User login
Long COVID: New Info on Who Is Most Likely to Get It
The COVID-19 pandemic may no longer be a global public health emergency, but millions continue to struggle with the aftermath: Long COVID. New research and clinical anecdotes suggest that certain individuals are more likely to be afflicted by the condition, nearly 4 years after the virus emerged.
, said doctors who specialize in treating the condition.
Many patients with long COVID struggle with debilitating fatigue, brain fog, and cognitive impairment. The condition is also characterized by a catalog of other symptoms that may be difficult to recognize as long COVID, experts said. That’s especially true when patients may not mention seemingly unrelated information, such as underlying health conditions that might make them more vulnerable. This makes screening for certain conditions and investigating every symptom especially important.
The severity of a patient’s initial infection is not the only determining factor for developing long COVID, experts said.
“Don’t judge the person based on how sick they were initially,” said Mark Bayley, MD, medical director of the Toronto Rehabilitation Institute at University Health Network and a professor with the Temerty Faculty of Medicine at the University of Toronto. “You have to evaluate every symptom as best you can to make sure you’re not missing anything else.”
Someone who only had a bad cough or felt really unwell for just a few days and recovered but started feeling rotten again later — “that’s the person that we are seeing for long COVID,” said Dr. Bayley.
While patients who become severely sick and require hospitalization have a higher risk of developing long COVID, this group size is small compared with the much larger number of people infected overall. As a result, despite the lower risk, those who only become mild to moderately sick make up the vast majority of patients in long COVID clinics.
A small Northwestern Medicine study found that 41% of patients with long COVID never tested positive for COVID-19 but were found to have antibodies that indicated exposure to the virus.
Doctors treating patients with long COVID should consider several risk factors, specialists said. They include:
- A history of asthma, eczema, or allergies
- Signs of autonomic nervous system dysfunction
- Preexisting immune system issues
- Chronic infections
- Diabetes
- Being slightly overweight
- A preexisting history of anxiety or depression
- Joint hypermobility ( being “double-jointed” with pain and other symptoms)
Screening for Allergies
Alba Azola, MD, assistant professor of Physical Medicine and Rehabilitation at Johns Hopkins Medicine, said a history of asthma, allergies, and eczema and an onset of new food allergies may be an important factor in long COVID that doctors should consider when evaluating at-risk patients.
It is important to identify this subgroup of patients because they respond to antihistamines and mast cell stabilizers, which not only relieve their allergy symptoms but may also help improve overall fatigue and their tolerance for basic activities like standing, Dr. Azola said.
A recently published systemic review of prospective cohort studies on long COVID also found that patients with preexisting allergic conditions like asthma or rhinitis may be linked to a higher risk of developing long COVID. The authors cautioned, however, that the evidence for the link is uncertain and more rigorous research is needed.
“It stands to reason that if your immune system tends to be a bit hyperactive that triggering it with a virus will make it worse,” said Dr. Bayley.
Signs of Dysautonomia, Joint Hypermobility
Patients should also be screened for signs and symptoms of dysautonomia, or autonomic nervous system disorder, such as postural orthostatic tachycardia syndrome (POTS) or another type of autonomic dysfunction, doctors said.
“There’s a whole list because the autonomic nervous system involves every part of your body, every system,” Dr. Azola said.
Issues with standing, vision, digestion, urination, and bowel movement, for example, appear to be multisystemic problems but may all be linked to autonomic dysfunction, she explained.
Patients who have POTS usually experience a worsening of symptoms after COVID infection, Dr. Azola said, adding that some patients may have even assumed their pre-COVID symptoms of POTS were normal.
She also screens for joint hypermobility or hypermobile Ehlers-Danlos syndrome, which affects connective tissue. Research has long shown a relationship between autonomic dysfunction, mast cell activation syndrome (repeated severe allergy symptoms that affect multiple systems), and the presence of hypermobility, Dr. Azola said. She added that gentle physical therapy can be helpful for patients with hypermobility issues.
Previous studies before and during the pandemic have also found that a substantial subset of patients with myalgic encephalomyelitis/chronic fatigue syndrome, which shares many similarities with long COVID, also have connective tissue/hypermobility disorders.
Depression, Anxiety, and Female Patients
People with a preexisting history of anxiety or depression also appear to be at a higher risk for long COVID, Dr. Bayley said, noting that patients with these conditions appear more vulnerable to brain fog and other difficulties brought on by COVID infection. Earlier research found biochemical evidence of brain inflammation that correlates with symptoms of anxiety in patients with long COVID.
“We know that depression is related to neurotransmitters like adrenaline and serotonin,” Dr. Bayley said. “The chronic inflammation that’s associated with COVID — this will make people feel more depressed because they’re not getting the neurotransmitters in their brain releasing at the right times.”
It may also put patients at a risk for anxiety due to fears of post-exertional malaise (PEM), where symptoms worsen after even very minor physical or mental exertion and can last days or weeks.
“You can see how that leads to a bit of a vicious cycle,” said Dr. Bayley, explaining that the cycle of fear and avoidance makes patients less active and deconditioned. But he added that learning to manage their activity can actually help mitigate PEM due to the anti-inflammatory effects of exercise, its positive impact on mood, and benefits to the immune and cardiovascular systems.
Meanwhile, a number of epidemiologic studies have found a higher prevalence of long COVID among women. Perimenopausal and menopausal women in particular appeared more prone, and at least one study reported that women under 50 years were five times more likely to develop post-COVID symptoms than men.
A recent small UK study that focused on COVID-19 hospitalizations found that women who had lower levels of inflammatory biomarkers at admission were more likely to experience certain long-term symptoms like muscle ache, low mood and anxiety, adding to earlier research linking female patients, long COVID, and neuropsychiatric symptoms.
History of Immune Dysfunction, Diabetes, Elevated Body Mass Index (BMI)
Immune dysfunction, a history of recurrent infections, or chronic sinus infections are also common among patients under Dr. Azola and her team’s care. Those who have arthritis or other autoimmune diseases such as lupus also appear more vulnerable, Dr. Bayley said, along with patients who have diabetes or a little overweight.
Recent research out of the University of Queensland found that being overweight can negatively affect the body’s immune response to the SARS-CoV-2 virus. Blood samples collected 13 months after infection, for example, found that individuals with a higher BMI had lower antibody activity and a reduced percentage of relevant B cells that help build antibodies to fight the virus. Being overweight did not affect the antibody response to the COVID-19 vaccines, however, giving further support for vaccination over infection-induced immunity as an important protective factor, researchers said.
Narrowing the Information Gap
The latest Centers for Centers for Disease Control and Prevention’s Household Pulse Survey estimates that 14% of all American adults have had long COVID at some point, with more than 5% of the entire adult population currently experiencing long COVID. With millions of Americans affected, experts and advocates highlight the importance of bridging the knowledge gap with primary care doctors.
Long COVID specialists said understanding these connections helps guide treatment plans and manage symptoms, such as finding the right medications, improving tolerance, optimizing sleep, applying cognitive strategies for brain fog, dietary changes, respiratory exercises to help with shortness of breath, and finding the fine line between what causes PEM and what doesn’t.
“Whenever you see a disease like this one, you always have to ask yourself, is there an alternative way of looking at this that might explain what we’re seeing?” said Dr. Bayley. “It remains to be said that all bets are still open and that we need to continue to be very broad thinking about this.”
A version of this article appeared on Medscape.com.
The COVID-19 pandemic may no longer be a global public health emergency, but millions continue to struggle with the aftermath: Long COVID. New research and clinical anecdotes suggest that certain individuals are more likely to be afflicted by the condition, nearly 4 years after the virus emerged.
, said doctors who specialize in treating the condition.
Many patients with long COVID struggle with debilitating fatigue, brain fog, and cognitive impairment. The condition is also characterized by a catalog of other symptoms that may be difficult to recognize as long COVID, experts said. That’s especially true when patients may not mention seemingly unrelated information, such as underlying health conditions that might make them more vulnerable. This makes screening for certain conditions and investigating every symptom especially important.
The severity of a patient’s initial infection is not the only determining factor for developing long COVID, experts said.
“Don’t judge the person based on how sick they were initially,” said Mark Bayley, MD, medical director of the Toronto Rehabilitation Institute at University Health Network and a professor with the Temerty Faculty of Medicine at the University of Toronto. “You have to evaluate every symptom as best you can to make sure you’re not missing anything else.”
Someone who only had a bad cough or felt really unwell for just a few days and recovered but started feeling rotten again later — “that’s the person that we are seeing for long COVID,” said Dr. Bayley.
While patients who become severely sick and require hospitalization have a higher risk of developing long COVID, this group size is small compared with the much larger number of people infected overall. As a result, despite the lower risk, those who only become mild to moderately sick make up the vast majority of patients in long COVID clinics.
A small Northwestern Medicine study found that 41% of patients with long COVID never tested positive for COVID-19 but were found to have antibodies that indicated exposure to the virus.
Doctors treating patients with long COVID should consider several risk factors, specialists said. They include:
- A history of asthma, eczema, or allergies
- Signs of autonomic nervous system dysfunction
- Preexisting immune system issues
- Chronic infections
- Diabetes
- Being slightly overweight
- A preexisting history of anxiety or depression
- Joint hypermobility ( being “double-jointed” with pain and other symptoms)
Screening for Allergies
Alba Azola, MD, assistant professor of Physical Medicine and Rehabilitation at Johns Hopkins Medicine, said a history of asthma, allergies, and eczema and an onset of new food allergies may be an important factor in long COVID that doctors should consider when evaluating at-risk patients.
It is important to identify this subgroup of patients because they respond to antihistamines and mast cell stabilizers, which not only relieve their allergy symptoms but may also help improve overall fatigue and their tolerance for basic activities like standing, Dr. Azola said.
A recently published systemic review of prospective cohort studies on long COVID also found that patients with preexisting allergic conditions like asthma or rhinitis may be linked to a higher risk of developing long COVID. The authors cautioned, however, that the evidence for the link is uncertain and more rigorous research is needed.
“It stands to reason that if your immune system tends to be a bit hyperactive that triggering it with a virus will make it worse,” said Dr. Bayley.
Signs of Dysautonomia, Joint Hypermobility
Patients should also be screened for signs and symptoms of dysautonomia, or autonomic nervous system disorder, such as postural orthostatic tachycardia syndrome (POTS) or another type of autonomic dysfunction, doctors said.
“There’s a whole list because the autonomic nervous system involves every part of your body, every system,” Dr. Azola said.
Issues with standing, vision, digestion, urination, and bowel movement, for example, appear to be multisystemic problems but may all be linked to autonomic dysfunction, she explained.
Patients who have POTS usually experience a worsening of symptoms after COVID infection, Dr. Azola said, adding that some patients may have even assumed their pre-COVID symptoms of POTS were normal.
She also screens for joint hypermobility or hypermobile Ehlers-Danlos syndrome, which affects connective tissue. Research has long shown a relationship between autonomic dysfunction, mast cell activation syndrome (repeated severe allergy symptoms that affect multiple systems), and the presence of hypermobility, Dr. Azola said. She added that gentle physical therapy can be helpful for patients with hypermobility issues.
Previous studies before and during the pandemic have also found that a substantial subset of patients with myalgic encephalomyelitis/chronic fatigue syndrome, which shares many similarities with long COVID, also have connective tissue/hypermobility disorders.
Depression, Anxiety, and Female Patients
People with a preexisting history of anxiety or depression also appear to be at a higher risk for long COVID, Dr. Bayley said, noting that patients with these conditions appear more vulnerable to brain fog and other difficulties brought on by COVID infection. Earlier research found biochemical evidence of brain inflammation that correlates with symptoms of anxiety in patients with long COVID.
“We know that depression is related to neurotransmitters like adrenaline and serotonin,” Dr. Bayley said. “The chronic inflammation that’s associated with COVID — this will make people feel more depressed because they’re not getting the neurotransmitters in their brain releasing at the right times.”
It may also put patients at a risk for anxiety due to fears of post-exertional malaise (PEM), where symptoms worsen after even very minor physical or mental exertion and can last days or weeks.
“You can see how that leads to a bit of a vicious cycle,” said Dr. Bayley, explaining that the cycle of fear and avoidance makes patients less active and deconditioned. But he added that learning to manage their activity can actually help mitigate PEM due to the anti-inflammatory effects of exercise, its positive impact on mood, and benefits to the immune and cardiovascular systems.
Meanwhile, a number of epidemiologic studies have found a higher prevalence of long COVID among women. Perimenopausal and menopausal women in particular appeared more prone, and at least one study reported that women under 50 years were five times more likely to develop post-COVID symptoms than men.
A recent small UK study that focused on COVID-19 hospitalizations found that women who had lower levels of inflammatory biomarkers at admission were more likely to experience certain long-term symptoms like muscle ache, low mood and anxiety, adding to earlier research linking female patients, long COVID, and neuropsychiatric symptoms.
History of Immune Dysfunction, Diabetes, Elevated Body Mass Index (BMI)
Immune dysfunction, a history of recurrent infections, or chronic sinus infections are also common among patients under Dr. Azola and her team’s care. Those who have arthritis or other autoimmune diseases such as lupus also appear more vulnerable, Dr. Bayley said, along with patients who have diabetes or a little overweight.
Recent research out of the University of Queensland found that being overweight can negatively affect the body’s immune response to the SARS-CoV-2 virus. Blood samples collected 13 months after infection, for example, found that individuals with a higher BMI had lower antibody activity and a reduced percentage of relevant B cells that help build antibodies to fight the virus. Being overweight did not affect the antibody response to the COVID-19 vaccines, however, giving further support for vaccination over infection-induced immunity as an important protective factor, researchers said.
Narrowing the Information Gap
The latest Centers for Centers for Disease Control and Prevention’s Household Pulse Survey estimates that 14% of all American adults have had long COVID at some point, with more than 5% of the entire adult population currently experiencing long COVID. With millions of Americans affected, experts and advocates highlight the importance of bridging the knowledge gap with primary care doctors.
Long COVID specialists said understanding these connections helps guide treatment plans and manage symptoms, such as finding the right medications, improving tolerance, optimizing sleep, applying cognitive strategies for brain fog, dietary changes, respiratory exercises to help with shortness of breath, and finding the fine line between what causes PEM and what doesn’t.
“Whenever you see a disease like this one, you always have to ask yourself, is there an alternative way of looking at this that might explain what we’re seeing?” said Dr. Bayley. “It remains to be said that all bets are still open and that we need to continue to be very broad thinking about this.”
A version of this article appeared on Medscape.com.
The COVID-19 pandemic may no longer be a global public health emergency, but millions continue to struggle with the aftermath: Long COVID. New research and clinical anecdotes suggest that certain individuals are more likely to be afflicted by the condition, nearly 4 years after the virus emerged.
, said doctors who specialize in treating the condition.
Many patients with long COVID struggle with debilitating fatigue, brain fog, and cognitive impairment. The condition is also characterized by a catalog of other symptoms that may be difficult to recognize as long COVID, experts said. That’s especially true when patients may not mention seemingly unrelated information, such as underlying health conditions that might make them more vulnerable. This makes screening for certain conditions and investigating every symptom especially important.
The severity of a patient’s initial infection is not the only determining factor for developing long COVID, experts said.
“Don’t judge the person based on how sick they were initially,” said Mark Bayley, MD, medical director of the Toronto Rehabilitation Institute at University Health Network and a professor with the Temerty Faculty of Medicine at the University of Toronto. “You have to evaluate every symptom as best you can to make sure you’re not missing anything else.”
Someone who only had a bad cough or felt really unwell for just a few days and recovered but started feeling rotten again later — “that’s the person that we are seeing for long COVID,” said Dr. Bayley.
While patients who become severely sick and require hospitalization have a higher risk of developing long COVID, this group size is small compared with the much larger number of people infected overall. As a result, despite the lower risk, those who only become mild to moderately sick make up the vast majority of patients in long COVID clinics.
A small Northwestern Medicine study found that 41% of patients with long COVID never tested positive for COVID-19 but were found to have antibodies that indicated exposure to the virus.
Doctors treating patients with long COVID should consider several risk factors, specialists said. They include:
- A history of asthma, eczema, or allergies
- Signs of autonomic nervous system dysfunction
- Preexisting immune system issues
- Chronic infections
- Diabetes
- Being slightly overweight
- A preexisting history of anxiety or depression
- Joint hypermobility ( being “double-jointed” with pain and other symptoms)
Screening for Allergies
Alba Azola, MD, assistant professor of Physical Medicine and Rehabilitation at Johns Hopkins Medicine, said a history of asthma, allergies, and eczema and an onset of new food allergies may be an important factor in long COVID that doctors should consider when evaluating at-risk patients.
It is important to identify this subgroup of patients because they respond to antihistamines and mast cell stabilizers, which not only relieve their allergy symptoms but may also help improve overall fatigue and their tolerance for basic activities like standing, Dr. Azola said.
A recently published systemic review of prospective cohort studies on long COVID also found that patients with preexisting allergic conditions like asthma or rhinitis may be linked to a higher risk of developing long COVID. The authors cautioned, however, that the evidence for the link is uncertain and more rigorous research is needed.
“It stands to reason that if your immune system tends to be a bit hyperactive that triggering it with a virus will make it worse,” said Dr. Bayley.
Signs of Dysautonomia, Joint Hypermobility
Patients should also be screened for signs and symptoms of dysautonomia, or autonomic nervous system disorder, such as postural orthostatic tachycardia syndrome (POTS) or another type of autonomic dysfunction, doctors said.
“There’s a whole list because the autonomic nervous system involves every part of your body, every system,” Dr. Azola said.
Issues with standing, vision, digestion, urination, and bowel movement, for example, appear to be multisystemic problems but may all be linked to autonomic dysfunction, she explained.
Patients who have POTS usually experience a worsening of symptoms after COVID infection, Dr. Azola said, adding that some patients may have even assumed their pre-COVID symptoms of POTS were normal.
She also screens for joint hypermobility or hypermobile Ehlers-Danlos syndrome, which affects connective tissue. Research has long shown a relationship between autonomic dysfunction, mast cell activation syndrome (repeated severe allergy symptoms that affect multiple systems), and the presence of hypermobility, Dr. Azola said. She added that gentle physical therapy can be helpful for patients with hypermobility issues.
Previous studies before and during the pandemic have also found that a substantial subset of patients with myalgic encephalomyelitis/chronic fatigue syndrome, which shares many similarities with long COVID, also have connective tissue/hypermobility disorders.
Depression, Anxiety, and Female Patients
People with a preexisting history of anxiety or depression also appear to be at a higher risk for long COVID, Dr. Bayley said, noting that patients with these conditions appear more vulnerable to brain fog and other difficulties brought on by COVID infection. Earlier research found biochemical evidence of brain inflammation that correlates with symptoms of anxiety in patients with long COVID.
“We know that depression is related to neurotransmitters like adrenaline and serotonin,” Dr. Bayley said. “The chronic inflammation that’s associated with COVID — this will make people feel more depressed because they’re not getting the neurotransmitters in their brain releasing at the right times.”
It may also put patients at a risk for anxiety due to fears of post-exertional malaise (PEM), where symptoms worsen after even very minor physical or mental exertion and can last days or weeks.
“You can see how that leads to a bit of a vicious cycle,” said Dr. Bayley, explaining that the cycle of fear and avoidance makes patients less active and deconditioned. But he added that learning to manage their activity can actually help mitigate PEM due to the anti-inflammatory effects of exercise, its positive impact on mood, and benefits to the immune and cardiovascular systems.
Meanwhile, a number of epidemiologic studies have found a higher prevalence of long COVID among women. Perimenopausal and menopausal women in particular appeared more prone, and at least one study reported that women under 50 years were five times more likely to develop post-COVID symptoms than men.
A recent small UK study that focused on COVID-19 hospitalizations found that women who had lower levels of inflammatory biomarkers at admission were more likely to experience certain long-term symptoms like muscle ache, low mood and anxiety, adding to earlier research linking female patients, long COVID, and neuropsychiatric symptoms.
History of Immune Dysfunction, Diabetes, Elevated Body Mass Index (BMI)
Immune dysfunction, a history of recurrent infections, or chronic sinus infections are also common among patients under Dr. Azola and her team’s care. Those who have arthritis or other autoimmune diseases such as lupus also appear more vulnerable, Dr. Bayley said, along with patients who have diabetes or a little overweight.
Recent research out of the University of Queensland found that being overweight can negatively affect the body’s immune response to the SARS-CoV-2 virus. Blood samples collected 13 months after infection, for example, found that individuals with a higher BMI had lower antibody activity and a reduced percentage of relevant B cells that help build antibodies to fight the virus. Being overweight did not affect the antibody response to the COVID-19 vaccines, however, giving further support for vaccination over infection-induced immunity as an important protective factor, researchers said.
Narrowing the Information Gap
The latest Centers for Centers for Disease Control and Prevention’s Household Pulse Survey estimates that 14% of all American adults have had long COVID at some point, with more than 5% of the entire adult population currently experiencing long COVID. With millions of Americans affected, experts and advocates highlight the importance of bridging the knowledge gap with primary care doctors.
Long COVID specialists said understanding these connections helps guide treatment plans and manage symptoms, such as finding the right medications, improving tolerance, optimizing sleep, applying cognitive strategies for brain fog, dietary changes, respiratory exercises to help with shortness of breath, and finding the fine line between what causes PEM and what doesn’t.
“Whenever you see a disease like this one, you always have to ask yourself, is there an alternative way of looking at this that might explain what we’re seeing?” said Dr. Bayley. “It remains to be said that all bets are still open and that we need to continue to be very broad thinking about this.”
A version of this article appeared on Medscape.com.
The Evolving Treatment Paradigm for Diffuse Large B-Cell Lymphoma

Non-Hodgkin lymphomas (NHLs) are cancers that arise in a type of white blood cell called the lymphocyte. NHLs are divided into B- and T-cell subtypes, as well as aggressive and indolent forms. Management varies widely depending on the disease type. We will focus on the most common type of NHL, diffuse large B-cell lymphoma (DLBCL), for which there have been significant treatment advances in recent years.
DLBCL is curable in about two-thirds of patients using chemoimmunotherapy. The longstanding frontline treatment for this disease has been R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone). In 2023, an antibody-drug conjugate against the B-cell surface protein CD79b, polatuzumab vedotin, was approved by the US Food and Drug Administration (FDA) in combination with R-CHP (rituximab, cyclophosphamide, doxorubicin, prednisone) for newly diagnosed DLBCL based on an improvement in progression-free survival at 2 years in patients with high-risk disease features enrolled in the POLARIX study.
For patients who do not respond to the initial treatment or in whom the disease recurs, the historical standard of care treatment strategy was high-dose chemotherapy followed by autologous stem cell transplant (ASCT). Unfortunately, this approach is not feasible or not successful in a significant percentage of patients with relapsed or refractory DLBCL.
A newer strategy for DLBCL is chimeric antigen receptor (CAR) T-cell therapy. In this treatment, T cells are collected from a patient and genetically modified to target a protein on the lymphoma cells called CD19. This type of treatment was initially approved in the third-line setting for DLBCL based on the ZUMA-1 (axi-cel), JULIET (tisa-cel), and TRANSCEND (liso-cel) clinical trials. More recently, in 2022, 2 of these agents received approval in the second-line setting in patients who relapse or are refractory to initial treatment within 1 year; axi-cel was approved based on the ZUMA-7 trial and liso-cel was approved based on the TRANSFORM trial.
Unfortunately, not all patients are eligible for ASCT and CAR T-cell therapy due to factors including age, comorbidities, and disease characteristics. Some patients prefer alternative therapies based on the potential side effects of CAR T-cell therapy and ASCT. Toxicities associated with CAR T-cell therapy include an inflammatory response called cytokine release syndrome and neurologic events.
For patients who are not eligible for or who relapse after ASCT or CAR T-cell therapy, several alternative treatment options are FDA approved. Novel strategies include polatuzumab vedotin with bendamustine and rituximab and tafasitamab plus lenalidomide. Tafasitamab is a monoclonal antibody against CD19 and lenalidomide is an oral anticancer agent originally approved for use in multiple myeloma. Lenalidomide is also effective and commonly used in other NHL subtypes.
In 2023, a new category of treatment called bispecific antibodies was approved in patients with DLBCL in whom the disease recurs after 2 lines of therapy. These drugs (epcoritamab and glofitamab) are a form of immunotherapy that connects B cells with T cells to enable a person’s own immune system to better fight the lymphoma. While these drugs can have similar toxicities as CAR T-cell therapy, the severity and incidence are much lower. In contrast to CAR T-cell therapy, which requires only 1 infusion, these drugs are given regularly in either subcutaneous or intravenous form for several months.
Two other FDA-approved treatment options for relapsed and refractory DLBCL are loncastuximab tesirine, an antibody-drug conjugate targeting CD19 with approval based on the results of the LOTIS-2 trial, and the oral selective inhibitor of nuclear export called selinexor, based on the results from the SADAL trial. Selinexor is a fully synthetic small-molecule compound, developed by means of a structure-based drug design process known as induced-fit docking. It binds to a cysteine residue in the nuclear export signal groove of exportin 1. Selinexor is approved for use in adults with relapsed or refractory DLBCL who have received at least 2 types of systemic therapy. Trials investigating these agents in combination with other novel treatments are ongoing.
The treatment landscape for DLBCL has changed markedly over the past several years. Therapies can be tailored for individual patients based on their disease status and characteristics, comorbidities, and treatment preferences. Research with novel strategies continues with the goal of a cure for all patients diagnosed with DLBCL.
Non-Hodgkin lymphomas (NHLs) are cancers that arise in a type of white blood cell called the lymphocyte. NHLs are divided into B- and T-cell subtypes, as well as aggressive and indolent forms. Management varies widely depending on the disease type. We will focus on the most common type of NHL, diffuse large B-cell lymphoma (DLBCL), for which there have been significant treatment advances in recent years.
DLBCL is curable in about two-thirds of patients using chemoimmunotherapy. The longstanding frontline treatment for this disease has been R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone). In 2023, an antibody-drug conjugate against the B-cell surface protein CD79b, polatuzumab vedotin, was approved by the US Food and Drug Administration (FDA) in combination with R-CHP (rituximab, cyclophosphamide, doxorubicin, prednisone) for newly diagnosed DLBCL based on an improvement in progression-free survival at 2 years in patients with high-risk disease features enrolled in the POLARIX study.
For patients who do not respond to the initial treatment or in whom the disease recurs, the historical standard of care treatment strategy was high-dose chemotherapy followed by autologous stem cell transplant (ASCT). Unfortunately, this approach is not feasible or not successful in a significant percentage of patients with relapsed or refractory DLBCL.
A newer strategy for DLBCL is chimeric antigen receptor (CAR) T-cell therapy. In this treatment, T cells are collected from a patient and genetically modified to target a protein on the lymphoma cells called CD19. This type of treatment was initially approved in the third-line setting for DLBCL based on the ZUMA-1 (axi-cel), JULIET (tisa-cel), and TRANSCEND (liso-cel) clinical trials. More recently, in 2022, 2 of these agents received approval in the second-line setting in patients who relapse or are refractory to initial treatment within 1 year; axi-cel was approved based on the ZUMA-7 trial and liso-cel was approved based on the TRANSFORM trial.
Unfortunately, not all patients are eligible for ASCT and CAR T-cell therapy due to factors including age, comorbidities, and disease characteristics. Some patients prefer alternative therapies based on the potential side effects of CAR T-cell therapy and ASCT. Toxicities associated with CAR T-cell therapy include an inflammatory response called cytokine release syndrome and neurologic events.
For patients who are not eligible for or who relapse after ASCT or CAR T-cell therapy, several alternative treatment options are FDA approved. Novel strategies include polatuzumab vedotin with bendamustine and rituximab and tafasitamab plus lenalidomide. Tafasitamab is a monoclonal antibody against CD19 and lenalidomide is an oral anticancer agent originally approved for use in multiple myeloma. Lenalidomide is also effective and commonly used in other NHL subtypes.
In 2023, a new category of treatment called bispecific antibodies was approved in patients with DLBCL in whom the disease recurs after 2 lines of therapy. These drugs (epcoritamab and glofitamab) are a form of immunotherapy that connects B cells with T cells to enable a person’s own immune system to better fight the lymphoma. While these drugs can have similar toxicities as CAR T-cell therapy, the severity and incidence are much lower. In contrast to CAR T-cell therapy, which requires only 1 infusion, these drugs are given regularly in either subcutaneous or intravenous form for several months.
Two other FDA-approved treatment options for relapsed and refractory DLBCL are loncastuximab tesirine, an antibody-drug conjugate targeting CD19 with approval based on the results of the LOTIS-2 trial, and the oral selective inhibitor of nuclear export called selinexor, based on the results from the SADAL trial. Selinexor is a fully synthetic small-molecule compound, developed by means of a structure-based drug design process known as induced-fit docking. It binds to a cysteine residue in the nuclear export signal groove of exportin 1. Selinexor is approved for use in adults with relapsed or refractory DLBCL who have received at least 2 types of systemic therapy. Trials investigating these agents in combination with other novel treatments are ongoing.
The treatment landscape for DLBCL has changed markedly over the past several years. Therapies can be tailored for individual patients based on their disease status and characteristics, comorbidities, and treatment preferences. Research with novel strategies continues with the goal of a cure for all patients diagnosed with DLBCL.
Non-Hodgkin lymphomas (NHLs) are cancers that arise in a type of white blood cell called the lymphocyte. NHLs are divided into B- and T-cell subtypes, as well as aggressive and indolent forms. Management varies widely depending on the disease type. We will focus on the most common type of NHL, diffuse large B-cell lymphoma (DLBCL), for which there have been significant treatment advances in recent years.
DLBCL is curable in about two-thirds of patients using chemoimmunotherapy. The longstanding frontline treatment for this disease has been R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone). In 2023, an antibody-drug conjugate against the B-cell surface protein CD79b, polatuzumab vedotin, was approved by the US Food and Drug Administration (FDA) in combination with R-CHP (rituximab, cyclophosphamide, doxorubicin, prednisone) for newly diagnosed DLBCL based on an improvement in progression-free survival at 2 years in patients with high-risk disease features enrolled in the POLARIX study.
For patients who do not respond to the initial treatment or in whom the disease recurs, the historical standard of care treatment strategy was high-dose chemotherapy followed by autologous stem cell transplant (ASCT). Unfortunately, this approach is not feasible or not successful in a significant percentage of patients with relapsed or refractory DLBCL.
A newer strategy for DLBCL is chimeric antigen receptor (CAR) T-cell therapy. In this treatment, T cells are collected from a patient and genetically modified to target a protein on the lymphoma cells called CD19. This type of treatment was initially approved in the third-line setting for DLBCL based on the ZUMA-1 (axi-cel), JULIET (tisa-cel), and TRANSCEND (liso-cel) clinical trials. More recently, in 2022, 2 of these agents received approval in the second-line setting in patients who relapse or are refractory to initial treatment within 1 year; axi-cel was approved based on the ZUMA-7 trial and liso-cel was approved based on the TRANSFORM trial.
Unfortunately, not all patients are eligible for ASCT and CAR T-cell therapy due to factors including age, comorbidities, and disease characteristics. Some patients prefer alternative therapies based on the potential side effects of CAR T-cell therapy and ASCT. Toxicities associated with CAR T-cell therapy include an inflammatory response called cytokine release syndrome and neurologic events.
For patients who are not eligible for or who relapse after ASCT or CAR T-cell therapy, several alternative treatment options are FDA approved. Novel strategies include polatuzumab vedotin with bendamustine and rituximab and tafasitamab plus lenalidomide. Tafasitamab is a monoclonal antibody against CD19 and lenalidomide is an oral anticancer agent originally approved for use in multiple myeloma. Lenalidomide is also effective and commonly used in other NHL subtypes.
In 2023, a new category of treatment called bispecific antibodies was approved in patients with DLBCL in whom the disease recurs after 2 lines of therapy. These drugs (epcoritamab and glofitamab) are a form of immunotherapy that connects B cells with T cells to enable a person’s own immune system to better fight the lymphoma. While these drugs can have similar toxicities as CAR T-cell therapy, the severity and incidence are much lower. In contrast to CAR T-cell therapy, which requires only 1 infusion, these drugs are given regularly in either subcutaneous or intravenous form for several months.
Two other FDA-approved treatment options for relapsed and refractory DLBCL are loncastuximab tesirine, an antibody-drug conjugate targeting CD19 with approval based on the results of the LOTIS-2 trial, and the oral selective inhibitor of nuclear export called selinexor, based on the results from the SADAL trial. Selinexor is a fully synthetic small-molecule compound, developed by means of a structure-based drug design process known as induced-fit docking. It binds to a cysteine residue in the nuclear export signal groove of exportin 1. Selinexor is approved for use in adults with relapsed or refractory DLBCL who have received at least 2 types of systemic therapy. Trials investigating these agents in combination with other novel treatments are ongoing.
The treatment landscape for DLBCL has changed markedly over the past several years. Therapies can be tailored for individual patients based on their disease status and characteristics, comorbidities, and treatment preferences. Research with novel strategies continues with the goal of a cure for all patients diagnosed with DLBCL.
Updates on Investigational Treatments for HR-Positive, HER2-Negative Breast Cancer

Results from TROPION-Breast01, EMBER, and OPERA were recently presented at ESMO Breast Cancer 2023.
A number of exciting updates on systemic therapies for the treatment of hormone receptor (HR)-positive, HER2-negative breast cancer were presented at the European Society for Medical Oncology (ESMO) Breast Cancer 2023, including novel endocrine agents and antibody-drug conjugates (ADC). We have highlighted 3 key studies, including the phase III study of datopotamab deruxtecan (Dato-DXd), the new trophoblast cell surface antigen 2 (TROP2)-directed ADC; the phase I study of imlunestrant, a selective estrogen receptor degrader (SERD); and phase I/II data evaluating OP-1250, a small molecule oral complete estrogen receptor antagonist (CERAN) and SERD.
TROPION-Breast01: Dato-DXd Improves Progression-Free Survival Compared With Systemic Chemotherapy
Study synopsis
Dato-DXd, an investigational TROP2 ADC, resulted in significantly improved progression-free survival (PFS) when compared with investigator’s choice chemotherapy (ICC) in individuals with inoperable or metastatic HR-positive, HER2-low or HER2-negative breast cancer, according to a randomized phase III trial.
Participants in the study had progressed on or were not eligible for endocrine therapy and had received 1 or 2 prior lines of systemic chemotherapy. Patients were randomized to receive either 6 mg/kg of Dato-DXd once every 3 weeks (n=365; median age 56), or ICC with eribulin, vinorelbine, capecitabine, or gemcitabine (n=367; median age 54) until progression or unacceptable toxicity. Blinded independent review assessed PFS and overall survival. Among the results:
In the blinded independent review, PFS was 6.9 months for Dato-DXd and 4.9 months for ICC (HR 0.63 [95% CI: 0.52, 0.76]; p<0.0001)
At 6 months, 53% of participants receiving Dato-DXd achieved PFS, compared with 39% in the systemic chemotherapy contingent
In the Dato-DXd group, treatment-related adverse events led to dose reductions in 23% and discontinuation in 3% of patients
In the systemic chemotherapy cohort, the dose reduction and discontinuation rates were 32% and 3%, respectively
At the time data were reported at ESMO, overall survival data were not mature but trending favorably for Dato-DXd
The investigators concluded that Dato-DXd is a promising novel treatment option for individuals with inoperable or metastatic HR-positive, HER2-low or HER2-negative breast cancer who have received prior chemotherapy.
EMBER: Imlunestrant Alone or With a Kinase Inhibitor: Early Safety and Efficacy Results Are Encouraging
Study synopsis
The SERD imlunestrant—used either alone or combined with a kinase inhibitor—showed favorable efficacy in individuals with estrogen receptor (ER)-positive, HER2-negative advanced breast cancer, according to the first set of clinical data reported from the phase 1a/b EMBER study.
Key eligibility criteria for phase 1b enrollment included prior sensitivity to endocrine therapy, ≤2 prior therapies, and a PIK3CA mutation (alpelisib arm only). Prior therapies included endocrine therapy (100%), CDK4/6 inhibitors (100%), hormonal therapy with fulvestrant (35%), and chemotherapy (17%). At baseline, 46% of patients had visceral disease and 46% had an ESR1 mutation. Participants received imlunestrant alone (n=114) or with the kinase inhibitors everolimus (n=42) or alpelisib (n=21). Investigators assessed each regimen’s safety profile, as well as the objective response rate and clinical benefit rate.
The safety profile of each regimen was similar to those seen with everolimus and alpelisib alone. No cardiac or ocular toxicities were observed. Regarding grade ≥3 treatment-related adverse events:
The imlunestrant alone group experienced fatigue (2%) and neutropenia (2%)
The imlunestrant + everolimus group experienced hypertriglyceridemia (5%) and aspartate aminotransferase increase (5%)
The imlunestrant + alpelisib cohort experienced rash (43%) and hyperglycemia (10%).
In the imlunestrant alone group, 2% of individuals had their doses reduced due to adverse events; none discontinued treatment
In the imlunestrant + everolimus cohort, 12% of patients experienced dose reduction due to everolimus and 2% due to both medications; 2% discontinued treatment due to everolimus
In the imlunestrant + alpelisib cohort, 24% of patients experienced dose reduction due to alpelisib and 14% due to both medications; 29% discontinued treatment due to alpelisib
Regarding efficacy:
The objective response rates in the imlunestrant alone, imlunestrant + everolimus, and imlunestrant + alpelisib groups were 9%, 21%, and 50%, respectively
The clinical benefit rates in the imlunestrant alone, imlunestrant + everolimus, and imlunestrant + alpelisib groups were 42%, 62%, and 62%, respectively
Investigators concluded that imlunestrant used alone or in combination with 1 of the 2 kinase inhibitors demonstrated robust efficacy in individuals with pretreated, ER-positive, HER2-negative advanced breast cancer.
OPERA: OP-1250 Paired With a CDK4/6 Inhibitor: Anti-Tumor Activity With No Dose-Limiting Toxicities
Study synopsis
OP-1250, a CERAN and SERD, continues to show promising results when paired with a CDK4/6 inhibitor. The combination of OP-1250 and the CDK4/6 inhibitor palbociclib appears to be well tolerated and has a similar safety profile to each drug when used alone, according to a phase I/II study involving 20 individuals with pretreated ER-positive, HER2-negative breast cancer.
Participants had advanced or metastatic ER-positive, HER2-negative breast cancer that progressed on ≤1 lines of endocrine therapy. Fourteen participants had received prior CDK4/6 inhibitor therapy, including 11 who were previously treated with palbociclib. Patients received escalating doses of OP-1250 with 125 mg of palbociclib orally daily for 21 of 28 days. OP-1250 doses were 30 mg (n=3), 60 mg (n=3), 90 mg (n=3), and 120 mg (n=11). Investigators assessed pharmacokinetics, drug-drug interactions, safety, and efficacy. Among the results observed to date:
Grade 3 neutropenia occurred in 55% of participants
There were no grade 4 treatment-related adverse events and no dose-limiting toxicities
OP-1250 exposure yielded similar results to what was seen in the previous monotherapy study
Palbociclib exposure was comparable to published monotherapy data when combined with OP-1250 for all dosages
Investigators observed antitumor activity, including partial responses
Researchers concluded that OP-1250 does not affect the pharmacokinetics of palbociclib, and there do not appear to be drug-drug interactions. Tumor response to this combination was encouraging and requires continued investigation.
Conclusions
These 3 studies presented at ESMO 2023 highlight exciting novel therapies for the treatment of HR-positive, HER2-low, and HER2-negative metastatic breast cancer. The EMBER and OPERA updates provide support for the safety and efficacy of these novel endocrine agents in combination with kinase inhibitors and CDK4/6 inhibitors, respectively, in patients with endocrine-sensitive disease, while the TROPION-01 study demonstrates the encouraging efficacy and safety of a second TROP-2-directed ADC in a more heavily pretreated population.
Results from TROPION-Breast01, EMBER, and OPERA were recently presented at ESMO Breast Cancer 2023.
A number of exciting updates on systemic therapies for the treatment of hormone receptor (HR)-positive, HER2-negative breast cancer were presented at the European Society for Medical Oncology (ESMO) Breast Cancer 2023, including novel endocrine agents and antibody-drug conjugates (ADC). We have highlighted 3 key studies, including the phase III study of datopotamab deruxtecan (Dato-DXd), the new trophoblast cell surface antigen 2 (TROP2)-directed ADC; the phase I study of imlunestrant, a selective estrogen receptor degrader (SERD); and phase I/II data evaluating OP-1250, a small molecule oral complete estrogen receptor antagonist (CERAN) and SERD.
TROPION-Breast01: Dato-DXd Improves Progression-Free Survival Compared With Systemic Chemotherapy
Study synopsis
Dato-DXd, an investigational TROP2 ADC, resulted in significantly improved progression-free survival (PFS) when compared with investigator’s choice chemotherapy (ICC) in individuals with inoperable or metastatic HR-positive, HER2-low or HER2-negative breast cancer, according to a randomized phase III trial.
Participants in the study had progressed on or were not eligible for endocrine therapy and had received 1 or 2 prior lines of systemic chemotherapy. Patients were randomized to receive either 6 mg/kg of Dato-DXd once every 3 weeks (n=365; median age 56), or ICC with eribulin, vinorelbine, capecitabine, or gemcitabine (n=367; median age 54) until progression or unacceptable toxicity. Blinded independent review assessed PFS and overall survival. Among the results:
In the blinded independent review, PFS was 6.9 months for Dato-DXd and 4.9 months for ICC (HR 0.63 [95% CI: 0.52, 0.76]; p<0.0001)
At 6 months, 53% of participants receiving Dato-DXd achieved PFS, compared with 39% in the systemic chemotherapy contingent
In the Dato-DXd group, treatment-related adverse events led to dose reductions in 23% and discontinuation in 3% of patients
In the systemic chemotherapy cohort, the dose reduction and discontinuation rates were 32% and 3%, respectively
At the time data were reported at ESMO, overall survival data were not mature but trending favorably for Dato-DXd
The investigators concluded that Dato-DXd is a promising novel treatment option for individuals with inoperable or metastatic HR-positive, HER2-low or HER2-negative breast cancer who have received prior chemotherapy.
EMBER: Imlunestrant Alone or With a Kinase Inhibitor: Early Safety and Efficacy Results Are Encouraging
Study synopsis
The SERD imlunestrant—used either alone or combined with a kinase inhibitor—showed favorable efficacy in individuals with estrogen receptor (ER)-positive, HER2-negative advanced breast cancer, according to the first set of clinical data reported from the phase 1a/b EMBER study.
Key eligibility criteria for phase 1b enrollment included prior sensitivity to endocrine therapy, ≤2 prior therapies, and a PIK3CA mutation (alpelisib arm only). Prior therapies included endocrine therapy (100%), CDK4/6 inhibitors (100%), hormonal therapy with fulvestrant (35%), and chemotherapy (17%). At baseline, 46% of patients had visceral disease and 46% had an ESR1 mutation. Participants received imlunestrant alone (n=114) or with the kinase inhibitors everolimus (n=42) or alpelisib (n=21). Investigators assessed each regimen’s safety profile, as well as the objective response rate and clinical benefit rate.
The safety profile of each regimen was similar to those seen with everolimus and alpelisib alone. No cardiac or ocular toxicities were observed. Regarding grade ≥3 treatment-related adverse events:
The imlunestrant alone group experienced fatigue (2%) and neutropenia (2%)
The imlunestrant + everolimus group experienced hypertriglyceridemia (5%) and aspartate aminotransferase increase (5%)
The imlunestrant + alpelisib cohort experienced rash (43%) and hyperglycemia (10%).
In the imlunestrant alone group, 2% of individuals had their doses reduced due to adverse events; none discontinued treatment
In the imlunestrant + everolimus cohort, 12% of patients experienced dose reduction due to everolimus and 2% due to both medications; 2% discontinued treatment due to everolimus
In the imlunestrant + alpelisib cohort, 24% of patients experienced dose reduction due to alpelisib and 14% due to both medications; 29% discontinued treatment due to alpelisib
Regarding efficacy:
The objective response rates in the imlunestrant alone, imlunestrant + everolimus, and imlunestrant + alpelisib groups were 9%, 21%, and 50%, respectively
The clinical benefit rates in the imlunestrant alone, imlunestrant + everolimus, and imlunestrant + alpelisib groups were 42%, 62%, and 62%, respectively
Investigators concluded that imlunestrant used alone or in combination with 1 of the 2 kinase inhibitors demonstrated robust efficacy in individuals with pretreated, ER-positive, HER2-negative advanced breast cancer.
OPERA: OP-1250 Paired With a CDK4/6 Inhibitor: Anti-Tumor Activity With No Dose-Limiting Toxicities
Study synopsis
OP-1250, a CERAN and SERD, continues to show promising results when paired with a CDK4/6 inhibitor. The combination of OP-1250 and the CDK4/6 inhibitor palbociclib appears to be well tolerated and has a similar safety profile to each drug when used alone, according to a phase I/II study involving 20 individuals with pretreated ER-positive, HER2-negative breast cancer.
Participants had advanced or metastatic ER-positive, HER2-negative breast cancer that progressed on ≤1 lines of endocrine therapy. Fourteen participants had received prior CDK4/6 inhibitor therapy, including 11 who were previously treated with palbociclib. Patients received escalating doses of OP-1250 with 125 mg of palbociclib orally daily for 21 of 28 days. OP-1250 doses were 30 mg (n=3), 60 mg (n=3), 90 mg (n=3), and 120 mg (n=11). Investigators assessed pharmacokinetics, drug-drug interactions, safety, and efficacy. Among the results observed to date:
Grade 3 neutropenia occurred in 55% of participants
There were no grade 4 treatment-related adverse events and no dose-limiting toxicities
OP-1250 exposure yielded similar results to what was seen in the previous monotherapy study
Palbociclib exposure was comparable to published monotherapy data when combined with OP-1250 for all dosages
Investigators observed antitumor activity, including partial responses
Researchers concluded that OP-1250 does not affect the pharmacokinetics of palbociclib, and there do not appear to be drug-drug interactions. Tumor response to this combination was encouraging and requires continued investigation.
Conclusions
These 3 studies presented at ESMO 2023 highlight exciting novel therapies for the treatment of HR-positive, HER2-low, and HER2-negative metastatic breast cancer. The EMBER and OPERA updates provide support for the safety and efficacy of these novel endocrine agents in combination with kinase inhibitors and CDK4/6 inhibitors, respectively, in patients with endocrine-sensitive disease, while the TROPION-01 study demonstrates the encouraging efficacy and safety of a second TROP-2-directed ADC in a more heavily pretreated population.
Results from TROPION-Breast01, EMBER, and OPERA were recently presented at ESMO Breast Cancer 2023.
A number of exciting updates on systemic therapies for the treatment of hormone receptor (HR)-positive, HER2-negative breast cancer were presented at the European Society for Medical Oncology (ESMO) Breast Cancer 2023, including novel endocrine agents and antibody-drug conjugates (ADC). We have highlighted 3 key studies, including the phase III study of datopotamab deruxtecan (Dato-DXd), the new trophoblast cell surface antigen 2 (TROP2)-directed ADC; the phase I study of imlunestrant, a selective estrogen receptor degrader (SERD); and phase I/II data evaluating OP-1250, a small molecule oral complete estrogen receptor antagonist (CERAN) and SERD.
TROPION-Breast01: Dato-DXd Improves Progression-Free Survival Compared With Systemic Chemotherapy
Study synopsis
Dato-DXd, an investigational TROP2 ADC, resulted in significantly improved progression-free survival (PFS) when compared with investigator’s choice chemotherapy (ICC) in individuals with inoperable or metastatic HR-positive, HER2-low or HER2-negative breast cancer, according to a randomized phase III trial.
Participants in the study had progressed on or were not eligible for endocrine therapy and had received 1 or 2 prior lines of systemic chemotherapy. Patients were randomized to receive either 6 mg/kg of Dato-DXd once every 3 weeks (n=365; median age 56), or ICC with eribulin, vinorelbine, capecitabine, or gemcitabine (n=367; median age 54) until progression or unacceptable toxicity. Blinded independent review assessed PFS and overall survival. Among the results:
In the blinded independent review, PFS was 6.9 months for Dato-DXd and 4.9 months for ICC (HR 0.63 [95% CI: 0.52, 0.76]; p<0.0001)
At 6 months, 53% of participants receiving Dato-DXd achieved PFS, compared with 39% in the systemic chemotherapy contingent
In the Dato-DXd group, treatment-related adverse events led to dose reductions in 23% and discontinuation in 3% of patients
In the systemic chemotherapy cohort, the dose reduction and discontinuation rates were 32% and 3%, respectively
At the time data were reported at ESMO, overall survival data were not mature but trending favorably for Dato-DXd
The investigators concluded that Dato-DXd is a promising novel treatment option for individuals with inoperable or metastatic HR-positive, HER2-low or HER2-negative breast cancer who have received prior chemotherapy.
EMBER: Imlunestrant Alone or With a Kinase Inhibitor: Early Safety and Efficacy Results Are Encouraging
Study synopsis
The SERD imlunestrant—used either alone or combined with a kinase inhibitor—showed favorable efficacy in individuals with estrogen receptor (ER)-positive, HER2-negative advanced breast cancer, according to the first set of clinical data reported from the phase 1a/b EMBER study.
Key eligibility criteria for phase 1b enrollment included prior sensitivity to endocrine therapy, ≤2 prior therapies, and a PIK3CA mutation (alpelisib arm only). Prior therapies included endocrine therapy (100%), CDK4/6 inhibitors (100%), hormonal therapy with fulvestrant (35%), and chemotherapy (17%). At baseline, 46% of patients had visceral disease and 46% had an ESR1 mutation. Participants received imlunestrant alone (n=114) or with the kinase inhibitors everolimus (n=42) or alpelisib (n=21). Investigators assessed each regimen’s safety profile, as well as the objective response rate and clinical benefit rate.
The safety profile of each regimen was similar to those seen with everolimus and alpelisib alone. No cardiac or ocular toxicities were observed. Regarding grade ≥3 treatment-related adverse events:
The imlunestrant alone group experienced fatigue (2%) and neutropenia (2%)
The imlunestrant + everolimus group experienced hypertriglyceridemia (5%) and aspartate aminotransferase increase (5%)
The imlunestrant + alpelisib cohort experienced rash (43%) and hyperglycemia (10%).
In the imlunestrant alone group, 2% of individuals had their doses reduced due to adverse events; none discontinued treatment
In the imlunestrant + everolimus cohort, 12% of patients experienced dose reduction due to everolimus and 2% due to both medications; 2% discontinued treatment due to everolimus
In the imlunestrant + alpelisib cohort, 24% of patients experienced dose reduction due to alpelisib and 14% due to both medications; 29% discontinued treatment due to alpelisib
Regarding efficacy:
The objective response rates in the imlunestrant alone, imlunestrant + everolimus, and imlunestrant + alpelisib groups were 9%, 21%, and 50%, respectively
The clinical benefit rates in the imlunestrant alone, imlunestrant + everolimus, and imlunestrant + alpelisib groups were 42%, 62%, and 62%, respectively
Investigators concluded that imlunestrant used alone or in combination with 1 of the 2 kinase inhibitors demonstrated robust efficacy in individuals with pretreated, ER-positive, HER2-negative advanced breast cancer.
OPERA: OP-1250 Paired With a CDK4/6 Inhibitor: Anti-Tumor Activity With No Dose-Limiting Toxicities
Study synopsis
OP-1250, a CERAN and SERD, continues to show promising results when paired with a CDK4/6 inhibitor. The combination of OP-1250 and the CDK4/6 inhibitor palbociclib appears to be well tolerated and has a similar safety profile to each drug when used alone, according to a phase I/II study involving 20 individuals with pretreated ER-positive, HER2-negative breast cancer.
Participants had advanced or metastatic ER-positive, HER2-negative breast cancer that progressed on ≤1 lines of endocrine therapy. Fourteen participants had received prior CDK4/6 inhibitor therapy, including 11 who were previously treated with palbociclib. Patients received escalating doses of OP-1250 with 125 mg of palbociclib orally daily for 21 of 28 days. OP-1250 doses were 30 mg (n=3), 60 mg (n=3), 90 mg (n=3), and 120 mg (n=11). Investigators assessed pharmacokinetics, drug-drug interactions, safety, and efficacy. Among the results observed to date:
Grade 3 neutropenia occurred in 55% of participants
There were no grade 4 treatment-related adverse events and no dose-limiting toxicities
OP-1250 exposure yielded similar results to what was seen in the previous monotherapy study
Palbociclib exposure was comparable to published monotherapy data when combined with OP-1250 for all dosages
Investigators observed antitumor activity, including partial responses
Researchers concluded that OP-1250 does not affect the pharmacokinetics of palbociclib, and there do not appear to be drug-drug interactions. Tumor response to this combination was encouraging and requires continued investigation.
Conclusions
These 3 studies presented at ESMO 2023 highlight exciting novel therapies for the treatment of HR-positive, HER2-low, and HER2-negative metastatic breast cancer. The EMBER and OPERA updates provide support for the safety and efficacy of these novel endocrine agents in combination with kinase inhibitors and CDK4/6 inhibitors, respectively, in patients with endocrine-sensitive disease, while the TROPION-01 study demonstrates the encouraging efficacy and safety of a second TROP-2-directed ADC in a more heavily pretreated population.
How to prescribe Zepbound
December marks the advent of the approval of tirzepatide (Zepbound) for on-label treatment of obesity. In November 2023, the US Food and Drug Administration (FDA) approved it for the treatment of obesity in adults.
In May 2022, the FDA approved Mounjaro, which is tirzepatide, for type 2 diabetes. Since then, many physicians, including myself, have prescribed it off-label for obesity. As an endocrinologist treating both obesity and diabetes, 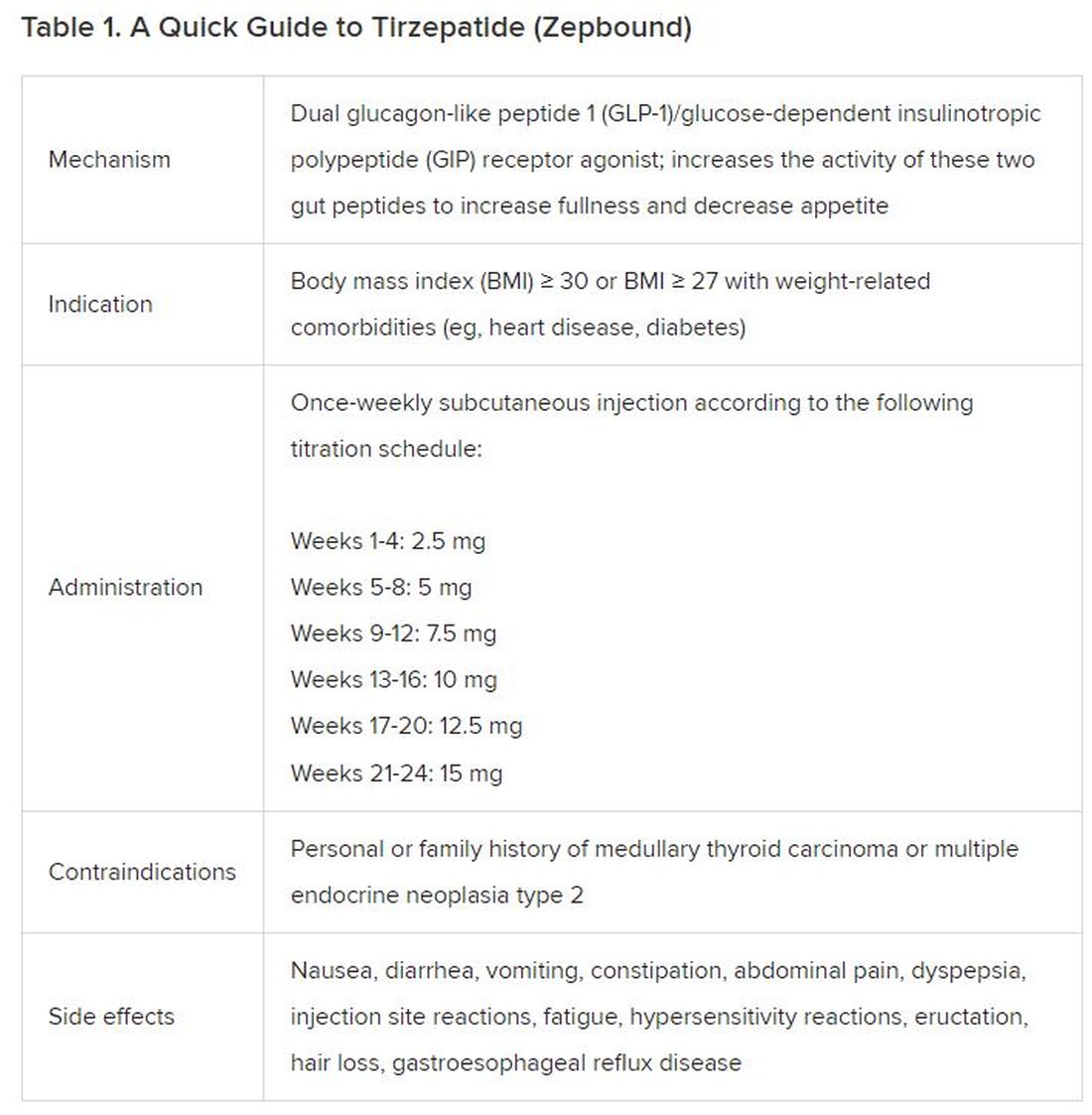
The Expertise
Because GLP-1 receptor agonists have been around since 2005, we’ve had over a decade of clinical experience with these medications. Table 2 provides more nuanced information on tirzepatide (as Zepbound, for obesity) based on our experiences with dulaglutide, liraglutide, semaglutide, and tirzepatide (as Mounjaro).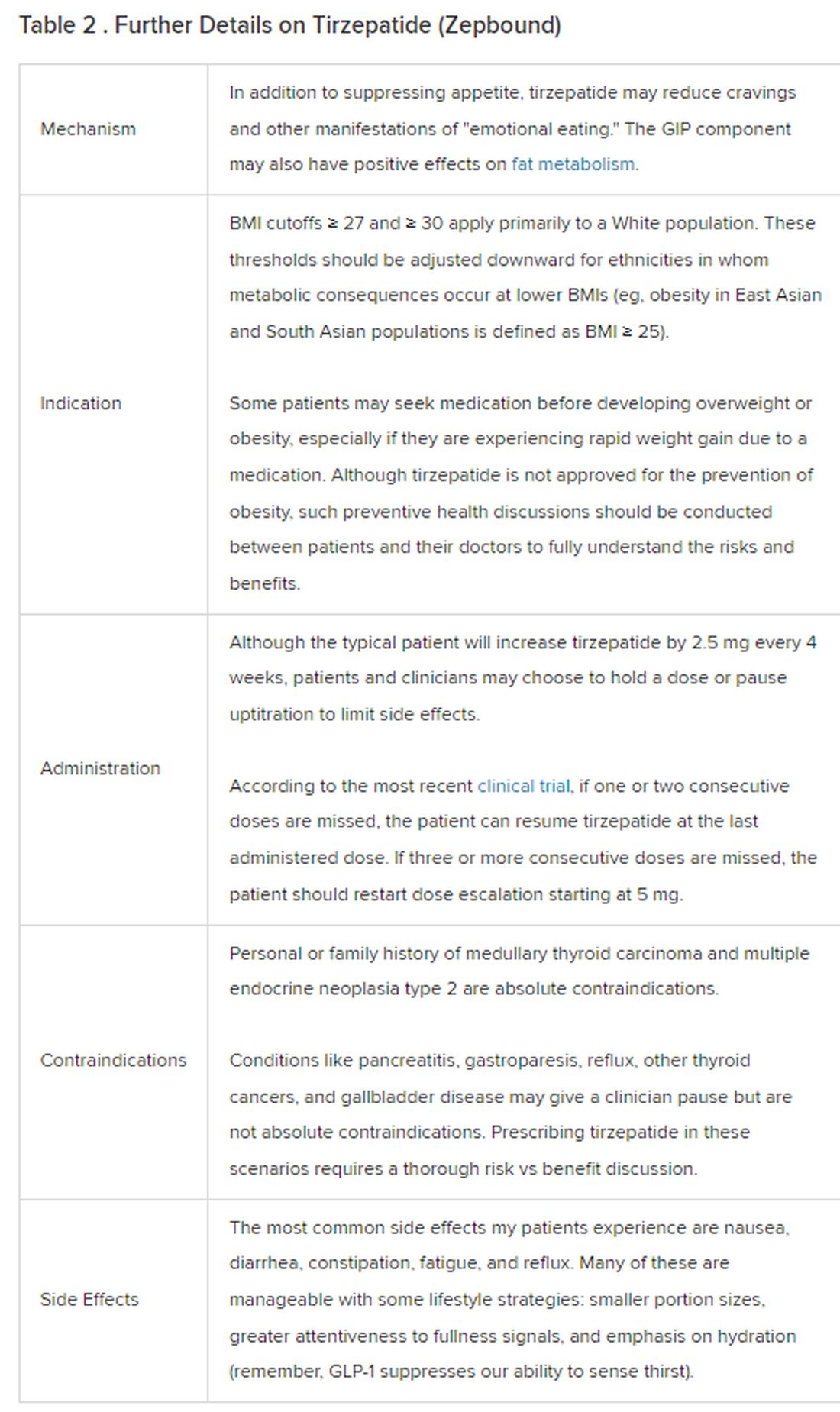
The Reality
In today’s increasingly complex healthcare system, the reality of providing high-quality obesity care is challenging. When discussing tirzepatide with patients, I use a 4 Cs schematic — comorbidities, cautions, costs, choices — to cover the most frequently asked questions.
Comorbidities
In trials, tirzepatide reduced A1c by about 2%. In one diabetes trial, tirzepatide reduced liver fat content significantly more than the comparator (insulin), and trials of tirzepatide in nonalcoholic steatohepatitis are ongoing. A prespecified meta-analysis of tirzepatide and cardiovascular disease estimated a 20% reduction in the risk for cardiovascular death, myocardial infarction, stroke, and hospitalized unstable angina. Tirzepatide as well as other GLP-1 agonists may be beneficial in alcohol use disorder. Prescribing tirzepatide to patients who have or are at risk of developing such comorbidities is an ideal way to target multiple metabolic diseases with one agent.
Cautions
The first principle of medicine is “do no harm.” Tirzepatide may be a poor option for individuals with a history of pancreatitis, gastroparesis, or severe gastroesophageal reflux disease. Because tirzepatide may interfere with the efficacy of estrogen-containing contraceptives during its uptitration phase, women should speak with their doctors about appropriate birth control options (eg, progestin-only, barrier methods). In clinical trials of tirzepatide, male participants were also advised to use reliable contraception. If patients are family-planning, tirzepatide should be discontinued 2 months (for women) and 4 months (for men) before conception, because its effects on fertility or pregnancy are currently unknown.
Costs
At a retail price of $1279 per month, Zepbound is only slightly more affordable than its main competitor, Wegovy (semaglutide 2.4 mg). Complex pharmacy negotiations may reduce this cost, but even with rebates, coupons, and commercial insurance, these costs still place tirzepatide out of reach for many patients. For patients who cannot access tirzepatide, clinicians should discuss more cost-feasible, evidence-based alternatives: for example, phentermine, phentermine-topiramate, naltrexone-bupropion, metformin, bupropion, or topiramate.
Choices
Patient preference drives much of today’s clinical decision-making. Some patients may be switching from semaglutide to tirzepatide, whether by choice or on the basis of physician recommendation. Although no head-to-head obesity trial exists, data from SURPASS-2 and SUSTAIN-FORTE can inform therapeutic equivalence:
- Semaglutide 1.0 mg to tirzepatide 2.5 mg will be a step-down; 5 mg will be a step-up
- Semaglutide 2.0 or 2.4 mg to tirzepatide 5 mg is probably equivalent
The decision to switch therapeutics may depend on weight loss goals, side effect tolerability, or insurance coverage. As with all medications, the use of tirzepatide should progress with shared decision-making, thorough discussions of risks vs benefits, and individualized regimens tailored to each patient’s needs.
The newly approved Zepbound is a valuable addition to our toolbox of obesity treatments. Patients and providers alike are excited for its potential as a highly effective antiobesity medication that can cause a degree of weight loss necessary to reverse comorbidities. The medical management of obesity with agents like tirzepatide holds great promise in addressing today’s obesity epidemic.
Dr. Tchang is Assistant Professor, Clinical Medicine, Division of Endocrinology, Diabetes, and Metabolism, Weill Cornell Medicine; Physician, Department of Medicine, Iris Cantor Women’s Health Center, Comprehensive Weight Control Center, New York, NY. She disclosed ties to Gelesis and Novo Nordisk.
A version of this article appeared on Medscape.com.
December marks the advent of the approval of tirzepatide (Zepbound) for on-label treatment of obesity. In November 2023, the US Food and Drug Administration (FDA) approved it for the treatment of obesity in adults.
In May 2022, the FDA approved Mounjaro, which is tirzepatide, for type 2 diabetes. Since then, many physicians, including myself, have prescribed it off-label for obesity. As an endocrinologist treating both obesity and diabetes,
The Expertise
Because GLP-1 receptor agonists have been around since 2005, we’ve had over a decade of clinical experience with these medications. Table 2 provides more nuanced information on tirzepatide (as Zepbound, for obesity) based on our experiences with dulaglutide, liraglutide, semaglutide, and tirzepatide (as Mounjaro).
The Reality
In today’s increasingly complex healthcare system, the reality of providing high-quality obesity care is challenging. When discussing tirzepatide with patients, I use a 4 Cs schematic — comorbidities, cautions, costs, choices — to cover the most frequently asked questions.
Comorbidities
In trials, tirzepatide reduced A1c by about 2%. In one diabetes trial, tirzepatide reduced liver fat content significantly more than the comparator (insulin), and trials of tirzepatide in nonalcoholic steatohepatitis are ongoing. A prespecified meta-analysis of tirzepatide and cardiovascular disease estimated a 20% reduction in the risk for cardiovascular death, myocardial infarction, stroke, and hospitalized unstable angina. Tirzepatide as well as other GLP-1 agonists may be beneficial in alcohol use disorder. Prescribing tirzepatide to patients who have or are at risk of developing such comorbidities is an ideal way to target multiple metabolic diseases with one agent.
Cautions
The first principle of medicine is “do no harm.” Tirzepatide may be a poor option for individuals with a history of pancreatitis, gastroparesis, or severe gastroesophageal reflux disease. Because tirzepatide may interfere with the efficacy of estrogen-containing contraceptives during its uptitration phase, women should speak with their doctors about appropriate birth control options (eg, progestin-only, barrier methods). In clinical trials of tirzepatide, male participants were also advised to use reliable contraception. If patients are family-planning, tirzepatide should be discontinued 2 months (for women) and 4 months (for men) before conception, because its effects on fertility or pregnancy are currently unknown.
Costs
At a retail price of $1279 per month, Zepbound is only slightly more affordable than its main competitor, Wegovy (semaglutide 2.4 mg). Complex pharmacy negotiations may reduce this cost, but even with rebates, coupons, and commercial insurance, these costs still place tirzepatide out of reach for many patients. For patients who cannot access tirzepatide, clinicians should discuss more cost-feasible, evidence-based alternatives: for example, phentermine, phentermine-topiramate, naltrexone-bupropion, metformin, bupropion, or topiramate.
Choices
Patient preference drives much of today’s clinical decision-making. Some patients may be switching from semaglutide to tirzepatide, whether by choice or on the basis of physician recommendation. Although no head-to-head obesity trial exists, data from SURPASS-2 and SUSTAIN-FORTE can inform therapeutic equivalence:
- Semaglutide 1.0 mg to tirzepatide 2.5 mg will be a step-down; 5 mg will be a step-up
- Semaglutide 2.0 or 2.4 mg to tirzepatide 5 mg is probably equivalent
The decision to switch therapeutics may depend on weight loss goals, side effect tolerability, or insurance coverage. As with all medications, the use of tirzepatide should progress with shared decision-making, thorough discussions of risks vs benefits, and individualized regimens tailored to each patient’s needs.
The newly approved Zepbound is a valuable addition to our toolbox of obesity treatments. Patients and providers alike are excited for its potential as a highly effective antiobesity medication that can cause a degree of weight loss necessary to reverse comorbidities. The medical management of obesity with agents like tirzepatide holds great promise in addressing today’s obesity epidemic.
Dr. Tchang is Assistant Professor, Clinical Medicine, Division of Endocrinology, Diabetes, and Metabolism, Weill Cornell Medicine; Physician, Department of Medicine, Iris Cantor Women’s Health Center, Comprehensive Weight Control Center, New York, NY. She disclosed ties to Gelesis and Novo Nordisk.
A version of this article appeared on Medscape.com.
December marks the advent of the approval of tirzepatide (Zepbound) for on-label treatment of obesity. In November 2023, the US Food and Drug Administration (FDA) approved it for the treatment of obesity in adults.
In May 2022, the FDA approved Mounjaro, which is tirzepatide, for type 2 diabetes. Since then, many physicians, including myself, have prescribed it off-label for obesity. As an endocrinologist treating both obesity and diabetes,
The Expertise
Because GLP-1 receptor agonists have been around since 2005, we’ve had over a decade of clinical experience with these medications. Table 2 provides more nuanced information on tirzepatide (as Zepbound, for obesity) based on our experiences with dulaglutide, liraglutide, semaglutide, and tirzepatide (as Mounjaro).
The Reality
In today’s increasingly complex healthcare system, the reality of providing high-quality obesity care is challenging. When discussing tirzepatide with patients, I use a 4 Cs schematic — comorbidities, cautions, costs, choices — to cover the most frequently asked questions.
Comorbidities
In trials, tirzepatide reduced A1c by about 2%. In one diabetes trial, tirzepatide reduced liver fat content significantly more than the comparator (insulin), and trials of tirzepatide in nonalcoholic steatohepatitis are ongoing. A prespecified meta-analysis of tirzepatide and cardiovascular disease estimated a 20% reduction in the risk for cardiovascular death, myocardial infarction, stroke, and hospitalized unstable angina. Tirzepatide as well as other GLP-1 agonists may be beneficial in alcohol use disorder. Prescribing tirzepatide to patients who have or are at risk of developing such comorbidities is an ideal way to target multiple metabolic diseases with one agent.
Cautions
The first principle of medicine is “do no harm.” Tirzepatide may be a poor option for individuals with a history of pancreatitis, gastroparesis, or severe gastroesophageal reflux disease. Because tirzepatide may interfere with the efficacy of estrogen-containing contraceptives during its uptitration phase, women should speak with their doctors about appropriate birth control options (eg, progestin-only, barrier methods). In clinical trials of tirzepatide, male participants were also advised to use reliable contraception. If patients are family-planning, tirzepatide should be discontinued 2 months (for women) and 4 months (for men) before conception, because its effects on fertility or pregnancy are currently unknown.
Costs
At a retail price of $1279 per month, Zepbound is only slightly more affordable than its main competitor, Wegovy (semaglutide 2.4 mg). Complex pharmacy negotiations may reduce this cost, but even with rebates, coupons, and commercial insurance, these costs still place tirzepatide out of reach for many patients. For patients who cannot access tirzepatide, clinicians should discuss more cost-feasible, evidence-based alternatives: for example, phentermine, phentermine-topiramate, naltrexone-bupropion, metformin, bupropion, or topiramate.
Choices
Patient preference drives much of today’s clinical decision-making. Some patients may be switching from semaglutide to tirzepatide, whether by choice or on the basis of physician recommendation. Although no head-to-head obesity trial exists, data from SURPASS-2 and SUSTAIN-FORTE can inform therapeutic equivalence:
- Semaglutide 1.0 mg to tirzepatide 2.5 mg will be a step-down; 5 mg will be a step-up
- Semaglutide 2.0 or 2.4 mg to tirzepatide 5 mg is probably equivalent
The decision to switch therapeutics may depend on weight loss goals, side effect tolerability, or insurance coverage. As with all medications, the use of tirzepatide should progress with shared decision-making, thorough discussions of risks vs benefits, and individualized regimens tailored to each patient’s needs.
The newly approved Zepbound is a valuable addition to our toolbox of obesity treatments. Patients and providers alike are excited for its potential as a highly effective antiobesity medication that can cause a degree of weight loss necessary to reverse comorbidities. The medical management of obesity with agents like tirzepatide holds great promise in addressing today’s obesity epidemic.
Dr. Tchang is Assistant Professor, Clinical Medicine, Division of Endocrinology, Diabetes, and Metabolism, Weill Cornell Medicine; Physician, Department of Medicine, Iris Cantor Women’s Health Center, Comprehensive Weight Control Center, New York, NY. She disclosed ties to Gelesis and Novo Nordisk.
A version of this article appeared on Medscape.com.
Uveitis Associated with Psoriatic Arthritis: Characteristics, Approaches, and Treatment


With the growing number of treatment options for psoriatic arthritis (PsA), therapeutic decision-making has shifted to an increasingly tailored and patient-centered approach. A number of factors contribute to the treatment decision-making process, including age, insurance restrictions, route of administration, side effect profile, comorbidities, and extra-articular manifestations of the disease. In this article, we discuss an extra-articular comorbidity, uveitis, which is frequently seen in patients with PsA. We discuss clinical characteristics of uveitis associated with PsA and describe how the presence of uveitis influences our treatment approach to PsA, based on existing data.
Uveitis refers broadly to inflammation of the uvea, the vascularized and pigmented layer of the eye composed of the iris, the ciliary body, and the choroid. While infection is a common cause of uveitis, many cases are noninfectious and are often associated with an underlying autoimmune or systemic inflammatory disorder. Uveitis is frequently reported in diseases in the spondyloarthritis (SpA) family, including axial spondyloarthritis (AxSpA) and reactive arthritis, as well as PsA. Exact estimates of the prevalence of uveitis in PsA vary widely from 7%-25%, depending on the particular cohort studied.1,2 In all forms of SpA, the anterior chamber of the uvea is the most likely to be affected.3 However, compared to patients with AxSpA, patients with PsA appear to have a higher rate of posterior involvement. In addition, patients with PsA appear to have higher frequencies of insidious, bilateral uveitis, as compared to the acute, unilateral, anterior uveitis that is most characteristic of AxSpA.4 Women with PsA may be more likely than men to experience uveitis, although this has not been a consistent finding.5
Patients with PsA who are human leukocyte antigen B27 (HLA-B27) positive may be at risk for more severe and refractory anterior uveitis compared to those who do not express the allele.5 Those who are HLA-B27 positive are also known to have higher rates of axial involvement. It has therefore been postulated that 2 phenotypes of uveitis may exist in PsA: patients who are HLA-B27 positive who have axial disease and severe, unilateral anterior uveitis reminiscent of other forms of SpA, and patients who are HLA-B27 negative, often women, with peripheral-predominant arthritis who are prone to the classic anterior uveitis but may also develop atypical bilateral, insidious, and/or posterior involvement.4 Specific characteristics of PsA may also provide information about the risk for developing uveitis. For example, dactylitis has been linked to a higher risk of developing uveitis in some, but not all, cohorts of patients with PsA, and the risk of uveitis in PsA has been found in many studies to correlate with longer duration of disease.6-8
The presence of uveitis signals a disruption in the blood-retina barrier and the subsequent entrance of inflammatory cells into the eye. An entire explanation of pathogenesis is beyond the scope of this article; however, it is worth noting that many of the inflammatory mediators of active uveitis mirror those of PsA. For instance, both the mesenchymal cells in enthesitis and the cells of the ciliary body express receptors for interleukin (IL)-23, suggesting a potential role of the signaling pathways involving this cytokine in both diseases.9 Another study found increased serum levels of IL-17, a known mediator of PsA disease, in patients with active uveitis.10 Despite these common pathogenesis links, there are limited data on the utility of certain existing PsA treatments on uveitis manifestations.
Our approach is always to manage uveitis associated with PsA in collaboration with a specialized and experienced ophthalmologist. Uncontrolled uveitis can be vision threatening and contribute to long-term morbidity associated with PsA, so timely recognition, evaluation, and appropriate treatment are important. Ocular glucocorticoid (GC) drops may be used as first-line therapy, particularly for anterior uveitis, to quickly quell inflammation. Escalation to systemic GCs for more severe or posteriorly localized disease may be considered carefully, given the known risk of worsening skin psoriasis (PsO) with GC withdrawal after a course of therapy. Use of GC-sparing therapy should be determined on a case-by-case basis. While generalized, noninfectious uveitis often resolves with GC treatment, the risk of uveitis recurrence in patients with PsA and the challenges of systemic GCs with PsO lead us to frequently consider GC-sparing therapy that addresses ocular, musculoskeletal, and cutaneous manifestations. Tumor necrosis factor inhibitors (TNF-I) are our typical first-line considerations for GC-sparing therapy in patients with PsA with inflammatory joint symptoms and uveitis, although nonbiologic therapy can be considered first-line therapy in select populations.
Data establishing the efficacy of TNF-I come largely from randomized controlled trials (RCTs) of adalimumab (ADA) compared to placebo in noninfectious uveitis.11 While these trials focused on idiopathic posterior or pan-uveitis, these data have been extrapolated to SpA-associated anterior uveitis, and large registry analyses have supported use of TNF-I in this population.12 When selecting a particular TNF-I in a patient with current or past uveitis, we frequently start with ADA, based on supportive, albeit uncontrolled, data suggesting a reduction in the risk of recurrence with this agent in patients with SpA and uveitis.12 For patients who are unable to tolerate subcutaneous injections, who fail ADA, or who we suspect will require higher, titratable dosing, we favor infliximab infusions. Other data suggest that golimumab and certolizumab are also reasonable alternatives.13,14 We do not generally use etanercept, as the limited data that are available suggest that it is less effective at reducing risk of uveitis recurrence.12 Methotrexate or leflunomide may be an appropriate first line choice for patients with peripheral-predominant PsA and uveitis, but it is important to note that these agents are not effective for axial disease.
Despite the mechanistic data implicating the role of IL-17 in uveitis associated with PsA, the IL-17A inhibitor, secukinumab, failed to show a reduction in uveitis recurrence, compared to placebo, in pooled analysis of RCTs of noninfectious uveitis.15 However, a phase 2 trial of intravenous secukinumab in noninfectious uveitis showed promise, possibly because this dosing regimen can achieve higher effective concentrations.16 It is not our current practice to use secukinumab or ixekizumab as a first-line therapy in patients with PsA and concurrent uveitis, owing to a lack of data supporting efficacy. A novel IL-17A/F inhibitor, bimekizumab (BKZ), has recently been used in several successful phase 3 trials in patients with both TNF-naïve and TNF-nonresponder SpA, including AxSpA and PsA.17 Interestingly, data from the phase 2 and 3 trials of BKZ found low incidence rates of uveitis in patients with SpA treated with BKZ compared to placebo, suggesting that BKZ might be more effective in uveitis than other IL-17 inhibitors, but these data need to be confirmed.
Successful use of Janus kinase (JAK) inhibitors in noninfectious uveitis, including cases associated with inflammatory arthritis, has been described in case reports as well as in current phase 2 trials.18 The dual IL-12/IL-23 inhibitor, ustekinumab, also showed initial promise in a small, nonrandomized, uncontrolled phase 1/2 study of the treatment of posterior uveitis, as well as success in few case reports of PsA-associated uveitis.19 However, a post-hoc analysis of extra-intestinal manifestations, including uveitis and iritis, in patients with inflammatory bowel disease treated with ustekinumab found no benefit in preventing or treating ocular disease compared to placebo.20 Given the paucity of available data, JAK inhibitors and the IL-12/IL-23 inhibitor, ustekinumab, are not part of our typical treatment algorithm for patients with PsA-associated uveitis.
In conclusion, uveitis is a frequent extra-articular comorbidity of PsA, and it may present differently than the typical acute onset, unilateral anterior uveitis seen in SpA. While uveitis may share many different immunologic threads with PsA, the most convincing data support the use of TNF-I as a GC-sparing agent in this setting, particularly ADA, infliximab, golimumab, or certolizumab. Our approach is generally to start with these agents or methotrexate when directed therapy is needed for uveitis in PsA. Further investigation into the use of the IL-17A/F inhibitor BKZ and JAK inhibitors, as well as tyrosine kinase 2 inhibitors, in PsA associated uveitis may yield additional options for our patients.21
1. De Vicente Delmas A, Sanchez-Bilbao L, Calvo-Rio V, et al. Uveitis in psoriatic arthritis: study of 406 patients in a single university center and literature review. RMD Open. 2023;9(1):e002781.
2. Rademacher J, Poddubnyy D, Pleyer U. Uveitis in spondyloarthritis. Ther Adv Musculoskelet Dis. 2020;12:1759720X20951733.
3. Zeboulon N, Dougados M, Gossec L. Prevalence and characteristics of uveitis in the spondyloarthropathies: a systematic literature review. Ann Rheum Dis. 2008;67(7):955-959.
4. Paiva ES, Macaluso DC, Edwards A, Rosenbaum JT. Characterisation of uveitis in patients with psoriatic arthritis. Ann Rheum Dis. 2000;59(1):67-70.
5. Fraga NA, Oliveira Mde F, Follador I, Rocha Bde O, Rego VR. Psoriasis and uveitis: a literature review. An Bras Dermatol. 2012;87(6):877-883.
6. Niccoli L, Nannini C, Cassara E, et al. Frequency of iridocyclitis in patients with early psoriatic arthritis: a prospective, follow up study. Int J Rheum Dis. 2012;15(4):414-418.
7. Yasar Bilge NS, Kalyoncu U, Atagunduz P, et al. Uveitis-related factors in patients with spondyloarthritis: TReasure Real-Life Results. Am J Ophthalmol. 2021;228:58-64.
8. Chia AYT, Ang GWX, Chan ASY, Chan W, Chong TKY, Leung YY. Managing psoriatic arthritis with inflammatory bowel disease and/or uveitis. Front Med (Lausanne). 2021;8:737256.
9. Reinhardt A, Yevsa T, Worbs T, et al. Interleukin-23-dependent gamma/delta T cells produce interleukin-17 and accumulate in the enthesis, aortic valve, and ciliary body in mice. Arthritis Rheumatol. 2016;68(10):2476-2486.
10. Jawad S, Liu B, Agron E, Nussenblatt RB, Sen HN. Elevated serum levels of interleukin-17A in uveitis patients. Ocul Immunol Inflamm. 2013;21(6):434-439.
11. Merrill PT, Vitale A, Zierhut M, et al. Efficacy of adalimumab in non-infectious uveitis across different etiologies: a post hoc analysis of the VISUAL I and VISUAL II Trials. Ocul Immunol Inflamm. 2021;29(7-8):1569-1575.
12. Lie E, Lindstrom U, Zverkova-Sandstrom T, et al. Tumour necrosis factor inhibitor treatment and occurrence of anterior uveitis in ankylosing spondylitis: results from the Swedish biologics register. Ann Rheum Dis. 2017;76(9):1515-1521.
13. van der Horst-Bruinsma I, van Bentum R, Verbraak FD, et al. The impact of certolizumab pegol treatment on the incidence of anterior uveitis flares in patients with axial spondyloarthritis: 48-week interim results from C-VIEW. RMD Open. 2020;6(1):e001161.
14. Calvo-Rio V, Blanco R, Santos-Gomez M, et al. Golimumab in refractory uveitis related to spondyloarthritis. Multicenter study of 15 patients. Semin Arthritis Rheum. 2016;46(1):95-101.
15. Dick AD, Tugal-Tutkun I, Foster S, et al. Secukinumab in the treatment of noninfectious uveitis: results of three randomized, controlled clinical trials. Ophthalmology. 2013;120(4):777-787.
16. Letko E, Yeh S, Foster CS, et al. Efficacy and safety of intravenous secukinumab in noninfectious uveitis requiring steroid-sparing immunosuppressive therapy. Ophthalmology. 2015;122(5):939-948.
17. van der Heijde D, Deodhar A, Baraliakos X, et al. Efficacy and safety of bimekizumab in axial spondyloarthritis: results of two parallel phase 3 randomised controlled trials. Ann Rheum Dis. 2023;82(4):515-526.
18. Dhillon S, Keam SJ. Filgotinib: first approval. Drugs. 2020;80(18):1987-1997.
19. Pepple KL, Lin P. Targeting interleukin-23 in the treatment of noninfectious uveitis. Ophthalmology. 2018;125(12):1977-1983.
20. Narula N, Aruljothy A, Wong ECL, et al. The impact of ustekinumab on extraintestinal manifestations of Crohn’s disease: a post hoc analysis of the UNITI studies. United European Gastroenterol J. 2021;9(5):581-589.
21. Rusinol L, Puig L. Tyk2 targeting in immune-mediated inflammatory diseases. Int J Mol Sci. 2023;24(4):3391.
With the growing number of treatment options for psoriatic arthritis (PsA), therapeutic decision-making has shifted to an increasingly tailored and patient-centered approach. A number of factors contribute to the treatment decision-making process, including age, insurance restrictions, route of administration, side effect profile, comorbidities, and extra-articular manifestations of the disease. In this article, we discuss an extra-articular comorbidity, uveitis, which is frequently seen in patients with PsA. We discuss clinical characteristics of uveitis associated with PsA and describe how the presence of uveitis influences our treatment approach to PsA, based on existing data.
Uveitis refers broadly to inflammation of the uvea, the vascularized and pigmented layer of the eye composed of the iris, the ciliary body, and the choroid. While infection is a common cause of uveitis, many cases are noninfectious and are often associated with an underlying autoimmune or systemic inflammatory disorder. Uveitis is frequently reported in diseases in the spondyloarthritis (SpA) family, including axial spondyloarthritis (AxSpA) and reactive arthritis, as well as PsA. Exact estimates of the prevalence of uveitis in PsA vary widely from 7%-25%, depending on the particular cohort studied.1,2 In all forms of SpA, the anterior chamber of the uvea is the most likely to be affected.3 However, compared to patients with AxSpA, patients with PsA appear to have a higher rate of posterior involvement. In addition, patients with PsA appear to have higher frequencies of insidious, bilateral uveitis, as compared to the acute, unilateral, anterior uveitis that is most characteristic of AxSpA.4 Women with PsA may be more likely than men to experience uveitis, although this has not been a consistent finding.5
Patients with PsA who are human leukocyte antigen B27 (HLA-B27) positive may be at risk for more severe and refractory anterior uveitis compared to those who do not express the allele.5 Those who are HLA-B27 positive are also known to have higher rates of axial involvement. It has therefore been postulated that 2 phenotypes of uveitis may exist in PsA: patients who are HLA-B27 positive who have axial disease and severe, unilateral anterior uveitis reminiscent of other forms of SpA, and patients who are HLA-B27 negative, often women, with peripheral-predominant arthritis who are prone to the classic anterior uveitis but may also develop atypical bilateral, insidious, and/or posterior involvement.4 Specific characteristics of PsA may also provide information about the risk for developing uveitis. For example, dactylitis has been linked to a higher risk of developing uveitis in some, but not all, cohorts of patients with PsA, and the risk of uveitis in PsA has been found in many studies to correlate with longer duration of disease.6-8
The presence of uveitis signals a disruption in the blood-retina barrier and the subsequent entrance of inflammatory cells into the eye. An entire explanation of pathogenesis is beyond the scope of this article; however, it is worth noting that many of the inflammatory mediators of active uveitis mirror those of PsA. For instance, both the mesenchymal cells in enthesitis and the cells of the ciliary body express receptors for interleukin (IL)-23, suggesting a potential role of the signaling pathways involving this cytokine in both diseases.9 Another study found increased serum levels of IL-17, a known mediator of PsA disease, in patients with active uveitis.10 Despite these common pathogenesis links, there are limited data on the utility of certain existing PsA treatments on uveitis manifestations.
Our approach is always to manage uveitis associated with PsA in collaboration with a specialized and experienced ophthalmologist. Uncontrolled uveitis can be vision threatening and contribute to long-term morbidity associated with PsA, so timely recognition, evaluation, and appropriate treatment are important. Ocular glucocorticoid (GC) drops may be used as first-line therapy, particularly for anterior uveitis, to quickly quell inflammation. Escalation to systemic GCs for more severe or posteriorly localized disease may be considered carefully, given the known risk of worsening skin psoriasis (PsO) with GC withdrawal after a course of therapy. Use of GC-sparing therapy should be determined on a case-by-case basis. While generalized, noninfectious uveitis often resolves with GC treatment, the risk of uveitis recurrence in patients with PsA and the challenges of systemic GCs with PsO lead us to frequently consider GC-sparing therapy that addresses ocular, musculoskeletal, and cutaneous manifestations. Tumor necrosis factor inhibitors (TNF-I) are our typical first-line considerations for GC-sparing therapy in patients with PsA with inflammatory joint symptoms and uveitis, although nonbiologic therapy can be considered first-line therapy in select populations.
Data establishing the efficacy of TNF-I come largely from randomized controlled trials (RCTs) of adalimumab (ADA) compared to placebo in noninfectious uveitis.11 While these trials focused on idiopathic posterior or pan-uveitis, these data have been extrapolated to SpA-associated anterior uveitis, and large registry analyses have supported use of TNF-I in this population.12 When selecting a particular TNF-I in a patient with current or past uveitis, we frequently start with ADA, based on supportive, albeit uncontrolled, data suggesting a reduction in the risk of recurrence with this agent in patients with SpA and uveitis.12 For patients who are unable to tolerate subcutaneous injections, who fail ADA, or who we suspect will require higher, titratable dosing, we favor infliximab infusions. Other data suggest that golimumab and certolizumab are also reasonable alternatives.13,14 We do not generally use etanercept, as the limited data that are available suggest that it is less effective at reducing risk of uveitis recurrence.12 Methotrexate or leflunomide may be an appropriate first line choice for patients with peripheral-predominant PsA and uveitis, but it is important to note that these agents are not effective for axial disease.
Despite the mechanistic data implicating the role of IL-17 in uveitis associated with PsA, the IL-17A inhibitor, secukinumab, failed to show a reduction in uveitis recurrence, compared to placebo, in pooled analysis of RCTs of noninfectious uveitis.15 However, a phase 2 trial of intravenous secukinumab in noninfectious uveitis showed promise, possibly because this dosing regimen can achieve higher effective concentrations.16 It is not our current practice to use secukinumab or ixekizumab as a first-line therapy in patients with PsA and concurrent uveitis, owing to a lack of data supporting efficacy. A novel IL-17A/F inhibitor, bimekizumab (BKZ), has recently been used in several successful phase 3 trials in patients with both TNF-naïve and TNF-nonresponder SpA, including AxSpA and PsA.17 Interestingly, data from the phase 2 and 3 trials of BKZ found low incidence rates of uveitis in patients with SpA treated with BKZ compared to placebo, suggesting that BKZ might be more effective in uveitis than other IL-17 inhibitors, but these data need to be confirmed.
Successful use of Janus kinase (JAK) inhibitors in noninfectious uveitis, including cases associated with inflammatory arthritis, has been described in case reports as well as in current phase 2 trials.18 The dual IL-12/IL-23 inhibitor, ustekinumab, also showed initial promise in a small, nonrandomized, uncontrolled phase 1/2 study of the treatment of posterior uveitis, as well as success in few case reports of PsA-associated uveitis.19 However, a post-hoc analysis of extra-intestinal manifestations, including uveitis and iritis, in patients with inflammatory bowel disease treated with ustekinumab found no benefit in preventing or treating ocular disease compared to placebo.20 Given the paucity of available data, JAK inhibitors and the IL-12/IL-23 inhibitor, ustekinumab, are not part of our typical treatment algorithm for patients with PsA-associated uveitis.
In conclusion, uveitis is a frequent extra-articular comorbidity of PsA, and it may present differently than the typical acute onset, unilateral anterior uveitis seen in SpA. While uveitis may share many different immunologic threads with PsA, the most convincing data support the use of TNF-I as a GC-sparing agent in this setting, particularly ADA, infliximab, golimumab, or certolizumab. Our approach is generally to start with these agents or methotrexate when directed therapy is needed for uveitis in PsA. Further investigation into the use of the IL-17A/F inhibitor BKZ and JAK inhibitors, as well as tyrosine kinase 2 inhibitors, in PsA associated uveitis may yield additional options for our patients.21
With the growing number of treatment options for psoriatic arthritis (PsA), therapeutic decision-making has shifted to an increasingly tailored and patient-centered approach. A number of factors contribute to the treatment decision-making process, including age, insurance restrictions, route of administration, side effect profile, comorbidities, and extra-articular manifestations of the disease. In this article, we discuss an extra-articular comorbidity, uveitis, which is frequently seen in patients with PsA. We discuss clinical characteristics of uveitis associated with PsA and describe how the presence of uveitis influences our treatment approach to PsA, based on existing data.
Uveitis refers broadly to inflammation of the uvea, the vascularized and pigmented layer of the eye composed of the iris, the ciliary body, and the choroid. While infection is a common cause of uveitis, many cases are noninfectious and are often associated with an underlying autoimmune or systemic inflammatory disorder. Uveitis is frequently reported in diseases in the spondyloarthritis (SpA) family, including axial spondyloarthritis (AxSpA) and reactive arthritis, as well as PsA. Exact estimates of the prevalence of uveitis in PsA vary widely from 7%-25%, depending on the particular cohort studied.1,2 In all forms of SpA, the anterior chamber of the uvea is the most likely to be affected.3 However, compared to patients with AxSpA, patients with PsA appear to have a higher rate of posterior involvement. In addition, patients with PsA appear to have higher frequencies of insidious, bilateral uveitis, as compared to the acute, unilateral, anterior uveitis that is most characteristic of AxSpA.4 Women with PsA may be more likely than men to experience uveitis, although this has not been a consistent finding.5
Patients with PsA who are human leukocyte antigen B27 (HLA-B27) positive may be at risk for more severe and refractory anterior uveitis compared to those who do not express the allele.5 Those who are HLA-B27 positive are also known to have higher rates of axial involvement. It has therefore been postulated that 2 phenotypes of uveitis may exist in PsA: patients who are HLA-B27 positive who have axial disease and severe, unilateral anterior uveitis reminiscent of other forms of SpA, and patients who are HLA-B27 negative, often women, with peripheral-predominant arthritis who are prone to the classic anterior uveitis but may also develop atypical bilateral, insidious, and/or posterior involvement.4 Specific characteristics of PsA may also provide information about the risk for developing uveitis. For example, dactylitis has been linked to a higher risk of developing uveitis in some, but not all, cohorts of patients with PsA, and the risk of uveitis in PsA has been found in many studies to correlate with longer duration of disease.6-8
The presence of uveitis signals a disruption in the blood-retina barrier and the subsequent entrance of inflammatory cells into the eye. An entire explanation of pathogenesis is beyond the scope of this article; however, it is worth noting that many of the inflammatory mediators of active uveitis mirror those of PsA. For instance, both the mesenchymal cells in enthesitis and the cells of the ciliary body express receptors for interleukin (IL)-23, suggesting a potential role of the signaling pathways involving this cytokine in both diseases.9 Another study found increased serum levels of IL-17, a known mediator of PsA disease, in patients with active uveitis.10 Despite these common pathogenesis links, there are limited data on the utility of certain existing PsA treatments on uveitis manifestations.
Our approach is always to manage uveitis associated with PsA in collaboration with a specialized and experienced ophthalmologist. Uncontrolled uveitis can be vision threatening and contribute to long-term morbidity associated with PsA, so timely recognition, evaluation, and appropriate treatment are important. Ocular glucocorticoid (GC) drops may be used as first-line therapy, particularly for anterior uveitis, to quickly quell inflammation. Escalation to systemic GCs for more severe or posteriorly localized disease may be considered carefully, given the known risk of worsening skin psoriasis (PsO) with GC withdrawal after a course of therapy. Use of GC-sparing therapy should be determined on a case-by-case basis. While generalized, noninfectious uveitis often resolves with GC treatment, the risk of uveitis recurrence in patients with PsA and the challenges of systemic GCs with PsO lead us to frequently consider GC-sparing therapy that addresses ocular, musculoskeletal, and cutaneous manifestations. Tumor necrosis factor inhibitors (TNF-I) are our typical first-line considerations for GC-sparing therapy in patients with PsA with inflammatory joint symptoms and uveitis, although nonbiologic therapy can be considered first-line therapy in select populations.
Data establishing the efficacy of TNF-I come largely from randomized controlled trials (RCTs) of adalimumab (ADA) compared to placebo in noninfectious uveitis.11 While these trials focused on idiopathic posterior or pan-uveitis, these data have been extrapolated to SpA-associated anterior uveitis, and large registry analyses have supported use of TNF-I in this population.12 When selecting a particular TNF-I in a patient with current or past uveitis, we frequently start with ADA, based on supportive, albeit uncontrolled, data suggesting a reduction in the risk of recurrence with this agent in patients with SpA and uveitis.12 For patients who are unable to tolerate subcutaneous injections, who fail ADA, or who we suspect will require higher, titratable dosing, we favor infliximab infusions. Other data suggest that golimumab and certolizumab are also reasonable alternatives.13,14 We do not generally use etanercept, as the limited data that are available suggest that it is less effective at reducing risk of uveitis recurrence.12 Methotrexate or leflunomide may be an appropriate first line choice for patients with peripheral-predominant PsA and uveitis, but it is important to note that these agents are not effective for axial disease.
Despite the mechanistic data implicating the role of IL-17 in uveitis associated with PsA, the IL-17A inhibitor, secukinumab, failed to show a reduction in uveitis recurrence, compared to placebo, in pooled analysis of RCTs of noninfectious uveitis.15 However, a phase 2 trial of intravenous secukinumab in noninfectious uveitis showed promise, possibly because this dosing regimen can achieve higher effective concentrations.16 It is not our current practice to use secukinumab or ixekizumab as a first-line therapy in patients with PsA and concurrent uveitis, owing to a lack of data supporting efficacy. A novel IL-17A/F inhibitor, bimekizumab (BKZ), has recently been used in several successful phase 3 trials in patients with both TNF-naïve and TNF-nonresponder SpA, including AxSpA and PsA.17 Interestingly, data from the phase 2 and 3 trials of BKZ found low incidence rates of uveitis in patients with SpA treated with BKZ compared to placebo, suggesting that BKZ might be more effective in uveitis than other IL-17 inhibitors, but these data need to be confirmed.
Successful use of Janus kinase (JAK) inhibitors in noninfectious uveitis, including cases associated with inflammatory arthritis, has been described in case reports as well as in current phase 2 trials.18 The dual IL-12/IL-23 inhibitor, ustekinumab, also showed initial promise in a small, nonrandomized, uncontrolled phase 1/2 study of the treatment of posterior uveitis, as well as success in few case reports of PsA-associated uveitis.19 However, a post-hoc analysis of extra-intestinal manifestations, including uveitis and iritis, in patients with inflammatory bowel disease treated with ustekinumab found no benefit in preventing or treating ocular disease compared to placebo.20 Given the paucity of available data, JAK inhibitors and the IL-12/IL-23 inhibitor, ustekinumab, are not part of our typical treatment algorithm for patients with PsA-associated uveitis.
In conclusion, uveitis is a frequent extra-articular comorbidity of PsA, and it may present differently than the typical acute onset, unilateral anterior uveitis seen in SpA. While uveitis may share many different immunologic threads with PsA, the most convincing data support the use of TNF-I as a GC-sparing agent in this setting, particularly ADA, infliximab, golimumab, or certolizumab. Our approach is generally to start with these agents or methotrexate when directed therapy is needed for uveitis in PsA. Further investigation into the use of the IL-17A/F inhibitor BKZ and JAK inhibitors, as well as tyrosine kinase 2 inhibitors, in PsA associated uveitis may yield additional options for our patients.21
1. De Vicente Delmas A, Sanchez-Bilbao L, Calvo-Rio V, et al. Uveitis in psoriatic arthritis: study of 406 patients in a single university center and literature review. RMD Open. 2023;9(1):e002781.
2. Rademacher J, Poddubnyy D, Pleyer U. Uveitis in spondyloarthritis. Ther Adv Musculoskelet Dis. 2020;12:1759720X20951733.
3. Zeboulon N, Dougados M, Gossec L. Prevalence and characteristics of uveitis in the spondyloarthropathies: a systematic literature review. Ann Rheum Dis. 2008;67(7):955-959.
4. Paiva ES, Macaluso DC, Edwards A, Rosenbaum JT. Characterisation of uveitis in patients with psoriatic arthritis. Ann Rheum Dis. 2000;59(1):67-70.
5. Fraga NA, Oliveira Mde F, Follador I, Rocha Bde O, Rego VR. Psoriasis and uveitis: a literature review. An Bras Dermatol. 2012;87(6):877-883.
6. Niccoli L, Nannini C, Cassara E, et al. Frequency of iridocyclitis in patients with early psoriatic arthritis: a prospective, follow up study. Int J Rheum Dis. 2012;15(4):414-418.
7. Yasar Bilge NS, Kalyoncu U, Atagunduz P, et al. Uveitis-related factors in patients with spondyloarthritis: TReasure Real-Life Results. Am J Ophthalmol. 2021;228:58-64.
8. Chia AYT, Ang GWX, Chan ASY, Chan W, Chong TKY, Leung YY. Managing psoriatic arthritis with inflammatory bowel disease and/or uveitis. Front Med (Lausanne). 2021;8:737256.
9. Reinhardt A, Yevsa T, Worbs T, et al. Interleukin-23-dependent gamma/delta T cells produce interleukin-17 and accumulate in the enthesis, aortic valve, and ciliary body in mice. Arthritis Rheumatol. 2016;68(10):2476-2486.
10. Jawad S, Liu B, Agron E, Nussenblatt RB, Sen HN. Elevated serum levels of interleukin-17A in uveitis patients. Ocul Immunol Inflamm. 2013;21(6):434-439.
11. Merrill PT, Vitale A, Zierhut M, et al. Efficacy of adalimumab in non-infectious uveitis across different etiologies: a post hoc analysis of the VISUAL I and VISUAL II Trials. Ocul Immunol Inflamm. 2021;29(7-8):1569-1575.
12. Lie E, Lindstrom U, Zverkova-Sandstrom T, et al. Tumour necrosis factor inhibitor treatment and occurrence of anterior uveitis in ankylosing spondylitis: results from the Swedish biologics register. Ann Rheum Dis. 2017;76(9):1515-1521.
13. van der Horst-Bruinsma I, van Bentum R, Verbraak FD, et al. The impact of certolizumab pegol treatment on the incidence of anterior uveitis flares in patients with axial spondyloarthritis: 48-week interim results from C-VIEW. RMD Open. 2020;6(1):e001161.
14. Calvo-Rio V, Blanco R, Santos-Gomez M, et al. Golimumab in refractory uveitis related to spondyloarthritis. Multicenter study of 15 patients. Semin Arthritis Rheum. 2016;46(1):95-101.
15. Dick AD, Tugal-Tutkun I, Foster S, et al. Secukinumab in the treatment of noninfectious uveitis: results of three randomized, controlled clinical trials. Ophthalmology. 2013;120(4):777-787.
16. Letko E, Yeh S, Foster CS, et al. Efficacy and safety of intravenous secukinumab in noninfectious uveitis requiring steroid-sparing immunosuppressive therapy. Ophthalmology. 2015;122(5):939-948.
17. van der Heijde D, Deodhar A, Baraliakos X, et al. Efficacy and safety of bimekizumab in axial spondyloarthritis: results of two parallel phase 3 randomised controlled trials. Ann Rheum Dis. 2023;82(4):515-526.
18. Dhillon S, Keam SJ. Filgotinib: first approval. Drugs. 2020;80(18):1987-1997.
19. Pepple KL, Lin P. Targeting interleukin-23 in the treatment of noninfectious uveitis. Ophthalmology. 2018;125(12):1977-1983.
20. Narula N, Aruljothy A, Wong ECL, et al. The impact of ustekinumab on extraintestinal manifestations of Crohn’s disease: a post hoc analysis of the UNITI studies. United European Gastroenterol J. 2021;9(5):581-589.
21. Rusinol L, Puig L. Tyk2 targeting in immune-mediated inflammatory diseases. Int J Mol Sci. 2023;24(4):3391.
1. De Vicente Delmas A, Sanchez-Bilbao L, Calvo-Rio V, et al. Uveitis in psoriatic arthritis: study of 406 patients in a single university center and literature review. RMD Open. 2023;9(1):e002781.
2. Rademacher J, Poddubnyy D, Pleyer U. Uveitis in spondyloarthritis. Ther Adv Musculoskelet Dis. 2020;12:1759720X20951733.
3. Zeboulon N, Dougados M, Gossec L. Prevalence and characteristics of uveitis in the spondyloarthropathies: a systematic literature review. Ann Rheum Dis. 2008;67(7):955-959.
4. Paiva ES, Macaluso DC, Edwards A, Rosenbaum JT. Characterisation of uveitis in patients with psoriatic arthritis. Ann Rheum Dis. 2000;59(1):67-70.
5. Fraga NA, Oliveira Mde F, Follador I, Rocha Bde O, Rego VR. Psoriasis and uveitis: a literature review. An Bras Dermatol. 2012;87(6):877-883.
6. Niccoli L, Nannini C, Cassara E, et al. Frequency of iridocyclitis in patients with early psoriatic arthritis: a prospective, follow up study. Int J Rheum Dis. 2012;15(4):414-418.
7. Yasar Bilge NS, Kalyoncu U, Atagunduz P, et al. Uveitis-related factors in patients with spondyloarthritis: TReasure Real-Life Results. Am J Ophthalmol. 2021;228:58-64.
8. Chia AYT, Ang GWX, Chan ASY, Chan W, Chong TKY, Leung YY. Managing psoriatic arthritis with inflammatory bowel disease and/or uveitis. Front Med (Lausanne). 2021;8:737256.
9. Reinhardt A, Yevsa T, Worbs T, et al. Interleukin-23-dependent gamma/delta T cells produce interleukin-17 and accumulate in the enthesis, aortic valve, and ciliary body in mice. Arthritis Rheumatol. 2016;68(10):2476-2486.
10. Jawad S, Liu B, Agron E, Nussenblatt RB, Sen HN. Elevated serum levels of interleukin-17A in uveitis patients. Ocul Immunol Inflamm. 2013;21(6):434-439.
11. Merrill PT, Vitale A, Zierhut M, et al. Efficacy of adalimumab in non-infectious uveitis across different etiologies: a post hoc analysis of the VISUAL I and VISUAL II Trials. Ocul Immunol Inflamm. 2021;29(7-8):1569-1575.
12. Lie E, Lindstrom U, Zverkova-Sandstrom T, et al. Tumour necrosis factor inhibitor treatment and occurrence of anterior uveitis in ankylosing spondylitis: results from the Swedish biologics register. Ann Rheum Dis. 2017;76(9):1515-1521.
13. van der Horst-Bruinsma I, van Bentum R, Verbraak FD, et al. The impact of certolizumab pegol treatment on the incidence of anterior uveitis flares in patients with axial spondyloarthritis: 48-week interim results from C-VIEW. RMD Open. 2020;6(1):e001161.
14. Calvo-Rio V, Blanco R, Santos-Gomez M, et al. Golimumab in refractory uveitis related to spondyloarthritis. Multicenter study of 15 patients. Semin Arthritis Rheum. 2016;46(1):95-101.
15. Dick AD, Tugal-Tutkun I, Foster S, et al. Secukinumab in the treatment of noninfectious uveitis: results of three randomized, controlled clinical trials. Ophthalmology. 2013;120(4):777-787.
16. Letko E, Yeh S, Foster CS, et al. Efficacy and safety of intravenous secukinumab in noninfectious uveitis requiring steroid-sparing immunosuppressive therapy. Ophthalmology. 2015;122(5):939-948.
17. van der Heijde D, Deodhar A, Baraliakos X, et al. Efficacy and safety of bimekizumab in axial spondyloarthritis: results of two parallel phase 3 randomised controlled trials. Ann Rheum Dis. 2023;82(4):515-526.
18. Dhillon S, Keam SJ. Filgotinib: first approval. Drugs. 2020;80(18):1987-1997.
19. Pepple KL, Lin P. Targeting interleukin-23 in the treatment of noninfectious uveitis. Ophthalmology. 2018;125(12):1977-1983.
20. Narula N, Aruljothy A, Wong ECL, et al. The impact of ustekinumab on extraintestinal manifestations of Crohn’s disease: a post hoc analysis of the UNITI studies. United European Gastroenterol J. 2021;9(5):581-589.
21. Rusinol L, Puig L. Tyk2 targeting in immune-mediated inflammatory diseases. Int J Mol Sci. 2023;24(4):3391.
Switching Patients From a Triptan to a Gepant for Acute Migraine Care and Effective Preventives

Dr. Rapoport: Most patients who come into my office today, even those whom I have
treated for the last 30 years for acute care of migraine attacks, are taking 1 of the 7
triptan medications available. They might be taking triptans as a tablet—the most
common form—as a nasal spray, or by injection; however, not all patients are suited for
triptans, and sometimes, the need arises to switch to a different class of medication for
treating migraine acutely.
What are the reasons patients switch from a triptan to a gepant?
For some patients, triptans are not working well enough or are causing adverse events.
Other patients have developed cardiac risk factors such as elevated blood pressure,
obesity, smoking, and/or lack of exercise. I am always concerned about constriction of
the coronary blood vessels. Patients who already have some cardiac risk factors and
those who have some actual cardiac disease or have had a previous heart attack
already have constriction of their blood vessels and are not candidates for triptans, as
they are contraindicated.
How do you switch a patient from a triptan to a gepant?
It is important to have some discussion with the patient before the switch. For example,
if a patient with no cardiac risk factors comes into the office asking about this new
medicine, I will ask them several questions about their triptan to ensure it works well
enough (ie, to ascertain if the patient’s migraines improve within 30 to 60 minutes and
are much better within 2 hours of taking the medication). I want to be sure that they do
not have any adverse events related to the triptan, such as chest pain, drowsiness, or
dizziness. I like to ensure that whatever they are taking works long enough—at least 24
hours, preferably 48 hours—so they no longer have a headache, especially the next
day. If the headache comes back the next day, they must re-treat. If I determine the
triptan is not working well for them or they have significant adverse events, I will move
on.
Gepants are small-molecule calcitonin gene-related peptide (CGRP) receptor
antagonists, which are pills that only last for 2 to 3 days in the body. There are 2
gepants for acute care and 2 for the prevention of migraine. The first gepant approved
by the US Food and Drug Administration (FDA) for acute care was ubrogepant
(Ubrelvy), which comes in 2 sizes, 50 mg or 100 mg tablets. I sometimes start with 50
mg, but for the more difficult migraine patient, I will start with 100 mg. If the medicine is
not doing a complete job within 2 hours, the patient may take a second dose, up to 200
mg. Some adverse events may include nausea or slight drowsiness. The patient should
avoid certain medicines such as antifungal medicines (eg, ketoconazole, itraconazole)
and certain antibiotics like clarithromycin.
Another gepant, rimegepant (Nurtec), comes in only 1 size, a 75-mg oral disintegrating
tablet, which can be used both for acute care of migraine and for prevention. Patients
can take a tablet as soon as their migraine attack begins, and they are not to repeat it
that day. If the headache does not go away in 2 hours, I want them to then take a triptan
and an anti-inflammatory drug (there is no contraindication to mix these drugs). I want
them to try it at least 1 more time, encouraging patients to take it early, right at the start
of the headache. If the medicine is still not working by the second or third time, they
should stop using it. Preventively, patients take 75 mg every other day, which can be
quite effective. Side effects are slight nausea and some abdominal pain or dyspepsia.
A third gepant is atogepant (Qulipta), which is only for migraine prevention. It comes in
10 mg, 30 mg, and 60 mg and is taken once every day as a preventive. It can cause
some drowsiness, constipation, and nausea.
Are there any other acute care drugs you recommend if triptans are not working?
Yes, there is another drug class called the ditans. These medications work very well but
have more adverse events associated with them than I like. A higher percentage of
patients seem to be pain-free in 2 hours when using a ditan; however, the only one
available, lasmiditan (Reyvow), has never been studied against a gepant, so I cannot
say if one is better than the other. Lasmitidan works similarly to a triptan by stimulating
serotonin 1F receptors but does not constrict blood vessels. Up to 15% of patients have
dizziness and up to 7% have drowsiness, so patients should not drive within 8 hours
after taking lasmiditan. This medication is available in 2 sizes, 100 mg and 200 mg. I
usually give patients a 200-mg dose, which is good enough for 24 hours. Ditans are a
Schedule V drug, meaning some patients might take more than they should because it
makes them feel good. It can be a challenging drug to get, but it is an excellent acute
care drug when none of the mentioned adverse events occur.
Which preventive drugs do you tend to prescribe your patients for migraine since
triptans are not preventive?
For many years, we have used some of the older preventives. Antidepressants can be
an option for preventive treatment of migraine. Amitriptyline, a tricyclic antidepressant, is
a pretty good medicine. However, it has a lot of adverse events associated with it,
including dry mouth, weight gain, and drowsiness, so patients who take this at night
often sleep better. The dose is 10 mg to 50 mg taken before bed. This drug is often
used, but I would not say I like to prescribe it as much as other medications, even
though amitriptyline is effective and likely to work by affecting the level of serotonin and
other chemicals in the brain. There is little evidence that other classes of
antidepressants, such as selective serotonin reuptake inhibitors and serotonin and
norepinephrine reuptake inhibitors, are effective for migraine prevention. Adverse
effects may include weight gain, fatigue, constipation, and dry mouth, making it difficult
for a patient to stick with treatment.
Beta blockers are another preventive medication option for migraine. Beta blockers are
best known as a medical treatment for cardiovascular conditions, such as hypertension,
stable or unstable angina, and congestive heart failure. Beta blockers prevent the stress
hormone adrenaline (epinephrine) from binding to beta receptors, slowing heart rate
and lowering blood pressure. A commonly used beta blocker is propranolol (Inderal),
which also comes in a long-acting preparation. Doses range from 60 mg to 180
mg. Other beta blockers effective for migraine prevention include metoprolol, nadolol,
and atenolol.
Many of my patients are young, healthy females who like to exercise. Most report that
their heart rate is slow, they get short of breath, and they cannot exercise as effectively
while on a beta blocker. It also takes about 2 months until this medication starts
working. Patients may feel as if they are having too many adverse events, so I start
them on a very low dose and build it up gradually for a month and see how they are
feeling.
Epilepsy medicines can also be used to prevent migraine. There are 2 common
epilepsy medications. Topiramate (Topamax) doses can range from 75 mg to 100 mg
and are sometimes higher. Topiramate is a good medicine, but there are many potential
adverse events: tingling in the extremities, difficulty finding words when speaking,
confusion, raised eye pressure, and others. Divalproex sodium (Depakote) is another
popular medication, available in 500 mg to 1000 mg doses. This medicine can cause
some endocrine problems in women and can also damage the spinal cords of a fetus,
so this drug should not be taken during early pregnancy.
Monoclonal antibodies against CGRP are a strong preventive medication and a new
class of drugs that were first approved by the FDA in 2018. They are designed to
prevent episodic migraine (up to 14 headache days per month), chronic migraine (15 or
more headache days per month) and seem to work when a patient has medication
overuse headaches. CGRP is a neuropeptide involved in many body processes,
including blood pressure regulation, tissue repair, wound healing, and inflammation, and
is a potent vasodilator. When CGRP is released in the brain, it affects the trigeminal
nerve, increasing pain transmission and sensitivities to touch and temperature. CGRP
also causes inflammation and pain that happen during a migraine; it makes headache
pain worse and causes headaches to last longer.
Some CGRP inhibitors block CGRP from binding to CGRP receptors, a key contributor
to the trigeminal nerve pain and inflammation of migraine, while some grab the CGRP
and prevent it from activating the receptor.
The 2 classes of these drugs are monoclonal antibodies against CGRP and small
molecule CGRP antagonists. Fortunately, CGRPs have long half-lives and work for 1 to
3 months. The CGRP monoclonal antibodies are large molecule drugs. There are 4
different types, and 2 of them are injected by the patient at home once a month. One
can be injected at home once a month or every 3 months. For the latter option, patients
need to triple up with 3 injections in one day, so they do not have to inject for 3 months.
The fourth CGRP is an intravenous infusion that can be administered in an infusion
center or at home. This one is more inconvenient, but it is a strong drug. The small
molecule CGRP antagonists are taken by mouth in pill form. All CGRPs have been
shown to decrease the number of headaches per month.
The main goal of preventive therapy is to lessen the impact of migraines on patients’
lives by reducing how often they occur, how severe they are, and how long they last.
Preventive therapy also decreases disability and improves patients’ functioning over
time. Preventive therapy can help keep the costs for migraine care down by reducing
the need for acute treatments and allowing the patient to keep working or taking care of
their kids. Furthermore, preventive medications can make acute migraine treatments
more effective and help avoid the overuse of acute medications.
Dr. Rapoport: Most patients who come into my office today, even those whom I have
treated for the last 30 years for acute care of migraine attacks, are taking 1 of the 7
triptan medications available. They might be taking triptans as a tablet—the most
common form—as a nasal spray, or by injection; however, not all patients are suited for
triptans, and sometimes, the need arises to switch to a different class of medication for
treating migraine acutely.
What are the reasons patients switch from a triptan to a gepant?
For some patients, triptans are not working well enough or are causing adverse events.
Other patients have developed cardiac risk factors such as elevated blood pressure,
obesity, smoking, and/or lack of exercise. I am always concerned about constriction of
the coronary blood vessels. Patients who already have some cardiac risk factors and
those who have some actual cardiac disease or have had a previous heart attack
already have constriction of their blood vessels and are not candidates for triptans, as
they are contraindicated.
How do you switch a patient from a triptan to a gepant?
It is important to have some discussion with the patient before the switch. For example,
if a patient with no cardiac risk factors comes into the office asking about this new
medicine, I will ask them several questions about their triptan to ensure it works well
enough (ie, to ascertain if the patient’s migraines improve within 30 to 60 minutes and
are much better within 2 hours of taking the medication). I want to be sure that they do
not have any adverse events related to the triptan, such as chest pain, drowsiness, or
dizziness. I like to ensure that whatever they are taking works long enough—at least 24
hours, preferably 48 hours—so they no longer have a headache, especially the next
day. If the headache comes back the next day, they must re-treat. If I determine the
triptan is not working well for them or they have significant adverse events, I will move
on.
Gepants are small-molecule calcitonin gene-related peptide (CGRP) receptor
antagonists, which are pills that only last for 2 to 3 days in the body. There are 2
gepants for acute care and 2 for the prevention of migraine. The first gepant approved
by the US Food and Drug Administration (FDA) for acute care was ubrogepant
(Ubrelvy), which comes in 2 sizes, 50 mg or 100 mg tablets. I sometimes start with 50
mg, but for the more difficult migraine patient, I will start with 100 mg. If the medicine is
not doing a complete job within 2 hours, the patient may take a second dose, up to 200
mg. Some adverse events may include nausea or slight drowsiness. The patient should
avoid certain medicines such as antifungal medicines (eg, ketoconazole, itraconazole)
and certain antibiotics like clarithromycin.
Another gepant, rimegepant (Nurtec), comes in only 1 size, a 75-mg oral disintegrating
tablet, which can be used both for acute care of migraine and for prevention. Patients
can take a tablet as soon as their migraine attack begins, and they are not to repeat it
that day. If the headache does not go away in 2 hours, I want them to then take a triptan
and an anti-inflammatory drug (there is no contraindication to mix these drugs). I want
them to try it at least 1 more time, encouraging patients to take it early, right at the start
of the headache. If the medicine is still not working by the second or third time, they
should stop using it. Preventively, patients take 75 mg every other day, which can be
quite effective. Side effects are slight nausea and some abdominal pain or dyspepsia.
A third gepant is atogepant (Qulipta), which is only for migraine prevention. It comes in
10 mg, 30 mg, and 60 mg and is taken once every day as a preventive. It can cause
some drowsiness, constipation, and nausea.
Are there any other acute care drugs you recommend if triptans are not working?
Yes, there is another drug class called the ditans. These medications work very well but
have more adverse events associated with them than I like. A higher percentage of
patients seem to be pain-free in 2 hours when using a ditan; however, the only one
available, lasmiditan (Reyvow), has never been studied against a gepant, so I cannot
say if one is better than the other. Lasmitidan works similarly to a triptan by stimulating
serotonin 1F receptors but does not constrict blood vessels. Up to 15% of patients have
dizziness and up to 7% have drowsiness, so patients should not drive within 8 hours
after taking lasmiditan. This medication is available in 2 sizes, 100 mg and 200 mg. I
usually give patients a 200-mg dose, which is good enough for 24 hours. Ditans are a
Schedule V drug, meaning some patients might take more than they should because it
makes them feel good. It can be a challenging drug to get, but it is an excellent acute
care drug when none of the mentioned adverse events occur.
Which preventive drugs do you tend to prescribe your patients for migraine since
triptans are not preventive?
For many years, we have used some of the older preventives. Antidepressants can be
an option for preventive treatment of migraine. Amitriptyline, a tricyclic antidepressant, is
a pretty good medicine. However, it has a lot of adverse events associated with it,
including dry mouth, weight gain, and drowsiness, so patients who take this at night
often sleep better. The dose is 10 mg to 50 mg taken before bed. This drug is often
used, but I would not say I like to prescribe it as much as other medications, even
though amitriptyline is effective and likely to work by affecting the level of serotonin and
other chemicals in the brain. There is little evidence that other classes of
antidepressants, such as selective serotonin reuptake inhibitors and serotonin and
norepinephrine reuptake inhibitors, are effective for migraine prevention. Adverse
effects may include weight gain, fatigue, constipation, and dry mouth, making it difficult
for a patient to stick with treatment.
Beta blockers are another preventive medication option for migraine. Beta blockers are
best known as a medical treatment for cardiovascular conditions, such as hypertension,
stable or unstable angina, and congestive heart failure. Beta blockers prevent the stress
hormone adrenaline (epinephrine) from binding to beta receptors, slowing heart rate
and lowering blood pressure. A commonly used beta blocker is propranolol (Inderal),
which also comes in a long-acting preparation. Doses range from 60 mg to 180
mg. Other beta blockers effective for migraine prevention include metoprolol, nadolol,
and atenolol.
Many of my patients are young, healthy females who like to exercise. Most report that
their heart rate is slow, they get short of breath, and they cannot exercise as effectively
while on a beta blocker. It also takes about 2 months until this medication starts
working. Patients may feel as if they are having too many adverse events, so I start
them on a very low dose and build it up gradually for a month and see how they are
feeling.
Epilepsy medicines can also be used to prevent migraine. There are 2 common
epilepsy medications. Topiramate (Topamax) doses can range from 75 mg to 100 mg
and are sometimes higher. Topiramate is a good medicine, but there are many potential
adverse events: tingling in the extremities, difficulty finding words when speaking,
confusion, raised eye pressure, and others. Divalproex sodium (Depakote) is another
popular medication, available in 500 mg to 1000 mg doses. This medicine can cause
some endocrine problems in women and can also damage the spinal cords of a fetus,
so this drug should not be taken during early pregnancy.
Monoclonal antibodies against CGRP are a strong preventive medication and a new
class of drugs that were first approved by the FDA in 2018. They are designed to
prevent episodic migraine (up to 14 headache days per month), chronic migraine (15 or
more headache days per month) and seem to work when a patient has medication
overuse headaches. CGRP is a neuropeptide involved in many body processes,
including blood pressure regulation, tissue repair, wound healing, and inflammation, and
is a potent vasodilator. When CGRP is released in the brain, it affects the trigeminal
nerve, increasing pain transmission and sensitivities to touch and temperature. CGRP
also causes inflammation and pain that happen during a migraine; it makes headache
pain worse and causes headaches to last longer.
Some CGRP inhibitors block CGRP from binding to CGRP receptors, a key contributor
to the trigeminal nerve pain and inflammation of migraine, while some grab the CGRP
and prevent it from activating the receptor.
The 2 classes of these drugs are monoclonal antibodies against CGRP and small
molecule CGRP antagonists. Fortunately, CGRPs have long half-lives and work for 1 to
3 months. The CGRP monoclonal antibodies are large molecule drugs. There are 4
different types, and 2 of them are injected by the patient at home once a month. One
can be injected at home once a month or every 3 months. For the latter option, patients
need to triple up with 3 injections in one day, so they do not have to inject for 3 months.
The fourth CGRP is an intravenous infusion that can be administered in an infusion
center or at home. This one is more inconvenient, but it is a strong drug. The small
molecule CGRP antagonists are taken by mouth in pill form. All CGRPs have been
shown to decrease the number of headaches per month.
The main goal of preventive therapy is to lessen the impact of migraines on patients’
lives by reducing how often they occur, how severe they are, and how long they last.
Preventive therapy also decreases disability and improves patients’ functioning over
time. Preventive therapy can help keep the costs for migraine care down by reducing
the need for acute treatments and allowing the patient to keep working or taking care of
their kids. Furthermore, preventive medications can make acute migraine treatments
more effective and help avoid the overuse of acute medications.
Dr. Rapoport: Most patients who come into my office today, even those whom I have
treated for the last 30 years for acute care of migraine attacks, are taking 1 of the 7
triptan medications available. They might be taking triptans as a tablet—the most
common form—as a nasal spray, or by injection; however, not all patients are suited for
triptans, and sometimes, the need arises to switch to a different class of medication for
treating migraine acutely.
What are the reasons patients switch from a triptan to a gepant?
For some patients, triptans are not working well enough or are causing adverse events.
Other patients have developed cardiac risk factors such as elevated blood pressure,
obesity, smoking, and/or lack of exercise. I am always concerned about constriction of
the coronary blood vessels. Patients who already have some cardiac risk factors and
those who have some actual cardiac disease or have had a previous heart attack
already have constriction of their blood vessels and are not candidates for triptans, as
they are contraindicated.
How do you switch a patient from a triptan to a gepant?
It is important to have some discussion with the patient before the switch. For example,
if a patient with no cardiac risk factors comes into the office asking about this new
medicine, I will ask them several questions about their triptan to ensure it works well
enough (ie, to ascertain if the patient’s migraines improve within 30 to 60 minutes and
are much better within 2 hours of taking the medication). I want to be sure that they do
not have any adverse events related to the triptan, such as chest pain, drowsiness, or
dizziness. I like to ensure that whatever they are taking works long enough—at least 24
hours, preferably 48 hours—so they no longer have a headache, especially the next
day. If the headache comes back the next day, they must re-treat. If I determine the
triptan is not working well for them or they have significant adverse events, I will move
on.
Gepants are small-molecule calcitonin gene-related peptide (CGRP) receptor
antagonists, which are pills that only last for 2 to 3 days in the body. There are 2
gepants for acute care and 2 for the prevention of migraine. The first gepant approved
by the US Food and Drug Administration (FDA) for acute care was ubrogepant
(Ubrelvy), which comes in 2 sizes, 50 mg or 100 mg tablets. I sometimes start with 50
mg, but for the more difficult migraine patient, I will start with 100 mg. If the medicine is
not doing a complete job within 2 hours, the patient may take a second dose, up to 200
mg. Some adverse events may include nausea or slight drowsiness. The patient should
avoid certain medicines such as antifungal medicines (eg, ketoconazole, itraconazole)
and certain antibiotics like clarithromycin.
Another gepant, rimegepant (Nurtec), comes in only 1 size, a 75-mg oral disintegrating
tablet, which can be used both for acute care of migraine and for prevention. Patients
can take a tablet as soon as their migraine attack begins, and they are not to repeat it
that day. If the headache does not go away in 2 hours, I want them to then take a triptan
and an anti-inflammatory drug (there is no contraindication to mix these drugs). I want
them to try it at least 1 more time, encouraging patients to take it early, right at the start
of the headache. If the medicine is still not working by the second or third time, they
should stop using it. Preventively, patients take 75 mg every other day, which can be
quite effective. Side effects are slight nausea and some abdominal pain or dyspepsia.
A third gepant is atogepant (Qulipta), which is only for migraine prevention. It comes in
10 mg, 30 mg, and 60 mg and is taken once every day as a preventive. It can cause
some drowsiness, constipation, and nausea.
Are there any other acute care drugs you recommend if triptans are not working?
Yes, there is another drug class called the ditans. These medications work very well but
have more adverse events associated with them than I like. A higher percentage of
patients seem to be pain-free in 2 hours when using a ditan; however, the only one
available, lasmiditan (Reyvow), has never been studied against a gepant, so I cannot
say if one is better than the other. Lasmitidan works similarly to a triptan by stimulating
serotonin 1F receptors but does not constrict blood vessels. Up to 15% of patients have
dizziness and up to 7% have drowsiness, so patients should not drive within 8 hours
after taking lasmiditan. This medication is available in 2 sizes, 100 mg and 200 mg. I
usually give patients a 200-mg dose, which is good enough for 24 hours. Ditans are a
Schedule V drug, meaning some patients might take more than they should because it
makes them feel good. It can be a challenging drug to get, but it is an excellent acute
care drug when none of the mentioned adverse events occur.
Which preventive drugs do you tend to prescribe your patients for migraine since
triptans are not preventive?
For many years, we have used some of the older preventives. Antidepressants can be
an option for preventive treatment of migraine. Amitriptyline, a tricyclic antidepressant, is
a pretty good medicine. However, it has a lot of adverse events associated with it,
including dry mouth, weight gain, and drowsiness, so patients who take this at night
often sleep better. The dose is 10 mg to 50 mg taken before bed. This drug is often
used, but I would not say I like to prescribe it as much as other medications, even
though amitriptyline is effective and likely to work by affecting the level of serotonin and
other chemicals in the brain. There is little evidence that other classes of
antidepressants, such as selective serotonin reuptake inhibitors and serotonin and
norepinephrine reuptake inhibitors, are effective for migraine prevention. Adverse
effects may include weight gain, fatigue, constipation, and dry mouth, making it difficult
for a patient to stick with treatment.
Beta blockers are another preventive medication option for migraine. Beta blockers are
best known as a medical treatment for cardiovascular conditions, such as hypertension,
stable or unstable angina, and congestive heart failure. Beta blockers prevent the stress
hormone adrenaline (epinephrine) from binding to beta receptors, slowing heart rate
and lowering blood pressure. A commonly used beta blocker is propranolol (Inderal),
which also comes in a long-acting preparation. Doses range from 60 mg to 180
mg. Other beta blockers effective for migraine prevention include metoprolol, nadolol,
and atenolol.
Many of my patients are young, healthy females who like to exercise. Most report that
their heart rate is slow, they get short of breath, and they cannot exercise as effectively
while on a beta blocker. It also takes about 2 months until this medication starts
working. Patients may feel as if they are having too many adverse events, so I start
them on a very low dose and build it up gradually for a month and see how they are
feeling.
Epilepsy medicines can also be used to prevent migraine. There are 2 common
epilepsy medications. Topiramate (Topamax) doses can range from 75 mg to 100 mg
and are sometimes higher. Topiramate is a good medicine, but there are many potential
adverse events: tingling in the extremities, difficulty finding words when speaking,
confusion, raised eye pressure, and others. Divalproex sodium (Depakote) is another
popular medication, available in 500 mg to 1000 mg doses. This medicine can cause
some endocrine problems in women and can also damage the spinal cords of a fetus,
so this drug should not be taken during early pregnancy.
Monoclonal antibodies against CGRP are a strong preventive medication and a new
class of drugs that were first approved by the FDA in 2018. They are designed to
prevent episodic migraine (up to 14 headache days per month), chronic migraine (15 or
more headache days per month) and seem to work when a patient has medication
overuse headaches. CGRP is a neuropeptide involved in many body processes,
including blood pressure regulation, tissue repair, wound healing, and inflammation, and
is a potent vasodilator. When CGRP is released in the brain, it affects the trigeminal
nerve, increasing pain transmission and sensitivities to touch and temperature. CGRP
also causes inflammation and pain that happen during a migraine; it makes headache
pain worse and causes headaches to last longer.
Some CGRP inhibitors block CGRP from binding to CGRP receptors, a key contributor
to the trigeminal nerve pain and inflammation of migraine, while some grab the CGRP
and prevent it from activating the receptor.
The 2 classes of these drugs are monoclonal antibodies against CGRP and small
molecule CGRP antagonists. Fortunately, CGRPs have long half-lives and work for 1 to
3 months. The CGRP monoclonal antibodies are large molecule drugs. There are 4
different types, and 2 of them are injected by the patient at home once a month. One
can be injected at home once a month or every 3 months. For the latter option, patients
need to triple up with 3 injections in one day, so they do not have to inject for 3 months.
The fourth CGRP is an intravenous infusion that can be administered in an infusion
center or at home. This one is more inconvenient, but it is a strong drug. The small
molecule CGRP antagonists are taken by mouth in pill form. All CGRPs have been
shown to decrease the number of headaches per month.
The main goal of preventive therapy is to lessen the impact of migraines on patients’
lives by reducing how often they occur, how severe they are, and how long they last.
Preventive therapy also decreases disability and improves patients’ functioning over
time. Preventive therapy can help keep the costs for migraine care down by reducing
the need for acute treatments and allowing the patient to keep working or taking care of
their kids. Furthermore, preventive medications can make acute migraine treatments
more effective and help avoid the overuse of acute medications.
How Medical Education Is Evolving in the Wake of the COVID-19 Pandemic

Question: What doubles every 2 months and takes more than a decade and a half to reach its ultimate destination?
Answer: Medical knowledge.
In 2011, researchers projected that by 2020, medical knowledge would double every 73 days. Also in 2011, investigators estimated that clinical research takes 17 years to translate from bench to bedside.
This “fast-slow” paradox became more relevant than ever in 2020, when the coronavirus pandemic brought the world to a near standstill. Stakeholders in undergraduate, postgraduate, and continuing medical education (CME) were suddenly faced with choices that had been discussed theoretically but not yet applied on a wide scale: How do we deliver education if in-person instruction is not an option?
Organized medicine and the clinical community made choices based on groundwork that had been laid prior to the pandemic. The medical community acted quickly out of necessity, implementing novel learning methods that are now being utilized and that need to be assessed in an ongoing manner.
The Backdrop
Medical education has long been dominated by an in-person, didactic model anchored in teacher-centered, classroom-based learning. This design has been firmly entrenched for more than 100 years, since the publication of the Flexner report in 1910, which established the standard of 4 years of medical education. Prior to 2020, many experts acknowledged that alternative practices and emerging technologies should play a role in medical education, but indecision abounded, perhaps because there was no real-world catalyst for reform. Thus, despite various attempts, the adoption of alternative forms of teaching moved slowly.
Pre-pandemic efforts
In 2017, the American Medication Association issued a report calling for “one of the most complete curricular reforms since the Flexner Report.” It urged leaders to “rethink nearly every facet of physician training,” including “greater emphasis on new technology.” The report also suggested a 14-month pre-rotation program focused on the core medical knowledge necessary to practice in a hospital setting, along with work in a primary care setting once every other week.
Before the pandemic, “blended learning” (digital and live) and “flipped classroom” approaches were assessed. A meta-analysis comparing a blended learning format to traditional classroom model programs found that blended learning resulted in better knowledge outcomes. In the flipped classroom approach, non-classroom individual or group activities replace in-class instruction after pre-class self-preparation with provided resources. A meta-analysis of 28 comparative studies showed that the flipped classroom approach resulted in improved learning compared to traditional methods. Additionally, bite-sized learning approaches have been implemented and evaluated, showing improvement in immediate knowledge recall.
Barriers to widespread implementation
Despite the need to increase medical knowledge dissemination and implement approaches proven to do so effectively, barriers to adoption are well documented. Obstacles include time limitations, inadequate technical skills, insufficient infrastructure, and a wide variety in and range of expertise of both learners and institutional strategies. There are also differences in effective techniques for teaching various topics based on the content. Some topics require knowledge-based training, whereas others fall more easily into skills-based training.
Additionally, when it comes to new evidence that needs to be translated to clinical evaluation and delivery, there is ongoing debate about the established peer-review process, which is rigorous but time-consuming vs the open-access publication process, which can disseminate information more quickly but is prone to error.
Proposed solutions
Proposed solutions to these barriers include improving educator skills, offering incentives for innovative content development, cultivating better institutional strategies, and achieving buy-in from all stakeholders. Also important is thoughtful adaptation of content to various electronic formats, such as audiovisual presentation of educational material, social media content, and gamification of content, as well as ongoing assessment of both education delivery and consumption—followed by rapid pivoting when necessary.
Despite these clearly identified challenges and thoughtful solutions, change was relatively slow until March 2020.
The Trigger
With medical knowledge expanding so rapidly, imagine if medical education moved slowly or came to a complete halt when a worldwide pandemic was declared, the effects would have been catastrophic. COVID pushed organized medicine and the healthcare community to accelerate the adoption of novel technological approaches to keep the medical knowledge pipeline flowing at a relatively reasonable— if not ideal—rate.
Challenges the pandemic produced, along with potential mitigation strategies, are outlined below.
Economic consequences: The pandemic resulted in lost income for training programs and decreased funding for graduate medical education.
Possible solution: Creating budget allowances to adopt new technologies
Impact on diversity, equity, and inclusion: COVID-19 amplified existing implicit and explicit biases in society, particularly in the field of medicine. Women trainees and individuals from disadvantaged backgrounds were disproportionately impacted.
Possible solution: Creating programs that increase awareness of the subtle nature of implicit bias and the outsized impact it can have on certain segments of the population, and offering resources to mitigate stressors such as childcare and access to technology solutions
Impact on mental health and wellness: Working through the pandemic was challenging professionally, and the pandemic also exposed individuals to stigma, loneliness, and behavioral health issues (eg, mood and sleeping disorders), which created challenges in personal lives as well. These challenges lasted well over 2 years and have a clear ongoing impact.
Possible solution: Providing accessible behavioral health resources, regularly assessing and addressing burnout, and regularly cycling trainees off of high-intensity rotations
Education delivery challenges: The sudden cancellation of in-person classes and training, from medical school lectures to rotations, created uncertainty. In-person rounds and bedside learning were significantly restricted. Moreover, as the need to perform clinical duties during the pandemic increased, time for teaching decreased. Some areas were more heavily impacted than others (eg, instruction around elective surgeries, outpatient medicine, and non-critical care training).
Possible solution: Digitizing education delivery and developing other innovative methods to compensate for a lack of face-to-face instruction
Sudden need for rapid information dissemination: The limits of traditional peer review were tested during the pandemic. Managing individuals infected with the novel coronavirus created a situation where the clinical community needed scientific information quickly, increasing the risk of misinformation.
Possible solution: Disseminating information as quickly as possible by leveraging public-private partnerships and government investment in high-quality science while maintaining peer review integrity to ensure rigorous evaluation
The Evidence
Early evidence is emerging about efforts undertaken during the pandemic to maintain adequate levels of preclinical learning, clinical training, and CME.
Preclinical learning: Virtual formats are generally accepted, and interactive discussion is preferred. But be aware of potential stressors.
A cross-sectional study involving 173 histology and pathology students at European University Cyprus found that preclinical medical education is possible via virtual learning. The pandemic forced respondents to adapt immediately to emergency remote teaching. Survey results found the concept was generally well accepted, though some stressors (eg, poor internet connection) impacted perception. Most histology and pathology students (58% and 68%, respectively) said they would prefer blended learning in the future, compared with all-live (39% and 28%, respectively), or all-virtual (4% and 5%, respectively) classrooms.
In a systematic review of 13 studies that compared digital learning with live classroom education for medical and nursing students, investigators from China found that standalone digital models are as effective as conventional modalities for improving knowledge and practice. Moreover, students preferred interactive discussion to a straight lecture format when participating online.
Clinical training: Virtual clerkships work, but a blended approach seems preferable.
In a study involving 16 third-year medical students in the general surgery clerkship at Cleveland Clinic, respondents reported their experience before and after participating in a case-based virtual surgery clerkship program. Students were significantly more confident that they could independently assess a surgical consult after taking the course. Average scores of curriculum-based surgical knowledge increased as well.
In an assessment of alternative approaches to clinical clerkships involving 42 students, investigators from China evaluated the impact of using simulated electronic health records (EHRs) for inpatient training and electronic problem-based learning and virtual interviews for outpatient training. Students using simulated EHRs felt it improved their ability to write in and summarize the record. Those who participated in electronic problem-based learning and virtual interviewing said their interviewing and counseling skills improved. However, students also noted traditional clinical clerkships are better for certain types of learning, suggesting that a blended approach is preferred.
CME: Virtual CME is accepted and improves performance, but barriers remain, including a preference for face-to-face networking.
Researchers reviewed 2,007 post-activity responses from clinicians who participated in online CME at a South Korean hospital. Of the 1332 participants who reported their satisfaction level, 85% reported being satisfied with the format and content. Among all respondents, nearly 9 in 10 said that the content would influence the way they practice. Of the 611 participants who responded to a follow-up survey 3 months later, 78% said they made changes in their clinical practice based on what they learned.
However, many clinicians prefer in-person CME. A Canadian-based memory clinic held 5 interprofessional education sessions and reported on participant experience; 3 of the sessions occurred live before March 2020 and 2 were held via videoconference once the pandemic was declared. Ratings of satisfaction, relevance, knowledge acquisition, and knowledge application were similar in both groups, but the virtual sessions were rated as less enjoyable and lacking in networking opportunities. In-person learning was preferred.
Primary care clinicians in Portugal evaluated a CME digital platform and reported several barriers, including time constraints, perceived excessive work, lack of digital competence, lack of motivation, and emotional factors.
The Future
Although challenges remain, changes due to the pandemic have been implemented in medical training and have shown preliminary success in certain domains. Medical education is rapidly evolving, and as we move further from the pandemic, diligent ongoing evaluation is needed to assess the best use of technology and various innovative teaching modalities. Keeping medical education learner-centered and instituting timely course correction if certain modalities of knowledge/skill delivery are found to be ineffective will be key to ensuring the robustness of training for future generations.
Question: What doubles every 2 months and takes more than a decade and a half to reach its ultimate destination?
Answer: Medical knowledge.
In 2011, researchers projected that by 2020, medical knowledge would double every 73 days. Also in 2011, investigators estimated that clinical research takes 17 years to translate from bench to bedside.
This “fast-slow” paradox became more relevant than ever in 2020, when the coronavirus pandemic brought the world to a near standstill. Stakeholders in undergraduate, postgraduate, and continuing medical education (CME) were suddenly faced with choices that had been discussed theoretically but not yet applied on a wide scale: How do we deliver education if in-person instruction is not an option?
Organized medicine and the clinical community made choices based on groundwork that had been laid prior to the pandemic. The medical community acted quickly out of necessity, implementing novel learning methods that are now being utilized and that need to be assessed in an ongoing manner.
The Backdrop
Medical education has long been dominated by an in-person, didactic model anchored in teacher-centered, classroom-based learning. This design has been firmly entrenched for more than 100 years, since the publication of the Flexner report in 1910, which established the standard of 4 years of medical education. Prior to 2020, many experts acknowledged that alternative practices and emerging technologies should play a role in medical education, but indecision abounded, perhaps because there was no real-world catalyst for reform. Thus, despite various attempts, the adoption of alternative forms of teaching moved slowly.
Pre-pandemic efforts
In 2017, the American Medication Association issued a report calling for “one of the most complete curricular reforms since the Flexner Report.” It urged leaders to “rethink nearly every facet of physician training,” including “greater emphasis on new technology.” The report also suggested a 14-month pre-rotation program focused on the core medical knowledge necessary to practice in a hospital setting, along with work in a primary care setting once every other week.
Before the pandemic, “blended learning” (digital and live) and “flipped classroom” approaches were assessed. A meta-analysis comparing a blended learning format to traditional classroom model programs found that blended learning resulted in better knowledge outcomes. In the flipped classroom approach, non-classroom individual or group activities replace in-class instruction after pre-class self-preparation with provided resources. A meta-analysis of 28 comparative studies showed that the flipped classroom approach resulted in improved learning compared to traditional methods. Additionally, bite-sized learning approaches have been implemented and evaluated, showing improvement in immediate knowledge recall.
Barriers to widespread implementation
Despite the need to increase medical knowledge dissemination and implement approaches proven to do so effectively, barriers to adoption are well documented. Obstacles include time limitations, inadequate technical skills, insufficient infrastructure, and a wide variety in and range of expertise of both learners and institutional strategies. There are also differences in effective techniques for teaching various topics based on the content. Some topics require knowledge-based training, whereas others fall more easily into skills-based training.
Additionally, when it comes to new evidence that needs to be translated to clinical evaluation and delivery, there is ongoing debate about the established peer-review process, which is rigorous but time-consuming vs the open-access publication process, which can disseminate information more quickly but is prone to error.
Proposed solutions
Proposed solutions to these barriers include improving educator skills, offering incentives for innovative content development, cultivating better institutional strategies, and achieving buy-in from all stakeholders. Also important is thoughtful adaptation of content to various electronic formats, such as audiovisual presentation of educational material, social media content, and gamification of content, as well as ongoing assessment of both education delivery and consumption—followed by rapid pivoting when necessary.
Despite these clearly identified challenges and thoughtful solutions, change was relatively slow until March 2020.
The Trigger
With medical knowledge expanding so rapidly, imagine if medical education moved slowly or came to a complete halt when a worldwide pandemic was declared, the effects would have been catastrophic. COVID pushed organized medicine and the healthcare community to accelerate the adoption of novel technological approaches to keep the medical knowledge pipeline flowing at a relatively reasonable— if not ideal—rate.
Challenges the pandemic produced, along with potential mitigation strategies, are outlined below.
Economic consequences: The pandemic resulted in lost income for training programs and decreased funding for graduate medical education.
Possible solution: Creating budget allowances to adopt new technologies
Impact on diversity, equity, and inclusion: COVID-19 amplified existing implicit and explicit biases in society, particularly in the field of medicine. Women trainees and individuals from disadvantaged backgrounds were disproportionately impacted.
Possible solution: Creating programs that increase awareness of the subtle nature of implicit bias and the outsized impact it can have on certain segments of the population, and offering resources to mitigate stressors such as childcare and access to technology solutions
Impact on mental health and wellness: Working through the pandemic was challenging professionally, and the pandemic also exposed individuals to stigma, loneliness, and behavioral health issues (eg, mood and sleeping disorders), which created challenges in personal lives as well. These challenges lasted well over 2 years and have a clear ongoing impact.
Possible solution: Providing accessible behavioral health resources, regularly assessing and addressing burnout, and regularly cycling trainees off of high-intensity rotations
Education delivery challenges: The sudden cancellation of in-person classes and training, from medical school lectures to rotations, created uncertainty. In-person rounds and bedside learning were significantly restricted. Moreover, as the need to perform clinical duties during the pandemic increased, time for teaching decreased. Some areas were more heavily impacted than others (eg, instruction around elective surgeries, outpatient medicine, and non-critical care training).
Possible solution: Digitizing education delivery and developing other innovative methods to compensate for a lack of face-to-face instruction
Sudden need for rapid information dissemination: The limits of traditional peer review were tested during the pandemic. Managing individuals infected with the novel coronavirus created a situation where the clinical community needed scientific information quickly, increasing the risk of misinformation.
Possible solution: Disseminating information as quickly as possible by leveraging public-private partnerships and government investment in high-quality science while maintaining peer review integrity to ensure rigorous evaluation
The Evidence
Early evidence is emerging about efforts undertaken during the pandemic to maintain adequate levels of preclinical learning, clinical training, and CME.
Preclinical learning: Virtual formats are generally accepted, and interactive discussion is preferred. But be aware of potential stressors.
A cross-sectional study involving 173 histology and pathology students at European University Cyprus found that preclinical medical education is possible via virtual learning. The pandemic forced respondents to adapt immediately to emergency remote teaching. Survey results found the concept was generally well accepted, though some stressors (eg, poor internet connection) impacted perception. Most histology and pathology students (58% and 68%, respectively) said they would prefer blended learning in the future, compared with all-live (39% and 28%, respectively), or all-virtual (4% and 5%, respectively) classrooms.
In a systematic review of 13 studies that compared digital learning with live classroom education for medical and nursing students, investigators from China found that standalone digital models are as effective as conventional modalities for improving knowledge and practice. Moreover, students preferred interactive discussion to a straight lecture format when participating online.
Clinical training: Virtual clerkships work, but a blended approach seems preferable.
In a study involving 16 third-year medical students in the general surgery clerkship at Cleveland Clinic, respondents reported their experience before and after participating in a case-based virtual surgery clerkship program. Students were significantly more confident that they could independently assess a surgical consult after taking the course. Average scores of curriculum-based surgical knowledge increased as well.
In an assessment of alternative approaches to clinical clerkships involving 42 students, investigators from China evaluated the impact of using simulated electronic health records (EHRs) for inpatient training and electronic problem-based learning and virtual interviews for outpatient training. Students using simulated EHRs felt it improved their ability to write in and summarize the record. Those who participated in electronic problem-based learning and virtual interviewing said their interviewing and counseling skills improved. However, students also noted traditional clinical clerkships are better for certain types of learning, suggesting that a blended approach is preferred.
CME: Virtual CME is accepted and improves performance, but barriers remain, including a preference for face-to-face networking.
Researchers reviewed 2,007 post-activity responses from clinicians who participated in online CME at a South Korean hospital. Of the 1332 participants who reported their satisfaction level, 85% reported being satisfied with the format and content. Among all respondents, nearly 9 in 10 said that the content would influence the way they practice. Of the 611 participants who responded to a follow-up survey 3 months later, 78% said they made changes in their clinical practice based on what they learned.
However, many clinicians prefer in-person CME. A Canadian-based memory clinic held 5 interprofessional education sessions and reported on participant experience; 3 of the sessions occurred live before March 2020 and 2 were held via videoconference once the pandemic was declared. Ratings of satisfaction, relevance, knowledge acquisition, and knowledge application were similar in both groups, but the virtual sessions were rated as less enjoyable and lacking in networking opportunities. In-person learning was preferred.
Primary care clinicians in Portugal evaluated a CME digital platform and reported several barriers, including time constraints, perceived excessive work, lack of digital competence, lack of motivation, and emotional factors.
The Future
Although challenges remain, changes due to the pandemic have been implemented in medical training and have shown preliminary success in certain domains. Medical education is rapidly evolving, and as we move further from the pandemic, diligent ongoing evaluation is needed to assess the best use of technology and various innovative teaching modalities. Keeping medical education learner-centered and instituting timely course correction if certain modalities of knowledge/skill delivery are found to be ineffective will be key to ensuring the robustness of training for future generations.
Question: What doubles every 2 months and takes more than a decade and a half to reach its ultimate destination?
Answer: Medical knowledge.
In 2011, researchers projected that by 2020, medical knowledge would double every 73 days. Also in 2011, investigators estimated that clinical research takes 17 years to translate from bench to bedside.
This “fast-slow” paradox became more relevant than ever in 2020, when the coronavirus pandemic brought the world to a near standstill. Stakeholders in undergraduate, postgraduate, and continuing medical education (CME) were suddenly faced with choices that had been discussed theoretically but not yet applied on a wide scale: How do we deliver education if in-person instruction is not an option?
Organized medicine and the clinical community made choices based on groundwork that had been laid prior to the pandemic. The medical community acted quickly out of necessity, implementing novel learning methods that are now being utilized and that need to be assessed in an ongoing manner.
The Backdrop
Medical education has long been dominated by an in-person, didactic model anchored in teacher-centered, classroom-based learning. This design has been firmly entrenched for more than 100 years, since the publication of the Flexner report in 1910, which established the standard of 4 years of medical education. Prior to 2020, many experts acknowledged that alternative practices and emerging technologies should play a role in medical education, but indecision abounded, perhaps because there was no real-world catalyst for reform. Thus, despite various attempts, the adoption of alternative forms of teaching moved slowly.
Pre-pandemic efforts
In 2017, the American Medication Association issued a report calling for “one of the most complete curricular reforms since the Flexner Report.” It urged leaders to “rethink nearly every facet of physician training,” including “greater emphasis on new technology.” The report also suggested a 14-month pre-rotation program focused on the core medical knowledge necessary to practice in a hospital setting, along with work in a primary care setting once every other week.
Before the pandemic, “blended learning” (digital and live) and “flipped classroom” approaches were assessed. A meta-analysis comparing a blended learning format to traditional classroom model programs found that blended learning resulted in better knowledge outcomes. In the flipped classroom approach, non-classroom individual or group activities replace in-class instruction after pre-class self-preparation with provided resources. A meta-analysis of 28 comparative studies showed that the flipped classroom approach resulted in improved learning compared to traditional methods. Additionally, bite-sized learning approaches have been implemented and evaluated, showing improvement in immediate knowledge recall.
Barriers to widespread implementation
Despite the need to increase medical knowledge dissemination and implement approaches proven to do so effectively, barriers to adoption are well documented. Obstacles include time limitations, inadequate technical skills, insufficient infrastructure, and a wide variety in and range of expertise of both learners and institutional strategies. There are also differences in effective techniques for teaching various topics based on the content. Some topics require knowledge-based training, whereas others fall more easily into skills-based training.
Additionally, when it comes to new evidence that needs to be translated to clinical evaluation and delivery, there is ongoing debate about the established peer-review process, which is rigorous but time-consuming vs the open-access publication process, which can disseminate information more quickly but is prone to error.
Proposed solutions
Proposed solutions to these barriers include improving educator skills, offering incentives for innovative content development, cultivating better institutional strategies, and achieving buy-in from all stakeholders. Also important is thoughtful adaptation of content to various electronic formats, such as audiovisual presentation of educational material, social media content, and gamification of content, as well as ongoing assessment of both education delivery and consumption—followed by rapid pivoting when necessary.
Despite these clearly identified challenges and thoughtful solutions, change was relatively slow until March 2020.
The Trigger
With medical knowledge expanding so rapidly, imagine if medical education moved slowly or came to a complete halt when a worldwide pandemic was declared, the effects would have been catastrophic. COVID pushed organized medicine and the healthcare community to accelerate the adoption of novel technological approaches to keep the medical knowledge pipeline flowing at a relatively reasonable— if not ideal—rate.
Challenges the pandemic produced, along with potential mitigation strategies, are outlined below.
Economic consequences: The pandemic resulted in lost income for training programs and decreased funding for graduate medical education.
Possible solution: Creating budget allowances to adopt new technologies
Impact on diversity, equity, and inclusion: COVID-19 amplified existing implicit and explicit biases in society, particularly in the field of medicine. Women trainees and individuals from disadvantaged backgrounds were disproportionately impacted.
Possible solution: Creating programs that increase awareness of the subtle nature of implicit bias and the outsized impact it can have on certain segments of the population, and offering resources to mitigate stressors such as childcare and access to technology solutions
Impact on mental health and wellness: Working through the pandemic was challenging professionally, and the pandemic also exposed individuals to stigma, loneliness, and behavioral health issues (eg, mood and sleeping disorders), which created challenges in personal lives as well. These challenges lasted well over 2 years and have a clear ongoing impact.
Possible solution: Providing accessible behavioral health resources, regularly assessing and addressing burnout, and regularly cycling trainees off of high-intensity rotations
Education delivery challenges: The sudden cancellation of in-person classes and training, from medical school lectures to rotations, created uncertainty. In-person rounds and bedside learning were significantly restricted. Moreover, as the need to perform clinical duties during the pandemic increased, time for teaching decreased. Some areas were more heavily impacted than others (eg, instruction around elective surgeries, outpatient medicine, and non-critical care training).
Possible solution: Digitizing education delivery and developing other innovative methods to compensate for a lack of face-to-face instruction
Sudden need for rapid information dissemination: The limits of traditional peer review were tested during the pandemic. Managing individuals infected with the novel coronavirus created a situation where the clinical community needed scientific information quickly, increasing the risk of misinformation.
Possible solution: Disseminating information as quickly as possible by leveraging public-private partnerships and government investment in high-quality science while maintaining peer review integrity to ensure rigorous evaluation
The Evidence
Early evidence is emerging about efforts undertaken during the pandemic to maintain adequate levels of preclinical learning, clinical training, and CME.
Preclinical learning: Virtual formats are generally accepted, and interactive discussion is preferred. But be aware of potential stressors.
A cross-sectional study involving 173 histology and pathology students at European University Cyprus found that preclinical medical education is possible via virtual learning. The pandemic forced respondents to adapt immediately to emergency remote teaching. Survey results found the concept was generally well accepted, though some stressors (eg, poor internet connection) impacted perception. Most histology and pathology students (58% and 68%, respectively) said they would prefer blended learning in the future, compared with all-live (39% and 28%, respectively), or all-virtual (4% and 5%, respectively) classrooms.
In a systematic review of 13 studies that compared digital learning with live classroom education for medical and nursing students, investigators from China found that standalone digital models are as effective as conventional modalities for improving knowledge and practice. Moreover, students preferred interactive discussion to a straight lecture format when participating online.
Clinical training: Virtual clerkships work, but a blended approach seems preferable.
In a study involving 16 third-year medical students in the general surgery clerkship at Cleveland Clinic, respondents reported their experience before and after participating in a case-based virtual surgery clerkship program. Students were significantly more confident that they could independently assess a surgical consult after taking the course. Average scores of curriculum-based surgical knowledge increased as well.
In an assessment of alternative approaches to clinical clerkships involving 42 students, investigators from China evaluated the impact of using simulated electronic health records (EHRs) for inpatient training and electronic problem-based learning and virtual interviews for outpatient training. Students using simulated EHRs felt it improved their ability to write in and summarize the record. Those who participated in electronic problem-based learning and virtual interviewing said their interviewing and counseling skills improved. However, students also noted traditional clinical clerkships are better for certain types of learning, suggesting that a blended approach is preferred.
CME: Virtual CME is accepted and improves performance, but barriers remain, including a preference for face-to-face networking.
Researchers reviewed 2,007 post-activity responses from clinicians who participated in online CME at a South Korean hospital. Of the 1332 participants who reported their satisfaction level, 85% reported being satisfied with the format and content. Among all respondents, nearly 9 in 10 said that the content would influence the way they practice. Of the 611 participants who responded to a follow-up survey 3 months later, 78% said they made changes in their clinical practice based on what they learned.
However, many clinicians prefer in-person CME. A Canadian-based memory clinic held 5 interprofessional education sessions and reported on participant experience; 3 of the sessions occurred live before March 2020 and 2 were held via videoconference once the pandemic was declared. Ratings of satisfaction, relevance, knowledge acquisition, and knowledge application were similar in both groups, but the virtual sessions were rated as less enjoyable and lacking in networking opportunities. In-person learning was preferred.
Primary care clinicians in Portugal evaluated a CME digital platform and reported several barriers, including time constraints, perceived excessive work, lack of digital competence, lack of motivation, and emotional factors.
The Future
Although challenges remain, changes due to the pandemic have been implemented in medical training and have shown preliminary success in certain domains. Medical education is rapidly evolving, and as we move further from the pandemic, diligent ongoing evaluation is needed to assess the best use of technology and various innovative teaching modalities. Keeping medical education learner-centered and instituting timely course correction if certain modalities of knowledge/skill delivery are found to be ineffective will be key to ensuring the robustness of training for future generations.
Can zoo poo help manage diabetic foot ulcers?
In a striking convergence of veterinary biology and medical science, researchers from the University of Sheffield (England) have unveiled findings that could potentially advance the treatment of diabetic foot ulcers, a condition affecting an estimated 18.6 million people worldwide. The unexpected ingredient in this potentially transformative therapy? Feces from endangered species, sourced from Yorkshire Wildlife Park, Doncaster, England.
The scourge of antibiotic resistance
Diabetic foot ulcers are a significant challenge in health care, not only because of their prevalence but also because of the alarming rise of antibiotic-resistant bacterial infections. Current antibiotic treatments frequently fail, leading to life-altering consequences like amputations and significant health care costs – estimated at one-third of the total direct costs of diabetes care. The critical need for alternative therapies has propelled scientists into a pressing search for novel antimicrobial agents.
A pioneering approach: zoo poo as bioactive goldmine
Led by Professor Graham Stafford, chair of molecular microbiology at the University of Sheffield, the research team began to explore a rather unorthodox resource: the fecal matter of endangered animals like Guinea baboons, lemurs, and Visayan pigs. While such a source might seem surprising at first glance, the rationale becomes clear when considering the nature of bacteriophages.
What are bacteriophages?
Bacteriophages, commonly known as phages, are viruses that selectively target and kill bacteria. Despite being the most prevalent biological entities on Earth, their therapeutic potential has remained largely untapped. What makes bacteriophages particularly interesting is their ability to kill antibiotic-resistant bacteria – a feature making them prime candidates for treating otherwise unmanageable diabetic foot ulcers. (Armstrong DG, et al; Fish R, et al).
Findings and future directions
Professor Stafford and his team discovered that the feces of several endangered animals harbored bacteriophages capable of killing bacterial strains resistant to antibiotics. The findings not only hold promise for a groundbreaking treatment but also provide another compelling reason to conserve endangered species: Their inherent biodiversity might contain cures for a range of infectious diseases.
While research is ongoing and clinical trials have not yet begun, the preliminary results are overwhelmingly promising.
We often look to complex technologies and synthetic materials for medical science breakthroughs, yet sometimes the most innovative solutions can be found in the most overlooked places. In this case, the feces of endangered species could turn out to be a vital asset in battling antibiotic resistance, thus affecting diabetic foot care in ways we never imagined possible.
The research conducted at the University of Sheffield also serves as a powerful argument for a One Health approach – a multidisciplinary field focusing on the interconnectedness of human, animal, and environmental health.
This intriguing work reaffirms the need for an interdisciplinary approach in tackling the world’s pressing health care challenges. The collaborative efforts between the University of Sheffield and Yorkshire Wildlife Park exemplify how academic research and conservation can come together to yield solutions for some of the most devastating and costly health conditions, while also underscoring the invaluable role that biodiversity plays in our collective well-being. Here’s to teaming up to act against amputation worldwide.
Dr. Armstrong is professor of surgery and director of limb preservation at University of Southern California, Los Angeles. He has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
In a striking convergence of veterinary biology and medical science, researchers from the University of Sheffield (England) have unveiled findings that could potentially advance the treatment of diabetic foot ulcers, a condition affecting an estimated 18.6 million people worldwide. The unexpected ingredient in this potentially transformative therapy? Feces from endangered species, sourced from Yorkshire Wildlife Park, Doncaster, England.
The scourge of antibiotic resistance
Diabetic foot ulcers are a significant challenge in health care, not only because of their prevalence but also because of the alarming rise of antibiotic-resistant bacterial infections. Current antibiotic treatments frequently fail, leading to life-altering consequences like amputations and significant health care costs – estimated at one-third of the total direct costs of diabetes care. The critical need for alternative therapies has propelled scientists into a pressing search for novel antimicrobial agents.
A pioneering approach: zoo poo as bioactive goldmine
Led by Professor Graham Stafford, chair of molecular microbiology at the University of Sheffield, the research team began to explore a rather unorthodox resource: the fecal matter of endangered animals like Guinea baboons, lemurs, and Visayan pigs. While such a source might seem surprising at first glance, the rationale becomes clear when considering the nature of bacteriophages.
What are bacteriophages?
Bacteriophages, commonly known as phages, are viruses that selectively target and kill bacteria. Despite being the most prevalent biological entities on Earth, their therapeutic potential has remained largely untapped. What makes bacteriophages particularly interesting is their ability to kill antibiotic-resistant bacteria – a feature making them prime candidates for treating otherwise unmanageable diabetic foot ulcers. (Armstrong DG, et al; Fish R, et al).
Findings and future directions
Professor Stafford and his team discovered that the feces of several endangered animals harbored bacteriophages capable of killing bacterial strains resistant to antibiotics. The findings not only hold promise for a groundbreaking treatment but also provide another compelling reason to conserve endangered species: Their inherent biodiversity might contain cures for a range of infectious diseases.
While research is ongoing and clinical trials have not yet begun, the preliminary results are overwhelmingly promising.
We often look to complex technologies and synthetic materials for medical science breakthroughs, yet sometimes the most innovative solutions can be found in the most overlooked places. In this case, the feces of endangered species could turn out to be a vital asset in battling antibiotic resistance, thus affecting diabetic foot care in ways we never imagined possible.
The research conducted at the University of Sheffield also serves as a powerful argument for a One Health approach – a multidisciplinary field focusing on the interconnectedness of human, animal, and environmental health.
This intriguing work reaffirms the need for an interdisciplinary approach in tackling the world’s pressing health care challenges. The collaborative efforts between the University of Sheffield and Yorkshire Wildlife Park exemplify how academic research and conservation can come together to yield solutions for some of the most devastating and costly health conditions, while also underscoring the invaluable role that biodiversity plays in our collective well-being. Here’s to teaming up to act against amputation worldwide.
Dr. Armstrong is professor of surgery and director of limb preservation at University of Southern California, Los Angeles. He has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
In a striking convergence of veterinary biology and medical science, researchers from the University of Sheffield (England) have unveiled findings that could potentially advance the treatment of diabetic foot ulcers, a condition affecting an estimated 18.6 million people worldwide. The unexpected ingredient in this potentially transformative therapy? Feces from endangered species, sourced from Yorkshire Wildlife Park, Doncaster, England.
The scourge of antibiotic resistance
Diabetic foot ulcers are a significant challenge in health care, not only because of their prevalence but also because of the alarming rise of antibiotic-resistant bacterial infections. Current antibiotic treatments frequently fail, leading to life-altering consequences like amputations and significant health care costs – estimated at one-third of the total direct costs of diabetes care. The critical need for alternative therapies has propelled scientists into a pressing search for novel antimicrobial agents.
A pioneering approach: zoo poo as bioactive goldmine
Led by Professor Graham Stafford, chair of molecular microbiology at the University of Sheffield, the research team began to explore a rather unorthodox resource: the fecal matter of endangered animals like Guinea baboons, lemurs, and Visayan pigs. While such a source might seem surprising at first glance, the rationale becomes clear when considering the nature of bacteriophages.
What are bacteriophages?
Bacteriophages, commonly known as phages, are viruses that selectively target and kill bacteria. Despite being the most prevalent biological entities on Earth, their therapeutic potential has remained largely untapped. What makes bacteriophages particularly interesting is their ability to kill antibiotic-resistant bacteria – a feature making them prime candidates for treating otherwise unmanageable diabetic foot ulcers. (Armstrong DG, et al; Fish R, et al).
Findings and future directions
Professor Stafford and his team discovered that the feces of several endangered animals harbored bacteriophages capable of killing bacterial strains resistant to antibiotics. The findings not only hold promise for a groundbreaking treatment but also provide another compelling reason to conserve endangered species: Their inherent biodiversity might contain cures for a range of infectious diseases.
While research is ongoing and clinical trials have not yet begun, the preliminary results are overwhelmingly promising.
We often look to complex technologies and synthetic materials for medical science breakthroughs, yet sometimes the most innovative solutions can be found in the most overlooked places. In this case, the feces of endangered species could turn out to be a vital asset in battling antibiotic resistance, thus affecting diabetic foot care in ways we never imagined possible.
The research conducted at the University of Sheffield also serves as a powerful argument for a One Health approach – a multidisciplinary field focusing on the interconnectedness of human, animal, and environmental health.
This intriguing work reaffirms the need for an interdisciplinary approach in tackling the world’s pressing health care challenges. The collaborative efforts between the University of Sheffield and Yorkshire Wildlife Park exemplify how academic research and conservation can come together to yield solutions for some of the most devastating and costly health conditions, while also underscoring the invaluable role that biodiversity plays in our collective well-being. Here’s to teaming up to act against amputation worldwide.
Dr. Armstrong is professor of surgery and director of limb preservation at University of Southern California, Los Angeles. He has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
CAR T-Cell Therapy: Promising Treatments in Development for DLBCL

There have been several recent developments in the treatment of B-cell lymphoma; however, one of the most significant advances has been the development of chimeric antigen receptor (CAR) T-cell therapy. CAR T-cell therapy is a type of personalized immunotherapy that can help cure some people with aggressive non-Hodgkin lymphoma (NHL), including diffuse large B-cell lymphoma (DLBCL), the most common form of aggressive NHL. CAR T-cell therapy has revolutionized the treatment of hematologic malignancies over the past 5 years, with impressive response rates and durable remissions for patients who previously had no viable options. This strategy is highly effective in patients with relapsed/refractory DLBCL, as well as mantle cell lymphoma, follicular lymphoma, acute lymphoblastic leukemia (ALL), and multiple myeloma, as evidenced by recent regulatory approvals.
In 2021, the FDA also approved lisocabtagene maraleucel (liso-cel), a new CAR T-cell therapy for the treatment of adults with relapsed or refractory (nonresponsive) large B-cell lymphoma (LBCL) have been treated with at least 2 prior lines of therapy. These products have design differences, including differences in the costimulatory domain, mechanism of gene/transgene delivery, ability for cryopreservation, and need for T-cell selection.
The CAR T-cell therapy axi-cel demonstrated superior results in the ZUMA-7 clinical trial, which compared CAR T-cell therapy directly to traditional chemotherapy with intended autologous stem cell transplant (ASCT). About 55% of patients were still alive 4 years after receiving axi-cel, compared with 46% of those who initially received the standard treatment for relapsed disease. Based on these results, axi-cel is now the preferred treatment for people whose DLBCL has recurred with 12 months of front-line treatment or who are resistant to standard initial treatment.
Additionally, the BELINDA trial was a randomized phase 3 trial that compared CAR T-cell therapy with liso-cel with second-line chemotherapy with planned ASCT. Like ZUMA-7, this study also demonstrated an improvement in progression-free survival (PFS) compared to standard treatment. As such, CAR T-cell therapy represents the new standard of care for second-line treatment in appropriate patients with refractory or early relapsing LBCL.
There have been several other recent studies on the use of CAR T-cell therapy for B-cell lymphoma. One study, published in Blood Advances (2023), found that receiving a greater number of therapies prior to CAR T-cell therapy is associated with poorer outcomes in patients with aggressive relapsed/refractory B-cell NHL. The study, which included 514 patients from 13 centers treated with CAR-T for aggressive B-cell NHL between 2015 and 2021, found that a greater number of lines of therapy before CAR-T apheresis and bridging therapy were predictive of inferior PFS and overall survival.
Another study compared 2 CD19-targeting CAR T-cell treatments, axi-cel and tisa-cel, with ASCT in the second line setting for LBCL. The study found that axi-cel was superior to ASCT, with longer median event-free survival and a higher response rate. However, tisa-cel was not found to be superior to ASCT. Further studies will be needed to definitively characterize the relative benefits of CAR-T cell therapies and standard second-line treatments for different subgroups of patients with LBCL.
An increasing number of effective targeted agents for DLBCL, including novel monoclonal antibodies (tafasitamab) and antibody-drug conjugates (polatuzumab vedotin and loncastuximab teserine), are being used in earlier lines of therapy. Additionally, 2 anti-CD20 bispecific antibodies (epcoritamab and glofitamab) have gained approval for relapsed/refractory DLBCL due to high response rates. Future studies will be needed to determine if treatment with these agents can produce durable remissions like that of CAR-T cell therapy.
There have been several recent developments in the treatment of B-cell lymphoma; however, one of the most significant advances has been the development of chimeric antigen receptor (CAR) T-cell therapy. CAR T-cell therapy is a type of personalized immunotherapy that can help cure some people with aggressive non-Hodgkin lymphoma (NHL), including diffuse large B-cell lymphoma (DLBCL), the most common form of aggressive NHL. CAR T-cell therapy has revolutionized the treatment of hematologic malignancies over the past 5 years, with impressive response rates and durable remissions for patients who previously had no viable options. This strategy is highly effective in patients with relapsed/refractory DLBCL, as well as mantle cell lymphoma, follicular lymphoma, acute lymphoblastic leukemia (ALL), and multiple myeloma, as evidenced by recent regulatory approvals.
In 2021, the FDA also approved lisocabtagene maraleucel (liso-cel), a new CAR T-cell therapy for the treatment of adults with relapsed or refractory (nonresponsive) large B-cell lymphoma (LBCL) have been treated with at least 2 prior lines of therapy. These products have design differences, including differences in the costimulatory domain, mechanism of gene/transgene delivery, ability for cryopreservation, and need for T-cell selection.
The CAR T-cell therapy axi-cel demonstrated superior results in the ZUMA-7 clinical trial, which compared CAR T-cell therapy directly to traditional chemotherapy with intended autologous stem cell transplant (ASCT). About 55% of patients were still alive 4 years after receiving axi-cel, compared with 46% of those who initially received the standard treatment for relapsed disease. Based on these results, axi-cel is now the preferred treatment for people whose DLBCL has recurred with 12 months of front-line treatment or who are resistant to standard initial treatment.
Additionally, the BELINDA trial was a randomized phase 3 trial that compared CAR T-cell therapy with liso-cel with second-line chemotherapy with planned ASCT. Like ZUMA-7, this study also demonstrated an improvement in progression-free survival (PFS) compared to standard treatment. As such, CAR T-cell therapy represents the new standard of care for second-line treatment in appropriate patients with refractory or early relapsing LBCL.
There have been several other recent studies on the use of CAR T-cell therapy for B-cell lymphoma. One study, published in Blood Advances (2023), found that receiving a greater number of therapies prior to CAR T-cell therapy is associated with poorer outcomes in patients with aggressive relapsed/refractory B-cell NHL. The study, which included 514 patients from 13 centers treated with CAR-T for aggressive B-cell NHL between 2015 and 2021, found that a greater number of lines of therapy before CAR-T apheresis and bridging therapy were predictive of inferior PFS and overall survival.
Another study compared 2 CD19-targeting CAR T-cell treatments, axi-cel and tisa-cel, with ASCT in the second line setting for LBCL. The study found that axi-cel was superior to ASCT, with longer median event-free survival and a higher response rate. However, tisa-cel was not found to be superior to ASCT. Further studies will be needed to definitively characterize the relative benefits of CAR-T cell therapies and standard second-line treatments for different subgroups of patients with LBCL.
An increasing number of effective targeted agents for DLBCL, including novel monoclonal antibodies (tafasitamab) and antibody-drug conjugates (polatuzumab vedotin and loncastuximab teserine), are being used in earlier lines of therapy. Additionally, 2 anti-CD20 bispecific antibodies (epcoritamab and glofitamab) have gained approval for relapsed/refractory DLBCL due to high response rates. Future studies will be needed to determine if treatment with these agents can produce durable remissions like that of CAR-T cell therapy.
There have been several recent developments in the treatment of B-cell lymphoma; however, one of the most significant advances has been the development of chimeric antigen receptor (CAR) T-cell therapy. CAR T-cell therapy is a type of personalized immunotherapy that can help cure some people with aggressive non-Hodgkin lymphoma (NHL), including diffuse large B-cell lymphoma (DLBCL), the most common form of aggressive NHL. CAR T-cell therapy has revolutionized the treatment of hematologic malignancies over the past 5 years, with impressive response rates and durable remissions for patients who previously had no viable options. This strategy is highly effective in patients with relapsed/refractory DLBCL, as well as mantle cell lymphoma, follicular lymphoma, acute lymphoblastic leukemia (ALL), and multiple myeloma, as evidenced by recent regulatory approvals.
In 2021, the FDA also approved lisocabtagene maraleucel (liso-cel), a new CAR T-cell therapy for the treatment of adults with relapsed or refractory (nonresponsive) large B-cell lymphoma (LBCL) have been treated with at least 2 prior lines of therapy. These products have design differences, including differences in the costimulatory domain, mechanism of gene/transgene delivery, ability for cryopreservation, and need for T-cell selection.
The CAR T-cell therapy axi-cel demonstrated superior results in the ZUMA-7 clinical trial, which compared CAR T-cell therapy directly to traditional chemotherapy with intended autologous stem cell transplant (ASCT). About 55% of patients were still alive 4 years after receiving axi-cel, compared with 46% of those who initially received the standard treatment for relapsed disease. Based on these results, axi-cel is now the preferred treatment for people whose DLBCL has recurred with 12 months of front-line treatment or who are resistant to standard initial treatment.
Additionally, the BELINDA trial was a randomized phase 3 trial that compared CAR T-cell therapy with liso-cel with second-line chemotherapy with planned ASCT. Like ZUMA-7, this study also demonstrated an improvement in progression-free survival (PFS) compared to standard treatment. As such, CAR T-cell therapy represents the new standard of care for second-line treatment in appropriate patients with refractory or early relapsing LBCL.
There have been several other recent studies on the use of CAR T-cell therapy for B-cell lymphoma. One study, published in Blood Advances (2023), found that receiving a greater number of therapies prior to CAR T-cell therapy is associated with poorer outcomes in patients with aggressive relapsed/refractory B-cell NHL. The study, which included 514 patients from 13 centers treated with CAR-T for aggressive B-cell NHL between 2015 and 2021, found that a greater number of lines of therapy before CAR-T apheresis and bridging therapy were predictive of inferior PFS and overall survival.
Another study compared 2 CD19-targeting CAR T-cell treatments, axi-cel and tisa-cel, with ASCT in the second line setting for LBCL. The study found that axi-cel was superior to ASCT, with longer median event-free survival and a higher response rate. However, tisa-cel was not found to be superior to ASCT. Further studies will be needed to definitively characterize the relative benefits of CAR-T cell therapies and standard second-line treatments for different subgroups of patients with LBCL.
An increasing number of effective targeted agents for DLBCL, including novel monoclonal antibodies (tafasitamab) and antibody-drug conjugates (polatuzumab vedotin and loncastuximab teserine), are being used in earlier lines of therapy. Additionally, 2 anti-CD20 bispecific antibodies (epcoritamab and glofitamab) have gained approval for relapsed/refractory DLBCL due to high response rates. Future studies will be needed to determine if treatment with these agents can produce durable remissions like that of CAR-T cell therapy.
Diversity in Multiple Sclerosis Care: How the Field of Underrepresented Minorities Has Evolved, and Where We Still See Areas for Improvement

The persistent notion that multiple sclerosis (MS) is predominantly a White patient’s disease has been challenged by scientific data and our clinical experience in the field. Recent research has shown a higher risk of MS in non-White populations than originally thought. This may be surprising, but new data are influencing the way we now approach MS in under-represented minorities, bringing this topic to the forefront of scientific interest.
The early conviction that “there is no MS in minorities” led to underdiagnosis and misdiagnosis of MS in those patients, which in turn deepened these patients’ distrust of physicians and reluctance to seek further medical care, very often delivered by non-minority providers. Inequities in social determinants of health, low health literacy, and lack of private insurance, along with structural racism in healthcare, has further hindered active engagement with an already marginalized patient population in their MS care. This lack of engagement and lack of minorities in scientific research has proved to be unfavorable for MS research as well, creating large and persistent knowledge gaps in understanding MS course, severity, and response to treatment specific to this group. A 2014 PubMed search found 52,000 publications on MS in English, but in only 136 of those were minority patients with MS (Black or Hispanic/Latino) the primary research focus. In 2019, the same search indicated that the subsequent 5 years produced only 30 more articles focusing solely on minority patients.
Research participation of underrepresented minorities is another area where we, as a field, continue to fail these patients. A review of participant enrollment in MS clinical trials that took place between 1993 and 2006 showed a significant decrease in the percentage of enrolled Black patients (from 7% to about 4%). This trend did not improve by the DEFINE treatment trial (2012), in which only 2% of enrolled patients were Black. Of the 1246 participants in the 2019 SUNBEAM MS study, only 2 were Black. Low numbers of minority patients in trials prevent us from drawing any reasonable conclusion as to the efficacy of disease-modifying agents in those patients and make the goal of personalized medicine for this group impossible.
The results of the research conducted on these groups are compelling and should be prompting further work. Not only do Black patients have a higher risk of MS, but there is also now convincing evidence that MS in minorities is more severe overall, causing early progression of disability and necessitating assistive gait devices such as a cane or wheelchair. Minority patients tend to have more extensive involvement of spinal cord and infra-tentorial brain structures during the disease, which could explain the increased likelihood of more severe disease and earlier disability. Minority patients were admitted to nursing homes at a younger age, with greater physical and cognitive impairment than nonminority patients. A study looking at MS mortality between 1999 and 2015 found that Black males with MS had the highest mortality rate before age 45, and Black females before age 53. MS mortality increased with age but peaked at age 55 to 64 for Black patients and 65 to 74 for White patients. Underrepresented minorities are also less likely to use community resources, case management, medical equipment, and home nursing services. When looking at other measures of disease impact on these patients, studies evaluating magnetic resonance imaging (MRI) data showed higher lesion volume in Black patients with MS, as well as a higher degree of brain demyelination and atrophy when compared with White patients.
Treatment strategies currently used for underrepresented minority patients, as well as estimations of medication efficacy, treatment responses, and adverse-event profiles are largely driven by data from clinical trials with only minimal representation of those patients. How can we propose a patient-tailored and individualized treatment plan without these crucial data? Given that, to this day, not a single trial has focused solely on underrepresented minorities, we are left with either post hoc exploratory subgroup analyses of existing trials or pragmatic, observational, and very often retrospective studies using chart analysis. Notwithstanding the methodological flaws of either approach, prior studies did suggest worse response to platform therapies in Black patients, but equal response to high-efficacy therapies when compared with White patients.
Definitive biological underpinnings of disparities in disease severity have not been identified. In recent years, the field of health outcomes research has suggested we move away from considering racial categories as biologically distinct and instead focus on long-overlooked sociodemographic and modifiable lifestyle
factors. The role of diet, exercise, body mass index, smoking, and vascular comorbidities as risk factors associated with worse MS outcomes has been previously shown; however, these factors have not been rigorously assessed in underrepresented populations with MS. Recent studies focused on uncovering what drives the differences in MS severity in underrepresented populations disagree on the role biological differences, socioeconomic disparities, and structural racism in both healthcare settings and society play in answering this question. While it is plausible that a combination of these factors might explain our observations, more research on larger, underserved patient populations and better-defined measures of socioeconomic differences are needed to answer this complex question.
The path of recognizing and correcting our mistakes is not simple but must be done, and our underrepresented minority patients depend on our swift action. There are many places where we as a field of experts can and must do better—in communities, healthcare systems, and society in general.
Increasing community health literacy around MS, rebuilding trust, and addressing structural racism on every level is important. Outreach and educational programs that include in-person meetings and leverage social media platforms can help empower patients and their families—and hopefully increase trust in healthcare providers. Devising targeted interventions focusing on modifiable factors of a healthy lifestyle such as diet and exercise can increase community engagement and strengthen the support system for our patients. Increasing diversity in our own field of physicians, nurses, and other healthcare providers can also aid in strengthening mutual relationships.
Improving access to comprehensive MS care for underrepresented minorities who very often also lack robust insurance coverage is paramount. Recipients of comprehensive care are more likely to participate in research, as these patients receive more well-rounded care and have a lower risk of mismanaged comorbidities. Their involvement in the treatment plan is higher, which also improves compliance with treatment. Patients in comprehensive care centers are more likely to receive newer treatment agents with better efficacy without hindrance of monitoring barriers, and they are likely to benefit from treatment strategies using newly approved agents soon after US Food and Drug Administration approval.
Increasing research participation and, ideally, conducting a clinical trial devoted solely to studying MS in underrepresented minorities is something for which we should actively strive. Identifying the main factors prohibiting enrollment and retention of a high number of minority participants in trials is critical to success. Multiple deterrents in day-to-day life, very often directly connected to economic hardship and racism, pose a very real threat to equitable trial participation. To even consider a successful trial for underrepresented minorities, we must do better in devising strategies and accommodations to help overcome those barriers.
The underserved minorities with MS deserve and need our attention and focus. These patients have largely been neglected and forgotten, but now are emerging at the forefront of our attention—where they belong.
The persistent notion that multiple sclerosis (MS) is predominantly a White patient’s disease has been challenged by scientific data and our clinical experience in the field. Recent research has shown a higher risk of MS in non-White populations than originally thought. This may be surprising, but new data are influencing the way we now approach MS in under-represented minorities, bringing this topic to the forefront of scientific interest.
The early conviction that “there is no MS in minorities” led to underdiagnosis and misdiagnosis of MS in those patients, which in turn deepened these patients’ distrust of physicians and reluctance to seek further medical care, very often delivered by non-minority providers. Inequities in social determinants of health, low health literacy, and lack of private insurance, along with structural racism in healthcare, has further hindered active engagement with an already marginalized patient population in their MS care. This lack of engagement and lack of minorities in scientific research has proved to be unfavorable for MS research as well, creating large and persistent knowledge gaps in understanding MS course, severity, and response to treatment specific to this group. A 2014 PubMed search found 52,000 publications on MS in English, but in only 136 of those were minority patients with MS (Black or Hispanic/Latino) the primary research focus. In 2019, the same search indicated that the subsequent 5 years produced only 30 more articles focusing solely on minority patients.
Research participation of underrepresented minorities is another area where we, as a field, continue to fail these patients. A review of participant enrollment in MS clinical trials that took place between 1993 and 2006 showed a significant decrease in the percentage of enrolled Black patients (from 7% to about 4%). This trend did not improve by the DEFINE treatment trial (2012), in which only 2% of enrolled patients were Black. Of the 1246 participants in the 2019 SUNBEAM MS study, only 2 were Black. Low numbers of minority patients in trials prevent us from drawing any reasonable conclusion as to the efficacy of disease-modifying agents in those patients and make the goal of personalized medicine for this group impossible.
The results of the research conducted on these groups are compelling and should be prompting further work. Not only do Black patients have a higher risk of MS, but there is also now convincing evidence that MS in minorities is more severe overall, causing early progression of disability and necessitating assistive gait devices such as a cane or wheelchair. Minority patients tend to have more extensive involvement of spinal cord and infra-tentorial brain structures during the disease, which could explain the increased likelihood of more severe disease and earlier disability. Minority patients were admitted to nursing homes at a younger age, with greater physical and cognitive impairment than nonminority patients. A study looking at MS mortality between 1999 and 2015 found that Black males with MS had the highest mortality rate before age 45, and Black females before age 53. MS mortality increased with age but peaked at age 55 to 64 for Black patients and 65 to 74 for White patients. Underrepresented minorities are also less likely to use community resources, case management, medical equipment, and home nursing services. When looking at other measures of disease impact on these patients, studies evaluating magnetic resonance imaging (MRI) data showed higher lesion volume in Black patients with MS, as well as a higher degree of brain demyelination and atrophy when compared with White patients.
Treatment strategies currently used for underrepresented minority patients, as well as estimations of medication efficacy, treatment responses, and adverse-event profiles are largely driven by data from clinical trials with only minimal representation of those patients. How can we propose a patient-tailored and individualized treatment plan without these crucial data? Given that, to this day, not a single trial has focused solely on underrepresented minorities, we are left with either post hoc exploratory subgroup analyses of existing trials or pragmatic, observational, and very often retrospective studies using chart analysis. Notwithstanding the methodological flaws of either approach, prior studies did suggest worse response to platform therapies in Black patients, but equal response to high-efficacy therapies when compared with White patients.
Definitive biological underpinnings of disparities in disease severity have not been identified. In recent years, the field of health outcomes research has suggested we move away from considering racial categories as biologically distinct and instead focus on long-overlooked sociodemographic and modifiable lifestyle
factors. The role of diet, exercise, body mass index, smoking, and vascular comorbidities as risk factors associated with worse MS outcomes has been previously shown; however, these factors have not been rigorously assessed in underrepresented populations with MS. Recent studies focused on uncovering what drives the differences in MS severity in underrepresented populations disagree on the role biological differences, socioeconomic disparities, and structural racism in both healthcare settings and society play in answering this question. While it is plausible that a combination of these factors might explain our observations, more research on larger, underserved patient populations and better-defined measures of socioeconomic differences are needed to answer this complex question.
The path of recognizing and correcting our mistakes is not simple but must be done, and our underrepresented minority patients depend on our swift action. There are many places where we as a field of experts can and must do better—in communities, healthcare systems, and society in general.
Increasing community health literacy around MS, rebuilding trust, and addressing structural racism on every level is important. Outreach and educational programs that include in-person meetings and leverage social media platforms can help empower patients and their families—and hopefully increase trust in healthcare providers. Devising targeted interventions focusing on modifiable factors of a healthy lifestyle such as diet and exercise can increase community engagement and strengthen the support system for our patients. Increasing diversity in our own field of physicians, nurses, and other healthcare providers can also aid in strengthening mutual relationships.
Improving access to comprehensive MS care for underrepresented minorities who very often also lack robust insurance coverage is paramount. Recipients of comprehensive care are more likely to participate in research, as these patients receive more well-rounded care and have a lower risk of mismanaged comorbidities. Their involvement in the treatment plan is higher, which also improves compliance with treatment. Patients in comprehensive care centers are more likely to receive newer treatment agents with better efficacy without hindrance of monitoring barriers, and they are likely to benefit from treatment strategies using newly approved agents soon after US Food and Drug Administration approval.
Increasing research participation and, ideally, conducting a clinical trial devoted solely to studying MS in underrepresented minorities is something for which we should actively strive. Identifying the main factors prohibiting enrollment and retention of a high number of minority participants in trials is critical to success. Multiple deterrents in day-to-day life, very often directly connected to economic hardship and racism, pose a very real threat to equitable trial participation. To even consider a successful trial for underrepresented minorities, we must do better in devising strategies and accommodations to help overcome those barriers.
The underserved minorities with MS deserve and need our attention and focus. These patients have largely been neglected and forgotten, but now are emerging at the forefront of our attention—where they belong.
The persistent notion that multiple sclerosis (MS) is predominantly a White patient’s disease has been challenged by scientific data and our clinical experience in the field. Recent research has shown a higher risk of MS in non-White populations than originally thought. This may be surprising, but new data are influencing the way we now approach MS in under-represented minorities, bringing this topic to the forefront of scientific interest.
The early conviction that “there is no MS in minorities” led to underdiagnosis and misdiagnosis of MS in those patients, which in turn deepened these patients’ distrust of physicians and reluctance to seek further medical care, very often delivered by non-minority providers. Inequities in social determinants of health, low health literacy, and lack of private insurance, along with structural racism in healthcare, has further hindered active engagement with an already marginalized patient population in their MS care. This lack of engagement and lack of minorities in scientific research has proved to be unfavorable for MS research as well, creating large and persistent knowledge gaps in understanding MS course, severity, and response to treatment specific to this group. A 2014 PubMed search found 52,000 publications on MS in English, but in only 136 of those were minority patients with MS (Black or Hispanic/Latino) the primary research focus. In 2019, the same search indicated that the subsequent 5 years produced only 30 more articles focusing solely on minority patients.
Research participation of underrepresented minorities is another area where we, as a field, continue to fail these patients. A review of participant enrollment in MS clinical trials that took place between 1993 and 2006 showed a significant decrease in the percentage of enrolled Black patients (from 7% to about 4%). This trend did not improve by the DEFINE treatment trial (2012), in which only 2% of enrolled patients were Black. Of the 1246 participants in the 2019 SUNBEAM MS study, only 2 were Black. Low numbers of minority patients in trials prevent us from drawing any reasonable conclusion as to the efficacy of disease-modifying agents in those patients and make the goal of personalized medicine for this group impossible.
The results of the research conducted on these groups are compelling and should be prompting further work. Not only do Black patients have a higher risk of MS, but there is also now convincing evidence that MS in minorities is more severe overall, causing early progression of disability and necessitating assistive gait devices such as a cane or wheelchair. Minority patients tend to have more extensive involvement of spinal cord and infra-tentorial brain structures during the disease, which could explain the increased likelihood of more severe disease and earlier disability. Minority patients were admitted to nursing homes at a younger age, with greater physical and cognitive impairment than nonminority patients. A study looking at MS mortality between 1999 and 2015 found that Black males with MS had the highest mortality rate before age 45, and Black females before age 53. MS mortality increased with age but peaked at age 55 to 64 for Black patients and 65 to 74 for White patients. Underrepresented minorities are also less likely to use community resources, case management, medical equipment, and home nursing services. When looking at other measures of disease impact on these patients, studies evaluating magnetic resonance imaging (MRI) data showed higher lesion volume in Black patients with MS, as well as a higher degree of brain demyelination and atrophy when compared with White patients.
Treatment strategies currently used for underrepresented minority patients, as well as estimations of medication efficacy, treatment responses, and adverse-event profiles are largely driven by data from clinical trials with only minimal representation of those patients. How can we propose a patient-tailored and individualized treatment plan without these crucial data? Given that, to this day, not a single trial has focused solely on underrepresented minorities, we are left with either post hoc exploratory subgroup analyses of existing trials or pragmatic, observational, and very often retrospective studies using chart analysis. Notwithstanding the methodological flaws of either approach, prior studies did suggest worse response to platform therapies in Black patients, but equal response to high-efficacy therapies when compared with White patients.
Definitive biological underpinnings of disparities in disease severity have not been identified. In recent years, the field of health outcomes research has suggested we move away from considering racial categories as biologically distinct and instead focus on long-overlooked sociodemographic and modifiable lifestyle
factors. The role of diet, exercise, body mass index, smoking, and vascular comorbidities as risk factors associated with worse MS outcomes has been previously shown; however, these factors have not been rigorously assessed in underrepresented populations with MS. Recent studies focused on uncovering what drives the differences in MS severity in underrepresented populations disagree on the role biological differences, socioeconomic disparities, and structural racism in both healthcare settings and society play in answering this question. While it is plausible that a combination of these factors might explain our observations, more research on larger, underserved patient populations and better-defined measures of socioeconomic differences are needed to answer this complex question.
The path of recognizing and correcting our mistakes is not simple but must be done, and our underrepresented minority patients depend on our swift action. There are many places where we as a field of experts can and must do better—in communities, healthcare systems, and society in general.
Increasing community health literacy around MS, rebuilding trust, and addressing structural racism on every level is important. Outreach and educational programs that include in-person meetings and leverage social media platforms can help empower patients and their families—and hopefully increase trust in healthcare providers. Devising targeted interventions focusing on modifiable factors of a healthy lifestyle such as diet and exercise can increase community engagement and strengthen the support system for our patients. Increasing diversity in our own field of physicians, nurses, and other healthcare providers can also aid in strengthening mutual relationships.
Improving access to comprehensive MS care for underrepresented minorities who very often also lack robust insurance coverage is paramount. Recipients of comprehensive care are more likely to participate in research, as these patients receive more well-rounded care and have a lower risk of mismanaged comorbidities. Their involvement in the treatment plan is higher, which also improves compliance with treatment. Patients in comprehensive care centers are more likely to receive newer treatment agents with better efficacy without hindrance of monitoring barriers, and they are likely to benefit from treatment strategies using newly approved agents soon after US Food and Drug Administration approval.
Increasing research participation and, ideally, conducting a clinical trial devoted solely to studying MS in underrepresented minorities is something for which we should actively strive. Identifying the main factors prohibiting enrollment and retention of a high number of minority participants in trials is critical to success. Multiple deterrents in day-to-day life, very often directly connected to economic hardship and racism, pose a very real threat to equitable trial participation. To even consider a successful trial for underrepresented minorities, we must do better in devising strategies and accommodations to help overcome those barriers.
The underserved minorities with MS deserve and need our attention and focus. These patients have largely been neglected and forgotten, but now are emerging at the forefront of our attention—where they belong.