User login
Know the 15% rule in scleroderma
MAUI, HAWAII – The 15% rule in scleroderma is a handy tool that raises awareness of the disease’s associated prevalence of various severe organ complications so clinicians can screen appropriately, Janet Pope, MD, said at the 2020 Rheumatology Winter Clinical Symposium.

Dr. Pope and colleagues in the Canadian Scleroderma Research Group developed the 15% rule because they recognized that scleroderma is rare enough that most physicians practicing outside of a few specialized centers don’t see many affected patients. The systemic autoimmune disease is marked by numerous possible expressions of vascular inflammation and malfunction, fibrosis, and autoimmunity in different organ systems.
“A lot of clinicians do not know how common this stuff is,” according to Dr. Pope, professor of medicine at the University of Western Ontario and head of the division of rheumatology at St. Joseph’s Health Center in London, Ont.
Basically, the 15% rule holds that, at any given time, a patient with scleroderma has roughly a 15% chance – or one in six – of having any of an extensive array of severe organ complications. That means a 15% chance of having prevalent clinically significant pulmonary hypertension as defined by a systolic pulmonary artery pressure of 45 mm Hg or more on Doppler echocardiography, a 15% likelihood of interstitial lung disease or clinically significant pulmonary fibrosis as suggested by a forced vital capacity less than 70% of predicted, a 15% prevalence of Sjögren’s syndrome, a 15% likelihood of having pulmonary artery hypertension upon right heart catheterization, a 15% chance of inflammatory arthritis, and a one-in-six chance of having a myopathy or myositis. Also, diastolic dysfunction, 15%. Ditto symptomatic arrhythmias.
“It’s a good little rule of thumb,” Dr. Pope commented.
The odds of having a current digital ulcer on any given day? Again, about 15%. In addition, scleroderma patients have a 15% lifetime risk of developing a complicated digital ulcer requiring hospitalization and/or amputation, she continued.
And while the prevalence of scleroderma renal crisis in the overall population with scleroderma is low, at 3%, in the subgroup with diffuse cutaneous systemic sclerosis, it climbs to 12%-15%.
Every rule has its exceptions. The 15% rule doesn’t apply to Raynaud’s phenomenon, which is present in nearly all patients with scleroderma, nor to gastroesophageal reflux disease or dysphagia, present in roughly 80% of patients.
Dr. Pope and coinvestigators developed the 15% rule pertaining to the prevalence of serious organ complications in scleroderma by conducting a systematic review of 69 published studies, each including a minimum of 50 scleroderma patients. The detailed results of the systematic review have been published.
Dr. Pope reported receiving research grants from and/or serving as a consultant to more than a dozen pharmaceutical companies.
MAUI, HAWAII – The 15% rule in scleroderma is a handy tool that raises awareness of the disease’s associated prevalence of various severe organ complications so clinicians can screen appropriately, Janet Pope, MD, said at the 2020 Rheumatology Winter Clinical Symposium.
Dr. Pope and colleagues in the Canadian Scleroderma Research Group developed the 15% rule because they recognized that scleroderma is rare enough that most physicians practicing outside of a few specialized centers don’t see many affected patients. The systemic autoimmune disease is marked by numerous possible expressions of vascular inflammation and malfunction, fibrosis, and autoimmunity in different organ systems.
“A lot of clinicians do not know how common this stuff is,” according to Dr. Pope, professor of medicine at the University of Western Ontario and head of the division of rheumatology at St. Joseph’s Health Center in London, Ont.
Basically, the 15% rule holds that, at any given time, a patient with scleroderma has roughly a 15% chance – or one in six – of having any of an extensive array of severe organ complications. That means a 15% chance of having prevalent clinically significant pulmonary hypertension as defined by a systolic pulmonary artery pressure of 45 mm Hg or more on Doppler echocardiography, a 15% likelihood of interstitial lung disease or clinically significant pulmonary fibrosis as suggested by a forced vital capacity less than 70% of predicted, a 15% prevalence of Sjögren’s syndrome, a 15% likelihood of having pulmonary artery hypertension upon right heart catheterization, a 15% chance of inflammatory arthritis, and a one-in-six chance of having a myopathy or myositis. Also, diastolic dysfunction, 15%. Ditto symptomatic arrhythmias.
“It’s a good little rule of thumb,” Dr. Pope commented.
The odds of having a current digital ulcer on any given day? Again, about 15%. In addition, scleroderma patients have a 15% lifetime risk of developing a complicated digital ulcer requiring hospitalization and/or amputation, she continued.
And while the prevalence of scleroderma renal crisis in the overall population with scleroderma is low, at 3%, in the subgroup with diffuse cutaneous systemic sclerosis, it climbs to 12%-15%.
Every rule has its exceptions. The 15% rule doesn’t apply to Raynaud’s phenomenon, which is present in nearly all patients with scleroderma, nor to gastroesophageal reflux disease or dysphagia, present in roughly 80% of patients.
Dr. Pope and coinvestigators developed the 15% rule pertaining to the prevalence of serious organ complications in scleroderma by conducting a systematic review of 69 published studies, each including a minimum of 50 scleroderma patients. The detailed results of the systematic review have been published.
Dr. Pope reported receiving research grants from and/or serving as a consultant to more than a dozen pharmaceutical companies.
MAUI, HAWAII – The 15% rule in scleroderma is a handy tool that raises awareness of the disease’s associated prevalence of various severe organ complications so clinicians can screen appropriately, Janet Pope, MD, said at the 2020 Rheumatology Winter Clinical Symposium.
Dr. Pope and colleagues in the Canadian Scleroderma Research Group developed the 15% rule because they recognized that scleroderma is rare enough that most physicians practicing outside of a few specialized centers don’t see many affected patients. The systemic autoimmune disease is marked by numerous possible expressions of vascular inflammation and malfunction, fibrosis, and autoimmunity in different organ systems.
“A lot of clinicians do not know how common this stuff is,” according to Dr. Pope, professor of medicine at the University of Western Ontario and head of the division of rheumatology at St. Joseph’s Health Center in London, Ont.
Basically, the 15% rule holds that, at any given time, a patient with scleroderma has roughly a 15% chance – or one in six – of having any of an extensive array of severe organ complications. That means a 15% chance of having prevalent clinically significant pulmonary hypertension as defined by a systolic pulmonary artery pressure of 45 mm Hg or more on Doppler echocardiography, a 15% likelihood of interstitial lung disease or clinically significant pulmonary fibrosis as suggested by a forced vital capacity less than 70% of predicted, a 15% prevalence of Sjögren’s syndrome, a 15% likelihood of having pulmonary artery hypertension upon right heart catheterization, a 15% chance of inflammatory arthritis, and a one-in-six chance of having a myopathy or myositis. Also, diastolic dysfunction, 15%. Ditto symptomatic arrhythmias.
“It’s a good little rule of thumb,” Dr. Pope commented.
The odds of having a current digital ulcer on any given day? Again, about 15%. In addition, scleroderma patients have a 15% lifetime risk of developing a complicated digital ulcer requiring hospitalization and/or amputation, she continued.
And while the prevalence of scleroderma renal crisis in the overall population with scleroderma is low, at 3%, in the subgroup with diffuse cutaneous systemic sclerosis, it climbs to 12%-15%.
Every rule has its exceptions. The 15% rule doesn’t apply to Raynaud’s phenomenon, which is present in nearly all patients with scleroderma, nor to gastroesophageal reflux disease or dysphagia, present in roughly 80% of patients.
Dr. Pope and coinvestigators developed the 15% rule pertaining to the prevalence of serious organ complications in scleroderma by conducting a systematic review of 69 published studies, each including a minimum of 50 scleroderma patients. The detailed results of the systematic review have been published.
Dr. Pope reported receiving research grants from and/or serving as a consultant to more than a dozen pharmaceutical companies.
REPORTING FROM RWCS 2020
Meta-analysis eyes impact of adherence to HCQ among SLE patients
Low serum levels of hydroxychloroquine (HCQ) among patients with systemic lupus erythematosus are associated with a threefold increased likelihood of physician- and patient-reported nonadherence to the medication. In addition, routine monitoring of HCQ levels are associated with improvements in adherence and disease activity.
Those are two key findings from a systematic review and meta-analysis published in Arthritis Care & Research.
“HCQ is recommended for all patients with systemic lupus erythematosus (SLE, or lupus) to reduce disease activity and improve damage-free-survival,” the authors, led by Shivani Garg, MD, of the University of Wisconsin–Madison, wrote in the article. “Yet, up to 83% of lupus patients are nonadherent to HCQ commonly because of poor understanding of benefits of HCQ, lack of motivation to continue therapy, and inflated concerns regarding side effects from HCQ use.”
For their analysis, the researchers drew from 17 published observational and interventional studies that measured HCQ levels and assessed adherence or Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) in adults with SLE. They used forest plots to compare pooled estimates of correlations between HCQ levels and reported nonadherence, or SLEDAI scores. Patient-reported nonadherence was defined as less than 80% medication adherence reported, and physician-reported adherence was estimated based on physicians’ interpretations of the previous month’s adherence as reported by patients during clinic visits.
The study population consisted of 1,223 patients. Dr. Garg and colleagues found a threefold higher odds of reported nonadherence in patients with low HCQ levels (odds ratio, 2.95; P less than .001). The mean SLEDAI score was 3.14 points higher in a group with below-threshold HCQ levels on a priori analysis (P = .053), and 1.4 points higher in a group with HCQ levels below 500 ng/mL (P = .039). Among all patients, those with HCQ levels 750 ng/mL or greater had a 58% lower risk of active disease, and their SLEDAI score was 3.2 points lower. “Our study support levels greater than or equal to 750 ng/mL to be clinically meaningful and statistically significant to identify disease flare (change in SLEDAI greater than or equal to 3 points) and predict active disease (SLEDAI greater than or equal to 6),” the authors wrote.

In an interview, Michelle A. Petri, MD, MPH, took issue with the HCQ goal of 750 ng/mL or greater recommended by the authors. “I think that was premature,” said Dr. Petri, professor of medicine at Johns Hopkins University, Baltimore. “We presented data at last year’s ACR [which showed] that the level needs to be higher than that to prevent thrombosis. But it is important to open the discussion that HCQ blood levels are not just for nonadherence. I believe they will help us to reduce retinopathy, and also to make sure the dose remains in an efficacious range, such as what is needed to prevent thrombosis.”
Dr. Petri, who also directs the Hopkins Lupus Center, said that the study’s overall conclusions confirms the need for blood testing for HCQ to identify nonadherence. “Everyone remembers the saying of the [former] Surgeon General Dr. C. Everett Koop: ‘Drugs can’t work if patients don’t take them!’ – in particular, blood levels which represent what the patient has taken in the last month. I call blood levels the ‘lupus A1C.’ ”
She added that HCQ blood levels have utility for nonadherence, prediction of retinopathy, and prevention of thrombosis. Such tests “are now much more widely available, including by some large national laboratories such as Quest Diagnostics, as well as by Exagen. No more excuses.” LabCorp plans to start offering HCQ blood level testing by the middle of 2020, she said.
In their manuscript, the study authors acknowledged certain limitations of their analysis, including the fact that there were only four studies that measured HCQ levels and nonadherence or SLEDAI. “Second, most of the studies that examined the correlation between reported adherence and HCQ blood levels were performed in Europe, and there was only one small U.S. study,” they wrote. “Therefore, generalizability for our findings could be limited because of differences in cultural beliefs, social issues, and insurance/medical coverage in populations from diverse countries.”
The study authors reported having no disclosures. Dr. Petri disclosed that she has conducted research on HCQ that was funded by the National Institutes of Health. She has also conducted research for Exagen.
SOURCE: Garg S et al. Arthritis Care Res. 2020 Jan 31. doi: 10.1002/acr.24155.
Low serum levels of hydroxychloroquine (HCQ) among patients with systemic lupus erythematosus are associated with a threefold increased likelihood of physician- and patient-reported nonadherence to the medication. In addition, routine monitoring of HCQ levels are associated with improvements in adherence and disease activity.
Those are two key findings from a systematic review and meta-analysis published in Arthritis Care & Research.
“HCQ is recommended for all patients with systemic lupus erythematosus (SLE, or lupus) to reduce disease activity and improve damage-free-survival,” the authors, led by Shivani Garg, MD, of the University of Wisconsin–Madison, wrote in the article. “Yet, up to 83% of lupus patients are nonadherent to HCQ commonly because of poor understanding of benefits of HCQ, lack of motivation to continue therapy, and inflated concerns regarding side effects from HCQ use.”
For their analysis, the researchers drew from 17 published observational and interventional studies that measured HCQ levels and assessed adherence or Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) in adults with SLE. They used forest plots to compare pooled estimates of correlations between HCQ levels and reported nonadherence, or SLEDAI scores. Patient-reported nonadherence was defined as less than 80% medication adherence reported, and physician-reported adherence was estimated based on physicians’ interpretations of the previous month’s adherence as reported by patients during clinic visits.
The study population consisted of 1,223 patients. Dr. Garg and colleagues found a threefold higher odds of reported nonadherence in patients with low HCQ levels (odds ratio, 2.95; P less than .001). The mean SLEDAI score was 3.14 points higher in a group with below-threshold HCQ levels on a priori analysis (P = .053), and 1.4 points higher in a group with HCQ levels below 500 ng/mL (P = .039). Among all patients, those with HCQ levels 750 ng/mL or greater had a 58% lower risk of active disease, and their SLEDAI score was 3.2 points lower. “Our study support levels greater than or equal to 750 ng/mL to be clinically meaningful and statistically significant to identify disease flare (change in SLEDAI greater than or equal to 3 points) and predict active disease (SLEDAI greater than or equal to 6),” the authors wrote.
In an interview, Michelle A. Petri, MD, MPH, took issue with the HCQ goal of 750 ng/mL or greater recommended by the authors. “I think that was premature,” said Dr. Petri, professor of medicine at Johns Hopkins University, Baltimore. “We presented data at last year’s ACR [which showed] that the level needs to be higher than that to prevent thrombosis. But it is important to open the discussion that HCQ blood levels are not just for nonadherence. I believe they will help us to reduce retinopathy, and also to make sure the dose remains in an efficacious range, such as what is needed to prevent thrombosis.”
Dr. Petri, who also directs the Hopkins Lupus Center, said that the study’s overall conclusions confirms the need for blood testing for HCQ to identify nonadherence. “Everyone remembers the saying of the [former] Surgeon General Dr. C. Everett Koop: ‘Drugs can’t work if patients don’t take them!’ – in particular, blood levels which represent what the patient has taken in the last month. I call blood levels the ‘lupus A1C.’ ”
She added that HCQ blood levels have utility for nonadherence, prediction of retinopathy, and prevention of thrombosis. Such tests “are now much more widely available, including by some large national laboratories such as Quest Diagnostics, as well as by Exagen. No more excuses.” LabCorp plans to start offering HCQ blood level testing by the middle of 2020, she said.
In their manuscript, the study authors acknowledged certain limitations of their analysis, including the fact that there were only four studies that measured HCQ levels and nonadherence or SLEDAI. “Second, most of the studies that examined the correlation between reported adherence and HCQ blood levels were performed in Europe, and there was only one small U.S. study,” they wrote. “Therefore, generalizability for our findings could be limited because of differences in cultural beliefs, social issues, and insurance/medical coverage in populations from diverse countries.”
The study authors reported having no disclosures. Dr. Petri disclosed that she has conducted research on HCQ that was funded by the National Institutes of Health. She has also conducted research for Exagen.
SOURCE: Garg S et al. Arthritis Care Res. 2020 Jan 31. doi: 10.1002/acr.24155.
Low serum levels of hydroxychloroquine (HCQ) among patients with systemic lupus erythematosus are associated with a threefold increased likelihood of physician- and patient-reported nonadherence to the medication. In addition, routine monitoring of HCQ levels are associated with improvements in adherence and disease activity.
Those are two key findings from a systematic review and meta-analysis published in Arthritis Care & Research.
“HCQ is recommended for all patients with systemic lupus erythematosus (SLE, or lupus) to reduce disease activity and improve damage-free-survival,” the authors, led by Shivani Garg, MD, of the University of Wisconsin–Madison, wrote in the article. “Yet, up to 83% of lupus patients are nonadherent to HCQ commonly because of poor understanding of benefits of HCQ, lack of motivation to continue therapy, and inflated concerns regarding side effects from HCQ use.”
For their analysis, the researchers drew from 17 published observational and interventional studies that measured HCQ levels and assessed adherence or Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) in adults with SLE. They used forest plots to compare pooled estimates of correlations between HCQ levels and reported nonadherence, or SLEDAI scores. Patient-reported nonadherence was defined as less than 80% medication adherence reported, and physician-reported adherence was estimated based on physicians’ interpretations of the previous month’s adherence as reported by patients during clinic visits.
The study population consisted of 1,223 patients. Dr. Garg and colleagues found a threefold higher odds of reported nonadherence in patients with low HCQ levels (odds ratio, 2.95; P less than .001). The mean SLEDAI score was 3.14 points higher in a group with below-threshold HCQ levels on a priori analysis (P = .053), and 1.4 points higher in a group with HCQ levels below 500 ng/mL (P = .039). Among all patients, those with HCQ levels 750 ng/mL or greater had a 58% lower risk of active disease, and their SLEDAI score was 3.2 points lower. “Our study support levels greater than or equal to 750 ng/mL to be clinically meaningful and statistically significant to identify disease flare (change in SLEDAI greater than or equal to 3 points) and predict active disease (SLEDAI greater than or equal to 6),” the authors wrote.
In an interview, Michelle A. Petri, MD, MPH, took issue with the HCQ goal of 750 ng/mL or greater recommended by the authors. “I think that was premature,” said Dr. Petri, professor of medicine at Johns Hopkins University, Baltimore. “We presented data at last year’s ACR [which showed] that the level needs to be higher than that to prevent thrombosis. But it is important to open the discussion that HCQ blood levels are not just for nonadherence. I believe they will help us to reduce retinopathy, and also to make sure the dose remains in an efficacious range, such as what is needed to prevent thrombosis.”
Dr. Petri, who also directs the Hopkins Lupus Center, said that the study’s overall conclusions confirms the need for blood testing for HCQ to identify nonadherence. “Everyone remembers the saying of the [former] Surgeon General Dr. C. Everett Koop: ‘Drugs can’t work if patients don’t take them!’ – in particular, blood levels which represent what the patient has taken in the last month. I call blood levels the ‘lupus A1C.’ ”
She added that HCQ blood levels have utility for nonadherence, prediction of retinopathy, and prevention of thrombosis. Such tests “are now much more widely available, including by some large national laboratories such as Quest Diagnostics, as well as by Exagen. No more excuses.” LabCorp plans to start offering HCQ blood level testing by the middle of 2020, she said.
In their manuscript, the study authors acknowledged certain limitations of their analysis, including the fact that there were only four studies that measured HCQ levels and nonadherence or SLEDAI. “Second, most of the studies that examined the correlation between reported adherence and HCQ blood levels were performed in Europe, and there was only one small U.S. study,” they wrote. “Therefore, generalizability for our findings could be limited because of differences in cultural beliefs, social issues, and insurance/medical coverage in populations from diverse countries.”
The study authors reported having no disclosures. Dr. Petri disclosed that she has conducted research on HCQ that was funded by the National Institutes of Health. She has also conducted research for Exagen.
SOURCE: Garg S et al. Arthritis Care Res. 2020 Jan 31. doi: 10.1002/acr.24155.
FROM ARTHRITIS CARE & RESEARCH
February 2020
Subacute cutaneous lupus erythematosus
Subacute cutaneous lupus erythematosus (SCLE) is a type of cutaneous lupus erythematosus that may occur independently of or in combination with systemic lupus erythematosus. About 10%-15% of patients with SCLE will develop systemic lupus erythematosus. White females are more typically affected.
SCLE lesions often present as scaly, annular, or polycyclic scaly patches and plaques with central clearing. They may appear psoriasiform. They heal without atrophy or scarring but may leave dyspigmentation. Follicular plugging is absent. Lesions generally occur on sun exposed areas such as the neck, V of the chest, and upper extremities. Up to 75% of patients may exhibit associated symptoms such as photosensitivity, oral ulcers, and arthritis. Less than 20% of patients will develop internal disease, including nephritis and pulmonary disease. Symptoms of Sjögren’s syndrome and SCLE may overlap in some patients, and will portend higher risk for internal disease.
The differential diagnosis includes eczema, psoriasis, dermatophytosis, granuloma annulare, and erythema annulare centrifugum. Histology reveals epidermal atrophy and keratinocyte apoptosis, with a superficial and perivascular lymphohistiocytic infiltrate in the upper dermis. Interface changes at the dermal-epidermal junction can be seen. Direct immunofluorescence of lesional skin is positive in one-third of cases, often revealing granular deposits of IgG and IgM at the dermal-epidermal junction and around hair follicles (called the lupus-band test). Serology in SCLE may reveal a positive antinuclear antigen test, as well as positive Ro/SSA antigen. Other lupus serologies such as La/SSB, dsDNA, antihistone, and Sm antibodies may be positive, but are less commonly seen.

Several drugs may cause SCLE, such as hydrochlorothiazide, terbinafine, ACE inhibitors, NSAIDs, calcium-channel blockers, interferons, anticonvulsants, griseofulvin, penicillamine, spironolactone, tumor necrosis factor–alpha inhibitors, and statins. Discontinuing the offending medications may clear the lesions, but not always.
Treatment includes sunscreen and avoidance of sun exposure. Potent topical corticosteroids are helpful. If systemic treatment is indicated, antimalarials are first line.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
Subacute cutaneous lupus erythematosus
Subacute cutaneous lupus erythematosus (SCLE) is a type of cutaneous lupus erythematosus that may occur independently of or in combination with systemic lupus erythematosus. About 10%-15% of patients with SCLE will develop systemic lupus erythematosus. White females are more typically affected.
SCLE lesions often present as scaly, annular, or polycyclic scaly patches and plaques with central clearing. They may appear psoriasiform. They heal without atrophy or scarring but may leave dyspigmentation. Follicular plugging is absent. Lesions generally occur on sun exposed areas such as the neck, V of the chest, and upper extremities. Up to 75% of patients may exhibit associated symptoms such as photosensitivity, oral ulcers, and arthritis. Less than 20% of patients will develop internal disease, including nephritis and pulmonary disease. Symptoms of Sjögren’s syndrome and SCLE may overlap in some patients, and will portend higher risk for internal disease.
The differential diagnosis includes eczema, psoriasis, dermatophytosis, granuloma annulare, and erythema annulare centrifugum. Histology reveals epidermal atrophy and keratinocyte apoptosis, with a superficial and perivascular lymphohistiocytic infiltrate in the upper dermis. Interface changes at the dermal-epidermal junction can be seen. Direct immunofluorescence of lesional skin is positive in one-third of cases, often revealing granular deposits of IgG and IgM at the dermal-epidermal junction and around hair follicles (called the lupus-band test). Serology in SCLE may reveal a positive antinuclear antigen test, as well as positive Ro/SSA antigen. Other lupus serologies such as La/SSB, dsDNA, antihistone, and Sm antibodies may be positive, but are less commonly seen.
Several drugs may cause SCLE, such as hydrochlorothiazide, terbinafine, ACE inhibitors, NSAIDs, calcium-channel blockers, interferons, anticonvulsants, griseofulvin, penicillamine, spironolactone, tumor necrosis factor–alpha inhibitors, and statins. Discontinuing the offending medications may clear the lesions, but not always.
Treatment includes sunscreen and avoidance of sun exposure. Potent topical corticosteroids are helpful. If systemic treatment is indicated, antimalarials are first line.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
Subacute cutaneous lupus erythematosus
Subacute cutaneous lupus erythematosus (SCLE) is a type of cutaneous lupus erythematosus that may occur independently of or in combination with systemic lupus erythematosus. About 10%-15% of patients with SCLE will develop systemic lupus erythematosus. White females are more typically affected.
SCLE lesions often present as scaly, annular, or polycyclic scaly patches and plaques with central clearing. They may appear psoriasiform. They heal without atrophy or scarring but may leave dyspigmentation. Follicular plugging is absent. Lesions generally occur on sun exposed areas such as the neck, V of the chest, and upper extremities. Up to 75% of patients may exhibit associated symptoms such as photosensitivity, oral ulcers, and arthritis. Less than 20% of patients will develop internal disease, including nephritis and pulmonary disease. Symptoms of Sjögren’s syndrome and SCLE may overlap in some patients, and will portend higher risk for internal disease.
The differential diagnosis includes eczema, psoriasis, dermatophytosis, granuloma annulare, and erythema annulare centrifugum. Histology reveals epidermal atrophy and keratinocyte apoptosis, with a superficial and perivascular lymphohistiocytic infiltrate in the upper dermis. Interface changes at the dermal-epidermal junction can be seen. Direct immunofluorescence of lesional skin is positive in one-third of cases, often revealing granular deposits of IgG and IgM at the dermal-epidermal junction and around hair follicles (called the lupus-band test). Serology in SCLE may reveal a positive antinuclear antigen test, as well as positive Ro/SSA antigen. Other lupus serologies such as La/SSB, dsDNA, antihistone, and Sm antibodies may be positive, but are less commonly seen.
Several drugs may cause SCLE, such as hydrochlorothiazide, terbinafine, ACE inhibitors, NSAIDs, calcium-channel blockers, interferons, anticonvulsants, griseofulvin, penicillamine, spironolactone, tumor necrosis factor–alpha inhibitors, and statins. Discontinuing the offending medications may clear the lesions, but not always.
Treatment includes sunscreen and avoidance of sun exposure. Potent topical corticosteroids are helpful. If systemic treatment is indicated, antimalarials are first line.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
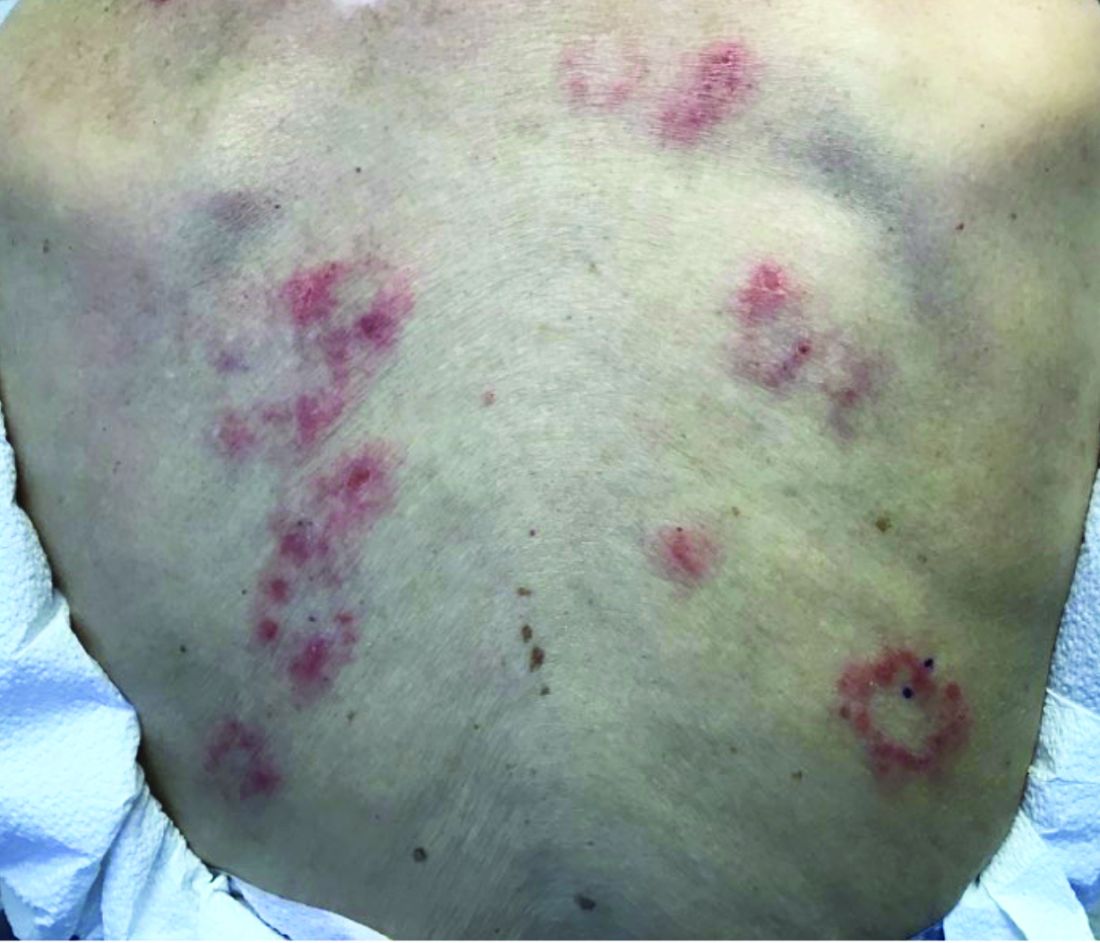
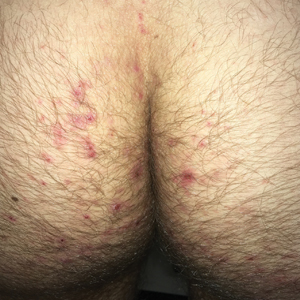
Persistent Pruritic Papules on the Buttocks
The Diagnosis: Dermatitis Herpetiformis
Dermatitis herpetiformis (DH), also known as Duhring disease, is a rare autoimmune disease. It is the cutaneous manifestation of gluten sensitivity with antibodies targeting epidermal transglutaminase.1 Symptoms of DH generally arise in the fourth decade of life, and children are less commonly affected,2 though the diagnosis should be considered at any age, as our patient was aged 19 years at the time of presentation. Dermatitis herpetiformis predominantly affects white individuals of Northern European heritage, and males more often are affected than females.2 There is a strong association with HLA-B8, HLA-DR3, and HLA-DQw2.3 It also is associated with other autoimmune disorders, including autoimmune thyroid disease, type 1 diabetes mellitus, and pernicious anemia.2
Clinically, DH is characterized by groups of intensely pruritic papulovesicles that are symmetrically located on the extensor surfaces of the upper and lower extremities, scalp, nuchal area, back, and buttocks (quiz image). Less often, the groin also may be involved, as it was in our patient (Figure 1).4 Lesions can be papulovesicular or bullous, though they often are excoriated, and primary lesions may be difficult to identify.2 The disease may have spontaneous remissions with frequent relapses. Most patients with DH have an asymptomatic gluten-sensitive enteropathy.3

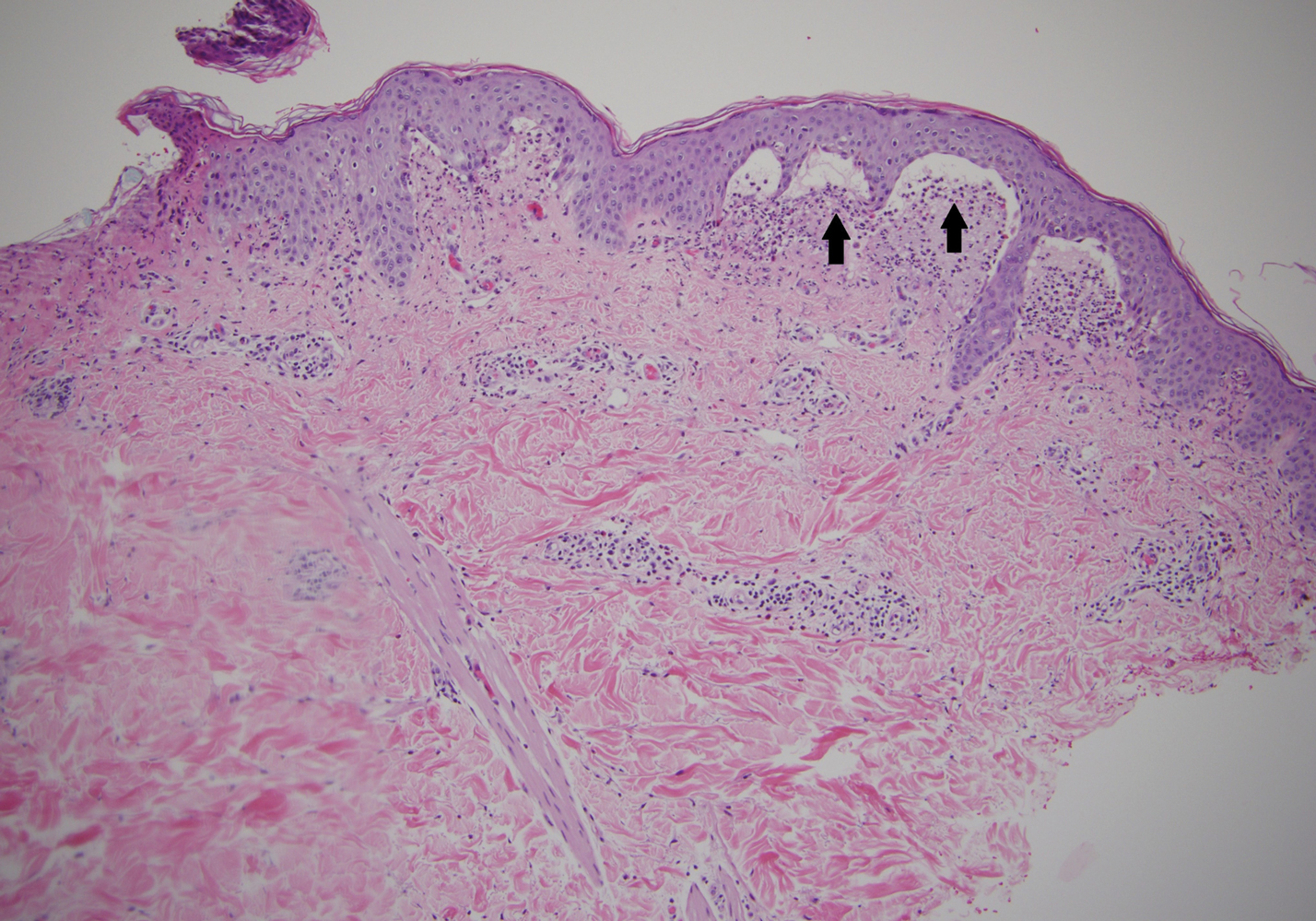
A punch biopsy of a representative nonexcoriated lesion from our patient showed the characteristic collections of neutrophils and fibrin at the tips of dermal papillae on hematoxylin and eosin staining (Figure 2). These findings are suggestive of DH.1 However, other bullous diseases, such as linear IgA dermatosis and epidermolysis bullosa acquisita, may have similar appearance on histology.1 Direct immunofluorescence (DIF) of perilesional skin is the gold standard for diagnosing DH, with a sensitivity and specificity close to 100%.1 Deposits of IgA generally are concentrated in previously involved skin or noninflamed perilesional skin; DIF of erythematous or lesional skin may be false negative.5 In our patient, DIF of perilesional uninvolved skin showed granular deposits of IgA at the dermoepidermal junction with accentuation in the dermal papillae (Figure 3), further suggestive of the diagnosis of DH.6

Patients with DH frequently will have specific IgA antibodies including anti-tissue transglutaminase (tTG), anti-epidermal transglutaminase, antiendomysial antibodies, and anti-synthetic deamidated gliadin peptides.1 Only some of these tests are widely available, and the anti-tTG enzyme-linked immunosorbent assay is the least expensive and easiest to perform. A positive anti-tTG antibody test has a sensitivity ranging from 47% to 95% and a specificity greater than 90%.1 Our patient tested positive for anti-tTG and anti-deamidated synthetic gliadin-derived peptides. A diagnostic algorithm based on current evidence suggests that in patients with clinical evidence of DH, typical DIF findings combined with positive anti-tTG antibodies confirms the diagnosis of DH, as was seen in our patient.1,7 In situations where histopathology and/or antibody testing are inconclusive, additional testing to include HLA antigen typing, duodenal biopsy, and supplemental skin biopsies may be performed to help confirm or exclude a diagnosis of DH. It is unnecessary to perform a duodenal biopsy in patients with a confirmed diagnosis of DH, as DH is considered the specific cutaneous manifestation of celiac disease, and the diagnosis of DH is a diagnosis of celiac disease.1
Although pruritic papules may be found in several conditions, clinical, histopathologic, and DIF findings can help to confirm the diagnosis. Allergic contact dermatitis is a type IV hypersensitivity reaction causing eruptions of varying morphology, generally manifesting as pruritic scaly plaques that can occur anywhere on the body exposed to the offending allergen. Key histopathologic features of allergic contact dermatitis are eosinophilic spongiosis and exocytosis of eosinophils and lymphocytes.8 Papular urticaria is a hypersensitivity disorder to insect bites that consists of pruritic papules on exposed areas of skin, typically in children younger than 10 years. Genital and axillary areas usually are spared. The diagnosis is clinical.9 Recurrent herpes simplex virus infection is a short-lived outbreak that generally improves within 10 days. Herpes simplex virus infections usually are comprised of small vesicular or ulcerative lesions. Tzanck smear, skin biopsy, direct fluorescent antibody, viral culture, and polymerase chain reaction are diagnostic methods for herpes simplex virus.10 Scabies lesions typically are pruritic, erythematous, often excoriated papules and burrows located most commonly in the webs of fingers, wrists, axillae, areolae, waist, and genitalia. Diagnosis can be confirmed with scabies preparation (skin scraping showing mites, eggs, or feces), dermoscopy showing mites and burrows in vivo, or biopsy.11
First-line treatment in patients with DH and celiac disease is a strict gluten-free diet (GFD).1 A GFD will resolve the cutaneous and gastrointestinal manifestations and is the only thing that will reduce the risk for lymphoma and other diseases associated with gluten-induced enteropathy.1,2 A GFD alone will provide symptomatic relief over several months; dapsone and sulfapyridine can provide rapid relief of the pruritus and skin manifestations and usually can be weaned or discontinued after several months of following a strict GFD.12 Patients on sulfone therapy require regular follow-up and monitoring due to the risk for hemolytic anemia and other adverse effects as well as to determine the appropriate time to discontinue the medication.12 Although some patients are able to tolerate reintroduction of gluten into their diets after a period of remission, most will experience recurrent dermatologic manifestations if they continue to consume gluten.3
Because of our patient's impending move out of the area, no oral medications were started, and he was instructed to follow a GFD and seek medical care at his new location. The patient was contacted 6 months later and reported resolution of all skin lesions with just a GFD. The patient continued to follow-up with a dermatologist and gastroenterologist at his new location.
- Antiga E, Caproni M. The diagnosis and treatment of dermatitis herpetiformis. Clin Cosmet Investig Dermatol. 2015;8:257-265.
- Bolognia JL, Jorizzo JL, Schaffer JV. Dermatitis herpetiformis and linear IgA bullous dermatosis. In: Bolognia JL, Schaffer JV, Cerroni L. Dermatology. Vol 1. 4th ed. Philadelphia, PA: Elsevier; 2017:527-537.
- James WD, Elston DM, Treat JR, et al. Chronic blistering dermatoses. In: James WD, Elston DM, Treat JR, et al, eds. Andrews' Diseases of the Skin: Clinical Dermatology. 13th ed. Philadelphia, PA: Elsevier; 2020:453-474.
- Bolotin D, Petronic-Rosic V. Dermatitis herpetiformis. part 1. epidemiology, pathogenesis, and clinical presentation. J Am Acad Dermatol. 2011;64:1017-1024.
- Zone JJ, Meyer LJ, Petersen MJ. Deposition of granular IgA relative to clinical lesions in dermatitis herpetiformis. Arch Dermatol. 1996;132:912-918.
- Lever WF, Elder DE. Lever's Histopathology of the Skin. Vol 1. Philadelphia, PA: Wolters Kluwer Health/Lippincott; 2009.
- Hull C. Dermatitis herpetiformis. https://www.uptodate.com/contents/dermatitis-herpetiformis. Published October 14, 2016. Updated September 25, 2019. Accessed December 10, 2019.
Yiannias J. Clinical features and diagnosis of allergic contact dermatitis. UpToDate. https://www.uptodate.com/contents/clinical-features-and-diagnosis-of-allergic-contact-dermatitissearch=allergic%20contact%20dermatitis&source=search_result&selectedTitle=2~150
&usage_type=default&display_rank=2#H27385290. Updated May 17, 2019. Accessed December 11, 2019.Goddard J, Stewart PH. Insect and other arthropod bites. UpToDate. https://www.uptodate.com/contents/insect-and-other-arthropod-bites?search=papular%2urticaria§ionRank=1&usage_type=default&anchor=H4&source=machineLearning&selectedTitle=1~24&display_rank=1#
H4. Updated October 31, 2019. Accessed December 11, 2019.Christine J, Wald A. Epidemiology, clinical manifestations, and diagnosis of herpes simplex virus type 1 infection. UpToDate. https://www.uptodate.com/contents/epidemiology-clinical-manifestations-and-diagnosis-of-herpes-simplex-virus-type-1-infection?search=herpes%20simplex&source=search_result&selectedTitle=1~150&usage_type=default&display_rank=1. Updated July 23, 2019. Accessed December 11, 2019.
- Goldstein B, Goldstein A. Scabies: epidemiology, clinical features, and diagnosis. UpToDate. https://www.uptodate.com/contents/scabies-epidemiology-clinical-features-and-diagnosis?search=scabies&source=search_result&selectedTitle=1~92&usagetype=default&display_rank=1. Updated August 2, 2019. Accessed December 11, 2019.
- Bolotin D, Petronic-Rosic V. Dermatitis herpetiformis. part 2. diagnosis, management, and prognosis. J Am Acad Dermatol. 2011;64:1027-1033.
The Diagnosis: Dermatitis Herpetiformis
Dermatitis herpetiformis (DH), also known as Duhring disease, is a rare autoimmune disease. It is the cutaneous manifestation of gluten sensitivity with antibodies targeting epidermal transglutaminase.1 Symptoms of DH generally arise in the fourth decade of life, and children are less commonly affected,2 though the diagnosis should be considered at any age, as our patient was aged 19 years at the time of presentation. Dermatitis herpetiformis predominantly affects white individuals of Northern European heritage, and males more often are affected than females.2 There is a strong association with HLA-B8, HLA-DR3, and HLA-DQw2.3 It also is associated with other autoimmune disorders, including autoimmune thyroid disease, type 1 diabetes mellitus, and pernicious anemia.2
Clinically, DH is characterized by groups of intensely pruritic papulovesicles that are symmetrically located on the extensor surfaces of the upper and lower extremities, scalp, nuchal area, back, and buttocks (quiz image). Less often, the groin also may be involved, as it was in our patient (Figure 1).4 Lesions can be papulovesicular or bullous, though they often are excoriated, and primary lesions may be difficult to identify.2 The disease may have spontaneous remissions with frequent relapses. Most patients with DH have an asymptomatic gluten-sensitive enteropathy.3
A punch biopsy of a representative nonexcoriated lesion from our patient showed the characteristic collections of neutrophils and fibrin at the tips of dermal papillae on hematoxylin and eosin staining (Figure 2). These findings are suggestive of DH.1 However, other bullous diseases, such as linear IgA dermatosis and epidermolysis bullosa acquisita, may have similar appearance on histology.1 Direct immunofluorescence (DIF) of perilesional skin is the gold standard for diagnosing DH, with a sensitivity and specificity close to 100%.1 Deposits of IgA generally are concentrated in previously involved skin or noninflamed perilesional skin; DIF of erythematous or lesional skin may be false negative.5 In our patient, DIF of perilesional uninvolved skin showed granular deposits of IgA at the dermoepidermal junction with accentuation in the dermal papillae (Figure 3), further suggestive of the diagnosis of DH.6
Patients with DH frequently will have specific IgA antibodies including anti-tissue transglutaminase (tTG), anti-epidermal transglutaminase, antiendomysial antibodies, and anti-synthetic deamidated gliadin peptides.1 Only some of these tests are widely available, and the anti-tTG enzyme-linked immunosorbent assay is the least expensive and easiest to perform. A positive anti-tTG antibody test has a sensitivity ranging from 47% to 95% and a specificity greater than 90%.1 Our patient tested positive for anti-tTG and anti-deamidated synthetic gliadin-derived peptides. A diagnostic algorithm based on current evidence suggests that in patients with clinical evidence of DH, typical DIF findings combined with positive anti-tTG antibodies confirms the diagnosis of DH, as was seen in our patient.1,7 In situations where histopathology and/or antibody testing are inconclusive, additional testing to include HLA antigen typing, duodenal biopsy, and supplemental skin biopsies may be performed to help confirm or exclude a diagnosis of DH. It is unnecessary to perform a duodenal biopsy in patients with a confirmed diagnosis of DH, as DH is considered the specific cutaneous manifestation of celiac disease, and the diagnosis of DH is a diagnosis of celiac disease.1
Although pruritic papules may be found in several conditions, clinical, histopathologic, and DIF findings can help to confirm the diagnosis. Allergic contact dermatitis is a type IV hypersensitivity reaction causing eruptions of varying morphology, generally manifesting as pruritic scaly plaques that can occur anywhere on the body exposed to the offending allergen. Key histopathologic features of allergic contact dermatitis are eosinophilic spongiosis and exocytosis of eosinophils and lymphocytes.8 Papular urticaria is a hypersensitivity disorder to insect bites that consists of pruritic papules on exposed areas of skin, typically in children younger than 10 years. Genital and axillary areas usually are spared. The diagnosis is clinical.9 Recurrent herpes simplex virus infection is a short-lived outbreak that generally improves within 10 days. Herpes simplex virus infections usually are comprised of small vesicular or ulcerative lesions. Tzanck smear, skin biopsy, direct fluorescent antibody, viral culture, and polymerase chain reaction are diagnostic methods for herpes simplex virus.10 Scabies lesions typically are pruritic, erythematous, often excoriated papules and burrows located most commonly in the webs of fingers, wrists, axillae, areolae, waist, and genitalia. Diagnosis can be confirmed with scabies preparation (skin scraping showing mites, eggs, or feces), dermoscopy showing mites and burrows in vivo, or biopsy.11
First-line treatment in patients with DH and celiac disease is a strict gluten-free diet (GFD).1 A GFD will resolve the cutaneous and gastrointestinal manifestations and is the only thing that will reduce the risk for lymphoma and other diseases associated with gluten-induced enteropathy.1,2 A GFD alone will provide symptomatic relief over several months; dapsone and sulfapyridine can provide rapid relief of the pruritus and skin manifestations and usually can be weaned or discontinued after several months of following a strict GFD.12 Patients on sulfone therapy require regular follow-up and monitoring due to the risk for hemolytic anemia and other adverse effects as well as to determine the appropriate time to discontinue the medication.12 Although some patients are able to tolerate reintroduction of gluten into their diets after a period of remission, most will experience recurrent dermatologic manifestations if they continue to consume gluten.3
Because of our patient's impending move out of the area, no oral medications were started, and he was instructed to follow a GFD and seek medical care at his new location. The patient was contacted 6 months later and reported resolution of all skin lesions with just a GFD. The patient continued to follow-up with a dermatologist and gastroenterologist at his new location.
The Diagnosis: Dermatitis Herpetiformis
Dermatitis herpetiformis (DH), also known as Duhring disease, is a rare autoimmune disease. It is the cutaneous manifestation of gluten sensitivity with antibodies targeting epidermal transglutaminase.1 Symptoms of DH generally arise in the fourth decade of life, and children are less commonly affected,2 though the diagnosis should be considered at any age, as our patient was aged 19 years at the time of presentation. Dermatitis herpetiformis predominantly affects white individuals of Northern European heritage, and males more often are affected than females.2 There is a strong association with HLA-B8, HLA-DR3, and HLA-DQw2.3 It also is associated with other autoimmune disorders, including autoimmune thyroid disease, type 1 diabetes mellitus, and pernicious anemia.2
Clinically, DH is characterized by groups of intensely pruritic papulovesicles that are symmetrically located on the extensor surfaces of the upper and lower extremities, scalp, nuchal area, back, and buttocks (quiz image). Less often, the groin also may be involved, as it was in our patient (Figure 1).4 Lesions can be papulovesicular or bullous, though they often are excoriated, and primary lesions may be difficult to identify.2 The disease may have spontaneous remissions with frequent relapses. Most patients with DH have an asymptomatic gluten-sensitive enteropathy.3
A punch biopsy of a representative nonexcoriated lesion from our patient showed the characteristic collections of neutrophils and fibrin at the tips of dermal papillae on hematoxylin and eosin staining (Figure 2). These findings are suggestive of DH.1 However, other bullous diseases, such as linear IgA dermatosis and epidermolysis bullosa acquisita, may have similar appearance on histology.1 Direct immunofluorescence (DIF) of perilesional skin is the gold standard for diagnosing DH, with a sensitivity and specificity close to 100%.1 Deposits of IgA generally are concentrated in previously involved skin or noninflamed perilesional skin; DIF of erythematous or lesional skin may be false negative.5 In our patient, DIF of perilesional uninvolved skin showed granular deposits of IgA at the dermoepidermal junction with accentuation in the dermal papillae (Figure 3), further suggestive of the diagnosis of DH.6
Patients with DH frequently will have specific IgA antibodies including anti-tissue transglutaminase (tTG), anti-epidermal transglutaminase, antiendomysial antibodies, and anti-synthetic deamidated gliadin peptides.1 Only some of these tests are widely available, and the anti-tTG enzyme-linked immunosorbent assay is the least expensive and easiest to perform. A positive anti-tTG antibody test has a sensitivity ranging from 47% to 95% and a specificity greater than 90%.1 Our patient tested positive for anti-tTG and anti-deamidated synthetic gliadin-derived peptides. A diagnostic algorithm based on current evidence suggests that in patients with clinical evidence of DH, typical DIF findings combined with positive anti-tTG antibodies confirms the diagnosis of DH, as was seen in our patient.1,7 In situations where histopathology and/or antibody testing are inconclusive, additional testing to include HLA antigen typing, duodenal biopsy, and supplemental skin biopsies may be performed to help confirm or exclude a diagnosis of DH. It is unnecessary to perform a duodenal biopsy in patients with a confirmed diagnosis of DH, as DH is considered the specific cutaneous manifestation of celiac disease, and the diagnosis of DH is a diagnosis of celiac disease.1
Although pruritic papules may be found in several conditions, clinical, histopathologic, and DIF findings can help to confirm the diagnosis. Allergic contact dermatitis is a type IV hypersensitivity reaction causing eruptions of varying morphology, generally manifesting as pruritic scaly plaques that can occur anywhere on the body exposed to the offending allergen. Key histopathologic features of allergic contact dermatitis are eosinophilic spongiosis and exocytosis of eosinophils and lymphocytes.8 Papular urticaria is a hypersensitivity disorder to insect bites that consists of pruritic papules on exposed areas of skin, typically in children younger than 10 years. Genital and axillary areas usually are spared. The diagnosis is clinical.9 Recurrent herpes simplex virus infection is a short-lived outbreak that generally improves within 10 days. Herpes simplex virus infections usually are comprised of small vesicular or ulcerative lesions. Tzanck smear, skin biopsy, direct fluorescent antibody, viral culture, and polymerase chain reaction are diagnostic methods for herpes simplex virus.10 Scabies lesions typically are pruritic, erythematous, often excoriated papules and burrows located most commonly in the webs of fingers, wrists, axillae, areolae, waist, and genitalia. Diagnosis can be confirmed with scabies preparation (skin scraping showing mites, eggs, or feces), dermoscopy showing mites and burrows in vivo, or biopsy.11
First-line treatment in patients with DH and celiac disease is a strict gluten-free diet (GFD).1 A GFD will resolve the cutaneous and gastrointestinal manifestations and is the only thing that will reduce the risk for lymphoma and other diseases associated with gluten-induced enteropathy.1,2 A GFD alone will provide symptomatic relief over several months; dapsone and sulfapyridine can provide rapid relief of the pruritus and skin manifestations and usually can be weaned or discontinued after several months of following a strict GFD.12 Patients on sulfone therapy require regular follow-up and monitoring due to the risk for hemolytic anemia and other adverse effects as well as to determine the appropriate time to discontinue the medication.12 Although some patients are able to tolerate reintroduction of gluten into their diets after a period of remission, most will experience recurrent dermatologic manifestations if they continue to consume gluten.3
Because of our patient's impending move out of the area, no oral medications were started, and he was instructed to follow a GFD and seek medical care at his new location. The patient was contacted 6 months later and reported resolution of all skin lesions with just a GFD. The patient continued to follow-up with a dermatologist and gastroenterologist at his new location.
- Antiga E, Caproni M. The diagnosis and treatment of dermatitis herpetiformis. Clin Cosmet Investig Dermatol. 2015;8:257-265.
- Bolognia JL, Jorizzo JL, Schaffer JV. Dermatitis herpetiformis and linear IgA bullous dermatosis. In: Bolognia JL, Schaffer JV, Cerroni L. Dermatology. Vol 1. 4th ed. Philadelphia, PA: Elsevier; 2017:527-537.
- James WD, Elston DM, Treat JR, et al. Chronic blistering dermatoses. In: James WD, Elston DM, Treat JR, et al, eds. Andrews' Diseases of the Skin: Clinical Dermatology. 13th ed. Philadelphia, PA: Elsevier; 2020:453-474.
- Bolotin D, Petronic-Rosic V. Dermatitis herpetiformis. part 1. epidemiology, pathogenesis, and clinical presentation. J Am Acad Dermatol. 2011;64:1017-1024.
- Zone JJ, Meyer LJ, Petersen MJ. Deposition of granular IgA relative to clinical lesions in dermatitis herpetiformis. Arch Dermatol. 1996;132:912-918.
- Lever WF, Elder DE. Lever's Histopathology of the Skin. Vol 1. Philadelphia, PA: Wolters Kluwer Health/Lippincott; 2009.
- Hull C. Dermatitis herpetiformis. https://www.uptodate.com/contents/dermatitis-herpetiformis. Published October 14, 2016. Updated September 25, 2019. Accessed December 10, 2019.
Yiannias J. Clinical features and diagnosis of allergic contact dermatitis. UpToDate. https://www.uptodate.com/contents/clinical-features-and-diagnosis-of-allergic-contact-dermatitissearch=allergic%20contact%20dermatitis&source=search_result&selectedTitle=2~150
&usage_type=default&display_rank=2#H27385290. Updated May 17, 2019. Accessed December 11, 2019.Goddard J, Stewart PH. Insect and other arthropod bites. UpToDate. https://www.uptodate.com/contents/insect-and-other-arthropod-bites?search=papular%2urticaria§ionRank=1&usage_type=default&anchor=H4&source=machineLearning&selectedTitle=1~24&display_rank=1#
H4. Updated October 31, 2019. Accessed December 11, 2019.Christine J, Wald A. Epidemiology, clinical manifestations, and diagnosis of herpes simplex virus type 1 infection. UpToDate. https://www.uptodate.com/contents/epidemiology-clinical-manifestations-and-diagnosis-of-herpes-simplex-virus-type-1-infection?search=herpes%20simplex&source=search_result&selectedTitle=1~150&usage_type=default&display_rank=1. Updated July 23, 2019. Accessed December 11, 2019.
- Goldstein B, Goldstein A. Scabies: epidemiology, clinical features, and diagnosis. UpToDate. https://www.uptodate.com/contents/scabies-epidemiology-clinical-features-and-diagnosis?search=scabies&source=search_result&selectedTitle=1~92&usagetype=default&display_rank=1. Updated August 2, 2019. Accessed December 11, 2019.
- Bolotin D, Petronic-Rosic V. Dermatitis herpetiformis. part 2. diagnosis, management, and prognosis. J Am Acad Dermatol. 2011;64:1027-1033.
- Antiga E, Caproni M. The diagnosis and treatment of dermatitis herpetiformis. Clin Cosmet Investig Dermatol. 2015;8:257-265.
- Bolognia JL, Jorizzo JL, Schaffer JV. Dermatitis herpetiformis and linear IgA bullous dermatosis. In: Bolognia JL, Schaffer JV, Cerroni L. Dermatology. Vol 1. 4th ed. Philadelphia, PA: Elsevier; 2017:527-537.
- James WD, Elston DM, Treat JR, et al. Chronic blistering dermatoses. In: James WD, Elston DM, Treat JR, et al, eds. Andrews' Diseases of the Skin: Clinical Dermatology. 13th ed. Philadelphia, PA: Elsevier; 2020:453-474.
- Bolotin D, Petronic-Rosic V. Dermatitis herpetiformis. part 1. epidemiology, pathogenesis, and clinical presentation. J Am Acad Dermatol. 2011;64:1017-1024.
- Zone JJ, Meyer LJ, Petersen MJ. Deposition of granular IgA relative to clinical lesions in dermatitis herpetiformis. Arch Dermatol. 1996;132:912-918.
- Lever WF, Elder DE. Lever's Histopathology of the Skin. Vol 1. Philadelphia, PA: Wolters Kluwer Health/Lippincott; 2009.
- Hull C. Dermatitis herpetiformis. https://www.uptodate.com/contents/dermatitis-herpetiformis. Published October 14, 2016. Updated September 25, 2019. Accessed December 10, 2019.
Yiannias J. Clinical features and diagnosis of allergic contact dermatitis. UpToDate. https://www.uptodate.com/contents/clinical-features-and-diagnosis-of-allergic-contact-dermatitissearch=allergic%20contact%20dermatitis&source=search_result&selectedTitle=2~150
&usage_type=default&display_rank=2#H27385290. Updated May 17, 2019. Accessed December 11, 2019.Goddard J, Stewart PH. Insect and other arthropod bites. UpToDate. https://www.uptodate.com/contents/insect-and-other-arthropod-bites?search=papular%2urticaria§ionRank=1&usage_type=default&anchor=H4&source=machineLearning&selectedTitle=1~24&display_rank=1#
H4. Updated October 31, 2019. Accessed December 11, 2019.Christine J, Wald A. Epidemiology, clinical manifestations, and diagnosis of herpes simplex virus type 1 infection. UpToDate. https://www.uptodate.com/contents/epidemiology-clinical-manifestations-and-diagnosis-of-herpes-simplex-virus-type-1-infection?search=herpes%20simplex&source=search_result&selectedTitle=1~150&usage_type=default&display_rank=1. Updated July 23, 2019. Accessed December 11, 2019.
- Goldstein B, Goldstein A. Scabies: epidemiology, clinical features, and diagnosis. UpToDate. https://www.uptodate.com/contents/scabies-epidemiology-clinical-features-and-diagnosis?search=scabies&source=search_result&selectedTitle=1~92&usagetype=default&display_rank=1. Updated August 2, 2019. Accessed December 11, 2019.
- Bolotin D, Petronic-Rosic V. Dermatitis herpetiformis. part 2. diagnosis, management, and prognosis. J Am Acad Dermatol. 2011;64:1027-1033.
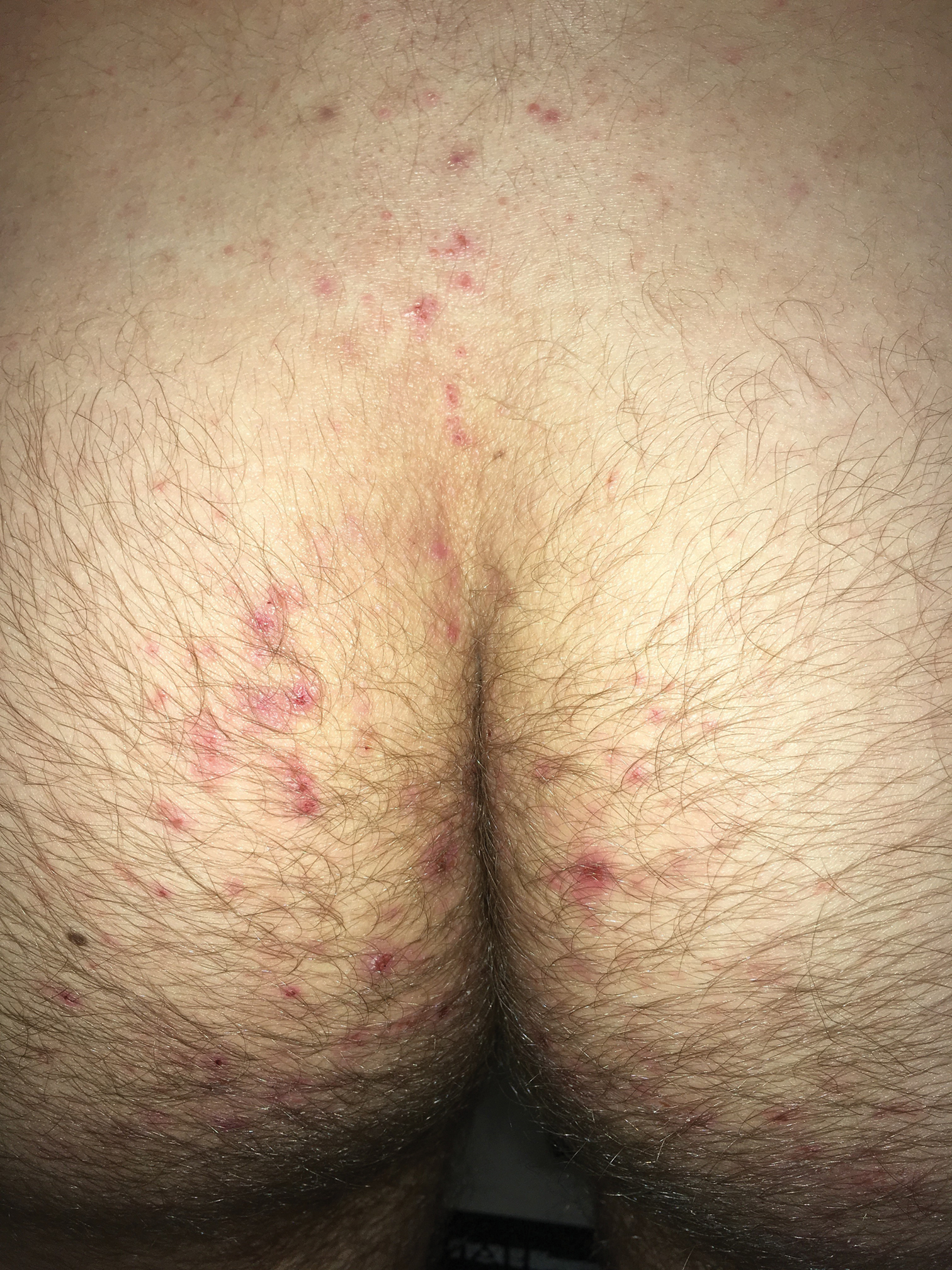
A 19-year-old man presented to the dermatology clinic with intermittent pruritic lesions that began on the bilateral buttocks when he was living in Reserve Officers' Training Corps dormitories several months prior. The eruption then spread to involve the penis, suprapubic area, periumbilical area, and flanks. The patient attempted to treat the lesions with topical antifungals prior to evaluation in the emergency department where he was treated with permethrin 5% on 2 separate occasions without any improvement. A medical history was normal, and he denied recent travel, animal contacts, or new medications. Physical examination revealed several 2- to 4-mm erythematous papules and superficial erosions with an ill-defined erythematous background most notable on the penis, suprapubic area, periumbilical area, flanks, and buttocks.
VEDOSS study describes predictors of progression to systemic sclerosis
ATLANTA – , according to recent results from the Very Early Diagnosis Of Systemic Sclerosis (VEDOSS) study presented at the annual meeting of the American College of Rheumatology.

“Our data show that thanks [to a] combination of the signs that characterize the various phases of the disease, patients can be diagnosed [with systemic sclerosis] in the very early stages,” first author Silvia Bellando-Randone, MD, PhD, assistant professor in the division of rheumatology at the University of Florence (Italy), said in her presentation.
Dr. Bellando-Randone and colleagues performed a longitudinal, observational study of 742 patients (mean 45.7 years old) at 42 centers in a cohort of mostly women (90%), nearly all of whom had had Raynaud’s phenomenon for longer than 36 months (97.5%). Patients were excluded if they had systemic sclerosis based on ACR 1980 classification criteria and/or ACR–European League Against Rheumatism 2013 criteria, had systemic sclerosis together with other connective-tissue diseases, or were unlikely to be present for three consecutive annual exams. Data collection began in March 2012 with follow-up of 5 years.
The researchers determined the positive predictive values (PPV) and negative predictive values (NPV) of clinical features, systemic sclerosis–specific antibodies, and nailfold video capillaroscopy (NVC) abnormalities on progression from Raynaud’s phenomenon to systemic sclerosis. Laboratory data collected at baseline included presence of antinuclear antibodies (ANA), anticentromere antibodies (ACA), anti-DNA topoisomerase I antibodies (anti-Scl-70), anti-U1RNP antibodies, anti-RNA polymerase III antibodies (ARA), N-terminal pro b-type natriuretic peptides (NT-proBNP), and C-reactive protein/erythrocyte sedimentation rate. Dr. Bellando-Randone and colleagues also collected clinical, pulmonary function, lung high-resolution CT, echocardiographic, and ECG data at baseline.
Predictions were based on these factors alone and in combination. Overall, 65% of patients had positive ANA. Other baseline characteristics present in patients that predicted systemic sclerosis included positive ACA/anti-Scl-70/ARA (32%), NVC abnormalities such as giant capillaries (25%), and puffy fingers (17%).
Using Kaplan-Meier analysis, the researchers found 7.4% of 401 patients who were ANA positive progressed to meet ACR-EULAR 2013 criteria, and the percentage of these patients increased to 29.3% at 3 years and 44.1% at 5 years. When the researchers considered disease-specific antibodies alone, 10.6% of 90 patients progressed from Raynaud’s phenomenon to systemic sclerosis within 1 year, 39.6% within 3 years, and 50.3% within 5 years. When the researchers analyzed disease-specific antibodies and NVC abnormalities together, 16% of 72 patients progressed to systemic sclerosis within 1 year, 61.7% within 3 years, and 77.4% within 5 years.
Puffy fingers also were a predictor of progression, and 14.4% of 69 patients with puffy fingers alone progressed from Raynaud’s phenomenon to systemic sclerosis at 1 year, 47.7% at 3 years, and 67.9% at 5 years. Considering puffy fingers and disease-specific antibodies together, 20% of 27 patients progressed at 1 year, 56.3% at 3 years, and 91.3% at 5 years. No patients with puffy fingers together and NVC abnormalities progressed to systemic sclerosis at 1 year, but 60.4% of 22 patients progressed at 3 years before plateauing at 5 years. For patients with NVC abnormalities alone, 7.1% progressed to systemic sclerosis from Raynaud’s phenomenon at 1 year, 39.4% at 3 years, and 52.7% at 5 years.
“Regarding capillaroscopy, we have to say that not all centers that participated were equally screened in capillaroscopy, and so we cannot assume the accuracy of this data,” she said.
Dr. Bellando-Randone noted that, apart from puffy fingers, disease-specific antibodies, and NVC abnormalities, patients were more likely to have a history of esophageal symptoms if they progressed to systemic sclerosis (37.3%), compared with patients who did not progress (23.6%; P = .003).
Puffy fingers alone were an independent predictor of systemic sclerosis (PPV, 78.9%; NPV, 45.1%) as well as in combination with disease-specific antibodies (PPV, 94.1%; NPV, 43.9%). The combination of disease-specific antibodies plus NVC abnormalities also independently predicted progression to systemic sclerosis (PPV, 82.2%; NPV, 50.4%). In a Cox multivariate analysis, disease-specific antibodies (relative risk, 5.4; 95% confidence interval, 3.7-7.9) and puffy fingers (RR, 3.0; 95% CI, 2.0-4.4) together were strongly predictive of progression from Raynaud’s phenomenon to systemic sclerosis (RR, 4.3; 95% CI, 2.6-7.3).
“This is really important for the risk stratification of patients [in] the very early stages of the disease, even if these data should be corroborated by larger data in larger studies in the future,” said Dr. Bellando-Randone.
The investigators reported having no conflicts of interest.
SOURCE: Bellando-Randone S et al. Arthritis Rheumatol. 2019;71(suppl 10). Abstract 2914.
ATLANTA – , according to recent results from the Very Early Diagnosis Of Systemic Sclerosis (VEDOSS) study presented at the annual meeting of the American College of Rheumatology.
“Our data show that thanks [to a] combination of the signs that characterize the various phases of the disease, patients can be diagnosed [with systemic sclerosis] in the very early stages,” first author Silvia Bellando-Randone, MD, PhD, assistant professor in the division of rheumatology at the University of Florence (Italy), said in her presentation.
Dr. Bellando-Randone and colleagues performed a longitudinal, observational study of 742 patients (mean 45.7 years old) at 42 centers in a cohort of mostly women (90%), nearly all of whom had had Raynaud’s phenomenon for longer than 36 months (97.5%). Patients were excluded if they had systemic sclerosis based on ACR 1980 classification criteria and/or ACR–European League Against Rheumatism 2013 criteria, had systemic sclerosis together with other connective-tissue diseases, or were unlikely to be present for three consecutive annual exams. Data collection began in March 2012 with follow-up of 5 years.
The researchers determined the positive predictive values (PPV) and negative predictive values (NPV) of clinical features, systemic sclerosis–specific antibodies, and nailfold video capillaroscopy (NVC) abnormalities on progression from Raynaud’s phenomenon to systemic sclerosis. Laboratory data collected at baseline included presence of antinuclear antibodies (ANA), anticentromere antibodies (ACA), anti-DNA topoisomerase I antibodies (anti-Scl-70), anti-U1RNP antibodies, anti-RNA polymerase III antibodies (ARA), N-terminal pro b-type natriuretic peptides (NT-proBNP), and C-reactive protein/erythrocyte sedimentation rate. Dr. Bellando-Randone and colleagues also collected clinical, pulmonary function, lung high-resolution CT, echocardiographic, and ECG data at baseline.
Predictions were based on these factors alone and in combination. Overall, 65% of patients had positive ANA. Other baseline characteristics present in patients that predicted systemic sclerosis included positive ACA/anti-Scl-70/ARA (32%), NVC abnormalities such as giant capillaries (25%), and puffy fingers (17%).
Using Kaplan-Meier analysis, the researchers found 7.4% of 401 patients who were ANA positive progressed to meet ACR-EULAR 2013 criteria, and the percentage of these patients increased to 29.3% at 3 years and 44.1% at 5 years. When the researchers considered disease-specific antibodies alone, 10.6% of 90 patients progressed from Raynaud’s phenomenon to systemic sclerosis within 1 year, 39.6% within 3 years, and 50.3% within 5 years. When the researchers analyzed disease-specific antibodies and NVC abnormalities together, 16% of 72 patients progressed to systemic sclerosis within 1 year, 61.7% within 3 years, and 77.4% within 5 years.
Puffy fingers also were a predictor of progression, and 14.4% of 69 patients with puffy fingers alone progressed from Raynaud’s phenomenon to systemic sclerosis at 1 year, 47.7% at 3 years, and 67.9% at 5 years. Considering puffy fingers and disease-specific antibodies together, 20% of 27 patients progressed at 1 year, 56.3% at 3 years, and 91.3% at 5 years. No patients with puffy fingers together and NVC abnormalities progressed to systemic sclerosis at 1 year, but 60.4% of 22 patients progressed at 3 years before plateauing at 5 years. For patients with NVC abnormalities alone, 7.1% progressed to systemic sclerosis from Raynaud’s phenomenon at 1 year, 39.4% at 3 years, and 52.7% at 5 years.
“Regarding capillaroscopy, we have to say that not all centers that participated were equally screened in capillaroscopy, and so we cannot assume the accuracy of this data,” she said.
Dr. Bellando-Randone noted that, apart from puffy fingers, disease-specific antibodies, and NVC abnormalities, patients were more likely to have a history of esophageal symptoms if they progressed to systemic sclerosis (37.3%), compared with patients who did not progress (23.6%; P = .003).
Puffy fingers alone were an independent predictor of systemic sclerosis (PPV, 78.9%; NPV, 45.1%) as well as in combination with disease-specific antibodies (PPV, 94.1%; NPV, 43.9%). The combination of disease-specific antibodies plus NVC abnormalities also independently predicted progression to systemic sclerosis (PPV, 82.2%; NPV, 50.4%). In a Cox multivariate analysis, disease-specific antibodies (relative risk, 5.4; 95% confidence interval, 3.7-7.9) and puffy fingers (RR, 3.0; 95% CI, 2.0-4.4) together were strongly predictive of progression from Raynaud’s phenomenon to systemic sclerosis (RR, 4.3; 95% CI, 2.6-7.3).
“This is really important for the risk stratification of patients [in] the very early stages of the disease, even if these data should be corroborated by larger data in larger studies in the future,” said Dr. Bellando-Randone.
The investigators reported having no conflicts of interest.
SOURCE: Bellando-Randone S et al. Arthritis Rheumatol. 2019;71(suppl 10). Abstract 2914.
ATLANTA – , according to recent results from the Very Early Diagnosis Of Systemic Sclerosis (VEDOSS) study presented at the annual meeting of the American College of Rheumatology.
“Our data show that thanks [to a] combination of the signs that characterize the various phases of the disease, patients can be diagnosed [with systemic sclerosis] in the very early stages,” first author Silvia Bellando-Randone, MD, PhD, assistant professor in the division of rheumatology at the University of Florence (Italy), said in her presentation.
Dr. Bellando-Randone and colleagues performed a longitudinal, observational study of 742 patients (mean 45.7 years old) at 42 centers in a cohort of mostly women (90%), nearly all of whom had had Raynaud’s phenomenon for longer than 36 months (97.5%). Patients were excluded if they had systemic sclerosis based on ACR 1980 classification criteria and/or ACR–European League Against Rheumatism 2013 criteria, had systemic sclerosis together with other connective-tissue diseases, or were unlikely to be present for three consecutive annual exams. Data collection began in March 2012 with follow-up of 5 years.
The researchers determined the positive predictive values (PPV) and negative predictive values (NPV) of clinical features, systemic sclerosis–specific antibodies, and nailfold video capillaroscopy (NVC) abnormalities on progression from Raynaud’s phenomenon to systemic sclerosis. Laboratory data collected at baseline included presence of antinuclear antibodies (ANA), anticentromere antibodies (ACA), anti-DNA topoisomerase I antibodies (anti-Scl-70), anti-U1RNP antibodies, anti-RNA polymerase III antibodies (ARA), N-terminal pro b-type natriuretic peptides (NT-proBNP), and C-reactive protein/erythrocyte sedimentation rate. Dr. Bellando-Randone and colleagues also collected clinical, pulmonary function, lung high-resolution CT, echocardiographic, and ECG data at baseline.
Predictions were based on these factors alone and in combination. Overall, 65% of patients had positive ANA. Other baseline characteristics present in patients that predicted systemic sclerosis included positive ACA/anti-Scl-70/ARA (32%), NVC abnormalities such as giant capillaries (25%), and puffy fingers (17%).
Using Kaplan-Meier analysis, the researchers found 7.4% of 401 patients who were ANA positive progressed to meet ACR-EULAR 2013 criteria, and the percentage of these patients increased to 29.3% at 3 years and 44.1% at 5 years. When the researchers considered disease-specific antibodies alone, 10.6% of 90 patients progressed from Raynaud’s phenomenon to systemic sclerosis within 1 year, 39.6% within 3 years, and 50.3% within 5 years. When the researchers analyzed disease-specific antibodies and NVC abnormalities together, 16% of 72 patients progressed to systemic sclerosis within 1 year, 61.7% within 3 years, and 77.4% within 5 years.
Puffy fingers also were a predictor of progression, and 14.4% of 69 patients with puffy fingers alone progressed from Raynaud’s phenomenon to systemic sclerosis at 1 year, 47.7% at 3 years, and 67.9% at 5 years. Considering puffy fingers and disease-specific antibodies together, 20% of 27 patients progressed at 1 year, 56.3% at 3 years, and 91.3% at 5 years. No patients with puffy fingers together and NVC abnormalities progressed to systemic sclerosis at 1 year, but 60.4% of 22 patients progressed at 3 years before plateauing at 5 years. For patients with NVC abnormalities alone, 7.1% progressed to systemic sclerosis from Raynaud’s phenomenon at 1 year, 39.4% at 3 years, and 52.7% at 5 years.
“Regarding capillaroscopy, we have to say that not all centers that participated were equally screened in capillaroscopy, and so we cannot assume the accuracy of this data,” she said.
Dr. Bellando-Randone noted that, apart from puffy fingers, disease-specific antibodies, and NVC abnormalities, patients were more likely to have a history of esophageal symptoms if they progressed to systemic sclerosis (37.3%), compared with patients who did not progress (23.6%; P = .003).
Puffy fingers alone were an independent predictor of systemic sclerosis (PPV, 78.9%; NPV, 45.1%) as well as in combination with disease-specific antibodies (PPV, 94.1%; NPV, 43.9%). The combination of disease-specific antibodies plus NVC abnormalities also independently predicted progression to systemic sclerosis (PPV, 82.2%; NPV, 50.4%). In a Cox multivariate analysis, disease-specific antibodies (relative risk, 5.4; 95% confidence interval, 3.7-7.9) and puffy fingers (RR, 3.0; 95% CI, 2.0-4.4) together were strongly predictive of progression from Raynaud’s phenomenon to systemic sclerosis (RR, 4.3; 95% CI, 2.6-7.3).
“This is really important for the risk stratification of patients [in] the very early stages of the disease, even if these data should be corroborated by larger data in larger studies in the future,” said Dr. Bellando-Randone.
The investigators reported having no conflicts of interest.
SOURCE: Bellando-Randone S et al. Arthritis Rheumatol. 2019;71(suppl 10). Abstract 2914.
REPORTING FROM ACR 2019
TULIP trials show clinical benefit of anifrolumab for SLE
ATLANTA –

In TULIP-1, which compared intravenous anifrolumab at doses of 300 or 150 mg and placebo given every 4 weeks for 48 weeks, the primary endpoint of SLE Responder Index (SRI) in the 300 mg versus the placebo group was not met, but in post hoc analyses, numeric improvements at thresholds associated with clinical benefit were observed for several secondary outcomes, Richard A. Furie, MD, a professor of medicine at the Hofstra University/Northwell, Hempstead, N.Y., reported during a plenary session at the annual meeting of the American College of Rheumatology.
The findings were published online Nov. 11 in Lancet Rheumatology.
TULIP-2 compared IV anifrolumab at a dose of 300 mg versus placebo every 4 weeks for 48 weeks and demonstrated the superiority of anifrolumab for multiple efficacy endpoints, including the primary study endpoint of British Isles Lupus Assessment Group (BILAG)-based Composite Lupus Assessment (BICLA), Eric F. Morand, MD, PhD, reported during a late-breaking abstract session at the meeting.
The double-blind, phase 3 TULIP trials each enrolled seropositive SLE patients with moderate to severe active disease despite standard-of-care therapy (SOC). All patients met ACR criteria, had a SLE Disease Activity Index (SLEDAI)-2K of 6 or greater, and BILAG index scoring showing one or more organ systems with grade A involvement or two or more with grade B. Both trials required stable SOC therapy throughout the study except for mandatory attempts at oral corticosteroid (OCS) tapering for patients who were receiving 10 mg/day or more of prednisone or its equivalent at study entry.
The trials followed a phase 2 trial, reported by Dr. Furie at the 2015 ACR meeting and published in Arthritis & Rheumatology in 2017, which showed “very robust” efficacy of anifrolumab in this setting.
“The burning question for the last 20 years has been, ‘Can type 1 interferon inhibitors actually reduce lupus clinical activity?’ ” Dr. Furie said. “The problem here [is that] you can inhibit interferon-alpha, but there are four other subtypes capable of binding to the interferon receptor.”
Anifrolumab, which was first studied in scleroderma, inhibits the interferon (IFN) receptor, thereby providing broader inhibition than strategies that specifically target interferon-alpha, he explained.
In the phase 2 trial, the primary composite endpoint of SRI response at day 169 and sustained reduction of OCS dose between days 85 and 169 was met by 51.5% of patients receiving 300 mg of anifrolumab versus 26.6% of those receiving placebo.
TULIP-1
The TULIP-1 trial, however, failed to show a significant difference in the primary endpoint of week 52 SRI, although initial analyses showed some numeric benefit with respect to BICLA, OCS dose reductions, and other organ-specific endpoints.
The percentage of SRI responders at week 52 in the double-blind trial was 36.2% in 180 patients who received 300 mg anifrolumab vs. 40.4% in 184 who received placebo (nominal P value = .41), and in a subgroup of patients who had high IFN gene signature (IFNGS) test results, the rates were 35.9% and 39.3% (nominal P value = 0.55), respectively.
Sustained OCS reduction to 7.5 mg/day or less occurred in 41% of anifrolumab and 32.1% of placebo group patients, and a 50% or greater reduction in Cutaneous Lupus Erythematosus Disease Activity Severity Index (CLASI) activity from baseline to week 12 occurred in 41.9% and 24.9%, respectively. The annualized flare rate to week 52 was 0.72 for anifrolumab and 0.60 for placebo.
BICLA response at week 52 was 37.1% with anifrolumab versus 27% with placebo, and a 50% or greater reduction in active joints from baseline to week 52 occurred in 47% versus 32.5% of patients in the groups, respectively.
The 150-mg dose, which was included to provide dose-response data, did not show efficacy in secondary outcomes.
“We see a delta of about 10 percentage points [for BICLA], and about a 15-percentage point change [in swollen and tender joint count] in favor of anifrolumab,” Dr. Furie said. “So why the big difference between phase 2 results and phase 3 results? Well, that led to a year-long interrogation of all the data ... [which revealed that] about 8% of patients were misclassified as nonresponders for [NSAID] use.”
The medication rules in the study automatically required any patient who used a restricted drug, including NSAIDs, to be classified as a nonresponder. That means a patient who took an NSAID for a headache at the beginning of the study, for example, would have been considered a nonresponder regardless of their outcome, he explained.
“This led to a review of all the restricted medication classification rules, and after unblinding, a meeting was convened with SLE experts and the sponsors to actually revise the medication rules just to make them clinically more appropriate. The key analyses were repeated post hoc,” he said.
The difference between the treatment and placebo groups in terms of the week 52 SRI didn’t change much in the post hoc analysis (46.9% vs. 43% of treatment and placebo patients, respectively, met the endpoint). Similarly, SRI rates in the IFNGS test–high subgroup were 48.2% and 41.8%, respectively.
However, more pronounced “shifts to the right,” indicating larger differences favoring anifrolumab over placebo, were seen for OCS dose reduction (48.8% vs. 32.1%), CLASI response (43.6% vs. 24.9%), and BICLA response (48.1% vs. 29.8%).
“For BICLA response, we see a fairly significant change ... with what appears to be a clinically significant delta (about 16 percentage points), and as far as the change in active joints, also very significant in my eyes,” he said.
Also of note, the time to BICLA response sustained to week 52 was improved with anifrolumab (hazard ratio, 1.93), and CLASI response differences emerged early, at about 12 weeks, he said.
The type 1 IFNGS was reduced by a median of 88% to 90% in the anifrolumab groups vs. with placebo, and modest changes in serologies were also noted.
Serious adverse events occurred in 13.9% and 10.8% of patients in the anifrolumab 300- and 150-mg arms, compared with 16.3% in the placebo arm. Herpes zoster was more common in the anifrolumab groups (5.6% for 300 mg and 5.4% for 150 mg vs. 1.6% for placebo).
“But other than that, no major standouts as far as the safety profile,” Dr. Furie said.
The findings, particularly after the medication rules were amended, suggest efficacy of anifrolumab for corticosteroid reductions, skin activity, BICLA, and joint scores, he said, noting that corticosteroid dose reductions are very important for patients, and that BICLA is “actually a very rigorous composite.”
Importantly, the findings also underscore the importance and impact of medication rules, and the critical role that endpoint selection plays in SLE trials.
“We’ve been seeing discordance lately between the SRI and BICLA ... so [there is] still a lot to learn,” he said. “And I think it’s important in evaluating the drug effect to look at the totality of the data.”
TULIP-2
BICLA response, the primary endpoint of TULIP-2, was achieved by 47.8% of 180 patients who received anifrolumab, compared with 31.5% of 182 who received placebo, said Dr. Morand, professor and head of the School of Clinical Sciences at Monash University, Melbourne.
“The effect size was 16.3 percentage points with an adjusted p value of 0.001. Therefore, the primary outcome of this trial was attained,” said Dr. Morand, who also is head of the Monash Health Rheumatology Unit. “Separation between the treatment arms occurred early and was maintained across the progression of the trial.”
Anifrolumab was also superior to placebo for key secondary endpoints, including OCS dose reduction to 7.5 mg/day or less (51.5% vs. 30.2%) and CLASI response (49.0% vs. 25.0%).
“Joint responses did not show a significant difference between the anifrolumab and placebo arms,” he said, adding that the annualized flare rate also did not differ significantly between the groups, but was numerically lower in anifrolumab-treated patients (0.43 vs. 0.64; rate ratio, 0.67; P = .081).
Numeric differences also favored anifrolumab for multiple secondary endpoints, including SRI responses, time to onset of BICLA-sustained response, and time to first flare, he noted.
Further, in patients with high baseline IFNGS, anifrolumab induced neutralization of IFNGS by week 12, with a median suppression of 88.0%, which persisted for the duration of the study; no such effect was seen in the placebo arm.
Serum anti–double stranded DNA also trended toward normalization with anifrolumab.
The safety profile of anifrolumab was similar to that seen in previous trials, including TULIP-1, with herpes zoster occurring more often in those receiving anifrolumab (7.2% vs. 1.1% in the placebo group), Dr. Morand said, noting that “all herpes zoster episodes were cutaneous, all responded to antiviral therapy, and none required [treatment] discontinuation.”
Serious adverse events, including pneumonia and SLE worsening, occurred less frequently in the anifrolumab arm (8.3% vs. 17.0%, respectively), as did adverse events leading to treatment discontinuation (2.8% and 7.1%). One death occurred in the anifrolumab group from community-acquired pneumonia, and few patients (0.6%) developed antidrug antibodies.
No new safety signals were identified, he said, noting that “the findings add to cumulative evidence identifying anifrolumab as a potential new treatment option for SLE.”
“In conclusion, TULIP-2 was a positive phase 3 trial in lupus, and there aren’t many times that that sentence has been spoken,” he said.
The TULIP-1 and -2 trials were sponsored by AstraZeneca. Dr. Furie And Dr. Morand both reported grant/research support and consulting fees from AstraZeneca, as well as speaker’s bureau participation for AstraZeneca.
SOURCES: Furie RA et al. Arthritis Rheumatol. 2019;71(suppl 10), Abstract 1763; Morand EF et al. Arthritis Rheumatol. 2019;71(suppl 10), Abstract L17.
ATLANTA –
In TULIP-1, which compared intravenous anifrolumab at doses of 300 or 150 mg and placebo given every 4 weeks for 48 weeks, the primary endpoint of SLE Responder Index (SRI) in the 300 mg versus the placebo group was not met, but in post hoc analyses, numeric improvements at thresholds associated with clinical benefit were observed for several secondary outcomes, Richard A. Furie, MD, a professor of medicine at the Hofstra University/Northwell, Hempstead, N.Y., reported during a plenary session at the annual meeting of the American College of Rheumatology.
The findings were published online Nov. 11 in Lancet Rheumatology.
TULIP-2 compared IV anifrolumab at a dose of 300 mg versus placebo every 4 weeks for 48 weeks and demonstrated the superiority of anifrolumab for multiple efficacy endpoints, including the primary study endpoint of British Isles Lupus Assessment Group (BILAG)-based Composite Lupus Assessment (BICLA), Eric F. Morand, MD, PhD, reported during a late-breaking abstract session at the meeting.
The double-blind, phase 3 TULIP trials each enrolled seropositive SLE patients with moderate to severe active disease despite standard-of-care therapy (SOC). All patients met ACR criteria, had a SLE Disease Activity Index (SLEDAI)-2K of 6 or greater, and BILAG index scoring showing one or more organ systems with grade A involvement or two or more with grade B. Both trials required stable SOC therapy throughout the study except for mandatory attempts at oral corticosteroid (OCS) tapering for patients who were receiving 10 mg/day or more of prednisone or its equivalent at study entry.
The trials followed a phase 2 trial, reported by Dr. Furie at the 2015 ACR meeting and published in Arthritis & Rheumatology in 2017, which showed “very robust” efficacy of anifrolumab in this setting.
“The burning question for the last 20 years has been, ‘Can type 1 interferon inhibitors actually reduce lupus clinical activity?’ ” Dr. Furie said. “The problem here [is that] you can inhibit interferon-alpha, but there are four other subtypes capable of binding to the interferon receptor.”
Anifrolumab, which was first studied in scleroderma, inhibits the interferon (IFN) receptor, thereby providing broader inhibition than strategies that specifically target interferon-alpha, he explained.
In the phase 2 trial, the primary composite endpoint of SRI response at day 169 and sustained reduction of OCS dose between days 85 and 169 was met by 51.5% of patients receiving 300 mg of anifrolumab versus 26.6% of those receiving placebo.
TULIP-1
The TULIP-1 trial, however, failed to show a significant difference in the primary endpoint of week 52 SRI, although initial analyses showed some numeric benefit with respect to BICLA, OCS dose reductions, and other organ-specific endpoints.
The percentage of SRI responders at week 52 in the double-blind trial was 36.2% in 180 patients who received 300 mg anifrolumab vs. 40.4% in 184 who received placebo (nominal P value = .41), and in a subgroup of patients who had high IFN gene signature (IFNGS) test results, the rates were 35.9% and 39.3% (nominal P value = 0.55), respectively.
Sustained OCS reduction to 7.5 mg/day or less occurred in 41% of anifrolumab and 32.1% of placebo group patients, and a 50% or greater reduction in Cutaneous Lupus Erythematosus Disease Activity Severity Index (CLASI) activity from baseline to week 12 occurred in 41.9% and 24.9%, respectively. The annualized flare rate to week 52 was 0.72 for anifrolumab and 0.60 for placebo.
BICLA response at week 52 was 37.1% with anifrolumab versus 27% with placebo, and a 50% or greater reduction in active joints from baseline to week 52 occurred in 47% versus 32.5% of patients in the groups, respectively.
The 150-mg dose, which was included to provide dose-response data, did not show efficacy in secondary outcomes.
“We see a delta of about 10 percentage points [for BICLA], and about a 15-percentage point change [in swollen and tender joint count] in favor of anifrolumab,” Dr. Furie said. “So why the big difference between phase 2 results and phase 3 results? Well, that led to a year-long interrogation of all the data ... [which revealed that] about 8% of patients were misclassified as nonresponders for [NSAID] use.”
The medication rules in the study automatically required any patient who used a restricted drug, including NSAIDs, to be classified as a nonresponder. That means a patient who took an NSAID for a headache at the beginning of the study, for example, would have been considered a nonresponder regardless of their outcome, he explained.
“This led to a review of all the restricted medication classification rules, and after unblinding, a meeting was convened with SLE experts and the sponsors to actually revise the medication rules just to make them clinically more appropriate. The key analyses were repeated post hoc,” he said.
The difference between the treatment and placebo groups in terms of the week 52 SRI didn’t change much in the post hoc analysis (46.9% vs. 43% of treatment and placebo patients, respectively, met the endpoint). Similarly, SRI rates in the IFNGS test–high subgroup were 48.2% and 41.8%, respectively.
However, more pronounced “shifts to the right,” indicating larger differences favoring anifrolumab over placebo, were seen for OCS dose reduction (48.8% vs. 32.1%), CLASI response (43.6% vs. 24.9%), and BICLA response (48.1% vs. 29.8%).
“For BICLA response, we see a fairly significant change ... with what appears to be a clinically significant delta (about 16 percentage points), and as far as the change in active joints, also very significant in my eyes,” he said.
Also of note, the time to BICLA response sustained to week 52 was improved with anifrolumab (hazard ratio, 1.93), and CLASI response differences emerged early, at about 12 weeks, he said.
The type 1 IFNGS was reduced by a median of 88% to 90% in the anifrolumab groups vs. with placebo, and modest changes in serologies were also noted.
Serious adverse events occurred in 13.9% and 10.8% of patients in the anifrolumab 300- and 150-mg arms, compared with 16.3% in the placebo arm. Herpes zoster was more common in the anifrolumab groups (5.6% for 300 mg and 5.4% for 150 mg vs. 1.6% for placebo).
“But other than that, no major standouts as far as the safety profile,” Dr. Furie said.
The findings, particularly after the medication rules were amended, suggest efficacy of anifrolumab for corticosteroid reductions, skin activity, BICLA, and joint scores, he said, noting that corticosteroid dose reductions are very important for patients, and that BICLA is “actually a very rigorous composite.”
Importantly, the findings also underscore the importance and impact of medication rules, and the critical role that endpoint selection plays in SLE trials.
“We’ve been seeing discordance lately between the SRI and BICLA ... so [there is] still a lot to learn,” he said. “And I think it’s important in evaluating the drug effect to look at the totality of the data.”
TULIP-2
BICLA response, the primary endpoint of TULIP-2, was achieved by 47.8% of 180 patients who received anifrolumab, compared with 31.5% of 182 who received placebo, said Dr. Morand, professor and head of the School of Clinical Sciences at Monash University, Melbourne.
“The effect size was 16.3 percentage points with an adjusted p value of 0.001. Therefore, the primary outcome of this trial was attained,” said Dr. Morand, who also is head of the Monash Health Rheumatology Unit. “Separation between the treatment arms occurred early and was maintained across the progression of the trial.”
Anifrolumab was also superior to placebo for key secondary endpoints, including OCS dose reduction to 7.5 mg/day or less (51.5% vs. 30.2%) and CLASI response (49.0% vs. 25.0%).
“Joint responses did not show a significant difference between the anifrolumab and placebo arms,” he said, adding that the annualized flare rate also did not differ significantly between the groups, but was numerically lower in anifrolumab-treated patients (0.43 vs. 0.64; rate ratio, 0.67; P = .081).
Numeric differences also favored anifrolumab for multiple secondary endpoints, including SRI responses, time to onset of BICLA-sustained response, and time to first flare, he noted.
Further, in patients with high baseline IFNGS, anifrolumab induced neutralization of IFNGS by week 12, with a median suppression of 88.0%, which persisted for the duration of the study; no such effect was seen in the placebo arm.
Serum anti–double stranded DNA also trended toward normalization with anifrolumab.
The safety profile of anifrolumab was similar to that seen in previous trials, including TULIP-1, with herpes zoster occurring more often in those receiving anifrolumab (7.2% vs. 1.1% in the placebo group), Dr. Morand said, noting that “all herpes zoster episodes were cutaneous, all responded to antiviral therapy, and none required [treatment] discontinuation.”
Serious adverse events, including pneumonia and SLE worsening, occurred less frequently in the anifrolumab arm (8.3% vs. 17.0%, respectively), as did adverse events leading to treatment discontinuation (2.8% and 7.1%). One death occurred in the anifrolumab group from community-acquired pneumonia, and few patients (0.6%) developed antidrug antibodies.
No new safety signals were identified, he said, noting that “the findings add to cumulative evidence identifying anifrolumab as a potential new treatment option for SLE.”
“In conclusion, TULIP-2 was a positive phase 3 trial in lupus, and there aren’t many times that that sentence has been spoken,” he said.
The TULIP-1 and -2 trials were sponsored by AstraZeneca. Dr. Furie And Dr. Morand both reported grant/research support and consulting fees from AstraZeneca, as well as speaker’s bureau participation for AstraZeneca.
SOURCES: Furie RA et al. Arthritis Rheumatol. 2019;71(suppl 10), Abstract 1763; Morand EF et al. Arthritis Rheumatol. 2019;71(suppl 10), Abstract L17.
ATLANTA –
In TULIP-1, which compared intravenous anifrolumab at doses of 300 or 150 mg and placebo given every 4 weeks for 48 weeks, the primary endpoint of SLE Responder Index (SRI) in the 300 mg versus the placebo group was not met, but in post hoc analyses, numeric improvements at thresholds associated with clinical benefit were observed for several secondary outcomes, Richard A. Furie, MD, a professor of medicine at the Hofstra University/Northwell, Hempstead, N.Y., reported during a plenary session at the annual meeting of the American College of Rheumatology.
The findings were published online Nov. 11 in Lancet Rheumatology.
TULIP-2 compared IV anifrolumab at a dose of 300 mg versus placebo every 4 weeks for 48 weeks and demonstrated the superiority of anifrolumab for multiple efficacy endpoints, including the primary study endpoint of British Isles Lupus Assessment Group (BILAG)-based Composite Lupus Assessment (BICLA), Eric F. Morand, MD, PhD, reported during a late-breaking abstract session at the meeting.
The double-blind, phase 3 TULIP trials each enrolled seropositive SLE patients with moderate to severe active disease despite standard-of-care therapy (SOC). All patients met ACR criteria, had a SLE Disease Activity Index (SLEDAI)-2K of 6 or greater, and BILAG index scoring showing one or more organ systems with grade A involvement or two or more with grade B. Both trials required stable SOC therapy throughout the study except for mandatory attempts at oral corticosteroid (OCS) tapering for patients who were receiving 10 mg/day or more of prednisone or its equivalent at study entry.
The trials followed a phase 2 trial, reported by Dr. Furie at the 2015 ACR meeting and published in Arthritis & Rheumatology in 2017, which showed “very robust” efficacy of anifrolumab in this setting.
“The burning question for the last 20 years has been, ‘Can type 1 interferon inhibitors actually reduce lupus clinical activity?’ ” Dr. Furie said. “The problem here [is that] you can inhibit interferon-alpha, but there are four other subtypes capable of binding to the interferon receptor.”
Anifrolumab, which was first studied in scleroderma, inhibits the interferon (IFN) receptor, thereby providing broader inhibition than strategies that specifically target interferon-alpha, he explained.
In the phase 2 trial, the primary composite endpoint of SRI response at day 169 and sustained reduction of OCS dose between days 85 and 169 was met by 51.5% of patients receiving 300 mg of anifrolumab versus 26.6% of those receiving placebo.
TULIP-1
The TULIP-1 trial, however, failed to show a significant difference in the primary endpoint of week 52 SRI, although initial analyses showed some numeric benefit with respect to BICLA, OCS dose reductions, and other organ-specific endpoints.
The percentage of SRI responders at week 52 in the double-blind trial was 36.2% in 180 patients who received 300 mg anifrolumab vs. 40.4% in 184 who received placebo (nominal P value = .41), and in a subgroup of patients who had high IFN gene signature (IFNGS) test results, the rates were 35.9% and 39.3% (nominal P value = 0.55), respectively.
Sustained OCS reduction to 7.5 mg/day or less occurred in 41% of anifrolumab and 32.1% of placebo group patients, and a 50% or greater reduction in Cutaneous Lupus Erythematosus Disease Activity Severity Index (CLASI) activity from baseline to week 12 occurred in 41.9% and 24.9%, respectively. The annualized flare rate to week 52 was 0.72 for anifrolumab and 0.60 for placebo.
BICLA response at week 52 was 37.1% with anifrolumab versus 27% with placebo, and a 50% or greater reduction in active joints from baseline to week 52 occurred in 47% versus 32.5% of patients in the groups, respectively.
The 150-mg dose, which was included to provide dose-response data, did not show efficacy in secondary outcomes.
“We see a delta of about 10 percentage points [for BICLA], and about a 15-percentage point change [in swollen and tender joint count] in favor of anifrolumab,” Dr. Furie said. “So why the big difference between phase 2 results and phase 3 results? Well, that led to a year-long interrogation of all the data ... [which revealed that] about 8% of patients were misclassified as nonresponders for [NSAID] use.”
The medication rules in the study automatically required any patient who used a restricted drug, including NSAIDs, to be classified as a nonresponder. That means a patient who took an NSAID for a headache at the beginning of the study, for example, would have been considered a nonresponder regardless of their outcome, he explained.
“This led to a review of all the restricted medication classification rules, and after unblinding, a meeting was convened with SLE experts and the sponsors to actually revise the medication rules just to make them clinically more appropriate. The key analyses were repeated post hoc,” he said.
The difference between the treatment and placebo groups in terms of the week 52 SRI didn’t change much in the post hoc analysis (46.9% vs. 43% of treatment and placebo patients, respectively, met the endpoint). Similarly, SRI rates in the IFNGS test–high subgroup were 48.2% and 41.8%, respectively.
However, more pronounced “shifts to the right,” indicating larger differences favoring anifrolumab over placebo, were seen for OCS dose reduction (48.8% vs. 32.1%), CLASI response (43.6% vs. 24.9%), and BICLA response (48.1% vs. 29.8%).
“For BICLA response, we see a fairly significant change ... with what appears to be a clinically significant delta (about 16 percentage points), and as far as the change in active joints, also very significant in my eyes,” he said.
Also of note, the time to BICLA response sustained to week 52 was improved with anifrolumab (hazard ratio, 1.93), and CLASI response differences emerged early, at about 12 weeks, he said.
The type 1 IFNGS was reduced by a median of 88% to 90% in the anifrolumab groups vs. with placebo, and modest changes in serologies were also noted.
Serious adverse events occurred in 13.9% and 10.8% of patients in the anifrolumab 300- and 150-mg arms, compared with 16.3% in the placebo arm. Herpes zoster was more common in the anifrolumab groups (5.6% for 300 mg and 5.4% for 150 mg vs. 1.6% for placebo).
“But other than that, no major standouts as far as the safety profile,” Dr. Furie said.
The findings, particularly after the medication rules were amended, suggest efficacy of anifrolumab for corticosteroid reductions, skin activity, BICLA, and joint scores, he said, noting that corticosteroid dose reductions are very important for patients, and that BICLA is “actually a very rigorous composite.”
Importantly, the findings also underscore the importance and impact of medication rules, and the critical role that endpoint selection plays in SLE trials.
“We’ve been seeing discordance lately between the SRI and BICLA ... so [there is] still a lot to learn,” he said. “And I think it’s important in evaluating the drug effect to look at the totality of the data.”
TULIP-2
BICLA response, the primary endpoint of TULIP-2, was achieved by 47.8% of 180 patients who received anifrolumab, compared with 31.5% of 182 who received placebo, said Dr. Morand, professor and head of the School of Clinical Sciences at Monash University, Melbourne.
“The effect size was 16.3 percentage points with an adjusted p value of 0.001. Therefore, the primary outcome of this trial was attained,” said Dr. Morand, who also is head of the Monash Health Rheumatology Unit. “Separation between the treatment arms occurred early and was maintained across the progression of the trial.”
Anifrolumab was also superior to placebo for key secondary endpoints, including OCS dose reduction to 7.5 mg/day or less (51.5% vs. 30.2%) and CLASI response (49.0% vs. 25.0%).
“Joint responses did not show a significant difference between the anifrolumab and placebo arms,” he said, adding that the annualized flare rate also did not differ significantly between the groups, but was numerically lower in anifrolumab-treated patients (0.43 vs. 0.64; rate ratio, 0.67; P = .081).
Numeric differences also favored anifrolumab for multiple secondary endpoints, including SRI responses, time to onset of BICLA-sustained response, and time to first flare, he noted.
Further, in patients with high baseline IFNGS, anifrolumab induced neutralization of IFNGS by week 12, with a median suppression of 88.0%, which persisted for the duration of the study; no such effect was seen in the placebo arm.
Serum anti–double stranded DNA also trended toward normalization with anifrolumab.
The safety profile of anifrolumab was similar to that seen in previous trials, including TULIP-1, with herpes zoster occurring more often in those receiving anifrolumab (7.2% vs. 1.1% in the placebo group), Dr. Morand said, noting that “all herpes zoster episodes were cutaneous, all responded to antiviral therapy, and none required [treatment] discontinuation.”
Serious adverse events, including pneumonia and SLE worsening, occurred less frequently in the anifrolumab arm (8.3% vs. 17.0%, respectively), as did adverse events leading to treatment discontinuation (2.8% and 7.1%). One death occurred in the anifrolumab group from community-acquired pneumonia, and few patients (0.6%) developed antidrug antibodies.
No new safety signals were identified, he said, noting that “the findings add to cumulative evidence identifying anifrolumab as a potential new treatment option for SLE.”
“In conclusion, TULIP-2 was a positive phase 3 trial in lupus, and there aren’t many times that that sentence has been spoken,” he said.
The TULIP-1 and -2 trials were sponsored by AstraZeneca. Dr. Furie And Dr. Morand both reported grant/research support and consulting fees from AstraZeneca, as well as speaker’s bureau participation for AstraZeneca.
SOURCES: Furie RA et al. Arthritis Rheumatol. 2019;71(suppl 10), Abstract 1763; Morand EF et al. Arthritis Rheumatol. 2019;71(suppl 10), Abstract L17.
REPORTING FROM ACR 2019
Glucocorticoids linked to damage accrual in SLE regardless of disease activity
, according to recent findings published in The Lancet Rheumatology.
“Disentangling the potential contribution of glucocorticoid use to organ damage in SLE is confounded, in most studies, by the fact that glucocorticoid use is usually associated with active disease. Our study showed that organ damage accrual occurred in a similar proportion of patients without disease activity as in the overall cohort, and that glucocorticoid use was a significant risk factor for damage,” wrote Diane Apostolopoulos, MBBS, of Monash University, Melbourne, and colleagues.
The longitudinal cohort study prospectively enrolled 1,707 patients with SLE from May 2013 to December 2016. Study participants were recruited from 13 institutions throughout Australia and Asia. The researchers defined glucocorticoid use as any exposure and cumulative exposure to prednisolone, in addition to mean time-adjusted daily prednisolone dose. Follow-up assessment occurred at least once every 6 months and varied depending on clinical necessity. At baseline, the researchers collected various demographic information, including smoking status, age, and education level, among others. The primary endpoint measured was organ damage accrual, which was assessed at baseline and annually thereafter. In addition, disease activity was evaluated in two multivariable models using Physician Global Assessment (PGA) and Systemic Lupus Erythematosus Disease Activity Index 2000 (SLEDAI-2K) scores.
After a median duration of 2.2 years, the researchers found that 82.3% of patients were exposed to prednisolone, and 14.9% of patients had experienced a damage accrual event. In the PGA model, mean time-adjusted PGA score (based on a 0- to 1-unit increase) was associated with damage accrual independent of clinical or serologic disease activity (hazard ratio, 1.05; P = .0012).
In the SLEDAI-2K model, baseline damage scores were independently associated with damage accrual (hazard ratio, 1.32; P = .0427).
In both models, ethnicity (Asian vs. non-Asian), age at study enrollment, and mean time-adjusted prednisolone dose were parameters independently associated with damage accrual.
“Novel findings from this study were that Asian ethnicity was protective when compared with any non-Asian ethnicity, and that antimalarial usage was not protective for damage accrual,” the researchers wrote.
Despite the novel results, the researchers acknowledged that the protective effects of antimalarials could have gone undetected because of the short duration of follow-up.
In a related editorial, Guillermo Ruiz-Irastorza, MD, PhD, of University of the Basque Country in Bizkaia, Spain, said that the study provides novel data highlighting that glucocorticoid use has the potential to negatively affect the clinical progression of patients with SLE (Lancet Rheumatol. 2019 Nov 25. doi: 10.1016/S2665-9913(19)30132-8).
One question that remains from the current study is how to effectively reduce glucocorticoid-related adverse events, while still managing the disease, he further explained.
“These findings suggest that unnecessary use of glucocorticoids should be avoided in the management of the disease where possible,” the investigators concluded.
The study was funded by UCB Pharma, GlaxoSmithKline, Janssen, Bristol-Myers Squibb, and AstraZeneca. The authors reported financial affiliations with Abbott, AbbVie, Astellas, Ayumi Pharmaceutical, Bristol-Myers Squibb, Novartis, Pfizer, and several others.
SOURCE: Apostolopoulos D et al. Lancet Rheumatol. 2019 Nov 25. doi: 10.1016/S2665-9913(19)30105-5.
, according to recent findings published in The Lancet Rheumatology.
“Disentangling the potential contribution of glucocorticoid use to organ damage in SLE is confounded, in most studies, by the fact that glucocorticoid use is usually associated with active disease. Our study showed that organ damage accrual occurred in a similar proportion of patients without disease activity as in the overall cohort, and that glucocorticoid use was a significant risk factor for damage,” wrote Diane Apostolopoulos, MBBS, of Monash University, Melbourne, and colleagues.
The longitudinal cohort study prospectively enrolled 1,707 patients with SLE from May 2013 to December 2016. Study participants were recruited from 13 institutions throughout Australia and Asia. The researchers defined glucocorticoid use as any exposure and cumulative exposure to prednisolone, in addition to mean time-adjusted daily prednisolone dose. Follow-up assessment occurred at least once every 6 months and varied depending on clinical necessity. At baseline, the researchers collected various demographic information, including smoking status, age, and education level, among others. The primary endpoint measured was organ damage accrual, which was assessed at baseline and annually thereafter. In addition, disease activity was evaluated in two multivariable models using Physician Global Assessment (PGA) and Systemic Lupus Erythematosus Disease Activity Index 2000 (SLEDAI-2K) scores.
After a median duration of 2.2 years, the researchers found that 82.3% of patients were exposed to prednisolone, and 14.9% of patients had experienced a damage accrual event. In the PGA model, mean time-adjusted PGA score (based on a 0- to 1-unit increase) was associated with damage accrual independent of clinical or serologic disease activity (hazard ratio, 1.05; P = .0012).
In the SLEDAI-2K model, baseline damage scores were independently associated with damage accrual (hazard ratio, 1.32; P = .0427).
In both models, ethnicity (Asian vs. non-Asian), age at study enrollment, and mean time-adjusted prednisolone dose were parameters independently associated with damage accrual.
“Novel findings from this study were that Asian ethnicity was protective when compared with any non-Asian ethnicity, and that antimalarial usage was not protective for damage accrual,” the researchers wrote.
Despite the novel results, the researchers acknowledged that the protective effects of antimalarials could have gone undetected because of the short duration of follow-up.
In a related editorial, Guillermo Ruiz-Irastorza, MD, PhD, of University of the Basque Country in Bizkaia, Spain, said that the study provides novel data highlighting that glucocorticoid use has the potential to negatively affect the clinical progression of patients with SLE (Lancet Rheumatol. 2019 Nov 25. doi: 10.1016/S2665-9913(19)30132-8).
One question that remains from the current study is how to effectively reduce glucocorticoid-related adverse events, while still managing the disease, he further explained.
“These findings suggest that unnecessary use of glucocorticoids should be avoided in the management of the disease where possible,” the investigators concluded.
The study was funded by UCB Pharma, GlaxoSmithKline, Janssen, Bristol-Myers Squibb, and AstraZeneca. The authors reported financial affiliations with Abbott, AbbVie, Astellas, Ayumi Pharmaceutical, Bristol-Myers Squibb, Novartis, Pfizer, and several others.
SOURCE: Apostolopoulos D et al. Lancet Rheumatol. 2019 Nov 25. doi: 10.1016/S2665-9913(19)30105-5.
, according to recent findings published in The Lancet Rheumatology.
“Disentangling the potential contribution of glucocorticoid use to organ damage in SLE is confounded, in most studies, by the fact that glucocorticoid use is usually associated with active disease. Our study showed that organ damage accrual occurred in a similar proportion of patients without disease activity as in the overall cohort, and that glucocorticoid use was a significant risk factor for damage,” wrote Diane Apostolopoulos, MBBS, of Monash University, Melbourne, and colleagues.
The longitudinal cohort study prospectively enrolled 1,707 patients with SLE from May 2013 to December 2016. Study participants were recruited from 13 institutions throughout Australia and Asia. The researchers defined glucocorticoid use as any exposure and cumulative exposure to prednisolone, in addition to mean time-adjusted daily prednisolone dose. Follow-up assessment occurred at least once every 6 months and varied depending on clinical necessity. At baseline, the researchers collected various demographic information, including smoking status, age, and education level, among others. The primary endpoint measured was organ damage accrual, which was assessed at baseline and annually thereafter. In addition, disease activity was evaluated in two multivariable models using Physician Global Assessment (PGA) and Systemic Lupus Erythematosus Disease Activity Index 2000 (SLEDAI-2K) scores.
After a median duration of 2.2 years, the researchers found that 82.3% of patients were exposed to prednisolone, and 14.9% of patients had experienced a damage accrual event. In the PGA model, mean time-adjusted PGA score (based on a 0- to 1-unit increase) was associated with damage accrual independent of clinical or serologic disease activity (hazard ratio, 1.05; P = .0012).
In the SLEDAI-2K model, baseline damage scores were independently associated with damage accrual (hazard ratio, 1.32; P = .0427).
In both models, ethnicity (Asian vs. non-Asian), age at study enrollment, and mean time-adjusted prednisolone dose were parameters independently associated with damage accrual.
“Novel findings from this study were that Asian ethnicity was protective when compared with any non-Asian ethnicity, and that antimalarial usage was not protective for damage accrual,” the researchers wrote.
Despite the novel results, the researchers acknowledged that the protective effects of antimalarials could have gone undetected because of the short duration of follow-up.
In a related editorial, Guillermo Ruiz-Irastorza, MD, PhD, of University of the Basque Country in Bizkaia, Spain, said that the study provides novel data highlighting that glucocorticoid use has the potential to negatively affect the clinical progression of patients with SLE (Lancet Rheumatol. 2019 Nov 25. doi: 10.1016/S2665-9913(19)30132-8).
One question that remains from the current study is how to effectively reduce glucocorticoid-related adverse events, while still managing the disease, he further explained.
“These findings suggest that unnecessary use of glucocorticoids should be avoided in the management of the disease where possible,” the investigators concluded.
The study was funded by UCB Pharma, GlaxoSmithKline, Janssen, Bristol-Myers Squibb, and AstraZeneca. The authors reported financial affiliations with Abbott, AbbVie, Astellas, Ayumi Pharmaceutical, Bristol-Myers Squibb, Novartis, Pfizer, and several others.
SOURCE: Apostolopoulos D et al. Lancet Rheumatol. 2019 Nov 25. doi: 10.1016/S2665-9913(19)30105-5.
FROM THE LANCET RHEUMATOLOGY
Fewer people are dying from systemic sclerosis at younger ages
ATLANTA – Patients with systemic sclerosis aged 44 years and younger in the United States now have mortality comparable to that of the general population in that age group, according to recent results presented at the annual meeting of the American College of Rheumatology.

“Mortality for scleroderma has steadily decreased in younger ages for the last 5 decades,” Ram R. Singh, MD, professor of medicine, pathology, and laboratory medicine at the University of California, Los Angeles, said in his presentation.
Using the Centers for Disease Control and Prevention’s National Vital Statistics System database, Dr. Singh and colleagues analyzed data of adults with systemic sclerosis (SSc) and identified 46,798 adults who died between 1968 and 2015. They divided the adults with and without SSc into three different age groups: 44 and younger, 45-64, and 65 and older. The researchers performed a joinpoint trend analysis, calculating the annual percent change (APC) and average APC as well as the age-standardized mortality rate (ASMR) in each age group.
In 1968, 466 deaths were attributed to SSc, compared with 1,195 deaths in 2015. Between 1968 and 2015, there was a 19% cumulative percentage increase in SSc-related deaths, compared with a 44% decrease in mortality not attributed to SSc; when the researchers analyzed the ratio of SSc-related ASMR to non-SSc-related ASMR, there was an increase of 112%, Dr. Singh said.
When analyzing the mortality of adults with SSc by age group during 1968-2015 using the CDC’s database, Dr. Singh and colleagues found 5,457 deaths in adults 44 and younger (11.7%), 18,395 deaths in adults aged 45-64 (39.3%), and 22,946 deaths in adults aged 65 and older (49.0%), compared with totals for the general population of 10.3 million deaths in adults aged 44 and younger (9.7%), 20.8 million deaths in adults 45-64 years (19.6%), and 74.8 million deaths in adults aged 65 and older (70.6%).
Over the 48-year period, there were three major trends in SSc-related ASMR, Dr. Singh noted. In the first trend period between 1968 and 1988, there was a 1.0% increase per year (95% confidence interval, 0.6%-1.4%). The second trend period, lasting until 2000, saw a 2.2% increase per year (95% CI, 1.6%-2.7%), while the SSc-related ASMR declined by 2.6% per year in the third trend period from 2001 to 2015 (95% CI, –3.1% to –2.2%).
The percentage of annual deaths for adults with SSc decreased between 1968 and 2015, from 23.4% to 5.7%, and the average APC was greater among adults aged 44 and younger with SSc (–2.2%; 95% CI, –2.4% to –2.0%) than for adults without SSc in the same age group (–1.5%; 95% CI, –1.9% to –1.1%).
There was a cumulative 60% decrease in the ASMR of adults with SSc aged 44 and younger between 1968 and 2015 from an ASMR of 1.0 per million (95% CI, 0.8%-1.2%) to an ASMR of 0.4 per million (0.3-0.5). Adults aged 45-64 years with SSc had a cumulative 20.3% decrease in ASMR over the same time period, with an ASMR of 5.9 per million in 1968 (95% CI, 5.2-6.7) and an ASMR of 4.7 per million in 2015 (95% CI, 4.2-5.2). However, adults aged 65 and older with SSc had a 187% cumulative increase in ASMR, with an ASMR of 5.4 per million in 1968 (95% CI, 4.4-6.5) and an ASMR of 15.5 per million in 2015 (95% CI, 14.3-16.6). Adults with non-SSc-related deaths between 1968 and 2015 had a 50.0% cumulative decrease in ASMR in the group aged 44 and under, a 48.0% cumulative decrease in the 45-year to 64-year-old group, and a 42.1% decrease in the 65-year-old or older group.
The ratio of SSc to non-SSc ASMRs between 1968 and 2015 in the group aged 44 and younger declined 20.0%, whereas there was a 53.1% cumulative increase in the 45- to 64-year-old group and a 395.4% cumulative increase in the 65-year-old and older group. In the oldest group, the APC increased by 3.9% each year for 33 years (95% CI, 3.7%-4.1%) before declining by 1.6% until 2015 (95% CI, –2.0 to –1.3). In contrast, the APC for adults 44 and younger never significantly increased over the 48 years, Dr. Singh noted.
“Increasing scleroderma mortality in older age could be due to improving survival and/or increasing age of onset of scleroderma,” he said.
Dr. Singh reported no conflicts of interest.
SOURCE: Yen E et al. Arthritis Rheumatol. 2019;71(suppl 10), Abstract 825.
ATLANTA – Patients with systemic sclerosis aged 44 years and younger in the United States now have mortality comparable to that of the general population in that age group, according to recent results presented at the annual meeting of the American College of Rheumatology.
“Mortality for scleroderma has steadily decreased in younger ages for the last 5 decades,” Ram R. Singh, MD, professor of medicine, pathology, and laboratory medicine at the University of California, Los Angeles, said in his presentation.
Using the Centers for Disease Control and Prevention’s National Vital Statistics System database, Dr. Singh and colleagues analyzed data of adults with systemic sclerosis (SSc) and identified 46,798 adults who died between 1968 and 2015. They divided the adults with and without SSc into three different age groups: 44 and younger, 45-64, and 65 and older. The researchers performed a joinpoint trend analysis, calculating the annual percent change (APC) and average APC as well as the age-standardized mortality rate (ASMR) in each age group.
In 1968, 466 deaths were attributed to SSc, compared with 1,195 deaths in 2015. Between 1968 and 2015, there was a 19% cumulative percentage increase in SSc-related deaths, compared with a 44% decrease in mortality not attributed to SSc; when the researchers analyzed the ratio of SSc-related ASMR to non-SSc-related ASMR, there was an increase of 112%, Dr. Singh said.
When analyzing the mortality of adults with SSc by age group during 1968-2015 using the CDC’s database, Dr. Singh and colleagues found 5,457 deaths in adults 44 and younger (11.7%), 18,395 deaths in adults aged 45-64 (39.3%), and 22,946 deaths in adults aged 65 and older (49.0%), compared with totals for the general population of 10.3 million deaths in adults aged 44 and younger (9.7%), 20.8 million deaths in adults 45-64 years (19.6%), and 74.8 million deaths in adults aged 65 and older (70.6%).
Over the 48-year period, there were three major trends in SSc-related ASMR, Dr. Singh noted. In the first trend period between 1968 and 1988, there was a 1.0% increase per year (95% confidence interval, 0.6%-1.4%). The second trend period, lasting until 2000, saw a 2.2% increase per year (95% CI, 1.6%-2.7%), while the SSc-related ASMR declined by 2.6% per year in the third trend period from 2001 to 2015 (95% CI, –3.1% to –2.2%).
The percentage of annual deaths for adults with SSc decreased between 1968 and 2015, from 23.4% to 5.7%, and the average APC was greater among adults aged 44 and younger with SSc (–2.2%; 95% CI, –2.4% to –2.0%) than for adults without SSc in the same age group (–1.5%; 95% CI, –1.9% to –1.1%).
There was a cumulative 60% decrease in the ASMR of adults with SSc aged 44 and younger between 1968 and 2015 from an ASMR of 1.0 per million (95% CI, 0.8%-1.2%) to an ASMR of 0.4 per million (0.3-0.5). Adults aged 45-64 years with SSc had a cumulative 20.3% decrease in ASMR over the same time period, with an ASMR of 5.9 per million in 1968 (95% CI, 5.2-6.7) and an ASMR of 4.7 per million in 2015 (95% CI, 4.2-5.2). However, adults aged 65 and older with SSc had a 187% cumulative increase in ASMR, with an ASMR of 5.4 per million in 1968 (95% CI, 4.4-6.5) and an ASMR of 15.5 per million in 2015 (95% CI, 14.3-16.6). Adults with non-SSc-related deaths between 1968 and 2015 had a 50.0% cumulative decrease in ASMR in the group aged 44 and under, a 48.0% cumulative decrease in the 45-year to 64-year-old group, and a 42.1% decrease in the 65-year-old or older group.
The ratio of SSc to non-SSc ASMRs between 1968 and 2015 in the group aged 44 and younger declined 20.0%, whereas there was a 53.1% cumulative increase in the 45- to 64-year-old group and a 395.4% cumulative increase in the 65-year-old and older group. In the oldest group, the APC increased by 3.9% each year for 33 years (95% CI, 3.7%-4.1%) before declining by 1.6% until 2015 (95% CI, –2.0 to –1.3). In contrast, the APC for adults 44 and younger never significantly increased over the 48 years, Dr. Singh noted.
“Increasing scleroderma mortality in older age could be due to improving survival and/or increasing age of onset of scleroderma,” he said.
Dr. Singh reported no conflicts of interest.
SOURCE: Yen E et al. Arthritis Rheumatol. 2019;71(suppl 10), Abstract 825.
ATLANTA – Patients with systemic sclerosis aged 44 years and younger in the United States now have mortality comparable to that of the general population in that age group, according to recent results presented at the annual meeting of the American College of Rheumatology.
“Mortality for scleroderma has steadily decreased in younger ages for the last 5 decades,” Ram R. Singh, MD, professor of medicine, pathology, and laboratory medicine at the University of California, Los Angeles, said in his presentation.
Using the Centers for Disease Control and Prevention’s National Vital Statistics System database, Dr. Singh and colleagues analyzed data of adults with systemic sclerosis (SSc) and identified 46,798 adults who died between 1968 and 2015. They divided the adults with and without SSc into three different age groups: 44 and younger, 45-64, and 65 and older. The researchers performed a joinpoint trend analysis, calculating the annual percent change (APC) and average APC as well as the age-standardized mortality rate (ASMR) in each age group.
In 1968, 466 deaths were attributed to SSc, compared with 1,195 deaths in 2015. Between 1968 and 2015, there was a 19% cumulative percentage increase in SSc-related deaths, compared with a 44% decrease in mortality not attributed to SSc; when the researchers analyzed the ratio of SSc-related ASMR to non-SSc-related ASMR, there was an increase of 112%, Dr. Singh said.
When analyzing the mortality of adults with SSc by age group during 1968-2015 using the CDC’s database, Dr. Singh and colleagues found 5,457 deaths in adults 44 and younger (11.7%), 18,395 deaths in adults aged 45-64 (39.3%), and 22,946 deaths in adults aged 65 and older (49.0%), compared with totals for the general population of 10.3 million deaths in adults aged 44 and younger (9.7%), 20.8 million deaths in adults 45-64 years (19.6%), and 74.8 million deaths in adults aged 65 and older (70.6%).
Over the 48-year period, there were three major trends in SSc-related ASMR, Dr. Singh noted. In the first trend period between 1968 and 1988, there was a 1.0% increase per year (95% confidence interval, 0.6%-1.4%). The second trend period, lasting until 2000, saw a 2.2% increase per year (95% CI, 1.6%-2.7%), while the SSc-related ASMR declined by 2.6% per year in the third trend period from 2001 to 2015 (95% CI, –3.1% to –2.2%).
The percentage of annual deaths for adults with SSc decreased between 1968 and 2015, from 23.4% to 5.7%, and the average APC was greater among adults aged 44 and younger with SSc (–2.2%; 95% CI, –2.4% to –2.0%) than for adults without SSc in the same age group (–1.5%; 95% CI, –1.9% to –1.1%).
There was a cumulative 60% decrease in the ASMR of adults with SSc aged 44 and younger between 1968 and 2015 from an ASMR of 1.0 per million (95% CI, 0.8%-1.2%) to an ASMR of 0.4 per million (0.3-0.5). Adults aged 45-64 years with SSc had a cumulative 20.3% decrease in ASMR over the same time period, with an ASMR of 5.9 per million in 1968 (95% CI, 5.2-6.7) and an ASMR of 4.7 per million in 2015 (95% CI, 4.2-5.2). However, adults aged 65 and older with SSc had a 187% cumulative increase in ASMR, with an ASMR of 5.4 per million in 1968 (95% CI, 4.4-6.5) and an ASMR of 15.5 per million in 2015 (95% CI, 14.3-16.6). Adults with non-SSc-related deaths between 1968 and 2015 had a 50.0% cumulative decrease in ASMR in the group aged 44 and under, a 48.0% cumulative decrease in the 45-year to 64-year-old group, and a 42.1% decrease in the 65-year-old or older group.
The ratio of SSc to non-SSc ASMRs between 1968 and 2015 in the group aged 44 and younger declined 20.0%, whereas there was a 53.1% cumulative increase in the 45- to 64-year-old group and a 395.4% cumulative increase in the 65-year-old and older group. In the oldest group, the APC increased by 3.9% each year for 33 years (95% CI, 3.7%-4.1%) before declining by 1.6% until 2015 (95% CI, –2.0 to –1.3). In contrast, the APC for adults 44 and younger never significantly increased over the 48 years, Dr. Singh noted.
“Increasing scleroderma mortality in older age could be due to improving survival and/or increasing age of onset of scleroderma,” he said.
Dr. Singh reported no conflicts of interest.
SOURCE: Yen E et al. Arthritis Rheumatol. 2019;71(suppl 10), Abstract 825.
REPORTING FROM ACR 2019
CVD risk in black SLE patients 18 times higher than in whites
ATLANTA – Black race was the single greatest predictor of cardiovascular disease (CVD) events in systemic lupus erythematosus, with black patients having an 18-fold higher risk than white patients from 2 years before to 8 years after diagnosis, according to a review of 336 patients in the Georgia Lupus Registry that was presented at the annual meeting of the American College of Rheumatology.
The greatest risk was in the first 2 years after diagnosis, which has been reported before in white patients, but not before in a mostly (75%) black cohort.
Lupus is known to strike earlier and be more aggressive in black patients, so “we were expecting racial disparities in incident CVD, but” the magnitude of the increased risk “was very surprising. This study [identifies] a population that needs more attention, more targeted CVD prevention. We have to intervene early and be on top of everything,” especially for black patients, said lead investigator Shivani Garg, MD, an assistant professor of rheumatology at the University of Wisconsin–Madison.
Lipids, blood pressure, and the other usual CVD risk factors, as well as lupus itself, have to be optimally controlled; glucocorticoid use limited as much as possible; and there needs to be improved adherence to hydroxychloroquine, which has been shown to reduce CVD events in lupus patients, she said in an interview.
The 336 patients, mostly women (87%) from the Atlanta area, were diagnosed during 2002-2004 at a mean age of 40 years. Dr. Garg and associates reviewed CVD events – ischemic heart disease, stroke, transient ischemic attack, and peripheral vascular disease – and death over 16 years, beginning 2 years before diagnosis.
About 22% of subjects had a CVD event, most commonly within 2 years after diagnosis. The risk was 500% higher in black patients overall (adjusted hazard ratio, 6.4; 95% confidence interval, 2.4-17.5; P = .0003), and markedly higher in the first 10 years (aHR, 18; 95% CI, 2.2-141; P less than .0001). The findings were not adjusted for socioeconomic factors.
In the first 12 years of the study, the mean age at lupus diagnosis was 46 years and the first CVD event occurred at an average of 48 years. From 12 to 16 years follow-up, the mean age of diagnosis was 38 years, and the first CVD event occurred at 52 years.
Age older than 65 years (aHR, 7.9; 95% CI, 2.2-29) and the presence of disease-associated antibodies (aHR, 2.1; 95% CI, 1.01-4.4) increased CVD risk, which wasn’t surprising, but another predictor – discoid lupus – was unexpected (aHR, 3.2; 95% CI, 1.5-6.8). “A lot of times, we’ve considered discoid rash to be a milder form, but these patients have some kind of chronic, smoldering inflammation that is leading to atherosclerosis,” Dr. Garg said.
At diagnosis, 84% of the subjects had lupus hematologic disorders, 69% immunologic disorders, and 14% a discoid rash. CVD risk factor data were not collected.
There was no external funding, and the investigators reported no disclosures.
SOURCE: Garg S et al. Arthritis Rheumatol. 2019;71(suppl 10), Abstract 805.
ATLANTA – Black race was the single greatest predictor of cardiovascular disease (CVD) events in systemic lupus erythematosus, with black patients having an 18-fold higher risk than white patients from 2 years before to 8 years after diagnosis, according to a review of 336 patients in the Georgia Lupus Registry that was presented at the annual meeting of the American College of Rheumatology.
The greatest risk was in the first 2 years after diagnosis, which has been reported before in white patients, but not before in a mostly (75%) black cohort.
Lupus is known to strike earlier and be more aggressive in black patients, so “we were expecting racial disparities in incident CVD, but” the magnitude of the increased risk “was very surprising. This study [identifies] a population that needs more attention, more targeted CVD prevention. We have to intervene early and be on top of everything,” especially for black patients, said lead investigator Shivani Garg, MD, an assistant professor of rheumatology at the University of Wisconsin–Madison.
Lipids, blood pressure, and the other usual CVD risk factors, as well as lupus itself, have to be optimally controlled; glucocorticoid use limited as much as possible; and there needs to be improved adherence to hydroxychloroquine, which has been shown to reduce CVD events in lupus patients, she said in an interview.
The 336 patients, mostly women (87%) from the Atlanta area, were diagnosed during 2002-2004 at a mean age of 40 years. Dr. Garg and associates reviewed CVD events – ischemic heart disease, stroke, transient ischemic attack, and peripheral vascular disease – and death over 16 years, beginning 2 years before diagnosis.
About 22% of subjects had a CVD event, most commonly within 2 years after diagnosis. The risk was 500% higher in black patients overall (adjusted hazard ratio, 6.4; 95% confidence interval, 2.4-17.5; P = .0003), and markedly higher in the first 10 years (aHR, 18; 95% CI, 2.2-141; P less than .0001). The findings were not adjusted for socioeconomic factors.
In the first 12 years of the study, the mean age at lupus diagnosis was 46 years and the first CVD event occurred at an average of 48 years. From 12 to 16 years follow-up, the mean age of diagnosis was 38 years, and the first CVD event occurred at 52 years.
Age older than 65 years (aHR, 7.9; 95% CI, 2.2-29) and the presence of disease-associated antibodies (aHR, 2.1; 95% CI, 1.01-4.4) increased CVD risk, which wasn’t surprising, but another predictor – discoid lupus – was unexpected (aHR, 3.2; 95% CI, 1.5-6.8). “A lot of times, we’ve considered discoid rash to be a milder form, but these patients have some kind of chronic, smoldering inflammation that is leading to atherosclerosis,” Dr. Garg said.
At diagnosis, 84% of the subjects had lupus hematologic disorders, 69% immunologic disorders, and 14% a discoid rash. CVD risk factor data were not collected.
There was no external funding, and the investigators reported no disclosures.
SOURCE: Garg S et al. Arthritis Rheumatol. 2019;71(suppl 10), Abstract 805.
ATLANTA – Black race was the single greatest predictor of cardiovascular disease (CVD) events in systemic lupus erythematosus, with black patients having an 18-fold higher risk than white patients from 2 years before to 8 years after diagnosis, according to a review of 336 patients in the Georgia Lupus Registry that was presented at the annual meeting of the American College of Rheumatology.
The greatest risk was in the first 2 years after diagnosis, which has been reported before in white patients, but not before in a mostly (75%) black cohort.
Lupus is known to strike earlier and be more aggressive in black patients, so “we were expecting racial disparities in incident CVD, but” the magnitude of the increased risk “was very surprising. This study [identifies] a population that needs more attention, more targeted CVD prevention. We have to intervene early and be on top of everything,” especially for black patients, said lead investigator Shivani Garg, MD, an assistant professor of rheumatology at the University of Wisconsin–Madison.
Lipids, blood pressure, and the other usual CVD risk factors, as well as lupus itself, have to be optimally controlled; glucocorticoid use limited as much as possible; and there needs to be improved adherence to hydroxychloroquine, which has been shown to reduce CVD events in lupus patients, she said in an interview.
The 336 patients, mostly women (87%) from the Atlanta area, were diagnosed during 2002-2004 at a mean age of 40 years. Dr. Garg and associates reviewed CVD events – ischemic heart disease, stroke, transient ischemic attack, and peripheral vascular disease – and death over 16 years, beginning 2 years before diagnosis.
About 22% of subjects had a CVD event, most commonly within 2 years after diagnosis. The risk was 500% higher in black patients overall (adjusted hazard ratio, 6.4; 95% confidence interval, 2.4-17.5; P = .0003), and markedly higher in the first 10 years (aHR, 18; 95% CI, 2.2-141; P less than .0001). The findings were not adjusted for socioeconomic factors.
In the first 12 years of the study, the mean age at lupus diagnosis was 46 years and the first CVD event occurred at an average of 48 years. From 12 to 16 years follow-up, the mean age of diagnosis was 38 years, and the first CVD event occurred at 52 years.
Age older than 65 years (aHR, 7.9; 95% CI, 2.2-29) and the presence of disease-associated antibodies (aHR, 2.1; 95% CI, 1.01-4.4) increased CVD risk, which wasn’t surprising, but another predictor – discoid lupus – was unexpected (aHR, 3.2; 95% CI, 1.5-6.8). “A lot of times, we’ve considered discoid rash to be a milder form, but these patients have some kind of chronic, smoldering inflammation that is leading to atherosclerosis,” Dr. Garg said.
At diagnosis, 84% of the subjects had lupus hematologic disorders, 69% immunologic disorders, and 14% a discoid rash. CVD risk factor data were not collected.
There was no external funding, and the investigators reported no disclosures.
SOURCE: Garg S et al. Arthritis Rheumatol. 2019;71(suppl 10), Abstract 805.
REPORTING FROM ACR 2019
Vitiligo: First-ever RCT is smashing success
MADRID – cream for the treatment of vitiligo, Amit G. Pandya, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.

“I have been waiting 30 years for the first clinical trial for vitiligo. I know many of you dermatologists have been waiting for something for vitiligo, so I’m happy to present the results of the first randomized, placebo-controlled, double-blind, prospective trial of a topical agent in history for vitiligo,” said Dr. Pandya, who was clearly overjoyed to present the final results of the 52-week trial.
Ruxolitinib is a Janus kinase (JAK) 1 and 2 inhibitor. Topical ruxolitinib is under study for vitiligo because this chronic autoimmune disease targeting melanocytes is now recognized as being driven by signaling through the JAK 1/2 pathways.
The interim 24-week results of the phase 2 trial, presented earlier in the year at the World Congress of Dermatology in Milan, showed significant repigmentation with ruxolitinib cream. Dr. Pandya’s key message at EADV 2019 was that continued treatment out to a year brought substantial further improvement, and with a benign safety profile indistinguishable from vehicle control.
“We see a tremendous difference between 6 months and 1 year,” said Dr. Pandya, professor of dermatology at the University of Texas, Dallas. “For the first time, we dare talk about F-VASI75 [Facial Vitiligo Area Scoring Index] and F-VASI90 responses. We don’t usually tell patients that they can get 75% or 90% of their color back, and yet the week-52 F-VASI75 rate was 51.5%, up from 30.3% at week 24. And the F-VASI90 response was 33.3%, versus 12.1% at week 24.”
F-VASI is measured using the patient’s hand, which is typically equivalent to about 1% of body surface area. The mean baseline F-VASI was 1.26% in this study of 157 mostly middle-aged adults with longstanding vitiligo of a mean 14-year duration. That’s fairly severe vitiligo, since the total face occupies only about 4% of total body surface area.
The primary study endpoint was achievement of greater than 50% repigmentation in the F-VASI, or an F-VASI50 response. Under double-blind conditions at 52 weeks in the group randomized to 1.5% ruxolitinib cream twice a day, the highest dose used in the trial, the F-VASI50 rate was 57.6%. That’s up from a week-24 F-VASI50 of 45.5%, and a week-34 response rate of 51.5%.
A key secondary endpoint was T-VASI50, reflecting the total body response.
“Patients don’t just want their face to be better, they want their chest, arms, elbows, knees, hands, and feet to be better,” the dermatologist commented.
The week-52 T-VASI50 rate was 36.4%, up substantially from 12.1% at week 24. And that week-52 T-VASI50 rate probably underestimates the full potential benefit. That’s because a safety-based study rule prohibited patients from applying the cream to more than 20% of their body surface area. Adverse effects reported for oral ruxolitinib, approved for treatment of myelofibrosis, polycythemia vera, and acute graft-versus-host disease, include thrombocytopenia and anemia.
“In this early study we didn’t want to take a chance of systemic absorption with serum levels that would potentially affect the bone marrow,” Dr. Pandya explained.
He noted that 57 study participants had a baseline T-VASI greater than 20% of their body surface area and thus weren’t able to treat all of their disease. In the 100 patients with a vitiligo-involved total body surface area of 20% or less, however, the week-52 T-VASI50 reached 45%, compared with 20% at week 24.
Another prespecified secondary endpoint was the proportion of patients who received a facial physician’s global assessment of clear or almost clear. About 21% of patients in the highest-dose group achieved this milestone at 52 weeks.
A phase 3, randomized, controlled trial of ruxolitinib cream is ongoing and should be completed next year. Dr. Pandya reported receiving research funding from and serving as a consultant to Incyte, the study sponsor. He has similar financial relationships with Pfizer, Aclaris Therapeutics, and the Immune Tolerance Network.
MADRID – cream for the treatment of vitiligo, Amit G. Pandya, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“I have been waiting 30 years for the first clinical trial for vitiligo. I know many of you dermatologists have been waiting for something for vitiligo, so I’m happy to present the results of the first randomized, placebo-controlled, double-blind, prospective trial of a topical agent in history for vitiligo,” said Dr. Pandya, who was clearly overjoyed to present the final results of the 52-week trial.
Ruxolitinib is a Janus kinase (JAK) 1 and 2 inhibitor. Topical ruxolitinib is under study for vitiligo because this chronic autoimmune disease targeting melanocytes is now recognized as being driven by signaling through the JAK 1/2 pathways.
The interim 24-week results of the phase 2 trial, presented earlier in the year at the World Congress of Dermatology in Milan, showed significant repigmentation with ruxolitinib cream. Dr. Pandya’s key message at EADV 2019 was that continued treatment out to a year brought substantial further improvement, and with a benign safety profile indistinguishable from vehicle control.
“We see a tremendous difference between 6 months and 1 year,” said Dr. Pandya, professor of dermatology at the University of Texas, Dallas. “For the first time, we dare talk about F-VASI75 [Facial Vitiligo Area Scoring Index] and F-VASI90 responses. We don’t usually tell patients that they can get 75% or 90% of their color back, and yet the week-52 F-VASI75 rate was 51.5%, up from 30.3% at week 24. And the F-VASI90 response was 33.3%, versus 12.1% at week 24.”
F-VASI is measured using the patient’s hand, which is typically equivalent to about 1% of body surface area. The mean baseline F-VASI was 1.26% in this study of 157 mostly middle-aged adults with longstanding vitiligo of a mean 14-year duration. That’s fairly severe vitiligo, since the total face occupies only about 4% of total body surface area.
The primary study endpoint was achievement of greater than 50% repigmentation in the F-VASI, or an F-VASI50 response. Under double-blind conditions at 52 weeks in the group randomized to 1.5% ruxolitinib cream twice a day, the highest dose used in the trial, the F-VASI50 rate was 57.6%. That’s up from a week-24 F-VASI50 of 45.5%, and a week-34 response rate of 51.5%.
A key secondary endpoint was T-VASI50, reflecting the total body response.
“Patients don’t just want their face to be better, they want their chest, arms, elbows, knees, hands, and feet to be better,” the dermatologist commented.
The week-52 T-VASI50 rate was 36.4%, up substantially from 12.1% at week 24. And that week-52 T-VASI50 rate probably underestimates the full potential benefit. That’s because a safety-based study rule prohibited patients from applying the cream to more than 20% of their body surface area. Adverse effects reported for oral ruxolitinib, approved for treatment of myelofibrosis, polycythemia vera, and acute graft-versus-host disease, include thrombocytopenia and anemia.
“In this early study we didn’t want to take a chance of systemic absorption with serum levels that would potentially affect the bone marrow,” Dr. Pandya explained.
He noted that 57 study participants had a baseline T-VASI greater than 20% of their body surface area and thus weren’t able to treat all of their disease. In the 100 patients with a vitiligo-involved total body surface area of 20% or less, however, the week-52 T-VASI50 reached 45%, compared with 20% at week 24.
Another prespecified secondary endpoint was the proportion of patients who received a facial physician’s global assessment of clear or almost clear. About 21% of patients in the highest-dose group achieved this milestone at 52 weeks.
A phase 3, randomized, controlled trial of ruxolitinib cream is ongoing and should be completed next year. Dr. Pandya reported receiving research funding from and serving as a consultant to Incyte, the study sponsor. He has similar financial relationships with Pfizer, Aclaris Therapeutics, and the Immune Tolerance Network.
MADRID – cream for the treatment of vitiligo, Amit G. Pandya, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“I have been waiting 30 years for the first clinical trial for vitiligo. I know many of you dermatologists have been waiting for something for vitiligo, so I’m happy to present the results of the first randomized, placebo-controlled, double-blind, prospective trial of a topical agent in history for vitiligo,” said Dr. Pandya, who was clearly overjoyed to present the final results of the 52-week trial.
Ruxolitinib is a Janus kinase (JAK) 1 and 2 inhibitor. Topical ruxolitinib is under study for vitiligo because this chronic autoimmune disease targeting melanocytes is now recognized as being driven by signaling through the JAK 1/2 pathways.
The interim 24-week results of the phase 2 trial, presented earlier in the year at the World Congress of Dermatology in Milan, showed significant repigmentation with ruxolitinib cream. Dr. Pandya’s key message at EADV 2019 was that continued treatment out to a year brought substantial further improvement, and with a benign safety profile indistinguishable from vehicle control.
“We see a tremendous difference between 6 months and 1 year,” said Dr. Pandya, professor of dermatology at the University of Texas, Dallas. “For the first time, we dare talk about F-VASI75 [Facial Vitiligo Area Scoring Index] and F-VASI90 responses. We don’t usually tell patients that they can get 75% or 90% of their color back, and yet the week-52 F-VASI75 rate was 51.5%, up from 30.3% at week 24. And the F-VASI90 response was 33.3%, versus 12.1% at week 24.”
F-VASI is measured using the patient’s hand, which is typically equivalent to about 1% of body surface area. The mean baseline F-VASI was 1.26% in this study of 157 mostly middle-aged adults with longstanding vitiligo of a mean 14-year duration. That’s fairly severe vitiligo, since the total face occupies only about 4% of total body surface area.
The primary study endpoint was achievement of greater than 50% repigmentation in the F-VASI, or an F-VASI50 response. Under double-blind conditions at 52 weeks in the group randomized to 1.5% ruxolitinib cream twice a day, the highest dose used in the trial, the F-VASI50 rate was 57.6%. That’s up from a week-24 F-VASI50 of 45.5%, and a week-34 response rate of 51.5%.
A key secondary endpoint was T-VASI50, reflecting the total body response.
“Patients don’t just want their face to be better, they want their chest, arms, elbows, knees, hands, and feet to be better,” the dermatologist commented.
The week-52 T-VASI50 rate was 36.4%, up substantially from 12.1% at week 24. And that week-52 T-VASI50 rate probably underestimates the full potential benefit. That’s because a safety-based study rule prohibited patients from applying the cream to more than 20% of their body surface area. Adverse effects reported for oral ruxolitinib, approved for treatment of myelofibrosis, polycythemia vera, and acute graft-versus-host disease, include thrombocytopenia and anemia.
“In this early study we didn’t want to take a chance of systemic absorption with serum levels that would potentially affect the bone marrow,” Dr. Pandya explained.
He noted that 57 study participants had a baseline T-VASI greater than 20% of their body surface area and thus weren’t able to treat all of their disease. In the 100 patients with a vitiligo-involved total body surface area of 20% or less, however, the week-52 T-VASI50 reached 45%, compared with 20% at week 24.
Another prespecified secondary endpoint was the proportion of patients who received a facial physician’s global assessment of clear or almost clear. About 21% of patients in the highest-dose group achieved this milestone at 52 weeks.
A phase 3, randomized, controlled trial of ruxolitinib cream is ongoing and should be completed next year. Dr. Pandya reported receiving research funding from and serving as a consultant to Incyte, the study sponsor. He has similar financial relationships with Pfizer, Aclaris Therapeutics, and the Immune Tolerance Network.
REPORTING FROM THE EADV CONGRESS