User login
Long-term side effects of CAR T cells mostly mild
SAN DIEGO – Longer-term follow-up of patients treated with CD19-targeted chimeric antigen receptor (CAR) T cells for hematologic malignancies indicates that the altered cells are generally safe, with most late events being mild in nature and possibly related to therapies delivered before or after CAR T cells, investigators reported.

Among patients treated with CD19-targeted CAR T cells for relapsed or refractory chronic lymphocytic leukemia (CLL) or non-Hodgkin lymphoma (NHL), the most frequent late adverse event was hypogammaglobulinemia, which occurred in 29 of 48 patients evaluated, reported Ana Cordeiro, MD, from the Fred Hutchinson Cancer Research Center in Seattle.
“Our results suggest that CD19 CAR T cells are safe,” Dr. Cordeiro said at the annual meeting of the American Society of Hematology. “However, continuing with prospective systematic and long-term follow-up of these patients is required for better understanding of these late effects.”
Dr. Cordeiro and colleagues studied a total of 60 patients who were enrolled in a phase 1/2 trial at their center of a CD19-targeted CAR T-cell construct and survived for at least 1 year.
The goal of the study was to describe complications that occurred or persisted beyond 90 days after CAR T-cell infusion.
The cohort included 43 patients treated for NHL and 17 treated for CLL. Patients with CLL were followed for a median of 27.5 months, and patients with NHL were followed for a median of 23.8 months.
As of September 2018, 47 patients were still alive, including 15 patients with CLL (88%) and 32 patients with NHL (74%). Of the 17 patients who died, 10 died from progressive disease (2 from CLL and 8 from NHL), and 3 patients died from nonrelapse causes associated with complications from subsequent allogeneic stem cell transplantation (allo-HCT), including 1 patient from graft-versus-host disease (GVHD) and infection, 1 from infection, and 1 from cerberovascular accident/thrombotic microangiopathy.
Of 38 patients who received additional therapies, 17 had subsequent CAR T-cell infusions under the same protocol, and 16 went on to allo-HCT. Treatments for the remaining five patients were not specified.
Of the 22 patients who did not receive additional treatment for their primary malignancies, 21 were in ongoing complete remission following a single CAR T-cell infusion after a median follow-up of 28 months. However, two patients in this group did require treatment for therapy-related myelodysplastic syndrome (t-MDS). The remaining patient had a small CLL clone at last follow-up.
Late adverse events included the following:
- Late significant cytopenias in three of 19 patients evaluated.
- Late hypogammaglobulinemia in 29 of 48 evaluated patients.
- A total of 138 late infections in 31 of the 60 patients.
- Subsequent malignancies in 10 of the 60 patients, including t-MDS, nonmelanoma skin cancer, and noninvasive bladder cancer.
- Late immune-related events in seven patients.
- Late neurogenic/psychiatric events, including one case each of transient ischemic attack at 3.8 months, encephalopathy and myoclonic seizure in the setting of chemotherapy, and a fatal cerebrovascular accident in the setting of allo-HCT and thrombotic microangiopathy. These patients did not have acute neurotoxicity after CAR T-cell therapy, Dr. Cordeiro noted. In addition, three patients experienced exacerbation of depression or anxiety following infusion.
- GVHD in nine patients at a median time from allo-HCT to first CAR T-cell infusion of 46.3 months (range, 6.7 months to 11 years).
Focusing on those patients who achieve complete remissions after CAR T-cell therapy could help investigators isolate late events that are most likely related to CAR T cells, Dr. Cordeiro said.
Dr. Cordeiro reported having no relevant conflicts of interest.
SOURCE: Cordeiro A et al. ASH 2018, Abstract 223.
SAN DIEGO – Longer-term follow-up of patients treated with CD19-targeted chimeric antigen receptor (CAR) T cells for hematologic malignancies indicates that the altered cells are generally safe, with most late events being mild in nature and possibly related to therapies delivered before or after CAR T cells, investigators reported.
Among patients treated with CD19-targeted CAR T cells for relapsed or refractory chronic lymphocytic leukemia (CLL) or non-Hodgkin lymphoma (NHL), the most frequent late adverse event was hypogammaglobulinemia, which occurred in 29 of 48 patients evaluated, reported Ana Cordeiro, MD, from the Fred Hutchinson Cancer Research Center in Seattle.
“Our results suggest that CD19 CAR T cells are safe,” Dr. Cordeiro said at the annual meeting of the American Society of Hematology. “However, continuing with prospective systematic and long-term follow-up of these patients is required for better understanding of these late effects.”
Dr. Cordeiro and colleagues studied a total of 60 patients who were enrolled in a phase 1/2 trial at their center of a CD19-targeted CAR T-cell construct and survived for at least 1 year.
The goal of the study was to describe complications that occurred or persisted beyond 90 days after CAR T-cell infusion.
The cohort included 43 patients treated for NHL and 17 treated for CLL. Patients with CLL were followed for a median of 27.5 months, and patients with NHL were followed for a median of 23.8 months.
As of September 2018, 47 patients were still alive, including 15 patients with CLL (88%) and 32 patients with NHL (74%). Of the 17 patients who died, 10 died from progressive disease (2 from CLL and 8 from NHL), and 3 patients died from nonrelapse causes associated with complications from subsequent allogeneic stem cell transplantation (allo-HCT), including 1 patient from graft-versus-host disease (GVHD) and infection, 1 from infection, and 1 from cerberovascular accident/thrombotic microangiopathy.
Of 38 patients who received additional therapies, 17 had subsequent CAR T-cell infusions under the same protocol, and 16 went on to allo-HCT. Treatments for the remaining five patients were not specified.
Of the 22 patients who did not receive additional treatment for their primary malignancies, 21 were in ongoing complete remission following a single CAR T-cell infusion after a median follow-up of 28 months. However, two patients in this group did require treatment for therapy-related myelodysplastic syndrome (t-MDS). The remaining patient had a small CLL clone at last follow-up.
Late adverse events included the following:
- Late significant cytopenias in three of 19 patients evaluated.
- Late hypogammaglobulinemia in 29 of 48 evaluated patients.
- A total of 138 late infections in 31 of the 60 patients.
- Subsequent malignancies in 10 of the 60 patients, including t-MDS, nonmelanoma skin cancer, and noninvasive bladder cancer.
- Late immune-related events in seven patients.
- Late neurogenic/psychiatric events, including one case each of transient ischemic attack at 3.8 months, encephalopathy and myoclonic seizure in the setting of chemotherapy, and a fatal cerebrovascular accident in the setting of allo-HCT and thrombotic microangiopathy. These patients did not have acute neurotoxicity after CAR T-cell therapy, Dr. Cordeiro noted. In addition, three patients experienced exacerbation of depression or anxiety following infusion.
- GVHD in nine patients at a median time from allo-HCT to first CAR T-cell infusion of 46.3 months (range, 6.7 months to 11 years).
Focusing on those patients who achieve complete remissions after CAR T-cell therapy could help investigators isolate late events that are most likely related to CAR T cells, Dr. Cordeiro said.
Dr. Cordeiro reported having no relevant conflicts of interest.
SOURCE: Cordeiro A et al. ASH 2018, Abstract 223.
SAN DIEGO – Longer-term follow-up of patients treated with CD19-targeted chimeric antigen receptor (CAR) T cells for hematologic malignancies indicates that the altered cells are generally safe, with most late events being mild in nature and possibly related to therapies delivered before or after CAR T cells, investigators reported.
Among patients treated with CD19-targeted CAR T cells for relapsed or refractory chronic lymphocytic leukemia (CLL) or non-Hodgkin lymphoma (NHL), the most frequent late adverse event was hypogammaglobulinemia, which occurred in 29 of 48 patients evaluated, reported Ana Cordeiro, MD, from the Fred Hutchinson Cancer Research Center in Seattle.
“Our results suggest that CD19 CAR T cells are safe,” Dr. Cordeiro said at the annual meeting of the American Society of Hematology. “However, continuing with prospective systematic and long-term follow-up of these patients is required for better understanding of these late effects.”
Dr. Cordeiro and colleagues studied a total of 60 patients who were enrolled in a phase 1/2 trial at their center of a CD19-targeted CAR T-cell construct and survived for at least 1 year.
The goal of the study was to describe complications that occurred or persisted beyond 90 days after CAR T-cell infusion.
The cohort included 43 patients treated for NHL and 17 treated for CLL. Patients with CLL were followed for a median of 27.5 months, and patients with NHL were followed for a median of 23.8 months.
As of September 2018, 47 patients were still alive, including 15 patients with CLL (88%) and 32 patients with NHL (74%). Of the 17 patients who died, 10 died from progressive disease (2 from CLL and 8 from NHL), and 3 patients died from nonrelapse causes associated with complications from subsequent allogeneic stem cell transplantation (allo-HCT), including 1 patient from graft-versus-host disease (GVHD) and infection, 1 from infection, and 1 from cerberovascular accident/thrombotic microangiopathy.
Of 38 patients who received additional therapies, 17 had subsequent CAR T-cell infusions under the same protocol, and 16 went on to allo-HCT. Treatments for the remaining five patients were not specified.
Of the 22 patients who did not receive additional treatment for their primary malignancies, 21 were in ongoing complete remission following a single CAR T-cell infusion after a median follow-up of 28 months. However, two patients in this group did require treatment for therapy-related myelodysplastic syndrome (t-MDS). The remaining patient had a small CLL clone at last follow-up.
Late adverse events included the following:
- Late significant cytopenias in three of 19 patients evaluated.
- Late hypogammaglobulinemia in 29 of 48 evaluated patients.
- A total of 138 late infections in 31 of the 60 patients.
- Subsequent malignancies in 10 of the 60 patients, including t-MDS, nonmelanoma skin cancer, and noninvasive bladder cancer.
- Late immune-related events in seven patients.
- Late neurogenic/psychiatric events, including one case each of transient ischemic attack at 3.8 months, encephalopathy and myoclonic seizure in the setting of chemotherapy, and a fatal cerebrovascular accident in the setting of allo-HCT and thrombotic microangiopathy. These patients did not have acute neurotoxicity after CAR T-cell therapy, Dr. Cordeiro noted. In addition, three patients experienced exacerbation of depression or anxiety following infusion.
- GVHD in nine patients at a median time from allo-HCT to first CAR T-cell infusion of 46.3 months (range, 6.7 months to 11 years).
Focusing on those patients who achieve complete remissions after CAR T-cell therapy could help investigators isolate late events that are most likely related to CAR T cells, Dr. Cordeiro said.
Dr. Cordeiro reported having no relevant conflicts of interest.
SOURCE: Cordeiro A et al. ASH 2018, Abstract 223.
REPORTING FROM ASH 2018
Key clinical point:
Major finding: The most frequent adverse event was hypogammaglobulinemia in 60% of evaluable patients.
Study details: Prospective observational study of 60 patients with relapsed/refractory CLL or NHL.
Disclosures: Dr. Cordeiro reported having no relevant conflicts of interest.
Source: Cordeiro A et al. ASH 2018, Abstract 223.
Lenalidomide maintenance improves MCL survival after ASCT
SAN DIEGO – For patients 65 years or younger with mantle cell lymphoma (MCL) who have undergone autologous stem cell transplantation (ASCT), maintenance therapy with lenalidomide (Revlimid) can significantly improve progression-free survival (PFS), suggest results of the phase, 3 randomized MCL0208 trial.

After a median follow-up of 39 months, the 3-year PFS in an intention-to-treat analysis was 80% for patients treated with ASCT and lenalidomide maintenance, compared with 64% for patients treated with ASCT alone, reported Marco Ladetto, MD, of Azienda Ospedaliera Nazionale SS. Antonio e Biagio e Cesare Arrigo in Alessandria, Italy.
“Lenalidomide maintenance after autologous stem cell transplant has substantial clinical activity in mantle cell lymphoma in terms of progression-free survival,” he said at the annual meeting of the American Society of Hematology. “Follow-up is still too short for meaningful overall survival considerations.”
Dr. Ladetto and his colleagues at centers in Italy and Portugal enrolled patients aged 18-65 years with previously untreated MCL stage III or IV, or stage II with bulky disease (5 cm or greater), and good performance status.
The patients first underwent induction with three cycles of R-CHOP (rituximab, cyclophosphamide, doxorubicin, and prednisone), which was followed by treatment with rituximab plus high-dose cyclophosphamide and two cycles of rituximab with high-dose cytarabine. Stem cells were collected after the first course of the latter regimen.
The patients then underwent conditioning with BEAM (carmustine, etoposide, cytarabine, melphalan) and ASCT.
Following ASCT, patients with complete or partial remissions were randomized either to maintenance therapy with lenalidomide 15 mg for 21 of 28 days for each cycle or to observation.
Of the 303 patients initially enrolled, 248 went on to ASCT, and 205 went on to randomization – 104 assigned to maintenance and 101 assigned to observation.
A total of 52 patients completed 2 years of maintenance: Of the rest, 2 patients died from toxicities (thrombotic thrombocytopenic purpura and pneumonia), 7 had disease progression, 41 dropped out for nonprogression reasons, and 2 patients were still in maintenance at the time of the data cutoff. In this arm, 6 of 8 patients with partial responses converted to complete responses by the end of maintenance. More than a quarter of patients (28%) received less than 25% of the planned lenalidomide dose.
In the observation arm, 1 patient died from pneumonia, 20 had disease progression, 3 were lost to follow-up, 6 were still under observation, and 71 completed observation. In this arm, 1 of 4 patients with a partial response converted to a complete response at the end of the observation period.
Despite suboptimal dosing in a large proportion of patients, the PFS primary endpoint showed significant benefit for lenalidomide, with an unstratified hazard ratio of 0.52 (P = .015) and a stratified HR of 0.51 (P = .013).
At a median follow-up of 39 months from randomization, 3-year overall survival (OS) rates were 93% with lenalidomide and 86% with observation, a difference that was not statistically significant.
Grade 3 or 4 hematologic toxicities occurred in 63% of patients in the lenalidomide arm, compared with 11% in the observation arm. The respective rates of granulocytopenia were 59% vs. 10%. Nonhematological grade 3 toxicity was comparable in the two arms except for grade 3 or 4 infections, which were more common with lenalidomide. Seven patients in the lenalidomide arm and three patients in the observation arm developed second cancers.
Dr. Ladetto noted that difficulties in delivering the planned dose of lenalidomide may have been caused by an already-stressed hematopoietic compartment; he commented that the question of the relative benefit of a fixed lenalidomide schedule or an until-progression approach still needs to be answered.
Additionally, the induction schedule used in the trial, while feasible, is not superior to “less cumbersome and possibly less toxic regimens,” he said.
The study was supported by the Italian Lymphoma Foundation (Fondazione Italiana Linfomi) with the European Mantle Cell Lymphoma Network. Dr. Ladetto reported honoraria from Roche, Celgene, Acerta, Janssen, AbbVie, and Sandoz, as well as off-label use of lenalidomide.
SOURCE: Ladetto M et al. ASH 2018, Abstract 401.
SAN DIEGO – For patients 65 years or younger with mantle cell lymphoma (MCL) who have undergone autologous stem cell transplantation (ASCT), maintenance therapy with lenalidomide (Revlimid) can significantly improve progression-free survival (PFS), suggest results of the phase, 3 randomized MCL0208 trial.
After a median follow-up of 39 months, the 3-year PFS in an intention-to-treat analysis was 80% for patients treated with ASCT and lenalidomide maintenance, compared with 64% for patients treated with ASCT alone, reported Marco Ladetto, MD, of Azienda Ospedaliera Nazionale SS. Antonio e Biagio e Cesare Arrigo in Alessandria, Italy.
“Lenalidomide maintenance after autologous stem cell transplant has substantial clinical activity in mantle cell lymphoma in terms of progression-free survival,” he said at the annual meeting of the American Society of Hematology. “Follow-up is still too short for meaningful overall survival considerations.”
Dr. Ladetto and his colleagues at centers in Italy and Portugal enrolled patients aged 18-65 years with previously untreated MCL stage III or IV, or stage II with bulky disease (5 cm or greater), and good performance status.
The patients first underwent induction with three cycles of R-CHOP (rituximab, cyclophosphamide, doxorubicin, and prednisone), which was followed by treatment with rituximab plus high-dose cyclophosphamide and two cycles of rituximab with high-dose cytarabine. Stem cells were collected after the first course of the latter regimen.
The patients then underwent conditioning with BEAM (carmustine, etoposide, cytarabine, melphalan) and ASCT.
Following ASCT, patients with complete or partial remissions were randomized either to maintenance therapy with lenalidomide 15 mg for 21 of 28 days for each cycle or to observation.
Of the 303 patients initially enrolled, 248 went on to ASCT, and 205 went on to randomization – 104 assigned to maintenance and 101 assigned to observation.
A total of 52 patients completed 2 years of maintenance: Of the rest, 2 patients died from toxicities (thrombotic thrombocytopenic purpura and pneumonia), 7 had disease progression, 41 dropped out for nonprogression reasons, and 2 patients were still in maintenance at the time of the data cutoff. In this arm, 6 of 8 patients with partial responses converted to complete responses by the end of maintenance. More than a quarter of patients (28%) received less than 25% of the planned lenalidomide dose.
In the observation arm, 1 patient died from pneumonia, 20 had disease progression, 3 were lost to follow-up, 6 were still under observation, and 71 completed observation. In this arm, 1 of 4 patients with a partial response converted to a complete response at the end of the observation period.
Despite suboptimal dosing in a large proportion of patients, the PFS primary endpoint showed significant benefit for lenalidomide, with an unstratified hazard ratio of 0.52 (P = .015) and a stratified HR of 0.51 (P = .013).
At a median follow-up of 39 months from randomization, 3-year overall survival (OS) rates were 93% with lenalidomide and 86% with observation, a difference that was not statistically significant.
Grade 3 or 4 hematologic toxicities occurred in 63% of patients in the lenalidomide arm, compared with 11% in the observation arm. The respective rates of granulocytopenia were 59% vs. 10%. Nonhematological grade 3 toxicity was comparable in the two arms except for grade 3 or 4 infections, which were more common with lenalidomide. Seven patients in the lenalidomide arm and three patients in the observation arm developed second cancers.
Dr. Ladetto noted that difficulties in delivering the planned dose of lenalidomide may have been caused by an already-stressed hematopoietic compartment; he commented that the question of the relative benefit of a fixed lenalidomide schedule or an until-progression approach still needs to be answered.
Additionally, the induction schedule used in the trial, while feasible, is not superior to “less cumbersome and possibly less toxic regimens,” he said.
The study was supported by the Italian Lymphoma Foundation (Fondazione Italiana Linfomi) with the European Mantle Cell Lymphoma Network. Dr. Ladetto reported honoraria from Roche, Celgene, Acerta, Janssen, AbbVie, and Sandoz, as well as off-label use of lenalidomide.
SOURCE: Ladetto M et al. ASH 2018, Abstract 401.
SAN DIEGO – For patients 65 years or younger with mantle cell lymphoma (MCL) who have undergone autologous stem cell transplantation (ASCT), maintenance therapy with lenalidomide (Revlimid) can significantly improve progression-free survival (PFS), suggest results of the phase, 3 randomized MCL0208 trial.
After a median follow-up of 39 months, the 3-year PFS in an intention-to-treat analysis was 80% for patients treated with ASCT and lenalidomide maintenance, compared with 64% for patients treated with ASCT alone, reported Marco Ladetto, MD, of Azienda Ospedaliera Nazionale SS. Antonio e Biagio e Cesare Arrigo in Alessandria, Italy.
“Lenalidomide maintenance after autologous stem cell transplant has substantial clinical activity in mantle cell lymphoma in terms of progression-free survival,” he said at the annual meeting of the American Society of Hematology. “Follow-up is still too short for meaningful overall survival considerations.”
Dr. Ladetto and his colleagues at centers in Italy and Portugal enrolled patients aged 18-65 years with previously untreated MCL stage III or IV, or stage II with bulky disease (5 cm or greater), and good performance status.
The patients first underwent induction with three cycles of R-CHOP (rituximab, cyclophosphamide, doxorubicin, and prednisone), which was followed by treatment with rituximab plus high-dose cyclophosphamide and two cycles of rituximab with high-dose cytarabine. Stem cells were collected after the first course of the latter regimen.
The patients then underwent conditioning with BEAM (carmustine, etoposide, cytarabine, melphalan) and ASCT.
Following ASCT, patients with complete or partial remissions were randomized either to maintenance therapy with lenalidomide 15 mg for 21 of 28 days for each cycle or to observation.
Of the 303 patients initially enrolled, 248 went on to ASCT, and 205 went on to randomization – 104 assigned to maintenance and 101 assigned to observation.
A total of 52 patients completed 2 years of maintenance: Of the rest, 2 patients died from toxicities (thrombotic thrombocytopenic purpura and pneumonia), 7 had disease progression, 41 dropped out for nonprogression reasons, and 2 patients were still in maintenance at the time of the data cutoff. In this arm, 6 of 8 patients with partial responses converted to complete responses by the end of maintenance. More than a quarter of patients (28%) received less than 25% of the planned lenalidomide dose.
In the observation arm, 1 patient died from pneumonia, 20 had disease progression, 3 were lost to follow-up, 6 were still under observation, and 71 completed observation. In this arm, 1 of 4 patients with a partial response converted to a complete response at the end of the observation period.
Despite suboptimal dosing in a large proportion of patients, the PFS primary endpoint showed significant benefit for lenalidomide, with an unstratified hazard ratio of 0.52 (P = .015) and a stratified HR of 0.51 (P = .013).
At a median follow-up of 39 months from randomization, 3-year overall survival (OS) rates were 93% with lenalidomide and 86% with observation, a difference that was not statistically significant.
Grade 3 or 4 hematologic toxicities occurred in 63% of patients in the lenalidomide arm, compared with 11% in the observation arm. The respective rates of granulocytopenia were 59% vs. 10%. Nonhematological grade 3 toxicity was comparable in the two arms except for grade 3 or 4 infections, which were more common with lenalidomide. Seven patients in the lenalidomide arm and three patients in the observation arm developed second cancers.
Dr. Ladetto noted that difficulties in delivering the planned dose of lenalidomide may have been caused by an already-stressed hematopoietic compartment; he commented that the question of the relative benefit of a fixed lenalidomide schedule or an until-progression approach still needs to be answered.
Additionally, the induction schedule used in the trial, while feasible, is not superior to “less cumbersome and possibly less toxic regimens,” he said.
The study was supported by the Italian Lymphoma Foundation (Fondazione Italiana Linfomi) with the European Mantle Cell Lymphoma Network. Dr. Ladetto reported honoraria from Roche, Celgene, Acerta, Janssen, AbbVie, and Sandoz, as well as off-label use of lenalidomide.
SOURCE: Ladetto M et al. ASH 2018, Abstract 401.
REPORTING FROM ASH 2018
Key clinical point:
Major finding: The 3-year PFS rate was 80% for patients on lenalidomide maintenance, compared with 64% for patients on observation alone.
Study details: An open-label, randomized, phase 3 trial with 205 patients randomized to lenalidomide or observation.
Disclosures: The study was supported by the Italian Lymphoma Foundation (Fondazione Italiana Linfomi) with the European Mantle Cell Lymphoma Network. Dr. Ladetto reported honoraria from Roche, Celgene, Acerta, Janssen, AbbVie, and Sandoz, as well as off-label use of lenalidomide.
Source: Ladetto M et al. ASH 2018, Abstract 401.
Checkmate 436: Two-drug combo is ‘promising’ for PMBCL
SAN DIEGO – Nivolumab plus brentuximab vedotin may be a new treatment option for patients with relapsed/refractory primary mediastinal large B-cell lymphoma (PMBCL), according to investigators from the CheckMate 436 trial.
Interim results from this phase 1/2 trial revealed an overall response rate of 70%, including a complete response rate of 27%.
“It’s very promising ... to see this level of activity in this advanced, relapsed/refractory population,” said Joseph E. Eid, MD, senior vice president of Bristol-Myers Squibb, which is sponsoring CheckMate 436 in collaboration with Seattle Genetics.
Dr. Eid noted that adverse events (AEs) observed with this regimen were consistent with the safety profiles of nivolumab and brentuximab vedotin alone.
These results were presented as a poster at the annual meeting of the American Society of Hematology.
Dr. Eid noted that patients with relapsed or refractory PMBCL have limited treatment options.
“The initial therapy works well in 70% to 80% of patients but the patients who fail don’t have good options,” he said.
Prior research has shown that PMBCL is often characterized by overexpression of the PD-1 ligands PD-L1 and PD-L2, and most PMBCL expresses CD30.
Dr. Eid said CheckMate 436 (NCT02581631) was designed to “take advantage” of these characteristics by employing the anti-PD-1 checkpoint inhibitor nivolumab and the anti-CD30 antibody-drug conjugate brentuximab vedotin.
The interim analysis of this trial included 30 patients with relapsed/refractory PMBCL. Their median age at enrollment was 35.5 and 57% of patients were female. More than half of the patients (60%) had refractory disease, 23% had relapsed disease, and 17% had both.
The median number of prior therapies was two and 13% of patients had prior autologous stem cell transplant.
The patients received nivolumab at 240 mg and brentuximab vedotin at 1.8 mg/kg every 3 weeks until progression or unacceptable toxicity.
At a median follow-up of 6.1 months, 10 patients were still on treatment. Reasons for discontinuation included maximum clinical benefit, disease progression, AEs unrelated to treatment, patient request, and other concerns.
The rate of treatment-related AEs was 83%. The most common of these were neutropenia (27%), peripheral neuropathy (20%), hyperthyroidism (13%), rash (10%), and thrombocytopenia (10%).
Grade 3-4 treatment-related AEs included neutropenia (27%), thrombocytopenia (7%), decreased neutrophil count (7%), hypersensitivity (3%), diarrhea (3%), and maculopapular rash (3%).
The rate of serious treatment-related AEs was 10%. This included grade 3-4 diarrhea and maculopapular rash and grade 5 acute kidney injury.
The acute kidney injury was the only fatal AE considered treatment related. There were three other deaths in the trial, but they were considered unrelated to treatment.
The complete response rate was 27% (n = 8), and the partial response rate was 43% (n = 13), for an overall response rate of 70% (n = 21).
“The early indication is that 70% response is a pretty good outcome in a relapsed/refractory population that, otherwise, their outcome is pretty dismal,” Dr. Eid said.
Ten percent of patients (n = 3) had stable disease, 13% (n = 4) progressed, and investigators were unable to determine the status for 7% of patients (n = 2).
The median time to response was 1.3 months, and the median time to complete response was 3.0 months. The median duration of response and complete response were not reached.
Overall and progression-free survival data are not yet mature.
Still, the investigators concluded that nivolumab and brentuximab vedotin “may provide a new treatment option” for patients with relapsed/refractory PMBCL.
This trial is supported by Bristol-Myers Squibb in collaboration with Seattle Genetics. Investigators reported relationships with Bristol-Myers Squibb, Seattle Genetics, and a range of other companies.
SOURCE: Moskowitz AJ et al. ASH 2018. Abstract 1691.
SAN DIEGO – Nivolumab plus brentuximab vedotin may be a new treatment option for patients with relapsed/refractory primary mediastinal large B-cell lymphoma (PMBCL), according to investigators from the CheckMate 436 trial.
Interim results from this phase 1/2 trial revealed an overall response rate of 70%, including a complete response rate of 27%.
“It’s very promising ... to see this level of activity in this advanced, relapsed/refractory population,” said Joseph E. Eid, MD, senior vice president of Bristol-Myers Squibb, which is sponsoring CheckMate 436 in collaboration with Seattle Genetics.
Dr. Eid noted that adverse events (AEs) observed with this regimen were consistent with the safety profiles of nivolumab and brentuximab vedotin alone.
These results were presented as a poster at the annual meeting of the American Society of Hematology.
Dr. Eid noted that patients with relapsed or refractory PMBCL have limited treatment options.
“The initial therapy works well in 70% to 80% of patients but the patients who fail don’t have good options,” he said.
Prior research has shown that PMBCL is often characterized by overexpression of the PD-1 ligands PD-L1 and PD-L2, and most PMBCL expresses CD30.
Dr. Eid said CheckMate 436 (NCT02581631) was designed to “take advantage” of these characteristics by employing the anti-PD-1 checkpoint inhibitor nivolumab and the anti-CD30 antibody-drug conjugate brentuximab vedotin.
The interim analysis of this trial included 30 patients with relapsed/refractory PMBCL. Their median age at enrollment was 35.5 and 57% of patients were female. More than half of the patients (60%) had refractory disease, 23% had relapsed disease, and 17% had both.
The median number of prior therapies was two and 13% of patients had prior autologous stem cell transplant.
The patients received nivolumab at 240 mg and brentuximab vedotin at 1.8 mg/kg every 3 weeks until progression or unacceptable toxicity.
At a median follow-up of 6.1 months, 10 patients were still on treatment. Reasons for discontinuation included maximum clinical benefit, disease progression, AEs unrelated to treatment, patient request, and other concerns.
The rate of treatment-related AEs was 83%. The most common of these were neutropenia (27%), peripheral neuropathy (20%), hyperthyroidism (13%), rash (10%), and thrombocytopenia (10%).
Grade 3-4 treatment-related AEs included neutropenia (27%), thrombocytopenia (7%), decreased neutrophil count (7%), hypersensitivity (3%), diarrhea (3%), and maculopapular rash (3%).
The rate of serious treatment-related AEs was 10%. This included grade 3-4 diarrhea and maculopapular rash and grade 5 acute kidney injury.
The acute kidney injury was the only fatal AE considered treatment related. There were three other deaths in the trial, but they were considered unrelated to treatment.
The complete response rate was 27% (n = 8), and the partial response rate was 43% (n = 13), for an overall response rate of 70% (n = 21).
“The early indication is that 70% response is a pretty good outcome in a relapsed/refractory population that, otherwise, their outcome is pretty dismal,” Dr. Eid said.
Ten percent of patients (n = 3) had stable disease, 13% (n = 4) progressed, and investigators were unable to determine the status for 7% of patients (n = 2).
The median time to response was 1.3 months, and the median time to complete response was 3.0 months. The median duration of response and complete response were not reached.
Overall and progression-free survival data are not yet mature.
Still, the investigators concluded that nivolumab and brentuximab vedotin “may provide a new treatment option” for patients with relapsed/refractory PMBCL.
This trial is supported by Bristol-Myers Squibb in collaboration with Seattle Genetics. Investigators reported relationships with Bristol-Myers Squibb, Seattle Genetics, and a range of other companies.
SOURCE: Moskowitz AJ et al. ASH 2018. Abstract 1691.
SAN DIEGO – Nivolumab plus brentuximab vedotin may be a new treatment option for patients with relapsed/refractory primary mediastinal large B-cell lymphoma (PMBCL), according to investigators from the CheckMate 436 trial.
Interim results from this phase 1/2 trial revealed an overall response rate of 70%, including a complete response rate of 27%.
“It’s very promising ... to see this level of activity in this advanced, relapsed/refractory population,” said Joseph E. Eid, MD, senior vice president of Bristol-Myers Squibb, which is sponsoring CheckMate 436 in collaboration with Seattle Genetics.
Dr. Eid noted that adverse events (AEs) observed with this regimen were consistent with the safety profiles of nivolumab and brentuximab vedotin alone.
These results were presented as a poster at the annual meeting of the American Society of Hematology.
Dr. Eid noted that patients with relapsed or refractory PMBCL have limited treatment options.
“The initial therapy works well in 70% to 80% of patients but the patients who fail don’t have good options,” he said.
Prior research has shown that PMBCL is often characterized by overexpression of the PD-1 ligands PD-L1 and PD-L2, and most PMBCL expresses CD30.
Dr. Eid said CheckMate 436 (NCT02581631) was designed to “take advantage” of these characteristics by employing the anti-PD-1 checkpoint inhibitor nivolumab and the anti-CD30 antibody-drug conjugate brentuximab vedotin.
The interim analysis of this trial included 30 patients with relapsed/refractory PMBCL. Their median age at enrollment was 35.5 and 57% of patients were female. More than half of the patients (60%) had refractory disease, 23% had relapsed disease, and 17% had both.
The median number of prior therapies was two and 13% of patients had prior autologous stem cell transplant.
The patients received nivolumab at 240 mg and brentuximab vedotin at 1.8 mg/kg every 3 weeks until progression or unacceptable toxicity.
At a median follow-up of 6.1 months, 10 patients were still on treatment. Reasons for discontinuation included maximum clinical benefit, disease progression, AEs unrelated to treatment, patient request, and other concerns.
The rate of treatment-related AEs was 83%. The most common of these were neutropenia (27%), peripheral neuropathy (20%), hyperthyroidism (13%), rash (10%), and thrombocytopenia (10%).
Grade 3-4 treatment-related AEs included neutropenia (27%), thrombocytopenia (7%), decreased neutrophil count (7%), hypersensitivity (3%), diarrhea (3%), and maculopapular rash (3%).
The rate of serious treatment-related AEs was 10%. This included grade 3-4 diarrhea and maculopapular rash and grade 5 acute kidney injury.
The acute kidney injury was the only fatal AE considered treatment related. There were three other deaths in the trial, but they were considered unrelated to treatment.
The complete response rate was 27% (n = 8), and the partial response rate was 43% (n = 13), for an overall response rate of 70% (n = 21).
“The early indication is that 70% response is a pretty good outcome in a relapsed/refractory population that, otherwise, their outcome is pretty dismal,” Dr. Eid said.
Ten percent of patients (n = 3) had stable disease, 13% (n = 4) progressed, and investigators were unable to determine the status for 7% of patients (n = 2).
The median time to response was 1.3 months, and the median time to complete response was 3.0 months. The median duration of response and complete response were not reached.
Overall and progression-free survival data are not yet mature.
Still, the investigators concluded that nivolumab and brentuximab vedotin “may provide a new treatment option” for patients with relapsed/refractory PMBCL.
This trial is supported by Bristol-Myers Squibb in collaboration with Seattle Genetics. Investigators reported relationships with Bristol-Myers Squibb, Seattle Genetics, and a range of other companies.
SOURCE: Moskowitz AJ et al. ASH 2018. Abstract 1691.
REPORTING FROM ASH 2018
Key clinical point:
Major finding: The overall response rate was 70%, including a complete response rate of 27%.
Study details: A phase 1/2 study of 30 patients.
Disclosures: This trial is supported by Bristol-Myers Squibb in collaboration with Seattle Genetics, and investigators reported relationships with a range of other companies.
Source: Moskowitz AJ et al. ASH 2018, Abstract 1691.
Survivors of childhood Hodgkin lymphoma face 14-fold risk of second cancers
Survivors of childhood Hodgkin lymphoma have a 14-fold greater risk for second cancers, compared with the general population, according to newly published data.
The subsequent malignant neoplasms (SMNs) tend to follow specific patterns depending on the patient’s age at treatment, sex, treatment modality, and body region treated.
And although the risk of SMNs appears to be somewhat lower for patients treated in more recent decades, it is still significantly elevated, compared with that of the general population, according to Anna S. Holmqvist, MD, PhD, from Lund University (Sweden), and her colleagues.
“A major goal of the current study was to develop evidence with which to guide the screening of survivors of HL for the development of [solid] SMNs,” the investigators wrote in Cancer.
They examined at data from the Late Effects Study Group, a multinational cohort of patients aged 16 years or younger who were treated for Hodgkin lymphoma and other cancers from 1955 to 1986.
The current report is the third update from an expanded cohort, including data on 1,136 patients with a median follow-up of 26.6 years. The median patient age at diagnosis was 11 years and the patients were followed for 23,212 person-years following the Hodgkin lymphoma diagnosis.
In all, 162 patients developed a total of 196 solid SMNs, including breast cancer in 54 patients, basal cell carcinoma in 34 patients, thyroid cancer in 30, colorectal cancer in 15, lung cancer in 11, other malignancies in 40, and disease site not available in 12 patients.
The cumulative incidence of any solid SMN 40 years after a diagnosis of Hodgkin lymphoma was 26.4%. The standardized incidence ratio for the entire cohort was 14.0, compared with the general population as derived from the Surveillance, Epidemiology and End Results database.
Predisposing factors for breast cancer in females included a Hodgkin lymphoma diagnosis from the ages of 10-16 years, and treatment with radiotherapy to the chest.
The patients at highest risk for subsequent development of lung cancer were males treated with chest radiotherapy before age 10 years. Those at highest risk for colorectal cancer were males and females who had received abdominal/pelvic radiotherapy and high-dose alkylating agents. Patients at highest risk for thyroid cancers were females who had been treated with radiotherapy to the neck before the age of 10.
The cumulative incidence for breast cancer by age 50 years for those at highest risk was 45.3%. The respective cumulative incidences for lung, colorectal, and thyroid cancers by age 50 were 4.2%, 9.5%, and 17.3%.
The investigators noted that patients treated more recently are likely to have received lower doses and volumes of radiotherapy, compared with patients treated in 1970s and earlier. “However, for the cohort of patients treated between 1955 and 1986, it is clear that continued surveillance for [solid] SMNs is essential because their risk continues to increase as these survivors enter their fourth and subsequent decades of life.”
No specific funding source for the study was reported. The authors made no financial disclosures.
SOURCE: Holmqvist AS et al. Cancer. 2018 Dec 17. doi: 10.1002/cncr.31807.
Survivors of childhood Hodgkin lymphoma have a 14-fold greater risk for second cancers, compared with the general population, according to newly published data.
The subsequent malignant neoplasms (SMNs) tend to follow specific patterns depending on the patient’s age at treatment, sex, treatment modality, and body region treated.
And although the risk of SMNs appears to be somewhat lower for patients treated in more recent decades, it is still significantly elevated, compared with that of the general population, according to Anna S. Holmqvist, MD, PhD, from Lund University (Sweden), and her colleagues.
“A major goal of the current study was to develop evidence with which to guide the screening of survivors of HL for the development of [solid] SMNs,” the investigators wrote in Cancer.
They examined at data from the Late Effects Study Group, a multinational cohort of patients aged 16 years or younger who were treated for Hodgkin lymphoma and other cancers from 1955 to 1986.
The current report is the third update from an expanded cohort, including data on 1,136 patients with a median follow-up of 26.6 years. The median patient age at diagnosis was 11 years and the patients were followed for 23,212 person-years following the Hodgkin lymphoma diagnosis.
In all, 162 patients developed a total of 196 solid SMNs, including breast cancer in 54 patients, basal cell carcinoma in 34 patients, thyroid cancer in 30, colorectal cancer in 15, lung cancer in 11, other malignancies in 40, and disease site not available in 12 patients.
The cumulative incidence of any solid SMN 40 years after a diagnosis of Hodgkin lymphoma was 26.4%. The standardized incidence ratio for the entire cohort was 14.0, compared with the general population as derived from the Surveillance, Epidemiology and End Results database.
Predisposing factors for breast cancer in females included a Hodgkin lymphoma diagnosis from the ages of 10-16 years, and treatment with radiotherapy to the chest.
The patients at highest risk for subsequent development of lung cancer were males treated with chest radiotherapy before age 10 years. Those at highest risk for colorectal cancer were males and females who had received abdominal/pelvic radiotherapy and high-dose alkylating agents. Patients at highest risk for thyroid cancers were females who had been treated with radiotherapy to the neck before the age of 10.
The cumulative incidence for breast cancer by age 50 years for those at highest risk was 45.3%. The respective cumulative incidences for lung, colorectal, and thyroid cancers by age 50 were 4.2%, 9.5%, and 17.3%.
The investigators noted that patients treated more recently are likely to have received lower doses and volumes of radiotherapy, compared with patients treated in 1970s and earlier. “However, for the cohort of patients treated between 1955 and 1986, it is clear that continued surveillance for [solid] SMNs is essential because their risk continues to increase as these survivors enter their fourth and subsequent decades of life.”
No specific funding source for the study was reported. The authors made no financial disclosures.
SOURCE: Holmqvist AS et al. Cancer. 2018 Dec 17. doi: 10.1002/cncr.31807.
Survivors of childhood Hodgkin lymphoma have a 14-fold greater risk for second cancers, compared with the general population, according to newly published data.
The subsequent malignant neoplasms (SMNs) tend to follow specific patterns depending on the patient’s age at treatment, sex, treatment modality, and body region treated.
And although the risk of SMNs appears to be somewhat lower for patients treated in more recent decades, it is still significantly elevated, compared with that of the general population, according to Anna S. Holmqvist, MD, PhD, from Lund University (Sweden), and her colleagues.
“A major goal of the current study was to develop evidence with which to guide the screening of survivors of HL for the development of [solid] SMNs,” the investigators wrote in Cancer.
They examined at data from the Late Effects Study Group, a multinational cohort of patients aged 16 years or younger who were treated for Hodgkin lymphoma and other cancers from 1955 to 1986.
The current report is the third update from an expanded cohort, including data on 1,136 patients with a median follow-up of 26.6 years. The median patient age at diagnosis was 11 years and the patients were followed for 23,212 person-years following the Hodgkin lymphoma diagnosis.
In all, 162 patients developed a total of 196 solid SMNs, including breast cancer in 54 patients, basal cell carcinoma in 34 patients, thyroid cancer in 30, colorectal cancer in 15, lung cancer in 11, other malignancies in 40, and disease site not available in 12 patients.
The cumulative incidence of any solid SMN 40 years after a diagnosis of Hodgkin lymphoma was 26.4%. The standardized incidence ratio for the entire cohort was 14.0, compared with the general population as derived from the Surveillance, Epidemiology and End Results database.
Predisposing factors for breast cancer in females included a Hodgkin lymphoma diagnosis from the ages of 10-16 years, and treatment with radiotherapy to the chest.
The patients at highest risk for subsequent development of lung cancer were males treated with chest radiotherapy before age 10 years. Those at highest risk for colorectal cancer were males and females who had received abdominal/pelvic radiotherapy and high-dose alkylating agents. Patients at highest risk for thyroid cancers were females who had been treated with radiotherapy to the neck before the age of 10.
The cumulative incidence for breast cancer by age 50 years for those at highest risk was 45.3%. The respective cumulative incidences for lung, colorectal, and thyroid cancers by age 50 were 4.2%, 9.5%, and 17.3%.
The investigators noted that patients treated more recently are likely to have received lower doses and volumes of radiotherapy, compared with patients treated in 1970s and earlier. “However, for the cohort of patients treated between 1955 and 1986, it is clear that continued surveillance for [solid] SMNs is essential because their risk continues to increase as these survivors enter their fourth and subsequent decades of life.”
No specific funding source for the study was reported. The authors made no financial disclosures.
SOURCE: Holmqvist AS et al. Cancer. 2018 Dec 17. doi: 10.1002/cncr.31807.
FROM CANCER
Key clinical point:
Major finding: The risk for a subsequent malignant neoplasm among survivors of childhood Hodgkin lymphoma was 14-fold higher than that of the general population.
Study details: The third update of data on a cohort of 1,136 childhood Hodgkin lymphoma survivors followed for a median of 26.6 years.
Disclosures: No specific funding source for the study was reported. The authors made no financial disclosures.
Source: Holmqvist AS et al. Cancer. 2018 Dec 17. doi: 10.1002/cncr.31807.
ECHELON-2: BV-CHP boosts survival in PTCL
SAN DIEGO – A newly approved treatment regimen provides a survival benefit over standard therapy for patients with CD30-positive peripheral T-cell lymphomas (PTCLs), according to new research presented at the annual meeting of the American Society of Hematology.
In the ECHELON-2 trial, patients who received brentuximab vedotin (BV) plus cyclophosphamide, doxorubicin, and prednisone (CHP) had superior progression-free survival (PFS) and overall survival (OS), compared with patients who received standard treatment with cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP).
These results supported the recent U.S. approval of BV in combination with CHP for adults with previously untreated, systemic anaplastic large cell lymphoma or other CD30-expressing PTCLs.
“ECHELON-2 is the first prospective trial in peripheral T-cell lymphoma to show an overall survival benefit over CHOP,” said Steven M. Horwitz, MD, of Memorial Sloan Kettering Cancer Center, with locations in New York and New Jersey.
Dr. Horwitz presented data from this trial at the ASH meeting. Results were simultaneously published in the Lancet (2018 Dec 3. doi: 10.1016/S0140-6736[18]32984-2).
ECHELON-2 (NCT01777152) enrolled 452 patients with previously untreated, CD30-positive PTCL. Subtypes included ALK-positive or ALK-negative systemic anaplastic large-cell lymphoma, PTCL not otherwise specified, angioimmunoblastic T-cell lymphoma, enteropathy-associated T-cell lymphoma, and adult T-cell leukemia/lymphoma.
Patients were randomized to receive BV-CHP plus placebo (n = 226) or CHOP plus placebo (n = 226) every 3 weeks for six to eight cycles.
At baseline, the median age was 58 in the BV-CHP arm and the CHOP arm. The majority of patients were male – 59% in the BV-CHP arm and 67% in the CHOP arm – and most patients had stage III/IV disease, 81% and 80%, respectively.
In all, 89% of patients in the BV-CHP arm and 81% in the CHOP arm completed six or more cycles of their assigned treatment.
The overall response rate was 83% in the BV-CHP arm and 72% in the CHOP arm (P = .0032). The complete response rates were 68% and 56%, respectively (P = .0066).
At a median follow-up of 36.2 months, the median PFS was 48.2 months in the BV-CHP arm and 20.8 months in the CHOP arm. The rate of death or progression was 42% in the BV-CHP arm and 55% in the CHOP arm (hazard ratio = 0.71, P = .011).
At a median follow-up of 42.1 months, the median OS was not reached in either treatment arm. The rate of death was 23% in the BV-CHP arm and 32% in the CHOP arm (HR = 0.66, P = .0244).
Dr. Horwitz noted that this study was not powered to determine differences in PFS or OS by PTCL subtypes.
BV-CHP had a safety profile comparable with that of CHOP, Dr. Horwitz said.
The rate of adverse events (AEs) was 99% in the BV-CHP arm and 98% in the CHOP arm. Grade 3 or higher AEs occurred in 66% and 65% of patients, respectively. Serious AEs occurred in 39% and 38%, respectively.
Three percent of patients in the BV-CHP arm and 4% of those in the CHOP arm had fatal AEs.
The study was funded by Seattle Genetics, Millennium Pharmaceuticals, and the National Institutes of Health. Dr. Horwitz reported relationships with Seattle Genetics, Millennium Pharmaceuticals, and other companies.
SOURCE: Horwitz S et al. ASH 2018, Abstract 997.
SAN DIEGO – A newly approved treatment regimen provides a survival benefit over standard therapy for patients with CD30-positive peripheral T-cell lymphomas (PTCLs), according to new research presented at the annual meeting of the American Society of Hematology.
In the ECHELON-2 trial, patients who received brentuximab vedotin (BV) plus cyclophosphamide, doxorubicin, and prednisone (CHP) had superior progression-free survival (PFS) and overall survival (OS), compared with patients who received standard treatment with cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP).
These results supported the recent U.S. approval of BV in combination with CHP for adults with previously untreated, systemic anaplastic large cell lymphoma or other CD30-expressing PTCLs.
“ECHELON-2 is the first prospective trial in peripheral T-cell lymphoma to show an overall survival benefit over CHOP,” said Steven M. Horwitz, MD, of Memorial Sloan Kettering Cancer Center, with locations in New York and New Jersey.
Dr. Horwitz presented data from this trial at the ASH meeting. Results were simultaneously published in the Lancet (2018 Dec 3. doi: 10.1016/S0140-6736[18]32984-2).
ECHELON-2 (NCT01777152) enrolled 452 patients with previously untreated, CD30-positive PTCL. Subtypes included ALK-positive or ALK-negative systemic anaplastic large-cell lymphoma, PTCL not otherwise specified, angioimmunoblastic T-cell lymphoma, enteropathy-associated T-cell lymphoma, and adult T-cell leukemia/lymphoma.
Patients were randomized to receive BV-CHP plus placebo (n = 226) or CHOP plus placebo (n = 226) every 3 weeks for six to eight cycles.
At baseline, the median age was 58 in the BV-CHP arm and the CHOP arm. The majority of patients were male – 59% in the BV-CHP arm and 67% in the CHOP arm – and most patients had stage III/IV disease, 81% and 80%, respectively.
In all, 89% of patients in the BV-CHP arm and 81% in the CHOP arm completed six or more cycles of their assigned treatment.
The overall response rate was 83% in the BV-CHP arm and 72% in the CHOP arm (P = .0032). The complete response rates were 68% and 56%, respectively (P = .0066).
At a median follow-up of 36.2 months, the median PFS was 48.2 months in the BV-CHP arm and 20.8 months in the CHOP arm. The rate of death or progression was 42% in the BV-CHP arm and 55% in the CHOP arm (hazard ratio = 0.71, P = .011).
At a median follow-up of 42.1 months, the median OS was not reached in either treatment arm. The rate of death was 23% in the BV-CHP arm and 32% in the CHOP arm (HR = 0.66, P = .0244).
Dr. Horwitz noted that this study was not powered to determine differences in PFS or OS by PTCL subtypes.
BV-CHP had a safety profile comparable with that of CHOP, Dr. Horwitz said.
The rate of adverse events (AEs) was 99% in the BV-CHP arm and 98% in the CHOP arm. Grade 3 or higher AEs occurred in 66% and 65% of patients, respectively. Serious AEs occurred in 39% and 38%, respectively.
Three percent of patients in the BV-CHP arm and 4% of those in the CHOP arm had fatal AEs.
The study was funded by Seattle Genetics, Millennium Pharmaceuticals, and the National Institutes of Health. Dr. Horwitz reported relationships with Seattle Genetics, Millennium Pharmaceuticals, and other companies.
SOURCE: Horwitz S et al. ASH 2018, Abstract 997.
SAN DIEGO – A newly approved treatment regimen provides a survival benefit over standard therapy for patients with CD30-positive peripheral T-cell lymphomas (PTCLs), according to new research presented at the annual meeting of the American Society of Hematology.
In the ECHELON-2 trial, patients who received brentuximab vedotin (BV) plus cyclophosphamide, doxorubicin, and prednisone (CHP) had superior progression-free survival (PFS) and overall survival (OS), compared with patients who received standard treatment with cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP).
These results supported the recent U.S. approval of BV in combination with CHP for adults with previously untreated, systemic anaplastic large cell lymphoma or other CD30-expressing PTCLs.
“ECHELON-2 is the first prospective trial in peripheral T-cell lymphoma to show an overall survival benefit over CHOP,” said Steven M. Horwitz, MD, of Memorial Sloan Kettering Cancer Center, with locations in New York and New Jersey.
Dr. Horwitz presented data from this trial at the ASH meeting. Results were simultaneously published in the Lancet (2018 Dec 3. doi: 10.1016/S0140-6736[18]32984-2).
ECHELON-2 (NCT01777152) enrolled 452 patients with previously untreated, CD30-positive PTCL. Subtypes included ALK-positive or ALK-negative systemic anaplastic large-cell lymphoma, PTCL not otherwise specified, angioimmunoblastic T-cell lymphoma, enteropathy-associated T-cell lymphoma, and adult T-cell leukemia/lymphoma.
Patients were randomized to receive BV-CHP plus placebo (n = 226) or CHOP plus placebo (n = 226) every 3 weeks for six to eight cycles.
At baseline, the median age was 58 in the BV-CHP arm and the CHOP arm. The majority of patients were male – 59% in the BV-CHP arm and 67% in the CHOP arm – and most patients had stage III/IV disease, 81% and 80%, respectively.
In all, 89% of patients in the BV-CHP arm and 81% in the CHOP arm completed six or more cycles of their assigned treatment.
The overall response rate was 83% in the BV-CHP arm and 72% in the CHOP arm (P = .0032). The complete response rates were 68% and 56%, respectively (P = .0066).
At a median follow-up of 36.2 months, the median PFS was 48.2 months in the BV-CHP arm and 20.8 months in the CHOP arm. The rate of death or progression was 42% in the BV-CHP arm and 55% in the CHOP arm (hazard ratio = 0.71, P = .011).
At a median follow-up of 42.1 months, the median OS was not reached in either treatment arm. The rate of death was 23% in the BV-CHP arm and 32% in the CHOP arm (HR = 0.66, P = .0244).
Dr. Horwitz noted that this study was not powered to determine differences in PFS or OS by PTCL subtypes.
BV-CHP had a safety profile comparable with that of CHOP, Dr. Horwitz said.
The rate of adverse events (AEs) was 99% in the BV-CHP arm and 98% in the CHOP arm. Grade 3 or higher AEs occurred in 66% and 65% of patients, respectively. Serious AEs occurred in 39% and 38%, respectively.
Three percent of patients in the BV-CHP arm and 4% of those in the CHOP arm had fatal AEs.
The study was funded by Seattle Genetics, Millennium Pharmaceuticals, and the National Institutes of Health. Dr. Horwitz reported relationships with Seattle Genetics, Millennium Pharmaceuticals, and other companies.
SOURCE: Horwitz S et al. ASH 2018, Abstract 997.
REPORTING FROM ASH 2018
Key clinical point:
Major finding: The rate of death or progression was 42% in the BV-CHP arm and 55% in the CHOP arm (hazard ratio = 0.71, P = .011), while the rate of death alone was 23% and 32%, respectively (HR = 0.66, P = .0244).
Study details: A phase 3 trial of 452 patients with peripheral T-cell lymphoma.
Disclosures: The study was funded by Seattle Genetics, Millennium Pharmaceuticals, and the National Institutes of Health. Dr. Horwitz reported relationships with Seattle Genetics, Millennium Pharmaceuticals, and other companies.
Source: Horwitz S et al. ASH 2018, Abstract 997.

CHMP recommends BV+AVD for Hodgkin lymphoma

The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended expanding the marketing authorization for brentuximab vedotin (BV).
The CHMP has recommended approval for BV (Adcetris) in combination with doxorubicin, vinblastine, and dacarbazine (AVD) to treat adults with previously untreated, CD30+, stage IV Hodgkin lymphoma (HL).
The CHMP’s recommendation will be reviewed by the European Commission (EC), which has the authority to approve medicines for use in the European Union, Norway, Iceland, and Liechtenstein.
The EC usually makes a decision within 67 days of a CHMP recommendation.
BV is already EC-approved to treat adults with:
- CD30+ HL at increased risk of relapse or progression following autologous stem cell transplant (ASCT)
- Relapsed or refractory, CD30+ HL following ASCT or following at least two prior therapies when ASCT or multi-agent chemotherapy is not a treatment option
- Relapsed or refractory systemic anaplastic large-cell lymphoma
- CD30+ cutaneous T-cell lymphoma after at least one prior systemic therapy.
Phase 3 trial
The CHMP’s recommendation to approve BV in combination with AVD is supported by the phase 3 ECHELON-1 trial (NCT01712490).
Result from ECHELON-1 were presented at the 2017 ASH Annual Meeting and simultaneously published in The New England Journal of Medicine.
In this trial, researchers compared BV plus AVD (BV+AVD) to doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD) as frontline treatment for 1334 patients with advanced HL.
The primary endpoint was modified progression-free survival (PFS), which was defined as time to progression, death, or evidence of non-complete response after completion of frontline therapy followed by subsequent anticancer therapy.
According to an independent review committee, BV+AVD provided a significant improvement in modified PFS compared to ABVD. The hazard ratio was 0.77 (P=0.035), which corresponds to a 23% reduction in the risk of progression, death, or the need for additional anticancer therapy.
The 2-year modified PFS rate was 82.1% in the BV+AVD arm and 77.2% in the ABVD arm.
There was no significant difference between the treatment arms when it came to response rates or overall survival.
The objective response rate was 86% in the BV+AVD arm and 83% in the ABVD arm (P=0.12). The complete response rate was 73% and 70%, respectively (P=0.22).
The interim 2-year overall survival rate was 97% in the BV+AVD arm and 95% in the ABVD arm (hazard ratio=0.72; P=0.19).
The overall incidence of adverse events (AEs) was 99% in the BV+AVD arm and 98% in the ABVD arm. The incidence of grade 3 or higher AEs was 83% and 66%, respectively, and the incidence of serious AEs was 43% and 27%, respectively.
Neutropenia, febrile neutropenia, and peripheral neuropathy were more common with BV+AVD, while pulmonary toxicity was more common with ABVD.
The ECHELON-1 trial was sponsored by Millennium Pharmaceuticals, Inc. (a Takeda company) in collaboration with Seattle Genetics, Inc.
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended expanding the marketing authorization for brentuximab vedotin (BV).
The CHMP has recommended approval for BV (Adcetris) in combination with doxorubicin, vinblastine, and dacarbazine (AVD) to treat adults with previously untreated, CD30+, stage IV Hodgkin lymphoma (HL).
The CHMP’s recommendation will be reviewed by the European Commission (EC), which has the authority to approve medicines for use in the European Union, Norway, Iceland, and Liechtenstein.
The EC usually makes a decision within 67 days of a CHMP recommendation.
BV is already EC-approved to treat adults with:
- CD30+ HL at increased risk of relapse or progression following autologous stem cell transplant (ASCT)
- Relapsed or refractory, CD30+ HL following ASCT or following at least two prior therapies when ASCT or multi-agent chemotherapy is not a treatment option
- Relapsed or refractory systemic anaplastic large-cell lymphoma
- CD30+ cutaneous T-cell lymphoma after at least one prior systemic therapy.
Phase 3 trial
The CHMP’s recommendation to approve BV in combination with AVD is supported by the phase 3 ECHELON-1 trial (NCT01712490).
Result from ECHELON-1 were presented at the 2017 ASH Annual Meeting and simultaneously published in The New England Journal of Medicine.
In this trial, researchers compared BV plus AVD (BV+AVD) to doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD) as frontline treatment for 1334 patients with advanced HL.
The primary endpoint was modified progression-free survival (PFS), which was defined as time to progression, death, or evidence of non-complete response after completion of frontline therapy followed by subsequent anticancer therapy.
According to an independent review committee, BV+AVD provided a significant improvement in modified PFS compared to ABVD. The hazard ratio was 0.77 (P=0.035), which corresponds to a 23% reduction in the risk of progression, death, or the need for additional anticancer therapy.
The 2-year modified PFS rate was 82.1% in the BV+AVD arm and 77.2% in the ABVD arm.
There was no significant difference between the treatment arms when it came to response rates or overall survival.
The objective response rate was 86% in the BV+AVD arm and 83% in the ABVD arm (P=0.12). The complete response rate was 73% and 70%, respectively (P=0.22).
The interim 2-year overall survival rate was 97% in the BV+AVD arm and 95% in the ABVD arm (hazard ratio=0.72; P=0.19).
The overall incidence of adverse events (AEs) was 99% in the BV+AVD arm and 98% in the ABVD arm. The incidence of grade 3 or higher AEs was 83% and 66%, respectively, and the incidence of serious AEs was 43% and 27%, respectively.
Neutropenia, febrile neutropenia, and peripheral neuropathy were more common with BV+AVD, while pulmonary toxicity was more common with ABVD.
The ECHELON-1 trial was sponsored by Millennium Pharmaceuticals, Inc. (a Takeda company) in collaboration with Seattle Genetics, Inc.
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended expanding the marketing authorization for brentuximab vedotin (BV).
The CHMP has recommended approval for BV (Adcetris) in combination with doxorubicin, vinblastine, and dacarbazine (AVD) to treat adults with previously untreated, CD30+, stage IV Hodgkin lymphoma (HL).
The CHMP’s recommendation will be reviewed by the European Commission (EC), which has the authority to approve medicines for use in the European Union, Norway, Iceland, and Liechtenstein.
The EC usually makes a decision within 67 days of a CHMP recommendation.
BV is already EC-approved to treat adults with:
- CD30+ HL at increased risk of relapse or progression following autologous stem cell transplant (ASCT)
- Relapsed or refractory, CD30+ HL following ASCT or following at least two prior therapies when ASCT or multi-agent chemotherapy is not a treatment option
- Relapsed or refractory systemic anaplastic large-cell lymphoma
- CD30+ cutaneous T-cell lymphoma after at least one prior systemic therapy.
Phase 3 trial
The CHMP’s recommendation to approve BV in combination with AVD is supported by the phase 3 ECHELON-1 trial (NCT01712490).
Result from ECHELON-1 were presented at the 2017 ASH Annual Meeting and simultaneously published in The New England Journal of Medicine.
In this trial, researchers compared BV plus AVD (BV+AVD) to doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD) as frontline treatment for 1334 patients with advanced HL.
The primary endpoint was modified progression-free survival (PFS), which was defined as time to progression, death, or evidence of non-complete response after completion of frontline therapy followed by subsequent anticancer therapy.
According to an independent review committee, BV+AVD provided a significant improvement in modified PFS compared to ABVD. The hazard ratio was 0.77 (P=0.035), which corresponds to a 23% reduction in the risk of progression, death, or the need for additional anticancer therapy.
The 2-year modified PFS rate was 82.1% in the BV+AVD arm and 77.2% in the ABVD arm.
There was no significant difference between the treatment arms when it came to response rates or overall survival.
The objective response rate was 86% in the BV+AVD arm and 83% in the ABVD arm (P=0.12). The complete response rate was 73% and 70%, respectively (P=0.22).
The interim 2-year overall survival rate was 97% in the BV+AVD arm and 95% in the ABVD arm (hazard ratio=0.72; P=0.19).
The overall incidence of adverse events (AEs) was 99% in the BV+AVD arm and 98% in the ABVD arm. The incidence of grade 3 or higher AEs was 83% and 66%, respectively, and the incidence of serious AEs was 43% and 27%, respectively.
Neutropenia, febrile neutropenia, and peripheral neuropathy were more common with BV+AVD, while pulmonary toxicity was more common with ABVD.
The ECHELON-1 trial was sponsored by Millennium Pharmaceuticals, Inc. (a Takeda company) in collaboration with Seattle Genetics, Inc.
Lymphodepletion improves efficacy of CAR T cells in HL

SAN DIEGO—A phase 1 study suggests lymphodepletion can improve the efficacy of CD30-directed chimeric antigen receptor (CAR) T-cell therapy in patients with Hodgkin lymphoma (HL).
Researchers observed improved responses in HL patients treated with fludarabine and cyclophosphamide prior to CD30.CAR T-cell therapy.
This lymphodepleting regimen was also associated with increased toxicity, compared to no lymphodepletion. However, researchers consider the regimen safe.
Carlos A. Ramos, MD, of Baylor College of Medicine in Houston, Texas, presented these results at the 2018 ASH Annual Meeting (abstract 680*).
Without lymphodepletion
Dr. Ramos first discussed a previous phase 1 trial (NCT01316146), which was published in The Journal of Clinical Investigation in 2017.
In this trial, he and his colleagues had tested CD30.CAR T-cell therapy in patients with relapsed/refractory, CD30+ HL or T-cell non-Hodgkin lymphoma. None of these patients underwent lymphodepletion.
There were no dose-limiting toxicities in this trial—including no neurotoxicity or cytokine release syndrome—but responses were “limited,” according to Dr. Ramos.
Three patients achieved a complete response (CR), three had stable disease, and three progressed.
“Although we saw no significant toxicities and some good clinical responses . . ., the bottom line is that the responses were still quite limited, with several patients having, at most, stable disease or progressive disease,” Dr. Ramos said.
With lymphodepletion
Results from the previous trial prompted Dr. Ramos and his colleagues to conduct the RELY-30 trial (NCT02917083) and investigate whether lymphodepletion would improve responses to CD30.CAR T-cell therapy.
Thus far, 11 patients have been treated on this trial. All had relapsed, CD30+ HL at baseline. Six patients are male, and their median age at baseline was 30 (range, 17-69).
The patients had a median of 5 prior treatments (range, 2-9). This included PD-1 inhibitors (n=10), brentuximab vedotin (n=8), and transplant (n=6).
All patients received lymphodepletion with cyclophosphamide at 500 mg/m2 and fludarabine at 30 mg/m2 daily for 3 days. They then received CD30.CAR T-cell therapy at 2×107 cells/m2 or 1×108 cells/m2.
Dr. Ramos noted that CD30.CAR T-cell expansion was dose-dependent and increased by lymphodepleting chemotherapy.
“The peak expansion is much higher [with lymphodepletion], probably in the order of two to three logs higher than what we see without lymphodepleting chemotherapy,” he said. “So chemotherapy makes a difference.”
Increased CD30.CAR T-cell expansion was associated with improved response. Of the nine evaluable patients, six achieved a CR, and three progressed.
Four complete responders were still in CR at last follow-up, one of them for more than a year. However, two complete responders ultimately progressed.
In addition to improved responses, the researchers observed increased toxicity in this trial. Dr. Ramos said some of these toxicities are “probably attributable” to the lymphodepleting chemotherapy.
Toxicities included grade 1 cytokine release syndrome (no tocilizumab required), maculopapular rash, transient cytopenias, nausea, vomiting, and alopecia.
Dr. Ramos said these results suggest adoptive transfer of CD30.CAR T cells is “safe, even with chemotherapy.”
He noted that the duration of response with this treatment is unknown, but trial enrollment and follow-up are ongoing.
RELY-30 was sponsored by Baylor College of Medicine. Dr. Ramos reported relationships with Novartis, Celgene, Bluebird Bio, and Tessa Therapeutics.
*Data in the abstract differ from the presentation.
SAN DIEGO—A phase 1 study suggests lymphodepletion can improve the efficacy of CD30-directed chimeric antigen receptor (CAR) T-cell therapy in patients with Hodgkin lymphoma (HL).
Researchers observed improved responses in HL patients treated with fludarabine and cyclophosphamide prior to CD30.CAR T-cell therapy.
This lymphodepleting regimen was also associated with increased toxicity, compared to no lymphodepletion. However, researchers consider the regimen safe.
Carlos A. Ramos, MD, of Baylor College of Medicine in Houston, Texas, presented these results at the 2018 ASH Annual Meeting (abstract 680*).
Without lymphodepletion
Dr. Ramos first discussed a previous phase 1 trial (NCT01316146), which was published in The Journal of Clinical Investigation in 2017.
In this trial, he and his colleagues had tested CD30.CAR T-cell therapy in patients with relapsed/refractory, CD30+ HL or T-cell non-Hodgkin lymphoma. None of these patients underwent lymphodepletion.
There were no dose-limiting toxicities in this trial—including no neurotoxicity or cytokine release syndrome—but responses were “limited,” according to Dr. Ramos.
Three patients achieved a complete response (CR), three had stable disease, and three progressed.
“Although we saw no significant toxicities and some good clinical responses . . ., the bottom line is that the responses were still quite limited, with several patients having, at most, stable disease or progressive disease,” Dr. Ramos said.
With lymphodepletion
Results from the previous trial prompted Dr. Ramos and his colleagues to conduct the RELY-30 trial (NCT02917083) and investigate whether lymphodepletion would improve responses to CD30.CAR T-cell therapy.
Thus far, 11 patients have been treated on this trial. All had relapsed, CD30+ HL at baseline. Six patients are male, and their median age at baseline was 30 (range, 17-69).
The patients had a median of 5 prior treatments (range, 2-9). This included PD-1 inhibitors (n=10), brentuximab vedotin (n=8), and transplant (n=6).
All patients received lymphodepletion with cyclophosphamide at 500 mg/m2 and fludarabine at 30 mg/m2 daily for 3 days. They then received CD30.CAR T-cell therapy at 2×107 cells/m2 or 1×108 cells/m2.
Dr. Ramos noted that CD30.CAR T-cell expansion was dose-dependent and increased by lymphodepleting chemotherapy.
“The peak expansion is much higher [with lymphodepletion], probably in the order of two to three logs higher than what we see without lymphodepleting chemotherapy,” he said. “So chemotherapy makes a difference.”
Increased CD30.CAR T-cell expansion was associated with improved response. Of the nine evaluable patients, six achieved a CR, and three progressed.
Four complete responders were still in CR at last follow-up, one of them for more than a year. However, two complete responders ultimately progressed.
In addition to improved responses, the researchers observed increased toxicity in this trial. Dr. Ramos said some of these toxicities are “probably attributable” to the lymphodepleting chemotherapy.
Toxicities included grade 1 cytokine release syndrome (no tocilizumab required), maculopapular rash, transient cytopenias, nausea, vomiting, and alopecia.
Dr. Ramos said these results suggest adoptive transfer of CD30.CAR T cells is “safe, even with chemotherapy.”
He noted that the duration of response with this treatment is unknown, but trial enrollment and follow-up are ongoing.
RELY-30 was sponsored by Baylor College of Medicine. Dr. Ramos reported relationships with Novartis, Celgene, Bluebird Bio, and Tessa Therapeutics.
*Data in the abstract differ from the presentation.
SAN DIEGO—A phase 1 study suggests lymphodepletion can improve the efficacy of CD30-directed chimeric antigen receptor (CAR) T-cell therapy in patients with Hodgkin lymphoma (HL).
Researchers observed improved responses in HL patients treated with fludarabine and cyclophosphamide prior to CD30.CAR T-cell therapy.
This lymphodepleting regimen was also associated with increased toxicity, compared to no lymphodepletion. However, researchers consider the regimen safe.
Carlos A. Ramos, MD, of Baylor College of Medicine in Houston, Texas, presented these results at the 2018 ASH Annual Meeting (abstract 680*).
Without lymphodepletion
Dr. Ramos first discussed a previous phase 1 trial (NCT01316146), which was published in The Journal of Clinical Investigation in 2017.
In this trial, he and his colleagues had tested CD30.CAR T-cell therapy in patients with relapsed/refractory, CD30+ HL or T-cell non-Hodgkin lymphoma. None of these patients underwent lymphodepletion.
There were no dose-limiting toxicities in this trial—including no neurotoxicity or cytokine release syndrome—but responses were “limited,” according to Dr. Ramos.
Three patients achieved a complete response (CR), three had stable disease, and three progressed.
“Although we saw no significant toxicities and some good clinical responses . . ., the bottom line is that the responses were still quite limited, with several patients having, at most, stable disease or progressive disease,” Dr. Ramos said.
With lymphodepletion
Results from the previous trial prompted Dr. Ramos and his colleagues to conduct the RELY-30 trial (NCT02917083) and investigate whether lymphodepletion would improve responses to CD30.CAR T-cell therapy.
Thus far, 11 patients have been treated on this trial. All had relapsed, CD30+ HL at baseline. Six patients are male, and their median age at baseline was 30 (range, 17-69).
The patients had a median of 5 prior treatments (range, 2-9). This included PD-1 inhibitors (n=10), brentuximab vedotin (n=8), and transplant (n=6).
All patients received lymphodepletion with cyclophosphamide at 500 mg/m2 and fludarabine at 30 mg/m2 daily for 3 days. They then received CD30.CAR T-cell therapy at 2×107 cells/m2 or 1×108 cells/m2.
Dr. Ramos noted that CD30.CAR T-cell expansion was dose-dependent and increased by lymphodepleting chemotherapy.
“The peak expansion is much higher [with lymphodepletion], probably in the order of two to three logs higher than what we see without lymphodepleting chemotherapy,” he said. “So chemotherapy makes a difference.”
Increased CD30.CAR T-cell expansion was associated with improved response. Of the nine evaluable patients, six achieved a CR, and three progressed.
Four complete responders were still in CR at last follow-up, one of them for more than a year. However, two complete responders ultimately progressed.
In addition to improved responses, the researchers observed increased toxicity in this trial. Dr. Ramos said some of these toxicities are “probably attributable” to the lymphodepleting chemotherapy.
Toxicities included grade 1 cytokine release syndrome (no tocilizumab required), maculopapular rash, transient cytopenias, nausea, vomiting, and alopecia.
Dr. Ramos said these results suggest adoptive transfer of CD30.CAR T cells is “safe, even with chemotherapy.”
He noted that the duration of response with this treatment is unknown, but trial enrollment and follow-up are ongoing.
RELY-30 was sponsored by Baylor College of Medicine. Dr. Ramos reported relationships with Novartis, Celgene, Bluebird Bio, and Tessa Therapeutics.
*Data in the abstract differ from the presentation.

Mutation confers resistance to venetoclax in CLL

SAN DIEGO—A recurrent mutation in BCL2, the therapeutic target of venetoclax, appears to be a major contributor to drug resistance in patients with chronic lymphocytic leukemia (CLL), investigators reported.
The mutation has been detected in some patients with CLL up to 2 years before resistance to venetoclax actually develops, according to Piers Blombery, MBBS, of the Peter MacCallum Cancer Center in Melbourne, Victoria, Australia.
“We have identified the first acquired BCL2 mutation developed in patients clinically treated with venetoclax,” he said during the late-breaking abstracts session at the 2018 ASH Annual Meeting.
The mutation, which the investigators have labeled BCL2 Gly101Val, “is a recurrent and frequent mediator of resistance and may be detected years before clinical relapse occurs,” Dr. Blombery added.
A paper on the mutation was published in Cancer Discovery to coincide with the presentation at ASH (abstract LBA-7).
Despite the demonstrated efficacy of venetoclax as continuous therapy in patients with relapsed or refractory CLL, the majority of patients experience disease progression, prompting the investigators to explore molecular mechanisms of secondary resistance.
To do this, they analyzed paired samples from 15 patients with CLL, enrolled in clinical trials of venetoclax, collected both before the start of venetoclax therapy and at the time of disease progression.
In seven patients, the investigators identified a novel mutation that showed up at the time of progression but was absent from the pre-venetoclax samples.
The mutation first became detectable from about 19 to 42 months after the start of therapy and preceded clinical progression by as much as 25 months, the investigators found.
They pinned the mutation down to the BH3-binding groove on BCL2, the same molecular site targeted by venetoclax. They found the mutation was not present in samples from 96 patients with venetoclax-naive CLL nor in any other B-cell malignancies.
Searches for references to the mutation in both a cancer database (COSMIC) and a population database (gnomAD) came up empty.
In other experiments, the investigators determined that cell lines overexpressing BCL2 Gly101Val are resistant to venetoclax, and, in the presence of venetoclax in vitro, BCL2 Gly101Val-expressing cells have a growth advantage compared with wild-type cells.
Additionally, they showed that the mutation results in impaired venetoclax binding in vitro.
“BCL2 Gly101Val is observed subclonally, implicating multiple mechanisms of venetoclax resistance in the same patient,” Dr. Blombery said.
He added that the identification of the resistance mutation is a strong rationale for using combination therapy to treat patients with relapsed or refractory CLL to help prevent or attenuate selection pressures that lead to resistance.
Dr. Blombery reported having no relevant disclosures. The investigators were supported by the Wilson Center for Lymphoma Genomics, Snowdome Foundation, National Health Medical Research Council, Leukemia and Lymphoma Society, Leukemia Foundation, Cancer Council of Victoria, and Australian Cancer Research Foundation.
SAN DIEGO—A recurrent mutation in BCL2, the therapeutic target of venetoclax, appears to be a major contributor to drug resistance in patients with chronic lymphocytic leukemia (CLL), investigators reported.
The mutation has been detected in some patients with CLL up to 2 years before resistance to venetoclax actually develops, according to Piers Blombery, MBBS, of the Peter MacCallum Cancer Center in Melbourne, Victoria, Australia.
“We have identified the first acquired BCL2 mutation developed in patients clinically treated with venetoclax,” he said during the late-breaking abstracts session at the 2018 ASH Annual Meeting.
The mutation, which the investigators have labeled BCL2 Gly101Val, “is a recurrent and frequent mediator of resistance and may be detected years before clinical relapse occurs,” Dr. Blombery added.
A paper on the mutation was published in Cancer Discovery to coincide with the presentation at ASH (abstract LBA-7).
Despite the demonstrated efficacy of venetoclax as continuous therapy in patients with relapsed or refractory CLL, the majority of patients experience disease progression, prompting the investigators to explore molecular mechanisms of secondary resistance.
To do this, they analyzed paired samples from 15 patients with CLL, enrolled in clinical trials of venetoclax, collected both before the start of venetoclax therapy and at the time of disease progression.
In seven patients, the investigators identified a novel mutation that showed up at the time of progression but was absent from the pre-venetoclax samples.
The mutation first became detectable from about 19 to 42 months after the start of therapy and preceded clinical progression by as much as 25 months, the investigators found.
They pinned the mutation down to the BH3-binding groove on BCL2, the same molecular site targeted by venetoclax. They found the mutation was not present in samples from 96 patients with venetoclax-naive CLL nor in any other B-cell malignancies.
Searches for references to the mutation in both a cancer database (COSMIC) and a population database (gnomAD) came up empty.
In other experiments, the investigators determined that cell lines overexpressing BCL2 Gly101Val are resistant to venetoclax, and, in the presence of venetoclax in vitro, BCL2 Gly101Val-expressing cells have a growth advantage compared with wild-type cells.
Additionally, they showed that the mutation results in impaired venetoclax binding in vitro.
“BCL2 Gly101Val is observed subclonally, implicating multiple mechanisms of venetoclax resistance in the same patient,” Dr. Blombery said.
He added that the identification of the resistance mutation is a strong rationale for using combination therapy to treat patients with relapsed or refractory CLL to help prevent or attenuate selection pressures that lead to resistance.
Dr. Blombery reported having no relevant disclosures. The investigators were supported by the Wilson Center for Lymphoma Genomics, Snowdome Foundation, National Health Medical Research Council, Leukemia and Lymphoma Society, Leukemia Foundation, Cancer Council of Victoria, and Australian Cancer Research Foundation.
SAN DIEGO—A recurrent mutation in BCL2, the therapeutic target of venetoclax, appears to be a major contributor to drug resistance in patients with chronic lymphocytic leukemia (CLL), investigators reported.
The mutation has been detected in some patients with CLL up to 2 years before resistance to venetoclax actually develops, according to Piers Blombery, MBBS, of the Peter MacCallum Cancer Center in Melbourne, Victoria, Australia.
“We have identified the first acquired BCL2 mutation developed in patients clinically treated with venetoclax,” he said during the late-breaking abstracts session at the 2018 ASH Annual Meeting.
The mutation, which the investigators have labeled BCL2 Gly101Val, “is a recurrent and frequent mediator of resistance and may be detected years before clinical relapse occurs,” Dr. Blombery added.
A paper on the mutation was published in Cancer Discovery to coincide with the presentation at ASH (abstract LBA-7).
Despite the demonstrated efficacy of venetoclax as continuous therapy in patients with relapsed or refractory CLL, the majority of patients experience disease progression, prompting the investigators to explore molecular mechanisms of secondary resistance.
To do this, they analyzed paired samples from 15 patients with CLL, enrolled in clinical trials of venetoclax, collected both before the start of venetoclax therapy and at the time of disease progression.
In seven patients, the investigators identified a novel mutation that showed up at the time of progression but was absent from the pre-venetoclax samples.
The mutation first became detectable from about 19 to 42 months after the start of therapy and preceded clinical progression by as much as 25 months, the investigators found.
They pinned the mutation down to the BH3-binding groove on BCL2, the same molecular site targeted by venetoclax. They found the mutation was not present in samples from 96 patients with venetoclax-naive CLL nor in any other B-cell malignancies.
Searches for references to the mutation in both a cancer database (COSMIC) and a population database (gnomAD) came up empty.
In other experiments, the investigators determined that cell lines overexpressing BCL2 Gly101Val are resistant to venetoclax, and, in the presence of venetoclax in vitro, BCL2 Gly101Val-expressing cells have a growth advantage compared with wild-type cells.
Additionally, they showed that the mutation results in impaired venetoclax binding in vitro.
“BCL2 Gly101Val is observed subclonally, implicating multiple mechanisms of venetoclax resistance in the same patient,” Dr. Blombery said.
He added that the identification of the resistance mutation is a strong rationale for using combination therapy to treat patients with relapsed or refractory CLL to help prevent or attenuate selection pressures that lead to resistance.
Dr. Blombery reported having no relevant disclosures. The investigators were supported by the Wilson Center for Lymphoma Genomics, Snowdome Foundation, National Health Medical Research Council, Leukemia and Lymphoma Society, Leukemia Foundation, Cancer Council of Victoria, and Australian Cancer Research Foundation.
Intravascular large B-cell lymphoma: an elusive diagnosis with challenging management
Intravascular large B-cell lymphoma (IVBCL) is an aggressive and systemically disseminated disease that affects the elderly, with a median age of diagnosis around 70 years and no gender predilection. It is a rare subtype of extranodal diffuse large B-cell lymphoma (DLBCL) characterized by selective growth of neoplastic cells within blood vessel lumen without any obvious extravascular tumor mass. Hence, an absence of marked lymphadenopathy and heterogeneous clinical presentation make it difficult to diagnose accurately and timely, with roughly half of the cases found postmortem in previous case reports.1,2 The exact incidence of this disease is not known, but more recently, the accuracy of diagnosis of this type of lymphoma has improved with random skin and bone marrow biopsy.1,2 We present here a clinical case of this disease with an atypical presentation followed by a detailed review of its clinical aspects.
Case presentation and summary
A 43-year-old white woman with a history of hypothyroidism and recurrent ovarian cysts presented to clinic with 3 months of loss of appetite, abdominal distension, pelvic pain, and progressive lower-extremity swelling. A physical examination was notable for marked abdominal distension, diffuse lower abdominal tenderness, and pitting lower-extremity edema. No skin rash or any other cutaneous abnormality was noted on exam. Laboratory test results revealed a lactate dehydrogenase (LDH) level of 1652 U/L and a CA-125 level of 50 U/mL (reference range, 0-35 U/mL). No significant beta-human chorionic gonadotropin and alpha-fetoprotein levels were detected. Computed-tomographic (CT) imaging revealed small bilateral pleural effusions and gallbladder wall thickening with abdominal wall edema, but it was otherwise unrevealing. An echocardiogram showed normal cardiac structure and function, with a left ventricular ejection fraction of 60%. No protein was detected in the patient’s urine, and thyroid function tests were unrevealing. Doppler ultrasound studies of her lower extremities and abdomen revealed no thrombosis. Given the patient’s continued pelvic pain, history of ovarian cysts, and elevation in CA-125, she underwent a laparoscopic total abdominal hysterectomy and bilateral salpingoopherectomy.
Histologic examination revealed neoplastic cells involving only the vascular lumina of the cervix, endomyometrium, bilateral fallopian tubes, and bilateral ovaries (Figure 1). Immunohistochemistry stains were positive for CD5, CD20, PAX-5, CD45, BCL-2, and BCL-6 and focally positive for CD10. Peripheral smear showed pseudo-Pelger–Huet cells with 5% atypical lymphoma cells (Figure 2). Complete staging with positron-emission and CT (PET–CT) imaging revealed no metabolic activity, and a bone marrow biopsy showed trilineage hematopoiesis with adequate maturation and less than 5% of the marrow involved with large B-cell lymphoma cells. A diagnosis of IVBCL was made.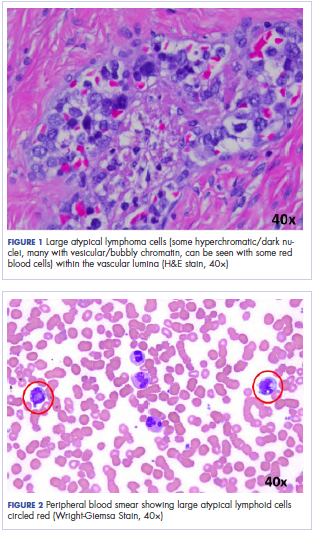
Further work-up to rule out involvement of the central nervous system (CNS) included magnetic-resonance imaging (MRI) of the brain and cerebrospinal fluid (CSF) cytology and flow cytometry, which were negative.
Our patient underwent treatment with 6 cycles of infusional, dose-adjusted R-EPOCH (rituximab, etoposide phosphate, prednisone, vincristine sulfate, cyclophosphamide, doxorubicin hydrochloride) and 6 doses of prophylactic intrathecal chemotherapy with alternating methotrexate and cytarabine (Ara-C), and initial and subsequent CSF sampling showed no disease involvement. Consolidation with high-dose chemotherapy with R-BEAM (rituximab, carmustine, etoposide, Ara-C [cytarabine], melphalan) followed by rescue autologous stem cell transplantation (ASCT) was performed, and the patient has remained in clinical and hematologic remission for the past 24 months.
Discussion
Clinical presentation
The clinical manifestation of this disease is highly variable, and virtually any organ can be involved. Besides causing constitutional symptoms, including fatigue, B symptoms, and decline in performance status, heterogeneity of the clinical presentation depends on the organ system involved. One of the exceptional features of this disease is the difference in clinical presentation based on the geographical origin of the patient.2-4
Western-variant IVBCL has a higher frequency of CNS and skin involvement, whereas Asian-variant IVBCL shows preferential involvement of bone marrow with hemophagocytosis, hepatosplenomegaly, and thrombocytopenia. However, these 2 clinical variants have no difference in clinical outcome, except with the cutaneous-variant kind.24 A retrospective case series of 38 Western-variant IVBCL cases showed that 55% of patients had B symptoms with poor performance status.3 Brain and skin were the organs that were most frequently involved, with 68% of patients having involvement of at least 1 of those organs. Ten patients in this case series had disease that was exclusively limited to the skin and described as a “cutaneous variant” of IVBCL.3
Similarly, a retrospective case series of 96 cases of Asian-variant IVBCL showed B symptoms in 76% of patients, with predominant bone marrow involvement in 75% of patients, accompanied by hemophagocytosis in 66% and hepatosplenomegaly and anemia/thrombocytopenia in 77% and 84% of the patients, respectively.4 This difference in clinical presentation might have existed as a result of ethnic difference associated with production of inflammatory cytokines, including interferon gamma, tumor necrosis factor-alpha, interlukin-1 beta, and soluble interlukin-2 receptor, with levels of soluble interlukin-2 receptor found to be significantly higher in Asian patients than non-Asian patients.2
Diagnosis
Involved organ biopsy is mandatory for establishing the diagnosis of IVBCL. Laboratory findings are nonspecific, with the most common abnormality being increased serum LDH and beta-2 microglobulin levels observed in 80% to 90% or more of patients. Despite its intravascular growth pattern, IVBCL was associated with peripheral blood involvement in only 5% to 9% of patients.1
Staging
Clinical staging work-up suggested for IVBCL patients by International Extranodal lymphoma study group in 2005 included physical examination (with emphasis on nervous system and skin), routine blood studies, peripheral blood smear, total body CT scan with contrast or PET–CT scan, MRI brain with contrast, CSF cytology, and bone marrow or organ biopsy.1 The role of fluorodeoxyglucose-PET scan is controversial but can be helpful to detect unexpected locations for biopsy and to assess treatment response.5,6
Morphology and immunophenotyping
In general, IVBCL histopathology shows large neoplastic lymphoid cells with large nuclei along with one or more nucleoli and scant cytoplasm within blood vessel lumen. Immunophenotypically, IVBCL cells mostly express nongerminal B-cell–associated markers with CD79a (100%), CD20 (96%), MUM-IRF4 (95%), CD5 (38%), and CD10 (12%) expressions. IVBCL cells have been demonstrated to lack cell surface protein CD29 and CD54 critical to transvascular migration. Similarly, aberrant expression of proteins such as CD11a and CXCR3 allows lymphoma cells to be attracted to endothelial cells, which might explain their intravascular confinement.7
Genetics
No pathognomic cytogenetic abnormalities have been reported in IVBCL to date, and the genetic features of this disease are not yet completely understood.2,7
Management
IVBCL is considered a stage IV disseminated disease with an International Prognostic Index score of high-intermediate to high in most cases. Half of the patients with IVBCL who were treated with anthracycline-based chemotherapy relapsed and died within 18 months of diagnosis. One third of the relapses involved the CNS, thereby highlighting the importance of prophylactic CNS-directed Intrathecal therapy in an induction treatment regimen.2-4 Ferreri and colleagues reported in their case series response rates of about 60%, with an overall survival (OS) of 3 years of 30% in patients who were treated with anthracycline-based chemotherapy. A multivariate analysis of the entire series showed cutaneous variant of the disease to be an independent favorable prognostic factor for OS.3
In the Murase and colleagues case series, the authors reported 67% response rates and a median OS of 13 months with CHOP (cyclophosphamide, doxorubicin hydrochloride, vincristine sulfate, prednisone) or CHOP-like regimens. Multivariate analysis showed older age, thrombocytopenia, and absence of anthracycline-based chemotherapy to be an independent negative prognostic factor for OS.4 Another retrospective analysis by Shimada and colleagues of 106 patients with IVBCL showed improved outcome with the addition of rituximab to CHOP-based chemotherapy (R-CHOP). Complete response rate (CR), 2-year progression-free survival, and OS were significantly higher for patients in rituximab-chemotherapy group than for those in the chemotherapy-alone group (CR, 82% vs 51%, respectively, P = .001; PFS, 56% vs 27%; OS, 66% vs 46%, P = .001), thereby establishing rituximab with CHOP-based therapy as induction therapy for IVBCL patients.8
The role of high-dose chemotherapy followed by ASCT could also be used as consolidation therapy to improve clinical outcomes as reported in 7 patients, showing durable remission after transplant in these 2 case series.3,4 Another retrospective analysis of 6 patients with IVBCL who were treated with 6 cycles of R-CHOP as induction therapy and consolidated with ASCT reported all patients to be alive and in complete remission after a median follow-up of 56 months.9 Based on the retrospective case series data by Kato and colleagues and considering that more than 80% of the patients with IVBCL were in the high-risk International Prognostic Index group, ASCT in first remission might be a useful treatment option for durable remission; however, because the median age for the diagnosis of IVBCL is about 70 years, ASCT may not be a realistic option for all patients.
Conclusions
IVBCL is a rare, aggressive, and distinct type of DLBCL with complex constellations of symptoms requiring strong clinical suspicion to establish this challenging diagnosis. Rituximab with anthracycline-based therapy along with prophylactic CNS-directed therapy followed by consolidative ASCT may lead to long-term remission. More research is needed into the genetic features of this disease to better understand its pathogenesis and potential targets for treatment.
1. Ponzoni M, Ferreri AJ, Campo E, et al. Definition, diagnosis, and management of intravascular large B-cell lymphoma: proposals and perspectives from an international consensus meeting. J Clin Oncol. 2007;25(21):3168-3173.
2. Shimada K, Kinoshita T, Naoe T, Nakamura S. Presentation and management of intravascular large B-cell lymphoma. Lancet Oncol. 2009;10(9):895-902.
3. Ferreri AJ, Campo E, Seymour JF, et al. Intravascular lymphoma: clinical presentation, natural history, management and prognostic factors in a series of 38 cases, with special emphasis on the ‘cutaneous variant’. Br J Haematol. 2004;127(2):173-183.
4. Murase T, Yamaguchi M, Suzuki R, et al. Intravascular large B-cell lymphoma (IVLBCL): a clinicopathologic study of 96 cases with special reference to the immunophenotypic heterogeneity of CD5. Blood. 2007;109(2):478-485.
5. Miura Y, Tsudo M. Fluorodeoxyglucose-PET/CT for diagnosis of intravascular large B-cell lymphoma. Mayo Clin Proc. 2010;85(8):e56-e57.
6. Shimada K, Kosugi H, Shimada S, et al. Evaluation of organ involvement in intravascular large B-cell lymphoma by 18F-fluorodeoxyglucose positron emission tomography. Int J Hematol. 2008;88(2):149-153.
7. Orwat DE, Batalis NI. Intravascular large B-cell lymphoma. Arch Pathol Lab Med. 2012;136(3):333-338.
8. Shimada K, Matsue K, Yamamoto K, et al. Retrospective analysis of intravascular large B-cell lymphoma treated with rituximab-containing chemotherapy as reported by the IVL study group in Japan. J Clin Oncol. 2008;26(19):3189-3195.
9. Kato K, Ohno Y, Kamimura T, et al. Long-term remission after high-dose chemotherapy followed by auto-SCT as consolidation for intravascular large B-cell lymphoma. Bone Marrow Transplant. 2014;49(12):1543-1544.
Intravascular large B-cell lymphoma (IVBCL) is an aggressive and systemically disseminated disease that affects the elderly, with a median age of diagnosis around 70 years and no gender predilection. It is a rare subtype of extranodal diffuse large B-cell lymphoma (DLBCL) characterized by selective growth of neoplastic cells within blood vessel lumen without any obvious extravascular tumor mass. Hence, an absence of marked lymphadenopathy and heterogeneous clinical presentation make it difficult to diagnose accurately and timely, with roughly half of the cases found postmortem in previous case reports.1,2 The exact incidence of this disease is not known, but more recently, the accuracy of diagnosis of this type of lymphoma has improved with random skin and bone marrow biopsy.1,2 We present here a clinical case of this disease with an atypical presentation followed by a detailed review of its clinical aspects.
Case presentation and summary
A 43-year-old white woman with a history of hypothyroidism and recurrent ovarian cysts presented to clinic with 3 months of loss of appetite, abdominal distension, pelvic pain, and progressive lower-extremity swelling. A physical examination was notable for marked abdominal distension, diffuse lower abdominal tenderness, and pitting lower-extremity edema. No skin rash or any other cutaneous abnormality was noted on exam. Laboratory test results revealed a lactate dehydrogenase (LDH) level of 1652 U/L and a CA-125 level of 50 U/mL (reference range, 0-35 U/mL). No significant beta-human chorionic gonadotropin and alpha-fetoprotein levels were detected. Computed-tomographic (CT) imaging revealed small bilateral pleural effusions and gallbladder wall thickening with abdominal wall edema, but it was otherwise unrevealing. An echocardiogram showed normal cardiac structure and function, with a left ventricular ejection fraction of 60%. No protein was detected in the patient’s urine, and thyroid function tests were unrevealing. Doppler ultrasound studies of her lower extremities and abdomen revealed no thrombosis. Given the patient’s continued pelvic pain, history of ovarian cysts, and elevation in CA-125, she underwent a laparoscopic total abdominal hysterectomy and bilateral salpingoopherectomy.
Histologic examination revealed neoplastic cells involving only the vascular lumina of the cervix, endomyometrium, bilateral fallopian tubes, and bilateral ovaries (Figure 1). Immunohistochemistry stains were positive for CD5, CD20, PAX-5, CD45, BCL-2, and BCL-6 and focally positive for CD10. Peripheral smear showed pseudo-Pelger–Huet cells with 5% atypical lymphoma cells (Figure 2). Complete staging with positron-emission and CT (PET–CT) imaging revealed no metabolic activity, and a bone marrow biopsy showed trilineage hematopoiesis with adequate maturation and less than 5% of the marrow involved with large B-cell lymphoma cells. A diagnosis of IVBCL was made.
Further work-up to rule out involvement of the central nervous system (CNS) included magnetic-resonance imaging (MRI) of the brain and cerebrospinal fluid (CSF) cytology and flow cytometry, which were negative.
Our patient underwent treatment with 6 cycles of infusional, dose-adjusted R-EPOCH (rituximab, etoposide phosphate, prednisone, vincristine sulfate, cyclophosphamide, doxorubicin hydrochloride) and 6 doses of prophylactic intrathecal chemotherapy with alternating methotrexate and cytarabine (Ara-C), and initial and subsequent CSF sampling showed no disease involvement. Consolidation with high-dose chemotherapy with R-BEAM (rituximab, carmustine, etoposide, Ara-C [cytarabine], melphalan) followed by rescue autologous stem cell transplantation (ASCT) was performed, and the patient has remained in clinical and hematologic remission for the past 24 months.
Discussion
Clinical presentation
The clinical manifestation of this disease is highly variable, and virtually any organ can be involved. Besides causing constitutional symptoms, including fatigue, B symptoms, and decline in performance status, heterogeneity of the clinical presentation depends on the organ system involved. One of the exceptional features of this disease is the difference in clinical presentation based on the geographical origin of the patient.2-4
Western-variant IVBCL has a higher frequency of CNS and skin involvement, whereas Asian-variant IVBCL shows preferential involvement of bone marrow with hemophagocytosis, hepatosplenomegaly, and thrombocytopenia. However, these 2 clinical variants have no difference in clinical outcome, except with the cutaneous-variant kind.24 A retrospective case series of 38 Western-variant IVBCL cases showed that 55% of patients had B symptoms with poor performance status.3 Brain and skin were the organs that were most frequently involved, with 68% of patients having involvement of at least 1 of those organs. Ten patients in this case series had disease that was exclusively limited to the skin and described as a “cutaneous variant” of IVBCL.3
Similarly, a retrospective case series of 96 cases of Asian-variant IVBCL showed B symptoms in 76% of patients, with predominant bone marrow involvement in 75% of patients, accompanied by hemophagocytosis in 66% and hepatosplenomegaly and anemia/thrombocytopenia in 77% and 84% of the patients, respectively.4 This difference in clinical presentation might have existed as a result of ethnic difference associated with production of inflammatory cytokines, including interferon gamma, tumor necrosis factor-alpha, interlukin-1 beta, and soluble interlukin-2 receptor, with levels of soluble interlukin-2 receptor found to be significantly higher in Asian patients than non-Asian patients.2
Diagnosis
Involved organ biopsy is mandatory for establishing the diagnosis of IVBCL. Laboratory findings are nonspecific, with the most common abnormality being increased serum LDH and beta-2 microglobulin levels observed in 80% to 90% or more of patients. Despite its intravascular growth pattern, IVBCL was associated with peripheral blood involvement in only 5% to 9% of patients.1
Staging
Clinical staging work-up suggested for IVBCL patients by International Extranodal lymphoma study group in 2005 included physical examination (with emphasis on nervous system and skin), routine blood studies, peripheral blood smear, total body CT scan with contrast or PET–CT scan, MRI brain with contrast, CSF cytology, and bone marrow or organ biopsy.1 The role of fluorodeoxyglucose-PET scan is controversial but can be helpful to detect unexpected locations for biopsy and to assess treatment response.5,6
Morphology and immunophenotyping
In general, IVBCL histopathology shows large neoplastic lymphoid cells with large nuclei along with one or more nucleoli and scant cytoplasm within blood vessel lumen. Immunophenotypically, IVBCL cells mostly express nongerminal B-cell–associated markers with CD79a (100%), CD20 (96%), MUM-IRF4 (95%), CD5 (38%), and CD10 (12%) expressions. IVBCL cells have been demonstrated to lack cell surface protein CD29 and CD54 critical to transvascular migration. Similarly, aberrant expression of proteins such as CD11a and CXCR3 allows lymphoma cells to be attracted to endothelial cells, which might explain their intravascular confinement.7
Genetics
No pathognomic cytogenetic abnormalities have been reported in IVBCL to date, and the genetic features of this disease are not yet completely understood.2,7
Management
IVBCL is considered a stage IV disseminated disease with an International Prognostic Index score of high-intermediate to high in most cases. Half of the patients with IVBCL who were treated with anthracycline-based chemotherapy relapsed and died within 18 months of diagnosis. One third of the relapses involved the CNS, thereby highlighting the importance of prophylactic CNS-directed Intrathecal therapy in an induction treatment regimen.2-4 Ferreri and colleagues reported in their case series response rates of about 60%, with an overall survival (OS) of 3 years of 30% in patients who were treated with anthracycline-based chemotherapy. A multivariate analysis of the entire series showed cutaneous variant of the disease to be an independent favorable prognostic factor for OS.3
In the Murase and colleagues case series, the authors reported 67% response rates and a median OS of 13 months with CHOP (cyclophosphamide, doxorubicin hydrochloride, vincristine sulfate, prednisone) or CHOP-like regimens. Multivariate analysis showed older age, thrombocytopenia, and absence of anthracycline-based chemotherapy to be an independent negative prognostic factor for OS.4 Another retrospective analysis by Shimada and colleagues of 106 patients with IVBCL showed improved outcome with the addition of rituximab to CHOP-based chemotherapy (R-CHOP). Complete response rate (CR), 2-year progression-free survival, and OS were significantly higher for patients in rituximab-chemotherapy group than for those in the chemotherapy-alone group (CR, 82% vs 51%, respectively, P = .001; PFS, 56% vs 27%; OS, 66% vs 46%, P = .001), thereby establishing rituximab with CHOP-based therapy as induction therapy for IVBCL patients.8
The role of high-dose chemotherapy followed by ASCT could also be used as consolidation therapy to improve clinical outcomes as reported in 7 patients, showing durable remission after transplant in these 2 case series.3,4 Another retrospective analysis of 6 patients with IVBCL who were treated with 6 cycles of R-CHOP as induction therapy and consolidated with ASCT reported all patients to be alive and in complete remission after a median follow-up of 56 months.9 Based on the retrospective case series data by Kato and colleagues and considering that more than 80% of the patients with IVBCL were in the high-risk International Prognostic Index group, ASCT in first remission might be a useful treatment option for durable remission; however, because the median age for the diagnosis of IVBCL is about 70 years, ASCT may not be a realistic option for all patients.
Conclusions
IVBCL is a rare, aggressive, and distinct type of DLBCL with complex constellations of symptoms requiring strong clinical suspicion to establish this challenging diagnosis. Rituximab with anthracycline-based therapy along with prophylactic CNS-directed therapy followed by consolidative ASCT may lead to long-term remission. More research is needed into the genetic features of this disease to better understand its pathogenesis and potential targets for treatment.
Intravascular large B-cell lymphoma (IVBCL) is an aggressive and systemically disseminated disease that affects the elderly, with a median age of diagnosis around 70 years and no gender predilection. It is a rare subtype of extranodal diffuse large B-cell lymphoma (DLBCL) characterized by selective growth of neoplastic cells within blood vessel lumen without any obvious extravascular tumor mass. Hence, an absence of marked lymphadenopathy and heterogeneous clinical presentation make it difficult to diagnose accurately and timely, with roughly half of the cases found postmortem in previous case reports.1,2 The exact incidence of this disease is not known, but more recently, the accuracy of diagnosis of this type of lymphoma has improved with random skin and bone marrow biopsy.1,2 We present here a clinical case of this disease with an atypical presentation followed by a detailed review of its clinical aspects.
Case presentation and summary
A 43-year-old white woman with a history of hypothyroidism and recurrent ovarian cysts presented to clinic with 3 months of loss of appetite, abdominal distension, pelvic pain, and progressive lower-extremity swelling. A physical examination was notable for marked abdominal distension, diffuse lower abdominal tenderness, and pitting lower-extremity edema. No skin rash or any other cutaneous abnormality was noted on exam. Laboratory test results revealed a lactate dehydrogenase (LDH) level of 1652 U/L and a CA-125 level of 50 U/mL (reference range, 0-35 U/mL). No significant beta-human chorionic gonadotropin and alpha-fetoprotein levels were detected. Computed-tomographic (CT) imaging revealed small bilateral pleural effusions and gallbladder wall thickening with abdominal wall edema, but it was otherwise unrevealing. An echocardiogram showed normal cardiac structure and function, with a left ventricular ejection fraction of 60%. No protein was detected in the patient’s urine, and thyroid function tests were unrevealing. Doppler ultrasound studies of her lower extremities and abdomen revealed no thrombosis. Given the patient’s continued pelvic pain, history of ovarian cysts, and elevation in CA-125, she underwent a laparoscopic total abdominal hysterectomy and bilateral salpingoopherectomy.
Histologic examination revealed neoplastic cells involving only the vascular lumina of the cervix, endomyometrium, bilateral fallopian tubes, and bilateral ovaries (Figure 1). Immunohistochemistry stains were positive for CD5, CD20, PAX-5, CD45, BCL-2, and BCL-6 and focally positive for CD10. Peripheral smear showed pseudo-Pelger–Huet cells with 5% atypical lymphoma cells (Figure 2). Complete staging with positron-emission and CT (PET–CT) imaging revealed no metabolic activity, and a bone marrow biopsy showed trilineage hematopoiesis with adequate maturation and less than 5% of the marrow involved with large B-cell lymphoma cells. A diagnosis of IVBCL was made.
Further work-up to rule out involvement of the central nervous system (CNS) included magnetic-resonance imaging (MRI) of the brain and cerebrospinal fluid (CSF) cytology and flow cytometry, which were negative.
Our patient underwent treatment with 6 cycles of infusional, dose-adjusted R-EPOCH (rituximab, etoposide phosphate, prednisone, vincristine sulfate, cyclophosphamide, doxorubicin hydrochloride) and 6 doses of prophylactic intrathecal chemotherapy with alternating methotrexate and cytarabine (Ara-C), and initial and subsequent CSF sampling showed no disease involvement. Consolidation with high-dose chemotherapy with R-BEAM (rituximab, carmustine, etoposide, Ara-C [cytarabine], melphalan) followed by rescue autologous stem cell transplantation (ASCT) was performed, and the patient has remained in clinical and hematologic remission for the past 24 months.
Discussion
Clinical presentation
The clinical manifestation of this disease is highly variable, and virtually any organ can be involved. Besides causing constitutional symptoms, including fatigue, B symptoms, and decline in performance status, heterogeneity of the clinical presentation depends on the organ system involved. One of the exceptional features of this disease is the difference in clinical presentation based on the geographical origin of the patient.2-4
Western-variant IVBCL has a higher frequency of CNS and skin involvement, whereas Asian-variant IVBCL shows preferential involvement of bone marrow with hemophagocytosis, hepatosplenomegaly, and thrombocytopenia. However, these 2 clinical variants have no difference in clinical outcome, except with the cutaneous-variant kind.24 A retrospective case series of 38 Western-variant IVBCL cases showed that 55% of patients had B symptoms with poor performance status.3 Brain and skin were the organs that were most frequently involved, with 68% of patients having involvement of at least 1 of those organs. Ten patients in this case series had disease that was exclusively limited to the skin and described as a “cutaneous variant” of IVBCL.3
Similarly, a retrospective case series of 96 cases of Asian-variant IVBCL showed B symptoms in 76% of patients, with predominant bone marrow involvement in 75% of patients, accompanied by hemophagocytosis in 66% and hepatosplenomegaly and anemia/thrombocytopenia in 77% and 84% of the patients, respectively.4 This difference in clinical presentation might have existed as a result of ethnic difference associated with production of inflammatory cytokines, including interferon gamma, tumor necrosis factor-alpha, interlukin-1 beta, and soluble interlukin-2 receptor, with levels of soluble interlukin-2 receptor found to be significantly higher in Asian patients than non-Asian patients.2
Diagnosis
Involved organ biopsy is mandatory for establishing the diagnosis of IVBCL. Laboratory findings are nonspecific, with the most common abnormality being increased serum LDH and beta-2 microglobulin levels observed in 80% to 90% or more of patients. Despite its intravascular growth pattern, IVBCL was associated with peripheral blood involvement in only 5% to 9% of patients.1
Staging
Clinical staging work-up suggested for IVBCL patients by International Extranodal lymphoma study group in 2005 included physical examination (with emphasis on nervous system and skin), routine blood studies, peripheral blood smear, total body CT scan with contrast or PET–CT scan, MRI brain with contrast, CSF cytology, and bone marrow or organ biopsy.1 The role of fluorodeoxyglucose-PET scan is controversial but can be helpful to detect unexpected locations for biopsy and to assess treatment response.5,6
Morphology and immunophenotyping
In general, IVBCL histopathology shows large neoplastic lymphoid cells with large nuclei along with one or more nucleoli and scant cytoplasm within blood vessel lumen. Immunophenotypically, IVBCL cells mostly express nongerminal B-cell–associated markers with CD79a (100%), CD20 (96%), MUM-IRF4 (95%), CD5 (38%), and CD10 (12%) expressions. IVBCL cells have been demonstrated to lack cell surface protein CD29 and CD54 critical to transvascular migration. Similarly, aberrant expression of proteins such as CD11a and CXCR3 allows lymphoma cells to be attracted to endothelial cells, which might explain their intravascular confinement.7
Genetics
No pathognomic cytogenetic abnormalities have been reported in IVBCL to date, and the genetic features of this disease are not yet completely understood.2,7
Management
IVBCL is considered a stage IV disseminated disease with an International Prognostic Index score of high-intermediate to high in most cases. Half of the patients with IVBCL who were treated with anthracycline-based chemotherapy relapsed and died within 18 months of diagnosis. One third of the relapses involved the CNS, thereby highlighting the importance of prophylactic CNS-directed Intrathecal therapy in an induction treatment regimen.2-4 Ferreri and colleagues reported in their case series response rates of about 60%, with an overall survival (OS) of 3 years of 30% in patients who were treated with anthracycline-based chemotherapy. A multivariate analysis of the entire series showed cutaneous variant of the disease to be an independent favorable prognostic factor for OS.3
In the Murase and colleagues case series, the authors reported 67% response rates and a median OS of 13 months with CHOP (cyclophosphamide, doxorubicin hydrochloride, vincristine sulfate, prednisone) or CHOP-like regimens. Multivariate analysis showed older age, thrombocytopenia, and absence of anthracycline-based chemotherapy to be an independent negative prognostic factor for OS.4 Another retrospective analysis by Shimada and colleagues of 106 patients with IVBCL showed improved outcome with the addition of rituximab to CHOP-based chemotherapy (R-CHOP). Complete response rate (CR), 2-year progression-free survival, and OS were significantly higher for patients in rituximab-chemotherapy group than for those in the chemotherapy-alone group (CR, 82% vs 51%, respectively, P = .001; PFS, 56% vs 27%; OS, 66% vs 46%, P = .001), thereby establishing rituximab with CHOP-based therapy as induction therapy for IVBCL patients.8
The role of high-dose chemotherapy followed by ASCT could also be used as consolidation therapy to improve clinical outcomes as reported in 7 patients, showing durable remission after transplant in these 2 case series.3,4 Another retrospective analysis of 6 patients with IVBCL who were treated with 6 cycles of R-CHOP as induction therapy and consolidated with ASCT reported all patients to be alive and in complete remission after a median follow-up of 56 months.9 Based on the retrospective case series data by Kato and colleagues and considering that more than 80% of the patients with IVBCL were in the high-risk International Prognostic Index group, ASCT in first remission might be a useful treatment option for durable remission; however, because the median age for the diagnosis of IVBCL is about 70 years, ASCT may not be a realistic option for all patients.
Conclusions
IVBCL is a rare, aggressive, and distinct type of DLBCL with complex constellations of symptoms requiring strong clinical suspicion to establish this challenging diagnosis. Rituximab with anthracycline-based therapy along with prophylactic CNS-directed therapy followed by consolidative ASCT may lead to long-term remission. More research is needed into the genetic features of this disease to better understand its pathogenesis and potential targets for treatment.
1. Ponzoni M, Ferreri AJ, Campo E, et al. Definition, diagnosis, and management of intravascular large B-cell lymphoma: proposals and perspectives from an international consensus meeting. J Clin Oncol. 2007;25(21):3168-3173.
2. Shimada K, Kinoshita T, Naoe T, Nakamura S. Presentation and management of intravascular large B-cell lymphoma. Lancet Oncol. 2009;10(9):895-902.
3. Ferreri AJ, Campo E, Seymour JF, et al. Intravascular lymphoma: clinical presentation, natural history, management and prognostic factors in a series of 38 cases, with special emphasis on the ‘cutaneous variant’. Br J Haematol. 2004;127(2):173-183.
4. Murase T, Yamaguchi M, Suzuki R, et al. Intravascular large B-cell lymphoma (IVLBCL): a clinicopathologic study of 96 cases with special reference to the immunophenotypic heterogeneity of CD5. Blood. 2007;109(2):478-485.
5. Miura Y, Tsudo M. Fluorodeoxyglucose-PET/CT for diagnosis of intravascular large B-cell lymphoma. Mayo Clin Proc. 2010;85(8):e56-e57.
6. Shimada K, Kosugi H, Shimada S, et al. Evaluation of organ involvement in intravascular large B-cell lymphoma by 18F-fluorodeoxyglucose positron emission tomography. Int J Hematol. 2008;88(2):149-153.
7. Orwat DE, Batalis NI. Intravascular large B-cell lymphoma. Arch Pathol Lab Med. 2012;136(3):333-338.
8. Shimada K, Matsue K, Yamamoto K, et al. Retrospective analysis of intravascular large B-cell lymphoma treated with rituximab-containing chemotherapy as reported by the IVL study group in Japan. J Clin Oncol. 2008;26(19):3189-3195.
9. Kato K, Ohno Y, Kamimura T, et al. Long-term remission after high-dose chemotherapy followed by auto-SCT as consolidation for intravascular large B-cell lymphoma. Bone Marrow Transplant. 2014;49(12):1543-1544.
1. Ponzoni M, Ferreri AJ, Campo E, et al. Definition, diagnosis, and management of intravascular large B-cell lymphoma: proposals and perspectives from an international consensus meeting. J Clin Oncol. 2007;25(21):3168-3173.
2. Shimada K, Kinoshita T, Naoe T, Nakamura S. Presentation and management of intravascular large B-cell lymphoma. Lancet Oncol. 2009;10(9):895-902.
3. Ferreri AJ, Campo E, Seymour JF, et al. Intravascular lymphoma: clinical presentation, natural history, management and prognostic factors in a series of 38 cases, with special emphasis on the ‘cutaneous variant’. Br J Haematol. 2004;127(2):173-183.
4. Murase T, Yamaguchi M, Suzuki R, et al. Intravascular large B-cell lymphoma (IVLBCL): a clinicopathologic study of 96 cases with special reference to the immunophenotypic heterogeneity of CD5. Blood. 2007;109(2):478-485.
5. Miura Y, Tsudo M. Fluorodeoxyglucose-PET/CT for diagnosis of intravascular large B-cell lymphoma. Mayo Clin Proc. 2010;85(8):e56-e57.
6. Shimada K, Kosugi H, Shimada S, et al. Evaluation of organ involvement in intravascular large B-cell lymphoma by 18F-fluorodeoxyglucose positron emission tomography. Int J Hematol. 2008;88(2):149-153.
7. Orwat DE, Batalis NI. Intravascular large B-cell lymphoma. Arch Pathol Lab Med. 2012;136(3):333-338.
8. Shimada K, Matsue K, Yamamoto K, et al. Retrospective analysis of intravascular large B-cell lymphoma treated with rituximab-containing chemotherapy as reported by the IVL study group in Japan. J Clin Oncol. 2008;26(19):3189-3195.
9. Kato K, Ohno Y, Kamimura T, et al. Long-term remission after high-dose chemotherapy followed by auto-SCT as consolidation for intravascular large B-cell lymphoma. Bone Marrow Transplant. 2014;49(12):1543-1544.
Phase 3 study confirms biosimilarity of PF-05280586 with rituximab
SAN DIEGO – The potential rituximab biosimilar drug PF-05280586 showed efficacy, safety, immunogenicity, pharmacokinetics, and pharmacodynamics similar to those of rituximab at up to 26 weeks in a randomized phase 3 study of treatment-naive patients with CD20-positive low tumor burden follicular lymphoma (LTB-FL).

The primary endpoint of overall response rate at 26 weeks was 75.5% in 196 patients randomized to receive PF-05280586, and 70.7% in 198 patients who received a rituximab reference product sourced from the European Union (MabThera; rituximab‑EU), Jeff Sharman, MD, reported at the annual meeting of the American Society of Hematology.
“This resulted in a difference between the two arms of 4.66%,” said Dr. Sharman of Willamette Valley Cancer Institute and Research Center, Springfield, Ore.
The 95% confidence interval for this difference ... was entirely contained within the prespecified equivalence margin, he said.
“Depth of response was a key secondary endpoint, and rates of complete response were 29.3% and 30.4%, respectively,” he said, noting that rates of partial response, stable response, and progressive disease were also similar between the two study arms.
Estimated 1-year progression-free survival (PFS) rates were also highly similar at 76.4% and 81.2% in the PF-05280586 and rituximab-EU arms.
Rapid depletion in CD19-positive B-cell counts was observed in both groups after initial dosing, with recovery by week 39 and a sustained increase until the end of week 52.
Treatment-emergent adverse events (TEAEs) occurred in 78.6% vs. 72.1% of patients in the PF‑05280586 vs. rituximab‑EU arms, respectively, and the rates of serious adverse events and grade 3 events were similar in the groups, as were rates of infusion interruptions or infusion-related reactions (IRRs), Dr. Sharman said.
IRRs occurred in about 25% of patients in each arm, and most were grade 1 or 2. Grade 3 IRRs occurred in 2.6% vs. 0.5% of patients in the groups, respectively, and no grade 4 IRRs occurred.
Rates of anti-drug antibodies were also similar in the two groups, as were serum drug concentrations – regardless of anti-drug antibody status, he noted.
Study subjects were adults with a mean age of 60 years and histologically confirmed CD20-positive grade 1-3a follicular lymphoma with no prior rituximab or system therapy for B-cell non-Hodgkin lymphoma (NHL). They had Ann Arbor disease stages II (26.9%), III (44.2%) or IV (28.9%), ECOG performance status of 0-1, and at least 1 measurable disease lesion identifiable on imaging.
Risk level as assessed by the Follicular Lymphoma International Prognostic Index–2 was low in 28.4%, medium in 66%, and high in 5.6% of patients.
Treatment with each agent was given at intravenous doses of 375 mg/m2 weekly for 4 weeks at days 1, 8, 15, and 22.
PF-05280586 is being developed by Pfizer, and in this 52-week double-blind study – the largest study to date of the early use of the potential rituximab biosimilar in patients with previously untreated CD20-positive LTB-FL – the primary endpoint was met, demonstrating its therapeutic equivalence with rituximab-EU for overall response rate at week 26, Dr. Sharman said.
“These results therefore confirm the biosimilarity of PF-05280586 with rituximab-EU,” he concluded.
Of note, the reporting of these findings comes on the heels of the first Food and Drug Administration approval of a biosimilar rituximab product for the treatment of NHL; Celltrion’s product Truxima (formerly CT-P10), a biosimilar of Genentech’s Rituxan (rituximab), was approved Nov. 28 to treat adults with CD20-positive, B-cell NHL, either as a single agent or in combination with chemotherapy.
The PF-0528056 study was sponsored by Pfizer. Dr. Sharman has been a consultant for, and/or received research funding and honoraria from Acerta, Pharmacyclics (an AbbVie Company), Pfizer, TG Therapeutics, Abbvie, and Genentech.
SOURCE: Sharman J et al. ASH 2018: Abstract 394.
SAN DIEGO – The potential rituximab biosimilar drug PF-05280586 showed efficacy, safety, immunogenicity, pharmacokinetics, and pharmacodynamics similar to those of rituximab at up to 26 weeks in a randomized phase 3 study of treatment-naive patients with CD20-positive low tumor burden follicular lymphoma (LTB-FL).
The primary endpoint of overall response rate at 26 weeks was 75.5% in 196 patients randomized to receive PF-05280586, and 70.7% in 198 patients who received a rituximab reference product sourced from the European Union (MabThera; rituximab‑EU), Jeff Sharman, MD, reported at the annual meeting of the American Society of Hematology.
“This resulted in a difference between the two arms of 4.66%,” said Dr. Sharman of Willamette Valley Cancer Institute and Research Center, Springfield, Ore.
The 95% confidence interval for this difference ... was entirely contained within the prespecified equivalence margin, he said.
“Depth of response was a key secondary endpoint, and rates of complete response were 29.3% and 30.4%, respectively,” he said, noting that rates of partial response, stable response, and progressive disease were also similar between the two study arms.
Estimated 1-year progression-free survival (PFS) rates were also highly similar at 76.4% and 81.2% in the PF-05280586 and rituximab-EU arms.
Rapid depletion in CD19-positive B-cell counts was observed in both groups after initial dosing, with recovery by week 39 and a sustained increase until the end of week 52.
Treatment-emergent adverse events (TEAEs) occurred in 78.6% vs. 72.1% of patients in the PF‑05280586 vs. rituximab‑EU arms, respectively, and the rates of serious adverse events and grade 3 events were similar in the groups, as were rates of infusion interruptions or infusion-related reactions (IRRs), Dr. Sharman said.
IRRs occurred in about 25% of patients in each arm, and most were grade 1 or 2. Grade 3 IRRs occurred in 2.6% vs. 0.5% of patients in the groups, respectively, and no grade 4 IRRs occurred.
Rates of anti-drug antibodies were also similar in the two groups, as were serum drug concentrations – regardless of anti-drug antibody status, he noted.
Study subjects were adults with a mean age of 60 years and histologically confirmed CD20-positive grade 1-3a follicular lymphoma with no prior rituximab or system therapy for B-cell non-Hodgkin lymphoma (NHL). They had Ann Arbor disease stages II (26.9%), III (44.2%) or IV (28.9%), ECOG performance status of 0-1, and at least 1 measurable disease lesion identifiable on imaging.
Risk level as assessed by the Follicular Lymphoma International Prognostic Index–2 was low in 28.4%, medium in 66%, and high in 5.6% of patients.
Treatment with each agent was given at intravenous doses of 375 mg/m2 weekly for 4 weeks at days 1, 8, 15, and 22.
PF-05280586 is being developed by Pfizer, and in this 52-week double-blind study – the largest study to date of the early use of the potential rituximab biosimilar in patients with previously untreated CD20-positive LTB-FL – the primary endpoint was met, demonstrating its therapeutic equivalence with rituximab-EU for overall response rate at week 26, Dr. Sharman said.
“These results therefore confirm the biosimilarity of PF-05280586 with rituximab-EU,” he concluded.
Of note, the reporting of these findings comes on the heels of the first Food and Drug Administration approval of a biosimilar rituximab product for the treatment of NHL; Celltrion’s product Truxima (formerly CT-P10), a biosimilar of Genentech’s Rituxan (rituximab), was approved Nov. 28 to treat adults with CD20-positive, B-cell NHL, either as a single agent or in combination with chemotherapy.
The PF-0528056 study was sponsored by Pfizer. Dr. Sharman has been a consultant for, and/or received research funding and honoraria from Acerta, Pharmacyclics (an AbbVie Company), Pfizer, TG Therapeutics, Abbvie, and Genentech.
SOURCE: Sharman J et al. ASH 2018: Abstract 394.
SAN DIEGO – The potential rituximab biosimilar drug PF-05280586 showed efficacy, safety, immunogenicity, pharmacokinetics, and pharmacodynamics similar to those of rituximab at up to 26 weeks in a randomized phase 3 study of treatment-naive patients with CD20-positive low tumor burden follicular lymphoma (LTB-FL).
The primary endpoint of overall response rate at 26 weeks was 75.5% in 196 patients randomized to receive PF-05280586, and 70.7% in 198 patients who received a rituximab reference product sourced from the European Union (MabThera; rituximab‑EU), Jeff Sharman, MD, reported at the annual meeting of the American Society of Hematology.
“This resulted in a difference between the two arms of 4.66%,” said Dr. Sharman of Willamette Valley Cancer Institute and Research Center, Springfield, Ore.
The 95% confidence interval for this difference ... was entirely contained within the prespecified equivalence margin, he said.
“Depth of response was a key secondary endpoint, and rates of complete response were 29.3% and 30.4%, respectively,” he said, noting that rates of partial response, stable response, and progressive disease were also similar between the two study arms.
Estimated 1-year progression-free survival (PFS) rates were also highly similar at 76.4% and 81.2% in the PF-05280586 and rituximab-EU arms.
Rapid depletion in CD19-positive B-cell counts was observed in both groups after initial dosing, with recovery by week 39 and a sustained increase until the end of week 52.
Treatment-emergent adverse events (TEAEs) occurred in 78.6% vs. 72.1% of patients in the PF‑05280586 vs. rituximab‑EU arms, respectively, and the rates of serious adverse events and grade 3 events were similar in the groups, as were rates of infusion interruptions or infusion-related reactions (IRRs), Dr. Sharman said.
IRRs occurred in about 25% of patients in each arm, and most were grade 1 or 2. Grade 3 IRRs occurred in 2.6% vs. 0.5% of patients in the groups, respectively, and no grade 4 IRRs occurred.
Rates of anti-drug antibodies were also similar in the two groups, as were serum drug concentrations – regardless of anti-drug antibody status, he noted.
Study subjects were adults with a mean age of 60 years and histologically confirmed CD20-positive grade 1-3a follicular lymphoma with no prior rituximab or system therapy for B-cell non-Hodgkin lymphoma (NHL). They had Ann Arbor disease stages II (26.9%), III (44.2%) or IV (28.9%), ECOG performance status of 0-1, and at least 1 measurable disease lesion identifiable on imaging.
Risk level as assessed by the Follicular Lymphoma International Prognostic Index–2 was low in 28.4%, medium in 66%, and high in 5.6% of patients.
Treatment with each agent was given at intravenous doses of 375 mg/m2 weekly for 4 weeks at days 1, 8, 15, and 22.
PF-05280586 is being developed by Pfizer, and in this 52-week double-blind study – the largest study to date of the early use of the potential rituximab biosimilar in patients with previously untreated CD20-positive LTB-FL – the primary endpoint was met, demonstrating its therapeutic equivalence with rituximab-EU for overall response rate at week 26, Dr. Sharman said.
“These results therefore confirm the biosimilarity of PF-05280586 with rituximab-EU,” he concluded.
Of note, the reporting of these findings comes on the heels of the first Food and Drug Administration approval of a biosimilar rituximab product for the treatment of NHL; Celltrion’s product Truxima (formerly CT-P10), a biosimilar of Genentech’s Rituxan (rituximab), was approved Nov. 28 to treat adults with CD20-positive, B-cell NHL, either as a single agent or in combination with chemotherapy.
The PF-0528056 study was sponsored by Pfizer. Dr. Sharman has been a consultant for, and/or received research funding and honoraria from Acerta, Pharmacyclics (an AbbVie Company), Pfizer, TG Therapeutics, Abbvie, and Genentech.
SOURCE: Sharman J et al. ASH 2018: Abstract 394.
REPORTING FROM ASH 2018
Key clinical point: PF-05280586 shows biosimilarity to rituximab at up to 26 weeks.
Major finding: ORR at 26 weeks was 75.5% vs. 70.7% with PF-05280586 vs. rituximab, respectively.
Study details: A phase 3 study of 394 patients.
Disclosures: This study was sponsored by Pfizer. Dr. Sharman has been a consultant for, and/or received research funding and honoraria from Acerta, Pharmacyclics (an AbbVie Company), Pfizer, TG Therapeutics, Abbvie, and Genentech.
Source: Sharman J et al. ASH 2018: Abstract 394.