User login
Ulcer on leg
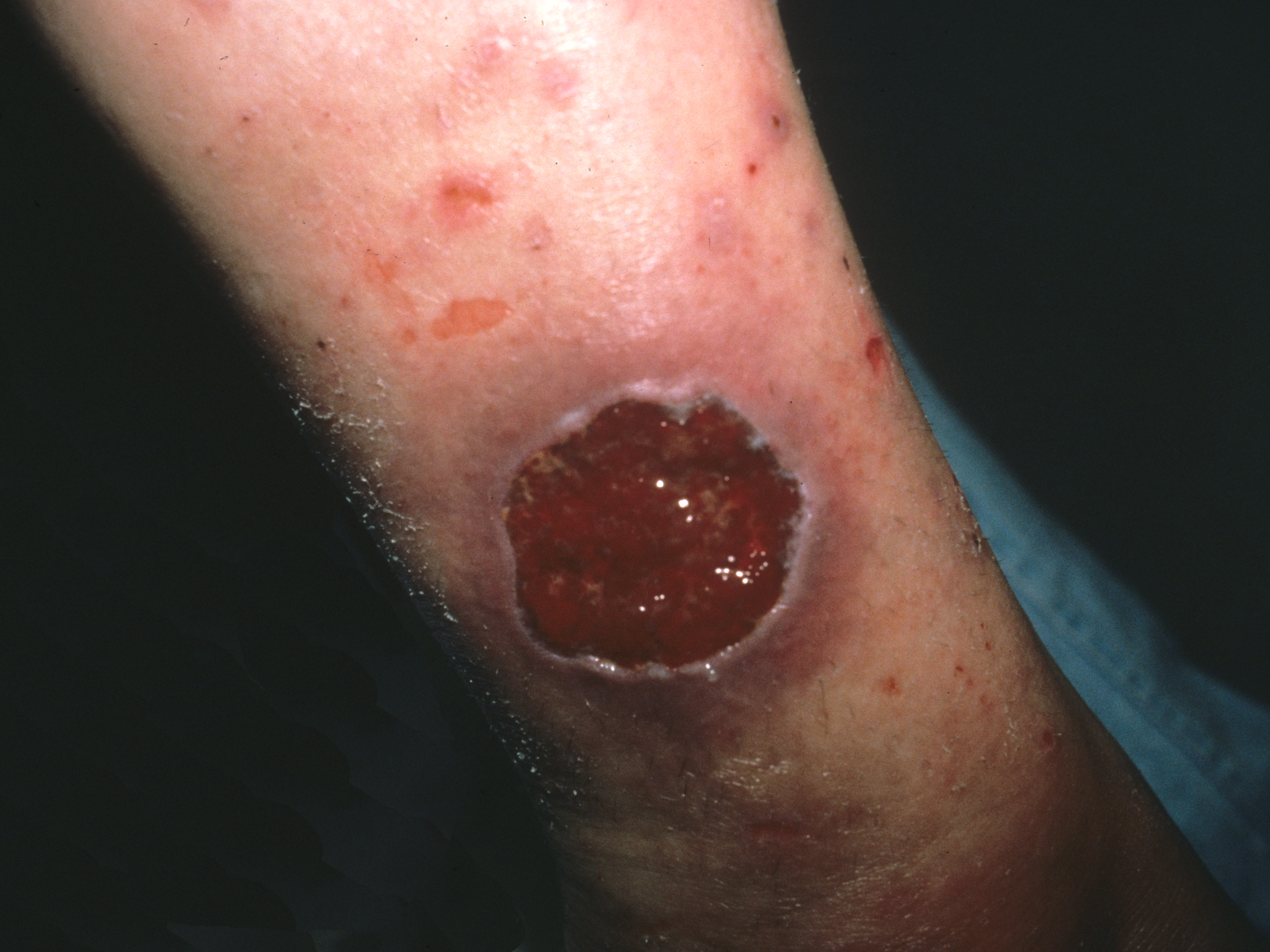
The family physician made the presumptive diagnosis of pyoderma gangrenosum. At least 50% of cases are associated with systemic diseases such as inflammatory bowel disease, hematologic malignancy, and arthritis. It’s estimated that 30% of patients with pyoderma gangrenosum experience pathergy (initiation at the site of trauma or injury). This patient’s history was consistent with pathergy.
Typically pyoderma gangrenosum presents with a deep, painful ulcer. The ulcer has a well-defined border, which is usually violet or blue. The ulcer edge is often undermined and the surrounding skin is erythematous and indurated. Pyoderma gangrenosum usually starts as small papules that coalesce; the central area then undergoes necrosis to form a single ulcer. The lesions are painful and the pain can be severe. Patients may have malaise, arthralgia, and myalgia. Pyoderma gangrenosum is most commonly seen on the legs, but can occur on any skin surface (including the genitalia) and around a stoma.
The physician performed a punch biopsy including the active ulcer and the border to rule out other causes of ulcerative skin lesions. The biopsy showed a neutrophilic infiltrate consistent with pyoderma gangrenosum.
Treatment options include:
- topical medications, such as potent steroid ointments, tacrolimus ointment, and silver sulfadiazine
- intralesional injections with corticosteroids
- systemic treatment with oral corticosteroids
- treatment with mycophenolate mofetil, oral tacrolimus, dapsone, azathioprine, or infliximab (each used successfully, according to published case reports).
Surgical debridement is contraindicated since pathergy will make the lesions worse.
In this case, the patient chose topical clobetasol ointment. She was lost to follow-up.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Flowers D, Usatine R. Pyoderma gangrenosum. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. The Color Atlas of Family Medicine. New York, NY: McGraw-Hill; 2009:735-739.
To learn more about The Color Atlas of Family Medicine, see:
* http://www.amazon.com/Color-Atlas-Family-Medicine/dp/0071474641
* http://www.mhprofessional.com/product.php?isbn=0071474641
The family physician made the presumptive diagnosis of pyoderma gangrenosum. At least 50% of cases are associated with systemic diseases such as inflammatory bowel disease, hematologic malignancy, and arthritis. It’s estimated that 30% of patients with pyoderma gangrenosum experience pathergy (initiation at the site of trauma or injury). This patient’s history was consistent with pathergy.
Typically pyoderma gangrenosum presents with a deep, painful ulcer. The ulcer has a well-defined border, which is usually violet or blue. The ulcer edge is often undermined and the surrounding skin is erythematous and indurated. Pyoderma gangrenosum usually starts as small papules that coalesce; the central area then undergoes necrosis to form a single ulcer. The lesions are painful and the pain can be severe. Patients may have malaise, arthralgia, and myalgia. Pyoderma gangrenosum is most commonly seen on the legs, but can occur on any skin surface (including the genitalia) and around a stoma.
The physician performed a punch biopsy including the active ulcer and the border to rule out other causes of ulcerative skin lesions. The biopsy showed a neutrophilic infiltrate consistent with pyoderma gangrenosum.
Treatment options include:
- topical medications, such as potent steroid ointments, tacrolimus ointment, and silver sulfadiazine
- intralesional injections with corticosteroids
- systemic treatment with oral corticosteroids
- treatment with mycophenolate mofetil, oral tacrolimus, dapsone, azathioprine, or infliximab (each used successfully, according to published case reports).
Surgical debridement is contraindicated since pathergy will make the lesions worse.
In this case, the patient chose topical clobetasol ointment. She was lost to follow-up.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Flowers D, Usatine R. Pyoderma gangrenosum. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. The Color Atlas of Family Medicine. New York, NY: McGraw-Hill; 2009:735-739.
To learn more about The Color Atlas of Family Medicine, see:
* http://www.amazon.com/Color-Atlas-Family-Medicine/dp/0071474641
* http://www.mhprofessional.com/product.php?isbn=0071474641
The family physician made the presumptive diagnosis of pyoderma gangrenosum. At least 50% of cases are associated with systemic diseases such as inflammatory bowel disease, hematologic malignancy, and arthritis. It’s estimated that 30% of patients with pyoderma gangrenosum experience pathergy (initiation at the site of trauma or injury). This patient’s history was consistent with pathergy.
Typically pyoderma gangrenosum presents with a deep, painful ulcer. The ulcer has a well-defined border, which is usually violet or blue. The ulcer edge is often undermined and the surrounding skin is erythematous and indurated. Pyoderma gangrenosum usually starts as small papules that coalesce; the central area then undergoes necrosis to form a single ulcer. The lesions are painful and the pain can be severe. Patients may have malaise, arthralgia, and myalgia. Pyoderma gangrenosum is most commonly seen on the legs, but can occur on any skin surface (including the genitalia) and around a stoma.
The physician performed a punch biopsy including the active ulcer and the border to rule out other causes of ulcerative skin lesions. The biopsy showed a neutrophilic infiltrate consistent with pyoderma gangrenosum.
Treatment options include:
- topical medications, such as potent steroid ointments, tacrolimus ointment, and silver sulfadiazine
- intralesional injections with corticosteroids
- systemic treatment with oral corticosteroids
- treatment with mycophenolate mofetil, oral tacrolimus, dapsone, azathioprine, or infliximab (each used successfully, according to published case reports).
Surgical debridement is contraindicated since pathergy will make the lesions worse.
In this case, the patient chose topical clobetasol ointment. She was lost to follow-up.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Flowers D, Usatine R. Pyoderma gangrenosum. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. The Color Atlas of Family Medicine. New York, NY: McGraw-Hill; 2009:735-739.
To learn more about The Color Atlas of Family Medicine, see:
* http://www.amazon.com/Color-Atlas-Family-Medicine/dp/0071474641
* http://www.mhprofessional.com/product.php?isbn=0071474641
Multiple facial bumps with weight loss
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
A 12-Year-old girl came into our hospital for treatment of multiple bumps that had developed around her eyes and other areas of her face 2 months earlier. She had difficulty opening her eyes and complained of gradual weight loss.
On examination, we noted numerous skin-colored, shiny, dome-shaped, coalescing papules and nodules with central umbilications that were distributed mostly on her periocular and perinasal areas (FIGURE). When we expressed the papules with forceps, they exuded a cheesy material. We also noticed crusting and signs of inflammation on her eyelids.
The systemic examination was unremarkable.
FIGURE
Opening her eyes was difficult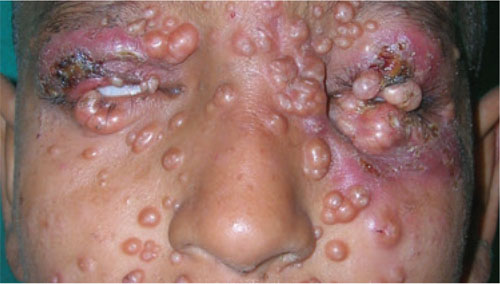
This 12-year-old patient had multiple dome-shaped, coalescing papules and nodules with central umbilications in the periocular and perinasal areas.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Giant molluscum contagiosum
Molluscum contagiosum (MC) is a relatively common, benign, viral cutaneous infection that primarily affects children, sexually active adults, and immunodeficient individuals. MC accounts for approximately 1% of all diagnosed skin disorders in the United States; internationally, however, the incidence is higher.1 The causative organism of MC is a member of the Poxviridae family2 and is thought to be transmitted by close personal contact, autoinoculation, and fomites.3
MC is clinically characterized by the presence of pearly white, dome-shaped papules or nodules with central dells. The lesions are typically located on the trunk, body folds, extremities, and genitalia (particularly when the infection is sexually acquired).2,3 Pruritus and an eczematous reaction can develop around the lesions.
MC is a recognized ocular complication of acquired immune deficiency syndrome (AIDS). Periocular MC can also occur after eyebrow shaping in beauty salons.4 In human immunodeficiency virus (HIV)-positive patients, lesions are usually widespread, tend to be large, and usually occur during the advanced stage of HIV infection.2,5
The differential includes carcinoma
When considering a diagnosis of MC, you’ll need to rule out the following causes of similar-looking papules and nodules:
Nodular basal cell carcinoma presents as a slow-growing, firm, shiny, pearly nodule with fine telangiectasia. It may also present as a cystic lesion that can be mistaken for inclusion cysts of the eyelid. If left untreated, the tumor may ulcerate.
Juvenile xanthogranulomas are rubbery, tan-orange papules or nodules. Patients may have one or several papules or nodules in the head and neck region; these lesions may appear elsewhere, as well.
Cryptococcosis may present as painless papules or pustules, which then become nodules that may ulcerate. The lesions may show central umbilications.
Keratoacanthoma begins as a firm, roundish, skin-colored or reddish papule that rapidly progresses to a dome-shaped nodule, with a smooth, shiny surface and a central crateriform ulceration or keratin plug. Patients typically have a solitary lesion that may appear on the face, neck, or dorsum of the upper extremities.
Penicillosis often presents with MC-like skin lesions, in addition to fever, anemia, hepatomegaly, lymphadenopathy, and productive cough.
History and lab work clinch the Dx
Diagnosis is made by the distinctive clinical appearance, but can be confirmed by skin biopsy demonstrating eosinophilic molluscum bodies packed into the cells of the spinous layer of the epidermis.3 Giemsa stain of the material obtained from a crushed papule will reveal the presence of pathognomonic “molluscum bodies” in the cells of the epidermis.2,3
Our patient’s Giemsa stain revealed molluscum bodies. And since it is always wise to rule out concomitant HIV infection in patients who have giant MC, we tested our patient. Her results were positive; she had a CD4+ count of 93 cells/mm3.
Many treatment options from which to choose
MC is usually self-limiting, although it can take several months—or even a few years—to resolve on its own6 (strength of recommendation [SOR]: B). However, most patients with MC should receive treatment to obtain relief from symptoms, prevent autoinoculation or transmission to close contacts, decrease occurrence of scarring, reduce secondary bacterial infections, and improve cosmesis.
Several treatment options are available, and most rely on destruction of the lesions. Manual extrusion is a simple but effective therapy6 (SOR: B). Cryotherapy and curettage are also effective treatment options5 (SOR: C). Pretreatment topical anesthesia is often helpful if these therapies are used in children.
Topical imiquimod2 (1%-5%) cream applied 3 to 7 times a week can be used to treat generalized MC infection or MC localized to the anogenital area6 (SOR: A). Some patients may improve with topical tretinoin therapy6 (SOR: C).
Chemical cauterization with 10% povidone iodine with 50% salicylic acid7 (SOR: B), 10% potassium hydroxide8 (SOR: B), cantharidin2 (SOR: C), or 25% to 50% trichloroacetic acid6 (SOR: C) is also effective. Treatment with flashlamp pulsed dye laser is a safe and efficient treatment modality9 (SOR: C). Cidofovir10 (1%-3%) cream or ointment, electron beam therapy, and photodynamic therapy have also been used with variable success rates6 (SOR: C).
MC is particularly difficult to treat in patients with poorly managed HIV and AIDS. Pairing proper antiretroviral therapy with lesion-destroying therapies is usually helpful for these patients.3
If you are caring for a patient with giant MC, you’ll need to stress the benign—but potentially contagious—nature of the disease. Tell the patient to wash his or her hands frequently, to avoid scratching the lesions, and to keep infected areas covered with clothing (when possible). In suspected sexually transmitted cases, the patient should adopt safe sexual practices or abstinence, if necessary. It is unclear whether condoms or other barrier methods provide adequate protection.1
Our patient transfers to the HIV clinic
We sequentially expressed the large lesions on our patient’s face and put her on a course of cefadroxil to control the secondary infection of MC. Her facial lesions gradually improved over 2 months.
We also referred the patient to our institution’s HIV clinic, where she was put on highly active antiretroviral therapy (HAART). We advised her mother to get tested for HIV, and she turned out to be HIV positive, as well.
CORRESPONDENCE Sudip Kumar Ghosh, MD, DNB, Department of Dermatology, Venereology, and Leprosy, R.G. Kar Medical College, 1 Khudiram Bose Sarani, Kolkata-700004, West Bengal, India; [email protected]
1. Taillac PP. Molluscum contagiosum: eMedicine, Emergency Medicine. Available at: . Accessed October 29, 2010.
2. Tom W, Friedlander SF. Poxvirus infections. In: Wolff K, Goldsmith LA, Katz SI, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: McGraw-Hill; 2008: 1899-1913.
3. Turchin I, Barankin B. Dermacase. Molluscum contagiosum. Can Fam Physician. 2006;52:1395-1407.
4. Ghosh SK, Bandyopadhyay D. Molluscum contagiosum after eyebrow shaping: a beauty salon hazard. Clin Exp Dermatol. 2009;34:e339-e340.
5. Gur I. The epidemiology of molluscum contagiosum in HIVseropositive patients: a unique entity or insignificant finding? Int J STD AIDS. 2008;19:503-506.
6. Mckenna DB, Benton EC. Molluscum contagiosum. In: Lebwohl MG, Heymann WR, Berth-Jones J, et al, eds. Treatment of Skin Disease: Comprehensive Therapeutic Strategies. 2nd ed. London: Mosby; 2002: 399-401.
7. Ohkuma M. Molluscum contagiosum treated with iodine solution and salicylic acid plaster. Int J Dermatol. 1990;29:443-445.
8. Mahajan BB, Pall A, Gupta RR. Topical 20% KOH—an effective therapeutic modality for molluscum contagiosum in children. Indian J Dermatol Venereol Leprol. 2003;69:175-177.
9. Binder B, Weger W, Komericki P, et al. Treatment of molluscum contagiosum with a pulsed dye laser: pilot study with 19 children. J Dtsch Dermatol Ges. 2008;6:121-125.
10. Watanabe T, Tamaki K. Cidofovir diphosphate inhibits molluscum contagiosum virus DNA polymerase activity. J Invest Dermatol. 2008;128:1327-1329.
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
A 12-Year-old girl came into our hospital for treatment of multiple bumps that had developed around her eyes and other areas of her face 2 months earlier. She had difficulty opening her eyes and complained of gradual weight loss.
On examination, we noted numerous skin-colored, shiny, dome-shaped, coalescing papules and nodules with central umbilications that were distributed mostly on her periocular and perinasal areas (FIGURE). When we expressed the papules with forceps, they exuded a cheesy material. We also noticed crusting and signs of inflammation on her eyelids.
The systemic examination was unremarkable.
FIGURE
Opening her eyes was difficult
This 12-year-old patient had multiple dome-shaped, coalescing papules and nodules with central umbilications in the periocular and perinasal areas.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Giant molluscum contagiosum
Molluscum contagiosum (MC) is a relatively common, benign, viral cutaneous infection that primarily affects children, sexually active adults, and immunodeficient individuals. MC accounts for approximately 1% of all diagnosed skin disorders in the United States; internationally, however, the incidence is higher.1 The causative organism of MC is a member of the Poxviridae family2 and is thought to be transmitted by close personal contact, autoinoculation, and fomites.3
MC is clinically characterized by the presence of pearly white, dome-shaped papules or nodules with central dells. The lesions are typically located on the trunk, body folds, extremities, and genitalia (particularly when the infection is sexually acquired).2,3 Pruritus and an eczematous reaction can develop around the lesions.
MC is a recognized ocular complication of acquired immune deficiency syndrome (AIDS). Periocular MC can also occur after eyebrow shaping in beauty salons.4 In human immunodeficiency virus (HIV)-positive patients, lesions are usually widespread, tend to be large, and usually occur during the advanced stage of HIV infection.2,5
The differential includes carcinoma
When considering a diagnosis of MC, you’ll need to rule out the following causes of similar-looking papules and nodules:
Nodular basal cell carcinoma presents as a slow-growing, firm, shiny, pearly nodule with fine telangiectasia. It may also present as a cystic lesion that can be mistaken for inclusion cysts of the eyelid. If left untreated, the tumor may ulcerate.
Juvenile xanthogranulomas are rubbery, tan-orange papules or nodules. Patients may have one or several papules or nodules in the head and neck region; these lesions may appear elsewhere, as well.
Cryptococcosis may present as painless papules or pustules, which then become nodules that may ulcerate. The lesions may show central umbilications.
Keratoacanthoma begins as a firm, roundish, skin-colored or reddish papule that rapidly progresses to a dome-shaped nodule, with a smooth, shiny surface and a central crateriform ulceration or keratin plug. Patients typically have a solitary lesion that may appear on the face, neck, or dorsum of the upper extremities.
Penicillosis often presents with MC-like skin lesions, in addition to fever, anemia, hepatomegaly, lymphadenopathy, and productive cough.
History and lab work clinch the Dx
Diagnosis is made by the distinctive clinical appearance, but can be confirmed by skin biopsy demonstrating eosinophilic molluscum bodies packed into the cells of the spinous layer of the epidermis.3 Giemsa stain of the material obtained from a crushed papule will reveal the presence of pathognomonic “molluscum bodies” in the cells of the epidermis.2,3
Our patient’s Giemsa stain revealed molluscum bodies. And since it is always wise to rule out concomitant HIV infection in patients who have giant MC, we tested our patient. Her results were positive; she had a CD4+ count of 93 cells/mm3.
Many treatment options from which to choose
MC is usually self-limiting, although it can take several months—or even a few years—to resolve on its own6 (strength of recommendation [SOR]: B). However, most patients with MC should receive treatment to obtain relief from symptoms, prevent autoinoculation or transmission to close contacts, decrease occurrence of scarring, reduce secondary bacterial infections, and improve cosmesis.
Several treatment options are available, and most rely on destruction of the lesions. Manual extrusion is a simple but effective therapy6 (SOR: B). Cryotherapy and curettage are also effective treatment options5 (SOR: C). Pretreatment topical anesthesia is often helpful if these therapies are used in children.
Topical imiquimod2 (1%-5%) cream applied 3 to 7 times a week can be used to treat generalized MC infection or MC localized to the anogenital area6 (SOR: A). Some patients may improve with topical tretinoin therapy6 (SOR: C).
Chemical cauterization with 10% povidone iodine with 50% salicylic acid7 (SOR: B), 10% potassium hydroxide8 (SOR: B), cantharidin2 (SOR: C), or 25% to 50% trichloroacetic acid6 (SOR: C) is also effective. Treatment with flashlamp pulsed dye laser is a safe and efficient treatment modality9 (SOR: C). Cidofovir10 (1%-3%) cream or ointment, electron beam therapy, and photodynamic therapy have also been used with variable success rates6 (SOR: C).
MC is particularly difficult to treat in patients with poorly managed HIV and AIDS. Pairing proper antiretroviral therapy with lesion-destroying therapies is usually helpful for these patients.3
If you are caring for a patient with giant MC, you’ll need to stress the benign—but potentially contagious—nature of the disease. Tell the patient to wash his or her hands frequently, to avoid scratching the lesions, and to keep infected areas covered with clothing (when possible). In suspected sexually transmitted cases, the patient should adopt safe sexual practices or abstinence, if necessary. It is unclear whether condoms or other barrier methods provide adequate protection.1
Our patient transfers to the HIV clinic
We sequentially expressed the large lesions on our patient’s face and put her on a course of cefadroxil to control the secondary infection of MC. Her facial lesions gradually improved over 2 months.
We also referred the patient to our institution’s HIV clinic, where she was put on highly active antiretroviral therapy (HAART). We advised her mother to get tested for HIV, and she turned out to be HIV positive, as well.
CORRESPONDENCE Sudip Kumar Ghosh, MD, DNB, Department of Dermatology, Venereology, and Leprosy, R.G. Kar Medical College, 1 Khudiram Bose Sarani, Kolkata-700004, West Bengal, India; [email protected]
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
A 12-Year-old girl came into our hospital for treatment of multiple bumps that had developed around her eyes and other areas of her face 2 months earlier. She had difficulty opening her eyes and complained of gradual weight loss.
On examination, we noted numerous skin-colored, shiny, dome-shaped, coalescing papules and nodules with central umbilications that were distributed mostly on her periocular and perinasal areas (FIGURE). When we expressed the papules with forceps, they exuded a cheesy material. We also noticed crusting and signs of inflammation on her eyelids.
The systemic examination was unremarkable.
FIGURE
Opening her eyes was difficult
This 12-year-old patient had multiple dome-shaped, coalescing papules and nodules with central umbilications in the periocular and perinasal areas.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Giant molluscum contagiosum
Molluscum contagiosum (MC) is a relatively common, benign, viral cutaneous infection that primarily affects children, sexually active adults, and immunodeficient individuals. MC accounts for approximately 1% of all diagnosed skin disorders in the United States; internationally, however, the incidence is higher.1 The causative organism of MC is a member of the Poxviridae family2 and is thought to be transmitted by close personal contact, autoinoculation, and fomites.3
MC is clinically characterized by the presence of pearly white, dome-shaped papules or nodules with central dells. The lesions are typically located on the trunk, body folds, extremities, and genitalia (particularly when the infection is sexually acquired).2,3 Pruritus and an eczematous reaction can develop around the lesions.
MC is a recognized ocular complication of acquired immune deficiency syndrome (AIDS). Periocular MC can also occur after eyebrow shaping in beauty salons.4 In human immunodeficiency virus (HIV)-positive patients, lesions are usually widespread, tend to be large, and usually occur during the advanced stage of HIV infection.2,5
The differential includes carcinoma
When considering a diagnosis of MC, you’ll need to rule out the following causes of similar-looking papules and nodules:
Nodular basal cell carcinoma presents as a slow-growing, firm, shiny, pearly nodule with fine telangiectasia. It may also present as a cystic lesion that can be mistaken for inclusion cysts of the eyelid. If left untreated, the tumor may ulcerate.
Juvenile xanthogranulomas are rubbery, tan-orange papules or nodules. Patients may have one or several papules or nodules in the head and neck region; these lesions may appear elsewhere, as well.
Cryptococcosis may present as painless papules or pustules, which then become nodules that may ulcerate. The lesions may show central umbilications.
Keratoacanthoma begins as a firm, roundish, skin-colored or reddish papule that rapidly progresses to a dome-shaped nodule, with a smooth, shiny surface and a central crateriform ulceration or keratin plug. Patients typically have a solitary lesion that may appear on the face, neck, or dorsum of the upper extremities.
Penicillosis often presents with MC-like skin lesions, in addition to fever, anemia, hepatomegaly, lymphadenopathy, and productive cough.
History and lab work clinch the Dx
Diagnosis is made by the distinctive clinical appearance, but can be confirmed by skin biopsy demonstrating eosinophilic molluscum bodies packed into the cells of the spinous layer of the epidermis.3 Giemsa stain of the material obtained from a crushed papule will reveal the presence of pathognomonic “molluscum bodies” in the cells of the epidermis.2,3
Our patient’s Giemsa stain revealed molluscum bodies. And since it is always wise to rule out concomitant HIV infection in patients who have giant MC, we tested our patient. Her results were positive; she had a CD4+ count of 93 cells/mm3.
Many treatment options from which to choose
MC is usually self-limiting, although it can take several months—or even a few years—to resolve on its own6 (strength of recommendation [SOR]: B). However, most patients with MC should receive treatment to obtain relief from symptoms, prevent autoinoculation or transmission to close contacts, decrease occurrence of scarring, reduce secondary bacterial infections, and improve cosmesis.
Several treatment options are available, and most rely on destruction of the lesions. Manual extrusion is a simple but effective therapy6 (SOR: B). Cryotherapy and curettage are also effective treatment options5 (SOR: C). Pretreatment topical anesthesia is often helpful if these therapies are used in children.
Topical imiquimod2 (1%-5%) cream applied 3 to 7 times a week can be used to treat generalized MC infection or MC localized to the anogenital area6 (SOR: A). Some patients may improve with topical tretinoin therapy6 (SOR: C).
Chemical cauterization with 10% povidone iodine with 50% salicylic acid7 (SOR: B), 10% potassium hydroxide8 (SOR: B), cantharidin2 (SOR: C), or 25% to 50% trichloroacetic acid6 (SOR: C) is also effective. Treatment with flashlamp pulsed dye laser is a safe and efficient treatment modality9 (SOR: C). Cidofovir10 (1%-3%) cream or ointment, electron beam therapy, and photodynamic therapy have also been used with variable success rates6 (SOR: C).
MC is particularly difficult to treat in patients with poorly managed HIV and AIDS. Pairing proper antiretroviral therapy with lesion-destroying therapies is usually helpful for these patients.3
If you are caring for a patient with giant MC, you’ll need to stress the benign—but potentially contagious—nature of the disease. Tell the patient to wash his or her hands frequently, to avoid scratching the lesions, and to keep infected areas covered with clothing (when possible). In suspected sexually transmitted cases, the patient should adopt safe sexual practices or abstinence, if necessary. It is unclear whether condoms or other barrier methods provide adequate protection.1
Our patient transfers to the HIV clinic
We sequentially expressed the large lesions on our patient’s face and put her on a course of cefadroxil to control the secondary infection of MC. Her facial lesions gradually improved over 2 months.
We also referred the patient to our institution’s HIV clinic, where she was put on highly active antiretroviral therapy (HAART). We advised her mother to get tested for HIV, and she turned out to be HIV positive, as well.
CORRESPONDENCE Sudip Kumar Ghosh, MD, DNB, Department of Dermatology, Venereology, and Leprosy, R.G. Kar Medical College, 1 Khudiram Bose Sarani, Kolkata-700004, West Bengal, India; [email protected]
1. Taillac PP. Molluscum contagiosum: eMedicine, Emergency Medicine. Available at: . Accessed October 29, 2010.
2. Tom W, Friedlander SF. Poxvirus infections. In: Wolff K, Goldsmith LA, Katz SI, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: McGraw-Hill; 2008: 1899-1913.
3. Turchin I, Barankin B. Dermacase. Molluscum contagiosum. Can Fam Physician. 2006;52:1395-1407.
4. Ghosh SK, Bandyopadhyay D. Molluscum contagiosum after eyebrow shaping: a beauty salon hazard. Clin Exp Dermatol. 2009;34:e339-e340.
5. Gur I. The epidemiology of molluscum contagiosum in HIVseropositive patients: a unique entity or insignificant finding? Int J STD AIDS. 2008;19:503-506.
6. Mckenna DB, Benton EC. Molluscum contagiosum. In: Lebwohl MG, Heymann WR, Berth-Jones J, et al, eds. Treatment of Skin Disease: Comprehensive Therapeutic Strategies. 2nd ed. London: Mosby; 2002: 399-401.
7. Ohkuma M. Molluscum contagiosum treated with iodine solution and salicylic acid plaster. Int J Dermatol. 1990;29:443-445.
8. Mahajan BB, Pall A, Gupta RR. Topical 20% KOH—an effective therapeutic modality for molluscum contagiosum in children. Indian J Dermatol Venereol Leprol. 2003;69:175-177.
9. Binder B, Weger W, Komericki P, et al. Treatment of molluscum contagiosum with a pulsed dye laser: pilot study with 19 children. J Dtsch Dermatol Ges. 2008;6:121-125.
10. Watanabe T, Tamaki K. Cidofovir diphosphate inhibits molluscum contagiosum virus DNA polymerase activity. J Invest Dermatol. 2008;128:1327-1329.
1. Taillac PP. Molluscum contagiosum: eMedicine, Emergency Medicine. Available at: . Accessed October 29, 2010.
2. Tom W, Friedlander SF. Poxvirus infections. In: Wolff K, Goldsmith LA, Katz SI, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: McGraw-Hill; 2008: 1899-1913.
3. Turchin I, Barankin B. Dermacase. Molluscum contagiosum. Can Fam Physician. 2006;52:1395-1407.
4. Ghosh SK, Bandyopadhyay D. Molluscum contagiosum after eyebrow shaping: a beauty salon hazard. Clin Exp Dermatol. 2009;34:e339-e340.
5. Gur I. The epidemiology of molluscum contagiosum in HIVseropositive patients: a unique entity or insignificant finding? Int J STD AIDS. 2008;19:503-506.
6. Mckenna DB, Benton EC. Molluscum contagiosum. In: Lebwohl MG, Heymann WR, Berth-Jones J, et al, eds. Treatment of Skin Disease: Comprehensive Therapeutic Strategies. 2nd ed. London: Mosby; 2002: 399-401.
7. Ohkuma M. Molluscum contagiosum treated with iodine solution and salicylic acid plaster. Int J Dermatol. 1990;29:443-445.
8. Mahajan BB, Pall A, Gupta RR. Topical 20% KOH—an effective therapeutic modality for molluscum contagiosum in children. Indian J Dermatol Venereol Leprol. 2003;69:175-177.
9. Binder B, Weger W, Komericki P, et al. Treatment of molluscum contagiosum with a pulsed dye laser: pilot study with 19 children. J Dtsch Dermatol Ges. 2008;6:121-125.
10. Watanabe T, Tamaki K. Cidofovir diphosphate inhibits molluscum contagiosum virus DNA polymerase activity. J Invest Dermatol. 2008;128:1327-1329.
Verrucous papule on thigh
A 21-year-old man came into our medical center to have a lesion on his thigh examined. He said the lesion developed a few months earlier at the site of minimal trauma. He noted that, over the previous few months, the lesion had progressively darkened and it bled sporadically. On examination, we noted a solitary 7.5-mm firm, blue-black verrucous papule over the right medial thigh (FIGURES 1A AND 1B). There were no other lesions.
The patient indicated that he had gotten sunburned many times in the past. He also said that he had an aunt who’d had a melanoma.
FIGURE 1
A lesion that bled sporadically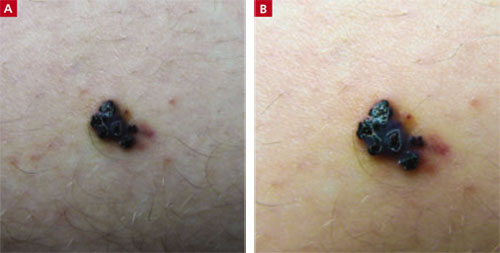
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Angiokeratoma
An angiokeratoma is a benign pink-red to blue-black variably sized papule or plaque that is typically 2 to 10 mm in diameter.1 Angiokeratomas are composed of a series of subepidermal dilated capillaries that have a characteristic hyperkeratotic surface and bleed easily.2 These lesions are rare, with a prevalence estimated to be 0.16% in the general population.3
The pathogenesis of angiokeratoma formation is unclear; however, multiple theories exist. The development of these lesions may be related to repeated trauma or friction at a particular site.4 Alternatively, increased venous blood pressure or primary degeneration of vascular elastic tissue could explain their development.5 While their cause is unclear, the initial event in the development of an angiokeratoma is believed to be the development of a vascular ectasia within the papillary dermis. The epidermal reaction appears to be a secondary phenomenon due to increased proliferative capacity on the surface of the vessels.5
The most common form—as seen in this case—is the solitary or sporadic angiokeratoma. It comprises 70% to 83% of all cases of angiokeratomas3 and usually develops on the lower extremities. Angiokeratomas typically arise during the first 2 decades of life,6 and are more common in men.3 Other types of angiokeratomas include angiokeratoma of Mibelli, angiokeratoma of Fordyce, angiokeratoma circumscriptum, and angiokeratoma corporis diffusum (Fabry’s disease).7,8
Angiokeratoma of Mibelli is characterized by pink to dark red papules or verrucoid nodules that occur most commonly in men7 and involve the bony prominences, such as the elbows.
Fordyce lesions involve the scrotum or vulva and are usually numerous and related to conditions with elevated venous pressure.
Angiokeratoma circumscriptum usually present as papules that commonly coalesce to form plaques.
Fabry’s disease, or angiokeratoma corporis diffusum, is an X-linked recessive disease related to a deficiency in alpha-galactosidase A. This leads to multiple, variably sized angiokeratomas occurring in childhood that are concentrated between the umbilicus and the knees. This disease invariably leads to involvement of other organs, which may result in renal failure, myocardial infarction, or cerebrovascular accidents.1,7
A mimicker of melanoma
An angiokeratoma is an uncommon, though important, mimicker of melanoma. (For more on other lesions that can be confused with melanoma, see “Nonmelanocytic melanoma mimickers”.)
Melanoma is the most aggressive and potentially life-threatening neoplasm in the differential diagnosis of an angiokeratoma. Risk factors for melanoma include increasing age, fair skin and hair color, tendency for freckling, number of moles (5 large or >50 small nevi doubles the risk of melanoma), a personal or first-degree family history of melanoma, and a history of intermittent sunburns.9-12
A number of nonmelanocytic lesions can be confused with melanoma. They include the following:
Actinic keratoses (AKs) are a type of keratinocytic neoplasm that typically develops on the sun-exposed skin of the elderly. An AK is typically 3 to 10 mm in size, pink to red in color, and has scaling secondary to local hyperkeratosis. If these lesions are left untreated, they can develop into squamous cell carcinomas (SCCs) at a rate of 0.24% annually.15,16 Thus, AKs are more often a concern for SCC than for melanoma. However, the pigmented variant of an AK can clinically and histologically raise concern for melanoma due to its pigmentation and microscopic evidence of melanin within keratinocytes and macrophages.15 If it is not possible to differentiate an AK from melanoma clinically or histologically, immunohistochemistry is often required to make the final diagnosis. For example, immunohistochemical staining with S-100 can be used to identify epidermal melanocytes and distinguish them from atypical keratinocytes.17
Basal cell carcinoma (BCC) is the most common skin cancer.18 While most BCCs are amelanocytic, 7% of BCCs are pigmented and present as irregularly pigmented nodules with irregular telangiectatic vessels on their surface. The center of a BCC may be depressed or ulcerated and may easily crust or bleed. Definitive diagnosis may be made histologically. A BCC typically consists of columns of basaloid cells with atypical nuclei, sparse cytoplasm, and peripheral cellular palisading.19 BCCs are easily differentiated from melanoma using immunohistochemistry, as they are negative for traditional melanocytic markers.17
Seborrheic keratoses (SKs) are among the most common skin lesions and represent a benign proliferation of immature keratinocytes. The appearance of an SK can vary from a smooth peppered appearance to a rough surface that may be irregularly pigmented, dry, and fissured. Given their range of presentation, it is common for SKs to be biopsied to evaluate for melanoma and occasionally BCC.20
Dermatofibromas (DFs) are common benign skin lesions that typically appear as pink-to brown-colored firm nodules that represent a localized response to skin injury and inflammation. DFs are typically 3 to 10 mm in diameter and are most commonly located on the anterior surface of the thigh. Histologic analysis of a DF reveals an acanthotic epidermis with a proliferation of spindle cells in the mid and lower dermis, with capillaries dispersed throughout. A common finding in DFs is the trapping of collagen within the spindle cell at the periphery of the lesion.21
How to diagnose angiokeratoma
The clinical presentation typically suffices in making the diagnosis of an angiokeratoma. If dermoscopy is performed, the characteristic findings include the presence of scale and purple lacunae13 (FIGURE 2). However, when there is suspicion of melanoma or the clinical diagnosis is in doubt, the entire lesion should be removed (with narrow margins) in order to obtain a definitive diagnosis. Histological findings consist of dilated subepidermal vessels associated with epidermal hyperkeratosis.3
FIGURE 2
A view from the dermatoscope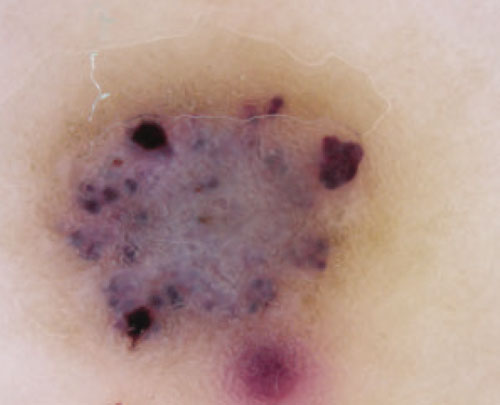
No need to treat, unless there are cosmetic concerns
If the diagnosis is straightforward and a biopsy is not needed, no treatment is necessary because simple angiokeratomas are benign entities. However, treatment may be considered for cosmetic purposes, or to prevent bothersome bleeding. Angiokeratomas may be removed via shave or standard excision, electrodessication and curettage, or destroyed with a laser. For Fabry’s disease, in which numerous angiokeratomas pose a cosmetic concern, laser therapy, including the use of an argon, copper, Nd:Yag, KTP 532-nm, or Candela V-beam laser, is preferred.14
In our patient’s case, we performed a 2-mm punch biopsy, which revealed that the lesion was an angiokeratoma. It was subsequently removed by shave biopsy with clear margins.
CORRESPONDENCE
Thomas M. Beachkofsky, MD, Capt, USAF, MC, Department of the Air Force, Wilford Hall Medical Center, 59 MDW/SG05D/ Dermatology, 2200 Bergquist Drive, Suite 1, Lackland AFB, TX 78236-9908; [email protected]
1. Karen JK, Hale EK, Ma L. Angiokeratoma corporis diffusum. Dermatol Online J. [Internet]. 2005;11:8. Available at: http://dermatology.cdlib.org/114/NYU/NYUtexts/0419054.html. Accessed September 24, 2010.
2. Schiller PI, Itin PH. Angiokeratomas: an update. Dermatology. 1996;193:275-282.
3. Zaballos P, Dauft C, Puig S, et al. Dermoscopy of solitary angiokeratomas: a morphological study. Arch Dermatol. 2007;143:318-325.
4. Kim JH, Nam TS, Kim SH. Solitary angiokeratoma developed in one area of lymphangioma circumscriptum. J Korean Med Sci. 1988;3:169-170.
5. Sion-Vardy N, Manor E, Puterman M, et al. Solitary angiokeratoma of the tongue. Med Oral Patol Oral Cir Bucal. 2008;13:12-14.
6. Vascular tumors and malformations In: Habif TP, Campbell JL, Dinulos JG, et al, eds. Skin Disease: Diagnosis and Treatment. New York, NY: Mosby; 2004:486–487.
7. Leis-Dosil VM, Alijo-Serrano F, Aviles-Izquierdo JA, et al. Angiokeratoma of the glans penis: clinical, histopathological and dermoscopic correlation. Dermatol Online J. [Internet]. 2007;13:19. Available from: http://dermatology.cdlib.org/132/case_presentations/angiokeratoma/dosil.html. Accessed September 24, 2010.
8. Erkek E, Basar MM, Bagci Y, et al. Fordyce angiokeratomas as clues to local venous hypertension. Arch Dermatol. 2005;141:1325-1326.
9. Rager EL, Bridgeford EP, Ollila DW. Cutaneous melanoma: update on prevention, screening, diagnosis, and treatment. Am Fam Physician. 2005;72:269-276.
10. Chudnovsky Y, Khavari PA, Adams AE. Melanoma genetics and the development of rational therapeutics. J Clin Invest. 2005;115:813-824.
11. Ortiz CA, Goodwin JS, Freeman JL. The effect of socioeconomic factors on incidence, stage at diagnosis, and survival of cutaneous melanoma. Med Sci Monit. 2005;11:163-172.
12. Abbasi NR, Shaw HM, Rigel DS, et al. Early diagnosis of cutaneous melanoma. JAMA. 2004;292:2771-2776.
13. Johr RH, Soyer P, Argenziano G, et al. Dermoscopy: The Essentials. New York, NY: Mosby; 2007:130.
14. Enjolras O. Vascular malformations. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. Philadelphia, Pa: Mosby; 2003: 1621–1622.
15. Peris K, Micantonio T, Piccolo D, et al. Dermoscopic features of actinic keratosis. J Dtsch Dermatol Ges. 2007;5:970-976.
16. McIntyre WJ, Downs MR, Bedwell SA. Treatment options for actinic keratosis. Am Fam Physician. 2007;76:667-671.
17. Kamil ZS, Tong LC, Habeeb AA, et al. Non-melanocytic mimics of melanoma: part 1: intraepidermal mimics. J Clin Pathol. 2009;62:120-127.
18. Wong CS, Strange RC, Lear JT. Basal cell carcinoma. BMJ. 2003;327:794-798.
19. Menzies SW. Dermoscopy of pigmented basal cell carcinoma. Clin Dermatol. 2002;20:268-269.
20. Braun RP, Rabinovitz H, Oliviero M, et al. Dermoscopic diagnosis of seborrheic keratosis. Clin Dermatol. 2002;20:270-272.
21. Agero AL, Taliercio S, Dusza SW, et al. Conventional and polarized dermoscopy features of dermatofibroma. Arch Dermatol. 2006;142:1431-1437.
A 21-year-old man came into our medical center to have a lesion on his thigh examined. He said the lesion developed a few months earlier at the site of minimal trauma. He noted that, over the previous few months, the lesion had progressively darkened and it bled sporadically. On examination, we noted a solitary 7.5-mm firm, blue-black verrucous papule over the right medial thigh (FIGURES 1A AND 1B). There were no other lesions.
The patient indicated that he had gotten sunburned many times in the past. He also said that he had an aunt who’d had a melanoma.
FIGURE 1
A lesion that bled sporadically
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Angiokeratoma
An angiokeratoma is a benign pink-red to blue-black variably sized papule or plaque that is typically 2 to 10 mm in diameter.1 Angiokeratomas are composed of a series of subepidermal dilated capillaries that have a characteristic hyperkeratotic surface and bleed easily.2 These lesions are rare, with a prevalence estimated to be 0.16% in the general population.3
The pathogenesis of angiokeratoma formation is unclear; however, multiple theories exist. The development of these lesions may be related to repeated trauma or friction at a particular site.4 Alternatively, increased venous blood pressure or primary degeneration of vascular elastic tissue could explain their development.5 While their cause is unclear, the initial event in the development of an angiokeratoma is believed to be the development of a vascular ectasia within the papillary dermis. The epidermal reaction appears to be a secondary phenomenon due to increased proliferative capacity on the surface of the vessels.5
The most common form—as seen in this case—is the solitary or sporadic angiokeratoma. It comprises 70% to 83% of all cases of angiokeratomas3 and usually develops on the lower extremities. Angiokeratomas typically arise during the first 2 decades of life,6 and are more common in men.3 Other types of angiokeratomas include angiokeratoma of Mibelli, angiokeratoma of Fordyce, angiokeratoma circumscriptum, and angiokeratoma corporis diffusum (Fabry’s disease).7,8
Angiokeratoma of Mibelli is characterized by pink to dark red papules or verrucoid nodules that occur most commonly in men7 and involve the bony prominences, such as the elbows.
Fordyce lesions involve the scrotum or vulva and are usually numerous and related to conditions with elevated venous pressure.
Angiokeratoma circumscriptum usually present as papules that commonly coalesce to form plaques.
Fabry’s disease, or angiokeratoma corporis diffusum, is an X-linked recessive disease related to a deficiency in alpha-galactosidase A. This leads to multiple, variably sized angiokeratomas occurring in childhood that are concentrated between the umbilicus and the knees. This disease invariably leads to involvement of other organs, which may result in renal failure, myocardial infarction, or cerebrovascular accidents.1,7
A mimicker of melanoma
An angiokeratoma is an uncommon, though important, mimicker of melanoma. (For more on other lesions that can be confused with melanoma, see “Nonmelanocytic melanoma mimickers”.)
Melanoma is the most aggressive and potentially life-threatening neoplasm in the differential diagnosis of an angiokeratoma. Risk factors for melanoma include increasing age, fair skin and hair color, tendency for freckling, number of moles (5 large or >50 small nevi doubles the risk of melanoma), a personal or first-degree family history of melanoma, and a history of intermittent sunburns.9-12
A number of nonmelanocytic lesions can be confused with melanoma. They include the following:
Actinic keratoses (AKs) are a type of keratinocytic neoplasm that typically develops on the sun-exposed skin of the elderly. An AK is typically 3 to 10 mm in size, pink to red in color, and has scaling secondary to local hyperkeratosis. If these lesions are left untreated, they can develop into squamous cell carcinomas (SCCs) at a rate of 0.24% annually.15,16 Thus, AKs are more often a concern for SCC than for melanoma. However, the pigmented variant of an AK can clinically and histologically raise concern for melanoma due to its pigmentation and microscopic evidence of melanin within keratinocytes and macrophages.15 If it is not possible to differentiate an AK from melanoma clinically or histologically, immunohistochemistry is often required to make the final diagnosis. For example, immunohistochemical staining with S-100 can be used to identify epidermal melanocytes and distinguish them from atypical keratinocytes.17
Basal cell carcinoma (BCC) is the most common skin cancer.18 While most BCCs are amelanocytic, 7% of BCCs are pigmented and present as irregularly pigmented nodules with irregular telangiectatic vessels on their surface. The center of a BCC may be depressed or ulcerated and may easily crust or bleed. Definitive diagnosis may be made histologically. A BCC typically consists of columns of basaloid cells with atypical nuclei, sparse cytoplasm, and peripheral cellular palisading.19 BCCs are easily differentiated from melanoma using immunohistochemistry, as they are negative for traditional melanocytic markers.17
Seborrheic keratoses (SKs) are among the most common skin lesions and represent a benign proliferation of immature keratinocytes. The appearance of an SK can vary from a smooth peppered appearance to a rough surface that may be irregularly pigmented, dry, and fissured. Given their range of presentation, it is common for SKs to be biopsied to evaluate for melanoma and occasionally BCC.20
Dermatofibromas (DFs) are common benign skin lesions that typically appear as pink-to brown-colored firm nodules that represent a localized response to skin injury and inflammation. DFs are typically 3 to 10 mm in diameter and are most commonly located on the anterior surface of the thigh. Histologic analysis of a DF reveals an acanthotic epidermis with a proliferation of spindle cells in the mid and lower dermis, with capillaries dispersed throughout. A common finding in DFs is the trapping of collagen within the spindle cell at the periphery of the lesion.21
How to diagnose angiokeratoma
The clinical presentation typically suffices in making the diagnosis of an angiokeratoma. If dermoscopy is performed, the characteristic findings include the presence of scale and purple lacunae13 (FIGURE 2). However, when there is suspicion of melanoma or the clinical diagnosis is in doubt, the entire lesion should be removed (with narrow margins) in order to obtain a definitive diagnosis. Histological findings consist of dilated subepidermal vessels associated with epidermal hyperkeratosis.3
FIGURE 2
A view from the dermatoscope
No need to treat, unless there are cosmetic concerns
If the diagnosis is straightforward and a biopsy is not needed, no treatment is necessary because simple angiokeratomas are benign entities. However, treatment may be considered for cosmetic purposes, or to prevent bothersome bleeding. Angiokeratomas may be removed via shave or standard excision, electrodessication and curettage, or destroyed with a laser. For Fabry’s disease, in which numerous angiokeratomas pose a cosmetic concern, laser therapy, including the use of an argon, copper, Nd:Yag, KTP 532-nm, or Candela V-beam laser, is preferred.14
In our patient’s case, we performed a 2-mm punch biopsy, which revealed that the lesion was an angiokeratoma. It was subsequently removed by shave biopsy with clear margins.
CORRESPONDENCE
Thomas M. Beachkofsky, MD, Capt, USAF, MC, Department of the Air Force, Wilford Hall Medical Center, 59 MDW/SG05D/ Dermatology, 2200 Bergquist Drive, Suite 1, Lackland AFB, TX 78236-9908; [email protected]
A 21-year-old man came into our medical center to have a lesion on his thigh examined. He said the lesion developed a few months earlier at the site of minimal trauma. He noted that, over the previous few months, the lesion had progressively darkened and it bled sporadically. On examination, we noted a solitary 7.5-mm firm, blue-black verrucous papule over the right medial thigh (FIGURES 1A AND 1B). There were no other lesions.
The patient indicated that he had gotten sunburned many times in the past. He also said that he had an aunt who’d had a melanoma.
FIGURE 1
A lesion that bled sporadically
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Angiokeratoma
An angiokeratoma is a benign pink-red to blue-black variably sized papule or plaque that is typically 2 to 10 mm in diameter.1 Angiokeratomas are composed of a series of subepidermal dilated capillaries that have a characteristic hyperkeratotic surface and bleed easily.2 These lesions are rare, with a prevalence estimated to be 0.16% in the general population.3
The pathogenesis of angiokeratoma formation is unclear; however, multiple theories exist. The development of these lesions may be related to repeated trauma or friction at a particular site.4 Alternatively, increased venous blood pressure or primary degeneration of vascular elastic tissue could explain their development.5 While their cause is unclear, the initial event in the development of an angiokeratoma is believed to be the development of a vascular ectasia within the papillary dermis. The epidermal reaction appears to be a secondary phenomenon due to increased proliferative capacity on the surface of the vessels.5
The most common form—as seen in this case—is the solitary or sporadic angiokeratoma. It comprises 70% to 83% of all cases of angiokeratomas3 and usually develops on the lower extremities. Angiokeratomas typically arise during the first 2 decades of life,6 and are more common in men.3 Other types of angiokeratomas include angiokeratoma of Mibelli, angiokeratoma of Fordyce, angiokeratoma circumscriptum, and angiokeratoma corporis diffusum (Fabry’s disease).7,8
Angiokeratoma of Mibelli is characterized by pink to dark red papules or verrucoid nodules that occur most commonly in men7 and involve the bony prominences, such as the elbows.
Fordyce lesions involve the scrotum or vulva and are usually numerous and related to conditions with elevated venous pressure.
Angiokeratoma circumscriptum usually present as papules that commonly coalesce to form plaques.
Fabry’s disease, or angiokeratoma corporis diffusum, is an X-linked recessive disease related to a deficiency in alpha-galactosidase A. This leads to multiple, variably sized angiokeratomas occurring in childhood that are concentrated between the umbilicus and the knees. This disease invariably leads to involvement of other organs, which may result in renal failure, myocardial infarction, or cerebrovascular accidents.1,7
A mimicker of melanoma
An angiokeratoma is an uncommon, though important, mimicker of melanoma. (For more on other lesions that can be confused with melanoma, see “Nonmelanocytic melanoma mimickers”.)
Melanoma is the most aggressive and potentially life-threatening neoplasm in the differential diagnosis of an angiokeratoma. Risk factors for melanoma include increasing age, fair skin and hair color, tendency for freckling, number of moles (5 large or >50 small nevi doubles the risk of melanoma), a personal or first-degree family history of melanoma, and a history of intermittent sunburns.9-12
A number of nonmelanocytic lesions can be confused with melanoma. They include the following:
Actinic keratoses (AKs) are a type of keratinocytic neoplasm that typically develops on the sun-exposed skin of the elderly. An AK is typically 3 to 10 mm in size, pink to red in color, and has scaling secondary to local hyperkeratosis. If these lesions are left untreated, they can develop into squamous cell carcinomas (SCCs) at a rate of 0.24% annually.15,16 Thus, AKs are more often a concern for SCC than for melanoma. However, the pigmented variant of an AK can clinically and histologically raise concern for melanoma due to its pigmentation and microscopic evidence of melanin within keratinocytes and macrophages.15 If it is not possible to differentiate an AK from melanoma clinically or histologically, immunohistochemistry is often required to make the final diagnosis. For example, immunohistochemical staining with S-100 can be used to identify epidermal melanocytes and distinguish them from atypical keratinocytes.17
Basal cell carcinoma (BCC) is the most common skin cancer.18 While most BCCs are amelanocytic, 7% of BCCs are pigmented and present as irregularly pigmented nodules with irregular telangiectatic vessels on their surface. The center of a BCC may be depressed or ulcerated and may easily crust or bleed. Definitive diagnosis may be made histologically. A BCC typically consists of columns of basaloid cells with atypical nuclei, sparse cytoplasm, and peripheral cellular palisading.19 BCCs are easily differentiated from melanoma using immunohistochemistry, as they are negative for traditional melanocytic markers.17
Seborrheic keratoses (SKs) are among the most common skin lesions and represent a benign proliferation of immature keratinocytes. The appearance of an SK can vary from a smooth peppered appearance to a rough surface that may be irregularly pigmented, dry, and fissured. Given their range of presentation, it is common for SKs to be biopsied to evaluate for melanoma and occasionally BCC.20
Dermatofibromas (DFs) are common benign skin lesions that typically appear as pink-to brown-colored firm nodules that represent a localized response to skin injury and inflammation. DFs are typically 3 to 10 mm in diameter and are most commonly located on the anterior surface of the thigh. Histologic analysis of a DF reveals an acanthotic epidermis with a proliferation of spindle cells in the mid and lower dermis, with capillaries dispersed throughout. A common finding in DFs is the trapping of collagen within the spindle cell at the periphery of the lesion.21
How to diagnose angiokeratoma
The clinical presentation typically suffices in making the diagnosis of an angiokeratoma. If dermoscopy is performed, the characteristic findings include the presence of scale and purple lacunae13 (FIGURE 2). However, when there is suspicion of melanoma or the clinical diagnosis is in doubt, the entire lesion should be removed (with narrow margins) in order to obtain a definitive diagnosis. Histological findings consist of dilated subepidermal vessels associated with epidermal hyperkeratosis.3
FIGURE 2
A view from the dermatoscope
No need to treat, unless there are cosmetic concerns
If the diagnosis is straightforward and a biopsy is not needed, no treatment is necessary because simple angiokeratomas are benign entities. However, treatment may be considered for cosmetic purposes, or to prevent bothersome bleeding. Angiokeratomas may be removed via shave or standard excision, electrodessication and curettage, or destroyed with a laser. For Fabry’s disease, in which numerous angiokeratomas pose a cosmetic concern, laser therapy, including the use of an argon, copper, Nd:Yag, KTP 532-nm, or Candela V-beam laser, is preferred.14
In our patient’s case, we performed a 2-mm punch biopsy, which revealed that the lesion was an angiokeratoma. It was subsequently removed by shave biopsy with clear margins.
CORRESPONDENCE
Thomas M. Beachkofsky, MD, Capt, USAF, MC, Department of the Air Force, Wilford Hall Medical Center, 59 MDW/SG05D/ Dermatology, 2200 Bergquist Drive, Suite 1, Lackland AFB, TX 78236-9908; [email protected]
1. Karen JK, Hale EK, Ma L. Angiokeratoma corporis diffusum. Dermatol Online J. [Internet]. 2005;11:8. Available at: http://dermatology.cdlib.org/114/NYU/NYUtexts/0419054.html. Accessed September 24, 2010.
2. Schiller PI, Itin PH. Angiokeratomas: an update. Dermatology. 1996;193:275-282.
3. Zaballos P, Dauft C, Puig S, et al. Dermoscopy of solitary angiokeratomas: a morphological study. Arch Dermatol. 2007;143:318-325.
4. Kim JH, Nam TS, Kim SH. Solitary angiokeratoma developed in one area of lymphangioma circumscriptum. J Korean Med Sci. 1988;3:169-170.
5. Sion-Vardy N, Manor E, Puterman M, et al. Solitary angiokeratoma of the tongue. Med Oral Patol Oral Cir Bucal. 2008;13:12-14.
6. Vascular tumors and malformations In: Habif TP, Campbell JL, Dinulos JG, et al, eds. Skin Disease: Diagnosis and Treatment. New York, NY: Mosby; 2004:486–487.
7. Leis-Dosil VM, Alijo-Serrano F, Aviles-Izquierdo JA, et al. Angiokeratoma of the glans penis: clinical, histopathological and dermoscopic correlation. Dermatol Online J. [Internet]. 2007;13:19. Available from: http://dermatology.cdlib.org/132/case_presentations/angiokeratoma/dosil.html. Accessed September 24, 2010.
8. Erkek E, Basar MM, Bagci Y, et al. Fordyce angiokeratomas as clues to local venous hypertension. Arch Dermatol. 2005;141:1325-1326.
9. Rager EL, Bridgeford EP, Ollila DW. Cutaneous melanoma: update on prevention, screening, diagnosis, and treatment. Am Fam Physician. 2005;72:269-276.
10. Chudnovsky Y, Khavari PA, Adams AE. Melanoma genetics and the development of rational therapeutics. J Clin Invest. 2005;115:813-824.
11. Ortiz CA, Goodwin JS, Freeman JL. The effect of socioeconomic factors on incidence, stage at diagnosis, and survival of cutaneous melanoma. Med Sci Monit. 2005;11:163-172.
12. Abbasi NR, Shaw HM, Rigel DS, et al. Early diagnosis of cutaneous melanoma. JAMA. 2004;292:2771-2776.
13. Johr RH, Soyer P, Argenziano G, et al. Dermoscopy: The Essentials. New York, NY: Mosby; 2007:130.
14. Enjolras O. Vascular malformations. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. Philadelphia, Pa: Mosby; 2003: 1621–1622.
15. Peris K, Micantonio T, Piccolo D, et al. Dermoscopic features of actinic keratosis. J Dtsch Dermatol Ges. 2007;5:970-976.
16. McIntyre WJ, Downs MR, Bedwell SA. Treatment options for actinic keratosis. Am Fam Physician. 2007;76:667-671.
17. Kamil ZS, Tong LC, Habeeb AA, et al. Non-melanocytic mimics of melanoma: part 1: intraepidermal mimics. J Clin Pathol. 2009;62:120-127.
18. Wong CS, Strange RC, Lear JT. Basal cell carcinoma. BMJ. 2003;327:794-798.
19. Menzies SW. Dermoscopy of pigmented basal cell carcinoma. Clin Dermatol. 2002;20:268-269.
20. Braun RP, Rabinovitz H, Oliviero M, et al. Dermoscopic diagnosis of seborrheic keratosis. Clin Dermatol. 2002;20:270-272.
21. Agero AL, Taliercio S, Dusza SW, et al. Conventional and polarized dermoscopy features of dermatofibroma. Arch Dermatol. 2006;142:1431-1437.
1. Karen JK, Hale EK, Ma L. Angiokeratoma corporis diffusum. Dermatol Online J. [Internet]. 2005;11:8. Available at: http://dermatology.cdlib.org/114/NYU/NYUtexts/0419054.html. Accessed September 24, 2010.
2. Schiller PI, Itin PH. Angiokeratomas: an update. Dermatology. 1996;193:275-282.
3. Zaballos P, Dauft C, Puig S, et al. Dermoscopy of solitary angiokeratomas: a morphological study. Arch Dermatol. 2007;143:318-325.
4. Kim JH, Nam TS, Kim SH. Solitary angiokeratoma developed in one area of lymphangioma circumscriptum. J Korean Med Sci. 1988;3:169-170.
5. Sion-Vardy N, Manor E, Puterman M, et al. Solitary angiokeratoma of the tongue. Med Oral Patol Oral Cir Bucal. 2008;13:12-14.
6. Vascular tumors and malformations In: Habif TP, Campbell JL, Dinulos JG, et al, eds. Skin Disease: Diagnosis and Treatment. New York, NY: Mosby; 2004:486–487.
7. Leis-Dosil VM, Alijo-Serrano F, Aviles-Izquierdo JA, et al. Angiokeratoma of the glans penis: clinical, histopathological and dermoscopic correlation. Dermatol Online J. [Internet]. 2007;13:19. Available from: http://dermatology.cdlib.org/132/case_presentations/angiokeratoma/dosil.html. Accessed September 24, 2010.
8. Erkek E, Basar MM, Bagci Y, et al. Fordyce angiokeratomas as clues to local venous hypertension. Arch Dermatol. 2005;141:1325-1326.
9. Rager EL, Bridgeford EP, Ollila DW. Cutaneous melanoma: update on prevention, screening, diagnosis, and treatment. Am Fam Physician. 2005;72:269-276.
10. Chudnovsky Y, Khavari PA, Adams AE. Melanoma genetics and the development of rational therapeutics. J Clin Invest. 2005;115:813-824.
11. Ortiz CA, Goodwin JS, Freeman JL. The effect of socioeconomic factors on incidence, stage at diagnosis, and survival of cutaneous melanoma. Med Sci Monit. 2005;11:163-172.
12. Abbasi NR, Shaw HM, Rigel DS, et al. Early diagnosis of cutaneous melanoma. JAMA. 2004;292:2771-2776.
13. Johr RH, Soyer P, Argenziano G, et al. Dermoscopy: The Essentials. New York, NY: Mosby; 2007:130.
14. Enjolras O. Vascular malformations. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. Philadelphia, Pa: Mosby; 2003: 1621–1622.
15. Peris K, Micantonio T, Piccolo D, et al. Dermoscopic features of actinic keratosis. J Dtsch Dermatol Ges. 2007;5:970-976.
16. McIntyre WJ, Downs MR, Bedwell SA. Treatment options for actinic keratosis. Am Fam Physician. 2007;76:667-671.
17. Kamil ZS, Tong LC, Habeeb AA, et al. Non-melanocytic mimics of melanoma: part 1: intraepidermal mimics. J Clin Pathol. 2009;62:120-127.
18. Wong CS, Strange RC, Lear JT. Basal cell carcinoma. BMJ. 2003;327:794-798.
19. Menzies SW. Dermoscopy of pigmented basal cell carcinoma. Clin Dermatol. 2002;20:268-269.
20. Braun RP, Rabinovitz H, Oliviero M, et al. Dermoscopic diagnosis of seborrheic keratosis. Clin Dermatol. 2002;20:270-272.
21. Agero AL, Taliercio S, Dusza SW, et al. Conventional and polarized dermoscopy features of dermatofibroma. Arch Dermatol. 2006;142:1431-1437.
Erythematous rash on face
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
A 27-year-old Caucasian woman came into our clinic with an erythematous, papulopustular rash on her face. The small papules and pustules formed a confluence around her mouth and on her chin; the vermilion border was spared (FIGURE). The patient said that the rash started as a dry scaly patch on the corner of her mouth, and it spread over the course of a few weeks.
The patient had a history of eczema for which she used mometasone furoate cream. Initially, she thought the rash was a flare-up of her eczema, so she used her steroid cream. After using the cream on her face for a month, the patient reported that the rash continued to worsen and spread. She said that the rash was mildly itchy and that when she opened her mouth, it was moderately painful.
FIGURE
Patient confused rash with eczema flare
WHAT IS YOUR DIAGNOSIS?
Diagnosis: Perioral dermatitis
Perioral dermatitis occurs in men and women of all ages and races, though it is more common in women between the ages of 16 and 45.1,2 Many agents have been implicated in the etiology of perioral dermatitis, including infectious pathogens, hormonal factors, and steroids. Moisturizing creams and cosmetics, such as foundation and blush, can cause occlusion of skin follicles, leading to proliferation of skin flora and the resultant papulopustular rash seen in perioral dermatitis.3
In a similar manner, fluorinated corticosteroids enable opportunistic fusobacteria to become pathogenic, leading to the condition. Other risk factors include premenstrual hormone changes, pregnancy, and the use of oral contraceptives, fluorinated toothpaste, inhaled steroids, or glucocorticoids.4
It’s easy to distinguish from these 3 conditions
The differential diagnosis includes contact dermatitis, atopic dermatitis, and rosacea.
- Contact dermatitis is similar to perioral dermatitis in that the patient may indicate that she started using a new skin product. In most cases, the pruritus associated with contact dermatitis will aid in differentiating the 2 diagnoses.
- Atopic dermatitis is more common in children and rarely has an adult onset. Often, there is a personal or family history of asthma or allergies. Distribution in adults is more typically on flexure surfaces, hands, and upper eyelids, and it is itchier than perioral dermatitis.
- Rosacea is often associated with flushing, and is exacerbated by the ingestion of hot food and drinks, alcohol (red wine), and exposure to sun. The distribution is typically on the forehead, cheeks, nose, and around the eyes—rather than around the mouth.
A rash that’s painful and mildly itchy
Perioral dermatitis has distinct clinical features that distinguish it from other facial dermatoses. The rash is classically described as tiny, dry, erythematous papulopustules in a pattern around the mouth, nasolabial folds, and chin, with sparing of the vermilion border.1,2 The clinical course is variable, but is often chronic, with flares. Typically, the rash is only mildly pruritic, but a burning or painful sensation is common. Intolerance to sunlight, drying agents (such as soaps), or irritants (such as cosmetics) is also common.3
Treatment: Discontinue steroids, start antibiotics
If left untreated, perioral dermatitis rarely resolves on its own and will have a fluctuating course, punctuated by flares, that will last for years. The prognosis is excellent, however, once appropriate therapy is instituted; recurrence after treatment is low.5
Cessation of topical steroids is a mainstay of treatment6,7 (strength of recommendation [SOR]: B). High-potency topical steroids can cause short-term improvement of the rash; removal will cause short-term worsening of symptoms. It’s best, then, to switch your patient to a less potent steroid, and then gradually discontinue the steroid3 (SOR: C). Doing so can help the patient to avoid a rebound flare and the temptation to restart the steroid for short-term relief.
It’s also a good idea to tell the patient to stop using other causative agents, such as moisturizing creams, blush, foundation, oral contraceptives, and fluorinated toothpaste6 (SOR: C).
Several antibiotic regimens are successful in the treatment of perioral dermatitis. Tetracycline 250 mg twice daily, minocycline 100 mg daily, or doxycycline 100 mg daily for 2 to 3 weeks is the initial treatment. The treatment course may last up to 6 weeks8 (SOR: B). For children, pregnant women, or patients with allergies to certain antibiotics, erythromycin 250 mg twice daily for up to 6 weeks is also an option7 (SOR: C).
Topical treatments can also be used, but often take longer and have been shown to be less effective than oral therapies. Metronidazole 0.75% gel applied twice daily for 14 weeks or 1% cream applied twice daily for 8 weeks has been shown to be useful9,10 (SOR: C). Erythromycin 2% gel applied twice daily for several months is also effective7 (SOR: C).
Discontinuing the steroid was a challenge for our patient
We told our patient to discontinue her steroid cream, and we started her on metronidazole gel. She returned with significant worsening of her rash, including swelling and erythema. We therefore prescribed a brief course of prednisone for short-term relief while she was started on oral doxycycline. After 6 weeks of oral doxycycline therapy, her rash resolved. At a 6-month follow-up, the patient had experienced no further recurrence of the rash.
CORRESPONDENCE Marc Babaoff, MD, MAHEC Family Medicine Residency Program, 118 W. T. Weaver Boulevard, Asheville, NC 28804; [email protected]
1. Fitzpatrick TB, Johnson RA, Wolff K. Color Atlas and Synopsis of Clinical Dermatology: Common and Serious Diseases. New York: McGraw Hill; 1997:16–17.
2. White G. Color Atlas of Dermatology. 3rd ed. London: Elsevier Science Ltd.; 2004:89.
3. Hafeez ZH. Perioral dermatitis: an update. Int J Dermatol. 2003;42:514-517.
4. Wilkinson DS, Kirton V, Wilkinson JD. Perioral dermatitis: a 12-year review. Br J Dermatol. 1979;101:245-257.
5. Kalkoff KW, Buck A. Etiology of perioral dermatitis [in German]. Hautarzt. 1977;28:74-77.
6. Hengge UR, Ruzicka T, Schwartz RA, et al. Adverse effects of topical glucocorticosteroids. J Am Acad Dermatol. 2006;54:1-15.
7. Weber K, Thurmayr R. Critical appraisal of reports on the treatment of perioral dermatitis. Dermatology. 2005;210:300-307.
8. Macdonald A, Feiwel M. Perioral dermatitis: aetiology and treatment with tetracycline. Br J Dermatol. 1972;87:315-319.
9. Miller SR, Shalita AR. Topical metronidazole gel (0.75%) for the treatment of perioral dermatitis in children. J Am Acad Dermatol. 1994;31:847-848.
10. Veien NK, Munkvad JM, Nielsen AO, et al. Topical metronidazole in the treatment of perioral dermatitis. J Am Acad Dermatol. 1991;24:258-260.
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
A 27-year-old Caucasian woman came into our clinic with an erythematous, papulopustular rash on her face. The small papules and pustules formed a confluence around her mouth and on her chin; the vermilion border was spared (FIGURE). The patient said that the rash started as a dry scaly patch on the corner of her mouth, and it spread over the course of a few weeks.
The patient had a history of eczema for which she used mometasone furoate cream. Initially, she thought the rash was a flare-up of her eczema, so she used her steroid cream. After using the cream on her face for a month, the patient reported that the rash continued to worsen and spread. She said that the rash was mildly itchy and that when she opened her mouth, it was moderately painful.
FIGURE
Patient confused rash with eczema flare
WHAT IS YOUR DIAGNOSIS?
Diagnosis: Perioral dermatitis
Perioral dermatitis occurs in men and women of all ages and races, though it is more common in women between the ages of 16 and 45.1,2 Many agents have been implicated in the etiology of perioral dermatitis, including infectious pathogens, hormonal factors, and steroids. Moisturizing creams and cosmetics, such as foundation and blush, can cause occlusion of skin follicles, leading to proliferation of skin flora and the resultant papulopustular rash seen in perioral dermatitis.3
In a similar manner, fluorinated corticosteroids enable opportunistic fusobacteria to become pathogenic, leading to the condition. Other risk factors include premenstrual hormone changes, pregnancy, and the use of oral contraceptives, fluorinated toothpaste, inhaled steroids, or glucocorticoids.4
It’s easy to distinguish from these 3 conditions
The differential diagnosis includes contact dermatitis, atopic dermatitis, and rosacea.
- Contact dermatitis is similar to perioral dermatitis in that the patient may indicate that she started using a new skin product. In most cases, the pruritus associated with contact dermatitis will aid in differentiating the 2 diagnoses.
- Atopic dermatitis is more common in children and rarely has an adult onset. Often, there is a personal or family history of asthma or allergies. Distribution in adults is more typically on flexure surfaces, hands, and upper eyelids, and it is itchier than perioral dermatitis.
- Rosacea is often associated with flushing, and is exacerbated by the ingestion of hot food and drinks, alcohol (red wine), and exposure to sun. The distribution is typically on the forehead, cheeks, nose, and around the eyes—rather than around the mouth.
A rash that’s painful and mildly itchy
Perioral dermatitis has distinct clinical features that distinguish it from other facial dermatoses. The rash is classically described as tiny, dry, erythematous papulopustules in a pattern around the mouth, nasolabial folds, and chin, with sparing of the vermilion border.1,2 The clinical course is variable, but is often chronic, with flares. Typically, the rash is only mildly pruritic, but a burning or painful sensation is common. Intolerance to sunlight, drying agents (such as soaps), or irritants (such as cosmetics) is also common.3
Treatment: Discontinue steroids, start antibiotics
If left untreated, perioral dermatitis rarely resolves on its own and will have a fluctuating course, punctuated by flares, that will last for years. The prognosis is excellent, however, once appropriate therapy is instituted; recurrence after treatment is low.5
Cessation of topical steroids is a mainstay of treatment6,7 (strength of recommendation [SOR]: B). High-potency topical steroids can cause short-term improvement of the rash; removal will cause short-term worsening of symptoms. It’s best, then, to switch your patient to a less potent steroid, and then gradually discontinue the steroid3 (SOR: C). Doing so can help the patient to avoid a rebound flare and the temptation to restart the steroid for short-term relief.
It’s also a good idea to tell the patient to stop using other causative agents, such as moisturizing creams, blush, foundation, oral contraceptives, and fluorinated toothpaste6 (SOR: C).
Several antibiotic regimens are successful in the treatment of perioral dermatitis. Tetracycline 250 mg twice daily, minocycline 100 mg daily, or doxycycline 100 mg daily for 2 to 3 weeks is the initial treatment. The treatment course may last up to 6 weeks8 (SOR: B). For children, pregnant women, or patients with allergies to certain antibiotics, erythromycin 250 mg twice daily for up to 6 weeks is also an option7 (SOR: C).
Topical treatments can also be used, but often take longer and have been shown to be less effective than oral therapies. Metronidazole 0.75% gel applied twice daily for 14 weeks or 1% cream applied twice daily for 8 weeks has been shown to be useful9,10 (SOR: C). Erythromycin 2% gel applied twice daily for several months is also effective7 (SOR: C).
Discontinuing the steroid was a challenge for our patient
We told our patient to discontinue her steroid cream, and we started her on metronidazole gel. She returned with significant worsening of her rash, including swelling and erythema. We therefore prescribed a brief course of prednisone for short-term relief while she was started on oral doxycycline. After 6 weeks of oral doxycycline therapy, her rash resolved. At a 6-month follow-up, the patient had experienced no further recurrence of the rash.
CORRESPONDENCE Marc Babaoff, MD, MAHEC Family Medicine Residency Program, 118 W. T. Weaver Boulevard, Asheville, NC 28804; [email protected]
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
A 27-year-old Caucasian woman came into our clinic with an erythematous, papulopustular rash on her face. The small papules and pustules formed a confluence around her mouth and on her chin; the vermilion border was spared (FIGURE). The patient said that the rash started as a dry scaly patch on the corner of her mouth, and it spread over the course of a few weeks.
The patient had a history of eczema for which she used mometasone furoate cream. Initially, she thought the rash was a flare-up of her eczema, so she used her steroid cream. After using the cream on her face for a month, the patient reported that the rash continued to worsen and spread. She said that the rash was mildly itchy and that when she opened her mouth, it was moderately painful.
FIGURE
Patient confused rash with eczema flare
WHAT IS YOUR DIAGNOSIS?
Diagnosis: Perioral dermatitis
Perioral dermatitis occurs in men and women of all ages and races, though it is more common in women between the ages of 16 and 45.1,2 Many agents have been implicated in the etiology of perioral dermatitis, including infectious pathogens, hormonal factors, and steroids. Moisturizing creams and cosmetics, such as foundation and blush, can cause occlusion of skin follicles, leading to proliferation of skin flora and the resultant papulopustular rash seen in perioral dermatitis.3
In a similar manner, fluorinated corticosteroids enable opportunistic fusobacteria to become pathogenic, leading to the condition. Other risk factors include premenstrual hormone changes, pregnancy, and the use of oral contraceptives, fluorinated toothpaste, inhaled steroids, or glucocorticoids.4
It’s easy to distinguish from these 3 conditions
The differential diagnosis includes contact dermatitis, atopic dermatitis, and rosacea.
- Contact dermatitis is similar to perioral dermatitis in that the patient may indicate that she started using a new skin product. In most cases, the pruritus associated with contact dermatitis will aid in differentiating the 2 diagnoses.
- Atopic dermatitis is more common in children and rarely has an adult onset. Often, there is a personal or family history of asthma or allergies. Distribution in adults is more typically on flexure surfaces, hands, and upper eyelids, and it is itchier than perioral dermatitis.
- Rosacea is often associated with flushing, and is exacerbated by the ingestion of hot food and drinks, alcohol (red wine), and exposure to sun. The distribution is typically on the forehead, cheeks, nose, and around the eyes—rather than around the mouth.
A rash that’s painful and mildly itchy
Perioral dermatitis has distinct clinical features that distinguish it from other facial dermatoses. The rash is classically described as tiny, dry, erythematous papulopustules in a pattern around the mouth, nasolabial folds, and chin, with sparing of the vermilion border.1,2 The clinical course is variable, but is often chronic, with flares. Typically, the rash is only mildly pruritic, but a burning or painful sensation is common. Intolerance to sunlight, drying agents (such as soaps), or irritants (such as cosmetics) is also common.3
Treatment: Discontinue steroids, start antibiotics
If left untreated, perioral dermatitis rarely resolves on its own and will have a fluctuating course, punctuated by flares, that will last for years. The prognosis is excellent, however, once appropriate therapy is instituted; recurrence after treatment is low.5
Cessation of topical steroids is a mainstay of treatment6,7 (strength of recommendation [SOR]: B). High-potency topical steroids can cause short-term improvement of the rash; removal will cause short-term worsening of symptoms. It’s best, then, to switch your patient to a less potent steroid, and then gradually discontinue the steroid3 (SOR: C). Doing so can help the patient to avoid a rebound flare and the temptation to restart the steroid for short-term relief.
It’s also a good idea to tell the patient to stop using other causative agents, such as moisturizing creams, blush, foundation, oral contraceptives, and fluorinated toothpaste6 (SOR: C).
Several antibiotic regimens are successful in the treatment of perioral dermatitis. Tetracycline 250 mg twice daily, minocycline 100 mg daily, or doxycycline 100 mg daily for 2 to 3 weeks is the initial treatment. The treatment course may last up to 6 weeks8 (SOR: B). For children, pregnant women, or patients with allergies to certain antibiotics, erythromycin 250 mg twice daily for up to 6 weeks is also an option7 (SOR: C).
Topical treatments can also be used, but often take longer and have been shown to be less effective than oral therapies. Metronidazole 0.75% gel applied twice daily for 14 weeks or 1% cream applied twice daily for 8 weeks has been shown to be useful9,10 (SOR: C). Erythromycin 2% gel applied twice daily for several months is also effective7 (SOR: C).
Discontinuing the steroid was a challenge for our patient
We told our patient to discontinue her steroid cream, and we started her on metronidazole gel. She returned with significant worsening of her rash, including swelling and erythema. We therefore prescribed a brief course of prednisone for short-term relief while she was started on oral doxycycline. After 6 weeks of oral doxycycline therapy, her rash resolved. At a 6-month follow-up, the patient had experienced no further recurrence of the rash.
CORRESPONDENCE Marc Babaoff, MD, MAHEC Family Medicine Residency Program, 118 W. T. Weaver Boulevard, Asheville, NC 28804; [email protected]
1. Fitzpatrick TB, Johnson RA, Wolff K. Color Atlas and Synopsis of Clinical Dermatology: Common and Serious Diseases. New York: McGraw Hill; 1997:16–17.
2. White G. Color Atlas of Dermatology. 3rd ed. London: Elsevier Science Ltd.; 2004:89.
3. Hafeez ZH. Perioral dermatitis: an update. Int J Dermatol. 2003;42:514-517.
4. Wilkinson DS, Kirton V, Wilkinson JD. Perioral dermatitis: a 12-year review. Br J Dermatol. 1979;101:245-257.
5. Kalkoff KW, Buck A. Etiology of perioral dermatitis [in German]. Hautarzt. 1977;28:74-77.
6. Hengge UR, Ruzicka T, Schwartz RA, et al. Adverse effects of topical glucocorticosteroids. J Am Acad Dermatol. 2006;54:1-15.
7. Weber K, Thurmayr R. Critical appraisal of reports on the treatment of perioral dermatitis. Dermatology. 2005;210:300-307.
8. Macdonald A, Feiwel M. Perioral dermatitis: aetiology and treatment with tetracycline. Br J Dermatol. 1972;87:315-319.
9. Miller SR, Shalita AR. Topical metronidazole gel (0.75%) for the treatment of perioral dermatitis in children. J Am Acad Dermatol. 1994;31:847-848.
10. Veien NK, Munkvad JM, Nielsen AO, et al. Topical metronidazole in the treatment of perioral dermatitis. J Am Acad Dermatol. 1991;24:258-260.
1. Fitzpatrick TB, Johnson RA, Wolff K. Color Atlas and Synopsis of Clinical Dermatology: Common and Serious Diseases. New York: McGraw Hill; 1997:16–17.
2. White G. Color Atlas of Dermatology. 3rd ed. London: Elsevier Science Ltd.; 2004:89.
3. Hafeez ZH. Perioral dermatitis: an update. Int J Dermatol. 2003;42:514-517.
4. Wilkinson DS, Kirton V, Wilkinson JD. Perioral dermatitis: a 12-year review. Br J Dermatol. 1979;101:245-257.
5. Kalkoff KW, Buck A. Etiology of perioral dermatitis [in German]. Hautarzt. 1977;28:74-77.
6. Hengge UR, Ruzicka T, Schwartz RA, et al. Adverse effects of topical glucocorticosteroids. J Am Acad Dermatol. 2006;54:1-15.
7. Weber K, Thurmayr R. Critical appraisal of reports on the treatment of perioral dermatitis. Dermatology. 2005;210:300-307.
8. Macdonald A, Feiwel M. Perioral dermatitis: aetiology and treatment with tetracycline. Br J Dermatol. 1972;87:315-319.
9. Miller SR, Shalita AR. Topical metronidazole gel (0.75%) for the treatment of perioral dermatitis in children. J Am Acad Dermatol. 1994;31:847-848.
10. Veien NK, Munkvad JM, Nielsen AO, et al. Topical metronidazole in the treatment of perioral dermatitis. J Am Acad Dermatol. 1991;24:258-260.
Bluish pigmentation of face and sclera
A 67-YEAR-OLD CAUCASIAN WOMAN came to the diabetes center for a routine check-up. She indicated that her face and eyes had become discolored, and that the discoloration had occurred gradually. She denied pruritus, erythema, edema, or pain at the site, and reported no vision changes, dry eyes, or other constitutional signs and symptoms. She did not smoke.
Th e patient’s past medical history was significant for type 2 diabetes, hypertension, depression, dyslipidemia, and rosacea. The medications she was taking included pioglitazone, metformin, coral calcium, glyburide, atorvastatin, fosinopril, and minocycline.
On physical exam, there was a bluish pigmentation of the left lateral forehead and cheek (FIGURE 1A) and bluish pigmentation of the sclera (FIGURE 1B). The rest of her exam was normal.
We were concerned about venous dilation, and ordered a computed tomography scan of the chest and an ultrasound of the neck. Both were normal, thus ruling out a thoracic outlet obstruction.
FIGURE 1
Unusual pigmentation of forehead and eyes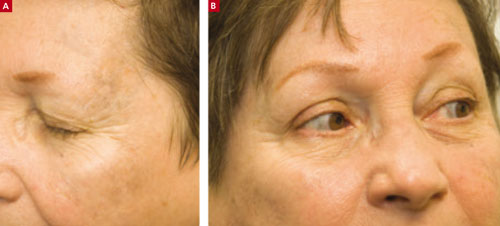
This 67-year-old woman said that the bluish discoloration of her face and eyes had come on gradually. She had no problems with her vision and no pain in her eyes.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Minocycline-induced hyperpigmentation
We referred the patient to a dermatologist, who diagnosed minocycline-induced hyperpigmentation of the skin and sclera.
Minocycline hydrochloride, a derivative of tetracycline, is a broad-spectrum antibiotic that is often used to treat rosacea (which our patient had) and acne vulgaris. Minocycline’s lipid-soluble properties enable penetration into sebaceous glands and subsequent clearing of acne in many cases.1
Despite minocycline’s therapeutic effect, it can lead to a non–dose-dependent pigmentation of the skin (typically a blue-gray pigmentation on the legs), nails, sclera, bones, oral mucosa, teeth, and thyroid.1,2
Rare serious adverse effects include systemic lupus erythematosus (antinuclear antibody positive, DNA antibody negative), pseudotumor cerebri syndrome, and an autoimmune drug reaction leading to hepatitis. Resolution of these conditions occurs slowly once minocycline is discontinued.1 However, scleral hyperpigmentation may be permanent. This pigmentation appears blue-gray.3
Hyperpigmentation falls into 4 categories
Although the etiology of the minocycline-induced hyperpigmentation is unclear, iron and melanin-staining granules identified in dermal dendrocytes and macrophages are likely responsible for producing the blue-gray pigmentation of the skin.2,4
The hyperpigmentation of minocycline is classified into 4 different types, based on clinical features, light and electron microscopy, and energy dispersive X-ray analysis:
Type I is a blue-gray discoloration that appears on the face at sites of inflammation or scarring.
Type II is characterized by blue-gray pigmentation of normal skin of the anterior lower legs.
In Type III, the skin has a muddy brown appearance at sun-exposed sites.2
Type IV occurs in scars, giving them a blue-gray appearance.5
Differential Dx includes Addison’s disease
Addison’s disease, hemochromatosis, and melanoma all make up the differential.
The hyperpigmentation of Addison’s disease is a characteristic brown or bronzing of the skin, most notably in the creases of the hands. Mucous membranes may develop a bluish pigmentation.
Patients with hemochromatosis develop bronzing of the skin caused by iron deposits in the dermis. This pigmentation is characterized by a generalized metallic or slate gray coloration of the face, neck, upper extremities, lower legs, and genitals. This coloration also appears in scars.
Melanoma was less likely in this case because of the uniformity of pigmentation on the skin and sclera, as well as the symmetrical pigmentation of the sclera. A skin biopsy would be helpful in ruling out melanoma.6
A biopsy of this lesion would show granules within macrophages that stain positive for iron and melanin, confirming the clinical diagnosis of minocycline-induced hyperpigmentation.2
Patients may not want to wait for discoloration to go away
Some patients find the benign hyperpigmentation cosmetically undesirable and may not want to wait to see if the discoloration goes away. The Q-switched ruby laser has been successful in removing minocycline-induced hyperpigmentation of the skin and oral mucosa.2
Our patient stopped the drug, and saw (some) improvement
Once the diagnosis was made, our patient stopped taking minocycline—which she’d been taking at a low daily dose for 4 years. She was started on metronidazole for her rosacea. Three years later, the patient had a slight clinical improvement in the bluish appearance of her face and sclera.
Minocycline is still a good option for the treatment of acne and rosacea. If you prescribe it, though, be sure to monitor for hyperpigmentation on a regular basis.
CORRESPONDENCE Jay Shubrook, DO, associate professor of family medicine, director of clinical research, Cornwell Center, O’Bleness Memorial Hospital, 65 Hospital Drive, Athens, OH 45701; [email protected].
1. Habif TP. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 4th ed. Philadelphia, Pa: Mosby Inc; 2004:181–182.
2. James W, Berger T, Elston D. Andrews’ Diseases of the Skin: Clinical Dermatology. 10th ed. Philadelphia, Pa: Elsevier Inc; 2006:125–126.
3. Fraundfelder FT, Randall JA. Minocycline-induced scleral pigmentation. Ophthalmology. 1997;104:936-938.
4. Altman DA, Fiveson DP, Lee MW. Minocycline hyperpigmentation: model for in situ phagocytic activity of factor XIIa dermal dendrocytes. J Cutan Pathol. 1992;19:340-345.
5. Mouton RW, Jordann HF, Schneider JW. A new type of minocycline-induced cutaneous hyperpigmentation. Clin Exper Dermatol. 2004;29:8-14.
6. Kasper DL, Braunwald E, Fauci AS, et al. Harrison’s Principles of Internal Medicine. 16th ed. New York, NY: McGraw-Hill Companies, Inc; 2005:2142, 2300.
A 67-YEAR-OLD CAUCASIAN WOMAN came to the diabetes center for a routine check-up. She indicated that her face and eyes had become discolored, and that the discoloration had occurred gradually. She denied pruritus, erythema, edema, or pain at the site, and reported no vision changes, dry eyes, or other constitutional signs and symptoms. She did not smoke.
Th e patient’s past medical history was significant for type 2 diabetes, hypertension, depression, dyslipidemia, and rosacea. The medications she was taking included pioglitazone, metformin, coral calcium, glyburide, atorvastatin, fosinopril, and minocycline.
On physical exam, there was a bluish pigmentation of the left lateral forehead and cheek (FIGURE 1A) and bluish pigmentation of the sclera (FIGURE 1B). The rest of her exam was normal.
We were concerned about venous dilation, and ordered a computed tomography scan of the chest and an ultrasound of the neck. Both were normal, thus ruling out a thoracic outlet obstruction.
FIGURE 1
Unusual pigmentation of forehead and eyes
This 67-year-old woman said that the bluish discoloration of her face and eyes had come on gradually. She had no problems with her vision and no pain in her eyes.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Minocycline-induced hyperpigmentation
We referred the patient to a dermatologist, who diagnosed minocycline-induced hyperpigmentation of the skin and sclera.
Minocycline hydrochloride, a derivative of tetracycline, is a broad-spectrum antibiotic that is often used to treat rosacea (which our patient had) and acne vulgaris. Minocycline’s lipid-soluble properties enable penetration into sebaceous glands and subsequent clearing of acne in many cases.1
Despite minocycline’s therapeutic effect, it can lead to a non–dose-dependent pigmentation of the skin (typically a blue-gray pigmentation on the legs), nails, sclera, bones, oral mucosa, teeth, and thyroid.1,2
Rare serious adverse effects include systemic lupus erythematosus (antinuclear antibody positive, DNA antibody negative), pseudotumor cerebri syndrome, and an autoimmune drug reaction leading to hepatitis. Resolution of these conditions occurs slowly once minocycline is discontinued.1 However, scleral hyperpigmentation may be permanent. This pigmentation appears blue-gray.3
Hyperpigmentation falls into 4 categories
Although the etiology of the minocycline-induced hyperpigmentation is unclear, iron and melanin-staining granules identified in dermal dendrocytes and macrophages are likely responsible for producing the blue-gray pigmentation of the skin.2,4
The hyperpigmentation of minocycline is classified into 4 different types, based on clinical features, light and electron microscopy, and energy dispersive X-ray analysis:
Type I is a blue-gray discoloration that appears on the face at sites of inflammation or scarring.
Type II is characterized by blue-gray pigmentation of normal skin of the anterior lower legs.
In Type III, the skin has a muddy brown appearance at sun-exposed sites.2
Type IV occurs in scars, giving them a blue-gray appearance.5
Differential Dx includes Addison’s disease
Addison’s disease, hemochromatosis, and melanoma all make up the differential.
The hyperpigmentation of Addison’s disease is a characteristic brown or bronzing of the skin, most notably in the creases of the hands. Mucous membranes may develop a bluish pigmentation.
Patients with hemochromatosis develop bronzing of the skin caused by iron deposits in the dermis. This pigmentation is characterized by a generalized metallic or slate gray coloration of the face, neck, upper extremities, lower legs, and genitals. This coloration also appears in scars.
Melanoma was less likely in this case because of the uniformity of pigmentation on the skin and sclera, as well as the symmetrical pigmentation of the sclera. A skin biopsy would be helpful in ruling out melanoma.6
A biopsy of this lesion would show granules within macrophages that stain positive for iron and melanin, confirming the clinical diagnosis of minocycline-induced hyperpigmentation.2
Patients may not want to wait for discoloration to go away
Some patients find the benign hyperpigmentation cosmetically undesirable and may not want to wait to see if the discoloration goes away. The Q-switched ruby laser has been successful in removing minocycline-induced hyperpigmentation of the skin and oral mucosa.2
Our patient stopped the drug, and saw (some) improvement
Once the diagnosis was made, our patient stopped taking minocycline—which she’d been taking at a low daily dose for 4 years. She was started on metronidazole for her rosacea. Three years later, the patient had a slight clinical improvement in the bluish appearance of her face and sclera.
Minocycline is still a good option for the treatment of acne and rosacea. If you prescribe it, though, be sure to monitor for hyperpigmentation on a regular basis.
CORRESPONDENCE Jay Shubrook, DO, associate professor of family medicine, director of clinical research, Cornwell Center, O’Bleness Memorial Hospital, 65 Hospital Drive, Athens, OH 45701; [email protected].
A 67-YEAR-OLD CAUCASIAN WOMAN came to the diabetes center for a routine check-up. She indicated that her face and eyes had become discolored, and that the discoloration had occurred gradually. She denied pruritus, erythema, edema, or pain at the site, and reported no vision changes, dry eyes, or other constitutional signs and symptoms. She did not smoke.
Th e patient’s past medical history was significant for type 2 diabetes, hypertension, depression, dyslipidemia, and rosacea. The medications she was taking included pioglitazone, metformin, coral calcium, glyburide, atorvastatin, fosinopril, and minocycline.
On physical exam, there was a bluish pigmentation of the left lateral forehead and cheek (FIGURE 1A) and bluish pigmentation of the sclera (FIGURE 1B). The rest of her exam was normal.
We were concerned about venous dilation, and ordered a computed tomography scan of the chest and an ultrasound of the neck. Both were normal, thus ruling out a thoracic outlet obstruction.
FIGURE 1
Unusual pigmentation of forehead and eyes
This 67-year-old woman said that the bluish discoloration of her face and eyes had come on gradually. She had no problems with her vision and no pain in her eyes.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Minocycline-induced hyperpigmentation
We referred the patient to a dermatologist, who diagnosed minocycline-induced hyperpigmentation of the skin and sclera.
Minocycline hydrochloride, a derivative of tetracycline, is a broad-spectrum antibiotic that is often used to treat rosacea (which our patient had) and acne vulgaris. Minocycline’s lipid-soluble properties enable penetration into sebaceous glands and subsequent clearing of acne in many cases.1
Despite minocycline’s therapeutic effect, it can lead to a non–dose-dependent pigmentation of the skin (typically a blue-gray pigmentation on the legs), nails, sclera, bones, oral mucosa, teeth, and thyroid.1,2
Rare serious adverse effects include systemic lupus erythematosus (antinuclear antibody positive, DNA antibody negative), pseudotumor cerebri syndrome, and an autoimmune drug reaction leading to hepatitis. Resolution of these conditions occurs slowly once minocycline is discontinued.1 However, scleral hyperpigmentation may be permanent. This pigmentation appears blue-gray.3
Hyperpigmentation falls into 4 categories
Although the etiology of the minocycline-induced hyperpigmentation is unclear, iron and melanin-staining granules identified in dermal dendrocytes and macrophages are likely responsible for producing the blue-gray pigmentation of the skin.2,4
The hyperpigmentation of minocycline is classified into 4 different types, based on clinical features, light and electron microscopy, and energy dispersive X-ray analysis:
Type I is a blue-gray discoloration that appears on the face at sites of inflammation or scarring.
Type II is characterized by blue-gray pigmentation of normal skin of the anterior lower legs.
In Type III, the skin has a muddy brown appearance at sun-exposed sites.2
Type IV occurs in scars, giving them a blue-gray appearance.5
Differential Dx includes Addison’s disease
Addison’s disease, hemochromatosis, and melanoma all make up the differential.
The hyperpigmentation of Addison’s disease is a characteristic brown or bronzing of the skin, most notably in the creases of the hands. Mucous membranes may develop a bluish pigmentation.
Patients with hemochromatosis develop bronzing of the skin caused by iron deposits in the dermis. This pigmentation is characterized by a generalized metallic or slate gray coloration of the face, neck, upper extremities, lower legs, and genitals. This coloration also appears in scars.
Melanoma was less likely in this case because of the uniformity of pigmentation on the skin and sclera, as well as the symmetrical pigmentation of the sclera. A skin biopsy would be helpful in ruling out melanoma.6
A biopsy of this lesion would show granules within macrophages that stain positive for iron and melanin, confirming the clinical diagnosis of minocycline-induced hyperpigmentation.2
Patients may not want to wait for discoloration to go away
Some patients find the benign hyperpigmentation cosmetically undesirable and may not want to wait to see if the discoloration goes away. The Q-switched ruby laser has been successful in removing minocycline-induced hyperpigmentation of the skin and oral mucosa.2
Our patient stopped the drug, and saw (some) improvement
Once the diagnosis was made, our patient stopped taking minocycline—which she’d been taking at a low daily dose for 4 years. She was started on metronidazole for her rosacea. Three years later, the patient had a slight clinical improvement in the bluish appearance of her face and sclera.
Minocycline is still a good option for the treatment of acne and rosacea. If you prescribe it, though, be sure to monitor for hyperpigmentation on a regular basis.
CORRESPONDENCE Jay Shubrook, DO, associate professor of family medicine, director of clinical research, Cornwell Center, O’Bleness Memorial Hospital, 65 Hospital Drive, Athens, OH 45701; [email protected].
1. Habif TP. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 4th ed. Philadelphia, Pa: Mosby Inc; 2004:181–182.
2. James W, Berger T, Elston D. Andrews’ Diseases of the Skin: Clinical Dermatology. 10th ed. Philadelphia, Pa: Elsevier Inc; 2006:125–126.
3. Fraundfelder FT, Randall JA. Minocycline-induced scleral pigmentation. Ophthalmology. 1997;104:936-938.
4. Altman DA, Fiveson DP, Lee MW. Minocycline hyperpigmentation: model for in situ phagocytic activity of factor XIIa dermal dendrocytes. J Cutan Pathol. 1992;19:340-345.
5. Mouton RW, Jordann HF, Schneider JW. A new type of minocycline-induced cutaneous hyperpigmentation. Clin Exper Dermatol. 2004;29:8-14.
6. Kasper DL, Braunwald E, Fauci AS, et al. Harrison’s Principles of Internal Medicine. 16th ed. New York, NY: McGraw-Hill Companies, Inc; 2005:2142, 2300.
1. Habif TP. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 4th ed. Philadelphia, Pa: Mosby Inc; 2004:181–182.
2. James W, Berger T, Elston D. Andrews’ Diseases of the Skin: Clinical Dermatology. 10th ed. Philadelphia, Pa: Elsevier Inc; 2006:125–126.
3. Fraundfelder FT, Randall JA. Minocycline-induced scleral pigmentation. Ophthalmology. 1997;104:936-938.
4. Altman DA, Fiveson DP, Lee MW. Minocycline hyperpigmentation: model for in situ phagocytic activity of factor XIIa dermal dendrocytes. J Cutan Pathol. 1992;19:340-345.
5. Mouton RW, Jordann HF, Schneider JW. A new type of minocycline-induced cutaneous hyperpigmentation. Clin Exper Dermatol. 2004;29:8-14.
6. Kasper DL, Braunwald E, Fauci AS, et al. Harrison’s Principles of Internal Medicine. 16th ed. New York, NY: McGraw-Hill Companies, Inc; 2005:2142, 2300.
Painful rash on face
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
A 68-year-old woman came into the clinic for treatment of a painful, erythematous skin rash over the bridge of her nose. She’d had the rash for 4 days, and it was spreading to the malar area and up around her eyelids and forehead (FIGURE 1). The patient said she felt “out of sorts,” was nauseous, and had a low fever. She said she’d recently had a sore throat. Her past medical history included hypertension.
The patient had elevated and indurated shiny skin plaques involving the nose, cheeks, eyelids, and forehead. She also had some blisters and crusty lesions. On palpation, the skin was hot and tender. The pharynx was unremarkable and the neck supple, with no palpable nodes.
FIGURE 1
Spreading rash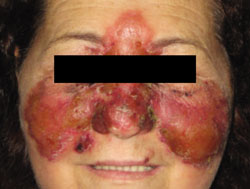
The 68-year-old patient said that she’d recently had a sore throat. She indicated that she felt nauseous and out of sorts.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Erysipelas
Erysipelas is an acute superficial cellulitis with lymphatic involvement. It is characterized by the abrupt onset of a warm, erythematous rash with a sharply demarcated, indurated, elevated margin. There are no suppurative foci; sometimes, however, there are bullae or vesicles.
In facial “butterfly” erysipelas (which this patient had), the plaques may involve the eyelids, cheeks, nose, and forehead. Upon palpation, the skin is hot and tender. As the process develops, the color becomes a dark, fiery red and vesicles appear at the advancing border and over the surface. Associated regional lymphadenopathy may be present. There is no necrosis.1
Most cases are caused by the Streptococcus species—usually group A (Streptococcus pyogenes)—and groups B, C, and G. (Erysipelas is occasionally caused by Staphylococcus aureus.) After prodromal symptoms that last for 4 to 48 hours and include malaise, chills, fever, anorexia, and vomiting, more red, tender firm spots develop.
Predisposing conditions for erysipelas include disruption to the skin barrier as a result of trauma, lymph stasis (prior radiation, mastectomy, saphenous vein harvest, lymphadenectomy), injection drug use, ulcers, wounds, dermatophytic infections, and edema. Toe intertrigo is perhaps the most common site for pathogen entry. The source of infection in facial erysipelas, however, is often the nasopharynx.2,3
Differential diagnosis includes cellulitis, rosacea
The differential diagnosis for erysipelas include cellulitis, rosacea, lupus erythematous “butterfly rash,” and seborrheic dermatitis.
Cellulitis is associated with skin erythema, edema, and warmth in the absence of underlying suppurative foci. Lymphangitis and inflammation of regional lymph nodes may occur. Vesicles, bullae, and ecchymoses (or petechiae) may also be present.4
Rosacea is a disorder that occurs in middle-aged and older adults, and is more common in women.5 It is characterized by symmetrical vascular dilation on the central face, including the nose, cheek, eyelids, and forehead. Facial erythema and telangiectasias—typically on the cheeks—are also present, as well as late papules and pustules.6 There are no comedones, which you would see with acne vulgaris.6 Rosacea is made worse by heat or sunlight, emotional stressors, and drinking alcohol or eating spicy foods.
Lupus erythematous “butterfly rash” is characterized by a macular confluent erythema over the cheeks and bridge of the nose with fine scaling, erosions, and crusts. It appears in approximately half of patients with systemic lupus erythematosus, usually after sun exposure.7,8
Seborrheic dermatitis is characterized by intermittent, active phases that manifest with burning, scaling, excess oil secretion, and itching. Seborrheic dermatitis usually appears over areas that are rich in sebaceous glands, such as the lateral sides of the nose and the nasolabial folds, eyebrows, glabella, and scalp.9
The exam holds the key to diagnosis
The diagnosis of erysipelas is based on clinical manifestations. Blood cultures, needle aspirations, and punch biopsies are not usually helpful. Blood cultures are positive in less than 5% of cases.4 Punch biopsy is positive in 20% to 30% of cases.4
Needle aspiration and skin biopsies may be considered for a patient with a severe infection that is not responding to treatment, or for a patient who has diabetes, a malignancy, or unusual predisposing associated factors, such as an immersion injury, an animal bite, neutropenia, or immunodeficiency.
Treatment centers on penicillin
With early diagnosis and proper treatment, the prognosis for erysipelas is excellent. Empiric antimicrobial therapy should include activity against beta-hemolytic Streptococcus. Penicillin is the treatment of choice4,10 (strength of recommendation [SOR]: A). Streptococcus strains are susceptible to penicillin and 99.5% are susceptible to clindamycin. A 7% macrolide resistance has been reported in the United States.4
Oral therapy is recommended for mild infections (or for those who have improved after intravenous [IV] antibiotics). Treat with penicillin V 500 mg PO 4 times daily or amoxicillin 500 mg PO 3 times daily. Other choices include clindamycin 300 mg PO for 7 to 10 days; azithromycin 500 mg PO for 1 day, followed by 250 mg PO daily for 4 days; or clarithromycin 250 mg PO twice a day for 7 to 10 days4,10 (SOR: A).
Consider hospitalization for severely ill patients, for those who are unable to tolerate oral medications, and for those who require parenteral antibiotic therapy for systemic symptoms and rapidly progressing erythema4 (SOR: A). Regimens include penicillin G 2 million to 4 million units IV every 4 to 6 hours; cefazolin 0.5 to 1 g IV every 8 hours; cefotaxime 1 to 2 g IV every 8 hours; and ceftriaxone 1 to 2 g IV every 24 hours.
If staphylococcal infection is suspected, a penicillinase-resistant semisynthetic penicillin or a first-generation cephalosporin, such as cefazolin, can be selected. For oral therapy, cephalexin 500 mg 4 times daily can be used4,11 (SOR: A). Macrolides (erythromycin 250 mg PO every 6 hours) are also an option4 (SOR: A). For presumed methicillin-resistant Staphylococcus aureus (MRSA), you can prescribe clindamycin 600 mg IV every 8 hours; vancomycin 15 mg/kg IV every 12 hours; or linezolid 600 mg every 12 hours. The total duration of antibiotic therapy may be extended to up to 14 days, and tailored to clinical improvement.4,10
Steroids can slightly reduce healing time and antibiotic duration in patients with erysipelas. You can prescribe prednisone 30 mg PO and taper over 8 days.4,12
Be sure to treat underlying predisposing conditions such as tinea pedis, venous eczema, and trauma sites. Encourage supportive measures, such as elevating the affected extremity, as well.
Improvement in a few days. Patients with erysipelas typically show improvement in 24 to 48 hours of beginning antibiotic therapy. Don’t be too quick to consider antibiotic failure in patients with worsening erythema after initiating antimicrobial therapy; this may be related to the destruction of pathogens that release enzymes (including deoxyribonuclease B [DNase B] and hyaluronidase) that increase local inflammation.
A good outcome for the patient
The patient was hospitalized and received cefazolin 1 g IV every 8 hours for 3 da ys. She was discharged on cephalexin 500 mg PO, every 6 hours for 7 days, with excellent results. FIGURE 2 shows the patient 4 days after she finished taking the cephalexin.
FIGURE 2
A few days, a big difference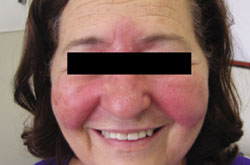
The patient received cefazolin 1 g IV every 8 hours for 3 days, followed by oral cephalexin 500 mg every 6 hours for 7 days. This photo shows the patient 4 days after she finished taking the cephalexin.
CORRESPONDENCE Felix B. Chang, MD, Leominster Community Health Connections Family Health Center, 14 Manning Ave, Suite 402, Leominster, MA 01453; [email protected].
1. Chang F, Lopes A. Erysipelas. In: Domino FJ, ed. The 5-Minute Clinical Consult 2008. 16th ed. Philadelphia, Pa: Lippincott Williams & Wilkins; 2008:448–449.
2. Jorup-Ronstrom C. Epidemiological, bacteriological and complicating features of erysipelas. Scand J Infect Dis. 1986;18:519-524.
3. Davis L, Cole J. Erysipelas. Dermatology-bacterial infections. Available at: http://www.imedicine.com/printtopic.asp?bookid=2&topic=129. Accessed at January 11, 2010.
4. Stevens DL, Bisno AL, Chambers HF. Infectious Diseases Society of America (ISDA) practice guidelines for diagnosis and management of skin and soft tissue infections. Clin Infect Dis. 2005;41:1373-1406.
5. Wilking J, Dahl M, Detmar M, et al. Standard classification of rosacea: report of the National Rosacea Society Expert Committee on the Classification and Staging of Rosacea. J Am Acad Dermatol. 2002;46:584-587.
6. Crawford GH, Pelle MT, James WD. Rosacea: I. Etiology, pathogenesis, and subtype classification. J Am Acad Dermatol. 2004;51:327-341.
7. Patel P, Werth V. Cutaneous lupus erythematosus: a review. Dermatol Clin. 2002;20:373-385.
8. Madhok R, Wu O. Systemic lupus erythematosus. Am Fam Physician. 2007;76:1351-1353.
9. Gupta AK, Bluhm R. Seborrheic dermatitis. J Eur Acad Dermatol Venereol. 2004;18:13-26.
10. Van Beneden CA, Facklam R, Lynfield R, et al. Erythromycin resistance among invasive group A streptococcal infections, United States, 1999-2001 [abstract 345]. In: Proceedings and Abstract of the 42nd Annual Meeting of the Infectious Diseases Society of America (Boston). Alexandria, Va: Infectious Diseases Society of America; 2004:102.
11. Stulberg DL, Penrod MA, Blatny RA. Common bacterial skin infections. Am Fam Physician. 2002;66:119-124.
12. Bergkvist PI, Sjobeck K. Relapse of erysipelas following treatment with prednisolone or placebo in addition to antibiotics: a 1-year follow up. Scand J Infect Dis. 1998;30:206-207.
CORRESPONDENCE Felix B. Chang, MD, Leominster Community Health Connections Family Health Center, 14 Manning Ave, Suite 402, Leominster, MA 01453; [email protected].
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
A 68-year-old woman came into the clinic for treatment of a painful, erythematous skin rash over the bridge of her nose. She’d had the rash for 4 days, and it was spreading to the malar area and up around her eyelids and forehead (FIGURE 1). The patient said she felt “out of sorts,” was nauseous, and had a low fever. She said she’d recently had a sore throat. Her past medical history included hypertension.
The patient had elevated and indurated shiny skin plaques involving the nose, cheeks, eyelids, and forehead. She also had some blisters and crusty lesions. On palpation, the skin was hot and tender. The pharynx was unremarkable and the neck supple, with no palpable nodes.
FIGURE 1
Spreading rash
The 68-year-old patient said that she’d recently had a sore throat. She indicated that she felt nauseous and out of sorts.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Erysipelas
Erysipelas is an acute superficial cellulitis with lymphatic involvement. It is characterized by the abrupt onset of a warm, erythematous rash with a sharply demarcated, indurated, elevated margin. There are no suppurative foci; sometimes, however, there are bullae or vesicles.
In facial “butterfly” erysipelas (which this patient had), the plaques may involve the eyelids, cheeks, nose, and forehead. Upon palpation, the skin is hot and tender. As the process develops, the color becomes a dark, fiery red and vesicles appear at the advancing border and over the surface. Associated regional lymphadenopathy may be present. There is no necrosis.1
Most cases are caused by the Streptococcus species—usually group A (Streptococcus pyogenes)—and groups B, C, and G. (Erysipelas is occasionally caused by Staphylococcus aureus.) After prodromal symptoms that last for 4 to 48 hours and include malaise, chills, fever, anorexia, and vomiting, more red, tender firm spots develop.
Predisposing conditions for erysipelas include disruption to the skin barrier as a result of trauma, lymph stasis (prior radiation, mastectomy, saphenous vein harvest, lymphadenectomy), injection drug use, ulcers, wounds, dermatophytic infections, and edema. Toe intertrigo is perhaps the most common site for pathogen entry. The source of infection in facial erysipelas, however, is often the nasopharynx.2,3
Differential diagnosis includes cellulitis, rosacea
The differential diagnosis for erysipelas include cellulitis, rosacea, lupus erythematous “butterfly rash,” and seborrheic dermatitis.
Cellulitis is associated with skin erythema, edema, and warmth in the absence of underlying suppurative foci. Lymphangitis and inflammation of regional lymph nodes may occur. Vesicles, bullae, and ecchymoses (or petechiae) may also be present.4
Rosacea is a disorder that occurs in middle-aged and older adults, and is more common in women.5 It is characterized by symmetrical vascular dilation on the central face, including the nose, cheek, eyelids, and forehead. Facial erythema and telangiectasias—typically on the cheeks—are also present, as well as late papules and pustules.6 There are no comedones, which you would see with acne vulgaris.6 Rosacea is made worse by heat or sunlight, emotional stressors, and drinking alcohol or eating spicy foods.
Lupus erythematous “butterfly rash” is characterized by a macular confluent erythema over the cheeks and bridge of the nose with fine scaling, erosions, and crusts. It appears in approximately half of patients with systemic lupus erythematosus, usually after sun exposure.7,8
Seborrheic dermatitis is characterized by intermittent, active phases that manifest with burning, scaling, excess oil secretion, and itching. Seborrheic dermatitis usually appears over areas that are rich in sebaceous glands, such as the lateral sides of the nose and the nasolabial folds, eyebrows, glabella, and scalp.9
The exam holds the key to diagnosis
The diagnosis of erysipelas is based on clinical manifestations. Blood cultures, needle aspirations, and punch biopsies are not usually helpful. Blood cultures are positive in less than 5% of cases.4 Punch biopsy is positive in 20% to 30% of cases.4
Needle aspiration and skin biopsies may be considered for a patient with a severe infection that is not responding to treatment, or for a patient who has diabetes, a malignancy, or unusual predisposing associated factors, such as an immersion injury, an animal bite, neutropenia, or immunodeficiency.
Treatment centers on penicillin
With early diagnosis and proper treatment, the prognosis for erysipelas is excellent. Empiric antimicrobial therapy should include activity against beta-hemolytic Streptococcus. Penicillin is the treatment of choice4,10 (strength of recommendation [SOR]: A). Streptococcus strains are susceptible to penicillin and 99.5% are susceptible to clindamycin. A 7% macrolide resistance has been reported in the United States.4
Oral therapy is recommended for mild infections (or for those who have improved after intravenous [IV] antibiotics). Treat with penicillin V 500 mg PO 4 times daily or amoxicillin 500 mg PO 3 times daily. Other choices include clindamycin 300 mg PO for 7 to 10 days; azithromycin 500 mg PO for 1 day, followed by 250 mg PO daily for 4 days; or clarithromycin 250 mg PO twice a day for 7 to 10 days4,10 (SOR: A).
Consider hospitalization for severely ill patients, for those who are unable to tolerate oral medications, and for those who require parenteral antibiotic therapy for systemic symptoms and rapidly progressing erythema4 (SOR: A). Regimens include penicillin G 2 million to 4 million units IV every 4 to 6 hours; cefazolin 0.5 to 1 g IV every 8 hours; cefotaxime 1 to 2 g IV every 8 hours; and ceftriaxone 1 to 2 g IV every 24 hours.
If staphylococcal infection is suspected, a penicillinase-resistant semisynthetic penicillin or a first-generation cephalosporin, such as cefazolin, can be selected. For oral therapy, cephalexin 500 mg 4 times daily can be used4,11 (SOR: A). Macrolides (erythromycin 250 mg PO every 6 hours) are also an option4 (SOR: A). For presumed methicillin-resistant Staphylococcus aureus (MRSA), you can prescribe clindamycin 600 mg IV every 8 hours; vancomycin 15 mg/kg IV every 12 hours; or linezolid 600 mg every 12 hours. The total duration of antibiotic therapy may be extended to up to 14 days, and tailored to clinical improvement.4,10
Steroids can slightly reduce healing time and antibiotic duration in patients with erysipelas. You can prescribe prednisone 30 mg PO and taper over 8 days.4,12
Be sure to treat underlying predisposing conditions such as tinea pedis, venous eczema, and trauma sites. Encourage supportive measures, such as elevating the affected extremity, as well.
Improvement in a few days. Patients with erysipelas typically show improvement in 24 to 48 hours of beginning antibiotic therapy. Don’t be too quick to consider antibiotic failure in patients with worsening erythema after initiating antimicrobial therapy; this may be related to the destruction of pathogens that release enzymes (including deoxyribonuclease B [DNase B] and hyaluronidase) that increase local inflammation.
A good outcome for the patient
The patient was hospitalized and received cefazolin 1 g IV every 8 hours for 3 da ys. She was discharged on cephalexin 500 mg PO, every 6 hours for 7 days, with excellent results. FIGURE 2 shows the patient 4 days after she finished taking the cephalexin.
FIGURE 2
A few days, a big difference
The patient received cefazolin 1 g IV every 8 hours for 3 days, followed by oral cephalexin 500 mg every 6 hours for 7 days. This photo shows the patient 4 days after she finished taking the cephalexin.
CORRESPONDENCE Felix B. Chang, MD, Leominster Community Health Connections Family Health Center, 14 Manning Ave, Suite 402, Leominster, MA 01453; [email protected].
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
A 68-year-old woman came into the clinic for treatment of a painful, erythematous skin rash over the bridge of her nose. She’d had the rash for 4 days, and it was spreading to the malar area and up around her eyelids and forehead (FIGURE 1). The patient said she felt “out of sorts,” was nauseous, and had a low fever. She said she’d recently had a sore throat. Her past medical history included hypertension.
The patient had elevated and indurated shiny skin plaques involving the nose, cheeks, eyelids, and forehead. She also had some blisters and crusty lesions. On palpation, the skin was hot and tender. The pharynx was unremarkable and the neck supple, with no palpable nodes.
FIGURE 1
Spreading rash
The 68-year-old patient said that she’d recently had a sore throat. She indicated that she felt nauseous and out of sorts.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Erysipelas
Erysipelas is an acute superficial cellulitis with lymphatic involvement. It is characterized by the abrupt onset of a warm, erythematous rash with a sharply demarcated, indurated, elevated margin. There are no suppurative foci; sometimes, however, there are bullae or vesicles.
In facial “butterfly” erysipelas (which this patient had), the plaques may involve the eyelids, cheeks, nose, and forehead. Upon palpation, the skin is hot and tender. As the process develops, the color becomes a dark, fiery red and vesicles appear at the advancing border and over the surface. Associated regional lymphadenopathy may be present. There is no necrosis.1
Most cases are caused by the Streptococcus species—usually group A (Streptococcus pyogenes)—and groups B, C, and G. (Erysipelas is occasionally caused by Staphylococcus aureus.) After prodromal symptoms that last for 4 to 48 hours and include malaise, chills, fever, anorexia, and vomiting, more red, tender firm spots develop.
Predisposing conditions for erysipelas include disruption to the skin barrier as a result of trauma, lymph stasis (prior radiation, mastectomy, saphenous vein harvest, lymphadenectomy), injection drug use, ulcers, wounds, dermatophytic infections, and edema. Toe intertrigo is perhaps the most common site for pathogen entry. The source of infection in facial erysipelas, however, is often the nasopharynx.2,3
Differential diagnosis includes cellulitis, rosacea
The differential diagnosis for erysipelas include cellulitis, rosacea, lupus erythematous “butterfly rash,” and seborrheic dermatitis.
Cellulitis is associated with skin erythema, edema, and warmth in the absence of underlying suppurative foci. Lymphangitis and inflammation of regional lymph nodes may occur. Vesicles, bullae, and ecchymoses (or petechiae) may also be present.4
Rosacea is a disorder that occurs in middle-aged and older adults, and is more common in women.5 It is characterized by symmetrical vascular dilation on the central face, including the nose, cheek, eyelids, and forehead. Facial erythema and telangiectasias—typically on the cheeks—are also present, as well as late papules and pustules.6 There are no comedones, which you would see with acne vulgaris.6 Rosacea is made worse by heat or sunlight, emotional stressors, and drinking alcohol or eating spicy foods.
Lupus erythematous “butterfly rash” is characterized by a macular confluent erythema over the cheeks and bridge of the nose with fine scaling, erosions, and crusts. It appears in approximately half of patients with systemic lupus erythematosus, usually after sun exposure.7,8
Seborrheic dermatitis is characterized by intermittent, active phases that manifest with burning, scaling, excess oil secretion, and itching. Seborrheic dermatitis usually appears over areas that are rich in sebaceous glands, such as the lateral sides of the nose and the nasolabial folds, eyebrows, glabella, and scalp.9
The exam holds the key to diagnosis
The diagnosis of erysipelas is based on clinical manifestations. Blood cultures, needle aspirations, and punch biopsies are not usually helpful. Blood cultures are positive in less than 5% of cases.4 Punch biopsy is positive in 20% to 30% of cases.4
Needle aspiration and skin biopsies may be considered for a patient with a severe infection that is not responding to treatment, or for a patient who has diabetes, a malignancy, or unusual predisposing associated factors, such as an immersion injury, an animal bite, neutropenia, or immunodeficiency.
Treatment centers on penicillin
With early diagnosis and proper treatment, the prognosis for erysipelas is excellent. Empiric antimicrobial therapy should include activity against beta-hemolytic Streptococcus. Penicillin is the treatment of choice4,10 (strength of recommendation [SOR]: A). Streptococcus strains are susceptible to penicillin and 99.5% are susceptible to clindamycin. A 7% macrolide resistance has been reported in the United States.4
Oral therapy is recommended for mild infections (or for those who have improved after intravenous [IV] antibiotics). Treat with penicillin V 500 mg PO 4 times daily or amoxicillin 500 mg PO 3 times daily. Other choices include clindamycin 300 mg PO for 7 to 10 days; azithromycin 500 mg PO for 1 day, followed by 250 mg PO daily for 4 days; or clarithromycin 250 mg PO twice a day for 7 to 10 days4,10 (SOR: A).
Consider hospitalization for severely ill patients, for those who are unable to tolerate oral medications, and for those who require parenteral antibiotic therapy for systemic symptoms and rapidly progressing erythema4 (SOR: A). Regimens include penicillin G 2 million to 4 million units IV every 4 to 6 hours; cefazolin 0.5 to 1 g IV every 8 hours; cefotaxime 1 to 2 g IV every 8 hours; and ceftriaxone 1 to 2 g IV every 24 hours.
If staphylococcal infection is suspected, a penicillinase-resistant semisynthetic penicillin or a first-generation cephalosporin, such as cefazolin, can be selected. For oral therapy, cephalexin 500 mg 4 times daily can be used4,11 (SOR: A). Macrolides (erythromycin 250 mg PO every 6 hours) are also an option4 (SOR: A). For presumed methicillin-resistant Staphylococcus aureus (MRSA), you can prescribe clindamycin 600 mg IV every 8 hours; vancomycin 15 mg/kg IV every 12 hours; or linezolid 600 mg every 12 hours. The total duration of antibiotic therapy may be extended to up to 14 days, and tailored to clinical improvement.4,10
Steroids can slightly reduce healing time and antibiotic duration in patients with erysipelas. You can prescribe prednisone 30 mg PO and taper over 8 days.4,12
Be sure to treat underlying predisposing conditions such as tinea pedis, venous eczema, and trauma sites. Encourage supportive measures, such as elevating the affected extremity, as well.
Improvement in a few days. Patients with erysipelas typically show improvement in 24 to 48 hours of beginning antibiotic therapy. Don’t be too quick to consider antibiotic failure in patients with worsening erythema after initiating antimicrobial therapy; this may be related to the destruction of pathogens that release enzymes (including deoxyribonuclease B [DNase B] and hyaluronidase) that increase local inflammation.
A good outcome for the patient
The patient was hospitalized and received cefazolin 1 g IV every 8 hours for 3 da ys. She was discharged on cephalexin 500 mg PO, every 6 hours for 7 days, with excellent results. FIGURE 2 shows the patient 4 days after she finished taking the cephalexin.
FIGURE 2
A few days, a big difference
The patient received cefazolin 1 g IV every 8 hours for 3 days, followed by oral cephalexin 500 mg every 6 hours for 7 days. This photo shows the patient 4 days after she finished taking the cephalexin.
CORRESPONDENCE Felix B. Chang, MD, Leominster Community Health Connections Family Health Center, 14 Manning Ave, Suite 402, Leominster, MA 01453; [email protected].
1. Chang F, Lopes A. Erysipelas. In: Domino FJ, ed. The 5-Minute Clinical Consult 2008. 16th ed. Philadelphia, Pa: Lippincott Williams & Wilkins; 2008:448–449.
2. Jorup-Ronstrom C. Epidemiological, bacteriological and complicating features of erysipelas. Scand J Infect Dis. 1986;18:519-524.
3. Davis L, Cole J. Erysipelas. Dermatology-bacterial infections. Available at: http://www.imedicine.com/printtopic.asp?bookid=2&topic=129. Accessed at January 11, 2010.
4. Stevens DL, Bisno AL, Chambers HF. Infectious Diseases Society of America (ISDA) practice guidelines for diagnosis and management of skin and soft tissue infections. Clin Infect Dis. 2005;41:1373-1406.
5. Wilking J, Dahl M, Detmar M, et al. Standard classification of rosacea: report of the National Rosacea Society Expert Committee on the Classification and Staging of Rosacea. J Am Acad Dermatol. 2002;46:584-587.
6. Crawford GH, Pelle MT, James WD. Rosacea: I. Etiology, pathogenesis, and subtype classification. J Am Acad Dermatol. 2004;51:327-341.
7. Patel P, Werth V. Cutaneous lupus erythematosus: a review. Dermatol Clin. 2002;20:373-385.
8. Madhok R, Wu O. Systemic lupus erythematosus. Am Fam Physician. 2007;76:1351-1353.
9. Gupta AK, Bluhm R. Seborrheic dermatitis. J Eur Acad Dermatol Venereol. 2004;18:13-26.
10. Van Beneden CA, Facklam R, Lynfield R, et al. Erythromycin resistance among invasive group A streptococcal infections, United States, 1999-2001 [abstract 345]. In: Proceedings and Abstract of the 42nd Annual Meeting of the Infectious Diseases Society of America (Boston). Alexandria, Va: Infectious Diseases Society of America; 2004:102.
11. Stulberg DL, Penrod MA, Blatny RA. Common bacterial skin infections. Am Fam Physician. 2002;66:119-124.
12. Bergkvist PI, Sjobeck K. Relapse of erysipelas following treatment with prednisolone or placebo in addition to antibiotics: a 1-year follow up. Scand J Infect Dis. 1998;30:206-207.
CORRESPONDENCE Felix B. Chang, MD, Leominster Community Health Connections Family Health Center, 14 Manning Ave, Suite 402, Leominster, MA 01453; [email protected].
1. Chang F, Lopes A. Erysipelas. In: Domino FJ, ed. The 5-Minute Clinical Consult 2008. 16th ed. Philadelphia, Pa: Lippincott Williams & Wilkins; 2008:448–449.
2. Jorup-Ronstrom C. Epidemiological, bacteriological and complicating features of erysipelas. Scand J Infect Dis. 1986;18:519-524.
3. Davis L, Cole J. Erysipelas. Dermatology-bacterial infections. Available at: http://www.imedicine.com/printtopic.asp?bookid=2&topic=129. Accessed at January 11, 2010.
4. Stevens DL, Bisno AL, Chambers HF. Infectious Diseases Society of America (ISDA) practice guidelines for diagnosis and management of skin and soft tissue infections. Clin Infect Dis. 2005;41:1373-1406.
5. Wilking J, Dahl M, Detmar M, et al. Standard classification of rosacea: report of the National Rosacea Society Expert Committee on the Classification and Staging of Rosacea. J Am Acad Dermatol. 2002;46:584-587.
6. Crawford GH, Pelle MT, James WD. Rosacea: I. Etiology, pathogenesis, and subtype classification. J Am Acad Dermatol. 2004;51:327-341.
7. Patel P, Werth V. Cutaneous lupus erythematosus: a review. Dermatol Clin. 2002;20:373-385.
8. Madhok R, Wu O. Systemic lupus erythematosus. Am Fam Physician. 2007;76:1351-1353.
9. Gupta AK, Bluhm R. Seborrheic dermatitis. J Eur Acad Dermatol Venereol. 2004;18:13-26.
10. Van Beneden CA, Facklam R, Lynfield R, et al. Erythromycin resistance among invasive group A streptococcal infections, United States, 1999-2001 [abstract 345]. In: Proceedings and Abstract of the 42nd Annual Meeting of the Infectious Diseases Society of America (Boston). Alexandria, Va: Infectious Diseases Society of America; 2004:102.
11. Stulberg DL, Penrod MA, Blatny RA. Common bacterial skin infections. Am Fam Physician. 2002;66:119-124.
12. Bergkvist PI, Sjobeck K. Relapse of erysipelas following treatment with prednisolone or placebo in addition to antibiotics: a 1-year follow up. Scand J Infect Dis. 1998;30:206-207.
CORRESPONDENCE Felix B. Chang, MD, Leominster Community Health Connections Family Health Center, 14 Manning Ave, Suite 402, Leominster, MA 01453; [email protected].
Pustular eruption on face
A 30-YEAR-OLD WOMAN came into our clinic for treatment of a facial rash. She said that she first noticed the rash (FIGURE) about 2 months earlier. Over the previous month, the eruption had worsened. Interestingly, the patient noted that she had started Bikram yoga (an intensive form of yoga performed in a room heated, in this case, to 105°F) 5 weeks prior to the onset of symptoms. She was taking the yoga classes 2 to 4 times a week and said that she experienced an exacerbation of her symptoms after each 1-hour session.
FIGURE
Yoga as a trigger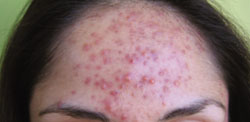
Shortly after she started taking Bikram yoga classes, this 30-year-old patient developed inflammatory papules and pustules. Her symptoms worsened after each 1-hour session. On physical exam, there were erythematous, inflammatory papules and pustules concentrated on her forehead. No comedones were present.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Rosacea
Our patient had rosacea, an inflammatory condition of the skin that typically affects the convex portions of the central face. This chronic cutaneous disorder usually starts after age 30 in both men and women, and is more prevalent in those with fairer skin.1 In fact, an epidemiologic study showed the prevalence to be as high as 10% in the Swedish population.2 The condition, which is not life threatening, can be controlled, although not cured. Its effect on appearance may have a negative impact on a patient’s quality of life.
The etiology and pathogenesis of rosacea are unknown. However, different pathogenic mechanisms have been discussed in the literature, including vasculature reactivity, dermal matrix degeneration, microbial organisms, and activities that cause flushing or blushing, such as spicy food, alcohol consumption, or emotional stressors. Rosacea flare-ups have also been linked to extremes in temperature, as was the case with our patient.
Due to the varied clinical manifestations, it is likely that genetics may also play a role in the development of rosacea.3,4
The differential. Rosacea can be confused with acne, systemic lupus erythematosus, and sarcoidosis.
A standardized approach to diagnosing rosacea
In 2002, an expert committee assembled by the National Rosacea Society established primary and secondary criteria for diagnosing rosacea.5 Diagnosis is based on the presence of 1 or more of the following signs in a central face distribution:
- flushing (transient erythema)
- persistent erythema
- papules and pustules
- telangiectasia.
Additionally, 1 or more of the following secondary features may also be present:
- burning or stinging
- elevated red inflammatory papules or plaques
- dry appearance
- edema
- ocular manifestations
- extrafacial rosacea
- phymatous changes (most commonly on the nose).
Rosacea comes in many forms
According to the expert committee assembled by the National Rosacea Society, the primary and secondary features (above) can be used to designate specific subtypes of rosacea.
Erythematotelangiectatic rosacea is generally characterized by flushing and persistent central facial erythema. However, a history of flushing alone is common among these patients.5 Flushing episodes usually last longer than 10 minutes3 and can be triggered by any vasodilating stimulus, like exercise, cold, heat, sunlight, hot beverages, or alcohol.6-8
Papulopustular rosacea is the form of rosacea that our patient had, and is known as classic rosacea or pink papular rosacea. It is characterized by persistent erythema in the central portion of the face with persistent or episodic papules and/or pustules. These inflammatory papules and pustules may also occur in the perioral, perinasal, or periocular areas.5 Edema may accompany inflammatory episodes, but is frequently subtle.9
This subtype may be confused with acne vulgaris. The key to differentiation is looking for comedones; they are present in acne vulgaris, but absent in papulopustular rosacea. However, both rosacea and acne may be present in the same patient, making diagnosis and treatment more difficult.
Phymatous rosacea usually involves the nose (rhinophyma), but can also affect the forehead, chin, cheeks, and ears. The distinct appearance of this subtype comes from enlargement, thickened skin, and irregular surface nodularities.5 Historically, rhinophyma has been associated with alcoholism, but there is no clear evidence of this association.10
Ocular rosacea affects the eyelids, conjunctiva, and cornea. Consider this diagnosis when there is 1 of more of the following findings: foreign body sensation, burning or stinging, dryness, itching, photosensitivity, blurred vision, conjunctival telangiectases, or periorbital edema.5
Corneal involvement can threaten sight, and up to 58% of rosacea patients may experience ocular manifestations.11 Therefore, it is imperative that you ask patients with rosacea if they’ve had any problems with their eyes, and that you examine the conjunctivae and eyelids.
Tx hinges on oral, topical agents—as well as avoidance
Erythematotelangiectatic and papulopustular rosacea have common therapies that include a long list of oral and topical agents. Agents that are most commonly used include oral tetracyclines, topical sodium sulfacetamide, azelaic acid, and metronidazole.12
Another approach to treatment is called the “avoidance policy,” where triggers for blushing and facial erythema are identified and then avoided. One survey by the National Rosacea Society study found that 78% of patients felt that avoiding triggers was at least somewhat effective in controlling their rosacea.13
Time for a different form of yoga?
Because our patient developed papulopustular rosacea after taking Bikram yoga classes, we advised her to avoid this particular form of exercise because of the heat. We told her that she could, however, participate in other forms of yoga, as long as they were not done in a hot environment.
Due to the severity and severe inflammatory nature of her eruption, we started the patient on oral minocycline 100 mg twice daily, and 3 topical medicines including sodium sulfacetamide/sulfur 10%/5% wash, followed by azelaic acid 20% cream every night, and metronidazole 1% gel every morning to the affected areas.
Our patient’s condition responded to treatment. The erythema on her face improved and the number of papules and pustules declined.
CORRESPONDENCE: Heather W. Wickless, MD, MPH, Durango Dermatology, 523-B South Camino del Rio, Durango, CO 81303; [email protected]
1. Gupta AK, Chaudhry MM. Rosacea and its management: an overview. J Eur Acad Dermatol Venereol. 2005;19:273-285.
2. Berg M, Liden S. An epidemiological study of rosacea. Acta Derm Venereol. 1989;69:419-423.
3. Crawford GH, Pelle MT, James WD. Rosacea: I. Etiology, pathogenesis, and subtype classification. J Am Acad Dermatol. 2004;51:327-341.
4. Diamantis S, Waldorf HA. Rosacea: clinical presentation and pathophysiology. J Drugs Dermatol. 2006;5:8-12.
5. Wilkin J, Dahl M, Detmar M, et al. Standard classification of rosacea: report of the National Rosacea Society Expert Committee on the Classification and Staging of Rosacea. J Am Acad Dermatol. 2002;46:584-587.
6. Wilkin JK. Oral thermal-induced flushing in erythematotelangiectatic rosacea. J Invest Dermatol. 1981;76:15-18.
7. Wilkin JK. Flushing reactions: consequences and mechanisms. Ann Intern Med. 1981;95:468-476.
8. Dupont C. The role of sunshine in rosacea. J Am Acad Dermatol. 1986;15:713-714.
9. Chen DM, Crosby DL. Periorbital edema as an initial presentation of rosacea. J Am Acad Dermatol. 1997;37:346-348.
10. Curnier A, Choudhary S. Rhinophyma: dispelling the myths. Plast Reconstr Surg. 2004;114:351-354.
11. Akpek EK, Merchant A, Pinar V, Foster CS. Ocular rosacea: patient characteristics and follow-up. Ophthalmology. 1997;104:1863-1867.
12. Gupta AK, Chaudhry MM. Rosacea and its management: an overview. J Eur Acad Dermatol Venereol. 2005;19:273-285.
13. National Rosacea Society. Survey finds that rosacea flare-ups are common but can be controlled. Spring 1999. Rosacea Rev. Available at: http://www.rosacea.org/rr/1999/spring/article_3.php. Accessed March 19, 2009.
A 30-YEAR-OLD WOMAN came into our clinic for treatment of a facial rash. She said that she first noticed the rash (FIGURE) about 2 months earlier. Over the previous month, the eruption had worsened. Interestingly, the patient noted that she had started Bikram yoga (an intensive form of yoga performed in a room heated, in this case, to 105°F) 5 weeks prior to the onset of symptoms. She was taking the yoga classes 2 to 4 times a week and said that she experienced an exacerbation of her symptoms after each 1-hour session.
FIGURE
Yoga as a trigger
Shortly after she started taking Bikram yoga classes, this 30-year-old patient developed inflammatory papules and pustules. Her symptoms worsened after each 1-hour session. On physical exam, there were erythematous, inflammatory papules and pustules concentrated on her forehead. No comedones were present.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Rosacea
Our patient had rosacea, an inflammatory condition of the skin that typically affects the convex portions of the central face. This chronic cutaneous disorder usually starts after age 30 in both men and women, and is more prevalent in those with fairer skin.1 In fact, an epidemiologic study showed the prevalence to be as high as 10% in the Swedish population.2 The condition, which is not life threatening, can be controlled, although not cured. Its effect on appearance may have a negative impact on a patient’s quality of life.
The etiology and pathogenesis of rosacea are unknown. However, different pathogenic mechanisms have been discussed in the literature, including vasculature reactivity, dermal matrix degeneration, microbial organisms, and activities that cause flushing or blushing, such as spicy food, alcohol consumption, or emotional stressors. Rosacea flare-ups have also been linked to extremes in temperature, as was the case with our patient.
Due to the varied clinical manifestations, it is likely that genetics may also play a role in the development of rosacea.3,4
The differential. Rosacea can be confused with acne, systemic lupus erythematosus, and sarcoidosis.
A standardized approach to diagnosing rosacea
In 2002, an expert committee assembled by the National Rosacea Society established primary and secondary criteria for diagnosing rosacea.5 Diagnosis is based on the presence of 1 or more of the following signs in a central face distribution:
- flushing (transient erythema)
- persistent erythema
- papules and pustules
- telangiectasia.
Additionally, 1 or more of the following secondary features may also be present:
- burning or stinging
- elevated red inflammatory papules or plaques
- dry appearance
- edema
- ocular manifestations
- extrafacial rosacea
- phymatous changes (most commonly on the nose).
Rosacea comes in many forms
According to the expert committee assembled by the National Rosacea Society, the primary and secondary features (above) can be used to designate specific subtypes of rosacea.
Erythematotelangiectatic rosacea is generally characterized by flushing and persistent central facial erythema. However, a history of flushing alone is common among these patients.5 Flushing episodes usually last longer than 10 minutes3 and can be triggered by any vasodilating stimulus, like exercise, cold, heat, sunlight, hot beverages, or alcohol.6-8
Papulopustular rosacea is the form of rosacea that our patient had, and is known as classic rosacea or pink papular rosacea. It is characterized by persistent erythema in the central portion of the face with persistent or episodic papules and/or pustules. These inflammatory papules and pustules may also occur in the perioral, perinasal, or periocular areas.5 Edema may accompany inflammatory episodes, but is frequently subtle.9
This subtype may be confused with acne vulgaris. The key to differentiation is looking for comedones; they are present in acne vulgaris, but absent in papulopustular rosacea. However, both rosacea and acne may be present in the same patient, making diagnosis and treatment more difficult.
Phymatous rosacea usually involves the nose (rhinophyma), but can also affect the forehead, chin, cheeks, and ears. The distinct appearance of this subtype comes from enlargement, thickened skin, and irregular surface nodularities.5 Historically, rhinophyma has been associated with alcoholism, but there is no clear evidence of this association.10
Ocular rosacea affects the eyelids, conjunctiva, and cornea. Consider this diagnosis when there is 1 of more of the following findings: foreign body sensation, burning or stinging, dryness, itching, photosensitivity, blurred vision, conjunctival telangiectases, or periorbital edema.5
Corneal involvement can threaten sight, and up to 58% of rosacea patients may experience ocular manifestations.11 Therefore, it is imperative that you ask patients with rosacea if they’ve had any problems with their eyes, and that you examine the conjunctivae and eyelids.
Tx hinges on oral, topical agents—as well as avoidance
Erythematotelangiectatic and papulopustular rosacea have common therapies that include a long list of oral and topical agents. Agents that are most commonly used include oral tetracyclines, topical sodium sulfacetamide, azelaic acid, and metronidazole.12
Another approach to treatment is called the “avoidance policy,” where triggers for blushing and facial erythema are identified and then avoided. One survey by the National Rosacea Society study found that 78% of patients felt that avoiding triggers was at least somewhat effective in controlling their rosacea.13
Time for a different form of yoga?
Because our patient developed papulopustular rosacea after taking Bikram yoga classes, we advised her to avoid this particular form of exercise because of the heat. We told her that she could, however, participate in other forms of yoga, as long as they were not done in a hot environment.
Due to the severity and severe inflammatory nature of her eruption, we started the patient on oral minocycline 100 mg twice daily, and 3 topical medicines including sodium sulfacetamide/sulfur 10%/5% wash, followed by azelaic acid 20% cream every night, and metronidazole 1% gel every morning to the affected areas.
Our patient’s condition responded to treatment. The erythema on her face improved and the number of papules and pustules declined.
CORRESPONDENCE: Heather W. Wickless, MD, MPH, Durango Dermatology, 523-B South Camino del Rio, Durango, CO 81303; [email protected]
A 30-YEAR-OLD WOMAN came into our clinic for treatment of a facial rash. She said that she first noticed the rash (FIGURE) about 2 months earlier. Over the previous month, the eruption had worsened. Interestingly, the patient noted that she had started Bikram yoga (an intensive form of yoga performed in a room heated, in this case, to 105°F) 5 weeks prior to the onset of symptoms. She was taking the yoga classes 2 to 4 times a week and said that she experienced an exacerbation of her symptoms after each 1-hour session.
FIGURE
Yoga as a trigger
Shortly after she started taking Bikram yoga classes, this 30-year-old patient developed inflammatory papules and pustules. Her symptoms worsened after each 1-hour session. On physical exam, there were erythematous, inflammatory papules and pustules concentrated on her forehead. No comedones were present.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Rosacea
Our patient had rosacea, an inflammatory condition of the skin that typically affects the convex portions of the central face. This chronic cutaneous disorder usually starts after age 30 in both men and women, and is more prevalent in those with fairer skin.1 In fact, an epidemiologic study showed the prevalence to be as high as 10% in the Swedish population.2 The condition, which is not life threatening, can be controlled, although not cured. Its effect on appearance may have a negative impact on a patient’s quality of life.
The etiology and pathogenesis of rosacea are unknown. However, different pathogenic mechanisms have been discussed in the literature, including vasculature reactivity, dermal matrix degeneration, microbial organisms, and activities that cause flushing or blushing, such as spicy food, alcohol consumption, or emotional stressors. Rosacea flare-ups have also been linked to extremes in temperature, as was the case with our patient.
Due to the varied clinical manifestations, it is likely that genetics may also play a role in the development of rosacea.3,4
The differential. Rosacea can be confused with acne, systemic lupus erythematosus, and sarcoidosis.
A standardized approach to diagnosing rosacea
In 2002, an expert committee assembled by the National Rosacea Society established primary and secondary criteria for diagnosing rosacea.5 Diagnosis is based on the presence of 1 or more of the following signs in a central face distribution:
- flushing (transient erythema)
- persistent erythema
- papules and pustules
- telangiectasia.
Additionally, 1 or more of the following secondary features may also be present:
- burning or stinging
- elevated red inflammatory papules or plaques
- dry appearance
- edema
- ocular manifestations
- extrafacial rosacea
- phymatous changes (most commonly on the nose).
Rosacea comes in many forms
According to the expert committee assembled by the National Rosacea Society, the primary and secondary features (above) can be used to designate specific subtypes of rosacea.
Erythematotelangiectatic rosacea is generally characterized by flushing and persistent central facial erythema. However, a history of flushing alone is common among these patients.5 Flushing episodes usually last longer than 10 minutes3 and can be triggered by any vasodilating stimulus, like exercise, cold, heat, sunlight, hot beverages, or alcohol.6-8
Papulopustular rosacea is the form of rosacea that our patient had, and is known as classic rosacea or pink papular rosacea. It is characterized by persistent erythema in the central portion of the face with persistent or episodic papules and/or pustules. These inflammatory papules and pustules may also occur in the perioral, perinasal, or periocular areas.5 Edema may accompany inflammatory episodes, but is frequently subtle.9
This subtype may be confused with acne vulgaris. The key to differentiation is looking for comedones; they are present in acne vulgaris, but absent in papulopustular rosacea. However, both rosacea and acne may be present in the same patient, making diagnosis and treatment more difficult.
Phymatous rosacea usually involves the nose (rhinophyma), but can also affect the forehead, chin, cheeks, and ears. The distinct appearance of this subtype comes from enlargement, thickened skin, and irregular surface nodularities.5 Historically, rhinophyma has been associated with alcoholism, but there is no clear evidence of this association.10
Ocular rosacea affects the eyelids, conjunctiva, and cornea. Consider this diagnosis when there is 1 of more of the following findings: foreign body sensation, burning or stinging, dryness, itching, photosensitivity, blurred vision, conjunctival telangiectases, or periorbital edema.5
Corneal involvement can threaten sight, and up to 58% of rosacea patients may experience ocular manifestations.11 Therefore, it is imperative that you ask patients with rosacea if they’ve had any problems with their eyes, and that you examine the conjunctivae and eyelids.
Tx hinges on oral, topical agents—as well as avoidance
Erythematotelangiectatic and papulopustular rosacea have common therapies that include a long list of oral and topical agents. Agents that are most commonly used include oral tetracyclines, topical sodium sulfacetamide, azelaic acid, and metronidazole.12
Another approach to treatment is called the “avoidance policy,” where triggers for blushing and facial erythema are identified and then avoided. One survey by the National Rosacea Society study found that 78% of patients felt that avoiding triggers was at least somewhat effective in controlling their rosacea.13
Time for a different form of yoga?
Because our patient developed papulopustular rosacea after taking Bikram yoga classes, we advised her to avoid this particular form of exercise because of the heat. We told her that she could, however, participate in other forms of yoga, as long as they were not done in a hot environment.
Due to the severity and severe inflammatory nature of her eruption, we started the patient on oral minocycline 100 mg twice daily, and 3 topical medicines including sodium sulfacetamide/sulfur 10%/5% wash, followed by azelaic acid 20% cream every night, and metronidazole 1% gel every morning to the affected areas.
Our patient’s condition responded to treatment. The erythema on her face improved and the number of papules and pustules declined.
CORRESPONDENCE: Heather W. Wickless, MD, MPH, Durango Dermatology, 523-B South Camino del Rio, Durango, CO 81303; [email protected]
1. Gupta AK, Chaudhry MM. Rosacea and its management: an overview. J Eur Acad Dermatol Venereol. 2005;19:273-285.
2. Berg M, Liden S. An epidemiological study of rosacea. Acta Derm Venereol. 1989;69:419-423.
3. Crawford GH, Pelle MT, James WD. Rosacea: I. Etiology, pathogenesis, and subtype classification. J Am Acad Dermatol. 2004;51:327-341.
4. Diamantis S, Waldorf HA. Rosacea: clinical presentation and pathophysiology. J Drugs Dermatol. 2006;5:8-12.
5. Wilkin J, Dahl M, Detmar M, et al. Standard classification of rosacea: report of the National Rosacea Society Expert Committee on the Classification and Staging of Rosacea. J Am Acad Dermatol. 2002;46:584-587.
6. Wilkin JK. Oral thermal-induced flushing in erythematotelangiectatic rosacea. J Invest Dermatol. 1981;76:15-18.
7. Wilkin JK. Flushing reactions: consequences and mechanisms. Ann Intern Med. 1981;95:468-476.
8. Dupont C. The role of sunshine in rosacea. J Am Acad Dermatol. 1986;15:713-714.
9. Chen DM, Crosby DL. Periorbital edema as an initial presentation of rosacea. J Am Acad Dermatol. 1997;37:346-348.
10. Curnier A, Choudhary S. Rhinophyma: dispelling the myths. Plast Reconstr Surg. 2004;114:351-354.
11. Akpek EK, Merchant A, Pinar V, Foster CS. Ocular rosacea: patient characteristics and follow-up. Ophthalmology. 1997;104:1863-1867.
12. Gupta AK, Chaudhry MM. Rosacea and its management: an overview. J Eur Acad Dermatol Venereol. 2005;19:273-285.
13. National Rosacea Society. Survey finds that rosacea flare-ups are common but can be controlled. Spring 1999. Rosacea Rev. Available at: http://www.rosacea.org/rr/1999/spring/article_3.php. Accessed March 19, 2009.
1. Gupta AK, Chaudhry MM. Rosacea and its management: an overview. J Eur Acad Dermatol Venereol. 2005;19:273-285.
2. Berg M, Liden S. An epidemiological study of rosacea. Acta Derm Venereol. 1989;69:419-423.
3. Crawford GH, Pelle MT, James WD. Rosacea: I. Etiology, pathogenesis, and subtype classification. J Am Acad Dermatol. 2004;51:327-341.
4. Diamantis S, Waldorf HA. Rosacea: clinical presentation and pathophysiology. J Drugs Dermatol. 2006;5:8-12.
5. Wilkin J, Dahl M, Detmar M, et al. Standard classification of rosacea: report of the National Rosacea Society Expert Committee on the Classification and Staging of Rosacea. J Am Acad Dermatol. 2002;46:584-587.
6. Wilkin JK. Oral thermal-induced flushing in erythematotelangiectatic rosacea. J Invest Dermatol. 1981;76:15-18.
7. Wilkin JK. Flushing reactions: consequences and mechanisms. Ann Intern Med. 1981;95:468-476.
8. Dupont C. The role of sunshine in rosacea. J Am Acad Dermatol. 1986;15:713-714.
9. Chen DM, Crosby DL. Periorbital edema as an initial presentation of rosacea. J Am Acad Dermatol. 1997;37:346-348.
10. Curnier A, Choudhary S. Rhinophyma: dispelling the myths. Plast Reconstr Surg. 2004;114:351-354.
11. Akpek EK, Merchant A, Pinar V, Foster CS. Ocular rosacea: patient characteristics and follow-up. Ophthalmology. 1997;104:1863-1867.
12. Gupta AK, Chaudhry MM. Rosacea and its management: an overview. J Eur Acad Dermatol Venereol. 2005;19:273-285.
13. National Rosacea Society. Survey finds that rosacea flare-ups are common but can be controlled. Spring 1999. Rosacea Rev. Available at: http://www.rosacea.org/rr/1999/spring/article_3.php. Accessed March 19, 2009.
A mediastinal mass
A 79-YEAR-OLD MAN came to our emergency department and asked that we check on a mediastinal mass that was first detected on a routine chest film 3 years earlier. The patient was asymptomatic; his medical history was unremarkable except for vitamin B12 deficiency. Physical examination revealed no abnormalities except for minor ataxia that was attributed to the lack of sufficient vitamin B12. A chest radiograph (FIGURE 1A) revealed a mass located in the upper right thorax with a slight deviation of the trachea to the left, consistent with previous x-ray findings. A computed tomography (CT) scan of the thorax (FIGURE 1B) showed a heterogeneous, multinodular mass in the anterior mediastinum with a maximal longitudinal diameter of 13.5 cm and a diagonal diameter of 9 cm. There was no obstruction or invasion of the trachea, esophagus, or mediastinal vessels.
FIGURE 1
X-ray and CT point to a diagnosis
The patient’s x-ray (A) revealed a mass in the upper right thorax with slight deviation of the trachea to the left (arrow). A CT scan of the thorax (B) revealed a heterogeneous, multinodular mass in the anterior mediastinum (asterisk). The mass did not obstruct or invade the trachea, esophagus, or mediastinal vessels.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU MANAGE THIS CONDITION?
Diagnosis: Retrosternal goiter
A retrosternal goiter was the most plausible diagnosis because of the patient’s advanced age, the asymptomatic behavior of the tumor, and its development over several years. The multilobular heterogenous appearance in the CT scan supported this diagnosis and made other disorders, such as thymoma and non-Hodgkin’s disease (to be discussed in a bit) very unlikely. In addition, the noninfiltrative behavior of the tumor suggested a benign mass, and CT images obtained from the neck (not shown) demonstrated that the mass originated from the right thyroid lobe without infiltrative or obstructive growth.
Benign mediastinal goiters are relatively common in adults, and the incidence increases with age.1,2 When a goiter affects adjacent structures or obstructs the trachea, common symptoms include dyspnea, wheezing, coughing, jugular vein compression, dysphagia, vocal cord palsy, phrenic nerve palsy, and Horner’s syndrome.
Differential diagnosis includes thymomas and thymic carcinomas
Masses in the anterior mediastinum comprise a variety of pathologic entities and are more likely to be malignant than masses in other mediastinal compartments.3 The most common lesions are thymomas and thymic carcinomas, lymphomas, germ cell tumors, intrathoracic thyroid or parathyroid lesions, and metastases. Up to 50% of mediastinal tumors are detected incidentally, and cause few—if any—symptoms.4,5
Thymoma and thymic carcinomas represent approximately 20% of the cases of primary anterior mediastinal masses, and can be invasive. In addition, myasthenia gravis or tumor-related syndromes are very suggestive of thymoma.6 CT or magnetic resonance imaging (MRI) studies may help to differentiate the mass in the anterior mediastinum, but histopathological analysis is required to establish a definitive diagnosis.
Lymphomas account for 10% to 20% of primary anterior mediastinal masses. While Hodgkin’s lymphoma predominantly occurs in the third and fourth decade, non-Hodgkin’s disease may affect people at any age.7,8 Most patients are symptomatic at the time of diagnosis, complaining of fever, weight loss, and night sweats. Diagnosis is suggested by a CT scan showing a lobulated mass, but confirmatory histology is necessary to guide treatment and may be accomplished by needle biopsy or lymph node excision on a different body site.
Germ cell tumors represent 15% of anterior mediastinal masses, with an even higher share in children, and include benign teratoma, seminomas, and nonseminomatous germ cell tumors.9 CT and MRI studies may help to clarify the relationship of the tumor tissue to surrounding structures. While teratomas are most likely to occur in children and young adults (and contain various amounts of tissue derived from the ectoderm, endoderm, and mesoderm), seminomas occur predominantly in young men between 20 and 40 years of age (and often have cellular heterogeneity and contain elements of embryonal malignancy).10,11 Seminomas are the most common malignant germ cell tumors, but need to be distinguished from nonseminomatous germ cell tumors; serum levels for alpha-fetoprotein and beta-human chorionic gonadotropin may help, but fine needle aspiration cytology is definitely needed.
Mediastinal cysts are common benign lesions of the mediastinum of pericardial, enteric, bronchogenic, or thymic origin, and are easily detected by CT or MRI studies. In addition, growth of parathyroid tissue can result in a retrosternal tumor, but such tumors are rarely of significant size.
Diagnosis hinges primarily on the CT scan
CT imaging is the method of choice to determine whether a mediastinal mass is thyroidal in origin and to define the extent of substernal goiter and the potential impingement on adjacent structures.12 These scans drive the need for and choice of therapy.13 An MRI of the thyroid gland may be helpful in distinguishing the thyroid from other tissue; ultrasonography is more accurate for defining thyroid anatomy in the anterior neck.
In the case of our patient, we used thyroid ultrasonography to confirm the CT findings. Further diagnostic tests included measurements of serum thyroid-stimulating hormone, triiodothyronine, and thyroxine, showing euthyroidism. Thyroid radionuclide imaging with 123-iodine was not performed in this case. While it can identify a substernal tumor as being thyroid tissue, the method can also be misleading because of impaired radioiodine uptake in some substernal goiters.14
Given our patient’s lack of pain, tenderness, or firmness in the cervical goiter and the results of his imaging studies, a fine-needle aspiration biopsy was not performed to rule out a malignant disorder like thyroid cancer.
Treatment ranges from watch and wait to surgery
Treatment of asymptomatic retrosternal goiter remains controversial. Levothyroxine suppression has a limited role in reducing the size or stopping the growth of the thyroid. Surgery is the method of choice in patients with obstructive symptoms, given the risk of progressive tracheal compression.
In the present case, we decided to watch and wait because of the patient’s age, history, and lack of any signs of obstruction of the trachea, esophagus, or mediastinal vessels. We recommended that the patient come back for a follow-up visit in 6 to 12 months.
CORRESPONDENCE: Christian S. Haas, MD, University Hospital Schleswig-Holstein–Campus Luebeck, Department of Medicine I, Ratzeburger Allee 160, 23538 Luebeck, Germany; [email protected]
1. Singh B, Lucente FE, Shaha AR. Substernal goiter: a clinical review. Am J Otolaryngol. 1994;15:409-416.
2. Strollo DC, Rosado de Christenson ML, Jett JR. Primary mediastinal tumors. Part 1: tumors of the anterior mediastinum. Chest. 1997;112:511-522.
3. Hoffman OA, Gillespie DJ, Aughenbaugh GL, et al. Primary mediastinal neoplasms (other than thymoma). Mayo Clin Proc. 1993;68:880-891.
4. Davis RD, Jr, Oldham HN, Jr, Sabiston DC, Jr. Primary cysts and neoplasms of the mediastinum: recent changes in clinical presentation, methods of diagnosis, management, and results. Ann Thorac Surg. 1987;44:229-237.
5. den Bakker MA, Oosterhuis JW. Tumours and tumour-like conditions of the thymus other than thymoma; a practical approach. Histopathology. 2009;54:69-89.
6. Gerein AN, Srivastava SP, Burgess J. Thymoma: a ten year review. Am J Surg. 1978;136:49-53.
7. van Heerden JA, Harison EG, Jr, Bernatz PE, et al. Mediastinal malignant lymphoma. Chest. 1970;57:518-529.
8. Hoppe RT. The non-Hodgkin’s lymphomas: pathology, staging, treatment. Curr Probl Cancer. 1987;11:363-447.
9. Luna MA, Valenquela-Tamariz J. Germ cell tumors of the mediastinum: postmortem findings. Am J Clin Pathol. 1976;65:450-454.
10. Moran CA, Suster S, Koss MN. Primary germ cell tumors of the mediastinum: III. Yolk sac tumor, embryonal carcinoma, choriocarcinoma, and combined nonteratomatous germ cell tumors of the mediastinum—a clinicopathologic and immunohistochemical study of 64 cases. Cancer. 1997;80:699-707.
11. Polansky SM, Barwick KW, Ravin CE. Primary mediastinal seminoma. AJR Am J Roentgenol. 1979;132:17-21.
12. Jennings A. Evaluation of substernal goiters using computed tomography and MR imaging. Endocrinol Metab Clin North Am. 2001;30:401-414, ix.
13. Wright CD, Mathisen DJ. Mediastinal tumors: diagnosis and treatment. World J Surg. 2001;25:204-209.
14. Park HM, Tarver RD, Siddiqui AR, et al. Efficacy of thyroid scintigraphy in the diagnosis of intrathoracic goiter. AJR Am J Roentgenol. 1987;148:527-529.
A 79-YEAR-OLD MAN came to our emergency department and asked that we check on a mediastinal mass that was first detected on a routine chest film 3 years earlier. The patient was asymptomatic; his medical history was unremarkable except for vitamin B12 deficiency. Physical examination revealed no abnormalities except for minor ataxia that was attributed to the lack of sufficient vitamin B12. A chest radiograph (FIGURE 1A) revealed a mass located in the upper right thorax with a slight deviation of the trachea to the left, consistent with previous x-ray findings. A computed tomography (CT) scan of the thorax (FIGURE 1B) showed a heterogeneous, multinodular mass in the anterior mediastinum with a maximal longitudinal diameter of 13.5 cm and a diagonal diameter of 9 cm. There was no obstruction or invasion of the trachea, esophagus, or mediastinal vessels.
FIGURE 1
X-ray and CT point to a diagnosis
The patient’s x-ray (A) revealed a mass in the upper right thorax with slight deviation of the trachea to the left (arrow). A CT scan of the thorax (B) revealed a heterogeneous, multinodular mass in the anterior mediastinum (asterisk). The mass did not obstruct or invade the trachea, esophagus, or mediastinal vessels.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU MANAGE THIS CONDITION?
Diagnosis: Retrosternal goiter
A retrosternal goiter was the most plausible diagnosis because of the patient’s advanced age, the asymptomatic behavior of the tumor, and its development over several years. The multilobular heterogenous appearance in the CT scan supported this diagnosis and made other disorders, such as thymoma and non-Hodgkin’s disease (to be discussed in a bit) very unlikely. In addition, the noninfiltrative behavior of the tumor suggested a benign mass, and CT images obtained from the neck (not shown) demonstrated that the mass originated from the right thyroid lobe without infiltrative or obstructive growth.
Benign mediastinal goiters are relatively common in adults, and the incidence increases with age.1,2 When a goiter affects adjacent structures or obstructs the trachea, common symptoms include dyspnea, wheezing, coughing, jugular vein compression, dysphagia, vocal cord palsy, phrenic nerve palsy, and Horner’s syndrome.
Differential diagnosis includes thymomas and thymic carcinomas
Masses in the anterior mediastinum comprise a variety of pathologic entities and are more likely to be malignant than masses in other mediastinal compartments.3 The most common lesions are thymomas and thymic carcinomas, lymphomas, germ cell tumors, intrathoracic thyroid or parathyroid lesions, and metastases. Up to 50% of mediastinal tumors are detected incidentally, and cause few—if any—symptoms.4,5
Thymoma and thymic carcinomas represent approximately 20% of the cases of primary anterior mediastinal masses, and can be invasive. In addition, myasthenia gravis or tumor-related syndromes are very suggestive of thymoma.6 CT or magnetic resonance imaging (MRI) studies may help to differentiate the mass in the anterior mediastinum, but histopathological analysis is required to establish a definitive diagnosis.
Lymphomas account for 10% to 20% of primary anterior mediastinal masses. While Hodgkin’s lymphoma predominantly occurs in the third and fourth decade, non-Hodgkin’s disease may affect people at any age.7,8 Most patients are symptomatic at the time of diagnosis, complaining of fever, weight loss, and night sweats. Diagnosis is suggested by a CT scan showing a lobulated mass, but confirmatory histology is necessary to guide treatment and may be accomplished by needle biopsy or lymph node excision on a different body site.
Germ cell tumors represent 15% of anterior mediastinal masses, with an even higher share in children, and include benign teratoma, seminomas, and nonseminomatous germ cell tumors.9 CT and MRI studies may help to clarify the relationship of the tumor tissue to surrounding structures. While teratomas are most likely to occur in children and young adults (and contain various amounts of tissue derived from the ectoderm, endoderm, and mesoderm), seminomas occur predominantly in young men between 20 and 40 years of age (and often have cellular heterogeneity and contain elements of embryonal malignancy).10,11 Seminomas are the most common malignant germ cell tumors, but need to be distinguished from nonseminomatous germ cell tumors; serum levels for alpha-fetoprotein and beta-human chorionic gonadotropin may help, but fine needle aspiration cytology is definitely needed.
Mediastinal cysts are common benign lesions of the mediastinum of pericardial, enteric, bronchogenic, or thymic origin, and are easily detected by CT or MRI studies. In addition, growth of parathyroid tissue can result in a retrosternal tumor, but such tumors are rarely of significant size.
Diagnosis hinges primarily on the CT scan
CT imaging is the method of choice to determine whether a mediastinal mass is thyroidal in origin and to define the extent of substernal goiter and the potential impingement on adjacent structures.12 These scans drive the need for and choice of therapy.13 An MRI of the thyroid gland may be helpful in distinguishing the thyroid from other tissue; ultrasonography is more accurate for defining thyroid anatomy in the anterior neck.
In the case of our patient, we used thyroid ultrasonography to confirm the CT findings. Further diagnostic tests included measurements of serum thyroid-stimulating hormone, triiodothyronine, and thyroxine, showing euthyroidism. Thyroid radionuclide imaging with 123-iodine was not performed in this case. While it can identify a substernal tumor as being thyroid tissue, the method can also be misleading because of impaired radioiodine uptake in some substernal goiters.14
Given our patient’s lack of pain, tenderness, or firmness in the cervical goiter and the results of his imaging studies, a fine-needle aspiration biopsy was not performed to rule out a malignant disorder like thyroid cancer.
Treatment ranges from watch and wait to surgery
Treatment of asymptomatic retrosternal goiter remains controversial. Levothyroxine suppression has a limited role in reducing the size or stopping the growth of the thyroid. Surgery is the method of choice in patients with obstructive symptoms, given the risk of progressive tracheal compression.
In the present case, we decided to watch and wait because of the patient’s age, history, and lack of any signs of obstruction of the trachea, esophagus, or mediastinal vessels. We recommended that the patient come back for a follow-up visit in 6 to 12 months.
CORRESPONDENCE: Christian S. Haas, MD, University Hospital Schleswig-Holstein–Campus Luebeck, Department of Medicine I, Ratzeburger Allee 160, 23538 Luebeck, Germany; [email protected]
A 79-YEAR-OLD MAN came to our emergency department and asked that we check on a mediastinal mass that was first detected on a routine chest film 3 years earlier. The patient was asymptomatic; his medical history was unremarkable except for vitamin B12 deficiency. Physical examination revealed no abnormalities except for minor ataxia that was attributed to the lack of sufficient vitamin B12. A chest radiograph (FIGURE 1A) revealed a mass located in the upper right thorax with a slight deviation of the trachea to the left, consistent with previous x-ray findings. A computed tomography (CT) scan of the thorax (FIGURE 1B) showed a heterogeneous, multinodular mass in the anterior mediastinum with a maximal longitudinal diameter of 13.5 cm and a diagonal diameter of 9 cm. There was no obstruction or invasion of the trachea, esophagus, or mediastinal vessels.
FIGURE 1
X-ray and CT point to a diagnosis
The patient’s x-ray (A) revealed a mass in the upper right thorax with slight deviation of the trachea to the left (arrow). A CT scan of the thorax (B) revealed a heterogeneous, multinodular mass in the anterior mediastinum (asterisk). The mass did not obstruct or invade the trachea, esophagus, or mediastinal vessels.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU MANAGE THIS CONDITION?
Diagnosis: Retrosternal goiter
A retrosternal goiter was the most plausible diagnosis because of the patient’s advanced age, the asymptomatic behavior of the tumor, and its development over several years. The multilobular heterogenous appearance in the CT scan supported this diagnosis and made other disorders, such as thymoma and non-Hodgkin’s disease (to be discussed in a bit) very unlikely. In addition, the noninfiltrative behavior of the tumor suggested a benign mass, and CT images obtained from the neck (not shown) demonstrated that the mass originated from the right thyroid lobe without infiltrative or obstructive growth.
Benign mediastinal goiters are relatively common in adults, and the incidence increases with age.1,2 When a goiter affects adjacent structures or obstructs the trachea, common symptoms include dyspnea, wheezing, coughing, jugular vein compression, dysphagia, vocal cord palsy, phrenic nerve palsy, and Horner’s syndrome.
Differential diagnosis includes thymomas and thymic carcinomas
Masses in the anterior mediastinum comprise a variety of pathologic entities and are more likely to be malignant than masses in other mediastinal compartments.3 The most common lesions are thymomas and thymic carcinomas, lymphomas, germ cell tumors, intrathoracic thyroid or parathyroid lesions, and metastases. Up to 50% of mediastinal tumors are detected incidentally, and cause few—if any—symptoms.4,5
Thymoma and thymic carcinomas represent approximately 20% of the cases of primary anterior mediastinal masses, and can be invasive. In addition, myasthenia gravis or tumor-related syndromes are very suggestive of thymoma.6 CT or magnetic resonance imaging (MRI) studies may help to differentiate the mass in the anterior mediastinum, but histopathological analysis is required to establish a definitive diagnosis.
Lymphomas account for 10% to 20% of primary anterior mediastinal masses. While Hodgkin’s lymphoma predominantly occurs in the third and fourth decade, non-Hodgkin’s disease may affect people at any age.7,8 Most patients are symptomatic at the time of diagnosis, complaining of fever, weight loss, and night sweats. Diagnosis is suggested by a CT scan showing a lobulated mass, but confirmatory histology is necessary to guide treatment and may be accomplished by needle biopsy or lymph node excision on a different body site.
Germ cell tumors represent 15% of anterior mediastinal masses, with an even higher share in children, and include benign teratoma, seminomas, and nonseminomatous germ cell tumors.9 CT and MRI studies may help to clarify the relationship of the tumor tissue to surrounding structures. While teratomas are most likely to occur in children and young adults (and contain various amounts of tissue derived from the ectoderm, endoderm, and mesoderm), seminomas occur predominantly in young men between 20 and 40 years of age (and often have cellular heterogeneity and contain elements of embryonal malignancy).10,11 Seminomas are the most common malignant germ cell tumors, but need to be distinguished from nonseminomatous germ cell tumors; serum levels for alpha-fetoprotein and beta-human chorionic gonadotropin may help, but fine needle aspiration cytology is definitely needed.
Mediastinal cysts are common benign lesions of the mediastinum of pericardial, enteric, bronchogenic, or thymic origin, and are easily detected by CT or MRI studies. In addition, growth of parathyroid tissue can result in a retrosternal tumor, but such tumors are rarely of significant size.
Diagnosis hinges primarily on the CT scan
CT imaging is the method of choice to determine whether a mediastinal mass is thyroidal in origin and to define the extent of substernal goiter and the potential impingement on adjacent structures.12 These scans drive the need for and choice of therapy.13 An MRI of the thyroid gland may be helpful in distinguishing the thyroid from other tissue; ultrasonography is more accurate for defining thyroid anatomy in the anterior neck.
In the case of our patient, we used thyroid ultrasonography to confirm the CT findings. Further diagnostic tests included measurements of serum thyroid-stimulating hormone, triiodothyronine, and thyroxine, showing euthyroidism. Thyroid radionuclide imaging with 123-iodine was not performed in this case. While it can identify a substernal tumor as being thyroid tissue, the method can also be misleading because of impaired radioiodine uptake in some substernal goiters.14
Given our patient’s lack of pain, tenderness, or firmness in the cervical goiter and the results of his imaging studies, a fine-needle aspiration biopsy was not performed to rule out a malignant disorder like thyroid cancer.
Treatment ranges from watch and wait to surgery
Treatment of asymptomatic retrosternal goiter remains controversial. Levothyroxine suppression has a limited role in reducing the size or stopping the growth of the thyroid. Surgery is the method of choice in patients with obstructive symptoms, given the risk of progressive tracheal compression.
In the present case, we decided to watch and wait because of the patient’s age, history, and lack of any signs of obstruction of the trachea, esophagus, or mediastinal vessels. We recommended that the patient come back for a follow-up visit in 6 to 12 months.
CORRESPONDENCE: Christian S. Haas, MD, University Hospital Schleswig-Holstein–Campus Luebeck, Department of Medicine I, Ratzeburger Allee 160, 23538 Luebeck, Germany; [email protected]
1. Singh B, Lucente FE, Shaha AR. Substernal goiter: a clinical review. Am J Otolaryngol. 1994;15:409-416.
2. Strollo DC, Rosado de Christenson ML, Jett JR. Primary mediastinal tumors. Part 1: tumors of the anterior mediastinum. Chest. 1997;112:511-522.
3. Hoffman OA, Gillespie DJ, Aughenbaugh GL, et al. Primary mediastinal neoplasms (other than thymoma). Mayo Clin Proc. 1993;68:880-891.
4. Davis RD, Jr, Oldham HN, Jr, Sabiston DC, Jr. Primary cysts and neoplasms of the mediastinum: recent changes in clinical presentation, methods of diagnosis, management, and results. Ann Thorac Surg. 1987;44:229-237.
5. den Bakker MA, Oosterhuis JW. Tumours and tumour-like conditions of the thymus other than thymoma; a practical approach. Histopathology. 2009;54:69-89.
6. Gerein AN, Srivastava SP, Burgess J. Thymoma: a ten year review. Am J Surg. 1978;136:49-53.
7. van Heerden JA, Harison EG, Jr, Bernatz PE, et al. Mediastinal malignant lymphoma. Chest. 1970;57:518-529.
8. Hoppe RT. The non-Hodgkin’s lymphomas: pathology, staging, treatment. Curr Probl Cancer. 1987;11:363-447.
9. Luna MA, Valenquela-Tamariz J. Germ cell tumors of the mediastinum: postmortem findings. Am J Clin Pathol. 1976;65:450-454.
10. Moran CA, Suster S, Koss MN. Primary germ cell tumors of the mediastinum: III. Yolk sac tumor, embryonal carcinoma, choriocarcinoma, and combined nonteratomatous germ cell tumors of the mediastinum—a clinicopathologic and immunohistochemical study of 64 cases. Cancer. 1997;80:699-707.
11. Polansky SM, Barwick KW, Ravin CE. Primary mediastinal seminoma. AJR Am J Roentgenol. 1979;132:17-21.
12. Jennings A. Evaluation of substernal goiters using computed tomography and MR imaging. Endocrinol Metab Clin North Am. 2001;30:401-414, ix.
13. Wright CD, Mathisen DJ. Mediastinal tumors: diagnosis and treatment. World J Surg. 2001;25:204-209.
14. Park HM, Tarver RD, Siddiqui AR, et al. Efficacy of thyroid scintigraphy in the diagnosis of intrathoracic goiter. AJR Am J Roentgenol. 1987;148:527-529.
1. Singh B, Lucente FE, Shaha AR. Substernal goiter: a clinical review. Am J Otolaryngol. 1994;15:409-416.
2. Strollo DC, Rosado de Christenson ML, Jett JR. Primary mediastinal tumors. Part 1: tumors of the anterior mediastinum. Chest. 1997;112:511-522.
3. Hoffman OA, Gillespie DJ, Aughenbaugh GL, et al. Primary mediastinal neoplasms (other than thymoma). Mayo Clin Proc. 1993;68:880-891.
4. Davis RD, Jr, Oldham HN, Jr, Sabiston DC, Jr. Primary cysts and neoplasms of the mediastinum: recent changes in clinical presentation, methods of diagnosis, management, and results. Ann Thorac Surg. 1987;44:229-237.
5. den Bakker MA, Oosterhuis JW. Tumours and tumour-like conditions of the thymus other than thymoma; a practical approach. Histopathology. 2009;54:69-89.
6. Gerein AN, Srivastava SP, Burgess J. Thymoma: a ten year review. Am J Surg. 1978;136:49-53.
7. van Heerden JA, Harison EG, Jr, Bernatz PE, et al. Mediastinal malignant lymphoma. Chest. 1970;57:518-529.
8. Hoppe RT. The non-Hodgkin’s lymphomas: pathology, staging, treatment. Curr Probl Cancer. 1987;11:363-447.
9. Luna MA, Valenquela-Tamariz J. Germ cell tumors of the mediastinum: postmortem findings. Am J Clin Pathol. 1976;65:450-454.
10. Moran CA, Suster S, Koss MN. Primary germ cell tumors of the mediastinum: III. Yolk sac tumor, embryonal carcinoma, choriocarcinoma, and combined nonteratomatous germ cell tumors of the mediastinum—a clinicopathologic and immunohistochemical study of 64 cases. Cancer. 1997;80:699-707.
11. Polansky SM, Barwick KW, Ravin CE. Primary mediastinal seminoma. AJR Am J Roentgenol. 1979;132:17-21.
12. Jennings A. Evaluation of substernal goiters using computed tomography and MR imaging. Endocrinol Metab Clin North Am. 2001;30:401-414, ix.
13. Wright CD, Mathisen DJ. Mediastinal tumors: diagnosis and treatment. World J Surg. 2001;25:204-209.
14. Park HM, Tarver RD, Siddiqui AR, et al. Efficacy of thyroid scintigraphy in the diagnosis of intrathoracic goiter. AJR Am J Roentgenol. 1987;148:527-529.
Diffuse rash and cough in elderly woman with a UTI
A 66-YEAR-OLD WOMAN came into the emergency department with a diffuse rash and a cough. She had a rash on the palms of her hands, which had developed the day before, but had improved a bit. She also had a rash on her feet, legs, and lower abdomen, which had developed that morning.
She said that over the previous 2 days she’d had a fever, dry cough, and some difficulty breathing. Her past medical history was significant for asthma, diabetes, hypertension, and osteoarthritis. Her medications included atenolol, celecoxib, metformin, pioglitazone, and an albuterol inhaler, as needed. In addition, she was on the ninth day of a 10-day course of nitrofurantoin for acute cystitis. She was allergic to ampicillin and erythromycin.
On physical exam, she had a fever of 101.5°F. On lung examination, she had diffuse wheezes and mild bibasilar crackles. Examination of her skin revealed a nonpainful, nonpruritic, erythematous, maculopapular rash located on the palms and legs (FIGURES 1A AND 1B), as well as on her lower abdomen. Chest radiograph showed mild opacification in the bases of the lungs (FIGURES 2A AND 2B). Her labs were significant for a white blood cell (WBC) count of 11.3 ×103/mm3.
What is your diagnosis?
FIGURE 1
Rash on hands, feet, legs, and lower abdomen
The patient had generalized palmar erythema with 1- to 2-mm papules (A). She also had an erythematous maculopapular rash that extended from the medial and dorsal aspects of her feet cranially to her lower abdomen (B).
FIGURE 2
Mild opacification in lung bases
A posterior-anterior chest radiograph revealed bilateral lower lung opacities that were greater on the left side than on the right (A). A lateral chest radiograph revealed a positive spine sign: failure of the vertebral bodies to become more radiolucent as one looks down the spinal column, suggesting a posterior-inferior lung infiltrate that opacifies the normally radiolucent vertebral bodies of the lower chest (B).
Diagnosis: nitrofurantoin-induced lung disease
Nitrofurantoin (Macrodantin, Macrobid) is frequently used as first-line treatment for uncomplicated urinary tract infections (UTIs) and as prophylaxis for recurrent UTIs. Although generally well tolerated, it has a rare but serious side effect profile, including aplastic anemia, peripheral neuropathy, liver toxicity, and—as in the case of this patient—pulmonary toxicity.1
Nitrofurantoin-induced lung disease has an incidence of 3 cases per 1000 patients.2 Not surprisingly, elderly women are often affected, as this population is particularly susceptible to UTIs (and their recurrence), and thus are prescribed nitrofurantoin.3 The pathophysiology of the acute and chronic form of this drug reaction is unknown, but may be either an immunologic hypersensitivity reaction or a direct cytotoxic effect by nitrofurantoin or its metabolites on the lung parenchyma.4
The acute presentation begins within 3 weeks of initiation of nitrofurantoin. The most common symptoms include fever, dry cough, and dyspnea. Less common symptoms include an erythematous maculopapular rash, fatigue, arthralgias/myalgias, and anorexia. Patients may experience bronchospasm and have audible wheezing on exam. Eight percent of patients will have an abnormal chest radiograph, most often showing diffuse interstitial infiltrates or pleural effusions. An elevated WBC count appears in 40% of patients, many having an eosinophilia, and an elevated erythrocyte sedimentation rate may be present.5 Pulmonary function tests (PFTs), if performed, will show a restrictive pattern and a V/Q scan will show a ventilation-perfusion mismatch.2
The chronic presentation is much less common than the acute form. It occurs in patients on daily nitrofurantoin and its onset is delayed months to years after initiation of therapy. Symptoms are insidious and include cough and gradually worsening dyspnea on exertion. Physical exam may reveal wheezing or crackles and chest radiograph can show interstitial or alveolar infiltrates, as well as pleural effusions. Laboratory evaluation is similar to the acute form, and PFT findings also reveal a restrictive pattern.
Computed tomography has variable findings, such as ground-glass opacities, interstitial fibrosis, consolidation, peribronchial thickening, and centrilobular nodules. Lung biopsies have shown varying pathologic features: nonspecific interstitial pneumonia, pulmonary fibrosis, bronchiolitis obliterans organizing pneumonia, and hypersensitivity pneumonitis. 6,7
Differential Dx includes anaphylaxis, pneumonia
The differential diagnosis for nitrofurantoin-induced lung disease includes anaphylaxis, asthma, bronchitis, and pneumonia. Drug anaphylaxis typically occurs within hours of administration and often has skin findings of urticaria, pruritus, and angioedema of the oral mucosa. Patients may present with dyspnea, wheezing, stridor, hypoxemia, and respiratory distress.8
Although asthma, bronchitis, and pneumonia can present with a fever, cough, and radiograph findings, they are not usually associated with a rash. Identifying and removing the offending agent—a medication—is key in differentiating this disease from other common lung ailments.
Stop the offending agent, consider corticosteroids
Immediately discontinuing nitrofurantoin is usually sufficient to resolve the symptoms of acute nitrofurantoin-induced lung disease, often within 24 to 48 hours. Chest radiograph and PFT changes resolve within weeks of discontinuation. (Reintroducing nitrofurantoin to the patient will produce similar symptoms with a more rapid onset.) Corticosteroids are often used to treat this lung injury, although their effectiveness remains unproven.
With the chronic reaction, nitrofurantoin therapy is stopped and most patients are started on corticosteroids, typically dosed at 20 to 40 mg per day with a prolonged taper over several months. With treatment, many patients recover from their chronic pulmonary reaction, although it may take months to years.3 Some patients may never fully recover and continue to have symptoms, radiographic findings, and PFT changes characteristic of pulmonary fibrosis.
More seriously, nitrofurantoin-induced lung disease may require hospitalization. Rare cases have required lung transplantation and others have resulted in death due to respiratory failure.1
A speedy recovery for our patient
We told our patient to discontinue the nitrofurantoin, and we opted not to start her on corticosteroids. Her symptoms resolved within 3 days of discontinuing the medication, and she had no return of her UTI symptoms. Her rash also resolved within 5 days.
CORRESPONDENCE: Drew C. Baird, MD, Department of Family and Community Medicine, Carl R. Darnall Army Medical Center, 36000 Darnall Loop, Fort Hood, TX 76544; [email protected]
1. Goemaere N, Grijm K, van Hal P, et al. Nitrofurantoin-induced pulmonary fibrosis: a case report. J Med Case Reports. 2008;2:169.-
2. Hainer BL, White AA. Nitrofurantoin pulmonary toxicity. J Fam Pract. 1981;13:817-823.
3. Bhullar S, Lele SM, Kraman S. Severe nitrofurantoin lung disease resolving without the use of steroids. J Postgrad Med. 2007;53:111-113.
4. Peall AF, Hodges A. Concomitant pulmonary and hepatic toxicity secondary to nitrofurantoin: a case report. J Med Case Reports. 2007;1:59.-
5. Holmberg L, Boman G, Bottiger LE, et al. Adverse reactions to nitrofurantoin. Analysis of 921 reports. Am J Med. 1980;69:733-738.
6. Mendez JL, Nadrous HF, Hartman TE, et al. Chronic nitrofurantoin-induced lung disease. Mayo Clin Proc. 2005;80:1298-1302.
7. Cameron RJ, Kolbe J, Wilsher ML, et al. Bronchiolitis obliterans organizing pneumonia associated with the use of nitrofurantoin. Thorax. 2000;55:249-251.
8. Tang AW. A practical guide to anaphylaxis. Am Fam Physician. 2003;68:1325-1332.
A 66-YEAR-OLD WOMAN came into the emergency department with a diffuse rash and a cough. She had a rash on the palms of her hands, which had developed the day before, but had improved a bit. She also had a rash on her feet, legs, and lower abdomen, which had developed that morning.
She said that over the previous 2 days she’d had a fever, dry cough, and some difficulty breathing. Her past medical history was significant for asthma, diabetes, hypertension, and osteoarthritis. Her medications included atenolol, celecoxib, metformin, pioglitazone, and an albuterol inhaler, as needed. In addition, she was on the ninth day of a 10-day course of nitrofurantoin for acute cystitis. She was allergic to ampicillin and erythromycin.
On physical exam, she had a fever of 101.5°F. On lung examination, she had diffuse wheezes and mild bibasilar crackles. Examination of her skin revealed a nonpainful, nonpruritic, erythematous, maculopapular rash located on the palms and legs (FIGURES 1A AND 1B), as well as on her lower abdomen. Chest radiograph showed mild opacification in the bases of the lungs (FIGURES 2A AND 2B). Her labs were significant for a white blood cell (WBC) count of 11.3 ×103/mm3.
What is your diagnosis?
FIGURE 1
Rash on hands, feet, legs, and lower abdomen
The patient had generalized palmar erythema with 1- to 2-mm papules (A). She also had an erythematous maculopapular rash that extended from the medial and dorsal aspects of her feet cranially to her lower abdomen (B).
FIGURE 2
Mild opacification in lung bases
A posterior-anterior chest radiograph revealed bilateral lower lung opacities that were greater on the left side than on the right (A). A lateral chest radiograph revealed a positive spine sign: failure of the vertebral bodies to become more radiolucent as one looks down the spinal column, suggesting a posterior-inferior lung infiltrate that opacifies the normally radiolucent vertebral bodies of the lower chest (B).
Diagnosis: nitrofurantoin-induced lung disease
Nitrofurantoin (Macrodantin, Macrobid) is frequently used as first-line treatment for uncomplicated urinary tract infections (UTIs) and as prophylaxis for recurrent UTIs. Although generally well tolerated, it has a rare but serious side effect profile, including aplastic anemia, peripheral neuropathy, liver toxicity, and—as in the case of this patient—pulmonary toxicity.1
Nitrofurantoin-induced lung disease has an incidence of 3 cases per 1000 patients.2 Not surprisingly, elderly women are often affected, as this population is particularly susceptible to UTIs (and their recurrence), and thus are prescribed nitrofurantoin.3 The pathophysiology of the acute and chronic form of this drug reaction is unknown, but may be either an immunologic hypersensitivity reaction or a direct cytotoxic effect by nitrofurantoin or its metabolites on the lung parenchyma.4
The acute presentation begins within 3 weeks of initiation of nitrofurantoin. The most common symptoms include fever, dry cough, and dyspnea. Less common symptoms include an erythematous maculopapular rash, fatigue, arthralgias/myalgias, and anorexia. Patients may experience bronchospasm and have audible wheezing on exam. Eight percent of patients will have an abnormal chest radiograph, most often showing diffuse interstitial infiltrates or pleural effusions. An elevated WBC count appears in 40% of patients, many having an eosinophilia, and an elevated erythrocyte sedimentation rate may be present.5 Pulmonary function tests (PFTs), if performed, will show a restrictive pattern and a V/Q scan will show a ventilation-perfusion mismatch.2
The chronic presentation is much less common than the acute form. It occurs in patients on daily nitrofurantoin and its onset is delayed months to years after initiation of therapy. Symptoms are insidious and include cough and gradually worsening dyspnea on exertion. Physical exam may reveal wheezing or crackles and chest radiograph can show interstitial or alveolar infiltrates, as well as pleural effusions. Laboratory evaluation is similar to the acute form, and PFT findings also reveal a restrictive pattern.
Computed tomography has variable findings, such as ground-glass opacities, interstitial fibrosis, consolidation, peribronchial thickening, and centrilobular nodules. Lung biopsies have shown varying pathologic features: nonspecific interstitial pneumonia, pulmonary fibrosis, bronchiolitis obliterans organizing pneumonia, and hypersensitivity pneumonitis. 6,7
Differential Dx includes anaphylaxis, pneumonia
The differential diagnosis for nitrofurantoin-induced lung disease includes anaphylaxis, asthma, bronchitis, and pneumonia. Drug anaphylaxis typically occurs within hours of administration and often has skin findings of urticaria, pruritus, and angioedema of the oral mucosa. Patients may present with dyspnea, wheezing, stridor, hypoxemia, and respiratory distress.8
Although asthma, bronchitis, and pneumonia can present with a fever, cough, and radiograph findings, they are not usually associated with a rash. Identifying and removing the offending agent—a medication—is key in differentiating this disease from other common lung ailments.
Stop the offending agent, consider corticosteroids
Immediately discontinuing nitrofurantoin is usually sufficient to resolve the symptoms of acute nitrofurantoin-induced lung disease, often within 24 to 48 hours. Chest radiograph and PFT changes resolve within weeks of discontinuation. (Reintroducing nitrofurantoin to the patient will produce similar symptoms with a more rapid onset.) Corticosteroids are often used to treat this lung injury, although their effectiveness remains unproven.
With the chronic reaction, nitrofurantoin therapy is stopped and most patients are started on corticosteroids, typically dosed at 20 to 40 mg per day with a prolonged taper over several months. With treatment, many patients recover from their chronic pulmonary reaction, although it may take months to years.3 Some patients may never fully recover and continue to have symptoms, radiographic findings, and PFT changes characteristic of pulmonary fibrosis.
More seriously, nitrofurantoin-induced lung disease may require hospitalization. Rare cases have required lung transplantation and others have resulted in death due to respiratory failure.1
A speedy recovery for our patient
We told our patient to discontinue the nitrofurantoin, and we opted not to start her on corticosteroids. Her symptoms resolved within 3 days of discontinuing the medication, and she had no return of her UTI symptoms. Her rash also resolved within 5 days.
CORRESPONDENCE: Drew C. Baird, MD, Department of Family and Community Medicine, Carl R. Darnall Army Medical Center, 36000 Darnall Loop, Fort Hood, TX 76544; [email protected]
A 66-YEAR-OLD WOMAN came into the emergency department with a diffuse rash and a cough. She had a rash on the palms of her hands, which had developed the day before, but had improved a bit. She also had a rash on her feet, legs, and lower abdomen, which had developed that morning.
She said that over the previous 2 days she’d had a fever, dry cough, and some difficulty breathing. Her past medical history was significant for asthma, diabetes, hypertension, and osteoarthritis. Her medications included atenolol, celecoxib, metformin, pioglitazone, and an albuterol inhaler, as needed. In addition, she was on the ninth day of a 10-day course of nitrofurantoin for acute cystitis. She was allergic to ampicillin and erythromycin.
On physical exam, she had a fever of 101.5°F. On lung examination, she had diffuse wheezes and mild bibasilar crackles. Examination of her skin revealed a nonpainful, nonpruritic, erythematous, maculopapular rash located on the palms and legs (FIGURES 1A AND 1B), as well as on her lower abdomen. Chest radiograph showed mild opacification in the bases of the lungs (FIGURES 2A AND 2B). Her labs were significant for a white blood cell (WBC) count of 11.3 ×103/mm3.
What is your diagnosis?
FIGURE 1
Rash on hands, feet, legs, and lower abdomen
The patient had generalized palmar erythema with 1- to 2-mm papules (A). She also had an erythematous maculopapular rash that extended from the medial and dorsal aspects of her feet cranially to her lower abdomen (B).
FIGURE 2
Mild opacification in lung bases
A posterior-anterior chest radiograph revealed bilateral lower lung opacities that were greater on the left side than on the right (A). A lateral chest radiograph revealed a positive spine sign: failure of the vertebral bodies to become more radiolucent as one looks down the spinal column, suggesting a posterior-inferior lung infiltrate that opacifies the normally radiolucent vertebral bodies of the lower chest (B).
Diagnosis: nitrofurantoin-induced lung disease
Nitrofurantoin (Macrodantin, Macrobid) is frequently used as first-line treatment for uncomplicated urinary tract infections (UTIs) and as prophylaxis for recurrent UTIs. Although generally well tolerated, it has a rare but serious side effect profile, including aplastic anemia, peripheral neuropathy, liver toxicity, and—as in the case of this patient—pulmonary toxicity.1
Nitrofurantoin-induced lung disease has an incidence of 3 cases per 1000 patients.2 Not surprisingly, elderly women are often affected, as this population is particularly susceptible to UTIs (and their recurrence), and thus are prescribed nitrofurantoin.3 The pathophysiology of the acute and chronic form of this drug reaction is unknown, but may be either an immunologic hypersensitivity reaction or a direct cytotoxic effect by nitrofurantoin or its metabolites on the lung parenchyma.4
The acute presentation begins within 3 weeks of initiation of nitrofurantoin. The most common symptoms include fever, dry cough, and dyspnea. Less common symptoms include an erythematous maculopapular rash, fatigue, arthralgias/myalgias, and anorexia. Patients may experience bronchospasm and have audible wheezing on exam. Eight percent of patients will have an abnormal chest radiograph, most often showing diffuse interstitial infiltrates or pleural effusions. An elevated WBC count appears in 40% of patients, many having an eosinophilia, and an elevated erythrocyte sedimentation rate may be present.5 Pulmonary function tests (PFTs), if performed, will show a restrictive pattern and a V/Q scan will show a ventilation-perfusion mismatch.2
The chronic presentation is much less common than the acute form. It occurs in patients on daily nitrofurantoin and its onset is delayed months to years after initiation of therapy. Symptoms are insidious and include cough and gradually worsening dyspnea on exertion. Physical exam may reveal wheezing or crackles and chest radiograph can show interstitial or alveolar infiltrates, as well as pleural effusions. Laboratory evaluation is similar to the acute form, and PFT findings also reveal a restrictive pattern.
Computed tomography has variable findings, such as ground-glass opacities, interstitial fibrosis, consolidation, peribronchial thickening, and centrilobular nodules. Lung biopsies have shown varying pathologic features: nonspecific interstitial pneumonia, pulmonary fibrosis, bronchiolitis obliterans organizing pneumonia, and hypersensitivity pneumonitis. 6,7
Differential Dx includes anaphylaxis, pneumonia
The differential diagnosis for nitrofurantoin-induced lung disease includes anaphylaxis, asthma, bronchitis, and pneumonia. Drug anaphylaxis typically occurs within hours of administration and often has skin findings of urticaria, pruritus, and angioedema of the oral mucosa. Patients may present with dyspnea, wheezing, stridor, hypoxemia, and respiratory distress.8
Although asthma, bronchitis, and pneumonia can present with a fever, cough, and radiograph findings, they are not usually associated with a rash. Identifying and removing the offending agent—a medication—is key in differentiating this disease from other common lung ailments.
Stop the offending agent, consider corticosteroids
Immediately discontinuing nitrofurantoin is usually sufficient to resolve the symptoms of acute nitrofurantoin-induced lung disease, often within 24 to 48 hours. Chest radiograph and PFT changes resolve within weeks of discontinuation. (Reintroducing nitrofurantoin to the patient will produce similar symptoms with a more rapid onset.) Corticosteroids are often used to treat this lung injury, although their effectiveness remains unproven.
With the chronic reaction, nitrofurantoin therapy is stopped and most patients are started on corticosteroids, typically dosed at 20 to 40 mg per day with a prolonged taper over several months. With treatment, many patients recover from their chronic pulmonary reaction, although it may take months to years.3 Some patients may never fully recover and continue to have symptoms, radiographic findings, and PFT changes characteristic of pulmonary fibrosis.
More seriously, nitrofurantoin-induced lung disease may require hospitalization. Rare cases have required lung transplantation and others have resulted in death due to respiratory failure.1
A speedy recovery for our patient
We told our patient to discontinue the nitrofurantoin, and we opted not to start her on corticosteroids. Her symptoms resolved within 3 days of discontinuing the medication, and she had no return of her UTI symptoms. Her rash also resolved within 5 days.
CORRESPONDENCE: Drew C. Baird, MD, Department of Family and Community Medicine, Carl R. Darnall Army Medical Center, 36000 Darnall Loop, Fort Hood, TX 76544; [email protected]
1. Goemaere N, Grijm K, van Hal P, et al. Nitrofurantoin-induced pulmonary fibrosis: a case report. J Med Case Reports. 2008;2:169.-
2. Hainer BL, White AA. Nitrofurantoin pulmonary toxicity. J Fam Pract. 1981;13:817-823.
3. Bhullar S, Lele SM, Kraman S. Severe nitrofurantoin lung disease resolving without the use of steroids. J Postgrad Med. 2007;53:111-113.
4. Peall AF, Hodges A. Concomitant pulmonary and hepatic toxicity secondary to nitrofurantoin: a case report. J Med Case Reports. 2007;1:59.-
5. Holmberg L, Boman G, Bottiger LE, et al. Adverse reactions to nitrofurantoin. Analysis of 921 reports. Am J Med. 1980;69:733-738.
6. Mendez JL, Nadrous HF, Hartman TE, et al. Chronic nitrofurantoin-induced lung disease. Mayo Clin Proc. 2005;80:1298-1302.
7. Cameron RJ, Kolbe J, Wilsher ML, et al. Bronchiolitis obliterans organizing pneumonia associated with the use of nitrofurantoin. Thorax. 2000;55:249-251.
8. Tang AW. A practical guide to anaphylaxis. Am Fam Physician. 2003;68:1325-1332.
1. Goemaere N, Grijm K, van Hal P, et al. Nitrofurantoin-induced pulmonary fibrosis: a case report. J Med Case Reports. 2008;2:169.-
2. Hainer BL, White AA. Nitrofurantoin pulmonary toxicity. J Fam Pract. 1981;13:817-823.
3. Bhullar S, Lele SM, Kraman S. Severe nitrofurantoin lung disease resolving without the use of steroids. J Postgrad Med. 2007;53:111-113.
4. Peall AF, Hodges A. Concomitant pulmonary and hepatic toxicity secondary to nitrofurantoin: a case report. J Med Case Reports. 2007;1:59.-
5. Holmberg L, Boman G, Bottiger LE, et al. Adverse reactions to nitrofurantoin. Analysis of 921 reports. Am J Med. 1980;69:733-738.
6. Mendez JL, Nadrous HF, Hartman TE, et al. Chronic nitrofurantoin-induced lung disease. Mayo Clin Proc. 2005;80:1298-1302.
7. Cameron RJ, Kolbe J, Wilsher ML, et al. Bronchiolitis obliterans organizing pneumonia associated with the use of nitrofurantoin. Thorax. 2000;55:249-251.
8. Tang AW. A practical guide to anaphylaxis. Am Fam Physician. 2003;68:1325-1332.
A progressive scalp lesion
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
A 47-YEAR-OLD AFRICAN AMERICAN WOMAN sought care at our clinic for multiple progressive scalp lesions. She said that she first noticed the lesions 13 years ago when her children were diagnosed with ringworm on the scalp. At that time, her physician thought that she, too, had tinea capitis, and she was treated with 6 weeks of griseofulvin. The lesions persisted, however.
She told us that the lesions were nonpruritic and that she didn’t have any other symptoms. The patient did not have a history of trauma or exposure of chemicals to the scalp, and she was not taking any prescription or over-the-counter medications.
Examination of her scalp revealed scattered irregularly shaped, nontender lesions that were centrally hypopigmented and peripherally hyperpigmented. She also had scarring and hair loss (FIGURE 1). She had no other lesions on her body.
FIGURE 1
Irregularly shaped scalp lesions
Our 47-year-old patient had multiple scattered scalp lesions that were nontender and centrally hypopigmented. scarring, alopecia, and surrounding areas of hyperpigmentation were also visible.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU MANAGE THIS CONDITION?
Diagnosis: Discoid lupus erythematosus
A punch biopsy and tissue pathology confirmed that our patient had discoid lupus erythematosus (DLE).
DLE is a common type of cutaneous lupus that is chronic and is typically associated with atrophy and scarring of the skin. The primary discoid lesion is a discrete erythematous papule or plaque with adherent scaling, follicular plugging, atrophic scarring, central hypopigmentation, and hyperpigmented borders.
DLE is a dermatosis that is localized in 80% of patients and occurs mainly on sun-exposed areas of the skin, such as the scalp, face, and ears. In 20% of cases it occurs on the extremities and upper trunk. Women are affected more than men, and it can affect any age group, although it is more common in individuals between the ages of 20 and 40.1
The etiology of DLE is unknown. What we do know is that less than 5% of patients with DLE eventually end up with systemic lupus erythematosus (SLE), while up to 25% of patients with SLE go on to develop chronic discoid lesions.2
Abnormalities in serology are not common in DLE. About 20% of patients with DLE show positive antinuclear antibody titers when tested. This is the case in the presence of widespread disease more so than in localized DLE. The presence of antinative (antidouble-stranded) or anti-Smith antibodies is usually suggestive of systemic symptoms and occurs in 5% of cases.3
Several conditions mimic DLE
The differential diagnosis for DLE includes the following:
Lichen plano pilaris. This condition has a predilection to the crown of the head and the frontal central region of the scalp; it is also associated with bilateral eyebrow hair loss. Patients may complain of pruritus, localized tenderness, and a burning sensation. The etiology is unknown; it is most often seen in middle-aged women with a chronic, progressive clinical course.4
Alopecia areata. Patients suddenly lose hair in patches. The hair grows back, and then falls out again. This asymptomatic condition is a tissue-restricted autoimmune disease of the hair follicle that most commonly occurs among children and young adults.5
Dissecting scalp cellulitis. This is a suppurating and cicatrizing disease of the scalp of unknown etiology. It typically affects the scalp vertex and occipital region, and is most common among young black men. The erythematous papules eventually discharge seropurulent material and form underlying intercommunicating sinuses with eventual scarring.6 Patients are likely to complain of pain and pruritus.
Tinea capitis. As noted earlier, this is commonly referred to as “ringworm” and is of a fungal, infectious etiology that typically affects children. Permanent scarring and alopecia are common in affected areas, and patients complain of itching and burning.
Diagnosis can be made on clinical grounds
While a clinical diagnosis of DLE can be made, a tissue biopsy of a new inflamed site is confirmatory. Histopathologic findings show hyperkeratosis, follicular plugging, thickening of the basement membrane, atrophic epidermis, and dermal perifollicular and periappendageal lymphocytic inflammatory infiltrate.
Direct immunofluorescence of lesions shows granular immunoglobulin and complement deposition at the dermal-epidermal junction.7,8
Early treatment is key
Early treatment may be helpful in preventing permanent scarring. Therapeutic options commonly used are oral antimalarials (strength of recommendation [SOR]: A) and topical (SOR: A) or intralesional (SOR: B) corticosteroids.9 Other topical agents, such as calcineurin inhibitors, retinoids, and imiquimod, have been found to be helpful in some cases.
Alternative systemic agents that appear to be useful include methotrexate, azathioprine, thalidomide, dapsone, and mycophenolate mofetil. Patients should be advised to avoid the sun and wear broad-spectrum sunscreen.7,10
Finally, the patient sees some improvement
We discussed the risks and benefits of the various treatments, and our patient elected to start hydroxychloroquine (Plaquenil), 200 to 400 mg/day orally (not to exceed 6.5 mg/kg per day). We referred her for a baseline ophthalmological exam and stressed that she needed a repeat exam every 6 to 12 months while she remained on the hydroxychloroquine. We also referred her to a rheumatologist.
After 4 months of treatment, she showed some improvement (FIGURE 2), with no side effects from the medication. The patient was subsequently lost to follow-up.
FIGURE 2
A visible improvement
After 4 months of treatment with hydroxychloroquine, the patient’s scalp lesions improved and there was evidence of hair growth.
CORRESPONDENCE: Ahunna Ahiarah, MD, UB Family Medicine, 1315 Jefferson Avenue, Buffalo, NY 14208; [email protected]
1. Tlacuilo-Parra A, Guevara-Gutierrez E, Gutierrez-Murillo F, et al. Pimecrolimus 1% cream for the treatment of discoid lupus erythematosus. Rheumatology. 2005;44:1564-1568.
2. Wouters CH, Diegenant C, Ceuppens JL, et al. The circulating lymphocyte profiles in patients with discoid lupus erythematosus and systemic lupus erythematosus suggest a pathogenetic relationship. Br J Dermatol. 2004;150:693-700.
3. Callen JP. Chronic cutaneous lupus erythematosus. Clinical, laboratory, therapeutic, and prognostic examination of 62 patients. Arch Dermatol. 1982;118:412-416.
4. Tandon YK, Somani N, Cevasco NC, et al. A histologic review of 27 patients with lichen planopilaris. J Am Acad Dermatol. 2008;59:91-98.
5. Gilhar A, Kalish RS. Alopecia areata: a tissue specific autoimmune disease of the hair follicle. Autoimmun Rev. 2006;5:64-69.
6. Monroe M, Crutchfield C. Dissecting cellulitis of the scalp. Dermatol Nurs. 2005;17:208.-
7. Callen JP. Collagen vascular diseases. J Am Acad Dermatol. 2004;51:427-439.
8. Lee LA. Lupus erythematosus. In: Sams WM Jr, Lynch PJ, eds. Principles and Practice of Dermatology. New York: Churchill Livingstone; 1996:581–598.
9. Callen JP. Update on the management of cutaneous lupus erythematosus. Br J Dermatol. 2004;151:731-736.
10. Jessop S, Whitelaw D, Jordaan F. Drugs for discoid lupus erythematosus. Cochrane Database Syst Rev. 2000;(2):CD002954.-
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
A 47-YEAR-OLD AFRICAN AMERICAN WOMAN sought care at our clinic for multiple progressive scalp lesions. She said that she first noticed the lesions 13 years ago when her children were diagnosed with ringworm on the scalp. At that time, her physician thought that she, too, had tinea capitis, and she was treated with 6 weeks of griseofulvin. The lesions persisted, however.
She told us that the lesions were nonpruritic and that she didn’t have any other symptoms. The patient did not have a history of trauma or exposure of chemicals to the scalp, and she was not taking any prescription or over-the-counter medications.
Examination of her scalp revealed scattered irregularly shaped, nontender lesions that were centrally hypopigmented and peripherally hyperpigmented. She also had scarring and hair loss (FIGURE 1). She had no other lesions on her body.
FIGURE 1
Irregularly shaped scalp lesions
Our 47-year-old patient had multiple scattered scalp lesions that were nontender and centrally hypopigmented. scarring, alopecia, and surrounding areas of hyperpigmentation were also visible.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU MANAGE THIS CONDITION?
Diagnosis: Discoid lupus erythematosus
A punch biopsy and tissue pathology confirmed that our patient had discoid lupus erythematosus (DLE).
DLE is a common type of cutaneous lupus that is chronic and is typically associated with atrophy and scarring of the skin. The primary discoid lesion is a discrete erythematous papule or plaque with adherent scaling, follicular plugging, atrophic scarring, central hypopigmentation, and hyperpigmented borders.
DLE is a dermatosis that is localized in 80% of patients and occurs mainly on sun-exposed areas of the skin, such as the scalp, face, and ears. In 20% of cases it occurs on the extremities and upper trunk. Women are affected more than men, and it can affect any age group, although it is more common in individuals between the ages of 20 and 40.1
The etiology of DLE is unknown. What we do know is that less than 5% of patients with DLE eventually end up with systemic lupus erythematosus (SLE), while up to 25% of patients with SLE go on to develop chronic discoid lesions.2
Abnormalities in serology are not common in DLE. About 20% of patients with DLE show positive antinuclear antibody titers when tested. This is the case in the presence of widespread disease more so than in localized DLE. The presence of antinative (antidouble-stranded) or anti-Smith antibodies is usually suggestive of systemic symptoms and occurs in 5% of cases.3
Several conditions mimic DLE
The differential diagnosis for DLE includes the following:
Lichen plano pilaris. This condition has a predilection to the crown of the head and the frontal central region of the scalp; it is also associated with bilateral eyebrow hair loss. Patients may complain of pruritus, localized tenderness, and a burning sensation. The etiology is unknown; it is most often seen in middle-aged women with a chronic, progressive clinical course.4
Alopecia areata. Patients suddenly lose hair in patches. The hair grows back, and then falls out again. This asymptomatic condition is a tissue-restricted autoimmune disease of the hair follicle that most commonly occurs among children and young adults.5
Dissecting scalp cellulitis. This is a suppurating and cicatrizing disease of the scalp of unknown etiology. It typically affects the scalp vertex and occipital region, and is most common among young black men. The erythematous papules eventually discharge seropurulent material and form underlying intercommunicating sinuses with eventual scarring.6 Patients are likely to complain of pain and pruritus.
Tinea capitis. As noted earlier, this is commonly referred to as “ringworm” and is of a fungal, infectious etiology that typically affects children. Permanent scarring and alopecia are common in affected areas, and patients complain of itching and burning.
Diagnosis can be made on clinical grounds
While a clinical diagnosis of DLE can be made, a tissue biopsy of a new inflamed site is confirmatory. Histopathologic findings show hyperkeratosis, follicular plugging, thickening of the basement membrane, atrophic epidermis, and dermal perifollicular and periappendageal lymphocytic inflammatory infiltrate.
Direct immunofluorescence of lesions shows granular immunoglobulin and complement deposition at the dermal-epidermal junction.7,8
Early treatment is key
Early treatment may be helpful in preventing permanent scarring. Therapeutic options commonly used are oral antimalarials (strength of recommendation [SOR]: A) and topical (SOR: A) or intralesional (SOR: B) corticosteroids.9 Other topical agents, such as calcineurin inhibitors, retinoids, and imiquimod, have been found to be helpful in some cases.
Alternative systemic agents that appear to be useful include methotrexate, azathioprine, thalidomide, dapsone, and mycophenolate mofetil. Patients should be advised to avoid the sun and wear broad-spectrum sunscreen.7,10
Finally, the patient sees some improvement
We discussed the risks and benefits of the various treatments, and our patient elected to start hydroxychloroquine (Plaquenil), 200 to 400 mg/day orally (not to exceed 6.5 mg/kg per day). We referred her for a baseline ophthalmological exam and stressed that she needed a repeat exam every 6 to 12 months while she remained on the hydroxychloroquine. We also referred her to a rheumatologist.
After 4 months of treatment, she showed some improvement (FIGURE 2), with no side effects from the medication. The patient was subsequently lost to follow-up.
FIGURE 2
A visible improvement
After 4 months of treatment with hydroxychloroquine, the patient’s scalp lesions improved and there was evidence of hair growth.
CORRESPONDENCE: Ahunna Ahiarah, MD, UB Family Medicine, 1315 Jefferson Avenue, Buffalo, NY 14208; [email protected]
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
A 47-YEAR-OLD AFRICAN AMERICAN WOMAN sought care at our clinic for multiple progressive scalp lesions. She said that she first noticed the lesions 13 years ago when her children were diagnosed with ringworm on the scalp. At that time, her physician thought that she, too, had tinea capitis, and she was treated with 6 weeks of griseofulvin. The lesions persisted, however.
She told us that the lesions were nonpruritic and that she didn’t have any other symptoms. The patient did not have a history of trauma or exposure of chemicals to the scalp, and she was not taking any prescription or over-the-counter medications.
Examination of her scalp revealed scattered irregularly shaped, nontender lesions that were centrally hypopigmented and peripherally hyperpigmented. She also had scarring and hair loss (FIGURE 1). She had no other lesions on her body.
FIGURE 1
Irregularly shaped scalp lesions
Our 47-year-old patient had multiple scattered scalp lesions that were nontender and centrally hypopigmented. scarring, alopecia, and surrounding areas of hyperpigmentation were also visible.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU MANAGE THIS CONDITION?
Diagnosis: Discoid lupus erythematosus
A punch biopsy and tissue pathology confirmed that our patient had discoid lupus erythematosus (DLE).
DLE is a common type of cutaneous lupus that is chronic and is typically associated with atrophy and scarring of the skin. The primary discoid lesion is a discrete erythematous papule or plaque with adherent scaling, follicular plugging, atrophic scarring, central hypopigmentation, and hyperpigmented borders.
DLE is a dermatosis that is localized in 80% of patients and occurs mainly on sun-exposed areas of the skin, such as the scalp, face, and ears. In 20% of cases it occurs on the extremities and upper trunk. Women are affected more than men, and it can affect any age group, although it is more common in individuals between the ages of 20 and 40.1
The etiology of DLE is unknown. What we do know is that less than 5% of patients with DLE eventually end up with systemic lupus erythematosus (SLE), while up to 25% of patients with SLE go on to develop chronic discoid lesions.2
Abnormalities in serology are not common in DLE. About 20% of patients with DLE show positive antinuclear antibody titers when tested. This is the case in the presence of widespread disease more so than in localized DLE. The presence of antinative (antidouble-stranded) or anti-Smith antibodies is usually suggestive of systemic symptoms and occurs in 5% of cases.3
Several conditions mimic DLE
The differential diagnosis for DLE includes the following:
Lichen plano pilaris. This condition has a predilection to the crown of the head and the frontal central region of the scalp; it is also associated with bilateral eyebrow hair loss. Patients may complain of pruritus, localized tenderness, and a burning sensation. The etiology is unknown; it is most often seen in middle-aged women with a chronic, progressive clinical course.4
Alopecia areata. Patients suddenly lose hair in patches. The hair grows back, and then falls out again. This asymptomatic condition is a tissue-restricted autoimmune disease of the hair follicle that most commonly occurs among children and young adults.5
Dissecting scalp cellulitis. This is a suppurating and cicatrizing disease of the scalp of unknown etiology. It typically affects the scalp vertex and occipital region, and is most common among young black men. The erythematous papules eventually discharge seropurulent material and form underlying intercommunicating sinuses with eventual scarring.6 Patients are likely to complain of pain and pruritus.
Tinea capitis. As noted earlier, this is commonly referred to as “ringworm” and is of a fungal, infectious etiology that typically affects children. Permanent scarring and alopecia are common in affected areas, and patients complain of itching and burning.
Diagnosis can be made on clinical grounds
While a clinical diagnosis of DLE can be made, a tissue biopsy of a new inflamed site is confirmatory. Histopathologic findings show hyperkeratosis, follicular plugging, thickening of the basement membrane, atrophic epidermis, and dermal perifollicular and periappendageal lymphocytic inflammatory infiltrate.
Direct immunofluorescence of lesions shows granular immunoglobulin and complement deposition at the dermal-epidermal junction.7,8
Early treatment is key
Early treatment may be helpful in preventing permanent scarring. Therapeutic options commonly used are oral antimalarials (strength of recommendation [SOR]: A) and topical (SOR: A) or intralesional (SOR: B) corticosteroids.9 Other topical agents, such as calcineurin inhibitors, retinoids, and imiquimod, have been found to be helpful in some cases.
Alternative systemic agents that appear to be useful include methotrexate, azathioprine, thalidomide, dapsone, and mycophenolate mofetil. Patients should be advised to avoid the sun and wear broad-spectrum sunscreen.7,10
Finally, the patient sees some improvement
We discussed the risks and benefits of the various treatments, and our patient elected to start hydroxychloroquine (Plaquenil), 200 to 400 mg/day orally (not to exceed 6.5 mg/kg per day). We referred her for a baseline ophthalmological exam and stressed that she needed a repeat exam every 6 to 12 months while she remained on the hydroxychloroquine. We also referred her to a rheumatologist.
After 4 months of treatment, she showed some improvement (FIGURE 2), with no side effects from the medication. The patient was subsequently lost to follow-up.
FIGURE 2
A visible improvement
After 4 months of treatment with hydroxychloroquine, the patient’s scalp lesions improved and there was evidence of hair growth.
CORRESPONDENCE: Ahunna Ahiarah, MD, UB Family Medicine, 1315 Jefferson Avenue, Buffalo, NY 14208; [email protected]
1. Tlacuilo-Parra A, Guevara-Gutierrez E, Gutierrez-Murillo F, et al. Pimecrolimus 1% cream for the treatment of discoid lupus erythematosus. Rheumatology. 2005;44:1564-1568.
2. Wouters CH, Diegenant C, Ceuppens JL, et al. The circulating lymphocyte profiles in patients with discoid lupus erythematosus and systemic lupus erythematosus suggest a pathogenetic relationship. Br J Dermatol. 2004;150:693-700.
3. Callen JP. Chronic cutaneous lupus erythematosus. Clinical, laboratory, therapeutic, and prognostic examination of 62 patients. Arch Dermatol. 1982;118:412-416.
4. Tandon YK, Somani N, Cevasco NC, et al. A histologic review of 27 patients with lichen planopilaris. J Am Acad Dermatol. 2008;59:91-98.
5. Gilhar A, Kalish RS. Alopecia areata: a tissue specific autoimmune disease of the hair follicle. Autoimmun Rev. 2006;5:64-69.
6. Monroe M, Crutchfield C. Dissecting cellulitis of the scalp. Dermatol Nurs. 2005;17:208.-
7. Callen JP. Collagen vascular diseases. J Am Acad Dermatol. 2004;51:427-439.
8. Lee LA. Lupus erythematosus. In: Sams WM Jr, Lynch PJ, eds. Principles and Practice of Dermatology. New York: Churchill Livingstone; 1996:581–598.
9. Callen JP. Update on the management of cutaneous lupus erythematosus. Br J Dermatol. 2004;151:731-736.
10. Jessop S, Whitelaw D, Jordaan F. Drugs for discoid lupus erythematosus. Cochrane Database Syst Rev. 2000;(2):CD002954.-
1. Tlacuilo-Parra A, Guevara-Gutierrez E, Gutierrez-Murillo F, et al. Pimecrolimus 1% cream for the treatment of discoid lupus erythematosus. Rheumatology. 2005;44:1564-1568.
2. Wouters CH, Diegenant C, Ceuppens JL, et al. The circulating lymphocyte profiles in patients with discoid lupus erythematosus and systemic lupus erythematosus suggest a pathogenetic relationship. Br J Dermatol. 2004;150:693-700.
3. Callen JP. Chronic cutaneous lupus erythematosus. Clinical, laboratory, therapeutic, and prognostic examination of 62 patients. Arch Dermatol. 1982;118:412-416.
4. Tandon YK, Somani N, Cevasco NC, et al. A histologic review of 27 patients with lichen planopilaris. J Am Acad Dermatol. 2008;59:91-98.
5. Gilhar A, Kalish RS. Alopecia areata: a tissue specific autoimmune disease of the hair follicle. Autoimmun Rev. 2006;5:64-69.
6. Monroe M, Crutchfield C. Dissecting cellulitis of the scalp. Dermatol Nurs. 2005;17:208.-
7. Callen JP. Collagen vascular diseases. J Am Acad Dermatol. 2004;51:427-439.
8. Lee LA. Lupus erythematosus. In: Sams WM Jr, Lynch PJ, eds. Principles and Practice of Dermatology. New York: Churchill Livingstone; 1996:581–598.
9. Callen JP. Update on the management of cutaneous lupus erythematosus. Br J Dermatol. 2004;151:731-736.
10. Jessop S, Whitelaw D, Jordaan F. Drugs for discoid lupus erythematosus. Cochrane Database Syst Rev. 2000;(2):CD002954.-
The Journal of Family Practice ©2010 Dowden Health Media