User login
EULAR takes SLE guidance to the next level

The goal was “to update the EULAR recommendations for the management of systemic lupus erythematosus [SLE], based on literature review and expert consensus,” said authors led by Antonis Fanouriakis, MD, PhD, of the rheumatology and clinical immunology unit at Attikon University Hospital, Athens.
The team accomplished their aim in 33 recommendations – about twice as many as in 2008 – covering goals of therapy, treatment, specific manifestations, and comorbidities (Ann Rheum Dis. 2019 Mar 29. doi: 10.1136/annrheumdis-2019-215089).
A lot has changed in the past 11 years, and the new guidelines reflect that. Biologics, for instance, were barely mentioned in 2008, except as a topic for future research. The new document makes a strong recommendation for add-on belimumab (Benlysta) to be considered in persistently active or flaring extrarenal disease, and rituximab (Rituxan) for organ-threatening, refractory disease.
The group now also makes a strong recommendation for hydroxychloroquine (Plaquenil) for all lupus patients, barring contraindications, at a dose not exceeding 5 mg per kg real body weight, with ophthalmologic screening performed at baseline, after 5 years, and yearly thereafter. It also calls for routine antiphospholipid antibody testing.
Calcineurin inhibitors weren’t mentioned at all in 2008, but they show up in the new document with a moderate recommendation as first-line topical options for skin disease, along with glucocorticoids. The authors also made a moderate recommendation for diagnostic kidney biopsy, calling it “essential” to catch renal involvement early; EULAR was less certain in 2008. Also new in 2019, and barely mentioned in 2008, there’s an entire section on hematologic manifestations, as well as advice on thalidomide for cutaneous disease.
For hematologic disease, the group makes a weak recommendation for pulsed intravenous methylprednisolone and/or intravenous immunoglobulin, with mycophenolate, azathioprine, or cyclosporine for maintenance. Cyclophosphamide, along with rituximab, are options for severe hematologic cases.
Cardiovascular disease, like biologics, was mentioned mostly in 2008 as a topic for future research; the new guidelines contain an entire section on the issue. There’s a strong recommendation for regular assessment of traditional and disease-related risk factors, including persistently active disease; increased disease duration; medium or high titers of antiphospholipid antibodies; renal involvement; and chronic glucocorticoid use. High-risk people, the document notes, “may be candidates for preventative strategies as in the general population,” including low-dose aspirin and statins.
The new guidance is also more certain about mycophenolate for renal disease, with a strong recommendation for use as an induction and maintenance agent, with azathioprine the other strong candidate for maintenance. EULAR also made a weak recommendation for mycophenolate with low-dose calcineurin inhibitors in severe nephrotic syndrome, in some circumstances.
For antiphospholipid antibody carriers, EULAR noted that a recent randomized, open-label trial comparing rivaroxaban against warfarin “was prematurely terminated due to an excess of thromboembolic events in the rivaroxaban arm. Thus, in patients with SLE-antiphospholipid syndrome, “use of novel oral anticoagulants for secondary prevention should be avoided.”
The group notes that management should aim at “remission of disease symptoms and signs, prevention of damage accrual, and minimization of drug side effects, as well as improvement of quality of life.” To that end, it said newly defined low disease-activity states, such as an SLE Disease Activity Index score of 3 or less on antimalarials, are useful to guide treatment, and have comparable rates of remission and flare prevention.
SOURCE: Fanouriakis A et al. Ann Rheum Dis. 2019 Mar 29. doi: 10.1136/annrheumdis-2019-215089
The goal was “to update the EULAR recommendations for the management of systemic lupus erythematosus [SLE], based on literature review and expert consensus,” said authors led by Antonis Fanouriakis, MD, PhD, of the rheumatology and clinical immunology unit at Attikon University Hospital, Athens.
The team accomplished their aim in 33 recommendations – about twice as many as in 2008 – covering goals of therapy, treatment, specific manifestations, and comorbidities (Ann Rheum Dis. 2019 Mar 29. doi: 10.1136/annrheumdis-2019-215089).
A lot has changed in the past 11 years, and the new guidelines reflect that. Biologics, for instance, were barely mentioned in 2008, except as a topic for future research. The new document makes a strong recommendation for add-on belimumab (Benlysta) to be considered in persistently active or flaring extrarenal disease, and rituximab (Rituxan) for organ-threatening, refractory disease.
The group now also makes a strong recommendation for hydroxychloroquine (Plaquenil) for all lupus patients, barring contraindications, at a dose not exceeding 5 mg per kg real body weight, with ophthalmologic screening performed at baseline, after 5 years, and yearly thereafter. It also calls for routine antiphospholipid antibody testing.
Calcineurin inhibitors weren’t mentioned at all in 2008, but they show up in the new document with a moderate recommendation as first-line topical options for skin disease, along with glucocorticoids. The authors also made a moderate recommendation for diagnostic kidney biopsy, calling it “essential” to catch renal involvement early; EULAR was less certain in 2008. Also new in 2019, and barely mentioned in 2008, there’s an entire section on hematologic manifestations, as well as advice on thalidomide for cutaneous disease.
For hematologic disease, the group makes a weak recommendation for pulsed intravenous methylprednisolone and/or intravenous immunoglobulin, with mycophenolate, azathioprine, or cyclosporine for maintenance. Cyclophosphamide, along with rituximab, are options for severe hematologic cases.
Cardiovascular disease, like biologics, was mentioned mostly in 2008 as a topic for future research; the new guidelines contain an entire section on the issue. There’s a strong recommendation for regular assessment of traditional and disease-related risk factors, including persistently active disease; increased disease duration; medium or high titers of antiphospholipid antibodies; renal involvement; and chronic glucocorticoid use. High-risk people, the document notes, “may be candidates for preventative strategies as in the general population,” including low-dose aspirin and statins.
The new guidance is also more certain about mycophenolate for renal disease, with a strong recommendation for use as an induction and maintenance agent, with azathioprine the other strong candidate for maintenance. EULAR also made a weak recommendation for mycophenolate with low-dose calcineurin inhibitors in severe nephrotic syndrome, in some circumstances.
For antiphospholipid antibody carriers, EULAR noted that a recent randomized, open-label trial comparing rivaroxaban against warfarin “was prematurely terminated due to an excess of thromboembolic events in the rivaroxaban arm. Thus, in patients with SLE-antiphospholipid syndrome, “use of novel oral anticoagulants for secondary prevention should be avoided.”
The group notes that management should aim at “remission of disease symptoms and signs, prevention of damage accrual, and minimization of drug side effects, as well as improvement of quality of life.” To that end, it said newly defined low disease-activity states, such as an SLE Disease Activity Index score of 3 or less on antimalarials, are useful to guide treatment, and have comparable rates of remission and flare prevention.
SOURCE: Fanouriakis A et al. Ann Rheum Dis. 2019 Mar 29. doi: 10.1136/annrheumdis-2019-215089
The goal was “to update the EULAR recommendations for the management of systemic lupus erythematosus [SLE], based on literature review and expert consensus,” said authors led by Antonis Fanouriakis, MD, PhD, of the rheumatology and clinical immunology unit at Attikon University Hospital, Athens.
The team accomplished their aim in 33 recommendations – about twice as many as in 2008 – covering goals of therapy, treatment, specific manifestations, and comorbidities (Ann Rheum Dis. 2019 Mar 29. doi: 10.1136/annrheumdis-2019-215089).
A lot has changed in the past 11 years, and the new guidelines reflect that. Biologics, for instance, were barely mentioned in 2008, except as a topic for future research. The new document makes a strong recommendation for add-on belimumab (Benlysta) to be considered in persistently active or flaring extrarenal disease, and rituximab (Rituxan) for organ-threatening, refractory disease.
The group now also makes a strong recommendation for hydroxychloroquine (Plaquenil) for all lupus patients, barring contraindications, at a dose not exceeding 5 mg per kg real body weight, with ophthalmologic screening performed at baseline, after 5 years, and yearly thereafter. It also calls for routine antiphospholipid antibody testing.
Calcineurin inhibitors weren’t mentioned at all in 2008, but they show up in the new document with a moderate recommendation as first-line topical options for skin disease, along with glucocorticoids. The authors also made a moderate recommendation for diagnostic kidney biopsy, calling it “essential” to catch renal involvement early; EULAR was less certain in 2008. Also new in 2019, and barely mentioned in 2008, there’s an entire section on hematologic manifestations, as well as advice on thalidomide for cutaneous disease.
For hematologic disease, the group makes a weak recommendation for pulsed intravenous methylprednisolone and/or intravenous immunoglobulin, with mycophenolate, azathioprine, or cyclosporine for maintenance. Cyclophosphamide, along with rituximab, are options for severe hematologic cases.
Cardiovascular disease, like biologics, was mentioned mostly in 2008 as a topic for future research; the new guidelines contain an entire section on the issue. There’s a strong recommendation for regular assessment of traditional and disease-related risk factors, including persistently active disease; increased disease duration; medium or high titers of antiphospholipid antibodies; renal involvement; and chronic glucocorticoid use. High-risk people, the document notes, “may be candidates for preventative strategies as in the general population,” including low-dose aspirin and statins.
The new guidance is also more certain about mycophenolate for renal disease, with a strong recommendation for use as an induction and maintenance agent, with azathioprine the other strong candidate for maintenance. EULAR also made a weak recommendation for mycophenolate with low-dose calcineurin inhibitors in severe nephrotic syndrome, in some circumstances.
For antiphospholipid antibody carriers, EULAR noted that a recent randomized, open-label trial comparing rivaroxaban against warfarin “was prematurely terminated due to an excess of thromboembolic events in the rivaroxaban arm. Thus, in patients with SLE-antiphospholipid syndrome, “use of novel oral anticoagulants for secondary prevention should be avoided.”
The group notes that management should aim at “remission of disease symptoms and signs, prevention of damage accrual, and minimization of drug side effects, as well as improvement of quality of life.” To that end, it said newly defined low disease-activity states, such as an SLE Disease Activity Index score of 3 or less on antimalarials, are useful to guide treatment, and have comparable rates of remission and flare prevention.
SOURCE: Fanouriakis A et al. Ann Rheum Dis. 2019 Mar 29. doi: 10.1136/annrheumdis-2019-215089
FROM ANNALS OF THE RHEUMATIC DISEASES
Tackling the challenges of pediatric localized scleroderma
MAUI, HAWAII – One of the most important steps to take when a child has received a biopsy-confirmed diagnosis of localized scleroderma is to sit down with the family and explain the rationale for the aggressive therapies to come, Anne M. Stevens, MD, PhD, said at the 2019 Rheumatology Winter Clinical Symposium.

It can be a tough sell at first, especially when a child has only a small red streak on the nose and perhaps a subtle linear lesion on the forehead or scalp. But the family has to come to understand that this is a serious, chronic, progressive fibrotic disease.
“Talk about what a big impact this disease can have on growth of a limb and the normal life of a child because of the cosmetic appearance. Explain that the length of treatment course is based on the long-term outcomes and quality of life. This discussion is usually sufficient” to convince people to give their children “these pretty serious medications,” said Dr. Stevens, professor of pediatrics and head of the division of pediatric rheumatology at the University of Washington, Seattle.
“The treatment goal is to control inflammation and prevent damage in these patients, who we like to catch very early, when it’s a subtle lesion,” she added.
The biggest problem
The biggest contributors to poor quality of life in patients with juvenile localized scleroderma are the extracutaneous manifestations, which occur in up to 50% of cases. Joint pain occurs in roughly 20% of patients, joint contractures due to fibrosis of skin and/or tendons in 30%, and myalgia with or without myositis in 15%. Muscle atrophy due to the deep component of the scleroderma can occur. Moreover, growth problems – especially leg or arm length discrepancies – happen in about 20% of patients in prospective studies. These growth problems may not be obvious until a child enters a growth spurt, at which point there is a limited ability to achieve improvement. That’s why Dr. Stevens recommends that every child with localized scleroderma should get a full joint exam at every visit, with measurement and photos of lesions and recording of all erythematous, violaceous, and waxy-hued areas. And if there are lesions on the head, annual eye exams are warranted.
The prevalence of juvenile localized scleroderma in the United States is about 3 per 100,000, with a mean age of onset of 8.2 years. That makes it 100-fold more common than pediatric systemic sclerosis.
The treatment ladder
There are no Food and Drug Administration–approved medications for localized scleroderma in children. It’s all off label. That being said, there is strong consensus among members of the Childhood Arthritis and Rheumatology Research Alliance that the first-line therapy is methotrexate at 15 mg/m2 or a maximum of 20 mg/week plus intravenous corticosteroids weaned over the course of 3-6 months. This is the treatment regimen with the best supporting evidence of safety and efficacy, including a single Italian randomized, double-blind, placebo-controlled clinical trial (Arthritis Rheum. 2011 Jul;63[7]:1998-2006) and an accompanying long-term, open-label follow-up study (J Am Acad Dermatol. 2012 Dec;67[6]:1151-6).
All of the other treatments she uses for juvenile localized scleroderma – mycophenolate mofetil (CellCept), abatacept (Orencia), tocilizumab (Actemra), and occasionally others – are backed only by a smattering of small case series. However, given the serious potential trajectory of this disease, that modest evidence base has been sufficient for her to receive insurance coverage approval of these agents.
In the randomized trial of first-line methotrexate, 48 of 65 patients treated with methotrexate plus steroid (74%) were responders. And among those 48 responders, 35 (73%) maintained a clinical remission for a mean of 25 months off-drug, while another 13 (27%) were in clinical remission on methotrexate. Twenty-eight patients developed side effects that were generally mild; no one required treatment discontinuation. At the 5-year mark, after an average of an initial 2 years on methotrexate, half of the patients were in a sustained clinical remission, which Dr. Stevens deemed “pretty good” considering the well established and manageable safety profile of the drug.
If a patient fails to respond to methotrexate plus corticosteroids within a few months or later experiences disease progression, Dr. Stevens’ second-line therapy is mycophenolate mofetil in conjunction with corticosteroids. Its use in arresting juvenile localized scleroderma is supported by two favorable published case series, the largest of which includes 10 patients (Rheumatology [Oxford]. 2009 Nov;48[11]:1410-3).
Dr. Stevens’ third-line therapy is intravenous abatacept at 10 mg/kg monthly along with intravenous methylprednisolone at 500 mg/week. There are five published case series, the most recent and largest of which included 13 adult patients, two of whom had en coup de sabre lesions (Acta Derm Venereol. 2018 Apr 16;98[4]:465-6). The biologic also shows promise in patients with advanced severe disease with deep tissue involvement (Semin Arthritis Rheum. 2017 Jun;46[6]:775-81). And abatacept has a plausible mechanism of action in localized scleroderma: French investigators have shown it induces regression of skin fibrosis in a mouse model of the disease (Ann Rheum Dis. 2016 Dec;75[12]:2142-9).
Her fourth-line strategy is the anti-interleukin-6 agent tocilizumab, again in conjunction with corticosteroids. In a translational study, tocilizumab has been shown to normalize dermal fibroblasts and collagen in patients with systemic sclerosis (Ann Rheum Dis. 2018 Sep;77[9]:1362-71). And there have been two promising small retrospective case series as well. A more definitive clinical trial is planned.
Dr. Stevens said that when starting a biologic agent in a child with localized scleroderma, she routinely adds methotrexate until the disease is under control.
Drugs supported by case reports and worth considering on an individual basis as a last resort are hydroxychloroquine, azathioprine, cyclosporine, and imatinib mesylate (Gleevec).
For mild, superficial lesions that don’t cross joints, ultraviolet light A phototherapy is a therapeutic option. It displayed significant benefit in a systematic review and meta-analysis of 19 studies comparing it to methotrexate, although the results with methotrexate were deemed superior (Semin Arthritis Rheum. 2018 Dec;48[3]:495-503).
The pros and cons of getting a baseline brain MRI
Children with localized scleroderma have increased rates of severe headache, peripheral neuropathy, complex partial seizures, and stroke. So it had been Dr. Stevens’ routine practice to obtain an initial brain MRI at the time of diagnosis. Of late, though, she has reconsidered that practice.
“The problem is that some patients with abnormal MRI lesions have no CNS disease at all, and there are also a fair number of patients with a normal MRI who have CNS symptoms. So in our practice we’re pulling back on doing screening MRIs because we don’t know what to do with the findings, and it just makes everybody worried,” she said.
However, if a child with localized scleroderma develops headaches, seizures, neuropathies, or other CNS symptoms, then by all means get an MRI, and if it shows findings such as brain atrophy, white matter lesions, calcifications, or leptomeningeal enhancement, consider treatment, she added.
Dr. Stevens reported receiving research funding from Kineta and Seattle Genetics.
MAUI, HAWAII – One of the most important steps to take when a child has received a biopsy-confirmed diagnosis of localized scleroderma is to sit down with the family and explain the rationale for the aggressive therapies to come, Anne M. Stevens, MD, PhD, said at the 2019 Rheumatology Winter Clinical Symposium.
It can be a tough sell at first, especially when a child has only a small red streak on the nose and perhaps a subtle linear lesion on the forehead or scalp. But the family has to come to understand that this is a serious, chronic, progressive fibrotic disease.
“Talk about what a big impact this disease can have on growth of a limb and the normal life of a child because of the cosmetic appearance. Explain that the length of treatment course is based on the long-term outcomes and quality of life. This discussion is usually sufficient” to convince people to give their children “these pretty serious medications,” said Dr. Stevens, professor of pediatrics and head of the division of pediatric rheumatology at the University of Washington, Seattle.
“The treatment goal is to control inflammation and prevent damage in these patients, who we like to catch very early, when it’s a subtle lesion,” she added.
The biggest problem
The biggest contributors to poor quality of life in patients with juvenile localized scleroderma are the extracutaneous manifestations, which occur in up to 50% of cases. Joint pain occurs in roughly 20% of patients, joint contractures due to fibrosis of skin and/or tendons in 30%, and myalgia with or without myositis in 15%. Muscle atrophy due to the deep component of the scleroderma can occur. Moreover, growth problems – especially leg or arm length discrepancies – happen in about 20% of patients in prospective studies. These growth problems may not be obvious until a child enters a growth spurt, at which point there is a limited ability to achieve improvement. That’s why Dr. Stevens recommends that every child with localized scleroderma should get a full joint exam at every visit, with measurement and photos of lesions and recording of all erythematous, violaceous, and waxy-hued areas. And if there are lesions on the head, annual eye exams are warranted.
The prevalence of juvenile localized scleroderma in the United States is about 3 per 100,000, with a mean age of onset of 8.2 years. That makes it 100-fold more common than pediatric systemic sclerosis.
The treatment ladder
There are no Food and Drug Administration–approved medications for localized scleroderma in children. It’s all off label. That being said, there is strong consensus among members of the Childhood Arthritis and Rheumatology Research Alliance that the first-line therapy is methotrexate at 15 mg/m2 or a maximum of 20 mg/week plus intravenous corticosteroids weaned over the course of 3-6 months. This is the treatment regimen with the best supporting evidence of safety and efficacy, including a single Italian randomized, double-blind, placebo-controlled clinical trial (Arthritis Rheum. 2011 Jul;63[7]:1998-2006) and an accompanying long-term, open-label follow-up study (J Am Acad Dermatol. 2012 Dec;67[6]:1151-6).
All of the other treatments she uses for juvenile localized scleroderma – mycophenolate mofetil (CellCept), abatacept (Orencia), tocilizumab (Actemra), and occasionally others – are backed only by a smattering of small case series. However, given the serious potential trajectory of this disease, that modest evidence base has been sufficient for her to receive insurance coverage approval of these agents.
In the randomized trial of first-line methotrexate, 48 of 65 patients treated with methotrexate plus steroid (74%) were responders. And among those 48 responders, 35 (73%) maintained a clinical remission for a mean of 25 months off-drug, while another 13 (27%) were in clinical remission on methotrexate. Twenty-eight patients developed side effects that were generally mild; no one required treatment discontinuation. At the 5-year mark, after an average of an initial 2 years on methotrexate, half of the patients were in a sustained clinical remission, which Dr. Stevens deemed “pretty good” considering the well established and manageable safety profile of the drug.
If a patient fails to respond to methotrexate plus corticosteroids within a few months or later experiences disease progression, Dr. Stevens’ second-line therapy is mycophenolate mofetil in conjunction with corticosteroids. Its use in arresting juvenile localized scleroderma is supported by two favorable published case series, the largest of which includes 10 patients (Rheumatology [Oxford]. 2009 Nov;48[11]:1410-3).
Dr. Stevens’ third-line therapy is intravenous abatacept at 10 mg/kg monthly along with intravenous methylprednisolone at 500 mg/week. There are five published case series, the most recent and largest of which included 13 adult patients, two of whom had en coup de sabre lesions (Acta Derm Venereol. 2018 Apr 16;98[4]:465-6). The biologic also shows promise in patients with advanced severe disease with deep tissue involvement (Semin Arthritis Rheum. 2017 Jun;46[6]:775-81). And abatacept has a plausible mechanism of action in localized scleroderma: French investigators have shown it induces regression of skin fibrosis in a mouse model of the disease (Ann Rheum Dis. 2016 Dec;75[12]:2142-9).
Her fourth-line strategy is the anti-interleukin-6 agent tocilizumab, again in conjunction with corticosteroids. In a translational study, tocilizumab has been shown to normalize dermal fibroblasts and collagen in patients with systemic sclerosis (Ann Rheum Dis. 2018 Sep;77[9]:1362-71). And there have been two promising small retrospective case series as well. A more definitive clinical trial is planned.
Dr. Stevens said that when starting a biologic agent in a child with localized scleroderma, she routinely adds methotrexate until the disease is under control.
Drugs supported by case reports and worth considering on an individual basis as a last resort are hydroxychloroquine, azathioprine, cyclosporine, and imatinib mesylate (Gleevec).
For mild, superficial lesions that don’t cross joints, ultraviolet light A phototherapy is a therapeutic option. It displayed significant benefit in a systematic review and meta-analysis of 19 studies comparing it to methotrexate, although the results with methotrexate were deemed superior (Semin Arthritis Rheum. 2018 Dec;48[3]:495-503).
The pros and cons of getting a baseline brain MRI
Children with localized scleroderma have increased rates of severe headache, peripheral neuropathy, complex partial seizures, and stroke. So it had been Dr. Stevens’ routine practice to obtain an initial brain MRI at the time of diagnosis. Of late, though, she has reconsidered that practice.
“The problem is that some patients with abnormal MRI lesions have no CNS disease at all, and there are also a fair number of patients with a normal MRI who have CNS symptoms. So in our practice we’re pulling back on doing screening MRIs because we don’t know what to do with the findings, and it just makes everybody worried,” she said.
However, if a child with localized scleroderma develops headaches, seizures, neuropathies, or other CNS symptoms, then by all means get an MRI, and if it shows findings such as brain atrophy, white matter lesions, calcifications, or leptomeningeal enhancement, consider treatment, she added.
Dr. Stevens reported receiving research funding from Kineta and Seattle Genetics.
MAUI, HAWAII – One of the most important steps to take when a child has received a biopsy-confirmed diagnosis of localized scleroderma is to sit down with the family and explain the rationale for the aggressive therapies to come, Anne M. Stevens, MD, PhD, said at the 2019 Rheumatology Winter Clinical Symposium.
It can be a tough sell at first, especially when a child has only a small red streak on the nose and perhaps a subtle linear lesion on the forehead or scalp. But the family has to come to understand that this is a serious, chronic, progressive fibrotic disease.
“Talk about what a big impact this disease can have on growth of a limb and the normal life of a child because of the cosmetic appearance. Explain that the length of treatment course is based on the long-term outcomes and quality of life. This discussion is usually sufficient” to convince people to give their children “these pretty serious medications,” said Dr. Stevens, professor of pediatrics and head of the division of pediatric rheumatology at the University of Washington, Seattle.
“The treatment goal is to control inflammation and prevent damage in these patients, who we like to catch very early, when it’s a subtle lesion,” she added.
The biggest problem
The biggest contributors to poor quality of life in patients with juvenile localized scleroderma are the extracutaneous manifestations, which occur in up to 50% of cases. Joint pain occurs in roughly 20% of patients, joint contractures due to fibrosis of skin and/or tendons in 30%, and myalgia with or without myositis in 15%. Muscle atrophy due to the deep component of the scleroderma can occur. Moreover, growth problems – especially leg or arm length discrepancies – happen in about 20% of patients in prospective studies. These growth problems may not be obvious until a child enters a growth spurt, at which point there is a limited ability to achieve improvement. That’s why Dr. Stevens recommends that every child with localized scleroderma should get a full joint exam at every visit, with measurement and photos of lesions and recording of all erythematous, violaceous, and waxy-hued areas. And if there are lesions on the head, annual eye exams are warranted.
The prevalence of juvenile localized scleroderma in the United States is about 3 per 100,000, with a mean age of onset of 8.2 years. That makes it 100-fold more common than pediatric systemic sclerosis.
The treatment ladder
There are no Food and Drug Administration–approved medications for localized scleroderma in children. It’s all off label. That being said, there is strong consensus among members of the Childhood Arthritis and Rheumatology Research Alliance that the first-line therapy is methotrexate at 15 mg/m2 or a maximum of 20 mg/week plus intravenous corticosteroids weaned over the course of 3-6 months. This is the treatment regimen with the best supporting evidence of safety and efficacy, including a single Italian randomized, double-blind, placebo-controlled clinical trial (Arthritis Rheum. 2011 Jul;63[7]:1998-2006) and an accompanying long-term, open-label follow-up study (J Am Acad Dermatol. 2012 Dec;67[6]:1151-6).
All of the other treatments she uses for juvenile localized scleroderma – mycophenolate mofetil (CellCept), abatacept (Orencia), tocilizumab (Actemra), and occasionally others – are backed only by a smattering of small case series. However, given the serious potential trajectory of this disease, that modest evidence base has been sufficient for her to receive insurance coverage approval of these agents.
In the randomized trial of first-line methotrexate, 48 of 65 patients treated with methotrexate plus steroid (74%) were responders. And among those 48 responders, 35 (73%) maintained a clinical remission for a mean of 25 months off-drug, while another 13 (27%) were in clinical remission on methotrexate. Twenty-eight patients developed side effects that were generally mild; no one required treatment discontinuation. At the 5-year mark, after an average of an initial 2 years on methotrexate, half of the patients were in a sustained clinical remission, which Dr. Stevens deemed “pretty good” considering the well established and manageable safety profile of the drug.
If a patient fails to respond to methotrexate plus corticosteroids within a few months or later experiences disease progression, Dr. Stevens’ second-line therapy is mycophenolate mofetil in conjunction with corticosteroids. Its use in arresting juvenile localized scleroderma is supported by two favorable published case series, the largest of which includes 10 patients (Rheumatology [Oxford]. 2009 Nov;48[11]:1410-3).
Dr. Stevens’ third-line therapy is intravenous abatacept at 10 mg/kg monthly along with intravenous methylprednisolone at 500 mg/week. There are five published case series, the most recent and largest of which included 13 adult patients, two of whom had en coup de sabre lesions (Acta Derm Venereol. 2018 Apr 16;98[4]:465-6). The biologic also shows promise in patients with advanced severe disease with deep tissue involvement (Semin Arthritis Rheum. 2017 Jun;46[6]:775-81). And abatacept has a plausible mechanism of action in localized scleroderma: French investigators have shown it induces regression of skin fibrosis in a mouse model of the disease (Ann Rheum Dis. 2016 Dec;75[12]:2142-9).
Her fourth-line strategy is the anti-interleukin-6 agent tocilizumab, again in conjunction with corticosteroids. In a translational study, tocilizumab has been shown to normalize dermal fibroblasts and collagen in patients with systemic sclerosis (Ann Rheum Dis. 2018 Sep;77[9]:1362-71). And there have been two promising small retrospective case series as well. A more definitive clinical trial is planned.
Dr. Stevens said that when starting a biologic agent in a child with localized scleroderma, she routinely adds methotrexate until the disease is under control.
Drugs supported by case reports and worth considering on an individual basis as a last resort are hydroxychloroquine, azathioprine, cyclosporine, and imatinib mesylate (Gleevec).
For mild, superficial lesions that don’t cross joints, ultraviolet light A phototherapy is a therapeutic option. It displayed significant benefit in a systematic review and meta-analysis of 19 studies comparing it to methotrexate, although the results with methotrexate were deemed superior (Semin Arthritis Rheum. 2018 Dec;48[3]:495-503).
The pros and cons of getting a baseline brain MRI
Children with localized scleroderma have increased rates of severe headache, peripheral neuropathy, complex partial seizures, and stroke. So it had been Dr. Stevens’ routine practice to obtain an initial brain MRI at the time of diagnosis. Of late, though, she has reconsidered that practice.
“The problem is that some patients with abnormal MRI lesions have no CNS disease at all, and there are also a fair number of patients with a normal MRI who have CNS symptoms. So in our practice we’re pulling back on doing screening MRIs because we don’t know what to do with the findings, and it just makes everybody worried,” she said.
However, if a child with localized scleroderma develops headaches, seizures, neuropathies, or other CNS symptoms, then by all means get an MRI, and if it shows findings such as brain atrophy, white matter lesions, calcifications, or leptomeningeal enhancement, consider treatment, she added.
Dr. Stevens reported receiving research funding from Kineta and Seattle Genetics.
REPORTING FROM RWCS 2019
FDA panel leans toward more robust breast implant surveillance
SILVER SPRING, MD. – A mandatory, comprehensive approach to collecting adverse event data from breast implant recipients was favored during a March 25 hearing by a Food and Drug Administration advisory panel that oversees surgical devices.
This additional data could offer more complete information during the informed consent process for breast implants and potentially validate a new, autoimmune-like syndrome – breast implant illness (BII).
On the first day of a scheduled 2-day hearing, the advisory panel held no votes and took no formal actions. After a day of expert presentations and comments from more than 40 members of the public – mostly personal stories from affected patients and from plastic surgeons who place breast implants, panel members discussed a handful of questions from the FDA about relevant data to collect to better define the risks posed to breast implant recipients from breast-implant associated anaplastic large cell lymphoma (BIA-ALCL) and BII.
The advisory panel meeting took place as reports recently appeared documenting the scope of BIA-ALCL (Plast Reconstr Surg. 2019 March;143[3S]:65S-73S) and how to diagnose and manage BIA-ALCL (Aesthetic Surg J. 2019 March;39[S1}:S3-S13), and the existence of BII (Plast Reconstr Surg. 2019 March;143[3S]:74S-81S).
During the day’s two public comment periods, the panel heard from several women who gave brief accounts of developing and dealing with BIA-ALCL or BII.
“We think it’s important that all breast implant patients be aware of the risk for BIA-ALCL,” said Binita Ashar, MD, director of the FDAs Division of Surgical Devices. The FDA “is asking the panel what further steps need to be taken to understand the BIA-ALCL risk,” said Dr. Ashar as she opened the meeting of the General and Plastic Surgery Devices Panel of the Medical Devices Advisory Committee.

While the agency, as well as the plastic surgery community, have acknowledged the existence of BIA-ALCL since 2011, only recently have good data emerged on the scope of the complication. During the hearing, Mark W. Clemens, MD, a plastic surgeon at MD Anderson Cancer Center in Houston, reported on his analysis of 457 unique cases of BIA-ALCL reported to the FDA since 2011. He found that the vast majority of cases had occurred in women who had received textured implants while a relatively small minority were linked with the placement of smooth implants.
Further scrutiny of the reported details of each case showed that none of the lymphomas were linked with a confirmed instance of “pure” smooth implant exposure. He also estimated the U.S. incidence of BIA-ALCL as roughly one case for every 20,000 implants. Complete, en bloc removal of the implant seems to be the most effective way to treat the cancer; most explanted patients have a good prognosis, he said.
Despite the apparent link between textured implants specifically and onset of BIA-ALCL, some panel members did not see a ban on textured implants as the answer.

Texturing the implant helps to stabilize the implant in position. Without texturing “we would need to use something else to stabilize the implant, or there would be a tsunami of reoperations,” said panel member Mary H. McGrath, MD, professor of surgery at the University of California, San Francisco. The main alternative to texturing for stabilizing implants is to wrap them in place using surgical mesh, but that approach may also cause problems.
“Instead of just taking textured implants off the market, we need to also look at their advantages. A critical issue is informed consent,” said panel member Marc E. Lippman, MD, a professor of medicine at Georgetown University, Washington. Banning smooth implants based on what’s known so far “would be an extraordinary over reaction,” he said during the first day’s session.
Current U.S. anecdotal experience suggests that a ban may not even be necessary because “plastic surgeons are more and more walking away from textured implants” because of the apparent link to BIA-ALCL, Dr. McGrath said.
BII has been a more recent and more controversial complication of breast implants. As recently as September 2018, Dr. Ashar said in a written statement that “the agency continues to believe that the weight of the currently available scientific evidence does not conclusively demonstrate an association between breast implants and connective tissue diseases,” the types of symptoms that characterize BII.
While the panel heard no new, conclusive evidence of a causal link between breast implants and the range of symptoms that some implant recipients report and is now collectively known as BII, several participants seemed convinced that the syndrome was real and needed better surveillance and study.
“It’s in the same family as chronic fatigue syndrome and fibromyalgia. It’s not a diagnosis, but a set of symptoms.” said Benjamin O. Anderson, MD, a surgical oncologist and professor of surgery at the University of Washington in Seattle and a panel member. “It’s a giant challenge. BII is a constellation of difficult symptoms. We need to think about how we ask patients, what are your symptoms?”

Frank R. Lewis Jr., MD, committee chair, said a more standardized measure of the most common BII symptoms is needed. “That may be exceedingly difficult, with as many as a hundred reported symptoms,” said Dr. Lewis, executive director, emeritus, of the American Board of Surgery in Philadelphia.
The hearing featured results from some of the most research projects aimed at fleshing out an understanding of BII.
Diana Zuckerman, PhD, president of the National Center for Health Research, reported data she and her associates collected in an online survey completed in late 2018 and early 2019 by 449 women who had approached the Center for help in getting health insurance coverage for medically-necessary explantation of their breast implants.

Their most common symptoms included joint, muscle or back pain, weakness or stiffness; fatigue; “brain fog;” and anxiety and depression. More than two-thirds of the respondents had a family history and 3% had a personal history of an autoimmune disease, and 61% said their symptoms improved after their implants were removed, Dr. Zuckerman reported during her presentation to the panel.
During the discussion, panel members seemed intent on expanding mandatory, routine surveillance to all breast implants placed in U.S. practice.
Andrea L. Pusic, MD, president of the Plastic Surgery Foundation, summarized the recent launch of the National Breast Implant Registry by the Foundation and its parent organization, the American Society of Plastic Surgeons. These organizations, and plastic surgeons in general, would be amenable to collecting the data the FDA deemed necessary to better track BIA-ALCL and BII, said Dr. Pusic, professor of surgery at Harvard Medical School and chief of plastic and reconstructive surgery at Brigham and Women’s Hospital in Boston.
“Plastic surgeons are willing to enter these data because we know they are important,” she told the FDA panel.
Dr. Ashar, Dr. Clemens, Dr. McGrath, Dr. Lippman, Dr. Anderson, Dr. Lewis, Dr. Zuckerman, and Dr. Pusic reported having no relevant commercial disclosures.
SILVER SPRING, MD. – A mandatory, comprehensive approach to collecting adverse event data from breast implant recipients was favored during a March 25 hearing by a Food and Drug Administration advisory panel that oversees surgical devices.
This additional data could offer more complete information during the informed consent process for breast implants and potentially validate a new, autoimmune-like syndrome – breast implant illness (BII).
On the first day of a scheduled 2-day hearing, the advisory panel held no votes and took no formal actions. After a day of expert presentations and comments from more than 40 members of the public – mostly personal stories from affected patients and from plastic surgeons who place breast implants, panel members discussed a handful of questions from the FDA about relevant data to collect to better define the risks posed to breast implant recipients from breast-implant associated anaplastic large cell lymphoma (BIA-ALCL) and BII.
The advisory panel meeting took place as reports recently appeared documenting the scope of BIA-ALCL (Plast Reconstr Surg. 2019 March;143[3S]:65S-73S) and how to diagnose and manage BIA-ALCL (Aesthetic Surg J. 2019 March;39[S1}:S3-S13), and the existence of BII (Plast Reconstr Surg. 2019 March;143[3S]:74S-81S).
During the day’s two public comment periods, the panel heard from several women who gave brief accounts of developing and dealing with BIA-ALCL or BII.
“We think it’s important that all breast implant patients be aware of the risk for BIA-ALCL,” said Binita Ashar, MD, director of the FDAs Division of Surgical Devices. The FDA “is asking the panel what further steps need to be taken to understand the BIA-ALCL risk,” said Dr. Ashar as she opened the meeting of the General and Plastic Surgery Devices Panel of the Medical Devices Advisory Committee.
While the agency, as well as the plastic surgery community, have acknowledged the existence of BIA-ALCL since 2011, only recently have good data emerged on the scope of the complication. During the hearing, Mark W. Clemens, MD, a plastic surgeon at MD Anderson Cancer Center in Houston, reported on his analysis of 457 unique cases of BIA-ALCL reported to the FDA since 2011. He found that the vast majority of cases had occurred in women who had received textured implants while a relatively small minority were linked with the placement of smooth implants.
Further scrutiny of the reported details of each case showed that none of the lymphomas were linked with a confirmed instance of “pure” smooth implant exposure. He also estimated the U.S. incidence of BIA-ALCL as roughly one case for every 20,000 implants. Complete, en bloc removal of the implant seems to be the most effective way to treat the cancer; most explanted patients have a good prognosis, he said.
Despite the apparent link between textured implants specifically and onset of BIA-ALCL, some panel members did not see a ban on textured implants as the answer.
Texturing the implant helps to stabilize the implant in position. Without texturing “we would need to use something else to stabilize the implant, or there would be a tsunami of reoperations,” said panel member Mary H. McGrath, MD, professor of surgery at the University of California, San Francisco. The main alternative to texturing for stabilizing implants is to wrap them in place using surgical mesh, but that approach may also cause problems.
“Instead of just taking textured implants off the market, we need to also look at their advantages. A critical issue is informed consent,” said panel member Marc E. Lippman, MD, a professor of medicine at Georgetown University, Washington. Banning smooth implants based on what’s known so far “would be an extraordinary over reaction,” he said during the first day’s session.
Current U.S. anecdotal experience suggests that a ban may not even be necessary because “plastic surgeons are more and more walking away from textured implants” because of the apparent link to BIA-ALCL, Dr. McGrath said.
BII has been a more recent and more controversial complication of breast implants. As recently as September 2018, Dr. Ashar said in a written statement that “the agency continues to believe that the weight of the currently available scientific evidence does not conclusively demonstrate an association between breast implants and connective tissue diseases,” the types of symptoms that characterize BII.
While the panel heard no new, conclusive evidence of a causal link between breast implants and the range of symptoms that some implant recipients report and is now collectively known as BII, several participants seemed convinced that the syndrome was real and needed better surveillance and study.
“It’s in the same family as chronic fatigue syndrome and fibromyalgia. It’s not a diagnosis, but a set of symptoms.” said Benjamin O. Anderson, MD, a surgical oncologist and professor of surgery at the University of Washington in Seattle and a panel member. “It’s a giant challenge. BII is a constellation of difficult symptoms. We need to think about how we ask patients, what are your symptoms?”
Frank R. Lewis Jr., MD, committee chair, said a more standardized measure of the most common BII symptoms is needed. “That may be exceedingly difficult, with as many as a hundred reported symptoms,” said Dr. Lewis, executive director, emeritus, of the American Board of Surgery in Philadelphia.
The hearing featured results from some of the most research projects aimed at fleshing out an understanding of BII.
Diana Zuckerman, PhD, president of the National Center for Health Research, reported data she and her associates collected in an online survey completed in late 2018 and early 2019 by 449 women who had approached the Center for help in getting health insurance coverage for medically-necessary explantation of their breast implants.
Their most common symptoms included joint, muscle or back pain, weakness or stiffness; fatigue; “brain fog;” and anxiety and depression. More than two-thirds of the respondents had a family history and 3% had a personal history of an autoimmune disease, and 61% said their symptoms improved after their implants were removed, Dr. Zuckerman reported during her presentation to the panel.
During the discussion, panel members seemed intent on expanding mandatory, routine surveillance to all breast implants placed in U.S. practice.
Andrea L. Pusic, MD, president of the Plastic Surgery Foundation, summarized the recent launch of the National Breast Implant Registry by the Foundation and its parent organization, the American Society of Plastic Surgeons. These organizations, and plastic surgeons in general, would be amenable to collecting the data the FDA deemed necessary to better track BIA-ALCL and BII, said Dr. Pusic, professor of surgery at Harvard Medical School and chief of plastic and reconstructive surgery at Brigham and Women’s Hospital in Boston.
“Plastic surgeons are willing to enter these data because we know they are important,” she told the FDA panel.
Dr. Ashar, Dr. Clemens, Dr. McGrath, Dr. Lippman, Dr. Anderson, Dr. Lewis, Dr. Zuckerman, and Dr. Pusic reported having no relevant commercial disclosures.
SILVER SPRING, MD. – A mandatory, comprehensive approach to collecting adverse event data from breast implant recipients was favored during a March 25 hearing by a Food and Drug Administration advisory panel that oversees surgical devices.
This additional data could offer more complete information during the informed consent process for breast implants and potentially validate a new, autoimmune-like syndrome – breast implant illness (BII).
On the first day of a scheduled 2-day hearing, the advisory panel held no votes and took no formal actions. After a day of expert presentations and comments from more than 40 members of the public – mostly personal stories from affected patients and from plastic surgeons who place breast implants, panel members discussed a handful of questions from the FDA about relevant data to collect to better define the risks posed to breast implant recipients from breast-implant associated anaplastic large cell lymphoma (BIA-ALCL) and BII.
The advisory panel meeting took place as reports recently appeared documenting the scope of BIA-ALCL (Plast Reconstr Surg. 2019 March;143[3S]:65S-73S) and how to diagnose and manage BIA-ALCL (Aesthetic Surg J. 2019 March;39[S1}:S3-S13), and the existence of BII (Plast Reconstr Surg. 2019 March;143[3S]:74S-81S).
During the day’s two public comment periods, the panel heard from several women who gave brief accounts of developing and dealing with BIA-ALCL or BII.
“We think it’s important that all breast implant patients be aware of the risk for BIA-ALCL,” said Binita Ashar, MD, director of the FDAs Division of Surgical Devices. The FDA “is asking the panel what further steps need to be taken to understand the BIA-ALCL risk,” said Dr. Ashar as she opened the meeting of the General and Plastic Surgery Devices Panel of the Medical Devices Advisory Committee.
While the agency, as well as the plastic surgery community, have acknowledged the existence of BIA-ALCL since 2011, only recently have good data emerged on the scope of the complication. During the hearing, Mark W. Clemens, MD, a plastic surgeon at MD Anderson Cancer Center in Houston, reported on his analysis of 457 unique cases of BIA-ALCL reported to the FDA since 2011. He found that the vast majority of cases had occurred in women who had received textured implants while a relatively small minority were linked with the placement of smooth implants.
Further scrutiny of the reported details of each case showed that none of the lymphomas were linked with a confirmed instance of “pure” smooth implant exposure. He also estimated the U.S. incidence of BIA-ALCL as roughly one case for every 20,000 implants. Complete, en bloc removal of the implant seems to be the most effective way to treat the cancer; most explanted patients have a good prognosis, he said.
Despite the apparent link between textured implants specifically and onset of BIA-ALCL, some panel members did not see a ban on textured implants as the answer.
Texturing the implant helps to stabilize the implant in position. Without texturing “we would need to use something else to stabilize the implant, or there would be a tsunami of reoperations,” said panel member Mary H. McGrath, MD, professor of surgery at the University of California, San Francisco. The main alternative to texturing for stabilizing implants is to wrap them in place using surgical mesh, but that approach may also cause problems.
“Instead of just taking textured implants off the market, we need to also look at their advantages. A critical issue is informed consent,” said panel member Marc E. Lippman, MD, a professor of medicine at Georgetown University, Washington. Banning smooth implants based on what’s known so far “would be an extraordinary over reaction,” he said during the first day’s session.
Current U.S. anecdotal experience suggests that a ban may not even be necessary because “plastic surgeons are more and more walking away from textured implants” because of the apparent link to BIA-ALCL, Dr. McGrath said.
BII has been a more recent and more controversial complication of breast implants. As recently as September 2018, Dr. Ashar said in a written statement that “the agency continues to believe that the weight of the currently available scientific evidence does not conclusively demonstrate an association between breast implants and connective tissue diseases,” the types of symptoms that characterize BII.
While the panel heard no new, conclusive evidence of a causal link between breast implants and the range of symptoms that some implant recipients report and is now collectively known as BII, several participants seemed convinced that the syndrome was real and needed better surveillance and study.
“It’s in the same family as chronic fatigue syndrome and fibromyalgia. It’s not a diagnosis, but a set of symptoms.” said Benjamin O. Anderson, MD, a surgical oncologist and professor of surgery at the University of Washington in Seattle and a panel member. “It’s a giant challenge. BII is a constellation of difficult symptoms. We need to think about how we ask patients, what are your symptoms?”
Frank R. Lewis Jr., MD, committee chair, said a more standardized measure of the most common BII symptoms is needed. “That may be exceedingly difficult, with as many as a hundred reported symptoms,” said Dr. Lewis, executive director, emeritus, of the American Board of Surgery in Philadelphia.
The hearing featured results from some of the most research projects aimed at fleshing out an understanding of BII.
Diana Zuckerman, PhD, president of the National Center for Health Research, reported data she and her associates collected in an online survey completed in late 2018 and early 2019 by 449 women who had approached the Center for help in getting health insurance coverage for medically-necessary explantation of their breast implants.
Their most common symptoms included joint, muscle or back pain, weakness or stiffness; fatigue; “brain fog;” and anxiety and depression. More than two-thirds of the respondents had a family history and 3% had a personal history of an autoimmune disease, and 61% said their symptoms improved after their implants were removed, Dr. Zuckerman reported during her presentation to the panel.
During the discussion, panel members seemed intent on expanding mandatory, routine surveillance to all breast implants placed in U.S. practice.
Andrea L. Pusic, MD, president of the Plastic Surgery Foundation, summarized the recent launch of the National Breast Implant Registry by the Foundation and its parent organization, the American Society of Plastic Surgeons. These organizations, and plastic surgeons in general, would be amenable to collecting the data the FDA deemed necessary to better track BIA-ALCL and BII, said Dr. Pusic, professor of surgery at Harvard Medical School and chief of plastic and reconstructive surgery at Brigham and Women’s Hospital in Boston.
“Plastic surgeons are willing to enter these data because we know they are important,” she told the FDA panel.
Dr. Ashar, Dr. Clemens, Dr. McGrath, Dr. Lippman, Dr. Anderson, Dr. Lewis, Dr. Zuckerman, and Dr. Pusic reported having no relevant commercial disclosures.
REPORTING FROM AN FDA ADVISORY COMMITTEE MEETING

Socioeconomic status affects scleroderma severity in African Americans
according to findings from an analysis of single-center cohort data over a 10-year period.

Indeed, among patients in the cohort of 402 scleroderma patients at MedStar Georgetown University Hospital in Washington, lower household income was predictive of higher mortality during follow-up, independent of race, according to first author Duncan F. Moore, MD, and his colleagues at the hospital.
Previous studies have demonstrated increased risk for scleroderma in African American patients, who also are more likely than non–African Americans to be diagnosed at a younger age and to have conditions including more diffuse cutaneous disease, more severe restrictive lung disease, more cardiac and renal involvement, and increased mortality, the authors wrote in Arthritis Care & Research.
“We did clearly show that African Americans have worse outcomes and severe pulmonary involvement, but I was surprised that there still was a major contribution of socioeconomic status affecting outcomes for all patients, even though only 10% of our patients were indigent and on medical assistance,” Virginia Steen, MD, senior author of the study and professor of rheumatology at Georgetown University, said in an interview. “I still feel strongly that there are likely genetic issues as to why African Americans have such severe disease. We are eager to learn more from the GRASP [Genome Research in African American Scleroderma Patients] study, which is specifically looking at the genetic issues in African American scleroderma patients,” she said.
Of the 402 scleroderma patients at MedStar Georgetown who were seen during 2006-2016, 202 were African American. A total of 186 African American and 184 non–African American patients in the study met the 2013 American College of Rheumatology/European League Against Rheumatism criteria for systemic sclerosis (SSc). Demographics including gender (87% female) and age (mean of 48 years) were similar between the groups.
Overall, the African American patients showed more severe lung disease, more pulmonary hypertension, and more severe cardiac involvement than did non–African American patients, and autoantibodies were significantly different between the groups.
During follow-up, mortality proved much higher among African Americans at 21%, compared with 11% in non–African Americans (P = .005). However, the unadjusted hazard ratio for death declined from 2.061 (P = .006) to a nonsignificant 1.256 after adjustment for socioeconomic variables.
All socioeconomic measures showed significant differences between the groups. African Americans were more likely to be single and disabled at the initial study visit and to have Medicaid, but they were less likely to be a homemaker, have private insurance, or have a college degree. African Americans’ $74,000 median household income (based on ZIP code) was also a statistically significant $23,000 less than non–African American patients. But the researchers noted that “for every additional $10,000 of household income, independent of race, the hazard of death during follow-up declined by 15.5%.”
Notable differences in antibodies appeared between the groups, with more African American patients having isolated nucleolar ANA, anti-U1RNP antibody, or other positive antinuclear antibodies without SSc-specific antibodies. African American patients also were less likely to have anticentromere or anti-RNA polymerase III antibodies.
The study findings were limited by several factors, including possible bias in the matching process and the use of only index values for socioeconomic variables, the researchers noted.
Regardless of relative socioeconomic and genetic influences, “it is clear that African Americans with scleroderma merit more intensive efforts to facilitate timely diagnosis and access to continued evaluation and suppressive treatment, particularly with respect to cardiopulmonary involvement,” they wrote.
Next steps for research, according to Dr. Steen, include studying clinical subsets of African American patients to try to identify factors to predict outcomes, including the nucleolar pattern ANA, overlap with lupus, history of hypertension, and the relationship with renal crisis.
“We are also looking at whether the African American patients are less responsive to mycophenolate than the non–African American patients. We definitely need to find ways to be more aggressive at identifying and treating African American patients early in their disease,” she added.
The researchers had no financial conflicts to disclose. Dr. Steen serves on the MDedge Rheumatology Editorial Advisory Board.
SOURCE: Moore DF et al. Arthritis Care Res. 2019 March 1. doi: 10.1002/acr.23861.
“Not only do patients who manifest the diffuse cutaneous subset of disease experience a more severe course, but so do affected persons of African American race,” Nadia D. Morgan, MBBS, and Allan C. Gelber, MD, wrote in an accompanying editorial. The effects of socioeconomic status should not be overlooked based on the current study, in which the inclusion of socioeconomic factors eliminated the significance of association between race and mortality among scleroderma patients, they wrote.

“Overall, and in the context of these published reports which underscore the disproportionate and adverse impact of scleroderma among African Americans, and in light of the ongoing efforts of the GRASP study, the current paper by Moore et al. emphasizes the importance of socioeconomic status, and of socioeconomic determinants of health, to account for differences in clinically relevant outcomes,” they wrote.

Dr. Gelber is affiliated with the division of rheumatology at Johns Hopkins University, Baltimore. Dr. Morgan, who was also with Johns Hopkins, died before publication of the editorial. They made no conflict of interest disclosures.
“Not only do patients who manifest the diffuse cutaneous subset of disease experience a more severe course, but so do affected persons of African American race,” Nadia D. Morgan, MBBS, and Allan C. Gelber, MD, wrote in an accompanying editorial. The effects of socioeconomic status should not be overlooked based on the current study, in which the inclusion of socioeconomic factors eliminated the significance of association between race and mortality among scleroderma patients, they wrote.
“Overall, and in the context of these published reports which underscore the disproportionate and adverse impact of scleroderma among African Americans, and in light of the ongoing efforts of the GRASP study, the current paper by Moore et al. emphasizes the importance of socioeconomic status, and of socioeconomic determinants of health, to account for differences in clinically relevant outcomes,” they wrote.
Dr. Gelber is affiliated with the division of rheumatology at Johns Hopkins University, Baltimore. Dr. Morgan, who was also with Johns Hopkins, died before publication of the editorial. They made no conflict of interest disclosures.
“Not only do patients who manifest the diffuse cutaneous subset of disease experience a more severe course, but so do affected persons of African American race,” Nadia D. Morgan, MBBS, and Allan C. Gelber, MD, wrote in an accompanying editorial. The effects of socioeconomic status should not be overlooked based on the current study, in which the inclusion of socioeconomic factors eliminated the significance of association between race and mortality among scleroderma patients, they wrote.
“Overall, and in the context of these published reports which underscore the disproportionate and adverse impact of scleroderma among African Americans, and in light of the ongoing efforts of the GRASP study, the current paper by Moore et al. emphasizes the importance of socioeconomic status, and of socioeconomic determinants of health, to account for differences in clinically relevant outcomes,” they wrote.
Dr. Gelber is affiliated with the division of rheumatology at Johns Hopkins University, Baltimore. Dr. Morgan, who was also with Johns Hopkins, died before publication of the editorial. They made no conflict of interest disclosures.
according to findings from an analysis of single-center cohort data over a 10-year period.
Indeed, among patients in the cohort of 402 scleroderma patients at MedStar Georgetown University Hospital in Washington, lower household income was predictive of higher mortality during follow-up, independent of race, according to first author Duncan F. Moore, MD, and his colleagues at the hospital.
Previous studies have demonstrated increased risk for scleroderma in African American patients, who also are more likely than non–African Americans to be diagnosed at a younger age and to have conditions including more diffuse cutaneous disease, more severe restrictive lung disease, more cardiac and renal involvement, and increased mortality, the authors wrote in Arthritis Care & Research.
“We did clearly show that African Americans have worse outcomes and severe pulmonary involvement, but I was surprised that there still was a major contribution of socioeconomic status affecting outcomes for all patients, even though only 10% of our patients were indigent and on medical assistance,” Virginia Steen, MD, senior author of the study and professor of rheumatology at Georgetown University, said in an interview. “I still feel strongly that there are likely genetic issues as to why African Americans have such severe disease. We are eager to learn more from the GRASP [Genome Research in African American Scleroderma Patients] study, which is specifically looking at the genetic issues in African American scleroderma patients,” she said.
Of the 402 scleroderma patients at MedStar Georgetown who were seen during 2006-2016, 202 were African American. A total of 186 African American and 184 non–African American patients in the study met the 2013 American College of Rheumatology/European League Against Rheumatism criteria for systemic sclerosis (SSc). Demographics including gender (87% female) and age (mean of 48 years) were similar between the groups.
Overall, the African American patients showed more severe lung disease, more pulmonary hypertension, and more severe cardiac involvement than did non–African American patients, and autoantibodies were significantly different between the groups.
During follow-up, mortality proved much higher among African Americans at 21%, compared with 11% in non–African Americans (P = .005). However, the unadjusted hazard ratio for death declined from 2.061 (P = .006) to a nonsignificant 1.256 after adjustment for socioeconomic variables.
All socioeconomic measures showed significant differences between the groups. African Americans were more likely to be single and disabled at the initial study visit and to have Medicaid, but they were less likely to be a homemaker, have private insurance, or have a college degree. African Americans’ $74,000 median household income (based on ZIP code) was also a statistically significant $23,000 less than non–African American patients. But the researchers noted that “for every additional $10,000 of household income, independent of race, the hazard of death during follow-up declined by 15.5%.”
Notable differences in antibodies appeared between the groups, with more African American patients having isolated nucleolar ANA, anti-U1RNP antibody, or other positive antinuclear antibodies without SSc-specific antibodies. African American patients also were less likely to have anticentromere or anti-RNA polymerase III antibodies.
The study findings were limited by several factors, including possible bias in the matching process and the use of only index values for socioeconomic variables, the researchers noted.
Regardless of relative socioeconomic and genetic influences, “it is clear that African Americans with scleroderma merit more intensive efforts to facilitate timely diagnosis and access to continued evaluation and suppressive treatment, particularly with respect to cardiopulmonary involvement,” they wrote.
Next steps for research, according to Dr. Steen, include studying clinical subsets of African American patients to try to identify factors to predict outcomes, including the nucleolar pattern ANA, overlap with lupus, history of hypertension, and the relationship with renal crisis.
“We are also looking at whether the African American patients are less responsive to mycophenolate than the non–African American patients. We definitely need to find ways to be more aggressive at identifying and treating African American patients early in their disease,” she added.
The researchers had no financial conflicts to disclose. Dr. Steen serves on the MDedge Rheumatology Editorial Advisory Board.
SOURCE: Moore DF et al. Arthritis Care Res. 2019 March 1. doi: 10.1002/acr.23861.
according to findings from an analysis of single-center cohort data over a 10-year period.
Indeed, among patients in the cohort of 402 scleroderma patients at MedStar Georgetown University Hospital in Washington, lower household income was predictive of higher mortality during follow-up, independent of race, according to first author Duncan F. Moore, MD, and his colleagues at the hospital.
Previous studies have demonstrated increased risk for scleroderma in African American patients, who also are more likely than non–African Americans to be diagnosed at a younger age and to have conditions including more diffuse cutaneous disease, more severe restrictive lung disease, more cardiac and renal involvement, and increased mortality, the authors wrote in Arthritis Care & Research.
“We did clearly show that African Americans have worse outcomes and severe pulmonary involvement, but I was surprised that there still was a major contribution of socioeconomic status affecting outcomes for all patients, even though only 10% of our patients were indigent and on medical assistance,” Virginia Steen, MD, senior author of the study and professor of rheumatology at Georgetown University, said in an interview. “I still feel strongly that there are likely genetic issues as to why African Americans have such severe disease. We are eager to learn more from the GRASP [Genome Research in African American Scleroderma Patients] study, which is specifically looking at the genetic issues in African American scleroderma patients,” she said.
Of the 402 scleroderma patients at MedStar Georgetown who were seen during 2006-2016, 202 were African American. A total of 186 African American and 184 non–African American patients in the study met the 2013 American College of Rheumatology/European League Against Rheumatism criteria for systemic sclerosis (SSc). Demographics including gender (87% female) and age (mean of 48 years) were similar between the groups.
Overall, the African American patients showed more severe lung disease, more pulmonary hypertension, and more severe cardiac involvement than did non–African American patients, and autoantibodies were significantly different between the groups.
During follow-up, mortality proved much higher among African Americans at 21%, compared with 11% in non–African Americans (P = .005). However, the unadjusted hazard ratio for death declined from 2.061 (P = .006) to a nonsignificant 1.256 after adjustment for socioeconomic variables.
All socioeconomic measures showed significant differences between the groups. African Americans were more likely to be single and disabled at the initial study visit and to have Medicaid, but they were less likely to be a homemaker, have private insurance, or have a college degree. African Americans’ $74,000 median household income (based on ZIP code) was also a statistically significant $23,000 less than non–African American patients. But the researchers noted that “for every additional $10,000 of household income, independent of race, the hazard of death during follow-up declined by 15.5%.”
Notable differences in antibodies appeared between the groups, with more African American patients having isolated nucleolar ANA, anti-U1RNP antibody, or other positive antinuclear antibodies without SSc-specific antibodies. African American patients also were less likely to have anticentromere or anti-RNA polymerase III antibodies.
The study findings were limited by several factors, including possible bias in the matching process and the use of only index values for socioeconomic variables, the researchers noted.
Regardless of relative socioeconomic and genetic influences, “it is clear that African Americans with scleroderma merit more intensive efforts to facilitate timely diagnosis and access to continued evaluation and suppressive treatment, particularly with respect to cardiopulmonary involvement,” they wrote.
Next steps for research, according to Dr. Steen, include studying clinical subsets of African American patients to try to identify factors to predict outcomes, including the nucleolar pattern ANA, overlap with lupus, history of hypertension, and the relationship with renal crisis.
“We are also looking at whether the African American patients are less responsive to mycophenolate than the non–African American patients. We definitely need to find ways to be more aggressive at identifying and treating African American patients early in their disease,” she added.
The researchers had no financial conflicts to disclose. Dr. Steen serves on the MDedge Rheumatology Editorial Advisory Board.
SOURCE: Moore DF et al. Arthritis Care Res. 2019 March 1. doi: 10.1002/acr.23861.
FROM ARTHRITIS CARE & RESEARCH
Resistant hypertension hits SLE patients hard
at a tertiary care center.

A patient with resistant hypertension either has blood pressure remaining above 140/90 mm Hg while taking three antihypertensive medications or requires the use of four or more antihypertensives to attain blood pressure control. Resistant hypertension, which was more likely to occur among blacks and patients with lower renal function, hypercholesterolemia, and increased inflammatory markers, increased the risk of death nearly threefold (hazard ratio, 2.91; P = .0005) when compared with those who didn’t have this condition.
The results of this analysis were published March 15 in Arthritis Care & Research (doi: 10.1002/acr.23880). We covered this study at the 2018 annual meeting of the American College of Rheumatology in Chicago before it was published in the journal. Read our previous story at the link above.
at a tertiary care center.
A patient with resistant hypertension either has blood pressure remaining above 140/90 mm Hg while taking three antihypertensive medications or requires the use of four or more antihypertensives to attain blood pressure control. Resistant hypertension, which was more likely to occur among blacks and patients with lower renal function, hypercholesterolemia, and increased inflammatory markers, increased the risk of death nearly threefold (hazard ratio, 2.91; P = .0005) when compared with those who didn’t have this condition.
The results of this analysis were published March 15 in Arthritis Care & Research (doi: 10.1002/acr.23880). We covered this study at the 2018 annual meeting of the American College of Rheumatology in Chicago before it was published in the journal. Read our previous story at the link above.
at a tertiary care center.
A patient with resistant hypertension either has blood pressure remaining above 140/90 mm Hg while taking three antihypertensive medications or requires the use of four or more antihypertensives to attain blood pressure control. Resistant hypertension, which was more likely to occur among blacks and patients with lower renal function, hypercholesterolemia, and increased inflammatory markers, increased the risk of death nearly threefold (hazard ratio, 2.91; P = .0005) when compared with those who didn’t have this condition.
The results of this analysis were published March 15 in Arthritis Care & Research (doi: 10.1002/acr.23880). We covered this study at the 2018 annual meeting of the American College of Rheumatology in Chicago before it was published in the journal. Read our previous story at the link above.
FROM ARTHRITIS CARE & RESEARCH
BTK inhibitor calms pemphigus vulgaris with low-dose steroids
WASHINGTON – An investigational molecule that blocks the downstream proinflammatory effects of B cells controlled disease activity and induced clinical remission in patients with pemphigus by 12 weeks.
At the end of a 24-week, open-label trial, a key driver of the sometimes-fatal blistering disease, Deedee Murrell, MD, said at the annual meeting of the American Academy of Dermatology.
The clinical efficacy plus a favorable safety profile supports the further development of the molecule, designed and manufactured by Principia Biopharma in San Francisco. The company is currently recruiting for a pivotal phase 3 trial of PRN1008 in 120 patients with moderate to severe pemphigus vulgaris.
Despite the recent approval of rituximab (Rituxan) for moderate to severe pemphigus, there remains an unmet need for a quick-acting, steroid-sparing, anti-inflammatory treatment, said Dr. Murrell, professor and head of the department of dermatology at the University of New South Wales, Sydney.
“We need something to use instead of high-dose steroids while we are waiting for rituximab to kick in, which can take 3 months,” and rituximab, which depletes B cells, puts patients at risk for infection, she said. “We need something that has rapid onset, is steroid sparing, safe for chronic administration, avoids B-cell depletion, and is convenient.”
Blocking the BTK receptor on B cells puts the brakes on the B-cell mediated inflammatory pathway, preventing activation of monocytes, macrophages, mast cells, basophils, and neutrophils. At the same time, however, it does not deplete the B-cell population, said Dr. Murrell, the lead investigator.
The BELIEVE study comprised 27 patients with mild to severe pemphigus of an average 6 years’ duration. Most (18) had relapsing disease; the remainder had newly diagnosed pemphigus. A majority (16) had severe disease, as measured by a score of 15 or more on the Pemphigus Disease Activity Index (PDAI). Almost all (23) were positive for antidesmoglein antibodies. Only one patient was negative for antibodies.
The mean corticosteroid dose at baseline was 14 mg/day, although that ranged from no steroids to 30 mg/day.
The study consisted of a 12-week treatment phase and a 12-week follow-up phase. During treatment, patients could take no more than 0.5 mg/kg of prednisone daily, although with 400 mg PRN1008 twice a day. They were allowed to undertake rescue immunosuppression if they experienced a disease flare.
The primary endpoint was disease control by day 29 as evidenced by no new lesions. Secondary endpoints were complete remission, minimization of prednisone, quality of life, antibody levels, and clinician measures including the PDAI and the Autoimmune Bullous Skin Disorder Intensity Score.
By the end of week 4, 54% of patients had achieved the primary endpoint. The benefit continued to expand, with 73% reaching that response by the end of week 12. During this period, the mean prednisone dose was 12 mg/day.
Among the 24 patients who completed the study, complete remission occurred in 17% by week 12. However, patients continued to respond through the follow-up period, even after the study medication was stopped. By week 24, 25% of these patients experienced a complete remission. At the point of remission, the mean steroid dose was 8 mg/day. The median duration of remission was 2 months after stopping PRN1008.
The PDAI fell by a median of 70% by week 12 and was maintained at that level by the end of week 24. The median level of antidesmoglein autoantibodies fell by up to 65%. Again, the improvement continued throughout the off-drug follow-up period. In subgroup analyses, PRN1008 was more effective in patients with moderate to severe disease than those with mild disease (80% response vs. 64%). It was equally effective in those with newly diagnosed disease (75% vs. 72%) and regardless of antibody level at baseline.
The adverse event profile was relatively benign. Most side effects were mild and transient, and included upper abdominal pain, headache, and nausea. There were two mild infections and one serious infection, which presented in a patient with a long-standing localized cellulitis that activated and was associated a high fever. It was culture negative and PRN1008 was restarted without issue.
There was also one serious adverse event and one death, both unrelated to the study drug. One patient developed a pancreatic cyst that was discovered on day 29. The patient dropped out of the study to have elective surgery. The death occurred in a patient who developed acute respiratory failure on day 8 of treatment, caused by an undiagnosed congenital pulmonary sequestration. The patient died of a brain embolism shortly after lung surgery.
Dr. Murrell designed the study and was an investigator. She reported a financial relationship with Principia, as well as with numerous other pharmaceutical companies.
SOURCE: Murrell D et al. AAD 2019, Session S034.
WASHINGTON – An investigational molecule that blocks the downstream proinflammatory effects of B cells controlled disease activity and induced clinical remission in patients with pemphigus by 12 weeks.
At the end of a 24-week, open-label trial, a key driver of the sometimes-fatal blistering disease, Deedee Murrell, MD, said at the annual meeting of the American Academy of Dermatology.
The clinical efficacy plus a favorable safety profile supports the further development of the molecule, designed and manufactured by Principia Biopharma in San Francisco. The company is currently recruiting for a pivotal phase 3 trial of PRN1008 in 120 patients with moderate to severe pemphigus vulgaris.
Despite the recent approval of rituximab (Rituxan) for moderate to severe pemphigus, there remains an unmet need for a quick-acting, steroid-sparing, anti-inflammatory treatment, said Dr. Murrell, professor and head of the department of dermatology at the University of New South Wales, Sydney.
“We need something to use instead of high-dose steroids while we are waiting for rituximab to kick in, which can take 3 months,” and rituximab, which depletes B cells, puts patients at risk for infection, she said. “We need something that has rapid onset, is steroid sparing, safe for chronic administration, avoids B-cell depletion, and is convenient.”
Blocking the BTK receptor on B cells puts the brakes on the B-cell mediated inflammatory pathway, preventing activation of monocytes, macrophages, mast cells, basophils, and neutrophils. At the same time, however, it does not deplete the B-cell population, said Dr. Murrell, the lead investigator.
The BELIEVE study comprised 27 patients with mild to severe pemphigus of an average 6 years’ duration. Most (18) had relapsing disease; the remainder had newly diagnosed pemphigus. A majority (16) had severe disease, as measured by a score of 15 or more on the Pemphigus Disease Activity Index (PDAI). Almost all (23) were positive for antidesmoglein antibodies. Only one patient was negative for antibodies.
The mean corticosteroid dose at baseline was 14 mg/day, although that ranged from no steroids to 30 mg/day.
The study consisted of a 12-week treatment phase and a 12-week follow-up phase. During treatment, patients could take no more than 0.5 mg/kg of prednisone daily, although with 400 mg PRN1008 twice a day. They were allowed to undertake rescue immunosuppression if they experienced a disease flare.
The primary endpoint was disease control by day 29 as evidenced by no new lesions. Secondary endpoints were complete remission, minimization of prednisone, quality of life, antibody levels, and clinician measures including the PDAI and the Autoimmune Bullous Skin Disorder Intensity Score.
By the end of week 4, 54% of patients had achieved the primary endpoint. The benefit continued to expand, with 73% reaching that response by the end of week 12. During this period, the mean prednisone dose was 12 mg/day.
Among the 24 patients who completed the study, complete remission occurred in 17% by week 12. However, patients continued to respond through the follow-up period, even after the study medication was stopped. By week 24, 25% of these patients experienced a complete remission. At the point of remission, the mean steroid dose was 8 mg/day. The median duration of remission was 2 months after stopping PRN1008.
The PDAI fell by a median of 70% by week 12 and was maintained at that level by the end of week 24. The median level of antidesmoglein autoantibodies fell by up to 65%. Again, the improvement continued throughout the off-drug follow-up period. In subgroup analyses, PRN1008 was more effective in patients with moderate to severe disease than those with mild disease (80% response vs. 64%). It was equally effective in those with newly diagnosed disease (75% vs. 72%) and regardless of antibody level at baseline.
The adverse event profile was relatively benign. Most side effects were mild and transient, and included upper abdominal pain, headache, and nausea. There were two mild infections and one serious infection, which presented in a patient with a long-standing localized cellulitis that activated and was associated a high fever. It was culture negative and PRN1008 was restarted without issue.
There was also one serious adverse event and one death, both unrelated to the study drug. One patient developed a pancreatic cyst that was discovered on day 29. The patient dropped out of the study to have elective surgery. The death occurred in a patient who developed acute respiratory failure on day 8 of treatment, caused by an undiagnosed congenital pulmonary sequestration. The patient died of a brain embolism shortly after lung surgery.
Dr. Murrell designed the study and was an investigator. She reported a financial relationship with Principia, as well as with numerous other pharmaceutical companies.
SOURCE: Murrell D et al. AAD 2019, Session S034.
WASHINGTON – An investigational molecule that blocks the downstream proinflammatory effects of B cells controlled disease activity and induced clinical remission in patients with pemphigus by 12 weeks.
At the end of a 24-week, open-label trial, a key driver of the sometimes-fatal blistering disease, Deedee Murrell, MD, said at the annual meeting of the American Academy of Dermatology.
The clinical efficacy plus a favorable safety profile supports the further development of the molecule, designed and manufactured by Principia Biopharma in San Francisco. The company is currently recruiting for a pivotal phase 3 trial of PRN1008 in 120 patients with moderate to severe pemphigus vulgaris.
Despite the recent approval of rituximab (Rituxan) for moderate to severe pemphigus, there remains an unmet need for a quick-acting, steroid-sparing, anti-inflammatory treatment, said Dr. Murrell, professor and head of the department of dermatology at the University of New South Wales, Sydney.
“We need something to use instead of high-dose steroids while we are waiting for rituximab to kick in, which can take 3 months,” and rituximab, which depletes B cells, puts patients at risk for infection, she said. “We need something that has rapid onset, is steroid sparing, safe for chronic administration, avoids B-cell depletion, and is convenient.”
Blocking the BTK receptor on B cells puts the brakes on the B-cell mediated inflammatory pathway, preventing activation of monocytes, macrophages, mast cells, basophils, and neutrophils. At the same time, however, it does not deplete the B-cell population, said Dr. Murrell, the lead investigator.
The BELIEVE study comprised 27 patients with mild to severe pemphigus of an average 6 years’ duration. Most (18) had relapsing disease; the remainder had newly diagnosed pemphigus. A majority (16) had severe disease, as measured by a score of 15 or more on the Pemphigus Disease Activity Index (PDAI). Almost all (23) were positive for antidesmoglein antibodies. Only one patient was negative for antibodies.
The mean corticosteroid dose at baseline was 14 mg/day, although that ranged from no steroids to 30 mg/day.
The study consisted of a 12-week treatment phase and a 12-week follow-up phase. During treatment, patients could take no more than 0.5 mg/kg of prednisone daily, although with 400 mg PRN1008 twice a day. They were allowed to undertake rescue immunosuppression if they experienced a disease flare.
The primary endpoint was disease control by day 29 as evidenced by no new lesions. Secondary endpoints were complete remission, minimization of prednisone, quality of life, antibody levels, and clinician measures including the PDAI and the Autoimmune Bullous Skin Disorder Intensity Score.
By the end of week 4, 54% of patients had achieved the primary endpoint. The benefit continued to expand, with 73% reaching that response by the end of week 12. During this period, the mean prednisone dose was 12 mg/day.
Among the 24 patients who completed the study, complete remission occurred in 17% by week 12. However, patients continued to respond through the follow-up period, even after the study medication was stopped. By week 24, 25% of these patients experienced a complete remission. At the point of remission, the mean steroid dose was 8 mg/day. The median duration of remission was 2 months after stopping PRN1008.
The PDAI fell by a median of 70% by week 12 and was maintained at that level by the end of week 24. The median level of antidesmoglein autoantibodies fell by up to 65%. Again, the improvement continued throughout the off-drug follow-up period. In subgroup analyses, PRN1008 was more effective in patients with moderate to severe disease than those with mild disease (80% response vs. 64%). It was equally effective in those with newly diagnosed disease (75% vs. 72%) and regardless of antibody level at baseline.
The adverse event profile was relatively benign. Most side effects were mild and transient, and included upper abdominal pain, headache, and nausea. There were two mild infections and one serious infection, which presented in a patient with a long-standing localized cellulitis that activated and was associated a high fever. It was culture negative and PRN1008 was restarted without issue.
There was also one serious adverse event and one death, both unrelated to the study drug. One patient developed a pancreatic cyst that was discovered on day 29. The patient dropped out of the study to have elective surgery. The death occurred in a patient who developed acute respiratory failure on day 8 of treatment, caused by an undiagnosed congenital pulmonary sequestration. The patient died of a brain embolism shortly after lung surgery.
Dr. Murrell designed the study and was an investigator. She reported a financial relationship with Principia, as well as with numerous other pharmaceutical companies.
SOURCE: Murrell D et al. AAD 2019, Session S034.
REPORTING FROM AAD 2019
Concurrent Keratoacanthomas and Nonsarcoidal Granulomatous Reactions in New and Preexisting Tattoos
To the Editor:
Cutaneous reactions to tattoos are common and histologically diverse. As outlined by Jacob,1 these reactions can be categorized into 4 main groups: inoculative/infective, hypersensitive, neoplastic, and coincidental. A thorough history and physical examination can aid in distinguishing the type of cutaneous reaction, but diagnosis often requires histopathologic clarification. We report the case of a patient who presented with painful indurated nodules within red ink areas of new and preexisting tattoos.
A 48-year-old woman with no prior medical conditions presented with tender pruritic nodules at the site of a new tattoo and within recently retouched tattoos of 5 months’ duration. The tattoos were done at an “organic” tattoo parlor 8 months prior to presentation. Simultaneously, the patient also developed induration and pain in 2 older tattoos that had been done 10 years prior and had not been retouched.
Physical examination revealed 2 smooth and serpiginous nodules nested perfectly within the new red tattoo on the left medial ankle (Figure 1A). Examination of the retouched tattoos on the dorsum of the right foot revealed 4 discrete nodules within the red, heart-shaped areas of the tattoos (Figure 2A). Additionally, the red-inked portions of an older tattoo on the left lateral calf that were outlined in red ink also were raised and indurated (Figure 3A), and a tattoo on the right volar wrist, also in red ink, was indurated and tender to palpation. The remainder of the physical examination was normal.
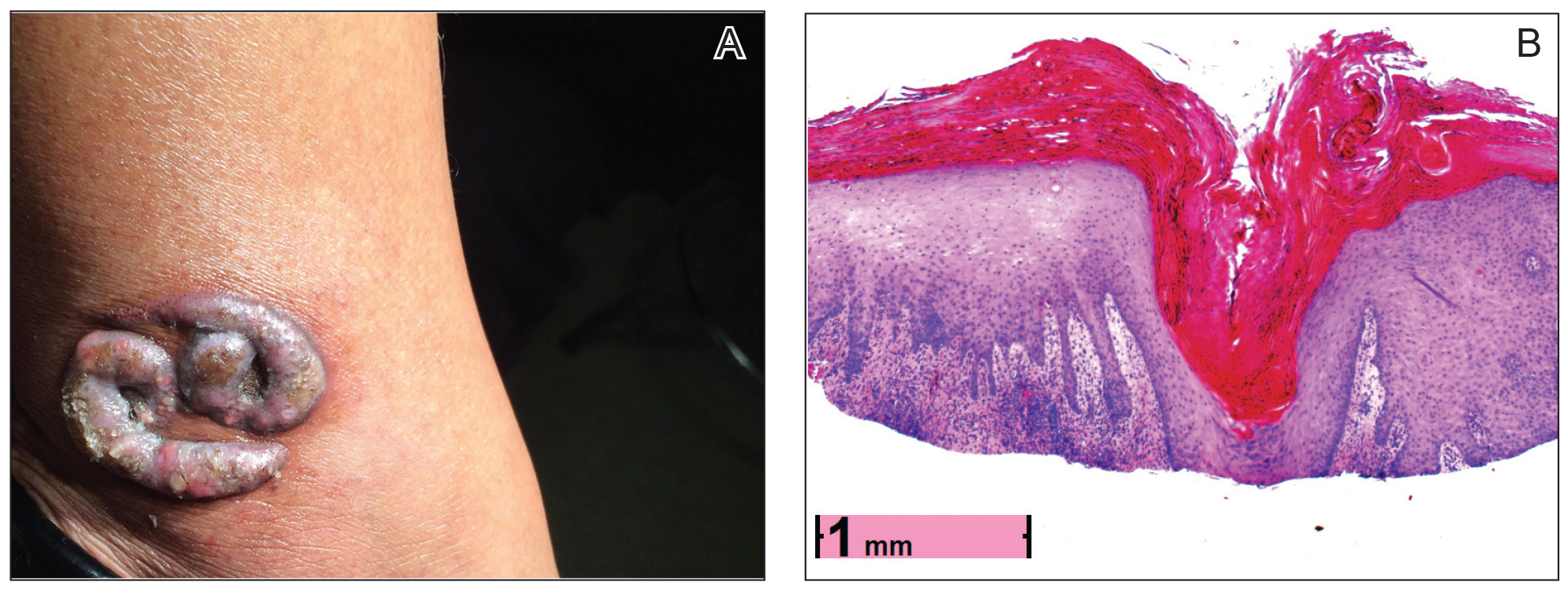

contiguous dilated follicular infundibula with atypical keratinocytes that had hyperchromatic nuclei, consistent with a keratoacanthoma, as well as a lymphocytic infiltrate in the dermis above a dense infiltrate of lymphocytes and histiocytes (H&E, original magnification ×2.5 [original magnification ×6.2]).
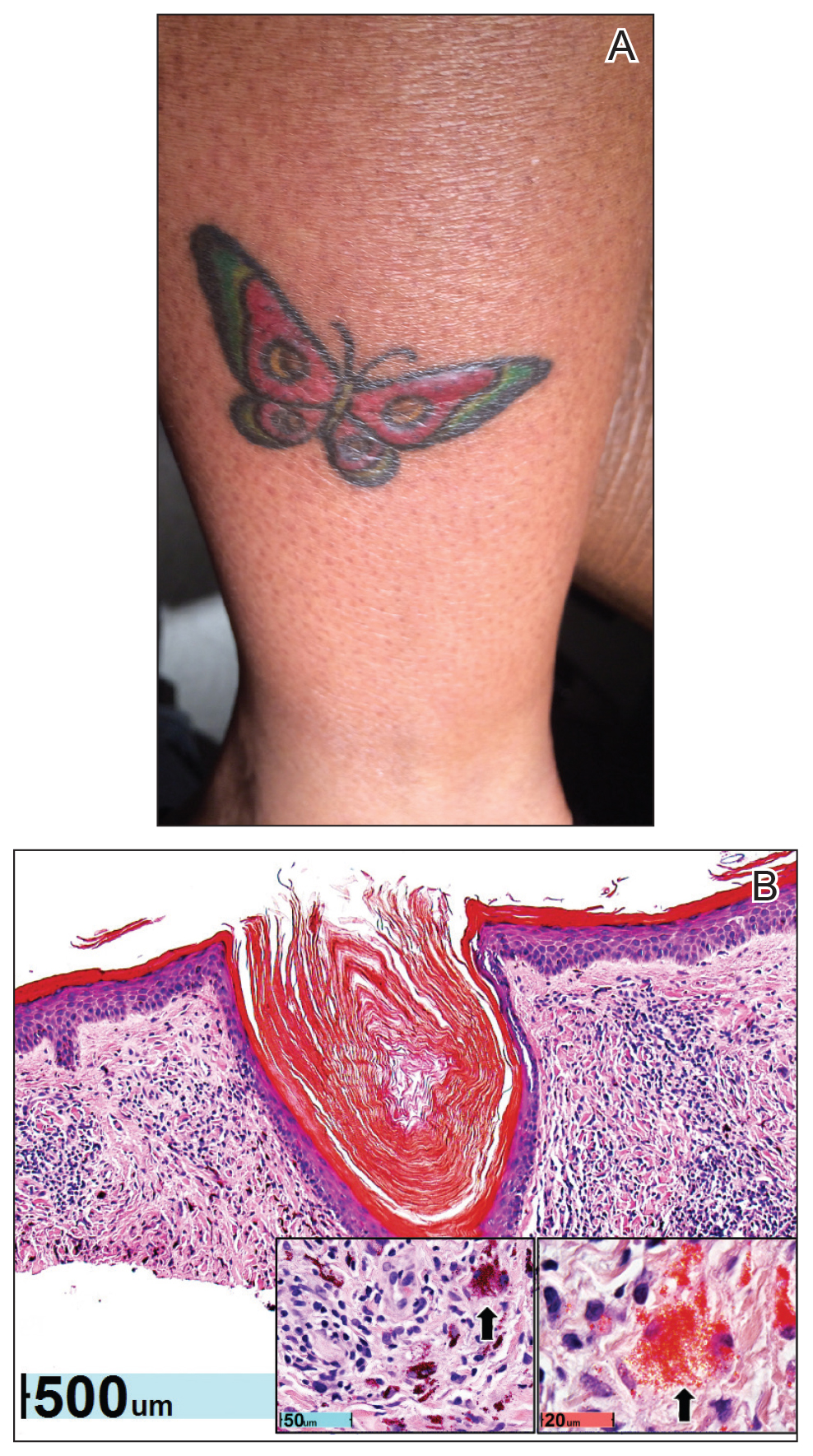
The lesions continued to enlarge and become increasingly painful despite trials of fluticasone propionate cream 0.05%, clobetasol propionate gel 0.05%, a 7-day course of oral levofloxacin, and a 10-day course of oral amoxicillin-clavulanate. Ultimately, a shave biopsy from the new tattoo on the left medial ankle revealed an early keratoacanthoma (KA)(Figure 1B). Subsequent shave biopsies of the retouched tattoos on the dorsal foot and the preexisting tattoo on the calf revealed KAs and a granulomatous reaction, respectively (Figures 2B and 3B). The left ankle KA was treated with 2 injections of 5-fluorouracil without improvement. The patient ultimately underwent Mohs micrographic surgery of the left ankle KA and underwent total excision with skin graft.
The development of KAs within tattoos is a known but poorly understood phenomenon.2 Keratoacanthomas are common keratinizing, squamous cell lesions of follicular origin distinguished by their eruptive onset, rapid growth, and spontaneous involution. They typically present as solitary isolated nodules arising in sun-exposed areas of patients of either sex, with a predilection for individuals of Fitzpatrick skin types I and II and in areas of prior trauma or sun damage.3
Histologically, the proliferative phase is defined by keratin-filled invagination of the epidermis into the dermis, with areas of hyperkeratosis, acanthosis, and mitotic activity within the strands and nodules. A high degree of nuclear atypia underlines the diagnostic difficulty in distinguishing KAs from squamous cell carcinomas (SCCs). A fully developed KA has less prominent cellular atypia and a characteristic buttressing lip of epithelium extending over the edges of an irregular, keratin-filled crater. In the final involution stage of KAs, granulation tissue and fibrosis predominate and apoptotic cells may be noted.4
The etiology of KAs remains controversial, but several factors have been correlated with their development, including UV light exposure, chemical carcinogenesis, genetic predisposition, viruses (namely human papillomavirus infection), immunosuppression, treatment with BRAF inhibitors, and trauma. Keratoacanthoma incidence also has been associated with chronic scarring diseases such as discoid lupus erythematous5 and lichen planus.6 Although solitary lesions are more typical, multiple generalized KAs can arise at once, as observed in generalized eruptive KA of Grzybowski, a rare condition, as well as in the multiple self-healing epitheliomas seen in Ferguson-Smith disease.
Because of the unusual histology of KAs and their tendency to spontaneously regress, it is not totally understood where they fall on the benign vs malignant spectrum. Some contest that KAs are benign and self-limited reactive proliferations, whereas others propose they are malignant variants of SCC.3,4,7,8 This debate is compounded by the difficulty in distinguishing KAs from SCC when specimen sampling is inadequate and given documentation that SCCs can develop within KAs over time.7 There also is some concern regarding the remote possibility of aggressive infiltration and even metastasis. One systematic review by Savage and Maize8 attempted to clarify the biologic behavior and malignant potential of KAs. Their review of 445 cases of KA with reported follow-up led to the conclusion that KAs exhibit a benign natural course with no reliable reports of death or metastasis. This finding was in stark contrast to 429 cases of SCC, of which 61 cases (14.2%) resulted in metastasis despite treatment.8
Our patient’s presentation was unique compared to others already reported in the literature because of the simultaneous development of nonsarcoidal granulomatous dermatitis within the older and nonretouched tattoos. Nonsarcoidal granulomatous dermatitis, which encompasses inflammatory skin diseases with histiocytes, is a reactive cutaneous proliferation that also has been reported to occur within tattoos.9,10 Granulomatous tattoo reactions can be further subdivided as foreign body type or sarcoidal type. Foreign body reactions are distinguished by the presence of pigment-containing multinucleated giant cells (as seen in our patient), whereas the sarcoidal type contains compact nodules of epithelioid histiocytes with few lymphocytes.4
The concurrent development of 2 clinically and histologically distinct entities suggests that a similar overlapping pathogenesis underlies each. One hypothesis is that the introduction of exogenous dyes may have instigated an inflammatory foreign body reaction, with the red ink acting as the unifying offender. The formation of granulomas in the preexisting tattoos is likely explained by an exaggerated immune response in the form of a type IV delayed hypersensitivity reaction triggered by reintroduction of the antigen—the red ink—in a presensitized host. Secondly, the parallel development of KAs within the new and retouched tattoos could be a result of the traumatic direct inoculation of the foreign material to which the body was presensitized and subsequent attempt by the skin to degrade and remove it.11
This case provides an example of the development of multiple KAs via a reactive process. Many other similar cases have been described in the literature, including case reports of KAs arising in areas of trauma such as thermal burns, vaccination sites, scars, skin grafts, arthropod bites, and tattoos.2-4,8 Together, the trauma and immune response may lead to localized inflammation and/or cellular hyperplasia, ultimately predisposing the individual to the development of dermoepidermal proliferation. Moreover, the exaggerated keratinocyte proliferation in KAs in response to trauma is reminiscent of the Köbner phenomenon. Other lesions that demonstrate köbnerization also have been reported to occur within new tattoos, including psoriasis, lichen planus, molluscum contagiosum, and verruca vulgaris.1,3
Although KAs are not always a consequence of trauma among humans, trauma-induced KA has been proven as a reliable phenomenon among animal models; an older study showed consistent KA development after animal skin was traumatized from the application of chemical carcinogens.12 Keratoacanthomas within areas of trauma seem to develop rapidly—within a week to a year after trauma—while the development of trauma-related nonmelanoma skin cancers appears to take longer, approximately 1 to 50 years later.13
More research is needed to clarify the pathophysiology of KAs and its precise relationship to trauma and immunology, but our case adds additional weight to the idea that some KAs are primarily reactive phenomena, sharing features of other reactive cutaneous proliferations such as foreign body granulomas.
- Jacob CI. Tattoo-associated dermatoses: a case report and review of the literature. Dermatol Surg. 2002;28:962-965.
- Fraga GR, Prossick TA. Tattoo-associated keratoacanthomas: a series of 8 patients with 11 keratoacanthomas. J Cutan Pathol. 2010;37:85-90.
- Goldsmith LA, Katz SL, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. New York, NY: McGraw-Hill; 2012.
- Elder DE, Elenitsas R, Johnson BL Jr, et al, eds. Lever’s Histopathology of the Skin. 9th ed. Philadelphia: Lippincott, 2005.
- Minicucci EM, Weber SA, Stolf HO, et al. Keratoacanthoma of the lower lip complicating discoid lupus erythematosus in a 14-year-old boy. Pediatr Dermatol. 2007;24:329-330.
- Giesecke LM, Reid CM, James CL, et al. Giant keratoacanthoma arising in hypertrophic lichen planus. Australas J Dermatol. 2003;44:267-269.
- Weedon DD, Malo J, Brooks D, et al. Squamous cell carcinoma arising in keratoacanthoma: a neglected phenomenon in the elderly. Am J Dermatopathol. 2010;32:423-426.
- Savage JA, Maize JC. Keratoacanthoma clinical behavior: a systematic review. Am J Dermatopathol. 2014;36:422-429.
- Schwartz RA, Mathias CG, Miller CH, et al. Granulomatous reaction to purple tattoo pigment. Contact Derm. 1987;16:198-202.
- Bagley MP, Schwartz RA, Lambert WC. Hyperplastic reaction developing within a tattoo. granulomatous tattoo reaction, probably to mercuric sulfide (cinnabar). Arch Dermatol. 1987;123:1557, 1560-1561.
- Kluger N, Plantier F, Moguelet P, et al. Tattoos: natural history and histopathology of cutaneous reactions. Ann Dermatol Venereol. 2011;138:146-154.
- Ghadially FN, Barton BW, Kerridge DF. The etiology of keratoacanthoma. Cancer. 1963;16:603-611.
- Kluger N, Koljonen V. Tattoos, inks, and cancer. Lancet Oncol. 2012;13:e161-168.
To the Editor:
Cutaneous reactions to tattoos are common and histologically diverse. As outlined by Jacob,1 these reactions can be categorized into 4 main groups: inoculative/infective, hypersensitive, neoplastic, and coincidental. A thorough history and physical examination can aid in distinguishing the type of cutaneous reaction, but diagnosis often requires histopathologic clarification. We report the case of a patient who presented with painful indurated nodules within red ink areas of new and preexisting tattoos.
A 48-year-old woman with no prior medical conditions presented with tender pruritic nodules at the site of a new tattoo and within recently retouched tattoos of 5 months’ duration. The tattoos were done at an “organic” tattoo parlor 8 months prior to presentation. Simultaneously, the patient also developed induration and pain in 2 older tattoos that had been done 10 years prior and had not been retouched.
Physical examination revealed 2 smooth and serpiginous nodules nested perfectly within the new red tattoo on the left medial ankle (Figure 1A). Examination of the retouched tattoos on the dorsum of the right foot revealed 4 discrete nodules within the red, heart-shaped areas of the tattoos (Figure 2A). Additionally, the red-inked portions of an older tattoo on the left lateral calf that were outlined in red ink also were raised and indurated (Figure 3A), and a tattoo on the right volar wrist, also in red ink, was indurated and tender to palpation. The remainder of the physical examination was normal.
contiguous dilated follicular infundibula with atypical keratinocytes that had hyperchromatic nuclei, consistent with a keratoacanthoma, as well as a lymphocytic infiltrate in the dermis above a dense infiltrate of lymphocytes and histiocytes (H&E, original magnification ×2.5 [original magnification ×6.2]).
The lesions continued to enlarge and become increasingly painful despite trials of fluticasone propionate cream 0.05%, clobetasol propionate gel 0.05%, a 7-day course of oral levofloxacin, and a 10-day course of oral amoxicillin-clavulanate. Ultimately, a shave biopsy from the new tattoo on the left medial ankle revealed an early keratoacanthoma (KA)(Figure 1B). Subsequent shave biopsies of the retouched tattoos on the dorsal foot and the preexisting tattoo on the calf revealed KAs and a granulomatous reaction, respectively (Figures 2B and 3B). The left ankle KA was treated with 2 injections of 5-fluorouracil without improvement. The patient ultimately underwent Mohs micrographic surgery of the left ankle KA and underwent total excision with skin graft.
The development of KAs within tattoos is a known but poorly understood phenomenon.2 Keratoacanthomas are common keratinizing, squamous cell lesions of follicular origin distinguished by their eruptive onset, rapid growth, and spontaneous involution. They typically present as solitary isolated nodules arising in sun-exposed areas of patients of either sex, with a predilection for individuals of Fitzpatrick skin types I and II and in areas of prior trauma or sun damage.3
Histologically, the proliferative phase is defined by keratin-filled invagination of the epidermis into the dermis, with areas of hyperkeratosis, acanthosis, and mitotic activity within the strands and nodules. A high degree of nuclear atypia underlines the diagnostic difficulty in distinguishing KAs from squamous cell carcinomas (SCCs). A fully developed KA has less prominent cellular atypia and a characteristic buttressing lip of epithelium extending over the edges of an irregular, keratin-filled crater. In the final involution stage of KAs, granulation tissue and fibrosis predominate and apoptotic cells may be noted.4
The etiology of KAs remains controversial, but several factors have been correlated with their development, including UV light exposure, chemical carcinogenesis, genetic predisposition, viruses (namely human papillomavirus infection), immunosuppression, treatment with BRAF inhibitors, and trauma. Keratoacanthoma incidence also has been associated with chronic scarring diseases such as discoid lupus erythematous5 and lichen planus.6 Although solitary lesions are more typical, multiple generalized KAs can arise at once, as observed in generalized eruptive KA of Grzybowski, a rare condition, as well as in the multiple self-healing epitheliomas seen in Ferguson-Smith disease.
Because of the unusual histology of KAs and their tendency to spontaneously regress, it is not totally understood where they fall on the benign vs malignant spectrum. Some contest that KAs are benign and self-limited reactive proliferations, whereas others propose they are malignant variants of SCC.3,4,7,8 This debate is compounded by the difficulty in distinguishing KAs from SCC when specimen sampling is inadequate and given documentation that SCCs can develop within KAs over time.7 There also is some concern regarding the remote possibility of aggressive infiltration and even metastasis. One systematic review by Savage and Maize8 attempted to clarify the biologic behavior and malignant potential of KAs. Their review of 445 cases of KA with reported follow-up led to the conclusion that KAs exhibit a benign natural course with no reliable reports of death or metastasis. This finding was in stark contrast to 429 cases of SCC, of which 61 cases (14.2%) resulted in metastasis despite treatment.8
Our patient’s presentation was unique compared to others already reported in the literature because of the simultaneous development of nonsarcoidal granulomatous dermatitis within the older and nonretouched tattoos. Nonsarcoidal granulomatous dermatitis, which encompasses inflammatory skin diseases with histiocytes, is a reactive cutaneous proliferation that also has been reported to occur within tattoos.9,10 Granulomatous tattoo reactions can be further subdivided as foreign body type or sarcoidal type. Foreign body reactions are distinguished by the presence of pigment-containing multinucleated giant cells (as seen in our patient), whereas the sarcoidal type contains compact nodules of epithelioid histiocytes with few lymphocytes.4
The concurrent development of 2 clinically and histologically distinct entities suggests that a similar overlapping pathogenesis underlies each. One hypothesis is that the introduction of exogenous dyes may have instigated an inflammatory foreign body reaction, with the red ink acting as the unifying offender. The formation of granulomas in the preexisting tattoos is likely explained by an exaggerated immune response in the form of a type IV delayed hypersensitivity reaction triggered by reintroduction of the antigen—the red ink—in a presensitized host. Secondly, the parallel development of KAs within the new and retouched tattoos could be a result of the traumatic direct inoculation of the foreign material to which the body was presensitized and subsequent attempt by the skin to degrade and remove it.11
This case provides an example of the development of multiple KAs via a reactive process. Many other similar cases have been described in the literature, including case reports of KAs arising in areas of trauma such as thermal burns, vaccination sites, scars, skin grafts, arthropod bites, and tattoos.2-4,8 Together, the trauma and immune response may lead to localized inflammation and/or cellular hyperplasia, ultimately predisposing the individual to the development of dermoepidermal proliferation. Moreover, the exaggerated keratinocyte proliferation in KAs in response to trauma is reminiscent of the Köbner phenomenon. Other lesions that demonstrate köbnerization also have been reported to occur within new tattoos, including psoriasis, lichen planus, molluscum contagiosum, and verruca vulgaris.1,3
Although KAs are not always a consequence of trauma among humans, trauma-induced KA has been proven as a reliable phenomenon among animal models; an older study showed consistent KA development after animal skin was traumatized from the application of chemical carcinogens.12 Keratoacanthomas within areas of trauma seem to develop rapidly—within a week to a year after trauma—while the development of trauma-related nonmelanoma skin cancers appears to take longer, approximately 1 to 50 years later.13
More research is needed to clarify the pathophysiology of KAs and its precise relationship to trauma and immunology, but our case adds additional weight to the idea that some KAs are primarily reactive phenomena, sharing features of other reactive cutaneous proliferations such as foreign body granulomas.
To the Editor:
Cutaneous reactions to tattoos are common and histologically diverse. As outlined by Jacob,1 these reactions can be categorized into 4 main groups: inoculative/infective, hypersensitive, neoplastic, and coincidental. A thorough history and physical examination can aid in distinguishing the type of cutaneous reaction, but diagnosis often requires histopathologic clarification. We report the case of a patient who presented with painful indurated nodules within red ink areas of new and preexisting tattoos.
A 48-year-old woman with no prior medical conditions presented with tender pruritic nodules at the site of a new tattoo and within recently retouched tattoos of 5 months’ duration. The tattoos were done at an “organic” tattoo parlor 8 months prior to presentation. Simultaneously, the patient also developed induration and pain in 2 older tattoos that had been done 10 years prior and had not been retouched.
Physical examination revealed 2 smooth and serpiginous nodules nested perfectly within the new red tattoo on the left medial ankle (Figure 1A). Examination of the retouched tattoos on the dorsum of the right foot revealed 4 discrete nodules within the red, heart-shaped areas of the tattoos (Figure 2A). Additionally, the red-inked portions of an older tattoo on the left lateral calf that were outlined in red ink also were raised and indurated (Figure 3A), and a tattoo on the right volar wrist, also in red ink, was indurated and tender to palpation. The remainder of the physical examination was normal.
contiguous dilated follicular infundibula with atypical keratinocytes that had hyperchromatic nuclei, consistent with a keratoacanthoma, as well as a lymphocytic infiltrate in the dermis above a dense infiltrate of lymphocytes and histiocytes (H&E, original magnification ×2.5 [original magnification ×6.2]).
The lesions continued to enlarge and become increasingly painful despite trials of fluticasone propionate cream 0.05%, clobetasol propionate gel 0.05%, a 7-day course of oral levofloxacin, and a 10-day course of oral amoxicillin-clavulanate. Ultimately, a shave biopsy from the new tattoo on the left medial ankle revealed an early keratoacanthoma (KA)(Figure 1B). Subsequent shave biopsies of the retouched tattoos on the dorsal foot and the preexisting tattoo on the calf revealed KAs and a granulomatous reaction, respectively (Figures 2B and 3B). The left ankle KA was treated with 2 injections of 5-fluorouracil without improvement. The patient ultimately underwent Mohs micrographic surgery of the left ankle KA and underwent total excision with skin graft.
The development of KAs within tattoos is a known but poorly understood phenomenon.2 Keratoacanthomas are common keratinizing, squamous cell lesions of follicular origin distinguished by their eruptive onset, rapid growth, and spontaneous involution. They typically present as solitary isolated nodules arising in sun-exposed areas of patients of either sex, with a predilection for individuals of Fitzpatrick skin types I and II and in areas of prior trauma or sun damage.3
Histologically, the proliferative phase is defined by keratin-filled invagination of the epidermis into the dermis, with areas of hyperkeratosis, acanthosis, and mitotic activity within the strands and nodules. A high degree of nuclear atypia underlines the diagnostic difficulty in distinguishing KAs from squamous cell carcinomas (SCCs). A fully developed KA has less prominent cellular atypia and a characteristic buttressing lip of epithelium extending over the edges of an irregular, keratin-filled crater. In the final involution stage of KAs, granulation tissue and fibrosis predominate and apoptotic cells may be noted.4
The etiology of KAs remains controversial, but several factors have been correlated with their development, including UV light exposure, chemical carcinogenesis, genetic predisposition, viruses (namely human papillomavirus infection), immunosuppression, treatment with BRAF inhibitors, and trauma. Keratoacanthoma incidence also has been associated with chronic scarring diseases such as discoid lupus erythematous5 and lichen planus.6 Although solitary lesions are more typical, multiple generalized KAs can arise at once, as observed in generalized eruptive KA of Grzybowski, a rare condition, as well as in the multiple self-healing epitheliomas seen in Ferguson-Smith disease.
Because of the unusual histology of KAs and their tendency to spontaneously regress, it is not totally understood where they fall on the benign vs malignant spectrum. Some contest that KAs are benign and self-limited reactive proliferations, whereas others propose they are malignant variants of SCC.3,4,7,8 This debate is compounded by the difficulty in distinguishing KAs from SCC when specimen sampling is inadequate and given documentation that SCCs can develop within KAs over time.7 There also is some concern regarding the remote possibility of aggressive infiltration and even metastasis. One systematic review by Savage and Maize8 attempted to clarify the biologic behavior and malignant potential of KAs. Their review of 445 cases of KA with reported follow-up led to the conclusion that KAs exhibit a benign natural course with no reliable reports of death or metastasis. This finding was in stark contrast to 429 cases of SCC, of which 61 cases (14.2%) resulted in metastasis despite treatment.8
Our patient’s presentation was unique compared to others already reported in the literature because of the simultaneous development of nonsarcoidal granulomatous dermatitis within the older and nonretouched tattoos. Nonsarcoidal granulomatous dermatitis, which encompasses inflammatory skin diseases with histiocytes, is a reactive cutaneous proliferation that also has been reported to occur within tattoos.9,10 Granulomatous tattoo reactions can be further subdivided as foreign body type or sarcoidal type. Foreign body reactions are distinguished by the presence of pigment-containing multinucleated giant cells (as seen in our patient), whereas the sarcoidal type contains compact nodules of epithelioid histiocytes with few lymphocytes.4
The concurrent development of 2 clinically and histologically distinct entities suggests that a similar overlapping pathogenesis underlies each. One hypothesis is that the introduction of exogenous dyes may have instigated an inflammatory foreign body reaction, with the red ink acting as the unifying offender. The formation of granulomas in the preexisting tattoos is likely explained by an exaggerated immune response in the form of a type IV delayed hypersensitivity reaction triggered by reintroduction of the antigen—the red ink—in a presensitized host. Secondly, the parallel development of KAs within the new and retouched tattoos could be a result of the traumatic direct inoculation of the foreign material to which the body was presensitized and subsequent attempt by the skin to degrade and remove it.11
This case provides an example of the development of multiple KAs via a reactive process. Many other similar cases have been described in the literature, including case reports of KAs arising in areas of trauma such as thermal burns, vaccination sites, scars, skin grafts, arthropod bites, and tattoos.2-4,8 Together, the trauma and immune response may lead to localized inflammation and/or cellular hyperplasia, ultimately predisposing the individual to the development of dermoepidermal proliferation. Moreover, the exaggerated keratinocyte proliferation in KAs in response to trauma is reminiscent of the Köbner phenomenon. Other lesions that demonstrate köbnerization also have been reported to occur within new tattoos, including psoriasis, lichen planus, molluscum contagiosum, and verruca vulgaris.1,3
Although KAs are not always a consequence of trauma among humans, trauma-induced KA has been proven as a reliable phenomenon among animal models; an older study showed consistent KA development after animal skin was traumatized from the application of chemical carcinogens.12 Keratoacanthomas within areas of trauma seem to develop rapidly—within a week to a year after trauma—while the development of trauma-related nonmelanoma skin cancers appears to take longer, approximately 1 to 50 years later.13
More research is needed to clarify the pathophysiology of KAs and its precise relationship to trauma and immunology, but our case adds additional weight to the idea that some KAs are primarily reactive phenomena, sharing features of other reactive cutaneous proliferations such as foreign body granulomas.
- Jacob CI. Tattoo-associated dermatoses: a case report and review of the literature. Dermatol Surg. 2002;28:962-965.
- Fraga GR, Prossick TA. Tattoo-associated keratoacanthomas: a series of 8 patients with 11 keratoacanthomas. J Cutan Pathol. 2010;37:85-90.
- Goldsmith LA, Katz SL, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. New York, NY: McGraw-Hill; 2012.
- Elder DE, Elenitsas R, Johnson BL Jr, et al, eds. Lever’s Histopathology of the Skin. 9th ed. Philadelphia: Lippincott, 2005.
- Minicucci EM, Weber SA, Stolf HO, et al. Keratoacanthoma of the lower lip complicating discoid lupus erythematosus in a 14-year-old boy. Pediatr Dermatol. 2007;24:329-330.
- Giesecke LM, Reid CM, James CL, et al. Giant keratoacanthoma arising in hypertrophic lichen planus. Australas J Dermatol. 2003;44:267-269.
- Weedon DD, Malo J, Brooks D, et al. Squamous cell carcinoma arising in keratoacanthoma: a neglected phenomenon in the elderly. Am J Dermatopathol. 2010;32:423-426.
- Savage JA, Maize JC. Keratoacanthoma clinical behavior: a systematic review. Am J Dermatopathol. 2014;36:422-429.
- Schwartz RA, Mathias CG, Miller CH, et al. Granulomatous reaction to purple tattoo pigment. Contact Derm. 1987;16:198-202.
- Bagley MP, Schwartz RA, Lambert WC. Hyperplastic reaction developing within a tattoo. granulomatous tattoo reaction, probably to mercuric sulfide (cinnabar). Arch Dermatol. 1987;123:1557, 1560-1561.
- Kluger N, Plantier F, Moguelet P, et al. Tattoos: natural history and histopathology of cutaneous reactions. Ann Dermatol Venereol. 2011;138:146-154.
- Ghadially FN, Barton BW, Kerridge DF. The etiology of keratoacanthoma. Cancer. 1963;16:603-611.
- Kluger N, Koljonen V. Tattoos, inks, and cancer. Lancet Oncol. 2012;13:e161-168.
- Jacob CI. Tattoo-associated dermatoses: a case report and review of the literature. Dermatol Surg. 2002;28:962-965.
- Fraga GR, Prossick TA. Tattoo-associated keratoacanthomas: a series of 8 patients with 11 keratoacanthomas. J Cutan Pathol. 2010;37:85-90.
- Goldsmith LA, Katz SL, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. New York, NY: McGraw-Hill; 2012.
- Elder DE, Elenitsas R, Johnson BL Jr, et al, eds. Lever’s Histopathology of the Skin. 9th ed. Philadelphia: Lippincott, 2005.
- Minicucci EM, Weber SA, Stolf HO, et al. Keratoacanthoma of the lower lip complicating discoid lupus erythematosus in a 14-year-old boy. Pediatr Dermatol. 2007;24:329-330.
- Giesecke LM, Reid CM, James CL, et al. Giant keratoacanthoma arising in hypertrophic lichen planus. Australas J Dermatol. 2003;44:267-269.
- Weedon DD, Malo J, Brooks D, et al. Squamous cell carcinoma arising in keratoacanthoma: a neglected phenomenon in the elderly. Am J Dermatopathol. 2010;32:423-426.
- Savage JA, Maize JC. Keratoacanthoma clinical behavior: a systematic review. Am J Dermatopathol. 2014;36:422-429.
- Schwartz RA, Mathias CG, Miller CH, et al. Granulomatous reaction to purple tattoo pigment. Contact Derm. 1987;16:198-202.
- Bagley MP, Schwartz RA, Lambert WC. Hyperplastic reaction developing within a tattoo. granulomatous tattoo reaction, probably to mercuric sulfide (cinnabar). Arch Dermatol. 1987;123:1557, 1560-1561.
- Kluger N, Plantier F, Moguelet P, et al. Tattoos: natural history and histopathology of cutaneous reactions. Ann Dermatol Venereol. 2011;138:146-154.
- Ghadially FN, Barton BW, Kerridge DF. The etiology of keratoacanthoma. Cancer. 1963;16:603-611.
- Kluger N, Koljonen V. Tattoos, inks, and cancer. Lancet Oncol. 2012;13:e161-168.
Practice Points
- Keratoacanthomas (KAs) are common keratinizing, squamous cell lesions of follicular origin distinguished by their eruptive onset, rapid growth, and spontaneous involution.
- The etiology of KAs remains controversial, but several factors have been correlated with their development, including UV light exposure, chemical carcinogenesis, genetic predisposition, viruses (namely human papillomavirus infection), immunosuppression, scarring disorders, and trauma (including tattoos).
- Because of the unusual histology of KAs and their tendency to spontaneously regress, it is not totally understood where they fall on the benign vs malignant spectrum. Our case adds additional weight to the idea that some KAs are primarily reactive phenomena sharing features of other reactive cutaneous proliferations such as foreign body granulomas.

List of medications linked to drug-induced lupus expands
leaving the overall number now standing at 118.
Among the 118 suspected drugs found in VigiBase, the WHO’s global deduplicated individual case safety reports (ICSR) database, 42 had not been previously reported in association with drug-induced lupus (DIL) and 76 had been previously reported in association with DIL in Medline. DIL was reported as a serious adverse event in 55.4% of cases, according to French researchers led by Laurent Arnaud, MD, PhD, of the department of rheumatology at Hôpitaux Universitaires de Strasbourg and Centre National de Références des Maladies Systémiques Rares, Strasbourg, France.
Dr. Arnaud and his colleagues conducted a case-noncase analysis for each drug associated with DIL in order to compare the proportion of specific adverse drug reactions (ADRs) reported for a single drug with the proportion of the same ADR for all other treatments in VigiBase, which receives reports from more than 130 country members of the WHO Programme for International Drug Monitoring and contains over 16 million deduplicated ICSRs recorded by pharmacovigilance centers since 1967. They searched for cases classified as systemic lupus erythematosus (SLE) and identified 12,166 ICSRs of DIL; from these they found 118 suspected drugs with significant pharmacovigilance signal from 8,163 ICSRs that mostly originated from the Americas (65%) and Europe (23%).
In line with what the study authors expected, the drugs associated with the highest number of DIL cases were the antitumor necrosis factor agents infliximab, adalimumab, and etanercept, and the drugs associated with the highest disproportional reporting of DIL were procainamide and hydralazine.
“This is an important finding because these are the two drugs associated with the highest risk of DIL in the literature, therefore confirming the reliability of our approach using a large pharmacovigilance database,” the researchers wrote in Annals of the Rheumatic Diseases.
Overall, DIL was considered definite for 9 drugs (procainamide, hydralazine, minocycline, quinidine, isoniazid, terbinafine, methyldopa, dihydralazine, and chlorpromazine), probable for 19 drugs, and possible for 45 drugs.
The median age of DIL onset was 49 years, which the authors noted was about 2 decades older than that of spontaneous SLE.
They also observed a marked predominance in females (female to male sex ratio, 4.3), a finding that contrasted with previous studies reporting a female to male sex ratio closer to 1:1.
Dr. Arnaud and his colleagues stated that their finding of a median delay between the reported start of suspected treatment and DIL occurrence of 172 days (interquartile range, 35-610 days) suggested that DIL mostly appears after a few months and usually within the first 2 years of treatment with the suspected drug.
“The analysis of the median reporting years for each suspected drug shows a clear evolution of suspected drugs during the past decades. This further underlines that the constantly changing spectrum of DIL should be monitored continuously, and further validates the interest of our approach using the WHO international pharmacovigilance database, the biggest database of this kind with over 16 million deduplicated ICSRs,” they wrote.
The researchers added that distinguishing DIL from SLE is important because its prognosis is usually good when the drug is withdrawn, but the spectrum of DIL is constantly evolving, with drugs once described as strongly linked to DIL now prescribed less frequently.
“The first case of DIL was reported in 1945 with sulfadiazine, while hydralazine DIL was first reported in 1953. Since then, pharmacopoeia has strongly evolved, and one could hypothesize that so has the spectrum of drugs that can induce DIL,” they wrote.
“The detailed list of suspected drugs may prove useful to physicians when confronted with potential DIL cases. Altogether, these findings may help in improving the identification of this constantly evolving disease,” they concluded.
The current study was limited by the lack of a uniform set of criteria for the diagnosis of DIL and by the level of reported details available in VigiBase.
The authors had no outside funding for the study and reported having no conflicts of interest.
SOURCE: Arnaud L et al. Ann Rheum Dis. 2019 Feb 4. doi: 10.1136/annrheumdis-2018-214598.
This new and updated list of possible lupus-inducing drugs includes a growing range of treatment categories, chemical structures, and pharmacologic actions. Yet it is still unclear what the common denominator is that links them.
Drug-induced lupus (DIL) is a peculiar adverse drug reaction that appears to be unrelated to any known property of the inducing agent, although cytokine modulating biologics are a possible exception. Nevertheless, the in vivo metabolism of dissimilar drugs to products with a common, reactive property may go some way to explaining how compounds with different pharmacologic and chemical structures could induce similar adverse reactions.
The findings by Arnaud et al. need better documentation than just positive pharmacovigilance signals. For example, a drug with a relatively high signal does not necessarily translate to a high propensity for causing lupus-like symptoms. It may be a reflection of high drug usage or an awareness of the report contributors for detecting new-onset systemic lupus erythematosus.
Regardless, this research serves to help and inform the medical community to increase the vigilance of previously unreported DIL and perhaps motivate the publication of novel, convincing case reports.
Robert L. Rubin, PhD, is with the University of New Mexico, Albuquerque. His comments are adapted from an editorial accompanying the report by Arnaud et al. (Ann Rheum Dis. 2019 Feb 13. doi: annrheumdis-2018-214785). He reported having no relevant disclosures.
This new and updated list of possible lupus-inducing drugs includes a growing range of treatment categories, chemical structures, and pharmacologic actions. Yet it is still unclear what the common denominator is that links them.
Drug-induced lupus (DIL) is a peculiar adverse drug reaction that appears to be unrelated to any known property of the inducing agent, although cytokine modulating biologics are a possible exception. Nevertheless, the in vivo metabolism of dissimilar drugs to products with a common, reactive property may go some way to explaining how compounds with different pharmacologic and chemical structures could induce similar adverse reactions.
The findings by Arnaud et al. need better documentation than just positive pharmacovigilance signals. For example, a drug with a relatively high signal does not necessarily translate to a high propensity for causing lupus-like symptoms. It may be a reflection of high drug usage or an awareness of the report contributors for detecting new-onset systemic lupus erythematosus.
Regardless, this research serves to help and inform the medical community to increase the vigilance of previously unreported DIL and perhaps motivate the publication of novel, convincing case reports.
Robert L. Rubin, PhD, is with the University of New Mexico, Albuquerque. His comments are adapted from an editorial accompanying the report by Arnaud et al. (Ann Rheum Dis. 2019 Feb 13. doi: annrheumdis-2018-214785). He reported having no relevant disclosures.
This new and updated list of possible lupus-inducing drugs includes a growing range of treatment categories, chemical structures, and pharmacologic actions. Yet it is still unclear what the common denominator is that links them.
Drug-induced lupus (DIL) is a peculiar adverse drug reaction that appears to be unrelated to any known property of the inducing agent, although cytokine modulating biologics are a possible exception. Nevertheless, the in vivo metabolism of dissimilar drugs to products with a common, reactive property may go some way to explaining how compounds with different pharmacologic and chemical structures could induce similar adverse reactions.
The findings by Arnaud et al. need better documentation than just positive pharmacovigilance signals. For example, a drug with a relatively high signal does not necessarily translate to a high propensity for causing lupus-like symptoms. It may be a reflection of high drug usage or an awareness of the report contributors for detecting new-onset systemic lupus erythematosus.
Regardless, this research serves to help and inform the medical community to increase the vigilance of previously unreported DIL and perhaps motivate the publication of novel, convincing case reports.
Robert L. Rubin, PhD, is with the University of New Mexico, Albuquerque. His comments are adapted from an editorial accompanying the report by Arnaud et al. (Ann Rheum Dis. 2019 Feb 13. doi: annrheumdis-2018-214785). He reported having no relevant disclosures.
leaving the overall number now standing at 118.
Among the 118 suspected drugs found in VigiBase, the WHO’s global deduplicated individual case safety reports (ICSR) database, 42 had not been previously reported in association with drug-induced lupus (DIL) and 76 had been previously reported in association with DIL in Medline. DIL was reported as a serious adverse event in 55.4% of cases, according to French researchers led by Laurent Arnaud, MD, PhD, of the department of rheumatology at Hôpitaux Universitaires de Strasbourg and Centre National de Références des Maladies Systémiques Rares, Strasbourg, France.
Dr. Arnaud and his colleagues conducted a case-noncase analysis for each drug associated with DIL in order to compare the proportion of specific adverse drug reactions (ADRs) reported for a single drug with the proportion of the same ADR for all other treatments in VigiBase, which receives reports from more than 130 country members of the WHO Programme for International Drug Monitoring and contains over 16 million deduplicated ICSRs recorded by pharmacovigilance centers since 1967. They searched for cases classified as systemic lupus erythematosus (SLE) and identified 12,166 ICSRs of DIL; from these they found 118 suspected drugs with significant pharmacovigilance signal from 8,163 ICSRs that mostly originated from the Americas (65%) and Europe (23%).
In line with what the study authors expected, the drugs associated with the highest number of DIL cases were the antitumor necrosis factor agents infliximab, adalimumab, and etanercept, and the drugs associated with the highest disproportional reporting of DIL were procainamide and hydralazine.
“This is an important finding because these are the two drugs associated with the highest risk of DIL in the literature, therefore confirming the reliability of our approach using a large pharmacovigilance database,” the researchers wrote in Annals of the Rheumatic Diseases.
Overall, DIL was considered definite for 9 drugs (procainamide, hydralazine, minocycline, quinidine, isoniazid, terbinafine, methyldopa, dihydralazine, and chlorpromazine), probable for 19 drugs, and possible for 45 drugs.
The median age of DIL onset was 49 years, which the authors noted was about 2 decades older than that of spontaneous SLE.
They also observed a marked predominance in females (female to male sex ratio, 4.3), a finding that contrasted with previous studies reporting a female to male sex ratio closer to 1:1.
Dr. Arnaud and his colleagues stated that their finding of a median delay between the reported start of suspected treatment and DIL occurrence of 172 days (interquartile range, 35-610 days) suggested that DIL mostly appears after a few months and usually within the first 2 years of treatment with the suspected drug.
“The analysis of the median reporting years for each suspected drug shows a clear evolution of suspected drugs during the past decades. This further underlines that the constantly changing spectrum of DIL should be monitored continuously, and further validates the interest of our approach using the WHO international pharmacovigilance database, the biggest database of this kind with over 16 million deduplicated ICSRs,” they wrote.
The researchers added that distinguishing DIL from SLE is important because its prognosis is usually good when the drug is withdrawn, but the spectrum of DIL is constantly evolving, with drugs once described as strongly linked to DIL now prescribed less frequently.
“The first case of DIL was reported in 1945 with sulfadiazine, while hydralazine DIL was first reported in 1953. Since then, pharmacopoeia has strongly evolved, and one could hypothesize that so has the spectrum of drugs that can induce DIL,” they wrote.
“The detailed list of suspected drugs may prove useful to physicians when confronted with potential DIL cases. Altogether, these findings may help in improving the identification of this constantly evolving disease,” they concluded.
The current study was limited by the lack of a uniform set of criteria for the diagnosis of DIL and by the level of reported details available in VigiBase.
The authors had no outside funding for the study and reported having no conflicts of interest.
SOURCE: Arnaud L et al. Ann Rheum Dis. 2019 Feb 4. doi: 10.1136/annrheumdis-2018-214598.
leaving the overall number now standing at 118.
Among the 118 suspected drugs found in VigiBase, the WHO’s global deduplicated individual case safety reports (ICSR) database, 42 had not been previously reported in association with drug-induced lupus (DIL) and 76 had been previously reported in association with DIL in Medline. DIL was reported as a serious adverse event in 55.4% of cases, according to French researchers led by Laurent Arnaud, MD, PhD, of the department of rheumatology at Hôpitaux Universitaires de Strasbourg and Centre National de Références des Maladies Systémiques Rares, Strasbourg, France.
Dr. Arnaud and his colleagues conducted a case-noncase analysis for each drug associated with DIL in order to compare the proportion of specific adverse drug reactions (ADRs) reported for a single drug with the proportion of the same ADR for all other treatments in VigiBase, which receives reports from more than 130 country members of the WHO Programme for International Drug Monitoring and contains over 16 million deduplicated ICSRs recorded by pharmacovigilance centers since 1967. They searched for cases classified as systemic lupus erythematosus (SLE) and identified 12,166 ICSRs of DIL; from these they found 118 suspected drugs with significant pharmacovigilance signal from 8,163 ICSRs that mostly originated from the Americas (65%) and Europe (23%).
In line with what the study authors expected, the drugs associated with the highest number of DIL cases were the antitumor necrosis factor agents infliximab, adalimumab, and etanercept, and the drugs associated with the highest disproportional reporting of DIL were procainamide and hydralazine.
“This is an important finding because these are the two drugs associated with the highest risk of DIL in the literature, therefore confirming the reliability of our approach using a large pharmacovigilance database,” the researchers wrote in Annals of the Rheumatic Diseases.
Overall, DIL was considered definite for 9 drugs (procainamide, hydralazine, minocycline, quinidine, isoniazid, terbinafine, methyldopa, dihydralazine, and chlorpromazine), probable for 19 drugs, and possible for 45 drugs.
The median age of DIL onset was 49 years, which the authors noted was about 2 decades older than that of spontaneous SLE.
They also observed a marked predominance in females (female to male sex ratio, 4.3), a finding that contrasted with previous studies reporting a female to male sex ratio closer to 1:1.
Dr. Arnaud and his colleagues stated that their finding of a median delay between the reported start of suspected treatment and DIL occurrence of 172 days (interquartile range, 35-610 days) suggested that DIL mostly appears after a few months and usually within the first 2 years of treatment with the suspected drug.
“The analysis of the median reporting years for each suspected drug shows a clear evolution of suspected drugs during the past decades. This further underlines that the constantly changing spectrum of DIL should be monitored continuously, and further validates the interest of our approach using the WHO international pharmacovigilance database, the biggest database of this kind with over 16 million deduplicated ICSRs,” they wrote.
The researchers added that distinguishing DIL from SLE is important because its prognosis is usually good when the drug is withdrawn, but the spectrum of DIL is constantly evolving, with drugs once described as strongly linked to DIL now prescribed less frequently.
“The first case of DIL was reported in 1945 with sulfadiazine, while hydralazine DIL was first reported in 1953. Since then, pharmacopoeia has strongly evolved, and one could hypothesize that so has the spectrum of drugs that can induce DIL,” they wrote.
“The detailed list of suspected drugs may prove useful to physicians when confronted with potential DIL cases. Altogether, these findings may help in improving the identification of this constantly evolving disease,” they concluded.
The current study was limited by the lack of a uniform set of criteria for the diagnosis of DIL and by the level of reported details available in VigiBase.
The authors had no outside funding for the study and reported having no conflicts of interest.
SOURCE: Arnaud L et al. Ann Rheum Dis. 2019 Feb 4. doi: 10.1136/annrheumdis-2018-214598.
FROM ANNALS OF THE RHEUMATIC DISEASES
New findings raise questions about the role of ANAs in SLE
Antinuclear antibodies (ANAs) have long been considered an important marker in rheumatologic conditions, particularly for the diagnosis and classification of patients with systemic lupus erythematosus, but recent findings are raising new questions about their role.

“We’ve measured ANAs for a long time – it’s a very important test in rheumatology,” David S. Pisetsky, MD, PhD, explained in an interview.
However, even though this test has been around for decades, “some interesting things have developed around it that have made a lot of people, including me, take a second look,” said Dr. Pisetsky, professor of medicine and immunology at Duke University, Durham, N.C.
He elaborated on those recent findings, which relate to the findings of ANA negativity in patients with an established diagnosis of systemic lupus erythematosus (SLE) and to variability among ANA test kit findings, during a presentation at the Winter Rheumatology Symposium sponsored by the American College of Rheumatology.
“Screening of patients during clinical trials for new treatments of SLE suggest that a significant number of people with lupus – 20%-30%, in fact – are ANA negative despite disease activity at the time the test is done,” he said.
For example, unpublished (but recently submitted) data from a phase 2 trial looking at the efficacy and safety of an interleukin-6 monoclonal antibody for the treatment of SLE showed that 23.8% of baseline samples from 183 SLE patients with positive historical ANA and clinically active lupus prior to randomization were ANA negative.
A particular concern with respect to such findings is that ANA positivity is typically a criterion for entry into clinical trials of therapies for lupus and prescription of medications approved for active lupus, Dr. Pisetsky said.
“On the other hand, about 20% of otherwise healthy people – especially women – can be ANA positive, so it’s always been problematic as a screening test due to these false positives, but these new findings suggest that in lupus a real concern is false negatives,” he said. “It’s quite a surprise.”
The findings raise questions about whether ANA negativity in SLE reflects the natural history of the disease, an effect of treatments, or a problem with the assays.
It appears an important problem relates to test kit variability, he said.
“There are lots of different ANA test kits. Their performance characteristics are very different. The performance of ANA tests is much more variable than people realize,” he said, citing data from an analysis that he and his colleagues conducted using 103 samples from a cohort of patients with established SLE.
In that 2017 study, an ANA enzyme-linked immunosorbent assay showed an ANA-negativity rate of 11.7% with zero indeterminate tests, whereas three different test kits showed ANA-negativity rates of 22.3% (with 8.7% of samples reported as indeterminate), 9.7% (with another 9.7% indeterminate), and 4.9% (with another 1.9% indeterminate), respectively. Multiplex testing showed a 13.6% ANA-negativity rate and an indeterminate rate of 7.8% (Ann Rheum Dis. 2018;77:911-3).
Only one sample tested negative for ANA on all three test kits, and disagreement about ANA negativity occurred in one-third of the samples, he said.
Anti–double-stranded DNA assays
Recent findings also raise questions about the use of assays that specifically assess for anti–double-stranded DNA (anti-dsDNA) antibodies, which are highly associated with SLE and have been used as a biomarker for the disease, Dr. Pisetsky said.
For example, a comparison of two anti-dsDNA assays showed discordant results with respect to negativity for anti-dsDNA antibodies in 64 of 181 samples from SLE patients. One assay showed a 70.7% rate of anti-dsDNA negativity and the other showed a 37.6% rate.
The concern regarding test variability relates to the issue of ANA positivity and eligibility for study enrollment and certain treatments; test variability can affect the diagnosis of patients with SLE because ANA positivity is an important finding in routine clinical care, and for anti-dsDNA, test variability can affect assessment of disease activity, he explained.
Tests may differ in a number of ways, such as in their specificity, sensitivity, avidity, and range of epitopes detected. Unfortunately, not enough is known at this point to make specific recommendations regarding best test kits, and while there are alternative technologies that could be useful for ANA testing, none has been validated for particular use in the assessment of trial eligibility, Dr. Pisetsky said.
Nonetheless, awareness of the test variability is important, especially when it comes to assessing patients for trial eligibility and prescribing medications, he added. “For practical, real-world utilization, people need to know about this.”
Dr. Pisetsky reported receiving ANA-related research support from Pfizer, conducting collaborative research with Bio-Rad and EuroImmun, and serving as an adviser to ImmunArray.
Antinuclear antibodies (ANAs) have long been considered an important marker in rheumatologic conditions, particularly for the diagnosis and classification of patients with systemic lupus erythematosus, but recent findings are raising new questions about their role.
“We’ve measured ANAs for a long time – it’s a very important test in rheumatology,” David S. Pisetsky, MD, PhD, explained in an interview.
However, even though this test has been around for decades, “some interesting things have developed around it that have made a lot of people, including me, take a second look,” said Dr. Pisetsky, professor of medicine and immunology at Duke University, Durham, N.C.
He elaborated on those recent findings, which relate to the findings of ANA negativity in patients with an established diagnosis of systemic lupus erythematosus (SLE) and to variability among ANA test kit findings, during a presentation at the Winter Rheumatology Symposium sponsored by the American College of Rheumatology.
“Screening of patients during clinical trials for new treatments of SLE suggest that a significant number of people with lupus – 20%-30%, in fact – are ANA negative despite disease activity at the time the test is done,” he said.
For example, unpublished (but recently submitted) data from a phase 2 trial looking at the efficacy and safety of an interleukin-6 monoclonal antibody for the treatment of SLE showed that 23.8% of baseline samples from 183 SLE patients with positive historical ANA and clinically active lupus prior to randomization were ANA negative.
A particular concern with respect to such findings is that ANA positivity is typically a criterion for entry into clinical trials of therapies for lupus and prescription of medications approved for active lupus, Dr. Pisetsky said.
“On the other hand, about 20% of otherwise healthy people – especially women – can be ANA positive, so it’s always been problematic as a screening test due to these false positives, but these new findings suggest that in lupus a real concern is false negatives,” he said. “It’s quite a surprise.”
The findings raise questions about whether ANA negativity in SLE reflects the natural history of the disease, an effect of treatments, or a problem with the assays.
It appears an important problem relates to test kit variability, he said.
“There are lots of different ANA test kits. Their performance characteristics are very different. The performance of ANA tests is much more variable than people realize,” he said, citing data from an analysis that he and his colleagues conducted using 103 samples from a cohort of patients with established SLE.
In that 2017 study, an ANA enzyme-linked immunosorbent assay showed an ANA-negativity rate of 11.7% with zero indeterminate tests, whereas three different test kits showed ANA-negativity rates of 22.3% (with 8.7% of samples reported as indeterminate), 9.7% (with another 9.7% indeterminate), and 4.9% (with another 1.9% indeterminate), respectively. Multiplex testing showed a 13.6% ANA-negativity rate and an indeterminate rate of 7.8% (Ann Rheum Dis. 2018;77:911-3).
Only one sample tested negative for ANA on all three test kits, and disagreement about ANA negativity occurred in one-third of the samples, he said.
Anti–double-stranded DNA assays
Recent findings also raise questions about the use of assays that specifically assess for anti–double-stranded DNA (anti-dsDNA) antibodies, which are highly associated with SLE and have been used as a biomarker for the disease, Dr. Pisetsky said.
For example, a comparison of two anti-dsDNA assays showed discordant results with respect to negativity for anti-dsDNA antibodies in 64 of 181 samples from SLE patients. One assay showed a 70.7% rate of anti-dsDNA negativity and the other showed a 37.6% rate.
The concern regarding test variability relates to the issue of ANA positivity and eligibility for study enrollment and certain treatments; test variability can affect the diagnosis of patients with SLE because ANA positivity is an important finding in routine clinical care, and for anti-dsDNA, test variability can affect assessment of disease activity, he explained.
Tests may differ in a number of ways, such as in their specificity, sensitivity, avidity, and range of epitopes detected. Unfortunately, not enough is known at this point to make specific recommendations regarding best test kits, and while there are alternative technologies that could be useful for ANA testing, none has been validated for particular use in the assessment of trial eligibility, Dr. Pisetsky said.
Nonetheless, awareness of the test variability is important, especially when it comes to assessing patients for trial eligibility and prescribing medications, he added. “For practical, real-world utilization, people need to know about this.”
Dr. Pisetsky reported receiving ANA-related research support from Pfizer, conducting collaborative research with Bio-Rad and EuroImmun, and serving as an adviser to ImmunArray.
Antinuclear antibodies (ANAs) have long been considered an important marker in rheumatologic conditions, particularly for the diagnosis and classification of patients with systemic lupus erythematosus, but recent findings are raising new questions about their role.
“We’ve measured ANAs for a long time – it’s a very important test in rheumatology,” David S. Pisetsky, MD, PhD, explained in an interview.
However, even though this test has been around for decades, “some interesting things have developed around it that have made a lot of people, including me, take a second look,” said Dr. Pisetsky, professor of medicine and immunology at Duke University, Durham, N.C.
He elaborated on those recent findings, which relate to the findings of ANA negativity in patients with an established diagnosis of systemic lupus erythematosus (SLE) and to variability among ANA test kit findings, during a presentation at the Winter Rheumatology Symposium sponsored by the American College of Rheumatology.
“Screening of patients during clinical trials for new treatments of SLE suggest that a significant number of people with lupus – 20%-30%, in fact – are ANA negative despite disease activity at the time the test is done,” he said.
For example, unpublished (but recently submitted) data from a phase 2 trial looking at the efficacy and safety of an interleukin-6 monoclonal antibody for the treatment of SLE showed that 23.8% of baseline samples from 183 SLE patients with positive historical ANA and clinically active lupus prior to randomization were ANA negative.
A particular concern with respect to such findings is that ANA positivity is typically a criterion for entry into clinical trials of therapies for lupus and prescription of medications approved for active lupus, Dr. Pisetsky said.
“On the other hand, about 20% of otherwise healthy people – especially women – can be ANA positive, so it’s always been problematic as a screening test due to these false positives, but these new findings suggest that in lupus a real concern is false negatives,” he said. “It’s quite a surprise.”
The findings raise questions about whether ANA negativity in SLE reflects the natural history of the disease, an effect of treatments, or a problem with the assays.
It appears an important problem relates to test kit variability, he said.
“There are lots of different ANA test kits. Their performance characteristics are very different. The performance of ANA tests is much more variable than people realize,” he said, citing data from an analysis that he and his colleagues conducted using 103 samples from a cohort of patients with established SLE.
In that 2017 study, an ANA enzyme-linked immunosorbent assay showed an ANA-negativity rate of 11.7% with zero indeterminate tests, whereas three different test kits showed ANA-negativity rates of 22.3% (with 8.7% of samples reported as indeterminate), 9.7% (with another 9.7% indeterminate), and 4.9% (with another 1.9% indeterminate), respectively. Multiplex testing showed a 13.6% ANA-negativity rate and an indeterminate rate of 7.8% (Ann Rheum Dis. 2018;77:911-3).
Only one sample tested negative for ANA on all three test kits, and disagreement about ANA negativity occurred in one-third of the samples, he said.
Anti–double-stranded DNA assays
Recent findings also raise questions about the use of assays that specifically assess for anti–double-stranded DNA (anti-dsDNA) antibodies, which are highly associated with SLE and have been used as a biomarker for the disease, Dr. Pisetsky said.
For example, a comparison of two anti-dsDNA assays showed discordant results with respect to negativity for anti-dsDNA antibodies in 64 of 181 samples from SLE patients. One assay showed a 70.7% rate of anti-dsDNA negativity and the other showed a 37.6% rate.
The concern regarding test variability relates to the issue of ANA positivity and eligibility for study enrollment and certain treatments; test variability can affect the diagnosis of patients with SLE because ANA positivity is an important finding in routine clinical care, and for anti-dsDNA, test variability can affect assessment of disease activity, he explained.
Tests may differ in a number of ways, such as in their specificity, sensitivity, avidity, and range of epitopes detected. Unfortunately, not enough is known at this point to make specific recommendations regarding best test kits, and while there are alternative technologies that could be useful for ANA testing, none has been validated for particular use in the assessment of trial eligibility, Dr. Pisetsky said.
Nonetheless, awareness of the test variability is important, especially when it comes to assessing patients for trial eligibility and prescribing medications, he added. “For practical, real-world utilization, people need to know about this.”
Dr. Pisetsky reported receiving ANA-related research support from Pfizer, conducting collaborative research with Bio-Rad and EuroImmun, and serving as an adviser to ImmunArray.
EXPERT ANALYSIS FROM THE WINTER RHEUMATOLOGY SYMPOSIUM
Novel SSc classification scheme aims to improve risk stratification
CHICAGO – A simple new classification scheme that combines autoantibody specificity and extent of skin involvement could improve risk stratification of patients with systemic sclerosis, according to researchers at University College London.
“The Le Roy et al. classification of SSc [systemic sclerosis] into limited and diffuse cutaneous subtype remains the most commonly used classification system for systemic sclerosis, but autoantibodies are much better predictors of organ involvement, and while more sophisticated approaches exist, this proposed simple classification using antibodies and skin subset is relevant to clinical practice and could help risk stratification,” Svetlana I. Nihtyanova, MD, said at the annual meeting of the American College of Rheumatology.
Dr. Nihtyanova, a clinical research fellow at University College London, reported how she and her colleagues at UCL divided 1,025 SSc patients into 12 subgroups based on skin subset and autoantibodies and then conducted Kaplan-Meier estimates of survival and cumulative incidence of organ complications to rank these 12 subgroups by endpoint estimates. They merged subgroups with similar ranking in multiple endpoints, ending up with seven groups in the final classification.
Group 1 comprised anti–centromere antibody–positive limited cutaneous SSc (lcSSc) patients and accounted for 29% of patients.
“This was the subgroup with the highest survival (72%) and the lowest incidence of pulmonary fibrosis (13%) and scleroderma renal crisis (no cases) at 20 years from onset,” she said, noting that the incidence of pulmonary hypertension in this group was similar to the average for the whole cohort.
Group 2 comprised all anti–RNA polymerase antibody–positive subjects and accounted for 11% of patients. This group had the highest incidence of scleroderma renal crisis (SRC; 32% at 20 years), but other organ complications and survival were similar to the cohort average.
Group 3 comprised Scl-70–positive lcSSc patients, and accounted for 11% of patients.
“Although incidence of pulmonary fibrosis in this group was the second highest (69% at 20 years), other complications were rare,” Dr. Nihtyanova said, adding that this group had the lowest incidence of pulmonary hypertension (6%) and the second lowest incidence of SRC (3%) at 20 years.
Group 4, conversely, included Scl-70–positive dcSSc patients and accounted for 11% of patients, who had a very poor prognosis; they had the highest incidence of pulmonary fibrosis (91%) and cardiac scleroderma (14%), and the worst survival (41%) at 20 years, she said.
Group 5 included all U3 RNP–positive patients, accounting for 5% of patients.
“Although survival in this group was not bad (70% at 20 years), the group had the highest pulmonary hypertension incidence (40%) and the second highest incidence of cardiac SSc (11%) at 20 years,” she noted.
Groups 6 and 7 (comprising 22% and 11% of study subjects, respectively) included lcSSc and diffuse cutaneous SSc (dcSSc) patients with other antibody specificities. Group 6 had low overall SRC and cardiac SSc risk, while other outcomes were similar to the cohort average. Group 7, however, had poor prognosis, with the second lowest survival (42% at 20 years) and above average rates of organ disease, particularly pulmonary fibrosis and SRC, she said.
Overall, estimated survival for the entire cohort was 60% at 20 years from onset, and in that time frame 44% developed significant pulmonary fibrosis, 25% pulmonary hypertension, 7% SRC, and 6% cardiac SSc. The patients had a mean age of 47 years at disease onset, and 16% were men. Diffuse cutaneous SSc was diagnosed in 35% of the subjects, she noted.
Dr. Nihtyanova reported having no disclosures.
SOURCE: Nihtyanova S et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 2935.
CHICAGO – A simple new classification scheme that combines autoantibody specificity and extent of skin involvement could improve risk stratification of patients with systemic sclerosis, according to researchers at University College London.
“The Le Roy et al. classification of SSc [systemic sclerosis] into limited and diffuse cutaneous subtype remains the most commonly used classification system for systemic sclerosis, but autoantibodies are much better predictors of organ involvement, and while more sophisticated approaches exist, this proposed simple classification using antibodies and skin subset is relevant to clinical practice and could help risk stratification,” Svetlana I. Nihtyanova, MD, said at the annual meeting of the American College of Rheumatology.
Dr. Nihtyanova, a clinical research fellow at University College London, reported how she and her colleagues at UCL divided 1,025 SSc patients into 12 subgroups based on skin subset and autoantibodies and then conducted Kaplan-Meier estimates of survival and cumulative incidence of organ complications to rank these 12 subgroups by endpoint estimates. They merged subgroups with similar ranking in multiple endpoints, ending up with seven groups in the final classification.
Group 1 comprised anti–centromere antibody–positive limited cutaneous SSc (lcSSc) patients and accounted for 29% of patients.
“This was the subgroup with the highest survival (72%) and the lowest incidence of pulmonary fibrosis (13%) and scleroderma renal crisis (no cases) at 20 years from onset,” she said, noting that the incidence of pulmonary hypertension in this group was similar to the average for the whole cohort.
Group 2 comprised all anti–RNA polymerase antibody–positive subjects and accounted for 11% of patients. This group had the highest incidence of scleroderma renal crisis (SRC; 32% at 20 years), but other organ complications and survival were similar to the cohort average.
Group 3 comprised Scl-70–positive lcSSc patients, and accounted for 11% of patients.
“Although incidence of pulmonary fibrosis in this group was the second highest (69% at 20 years), other complications were rare,” Dr. Nihtyanova said, adding that this group had the lowest incidence of pulmonary hypertension (6%) and the second lowest incidence of SRC (3%) at 20 years.
Group 4, conversely, included Scl-70–positive dcSSc patients and accounted for 11% of patients, who had a very poor prognosis; they had the highest incidence of pulmonary fibrosis (91%) and cardiac scleroderma (14%), and the worst survival (41%) at 20 years, she said.
Group 5 included all U3 RNP–positive patients, accounting for 5% of patients.
“Although survival in this group was not bad (70% at 20 years), the group had the highest pulmonary hypertension incidence (40%) and the second highest incidence of cardiac SSc (11%) at 20 years,” she noted.
Groups 6 and 7 (comprising 22% and 11% of study subjects, respectively) included lcSSc and diffuse cutaneous SSc (dcSSc) patients with other antibody specificities. Group 6 had low overall SRC and cardiac SSc risk, while other outcomes were similar to the cohort average. Group 7, however, had poor prognosis, with the second lowest survival (42% at 20 years) and above average rates of organ disease, particularly pulmonary fibrosis and SRC, she said.
Overall, estimated survival for the entire cohort was 60% at 20 years from onset, and in that time frame 44% developed significant pulmonary fibrosis, 25% pulmonary hypertension, 7% SRC, and 6% cardiac SSc. The patients had a mean age of 47 years at disease onset, and 16% were men. Diffuse cutaneous SSc was diagnosed in 35% of the subjects, she noted.
Dr. Nihtyanova reported having no disclosures.
SOURCE: Nihtyanova S et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 2935.
CHICAGO – A simple new classification scheme that combines autoantibody specificity and extent of skin involvement could improve risk stratification of patients with systemic sclerosis, according to researchers at University College London.
“The Le Roy et al. classification of SSc [systemic sclerosis] into limited and diffuse cutaneous subtype remains the most commonly used classification system for systemic sclerosis, but autoantibodies are much better predictors of organ involvement, and while more sophisticated approaches exist, this proposed simple classification using antibodies and skin subset is relevant to clinical practice and could help risk stratification,” Svetlana I. Nihtyanova, MD, said at the annual meeting of the American College of Rheumatology.
Dr. Nihtyanova, a clinical research fellow at University College London, reported how she and her colleagues at UCL divided 1,025 SSc patients into 12 subgroups based on skin subset and autoantibodies and then conducted Kaplan-Meier estimates of survival and cumulative incidence of organ complications to rank these 12 subgroups by endpoint estimates. They merged subgroups with similar ranking in multiple endpoints, ending up with seven groups in the final classification.
Group 1 comprised anti–centromere antibody–positive limited cutaneous SSc (lcSSc) patients and accounted for 29% of patients.
“This was the subgroup with the highest survival (72%) and the lowest incidence of pulmonary fibrosis (13%) and scleroderma renal crisis (no cases) at 20 years from onset,” she said, noting that the incidence of pulmonary hypertension in this group was similar to the average for the whole cohort.
Group 2 comprised all anti–RNA polymerase antibody–positive subjects and accounted for 11% of patients. This group had the highest incidence of scleroderma renal crisis (SRC; 32% at 20 years), but other organ complications and survival were similar to the cohort average.
Group 3 comprised Scl-70–positive lcSSc patients, and accounted for 11% of patients.
“Although incidence of pulmonary fibrosis in this group was the second highest (69% at 20 years), other complications were rare,” Dr. Nihtyanova said, adding that this group had the lowest incidence of pulmonary hypertension (6%) and the second lowest incidence of SRC (3%) at 20 years.
Group 4, conversely, included Scl-70–positive dcSSc patients and accounted for 11% of patients, who had a very poor prognosis; they had the highest incidence of pulmonary fibrosis (91%) and cardiac scleroderma (14%), and the worst survival (41%) at 20 years, she said.
Group 5 included all U3 RNP–positive patients, accounting for 5% of patients.
“Although survival in this group was not bad (70% at 20 years), the group had the highest pulmonary hypertension incidence (40%) and the second highest incidence of cardiac SSc (11%) at 20 years,” she noted.
Groups 6 and 7 (comprising 22% and 11% of study subjects, respectively) included lcSSc and diffuse cutaneous SSc (dcSSc) patients with other antibody specificities. Group 6 had low overall SRC and cardiac SSc risk, while other outcomes were similar to the cohort average. Group 7, however, had poor prognosis, with the second lowest survival (42% at 20 years) and above average rates of organ disease, particularly pulmonary fibrosis and SRC, she said.
Overall, estimated survival for the entire cohort was 60% at 20 years from onset, and in that time frame 44% developed significant pulmonary fibrosis, 25% pulmonary hypertension, 7% SRC, and 6% cardiac SSc. The patients had a mean age of 47 years at disease onset, and 16% were men. Diffuse cutaneous SSc was diagnosed in 35% of the subjects, she noted.
Dr. Nihtyanova reported having no disclosures.
SOURCE: Nihtyanova S et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 2935.
REPORTING FROM THE ACR ANNUAL MEETING
Key clinical point:
Major finding: The classification scheme for SSc risk stratification identified seven distinct SSc subgroups.
Study details: Development and testing of a novel risk classification scheme in 1,025 SSc patients.
Disclosures: Dr. Nihtyanova reported having no disclosures.
Source: Nihtyanova S et al. Arthritis Rheumatol. 2018;70(Suppl 10):Abstract 2935.