User login
The Risk and Treatment for Wilms Tumors
Q) In school, they always emphasized the abdominal exam to rule out Wilms tumors. Are Wilms tumors still with us? Has treatment and evaluation changed?
Wilms tumor is a renal cancer found most commonly in children younger than 9 and represents approximately 7% of all malignancies in children.8,9 It can occur in one or both kidneys, with earlier diagnosis noted with bilateral involvement. Risk is highest among non-Hispanic white persons and African-Americans and lowest among Asians.8
Wilms tumor develops due to a genetic mutation in the WT1 gene located on the 11p13 chromosome. Defects are also noted on the 11p15 chromosome and the p53 tumor suppressor gene.10 Urbach et al recently identified a relationship between the LIN28 gene and Wilms tumor.11 Tumors develop when embryonic renal cells that should cease growing at the time of birth continue to grow in the postnatal period. Wilms tumor can be familial or sporadic. It can also be associated with various congenital anomalies manifested within various syndromes (see Table 2), as well as isolated genitourinary abnormalities, especially in boys.10

Most children present with a palpable, smooth, firm, generally painless mass in the abdomen; those who have bilateral renal involvement usually present earlier than those with unilateral involvement. Palpation of the abdomen during examination, if vigorous, can result in rupture of the renal capsule and tumor spillage. Additional symptoms include hematuria, fever, and hypertension. Referral to pediatric oncology is imperative.12
Definitive diagnosis is made by histologic evaluation following biopsy or surgical excision.13 Other possible diagnostic tests include but are not limited to abdominal ultrasound or CT; chest CT (to rule out metastatic lung disease); urinalysis (to evaluate for hematuria and proteinuria); liver function studies (to evaluate for hepatic involvement); and laboratory studies to measure coagulation, serum calcium, blood urea nitrogen, creatinine, and complete blood count.
Histologic examination for staging (I-V) occurs following surgical excision of the tumor. There are two staging systems available: the National Wilms Tumor Study, based on postoperative tumor evaluation, and the International Society of Pediatric Oncology, based on postchemotherapy evaluation.13
Treatment options include surgical excision (including complete nephrectomy of the affected kidney), chemotherapy based on tumor staging, and internal and/or external radiation therapy.13
Susan E. Brown, MS, ARNP,
ACNP-BC, CCRN
Great River Nephrology,
West Burlington, Iowa
REFERENCES
1. United States Renal Data System. Annual data report: atlas of chronic kidney disease and end-stage renal disease in the United States (2012). www.usrds.org/2012/view/v1_01.aspx. Accessed October 19, 2014.
2. Kopp JB, Nelson GW, Sampath K, et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy.
J Am Soc Nephrol. 2011;22(11):2129-2137.
3. Parsa A, Kao L, Xie D, et al; AASK and CRIC Study Investigators. APOL1 risk variants, race and progression of chronic kidney disease.
N Engl J Med. 2013;369:2183-2196.
4. Foster MC, Coresh J, Fornage M, et al. APOL1 variants associate with increased risk of CKD among African Americans. J Am Soc Nephrol. 2013;24(9):1484-1491.
5. Lipkowitz MS, Freedman BI, Langefeld CD, et al; AASK Investigators. Apolipoprotein L1 gene variants associate with hypertension-attributed nephropathy and the rate of kidney function decline in African Americans. Kidney Int. 2013;83(1):114–120.
6. Reeves-Daniel AM, DePalma JA, Bleyer AJ, et al. The APOL1 gene and allograft survival after kidney transplantation. Am J Transplant. 2011;11(5):1025-1030.
7. Partners Healthcare Personalized Medicine. Order APOL1 genotyping test for non-diabetic nephropathy kidney disease. http://personal izedmedicine.partners.org/Laboratory-For-Molecular-Medicine/Ordering/Kidney-Dis ease/APOL1-Gene-Sequencing.aspx. Accessed October 19, 2014.
8. Grovas A, Fremgen A, Rauck A, et al. The National Cancer Data Base report on patterns of childhood cancers in the United States. Cancer. 1997;80(12):2321-2332.
9. Johns Hopkins Medicine. Wilm’s tumor. www.hopkinsmedicine.org/kimmel_cancer_center/centers/pediatric_oncology/cancer_types/wilms_tumor.html. Accessed October 19, 2014.
10. Dome JS, Huff V. Wilms tumor overview. In: Pagon RA, Adam MP, Ardinger HH, et al (eds). GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle; 1993-2014. www.ncbi.nlm.nih.gov/books/NBK1294/. Accessed October 19, 2014.
11. Urbach A, Yermalovich A, Zhang J, et al. Lin28 sustains early renal progenitors and induces Wilms tumor. Genes & Dev. 2014;28:971-982.
12. Fernandez C, Geller JI, Ehrlich PF, et al. Renal tumors. In: Pizzo P, Poplack D (eds). Principles and Practice of Pediatric Oncology. 6th ed, St Louis, MO: Lippincott Williams & Wilkins. 2011; 861.
13. Metzger ML, Dome JS. Current therapy for Wilms’ tumor. Oncologist. 2005;10(10):815-826.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Susan E. Brown, MS, ARNP, ACNP-BC, CCRN, who practices at Great River Nephrology in West Burlington, Iowa.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Susan E. Brown, MS, ARNP, ACNP-BC, CCRN, who practices at Great River Nephrology in West Burlington, Iowa.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Susan E. Brown, MS, ARNP, ACNP-BC, CCRN, who practices at Great River Nephrology in West Burlington, Iowa.
Q) In school, they always emphasized the abdominal exam to rule out Wilms tumors. Are Wilms tumors still with us? Has treatment and evaluation changed?
Wilms tumor is a renal cancer found most commonly in children younger than 9 and represents approximately 7% of all malignancies in children.8,9 It can occur in one or both kidneys, with earlier diagnosis noted with bilateral involvement. Risk is highest among non-Hispanic white persons and African-Americans and lowest among Asians.8
Wilms tumor develops due to a genetic mutation in the WT1 gene located on the 11p13 chromosome. Defects are also noted on the 11p15 chromosome and the p53 tumor suppressor gene.10 Urbach et al recently identified a relationship between the LIN28 gene and Wilms tumor.11 Tumors develop when embryonic renal cells that should cease growing at the time of birth continue to grow in the postnatal period. Wilms tumor can be familial or sporadic. It can also be associated with various congenital anomalies manifested within various syndromes (see Table 2), as well as isolated genitourinary abnormalities, especially in boys.10
Most children present with a palpable, smooth, firm, generally painless mass in the abdomen; those who have bilateral renal involvement usually present earlier than those with unilateral involvement. Palpation of the abdomen during examination, if vigorous, can result in rupture of the renal capsule and tumor spillage. Additional symptoms include hematuria, fever, and hypertension. Referral to pediatric oncology is imperative.12
Definitive diagnosis is made by histologic evaluation following biopsy or surgical excision.13 Other possible diagnostic tests include but are not limited to abdominal ultrasound or CT; chest CT (to rule out metastatic lung disease); urinalysis (to evaluate for hematuria and proteinuria); liver function studies (to evaluate for hepatic involvement); and laboratory studies to measure coagulation, serum calcium, blood urea nitrogen, creatinine, and complete blood count.
Histologic examination for staging (I-V) occurs following surgical excision of the tumor. There are two staging systems available: the National Wilms Tumor Study, based on postoperative tumor evaluation, and the International Society of Pediatric Oncology, based on postchemotherapy evaluation.13
Treatment options include surgical excision (including complete nephrectomy of the affected kidney), chemotherapy based on tumor staging, and internal and/or external radiation therapy.13
Susan E. Brown, MS, ARNP,
ACNP-BC, CCRN
Great River Nephrology,
West Burlington, Iowa
REFERENCES
1. United States Renal Data System. Annual data report: atlas of chronic kidney disease and end-stage renal disease in the United States (2012). www.usrds.org/2012/view/v1_01.aspx. Accessed October 19, 2014.
2. Kopp JB, Nelson GW, Sampath K, et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy.
J Am Soc Nephrol. 2011;22(11):2129-2137.
3. Parsa A, Kao L, Xie D, et al; AASK and CRIC Study Investigators. APOL1 risk variants, race and progression of chronic kidney disease.
N Engl J Med. 2013;369:2183-2196.
4. Foster MC, Coresh J, Fornage M, et al. APOL1 variants associate with increased risk of CKD among African Americans. J Am Soc Nephrol. 2013;24(9):1484-1491.
5. Lipkowitz MS, Freedman BI, Langefeld CD, et al; AASK Investigators. Apolipoprotein L1 gene variants associate with hypertension-attributed nephropathy and the rate of kidney function decline in African Americans. Kidney Int. 2013;83(1):114–120.
6. Reeves-Daniel AM, DePalma JA, Bleyer AJ, et al. The APOL1 gene and allograft survival after kidney transplantation. Am J Transplant. 2011;11(5):1025-1030.
7. Partners Healthcare Personalized Medicine. Order APOL1 genotyping test for non-diabetic nephropathy kidney disease. http://personal izedmedicine.partners.org/Laboratory-For-Molecular-Medicine/Ordering/Kidney-Dis ease/APOL1-Gene-Sequencing.aspx. Accessed October 19, 2014.
8. Grovas A, Fremgen A, Rauck A, et al. The National Cancer Data Base report on patterns of childhood cancers in the United States. Cancer. 1997;80(12):2321-2332.
9. Johns Hopkins Medicine. Wilm’s tumor. www.hopkinsmedicine.org/kimmel_cancer_center/centers/pediatric_oncology/cancer_types/wilms_tumor.html. Accessed October 19, 2014.
10. Dome JS, Huff V. Wilms tumor overview. In: Pagon RA, Adam MP, Ardinger HH, et al (eds). GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle; 1993-2014. www.ncbi.nlm.nih.gov/books/NBK1294/. Accessed October 19, 2014.
11. Urbach A, Yermalovich A, Zhang J, et al. Lin28 sustains early renal progenitors and induces Wilms tumor. Genes & Dev. 2014;28:971-982.
12. Fernandez C, Geller JI, Ehrlich PF, et al. Renal tumors. In: Pizzo P, Poplack D (eds). Principles and Practice of Pediatric Oncology. 6th ed, St Louis, MO: Lippincott Williams & Wilkins. 2011; 861.
13. Metzger ML, Dome JS. Current therapy for Wilms’ tumor. Oncologist. 2005;10(10):815-826.
Q) In school, they always emphasized the abdominal exam to rule out Wilms tumors. Are Wilms tumors still with us? Has treatment and evaluation changed?
Wilms tumor is a renal cancer found most commonly in children younger than 9 and represents approximately 7% of all malignancies in children.8,9 It can occur in one or both kidneys, with earlier diagnosis noted with bilateral involvement. Risk is highest among non-Hispanic white persons and African-Americans and lowest among Asians.8
Wilms tumor develops due to a genetic mutation in the WT1 gene located on the 11p13 chromosome. Defects are also noted on the 11p15 chromosome and the p53 tumor suppressor gene.10 Urbach et al recently identified a relationship between the LIN28 gene and Wilms tumor.11 Tumors develop when embryonic renal cells that should cease growing at the time of birth continue to grow in the postnatal period. Wilms tumor can be familial or sporadic. It can also be associated with various congenital anomalies manifested within various syndromes (see Table 2), as well as isolated genitourinary abnormalities, especially in boys.10
Most children present with a palpable, smooth, firm, generally painless mass in the abdomen; those who have bilateral renal involvement usually present earlier than those with unilateral involvement. Palpation of the abdomen during examination, if vigorous, can result in rupture of the renal capsule and tumor spillage. Additional symptoms include hematuria, fever, and hypertension. Referral to pediatric oncology is imperative.12
Definitive diagnosis is made by histologic evaluation following biopsy or surgical excision.13 Other possible diagnostic tests include but are not limited to abdominal ultrasound or CT; chest CT (to rule out metastatic lung disease); urinalysis (to evaluate for hematuria and proteinuria); liver function studies (to evaluate for hepatic involvement); and laboratory studies to measure coagulation, serum calcium, blood urea nitrogen, creatinine, and complete blood count.
Histologic examination for staging (I-V) occurs following surgical excision of the tumor. There are two staging systems available: the National Wilms Tumor Study, based on postoperative tumor evaluation, and the International Society of Pediatric Oncology, based on postchemotherapy evaluation.13
Treatment options include surgical excision (including complete nephrectomy of the affected kidney), chemotherapy based on tumor staging, and internal and/or external radiation therapy.13
Susan E. Brown, MS, ARNP,
ACNP-BC, CCRN
Great River Nephrology,
West Burlington, Iowa
REFERENCES
1. United States Renal Data System. Annual data report: atlas of chronic kidney disease and end-stage renal disease in the United States (2012). www.usrds.org/2012/view/v1_01.aspx. Accessed October 19, 2014.
2. Kopp JB, Nelson GW, Sampath K, et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy.
J Am Soc Nephrol. 2011;22(11):2129-2137.
3. Parsa A, Kao L, Xie D, et al; AASK and CRIC Study Investigators. APOL1 risk variants, race and progression of chronic kidney disease.
N Engl J Med. 2013;369:2183-2196.
4. Foster MC, Coresh J, Fornage M, et al. APOL1 variants associate with increased risk of CKD among African Americans. J Am Soc Nephrol. 2013;24(9):1484-1491.
5. Lipkowitz MS, Freedman BI, Langefeld CD, et al; AASK Investigators. Apolipoprotein L1 gene variants associate with hypertension-attributed nephropathy and the rate of kidney function decline in African Americans. Kidney Int. 2013;83(1):114–120.
6. Reeves-Daniel AM, DePalma JA, Bleyer AJ, et al. The APOL1 gene and allograft survival after kidney transplantation. Am J Transplant. 2011;11(5):1025-1030.
7. Partners Healthcare Personalized Medicine. Order APOL1 genotyping test for non-diabetic nephropathy kidney disease. http://personal izedmedicine.partners.org/Laboratory-For-Molecular-Medicine/Ordering/Kidney-Dis ease/APOL1-Gene-Sequencing.aspx. Accessed October 19, 2014.
8. Grovas A, Fremgen A, Rauck A, et al. The National Cancer Data Base report on patterns of childhood cancers in the United States. Cancer. 1997;80(12):2321-2332.
9. Johns Hopkins Medicine. Wilm’s tumor. www.hopkinsmedicine.org/kimmel_cancer_center/centers/pediatric_oncology/cancer_types/wilms_tumor.html. Accessed October 19, 2014.
10. Dome JS, Huff V. Wilms tumor overview. In: Pagon RA, Adam MP, Ardinger HH, et al (eds). GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle; 1993-2014. www.ncbi.nlm.nih.gov/books/NBK1294/. Accessed October 19, 2014.
11. Urbach A, Yermalovich A, Zhang J, et al. Lin28 sustains early renal progenitors and induces Wilms tumor. Genes & Dev. 2014;28:971-982.
12. Fernandez C, Geller JI, Ehrlich PF, et al. Renal tumors. In: Pizzo P, Poplack D (eds). Principles and Practice of Pediatric Oncology. 6th ed, St Louis, MO: Lippincott Williams & Wilkins. 2011; 861.
13. Metzger ML, Dome JS. Current therapy for Wilms’ tumor. Oncologist. 2005;10(10):815-826.
Targeting the Kidneys to Improve Glycemic Control
A 37-year-old woman with a history of papillary carcinoma (status post total thyroidectomy 12 years ago, with negative recurrence) presents for a check-up. She also has polycystic ovarian syndrome (PCOS) with obesity and is taking metformin XR (one 500-mg tablet bid). Her visit is uneventful, and she leaves the office with an order for labwork.
Results indicate normal thyroid function and negative thyroglobulin. However, her serum glucose level is 350 mg/dL, so the patient is called and informed of the result. She denies polyphagia, polydipsia, and polyuria. Repeat blood work confirms overt hyperglycemia (320 mg/dL) with an A1C of 13%, undetectable C-peptide, and negative glutamic acid decarboxylase 65 (GAD65) and islet cell antibodies.
She is advised to increase her metformin dose (to two 500-mg tablets bid) and is started on insulin detemir (20 U every evening), with instructions to increase the latter by three units every two to three days until a target fasting glucose level of 100 to 140 mg/dL is achieved. She is also advised to follow a low-carbohydrate diet and increase her exercise.
The patient returns in two weeks for follow-up. She remains asymptomatic and has now increased her insulin detemir to 34 U bid (she started splitting the dosage after it reached 50 U/d). However, her glucose is still in the low 200s in the morning and the high 200s during the day (after lunch and dinner).
Her overt hyperglycemia is most likely a result of her longstanding insulin resistance, essential lack of b-cell function, and PCOS-associated obesity. Once diabetes from autoimmunity is ruled out by laboratory findings (negative antibodies) and clinical assessment (classic metabolic syndrome features), we focus on her glycemic control.
Even with nearly 70 U/d of insulin, the patient’s glycemic improvement is disappointing, suggesting significant insulin resistance and glucose toxicity. Living in an era with numerous classes of antidiabetic medications, we have lengthy discussions on treatment options. Canagliflozin, recently (at the time) approved, is included. The patient is interested in this new medication, and it is a reasonable choice to get her out of the glucotoxic phase.
After a discussion of benefits and potential adverse effects, she is placed on canagliflozin 100 mg/d. Her glucose log in one week shows fasting glucose values in the range of 140 to 160 mg/dL and postprandial glucose values in the 180s. As a result, she lowers her insulin to 25 U bid. Her renal panel shows a potassium level of 4.3 mEq/L (reference range, 3.5 to 5.3) and a glomerular filtration rate (GFR) of 103 mL/min/1.73 m2. She is advised to further increase her canagliflozin to 300 mg and slowly titrate her insulin down as needed, with a target fasting glucose level of 80 to 110 mg/dL and a postprandial target of 100 to 140 mg/dL.
What are SGLT2 inhibitors, and how do they work?
What are SGLT2 inhibitors, and how do they work?
Sodium-GLucose co-Transporter 2 (SGLT2) inhibitors are a new class of antihyperglycemic agent. The first, canagliflozin, was approved by the FDA in March 2013, followed by dapagliflozin (January 2014) and empagliflozin (August 2014).
As glucose is filtered through the nephrons of the kidney, about 90% is reabsorbed via SGLT2 in the proximal tubule (SGLT1 is responsible for the remaining 10%) so that glucose calories are not eliminated through urine.1 In a healthy person, the renal glucose threshold is about 180 mg/dL.1 When blood glucose exceeds this level, glucose is excreted into the urine. However, in diabetic patients, this threshold is higher due to the up-regulation of SGLT2s (and other glucose transporters), which worsens hyperglycemia.1 SGLT2 inhibitors will reset the threshold, which in turn will increase glucosuria and thereby lower serum glucose.1
SGLT2 inhibitors lower A1C by about 0.7% to 0.8%.2 Independent of other mechanisms such as the degree of b-cell function or insulin resistance, these agents can be used regardless of the duration of diabetes3 if the GFR is intact (≥ 45 mL/min/1.73 m2 for canagliflozin and empagliflozin, ≥ 60 mL/min/ 1.73 m2 for dapagliflozin).4,5
What are the risks and benefits associated with these agents?
What are the risks and benefits associated with these agents?
Modest weight loss is seen with the use of SGLT2 inhibitors. Initial weight loss is believed to be related to volume loss, but more sustained weight loss is thought to be from loss of fat mass.6 This is not surprising, as excreting glucose means excreting calories through urine.
Risk for hypoglycemia is extremely low, which makes this therapeutic class an attractive option. However, caution should be exercised when SGLT2 inhibitors are combined with other agents known to cause hypoglycemia (sulfonylureas and insulin).6
The most common adverse effect is genital mycotic infection. Women with a history of recurrent genital mycotic infection and uncircumcised men are at the greatest risk.6
Due to increased glycosuria, which results in an osmotic diuresis, modest blood pressure improvement has been seen (3 to 4 mm Hg systolic and 1 to 2 mm Hg diastolic7,8) in patients taking SGLT2 inhibitors, which is an additional benefit for hypertensive diabetic patients.6 On the other hand, use of SGLT2 inhibitors can also cause dehydration and volume depletion and can raise serum creatinine in patients who are already taking diuretics (particularly loop diuretics).6 Drug tolerance and adherence can be improved by advising patients to expect transient increased urination (approximately 135 to 350 mL/d increase from baseline5,9) and emphasizing the importance of good hydration and maintaining good genital hygiene.
A slight increase in LDL cholesterol was seen in clinical trials of the SGLT2 inhibitors, although this phenomenon is poorly understood. However, HDL cholesterol increased as well, maintaining the LDL:HDL ratio.6 No long-term cardiovascular outcome data are available at this time; as with any new antidiabetic medication, postmarketing studies, as required by the FDA, are currently ongoing.6
What are the options in this therapeutic category, and how are they distinct?
What are the options in this therapeutic category, and how are they distinct?
As mentioned previously, there are currently three SGLT2 inhibitors on the market: canagliflozin, dapagliflozin, and empagliflozin. There are subtle clinical differences among these three agents, which might direct the clinician’s choice.
First, canagliflozin is available in dosages of 100 and 300 mg. The starting dosage is 100 mg, which can be titrated to 300 mg in patients with a GFR ≥ 60 mL/min/1.73 m2 who require a greater glucose-lowering effect. Those with a GFR < 60 mL/min/1.73 m2 but ≥ 45 mL/min/1.73 m2 are limited to the 100-mg dosage. Dapagliflozin is available in 5-mg and 10-mg dosages, the former being the starting dosage. But dapagliflozin is not recommended in patients whose GFR is < 60 mL/min/1.73 m2.4
Empagliflozin is available in dosages of 10 and 25 mg. The starting dosage of 10 mg can be increased to 25 mg if the patient has not achieved his/her target glucose level. Either can be used in patients with a GFR ≥ 45 mL/min/1.73 m2.5
Second, hyperkalemia was seen in patients taking canagliflozin but not in those taking dapagliflozin or empagliflozin. Therefore, serum potassium should be monitored and caution used, especially when patients are being treated with potassium-sparing diuretics and/or ACE inhibitors or angiotensin II receptor blockers.6
Third, dapagliflozin carries a warning for bladder cancer, as higher rates of newly diagnosed bladder cancer were seen with this drug compared with placebo or comparator drugs (0.17% vs 0.03%, respectively).4 However, this finding may have resulted from a randomization imbalance of patients in the study, and further research is needed to clarify this risk.6 It is not recommended that dapagliflozin be used in patients with active or a history of bladder cancer at this time.
With these agents, there is a paradoxical rise in glucagon that increases endogenous glucose production from the liver.10 The mechanism is poorly understood, but it might be due to the body’s compensatory (survival) mechanism to “make up” the loss of glucose through urine by increasing hepatic gluconeogenesis.
Using an incretin agent, such as dipeptidyl peptidase 4 (DPP-4) inhibitors or glucagon-like peptide 1 (GLP-1) receptor agonists, in conjunction with an SGLT2 inhibitor, has been suggested as a way to potentiate the glucose-lowering effect, as it may attenuate the paradoxical rise in glucagon.10 Since the incretin class is weight neutral (DPP-4 inhibitors) or associated with weight loss (GLP-1 agonists), using incretins with SGLT2 inhibitors might produce more significant weight loss, which has numerous additional benefits for diabetic patients.
SGLT2 inhibitors are currently approved as an adjunct to diet and exercise for patients with type 2 diabetes. They are not approved for those with type 1 diabetes, although the mechanism of action of these drugs (which is independent of the b-cell function) might make them effective in this population. Active pilot studies of this patient population are in progress.11
Conclusion
In summary, SGLT2 inhibitors are an exciting new class of antidiabetic medication that offers a unique mechanism to lower serum glucose. It is the only medication that will actually remove glucose from the body; by contrast, all other existing antidiabetic medications move glucose within the body (to liver, fat, muscle, etc).
There is no curative medication for diabetes. But with an increasing diabetic population and an emphasis on individualizing antihyperglycemic regimens, we always welcome medications with novel mechanisms of action. Due to SLGT2 inhibitors’ recent approval, however, short-term and long-term adverse effects are unknown, and ongoing postmarketing surveillance should be closely followed.
References
1. Abdul-Ghani MA, DeFronzo RA. Inhibition of renal glucose reabsorption: a novel strategy for achieving glucose control in type 2 diabetes mellitus. Endocr Pract. 2008;14:782-790.
2. Berhan A, Barker A. Sodium glucose co-transport 2 inhibitors in the treatment of type 2 diabetes mellitus: a meta-analysis of randomized double-blind controlled trials. BMC Endocr Disord. 2013;13(1):58.
3. Wilding JP, Norwood P, T’joen C, et al. A study of dapagliflozin in patients with type 2 diabetes receiving high doses of insulin plus insulin sensitizers. Diabetes Care. 2009;32:1656-1662.
4. Taylor JR. Dapagliflozin offers differences from other SGLT2 inhibitors. Endocrine Today. May 2014.
5. Jardiance [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals, Inc; 2014.
6. Bakris G, Fonseca VA, Peters AL, Wysham CH. Clinical perspectives on the role of the kidney in the pathophysiology of T2DM: emerging options for treatment [video series]. 2013. www.thedoctorschannel.com/view/the-kid ney-in-t2dm-cme-part-1/. Accessed September 12, 2014.
7. Vercruysse F. Efficacy and safety of canagliflozin in subjects with type 2 diabetes mellitus inadequately controlled with metformin plus sulphonylurea over 52 weeks [abstract 934]. Presented at the 49th European Association for the Study of Diabetes Annual Meeting: Barcelona; September 24, 2013.
8. Hach T. Empagliflozin improves glycaemic parameters and cardiovascular risk factors in patients with type 2 diabetes: pooled data from four pivotal phase III trials [abstract 943]. Presented at the 49th European Association for the Study of Diabetes Annual Meeting: Barcelona; September 24, 2013.
9. List JF, Woo V, Morales E, et al. Sodium-glucose co-transport inhibition with dapagliflozin in type 2 diabetes mellitus. Diabetes Care. 2009;32(4):650-657.
10. Merovci A, Solis-Herrera C, Daniele G, et al. Dapagliflozin improves muscle insulin sensitivity but enhances endogenous glucose production. J Clin Invest. 2014;124(5):2287.
11. Perkins BA, Cherney DZ, Partridge H, et al. Sodium-glucose cotransporter 2 inhibition and glycemic control in type 1 diabetes: results of an 8-week open-label proof-of-concept trial. Diabetes Care. 2014;37(5):1480-1483.
Ji Hyun Chun practices at Endocrinology Associates in Scottsdale, Arizona, and is a faculty member in the Arizona School of Health Sciences at AT Still University.
Clinician Reviews in partnership with![]()
Ji Hyun Chun practices at Endocrinology Associates in Scottsdale, Arizona, and is a faculty member in the Arizona School of Health Sciences at AT Still University.
Clinician Reviews in partnership with![]()
Ji Hyun Chun practices at Endocrinology Associates in Scottsdale, Arizona, and is a faculty member in the Arizona School of Health Sciences at AT Still University.
Clinician Reviews in partnership with![]()
A 37-year-old woman with a history of papillary carcinoma (status post total thyroidectomy 12 years ago, with negative recurrence) presents for a check-up. She also has polycystic ovarian syndrome (PCOS) with obesity and is taking metformin XR (one 500-mg tablet bid). Her visit is uneventful, and she leaves the office with an order for labwork.
Results indicate normal thyroid function and negative thyroglobulin. However, her serum glucose level is 350 mg/dL, so the patient is called and informed of the result. She denies polyphagia, polydipsia, and polyuria. Repeat blood work confirms overt hyperglycemia (320 mg/dL) with an A1C of 13%, undetectable C-peptide, and negative glutamic acid decarboxylase 65 (GAD65) and islet cell antibodies.
She is advised to increase her metformin dose (to two 500-mg tablets bid) and is started on insulin detemir (20 U every evening), with instructions to increase the latter by three units every two to three days until a target fasting glucose level of 100 to 140 mg/dL is achieved. She is also advised to follow a low-carbohydrate diet and increase her exercise.
The patient returns in two weeks for follow-up. She remains asymptomatic and has now increased her insulin detemir to 34 U bid (she started splitting the dosage after it reached 50 U/d). However, her glucose is still in the low 200s in the morning and the high 200s during the day (after lunch and dinner).
Her overt hyperglycemia is most likely a result of her longstanding insulin resistance, essential lack of b-cell function, and PCOS-associated obesity. Once diabetes from autoimmunity is ruled out by laboratory findings (negative antibodies) and clinical assessment (classic metabolic syndrome features), we focus on her glycemic control.
Even with nearly 70 U/d of insulin, the patient’s glycemic improvement is disappointing, suggesting significant insulin resistance and glucose toxicity. Living in an era with numerous classes of antidiabetic medications, we have lengthy discussions on treatment options. Canagliflozin, recently (at the time) approved, is included. The patient is interested in this new medication, and it is a reasonable choice to get her out of the glucotoxic phase.
After a discussion of benefits and potential adverse effects, she is placed on canagliflozin 100 mg/d. Her glucose log in one week shows fasting glucose values in the range of 140 to 160 mg/dL and postprandial glucose values in the 180s. As a result, she lowers her insulin to 25 U bid. Her renal panel shows a potassium level of 4.3 mEq/L (reference range, 3.5 to 5.3) and a glomerular filtration rate (GFR) of 103 mL/min/1.73 m2. She is advised to further increase her canagliflozin to 300 mg and slowly titrate her insulin down as needed, with a target fasting glucose level of 80 to 110 mg/dL and a postprandial target of 100 to 140 mg/dL.
What are SGLT2 inhibitors, and how do they work?
What are SGLT2 inhibitors, and how do they work?
Sodium-GLucose co-Transporter 2 (SGLT2) inhibitors are a new class of antihyperglycemic agent. The first, canagliflozin, was approved by the FDA in March 2013, followed by dapagliflozin (January 2014) and empagliflozin (August 2014).
As glucose is filtered through the nephrons of the kidney, about 90% is reabsorbed via SGLT2 in the proximal tubule (SGLT1 is responsible for the remaining 10%) so that glucose calories are not eliminated through urine.1 In a healthy person, the renal glucose threshold is about 180 mg/dL.1 When blood glucose exceeds this level, glucose is excreted into the urine. However, in diabetic patients, this threshold is higher due to the up-regulation of SGLT2s (and other glucose transporters), which worsens hyperglycemia.1 SGLT2 inhibitors will reset the threshold, which in turn will increase glucosuria and thereby lower serum glucose.1
SGLT2 inhibitors lower A1C by about 0.7% to 0.8%.2 Independent of other mechanisms such as the degree of b-cell function or insulin resistance, these agents can be used regardless of the duration of diabetes3 if the GFR is intact (≥ 45 mL/min/1.73 m2 for canagliflozin and empagliflozin, ≥ 60 mL/min/ 1.73 m2 for dapagliflozin).4,5
What are the risks and benefits associated with these agents?
What are the risks and benefits associated with these agents?
Modest weight loss is seen with the use of SGLT2 inhibitors. Initial weight loss is believed to be related to volume loss, but more sustained weight loss is thought to be from loss of fat mass.6 This is not surprising, as excreting glucose means excreting calories through urine.
Risk for hypoglycemia is extremely low, which makes this therapeutic class an attractive option. However, caution should be exercised when SGLT2 inhibitors are combined with other agents known to cause hypoglycemia (sulfonylureas and insulin).6
The most common adverse effect is genital mycotic infection. Women with a history of recurrent genital mycotic infection and uncircumcised men are at the greatest risk.6
Due to increased glycosuria, which results in an osmotic diuresis, modest blood pressure improvement has been seen (3 to 4 mm Hg systolic and 1 to 2 mm Hg diastolic7,8) in patients taking SGLT2 inhibitors, which is an additional benefit for hypertensive diabetic patients.6 On the other hand, use of SGLT2 inhibitors can also cause dehydration and volume depletion and can raise serum creatinine in patients who are already taking diuretics (particularly loop diuretics).6 Drug tolerance and adherence can be improved by advising patients to expect transient increased urination (approximately 135 to 350 mL/d increase from baseline5,9) and emphasizing the importance of good hydration and maintaining good genital hygiene.
A slight increase in LDL cholesterol was seen in clinical trials of the SGLT2 inhibitors, although this phenomenon is poorly understood. However, HDL cholesterol increased as well, maintaining the LDL:HDL ratio.6 No long-term cardiovascular outcome data are available at this time; as with any new antidiabetic medication, postmarketing studies, as required by the FDA, are currently ongoing.6
What are the options in this therapeutic category, and how are they distinct?
What are the options in this therapeutic category, and how are they distinct?
As mentioned previously, there are currently three SGLT2 inhibitors on the market: canagliflozin, dapagliflozin, and empagliflozin. There are subtle clinical differences among these three agents, which might direct the clinician’s choice.
First, canagliflozin is available in dosages of 100 and 300 mg. The starting dosage is 100 mg, which can be titrated to 300 mg in patients with a GFR ≥ 60 mL/min/1.73 m2 who require a greater glucose-lowering effect. Those with a GFR < 60 mL/min/1.73 m2 but ≥ 45 mL/min/1.73 m2 are limited to the 100-mg dosage. Dapagliflozin is available in 5-mg and 10-mg dosages, the former being the starting dosage. But dapagliflozin is not recommended in patients whose GFR is < 60 mL/min/1.73 m2.4
Empagliflozin is available in dosages of 10 and 25 mg. The starting dosage of 10 mg can be increased to 25 mg if the patient has not achieved his/her target glucose level. Either can be used in patients with a GFR ≥ 45 mL/min/1.73 m2.5
Second, hyperkalemia was seen in patients taking canagliflozin but not in those taking dapagliflozin or empagliflozin. Therefore, serum potassium should be monitored and caution used, especially when patients are being treated with potassium-sparing diuretics and/or ACE inhibitors or angiotensin II receptor blockers.6
Third, dapagliflozin carries a warning for bladder cancer, as higher rates of newly diagnosed bladder cancer were seen with this drug compared with placebo or comparator drugs (0.17% vs 0.03%, respectively).4 However, this finding may have resulted from a randomization imbalance of patients in the study, and further research is needed to clarify this risk.6 It is not recommended that dapagliflozin be used in patients with active or a history of bladder cancer at this time.
With these agents, there is a paradoxical rise in glucagon that increases endogenous glucose production from the liver.10 The mechanism is poorly understood, but it might be due to the body’s compensatory (survival) mechanism to “make up” the loss of glucose through urine by increasing hepatic gluconeogenesis.
Using an incretin agent, such as dipeptidyl peptidase 4 (DPP-4) inhibitors or glucagon-like peptide 1 (GLP-1) receptor agonists, in conjunction with an SGLT2 inhibitor, has been suggested as a way to potentiate the glucose-lowering effect, as it may attenuate the paradoxical rise in glucagon.10 Since the incretin class is weight neutral (DPP-4 inhibitors) or associated with weight loss (GLP-1 agonists), using incretins with SGLT2 inhibitors might produce more significant weight loss, which has numerous additional benefits for diabetic patients.
SGLT2 inhibitors are currently approved as an adjunct to diet and exercise for patients with type 2 diabetes. They are not approved for those with type 1 diabetes, although the mechanism of action of these drugs (which is independent of the b-cell function) might make them effective in this population. Active pilot studies of this patient population are in progress.11
Conclusion
In summary, SGLT2 inhibitors are an exciting new class of antidiabetic medication that offers a unique mechanism to lower serum glucose. It is the only medication that will actually remove glucose from the body; by contrast, all other existing antidiabetic medications move glucose within the body (to liver, fat, muscle, etc).
There is no curative medication for diabetes. But with an increasing diabetic population and an emphasis on individualizing antihyperglycemic regimens, we always welcome medications with novel mechanisms of action. Due to SLGT2 inhibitors’ recent approval, however, short-term and long-term adverse effects are unknown, and ongoing postmarketing surveillance should be closely followed.
References
1. Abdul-Ghani MA, DeFronzo RA. Inhibition of renal glucose reabsorption: a novel strategy for achieving glucose control in type 2 diabetes mellitus. Endocr Pract. 2008;14:782-790.
2. Berhan A, Barker A. Sodium glucose co-transport 2 inhibitors in the treatment of type 2 diabetes mellitus: a meta-analysis of randomized double-blind controlled trials. BMC Endocr Disord. 2013;13(1):58.
3. Wilding JP, Norwood P, T’joen C, et al. A study of dapagliflozin in patients with type 2 diabetes receiving high doses of insulin plus insulin sensitizers. Diabetes Care. 2009;32:1656-1662.
4. Taylor JR. Dapagliflozin offers differences from other SGLT2 inhibitors. Endocrine Today. May 2014.
5. Jardiance [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals, Inc; 2014.
6. Bakris G, Fonseca VA, Peters AL, Wysham CH. Clinical perspectives on the role of the kidney in the pathophysiology of T2DM: emerging options for treatment [video series]. 2013. www.thedoctorschannel.com/view/the-kid ney-in-t2dm-cme-part-1/. Accessed September 12, 2014.
7. Vercruysse F. Efficacy and safety of canagliflozin in subjects with type 2 diabetes mellitus inadequately controlled with metformin plus sulphonylurea over 52 weeks [abstract 934]. Presented at the 49th European Association for the Study of Diabetes Annual Meeting: Barcelona; September 24, 2013.
8. Hach T. Empagliflozin improves glycaemic parameters and cardiovascular risk factors in patients with type 2 diabetes: pooled data from four pivotal phase III trials [abstract 943]. Presented at the 49th European Association for the Study of Diabetes Annual Meeting: Barcelona; September 24, 2013.
9. List JF, Woo V, Morales E, et al. Sodium-glucose co-transport inhibition with dapagliflozin in type 2 diabetes mellitus. Diabetes Care. 2009;32(4):650-657.
10. Merovci A, Solis-Herrera C, Daniele G, et al. Dapagliflozin improves muscle insulin sensitivity but enhances endogenous glucose production. J Clin Invest. 2014;124(5):2287.
11. Perkins BA, Cherney DZ, Partridge H, et al. Sodium-glucose cotransporter 2 inhibition and glycemic control in type 1 diabetes: results of an 8-week open-label proof-of-concept trial. Diabetes Care. 2014;37(5):1480-1483.
A 37-year-old woman with a history of papillary carcinoma (status post total thyroidectomy 12 years ago, with negative recurrence) presents for a check-up. She also has polycystic ovarian syndrome (PCOS) with obesity and is taking metformin XR (one 500-mg tablet bid). Her visit is uneventful, and she leaves the office with an order for labwork.
Results indicate normal thyroid function and negative thyroglobulin. However, her serum glucose level is 350 mg/dL, so the patient is called and informed of the result. She denies polyphagia, polydipsia, and polyuria. Repeat blood work confirms overt hyperglycemia (320 mg/dL) with an A1C of 13%, undetectable C-peptide, and negative glutamic acid decarboxylase 65 (GAD65) and islet cell antibodies.
She is advised to increase her metformin dose (to two 500-mg tablets bid) and is started on insulin detemir (20 U every evening), with instructions to increase the latter by three units every two to three days until a target fasting glucose level of 100 to 140 mg/dL is achieved. She is also advised to follow a low-carbohydrate diet and increase her exercise.
The patient returns in two weeks for follow-up. She remains asymptomatic and has now increased her insulin detemir to 34 U bid (she started splitting the dosage after it reached 50 U/d). However, her glucose is still in the low 200s in the morning and the high 200s during the day (after lunch and dinner).
Her overt hyperglycemia is most likely a result of her longstanding insulin resistance, essential lack of b-cell function, and PCOS-associated obesity. Once diabetes from autoimmunity is ruled out by laboratory findings (negative antibodies) and clinical assessment (classic metabolic syndrome features), we focus on her glycemic control.
Even with nearly 70 U/d of insulin, the patient’s glycemic improvement is disappointing, suggesting significant insulin resistance and glucose toxicity. Living in an era with numerous classes of antidiabetic medications, we have lengthy discussions on treatment options. Canagliflozin, recently (at the time) approved, is included. The patient is interested in this new medication, and it is a reasonable choice to get her out of the glucotoxic phase.
After a discussion of benefits and potential adverse effects, she is placed on canagliflozin 100 mg/d. Her glucose log in one week shows fasting glucose values in the range of 140 to 160 mg/dL and postprandial glucose values in the 180s. As a result, she lowers her insulin to 25 U bid. Her renal panel shows a potassium level of 4.3 mEq/L (reference range, 3.5 to 5.3) and a glomerular filtration rate (GFR) of 103 mL/min/1.73 m2. She is advised to further increase her canagliflozin to 300 mg and slowly titrate her insulin down as needed, with a target fasting glucose level of 80 to 110 mg/dL and a postprandial target of 100 to 140 mg/dL.
What are SGLT2 inhibitors, and how do they work?
What are SGLT2 inhibitors, and how do they work?
Sodium-GLucose co-Transporter 2 (SGLT2) inhibitors are a new class of antihyperglycemic agent. The first, canagliflozin, was approved by the FDA in March 2013, followed by dapagliflozin (January 2014) and empagliflozin (August 2014).
As glucose is filtered through the nephrons of the kidney, about 90% is reabsorbed via SGLT2 in the proximal tubule (SGLT1 is responsible for the remaining 10%) so that glucose calories are not eliminated through urine.1 In a healthy person, the renal glucose threshold is about 180 mg/dL.1 When blood glucose exceeds this level, glucose is excreted into the urine. However, in diabetic patients, this threshold is higher due to the up-regulation of SGLT2s (and other glucose transporters), which worsens hyperglycemia.1 SGLT2 inhibitors will reset the threshold, which in turn will increase glucosuria and thereby lower serum glucose.1
SGLT2 inhibitors lower A1C by about 0.7% to 0.8%.2 Independent of other mechanisms such as the degree of b-cell function or insulin resistance, these agents can be used regardless of the duration of diabetes3 if the GFR is intact (≥ 45 mL/min/1.73 m2 for canagliflozin and empagliflozin, ≥ 60 mL/min/ 1.73 m2 for dapagliflozin).4,5
What are the risks and benefits associated with these agents?
What are the risks and benefits associated with these agents?
Modest weight loss is seen with the use of SGLT2 inhibitors. Initial weight loss is believed to be related to volume loss, but more sustained weight loss is thought to be from loss of fat mass.6 This is not surprising, as excreting glucose means excreting calories through urine.
Risk for hypoglycemia is extremely low, which makes this therapeutic class an attractive option. However, caution should be exercised when SGLT2 inhibitors are combined with other agents known to cause hypoglycemia (sulfonylureas and insulin).6
The most common adverse effect is genital mycotic infection. Women with a history of recurrent genital mycotic infection and uncircumcised men are at the greatest risk.6
Due to increased glycosuria, which results in an osmotic diuresis, modest blood pressure improvement has been seen (3 to 4 mm Hg systolic and 1 to 2 mm Hg diastolic7,8) in patients taking SGLT2 inhibitors, which is an additional benefit for hypertensive diabetic patients.6 On the other hand, use of SGLT2 inhibitors can also cause dehydration and volume depletion and can raise serum creatinine in patients who are already taking diuretics (particularly loop diuretics).6 Drug tolerance and adherence can be improved by advising patients to expect transient increased urination (approximately 135 to 350 mL/d increase from baseline5,9) and emphasizing the importance of good hydration and maintaining good genital hygiene.
A slight increase in LDL cholesterol was seen in clinical trials of the SGLT2 inhibitors, although this phenomenon is poorly understood. However, HDL cholesterol increased as well, maintaining the LDL:HDL ratio.6 No long-term cardiovascular outcome data are available at this time; as with any new antidiabetic medication, postmarketing studies, as required by the FDA, are currently ongoing.6
What are the options in this therapeutic category, and how are they distinct?
What are the options in this therapeutic category, and how are they distinct?
As mentioned previously, there are currently three SGLT2 inhibitors on the market: canagliflozin, dapagliflozin, and empagliflozin. There are subtle clinical differences among these three agents, which might direct the clinician’s choice.
First, canagliflozin is available in dosages of 100 and 300 mg. The starting dosage is 100 mg, which can be titrated to 300 mg in patients with a GFR ≥ 60 mL/min/1.73 m2 who require a greater glucose-lowering effect. Those with a GFR < 60 mL/min/1.73 m2 but ≥ 45 mL/min/1.73 m2 are limited to the 100-mg dosage. Dapagliflozin is available in 5-mg and 10-mg dosages, the former being the starting dosage. But dapagliflozin is not recommended in patients whose GFR is < 60 mL/min/1.73 m2.4
Empagliflozin is available in dosages of 10 and 25 mg. The starting dosage of 10 mg can be increased to 25 mg if the patient has not achieved his/her target glucose level. Either can be used in patients with a GFR ≥ 45 mL/min/1.73 m2.5
Second, hyperkalemia was seen in patients taking canagliflozin but not in those taking dapagliflozin or empagliflozin. Therefore, serum potassium should be monitored and caution used, especially when patients are being treated with potassium-sparing diuretics and/or ACE inhibitors or angiotensin II receptor blockers.6
Third, dapagliflozin carries a warning for bladder cancer, as higher rates of newly diagnosed bladder cancer were seen with this drug compared with placebo or comparator drugs (0.17% vs 0.03%, respectively).4 However, this finding may have resulted from a randomization imbalance of patients in the study, and further research is needed to clarify this risk.6 It is not recommended that dapagliflozin be used in patients with active or a history of bladder cancer at this time.
With these agents, there is a paradoxical rise in glucagon that increases endogenous glucose production from the liver.10 The mechanism is poorly understood, but it might be due to the body’s compensatory (survival) mechanism to “make up” the loss of glucose through urine by increasing hepatic gluconeogenesis.
Using an incretin agent, such as dipeptidyl peptidase 4 (DPP-4) inhibitors or glucagon-like peptide 1 (GLP-1) receptor agonists, in conjunction with an SGLT2 inhibitor, has been suggested as a way to potentiate the glucose-lowering effect, as it may attenuate the paradoxical rise in glucagon.10 Since the incretin class is weight neutral (DPP-4 inhibitors) or associated with weight loss (GLP-1 agonists), using incretins with SGLT2 inhibitors might produce more significant weight loss, which has numerous additional benefits for diabetic patients.
SGLT2 inhibitors are currently approved as an adjunct to diet and exercise for patients with type 2 diabetes. They are not approved for those with type 1 diabetes, although the mechanism of action of these drugs (which is independent of the b-cell function) might make them effective in this population. Active pilot studies of this patient population are in progress.11
Conclusion
In summary, SGLT2 inhibitors are an exciting new class of antidiabetic medication that offers a unique mechanism to lower serum glucose. It is the only medication that will actually remove glucose from the body; by contrast, all other existing antidiabetic medications move glucose within the body (to liver, fat, muscle, etc).
There is no curative medication for diabetes. But with an increasing diabetic population and an emphasis on individualizing antihyperglycemic regimens, we always welcome medications with novel mechanisms of action. Due to SLGT2 inhibitors’ recent approval, however, short-term and long-term adverse effects are unknown, and ongoing postmarketing surveillance should be closely followed.
References
1. Abdul-Ghani MA, DeFronzo RA. Inhibition of renal glucose reabsorption: a novel strategy for achieving glucose control in type 2 diabetes mellitus. Endocr Pract. 2008;14:782-790.
2. Berhan A, Barker A. Sodium glucose co-transport 2 inhibitors in the treatment of type 2 diabetes mellitus: a meta-analysis of randomized double-blind controlled trials. BMC Endocr Disord. 2013;13(1):58.
3. Wilding JP, Norwood P, T’joen C, et al. A study of dapagliflozin in patients with type 2 diabetes receiving high doses of insulin plus insulin sensitizers. Diabetes Care. 2009;32:1656-1662.
4. Taylor JR. Dapagliflozin offers differences from other SGLT2 inhibitors. Endocrine Today. May 2014.
5. Jardiance [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals, Inc; 2014.
6. Bakris G, Fonseca VA, Peters AL, Wysham CH. Clinical perspectives on the role of the kidney in the pathophysiology of T2DM: emerging options for treatment [video series]. 2013. www.thedoctorschannel.com/view/the-kid ney-in-t2dm-cme-part-1/. Accessed September 12, 2014.
7. Vercruysse F. Efficacy and safety of canagliflozin in subjects with type 2 diabetes mellitus inadequately controlled with metformin plus sulphonylurea over 52 weeks [abstract 934]. Presented at the 49th European Association for the Study of Diabetes Annual Meeting: Barcelona; September 24, 2013.
8. Hach T. Empagliflozin improves glycaemic parameters and cardiovascular risk factors in patients with type 2 diabetes: pooled data from four pivotal phase III trials [abstract 943]. Presented at the 49th European Association for the Study of Diabetes Annual Meeting: Barcelona; September 24, 2013.
9. List JF, Woo V, Morales E, et al. Sodium-glucose co-transport inhibition with dapagliflozin in type 2 diabetes mellitus. Diabetes Care. 2009;32(4):650-657.
10. Merovci A, Solis-Herrera C, Daniele G, et al. Dapagliflozin improves muscle insulin sensitivity but enhances endogenous glucose production. J Clin Invest. 2014;124(5):2287.
11. Perkins BA, Cherney DZ, Partridge H, et al. Sodium-glucose cotransporter 2 inhibition and glycemic control in type 1 diabetes: results of an 8-week open-label proof-of-concept trial. Diabetes Care. 2014;37(5):1480-1483.
Overcorrection of Hyponatremia
Q) A clinic patient of mine was recently admitted to the hospital with hyponatremia (serum sodium, 115 mEq/L). She was treated with 2 L of normal saline and discharged home 48 hours later, at her baseline mental status with a serum sodium level of 132 mEq/L. Two days later, she was readmitted for mental status changes, and MRI showed brain swelling. The neurologist stated this was a result of the initial treatment for her hyponatremia. How is this possible?
The cause-and-effect relationship between rapid correction of chronic hyponatremia and subsequent development of neurologic problems was discovered in the late 1970s. Central pontine and extrapontine myelinolysis (known as osmotic demyelination syndrome or ODS) is a neurologic condition that can occur from rapid sodium correction. It is diagnosed by MRI, which shows hyperintense lesions on T2-weighted images. Clinical signs include upper motor neuron signs, pseudobulbar palsy, spastic quadriparesis, and mental status changes ranging from mild confusion to coma.2
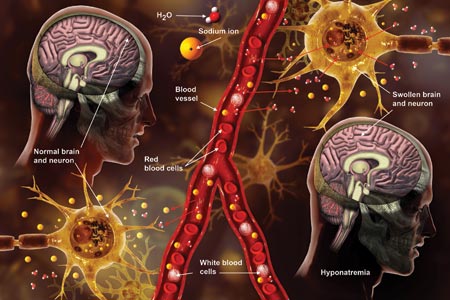
Treatment for hyponatremia should be guided by symptom management.2,3 If a patient is asymptomatic, a simple and effective strategy is to keep NPO for 24 hours, except for medications. Simple food and fluid restriction will likely increase the serum sodium level because of obligate solute losses and urinary electrolyte free water loss.2,4 While the first instinct is to feed these patients, as they often appear malnourished, this can cause a solute load leading to a too-rapid sodium correction. After 24 hours, if intake restriction is not effective, use 0.5% normal saline but with limited dosing orders, as usual saline dosing can cause too rapid a correction.2
For symptomatic patients (confusion, seizures, coma), the goal is to initially elevate sodium by 1 to 2 mEq/L per hour for the first two to three hours. Do not exceed 10 mEq/L in 24 hours or 18 mEq/L in 48 hours. Exceeding these limits puts patients at high risk for ODS. In fact, even when staying within these parameters, there is some risk for overcorrection. It is always better to go slowly.2,3
In the patient with hyponatremia due to low solute intake (eg, beer potomania), diuresis can start spontaneously after a period of food and fluid restriction. It can also be initiated with just a small amount of solute. For example, administering an IV antibiotic with a base solution of 100 mL of normal saline or a “banana bag” (an IV solution containing 0.5 to 1 L of normal saline with multivitamins/minerals that cause the fluid to be yellow) can produce several liters of diuresis.2 Once you open the floodgate, you can unintentionally cause too-rapid correction that could lead to ODS.
In chronic hyponatremic patients, low antidiuretic hormone (ADH) levels are often found; thus when a solute is introduced, there is little ADH in the system to protect against excessive water loss and electrolyte imbalance. At the same time, excessive water loss can translate to higher sodium levels and increase the risk for cerebral edema. If rapid diuresis occurs, an infusion of D5W (5% dextrose in water) to match the rate of urine output may prevent a rapid serum sodium level rise. Frequent monitoring of serum sodium levels is often necessary. In instances where ODS is already present, there are case studies of improved neurologic outcomes with reduction of serum sodium levels.2,3
While the treatment of hyponatremia at first glance seems straightforward—replace that which is lost—it can actually transform a seemingly simple problem into a complicated clinical course requiring intensive care, due to the need for frequent monitoring and intervention.
Kristina Unterseher, MSN, FNP, CNN-NP
Peacehealth St. John
Medical Center
Longview, WA
REFERENCES
1. Hilden T, Swensen TL. Electrolyte disturbances in beer drinkers: a specific “hypo-osmolaity syndrome.” Lancet. 1975;2(7928):245-246.
2. Sanghvi SR, Kellerman PS, Nanovic L. Beer potomania: an unusual cause of hyponatremia at high risk of complications from rapid correction. Am J Kidney Dis. 2007;50(4):673-680.
3. Bhattarai N, Poonam K, Panda M. Beer potomania: a case report. BMJ Case Rep. 2010; 2010: bcr10.2009.2414.
4. Campbell M. Hyponatremia and central pontine myelinolysis as a result of beer potomania: a case report. Prim Care Companion J Clin Psychiatry. 2010;12(4):PCC.09100936.
5. Thaler SM, Teitelbaum I, Beri T. “Beer potomania” in non-beer drinkers: effect of low dietary solute intake. Am J Kidney Dis. 1998;31(6):1028-1031.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Kristina Unterseher, MSN, FNP, CNN-NP, who practices at Peacehealth St. John Medical Center in Longview, Washington.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Kristina Unterseher, MSN, FNP, CNN-NP, who practices at Peacehealth St. John Medical Center in Longview, Washington.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Kristina Unterseher, MSN, FNP, CNN-NP, who practices at Peacehealth St. John Medical Center in Longview, Washington.
Q) A clinic patient of mine was recently admitted to the hospital with hyponatremia (serum sodium, 115 mEq/L). She was treated with 2 L of normal saline and discharged home 48 hours later, at her baseline mental status with a serum sodium level of 132 mEq/L. Two days later, she was readmitted for mental status changes, and MRI showed brain swelling. The neurologist stated this was a result of the initial treatment for her hyponatremia. How is this possible?
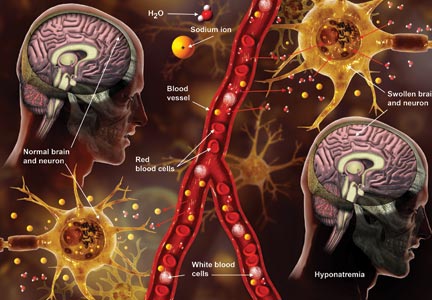
The cause-and-effect relationship between rapid correction of chronic hyponatremia and subsequent development of neurologic problems was discovered in the late 1970s. Central pontine and extrapontine myelinolysis (known as osmotic demyelination syndrome or ODS) is a neurologic condition that can occur from rapid sodium correction. It is diagnosed by MRI, which shows hyperintense lesions on T2-weighted images. Clinical signs include upper motor neuron signs, pseudobulbar palsy, spastic quadriparesis, and mental status changes ranging from mild confusion to coma.2
Treatment for hyponatremia should be guided by symptom management.2,3 If a patient is asymptomatic, a simple and effective strategy is to keep NPO for 24 hours, except for medications. Simple food and fluid restriction will likely increase the serum sodium level because of obligate solute losses and urinary electrolyte free water loss.2,4 While the first instinct is to feed these patients, as they often appear malnourished, this can cause a solute load leading to a too-rapid sodium correction. After 24 hours, if intake restriction is not effective, use 0.5% normal saline but with limited dosing orders, as usual saline dosing can cause too rapid a correction.2
For symptomatic patients (confusion, seizures, coma), the goal is to initially elevate sodium by 1 to 2 mEq/L per hour for the first two to three hours. Do not exceed 10 mEq/L in 24 hours or 18 mEq/L in 48 hours. Exceeding these limits puts patients at high risk for ODS. In fact, even when staying within these parameters, there is some risk for overcorrection. It is always better to go slowly.2,3
In the patient with hyponatremia due to low solute intake (eg, beer potomania), diuresis can start spontaneously after a period of food and fluid restriction. It can also be initiated with just a small amount of solute. For example, administering an IV antibiotic with a base solution of 100 mL of normal saline or a “banana bag” (an IV solution containing 0.5 to 1 L of normal saline with multivitamins/minerals that cause the fluid to be yellow) can produce several liters of diuresis.2 Once you open the floodgate, you can unintentionally cause too-rapid correction that could lead to ODS.
In chronic hyponatremic patients, low antidiuretic hormone (ADH) levels are often found; thus when a solute is introduced, there is little ADH in the system to protect against excessive water loss and electrolyte imbalance. At the same time, excessive water loss can translate to higher sodium levels and increase the risk for cerebral edema. If rapid diuresis occurs, an infusion of D5W (5% dextrose in water) to match the rate of urine output may prevent a rapid serum sodium level rise. Frequent monitoring of serum sodium levels is often necessary. In instances where ODS is already present, there are case studies of improved neurologic outcomes with reduction of serum sodium levels.2,3
While the treatment of hyponatremia at first glance seems straightforward—replace that which is lost—it can actually transform a seemingly simple problem into a complicated clinical course requiring intensive care, due to the need for frequent monitoring and intervention.
Kristina Unterseher, MSN, FNP, CNN-NP
Peacehealth St. John
Medical Center
Longview, WA
REFERENCES
1. Hilden T, Swensen TL. Electrolyte disturbances in beer drinkers: a specific “hypo-osmolaity syndrome.” Lancet. 1975;2(7928):245-246.
2. Sanghvi SR, Kellerman PS, Nanovic L. Beer potomania: an unusual cause of hyponatremia at high risk of complications from rapid correction. Am J Kidney Dis. 2007;50(4):673-680.
3. Bhattarai N, Poonam K, Panda M. Beer potomania: a case report. BMJ Case Rep. 2010; 2010: bcr10.2009.2414.
4. Campbell M. Hyponatremia and central pontine myelinolysis as a result of beer potomania: a case report. Prim Care Companion J Clin Psychiatry. 2010;12(4):PCC.09100936.
5. Thaler SM, Teitelbaum I, Beri T. “Beer potomania” in non-beer drinkers: effect of low dietary solute intake. Am J Kidney Dis. 1998;31(6):1028-1031.
Q) A clinic patient of mine was recently admitted to the hospital with hyponatremia (serum sodium, 115 mEq/L). She was treated with 2 L of normal saline and discharged home 48 hours later, at her baseline mental status with a serum sodium level of 132 mEq/L. Two days later, she was readmitted for mental status changes, and MRI showed brain swelling. The neurologist stated this was a result of the initial treatment for her hyponatremia. How is this possible?
The cause-and-effect relationship between rapid correction of chronic hyponatremia and subsequent development of neurologic problems was discovered in the late 1970s. Central pontine and extrapontine myelinolysis (known as osmotic demyelination syndrome or ODS) is a neurologic condition that can occur from rapid sodium correction. It is diagnosed by MRI, which shows hyperintense lesions on T2-weighted images. Clinical signs include upper motor neuron signs, pseudobulbar palsy, spastic quadriparesis, and mental status changes ranging from mild confusion to coma.2
Treatment for hyponatremia should be guided by symptom management.2,3 If a patient is asymptomatic, a simple and effective strategy is to keep NPO for 24 hours, except for medications. Simple food and fluid restriction will likely increase the serum sodium level because of obligate solute losses and urinary electrolyte free water loss.2,4 While the first instinct is to feed these patients, as they often appear malnourished, this can cause a solute load leading to a too-rapid sodium correction. After 24 hours, if intake restriction is not effective, use 0.5% normal saline but with limited dosing orders, as usual saline dosing can cause too rapid a correction.2
For symptomatic patients (confusion, seizures, coma), the goal is to initially elevate sodium by 1 to 2 mEq/L per hour for the first two to three hours. Do not exceed 10 mEq/L in 24 hours or 18 mEq/L in 48 hours. Exceeding these limits puts patients at high risk for ODS. In fact, even when staying within these parameters, there is some risk for overcorrection. It is always better to go slowly.2,3
In the patient with hyponatremia due to low solute intake (eg, beer potomania), diuresis can start spontaneously after a period of food and fluid restriction. It can also be initiated with just a small amount of solute. For example, administering an IV antibiotic with a base solution of 100 mL of normal saline or a “banana bag” (an IV solution containing 0.5 to 1 L of normal saline with multivitamins/minerals that cause the fluid to be yellow) can produce several liters of diuresis.2 Once you open the floodgate, you can unintentionally cause too-rapid correction that could lead to ODS.
In chronic hyponatremic patients, low antidiuretic hormone (ADH) levels are often found; thus when a solute is introduced, there is little ADH in the system to protect against excessive water loss and electrolyte imbalance. At the same time, excessive water loss can translate to higher sodium levels and increase the risk for cerebral edema. If rapid diuresis occurs, an infusion of D5W (5% dextrose in water) to match the rate of urine output may prevent a rapid serum sodium level rise. Frequent monitoring of serum sodium levels is often necessary. In instances where ODS is already present, there are case studies of improved neurologic outcomes with reduction of serum sodium levels.2,3
While the treatment of hyponatremia at first glance seems straightforward—replace that which is lost—it can actually transform a seemingly simple problem into a complicated clinical course requiring intensive care, due to the need for frequent monitoring and intervention.
Kristina Unterseher, MSN, FNP, CNN-NP
Peacehealth St. John
Medical Center
Longview, WA
REFERENCES
1. Hilden T, Swensen TL. Electrolyte disturbances in beer drinkers: a specific “hypo-osmolaity syndrome.” Lancet. 1975;2(7928):245-246.
2. Sanghvi SR, Kellerman PS, Nanovic L. Beer potomania: an unusual cause of hyponatremia at high risk of complications from rapid correction. Am J Kidney Dis. 2007;50(4):673-680.
3. Bhattarai N, Poonam K, Panda M. Beer potomania: a case report. BMJ Case Rep. 2010; 2010: bcr10.2009.2414.
4. Campbell M. Hyponatremia and central pontine myelinolysis as a result of beer potomania: a case report. Prim Care Companion J Clin Psychiatry. 2010;12(4):PCC.09100936.
5. Thaler SM, Teitelbaum I, Beri T. “Beer potomania” in non-beer drinkers: effect of low dietary solute intake. Am J Kidney Dis. 1998;31(6):1028-1031.
Hyponatremia: Beer Potomania
Q) Recently, we had a patient admitted for hyponatremia with
a serum sodium level of 117 mEq/L. One of the hospitalists mentioned “beer potomania” in the differential. Not wanting to look dumb, I just agreed. What is beer potomania, and how is it related to low serum sodium?
Potomania is the excessive consumption of alcoholic beverages; beer potomania is used to refer to a dilutional hyponatremia caused by excessive consumption of beer.1 First recognized in 1971, this cause of hyponatremia is not the most common but should be in the differential if the patient is a heavy alcohol imbiber who presents with encephalopathy and low serum sodium.
When considering this diagnosis, keep in mind that hyponatremia is common among chronic alcoholics and can be due to conditions such as cirrhosis, congestive heart failure, syndrome of inappropriate antidiuretic hormone (SIADH) secretion, and hypovolemia. Less common but still belonging in the differential are pseudohyponatremia secondary to alcohol-induced severe hypertriglyceridemia and cerebral salt wasting syndrome.2,3
Beer potomania usually manifests as altered mental status, weakness, and gait disturbance with an average serum sodium concentration of 108 mEq/L.3 Other abnormal lab results consistent with this diagnosis include hypokalemia (mean potassium, 3 mEq/L) and low blood urea nitrogen and urine sodium levels.2,3 Another fairly consistent finding is a recent personal history of binge drinking (more than about 5 L, or 14 cans of beer, in 24 hours) and/or history of illness (vomiting, diarrhea) that predisposed the patient to a rapid drop in serum sodium levels.2
Based on the information presented thus far, you may ask, “Why haven’t I seen this diagnosed more often? There are a lot of beer bingers out there!” Good question. Let’s review the pathophysiology of beer potomania. When patients have poor protein and solute (food, electrolytes) intake, they can experience water intoxication with smaller-than-usual volumes of fluid. The kidneys need a certain amount of solute to facilitate free water clearance (the ability to clear excess fluid from the body). A lack of adequate solute results in a buildup of free water in the vascular system, leading to a dilutional hyponatremia.3
Free water clearance is dependent on both solute excretion and the ability to dilute urine. Someone consuming an average diet will excrete 600 to 900 mOsm/d of solute. This osmolar load in-cludes urea generated from protein (10 g of protein produces about 50 mOsm of urea), along with dietary sodium and potassium. The maximum capacity for urinary dilution is 50 mOsm/L. In a nutritionally sound person, a lot of fluid—about 20 L—would be required to overwhelm the body’s capacity for urinary dilution.2
However, when you don’t eat, the body starts to break down tissue to create energy to survive. This catabolism creates 100 to 150 mOsm/d of urea, allowing you to continue to appropriately excrete a moderate amount of fluid in spite of poor solute intake ... as long as you are not drinking excessive amounts of water.5
Alcoholics get a moderate amount of their calories via beer consumption and do not experience this endogenous protein breakdown or its resultant low urea/solute level. With low solute intake, dramatically lower fluid intake (about 14 cans of beer) will overwhelm the kidneys’ ability to clear excess free water in the body.2 Fortunately, most heavy beer drinkers continue to eat at least modestly, which is sufficient to avoid this rare type of hyponatremia. Chronic alcoholics who go on a drinking binge beyond their normal baseline alcohol consumption, or who develop a flulike illness that causes electrolyte depletion (via diarrhea or vomiting), are at higher risk for beer potomania.
Kristina Unterseher, MSN, FNP, CNN-NP
Peacehealth St. John
Medical Center
Longview, WA
REFERENCES
1. Hilden T, Swensen TL. Electrolyte disturbances in beer drinkers: a specific “hypo-osmolaity syndrome.” Lancet. 1975;2(7928):245-246.
2. Sanghvi SR, Kellerman PS, Nanovic L. Beer potomania: an unusual cause of hyponatremia at high risk of complications from rapid correction. Am J Kidney Dis. 2007;50(4):673-680.
3. Bhattarai N, Poonam K, Panda M. Beer potomania: a case report. BMJ Case Rep. 2010; 2010: bcr10.2009.2414.
4. Campbell M. Hyponatremia and central pontine myelinolysis as a result of beer potomania: a case report. Prim Care Companion J Clin Psychiatry. 2010;12(4):PCC.09100936.
5. Thaler SM, Teitelbaum I, Beri T. “Beer potomania” in non-beer drinkers: effect of low dietary solute intake. Am J Kidney Dis. 1998;31(6):1028-1031.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Kristina Unterseher, MSN, FNP, CNN-NP, who practices at Peacehealth St. John Medical Center in Longview, Washington.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Kristina Unterseher, MSN, FNP, CNN-NP, who practices at Peacehealth St. John Medical Center in Longview, Washington.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Kristina Unterseher, MSN, FNP, CNN-NP, who practices at Peacehealth St. John Medical Center in Longview, Washington.
Q) Recently, we had a patient admitted for hyponatremia with
a serum sodium level of 117 mEq/L. One of the hospitalists mentioned “beer potomania” in the differential. Not wanting to look dumb, I just agreed. What is beer potomania, and how is it related to low serum sodium?
Potomania is the excessive consumption of alcoholic beverages; beer potomania is used to refer to a dilutional hyponatremia caused by excessive consumption of beer.1 First recognized in 1971, this cause of hyponatremia is not the most common but should be in the differential if the patient is a heavy alcohol imbiber who presents with encephalopathy and low serum sodium.
When considering this diagnosis, keep in mind that hyponatremia is common among chronic alcoholics and can be due to conditions such as cirrhosis, congestive heart failure, syndrome of inappropriate antidiuretic hormone (SIADH) secretion, and hypovolemia. Less common but still belonging in the differential are pseudohyponatremia secondary to alcohol-induced severe hypertriglyceridemia and cerebral salt wasting syndrome.2,3
Beer potomania usually manifests as altered mental status, weakness, and gait disturbance with an average serum sodium concentration of 108 mEq/L.3 Other abnormal lab results consistent with this diagnosis include hypokalemia (mean potassium, 3 mEq/L) and low blood urea nitrogen and urine sodium levels.2,3 Another fairly consistent finding is a recent personal history of binge drinking (more than about 5 L, or 14 cans of beer, in 24 hours) and/or history of illness (vomiting, diarrhea) that predisposed the patient to a rapid drop in serum sodium levels.2
Based on the information presented thus far, you may ask, “Why haven’t I seen this diagnosed more often? There are a lot of beer bingers out there!” Good question. Let’s review the pathophysiology of beer potomania. When patients have poor protein and solute (food, electrolytes) intake, they can experience water intoxication with smaller-than-usual volumes of fluid. The kidneys need a certain amount of solute to facilitate free water clearance (the ability to clear excess fluid from the body). A lack of adequate solute results in a buildup of free water in the vascular system, leading to a dilutional hyponatremia.3
Free water clearance is dependent on both solute excretion and the ability to dilute urine. Someone consuming an average diet will excrete 600 to 900 mOsm/d of solute. This osmolar load in-cludes urea generated from protein (10 g of protein produces about 50 mOsm of urea), along with dietary sodium and potassium. The maximum capacity for urinary dilution is 50 mOsm/L. In a nutritionally sound person, a lot of fluid—about 20 L—would be required to overwhelm the body’s capacity for urinary dilution.2
However, when you don’t eat, the body starts to break down tissue to create energy to survive. This catabolism creates 100 to 150 mOsm/d of urea, allowing you to continue to appropriately excrete a moderate amount of fluid in spite of poor solute intake ... as long as you are not drinking excessive amounts of water.5
Alcoholics get a moderate amount of their calories via beer consumption and do not experience this endogenous protein breakdown or its resultant low urea/solute level. With low solute intake, dramatically lower fluid intake (about 14 cans of beer) will overwhelm the kidneys’ ability to clear excess free water in the body.2 Fortunately, most heavy beer drinkers continue to eat at least modestly, which is sufficient to avoid this rare type of hyponatremia. Chronic alcoholics who go on a drinking binge beyond their normal baseline alcohol consumption, or who develop a flulike illness that causes electrolyte depletion (via diarrhea or vomiting), are at higher risk for beer potomania.
Kristina Unterseher, MSN, FNP, CNN-NP
Peacehealth St. John
Medical Center
Longview, WA
REFERENCES
1. Hilden T, Swensen TL. Electrolyte disturbances in beer drinkers: a specific “hypo-osmolaity syndrome.” Lancet. 1975;2(7928):245-246.
2. Sanghvi SR, Kellerman PS, Nanovic L. Beer potomania: an unusual cause of hyponatremia at high risk of complications from rapid correction. Am J Kidney Dis. 2007;50(4):673-680.
3. Bhattarai N, Poonam K, Panda M. Beer potomania: a case report. BMJ Case Rep. 2010; 2010: bcr10.2009.2414.
4. Campbell M. Hyponatremia and central pontine myelinolysis as a result of beer potomania: a case report. Prim Care Companion J Clin Psychiatry. 2010;12(4):PCC.09100936.
5. Thaler SM, Teitelbaum I, Beri T. “Beer potomania” in non-beer drinkers: effect of low dietary solute intake. Am J Kidney Dis. 1998;31(6):1028-1031.
Q) Recently, we had a patient admitted for hyponatremia with
a serum sodium level of 117 mEq/L. One of the hospitalists mentioned “beer potomania” in the differential. Not wanting to look dumb, I just agreed. What is beer potomania, and how is it related to low serum sodium?
Potomania is the excessive consumption of alcoholic beverages; beer potomania is used to refer to a dilutional hyponatremia caused by excessive consumption of beer.1 First recognized in 1971, this cause of hyponatremia is not the most common but should be in the differential if the patient is a heavy alcohol imbiber who presents with encephalopathy and low serum sodium.
When considering this diagnosis, keep in mind that hyponatremia is common among chronic alcoholics and can be due to conditions such as cirrhosis, congestive heart failure, syndrome of inappropriate antidiuretic hormone (SIADH) secretion, and hypovolemia. Less common but still belonging in the differential are pseudohyponatremia secondary to alcohol-induced severe hypertriglyceridemia and cerebral salt wasting syndrome.2,3
Beer potomania usually manifests as altered mental status, weakness, and gait disturbance with an average serum sodium concentration of 108 mEq/L.3 Other abnormal lab results consistent with this diagnosis include hypokalemia (mean potassium, 3 mEq/L) and low blood urea nitrogen and urine sodium levels.2,3 Another fairly consistent finding is a recent personal history of binge drinking (more than about 5 L, or 14 cans of beer, in 24 hours) and/or history of illness (vomiting, diarrhea) that predisposed the patient to a rapid drop in serum sodium levels.2
Based on the information presented thus far, you may ask, “Why haven’t I seen this diagnosed more often? There are a lot of beer bingers out there!” Good question. Let’s review the pathophysiology of beer potomania. When patients have poor protein and solute (food, electrolytes) intake, they can experience water intoxication with smaller-than-usual volumes of fluid. The kidneys need a certain amount of solute to facilitate free water clearance (the ability to clear excess fluid from the body). A lack of adequate solute results in a buildup of free water in the vascular system, leading to a dilutional hyponatremia.3
Free water clearance is dependent on both solute excretion and the ability to dilute urine. Someone consuming an average diet will excrete 600 to 900 mOsm/d of solute. This osmolar load in-cludes urea generated from protein (10 g of protein produces about 50 mOsm of urea), along with dietary sodium and potassium. The maximum capacity for urinary dilution is 50 mOsm/L. In a nutritionally sound person, a lot of fluid—about 20 L—would be required to overwhelm the body’s capacity for urinary dilution.2
However, when you don’t eat, the body starts to break down tissue to create energy to survive. This catabolism creates 100 to 150 mOsm/d of urea, allowing you to continue to appropriately excrete a moderate amount of fluid in spite of poor solute intake ... as long as you are not drinking excessive amounts of water.5
Alcoholics get a moderate amount of their calories via beer consumption and do not experience this endogenous protein breakdown or its resultant low urea/solute level. With low solute intake, dramatically lower fluid intake (about 14 cans of beer) will overwhelm the kidneys’ ability to clear excess free water in the body.2 Fortunately, most heavy beer drinkers continue to eat at least modestly, which is sufficient to avoid this rare type of hyponatremia. Chronic alcoholics who go on a drinking binge beyond their normal baseline alcohol consumption, or who develop a flulike illness that causes electrolyte depletion (via diarrhea or vomiting), are at higher risk for beer potomania.
Kristina Unterseher, MSN, FNP, CNN-NP
Peacehealth St. John
Medical Center
Longview, WA
REFERENCES
1. Hilden T, Swensen TL. Electrolyte disturbances in beer drinkers: a specific “hypo-osmolaity syndrome.” Lancet. 1975;2(7928):245-246.
2. Sanghvi SR, Kellerman PS, Nanovic L. Beer potomania: an unusual cause of hyponatremia at high risk of complications from rapid correction. Am J Kidney Dis. 2007;50(4):673-680.
3. Bhattarai N, Poonam K, Panda M. Beer potomania: a case report. BMJ Case Rep. 2010; 2010: bcr10.2009.2414.
4. Campbell M. Hyponatremia and central pontine myelinolysis as a result of beer potomania: a case report. Prim Care Companion J Clin Psychiatry. 2010;12(4):PCC.09100936.
5. Thaler SM, Teitelbaum I, Beri T. “Beer potomania” in non-beer drinkers: effect of low dietary solute intake. Am J Kidney Dis. 1998;31(6):1028-1031.
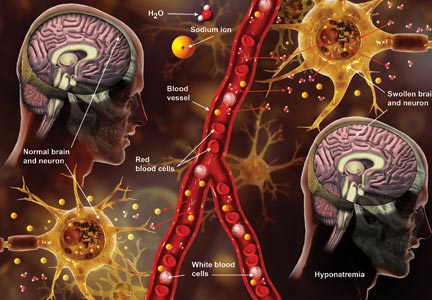
Subacute Thyroiditis
Jerry, a 48-year-old white man, is referred to endocrinology for abnormal results of thyroid tests performed four weeks ago (see table for values). Two months ago, Jerry developed an upper respiratory infection (URI) with fever, odynophagia, and anterior neck discomfort. His symptoms resolved after two weeks; however, he has since developed fatigue and nervousness.
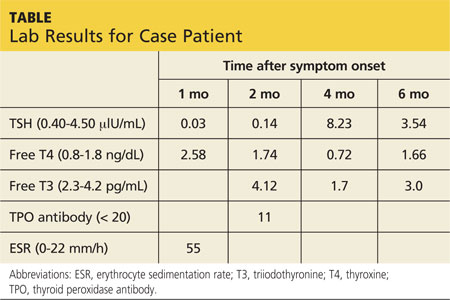
The remaining review of systems is unremarkable. Medical history is negative. Jerry denies any factors that can affect thyroid function: He does not take thyroid medication, OTC thyroid supplements, amiodarone, lithium, or interferon-α, does not have high iodine intake, and has not undergone head/neck irradiation. There is no personal or family history of thyroid disease, organ-specific autoimmune disease (ie, vitiligo, myasthenia gravis, or Sjögren syndrome) or systemic autoimmune disease (rheumatoid arthritis, systemic lupus erythematosus, or progressive systemic sclerosis).
Vital signs are stable. On physical examination, his thyroid gland is firm, with slight enlargement of the left lobe and mild tenderness. There are no palpable nodules or cervical adenopathy. The remainder of the exam is unremarkable.
Lab studies (see table) reveal an elevated erythrocyte sedimentation rate (ESR) and suppressed TSH, with normal free thyroxine (T4) and free triiodothyronine (T3) levels. His thyroid peroxidase antibody (Anti-TPO) is negative. Radioactive iodine uptake (RAIU) reveals a low 24-hour uptake of 4% (normal, 5% to 30%).
Jerry is given the presumptive diagnosis of subacute thyroiditis (SAT). He is advised that the condition will progress through multiple phases—from the initial thyrotoxicosis to euthyroidism
to transient hypothyroid—before resolution and is educated on the symptoms and signs to watch for. Since he presented in a euthyroid phase, with only mild anterior neck tenderness, no treatment is indicated. He is instructed to follow up for thyroid function testing in four to six weeks and to call with any symptomatic changes.
Two months later, Jerry returns with complaints of ongoing fatigue, unintentional weight gain, and “mental fog.” Physical exam findings are unremarkable except for a small, firm thyroid gland without the tenderness elicited previously. Labwork reveals an elevated TSH with low free T4 and free T3. He is again counseled regarding the natural history of SAT and reassured that his symptoms will abate as his thyroid hormone levels normalize. He is advised to continue the plan of follow-up testing every four to six weeks.
Approximately eight weeks later, Jerry’s thyroid function studies indicate normal levels, and he is notified of the results. Jerry comments that his symptoms have completely resolved and he is back to feeling like his usual self. He is discharged to follow-up as needed.
What is subacute thyroiditis?
WHAT IS SUBACUTE THYROIDITIS?
Subacute thyroiditis is also known as de Quervain thyroiditis or granulomatous giant cell thyroiditis.1,2 The most common cause of thyroid pain, it is a self-limited inflammatory disorder in which a painful tender goiter is associated with malaise, fever, and transient thyroid dysfunction.2,3 As with other thyroid disorders, SAT occurs most frequently in women ages 40 to 50.2,3 Thought to be of viral origin, it usually occurs after a URI and commonly correlates with the peak incidence of viral infections (spring/fall).2,3
The disruptive process begins with inflammatory destruction of thyroid follicles.2 This causes leakage of stored colloid, which is broken down, releasing unregulated T4 and T3 into the circulation and resulting in a thyrotoxicosis that typically lasts six weeks.1,2,4 Thyroid cells are incapable of producing new thyroid hormone during this time, so as excess circulating hormone is utilized, T4 and T3 levels become normal, then deficient, and the patient transitions through a period of euthyroidism to transient hypothyroidism.1,2,4 As the disruption of thyroid parenchyma abates, recovery ensues. The follicles regenerate, colloid is repleted, and normal thyroid function is restored.1-4
SAT typically lasts four to six months, although painful thyromegaly may persist for one year after resolution of thyroid dysfunction.2 Throughout the course of SAT, thyroid test results can be confusing, and misdiagnosis of hyperthyroidism or hypothyroidism may occur unless each phase of SAT is recognized.
Phases of SAT >>
PRODROME
The precursor URI is followed in days or weeks by the clinical manifestations of SAT. These typically include myalgia, pharyngitis, low-grade fever, and fatigue.2
There may be pain of varying degrees in part or all of one or both lobes; the pain often migrates to the entire gland and may radiate to the angle of the jaw or the ear of the affected side(s). Moving the head, swallowing, or coughing aggravates the pain.2
The hallmark of SAT is a markedly elevated ESR (often > 100 mm/h).1-3 Leukocyte count is normal (50% of cases) or only slightly elevated (50%).2
THYROTOXIC PHASE
Fifty percent of patients have mild to moderate symptoms of hyperthyroidism, including nervousness, weight loss, heat intolerance, or palpitations; hoarseness or dysphagia may be present.2 Signs include tremors or tachycardia. The thyroid gland may reveal slight to moderate unilateral enlargement, usually firm in the involved area, and tenderness may be mild, moderate, or severe.2 Cervical lymphadenopathy is absent.2 Serum T4 and T3 levels are elevated, and TSH is suppressed.1-4
Thyroid antibodies (antithyroid peroxidase antibodies [Anti-TPO or TPOAb] or antithyroglobulin antibodies [Anti-TG or TgAb]) have been found in 42% to 62% of patients with SAT.2 These transitory immunologic markers develop several weeks after the onset and appear to be a physiologic response to the inflammatory insult to the gland.2 In most patients, the antibody titer gradually decreases, then disappears as the disease resolves.2-4
The 24-hour RAIU is low
(< 5%) in the toxic phase of SAT, and thyroid scan will reveal a patchy and irregular distribution of the tracer.2,3 The thyrotoxicosis during this early phase is caused by the inflammatory release of preformed thyroid hormones (not hyperfunctioning in the gland), resulting in a “low-uptake thyrotoxicosis.”2 This differentiates SAT from the elevated uptake seen in Graves disease (> 30% at 24 hours).2
TRANSIENT HYPOTHYROIDISM PHASE
As circulating T4 and T3 are utilized but follicular function remains temporarily impaired, levels decline, resulting in a period of euthyroidism followed by hypothyroidism. TSH levels, previously suppressed in the thyrotoxic phase, now become elevated. This transient hypothyroidism occurs in two-thirds of patients, and the presentation varies from subclinical to pronounced.2
RECOVERY PHASE
After several weeks or months, all thyroid function studies return to normal and complete recovery commonly ensues. SAT rarely recurs, most likely due to immunity to the precipitating virus.1,2,4
Management of SAT >>
MANAGEMENT
Thyroid function should be monitored by testing every two to four weeks, dependent on the severity of the patient’s symptoms and rate of progression.1 Often, no treatment is required.1,2
Symptomatic relief of mild thyroid pain can be achieved with NSAIDs or aspirin (2 to 3 g/d). Severe symptoms can be treated with short-term prednisone, which should be tapered and discontinued.1-3 Steroids suppress the inflammatory response, and the dramatic relief of thyroid pain within 24 hours can be diagnostic of SAT.2
During the thyrotoxic phase, β-blockers (propranolol) can alleviate adrenergic symptoms, with the dose tapered once the patient is euthyroid.1-3 Antithyroid medications that directly inhibit thyroid hormone synthesis (eg, methimazole or propylthiouracil) are ineffective due to the lack of T4 and T3 production in the follicular cells after the inflammatory response.2,3
During the transient hypothyroid phase, thyroid hormone replacement may be indicated if the TSH level is markedly elevated or the phase refractory. However, levothyroxine therapy should be low dose (< 100 μg) and not be considered lifelong.2,3
DIFFERENTIAL DIAGNOSIS
During the prodrome, SAT is often misdiagnosed as pharyngitis. Acute suppurative thyroiditis initially may mimic SAT, but the febrile and leukocytic responses are greater, and localized edema, erythema, and tenderness become more evident as the condition progresses.
Painless or silent thyroiditis is distinguished from SAT by the lack of pain or tenderness and a normal ESR in the presence of a similar pattern of thyroid dysfunction. Graves disease presents with symptoms similar to the thyrotoxic phase of SAT, but T3 is usually disproportionately elevated compared to T4, RAIU is elevated, and thyroid antibodies are prevalent.2
CONCLUSION
Primary care providers may encounter SAT at some point, and a level of clinical suspicion must be maintained. Referral to endocrinology may be warranted in some cases; however, textbook cases can often be followed in primary care. Patient education is the foundation of SAT care. Symptomatic treatments may be employed as needed. Fortunately, for most patients, this self-limited disease state rarely leads to complications.
REFERENCES
1. Cooper DS. The thyroid gland. In: Gardner D, Shobeck D (eds). Greenspan’s Basic and Clinical Endocrinology. 9th ed. China: McGraw-Hill; 2011:163-226.
2. Guimaraes VC. Subacute and Riedel’s thyroiditis. In: Jameson JL, De Groot LJ (eds). Endocrinology Adult and Pediatric. 6th ed. Philadelphia: Saunders; 2010:1595-1600.
3. Jameson JL. Disorders of the thyroid gland. In: Jameson JL (ed). Harrison’s Endocrinology. 2nd ed. China: McGraw-Hill; 2010: 62-98.
4. Smallridge RC. Thyroiditis. In: McDermott MT (ed). Endocrine Secrets. 6th ed. Philadelphia, PA: Elsevier Saunders; 2013:289-293.
Jerry, a 48-year-old white man, is referred to endocrinology for abnormal results of thyroid tests performed four weeks ago (see table for values). Two months ago, Jerry developed an upper respiratory infection (URI) with fever, odynophagia, and anterior neck discomfort. His symptoms resolved after two weeks; however, he has since developed fatigue and nervousness.
The remaining review of systems is unremarkable. Medical history is negative. Jerry denies any factors that can affect thyroid function: He does not take thyroid medication, OTC thyroid supplements, amiodarone, lithium, or interferon-α, does not have high iodine intake, and has not undergone head/neck irradiation. There is no personal or family history of thyroid disease, organ-specific autoimmune disease (ie, vitiligo, myasthenia gravis, or Sjögren syndrome) or systemic autoimmune disease (rheumatoid arthritis, systemic lupus erythematosus, or progressive systemic sclerosis).
Vital signs are stable. On physical examination, his thyroid gland is firm, with slight enlargement of the left lobe and mild tenderness. There are no palpable nodules or cervical adenopathy. The remainder of the exam is unremarkable.
Lab studies (see table) reveal an elevated erythrocyte sedimentation rate (ESR) and suppressed TSH, with normal free thyroxine (T4) and free triiodothyronine (T3) levels. His thyroid peroxidase antibody (Anti-TPO) is negative. Radioactive iodine uptake (RAIU) reveals a low 24-hour uptake of 4% (normal, 5% to 30%).
Jerry is given the presumptive diagnosis of subacute thyroiditis (SAT). He is advised that the condition will progress through multiple phases—from the initial thyrotoxicosis to euthyroidism
to transient hypothyroid—before resolution and is educated on the symptoms and signs to watch for. Since he presented in a euthyroid phase, with only mild anterior neck tenderness, no treatment is indicated. He is instructed to follow up for thyroid function testing in four to six weeks and to call with any symptomatic changes.
Two months later, Jerry returns with complaints of ongoing fatigue, unintentional weight gain, and “mental fog.” Physical exam findings are unremarkable except for a small, firm thyroid gland without the tenderness elicited previously. Labwork reveals an elevated TSH with low free T4 and free T3. He is again counseled regarding the natural history of SAT and reassured that his symptoms will abate as his thyroid hormone levels normalize. He is advised to continue the plan of follow-up testing every four to six weeks.
Approximately eight weeks later, Jerry’s thyroid function studies indicate normal levels, and he is notified of the results. Jerry comments that his symptoms have completely resolved and he is back to feeling like his usual self. He is discharged to follow-up as needed.
What is subacute thyroiditis?
WHAT IS SUBACUTE THYROIDITIS?
Subacute thyroiditis is also known as de Quervain thyroiditis or granulomatous giant cell thyroiditis.1,2 The most common cause of thyroid pain, it is a self-limited inflammatory disorder in which a painful tender goiter is associated with malaise, fever, and transient thyroid dysfunction.2,3 As with other thyroid disorders, SAT occurs most frequently in women ages 40 to 50.2,3 Thought to be of viral origin, it usually occurs after a URI and commonly correlates with the peak incidence of viral infections (spring/fall).2,3
The disruptive process begins with inflammatory destruction of thyroid follicles.2 This causes leakage of stored colloid, which is broken down, releasing unregulated T4 and T3 into the circulation and resulting in a thyrotoxicosis that typically lasts six weeks.1,2,4 Thyroid cells are incapable of producing new thyroid hormone during this time, so as excess circulating hormone is utilized, T4 and T3 levels become normal, then deficient, and the patient transitions through a period of euthyroidism to transient hypothyroidism.1,2,4 As the disruption of thyroid parenchyma abates, recovery ensues. The follicles regenerate, colloid is repleted, and normal thyroid function is restored.1-4
SAT typically lasts four to six months, although painful thyromegaly may persist for one year after resolution of thyroid dysfunction.2 Throughout the course of SAT, thyroid test results can be confusing, and misdiagnosis of hyperthyroidism or hypothyroidism may occur unless each phase of SAT is recognized.
Phases of SAT >>
PRODROME
The precursor URI is followed in days or weeks by the clinical manifestations of SAT. These typically include myalgia, pharyngitis, low-grade fever, and fatigue.2
There may be pain of varying degrees in part or all of one or both lobes; the pain often migrates to the entire gland and may radiate to the angle of the jaw or the ear of the affected side(s). Moving the head, swallowing, or coughing aggravates the pain.2
The hallmark of SAT is a markedly elevated ESR (often > 100 mm/h).1-3 Leukocyte count is normal (50% of cases) or only slightly elevated (50%).2
THYROTOXIC PHASE
Fifty percent of patients have mild to moderate symptoms of hyperthyroidism, including nervousness, weight loss, heat intolerance, or palpitations; hoarseness or dysphagia may be present.2 Signs include tremors or tachycardia. The thyroid gland may reveal slight to moderate unilateral enlargement, usually firm in the involved area, and tenderness may be mild, moderate, or severe.2 Cervical lymphadenopathy is absent.2 Serum T4 and T3 levels are elevated, and TSH is suppressed.1-4
Thyroid antibodies (antithyroid peroxidase antibodies [Anti-TPO or TPOAb] or antithyroglobulin antibodies [Anti-TG or TgAb]) have been found in 42% to 62% of patients with SAT.2 These transitory immunologic markers develop several weeks after the onset and appear to be a physiologic response to the inflammatory insult to the gland.2 In most patients, the antibody titer gradually decreases, then disappears as the disease resolves.2-4
The 24-hour RAIU is low
(< 5%) in the toxic phase of SAT, and thyroid scan will reveal a patchy and irregular distribution of the tracer.2,3 The thyrotoxicosis during this early phase is caused by the inflammatory release of preformed thyroid hormones (not hyperfunctioning in the gland), resulting in a “low-uptake thyrotoxicosis.”2 This differentiates SAT from the elevated uptake seen in Graves disease (> 30% at 24 hours).2
TRANSIENT HYPOTHYROIDISM PHASE
As circulating T4 and T3 are utilized but follicular function remains temporarily impaired, levels decline, resulting in a period of euthyroidism followed by hypothyroidism. TSH levels, previously suppressed in the thyrotoxic phase, now become elevated. This transient hypothyroidism occurs in two-thirds of patients, and the presentation varies from subclinical to pronounced.2
RECOVERY PHASE
After several weeks or months, all thyroid function studies return to normal and complete recovery commonly ensues. SAT rarely recurs, most likely due to immunity to the precipitating virus.1,2,4
Management of SAT >>
MANAGEMENT
Thyroid function should be monitored by testing every two to four weeks, dependent on the severity of the patient’s symptoms and rate of progression.1 Often, no treatment is required.1,2
Symptomatic relief of mild thyroid pain can be achieved with NSAIDs or aspirin (2 to 3 g/d). Severe symptoms can be treated with short-term prednisone, which should be tapered and discontinued.1-3 Steroids suppress the inflammatory response, and the dramatic relief of thyroid pain within 24 hours can be diagnostic of SAT.2
During the thyrotoxic phase, β-blockers (propranolol) can alleviate adrenergic symptoms, with the dose tapered once the patient is euthyroid.1-3 Antithyroid medications that directly inhibit thyroid hormone synthesis (eg, methimazole or propylthiouracil) are ineffective due to the lack of T4 and T3 production in the follicular cells after the inflammatory response.2,3
During the transient hypothyroid phase, thyroid hormone replacement may be indicated if the TSH level is markedly elevated or the phase refractory. However, levothyroxine therapy should be low dose (< 100 μg) and not be considered lifelong.2,3
DIFFERENTIAL DIAGNOSIS
During the prodrome, SAT is often misdiagnosed as pharyngitis. Acute suppurative thyroiditis initially may mimic SAT, but the febrile and leukocytic responses are greater, and localized edema, erythema, and tenderness become more evident as the condition progresses.
Painless or silent thyroiditis is distinguished from SAT by the lack of pain or tenderness and a normal ESR in the presence of a similar pattern of thyroid dysfunction. Graves disease presents with symptoms similar to the thyrotoxic phase of SAT, but T3 is usually disproportionately elevated compared to T4, RAIU is elevated, and thyroid antibodies are prevalent.2
CONCLUSION
Primary care providers may encounter SAT at some point, and a level of clinical suspicion must be maintained. Referral to endocrinology may be warranted in some cases; however, textbook cases can often be followed in primary care. Patient education is the foundation of SAT care. Symptomatic treatments may be employed as needed. Fortunately, for most patients, this self-limited disease state rarely leads to complications.
REFERENCES
1. Cooper DS. The thyroid gland. In: Gardner D, Shobeck D (eds). Greenspan’s Basic and Clinical Endocrinology. 9th ed. China: McGraw-Hill; 2011:163-226.
2. Guimaraes VC. Subacute and Riedel’s thyroiditis. In: Jameson JL, De Groot LJ (eds). Endocrinology Adult and Pediatric. 6th ed. Philadelphia: Saunders; 2010:1595-1600.
3. Jameson JL. Disorders of the thyroid gland. In: Jameson JL (ed). Harrison’s Endocrinology. 2nd ed. China: McGraw-Hill; 2010: 62-98.
4. Smallridge RC. Thyroiditis. In: McDermott MT (ed). Endocrine Secrets. 6th ed. Philadelphia, PA: Elsevier Saunders; 2013:289-293.
Jerry, a 48-year-old white man, is referred to endocrinology for abnormal results of thyroid tests performed four weeks ago (see table for values). Two months ago, Jerry developed an upper respiratory infection (URI) with fever, odynophagia, and anterior neck discomfort. His symptoms resolved after two weeks; however, he has since developed fatigue and nervousness.
The remaining review of systems is unremarkable. Medical history is negative. Jerry denies any factors that can affect thyroid function: He does not take thyroid medication, OTC thyroid supplements, amiodarone, lithium, or interferon-α, does not have high iodine intake, and has not undergone head/neck irradiation. There is no personal or family history of thyroid disease, organ-specific autoimmune disease (ie, vitiligo, myasthenia gravis, or Sjögren syndrome) or systemic autoimmune disease (rheumatoid arthritis, systemic lupus erythematosus, or progressive systemic sclerosis).
Vital signs are stable. On physical examination, his thyroid gland is firm, with slight enlargement of the left lobe and mild tenderness. There are no palpable nodules or cervical adenopathy. The remainder of the exam is unremarkable.
Lab studies (see table) reveal an elevated erythrocyte sedimentation rate (ESR) and suppressed TSH, with normal free thyroxine (T4) and free triiodothyronine (T3) levels. His thyroid peroxidase antibody (Anti-TPO) is negative. Radioactive iodine uptake (RAIU) reveals a low 24-hour uptake of 4% (normal, 5% to 30%).
Jerry is given the presumptive diagnosis of subacute thyroiditis (SAT). He is advised that the condition will progress through multiple phases—from the initial thyrotoxicosis to euthyroidism
to transient hypothyroid—before resolution and is educated on the symptoms and signs to watch for. Since he presented in a euthyroid phase, with only mild anterior neck tenderness, no treatment is indicated. He is instructed to follow up for thyroid function testing in four to six weeks and to call with any symptomatic changes.
Two months later, Jerry returns with complaints of ongoing fatigue, unintentional weight gain, and “mental fog.” Physical exam findings are unremarkable except for a small, firm thyroid gland without the tenderness elicited previously. Labwork reveals an elevated TSH with low free T4 and free T3. He is again counseled regarding the natural history of SAT and reassured that his symptoms will abate as his thyroid hormone levels normalize. He is advised to continue the plan of follow-up testing every four to six weeks.
Approximately eight weeks later, Jerry’s thyroid function studies indicate normal levels, and he is notified of the results. Jerry comments that his symptoms have completely resolved and he is back to feeling like his usual self. He is discharged to follow-up as needed.
What is subacute thyroiditis?
WHAT IS SUBACUTE THYROIDITIS?
Subacute thyroiditis is also known as de Quervain thyroiditis or granulomatous giant cell thyroiditis.1,2 The most common cause of thyroid pain, it is a self-limited inflammatory disorder in which a painful tender goiter is associated with malaise, fever, and transient thyroid dysfunction.2,3 As with other thyroid disorders, SAT occurs most frequently in women ages 40 to 50.2,3 Thought to be of viral origin, it usually occurs after a URI and commonly correlates with the peak incidence of viral infections (spring/fall).2,3
The disruptive process begins with inflammatory destruction of thyroid follicles.2 This causes leakage of stored colloid, which is broken down, releasing unregulated T4 and T3 into the circulation and resulting in a thyrotoxicosis that typically lasts six weeks.1,2,4 Thyroid cells are incapable of producing new thyroid hormone during this time, so as excess circulating hormone is utilized, T4 and T3 levels become normal, then deficient, and the patient transitions through a period of euthyroidism to transient hypothyroidism.1,2,4 As the disruption of thyroid parenchyma abates, recovery ensues. The follicles regenerate, colloid is repleted, and normal thyroid function is restored.1-4
SAT typically lasts four to six months, although painful thyromegaly may persist for one year after resolution of thyroid dysfunction.2 Throughout the course of SAT, thyroid test results can be confusing, and misdiagnosis of hyperthyroidism or hypothyroidism may occur unless each phase of SAT is recognized.
Phases of SAT >>
PRODROME
The precursor URI is followed in days or weeks by the clinical manifestations of SAT. These typically include myalgia, pharyngitis, low-grade fever, and fatigue.2
There may be pain of varying degrees in part or all of one or both lobes; the pain often migrates to the entire gland and may radiate to the angle of the jaw or the ear of the affected side(s). Moving the head, swallowing, or coughing aggravates the pain.2
The hallmark of SAT is a markedly elevated ESR (often > 100 mm/h).1-3 Leukocyte count is normal (50% of cases) or only slightly elevated (50%).2
THYROTOXIC PHASE
Fifty percent of patients have mild to moderate symptoms of hyperthyroidism, including nervousness, weight loss, heat intolerance, or palpitations; hoarseness or dysphagia may be present.2 Signs include tremors or tachycardia. The thyroid gland may reveal slight to moderate unilateral enlargement, usually firm in the involved area, and tenderness may be mild, moderate, or severe.2 Cervical lymphadenopathy is absent.2 Serum T4 and T3 levels are elevated, and TSH is suppressed.1-4
Thyroid antibodies (antithyroid peroxidase antibodies [Anti-TPO or TPOAb] or antithyroglobulin antibodies [Anti-TG or TgAb]) have been found in 42% to 62% of patients with SAT.2 These transitory immunologic markers develop several weeks after the onset and appear to be a physiologic response to the inflammatory insult to the gland.2 In most patients, the antibody titer gradually decreases, then disappears as the disease resolves.2-4
The 24-hour RAIU is low
(< 5%) in the toxic phase of SAT, and thyroid scan will reveal a patchy and irregular distribution of the tracer.2,3 The thyrotoxicosis during this early phase is caused by the inflammatory release of preformed thyroid hormones (not hyperfunctioning in the gland), resulting in a “low-uptake thyrotoxicosis.”2 This differentiates SAT from the elevated uptake seen in Graves disease (> 30% at 24 hours).2
TRANSIENT HYPOTHYROIDISM PHASE
As circulating T4 and T3 are utilized but follicular function remains temporarily impaired, levels decline, resulting in a period of euthyroidism followed by hypothyroidism. TSH levels, previously suppressed in the thyrotoxic phase, now become elevated. This transient hypothyroidism occurs in two-thirds of patients, and the presentation varies from subclinical to pronounced.2
RECOVERY PHASE
After several weeks or months, all thyroid function studies return to normal and complete recovery commonly ensues. SAT rarely recurs, most likely due to immunity to the precipitating virus.1,2,4
Management of SAT >>
MANAGEMENT
Thyroid function should be monitored by testing every two to four weeks, dependent on the severity of the patient’s symptoms and rate of progression.1 Often, no treatment is required.1,2
Symptomatic relief of mild thyroid pain can be achieved with NSAIDs or aspirin (2 to 3 g/d). Severe symptoms can be treated with short-term prednisone, which should be tapered and discontinued.1-3 Steroids suppress the inflammatory response, and the dramatic relief of thyroid pain within 24 hours can be diagnostic of SAT.2
During the thyrotoxic phase, β-blockers (propranolol) can alleviate adrenergic symptoms, with the dose tapered once the patient is euthyroid.1-3 Antithyroid medications that directly inhibit thyroid hormone synthesis (eg, methimazole or propylthiouracil) are ineffective due to the lack of T4 and T3 production in the follicular cells after the inflammatory response.2,3
During the transient hypothyroid phase, thyroid hormone replacement may be indicated if the TSH level is markedly elevated or the phase refractory. However, levothyroxine therapy should be low dose (< 100 μg) and not be considered lifelong.2,3
DIFFERENTIAL DIAGNOSIS
During the prodrome, SAT is often misdiagnosed as pharyngitis. Acute suppurative thyroiditis initially may mimic SAT, but the febrile and leukocytic responses are greater, and localized edema, erythema, and tenderness become more evident as the condition progresses.
Painless or silent thyroiditis is distinguished from SAT by the lack of pain or tenderness and a normal ESR in the presence of a similar pattern of thyroid dysfunction. Graves disease presents with symptoms similar to the thyrotoxic phase of SAT, but T3 is usually disproportionately elevated compared to T4, RAIU is elevated, and thyroid antibodies are prevalent.2
CONCLUSION
Primary care providers may encounter SAT at some point, and a level of clinical suspicion must be maintained. Referral to endocrinology may be warranted in some cases; however, textbook cases can often be followed in primary care. Patient education is the foundation of SAT care. Symptomatic treatments may be employed as needed. Fortunately, for most patients, this self-limited disease state rarely leads to complications.
REFERENCES
1. Cooper DS. The thyroid gland. In: Gardner D, Shobeck D (eds). Greenspan’s Basic and Clinical Endocrinology. 9th ed. China: McGraw-Hill; 2011:163-226.
2. Guimaraes VC. Subacute and Riedel’s thyroiditis. In: Jameson JL, De Groot LJ (eds). Endocrinology Adult and Pediatric. 6th ed. Philadelphia: Saunders; 2010:1595-1600.
3. Jameson JL. Disorders of the thyroid gland. In: Jameson JL (ed). Harrison’s Endocrinology. 2nd ed. China: McGraw-Hill; 2010: 62-98.
4. Smallridge RC. Thyroiditis. In: McDermott MT (ed). Endocrine Secrets. 6th ed. Philadelphia, PA: Elsevier Saunders; 2013:289-293.
Updates on Kidney Donation
Q) A good friend was diagnosed with chronic kidney disease (CKD) and is presently undergoing workup for a transplant. He is 60 and otherwise healthy; his glomerular filtration rate (GFR) is 14, and he has no uremic symptoms. If I volunteer to give him a kidney, are there any long-term risks for me?
Kidney failure, dialysis, and kidney transplant are terms that can invoke stress and uncertainty in patients with end-stage renal disease (ESRD) and among their family members and friends. In addition to adjusting to the changes wrought by ESRD, patients may also be burdened by the prospect of a family member or friend donating a kidney to them and the concern that the donation will lead to complications for their donor. Family members or friends who volunteer may also experience stress, uncertain of their own risk for ESRD in the future.
Past research improperly compared relative risk for ESRD in donors with that in the general population (without accounting for higher propensity for complications in donors with preexisting conditions). In an effort to correct this misperception, a study recently published in JAMA compared the risk for ESRD in donors with that in a healthy group of nondonors.1 The nondonor pool was taken from the National Health and Nutrition Examination Survey (NHANES III), which assesses the health and nutritional status of adults and children in the United States.
The JAMA study included a cohort of 96,217 kidney donors in the US in a 17-year period and a cohort of 20,024 participants in a six-year period of the NHANES III trial. This data was then compared to Centers for Medicare & Medicaid Services (CMS) data to determine the development of ESRD in kidney donors. ESRD was defined by CMS as the initiation of dialysis, placement on the kidney transplant waiting list, or receipt of a living or deceased donor kidney transplant.
In addition to comparing risk for ESRD in kidney donors with that of a healthy population of nondonors, the researchers also stratified their results demographically. Thus, the lifetime rate of kidney failure in donors is 90 per 10,000, compared with 326 per 10,000 in the general population of nondonors. In healthy nondonors, the risk for kidney failure was 14 per 10,000. After 15 years, the risk for kidney failure associated with donating a kidney was 51 per 10,000 in African-American donors and 23 per 10,000 in white donors. So while the study did reveal an increased risk associated with kidney donation, the degree of risk is considered small.
These findings demonstrate the importance of understanding the facts surrounding inherent risk for ESRD in kidney donation. Overall, a donor’s lifetime risk is considered minuscule. So, to answer the question, yes, there is a slight increase in risk for kidney failure if you donate to your friend. That said, the risk is 0.014 x a standardized risk of 1. This increases at 15 years to 0.51 for African-American and 0.23 for white donors. With such tiny increases, you can safely feel good about donating a kidney to your friend.
Donna Reesman, MSN, CNP
VP Clinical & Quality Management
St Clair Specialty Physicians Detroit
REFERENCES
1. Muzaale AD, Massie AB, Wang MC, et al. Risk of end-stage renal disease following live kidney donation. JAMA. 2014;311(6):579-586.
2. CDC. HIV in the United States: at a glance (2013). www.cdc.gov/hiv/statistics/basics/ataglance.html. Accessed June 16, 2014.
3. Frassetto LA, Tan-Tam C, Stock PG. Renal transplantation in patients with HIV. Nat Rev Nephrol. 2009;5(10):582-589.
4. Malani PN. New law allows organ transplants from deceased HIV-infected donors to HIV-infected recipients. JAMA. 2013;310(23): 2492-2493.
5. Muller E, Kahn D, Mendelson M. Renal transplantation between HIV-positive donors and recipients. N Engl J Med. 2010;362(24):2336-2337.
6. Mariani LH, Berns JS. Viral nephropathies. In: Gilbert SJ, Weiner DE, eds. National Kidney Foundation’s Primer on Kidney Diseases. 6th ed. Elsevier; 2014:253-261.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP.
Q) A good friend was diagnosed with chronic kidney disease (CKD) and is presently undergoing workup for a transplant. He is 60 and otherwise healthy; his glomerular filtration rate (GFR) is 14, and he has no uremic symptoms. If I volunteer to give him a kidney, are there any long-term risks for me?
Kidney failure, dialysis, and kidney transplant are terms that can invoke stress and uncertainty in patients with end-stage renal disease (ESRD) and among their family members and friends. In addition to adjusting to the changes wrought by ESRD, patients may also be burdened by the prospect of a family member or friend donating a kidney to them and the concern that the donation will lead to complications for their donor. Family members or friends who volunteer may also experience stress, uncertain of their own risk for ESRD in the future.
Past research improperly compared relative risk for ESRD in donors with that in the general population (without accounting for higher propensity for complications in donors with preexisting conditions). In an effort to correct this misperception, a study recently published in JAMA compared the risk for ESRD in donors with that in a healthy group of nondonors.1 The nondonor pool was taken from the National Health and Nutrition Examination Survey (NHANES III), which assesses the health and nutritional status of adults and children in the United States.
The JAMA study included a cohort of 96,217 kidney donors in the US in a 17-year period and a cohort of 20,024 participants in a six-year period of the NHANES III trial. This data was then compared to Centers for Medicare & Medicaid Services (CMS) data to determine the development of ESRD in kidney donors. ESRD was defined by CMS as the initiation of dialysis, placement on the kidney transplant waiting list, or receipt of a living or deceased donor kidney transplant.
In addition to comparing risk for ESRD in kidney donors with that of a healthy population of nondonors, the researchers also stratified their results demographically. Thus, the lifetime rate of kidney failure in donors is 90 per 10,000, compared with 326 per 10,000 in the general population of nondonors. In healthy nondonors, the risk for kidney failure was 14 per 10,000. After 15 years, the risk for kidney failure associated with donating a kidney was 51 per 10,000 in African-American donors and 23 per 10,000 in white donors. So while the study did reveal an increased risk associated with kidney donation, the degree of risk is considered small.
These findings demonstrate the importance of understanding the facts surrounding inherent risk for ESRD in kidney donation. Overall, a donor’s lifetime risk is considered minuscule. So, to answer the question, yes, there is a slight increase in risk for kidney failure if you donate to your friend. That said, the risk is 0.014 x a standardized risk of 1. This increases at 15 years to 0.51 for African-American and 0.23 for white donors. With such tiny increases, you can safely feel good about donating a kidney to your friend.
Donna Reesman, MSN, CNP
VP Clinical & Quality Management
St Clair Specialty Physicians Detroit
REFERENCES
1. Muzaale AD, Massie AB, Wang MC, et al. Risk of end-stage renal disease following live kidney donation. JAMA. 2014;311(6):579-586.
2. CDC. HIV in the United States: at a glance (2013). www.cdc.gov/hiv/statistics/basics/ataglance.html. Accessed June 16, 2014.
3. Frassetto LA, Tan-Tam C, Stock PG. Renal transplantation in patients with HIV. Nat Rev Nephrol. 2009;5(10):582-589.
4. Malani PN. New law allows organ transplants from deceased HIV-infected donors to HIV-infected recipients. JAMA. 2013;310(23): 2492-2493.
5. Muller E, Kahn D, Mendelson M. Renal transplantation between HIV-positive donors and recipients. N Engl J Med. 2010;362(24):2336-2337.
6. Mariani LH, Berns JS. Viral nephropathies. In: Gilbert SJ, Weiner DE, eds. National Kidney Foundation’s Primer on Kidney Diseases. 6th ed. Elsevier; 2014:253-261.
Q) A good friend was diagnosed with chronic kidney disease (CKD) and is presently undergoing workup for a transplant. He is 60 and otherwise healthy; his glomerular filtration rate (GFR) is 14, and he has no uremic symptoms. If I volunteer to give him a kidney, are there any long-term risks for me?
Kidney failure, dialysis, and kidney transplant are terms that can invoke stress and uncertainty in patients with end-stage renal disease (ESRD) and among their family members and friends. In addition to adjusting to the changes wrought by ESRD, patients may also be burdened by the prospect of a family member or friend donating a kidney to them and the concern that the donation will lead to complications for their donor. Family members or friends who volunteer may also experience stress, uncertain of their own risk for ESRD in the future.
Past research improperly compared relative risk for ESRD in donors with that in the general population (without accounting for higher propensity for complications in donors with preexisting conditions). In an effort to correct this misperception, a study recently published in JAMA compared the risk for ESRD in donors with that in a healthy group of nondonors.1 The nondonor pool was taken from the National Health and Nutrition Examination Survey (NHANES III), which assesses the health and nutritional status of adults and children in the United States.
The JAMA study included a cohort of 96,217 kidney donors in the US in a 17-year period and a cohort of 20,024 participants in a six-year period of the NHANES III trial. This data was then compared to Centers for Medicare & Medicaid Services (CMS) data to determine the development of ESRD in kidney donors. ESRD was defined by CMS as the initiation of dialysis, placement on the kidney transplant waiting list, or receipt of a living or deceased donor kidney transplant.
In addition to comparing risk for ESRD in kidney donors with that of a healthy population of nondonors, the researchers also stratified their results demographically. Thus, the lifetime rate of kidney failure in donors is 90 per 10,000, compared with 326 per 10,000 in the general population of nondonors. In healthy nondonors, the risk for kidney failure was 14 per 10,000. After 15 years, the risk for kidney failure associated with donating a kidney was 51 per 10,000 in African-American donors and 23 per 10,000 in white donors. So while the study did reveal an increased risk associated with kidney donation, the degree of risk is considered small.
These findings demonstrate the importance of understanding the facts surrounding inherent risk for ESRD in kidney donation. Overall, a donor’s lifetime risk is considered minuscule. So, to answer the question, yes, there is a slight increase in risk for kidney failure if you donate to your friend. That said, the risk is 0.014 x a standardized risk of 1. This increases at 15 years to 0.51 for African-American and 0.23 for white donors. With such tiny increases, you can safely feel good about donating a kidney to your friend.
Donna Reesman, MSN, CNP
VP Clinical & Quality Management
St Clair Specialty Physicians Detroit
REFERENCES
1. Muzaale AD, Massie AB, Wang MC, et al. Risk of end-stage renal disease following live kidney donation. JAMA. 2014;311(6):579-586.
2. CDC. HIV in the United States: at a glance (2013). www.cdc.gov/hiv/statistics/basics/ataglance.html. Accessed June 16, 2014.
3. Frassetto LA, Tan-Tam C, Stock PG. Renal transplantation in patients with HIV. Nat Rev Nephrol. 2009;5(10):582-589.
4. Malani PN. New law allows organ transplants from deceased HIV-infected donors to HIV-infected recipients. JAMA. 2013;310(23): 2492-2493.
5. Muller E, Kahn D, Mendelson M. Renal transplantation between HIV-positive donors and recipients. N Engl J Med. 2010;362(24):2336-2337.
6. Mariani LH, Berns JS. Viral nephropathies. In: Gilbert SJ, Weiner DE, eds. National Kidney Foundation’s Primer on Kidney Diseases. 6th ed. Elsevier; 2014:253-261.
Kidney Donation & HIV
Q) Now that patients are living with HIV/AIDS, can they donate kidneys or receive a kidney transplant?
Kidney disease often has multiple causes, including hypertension, diabetes, inherited conditions, and viral illnesses. The latter include primarily HIV, hepatitis C, and hepatitis B. With advances in the treatment of viral illnesses, the question of whether patients with these viruses can donate or receive a kidney transplant is being discussed not only in the United States but also worldwide.
The most recent CDC figures estimate that more than 1.1 million people in the US are living with HIV, of whom one in six (or nearly 16%) are undiagnosed. There are approximately 50,000 new infections reported annually.2
The Organ Transplant Amendments Act of 1988 banned HIV-positive people from donating organs. However, with the introduction of highly active antiretroviral therapy (HAART, now often referred to as active antiretroviral therapy) and the effective prophylaxis and management of opportunistic infections, mortality has been reduced. HIV/AIDS is often seen as a chronic disease and not the death sentence it once was.3 Since the development of HAART, there have been successful transplants to HIV-positive recipients from non–HIV-infected donors.
In November 2013, President Obama signed the HIV Organ Policy Equity (HOPE) Act, which lifted the ban on using organs from HIV-infected donors. The legislation directs the Department of Health and Human Services and the Organ Procurement and Transplantation Network to develop standards to make these transplants possible.4
Although there have not been any documented cases of transplants from HIV-infected donors to HIV-infected recipients in this country, such transplants have been very successful in South Africa.5 There, to qualify for kidney transplant, all recipients must have proven adherence, virologic suppression, and immune constitution. Donor suitability is defined as HIV infection (confirmed with the use of enzyme-linked immunosorbent assay), absence of proteinuria, and a normal kidney as assessed with post hoc renal biopsy.5
One of the chief concerns has been the effect of further immunosuppression on the recipients and the possibility of disease progression. Although the sample size is limited (four transplants), data from the available cases indicate no evidence of organ rejection at 12 months post-transplantation. In addition, the recipients’ CD4 counts remained lower than baseline due to immunosuppressive therapy. All four patients maintained a viral load of less than 50 copies, which suggested that any virus transplanted along with the kidney had not affected control of HIV infection.5 However, it should be noted that many of the agents used for posttransplant maintenance immunosuppression (mycophenolate mofetil, cyclosporine, tacrolimus, and sirolimus) have antiretroviral properties.3
HIV patients in the US must meet the following criteria to be listed for a transplant:
• Diagnosis of ESRD with at least a five-year life-expectancy
• CD4 count of > 200 cells/ μL for at least six months
• Undetectable HIV viremia (< 50 HIV-1 RNA copies/mL)
• Demonstrated adherence to stable antiviral regimen for at least six months
• Absence of AIDS-defining illness following successful immune reconstitution6
A prospective trial of 150 patients in 19 US transplant centers who met the above criteria demonstrated patient survival and graft survival rates comparable to those in patients ages 65 and older.6
While awaiting the donation, HIV patients can continue hemodialysis and peritoneal dialysis. With the improved antiviral drugs, HIV patients have a survival rate similar to the non–HIV-infected population.
Transplantation is the goal and certainly the hope of many advanced-stage kidney patients, but in reality, the need far exceeds the resources. The HOPE Act opens the door for many patients who were previously excluded from the possibility of a life without dialysis. Taking care of these patients will be a team effort, encompassing HIV and infectious disease specialists, pharmacists, nephrologists, transplant surgeons and coordinators, and primary care providers—including, of course, advanced practitioners.
Shelly Levinstein, MSN, CRNP
Nephrology Associates of York
York, PA
REFERENCES
1. Muzaale AD, Massie AB, Wang MC, et al. Risk of end-stage renal disease following live kidney donation. JAMA. 2014;311(6):579-586.
2. CDC. HIV in the United States: at a glance (2013). www.cdc.gov/hiv/statistics/basics/ataglance.html. Accessed June 16, 2014.
3. Frassetto LA, Tan-Tam C, Stock PG. Renal transplantation in patients with HIV. Nat Rev Nephrol. 2009;5(10):582-589.
4. Malani PN. New law allows organ transplants from deceased HIV-infected donors to HIV-infected recipients. JAMA. 2013;310(23): 2492-2493.
5. Muller E, Kahn D, Mendelson M. Renal transplantation between HIV-positive donors and recipients. N Engl J Med. 2010;362(24):2336-2337.
6. Mariani LH, Berns JS. Viral nephropathies. In: Gilbert SJ, Weiner DE, eds. National Kidney Foundation’s Primer on Kidney Diseases. 6th ed. Elsevier; 2014:253-261.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP.
Q) Now that patients are living with HIV/AIDS, can they donate kidneys or receive a kidney transplant?
Kidney disease often has multiple causes, including hypertension, diabetes, inherited conditions, and viral illnesses. The latter include primarily HIV, hepatitis C, and hepatitis B. With advances in the treatment of viral illnesses, the question of whether patients with these viruses can donate or receive a kidney transplant is being discussed not only in the United States but also worldwide.
The most recent CDC figures estimate that more than 1.1 million people in the US are living with HIV, of whom one in six (or nearly 16%) are undiagnosed. There are approximately 50,000 new infections reported annually.2
The Organ Transplant Amendments Act of 1988 banned HIV-positive people from donating organs. However, with the introduction of highly active antiretroviral therapy (HAART, now often referred to as active antiretroviral therapy) and the effective prophylaxis and management of opportunistic infections, mortality has been reduced. HIV/AIDS is often seen as a chronic disease and not the death sentence it once was.3 Since the development of HAART, there have been successful transplants to HIV-positive recipients from non–HIV-infected donors.
In November 2013, President Obama signed the HIV Organ Policy Equity (HOPE) Act, which lifted the ban on using organs from HIV-infected donors. The legislation directs the Department of Health and Human Services and the Organ Procurement and Transplantation Network to develop standards to make these transplants possible.4
Although there have not been any documented cases of transplants from HIV-infected donors to HIV-infected recipients in this country, such transplants have been very successful in South Africa.5 There, to qualify for kidney transplant, all recipients must have proven adherence, virologic suppression, and immune constitution. Donor suitability is defined as HIV infection (confirmed with the use of enzyme-linked immunosorbent assay), absence of proteinuria, and a normal kidney as assessed with post hoc renal biopsy.5
One of the chief concerns has been the effect of further immunosuppression on the recipients and the possibility of disease progression. Although the sample size is limited (four transplants), data from the available cases indicate no evidence of organ rejection at 12 months post-transplantation. In addition, the recipients’ CD4 counts remained lower than baseline due to immunosuppressive therapy. All four patients maintained a viral load of less than 50 copies, which suggested that any virus transplanted along with the kidney had not affected control of HIV infection.5 However, it should be noted that many of the agents used for posttransplant maintenance immunosuppression (mycophenolate mofetil, cyclosporine, tacrolimus, and sirolimus) have antiretroviral properties.3
HIV patients in the US must meet the following criteria to be listed for a transplant:
• Diagnosis of ESRD with at least a five-year life-expectancy
• CD4 count of > 200 cells/ μL for at least six months
• Undetectable HIV viremia (< 50 HIV-1 RNA copies/mL)
• Demonstrated adherence to stable antiviral regimen for at least six months
• Absence of AIDS-defining illness following successful immune reconstitution6
A prospective trial of 150 patients in 19 US transplant centers who met the above criteria demonstrated patient survival and graft survival rates comparable to those in patients ages 65 and older.6
While awaiting the donation, HIV patients can continue hemodialysis and peritoneal dialysis. With the improved antiviral drugs, HIV patients have a survival rate similar to the non–HIV-infected population.
Transplantation is the goal and certainly the hope of many advanced-stage kidney patients, but in reality, the need far exceeds the resources. The HOPE Act opens the door for many patients who were previously excluded from the possibility of a life without dialysis. Taking care of these patients will be a team effort, encompassing HIV and infectious disease specialists, pharmacists, nephrologists, transplant surgeons and coordinators, and primary care providers—including, of course, advanced practitioners.
Shelly Levinstein, MSN, CRNP
Nephrology Associates of York
York, PA
REFERENCES
1. Muzaale AD, Massie AB, Wang MC, et al. Risk of end-stage renal disease following live kidney donation. JAMA. 2014;311(6):579-586.
2. CDC. HIV in the United States: at a glance (2013). www.cdc.gov/hiv/statistics/basics/ataglance.html. Accessed June 16, 2014.
3. Frassetto LA, Tan-Tam C, Stock PG. Renal transplantation in patients with HIV. Nat Rev Nephrol. 2009;5(10):582-589.
4. Malani PN. New law allows organ transplants from deceased HIV-infected donors to HIV-infected recipients. JAMA. 2013;310(23): 2492-2493.
5. Muller E, Kahn D, Mendelson M. Renal transplantation between HIV-positive donors and recipients. N Engl J Med. 2010;362(24):2336-2337.
6. Mariani LH, Berns JS. Viral nephropathies. In: Gilbert SJ, Weiner DE, eds. National Kidney Foundation’s Primer on Kidney Diseases. 6th ed. Elsevier; 2014:253-261.
Q) Now that patients are living with HIV/AIDS, can they donate kidneys or receive a kidney transplant?
Kidney disease often has multiple causes, including hypertension, diabetes, inherited conditions, and viral illnesses. The latter include primarily HIV, hepatitis C, and hepatitis B. With advances in the treatment of viral illnesses, the question of whether patients with these viruses can donate or receive a kidney transplant is being discussed not only in the United States but also worldwide.
The most recent CDC figures estimate that more than 1.1 million people in the US are living with HIV, of whom one in six (or nearly 16%) are undiagnosed. There are approximately 50,000 new infections reported annually.2
The Organ Transplant Amendments Act of 1988 banned HIV-positive people from donating organs. However, with the introduction of highly active antiretroviral therapy (HAART, now often referred to as active antiretroviral therapy) and the effective prophylaxis and management of opportunistic infections, mortality has been reduced. HIV/AIDS is often seen as a chronic disease and not the death sentence it once was.3 Since the development of HAART, there have been successful transplants to HIV-positive recipients from non–HIV-infected donors.
In November 2013, President Obama signed the HIV Organ Policy Equity (HOPE) Act, which lifted the ban on using organs from HIV-infected donors. The legislation directs the Department of Health and Human Services and the Organ Procurement and Transplantation Network to develop standards to make these transplants possible.4
Although there have not been any documented cases of transplants from HIV-infected donors to HIV-infected recipients in this country, such transplants have been very successful in South Africa.5 There, to qualify for kidney transplant, all recipients must have proven adherence, virologic suppression, and immune constitution. Donor suitability is defined as HIV infection (confirmed with the use of enzyme-linked immunosorbent assay), absence of proteinuria, and a normal kidney as assessed with post hoc renal biopsy.5
One of the chief concerns has been the effect of further immunosuppression on the recipients and the possibility of disease progression. Although the sample size is limited (four transplants), data from the available cases indicate no evidence of organ rejection at 12 months post-transplantation. In addition, the recipients’ CD4 counts remained lower than baseline due to immunosuppressive therapy. All four patients maintained a viral load of less than 50 copies, which suggested that any virus transplanted along with the kidney had not affected control of HIV infection.5 However, it should be noted that many of the agents used for posttransplant maintenance immunosuppression (mycophenolate mofetil, cyclosporine, tacrolimus, and sirolimus) have antiretroviral properties.3
HIV patients in the US must meet the following criteria to be listed for a transplant:
• Diagnosis of ESRD with at least a five-year life-expectancy
• CD4 count of > 200 cells/ μL for at least six months
• Undetectable HIV viremia (< 50 HIV-1 RNA copies/mL)
• Demonstrated adherence to stable antiviral regimen for at least six months
• Absence of AIDS-defining illness following successful immune reconstitution6
A prospective trial of 150 patients in 19 US transplant centers who met the above criteria demonstrated patient survival and graft survival rates comparable to those in patients ages 65 and older.6
While awaiting the donation, HIV patients can continue hemodialysis and peritoneal dialysis. With the improved antiviral drugs, HIV patients have a survival rate similar to the non–HIV-infected population.
Transplantation is the goal and certainly the hope of many advanced-stage kidney patients, but in reality, the need far exceeds the resources. The HOPE Act opens the door for many patients who were previously excluded from the possibility of a life without dialysis. Taking care of these patients will be a team effort, encompassing HIV and infectious disease specialists, pharmacists, nephrologists, transplant surgeons and coordinators, and primary care providers—including, of course, advanced practitioners.
Shelly Levinstein, MSN, CRNP
Nephrology Associates of York
York, PA
REFERENCES
1. Muzaale AD, Massie AB, Wang MC, et al. Risk of end-stage renal disease following live kidney donation. JAMA. 2014;311(6):579-586.
2. CDC. HIV in the United States: at a glance (2013). www.cdc.gov/hiv/statistics/basics/ataglance.html. Accessed June 16, 2014.
3. Frassetto LA, Tan-Tam C, Stock PG. Renal transplantation in patients with HIV. Nat Rev Nephrol. 2009;5(10):582-589.
4. Malani PN. New law allows organ transplants from deceased HIV-infected donors to HIV-infected recipients. JAMA. 2013;310(23): 2492-2493.
5. Muller E, Kahn D, Mendelson M. Renal transplantation between HIV-positive donors and recipients. N Engl J Med. 2010;362(24):2336-2337.
6. Mariani LH, Berns JS. Viral nephropathies. In: Gilbert SJ, Weiner DE, eds. National Kidney Foundation’s Primer on Kidney Diseases. 6th ed. Elsevier; 2014:253-261.
Androgen Deficiency Syndrome: A Rational Approach to Male Hypogonadism
During a routine physical examination, a 65-year-old man wants to find out if he has “Low T.” He complains of fatigue, decreased libido, and erectile dysfunction (ED) for the past five years. He has a history of type 2 diabetes, hypertension, hyperlipidemia, obstructive sleep apnea, and chronic low back pain. His current medications include metformin, glipizide, lisinopril, atorvastatin, and hydrocodone for back pain. Given these clinical features, the next step will be to find out if he has hypogonadism (androgen deficiency).
The Endocrine Society defines hypogonadism as a clinical syndrome in which the testes produce insufficient testosterone as a consequence of an interruption of the hypothalamic-pituitary-testicular axis. Although prevalence is high in older men, the Endocrine Society does not recommend screening the general population for hypogonadism.1 Rather, screening should be limited to patients with clinical conditions associated with high prevalence of hypogonadism. Of note, approximately 30% of adults with type 2 diabetes have a subnormal testosterone concentration.2
Q: What is pertinent in the history?
The first step in evaluation of hypogonadism is a detailed history. Signs and symptoms such as decreased libido, hot flashes, decreased shaving frequency, breast enlargement/tenderness, and decreased testicular size are highly suggestive of hypogonadism. Other, less specific signs and symptoms include dysthymia, poor concentration, sleep disturbances, fatigue, reduction in muscle strength, and diminished work performance.
If these signs and symptoms are present, the likelihood of hypogonadism is high and further evaluation is needed.1,3 Note any history of alcoholism, liver problems, and testicular trauma or surgery.
A detailed medication history is also important. Some medications, such as opiates, can affect the release of gonadotropins. Among men taking long-term opiates for chronic noncancer pain, the prevalence of hypogonadism is 75%.4 Other drugs, such as spironolactone, can block the androgen effect and lead to hypogonadism.1
Recent reports have suggested an association between testosterone replacement therapy and increased cardiovascular events, making a detailed cardiovascular history essential.5,6 One study found that men ages 75 and older with limited mobility and other comorbidities who used testosterone gel had an increased risk for cardiovascular events.7 Therefore, clinicians need to be cognizant of this risk when considering testosterone therapy for their patients.
On the next page: Physical exam, lab tests, and treatments >>
Q: What does the physical exam reveal?
In hypogonadotropic hy pogonadism, physical examination does not usually provide much information, as compared to congenital hypogonadal syndromes (eg, Klinefelter and Kallmann syndromes). However, small testicular volume and/or gynecomastia would indicate hypogonadism.
Q: What lab tests should be ordered?
Serum total and free testosterone should be measured, preferably by liquid gas chromatography. The sample should be drawn before 10 am to limit the effects of diurnal variation. If the total testosterone is less than
300 ng/dL, a second morning sample should be drawn and tested. Serum prolactin, follicle-stimulating hormone (FSH) and luteinizing hormone (LH), complete blood count, prostate-specific antigen (PSA), comprehensive metabolic panel, and ferritin should also be measured.
There is generally little benefit to testosterone therapy when total testosterone is greater than 350 ng/dL.8 The level of testosterone at which hypogonadal symptoms manifest and testosterone replacement provides improvement is yet to be determined. Buvat et al suggest that men with total testosterone levels less than 230 ng/dL usually benefit from therapy.8 If the total testosterone level is less than 150 ng/dL in the setting of secondary hypogonadism (low to low-normal LH/FSH) or if prolactin is elevated, MRI of the sella is recommended to rule out pituitary adenoma.1
Q: Once the diagnosis is confirmed, what treatment should you recommend?
The goal of therapy for confirmed hypogonadism is to normalize the testosterone level. Testosterone replacement therapy may help to improve libido, fatigue, muscle strength, and bone density. However, in the elderly (particularly those older than 70), these therapeutic benefits have not been proven. Therefore, before initiating therapy, the clinician should discuss in detail the risks versus the benefits of testosterone replacement for a particular patient.
Simple lifestyle modifications, such as weight loss and exercise, have been shown to increase total and free testosterone levels.3,8 For patients with obstructive sleep apnea (OSA), a known risk factor for hypogonadism, compliance with CPAP therapy has been associated with modest improvement in testosterone level. If it is appropriate for the patient to discontinue use of certain medications, such as opiates, he or she may experience an improvement in testosterone level as a result.
If the patient’s testosterone levels remain low after these changes have been implemented, consider testosterone therapy. Testosterone products currently available in the United States include transdermal preparations (gel, patch), intramuscular injection, and subcutaneous pellets.
On the next page: Contraindications, adverse effects, and follow-up >>
Q: What are the contraindications to testosterone therapy?
Testosterone therapy is contraindicated in patients with metastatic prostate cancer and breast cancer. An unevaluated prostate nodule, indurated prostate, PSA greater than 4 ng/mL, elevated hematocrit (>50%), severe lower urinary tract symptoms, poorly controlled congestive heart failure, and untreated severe OSA are associated with moderate to high risk for adverse outcomes; the Endocrine Society has recommended against using testosterone in affected patients.1
Q: What are the adverse effects of testosterone replacement therapy?
Testosterone replacement may worsen symptoms of benign prostatic hyperplasia (ie, urinary urgency, hesitancy, and frequency). Also, testosterone replacement can lead to marked elevation of hemoglobin and hematocrit levels.
Increased cardiovascular events have been associated with androgen replacement, especially in men with prior coronary artery disease. A positive cardiovascular history necessitates discussion with the patient regarding the risks versus the benefits of testosterone replacement therapy.5 In a recent study of obese, hypogonadal men with severe OSA, testosterone therapy was associated with transient worsening of sleep apnea.9
Q: What does monitoring/ follow-up entail?
In patients with long-standing hypogonadism, a lower starting dose of testosterone is recommended, which can be gradually increased. After starting testosterone therapy, patients should be monitored in the first three to six months for total testosterone, PSA, and hematocrit and for improvement of symptoms (ie, fatigue, ED, decreased libido) or worsening of benign prostatic hyperplasia signs/symptoms.
For men ages 40 and older, if the baseline PSA is greater than 0.6 ng/mL, a digital rectal exam (DRE) is recommended prior to initiation of therapy and should be followed in accordance with prostate cancer screening guidelines.1
Patients placed on testosterone cypionate/enanthate IM injections should have their testosterone checked at a midpoint between their injections, with the target testosterone level between 400 and 700 ng/dL.1 For those using gel or transdermal preparations, a morning total testosterone level should be measured.
Urology consultation is recommended if the PSA concentration rises by 1.4 ng/dL within 12 months, if the American Urological Association/International Prostate Symptom Score is greater than 19, or if there is an abnormal DRE.1,8 Treatment with testosterone should be postponed or withheld if the patient’s hematocrit is greater than 54% but may be resumed when it has decreased to normal levels.1
On the next page: References >>
REFERENCES
1. Bhasin S, Cunningham GR, Hayes FJ, et al. Testosterone therapy in men with androgen deficiency syndromes: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2010;95(6):2536-2559.
2. Dandona P, Dhindsa S. Update: hypogonadotropic hypogonadism in type 2 diabetes and obesity. J Clin Endocrinol Metab. 2011;96(9): 2643-2651.
3. Tajar A, Forti G, O’Neill TW, et al. Characteristics of secondary, primary, and compensated hypogonadism in aging men: evidence from the European Male Ageing Study. J Clin Endocrinol Metab. 2010;95(4):1810-1818.
4. Fraser LA, Morrison D, Morley-Forster P, et al. Oral opioids for chronic non-cancer pain: higher prevalence of hypogonadism in men than in women. Exp Clin Endocrinol Diabetes. 2009;117(1):38-43.
5. Vigen R, O’Donnell CI, Baron AE, et al. Association of testosterone therapy with mortality, myocardial infarction, and stroke in men with low testosterone levels. JAMA. 2013;310(17): 1829-1836.
6. Finkle WD, Greenland S, Ridgeway GK, et al. Increased risk of non-fatal myocardial infarction following testosterone therapy prescription in men. PloS One. 2014;9(1): e85805.
7. Basaria S, Coviello AD, Travison TG, et al. Adverse events associated with testosterone administration. N Engl J Med. 2010;363(2):109-122.
8. Buvat J, Maggi M, Guay A, Torres LO. Testosterone deficiency in men: systematic review and standard operating procedures for diagnosis and treatment. J Sex Med. 2013;10(1): 245-284.
9. Hoyos CM, Killick R, Yee BJ, et al. Effects of testosterone therapy on sleep and breathing in obese men with severe obstructive sleep apnoea: a randomized placebo-controlled trial. Clin Endocrinol (Oxf). 2012;77(4):
599-607.
| Clinician Reviews in partnership with |
Sheila B. Pinkson practices at Audie L. Murphy VA Hospital in San Antonio and is an Adjunct Assistant Professor in Endocrinology at the University of Texas Health Science Center at San Antonio (UTHSCSA). Devjit Tripathy is a staff physician at Audie L. Murphy VA Hospital in San Antonio and an Associate Professor in the Endocrinology/Diabetes Division at UTHSCSA.
| Clinician Reviews in partnership with |
Sheila B. Pinkson practices at Audie L. Murphy VA Hospital in San Antonio and is an Adjunct Assistant Professor in Endocrinology at the University of Texas Health Science Center at San Antonio (UTHSCSA). Devjit Tripathy is a staff physician at Audie L. Murphy VA Hospital in San Antonio and an Associate Professor in the Endocrinology/Diabetes Division at UTHSCSA.
| Clinician Reviews in partnership with |
Sheila B. Pinkson practices at Audie L. Murphy VA Hospital in San Antonio and is an Adjunct Assistant Professor in Endocrinology at the University of Texas Health Science Center at San Antonio (UTHSCSA). Devjit Tripathy is a staff physician at Audie L. Murphy VA Hospital in San Antonio and an Associate Professor in the Endocrinology/Diabetes Division at UTHSCSA.
During a routine physical examination, a 65-year-old man wants to find out if he has “Low T.” He complains of fatigue, decreased libido, and erectile dysfunction (ED) for the past five years. He has a history of type 2 diabetes, hypertension, hyperlipidemia, obstructive sleep apnea, and chronic low back pain. His current medications include metformin, glipizide, lisinopril, atorvastatin, and hydrocodone for back pain. Given these clinical features, the next step will be to find out if he has hypogonadism (androgen deficiency).
The Endocrine Society defines hypogonadism as a clinical syndrome in which the testes produce insufficient testosterone as a consequence of an interruption of the hypothalamic-pituitary-testicular axis. Although prevalence is high in older men, the Endocrine Society does not recommend screening the general population for hypogonadism.1 Rather, screening should be limited to patients with clinical conditions associated with high prevalence of hypogonadism. Of note, approximately 30% of adults with type 2 diabetes have a subnormal testosterone concentration.2
Q: What is pertinent in the history?
The first step in evaluation of hypogonadism is a detailed history. Signs and symptoms such as decreased libido, hot flashes, decreased shaving frequency, breast enlargement/tenderness, and decreased testicular size are highly suggestive of hypogonadism. Other, less specific signs and symptoms include dysthymia, poor concentration, sleep disturbances, fatigue, reduction in muscle strength, and diminished work performance.
If these signs and symptoms are present, the likelihood of hypogonadism is high and further evaluation is needed.1,3 Note any history of alcoholism, liver problems, and testicular trauma or surgery.
A detailed medication history is also important. Some medications, such as opiates, can affect the release of gonadotropins. Among men taking long-term opiates for chronic noncancer pain, the prevalence of hypogonadism is 75%.4 Other drugs, such as spironolactone, can block the androgen effect and lead to hypogonadism.1
Recent reports have suggested an association between testosterone replacement therapy and increased cardiovascular events, making a detailed cardiovascular history essential.5,6 One study found that men ages 75 and older with limited mobility and other comorbidities who used testosterone gel had an increased risk for cardiovascular events.7 Therefore, clinicians need to be cognizant of this risk when considering testosterone therapy for their patients.
On the next page: Physical exam, lab tests, and treatments >>
Q: What does the physical exam reveal?
In hypogonadotropic hy pogonadism, physical examination does not usually provide much information, as compared to congenital hypogonadal syndromes (eg, Klinefelter and Kallmann syndromes). However, small testicular volume and/or gynecomastia would indicate hypogonadism.
Q: What lab tests should be ordered?
Serum total and free testosterone should be measured, preferably by liquid gas chromatography. The sample should be drawn before 10 am to limit the effects of diurnal variation. If the total testosterone is less than
300 ng/dL, a second morning sample should be drawn and tested. Serum prolactin, follicle-stimulating hormone (FSH) and luteinizing hormone (LH), complete blood count, prostate-specific antigen (PSA), comprehensive metabolic panel, and ferritin should also be measured.
There is generally little benefit to testosterone therapy when total testosterone is greater than 350 ng/dL.8 The level of testosterone at which hypogonadal symptoms manifest and testosterone replacement provides improvement is yet to be determined. Buvat et al suggest that men with total testosterone levels less than 230 ng/dL usually benefit from therapy.8 If the total testosterone level is less than 150 ng/dL in the setting of secondary hypogonadism (low to low-normal LH/FSH) or if prolactin is elevated, MRI of the sella is recommended to rule out pituitary adenoma.1
Q: Once the diagnosis is confirmed, what treatment should you recommend?
The goal of therapy for confirmed hypogonadism is to normalize the testosterone level. Testosterone replacement therapy may help to improve libido, fatigue, muscle strength, and bone density. However, in the elderly (particularly those older than 70), these therapeutic benefits have not been proven. Therefore, before initiating therapy, the clinician should discuss in detail the risks versus the benefits of testosterone replacement for a particular patient.
Simple lifestyle modifications, such as weight loss and exercise, have been shown to increase total and free testosterone levels.3,8 For patients with obstructive sleep apnea (OSA), a known risk factor for hypogonadism, compliance with CPAP therapy has been associated with modest improvement in testosterone level. If it is appropriate for the patient to discontinue use of certain medications, such as opiates, he or she may experience an improvement in testosterone level as a result.
If the patient’s testosterone levels remain low after these changes have been implemented, consider testosterone therapy. Testosterone products currently available in the United States include transdermal preparations (gel, patch), intramuscular injection, and subcutaneous pellets.
On the next page: Contraindications, adverse effects, and follow-up >>
Q: What are the contraindications to testosterone therapy?
Testosterone therapy is contraindicated in patients with metastatic prostate cancer and breast cancer. An unevaluated prostate nodule, indurated prostate, PSA greater than 4 ng/mL, elevated hematocrit (>50%), severe lower urinary tract symptoms, poorly controlled congestive heart failure, and untreated severe OSA are associated with moderate to high risk for adverse outcomes; the Endocrine Society has recommended against using testosterone in affected patients.1
Q: What are the adverse effects of testosterone replacement therapy?
Testosterone replacement may worsen symptoms of benign prostatic hyperplasia (ie, urinary urgency, hesitancy, and frequency). Also, testosterone replacement can lead to marked elevation of hemoglobin and hematocrit levels.
Increased cardiovascular events have been associated with androgen replacement, especially in men with prior coronary artery disease. A positive cardiovascular history necessitates discussion with the patient regarding the risks versus the benefits of testosterone replacement therapy.5 In a recent study of obese, hypogonadal men with severe OSA, testosterone therapy was associated with transient worsening of sleep apnea.9
Q: What does monitoring/ follow-up entail?
In patients with long-standing hypogonadism, a lower starting dose of testosterone is recommended, which can be gradually increased. After starting testosterone therapy, patients should be monitored in the first three to six months for total testosterone, PSA, and hematocrit and for improvement of symptoms (ie, fatigue, ED, decreased libido) or worsening of benign prostatic hyperplasia signs/symptoms.
For men ages 40 and older, if the baseline PSA is greater than 0.6 ng/mL, a digital rectal exam (DRE) is recommended prior to initiation of therapy and should be followed in accordance with prostate cancer screening guidelines.1
Patients placed on testosterone cypionate/enanthate IM injections should have their testosterone checked at a midpoint between their injections, with the target testosterone level between 400 and 700 ng/dL.1 For those using gel or transdermal preparations, a morning total testosterone level should be measured.
Urology consultation is recommended if the PSA concentration rises by 1.4 ng/dL within 12 months, if the American Urological Association/International Prostate Symptom Score is greater than 19, or if there is an abnormal DRE.1,8 Treatment with testosterone should be postponed or withheld if the patient’s hematocrit is greater than 54% but may be resumed when it has decreased to normal levels.1
On the next page: References >>
REFERENCES
1. Bhasin S, Cunningham GR, Hayes FJ, et al. Testosterone therapy in men with androgen deficiency syndromes: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2010;95(6):2536-2559.
2. Dandona P, Dhindsa S. Update: hypogonadotropic hypogonadism in type 2 diabetes and obesity. J Clin Endocrinol Metab. 2011;96(9): 2643-2651.
3. Tajar A, Forti G, O’Neill TW, et al. Characteristics of secondary, primary, and compensated hypogonadism in aging men: evidence from the European Male Ageing Study. J Clin Endocrinol Metab. 2010;95(4):1810-1818.
4. Fraser LA, Morrison D, Morley-Forster P, et al. Oral opioids for chronic non-cancer pain: higher prevalence of hypogonadism in men than in women. Exp Clin Endocrinol Diabetes. 2009;117(1):38-43.
5. Vigen R, O’Donnell CI, Baron AE, et al. Association of testosterone therapy with mortality, myocardial infarction, and stroke in men with low testosterone levels. JAMA. 2013;310(17): 1829-1836.
6. Finkle WD, Greenland S, Ridgeway GK, et al. Increased risk of non-fatal myocardial infarction following testosterone therapy prescription in men. PloS One. 2014;9(1): e85805.
7. Basaria S, Coviello AD, Travison TG, et al. Adverse events associated with testosterone administration. N Engl J Med. 2010;363(2):109-122.
8. Buvat J, Maggi M, Guay A, Torres LO. Testosterone deficiency in men: systematic review and standard operating procedures for diagnosis and treatment. J Sex Med. 2013;10(1): 245-284.
9. Hoyos CM, Killick R, Yee BJ, et al. Effects of testosterone therapy on sleep and breathing in obese men with severe obstructive sleep apnoea: a randomized placebo-controlled trial. Clin Endocrinol (Oxf). 2012;77(4):
599-607.
During a routine physical examination, a 65-year-old man wants to find out if he has “Low T.” He complains of fatigue, decreased libido, and erectile dysfunction (ED) for the past five years. He has a history of type 2 diabetes, hypertension, hyperlipidemia, obstructive sleep apnea, and chronic low back pain. His current medications include metformin, glipizide, lisinopril, atorvastatin, and hydrocodone for back pain. Given these clinical features, the next step will be to find out if he has hypogonadism (androgen deficiency).
The Endocrine Society defines hypogonadism as a clinical syndrome in which the testes produce insufficient testosterone as a consequence of an interruption of the hypothalamic-pituitary-testicular axis. Although prevalence is high in older men, the Endocrine Society does not recommend screening the general population for hypogonadism.1 Rather, screening should be limited to patients with clinical conditions associated with high prevalence of hypogonadism. Of note, approximately 30% of adults with type 2 diabetes have a subnormal testosterone concentration.2
Q: What is pertinent in the history?
The first step in evaluation of hypogonadism is a detailed history. Signs and symptoms such as decreased libido, hot flashes, decreased shaving frequency, breast enlargement/tenderness, and decreased testicular size are highly suggestive of hypogonadism. Other, less specific signs and symptoms include dysthymia, poor concentration, sleep disturbances, fatigue, reduction in muscle strength, and diminished work performance.
If these signs and symptoms are present, the likelihood of hypogonadism is high and further evaluation is needed.1,3 Note any history of alcoholism, liver problems, and testicular trauma or surgery.
A detailed medication history is also important. Some medications, such as opiates, can affect the release of gonadotropins. Among men taking long-term opiates for chronic noncancer pain, the prevalence of hypogonadism is 75%.4 Other drugs, such as spironolactone, can block the androgen effect and lead to hypogonadism.1
Recent reports have suggested an association between testosterone replacement therapy and increased cardiovascular events, making a detailed cardiovascular history essential.5,6 One study found that men ages 75 and older with limited mobility and other comorbidities who used testosterone gel had an increased risk for cardiovascular events.7 Therefore, clinicians need to be cognizant of this risk when considering testosterone therapy for their patients.
On the next page: Physical exam, lab tests, and treatments >>
Q: What does the physical exam reveal?
In hypogonadotropic hy pogonadism, physical examination does not usually provide much information, as compared to congenital hypogonadal syndromes (eg, Klinefelter and Kallmann syndromes). However, small testicular volume and/or gynecomastia would indicate hypogonadism.
Q: What lab tests should be ordered?
Serum total and free testosterone should be measured, preferably by liquid gas chromatography. The sample should be drawn before 10 am to limit the effects of diurnal variation. If the total testosterone is less than
300 ng/dL, a second morning sample should be drawn and tested. Serum prolactin, follicle-stimulating hormone (FSH) and luteinizing hormone (LH), complete blood count, prostate-specific antigen (PSA), comprehensive metabolic panel, and ferritin should also be measured.
There is generally little benefit to testosterone therapy when total testosterone is greater than 350 ng/dL.8 The level of testosterone at which hypogonadal symptoms manifest and testosterone replacement provides improvement is yet to be determined. Buvat et al suggest that men with total testosterone levels less than 230 ng/dL usually benefit from therapy.8 If the total testosterone level is less than 150 ng/dL in the setting of secondary hypogonadism (low to low-normal LH/FSH) or if prolactin is elevated, MRI of the sella is recommended to rule out pituitary adenoma.1
Q: Once the diagnosis is confirmed, what treatment should you recommend?
The goal of therapy for confirmed hypogonadism is to normalize the testosterone level. Testosterone replacement therapy may help to improve libido, fatigue, muscle strength, and bone density. However, in the elderly (particularly those older than 70), these therapeutic benefits have not been proven. Therefore, before initiating therapy, the clinician should discuss in detail the risks versus the benefits of testosterone replacement for a particular patient.
Simple lifestyle modifications, such as weight loss and exercise, have been shown to increase total and free testosterone levels.3,8 For patients with obstructive sleep apnea (OSA), a known risk factor for hypogonadism, compliance with CPAP therapy has been associated with modest improvement in testosterone level. If it is appropriate for the patient to discontinue use of certain medications, such as opiates, he or she may experience an improvement in testosterone level as a result.
If the patient’s testosterone levels remain low after these changes have been implemented, consider testosterone therapy. Testosterone products currently available in the United States include transdermal preparations (gel, patch), intramuscular injection, and subcutaneous pellets.
On the next page: Contraindications, adverse effects, and follow-up >>
Q: What are the contraindications to testosterone therapy?
Testosterone therapy is contraindicated in patients with metastatic prostate cancer and breast cancer. An unevaluated prostate nodule, indurated prostate, PSA greater than 4 ng/mL, elevated hematocrit (>50%), severe lower urinary tract symptoms, poorly controlled congestive heart failure, and untreated severe OSA are associated with moderate to high risk for adverse outcomes; the Endocrine Society has recommended against using testosterone in affected patients.1
Q: What are the adverse effects of testosterone replacement therapy?
Testosterone replacement may worsen symptoms of benign prostatic hyperplasia (ie, urinary urgency, hesitancy, and frequency). Also, testosterone replacement can lead to marked elevation of hemoglobin and hematocrit levels.
Increased cardiovascular events have been associated with androgen replacement, especially in men with prior coronary artery disease. A positive cardiovascular history necessitates discussion with the patient regarding the risks versus the benefits of testosterone replacement therapy.5 In a recent study of obese, hypogonadal men with severe OSA, testosterone therapy was associated with transient worsening of sleep apnea.9
Q: What does monitoring/ follow-up entail?
In patients with long-standing hypogonadism, a lower starting dose of testosterone is recommended, which can be gradually increased. After starting testosterone therapy, patients should be monitored in the first three to six months for total testosterone, PSA, and hematocrit and for improvement of symptoms (ie, fatigue, ED, decreased libido) or worsening of benign prostatic hyperplasia signs/symptoms.
For men ages 40 and older, if the baseline PSA is greater than 0.6 ng/mL, a digital rectal exam (DRE) is recommended prior to initiation of therapy and should be followed in accordance with prostate cancer screening guidelines.1
Patients placed on testosterone cypionate/enanthate IM injections should have their testosterone checked at a midpoint between their injections, with the target testosterone level between 400 and 700 ng/dL.1 For those using gel or transdermal preparations, a morning total testosterone level should be measured.
Urology consultation is recommended if the PSA concentration rises by 1.4 ng/dL within 12 months, if the American Urological Association/International Prostate Symptom Score is greater than 19, or if there is an abnormal DRE.1,8 Treatment with testosterone should be postponed or withheld if the patient’s hematocrit is greater than 54% but may be resumed when it has decreased to normal levels.1
On the next page: References >>
REFERENCES
1. Bhasin S, Cunningham GR, Hayes FJ, et al. Testosterone therapy in men with androgen deficiency syndromes: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2010;95(6):2536-2559.
2. Dandona P, Dhindsa S. Update: hypogonadotropic hypogonadism in type 2 diabetes and obesity. J Clin Endocrinol Metab. 2011;96(9): 2643-2651.
3. Tajar A, Forti G, O’Neill TW, et al. Characteristics of secondary, primary, and compensated hypogonadism in aging men: evidence from the European Male Ageing Study. J Clin Endocrinol Metab. 2010;95(4):1810-1818.
4. Fraser LA, Morrison D, Morley-Forster P, et al. Oral opioids for chronic non-cancer pain: higher prevalence of hypogonadism in men than in women. Exp Clin Endocrinol Diabetes. 2009;117(1):38-43.
5. Vigen R, O’Donnell CI, Baron AE, et al. Association of testosterone therapy with mortality, myocardial infarction, and stroke in men with low testosterone levels. JAMA. 2013;310(17): 1829-1836.
6. Finkle WD, Greenland S, Ridgeway GK, et al. Increased risk of non-fatal myocardial infarction following testosterone therapy prescription in men. PloS One. 2014;9(1): e85805.
7. Basaria S, Coviello AD, Travison TG, et al. Adverse events associated with testosterone administration. N Engl J Med. 2010;363(2):109-122.
8. Buvat J, Maggi M, Guay A, Torres LO. Testosterone deficiency in men: systematic review and standard operating procedures for diagnosis and treatment. J Sex Med. 2013;10(1): 245-284.
9. Hoyos CM, Killick R, Yee BJ, et al. Effects of testosterone therapy on sleep and breathing in obese men with severe obstructive sleep apnoea: a randomized placebo-controlled trial. Clin Endocrinol (Oxf). 2012;77(4):
599-607.
Anemia, A1C, and Rhabdomyolysis
Q) Does anemia in CKD patients affect their A1C? Is A1C accurate in CKD patients?
Tight glycemic control is imperative for patients with chronic kidney disease (CKD), but the management of diabetes in CKD can be complex due to factors including anemia and changes in glucose and insulin homeostasis.
A1C is directly proportionate to the ambient blood glucose concentration and in the general diabetic population has proven to be a reliable marker.1 However, it may not be valid in patients with diabetes and CKD. Reduced red blood cell (RBC) lifespan, rapid hemolysis, and iron deficiency may lead to falsely decreased results.2 Decreased RBC survival results from an increase in hemoglobin turnover, which decreases glycemic exposure time.1 This process then lowers the amount of nonenzymatic glucose binding to hemoglobin.1 Folate deficiency caused by impaired intestinal absorption in CKD also affects RBC survival.3 Falsely increased results may be related to carbamylation of the hemoglobin and acidosis, both of which are influenced by uremia.2
Special considerations should be made for dialysis patients with diabetes. In hemodialysis patients, A1C may be falsely decreased due to blood loss, RBC transfusion, and erythropoietin therapy.3 Observational studies have shown that erythropoietin therapy is associated with lower A1C due to the increased number of immature RBCs that have a decreased glycemic exposure time.1 In peritoneal dialysis patients, A1C may increase after the start of therapy as a result of dialysate absorption.3
Research suggests that glycated albumin (GA) provides a short-term index of glycemic control (typically two to three weeks) and is not influenced by albumin concentration, RBC lifespan, or erythropoietin administration.1 A clear consensus on optimal levels of GA has not been established, but GA may be a more reliable marker of glycemic control in patients with diabetes and CKD. Further research is needed to establish a target GA level that predicts the best prognosis for patients with both conditions.1
A1C is the most reliable marker at this time, but special considerations should be made for the patient with CKD. Rather than focus on a single measurement, clinicians need to consider the patient’s symptoms and results from all labwork, along with A1C, to best evaluate glycemic control.4 Further research is needed in patients with diabetes and CKD to explore other reliable markers to help maintain tight glycemic control.
Continued on next page >>
Q) One of my patients developed severe leg cramps while taking statins. I felt it was questionable rhabdomyolysis and stopped the medication; the leg pain went away. Is there a way to know if the rhabdomyolysis is progressive?
Rhabdomyolysis is a serious condition caused by the breakdown of muscle tissue that leads to the release of myoglobin into the bloodstream. This condition can lead to severe kidney failure and death.
Previously, there has been no easy method to predict progressive rhabdomyolysis. But researchers from Brigham and Women’s Hospital recently developed the Rhabdomyolysis Risk Calculator, a prediction score that can help determine whether a patient with rhabdomyolysis is at risk for severe kidney failure or death.
The researchers conducted a retrospective cohort study of 2,371 patients admitted between 2000 and 2011 and examined variables that may be associated with kidney failure and death.5 They identified independent predictors for these outcomes, including age; gender; initial levels of phosphate, calcium, creatinine, carbon dioxide, and creatine kinase; and etiology of rhabdomyolysis (myositis, exercise, statin use, or seizure).5
This tool can assist providers in developing a patient-specific treatment plan. However, further research is needed to validate the current variables, verify the risk prediction score in other populations, and examine its ability to guide individualized treatment plans.
The Rhabdomyolysis Risk Calculator is available at www.brighamandwomens.org/research/rhabdo/default.aspx
Kristy Washinger, MSN, CRNP
Nephrology Associates of Central PA
Camp Hill, PA
REFERENCES
1. Vos FE, Schollum JB, Walker RJ. Glycated albumin is the preferred marker for assessing glycaemic control in advanced chronic kidney disease. Nephrol Dial Transplant Plus. 2011; 4(6):368-375.
2. National Kidney Foundation Kidney Disease Outcomes Quality Initiative. Clinical practice guidelines and clinical practice recommendations for diabetes and chronic kidney disease. Guideline 2: management of hyperglycemia and general diabetes care in chronic kidney disease. www.kidney.org/professionals/kdoqi/guideline_diabetes/guide2.htm. Accessed April 15, 2014.
3. Sharif A, Baboolal K. Diagnostic application of the A1c assay in renal disease. J Am Soc Nephrol. 2010;21(3):383-385.
4. American Diabetes Association. Standards of medical care in diabetes—2013. Diabetes Care. 2013;36(suppl 1):S11-S66.
5. McMahon GM, Zeng X, Walker SS. A risk prediction score for kidney failure or mortality in rhabdomyolysis. JAMA Intern Med. 2013;173(19):1821-1828.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Kristy Washinger, MSN, CRNP, who practices at Nephrology Associates of Central PA in Camp Hill, Pennsylvania.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Kristy Washinger, MSN, CRNP, who practices at Nephrology Associates of Central PA in Camp Hill, Pennsylvania.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Kristy Washinger, MSN, CRNP, who practices at Nephrology Associates of Central PA in Camp Hill, Pennsylvania.
Q) Does anemia in CKD patients affect their A1C? Is A1C accurate in CKD patients?
Tight glycemic control is imperative for patients with chronic kidney disease (CKD), but the management of diabetes in CKD can be complex due to factors including anemia and changes in glucose and insulin homeostasis.
A1C is directly proportionate to the ambient blood glucose concentration and in the general diabetic population has proven to be a reliable marker.1 However, it may not be valid in patients with diabetes and CKD. Reduced red blood cell (RBC) lifespan, rapid hemolysis, and iron deficiency may lead to falsely decreased results.2 Decreased RBC survival results from an increase in hemoglobin turnover, which decreases glycemic exposure time.1 This process then lowers the amount of nonenzymatic glucose binding to hemoglobin.1 Folate deficiency caused by impaired intestinal absorption in CKD also affects RBC survival.3 Falsely increased results may be related to carbamylation of the hemoglobin and acidosis, both of which are influenced by uremia.2
Special considerations should be made for dialysis patients with diabetes. In hemodialysis patients, A1C may be falsely decreased due to blood loss, RBC transfusion, and erythropoietin therapy.3 Observational studies have shown that erythropoietin therapy is associated with lower A1C due to the increased number of immature RBCs that have a decreased glycemic exposure time.1 In peritoneal dialysis patients, A1C may increase after the start of therapy as a result of dialysate absorption.3
Research suggests that glycated albumin (GA) provides a short-term index of glycemic control (typically two to three weeks) and is not influenced by albumin concentration, RBC lifespan, or erythropoietin administration.1 A clear consensus on optimal levels of GA has not been established, but GA may be a more reliable marker of glycemic control in patients with diabetes and CKD. Further research is needed to establish a target GA level that predicts the best prognosis for patients with both conditions.1
A1C is the most reliable marker at this time, but special considerations should be made for the patient with CKD. Rather than focus on a single measurement, clinicians need to consider the patient’s symptoms and results from all labwork, along with A1C, to best evaluate glycemic control.4 Further research is needed in patients with diabetes and CKD to explore other reliable markers to help maintain tight glycemic control.
Continued on next page >>
Q) One of my patients developed severe leg cramps while taking statins. I felt it was questionable rhabdomyolysis and stopped the medication; the leg pain went away. Is there a way to know if the rhabdomyolysis is progressive?
Rhabdomyolysis is a serious condition caused by the breakdown of muscle tissue that leads to the release of myoglobin into the bloodstream. This condition can lead to severe kidney failure and death.
Previously, there has been no easy method to predict progressive rhabdomyolysis. But researchers from Brigham and Women’s Hospital recently developed the Rhabdomyolysis Risk Calculator, a prediction score that can help determine whether a patient with rhabdomyolysis is at risk for severe kidney failure or death.
The researchers conducted a retrospective cohort study of 2,371 patients admitted between 2000 and 2011 and examined variables that may be associated with kidney failure and death.5 They identified independent predictors for these outcomes, including age; gender; initial levels of phosphate, calcium, creatinine, carbon dioxide, and creatine kinase; and etiology of rhabdomyolysis (myositis, exercise, statin use, or seizure).5
This tool can assist providers in developing a patient-specific treatment plan. However, further research is needed to validate the current variables, verify the risk prediction score in other populations, and examine its ability to guide individualized treatment plans.
The Rhabdomyolysis Risk Calculator is available at www.brighamandwomens.org/research/rhabdo/default.aspx
Kristy Washinger, MSN, CRNP
Nephrology Associates of Central PA
Camp Hill, PA
REFERENCES
1. Vos FE, Schollum JB, Walker RJ. Glycated albumin is the preferred marker for assessing glycaemic control in advanced chronic kidney disease. Nephrol Dial Transplant Plus. 2011; 4(6):368-375.
2. National Kidney Foundation Kidney Disease Outcomes Quality Initiative. Clinical practice guidelines and clinical practice recommendations for diabetes and chronic kidney disease. Guideline 2: management of hyperglycemia and general diabetes care in chronic kidney disease. www.kidney.org/professionals/kdoqi/guideline_diabetes/guide2.htm. Accessed April 15, 2014.
3. Sharif A, Baboolal K. Diagnostic application of the A1c assay in renal disease. J Am Soc Nephrol. 2010;21(3):383-385.
4. American Diabetes Association. Standards of medical care in diabetes—2013. Diabetes Care. 2013;36(suppl 1):S11-S66.
5. McMahon GM, Zeng X, Walker SS. A risk prediction score for kidney failure or mortality in rhabdomyolysis. JAMA Intern Med. 2013;173(19):1821-1828.
Q) Does anemia in CKD patients affect their A1C? Is A1C accurate in CKD patients?
Tight glycemic control is imperative for patients with chronic kidney disease (CKD), but the management of diabetes in CKD can be complex due to factors including anemia and changes in glucose and insulin homeostasis.
A1C is directly proportionate to the ambient blood glucose concentration and in the general diabetic population has proven to be a reliable marker.1 However, it may not be valid in patients with diabetes and CKD. Reduced red blood cell (RBC) lifespan, rapid hemolysis, and iron deficiency may lead to falsely decreased results.2 Decreased RBC survival results from an increase in hemoglobin turnover, which decreases glycemic exposure time.1 This process then lowers the amount of nonenzymatic glucose binding to hemoglobin.1 Folate deficiency caused by impaired intestinal absorption in CKD also affects RBC survival.3 Falsely increased results may be related to carbamylation of the hemoglobin and acidosis, both of which are influenced by uremia.2
Special considerations should be made for dialysis patients with diabetes. In hemodialysis patients, A1C may be falsely decreased due to blood loss, RBC transfusion, and erythropoietin therapy.3 Observational studies have shown that erythropoietin therapy is associated with lower A1C due to the increased number of immature RBCs that have a decreased glycemic exposure time.1 In peritoneal dialysis patients, A1C may increase after the start of therapy as a result of dialysate absorption.3
Research suggests that glycated albumin (GA) provides a short-term index of glycemic control (typically two to three weeks) and is not influenced by albumin concentration, RBC lifespan, or erythropoietin administration.1 A clear consensus on optimal levels of GA has not been established, but GA may be a more reliable marker of glycemic control in patients with diabetes and CKD. Further research is needed to establish a target GA level that predicts the best prognosis for patients with both conditions.1
A1C is the most reliable marker at this time, but special considerations should be made for the patient with CKD. Rather than focus on a single measurement, clinicians need to consider the patient’s symptoms and results from all labwork, along with A1C, to best evaluate glycemic control.4 Further research is needed in patients with diabetes and CKD to explore other reliable markers to help maintain tight glycemic control.
Continued on next page >>
Q) One of my patients developed severe leg cramps while taking statins. I felt it was questionable rhabdomyolysis and stopped the medication; the leg pain went away. Is there a way to know if the rhabdomyolysis is progressive?
Rhabdomyolysis is a serious condition caused by the breakdown of muscle tissue that leads to the release of myoglobin into the bloodstream. This condition can lead to severe kidney failure and death.
Previously, there has been no easy method to predict progressive rhabdomyolysis. But researchers from Brigham and Women’s Hospital recently developed the Rhabdomyolysis Risk Calculator, a prediction score that can help determine whether a patient with rhabdomyolysis is at risk for severe kidney failure or death.
The researchers conducted a retrospective cohort study of 2,371 patients admitted between 2000 and 2011 and examined variables that may be associated with kidney failure and death.5 They identified independent predictors for these outcomes, including age; gender; initial levels of phosphate, calcium, creatinine, carbon dioxide, and creatine kinase; and etiology of rhabdomyolysis (myositis, exercise, statin use, or seizure).5
This tool can assist providers in developing a patient-specific treatment plan. However, further research is needed to validate the current variables, verify the risk prediction score in other populations, and examine its ability to guide individualized treatment plans.
The Rhabdomyolysis Risk Calculator is available at www.brighamandwomens.org/research/rhabdo/default.aspx
Kristy Washinger, MSN, CRNP
Nephrology Associates of Central PA
Camp Hill, PA
REFERENCES
1. Vos FE, Schollum JB, Walker RJ. Glycated albumin is the preferred marker for assessing glycaemic control in advanced chronic kidney disease. Nephrol Dial Transplant Plus. 2011; 4(6):368-375.
2. National Kidney Foundation Kidney Disease Outcomes Quality Initiative. Clinical practice guidelines and clinical practice recommendations for diabetes and chronic kidney disease. Guideline 2: management of hyperglycemia and general diabetes care in chronic kidney disease. www.kidney.org/professionals/kdoqi/guideline_diabetes/guide2.htm. Accessed April 15, 2014.
3. Sharif A, Baboolal K. Diagnostic application of the A1c assay in renal disease. J Am Soc Nephrol. 2010;21(3):383-385.
4. American Diabetes Association. Standards of medical care in diabetes—2013. Diabetes Care. 2013;36(suppl 1):S11-S66.
5. McMahon GM, Zeng X, Walker SS. A risk prediction score for kidney failure or mortality in rhabdomyolysis. JAMA Intern Med. 2013;173(19):1821-1828.
Diabetic Amyotrophy: A Rare but Striking Neuropathy
A 45-year-old man, RT, with a six-month history of poorly controlled type 2 diabetes presents for evaluation of increased weakness and pain in the left lower extremity. The symptoms developed in the past three weeks. Previously able to ambulate without assistance, he purchased a cane yesterday due to concerns about falling.
RT reports poor adherence to his diabetes medications. His fingerstick blood sugars have ranged from 200 to 380 mg/dL over the past month. His weight has been stable; his BMI is 34. Review of other systems is negative. Vital signs include a blood pressure of 125/82 mm Hg; pulse, 74 beats/min; and respiratory rate, 16 breaths/min.
Physical examination is notable for muscle atrophy and tenderness to compression in the left quadriceps. Straight leg raise does not elicit pain bilaterally. Muscle strength is 4-/5 in the left hip with pain elicited on hip flexion, 4-/5 in the left knee, and 5/5 in the left ankle. Muscle strength is 4+/5 in the right hip, 5/5 in the right knee, and 5/5 in the right ankle. Muscle strength in both upper extremities is 5/5. Patellar deep tendon reflexes (DTRs) and ankle DTRs are absent bilaterally. Biceps and triceps DTRs are each 2+ bilaterally. Gait is slow and unsteady with use of the cane. Cranial nerves I-XII are intact. Sensation to sharp and dull testing is normal in both the upper and lower extremities.
Labwork reveals an A1C of 10.8%. The patient’s thyroid function studies, creatine kinase, and vitamin B12 level are all in normal range. The serum creatinine is 1.2 mg/dL, and eGFR (estimated glomerular filtration rate) is 58 mL/min/1.73 m2. Liver enzymes are normal, and complete blood count and other chemistry panels are unremarkable.
RT is referred to neurology. MRI of the thoracic and lumbar spine shows no mass lesions or disc disease. Electromyography reveals findings consistent with denervation and axonal damage in the proximal muscles in both lower extremities (left > right).
RT is diagnosed with diabetic amyotrophy and begins physical therapy three days a week. He achieves aggressive improvement in blood sugar control, and after three months, his A1C has improved to 7%. Although still using a cane, he reports improved muscle strength in the lower extremities and better gait stability.
Continued on next page >>
PREVALENCE AND TYPES OF DIABETIC PERIPHERAL NEUROPATHY
According to the CDC, 25.8 million children and adults in the United States (8.3% of the population) have diabetes. Approximately 60% to 70% of them have mild to severe neuropathy.1
Distal symmetric neuropathy is the most common form of diabetic peripheral neuropathy, accounting for more than 50% of cases. It is characterized by distal onset, predominately sensory polyneuropathy, and slow proximal progression.2
In contrast, diabetic amyotrophy is very rare, accounting for only 1% of all cases of neuropathy in diabetes. Prevalence is higher in those with type 2 versus type 1 diabetes (1.1% and 0.3%, respectively).3,4 The most commonly misdiagnosed of the asymmetric diabetic neuropathies, diabetic amyotrophy is characterized by acute, progressive, asymmetrical weakness and pain in the muscles of the proximal lower extremities.5 It is also been referred to as proximal diabetic neuropathy, ischemic mononeuropathy multiplex, diabetic femoral neuropathy, Bruns-Garland syndrome, and diabetic lumbosacral polyradiculopathy.5
LOCALIZATION AND PATHOGENESIS
The site of the lesion in diabetic amyotrophy remains controversial; it is theorized that diabetic amyotrophy may result from involvement of multiple sites, such as lumbosacral anterior horn cells, motor roots, plexus, or motor axons to the muscles of the proximal lower limbs.4
The pathogenesis remains unknown. One theory is that hyperglycemia may cause metabolic derangements in nerve conduction. Another is that there is ischemic damage followed by axonal degeneration. Immune-mediated inflammatory processes, such as microvasculitis, have also been proposed as causes.4,6
CLINICAL FEATURES
Diabetic amyotrophy is characterized by relatively rapid, progressive asymmetrical weakness and pain in the muscles in the proximal lower extremities; it develops over weeks to months and may continue for more than one year.2,6 It typically begins unilaterally and can progress bilaterally—normally without impairment in sensation. Patients commonly experience pain in the hip, buttock, or thigh, as well as difficulty walking, standing, or climbing stairs. Occasionally, the condition is painless and can be associated with weight loss. It causes significant acute disability, with the degree of recovery variable.2,4
Diabetic amyotrophy often presents either at diagnosis of diabetes or shortly thereafter. It most commonly affects men ages 40 to 50 and older, with higher incidence in type 2 diabetes.2,5
Physical exam findings include proximal muscle weakness and atrophy in the quadriceps, hamstring, gluteal, hip adductors/abductors, and iliopsoas muscles.4,5 Typically, there is no sensory impairment; however, mild sensory loss may be observed in patients with coexisting chronic distal sensorimotor polyneuropathy.2,4 The patellar tendon reflexes are typically diminished or absent, and the ankle reflexes may be normal or diminished.4
Continued on next page >>
DIAGNOSTIC WORK-UP AND DIFFERENTIAL DIAGNOSIS
Although the diagnosis of diabetic amyotrophy is made primarily through detailed history taking and neurologic examination, other studies—electromyography, nerve conduction, imaging and labs, and nerve biopsy—may provide confirmation. Referral to neurology should also be considered.
The differential diagnosis is extensive and includes myopathies, muscular dystrophies, intervertebral disc disease, spinal stenosis, polyradiculopathies due to porphyria, amyloid, heavy metal poisoning, anterior horn cell diseases (eg, poliomyelitis), neoplasms, chronic inflammatory demyelinating polyneuropathy, Guillain-Barré syndrome, monoclonal gammopathy, inflammatory vasculitis, hypothyroidism, vitamin B6 or B12 deficiencies, syphilis, AIDS, Lyme disease, and Charcot-Marie-Tooth disease.2,5-7 Diabetic neuropathic cachexia should also be considered in the differential, as it presents with weight loss and lower limb pain but no weakness.5
Lab evaluation should begin with analysis of fasting plasma glucose, complete blood count, comprehensive metabolic profile, A1C, erythrocyte sedimentation rate (ESR), creatine kinase, vitamin B12, and thyroid-stimulating hormone levels.7 Elevations in ESR and positive rheumatoid factor and antinuclear antibody can occur in patients with diabetic amyotrophy and are suggestive of a coexisting autoimmune disorder.6 Serum creatine kinase and thyroid function studies are normal.4 Additional lab tests, if clinically indicated, include paraneoplastic panel to evaluate for occult malignancy, antimyelin-associated glycoprotein antibodies, antiganglioside antibodies, cryoglobulins, cerebrospinal fluid analysis, porphyrin titers, and testing for heavy metals.7
Electrodiagnostic studies are recommended if the diagnosis of diabetic amyotrophy remains unclear following history taking, physical examination, and preliminary testing. Electromyography and nerve conduction studies typically reveal findings consistent with denervation and axonal damage in proximal muscles of the lower extremities.4 If demyelination is observed, a diagnosis of chronic demyelinating polyneuropathy should be considered.5
Nerve biopsy is considered if the diagnosis remains unclear after laboratory and electrodiagnostic testing or when confirmation of the diagnosis is needed before starting aggressive treatment. The sural and superficial peroneal nerves are preferred for biopsy. In cases of diabetic amyotrophy, sural nerve biopsy reveals significant fiber loss in an asymmetric fashion, resembling focal ischemia.5
MRI or CT scan of the lumbosacral spine is employed to exclude mass lesions and structural disorders such as spinal stenosis and disc disease.4 Cerebrospinal fluid is typically acellular, with a mildly elevated protein level of 60 to 100 mg/dL (but occasionally as high as 400 mg/dL).5
Continued on next page >>
PROGNOSIS AND MANAGEMENT
The course of diabetic amyotrophy is variable. There is often gradual but incomplete restoration in muscle strength in correlation with aggressive glycemic control and physical therapy.2 The majority of patients have residual muscle weakness, absent patellar and/or ankle DTRs, exercise-related pain, stiffness, and difficulty walking or climbing stairs. Full recovery of strength only occurs in 10% to 20% of patients.6
Treatment with IV immunoglobulin or other immunosuppressive drugs is controversial. According to a Cochrane review of immunotherapy for diabetic amyotrophy, only one completed controlled trial using IV methylprednisolone was found. There is currently no evidence to support use of immunoglobulins to halt progression and improve symptoms.8
Neuropathic pain may be difficult to control. The severe pain associated with diabetic amyotrophy begins to diminish several months after onset, but residual pain may persist for several years. Pregabalin, duloxetine, tricyclic antidepressants, antiepileptic drugs, and narcotic analgesics can be helpful.2,4 High doses of corticosteroids may lead to improvement of severe pain in some patients with diabetic amyotrophy.5
References >>
REFERENCES
1. CDC. National diabetes fact sheet: national estimates and general information on diabetes and prediabetes in the United States, 2011. Atlanta, GA: US Department of Health and Human Services, Centers for Disease Control and Prevention, 2011.
2. Nagsayi S, Somasekhar C, James CM. Diagnosis and management of diabetic amyotrophy. Geriatric Med. 2010;40:327-329.
3. Pasnoor M, Dimachkie MM, Kluding P, Barohn RJ. Diabetic neuropathy part 1: overview and symmetric phenotypes. Neurol Clin. 2013;31(2):425-445.
4. Sander HW, Chokroverty S. Diabetic amyotrophy: current concepts. Semin Neurol. 1996;16(2):173-177.
5. Pasnoor M, Dimachkie MM, Barohn RJ. Diabetic neuropathy part 2: proximal and asymmetric phenotypes. Neurol Clin. 2013;31(2): 447-462.
6. Idiculla J, Shirazi N, Opacka-Juffry J, Ganapathi. Diabetic amyotrophy: a brief review. Natl Med J India. 2004;17(4):
200-202.
7. Azhary H, Farooq M, Bhanushali M, Majid A. Peripheral neuropathy: differential diagnosis and management. Am Fam Physician. 2010;81(7):887-892.
8. Chan YC, Lo YL, Chan ES. Immunotherapy for diabetic amyotrophy. Cochrane Database Syst Rev. 2012;13(6):2-6.
A 45-year-old man, RT, with a six-month history of poorly controlled type 2 diabetes presents for evaluation of increased weakness and pain in the left lower extremity. The symptoms developed in the past three weeks. Previously able to ambulate without assistance, he purchased a cane yesterday due to concerns about falling.
RT reports poor adherence to his diabetes medications. His fingerstick blood sugars have ranged from 200 to 380 mg/dL over the past month. His weight has been stable; his BMI is 34. Review of other systems is negative. Vital signs include a blood pressure of 125/82 mm Hg; pulse, 74 beats/min; and respiratory rate, 16 breaths/min.
Physical examination is notable for muscle atrophy and tenderness to compression in the left quadriceps. Straight leg raise does not elicit pain bilaterally. Muscle strength is 4-/5 in the left hip with pain elicited on hip flexion, 4-/5 in the left knee, and 5/5 in the left ankle. Muscle strength is 4+/5 in the right hip, 5/5 in the right knee, and 5/5 in the right ankle. Muscle strength in both upper extremities is 5/5. Patellar deep tendon reflexes (DTRs) and ankle DTRs are absent bilaterally. Biceps and triceps DTRs are each 2+ bilaterally. Gait is slow and unsteady with use of the cane. Cranial nerves I-XII are intact. Sensation to sharp and dull testing is normal in both the upper and lower extremities.
Labwork reveals an A1C of 10.8%. The patient’s thyroid function studies, creatine kinase, and vitamin B12 level are all in normal range. The serum creatinine is 1.2 mg/dL, and eGFR (estimated glomerular filtration rate) is 58 mL/min/1.73 m2. Liver enzymes are normal, and complete blood count and other chemistry panels are unremarkable.
RT is referred to neurology. MRI of the thoracic and lumbar spine shows no mass lesions or disc disease. Electromyography reveals findings consistent with denervation and axonal damage in the proximal muscles in both lower extremities (left > right).
RT is diagnosed with diabetic amyotrophy and begins physical therapy three days a week. He achieves aggressive improvement in blood sugar control, and after three months, his A1C has improved to 7%. Although still using a cane, he reports improved muscle strength in the lower extremities and better gait stability.
Continued on next page >>
PREVALENCE AND TYPES OF DIABETIC PERIPHERAL NEUROPATHY
According to the CDC, 25.8 million children and adults in the United States (8.3% of the population) have diabetes. Approximately 60% to 70% of them have mild to severe neuropathy.1
Distal symmetric neuropathy is the most common form of diabetic peripheral neuropathy, accounting for more than 50% of cases. It is characterized by distal onset, predominately sensory polyneuropathy, and slow proximal progression.2
In contrast, diabetic amyotrophy is very rare, accounting for only 1% of all cases of neuropathy in diabetes. Prevalence is higher in those with type 2 versus type 1 diabetes (1.1% and 0.3%, respectively).3,4 The most commonly misdiagnosed of the asymmetric diabetic neuropathies, diabetic amyotrophy is characterized by acute, progressive, asymmetrical weakness and pain in the muscles of the proximal lower extremities.5 It is also been referred to as proximal diabetic neuropathy, ischemic mononeuropathy multiplex, diabetic femoral neuropathy, Bruns-Garland syndrome, and diabetic lumbosacral polyradiculopathy.5
LOCALIZATION AND PATHOGENESIS
The site of the lesion in diabetic amyotrophy remains controversial; it is theorized that diabetic amyotrophy may result from involvement of multiple sites, such as lumbosacral anterior horn cells, motor roots, plexus, or motor axons to the muscles of the proximal lower limbs.4
The pathogenesis remains unknown. One theory is that hyperglycemia may cause metabolic derangements in nerve conduction. Another is that there is ischemic damage followed by axonal degeneration. Immune-mediated inflammatory processes, such as microvasculitis, have also been proposed as causes.4,6
CLINICAL FEATURES
Diabetic amyotrophy is characterized by relatively rapid, progressive asymmetrical weakness and pain in the muscles in the proximal lower extremities; it develops over weeks to months and may continue for more than one year.2,6 It typically begins unilaterally and can progress bilaterally—normally without impairment in sensation. Patients commonly experience pain in the hip, buttock, or thigh, as well as difficulty walking, standing, or climbing stairs. Occasionally, the condition is painless and can be associated with weight loss. It causes significant acute disability, with the degree of recovery variable.2,4
Diabetic amyotrophy often presents either at diagnosis of diabetes or shortly thereafter. It most commonly affects men ages 40 to 50 and older, with higher incidence in type 2 diabetes.2,5
Physical exam findings include proximal muscle weakness and atrophy in the quadriceps, hamstring, gluteal, hip adductors/abductors, and iliopsoas muscles.4,5 Typically, there is no sensory impairment; however, mild sensory loss may be observed in patients with coexisting chronic distal sensorimotor polyneuropathy.2,4 The patellar tendon reflexes are typically diminished or absent, and the ankle reflexes may be normal or diminished.4
Continued on next page >>
DIAGNOSTIC WORK-UP AND DIFFERENTIAL DIAGNOSIS
Although the diagnosis of diabetic amyotrophy is made primarily through detailed history taking and neurologic examination, other studies—electromyography, nerve conduction, imaging and labs, and nerve biopsy—may provide confirmation. Referral to neurology should also be considered.
The differential diagnosis is extensive and includes myopathies, muscular dystrophies, intervertebral disc disease, spinal stenosis, polyradiculopathies due to porphyria, amyloid, heavy metal poisoning, anterior horn cell diseases (eg, poliomyelitis), neoplasms, chronic inflammatory demyelinating polyneuropathy, Guillain-Barré syndrome, monoclonal gammopathy, inflammatory vasculitis, hypothyroidism, vitamin B6 or B12 deficiencies, syphilis, AIDS, Lyme disease, and Charcot-Marie-Tooth disease.2,5-7 Diabetic neuropathic cachexia should also be considered in the differential, as it presents with weight loss and lower limb pain but no weakness.5
Lab evaluation should begin with analysis of fasting plasma glucose, complete blood count, comprehensive metabolic profile, A1C, erythrocyte sedimentation rate (ESR), creatine kinase, vitamin B12, and thyroid-stimulating hormone levels.7 Elevations in ESR and positive rheumatoid factor and antinuclear antibody can occur in patients with diabetic amyotrophy and are suggestive of a coexisting autoimmune disorder.6 Serum creatine kinase and thyroid function studies are normal.4 Additional lab tests, if clinically indicated, include paraneoplastic panel to evaluate for occult malignancy, antimyelin-associated glycoprotein antibodies, antiganglioside antibodies, cryoglobulins, cerebrospinal fluid analysis, porphyrin titers, and testing for heavy metals.7
Electrodiagnostic studies are recommended if the diagnosis of diabetic amyotrophy remains unclear following history taking, physical examination, and preliminary testing. Electromyography and nerve conduction studies typically reveal findings consistent with denervation and axonal damage in proximal muscles of the lower extremities.4 If demyelination is observed, a diagnosis of chronic demyelinating polyneuropathy should be considered.5
Nerve biopsy is considered if the diagnosis remains unclear after laboratory and electrodiagnostic testing or when confirmation of the diagnosis is needed before starting aggressive treatment. The sural and superficial peroneal nerves are preferred for biopsy. In cases of diabetic amyotrophy, sural nerve biopsy reveals significant fiber loss in an asymmetric fashion, resembling focal ischemia.5
MRI or CT scan of the lumbosacral spine is employed to exclude mass lesions and structural disorders such as spinal stenosis and disc disease.4 Cerebrospinal fluid is typically acellular, with a mildly elevated protein level of 60 to 100 mg/dL (but occasionally as high as 400 mg/dL).5
Continued on next page >>
PROGNOSIS AND MANAGEMENT
The course of diabetic amyotrophy is variable. There is often gradual but incomplete restoration in muscle strength in correlation with aggressive glycemic control and physical therapy.2 The majority of patients have residual muscle weakness, absent patellar and/or ankle DTRs, exercise-related pain, stiffness, and difficulty walking or climbing stairs. Full recovery of strength only occurs in 10% to 20% of patients.6
Treatment with IV immunoglobulin or other immunosuppressive drugs is controversial. According to a Cochrane review of immunotherapy for diabetic amyotrophy, only one completed controlled trial using IV methylprednisolone was found. There is currently no evidence to support use of immunoglobulins to halt progression and improve symptoms.8
Neuropathic pain may be difficult to control. The severe pain associated with diabetic amyotrophy begins to diminish several months after onset, but residual pain may persist for several years. Pregabalin, duloxetine, tricyclic antidepressants, antiepileptic drugs, and narcotic analgesics can be helpful.2,4 High doses of corticosteroids may lead to improvement of severe pain in some patients with diabetic amyotrophy.5
References >>
REFERENCES
1. CDC. National diabetes fact sheet: national estimates and general information on diabetes and prediabetes in the United States, 2011. Atlanta, GA: US Department of Health and Human Services, Centers for Disease Control and Prevention, 2011.
2. Nagsayi S, Somasekhar C, James CM. Diagnosis and management of diabetic amyotrophy. Geriatric Med. 2010;40:327-329.
3. Pasnoor M, Dimachkie MM, Kluding P, Barohn RJ. Diabetic neuropathy part 1: overview and symmetric phenotypes. Neurol Clin. 2013;31(2):425-445.
4. Sander HW, Chokroverty S. Diabetic amyotrophy: current concepts. Semin Neurol. 1996;16(2):173-177.
5. Pasnoor M, Dimachkie MM, Barohn RJ. Diabetic neuropathy part 2: proximal and asymmetric phenotypes. Neurol Clin. 2013;31(2): 447-462.
6. Idiculla J, Shirazi N, Opacka-Juffry J, Ganapathi. Diabetic amyotrophy: a brief review. Natl Med J India. 2004;17(4):
200-202.
7. Azhary H, Farooq M, Bhanushali M, Majid A. Peripheral neuropathy: differential diagnosis and management. Am Fam Physician. 2010;81(7):887-892.
8. Chan YC, Lo YL, Chan ES. Immunotherapy for diabetic amyotrophy. Cochrane Database Syst Rev. 2012;13(6):2-6.
A 45-year-old man, RT, with a six-month history of poorly controlled type 2 diabetes presents for evaluation of increased weakness and pain in the left lower extremity. The symptoms developed in the past three weeks. Previously able to ambulate without assistance, he purchased a cane yesterday due to concerns about falling.
RT reports poor adherence to his diabetes medications. His fingerstick blood sugars have ranged from 200 to 380 mg/dL over the past month. His weight has been stable; his BMI is 34. Review of other systems is negative. Vital signs include a blood pressure of 125/82 mm Hg; pulse, 74 beats/min; and respiratory rate, 16 breaths/min.
Physical examination is notable for muscle atrophy and tenderness to compression in the left quadriceps. Straight leg raise does not elicit pain bilaterally. Muscle strength is 4-/5 in the left hip with pain elicited on hip flexion, 4-/5 in the left knee, and 5/5 in the left ankle. Muscle strength is 4+/5 in the right hip, 5/5 in the right knee, and 5/5 in the right ankle. Muscle strength in both upper extremities is 5/5. Patellar deep tendon reflexes (DTRs) and ankle DTRs are absent bilaterally. Biceps and triceps DTRs are each 2+ bilaterally. Gait is slow and unsteady with use of the cane. Cranial nerves I-XII are intact. Sensation to sharp and dull testing is normal in both the upper and lower extremities.
Labwork reveals an A1C of 10.8%. The patient’s thyroid function studies, creatine kinase, and vitamin B12 level are all in normal range. The serum creatinine is 1.2 mg/dL, and eGFR (estimated glomerular filtration rate) is 58 mL/min/1.73 m2. Liver enzymes are normal, and complete blood count and other chemistry panels are unremarkable.
RT is referred to neurology. MRI of the thoracic and lumbar spine shows no mass lesions or disc disease. Electromyography reveals findings consistent with denervation and axonal damage in the proximal muscles in both lower extremities (left > right).
RT is diagnosed with diabetic amyotrophy and begins physical therapy three days a week. He achieves aggressive improvement in blood sugar control, and after three months, his A1C has improved to 7%. Although still using a cane, he reports improved muscle strength in the lower extremities and better gait stability.
Continued on next page >>
PREVALENCE AND TYPES OF DIABETIC PERIPHERAL NEUROPATHY
According to the CDC, 25.8 million children and adults in the United States (8.3% of the population) have diabetes. Approximately 60% to 70% of them have mild to severe neuropathy.1
Distal symmetric neuropathy is the most common form of diabetic peripheral neuropathy, accounting for more than 50% of cases. It is characterized by distal onset, predominately sensory polyneuropathy, and slow proximal progression.2
In contrast, diabetic amyotrophy is very rare, accounting for only 1% of all cases of neuropathy in diabetes. Prevalence is higher in those with type 2 versus type 1 diabetes (1.1% and 0.3%, respectively).3,4 The most commonly misdiagnosed of the asymmetric diabetic neuropathies, diabetic amyotrophy is characterized by acute, progressive, asymmetrical weakness and pain in the muscles of the proximal lower extremities.5 It is also been referred to as proximal diabetic neuropathy, ischemic mononeuropathy multiplex, diabetic femoral neuropathy, Bruns-Garland syndrome, and diabetic lumbosacral polyradiculopathy.5
LOCALIZATION AND PATHOGENESIS
The site of the lesion in diabetic amyotrophy remains controversial; it is theorized that diabetic amyotrophy may result from involvement of multiple sites, such as lumbosacral anterior horn cells, motor roots, plexus, or motor axons to the muscles of the proximal lower limbs.4
The pathogenesis remains unknown. One theory is that hyperglycemia may cause metabolic derangements in nerve conduction. Another is that there is ischemic damage followed by axonal degeneration. Immune-mediated inflammatory processes, such as microvasculitis, have also been proposed as causes.4,6
CLINICAL FEATURES
Diabetic amyotrophy is characterized by relatively rapid, progressive asymmetrical weakness and pain in the muscles in the proximal lower extremities; it develops over weeks to months and may continue for more than one year.2,6 It typically begins unilaterally and can progress bilaterally—normally without impairment in sensation. Patients commonly experience pain in the hip, buttock, or thigh, as well as difficulty walking, standing, or climbing stairs. Occasionally, the condition is painless and can be associated with weight loss. It causes significant acute disability, with the degree of recovery variable.2,4
Diabetic amyotrophy often presents either at diagnosis of diabetes or shortly thereafter. It most commonly affects men ages 40 to 50 and older, with higher incidence in type 2 diabetes.2,5
Physical exam findings include proximal muscle weakness and atrophy in the quadriceps, hamstring, gluteal, hip adductors/abductors, and iliopsoas muscles.4,5 Typically, there is no sensory impairment; however, mild sensory loss may be observed in patients with coexisting chronic distal sensorimotor polyneuropathy.2,4 The patellar tendon reflexes are typically diminished or absent, and the ankle reflexes may be normal or diminished.4
Continued on next page >>
DIAGNOSTIC WORK-UP AND DIFFERENTIAL DIAGNOSIS
Although the diagnosis of diabetic amyotrophy is made primarily through detailed history taking and neurologic examination, other studies—electromyography, nerve conduction, imaging and labs, and nerve biopsy—may provide confirmation. Referral to neurology should also be considered.
The differential diagnosis is extensive and includes myopathies, muscular dystrophies, intervertebral disc disease, spinal stenosis, polyradiculopathies due to porphyria, amyloid, heavy metal poisoning, anterior horn cell diseases (eg, poliomyelitis), neoplasms, chronic inflammatory demyelinating polyneuropathy, Guillain-Barré syndrome, monoclonal gammopathy, inflammatory vasculitis, hypothyroidism, vitamin B6 or B12 deficiencies, syphilis, AIDS, Lyme disease, and Charcot-Marie-Tooth disease.2,5-7 Diabetic neuropathic cachexia should also be considered in the differential, as it presents with weight loss and lower limb pain but no weakness.5
Lab evaluation should begin with analysis of fasting plasma glucose, complete blood count, comprehensive metabolic profile, A1C, erythrocyte sedimentation rate (ESR), creatine kinase, vitamin B12, and thyroid-stimulating hormone levels.7 Elevations in ESR and positive rheumatoid factor and antinuclear antibody can occur in patients with diabetic amyotrophy and are suggestive of a coexisting autoimmune disorder.6 Serum creatine kinase and thyroid function studies are normal.4 Additional lab tests, if clinically indicated, include paraneoplastic panel to evaluate for occult malignancy, antimyelin-associated glycoprotein antibodies, antiganglioside antibodies, cryoglobulins, cerebrospinal fluid analysis, porphyrin titers, and testing for heavy metals.7
Electrodiagnostic studies are recommended if the diagnosis of diabetic amyotrophy remains unclear following history taking, physical examination, and preliminary testing. Electromyography and nerve conduction studies typically reveal findings consistent with denervation and axonal damage in proximal muscles of the lower extremities.4 If demyelination is observed, a diagnosis of chronic demyelinating polyneuropathy should be considered.5
Nerve biopsy is considered if the diagnosis remains unclear after laboratory and electrodiagnostic testing or when confirmation of the diagnosis is needed before starting aggressive treatment. The sural and superficial peroneal nerves are preferred for biopsy. In cases of diabetic amyotrophy, sural nerve biopsy reveals significant fiber loss in an asymmetric fashion, resembling focal ischemia.5
MRI or CT scan of the lumbosacral spine is employed to exclude mass lesions and structural disorders such as spinal stenosis and disc disease.4 Cerebrospinal fluid is typically acellular, with a mildly elevated protein level of 60 to 100 mg/dL (but occasionally as high as 400 mg/dL).5
Continued on next page >>
PROGNOSIS AND MANAGEMENT
The course of diabetic amyotrophy is variable. There is often gradual but incomplete restoration in muscle strength in correlation with aggressive glycemic control and physical therapy.2 The majority of patients have residual muscle weakness, absent patellar and/or ankle DTRs, exercise-related pain, stiffness, and difficulty walking or climbing stairs. Full recovery of strength only occurs in 10% to 20% of patients.6
Treatment with IV immunoglobulin or other immunosuppressive drugs is controversial. According to a Cochrane review of immunotherapy for diabetic amyotrophy, only one completed controlled trial using IV methylprednisolone was found. There is currently no evidence to support use of immunoglobulins to halt progression and improve symptoms.8
Neuropathic pain may be difficult to control. The severe pain associated with diabetic amyotrophy begins to diminish several months after onset, but residual pain may persist for several years. Pregabalin, duloxetine, tricyclic antidepressants, antiepileptic drugs, and narcotic analgesics can be helpful.2,4 High doses of corticosteroids may lead to improvement of severe pain in some patients with diabetic amyotrophy.5
References >>
REFERENCES
1. CDC. National diabetes fact sheet: national estimates and general information on diabetes and prediabetes in the United States, 2011. Atlanta, GA: US Department of Health and Human Services, Centers for Disease Control and Prevention, 2011.
2. Nagsayi S, Somasekhar C, James CM. Diagnosis and management of diabetic amyotrophy. Geriatric Med. 2010;40:327-329.
3. Pasnoor M, Dimachkie MM, Kluding P, Barohn RJ. Diabetic neuropathy part 1: overview and symmetric phenotypes. Neurol Clin. 2013;31(2):425-445.
4. Sander HW, Chokroverty S. Diabetic amyotrophy: current concepts. Semin Neurol. 1996;16(2):173-177.
5. Pasnoor M, Dimachkie MM, Barohn RJ. Diabetic neuropathy part 2: proximal and asymmetric phenotypes. Neurol Clin. 2013;31(2): 447-462.
6. Idiculla J, Shirazi N, Opacka-Juffry J, Ganapathi. Diabetic amyotrophy: a brief review. Natl Med J India. 2004;17(4):
200-202.
7. Azhary H, Farooq M, Bhanushali M, Majid A. Peripheral neuropathy: differential diagnosis and management. Am Fam Physician. 2010;81(7):887-892.
8. Chan YC, Lo YL, Chan ES. Immunotherapy for diabetic amyotrophy. Cochrane Database Syst Rev. 2012;13(6):2-6.