User login
From neuroplasticity to psychoplasticity: Psilocybin may reverse personality disorders and political fanaticism
One of psychiatry’s long-standing dogmas is that personality disorders are enduring, unchangeable, and not amenable to treatment with potent psychotropics or intensive psychotherapy. I propose that this dogma may soon be shattered.
Several other dogmas in psychiatry have been demolished over the past several decades:
- that “insanity” is completely irreversible and requires lifetime institutionalization. The serendipitous discovery of chlorpromazine1 annihilated this centuries-old dogma
- that chronic, severe, refractory depression (with ongoing suicidal urges) that fails to improve with pharmacotherapy or electroconvulsive therapy (ECT) is hopeless and untreatable, until ketamine not only pulverized this dogma, but did it with lightning speed, dazzling us all2
- that dissociative agents such as ketamine are dangerous and condemnable drugs of abuse, until the therapeutic effect of ketamine slayed that dragon3
- that ECT “fries” the brain (as malevolently propagated by antipsychiatry cults), which was completely disproven by neuroimaging studies that show the hippocampus (which shrinks during depression) actually grows by >10% after a few ECT sessions4
- that psychotherapy is not a “real” treatment because talking cannot reverse a psychiatric brain disorder, until studies showed significant neuroplasticity with psychotherapy and decrease in inflammatory biomarkers with cognitive-behavioral therapy (CBT)5
- that persons with refractory hallucinations and delusions are doomed to a life of disability, until clozapine torpedoed that pessimistic dogma6
- that hallucinogens/psychedelics are dangerous and should be banned, until a jarring paradigm shift occurred with the discovery of psilocybin’s transformative effects, and the remarkable therapeutic effects of its mystical trips.7
Psilocybin’s therapeutic effects
Psilocybin has already proved to have a strong and lasting effect on depression and promises to have therapeutic benefits for patients with substance use disorders, posttraumatic stress disorder (PTSD), and anxiety.8 In addition, when the multiple psychological and neurobiological effects of psilocybin (and of other psychedelics) are examined, I see a very promising path to amelioration of severe personality disorders such as psychopathy, antisocial behavior, and narcissism. The mechanism(s) of action of psilocybin on the human brain are drastically different from any man-made psychotropic agent. As a psychiatric neuroscientist, I envision the neurologic impact of psilocybin to be conducive to a complete transformation of a patient’s view of themself, other people, and the meaning of life. It is reminiscent of religious conversion.
The psychological effects of psilocybin in humans have been described as follows:
- emotional breakthrough9
- increased psychological flexibility,10,11 a very cortical effect
- mystical experience,12 which results in sudden and significant changes in behavior and perception and includes the following dimensions: sacredness, noetic quality, deeply felt positive mood, ineffability, paradoxicality, and transcendence of time and space13
- oceanic boundlessness, feeling “one with the universe”14
- universal interconnectedness, insightfulness, blissful state, spiritual experience14
- ego dissolution,15 with loss of one’s personal identity
- increased neuroplasticity16
- changes in cognition and increase in insight.17
The neurobiological effects of psilocybin are mediated by serotonin 5HT2A agonism and include the following18:
- reduction in the activity of the medial prefrontal cortex, which regulates memory, attention, inhibitory control, and habit
- a decrease in the connectivity between the medial prefrontal cortex and the posterior cingulate cortex, which regulates memory and emotions
- reducing the default mode network, which is active during rest, stimulating internal thoughts and reminiscing about previous feelings and events, sometimes including ruminations. Psilocybin reverses those processes to thinking about others, not just the self, and becoming more open-minded about the world and other people. This can be therapeutic for depression, which is often associated with negative ruminations but also with entrenched habits (addictive behaviors), anxiety, PTSD, and obsessive-compulsive disorders
- increased global functional connectivity among various brain networks, leading to stronger functional integration of behavior
- collapse of major cortical oscillatory rhythms such as alpha and others that perpetuate “prior” beliefs
- extensive neuroplasticity and recalibration of thought processes and decomposition of pathological beliefs, referred to as REBUS (relaxed beliefs under psychedelics).
The bottom line is psilocybin and other psychedelics can dramatically alter, reshape, and relax rigid beliefs and personality traits by decreasing “neuroticism” and increasing “extraversion,” insightfulness, openness, and possibly conscientiousness.19 Although no studies of psychedelics in psychopathic, antisocial, or narcissistic personality disorders have been conducted, it is very reasonable to speculate that psilocybin may reverse traits of these disorders such as callousness, lack of empathy, and pathological self-centeredness.
Going further, a preliminary report suggests psilocybin can modify political views by decreasing authoritarianism and increasing libertarianism.20,21 In the current political zeitgeist, could psychedelics such as psilocybin reduce or even eliminate political extremism and visceral hatred on all sides? It would be remarkable research to carry out to heal a politically divided populace.The dogma of untreatable personality disorders or hopelessly entrenched political extremism is on the chopping block, and psychedelics offer hope to splinter those beliefs by concurrently remodeling brain tissue (neuroplasticity) and rectifying the mindset (psychoplasticity).
1. Delay J, Deniker P. Neuroleptic effects of chlorpromazine in therapeutics of neuropsychiatry. J Clin Exp Psychopathol. 1955;16(2):104-112.
2. Walsh Z, Mollaahmetoglu OM, Rootman, J, et al. Ketamine for the treatment of mental health and substance use disorders: comprehensive systematic review. BJPsych Open. 2021;8(1):e19. doi:10.1192/bjo.2021.1061
3. Lener MS, Kadriu B, Zarate CA Jr. Ketamine and beyond: investigations into the potential of glutamatergic agents to treat depression. Drugs. 2017;77(4):381-401.
4. Ayers B, Leaver A, Woods RP, et al. Structural plasticity of the hippocampus and amygdala induced by electroconvulsive therapy in major depression. Biol Psychiatry. 2016;79(4):282-292.
5. Cao B, Li R, Ding L, Xu J, et al. Does cognitive behaviour therapy affect peripheral inflammation of depression? A protocol for the systematic review and meta-analysis. BMJ Open. 2021;11(12):e048162. doi:10.1136/bmjopen-2020-048162
6. Wagner E, Siafis S, Fernando P, et al. Efficacy and safety of clozapine in psychotic disorders—a systematic quantitative meta-review. Transl Psychiatry. 2021;11(1):487.
7. Daws RE, Timmermann C, Giribaldi B, et al. Increased global integration in the brain after psilocybin therapy for depression. Nat Med. 2022;28(4):844-851.
8. Pearson C, Siegel J, Gold JA. Psilocybin-assisted psychotherapy for depression: emerging research on a psychedelic compound with a rich history. J Neurol Sci. 2022;434:120096. doi:10.1016/j.jns.2021.120096
9. Roseman L, Haijen E, Idialu-Ikato K, et al. Emotional breakthrough and psychedelics: validation of the Emotional Breakthrough Inventory. J Psychopharmacol. 2019;33(9):1076-1087.
10. Davis AK, Barrett FS, Griffiths RR. Psychological flexibility mediates the relations between acute psychedelic effects and subjective decreases in depression and anxiety. J Contextual Behav Sci. 2020;15:39-45.
11. Hayes SC, Luoma JB, Bond FW, et al. Acceptance and commitment therapy: model, processes and outcomes. Behav Res Ther. 2006;44(1):1-25.
12. Ross S, Bossis A, Guss J, et al. Rapid and sustained symptom reduction following psilocybin treatment for anxiety and depression in patients with life-threatening cancer: a randomized controlled trial. J Psychopharmacol. 2016;30(12):1165-1180.
13. Stace WT. Mysticism and Philosophy. Macmillan Pub Ltd; 1960:37.
14. Barrett FS, Griffiths RR. Classic hallucinogens and mystical experiences: phenomenology and neural correlates. Curr Top Behav Neurosci. 2018;36:393-430.
15. Nour MM, Evans L, Nutt D, et al. Ego-dissolution and psychedelics: validation of the Ego-Dissolution Inventory (EDI). Front Hum Neurosci. 2016;10:269. doi:10.3389/fnhum.2016.00269
16. Olson DE. The subjective effects of psychedelics may not be necessary for their enduring therapeutic effects. ACS Pharmacol Transl Sci. 2020;4(2):563-567.
17. Carhart-Harris RL, Bolstridge M, Day CMJ, et al. Psilocybin with psychological support for treatment-resistant depression: six-month follow-up. Psychopharmacology (Berl). 2018;235(2):399-408.
18. Carhart-Harris RL. How do psychedelics work? Curr Opin Psychiatry. 2019;32(1):16-21.
19. Erritzoe D, Roseman L, Nour MM, et al. Effects of psilocybin therapy on personality structure. Acta Psychiatr Scand. 2018;138(5):368-378.
20. Lyons T, Carhart-Harris RL. Increased nature relatedness and decreased authoritarian political views after psilocybin for treatment-resistant depression. J Psychopharmacol. 2018;32(7):811-819.
21. Nour MM, Evans L, Carhart-Harris RL. Psychedelics, personality and political perspectives. J Psychoactive Drugs. 2017;49(3):182-191.
One of psychiatry’s long-standing dogmas is that personality disorders are enduring, unchangeable, and not amenable to treatment with potent psychotropics or intensive psychotherapy. I propose that this dogma may soon be shattered.
Several other dogmas in psychiatry have been demolished over the past several decades:
- that “insanity” is completely irreversible and requires lifetime institutionalization. The serendipitous discovery of chlorpromazine1 annihilated this centuries-old dogma
- that chronic, severe, refractory depression (with ongoing suicidal urges) that fails to improve with pharmacotherapy or electroconvulsive therapy (ECT) is hopeless and untreatable, until ketamine not only pulverized this dogma, but did it with lightning speed, dazzling us all2
- that dissociative agents such as ketamine are dangerous and condemnable drugs of abuse, until the therapeutic effect of ketamine slayed that dragon3
- that ECT “fries” the brain (as malevolently propagated by antipsychiatry cults), which was completely disproven by neuroimaging studies that show the hippocampus (which shrinks during depression) actually grows by >10% after a few ECT sessions4
- that psychotherapy is not a “real” treatment because talking cannot reverse a psychiatric brain disorder, until studies showed significant neuroplasticity with psychotherapy and decrease in inflammatory biomarkers with cognitive-behavioral therapy (CBT)5
- that persons with refractory hallucinations and delusions are doomed to a life of disability, until clozapine torpedoed that pessimistic dogma6
- that hallucinogens/psychedelics are dangerous and should be banned, until a jarring paradigm shift occurred with the discovery of psilocybin’s transformative effects, and the remarkable therapeutic effects of its mystical trips.7
Psilocybin’s therapeutic effects
Psilocybin has already proved to have a strong and lasting effect on depression and promises to have therapeutic benefits for patients with substance use disorders, posttraumatic stress disorder (PTSD), and anxiety.8 In addition, when the multiple psychological and neurobiological effects of psilocybin (and of other psychedelics) are examined, I see a very promising path to amelioration of severe personality disorders such as psychopathy, antisocial behavior, and narcissism. The mechanism(s) of action of psilocybin on the human brain are drastically different from any man-made psychotropic agent. As a psychiatric neuroscientist, I envision the neurologic impact of psilocybin to be conducive to a complete transformation of a patient’s view of themself, other people, and the meaning of life. It is reminiscent of religious conversion.
The psychological effects of psilocybin in humans have been described as follows:
- emotional breakthrough9
- increased psychological flexibility,10,11 a very cortical effect
- mystical experience,12 which results in sudden and significant changes in behavior and perception and includes the following dimensions: sacredness, noetic quality, deeply felt positive mood, ineffability, paradoxicality, and transcendence of time and space13
- oceanic boundlessness, feeling “one with the universe”14
- universal interconnectedness, insightfulness, blissful state, spiritual experience14
- ego dissolution,15 with loss of one’s personal identity
- increased neuroplasticity16
- changes in cognition and increase in insight.17
The neurobiological effects of psilocybin are mediated by serotonin 5HT2A agonism and include the following18:
- reduction in the activity of the medial prefrontal cortex, which regulates memory, attention, inhibitory control, and habit
- a decrease in the connectivity between the medial prefrontal cortex and the posterior cingulate cortex, which regulates memory and emotions
- reducing the default mode network, which is active during rest, stimulating internal thoughts and reminiscing about previous feelings and events, sometimes including ruminations. Psilocybin reverses those processes to thinking about others, not just the self, and becoming more open-minded about the world and other people. This can be therapeutic for depression, which is often associated with negative ruminations but also with entrenched habits (addictive behaviors), anxiety, PTSD, and obsessive-compulsive disorders
- increased global functional connectivity among various brain networks, leading to stronger functional integration of behavior
- collapse of major cortical oscillatory rhythms such as alpha and others that perpetuate “prior” beliefs
- extensive neuroplasticity and recalibration of thought processes and decomposition of pathological beliefs, referred to as REBUS (relaxed beliefs under psychedelics).
The bottom line is psilocybin and other psychedelics can dramatically alter, reshape, and relax rigid beliefs and personality traits by decreasing “neuroticism” and increasing “extraversion,” insightfulness, openness, and possibly conscientiousness.19 Although no studies of psychedelics in psychopathic, antisocial, or narcissistic personality disorders have been conducted, it is very reasonable to speculate that psilocybin may reverse traits of these disorders such as callousness, lack of empathy, and pathological self-centeredness.
Going further, a preliminary report suggests psilocybin can modify political views by decreasing authoritarianism and increasing libertarianism.20,21 In the current political zeitgeist, could psychedelics such as psilocybin reduce or even eliminate political extremism and visceral hatred on all sides? It would be remarkable research to carry out to heal a politically divided populace.The dogma of untreatable personality disorders or hopelessly entrenched political extremism is on the chopping block, and psychedelics offer hope to splinter those beliefs by concurrently remodeling brain tissue (neuroplasticity) and rectifying the mindset (psychoplasticity).
One of psychiatry’s long-standing dogmas is that personality disorders are enduring, unchangeable, and not amenable to treatment with potent psychotropics or intensive psychotherapy. I propose that this dogma may soon be shattered.
Several other dogmas in psychiatry have been demolished over the past several decades:
- that “insanity” is completely irreversible and requires lifetime institutionalization. The serendipitous discovery of chlorpromazine1 annihilated this centuries-old dogma
- that chronic, severe, refractory depression (with ongoing suicidal urges) that fails to improve with pharmacotherapy or electroconvulsive therapy (ECT) is hopeless and untreatable, until ketamine not only pulverized this dogma, but did it with lightning speed, dazzling us all2
- that dissociative agents such as ketamine are dangerous and condemnable drugs of abuse, until the therapeutic effect of ketamine slayed that dragon3
- that ECT “fries” the brain (as malevolently propagated by antipsychiatry cults), which was completely disproven by neuroimaging studies that show the hippocampus (which shrinks during depression) actually grows by >10% after a few ECT sessions4
- that psychotherapy is not a “real” treatment because talking cannot reverse a psychiatric brain disorder, until studies showed significant neuroplasticity with psychotherapy and decrease in inflammatory biomarkers with cognitive-behavioral therapy (CBT)5
- that persons with refractory hallucinations and delusions are doomed to a life of disability, until clozapine torpedoed that pessimistic dogma6
- that hallucinogens/psychedelics are dangerous and should be banned, until a jarring paradigm shift occurred with the discovery of psilocybin’s transformative effects, and the remarkable therapeutic effects of its mystical trips.7
Psilocybin’s therapeutic effects
Psilocybin has already proved to have a strong and lasting effect on depression and promises to have therapeutic benefits for patients with substance use disorders, posttraumatic stress disorder (PTSD), and anxiety.8 In addition, when the multiple psychological and neurobiological effects of psilocybin (and of other psychedelics) are examined, I see a very promising path to amelioration of severe personality disorders such as psychopathy, antisocial behavior, and narcissism. The mechanism(s) of action of psilocybin on the human brain are drastically different from any man-made psychotropic agent. As a psychiatric neuroscientist, I envision the neurologic impact of psilocybin to be conducive to a complete transformation of a patient’s view of themself, other people, and the meaning of life. It is reminiscent of religious conversion.
The psychological effects of psilocybin in humans have been described as follows:
- emotional breakthrough9
- increased psychological flexibility,10,11 a very cortical effect
- mystical experience,12 which results in sudden and significant changes in behavior and perception and includes the following dimensions: sacredness, noetic quality, deeply felt positive mood, ineffability, paradoxicality, and transcendence of time and space13
- oceanic boundlessness, feeling “one with the universe”14
- universal interconnectedness, insightfulness, blissful state, spiritual experience14
- ego dissolution,15 with loss of one’s personal identity
- increased neuroplasticity16
- changes in cognition and increase in insight.17
The neurobiological effects of psilocybin are mediated by serotonin 5HT2A agonism and include the following18:
- reduction in the activity of the medial prefrontal cortex, which regulates memory, attention, inhibitory control, and habit
- a decrease in the connectivity between the medial prefrontal cortex and the posterior cingulate cortex, which regulates memory and emotions
- reducing the default mode network, which is active during rest, stimulating internal thoughts and reminiscing about previous feelings and events, sometimes including ruminations. Psilocybin reverses those processes to thinking about others, not just the self, and becoming more open-minded about the world and other people. This can be therapeutic for depression, which is often associated with negative ruminations but also with entrenched habits (addictive behaviors), anxiety, PTSD, and obsessive-compulsive disorders
- increased global functional connectivity among various brain networks, leading to stronger functional integration of behavior
- collapse of major cortical oscillatory rhythms such as alpha and others that perpetuate “prior” beliefs
- extensive neuroplasticity and recalibration of thought processes and decomposition of pathological beliefs, referred to as REBUS (relaxed beliefs under psychedelics).
The bottom line is psilocybin and other psychedelics can dramatically alter, reshape, and relax rigid beliefs and personality traits by decreasing “neuroticism” and increasing “extraversion,” insightfulness, openness, and possibly conscientiousness.19 Although no studies of psychedelics in psychopathic, antisocial, or narcissistic personality disorders have been conducted, it is very reasonable to speculate that psilocybin may reverse traits of these disorders such as callousness, lack of empathy, and pathological self-centeredness.
Going further, a preliminary report suggests psilocybin can modify political views by decreasing authoritarianism and increasing libertarianism.20,21 In the current political zeitgeist, could psychedelics such as psilocybin reduce or even eliminate political extremism and visceral hatred on all sides? It would be remarkable research to carry out to heal a politically divided populace.The dogma of untreatable personality disorders or hopelessly entrenched political extremism is on the chopping block, and psychedelics offer hope to splinter those beliefs by concurrently remodeling brain tissue (neuroplasticity) and rectifying the mindset (psychoplasticity).
1. Delay J, Deniker P. Neuroleptic effects of chlorpromazine in therapeutics of neuropsychiatry. J Clin Exp Psychopathol. 1955;16(2):104-112.
2. Walsh Z, Mollaahmetoglu OM, Rootman, J, et al. Ketamine for the treatment of mental health and substance use disorders: comprehensive systematic review. BJPsych Open. 2021;8(1):e19. doi:10.1192/bjo.2021.1061
3. Lener MS, Kadriu B, Zarate CA Jr. Ketamine and beyond: investigations into the potential of glutamatergic agents to treat depression. Drugs. 2017;77(4):381-401.
4. Ayers B, Leaver A, Woods RP, et al. Structural plasticity of the hippocampus and amygdala induced by electroconvulsive therapy in major depression. Biol Psychiatry. 2016;79(4):282-292.
5. Cao B, Li R, Ding L, Xu J, et al. Does cognitive behaviour therapy affect peripheral inflammation of depression? A protocol for the systematic review and meta-analysis. BMJ Open. 2021;11(12):e048162. doi:10.1136/bmjopen-2020-048162
6. Wagner E, Siafis S, Fernando P, et al. Efficacy and safety of clozapine in psychotic disorders—a systematic quantitative meta-review. Transl Psychiatry. 2021;11(1):487.
7. Daws RE, Timmermann C, Giribaldi B, et al. Increased global integration in the brain after psilocybin therapy for depression. Nat Med. 2022;28(4):844-851.
8. Pearson C, Siegel J, Gold JA. Psilocybin-assisted psychotherapy for depression: emerging research on a psychedelic compound with a rich history. J Neurol Sci. 2022;434:120096. doi:10.1016/j.jns.2021.120096
9. Roseman L, Haijen E, Idialu-Ikato K, et al. Emotional breakthrough and psychedelics: validation of the Emotional Breakthrough Inventory. J Psychopharmacol. 2019;33(9):1076-1087.
10. Davis AK, Barrett FS, Griffiths RR. Psychological flexibility mediates the relations between acute psychedelic effects and subjective decreases in depression and anxiety. J Contextual Behav Sci. 2020;15:39-45.
11. Hayes SC, Luoma JB, Bond FW, et al. Acceptance and commitment therapy: model, processes and outcomes. Behav Res Ther. 2006;44(1):1-25.
12. Ross S, Bossis A, Guss J, et al. Rapid and sustained symptom reduction following psilocybin treatment for anxiety and depression in patients with life-threatening cancer: a randomized controlled trial. J Psychopharmacol. 2016;30(12):1165-1180.
13. Stace WT. Mysticism and Philosophy. Macmillan Pub Ltd; 1960:37.
14. Barrett FS, Griffiths RR. Classic hallucinogens and mystical experiences: phenomenology and neural correlates. Curr Top Behav Neurosci. 2018;36:393-430.
15. Nour MM, Evans L, Nutt D, et al. Ego-dissolution and psychedelics: validation of the Ego-Dissolution Inventory (EDI). Front Hum Neurosci. 2016;10:269. doi:10.3389/fnhum.2016.00269
16. Olson DE. The subjective effects of psychedelics may not be necessary for their enduring therapeutic effects. ACS Pharmacol Transl Sci. 2020;4(2):563-567.
17. Carhart-Harris RL, Bolstridge M, Day CMJ, et al. Psilocybin with psychological support for treatment-resistant depression: six-month follow-up. Psychopharmacology (Berl). 2018;235(2):399-408.
18. Carhart-Harris RL. How do psychedelics work? Curr Opin Psychiatry. 2019;32(1):16-21.
19. Erritzoe D, Roseman L, Nour MM, et al. Effects of psilocybin therapy on personality structure. Acta Psychiatr Scand. 2018;138(5):368-378.
20. Lyons T, Carhart-Harris RL. Increased nature relatedness and decreased authoritarian political views after psilocybin for treatment-resistant depression. J Psychopharmacol. 2018;32(7):811-819.
21. Nour MM, Evans L, Carhart-Harris RL. Psychedelics, personality and political perspectives. J Psychoactive Drugs. 2017;49(3):182-191.
1. Delay J, Deniker P. Neuroleptic effects of chlorpromazine in therapeutics of neuropsychiatry. J Clin Exp Psychopathol. 1955;16(2):104-112.
2. Walsh Z, Mollaahmetoglu OM, Rootman, J, et al. Ketamine for the treatment of mental health and substance use disorders: comprehensive systematic review. BJPsych Open. 2021;8(1):e19. doi:10.1192/bjo.2021.1061
3. Lener MS, Kadriu B, Zarate CA Jr. Ketamine and beyond: investigations into the potential of glutamatergic agents to treat depression. Drugs. 2017;77(4):381-401.
4. Ayers B, Leaver A, Woods RP, et al. Structural plasticity of the hippocampus and amygdala induced by electroconvulsive therapy in major depression. Biol Psychiatry. 2016;79(4):282-292.
5. Cao B, Li R, Ding L, Xu J, et al. Does cognitive behaviour therapy affect peripheral inflammation of depression? A protocol for the systematic review and meta-analysis. BMJ Open. 2021;11(12):e048162. doi:10.1136/bmjopen-2020-048162
6. Wagner E, Siafis S, Fernando P, et al. Efficacy and safety of clozapine in psychotic disorders—a systematic quantitative meta-review. Transl Psychiatry. 2021;11(1):487.
7. Daws RE, Timmermann C, Giribaldi B, et al. Increased global integration in the brain after psilocybin therapy for depression. Nat Med. 2022;28(4):844-851.
8. Pearson C, Siegel J, Gold JA. Psilocybin-assisted psychotherapy for depression: emerging research on a psychedelic compound with a rich history. J Neurol Sci. 2022;434:120096. doi:10.1016/j.jns.2021.120096
9. Roseman L, Haijen E, Idialu-Ikato K, et al. Emotional breakthrough and psychedelics: validation of the Emotional Breakthrough Inventory. J Psychopharmacol. 2019;33(9):1076-1087.
10. Davis AK, Barrett FS, Griffiths RR. Psychological flexibility mediates the relations between acute psychedelic effects and subjective decreases in depression and anxiety. J Contextual Behav Sci. 2020;15:39-45.
11. Hayes SC, Luoma JB, Bond FW, et al. Acceptance and commitment therapy: model, processes and outcomes. Behav Res Ther. 2006;44(1):1-25.
12. Ross S, Bossis A, Guss J, et al. Rapid and sustained symptom reduction following psilocybin treatment for anxiety and depression in patients with life-threatening cancer: a randomized controlled trial. J Psychopharmacol. 2016;30(12):1165-1180.
13. Stace WT. Mysticism and Philosophy. Macmillan Pub Ltd; 1960:37.
14. Barrett FS, Griffiths RR. Classic hallucinogens and mystical experiences: phenomenology and neural correlates. Curr Top Behav Neurosci. 2018;36:393-430.
15. Nour MM, Evans L, Nutt D, et al. Ego-dissolution and psychedelics: validation of the Ego-Dissolution Inventory (EDI). Front Hum Neurosci. 2016;10:269. doi:10.3389/fnhum.2016.00269
16. Olson DE. The subjective effects of psychedelics may not be necessary for their enduring therapeutic effects. ACS Pharmacol Transl Sci. 2020;4(2):563-567.
17. Carhart-Harris RL, Bolstridge M, Day CMJ, et al. Psilocybin with psychological support for treatment-resistant depression: six-month follow-up. Psychopharmacology (Berl). 2018;235(2):399-408.
18. Carhart-Harris RL. How do psychedelics work? Curr Opin Psychiatry. 2019;32(1):16-21.
19. Erritzoe D, Roseman L, Nour MM, et al. Effects of psilocybin therapy on personality structure. Acta Psychiatr Scand. 2018;138(5):368-378.
20. Lyons T, Carhart-Harris RL. Increased nature relatedness and decreased authoritarian political views after psilocybin for treatment-resistant depression. J Psychopharmacol. 2018;32(7):811-819.
21. Nour MM, Evans L, Carhart-Harris RL. Psychedelics, personality and political perspectives. J Psychoactive Drugs. 2017;49(3):182-191.

Mifepristone for the treatment of miscarriage and fetal demise
In the uterus, coordinated myometrial cell contraction is not triggered by neural activation; instead, myometrial cells work together as a contractile syncytium through cell-to-cell gap junction connections permitting the intercellular sharing of small molecules, which in turn facilitates activation of the actin-myosin contractile apparatus and coordinated uterine contraction. In myometrial cells, connexin 43 (Cx43) is the main gap junction protein. Cx43 permits the passage of small hydrophilic molecules (ATP) and ions (calcium) cell to cell. Estradiol increases Cx43 synthesis in human myometrial cells.1 Progesterone decreases Cx43 synthesis effectively isolating myometrial cells, reducing cell-to-cell sharing of chemicals that stimulate contraction, blocking coordinated uterine contraction.2 Progesterone suppression of Cx43 synthesis helps to prevent premature uterine contraction during pregnancy. At term, decreases in progesterone levels result in an increase in Cx43 synthesis, facilitating the onset of effective labor. In myometrial cells, antiprogestins, including mifepristone, increase the number of gap junction connections, facilitating a coordinated contractile signal in response to misoprostol or oxytocin.3,4
It takes time for antiprogestins to stimulate myometrial cell production of Cx43. In the rat myometrium the administration of mifepristone results in a 2.5-fold increase of Cx43 mRNA transcripts within 9 hours and a 5.6-fold increase in 24 hours.3 Hence, most mifepristone treatment protocols involve administering mifepristone and waiting 24 to 48 hours before administering an agent that stimulates myometrial contraction, such as misoprostol. Antiprogestins also increase the sensitivity of myometrial cells to oxytocin stimulation of uterine contractions by increasing Cx43 concentration.4
Progesterone also regulates other important biological processes in the cervix, decidua, placenta, and cervix. Antiprogestins can facilitate cervical ripening and disrupt decidual function, interfering with the attachment of pregnancy tissue.5 In the cervix, antiprogestins increase matrix metalloproteinase expression, disrupting collagen organization, decreasing cervical tensile strength and leading to cervical ripening.6
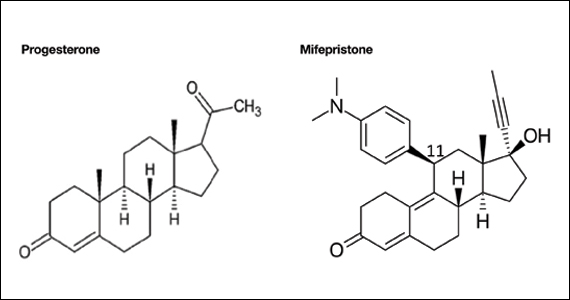
Pharmacology of mifepristone
Mifepristone is an antiprogestin and antiglucocorticoid with high-affinity binding to both the progesterone and glucocorticoid receptors (FIGURE 1). The phenylaminodimethyl group at C-11 of mifepristone changes the positional equilibrium of helix 12 of the progesterone receptor, reducing the ability of the receptor to bind required co-activators, limiting receptor binding to DNA, resulting in an antiprogesterone effect.7 At the low, single-dose used for treatment of miscarriage and fetal demise (200 mg one dose), mifepristone is an antiprogestin. At the high, daily dose used for the treatment of hyperglycemia caused by Cushing disease (≥ 300 mg daily), mifepristone is also an antiglucocorticoid.
Although mifepristone is a powerful antiglucocorticoid, in patients with an intact hypothalamic-pituitary-adrenal axis, mifepristone does not cause adrenal insufficiency. In people with an intact hypothalamic-pituitary-adrenal axis, daily administration of mifepristone (≥ 200 mg) for 7 days or longer results in an increase in pituitary secretion of ACTH and adrenal secretion of cortisol, largely overcoming the antiglucocorticoid action of mifepristone.8-10 This compensatory increase in ACTH and cortisol is not possible in patients who have had a hypophysectomy or bilateral adrenalectomy or have adrenal suppression due to long-term treatment with high doses of glucocorticoids. Mifepristone is contraindicated for patients with these conditions because it may cause glucocorticoid insufficiency by blocking glucocorticoid receptors.
The terminal half-life of mifepristone is 18 hours.11 Following oral administration of a single dose of mifepristone 200 mg the peak circulating concentration is reached in 90 minutes. Mifepristone is metabolized by CYP3A4 and is also a strong inhibitor of CYP3A4. Contraindications to the use of mifepristone include adrenal failure, porphyria, hemorrhagic diseases, anticoagulation, an IUD in the uterus, ectopic pregnancy, long-term glucocorticoid administration, and an undiagnosed adnexal mass.
Continue to: Mifepristone-misoprostol for the treatment of early missed miscarriage with a gestational sac...
Mifepristone-misoprostol for the treatment of early missed miscarriage with a gestational sac
For patients with a miscarriage, the treatment options to resolve the pregnancy loss are expectant management, medication, or surgery.12 Joint decision-making is recommended to establish a management plan that supports the patient’s values. Expectant management is most likely to result in a multi-week process to achieve completion of the miscarriage. A surgical procedure is most likely to result in rapid resolution of the miscarriage with the greatest rate of success. Surgical evacuation of the uterus may be the preferred option for patients who have excessive uterine bleeding or concerning vital signs. Both medical and surgical management are more likely than expectant management to successfully resolve the miscarriage.13
In the past, the standard approach to medication management of a miscarriage was the administration of one or more doses of misoprostol, a synthetic prostaglandin E1. However, two large trials have reported that the dual-medication sequence of mifepristone followed 24 to 48 hours later by misoprostol is more effective than misoprostol alone for resolving a miscarriage.14,15 This is probably due to mifepristone making the uterus more responsive to the effects of misoprostol.
Schreiber and colleagues14 reported a study of 300 patients with an anembryonic gestation or embryonic demise, between 5 and 12 completed weeks of gestation, who were randomly assigned to treatment with mifepristone (200 mg) followed in 24 to 48 hours with vaginal misoprostol (800 µg) or vaginal misoprostol (800 µg) alone. Ultrasonography was performed 1 to 4 days after misoprostol administration. Successful treatment was defined as expulsion of the gestational sac plus no additional surgical or medical intervention within 30 days after treatment. In this study, the dual-medication regimen of mifepristone-misoprostol was more successful than misoprostol alone in resolving the miscarriage, 84% and 67%, respectively (relative risk [RR], 1.25; 95% confidence interval [CI], 1.09–1.43).
Surgical evacuation of the uterus occurred less often with mifepristone-misoprostol treatment than with misoprostol monotherapy—9% and 24%, respectively (RR, 0.37; 95% CI, 0.21–0.68). Pelvic infection occurred in 2 patients (1.3%) in each group. Uterine bleeding managed with blood transfusion occurred in 3 patients who received mifepristone-misoprostol and 1 patient who received misoprostol alone. In this study, clinical factors including active bleeding, parity, and gestational age did not influence treatment success with the mifepristone-misoprostol regimen.16 The mifepristone-misoprostol regimen was reported to be more cost-effective than misoprostol alone.17
Chu and colleagues15 reported a study of medication treatment of missed miscarriage that included more than 700 patients randomly assigned to treatment with mifepristone-misoprostol or placebo-misoprostol. Missed miscarriage was diagnosed by an ultrasound demonstrating a gestational sac and a nonviable pregnancy. The doses of mifepristone and misoprostol were 200 mg and 800 µg, respectively. In this study the misoprostol was administered 48 hours following mifepristone or placebo using a vaginal, oral, or buccal route, but 90% of patients used the vaginal route. Treatment was considered successful if the patient passed the gestational sac as determined by an ultrasound performed 7 days after entry into the study. If the gestational sac was passed, the patients were asked to do a urine pregnancy test 3 weeks after entering the study to conclude their care episode. If patients did not pass the gestational sac, they were offered a second dose of misoprostol or surgical evacuation. In this study, mifepristone-misoprostol resulted in fewer patients who did not pass the gestational sac within 7 days after entry into the study than placebo (mifepristone-misoprostol, 17% vs placebo-misoprostol, 24% (P=.043). Surgical intervention was performed in 25% of patients treated with placebo-misoprostol and 17% of patients treated with mifepristone-misoprostol (RR, 0.73; 95% CI, 0.53–0.95; P=.021). A cost-effectiveness analysis of the trial results reported that the combination of mifepristone-misoprostol was less costly than misoprostol alone for the management of missed miscarriages.18
Misoprostol can be administered by an oral, buccal, rectal, or vaginal route.19,20 Vaginal administration results in higher circulating concentrations of misoprostol than buccal administration, but both routes of administration produce similar mean uterine tone and mean uterine activity as measured by an intrauterine pressure transducer over 5 hours.21 Hence, at our institution, we most often use buccal administration of misoprostol. To assess effectiveness of mifepristone-misoprostol treatment, 1 week after treatment with a pelvic ultrasound to detect expulsion of the gestational sac. Alternatively, a urine pregnancy test can be performed 3 weeks following medication treatment. The mifepristone-misoprostol regimen is not approved by the US Food and Drug Administration for the treatment of miscarriage.
Continue to: Mifepristone-misoprostol for the treatment of fetal demise...
Mifepristone-misoprostol for the treatment of fetal demise
Fetal loss in the second or third trimesters is a devastating experience for most patients, painfully echoing in the heart and mind for years. Empathic and effective treatment of fetal loss may reduce the adverse impact of the event. Multiple studies have reported that combinations of mifepristone and misoprostol reduced the time from initiation of labor contractions to birth compared with misoprostol alone.22-28 In addition, the combination of mifepristone-misoprostol reduced the amount of misoprostol needed to achieve delivery.22,23
In one clinical trial, 66 patients with fetal demise between 14 and 28 weeks’ gestation were randomized to receive mifepristone 200 mg or placebo.22 Twenty-four to 48 hours later, misoprostol for induction of labor was initiated. Among the patients from 14 to 23 completed weeks of gestation, the misoprostol dose was 400 µg vaginally every 6 hours. For patients from 24 to 28 weeks gestation, the misoprostol dose was 200 µg vaginally every 4 hours. The median times from initiation of misoprostol to birth for the patients in the mifepristone and placebo groups were 6.8 hours and 10.5 hours (P=.002).
Compared with the patients in the placebo-misoprostol group, the patients in the mifepristone-misoprostol group required fewer doses of misoprostol (2.1 vs 3.4; P=.002) and a lower total dose of misoprostol (768 µg vs 1,182 µg; P=.003). All patients in the mifepristone group delivered within 24 hours. By contrast, 13% of the patients in the placebo group delivered more than 24 hours after the initiation of misoprostol treatment. Five patients were readmitted with retained products of conception needing suction curettage—4 in the placebo group and 1 in the mifepristone group.22
In a second clinical trial, 110 patients with fetal demise after 20 weeks of gestation were randomized to receive mifepristone 200 mg or placebo.23 Thirty-six to 48 hours later, misoprostol for induction of labor was initiated. Among the patients from 20 to 25 completed weeks of gestation, the misoprostol dose was 100 µg vaginally every 6 hours for a maximum of 4 doses. For patients ≥26 weeks gestation, the misoprostol dose was 50 µg vaginally every 4 hours for a maximum of 6 doses. The median times from initiation of misoprostol to birth for the patients in the mifepristone and placebo groups were 9.8 hours and 16.3 hours. (P=.001).
Compared with the patients in the placebo-misoprostol group, the patients in the mifepristone-misoprostol group required a lower total dose of misoprostol (110 µg vs 198 µg, P<.001).
Delivery within 24 hours following initiation of misoprostol occurred in 93% and 73% of the patients in the mifepristone and placebo groups, respectively (P<.001). Compared with patients in the mifepristone group, shivering occurred more frequently among the patients in the placebo group (7.5% vs 19.2%; P=.09), likely because they received greater doses of misoprostol.23
Miscarriage and fetal demise frequently cause patients to experience a range of emotions including denial, numbness, grief, anger, guilt, and depression. It may take months or years for people to progress to a tentative acceptance of the loss, refocusing on future aspirations. Empathic care and timely and effective medical intervention to resolve the pregnancy loss optimize outcomes. For medication treatment of miscarriage and fetal demise, mifepristone is an important agent because it improves the success rate for resolution of miscarriage without surgery and it shortens the time of labor for inductions for fetal demise. Obstetrician-gynecologists are the specialists leading advances in treatment of miscarriage and fetal demise. I encourage you to use mifepristone in your care of appropriate patients with miscarriage and fetal demise. ●
- Andersen J, Grine E, Eng L, et al. Expression of connexin-43 in human myometrium and leiomyoma. Am J Obstet Gynecol. 1993;169:1266-1276. doi: 10.1016/0002-9378(93)90293-r.
- Ou CW, Orsino A, Lye SJ. Expression of connexin-43 and connexin-26 in the rat myometrium during pregnancy and labor is differentially regulated by mechanical and hormonal signals. Endocrinology. 1997;138:5398-5407. doi: 10.1210 /endo.138.12.5624.
- Petrocelli T, Lye SJ. Regulation of transcripts encoding the myometrial gap junction protein, connexin-43, by estrogen and progesterone. Endocrinology. 1993;133:284-290. doi: 10.1210 /endo.133.1.8391423.
- Chwalisz K, Fahrenholz F, Hackenberg M, et al. The progesterone antagonist onapristone increases the effectiveness of oxytocin to produce delivery without changing the myometrial oxytocin receptor concentration. Am J Obstet Gynecol. 1991;165:1760-1770. doi: 10.1016/0002 -9378(91)90030-u.
- Large MJ, DeMayo FJ. The regulation of embryo implantation and endometrial decidualization by progesterone receptor signaling. Mol Cell Endocrinol. 2012;358:155-165. doi: 10.1016 /j.mce.2011.07.027.
- Clark K, Ji H, Feltovich H, et al. Mifepristone-induced cervical ripening: structural, biomechanical and molecular events. Am J Obstet Gynecol. 2006;194:1391-1398. doi: 10.1016 /j.ajog.2005.11.026.
- Raaijmakers HCA, Versteegh JE, Uitdehaag JCM. T he x-ray structure of RU486 bound to the progesterone receptor in a destabilized agonist conformation. J Biol Chem. 2009;284:19572-19579. doi: 10.1074/jbc.M109.007872.
- Yuen KCJ, Moraitis A, Nguyen D. Evaluation of evidence of adrenal insufficiency in trials of normocortisolemic patients treated with mifepristone. J Endocr Soc. 2017;1:237-246. doi: 10.1210 /js.2016-1097.
- Spitz IM, Grunberg SM, Chabbert-Buffet N, et al. Management of patients receiving long-term treatment with mifepristone. Fertil Steril. 2005;84:1719-1726. doi: 10.1016 /j.fertnstert.2005.05.056.
- Bertagna X, Escourolle H, Pinquier JL, et al. Administration of RU 486 for 8 days in normal volunteers: antiglucocorticoid effect with no evidence of peripheral cortisol deprivation. J Clin Endocrinol Metab. 1994;78:375-380. doi: 10.1210 /jcem.78.2.8106625.
- Mifeprex [package insert]. New York, NY: Danco Laboratories; March 2016.
- Early pregnancy loss. ACOG Practice Bulletin No 200. American College of Obstetricians and Gynecologists. Obstet Gynecol. 2018;132:e197-e207. doi: /AOG.0000000000002899. 10.1097
- Chu J, Devall AJ, Hardy P, et al. What is the best method for managing early miscarriage? BMJ. 2020;368:l6483. doi: 10.1136/bmj.l6438.
- Schreiber C, Creinin MD, Atrio J, et al. Mifepristone pretreatment for the medical management of early pregnancy loss. N Engl J Med. 2018;378:2161-2170. doi: 10.1056 /NEJMoa1715726.
- Chu JJ, Devall AJ, Beeson LE, et al. Mifepristone and misoprostol versus misoprostol alone for the management of missed miscarriage (MifeMiso): a randomised, double-blind, placebo-controlled trial. Lancet. 2020;396:770-778. doi: 10.1016 /S0140-6736(20)31788-8.
- Sonalkar S, Koelper N, Creinin MD, et al. Management of early pregnancy loss with mifepristone and misoprostol: clinical predictors of treatment success from a randomized trial. Am J Obstet Gynecol. 2020;223:551.e1-e7. doi: 10.1016/j. ajog.2020.04.006. 17.
- Nagendra D, Koelper N, Loza-Avalos SE, et al. Cost-effectiveness of mifepristone pretreatment for the medical management of nonviable early pregnancy: secondary analysis of a randomized clinical trial. JAMA Netw Open. 2020;3:e201594. doi: 10.1001/jamanetworkopen.2020.1594.
- Okeke-Ogwulu CB, Williams EV, Chu JJ, et al. Cost-effectiveness of mifepristone and misoprostol versus misoprostol alone for the management of missed miscarriage: an economic evaluation based on the MifeMiso trial. BJOG. 2021;128: 1534-1545. doi: 10.1111/1471-0528.16737.
- Tang OS, Schweer H, Seyberth HW, et al. Pharmacokinetics of different routes of administration of misoprostol. Hum Reprod. 2002;17:332336. doi: 10.1093/humrep/17.2.332.
- Schaff EA, DiCenzo R, Fielding SL. Comparison of misoprostol plasma concentrations following buccal and sublingual administration. Contraception. 2005;71:22-25. doi: 10.1016 /j.contraception.2004.06.014.
- Meckstroth KR, Whitaker AK, Bertisch S, et al. Misoprostol administered by epithelial routes: drug absorption and uterine response. Obstet Gynecol. 2006;108:582-590. doi: 10.1097/01 .AOG.0000230398.32794.9d.
- Allanson ER, Copson S, Spilsbury K, et al. Pretreatment with mifepristone compared with misoprostol alone for delivery after fetal death between 14 and 28 weeks of gestation. Obstet Gynecol. 2021;137:801-809. doi: 10.1097 /AOG.0000000000004344.
- Chaudhuri P, Datta S. Mifepristone and misoprostol compared with misoprostol alone for induction of labor in intrauterine fetal death: a randomized trial. J Obstet Gynaecol Res. 2015;41:1884-1890. doi: 10.1111/jog.12815.
- Fyfe R, Murray H. Comparison of induction of labour regimens for termination of pregnancy with and without mifepristone, from 20 to 41 weeks gestation. Aust N Z J Obstet Gynaecol. 2017;57:604-608. doi: 10.1111 /ajo.12648.
- Panda S, Jha V, Singh S. Role of combination of mifepristone and misoprostol verses misoprostol alone in induction of labour in late intrauterine fetal death: a prospective study. J Family Reprod Health. 2013;7:177-179.
- Vayrynen W, Heikinheimo O, Nuutila M. Misoprostol-only versus mifepristone plus misoprostol in induction of labor following intrauterine fetal death. Acta Obstet Gynecol Scand. 2007;86: 701-705. doi: 10.1080/00016340701379853.
- Sharma D, Singhal SR, Poonam AP. Comparison of mifepristone combination with misoprostol and misoprostol alone in the management of intrauterine death. Taiwan J Obstet Gynecol. 2011;50:322-325. doi: 10.1016/j.tjog.2011.07.007.
- Stibbe KJM, de Weerd S. Induction of delivery by mifepristone and misoprostol in termination of pregnancy and intrauterine fetal death: 2nd and 3rd trimester induction of labour. Arch Gynecol Obstet. 2012;286:795-796. doi: 10.1007 /s00404-012-2289-3.

Robert L. Barbieri, MD
Editor in Chief, OBG Management
Chair Emeritus, Department of Obstetrics and Gynecology
Brigham and Women’s Hospital
Kate Macy Ladd Distinguished Professor of Obstetrics,
Gynecology and Reproductive Biology
Harvard Medical School
Boston, Massachusetts
Dr. Barbieri reports no financial relationships relevant to this article.
Robert L. Barbieri, MD
Editor in Chief, OBG Management
Chair Emeritus, Department of Obstetrics and Gynecology
Brigham and Women’s Hospital
Kate Macy Ladd Distinguished Professor of Obstetrics,
Gynecology and Reproductive Biology
Harvard Medical School
Boston, Massachusetts
Dr. Barbieri reports no financial relationships relevant to this article.
Robert L. Barbieri, MD
Editor in Chief, OBG Management
Chair Emeritus, Department of Obstetrics and Gynecology
Brigham and Women’s Hospital
Kate Macy Ladd Distinguished Professor of Obstetrics,
Gynecology and Reproductive Biology
Harvard Medical School
Boston, Massachusetts
Dr. Barbieri reports no financial relationships relevant to this article.
In the uterus, coordinated myometrial cell contraction is not triggered by neural activation; instead, myometrial cells work together as a contractile syncytium through cell-to-cell gap junction connections permitting the intercellular sharing of small molecules, which in turn facilitates activation of the actin-myosin contractile apparatus and coordinated uterine contraction. In myometrial cells, connexin 43 (Cx43) is the main gap junction protein. Cx43 permits the passage of small hydrophilic molecules (ATP) and ions (calcium) cell to cell. Estradiol increases Cx43 synthesis in human myometrial cells.1 Progesterone decreases Cx43 synthesis effectively isolating myometrial cells, reducing cell-to-cell sharing of chemicals that stimulate contraction, blocking coordinated uterine contraction.2 Progesterone suppression of Cx43 synthesis helps to prevent premature uterine contraction during pregnancy. At term, decreases in progesterone levels result in an increase in Cx43 synthesis, facilitating the onset of effective labor. In myometrial cells, antiprogestins, including mifepristone, increase the number of gap junction connections, facilitating a coordinated contractile signal in response to misoprostol or oxytocin.3,4
It takes time for antiprogestins to stimulate myometrial cell production of Cx43. In the rat myometrium the administration of mifepristone results in a 2.5-fold increase of Cx43 mRNA transcripts within 9 hours and a 5.6-fold increase in 24 hours.3 Hence, most mifepristone treatment protocols involve administering mifepristone and waiting 24 to 48 hours before administering an agent that stimulates myometrial contraction, such as misoprostol. Antiprogestins also increase the sensitivity of myometrial cells to oxytocin stimulation of uterine contractions by increasing Cx43 concentration.4
Progesterone also regulates other important biological processes in the cervix, decidua, placenta, and cervix. Antiprogestins can facilitate cervical ripening and disrupt decidual function, interfering with the attachment of pregnancy tissue.5 In the cervix, antiprogestins increase matrix metalloproteinase expression, disrupting collagen organization, decreasing cervical tensile strength and leading to cervical ripening.6
Pharmacology of mifepristone
Mifepristone is an antiprogestin and antiglucocorticoid with high-affinity binding to both the progesterone and glucocorticoid receptors (FIGURE 1). The phenylaminodimethyl group at C-11 of mifepristone changes the positional equilibrium of helix 12 of the progesterone receptor, reducing the ability of the receptor to bind required co-activators, limiting receptor binding to DNA, resulting in an antiprogesterone effect.7 At the low, single-dose used for treatment of miscarriage and fetal demise (200 mg one dose), mifepristone is an antiprogestin. At the high, daily dose used for the treatment of hyperglycemia caused by Cushing disease (≥ 300 mg daily), mifepristone is also an antiglucocorticoid.
Although mifepristone is a powerful antiglucocorticoid, in patients with an intact hypothalamic-pituitary-adrenal axis, mifepristone does not cause adrenal insufficiency. In people with an intact hypothalamic-pituitary-adrenal axis, daily administration of mifepristone (≥ 200 mg) for 7 days or longer results in an increase in pituitary secretion of ACTH and adrenal secretion of cortisol, largely overcoming the antiglucocorticoid action of mifepristone.8-10 This compensatory increase in ACTH and cortisol is not possible in patients who have had a hypophysectomy or bilateral adrenalectomy or have adrenal suppression due to long-term treatment with high doses of glucocorticoids. Mifepristone is contraindicated for patients with these conditions because it may cause glucocorticoid insufficiency by blocking glucocorticoid receptors.
The terminal half-life of mifepristone is 18 hours.11 Following oral administration of a single dose of mifepristone 200 mg the peak circulating concentration is reached in 90 minutes. Mifepristone is metabolized by CYP3A4 and is also a strong inhibitor of CYP3A4. Contraindications to the use of mifepristone include adrenal failure, porphyria, hemorrhagic diseases, anticoagulation, an IUD in the uterus, ectopic pregnancy, long-term glucocorticoid administration, and an undiagnosed adnexal mass.
Continue to: Mifepristone-misoprostol for the treatment of early missed miscarriage with a gestational sac...
Mifepristone-misoprostol for the treatment of early missed miscarriage with a gestational sac
For patients with a miscarriage, the treatment options to resolve the pregnancy loss are expectant management, medication, or surgery.12 Joint decision-making is recommended to establish a management plan that supports the patient’s values. Expectant management is most likely to result in a multi-week process to achieve completion of the miscarriage. A surgical procedure is most likely to result in rapid resolution of the miscarriage with the greatest rate of success. Surgical evacuation of the uterus may be the preferred option for patients who have excessive uterine bleeding or concerning vital signs. Both medical and surgical management are more likely than expectant management to successfully resolve the miscarriage.13
In the past, the standard approach to medication management of a miscarriage was the administration of one or more doses of misoprostol, a synthetic prostaglandin E1. However, two large trials have reported that the dual-medication sequence of mifepristone followed 24 to 48 hours later by misoprostol is more effective than misoprostol alone for resolving a miscarriage.14,15 This is probably due to mifepristone making the uterus more responsive to the effects of misoprostol.
Schreiber and colleagues14 reported a study of 300 patients with an anembryonic gestation or embryonic demise, between 5 and 12 completed weeks of gestation, who were randomly assigned to treatment with mifepristone (200 mg) followed in 24 to 48 hours with vaginal misoprostol (800 µg) or vaginal misoprostol (800 µg) alone. Ultrasonography was performed 1 to 4 days after misoprostol administration. Successful treatment was defined as expulsion of the gestational sac plus no additional surgical or medical intervention within 30 days after treatment. In this study, the dual-medication regimen of mifepristone-misoprostol was more successful than misoprostol alone in resolving the miscarriage, 84% and 67%, respectively (relative risk [RR], 1.25; 95% confidence interval [CI], 1.09–1.43).
Surgical evacuation of the uterus occurred less often with mifepristone-misoprostol treatment than with misoprostol monotherapy—9% and 24%, respectively (RR, 0.37; 95% CI, 0.21–0.68). Pelvic infection occurred in 2 patients (1.3%) in each group. Uterine bleeding managed with blood transfusion occurred in 3 patients who received mifepristone-misoprostol and 1 patient who received misoprostol alone. In this study, clinical factors including active bleeding, parity, and gestational age did not influence treatment success with the mifepristone-misoprostol regimen.16 The mifepristone-misoprostol regimen was reported to be more cost-effective than misoprostol alone.17
Chu and colleagues15 reported a study of medication treatment of missed miscarriage that included more than 700 patients randomly assigned to treatment with mifepristone-misoprostol or placebo-misoprostol. Missed miscarriage was diagnosed by an ultrasound demonstrating a gestational sac and a nonviable pregnancy. The doses of mifepristone and misoprostol were 200 mg and 800 µg, respectively. In this study the misoprostol was administered 48 hours following mifepristone or placebo using a vaginal, oral, or buccal route, but 90% of patients used the vaginal route. Treatment was considered successful if the patient passed the gestational sac as determined by an ultrasound performed 7 days after entry into the study. If the gestational sac was passed, the patients were asked to do a urine pregnancy test 3 weeks after entering the study to conclude their care episode. If patients did not pass the gestational sac, they were offered a second dose of misoprostol or surgical evacuation. In this study, mifepristone-misoprostol resulted in fewer patients who did not pass the gestational sac within 7 days after entry into the study than placebo (mifepristone-misoprostol, 17% vs placebo-misoprostol, 24% (P=.043). Surgical intervention was performed in 25% of patients treated with placebo-misoprostol and 17% of patients treated with mifepristone-misoprostol (RR, 0.73; 95% CI, 0.53–0.95; P=.021). A cost-effectiveness analysis of the trial results reported that the combination of mifepristone-misoprostol was less costly than misoprostol alone for the management of missed miscarriages.18
Misoprostol can be administered by an oral, buccal, rectal, or vaginal route.19,20 Vaginal administration results in higher circulating concentrations of misoprostol than buccal administration, but both routes of administration produce similar mean uterine tone and mean uterine activity as measured by an intrauterine pressure transducer over 5 hours.21 Hence, at our institution, we most often use buccal administration of misoprostol. To assess effectiveness of mifepristone-misoprostol treatment, 1 week after treatment with a pelvic ultrasound to detect expulsion of the gestational sac. Alternatively, a urine pregnancy test can be performed 3 weeks following medication treatment. The mifepristone-misoprostol regimen is not approved by the US Food and Drug Administration for the treatment of miscarriage.
Continue to: Mifepristone-misoprostol for the treatment of fetal demise...
Mifepristone-misoprostol for the treatment of fetal demise
Fetal loss in the second or third trimesters is a devastating experience for most patients, painfully echoing in the heart and mind for years. Empathic and effective treatment of fetal loss may reduce the adverse impact of the event. Multiple studies have reported that combinations of mifepristone and misoprostol reduced the time from initiation of labor contractions to birth compared with misoprostol alone.22-28 In addition, the combination of mifepristone-misoprostol reduced the amount of misoprostol needed to achieve delivery.22,23
In one clinical trial, 66 patients with fetal demise between 14 and 28 weeks’ gestation were randomized to receive mifepristone 200 mg or placebo.22 Twenty-four to 48 hours later, misoprostol for induction of labor was initiated. Among the patients from 14 to 23 completed weeks of gestation, the misoprostol dose was 400 µg vaginally every 6 hours. For patients from 24 to 28 weeks gestation, the misoprostol dose was 200 µg vaginally every 4 hours. The median times from initiation of misoprostol to birth for the patients in the mifepristone and placebo groups were 6.8 hours and 10.5 hours (P=.002).
Compared with the patients in the placebo-misoprostol group, the patients in the mifepristone-misoprostol group required fewer doses of misoprostol (2.1 vs 3.4; P=.002) and a lower total dose of misoprostol (768 µg vs 1,182 µg; P=.003). All patients in the mifepristone group delivered within 24 hours. By contrast, 13% of the patients in the placebo group delivered more than 24 hours after the initiation of misoprostol treatment. Five patients were readmitted with retained products of conception needing suction curettage—4 in the placebo group and 1 in the mifepristone group.22
In a second clinical trial, 110 patients with fetal demise after 20 weeks of gestation were randomized to receive mifepristone 200 mg or placebo.23 Thirty-six to 48 hours later, misoprostol for induction of labor was initiated. Among the patients from 20 to 25 completed weeks of gestation, the misoprostol dose was 100 µg vaginally every 6 hours for a maximum of 4 doses. For patients ≥26 weeks gestation, the misoprostol dose was 50 µg vaginally every 4 hours for a maximum of 6 doses. The median times from initiation of misoprostol to birth for the patients in the mifepristone and placebo groups were 9.8 hours and 16.3 hours. (P=.001).
Compared with the patients in the placebo-misoprostol group, the patients in the mifepristone-misoprostol group required a lower total dose of misoprostol (110 µg vs 198 µg, P<.001).
Delivery within 24 hours following initiation of misoprostol occurred in 93% and 73% of the patients in the mifepristone and placebo groups, respectively (P<.001). Compared with patients in the mifepristone group, shivering occurred more frequently among the patients in the placebo group (7.5% vs 19.2%; P=.09), likely because they received greater doses of misoprostol.23
Miscarriage and fetal demise frequently cause patients to experience a range of emotions including denial, numbness, grief, anger, guilt, and depression. It may take months or years for people to progress to a tentative acceptance of the loss, refocusing on future aspirations. Empathic care and timely and effective medical intervention to resolve the pregnancy loss optimize outcomes. For medication treatment of miscarriage and fetal demise, mifepristone is an important agent because it improves the success rate for resolution of miscarriage without surgery and it shortens the time of labor for inductions for fetal demise. Obstetrician-gynecologists are the specialists leading advances in treatment of miscarriage and fetal demise. I encourage you to use mifepristone in your care of appropriate patients with miscarriage and fetal demise. ●
In the uterus, coordinated myometrial cell contraction is not triggered by neural activation; instead, myometrial cells work together as a contractile syncytium through cell-to-cell gap junction connections permitting the intercellular sharing of small molecules, which in turn facilitates activation of the actin-myosin contractile apparatus and coordinated uterine contraction. In myometrial cells, connexin 43 (Cx43) is the main gap junction protein. Cx43 permits the passage of small hydrophilic molecules (ATP) and ions (calcium) cell to cell. Estradiol increases Cx43 synthesis in human myometrial cells.1 Progesterone decreases Cx43 synthesis effectively isolating myometrial cells, reducing cell-to-cell sharing of chemicals that stimulate contraction, blocking coordinated uterine contraction.2 Progesterone suppression of Cx43 synthesis helps to prevent premature uterine contraction during pregnancy. At term, decreases in progesterone levels result in an increase in Cx43 synthesis, facilitating the onset of effective labor. In myometrial cells, antiprogestins, including mifepristone, increase the number of gap junction connections, facilitating a coordinated contractile signal in response to misoprostol or oxytocin.3,4
It takes time for antiprogestins to stimulate myometrial cell production of Cx43. In the rat myometrium the administration of mifepristone results in a 2.5-fold increase of Cx43 mRNA transcripts within 9 hours and a 5.6-fold increase in 24 hours.3 Hence, most mifepristone treatment protocols involve administering mifepristone and waiting 24 to 48 hours before administering an agent that stimulates myometrial contraction, such as misoprostol. Antiprogestins also increase the sensitivity of myometrial cells to oxytocin stimulation of uterine contractions by increasing Cx43 concentration.4
Progesterone also regulates other important biological processes in the cervix, decidua, placenta, and cervix. Antiprogestins can facilitate cervical ripening and disrupt decidual function, interfering with the attachment of pregnancy tissue.5 In the cervix, antiprogestins increase matrix metalloproteinase expression, disrupting collagen organization, decreasing cervical tensile strength and leading to cervical ripening.6
Pharmacology of mifepristone
Mifepristone is an antiprogestin and antiglucocorticoid with high-affinity binding to both the progesterone and glucocorticoid receptors (FIGURE 1). The phenylaminodimethyl group at C-11 of mifepristone changes the positional equilibrium of helix 12 of the progesterone receptor, reducing the ability of the receptor to bind required co-activators, limiting receptor binding to DNA, resulting in an antiprogesterone effect.7 At the low, single-dose used for treatment of miscarriage and fetal demise (200 mg one dose), mifepristone is an antiprogestin. At the high, daily dose used for the treatment of hyperglycemia caused by Cushing disease (≥ 300 mg daily), mifepristone is also an antiglucocorticoid.
Although mifepristone is a powerful antiglucocorticoid, in patients with an intact hypothalamic-pituitary-adrenal axis, mifepristone does not cause adrenal insufficiency. In people with an intact hypothalamic-pituitary-adrenal axis, daily administration of mifepristone (≥ 200 mg) for 7 days or longer results in an increase in pituitary secretion of ACTH and adrenal secretion of cortisol, largely overcoming the antiglucocorticoid action of mifepristone.8-10 This compensatory increase in ACTH and cortisol is not possible in patients who have had a hypophysectomy or bilateral adrenalectomy or have adrenal suppression due to long-term treatment with high doses of glucocorticoids. Mifepristone is contraindicated for patients with these conditions because it may cause glucocorticoid insufficiency by blocking glucocorticoid receptors.
The terminal half-life of mifepristone is 18 hours.11 Following oral administration of a single dose of mifepristone 200 mg the peak circulating concentration is reached in 90 minutes. Mifepristone is metabolized by CYP3A4 and is also a strong inhibitor of CYP3A4. Contraindications to the use of mifepristone include adrenal failure, porphyria, hemorrhagic diseases, anticoagulation, an IUD in the uterus, ectopic pregnancy, long-term glucocorticoid administration, and an undiagnosed adnexal mass.
Continue to: Mifepristone-misoprostol for the treatment of early missed miscarriage with a gestational sac...
Mifepristone-misoprostol for the treatment of early missed miscarriage with a gestational sac
For patients with a miscarriage, the treatment options to resolve the pregnancy loss are expectant management, medication, or surgery.12 Joint decision-making is recommended to establish a management plan that supports the patient’s values. Expectant management is most likely to result in a multi-week process to achieve completion of the miscarriage. A surgical procedure is most likely to result in rapid resolution of the miscarriage with the greatest rate of success. Surgical evacuation of the uterus may be the preferred option for patients who have excessive uterine bleeding or concerning vital signs. Both medical and surgical management are more likely than expectant management to successfully resolve the miscarriage.13
In the past, the standard approach to medication management of a miscarriage was the administration of one or more doses of misoprostol, a synthetic prostaglandin E1. However, two large trials have reported that the dual-medication sequence of mifepristone followed 24 to 48 hours later by misoprostol is more effective than misoprostol alone for resolving a miscarriage.14,15 This is probably due to mifepristone making the uterus more responsive to the effects of misoprostol.
Schreiber and colleagues14 reported a study of 300 patients with an anembryonic gestation or embryonic demise, between 5 and 12 completed weeks of gestation, who were randomly assigned to treatment with mifepristone (200 mg) followed in 24 to 48 hours with vaginal misoprostol (800 µg) or vaginal misoprostol (800 µg) alone. Ultrasonography was performed 1 to 4 days after misoprostol administration. Successful treatment was defined as expulsion of the gestational sac plus no additional surgical or medical intervention within 30 days after treatment. In this study, the dual-medication regimen of mifepristone-misoprostol was more successful than misoprostol alone in resolving the miscarriage, 84% and 67%, respectively (relative risk [RR], 1.25; 95% confidence interval [CI], 1.09–1.43).
Surgical evacuation of the uterus occurred less often with mifepristone-misoprostol treatment than with misoprostol monotherapy—9% and 24%, respectively (RR, 0.37; 95% CI, 0.21–0.68). Pelvic infection occurred in 2 patients (1.3%) in each group. Uterine bleeding managed with blood transfusion occurred in 3 patients who received mifepristone-misoprostol and 1 patient who received misoprostol alone. In this study, clinical factors including active bleeding, parity, and gestational age did not influence treatment success with the mifepristone-misoprostol regimen.16 The mifepristone-misoprostol regimen was reported to be more cost-effective than misoprostol alone.17
Chu and colleagues15 reported a study of medication treatment of missed miscarriage that included more than 700 patients randomly assigned to treatment with mifepristone-misoprostol or placebo-misoprostol. Missed miscarriage was diagnosed by an ultrasound demonstrating a gestational sac and a nonviable pregnancy. The doses of mifepristone and misoprostol were 200 mg and 800 µg, respectively. In this study the misoprostol was administered 48 hours following mifepristone or placebo using a vaginal, oral, or buccal route, but 90% of patients used the vaginal route. Treatment was considered successful if the patient passed the gestational sac as determined by an ultrasound performed 7 days after entry into the study. If the gestational sac was passed, the patients were asked to do a urine pregnancy test 3 weeks after entering the study to conclude their care episode. If patients did not pass the gestational sac, they were offered a second dose of misoprostol or surgical evacuation. In this study, mifepristone-misoprostol resulted in fewer patients who did not pass the gestational sac within 7 days after entry into the study than placebo (mifepristone-misoprostol, 17% vs placebo-misoprostol, 24% (P=.043). Surgical intervention was performed in 25% of patients treated with placebo-misoprostol and 17% of patients treated with mifepristone-misoprostol (RR, 0.73; 95% CI, 0.53–0.95; P=.021). A cost-effectiveness analysis of the trial results reported that the combination of mifepristone-misoprostol was less costly than misoprostol alone for the management of missed miscarriages.18
Misoprostol can be administered by an oral, buccal, rectal, or vaginal route.19,20 Vaginal administration results in higher circulating concentrations of misoprostol than buccal administration, but both routes of administration produce similar mean uterine tone and mean uterine activity as measured by an intrauterine pressure transducer over 5 hours.21 Hence, at our institution, we most often use buccal administration of misoprostol. To assess effectiveness of mifepristone-misoprostol treatment, 1 week after treatment with a pelvic ultrasound to detect expulsion of the gestational sac. Alternatively, a urine pregnancy test can be performed 3 weeks following medication treatment. The mifepristone-misoprostol regimen is not approved by the US Food and Drug Administration for the treatment of miscarriage.
Continue to: Mifepristone-misoprostol for the treatment of fetal demise...
Mifepristone-misoprostol for the treatment of fetal demise
Fetal loss in the second or third trimesters is a devastating experience for most patients, painfully echoing in the heart and mind for years. Empathic and effective treatment of fetal loss may reduce the adverse impact of the event. Multiple studies have reported that combinations of mifepristone and misoprostol reduced the time from initiation of labor contractions to birth compared with misoprostol alone.22-28 In addition, the combination of mifepristone-misoprostol reduced the amount of misoprostol needed to achieve delivery.22,23
In one clinical trial, 66 patients with fetal demise between 14 and 28 weeks’ gestation were randomized to receive mifepristone 200 mg or placebo.22 Twenty-four to 48 hours later, misoprostol for induction of labor was initiated. Among the patients from 14 to 23 completed weeks of gestation, the misoprostol dose was 400 µg vaginally every 6 hours. For patients from 24 to 28 weeks gestation, the misoprostol dose was 200 µg vaginally every 4 hours. The median times from initiation of misoprostol to birth for the patients in the mifepristone and placebo groups were 6.8 hours and 10.5 hours (P=.002).
Compared with the patients in the placebo-misoprostol group, the patients in the mifepristone-misoprostol group required fewer doses of misoprostol (2.1 vs 3.4; P=.002) and a lower total dose of misoprostol (768 µg vs 1,182 µg; P=.003). All patients in the mifepristone group delivered within 24 hours. By contrast, 13% of the patients in the placebo group delivered more than 24 hours after the initiation of misoprostol treatment. Five patients were readmitted with retained products of conception needing suction curettage—4 in the placebo group and 1 in the mifepristone group.22
In a second clinical trial, 110 patients with fetal demise after 20 weeks of gestation were randomized to receive mifepristone 200 mg or placebo.23 Thirty-six to 48 hours later, misoprostol for induction of labor was initiated. Among the patients from 20 to 25 completed weeks of gestation, the misoprostol dose was 100 µg vaginally every 6 hours for a maximum of 4 doses. For patients ≥26 weeks gestation, the misoprostol dose was 50 µg vaginally every 4 hours for a maximum of 6 doses. The median times from initiation of misoprostol to birth for the patients in the mifepristone and placebo groups were 9.8 hours and 16.3 hours. (P=.001).
Compared with the patients in the placebo-misoprostol group, the patients in the mifepristone-misoprostol group required a lower total dose of misoprostol (110 µg vs 198 µg, P<.001).
Delivery within 24 hours following initiation of misoprostol occurred in 93% and 73% of the patients in the mifepristone and placebo groups, respectively (P<.001). Compared with patients in the mifepristone group, shivering occurred more frequently among the patients in the placebo group (7.5% vs 19.2%; P=.09), likely because they received greater doses of misoprostol.23
Miscarriage and fetal demise frequently cause patients to experience a range of emotions including denial, numbness, grief, anger, guilt, and depression. It may take months or years for people to progress to a tentative acceptance of the loss, refocusing on future aspirations. Empathic care and timely and effective medical intervention to resolve the pregnancy loss optimize outcomes. For medication treatment of miscarriage and fetal demise, mifepristone is an important agent because it improves the success rate for resolution of miscarriage without surgery and it shortens the time of labor for inductions for fetal demise. Obstetrician-gynecologists are the specialists leading advances in treatment of miscarriage and fetal demise. I encourage you to use mifepristone in your care of appropriate patients with miscarriage and fetal demise. ●
- Andersen J, Grine E, Eng L, et al. Expression of connexin-43 in human myometrium and leiomyoma. Am J Obstet Gynecol. 1993;169:1266-1276. doi: 10.1016/0002-9378(93)90293-r.
- Ou CW, Orsino A, Lye SJ. Expression of connexin-43 and connexin-26 in the rat myometrium during pregnancy and labor is differentially regulated by mechanical and hormonal signals. Endocrinology. 1997;138:5398-5407. doi: 10.1210 /endo.138.12.5624.
- Petrocelli T, Lye SJ. Regulation of transcripts encoding the myometrial gap junction protein, connexin-43, by estrogen and progesterone. Endocrinology. 1993;133:284-290. doi: 10.1210 /endo.133.1.8391423.
- Chwalisz K, Fahrenholz F, Hackenberg M, et al. The progesterone antagonist onapristone increases the effectiveness of oxytocin to produce delivery without changing the myometrial oxytocin receptor concentration. Am J Obstet Gynecol. 1991;165:1760-1770. doi: 10.1016/0002 -9378(91)90030-u.
- Large MJ, DeMayo FJ. The regulation of embryo implantation and endometrial decidualization by progesterone receptor signaling. Mol Cell Endocrinol. 2012;358:155-165. doi: 10.1016 /j.mce.2011.07.027.
- Clark K, Ji H, Feltovich H, et al. Mifepristone-induced cervical ripening: structural, biomechanical and molecular events. Am J Obstet Gynecol. 2006;194:1391-1398. doi: 10.1016 /j.ajog.2005.11.026.
- Raaijmakers HCA, Versteegh JE, Uitdehaag JCM. T he x-ray structure of RU486 bound to the progesterone receptor in a destabilized agonist conformation. J Biol Chem. 2009;284:19572-19579. doi: 10.1074/jbc.M109.007872.
- Yuen KCJ, Moraitis A, Nguyen D. Evaluation of evidence of adrenal insufficiency in trials of normocortisolemic patients treated with mifepristone. J Endocr Soc. 2017;1:237-246. doi: 10.1210 /js.2016-1097.
- Spitz IM, Grunberg SM, Chabbert-Buffet N, et al. Management of patients receiving long-term treatment with mifepristone. Fertil Steril. 2005;84:1719-1726. doi: 10.1016 /j.fertnstert.2005.05.056.
- Bertagna X, Escourolle H, Pinquier JL, et al. Administration of RU 486 for 8 days in normal volunteers: antiglucocorticoid effect with no evidence of peripheral cortisol deprivation. J Clin Endocrinol Metab. 1994;78:375-380. doi: 10.1210 /jcem.78.2.8106625.
- Mifeprex [package insert]. New York, NY: Danco Laboratories; March 2016.
- Early pregnancy loss. ACOG Practice Bulletin No 200. American College of Obstetricians and Gynecologists. Obstet Gynecol. 2018;132:e197-e207. doi: /AOG.0000000000002899. 10.1097
- Chu J, Devall AJ, Hardy P, et al. What is the best method for managing early miscarriage? BMJ. 2020;368:l6483. doi: 10.1136/bmj.l6438.
- Schreiber C, Creinin MD, Atrio J, et al. Mifepristone pretreatment for the medical management of early pregnancy loss. N Engl J Med. 2018;378:2161-2170. doi: 10.1056 /NEJMoa1715726.
- Chu JJ, Devall AJ, Beeson LE, et al. Mifepristone and misoprostol versus misoprostol alone for the management of missed miscarriage (MifeMiso): a randomised, double-blind, placebo-controlled trial. Lancet. 2020;396:770-778. doi: 10.1016 /S0140-6736(20)31788-8.
- Sonalkar S, Koelper N, Creinin MD, et al. Management of early pregnancy loss with mifepristone and misoprostol: clinical predictors of treatment success from a randomized trial. Am J Obstet Gynecol. 2020;223:551.e1-e7. doi: 10.1016/j. ajog.2020.04.006. 17.
- Nagendra D, Koelper N, Loza-Avalos SE, et al. Cost-effectiveness of mifepristone pretreatment for the medical management of nonviable early pregnancy: secondary analysis of a randomized clinical trial. JAMA Netw Open. 2020;3:e201594. doi: 10.1001/jamanetworkopen.2020.1594.
- Okeke-Ogwulu CB, Williams EV, Chu JJ, et al. Cost-effectiveness of mifepristone and misoprostol versus misoprostol alone for the management of missed miscarriage: an economic evaluation based on the MifeMiso trial. BJOG. 2021;128: 1534-1545. doi: 10.1111/1471-0528.16737.
- Tang OS, Schweer H, Seyberth HW, et al. Pharmacokinetics of different routes of administration of misoprostol. Hum Reprod. 2002;17:332336. doi: 10.1093/humrep/17.2.332.
- Schaff EA, DiCenzo R, Fielding SL. Comparison of misoprostol plasma concentrations following buccal and sublingual administration. Contraception. 2005;71:22-25. doi: 10.1016 /j.contraception.2004.06.014.
- Meckstroth KR, Whitaker AK, Bertisch S, et al. Misoprostol administered by epithelial routes: drug absorption and uterine response. Obstet Gynecol. 2006;108:582-590. doi: 10.1097/01 .AOG.0000230398.32794.9d.
- Allanson ER, Copson S, Spilsbury K, et al. Pretreatment with mifepristone compared with misoprostol alone for delivery after fetal death between 14 and 28 weeks of gestation. Obstet Gynecol. 2021;137:801-809. doi: 10.1097 /AOG.0000000000004344.
- Chaudhuri P, Datta S. Mifepristone and misoprostol compared with misoprostol alone for induction of labor in intrauterine fetal death: a randomized trial. J Obstet Gynaecol Res. 2015;41:1884-1890. doi: 10.1111/jog.12815.
- Fyfe R, Murray H. Comparison of induction of labour regimens for termination of pregnancy with and without mifepristone, from 20 to 41 weeks gestation. Aust N Z J Obstet Gynaecol. 2017;57:604-608. doi: 10.1111 /ajo.12648.
- Panda S, Jha V, Singh S. Role of combination of mifepristone and misoprostol verses misoprostol alone in induction of labour in late intrauterine fetal death: a prospective study. J Family Reprod Health. 2013;7:177-179.
- Vayrynen W, Heikinheimo O, Nuutila M. Misoprostol-only versus mifepristone plus misoprostol in induction of labor following intrauterine fetal death. Acta Obstet Gynecol Scand. 2007;86: 701-705. doi: 10.1080/00016340701379853.
- Sharma D, Singhal SR, Poonam AP. Comparison of mifepristone combination with misoprostol and misoprostol alone in the management of intrauterine death. Taiwan J Obstet Gynecol. 2011;50:322-325. doi: 10.1016/j.tjog.2011.07.007.
- Stibbe KJM, de Weerd S. Induction of delivery by mifepristone and misoprostol in termination of pregnancy and intrauterine fetal death: 2nd and 3rd trimester induction of labour. Arch Gynecol Obstet. 2012;286:795-796. doi: 10.1007 /s00404-012-2289-3.
- Andersen J, Grine E, Eng L, et al. Expression of connexin-43 in human myometrium and leiomyoma. Am J Obstet Gynecol. 1993;169:1266-1276. doi: 10.1016/0002-9378(93)90293-r.
- Ou CW, Orsino A, Lye SJ. Expression of connexin-43 and connexin-26 in the rat myometrium during pregnancy and labor is differentially regulated by mechanical and hormonal signals. Endocrinology. 1997;138:5398-5407. doi: 10.1210 /endo.138.12.5624.
- Petrocelli T, Lye SJ. Regulation of transcripts encoding the myometrial gap junction protein, connexin-43, by estrogen and progesterone. Endocrinology. 1993;133:284-290. doi: 10.1210 /endo.133.1.8391423.
- Chwalisz K, Fahrenholz F, Hackenberg M, et al. The progesterone antagonist onapristone increases the effectiveness of oxytocin to produce delivery without changing the myometrial oxytocin receptor concentration. Am J Obstet Gynecol. 1991;165:1760-1770. doi: 10.1016/0002 -9378(91)90030-u.
- Large MJ, DeMayo FJ. The regulation of embryo implantation and endometrial decidualization by progesterone receptor signaling. Mol Cell Endocrinol. 2012;358:155-165. doi: 10.1016 /j.mce.2011.07.027.
- Clark K, Ji H, Feltovich H, et al. Mifepristone-induced cervical ripening: structural, biomechanical and molecular events. Am J Obstet Gynecol. 2006;194:1391-1398. doi: 10.1016 /j.ajog.2005.11.026.
- Raaijmakers HCA, Versteegh JE, Uitdehaag JCM. T he x-ray structure of RU486 bound to the progesterone receptor in a destabilized agonist conformation. J Biol Chem. 2009;284:19572-19579. doi: 10.1074/jbc.M109.007872.
- Yuen KCJ, Moraitis A, Nguyen D. Evaluation of evidence of adrenal insufficiency in trials of normocortisolemic patients treated with mifepristone. J Endocr Soc. 2017;1:237-246. doi: 10.1210 /js.2016-1097.
- Spitz IM, Grunberg SM, Chabbert-Buffet N, et al. Management of patients receiving long-term treatment with mifepristone. Fertil Steril. 2005;84:1719-1726. doi: 10.1016 /j.fertnstert.2005.05.056.
- Bertagna X, Escourolle H, Pinquier JL, et al. Administration of RU 486 for 8 days in normal volunteers: antiglucocorticoid effect with no evidence of peripheral cortisol deprivation. J Clin Endocrinol Metab. 1994;78:375-380. doi: 10.1210 /jcem.78.2.8106625.
- Mifeprex [package insert]. New York, NY: Danco Laboratories; March 2016.
- Early pregnancy loss. ACOG Practice Bulletin No 200. American College of Obstetricians and Gynecologists. Obstet Gynecol. 2018;132:e197-e207. doi: /AOG.0000000000002899. 10.1097
- Chu J, Devall AJ, Hardy P, et al. What is the best method for managing early miscarriage? BMJ. 2020;368:l6483. doi: 10.1136/bmj.l6438.
- Schreiber C, Creinin MD, Atrio J, et al. Mifepristone pretreatment for the medical management of early pregnancy loss. N Engl J Med. 2018;378:2161-2170. doi: 10.1056 /NEJMoa1715726.
- Chu JJ, Devall AJ, Beeson LE, et al. Mifepristone and misoprostol versus misoprostol alone for the management of missed miscarriage (MifeMiso): a randomised, double-blind, placebo-controlled trial. Lancet. 2020;396:770-778. doi: 10.1016 /S0140-6736(20)31788-8.
- Sonalkar S, Koelper N, Creinin MD, et al. Management of early pregnancy loss with mifepristone and misoprostol: clinical predictors of treatment success from a randomized trial. Am J Obstet Gynecol. 2020;223:551.e1-e7. doi: 10.1016/j. ajog.2020.04.006. 17.
- Nagendra D, Koelper N, Loza-Avalos SE, et al. Cost-effectiveness of mifepristone pretreatment for the medical management of nonviable early pregnancy: secondary analysis of a randomized clinical trial. JAMA Netw Open. 2020;3:e201594. doi: 10.1001/jamanetworkopen.2020.1594.
- Okeke-Ogwulu CB, Williams EV, Chu JJ, et al. Cost-effectiveness of mifepristone and misoprostol versus misoprostol alone for the management of missed miscarriage: an economic evaluation based on the MifeMiso trial. BJOG. 2021;128: 1534-1545. doi: 10.1111/1471-0528.16737.
- Tang OS, Schweer H, Seyberth HW, et al. Pharmacokinetics of different routes of administration of misoprostol. Hum Reprod. 2002;17:332336. doi: 10.1093/humrep/17.2.332.
- Schaff EA, DiCenzo R, Fielding SL. Comparison of misoprostol plasma concentrations following buccal and sublingual administration. Contraception. 2005;71:22-25. doi: 10.1016 /j.contraception.2004.06.014.
- Meckstroth KR, Whitaker AK, Bertisch S, et al. Misoprostol administered by epithelial routes: drug absorption and uterine response. Obstet Gynecol. 2006;108:582-590. doi: 10.1097/01 .AOG.0000230398.32794.9d.
- Allanson ER, Copson S, Spilsbury K, et al. Pretreatment with mifepristone compared with misoprostol alone for delivery after fetal death between 14 and 28 weeks of gestation. Obstet Gynecol. 2021;137:801-809. doi: 10.1097 /AOG.0000000000004344.
- Chaudhuri P, Datta S. Mifepristone and misoprostol compared with misoprostol alone for induction of labor in intrauterine fetal death: a randomized trial. J Obstet Gynaecol Res. 2015;41:1884-1890. doi: 10.1111/jog.12815.
- Fyfe R, Murray H. Comparison of induction of labour regimens for termination of pregnancy with and without mifepristone, from 20 to 41 weeks gestation. Aust N Z J Obstet Gynaecol. 2017;57:604-608. doi: 10.1111 /ajo.12648.
- Panda S, Jha V, Singh S. Role of combination of mifepristone and misoprostol verses misoprostol alone in induction of labour in late intrauterine fetal death: a prospective study. J Family Reprod Health. 2013;7:177-179.
- Vayrynen W, Heikinheimo O, Nuutila M. Misoprostol-only versus mifepristone plus misoprostol in induction of labor following intrauterine fetal death. Acta Obstet Gynecol Scand. 2007;86: 701-705. doi: 10.1080/00016340701379853.
- Sharma D, Singhal SR, Poonam AP. Comparison of mifepristone combination with misoprostol and misoprostol alone in the management of intrauterine death. Taiwan J Obstet Gynecol. 2011;50:322-325. doi: 10.1016/j.tjog.2011.07.007.
- Stibbe KJM, de Weerd S. Induction of delivery by mifepristone and misoprostol in termination of pregnancy and intrauterine fetal death: 2nd and 3rd trimester induction of labour. Arch Gynecol Obstet. 2012;286:795-796. doi: 10.1007 /s00404-012-2289-3.
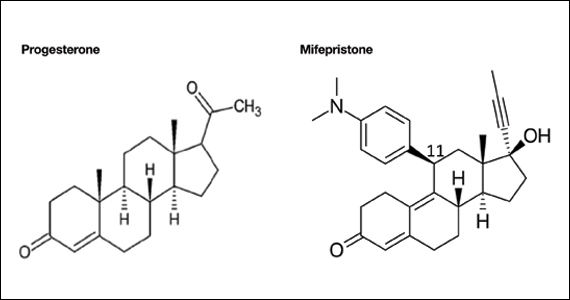
Reversing depression: A plethora of therapeutic strategies and mechanisms
Despite much progress, major depressive disorder (MDD) continues to be a challenging and life-threatening neuropsychiatric disorder. It is highly prevalent and afflicts tens of millions of Americans.
It is also ranked as the No. 1 disabling medical (not just psychiatric) condition by the World Health Organization.1 A significant proportion of patients with MDD do not respond adequately to several rounds of antidepressant medications,2 and many are labeled as having “treatment-resistant depression” (TRD).
In a previous article, I provocatively proposed that TRD is a myth.3 What I meant is that in a heterogeneous syndrome such as depression, failure to respond to 1, 2, or even 3 antidepressants should not imply TRD, because there is a “right treatment” that has not yet been identified for a given depressed patient. Most of those labeled as TRD have simply not yet received the pharmacotherapy or somatic therapy with the requisite mechanism of action for their variant of depression within a heterogeneous syndrome. IV ketamine, which, astonishingly, often reverses severe TRD of chronic duration within a few hours, is a prime example of why the term TRD is often used prematurely. Ketamine’s mechanism of action (immediate neuroplasticity via glutamate N-methyl-
Some clinicians may not be aware of the abundance of mechanisms of action currently available for the treatment of MDD as well as bipolar depression. Many practitioners, in both psychiatry and primary care, usually start the treatment of depression with a selective serotonin reuptake inhibitor, and if that does not produce a response or remission, they might switch to a serotonin-norepinephrine reuptake inhibitor. If that does not control the patient’s depressive symptoms, they start entertaining the notion that the patient may have TRD, not realizing that they have barely scratched the surface of the many therapeutic options and mechanisms of action, one of which could be the “best match” for a given patient.4
There will come a day when “precision psychiatry” finally arrives, and specific biomarkers will be developed to identify the “right” treatment for each patient within the heterogenous syndrome of depression.5 Until that day arrives, the treatment of depression will continue to be a process of trial and error, and hit or miss. But research will eventually discover genetic, neurochemical, neurophysiological, neuroimaging, or neuroimmune biomarkers that will rapidly guide clinicians to the correct treatment. This is critical to avoid inordinate delays in achieving remission and avert the ever-present risk of suicidal behavior.
The Table6 provides an overview of the numerous treatments currently available to manage depression. All increase brain-derived neurotrophic factor and restore healthy neuroplasticity and neurogenesis, which are impaired in MDD and currently believed to be a final common pathway for all depression treatments.7

These 41 therapeutic approaches to treating MDD or bipolar depression reflect the heterogeneity of mechanisms of action to address an equally heterogeneous syndrome. This implies that clinicians have a wide array of on-label options to manage patients with depression, aiming for remission, not just a good response, which typically is defined as a ≥50% reduction in total score on one of the validated rating scales used to quantify depression severity, such as the Montgomery-Åsberg Depression Rating Scale, Hamilton Depression Rating Scale, or Calgary Depression Scale for Schizophrenia.
Continue to: When several FDA-approved pharmacotherapies...
When several FDA-approved pharmacotherapies fall short and produce a suboptimal response, clinicians can resort to other treatment options known to have a higher efficacy than oral antidepressants. These include electroconvulsive therapy, repetitive transcranial magnetic stimulation, and vagus nerve stimulation. Other on-label options include adjunctive therapy with one of the approved second-generation antipsychotic agents or with adjunctive esketamine.
But if the patient still does not improve, one of many emerging off-label treatment options may work. One of the exciting new discoveries is the hallucinogen psilocybin, whose mechanism of action is truly unique. Unlike standard antidepressant medications, which modulate neurotransmitters, psilocybin increases the brain’s network flexibility, decreases the modularity of several key brain networks (especially the default-brain network, or DMN), and alters the dark and distorted mental perspective of depression to a much healthier and optimistic outlook about the self and the world.8 Such novel breakthroughs in the treatment of severe depression will shed some unprecedented insights into the core neurobiology of depression, and may lead to early intervention and prevention.
As the saying goes, all roads lead to Rome. Psychiatric clinicians should rejoice that there are abundant approaches and therapeutic mechanisms to relieve their severely melancholic (and often suicidal) patients from the grips of this disabling and life-altering brain syndrome.
1. World Health Organization. Depression: let’s talk says WHO, as depression tops list of causes of ill health. March 30, 2017. Accessed July 5, 2022. www.who.int/news/item/30-03-2017--depression-let-s-talk-says-who-as-depression-tops-list-of-causes-of-ill-health
2. Trivedi MH, Fava M, Wisniewski SR, et al. Medication augmentation after the failure of SSRIs for depression. N Eng J Med. 2006;354(12)1243-1252.
3. Nasrallah HA. Treatment resistance is a myth! Current Psychiatry. 2021;20(3):14-16,28.
4. Nasrallah HA. 10 Recent paradigm shifts in the neurobiology and treatment of depression. Current Psychiatry. 2015;14(2):10-13.
5. Nasrallah HA. Biomarkers in neuropsychiatric disorders: translating research to clinical applications. Biomarkers in Neuropsychiatry. 2019;1:100001. doi:10.1016/j.bionps.2019.100001
6. Procyshyn RM, Bezchlibnyk-Butler KZ, Jeffries JJ. Clinical Handbook of Psychotropic Drugs. 23rd ed. Hogrefe; 2019.
7. Tartt AN, Mariani, MB, Hen R, et al. Dysregulation of adult hippocampal neuroplasticity in major depression: pathogenesis and therapeutic implications. Mol Psychiatry. 2022;27(6):2689-2699.
8. Lowe H, Toyang N, Steele B, et al. The therapeutic potential of psilocybin. Molecules. 2021;26(10):2948. doi: 10.3390/molecules26102948
Despite much progress, major depressive disorder (MDD) continues to be a challenging and life-threatening neuropsychiatric disorder. It is highly prevalent and afflicts tens of millions of Americans.
It is also ranked as the No. 1 disabling medical (not just psychiatric) condition by the World Health Organization.1 A significant proportion of patients with MDD do not respond adequately to several rounds of antidepressant medications,2 and many are labeled as having “treatment-resistant depression” (TRD).
In a previous article, I provocatively proposed that TRD is a myth.3 What I meant is that in a heterogeneous syndrome such as depression, failure to respond to 1, 2, or even 3 antidepressants should not imply TRD, because there is a “right treatment” that has not yet been identified for a given depressed patient. Most of those labeled as TRD have simply not yet received the pharmacotherapy or somatic therapy with the requisite mechanism of action for their variant of depression within a heterogeneous syndrome. IV ketamine, which, astonishingly, often reverses severe TRD of chronic duration within a few hours, is a prime example of why the term TRD is often used prematurely. Ketamine’s mechanism of action (immediate neuroplasticity via glutamate N-methyl-
Some clinicians may not be aware of the abundance of mechanisms of action currently available for the treatment of MDD as well as bipolar depression. Many practitioners, in both psychiatry and primary care, usually start the treatment of depression with a selective serotonin reuptake inhibitor, and if that does not produce a response or remission, they might switch to a serotonin-norepinephrine reuptake inhibitor. If that does not control the patient’s depressive symptoms, they start entertaining the notion that the patient may have TRD, not realizing that they have barely scratched the surface of the many therapeutic options and mechanisms of action, one of which could be the “best match” for a given patient.4
There will come a day when “precision psychiatry” finally arrives, and specific biomarkers will be developed to identify the “right” treatment for each patient within the heterogenous syndrome of depression.5 Until that day arrives, the treatment of depression will continue to be a process of trial and error, and hit or miss. But research will eventually discover genetic, neurochemical, neurophysiological, neuroimaging, or neuroimmune biomarkers that will rapidly guide clinicians to the correct treatment. This is critical to avoid inordinate delays in achieving remission and avert the ever-present risk of suicidal behavior.
The Table6 provides an overview of the numerous treatments currently available to manage depression. All increase brain-derived neurotrophic factor and restore healthy neuroplasticity and neurogenesis, which are impaired in MDD and currently believed to be a final common pathway for all depression treatments.7
These 41 therapeutic approaches to treating MDD or bipolar depression reflect the heterogeneity of mechanisms of action to address an equally heterogeneous syndrome. This implies that clinicians have a wide array of on-label options to manage patients with depression, aiming for remission, not just a good response, which typically is defined as a ≥50% reduction in total score on one of the validated rating scales used to quantify depression severity, such as the Montgomery-Åsberg Depression Rating Scale, Hamilton Depression Rating Scale, or Calgary Depression Scale for Schizophrenia.
Continue to: When several FDA-approved pharmacotherapies...
When several FDA-approved pharmacotherapies fall short and produce a suboptimal response, clinicians can resort to other treatment options known to have a higher efficacy than oral antidepressants. These include electroconvulsive therapy, repetitive transcranial magnetic stimulation, and vagus nerve stimulation. Other on-label options include adjunctive therapy with one of the approved second-generation antipsychotic agents or with adjunctive esketamine.
But if the patient still does not improve, one of many emerging off-label treatment options may work. One of the exciting new discoveries is the hallucinogen psilocybin, whose mechanism of action is truly unique. Unlike standard antidepressant medications, which modulate neurotransmitters, psilocybin increases the brain’s network flexibility, decreases the modularity of several key brain networks (especially the default-brain network, or DMN), and alters the dark and distorted mental perspective of depression to a much healthier and optimistic outlook about the self and the world.8 Such novel breakthroughs in the treatment of severe depression will shed some unprecedented insights into the core neurobiology of depression, and may lead to early intervention and prevention.
As the saying goes, all roads lead to Rome. Psychiatric clinicians should rejoice that there are abundant approaches and therapeutic mechanisms to relieve their severely melancholic (and often suicidal) patients from the grips of this disabling and life-altering brain syndrome.
Despite much progress, major depressive disorder (MDD) continues to be a challenging and life-threatening neuropsychiatric disorder. It is highly prevalent and afflicts tens of millions of Americans.
It is also ranked as the No. 1 disabling medical (not just psychiatric) condition by the World Health Organization.1 A significant proportion of patients with MDD do not respond adequately to several rounds of antidepressant medications,2 and many are labeled as having “treatment-resistant depression” (TRD).
In a previous article, I provocatively proposed that TRD is a myth.3 What I meant is that in a heterogeneous syndrome such as depression, failure to respond to 1, 2, or even 3 antidepressants should not imply TRD, because there is a “right treatment” that has not yet been identified for a given depressed patient. Most of those labeled as TRD have simply not yet received the pharmacotherapy or somatic therapy with the requisite mechanism of action for their variant of depression within a heterogeneous syndrome. IV ketamine, which, astonishingly, often reverses severe TRD of chronic duration within a few hours, is a prime example of why the term TRD is often used prematurely. Ketamine’s mechanism of action (immediate neuroplasticity via glutamate N-methyl-
Some clinicians may not be aware of the abundance of mechanisms of action currently available for the treatment of MDD as well as bipolar depression. Many practitioners, in both psychiatry and primary care, usually start the treatment of depression with a selective serotonin reuptake inhibitor, and if that does not produce a response or remission, they might switch to a serotonin-norepinephrine reuptake inhibitor. If that does not control the patient’s depressive symptoms, they start entertaining the notion that the patient may have TRD, not realizing that they have barely scratched the surface of the many therapeutic options and mechanisms of action, one of which could be the “best match” for a given patient.4
There will come a day when “precision psychiatry” finally arrives, and specific biomarkers will be developed to identify the “right” treatment for each patient within the heterogenous syndrome of depression.5 Until that day arrives, the treatment of depression will continue to be a process of trial and error, and hit or miss. But research will eventually discover genetic, neurochemical, neurophysiological, neuroimaging, or neuroimmune biomarkers that will rapidly guide clinicians to the correct treatment. This is critical to avoid inordinate delays in achieving remission and avert the ever-present risk of suicidal behavior.
The Table6 provides an overview of the numerous treatments currently available to manage depression. All increase brain-derived neurotrophic factor and restore healthy neuroplasticity and neurogenesis, which are impaired in MDD and currently believed to be a final common pathway for all depression treatments.7
These 41 therapeutic approaches to treating MDD or bipolar depression reflect the heterogeneity of mechanisms of action to address an equally heterogeneous syndrome. This implies that clinicians have a wide array of on-label options to manage patients with depression, aiming for remission, not just a good response, which typically is defined as a ≥50% reduction in total score on one of the validated rating scales used to quantify depression severity, such as the Montgomery-Åsberg Depression Rating Scale, Hamilton Depression Rating Scale, or Calgary Depression Scale for Schizophrenia.
Continue to: When several FDA-approved pharmacotherapies...
When several FDA-approved pharmacotherapies fall short and produce a suboptimal response, clinicians can resort to other treatment options known to have a higher efficacy than oral antidepressants. These include electroconvulsive therapy, repetitive transcranial magnetic stimulation, and vagus nerve stimulation. Other on-label options include adjunctive therapy with one of the approved second-generation antipsychotic agents or with adjunctive esketamine.
But if the patient still does not improve, one of many emerging off-label treatment options may work. One of the exciting new discoveries is the hallucinogen psilocybin, whose mechanism of action is truly unique. Unlike standard antidepressant medications, which modulate neurotransmitters, psilocybin increases the brain’s network flexibility, decreases the modularity of several key brain networks (especially the default-brain network, or DMN), and alters the dark and distorted mental perspective of depression to a much healthier and optimistic outlook about the self and the world.8 Such novel breakthroughs in the treatment of severe depression will shed some unprecedented insights into the core neurobiology of depression, and may lead to early intervention and prevention.
As the saying goes, all roads lead to Rome. Psychiatric clinicians should rejoice that there are abundant approaches and therapeutic mechanisms to relieve their severely melancholic (and often suicidal) patients from the grips of this disabling and life-altering brain syndrome.
1. World Health Organization. Depression: let’s talk says WHO, as depression tops list of causes of ill health. March 30, 2017. Accessed July 5, 2022. www.who.int/news/item/30-03-2017--depression-let-s-talk-says-who-as-depression-tops-list-of-causes-of-ill-health
2. Trivedi MH, Fava M, Wisniewski SR, et al. Medication augmentation after the failure of SSRIs for depression. N Eng J Med. 2006;354(12)1243-1252.
3. Nasrallah HA. Treatment resistance is a myth! Current Psychiatry. 2021;20(3):14-16,28.
4. Nasrallah HA. 10 Recent paradigm shifts in the neurobiology and treatment of depression. Current Psychiatry. 2015;14(2):10-13.
5. Nasrallah HA. Biomarkers in neuropsychiatric disorders: translating research to clinical applications. Biomarkers in Neuropsychiatry. 2019;1:100001. doi:10.1016/j.bionps.2019.100001
6. Procyshyn RM, Bezchlibnyk-Butler KZ, Jeffries JJ. Clinical Handbook of Psychotropic Drugs. 23rd ed. Hogrefe; 2019.
7. Tartt AN, Mariani, MB, Hen R, et al. Dysregulation of adult hippocampal neuroplasticity in major depression: pathogenesis and therapeutic implications. Mol Psychiatry. 2022;27(6):2689-2699.
8. Lowe H, Toyang N, Steele B, et al. The therapeutic potential of psilocybin. Molecules. 2021;26(10):2948. doi: 10.3390/molecules26102948
1. World Health Organization. Depression: let’s talk says WHO, as depression tops list of causes of ill health. March 30, 2017. Accessed July 5, 2022. www.who.int/news/item/30-03-2017--depression-let-s-talk-says-who-as-depression-tops-list-of-causes-of-ill-health
2. Trivedi MH, Fava M, Wisniewski SR, et al. Medication augmentation after the failure of SSRIs for depression. N Eng J Med. 2006;354(12)1243-1252.
3. Nasrallah HA. Treatment resistance is a myth! Current Psychiatry. 2021;20(3):14-16,28.
4. Nasrallah HA. 10 Recent paradigm shifts in the neurobiology and treatment of depression. Current Psychiatry. 2015;14(2):10-13.
5. Nasrallah HA. Biomarkers in neuropsychiatric disorders: translating research to clinical applications. Biomarkers in Neuropsychiatry. 2019;1:100001. doi:10.1016/j.bionps.2019.100001
6. Procyshyn RM, Bezchlibnyk-Butler KZ, Jeffries JJ. Clinical Handbook of Psychotropic Drugs. 23rd ed. Hogrefe; 2019.
7. Tartt AN, Mariani, MB, Hen R, et al. Dysregulation of adult hippocampal neuroplasticity in major depression: pathogenesis and therapeutic implications. Mol Psychiatry. 2022;27(6):2689-2699.
8. Lowe H, Toyang N, Steele B, et al. The therapeutic potential of psilocybin. Molecules. 2021;26(10):2948. doi: 10.3390/molecules26102948
Misoprostol: Clinical pharmacology in obstetrics and gynecology
Oxytocin and prostaglandins are critically important regulators of uterine contraction. Obstetrician-gynecologists commonly prescribe oxytocin and prostaglandin agonists (misoprostol, dinoprostone) to stimulate uterine contraction for the induction of labor, prevention and treatment of postpartum hemorrhage, and treatment of miscarriage and fetal demise. The focus of this editorial is the clinical pharmacology of misoprostol.
Misoprostol is approved by the US Food and Drug Administration (FDA) for the prevention and treatment of nonsteroidal anti-inflammatory drug–induced gastric ulcers and for patients at high risk for gastric ulcers, including those with a history of gastric ulcers. The approved misoprostol route and dose for this indication is oral administration of 200 µg four times daily with food.1 Recent food intake and antacid use reduces the absorption of orally administered misoprostol. There are no FDA-approved indications for the use of misoprostol as a single agent in obstetrics and gynecology. The FDA has approved the combination of mifepristone and misoprostol for medication abortion in the first trimester. In contrast to misoprostol, PGE2 (dinoprostone) is approved by the FDA as a vaginal insert containing 10 mg of dinoprostone for the initiation and/or continuation of cervical ripening in patients at or near term in whom there is a medical or obstetric indication for induction of labor (Cervidil; Ferring Pharmaceuticals Inc, Parsippany, New Jersey).2
Pharmacology of misoprostol
Misoprostol is a prostaglandin E1 (PGE1) agonist analogue. Prostaglandin E1 (alprostadil) is rapidly metabolized, has a half-life in the range of minutes and is not orally active, requiring administration by intravenous infusion or injection. It is indicated to maintain a patent ductus arteriosus in newborns with ductal-dependent circulation and to treat erectile dysfunction.3 In contrast to PGE1, misoprostol has a methyl ester group at carbon-1 (C-1) that increases potency and duration of action. Misoprostol also has no hydroxyl group at C-15, replacing that moiety with the addition of both a methyl- and hydroxyl- group at C-16 (FIGURE). These molecular changes improve oral activity and increase duration of action.4 Pure misoprostol is a viscous oil. It is formulated into tables by dispersing the oil on hydroxypropyl methyl cellulose before compounding into tablets. Unlike naturally occurring prostaglandins (PGE1), misoprostol tablets are stabile at room temperature for years.4
Following absorption, the methyl ester at C-1 is enzymatically cleaved, yielding misoprostol acid, the active drug.4 Misoprostol binds to the E prostanoid receptor 3 (EP-3).5 Activation of myometrial EP-3 receptor induces an increase in intracellular phosphoinositol turnover and calcium mobilization, resulting in an increase in intracellular-free calcium, triggering actin-myosin contractility.6 The increase in free calcium is propagated cell-to-cell through gap junctions that link the myometrial cells to facilitate the generation of a coordinated contraction.
Misoprostol: Various routes of administration are not equal
Misoprostol can be given by an oral, buccal, vaginal, or rectal route of administration. To study the effect of the route of administration on uterine tone and contractility, investigators randomly assigned patients at 8 to 11 weeks’ gestation to receive misoprostol 400 µg as a single dose by the oral or vaginal route. Uterine tone and contractility were measured using an intrauterine pressure transducer. Compared to vaginal administration, oral administration of misprostol was associated with rapid attainment of peak plasma level at 30 minutes, followed by a decline in concentration by 60 minutes. This rapid onset and rapid offset of plasma concentration was paralleled by the onset of uterine tone within 8 minutes, but surprisingly no sustained uterine contractions.7 By contrast, following vaginal administration of misoprostol, serum levels rose slowly and peaked in 1 to 2 hours. Uterine tone increased within 21 minutes, and sustained uterine contractions were recorded for 4 hours.7 The rapid rise and fall in plasma misoprostol following oral administration and the more sustained plasma misoprostol concentration over 4 hours has been previously reported.8 In a second study involving patients 8 to 11 weeks’ gestation, the effect of a single dose of misoprostol 400 µg by an oral or vaginal route on uterine contractility was compared using an intrauterine pressure transducer.9 Confirming previous results, the time from misoprostol administration to increased uterine tone was more rapid with oral than with vaginal administration (8 min vs 19 min). Over the course of 4 hours, uterine contraction activity was greater with vaginal than with oral administration (454 vs 166 Montevideo units).9
Both studies reported that oral administration of misoprostol resulted in more rapid onset and offset of action than vaginal administration. Oral administration of a single dose of misoprostol 400 µg did not result in sustained uterine contractions in most patients in the first trimester. Vaginal administration produced a slower onset of increased uterine tone but sustained uterine contractions over 4 hours. Compared with vaginal administration of misoprostol, the rapid onset and offset of action of oral misoprostol may reduce the rate of tachysystole and changes in fetal heart rate observed with vaginal administration.10
An important finding is that buccal and vaginal administration of misoprostol have similar effects on uterine tone in the first trimester.11 To study the effect of buccal and vaginal administration of misoprostol on uterine tone, patients 6 to 13 weeks’ gestation were randomly allocated to receive a single dose of misoprostol 400 µg by a buccal or vaginal route.11 Uterine activity over 5 hours following administration was assessed using an intrauterine pressure transducer. Uterine tone 20 to 30 minutes after buccal or vaginal administration of misoprostol (400 µg) was 27 and 28 mm Hg, respectively. Peak uterine tone, as measured by an intrauterine pressure transducer, for buccal and vaginal administration of misoprostol was 49 mm Hg and 54 mm Hg, respectively. Total Alexandria units (AU) over 5 hours following buccal or vaginal administration was 6,537 AU and 6,090 AU, respectively.11
An AU is calculated as the average amplitude of the contractions (mm Hg) multiplied by the average duration of the contractions (min) multiplied by average frequency of contraction over 10 minutes.12 By contrast, a Montevideo unit does not include an assessment of contraction duration and is calculated as average amplitude of contractions (mm Hg) multiplied by frequency of uterine contractions over 10 minutes.12
In contrast to buccal or vaginal administration, rectal administration of misoprostol resulted in much lower peak uterine tone and contractility as measured by a pressure transducer. Uterine tone 20 to 30 minutes after vaginal and rectal administration of misoprostol (400 µg) was 28 and 19 mm Hg, respectively.11 Peak uterine tone, as measured by an intrauterine pressure transducer, for vaginal and rectal administration of misoprostol was 54 and 31 mm Hg, respectively. AUs over 5 hours following vaginal and rectal administration was 6,090 AU and 2,768 AU, respectively.11 Compared with buccal and vaginal administration of misoprostol, rectal administration produced less sustained uterine contractions in the first trimester of pregnancy. To achieve maximal sustained uterine contractions, buccal and vaginal routes of administration are superior to oral and rectal administration.
Continue to: Misoprostol and cervical ripening...
Misoprostol and cervical ripening
Misoprostol is commonly used to soften and ripen the cervix. Some of the cervical ripening effects of misoprostol are likely due to increased uterine tone. In addition, misoprostol may have a direct effect on the collagen structure of the cervix. To study the effect of misoprostol on the cervix, pregnant patients in the first trimester were randomly assigned to receive misoprostol 200 µg by vaginal self-administration, isosorbide mononitrate (IMN) 40 mg by vaginal self-administration or no treatment the evening prior to pregnancy termination.13 The following day, before uterine evacuation, a cervical biopsy was obtained for electron microscopy studies and immunohistochemistry to assess the presence of enzymes involved in collagen degradation, including matrix metalloproteinase 1 (MMP-1) and matrix metalloproteinase 9 (MMP-9). Electron microscopy demonstrated that pretreatment with misoprostol resulted in a pronounced splitting and disorganization of collagen fibers.13 Compared with misoprostol treatment, IMN produced less splitting and disorganization of collagen fibers, and in the no treatment group, no marked changes in the collagen framework were observed.
Compared with no treatment, misoprostol and IMN pretreatment were associated with marked increases in MMP-1 and MMP-9 as assessed by immunohistochemistry. Misoprostol pretreatment also resulted in a significant increase in interleukin-8 concentration compared with IMN pretreatment and no treatment (8.8 vs 2.7 vs 2.4 pg/mg tissue), respectively.13 Other investigators have also reported that misoprostol increased cervical leukocyte influx and collagen disrupting enzymes MMP-8 and MMP-9.14,15
An open-label clinical trial compared the efficacy of misoprostol versus Foley catheter for labor induction at term in 1,859 patients ≥ 37 weeks’ gestation with a Bishop score <6.16 Patients were randomly allocated to misoprostol (50 µg orally every 4 hours up to 3 times in 24 hours) versus placement of a 16 F or 18 F Foley catheter introduced through the cervix, filled with 30 mL of sodium chloride or water. The investigators reported that oral misoprostol and Foley catheter cervical ripening had similar safety and effectiveness for cervical ripening as a prelude to induction of labor, including no statistically significant differences in 5-minute Apgar score <7, umbilical cord artery pH ≤ 7.05, postpartum hemorrhage, or cesarean birth rate.16
Bottom line
Misoprostol and oxytocin are commonly prescribed in obstetric practice for cervical ripening and induction of labor, respectively. The dose and route of administration of misoprostol influences the effect on the uterus. For cervical ripening, where rapid onset and offset may help to reduce the risk of uterine tachysystole and worrisome fetal heart rate changes, low-dose (50 µg) oral administration of misoprostol may be a preferred dose and route. For the treatment of miscarriage and fetal demise, to stimulate sustained uterine contractions over many hours, buccal and vaginal administration of misoprostol are preferred. Rectal administration is generally inferior to buccal and vaginal administration for stimulating sustained uterine contractions and its uses should be limited. ●
Common side effects of misoprostol are abdominal cramping, diarrhea, nausea, vomiting, headache, and fever. Elevated temperature following misoprostol administration is a concerning side effect that may require further investigation to rule out an infection, especially if the elevated temperature persists for > 4 hours. The preoptic area of the anterior hypothalamus (POAH) plays a major role in thermoregulation. When an infection causes an increase in endogenous pyrogens, including interleukin-1β, interleukin-6 and tumor necrosis factor, prostaglandins are generated in the region of the POAH, increasing the thermoregulatory set point, triggering cutaneous vasoconstriction and shivering and non-shivering thermogenesis.1 Misoprostol, especially at doses >400 µg commonly causes both patient-reported chills and temperature elevation >38° C.
In a study comparing misoprostol and oxytocin for the management of the third stage of labor, 597 patients were randomly allocated to receive oxytocin 10 units by intramuscular injection or misoprostol 400 µg or 600 µg by the oral route.2 Patient-reported shivering occurred in 13%, 19%, and 28% of patients receiving oxytocin, misoprostol 400 µg and misoprostol 800 µg, respectively. A recorded temperature >38° C occurred within 1 hour of medication administration in approximately 3%, 2%, and 7.5% of patients receiving oxytocin, misoprostol 400 µg, and misoprostol 800 µg, respectively. In another study, 453 patients scheduled for a cesarean birth were randomly allocated to receive 1 of 3 doses of rectal misoprostol 200 μg, 400 μg, or 600 μg before incision. Fever was detected in 2.6%, 9.9%, and 5.1% of the patients receiving misoprostol 200 μg, 400 μg, or 600 μg, respectively.3
References
1. Aronoff DM, Neilson EG. Antipyretics: mechanisms of action and clinical use in fever suppression. Am J Med. 2001;111:304-315. doi: 10.1016/s0002-9343(01)00834-8.
2. Lumbiganon P, Hofmeyr J, Gumezoglu AM, et al. Misoprostol dose-related shivering and pyrexia in the third stage of labor. WHO Collaborative Trial of Misoprostol in the Management of the Third Stage of Labor. Br J Obstet Gynaecol. 1999;106:304-308. doi: 10.1111/j.1471-0528.1999.tb08266.x.
3. Sweed M, El-Said M, Abou-Gamrah AA, et al. Comparison between 200, 400 and 600 microgram rectal misoprostol before cesarean section: a randomized clinical trial. J Obstet Gynaecol Res. 2019;45:585-591. doi: 10.1111 /jog.13883.
- Cytotec [package insert]. Chicago, IL: GD Searle & Co. https://www.accessdata.fda.gov/drugsatfda_docs/label/2002/19268slr037.pdf. Accessed June 20, 2022.
- Cervidil [package insert]. St Louis, MO: Forrest Pharmaceuticals Inc.; May 2006. Accessed June 20, 2022.
- Caverject [package insert]. New York, NY: Pfizer Inc.; March 2014. Accessed June 20, 2022.
- Collins PW. Misoprostol: discovery, development and clinical applications. Med Res Rev. 1990;10:149-172. doi: 10.1002/med.2610100202.
- Audit M, White KI, Breton B, et al. Crystal structure of misoprostol bound to the labor inducer prostaglandin E2 receptor. Nat Chem Biol. 2019;15:11-17. doi: 10.1038/s41589-018-0160-y.
- Pallliser KH, Hirst JJ, Ooi G, et al. Prostaglandin E and F receptor expression and myometrial sensitivity in labor onset in the sheep. Biol Reprod. 2005;72:937-943. doi: 10.1095/biolreprod.104.035311.
- Gemzell-Danilesson K, Marions L, Rodriguez A, et al. Comparison between oral and vaginal administration of misoprostol on uterine contractility. Obstet Gynecol. 1999;93:275-280. doi: 10.1016/s0029-7844(98)00436-0.
- Zieman M, Fong SK, Benowitz NL, et al. Absorption kinetics of misoprostol with oral or vaginal administration. Obstet Gynecol. 1997;90:88-92. doi: 10.1016/S0029-7844(97)00111-7.
- Aronsson A, Bygdeman M, Gemzell-Danielsson K. Effects of misoprostol on uterine contractility following different routes of administration. Hum Reprod. 2004;19:81-84. doi: 10.1093/humrep/deh005.
- Young DC, Delaney T, Armson BA, et al. Oral misoprostol, low dose vaginal misoprostol and vaginal dinoprostone for labor induction: randomized controlled trial. PLOS One. 2020;15:e0227245. doi: 10.1371/journal.pone.0227245.
- Meckstroth KR, Whitaker AK, Bertisch S, et al. Misoprostol administered by epithelial routes. Drug absorption and uterine response. Obstet Gynecol. 2006;108:582-590. doi: 10.1097/01.AOG.0000230398.32794.9d.
- el-Sahwi S, Gaafar AA, Toppozada HK. A new unit for evaluation of uterine activity. Am J Obstet Gynecol. 1967;98:900-903. doi: 10.1016/0002-9378(67)90074-9.
- Vukas N, Ekerhovd E, Abrahamsson G, et al. Cervical priming in the first trimester: morphological and biochemical effects of misoprostol and isosorbide mononitrate. Acta Obstet Gyecol. 2009;88:43-51. doi: 10.1080/00016340802585440.
- Aronsson A, Ulfgren AK, Stabi B, et al. The effect of orally and vaginally administered misoprostol on inflammatory mediators and cervical ripening during early pregnancy. Contraception. 2005;72:33-39. doi: 10.1016/j.contraception.2005.02.012.
- Denison FC, Riley SC, Elliott CL, et al. The effect of mifepristone administration on leukocyte populations, matrix metalloproteinases and inflammatory mediators in the first trimester cervix. Mol Hum Reprod. 2000;6:541-548. doi: 10.1093/molehr/6.6.541.
- ten Eikelder MLG, Rengerink KO, Jozwiak M, et al. Induction of labour at term with oral misoprostol versus a Foley catheter (PROBAAT-II): a multicentre randomised controlled non-inferiority trial. Lancet. 2016;387:1619-1628. doi: 10.1016 /S0140-6736(16)00084-2.

Robert L. Barbieri, MD
Editor in Chief, OBG Management
Chair Emeritus, Department of Obstetrics and Gynecology
Brigham and Women’s Hospital
Kate Macy Ladd Distinguished Professor of Obstetrics,
Gynecology and Reproductive Biology
Harvard Medical School
Boston, Massachusetts
Dr. Barbieri reports no financial relationships relevant to this article.
Robert L. Barbieri, MD
Editor in Chief, OBG Management
Chair Emeritus, Department of Obstetrics and Gynecology
Brigham and Women’s Hospital
Kate Macy Ladd Distinguished Professor of Obstetrics,
Gynecology and Reproductive Biology
Harvard Medical School
Boston, Massachusetts
Dr. Barbieri reports no financial relationships relevant to this article.
Robert L. Barbieri, MD
Editor in Chief, OBG Management
Chair Emeritus, Department of Obstetrics and Gynecology
Brigham and Women’s Hospital
Kate Macy Ladd Distinguished Professor of Obstetrics,
Gynecology and Reproductive Biology
Harvard Medical School
Boston, Massachusetts
Dr. Barbieri reports no financial relationships relevant to this article.
Oxytocin and prostaglandins are critically important regulators of uterine contraction. Obstetrician-gynecologists commonly prescribe oxytocin and prostaglandin agonists (misoprostol, dinoprostone) to stimulate uterine contraction for the induction of labor, prevention and treatment of postpartum hemorrhage, and treatment of miscarriage and fetal demise. The focus of this editorial is the clinical pharmacology of misoprostol.
Misoprostol is approved by the US Food and Drug Administration (FDA) for the prevention and treatment of nonsteroidal anti-inflammatory drug–induced gastric ulcers and for patients at high risk for gastric ulcers, including those with a history of gastric ulcers. The approved misoprostol route and dose for this indication is oral administration of 200 µg four times daily with food.1 Recent food intake and antacid use reduces the absorption of orally administered misoprostol. There are no FDA-approved indications for the use of misoprostol as a single agent in obstetrics and gynecology. The FDA has approved the combination of mifepristone and misoprostol for medication abortion in the first trimester. In contrast to misoprostol, PGE2 (dinoprostone) is approved by the FDA as a vaginal insert containing 10 mg of dinoprostone for the initiation and/or continuation of cervical ripening in patients at or near term in whom there is a medical or obstetric indication for induction of labor (Cervidil; Ferring Pharmaceuticals Inc, Parsippany, New Jersey).2
Pharmacology of misoprostol
Misoprostol is a prostaglandin E1 (PGE1) agonist analogue. Prostaglandin E1 (alprostadil) is rapidly metabolized, has a half-life in the range of minutes and is not orally active, requiring administration by intravenous infusion or injection. It is indicated to maintain a patent ductus arteriosus in newborns with ductal-dependent circulation and to treat erectile dysfunction.3 In contrast to PGE1, misoprostol has a methyl ester group at carbon-1 (C-1) that increases potency and duration of action. Misoprostol also has no hydroxyl group at C-15, replacing that moiety with the addition of both a methyl- and hydroxyl- group at C-16 (FIGURE). These molecular changes improve oral activity and increase duration of action.4 Pure misoprostol is a viscous oil. It is formulated into tables by dispersing the oil on hydroxypropyl methyl cellulose before compounding into tablets. Unlike naturally occurring prostaglandins (PGE1), misoprostol tablets are stabile at room temperature for years.4
Following absorption, the methyl ester at C-1 is enzymatically cleaved, yielding misoprostol acid, the active drug.4 Misoprostol binds to the E prostanoid receptor 3 (EP-3).5 Activation of myometrial EP-3 receptor induces an increase in intracellular phosphoinositol turnover and calcium mobilization, resulting in an increase in intracellular-free calcium, triggering actin-myosin contractility.6 The increase in free calcium is propagated cell-to-cell through gap junctions that link the myometrial cells to facilitate the generation of a coordinated contraction.
Misoprostol: Various routes of administration are not equal
Misoprostol can be given by an oral, buccal, vaginal, or rectal route of administration. To study the effect of the route of administration on uterine tone and contractility, investigators randomly assigned patients at 8 to 11 weeks’ gestation to receive misoprostol 400 µg as a single dose by the oral or vaginal route. Uterine tone and contractility were measured using an intrauterine pressure transducer. Compared to vaginal administration, oral administration of misprostol was associated with rapid attainment of peak plasma level at 30 minutes, followed by a decline in concentration by 60 minutes. This rapid onset and rapid offset of plasma concentration was paralleled by the onset of uterine tone within 8 minutes, but surprisingly no sustained uterine contractions.7 By contrast, following vaginal administration of misoprostol, serum levels rose slowly and peaked in 1 to 2 hours. Uterine tone increased within 21 minutes, and sustained uterine contractions were recorded for 4 hours.7 The rapid rise and fall in plasma misoprostol following oral administration and the more sustained plasma misoprostol concentration over 4 hours has been previously reported.8 In a second study involving patients 8 to 11 weeks’ gestation, the effect of a single dose of misoprostol 400 µg by an oral or vaginal route on uterine contractility was compared using an intrauterine pressure transducer.9 Confirming previous results, the time from misoprostol administration to increased uterine tone was more rapid with oral than with vaginal administration (8 min vs 19 min). Over the course of 4 hours, uterine contraction activity was greater with vaginal than with oral administration (454 vs 166 Montevideo units).9
Both studies reported that oral administration of misoprostol resulted in more rapid onset and offset of action than vaginal administration. Oral administration of a single dose of misoprostol 400 µg did not result in sustained uterine contractions in most patients in the first trimester. Vaginal administration produced a slower onset of increased uterine tone but sustained uterine contractions over 4 hours. Compared with vaginal administration of misoprostol, the rapid onset and offset of action of oral misoprostol may reduce the rate of tachysystole and changes in fetal heart rate observed with vaginal administration.10
An important finding is that buccal and vaginal administration of misoprostol have similar effects on uterine tone in the first trimester.11 To study the effect of buccal and vaginal administration of misoprostol on uterine tone, patients 6 to 13 weeks’ gestation were randomly allocated to receive a single dose of misoprostol 400 µg by a buccal or vaginal route.11 Uterine activity over 5 hours following administration was assessed using an intrauterine pressure transducer. Uterine tone 20 to 30 minutes after buccal or vaginal administration of misoprostol (400 µg) was 27 and 28 mm Hg, respectively. Peak uterine tone, as measured by an intrauterine pressure transducer, for buccal and vaginal administration of misoprostol was 49 mm Hg and 54 mm Hg, respectively. Total Alexandria units (AU) over 5 hours following buccal or vaginal administration was 6,537 AU and 6,090 AU, respectively.11
An AU is calculated as the average amplitude of the contractions (mm Hg) multiplied by the average duration of the contractions (min) multiplied by average frequency of contraction over 10 minutes.12 By contrast, a Montevideo unit does not include an assessment of contraction duration and is calculated as average amplitude of contractions (mm Hg) multiplied by frequency of uterine contractions over 10 minutes.12
In contrast to buccal or vaginal administration, rectal administration of misoprostol resulted in much lower peak uterine tone and contractility as measured by a pressure transducer. Uterine tone 20 to 30 minutes after vaginal and rectal administration of misoprostol (400 µg) was 28 and 19 mm Hg, respectively.11 Peak uterine tone, as measured by an intrauterine pressure transducer, for vaginal and rectal administration of misoprostol was 54 and 31 mm Hg, respectively. AUs over 5 hours following vaginal and rectal administration was 6,090 AU and 2,768 AU, respectively.11 Compared with buccal and vaginal administration of misoprostol, rectal administration produced less sustained uterine contractions in the first trimester of pregnancy. To achieve maximal sustained uterine contractions, buccal and vaginal routes of administration are superior to oral and rectal administration.
Continue to: Misoprostol and cervical ripening...
Misoprostol and cervical ripening
Misoprostol is commonly used to soften and ripen the cervix. Some of the cervical ripening effects of misoprostol are likely due to increased uterine tone. In addition, misoprostol may have a direct effect on the collagen structure of the cervix. To study the effect of misoprostol on the cervix, pregnant patients in the first trimester were randomly assigned to receive misoprostol 200 µg by vaginal self-administration, isosorbide mononitrate (IMN) 40 mg by vaginal self-administration or no treatment the evening prior to pregnancy termination.13 The following day, before uterine evacuation, a cervical biopsy was obtained for electron microscopy studies and immunohistochemistry to assess the presence of enzymes involved in collagen degradation, including matrix metalloproteinase 1 (MMP-1) and matrix metalloproteinase 9 (MMP-9). Electron microscopy demonstrated that pretreatment with misoprostol resulted in a pronounced splitting and disorganization of collagen fibers.13 Compared with misoprostol treatment, IMN produced less splitting and disorganization of collagen fibers, and in the no treatment group, no marked changes in the collagen framework were observed.
Compared with no treatment, misoprostol and IMN pretreatment were associated with marked increases in MMP-1 and MMP-9 as assessed by immunohistochemistry. Misoprostol pretreatment also resulted in a significant increase in interleukin-8 concentration compared with IMN pretreatment and no treatment (8.8 vs 2.7 vs 2.4 pg/mg tissue), respectively.13 Other investigators have also reported that misoprostol increased cervical leukocyte influx and collagen disrupting enzymes MMP-8 and MMP-9.14,15
An open-label clinical trial compared the efficacy of misoprostol versus Foley catheter for labor induction at term in 1,859 patients ≥ 37 weeks’ gestation with a Bishop score <6.16 Patients were randomly allocated to misoprostol (50 µg orally every 4 hours up to 3 times in 24 hours) versus placement of a 16 F or 18 F Foley catheter introduced through the cervix, filled with 30 mL of sodium chloride or water. The investigators reported that oral misoprostol and Foley catheter cervical ripening had similar safety and effectiveness for cervical ripening as a prelude to induction of labor, including no statistically significant differences in 5-minute Apgar score <7, umbilical cord artery pH ≤ 7.05, postpartum hemorrhage, or cesarean birth rate.16
Bottom line
Misoprostol and oxytocin are commonly prescribed in obstetric practice for cervical ripening and induction of labor, respectively. The dose and route of administration of misoprostol influences the effect on the uterus. For cervical ripening, where rapid onset and offset may help to reduce the risk of uterine tachysystole and worrisome fetal heart rate changes, low-dose (50 µg) oral administration of misoprostol may be a preferred dose and route. For the treatment of miscarriage and fetal demise, to stimulate sustained uterine contractions over many hours, buccal and vaginal administration of misoprostol are preferred. Rectal administration is generally inferior to buccal and vaginal administration for stimulating sustained uterine contractions and its uses should be limited. ●
Common side effects of misoprostol are abdominal cramping, diarrhea, nausea, vomiting, headache, and fever. Elevated temperature following misoprostol administration is a concerning side effect that may require further investigation to rule out an infection, especially if the elevated temperature persists for > 4 hours. The preoptic area of the anterior hypothalamus (POAH) plays a major role in thermoregulation. When an infection causes an increase in endogenous pyrogens, including interleukin-1β, interleukin-6 and tumor necrosis factor, prostaglandins are generated in the region of the POAH, increasing the thermoregulatory set point, triggering cutaneous vasoconstriction and shivering and non-shivering thermogenesis.1 Misoprostol, especially at doses >400 µg commonly causes both patient-reported chills and temperature elevation >38° C.
In a study comparing misoprostol and oxytocin for the management of the third stage of labor, 597 patients were randomly allocated to receive oxytocin 10 units by intramuscular injection or misoprostol 400 µg or 600 µg by the oral route.2 Patient-reported shivering occurred in 13%, 19%, and 28% of patients receiving oxytocin, misoprostol 400 µg and misoprostol 800 µg, respectively. A recorded temperature >38° C occurred within 1 hour of medication administration in approximately 3%, 2%, and 7.5% of patients receiving oxytocin, misoprostol 400 µg, and misoprostol 800 µg, respectively. In another study, 453 patients scheduled for a cesarean birth were randomly allocated to receive 1 of 3 doses of rectal misoprostol 200 μg, 400 μg, or 600 μg before incision. Fever was detected in 2.6%, 9.9%, and 5.1% of the patients receiving misoprostol 200 μg, 400 μg, or 600 μg, respectively.3
References
1. Aronoff DM, Neilson EG. Antipyretics: mechanisms of action and clinical use in fever suppression. Am J Med. 2001;111:304-315. doi: 10.1016/s0002-9343(01)00834-8.
2. Lumbiganon P, Hofmeyr J, Gumezoglu AM, et al. Misoprostol dose-related shivering and pyrexia in the third stage of labor. WHO Collaborative Trial of Misoprostol in the Management of the Third Stage of Labor. Br J Obstet Gynaecol. 1999;106:304-308. doi: 10.1111/j.1471-0528.1999.tb08266.x.
3. Sweed M, El-Said M, Abou-Gamrah AA, et al. Comparison between 200, 400 and 600 microgram rectal misoprostol before cesarean section: a randomized clinical trial. J Obstet Gynaecol Res. 2019;45:585-591. doi: 10.1111 /jog.13883.
Oxytocin and prostaglandins are critically important regulators of uterine contraction. Obstetrician-gynecologists commonly prescribe oxytocin and prostaglandin agonists (misoprostol, dinoprostone) to stimulate uterine contraction for the induction of labor, prevention and treatment of postpartum hemorrhage, and treatment of miscarriage and fetal demise. The focus of this editorial is the clinical pharmacology of misoprostol.
Misoprostol is approved by the US Food and Drug Administration (FDA) for the prevention and treatment of nonsteroidal anti-inflammatory drug–induced gastric ulcers and for patients at high risk for gastric ulcers, including those with a history of gastric ulcers. The approved misoprostol route and dose for this indication is oral administration of 200 µg four times daily with food.1 Recent food intake and antacid use reduces the absorption of orally administered misoprostol. There are no FDA-approved indications for the use of misoprostol as a single agent in obstetrics and gynecology. The FDA has approved the combination of mifepristone and misoprostol for medication abortion in the first trimester. In contrast to misoprostol, PGE2 (dinoprostone) is approved by the FDA as a vaginal insert containing 10 mg of dinoprostone for the initiation and/or continuation of cervical ripening in patients at or near term in whom there is a medical or obstetric indication for induction of labor (Cervidil; Ferring Pharmaceuticals Inc, Parsippany, New Jersey).2
Pharmacology of misoprostol
Misoprostol is a prostaglandin E1 (PGE1) agonist analogue. Prostaglandin E1 (alprostadil) is rapidly metabolized, has a half-life in the range of minutes and is not orally active, requiring administration by intravenous infusion or injection. It is indicated to maintain a patent ductus arteriosus in newborns with ductal-dependent circulation and to treat erectile dysfunction.3 In contrast to PGE1, misoprostol has a methyl ester group at carbon-1 (C-1) that increases potency and duration of action. Misoprostol also has no hydroxyl group at C-15, replacing that moiety with the addition of both a methyl- and hydroxyl- group at C-16 (FIGURE). These molecular changes improve oral activity and increase duration of action.4 Pure misoprostol is a viscous oil. It is formulated into tables by dispersing the oil on hydroxypropyl methyl cellulose before compounding into tablets. Unlike naturally occurring prostaglandins (PGE1), misoprostol tablets are stabile at room temperature for years.4
Following absorption, the methyl ester at C-1 is enzymatically cleaved, yielding misoprostol acid, the active drug.4 Misoprostol binds to the E prostanoid receptor 3 (EP-3).5 Activation of myometrial EP-3 receptor induces an increase in intracellular phosphoinositol turnover and calcium mobilization, resulting in an increase in intracellular-free calcium, triggering actin-myosin contractility.6 The increase in free calcium is propagated cell-to-cell through gap junctions that link the myometrial cells to facilitate the generation of a coordinated contraction.
Misoprostol: Various routes of administration are not equal
Misoprostol can be given by an oral, buccal, vaginal, or rectal route of administration. To study the effect of the route of administration on uterine tone and contractility, investigators randomly assigned patients at 8 to 11 weeks’ gestation to receive misoprostol 400 µg as a single dose by the oral or vaginal route. Uterine tone and contractility were measured using an intrauterine pressure transducer. Compared to vaginal administration, oral administration of misprostol was associated with rapid attainment of peak plasma level at 30 minutes, followed by a decline in concentration by 60 minutes. This rapid onset and rapid offset of plasma concentration was paralleled by the onset of uterine tone within 8 minutes, but surprisingly no sustained uterine contractions.7 By contrast, following vaginal administration of misoprostol, serum levels rose slowly and peaked in 1 to 2 hours. Uterine tone increased within 21 minutes, and sustained uterine contractions were recorded for 4 hours.7 The rapid rise and fall in plasma misoprostol following oral administration and the more sustained plasma misoprostol concentration over 4 hours has been previously reported.8 In a second study involving patients 8 to 11 weeks’ gestation, the effect of a single dose of misoprostol 400 µg by an oral or vaginal route on uterine contractility was compared using an intrauterine pressure transducer.9 Confirming previous results, the time from misoprostol administration to increased uterine tone was more rapid with oral than with vaginal administration (8 min vs 19 min). Over the course of 4 hours, uterine contraction activity was greater with vaginal than with oral administration (454 vs 166 Montevideo units).9
Both studies reported that oral administration of misoprostol resulted in more rapid onset and offset of action than vaginal administration. Oral administration of a single dose of misoprostol 400 µg did not result in sustained uterine contractions in most patients in the first trimester. Vaginal administration produced a slower onset of increased uterine tone but sustained uterine contractions over 4 hours. Compared with vaginal administration of misoprostol, the rapid onset and offset of action of oral misoprostol may reduce the rate of tachysystole and changes in fetal heart rate observed with vaginal administration.10
An important finding is that buccal and vaginal administration of misoprostol have similar effects on uterine tone in the first trimester.11 To study the effect of buccal and vaginal administration of misoprostol on uterine tone, patients 6 to 13 weeks’ gestation were randomly allocated to receive a single dose of misoprostol 400 µg by a buccal or vaginal route.11 Uterine activity over 5 hours following administration was assessed using an intrauterine pressure transducer. Uterine tone 20 to 30 minutes after buccal or vaginal administration of misoprostol (400 µg) was 27 and 28 mm Hg, respectively. Peak uterine tone, as measured by an intrauterine pressure transducer, for buccal and vaginal administration of misoprostol was 49 mm Hg and 54 mm Hg, respectively. Total Alexandria units (AU) over 5 hours following buccal or vaginal administration was 6,537 AU and 6,090 AU, respectively.11
An AU is calculated as the average amplitude of the contractions (mm Hg) multiplied by the average duration of the contractions (min) multiplied by average frequency of contraction over 10 minutes.12 By contrast, a Montevideo unit does not include an assessment of contraction duration and is calculated as average amplitude of contractions (mm Hg) multiplied by frequency of uterine contractions over 10 minutes.12
In contrast to buccal or vaginal administration, rectal administration of misoprostol resulted in much lower peak uterine tone and contractility as measured by a pressure transducer. Uterine tone 20 to 30 minutes after vaginal and rectal administration of misoprostol (400 µg) was 28 and 19 mm Hg, respectively.11 Peak uterine tone, as measured by an intrauterine pressure transducer, for vaginal and rectal administration of misoprostol was 54 and 31 mm Hg, respectively. AUs over 5 hours following vaginal and rectal administration was 6,090 AU and 2,768 AU, respectively.11 Compared with buccal and vaginal administration of misoprostol, rectal administration produced less sustained uterine contractions in the first trimester of pregnancy. To achieve maximal sustained uterine contractions, buccal and vaginal routes of administration are superior to oral and rectal administration.
Continue to: Misoprostol and cervical ripening...
Misoprostol and cervical ripening
Misoprostol is commonly used to soften and ripen the cervix. Some of the cervical ripening effects of misoprostol are likely due to increased uterine tone. In addition, misoprostol may have a direct effect on the collagen structure of the cervix. To study the effect of misoprostol on the cervix, pregnant patients in the first trimester were randomly assigned to receive misoprostol 200 µg by vaginal self-administration, isosorbide mononitrate (IMN) 40 mg by vaginal self-administration or no treatment the evening prior to pregnancy termination.13 The following day, before uterine evacuation, a cervical biopsy was obtained for electron microscopy studies and immunohistochemistry to assess the presence of enzymes involved in collagen degradation, including matrix metalloproteinase 1 (MMP-1) and matrix metalloproteinase 9 (MMP-9). Electron microscopy demonstrated that pretreatment with misoprostol resulted in a pronounced splitting and disorganization of collagen fibers.13 Compared with misoprostol treatment, IMN produced less splitting and disorganization of collagen fibers, and in the no treatment group, no marked changes in the collagen framework were observed.
Compared with no treatment, misoprostol and IMN pretreatment were associated with marked increases in MMP-1 and MMP-9 as assessed by immunohistochemistry. Misoprostol pretreatment also resulted in a significant increase in interleukin-8 concentration compared with IMN pretreatment and no treatment (8.8 vs 2.7 vs 2.4 pg/mg tissue), respectively.13 Other investigators have also reported that misoprostol increased cervical leukocyte influx and collagen disrupting enzymes MMP-8 and MMP-9.14,15
An open-label clinical trial compared the efficacy of misoprostol versus Foley catheter for labor induction at term in 1,859 patients ≥ 37 weeks’ gestation with a Bishop score <6.16 Patients were randomly allocated to misoprostol (50 µg orally every 4 hours up to 3 times in 24 hours) versus placement of a 16 F or 18 F Foley catheter introduced through the cervix, filled with 30 mL of sodium chloride or water. The investigators reported that oral misoprostol and Foley catheter cervical ripening had similar safety and effectiveness for cervical ripening as a prelude to induction of labor, including no statistically significant differences in 5-minute Apgar score <7, umbilical cord artery pH ≤ 7.05, postpartum hemorrhage, or cesarean birth rate.16
Bottom line
Misoprostol and oxytocin are commonly prescribed in obstetric practice for cervical ripening and induction of labor, respectively. The dose and route of administration of misoprostol influences the effect on the uterus. For cervical ripening, where rapid onset and offset may help to reduce the risk of uterine tachysystole and worrisome fetal heart rate changes, low-dose (50 µg) oral administration of misoprostol may be a preferred dose and route. For the treatment of miscarriage and fetal demise, to stimulate sustained uterine contractions over many hours, buccal and vaginal administration of misoprostol are preferred. Rectal administration is generally inferior to buccal and vaginal administration for stimulating sustained uterine contractions and its uses should be limited. ●
Common side effects of misoprostol are abdominal cramping, diarrhea, nausea, vomiting, headache, and fever. Elevated temperature following misoprostol administration is a concerning side effect that may require further investigation to rule out an infection, especially if the elevated temperature persists for > 4 hours. The preoptic area of the anterior hypothalamus (POAH) plays a major role in thermoregulation. When an infection causes an increase in endogenous pyrogens, including interleukin-1β, interleukin-6 and tumor necrosis factor, prostaglandins are generated in the region of the POAH, increasing the thermoregulatory set point, triggering cutaneous vasoconstriction and shivering and non-shivering thermogenesis.1 Misoprostol, especially at doses >400 µg commonly causes both patient-reported chills and temperature elevation >38° C.
In a study comparing misoprostol and oxytocin for the management of the third stage of labor, 597 patients were randomly allocated to receive oxytocin 10 units by intramuscular injection or misoprostol 400 µg or 600 µg by the oral route.2 Patient-reported shivering occurred in 13%, 19%, and 28% of patients receiving oxytocin, misoprostol 400 µg and misoprostol 800 µg, respectively. A recorded temperature >38° C occurred within 1 hour of medication administration in approximately 3%, 2%, and 7.5% of patients receiving oxytocin, misoprostol 400 µg, and misoprostol 800 µg, respectively. In another study, 453 patients scheduled for a cesarean birth were randomly allocated to receive 1 of 3 doses of rectal misoprostol 200 μg, 400 μg, or 600 μg before incision. Fever was detected in 2.6%, 9.9%, and 5.1% of the patients receiving misoprostol 200 μg, 400 μg, or 600 μg, respectively.3
References
1. Aronoff DM, Neilson EG. Antipyretics: mechanisms of action and clinical use in fever suppression. Am J Med. 2001;111:304-315. doi: 10.1016/s0002-9343(01)00834-8.
2. Lumbiganon P, Hofmeyr J, Gumezoglu AM, et al. Misoprostol dose-related shivering and pyrexia in the third stage of labor. WHO Collaborative Trial of Misoprostol in the Management of the Third Stage of Labor. Br J Obstet Gynaecol. 1999;106:304-308. doi: 10.1111/j.1471-0528.1999.tb08266.x.
3. Sweed M, El-Said M, Abou-Gamrah AA, et al. Comparison between 200, 400 and 600 microgram rectal misoprostol before cesarean section: a randomized clinical trial. J Obstet Gynaecol Res. 2019;45:585-591. doi: 10.1111 /jog.13883.
- Cytotec [package insert]. Chicago, IL: GD Searle & Co. https://www.accessdata.fda.gov/drugsatfda_docs/label/2002/19268slr037.pdf. Accessed June 20, 2022.
- Cervidil [package insert]. St Louis, MO: Forrest Pharmaceuticals Inc.; May 2006. Accessed June 20, 2022.
- Caverject [package insert]. New York, NY: Pfizer Inc.; March 2014. Accessed June 20, 2022.
- Collins PW. Misoprostol: discovery, development and clinical applications. Med Res Rev. 1990;10:149-172. doi: 10.1002/med.2610100202.
- Audit M, White KI, Breton B, et al. Crystal structure of misoprostol bound to the labor inducer prostaglandin E2 receptor. Nat Chem Biol. 2019;15:11-17. doi: 10.1038/s41589-018-0160-y.
- Pallliser KH, Hirst JJ, Ooi G, et al. Prostaglandin E and F receptor expression and myometrial sensitivity in labor onset in the sheep. Biol Reprod. 2005;72:937-943. doi: 10.1095/biolreprod.104.035311.
- Gemzell-Danilesson K, Marions L, Rodriguez A, et al. Comparison between oral and vaginal administration of misoprostol on uterine contractility. Obstet Gynecol. 1999;93:275-280. doi: 10.1016/s0029-7844(98)00436-0.
- Zieman M, Fong SK, Benowitz NL, et al. Absorption kinetics of misoprostol with oral or vaginal administration. Obstet Gynecol. 1997;90:88-92. doi: 10.1016/S0029-7844(97)00111-7.
- Aronsson A, Bygdeman M, Gemzell-Danielsson K. Effects of misoprostol on uterine contractility following different routes of administration. Hum Reprod. 2004;19:81-84. doi: 10.1093/humrep/deh005.
- Young DC, Delaney T, Armson BA, et al. Oral misoprostol, low dose vaginal misoprostol and vaginal dinoprostone for labor induction: randomized controlled trial. PLOS One. 2020;15:e0227245. doi: 10.1371/journal.pone.0227245.
- Meckstroth KR, Whitaker AK, Bertisch S, et al. Misoprostol administered by epithelial routes. Drug absorption and uterine response. Obstet Gynecol. 2006;108:582-590. doi: 10.1097/01.AOG.0000230398.32794.9d.
- el-Sahwi S, Gaafar AA, Toppozada HK. A new unit for evaluation of uterine activity. Am J Obstet Gynecol. 1967;98:900-903. doi: 10.1016/0002-9378(67)90074-9.
- Vukas N, Ekerhovd E, Abrahamsson G, et al. Cervical priming in the first trimester: morphological and biochemical effects of misoprostol and isosorbide mononitrate. Acta Obstet Gyecol. 2009;88:43-51. doi: 10.1080/00016340802585440.
- Aronsson A, Ulfgren AK, Stabi B, et al. The effect of orally and vaginally administered misoprostol on inflammatory mediators and cervical ripening during early pregnancy. Contraception. 2005;72:33-39. doi: 10.1016/j.contraception.2005.02.012.
- Denison FC, Riley SC, Elliott CL, et al. The effect of mifepristone administration on leukocyte populations, matrix metalloproteinases and inflammatory mediators in the first trimester cervix. Mol Hum Reprod. 2000;6:541-548. doi: 10.1093/molehr/6.6.541.
- ten Eikelder MLG, Rengerink KO, Jozwiak M, et al. Induction of labour at term with oral misoprostol versus a Foley catheter (PROBAAT-II): a multicentre randomised controlled non-inferiority trial. Lancet. 2016;387:1619-1628. doi: 10.1016 /S0140-6736(16)00084-2.
- Cytotec [package insert]. Chicago, IL: GD Searle & Co. https://www.accessdata.fda.gov/drugsatfda_docs/label/2002/19268slr037.pdf. Accessed June 20, 2022.
- Cervidil [package insert]. St Louis, MO: Forrest Pharmaceuticals Inc.; May 2006. Accessed June 20, 2022.
- Caverject [package insert]. New York, NY: Pfizer Inc.; March 2014. Accessed June 20, 2022.
- Collins PW. Misoprostol: discovery, development and clinical applications. Med Res Rev. 1990;10:149-172. doi: 10.1002/med.2610100202.
- Audit M, White KI, Breton B, et al. Crystal structure of misoprostol bound to the labor inducer prostaglandin E2 receptor. Nat Chem Biol. 2019;15:11-17. doi: 10.1038/s41589-018-0160-y.
- Pallliser KH, Hirst JJ, Ooi G, et al. Prostaglandin E and F receptor expression and myometrial sensitivity in labor onset in the sheep. Biol Reprod. 2005;72:937-943. doi: 10.1095/biolreprod.104.035311.
- Gemzell-Danilesson K, Marions L, Rodriguez A, et al. Comparison between oral and vaginal administration of misoprostol on uterine contractility. Obstet Gynecol. 1999;93:275-280. doi: 10.1016/s0029-7844(98)00436-0.
- Zieman M, Fong SK, Benowitz NL, et al. Absorption kinetics of misoprostol with oral or vaginal administration. Obstet Gynecol. 1997;90:88-92. doi: 10.1016/S0029-7844(97)00111-7.
- Aronsson A, Bygdeman M, Gemzell-Danielsson K. Effects of misoprostol on uterine contractility following different routes of administration. Hum Reprod. 2004;19:81-84. doi: 10.1093/humrep/deh005.
- Young DC, Delaney T, Armson BA, et al. Oral misoprostol, low dose vaginal misoprostol and vaginal dinoprostone for labor induction: randomized controlled trial. PLOS One. 2020;15:e0227245. doi: 10.1371/journal.pone.0227245.
- Meckstroth KR, Whitaker AK, Bertisch S, et al. Misoprostol administered by epithelial routes. Drug absorption and uterine response. Obstet Gynecol. 2006;108:582-590. doi: 10.1097/01.AOG.0000230398.32794.9d.
- el-Sahwi S, Gaafar AA, Toppozada HK. A new unit for evaluation of uterine activity. Am J Obstet Gynecol. 1967;98:900-903. doi: 10.1016/0002-9378(67)90074-9.
- Vukas N, Ekerhovd E, Abrahamsson G, et al. Cervical priming in the first trimester: morphological and biochemical effects of misoprostol and isosorbide mononitrate. Acta Obstet Gyecol. 2009;88:43-51. doi: 10.1080/00016340802585440.
- Aronsson A, Ulfgren AK, Stabi B, et al. The effect of orally and vaginally administered misoprostol on inflammatory mediators and cervical ripening during early pregnancy. Contraception. 2005;72:33-39. doi: 10.1016/j.contraception.2005.02.012.
- Denison FC, Riley SC, Elliott CL, et al. The effect of mifepristone administration on leukocyte populations, matrix metalloproteinases and inflammatory mediators in the first trimester cervix. Mol Hum Reprod. 2000;6:541-548. doi: 10.1093/molehr/6.6.541.
- ten Eikelder MLG, Rengerink KO, Jozwiak M, et al. Induction of labour at term with oral misoprostol versus a Foley catheter (PROBAAT-II): a multicentre randomised controlled non-inferiority trial. Lancet. 2016;387:1619-1628. doi: 10.1016 /S0140-6736(16)00084-2.