User login
Influenza: U.S. activity was low this summer
Influenza activity in the United States was typically low over the summer months, with influenza A(H3N2) viruses predominating, according to the Centers for Disease Control and Prevention.
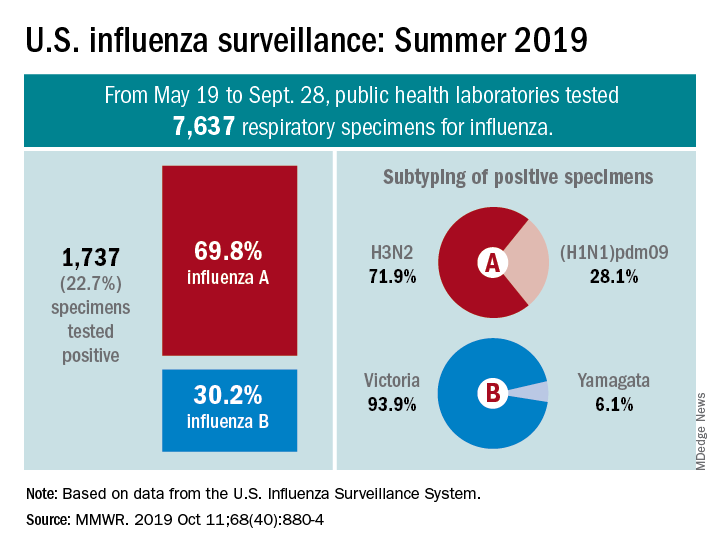
From May 19 to Sept. 28, 2019, weekly flu activity – measured by the percentage of outpatient visits to health care professionals for influenza-like illness (ILI) – was below the national baseline of 2.2%, ranging from 0.7% to 1.4%. Since mid-August, however, when the rate was last 0.7%, it has been climbing slowly but steadily and was up to 1.3% for the week ending Sept. 28, CDC data show.
The various public health laboratories of the U.S. Influenza Surveillance System tested over 7,600 respiratory samples from May 19 to Sept. 28, and 22.7% were positive for influenza viruses, Scott Epperson, DVM, and associates at the CDC’s influenza division said Oct. 10 in the MMWR.
Of the 1,737 samples found to be positive, 69.8% were influenza A and 30.2% were influenza B. The subtype split among specimens positive for Influenza A was 71.9% A(H3N2) and 28.1% A(H1N1)pdm09, while the samples positive for influenza B went 93.9% B/Victoria and 6.1% B/Yamagata, they reported.
Over the same time period in the Southern Hemisphere, “seasonal influenza viruses circulated widely, with influenza A(H3) predominating in many regions; however, influenza A(H1N1)pdm09 and influenza B viruses were predominant in some countries,” the CDC investigators noted.
They also reported the World Health Organization recommendations for the Southern Hemisphere’s 2020 flu vaccines. Components of the egg-based trivalent vaccine are an A/Brisbane/02/2018(H1N1)pdm09-like virus, an A/South Australia/34/2019(H3N2)-like virus, and a B/Washington/02/2019-like virus(B/Victoria lineage). The recommended quadrivalent vaccine adds a B/Phuket/3073/2013-like virus(B/Yamagata lineage), they wrote.
“It is too early in the season to know which viruses will circulate in the United States later this fall and winter or how severe the season might be; however, regardless of what is circulating, the best protection against influenza is an influenza vaccination,” Dr. Epperson and associates wrote.
SOURCE: Epperson S et al. MMWR. 2019 Oct 11;68(40):880-4.
Influenza activity in the United States was typically low over the summer months, with influenza A(H3N2) viruses predominating, according to the Centers for Disease Control and Prevention.
From May 19 to Sept. 28, 2019, weekly flu activity – measured by the percentage of outpatient visits to health care professionals for influenza-like illness (ILI) – was below the national baseline of 2.2%, ranging from 0.7% to 1.4%. Since mid-August, however, when the rate was last 0.7%, it has been climbing slowly but steadily and was up to 1.3% for the week ending Sept. 28, CDC data show.
The various public health laboratories of the U.S. Influenza Surveillance System tested over 7,600 respiratory samples from May 19 to Sept. 28, and 22.7% were positive for influenza viruses, Scott Epperson, DVM, and associates at the CDC’s influenza division said Oct. 10 in the MMWR.
Of the 1,737 samples found to be positive, 69.8% were influenza A and 30.2% were influenza B. The subtype split among specimens positive for Influenza A was 71.9% A(H3N2) and 28.1% A(H1N1)pdm09, while the samples positive for influenza B went 93.9% B/Victoria and 6.1% B/Yamagata, they reported.
Over the same time period in the Southern Hemisphere, “seasonal influenza viruses circulated widely, with influenza A(H3) predominating in many regions; however, influenza A(H1N1)pdm09 and influenza B viruses were predominant in some countries,” the CDC investigators noted.
They also reported the World Health Organization recommendations for the Southern Hemisphere’s 2020 flu vaccines. Components of the egg-based trivalent vaccine are an A/Brisbane/02/2018(H1N1)pdm09-like virus, an A/South Australia/34/2019(H3N2)-like virus, and a B/Washington/02/2019-like virus(B/Victoria lineage). The recommended quadrivalent vaccine adds a B/Phuket/3073/2013-like virus(B/Yamagata lineage), they wrote.
“It is too early in the season to know which viruses will circulate in the United States later this fall and winter or how severe the season might be; however, regardless of what is circulating, the best protection against influenza is an influenza vaccination,” Dr. Epperson and associates wrote.
SOURCE: Epperson S et al. MMWR. 2019 Oct 11;68(40):880-4.
Influenza activity in the United States was typically low over the summer months, with influenza A(H3N2) viruses predominating, according to the Centers for Disease Control and Prevention.
From May 19 to Sept. 28, 2019, weekly flu activity – measured by the percentage of outpatient visits to health care professionals for influenza-like illness (ILI) – was below the national baseline of 2.2%, ranging from 0.7% to 1.4%. Since mid-August, however, when the rate was last 0.7%, it has been climbing slowly but steadily and was up to 1.3% for the week ending Sept. 28, CDC data show.
The various public health laboratories of the U.S. Influenza Surveillance System tested over 7,600 respiratory samples from May 19 to Sept. 28, and 22.7% were positive for influenza viruses, Scott Epperson, DVM, and associates at the CDC’s influenza division said Oct. 10 in the MMWR.
Of the 1,737 samples found to be positive, 69.8% were influenza A and 30.2% were influenza B. The subtype split among specimens positive for Influenza A was 71.9% A(H3N2) and 28.1% A(H1N1)pdm09, while the samples positive for influenza B went 93.9% B/Victoria and 6.1% B/Yamagata, they reported.
Over the same time period in the Southern Hemisphere, “seasonal influenza viruses circulated widely, with influenza A(H3) predominating in many regions; however, influenza A(H1N1)pdm09 and influenza B viruses were predominant in some countries,” the CDC investigators noted.
They also reported the World Health Organization recommendations for the Southern Hemisphere’s 2020 flu vaccines. Components of the egg-based trivalent vaccine are an A/Brisbane/02/2018(H1N1)pdm09-like virus, an A/South Australia/34/2019(H3N2)-like virus, and a B/Washington/02/2019-like virus(B/Victoria lineage). The recommended quadrivalent vaccine adds a B/Phuket/3073/2013-like virus(B/Yamagata lineage), they wrote.
“It is too early in the season to know which viruses will circulate in the United States later this fall and winter or how severe the season might be; however, regardless of what is circulating, the best protection against influenza is an influenza vaccination,” Dr. Epperson and associates wrote.
SOURCE: Epperson S et al. MMWR. 2019 Oct 11;68(40):880-4.
FROM MMWR
New York declares end to 2018 measles outbreak
New York State has reported the end of all active measles cases related to the initial outbreak in 2018, but the state is now responding to new, unrelated cases in four counties, according to the Centers for Disease Control and Prevention.
The new cases – two in Nassau County and one each in Monroe, Putnam, and Rockland counties – are “related to measles exposures from international travel but not affiliated with the 2018 outbreak,” the New York State Department of Health said in a written statement. Officials in Rockland County had declared its 2018 measles outbreak, which involved 312 cases in 2018 and 2019, over on Sept. 25.
. Of those cases, 1,163 (93%) were associated with 22 outbreaks, with the two largest occurring in New York City and Rockland County. “These two almost year-long outbreaks placed the United States at risk for losing measles elimination status,” the CDC said in a separate report, but “robust responses … ended transmission before the 1-year mark.”
New York State has reported the end of all active measles cases related to the initial outbreak in 2018, but the state is now responding to new, unrelated cases in four counties, according to the Centers for Disease Control and Prevention.
The new cases – two in Nassau County and one each in Monroe, Putnam, and Rockland counties – are “related to measles exposures from international travel but not affiliated with the 2018 outbreak,” the New York State Department of Health said in a written statement. Officials in Rockland County had declared its 2018 measles outbreak, which involved 312 cases in 2018 and 2019, over on Sept. 25.
. Of those cases, 1,163 (93%) were associated with 22 outbreaks, with the two largest occurring in New York City and Rockland County. “These two almost year-long outbreaks placed the United States at risk for losing measles elimination status,” the CDC said in a separate report, but “robust responses … ended transmission before the 1-year mark.”
New York State has reported the end of all active measles cases related to the initial outbreak in 2018, but the state is now responding to new, unrelated cases in four counties, according to the Centers for Disease Control and Prevention.
The new cases – two in Nassau County and one each in Monroe, Putnam, and Rockland counties – are “related to measles exposures from international travel but not affiliated with the 2018 outbreak,” the New York State Department of Health said in a written statement. Officials in Rockland County had declared its 2018 measles outbreak, which involved 312 cases in 2018 and 2019, over on Sept. 25.
. Of those cases, 1,163 (93%) were associated with 22 outbreaks, with the two largest occurring in New York City and Rockland County. “These two almost year-long outbreaks placed the United States at risk for losing measles elimination status,” the CDC said in a separate report, but “robust responses … ended transmission before the 1-year mark.”
Twin births down among women 30 and older
according to the National Center for Health Statistics.
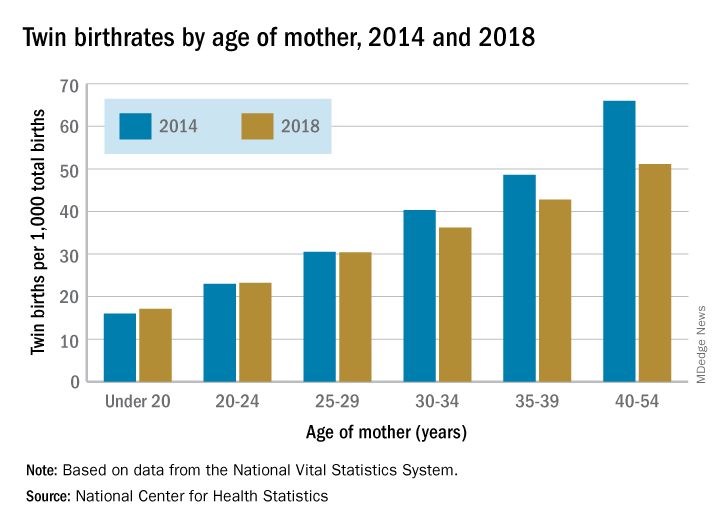
The twin birthrate, which had increased by 79% during 1980-2014, fell by 4% during 2014-2018, but that decline was “not universal across maternal age and race and Hispanic-origin groups,” the NCHS investigators said.
Twin birthrates fell by at least 10% for mothers aged 30 years and older from 2014 to 2018 but held steady for women in their twenties. Over that same period, the twin birthrate fell by a significant 7% among non-Hispanic white women (36.7 to 34.3 per 1,000 total births) but increased just slightly for non-Hispanic black women (40.0 to 40.5 per 1,000) and Hispanic women (24.1 to 24.4), the investigators reported.
For women 30 years and older, the drops in twin births got larger as age increased and were significant for each age group. The rate for women aged 30-34 years fell 10% as it went from 40.3 per 1,000 total births in 2014 to 36.2 per 1,000. The decrease was 12% (from 48.6 per 1,000 to 42.8) for women aged 35-39 and 23% (from 66.0 to 51.1) for those aged 40 years and older, they said based on data from the National Vital Statistics System.
The rates were basically unchanged for women in their 20s, from 23.0 to 23.2 in 20- to 24-year-olds and 30.5 to 30.4 in 25- to 29-year-olds – but there was a significant increase for the youngest group with rates among those younger than 20 years going from 16.0 to 17.1 per 1,000, the report showed.
according to the National Center for Health Statistics.
The twin birthrate, which had increased by 79% during 1980-2014, fell by 4% during 2014-2018, but that decline was “not universal across maternal age and race and Hispanic-origin groups,” the NCHS investigators said.
Twin birthrates fell by at least 10% for mothers aged 30 years and older from 2014 to 2018 but held steady for women in their twenties. Over that same period, the twin birthrate fell by a significant 7% among non-Hispanic white women (36.7 to 34.3 per 1,000 total births) but increased just slightly for non-Hispanic black women (40.0 to 40.5 per 1,000) and Hispanic women (24.1 to 24.4), the investigators reported.
For women 30 years and older, the drops in twin births got larger as age increased and were significant for each age group. The rate for women aged 30-34 years fell 10% as it went from 40.3 per 1,000 total births in 2014 to 36.2 per 1,000. The decrease was 12% (from 48.6 per 1,000 to 42.8) for women aged 35-39 and 23% (from 66.0 to 51.1) for those aged 40 years and older, they said based on data from the National Vital Statistics System.
The rates were basically unchanged for women in their 20s, from 23.0 to 23.2 in 20- to 24-year-olds and 30.5 to 30.4 in 25- to 29-year-olds – but there was a significant increase for the youngest group with rates among those younger than 20 years going from 16.0 to 17.1 per 1,000, the report showed.
according to the National Center for Health Statistics.
The twin birthrate, which had increased by 79% during 1980-2014, fell by 4% during 2014-2018, but that decline was “not universal across maternal age and race and Hispanic-origin groups,” the NCHS investigators said.
Twin birthrates fell by at least 10% for mothers aged 30 years and older from 2014 to 2018 but held steady for women in their twenties. Over that same period, the twin birthrate fell by a significant 7% among non-Hispanic white women (36.7 to 34.3 per 1,000 total births) but increased just slightly for non-Hispanic black women (40.0 to 40.5 per 1,000) and Hispanic women (24.1 to 24.4), the investigators reported.
For women 30 years and older, the drops in twin births got larger as age increased and were significant for each age group. The rate for women aged 30-34 years fell 10% as it went from 40.3 per 1,000 total births in 2014 to 36.2 per 1,000. The decrease was 12% (from 48.6 per 1,000 to 42.8) for women aged 35-39 and 23% (from 66.0 to 51.1) for those aged 40 years and older, they said based on data from the National Vital Statistics System.
The rates were basically unchanged for women in their 20s, from 23.0 to 23.2 in 20- to 24-year-olds and 30.5 to 30.4 in 25- to 29-year-olds – but there was a significant increase for the youngest group with rates among those younger than 20 years going from 16.0 to 17.1 per 1,000, the report showed.
FDA approves benralizumab autoinjector for eosinophilic asthma
according to a press release from AstraZeneca. Benralizumab is already approved as add-on maintenance for this form of asthma, but not for other eosinophilic conditions or for acute bronchospasm or status asthmaticus.

The autoinjector “pen” was tested for usability and pharmacokinetic exposure in two studies, the phase 3 GRECO trial and the phase 1 AMES trial, respectively. The multicenter, open-label GRECO trial was designed to assess patient- or caregiver-reported functionality, and it found that 97% of at-home administrations were successful at week 12 and week 16. The multicenter, randomized, open-label, parallel-group AMES trial compared pharmacokinetic exposure with the subcutaneous administration using either prefilled syringe or prefilled autoinjector; it found that the eosinophils were rapidly depleted in patients with use of either device.
The safety profiles in both trials were comparable to those seen in previous trials. Hypersensitivity reactions have been sometimes observed in the hours following administration of benralizumab; discontinuation is advised in case of any hypersensitivity reaction. The therapy should not be used to treat acute asthma symptoms, such as exacerbations, or bronchospasm, and any reduction in corticosteroid therapy should be gradual and performed under careful supervision of a health care professional. Although benralizumab’s effects on helminth infections are currently unknown, care should be taken with preexisting or incident infections.
Full prescribing information can be found on the AstraZeneca website.
according to a press release from AstraZeneca. Benralizumab is already approved as add-on maintenance for this form of asthma, but not for other eosinophilic conditions or for acute bronchospasm or status asthmaticus.
The autoinjector “pen” was tested for usability and pharmacokinetic exposure in two studies, the phase 3 GRECO trial and the phase 1 AMES trial, respectively. The multicenter, open-label GRECO trial was designed to assess patient- or caregiver-reported functionality, and it found that 97% of at-home administrations were successful at week 12 and week 16. The multicenter, randomized, open-label, parallel-group AMES trial compared pharmacokinetic exposure with the subcutaneous administration using either prefilled syringe or prefilled autoinjector; it found that the eosinophils were rapidly depleted in patients with use of either device.
The safety profiles in both trials were comparable to those seen in previous trials. Hypersensitivity reactions have been sometimes observed in the hours following administration of benralizumab; discontinuation is advised in case of any hypersensitivity reaction. The therapy should not be used to treat acute asthma symptoms, such as exacerbations, or bronchospasm, and any reduction in corticosteroid therapy should be gradual and performed under careful supervision of a health care professional. Although benralizumab’s effects on helminth infections are currently unknown, care should be taken with preexisting or incident infections.
Full prescribing information can be found on the AstraZeneca website.
according to a press release from AstraZeneca. Benralizumab is already approved as add-on maintenance for this form of asthma, but not for other eosinophilic conditions or for acute bronchospasm or status asthmaticus.
The autoinjector “pen” was tested for usability and pharmacokinetic exposure in two studies, the phase 3 GRECO trial and the phase 1 AMES trial, respectively. The multicenter, open-label GRECO trial was designed to assess patient- or caregiver-reported functionality, and it found that 97% of at-home administrations were successful at week 12 and week 16. The multicenter, randomized, open-label, parallel-group AMES trial compared pharmacokinetic exposure with the subcutaneous administration using either prefilled syringe or prefilled autoinjector; it found that the eosinophils were rapidly depleted in patients with use of either device.
The safety profiles in both trials were comparable to those seen in previous trials. Hypersensitivity reactions have been sometimes observed in the hours following administration of benralizumab; discontinuation is advised in case of any hypersensitivity reaction. The therapy should not be used to treat acute asthma symptoms, such as exacerbations, or bronchospasm, and any reduction in corticosteroid therapy should be gradual and performed under careful supervision of a health care professional. Although benralizumab’s effects on helminth infections are currently unknown, care should be taken with preexisting or incident infections.
Full prescribing information can be found on the AstraZeneca website.
Vaping-associated lung injury cases exceed 1,000
More than 1,000 cases of vaping-associated lung injury have been reported in 48 states and the U.S. Virgin Islands, according to a telebriefing by the Centers for Disease Control and Prevention.

As of Oct. 1, there have been 1,080 confirmed and probable cases of lung injury associated with the use of e-cigarettes, or vaping, said Anne Schuchat, MD, principal deputy director of the CDC. The latest figures were also reported in a statement issued by the CDC.
Dr. Schuchat said 18 related deaths in 15 states have been confirmed, and additional deaths are under investigation.
“As we have continued to get data for additional cases, the trends we reported last week persist,” Dr. Schuchat said (MMWR. 2019 Sep 27;68[39];860-4).
“Most patients reported a history of using THC [tetrahydrocannabinol]-containing products, and most patients are male and young people.” Of the 1,080 cases identified, approximately 70% are male, roughly 80% are younger than 35 years of age, and 37% are under 21 years of age. The patients’ median age is 23 years (range, 13-75 years). Among patients who have died, the median age is 50 years (range, 27-71 years).
The CDC now has information from 578 patients on the substances used in vaping products in the 90 days before symptom onset. About 78% of these patients reported using THC-containing products, and 37% reported exclusive use of THC-containing products. Roughly 58% of patients reported using nicotine-containing products, and 17% reported exclusive use of nicotine-containing products.
“I wish we had more answers regarding the specific harmful products or components that are causing these illnesses,” Dr. Schuchat said. She noted that THC-containing products appear to be the most commonly used, but these products don’t appear to be the only culprit. Additionally, in a report released recently in the New England Journal of Medicine (2019 Sep 9. doi: 10.1056/NEJMoa1911614), THC-containing products bought “off the street” were commonly used by patients with lung injuries. However, the CDC can’t say for certain if it’s safer for consumers to buy THC-containing products from a licensed dispensary.
The CDC has deployed staff to several states to help investigate the lung injuries, reached out to the clinical community to increase awareness of the injuries, and worked with clinicians and medical examiners to review assessments of patients who have developed these injuries, including those who have died. The CDC has also convened clinical professional societies to “help strengthen the detection, reporting, and management of cases,” Dr. Schuchat said.
In addition, the CDC has joined with the Food and Drug Administration and other public health partners to develop a laboratory plan for “continued testing of products, aerosol testing of substances produced by the products, and clinical pathology lung specimens from patients,” Dr. Schuchat said.
The FDA is also working to gather more information about vaping-associated lung injuries. The FDA is trying to obtain “critical details” about the specific products or substances that may be involved, said Judy McMeekin, PharmD, deputy associate commissioner for regulatory affairs at the FDA.
“There does not currently appear to be one product or substance involved in all of the cases,” Dr. McMeekin said. “We are leaving no stone unturned and following all potential leads regarding any particular product, constituent, or compound that may be at issue.”
The FDA has collected more than 440 samples of vaping devices and products from 18 states. The agency is still analyzing these samples, but a preliminary analysis has shown that some products contain THC concentrations ranging from 14% to 76%, and some products contain a combination of THC and vitamin E acetate ranging from 31% to 88%.
For information about the collection of vaping products for possible testing by the FDA, email [email protected]. For information about collection and submission of clinical specimens for possible testing by the CDC, see the Healthcare Provider webpage.
Clinicians and health officials who have questions about this outbreak can email [email protected]. All others with questions about this outbreak can contact CDC-INFO at 800-232-4636 or submit information at the Contact CDC-INFO page.
More than 1,000 cases of vaping-associated lung injury have been reported in 48 states and the U.S. Virgin Islands, according to a telebriefing by the Centers for Disease Control and Prevention.
As of Oct. 1, there have been 1,080 confirmed and probable cases of lung injury associated with the use of e-cigarettes, or vaping, said Anne Schuchat, MD, principal deputy director of the CDC. The latest figures were also reported in a statement issued by the CDC.
Dr. Schuchat said 18 related deaths in 15 states have been confirmed, and additional deaths are under investigation.
“As we have continued to get data for additional cases, the trends we reported last week persist,” Dr. Schuchat said (MMWR. 2019 Sep 27;68[39];860-4).
“Most patients reported a history of using THC [tetrahydrocannabinol]-containing products, and most patients are male and young people.” Of the 1,080 cases identified, approximately 70% are male, roughly 80% are younger than 35 years of age, and 37% are under 21 years of age. The patients’ median age is 23 years (range, 13-75 years). Among patients who have died, the median age is 50 years (range, 27-71 years).
The CDC now has information from 578 patients on the substances used in vaping products in the 90 days before symptom onset. About 78% of these patients reported using THC-containing products, and 37% reported exclusive use of THC-containing products. Roughly 58% of patients reported using nicotine-containing products, and 17% reported exclusive use of nicotine-containing products.
“I wish we had more answers regarding the specific harmful products or components that are causing these illnesses,” Dr. Schuchat said. She noted that THC-containing products appear to be the most commonly used, but these products don’t appear to be the only culprit. Additionally, in a report released recently in the New England Journal of Medicine (2019 Sep 9. doi: 10.1056/NEJMoa1911614), THC-containing products bought “off the street” were commonly used by patients with lung injuries. However, the CDC can’t say for certain if it’s safer for consumers to buy THC-containing products from a licensed dispensary.
The CDC has deployed staff to several states to help investigate the lung injuries, reached out to the clinical community to increase awareness of the injuries, and worked with clinicians and medical examiners to review assessments of patients who have developed these injuries, including those who have died. The CDC has also convened clinical professional societies to “help strengthen the detection, reporting, and management of cases,” Dr. Schuchat said.
In addition, the CDC has joined with the Food and Drug Administration and other public health partners to develop a laboratory plan for “continued testing of products, aerosol testing of substances produced by the products, and clinical pathology lung specimens from patients,” Dr. Schuchat said.
The FDA is also working to gather more information about vaping-associated lung injuries. The FDA is trying to obtain “critical details” about the specific products or substances that may be involved, said Judy McMeekin, PharmD, deputy associate commissioner for regulatory affairs at the FDA.
“There does not currently appear to be one product or substance involved in all of the cases,” Dr. McMeekin said. “We are leaving no stone unturned and following all potential leads regarding any particular product, constituent, or compound that may be at issue.”
The FDA has collected more than 440 samples of vaping devices and products from 18 states. The agency is still analyzing these samples, but a preliminary analysis has shown that some products contain THC concentrations ranging from 14% to 76%, and some products contain a combination of THC and vitamin E acetate ranging from 31% to 88%.
For information about the collection of vaping products for possible testing by the FDA, email [email protected]. For information about collection and submission of clinical specimens for possible testing by the CDC, see the Healthcare Provider webpage.
Clinicians and health officials who have questions about this outbreak can email [email protected]. All others with questions about this outbreak can contact CDC-INFO at 800-232-4636 or submit information at the Contact CDC-INFO page.
More than 1,000 cases of vaping-associated lung injury have been reported in 48 states and the U.S. Virgin Islands, according to a telebriefing by the Centers for Disease Control and Prevention.
As of Oct. 1, there have been 1,080 confirmed and probable cases of lung injury associated with the use of e-cigarettes, or vaping, said Anne Schuchat, MD, principal deputy director of the CDC. The latest figures were also reported in a statement issued by the CDC.
Dr. Schuchat said 18 related deaths in 15 states have been confirmed, and additional deaths are under investigation.
“As we have continued to get data for additional cases, the trends we reported last week persist,” Dr. Schuchat said (MMWR. 2019 Sep 27;68[39];860-4).
“Most patients reported a history of using THC [tetrahydrocannabinol]-containing products, and most patients are male and young people.” Of the 1,080 cases identified, approximately 70% are male, roughly 80% are younger than 35 years of age, and 37% are under 21 years of age. The patients’ median age is 23 years (range, 13-75 years). Among patients who have died, the median age is 50 years (range, 27-71 years).
The CDC now has information from 578 patients on the substances used in vaping products in the 90 days before symptom onset. About 78% of these patients reported using THC-containing products, and 37% reported exclusive use of THC-containing products. Roughly 58% of patients reported using nicotine-containing products, and 17% reported exclusive use of nicotine-containing products.
“I wish we had more answers regarding the specific harmful products or components that are causing these illnesses,” Dr. Schuchat said. She noted that THC-containing products appear to be the most commonly used, but these products don’t appear to be the only culprit. Additionally, in a report released recently in the New England Journal of Medicine (2019 Sep 9. doi: 10.1056/NEJMoa1911614), THC-containing products bought “off the street” were commonly used by patients with lung injuries. However, the CDC can’t say for certain if it’s safer for consumers to buy THC-containing products from a licensed dispensary.
The CDC has deployed staff to several states to help investigate the lung injuries, reached out to the clinical community to increase awareness of the injuries, and worked with clinicians and medical examiners to review assessments of patients who have developed these injuries, including those who have died. The CDC has also convened clinical professional societies to “help strengthen the detection, reporting, and management of cases,” Dr. Schuchat said.
In addition, the CDC has joined with the Food and Drug Administration and other public health partners to develop a laboratory plan for “continued testing of products, aerosol testing of substances produced by the products, and clinical pathology lung specimens from patients,” Dr. Schuchat said.
The FDA is also working to gather more information about vaping-associated lung injuries. The FDA is trying to obtain “critical details” about the specific products or substances that may be involved, said Judy McMeekin, PharmD, deputy associate commissioner for regulatory affairs at the FDA.
“There does not currently appear to be one product or substance involved in all of the cases,” Dr. McMeekin said. “We are leaving no stone unturned and following all potential leads regarding any particular product, constituent, or compound that may be at issue.”
The FDA has collected more than 440 samples of vaping devices and products from 18 states. The agency is still analyzing these samples, but a preliminary analysis has shown that some products contain THC concentrations ranging from 14% to 76%, and some products contain a combination of THC and vitamin E acetate ranging from 31% to 88%.
For information about the collection of vaping products for possible testing by the FDA, email [email protected]. For information about collection and submission of clinical specimens for possible testing by the CDC, see the Healthcare Provider webpage.
Clinicians and health officials who have questions about this outbreak can email [email protected]. All others with questions about this outbreak can contact CDC-INFO at 800-232-4636 or submit information at the Contact CDC-INFO page.
FDA approves Descovy as HIV PrEP for men and transgender women who have sex with men
The decision, backing the earlier recommendation of the FDA’s Antimicrobial Drugs Advisory Committee, was based upon results from DISCOVER, a pivotal, multiyear, global phase 3 clinical trial that evaluated the safety and efficacy of Descovy (emtricitabine 200 mg and tenofovir alafenamide 25-mg tablets for PrEP, compared with Truvada (emtricitabine 200 mg and tenofovir disoproxil fumarate 300-mg tablets).
DISCOVER included more than 5,300 adult cisgender men who have sex with men or transgender women who have sex with men.
In the trial, Descovy achieved noninferiority to Truvada.
Descovy has a Boxed Warning in its U.S. product label regarding the risk of posttreatment acute exacerbation of hepatitis B, according to the company.
The Descovy label also includes a Boxed Warning regarding the risk of drug resistance with PrEP use in undiagnosed early HIV-1 infection. The effectiveness of Descovy for PrEP in individuals at risk of HIV-1 from receptive vaginal sex was not tested, and thus cisgender women at risk for infection from vaginal sex were not included in the population for which the drug was approved.
The Descovy label and safety information is available here.
The FDA version of the announcement is available here.
The decision, backing the earlier recommendation of the FDA’s Antimicrobial Drugs Advisory Committee, was based upon results from DISCOVER, a pivotal, multiyear, global phase 3 clinical trial that evaluated the safety and efficacy of Descovy (emtricitabine 200 mg and tenofovir alafenamide 25-mg tablets for PrEP, compared with Truvada (emtricitabine 200 mg and tenofovir disoproxil fumarate 300-mg tablets).
DISCOVER included more than 5,300 adult cisgender men who have sex with men or transgender women who have sex with men.
In the trial, Descovy achieved noninferiority to Truvada.
Descovy has a Boxed Warning in its U.S. product label regarding the risk of posttreatment acute exacerbation of hepatitis B, according to the company.
The Descovy label also includes a Boxed Warning regarding the risk of drug resistance with PrEP use in undiagnosed early HIV-1 infection. The effectiveness of Descovy for PrEP in individuals at risk of HIV-1 from receptive vaginal sex was not tested, and thus cisgender women at risk for infection from vaginal sex were not included in the population for which the drug was approved.
The Descovy label and safety information is available here.
The FDA version of the announcement is available here.
The decision, backing the earlier recommendation of the FDA’s Antimicrobial Drugs Advisory Committee, was based upon results from DISCOVER, a pivotal, multiyear, global phase 3 clinical trial that evaluated the safety and efficacy of Descovy (emtricitabine 200 mg and tenofovir alafenamide 25-mg tablets for PrEP, compared with Truvada (emtricitabine 200 mg and tenofovir disoproxil fumarate 300-mg tablets).
DISCOVER included more than 5,300 adult cisgender men who have sex with men or transgender women who have sex with men.
In the trial, Descovy achieved noninferiority to Truvada.
Descovy has a Boxed Warning in its U.S. product label regarding the risk of posttreatment acute exacerbation of hepatitis B, according to the company.
The Descovy label also includes a Boxed Warning regarding the risk of drug resistance with PrEP use in undiagnosed early HIV-1 infection. The effectiveness of Descovy for PrEP in individuals at risk of HIV-1 from receptive vaginal sex was not tested, and thus cisgender women at risk for infection from vaginal sex were not included in the population for which the drug was approved.
The Descovy label and safety information is available here.
The FDA version of the announcement is available here.
FDA approves rituximab to treat children with rare vasculitis
The Food and Drug Administration approved rituximab (Rituxan) by injection to treat granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA) in children 2 years of age and older in combination with glucocorticoid treatment, according to an FDA news release.
These rare forms of vasculitis damage small blood vessels through inflammation and can lead to serious organ failure, including lungs and kidneys.
The Genentech drug received priority review and an orphan drug designation based on the results of a pediatric clinical trial of 25 patients aged 6-17 years with active GPA or MPA who were treated with rituximab in an international multicenter, open-label, uncontrolled study. Patients in the trial were also given methylprednisolone prior to starting treatment.
The trial consisted of a 6-month remission induction phase where patients were treated only with rituximab and glucocorticoids. In addition, patients who had not achieved remission could receive additional treatment, including other therapies, at the discretion of the investigator, according to the FDA. By 6 months, 14 of the patients were in remission, and after 18 months, all 25 patients were in remission.
Rituximab contains a boxed warning regarding increased risks of fatal infusion reactions, potentially fatal severe skin and mouth reactions, hepatitis B virus reactivation that may cause serious or lethal liver problems, and progressive multifocal leukoencephalopathy, a rare, potentially lethal brain infection.
The trial was conducted and sponsored by F. Hoffmann-La Roche, which owns Genentech.
The Food and Drug Administration approved rituximab (Rituxan) by injection to treat granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA) in children 2 years of age and older in combination with glucocorticoid treatment, according to an FDA news release.
These rare forms of vasculitis damage small blood vessels through inflammation and can lead to serious organ failure, including lungs and kidneys.
The Genentech drug received priority review and an orphan drug designation based on the results of a pediatric clinical trial of 25 patients aged 6-17 years with active GPA or MPA who were treated with rituximab in an international multicenter, open-label, uncontrolled study. Patients in the trial were also given methylprednisolone prior to starting treatment.
The trial consisted of a 6-month remission induction phase where patients were treated only with rituximab and glucocorticoids. In addition, patients who had not achieved remission could receive additional treatment, including other therapies, at the discretion of the investigator, according to the FDA. By 6 months, 14 of the patients were in remission, and after 18 months, all 25 patients were in remission.
Rituximab contains a boxed warning regarding increased risks of fatal infusion reactions, potentially fatal severe skin and mouth reactions, hepatitis B virus reactivation that may cause serious or lethal liver problems, and progressive multifocal leukoencephalopathy, a rare, potentially lethal brain infection.
The trial was conducted and sponsored by F. Hoffmann-La Roche, which owns Genentech.
The Food and Drug Administration approved rituximab (Rituxan) by injection to treat granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA) in children 2 years of age and older in combination with glucocorticoid treatment, according to an FDA news release.
These rare forms of vasculitis damage small blood vessels through inflammation and can lead to serious organ failure, including lungs and kidneys.
The Genentech drug received priority review and an orphan drug designation based on the results of a pediatric clinical trial of 25 patients aged 6-17 years with active GPA or MPA who were treated with rituximab in an international multicenter, open-label, uncontrolled study. Patients in the trial were also given methylprednisolone prior to starting treatment.
The trial consisted of a 6-month remission induction phase where patients were treated only with rituximab and glucocorticoids. In addition, patients who had not achieved remission could receive additional treatment, including other therapies, at the discretion of the investigator, according to the FDA. By 6 months, 14 of the patients were in remission, and after 18 months, all 25 patients were in remission.
Rituximab contains a boxed warning regarding increased risks of fatal infusion reactions, potentially fatal severe skin and mouth reactions, hepatitis B virus reactivation that may cause serious or lethal liver problems, and progressive multifocal leukoencephalopathy, a rare, potentially lethal brain infection.
The trial was conducted and sponsored by F. Hoffmann-La Roche, which owns Genentech.
CDC reports most vaping lung disease linked to THC-containing cartridges
and most products used were prepackaged, prefilled cartridges, according to new data released by the Centers for Disease Control and Prevention.

The majority of these products (66%) were THC-containing cartridges marketed under the brand name Dank. Dank cartridges are available at legal dispensaries and online in areas where they are legal. The Dank company posted a statement on its website warning buyers about fake cartridges and showing images of genuine cartridges. However, 89% of the cartridges were obtained on the street, from dealers, online, or from friends or social contacts, Jennifer Layden, MD, of the Illinois Department of Public Health said during a CDC telebriefing.
The illness was first recognized in Wisconsin and Illinois. Marijuana is illegal in Wisconsin; Illinois licensed recreational marijuana in 2009.
Other commonalties among cases have also emerged, Anne Schuchat, MD, deputy director of CDC, said during the call. More than two-thirds of the 805 confirmed or probable cases were male, and the median age was 23 years. The illness crosses age barriers, she said. About 62% were 18-24 years of age, and 54% under age 25. However, among the 12 deaths so far reported, the median age was 50 years. The age range was wide, from 27 to 71 years. Dr. Schuchat said data about medical comorbidities potentially linking the deaths is not yet available, although it is part of the ongoing investigation.
Other clinical commonalities included intensive use of THC-containing products and, in a small number of cases, concomitant use of benzodiazepenes, opioids, and narcotics.
Cases have now emerged in 46 states and in the U.S. Virgin Islands, although the number reported each week is dropping. However, this decrease may not represent a drop in newly occurring cases, but instead reflect delays in clinical recognition or reporting to local health departments, Dr. Schuchat said.
Regardless of the recent decline in reported cases, she said, the epidemic is serious, far reaching, and ongoing.
“I want to stress that this is a serious, life-threatening disease occurring mostly in otherwise healthy young people. These illnesses and deaths are occurring in the context of a dynamic marketplace with mix of products with mixes of ingredients, including potentially illicit substances. Users don’t know what’s in them and cannot tell from the ingredients listed on the packaging.”
Dr. Schuchat drew her data from two reports issued in the Morbidity and Mortality Weekly Report: a national case update by Peter A. Briss, MD, chair of CDC’s Lung Injury Response Epidemiology/Surveillance Group, and colleagues, and a regional report coauthored by Dr. Layden of cases in Illinois and Wisconsin.
In the national report, 514 patients self-reported their history of e-cigarette and vaping use. Among those, 395 (76.9%) reported using THC-containing products, and 292 (56.8%) reported using nicotine-containing products in the 30 days preceding symptom onset. Almost half (210; 40.9%) reported using both THC- and nicotine-containing products.
But there appeared to be no clear pattern of use, said Dr. Briss, who also participated in the briefing. More than a third (185; 36.0%) reported exclusive use of THC-containing products, and 82 (16.0%) reported exclusive use of nicotine-containing products.
The regional report added additional details.
Among the 86 patients who self-reported details, there were 234 unique cases of e-cigarette or THC vaping in 87 brands.
“Patients reported using numerous products and brands,” Dr. Layden noted. “Those who reported using THC products used an average of 2.1 different products and those who reported using nicotine products used about 1.3 different ones. Some patients reported using up to seven different brands, and these were used at least daily and sometimes numerous times in the day.”
According to the MMWR regional report, among the urinary THC screens obtained for 32 patients, “29 (91%) were positive for THC. One of these patients reported smoking combustible marijuana. Urinary THC levels for four patients who reported using THC-containing products exceeded 400 ng/ml, indicating intensive use of THC or THC-containing products.”
About 40% of THC users and 65% of nicotine-product users reported using the product at least five times a day; 52% said they used combustible marijuana in addition to the vapes, and 24% reported also smoking combustible tobacco.
There was a very low level of concomitant drug use. Two patients reported using LSD; one reported misusing dextroamphetamine-amphetamine (Adderall), and one reported misusing oxycodone. Two tested positive for benzodiazepines and opioids, and one each for only benzodiazepines, only opioids, only amphetamines. One patient screened positive for unidentified narcotics.
and most products used were prepackaged, prefilled cartridges, according to new data released by the Centers for Disease Control and Prevention.
The majority of these products (66%) were THC-containing cartridges marketed under the brand name Dank. Dank cartridges are available at legal dispensaries and online in areas where they are legal. The Dank company posted a statement on its website warning buyers about fake cartridges and showing images of genuine cartridges. However, 89% of the cartridges were obtained on the street, from dealers, online, or from friends or social contacts, Jennifer Layden, MD, of the Illinois Department of Public Health said during a CDC telebriefing.
The illness was first recognized in Wisconsin and Illinois. Marijuana is illegal in Wisconsin; Illinois licensed recreational marijuana in 2009.
Other commonalties among cases have also emerged, Anne Schuchat, MD, deputy director of CDC, said during the call. More than two-thirds of the 805 confirmed or probable cases were male, and the median age was 23 years. The illness crosses age barriers, she said. About 62% were 18-24 years of age, and 54% under age 25. However, among the 12 deaths so far reported, the median age was 50 years. The age range was wide, from 27 to 71 years. Dr. Schuchat said data about medical comorbidities potentially linking the deaths is not yet available, although it is part of the ongoing investigation.
Other clinical commonalities included intensive use of THC-containing products and, in a small number of cases, concomitant use of benzodiazepenes, opioids, and narcotics.
Cases have now emerged in 46 states and in the U.S. Virgin Islands, although the number reported each week is dropping. However, this decrease may not represent a drop in newly occurring cases, but instead reflect delays in clinical recognition or reporting to local health departments, Dr. Schuchat said.
Regardless of the recent decline in reported cases, she said, the epidemic is serious, far reaching, and ongoing.
“I want to stress that this is a serious, life-threatening disease occurring mostly in otherwise healthy young people. These illnesses and deaths are occurring in the context of a dynamic marketplace with mix of products with mixes of ingredients, including potentially illicit substances. Users don’t know what’s in them and cannot tell from the ingredients listed on the packaging.”
Dr. Schuchat drew her data from two reports issued in the Morbidity and Mortality Weekly Report: a national case update by Peter A. Briss, MD, chair of CDC’s Lung Injury Response Epidemiology/Surveillance Group, and colleagues, and a regional report coauthored by Dr. Layden of cases in Illinois and Wisconsin.
In the national report, 514 patients self-reported their history of e-cigarette and vaping use. Among those, 395 (76.9%) reported using THC-containing products, and 292 (56.8%) reported using nicotine-containing products in the 30 days preceding symptom onset. Almost half (210; 40.9%) reported using both THC- and nicotine-containing products.
But there appeared to be no clear pattern of use, said Dr. Briss, who also participated in the briefing. More than a third (185; 36.0%) reported exclusive use of THC-containing products, and 82 (16.0%) reported exclusive use of nicotine-containing products.
The regional report added additional details.
Among the 86 patients who self-reported details, there were 234 unique cases of e-cigarette or THC vaping in 87 brands.
“Patients reported using numerous products and brands,” Dr. Layden noted. “Those who reported using THC products used an average of 2.1 different products and those who reported using nicotine products used about 1.3 different ones. Some patients reported using up to seven different brands, and these were used at least daily and sometimes numerous times in the day.”
According to the MMWR regional report, among the urinary THC screens obtained for 32 patients, “29 (91%) were positive for THC. One of these patients reported smoking combustible marijuana. Urinary THC levels for four patients who reported using THC-containing products exceeded 400 ng/ml, indicating intensive use of THC or THC-containing products.”
About 40% of THC users and 65% of nicotine-product users reported using the product at least five times a day; 52% said they used combustible marijuana in addition to the vapes, and 24% reported also smoking combustible tobacco.
There was a very low level of concomitant drug use. Two patients reported using LSD; one reported misusing dextroamphetamine-amphetamine (Adderall), and one reported misusing oxycodone. Two tested positive for benzodiazepines and opioids, and one each for only benzodiazepines, only opioids, only amphetamines. One patient screened positive for unidentified narcotics.
and most products used were prepackaged, prefilled cartridges, according to new data released by the Centers for Disease Control and Prevention.
The majority of these products (66%) were THC-containing cartridges marketed under the brand name Dank. Dank cartridges are available at legal dispensaries and online in areas where they are legal. The Dank company posted a statement on its website warning buyers about fake cartridges and showing images of genuine cartridges. However, 89% of the cartridges were obtained on the street, from dealers, online, or from friends or social contacts, Jennifer Layden, MD, of the Illinois Department of Public Health said during a CDC telebriefing.
The illness was first recognized in Wisconsin and Illinois. Marijuana is illegal in Wisconsin; Illinois licensed recreational marijuana in 2009.
Other commonalties among cases have also emerged, Anne Schuchat, MD, deputy director of CDC, said during the call. More than two-thirds of the 805 confirmed or probable cases were male, and the median age was 23 years. The illness crosses age barriers, she said. About 62% were 18-24 years of age, and 54% under age 25. However, among the 12 deaths so far reported, the median age was 50 years. The age range was wide, from 27 to 71 years. Dr. Schuchat said data about medical comorbidities potentially linking the deaths is not yet available, although it is part of the ongoing investigation.
Other clinical commonalities included intensive use of THC-containing products and, in a small number of cases, concomitant use of benzodiazepenes, opioids, and narcotics.
Cases have now emerged in 46 states and in the U.S. Virgin Islands, although the number reported each week is dropping. However, this decrease may not represent a drop in newly occurring cases, but instead reflect delays in clinical recognition or reporting to local health departments, Dr. Schuchat said.
Regardless of the recent decline in reported cases, she said, the epidemic is serious, far reaching, and ongoing.
“I want to stress that this is a serious, life-threatening disease occurring mostly in otherwise healthy young people. These illnesses and deaths are occurring in the context of a dynamic marketplace with mix of products with mixes of ingredients, including potentially illicit substances. Users don’t know what’s in them and cannot tell from the ingredients listed on the packaging.”
Dr. Schuchat drew her data from two reports issued in the Morbidity and Mortality Weekly Report: a national case update by Peter A. Briss, MD, chair of CDC’s Lung Injury Response Epidemiology/Surveillance Group, and colleagues, and a regional report coauthored by Dr. Layden of cases in Illinois and Wisconsin.
In the national report, 514 patients self-reported their history of e-cigarette and vaping use. Among those, 395 (76.9%) reported using THC-containing products, and 292 (56.8%) reported using nicotine-containing products in the 30 days preceding symptom onset. Almost half (210; 40.9%) reported using both THC- and nicotine-containing products.
But there appeared to be no clear pattern of use, said Dr. Briss, who also participated in the briefing. More than a third (185; 36.0%) reported exclusive use of THC-containing products, and 82 (16.0%) reported exclusive use of nicotine-containing products.
The regional report added additional details.
Among the 86 patients who self-reported details, there were 234 unique cases of e-cigarette or THC vaping in 87 brands.
“Patients reported using numerous products and brands,” Dr. Layden noted. “Those who reported using THC products used an average of 2.1 different products and those who reported using nicotine products used about 1.3 different ones. Some patients reported using up to seven different brands, and these were used at least daily and sometimes numerous times in the day.”
According to the MMWR regional report, among the urinary THC screens obtained for 32 patients, “29 (91%) were positive for THC. One of these patients reported smoking combustible marijuana. Urinary THC levels for four patients who reported using THC-containing products exceeded 400 ng/ml, indicating intensive use of THC or THC-containing products.”
About 40% of THC users and 65% of nicotine-product users reported using the product at least five times a day; 52% said they used combustible marijuana in addition to the vapes, and 24% reported also smoking combustible tobacco.
There was a very low level of concomitant drug use. Two patients reported using LSD; one reported misusing dextroamphetamine-amphetamine (Adderall), and one reported misusing oxycodone. Two tested positive for benzodiazepines and opioids, and one each for only benzodiazepines, only opioids, only amphetamines. One patient screened positive for unidentified narcotics.
FDA expands Dysport’s upper-limb spasticity indication to children
The Food and Drug Administration has expanded the indication of abobotulinumtoxinA (Dysport) for upper-limb spasticity to include patients aged 2 years and older, according to a release from Ipsen. This botulinum toxin product received approval for this indication in adults in 2015 and approval for lower-limb spasticity in patients aged 2 years and older in 2016. Notably, Orphan Drug Exclusivity prevents it from being indicated for patients with cerebral palsy because another botulinum toxin product, onabotulinumtoxinA (Botox), already was approved for the indication in June 2019.
Spasticity affects the muscles and joints of extremities, especially in growing children, and is usually caused by nerve damage, such as head trauma or spinal cord injury. The degree of spasticity can vary from mild muscle stiffness to severe, painful, and uncontrollable muscle spasms.
AbobotulinumtoxinA was evaluated for upper-limb spasticity in a phase 3, randomized, double-blind, low-dose controlled, multicenter study; the study enrolled 210 children aged 2-17 years with the condition and a Modified Ashworth Scale grade 2 or greater for elbow and wrist flexors. The children were randomized 1:1:1 to injections of either 8 units/kg, 16 units/kg, or 2 units/kg into the elbow flexors and wrist flexors. At 6 weeks, there were statistically significant improvements in Modified Ashworth Scale grade, the primary endpoint, with least-square mean changes from baseline of –2.0, –2.3, and –1.6, respectively.
AbobotulinumtoxinA and all other botulinum toxin products carry a boxed warning, the most serious warning the FDA issues. This warning refers to risk of botulism-like symptoms caused by the botulinum toxin spreading away from the injection area; these symptoms can included sometimes life-threatening difficulty swallowing or breathing. AbobotulinumtoxinA is contraindicated in patients with known hypersensitivity to any botulinum toxin or any of the components, those with presence of infection at proposed injection site(s), and those with known allergy to cow’s milk protein. It is also important to note that botulinum toxin preparations are not interchangeable; the potency units of one are not the same as those of another. Full prescribing information can be found on the Ipsen website.
The Food and Drug Administration has expanded the indication of abobotulinumtoxinA (Dysport) for upper-limb spasticity to include patients aged 2 years and older, according to a release from Ipsen. This botulinum toxin product received approval for this indication in adults in 2015 and approval for lower-limb spasticity in patients aged 2 years and older in 2016. Notably, Orphan Drug Exclusivity prevents it from being indicated for patients with cerebral palsy because another botulinum toxin product, onabotulinumtoxinA (Botox), already was approved for the indication in June 2019.
Spasticity affects the muscles and joints of extremities, especially in growing children, and is usually caused by nerve damage, such as head trauma or spinal cord injury. The degree of spasticity can vary from mild muscle stiffness to severe, painful, and uncontrollable muscle spasms.
AbobotulinumtoxinA was evaluated for upper-limb spasticity in a phase 3, randomized, double-blind, low-dose controlled, multicenter study; the study enrolled 210 children aged 2-17 years with the condition and a Modified Ashworth Scale grade 2 or greater for elbow and wrist flexors. The children were randomized 1:1:1 to injections of either 8 units/kg, 16 units/kg, or 2 units/kg into the elbow flexors and wrist flexors. At 6 weeks, there were statistically significant improvements in Modified Ashworth Scale grade, the primary endpoint, with least-square mean changes from baseline of –2.0, –2.3, and –1.6, respectively.
AbobotulinumtoxinA and all other botulinum toxin products carry a boxed warning, the most serious warning the FDA issues. This warning refers to risk of botulism-like symptoms caused by the botulinum toxin spreading away from the injection area; these symptoms can included sometimes life-threatening difficulty swallowing or breathing. AbobotulinumtoxinA is contraindicated in patients with known hypersensitivity to any botulinum toxin or any of the components, those with presence of infection at proposed injection site(s), and those with known allergy to cow’s milk protein. It is also important to note that botulinum toxin preparations are not interchangeable; the potency units of one are not the same as those of another. Full prescribing information can be found on the Ipsen website.
The Food and Drug Administration has expanded the indication of abobotulinumtoxinA (Dysport) for upper-limb spasticity to include patients aged 2 years and older, according to a release from Ipsen. This botulinum toxin product received approval for this indication in adults in 2015 and approval for lower-limb spasticity in patients aged 2 years and older in 2016. Notably, Orphan Drug Exclusivity prevents it from being indicated for patients with cerebral palsy because another botulinum toxin product, onabotulinumtoxinA (Botox), already was approved for the indication in June 2019.
Spasticity affects the muscles and joints of extremities, especially in growing children, and is usually caused by nerve damage, such as head trauma or spinal cord injury. The degree of spasticity can vary from mild muscle stiffness to severe, painful, and uncontrollable muscle spasms.
AbobotulinumtoxinA was evaluated for upper-limb spasticity in a phase 3, randomized, double-blind, low-dose controlled, multicenter study; the study enrolled 210 children aged 2-17 years with the condition and a Modified Ashworth Scale grade 2 or greater for elbow and wrist flexors. The children were randomized 1:1:1 to injections of either 8 units/kg, 16 units/kg, or 2 units/kg into the elbow flexors and wrist flexors. At 6 weeks, there were statistically significant improvements in Modified Ashworth Scale grade, the primary endpoint, with least-square mean changes from baseline of –2.0, –2.3, and –1.6, respectively.
AbobotulinumtoxinA and all other botulinum toxin products carry a boxed warning, the most serious warning the FDA issues. This warning refers to risk of botulism-like symptoms caused by the botulinum toxin spreading away from the injection area; these symptoms can included sometimes life-threatening difficulty swallowing or breathing. AbobotulinumtoxinA is contraindicated in patients with known hypersensitivity to any botulinum toxin or any of the components, those with presence of infection at proposed injection site(s), and those with known allergy to cow’s milk protein. It is also important to note that botulinum toxin preparations are not interchangeable; the potency units of one are not the same as those of another. Full prescribing information can be found on the Ipsen website.
Daratumumab approved in combo with VTd for transplant-eligible multiple myeloma
The Food and Drug Administration has approved daratumumab in combination with certain therapies for newly diagnosed patients with multiple myeloma who are eligible for autologous stem cell transplant.
The approval specifies combination of this CD38-directed antibody with bortezomib (Velcade), thalidomide, and dexamethasone (VTd), according to an announcement from Janssen.
The approval is based on results from the CASSIOPEIA study. The first part of the study randomized 1,085 patients (median age, 58 years) and showed that, compared with VTd alone, the daratumumab-VTd combination had significantly better postconsolidation stringent complete response (29% vs. 20%; odds ratio, 1.60; 95% confidence interval, 1.21-2.12; P = .001) and a 53% reduction in risk of disease progression or death (hazard ratio, 0.47; 95% CI, 0.33-0.67; P = .0001).
The most frequent adverse reactions with 5% greater frequency in the daratumumab-VTd group were infusion reactions (including anaphylaxis), nausea, pyrexia, upper respiratory tract infection, and bronchitis. Full prescribing information, including contraindications and warnings, can be found on the Janssen website.
Daratumumab was initially approved in 2015, and in June 2019, it received approval, in combination with lenalidomide and dexamethasone, for treatment of patients with newly diagnosed multiple myeloma who are ineligible for autologous stem cell transplant.
The Food and Drug Administration has approved daratumumab in combination with certain therapies for newly diagnosed patients with multiple myeloma who are eligible for autologous stem cell transplant.
The approval specifies combination of this CD38-directed antibody with bortezomib (Velcade), thalidomide, and dexamethasone (VTd), according to an announcement from Janssen.
The approval is based on results from the CASSIOPEIA study. The first part of the study randomized 1,085 patients (median age, 58 years) and showed that, compared with VTd alone, the daratumumab-VTd combination had significantly better postconsolidation stringent complete response (29% vs. 20%; odds ratio, 1.60; 95% confidence interval, 1.21-2.12; P = .001) and a 53% reduction in risk of disease progression or death (hazard ratio, 0.47; 95% CI, 0.33-0.67; P = .0001).
The most frequent adverse reactions with 5% greater frequency in the daratumumab-VTd group were infusion reactions (including anaphylaxis), nausea, pyrexia, upper respiratory tract infection, and bronchitis. Full prescribing information, including contraindications and warnings, can be found on the Janssen website.
Daratumumab was initially approved in 2015, and in June 2019, it received approval, in combination with lenalidomide and dexamethasone, for treatment of patients with newly diagnosed multiple myeloma who are ineligible for autologous stem cell transplant.
The Food and Drug Administration has approved daratumumab in combination with certain therapies for newly diagnosed patients with multiple myeloma who are eligible for autologous stem cell transplant.
The approval specifies combination of this CD38-directed antibody with bortezomib (Velcade), thalidomide, and dexamethasone (VTd), according to an announcement from Janssen.
The approval is based on results from the CASSIOPEIA study. The first part of the study randomized 1,085 patients (median age, 58 years) and showed that, compared with VTd alone, the daratumumab-VTd combination had significantly better postconsolidation stringent complete response (29% vs. 20%; odds ratio, 1.60; 95% confidence interval, 1.21-2.12; P = .001) and a 53% reduction in risk of disease progression or death (hazard ratio, 0.47; 95% CI, 0.33-0.67; P = .0001).
The most frequent adverse reactions with 5% greater frequency in the daratumumab-VTd group were infusion reactions (including anaphylaxis), nausea, pyrexia, upper respiratory tract infection, and bronchitis. Full prescribing information, including contraindications and warnings, can be found on the Janssen website.
Daratumumab was initially approved in 2015, and in June 2019, it received approval, in combination with lenalidomide and dexamethasone, for treatment of patients with newly diagnosed multiple myeloma who are ineligible for autologous stem cell transplant.