User login
Low disease activity a valid target in early SLE, investigators say
, investigators in a small retrospective study have concluded.
Both complete remission (CR) and achievement of a lupus low disease activity state (LLDAS) were independently associated with lower accrual of early damage after 6 months of treatment, according to results of the study, reported in Arthritis Care & Research.
Patients maintaining LLDAS over the next 12 months developed considerably less damage than did those who never achieved LLDAS or who did not maintain LLDAS over time, according to the investigators, led by Alberto Floris, MD, and Matteo Piga, MD, of the AOU University Clinic of Cagliari (Italy).
Based on these findings, maintenance of both CR and LLDAS should be targeted for damage prevention, according to the authors. “Significant differences did not emerge between persistent LLDAS and CR over 12 months in preventing early damage,” they said in their report.
Their retrospective, single-center analysis included 116 adult patients with newly diagnosed SLE who had an SLE Disease Activity Index 2000 (SLEDAI-2K) score of at least 6 at baseline and started treatment at enrollment. The mean age of this patient cohort was approximately 35 years, and about 90% were female.
After 6 months of follow-up, 42% of the cohort achieved LLDAS, defined in part as a SLEDAI-2K of 4 or less with no major organ activity or new disease activity, while 21.6% met CR criteria, which included a SLEDAI-2K of 0.
Achieving LLDAS at that 6-month time point was independently associated with lower damage accrual at 18 months (odds ratio, 0.25; 95% confidence interval, 0.06-0.99; P = .049); likewise, a 6-month CR translated into less damage at 18 months, according to multivariate analysis (OR, 0.07; 95% CI, 0.01-0.59; P = .015).
Those patients in LLDAS at 6 months who maintained that target for another 12 months had significantly less damage when compared with those who lost response or never achieved it, the investigators added.
“Losing the early attained LLDAS and CR may frustrate prevention of damage, and therefore, LLDAS and CR maintenance should be targeted since the first stage of SLE management,” the investigators said in their report.
About 54% of the patients in LLDAS at 6 months achieved CR at an 18-month follow-up time point, while about 21% remained in LLDAS and 25% went on to develop active disease, the investigators said. Among those in CR at 6 months, 60% maintained it at 18 months, while 40% worsened to either LLDAS or active disease.
Dr. Floris, Dr. Piga, and coauthors reported no funding or competing interests related to their research.
SOURCE: Floris A et al. Arthritis Care Res. 2019 Oct 10. doi: 10.1002/acr.24086.
, investigators in a small retrospective study have concluded.
Both complete remission (CR) and achievement of a lupus low disease activity state (LLDAS) were independently associated with lower accrual of early damage after 6 months of treatment, according to results of the study, reported in Arthritis Care & Research.
Patients maintaining LLDAS over the next 12 months developed considerably less damage than did those who never achieved LLDAS or who did not maintain LLDAS over time, according to the investigators, led by Alberto Floris, MD, and Matteo Piga, MD, of the AOU University Clinic of Cagliari (Italy).
Based on these findings, maintenance of both CR and LLDAS should be targeted for damage prevention, according to the authors. “Significant differences did not emerge between persistent LLDAS and CR over 12 months in preventing early damage,” they said in their report.
Their retrospective, single-center analysis included 116 adult patients with newly diagnosed SLE who had an SLE Disease Activity Index 2000 (SLEDAI-2K) score of at least 6 at baseline and started treatment at enrollment. The mean age of this patient cohort was approximately 35 years, and about 90% were female.
After 6 months of follow-up, 42% of the cohort achieved LLDAS, defined in part as a SLEDAI-2K of 4 or less with no major organ activity or new disease activity, while 21.6% met CR criteria, which included a SLEDAI-2K of 0.
Achieving LLDAS at that 6-month time point was independently associated with lower damage accrual at 18 months (odds ratio, 0.25; 95% confidence interval, 0.06-0.99; P = .049); likewise, a 6-month CR translated into less damage at 18 months, according to multivariate analysis (OR, 0.07; 95% CI, 0.01-0.59; P = .015).
Those patients in LLDAS at 6 months who maintained that target for another 12 months had significantly less damage when compared with those who lost response or never achieved it, the investigators added.
“Losing the early attained LLDAS and CR may frustrate prevention of damage, and therefore, LLDAS and CR maintenance should be targeted since the first stage of SLE management,” the investigators said in their report.
About 54% of the patients in LLDAS at 6 months achieved CR at an 18-month follow-up time point, while about 21% remained in LLDAS and 25% went on to develop active disease, the investigators said. Among those in CR at 6 months, 60% maintained it at 18 months, while 40% worsened to either LLDAS or active disease.
Dr. Floris, Dr. Piga, and coauthors reported no funding or competing interests related to their research.
SOURCE: Floris A et al. Arthritis Care Res. 2019 Oct 10. doi: 10.1002/acr.24086.
, investigators in a small retrospective study have concluded.
Both complete remission (CR) and achievement of a lupus low disease activity state (LLDAS) were independently associated with lower accrual of early damage after 6 months of treatment, according to results of the study, reported in Arthritis Care & Research.
Patients maintaining LLDAS over the next 12 months developed considerably less damage than did those who never achieved LLDAS or who did not maintain LLDAS over time, according to the investigators, led by Alberto Floris, MD, and Matteo Piga, MD, of the AOU University Clinic of Cagliari (Italy).
Based on these findings, maintenance of both CR and LLDAS should be targeted for damage prevention, according to the authors. “Significant differences did not emerge between persistent LLDAS and CR over 12 months in preventing early damage,” they said in their report.
Their retrospective, single-center analysis included 116 adult patients with newly diagnosed SLE who had an SLE Disease Activity Index 2000 (SLEDAI-2K) score of at least 6 at baseline and started treatment at enrollment. The mean age of this patient cohort was approximately 35 years, and about 90% were female.
After 6 months of follow-up, 42% of the cohort achieved LLDAS, defined in part as a SLEDAI-2K of 4 or less with no major organ activity or new disease activity, while 21.6% met CR criteria, which included a SLEDAI-2K of 0.
Achieving LLDAS at that 6-month time point was independently associated with lower damage accrual at 18 months (odds ratio, 0.25; 95% confidence interval, 0.06-0.99; P = .049); likewise, a 6-month CR translated into less damage at 18 months, according to multivariate analysis (OR, 0.07; 95% CI, 0.01-0.59; P = .015).
Those patients in LLDAS at 6 months who maintained that target for another 12 months had significantly less damage when compared with those who lost response or never achieved it, the investigators added.
“Losing the early attained LLDAS and CR may frustrate prevention of damage, and therefore, LLDAS and CR maintenance should be targeted since the first stage of SLE management,” the investigators said in their report.
About 54% of the patients in LLDAS at 6 months achieved CR at an 18-month follow-up time point, while about 21% remained in LLDAS and 25% went on to develop active disease, the investigators said. Among those in CR at 6 months, 60% maintained it at 18 months, while 40% worsened to either LLDAS or active disease.
Dr. Floris, Dr. Piga, and coauthors reported no funding or competing interests related to their research.
SOURCE: Floris A et al. Arthritis Care Res. 2019 Oct 10. doi: 10.1002/acr.24086.
FROM ARTHRITIS CARE & RESEARCH
Systemic sclerosis raises risk of breast cancer, lung cancer, melanoma
in a population-linked cohort study published in Arthritis Care & Research.
Kathleen Morrisroe, MBBS, PhD, of St. Vincent’s Hospital Melbourne and colleagues matched deidentified patient data in the Australian Scleroderma Cohort Study (ASCS) with patients’ respective state cancer registry data between January 2008 and December 2015. The researchers also used the Australian Medical Benefit Schedule (MBS) to track health care costs for hospital admissions, presentations to the ED, other health visits, pathology, and imaging, as well as other associated costs for care, in each state. Based on this information, Dr. Morrisroe and colleagues calculated standardized incidence ratios (SIR) and standardized mortality ratios (SMR) for these patients by comparing them with the general population in Australia.
The results included 1,727 patients with systemic sclerosis (SSc) and cancer in the cohort, which consisted of mostly white (92.1%) women (85.9%) who had limited cutaneous SSc (73.9%). They were a mean of 46.6 years old when they were diagnosed with SSc and had a mean disease duration of 10.9 years. The incidence of cancer was 1.3% per year, and the overall prevalence for the cohort was 14.2%, which was higher than the general Australian population (SIR, 2.15; 95% confidence interval, 1.84-2.49). Breast cancer, melanoma, hematologic cancer, and lung cancer were the most common types of cancers found in the cohort, with early breast cancer (SIR, 3.07; 95% CI, 1.47-5.64), lung cancer (SIR, 3.07; 95% CI, 1.21-3.44), and early melanoma (SIR, 3.40; 95% CI, 1.10-7.93) having a higher incidence than the general population.
Patients with RNA polymerase III (RNAP) autoantibody had a higher incidence of early onset cancer (odds ratio, 2.9; P = .044), defined as a cancer diagnosis within 5 years of SSc diagnosis. Interstitial lung disease was also linked to an increased risk of lung cancer (OR, 2.83; P = .031), which persisted after the researchers performed a multivariate analysis.
Another factor that increased the overall risk of cancer was calcium channel blockers (OR, 1.47; P = .016), which also increased the risk of breast (OR, 1.61; P = .051) and melanoma-specific cancers (OR, 2.01; P = .042), a finding the researchers said was “unexpected, but has been reported in the literature with conflicting results.”
“This association is hypothesized to be related to the role of calcium in cell apoptosis, such as activation of the caspase pathway, induction of endonuclease activity and mitochondrial permeation,” Dr. Morrisroe and colleagues wrote.
SSc patients had more than a doubling of risk of mortality with incident cancer in comparison with SSc patients who did not have cancer (hazard ratio, 2.85; 95% CI, 1.51-5.37; P = .001). The average cost of health care annually for an SSc patient with cancer was AUD $1,496 (P less than .001), the researchers said.
This study was funded in part by Scleroderma Australia, Arthritis Australia, Actelion Australia, Bayer, CSL Biotherapies, GlaxoSmithKline Australia, and Pfizer. Dr. Morrisroe reported receiving support from Arthritis Australia and Royal Australasian College of Physicians Research Establishment Fellowships. Another author reported receiving a fellowship from the National Health and Medical Research Council of Australia. The other authors reported no relevant conflicts of interest.
SOURCE: Morrisroe K et al. Arthritis Care Res. 2019 Sep 20. doi: 10.1002/acr.24076
in a population-linked cohort study published in Arthritis Care & Research.
Kathleen Morrisroe, MBBS, PhD, of St. Vincent’s Hospital Melbourne and colleagues matched deidentified patient data in the Australian Scleroderma Cohort Study (ASCS) with patients’ respective state cancer registry data between January 2008 and December 2015. The researchers also used the Australian Medical Benefit Schedule (MBS) to track health care costs for hospital admissions, presentations to the ED, other health visits, pathology, and imaging, as well as other associated costs for care, in each state. Based on this information, Dr. Morrisroe and colleagues calculated standardized incidence ratios (SIR) and standardized mortality ratios (SMR) for these patients by comparing them with the general population in Australia.
The results included 1,727 patients with systemic sclerosis (SSc) and cancer in the cohort, which consisted of mostly white (92.1%) women (85.9%) who had limited cutaneous SSc (73.9%). They were a mean of 46.6 years old when they were diagnosed with SSc and had a mean disease duration of 10.9 years. The incidence of cancer was 1.3% per year, and the overall prevalence for the cohort was 14.2%, which was higher than the general Australian population (SIR, 2.15; 95% confidence interval, 1.84-2.49). Breast cancer, melanoma, hematologic cancer, and lung cancer were the most common types of cancers found in the cohort, with early breast cancer (SIR, 3.07; 95% CI, 1.47-5.64), lung cancer (SIR, 3.07; 95% CI, 1.21-3.44), and early melanoma (SIR, 3.40; 95% CI, 1.10-7.93) having a higher incidence than the general population.
Patients with RNA polymerase III (RNAP) autoantibody had a higher incidence of early onset cancer (odds ratio, 2.9; P = .044), defined as a cancer diagnosis within 5 years of SSc diagnosis. Interstitial lung disease was also linked to an increased risk of lung cancer (OR, 2.83; P = .031), which persisted after the researchers performed a multivariate analysis.
Another factor that increased the overall risk of cancer was calcium channel blockers (OR, 1.47; P = .016), which also increased the risk of breast (OR, 1.61; P = .051) and melanoma-specific cancers (OR, 2.01; P = .042), a finding the researchers said was “unexpected, but has been reported in the literature with conflicting results.”
“This association is hypothesized to be related to the role of calcium in cell apoptosis, such as activation of the caspase pathway, induction of endonuclease activity and mitochondrial permeation,” Dr. Morrisroe and colleagues wrote.
SSc patients had more than a doubling of risk of mortality with incident cancer in comparison with SSc patients who did not have cancer (hazard ratio, 2.85; 95% CI, 1.51-5.37; P = .001). The average cost of health care annually for an SSc patient with cancer was AUD $1,496 (P less than .001), the researchers said.
This study was funded in part by Scleroderma Australia, Arthritis Australia, Actelion Australia, Bayer, CSL Biotherapies, GlaxoSmithKline Australia, and Pfizer. Dr. Morrisroe reported receiving support from Arthritis Australia and Royal Australasian College of Physicians Research Establishment Fellowships. Another author reported receiving a fellowship from the National Health and Medical Research Council of Australia. The other authors reported no relevant conflicts of interest.
SOURCE: Morrisroe K et al. Arthritis Care Res. 2019 Sep 20. doi: 10.1002/acr.24076
in a population-linked cohort study published in Arthritis Care & Research.
Kathleen Morrisroe, MBBS, PhD, of St. Vincent’s Hospital Melbourne and colleagues matched deidentified patient data in the Australian Scleroderma Cohort Study (ASCS) with patients’ respective state cancer registry data between January 2008 and December 2015. The researchers also used the Australian Medical Benefit Schedule (MBS) to track health care costs for hospital admissions, presentations to the ED, other health visits, pathology, and imaging, as well as other associated costs for care, in each state. Based on this information, Dr. Morrisroe and colleagues calculated standardized incidence ratios (SIR) and standardized mortality ratios (SMR) for these patients by comparing them with the general population in Australia.
The results included 1,727 patients with systemic sclerosis (SSc) and cancer in the cohort, which consisted of mostly white (92.1%) women (85.9%) who had limited cutaneous SSc (73.9%). They were a mean of 46.6 years old when they were diagnosed with SSc and had a mean disease duration of 10.9 years. The incidence of cancer was 1.3% per year, and the overall prevalence for the cohort was 14.2%, which was higher than the general Australian population (SIR, 2.15; 95% confidence interval, 1.84-2.49). Breast cancer, melanoma, hematologic cancer, and lung cancer were the most common types of cancers found in the cohort, with early breast cancer (SIR, 3.07; 95% CI, 1.47-5.64), lung cancer (SIR, 3.07; 95% CI, 1.21-3.44), and early melanoma (SIR, 3.40; 95% CI, 1.10-7.93) having a higher incidence than the general population.
Patients with RNA polymerase III (RNAP) autoantibody had a higher incidence of early onset cancer (odds ratio, 2.9; P = .044), defined as a cancer diagnosis within 5 years of SSc diagnosis. Interstitial lung disease was also linked to an increased risk of lung cancer (OR, 2.83; P = .031), which persisted after the researchers performed a multivariate analysis.
Another factor that increased the overall risk of cancer was calcium channel blockers (OR, 1.47; P = .016), which also increased the risk of breast (OR, 1.61; P = .051) and melanoma-specific cancers (OR, 2.01; P = .042), a finding the researchers said was “unexpected, but has been reported in the literature with conflicting results.”
“This association is hypothesized to be related to the role of calcium in cell apoptosis, such as activation of the caspase pathway, induction of endonuclease activity and mitochondrial permeation,” Dr. Morrisroe and colleagues wrote.
SSc patients had more than a doubling of risk of mortality with incident cancer in comparison with SSc patients who did not have cancer (hazard ratio, 2.85; 95% CI, 1.51-5.37; P = .001). The average cost of health care annually for an SSc patient with cancer was AUD $1,496 (P less than .001), the researchers said.
This study was funded in part by Scleroderma Australia, Arthritis Australia, Actelion Australia, Bayer, CSL Biotherapies, GlaxoSmithKline Australia, and Pfizer. Dr. Morrisroe reported receiving support from Arthritis Australia and Royal Australasian College of Physicians Research Establishment Fellowships. Another author reported receiving a fellowship from the National Health and Medical Research Council of Australia. The other authors reported no relevant conflicts of interest.
SOURCE: Morrisroe K et al. Arthritis Care Res. 2019 Sep 20. doi: 10.1002/acr.24076
FROM ARTHRITIS CARE & RESEARCH
Zostavax proves safe, effective in patients with nonactive SLE
A live-attenuated herpes zoster vaccine can be used in individuals with systemic lupus erythematosus (SLE) if they are not intensively immunosuppressed and their condition is dormant, research suggests.
A paper published in Annals of the Rheumatic Diseases reported the outcomes of a randomized, placebo-controlled trial of the Zostavax herpes zoster vaccine in 90 adults with clinically stable SLE. Participants had to have been on a stable dose of immunosuppressive agents for at least 6 months and have a history of chicken pox or herpes zoster infection.
Chi Chiu Mok, MD, of the Tuen Mun Hospital in Hong Kong and coauthors wrote that herpes zoster reactivation has been reported to occur in 6.4 to 91.4 individuals with SLE per 1,000 patient-years, with consequences including postherpetic neuralgia and even death from disseminated infection. But because Zostavax is live-attenuated, it has not been widely used in immunocompromised people.
After a single subcutaneous dose of either the vaccine or placebo, researchers saw a significant increase in anti–varicella zoster virus (VZV) IgG antibodies in vaccinated individuals over 6 weeks. The magnitude of the increase in anti-VZV IgG seen in vaccinated individuals was on par with that previously seen in vaccinated healthy controls, although the authors noted that the absolute increase in values was lower.
“While the reason is not apparent, one contributing factor is the high rate of previous exposure to VZV infection in most participants, which could have led to a higher baseline anti-IgG anti-VZV value that limited its rise after vaccination,” the authors wrote.
In contrast, IgG reactivity declined in those who received the placebo injection, and the difference between the two groups was statistically significant after adjustment for baseline antibody titers.
The study also looked at the cell-mediated immune response to the vaccine and found the number of interferon-gamma secreting CD4+ T-cell spots increased in the vaccinated patients but decreased in the placebo arm, and by week 6 it was significantly higher in the treated group. The increase in the vaccine-treated patients was again similar to that previously seen in healthy controls.
However, prednisolone use at baseline may have attenuated the vaccine response. Vaccinated patients who were treated with prednisolone at baseline had a lower increase in T-cell spots and lower anti-VZV IgG reactivity after the vaccination than did those not taking prednisolone, although the difference between the two groups was not statistically significant. The study did not see any effect of age, sex, baseline lymphocyte count, disease activity scores, and other factors on response to the vaccine.
None of the patients who received the vaccine withdrew from the study because of serious adverse events. The most common adverse events reported were injection-site redness and pain, which were more common in the vaccine-treated group than in the placebo group. However these symptoms were mild and resolved by themselves after a few days. Two patients in the vaccine group and one in the placebo group experienced mild or moderate SLE flares.
The authors commented that this was the first randomized, controlled trial examining the safety and immune response of a live-attenuated herpes zoster vaccine in individuals with SLE and this trial showed it was safe and well tolerated in those with stable disease who were not on intensive immunosuppressive therapy.
“Despite the increased risk of HZ [herpes zoster] infection, SLE had the lowest HZ vaccination rates among age-eligible subjects, probably because of the concern of vaccine safety, the principle of contraindication to live-attenuated vaccines in immunocompromised hosts, as well as the current ambiguous guidelines for HZ vaccination in SLE,” they wrote.
But they also stressed that their results did not apply to patients with active disease or on more intensive immunosuppression and that longer-term data on the persistence of vaccine immunogenicity was still being collected.
The study was funded by the Hong Kong Research Fund Secretariat. No conflicts of interest were declared.
SOURCE: Mok CC et al. Ann Rheum Dis. 2019 Sep 17. doi: 10.1136/annrheumdis-2019-215925
Probably like me you have seen a bit of zoster in our patients with SLE, and rarely we get severe outbreaks in multiple dermatomes or in the eyes or other vulnerable areas in patients on immune suppression. So I think of Zostavax the way I think of shingles per se: The more immune compromised you are, the higher the risk of something bad happening … maybe. But we do know with Zostavax the risk is small.

Shingrix is a lot more effective than Zostavax and does not have the same issue of potentially causing the thing it prevents. But the most likely reason it works so well is that it has an adjuvant. We are generally a lot more concerned about injecting adjuvants in autoimmune patients here in the United States than they are in Europe where they have more experience with that, but this one is apparently a new adjuvant and has never been used in autoimmune patients, who were excluded from the trials of Shingrix. And a fair number of nonautoimmune patients get autoimmune-like symptoms in the Shingrix trials such as myalgias and fevers. I don’t think we have full confidence yet until we figure out just how worried we ought to be about that. In other words, if Shingrix only causes mild/moderate transient flares, then our patients might rationally consider that a fair trade for lifelong protection.
I think in some patients this is an easier decision than others. If somebody is 50 years old and healthy, hasn’t had nephritis or anything bad before (or not in the last 10 years), and is on no immune suppressant or just using stable, modest doses of such therapies, you would probably recommend doing something to avoid getting zoster. And here you can explain the choice to the patient: Zostavax provides good protection but less than Shingrix, is unlikely to make the patient flare, has very low risk of live vaccine causing much trouble in a generally healthy person; Shingrix is more effective overall, has caused some autoimmune symptoms in healthy people, and has unclear risk for a flare in a patient with a diagnosis (but that can be monitored).
For the sicker patients, we just have to weigh the risk of a natural zoster outbreak against the risk of a flare and the risk of disseminated zoster from the Zostavax, which is a pretty small risk but it is there. It’s a discussion you need to have in advance with each patient. Maybe with some patients, it is best to wait for an optimal time for either choice, when there’s not too much disease and not too much immune-compromising medication.
An unsolved issue for herpes zoster vaccination is age. Greater knowledge about how to best vaccinate would go a long way toward bolstering confidence in using the vaccines in patients a bit younger than 50 years given that zoster does occur in lupus patients at that age.
Joan Merrill, MD, is OMRF Professor of Medicine at the University of Oklahoma Health Sciences Center and a member of the Arthritis & Clinical Immunology Research Program at the Oklahoma Medical Research Foundation, both in Oklahoma City. She is a member of the editorial advisory board of Rheumatology News.
Probably like me you have seen a bit of zoster in our patients with SLE, and rarely we get severe outbreaks in multiple dermatomes or in the eyes or other vulnerable areas in patients on immune suppression. So I think of Zostavax the way I think of shingles per se: The more immune compromised you are, the higher the risk of something bad happening … maybe. But we do know with Zostavax the risk is small.
Shingrix is a lot more effective than Zostavax and does not have the same issue of potentially causing the thing it prevents. But the most likely reason it works so well is that it has an adjuvant. We are generally a lot more concerned about injecting adjuvants in autoimmune patients here in the United States than they are in Europe where they have more experience with that, but this one is apparently a new adjuvant and has never been used in autoimmune patients, who were excluded from the trials of Shingrix. And a fair number of nonautoimmune patients get autoimmune-like symptoms in the Shingrix trials such as myalgias and fevers. I don’t think we have full confidence yet until we figure out just how worried we ought to be about that. In other words, if Shingrix only causes mild/moderate transient flares, then our patients might rationally consider that a fair trade for lifelong protection.
I think in some patients this is an easier decision than others. If somebody is 50 years old and healthy, hasn’t had nephritis or anything bad before (or not in the last 10 years), and is on no immune suppressant or just using stable, modest doses of such therapies, you would probably recommend doing something to avoid getting zoster. And here you can explain the choice to the patient: Zostavax provides good protection but less than Shingrix, is unlikely to make the patient flare, has very low risk of live vaccine causing much trouble in a generally healthy person; Shingrix is more effective overall, has caused some autoimmune symptoms in healthy people, and has unclear risk for a flare in a patient with a diagnosis (but that can be monitored).
For the sicker patients, we just have to weigh the risk of a natural zoster outbreak against the risk of a flare and the risk of disseminated zoster from the Zostavax, which is a pretty small risk but it is there. It’s a discussion you need to have in advance with each patient. Maybe with some patients, it is best to wait for an optimal time for either choice, when there’s not too much disease and not too much immune-compromising medication.
An unsolved issue for herpes zoster vaccination is age. Greater knowledge about how to best vaccinate would go a long way toward bolstering confidence in using the vaccines in patients a bit younger than 50 years given that zoster does occur in lupus patients at that age.
Joan Merrill, MD, is OMRF Professor of Medicine at the University of Oklahoma Health Sciences Center and a member of the Arthritis & Clinical Immunology Research Program at the Oklahoma Medical Research Foundation, both in Oklahoma City. She is a member of the editorial advisory board of Rheumatology News.
Probably like me you have seen a bit of zoster in our patients with SLE, and rarely we get severe outbreaks in multiple dermatomes or in the eyes or other vulnerable areas in patients on immune suppression. So I think of Zostavax the way I think of shingles per se: The more immune compromised you are, the higher the risk of something bad happening … maybe. But we do know with Zostavax the risk is small.
Shingrix is a lot more effective than Zostavax and does not have the same issue of potentially causing the thing it prevents. But the most likely reason it works so well is that it has an adjuvant. We are generally a lot more concerned about injecting adjuvants in autoimmune patients here in the United States than they are in Europe where they have more experience with that, but this one is apparently a new adjuvant and has never been used in autoimmune patients, who were excluded from the trials of Shingrix. And a fair number of nonautoimmune patients get autoimmune-like symptoms in the Shingrix trials such as myalgias and fevers. I don’t think we have full confidence yet until we figure out just how worried we ought to be about that. In other words, if Shingrix only causes mild/moderate transient flares, then our patients might rationally consider that a fair trade for lifelong protection.
I think in some patients this is an easier decision than others. If somebody is 50 years old and healthy, hasn’t had nephritis or anything bad before (or not in the last 10 years), and is on no immune suppressant or just using stable, modest doses of such therapies, you would probably recommend doing something to avoid getting zoster. And here you can explain the choice to the patient: Zostavax provides good protection but less than Shingrix, is unlikely to make the patient flare, has very low risk of live vaccine causing much trouble in a generally healthy person; Shingrix is more effective overall, has caused some autoimmune symptoms in healthy people, and has unclear risk for a flare in a patient with a diagnosis (but that can be monitored).
For the sicker patients, we just have to weigh the risk of a natural zoster outbreak against the risk of a flare and the risk of disseminated zoster from the Zostavax, which is a pretty small risk but it is there. It’s a discussion you need to have in advance with each patient. Maybe with some patients, it is best to wait for an optimal time for either choice, when there’s not too much disease and not too much immune-compromising medication.
An unsolved issue for herpes zoster vaccination is age. Greater knowledge about how to best vaccinate would go a long way toward bolstering confidence in using the vaccines in patients a bit younger than 50 years given that zoster does occur in lupus patients at that age.
Joan Merrill, MD, is OMRF Professor of Medicine at the University of Oklahoma Health Sciences Center and a member of the Arthritis & Clinical Immunology Research Program at the Oklahoma Medical Research Foundation, both in Oklahoma City. She is a member of the editorial advisory board of Rheumatology News.
A live-attenuated herpes zoster vaccine can be used in individuals with systemic lupus erythematosus (SLE) if they are not intensively immunosuppressed and their condition is dormant, research suggests.
A paper published in Annals of the Rheumatic Diseases reported the outcomes of a randomized, placebo-controlled trial of the Zostavax herpes zoster vaccine in 90 adults with clinically stable SLE. Participants had to have been on a stable dose of immunosuppressive agents for at least 6 months and have a history of chicken pox or herpes zoster infection.
Chi Chiu Mok, MD, of the Tuen Mun Hospital in Hong Kong and coauthors wrote that herpes zoster reactivation has been reported to occur in 6.4 to 91.4 individuals with SLE per 1,000 patient-years, with consequences including postherpetic neuralgia and even death from disseminated infection. But because Zostavax is live-attenuated, it has not been widely used in immunocompromised people.
After a single subcutaneous dose of either the vaccine or placebo, researchers saw a significant increase in anti–varicella zoster virus (VZV) IgG antibodies in vaccinated individuals over 6 weeks. The magnitude of the increase in anti-VZV IgG seen in vaccinated individuals was on par with that previously seen in vaccinated healthy controls, although the authors noted that the absolute increase in values was lower.
“While the reason is not apparent, one contributing factor is the high rate of previous exposure to VZV infection in most participants, which could have led to a higher baseline anti-IgG anti-VZV value that limited its rise after vaccination,” the authors wrote.
In contrast, IgG reactivity declined in those who received the placebo injection, and the difference between the two groups was statistically significant after adjustment for baseline antibody titers.
The study also looked at the cell-mediated immune response to the vaccine and found the number of interferon-gamma secreting CD4+ T-cell spots increased in the vaccinated patients but decreased in the placebo arm, and by week 6 it was significantly higher in the treated group. The increase in the vaccine-treated patients was again similar to that previously seen in healthy controls.
However, prednisolone use at baseline may have attenuated the vaccine response. Vaccinated patients who were treated with prednisolone at baseline had a lower increase in T-cell spots and lower anti-VZV IgG reactivity after the vaccination than did those not taking prednisolone, although the difference between the two groups was not statistically significant. The study did not see any effect of age, sex, baseline lymphocyte count, disease activity scores, and other factors on response to the vaccine.
None of the patients who received the vaccine withdrew from the study because of serious adverse events. The most common adverse events reported were injection-site redness and pain, which were more common in the vaccine-treated group than in the placebo group. However these symptoms were mild and resolved by themselves after a few days. Two patients in the vaccine group and one in the placebo group experienced mild or moderate SLE flares.
The authors commented that this was the first randomized, controlled trial examining the safety and immune response of a live-attenuated herpes zoster vaccine in individuals with SLE and this trial showed it was safe and well tolerated in those with stable disease who were not on intensive immunosuppressive therapy.
“Despite the increased risk of HZ [herpes zoster] infection, SLE had the lowest HZ vaccination rates among age-eligible subjects, probably because of the concern of vaccine safety, the principle of contraindication to live-attenuated vaccines in immunocompromised hosts, as well as the current ambiguous guidelines for HZ vaccination in SLE,” they wrote.
But they also stressed that their results did not apply to patients with active disease or on more intensive immunosuppression and that longer-term data on the persistence of vaccine immunogenicity was still being collected.
The study was funded by the Hong Kong Research Fund Secretariat. No conflicts of interest were declared.
SOURCE: Mok CC et al. Ann Rheum Dis. 2019 Sep 17. doi: 10.1136/annrheumdis-2019-215925
A live-attenuated herpes zoster vaccine can be used in individuals with systemic lupus erythematosus (SLE) if they are not intensively immunosuppressed and their condition is dormant, research suggests.
A paper published in Annals of the Rheumatic Diseases reported the outcomes of a randomized, placebo-controlled trial of the Zostavax herpes zoster vaccine in 90 adults with clinically stable SLE. Participants had to have been on a stable dose of immunosuppressive agents for at least 6 months and have a history of chicken pox or herpes zoster infection.
Chi Chiu Mok, MD, of the Tuen Mun Hospital in Hong Kong and coauthors wrote that herpes zoster reactivation has been reported to occur in 6.4 to 91.4 individuals with SLE per 1,000 patient-years, with consequences including postherpetic neuralgia and even death from disseminated infection. But because Zostavax is live-attenuated, it has not been widely used in immunocompromised people.
After a single subcutaneous dose of either the vaccine or placebo, researchers saw a significant increase in anti–varicella zoster virus (VZV) IgG antibodies in vaccinated individuals over 6 weeks. The magnitude of the increase in anti-VZV IgG seen in vaccinated individuals was on par with that previously seen in vaccinated healthy controls, although the authors noted that the absolute increase in values was lower.
“While the reason is not apparent, one contributing factor is the high rate of previous exposure to VZV infection in most participants, which could have led to a higher baseline anti-IgG anti-VZV value that limited its rise after vaccination,” the authors wrote.
In contrast, IgG reactivity declined in those who received the placebo injection, and the difference between the two groups was statistically significant after adjustment for baseline antibody titers.
The study also looked at the cell-mediated immune response to the vaccine and found the number of interferon-gamma secreting CD4+ T-cell spots increased in the vaccinated patients but decreased in the placebo arm, and by week 6 it was significantly higher in the treated group. The increase in the vaccine-treated patients was again similar to that previously seen in healthy controls.
However, prednisolone use at baseline may have attenuated the vaccine response. Vaccinated patients who were treated with prednisolone at baseline had a lower increase in T-cell spots and lower anti-VZV IgG reactivity after the vaccination than did those not taking prednisolone, although the difference between the two groups was not statistically significant. The study did not see any effect of age, sex, baseline lymphocyte count, disease activity scores, and other factors on response to the vaccine.
None of the patients who received the vaccine withdrew from the study because of serious adverse events. The most common adverse events reported were injection-site redness and pain, which were more common in the vaccine-treated group than in the placebo group. However these symptoms were mild and resolved by themselves after a few days. Two patients in the vaccine group and one in the placebo group experienced mild or moderate SLE flares.
The authors commented that this was the first randomized, controlled trial examining the safety and immune response of a live-attenuated herpes zoster vaccine in individuals with SLE and this trial showed it was safe and well tolerated in those with stable disease who were not on intensive immunosuppressive therapy.
“Despite the increased risk of HZ [herpes zoster] infection, SLE had the lowest HZ vaccination rates among age-eligible subjects, probably because of the concern of vaccine safety, the principle of contraindication to live-attenuated vaccines in immunocompromised hosts, as well as the current ambiguous guidelines for HZ vaccination in SLE,” they wrote.
But they also stressed that their results did not apply to patients with active disease or on more intensive immunosuppression and that longer-term data on the persistence of vaccine immunogenicity was still being collected.
The study was funded by the Hong Kong Research Fund Secretariat. No conflicts of interest were declared.
SOURCE: Mok CC et al. Ann Rheum Dis. 2019 Sep 17. doi: 10.1136/annrheumdis-2019-215925
FROM ANNALS OF THE RHEUMATIC DISEASES
Study aims to define symptoms of Sjögren’s syndrome secondary to SLE
Sjögren’s syndrome secondary to systemic lupus erythematosus rises in frequency with age, affects nearly one-quarter of all people with SLE, and is marked by a systemic inflammatory state with high levels of proinflammatory cytokines.
Those are key findings from a Swedish study that set out to evaluate the subjective and objective symptoms of secondary Sjögren’s syndrome (sSS) from a large cohort of SLE patients and matched controls.
“The diagnosis SS is a clinical entity, based on dryness of eyes and mouth due to destructive inflammation in the exocrine glands, especially tear and salivary glands,” researchers led by Guillermo Ruacho, DMD, and Marika Kvarnström, MD, PhD, of the Karolinska Institute, wrote in a study published in the Journal of Rheumatology (doi: 10.3899/jrheum.190250). “SS can exist [as] isolated, primary SS (pSS) or together with other rheumatic diseases, referred to as secondary SS (sSS). A major difference according to the 2002 Revised American-European Consensus Criteria (AECC) is the classification where the serologic item (SSA/SSB antibodies) is included for pSS, but not for sSS (Ann Rheum Dis. 2002;61:554-8). In SLE, these autoantibodies are common, usually stable over time, and they appear early, even several years before disease onset.”
The researchers evaluated 504 consecutive SLE patients and 319 controls from the general population, who were matched for age and gender to the first 319 SLE patients. They used AECC to define SLE-sSS and conducted a thorough clinical investigation of all patients, including analysis of autoantibodies and 20 selected cytokines.
The researchers found that SLE-sSS occurred in 23% of the SLE patients. In comparison with SLE patients who did not have sSS, those in the SLE-sSS group were an average of 9 years older, more likely to be female (96% vs. 84%, respectively), and more likely to have leukopenia (57% vs. 45%), yet less likely to have nephritis (32% vs. 43%). Of 20 proinflammatory cytokines investigated, 6 were higher in the SLE-sSS group: TNF-alpha, IL-6, MCP-4, MIP-1beta, IL-12/IL-23p40, and IP-10. Other clinical measures higher in the SLE-sSS group were total IgG, anti-SSA/Ro52, anti-SSA/Ro60, anti-SSB/La antibodies, and rheumatoid factor (IgM and IgA; P less than .05 for all comparisons).
“To our knowledge this is the first study to investigate if systemic inflammation, as measured by cytokine levels, differs between SLE-sSS and SLE-nonsSS,” the researchers wrote. “In clinical practice, it is often difficult to delineate pSS from SLE-sSS. Organ manifestations commonly reported in pSS are fever, lymphadenopathy, parotid gland enlargement, Raynaud’s phenomenon, interstitial lung disease, peripheral neuropathy, and vasculitis. All these clinical features, except parotid gland enlargement, were investigated in the present study, but only peripheral neuropathy differed and was more frequent in SLE-sSS than in SLE-nonsSS.”
They acknowledged certain limitations of the study, including the fact that they did not measure saliva and tear production in controls without sicca symptoms.
The study was supported by funds from Swedish local and national governments, medical societies, foundations, and patient advocacy groups, One author is an employee at AstraZeneca, which provided reagents for the cytokine analyses but had no impact on the analyses, the authors said.
SOURCE: Ruacho G et al. J Rheumatol. 2019 Sep 1. doi: 10.3899/jrheum.190250.
Sjögren’s syndrome secondary to systemic lupus erythematosus rises in frequency with age, affects nearly one-quarter of all people with SLE, and is marked by a systemic inflammatory state with high levels of proinflammatory cytokines.
Those are key findings from a Swedish study that set out to evaluate the subjective and objective symptoms of secondary Sjögren’s syndrome (sSS) from a large cohort of SLE patients and matched controls.
“The diagnosis SS is a clinical entity, based on dryness of eyes and mouth due to destructive inflammation in the exocrine glands, especially tear and salivary glands,” researchers led by Guillermo Ruacho, DMD, and Marika Kvarnström, MD, PhD, of the Karolinska Institute, wrote in a study published in the Journal of Rheumatology (doi: 10.3899/jrheum.190250). “SS can exist [as] isolated, primary SS (pSS) or together with other rheumatic diseases, referred to as secondary SS (sSS). A major difference according to the 2002 Revised American-European Consensus Criteria (AECC) is the classification where the serologic item (SSA/SSB antibodies) is included for pSS, but not for sSS (Ann Rheum Dis. 2002;61:554-8). In SLE, these autoantibodies are common, usually stable over time, and they appear early, even several years before disease onset.”
The researchers evaluated 504 consecutive SLE patients and 319 controls from the general population, who were matched for age and gender to the first 319 SLE patients. They used AECC to define SLE-sSS and conducted a thorough clinical investigation of all patients, including analysis of autoantibodies and 20 selected cytokines.
The researchers found that SLE-sSS occurred in 23% of the SLE patients. In comparison with SLE patients who did not have sSS, those in the SLE-sSS group were an average of 9 years older, more likely to be female (96% vs. 84%, respectively), and more likely to have leukopenia (57% vs. 45%), yet less likely to have nephritis (32% vs. 43%). Of 20 proinflammatory cytokines investigated, 6 were higher in the SLE-sSS group: TNF-alpha, IL-6, MCP-4, MIP-1beta, IL-12/IL-23p40, and IP-10. Other clinical measures higher in the SLE-sSS group were total IgG, anti-SSA/Ro52, anti-SSA/Ro60, anti-SSB/La antibodies, and rheumatoid factor (IgM and IgA; P less than .05 for all comparisons).
“To our knowledge this is the first study to investigate if systemic inflammation, as measured by cytokine levels, differs between SLE-sSS and SLE-nonsSS,” the researchers wrote. “In clinical practice, it is often difficult to delineate pSS from SLE-sSS. Organ manifestations commonly reported in pSS are fever, lymphadenopathy, parotid gland enlargement, Raynaud’s phenomenon, interstitial lung disease, peripheral neuropathy, and vasculitis. All these clinical features, except parotid gland enlargement, were investigated in the present study, but only peripheral neuropathy differed and was more frequent in SLE-sSS than in SLE-nonsSS.”
They acknowledged certain limitations of the study, including the fact that they did not measure saliva and tear production in controls without sicca symptoms.
The study was supported by funds from Swedish local and national governments, medical societies, foundations, and patient advocacy groups, One author is an employee at AstraZeneca, which provided reagents for the cytokine analyses but had no impact on the analyses, the authors said.
SOURCE: Ruacho G et al. J Rheumatol. 2019 Sep 1. doi: 10.3899/jrheum.190250.
Sjögren’s syndrome secondary to systemic lupus erythematosus rises in frequency with age, affects nearly one-quarter of all people with SLE, and is marked by a systemic inflammatory state with high levels of proinflammatory cytokines.
Those are key findings from a Swedish study that set out to evaluate the subjective and objective symptoms of secondary Sjögren’s syndrome (sSS) from a large cohort of SLE patients and matched controls.
“The diagnosis SS is a clinical entity, based on dryness of eyes and mouth due to destructive inflammation in the exocrine glands, especially tear and salivary glands,” researchers led by Guillermo Ruacho, DMD, and Marika Kvarnström, MD, PhD, of the Karolinska Institute, wrote in a study published in the Journal of Rheumatology (doi: 10.3899/jrheum.190250). “SS can exist [as] isolated, primary SS (pSS) or together with other rheumatic diseases, referred to as secondary SS (sSS). A major difference according to the 2002 Revised American-European Consensus Criteria (AECC) is the classification where the serologic item (SSA/SSB antibodies) is included for pSS, but not for sSS (Ann Rheum Dis. 2002;61:554-8). In SLE, these autoantibodies are common, usually stable over time, and they appear early, even several years before disease onset.”
The researchers evaluated 504 consecutive SLE patients and 319 controls from the general population, who were matched for age and gender to the first 319 SLE patients. They used AECC to define SLE-sSS and conducted a thorough clinical investigation of all patients, including analysis of autoantibodies and 20 selected cytokines.
The researchers found that SLE-sSS occurred in 23% of the SLE patients. In comparison with SLE patients who did not have sSS, those in the SLE-sSS group were an average of 9 years older, more likely to be female (96% vs. 84%, respectively), and more likely to have leukopenia (57% vs. 45%), yet less likely to have nephritis (32% vs. 43%). Of 20 proinflammatory cytokines investigated, 6 were higher in the SLE-sSS group: TNF-alpha, IL-6, MCP-4, MIP-1beta, IL-12/IL-23p40, and IP-10. Other clinical measures higher in the SLE-sSS group were total IgG, anti-SSA/Ro52, anti-SSA/Ro60, anti-SSB/La antibodies, and rheumatoid factor (IgM and IgA; P less than .05 for all comparisons).
“To our knowledge this is the first study to investigate if systemic inflammation, as measured by cytokine levels, differs between SLE-sSS and SLE-nonsSS,” the researchers wrote. “In clinical practice, it is often difficult to delineate pSS from SLE-sSS. Organ manifestations commonly reported in pSS are fever, lymphadenopathy, parotid gland enlargement, Raynaud’s phenomenon, interstitial lung disease, peripheral neuropathy, and vasculitis. All these clinical features, except parotid gland enlargement, were investigated in the present study, but only peripheral neuropathy differed and was more frequent in SLE-sSS than in SLE-nonsSS.”
They acknowledged certain limitations of the study, including the fact that they did not measure saliva and tear production in controls without sicca symptoms.
The study was supported by funds from Swedish local and national governments, medical societies, foundations, and patient advocacy groups, One author is an employee at AstraZeneca, which provided reagents for the cytokine analyses but had no impact on the analyses, the authors said.
SOURCE: Ruacho G et al. J Rheumatol. 2019 Sep 1. doi: 10.3899/jrheum.190250.
FROM THE JOURNAL OF RHEUMATOLOGY
Painful and Pruritic Erosions on the Back
The Diagnosis: Bullous Systemic Lupus Erythematosus
Bullous systemic lupus erythematosus (BSLE) is a rare blistering disease that affects patients with systemic lupus erythematosus (SLE). Our patient had a several-year history of SLE and was being managed by a rheumatologist. She was taking hydroxychloroquine at the time of the flare. Although BSLE tends to present in those with SLE that has already been diagnosed, BSLE has been reported as a possible initial manifestation of SLE.1
Bullous systemic lupus erythematosus is estimated to occur in less than 5% of patients with SLE and is more common in black women between the second and third decades of life,2 though it also can be seen in the pediatric population.3 The lesions of BSLE usually present as subepidermal blisters often located on the face, neck, and arms on an erythematous or possibly urticarial base. Although non-BSLE vesiculobullous eruptions may be seen in patients with SLE, BSLE is differentiated from these other eruptions by its appearance on sun-exposed and non-sun-exposed areas of the body, while other vesiculobullous eruptions associated with SLE typically are limited to sun-exposed sites.4
Due to its clinical presentation overlapping with several vesiculobullous conditions, a set of diagnostic criteria have been suggested for BSLE, including the following: (1) fulfillment of the American Rheumatism Association's criteria for SLE5; (2) a new-onset vesiculobullous eruption, primarily on sun-exposed skin; (3) histology showing a subepidermal blister with a predominantly neutrophilic infiltrate; (4) presence of IgG, IgA, IgM, and C3 at the basement membrane zone; (5) evidence of antibodies to type VII collagen; and (6) immunoelectron microscopy showing codistribution of immunoglobulin deposits with anchoring fibrils/type VII collagen. To meet the diagnosis of type I BSLE, all 6 criteria must be satisfied. To meet the diagnosis of type II BSLE, only criteria 1 to 4 need to be satisfied.6
Patients with BSLE may be presumed to have a different but clinically similar vesiculobullous condition (eg, bullous pemphigoid, cutaneous manifestations of SLE) and may be started on systemic corticosteroids. However, BSLE patients often do not show great improvement while on corticosteroids and may even flare shortly after beginning systemic corticosteroid treatment. The current treatment of choice for BSLE is dapsone, a sulfa drug that is thought to exhibit its anti-inflammatory properties via the inhibition of the alternative pathway of the complement system and through the inhibition of polymorphonuclear leukocyte functions.7 A response to dapsone helps differentiate BSLE from histopathologically and immunopathologically identical conditions such as epidermolysis bullosa acquisita.4 Bullous systemic lupus erythematosus can be differentiated from dermatitis herpetiformis with the presence of antigliadin and antitissue transglutaminase antibodies, which are found in the latter. Additionally, BSLE may show the presence of IgG and IgM deposition in addition to IgA deposition, as opposed to dermatitis herpetiformis where only IgA is found.8 The presence of these additional antibody depositions also help differentiate BSLE from linear IgA bullous dermatosis (LABD), as LABD will only have IgA depositions and often presents with an annular, crown of jewels-like appearance. Finally, there is a well-described phenomenon of LABD being drug induced, particularly after a course of vancomycin,9 and such an association with vancomycin has not been documented for BSLE.
Our patient was diagnosed with BSLE following the flare approximately 1.5 years prior to the current presentation. She had been started on dapsone 75 mg daily at that time and was taking 75 mg at the time of presentation. She was admitted and treated as an inpatient with high-dose (1 mg/kg) intravenous prednisone due to the extensive current flare.
- Fujimoto W, Hamada T, Yamada J, et al. Bullous systemic lupus erythematosus as an initial manifestation of SLE. J Dermatol. 2005;32:1021-1027.
- Miziara ID, Mahmoud A, Chagury AA, et al. Bullous systemic lupus erythematosus: case report. Int Arch Otorhinolaryngol. 2013;17:344-346.
- Tincopa M, Puttgen KB, Sule S, et al. Bullous lupus: an unusual initial presentation of systemic lupus erythematosus in an adolescent girl. Pediatr Dermatol. 2010;27:373-376.
- Grover C, Khurana A, Sharma S, et al. Bullous systemic lupus erythematosus. Indian J Dermatol. 2013;58:492.
- Aringer M, Costenbader K, Daikh D, et al. 2019 European League Against Rheumatism/American College of RheumatologyClassification Criteria for Systemic Lupus Erythematosus [published online August 6, 2019]. Arthritis Rheumatol. 2019;71:1400-1412.
- Gammon WR, Briggaman RA. Bullous SLE: a phenotypically distinctive but immunologically heterogeneous bullous disorder. J Invest Dermatol. 1993;100:28S-34S.
- Duan L, Chen L, Zhong S, et al. Treatment of bullous systemic lupus erythematosus. J Immunol Res. 2015;2015:6.
- Barbosa WS, Rodarte CM, Guerra JG, et al. Bullous systemic lupus erythematosus: differential diagnosis with dermatitis herpetiformis. An Bras Dermatol. 2011;86(4 suppl 1):S92-S95.
- Yordanova I, Valtchev V, Gospodinov D, et al. IgA linear bullous dermatosis in childhood. J IMAB. 2015;21:1012-1014.
The Diagnosis: Bullous Systemic Lupus Erythematosus
Bullous systemic lupus erythematosus (BSLE) is a rare blistering disease that affects patients with systemic lupus erythematosus (SLE). Our patient had a several-year history of SLE and was being managed by a rheumatologist. She was taking hydroxychloroquine at the time of the flare. Although BSLE tends to present in those with SLE that has already been diagnosed, BSLE has been reported as a possible initial manifestation of SLE.1
Bullous systemic lupus erythematosus is estimated to occur in less than 5% of patients with SLE and is more common in black women between the second and third decades of life,2 though it also can be seen in the pediatric population.3 The lesions of BSLE usually present as subepidermal blisters often located on the face, neck, and arms on an erythematous or possibly urticarial base. Although non-BSLE vesiculobullous eruptions may be seen in patients with SLE, BSLE is differentiated from these other eruptions by its appearance on sun-exposed and non-sun-exposed areas of the body, while other vesiculobullous eruptions associated with SLE typically are limited to sun-exposed sites.4
Due to its clinical presentation overlapping with several vesiculobullous conditions, a set of diagnostic criteria have been suggested for BSLE, including the following: (1) fulfillment of the American Rheumatism Association's criteria for SLE5; (2) a new-onset vesiculobullous eruption, primarily on sun-exposed skin; (3) histology showing a subepidermal blister with a predominantly neutrophilic infiltrate; (4) presence of IgG, IgA, IgM, and C3 at the basement membrane zone; (5) evidence of antibodies to type VII collagen; and (6) immunoelectron microscopy showing codistribution of immunoglobulin deposits with anchoring fibrils/type VII collagen. To meet the diagnosis of type I BSLE, all 6 criteria must be satisfied. To meet the diagnosis of type II BSLE, only criteria 1 to 4 need to be satisfied.6
Patients with BSLE may be presumed to have a different but clinically similar vesiculobullous condition (eg, bullous pemphigoid, cutaneous manifestations of SLE) and may be started on systemic corticosteroids. However, BSLE patients often do not show great improvement while on corticosteroids and may even flare shortly after beginning systemic corticosteroid treatment. The current treatment of choice for BSLE is dapsone, a sulfa drug that is thought to exhibit its anti-inflammatory properties via the inhibition of the alternative pathway of the complement system and through the inhibition of polymorphonuclear leukocyte functions.7 A response to dapsone helps differentiate BSLE from histopathologically and immunopathologically identical conditions such as epidermolysis bullosa acquisita.4 Bullous systemic lupus erythematosus can be differentiated from dermatitis herpetiformis with the presence of antigliadin and antitissue transglutaminase antibodies, which are found in the latter. Additionally, BSLE may show the presence of IgG and IgM deposition in addition to IgA deposition, as opposed to dermatitis herpetiformis where only IgA is found.8 The presence of these additional antibody depositions also help differentiate BSLE from linear IgA bullous dermatosis (LABD), as LABD will only have IgA depositions and often presents with an annular, crown of jewels-like appearance. Finally, there is a well-described phenomenon of LABD being drug induced, particularly after a course of vancomycin,9 and such an association with vancomycin has not been documented for BSLE.
Our patient was diagnosed with BSLE following the flare approximately 1.5 years prior to the current presentation. She had been started on dapsone 75 mg daily at that time and was taking 75 mg at the time of presentation. She was admitted and treated as an inpatient with high-dose (1 mg/kg) intravenous prednisone due to the extensive current flare.
The Diagnosis: Bullous Systemic Lupus Erythematosus
Bullous systemic lupus erythematosus (BSLE) is a rare blistering disease that affects patients with systemic lupus erythematosus (SLE). Our patient had a several-year history of SLE and was being managed by a rheumatologist. She was taking hydroxychloroquine at the time of the flare. Although BSLE tends to present in those with SLE that has already been diagnosed, BSLE has been reported as a possible initial manifestation of SLE.1
Bullous systemic lupus erythematosus is estimated to occur in less than 5% of patients with SLE and is more common in black women between the second and third decades of life,2 though it also can be seen in the pediatric population.3 The lesions of BSLE usually present as subepidermal blisters often located on the face, neck, and arms on an erythematous or possibly urticarial base. Although non-BSLE vesiculobullous eruptions may be seen in patients with SLE, BSLE is differentiated from these other eruptions by its appearance on sun-exposed and non-sun-exposed areas of the body, while other vesiculobullous eruptions associated with SLE typically are limited to sun-exposed sites.4
Due to its clinical presentation overlapping with several vesiculobullous conditions, a set of diagnostic criteria have been suggested for BSLE, including the following: (1) fulfillment of the American Rheumatism Association's criteria for SLE5; (2) a new-onset vesiculobullous eruption, primarily on sun-exposed skin; (3) histology showing a subepidermal blister with a predominantly neutrophilic infiltrate; (4) presence of IgG, IgA, IgM, and C3 at the basement membrane zone; (5) evidence of antibodies to type VII collagen; and (6) immunoelectron microscopy showing codistribution of immunoglobulin deposits with anchoring fibrils/type VII collagen. To meet the diagnosis of type I BSLE, all 6 criteria must be satisfied. To meet the diagnosis of type II BSLE, only criteria 1 to 4 need to be satisfied.6
Patients with BSLE may be presumed to have a different but clinically similar vesiculobullous condition (eg, bullous pemphigoid, cutaneous manifestations of SLE) and may be started on systemic corticosteroids. However, BSLE patients often do not show great improvement while on corticosteroids and may even flare shortly after beginning systemic corticosteroid treatment. The current treatment of choice for BSLE is dapsone, a sulfa drug that is thought to exhibit its anti-inflammatory properties via the inhibition of the alternative pathway of the complement system and through the inhibition of polymorphonuclear leukocyte functions.7 A response to dapsone helps differentiate BSLE from histopathologically and immunopathologically identical conditions such as epidermolysis bullosa acquisita.4 Bullous systemic lupus erythematosus can be differentiated from dermatitis herpetiformis with the presence of antigliadin and antitissue transglutaminase antibodies, which are found in the latter. Additionally, BSLE may show the presence of IgG and IgM deposition in addition to IgA deposition, as opposed to dermatitis herpetiformis where only IgA is found.8 The presence of these additional antibody depositions also help differentiate BSLE from linear IgA bullous dermatosis (LABD), as LABD will only have IgA depositions and often presents with an annular, crown of jewels-like appearance. Finally, there is a well-described phenomenon of LABD being drug induced, particularly after a course of vancomycin,9 and such an association with vancomycin has not been documented for BSLE.
Our patient was diagnosed with BSLE following the flare approximately 1.5 years prior to the current presentation. She had been started on dapsone 75 mg daily at that time and was taking 75 mg at the time of presentation. She was admitted and treated as an inpatient with high-dose (1 mg/kg) intravenous prednisone due to the extensive current flare.
- Fujimoto W, Hamada T, Yamada J, et al. Bullous systemic lupus erythematosus as an initial manifestation of SLE. J Dermatol. 2005;32:1021-1027.
- Miziara ID, Mahmoud A, Chagury AA, et al. Bullous systemic lupus erythematosus: case report. Int Arch Otorhinolaryngol. 2013;17:344-346.
- Tincopa M, Puttgen KB, Sule S, et al. Bullous lupus: an unusual initial presentation of systemic lupus erythematosus in an adolescent girl. Pediatr Dermatol. 2010;27:373-376.
- Grover C, Khurana A, Sharma S, et al. Bullous systemic lupus erythematosus. Indian J Dermatol. 2013;58:492.
- Aringer M, Costenbader K, Daikh D, et al. 2019 European League Against Rheumatism/American College of RheumatologyClassification Criteria for Systemic Lupus Erythematosus [published online August 6, 2019]. Arthritis Rheumatol. 2019;71:1400-1412.
- Gammon WR, Briggaman RA. Bullous SLE: a phenotypically distinctive but immunologically heterogeneous bullous disorder. J Invest Dermatol. 1993;100:28S-34S.
- Duan L, Chen L, Zhong S, et al. Treatment of bullous systemic lupus erythematosus. J Immunol Res. 2015;2015:6.
- Barbosa WS, Rodarte CM, Guerra JG, et al. Bullous systemic lupus erythematosus: differential diagnosis with dermatitis herpetiformis. An Bras Dermatol. 2011;86(4 suppl 1):S92-S95.
- Yordanova I, Valtchev V, Gospodinov D, et al. IgA linear bullous dermatosis in childhood. J IMAB. 2015;21:1012-1014.
- Fujimoto W, Hamada T, Yamada J, et al. Bullous systemic lupus erythematosus as an initial manifestation of SLE. J Dermatol. 2005;32:1021-1027.
- Miziara ID, Mahmoud A, Chagury AA, et al. Bullous systemic lupus erythematosus: case report. Int Arch Otorhinolaryngol. 2013;17:344-346.
- Tincopa M, Puttgen KB, Sule S, et al. Bullous lupus: an unusual initial presentation of systemic lupus erythematosus in an adolescent girl. Pediatr Dermatol. 2010;27:373-376.
- Grover C, Khurana A, Sharma S, et al. Bullous systemic lupus erythematosus. Indian J Dermatol. 2013;58:492.
- Aringer M, Costenbader K, Daikh D, et al. 2019 European League Against Rheumatism/American College of RheumatologyClassification Criteria for Systemic Lupus Erythematosus [published online August 6, 2019]. Arthritis Rheumatol. 2019;71:1400-1412.
- Gammon WR, Briggaman RA. Bullous SLE: a phenotypically distinctive but immunologically heterogeneous bullous disorder. J Invest Dermatol. 1993;100:28S-34S.
- Duan L, Chen L, Zhong S, et al. Treatment of bullous systemic lupus erythematosus. J Immunol Res. 2015;2015:6.
- Barbosa WS, Rodarte CM, Guerra JG, et al. Bullous systemic lupus erythematosus: differential diagnosis with dermatitis herpetiformis. An Bras Dermatol. 2011;86(4 suppl 1):S92-S95.
- Yordanova I, Valtchev V, Gospodinov D, et al. IgA linear bullous dermatosis in childhood. J IMAB. 2015;21:1012-1014.
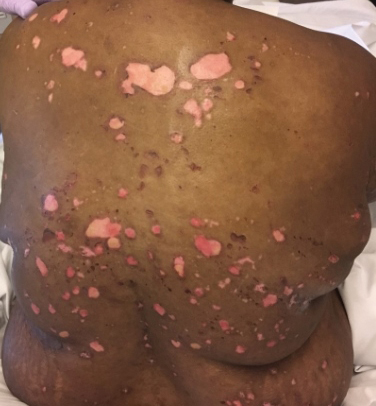
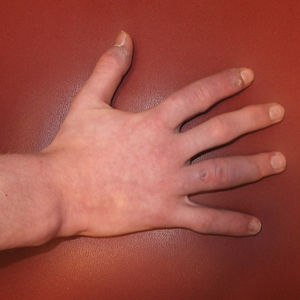
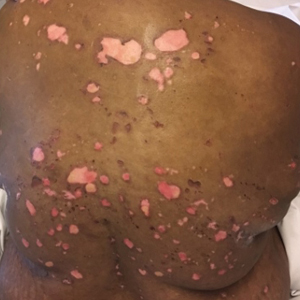
A 51-year-old black woman presented to the dermatology clinic with painful and pruritic erosions on the back, abdomen, neck, and arms of approximately 2 months' duration. The lesions started on the back and spread in a cephalocaudal manner. The patient denied any new changes in medication. Physical examination revealed large erosions with mild weeping of serosanguineous fluid on the back, abdomen, neck, and upper extremities. A few tense bullae were present on the dorsal aspect of the right hand. She had experienced a similar flare approximately 1.5 years prior to the current presentation. At that time, 2 shave biopsies from vesiculobullous lesions on the right side of the neck were sent for hematoxylin and eosin staining and direct immunofluorescence. Biopsy results showed a subepidermal blister that extended along the course of the hair follicle and was associated with an infiltrate of neutrophilic granulocytes that also extended along the course of the hair follicle. Direct immunofluorescence showed IgG and C3 deposition in the basement membrane zone extending along the floor of the blister where the epidermis was separated from the dermis.
FDA approves nintedanib for scleroderma interstitial lung disease
The Food and Drug Administration has approved nintedanib (Ofev) for the rare but sometimes deadly form of interstitial lung disease that’s caused by systemic sclerosis, or scleroderma.

Although scleroderma itself is rare, half of those patients present with scleroderma-related interstitial lung disease (SSc-ILD), and it remains the leading cause of death in scleroderma patients because it can lead to loss of pulmonary function. Nintedanib appears to slow the progress of SSc-ILD and is the first treatment approved for it, according to a news release from the FDA.
The approval is based on a randomized, double-blind, placebo-controlled trial of 576 patients aged 20-79 years with SSc-ILD. The primary efficacy endpoint was forced vital capacity, and patients on nintedanib showed less decline than did those on placebo.
The most frequent serious adverse event reported in this trial was pneumonia (2.8% with nintedanib vs. 0.3% with placebo). Adverse reactions that led to permanent dose reductions occurred in 34% of nintedanib patients and 4% of placebo-treated patients; the most common of these was diarrhea.
The full prescribing information, which is available on the FDA website, includes warnings for patients with moderate to severe hepatic impairment, elevated liver enzymes, and drug-induced liver injury, as well as those with gastrointestinal disorders. Nintedanib may cause embryo-fetal toxicity, so women of childbearing age should be counseled to avoid pregnancy while taking this drug.
Nintedanib received both Priority Review and Orphan Drug designation. The former meant the FDA intends to take action on the application within 6 months because the agency has determined that, if approved, it would have important effects on treatment of a serious condition. The latter provides incentives to assist and encourage development of drugs for rare diseases. The drug was approved in 2014 for adult patients with idiopathic pulmonary fibrosis, another interstitial lung disease.
The full release is available on the FDA website.
The Food and Drug Administration has approved nintedanib (Ofev) for the rare but sometimes deadly form of interstitial lung disease that’s caused by systemic sclerosis, or scleroderma.
Although scleroderma itself is rare, half of those patients present with scleroderma-related interstitial lung disease (SSc-ILD), and it remains the leading cause of death in scleroderma patients because it can lead to loss of pulmonary function. Nintedanib appears to slow the progress of SSc-ILD and is the first treatment approved for it, according to a news release from the FDA.
The approval is based on a randomized, double-blind, placebo-controlled trial of 576 patients aged 20-79 years with SSc-ILD. The primary efficacy endpoint was forced vital capacity, and patients on nintedanib showed less decline than did those on placebo.
The most frequent serious adverse event reported in this trial was pneumonia (2.8% with nintedanib vs. 0.3% with placebo). Adverse reactions that led to permanent dose reductions occurred in 34% of nintedanib patients and 4% of placebo-treated patients; the most common of these was diarrhea.
The full prescribing information, which is available on the FDA website, includes warnings for patients with moderate to severe hepatic impairment, elevated liver enzymes, and drug-induced liver injury, as well as those with gastrointestinal disorders. Nintedanib may cause embryo-fetal toxicity, so women of childbearing age should be counseled to avoid pregnancy while taking this drug.
Nintedanib received both Priority Review and Orphan Drug designation. The former meant the FDA intends to take action on the application within 6 months because the agency has determined that, if approved, it would have important effects on treatment of a serious condition. The latter provides incentives to assist and encourage development of drugs for rare diseases. The drug was approved in 2014 for adult patients with idiopathic pulmonary fibrosis, another interstitial lung disease.
The full release is available on the FDA website.
The Food and Drug Administration has approved nintedanib (Ofev) for the rare but sometimes deadly form of interstitial lung disease that’s caused by systemic sclerosis, or scleroderma.
Although scleroderma itself is rare, half of those patients present with scleroderma-related interstitial lung disease (SSc-ILD), and it remains the leading cause of death in scleroderma patients because it can lead to loss of pulmonary function. Nintedanib appears to slow the progress of SSc-ILD and is the first treatment approved for it, according to a news release from the FDA.
The approval is based on a randomized, double-blind, placebo-controlled trial of 576 patients aged 20-79 years with SSc-ILD. The primary efficacy endpoint was forced vital capacity, and patients on nintedanib showed less decline than did those on placebo.
The most frequent serious adverse event reported in this trial was pneumonia (2.8% with nintedanib vs. 0.3% with placebo). Adverse reactions that led to permanent dose reductions occurred in 34% of nintedanib patients and 4% of placebo-treated patients; the most common of these was diarrhea.
The full prescribing information, which is available on the FDA website, includes warnings for patients with moderate to severe hepatic impairment, elevated liver enzymes, and drug-induced liver injury, as well as those with gastrointestinal disorders. Nintedanib may cause embryo-fetal toxicity, so women of childbearing age should be counseled to avoid pregnancy while taking this drug.
Nintedanib received both Priority Review and Orphan Drug designation. The former meant the FDA intends to take action on the application within 6 months because the agency has determined that, if approved, it would have important effects on treatment of a serious condition. The latter provides incentives to assist and encourage development of drugs for rare diseases. The drug was approved in 2014 for adult patients with idiopathic pulmonary fibrosis, another interstitial lung disease.
The full release is available on the FDA website.
In vasculitis, the skin tells the story
MILAN – , Robert Micheletti, MD, said at the World Congress of Dermatology.

In granulomatous vasculitis, histiocytes and giant cells can play a significant role, explained Dr. Micheletti, director of the cutaneous vasculitis clinic at the University of Pennsylvania, Philadelphia. The condition may be secondary to an autoimmune disease such as lupus erythematosus or RA; a granulomatous disease such as Crohn’s disease or sarcoidosis; infections such as tuberculosis, a fungal disease, or herpes or zoster viruses, or lymphoma, Dr. Micheletti said.
However, a primary systemic vasculitis such as granulomatosis with polyangiitis (GPA; formerly known as Wegener’s polyangiitis) or eosinophilic granulomatosis with polyangiitis (EGPA; also known as Churg-Strauss vasculitis), giant cell arteritis, or Takayasu arteritis may also be responsible, he said. Occasionally, the culprit can also be a drug-induced vasculitis.
The physical examination gives clues to the size of involved vessels, which in turn helps to classify the vasculitis, Dr. Micheletti said.
When vasculitis affects small vessels, the skin findings will be palpable purpura, urticarial papules, vesicles, and petechiae, he said, adding that “The small vessel involvement accounts for the small size of the lesions, and complement cascade and inflammation account for the palpability of the lesions and the symptomatology.” As red blood cells extravasate from the affected vessels, nonblanching purpura develop, and gravity’s effect on the deposition of immune complex material dictates how lesions are distributed.
“Manifestations more typical of medium vessel vasculitis include subcutaneous nodules, livedo reticularis, retiform purpura, larger hemorrhagic bullae, and more significant ulceration and necrosis,” he said. “If such lesions are seen, suspect medium-vessel vasculitis or vasculitis overlapping small and medium vessels.” Cutaneous or systemic polyarteritis nodosa, antineutrophilic cytoplasmic autoantibody (ANCA)–associated vasculitis, and cryoglobulinemic vasculitis are examples, he added.
The particularities of renal manifestations of vasculitis also offer clues to the vessels involved. When a vasculitis patient has glomerulonephritis, suspect small-vessel involvement, Dr. Micheletti said. However, vasculitis affecting medium-sized vessels will cause renovascular hypertension and, potentially renal arterial aneurysms.
Nerves are typically spared in small-vessel vasculitis, while wrist or foot drop can be seen in mononeuritis multiplex.
Recently, the Diagnostic and Classification Criteria in Vasculitis Study (DCVAS) looked at more than 6,800 patients at over 130 sites around the world, proposing new classification criteria for ANCA-associated vasculitis (AAV) and large-vessel vasculitis. The study found that skin findings are common in AAV, with 30%-50% of cases presenting initially with skin lesions. Petechiae and/or purpura are the most common of the skin manifestations, he said. By contrast, for EGPA, allergic and nonspecific findings were the most common findings.
Although skin biopsy can confirm the diagnosis in up to 94% of AAV cases, it’s underutilized and performed in less than half (24%-44%) of cases, Dr. Micheletti said. The study’s findings “demonstrate the importance of a good skin exam, as well as its utility for diagnosis” of vasculitis, he said.
An additional finding form the DCVAS study was that skin lesions can give clues to severity of vasculitis: “Among 1,184 patients with ANCA-associated vasculitis, those with cutaneous involvement were more likely to have systemic manifestations of disease, more likely to have such severe manifestations as glomerulonephritis, alveolar hemorrhage, and mononeuritis,” said Dr. Micheletti, with a hazard ratio of 2.0 among those individuals who had EGPA or GPA.
“Skin findings have diagnostic and, potentially, prognostic importance,” he said. “Use the physician exam and your clinical acumen to your advantage,” but always confirm vasculitis with a biopsy. “Clinicopathologic correlation is key.” A simple urinalysis will screen for renal involvement, and is of “paramount importance,” he added.
Dr. Micheletti reported that he had no relevant disclosures.
MILAN – , Robert Micheletti, MD, said at the World Congress of Dermatology.
In granulomatous vasculitis, histiocytes and giant cells can play a significant role, explained Dr. Micheletti, director of the cutaneous vasculitis clinic at the University of Pennsylvania, Philadelphia. The condition may be secondary to an autoimmune disease such as lupus erythematosus or RA; a granulomatous disease such as Crohn’s disease or sarcoidosis; infections such as tuberculosis, a fungal disease, or herpes or zoster viruses, or lymphoma, Dr. Micheletti said.
However, a primary systemic vasculitis such as granulomatosis with polyangiitis (GPA; formerly known as Wegener’s polyangiitis) or eosinophilic granulomatosis with polyangiitis (EGPA; also known as Churg-Strauss vasculitis), giant cell arteritis, or Takayasu arteritis may also be responsible, he said. Occasionally, the culprit can also be a drug-induced vasculitis.
The physical examination gives clues to the size of involved vessels, which in turn helps to classify the vasculitis, Dr. Micheletti said.
When vasculitis affects small vessels, the skin findings will be palpable purpura, urticarial papules, vesicles, and petechiae, he said, adding that “The small vessel involvement accounts for the small size of the lesions, and complement cascade and inflammation account for the palpability of the lesions and the symptomatology.” As red blood cells extravasate from the affected vessels, nonblanching purpura develop, and gravity’s effect on the deposition of immune complex material dictates how lesions are distributed.
“Manifestations more typical of medium vessel vasculitis include subcutaneous nodules, livedo reticularis, retiform purpura, larger hemorrhagic bullae, and more significant ulceration and necrosis,” he said. “If such lesions are seen, suspect medium-vessel vasculitis or vasculitis overlapping small and medium vessels.” Cutaneous or systemic polyarteritis nodosa, antineutrophilic cytoplasmic autoantibody (ANCA)–associated vasculitis, and cryoglobulinemic vasculitis are examples, he added.
The particularities of renal manifestations of vasculitis also offer clues to the vessels involved. When a vasculitis patient has glomerulonephritis, suspect small-vessel involvement, Dr. Micheletti said. However, vasculitis affecting medium-sized vessels will cause renovascular hypertension and, potentially renal arterial aneurysms.
Nerves are typically spared in small-vessel vasculitis, while wrist or foot drop can be seen in mononeuritis multiplex.
Recently, the Diagnostic and Classification Criteria in Vasculitis Study (DCVAS) looked at more than 6,800 patients at over 130 sites around the world, proposing new classification criteria for ANCA-associated vasculitis (AAV) and large-vessel vasculitis. The study found that skin findings are common in AAV, with 30%-50% of cases presenting initially with skin lesions. Petechiae and/or purpura are the most common of the skin manifestations, he said. By contrast, for EGPA, allergic and nonspecific findings were the most common findings.
Although skin biopsy can confirm the diagnosis in up to 94% of AAV cases, it’s underutilized and performed in less than half (24%-44%) of cases, Dr. Micheletti said. The study’s findings “demonstrate the importance of a good skin exam, as well as its utility for diagnosis” of vasculitis, he said.
An additional finding form the DCVAS study was that skin lesions can give clues to severity of vasculitis: “Among 1,184 patients with ANCA-associated vasculitis, those with cutaneous involvement were more likely to have systemic manifestations of disease, more likely to have such severe manifestations as glomerulonephritis, alveolar hemorrhage, and mononeuritis,” said Dr. Micheletti, with a hazard ratio of 2.0 among those individuals who had EGPA or GPA.
“Skin findings have diagnostic and, potentially, prognostic importance,” he said. “Use the physician exam and your clinical acumen to your advantage,” but always confirm vasculitis with a biopsy. “Clinicopathologic correlation is key.” A simple urinalysis will screen for renal involvement, and is of “paramount importance,” he added.
Dr. Micheletti reported that he had no relevant disclosures.
MILAN – , Robert Micheletti, MD, said at the World Congress of Dermatology.
In granulomatous vasculitis, histiocytes and giant cells can play a significant role, explained Dr. Micheletti, director of the cutaneous vasculitis clinic at the University of Pennsylvania, Philadelphia. The condition may be secondary to an autoimmune disease such as lupus erythematosus or RA; a granulomatous disease such as Crohn’s disease or sarcoidosis; infections such as tuberculosis, a fungal disease, or herpes or zoster viruses, or lymphoma, Dr. Micheletti said.
However, a primary systemic vasculitis such as granulomatosis with polyangiitis (GPA; formerly known as Wegener’s polyangiitis) or eosinophilic granulomatosis with polyangiitis (EGPA; also known as Churg-Strauss vasculitis), giant cell arteritis, or Takayasu arteritis may also be responsible, he said. Occasionally, the culprit can also be a drug-induced vasculitis.
The physical examination gives clues to the size of involved vessels, which in turn helps to classify the vasculitis, Dr. Micheletti said.
When vasculitis affects small vessels, the skin findings will be palpable purpura, urticarial papules, vesicles, and petechiae, he said, adding that “The small vessel involvement accounts for the small size of the lesions, and complement cascade and inflammation account for the palpability of the lesions and the symptomatology.” As red blood cells extravasate from the affected vessels, nonblanching purpura develop, and gravity’s effect on the deposition of immune complex material dictates how lesions are distributed.
“Manifestations more typical of medium vessel vasculitis include subcutaneous nodules, livedo reticularis, retiform purpura, larger hemorrhagic bullae, and more significant ulceration and necrosis,” he said. “If such lesions are seen, suspect medium-vessel vasculitis or vasculitis overlapping small and medium vessels.” Cutaneous or systemic polyarteritis nodosa, antineutrophilic cytoplasmic autoantibody (ANCA)–associated vasculitis, and cryoglobulinemic vasculitis are examples, he added.
The particularities of renal manifestations of vasculitis also offer clues to the vessels involved. When a vasculitis patient has glomerulonephritis, suspect small-vessel involvement, Dr. Micheletti said. However, vasculitis affecting medium-sized vessels will cause renovascular hypertension and, potentially renal arterial aneurysms.
Nerves are typically spared in small-vessel vasculitis, while wrist or foot drop can be seen in mononeuritis multiplex.
Recently, the Diagnostic and Classification Criteria in Vasculitis Study (DCVAS) looked at more than 6,800 patients at over 130 sites around the world, proposing new classification criteria for ANCA-associated vasculitis (AAV) and large-vessel vasculitis. The study found that skin findings are common in AAV, with 30%-50% of cases presenting initially with skin lesions. Petechiae and/or purpura are the most common of the skin manifestations, he said. By contrast, for EGPA, allergic and nonspecific findings were the most common findings.
Although skin biopsy can confirm the diagnosis in up to 94% of AAV cases, it’s underutilized and performed in less than half (24%-44%) of cases, Dr. Micheletti said. The study’s findings “demonstrate the importance of a good skin exam, as well as its utility for diagnosis” of vasculitis, he said.
An additional finding form the DCVAS study was that skin lesions can give clues to severity of vasculitis: “Among 1,184 patients with ANCA-associated vasculitis, those with cutaneous involvement were more likely to have systemic manifestations of disease, more likely to have such severe manifestations as glomerulonephritis, alveolar hemorrhage, and mononeuritis,” said Dr. Micheletti, with a hazard ratio of 2.0 among those individuals who had EGPA or GPA.
“Skin findings have diagnostic and, potentially, prognostic importance,” he said. “Use the physician exam and your clinical acumen to your advantage,” but always confirm vasculitis with a biopsy. “Clinicopathologic correlation is key.” A simple urinalysis will screen for renal involvement, and is of “paramount importance,” he added.
Dr. Micheletti reported that he had no relevant disclosures.
AT WCD2019
International lupus community sets out top barriers to improving lupus outcomes
The heterogeneity of lupus and the subsequent lack of a clear disease definition have been identified by an international group of experts as the primary barriers hindering timely diagnosis, improved treatment options, and appropriate access to care.

A report published in Lupus Science & Medicine titled “Global Consensus Building and Prioritization of Fundamental Lupus Challenges: The ALPHA Project” describes the results of a first-ever global consensus on key barriers to advances in lupus care, including a lack of validated biomarkers and flawed clinical trial design.
A lack of access to medical professionals familiar with lupus, challenges in managing lupus because of social determinants, and lack of treatment adherence were also considered to be barriers to improving the outcomes of people living with lupus.
First author Susan Manzi, MD, codirector of the Lupus Center of Excellence at Allegheny Health Network, Pittsburgh, and her colleagues said that, in contrast to other autoimmune diseases such as rheumatoid arthritis and psoriasis, the field of lupus has struggled with establishing a clear pathway for lupus drug development because of “persistent challenges in understanding the biology of the disease, defining clinical trial entry criteria and end points, developing instruments to measure changes in clinical activity, and controlling background medications.”
The authors noted that the intention of the Addressing Lupus Pillars for Health Advancement (ALPHA) Project was to build on the work of other initiatives, including some that were international in scope or were still ongoing.
“The ALPHA project was founded as the first step in an ongoing commitment to identify, prioritize, and implement strategies to address the most pressing challenges that limit progress in lupus across the continuum,” they wrote. In a joint initiative, the Lupus Foundation of America (LFA) and the Tufts Center for the Study of Drug Development (Tufts CSDD) set up a Global Advisory Committee (GAC) that included 13 lupus experts from the United States, Australia, United Kingdom, Germany, and South Korea to guide and oversee the study. Members had extensive knowledge of the disease, with specific expertise in rheumatology, dermatology, immunology, nephrology, and pediatrics.
Next, in-depth interviews were conducted with 17 experts who were well respected in the lupus scientific and care communities and represented all stakeholders. Using information garnered from these interviews, the LFA, Tufts CSDD, and GAC collaborated to develop a survey that included 23 questions addressing attitudes and perceptions about lupus as well as the prioritization of the most pressing challenges to improving diagnosis, care, treatment, and research.
The online survey was sent to 366 candidates, from whom the researchers received 127 completed responses. Of these, 82 (65%) were clinician-researcher-scientists and 14 (11%) worked in industry/biotechnology, 13 (10%) were researcher-scientists, and 12 (9%) were clinicians; 5% marked “other.”
The research team used a weighting system to prioritize barriers ranked by respondents, whereby higher ratings represented the challenges of highest impact (a score of 9 was highest rating, with 1 the lowest).
Survey respondents ranked the following as the top barriers to improving outcomes in lupus:
- A lack of diagnostic, predictive, and prognostic biomarkers for lupus (weighted prioritization score of 7.294) and lack of biomarkers to predict drug response in clinical trials (weighted prioritization score of 6.614).
- Flawed clinical trial design (weighted prioritization score of 6.370).
- Lack of access to clinicians familiar with lupus (weighted prioritization score of 6.873), and limited awareness of lupus among nonexpert medical professionals (weighted prioritization score of 5.800).
- Barriers to effective management of lupus because of social determinants of care in predominantly lower socioeconomic status areas (weighted prioritization score of 6.937).
- A lack of treatment adherence (weighted prioritization score of 6.717).
“A strong consensus built throughout the study, as themes and insights gathered from the in-depth interviews were highly consistent with those collected in the survey,” the researchers noted.
They said it was not surprising that the development of biomarkers had received a high ranking, as advances in this area would help accelerate drug development and precision medicine as well as more practical aspects of clinical care.
The research team acknowledged that substantial funds would be needed to address the top priorities identified in the study, and some of the issues may be more easily addressed than others.
“In the past decade, the overall funding landscape for lupus has been on a decline, particularly through the National Institutes of Health – the largest public funder of lupus research in the world – during a time in which arguably, lupus research has been prolific,” they wrote.
They concluded that comprehensive measures were needed to transform the lupus research and health care landscape.
“Lupus experts must convene to determine feasible and coordinated approaches for addressing long-standing barriers across the global lupus community,” they stressed.
The next part of the project will involve an international stakeholder meeting to develop a global road map of specific recommendations to address identified barriers, which “may include multipronged strategies using regulatory and advocacy approaches, scientific consensus building, communication efforts, among other possible tactics,” they added.
The ALPHA Project was launched in partnership with founding partner EMD Serono Research & Development (a business of Merck KGaA) and through additional support by GlaxoSmithKline. Many authors of the report had financial connections to the pharmaceutical industry.
SOURCE: Manzi S et al. Lupus Sci Med. 2019;6:e000342. doi: 10.1136/lupus-2019-000342.
The heterogeneity of lupus and the subsequent lack of a clear disease definition have been identified by an international group of experts as the primary barriers hindering timely diagnosis, improved treatment options, and appropriate access to care.
A report published in Lupus Science & Medicine titled “Global Consensus Building and Prioritization of Fundamental Lupus Challenges: The ALPHA Project” describes the results of a first-ever global consensus on key barriers to advances in lupus care, including a lack of validated biomarkers and flawed clinical trial design.
A lack of access to medical professionals familiar with lupus, challenges in managing lupus because of social determinants, and lack of treatment adherence were also considered to be barriers to improving the outcomes of people living with lupus.
First author Susan Manzi, MD, codirector of the Lupus Center of Excellence at Allegheny Health Network, Pittsburgh, and her colleagues said that, in contrast to other autoimmune diseases such as rheumatoid arthritis and psoriasis, the field of lupus has struggled with establishing a clear pathway for lupus drug development because of “persistent challenges in understanding the biology of the disease, defining clinical trial entry criteria and end points, developing instruments to measure changes in clinical activity, and controlling background medications.”
The authors noted that the intention of the Addressing Lupus Pillars for Health Advancement (ALPHA) Project was to build on the work of other initiatives, including some that were international in scope or were still ongoing.
“The ALPHA project was founded as the first step in an ongoing commitment to identify, prioritize, and implement strategies to address the most pressing challenges that limit progress in lupus across the continuum,” they wrote. In a joint initiative, the Lupus Foundation of America (LFA) and the Tufts Center for the Study of Drug Development (Tufts CSDD) set up a Global Advisory Committee (GAC) that included 13 lupus experts from the United States, Australia, United Kingdom, Germany, and South Korea to guide and oversee the study. Members had extensive knowledge of the disease, with specific expertise in rheumatology, dermatology, immunology, nephrology, and pediatrics.
Next, in-depth interviews were conducted with 17 experts who were well respected in the lupus scientific and care communities and represented all stakeholders. Using information garnered from these interviews, the LFA, Tufts CSDD, and GAC collaborated to develop a survey that included 23 questions addressing attitudes and perceptions about lupus as well as the prioritization of the most pressing challenges to improving diagnosis, care, treatment, and research.
The online survey was sent to 366 candidates, from whom the researchers received 127 completed responses. Of these, 82 (65%) were clinician-researcher-scientists and 14 (11%) worked in industry/biotechnology, 13 (10%) were researcher-scientists, and 12 (9%) were clinicians; 5% marked “other.”
The research team used a weighting system to prioritize barriers ranked by respondents, whereby higher ratings represented the challenges of highest impact (a score of 9 was highest rating, with 1 the lowest).
Survey respondents ranked the following as the top barriers to improving outcomes in lupus:
- A lack of diagnostic, predictive, and prognostic biomarkers for lupus (weighted prioritization score of 7.294) and lack of biomarkers to predict drug response in clinical trials (weighted prioritization score of 6.614).
- Flawed clinical trial design (weighted prioritization score of 6.370).
- Lack of access to clinicians familiar with lupus (weighted prioritization score of 6.873), and limited awareness of lupus among nonexpert medical professionals (weighted prioritization score of 5.800).
- Barriers to effective management of lupus because of social determinants of care in predominantly lower socioeconomic status areas (weighted prioritization score of 6.937).
- A lack of treatment adherence (weighted prioritization score of 6.717).
“A strong consensus built throughout the study, as themes and insights gathered from the in-depth interviews were highly consistent with those collected in the survey,” the researchers noted.
They said it was not surprising that the development of biomarkers had received a high ranking, as advances in this area would help accelerate drug development and precision medicine as well as more practical aspects of clinical care.
The research team acknowledged that substantial funds would be needed to address the top priorities identified in the study, and some of the issues may be more easily addressed than others.
“In the past decade, the overall funding landscape for lupus has been on a decline, particularly through the National Institutes of Health – the largest public funder of lupus research in the world – during a time in which arguably, lupus research has been prolific,” they wrote.
They concluded that comprehensive measures were needed to transform the lupus research and health care landscape.
“Lupus experts must convene to determine feasible and coordinated approaches for addressing long-standing barriers across the global lupus community,” they stressed.
The next part of the project will involve an international stakeholder meeting to develop a global road map of specific recommendations to address identified barriers, which “may include multipronged strategies using regulatory and advocacy approaches, scientific consensus building, communication efforts, among other possible tactics,” they added.
The ALPHA Project was launched in partnership with founding partner EMD Serono Research & Development (a business of Merck KGaA) and through additional support by GlaxoSmithKline. Many authors of the report had financial connections to the pharmaceutical industry.
SOURCE: Manzi S et al. Lupus Sci Med. 2019;6:e000342. doi: 10.1136/lupus-2019-000342.
The heterogeneity of lupus and the subsequent lack of a clear disease definition have been identified by an international group of experts as the primary barriers hindering timely diagnosis, improved treatment options, and appropriate access to care.
A report published in Lupus Science & Medicine titled “Global Consensus Building and Prioritization of Fundamental Lupus Challenges: The ALPHA Project” describes the results of a first-ever global consensus on key barriers to advances in lupus care, including a lack of validated biomarkers and flawed clinical trial design.
A lack of access to medical professionals familiar with lupus, challenges in managing lupus because of social determinants, and lack of treatment adherence were also considered to be barriers to improving the outcomes of people living with lupus.
First author Susan Manzi, MD, codirector of the Lupus Center of Excellence at Allegheny Health Network, Pittsburgh, and her colleagues said that, in contrast to other autoimmune diseases such as rheumatoid arthritis and psoriasis, the field of lupus has struggled with establishing a clear pathway for lupus drug development because of “persistent challenges in understanding the biology of the disease, defining clinical trial entry criteria and end points, developing instruments to measure changes in clinical activity, and controlling background medications.”
The authors noted that the intention of the Addressing Lupus Pillars for Health Advancement (ALPHA) Project was to build on the work of other initiatives, including some that were international in scope or were still ongoing.
“The ALPHA project was founded as the first step in an ongoing commitment to identify, prioritize, and implement strategies to address the most pressing challenges that limit progress in lupus across the continuum,” they wrote. In a joint initiative, the Lupus Foundation of America (LFA) and the Tufts Center for the Study of Drug Development (Tufts CSDD) set up a Global Advisory Committee (GAC) that included 13 lupus experts from the United States, Australia, United Kingdom, Germany, and South Korea to guide and oversee the study. Members had extensive knowledge of the disease, with specific expertise in rheumatology, dermatology, immunology, nephrology, and pediatrics.
Next, in-depth interviews were conducted with 17 experts who were well respected in the lupus scientific and care communities and represented all stakeholders. Using information garnered from these interviews, the LFA, Tufts CSDD, and GAC collaborated to develop a survey that included 23 questions addressing attitudes and perceptions about lupus as well as the prioritization of the most pressing challenges to improving diagnosis, care, treatment, and research.
The online survey was sent to 366 candidates, from whom the researchers received 127 completed responses. Of these, 82 (65%) were clinician-researcher-scientists and 14 (11%) worked in industry/biotechnology, 13 (10%) were researcher-scientists, and 12 (9%) were clinicians; 5% marked “other.”
The research team used a weighting system to prioritize barriers ranked by respondents, whereby higher ratings represented the challenges of highest impact (a score of 9 was highest rating, with 1 the lowest).
Survey respondents ranked the following as the top barriers to improving outcomes in lupus:
- A lack of diagnostic, predictive, and prognostic biomarkers for lupus (weighted prioritization score of 7.294) and lack of biomarkers to predict drug response in clinical trials (weighted prioritization score of 6.614).
- Flawed clinical trial design (weighted prioritization score of 6.370).
- Lack of access to clinicians familiar with lupus (weighted prioritization score of 6.873), and limited awareness of lupus among nonexpert medical professionals (weighted prioritization score of 5.800).
- Barriers to effective management of lupus because of social determinants of care in predominantly lower socioeconomic status areas (weighted prioritization score of 6.937).
- A lack of treatment adherence (weighted prioritization score of 6.717).
“A strong consensus built throughout the study, as themes and insights gathered from the in-depth interviews were highly consistent with those collected in the survey,” the researchers noted.
They said it was not surprising that the development of biomarkers had received a high ranking, as advances in this area would help accelerate drug development and precision medicine as well as more practical aspects of clinical care.
The research team acknowledged that substantial funds would be needed to address the top priorities identified in the study, and some of the issues may be more easily addressed than others.
“In the past decade, the overall funding landscape for lupus has been on a decline, particularly through the National Institutes of Health – the largest public funder of lupus research in the world – during a time in which arguably, lupus research has been prolific,” they wrote.
They concluded that comprehensive measures were needed to transform the lupus research and health care landscape.
“Lupus experts must convene to determine feasible and coordinated approaches for addressing long-standing barriers across the global lupus community,” they stressed.
The next part of the project will involve an international stakeholder meeting to develop a global road map of specific recommendations to address identified barriers, which “may include multipronged strategies using regulatory and advocacy approaches, scientific consensus building, communication efforts, among other possible tactics,” they added.
The ALPHA Project was launched in partnership with founding partner EMD Serono Research & Development (a business of Merck KGaA) and through additional support by GlaxoSmithKline. Many authors of the report had financial connections to the pharmaceutical industry.
SOURCE: Manzi S et al. Lupus Sci Med. 2019;6:e000342. doi: 10.1136/lupus-2019-000342.
REPORTING FROM LUPUS SCIENCE & MEDICINE
FDA approves Otezla for treatment of Behçet’s-associated oral ulcers
The Food and Drug Administration has expanded the indication for apremilast (Otezla) to include the treatment of oral ulcers associated with Behçet’s disease in adults, according to an announcement from the manufacturer, Celgene.

FDA approval was based on results of the randomized, placebo-controlled, double-blind, phase 3 RELIEF trial, in which 207 patients with Behçet’s disease with active ulcers underwent treatment for 12 weeks with 30 mg apremilast or placebo. When measured on a visual analog scale, the reduction in pain from oral ulcers after 12 weeks in patients receiving apremilast was 42.7 points, compared with 18.7 points in the placebo group. Just over 50% of apremilast patients achieved complete response by week 12, compared with 22.3% in the placebo group.
The most common adverse events associated with apremilast during RELIEF were diarrhea, nausea, headache, and upper respiratory infection. This was consistent with apremilast’s known safety profile.
Apremilast is also indicated for treatment of patients with moderate to severe plaque psoriasis who are candidates for phototherapy or systemic therapy, and for patients with active psoriatic arthritis.
“Oral ulcers are a recurring and debilitating manifestation that affects nearly everyone living with Behçet’s disease and have an important negative impact on the quality of life for these patients. In the clinical trial, Otezla demonstrated improvements in measures of oral ulcers at week 12. Otezla has the potential to be a needed treatment option for U.S. patients and their physicians, who previously had limited options available,” Yusuf Yazici, MD, clinical associate professor in the department of medicine at New York University, said in the announcement.
The Food and Drug Administration has expanded the indication for apremilast (Otezla) to include the treatment of oral ulcers associated with Behçet’s disease in adults, according to an announcement from the manufacturer, Celgene.
FDA approval was based on results of the randomized, placebo-controlled, double-blind, phase 3 RELIEF trial, in which 207 patients with Behçet’s disease with active ulcers underwent treatment for 12 weeks with 30 mg apremilast or placebo. When measured on a visual analog scale, the reduction in pain from oral ulcers after 12 weeks in patients receiving apremilast was 42.7 points, compared with 18.7 points in the placebo group. Just over 50% of apremilast patients achieved complete response by week 12, compared with 22.3% in the placebo group.
The most common adverse events associated with apremilast during RELIEF were diarrhea, nausea, headache, and upper respiratory infection. This was consistent with apremilast’s known safety profile.
Apremilast is also indicated for treatment of patients with moderate to severe plaque psoriasis who are candidates for phototherapy or systemic therapy, and for patients with active psoriatic arthritis.
“Oral ulcers are a recurring and debilitating manifestation that affects nearly everyone living with Behçet’s disease and have an important negative impact on the quality of life for these patients. In the clinical trial, Otezla demonstrated improvements in measures of oral ulcers at week 12. Otezla has the potential to be a needed treatment option for U.S. patients and their physicians, who previously had limited options available,” Yusuf Yazici, MD, clinical associate professor in the department of medicine at New York University, said in the announcement.
The Food and Drug Administration has expanded the indication for apremilast (Otezla) to include the treatment of oral ulcers associated with Behçet’s disease in adults, according to an announcement from the manufacturer, Celgene.
FDA approval was based on results of the randomized, placebo-controlled, double-blind, phase 3 RELIEF trial, in which 207 patients with Behçet’s disease with active ulcers underwent treatment for 12 weeks with 30 mg apremilast or placebo. When measured on a visual analog scale, the reduction in pain from oral ulcers after 12 weeks in patients receiving apremilast was 42.7 points, compared with 18.7 points in the placebo group. Just over 50% of apremilast patients achieved complete response by week 12, compared with 22.3% in the placebo group.
The most common adverse events associated with apremilast during RELIEF were diarrhea, nausea, headache, and upper respiratory infection. This was consistent with apremilast’s known safety profile.
Apremilast is also indicated for treatment of patients with moderate to severe plaque psoriasis who are candidates for phototherapy or systemic therapy, and for patients with active psoriatic arthritis.
“Oral ulcers are a recurring and debilitating manifestation that affects nearly everyone living with Behçet’s disease and have an important negative impact on the quality of life for these patients. In the clinical trial, Otezla demonstrated improvements in measures of oral ulcers at week 12. Otezla has the potential to be a needed treatment option for U.S. patients and their physicians, who previously had limited options available,” Yusuf Yazici, MD, clinical associate professor in the department of medicine at New York University, said in the announcement.
A Unique Presentation of Lupus Erythematosus Tumidus in an Adolescent Boy
To the Editor:
Lupus erythematosus tumidus (LET) is a rarely diagnosed condition that was first described in 1909 by Hoffmann.1 Limited cases have been reported in the literature, with few documenting the disease in children.2 We report a unique clinical case of LET in a 14-year-old adolescent boy that was distributed solely on the hands. With slight heterogeneity in regards to clinical presentation and histopathology, there is a need for further exploration with regard to LET.
A 14-year-old adolescent boy presented to the dermatology clinic with progressive bilateral edema of 1 year’s duration with plaques and some scaling on the dorsal aspects of the digits and the nail bases predominantly on the right hand (Figure 1) and to a lesser extent on the left hand. The edema, erythema, and tenderness started in the right fifth digit; soon after the edema appeared, plaques began to form at the base of each nail bed, and the edema and erythema progressively spread to the other digits. He denied worsening of symptoms when exposed to cold temperatures. A complete review of systems was negative. The differential diagnoses included chilblain lupus erythematosus, perniosis, dermatomyositis, and polymorphous light eruption. A punch biopsy from the right fourth digit was performed.
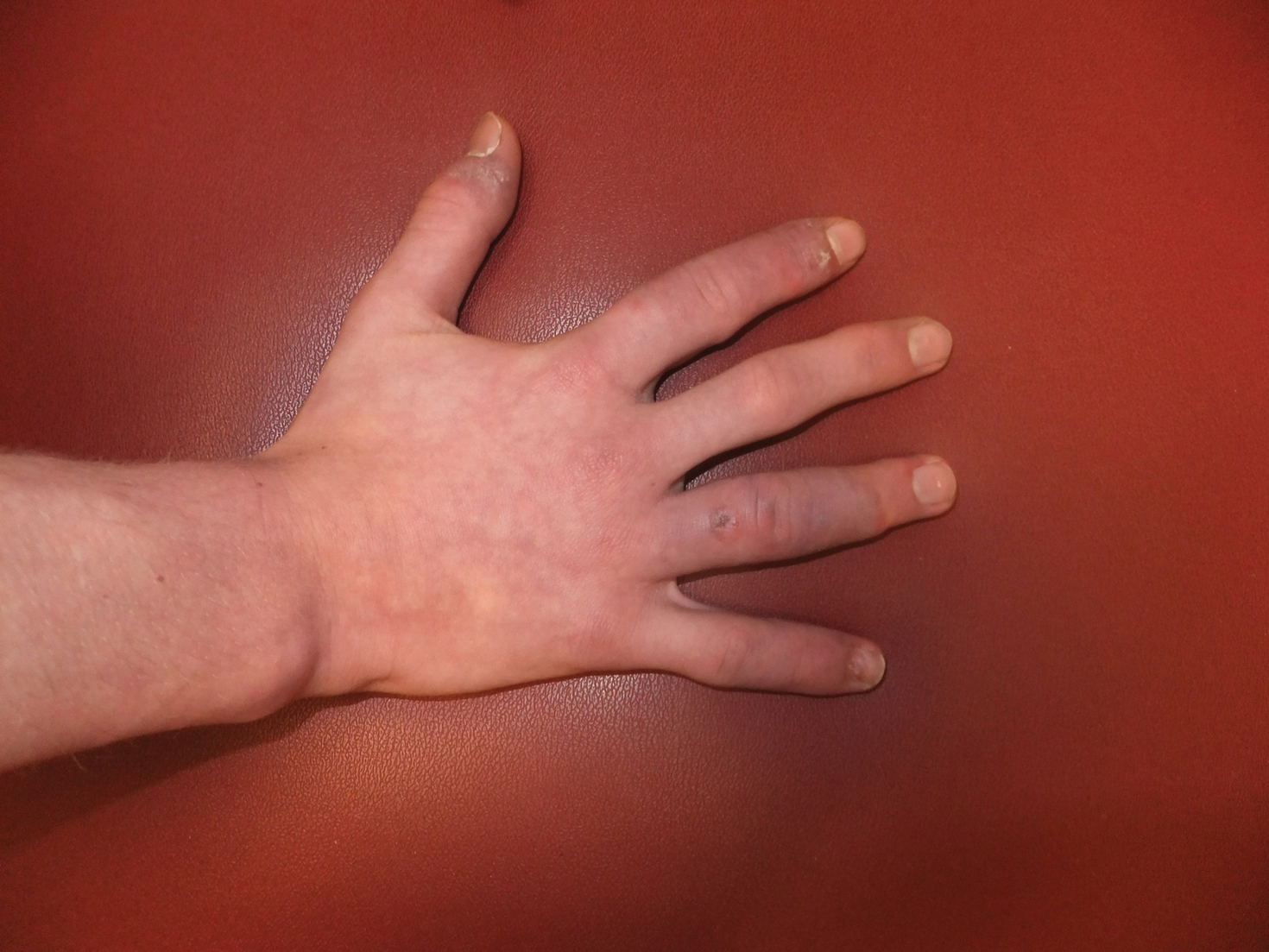
The biopsy showed superficial and deep perivascular and periadnexal mononuclear inflammation with large amounts of interstitial mucin deposition (Figure 2). The epidermis exhibited a loose orthokeratotic scale with no signs of interface damage. A diagnosis of perniosis was entertained but was ruled out due to the lack of papillary dermal edema and large amounts of mucin. With the lack of interface change and large amounts of mucin, a diagnosis of LET was favored over chilblain lupus erythematosus, as the latter diagnosis typically demonstrates interface change. The patient was started on hydroxychloroquine 200 mg twice daily and a short course of prednisone, and improvement of the lesions/plaques was noted at follow-up 6 weeks later. Continued improvement was noted 2 years after the initial presentation. His condition recurred when the hydroxychloroquine dosage was reduced to 200 mg once daily after 1 year. The patient did not report any adverse sequelae to treatment.
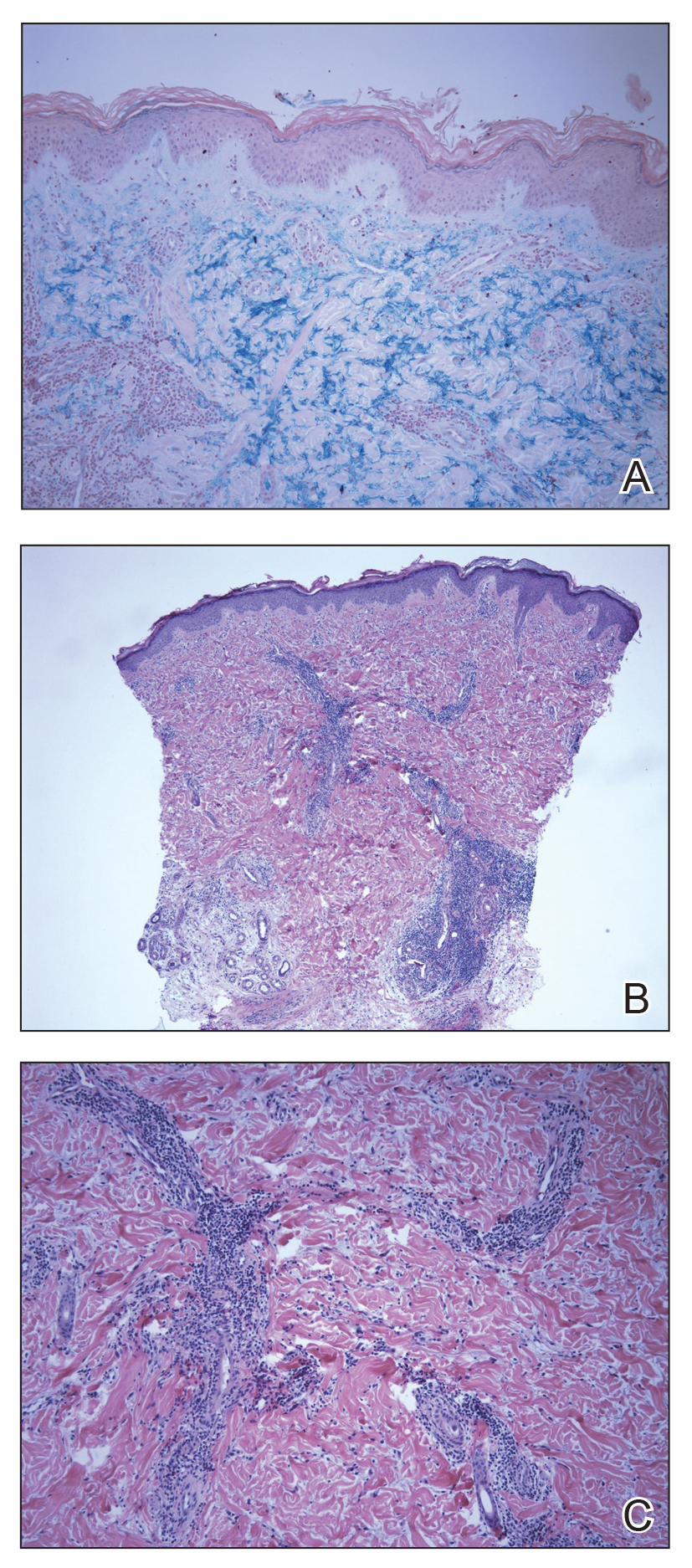
Histopathologic findings of superficial and deep perivascular and periadnexal lymphocytic infiltrates and interstitial dermal deposition of mucin in LET have remained consistent in the literature. Direct immunofluorescence has not revealed any complement or immunoglobulin deposition on the basement membrane.3,4 The epidermal characteristics are not as uniform, with the majority of cases in one review showing no epidermal changes and a minority showing minimal epidermal changes (eg, epidermal atrophy, hyperkeratosis, parakeratosis, acanthosis, spongiosis).5 When working up patients for LET, blood work usually is unremarkable, as LET rarely is associated with antinuclear antibodies or anti-Ro, anti-La, and anti-DNA antibodies.3,4 Lupus erythematosus tumidus generally is an independent process, but it has been reported to coexist with discoid lupus erythematosus and systemic lupus erythematosus in rare cases.6
The lesions of LET have been consistently described in the literature as photosensitive, erythematous, non-scarring, annular plaques and papules commonly occurring on the head/neck and other sun-exposed areas that do not cause hypopigmentation.3 Treatment of LET consists of systemic treatment with antimalarial drugs, sunscreens, and topical steroids for flares.
Lupus erythematosus tumidus is rare in children, with few case reports noted in the literature. Sonntag et al2 documented the disease in 3 children ranging from 3 to 8 years of age. Furthermore, Ruiz and Sanchez7 reported a case of LET in a 16-year-old adolescent girl. Our case is unique in that the lesions only occurred on the hands, whereas most case reports document distribution of the lesions on the head, neck, face, arms, back, and chest. Our patient’s age and the location of the lesions make it a unique clinical presentation of LET.
Reports in the literature show evidence of heterogeneity in the presentation, classification, and some of the histopathologic features of LET; however, there are minimal data on childhood LET. Further research and investigations are needed to more precisely define this condition.
Acknowledgment
The authors acknowledge Richard Schwartz, MD (Akron, Ohio), for reading the biopsy reports and assisting with photomicrographs.
- Hoffmann E. Demonstrationen: lupus erythematosus tumidus. Derm Zeitschr. 1909;16:159-160.
- Sonntag M, Lehmann P, Megahed M, et al. Lupus erythematosus tumidus in childhood. Dermatology. 2003;207:188-192.
- Schmitt V, Meuth AM, Amler S, et al. Lupus erythematosus tumidus is a separate subtype of cutaneous lupus erythematosus. Br J Dermatol. 2010;162:64-73.
- Vieira V, Del Pozo J, Yebra-Pimentel MT, et al. Lupus erythematosus tumidus: a series of 26 cases. Int J Dermatol. 2006;45:512-517.
- Kuhn A, Richter-Hintz D, Oslislo C, et al. Lupus erythematosus tumidus—a neglected subset of cutaneous lupus erythematosus: report of 40 cases. Arch Dermatol. 2000;136:1033-1041.
- Chen X, Wang S, Li L. A case report of lupus erythematosus tumidus converted from discoid lupus erythematosus. Medicine (Baltimore). 2018;97:e0375.
- Ruiz H, Sanchez J. Tumid lupus erythematosus. Am J Dermatopathol. 1999;21:356-360.
To the Editor:
Lupus erythematosus tumidus (LET) is a rarely diagnosed condition that was first described in 1909 by Hoffmann.1 Limited cases have been reported in the literature, with few documenting the disease in children.2 We report a unique clinical case of LET in a 14-year-old adolescent boy that was distributed solely on the hands. With slight heterogeneity in regards to clinical presentation and histopathology, there is a need for further exploration with regard to LET.
A 14-year-old adolescent boy presented to the dermatology clinic with progressive bilateral edema of 1 year’s duration with plaques and some scaling on the dorsal aspects of the digits and the nail bases predominantly on the right hand (Figure 1) and to a lesser extent on the left hand. The edema, erythema, and tenderness started in the right fifth digit; soon after the edema appeared, plaques began to form at the base of each nail bed, and the edema and erythema progressively spread to the other digits. He denied worsening of symptoms when exposed to cold temperatures. A complete review of systems was negative. The differential diagnoses included chilblain lupus erythematosus, perniosis, dermatomyositis, and polymorphous light eruption. A punch biopsy from the right fourth digit was performed.
The biopsy showed superficial and deep perivascular and periadnexal mononuclear inflammation with large amounts of interstitial mucin deposition (Figure 2). The epidermis exhibited a loose orthokeratotic scale with no signs of interface damage. A diagnosis of perniosis was entertained but was ruled out due to the lack of papillary dermal edema and large amounts of mucin. With the lack of interface change and large amounts of mucin, a diagnosis of LET was favored over chilblain lupus erythematosus, as the latter diagnosis typically demonstrates interface change. The patient was started on hydroxychloroquine 200 mg twice daily and a short course of prednisone, and improvement of the lesions/plaques was noted at follow-up 6 weeks later. Continued improvement was noted 2 years after the initial presentation. His condition recurred when the hydroxychloroquine dosage was reduced to 200 mg once daily after 1 year. The patient did not report any adverse sequelae to treatment.
Histopathologic findings of superficial and deep perivascular and periadnexal lymphocytic infiltrates and interstitial dermal deposition of mucin in LET have remained consistent in the literature. Direct immunofluorescence has not revealed any complement or immunoglobulin deposition on the basement membrane.3,4 The epidermal characteristics are not as uniform, with the majority of cases in one review showing no epidermal changes and a minority showing minimal epidermal changes (eg, epidermal atrophy, hyperkeratosis, parakeratosis, acanthosis, spongiosis).5 When working up patients for LET, blood work usually is unremarkable, as LET rarely is associated with antinuclear antibodies or anti-Ro, anti-La, and anti-DNA antibodies.3,4 Lupus erythematosus tumidus generally is an independent process, but it has been reported to coexist with discoid lupus erythematosus and systemic lupus erythematosus in rare cases.6
The lesions of LET have been consistently described in the literature as photosensitive, erythematous, non-scarring, annular plaques and papules commonly occurring on the head/neck and other sun-exposed areas that do not cause hypopigmentation.3 Treatment of LET consists of systemic treatment with antimalarial drugs, sunscreens, and topical steroids for flares.
Lupus erythematosus tumidus is rare in children, with few case reports noted in the literature. Sonntag et al2 documented the disease in 3 children ranging from 3 to 8 years of age. Furthermore, Ruiz and Sanchez7 reported a case of LET in a 16-year-old adolescent girl. Our case is unique in that the lesions only occurred on the hands, whereas most case reports document distribution of the lesions on the head, neck, face, arms, back, and chest. Our patient’s age and the location of the lesions make it a unique clinical presentation of LET.
Reports in the literature show evidence of heterogeneity in the presentation, classification, and some of the histopathologic features of LET; however, there are minimal data on childhood LET. Further research and investigations are needed to more precisely define this condition.
Acknowledgment
The authors acknowledge Richard Schwartz, MD (Akron, Ohio), for reading the biopsy reports and assisting with photomicrographs.
To the Editor:
Lupus erythematosus tumidus (LET) is a rarely diagnosed condition that was first described in 1909 by Hoffmann.1 Limited cases have been reported in the literature, with few documenting the disease in children.2 We report a unique clinical case of LET in a 14-year-old adolescent boy that was distributed solely on the hands. With slight heterogeneity in regards to clinical presentation and histopathology, there is a need for further exploration with regard to LET.
A 14-year-old adolescent boy presented to the dermatology clinic with progressive bilateral edema of 1 year’s duration with plaques and some scaling on the dorsal aspects of the digits and the nail bases predominantly on the right hand (Figure 1) and to a lesser extent on the left hand. The edema, erythema, and tenderness started in the right fifth digit; soon after the edema appeared, plaques began to form at the base of each nail bed, and the edema and erythema progressively spread to the other digits. He denied worsening of symptoms when exposed to cold temperatures. A complete review of systems was negative. The differential diagnoses included chilblain lupus erythematosus, perniosis, dermatomyositis, and polymorphous light eruption. A punch biopsy from the right fourth digit was performed.
The biopsy showed superficial and deep perivascular and periadnexal mononuclear inflammation with large amounts of interstitial mucin deposition (Figure 2). The epidermis exhibited a loose orthokeratotic scale with no signs of interface damage. A diagnosis of perniosis was entertained but was ruled out due to the lack of papillary dermal edema and large amounts of mucin. With the lack of interface change and large amounts of mucin, a diagnosis of LET was favored over chilblain lupus erythematosus, as the latter diagnosis typically demonstrates interface change. The patient was started on hydroxychloroquine 200 mg twice daily and a short course of prednisone, and improvement of the lesions/plaques was noted at follow-up 6 weeks later. Continued improvement was noted 2 years after the initial presentation. His condition recurred when the hydroxychloroquine dosage was reduced to 200 mg once daily after 1 year. The patient did not report any adverse sequelae to treatment.
Histopathologic findings of superficial and deep perivascular and periadnexal lymphocytic infiltrates and interstitial dermal deposition of mucin in LET have remained consistent in the literature. Direct immunofluorescence has not revealed any complement or immunoglobulin deposition on the basement membrane.3,4 The epidermal characteristics are not as uniform, with the majority of cases in one review showing no epidermal changes and a minority showing minimal epidermal changes (eg, epidermal atrophy, hyperkeratosis, parakeratosis, acanthosis, spongiosis).5 When working up patients for LET, blood work usually is unremarkable, as LET rarely is associated with antinuclear antibodies or anti-Ro, anti-La, and anti-DNA antibodies.3,4 Lupus erythematosus tumidus generally is an independent process, but it has been reported to coexist with discoid lupus erythematosus and systemic lupus erythematosus in rare cases.6
The lesions of LET have been consistently described in the literature as photosensitive, erythematous, non-scarring, annular plaques and papules commonly occurring on the head/neck and other sun-exposed areas that do not cause hypopigmentation.3 Treatment of LET consists of systemic treatment with antimalarial drugs, sunscreens, and topical steroids for flares.
Lupus erythematosus tumidus is rare in children, with few case reports noted in the literature. Sonntag et al2 documented the disease in 3 children ranging from 3 to 8 years of age. Furthermore, Ruiz and Sanchez7 reported a case of LET in a 16-year-old adolescent girl. Our case is unique in that the lesions only occurred on the hands, whereas most case reports document distribution of the lesions on the head, neck, face, arms, back, and chest. Our patient’s age and the location of the lesions make it a unique clinical presentation of LET.
Reports in the literature show evidence of heterogeneity in the presentation, classification, and some of the histopathologic features of LET; however, there are minimal data on childhood LET. Further research and investigations are needed to more precisely define this condition.
Acknowledgment
The authors acknowledge Richard Schwartz, MD (Akron, Ohio), for reading the biopsy reports and assisting with photomicrographs.
- Hoffmann E. Demonstrationen: lupus erythematosus tumidus. Derm Zeitschr. 1909;16:159-160.
- Sonntag M, Lehmann P, Megahed M, et al. Lupus erythematosus tumidus in childhood. Dermatology. 2003;207:188-192.
- Schmitt V, Meuth AM, Amler S, et al. Lupus erythematosus tumidus is a separate subtype of cutaneous lupus erythematosus. Br J Dermatol. 2010;162:64-73.
- Vieira V, Del Pozo J, Yebra-Pimentel MT, et al. Lupus erythematosus tumidus: a series of 26 cases. Int J Dermatol. 2006;45:512-517.
- Kuhn A, Richter-Hintz D, Oslislo C, et al. Lupus erythematosus tumidus—a neglected subset of cutaneous lupus erythematosus: report of 40 cases. Arch Dermatol. 2000;136:1033-1041.
- Chen X, Wang S, Li L. A case report of lupus erythematosus tumidus converted from discoid lupus erythematosus. Medicine (Baltimore). 2018;97:e0375.
- Ruiz H, Sanchez J. Tumid lupus erythematosus. Am J Dermatopathol. 1999;21:356-360.
- Hoffmann E. Demonstrationen: lupus erythematosus tumidus. Derm Zeitschr. 1909;16:159-160.
- Sonntag M, Lehmann P, Megahed M, et al. Lupus erythematosus tumidus in childhood. Dermatology. 2003;207:188-192.
- Schmitt V, Meuth AM, Amler S, et al. Lupus erythematosus tumidus is a separate subtype of cutaneous lupus erythematosus. Br J Dermatol. 2010;162:64-73.
- Vieira V, Del Pozo J, Yebra-Pimentel MT, et al. Lupus erythematosus tumidus: a series of 26 cases. Int J Dermatol. 2006;45:512-517.
- Kuhn A, Richter-Hintz D, Oslislo C, et al. Lupus erythematosus tumidus—a neglected subset of cutaneous lupus erythematosus: report of 40 cases. Arch Dermatol. 2000;136:1033-1041.
- Chen X, Wang S, Li L. A case report of lupus erythematosus tumidus converted from discoid lupus erythematosus. Medicine (Baltimore). 2018;97:e0375.
- Ruiz H, Sanchez J. Tumid lupus erythematosus. Am J Dermatopathol. 1999;21:356-360.
Practice Points
- Lupus erythematosus tumidus rarely occurs in the pediatric population.
- Lupus erythematosus tumidus is a unique subset of lupus associated with lack of interface change on histology and large amounts of mucin.
- Lesions typically present on the face and trunk but can very rarely present on the extremities and hands.