneous lesions, it may not always be enough. It is possible that there is a subpopulation of patients that may not respond to the simple withdrawal of cocaine. It also should be mentioned that there was no urine toxicology screen obtained to support our patient’s reported cocaine cessation. Therefore, it is possible that his worsening condition was secondary to continued cocaine use. However, the patient successfully responded to the combination of vardenafil and prednisone, regardless of whether his condition persisted due to continued use of cocaine or not. This case suggests the possibility of a new treatment option for levamisole-induced vasculitis for patients who continue to use levamisole despite instruction for cessation or for patients with refractory disease.
Conclusion
A trial of prednisone and vardenafil should be considered for patients with refractory levamisole-induced vasculitis. Further studies and discussions of disease course are needed to identify the best treatment of this skin condition, especially for patients with refractory lesions.
- Scheinfeld N, Rosenberg JD, Weinberg JM. Levamisole in dermatology: a review. Am J Clin Dermatol. 2004;5:97-104.
- Rongioletti F, Ghio L, Ginevri F, et al. Purpura of the ears: a distinctive vasculopathy with circulating autoantibodies complicating long-term treatment with levamisole in children. Br J Dermatol. 1999;140:948-951.
- National Drug Threat Assessment 2011. US Department of Justice National Drug Intelligence Center website. https://www.justice.gov/archive/ndic/pubs44/44849/44849p.pdf. Published August 2011. Accessed August 7, 2016.
- Buchanan JA, Heard K, Burbach C, et al. Prevalence of levamisole in urine toxicology screens positive for cocaine in an inner-city hospital. JAMA. 2011;305:1657-1658.
- Gross RL, Brucker J, Bahce-Altuntas A, et al. A novel cutaneous vasculitis syndrome induced by levamisole-contaminated cocaine. Clin Rheumatol. 2011;30:1385-1392.
- Waller JM, Feramisco JD, Alberta-Wszolek L, et al. Cocaine-associated retiform purpura and neutropenia: is levamisole the culprit? J Am Acad Dermatol. 2010;63:530-535.
- Poon SH, Baliog CR, Sams RN, et al. Syndrome of cocaine-levamisole-induced cutaneous vasculitis and immune-mediated leukopenia. Semin Arthritis Rheum. 2011;41:434-444.
- Brewer JD, Meves A, Bostwick JM, et al. Cocaine abuse: dermatologic manifestations and therapeutic approaches. J Am Acad Dermatol. 2008;59:483-487.
- Walsh NMG, Green PJ, Burlingame RW, et al. Cocaine-related retiform purpura: evidence to incriminate the adulterant, levamisole. J Cutan Pathol. 2010;37:1212-1219.
- Chung C, Tumeh PC, Birnbaum R, et al. Characteristic purpura of the ears, vasculitis, and neutropenia—a potential public health epidemic associated with levamisole adultered cocaine. J Am Acad Dermatol. 2011;65:722-725.
- Kahn TA, Cuchacovich R, Espinoza LR, et al. Vasculopathy, hematological, and immune abnormalities associated with levamisole-contaminated cocaine use. Semin Arthritis Rheum. 2011;41:445-454.
- Graf J, Lynch K, Yeh CL, et al. Purpura, cutaneous necrosis, and antineutrophil cytoplasmic antibodies associated with levamisole-adulterated cocaine. Arthritis Rheum. 2011;63:3998-4001.
- Farmer RW, Malhotra PS, Mays MP, et al. Necrotizing peripheral vasculitis/vasculopathy following the use of cocaine laced with levamisole. J Burn Care Res. 2012;33:e6-e11.
- Ching JA, Smith DJ Jr. Levamisole-induced skin necrosis of skin, soft tissue, and bone: case report and review of literature. J Burn Care Res. 2012;33:e1-e5.
- Buchanan JA, Vogel JA, Eberhardt AM. Levamisole-induced occlusive necrotizing vasculitis of the ears after use of cocaine contaminated with levamisole. J Med Toxicol. 2011;7:83-84.
- Graff N, Whitworth K, Trigger C. Purpuric skin eruption in an illicit drug user: levamisole-induced vasculitis. Am J Emer Med. 2016;34:1321.
- Schwartz BG, Kloner RA. Drug interactions with phosphodiesterase-5 inhibitors used for the treatment of erectile dysfunction or pulmonary hypertension. Circulation. 2010;122:88-95.
Levamisole is an immunomodulatory drug that had been used to treat various medical conditions, including parasitic infections, nephrotic syndrome, and colorectal cancer,1 before being withdrawn from the US market in 2000. The most common reasons for levamisole discontinuation were leukopenia and rashes (1%–2%),1 many of which included leg ulcers and necrotizing purpura of the ears.1,2 The drug is currently available only as a deworming agent in veterinary medicine.
Since 2007, increasing amounts of levamisole have been used as an adulterant in cocaine. In 2007, less than 10% of cocaine was contaminated with levamisole, with an increase to 77% by 2010.3 In addition, 78% of 249 urine toxicology screens that were positive for cocaine in an inner city hospital also tested positive for levamisole.4 Levamisole-cut cocaine has become a concern because it is associated with a life-threatening syndrome involving a necrotizing purpuric rash, autoantibody production, and leukopenia.5
Levamisole-induced vasculitis is an independent entity from cocaine-induced vasculitis, which is associated with skin findings ranging from palpable purpura and chronic ulcers to digital infarction secondary to its vasospastic activity.6-8 Cocaine-induced vasculopathy has been related to cytoplasmic antineutrophil cytoplasmic antibody positivity and often resembles Wegener granulomatosis.6 Although both cocaine and levamisole have reportedly caused acrally distributed purpura and vasculopathy, levamisole is specifically associated with retiform purpura, ear involvement, and leukopenia.6,9 In addition, levamisole-induced skin reactions have been linked to specific antibodies, including antinuclear, antiphospholipid, and perinuclear antineutrophil cytoplasmic antibody (p-ANCA).2,5-7,9-14
We present a case of refractory levamisole-induced vasculitis and review its clinical presentation, diagnostic approach, laboratory findings, histology, and management. Furthermore, we discuss the possibility of a new treatment option for levamisole-induced vasculitis for patients with refractory disease or for patients who continue to use levamisole.
Case Report
A 49-year-old man with a history of polysubstance abuse presented with intermittent fevers and painful swollen ears as well as joint pain of 3 weeks’ duration. One week after the lesions developed on the ears, similar lesions were seen on the legs, arms, and trunk. He admitted to cocaine use 3 weeks prior to presentation when the symptoms began.
On physical examination, violaceous patches with necrotic bleeding edges and overlying black eschars were noted on the helices, antihelices, and ear lobules bilaterally (Figure 1). Retiform, purpuric to dark brown patches, some with signs of epidermal necrosis, were scattered on the arms, legs, and chest (Figure 2).
Laboratory examination revealed renal failure, anemia of chronic disease, and thrombocytosis (Table). The patient also screened positive for lupus anticoagulant and antinuclear antibodies and had elevated p-ANCA and anti–double-stranded DNA (Table). He also had an elevated sedimentation rate (109 mm/h [reference range, 0–20 mm/h]) and C-reactive protein level (11.3 mg/dL [reference range, 0–1.0 mg/dL])(Table). Urine toxicology was positive for cocaine.
A punch biopsy of the left thigh was performed on the edge of a retiform purpuric patch. Histopathologic examination revealed epidermal necrosis with subjacent intraluminal vascular thrombi along with extravasated red blood cells and neutrophilic debris (leukocytoclasis) and fibrin in and around vessel walls, consistent with vasculitis (Figure 3).
The patient was admitted to the hospital for pain management and wound care. Despite cocaine cessation and oral prednisone taper, the lesions on the legs worsened over the next several weeks. His condition was further complicated by wound infections, nonhealing ulcers, and subjective fevers and chills requiring frequent hospitalization. The patient was managed by the dermatology department as an outpatient and in clinic between hospital visits. He was treated with antibiotics, ulcer debridement, compression wraps, and aspirin (81 mg once daily) with moderate improvement.
Ten weeks after the first visit, the patient returned with worsening and recurrent leg and ear lesions. He denied any cocaine use since the initial hospital admission; however, a toxicology screen was never obtained. It was decided that the patient would need additional treatment along with traditional trigger (cocaine) avoidance and wound care. Combined treatment with aspirin (81 mg once daily), oral prednisone (40 mg once daily), and vardenafil hydrochloride (20 mg twice weekly) was initiated. At the end of week 1, the patient began to exhibit signs of improvement, which continued over the next 4 weeks. He was then lost to follow-up.
Comment
Our patient presented with severe necrotizing cutaneous vasculitis, likely secondary to levamisole exposure. Some of our patient’s cutaneous findings may be explained exclusively on the basis of cocaine exposure, but the characteristic lesion distribution and histopathologic findings along with the evidence of autoantibody positivity and concurrent arthralgias make the combination of levamisole and cocaine a more likely cause. Similar skin lesions were first described in children treated with levamisole for nephrotic syndrome.2 The most common site of clinical involvement in these children was the ears, as seen in our patient. Our patient tested positive for p-ANCA, which is the most commonly reported autoantibody associated with this patient population. Sixty-one percent (20/33) of patients with levamisole-induced vasculitis from 2 separate reviews showed p-ANCA positivity.7,10
On histopathology, our patient’s skin biopsy findings were consistent with those of prior reports of levamisole-induced vasculitis, which describe patterns of thrombotic vasculitis, leukocytoclasis, and fibrin deposition or occlusive disease.2,6,7,9-14 Mixed histologic findings of vasculitis and thrombosis, usually with varying ages of thrombi, are characteristic of levamisole-induced purpura. In addition, the disease can present nonspecifically with pure microvascular thrombosis without vasculitis, especially later in the course.9
The recommended management of levamisole-induced vasculitis currently involves the withdrawal of the culprit adulterated cocaine along with supportive treatment. Spontaneous and complete clinical resolution of lesions has been reported within 2 to 3 weeks and serology normalization within 2 to 14 months of levamisole cessation.2,6 A 2011 review of patients with levamisole-induced vasculitis reported 66% (19/29) of cases with either full cutaneous resolution after levamisole withdrawal or recurrence with resumed use, supporting a causal relationship.7 Walsh et al9 described 2 patients with recurrent and exacerbated retiform purpura following cocaine binges. Both of these patients had urine samples that tested positive for levamisole.9 In more severe cases, medications shown to be effective include colchicine, polidocanol, antibiotics, methotrexate, anticoagulants, and most commonly systemic corticosteroids.7,10,11,15 Nonsteroidal anti-inflammatory drugs were successful in treating lesions in 2 patients with concurrent arthralgia.7 Rarely, patients have required surgical debridement or skin grafting due to advanced disease at initial presentation.9,12-14 One of the most severe cases of levamisole-induced vasculitis reported in the literature involved 52% of the patient’s total body surface area with skin, soft tissue, and bony necrosis requiring nasal amputation, upper lip excision, skin grafting, and extremity amputation.14 Another severe case with widespread skin involvement was recently reported.16
For unclear reasons, our patient continued to develop cutaneous lesions despite self-reported cocaine cessation. Complete resolution required the combination of vardenafil, prednisone, and aspirin, along with debridement and wound care. Vardenafil, a selective phosphodiesterase 5 inhibitor, enhances the effect of nitrous oxide by increasing levels of cyclic guanosine monophosphate,17 which results in smooth muscle relaxation and vasodilatation. The primary indication for vardenafil is the treatment of erectile dysfunction, but it often is used off label in diseases that may benefit from vasodilatation. Because of its mechanism of action, it is understandable that a vasodilator such as vardenafil could be therapeutic in a condition associated with thrombosis. Moreover, the autoinflammatory nature of levamisole-induced vasculitis makes corticosteroid treatment effective. Given the 10-week delay in improvement, we suspect that it was the combination of treatment or an individual agent that led to our patient’s eventual recovery.
There are few reports in the literature focusing on optimal treatment of levamisole-induced vasculitis and none that mention alternative management for patients who continue to develop new lesions despite cocaine avoidance. Although the discontinuation of levamisole seems to be imperative for resolution of cutaneous lesions, it may not always be enough. It is possible that there is a subpopulation of patients that may not respond to the simple withdrawal of cocaine. It also should be mentioned that there was no urine toxicology screen obtained to support our patient’s reported cocaine cessation. Therefore, it is possible that his worsening condition was secondary to continued cocaine use. However, the patient successfully responded to the combination of vardenafil and prednisone, regardless of whether his condition persisted due to continued use of cocaine or not. This case suggests the possibility of a new treatment option for levamisole-induced vasculitis for patients who continue to use levamisole despite instruction for cessation or for patients with refractory disease.
Conclusion
A trial of prednisone and vardenafil should be considered for patients with refractory levamisole-induced vasculitis. Further studies and discussions of disease course are needed to identify the best treatment of this skin condition, especially for patients with refractory lesions.
Levamisole is an immunomodulatory drug that had been used to treat various medical conditions, including parasitic infections, nephrotic syndrome, and colorectal cancer,1 before being withdrawn from the US market in 2000. The most common reasons for levamisole discontinuation were leukopenia and rashes (1%–2%),1 many of which included leg ulcers and necrotizing purpura of the ears.1,2 The drug is currently available only as a deworming agent in veterinary medicine.
Since 2007, increasing amounts of levamisole have been used as an adulterant in cocaine. In 2007, less than 10% of cocaine was contaminated with levamisole, with an increase to 77% by 2010.3 In addition, 78% of 249 urine toxicology screens that were positive for cocaine in an inner city hospital also tested positive for levamisole.4 Levamisole-cut cocaine has become a concern because it is associated with a life-threatening syndrome involving a necrotizing purpuric rash, autoantibody production, and leukopenia.5
Levamisole-induced vasculitis is an independent entity from cocaine-induced vasculitis, which is associated with skin findings ranging from palpable purpura and chronic ulcers to digital infarction secondary to its vasospastic activity.6-8 Cocaine-induced vasculopathy has been related to cytoplasmic antineutrophil cytoplasmic antibody positivity and often resembles Wegener granulomatosis.6 Although both cocaine and levamisole have reportedly caused acrally distributed purpura and vasculopathy, levamisole is specifically associated with retiform purpura, ear involvement, and leukopenia.6,9 In addition, levamisole-induced skin reactions have been linked to specific antibodies, including antinuclear, antiphospholipid, and perinuclear antineutrophil cytoplasmic antibody (p-ANCA).2,5-7,9-14
We present a case of refractory levamisole-induced vasculitis and review its clinical presentation, diagnostic approach, laboratory findings, histology, and management. Furthermore, we discuss the possibility of a new treatment option for levamisole-induced vasculitis for patients with refractory disease or for patients who continue to use levamisole.
Case Report
A 49-year-old man with a history of polysubstance abuse presented with intermittent fevers and painful swollen ears as well as joint pain of 3 weeks’ duration. One week after the lesions developed on the ears, similar lesions were seen on the legs, arms, and trunk. He admitted to cocaine use 3 weeks prior to presentation when the symptoms began.
On physical examination, violaceous patches with necrotic bleeding edges and overlying black eschars were noted on the helices, antihelices, and ear lobules bilaterally (Figure 1). Retiform, purpuric to dark brown patches, some with signs of epidermal necrosis, were scattered on the arms, legs, and chest (Figure 2).
Laboratory examination revealed renal failure, anemia of chronic disease, and thrombocytosis (Table). The patient also screened positive for lupus anticoagulant and antinuclear antibodies and had elevated p-ANCA and anti–double-stranded DNA (Table). He also had an elevated sedimentation rate (109 mm/h [reference range, 0–20 mm/h]) and C-reactive protein level (11.3 mg/dL [reference range, 0–1.0 mg/dL])(Table). Urine toxicology was positive for cocaine.
A punch biopsy of the left thigh was performed on the edge of a retiform purpuric patch. Histopathologic examination revealed epidermal necrosis with subjacent intraluminal vascular thrombi along with extravasated red blood cells and neutrophilic debris (leukocytoclasis) and fibrin in and around vessel walls, consistent with vasculitis (Figure 3).
The patient was admitted to the hospital for pain management and wound care. Despite cocaine cessation and oral prednisone taper, the lesions on the legs worsened over the next several weeks. His condition was further complicated by wound infections, nonhealing ulcers, and subjective fevers and chills requiring frequent hospitalization. The patient was managed by the dermatology department as an outpatient and in clinic between hospital visits. He was treated with antibiotics, ulcer debridement, compression wraps, and aspirin (81 mg once daily) with moderate improvement.
Ten weeks after the first visit, the patient returned with worsening and recurrent leg and ear lesions. He denied any cocaine use since the initial hospital admission; however, a toxicology screen was never obtained. It was decided that the patient would need additional treatment along with traditional trigger (cocaine) avoidance and wound care. Combined treatment with aspirin (81 mg once daily), oral prednisone (40 mg once daily), and vardenafil hydrochloride (20 mg twice weekly) was initiated. At the end of week 1, the patient began to exhibit signs of improvement, which continued over the next 4 weeks. He was then lost to follow-up.
Comment
Our patient presented with severe necrotizing cutaneous vasculitis, likely secondary to levamisole exposure. Some of our patient’s cutaneous findings may be explained exclusively on the basis of cocaine exposure, but the characteristic lesion distribution and histopathologic findings along with the evidence of autoantibody positivity and concurrent arthralgias make the combination of levamisole and cocaine a more likely cause. Similar skin lesions were first described in children treated with levamisole for nephrotic syndrome.2 The most common site of clinical involvement in these children was the ears, as seen in our patient. Our patient tested positive for p-ANCA, which is the most commonly reported autoantibody associated with this patient population. Sixty-one percent (20/33) of patients with levamisole-induced vasculitis from 2 separate reviews showed p-ANCA positivity.7,10
On histopathology, our patient’s skin biopsy findings were consistent with those of prior reports of levamisole-induced vasculitis, which describe patterns of thrombotic vasculitis, leukocytoclasis, and fibrin deposition or occlusive disease.2,6,7,9-14 Mixed histologic findings of vasculitis and thrombosis, usually with varying ages of thrombi, are characteristic of levamisole-induced purpura. In addition, the disease can present nonspecifically with pure microvascular thrombosis without vasculitis, especially later in the course.9
The recommended management of levamisole-induced vasculitis currently involves the withdrawal of the culprit adulterated cocaine along with supportive treatment. Spontaneous and complete clinical resolution of lesions has been reported within 2 to 3 weeks and serology normalization within 2 to 14 months of levamisole cessation.2,6 A 2011 review of patients with levamisole-induced vasculitis reported 66% (19/29) of cases with either full cutaneous resolution after levamisole withdrawal or recurrence with resumed use, supporting a causal relationship.7 Walsh et al9 described 2 patients with recurrent and exacerbated retiform purpura following cocaine binges. Both of these patients had urine samples that tested positive for levamisole.9 In more severe cases, medications shown to be effective include colchicine, polidocanol, antibiotics, methotrexate, anticoagulants, and most commonly systemic corticosteroids.7,10,11,15 Nonsteroidal anti-inflammatory drugs were successful in treating lesions in 2 patients with concurrent arthralgia.7 Rarely, patients have required surgical debridement or skin grafting due to advanced disease at initial presentation.9,12-14 One of the most severe cases of levamisole-induced vasculitis reported in the literature involved 52% of the patient’s total body surface area with skin, soft tissue, and bony necrosis requiring nasal amputation, upper lip excision, skin grafting, and extremity amputation.14 Another severe case with widespread skin involvement was recently reported.16
For unclear reasons, our patient continued to develop cutaneous lesions despite self-reported cocaine cessation. Complete resolution required the combination of vardenafil, prednisone, and aspirin, along with debridement and wound care. Vardenafil, a selective phosphodiesterase 5 inhibitor, enhances the effect of nitrous oxide by increasing levels of cyclic guanosine monophosphate,17 which results in smooth muscle relaxation and vasodilatation. The primary indication for vardenafil is the treatment of erectile dysfunction, but it often is used off label in diseases that may benefit from vasodilatation. Because of its mechanism of action, it is understandable that a vasodilator such as vardenafil could be therapeutic in a condition associated with thrombosis. Moreover, the autoinflammatory nature of levamisole-induced vasculitis makes corticosteroid treatment effective. Given the 10-week delay in improvement, we suspect that it was the combination of treatment or an individual agent that led to our patient’s eventual recovery.
There are few reports in the literature focusing on optimal treatment of levamisole-induced vasculitis and none that mention alternative management for patients who continue to develop new lesions despite cocaine avoidance. Although the discontinuation of levamisole seems to be imperative for resolution of cutaneous lesions, it may not always be enough. It is possible that there is a subpopulation of patients that may not respond to the simple withdrawal of cocaine. It also should be mentioned that there was no urine toxicology screen obtained to support our patient’s reported cocaine cessation. Therefore, it is possible that his worsening condition was secondary to continued cocaine use. However, the patient successfully responded to the combination of vardenafil and prednisone, regardless of whether his condition persisted due to continued use of cocaine or not. This case suggests the possibility of a new treatment option for levamisole-induced vasculitis for patients who continue to use levamisole despite instruction for cessation or for patients with refractory disease.
Conclusion
A trial of prednisone and vardenafil should be considered for patients with refractory levamisole-induced vasculitis. Further studies and discussions of disease course are needed to identify the best treatment of this skin condition, especially for patients with refractory lesions.
- Scheinfeld N, Rosenberg JD, Weinberg JM. Levamisole in dermatology: a review. Am J Clin Dermatol. 2004;5:97-104.
- Rongioletti F, Ghio L, Ginevri F, et al. Purpura of the ears: a distinctive vasculopathy with circulating autoantibodies complicating long-term treatment with levamisole in children. Br J Dermatol. 1999;140:948-951.
- National Drug Threat Assessment 2011. US Department of Justice National Drug Intelligence Center website. https://www.justice.gov/archive/ndic/pubs44/44849/44849p.pdf. Published August 2011. Accessed August 7, 2016.
- Buchanan JA, Heard K, Burbach C, et al. Prevalence of levamisole in urine toxicology screens positive for cocaine in an inner-city hospital. JAMA. 2011;305:1657-1658.
- Gross RL, Brucker J, Bahce-Altuntas A, et al. A novel cutaneous vasculitis syndrome induced by levamisole-contaminated cocaine. Clin Rheumatol. 2011;30:1385-1392.
- Waller JM, Feramisco JD, Alberta-Wszolek L, et al. Cocaine-associated retiform purpura and neutropenia: is levamisole the culprit? J Am Acad Dermatol. 2010;63:530-535.
- Poon SH, Baliog CR, Sams RN, et al. Syndrome of cocaine-levamisole-induced cutaneous vasculitis and immune-mediated leukopenia. Semin Arthritis Rheum. 2011;41:434-444.
- Brewer JD, Meves A, Bostwick JM, et al. Cocaine abuse: dermatologic manifestations and therapeutic approaches. J Am Acad Dermatol. 2008;59:483-487.
- Walsh NMG, Green PJ, Burlingame RW, et al. Cocaine-related retiform purpura: evidence to incriminate the adulterant, levamisole. J Cutan Pathol. 2010;37:1212-1219.
- Chung C, Tumeh PC, Birnbaum R, et al. Characteristic purpura of the ears, vasculitis, and neutropenia—a potential public health epidemic associated with levamisole adultered cocaine. J Am Acad Dermatol. 2011;65:722-725.
- Kahn TA, Cuchacovich R, Espinoza LR, et al. Vasculopathy, hematological, and immune abnormalities associated with levamisole-contaminated cocaine use. Semin Arthritis Rheum. 2011;41:445-454.
- Graf J, Lynch K, Yeh CL, et al. Purpura, cutaneous necrosis, and antineutrophil cytoplasmic antibodies associated with levamisole-adulterated cocaine. Arthritis Rheum. 2011;63:3998-4001.
- Farmer RW, Malhotra PS, Mays MP, et al. Necrotizing peripheral vasculitis/vasculopathy following the use of cocaine laced with levamisole. J Burn Care Res. 2012;33:e6-e11.
- Ching JA, Smith DJ Jr. Levamisole-induced skin necrosis of skin, soft tissue, and bone: case report and review of literature. J Burn Care Res. 2012;33:e1-e5.
- Buchanan JA, Vogel JA, Eberhardt AM. Levamisole-induced occlusive necrotizing vasculitis of the ears after use of cocaine contaminated with levamisole. J Med Toxicol. 2011;7:83-84.
- Graff N, Whitworth K, Trigger C. Purpuric skin eruption in an illicit drug user: levamisole-induced vasculitis. Am J Emer Med. 2016;34:1321.
- Schwartz BG, Kloner RA. Drug interactions with phosphodiesterase-5 inhibitors used for the treatment of erectile dysfunction or pulmonary hypertension. Circulation. 2010;122:88-95.
- Scheinfeld N, Rosenberg JD, Weinberg JM. Levamisole in dermatology: a review. Am J Clin Dermatol. 2004;5:97-104.
- Rongioletti F, Ghio L, Ginevri F, et al. Purpura of the ears: a distinctive vasculopathy with circulating autoantibodies complicating long-term treatment with levamisole in children. Br J Dermatol. 1999;140:948-951.
- National Drug Threat Assessment 2011. US Department of Justice National Drug Intelligence Center website. https://www.justice.gov/archive/ndic/pubs44/44849/44849p.pdf. Published August 2011. Accessed August 7, 2016.
- Buchanan JA, Heard K, Burbach C, et al. Prevalence of levamisole in urine toxicology screens positive for cocaine in an inner-city hospital. JAMA. 2011;305:1657-1658.
- Gross RL, Brucker J, Bahce-Altuntas A, et al. A novel cutaneous vasculitis syndrome induced by levamisole-contaminated cocaine. Clin Rheumatol. 2011;30:1385-1392.
- Waller JM, Feramisco JD, Alberta-Wszolek L, et al. Cocaine-associated retiform purpura and neutropenia: is levamisole the culprit? J Am Acad Dermatol. 2010;63:530-535.
- Poon SH, Baliog CR, Sams RN, et al. Syndrome of cocaine-levamisole-induced cutaneous vasculitis and immune-mediated leukopenia. Semin Arthritis Rheum. 2011;41:434-444.
- Brewer JD, Meves A, Bostwick JM, et al. Cocaine abuse: dermatologic manifestations and therapeutic approaches. J Am Acad Dermatol. 2008;59:483-487.
- Walsh NMG, Green PJ, Burlingame RW, et al. Cocaine-related retiform purpura: evidence to incriminate the adulterant, levamisole. J Cutan Pathol. 2010;37:1212-1219.
- Chung C, Tumeh PC, Birnbaum R, et al. Characteristic purpura of the ears, vasculitis, and neutropenia—a potential public health epidemic associated with levamisole adultered cocaine. J Am Acad Dermatol. 2011;65:722-725.
- Kahn TA, Cuchacovich R, Espinoza LR, et al. Vasculopathy, hematological, and immune abnormalities associated with levamisole-contaminated cocaine use. Semin Arthritis Rheum. 2011;41:445-454.
- Graf J, Lynch K, Yeh CL, et al. Purpura, cutaneous necrosis, and antineutrophil cytoplasmic antibodies associated with levamisole-adulterated cocaine. Arthritis Rheum. 2011;63:3998-4001.
- Farmer RW, Malhotra PS, Mays MP, et al. Necrotizing peripheral vasculitis/vasculopathy following the use of cocaine laced with levamisole. J Burn Care Res. 2012;33:e6-e11.
- Ching JA, Smith DJ Jr. Levamisole-induced skin necrosis of skin, soft tissue, and bone: case report and review of literature. J Burn Care Res. 2012;33:e1-e5.
- Buchanan JA, Vogel JA, Eberhardt AM. Levamisole-induced occlusive necrotizing vasculitis of the ears after use of cocaine contaminated with levamisole. J Med Toxicol. 2011;7:83-84.
- Graff N, Whitworth K, Trigger C. Purpuric skin eruption in an illicit drug user: levamisole-induced vasculitis. Am J Emer Med. 2016;34:1321.
- Schwartz BG, Kloner RA. Drug interactions with phosphodiesterase-5 inhibitors used for the treatment of erectile dysfunction or pulmonary hypertension. Circulation. 2010;122:88-95.
Practice Points
- Levamisole is an immunomodulatory drug that, before being withdrawn from the US market in 2000, was previously used to treat various medical conditions.
- A majority of the cocaine in the United States is contaminated with levamisole, which is added as an adulterant or bulking agent.
- Levamisole-cut cocaine is a concern because it is associated with a life-threatening syndrome involving a necrotizing purpuric rash, autoantibody production, and leukopenia.
- Although treatment of levamisole toxicity is primarily supportive and includes cessation of levamisole-cut cocaine, a trial of prednisone and vardenafil hydrochloride can be considered for refractory levamisole-induced vasculopathy or for patients who continue to use the drug.
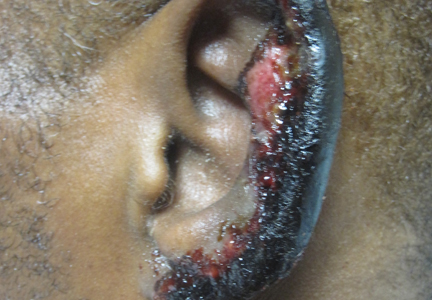
Subungual Onycholemmal Cyst of the Toenail Mimicking Subungual Melanoma
Case Report
A 23-year-old woman presented with a horizontal split along the midline of the right great toenail associated with some tenderness of 2 to 3 months’ duration. Approximately 5 years prior, she noticed a bluish-colored area under the nail that had been steadily increasing in size. She denied a history of trauma, drainage, or bleeding. There was no history of other nail abnormalities. Her medications and personal, family, and social history were noncontributory.
Physical examination of the right great toenail revealed a horizontal split of the nail plate with a bluish hue visible under the nail plate (Figure 1A). The remaining toenails and fingernails were normal. A punch biopsy of the nail bed was performed with a presumptive clinical diagnosis of subungual melanoma versus melanocytic nevus versus cyst (Figure 1B). Nail plate avulsion revealed a blackened nail bed dotted with areas of bluish color and a red friable nodule present focally. Upon further inspection, extension was apparent into the distal matrix.
Histopathologic examination revealed a cystic structure with an epithelial lining mostly reminiscent of an isthmus catagen cyst admixed with the presence of both an intermittent focal granular layer and an eosinophilic cuticle surrounding pink laminated keratin, most consistent with a diagnosis of subungual onycholemmal cyst (SOC)(Figure 2). A reexcision was performed with removal of half of the nail bed, including a portion of the distal matrix extending inferiorly to the bone. Variably sized, epithelium-lined, keratin-filled cystic structures emanated from the nail bed epithelium. There were foci of hemorrhage and granulation tissue secondary to cyst rupture (Figure 3). The defect healed by secondary intention. No clinical evidence of recurrence was seen at 6-month follow-up.

Subungual onycholemmal cysts, also known as subungual epidermoid cysts or subungual epidermoid inclusions, are rare and distinctive nail abnormalities occurring in the dermis of the nail bed. We present a case of an SOC in a toenail mimicking subungual malignant melanoma.
Originally described by Samman1 in 1959, SOCs were attributed to trauma to the nail with resultant implantation of the epidermis into the deeper tissue. Lewin2,3 examined 90 postmortem fingernail and nail bed samples and found 8 subungual epidermoid cysts associated with clubbing of the fingernails. He postulated that the early pathogenesis of clubbing involved dermal fibroblast proliferation in the nail bed, leading to sequestration of nail bed epithelium into the dermis with resultant cyst formation. Microscopic subungual cysts also were identified in normal-appearing nails without evidence of trauma, thought to have arisen from the tips of the nail bed rete ridges by a process of bulbous proliferation rather than sequestration. These findings in normal nails suggest that SOCs may represent a more common entity than previously recognized.
It is imperative to recognize the presence of nail unit tumors early because of the risk for permanent nail plate dystrophy and the possibility of a malignant tumor.4,5 Subungual onycholemmal cysts may present with a wide spectrum of clinical findings including marked subungual hyperkeratosis, onychodystrophy, ridging, nail bed pigmentation, clubbing, thickening, or less often a normal-appearing nail. Based on reported cases, several trends are evident. Although nail dystrophy is most often asymptomatic, pain is not uncommon.5,6 It most commonly involves single digits, predominantly thumbs and great toenails.7,8 This predilection suggests that trauma or other local factors may be involved in its pathogenesis. Of note, trauma to the nail may occur years before the development of the lesions or it may not be recalled at all.
Diagnosis requires a degree of clinical suspicion and a nail bed biopsy with partial or total nail plate avulsion to visualize the pathologic portion of the nail bed. Because surgical intervention may lead to the implantation of epithelium, recurrences after nail biopsy or excision may occur.
In contrast to epidermal inclusion cysts arising in the skin, most SOCs do not have a granular layer.9 Hair and nails represent analogous differentiation products of the ectoderm. The nail matrix is homologous to portions of the hair matrix, while the nail bed epithelium is comparable to the outer root sheath of the hair follicle.7 Subungual onycholemmal cysts originate from the nail bed epithelium, which keratinizes in the absence of a granular layer, similar to the follicular isthmus outer root sheath. Thus, SOCs are comparable to the outer root sheath–derived isthmus-catagen cysts because of their abrupt central keratinization.8
Subungual onycholemmal cysts also must be distinguished from slowly growing malignant tumors of the nail bed epithelium, referred to as onycholemmal carcinomas by Alessi et al.10 This entity characteristically presents in elderly patients as a slowly growing, circumscribed, subungual discoloration that may ulcerate, destroying the nail apparatus and penetrating the phalangeal bone. On histopathology, it is characterized by small cysts filled with eosinophilic keratin devoid of a granular layer and lined by atypical squamous epithelium accompanied by solid nests and strands of atypical keratinocytes within the dermis.11 When a cystic component and clear cells predominate, the designation of malignant proliferating onycholemmal cyst has been applied. Its infiltrative growth pattern with destruction of the underlying bone makes it an important entity to exclude when considering the differential diagnosis of tumors of the nail bed.
Subungual melanomas comprise only 1% to 3% of malignant melanomas and 85% are initially misdiagnosed due to their rarity and nonspecific variable presentation. Aside from clinical evidence of Hutchinson sign in the early stages in almost all cases, accurate diagnosis of subungual melanoma and differentiation from SOCs relies on histopathology. A biopsy is necessary to make the diagnosis, but even microscopic findings may be nonspecific during the early stages.
Conclusion
We report a case of a 23-year-old woman with horizontal ridging and tenderness of the right great toenail associated with pigmentation of 5 years’ duration due to an SOC. The etiology of these subungual cysts, with or without nail abnormalities, still remains unclear. Its predilection for the thumbs and great toenails suggests that trauma or other local factors may be involved in its pathogenesis. Because of the rarity of this entity, there are no guidelines for surgical treatment. Subungual onycholemmal cysts may be an underrecognized and more common entity that must be considered when discussing tumors of the nail unit.
- Samman PD. The human toe nail. its genesis and blood supply. Br J Dermatol. 1959;71:296-302.
- Lewin K. The normal fingernail. Br J Dermatol. 1965;77:421-430.
- Lewin K. Subungual epidermoid inclusions. Br J Dermatol. 1969;81:671-675.
- Dominguez-Cherit J, Chanussot-Deprez C, Maria-Sarti H, et al. Nail unit tumors: a study of 234 patients in the dermatology department of the “Dr. Manuel Gea González” General Hospital in Mexico City. Dermatol Surg. 2008;34:1363-1371.
- Sáez-de-Ocariz MM, Domínguez-Cherit J, García-Corona C. Subungual epidermoid cysts. Int J Dermatol. 2001;40:524-526.
- Molly DO, Herbert K. Subungual epidermoid cyst. J Hand Surg Br. 2006;31:345.
- Telang GH, Jellinek N. Multiple calcified subungual epidermoid inclusions. J Am Acad Dermatol. 2007;56:336-339.
- Fanti PA, Tosti A. Subungual epidermoid inclusions: report of 8 cases. Dermatologica. 1989;178:209-212.
- Takiyoshi N, Nakano H, Matsuzaki T, et al. An eclipse in the subungual space: a diagnostic sign for a subungual epidermal cyst? Br J Dermatol. 2009;161:962-963.
- Alessi E, Coggi A, Gianotti R, et al. Onycholemmal carcinoma. Am J Dermatopathol. 2004;26:397-402.
- Inaoki M, Makino E, Adachi M, et al. Onycholemmal carcinoma. J Cutan Pathol. 2006;33:577-580.
Case Report
A 23-year-old woman presented with a horizontal split along the midline of the right great toenail associated with some tenderness of 2 to 3 months’ duration. Approximately 5 years prior, she noticed a bluish-colored area under the nail that had been steadily increasing in size. She denied a history of trauma, drainage, or bleeding. There was no history of other nail abnormalities. Her medications and personal, family, and social history were noncontributory.
Physical examination of the right great toenail revealed a horizontal split of the nail plate with a bluish hue visible under the nail plate (Figure 1A). The remaining toenails and fingernails were normal. A punch biopsy of the nail bed was performed with a presumptive clinical diagnosis of subungual melanoma versus melanocytic nevus versus cyst (Figure 1B). Nail plate avulsion revealed a blackened nail bed dotted with areas of bluish color and a red friable nodule present focally. Upon further inspection, extension was apparent into the distal matrix.
Histopathologic examination revealed a cystic structure with an epithelial lining mostly reminiscent of an isthmus catagen cyst admixed with the presence of both an intermittent focal granular layer and an eosinophilic cuticle surrounding pink laminated keratin, most consistent with a diagnosis of subungual onycholemmal cyst (SOC)(Figure 2). A reexcision was performed with removal of half of the nail bed, including a portion of the distal matrix extending inferiorly to the bone. Variably sized, epithelium-lined, keratin-filled cystic structures emanated from the nail bed epithelium. There were foci of hemorrhage and granulation tissue secondary to cyst rupture (Figure 3). The defect healed by secondary intention. No clinical evidence of recurrence was seen at 6-month follow-up.
Subungual onycholemmal cysts, also known as subungual epidermoid cysts or subungual epidermoid inclusions, are rare and distinctive nail abnormalities occurring in the dermis of the nail bed. We present a case of an SOC in a toenail mimicking subungual malignant melanoma.
Originally described by Samman1 in 1959, SOCs were attributed to trauma to the nail with resultant implantation of the epidermis into the deeper tissue. Lewin2,3 examined 90 postmortem fingernail and nail bed samples and found 8 subungual epidermoid cysts associated with clubbing of the fingernails. He postulated that the early pathogenesis of clubbing involved dermal fibroblast proliferation in the nail bed, leading to sequestration of nail bed epithelium into the dermis with resultant cyst formation. Microscopic subungual cysts also were identified in normal-appearing nails without evidence of trauma, thought to have arisen from the tips of the nail bed rete ridges by a process of bulbous proliferation rather than sequestration. These findings in normal nails suggest that SOCs may represent a more common entity than previously recognized.
It is imperative to recognize the presence of nail unit tumors early because of the risk for permanent nail plate dystrophy and the possibility of a malignant tumor.4,5 Subungual onycholemmal cysts may present with a wide spectrum of clinical findings including marked subungual hyperkeratosis, onychodystrophy, ridging, nail bed pigmentation, clubbing, thickening, or less often a normal-appearing nail. Based on reported cases, several trends are evident. Although nail dystrophy is most often asymptomatic, pain is not uncommon.5,6 It most commonly involves single digits, predominantly thumbs and great toenails.7,8 This predilection suggests that trauma or other local factors may be involved in its pathogenesis. Of note, trauma to the nail may occur years before the development of the lesions or it may not be recalled at all.
Diagnosis requires a degree of clinical suspicion and a nail bed biopsy with partial or total nail plate avulsion to visualize the pathologic portion of the nail bed. Because surgical intervention may lead to the implantation of epithelium, recurrences after nail biopsy or excision may occur.
In contrast to epidermal inclusion cysts arising in the skin, most SOCs do not have a granular layer.9 Hair and nails represent analogous differentiation products of the ectoderm. The nail matrix is homologous to portions of the hair matrix, while the nail bed epithelium is comparable to the outer root sheath of the hair follicle.7 Subungual onycholemmal cysts originate from the nail bed epithelium, which keratinizes in the absence of a granular layer, similar to the follicular isthmus outer root sheath. Thus, SOCs are comparable to the outer root sheath–derived isthmus-catagen cysts because of their abrupt central keratinization.8
Subungual onycholemmal cysts also must be distinguished from slowly growing malignant tumors of the nail bed epithelium, referred to as onycholemmal carcinomas by Alessi et al.10 This entity characteristically presents in elderly patients as a slowly growing, circumscribed, subungual discoloration that may ulcerate, destroying the nail apparatus and penetrating the phalangeal bone. On histopathology, it is characterized by small cysts filled with eosinophilic keratin devoid of a granular layer and lined by atypical squamous epithelium accompanied by solid nests and strands of atypical keratinocytes within the dermis.11 When a cystic component and clear cells predominate, the designation of malignant proliferating onycholemmal cyst has been applied. Its infiltrative growth pattern with destruction of the underlying bone makes it an important entity to exclude when considering the differential diagnosis of tumors of the nail bed.
Subungual melanomas comprise only 1% to 3% of malignant melanomas and 85% are initially misdiagnosed due to their rarity and nonspecific variable presentation. Aside from clinical evidence of Hutchinson sign in the early stages in almost all cases, accurate diagnosis of subungual melanoma and differentiation from SOCs relies on histopathology. A biopsy is necessary to make the diagnosis, but even microscopic findings may be nonspecific during the early stages.
Conclusion
We report a case of a 23-year-old woman with horizontal ridging and tenderness of the right great toenail associated with pigmentation of 5 years’ duration due to an SOC. The etiology of these subungual cysts, with or without nail abnormalities, still remains unclear. Its predilection for the thumbs and great toenails suggests that trauma or other local factors may be involved in its pathogenesis. Because of the rarity of this entity, there are no guidelines for surgical treatment. Subungual onycholemmal cysts may be an underrecognized and more common entity that must be considered when discussing tumors of the nail unit.
Case Report
A 23-year-old woman presented with a horizontal split along the midline of the right great toenail associated with some tenderness of 2 to 3 months’ duration. Approximately 5 years prior, she noticed a bluish-colored area under the nail that had been steadily increasing in size. She denied a history of trauma, drainage, or bleeding. There was no history of other nail abnormalities. Her medications and personal, family, and social history were noncontributory.
Physical examination of the right great toenail revealed a horizontal split of the nail plate with a bluish hue visible under the nail plate (Figure 1A). The remaining toenails and fingernails were normal. A punch biopsy of the nail bed was performed with a presumptive clinical diagnosis of subungual melanoma versus melanocytic nevus versus cyst (Figure 1B). Nail plate avulsion revealed a blackened nail bed dotted with areas of bluish color and a red friable nodule present focally. Upon further inspection, extension was apparent into the distal matrix.
Histopathologic examination revealed a cystic structure with an epithelial lining mostly reminiscent of an isthmus catagen cyst admixed with the presence of both an intermittent focal granular layer and an eosinophilic cuticle surrounding pink laminated keratin, most consistent with a diagnosis of subungual onycholemmal cyst (SOC)(Figure 2). A reexcision was performed with removal of half of the nail bed, including a portion of the distal matrix extending inferiorly to the bone. Variably sized, epithelium-lined, keratin-filled cystic structures emanated from the nail bed epithelium. There were foci of hemorrhage and granulation tissue secondary to cyst rupture (Figure 3). The defect healed by secondary intention. No clinical evidence of recurrence was seen at 6-month follow-up.
Subungual onycholemmal cysts, also known as subungual epidermoid cysts or subungual epidermoid inclusions, are rare and distinctive nail abnormalities occurring in the dermis of the nail bed. We present a case of an SOC in a toenail mimicking subungual malignant melanoma.
Originally described by Samman1 in 1959, SOCs were attributed to trauma to the nail with resultant implantation of the epidermis into the deeper tissue. Lewin2,3 examined 90 postmortem fingernail and nail bed samples and found 8 subungual epidermoid cysts associated with clubbing of the fingernails. He postulated that the early pathogenesis of clubbing involved dermal fibroblast proliferation in the nail bed, leading to sequestration of nail bed epithelium into the dermis with resultant cyst formation. Microscopic subungual cysts also were identified in normal-appearing nails without evidence of trauma, thought to have arisen from the tips of the nail bed rete ridges by a process of bulbous proliferation rather than sequestration. These findings in normal nails suggest that SOCs may represent a more common entity than previously recognized.
It is imperative to recognize the presence of nail unit tumors early because of the risk for permanent nail plate dystrophy and the possibility of a malignant tumor.4,5 Subungual onycholemmal cysts may present with a wide spectrum of clinical findings including marked subungual hyperkeratosis, onychodystrophy, ridging, nail bed pigmentation, clubbing, thickening, or less often a normal-appearing nail. Based on reported cases, several trends are evident. Although nail dystrophy is most often asymptomatic, pain is not uncommon.5,6 It most commonly involves single digits, predominantly thumbs and great toenails.7,8 This predilection suggests that trauma or other local factors may be involved in its pathogenesis. Of note, trauma to the nail may occur years before the development of the lesions or it may not be recalled at all.
Diagnosis requires a degree of clinical suspicion and a nail bed biopsy with partial or total nail plate avulsion to visualize the pathologic portion of the nail bed. Because surgical intervention may lead to the implantation of epithelium, recurrences after nail biopsy or excision may occur.
In contrast to epidermal inclusion cysts arising in the skin, most SOCs do not have a granular layer.9 Hair and nails represent analogous differentiation products of the ectoderm. The nail matrix is homologous to portions of the hair matrix, while the nail bed epithelium is comparable to the outer root sheath of the hair follicle.7 Subungual onycholemmal cysts originate from the nail bed epithelium, which keratinizes in the absence of a granular layer, similar to the follicular isthmus outer root sheath. Thus, SOCs are comparable to the outer root sheath–derived isthmus-catagen cysts because of their abrupt central keratinization.8
Subungual onycholemmal cysts also must be distinguished from slowly growing malignant tumors of the nail bed epithelium, referred to as onycholemmal carcinomas by Alessi et al.10 This entity characteristically presents in elderly patients as a slowly growing, circumscribed, subungual discoloration that may ulcerate, destroying the nail apparatus and penetrating the phalangeal bone. On histopathology, it is characterized by small cysts filled with eosinophilic keratin devoid of a granular layer and lined by atypical squamous epithelium accompanied by solid nests and strands of atypical keratinocytes within the dermis.11 When a cystic component and clear cells predominate, the designation of malignant proliferating onycholemmal cyst has been applied. Its infiltrative growth pattern with destruction of the underlying bone makes it an important entity to exclude when considering the differential diagnosis of tumors of the nail bed.
Subungual melanomas comprise only 1% to 3% of malignant melanomas and 85% are initially misdiagnosed due to their rarity and nonspecific variable presentation. Aside from clinical evidence of Hutchinson sign in the early stages in almost all cases, accurate diagnosis of subungual melanoma and differentiation from SOCs relies on histopathology. A biopsy is necessary to make the diagnosis, but even microscopic findings may be nonspecific during the early stages.
Conclusion
We report a case of a 23-year-old woman with horizontal ridging and tenderness of the right great toenail associated with pigmentation of 5 years’ duration due to an SOC. The etiology of these subungual cysts, with or without nail abnormalities, still remains unclear. Its predilection for the thumbs and great toenails suggests that trauma or other local factors may be involved in its pathogenesis. Because of the rarity of this entity, there are no guidelines for surgical treatment. Subungual onycholemmal cysts may be an underrecognized and more common entity that must be considered when discussing tumors of the nail unit.
- Samman PD. The human toe nail. its genesis and blood supply. Br J Dermatol. 1959;71:296-302.
- Lewin K. The normal fingernail. Br J Dermatol. 1965;77:421-430.
- Lewin K. Subungual epidermoid inclusions. Br J Dermatol. 1969;81:671-675.
- Dominguez-Cherit J, Chanussot-Deprez C, Maria-Sarti H, et al. Nail unit tumors: a study of 234 patients in the dermatology department of the “Dr. Manuel Gea González” General Hospital in Mexico City. Dermatol Surg. 2008;34:1363-1371.
- Sáez-de-Ocariz MM, Domínguez-Cherit J, García-Corona C. Subungual epidermoid cysts. Int J Dermatol. 2001;40:524-526.
- Molly DO, Herbert K. Subungual epidermoid cyst. J Hand Surg Br. 2006;31:345.
- Telang GH, Jellinek N. Multiple calcified subungual epidermoid inclusions. J Am Acad Dermatol. 2007;56:336-339.
- Fanti PA, Tosti A. Subungual epidermoid inclusions: report of 8 cases. Dermatologica. 1989;178:209-212.
- Takiyoshi N, Nakano H, Matsuzaki T, et al. An eclipse in the subungual space: a diagnostic sign for a subungual epidermal cyst? Br J Dermatol. 2009;161:962-963.
- Alessi E, Coggi A, Gianotti R, et al. Onycholemmal carcinoma. Am J Dermatopathol. 2004;26:397-402.
- Inaoki M, Makino E, Adachi M, et al. Onycholemmal carcinoma. J Cutan Pathol. 2006;33:577-580.
- Samman PD. The human toe nail. its genesis and blood supply. Br J Dermatol. 1959;71:296-302.
- Lewin K. The normal fingernail. Br J Dermatol. 1965;77:421-430.
- Lewin K. Subungual epidermoid inclusions. Br J Dermatol. 1969;81:671-675.
- Dominguez-Cherit J, Chanussot-Deprez C, Maria-Sarti H, et al. Nail unit tumors: a study of 234 patients in the dermatology department of the “Dr. Manuel Gea González” General Hospital in Mexico City. Dermatol Surg. 2008;34:1363-1371.
- Sáez-de-Ocariz MM, Domínguez-Cherit J, García-Corona C. Subungual epidermoid cysts. Int J Dermatol. 2001;40:524-526.
- Molly DO, Herbert K. Subungual epidermoid cyst. J Hand Surg Br. 2006;31:345.
- Telang GH, Jellinek N. Multiple calcified subungual epidermoid inclusions. J Am Acad Dermatol. 2007;56:336-339.
- Fanti PA, Tosti A. Subungual epidermoid inclusions: report of 8 cases. Dermatologica. 1989;178:209-212.
- Takiyoshi N, Nakano H, Matsuzaki T, et al. An eclipse in the subungual space: a diagnostic sign for a subungual epidermal cyst? Br J Dermatol. 2009;161:962-963.
- Alessi E, Coggi A, Gianotti R, et al. Onycholemmal carcinoma. Am J Dermatopathol. 2004;26:397-402.
- Inaoki M, Makino E, Adachi M, et al. Onycholemmal carcinoma. J Cutan Pathol. 2006;33:577-580.
Practice Points
- Trauma to the nail may occur years before the development of subungual onycholemmal cysts or it may not be recalled at all.
- Diagnosis requires a degree of clinical suspicion and a nail bed biopsy.
- Subungual onycholemmal cysts must be distinguished from slowly growing malignant tumors of the nail bed epithelium.
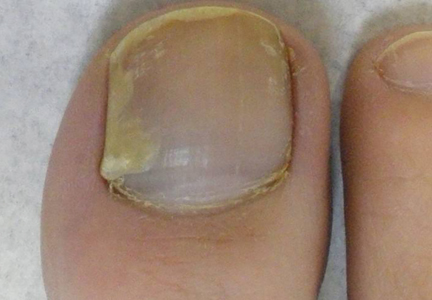
An Unusual Infection of Breast Tissue
A 50-year-old white woman who worked in a nail salon and had a history of Crohn disease (asymptomatic, off treatment for several years) presented to her primary care physician with right breast erythema and pain for the previous 3 days. The symptoms started as a red streak in the upper half of the breast with no reported history of a recent trauma to the breast and no history of prior surgeries. The patient reported having no nipple discharge or any associated constitutional symptoms.
On initial examination, the patient was found to have intact skin with 3 cm of localized redness. She was diagnosed with right breast mastitis and given oral erythromycin due to a history of allergy to penicillin. Four days later, while she was still taking oral antibiotics, her symptoms worsened. She was referred to the breast clinic and reevaluated by the breast surgeon, who reported findings of right breast tenderness and induration localized to the upper inner quadrant with no signs of fluctuation to suggest an abscess. A breast ultrasound showed inflammatory changes of the right breast around the perinipple-areola complex area. Fine-needle aspiration (FNA) was performed in the surgery clinic. Additional laboratory tests revealed no leukocytosis on routine blood work. She was hospitalized and received IV vancomycin and clindamycin. Clindamycin was given to cover anaerobes along with vancomycin, which is more reliable to cover staphylococcus, including methicillin-resistant Staphylococcus aureus empirically. The bacterial culture showed no growth, and the reported findings of the FNA were consistent with inflammatory process without evidence of malignancy. The erythema improved in a few days, and the patient was discharged home on oral linezolid, as she declined further IV antibiotics.
At the follow-up surgical clinic visit, the induration was the same, and the erythema had progressed to involve a larger area of the breast. Repeat ultrasound showed an edematous area within the retro-areolar region. Repeat FNA was performed in the clinic, and 3 mL of bloody fluid was obtained. The bacterial culture again showed no growth, and the cytologic findings revealed an inflammatory process with more exudates but no evidence of malignancy. Due to lack of expected clinical improvement with the treatment for presumptive acute bacterial infection, the decision was made to proceed with excisional biopsy to rule out malignances and other potential etiologies of an infectious/inflammatory process.
Excision of the involved area, consisting of inner upper quadrantectomy and terminal duct excision, was performed due to the operative findings of inflamed, necrotic tissue and the presence of multiple abscesses. The pathology report confirmed the presence of microabscesses and mastitis with periductal inflammation. Microbiology data revealed a negative bacterial culture and negative acid-fast bacilli (AFB) smear. However, the AFB culture from MB/BacT (a liquid media by BioMérieux, Marcy-l’Étoile, France) grew an atypical mycobacterium.
An infectious disease consultation was requested for further guidance. The AFB isolate was sent to the University of Texas Health Science Center, where the atypical mycobacterium was later identified as Mycobacteriumchelonae-abscessus complex. This was determined to be the Mycobacterium abscessus species. The susceptibility test report revealed the organism was sensitive to amikacin, cefoxitin, clarithromycin, and linezolid and demonstrated intermediate sensitivity to quinolones. The organism was resistant to doxycycline and sulfamethoxazole.
During evaluation in the infectious disease clinic, the patient reported night sweats. The examination of her right breast revealed a well-healed surgical scar with surrounding erythema and induration. Blood cultures for AFB taken later were negative. The patient was married with no risk factors for HIV but consented to a test, which also was negative.The patient was started on oral clarithromycin and ciprofloxacin, pending the sensitivity of the mycobacterium.
Due to the slow clinical improvement on oral antibiotics after surgery, intolerance to ciprofloxacin (severe nausea), and the sensitivity pattern, the decision was made to add IV cefoxitin (minimum inhibitory concentration [MIC] < 64) and continue with oral clarithromycin (MIC < 0.1).
During her 2-week follow-up visit, the patient reported significant improvement in pain, swelling, and induration of her breast, confirmed by physical examination. At the 12-week follow-up visit, the patient’s erythema and induration had resolved completely. The IV cefoxitin was discontinued, and her oral clarithromycin was renewed for another 3 months. The decision to treat with antibiotics after surgical removal of necrotic tissue was based on previous cases and the need to ensure complete eradication of the infection prior to breast reconstruction.
The patient remained asymptomatic at her 6-month follow-up visit after completion of the antibiotics course. No recurrence of the atypical mycobacterium infection was detected. A year later, she underwent reconstruction of her right breast.
Discussion
Mycobacterium abscessus is a human pathogen commonly found in the soil, water, or sewerage. The organism is a nontuberculous mycobacterium characterized by rapid growth and a lack of pigmentation on the gram-positive rods. A review of the literature revealed multiple cases of M abscessus infection of the skin, but only one reference was made concerning breast tissues in which rapidly growing M abscessus initially was misdiagnosed as fibrocystic disease. Yasar and colleagues reported a woman with a history of fibrocystic breast disease who presented with a breast abscess later identified as M abscessus.1 She was treated with amikacin, linezolid, and clarithromycin. The authors concluded that antimycobacterial therapy for M abscessus infection remains poorly established, and surgical therapy is often required in treating this atypical mycobacterium infection.
Two previous reports have referred to M abscessus infection in the breast tissue; however, this current case is unique. The authors believe this case study to be noteworthy in its description of an acute presentation in a woman with neither trauma nor history of breast disease, such as fibrocystic breasts. This unusual presentation makes the diagnosis of M abscessus infection more difficult to diagnose and treat in a timely manner.
Reports of M abscessus infection have been documented in skin and soft tissues. These cases involved prior trauma to the skin, such as acupuncture, filler injections, surgical procedures, or other traumatic events. Painful nodules and plaque formation also were reported with a culture showing polymorphonuclear microabscesses and granulomatous inflammation in the dermis and subcutaneous fat tissue, identified as M abscessus.2,3
Another study reported a case of M abscessus infection after a patient had a tattoo.4 Possible of infections from tattoos may be localized or systemic. Because more people are being tattooed and developing skin infections resistant to standard antibiotic treatment, M abscessus infection must be considered in the differential diagnosis of these infections.
Conclusion
Mycobacterium abscessus infection is usually seen in immunocompromised hosts or those with trauma. However, as more cases of M abscessus are seen in skin and soft-tissue infections because of more cosmetic injections, body art, or minor surgical procedures, clinicians must have a high degree of suspicion for this pathogen, especially if the patient does not respond to standard antibacterial therapy. Although amikacin and clarithromycin are 2 antimicrobial agents that have shown effectiveness against this pathogen, obtaining a skin biopsy along with mycobacterial culture and sensitivity testing is determining the proper agents for successful treatment. The importance of early recognition and proper antibiotic therapy is crucial to avoid delay in diagnosis and to decrease potential tissue loss.
1. Yasar KK, Pehlivanoglu F Sengoz G, Cabioglu N. Successfully treated Mycobacterium abscessus mastitis: a rare cause of breast masses. Indian J Med Microbiol. 2011;29(4):425-427.
2. Lee WJ, Kang SM, Sung H, et al. Non-tuberculous mycobacterial infections of the skin: a retrospective study of 29 cases. J Dermatol. 2010;37(11):965-972.
3. Kwon YH, Lee G-Y, Kim W-S, Kim KJ. A case of skin and soft tissue infection caused by Mycobacterium abscessus. Ann Dermatol. 2009;21(1):84-87.
4. Bechara C, Macheras E, Heym B, Pages A, Auffret N. Mycobacterium abscessus skin infection after tattooing: first case report and review of the literature. Dermatology. 2010;221(1):1-4.
A 50-year-old white woman who worked in a nail salon and had a history of Crohn disease (asymptomatic, off treatment for several years) presented to her primary care physician with right breast erythema and pain for the previous 3 days. The symptoms started as a red streak in the upper half of the breast with no reported history of a recent trauma to the breast and no history of prior surgeries. The patient reported having no nipple discharge or any associated constitutional symptoms.
On initial examination, the patient was found to have intact skin with 3 cm of localized redness. She was diagnosed with right breast mastitis and given oral erythromycin due to a history of allergy to penicillin. Four days later, while she was still taking oral antibiotics, her symptoms worsened. She was referred to the breast clinic and reevaluated by the breast surgeon, who reported findings of right breast tenderness and induration localized to the upper inner quadrant with no signs of fluctuation to suggest an abscess. A breast ultrasound showed inflammatory changes of the right breast around the perinipple-areola complex area. Fine-needle aspiration (FNA) was performed in the surgery clinic. Additional laboratory tests revealed no leukocytosis on routine blood work. She was hospitalized and received IV vancomycin and clindamycin. Clindamycin was given to cover anaerobes along with vancomycin, which is more reliable to cover staphylococcus, including methicillin-resistant Staphylococcus aureus empirically. The bacterial culture showed no growth, and the reported findings of the FNA were consistent with inflammatory process without evidence of malignancy. The erythema improved in a few days, and the patient was discharged home on oral linezolid, as she declined further IV antibiotics.
At the follow-up surgical clinic visit, the induration was the same, and the erythema had progressed to involve a larger area of the breast. Repeat ultrasound showed an edematous area within the retro-areolar region. Repeat FNA was performed in the clinic, and 3 mL of bloody fluid was obtained. The bacterial culture again showed no growth, and the cytologic findings revealed an inflammatory process with more exudates but no evidence of malignancy. Due to lack of expected clinical improvement with the treatment for presumptive acute bacterial infection, the decision was made to proceed with excisional biopsy to rule out malignances and other potential etiologies of an infectious/inflammatory process.
Excision of the involved area, consisting of inner upper quadrantectomy and terminal duct excision, was performed due to the operative findings of inflamed, necrotic tissue and the presence of multiple abscesses. The pathology report confirmed the presence of microabscesses and mastitis with periductal inflammation. Microbiology data revealed a negative bacterial culture and negative acid-fast bacilli (AFB) smear. However, the AFB culture from MB/BacT (a liquid media by BioMérieux, Marcy-l’Étoile, France) grew an atypical mycobacterium.
An infectious disease consultation was requested for further guidance. The AFB isolate was sent to the University of Texas Health Science Center, where the atypical mycobacterium was later identified as Mycobacteriumchelonae-abscessus complex. This was determined to be the Mycobacterium abscessus species. The susceptibility test report revealed the organism was sensitive to amikacin, cefoxitin, clarithromycin, and linezolid and demonstrated intermediate sensitivity to quinolones. The organism was resistant to doxycycline and sulfamethoxazole.
During evaluation in the infectious disease clinic, the patient reported night sweats. The examination of her right breast revealed a well-healed surgical scar with surrounding erythema and induration. Blood cultures for AFB taken later were negative. The patient was married with no risk factors for HIV but consented to a test, which also was negative.The patient was started on oral clarithromycin and ciprofloxacin, pending the sensitivity of the mycobacterium.
Due to the slow clinical improvement on oral antibiotics after surgery, intolerance to ciprofloxacin (severe nausea), and the sensitivity pattern, the decision was made to add IV cefoxitin (minimum inhibitory concentration [MIC] < 64) and continue with oral clarithromycin (MIC < 0.1).
During her 2-week follow-up visit, the patient reported significant improvement in pain, swelling, and induration of her breast, confirmed by physical examination. At the 12-week follow-up visit, the patient’s erythema and induration had resolved completely. The IV cefoxitin was discontinued, and her oral clarithromycin was renewed for another 3 months. The decision to treat with antibiotics after surgical removal of necrotic tissue was based on previous cases and the need to ensure complete eradication of the infection prior to breast reconstruction.
The patient remained asymptomatic at her 6-month follow-up visit after completion of the antibiotics course. No recurrence of the atypical mycobacterium infection was detected. A year later, she underwent reconstruction of her right breast.
Discussion
Mycobacterium abscessus is a human pathogen commonly found in the soil, water, or sewerage. The organism is a nontuberculous mycobacterium characterized by rapid growth and a lack of pigmentation on the gram-positive rods. A review of the literature revealed multiple cases of M abscessus infection of the skin, but only one reference was made concerning breast tissues in which rapidly growing M abscessus initially was misdiagnosed as fibrocystic disease. Yasar and colleagues reported a woman with a history of fibrocystic breast disease who presented with a breast abscess later identified as M abscessus.1 She was treated with amikacin, linezolid, and clarithromycin. The authors concluded that antimycobacterial therapy for M abscessus infection remains poorly established, and surgical therapy is often required in treating this atypical mycobacterium infection.
Two previous reports have referred to M abscessus infection in the breast tissue; however, this current case is unique. The authors believe this case study to be noteworthy in its description of an acute presentation in a woman with neither trauma nor history of breast disease, such as fibrocystic breasts. This unusual presentation makes the diagnosis of M abscessus infection more difficult to diagnose and treat in a timely manner.
Reports of M abscessus infection have been documented in skin and soft tissues. These cases involved prior trauma to the skin, such as acupuncture, filler injections, surgical procedures, or other traumatic events. Painful nodules and plaque formation also were reported with a culture showing polymorphonuclear microabscesses and granulomatous inflammation in the dermis and subcutaneous fat tissue, identified as M abscessus.2,3
Another study reported a case of M abscessus infection after a patient had a tattoo.4 Possible of infections from tattoos may be localized or systemic. Because more people are being tattooed and developing skin infections resistant to standard antibiotic treatment, M abscessus infection must be considered in the differential diagnosis of these infections.
Conclusion
Mycobacterium abscessus infection is usually seen in immunocompromised hosts or those with trauma. However, as more cases of M abscessus are seen in skin and soft-tissue infections because of more cosmetic injections, body art, or minor surgical procedures, clinicians must have a high degree of suspicion for this pathogen, especially if the patient does not respond to standard antibacterial therapy. Although amikacin and clarithromycin are 2 antimicrobial agents that have shown effectiveness against this pathogen, obtaining a skin biopsy along with mycobacterial culture and sensitivity testing is determining the proper agents for successful treatment. The importance of early recognition and proper antibiotic therapy is crucial to avoid delay in diagnosis and to decrease potential tissue loss.
A 50-year-old white woman who worked in a nail salon and had a history of Crohn disease (asymptomatic, off treatment for several years) presented to her primary care physician with right breast erythema and pain for the previous 3 days. The symptoms started as a red streak in the upper half of the breast with no reported history of a recent trauma to the breast and no history of prior surgeries. The patient reported having no nipple discharge or any associated constitutional symptoms.
On initial examination, the patient was found to have intact skin with 3 cm of localized redness. She was diagnosed with right breast mastitis and given oral erythromycin due to a history of allergy to penicillin. Four days later, while she was still taking oral antibiotics, her symptoms worsened. She was referred to the breast clinic and reevaluated by the breast surgeon, who reported findings of right breast tenderness and induration localized to the upper inner quadrant with no signs of fluctuation to suggest an abscess. A breast ultrasound showed inflammatory changes of the right breast around the perinipple-areola complex area. Fine-needle aspiration (FNA) was performed in the surgery clinic. Additional laboratory tests revealed no leukocytosis on routine blood work. She was hospitalized and received IV vancomycin and clindamycin. Clindamycin was given to cover anaerobes along with vancomycin, which is more reliable to cover staphylococcus, including methicillin-resistant Staphylococcus aureus empirically. The bacterial culture showed no growth, and the reported findings of the FNA were consistent with inflammatory process without evidence of malignancy. The erythema improved in a few days, and the patient was discharged home on oral linezolid, as she declined further IV antibiotics.
At the follow-up surgical clinic visit, the induration was the same, and the erythema had progressed to involve a larger area of the breast. Repeat ultrasound showed an edematous area within the retro-areolar region. Repeat FNA was performed in the clinic, and 3 mL of bloody fluid was obtained. The bacterial culture again showed no growth, and the cytologic findings revealed an inflammatory process with more exudates but no evidence of malignancy. Due to lack of expected clinical improvement with the treatment for presumptive acute bacterial infection, the decision was made to proceed with excisional biopsy to rule out malignances and other potential etiologies of an infectious/inflammatory process.
Excision of the involved area, consisting of inner upper quadrantectomy and terminal duct excision, was performed due to the operative findings of inflamed, necrotic tissue and the presence of multiple abscesses. The pathology report confirmed the presence of microabscesses and mastitis with periductal inflammation. Microbiology data revealed a negative bacterial culture and negative acid-fast bacilli (AFB) smear. However, the AFB culture from MB/BacT (a liquid media by BioMérieux, Marcy-l’Étoile, France) grew an atypical mycobacterium.
An infectious disease consultation was requested for further guidance. The AFB isolate was sent to the University of Texas Health Science Center, where the atypical mycobacterium was later identified as Mycobacteriumchelonae-abscessus complex. This was determined to be the Mycobacterium abscessus species. The susceptibility test report revealed the organism was sensitive to amikacin, cefoxitin, clarithromycin, and linezolid and demonstrated intermediate sensitivity to quinolones. The organism was resistant to doxycycline and sulfamethoxazole.
During evaluation in the infectious disease clinic, the patient reported night sweats. The examination of her right breast revealed a well-healed surgical scar with surrounding erythema and induration. Blood cultures for AFB taken later were negative. The patient was married with no risk factors for HIV but consented to a test, which also was negative.The patient was started on oral clarithromycin and ciprofloxacin, pending the sensitivity of the mycobacterium.
Due to the slow clinical improvement on oral antibiotics after surgery, intolerance to ciprofloxacin (severe nausea), and the sensitivity pattern, the decision was made to add IV cefoxitin (minimum inhibitory concentration [MIC] < 64) and continue with oral clarithromycin (MIC < 0.1).
During her 2-week follow-up visit, the patient reported significant improvement in pain, swelling, and induration of her breast, confirmed by physical examination. At the 12-week follow-up visit, the patient’s erythema and induration had resolved completely. The IV cefoxitin was discontinued, and her oral clarithromycin was renewed for another 3 months. The decision to treat with antibiotics after surgical removal of necrotic tissue was based on previous cases and the need to ensure complete eradication of the infection prior to breast reconstruction.
The patient remained asymptomatic at her 6-month follow-up visit after completion of the antibiotics course. No recurrence of the atypical mycobacterium infection was detected. A year later, she underwent reconstruction of her right breast.
Discussion
Mycobacterium abscessus is a human pathogen commonly found in the soil, water, or sewerage. The organism is a nontuberculous mycobacterium characterized by rapid growth and a lack of pigmentation on the gram-positive rods. A review of the literature revealed multiple cases of M abscessus infection of the skin, but only one reference was made concerning breast tissues in which rapidly growing M abscessus initially was misdiagnosed as fibrocystic disease. Yasar and colleagues reported a woman with a history of fibrocystic breast disease who presented with a breast abscess later identified as M abscessus.1 She was treated with amikacin, linezolid, and clarithromycin. The authors concluded that antimycobacterial therapy for M abscessus infection remains poorly established, and surgical therapy is often required in treating this atypical mycobacterium infection.
Two previous reports have referred to M abscessus infection in the breast tissue; however, this current case is unique. The authors believe this case study to be noteworthy in its description of an acute presentation in a woman with neither trauma nor history of breast disease, such as fibrocystic breasts. This unusual presentation makes the diagnosis of M abscessus infection more difficult to diagnose and treat in a timely manner.
Reports of M abscessus infection have been documented in skin and soft tissues. These cases involved prior trauma to the skin, such as acupuncture, filler injections, surgical procedures, or other traumatic events. Painful nodules and plaque formation also were reported with a culture showing polymorphonuclear microabscesses and granulomatous inflammation in the dermis and subcutaneous fat tissue, identified as M abscessus.2,3
Another study reported a case of M abscessus infection after a patient had a tattoo.4 Possible of infections from tattoos may be localized or systemic. Because more people are being tattooed and developing skin infections resistant to standard antibiotic treatment, M abscessus infection must be considered in the differential diagnosis of these infections.
Conclusion
Mycobacterium abscessus infection is usually seen in immunocompromised hosts or those with trauma. However, as more cases of M abscessus are seen in skin and soft-tissue infections because of more cosmetic injections, body art, or minor surgical procedures, clinicians must have a high degree of suspicion for this pathogen, especially if the patient does not respond to standard antibacterial therapy. Although amikacin and clarithromycin are 2 antimicrobial agents that have shown effectiveness against this pathogen, obtaining a skin biopsy along with mycobacterial culture and sensitivity testing is determining the proper agents for successful treatment. The importance of early recognition and proper antibiotic therapy is crucial to avoid delay in diagnosis and to decrease potential tissue loss.
1. Yasar KK, Pehlivanoglu F Sengoz G, Cabioglu N. Successfully treated Mycobacterium abscessus mastitis: a rare cause of breast masses. Indian J Med Microbiol. 2011;29(4):425-427.
2. Lee WJ, Kang SM, Sung H, et al. Non-tuberculous mycobacterial infections of the skin: a retrospective study of 29 cases. J Dermatol. 2010;37(11):965-972.
3. Kwon YH, Lee G-Y, Kim W-S, Kim KJ. A case of skin and soft tissue infection caused by Mycobacterium abscessus. Ann Dermatol. 2009;21(1):84-87.
4. Bechara C, Macheras E, Heym B, Pages A, Auffret N. Mycobacterium abscessus skin infection after tattooing: first case report and review of the literature. Dermatology. 2010;221(1):1-4.
1. Yasar KK, Pehlivanoglu F Sengoz G, Cabioglu N. Successfully treated Mycobacterium abscessus mastitis: a rare cause of breast masses. Indian J Med Microbiol. 2011;29(4):425-427.
2. Lee WJ, Kang SM, Sung H, et al. Non-tuberculous mycobacterial infections of the skin: a retrospective study of 29 cases. J Dermatol. 2010;37(11):965-972.
3. Kwon YH, Lee G-Y, Kim W-S, Kim KJ. A case of skin and soft tissue infection caused by Mycobacterium abscessus. Ann Dermatol. 2009;21(1):84-87.
4. Bechara C, Macheras E, Heym B, Pages A, Auffret N. Mycobacterium abscessus skin infection after tattooing: first case report and review of the literature. Dermatology. 2010;221(1):1-4.
Subungual Exostosis
Case Report
A 41-year-old man with no dermatologic history presented for a skin examination. During a full-body skin examination, a lesion was identified on the right third toe that was partially visible underneath the nail plate. The patient stated that the lesion had been present for many years and did not appear to be growing but did cause occasional pain. On examination a 1-cm verrucous, hyperkeratotic, tan papule was noted at the distal end of the nail bed causing partial onycholysis (Figure 1). It was not tender to palpation.
A shave biopsy was obtained of the visible portion of the lesion, which revealed hyperkeratosis, acanthosis, and a population of dermal spindle cells in a myxoid stroma that could not be definitively identified. Special stains were nondiagnostic. The patient was referred to dermatologic surgery for rebiopsy of the lesion after removal of the nail plate. Mature bone was seen embedded in the dermis (Figure 2), and a diagnosis of subungual exostosis was made. Radiography of the digit confirmed a bony excrescence from the tuft of the toe, and the patient was referred to orthopedic surgery for definitive excision. There was no evidence of recurrence at 1-year follow-up.
Comment
Subungual exostosis is an uncommon benign bone tumor located beneath or adjacent to the nail bed on the dorsal aspect of the distal phalanx.1 Although it can occur on any digit, 70% to 80% of cases have arisen on the distal phalanx of the hallux.2 Both sexes are equally susceptible. The majority of lesions occur during the second or third decades of life and usually are asymptomatic unless there is trauma or infection. Growth of the lesion over time can cause lifting or deformity of the nail plate and can cause slight discomfort while walking if located on the great toe.3 Common differential diagnoses include osteochondroma, wart, fibroma, paronychia, myositis ossificans, and pyogenic granuloma.3,4 Diagnosis can be confirmed with radiography, which should be performed prior to any biopsy or invasive procedure. In our patient, initial radiography could have obviated the need for 2 biopsies prior to definitive excision. Histopathologic evaluation typically reveals mature trabecular bone (Figure 2) surrounded by a fibrocartilage cap.
Subungual exostosis begins as an area of proliferating fibrous tissue with cartilaginous metaplasia located beneath or adjacent to the nail bed on the dorsal aspect of the distal phalanx.1 This cartilage undergoes enchondral ossification and is converted to trabecular bone. As the lesion grows and matures, the cartilaginous cap blends imperceptibly with the nail bed and comes into continuity with the underlying distal phalanx.1,3 This process continues until the lesion fuses completely with the distal phalanx.1 Although the cause of subungual exostosis has not been clearly established, chronic irritation, trauma, and chronic infections are considered causative factors of fibrocartilaginous metaplasia.4
The most commonly accepted treatment of subungual exostosis is a localized excision. Partial or total removal of the nail has traditionally be advocated to ensure complete excision of the exostosis, a nail-sparing technique that has been shown to enhance cosmetic results.3 Incomplete excision and incomplete maturation of the lesion have been reported to be responsible for almost 50% of recurrences.3 This high recurrence rate is due to difficulty in ensuring a total excision because the gradual merging of the fibrocartilage cap with the overlying nail bed makes it impossible to develop a cleavage plane5; as a result, it has been suggested that excision should only be attempted after maturation of the tumor so the cleavage plane can fully develop. Other studies claim that delaying treatment can result in elevation and deformity of the nail, pain, and secondary periungual infection.3
Conclusion
Subungual exostosis is a benign bony tumor of the distal phalanx that can cause pain and onycholysis. Radiography of the affected digit is a noninvasive way to confirm the diagnosis and should be part of the initial workup of any suspicious subungual tumor. Once identified, complete removal of the exostosis by excision has been shown to be an effective treatment with few complications.
- Letts M, Davidson D, Nizalik E. Subungual exostosis: diagnosis and treatment in children. J Trauma. 1998;44:346-349.
- Starnes A, Crosby K, Rowe DJ, et al. Subungual exostosis: a simple surgical technique. Dermatol Surg. 2012;38:258-260.
- Lokiec F, Ezra E, Krasin E, et al. A simple and efficient surgical technique for subungual exostosis. J Pediatr Orthop. 2001;21:76-79.
- Turan H, Uslu M, Erdem H. A case of subungual exostosis. Indian J Dermatol Venereol Leprol. 2012;78:186.
- Miller-Breslow A, Dorfman HD. Dupuytren’s (subungual) exostosis. Am J Surg Pathol. 1988;12:368-378.
Case Report
A 41-year-old man with no dermatologic history presented for a skin examination. During a full-body skin examination, a lesion was identified on the right third toe that was partially visible underneath the nail plate. The patient stated that the lesion had been present for many years and did not appear to be growing but did cause occasional pain. On examination a 1-cm verrucous, hyperkeratotic, tan papule was noted at the distal end of the nail bed causing partial onycholysis (Figure 1). It was not tender to palpation.
A shave biopsy was obtained of the visible portion of the lesion, which revealed hyperkeratosis, acanthosis, and a population of dermal spindle cells in a myxoid stroma that could not be definitively identified. Special stains were nondiagnostic. The patient was referred to dermatologic surgery for rebiopsy of the lesion after removal of the nail plate. Mature bone was seen embedded in the dermis (Figure 2), and a diagnosis of subungual exostosis was made. Radiography of the digit confirmed a bony excrescence from the tuft of the toe, and the patient was referred to orthopedic surgery for definitive excision. There was no evidence of recurrence at 1-year follow-up.
Comment
Subungual exostosis is an uncommon benign bone tumor located beneath or adjacent to the nail bed on the dorsal aspect of the distal phalanx.1 Although it can occur on any digit, 70% to 80% of cases have arisen on the distal phalanx of the hallux.2 Both sexes are equally susceptible. The majority of lesions occur during the second or third decades of life and usually are asymptomatic unless there is trauma or infection. Growth of the lesion over time can cause lifting or deformity of the nail plate and can cause slight discomfort while walking if located on the great toe.3 Common differential diagnoses include osteochondroma, wart, fibroma, paronychia, myositis ossificans, and pyogenic granuloma.3,4 Diagnosis can be confirmed with radiography, which should be performed prior to any biopsy or invasive procedure. In our patient, initial radiography could have obviated the need for 2 biopsies prior to definitive excision. Histopathologic evaluation typically reveals mature trabecular bone (Figure 2) surrounded by a fibrocartilage cap.
Subungual exostosis begins as an area of proliferating fibrous tissue with cartilaginous metaplasia located beneath or adjacent to the nail bed on the dorsal aspect of the distal phalanx.1 This cartilage undergoes enchondral ossification and is converted to trabecular bone. As the lesion grows and matures, the cartilaginous cap blends imperceptibly with the nail bed and comes into continuity with the underlying distal phalanx.1,3 This process continues until the lesion fuses completely with the distal phalanx.1 Although the cause of subungual exostosis has not been clearly established, chronic irritation, trauma, and chronic infections are considered causative factors of fibrocartilaginous metaplasia.4
The most commonly accepted treatment of subungual exostosis is a localized excision. Partial or total removal of the nail has traditionally be advocated to ensure complete excision of the exostosis, a nail-sparing technique that has been shown to enhance cosmetic results.3 Incomplete excision and incomplete maturation of the lesion have been reported to be responsible for almost 50% of recurrences.3 This high recurrence rate is due to difficulty in ensuring a total excision because the gradual merging of the fibrocartilage cap with the overlying nail bed makes it impossible to develop a cleavage plane5; as a result, it has been suggested that excision should only be attempted after maturation of the tumor so the cleavage plane can fully develop. Other studies claim that delaying treatment can result in elevation and deformity of the nail, pain, and secondary periungual infection.3
Conclusion
Subungual exostosis is a benign bony tumor of the distal phalanx that can cause pain and onycholysis. Radiography of the affected digit is a noninvasive way to confirm the diagnosis and should be part of the initial workup of any suspicious subungual tumor. Once identified, complete removal of the exostosis by excision has been shown to be an effective treatment with few complications.
Case Report
A 41-year-old man with no dermatologic history presented for a skin examination. During a full-body skin examination, a lesion was identified on the right third toe that was partially visible underneath the nail plate. The patient stated that the lesion had been present for many years and did not appear to be growing but did cause occasional pain. On examination a 1-cm verrucous, hyperkeratotic, tan papule was noted at the distal end of the nail bed causing partial onycholysis (Figure 1). It was not tender to palpation.
A shave biopsy was obtained of the visible portion of the lesion, which revealed hyperkeratosis, acanthosis, and a population of dermal spindle cells in a myxoid stroma that could not be definitively identified. Special stains were nondiagnostic. The patient was referred to dermatologic surgery for rebiopsy of the lesion after removal of the nail plate. Mature bone was seen embedded in the dermis (Figure 2), and a diagnosis of subungual exostosis was made. Radiography of the digit confirmed a bony excrescence from the tuft of the toe, and the patient was referred to orthopedic surgery for definitive excision. There was no evidence of recurrence at 1-year follow-up.
Comment
Subungual exostosis is an uncommon benign bone tumor located beneath or adjacent to the nail bed on the dorsal aspect of the distal phalanx.1 Although it can occur on any digit, 70% to 80% of cases have arisen on the distal phalanx of the hallux.2 Both sexes are equally susceptible. The majority of lesions occur during the second or third decades of life and usually are asymptomatic unless there is trauma or infection. Growth of the lesion over time can cause lifting or deformity of the nail plate and can cause slight discomfort while walking if located on the great toe.3 Common differential diagnoses include osteochondroma, wart, fibroma, paronychia, myositis ossificans, and pyogenic granuloma.3,4 Diagnosis can be confirmed with radiography, which should be performed prior to any biopsy or invasive procedure. In our patient, initial radiography could have obviated the need for 2 biopsies prior to definitive excision. Histopathologic evaluation typically reveals mature trabecular bone (Figure 2) surrounded by a fibrocartilage cap.
Subungual exostosis begins as an area of proliferating fibrous tissue with cartilaginous metaplasia located beneath or adjacent to the nail bed on the dorsal aspect of the distal phalanx.1 This cartilage undergoes enchondral ossification and is converted to trabecular bone. As the lesion grows and matures, the cartilaginous cap blends imperceptibly with the nail bed and comes into continuity with the underlying distal phalanx.1,3 This process continues until the lesion fuses completely with the distal phalanx.1 Although the cause of subungual exostosis has not been clearly established, chronic irritation, trauma, and chronic infections are considered causative factors of fibrocartilaginous metaplasia.4
The most commonly accepted treatment of subungual exostosis is a localized excision. Partial or total removal of the nail has traditionally be advocated to ensure complete excision of the exostosis, a nail-sparing technique that has been shown to enhance cosmetic results.3 Incomplete excision and incomplete maturation of the lesion have been reported to be responsible for almost 50% of recurrences.3 This high recurrence rate is due to difficulty in ensuring a total excision because the gradual merging of the fibrocartilage cap with the overlying nail bed makes it impossible to develop a cleavage plane5; as a result, it has been suggested that excision should only be attempted after maturation of the tumor so the cleavage plane can fully develop. Other studies claim that delaying treatment can result in elevation and deformity of the nail, pain, and secondary periungual infection.3
Conclusion
Subungual exostosis is a benign bony tumor of the distal phalanx that can cause pain and onycholysis. Radiography of the affected digit is a noninvasive way to confirm the diagnosis and should be part of the initial workup of any suspicious subungual tumor. Once identified, complete removal of the exostosis by excision has been shown to be an effective treatment with few complications.
- Letts M, Davidson D, Nizalik E. Subungual exostosis: diagnosis and treatment in children. J Trauma. 1998;44:346-349.
- Starnes A, Crosby K, Rowe DJ, et al. Subungual exostosis: a simple surgical technique. Dermatol Surg. 2012;38:258-260.
- Lokiec F, Ezra E, Krasin E, et al. A simple and efficient surgical technique for subungual exostosis. J Pediatr Orthop. 2001;21:76-79.
- Turan H, Uslu M, Erdem H. A case of subungual exostosis. Indian J Dermatol Venereol Leprol. 2012;78:186.
- Miller-Breslow A, Dorfman HD. Dupuytren’s (subungual) exostosis. Am J Surg Pathol. 1988;12:368-378.
- Letts M, Davidson D, Nizalik E. Subungual exostosis: diagnosis and treatment in children. J Trauma. 1998;44:346-349.
- Starnes A, Crosby K, Rowe DJ, et al. Subungual exostosis: a simple surgical technique. Dermatol Surg. 2012;38:258-260.
- Lokiec F, Ezra E, Krasin E, et al. A simple and efficient surgical technique for subungual exostosis. J Pediatr Orthop. 2001;21:76-79.
- Turan H, Uslu M, Erdem H. A case of subungual exostosis. Indian J Dermatol Venereol Leprol. 2012;78:186.
- Miller-Breslow A, Dorfman HD. Dupuytren’s (subungual) exostosis. Am J Surg Pathol. 1988;12:368-378.
Practice Points
- Subungual exostosis is a benign tumor that is most common on the hallux.
- Plain radiographs can identify an exostosis and should be part of the initial workup of any subungual tumor.
- Surgical excision is an effective and well-tolerated treatment of subungual exostosis.
An Unusual Cause of Syncope With T-Wave Abnormalities
Case
A 34-year-old man presented to our ED via emergency medical services (EMS) following a syncopal episode. The patient stated that as he was getting ready for work earlier that morning, he experienced sudden lightheadedness and passed out, whereupon his wife immediately called EMS. The patient denied any previous history of syncope, but said he had been experiencing frequent episodes of nausea and vomiting over the past week. He also complained of a mild occipital headache that acetaminophen had failed to relieve.
The patient had been seen at a different ED 3 days earlier for nausea and vomiting. After evaluating the patient, the emergency physician (EP) at this facility felt the most likely cause of the patient’s gastrointestinal issues was related to hydralazine, his antihypertensive medication, and advised the patient to discontinue its use.
During evaluation at our ED, the patient denied fever, chills, neck stiffness, numbness, weakness, tingling of the extremities, or difficulty walking. He also denied chest pain, shortness of breath, or urinary symptoms. The patient’s medical history was significant only for hypertension; he had not taken any antihypertensive or other medications for the past 3 days, as previously instructed by the EP at the other ED. The patient denied alcohol or drug abuse.
On physical examination, the patient’s vital signs were: temperature, 98.6°F; heart rate, 58 beats/minute; blood pressure, 130/90 mm Hg; and respiratory rate, 16 breaths/minute. Oxygen saturation was 100% on room air. Examination of the head was normal and without evidence of trauma. Both pupils measured 4 mm and were equally round and reactive to light; the patient’s extraocular movements were intact. The remainder of the head, eyes, ears, nose, and throat examination was normal. The neck was supple, without masses or meningeal signs. The cardiopulmonary and abdominal examinations were all normal. On neurological examination, the patient was awake, alert, and oriented to person, place, and time. Cranial nerves II through XII were intact, and the patient had 5/5 motor strength in all four extremities and a normal gait.
Because we were concerned about the patient’s unexplained syncopal episode, we ordered laboratory tests, including a complete blood count (CBC), evaluation of electrolytes and glucose levels, and kidney function. In addition, we also ordered an electrocardiogram (ECG) and a noncontrast computed tomography (CT) scan of the head. All laboratory test results were within normal range. The ECG, however, demonstrated sinus bradycardia (approximately 58 beats/minute), a normal PR and QRS interval, a normal axis, and an incomplete right bundle branch block with tall, large, splayed upright T waves in the precordial leads (Figure). Based on the abnormal ECG results, we ordered serum cardiac marker studies, the values of which were all within normal range. The noncontrast CT scan of the head revealed a low-density posterior fossa mass compressing the fourth ventricle with secondary hydrocephalus.
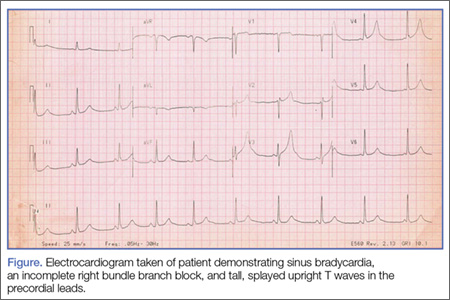
The patient was placed with his head in an upright position and given 1 g/kg mannitol and 10 mg dexamethasone intravenously (IV). Neurosurgery services were consulted, and the patient underwent surgery the following morning. Surgery confirmed the presence of a hemangioblastoma. The hemangioblastoma was successfully excised, and the patient had an uneventful recovery. Interestingly, the significant T-wave changes in the precordial leads were no longer present postoperatively.
Discussion
Syncope and near-syncope are common reasons for ED visits. Syncope is a syndrome characterized by a transient, self-limited episode of loss of consciousness occurring as a result of a brief interruption of the oxygen supply to the brain.1 This interruption is almost always due to a transient cessation of blood flow.1 In true syncope (as opposed to seizures or hypoglycemia), the episode is characterized by a rapid onset of loss of consciousness—with or without warning symptoms.1 It is important to determine the cause of syncope, because 7% to 23% of such patients will suffer serious outcomes within 7 to 30 days of their ED visit—either within a hospital setting or at home.2
Etiology
There are many causes of syncope. In most cases, the etiology falls under one of three broad categories: neurally mediated (or reflex mediated), orthostatic hypotensive-mediated, or cardiovascular (CV)-mediated. Less common causes of syncope include cerebrovascular injury.1 The Table outlines both common and uncommon causes of syncope.
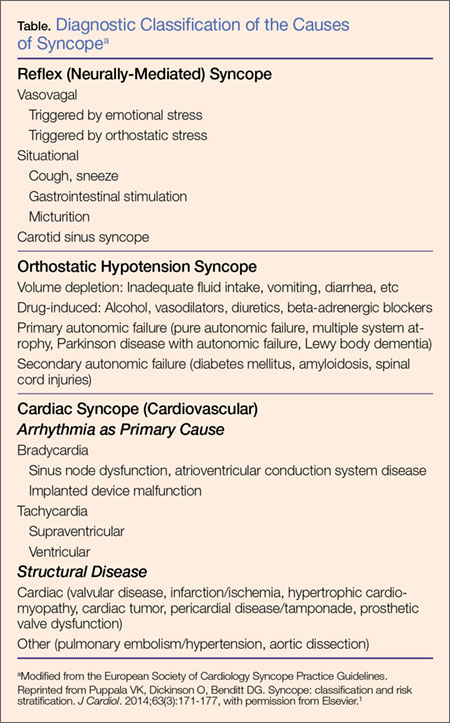
On presentation, our patient had several possible causes for his syncopal episode: an abnormal ECG (CV); multiple episodes of emesis (volume depletion); and headache (cerebrovascular). The EP worked up all three of these signs and symptoms simultaneously as is the appropriate protocol when evaluating an ED patient presenting with undifferentiated syncope.
Signs and Symptoms
Patients with undiagnosed brain tumors normally present with headache, seizures, nausea, vomiting, focal neurological deficits, or an altered mental status.3 Syncope is a very rare manifestation of a brain tumor3; however, our patient did complain of headache, nausea, and vomiting.
In addition to the unusual cause of the syncope, the abnormally large upright T waves make this case even more notable. T-wave changes are the most common ECG abnormality, seen in about 50% of abnormal tracings reviewed in a hospital population and in 2.4% of all ECGs.4
In general, T-wave changes are a result of local changes in the duration of repolarization. T-wave inversion is the most common T-wave abnormality and is typically observed in the setting of ischemia, post-ingestion of food, following an episode of tachycardia or anxiety, and autonomic dysfunction.5 However, in patients who have a cerebral etiology (usually hemorrhage), the T-wave changes may be either upright (as in our case) or inverted.5 Historically, subarachnoid hemorrhage (SAH) has been associated with ST-segment elevation and T-wave inversion. Hypothalamic stimulation and autonomic dysfunction have been linked to abnormal T-waves, but this has not been conclusively proven to be the cause of the abnormality.6 For all of the aforementioned reasons, the specificity for a given cause of T-wave changes is exceedingly low.5
Hyperacute T-wave amplitude, with prominent symmetrical T waves in at least two continuous leads, may be the earliest sign of acute transmural myocardial infarction (MI).7 It usually persists for only a brief time before other ECG findings of acute MI are observed. Other common causes of hyperacute T waves include hyperkalemia (usually narrow-based, and peaked), early repolarization, left ventricular hypertrophy, and acute myocarditis.8 Less common causes of prominent T waves include pre-excitation syndromes, pericarditis, and scorpion stings.9,10
Summary
It is unclear why our patient, who had a hemangioblastoma, demonstrated hyperacute T-wave abnormality on ECG. The abnormal upright T waves may have occurred secondary to the same theories regarding SAH, hypothalamic stimulation, or autonomic dysfunction. Regardless of the underlying etiology, this case serves as a reminder to the EP that not all T-wave changes on ECG are cardiac in origin.
1. Puppala VK, Dickinson O, Benditt DG. Syncope: classification and risk stratification. J Cardiol. 2014;63(3):171-177.
2. Thiruganasambandamoorthy V, Stiell IG, Sivilotti ML, et al. Risk stratification of adult emergency department syncope patients to predict short-term serious outcomes after discharge (RiSEDS) study. BMC Emerg Med. 2014;14(1):8.
3. van der Sluijs BM, Renier WO, Kappelle AC. Brain tumor as a rare cause of cardiac syncope. J Neurooncol. 2004;67(1-2):241-244.
4. Friedberg CK, Zagar A. Nonspecific ST and T-wave changes. Circulation. 1961;23:665-661.
5. Fisch C. T wave abnormalities due to extracardiac “functional” causes. ACC Curr J Rev. 1997;6(2):101-104.
6. Chatterjee S. ECG changes in subarachnoid hemorrhage: a synopsis. Neth Heart J. 2011;19(1):31-47.
7. Vojáčeka, J, Janskýb P, Janotac T. Third universal definition of myocardial infarction. Cor Vasa. 2013;55:e228-e235.
8. Levis JT. ECG diagnosis: hyperacute T waves. Perm J. 2015;19(3):79.
9. Somers MP, Brady WJ, Perron AD, Mattu A. The prominent T wave: electrocardiographic differential diagnosis. Am J Emerg Med. 2002;20(3):243-251.
10. Kumar MR, Bharath RV, Subrahmanyam BV, Rammohan P, Agrawal A. Scorpion envenomation and its management in adults. Sahel Med J. 2013;16(2):60-63.
Case
A 34-year-old man presented to our ED via emergency medical services (EMS) following a syncopal episode. The patient stated that as he was getting ready for work earlier that morning, he experienced sudden lightheadedness and passed out, whereupon his wife immediately called EMS. The patient denied any previous history of syncope, but said he had been experiencing frequent episodes of nausea and vomiting over the past week. He also complained of a mild occipital headache that acetaminophen had failed to relieve.
The patient had been seen at a different ED 3 days earlier for nausea and vomiting. After evaluating the patient, the emergency physician (EP) at this facility felt the most likely cause of the patient’s gastrointestinal issues was related to hydralazine, his antihypertensive medication, and advised the patient to discontinue its use.
During evaluation at our ED, the patient denied fever, chills, neck stiffness, numbness, weakness, tingling of the extremities, or difficulty walking. He also denied chest pain, shortness of breath, or urinary symptoms. The patient’s medical history was significant only for hypertension; he had not taken any antihypertensive or other medications for the past 3 days, as previously instructed by the EP at the other ED. The patient denied alcohol or drug abuse.
On physical examination, the patient’s vital signs were: temperature, 98.6°F; heart rate, 58 beats/minute; blood pressure, 130/90 mm Hg; and respiratory rate, 16 breaths/minute. Oxygen saturation was 100% on room air. Examination of the head was normal and without evidence of trauma. Both pupils measured 4 mm and were equally round and reactive to light; the patient’s extraocular movements were intact. The remainder of the head, eyes, ears, nose, and throat examination was normal. The neck was supple, without masses or meningeal signs. The cardiopulmonary and abdominal examinations were all normal. On neurological examination, the patient was awake, alert, and oriented to person, place, and time. Cranial nerves II through XII were intact, and the patient had 5/5 motor strength in all four extremities and a normal gait.
Because we were concerned about the patient’s unexplained syncopal episode, we ordered laboratory tests, including a complete blood count (CBC), evaluation of electrolytes and glucose levels, and kidney function. In addition, we also ordered an electrocardiogram (ECG) and a noncontrast computed tomography (CT) scan of the head. All laboratory test results were within normal range. The ECG, however, demonstrated sinus bradycardia (approximately 58 beats/minute), a normal PR and QRS interval, a normal axis, and an incomplete right bundle branch block with tall, large, splayed upright T waves in the precordial leads (Figure). Based on the abnormal ECG results, we ordered serum cardiac marker studies, the values of which were all within normal range. The noncontrast CT scan of the head revealed a low-density posterior fossa mass compressing the fourth ventricle with secondary hydrocephalus.
The patient was placed with his head in an upright position and given 1 g/kg mannitol and 10 mg dexamethasone intravenously (IV). Neurosurgery services were consulted, and the patient underwent surgery the following morning. Surgery confirmed the presence of a hemangioblastoma. The hemangioblastoma was successfully excised, and the patient had an uneventful recovery. Interestingly, the significant T-wave changes in the precordial leads were no longer present postoperatively.
Discussion
Syncope and near-syncope are common reasons for ED visits. Syncope is a syndrome characterized by a transient, self-limited episode of loss of consciousness occurring as a result of a brief interruption of the oxygen supply to the brain.1 This interruption is almost always due to a transient cessation of blood flow.1 In true syncope (as opposed to seizures or hypoglycemia), the episode is characterized by a rapid onset of loss of consciousness—with or without warning symptoms.1 It is important to determine the cause of syncope, because 7% to 23% of such patients will suffer serious outcomes within 7 to 30 days of their ED visit—either within a hospital setting or at home.2
Etiology
There are many causes of syncope. In most cases, the etiology falls under one of three broad categories: neurally mediated (or reflex mediated), orthostatic hypotensive-mediated, or cardiovascular (CV)-mediated. Less common causes of syncope include cerebrovascular injury.1 The Table outlines both common and uncommon causes of syncope.
On presentation, our patient had several possible causes for his syncopal episode: an abnormal ECG (CV); multiple episodes of emesis (volume depletion); and headache (cerebrovascular). The EP worked up all three of these signs and symptoms simultaneously as is the appropriate protocol when evaluating an ED patient presenting with undifferentiated syncope.
Signs and Symptoms
Patients with undiagnosed brain tumors normally present with headache, seizures, nausea, vomiting, focal neurological deficits, or an altered mental status.3 Syncope is a very rare manifestation of a brain tumor3; however, our patient did complain of headache, nausea, and vomiting.
In addition to the unusual cause of the syncope, the abnormally large upright T waves make this case even more notable. T-wave changes are the most common ECG abnormality, seen in about 50% of abnormal tracings reviewed in a hospital population and in 2.4% of all ECGs.4
In general, T-wave changes are a result of local changes in the duration of repolarization. T-wave inversion is the most common T-wave abnormality and is typically observed in the setting of ischemia, post-ingestion of food, following an episode of tachycardia or anxiety, and autonomic dysfunction.5 However, in patients who have a cerebral etiology (usually hemorrhage), the T-wave changes may be either upright (as in our case) or inverted.5 Historically, subarachnoid hemorrhage (SAH) has been associated with ST-segment elevation and T-wave inversion. Hypothalamic stimulation and autonomic dysfunction have been linked to abnormal T-waves, but this has not been conclusively proven to be the cause of the abnormality.6 For all of the aforementioned reasons, the specificity for a given cause of T-wave changes is exceedingly low.5
Hyperacute T-wave amplitude, with prominent symmetrical T waves in at least two continuous leads, may be the earliest sign of acute transmural myocardial infarction (MI).7 It usually persists for only a brief time before other ECG findings of acute MI are observed. Other common causes of hyperacute T waves include hyperkalemia (usually narrow-based, and peaked), early repolarization, left ventricular hypertrophy, and acute myocarditis.8 Less common causes of prominent T waves include pre-excitation syndromes, pericarditis, and scorpion stings.9,10
Summary
It is unclear why our patient, who had a hemangioblastoma, demonstrated hyperacute T-wave abnormality on ECG. The abnormal upright T waves may have occurred secondary to the same theories regarding SAH, hypothalamic stimulation, or autonomic dysfunction. Regardless of the underlying etiology, this case serves as a reminder to the EP that not all T-wave changes on ECG are cardiac in origin.
Case
A 34-year-old man presented to our ED via emergency medical services (EMS) following a syncopal episode. The patient stated that as he was getting ready for work earlier that morning, he experienced sudden lightheadedness and passed out, whereupon his wife immediately called EMS. The patient denied any previous history of syncope, but said he had been experiencing frequent episodes of nausea and vomiting over the past week. He also complained of a mild occipital headache that acetaminophen had failed to relieve.
The patient had been seen at a different ED 3 days earlier for nausea and vomiting. After evaluating the patient, the emergency physician (EP) at this facility felt the most likely cause of the patient’s gastrointestinal issues was related to hydralazine, his antihypertensive medication, and advised the patient to discontinue its use.
During evaluation at our ED, the patient denied fever, chills, neck stiffness, numbness, weakness, tingling of the extremities, or difficulty walking. He also denied chest pain, shortness of breath, or urinary symptoms. The patient’s medical history was significant only for hypertension; he had not taken any antihypertensive or other medications for the past 3 days, as previously instructed by the EP at the other ED. The patient denied alcohol or drug abuse.
On physical examination, the patient’s vital signs were: temperature, 98.6°F; heart rate, 58 beats/minute; blood pressure, 130/90 mm Hg; and respiratory rate, 16 breaths/minute. Oxygen saturation was 100% on room air. Examination of the head was normal and without evidence of trauma. Both pupils measured 4 mm and were equally round and reactive to light; the patient’s extraocular movements were intact. The remainder of the head, eyes, ears, nose, and throat examination was normal. The neck was supple, without masses or meningeal signs. The cardiopulmonary and abdominal examinations were all normal. On neurological examination, the patient was awake, alert, and oriented to person, place, and time. Cranial nerves II through XII were intact, and the patient had 5/5 motor strength in all four extremities and a normal gait.
Because we were concerned about the patient’s unexplained syncopal episode, we ordered laboratory tests, including a complete blood count (CBC), evaluation of electrolytes and glucose levels, and kidney function. In addition, we also ordered an electrocardiogram (ECG) and a noncontrast computed tomography (CT) scan of the head. All laboratory test results were within normal range. The ECG, however, demonstrated sinus bradycardia (approximately 58 beats/minute), a normal PR and QRS interval, a normal axis, and an incomplete right bundle branch block with tall, large, splayed upright T waves in the precordial leads (Figure). Based on the abnormal ECG results, we ordered serum cardiac marker studies, the values of which were all within normal range. The noncontrast CT scan of the head revealed a low-density posterior fossa mass compressing the fourth ventricle with secondary hydrocephalus.
The patient was placed with his head in an upright position and given 1 g/kg mannitol and 10 mg dexamethasone intravenously (IV). Neurosurgery services were consulted, and the patient underwent surgery the following morning. Surgery confirmed the presence of a hemangioblastoma. The hemangioblastoma was successfully excised, and the patient had an uneventful recovery. Interestingly, the significant T-wave changes in the precordial leads were no longer present postoperatively.
Discussion
Syncope and near-syncope are common reasons for ED visits. Syncope is a syndrome characterized by a transient, self-limited episode of loss of consciousness occurring as a result of a brief interruption of the oxygen supply to the brain.1 This interruption is almost always due to a transient cessation of blood flow.1 In true syncope (as opposed to seizures or hypoglycemia), the episode is characterized by a rapid onset of loss of consciousness—with or without warning symptoms.1 It is important to determine the cause of syncope, because 7% to 23% of such patients will suffer serious outcomes within 7 to 30 days of their ED visit—either within a hospital setting or at home.2
Etiology
There are many causes of syncope. In most cases, the etiology falls under one of three broad categories: neurally mediated (or reflex mediated), orthostatic hypotensive-mediated, or cardiovascular (CV)-mediated. Less common causes of syncope include cerebrovascular injury.1 The Table outlines both common and uncommon causes of syncope.
On presentation, our patient had several possible causes for his syncopal episode: an abnormal ECG (CV); multiple episodes of emesis (volume depletion); and headache (cerebrovascular). The EP worked up all three of these signs and symptoms simultaneously as is the appropriate protocol when evaluating an ED patient presenting with undifferentiated syncope.
Signs and Symptoms
Patients with undiagnosed brain tumors normally present with headache, seizures, nausea, vomiting, focal neurological deficits, or an altered mental status.3 Syncope is a very rare manifestation of a brain tumor3; however, our patient did complain of headache, nausea, and vomiting.
In addition to the unusual cause of the syncope, the abnormally large upright T waves make this case even more notable. T-wave changes are the most common ECG abnormality, seen in about 50% of abnormal tracings reviewed in a hospital population and in 2.4% of all ECGs.4
In general, T-wave changes are a result of local changes in the duration of repolarization. T-wave inversion is the most common T-wave abnormality and is typically observed in the setting of ischemia, post-ingestion of food, following an episode of tachycardia or anxiety, and autonomic dysfunction.5 However, in patients who have a cerebral etiology (usually hemorrhage), the T-wave changes may be either upright (as in our case) or inverted.5 Historically, subarachnoid hemorrhage (SAH) has been associated with ST-segment elevation and T-wave inversion. Hypothalamic stimulation and autonomic dysfunction have been linked to abnormal T-waves, but this has not been conclusively proven to be the cause of the abnormality.6 For all of the aforementioned reasons, the specificity for a given cause of T-wave changes is exceedingly low.5
Hyperacute T-wave amplitude, with prominent symmetrical T waves in at least two continuous leads, may be the earliest sign of acute transmural myocardial infarction (MI).7 It usually persists for only a brief time before other ECG findings of acute MI are observed. Other common causes of hyperacute T waves include hyperkalemia (usually narrow-based, and peaked), early repolarization, left ventricular hypertrophy, and acute myocarditis.8 Less common causes of prominent T waves include pre-excitation syndromes, pericarditis, and scorpion stings.9,10
Summary
It is unclear why our patient, who had a hemangioblastoma, demonstrated hyperacute T-wave abnormality on ECG. The abnormal upright T waves may have occurred secondary to the same theories regarding SAH, hypothalamic stimulation, or autonomic dysfunction. Regardless of the underlying etiology, this case serves as a reminder to the EP that not all T-wave changes on ECG are cardiac in origin.
1. Puppala VK, Dickinson O, Benditt DG. Syncope: classification and risk stratification. J Cardiol. 2014;63(3):171-177.
2. Thiruganasambandamoorthy V, Stiell IG, Sivilotti ML, et al. Risk stratification of adult emergency department syncope patients to predict short-term serious outcomes after discharge (RiSEDS) study. BMC Emerg Med. 2014;14(1):8.
3. van der Sluijs BM, Renier WO, Kappelle AC. Brain tumor as a rare cause of cardiac syncope. J Neurooncol. 2004;67(1-2):241-244.
4. Friedberg CK, Zagar A. Nonspecific ST and T-wave changes. Circulation. 1961;23:665-661.
5. Fisch C. T wave abnormalities due to extracardiac “functional” causes. ACC Curr J Rev. 1997;6(2):101-104.
6. Chatterjee S. ECG changes in subarachnoid hemorrhage: a synopsis. Neth Heart J. 2011;19(1):31-47.
7. Vojáčeka, J, Janskýb P, Janotac T. Third universal definition of myocardial infarction. Cor Vasa. 2013;55:e228-e235.
8. Levis JT. ECG diagnosis: hyperacute T waves. Perm J. 2015;19(3):79.
9. Somers MP, Brady WJ, Perron AD, Mattu A. The prominent T wave: electrocardiographic differential diagnosis. Am J Emerg Med. 2002;20(3):243-251.
10. Kumar MR, Bharath RV, Subrahmanyam BV, Rammohan P, Agrawal A. Scorpion envenomation and its management in adults. Sahel Med J. 2013;16(2):60-63.
1. Puppala VK, Dickinson O, Benditt DG. Syncope: classification and risk stratification. J Cardiol. 2014;63(3):171-177.
2. Thiruganasambandamoorthy V, Stiell IG, Sivilotti ML, et al. Risk stratification of adult emergency department syncope patients to predict short-term serious outcomes after discharge (RiSEDS) study. BMC Emerg Med. 2014;14(1):8.
3. van der Sluijs BM, Renier WO, Kappelle AC. Brain tumor as a rare cause of cardiac syncope. J Neurooncol. 2004;67(1-2):241-244.
4. Friedberg CK, Zagar A. Nonspecific ST and T-wave changes. Circulation. 1961;23:665-661.
5. Fisch C. T wave abnormalities due to extracardiac “functional” causes. ACC Curr J Rev. 1997;6(2):101-104.
6. Chatterjee S. ECG changes in subarachnoid hemorrhage: a synopsis. Neth Heart J. 2011;19(1):31-47.
7. Vojáčeka, J, Janskýb P, Janotac T. Third universal definition of myocardial infarction. Cor Vasa. 2013;55:e228-e235.
8. Levis JT. ECG diagnosis: hyperacute T waves. Perm J. 2015;19(3):79.
9. Somers MP, Brady WJ, Perron AD, Mattu A. The prominent T wave: electrocardiographic differential diagnosis. Am J Emerg Med. 2002;20(3):243-251.
10. Kumar MR, Bharath RV, Subrahmanyam BV, Rammohan P, Agrawal A. Scorpion envenomation and its management in adults. Sahel Med J. 2013;16(2):60-63.
A Rare Case of Traumatic Tension Pneumo-orbitum
Traumatic eye injuries ranging from mild corneal abrasions to penetrating globe injuries are commonly seen in the ED, and ocular trauma accounts for nearly 1% of all complaints in the ED.1 Up to 29% of facial fractures have associated eye injuries.2 Emergency physicians (EPs) must be aware of possible eye injuries, including traumatic vision loss, and the indicators for emergent interventions.
Tension pneumo-orbitum following facial trauma is rarely reported. We present a case of orbital compartment syndrome (OCS) in an elderly woman who sustained an orbital floor fracture and required emergent lateral canthotomy to preserve vision.
Case
A 76-year-old woman presented to the ED for evaluation of pain, swelling, and loss of vision in the right eye. She said she had been sitting in a chair tying her shoes when she lost her balance and fell forward, striking her head and the right side of her face against the floor. She experienced no loss of consciousness and denied any neck pain, jaw pain, or dizziness. She also denied any chest pain, shortness of breath, weakness, or loss of function in either her arms or legs. She did, however, note a small nosebleed that had stopped before she arrived at the ED. The patient’s primary complaint was a possible nasal bone fracture.
Her medical history was significant for hypertension and coronary artery disease. Her medications include amitriptyline, an antihistamine, aspirin, clopidogrel, diltiazem, folic acid, furosemide, hydralazine, levothyroxine, prednisone, and zolpidem. She stated that she was allergic to amoxicillin and sulfa drugs.
The patient’s vital signs at presentation were: blood pressure (BP), 193/82 mm Hg; heart rate, 71 beats/minute; respiratory rate, 16 breaths/minute; and temperature, 97°F. She was alert, oriented, and in no distress. Her head and neck examination showed no scalp lacerations or swelling. There was, however, significant swelling and ecchymosis around the right eye and swelling and ecchymosis around the nose, with dried blood in both nares. No septal hematoma was present. The patient had tenderness to palpation over the infraorbital area and nose. No gross facial instability was present, and Battle sign was not appreciated. No jaw or dental abnormalities were noted.
The patient’s right pupil was fixed and dilated, and she could not perceive light. She did have upward and lateral movement of the eye, but was unable to look down. A minimal amount of proptosis was noted. Her intraocular pressure (IOP) was elevated at 54 mm Hg (normal range, 10-20 mm Hg). The remainder of the examination, including the neurological examination, was unremarkable.
The patient received emergent head and facial computed tomography (CT) scans. The head CT showed no acute intracranial hemorrhage, mass, or infarct. The facial CT was read as a right orbital floor fracture with intraorbital air, and a right maxillary sinus hematoma. Laboratory evaluation revealed a hematocrit of 38% and a platelet count of 544,000/mcL (normal range, 150,000-450,000/mcL). The prothrombin time was 10.9 seconds (normal range, 11-13.5 seconds); the international normalized ratio was 0.8 (normal range, 0.8-1.1); and the partial thromboplastin time was 22.5 seconds (normal range, 25-35 seconds).
Because the patient was at risk for permanent visual impairment due to increased IOP from the injury, a lateral canthotomy was immediately performed. A small amount of air was released, and the proptosis was notably reduced.
At this point, the ophthalmologist arrived and used an 18-gauge needle to explore the retrobulbar space. Two pockets of air were released, which markedly reduced the tactile pressure of the globe. Repeat tonography of the globe was 28 mm Hg. The wound was left open to drain, and the patient was started on azithromycin. She was discharged home to follow up with ophthalmology.
The patient presented to the ED 2 months later for an unrelated condition. At that time, she reported a complete return of her vision with no deficits and no noticeable scarring around the eye.
Discussion
The orbit is an enclosed space, bordered by bone laterally and posteriorly—the orbital septa superiorly and inferiorly, and the globe anteriorly.3 The lateral canthus is a combined tendon-ligament that helps attach the tarsal plates of the lids and the orbicularis oculi muscles to the lateral orbital wall and zygoma, which forms the posterior orbital wall.3,4 The lateral canthal tendon is located beneath the lateral canthus and is comprised of the inferior and superior crus, which attaches to the inner aspect of the lateral orbital wall, forming a structure called Whitnall’s tubercle.3,4
Other than globe injuries, the most common findings in patients with orbital trauma are periocular lacerations (96%), orbital fractures (16%), and retrobulbar hemorrhage (8%).5 The most common cause of retrobulbar hemorrhage is ocular trauma, but it is also observed in facial fractures, orbital surgery, retrobulbar injections, venous anomalies, atherosclerosis, intraorbital aneurysm of the ophthalmic artery, lacerated ophthalmic artery, hypertension, hemophilia, leukemia, von Willebrand disease, and straining.3,6,7
In retrobulbar hemorrhage, an increased pressure in the orbital space can lead to optic nerve compression and vascular compromise.6 Important alternative diagnoses to consider include orbital cellulitis, orbital fracture, and globe rupture.3 Retrobulbar hemorrhage should be suspected in the clinical setting of exophthalmos, proptosis, diffuse subconjunctival hemorrhage, pain, visual loss or diplopia, periorbital edema, partial or complete ophthalmoplegia, resistance to retropulsion, increased IOP, a blanched ophthalmic artery on funduscopic examination, and an afferent pupillary defect.3,4,8,9 Less commonly, periorbital crepitus and infraorbital hypoesthesia can be appreciated.3
If a patient with a retrobulbar hemorrhage is experiencing diminished vision, an emergent lateral canthotomy should be attempted. Retrobulbar hemorrhage can be difficult to diagnose in the setting of trauma. There can be damage to the optic nerve with associated edema and vision loss that is not associated with a retrobulbar hemorrhage and does not require a lateral canthotomy.3,7 A dedicated CT scan of the orbits can aid in the diagnosis, but treatment should not be delayed.8
Patients with retrobulbar hemorrhage may initially present to the ED with intact visual acuity, but as the pressure behind the globe increases, vision will diminish.3 Although the medical literature has not established a definitive timeframe, it is believed that permanent visual compromise develops between 1 to 3 hours after ischemia develops.6 Animal studies show that visual loss due to central retinal artery ischemia may be reversible up to 100 minutes.3
Not all cases of retrobulbar hemorrhage are associated with vision loss. In patients without diminished vision, conservative treatments such as bed rest, elevation of the head of the bed, ice packs, analgesia, lowering BP, and sedatives should be attempted first.5 Acetazolamide and mannitol can also be considered in consultation with an ophthalmologist.
Pneumo-orbitum
The presence of pneumo-orbitum should alert the clinician to either a communication with a paranasal sinus, a gas-forming organism, or (rarely) Munchausen syndrome.10 Unlike most case presentations, most causes of pneumo-orbitum do not involve OCS and are self-limited.11 Traumatic pneumo-orbitum without OCS has been reported in the literature.12-15 However, traumatic tension pneumo-orbitum is rare.12-16 One case report involved an elderly man with an orbital floor fracture who developed recurrent tension pneumo-orbitum after blowing his nose while intoxicated.12 Another case involved a boy with tension pneumo-orbitum that required surgical decompression.16
In a patient who has experienced trauma, the combination of proptosis, elevated IOP, and vision loss likely represent a retrobulbar hematoma or OCS. A lateral canthotomy can help relieve IOP from either condition.17,18 Orbital compartment syndrome can be caused by edema, emphysema, and caroticocavernous fistula, leading to increased orbital pressure and decreased perfusion.17,18
In a review of 10 trauma patients with OCS, all cases were intubated due to the severity of the head trauma, and all had OCS due to edema.17 In a review of eight trauma patients with OCS, all had eye pain, reduced visual acuity, and proptosis.18 Most of the patients had periorbital edema, ophthalmoparesis, a relative afferent pupillary defect (as compared to a fixed and dilated pupil), and chemosis.18,19 All of the patients with OCS required cantholysis or a lateral canthotomy.18
Lateral Canthotomy
Although EPs rarely perform lateral canthotomy, knowledge of this procedure is important, because it can prevent vision loss in the appropriate clinical setting. To perform a lateral canthotomy, the area around the affected eye is cleaned with saline irrigation.4 One percent or 2% lidocaine with epinephrine is then injected into the lateral canthus of the affected eye.4,10 A straight hemostat is applied between the upper and lower lids, producing a crush injury along the site of local anesthesia for 1 to 2 minutes.3,4,10 This is done to reduce the risk of bleeding by devitalizing the tissue.4 Straight scissors are then used to make a 1-cm horizontal incision from the lateral canthal tendon to the lateral orbital rim.4 This initial incision exposes the orbicularis muscle, orbital septum, palpebral conjunctiva, and an area called Eisler’s pocket that sits anterior to the lateral canthal tendon.3
Cantholysis can then be performed by blunt dissection.10 The inferior crus of the lateral canthus is identified either visually or by palpation, and a 1- to 2-cm inferior-posterior cut of the inferior crus accomplishes the lateral canthotomy.3-4 After cutting the inferior crus, the lower lid should be pulled away easily, and if this does not occur, repeated attempts at cutting the inferior crus should be made.3 Pulling the lower eyelid down and away from the lateral orbital rim separates the skin and conjunctiva, aiding in visualization.4
After cutting the inferior crus, only a small amount of blood or air typically is expressed, but this is usually enough to prevent vision loss.3 When the procedure is performed correctly, the practitioner should be able to palpate a difference in the pressure of the globe, and tonography will show a reduced IOP. If the ocular pressure is still significantly elevated, the physician can proceed to cut the superior canthus of the lateral canthal tendon in a manner similar to cutting the inferior crus of the tendon.4 After the procedure is performed, urgent ophthalmologic consultation is required.
The risks of performing a lateral canthotomy include mechanical injury, hemorrhage, and infection.4 The incision from a lateral canthotomy generally does not need suturing and will heal without significant scarring.4 If the scissors are aimed superiorly instead of inferiorly for the inferior crus of the lateral canthal tendon, there is risk of injuring the levator aponeurosis leading to ptosis, as well as a small risk of injury to the lacrimal gland and lacrimal artery.3
Conclusion
Our patient demonstrates a case of traumatic OCS, a vision-threatening medical condition that requires rapid diagnosis and lateral canthotomy to lower IOP and reduce the risk of permanent vision loss. While an orbital CT scan may assist in confirming the diagnosis, treatment of IOP should not be delayed.
1. McCaig LF, Burt CW. National Hospital Ambulatory Medical Care Survey: 2001 Emergency Department Summary. Advance Data from Vital Health and Statistics; No. 335. https://www.cdc.gov/nchs/data/ad/ad335.pdf. Accessed July 22, 2016.
2. Knoop KJ, Dennis WR. Eye trauma. In: Wolfson AB, Hendy GW, Hendy PL, et al (eds). Harwood-Nuss’ Clinical Practice of Emergency Medicine. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2005.
3. Vassallo S, Hartstein M, Howard D, Stetz J. Traumatic retrobulbar hemorrhage: emergent decompression by lateral canthotomy and cantholysis. J Emerg Med. 2002;22(3):251-256.
4. Roberts JR, Hedges JR (eds). Clinical Procedures in Emergency Medicine, 4th ed. Philadelphia, PA: Saunders; 2004.
5. Hatton MP, Thakker MM, Ray S. Orbital and adnexal trauma associated with open-globe injuries. Ophthal Plast Reconstr Surg. 2002;18(6):458-461.
6. Suner S, Simmons W, Savitt DL. A porcine model for instruction of lateral canthotomy. Acad Emerg Med. 2000;7(7):837-838.
7. Goodall KL, Brahma A, Bates A, Leatherbarrow B. Lateral canthotomy and inferior cantholysis: an effective method of urgent orbital decompression for sight threatening acute retrobulbar hemorrhage. Injury. 1999;30(7):485-490.
8. Gerbino G, Ramieri GA, Nasi A. Diagnosis and treatment of retrobulbar haematomas following blunt orbital trauma: a description of eight cases. Int J Oral Maxillofac Surg. 2005;34(2):127-131.
9. Machado RA, Silveira RL, Borges HO, Filho AM, de Oliveira GM. Retrobulbar hemorrhage: A case report. J Contemp Dent Pract. 2006;7(2):130-136.
10. Winans JM, House LR, Robinson HE. Self-induced orbital emphysema as a presenting sign of Munchausen’s syndrome. Laryngoscope. 1983;93(9):1209-1211.
11. Zimmer-Galler IE, Bartley GB. Orbital emphysema: case reports and review of the literature. Mayo Clin Proc. 1994;69(2):115-121.
12. Ahnood D, Toft PB. Recurrent orbital compartment syndrome caused by a blow-out fracture and accumulation of air; management by orbital punctures. Acta Ophthalmol. 2012;90(12):199-200.
13. Martin PW, Williams AC. Supraorbital emphysema: report of a case. J Oral Surg. 1972;30(12):901-902.
14. Per BL, Sanders BB. Post-traumatic intraorbital pneumatocele--a rare case of unilateral exophthalmos. Br J Radio. 1971;44(519):214-215.
15. Haller ML, Brackup AH, Shiffman F. Intraorbital aerocele. Arch Ophthalmol. 1980;98(9):1612-1613.
16. Chaudhry IA, Al-Amri A, Shamsi FA, Al-Rashed W. Visual recovery after evacuation of orbital emphysema. Orbit. 2007;26(4):283-285.
17. Perry M. Acute proptosis in trauma: retrobulbar hemorrhage or orbital compartment syndrome—does it really matter? J Oral Maxillofac Surg. 2008;66(9):1913-1920.
18. Sun MT, Chan WO, Selva D. Traumatic orbital compartment syndrome: importance of the lateral canthotomy and cantholysis. Emerg Med Australas. 2014;26(3):274-278.
19. Belliveau MJ, Johnson D. Orbital compartment syndrome after head trauma. Lancet Neurol. 2015;14(2):136-137.
Traumatic eye injuries ranging from mild corneal abrasions to penetrating globe injuries are commonly seen in the ED, and ocular trauma accounts for nearly 1% of all complaints in the ED.1 Up to 29% of facial fractures have associated eye injuries.2 Emergency physicians (EPs) must be aware of possible eye injuries, including traumatic vision loss, and the indicators for emergent interventions.
Tension pneumo-orbitum following facial trauma is rarely reported. We present a case of orbital compartment syndrome (OCS) in an elderly woman who sustained an orbital floor fracture and required emergent lateral canthotomy to preserve vision.
Case
A 76-year-old woman presented to the ED for evaluation of pain, swelling, and loss of vision in the right eye. She said she had been sitting in a chair tying her shoes when she lost her balance and fell forward, striking her head and the right side of her face against the floor. She experienced no loss of consciousness and denied any neck pain, jaw pain, or dizziness. She also denied any chest pain, shortness of breath, weakness, or loss of function in either her arms or legs. She did, however, note a small nosebleed that had stopped before she arrived at the ED. The patient’s primary complaint was a possible nasal bone fracture.
Her medical history was significant for hypertension and coronary artery disease. Her medications include amitriptyline, an antihistamine, aspirin, clopidogrel, diltiazem, folic acid, furosemide, hydralazine, levothyroxine, prednisone, and zolpidem. She stated that she was allergic to amoxicillin and sulfa drugs.
The patient’s vital signs at presentation were: blood pressure (BP), 193/82 mm Hg; heart rate, 71 beats/minute; respiratory rate, 16 breaths/minute; and temperature, 97°F. She was alert, oriented, and in no distress. Her head and neck examination showed no scalp lacerations or swelling. There was, however, significant swelling and ecchymosis around the right eye and swelling and ecchymosis around the nose, with dried blood in both nares. No septal hematoma was present. The patient had tenderness to palpation over the infraorbital area and nose. No gross facial instability was present, and Battle sign was not appreciated. No jaw or dental abnormalities were noted.
The patient’s right pupil was fixed and dilated, and she could not perceive light. She did have upward and lateral movement of the eye, but was unable to look down. A minimal amount of proptosis was noted. Her intraocular pressure (IOP) was elevated at 54 mm Hg (normal range, 10-20 mm Hg). The remainder of the examination, including the neurological examination, was unremarkable.
The patient received emergent head and facial computed tomography (CT) scans. The head CT showed no acute intracranial hemorrhage, mass, or infarct. The facial CT was read as a right orbital floor fracture with intraorbital air, and a right maxillary sinus hematoma. Laboratory evaluation revealed a hematocrit of 38% and a platelet count of 544,000/mcL (normal range, 150,000-450,000/mcL). The prothrombin time was 10.9 seconds (normal range, 11-13.5 seconds); the international normalized ratio was 0.8 (normal range, 0.8-1.1); and the partial thromboplastin time was 22.5 seconds (normal range, 25-35 seconds).
Because the patient was at risk for permanent visual impairment due to increased IOP from the injury, a lateral canthotomy was immediately performed. A small amount of air was released, and the proptosis was notably reduced.
At this point, the ophthalmologist arrived and used an 18-gauge needle to explore the retrobulbar space. Two pockets of air were released, which markedly reduced the tactile pressure of the globe. Repeat tonography of the globe was 28 mm Hg. The wound was left open to drain, and the patient was started on azithromycin. She was discharged home to follow up with ophthalmology.
The patient presented to the ED 2 months later for an unrelated condition. At that time, she reported a complete return of her vision with no deficits and no noticeable scarring around the eye.
Discussion
The orbit is an enclosed space, bordered by bone laterally and posteriorly—the orbital septa superiorly and inferiorly, and the globe anteriorly.3 The lateral canthus is a combined tendon-ligament that helps attach the tarsal plates of the lids and the orbicularis oculi muscles to the lateral orbital wall and zygoma, which forms the posterior orbital wall.3,4 The lateral canthal tendon is located beneath the lateral canthus and is comprised of the inferior and superior crus, which attaches to the inner aspect of the lateral orbital wall, forming a structure called Whitnall’s tubercle.3,4
Other than globe injuries, the most common findings in patients with orbital trauma are periocular lacerations (96%), orbital fractures (16%), and retrobulbar hemorrhage (8%).5 The most common cause of retrobulbar hemorrhage is ocular trauma, but it is also observed in facial fractures, orbital surgery, retrobulbar injections, venous anomalies, atherosclerosis, intraorbital aneurysm of the ophthalmic artery, lacerated ophthalmic artery, hypertension, hemophilia, leukemia, von Willebrand disease, and straining.3,6,7
In retrobulbar hemorrhage, an increased pressure in the orbital space can lead to optic nerve compression and vascular compromise.6 Important alternative diagnoses to consider include orbital cellulitis, orbital fracture, and globe rupture.3 Retrobulbar hemorrhage should be suspected in the clinical setting of exophthalmos, proptosis, diffuse subconjunctival hemorrhage, pain, visual loss or diplopia, periorbital edema, partial or complete ophthalmoplegia, resistance to retropulsion, increased IOP, a blanched ophthalmic artery on funduscopic examination, and an afferent pupillary defect.3,4,8,9 Less commonly, periorbital crepitus and infraorbital hypoesthesia can be appreciated.3
If a patient with a retrobulbar hemorrhage is experiencing diminished vision, an emergent lateral canthotomy should be attempted. Retrobulbar hemorrhage can be difficult to diagnose in the setting of trauma. There can be damage to the optic nerve with associated edema and vision loss that is not associated with a retrobulbar hemorrhage and does not require a lateral canthotomy.3,7 A dedicated CT scan of the orbits can aid in the diagnosis, but treatment should not be delayed.8
Patients with retrobulbar hemorrhage may initially present to the ED with intact visual acuity, but as the pressure behind the globe increases, vision will diminish.3 Although the medical literature has not established a definitive timeframe, it is believed that permanent visual compromise develops between 1 to 3 hours after ischemia develops.6 Animal studies show that visual loss due to central retinal artery ischemia may be reversible up to 100 minutes.3
Not all cases of retrobulbar hemorrhage are associated with vision loss. In patients without diminished vision, conservative treatments such as bed rest, elevation of the head of the bed, ice packs, analgesia, lowering BP, and sedatives should be attempted first.5 Acetazolamide and mannitol can also be considered in consultation with an ophthalmologist.
Pneumo-orbitum
The presence of pneumo-orbitum should alert the clinician to either a communication with a paranasal sinus, a gas-forming organism, or (rarely) Munchausen syndrome.10 Unlike most case presentations, most causes of pneumo-orbitum do not involve OCS and are self-limited.11 Traumatic pneumo-orbitum without OCS has been reported in the literature.12-15 However, traumatic tension pneumo-orbitum is rare.12-16 One case report involved an elderly man with an orbital floor fracture who developed recurrent tension pneumo-orbitum after blowing his nose while intoxicated.12 Another case involved a boy with tension pneumo-orbitum that required surgical decompression.16
In a patient who has experienced trauma, the combination of proptosis, elevated IOP, and vision loss likely represent a retrobulbar hematoma or OCS. A lateral canthotomy can help relieve IOP from either condition.17,18 Orbital compartment syndrome can be caused by edema, emphysema, and caroticocavernous fistula, leading to increased orbital pressure and decreased perfusion.17,18
In a review of 10 trauma patients with OCS, all cases were intubated due to the severity of the head trauma, and all had OCS due to edema.17 In a review of eight trauma patients with OCS, all had eye pain, reduced visual acuity, and proptosis.18 Most of the patients had periorbital edema, ophthalmoparesis, a relative afferent pupillary defect (as compared to a fixed and dilated pupil), and chemosis.18,19 All of the patients with OCS required cantholysis or a lateral canthotomy.18
Lateral Canthotomy
Although EPs rarely perform lateral canthotomy, knowledge of this procedure is important, because it can prevent vision loss in the appropriate clinical setting. To perform a lateral canthotomy, the area around the affected eye is cleaned with saline irrigation.4 One percent or 2% lidocaine with epinephrine is then injected into the lateral canthus of the affected eye.4,10 A straight hemostat is applied between the upper and lower lids, producing a crush injury along the site of local anesthesia for 1 to 2 minutes.3,4,10 This is done to reduce the risk of bleeding by devitalizing the tissue.4 Straight scissors are then used to make a 1-cm horizontal incision from the lateral canthal tendon to the lateral orbital rim.4 This initial incision exposes the orbicularis muscle, orbital septum, palpebral conjunctiva, and an area called Eisler’s pocket that sits anterior to the lateral canthal tendon.3
Cantholysis can then be performed by blunt dissection.10 The inferior crus of the lateral canthus is identified either visually or by palpation, and a 1- to 2-cm inferior-posterior cut of the inferior crus accomplishes the lateral canthotomy.3-4 After cutting the inferior crus, the lower lid should be pulled away easily, and if this does not occur, repeated attempts at cutting the inferior crus should be made.3 Pulling the lower eyelid down and away from the lateral orbital rim separates the skin and conjunctiva, aiding in visualization.4
After cutting the inferior crus, only a small amount of blood or air typically is expressed, but this is usually enough to prevent vision loss.3 When the procedure is performed correctly, the practitioner should be able to palpate a difference in the pressure of the globe, and tonography will show a reduced IOP. If the ocular pressure is still significantly elevated, the physician can proceed to cut the superior canthus of the lateral canthal tendon in a manner similar to cutting the inferior crus of the tendon.4 After the procedure is performed, urgent ophthalmologic consultation is required.
The risks of performing a lateral canthotomy include mechanical injury, hemorrhage, and infection.4 The incision from a lateral canthotomy generally does not need suturing and will heal without significant scarring.4 If the scissors are aimed superiorly instead of inferiorly for the inferior crus of the lateral canthal tendon, there is risk of injuring the levator aponeurosis leading to ptosis, as well as a small risk of injury to the lacrimal gland and lacrimal artery.3
Conclusion
Our patient demonstrates a case of traumatic OCS, a vision-threatening medical condition that requires rapid diagnosis and lateral canthotomy to lower IOP and reduce the risk of permanent vision loss. While an orbital CT scan may assist in confirming the diagnosis, treatment of IOP should not be delayed.
Traumatic eye injuries ranging from mild corneal abrasions to penetrating globe injuries are commonly seen in the ED, and ocular trauma accounts for nearly 1% of all complaints in the ED.1 Up to 29% of facial fractures have associated eye injuries.2 Emergency physicians (EPs) must be aware of possible eye injuries, including traumatic vision loss, and the indicators for emergent interventions.
Tension pneumo-orbitum following facial trauma is rarely reported. We present a case of orbital compartment syndrome (OCS) in an elderly woman who sustained an orbital floor fracture and required emergent lateral canthotomy to preserve vision.
Case
A 76-year-old woman presented to the ED for evaluation of pain, swelling, and loss of vision in the right eye. She said she had been sitting in a chair tying her shoes when she lost her balance and fell forward, striking her head and the right side of her face against the floor. She experienced no loss of consciousness and denied any neck pain, jaw pain, or dizziness. She also denied any chest pain, shortness of breath, weakness, or loss of function in either her arms or legs. She did, however, note a small nosebleed that had stopped before she arrived at the ED. The patient’s primary complaint was a possible nasal bone fracture.
Her medical history was significant for hypertension and coronary artery disease. Her medications include amitriptyline, an antihistamine, aspirin, clopidogrel, diltiazem, folic acid, furosemide, hydralazine, levothyroxine, prednisone, and zolpidem. She stated that she was allergic to amoxicillin and sulfa drugs.
The patient’s vital signs at presentation were: blood pressure (BP), 193/82 mm Hg; heart rate, 71 beats/minute; respiratory rate, 16 breaths/minute; and temperature, 97°F. She was alert, oriented, and in no distress. Her head and neck examination showed no scalp lacerations or swelling. There was, however, significant swelling and ecchymosis around the right eye and swelling and ecchymosis around the nose, with dried blood in both nares. No septal hematoma was present. The patient had tenderness to palpation over the infraorbital area and nose. No gross facial instability was present, and Battle sign was not appreciated. No jaw or dental abnormalities were noted.
The patient’s right pupil was fixed and dilated, and she could not perceive light. She did have upward and lateral movement of the eye, but was unable to look down. A minimal amount of proptosis was noted. Her intraocular pressure (IOP) was elevated at 54 mm Hg (normal range, 10-20 mm Hg). The remainder of the examination, including the neurological examination, was unremarkable.
The patient received emergent head and facial computed tomography (CT) scans. The head CT showed no acute intracranial hemorrhage, mass, or infarct. The facial CT was read as a right orbital floor fracture with intraorbital air, and a right maxillary sinus hematoma. Laboratory evaluation revealed a hematocrit of 38% and a platelet count of 544,000/mcL (normal range, 150,000-450,000/mcL). The prothrombin time was 10.9 seconds (normal range, 11-13.5 seconds); the international normalized ratio was 0.8 (normal range, 0.8-1.1); and the partial thromboplastin time was 22.5 seconds (normal range, 25-35 seconds).
Because the patient was at risk for permanent visual impairment due to increased IOP from the injury, a lateral canthotomy was immediately performed. A small amount of air was released, and the proptosis was notably reduced.
At this point, the ophthalmologist arrived and used an 18-gauge needle to explore the retrobulbar space. Two pockets of air were released, which markedly reduced the tactile pressure of the globe. Repeat tonography of the globe was 28 mm Hg. The wound was left open to drain, and the patient was started on azithromycin. She was discharged home to follow up with ophthalmology.
The patient presented to the ED 2 months later for an unrelated condition. At that time, she reported a complete return of her vision with no deficits and no noticeable scarring around the eye.
Discussion
The orbit is an enclosed space, bordered by bone laterally and posteriorly—the orbital septa superiorly and inferiorly, and the globe anteriorly.3 The lateral canthus is a combined tendon-ligament that helps attach the tarsal plates of the lids and the orbicularis oculi muscles to the lateral orbital wall and zygoma, which forms the posterior orbital wall.3,4 The lateral canthal tendon is located beneath the lateral canthus and is comprised of the inferior and superior crus, which attaches to the inner aspect of the lateral orbital wall, forming a structure called Whitnall’s tubercle.3,4
Other than globe injuries, the most common findings in patients with orbital trauma are periocular lacerations (96%), orbital fractures (16%), and retrobulbar hemorrhage (8%).5 The most common cause of retrobulbar hemorrhage is ocular trauma, but it is also observed in facial fractures, orbital surgery, retrobulbar injections, venous anomalies, atherosclerosis, intraorbital aneurysm of the ophthalmic artery, lacerated ophthalmic artery, hypertension, hemophilia, leukemia, von Willebrand disease, and straining.3,6,7
In retrobulbar hemorrhage, an increased pressure in the orbital space can lead to optic nerve compression and vascular compromise.6 Important alternative diagnoses to consider include orbital cellulitis, orbital fracture, and globe rupture.3 Retrobulbar hemorrhage should be suspected in the clinical setting of exophthalmos, proptosis, diffuse subconjunctival hemorrhage, pain, visual loss or diplopia, periorbital edema, partial or complete ophthalmoplegia, resistance to retropulsion, increased IOP, a blanched ophthalmic artery on funduscopic examination, and an afferent pupillary defect.3,4,8,9 Less commonly, periorbital crepitus and infraorbital hypoesthesia can be appreciated.3
If a patient with a retrobulbar hemorrhage is experiencing diminished vision, an emergent lateral canthotomy should be attempted. Retrobulbar hemorrhage can be difficult to diagnose in the setting of trauma. There can be damage to the optic nerve with associated edema and vision loss that is not associated with a retrobulbar hemorrhage and does not require a lateral canthotomy.3,7 A dedicated CT scan of the orbits can aid in the diagnosis, but treatment should not be delayed.8
Patients with retrobulbar hemorrhage may initially present to the ED with intact visual acuity, but as the pressure behind the globe increases, vision will diminish.3 Although the medical literature has not established a definitive timeframe, it is believed that permanent visual compromise develops between 1 to 3 hours after ischemia develops.6 Animal studies show that visual loss due to central retinal artery ischemia may be reversible up to 100 minutes.3
Not all cases of retrobulbar hemorrhage are associated with vision loss. In patients without diminished vision, conservative treatments such as bed rest, elevation of the head of the bed, ice packs, analgesia, lowering BP, and sedatives should be attempted first.5 Acetazolamide and mannitol can also be considered in consultation with an ophthalmologist.
Pneumo-orbitum
The presence of pneumo-orbitum should alert the clinician to either a communication with a paranasal sinus, a gas-forming organism, or (rarely) Munchausen syndrome.10 Unlike most case presentations, most causes of pneumo-orbitum do not involve OCS and are self-limited.11 Traumatic pneumo-orbitum without OCS has been reported in the literature.12-15 However, traumatic tension pneumo-orbitum is rare.12-16 One case report involved an elderly man with an orbital floor fracture who developed recurrent tension pneumo-orbitum after blowing his nose while intoxicated.12 Another case involved a boy with tension pneumo-orbitum that required surgical decompression.16
In a patient who has experienced trauma, the combination of proptosis, elevated IOP, and vision loss likely represent a retrobulbar hematoma or OCS. A lateral canthotomy can help relieve IOP from either condition.17,18 Orbital compartment syndrome can be caused by edema, emphysema, and caroticocavernous fistula, leading to increased orbital pressure and decreased perfusion.17,18
In a review of 10 trauma patients with OCS, all cases were intubated due to the severity of the head trauma, and all had OCS due to edema.17 In a review of eight trauma patients with OCS, all had eye pain, reduced visual acuity, and proptosis.18 Most of the patients had periorbital edema, ophthalmoparesis, a relative afferent pupillary defect (as compared to a fixed and dilated pupil), and chemosis.18,19 All of the patients with OCS required cantholysis or a lateral canthotomy.18
Lateral Canthotomy
Although EPs rarely perform lateral canthotomy, knowledge of this procedure is important, because it can prevent vision loss in the appropriate clinical setting. To perform a lateral canthotomy, the area around the affected eye is cleaned with saline irrigation.4 One percent or 2% lidocaine with epinephrine is then injected into the lateral canthus of the affected eye.4,10 A straight hemostat is applied between the upper and lower lids, producing a crush injury along the site of local anesthesia for 1 to 2 minutes.3,4,10 This is done to reduce the risk of bleeding by devitalizing the tissue.4 Straight scissors are then used to make a 1-cm horizontal incision from the lateral canthal tendon to the lateral orbital rim.4 This initial incision exposes the orbicularis muscle, orbital septum, palpebral conjunctiva, and an area called Eisler’s pocket that sits anterior to the lateral canthal tendon.3
Cantholysis can then be performed by blunt dissection.10 The inferior crus of the lateral canthus is identified either visually or by palpation, and a 1- to 2-cm inferior-posterior cut of the inferior crus accomplishes the lateral canthotomy.3-4 After cutting the inferior crus, the lower lid should be pulled away easily, and if this does not occur, repeated attempts at cutting the inferior crus should be made.3 Pulling the lower eyelid down and away from the lateral orbital rim separates the skin and conjunctiva, aiding in visualization.4
After cutting the inferior crus, only a small amount of blood or air typically is expressed, but this is usually enough to prevent vision loss.3 When the procedure is performed correctly, the practitioner should be able to palpate a difference in the pressure of the globe, and tonography will show a reduced IOP. If the ocular pressure is still significantly elevated, the physician can proceed to cut the superior canthus of the lateral canthal tendon in a manner similar to cutting the inferior crus of the tendon.4 After the procedure is performed, urgent ophthalmologic consultation is required.
The risks of performing a lateral canthotomy include mechanical injury, hemorrhage, and infection.4 The incision from a lateral canthotomy generally does not need suturing and will heal without significant scarring.4 If the scissors are aimed superiorly instead of inferiorly for the inferior crus of the lateral canthal tendon, there is risk of injuring the levator aponeurosis leading to ptosis, as well as a small risk of injury to the lacrimal gland and lacrimal artery.3
Conclusion
Our patient demonstrates a case of traumatic OCS, a vision-threatening medical condition that requires rapid diagnosis and lateral canthotomy to lower IOP and reduce the risk of permanent vision loss. While an orbital CT scan may assist in confirming the diagnosis, treatment of IOP should not be delayed.
1. McCaig LF, Burt CW. National Hospital Ambulatory Medical Care Survey: 2001 Emergency Department Summary. Advance Data from Vital Health and Statistics; No. 335. https://www.cdc.gov/nchs/data/ad/ad335.pdf. Accessed July 22, 2016.
2. Knoop KJ, Dennis WR. Eye trauma. In: Wolfson AB, Hendy GW, Hendy PL, et al (eds). Harwood-Nuss’ Clinical Practice of Emergency Medicine. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2005.
3. Vassallo S, Hartstein M, Howard D, Stetz J. Traumatic retrobulbar hemorrhage: emergent decompression by lateral canthotomy and cantholysis. J Emerg Med. 2002;22(3):251-256.
4. Roberts JR, Hedges JR (eds). Clinical Procedures in Emergency Medicine, 4th ed. Philadelphia, PA: Saunders; 2004.
5. Hatton MP, Thakker MM, Ray S. Orbital and adnexal trauma associated with open-globe injuries. Ophthal Plast Reconstr Surg. 2002;18(6):458-461.
6. Suner S, Simmons W, Savitt DL. A porcine model for instruction of lateral canthotomy. Acad Emerg Med. 2000;7(7):837-838.
7. Goodall KL, Brahma A, Bates A, Leatherbarrow B. Lateral canthotomy and inferior cantholysis: an effective method of urgent orbital decompression for sight threatening acute retrobulbar hemorrhage. Injury. 1999;30(7):485-490.
8. Gerbino G, Ramieri GA, Nasi A. Diagnosis and treatment of retrobulbar haematomas following blunt orbital trauma: a description of eight cases. Int J Oral Maxillofac Surg. 2005;34(2):127-131.
9. Machado RA, Silveira RL, Borges HO, Filho AM, de Oliveira GM. Retrobulbar hemorrhage: A case report. J Contemp Dent Pract. 2006;7(2):130-136.
10. Winans JM, House LR, Robinson HE. Self-induced orbital emphysema as a presenting sign of Munchausen’s syndrome. Laryngoscope. 1983;93(9):1209-1211.
11. Zimmer-Galler IE, Bartley GB. Orbital emphysema: case reports and review of the literature. Mayo Clin Proc. 1994;69(2):115-121.
12. Ahnood D, Toft PB. Recurrent orbital compartment syndrome caused by a blow-out fracture and accumulation of air; management by orbital punctures. Acta Ophthalmol. 2012;90(12):199-200.
13. Martin PW, Williams AC. Supraorbital emphysema: report of a case. J Oral Surg. 1972;30(12):901-902.
14. Per BL, Sanders BB. Post-traumatic intraorbital pneumatocele--a rare case of unilateral exophthalmos. Br J Radio. 1971;44(519):214-215.
15. Haller ML, Brackup AH, Shiffman F. Intraorbital aerocele. Arch Ophthalmol. 1980;98(9):1612-1613.
16. Chaudhry IA, Al-Amri A, Shamsi FA, Al-Rashed W. Visual recovery after evacuation of orbital emphysema. Orbit. 2007;26(4):283-285.
17. Perry M. Acute proptosis in trauma: retrobulbar hemorrhage or orbital compartment syndrome—does it really matter? J Oral Maxillofac Surg. 2008;66(9):1913-1920.
18. Sun MT, Chan WO, Selva D. Traumatic orbital compartment syndrome: importance of the lateral canthotomy and cantholysis. Emerg Med Australas. 2014;26(3):274-278.
19. Belliveau MJ, Johnson D. Orbital compartment syndrome after head trauma. Lancet Neurol. 2015;14(2):136-137.
1. McCaig LF, Burt CW. National Hospital Ambulatory Medical Care Survey: 2001 Emergency Department Summary. Advance Data from Vital Health and Statistics; No. 335. https://www.cdc.gov/nchs/data/ad/ad335.pdf. Accessed July 22, 2016.
2. Knoop KJ, Dennis WR. Eye trauma. In: Wolfson AB, Hendy GW, Hendy PL, et al (eds). Harwood-Nuss’ Clinical Practice of Emergency Medicine. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2005.
3. Vassallo S, Hartstein M, Howard D, Stetz J. Traumatic retrobulbar hemorrhage: emergent decompression by lateral canthotomy and cantholysis. J Emerg Med. 2002;22(3):251-256.
4. Roberts JR, Hedges JR (eds). Clinical Procedures in Emergency Medicine, 4th ed. Philadelphia, PA: Saunders; 2004.
5. Hatton MP, Thakker MM, Ray S. Orbital and adnexal trauma associated with open-globe injuries. Ophthal Plast Reconstr Surg. 2002;18(6):458-461.
6. Suner S, Simmons W, Savitt DL. A porcine model for instruction of lateral canthotomy. Acad Emerg Med. 2000;7(7):837-838.
7. Goodall KL, Brahma A, Bates A, Leatherbarrow B. Lateral canthotomy and inferior cantholysis: an effective method of urgent orbital decompression for sight threatening acute retrobulbar hemorrhage. Injury. 1999;30(7):485-490.
8. Gerbino G, Ramieri GA, Nasi A. Diagnosis and treatment of retrobulbar haematomas following blunt orbital trauma: a description of eight cases. Int J Oral Maxillofac Surg. 2005;34(2):127-131.
9. Machado RA, Silveira RL, Borges HO, Filho AM, de Oliveira GM. Retrobulbar hemorrhage: A case report. J Contemp Dent Pract. 2006;7(2):130-136.
10. Winans JM, House LR, Robinson HE. Self-induced orbital emphysema as a presenting sign of Munchausen’s syndrome. Laryngoscope. 1983;93(9):1209-1211.
11. Zimmer-Galler IE, Bartley GB. Orbital emphysema: case reports and review of the literature. Mayo Clin Proc. 1994;69(2):115-121.
12. Ahnood D, Toft PB. Recurrent orbital compartment syndrome caused by a blow-out fracture and accumulation of air; management by orbital punctures. Acta Ophthalmol. 2012;90(12):199-200.
13. Martin PW, Williams AC. Supraorbital emphysema: report of a case. J Oral Surg. 1972;30(12):901-902.
14. Per BL, Sanders BB. Post-traumatic intraorbital pneumatocele--a rare case of unilateral exophthalmos. Br J Radio. 1971;44(519):214-215.
15. Haller ML, Brackup AH, Shiffman F. Intraorbital aerocele. Arch Ophthalmol. 1980;98(9):1612-1613.
16. Chaudhry IA, Al-Amri A, Shamsi FA, Al-Rashed W. Visual recovery after evacuation of orbital emphysema. Orbit. 2007;26(4):283-285.
17. Perry M. Acute proptosis in trauma: retrobulbar hemorrhage or orbital compartment syndrome—does it really matter? J Oral Maxillofac Surg. 2008;66(9):1913-1920.
18. Sun MT, Chan WO, Selva D. Traumatic orbital compartment syndrome: importance of the lateral canthotomy and cantholysis. Emerg Med Australas. 2014;26(3):274-278.
19. Belliveau MJ, Johnson D. Orbital compartment syndrome after head trauma. Lancet Neurol. 2015;14(2):136-137.

Aleukemic acute lymphoblastic leukemia with unusual clinical features
Acute lymphoblastic leukemia is a neoplastic proliferation of lymphoblasts in the bone marrow. Normal hematopoiesis is affected, and symptoms from anemia (fatigue, breathlessness), leukopenia (recurrent infections) or thrombocytopenia (easy bruising, mucosal bleeding) are typically described in ALL. Hepatosplenomegaly and B-symptoms (fever, weight loss, and night sweats) are frequently seen. Presence of lymphoblasts in the peripheral smear is indicative of ALL, and a bone marrow biopsy finding of >25% lymphoblasts is confirmatory. Absence of peripheral lymphoblasts in a patient with acute leukemia is known as aleukemic leukemia. Aleukemic leukemia is uncommon, and most cases have described skin lesions from lymphoblast infiltration (leukemia cutis) in addition to bone marrow involvement.1 We report a case of aleukemic ALL in an adult presenting with unusual clinical features including bone pain, osteolytic lesions, hypercalcemia, and normal blood counts. To our knowledge, this is fifth such case ever reported in an adult patient.
Click on the PDF icon at the top of this introduction to read the full article.
Acute lymphoblastic leukemia is a neoplastic proliferation of lymphoblasts in the bone marrow. Normal hematopoiesis is affected, and symptoms from anemia (fatigue, breathlessness), leukopenia (recurrent infections) or thrombocytopenia (easy bruising, mucosal bleeding) are typically described in ALL. Hepatosplenomegaly and B-symptoms (fever, weight loss, and night sweats) are frequently seen. Presence of lymphoblasts in the peripheral smear is indicative of ALL, and a bone marrow biopsy finding of >25% lymphoblasts is confirmatory. Absence of peripheral lymphoblasts in a patient with acute leukemia is known as aleukemic leukemia. Aleukemic leukemia is uncommon, and most cases have described skin lesions from lymphoblast infiltration (leukemia cutis) in addition to bone marrow involvement.1 We report a case of aleukemic ALL in an adult presenting with unusual clinical features including bone pain, osteolytic lesions, hypercalcemia, and normal blood counts. To our knowledge, this is fifth such case ever reported in an adult patient.
Click on the PDF icon at the top of this introduction to read the full article.
Acute lymphoblastic leukemia is a neoplastic proliferation of lymphoblasts in the bone marrow. Normal hematopoiesis is affected, and symptoms from anemia (fatigue, breathlessness), leukopenia (recurrent infections) or thrombocytopenia (easy bruising, mucosal bleeding) are typically described in ALL. Hepatosplenomegaly and B-symptoms (fever, weight loss, and night sweats) are frequently seen. Presence of lymphoblasts in the peripheral smear is indicative of ALL, and a bone marrow biopsy finding of >25% lymphoblasts is confirmatory. Absence of peripheral lymphoblasts in a patient with acute leukemia is known as aleukemic leukemia. Aleukemic leukemia is uncommon, and most cases have described skin lesions from lymphoblast infiltration (leukemia cutis) in addition to bone marrow involvement.1 We report a case of aleukemic ALL in an adult presenting with unusual clinical features including bone pain, osteolytic lesions, hypercalcemia, and normal blood counts. To our knowledge, this is fifth such case ever reported in an adult patient.
Click on the PDF icon at the top of this introduction to read the full article.
Severe pruritus • crusted lesions affecting face, extremities, and trunk • hepatitis C virus carrier • Dx?
THE CASE
An 85-year-old woman sought care at our outpatient clinic for a 9-month history of severe pruritus and crusted lesions on her face, extremities, and trunk. She had been diagnosed with hepatitis C virus (HCV) infection one year ago and was not taking any medication. The patient, who had been living with her family, had visited various clinics for her complaints and was diagnosed as having contact dermatitis and senile pruritus. She was prescribed topical mometasone furoate and moisturizers.
After 6 months of using this therapy, widespread grey-white plaques and minimal excoriation appeared on her face, scalp, and trunk. This was diagnosed as psoriasis, and the patient was prescribed topical corticosteroids, which she used for 9 months until she came to our clinic. She said the lesions regressed minimally with the topical corticosteroids, but did not fully clear.
Dermatologic examination revealed widespread erythema and grey-white, cohesive, thick, pruritic plaques on her scalp, face, trunk, and bilateral extremities (FIGURE 1). A punch biopsy specimen was taken from the border of a plaque on her trunk.
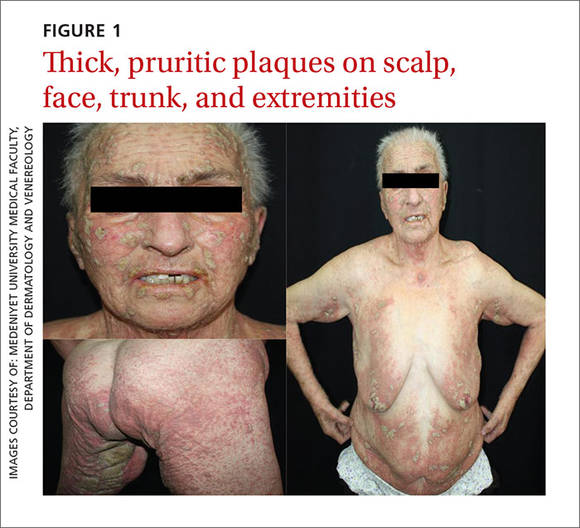
THE DIAGNOSIS
A complete blood cell count and wide biochemistry panel, including tumor markers and viral serology for human immunodeficiency virus (HIV), were normal. The patient had lymphadenopathy in her posterior cervical, bilateral preauricular, and bilateral inguinal regions.
Histopathologic examination revealed hyperkeratosis, acanthosis, and spongiotic edema in the epidermis, and vesiculation and mites in the stratum corneum. The dermal changes consisted of perivascular and diffuse cell infiltrates that were mainly mononuclear cells and eosinophilic granulocytes.
Based on the dermatologic examination and the histopathologic findings, we diagnosed the patient with crusted (Norwegian) scabies.
DISCUSSION
Crusted (Norwegian) scabies is a rare, highly contagious form of scabies that is characterized by the presence of millions of Sarcoptes scabiei var hominis mites in the epidermis.1 This variant of scabies can affect individuals of any age, gender, or race.2 It was first described by Boeck and Danielssen in 1848 in Norway and was named Norwegian scabies by von Hebra in 1862.3 In 2010, more than 200 cases of crusted scabies were reported in the literature.4
Crusted scabies is usually seen in immunocompromised patients, such as the elderly, those who’ve had solid organ transplantation, and those with HIV, malignancy, or malnutrition. Crusted scabies may also occur in patients with decreased sensory function (such as those with leprosy) or decreased ability to scratch, intellectual disabilities, and in those who use biologic agents or systemic/topical corticosteroids.4-8
Crusted scabies is associated with increased morbidity and mortality, especially in children and the elderly, because of complications such as secondary bacterial infections and sepsis.1,3 Widespread inflammation may also cause erythroderma, which can lead to metabolic disorders.
Distinguish it from other pruritic papulosquamous diseases
The differential diagnosis for crusted scabies includes psoriasis, eczema, cutaneous lymphoma, Darier disease, and adverse drug reactions.9 Crusted scabies can be differentiated from these other diagnoses by its clinical presentation and histopathological examination.
Crusted scabies is characterized by hyperkeratosis and wart-like crusts that are due to extreme proliferation of mites in the stratum corneum of the epidermis.2 Lesions are usually localized on acral sites (especially the hands), although the entire body, including the face and the scalp, can be involved.1 Psoriasiform or bullous pemphigoid-like eruptions have also been reported in the literature.5,9
Our patient presented with widespread erythema and psoriasiform grey-white crusts on her scalp, face, chest, periareolar region, and extremities. In addition, she did not have an immunosuppressant disease or medication history.
However, the fact that our patient was using topical corticosteroids for so long explained the extent of her condition. Topical corticosteroids have been linked to scabies incognito.10 Topical or systemic corticosteroid use for long periods of time may alter the skin immune system by suppressing cellular immunity, thereby reducing the inflammatory response. This may lead to progression of the regular variant of scabies to crusted scabies, as our patient had.
Topical treatments, oral ivermectin proven to be effective
Topical keratolytics, permethrin 5%, lindane 1%, crotamiton 10%, sulfur ointment (5%-10%), malathion 0.5%, benzyl benzoate (10%-25%), oral ivermectin (2 doses of 200 mcg/kg/dose), and systemic antihistamines are appropriate therapies.3 While oral ivermectin is effective, it is not available in Turkey.
Because of our patient’s hepatic disorder, we opted for a topical, rather than a systemic, treatment and recommended repeated applications of topical permethrin. Repeated treatment with topical permethrin is often sufficient in patients who are unable to take systemic therapy. In fact, Binic et al4 reported a case in which an elderly patient with crusted scabies (who had previously been treated with systemic and topical corticosteroids) responded well to repeated topical treatment with lindane 1%, 25% benzyl benzoate, and 10% precipitated sulfur.
Our patient. We prescribed topical 5% permethrin lotion for our patient to apply to her entire body 4 times a week and advised her to wash her clothing and bed linens at 140° F. She was scheduled for biweekly check-ups. We also advised the patient’s family to use the same topical therapy 2 times per week because crusted scabies is highly contagious. One month later, our patient’s lesions had resolved (FIGURE 2).
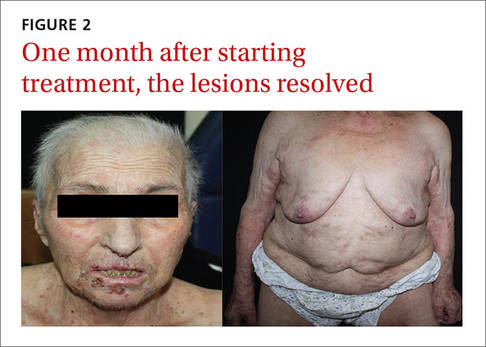
THE TAKEAWAY
Early diagnosis and treatment of crusted scabies is important, both for the treatment of the patient and to stop the spread of the disease. Although rare, crusted scabies should be included in the differential diagnosis of long-term pruritic papulosquamous diseases, and the possibility of an atypical presentation in all patients should be considered—whether their immunity is compromised or not. Scabies should also be considered in patients with a positive family history of the disease and in those with chronic pruritus that is unresponsive to topical therapies.
1. Burkhart CN, Burkhart CG, Morrell DS. Infestations. In: Bolognia JL, Jorizzo JL, Schaffer JV, et al, eds. Dermatology. 3rd ed. Philadelphia, PA: Mosby Elsevier; 2012;1423-1426.
2. Subramaniam G, Kaliaperumal K, Duraipandian J, et al. Norwegian scabies in a malnourished young adult: a case report. J Infect Dev Ctries. 2010;4:349-351.
3. Karthikeyan K. Crusted scabies. Indian J Dermatol Venereol Leprol. 2009;75:340-347.
4. Binic I, Jankovic A, Jovanovic D, et al. Crusted (Norwegian) scabies following systemic and topical corticosteroid therapy. J Korean Med Sci. 2010;25:188-191.
5. Ramachandran V, Shankar EM, Devaleenal B, et al. Atypically distributed cutaneous lesions of Norwegian scabies in an HIV-positive man in South India: a case report. J Med Case Rep. 2008;2:82.
6. Lai YC, Teng CJ, Chen PC, et al. Unusual scalp crusted scabies in an adult T-cell leukemia/lymphoma patient. Ups J Med Sci. 2011;116:77-78.
7. Saillard C, Darrieux L, Safa G. Crusted scabies complicates etanercept therapy in a patient with severe psoriasis. J Am Acad Dermatol. 2013;68:e138-e139.
8. Marlière V, Roul S, Labrèze C, et al. Crusted (Norwegian) scabies induced by use of topical corticosteroids and treated successfully with ivermectin. J Pediatr. 1999;135:122-124.
9. Goyal NN, Wong GA. Psoriasis or crusted scabies. Clin Exp Dermatol. 2008;33:211-212.
10. Kim KJ, Roh KH, Choi JH, et al. Scabies incognito presenting as urticaria pigmentosa in an infant. Pediatr Dermatol. 2002;19:409-411.
THE CASE
An 85-year-old woman sought care at our outpatient clinic for a 9-month history of severe pruritus and crusted lesions on her face, extremities, and trunk. She had been diagnosed with hepatitis C virus (HCV) infection one year ago and was not taking any medication. The patient, who had been living with her family, had visited various clinics for her complaints and was diagnosed as having contact dermatitis and senile pruritus. She was prescribed topical mometasone furoate and moisturizers.
After 6 months of using this therapy, widespread grey-white plaques and minimal excoriation appeared on her face, scalp, and trunk. This was diagnosed as psoriasis, and the patient was prescribed topical corticosteroids, which she used for 9 months until she came to our clinic. She said the lesions regressed minimally with the topical corticosteroids, but did not fully clear.
Dermatologic examination revealed widespread erythema and grey-white, cohesive, thick, pruritic plaques on her scalp, face, trunk, and bilateral extremities (FIGURE 1). A punch biopsy specimen was taken from the border of a plaque on her trunk.
THE DIAGNOSIS
A complete blood cell count and wide biochemistry panel, including tumor markers and viral serology for human immunodeficiency virus (HIV), were normal. The patient had lymphadenopathy in her posterior cervical, bilateral preauricular, and bilateral inguinal regions.
Histopathologic examination revealed hyperkeratosis, acanthosis, and spongiotic edema in the epidermis, and vesiculation and mites in the stratum corneum. The dermal changes consisted of perivascular and diffuse cell infiltrates that were mainly mononuclear cells and eosinophilic granulocytes.
Based on the dermatologic examination and the histopathologic findings, we diagnosed the patient with crusted (Norwegian) scabies.
DISCUSSION
Crusted (Norwegian) scabies is a rare, highly contagious form of scabies that is characterized by the presence of millions of Sarcoptes scabiei var hominis mites in the epidermis.1 This variant of scabies can affect individuals of any age, gender, or race.2 It was first described by Boeck and Danielssen in 1848 in Norway and was named Norwegian scabies by von Hebra in 1862.3 In 2010, more than 200 cases of crusted scabies were reported in the literature.4
Crusted scabies is usually seen in immunocompromised patients, such as the elderly, those who’ve had solid organ transplantation, and those with HIV, malignancy, or malnutrition. Crusted scabies may also occur in patients with decreased sensory function (such as those with leprosy) or decreased ability to scratch, intellectual disabilities, and in those who use biologic agents or systemic/topical corticosteroids.4-8
Crusted scabies is associated with increased morbidity and mortality, especially in children and the elderly, because of complications such as secondary bacterial infections and sepsis.1,3 Widespread inflammation may also cause erythroderma, which can lead to metabolic disorders.
Distinguish it from other pruritic papulosquamous diseases
The differential diagnosis for crusted scabies includes psoriasis, eczema, cutaneous lymphoma, Darier disease, and adverse drug reactions.9 Crusted scabies can be differentiated from these other diagnoses by its clinical presentation and histopathological examination.
Crusted scabies is characterized by hyperkeratosis and wart-like crusts that are due to extreme proliferation of mites in the stratum corneum of the epidermis.2 Lesions are usually localized on acral sites (especially the hands), although the entire body, including the face and the scalp, can be involved.1 Psoriasiform or bullous pemphigoid-like eruptions have also been reported in the literature.5,9
Our patient presented with widespread erythema and psoriasiform grey-white crusts on her scalp, face, chest, periareolar region, and extremities. In addition, she did not have an immunosuppressant disease or medication history.
However, the fact that our patient was using topical corticosteroids for so long explained the extent of her condition. Topical corticosteroids have been linked to scabies incognito.10 Topical or systemic corticosteroid use for long periods of time may alter the skin immune system by suppressing cellular immunity, thereby reducing the inflammatory response. This may lead to progression of the regular variant of scabies to crusted scabies, as our patient had.
Topical treatments, oral ivermectin proven to be effective
Topical keratolytics, permethrin 5%, lindane 1%, crotamiton 10%, sulfur ointment (5%-10%), malathion 0.5%, benzyl benzoate (10%-25%), oral ivermectin (2 doses of 200 mcg/kg/dose), and systemic antihistamines are appropriate therapies.3 While oral ivermectin is effective, it is not available in Turkey.
Because of our patient’s hepatic disorder, we opted for a topical, rather than a systemic, treatment and recommended repeated applications of topical permethrin. Repeated treatment with topical permethrin is often sufficient in patients who are unable to take systemic therapy. In fact, Binic et al4 reported a case in which an elderly patient with crusted scabies (who had previously been treated with systemic and topical corticosteroids) responded well to repeated topical treatment with lindane 1%, 25% benzyl benzoate, and 10% precipitated sulfur.
Our patient. We prescribed topical 5% permethrin lotion for our patient to apply to her entire body 4 times a week and advised her to wash her clothing and bed linens at 140° F. She was scheduled for biweekly check-ups. We also advised the patient’s family to use the same topical therapy 2 times per week because crusted scabies is highly contagious. One month later, our patient’s lesions had resolved (FIGURE 2).
THE TAKEAWAY
Early diagnosis and treatment of crusted scabies is important, both for the treatment of the patient and to stop the spread of the disease. Although rare, crusted scabies should be included in the differential diagnosis of long-term pruritic papulosquamous diseases, and the possibility of an atypical presentation in all patients should be considered—whether their immunity is compromised or not. Scabies should also be considered in patients with a positive family history of the disease and in those with chronic pruritus that is unresponsive to topical therapies.
THE CASE
An 85-year-old woman sought care at our outpatient clinic for a 9-month history of severe pruritus and crusted lesions on her face, extremities, and trunk. She had been diagnosed with hepatitis C virus (HCV) infection one year ago and was not taking any medication. The patient, who had been living with her family, had visited various clinics for her complaints and was diagnosed as having contact dermatitis and senile pruritus. She was prescribed topical mometasone furoate and moisturizers.
After 6 months of using this therapy, widespread grey-white plaques and minimal excoriation appeared on her face, scalp, and trunk. This was diagnosed as psoriasis, and the patient was prescribed topical corticosteroids, which she used for 9 months until she came to our clinic. She said the lesions regressed minimally with the topical corticosteroids, but did not fully clear.
Dermatologic examination revealed widespread erythema and grey-white, cohesive, thick, pruritic plaques on her scalp, face, trunk, and bilateral extremities (FIGURE 1). A punch biopsy specimen was taken from the border of a plaque on her trunk.
THE DIAGNOSIS
A complete blood cell count and wide biochemistry panel, including tumor markers and viral serology for human immunodeficiency virus (HIV), were normal. The patient had lymphadenopathy in her posterior cervical, bilateral preauricular, and bilateral inguinal regions.
Histopathologic examination revealed hyperkeratosis, acanthosis, and spongiotic edema in the epidermis, and vesiculation and mites in the stratum corneum. The dermal changes consisted of perivascular and diffuse cell infiltrates that were mainly mononuclear cells and eosinophilic granulocytes.
Based on the dermatologic examination and the histopathologic findings, we diagnosed the patient with crusted (Norwegian) scabies.
DISCUSSION
Crusted (Norwegian) scabies is a rare, highly contagious form of scabies that is characterized by the presence of millions of Sarcoptes scabiei var hominis mites in the epidermis.1 This variant of scabies can affect individuals of any age, gender, or race.2 It was first described by Boeck and Danielssen in 1848 in Norway and was named Norwegian scabies by von Hebra in 1862.3 In 2010, more than 200 cases of crusted scabies were reported in the literature.4
Crusted scabies is usually seen in immunocompromised patients, such as the elderly, those who’ve had solid organ transplantation, and those with HIV, malignancy, or malnutrition. Crusted scabies may also occur in patients with decreased sensory function (such as those with leprosy) or decreased ability to scratch, intellectual disabilities, and in those who use biologic agents or systemic/topical corticosteroids.4-8
Crusted scabies is associated with increased morbidity and mortality, especially in children and the elderly, because of complications such as secondary bacterial infections and sepsis.1,3 Widespread inflammation may also cause erythroderma, which can lead to metabolic disorders.
Distinguish it from other pruritic papulosquamous diseases
The differential diagnosis for crusted scabies includes psoriasis, eczema, cutaneous lymphoma, Darier disease, and adverse drug reactions.9 Crusted scabies can be differentiated from these other diagnoses by its clinical presentation and histopathological examination.
Crusted scabies is characterized by hyperkeratosis and wart-like crusts that are due to extreme proliferation of mites in the stratum corneum of the epidermis.2 Lesions are usually localized on acral sites (especially the hands), although the entire body, including the face and the scalp, can be involved.1 Psoriasiform or bullous pemphigoid-like eruptions have also been reported in the literature.5,9
Our patient presented with widespread erythema and psoriasiform grey-white crusts on her scalp, face, chest, periareolar region, and extremities. In addition, she did not have an immunosuppressant disease or medication history.
However, the fact that our patient was using topical corticosteroids for so long explained the extent of her condition. Topical corticosteroids have been linked to scabies incognito.10 Topical or systemic corticosteroid use for long periods of time may alter the skin immune system by suppressing cellular immunity, thereby reducing the inflammatory response. This may lead to progression of the regular variant of scabies to crusted scabies, as our patient had.
Topical treatments, oral ivermectin proven to be effective
Topical keratolytics, permethrin 5%, lindane 1%, crotamiton 10%, sulfur ointment (5%-10%), malathion 0.5%, benzyl benzoate (10%-25%), oral ivermectin (2 doses of 200 mcg/kg/dose), and systemic antihistamines are appropriate therapies.3 While oral ivermectin is effective, it is not available in Turkey.
Because of our patient’s hepatic disorder, we opted for a topical, rather than a systemic, treatment and recommended repeated applications of topical permethrin. Repeated treatment with topical permethrin is often sufficient in patients who are unable to take systemic therapy. In fact, Binic et al4 reported a case in which an elderly patient with crusted scabies (who had previously been treated with systemic and topical corticosteroids) responded well to repeated topical treatment with lindane 1%, 25% benzyl benzoate, and 10% precipitated sulfur.
Our patient. We prescribed topical 5% permethrin lotion for our patient to apply to her entire body 4 times a week and advised her to wash her clothing and bed linens at 140° F. She was scheduled for biweekly check-ups. We also advised the patient’s family to use the same topical therapy 2 times per week because crusted scabies is highly contagious. One month later, our patient’s lesions had resolved (FIGURE 2).
THE TAKEAWAY
Early diagnosis and treatment of crusted scabies is important, both for the treatment of the patient and to stop the spread of the disease. Although rare, crusted scabies should be included in the differential diagnosis of long-term pruritic papulosquamous diseases, and the possibility of an atypical presentation in all patients should be considered—whether their immunity is compromised or not. Scabies should also be considered in patients with a positive family history of the disease and in those with chronic pruritus that is unresponsive to topical therapies.
1. Burkhart CN, Burkhart CG, Morrell DS. Infestations. In: Bolognia JL, Jorizzo JL, Schaffer JV, et al, eds. Dermatology. 3rd ed. Philadelphia, PA: Mosby Elsevier; 2012;1423-1426.
2. Subramaniam G, Kaliaperumal K, Duraipandian J, et al. Norwegian scabies in a malnourished young adult: a case report. J Infect Dev Ctries. 2010;4:349-351.
3. Karthikeyan K. Crusted scabies. Indian J Dermatol Venereol Leprol. 2009;75:340-347.
4. Binic I, Jankovic A, Jovanovic D, et al. Crusted (Norwegian) scabies following systemic and topical corticosteroid therapy. J Korean Med Sci. 2010;25:188-191.
5. Ramachandran V, Shankar EM, Devaleenal B, et al. Atypically distributed cutaneous lesions of Norwegian scabies in an HIV-positive man in South India: a case report. J Med Case Rep. 2008;2:82.
6. Lai YC, Teng CJ, Chen PC, et al. Unusual scalp crusted scabies in an adult T-cell leukemia/lymphoma patient. Ups J Med Sci. 2011;116:77-78.
7. Saillard C, Darrieux L, Safa G. Crusted scabies complicates etanercept therapy in a patient with severe psoriasis. J Am Acad Dermatol. 2013;68:e138-e139.
8. Marlière V, Roul S, Labrèze C, et al. Crusted (Norwegian) scabies induced by use of topical corticosteroids and treated successfully with ivermectin. J Pediatr. 1999;135:122-124.
9. Goyal NN, Wong GA. Psoriasis or crusted scabies. Clin Exp Dermatol. 2008;33:211-212.
10. Kim KJ, Roh KH, Choi JH, et al. Scabies incognito presenting as urticaria pigmentosa in an infant. Pediatr Dermatol. 2002;19:409-411.
1. Burkhart CN, Burkhart CG, Morrell DS. Infestations. In: Bolognia JL, Jorizzo JL, Schaffer JV, et al, eds. Dermatology. 3rd ed. Philadelphia, PA: Mosby Elsevier; 2012;1423-1426.
2. Subramaniam G, Kaliaperumal K, Duraipandian J, et al. Norwegian scabies in a malnourished young adult: a case report. J Infect Dev Ctries. 2010;4:349-351.
3. Karthikeyan K. Crusted scabies. Indian J Dermatol Venereol Leprol. 2009;75:340-347.
4. Binic I, Jankovic A, Jovanovic D, et al. Crusted (Norwegian) scabies following systemic and topical corticosteroid therapy. J Korean Med Sci. 2010;25:188-191.
5. Ramachandran V, Shankar EM, Devaleenal B, et al. Atypically distributed cutaneous lesions of Norwegian scabies in an HIV-positive man in South India: a case report. J Med Case Rep. 2008;2:82.
6. Lai YC, Teng CJ, Chen PC, et al. Unusual scalp crusted scabies in an adult T-cell leukemia/lymphoma patient. Ups J Med Sci. 2011;116:77-78.
7. Saillard C, Darrieux L, Safa G. Crusted scabies complicates etanercept therapy in a patient with severe psoriasis. J Am Acad Dermatol. 2013;68:e138-e139.
8. Marlière V, Roul S, Labrèze C, et al. Crusted (Norwegian) scabies induced by use of topical corticosteroids and treated successfully with ivermectin. J Pediatr. 1999;135:122-124.
9. Goyal NN, Wong GA. Psoriasis or crusted scabies. Clin Exp Dermatol. 2008;33:211-212.
10. Kim KJ, Roh KH, Choi JH, et al. Scabies incognito presenting as urticaria pigmentosa in an infant. Pediatr Dermatol. 2002;19:409-411.
Swollen right hand and forearm • minor trauma to hand • previous diagnosis of cellulitis • Dx?
THE CASE
A 63-year-old woman with a history of hyperlipidemia presented to our hospital with a swollen right hand. The patient noted that she had closed her hand in a car door one week earlier, causing minor trauma to the right third metacarpophalangeal joint. Shortly after injuring her hand, she’d sought care at an outpatient facility, where she was given a diagnosis of cellulitis and a prescription for an oral antibiotic. The swelling, however, worsened, prompting her visit to our hospital. She was admitted for further work-up and started on intravenous (IV) antibiotics.
Her family history included a sister with deep vein thrombosis (DVT) and a maternal aunt with breast cancer. The patient denied oral contraceptive use or a personal history of malignancy. On physical examination, her right hand and forearm were swollen, tender, and erythematous.
THE DIAGNOSIS
Laboratory data showed a normal complete blood count and complete metabolic panel. The patient’s sedimentation rate and C-reactive protein level were elevated at 65 mm/hr and 110 mg/L, respectively. The patient was not improving on IV antibiotics, so we performed a right upper extremity venous duplex ultrasound. The ultrasound showed an occlusive thrombus in the right ulnar and radial veins (FIGURES 1 AND 2), and we diagnosed the patient with upper extremity deep vein thrombosis (UEDVT). (The timing of the diagnosis, relative to the injury to the patient’s hand, appeared to have been coincidental.)
Further work-up revealed normal complement C3 and C4 tests, as well as negative antinuclear, anti-double stranded DNA, anti-Smith, and anticardiolipin antibody tests. Similarly, a factor II DNA analysis was negative. However, the patient was positive for the factor V Leiden heterozygous mutation.
| 
|
DISCUSSION
More than 350,000 people are diagnosed with DVT or pulmonary embolism (PE) in the United States each year.1 Up to 4% of all DVTs involve the upper extremities.2 Secondary UEDVT, which occurs in patients with central venous catheters, malignancies, and thrombophilia, accounts for the majority of UEDVT cases; primary UEDVT is less common.3
Large epidemiologic studies have demonstrated that hypercoagulability is a risk factor for lower extremity DVT, but few data exist on the role of coagulation abnormalities in patients with primary UEDVT. The prevalence of clotting abnormalities in patients with primary UEDVT ranges from 8% to 43%.4,5 Factor V Leiden is the most common cause of inherited thrombophilia. Patients with heterozygous factor V Leiden mutation have a 7-fold increased risk of venous thrombosis.6
Héron and colleagues7 reported that 16 of 51 patients with at least one clotting abnormality had primary UEDVT. Factor V Leiden was found in 5 of those patients (20%). Interestingly, 3 of the 5 carriers of the factor V Leiden mutation were older than 45 years. Our patient was 63, which was consistent with these findings.
Malignancy is also an important risk factor for UEDVT.8,9 In patients with a DVT in an unusual location, age- and sex-appropriate cancer screenings are strongly recommended. (Our patient had undergone a colonoscopy 8 years earlier, which was normal. She’d also had a recent mammogram and Pap smear, which were normal, as well.)
It’s often difficult to distinguish between cellulitis and UEDVT
The differential diagnosis for UEDVT includes effort thrombosis (also known as Paget-Schroetter syndrome) and cellulitis.
Effort thrombosis usually occurs in young, otherwise healthy individuals and almost exclusively in the axillary and subclavian veins. Our patient’s age and a venous duplex ultrasound ruled out any thrombosis in these locations.
Distinguishing cellulitis from UEDVT based on clinical features can be difficult. In both conditions, the limb is swollen and painful and the skin is warm and erythematous. As a result, each condition is often misdiagnosed as the other.
But there are features that distinguish the 2. In patients with UEDVT, you’re likely to see limb pain and a palpable cord (a hard, thickened palpable vein along the line of the deep veins). On the other hand, patients with cellulitis tend to have more systemic symptoms, such as fever, chills, and swollen lymph nodes, as well as skin breakdown, ulcers, and pus.
The American College of Chest Physicians (ACCP) recommends that the initial evaluation for patients with suspected UEDVT be a combined modality ultrasound (compression with either Doppler or color Doppler) rather than D-dimer or venography.10 Quickly arriving at a proper diagnosis is critical, given that up to one-third of patients with UEDVT will develop a PE.7 Other complications include superior vena cava syndrome, septic thrombophlebitis, thoracic duct obstruction, and brachial plexopathy.11
Treat with anticoagulants for no longer than 3 months
The ACCP also recommends that patients who have UEDVT that isn’t associated with a central venous catheter or with cancer be treated with anticoagulation for no longer than 3 months.10
Our patient was started on enoxaparin and warfarin. After 5 days at our hospital, she was taken off the enoxaparin and discharged home on warfarin 5 mg/d. The swelling completely resolved one week later.
THE TAKEAWAY
Ulnar and radial DVT in a patient with factor V Leiden mutation is a rare condition. UEDVT should be included in the differential diagnosis for cellulitis whenever the diagnosis is uncertain or the patient doesn’t respond to antibiotics. Factor V Leiden mutation appears to be a risk factor in UEDVT and testing for it should be considered.
1. Office of the Surgeon General (US); National Heart, Lung, and Blood Institute (US). The Surgeon General’s Call to Action to Prevent Deep Vein Thrombosis and Pulmonary Embolism. Available at: http://www.ncbi.nlm.nih.gov/books/NBK44178/. Rockville, MD: Office of the Surgeon General (US); 2008.
2. Sawyer GA, Hayda R. Upper-extremity deep venous thrombosis following humeral shaft fracture. Orthopedics. 2011;34:141.
3. Leebeek FW, Stadhouders NA, van Stein D, et al. Hypercoagulability states in upper-extremity deep venous thrombosis. Am J Hematol. 2001;67:15-19.
4. Prandoni P, Polistena P, Bernardi E, et al. Upper-extremity deep vein thrombosis. Risk factors, diagnosis, and complications. Arch Intern Med. 1997;157:57-62.
5. Ruggeri M, Castaman G, Tosetto A, et al. Low prevalence of thrombophilic coagulation defects in patients with deep vein thrombosis of the upper limbs. Blood Coagul Fibrinolysis. 1997;8:191-194.
6. Ornstein DL, Cushman M. Cardiology patient page. Factor V Leiden. Circulation. 2003;107:e94-e97.
7. Héron E, Lozinguez O, Alhenc-Gelas M, et al. Hypercoagulable states in primary upper-extremity deep vein thrombosis. Arch Intern Med. 2000;160:382-386.
8. Linnemann B, Meister F, Schwonberg J, et al; MAISTHRO registry. Hereditary and acquired thrombophilia in patients with upper extremity deep-vein thrombosis. Results from the MAISTHRO registry. Thromb Haemost. 2008;100:440-446.
9. Monreal M, Lafoz E, Ruiz J, et al. Upper-extremity deep venous thrombosis and pulmonary embolism. A prospective study. Chest. 1991;99:280-283.
10. Holbrook A, Schulman S, Witt DM, et al; American College of Chest Physicians. Evidence-based management of anticoagulant therapy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141:e152S-e184S.
11. Becker DM, Philbrick JT, Walker FB 4th. Axillary and subclavian venous thrombosis. Prognosis and treatment. Arch Intern Med. 1991;151:1934-1943.
THE CASE
A 63-year-old woman with a history of hyperlipidemia presented to our hospital with a swollen right hand. The patient noted that she had closed her hand in a car door one week earlier, causing minor trauma to the right third metacarpophalangeal joint. Shortly after injuring her hand, she’d sought care at an outpatient facility, where she was given a diagnosis of cellulitis and a prescription for an oral antibiotic. The swelling, however, worsened, prompting her visit to our hospital. She was admitted for further work-up and started on intravenous (IV) antibiotics.
Her family history included a sister with deep vein thrombosis (DVT) and a maternal aunt with breast cancer. The patient denied oral contraceptive use or a personal history of malignancy. On physical examination, her right hand and forearm were swollen, tender, and erythematous.
THE DIAGNOSIS
Laboratory data showed a normal complete blood count and complete metabolic panel. The patient’s sedimentation rate and C-reactive protein level were elevated at 65 mm/hr and 110 mg/L, respectively. The patient was not improving on IV antibiotics, so we performed a right upper extremity venous duplex ultrasound. The ultrasound showed an occlusive thrombus in the right ulnar and radial veins (FIGURES 1 AND 2), and we diagnosed the patient with upper extremity deep vein thrombosis (UEDVT). (The timing of the diagnosis, relative to the injury to the patient’s hand, appeared to have been coincidental.)
Further work-up revealed normal complement C3 and C4 tests, as well as negative antinuclear, anti-double stranded DNA, anti-Smith, and anticardiolipin antibody tests. Similarly, a factor II DNA analysis was negative. However, the patient was positive for the factor V Leiden heterozygous mutation.
|
|
|
DISCUSSION
More than 350,000 people are diagnosed with DVT or pulmonary embolism (PE) in the United States each year.1 Up to 4% of all DVTs involve the upper extremities.2 Secondary UEDVT, which occurs in patients with central venous catheters, malignancies, and thrombophilia, accounts for the majority of UEDVT cases; primary UEDVT is less common.3
Large epidemiologic studies have demonstrated that hypercoagulability is a risk factor for lower extremity DVT, but few data exist on the role of coagulation abnormalities in patients with primary UEDVT. The prevalence of clotting abnormalities in patients with primary UEDVT ranges from 8% to 43%.4,5 Factor V Leiden is the most common cause of inherited thrombophilia. Patients with heterozygous factor V Leiden mutation have a 7-fold increased risk of venous thrombosis.6
Héron and colleagues7 reported that 16 of 51 patients with at least one clotting abnormality had primary UEDVT. Factor V Leiden was found in 5 of those patients (20%). Interestingly, 3 of the 5 carriers of the factor V Leiden mutation were older than 45 years. Our patient was 63, which was consistent with these findings.
Malignancy is also an important risk factor for UEDVT.8,9 In patients with a DVT in an unusual location, age- and sex-appropriate cancer screenings are strongly recommended. (Our patient had undergone a colonoscopy 8 years earlier, which was normal. She’d also had a recent mammogram and Pap smear, which were normal, as well.)
It’s often difficult to distinguish between cellulitis and UEDVT
The differential diagnosis for UEDVT includes effort thrombosis (also known as Paget-Schroetter syndrome) and cellulitis.
Effort thrombosis usually occurs in young, otherwise healthy individuals and almost exclusively in the axillary and subclavian veins. Our patient’s age and a venous duplex ultrasound ruled out any thrombosis in these locations.
Distinguishing cellulitis from UEDVT based on clinical features can be difficult. In both conditions, the limb is swollen and painful and the skin is warm and erythematous. As a result, each condition is often misdiagnosed as the other.
But there are features that distinguish the 2. In patients with UEDVT, you’re likely to see limb pain and a palpable cord (a hard, thickened palpable vein along the line of the deep veins). On the other hand, patients with cellulitis tend to have more systemic symptoms, such as fever, chills, and swollen lymph nodes, as well as skin breakdown, ulcers, and pus.
The American College of Chest Physicians (ACCP) recommends that the initial evaluation for patients with suspected UEDVT be a combined modality ultrasound (compression with either Doppler or color Doppler) rather than D-dimer or venography.10 Quickly arriving at a proper diagnosis is critical, given that up to one-third of patients with UEDVT will develop a PE.7 Other complications include superior vena cava syndrome, septic thrombophlebitis, thoracic duct obstruction, and brachial plexopathy.11
Treat with anticoagulants for no longer than 3 months
The ACCP also recommends that patients who have UEDVT that isn’t associated with a central venous catheter or with cancer be treated with anticoagulation for no longer than 3 months.10
Our patient was started on enoxaparin and warfarin. After 5 days at our hospital, she was taken off the enoxaparin and discharged home on warfarin 5 mg/d. The swelling completely resolved one week later.
THE TAKEAWAY
Ulnar and radial DVT in a patient with factor V Leiden mutation is a rare condition. UEDVT should be included in the differential diagnosis for cellulitis whenever the diagnosis is uncertain or the patient doesn’t respond to antibiotics. Factor V Leiden mutation appears to be a risk factor in UEDVT and testing for it should be considered.
THE CASE
A 63-year-old woman with a history of hyperlipidemia presented to our hospital with a swollen right hand. The patient noted that she had closed her hand in a car door one week earlier, causing minor trauma to the right third metacarpophalangeal joint. Shortly after injuring her hand, she’d sought care at an outpatient facility, where she was given a diagnosis of cellulitis and a prescription for an oral antibiotic. The swelling, however, worsened, prompting her visit to our hospital. She was admitted for further work-up and started on intravenous (IV) antibiotics.
Her family history included a sister with deep vein thrombosis (DVT) and a maternal aunt with breast cancer. The patient denied oral contraceptive use or a personal history of malignancy. On physical examination, her right hand and forearm were swollen, tender, and erythematous.
THE DIAGNOSIS
Laboratory data showed a normal complete blood count and complete metabolic panel. The patient’s sedimentation rate and C-reactive protein level were elevated at 65 mm/hr and 110 mg/L, respectively. The patient was not improving on IV antibiotics, so we performed a right upper extremity venous duplex ultrasound. The ultrasound showed an occlusive thrombus in the right ulnar and radial veins (FIGURES 1 AND 2), and we diagnosed the patient with upper extremity deep vein thrombosis (UEDVT). (The timing of the diagnosis, relative to the injury to the patient’s hand, appeared to have been coincidental.)
Further work-up revealed normal complement C3 and C4 tests, as well as negative antinuclear, anti-double stranded DNA, anti-Smith, and anticardiolipin antibody tests. Similarly, a factor II DNA analysis was negative. However, the patient was positive for the factor V Leiden heterozygous mutation.
|
|
|
DISCUSSION
More than 350,000 people are diagnosed with DVT or pulmonary embolism (PE) in the United States each year.1 Up to 4% of all DVTs involve the upper extremities.2 Secondary UEDVT, which occurs in patients with central venous catheters, malignancies, and thrombophilia, accounts for the majority of UEDVT cases; primary UEDVT is less common.3
Large epidemiologic studies have demonstrated that hypercoagulability is a risk factor for lower extremity DVT, but few data exist on the role of coagulation abnormalities in patients with primary UEDVT. The prevalence of clotting abnormalities in patients with primary UEDVT ranges from 8% to 43%.4,5 Factor V Leiden is the most common cause of inherited thrombophilia. Patients with heterozygous factor V Leiden mutation have a 7-fold increased risk of venous thrombosis.6
Héron and colleagues7 reported that 16 of 51 patients with at least one clotting abnormality had primary UEDVT. Factor V Leiden was found in 5 of those patients (20%). Interestingly, 3 of the 5 carriers of the factor V Leiden mutation were older than 45 years. Our patient was 63, which was consistent with these findings.
Malignancy is also an important risk factor for UEDVT.8,9 In patients with a DVT in an unusual location, age- and sex-appropriate cancer screenings are strongly recommended. (Our patient had undergone a colonoscopy 8 years earlier, which was normal. She’d also had a recent mammogram and Pap smear, which were normal, as well.)
It’s often difficult to distinguish between cellulitis and UEDVT
The differential diagnosis for UEDVT includes effort thrombosis (also known as Paget-Schroetter syndrome) and cellulitis.
Effort thrombosis usually occurs in young, otherwise healthy individuals and almost exclusively in the axillary and subclavian veins. Our patient’s age and a venous duplex ultrasound ruled out any thrombosis in these locations.
Distinguishing cellulitis from UEDVT based on clinical features can be difficult. In both conditions, the limb is swollen and painful and the skin is warm and erythematous. As a result, each condition is often misdiagnosed as the other.
But there are features that distinguish the 2. In patients with UEDVT, you’re likely to see limb pain and a palpable cord (a hard, thickened palpable vein along the line of the deep veins). On the other hand, patients with cellulitis tend to have more systemic symptoms, such as fever, chills, and swollen lymph nodes, as well as skin breakdown, ulcers, and pus.
The American College of Chest Physicians (ACCP) recommends that the initial evaluation for patients with suspected UEDVT be a combined modality ultrasound (compression with either Doppler or color Doppler) rather than D-dimer or venography.10 Quickly arriving at a proper diagnosis is critical, given that up to one-third of patients with UEDVT will develop a PE.7 Other complications include superior vena cava syndrome, septic thrombophlebitis, thoracic duct obstruction, and brachial plexopathy.11
Treat with anticoagulants for no longer than 3 months
The ACCP also recommends that patients who have UEDVT that isn’t associated with a central venous catheter or with cancer be treated with anticoagulation for no longer than 3 months.10
Our patient was started on enoxaparin and warfarin. After 5 days at our hospital, she was taken off the enoxaparin and discharged home on warfarin 5 mg/d. The swelling completely resolved one week later.
THE TAKEAWAY
Ulnar and radial DVT in a patient with factor V Leiden mutation is a rare condition. UEDVT should be included in the differential diagnosis for cellulitis whenever the diagnosis is uncertain or the patient doesn’t respond to antibiotics. Factor V Leiden mutation appears to be a risk factor in UEDVT and testing for it should be considered.
1. Office of the Surgeon General (US); National Heart, Lung, and Blood Institute (US). The Surgeon General’s Call to Action to Prevent Deep Vein Thrombosis and Pulmonary Embolism. Available at: http://www.ncbi.nlm.nih.gov/books/NBK44178/. Rockville, MD: Office of the Surgeon General (US); 2008.
2. Sawyer GA, Hayda R. Upper-extremity deep venous thrombosis following humeral shaft fracture. Orthopedics. 2011;34:141.
3. Leebeek FW, Stadhouders NA, van Stein D, et al. Hypercoagulability states in upper-extremity deep venous thrombosis. Am J Hematol. 2001;67:15-19.
4. Prandoni P, Polistena P, Bernardi E, et al. Upper-extremity deep vein thrombosis. Risk factors, diagnosis, and complications. Arch Intern Med. 1997;157:57-62.
5. Ruggeri M, Castaman G, Tosetto A, et al. Low prevalence of thrombophilic coagulation defects in patients with deep vein thrombosis of the upper limbs. Blood Coagul Fibrinolysis. 1997;8:191-194.
6. Ornstein DL, Cushman M. Cardiology patient page. Factor V Leiden. Circulation. 2003;107:e94-e97.
7. Héron E, Lozinguez O, Alhenc-Gelas M, et al. Hypercoagulable states in primary upper-extremity deep vein thrombosis. Arch Intern Med. 2000;160:382-386.
8. Linnemann B, Meister F, Schwonberg J, et al; MAISTHRO registry. Hereditary and acquired thrombophilia in patients with upper extremity deep-vein thrombosis. Results from the MAISTHRO registry. Thromb Haemost. 2008;100:440-446.
9. Monreal M, Lafoz E, Ruiz J, et al. Upper-extremity deep venous thrombosis and pulmonary embolism. A prospective study. Chest. 1991;99:280-283.
10. Holbrook A, Schulman S, Witt DM, et al; American College of Chest Physicians. Evidence-based management of anticoagulant therapy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141:e152S-e184S.
11. Becker DM, Philbrick JT, Walker FB 4th. Axillary and subclavian venous thrombosis. Prognosis and treatment. Arch Intern Med. 1991;151:1934-1943.
1. Office of the Surgeon General (US); National Heart, Lung, and Blood Institute (US). The Surgeon General’s Call to Action to Prevent Deep Vein Thrombosis and Pulmonary Embolism. Available at: http://www.ncbi.nlm.nih.gov/books/NBK44178/. Rockville, MD: Office of the Surgeon General (US); 2008.
2. Sawyer GA, Hayda R. Upper-extremity deep venous thrombosis following humeral shaft fracture. Orthopedics. 2011;34:141.
3. Leebeek FW, Stadhouders NA, van Stein D, et al. Hypercoagulability states in upper-extremity deep venous thrombosis. Am J Hematol. 2001;67:15-19.
4. Prandoni P, Polistena P, Bernardi E, et al. Upper-extremity deep vein thrombosis. Risk factors, diagnosis, and complications. Arch Intern Med. 1997;157:57-62.
5. Ruggeri M, Castaman G, Tosetto A, et al. Low prevalence of thrombophilic coagulation defects in patients with deep vein thrombosis of the upper limbs. Blood Coagul Fibrinolysis. 1997;8:191-194.
6. Ornstein DL, Cushman M. Cardiology patient page. Factor V Leiden. Circulation. 2003;107:e94-e97.
7. Héron E, Lozinguez O, Alhenc-Gelas M, et al. Hypercoagulable states in primary upper-extremity deep vein thrombosis. Arch Intern Med. 2000;160:382-386.
8. Linnemann B, Meister F, Schwonberg J, et al; MAISTHRO registry. Hereditary and acquired thrombophilia in patients with upper extremity deep-vein thrombosis. Results from the MAISTHRO registry. Thromb Haemost. 2008;100:440-446.
9. Monreal M, Lafoz E, Ruiz J, et al. Upper-extremity deep venous thrombosis and pulmonary embolism. A prospective study. Chest. 1991;99:280-283.
10. Holbrook A, Schulman S, Witt DM, et al; American College of Chest Physicians. Evidence-based management of anticoagulant therapy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141:e152S-e184S.
11. Becker DM, Philbrick JT, Walker FB 4th. Axillary and subclavian venous thrombosis. Prognosis and treatment. Arch Intern Med. 1991;151:1934-1943.