User login
Strategies for managing drug-induced tardive dyskinesia
Fifteen years ago, Mr. L, age 40, was given a diagnosis of schizophrenia, which has been treated with haloperidol, 10 mg/d. Approximately 1 year ago, he began experiencing consistent lip smacking, a sign of tardive dyskinesia. Vitamin E was added to the treatment regimen, after which the tardive dyskinesia symptoms resolved.
A few months later, however, those symptoms returned and became worse. In addition to lip smacking, Mr. L now also describes involuntary bilateral twitching and muscle spasms in both legs.
Haloperidol and vitamin E are discontinued and Mr. L is switched to olanzapine, 20 mg/d. Although olanzapine is effective for Mr. L's symptoms of schizophrenia, tardive dyskinesia persists, and he gains 60 pounds and develops diabetes. Olanzapine is discontinued and he begins a trial of risperidone, 4 mg/d.
While on risperidone, blood sugar control, measured by hemoglobin A1c, and insulin resistance improve, but Mr. L continues to have symptoms of tardive dyskinesia. Vitamin E is added again, but is ineffective. The treatment team switches Mr. L to clozapine but symptoms of tardive dyskinesia do not improve.

Extrapyramidal side effects are common with first-generation antipsychotics (FGA) such as haloperidol. Types of antipsychotic-induced movement disorders include dystonias, akathisias, pseudoparkinsonism, and tardive dyskinesia. Of these, tardive dyskinesia is the most concerning because it often is difficult to treat and may be irreversible.
Tardive dyskinesia involves abnormal, involuntary movements, usually involving the face and, sometimes, the limbs. Common symptoms include lip smacking, tongue protrusions, and puffing the cheeks1; severe tardive dyskinesia may affect the larynx and diaphragm, which can be life-threatening. The incidence of tardive dyskinesia is approximately 5% after the first year of FGA treatment and 1% with second-generation antipsychotics (SGAs).2 The risk increases with higher doses and longer duration of treatment, with a prevalence of 20% to 25% with long-term FGA use.3
Treatment strategies
There are no FDA-approved drugs for tardive dyskinesia (Table).4-6 The best strategy is to prevent tardive dyskinesia with judicious use of an antipsychotic. If a patient taking a FGA develops tardive dyskinesia, the first-line treatment is to switch to a SGA. Risperidone, olanzapine, quetiapine, and clozapine have a low risk of tardive dyskinesia. Newer agents, such as lurasidone, asenapine, iloperidone, and aripiprazole, might have a lower risk of tardive dyskinesia, possibly because of differences in dopamine blockage between these agents and FGAs. Clozapine is least likely to cause tardive dyskinesia, but it often is used as a last resort because of the risk of agranulocytosis and the need for frequent tests to measurewhite blood cells.1,4
Other treatments include melatonin, donepezil, vitamin B6, and vitamin E.4 These agents can reduce symptoms, but no large clinical trials have proved that the are efficacious. Last-resort treatments include suppressive treatment using FGAs several times a day, because the constant dopamine blockade may stop symptoms for a short time; this approach is not recommended because it can exacerbate symptoms of tardive dyskinesia.4
Other suppressive treatments used in severe or refractory cases include reserpineand tetrabenazine, which are used off-label and work by blocking monoamine transporters. This blockage results in a reduction in neurotransmitters such as dopamine, which have been implicated in the development of tardive dyskinesia. Compared with tetrabenazine, reserpine has a higher affinity for cells in the periphery and therefore causes side effects such as hypotension and diarrhea.7
Tetrabenazine is indicated for chorea associated with Huntington's disease and is used off-label for treating tardive dyskinesia. Tetrabenazine is thought to work by inhibiting human vesicular monoamine transporters. Blocking these transporters prevents monoamines such as dopamine from entering synaptic vesicles.8 Because of its side-effect profile, lack of large clinical trials, and high cost, tetrabenazine is used as a last-line treatment in severe cases of tardive dyskinesia. Adverse effects include somnolence (31%), insomnia (22%), depression (19%), and akathisia (19%). Tetrabenazine carries a black-box warning for depression and suicidalityand is contraindicated in patients with untreated or inadequately treated depression or who are suicidal.8 Assessing the patient's mental state is important when using this medication.
A review by Chen et al7 found that 9 of 11 studies had positive results for tetrabenazine treatment for tardive dyskinesia. Most of the studies were small and retrospective. The biggest prospective blinded study was a videotaped study by Ondo et al of 20 patients with tardive dyskinesia.9 At least 30 days before beginning the study patients discontinued the medication that caused their tardive dyskinesia and any treatments for tardive dyskinesia. Each patient was videotaped before starting tetrabenazine and an average of 20.3 weeks after starting the drug. Investigators' scores showed an average of 54.2% improvement in movement scores and participants' subjective scores reported an average of 60.4% improvement. One patient withdrew because of somnolence. The remaining 19 patients did not experience more than mild side effects and continued treatment with tetrabenazine after study completion.9
Treatment recommendations
Tardive dyskinesia is a difficult condition to treat; it is best, therefore, to prevent its onset by using the minimally effective antipsychotic dose, by preferential use of an SGA rather than a FGA, and by regular screening for tardive dyskinesia using a standardized rating scale such as the Abnormal Involuntary Movement Scale. Symptoms associated with tardive dyskinesia are more likely to resolve if caught early. If a patient develops tardive dyskinesia while taking a FGA, switching to a SGA may alleviate the symptoms.
Several medications can be used off-label to relieve symptoms, including vitamin E and tetrabenazine, which both have the most-although not considerable-literature-based support. Although some studies show benefit with tetrabenazine for tardive dyskinesia, larger clinical trials are needed to more strongly support its use. Tetrabenazine might be a good option for patients with refractory tardive dyskinesia but, given the associated risk of suicidality and depressive symptoms, careful monitoring of suicide risk is essential and may preclude its use for tardive dyskinesia in patients who are experiencing depressive symptoms.
Related Resources
• National Organization for Rare Disorders. Tardive dyskinesia. www.rarediseases.org/rare-disease-information/rare-diseases/byID/493/viewFullReport.
• Caroff SN, Miller DD, Campbell CE. Is there a rational management strategy for tardive dyskinesia? Current Psychiatry. 2011;10(10):22-32.
Drug Brand Names
Aripiprazole • Abilify Lurasidone • Latuda
Asenapine • Saphris Olanzapine • Zyprexa
Clozapine • Clozaril Quetiapine • Seroquel
Donepezil • Aricept Reserpine • Serpasil
Haloperidol • Haldol Risperidone • Risperdal
Iloperidone • Fanapt Tetrabenazine • Xenazine
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Aia PG, Revuelta GJ, Cloud LJ, et al. Tardive dyskinesia. Curr Treat Options Neurol. 2011;13(3):231-241.
2. Correll CU, Leucht S, Kane JM. Lower risk for tardive dyskinesia associated with second-generation antipsychotics: a systematic review of 1-year studies. Am J Psychiatry. 2004;161(3):414-425.
3. Crimson ML, Argo TR, Buckley PF. Schizophrenia. In: DiPiro JT, Talbert RL, Yee GC, et al, eds. Pharmacotherapy: a pathophysiologic approach. 8th ed. New York, NY: McGraw- Hill; 2011:1147-1172.
4. Margolese HC, Chouinard G, Kolivakis TT, et al. Tardive dyskinesia in the era of typical and atypical antipsychotics. Part 2: incidence and management strategies in patients with schizophrenia. Can J Psychiatry. 2005;50(11):703-714.
5. Facts & Comparisons. http://online.factsandcomparisons.com/index.aspx. Accessed November 4, 2012.
6. Natural Standard. http://www.naturalstandard.com/index.asp. Accessed November 4, 2012.
7. Chen JJ, Ondo WG, Dashtipour K, et al. Tetrabenazine for the treatment of hyperkinetic movement disorders: a review of the literature. Clin Ther. 2012;34(7):1487-1504.
8. Xanazine [package insert]. Deerfield, IL: Lundbeck Inc; 2012.
9. Ondo WG, Hanna PA, Jankovic J. Tetrabenazine treatment for tardive dyskinesia: assessment by randomized videotape protocol. Am J Psychiatry. 1999;156(8):1279-1281.
Fifteen years ago, Mr. L, age 40, was given a diagnosis of schizophrenia, which has been treated with haloperidol, 10 mg/d. Approximately 1 year ago, he began experiencing consistent lip smacking, a sign of tardive dyskinesia. Vitamin E was added to the treatment regimen, after which the tardive dyskinesia symptoms resolved.
A few months later, however, those symptoms returned and became worse. In addition to lip smacking, Mr. L now also describes involuntary bilateral twitching and muscle spasms in both legs.
Haloperidol and vitamin E are discontinued and Mr. L is switched to olanzapine, 20 mg/d. Although olanzapine is effective for Mr. L's symptoms of schizophrenia, tardive dyskinesia persists, and he gains 60 pounds and develops diabetes. Olanzapine is discontinued and he begins a trial of risperidone, 4 mg/d.
While on risperidone, blood sugar control, measured by hemoglobin A1c, and insulin resistance improve, but Mr. L continues to have symptoms of tardive dyskinesia. Vitamin E is added again, but is ineffective. The treatment team switches Mr. L to clozapine but symptoms of tardive dyskinesia do not improve.
Extrapyramidal side effects are common with first-generation antipsychotics (FGA) such as haloperidol. Types of antipsychotic-induced movement disorders include dystonias, akathisias, pseudoparkinsonism, and tardive dyskinesia. Of these, tardive dyskinesia is the most concerning because it often is difficult to treat and may be irreversible.
Tardive dyskinesia involves abnormal, involuntary movements, usually involving the face and, sometimes, the limbs. Common symptoms include lip smacking, tongue protrusions, and puffing the cheeks1; severe tardive dyskinesia may affect the larynx and diaphragm, which can be life-threatening. The incidence of tardive dyskinesia is approximately 5% after the first year of FGA treatment and 1% with second-generation antipsychotics (SGAs).2 The risk increases with higher doses and longer duration of treatment, with a prevalence of 20% to 25% with long-term FGA use.3
Treatment strategies
There are no FDA-approved drugs for tardive dyskinesia (Table).4-6 The best strategy is to prevent tardive dyskinesia with judicious use of an antipsychotic. If a patient taking a FGA develops tardive dyskinesia, the first-line treatment is to switch to a SGA. Risperidone, olanzapine, quetiapine, and clozapine have a low risk of tardive dyskinesia. Newer agents, such as lurasidone, asenapine, iloperidone, and aripiprazole, might have a lower risk of tardive dyskinesia, possibly because of differences in dopamine blockage between these agents and FGAs. Clozapine is least likely to cause tardive dyskinesia, but it often is used as a last resort because of the risk of agranulocytosis and the need for frequent tests to measurewhite blood cells.1,4
Other treatments include melatonin, donepezil, vitamin B6, and vitamin E.4 These agents can reduce symptoms, but no large clinical trials have proved that the are efficacious. Last-resort treatments include suppressive treatment using FGAs several times a day, because the constant dopamine blockade may stop symptoms for a short time; this approach is not recommended because it can exacerbate symptoms of tardive dyskinesia.4
Other suppressive treatments used in severe or refractory cases include reserpineand tetrabenazine, which are used off-label and work by blocking monoamine transporters. This blockage results in a reduction in neurotransmitters such as dopamine, which have been implicated in the development of tardive dyskinesia. Compared with tetrabenazine, reserpine has a higher affinity for cells in the periphery and therefore causes side effects such as hypotension and diarrhea.7
Tetrabenazine is indicated for chorea associated with Huntington's disease and is used off-label for treating tardive dyskinesia. Tetrabenazine is thought to work by inhibiting human vesicular monoamine transporters. Blocking these transporters prevents monoamines such as dopamine from entering synaptic vesicles.8 Because of its side-effect profile, lack of large clinical trials, and high cost, tetrabenazine is used as a last-line treatment in severe cases of tardive dyskinesia. Adverse effects include somnolence (31%), insomnia (22%), depression (19%), and akathisia (19%). Tetrabenazine carries a black-box warning for depression and suicidalityand is contraindicated in patients with untreated or inadequately treated depression or who are suicidal.8 Assessing the patient's mental state is important when using this medication.
A review by Chen et al7 found that 9 of 11 studies had positive results for tetrabenazine treatment for tardive dyskinesia. Most of the studies were small and retrospective. The biggest prospective blinded study was a videotaped study by Ondo et al of 20 patients with tardive dyskinesia.9 At least 30 days before beginning the study patients discontinued the medication that caused their tardive dyskinesia and any treatments for tardive dyskinesia. Each patient was videotaped before starting tetrabenazine and an average of 20.3 weeks after starting the drug. Investigators' scores showed an average of 54.2% improvement in movement scores and participants' subjective scores reported an average of 60.4% improvement. One patient withdrew because of somnolence. The remaining 19 patients did not experience more than mild side effects and continued treatment with tetrabenazine after study completion.9
Treatment recommendations
Tardive dyskinesia is a difficult condition to treat; it is best, therefore, to prevent its onset by using the minimally effective antipsychotic dose, by preferential use of an SGA rather than a FGA, and by regular screening for tardive dyskinesia using a standardized rating scale such as the Abnormal Involuntary Movement Scale. Symptoms associated with tardive dyskinesia are more likely to resolve if caught early. If a patient develops tardive dyskinesia while taking a FGA, switching to a SGA may alleviate the symptoms.
Several medications can be used off-label to relieve symptoms, including vitamin E and tetrabenazine, which both have the most-although not considerable-literature-based support. Although some studies show benefit with tetrabenazine for tardive dyskinesia, larger clinical trials are needed to more strongly support its use. Tetrabenazine might be a good option for patients with refractory tardive dyskinesia but, given the associated risk of suicidality and depressive symptoms, careful monitoring of suicide risk is essential and may preclude its use for tardive dyskinesia in patients who are experiencing depressive symptoms.
Related Resources
• National Organization for Rare Disorders. Tardive dyskinesia. www.rarediseases.org/rare-disease-information/rare-diseases/byID/493/viewFullReport.
• Caroff SN, Miller DD, Campbell CE. Is there a rational management strategy for tardive dyskinesia? Current Psychiatry. 2011;10(10):22-32.
Drug Brand Names
Aripiprazole • Abilify Lurasidone • Latuda
Asenapine • Saphris Olanzapine • Zyprexa
Clozapine • Clozaril Quetiapine • Seroquel
Donepezil • Aricept Reserpine • Serpasil
Haloperidol • Haldol Risperidone • Risperdal
Iloperidone • Fanapt Tetrabenazine • Xenazine
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Fifteen years ago, Mr. L, age 40, was given a diagnosis of schizophrenia, which has been treated with haloperidol, 10 mg/d. Approximately 1 year ago, he began experiencing consistent lip smacking, a sign of tardive dyskinesia. Vitamin E was added to the treatment regimen, after which the tardive dyskinesia symptoms resolved.
A few months later, however, those symptoms returned and became worse. In addition to lip smacking, Mr. L now also describes involuntary bilateral twitching and muscle spasms in both legs.
Haloperidol and vitamin E are discontinued and Mr. L is switched to olanzapine, 20 mg/d. Although olanzapine is effective for Mr. L's symptoms of schizophrenia, tardive dyskinesia persists, and he gains 60 pounds and develops diabetes. Olanzapine is discontinued and he begins a trial of risperidone, 4 mg/d.
While on risperidone, blood sugar control, measured by hemoglobin A1c, and insulin resistance improve, but Mr. L continues to have symptoms of tardive dyskinesia. Vitamin E is added again, but is ineffective. The treatment team switches Mr. L to clozapine but symptoms of tardive dyskinesia do not improve.
Extrapyramidal side effects are common with first-generation antipsychotics (FGA) such as haloperidol. Types of antipsychotic-induced movement disorders include dystonias, akathisias, pseudoparkinsonism, and tardive dyskinesia. Of these, tardive dyskinesia is the most concerning because it often is difficult to treat and may be irreversible.
Tardive dyskinesia involves abnormal, involuntary movements, usually involving the face and, sometimes, the limbs. Common symptoms include lip smacking, tongue protrusions, and puffing the cheeks1; severe tardive dyskinesia may affect the larynx and diaphragm, which can be life-threatening. The incidence of tardive dyskinesia is approximately 5% after the first year of FGA treatment and 1% with second-generation antipsychotics (SGAs).2 The risk increases with higher doses and longer duration of treatment, with a prevalence of 20% to 25% with long-term FGA use.3
Treatment strategies
There are no FDA-approved drugs for tardive dyskinesia (Table).4-6 The best strategy is to prevent tardive dyskinesia with judicious use of an antipsychotic. If a patient taking a FGA develops tardive dyskinesia, the first-line treatment is to switch to a SGA. Risperidone, olanzapine, quetiapine, and clozapine have a low risk of tardive dyskinesia. Newer agents, such as lurasidone, asenapine, iloperidone, and aripiprazole, might have a lower risk of tardive dyskinesia, possibly because of differences in dopamine blockage between these agents and FGAs. Clozapine is least likely to cause tardive dyskinesia, but it often is used as a last resort because of the risk of agranulocytosis and the need for frequent tests to measurewhite blood cells.1,4
Other treatments include melatonin, donepezil, vitamin B6, and vitamin E.4 These agents can reduce symptoms, but no large clinical trials have proved that the are efficacious. Last-resort treatments include suppressive treatment using FGAs several times a day, because the constant dopamine blockade may stop symptoms for a short time; this approach is not recommended because it can exacerbate symptoms of tardive dyskinesia.4
Other suppressive treatments used in severe or refractory cases include reserpineand tetrabenazine, which are used off-label and work by blocking monoamine transporters. This blockage results in a reduction in neurotransmitters such as dopamine, which have been implicated in the development of tardive dyskinesia. Compared with tetrabenazine, reserpine has a higher affinity for cells in the periphery and therefore causes side effects such as hypotension and diarrhea.7
Tetrabenazine is indicated for chorea associated with Huntington's disease and is used off-label for treating tardive dyskinesia. Tetrabenazine is thought to work by inhibiting human vesicular monoamine transporters. Blocking these transporters prevents monoamines such as dopamine from entering synaptic vesicles.8 Because of its side-effect profile, lack of large clinical trials, and high cost, tetrabenazine is used as a last-line treatment in severe cases of tardive dyskinesia. Adverse effects include somnolence (31%), insomnia (22%), depression (19%), and akathisia (19%). Tetrabenazine carries a black-box warning for depression and suicidalityand is contraindicated in patients with untreated or inadequately treated depression or who are suicidal.8 Assessing the patient's mental state is important when using this medication.
A review by Chen et al7 found that 9 of 11 studies had positive results for tetrabenazine treatment for tardive dyskinesia. Most of the studies were small and retrospective. The biggest prospective blinded study was a videotaped study by Ondo et al of 20 patients with tardive dyskinesia.9 At least 30 days before beginning the study patients discontinued the medication that caused their tardive dyskinesia and any treatments for tardive dyskinesia. Each patient was videotaped before starting tetrabenazine and an average of 20.3 weeks after starting the drug. Investigators' scores showed an average of 54.2% improvement in movement scores and participants' subjective scores reported an average of 60.4% improvement. One patient withdrew because of somnolence. The remaining 19 patients did not experience more than mild side effects and continued treatment with tetrabenazine after study completion.9
Treatment recommendations
Tardive dyskinesia is a difficult condition to treat; it is best, therefore, to prevent its onset by using the minimally effective antipsychotic dose, by preferential use of an SGA rather than a FGA, and by regular screening for tardive dyskinesia using a standardized rating scale such as the Abnormal Involuntary Movement Scale. Symptoms associated with tardive dyskinesia are more likely to resolve if caught early. If a patient develops tardive dyskinesia while taking a FGA, switching to a SGA may alleviate the symptoms.
Several medications can be used off-label to relieve symptoms, including vitamin E and tetrabenazine, which both have the most-although not considerable-literature-based support. Although some studies show benefit with tetrabenazine for tardive dyskinesia, larger clinical trials are needed to more strongly support its use. Tetrabenazine might be a good option for patients with refractory tardive dyskinesia but, given the associated risk of suicidality and depressive symptoms, careful monitoring of suicide risk is essential and may preclude its use for tardive dyskinesia in patients who are experiencing depressive symptoms.
Related Resources
• National Organization for Rare Disorders. Tardive dyskinesia. www.rarediseases.org/rare-disease-information/rare-diseases/byID/493/viewFullReport.
• Caroff SN, Miller DD, Campbell CE. Is there a rational management strategy for tardive dyskinesia? Current Psychiatry. 2011;10(10):22-32.
Drug Brand Names
Aripiprazole • Abilify Lurasidone • Latuda
Asenapine • Saphris Olanzapine • Zyprexa
Clozapine • Clozaril Quetiapine • Seroquel
Donepezil • Aricept Reserpine • Serpasil
Haloperidol • Haldol Risperidone • Risperdal
Iloperidone • Fanapt Tetrabenazine • Xenazine
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Aia PG, Revuelta GJ, Cloud LJ, et al. Tardive dyskinesia. Curr Treat Options Neurol. 2011;13(3):231-241.
2. Correll CU, Leucht S, Kane JM. Lower risk for tardive dyskinesia associated with second-generation antipsychotics: a systematic review of 1-year studies. Am J Psychiatry. 2004;161(3):414-425.
3. Crimson ML, Argo TR, Buckley PF. Schizophrenia. In: DiPiro JT, Talbert RL, Yee GC, et al, eds. Pharmacotherapy: a pathophysiologic approach. 8th ed. New York, NY: McGraw- Hill; 2011:1147-1172.
4. Margolese HC, Chouinard G, Kolivakis TT, et al. Tardive dyskinesia in the era of typical and atypical antipsychotics. Part 2: incidence and management strategies in patients with schizophrenia. Can J Psychiatry. 2005;50(11):703-714.
5. Facts & Comparisons. http://online.factsandcomparisons.com/index.aspx. Accessed November 4, 2012.
6. Natural Standard. http://www.naturalstandard.com/index.asp. Accessed November 4, 2012.
7. Chen JJ, Ondo WG, Dashtipour K, et al. Tetrabenazine for the treatment of hyperkinetic movement disorders: a review of the literature. Clin Ther. 2012;34(7):1487-1504.
8. Xanazine [package insert]. Deerfield, IL: Lundbeck Inc; 2012.
9. Ondo WG, Hanna PA, Jankovic J. Tetrabenazine treatment for tardive dyskinesia: assessment by randomized videotape protocol. Am J Psychiatry. 1999;156(8):1279-1281.
1. Aia PG, Revuelta GJ, Cloud LJ, et al. Tardive dyskinesia. Curr Treat Options Neurol. 2011;13(3):231-241.
2. Correll CU, Leucht S, Kane JM. Lower risk for tardive dyskinesia associated with second-generation antipsychotics: a systematic review of 1-year studies. Am J Psychiatry. 2004;161(3):414-425.
3. Crimson ML, Argo TR, Buckley PF. Schizophrenia. In: DiPiro JT, Talbert RL, Yee GC, et al, eds. Pharmacotherapy: a pathophysiologic approach. 8th ed. New York, NY: McGraw- Hill; 2011:1147-1172.
4. Margolese HC, Chouinard G, Kolivakis TT, et al. Tardive dyskinesia in the era of typical and atypical antipsychotics. Part 2: incidence and management strategies in patients with schizophrenia. Can J Psychiatry. 2005;50(11):703-714.
5. Facts & Comparisons. http://online.factsandcomparisons.com/index.aspx. Accessed November 4, 2012.
6. Natural Standard. http://www.naturalstandard.com/index.asp. Accessed November 4, 2012.
7. Chen JJ, Ondo WG, Dashtipour K, et al. Tetrabenazine for the treatment of hyperkinetic movement disorders: a review of the literature. Clin Ther. 2012;34(7):1487-1504.
8. Xanazine [package insert]. Deerfield, IL: Lundbeck Inc; 2012.
9. Ondo WG, Hanna PA, Jankovic J. Tetrabenazine treatment for tardive dyskinesia: assessment by randomized videotape protocol. Am J Psychiatry. 1999;156(8):1279-1281.
Is DXA Valid for Kidney Patients?
Q) A 53-year-old dialysis patient in my clinic says her nephrologist told her she did not need a DXA scan because it was not valid for kidney patients. Why not?
Osteoporosis is a condition of reduced bone mass, causing decreased bone strength and leading to increased risk for fractures. The World Health Organization definition of osteoporosis is based on bone mineral density (BMD). While there is some overlap between idiopathic osteoporosis and chronic kidney disease–mineral bone disorder (CKD-MBD), both conditions have different pathophysiologic backgrounds and require different treatments.1
There is not an accurate correlation between BMD as measured by DXA and the type of CKD-associated bone disease.2 Patients with CKD typically have lower bone density than the general population, making the interpretation of DXA (dual x-ray absorptiometry) scans more complicated. This is due to focal areas of osteosclerosis, the presence of extraskeletal calcifications, and the variable presence of osteomalacia.
The gold standard for assessment and diagnosis of bone disease in CKD patients is the iliac crest bone biopsy. However, it is not frequently performed due to the painful and invasive nature of the procedure and the limitations in access to centers where it is performed and to experienced pathologists.
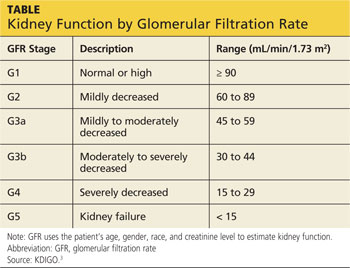
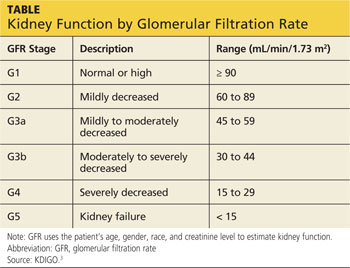
KDIGO (Kidney Disease Improving Global Outcomes) guidelines3 recommend that in patients with CKD stages 3 to 5D (see chart for explanation), measurements of serum parathyroid hormone or bone-specific alkaline phosphatase be used to evaluate bone disease, because markedly high or low values predict underlying bone turnover.
Shelly Levinstein, MSN, CRNP
Nephrology Associates of York
York, PA
REFERENCES
1. Toussaint N, Elder G, Kerr P. Bisphosphonates in chronic kidney disease; balancing potential benefits and adverse effects on bone and soft tissue. Clin J Am Soc Nephrol. 2009;4:221-233.
2. Tanenbaum N, Quarles LD. Bone disorders in chronic kidney disease. In: Greenberg A, Cheung AK, eds. Primer on Kidney Diseases. 5th ed. Philadelphia, PA: Elsevier Saunders; 2009:487-498.
3. Moe S, Drueke T, Cunningham J, et al; Kidney Disease: Improving Global Outcomes (KDIGO). Definition, evaluation, and classification of renal osteodystrophy: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2006;69:1945-1953.
4. Gilbert S, Weiner DE. National Kidney Foundation’s Primer on Kidney Diseases. 6th ed. Philadelphia, PA: Elsevier Saunders; 2013:330.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Shelly Levinstein, MSN, CRNP, who practices at Nephrology Associates of York in Pennsylvania.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Shelly Levinstein, MSN, CRNP, who practices at Nephrology Associates of York in Pennsylvania.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Shelly Levinstein, MSN, CRNP, who practices at Nephrology Associates of York in Pennsylvania.
Q) A 53-year-old dialysis patient in my clinic says her nephrologist told her she did not need a DXA scan because it was not valid for kidney patients. Why not?
Osteoporosis is a condition of reduced bone mass, causing decreased bone strength and leading to increased risk for fractures. The World Health Organization definition of osteoporosis is based on bone mineral density (BMD). While there is some overlap between idiopathic osteoporosis and chronic kidney disease–mineral bone disorder (CKD-MBD), both conditions have different pathophysiologic backgrounds and require different treatments.1
There is not an accurate correlation between BMD as measured by DXA and the type of CKD-associated bone disease.2 Patients with CKD typically have lower bone density than the general population, making the interpretation of DXA (dual x-ray absorptiometry) scans more complicated. This is due to focal areas of osteosclerosis, the presence of extraskeletal calcifications, and the variable presence of osteomalacia.
The gold standard for assessment and diagnosis of bone disease in CKD patients is the iliac crest bone biopsy. However, it is not frequently performed due to the painful and invasive nature of the procedure and the limitations in access to centers where it is performed and to experienced pathologists.
KDIGO (Kidney Disease Improving Global Outcomes) guidelines3 recommend that in patients with CKD stages 3 to 5D (see chart for explanation), measurements of serum parathyroid hormone or bone-specific alkaline phosphatase be used to evaluate bone disease, because markedly high or low values predict underlying bone turnover.
Shelly Levinstein, MSN, CRNP
Nephrology Associates of York
York, PA
REFERENCES
1. Toussaint N, Elder G, Kerr P. Bisphosphonates in chronic kidney disease; balancing potential benefits and adverse effects on bone and soft tissue. Clin J Am Soc Nephrol. 2009;4:221-233.
2. Tanenbaum N, Quarles LD. Bone disorders in chronic kidney disease. In: Greenberg A, Cheung AK, eds. Primer on Kidney Diseases. 5th ed. Philadelphia, PA: Elsevier Saunders; 2009:487-498.
3. Moe S, Drueke T, Cunningham J, et al; Kidney Disease: Improving Global Outcomes (KDIGO). Definition, evaluation, and classification of renal osteodystrophy: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2006;69:1945-1953.
4. Gilbert S, Weiner DE. National Kidney Foundation’s Primer on Kidney Diseases. 6th ed. Philadelphia, PA: Elsevier Saunders; 2013:330.
Q) A 53-year-old dialysis patient in my clinic says her nephrologist told her she did not need a DXA scan because it was not valid for kidney patients. Why not?
Osteoporosis is a condition of reduced bone mass, causing decreased bone strength and leading to increased risk for fractures. The World Health Organization definition of osteoporosis is based on bone mineral density (BMD). While there is some overlap between idiopathic osteoporosis and chronic kidney disease–mineral bone disorder (CKD-MBD), both conditions have different pathophysiologic backgrounds and require different treatments.1
There is not an accurate correlation between BMD as measured by DXA and the type of CKD-associated bone disease.2 Patients with CKD typically have lower bone density than the general population, making the interpretation of DXA (dual x-ray absorptiometry) scans more complicated. This is due to focal areas of osteosclerosis, the presence of extraskeletal calcifications, and the variable presence of osteomalacia.
The gold standard for assessment and diagnosis of bone disease in CKD patients is the iliac crest bone biopsy. However, it is not frequently performed due to the painful and invasive nature of the procedure and the limitations in access to centers where it is performed and to experienced pathologists.
KDIGO (Kidney Disease Improving Global Outcomes) guidelines3 recommend that in patients with CKD stages 3 to 5D (see chart for explanation), measurements of serum parathyroid hormone or bone-specific alkaline phosphatase be used to evaluate bone disease, because markedly high or low values predict underlying bone turnover.
Shelly Levinstein, MSN, CRNP
Nephrology Associates of York
York, PA
REFERENCES
1. Toussaint N, Elder G, Kerr P. Bisphosphonates in chronic kidney disease; balancing potential benefits and adverse effects on bone and soft tissue. Clin J Am Soc Nephrol. 2009;4:221-233.
2. Tanenbaum N, Quarles LD. Bone disorders in chronic kidney disease. In: Greenberg A, Cheung AK, eds. Primer on Kidney Diseases. 5th ed. Philadelphia, PA: Elsevier Saunders; 2009:487-498.
3. Moe S, Drueke T, Cunningham J, et al; Kidney Disease: Improving Global Outcomes (KDIGO). Definition, evaluation, and classification of renal osteodystrophy: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2006;69:1945-1953.
4. Gilbert S, Weiner DE. National Kidney Foundation’s Primer on Kidney Diseases. 6th ed. Philadelphia, PA: Elsevier Saunders; 2013:330.
How to Treat Bone Loss in Kidney Patients
Q) Since bone loss is common in patients with kidney disease, how should it be treated?
In elderly CKD patients, it is recognized that osteoporosis and CKD-MBD may co-exist—which only makes the bone-loss issue more problematic. Therefore, diagnosis and management are crucial to effective treatment.
Although osteoporosis is recognized and treated in CKD stages 1 to 3a, the interpretation of BMD levels in the osteoporotic range is controversial in advanced kidney disease (GFR < 35).1 There are limited data for treatment of patients with CKD-MBD.
Studies of bisphosphonate use in postmenopausal women and in patients with glucocorticoid-induced osteoporosis have generally excluded those with renal impairment. For these patients, treatment of low BMD using standard therapies for osteoporosis is not without potential for harm, due to the possibility of worsening low bone turnover, osteomalacia, mixed uremic osteodystrophy, and exacerbated hyperparathyroidism.
The choice of pharmaceutical treatment should be based on whether you are treating CKD-MBD or osteoporosis. A large percentage of CKD patients have adynamic bone disease with low bone resorption. In patients with low bone resorption, treatment entails choosing a drug that stimulates bone formation and not those that inhibit bone resorption. The benefit of bisphosphonate therapy is seen in patients with high bone resorption.4
In CKD stages 1 to 3, one must evaluate laboratory features of low BMD, including serum levels of calcium, phosphate, parathyroid hormone, alkaline phosphatase, and vitamin D. If all are found to be normal, bisphosphonate use in CKD stages 1 to 3 is usually safe.1
A bone biopsy is recommended for patients with advanced CKD stages 4 to 5/5D, with therapy individualized per disease process.1
Shelly Levinstein, MSN, CRNP
Nephrology Associates of York
York, PA
REFERENCES
1. Toussaint N, Elder G, Kerr P. Bisphosphonates in chronic kidney disease; balancing potential benefits and adverse effects on bone and soft tissue. Clin J Am Soc Nephrol. 2009;4:221-233.
2. Tanenbaum N, Quarles LD. Bone disorders in chronic kidney disease. In: Greenberg A, Cheung AK, eds. Primer on Kidney Diseases. 5th ed. Philadelphia, PA: Elsevier Saunders; 2009:487-498.
3. Moe S, Drueke T, Cunningham J, et al; Kidney Disease: Improving Global Outcomes (KDIGO). Definition, evaluation, and classification of renal osteodystrophy: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2006;69:1945-1953.
4. Gilbert S, Weiner DE. National Kidney Foundation’s Primer on Kidney Diseases. 6th ed. Philadelphia, PA: Elsevier Saunders; 2013:330.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Shelly Levinstein, MSN, CRNP, who practices at Nephrology Associates of York in Pennsylvania.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Shelly Levinstein, MSN, CRNP, who practices at Nephrology Associates of York in Pennsylvania.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Shelly Levinstein, MSN, CRNP, who practices at Nephrology Associates of York in Pennsylvania.
Q) Since bone loss is common in patients with kidney disease, how should it be treated?
In elderly CKD patients, it is recognized that osteoporosis and CKD-MBD may co-exist—which only makes the bone-loss issue more problematic. Therefore, diagnosis and management are crucial to effective treatment.
Although osteoporosis is recognized and treated in CKD stages 1 to 3a, the interpretation of BMD levels in the osteoporotic range is controversial in advanced kidney disease (GFR < 35).1 There are limited data for treatment of patients with CKD-MBD.
Studies of bisphosphonate use in postmenopausal women and in patients with glucocorticoid-induced osteoporosis have generally excluded those with renal impairment. For these patients, treatment of low BMD using standard therapies for osteoporosis is not without potential for harm, due to the possibility of worsening low bone turnover, osteomalacia, mixed uremic osteodystrophy, and exacerbated hyperparathyroidism.
The choice of pharmaceutical treatment should be based on whether you are treating CKD-MBD or osteoporosis. A large percentage of CKD patients have adynamic bone disease with low bone resorption. In patients with low bone resorption, treatment entails choosing a drug that stimulates bone formation and not those that inhibit bone resorption. The benefit of bisphosphonate therapy is seen in patients with high bone resorption.4
In CKD stages 1 to 3, one must evaluate laboratory features of low BMD, including serum levels of calcium, phosphate, parathyroid hormone, alkaline phosphatase, and vitamin D. If all are found to be normal, bisphosphonate use in CKD stages 1 to 3 is usually safe.1
A bone biopsy is recommended for patients with advanced CKD stages 4 to 5/5D, with therapy individualized per disease process.1
Shelly Levinstein, MSN, CRNP
Nephrology Associates of York
York, PA
REFERENCES
1. Toussaint N, Elder G, Kerr P. Bisphosphonates in chronic kidney disease; balancing potential benefits and adverse effects on bone and soft tissue. Clin J Am Soc Nephrol. 2009;4:221-233.
2. Tanenbaum N, Quarles LD. Bone disorders in chronic kidney disease. In: Greenberg A, Cheung AK, eds. Primer on Kidney Diseases. 5th ed. Philadelphia, PA: Elsevier Saunders; 2009:487-498.
3. Moe S, Drueke T, Cunningham J, et al; Kidney Disease: Improving Global Outcomes (KDIGO). Definition, evaluation, and classification of renal osteodystrophy: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2006;69:1945-1953.
4. Gilbert S, Weiner DE. National Kidney Foundation’s Primer on Kidney Diseases. 6th ed. Philadelphia, PA: Elsevier Saunders; 2013:330.
Q) Since bone loss is common in patients with kidney disease, how should it be treated?
In elderly CKD patients, it is recognized that osteoporosis and CKD-MBD may co-exist—which only makes the bone-loss issue more problematic. Therefore, diagnosis and management are crucial to effective treatment.
Although osteoporosis is recognized and treated in CKD stages 1 to 3a, the interpretation of BMD levels in the osteoporotic range is controversial in advanced kidney disease (GFR < 35).1 There are limited data for treatment of patients with CKD-MBD.
Studies of bisphosphonate use in postmenopausal women and in patients with glucocorticoid-induced osteoporosis have generally excluded those with renal impairment. For these patients, treatment of low BMD using standard therapies for osteoporosis is not without potential for harm, due to the possibility of worsening low bone turnover, osteomalacia, mixed uremic osteodystrophy, and exacerbated hyperparathyroidism.
The choice of pharmaceutical treatment should be based on whether you are treating CKD-MBD or osteoporosis. A large percentage of CKD patients have adynamic bone disease with low bone resorption. In patients with low bone resorption, treatment entails choosing a drug that stimulates bone formation and not those that inhibit bone resorption. The benefit of bisphosphonate therapy is seen in patients with high bone resorption.4
In CKD stages 1 to 3, one must evaluate laboratory features of low BMD, including serum levels of calcium, phosphate, parathyroid hormone, alkaline phosphatase, and vitamin D. If all are found to be normal, bisphosphonate use in CKD stages 1 to 3 is usually safe.1
A bone biopsy is recommended for patients with advanced CKD stages 4 to 5/5D, with therapy individualized per disease process.1
Shelly Levinstein, MSN, CRNP
Nephrology Associates of York
York, PA
REFERENCES
1. Toussaint N, Elder G, Kerr P. Bisphosphonates in chronic kidney disease; balancing potential benefits and adverse effects on bone and soft tissue. Clin J Am Soc Nephrol. 2009;4:221-233.
2. Tanenbaum N, Quarles LD. Bone disorders in chronic kidney disease. In: Greenberg A, Cheung AK, eds. Primer on Kidney Diseases. 5th ed. Philadelphia, PA: Elsevier Saunders; 2009:487-498.
3. Moe S, Drueke T, Cunningham J, et al; Kidney Disease: Improving Global Outcomes (KDIGO). Definition, evaluation, and classification of renal osteodystrophy: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2006;69:1945-1953.
4. Gilbert S, Weiner DE. National Kidney Foundation’s Primer on Kidney Diseases. 6th ed. Philadelphia, PA: Elsevier Saunders; 2013:330.
Hyperprolactinemia: Causes and Treatments
A 31-year-old woman is referred by her Ob-Gyn for elevated prolactin. She initially presented with a three-month history of amenorrhea, a negative home pregnancy test, and 100% compliance with condom use. She denies hirsutism and acne but admits to thin milky nipple discharge upon squeezing (but not spontaneous).
Two weeks ago, her Ob-Gyn ordered labs; results were negative for serum beta human chorionic gonadotropin and within normal ranges for thyroid-stimulating hormone (TSH), luteinizing hormone, follicle-stimulating hormone, estradiol, free and total testosterone, dehydroepiandrosterone sulfate (DHEAs), complete chemistry panel, and complete blood count. Her serum prolactin level was 110 ng/mL (normal, 3 to 27 ng/mL).
Q: How is prolactin physiologically regulated?
The primary role of prolactin, which is produced by lactotroph cells in the anterior pituitary gland, is to stimulate lactation and breast development. Prolactin is regulated by dopamine (also known as prolactin inhibitory hormone), which is secreted from the hypothalamus via an inhibitory pathway unique to the hypothalamus-pituitary hormone system. Dopamine essentially suppresses prolactin.
Other hormones can have a stimulatory effect on the anterior pituitary gland and thus increase prolactin levels. Estrogen can induce lactotroph hyperplasia and elevated prolactin; however, this is only clinically relevant in the context of estrogen surge during pregnancy. (Estrogen therapy, such as oral contraception or hormone replacement therapy, on the other hand, is targeted to “normal” estrogen levels.) Thyrotropin-releasing hormone (TRH) from the hypothalamus also stimulates the anterior pituitary gland, so patients with inadequately treated or untreated primary hypothyroidism will have mildly elevated prolactin.
Neurogenic stimuli of the chest wall, through nipple suckling or varicella zoster infection (shingles), can also increase prolactin secretion. And since prolactin is eliminated by the liver (75%) and the kidney (25%), significant liver disease and/or renal insufficiency can raise prolactin levels, due to decreased clearance.
What are the possible etiologies for elevated prolactin? See answer on the next page...
Q: What are the possible etiologies for elevated prolactin?
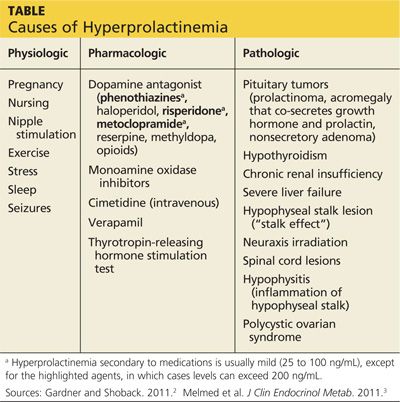
The causes of hyperprolactinemia fall into three categories: physiologic, pharmacologic, and pathologic.2 The table provides examples from each category.
A nonsecretory pituitary adenoma or any lesion in the brain that would disrupt the hypophyseal stalk may interfere with dopamine’s inhibitory control and thereby increase prolactin. This is called the stalk effect. It is important to note that not all MRI-proven pituitary adenomas are prolactin secreting, even in the presence of hyperprolactinemia. According to an autopsy series, about 12% of the general population had pituitary microadenoma.3
There is rough correlation between prolactinoma size and level of prolactin. Large nonsecretory pituitary adenomas have prolactin levels less than 150 ng/mL. Microprolactinomas (< 1 cm) are usually in the range of 100 to 250 ng/mL, while macroprolactinomas (> 1 cm) are generally
≥ 250 ng/mL. If the tumor is very large and invades the cavernous sinus, prolactin can measure in the 1,000s.3
Polycystic ovarian syndrome (PCOS) is a common disorder affecting women of reproductive age and the most common cause of underlying ovulatory problems. Patients with PCOS can have mildly elevated prolactin; the exact mechanism of hyperprolactinemia in PCOS is unknown. One theory is that constant high levels of estrogen experienced in PCOS would stimulate prolactin production. It is important to rule out other causes of hyperprolactinemia before making the diagnosis of PCOS.
What is the clinical significance of elevated prolactin? Why do we have to work up and treat it? See answer on the next page...
Q: What is the clinical significance of elevated prolactin? Why do we have to work up and treat it?
By physiologic mechanisms not completely understood, hyperprolactinemia can interrupt the gonadal axis, leading to hypogonadism. In women, it can cause irregular menstrual cycles, oligomenorrhea, amenorrhea, and infertility. In men, it can lower testosterone levels. Long-term effects include declining bone mineral density due to insufficient estrogen in women or testosterone in men.
With macroadenoma, the size of the tumor can have a mass effect such as headache and visual defect by compressing the optic chiasm (bitemporal hemianopsia), which may lead to permanent vision loss if left untreated. Referral to an ophthalmologist may be necessary for formal visual field examination.
How is hyperprolactinemia treated? See answer on the next page...
Q: How is hyperprolactinemia treated?
There are three options for treatment: medication, surgery, and radiation.
Dopamine agonists (bromocriptine, cabergoline) are effective in normalizing prolactin and reducing the size of the tumor in the majority of cases. However, some patients may require long-term treatment. Bromocriptine has been used since the late 1970s, but, due to better tolerance and less frequent dosing, cabergoline is the preferred agent.3
Transsphenoidal surgery is indicated for patients who are intolerant to medication, who have a medication-resistant tumor or significant mass effect, or who prefer definitive treatment. Women of childbearing age with a macroadenoma might consider surgery due to the risk for tumor expansion during pregnancy (estrogen effect) and risk for pituitary apoplexy (hemorrhage or infarct of the pituitary gland). Surgical risk is usually low with a neurosurgeon who has extensive experience.
Radiation can be considered for large tumors that are resistant to medication. It can be used as adjunctive therapy to surgery, since reducing the size of the tumor can make the surgical field smaller. In some medication-resistant tumors, radiation can raise sensitivity to medication.
What does follow-up entail? See next page for answer...
Q: What does follow-up entail?
Once medication is initiated or dosage is adjusted, have the patient follow up in one month and recheck the prolactin level to assess responsiveness to medication (as well as medication adherence). When a therapeutic prolactin level is achieved, recheck the prolactin and have the patient follow up at three and six months and then every six months thereafter.3
MRI of the pituitary gland should be performed at baseline, then in six months to assess tumor response to medication, and then at 12 and 24 months.3 If tumor regression has stabilized or if the tumor has shrunk to a nondetectable size, consider discontinuing the dopamine agonist. If medication is discontinued, recheck prolactin every three months for the first year; if it remains in normal reference range, simply check serum prolactin annually.3
See next page for summary.

See next page for references.
REFERENCES
1. Jameson JL. Harrison’s Endocrinology. 18th ed. China: McGraw-Hill; 2010.
2. Gardner D, Shoback D. Greenspan’s Basic & Clinical Endocrinology. 9th ed. China: McGraw-Hill; 2011.
3. Melmed S, Casanueva FF, Hoffman AR, et al. Diagnosis and treatment of hyperprolactinemia: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2011;96(2):273-288.
A 31-year-old woman is referred by her Ob-Gyn for elevated prolactin. She initially presented with a three-month history of amenorrhea, a negative home pregnancy test, and 100% compliance with condom use. She denies hirsutism and acne but admits to thin milky nipple discharge upon squeezing (but not spontaneous).
Two weeks ago, her Ob-Gyn ordered labs; results were negative for serum beta human chorionic gonadotropin and within normal ranges for thyroid-stimulating hormone (TSH), luteinizing hormone, follicle-stimulating hormone, estradiol, free and total testosterone, dehydroepiandrosterone sulfate (DHEAs), complete chemistry panel, and complete blood count. Her serum prolactin level was 110 ng/mL (normal, 3 to 27 ng/mL).
Q: How is prolactin physiologically regulated?
The primary role of prolactin, which is produced by lactotroph cells in the anterior pituitary gland, is to stimulate lactation and breast development. Prolactin is regulated by dopamine (also known as prolactin inhibitory hormone), which is secreted from the hypothalamus via an inhibitory pathway unique to the hypothalamus-pituitary hormone system. Dopamine essentially suppresses prolactin.
Other hormones can have a stimulatory effect on the anterior pituitary gland and thus increase prolactin levels. Estrogen can induce lactotroph hyperplasia and elevated prolactin; however, this is only clinically relevant in the context of estrogen surge during pregnancy. (Estrogen therapy, such as oral contraception or hormone replacement therapy, on the other hand, is targeted to “normal” estrogen levels.) Thyrotropin-releasing hormone (TRH) from the hypothalamus also stimulates the anterior pituitary gland, so patients with inadequately treated or untreated primary hypothyroidism will have mildly elevated prolactin.
Neurogenic stimuli of the chest wall, through nipple suckling or varicella zoster infection (shingles), can also increase prolactin secretion. And since prolactin is eliminated by the liver (75%) and the kidney (25%), significant liver disease and/or renal insufficiency can raise prolactin levels, due to decreased clearance.
What are the possible etiologies for elevated prolactin? See answer on the next page...
Q: What are the possible etiologies for elevated prolactin?
The causes of hyperprolactinemia fall into three categories: physiologic, pharmacologic, and pathologic.2 The table provides examples from each category.
A nonsecretory pituitary adenoma or any lesion in the brain that would disrupt the hypophyseal stalk may interfere with dopamine’s inhibitory control and thereby increase prolactin. This is called the stalk effect. It is important to note that not all MRI-proven pituitary adenomas are prolactin secreting, even in the presence of hyperprolactinemia. According to an autopsy series, about 12% of the general population had pituitary microadenoma.3
There is rough correlation between prolactinoma size and level of prolactin. Large nonsecretory pituitary adenomas have prolactin levels less than 150 ng/mL. Microprolactinomas (< 1 cm) are usually in the range of 100 to 250 ng/mL, while macroprolactinomas (> 1 cm) are generally
≥ 250 ng/mL. If the tumor is very large and invades the cavernous sinus, prolactin can measure in the 1,000s.3
Polycystic ovarian syndrome (PCOS) is a common disorder affecting women of reproductive age and the most common cause of underlying ovulatory problems. Patients with PCOS can have mildly elevated prolactin; the exact mechanism of hyperprolactinemia in PCOS is unknown. One theory is that constant high levels of estrogen experienced in PCOS would stimulate prolactin production. It is important to rule out other causes of hyperprolactinemia before making the diagnosis of PCOS.
What is the clinical significance of elevated prolactin? Why do we have to work up and treat it? See answer on the next page...
Q: What is the clinical significance of elevated prolactin? Why do we have to work up and treat it?
By physiologic mechanisms not completely understood, hyperprolactinemia can interrupt the gonadal axis, leading to hypogonadism. In women, it can cause irregular menstrual cycles, oligomenorrhea, amenorrhea, and infertility. In men, it can lower testosterone levels. Long-term effects include declining bone mineral density due to insufficient estrogen in women or testosterone in men.
With macroadenoma, the size of the tumor can have a mass effect such as headache and visual defect by compressing the optic chiasm (bitemporal hemianopsia), which may lead to permanent vision loss if left untreated. Referral to an ophthalmologist may be necessary for formal visual field examination.
How is hyperprolactinemia treated? See answer on the next page...
Q: How is hyperprolactinemia treated?
There are three options for treatment: medication, surgery, and radiation.
Dopamine agonists (bromocriptine, cabergoline) are effective in normalizing prolactin and reducing the size of the tumor in the majority of cases. However, some patients may require long-term treatment. Bromocriptine has been used since the late 1970s, but, due to better tolerance and less frequent dosing, cabergoline is the preferred agent.3
Transsphenoidal surgery is indicated for patients who are intolerant to medication, who have a medication-resistant tumor or significant mass effect, or who prefer definitive treatment. Women of childbearing age with a macroadenoma might consider surgery due to the risk for tumor expansion during pregnancy (estrogen effect) and risk for pituitary apoplexy (hemorrhage or infarct of the pituitary gland). Surgical risk is usually low with a neurosurgeon who has extensive experience.
Radiation can be considered for large tumors that are resistant to medication. It can be used as adjunctive therapy to surgery, since reducing the size of the tumor can make the surgical field smaller. In some medication-resistant tumors, radiation can raise sensitivity to medication.
What does follow-up entail? See next page for answer...
Q: What does follow-up entail?
Once medication is initiated or dosage is adjusted, have the patient follow up in one month and recheck the prolactin level to assess responsiveness to medication (as well as medication adherence). When a therapeutic prolactin level is achieved, recheck the prolactin and have the patient follow up at three and six months and then every six months thereafter.3
MRI of the pituitary gland should be performed at baseline, then in six months to assess tumor response to medication, and then at 12 and 24 months.3 If tumor regression has stabilized or if the tumor has shrunk to a nondetectable size, consider discontinuing the dopamine agonist. If medication is discontinued, recheck prolactin every three months for the first year; if it remains in normal reference range, simply check serum prolactin annually.3
See next page for summary.
See next page for references.
REFERENCES
1. Jameson JL. Harrison’s Endocrinology. 18th ed. China: McGraw-Hill; 2010.
2. Gardner D, Shoback D. Greenspan’s Basic & Clinical Endocrinology. 9th ed. China: McGraw-Hill; 2011.
3. Melmed S, Casanueva FF, Hoffman AR, et al. Diagnosis and treatment of hyperprolactinemia: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2011;96(2):273-288.
A 31-year-old woman is referred by her Ob-Gyn for elevated prolactin. She initially presented with a three-month history of amenorrhea, a negative home pregnancy test, and 100% compliance with condom use. She denies hirsutism and acne but admits to thin milky nipple discharge upon squeezing (but not spontaneous).
Two weeks ago, her Ob-Gyn ordered labs; results were negative for serum beta human chorionic gonadotropin and within normal ranges for thyroid-stimulating hormone (TSH), luteinizing hormone, follicle-stimulating hormone, estradiol, free and total testosterone, dehydroepiandrosterone sulfate (DHEAs), complete chemistry panel, and complete blood count. Her serum prolactin level was 110 ng/mL (normal, 3 to 27 ng/mL).
Q: How is prolactin physiologically regulated?
The primary role of prolactin, which is produced by lactotroph cells in the anterior pituitary gland, is to stimulate lactation and breast development. Prolactin is regulated by dopamine (also known as prolactin inhibitory hormone), which is secreted from the hypothalamus via an inhibitory pathway unique to the hypothalamus-pituitary hormone system. Dopamine essentially suppresses prolactin.
Other hormones can have a stimulatory effect on the anterior pituitary gland and thus increase prolactin levels. Estrogen can induce lactotroph hyperplasia and elevated prolactin; however, this is only clinically relevant in the context of estrogen surge during pregnancy. (Estrogen therapy, such as oral contraception or hormone replacement therapy, on the other hand, is targeted to “normal” estrogen levels.) Thyrotropin-releasing hormone (TRH) from the hypothalamus also stimulates the anterior pituitary gland, so patients with inadequately treated or untreated primary hypothyroidism will have mildly elevated prolactin.
Neurogenic stimuli of the chest wall, through nipple suckling or varicella zoster infection (shingles), can also increase prolactin secretion. And since prolactin is eliminated by the liver (75%) and the kidney (25%), significant liver disease and/or renal insufficiency can raise prolactin levels, due to decreased clearance.
What are the possible etiologies for elevated prolactin? See answer on the next page...
Q: What are the possible etiologies for elevated prolactin?
The causes of hyperprolactinemia fall into three categories: physiologic, pharmacologic, and pathologic.2 The table provides examples from each category.
A nonsecretory pituitary adenoma or any lesion in the brain that would disrupt the hypophyseal stalk may interfere with dopamine’s inhibitory control and thereby increase prolactin. This is called the stalk effect. It is important to note that not all MRI-proven pituitary adenomas are prolactin secreting, even in the presence of hyperprolactinemia. According to an autopsy series, about 12% of the general population had pituitary microadenoma.3
There is rough correlation between prolactinoma size and level of prolactin. Large nonsecretory pituitary adenomas have prolactin levels less than 150 ng/mL. Microprolactinomas (< 1 cm) are usually in the range of 100 to 250 ng/mL, while macroprolactinomas (> 1 cm) are generally
≥ 250 ng/mL. If the tumor is very large and invades the cavernous sinus, prolactin can measure in the 1,000s.3
Polycystic ovarian syndrome (PCOS) is a common disorder affecting women of reproductive age and the most common cause of underlying ovulatory problems. Patients with PCOS can have mildly elevated prolactin; the exact mechanism of hyperprolactinemia in PCOS is unknown. One theory is that constant high levels of estrogen experienced in PCOS would stimulate prolactin production. It is important to rule out other causes of hyperprolactinemia before making the diagnosis of PCOS.
What is the clinical significance of elevated prolactin? Why do we have to work up and treat it? See answer on the next page...
Q: What is the clinical significance of elevated prolactin? Why do we have to work up and treat it?
By physiologic mechanisms not completely understood, hyperprolactinemia can interrupt the gonadal axis, leading to hypogonadism. In women, it can cause irregular menstrual cycles, oligomenorrhea, amenorrhea, and infertility. In men, it can lower testosterone levels. Long-term effects include declining bone mineral density due to insufficient estrogen in women or testosterone in men.
With macroadenoma, the size of the tumor can have a mass effect such as headache and visual defect by compressing the optic chiasm (bitemporal hemianopsia), which may lead to permanent vision loss if left untreated. Referral to an ophthalmologist may be necessary for formal visual field examination.
How is hyperprolactinemia treated? See answer on the next page...
Q: How is hyperprolactinemia treated?
There are three options for treatment: medication, surgery, and radiation.
Dopamine agonists (bromocriptine, cabergoline) are effective in normalizing prolactin and reducing the size of the tumor in the majority of cases. However, some patients may require long-term treatment. Bromocriptine has been used since the late 1970s, but, due to better tolerance and less frequent dosing, cabergoline is the preferred agent.3
Transsphenoidal surgery is indicated for patients who are intolerant to medication, who have a medication-resistant tumor or significant mass effect, or who prefer definitive treatment. Women of childbearing age with a macroadenoma might consider surgery due to the risk for tumor expansion during pregnancy (estrogen effect) and risk for pituitary apoplexy (hemorrhage or infarct of the pituitary gland). Surgical risk is usually low with a neurosurgeon who has extensive experience.
Radiation can be considered for large tumors that are resistant to medication. It can be used as adjunctive therapy to surgery, since reducing the size of the tumor can make the surgical field smaller. In some medication-resistant tumors, radiation can raise sensitivity to medication.
What does follow-up entail? See next page for answer...
Q: What does follow-up entail?
Once medication is initiated or dosage is adjusted, have the patient follow up in one month and recheck the prolactin level to assess responsiveness to medication (as well as medication adherence). When a therapeutic prolactin level is achieved, recheck the prolactin and have the patient follow up at three and six months and then every six months thereafter.3
MRI of the pituitary gland should be performed at baseline, then in six months to assess tumor response to medication, and then at 12 and 24 months.3 If tumor regression has stabilized or if the tumor has shrunk to a nondetectable size, consider discontinuing the dopamine agonist. If medication is discontinued, recheck prolactin every three months for the first year; if it remains in normal reference range, simply check serum prolactin annually.3
See next page for summary.
See next page for references.
REFERENCES
1. Jameson JL. Harrison’s Endocrinology. 18th ed. China: McGraw-Hill; 2010.
2. Gardner D, Shoback D. Greenspan’s Basic & Clinical Endocrinology. 9th ed. China: McGraw-Hill; 2011.
3. Melmed S, Casanueva FF, Hoffman AR, et al. Diagnosis and treatment of hyperprolactinemia: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2011;96(2):273-288.
The Modern Realities of Kidney Stones: Diagnosis
Q) I am new to practice and working in urgent care. I was discussing the diagnosis of kidney stones with my supervising physician. He said he does an intravenous pyelogram (IVP) to diagnose stones. He is a little “old school,” and I’m not sure he is right. What is “state of the art” in the work-up and acute treatment for kidney stones?
An IVP involves taking a series of x-rays following the injection of dye into a patient’s vein. As the dye moves through the bloodstream, the anatomy of the urinary system can be better visualized and the stone location identified, as the dye tends to accumulate at areas of obstruction. The downside to this test is that contrast can cause allergic reactions in some patients and can only be used in those with normal renal function. Also, a radiologist is required to be present during the procedure, and the test can take a long time to complete if a severe blockage is present.4
Today, noncontrast CT is considered the gold standard for imaging renal calculi because it is fast, safe (no worries for those with contrast allergy or renal impairment), and nearly 100% accurate.5
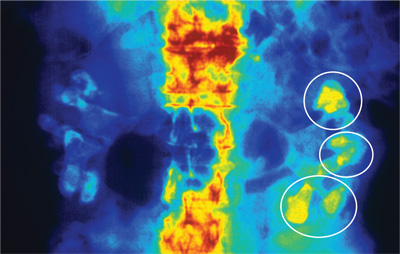
There are multiple options for treating kidney stones, though some are more invasive than others. For stones 2 cm or less identified in the upper or middle calyx and renal pelvis, extracorporeal shock wave lithotripsy (ESWL) is the treatment of choice.5,6 The goal of ESWL is to break the stone into small particles that can then be expelled through the urinary system. Adjunct measures, including mechanical percussion, diuresis, and inversion therapy, are often used following lithotripsy to facilitate passage of stone fragments. Medications such as calcium blockers and α-receptor blockers are also used to improve outcomes after lithotripsy. In the past, obese patients had less success with lithotripsy; however, technological advances have improved outcomes in depths up to 17 cm from skin to stone.6
For stones in the lower pole of the kidney (and depending on the size of the stone), ESWL, percutaneous nephrolitholapaxy, retrograde flexible ureteronephroscopy, and partial nephrectomy are options for treatment.3,7 Using an intravenous urogram, measurements and angles are calculated to help determine which procedure would be best for a particular patient.7 In simple terms, narrow angles and longer tube distances make it more difficult for stone particles to exit the urinary system. Therefore, if these problems are identified, an invasive approach may be needed to remove a stone. Other important considerations include stone size, patient symptoms, evidence of infection, or obstruction.7
ESWL is an attractive option for stone removal because it is noninvasive, has a reasonable safety profile, and is less costly than other, more invasive measures. However, advances in endoscopic instrument design are reducing complications previously associated with more invasive approaches, while improving long-term stone-free outcome rates. There may be increased utilization of procedures such as percutaneous nephrolitholapaxy and retrograde flexible ureteronephroscopy in the future.
Kristina Unterseher, CNN-NP
PeaceHealth
St. John Medical Group
Longview, WA
REFERENCES
1. Fink HA, Wilt TJ, Eidman KE, et al. Medical management to prevent recurrent nephrolithiasis in adults: a systematic review for an American College of Physicians Clinical Guideline. Ann Intern Med. 2013;158(7):535-543.
2. Hiatt RA, Ettinger B, Caan B, et al. Randomized controlled trial of a low animal protein, high fiber diet in the prevention of recurrent calcium oxalate kidney stones. Am J Epidemiol. 1996;144(1):25-33.
3. Moe OW. Kidney stones: pathophysiology and medical management. Lancet. 2006; 367(9507):333-344.
4. American College of Radiology and Radiological Society of North America. Intravenous pyelogram (2013). www.radiologyinfo.org/en/info.cfm?pg=ivp. Accessed December 16, 2013.
5. Boyce CJ, Pickhardt PJ, Lawrence EM, et al. Prevalence of urolithiasis in asymptomatic adults: objective determination using low dose noncontrast computerized tomography. J Urol. 2010;183(3):1017-1021.
6. Christian C, Thorsten B. The preferred treatment for upper tract stones is extracorporeal shock wave lithotripsy (ESWL) or ureteroscopic: pro ESWL. Urology. 2009;74(2):259-262.
7. Bourdoumis A, Papatsoris AG, Chrisofos M, Deliveliotis C. Lower pole stone management. Med Surg Urol. 2012. www.omicsonline.org/lower-pole-stone-management%20-2168-9857.S4-004.php?aid=7058?abstract _id=7058. Accessed December 16, 2013.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Kristina Unterseher, CNN-NP, who practices at PeaceHealth St. John Medical Group in Longview, Washington.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Kristina Unterseher, CNN-NP, who practices at PeaceHealth St. John Medical Group in Longview, Washington.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Kristina Unterseher, CNN-NP, who practices at PeaceHealth St. John Medical Group in Longview, Washington.
Q) I am new to practice and working in urgent care. I was discussing the diagnosis of kidney stones with my supervising physician. He said he does an intravenous pyelogram (IVP) to diagnose stones. He is a little “old school,” and I’m not sure he is right. What is “state of the art” in the work-up and acute treatment for kidney stones?
An IVP involves taking a series of x-rays following the injection of dye into a patient’s vein. As the dye moves through the bloodstream, the anatomy of the urinary system can be better visualized and the stone location identified, as the dye tends to accumulate at areas of obstruction. The downside to this test is that contrast can cause allergic reactions in some patients and can only be used in those with normal renal function. Also, a radiologist is required to be present during the procedure, and the test can take a long time to complete if a severe blockage is present.4
Today, noncontrast CT is considered the gold standard for imaging renal calculi because it is fast, safe (no worries for those with contrast allergy or renal impairment), and nearly 100% accurate.5
There are multiple options for treating kidney stones, though some are more invasive than others. For stones 2 cm or less identified in the upper or middle calyx and renal pelvis, extracorporeal shock wave lithotripsy (ESWL) is the treatment of choice.5,6 The goal of ESWL is to break the stone into small particles that can then be expelled through the urinary system. Adjunct measures, including mechanical percussion, diuresis, and inversion therapy, are often used following lithotripsy to facilitate passage of stone fragments. Medications such as calcium blockers and α-receptor blockers are also used to improve outcomes after lithotripsy. In the past, obese patients had less success with lithotripsy; however, technological advances have improved outcomes in depths up to 17 cm from skin to stone.6
For stones in the lower pole of the kidney (and depending on the size of the stone), ESWL, percutaneous nephrolitholapaxy, retrograde flexible ureteronephroscopy, and partial nephrectomy are options for treatment.3,7 Using an intravenous urogram, measurements and angles are calculated to help determine which procedure would be best for a particular patient.7 In simple terms, narrow angles and longer tube distances make it more difficult for stone particles to exit the urinary system. Therefore, if these problems are identified, an invasive approach may be needed to remove a stone. Other important considerations include stone size, patient symptoms, evidence of infection, or obstruction.7
ESWL is an attractive option for stone removal because it is noninvasive, has a reasonable safety profile, and is less costly than other, more invasive measures. However, advances in endoscopic instrument design are reducing complications previously associated with more invasive approaches, while improving long-term stone-free outcome rates. There may be increased utilization of procedures such as percutaneous nephrolitholapaxy and retrograde flexible ureteronephroscopy in the future.
Kristina Unterseher, CNN-NP
PeaceHealth
St. John Medical Group
Longview, WA
REFERENCES
1. Fink HA, Wilt TJ, Eidman KE, et al. Medical management to prevent recurrent nephrolithiasis in adults: a systematic review for an American College of Physicians Clinical Guideline. Ann Intern Med. 2013;158(7):535-543.
2. Hiatt RA, Ettinger B, Caan B, et al. Randomized controlled trial of a low animal protein, high fiber diet in the prevention of recurrent calcium oxalate kidney stones. Am J Epidemiol. 1996;144(1):25-33.
3. Moe OW. Kidney stones: pathophysiology and medical management. Lancet. 2006; 367(9507):333-344.
4. American College of Radiology and Radiological Society of North America. Intravenous pyelogram (2013). www.radiologyinfo.org/en/info.cfm?pg=ivp. Accessed December 16, 2013.
5. Boyce CJ, Pickhardt PJ, Lawrence EM, et al. Prevalence of urolithiasis in asymptomatic adults: objective determination using low dose noncontrast computerized tomography. J Urol. 2010;183(3):1017-1021.
6. Christian C, Thorsten B. The preferred treatment for upper tract stones is extracorporeal shock wave lithotripsy (ESWL) or ureteroscopic: pro ESWL. Urology. 2009;74(2):259-262.
7. Bourdoumis A, Papatsoris AG, Chrisofos M, Deliveliotis C. Lower pole stone management. Med Surg Urol. 2012. www.omicsonline.org/lower-pole-stone-management%20-2168-9857.S4-004.php?aid=7058?abstract _id=7058. Accessed December 16, 2013.
Q) I am new to practice and working in urgent care. I was discussing the diagnosis of kidney stones with my supervising physician. He said he does an intravenous pyelogram (IVP) to diagnose stones. He is a little “old school,” and I’m not sure he is right. What is “state of the art” in the work-up and acute treatment for kidney stones?
An IVP involves taking a series of x-rays following the injection of dye into a patient’s vein. As the dye moves through the bloodstream, the anatomy of the urinary system can be better visualized and the stone location identified, as the dye tends to accumulate at areas of obstruction. The downside to this test is that contrast can cause allergic reactions in some patients and can only be used in those with normal renal function. Also, a radiologist is required to be present during the procedure, and the test can take a long time to complete if a severe blockage is present.4
Today, noncontrast CT is considered the gold standard for imaging renal calculi because it is fast, safe (no worries for those with contrast allergy or renal impairment), and nearly 100% accurate.5
There are multiple options for treating kidney stones, though some are more invasive than others. For stones 2 cm or less identified in the upper or middle calyx and renal pelvis, extracorporeal shock wave lithotripsy (ESWL) is the treatment of choice.5,6 The goal of ESWL is to break the stone into small particles that can then be expelled through the urinary system. Adjunct measures, including mechanical percussion, diuresis, and inversion therapy, are often used following lithotripsy to facilitate passage of stone fragments. Medications such as calcium blockers and α-receptor blockers are also used to improve outcomes after lithotripsy. In the past, obese patients had less success with lithotripsy; however, technological advances have improved outcomes in depths up to 17 cm from skin to stone.6
For stones in the lower pole of the kidney (and depending on the size of the stone), ESWL, percutaneous nephrolitholapaxy, retrograde flexible ureteronephroscopy, and partial nephrectomy are options for treatment.3,7 Using an intravenous urogram, measurements and angles are calculated to help determine which procedure would be best for a particular patient.7 In simple terms, narrow angles and longer tube distances make it more difficult for stone particles to exit the urinary system. Therefore, if these problems are identified, an invasive approach may be needed to remove a stone. Other important considerations include stone size, patient symptoms, evidence of infection, or obstruction.7
ESWL is an attractive option for stone removal because it is noninvasive, has a reasonable safety profile, and is less costly than other, more invasive measures. However, advances in endoscopic instrument design are reducing complications previously associated with more invasive approaches, while improving long-term stone-free outcome rates. There may be increased utilization of procedures such as percutaneous nephrolitholapaxy and retrograde flexible ureteronephroscopy in the future.
Kristina Unterseher, CNN-NP
PeaceHealth
St. John Medical Group
Longview, WA
REFERENCES
1. Fink HA, Wilt TJ, Eidman KE, et al. Medical management to prevent recurrent nephrolithiasis in adults: a systematic review for an American College of Physicians Clinical Guideline. Ann Intern Med. 2013;158(7):535-543.
2. Hiatt RA, Ettinger B, Caan B, et al. Randomized controlled trial of a low animal protein, high fiber diet in the prevention of recurrent calcium oxalate kidney stones. Am J Epidemiol. 1996;144(1):25-33.
3. Moe OW. Kidney stones: pathophysiology and medical management. Lancet. 2006; 367(9507):333-344.
4. American College of Radiology and Radiological Society of North America. Intravenous pyelogram (2013). www.radiologyinfo.org/en/info.cfm?pg=ivp. Accessed December 16, 2013.
5. Boyce CJ, Pickhardt PJ, Lawrence EM, et al. Prevalence of urolithiasis in asymptomatic adults: objective determination using low dose noncontrast computerized tomography. J Urol. 2010;183(3):1017-1021.
6. Christian C, Thorsten B. The preferred treatment for upper tract stones is extracorporeal shock wave lithotripsy (ESWL) or ureteroscopic: pro ESWL. Urology. 2009;74(2):259-262.
7. Bourdoumis A, Papatsoris AG, Chrisofos M, Deliveliotis C. Lower pole stone management. Med Surg Urol. 2012. www.omicsonline.org/lower-pole-stone-management%20-2168-9857.S4-004.php?aid=7058?abstract _id=7058. Accessed December 16, 2013.
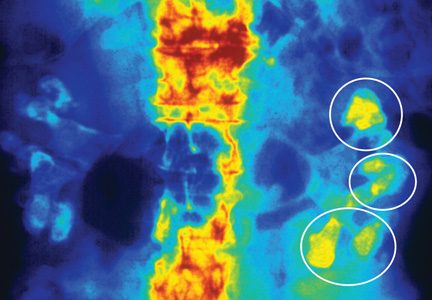
The Modern Realities of Kidney Stones: Preventing Reoccurrence
Q) A patient recently came in after an episode of kidney stones. He said he had never experienced such pain before (and this is a former Army Ranger!) and asked if there was anything he could do to keep it from happening again. I told him what I had learned in school (lots of fluids, no organ meats), but is there anything new?
Your patient has some reason for concern. For people who have had a symptomatic kidney stone, the likelihood of developing another within five years is 35% to 50% if no preventive action is taken.1 Certain factors—including family history, younger age at onset, and predisposing medical conditions (eg, hyperparathyroidism, diabetes, obesity, gout)—increase risk for recurrence.2,3
In the past, patients were often advised to restrict their dietary calcium intake to prevent calcium oxalate and/or calcium phosphate stones. However, more recent research has proven the opposite to be true: People with lower dietary calcium intake can be at greater risk for kidney stones.1-3 Therefore, encourage patients to consume about 800 to 1,200 mg/d of dietary calcium. Oral supplementation does not seem to yield the same protective benefits as dietary calcium. This may be related to absorption.1
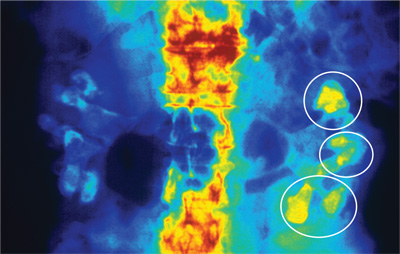
Diets high in oxalates (eg, chocolate, nuts, spinach) can increase risk for stone formation, particularly in patients who have bowel diseases that cause inflammation or a history of a bowel resection.2 Animal protein in the diet can cause hypercalcinuria and increased uric acid levels, which is particularly problematic for individuals with gout or inflammatory arthritis. High-sodium diets can cause higher urinary calcium oxalate levels, while diets high in phosphorus (particularly dark cola soft drinks) can increase risk for stone formation. Advise your patient to avoid foods high in oxalates, animal proteins, sodium, and phosphorus.2
Dehydration, either due to exercise or poor fluid intake, can result in concentrated urine, which facilitates stone formation. While opinions differ on the benefits of certain dietary restrictions, most research supports the idea that generous fluid intake is the most successful intervention in preventing recurrence of stone formation (regardless of underlying cause). Diluting the urine decreases the concentration of solutes responsible for stone formation.1-3
If the conservative measures of dietary restriction and adequate hydration fail, medications may be beneficial, depending on stone type or underlying metabolic condition. Thiazide diuretics can help lower urinary calcium by enhancing reabsorption of calcium from the distal convoluted tubule and sodium excretion; however, they should be used cautiously due to the risk for adverse effects such as dizziness and lightheadedness.1,3 Allopurinol can lower uric acid levels, decreasing recurrence of both uric acid and calcium oxalate stones. Hypocitraturia is prevalent in 20% to 60% of persons with stones; prescribing potassium citrate can inhibit crystal growth of calcium phosphate and calcium oxalate in urine.2
To help patients prevent stone recurrence, perform a comprehensive assessment of their dietary and lifestyle habits and medical history to identify possible contributing factors. Educate patients on adequate dietary calcium intake, generous water intake to keep urine dilute, and avoidance of dietary triggers.
Nephrolithiasis should be considered a manifestation of another underlying problem. If a patient presents with a kidney stone, attempt to identify the cause—not only to try to prevent recurrence, but also to identify a previously unrecognized disease process.
Kristina Unterseher, CNN-NP
PeaceHealth
St. John Medical Group
Longview, WA
REFERENCES
1. Fink HA, Wilt TJ, Eidman KE, et al. Medical management to prevent recurrent nephrolithiasis in adults: a systematic review for an American College of Physicians Clinical Guideline. Ann Intern Med. 2013;158(7):535-543.
2. Hiatt RA, Ettinger B, Caan B, et al. Randomized controlled trial of a low animal protein, high fiber diet in the prevention of recurrent calcium oxalate kidney stones. Am J Epidemiol. 1996;144(1):25-33.
3. Moe OW. Kidney stones: pathophysiology and medical management. Lancet. 2006; 367(9507):333-344.
4. American College of Radiology and Radiological Society of North America. Intravenous pyelogram (2013). www.radiologyinfo.org/en/info.cfm?pg=ivp. Accessed December 16, 2013.
5. Boyce CJ, Pickhardt PJ, Lawrence EM, et al. Prevalence of urolithiasis in asymptomatic adults: objective determination using low dose noncontrast computerized tomography. J Urol. 2010;183(3):1017-1021.
6. Christian C, Thorsten B. The preferred treatment for upper tract stones is extracorporeal shock wave lithotripsy (ESWL) or ureteroscopic: pro ESWL. Urology. 2009;74(2):259-262.
7. Bourdoumis A, Papatsoris AG, Chrisofos M, Deliveliotis C. Lower pole stone management. Med Surg Urol. 2012. www.omicsonline.org/lower-pole-stone-management%20-2168-9857.S4-004.php?aid=7058?abstract _id=7058. Accessed December 16, 2013.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Kristina Unterseher, CNN-NP, who practices at PeaceHealth St. John Medical Group in Longview, Washington.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Kristina Unterseher, CNN-NP, who practices at PeaceHealth St. John Medical Group in Longview, Washington.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Kristina Unterseher, CNN-NP, who practices at PeaceHealth St. John Medical Group in Longview, Washington.
Q) A patient recently came in after an episode of kidney stones. He said he had never experienced such pain before (and this is a former Army Ranger!) and asked if there was anything he could do to keep it from happening again. I told him what I had learned in school (lots of fluids, no organ meats), but is there anything new?
Your patient has some reason for concern. For people who have had a symptomatic kidney stone, the likelihood of developing another within five years is 35% to 50% if no preventive action is taken.1 Certain factors—including family history, younger age at onset, and predisposing medical conditions (eg, hyperparathyroidism, diabetes, obesity, gout)—increase risk for recurrence.2,3
In the past, patients were often advised to restrict their dietary calcium intake to prevent calcium oxalate and/or calcium phosphate stones. However, more recent research has proven the opposite to be true: People with lower dietary calcium intake can be at greater risk for kidney stones.1-3 Therefore, encourage patients to consume about 800 to 1,200 mg/d of dietary calcium. Oral supplementation does not seem to yield the same protective benefits as dietary calcium. This may be related to absorption.1
Diets high in oxalates (eg, chocolate, nuts, spinach) can increase risk for stone formation, particularly in patients who have bowel diseases that cause inflammation or a history of a bowel resection.2 Animal protein in the diet can cause hypercalcinuria and increased uric acid levels, which is particularly problematic for individuals with gout or inflammatory arthritis. High-sodium diets can cause higher urinary calcium oxalate levels, while diets high in phosphorus (particularly dark cola soft drinks) can increase risk for stone formation. Advise your patient to avoid foods high in oxalates, animal proteins, sodium, and phosphorus.2
Dehydration, either due to exercise or poor fluid intake, can result in concentrated urine, which facilitates stone formation. While opinions differ on the benefits of certain dietary restrictions, most research supports the idea that generous fluid intake is the most successful intervention in preventing recurrence of stone formation (regardless of underlying cause). Diluting the urine decreases the concentration of solutes responsible for stone formation.1-3
If the conservative measures of dietary restriction and adequate hydration fail, medications may be beneficial, depending on stone type or underlying metabolic condition. Thiazide diuretics can help lower urinary calcium by enhancing reabsorption of calcium from the distal convoluted tubule and sodium excretion; however, they should be used cautiously due to the risk for adverse effects such as dizziness and lightheadedness.1,3 Allopurinol can lower uric acid levels, decreasing recurrence of both uric acid and calcium oxalate stones. Hypocitraturia is prevalent in 20% to 60% of persons with stones; prescribing potassium citrate can inhibit crystal growth of calcium phosphate and calcium oxalate in urine.2
To help patients prevent stone recurrence, perform a comprehensive assessment of their dietary and lifestyle habits and medical history to identify possible contributing factors. Educate patients on adequate dietary calcium intake, generous water intake to keep urine dilute, and avoidance of dietary triggers.
Nephrolithiasis should be considered a manifestation of another underlying problem. If a patient presents with a kidney stone, attempt to identify the cause—not only to try to prevent recurrence, but also to identify a previously unrecognized disease process.
Kristina Unterseher, CNN-NP
PeaceHealth
St. John Medical Group
Longview, WA
REFERENCES
1. Fink HA, Wilt TJ, Eidman KE, et al. Medical management to prevent recurrent nephrolithiasis in adults: a systematic review for an American College of Physicians Clinical Guideline. Ann Intern Med. 2013;158(7):535-543.
2. Hiatt RA, Ettinger B, Caan B, et al. Randomized controlled trial of a low animal protein, high fiber diet in the prevention of recurrent calcium oxalate kidney stones. Am J Epidemiol. 1996;144(1):25-33.
3. Moe OW. Kidney stones: pathophysiology and medical management. Lancet. 2006; 367(9507):333-344.
4. American College of Radiology and Radiological Society of North America. Intravenous pyelogram (2013). www.radiologyinfo.org/en/info.cfm?pg=ivp. Accessed December 16, 2013.
5. Boyce CJ, Pickhardt PJ, Lawrence EM, et al. Prevalence of urolithiasis in asymptomatic adults: objective determination using low dose noncontrast computerized tomography. J Urol. 2010;183(3):1017-1021.
6. Christian C, Thorsten B. The preferred treatment for upper tract stones is extracorporeal shock wave lithotripsy (ESWL) or ureteroscopic: pro ESWL. Urology. 2009;74(2):259-262.
7. Bourdoumis A, Papatsoris AG, Chrisofos M, Deliveliotis C. Lower pole stone management. Med Surg Urol. 2012. www.omicsonline.org/lower-pole-stone-management%20-2168-9857.S4-004.php?aid=7058?abstract _id=7058. Accessed December 16, 2013.
Q) A patient recently came in after an episode of kidney stones. He said he had never experienced such pain before (and this is a former Army Ranger!) and asked if there was anything he could do to keep it from happening again. I told him what I had learned in school (lots of fluids, no organ meats), but is there anything new?
Your patient has some reason for concern. For people who have had a symptomatic kidney stone, the likelihood of developing another within five years is 35% to 50% if no preventive action is taken.1 Certain factors—including family history, younger age at onset, and predisposing medical conditions (eg, hyperparathyroidism, diabetes, obesity, gout)—increase risk for recurrence.2,3
In the past, patients were often advised to restrict their dietary calcium intake to prevent calcium oxalate and/or calcium phosphate stones. However, more recent research has proven the opposite to be true: People with lower dietary calcium intake can be at greater risk for kidney stones.1-3 Therefore, encourage patients to consume about 800 to 1,200 mg/d of dietary calcium. Oral supplementation does not seem to yield the same protective benefits as dietary calcium. This may be related to absorption.1
Diets high in oxalates (eg, chocolate, nuts, spinach) can increase risk for stone formation, particularly in patients who have bowel diseases that cause inflammation or a history of a bowel resection.2 Animal protein in the diet can cause hypercalcinuria and increased uric acid levels, which is particularly problematic for individuals with gout or inflammatory arthritis. High-sodium diets can cause higher urinary calcium oxalate levels, while diets high in phosphorus (particularly dark cola soft drinks) can increase risk for stone formation. Advise your patient to avoid foods high in oxalates, animal proteins, sodium, and phosphorus.2
Dehydration, either due to exercise or poor fluid intake, can result in concentrated urine, which facilitates stone formation. While opinions differ on the benefits of certain dietary restrictions, most research supports the idea that generous fluid intake is the most successful intervention in preventing recurrence of stone formation (regardless of underlying cause). Diluting the urine decreases the concentration of solutes responsible for stone formation.1-3
If the conservative measures of dietary restriction and adequate hydration fail, medications may be beneficial, depending on stone type or underlying metabolic condition. Thiazide diuretics can help lower urinary calcium by enhancing reabsorption of calcium from the distal convoluted tubule and sodium excretion; however, they should be used cautiously due to the risk for adverse effects such as dizziness and lightheadedness.1,3 Allopurinol can lower uric acid levels, decreasing recurrence of both uric acid and calcium oxalate stones. Hypocitraturia is prevalent in 20% to 60% of persons with stones; prescribing potassium citrate can inhibit crystal growth of calcium phosphate and calcium oxalate in urine.2
To help patients prevent stone recurrence, perform a comprehensive assessment of their dietary and lifestyle habits and medical history to identify possible contributing factors. Educate patients on adequate dietary calcium intake, generous water intake to keep urine dilute, and avoidance of dietary triggers.
Nephrolithiasis should be considered a manifestation of another underlying problem. If a patient presents with a kidney stone, attempt to identify the cause—not only to try to prevent recurrence, but also to identify a previously unrecognized disease process.
Kristina Unterseher, CNN-NP
PeaceHealth
St. John Medical Group
Longview, WA
REFERENCES
1. Fink HA, Wilt TJ, Eidman KE, et al. Medical management to prevent recurrent nephrolithiasis in adults: a systematic review for an American College of Physicians Clinical Guideline. Ann Intern Med. 2013;158(7):535-543.
2. Hiatt RA, Ettinger B, Caan B, et al. Randomized controlled trial of a low animal protein, high fiber diet in the prevention of recurrent calcium oxalate kidney stones. Am J Epidemiol. 1996;144(1):25-33.
3. Moe OW. Kidney stones: pathophysiology and medical management. Lancet. 2006; 367(9507):333-344.
4. American College of Radiology and Radiological Society of North America. Intravenous pyelogram (2013). www.radiologyinfo.org/en/info.cfm?pg=ivp. Accessed December 16, 2013.
5. Boyce CJ, Pickhardt PJ, Lawrence EM, et al. Prevalence of urolithiasis in asymptomatic adults: objective determination using low dose noncontrast computerized tomography. J Urol. 2010;183(3):1017-1021.
6. Christian C, Thorsten B. The preferred treatment for upper tract stones is extracorporeal shock wave lithotripsy (ESWL) or ureteroscopic: pro ESWL. Urology. 2009;74(2):259-262.
7. Bourdoumis A, Papatsoris AG, Chrisofos M, Deliveliotis C. Lower pole stone management. Med Surg Urol. 2012. www.omicsonline.org/lower-pole-stone-management%20-2168-9857.S4-004.php?aid=7058?abstract _id=7058. Accessed December 16, 2013.
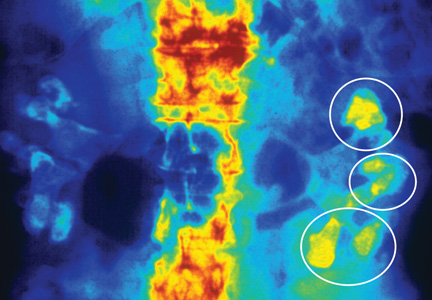
Hereditary Hemochromatosis as a Cause of Hypogonadism
JR, a 34-year-old Caucasian man, was in his normal state of good health until several months ago, when he developed fatigue, low libido, and insomnia. He reports normal erectile function, adding that he fathered a child at age 24. His medical history and remaining review of systems are negative. Physical exam is unremarkable. His BMI is 23.
Labwork reveals low free and total testosterone levels with low FSH and LH levels. Thyroid-stimulating hormone, free T4, and prolactin levels are within normal range, comprehensive metabolic panel is unremarkable, and pituitary MRI is negative. The complete blood count reveals slightly elevated hemoglobin and hematocrit, prompting ordering of iron studies that reveal elevated ferritin and serum iron levels and elevated percent transferrin saturation. Lab values are shown in the table.
Based on his elevated ferritin and transferrin saturation levels, JR undergoes genetic testing for hereditary hemochromatosis (HH) with C282Y and H63D mutation analysis. He is found to have the homozygous C282Y genotype (C282Y/C282Y) for HH.
JR establishes care with a hematologist and is advised to receive therapeutic phlebotomy until his ferritin level is between 10 and 50 ng/mL. An abdominal ultrasound, ordered to screen for hepatomegaly, yields normal results. JR elects not to receive testosterone replacement therapy.
Three months later, labwork reveals a free testosterone level of 120 pg/mL with normal hemoglobin and hematocrit levels, normal transaminases, a ferritin level of 28 ng/mL, and a percent transferrin saturation of 31%. Additional values are shown in the table.
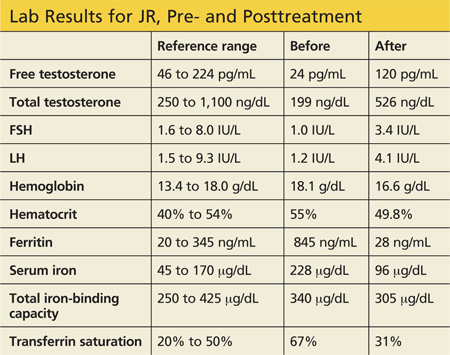
BACKGROUND AND GENETICS
Hereditary hemochromatosis is an autosomal recessive iron storage disorder in which intestinal iron absorption is markedly increased. This results in iron overload and excessive iron deposition in numerous tissues, glands, and organs.1
In patients with HH, a genetic defect causes abnormal expression of the HFE protein that regulates hepcidin production. Hepcidin is an iron regulatory hormone, secreted by hepatocytes, that decreases intestinal iron absorption in response to excess iron.2 Ninety percent of individuals affected by HH are homozygous for mutation at amino acid position 282 on the HFE gene, which causes an inappropriate decrease in hepcidin expression in response to elevated iron levels.2,3 Only 10% of individuals homozygous for the C282Y mutation actually develop clinically apparent end-organ damage.2
Being a carrier (heterozygous) for the C282Y mutation confers significantly lower risk for iron overload. The second most common mutation in the HFE gene, H63D, is associated with a milder phenotype. Those with compound heterozygosity for C282Y/H63D or homozygosity for H63D typically experience either mild or no detectable symptoms.1,3
There can be mutations in other genes involved in iron metabolism, but these represent more rare forms of hemochromatosis. Conditions such as thalassemia, sideroblastic anemia, porphyria cutanea tarda, and chronic liver disease may also be associated with iron overload.1,3
HH is most common in white populations of northern European descent. Multiple factors—including dietary iron intake, alcohol consumption, blood donation, blood loss associated with menstruation, and pregnancy—affect the expression of clinical features of hemochromatosis. Men are 24 times more likely than women to express clinical features of hemochromatosis.2 Approximately 70% of affected patients develop symptoms between ages 40 and 60.1
CLINICAL MANIFESTATIONS
The liver is typically the first organ affected, and hepatomegaly is present in 95% of symptomatic patients, even in the presence of normal transaminase levels.1 A bronzed, metallic, or slate gray skin coloration can occur due to increased iron deposition in the dermis. Arthralgias in the hands, wrists, hips, knees, and ankles are present in up to 50% of patients with hemochromatosis. Cardiac manifestations include restrictive cardiomyopathy, congestive heart failure, and arrhythmias.1
Iron deposition in the beta cells of the pancreas causes diabetes1 and in the pituitary causes hypogonadotropic hypogonadism in both men and women, resulting in decreased libido, amenorrhea, testicular atrophy, gynecomastia, and reduced body hair. Primary testicular dysfunction may occur due to iron deposition in the testicles.1,4 In the thyroid gland, iron deposition can lead to abnormal function. Secondary hypothyroidism is rare in the setting of iron overload, although iron deposition occasionally occurs in pituitary thyrotrophs (usually only to a mild degree). Adrenal insufficiency and hypoparathyroidism may also result from iron overload.5
CLINICAL STUDIES TO ASSESS IRON STORES
When assessing tissue iron stores, it is important to measure the serum iron level, total iron-binding capacity, and ferritin in the fasting state.2 This information can be used to calculate the percent transferrin saturation. If the serum ferritin is elevated (> 300 ng/mL in men and > 200 ng/mL in women) and/or the transferrin saturation is greater than 45%, referral to hematology or hepatology is recommended, along with genetic testing for hemochromatosis.1,2
Once the diagnosis of hemochromatosis has been confirmed, CT or MRI can be used to assess for increased density of the liver.1 Liver biopsy can determine the degree of fibrosis and is often considered in patients with more extreme elevations of serum ferritin levels and/or hepatomegaly. Liver biopsy is the only reliable method for determining whether hepatic cirrhosis, which increases risk for hepatocellular carcinoma, is present.1
TREATMENT
All patients with homozygous HH and evidence of iron overload require treatment, regardless of symptoms. Phlebotomy is the standard of care, due to its low cost and relative safety. Chelating agents are a second-line option when contraindications to phlebotomy (eg, anemia) exist.1,2
Alcohol consumption, especially in the presence of iron overload or liver disease, should be avoided, as it can increase risk for cirrhosis by nearly tenfold.1 Dietary modification is typically unnecessary, aside from the avoidance of iron and vitamin C supplementation.2 Patients should also eliminate raw shellfish from their diet, as they may carry bacteria that can cause potentially fatal infection (since high iron levels impair hepcidin bactericidal activity).2
The management of hepatic failure, cardiac failure, and diabetes in patients with HH is similar to conventional management of these conditions.1 With phlebotomy, the liver and spleen often decrease in size, liver function improves, skin pigmentation lightens, cardiac failure may be reversed, and diabetes control often improves.1,2 Testosterone levels may normalize after phlebotomy, especially if HH is diagnosed in the early stages. In more advanced cases, testosterone replacement therapy in combination with aggressive phlebotomy may be necessary.4
CONCLUSION
A high index of suspicion is required to diagnose hemochromatosis early. HH should be considered in the differential diagnosis for patients with hypogonadotropic hypogonadism, abnormal iron studies, elevated transaminase levels, and a family history of hemochromatosis.
Once the diagnosis is established, all first-degree relatives should be screened.1 Early therapy is crucial to prevent complications from iron overload.
REFERENCES
1. Powell LW. Hemochromatosis. In: Fauci AS, Braunwald E, Kasper DL, et al (eds). Harrison’s Principles of Internal Medicine. 17th ed. New York, NY: McGraw-Hill; 2008:2429-2433.
2. Crownover BK, Carlton JC. Hereditary hemochromatosis. Am Fam Phys. 2013;87(3):183-190.
3. Camaschella C. Understanding iron homeostasis through genetic analysis of hemochromatosis and related disorders. Blood. 2005;106(12):3710-3717.
4. McDermott JH, Walsh CH. Hypogonadism in hereditary hemochromatosis. J Clin Endocrinol Metab. 2005;90(4):2451-2455.
5. Hudec M, Grigerova M, Walsh CH. Secondary hypothyroidism in hereditary hemochromatosis: recovery after iron depletion. Thyroid. 2008;18(2):255-257.
JR, a 34-year-old Caucasian man, was in his normal state of good health until several months ago, when he developed fatigue, low libido, and insomnia. He reports normal erectile function, adding that he fathered a child at age 24. His medical history and remaining review of systems are negative. Physical exam is unremarkable. His BMI is 23.
Labwork reveals low free and total testosterone levels with low FSH and LH levels. Thyroid-stimulating hormone, free T4, and prolactin levels are within normal range, comprehensive metabolic panel is unremarkable, and pituitary MRI is negative. The complete blood count reveals slightly elevated hemoglobin and hematocrit, prompting ordering of iron studies that reveal elevated ferritin and serum iron levels and elevated percent transferrin saturation. Lab values are shown in the table.
Based on his elevated ferritin and transferrin saturation levels, JR undergoes genetic testing for hereditary hemochromatosis (HH) with C282Y and H63D mutation analysis. He is found to have the homozygous C282Y genotype (C282Y/C282Y) for HH.
JR establishes care with a hematologist and is advised to receive therapeutic phlebotomy until his ferritin level is between 10 and 50 ng/mL. An abdominal ultrasound, ordered to screen for hepatomegaly, yields normal results. JR elects not to receive testosterone replacement therapy.
Three months later, labwork reveals a free testosterone level of 120 pg/mL with normal hemoglobin and hematocrit levels, normal transaminases, a ferritin level of 28 ng/mL, and a percent transferrin saturation of 31%. Additional values are shown in the table.
BACKGROUND AND GENETICS
Hereditary hemochromatosis is an autosomal recessive iron storage disorder in which intestinal iron absorption is markedly increased. This results in iron overload and excessive iron deposition in numerous tissues, glands, and organs.1
In patients with HH, a genetic defect causes abnormal expression of the HFE protein that regulates hepcidin production. Hepcidin is an iron regulatory hormone, secreted by hepatocytes, that decreases intestinal iron absorption in response to excess iron.2 Ninety percent of individuals affected by HH are homozygous for mutation at amino acid position 282 on the HFE gene, which causes an inappropriate decrease in hepcidin expression in response to elevated iron levels.2,3 Only 10% of individuals homozygous for the C282Y mutation actually develop clinically apparent end-organ damage.2
Being a carrier (heterozygous) for the C282Y mutation confers significantly lower risk for iron overload. The second most common mutation in the HFE gene, H63D, is associated with a milder phenotype. Those with compound heterozygosity for C282Y/H63D or homozygosity for H63D typically experience either mild or no detectable symptoms.1,3
There can be mutations in other genes involved in iron metabolism, but these represent more rare forms of hemochromatosis. Conditions such as thalassemia, sideroblastic anemia, porphyria cutanea tarda, and chronic liver disease may also be associated with iron overload.1,3
HH is most common in white populations of northern European descent. Multiple factors—including dietary iron intake, alcohol consumption, blood donation, blood loss associated with menstruation, and pregnancy—affect the expression of clinical features of hemochromatosis. Men are 24 times more likely than women to express clinical features of hemochromatosis.2 Approximately 70% of affected patients develop symptoms between ages 40 and 60.1
CLINICAL MANIFESTATIONS
The liver is typically the first organ affected, and hepatomegaly is present in 95% of symptomatic patients, even in the presence of normal transaminase levels.1 A bronzed, metallic, or slate gray skin coloration can occur due to increased iron deposition in the dermis. Arthralgias in the hands, wrists, hips, knees, and ankles are present in up to 50% of patients with hemochromatosis. Cardiac manifestations include restrictive cardiomyopathy, congestive heart failure, and arrhythmias.1
Iron deposition in the beta cells of the pancreas causes diabetes1 and in the pituitary causes hypogonadotropic hypogonadism in both men and women, resulting in decreased libido, amenorrhea, testicular atrophy, gynecomastia, and reduced body hair. Primary testicular dysfunction may occur due to iron deposition in the testicles.1,4 In the thyroid gland, iron deposition can lead to abnormal function. Secondary hypothyroidism is rare in the setting of iron overload, although iron deposition occasionally occurs in pituitary thyrotrophs (usually only to a mild degree). Adrenal insufficiency and hypoparathyroidism may also result from iron overload.5
CLINICAL STUDIES TO ASSESS IRON STORES
When assessing tissue iron stores, it is important to measure the serum iron level, total iron-binding capacity, and ferritin in the fasting state.2 This information can be used to calculate the percent transferrin saturation. If the serum ferritin is elevated (> 300 ng/mL in men and > 200 ng/mL in women) and/or the transferrin saturation is greater than 45%, referral to hematology or hepatology is recommended, along with genetic testing for hemochromatosis.1,2
Once the diagnosis of hemochromatosis has been confirmed, CT or MRI can be used to assess for increased density of the liver.1 Liver biopsy can determine the degree of fibrosis and is often considered in patients with more extreme elevations of serum ferritin levels and/or hepatomegaly. Liver biopsy is the only reliable method for determining whether hepatic cirrhosis, which increases risk for hepatocellular carcinoma, is present.1
TREATMENT
All patients with homozygous HH and evidence of iron overload require treatment, regardless of symptoms. Phlebotomy is the standard of care, due to its low cost and relative safety. Chelating agents are a second-line option when contraindications to phlebotomy (eg, anemia) exist.1,2
Alcohol consumption, especially in the presence of iron overload or liver disease, should be avoided, as it can increase risk for cirrhosis by nearly tenfold.1 Dietary modification is typically unnecessary, aside from the avoidance of iron and vitamin C supplementation.2 Patients should also eliminate raw shellfish from their diet, as they may carry bacteria that can cause potentially fatal infection (since high iron levels impair hepcidin bactericidal activity).2
The management of hepatic failure, cardiac failure, and diabetes in patients with HH is similar to conventional management of these conditions.1 With phlebotomy, the liver and spleen often decrease in size, liver function improves, skin pigmentation lightens, cardiac failure may be reversed, and diabetes control often improves.1,2 Testosterone levels may normalize after phlebotomy, especially if HH is diagnosed in the early stages. In more advanced cases, testosterone replacement therapy in combination with aggressive phlebotomy may be necessary.4
CONCLUSION
A high index of suspicion is required to diagnose hemochromatosis early. HH should be considered in the differential diagnosis for patients with hypogonadotropic hypogonadism, abnormal iron studies, elevated transaminase levels, and a family history of hemochromatosis.
Once the diagnosis is established, all first-degree relatives should be screened.1 Early therapy is crucial to prevent complications from iron overload.
REFERENCES
1. Powell LW. Hemochromatosis. In: Fauci AS, Braunwald E, Kasper DL, et al (eds). Harrison’s Principles of Internal Medicine. 17th ed. New York, NY: McGraw-Hill; 2008:2429-2433.
2. Crownover BK, Carlton JC. Hereditary hemochromatosis. Am Fam Phys. 2013;87(3):183-190.
3. Camaschella C. Understanding iron homeostasis through genetic analysis of hemochromatosis and related disorders. Blood. 2005;106(12):3710-3717.
4. McDermott JH, Walsh CH. Hypogonadism in hereditary hemochromatosis. J Clin Endocrinol Metab. 2005;90(4):2451-2455.
5. Hudec M, Grigerova M, Walsh CH. Secondary hypothyroidism in hereditary hemochromatosis: recovery after iron depletion. Thyroid. 2008;18(2):255-257.
JR, a 34-year-old Caucasian man, was in his normal state of good health until several months ago, when he developed fatigue, low libido, and insomnia. He reports normal erectile function, adding that he fathered a child at age 24. His medical history and remaining review of systems are negative. Physical exam is unremarkable. His BMI is 23.
Labwork reveals low free and total testosterone levels with low FSH and LH levels. Thyroid-stimulating hormone, free T4, and prolactin levels are within normal range, comprehensive metabolic panel is unremarkable, and pituitary MRI is negative. The complete blood count reveals slightly elevated hemoglobin and hematocrit, prompting ordering of iron studies that reveal elevated ferritin and serum iron levels and elevated percent transferrin saturation. Lab values are shown in the table.
Based on his elevated ferritin and transferrin saturation levels, JR undergoes genetic testing for hereditary hemochromatosis (HH) with C282Y and H63D mutation analysis. He is found to have the homozygous C282Y genotype (C282Y/C282Y) for HH.
JR establishes care with a hematologist and is advised to receive therapeutic phlebotomy until his ferritin level is between 10 and 50 ng/mL. An abdominal ultrasound, ordered to screen for hepatomegaly, yields normal results. JR elects not to receive testosterone replacement therapy.
Three months later, labwork reveals a free testosterone level of 120 pg/mL with normal hemoglobin and hematocrit levels, normal transaminases, a ferritin level of 28 ng/mL, and a percent transferrin saturation of 31%. Additional values are shown in the table.
BACKGROUND AND GENETICS
Hereditary hemochromatosis is an autosomal recessive iron storage disorder in which intestinal iron absorption is markedly increased. This results in iron overload and excessive iron deposition in numerous tissues, glands, and organs.1
In patients with HH, a genetic defect causes abnormal expression of the HFE protein that regulates hepcidin production. Hepcidin is an iron regulatory hormone, secreted by hepatocytes, that decreases intestinal iron absorption in response to excess iron.2 Ninety percent of individuals affected by HH are homozygous for mutation at amino acid position 282 on the HFE gene, which causes an inappropriate decrease in hepcidin expression in response to elevated iron levels.2,3 Only 10% of individuals homozygous for the C282Y mutation actually develop clinically apparent end-organ damage.2
Being a carrier (heterozygous) for the C282Y mutation confers significantly lower risk for iron overload. The second most common mutation in the HFE gene, H63D, is associated with a milder phenotype. Those with compound heterozygosity for C282Y/H63D or homozygosity for H63D typically experience either mild or no detectable symptoms.1,3
There can be mutations in other genes involved in iron metabolism, but these represent more rare forms of hemochromatosis. Conditions such as thalassemia, sideroblastic anemia, porphyria cutanea tarda, and chronic liver disease may also be associated with iron overload.1,3
HH is most common in white populations of northern European descent. Multiple factors—including dietary iron intake, alcohol consumption, blood donation, blood loss associated with menstruation, and pregnancy—affect the expression of clinical features of hemochromatosis. Men are 24 times more likely than women to express clinical features of hemochromatosis.2 Approximately 70% of affected patients develop symptoms between ages 40 and 60.1
CLINICAL MANIFESTATIONS
The liver is typically the first organ affected, and hepatomegaly is present in 95% of symptomatic patients, even in the presence of normal transaminase levels.1 A bronzed, metallic, or slate gray skin coloration can occur due to increased iron deposition in the dermis. Arthralgias in the hands, wrists, hips, knees, and ankles are present in up to 50% of patients with hemochromatosis. Cardiac manifestations include restrictive cardiomyopathy, congestive heart failure, and arrhythmias.1
Iron deposition in the beta cells of the pancreas causes diabetes1 and in the pituitary causes hypogonadotropic hypogonadism in both men and women, resulting in decreased libido, amenorrhea, testicular atrophy, gynecomastia, and reduced body hair. Primary testicular dysfunction may occur due to iron deposition in the testicles.1,4 In the thyroid gland, iron deposition can lead to abnormal function. Secondary hypothyroidism is rare in the setting of iron overload, although iron deposition occasionally occurs in pituitary thyrotrophs (usually only to a mild degree). Adrenal insufficiency and hypoparathyroidism may also result from iron overload.5
CLINICAL STUDIES TO ASSESS IRON STORES
When assessing tissue iron stores, it is important to measure the serum iron level, total iron-binding capacity, and ferritin in the fasting state.2 This information can be used to calculate the percent transferrin saturation. If the serum ferritin is elevated (> 300 ng/mL in men and > 200 ng/mL in women) and/or the transferrin saturation is greater than 45%, referral to hematology or hepatology is recommended, along with genetic testing for hemochromatosis.1,2
Once the diagnosis of hemochromatosis has been confirmed, CT or MRI can be used to assess for increased density of the liver.1 Liver biopsy can determine the degree of fibrosis and is often considered in patients with more extreme elevations of serum ferritin levels and/or hepatomegaly. Liver biopsy is the only reliable method for determining whether hepatic cirrhosis, which increases risk for hepatocellular carcinoma, is present.1
TREATMENT
All patients with homozygous HH and evidence of iron overload require treatment, regardless of symptoms. Phlebotomy is the standard of care, due to its low cost and relative safety. Chelating agents are a second-line option when contraindications to phlebotomy (eg, anemia) exist.1,2
Alcohol consumption, especially in the presence of iron overload or liver disease, should be avoided, as it can increase risk for cirrhosis by nearly tenfold.1 Dietary modification is typically unnecessary, aside from the avoidance of iron and vitamin C supplementation.2 Patients should also eliminate raw shellfish from their diet, as they may carry bacteria that can cause potentially fatal infection (since high iron levels impair hepcidin bactericidal activity).2
The management of hepatic failure, cardiac failure, and diabetes in patients with HH is similar to conventional management of these conditions.1 With phlebotomy, the liver and spleen often decrease in size, liver function improves, skin pigmentation lightens, cardiac failure may be reversed, and diabetes control often improves.1,2 Testosterone levels may normalize after phlebotomy, especially if HH is diagnosed in the early stages. In more advanced cases, testosterone replacement therapy in combination with aggressive phlebotomy may be necessary.4
CONCLUSION
A high index of suspicion is required to diagnose hemochromatosis early. HH should be considered in the differential diagnosis for patients with hypogonadotropic hypogonadism, abnormal iron studies, elevated transaminase levels, and a family history of hemochromatosis.
Once the diagnosis is established, all first-degree relatives should be screened.1 Early therapy is crucial to prevent complications from iron overload.
REFERENCES
1. Powell LW. Hemochromatosis. In: Fauci AS, Braunwald E, Kasper DL, et al (eds). Harrison’s Principles of Internal Medicine. 17th ed. New York, NY: McGraw-Hill; 2008:2429-2433.
2. Crownover BK, Carlton JC. Hereditary hemochromatosis. Am Fam Phys. 2013;87(3):183-190.
3. Camaschella C. Understanding iron homeostasis through genetic analysis of hemochromatosis and related disorders. Blood. 2005;106(12):3710-3717.
4. McDermott JH, Walsh CH. Hypogonadism in hereditary hemochromatosis. J Clin Endocrinol Metab. 2005;90(4):2451-2455.
5. Hudec M, Grigerova M, Walsh CH. Secondary hypothyroidism in hereditary hemochromatosis: recovery after iron depletion. Thyroid. 2008;18(2):255-257.
Pregnancy: Hypertension and Risk for Kidney Disease
Q If a pregnant woman has mild hypertension (eg, 140/90 mm Hg) but no albuminuria, is she at higher risk for kidney disease as she ages?
Gestational hypertension and preeclampsia are two types of hypertension that occur during pregnancy. Both occur after 20 weeks’ gestation and include a blood pressure reading greater than 140/90 mm Hg and no maternal history of hypertension. Proteinuria does not occur in gestational hypertension as it does in preeclampsia (proteinuria ≥ 300 mg in a 24-h urine collection).
One study evaluated more than 26,000 Taiwanese women with hypertension during pregnancy and compared them to more than 200,000 women without hypertension during pregnancy. It was found that hypertension during pregnancy increased the risk for chronic kidney disease (CKD) and end-stage renal disease (ESRD) later in life. Preeclampsia and eclampsia were even more likely to increase the risk for ESRD, compared with gestational hypertension.1
Preeclampsia occurs after 20 weeks’ gestation and includes elevated blood pressure (≥ 140 mm Hg systolic or ≥ 90 mm Hg diastolic) and proteinuria (≥ 0.3 g in 24 h). Severe preeclampsia develops with the addition of worsening hypertension and proteinuria, oliguria less than 500 mL in 24 h, thrombocytopenia, hemolysis, elevated liver enzymes, low platelets, pulmonary edema, and fetal growth restriction. Eclampsia is when seizures occur in addition to preeclampsia.2 Gestational hypertension has also been linked with a higher risk for ischemic heart disease, myocardial infarcts and death, heart failure, ischem-
ic stroke, kidney disease, and diabetes.3
Because of the association between hypertension during pregnancy and subsequent development of CKD, it is very important that mothers who have hypertension during pregnancy continue to have their renal function monitored after delivery.
Mandy Trolinger, MS, RD, PA-C
Denver Nephrology
Denver, CO
Personal Note: Mandy is a two-time kidney transplant recipient. She delivered a healthy baby boy in 2012.
REFERENCES
1. Wang I-K, Muo C-H, Liang C-C, et al. Association between hypertensive disorders during pregnancy and end-stage renal disease: a population-based study. CMAJ. 2013;85: 207-213.
2. McPhee SJ, Papadakis MA, Tierney LM, et al. Current Medical Diagnosis and Treatment. 47th ed. New York, NY: McGraw-Hill/Lange; 2008.
3. Männistö T, Mendola P, Vääräsmäki M, et al. Elevated blood pressure in pregnancy and subsequent chronic disease risk. Circulation. 2013;127:681-690.
4. Nevis IF, Reitsma A, Dominic A, et al. Pregnancy outcomes in women with chronic kidney disease: a systematic review. Clin J Am Soc Nephrol. 2011;6:2587-2598.
5. Hou S. Pregnancy in chronic renal insufficiency and end-stage renal disease. Am J Kidney Dis. 1999;33:235-252.
6. Davison JM. Dialysis, transplantation, and pregnancy. Am J Kidney Dis. 1991;17:127-132.
7. Asamiya Y, Otsubo S, Matsuda Y, et al. The importance of low blood urea nitrogen levels in pregnant patients undergoing hemodialysis to optimize birth weight and gestational age. Kidney Int. 2009;75:1217-1222.
8. Giatras I, Levy DP, Malone FD, et al. Pregnancy during dialysis: case report and management guidelines. Nephrol Dial Transplant. 1998;13:3266-3272.
9. McKay DB, Josephson MA. Pregnancy in recipients of solid organs—effects on mother and child. N Engl J Med. 2006;354:1281-1293.
10. Sifontis NM, Coscia LA, Constantinescu S, et al. Pregnancy outcomes in solid organ transplant recipients with exposure to mycophenolate mofetil or sirolimus. Transplantation. 2006;82:1698-1702.
11. Kainz A, Harabacz I, Cowlrick IS, et al. Review of the course and outcome of 100 pregnancies in 84 women treated with tacrolimus. Transplantation. 2000;70:1718-1721.
12. Josephson MA. Pregnancy in renal transplant recipients: more questions answered, still more asked. Clin J Am Soc Nephrol. 2013;8: 182-183.
Q If a pregnant woman has mild hypertension (eg, 140/90 mm Hg) but no albuminuria, is she at higher risk for kidney disease as she ages?
Gestational hypertension and preeclampsia are two types of hypertension that occur during pregnancy. Both occur after 20 weeks’ gestation and include a blood pressure reading greater than 140/90 mm Hg and no maternal history of hypertension. Proteinuria does not occur in gestational hypertension as it does in preeclampsia (proteinuria ≥ 300 mg in a 24-h urine collection).
One study evaluated more than 26,000 Taiwanese women with hypertension during pregnancy and compared them to more than 200,000 women without hypertension during pregnancy. It was found that hypertension during pregnancy increased the risk for chronic kidney disease (CKD) and end-stage renal disease (ESRD) later in life. Preeclampsia and eclampsia were even more likely to increase the risk for ESRD, compared with gestational hypertension.1
Preeclampsia occurs after 20 weeks’ gestation and includes elevated blood pressure (≥ 140 mm Hg systolic or ≥ 90 mm Hg diastolic) and proteinuria (≥ 0.3 g in 24 h). Severe preeclampsia develops with the addition of worsening hypertension and proteinuria, oliguria less than 500 mL in 24 h, thrombocytopenia, hemolysis, elevated liver enzymes, low platelets, pulmonary edema, and fetal growth restriction. Eclampsia is when seizures occur in addition to preeclampsia.2 Gestational hypertension has also been linked with a higher risk for ischemic heart disease, myocardial infarcts and death, heart failure, ischem-
ic stroke, kidney disease, and diabetes.3
Because of the association between hypertension during pregnancy and subsequent development of CKD, it is very important that mothers who have hypertension during pregnancy continue to have their renal function monitored after delivery.
Mandy Trolinger, MS, RD, PA-C
Denver Nephrology
Denver, CO
Personal Note: Mandy is a two-time kidney transplant recipient. She delivered a healthy baby boy in 2012.
REFERENCES
1. Wang I-K, Muo C-H, Liang C-C, et al. Association between hypertensive disorders during pregnancy and end-stage renal disease: a population-based study. CMAJ. 2013;85: 207-213.
2. McPhee SJ, Papadakis MA, Tierney LM, et al. Current Medical Diagnosis and Treatment. 47th ed. New York, NY: McGraw-Hill/Lange; 2008.
3. Männistö T, Mendola P, Vääräsmäki M, et al. Elevated blood pressure in pregnancy and subsequent chronic disease risk. Circulation. 2013;127:681-690.
4. Nevis IF, Reitsma A, Dominic A, et al. Pregnancy outcomes in women with chronic kidney disease: a systematic review. Clin J Am Soc Nephrol. 2011;6:2587-2598.
5. Hou S. Pregnancy in chronic renal insufficiency and end-stage renal disease. Am J Kidney Dis. 1999;33:235-252.
6. Davison JM. Dialysis, transplantation, and pregnancy. Am J Kidney Dis. 1991;17:127-132.
7. Asamiya Y, Otsubo S, Matsuda Y, et al. The importance of low blood urea nitrogen levels in pregnant patients undergoing hemodialysis to optimize birth weight and gestational age. Kidney Int. 2009;75:1217-1222.
8. Giatras I, Levy DP, Malone FD, et al. Pregnancy during dialysis: case report and management guidelines. Nephrol Dial Transplant. 1998;13:3266-3272.
9. McKay DB, Josephson MA. Pregnancy in recipients of solid organs—effects on mother and child. N Engl J Med. 2006;354:1281-1293.
10. Sifontis NM, Coscia LA, Constantinescu S, et al. Pregnancy outcomes in solid organ transplant recipients with exposure to mycophenolate mofetil or sirolimus. Transplantation. 2006;82:1698-1702.
11. Kainz A, Harabacz I, Cowlrick IS, et al. Review of the course and outcome of 100 pregnancies in 84 women treated with tacrolimus. Transplantation. 2000;70:1718-1721.
12. Josephson MA. Pregnancy in renal transplant recipients: more questions answered, still more asked. Clin J Am Soc Nephrol. 2013;8: 182-183.
Q If a pregnant woman has mild hypertension (eg, 140/90 mm Hg) but no albuminuria, is she at higher risk for kidney disease as she ages?
Gestational hypertension and preeclampsia are two types of hypertension that occur during pregnancy. Both occur after 20 weeks’ gestation and include a blood pressure reading greater than 140/90 mm Hg and no maternal history of hypertension. Proteinuria does not occur in gestational hypertension as it does in preeclampsia (proteinuria ≥ 300 mg in a 24-h urine collection).
One study evaluated more than 26,000 Taiwanese women with hypertension during pregnancy and compared them to more than 200,000 women without hypertension during pregnancy. It was found that hypertension during pregnancy increased the risk for chronic kidney disease (CKD) and end-stage renal disease (ESRD) later in life. Preeclampsia and eclampsia were even more likely to increase the risk for ESRD, compared with gestational hypertension.1
Preeclampsia occurs after 20 weeks’ gestation and includes elevated blood pressure (≥ 140 mm Hg systolic or ≥ 90 mm Hg diastolic) and proteinuria (≥ 0.3 g in 24 h). Severe preeclampsia develops with the addition of worsening hypertension and proteinuria, oliguria less than 500 mL in 24 h, thrombocytopenia, hemolysis, elevated liver enzymes, low platelets, pulmonary edema, and fetal growth restriction. Eclampsia is when seizures occur in addition to preeclampsia.2 Gestational hypertension has also been linked with a higher risk for ischemic heart disease, myocardial infarcts and death, heart failure, ischem-
ic stroke, kidney disease, and diabetes.3
Because of the association between hypertension during pregnancy and subsequent development of CKD, it is very important that mothers who have hypertension during pregnancy continue to have their renal function monitored after delivery.
Mandy Trolinger, MS, RD, PA-C
Denver Nephrology
Denver, CO
Personal Note: Mandy is a two-time kidney transplant recipient. She delivered a healthy baby boy in 2012.
REFERENCES
1. Wang I-K, Muo C-H, Liang C-C, et al. Association between hypertensive disorders during pregnancy and end-stage renal disease: a population-based study. CMAJ. 2013;85: 207-213.
2. McPhee SJ, Papadakis MA, Tierney LM, et al. Current Medical Diagnosis and Treatment. 47th ed. New York, NY: McGraw-Hill/Lange; 2008.
3. Männistö T, Mendola P, Vääräsmäki M, et al. Elevated blood pressure in pregnancy and subsequent chronic disease risk. Circulation. 2013;127:681-690.
4. Nevis IF, Reitsma A, Dominic A, et al. Pregnancy outcomes in women with chronic kidney disease: a systematic review. Clin J Am Soc Nephrol. 2011;6:2587-2598.
5. Hou S. Pregnancy in chronic renal insufficiency and end-stage renal disease. Am J Kidney Dis. 1999;33:235-252.
6. Davison JM. Dialysis, transplantation, and pregnancy. Am J Kidney Dis. 1991;17:127-132.
7. Asamiya Y, Otsubo S, Matsuda Y, et al. The importance of low blood urea nitrogen levels in pregnant patients undergoing hemodialysis to optimize birth weight and gestational age. Kidney Int. 2009;75:1217-1222.
8. Giatras I, Levy DP, Malone FD, et al. Pregnancy during dialysis: case report and management guidelines. Nephrol Dial Transplant. 1998;13:3266-3272.
9. McKay DB, Josephson MA. Pregnancy in recipients of solid organs—effects on mother and child. N Engl J Med. 2006;354:1281-1293.
10. Sifontis NM, Coscia LA, Constantinescu S, et al. Pregnancy outcomes in solid organ transplant recipients with exposure to mycophenolate mofetil or sirolimus. Transplantation. 2006;82:1698-1702.
11. Kainz A, Harabacz I, Cowlrick IS, et al. Review of the course and outcome of 100 pregnancies in 84 women treated with tacrolimus. Transplantation. 2000;70:1718-1721.
12. Josephson MA. Pregnancy in renal transplant recipients: more questions answered, still more asked. Clin J Am Soc Nephrol. 2013;8: 182-183.
Pregnancy: CKD, Dialysis, and Transplant Patients
Q) I was having a “discussion” over lunch about CKD, pregnancy, and transplant. I said that dialysis patients cannot get pregnant, but someone said I was wrong. A friend said that transplant patients should not get pregnant because of the toxicity of the immunosuppressant medications they take, but another practitioner said that was in the “olden days.” What is the current state of the CKD, dialysis, and transplant patient and pregnancy?
The first healthy baby delivered by a pregnant kidney transplant patient was born in 1958. With the advances in treatment of kidney disease, we are now seeing more pregnancies in these patients. However, they are still considered high risk and should be monitored by a transplant nephrologist and a high-risk obstetrician.4
CKD patients (not on dialysis) are at increased risk for pregnancy complications. Maternal risks include gestational hypertension, preeclampsia/eclampsia, ESRD, or death. Fetal complications include prematurity, small-for-gestational-age babies, and stillbirth. It is very important to control hypertension because it is directly linked to fetal outcome; however, not all blood pressure medications are safe in pregnancy. ACE inhibitors, for example, are teratogenic and absolutely contraindicated.4
Fertility is decreased in dialysis patients; however, pregnancy occurs in 0.3% to 1.5% of women of childbearing age on dialysis. Pregnancy should be confirmed with an ultrasound because serum β-human chorionic gonadotropin can be falsely elevated in ESRD.5
It also can be difficult to monitor pregnancy weight gain due to fluid gains between dialysis treatments. Dialysis prescriptions should be increased either in time or frequency (or both), with a goal of keeping the blood urea nitrogen concentration below 50 mg/dL to improve maternal and fetal outcomes.6,7 Other important factors to control are metabolic acidosis, hypocalcemia, and anemia (increased erythropoietin-stimulating agents may be needed). Frequent uterine and fetal monitoring is also indicated to prevent preterm labor due to the dialysis process. Preeclampsia, prematurity, low birth weight, and hypertension are the most common risks in these pregnancies.8
Renal transplantation often returns fertility to normal and allows pregnancy to occur; however, it is recommended that female patients wait until one year post-transplant if the transplanted kidney is from a living related donor (two years if from a deceased donor), have a serum creatinine level less than 1.5 mg/dL, and a urinary protein level less than 500 mg/d.9 The immunosuppression regimen usually needs adjustment because certain immunosuppressants are contraindicated in pregnancy and have been linked to teratogenic effects; however, data is still limited in this area.10,11 Pregnant transplant recipients are at higher risk for preeclampsia, gestational diabetes, preterm delivery, small-for-gestational-age babies, miscarriage, stillbirth, neonatal death, and congenital abnormalities.12
The National Transplantation Pregnancy Registry (www.ntpr.giftoflifeinstitute.org/) is an ongoing registry in the United States that reports on transplant pregnancies and their outcomes. Data collected by the registry show that there have been many healthy pregnancies without any adverse maternal or fetal outcomes among transplant recipients.
Mandy Trolinger, MS, RD, PA-C
Denver Nephrology
Denver, CO
Personal Note: Mandy is a two-time kidney transplant recipient. She delivered a healthy baby boy in 2012.
REFERENCES
1. Wang I-K, Muo C-H, Liang C-C, et al. Association between hypertensive disorders during pregnancy and end-stage renal disease: a population-based study. CMAJ. 2013;85: 207-213.
2. McPhee SJ, Papadakis MA, Tierney LM, et al. Current Medical Diagnosis and Treatment. 47th ed. New York, NY: McGraw-Hill/Lange; 2008.
3. Männistö T, Mendola P, Vääräsmäki M, et al. Elevated blood pressure in pregnancy and subsequent chronic disease risk. Circulation. 2013;127:681-690.
4. Nevis IF, Reitsma A, Dominic A, et al. Pregnancy outcomes in women with chronic kidney disease: a systematic review. Clin J Am Soc Nephrol. 2011;6:2587-2598.
5. Hou S. Pregnancy in chronic renal insufficiency and end-stage renal disease. Am J Kidney Dis. 1999;33:235-252.
6. Davison JM. Dialysis, transplantation, and pregnancy. Am J Kidney Dis. 1991;17:127-132.
7. Asamiya Y, Otsubo S, Matsuda Y, et al. The importance of low blood urea nitrogen levels in pregnant patients undergoing hemodialysis to optimize birth weight and gestational age. Kidney Int. 2009;75:1217-1222.
8. Giatras I, Levy DP, Malone FD, et al. Pregnancy during dialysis: case report and management guidelines. Nephrol Dial Transplant. 1998;13:3266-3272.
9. McKay DB, Josephson MA. Pregnancy in recipients of solid organs—effects on mother and child. N Engl J Med. 2006;354:1281-1293.
10. Sifontis NM, Coscia LA, Constantinescu S, et al. Pregnancy outcomes in solid organ transplant recipients with exposure to mycophenolate mofetil or sirolimus. Transplantation. 2006;82:1698-1702.
11. Kainz A, Harabacz I, Cowlrick IS, et al. Review of the course and outcome of 100 pregnancies in 84 women treated with tacrolimus. Transplantation. 2000;70:1718-1721.
12. Josephson MA. Pregnancy in renal transplant recipients: more questions answered, still more asked. Clin J Am Soc Nephrol. 2013;8: 182-183.
Q) I was having a “discussion” over lunch about CKD, pregnancy, and transplant. I said that dialysis patients cannot get pregnant, but someone said I was wrong. A friend said that transplant patients should not get pregnant because of the toxicity of the immunosuppressant medications they take, but another practitioner said that was in the “olden days.” What is the current state of the CKD, dialysis, and transplant patient and pregnancy?
The first healthy baby delivered by a pregnant kidney transplant patient was born in 1958. With the advances in treatment of kidney disease, we are now seeing more pregnancies in these patients. However, they are still considered high risk and should be monitored by a transplant nephrologist and a high-risk obstetrician.4
CKD patients (not on dialysis) are at increased risk for pregnancy complications. Maternal risks include gestational hypertension, preeclampsia/eclampsia, ESRD, or death. Fetal complications include prematurity, small-for-gestational-age babies, and stillbirth. It is very important to control hypertension because it is directly linked to fetal outcome; however, not all blood pressure medications are safe in pregnancy. ACE inhibitors, for example, are teratogenic and absolutely contraindicated.4
Fertility is decreased in dialysis patients; however, pregnancy occurs in 0.3% to 1.5% of women of childbearing age on dialysis. Pregnancy should be confirmed with an ultrasound because serum β-human chorionic gonadotropin can be falsely elevated in ESRD.5
It also can be difficult to monitor pregnancy weight gain due to fluid gains between dialysis treatments. Dialysis prescriptions should be increased either in time or frequency (or both), with a goal of keeping the blood urea nitrogen concentration below 50 mg/dL to improve maternal and fetal outcomes.6,7 Other important factors to control are metabolic acidosis, hypocalcemia, and anemia (increased erythropoietin-stimulating agents may be needed). Frequent uterine and fetal monitoring is also indicated to prevent preterm labor due to the dialysis process. Preeclampsia, prematurity, low birth weight, and hypertension are the most common risks in these pregnancies.8
Renal transplantation often returns fertility to normal and allows pregnancy to occur; however, it is recommended that female patients wait until one year post-transplant if the transplanted kidney is from a living related donor (two years if from a deceased donor), have a serum creatinine level less than 1.5 mg/dL, and a urinary protein level less than 500 mg/d.9 The immunosuppression regimen usually needs adjustment because certain immunosuppressants are contraindicated in pregnancy and have been linked to teratogenic effects; however, data is still limited in this area.10,11 Pregnant transplant recipients are at higher risk for preeclampsia, gestational diabetes, preterm delivery, small-for-gestational-age babies, miscarriage, stillbirth, neonatal death, and congenital abnormalities.12
The National Transplantation Pregnancy Registry (www.ntpr.giftoflifeinstitute.org/) is an ongoing registry in the United States that reports on transplant pregnancies and their outcomes. Data collected by the registry show that there have been many healthy pregnancies without any adverse maternal or fetal outcomes among transplant recipients.
Mandy Trolinger, MS, RD, PA-C
Denver Nephrology
Denver, CO
Personal Note: Mandy is a two-time kidney transplant recipient. She delivered a healthy baby boy in 2012.
REFERENCES
1. Wang I-K, Muo C-H, Liang C-C, et al. Association between hypertensive disorders during pregnancy and end-stage renal disease: a population-based study. CMAJ. 2013;85: 207-213.
2. McPhee SJ, Papadakis MA, Tierney LM, et al. Current Medical Diagnosis and Treatment. 47th ed. New York, NY: McGraw-Hill/Lange; 2008.
3. Männistö T, Mendola P, Vääräsmäki M, et al. Elevated blood pressure in pregnancy and subsequent chronic disease risk. Circulation. 2013;127:681-690.
4. Nevis IF, Reitsma A, Dominic A, et al. Pregnancy outcomes in women with chronic kidney disease: a systematic review. Clin J Am Soc Nephrol. 2011;6:2587-2598.
5. Hou S. Pregnancy in chronic renal insufficiency and end-stage renal disease. Am J Kidney Dis. 1999;33:235-252.
6. Davison JM. Dialysis, transplantation, and pregnancy. Am J Kidney Dis. 1991;17:127-132.
7. Asamiya Y, Otsubo S, Matsuda Y, et al. The importance of low blood urea nitrogen levels in pregnant patients undergoing hemodialysis to optimize birth weight and gestational age. Kidney Int. 2009;75:1217-1222.
8. Giatras I, Levy DP, Malone FD, et al. Pregnancy during dialysis: case report and management guidelines. Nephrol Dial Transplant. 1998;13:3266-3272.
9. McKay DB, Josephson MA. Pregnancy in recipients of solid organs—effects on mother and child. N Engl J Med. 2006;354:1281-1293.
10. Sifontis NM, Coscia LA, Constantinescu S, et al. Pregnancy outcomes in solid organ transplant recipients with exposure to mycophenolate mofetil or sirolimus. Transplantation. 2006;82:1698-1702.
11. Kainz A, Harabacz I, Cowlrick IS, et al. Review of the course and outcome of 100 pregnancies in 84 women treated with tacrolimus. Transplantation. 2000;70:1718-1721.
12. Josephson MA. Pregnancy in renal transplant recipients: more questions answered, still more asked. Clin J Am Soc Nephrol. 2013;8: 182-183.
Q) I was having a “discussion” over lunch about CKD, pregnancy, and transplant. I said that dialysis patients cannot get pregnant, but someone said I was wrong. A friend said that transplant patients should not get pregnant because of the toxicity of the immunosuppressant medications they take, but another practitioner said that was in the “olden days.” What is the current state of the CKD, dialysis, and transplant patient and pregnancy?
The first healthy baby delivered by a pregnant kidney transplant patient was born in 1958. With the advances in treatment of kidney disease, we are now seeing more pregnancies in these patients. However, they are still considered high risk and should be monitored by a transplant nephrologist and a high-risk obstetrician.4
CKD patients (not on dialysis) are at increased risk for pregnancy complications. Maternal risks include gestational hypertension, preeclampsia/eclampsia, ESRD, or death. Fetal complications include prematurity, small-for-gestational-age babies, and stillbirth. It is very important to control hypertension because it is directly linked to fetal outcome; however, not all blood pressure medications are safe in pregnancy. ACE inhibitors, for example, are teratogenic and absolutely contraindicated.4
Fertility is decreased in dialysis patients; however, pregnancy occurs in 0.3% to 1.5% of women of childbearing age on dialysis. Pregnancy should be confirmed with an ultrasound because serum β-human chorionic gonadotropin can be falsely elevated in ESRD.5
It also can be difficult to monitor pregnancy weight gain due to fluid gains between dialysis treatments. Dialysis prescriptions should be increased either in time or frequency (or both), with a goal of keeping the blood urea nitrogen concentration below 50 mg/dL to improve maternal and fetal outcomes.6,7 Other important factors to control are metabolic acidosis, hypocalcemia, and anemia (increased erythropoietin-stimulating agents may be needed). Frequent uterine and fetal monitoring is also indicated to prevent preterm labor due to the dialysis process. Preeclampsia, prematurity, low birth weight, and hypertension are the most common risks in these pregnancies.8
Renal transplantation often returns fertility to normal and allows pregnancy to occur; however, it is recommended that female patients wait until one year post-transplant if the transplanted kidney is from a living related donor (two years if from a deceased donor), have a serum creatinine level less than 1.5 mg/dL, and a urinary protein level less than 500 mg/d.9 The immunosuppression regimen usually needs adjustment because certain immunosuppressants are contraindicated in pregnancy and have been linked to teratogenic effects; however, data is still limited in this area.10,11 Pregnant transplant recipients are at higher risk for preeclampsia, gestational diabetes, preterm delivery, small-for-gestational-age babies, miscarriage, stillbirth, neonatal death, and congenital abnormalities.12
The National Transplantation Pregnancy Registry (www.ntpr.giftoflifeinstitute.org/) is an ongoing registry in the United States that reports on transplant pregnancies and their outcomes. Data collected by the registry show that there have been many healthy pregnancies without any adverse maternal or fetal outcomes among transplant recipients.
Mandy Trolinger, MS, RD, PA-C
Denver Nephrology
Denver, CO
Personal Note: Mandy is a two-time kidney transplant recipient. She delivered a healthy baby boy in 2012.
REFERENCES
1. Wang I-K, Muo C-H, Liang C-C, et al. Association between hypertensive disorders during pregnancy and end-stage renal disease: a population-based study. CMAJ. 2013;85: 207-213.
2. McPhee SJ, Papadakis MA, Tierney LM, et al. Current Medical Diagnosis and Treatment. 47th ed. New York, NY: McGraw-Hill/Lange; 2008.
3. Männistö T, Mendola P, Vääräsmäki M, et al. Elevated blood pressure in pregnancy and subsequent chronic disease risk. Circulation. 2013;127:681-690.
4. Nevis IF, Reitsma A, Dominic A, et al. Pregnancy outcomes in women with chronic kidney disease: a systematic review. Clin J Am Soc Nephrol. 2011;6:2587-2598.
5. Hou S. Pregnancy in chronic renal insufficiency and end-stage renal disease. Am J Kidney Dis. 1999;33:235-252.
6. Davison JM. Dialysis, transplantation, and pregnancy. Am J Kidney Dis. 1991;17:127-132.
7. Asamiya Y, Otsubo S, Matsuda Y, et al. The importance of low blood urea nitrogen levels in pregnant patients undergoing hemodialysis to optimize birth weight and gestational age. Kidney Int. 2009;75:1217-1222.
8. Giatras I, Levy DP, Malone FD, et al. Pregnancy during dialysis: case report and management guidelines. Nephrol Dial Transplant. 1998;13:3266-3272.
9. McKay DB, Josephson MA. Pregnancy in recipients of solid organs—effects on mother and child. N Engl J Med. 2006;354:1281-1293.
10. Sifontis NM, Coscia LA, Constantinescu S, et al. Pregnancy outcomes in solid organ transplant recipients with exposure to mycophenolate mofetil or sirolimus. Transplantation. 2006;82:1698-1702.
11. Kainz A, Harabacz I, Cowlrick IS, et al. Review of the course and outcome of 100 pregnancies in 84 women treated with tacrolimus. Transplantation. 2000;70:1718-1721.
12. Josephson MA. Pregnancy in renal transplant recipients: more questions answered, still more asked. Clin J Am Soc Nephrol. 2013;8: 182-183.
Managing Gestational Diabetes: Let’s Nip It in The Bud
One of the most common complications of pregnancy is gestational diabetes mellitus (GDM). It is defined as glucose intolerance with first onset during pregnancy.1 In 2011, the incidence of GDM in the United States was between 2% and 10% of all pregnancies. Potential complications associated with GDM include macrosomia, pre-eclampsia, preterm birth, increased risk for cesarean section, neonatal hypoglycemia, shoulder dystocia, and polyhydramnios. Women with a history of gestational diabetes have a 35% to 60% likelihood of developing type 2 diabetes over the following 10 to 20 years.2
Q: When should screening for GDM occur?
According to the American Diabetes Association’s (ADA) 2012 Clinical Practice Recommendations, a pregnant woman should be screened for undiagnosed type 2 diabetes at her first prenatal visit if she has certain risk factors.3 These include, but are not limited to, family history of diabetes, overweight/obesity, sedentary lifestyle, elevated blood pressure and/or cholesterol, impaired fasting glucose or impaired glucose tolerance, or certain ethnic backgrounds (eg, Hispanic, Native American, and non-Hispanic black).4 In 2011, the ADA revised its recommendations for GDM screening and diagnosis to be in accordance with those from the International Association of Diabetes and Pregnancy Study Groups (IADPSG), an international consensus group with representatives from multiple obstetric and diabetes organizations, including ADA.
Q: How is GDM diagnosed?
Current recommendations stipulate that women with no previous history of diabetes or prediabetes undergo one-step testing: a 75-g glucose tolerance test (GTT) at 24 to 28 weeks’ gestation.5,6 For women with a prior history of GDM, screening is recommended earlier in the pregnancy. The GTT should be performed after an overnight fast of at least eight hours.3 An elevation of any one of the values above normal reference range is consistent with the diagnosis of GDM. (Previously, the diagnostic criteria required two abnormal values.) Multiple international studies using the new criteria have estimated an increased incidence of gestational diabetes in up to 18% of pregnancies.5,6
Some organizations have not endorsed the IADPSG/ADA diagnostic criteria at this time; as a result, many practitioners continue to use two-step testing for diagnosing GDM. To do the two-step testing, a 50-g glucose load is given, followed by a blood glucose reading one hour later. If the one-hour reading is within normal range, no further testing is warranted and the patient does not have gestational diabetes. If the test is abnormal, she must undergo a fasting three-hour GTT using a 100-g glucose load.
Q: What advice should a woman get once she’s diagnosed with GDM?
As soon as a woman is diagnosed with GDM, she should be referred for a gestational diabetes education class and nutrition counseling. Specifically, she should learn what it means for her to have GDM, implications for her and her baby, and the importance of eating a healthy diet (not the proverbial concept of “eating for two”), physical activity, self-monitoring blood glucose, and adherence to any prescribed medications.
Probably the most important aspect of education is nutrition counseling. It is known that smaller meals consumed more frequently throughout the day reduce spikes in blood glucose levels. One suggestion is to eat three small meals and three low-carbohydrate (15 g) snacks each day. Meals and snacks are generally established based on fixed carbohydrate amounts. A certified diabetes educator or registered dietitian (RD) can recommend healthy meal and snack ideas that are tasty, promote satiety, and minimize spikes in glucose levels.
Q: What are the current treatment options for GDM?
During the process of receiving GDM education, the patient should be prescribed a glucometer, along with specific glucose targets. Blood glucose should be checked multiple times a day, preferably fasting and postprandial measurements. Medical practices vary in their preferred glucose targets; some individuals require tighter control than others. The ADA suggests the following targets:
• Before a meal (preprandial):
95 mg/dL or less.
• One hour after a meal (postprandial): 140 mg/dL or less.
• Two hours after a meal (postprandial): 120 mg/dL or less. 7
If blood glucose levels remain within normal range, it is possible to control gestational diabetes with dietary modification and physical activity. If readings are consistently elevated, then the patient must be started on medication. There are currently no FDA-approved oral medications to treat gestational diabetes. Glyburide is commonly used, although it is not FDA approved for this indication. More studies to establish its safety are likely needed for FDA approval.8
If pharmaceutical treatment is warranted, insulin is the safest and most effective agent. It is the only medication that is FDA approved for treatment of GDM. Levemir (insulin detemir [rDNA origin] injection) gained FDA approval for use in pregnancy in 2012, so it has become more widespread than NPH for basal insulin usage.9
Although it is usually managed by an endocrinologist or perinatologist, an experienced obstetrician could also manage GDM. Often, the patient is referred to an endocrinologist. The endocrine provider, along with the diabetes educator and RD, focus on nutrition counseling and diabetes management so the obstetrician can focus on maternal and fetal health.
Q: What is the recommended follow-up?
Since embryonic and fetal development occurs at such a rapid rate, time is of the essence for getting a patient’s blood glucose to goal. While treating diabetes in general can be challenging, this is usually not the case with GDM. Most women with GDM are motivated to take care of themselves for the well-being of their developing baby. The influence of a baby developing inside a mother is so strong that diabetic women who become pregnant often take better care of themselves than they do when they are not pregnant.
The patient’s daily responsibilities should include eating a healthy and diet checking her blood glucose levels throughout the day. These readings must be recorded. Clinic visits should occur often, with emailing of glucose readings between visits as needed. The frequency of visits varies among practices, depending on the patient’s level of glucose control and intensity of the treatment regimen.
Q: Why is postpartum testing important?
After delivery, most cases of GDM usually resolve. However, approximately 5% to 10% of women with gestational diabetes are found to have diabetes immediately after pregnancy.2 To evaluate for persistent diabetes, a two-hour GTT should be done at six weeks’ postpartum. Although an A1C can now be used to diagnose diabetes, the ADA does not recommend checking it for this purpose.3
If the two-hour GTT result is normal, a woman should be screened for diabetes every three years for the rest of her life.3 If a diagnosis of impaired fasting glucose or impaired glucose tolerance is made, then she should be tested for diabetes on an annual basis or in the interim if she develops classic symptoms of hyperglycemia.3 If diabetes is diagnosed, she should be treated accordingly as a type 2 diabetic patient.
At this time, the patient should be counseled on lifestyle interventions and consider starting metformin therapy if appropriate. Diabetes education classes are available for prediabetes. To maintain good health and prevent/delay onset of type 2 diabetes, here are some tips to follow:
• The same diet as during pregnancy does not have to be followed, although healthy eating habits are always a good idea.
• Physical activity (approximately 30 min five times a week) will help shed weight gained during pregnancy.
• Breastfeeding promotes weight loss.10
• Patients should aim for weight loss of 7% of body weight.3
• Continue annual physical exams, keeping an eye on blood pressure, weight, and cholesterol levels.
It’s reasonable for the patient to check glucose levels occasionally after delivery. If elevated readings occur, the patient can make an appointment with her primary care provider or endocrinologist.
References
1. American Association for Clinical Chemistry. A New Definition of Gestational Diabetes. www.aacc.org/publications/cln/2010/may/Pages/CoverStory2May2010.aspx. Accessed June 30, 2013.
2. National Diabetes Statistics, 2011. www.diabetes.niddk.nih.gov/dm/pubs/statistics/#Gestational. Accessed July 22, 2013.
3. American Diabetes Association. 2012 Clinical Practice Recommendations. Diabetes Care. 2012;35(suppl 1). http://professional.diabetes.org/SlideLibrary/media/4839/ADA%20Standards%20of%20Medical%20Care%202012%20FINAL.ppt. Accessed June 24, 2013.
4. American Diabetes Association. Diabetes basics: your risk. www.diabetes.org/diabetes-basics/prevention/risk-factors. Accessed August 13, 2013.
5. American Diabetes Association. Diabetes Basics: What is Gestational Diabetes? www.diabetes.org/diabetes-basics/gestational/what-is-gestational-diabetes.html. Accessed August 13, 2013.
6. Johnson K. New criteria for gestational diabetes increase diagnoses (December 5, 2011). www.medscape.com/viewarticle/754733. Accessed August 13, 2013.
7. American Diabetes Association. Diabetes basics: how to treat gestational diabetes. www.diabetes.org/diabetes-basics/gestational/how-to-treat-gestational.html. Accessed August 13, 2013.
8. Moore TR. Glyburide for the treatment of gestational diabetes: a critical appraisal. Diabetes Care. 2007;30(suppl 2). http://care.diabetesjournals.org/content/30/Supplement_2/S209.full. Accessed August 13, 2013.
9. Lowes R. Levemir assigned more reassuring pregnancy risk category (April 2, 2012). www.medscape.com/viewarticle/761349. Accessed August 13, 2013.
10. Buchanan TA, Xiang AH, Page KA. Gestational diabetes mellitus: risks and management during and after pregnancy. Nat Rev Endocrinol. 2012;8(11):639-649.
One of the most common complications of pregnancy is gestational diabetes mellitus (GDM). It is defined as glucose intolerance with first onset during pregnancy.1 In 2011, the incidence of GDM in the United States was between 2% and 10% of all pregnancies. Potential complications associated with GDM include macrosomia, pre-eclampsia, preterm birth, increased risk for cesarean section, neonatal hypoglycemia, shoulder dystocia, and polyhydramnios. Women with a history of gestational diabetes have a 35% to 60% likelihood of developing type 2 diabetes over the following 10 to 20 years.2
Q: When should screening for GDM occur?
According to the American Diabetes Association’s (ADA) 2012 Clinical Practice Recommendations, a pregnant woman should be screened for undiagnosed type 2 diabetes at her first prenatal visit if she has certain risk factors.3 These include, but are not limited to, family history of diabetes, overweight/obesity, sedentary lifestyle, elevated blood pressure and/or cholesterol, impaired fasting glucose or impaired glucose tolerance, or certain ethnic backgrounds (eg, Hispanic, Native American, and non-Hispanic black).4 In 2011, the ADA revised its recommendations for GDM screening and diagnosis to be in accordance with those from the International Association of Diabetes and Pregnancy Study Groups (IADPSG), an international consensus group with representatives from multiple obstetric and diabetes organizations, including ADA.
Q: How is GDM diagnosed?
Current recommendations stipulate that women with no previous history of diabetes or prediabetes undergo one-step testing: a 75-g glucose tolerance test (GTT) at 24 to 28 weeks’ gestation.5,6 For women with a prior history of GDM, screening is recommended earlier in the pregnancy. The GTT should be performed after an overnight fast of at least eight hours.3 An elevation of any one of the values above normal reference range is consistent with the diagnosis of GDM. (Previously, the diagnostic criteria required two abnormal values.) Multiple international studies using the new criteria have estimated an increased incidence of gestational diabetes in up to 18% of pregnancies.5,6
Some organizations have not endorsed the IADPSG/ADA diagnostic criteria at this time; as a result, many practitioners continue to use two-step testing for diagnosing GDM. To do the two-step testing, a 50-g glucose load is given, followed by a blood glucose reading one hour later. If the one-hour reading is within normal range, no further testing is warranted and the patient does not have gestational diabetes. If the test is abnormal, she must undergo a fasting three-hour GTT using a 100-g glucose load.
Q: What advice should a woman get once she’s diagnosed with GDM?
As soon as a woman is diagnosed with GDM, she should be referred for a gestational diabetes education class and nutrition counseling. Specifically, she should learn what it means for her to have GDM, implications for her and her baby, and the importance of eating a healthy diet (not the proverbial concept of “eating for two”), physical activity, self-monitoring blood glucose, and adherence to any prescribed medications.
Probably the most important aspect of education is nutrition counseling. It is known that smaller meals consumed more frequently throughout the day reduce spikes in blood glucose levels. One suggestion is to eat three small meals and three low-carbohydrate (15 g) snacks each day. Meals and snacks are generally established based on fixed carbohydrate amounts. A certified diabetes educator or registered dietitian (RD) can recommend healthy meal and snack ideas that are tasty, promote satiety, and minimize spikes in glucose levels.
Q: What are the current treatment options for GDM?
During the process of receiving GDM education, the patient should be prescribed a glucometer, along with specific glucose targets. Blood glucose should be checked multiple times a day, preferably fasting and postprandial measurements. Medical practices vary in their preferred glucose targets; some individuals require tighter control than others. The ADA suggests the following targets:
• Before a meal (preprandial):
95 mg/dL or less.
• One hour after a meal (postprandial): 140 mg/dL or less.
• Two hours after a meal (postprandial): 120 mg/dL or less. 7
If blood glucose levels remain within normal range, it is possible to control gestational diabetes with dietary modification and physical activity. If readings are consistently elevated, then the patient must be started on medication. There are currently no FDA-approved oral medications to treat gestational diabetes. Glyburide is commonly used, although it is not FDA approved for this indication. More studies to establish its safety are likely needed for FDA approval.8
If pharmaceutical treatment is warranted, insulin is the safest and most effective agent. It is the only medication that is FDA approved for treatment of GDM. Levemir (insulin detemir [rDNA origin] injection) gained FDA approval for use in pregnancy in 2012, so it has become more widespread than NPH for basal insulin usage.9
Although it is usually managed by an endocrinologist or perinatologist, an experienced obstetrician could also manage GDM. Often, the patient is referred to an endocrinologist. The endocrine provider, along with the diabetes educator and RD, focus on nutrition counseling and diabetes management so the obstetrician can focus on maternal and fetal health.
Q: What is the recommended follow-up?
Since embryonic and fetal development occurs at such a rapid rate, time is of the essence for getting a patient’s blood glucose to goal. While treating diabetes in general can be challenging, this is usually not the case with GDM. Most women with GDM are motivated to take care of themselves for the well-being of their developing baby. The influence of a baby developing inside a mother is so strong that diabetic women who become pregnant often take better care of themselves than they do when they are not pregnant.
The patient’s daily responsibilities should include eating a healthy and diet checking her blood glucose levels throughout the day. These readings must be recorded. Clinic visits should occur often, with emailing of glucose readings between visits as needed. The frequency of visits varies among practices, depending on the patient’s level of glucose control and intensity of the treatment regimen.
Q: Why is postpartum testing important?
After delivery, most cases of GDM usually resolve. However, approximately 5% to 10% of women with gestational diabetes are found to have diabetes immediately after pregnancy.2 To evaluate for persistent diabetes, a two-hour GTT should be done at six weeks’ postpartum. Although an A1C can now be used to diagnose diabetes, the ADA does not recommend checking it for this purpose.3
If the two-hour GTT result is normal, a woman should be screened for diabetes every three years for the rest of her life.3 If a diagnosis of impaired fasting glucose or impaired glucose tolerance is made, then she should be tested for diabetes on an annual basis or in the interim if she develops classic symptoms of hyperglycemia.3 If diabetes is diagnosed, she should be treated accordingly as a type 2 diabetic patient.
At this time, the patient should be counseled on lifestyle interventions and consider starting metformin therapy if appropriate. Diabetes education classes are available for prediabetes. To maintain good health and prevent/delay onset of type 2 diabetes, here are some tips to follow:
• The same diet as during pregnancy does not have to be followed, although healthy eating habits are always a good idea.
• Physical activity (approximately 30 min five times a week) will help shed weight gained during pregnancy.
• Breastfeeding promotes weight loss.10
• Patients should aim for weight loss of 7% of body weight.3
• Continue annual physical exams, keeping an eye on blood pressure, weight, and cholesterol levels.
It’s reasonable for the patient to check glucose levels occasionally after delivery. If elevated readings occur, the patient can make an appointment with her primary care provider or endocrinologist.
References
1. American Association for Clinical Chemistry. A New Definition of Gestational Diabetes. www.aacc.org/publications/cln/2010/may/Pages/CoverStory2May2010.aspx. Accessed June 30, 2013.
2. National Diabetes Statistics, 2011. www.diabetes.niddk.nih.gov/dm/pubs/statistics/#Gestational. Accessed July 22, 2013.
3. American Diabetes Association. 2012 Clinical Practice Recommendations. Diabetes Care. 2012;35(suppl 1). http://professional.diabetes.org/SlideLibrary/media/4839/ADA%20Standards%20of%20Medical%20Care%202012%20FINAL.ppt. Accessed June 24, 2013.
4. American Diabetes Association. Diabetes basics: your risk. www.diabetes.org/diabetes-basics/prevention/risk-factors. Accessed August 13, 2013.
5. American Diabetes Association. Diabetes Basics: What is Gestational Diabetes? www.diabetes.org/diabetes-basics/gestational/what-is-gestational-diabetes.html. Accessed August 13, 2013.
6. Johnson K. New criteria for gestational diabetes increase diagnoses (December 5, 2011). www.medscape.com/viewarticle/754733. Accessed August 13, 2013.
7. American Diabetes Association. Diabetes basics: how to treat gestational diabetes. www.diabetes.org/diabetes-basics/gestational/how-to-treat-gestational.html. Accessed August 13, 2013.
8. Moore TR. Glyburide for the treatment of gestational diabetes: a critical appraisal. Diabetes Care. 2007;30(suppl 2). http://care.diabetesjournals.org/content/30/Supplement_2/S209.full. Accessed August 13, 2013.
9. Lowes R. Levemir assigned more reassuring pregnancy risk category (April 2, 2012). www.medscape.com/viewarticle/761349. Accessed August 13, 2013.
10. Buchanan TA, Xiang AH, Page KA. Gestational diabetes mellitus: risks and management during and after pregnancy. Nat Rev Endocrinol. 2012;8(11):639-649.
One of the most common complications of pregnancy is gestational diabetes mellitus (GDM). It is defined as glucose intolerance with first onset during pregnancy.1 In 2011, the incidence of GDM in the United States was between 2% and 10% of all pregnancies. Potential complications associated with GDM include macrosomia, pre-eclampsia, preterm birth, increased risk for cesarean section, neonatal hypoglycemia, shoulder dystocia, and polyhydramnios. Women with a history of gestational diabetes have a 35% to 60% likelihood of developing type 2 diabetes over the following 10 to 20 years.2
Q: When should screening for GDM occur?
According to the American Diabetes Association’s (ADA) 2012 Clinical Practice Recommendations, a pregnant woman should be screened for undiagnosed type 2 diabetes at her first prenatal visit if she has certain risk factors.3 These include, but are not limited to, family history of diabetes, overweight/obesity, sedentary lifestyle, elevated blood pressure and/or cholesterol, impaired fasting glucose or impaired glucose tolerance, or certain ethnic backgrounds (eg, Hispanic, Native American, and non-Hispanic black).4 In 2011, the ADA revised its recommendations for GDM screening and diagnosis to be in accordance with those from the International Association of Diabetes and Pregnancy Study Groups (IADPSG), an international consensus group with representatives from multiple obstetric and diabetes organizations, including ADA.
Q: How is GDM diagnosed?
Current recommendations stipulate that women with no previous history of diabetes or prediabetes undergo one-step testing: a 75-g glucose tolerance test (GTT) at 24 to 28 weeks’ gestation.5,6 For women with a prior history of GDM, screening is recommended earlier in the pregnancy. The GTT should be performed after an overnight fast of at least eight hours.3 An elevation of any one of the values above normal reference range is consistent with the diagnosis of GDM. (Previously, the diagnostic criteria required two abnormal values.) Multiple international studies using the new criteria have estimated an increased incidence of gestational diabetes in up to 18% of pregnancies.5,6
Some organizations have not endorsed the IADPSG/ADA diagnostic criteria at this time; as a result, many practitioners continue to use two-step testing for diagnosing GDM. To do the two-step testing, a 50-g glucose load is given, followed by a blood glucose reading one hour later. If the one-hour reading is within normal range, no further testing is warranted and the patient does not have gestational diabetes. If the test is abnormal, she must undergo a fasting three-hour GTT using a 100-g glucose load.
Q: What advice should a woman get once she’s diagnosed with GDM?
As soon as a woman is diagnosed with GDM, she should be referred for a gestational diabetes education class and nutrition counseling. Specifically, she should learn what it means for her to have GDM, implications for her and her baby, and the importance of eating a healthy diet (not the proverbial concept of “eating for two”), physical activity, self-monitoring blood glucose, and adherence to any prescribed medications.
Probably the most important aspect of education is nutrition counseling. It is known that smaller meals consumed more frequently throughout the day reduce spikes in blood glucose levels. One suggestion is to eat three small meals and three low-carbohydrate (15 g) snacks each day. Meals and snacks are generally established based on fixed carbohydrate amounts. A certified diabetes educator or registered dietitian (RD) can recommend healthy meal and snack ideas that are tasty, promote satiety, and minimize spikes in glucose levels.
Q: What are the current treatment options for GDM?
During the process of receiving GDM education, the patient should be prescribed a glucometer, along with specific glucose targets. Blood glucose should be checked multiple times a day, preferably fasting and postprandial measurements. Medical practices vary in their preferred glucose targets; some individuals require tighter control than others. The ADA suggests the following targets:
• Before a meal (preprandial):
95 mg/dL or less.
• One hour after a meal (postprandial): 140 mg/dL or less.
• Two hours after a meal (postprandial): 120 mg/dL or less. 7
If blood glucose levels remain within normal range, it is possible to control gestational diabetes with dietary modification and physical activity. If readings are consistently elevated, then the patient must be started on medication. There are currently no FDA-approved oral medications to treat gestational diabetes. Glyburide is commonly used, although it is not FDA approved for this indication. More studies to establish its safety are likely needed for FDA approval.8
If pharmaceutical treatment is warranted, insulin is the safest and most effective agent. It is the only medication that is FDA approved for treatment of GDM. Levemir (insulin detemir [rDNA origin] injection) gained FDA approval for use in pregnancy in 2012, so it has become more widespread than NPH for basal insulin usage.9
Although it is usually managed by an endocrinologist or perinatologist, an experienced obstetrician could also manage GDM. Often, the patient is referred to an endocrinologist. The endocrine provider, along with the diabetes educator and RD, focus on nutrition counseling and diabetes management so the obstetrician can focus on maternal and fetal health.
Q: What is the recommended follow-up?
Since embryonic and fetal development occurs at such a rapid rate, time is of the essence for getting a patient’s blood glucose to goal. While treating diabetes in general can be challenging, this is usually not the case with GDM. Most women with GDM are motivated to take care of themselves for the well-being of their developing baby. The influence of a baby developing inside a mother is so strong that diabetic women who become pregnant often take better care of themselves than they do when they are not pregnant.
The patient’s daily responsibilities should include eating a healthy and diet checking her blood glucose levels throughout the day. These readings must be recorded. Clinic visits should occur often, with emailing of glucose readings between visits as needed. The frequency of visits varies among practices, depending on the patient’s level of glucose control and intensity of the treatment regimen.
Q: Why is postpartum testing important?
After delivery, most cases of GDM usually resolve. However, approximately 5% to 10% of women with gestational diabetes are found to have diabetes immediately after pregnancy.2 To evaluate for persistent diabetes, a two-hour GTT should be done at six weeks’ postpartum. Although an A1C can now be used to diagnose diabetes, the ADA does not recommend checking it for this purpose.3
If the two-hour GTT result is normal, a woman should be screened for diabetes every three years for the rest of her life.3 If a diagnosis of impaired fasting glucose or impaired glucose tolerance is made, then she should be tested for diabetes on an annual basis or in the interim if she develops classic symptoms of hyperglycemia.3 If diabetes is diagnosed, she should be treated accordingly as a type 2 diabetic patient.
At this time, the patient should be counseled on lifestyle interventions and consider starting metformin therapy if appropriate. Diabetes education classes are available for prediabetes. To maintain good health and prevent/delay onset of type 2 diabetes, here are some tips to follow:
• The same diet as during pregnancy does not have to be followed, although healthy eating habits are always a good idea.
• Physical activity (approximately 30 min five times a week) will help shed weight gained during pregnancy.
• Breastfeeding promotes weight loss.10
• Patients should aim for weight loss of 7% of body weight.3
• Continue annual physical exams, keeping an eye on blood pressure, weight, and cholesterol levels.
It’s reasonable for the patient to check glucose levels occasionally after delivery. If elevated readings occur, the patient can make an appointment with her primary care provider or endocrinologist.
References
1. American Association for Clinical Chemistry. A New Definition of Gestational Diabetes. www.aacc.org/publications/cln/2010/may/Pages/CoverStory2May2010.aspx. Accessed June 30, 2013.
2. National Diabetes Statistics, 2011. www.diabetes.niddk.nih.gov/dm/pubs/statistics/#Gestational. Accessed July 22, 2013.
3. American Diabetes Association. 2012 Clinical Practice Recommendations. Diabetes Care. 2012;35(suppl 1). http://professional.diabetes.org/SlideLibrary/media/4839/ADA%20Standards%20of%20Medical%20Care%202012%20FINAL.ppt. Accessed June 24, 2013.
4. American Diabetes Association. Diabetes basics: your risk. www.diabetes.org/diabetes-basics/prevention/risk-factors. Accessed August 13, 2013.
5. American Diabetes Association. Diabetes Basics: What is Gestational Diabetes? www.diabetes.org/diabetes-basics/gestational/what-is-gestational-diabetes.html. Accessed August 13, 2013.
6. Johnson K. New criteria for gestational diabetes increase diagnoses (December 5, 2011). www.medscape.com/viewarticle/754733. Accessed August 13, 2013.
7. American Diabetes Association. Diabetes basics: how to treat gestational diabetes. www.diabetes.org/diabetes-basics/gestational/how-to-treat-gestational.html. Accessed August 13, 2013.
8. Moore TR. Glyburide for the treatment of gestational diabetes: a critical appraisal. Diabetes Care. 2007;30(suppl 2). http://care.diabetesjournals.org/content/30/Supplement_2/S209.full. Accessed August 13, 2013.
9. Lowes R. Levemir assigned more reassuring pregnancy risk category (April 2, 2012). www.medscape.com/viewarticle/761349. Accessed August 13, 2013.
10. Buchanan TA, Xiang AH, Page KA. Gestational diabetes mellitus: risks and management during and after pregnancy. Nat Rev Endocrinol. 2012;8(11):639-649.