User login
Up in Arms: Bilateral Luxatio Erecta Fracture-Dislocations
Unilateral inferior shoulder dislocation (luxatio erecta) is uncommon, accounting for only 0.5% of all shoulder dislocations.1 Bilateral luxatio erecta is extremely rare, having been described fewer than 20 times in the literature. The most common etiology is hyperabduction causing the humerus to lever on the acromion; less common is axial loading onto a fully abducted arm and an extended elbow.2 Hyperabduction can occur when a person grabs an object in an attempt to stop a fall, as occurred in the present case. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 58-year-old man with a trauma injury presented to our emergency department. For his open right elbow fracture, emergency medical services had given him fentanyl en route, and when he arrived he was less responsive. As the patient reported, he had been on a scaffold 16 feet high when it began to give way. He jumped for another scaffold, 3 to 4 feet away, but came up short and, in an attempt to stop himself from falling, grabbed onto it with arms extended and above his head. His hands and arms were immediately pulled up in full extension. When both shoulders became dislocated, he could not hold on and fell to the ground, landing on a buttock. He did not lose consciousness.
Physical examination revealed both arms abducted at the shoulder, and elbows extended (Figure 1). 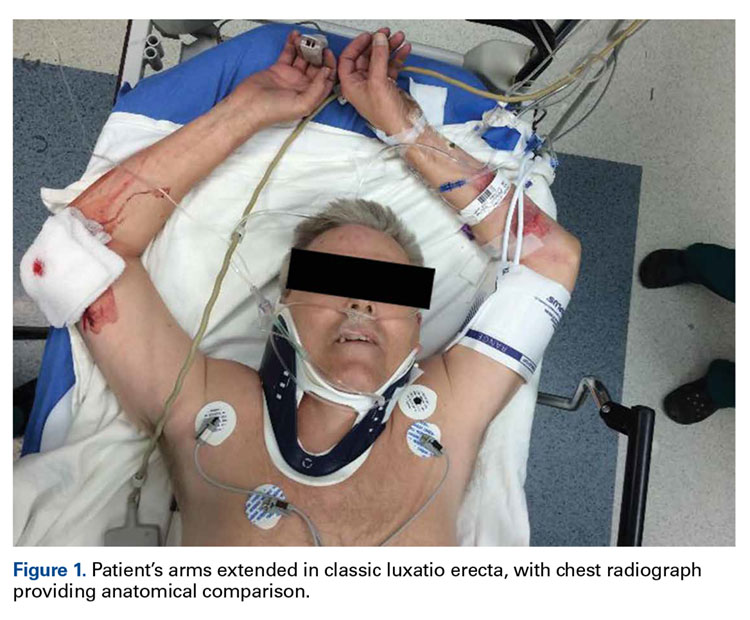
Radiographs confirmed the diagnosis and showed bilateral nondisplaced proximal humeral fractures of the greater tuberosity (Figure 2). 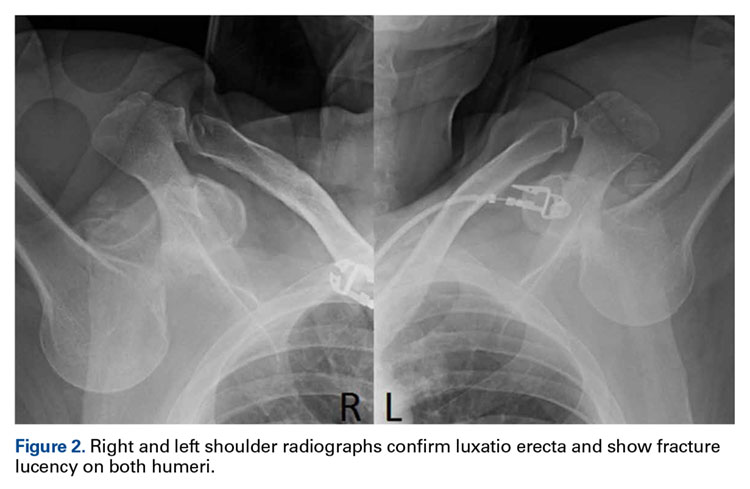
For the shoulder reductions, we administered propofol for conscious sedation and fentanyl for analgesia. Then, a sheet was wrapped supraclavicular and pulled across the torso inferiorly to allow countertraction when pulling the arm superiorly on the axial line. Another countertraction sheet was placed on the opposite side. For each arm, the countertraction was pulled inferiorly when the arm was pulled superiorly, both on the longitudinal plane. The arm was then gently rotated in adduction until reduction was achieved.
The right shoulder reduced relatively easily. The left shoulder reduced into an anterior dislocation—a relatively uncommon outcome in in-line traction attempts.3 (Reduction into anterior dislocation can also be a desired result in a specific technique of 2-step reduction, as described by Nho and colleagues.4) The patient’s anterior dislocation was then easily reduced into anatomical position with use of the Kocher technique of arm adduction with elbow flexion, followed by external rotation, and then finally into anatomical position with internal rotation.5 Both arms were then immobilized in full adduction with bilateral slings. The patient was admitted for further treatment of multiple fractures of the arms and vertebrae.
He was discharged in bilateral shoulder slings to an extended-care facility for physical therapy. One month after discharge, he could not elevate his arms and had minimal use of them. Two weeks later, magnetic resonance imaging showed a “comminuted greater tuberosity fracture with new displacement of fragments involving the attachment of the supraspinatus and infraspinatus; posterior subluxation of the glenohumeral joint with evidence of posterior and anterior labral tears; and large glenohumeral joint effusion.” The patient opted for surgical repair and underwent left shoulder arthroscopy with extensive débridement, open rotator cuff repair, open greater tuberosity reduction and internal fixation, and open biceps tenodesis. He was then discharged back to an extended-care facility to continue rehabilitation. One and a half months after surgery, he started the physical therapy phase of the massive rotator cuff repair protocol. He declined reverse total shoulder arthroplasty (RTSA).
Four and a half months after injury (3 months after surgery), the left shoulder demonstrated 20° of flexion and 70° to 110° of abduction (external rotation not tested), and the right shoulder demonstrated 30° of flexion and 70° to 110° of abduction (external rotation not tested). He had no instability and no lag with good external rotation.
Six months after injury, the patient still could not lift his arms above his head. He likely would not be able to do so without RTSA, which he again declined. He continued physical therapy and clinical follow-ups.
Discussion
Although inferior shoulder dislocations are rare, they carry a higher rate of complications, most of which our patient experienced. Our patient had bilateral humeral head fractures, which occur in 80% of cases.6 Postreduction CT showed the degree of his fractures (Figure 3).
Our patient also had reduced sensation in the axillary nerve distribution, which occurs in 60% of inferior dislocations.6 Axillary nerve injuries produce numbness in the lateral arm or posterior shoulder and weakness with shoulder flexion, abduction, and external rotation.7 In our patient’s case, sensation returned after reduction, which is typical (most patients have a positive prognosis).8 As the shoulder dislocates inferiorly, the humeral head tears the glenohumeral capsule inferiorly, which can damage the axillary artery. This artery becomes the brachial and eventually the radial and ulnar arteries, which can have decreased or absent pulses with injury.
Inferior dislocations are also associated with abundant soft-tissue injuries, including torn rotator cuff, shoulder capsule avulsion, and disruption of adjacent muscles (supraspinatus, infraspinatus, teres minor, subscapularis, pectoralis major).9Luxatio erecta is relatively easy to diagnose given the unmistakable arm positioning. The key for the physician is first to assess for the many possible complications, then to administer the proper sedation and analgesia for reduction, and finally to reassess for complications.
Am J Orthop. 2016;45(6):E328-E330. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
1. Camarda L, Martorana U, D’Arienzo M. A case of bilateral luxatio erecta. J Orthop Traumatol. 2009;10(2):97-99.
2. Musmeci E, Gaspari D, Sandri A, Regis D, Bartolozzi P. Bilateral luxatio erecta humeri associated with a unilateral brachial plexus and bilateral rotator cuff injuries: a case report. J Orthop Trauma. 2008;22(7):498-500.
3. Lam AC, Shih RD. Luxatio erecta complicated by anterior shoulder dislocation during reduction. West J Emerg Med. 2010;11(1):28-30.
4. Nho SJ, Dodson CC, Bardzik KF, Brophy RH, Domb BG, MacGillivray JD. The two-step maneuver for closed reduction of inferior glenohumeral dislocation (luxatio erecta to anterior dislocation to reduction). J Orthop Trauma. 2006;20(5):354-357.
5. Beattie TF, Steedman DJ, McGowan A, Robertson CE. A comparison of the Milch and Kocher techniques for acute anterior dislocation of the shoulder. Injury. 1986;17(5):349-352.
6. Mallon WJ, Bassett FH 3rd, Goldner RD. Luxatio erecta: the inferior glenohumeral dislocation. J Orthop Trauma. 1990;4(1):19-24.
7. Miller T. Peripheral nerve injuries at the shoulder. J Manipulative Physiol Ther. 1998;6(4):170-183.
8. Groh GI, Wirth MA, Rockwood CA Jr. Results of treatment of luxatio erecta (inferior shoulder dislocation). J Shoulder Elbow Surg. 2010;19(3):423-426.
9. Garcia R, Ponsky T, Brody F, Long J. Bilateral luxatio erecta complicated by venous thrombosis. J Trauma. 2006;60(5):1132-1134.
Unilateral inferior shoulder dislocation (luxatio erecta) is uncommon, accounting for only 0.5% of all shoulder dislocations.1 Bilateral luxatio erecta is extremely rare, having been described fewer than 20 times in the literature. The most common etiology is hyperabduction causing the humerus to lever on the acromion; less common is axial loading onto a fully abducted arm and an extended elbow.2 Hyperabduction can occur when a person grabs an object in an attempt to stop a fall, as occurred in the present case. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 58-year-old man with a trauma injury presented to our emergency department. For his open right elbow fracture, emergency medical services had given him fentanyl en route, and when he arrived he was less responsive. As the patient reported, he had been on a scaffold 16 feet high when it began to give way. He jumped for another scaffold, 3 to 4 feet away, but came up short and, in an attempt to stop himself from falling, grabbed onto it with arms extended and above his head. His hands and arms were immediately pulled up in full extension. When both shoulders became dislocated, he could not hold on and fell to the ground, landing on a buttock. He did not lose consciousness.
Physical examination revealed both arms abducted at the shoulder, and elbows extended (Figure 1).
Radiographs confirmed the diagnosis and showed bilateral nondisplaced proximal humeral fractures of the greater tuberosity (Figure 2).
For the shoulder reductions, we administered propofol for conscious sedation and fentanyl for analgesia. Then, a sheet was wrapped supraclavicular and pulled across the torso inferiorly to allow countertraction when pulling the arm superiorly on the axial line. Another countertraction sheet was placed on the opposite side. For each arm, the countertraction was pulled inferiorly when the arm was pulled superiorly, both on the longitudinal plane. The arm was then gently rotated in adduction until reduction was achieved.
The right shoulder reduced relatively easily. The left shoulder reduced into an anterior dislocation—a relatively uncommon outcome in in-line traction attempts.3 (Reduction into anterior dislocation can also be a desired result in a specific technique of 2-step reduction, as described by Nho and colleagues.4) The patient’s anterior dislocation was then easily reduced into anatomical position with use of the Kocher technique of arm adduction with elbow flexion, followed by external rotation, and then finally into anatomical position with internal rotation.5 Both arms were then immobilized in full adduction with bilateral slings. The patient was admitted for further treatment of multiple fractures of the arms and vertebrae.
He was discharged in bilateral shoulder slings to an extended-care facility for physical therapy. One month after discharge, he could not elevate his arms and had minimal use of them. Two weeks later, magnetic resonance imaging showed a “comminuted greater tuberosity fracture with new displacement of fragments involving the attachment of the supraspinatus and infraspinatus; posterior subluxation of the glenohumeral joint with evidence of posterior and anterior labral tears; and large glenohumeral joint effusion.” The patient opted for surgical repair and underwent left shoulder arthroscopy with extensive débridement, open rotator cuff repair, open greater tuberosity reduction and internal fixation, and open biceps tenodesis. He was then discharged back to an extended-care facility to continue rehabilitation. One and a half months after surgery, he started the physical therapy phase of the massive rotator cuff repair protocol. He declined reverse total shoulder arthroplasty (RTSA).
Four and a half months after injury (3 months after surgery), the left shoulder demonstrated 20° of flexion and 70° to 110° of abduction (external rotation not tested), and the right shoulder demonstrated 30° of flexion and 70° to 110° of abduction (external rotation not tested). He had no instability and no lag with good external rotation.
Six months after injury, the patient still could not lift his arms above his head. He likely would not be able to do so without RTSA, which he again declined. He continued physical therapy and clinical follow-ups.
Discussion
Although inferior shoulder dislocations are rare, they carry a higher rate of complications, most of which our patient experienced. Our patient had bilateral humeral head fractures, which occur in 80% of cases.6 Postreduction CT showed the degree of his fractures (Figure 3).
Our patient also had reduced sensation in the axillary nerve distribution, which occurs in 60% of inferior dislocations.6 Axillary nerve injuries produce numbness in the lateral arm or posterior shoulder and weakness with shoulder flexion, abduction, and external rotation.7 In our patient’s case, sensation returned after reduction, which is typical (most patients have a positive prognosis).8 As the shoulder dislocates inferiorly, the humeral head tears the glenohumeral capsule inferiorly, which can damage the axillary artery. This artery becomes the brachial and eventually the radial and ulnar arteries, which can have decreased or absent pulses with injury.
Inferior dislocations are also associated with abundant soft-tissue injuries, including torn rotator cuff, shoulder capsule avulsion, and disruption of adjacent muscles (supraspinatus, infraspinatus, teres minor, subscapularis, pectoralis major).9Luxatio erecta is relatively easy to diagnose given the unmistakable arm positioning. The key for the physician is first to assess for the many possible complications, then to administer the proper sedation and analgesia for reduction, and finally to reassess for complications.
Am J Orthop. 2016;45(6):E328-E330. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
Unilateral inferior shoulder dislocation (luxatio erecta) is uncommon, accounting for only 0.5% of all shoulder dislocations.1 Bilateral luxatio erecta is extremely rare, having been described fewer than 20 times in the literature. The most common etiology is hyperabduction causing the humerus to lever on the acromion; less common is axial loading onto a fully abducted arm and an extended elbow.2 Hyperabduction can occur when a person grabs an object in an attempt to stop a fall, as occurred in the present case. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 58-year-old man with a trauma injury presented to our emergency department. For his open right elbow fracture, emergency medical services had given him fentanyl en route, and when he arrived he was less responsive. As the patient reported, he had been on a scaffold 16 feet high when it began to give way. He jumped for another scaffold, 3 to 4 feet away, but came up short and, in an attempt to stop himself from falling, grabbed onto it with arms extended and above his head. His hands and arms were immediately pulled up in full extension. When both shoulders became dislocated, he could not hold on and fell to the ground, landing on a buttock. He did not lose consciousness.
Physical examination revealed both arms abducted at the shoulder, and elbows extended (Figure 1).
Radiographs confirmed the diagnosis and showed bilateral nondisplaced proximal humeral fractures of the greater tuberosity (Figure 2).
For the shoulder reductions, we administered propofol for conscious sedation and fentanyl for analgesia. Then, a sheet was wrapped supraclavicular and pulled across the torso inferiorly to allow countertraction when pulling the arm superiorly on the axial line. Another countertraction sheet was placed on the opposite side. For each arm, the countertraction was pulled inferiorly when the arm was pulled superiorly, both on the longitudinal plane. The arm was then gently rotated in adduction until reduction was achieved.
The right shoulder reduced relatively easily. The left shoulder reduced into an anterior dislocation—a relatively uncommon outcome in in-line traction attempts.3 (Reduction into anterior dislocation can also be a desired result in a specific technique of 2-step reduction, as described by Nho and colleagues.4) The patient’s anterior dislocation was then easily reduced into anatomical position with use of the Kocher technique of arm adduction with elbow flexion, followed by external rotation, and then finally into anatomical position with internal rotation.5 Both arms were then immobilized in full adduction with bilateral slings. The patient was admitted for further treatment of multiple fractures of the arms and vertebrae.
He was discharged in bilateral shoulder slings to an extended-care facility for physical therapy. One month after discharge, he could not elevate his arms and had minimal use of them. Two weeks later, magnetic resonance imaging showed a “comminuted greater tuberosity fracture with new displacement of fragments involving the attachment of the supraspinatus and infraspinatus; posterior subluxation of the glenohumeral joint with evidence of posterior and anterior labral tears; and large glenohumeral joint effusion.” The patient opted for surgical repair and underwent left shoulder arthroscopy with extensive débridement, open rotator cuff repair, open greater tuberosity reduction and internal fixation, and open biceps tenodesis. He was then discharged back to an extended-care facility to continue rehabilitation. One and a half months after surgery, he started the physical therapy phase of the massive rotator cuff repair protocol. He declined reverse total shoulder arthroplasty (RTSA).
Four and a half months after injury (3 months after surgery), the left shoulder demonstrated 20° of flexion and 70° to 110° of abduction (external rotation not tested), and the right shoulder demonstrated 30° of flexion and 70° to 110° of abduction (external rotation not tested). He had no instability and no lag with good external rotation.
Six months after injury, the patient still could not lift his arms above his head. He likely would not be able to do so without RTSA, which he again declined. He continued physical therapy and clinical follow-ups.
Discussion
Although inferior shoulder dislocations are rare, they carry a higher rate of complications, most of which our patient experienced. Our patient had bilateral humeral head fractures, which occur in 80% of cases.6 Postreduction CT showed the degree of his fractures (Figure 3).
Our patient also had reduced sensation in the axillary nerve distribution, which occurs in 60% of inferior dislocations.6 Axillary nerve injuries produce numbness in the lateral arm or posterior shoulder and weakness with shoulder flexion, abduction, and external rotation.7 In our patient’s case, sensation returned after reduction, which is typical (most patients have a positive prognosis).8 As the shoulder dislocates inferiorly, the humeral head tears the glenohumeral capsule inferiorly, which can damage the axillary artery. This artery becomes the brachial and eventually the radial and ulnar arteries, which can have decreased or absent pulses with injury.
Inferior dislocations are also associated with abundant soft-tissue injuries, including torn rotator cuff, shoulder capsule avulsion, and disruption of adjacent muscles (supraspinatus, infraspinatus, teres minor, subscapularis, pectoralis major).9Luxatio erecta is relatively easy to diagnose given the unmistakable arm positioning. The key for the physician is first to assess for the many possible complications, then to administer the proper sedation and analgesia for reduction, and finally to reassess for complications.
Am J Orthop. 2016;45(6):E328-E330. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
1. Camarda L, Martorana U, D’Arienzo M. A case of bilateral luxatio erecta. J Orthop Traumatol. 2009;10(2):97-99.
2. Musmeci E, Gaspari D, Sandri A, Regis D, Bartolozzi P. Bilateral luxatio erecta humeri associated with a unilateral brachial plexus and bilateral rotator cuff injuries: a case report. J Orthop Trauma. 2008;22(7):498-500.
3. Lam AC, Shih RD. Luxatio erecta complicated by anterior shoulder dislocation during reduction. West J Emerg Med. 2010;11(1):28-30.
4. Nho SJ, Dodson CC, Bardzik KF, Brophy RH, Domb BG, MacGillivray JD. The two-step maneuver for closed reduction of inferior glenohumeral dislocation (luxatio erecta to anterior dislocation to reduction). J Orthop Trauma. 2006;20(5):354-357.
5. Beattie TF, Steedman DJ, McGowan A, Robertson CE. A comparison of the Milch and Kocher techniques for acute anterior dislocation of the shoulder. Injury. 1986;17(5):349-352.
6. Mallon WJ, Bassett FH 3rd, Goldner RD. Luxatio erecta: the inferior glenohumeral dislocation. J Orthop Trauma. 1990;4(1):19-24.
7. Miller T. Peripheral nerve injuries at the shoulder. J Manipulative Physiol Ther. 1998;6(4):170-183.
8. Groh GI, Wirth MA, Rockwood CA Jr. Results of treatment of luxatio erecta (inferior shoulder dislocation). J Shoulder Elbow Surg. 2010;19(3):423-426.
9. Garcia R, Ponsky T, Brody F, Long J. Bilateral luxatio erecta complicated by venous thrombosis. J Trauma. 2006;60(5):1132-1134.
1. Camarda L, Martorana U, D’Arienzo M. A case of bilateral luxatio erecta. J Orthop Traumatol. 2009;10(2):97-99.
2. Musmeci E, Gaspari D, Sandri A, Regis D, Bartolozzi P. Bilateral luxatio erecta humeri associated with a unilateral brachial plexus and bilateral rotator cuff injuries: a case report. J Orthop Trauma. 2008;22(7):498-500.
3. Lam AC, Shih RD. Luxatio erecta complicated by anterior shoulder dislocation during reduction. West J Emerg Med. 2010;11(1):28-30.
4. Nho SJ, Dodson CC, Bardzik KF, Brophy RH, Domb BG, MacGillivray JD. The two-step maneuver for closed reduction of inferior glenohumeral dislocation (luxatio erecta to anterior dislocation to reduction). J Orthop Trauma. 2006;20(5):354-357.
5. Beattie TF, Steedman DJ, McGowan A, Robertson CE. A comparison of the Milch and Kocher techniques for acute anterior dislocation of the shoulder. Injury. 1986;17(5):349-352.
6. Mallon WJ, Bassett FH 3rd, Goldner RD. Luxatio erecta: the inferior glenohumeral dislocation. J Orthop Trauma. 1990;4(1):19-24.
7. Miller T. Peripheral nerve injuries at the shoulder. J Manipulative Physiol Ther. 1998;6(4):170-183.
8. Groh GI, Wirth MA, Rockwood CA Jr. Results of treatment of luxatio erecta (inferior shoulder dislocation). J Shoulder Elbow Surg. 2010;19(3):423-426.
9. Garcia R, Ponsky T, Brody F, Long J. Bilateral luxatio erecta complicated by venous thrombosis. J Trauma. 2006;60(5):1132-1134.

Intrauterine Device Migration
Although intrauterine devices (IUDs) are a mainstay of reversible contraception, they do carry the risk of complications, including septic abortion, abscess formation, ectopic pregnancy, bleeding, and uterine perforation.1 Although perforation is a relatively rare complication, occurring in 0.3 to 2.6 per 1,000 insertions for levonorgestrel-releasing intrauterine systems and 0.3 to 2.2 per 1,000 insertions for copper IUDs, it can lead to serious complications, including IUD migration to various sites.2 Most patients with uterine perforation and IUD migration present with abdominal pain and bleeding; however, 30% of patients are asymptomatic.3
This article presents the case of a young woman who was diagnosed with IUD migration into the abdominal cavity. I discuss the management of this uncommon complication, and stress the importance of adequate education for both patients and health care providers regarding proper surveillance.
Case
A 33-year-old woman (gravida 4, para 4, live 4) presented to our ED for evaluation of rectal bleeding that she had experienced intermittently over the past 2 years. She reported that the first occurrence had been 2 years ago, starting a few weeks after she had a cesarean delivery. The patient described the initial episode as bright red blood mixed with stool. She stated that subsequent episodes had been intermittent, felt as if she were “passing rocks” through her abdomen and rectum, and were accompanied by streaks of blood covering her stool. The day before the patient presented to the ED, she had experienced a second episode of a large bowel movement mixed with blood and accompanied by weakness, which prompted her to seek treatment.
A review of the patient’s symptoms revealed abdominal pain and weakness. She denied any bleeding disorders, fever, chills, sick contacts, anal trauma, presyncope, syncope, nausea, vomiting, diarrhea, or constipation. She further denied any prescription-medication use, illicit drug use, or smoking, but admitted to occasional alcohol use. Her last menstrual period had been 3 weeks prior to presentation. She denied any history of cancer or abnormal Pap smears. Her gynecologic history was significant for chlamydia and trichomoniasis, for which she had been treated. The patient’s surgical history was pertinent for umbilical hernia repair with surgical mesh.
On physical examination, the patient was mildly hypotensive (blood pressure, 97/78 mm Hg) but had a normal heart rate. She had mild conjunctival pallor. The abdominal examination exhibited normoactive bowel sounds with diffuse lower abdominal tenderness to deep palpation, but without rebound, guarding, or distension. Rectal examination revealed a small internal hemorrhoid at the 6 o’clock position (no active bleeding) and an external hemorrhoid with some tenderness to palpation; the external hemorrhoid was not thrombosed, had no signs of infection, and was the same color as the surrounding skin.
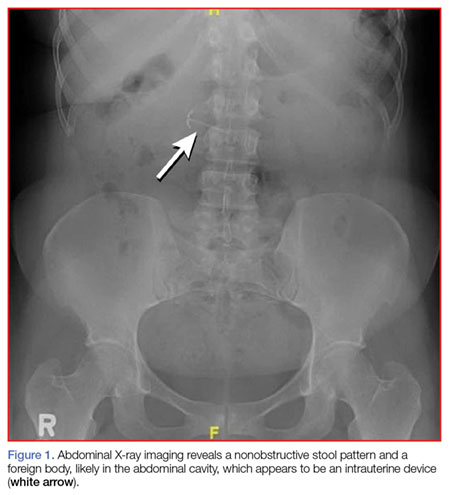
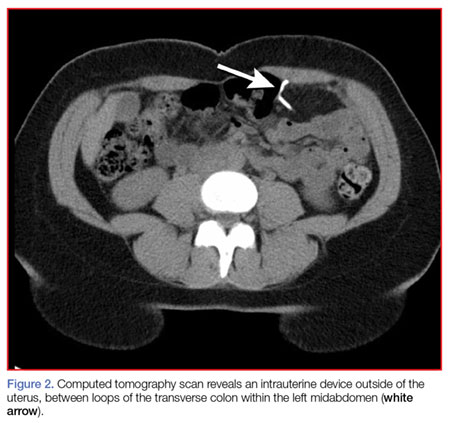
A fecal occult blood screen was negative, and a serum pregnancy test was also negative. Complete blood count, basic metabolic profile, and urinalysis were all unremarkable and within normal ranges. Abdominal X-ray revealed a nonobstructive stool pattern and a foreign body, likely in the abdominal cavity, which appeared to be an IUD (Figure 1). Computed tomography (CT) scans of the abdomen and pelvis without contrast were performed to accurately locate the foreign body and to assess for any complications. The CT scans revealed an IUD outside of the uterus, between loops of the transverse colon within the left midabdomen (Figure 2). There were no signs of infection, fluid, or free air. There were also findings of colonic diverticula and narrowed lumen, which were suggestive of diverticulosis.
The patient stated that the IUD had been placed several months after the vaginal birth of her third child. She continued to have normal menstrual periods with the IUD in place. Seven years later, she became pregnant with her fourth child, who was delivered via cesarean, secondary to fetal malpositioning. The IUD was not removed during the cesarean delivery.
Based on the CT scan findings, gynecology services was consulted, and the gynecologist recommended immediate follow-up in a gynecology clinic. The patient was discharged on a bowel regimen. She was assessed in a gynecology clinic 4 days later, where she was found to have a mobile retroverted uterus without tenderness or signs of infection. She underwent exploratory laparoscopy, during which the IUD was removed from the omentum in the left upper abdomen without complications.
Discussion
The IUD has had great acceptance among women since the 1960s. According to the World Health Organization, approximately 14.3% of women used an IUD in 2009.4 Although complications are rare, the most serious are perforation of the uterus and migration of the IUD into adjacent organs.1
Risk factors of uterine perforation include clinician inexperience in IUD placement, an immobile uterus, a retroverted uterus, and the presence of a myometrial defect.4 Heinemann et al2 also suggested that breastfeeding and IUD placement soon after a delivery (≤36 weeks) are independent risk factors, and the presence of both factors has an additive increase in risk of perforation.
Primary rupture of the uterus has been reported at the time of IUD insertion, but secondary or delayed rupture is more common and seems to be due to the spasms of the uterus.5 Although 85% of perforations do not affect other organs, the remaining 15% lead to complications in the adjacent visceral organs.6 The most frequent sites of migration are to the omentum (26.7%), pouch of Douglas (21.5%), large bowel (10.4%), myometrium (7.4%), broad ligament (6.7%), abdominal cavity (5.2%), adhesion to ileal loop serosa (4.4%) or large bowel serosa (3.7%), and mesentery (3%).7 Rare sites are to the appendix, abdominal wall, ovary, and bladder.7
Intrauterine device migration should be suspected in patients who become pregnant after IUD placement (as was the case for our patient), when the “threads” or string cannot be located while attempting to remove an IUD, or when a patient has an “expulsed” IUD without observation of the device thereafter. Even though expulsion of the device happens in approximately 8 per 1,000 insertions, uterine perforation is also a possibility in the case of a “lost” IUD.8 When a lost IUD is suspected, a pelvic examination should be performed to assess for threads or string location. If unsuccessful, ultrasound or plain abdominal radiographic imaging may be used to locate the IUD. Once IUD migration has been confirmed, cross-sectional imaging such as CT scans or magnetic resonance imaging (MRI) is suggested to rule out adjacent organ involvement before considering surgical removal.4 If colonic involvement is suspected, colonoscopy can be used to confirm the diagnosis before operative removal.4
Although management of a migrated IUD in an asymptomatic patient is controversial, there appears to be a consensus that all extrauterine devices should be removed unless the patient’s surgical risk is excessive.1,5,9 Retrieval of an IUD can be performed by laparotomy or laparoscopy.10,11
To avoid these complications and interventions, IUDs should be inserted by an appropriately trained professional, after proper patient selection. These devices should be monitored by periodic examinations, either by medical professionals or by well-informed patients. This can be done by either checking for the threads or string in the cervical opening or by ultrasound imaging to confirm the location of the IUD.
Conclusion
Although many patients with uterine perforation and IUD migration present with symptoms, approximately 30% are asymptomatic.3 If a patient has a lost IUD and the threads or string is not visible during pelvic examination, appropriate work-up, including transvaginal or transabdominal ultrasound or radiographs, should be obtained to confirm the position of the IUD. If IUD migration is suspected, cross-sectional imaging, such as CT scans or MRI, is recommended to rule out adjacent organ involvement before considering surgical removal.4
Even though only 15% of migrated IUDs lead to complications in the adjacent visceral organs,6 surgical removal of the IUD is advised regardless of the presence of symptoms or identified complications. Importantly, to prevent the delayed diagnosis and morbidity of IUD migration, patients with IUDs should be educated about the possibility of migration and the importance of regular self-examination for missing threads or string.
1. Hoşcan MB, Koşar A, Gümüştaş U, Güney M. Intravesical migration of intrauterine device resulting in pregnancy. Int J Urol. 2006;13(3):301-302.
2. Heinemann K, Reed S, Moehner S, Minh TD. Risk of uterine perforation with levonorgestrel-releasing and copper intrauterine devices in the European Active Surveillance Study on Intrauterine Devices. Contraception. 2015;91(4):274-279.
3. Singh SP, Mangla D, Chawan J, Haq AU. Asymptomatic presentation of silent uterine perforation by Cu-T 380A: a case report with review of literature. Int J Reprod Contracept Obstet Gynecol. 2014;3(4):1157-1159.
4. Akpinar F, Ozgur EN, Yilmaz S, Ustaoglu O. Sigmoid colon migration of an intrauterine device. Case Rep Obstet Gynecol. 2014;2014:207659.
5. Rahnemai-Azar AA, Apfel T, Naghshizadian R, Cosgrove JM, Farkas DT. Laparoscopic removal of migrated intrauterine device embedded in intestine. JSLS. 2014;18(3).
6. Zakin D, Stern WZ, Rosenblatt R. Complete and partial uterine perforation and embedding following insertion of intrauterine devices. II. Diagnostic methods, prevention, and management. Obstet Gynecol Surv. 1981;36(8):401-417.
7. Gill RS, Mok D, Hudson M, Shi X, Birch DW, Karmali S. Laparoscopic removal of an intra-abdominal intrauterine device: case and systematic review. Contraception. 2012;85(1):15-18.
8. Paterson H, Ashton J, Harrison-Woolrych M. A nationwide cohort study of the use of the levonorgestrel intrauterine device in New Zealand adolescents. Contraception. 2009;79(6):433-438.
9. Gorsline JC, Osborne NG. Management of the missing intrauterine contraceptive device: report of a case. Am J Obstet Gynecol. 1985;153(2):228-229.
10. Mederos R, Humaran L, Minervini D. Surgical removal of an intrauterine device perforating the sigmoid colon: a case report. Int J Surg. 2008;6(6):e60-e62.
11. Chi E, Rosenfeld D, Sokol TP. Laparoscopic removal of an intrauterine device perforating the sigmoid colon: a case report and review of the literature. Am Surg. 2005;71(12):1055-1057.
Although intrauterine devices (IUDs) are a mainstay of reversible contraception, they do carry the risk of complications, including septic abortion, abscess formation, ectopic pregnancy, bleeding, and uterine perforation.1 Although perforation is a relatively rare complication, occurring in 0.3 to 2.6 per 1,000 insertions for levonorgestrel-releasing intrauterine systems and 0.3 to 2.2 per 1,000 insertions for copper IUDs, it can lead to serious complications, including IUD migration to various sites.2 Most patients with uterine perforation and IUD migration present with abdominal pain and bleeding; however, 30% of patients are asymptomatic.3
This article presents the case of a young woman who was diagnosed with IUD migration into the abdominal cavity. I discuss the management of this uncommon complication, and stress the importance of adequate education for both patients and health care providers regarding proper surveillance.
Case
A 33-year-old woman (gravida 4, para 4, live 4) presented to our ED for evaluation of rectal bleeding that she had experienced intermittently over the past 2 years. She reported that the first occurrence had been 2 years ago, starting a few weeks after she had a cesarean delivery. The patient described the initial episode as bright red blood mixed with stool. She stated that subsequent episodes had been intermittent, felt as if she were “passing rocks” through her abdomen and rectum, and were accompanied by streaks of blood covering her stool. The day before the patient presented to the ED, she had experienced a second episode of a large bowel movement mixed with blood and accompanied by weakness, which prompted her to seek treatment.
A review of the patient’s symptoms revealed abdominal pain and weakness. She denied any bleeding disorders, fever, chills, sick contacts, anal trauma, presyncope, syncope, nausea, vomiting, diarrhea, or constipation. She further denied any prescription-medication use, illicit drug use, or smoking, but admitted to occasional alcohol use. Her last menstrual period had been 3 weeks prior to presentation. She denied any history of cancer or abnormal Pap smears. Her gynecologic history was significant for chlamydia and trichomoniasis, for which she had been treated. The patient’s surgical history was pertinent for umbilical hernia repair with surgical mesh.
On physical examination, the patient was mildly hypotensive (blood pressure, 97/78 mm Hg) but had a normal heart rate. She had mild conjunctival pallor. The abdominal examination exhibited normoactive bowel sounds with diffuse lower abdominal tenderness to deep palpation, but without rebound, guarding, or distension. Rectal examination revealed a small internal hemorrhoid at the 6 o’clock position (no active bleeding) and an external hemorrhoid with some tenderness to palpation; the external hemorrhoid was not thrombosed, had no signs of infection, and was the same color as the surrounding skin.
A fecal occult blood screen was negative, and a serum pregnancy test was also negative. Complete blood count, basic metabolic profile, and urinalysis were all unremarkable and within normal ranges. Abdominal X-ray revealed a nonobstructive stool pattern and a foreign body, likely in the abdominal cavity, which appeared to be an IUD (Figure 1). Computed tomography (CT) scans of the abdomen and pelvis without contrast were performed to accurately locate the foreign body and to assess for any complications. The CT scans revealed an IUD outside of the uterus, between loops of the transverse colon within the left midabdomen (Figure 2). There were no signs of infection, fluid, or free air. There were also findings of colonic diverticula and narrowed lumen, which were suggestive of diverticulosis.
The patient stated that the IUD had been placed several months after the vaginal birth of her third child. She continued to have normal menstrual periods with the IUD in place. Seven years later, she became pregnant with her fourth child, who was delivered via cesarean, secondary to fetal malpositioning. The IUD was not removed during the cesarean delivery.
Based on the CT scan findings, gynecology services was consulted, and the gynecologist recommended immediate follow-up in a gynecology clinic. The patient was discharged on a bowel regimen. She was assessed in a gynecology clinic 4 days later, where she was found to have a mobile retroverted uterus without tenderness or signs of infection. She underwent exploratory laparoscopy, during which the IUD was removed from the omentum in the left upper abdomen without complications.
Discussion
The IUD has had great acceptance among women since the 1960s. According to the World Health Organization, approximately 14.3% of women used an IUD in 2009.4 Although complications are rare, the most serious are perforation of the uterus and migration of the IUD into adjacent organs.1
Risk factors of uterine perforation include clinician inexperience in IUD placement, an immobile uterus, a retroverted uterus, and the presence of a myometrial defect.4 Heinemann et al2 also suggested that breastfeeding and IUD placement soon after a delivery (≤36 weeks) are independent risk factors, and the presence of both factors has an additive increase in risk of perforation.
Primary rupture of the uterus has been reported at the time of IUD insertion, but secondary or delayed rupture is more common and seems to be due to the spasms of the uterus.5 Although 85% of perforations do not affect other organs, the remaining 15% lead to complications in the adjacent visceral organs.6 The most frequent sites of migration are to the omentum (26.7%), pouch of Douglas (21.5%), large bowel (10.4%), myometrium (7.4%), broad ligament (6.7%), abdominal cavity (5.2%), adhesion to ileal loop serosa (4.4%) or large bowel serosa (3.7%), and mesentery (3%).7 Rare sites are to the appendix, abdominal wall, ovary, and bladder.7
Intrauterine device migration should be suspected in patients who become pregnant after IUD placement (as was the case for our patient), when the “threads” or string cannot be located while attempting to remove an IUD, or when a patient has an “expulsed” IUD without observation of the device thereafter. Even though expulsion of the device happens in approximately 8 per 1,000 insertions, uterine perforation is also a possibility in the case of a “lost” IUD.8 When a lost IUD is suspected, a pelvic examination should be performed to assess for threads or string location. If unsuccessful, ultrasound or plain abdominal radiographic imaging may be used to locate the IUD. Once IUD migration has been confirmed, cross-sectional imaging such as CT scans or magnetic resonance imaging (MRI) is suggested to rule out adjacent organ involvement before considering surgical removal.4 If colonic involvement is suspected, colonoscopy can be used to confirm the diagnosis before operative removal.4
Although management of a migrated IUD in an asymptomatic patient is controversial, there appears to be a consensus that all extrauterine devices should be removed unless the patient’s surgical risk is excessive.1,5,9 Retrieval of an IUD can be performed by laparotomy or laparoscopy.10,11
To avoid these complications and interventions, IUDs should be inserted by an appropriately trained professional, after proper patient selection. These devices should be monitored by periodic examinations, either by medical professionals or by well-informed patients. This can be done by either checking for the threads or string in the cervical opening or by ultrasound imaging to confirm the location of the IUD.
Conclusion
Although many patients with uterine perforation and IUD migration present with symptoms, approximately 30% are asymptomatic.3 If a patient has a lost IUD and the threads or string is not visible during pelvic examination, appropriate work-up, including transvaginal or transabdominal ultrasound or radiographs, should be obtained to confirm the position of the IUD. If IUD migration is suspected, cross-sectional imaging, such as CT scans or MRI, is recommended to rule out adjacent organ involvement before considering surgical removal.4
Even though only 15% of migrated IUDs lead to complications in the adjacent visceral organs,6 surgical removal of the IUD is advised regardless of the presence of symptoms or identified complications. Importantly, to prevent the delayed diagnosis and morbidity of IUD migration, patients with IUDs should be educated about the possibility of migration and the importance of regular self-examination for missing threads or string.
Although intrauterine devices (IUDs) are a mainstay of reversible contraception, they do carry the risk of complications, including septic abortion, abscess formation, ectopic pregnancy, bleeding, and uterine perforation.1 Although perforation is a relatively rare complication, occurring in 0.3 to 2.6 per 1,000 insertions for levonorgestrel-releasing intrauterine systems and 0.3 to 2.2 per 1,000 insertions for copper IUDs, it can lead to serious complications, including IUD migration to various sites.2 Most patients with uterine perforation and IUD migration present with abdominal pain and bleeding; however, 30% of patients are asymptomatic.3
This article presents the case of a young woman who was diagnosed with IUD migration into the abdominal cavity. I discuss the management of this uncommon complication, and stress the importance of adequate education for both patients and health care providers regarding proper surveillance.
Case
A 33-year-old woman (gravida 4, para 4, live 4) presented to our ED for evaluation of rectal bleeding that she had experienced intermittently over the past 2 years. She reported that the first occurrence had been 2 years ago, starting a few weeks after she had a cesarean delivery. The patient described the initial episode as bright red blood mixed with stool. She stated that subsequent episodes had been intermittent, felt as if she were “passing rocks” through her abdomen and rectum, and were accompanied by streaks of blood covering her stool. The day before the patient presented to the ED, she had experienced a second episode of a large bowel movement mixed with blood and accompanied by weakness, which prompted her to seek treatment.
A review of the patient’s symptoms revealed abdominal pain and weakness. She denied any bleeding disorders, fever, chills, sick contacts, anal trauma, presyncope, syncope, nausea, vomiting, diarrhea, or constipation. She further denied any prescription-medication use, illicit drug use, or smoking, but admitted to occasional alcohol use. Her last menstrual period had been 3 weeks prior to presentation. She denied any history of cancer or abnormal Pap smears. Her gynecologic history was significant for chlamydia and trichomoniasis, for which she had been treated. The patient’s surgical history was pertinent for umbilical hernia repair with surgical mesh.
On physical examination, the patient was mildly hypotensive (blood pressure, 97/78 mm Hg) but had a normal heart rate. She had mild conjunctival pallor. The abdominal examination exhibited normoactive bowel sounds with diffuse lower abdominal tenderness to deep palpation, but without rebound, guarding, or distension. Rectal examination revealed a small internal hemorrhoid at the 6 o’clock position (no active bleeding) and an external hemorrhoid with some tenderness to palpation; the external hemorrhoid was not thrombosed, had no signs of infection, and was the same color as the surrounding skin.
A fecal occult blood screen was negative, and a serum pregnancy test was also negative. Complete blood count, basic metabolic profile, and urinalysis were all unremarkable and within normal ranges. Abdominal X-ray revealed a nonobstructive stool pattern and a foreign body, likely in the abdominal cavity, which appeared to be an IUD (Figure 1). Computed tomography (CT) scans of the abdomen and pelvis without contrast were performed to accurately locate the foreign body and to assess for any complications. The CT scans revealed an IUD outside of the uterus, between loops of the transverse colon within the left midabdomen (Figure 2). There were no signs of infection, fluid, or free air. There were also findings of colonic diverticula and narrowed lumen, which were suggestive of diverticulosis.
The patient stated that the IUD had been placed several months after the vaginal birth of her third child. She continued to have normal menstrual periods with the IUD in place. Seven years later, she became pregnant with her fourth child, who was delivered via cesarean, secondary to fetal malpositioning. The IUD was not removed during the cesarean delivery.
Based on the CT scan findings, gynecology services was consulted, and the gynecologist recommended immediate follow-up in a gynecology clinic. The patient was discharged on a bowel regimen. She was assessed in a gynecology clinic 4 days later, where she was found to have a mobile retroverted uterus without tenderness or signs of infection. She underwent exploratory laparoscopy, during which the IUD was removed from the omentum in the left upper abdomen without complications.
Discussion
The IUD has had great acceptance among women since the 1960s. According to the World Health Organization, approximately 14.3% of women used an IUD in 2009.4 Although complications are rare, the most serious are perforation of the uterus and migration of the IUD into adjacent organs.1
Risk factors of uterine perforation include clinician inexperience in IUD placement, an immobile uterus, a retroverted uterus, and the presence of a myometrial defect.4 Heinemann et al2 also suggested that breastfeeding and IUD placement soon after a delivery (≤36 weeks) are independent risk factors, and the presence of both factors has an additive increase in risk of perforation.
Primary rupture of the uterus has been reported at the time of IUD insertion, but secondary or delayed rupture is more common and seems to be due to the spasms of the uterus.5 Although 85% of perforations do not affect other organs, the remaining 15% lead to complications in the adjacent visceral organs.6 The most frequent sites of migration are to the omentum (26.7%), pouch of Douglas (21.5%), large bowel (10.4%), myometrium (7.4%), broad ligament (6.7%), abdominal cavity (5.2%), adhesion to ileal loop serosa (4.4%) or large bowel serosa (3.7%), and mesentery (3%).7 Rare sites are to the appendix, abdominal wall, ovary, and bladder.7
Intrauterine device migration should be suspected in patients who become pregnant after IUD placement (as was the case for our patient), when the “threads” or string cannot be located while attempting to remove an IUD, or when a patient has an “expulsed” IUD without observation of the device thereafter. Even though expulsion of the device happens in approximately 8 per 1,000 insertions, uterine perforation is also a possibility in the case of a “lost” IUD.8 When a lost IUD is suspected, a pelvic examination should be performed to assess for threads or string location. If unsuccessful, ultrasound or plain abdominal radiographic imaging may be used to locate the IUD. Once IUD migration has been confirmed, cross-sectional imaging such as CT scans or magnetic resonance imaging (MRI) is suggested to rule out adjacent organ involvement before considering surgical removal.4 If colonic involvement is suspected, colonoscopy can be used to confirm the diagnosis before operative removal.4
Although management of a migrated IUD in an asymptomatic patient is controversial, there appears to be a consensus that all extrauterine devices should be removed unless the patient’s surgical risk is excessive.1,5,9 Retrieval of an IUD can be performed by laparotomy or laparoscopy.10,11
To avoid these complications and interventions, IUDs should be inserted by an appropriately trained professional, after proper patient selection. These devices should be monitored by periodic examinations, either by medical professionals or by well-informed patients. This can be done by either checking for the threads or string in the cervical opening or by ultrasound imaging to confirm the location of the IUD.
Conclusion
Although many patients with uterine perforation and IUD migration present with symptoms, approximately 30% are asymptomatic.3 If a patient has a lost IUD and the threads or string is not visible during pelvic examination, appropriate work-up, including transvaginal or transabdominal ultrasound or radiographs, should be obtained to confirm the position of the IUD. If IUD migration is suspected, cross-sectional imaging, such as CT scans or MRI, is recommended to rule out adjacent organ involvement before considering surgical removal.4
Even though only 15% of migrated IUDs lead to complications in the adjacent visceral organs,6 surgical removal of the IUD is advised regardless of the presence of symptoms or identified complications. Importantly, to prevent the delayed diagnosis and morbidity of IUD migration, patients with IUDs should be educated about the possibility of migration and the importance of regular self-examination for missing threads or string.
1. Hoşcan MB, Koşar A, Gümüştaş U, Güney M. Intravesical migration of intrauterine device resulting in pregnancy. Int J Urol. 2006;13(3):301-302.
2. Heinemann K, Reed S, Moehner S, Minh TD. Risk of uterine perforation with levonorgestrel-releasing and copper intrauterine devices in the European Active Surveillance Study on Intrauterine Devices. Contraception. 2015;91(4):274-279.
3. Singh SP, Mangla D, Chawan J, Haq AU. Asymptomatic presentation of silent uterine perforation by Cu-T 380A: a case report with review of literature. Int J Reprod Contracept Obstet Gynecol. 2014;3(4):1157-1159.
4. Akpinar F, Ozgur EN, Yilmaz S, Ustaoglu O. Sigmoid colon migration of an intrauterine device. Case Rep Obstet Gynecol. 2014;2014:207659.
5. Rahnemai-Azar AA, Apfel T, Naghshizadian R, Cosgrove JM, Farkas DT. Laparoscopic removal of migrated intrauterine device embedded in intestine. JSLS. 2014;18(3).
6. Zakin D, Stern WZ, Rosenblatt R. Complete and partial uterine perforation and embedding following insertion of intrauterine devices. II. Diagnostic methods, prevention, and management. Obstet Gynecol Surv. 1981;36(8):401-417.
7. Gill RS, Mok D, Hudson M, Shi X, Birch DW, Karmali S. Laparoscopic removal of an intra-abdominal intrauterine device: case and systematic review. Contraception. 2012;85(1):15-18.
8. Paterson H, Ashton J, Harrison-Woolrych M. A nationwide cohort study of the use of the levonorgestrel intrauterine device in New Zealand adolescents. Contraception. 2009;79(6):433-438.
9. Gorsline JC, Osborne NG. Management of the missing intrauterine contraceptive device: report of a case. Am J Obstet Gynecol. 1985;153(2):228-229.
10. Mederos R, Humaran L, Minervini D. Surgical removal of an intrauterine device perforating the sigmoid colon: a case report. Int J Surg. 2008;6(6):e60-e62.
11. Chi E, Rosenfeld D, Sokol TP. Laparoscopic removal of an intrauterine device perforating the sigmoid colon: a case report and review of the literature. Am Surg. 2005;71(12):1055-1057.
1. Hoşcan MB, Koşar A, Gümüştaş U, Güney M. Intravesical migration of intrauterine device resulting in pregnancy. Int J Urol. 2006;13(3):301-302.
2. Heinemann K, Reed S, Moehner S, Minh TD. Risk of uterine perforation with levonorgestrel-releasing and copper intrauterine devices in the European Active Surveillance Study on Intrauterine Devices. Contraception. 2015;91(4):274-279.
3. Singh SP, Mangla D, Chawan J, Haq AU. Asymptomatic presentation of silent uterine perforation by Cu-T 380A: a case report with review of literature. Int J Reprod Contracept Obstet Gynecol. 2014;3(4):1157-1159.
4. Akpinar F, Ozgur EN, Yilmaz S, Ustaoglu O. Sigmoid colon migration of an intrauterine device. Case Rep Obstet Gynecol. 2014;2014:207659.
5. Rahnemai-Azar AA, Apfel T, Naghshizadian R, Cosgrove JM, Farkas DT. Laparoscopic removal of migrated intrauterine device embedded in intestine. JSLS. 2014;18(3).
6. Zakin D, Stern WZ, Rosenblatt R. Complete and partial uterine perforation and embedding following insertion of intrauterine devices. II. Diagnostic methods, prevention, and management. Obstet Gynecol Surv. 1981;36(8):401-417.
7. Gill RS, Mok D, Hudson M, Shi X, Birch DW, Karmali S. Laparoscopic removal of an intra-abdominal intrauterine device: case and systematic review. Contraception. 2012;85(1):15-18.
8. Paterson H, Ashton J, Harrison-Woolrych M. A nationwide cohort study of the use of the levonorgestrel intrauterine device in New Zealand adolescents. Contraception. 2009;79(6):433-438.
9. Gorsline JC, Osborne NG. Management of the missing intrauterine contraceptive device: report of a case. Am J Obstet Gynecol. 1985;153(2):228-229.
10. Mederos R, Humaran L, Minervini D. Surgical removal of an intrauterine device perforating the sigmoid colon: a case report. Int J Surg. 2008;6(6):e60-e62.
11. Chi E, Rosenfeld D, Sokol TP. Laparoscopic removal of an intrauterine device perforating the sigmoid colon: a case report and review of the literature. Am Surg. 2005;71(12):1055-1057.

Dengue Fever: Two Unexpected Findings
Dengue fever is the most commonly transmitted arboviral disease in the world, affecting an estimated 2.5 billion people who live in areas endemic to the virus. This exposure yields an annual incidence of 100 million cases of dengue, which translates into 250,000 cases of hemorrhagic fever. With an expanding geographic distribution and increasing number of epidemics, the World Health Organization (WHO) has classified dengue as a major public health concern.1 Enhanced globalization and changing climate patterns have resulted in a dramatic increase in the incidence of dengue in both North and Central America. Aggregate North and Central American data from 2010 to the present revealed over 1.7 million cases of dengue, nearly 80,000 of which were severe, and 747 deaths.2 Based on these statistics, dengue fever should be considered in the differential diagnosis of febrile ED patients in the developed world who had a history of recent travel. We present two cases that highlight the complexity of diagnosis and novel complications associated with dengue fever.
Case Reports
Case 1
A 24-year-old man presented to the ED with a 4-day history of intermittent fever of up to 102.02°F, which was accompanied by chills, myalgia, and rigors. The patient stated that he had visited Vietnam, Thailand, Indonesia, and Malaysia 8 days prior to presentation, and had experienced mosquito bites daily throughout his travels. He further noted that his symptoms had improved on day 3 of his illness, but acutely worsened on day 4, which prompted him to visit the ED. The patient’s primary complaint was a severe retro-orbital headache, fever, and one episode of epistaxis.
On physical examination, the patient had conjunctivitis and hepatosplenomegaly, but otherwise appeared well. His laboratory evaluation was significant for leukopenia (white blood cell [WBC] count, 2.40 x 109/L), thrombocytopenia (platelet count, 123 x 109/L), and a positive mononuclear spot test. Both dengue immunoglobulin G (IgG) and immunoglobulin M (IgM) tests sent from the ED were negative. Based on the patient’s thrombocytopenia and epistaxis, as well as concerns that the patient was entering into the critical phase of dengue fever, he was admitted to the inpatient hospital for observation.
The patient’s course improved during his stay with symptomatic treatment and blood-count monitoring, and he was discharged home on hospital day 3. He followed up at our hospital travel clinic the day after discharge; a repeat dengue IgM test taken during this visit came back positive.
Case 2
A 51-year-old man presented to the ED with a 3-day history of intermittent fever and diffuse myalgia. He reported chills, night sweats, and the feeling of abdominal fullness. He denied nausea, vomiting, or changes in the character of his stool. He had no known sick contacts, but reported he had traveled from the Philippines 3 days prior to presentation and that his symptoms had developed en route to the United States. The patient also denied any known tick, mosquito, or animal exposures. He said he had treated his symptoms with acetaminophen and nonsteroidal anti-inflammatory drugs. Prior to his arrival at the ED, he had twice presented to a walk-in clinic earlier that day. Repeated laboratory testing at the ED showed a decrease in WBC count from 42.0 x 109/L to 31.0 x 109/L, as well as a declining platelet count from 123 x 109/L to 87 x 109/L. On physical examination, the patient was ill-appearing, diaphoretic, and had a temperature of 100.6°F. His vital signs were otherwise within normal limits.
With the exception of a mild diffuse petechial rash on the patient’s thighs bilaterally, the physical examination was unrevealing. A tourniquet test (TT) to assess capillary fragility was performed at bedside, and yielded a positive result (Figure 1). Work-up further demonstrated a declining WBC of 2.70 x 109/L and declining platelet count of 65 x 109/L.
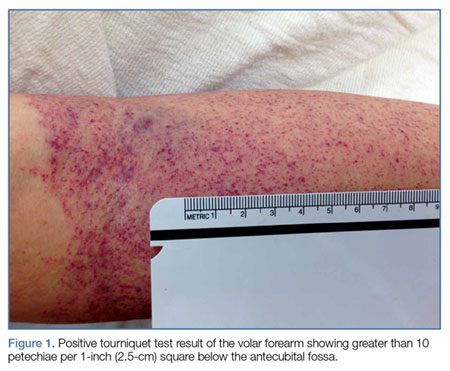
A polymerase chain reaction (PCR) test confirmed a diagnosis of dengue, with a positive dengue type-4 (DEN-4) serotype detection. Supportive care was initiated, and the patient was admitted to the inpatient hospital for continued treatment. He was discharged home on hospital day 5; however, he returned to the ED later that day with increasing headache and left flank pain. Work-up included axial and coronal computed tomography scans of the abdomen and pelvis, which revealed hematuria and a left upper pole renal infarction surrounded by mild perinephric fat stranding (Figure 2a and 2b) with maintenance of left renal artery/vein patency.
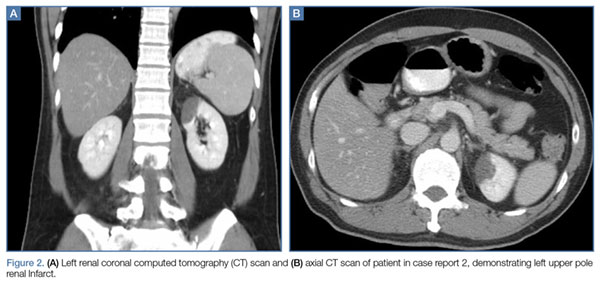
The patient was admitted to an inpatient floor, where symptomatic management was employed. He underwent unrevealing bubble echocardiography and lower extremity Doppler ultrasound imaging, and anticoagulation therapy was initiated per a consultation with hematology services. The patient was discharged home in improved, stable condition on hospital day 8.
Discussion
Dengue virus is a single-stranded, nonsegmented RNA virus in the Flaviviridae family. Four major subtypes exist: DEN-1, DEN-2, DEN-3, and DEN-4. Lifelong serotype-specific immunity is conferred following infection. The virus is transmitted by the female Aedes aegypti mosquito, which is found worldwide but has a predilection for tropical and subtropical regions. The Aedes aegypti mosquito remains an effective vector secondary to its diurnal feeding habit and nearly imperceptible bite.1,3
The viral incubation period for dengue is typically 3 to 7 days4; therefore, dengue is highly unlikely in patients whose symptoms begin more than 2 weeks after departure from an endemic area. Replication primarily occurs in the regional lymph nodes and disseminates through the lymphatic system and bloodstream.1
The 1997 WHO guidelines previously classified dengue into three categories: undifferentiated fever, dengue fever, and dengue hemorrhagic fever (which was further classified by four severity grades, with grades III and IV defined as dengue shock syndrome). However, changes in epidemiology of the disease and reports of difficulty applying the criteria in the clinical setting led to reclassification of dengue on a continuum from dengue to severe dengue in the WHO’s updated 2009 guidelines.4
Signs and Symptoms
The ramifications of dengue infection can range from asymptomatic (typically in young, immunocompetent patients) to lethal. Key symptoms of dengue fever include nausea, vomiting, fever, respiratory symptoms, morbilliform or maculopapular rash, and headache or retro-orbital pain. In addition, arthralgia (hence the colloquial name for dengue of “breakbone fever”), myalgia, and conjunctivitis may exist.3,4 Fever usually lasts 5 to 7 days and can be biphasic, with a return of symptoms after the initial resolution as seen in case report 1.4 Severe dengue is characterized by capillary leakage, hemorrhage, or end-organ damage.3-5 The most common bleeding sites are the skin, nose, and gums.
Diagnosis
Bedside evaluation for dengue can be performed with the TT—one of the WHO’s case definitions for dengue.6 This is accomplished by placing a manual blood pressure (BP) cuff on the arm and inflating it to halfway between systolic and diastolic BP for 5 minutes. The test is positive for dengue if more than 10 petechiae appear per 1-inch (2.5-cm) square below the antecubital fossa.7 Of note, the test has poor sensitivity (51.6%, 95% confidence interval [CI], 33-69), but good specificity (82.4%, 95% CI, 76-87).7,8 A positive TT combined with leukopenia increases the sensitivity to 93.9%, [95% CI, 89-96].7 While not specific to dengue infection, in the right clinical scenario, the TT is a simple bedside test to help confirm the diagnosis and is extremely useful in resource-limited settings.
During the initial days of illness, the virus may be detected by PCR, as viremia and fever usually correlate. Once defervescence occurs, IgM and then IgG antibodies become detectable. When using these antibody tests to evaluate for dengue, clinicians should be aware of cross-reactivity with other flavivirus infections, such as yellow fever or Japanese encephalitis (including immunological cross-reactivity).1 New diagnostic modalities include enzyme immunoassays that can detect dengue viral RNA within 24 to 48 hours, and viral antigen-detection kits, which can yield results in less than 1 hour.4
Aside from advanced laboratory testing, worsening thrombocytopenia in light of a rising hematocrit can be highly suggestive of dengue. Leukopenia with lymphopenia and mild elevation of hepatic enzymes (typically 2 to 5 times the upper limits of the normal reference range) are also often seen in active infections.1 The occurrence of these signs in conjunction with a rapid reduction in the platelets often signals transition to the critical phase of plasma leakage.1,4
Treatment
Treatment of dengue consists of supportive care and transfusion when necessary. The WHO recommends strict observation of patients with suspected dengue who have warning signs of severe disease (eg, abdominal pain, persistent vomiting, mucosal bleeding, lethargy, hepatomegaly, rapid increase in hematocrit with concomitant drop in platelet count). Inpatient treatment centers on judicious fluid management, trending blood count parameters, and monitoring for signs of plasma leakage and hemorrhage. Fluid resuscitation is titrated to optimize central and peripheral circulation and end-organ perfusion. Blood-product administration should be reserved for suspected or severe bleeding.4
While dengue fever was the final diagnosis in both of our case presentations, these cases also highlight key diagnostic and treatment dilemmas associated with dengue. The patient in the first case report demonstrated the characteristic biphasic fever seen with dengue—resolution of symptoms on day 3, but then return of fever and symptoms on day 4. Often the dengue-specific antibodies are not formed until after the resolution of fever. This patient represents a classic example of dengue as the serologic studies sent on day 4 of the patient’s illness were negative but then turned positive on day 7, illustrating the need for high clinical suspicion and underscoring the importance of initiating treatment despite laboratory confirmation.
Further, regarding the patient in the second case, though proteinuria, hematuria, acute renal failure, and glomerulonephritis are previously described renal complications of dengue,9 a thorough literature search yielded no prior published accounts of renal infarction. Given the patient’s previous healthy status and the lack of other hypothesis as to the mechanism of injury, we suspect this patient’s renal infarction was due to the transient hypercoagulability characteristic of dengue and responsible for other clinical manifestations of the disease.
Conclusion
In addition to more prevalent illnesses such as malaria, acute traveler’s diarrhea, and respiratory tract infections, dengue fever should be included in the differential diagnosis when evaluating a febrile patient who has a history of recent travel to countries where dengue is endemic. A high clinical suspicion, combined with a thorough history and physical examination, is essential to making the diagnosis.
Both of our case reports demonstrate some of the diagnostic limitations in the acute setting, and the breadth of clinical complications that can occur in this complex disease. With the increasing prevalence of dengue fever in North and Central America, it is likely that patients with the disease will present to EDs in the United States. Early diagnosis and awareness of potential complications can lead to timely initiation of life-saving supportive care.
1. Wilder-Smith A, Schwartz E. Dengue in travelers. N Engl J Med. 2005;353(9):924-932. dpo:10.1056/NEJMra041927
2. Pan American Health Organization, World Health Organization. Number of reported cases of dengue and severe dengue (SD), Region of the Americas (by country and subregion). Washington, DC: Pan American Health Organization. http://www.paho.org/hq/index.php?option=com_docman&task=doc_view&Itemid=&gid=35610&lang=es. Updated August 5, 2016. Accessed August 17, 2016.
3. Whitehorn J, Farrar J. Dengue. Clin Med (Lond). 2011;11(5):483-487.
4. World Health Organization. Dengue: Guidelines for Diagnosis, Treatment, Prevention and Control. New Edition. Geneva, Switzerland: World Health Organization; 2009. http://www.who.int/tdr/publications/documents/dengue-diagnosis.pdf. Accessed August 17, 2016.
5. Halstead SB. Dengue. Lancet. 2007;370(9599):1644-1652. doi:10.1016/S0140-6736(07)61687-0.
6. Centers for Disease Control and Prevention. Dengue Clinical Case Management E-learning. http://www.cdc.gov/dengue/training/cme/ccm/page73112.html; http://www.cdc.gov/dengue/training/cme/ccm/Tourniquet%20Test_F.pdf. Accessed August 17, 2016.
7. Gregory CJ, Lorenzi OD, Colón L, et al. Utility of the tourniquet test and the white blood cell count to differentiate dengue among acute febrile illnesses in the emergency room. PLoS Negl Trop Dis. 2011;5(12):e1400.
8. Mayxay M, Phetsouvanh R, Moore CE, et al. Predictive diagnostic value of the tourniquet test for the diagnosis of dengue infection in adults. Trop Med Int Health. 2011;16(1):127-133.
9. Lizarraga KJ, Nayer A. Dengue-associated kidney disease. J Nephropathol. 2014;3(2):57-62.
Dengue fever is the most commonly transmitted arboviral disease in the world, affecting an estimated 2.5 billion people who live in areas endemic to the virus. This exposure yields an annual incidence of 100 million cases of dengue, which translates into 250,000 cases of hemorrhagic fever. With an expanding geographic distribution and increasing number of epidemics, the World Health Organization (WHO) has classified dengue as a major public health concern.1 Enhanced globalization and changing climate patterns have resulted in a dramatic increase in the incidence of dengue in both North and Central America. Aggregate North and Central American data from 2010 to the present revealed over 1.7 million cases of dengue, nearly 80,000 of which were severe, and 747 deaths.2 Based on these statistics, dengue fever should be considered in the differential diagnosis of febrile ED patients in the developed world who had a history of recent travel. We present two cases that highlight the complexity of diagnosis and novel complications associated with dengue fever.
Case Reports
Case 1
A 24-year-old man presented to the ED with a 4-day history of intermittent fever of up to 102.02°F, which was accompanied by chills, myalgia, and rigors. The patient stated that he had visited Vietnam, Thailand, Indonesia, and Malaysia 8 days prior to presentation, and had experienced mosquito bites daily throughout his travels. He further noted that his symptoms had improved on day 3 of his illness, but acutely worsened on day 4, which prompted him to visit the ED. The patient’s primary complaint was a severe retro-orbital headache, fever, and one episode of epistaxis.
On physical examination, the patient had conjunctivitis and hepatosplenomegaly, but otherwise appeared well. His laboratory evaluation was significant for leukopenia (white blood cell [WBC] count, 2.40 x 109/L), thrombocytopenia (platelet count, 123 x 109/L), and a positive mononuclear spot test. Both dengue immunoglobulin G (IgG) and immunoglobulin M (IgM) tests sent from the ED were negative. Based on the patient’s thrombocytopenia and epistaxis, as well as concerns that the patient was entering into the critical phase of dengue fever, he was admitted to the inpatient hospital for observation.
The patient’s course improved during his stay with symptomatic treatment and blood-count monitoring, and he was discharged home on hospital day 3. He followed up at our hospital travel clinic the day after discharge; a repeat dengue IgM test taken during this visit came back positive.
Case 2
A 51-year-old man presented to the ED with a 3-day history of intermittent fever and diffuse myalgia. He reported chills, night sweats, and the feeling of abdominal fullness. He denied nausea, vomiting, or changes in the character of his stool. He had no known sick contacts, but reported he had traveled from the Philippines 3 days prior to presentation and that his symptoms had developed en route to the United States. The patient also denied any known tick, mosquito, or animal exposures. He said he had treated his symptoms with acetaminophen and nonsteroidal anti-inflammatory drugs. Prior to his arrival at the ED, he had twice presented to a walk-in clinic earlier that day. Repeated laboratory testing at the ED showed a decrease in WBC count from 42.0 x 109/L to 31.0 x 109/L, as well as a declining platelet count from 123 x 109/L to 87 x 109/L. On physical examination, the patient was ill-appearing, diaphoretic, and had a temperature of 100.6°F. His vital signs were otherwise within normal limits.
With the exception of a mild diffuse petechial rash on the patient’s thighs bilaterally, the physical examination was unrevealing. A tourniquet test (TT) to assess capillary fragility was performed at bedside, and yielded a positive result (Figure 1). Work-up further demonstrated a declining WBC of 2.70 x 109/L and declining platelet count of 65 x 109/L.
A polymerase chain reaction (PCR) test confirmed a diagnosis of dengue, with a positive dengue type-4 (DEN-4) serotype detection. Supportive care was initiated, and the patient was admitted to the inpatient hospital for continued treatment. He was discharged home on hospital day 5; however, he returned to the ED later that day with increasing headache and left flank pain. Work-up included axial and coronal computed tomography scans of the abdomen and pelvis, which revealed hematuria and a left upper pole renal infarction surrounded by mild perinephric fat stranding (Figure 2a and 2b) with maintenance of left renal artery/vein patency.
The patient was admitted to an inpatient floor, where symptomatic management was employed. He underwent unrevealing bubble echocardiography and lower extremity Doppler ultrasound imaging, and anticoagulation therapy was initiated per a consultation with hematology services. The patient was discharged home in improved, stable condition on hospital day 8.
Discussion
Dengue virus is a single-stranded, nonsegmented RNA virus in the Flaviviridae family. Four major subtypes exist: DEN-1, DEN-2, DEN-3, and DEN-4. Lifelong serotype-specific immunity is conferred following infection. The virus is transmitted by the female Aedes aegypti mosquito, which is found worldwide but has a predilection for tropical and subtropical regions. The Aedes aegypti mosquito remains an effective vector secondary to its diurnal feeding habit and nearly imperceptible bite.1,3
The viral incubation period for dengue is typically 3 to 7 days4; therefore, dengue is highly unlikely in patients whose symptoms begin more than 2 weeks after departure from an endemic area. Replication primarily occurs in the regional lymph nodes and disseminates through the lymphatic system and bloodstream.1
The 1997 WHO guidelines previously classified dengue into three categories: undifferentiated fever, dengue fever, and dengue hemorrhagic fever (which was further classified by four severity grades, with grades III and IV defined as dengue shock syndrome). However, changes in epidemiology of the disease and reports of difficulty applying the criteria in the clinical setting led to reclassification of dengue on a continuum from dengue to severe dengue in the WHO’s updated 2009 guidelines.4
Signs and Symptoms
The ramifications of dengue infection can range from asymptomatic (typically in young, immunocompetent patients) to lethal. Key symptoms of dengue fever include nausea, vomiting, fever, respiratory symptoms, morbilliform or maculopapular rash, and headache or retro-orbital pain. In addition, arthralgia (hence the colloquial name for dengue of “breakbone fever”), myalgia, and conjunctivitis may exist.3,4 Fever usually lasts 5 to 7 days and can be biphasic, with a return of symptoms after the initial resolution as seen in case report 1.4 Severe dengue is characterized by capillary leakage, hemorrhage, or end-organ damage.3-5 The most common bleeding sites are the skin, nose, and gums.
Diagnosis
Bedside evaluation for dengue can be performed with the TT—one of the WHO’s case definitions for dengue.6 This is accomplished by placing a manual blood pressure (BP) cuff on the arm and inflating it to halfway between systolic and diastolic BP for 5 minutes. The test is positive for dengue if more than 10 petechiae appear per 1-inch (2.5-cm) square below the antecubital fossa.7 Of note, the test has poor sensitivity (51.6%, 95% confidence interval [CI], 33-69), but good specificity (82.4%, 95% CI, 76-87).7,8 A positive TT combined with leukopenia increases the sensitivity to 93.9%, [95% CI, 89-96].7 While not specific to dengue infection, in the right clinical scenario, the TT is a simple bedside test to help confirm the diagnosis and is extremely useful in resource-limited settings.
During the initial days of illness, the virus may be detected by PCR, as viremia and fever usually correlate. Once defervescence occurs, IgM and then IgG antibodies become detectable. When using these antibody tests to evaluate for dengue, clinicians should be aware of cross-reactivity with other flavivirus infections, such as yellow fever or Japanese encephalitis (including immunological cross-reactivity).1 New diagnostic modalities include enzyme immunoassays that can detect dengue viral RNA within 24 to 48 hours, and viral antigen-detection kits, which can yield results in less than 1 hour.4
Aside from advanced laboratory testing, worsening thrombocytopenia in light of a rising hematocrit can be highly suggestive of dengue. Leukopenia with lymphopenia and mild elevation of hepatic enzymes (typically 2 to 5 times the upper limits of the normal reference range) are also often seen in active infections.1 The occurrence of these signs in conjunction with a rapid reduction in the platelets often signals transition to the critical phase of plasma leakage.1,4
Treatment
Treatment of dengue consists of supportive care and transfusion when necessary. The WHO recommends strict observation of patients with suspected dengue who have warning signs of severe disease (eg, abdominal pain, persistent vomiting, mucosal bleeding, lethargy, hepatomegaly, rapid increase in hematocrit with concomitant drop in platelet count). Inpatient treatment centers on judicious fluid management, trending blood count parameters, and monitoring for signs of plasma leakage and hemorrhage. Fluid resuscitation is titrated to optimize central and peripheral circulation and end-organ perfusion. Blood-product administration should be reserved for suspected or severe bleeding.4
While dengue fever was the final diagnosis in both of our case presentations, these cases also highlight key diagnostic and treatment dilemmas associated with dengue. The patient in the first case report demonstrated the characteristic biphasic fever seen with dengue—resolution of symptoms on day 3, but then return of fever and symptoms on day 4. Often the dengue-specific antibodies are not formed until after the resolution of fever. This patient represents a classic example of dengue as the serologic studies sent on day 4 of the patient’s illness were negative but then turned positive on day 7, illustrating the need for high clinical suspicion and underscoring the importance of initiating treatment despite laboratory confirmation.
Further, regarding the patient in the second case, though proteinuria, hematuria, acute renal failure, and glomerulonephritis are previously described renal complications of dengue,9 a thorough literature search yielded no prior published accounts of renal infarction. Given the patient’s previous healthy status and the lack of other hypothesis as to the mechanism of injury, we suspect this patient’s renal infarction was due to the transient hypercoagulability characteristic of dengue and responsible for other clinical manifestations of the disease.
Conclusion
In addition to more prevalent illnesses such as malaria, acute traveler’s diarrhea, and respiratory tract infections, dengue fever should be included in the differential diagnosis when evaluating a febrile patient who has a history of recent travel to countries where dengue is endemic. A high clinical suspicion, combined with a thorough history and physical examination, is essential to making the diagnosis.
Both of our case reports demonstrate some of the diagnostic limitations in the acute setting, and the breadth of clinical complications that can occur in this complex disease. With the increasing prevalence of dengue fever in North and Central America, it is likely that patients with the disease will present to EDs in the United States. Early diagnosis and awareness of potential complications can lead to timely initiation of life-saving supportive care.
Dengue fever is the most commonly transmitted arboviral disease in the world, affecting an estimated 2.5 billion people who live in areas endemic to the virus. This exposure yields an annual incidence of 100 million cases of dengue, which translates into 250,000 cases of hemorrhagic fever. With an expanding geographic distribution and increasing number of epidemics, the World Health Organization (WHO) has classified dengue as a major public health concern.1 Enhanced globalization and changing climate patterns have resulted in a dramatic increase in the incidence of dengue in both North and Central America. Aggregate North and Central American data from 2010 to the present revealed over 1.7 million cases of dengue, nearly 80,000 of which were severe, and 747 deaths.2 Based on these statistics, dengue fever should be considered in the differential diagnosis of febrile ED patients in the developed world who had a history of recent travel. We present two cases that highlight the complexity of diagnosis and novel complications associated with dengue fever.
Case Reports
Case 1
A 24-year-old man presented to the ED with a 4-day history of intermittent fever of up to 102.02°F, which was accompanied by chills, myalgia, and rigors. The patient stated that he had visited Vietnam, Thailand, Indonesia, and Malaysia 8 days prior to presentation, and had experienced mosquito bites daily throughout his travels. He further noted that his symptoms had improved on day 3 of his illness, but acutely worsened on day 4, which prompted him to visit the ED. The patient’s primary complaint was a severe retro-orbital headache, fever, and one episode of epistaxis.
On physical examination, the patient had conjunctivitis and hepatosplenomegaly, but otherwise appeared well. His laboratory evaluation was significant for leukopenia (white blood cell [WBC] count, 2.40 x 109/L), thrombocytopenia (platelet count, 123 x 109/L), and a positive mononuclear spot test. Both dengue immunoglobulin G (IgG) and immunoglobulin M (IgM) tests sent from the ED were negative. Based on the patient’s thrombocytopenia and epistaxis, as well as concerns that the patient was entering into the critical phase of dengue fever, he was admitted to the inpatient hospital for observation.
The patient’s course improved during his stay with symptomatic treatment and blood-count monitoring, and he was discharged home on hospital day 3. He followed up at our hospital travel clinic the day after discharge; a repeat dengue IgM test taken during this visit came back positive.
Case 2
A 51-year-old man presented to the ED with a 3-day history of intermittent fever and diffuse myalgia. He reported chills, night sweats, and the feeling of abdominal fullness. He denied nausea, vomiting, or changes in the character of his stool. He had no known sick contacts, but reported he had traveled from the Philippines 3 days prior to presentation and that his symptoms had developed en route to the United States. The patient also denied any known tick, mosquito, or animal exposures. He said he had treated his symptoms with acetaminophen and nonsteroidal anti-inflammatory drugs. Prior to his arrival at the ED, he had twice presented to a walk-in clinic earlier that day. Repeated laboratory testing at the ED showed a decrease in WBC count from 42.0 x 109/L to 31.0 x 109/L, as well as a declining platelet count from 123 x 109/L to 87 x 109/L. On physical examination, the patient was ill-appearing, diaphoretic, and had a temperature of 100.6°F. His vital signs were otherwise within normal limits.
With the exception of a mild diffuse petechial rash on the patient’s thighs bilaterally, the physical examination was unrevealing. A tourniquet test (TT) to assess capillary fragility was performed at bedside, and yielded a positive result (Figure 1). Work-up further demonstrated a declining WBC of 2.70 x 109/L and declining platelet count of 65 x 109/L.
A polymerase chain reaction (PCR) test confirmed a diagnosis of dengue, with a positive dengue type-4 (DEN-4) serotype detection. Supportive care was initiated, and the patient was admitted to the inpatient hospital for continued treatment. He was discharged home on hospital day 5; however, he returned to the ED later that day with increasing headache and left flank pain. Work-up included axial and coronal computed tomography scans of the abdomen and pelvis, which revealed hematuria and a left upper pole renal infarction surrounded by mild perinephric fat stranding (Figure 2a and 2b) with maintenance of left renal artery/vein patency.
The patient was admitted to an inpatient floor, where symptomatic management was employed. He underwent unrevealing bubble echocardiography and lower extremity Doppler ultrasound imaging, and anticoagulation therapy was initiated per a consultation with hematology services. The patient was discharged home in improved, stable condition on hospital day 8.
Discussion
Dengue virus is a single-stranded, nonsegmented RNA virus in the Flaviviridae family. Four major subtypes exist: DEN-1, DEN-2, DEN-3, and DEN-4. Lifelong serotype-specific immunity is conferred following infection. The virus is transmitted by the female Aedes aegypti mosquito, which is found worldwide but has a predilection for tropical and subtropical regions. The Aedes aegypti mosquito remains an effective vector secondary to its diurnal feeding habit and nearly imperceptible bite.1,3
The viral incubation period for dengue is typically 3 to 7 days4; therefore, dengue is highly unlikely in patients whose symptoms begin more than 2 weeks after departure from an endemic area. Replication primarily occurs in the regional lymph nodes and disseminates through the lymphatic system and bloodstream.1
The 1997 WHO guidelines previously classified dengue into three categories: undifferentiated fever, dengue fever, and dengue hemorrhagic fever (which was further classified by four severity grades, with grades III and IV defined as dengue shock syndrome). However, changes in epidemiology of the disease and reports of difficulty applying the criteria in the clinical setting led to reclassification of dengue on a continuum from dengue to severe dengue in the WHO’s updated 2009 guidelines.4
Signs and Symptoms
The ramifications of dengue infection can range from asymptomatic (typically in young, immunocompetent patients) to lethal. Key symptoms of dengue fever include nausea, vomiting, fever, respiratory symptoms, morbilliform or maculopapular rash, and headache or retro-orbital pain. In addition, arthralgia (hence the colloquial name for dengue of “breakbone fever”), myalgia, and conjunctivitis may exist.3,4 Fever usually lasts 5 to 7 days and can be biphasic, with a return of symptoms after the initial resolution as seen in case report 1.4 Severe dengue is characterized by capillary leakage, hemorrhage, or end-organ damage.3-5 The most common bleeding sites are the skin, nose, and gums.
Diagnosis
Bedside evaluation for dengue can be performed with the TT—one of the WHO’s case definitions for dengue.6 This is accomplished by placing a manual blood pressure (BP) cuff on the arm and inflating it to halfway between systolic and diastolic BP for 5 minutes. The test is positive for dengue if more than 10 petechiae appear per 1-inch (2.5-cm) square below the antecubital fossa.7 Of note, the test has poor sensitivity (51.6%, 95% confidence interval [CI], 33-69), but good specificity (82.4%, 95% CI, 76-87).7,8 A positive TT combined with leukopenia increases the sensitivity to 93.9%, [95% CI, 89-96].7 While not specific to dengue infection, in the right clinical scenario, the TT is a simple bedside test to help confirm the diagnosis and is extremely useful in resource-limited settings.
During the initial days of illness, the virus may be detected by PCR, as viremia and fever usually correlate. Once defervescence occurs, IgM and then IgG antibodies become detectable. When using these antibody tests to evaluate for dengue, clinicians should be aware of cross-reactivity with other flavivirus infections, such as yellow fever or Japanese encephalitis (including immunological cross-reactivity).1 New diagnostic modalities include enzyme immunoassays that can detect dengue viral RNA within 24 to 48 hours, and viral antigen-detection kits, which can yield results in less than 1 hour.4
Aside from advanced laboratory testing, worsening thrombocytopenia in light of a rising hematocrit can be highly suggestive of dengue. Leukopenia with lymphopenia and mild elevation of hepatic enzymes (typically 2 to 5 times the upper limits of the normal reference range) are also often seen in active infections.1 The occurrence of these signs in conjunction with a rapid reduction in the platelets often signals transition to the critical phase of plasma leakage.1,4
Treatment
Treatment of dengue consists of supportive care and transfusion when necessary. The WHO recommends strict observation of patients with suspected dengue who have warning signs of severe disease (eg, abdominal pain, persistent vomiting, mucosal bleeding, lethargy, hepatomegaly, rapid increase in hematocrit with concomitant drop in platelet count). Inpatient treatment centers on judicious fluid management, trending blood count parameters, and monitoring for signs of plasma leakage and hemorrhage. Fluid resuscitation is titrated to optimize central and peripheral circulation and end-organ perfusion. Blood-product administration should be reserved for suspected or severe bleeding.4
While dengue fever was the final diagnosis in both of our case presentations, these cases also highlight key diagnostic and treatment dilemmas associated with dengue. The patient in the first case report demonstrated the characteristic biphasic fever seen with dengue—resolution of symptoms on day 3, but then return of fever and symptoms on day 4. Often the dengue-specific antibodies are not formed until after the resolution of fever. This patient represents a classic example of dengue as the serologic studies sent on day 4 of the patient’s illness were negative but then turned positive on day 7, illustrating the need for high clinical suspicion and underscoring the importance of initiating treatment despite laboratory confirmation.
Further, regarding the patient in the second case, though proteinuria, hematuria, acute renal failure, and glomerulonephritis are previously described renal complications of dengue,9 a thorough literature search yielded no prior published accounts of renal infarction. Given the patient’s previous healthy status and the lack of other hypothesis as to the mechanism of injury, we suspect this patient’s renal infarction was due to the transient hypercoagulability characteristic of dengue and responsible for other clinical manifestations of the disease.
Conclusion
In addition to more prevalent illnesses such as malaria, acute traveler’s diarrhea, and respiratory tract infections, dengue fever should be included in the differential diagnosis when evaluating a febrile patient who has a history of recent travel to countries where dengue is endemic. A high clinical suspicion, combined with a thorough history and physical examination, is essential to making the diagnosis.
Both of our case reports demonstrate some of the diagnostic limitations in the acute setting, and the breadth of clinical complications that can occur in this complex disease. With the increasing prevalence of dengue fever in North and Central America, it is likely that patients with the disease will present to EDs in the United States. Early diagnosis and awareness of potential complications can lead to timely initiation of life-saving supportive care.
1. Wilder-Smith A, Schwartz E. Dengue in travelers. N Engl J Med. 2005;353(9):924-932. dpo:10.1056/NEJMra041927
2. Pan American Health Organization, World Health Organization. Number of reported cases of dengue and severe dengue (SD), Region of the Americas (by country and subregion). Washington, DC: Pan American Health Organization. http://www.paho.org/hq/index.php?option=com_docman&task=doc_view&Itemid=&gid=35610&lang=es. Updated August 5, 2016. Accessed August 17, 2016.
3. Whitehorn J, Farrar J. Dengue. Clin Med (Lond). 2011;11(5):483-487.
4. World Health Organization. Dengue: Guidelines for Diagnosis, Treatment, Prevention and Control. New Edition. Geneva, Switzerland: World Health Organization; 2009. http://www.who.int/tdr/publications/documents/dengue-diagnosis.pdf. Accessed August 17, 2016.
5. Halstead SB. Dengue. Lancet. 2007;370(9599):1644-1652. doi:10.1016/S0140-6736(07)61687-0.
6. Centers for Disease Control and Prevention. Dengue Clinical Case Management E-learning. http://www.cdc.gov/dengue/training/cme/ccm/page73112.html; http://www.cdc.gov/dengue/training/cme/ccm/Tourniquet%20Test_F.pdf. Accessed August 17, 2016.
7. Gregory CJ, Lorenzi OD, Colón L, et al. Utility of the tourniquet test and the white blood cell count to differentiate dengue among acute febrile illnesses in the emergency room. PLoS Negl Trop Dis. 2011;5(12):e1400.
8. Mayxay M, Phetsouvanh R, Moore CE, et al. Predictive diagnostic value of the tourniquet test for the diagnosis of dengue infection in adults. Trop Med Int Health. 2011;16(1):127-133.
9. Lizarraga KJ, Nayer A. Dengue-associated kidney disease. J Nephropathol. 2014;3(2):57-62.
1. Wilder-Smith A, Schwartz E. Dengue in travelers. N Engl J Med. 2005;353(9):924-932. dpo:10.1056/NEJMra041927
2. Pan American Health Organization, World Health Organization. Number of reported cases of dengue and severe dengue (SD), Region of the Americas (by country and subregion). Washington, DC: Pan American Health Organization. http://www.paho.org/hq/index.php?option=com_docman&task=doc_view&Itemid=&gid=35610&lang=es. Updated August 5, 2016. Accessed August 17, 2016.
3. Whitehorn J, Farrar J. Dengue. Clin Med (Lond). 2011;11(5):483-487.
4. World Health Organization. Dengue: Guidelines for Diagnosis, Treatment, Prevention and Control. New Edition. Geneva, Switzerland: World Health Organization; 2009. http://www.who.int/tdr/publications/documents/dengue-diagnosis.pdf. Accessed August 17, 2016.
5. Halstead SB. Dengue. Lancet. 2007;370(9599):1644-1652. doi:10.1016/S0140-6736(07)61687-0.
6. Centers for Disease Control and Prevention. Dengue Clinical Case Management E-learning. http://www.cdc.gov/dengue/training/cme/ccm/page73112.html; http://www.cdc.gov/dengue/training/cme/ccm/Tourniquet%20Test_F.pdf. Accessed August 17, 2016.
7. Gregory CJ, Lorenzi OD, Colón L, et al. Utility of the tourniquet test and the white blood cell count to differentiate dengue among acute febrile illnesses in the emergency room. PLoS Negl Trop Dis. 2011;5(12):e1400.
8. Mayxay M, Phetsouvanh R, Moore CE, et al. Predictive diagnostic value of the tourniquet test for the diagnosis of dengue infection in adults. Trop Med Int Health. 2011;16(1):127-133.
9. Lizarraga KJ, Nayer A. Dengue-associated kidney disease. J Nephropathol. 2014;3(2):57-62.
Depigmented patches, mild scaling on newborn • Dx?
THE CASE
A 21-year-old G3P2 mother gave birth to an African American girl via vaginal delivery. Labor had been induced due to gestational hypertension at term. She’d also had a stillborn at term at the age of 16 followed by a second live term birth 3 years ago. During this most recent pregnancy, she’d received adequate prenatal care and had been treated for chlamydia with a single dose of oral azithromycin 1 g.
The newborn had an Apgar score of 9 out of 9 and weighed 6.7 pounds at birth. During a physical examination in the nursery, the infant was found to have large areas of smooth depigmentation on her forehead, right forearm, lower abdomen, and left thigh, with surrounding areas of thickened skin that had mild scaling and hyperpigmentation (FIGURE). The depigmented areas involved approximately 15% of the newborn’s body. The father and paternal grandmother, who were present at the time of delivery, also had depigmented areas of their skin.
The newborn’s tongue was pink and her mucus membranes were moist. No macules or patches were noted on either the oral or vaginal mucosa. Cardiac, pulmonary, and ocular examinations (including evaluation of the retina by ophthalmoscopy) were normal. There was no nystagmus or strabismus. The newborn’s extremities were normal, symmetric, and moveable, and she was easily consoled.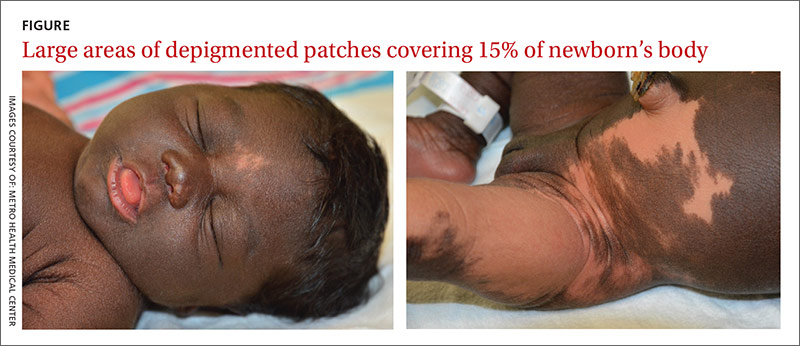
THE DIAGNOSIS
We diagnosed the newborn with piebaldism based on her appearance. Piebaldism consists of hypopigmented/depigmented areas and is a clinical diagnosis; no testing is required.
Concerned about the areas of hyperpigmentation, we decided to get a dermatologist’s opinion. The dermatology team briefly considered the diagnosis of a large melanocytic nevus with sparing of some areas, but a skin biopsy of a hyperpigmented area on the left leg came back with a normal number of melanocytes.
DISCUSSION
Piebaldism is a rare autosomal dominant disorder characterized by the congenital absence of melanocytes in affected areas of the skin and hair due to mutations of the c-kit gene. The c-kit gene affects the differentiation and migration of melanoblasts from the neural crest during embryonic life.1 The incidence of piebaldism is estimated to be less than one in 20,000.2 Both males and females are equally affected, and no race is spared.2,3
Affected individuals present with a white forelock and relatively stable, persistent depigmentation of skin with a characteristic distribution from birth.3 A white forelock arising from a triangular, elongated, or diamond-shaped midline or depigmented macule on the forehead may be the only manifestation in 80% to 90% of cases.3 The characteristic distribution of depigmented macules includes a central macule on the forehead, the anterior abdomen extending to the chest, the lateral trunk sparing the dorsal spine, and the mid-arms and legs sparing the hands and feet.2
Depigmented macules are rectangular, rhomboid, or irregular in shape and usually have a symmetrical distribution. Typically, islands of hyperpigmentation are present within and at the border of depigmented areas.4 Piebaldism is associated in rare instances with neurofibromatosis type 1, Hirschsprung’s disease, hearing loss, and Waardenburg syndrome.4
Histologically, melanocytes are absent or considerably reduced in the depigmented areas and are normal in number in the hyperpigmented areas.5
The differential diagnosis of piebaldism includes mosaicism, albinism, and vitiligo.
Cutaneous mosaicism stems from a gene mutation that occurs during embryogenesis and the lesions are distributed along certain patterns and forms. A chromosomal analysis of our patient showed a normal female karyotype that excluded mosaicism.
Albinism is a genetically inherited disorder characterized by partial or complete absence of melanin production in the skin, hair, and eyes.6 Eye and fundus examinations were normal in our patient, which excluded albinism.
Vitiligo is rarely present at birth but is usually acquired later in life. It results from an immune-mediated destruction of melanocytes and is not genetically inherited, although familial incidence has been reported.7
There are no effective therapies
A combination of dermabrasion and grafting of pigmented skin into depigmented areas, with or without phototherapy, has been used in select patients, although no solid data are available on its effectiveness.3 The lack of effective and safe therapies can make treatment challenging. Piebaldism is usually not medically harmful, but the emotional and psychological effects on the family and the patient as they grow up can be devastating. Therefore, supportive counseling is recommended.
Our patient. Supportive counseling and a follow-up appointment with a dermatologist was planned for our patient and her family.
THE TAKEAWAY
The clinical diagnosis of piebaldism is straightforward based on the presence of a white forelock in the frontal region, the appearance of depigmented macules since birth that stay relatively stable, and the presence of a similar pattern of depigmented macules in other family members. Histologic or genetic testing is not necessary to establish the diagnosis. Rarely, cases of piebaldism are associated with hearing loss, necessitating a hearing assessment and an audiology exam. Unfortunately, there are no effective treatments for piebaldism.
1. Ward KA, Moss C, Sanders DS. Human piebaldism: relationship between phenotype and site of kit gene mutation. Br J Dermatol. 1995;132:929-935.
2. Agarwal S, Ojha A. Piebaldism: A brief report and review of the literature. Indian Dermatol Online J. 2012;3:144-147.
3. Oiso N, Fukai K, Kawada A, et al. Piebaldism. J Dermatol. 2013;40:330-335.
4. Spritz RA, Itin PH, Gutmann DH. Piebaldism and neurofibromatosis type 1: horses of very different colors. J Invest Dermatol. 2004;122:xxxiv-xxxv.
5. Makino T, Yanagihara M, Oiso N, et al. Repigmentation of the epidermis around the acrosyringium in piebald skin: an ultrastructural examination. Br J Dermatol. 2013;168:910-912.
6. Karaman A. Oculocutaneous albinism type 1A: a case report. Dermatol Online J. 2008;14:13.
7. Plensdorf S, Martinez J. Common pigmentation disorders. Am Fam Physician. 2009;79:109-116.
THE CASE
A 21-year-old G3P2 mother gave birth to an African American girl via vaginal delivery. Labor had been induced due to gestational hypertension at term. She’d also had a stillborn at term at the age of 16 followed by a second live term birth 3 years ago. During this most recent pregnancy, she’d received adequate prenatal care and had been treated for chlamydia with a single dose of oral azithromycin 1 g.
The newborn had an Apgar score of 9 out of 9 and weighed 6.7 pounds at birth. During a physical examination in the nursery, the infant was found to have large areas of smooth depigmentation on her forehead, right forearm, lower abdomen, and left thigh, with surrounding areas of thickened skin that had mild scaling and hyperpigmentation (FIGURE). The depigmented areas involved approximately 15% of the newborn’s body. The father and paternal grandmother, who were present at the time of delivery, also had depigmented areas of their skin.
The newborn’s tongue was pink and her mucus membranes were moist. No macules or patches were noted on either the oral or vaginal mucosa. Cardiac, pulmonary, and ocular examinations (including evaluation of the retina by ophthalmoscopy) were normal. There was no nystagmus or strabismus. The newborn’s extremities were normal, symmetric, and moveable, and she was easily consoled.
THE DIAGNOSIS
We diagnosed the newborn with piebaldism based on her appearance. Piebaldism consists of hypopigmented/depigmented areas and is a clinical diagnosis; no testing is required.
Concerned about the areas of hyperpigmentation, we decided to get a dermatologist’s opinion. The dermatology team briefly considered the diagnosis of a large melanocytic nevus with sparing of some areas, but a skin biopsy of a hyperpigmented area on the left leg came back with a normal number of melanocytes.
DISCUSSION
Piebaldism is a rare autosomal dominant disorder characterized by the congenital absence of melanocytes in affected areas of the skin and hair due to mutations of the c-kit gene. The c-kit gene affects the differentiation and migration of melanoblasts from the neural crest during embryonic life.1 The incidence of piebaldism is estimated to be less than one in 20,000.2 Both males and females are equally affected, and no race is spared.2,3
Affected individuals present with a white forelock and relatively stable, persistent depigmentation of skin with a characteristic distribution from birth.3 A white forelock arising from a triangular, elongated, or diamond-shaped midline or depigmented macule on the forehead may be the only manifestation in 80% to 90% of cases.3 The characteristic distribution of depigmented macules includes a central macule on the forehead, the anterior abdomen extending to the chest, the lateral trunk sparing the dorsal spine, and the mid-arms and legs sparing the hands and feet.2
Depigmented macules are rectangular, rhomboid, or irregular in shape and usually have a symmetrical distribution. Typically, islands of hyperpigmentation are present within and at the border of depigmented areas.4 Piebaldism is associated in rare instances with neurofibromatosis type 1, Hirschsprung’s disease, hearing loss, and Waardenburg syndrome.4
Histologically, melanocytes are absent or considerably reduced in the depigmented areas and are normal in number in the hyperpigmented areas.5
The differential diagnosis of piebaldism includes mosaicism, albinism, and vitiligo.
Cutaneous mosaicism stems from a gene mutation that occurs during embryogenesis and the lesions are distributed along certain patterns and forms. A chromosomal analysis of our patient showed a normal female karyotype that excluded mosaicism.
Albinism is a genetically inherited disorder characterized by partial or complete absence of melanin production in the skin, hair, and eyes.6 Eye and fundus examinations were normal in our patient, which excluded albinism.
Vitiligo is rarely present at birth but is usually acquired later in life. It results from an immune-mediated destruction of melanocytes and is not genetically inherited, although familial incidence has been reported.7
There are no effective therapies
A combination of dermabrasion and grafting of pigmented skin into depigmented areas, with or without phototherapy, has been used in select patients, although no solid data are available on its effectiveness.3 The lack of effective and safe therapies can make treatment challenging. Piebaldism is usually not medically harmful, but the emotional and psychological effects on the family and the patient as they grow up can be devastating. Therefore, supportive counseling is recommended.
Our patient. Supportive counseling and a follow-up appointment with a dermatologist was planned for our patient and her family.
THE TAKEAWAY
The clinical diagnosis of piebaldism is straightforward based on the presence of a white forelock in the frontal region, the appearance of depigmented macules since birth that stay relatively stable, and the presence of a similar pattern of depigmented macules in other family members. Histologic or genetic testing is not necessary to establish the diagnosis. Rarely, cases of piebaldism are associated with hearing loss, necessitating a hearing assessment and an audiology exam. Unfortunately, there are no effective treatments for piebaldism.
THE CASE
A 21-year-old G3P2 mother gave birth to an African American girl via vaginal delivery. Labor had been induced due to gestational hypertension at term. She’d also had a stillborn at term at the age of 16 followed by a second live term birth 3 years ago. During this most recent pregnancy, she’d received adequate prenatal care and had been treated for chlamydia with a single dose of oral azithromycin 1 g.
The newborn had an Apgar score of 9 out of 9 and weighed 6.7 pounds at birth. During a physical examination in the nursery, the infant was found to have large areas of smooth depigmentation on her forehead, right forearm, lower abdomen, and left thigh, with surrounding areas of thickened skin that had mild scaling and hyperpigmentation (FIGURE). The depigmented areas involved approximately 15% of the newborn’s body. The father and paternal grandmother, who were present at the time of delivery, also had depigmented areas of their skin.
The newborn’s tongue was pink and her mucus membranes were moist. No macules or patches were noted on either the oral or vaginal mucosa. Cardiac, pulmonary, and ocular examinations (including evaluation of the retina by ophthalmoscopy) were normal. There was no nystagmus or strabismus. The newborn’s extremities were normal, symmetric, and moveable, and she was easily consoled.
THE DIAGNOSIS
We diagnosed the newborn with piebaldism based on her appearance. Piebaldism consists of hypopigmented/depigmented areas and is a clinical diagnosis; no testing is required.
Concerned about the areas of hyperpigmentation, we decided to get a dermatologist’s opinion. The dermatology team briefly considered the diagnosis of a large melanocytic nevus with sparing of some areas, but a skin biopsy of a hyperpigmented area on the left leg came back with a normal number of melanocytes.
DISCUSSION
Piebaldism is a rare autosomal dominant disorder characterized by the congenital absence of melanocytes in affected areas of the skin and hair due to mutations of the c-kit gene. The c-kit gene affects the differentiation and migration of melanoblasts from the neural crest during embryonic life.1 The incidence of piebaldism is estimated to be less than one in 20,000.2 Both males and females are equally affected, and no race is spared.2,3
Affected individuals present with a white forelock and relatively stable, persistent depigmentation of skin with a characteristic distribution from birth.3 A white forelock arising from a triangular, elongated, or diamond-shaped midline or depigmented macule on the forehead may be the only manifestation in 80% to 90% of cases.3 The characteristic distribution of depigmented macules includes a central macule on the forehead, the anterior abdomen extending to the chest, the lateral trunk sparing the dorsal spine, and the mid-arms and legs sparing the hands and feet.2
Depigmented macules are rectangular, rhomboid, or irregular in shape and usually have a symmetrical distribution. Typically, islands of hyperpigmentation are present within and at the border of depigmented areas.4 Piebaldism is associated in rare instances with neurofibromatosis type 1, Hirschsprung’s disease, hearing loss, and Waardenburg syndrome.4
Histologically, melanocytes are absent or considerably reduced in the depigmented areas and are normal in number in the hyperpigmented areas.5
The differential diagnosis of piebaldism includes mosaicism, albinism, and vitiligo.
Cutaneous mosaicism stems from a gene mutation that occurs during embryogenesis and the lesions are distributed along certain patterns and forms. A chromosomal analysis of our patient showed a normal female karyotype that excluded mosaicism.
Albinism is a genetically inherited disorder characterized by partial or complete absence of melanin production in the skin, hair, and eyes.6 Eye and fundus examinations were normal in our patient, which excluded albinism.
Vitiligo is rarely present at birth but is usually acquired later in life. It results from an immune-mediated destruction of melanocytes and is not genetically inherited, although familial incidence has been reported.7
There are no effective therapies
A combination of dermabrasion and grafting of pigmented skin into depigmented areas, with or without phototherapy, has been used in select patients, although no solid data are available on its effectiveness.3 The lack of effective and safe therapies can make treatment challenging. Piebaldism is usually not medically harmful, but the emotional and psychological effects on the family and the patient as they grow up can be devastating. Therefore, supportive counseling is recommended.
Our patient. Supportive counseling and a follow-up appointment with a dermatologist was planned for our patient and her family.
THE TAKEAWAY
The clinical diagnosis of piebaldism is straightforward based on the presence of a white forelock in the frontal region, the appearance of depigmented macules since birth that stay relatively stable, and the presence of a similar pattern of depigmented macules in other family members. Histologic or genetic testing is not necessary to establish the diagnosis. Rarely, cases of piebaldism are associated with hearing loss, necessitating a hearing assessment and an audiology exam. Unfortunately, there are no effective treatments for piebaldism.
1. Ward KA, Moss C, Sanders DS. Human piebaldism: relationship between phenotype and site of kit gene mutation. Br J Dermatol. 1995;132:929-935.
2. Agarwal S, Ojha A. Piebaldism: A brief report and review of the literature. Indian Dermatol Online J. 2012;3:144-147.
3. Oiso N, Fukai K, Kawada A, et al. Piebaldism. J Dermatol. 2013;40:330-335.
4. Spritz RA, Itin PH, Gutmann DH. Piebaldism and neurofibromatosis type 1: horses of very different colors. J Invest Dermatol. 2004;122:xxxiv-xxxv.
5. Makino T, Yanagihara M, Oiso N, et al. Repigmentation of the epidermis around the acrosyringium in piebald skin: an ultrastructural examination. Br J Dermatol. 2013;168:910-912.
6. Karaman A. Oculocutaneous albinism type 1A: a case report. Dermatol Online J. 2008;14:13.
7. Plensdorf S, Martinez J. Common pigmentation disorders. Am Fam Physician. 2009;79:109-116.
1. Ward KA, Moss C, Sanders DS. Human piebaldism: relationship between phenotype and site of kit gene mutation. Br J Dermatol. 1995;132:929-935.
2. Agarwal S, Ojha A. Piebaldism: A brief report and review of the literature. Indian Dermatol Online J. 2012;3:144-147.
3. Oiso N, Fukai K, Kawada A, et al. Piebaldism. J Dermatol. 2013;40:330-335.
4. Spritz RA, Itin PH, Gutmann DH. Piebaldism and neurofibromatosis type 1: horses of very different colors. J Invest Dermatol. 2004;122:xxxiv-xxxv.
5. Makino T, Yanagihara M, Oiso N, et al. Repigmentation of the epidermis around the acrosyringium in piebald skin: an ultrastructural examination. Br J Dermatol. 2013;168:910-912.
6. Karaman A. Oculocutaneous albinism type 1A: a case report. Dermatol Online J. 2008;14:13.
7. Plensdorf S, Martinez J. Common pigmentation disorders. Am Fam Physician. 2009;79:109-116.

An Unusual Case of Folliculitis Spinulosa Decalvans
Case Report
A 24-year-old man was referred to the dermatology department for evaluation of pustules, atrophic scars, and alopecia on the scalp of 6 years’ duration. Six years prior, erythema, scaling, and follicular keratotic papules had appeared on the superciliary arches, and he started to lose hair from the eyebrows. Three months later, he developed mildly pruritic and painful scaling and pustules on the scalp. These lesions resolved with atrophic scarring accompanied by alopecia. One year later, follicular keratotic papules developed on the cheeks, chest, abdomen, back, lateral upper arms, thighs, and axillae. Two years later, direct microscopy of the lesions on the scalp and fungal culture were negative. After 2 weeks of treatment with roxithromycin (0.15 g twice daily), the scalp pustules dried out and resolved; however, they recurred when the patient stopped taking the medication. Six months later, he was started on isotretinoin treatment (10 mg once daily) for half a year, but no improvement was seen. His parents were nonconsanguineous, and no other family members were affected.
Dermatologic examination revealed large areas of atrophic scarring and alopecia on the scalp. Only a few solitary hairs remained on the top of the head, with the follicles surrounded by keratotic papules, pustules, and black scabs. There was sparse hair on the forehead and temples and scattered hair clusters in the occipital region near the hairline. These follicles also were associated with keratotic papules (Figure 1A). Erythema, scales, and follicular keratotic papules of the superciliary arches with sparse eyebrows and axillary hairs were noted. Follicular keratotic papules also were observed on the cheeks, axillae, chest, abdomen, back, lateral upper arms, and thighs. Dental examination revealed a large space between the upper anterior teeth and the lower anterior teeth. The upper anterior teeth were anteverted, there was congenital absence of right lower central incisors, and the anterior teeth were in deep overbite and overjet (Figure 1B). There was gingival atrophy and calculus dentalis in the upper and lower teeth. He had a fissured tongue with atrophic filiform papillae (Figure 1C).
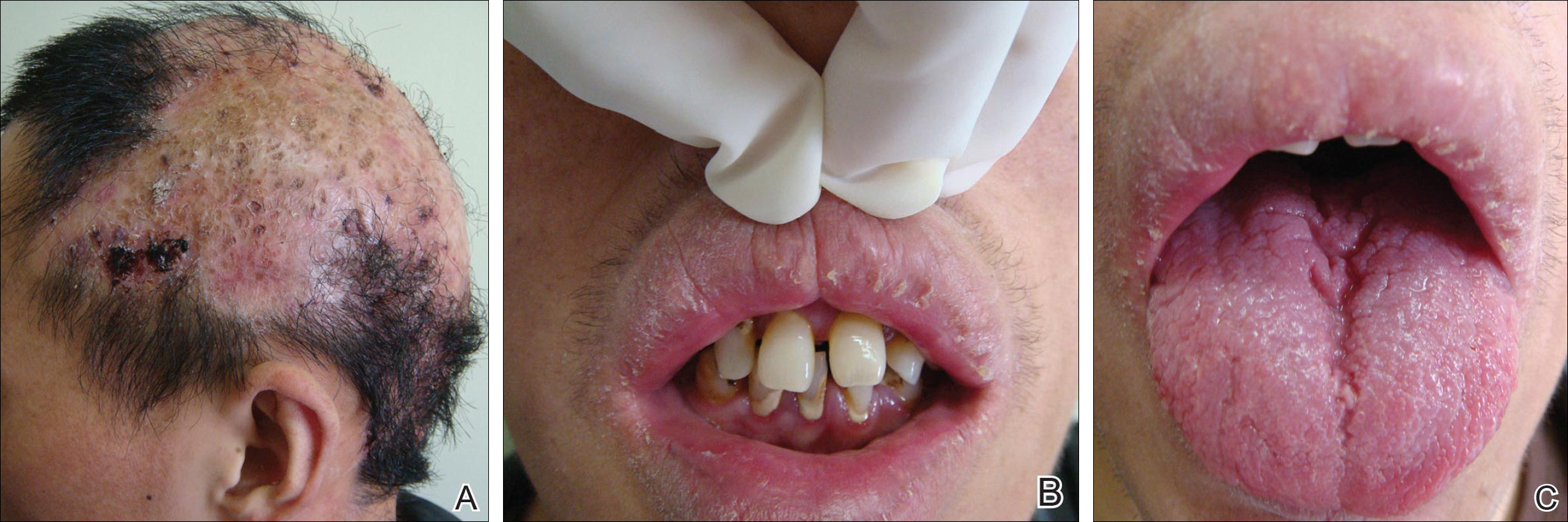
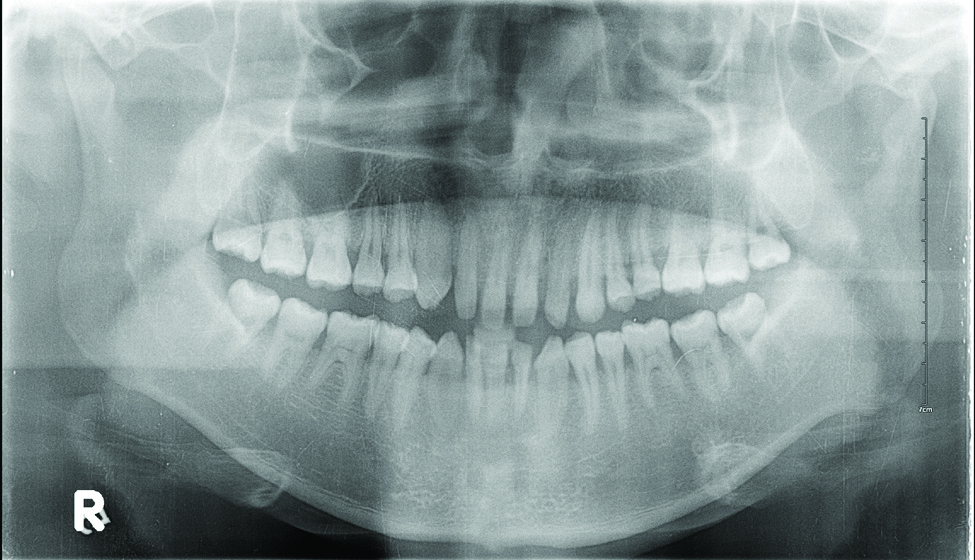
Laboratory testing of the blood, urine, stool, hepatic and renal function, and serum vitamin B2 and B12 levelswere all within reference range. A panoramic radiograph of the occlusal surface showed congenital absence of right lower central incisors (Figure 2), and a lateral projection of a cranial radiograph confirmed that the anterior teeth were in deep overbite and overjet. Direct microscopy and fungal culture of material collected from the dorsal tongue were negative. Direct microscopy and fungal culture of diseased hairs also were negative. A rapid plasma reagin test, Treponema pallidum hemagglutination assay, and human immunodeficiency virus test were negative. Staphylococcus aureus was isolated from the scalp pustules, and in vitro drug susceptibility testing showed that it was sensitive to clarithromycin and moxifloxacin. Pathological examination of a biopsy of the occipital skin lesions showed a thickened epidermal spinous layer and massive infiltration of plasma cells, neutrophils, and multinucleated giant cells around the hair follicles (Figure 3). Pathological examination of the skin lesions on the superciliary arch also showed infiltration of inflammatory cells in the dermis around the hair follicles.
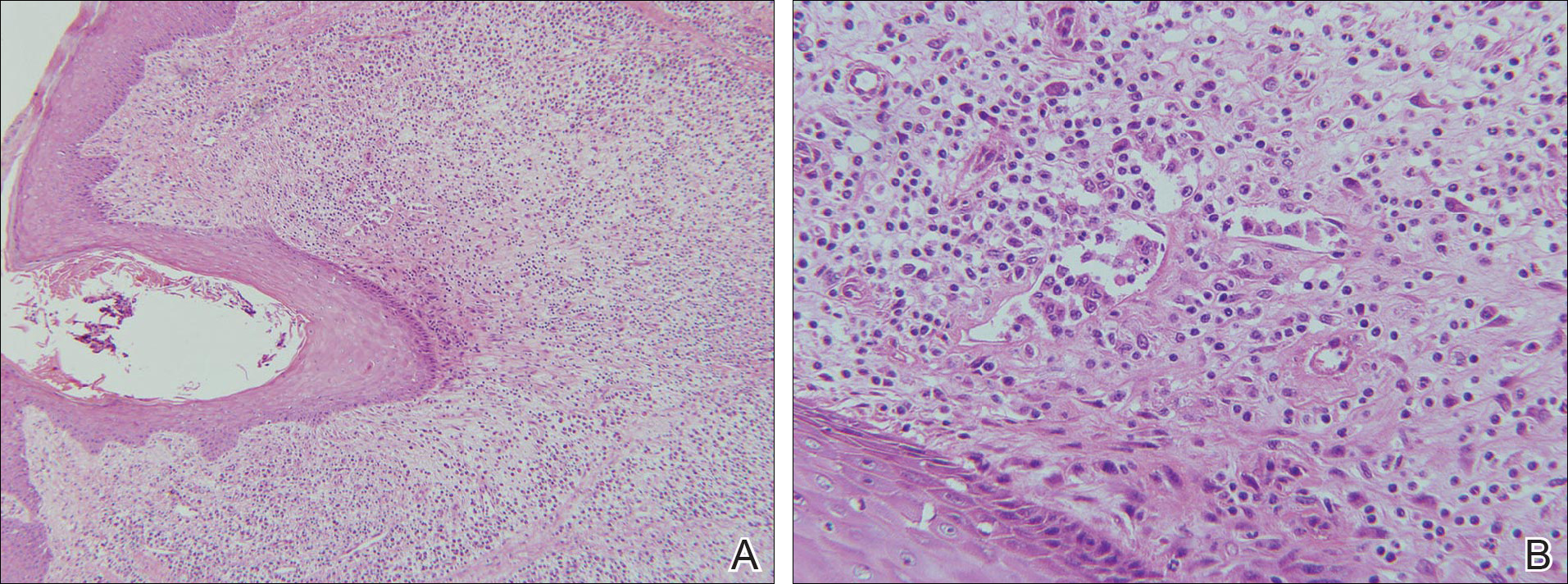
Based on these findings, a diagnosis of folliculitis spinulosa decalvans (FSD) was made and the patient was started on clarithromycin (0.25 g twice daily), metronidazole (0.2 g 3 times daily), viaminate (50 mg 3 times daily), and fusidic acid cream (coating the affected area twice daily). When he returned for follow-up 1 month later, the pustules had disappeared and the black scabs had fallen off, leaving atrophic scars. The long-term efficacy of this regimen is still under observation.
Comment
Folliculitis spinulosa decalvans, along with keratosis follicularis spinulosa decalvans (KFSD), keratosis pilaris atrophicans faciei, and atrophoderma vermiculatum, belongs to a group of diseases that includes keratosis pilaris atrophicans. In 1994, Oranje et al1 suggested the term folliculitis spinulosa decalvans, with signs including persistent pustules, characteristic keratotic papules, and scarring alopecia of the scalp, which may be exacerbated at puberty. Staphylococcus aureus was isolated from the pustules in one study2; however, in another study, repeated cultures were negative.3 Although the main inheritance pattern of KFSD is X-linked, autosomal-dominant inheritance is more common in FSD. Furthermore, there are certain differences in the clinical manifestations of these 2 conditions. Therefore, it remains controversial if FSD is an independent disease or merely a subtype of KFSD.
Our patient’s symptoms manifested after puberty, primarily pustules as well as atrophic and scarring alopecia of the scalp and follicular keratotic papules on the head, face, trunk, lateral upper arms, and thighs. Pathologic examination showed massive infiltration of plasma cells, neutrophils, and multinucleated giant cells around the hair follicles. The clinical and histopathologic findings met the diagnostic criteria for FSD.
Folliculitis spinulosa decalvans is a rare clinical condition with few cases reported.3-5 In addition to the aforementioned characteristic clinical manifestations, our patient also had dental anomalies, a fissured tongue, and atrophy of the tongue papillae, which are not known to be associated with FSD. Dental anomalies are characteristic of patients with Down syndrome, ectodermal dysplasia, Papillon-Lefèvre syndrome, and other conditions.6 Fissured tongue is a normal variant that occurs in 5% to 11% of individuals. It also is a classic but nonspecific feature of Melkersson-Rosenthal syndrome and may occur in psoriasis, Down syndrome, acromegaly, and Sjögren syndrome.7 Atrophy of the tongue papillae is associated with anemia, pellagra, Sjögren syndrome, candidiasis, and other conditions.8 Because there are no known reports of associations between FSD and any of these oral manifestations, it is possible that they were unrelated to FSD in our patient.
Folliculitis spinulosa decalvans usually is recurrent and there is no consistently effective treatment for it. Kunte et al4 reported that dapsone (100 mg/d) led to resolution of scalp inflammation and pustules within 1 month. Romine et al2 reported that a 3-week course of dichloroxacillin (250 mg 4 times daily) induced disappearance of pustules around the hair follicles. However, Hallai et al5 reported a patient who was resistant to isotretinoin treatment. In our case, after 1 month of treatment with clarithromycin, metronidazole, viaminate, and fusidic acid cream, the pustules had resolved and the black scabs had fallen off, leaving atrophic scars. The long-term efficacy of this regimen is still under observation.
Conclusion
We report a case of FSD with dental anomalies, a fissured tongue, and atrophy of tongue papillae, none of which have previously been reported in association with FSD. We, therefore, believe that our patient’s oral manifestations are unrelated to FSD.
- Oranje AP, van Osch LD, Oosterwijk JC. Keratosis pilaris atrophicans. one heterogeneous disease or a symptom in different clinical entities? Arch Dermatol. 1994;13:500-502.
- Romine KA, Rothschild JG, Hansen RC. Cicatricial alopecia and keratosis pilaris. keratosis follicularis spinulosa decalvans. Arch Dermatol. 1997;13:381-384.
- Di Lernia V, Ricci C. Folliculitis spinulosa decalvans: an uncommon entity within the keratosis pilaris atrophicans spectrum. Pediatr Dermatol. 2006;23:255-258.
- Kunte C, Loeser C, Wolff H. Folliculitis spinulosa decalvans: successful therapy with dapsone. J Am Acad Dermatol. 1998;39(5, pt 2):891-892.
- Hallai N, Thompson I, Williams P, et al. Folliculitis spinulosa decalvans: failure to respond to oral isotretinoin. J Eur Acad Dermatol Venereol. 2006;20:223-224.
- Scully C, Hegarty A. The oral cavity and lips. In: Burns T, Breathnach S, Cox N, et al. Rook’s Textbook of Dermatology. 8th ed. Oxford, England: Wiley-Blackwell; 2010:69.7-69.10.
- Wolff K, Goldsmith LA, Katz SI, et al. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: McGraw-Hill Companies; 2007:643.
- Mulliken RA, Casner MJ. Oral manifestations of systemic disease. Emerg Med Clin North Am. 2000;18:565-575.
Case Report
A 24-year-old man was referred to the dermatology department for evaluation of pustules, atrophic scars, and alopecia on the scalp of 6 years’ duration. Six years prior, erythema, scaling, and follicular keratotic papules had appeared on the superciliary arches, and he started to lose hair from the eyebrows. Three months later, he developed mildly pruritic and painful scaling and pustules on the scalp. These lesions resolved with atrophic scarring accompanied by alopecia. One year later, follicular keratotic papules developed on the cheeks, chest, abdomen, back, lateral upper arms, thighs, and axillae. Two years later, direct microscopy of the lesions on the scalp and fungal culture were negative. After 2 weeks of treatment with roxithromycin (0.15 g twice daily), the scalp pustules dried out and resolved; however, they recurred when the patient stopped taking the medication. Six months later, he was started on isotretinoin treatment (10 mg once daily) for half a year, but no improvement was seen. His parents were nonconsanguineous, and no other family members were affected.
Dermatologic examination revealed large areas of atrophic scarring and alopecia on the scalp. Only a few solitary hairs remained on the top of the head, with the follicles surrounded by keratotic papules, pustules, and black scabs. There was sparse hair on the forehead and temples and scattered hair clusters in the occipital region near the hairline. These follicles also were associated with keratotic papules (Figure 1A). Erythema, scales, and follicular keratotic papules of the superciliary arches with sparse eyebrows and axillary hairs were noted. Follicular keratotic papules also were observed on the cheeks, axillae, chest, abdomen, back, lateral upper arms, and thighs. Dental examination revealed a large space between the upper anterior teeth and the lower anterior teeth. The upper anterior teeth were anteverted, there was congenital absence of right lower central incisors, and the anterior teeth were in deep overbite and overjet (Figure 1B). There was gingival atrophy and calculus dentalis in the upper and lower teeth. He had a fissured tongue with atrophic filiform papillae (Figure 1C).
Laboratory testing of the blood, urine, stool, hepatic and renal function, and serum vitamin B2 and B12 levelswere all within reference range. A panoramic radiograph of the occlusal surface showed congenital absence of right lower central incisors (Figure 2), and a lateral projection of a cranial radiograph confirmed that the anterior teeth were in deep overbite and overjet. Direct microscopy and fungal culture of material collected from the dorsal tongue were negative. Direct microscopy and fungal culture of diseased hairs also were negative. A rapid plasma reagin test, Treponema pallidum hemagglutination assay, and human immunodeficiency virus test were negative. Staphylococcus aureus was isolated from the scalp pustules, and in vitro drug susceptibility testing showed that it was sensitive to clarithromycin and moxifloxacin. Pathological examination of a biopsy of the occipital skin lesions showed a thickened epidermal spinous layer and massive infiltration of plasma cells, neutrophils, and multinucleated giant cells around the hair follicles (Figure 3). Pathological examination of the skin lesions on the superciliary arch also showed infiltration of inflammatory cells in the dermis around the hair follicles.
Based on these findings, a diagnosis of folliculitis spinulosa decalvans (FSD) was made and the patient was started on clarithromycin (0.25 g twice daily), metronidazole (0.2 g 3 times daily), viaminate (50 mg 3 times daily), and fusidic acid cream (coating the affected area twice daily). When he returned for follow-up 1 month later, the pustules had disappeared and the black scabs had fallen off, leaving atrophic scars. The long-term efficacy of this regimen is still under observation.
Comment
Folliculitis spinulosa decalvans, along with keratosis follicularis spinulosa decalvans (KFSD), keratosis pilaris atrophicans faciei, and atrophoderma vermiculatum, belongs to a group of diseases that includes keratosis pilaris atrophicans. In 1994, Oranje et al1 suggested the term folliculitis spinulosa decalvans, with signs including persistent pustules, characteristic keratotic papules, and scarring alopecia of the scalp, which may be exacerbated at puberty. Staphylococcus aureus was isolated from the pustules in one study2; however, in another study, repeated cultures were negative.3 Although the main inheritance pattern of KFSD is X-linked, autosomal-dominant inheritance is more common in FSD. Furthermore, there are certain differences in the clinical manifestations of these 2 conditions. Therefore, it remains controversial if FSD is an independent disease or merely a subtype of KFSD.
Our patient’s symptoms manifested after puberty, primarily pustules as well as atrophic and scarring alopecia of the scalp and follicular keratotic papules on the head, face, trunk, lateral upper arms, and thighs. Pathologic examination showed massive infiltration of plasma cells, neutrophils, and multinucleated giant cells around the hair follicles. The clinical and histopathologic findings met the diagnostic criteria for FSD.
Folliculitis spinulosa decalvans is a rare clinical condition with few cases reported.3-5 In addition to the aforementioned characteristic clinical manifestations, our patient also had dental anomalies, a fissured tongue, and atrophy of the tongue papillae, which are not known to be associated with FSD. Dental anomalies are characteristic of patients with Down syndrome, ectodermal dysplasia, Papillon-Lefèvre syndrome, and other conditions.6 Fissured tongue is a normal variant that occurs in 5% to 11% of individuals. It also is a classic but nonspecific feature of Melkersson-Rosenthal syndrome and may occur in psoriasis, Down syndrome, acromegaly, and Sjögren syndrome.7 Atrophy of the tongue papillae is associated with anemia, pellagra, Sjögren syndrome, candidiasis, and other conditions.8 Because there are no known reports of associations between FSD and any of these oral manifestations, it is possible that they were unrelated to FSD in our patient.
Folliculitis spinulosa decalvans usually is recurrent and there is no consistently effective treatment for it. Kunte et al4 reported that dapsone (100 mg/d) led to resolution of scalp inflammation and pustules within 1 month. Romine et al2 reported that a 3-week course of dichloroxacillin (250 mg 4 times daily) induced disappearance of pustules around the hair follicles. However, Hallai et al5 reported a patient who was resistant to isotretinoin treatment. In our case, after 1 month of treatment with clarithromycin, metronidazole, viaminate, and fusidic acid cream, the pustules had resolved and the black scabs had fallen off, leaving atrophic scars. The long-term efficacy of this regimen is still under observation.
Conclusion
We report a case of FSD with dental anomalies, a fissured tongue, and atrophy of tongue papillae, none of which have previously been reported in association with FSD. We, therefore, believe that our patient’s oral manifestations are unrelated to FSD.
Case Report
A 24-year-old man was referred to the dermatology department for evaluation of pustules, atrophic scars, and alopecia on the scalp of 6 years’ duration. Six years prior, erythema, scaling, and follicular keratotic papules had appeared on the superciliary arches, and he started to lose hair from the eyebrows. Three months later, he developed mildly pruritic and painful scaling and pustules on the scalp. These lesions resolved with atrophic scarring accompanied by alopecia. One year later, follicular keratotic papules developed on the cheeks, chest, abdomen, back, lateral upper arms, thighs, and axillae. Two years later, direct microscopy of the lesions on the scalp and fungal culture were negative. After 2 weeks of treatment with roxithromycin (0.15 g twice daily), the scalp pustules dried out and resolved; however, they recurred when the patient stopped taking the medication. Six months later, he was started on isotretinoin treatment (10 mg once daily) for half a year, but no improvement was seen. His parents were nonconsanguineous, and no other family members were affected.
Dermatologic examination revealed large areas of atrophic scarring and alopecia on the scalp. Only a few solitary hairs remained on the top of the head, with the follicles surrounded by keratotic papules, pustules, and black scabs. There was sparse hair on the forehead and temples and scattered hair clusters in the occipital region near the hairline. These follicles also were associated with keratotic papules (Figure 1A). Erythema, scales, and follicular keratotic papules of the superciliary arches with sparse eyebrows and axillary hairs were noted. Follicular keratotic papules also were observed on the cheeks, axillae, chest, abdomen, back, lateral upper arms, and thighs. Dental examination revealed a large space between the upper anterior teeth and the lower anterior teeth. The upper anterior teeth were anteverted, there was congenital absence of right lower central incisors, and the anterior teeth were in deep overbite and overjet (Figure 1B). There was gingival atrophy and calculus dentalis in the upper and lower teeth. He had a fissured tongue with atrophic filiform papillae (Figure 1C).
Laboratory testing of the blood, urine, stool, hepatic and renal function, and serum vitamin B2 and B12 levelswere all within reference range. A panoramic radiograph of the occlusal surface showed congenital absence of right lower central incisors (Figure 2), and a lateral projection of a cranial radiograph confirmed that the anterior teeth were in deep overbite and overjet. Direct microscopy and fungal culture of material collected from the dorsal tongue were negative. Direct microscopy and fungal culture of diseased hairs also were negative. A rapid plasma reagin test, Treponema pallidum hemagglutination assay, and human immunodeficiency virus test were negative. Staphylococcus aureus was isolated from the scalp pustules, and in vitro drug susceptibility testing showed that it was sensitive to clarithromycin and moxifloxacin. Pathological examination of a biopsy of the occipital skin lesions showed a thickened epidermal spinous layer and massive infiltration of plasma cells, neutrophils, and multinucleated giant cells around the hair follicles (Figure 3). Pathological examination of the skin lesions on the superciliary arch also showed infiltration of inflammatory cells in the dermis around the hair follicles.
Based on these findings, a diagnosis of folliculitis spinulosa decalvans (FSD) was made and the patient was started on clarithromycin (0.25 g twice daily), metronidazole (0.2 g 3 times daily), viaminate (50 mg 3 times daily), and fusidic acid cream (coating the affected area twice daily). When he returned for follow-up 1 month later, the pustules had disappeared and the black scabs had fallen off, leaving atrophic scars. The long-term efficacy of this regimen is still under observation.
Comment
Folliculitis spinulosa decalvans, along with keratosis follicularis spinulosa decalvans (KFSD), keratosis pilaris atrophicans faciei, and atrophoderma vermiculatum, belongs to a group of diseases that includes keratosis pilaris atrophicans. In 1994, Oranje et al1 suggested the term folliculitis spinulosa decalvans, with signs including persistent pustules, characteristic keratotic papules, and scarring alopecia of the scalp, which may be exacerbated at puberty. Staphylococcus aureus was isolated from the pustules in one study2; however, in another study, repeated cultures were negative.3 Although the main inheritance pattern of KFSD is X-linked, autosomal-dominant inheritance is more common in FSD. Furthermore, there are certain differences in the clinical manifestations of these 2 conditions. Therefore, it remains controversial if FSD is an independent disease or merely a subtype of KFSD.
Our patient’s symptoms manifested after puberty, primarily pustules as well as atrophic and scarring alopecia of the scalp and follicular keratotic papules on the head, face, trunk, lateral upper arms, and thighs. Pathologic examination showed massive infiltration of plasma cells, neutrophils, and multinucleated giant cells around the hair follicles. The clinical and histopathologic findings met the diagnostic criteria for FSD.
Folliculitis spinulosa decalvans is a rare clinical condition with few cases reported.3-5 In addition to the aforementioned characteristic clinical manifestations, our patient also had dental anomalies, a fissured tongue, and atrophy of the tongue papillae, which are not known to be associated with FSD. Dental anomalies are characteristic of patients with Down syndrome, ectodermal dysplasia, Papillon-Lefèvre syndrome, and other conditions.6 Fissured tongue is a normal variant that occurs in 5% to 11% of individuals. It also is a classic but nonspecific feature of Melkersson-Rosenthal syndrome and may occur in psoriasis, Down syndrome, acromegaly, and Sjögren syndrome.7 Atrophy of the tongue papillae is associated with anemia, pellagra, Sjögren syndrome, candidiasis, and other conditions.8 Because there are no known reports of associations between FSD and any of these oral manifestations, it is possible that they were unrelated to FSD in our patient.
Folliculitis spinulosa decalvans usually is recurrent and there is no consistently effective treatment for it. Kunte et al4 reported that dapsone (100 mg/d) led to resolution of scalp inflammation and pustules within 1 month. Romine et al2 reported that a 3-week course of dichloroxacillin (250 mg 4 times daily) induced disappearance of pustules around the hair follicles. However, Hallai et al5 reported a patient who was resistant to isotretinoin treatment. In our case, after 1 month of treatment with clarithromycin, metronidazole, viaminate, and fusidic acid cream, the pustules had resolved and the black scabs had fallen off, leaving atrophic scars. The long-term efficacy of this regimen is still under observation.
Conclusion
We report a case of FSD with dental anomalies, a fissured tongue, and atrophy of tongue papillae, none of which have previously been reported in association with FSD. We, therefore, believe that our patient’s oral manifestations are unrelated to FSD.
- Oranje AP, van Osch LD, Oosterwijk JC. Keratosis pilaris atrophicans. one heterogeneous disease or a symptom in different clinical entities? Arch Dermatol. 1994;13:500-502.
- Romine KA, Rothschild JG, Hansen RC. Cicatricial alopecia and keratosis pilaris. keratosis follicularis spinulosa decalvans. Arch Dermatol. 1997;13:381-384.
- Di Lernia V, Ricci C. Folliculitis spinulosa decalvans: an uncommon entity within the keratosis pilaris atrophicans spectrum. Pediatr Dermatol. 2006;23:255-258.
- Kunte C, Loeser C, Wolff H. Folliculitis spinulosa decalvans: successful therapy with dapsone. J Am Acad Dermatol. 1998;39(5, pt 2):891-892.
- Hallai N, Thompson I, Williams P, et al. Folliculitis spinulosa decalvans: failure to respond to oral isotretinoin. J Eur Acad Dermatol Venereol. 2006;20:223-224.
- Scully C, Hegarty A. The oral cavity and lips. In: Burns T, Breathnach S, Cox N, et al. Rook’s Textbook of Dermatology. 8th ed. Oxford, England: Wiley-Blackwell; 2010:69.7-69.10.
- Wolff K, Goldsmith LA, Katz SI, et al. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: McGraw-Hill Companies; 2007:643.
- Mulliken RA, Casner MJ. Oral manifestations of systemic disease. Emerg Med Clin North Am. 2000;18:565-575.
- Oranje AP, van Osch LD, Oosterwijk JC. Keratosis pilaris atrophicans. one heterogeneous disease or a symptom in different clinical entities? Arch Dermatol. 1994;13:500-502.
- Romine KA, Rothschild JG, Hansen RC. Cicatricial alopecia and keratosis pilaris. keratosis follicularis spinulosa decalvans. Arch Dermatol. 1997;13:381-384.
- Di Lernia V, Ricci C. Folliculitis spinulosa decalvans: an uncommon entity within the keratosis pilaris atrophicans spectrum. Pediatr Dermatol. 2006;23:255-258.
- Kunte C, Loeser C, Wolff H. Folliculitis spinulosa decalvans: successful therapy with dapsone. J Am Acad Dermatol. 1998;39(5, pt 2):891-892.
- Hallai N, Thompson I, Williams P, et al. Folliculitis spinulosa decalvans: failure to respond to oral isotretinoin. J Eur Acad Dermatol Venereol. 2006;20:223-224.
- Scully C, Hegarty A. The oral cavity and lips. In: Burns T, Breathnach S, Cox N, et al. Rook’s Textbook of Dermatology. 8th ed. Oxford, England: Wiley-Blackwell; 2010:69.7-69.10.
- Wolff K, Goldsmith LA, Katz SI, et al. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: McGraw-Hill Companies; 2007:643.
- Mulliken RA, Casner MJ. Oral manifestations of systemic disease. Emerg Med Clin North Am. 2000;18:565-575.
Practice Points
- Folliculitis spinulosa decalvans (FSD) presents with persistent pustules, characteristic keratotic papules, and scarring alopecia of the scalp.
- In the case described here, oral manifestations also were present but are not characteristic of FSD.
Nevus Spilus: Is the Presence of Hair Associated With an Increased Risk for Melanoma?
The term nevus spilus (NS), also known as speckled lentiginous nevus, was first used in the 19th century to describe lesions with background café au lait–like lentiginous melanocytic hyperplasia speckled with small, 1- to 3-mm, darker foci. The dark spots reflect lentigines; junctional, compound, and intradermal nevus cell nests; and more rarely Spitz and blue nevi. Both macular and papular subtypes have been described.1 This birthmark is quite common, occurring in 1.3% to 2.3% of the adult population worldwide.2 Hypertrichosis has been described in NS.3-9 Two subsequent cases of malignant melanoma in hairy NS suggested that lesions may be particularly prone to malignant degeneration.4,8 We report an additional case of hairy NS that was not associated with melanoma and consider whether dermatologists should warn their patients about this association.
Case Report
A 26-year-old woman presented with a stable 7×8-cm, tan-brown, macular, pigmented birthmark studded with darker 1- to 2-mm, irregular, brown-black and blue, confettilike macules on the left proximal lateral thigh that had been present since birth (Figure 1). Dark terminal hairs were present, arising from both the darker and lighter pigmented areas but not the surrounding normal skin.
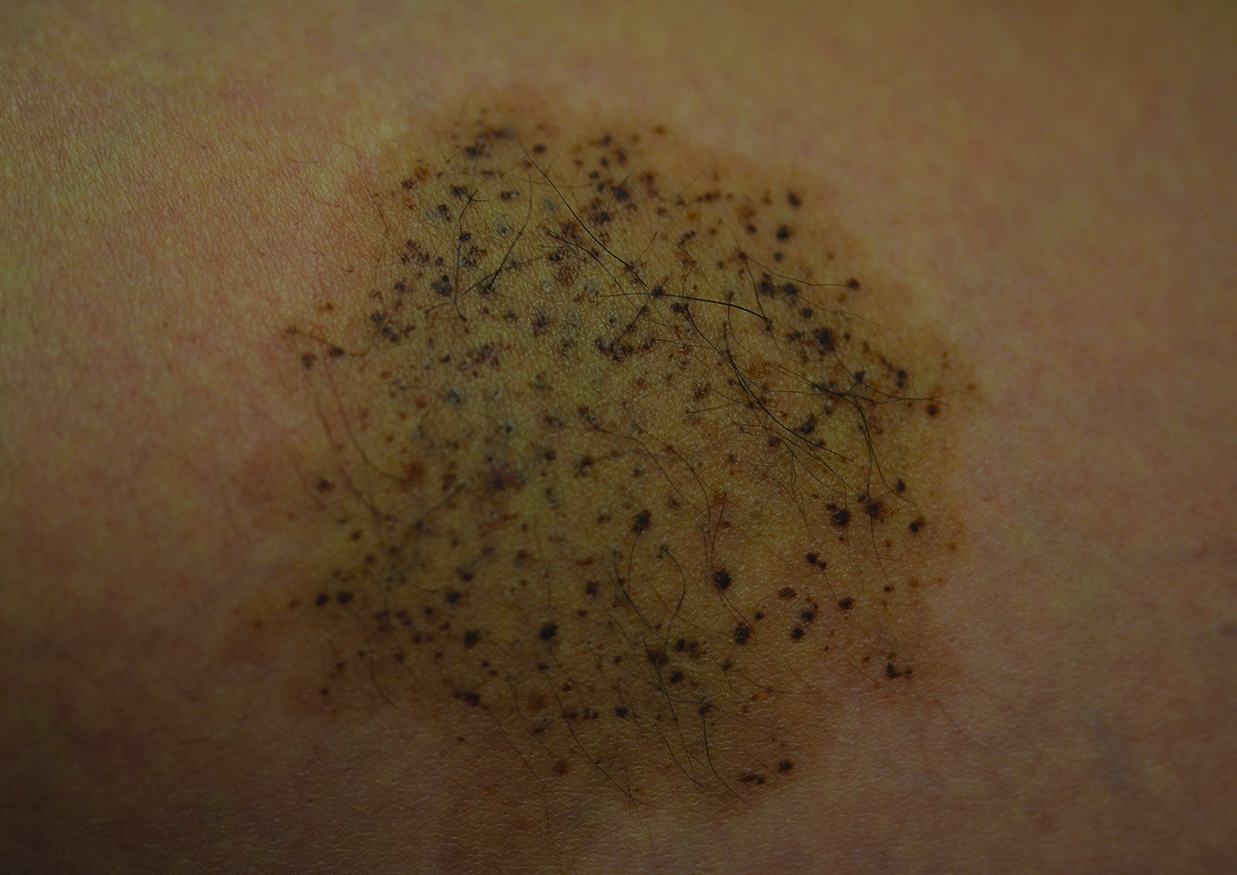
A 4-mm punch biopsy from one of the dark blue macules demonstrated uniform lentiginous melanocytic hyperplasia and nevus cell nests adjacent to the sweat glands extending into the mid dermis (Figure 2). No clinical evidence of malignant degeneration was present.
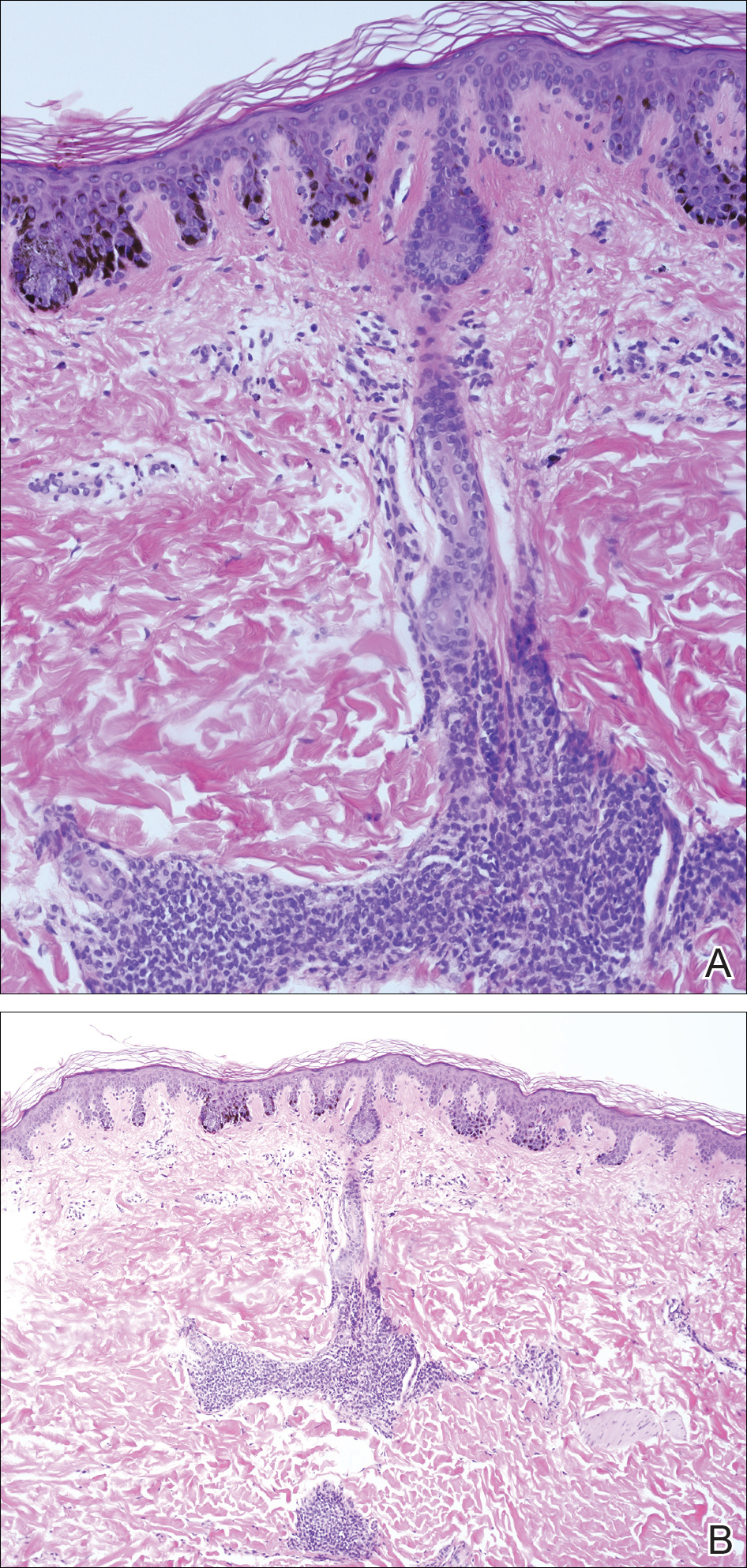
Comment
The risk for melanoma is increased in classic nonspeckled congenital nevi and the risk correlates with the size of the lesion and most probably the number of nevus cells in the lesion that increase the risk for a random mutation.8,10,11 It is likely that NS with or without hair presages a small increased risk for melanoma,6,9,12 which is not surprising because NS is a subtype of congenital melanocytic nevus (CMN), a condition that is present at birth and results from a proliferation of melanocytes.6 Nevus spilus, however, appears to have a notably lower risk for malignant degeneration than other classic CMN of the same size. The following support for this hypothesis is offered: First, CMN have nevus cells broadly filling the dermis that extend more deeply into the dermis than NS (Figure 2A).10 In our estimation, CMN have at least 100 times the number of nevus cells per square centimeter compared to NS. The potential for malignant degeneration of any one melanocyte is greater when more are present. Second, although some NS lesions evolve, classic CMN are universally more proliferative than NS.10,13 The involved skin in CMN thickens over time with increased numbers of melanocytes and marked overgrowth of adjacent tissue. Melanocytes in a proliferative phase may be more likely to undergo malignant degeneration.10
A PubMed search of articles indexed for MEDLINE using the search term nevus spilus and melanoma yielded 2 cases4,8 of melanoma arising among 15 cases of hairy NS in the literature, which led to the suggestion that the presence of hair could be associated with an increased risk for malignant degeneration in NS (Table). This apparent high incidence of melanoma most likely reflects referral/publication bias rather than a statistically significant association. In fact, the clinical lesion most clinically similar to hairy NS is Becker nevus, with tan macules demonstrating lentiginous melanocytic hyperplasia associated with numerous coarse terminal hairs. There is no indication that Becker nevi have a considerable premalignant potential, though one case of melanoma arising in a Becker nevus has been reported.9 There is no evidence to suggest that classic CMN with hypertrichosis has a greater premalignant potential than similar lesions without hypertrichosis.

We noticed the presence of hair in our patient’s lesion only after reports in the literature caused us to look for this phenomenon.9 This occurrence may actually be quite common. We do not recommend prophylactic excision of NS and believe the risk for malignant degeneration is low in NS with or without hair, though larger NS (>4 cm), especially giant, zosteriform, or segmental lesions, may have a greater risk.1,6,9,10 It is prudent for physicians to carefully examine NS and sample suspicious foci, especially when patients describe a lesion as changing.
- Vidaurri-de la Cruz H, Happle R. Two distinct types of speckled lentiginous nevi characterized by macular versus papular speckles. Dermatology. 2006;212:53-58.
- Ly L, Christie M, Swain S, et al. Melanoma(s) arising in large segmental speckled lentiginous nevi: a case series. J Am Acad Dermatol. 2011;64:1190-1193.
- Prose NS, Heilman E, Felman YM, et al. Multiple benign juvenile melanoma. J Am Acad Dermatol. 1983;9:236-242.
- Grinspan D, Casala A, Abulafia J, et al. Melanoma on dysplastic nevus spilus. Int J Dermatol. 1997;36:499-502 .
- Langenbach N, Pfau A, Landthaler M, et al. Naevi spili, café-au-lait spots and melanocytic naevi aggregated alongside Blaschko’s lines, with a review of segmental melanocytic lesions. Acta Derm Venereol. 1998;78:378-380.
- Schaffer JV, Orlow SJ, Lazova R, et al. Speckled lentiginous nevus: within the spectrum of congenital melanocytic nevi. Arch Dermatol. 2001;137:172-178.
- Saraswat A, Dogra S, Bansali A, et al. Phakomatosis pigmentokeratotica associated with hypophosphataemic vitamin D–resistant rickets: improvement in phosphate homeostasis after partial laser ablation. Br J Dermatol. 2003;148:1074-1076.
- Zeren-Bilgin
i , Gür S, Aydın O, et al. Melanoma arising in a hairy nevus spilus. Int J Dermatol. 2006;45:1362-1364. - Singh S, Jain N, Khanna N, et al. Hairy nevus spilus: a case series. Pediatr Dermatol. 2013;30:100-104.
- Price HN, Schaffer JV. Congenital melanocytic nevi—when to worry and how to treat: facts and controversies. Clin Dermatol. 2010;28:293-302.
- Alikhan Ali, Ibrahimi OA, Eisen DB. Congenital melanocytic nevi: where are we now? J Am Acad Dermatol. 2012;67:495.e1-495.e17.
- Haenssle HA, Kaune KM, Buhl T, et al. Melanoma arising in segmental nevus spilus: detection by sequential digital dermatoscopy. J Am Acad Dermatol. 2009;61:337-341.
- Cohen LM. Nevus spilus: congenital or acquired? Arch Dermatol. 2001;137:215-216.
The term nevus spilus (NS), also known as speckled lentiginous nevus, was first used in the 19th century to describe lesions with background café au lait–like lentiginous melanocytic hyperplasia speckled with small, 1- to 3-mm, darker foci. The dark spots reflect lentigines; junctional, compound, and intradermal nevus cell nests; and more rarely Spitz and blue nevi. Both macular and papular subtypes have been described.1 This birthmark is quite common, occurring in 1.3% to 2.3% of the adult population worldwide.2 Hypertrichosis has been described in NS.3-9 Two subsequent cases of malignant melanoma in hairy NS suggested that lesions may be particularly prone to malignant degeneration.4,8 We report an additional case of hairy NS that was not associated with melanoma and consider whether dermatologists should warn their patients about this association.
Case Report
A 26-year-old woman presented with a stable 7×8-cm, tan-brown, macular, pigmented birthmark studded with darker 1- to 2-mm, irregular, brown-black and blue, confettilike macules on the left proximal lateral thigh that had been present since birth (Figure 1). Dark terminal hairs were present, arising from both the darker and lighter pigmented areas but not the surrounding normal skin.
A 4-mm punch biopsy from one of the dark blue macules demonstrated uniform lentiginous melanocytic hyperplasia and nevus cell nests adjacent to the sweat glands extending into the mid dermis (Figure 2). No clinical evidence of malignant degeneration was present.
Comment
The risk for melanoma is increased in classic nonspeckled congenital nevi and the risk correlates with the size of the lesion and most probably the number of nevus cells in the lesion that increase the risk for a random mutation.8,10,11 It is likely that NS with or without hair presages a small increased risk for melanoma,6,9,12 which is not surprising because NS is a subtype of congenital melanocytic nevus (CMN), a condition that is present at birth and results from a proliferation of melanocytes.6 Nevus spilus, however, appears to have a notably lower risk for malignant degeneration than other classic CMN of the same size. The following support for this hypothesis is offered: First, CMN have nevus cells broadly filling the dermis that extend more deeply into the dermis than NS (Figure 2A).10 In our estimation, CMN have at least 100 times the number of nevus cells per square centimeter compared to NS. The potential for malignant degeneration of any one melanocyte is greater when more are present. Second, although some NS lesions evolve, classic CMN are universally more proliferative than NS.10,13 The involved skin in CMN thickens over time with increased numbers of melanocytes and marked overgrowth of adjacent tissue. Melanocytes in a proliferative phase may be more likely to undergo malignant degeneration.10
A PubMed search of articles indexed for MEDLINE using the search term nevus spilus and melanoma yielded 2 cases4,8 of melanoma arising among 15 cases of hairy NS in the literature, which led to the suggestion that the presence of hair could be associated with an increased risk for malignant degeneration in NS (Table). This apparent high incidence of melanoma most likely reflects referral/publication bias rather than a statistically significant association. In fact, the clinical lesion most clinically similar to hairy NS is Becker nevus, with tan macules demonstrating lentiginous melanocytic hyperplasia associated with numerous coarse terminal hairs. There is no indication that Becker nevi have a considerable premalignant potential, though one case of melanoma arising in a Becker nevus has been reported.9 There is no evidence to suggest that classic CMN with hypertrichosis has a greater premalignant potential than similar lesions without hypertrichosis.
We noticed the presence of hair in our patient’s lesion only after reports in the literature caused us to look for this phenomenon.9 This occurrence may actually be quite common. We do not recommend prophylactic excision of NS and believe the risk for malignant degeneration is low in NS with or without hair, though larger NS (>4 cm), especially giant, zosteriform, or segmental lesions, may have a greater risk.1,6,9,10 It is prudent for physicians to carefully examine NS and sample suspicious foci, especially when patients describe a lesion as changing.
The term nevus spilus (NS), also known as speckled lentiginous nevus, was first used in the 19th century to describe lesions with background café au lait–like lentiginous melanocytic hyperplasia speckled with small, 1- to 3-mm, darker foci. The dark spots reflect lentigines; junctional, compound, and intradermal nevus cell nests; and more rarely Spitz and blue nevi. Both macular and papular subtypes have been described.1 This birthmark is quite common, occurring in 1.3% to 2.3% of the adult population worldwide.2 Hypertrichosis has been described in NS.3-9 Two subsequent cases of malignant melanoma in hairy NS suggested that lesions may be particularly prone to malignant degeneration.4,8 We report an additional case of hairy NS that was not associated with melanoma and consider whether dermatologists should warn their patients about this association.
Case Report
A 26-year-old woman presented with a stable 7×8-cm, tan-brown, macular, pigmented birthmark studded with darker 1- to 2-mm, irregular, brown-black and blue, confettilike macules on the left proximal lateral thigh that had been present since birth (Figure 1). Dark terminal hairs were present, arising from both the darker and lighter pigmented areas but not the surrounding normal skin.
A 4-mm punch biopsy from one of the dark blue macules demonstrated uniform lentiginous melanocytic hyperplasia and nevus cell nests adjacent to the sweat glands extending into the mid dermis (Figure 2). No clinical evidence of malignant degeneration was present.
Comment
The risk for melanoma is increased in classic nonspeckled congenital nevi and the risk correlates with the size of the lesion and most probably the number of nevus cells in the lesion that increase the risk for a random mutation.8,10,11 It is likely that NS with or without hair presages a small increased risk for melanoma,6,9,12 which is not surprising because NS is a subtype of congenital melanocytic nevus (CMN), a condition that is present at birth and results from a proliferation of melanocytes.6 Nevus spilus, however, appears to have a notably lower risk for malignant degeneration than other classic CMN of the same size. The following support for this hypothesis is offered: First, CMN have nevus cells broadly filling the dermis that extend more deeply into the dermis than NS (Figure 2A).10 In our estimation, CMN have at least 100 times the number of nevus cells per square centimeter compared to NS. The potential for malignant degeneration of any one melanocyte is greater when more are present. Second, although some NS lesions evolve, classic CMN are universally more proliferative than NS.10,13 The involved skin in CMN thickens over time with increased numbers of melanocytes and marked overgrowth of adjacent tissue. Melanocytes in a proliferative phase may be more likely to undergo malignant degeneration.10
A PubMed search of articles indexed for MEDLINE using the search term nevus spilus and melanoma yielded 2 cases4,8 of melanoma arising among 15 cases of hairy NS in the literature, which led to the suggestion that the presence of hair could be associated with an increased risk for malignant degeneration in NS (Table). This apparent high incidence of melanoma most likely reflects referral/publication bias rather than a statistically significant association. In fact, the clinical lesion most clinically similar to hairy NS is Becker nevus, with tan macules demonstrating lentiginous melanocytic hyperplasia associated with numerous coarse terminal hairs. There is no indication that Becker nevi have a considerable premalignant potential, though one case of melanoma arising in a Becker nevus has been reported.9 There is no evidence to suggest that classic CMN with hypertrichosis has a greater premalignant potential than similar lesions without hypertrichosis.
We noticed the presence of hair in our patient’s lesion only after reports in the literature caused us to look for this phenomenon.9 This occurrence may actually be quite common. We do not recommend prophylactic excision of NS and believe the risk for malignant degeneration is low in NS with or without hair, though larger NS (>4 cm), especially giant, zosteriform, or segmental lesions, may have a greater risk.1,6,9,10 It is prudent for physicians to carefully examine NS and sample suspicious foci, especially when patients describe a lesion as changing.
- Vidaurri-de la Cruz H, Happle R. Two distinct types of speckled lentiginous nevi characterized by macular versus papular speckles. Dermatology. 2006;212:53-58.
- Ly L, Christie M, Swain S, et al. Melanoma(s) arising in large segmental speckled lentiginous nevi: a case series. J Am Acad Dermatol. 2011;64:1190-1193.
- Prose NS, Heilman E, Felman YM, et al. Multiple benign juvenile melanoma. J Am Acad Dermatol. 1983;9:236-242.
- Grinspan D, Casala A, Abulafia J, et al. Melanoma on dysplastic nevus spilus. Int J Dermatol. 1997;36:499-502 .
- Langenbach N, Pfau A, Landthaler M, et al. Naevi spili, café-au-lait spots and melanocytic naevi aggregated alongside Blaschko’s lines, with a review of segmental melanocytic lesions. Acta Derm Venereol. 1998;78:378-380.
- Schaffer JV, Orlow SJ, Lazova R, et al. Speckled lentiginous nevus: within the spectrum of congenital melanocytic nevi. Arch Dermatol. 2001;137:172-178.
- Saraswat A, Dogra S, Bansali A, et al. Phakomatosis pigmentokeratotica associated with hypophosphataemic vitamin D–resistant rickets: improvement in phosphate homeostasis after partial laser ablation. Br J Dermatol. 2003;148:1074-1076.
- Zeren-Bilgin
i , Gür S, Aydın O, et al. Melanoma arising in a hairy nevus spilus. Int J Dermatol. 2006;45:1362-1364. - Singh S, Jain N, Khanna N, et al. Hairy nevus spilus: a case series. Pediatr Dermatol. 2013;30:100-104.
- Price HN, Schaffer JV. Congenital melanocytic nevi—when to worry and how to treat: facts and controversies. Clin Dermatol. 2010;28:293-302.
- Alikhan Ali, Ibrahimi OA, Eisen DB. Congenital melanocytic nevi: where are we now? J Am Acad Dermatol. 2012;67:495.e1-495.e17.
- Haenssle HA, Kaune KM, Buhl T, et al. Melanoma arising in segmental nevus spilus: detection by sequential digital dermatoscopy. J Am Acad Dermatol. 2009;61:337-341.
- Cohen LM. Nevus spilus: congenital or acquired? Arch Dermatol. 2001;137:215-216.
- Vidaurri-de la Cruz H, Happle R. Two distinct types of speckled lentiginous nevi characterized by macular versus papular speckles. Dermatology. 2006;212:53-58.
- Ly L, Christie M, Swain S, et al. Melanoma(s) arising in large segmental speckled lentiginous nevi: a case series. J Am Acad Dermatol. 2011;64:1190-1193.
- Prose NS, Heilman E, Felman YM, et al. Multiple benign juvenile melanoma. J Am Acad Dermatol. 1983;9:236-242.
- Grinspan D, Casala A, Abulafia J, et al. Melanoma on dysplastic nevus spilus. Int J Dermatol. 1997;36:499-502 .
- Langenbach N, Pfau A, Landthaler M, et al. Naevi spili, café-au-lait spots and melanocytic naevi aggregated alongside Blaschko’s lines, with a review of segmental melanocytic lesions. Acta Derm Venereol. 1998;78:378-380.
- Schaffer JV, Orlow SJ, Lazova R, et al. Speckled lentiginous nevus: within the spectrum of congenital melanocytic nevi. Arch Dermatol. 2001;137:172-178.
- Saraswat A, Dogra S, Bansali A, et al. Phakomatosis pigmentokeratotica associated with hypophosphataemic vitamin D–resistant rickets: improvement in phosphate homeostasis after partial laser ablation. Br J Dermatol. 2003;148:1074-1076.
- Zeren-Bilgin
i , Gür S, Aydın O, et al. Melanoma arising in a hairy nevus spilus. Int J Dermatol. 2006;45:1362-1364. - Singh S, Jain N, Khanna N, et al. Hairy nevus spilus: a case series. Pediatr Dermatol. 2013;30:100-104.
- Price HN, Schaffer JV. Congenital melanocytic nevi—when to worry and how to treat: facts and controversies. Clin Dermatol. 2010;28:293-302.
- Alikhan Ali, Ibrahimi OA, Eisen DB. Congenital melanocytic nevi: where are we now? J Am Acad Dermatol. 2012;67:495.e1-495.e17.
- Haenssle HA, Kaune KM, Buhl T, et al. Melanoma arising in segmental nevus spilus: detection by sequential digital dermatoscopy. J Am Acad Dermatol. 2009;61:337-341.
- Cohen LM. Nevus spilus: congenital or acquired? Arch Dermatol. 2001;137:215-216.
Practice Points
- Nevus spilus (NS) appears as a café au lait macule studded with darker brown “moles.”
- Although melanoma has been described in NS, it is rare.
- There is no evidence that hairy NS are predisposed to melanoma.
Can Serum Free Light Chains Be Used for the Early Diagnosis of Monoclonal Immunoglobulin-Secreting B-Cell and Plasma-Cell Diseases? (FULL)
Patients who are undergoing multiple myeloma screening with serum protein electrophoresis and immunofixation, especially those with renal failure, also should receive serum free light chain testing to increase specificity and reduce false-negatives.
Multiple myeloma (MM) is a devastating disease with an estimated 26,850 new cases in 2015 according to Surveillance, Epidemiology, and End Results data and no definitive chemotherapeutic cure.1 In 97% of cases, MM is defined by monoclonal hypergammaglobulinemia, in which a malignant plasma cell clone secretes a monoclonal globulin; the remaining cases are nonsecretors.2 Each pathologically produced clonal globulin contains 2 heavy chains attached by disulfide linkage and 2 light chains. Unchecked plasma cell production is what later causes the symptoms of renal failure, bone destruction, and anemia.
The rate of MM is disproportionately high in the veteran population, and the VA health care system provides care for many of these patients. The higher rate is likely secondary to the predominantly male population, which has higher MM rates, and has been linked to Agent Orange exposure in Vietnam. As MM is not easy to diagnose, any algorithm or testing method would be of great benefit to this population.
The gold standard for MM detection remains serum protein electrophoresis (SPEP) with immunofixation (IFE), but other detection methods have been emerging. The method of serum free light chain (SFLC) assay has become more readily available, and its incorporation into diagnostic guidelines has become more apparent but is not universal.3
In the case series reported in this article, SPEP/IFE and SFLC assays were used to test 207 patients from the VA New York Harbor Healthcare System (VANYHHS). All these patients had a clinical context for MM testing.
Methods
In this retrospective study, the authors reviewed the charts of VANYHHS patients who were being treated for conditions that prompted SPEP/IFE and λ and κ SFLC analysis between December 2013 and March 2014. The study was exempt from institutional review board approval.
The SPEP/IFE analysis was performed with an automated electrophoresis machine (Sebia Electrophoresis), and the SFLC analysis was performed with an automated SFLC assay (Freelite). Sensitivity, specificity, and positive and negative predictive values were calculated using SPEP/IFE as the gold standard and SFLC κ-to-λ ratio asthe test method. Patients with a positive κ-to-λ ratio but negative SPEP were considered false-positives. These patients’ SFLC analyses were further analyzed in an effort to evaluate use of the κ-to-λ ratio as an early tumor marker.
The κ reference range used was 3.3 to 19.4 mg/L, and the λ reference range used was 5.7 to 26.3 mg/L.4 The traditional reference range for the κ-to-λ ratio is 0.26 to 1.65.5
Results
Of the 207 patients in this study, 205 were men. Mean age was 69 years (range, 28-97 years). Mean serum urea nitrogen level was 8.75 mmol/L (range, 2.86-38.21 mmol/L), and mean creatinine level was 140.59 μmol/L (range, 44.21-1503.14 μmol/L). Mean κ was 49.82 mg/L (range, 4.6-700.96 mg/L), and mean λ was 54.27 mg/L (range, 3-1,750 mg/L). Table 1 compares the SPEP and SFLC data. Sensitivity was 67%, specificity was 85%, positive predictive value was 58%, and negative predictive value was 89%. Concordance of the 2 methods was 80%. The false-positive group was followed up 16 months later to check for diagnosis of disease. Two of the 24 patients in this quadrant were later diagnosed with MM (Table 1). 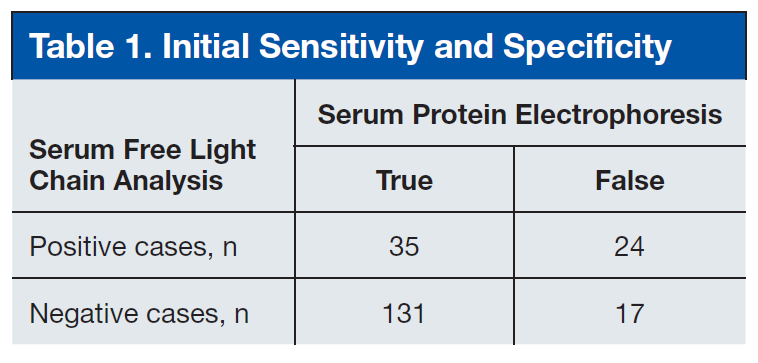
One of the patients with MM was an 82-year-old African American man with a history of hypertension, diabetes, and prostate cancer (Gleason 4 + 4 = 8/10). He presented to VANYHHS after a fall in which he sustained a pathologic fracture of the left acromion. Recurrent prostate cancer was initially suspected, and nuclear bone scintigraphy revealed increased uptake in the left shoulder and the posterior ninth rib. Results of computed tomography-guided biopsy showed the rib lesion packed with plasma cells and consistent with MM. Immunohistochemical analysis was positive for CD138 and κ in the malignant plasma cells. Initial SPEP performed before the biopsy showed an acute phase reaction with hypogammaglobulinemia, and SPEP after the biopsy showed an increased α-2 band but no monoclonal gammaglobulinopathy. The initial κ of 42.18 mg/L (κ-to-λ ratio, 4.01) was up to 67.53 mg/L 4 months later.
The other patient with MM was a 91-year-old man who had coronary artery disease after undergoing coronary artery bypass grafting in 1993, sick sinus syndrome after pacemaker implantation, hypertension, and anemia. He initially presented to the geriatrics clinic with polyneuropathy, which prompted SPEP and SFLC analysis. SPEP results showed a normal electrophoretic pattern, but κ increased to 47.52 mg/L (κ-to-λ ratio, 2.63). The decision was made to monitor the patient in the hematology clinic. Subsequent κ chain analysis revealed an increase to 59.50 mg/L. A repeat SPEP, performed 1 year after the first SPEP, revealed monoclonal immunoglobulin A on IFE.
Of the 24 patients with false-positive results, 16 had moderate-to-severe kidney disease (stage IIIa-IV).6All patients in this quadrant were men; their mean age was 75 years, and their mean creatinine level was 182.15 μmol/L. Further laboratory data are listed in Table 2. 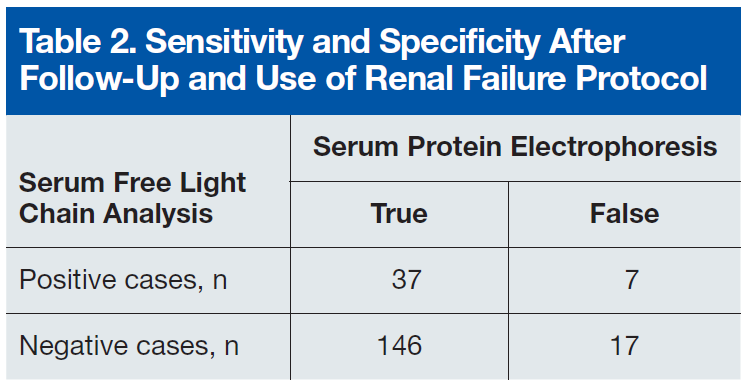
The patient whose biopsy results led to an MM diagnosis and the patient whose IFE led to a gammopathy diagnosis both maintained a glomerular filtration rate within normal limits. The Figure shows the κ-to-λ ratios of this quadrant logarithmically. 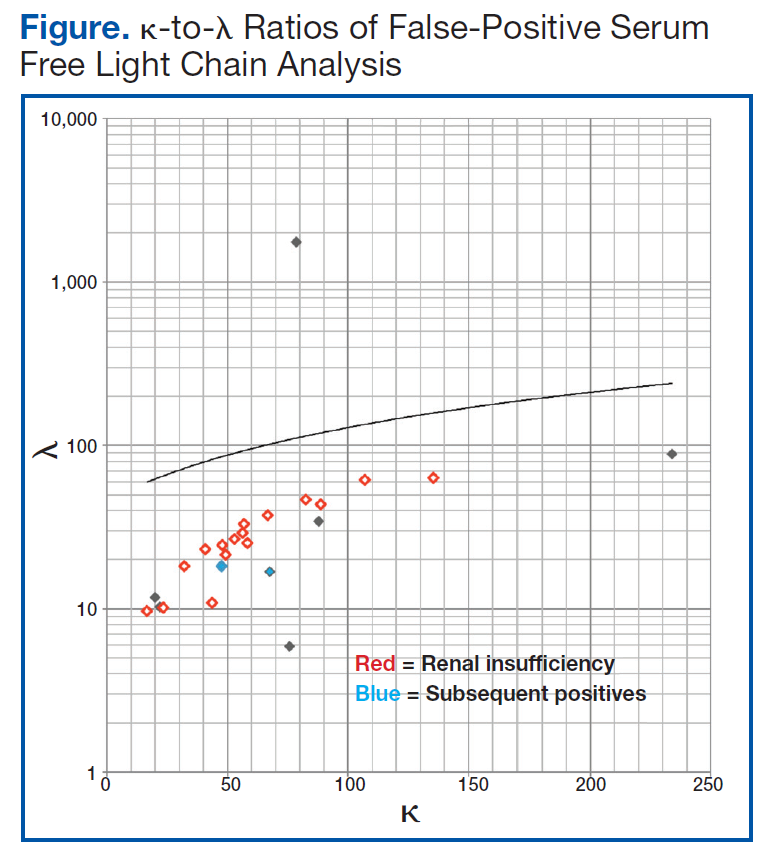
Discussion
Use of SFLC analysis as a supplement to serum and urine protein electrophoresis has been investigated and accepted in the recent literature.3,4,7,8 Use of light chains as a method of earlier or alternative detection has not been proved. In the present study of 207 patients, comparisons showed that more traditional MM detection methods and SFLC analysis are largely concordant. The 2 patients with MM and negative electrophoretic patterns provided a clear indication of the potential benefit of SFLC analysis in the diagnosis of secretory and nonsecretory myeloma.
In 2014, Kim and colleagues compared 2 SFLC assays (Freelite, N Latex) to each other and to SPEP in a 120-patient population.9 The Freelite results in their study correlated closely with VA population findings (κ-to-λ ratio sensitivity and specificity: 72.2% and 93.6%, respectively). N Latex, the newer SFLC assay, had lower sensitivity (64.6%) and higher specificity (100%). With application of the extended reference range (0.37-3.1) proposed by Hutchison and colleagues for use in patients with renal failure, SFLC becomes a more statistically powerful tool.5
The patients who tested false-positive had higher mean creatinine levels, and 16 had renal insufficiency. The 2 false-positive patients were later found to have clinical myeloma and were within the normal range of renal function. Of the 16 patients with an abnormal κ-to-λ ratio and renal failure, 15 would be within the revised normal reference range, leaving 9 false-positives, 2 of whom eventually were found to have disease. With the application of the extended light chain range (as per Hutchison) for those patients with renal failure, 15 of the original 24 false-positives became true-negatives. Two of the false-positives become true-positives after they were subsequently diagnosed. Therefore, SFLC analysis detected disease in 22% of the revised false-positives when SPEP could not.
Table 2 lists the revised data after follow-up and renal failure correction. The strongest aspect of SFLC analysis remains its 95% specificity; its 69% sensitivity remains relatively constant. The test’s positive predictive value is 84%, and its negative predictive value is 90%. In veteran and other at-risk populations, SFLC analysis proves to be a very powerful tool on its own.
Conclusion
Both patient cases described in this article demonstrate the usefulness of SFLC analysis as an adjunct to SPEP. The authors propose SFLC testing for all patients who are undergoing MM screening with SPEP/IFE. In patients with renal failure, the expanded reference range seems to reduce erroneous false-positive results. Patients who have abnormal ratios should be followed up in clinic with repeat MM testing. It seems clear that, at the very least, SFLC analysis is a necessary adjunct to SPEP testing. However, SFLC stands on its own merit as well.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Click here to read the digital edition.
1. National Cancer Institute, Surveillance, Epidemiology, and End Results (SEER) Program. SEER website. http://seer.cancer.gov/statfacts/html/mulmy.html. Accessed July 11, 2016.
2. Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78(1):21-33.
3. Dimopoulos M, Kyle R, Fermand JP, et al; International Myeloma Workshop Consensus Panel 3. Consensus recommendations for standard investigative workup: report of the International Myeloma Workshop Consensus Panel 3. Blood. 2011;117(18):4701-4705.
4. Katzmann JA, Clark RJ, Abraham RS, et al. Serum reference intervals and diagnostic ranges for free kappa and free lambda immunoglobulin light chains: relative sensitivity for detection of monoclonal light chains. Clin Chem. 2002;48(9):1437-1444.
5. Hutchison CA, Plant T, Drayson M, et al. Serum free light chain measurement
aids the diagnosis of myeloma in patients with severe renal failure.
BMC Nephrol. 2008;9:11.
6. Levey AS, Stevens LA, Schmid CH, et al; CKD-EPI (Chronic Kidney Disease
Epidemiology Collaboration). A new equation to estimate glomerular filtration
rate. Ann Intern Med. 2009;150(9):604-612.
7. McTaggart MP, Lindsay J, Kearney EM. Replacing urine protein electrophoresis
with serum free light chain analysis as a first-line test for detecting plasma
cell disorders offers increased diagnostic accuracy and potential health benefit
to patients. Am J Clin Pathol. 2013;140(6):890-897.
8. Abadie JM, Bankson DD. Assessment of serum free light chain assays for
plasma cell disorder screening in a Veterans Affairs population. Ann Clin Lab
Sci. 2006;36(2):157-162.
9. Kim HS, Kim HS, Shin KS, et al. Clinical comparisons of two free light chain
assays to immunofixation electrophoresis for detecting monoclonal gammopathy.
Biomed Res Int. 2014;2014:647238.
Note: Page numbers differ between the print issue and digital edition.
Patients who are undergoing multiple myeloma screening with serum protein electrophoresis and immunofixation, especially those with renal failure, also should receive serum free light chain testing to increase specificity and reduce false-negatives.
Multiple myeloma (MM) is a devastating disease with an estimated 26,850 new cases in 2015 according to Surveillance, Epidemiology, and End Results data and no definitive chemotherapeutic cure.1 In 97% of cases, MM is defined by monoclonal hypergammaglobulinemia, in which a malignant plasma cell clone secretes a monoclonal globulin; the remaining cases are nonsecretors.2 Each pathologically produced clonal globulin contains 2 heavy chains attached by disulfide linkage and 2 light chains. Unchecked plasma cell production is what later causes the symptoms of renal failure, bone destruction, and anemia.
The rate of MM is disproportionately high in the veteran population, and the VA health care system provides care for many of these patients. The higher rate is likely secondary to the predominantly male population, which has higher MM rates, and has been linked to Agent Orange exposure in Vietnam. As MM is not easy to diagnose, any algorithm or testing method would be of great benefit to this population.
The gold standard for MM detection remains serum protein electrophoresis (SPEP) with immunofixation (IFE), but other detection methods have been emerging. The method of serum free light chain (SFLC) assay has become more readily available, and its incorporation into diagnostic guidelines has become more apparent but is not universal.3
In the case series reported in this article, SPEP/IFE and SFLC assays were used to test 207 patients from the VA New York Harbor Healthcare System (VANYHHS). All these patients had a clinical context for MM testing.
Methods
In this retrospective study, the authors reviewed the charts of VANYHHS patients who were being treated for conditions that prompted SPEP/IFE and λ and κ SFLC analysis between December 2013 and March 2014. The study was exempt from institutional review board approval.
The SPEP/IFE analysis was performed with an automated electrophoresis machine (Sebia Electrophoresis), and the SFLC analysis was performed with an automated SFLC assay (Freelite). Sensitivity, specificity, and positive and negative predictive values were calculated using SPEP/IFE as the gold standard and SFLC κ-to-λ ratio asthe test method. Patients with a positive κ-to-λ ratio but negative SPEP were considered false-positives. These patients’ SFLC analyses were further analyzed in an effort to evaluate use of the κ-to-λ ratio as an early tumor marker.
The κ reference range used was 3.3 to 19.4 mg/L, and the λ reference range used was 5.7 to 26.3 mg/L.4 The traditional reference range for the κ-to-λ ratio is 0.26 to 1.65.5
Results
Of the 207 patients in this study, 205 were men. Mean age was 69 years (range, 28-97 years). Mean serum urea nitrogen level was 8.75 mmol/L (range, 2.86-38.21 mmol/L), and mean creatinine level was 140.59 μmol/L (range, 44.21-1503.14 μmol/L). Mean κ was 49.82 mg/L (range, 4.6-700.96 mg/L), and mean λ was 54.27 mg/L (range, 3-1,750 mg/L). Table 1 compares the SPEP and SFLC data. Sensitivity was 67%, specificity was 85%, positive predictive value was 58%, and negative predictive value was 89%. Concordance of the 2 methods was 80%. The false-positive group was followed up 16 months later to check for diagnosis of disease. Two of the 24 patients in this quadrant were later diagnosed with MM (Table 1).
One of the patients with MM was an 82-year-old African American man with a history of hypertension, diabetes, and prostate cancer (Gleason 4 + 4 = 8/10). He presented to VANYHHS after a fall in which he sustained a pathologic fracture of the left acromion. Recurrent prostate cancer was initially suspected, and nuclear bone scintigraphy revealed increased uptake in the left shoulder and the posterior ninth rib. Results of computed tomography-guided biopsy showed the rib lesion packed with plasma cells and consistent with MM. Immunohistochemical analysis was positive for CD138 and κ in the malignant plasma cells. Initial SPEP performed before the biopsy showed an acute phase reaction with hypogammaglobulinemia, and SPEP after the biopsy showed an increased α-2 band but no monoclonal gammaglobulinopathy. The initial κ of 42.18 mg/L (κ-to-λ ratio, 4.01) was up to 67.53 mg/L 4 months later.
The other patient with MM was a 91-year-old man who had coronary artery disease after undergoing coronary artery bypass grafting in 1993, sick sinus syndrome after pacemaker implantation, hypertension, and anemia. He initially presented to the geriatrics clinic with polyneuropathy, which prompted SPEP and SFLC analysis. SPEP results showed a normal electrophoretic pattern, but κ increased to 47.52 mg/L (κ-to-λ ratio, 2.63). The decision was made to monitor the patient in the hematology clinic. Subsequent κ chain analysis revealed an increase to 59.50 mg/L. A repeat SPEP, performed 1 year after the first SPEP, revealed monoclonal immunoglobulin A on IFE.
Of the 24 patients with false-positive results, 16 had moderate-to-severe kidney disease (stage IIIa-IV).6All patients in this quadrant were men; their mean age was 75 years, and their mean creatinine level was 182.15 μmol/L. Further laboratory data are listed in Table 2.
The patient whose biopsy results led to an MM diagnosis and the patient whose IFE led to a gammopathy diagnosis both maintained a glomerular filtration rate within normal limits. The Figure shows the κ-to-λ ratios of this quadrant logarithmically.
Discussion
Use of SFLC analysis as a supplement to serum and urine protein electrophoresis has been investigated and accepted in the recent literature.3,4,7,8 Use of light chains as a method of earlier or alternative detection has not been proved. In the present study of 207 patients, comparisons showed that more traditional MM detection methods and SFLC analysis are largely concordant. The 2 patients with MM and negative electrophoretic patterns provided a clear indication of the potential benefit of SFLC analysis in the diagnosis of secretory and nonsecretory myeloma.
In 2014, Kim and colleagues compared 2 SFLC assays (Freelite, N Latex) to each other and to SPEP in a 120-patient population.9 The Freelite results in their study correlated closely with VA population findings (κ-to-λ ratio sensitivity and specificity: 72.2% and 93.6%, respectively). N Latex, the newer SFLC assay, had lower sensitivity (64.6%) and higher specificity (100%). With application of the extended reference range (0.37-3.1) proposed by Hutchison and colleagues for use in patients with renal failure, SFLC becomes a more statistically powerful tool.5
The patients who tested false-positive had higher mean creatinine levels, and 16 had renal insufficiency. The 2 false-positive patients were later found to have clinical myeloma and were within the normal range of renal function. Of the 16 patients with an abnormal κ-to-λ ratio and renal failure, 15 would be within the revised normal reference range, leaving 9 false-positives, 2 of whom eventually were found to have disease. With the application of the extended light chain range (as per Hutchison) for those patients with renal failure, 15 of the original 24 false-positives became true-negatives. Two of the false-positives become true-positives after they were subsequently diagnosed. Therefore, SFLC analysis detected disease in 22% of the revised false-positives when SPEP could not.
Table 2 lists the revised data after follow-up and renal failure correction. The strongest aspect of SFLC analysis remains its 95% specificity; its 69% sensitivity remains relatively constant. The test’s positive predictive value is 84%, and its negative predictive value is 90%. In veteran and other at-risk populations, SFLC analysis proves to be a very powerful tool on its own.
Conclusion
Both patient cases described in this article demonstrate the usefulness of SFLC analysis as an adjunct to SPEP. The authors propose SFLC testing for all patients who are undergoing MM screening with SPEP/IFE. In patients with renal failure, the expanded reference range seems to reduce erroneous false-positive results. Patients who have abnormal ratios should be followed up in clinic with repeat MM testing. It seems clear that, at the very least, SFLC analysis is a necessary adjunct to SPEP testing. However, SFLC stands on its own merit as well.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Click here to read the digital edition.
Patients who are undergoing multiple myeloma screening with serum protein electrophoresis and immunofixation, especially those with renal failure, also should receive serum free light chain testing to increase specificity and reduce false-negatives.
Multiple myeloma (MM) is a devastating disease with an estimated 26,850 new cases in 2015 according to Surveillance, Epidemiology, and End Results data and no definitive chemotherapeutic cure.1 In 97% of cases, MM is defined by monoclonal hypergammaglobulinemia, in which a malignant plasma cell clone secretes a monoclonal globulin; the remaining cases are nonsecretors.2 Each pathologically produced clonal globulin contains 2 heavy chains attached by disulfide linkage and 2 light chains. Unchecked plasma cell production is what later causes the symptoms of renal failure, bone destruction, and anemia.
The rate of MM is disproportionately high in the veteran population, and the VA health care system provides care for many of these patients. The higher rate is likely secondary to the predominantly male population, which has higher MM rates, and has been linked to Agent Orange exposure in Vietnam. As MM is not easy to diagnose, any algorithm or testing method would be of great benefit to this population.
The gold standard for MM detection remains serum protein electrophoresis (SPEP) with immunofixation (IFE), but other detection methods have been emerging. The method of serum free light chain (SFLC) assay has become more readily available, and its incorporation into diagnostic guidelines has become more apparent but is not universal.3
In the case series reported in this article, SPEP/IFE and SFLC assays were used to test 207 patients from the VA New York Harbor Healthcare System (VANYHHS). All these patients had a clinical context for MM testing.
Methods
In this retrospective study, the authors reviewed the charts of VANYHHS patients who were being treated for conditions that prompted SPEP/IFE and λ and κ SFLC analysis between December 2013 and March 2014. The study was exempt from institutional review board approval.
The SPEP/IFE analysis was performed with an automated electrophoresis machine (Sebia Electrophoresis), and the SFLC analysis was performed with an automated SFLC assay (Freelite). Sensitivity, specificity, and positive and negative predictive values were calculated using SPEP/IFE as the gold standard and SFLC κ-to-λ ratio asthe test method. Patients with a positive κ-to-λ ratio but negative SPEP were considered false-positives. These patients’ SFLC analyses were further analyzed in an effort to evaluate use of the κ-to-λ ratio as an early tumor marker.
The κ reference range used was 3.3 to 19.4 mg/L, and the λ reference range used was 5.7 to 26.3 mg/L.4 The traditional reference range for the κ-to-λ ratio is 0.26 to 1.65.5
Results
Of the 207 patients in this study, 205 were men. Mean age was 69 years (range, 28-97 years). Mean serum urea nitrogen level was 8.75 mmol/L (range, 2.86-38.21 mmol/L), and mean creatinine level was 140.59 μmol/L (range, 44.21-1503.14 μmol/L). Mean κ was 49.82 mg/L (range, 4.6-700.96 mg/L), and mean λ was 54.27 mg/L (range, 3-1,750 mg/L). Table 1 compares the SPEP and SFLC data. Sensitivity was 67%, specificity was 85%, positive predictive value was 58%, and negative predictive value was 89%. Concordance of the 2 methods was 80%. The false-positive group was followed up 16 months later to check for diagnosis of disease. Two of the 24 patients in this quadrant were later diagnosed with MM (Table 1).
One of the patients with MM was an 82-year-old African American man with a history of hypertension, diabetes, and prostate cancer (Gleason 4 + 4 = 8/10). He presented to VANYHHS after a fall in which he sustained a pathologic fracture of the left acromion. Recurrent prostate cancer was initially suspected, and nuclear bone scintigraphy revealed increased uptake in the left shoulder and the posterior ninth rib. Results of computed tomography-guided biopsy showed the rib lesion packed with plasma cells and consistent with MM. Immunohistochemical analysis was positive for CD138 and κ in the malignant plasma cells. Initial SPEP performed before the biopsy showed an acute phase reaction with hypogammaglobulinemia, and SPEP after the biopsy showed an increased α-2 band but no monoclonal gammaglobulinopathy. The initial κ of 42.18 mg/L (κ-to-λ ratio, 4.01) was up to 67.53 mg/L 4 months later.
The other patient with MM was a 91-year-old man who had coronary artery disease after undergoing coronary artery bypass grafting in 1993, sick sinus syndrome after pacemaker implantation, hypertension, and anemia. He initially presented to the geriatrics clinic with polyneuropathy, which prompted SPEP and SFLC analysis. SPEP results showed a normal electrophoretic pattern, but κ increased to 47.52 mg/L (κ-to-λ ratio, 2.63). The decision was made to monitor the patient in the hematology clinic. Subsequent κ chain analysis revealed an increase to 59.50 mg/L. A repeat SPEP, performed 1 year after the first SPEP, revealed monoclonal immunoglobulin A on IFE.
Of the 24 patients with false-positive results, 16 had moderate-to-severe kidney disease (stage IIIa-IV).6All patients in this quadrant were men; their mean age was 75 years, and their mean creatinine level was 182.15 μmol/L. Further laboratory data are listed in Table 2.
The patient whose biopsy results led to an MM diagnosis and the patient whose IFE led to a gammopathy diagnosis both maintained a glomerular filtration rate within normal limits. The Figure shows the κ-to-λ ratios of this quadrant logarithmically.
Discussion
Use of SFLC analysis as a supplement to serum and urine protein electrophoresis has been investigated and accepted in the recent literature.3,4,7,8 Use of light chains as a method of earlier or alternative detection has not been proved. In the present study of 207 patients, comparisons showed that more traditional MM detection methods and SFLC analysis are largely concordant. The 2 patients with MM and negative electrophoretic patterns provided a clear indication of the potential benefit of SFLC analysis in the diagnosis of secretory and nonsecretory myeloma.
In 2014, Kim and colleagues compared 2 SFLC assays (Freelite, N Latex) to each other and to SPEP in a 120-patient population.9 The Freelite results in their study correlated closely with VA population findings (κ-to-λ ratio sensitivity and specificity: 72.2% and 93.6%, respectively). N Latex, the newer SFLC assay, had lower sensitivity (64.6%) and higher specificity (100%). With application of the extended reference range (0.37-3.1) proposed by Hutchison and colleagues for use in patients with renal failure, SFLC becomes a more statistically powerful tool.5
The patients who tested false-positive had higher mean creatinine levels, and 16 had renal insufficiency. The 2 false-positive patients were later found to have clinical myeloma and were within the normal range of renal function. Of the 16 patients with an abnormal κ-to-λ ratio and renal failure, 15 would be within the revised normal reference range, leaving 9 false-positives, 2 of whom eventually were found to have disease. With the application of the extended light chain range (as per Hutchison) for those patients with renal failure, 15 of the original 24 false-positives became true-negatives. Two of the false-positives become true-positives after they were subsequently diagnosed. Therefore, SFLC analysis detected disease in 22% of the revised false-positives when SPEP could not.
Table 2 lists the revised data after follow-up and renal failure correction. The strongest aspect of SFLC analysis remains its 95% specificity; its 69% sensitivity remains relatively constant. The test’s positive predictive value is 84%, and its negative predictive value is 90%. In veteran and other at-risk populations, SFLC analysis proves to be a very powerful tool on its own.
Conclusion
Both patient cases described in this article demonstrate the usefulness of SFLC analysis as an adjunct to SPEP. The authors propose SFLC testing for all patients who are undergoing MM screening with SPEP/IFE. In patients with renal failure, the expanded reference range seems to reduce erroneous false-positive results. Patients who have abnormal ratios should be followed up in clinic with repeat MM testing. It seems clear that, at the very least, SFLC analysis is a necessary adjunct to SPEP testing. However, SFLC stands on its own merit as well.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Click here to read the digital edition.
1. National Cancer Institute, Surveillance, Epidemiology, and End Results (SEER) Program. SEER website. http://seer.cancer.gov/statfacts/html/mulmy.html. Accessed July 11, 2016.
2. Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78(1):21-33.
3. Dimopoulos M, Kyle R, Fermand JP, et al; International Myeloma Workshop Consensus Panel 3. Consensus recommendations for standard investigative workup: report of the International Myeloma Workshop Consensus Panel 3. Blood. 2011;117(18):4701-4705.
4. Katzmann JA, Clark RJ, Abraham RS, et al. Serum reference intervals and diagnostic ranges for free kappa and free lambda immunoglobulin light chains: relative sensitivity for detection of monoclonal light chains. Clin Chem. 2002;48(9):1437-1444.
5. Hutchison CA, Plant T, Drayson M, et al. Serum free light chain measurement
aids the diagnosis of myeloma in patients with severe renal failure.
BMC Nephrol. 2008;9:11.
6. Levey AS, Stevens LA, Schmid CH, et al; CKD-EPI (Chronic Kidney Disease
Epidemiology Collaboration). A new equation to estimate glomerular filtration
rate. Ann Intern Med. 2009;150(9):604-612.
7. McTaggart MP, Lindsay J, Kearney EM. Replacing urine protein electrophoresis
with serum free light chain analysis as a first-line test for detecting plasma
cell disorders offers increased diagnostic accuracy and potential health benefit
to patients. Am J Clin Pathol. 2013;140(6):890-897.
8. Abadie JM, Bankson DD. Assessment of serum free light chain assays for
plasma cell disorder screening in a Veterans Affairs population. Ann Clin Lab
Sci. 2006;36(2):157-162.
9. Kim HS, Kim HS, Shin KS, et al. Clinical comparisons of two free light chain
assays to immunofixation electrophoresis for detecting monoclonal gammopathy.
Biomed Res Int. 2014;2014:647238.
Note: Page numbers differ between the print issue and digital edition.
1. National Cancer Institute, Surveillance, Epidemiology, and End Results (SEER) Program. SEER website. http://seer.cancer.gov/statfacts/html/mulmy.html. Accessed July 11, 2016.
2. Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78(1):21-33.
3. Dimopoulos M, Kyle R, Fermand JP, et al; International Myeloma Workshop Consensus Panel 3. Consensus recommendations for standard investigative workup: report of the International Myeloma Workshop Consensus Panel 3. Blood. 2011;117(18):4701-4705.
4. Katzmann JA, Clark RJ, Abraham RS, et al. Serum reference intervals and diagnostic ranges for free kappa and free lambda immunoglobulin light chains: relative sensitivity for detection of monoclonal light chains. Clin Chem. 2002;48(9):1437-1444.
5. Hutchison CA, Plant T, Drayson M, et al. Serum free light chain measurement
aids the diagnosis of myeloma in patients with severe renal failure.
BMC Nephrol. 2008;9:11.
6. Levey AS, Stevens LA, Schmid CH, et al; CKD-EPI (Chronic Kidney Disease
Epidemiology Collaboration). A new equation to estimate glomerular filtration
rate. Ann Intern Med. 2009;150(9):604-612.
7. McTaggart MP, Lindsay J, Kearney EM. Replacing urine protein electrophoresis
with serum free light chain analysis as a first-line test for detecting plasma
cell disorders offers increased diagnostic accuracy and potential health benefit
to patients. Am J Clin Pathol. 2013;140(6):890-897.
8. Abadie JM, Bankson DD. Assessment of serum free light chain assays for
plasma cell disorder screening in a Veterans Affairs population. Ann Clin Lab
Sci. 2006;36(2):157-162.
9. Kim HS, Kim HS, Shin KS, et al. Clinical comparisons of two free light chain
assays to immunofixation electrophoresis for detecting monoclonal gammopathy.
Biomed Res Int. 2014;2014:647238.
Note: Page numbers differ between the print issue and digital edition.
1,25-dihydroxyvitamin D hypercalcemia and imatinib hepatotoxicity in a patient with GIST
Click on the PDF icon at the top of this introduction to read the full article.
Click on the PDF icon at the top of this introduction to read the full article.
Click on the PDF icon at the top of this introduction to read the full article.
High-grade leiomyosarcoma of the transverse colon presenting with bowel perforation
Stromal or mesenchymal tumors account for about 1% of gastrointestinal (GI) tract neoplasms and are divided into 2 main categories: the gastrointestinal stromal tumors (GISTs; 60%- 90% of mesencymal tumors), and the non-GIST neoplasms (10%-30% of mesencymal tumors).1 The non-GIST neoplasms consist of a heterogenous group of soft-tissue tumors, identical to soft-tissue tumors elsewhere in the body.
Click on the PDF icon at the top of this introduction to read the full article.
Stromal or mesenchymal tumors account for about 1% of gastrointestinal (GI) tract neoplasms and are divided into 2 main categories: the gastrointestinal stromal tumors (GISTs; 60%- 90% of mesencymal tumors), and the non-GIST neoplasms (10%-30% of mesencymal tumors).1 The non-GIST neoplasms consist of a heterogenous group of soft-tissue tumors, identical to soft-tissue tumors elsewhere in the body.
Click on the PDF icon at the top of this introduction to read the full article.
Stromal or mesenchymal tumors account for about 1% of gastrointestinal (GI) tract neoplasms and are divided into 2 main categories: the gastrointestinal stromal tumors (GISTs; 60%- 90% of mesencymal tumors), and the non-GIST neoplasms (10%-30% of mesencymal tumors).1 The non-GIST neoplasms consist of a heterogenous group of soft-tissue tumors, identical to soft-tissue tumors elsewhere in the body.
Click on the PDF icon at the top of this introduction to read the full article.