User login
Delayed diagnosis of breast cancer: $15M award
Delayed diagnosis of breast cancer: $15M award
A woman in her mid-50s had been seen by a breast surgeon for 16 years for regular mammograms and sonograms. In May 2009, the breast surgeon misinterpreted a mammogram as negative, as did a radiologist who re-read the mammogram weeks later. In December 2010, the patient returned to the breast surgeon with nipple discharge. No further testing was conducted. In October 2011, the patient was found to have Stage IIIA breast cancer involving 4 lymph nodes. She underwent left radical mastectomy, chemotherapy, radiation therapy, and breast reconstruction. At time of trial, the cancer had invaded her vertebrae, was Stage IV, and most likely incurable.
PATIENT'S CLAIM: Although the surgeon admittedly did not possess the qualifications required under the Mammography Quality Standards Act, he interpreted about 5,000 mammograms per year in his office. In this case, he failed to detect a small breast tumor in May 2009. He also failed to perform testing when the patient reported nipple discharge. A more timely diagnosis of breast cancer at Stage I would have provided a 90% chance of long-term survival.
DEFENDANTS' DEFENSE: The defense held the radiologist fully liable because the surgeon was not a qualified interpreter of mammography, therefore relying on the radiologist’s interpretation. The radiologist was legally responsible for the missed diagnosis.
VERDICT: A $15M New York verdict was reached, finding the breast surgeon 75% at fault and the radiologist 25%. The radiologist settled before the trial (the jury was not informed of this). The breast surgeon was responsible for $11.25M. The defense indicated intent to appeal.
Alleged failure to evacuate uterus after cesarean delivery
A 37-year-old woman underwent cesarean delivery (CD) performed by 2 ObGyns. After delivery, she began to hemorrhage and the uterus became atonic. Hysterectomy was performed but the bleeding did not stop. The ObGyns called in 3 other ObGyns. During exploratory laparotomy, the bleeding was halted.
PATIENT'S CLAIM: She and her husband had hoped to have more children but the hysterectomy precluded that. She sued all 5 ObGyns, alleging that the delivering ObGyns failed to properly perform the CD and that each physician failed to properly perform the laparotomy, causing a large scar. The claim was discontinued against the 3 surgical ObGyns; trial addressed the 2 delivering ObGyns.
The patient’s expert ObGyn remarked that the hemorrhage was caused by a small placental remnant that remained in the uterus as a result of inadequate evacuation following delivery. The presence of the remnant was indicated by the uterine atony and should have prompted immediate investigation. The physicians’ notes did not document exploration of the uterus prior to closure.
PHYSICIAN'S DEFENSE: The defense’s expert contended that atony would not be a result of a small remnant of placenta. The patient’s uterus was properly evacuated, the hemorrhage was an unforeseeable complication, and the ObGyns properly addressed the hemorrhage.
VERDICT: A New York defense verdict was returned.
Alleged bowel injury during hysterectomy
Two days after a woman underwent a hysterectomy performed by her ObGyn, she went to the emergency department with increasing pain. Her ObGyn admitted her to the hospital. A general surgeon performed an exploratory laparotomy the next day that revealed an abscess; a 1-cm perforation of the patient’s bowel was surgically repaired. The patient had a difficult recovery. She developed pneumonia and respiratory failure. She underwent multiple repair surgeries for recurrent abscesses and fistulas because the wound was slow to heal.
PATIENT'S CLAIM: The ObGyn’s surgical technique was negligent. He injured the bowel when inserting a trocar and did not identify the injury in a timely manner. The expert witness commented that such an injury can sometimes be a surgical complication, but not in this case: the ObGyn rushed the procedure because he had another patient waiting for CD at another hospital.
PHYSICIAN'S DEFENSE: The ObGyn denied negligence and contended that the trocar used in surgery was too blunt to have caused a perforation. It would have been obvious to the ObGyn during surgery if a perforation had occurred. The perforation developed days after surgery within an abscess.
VERDICT: A Mississippi defense verdict was returned.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Delayed diagnosis of breast cancer: $15M award
A woman in her mid-50s had been seen by a breast surgeon for 16 years for regular mammograms and sonograms. In May 2009, the breast surgeon misinterpreted a mammogram as negative, as did a radiologist who re-read the mammogram weeks later. In December 2010, the patient returned to the breast surgeon with nipple discharge. No further testing was conducted. In October 2011, the patient was found to have Stage IIIA breast cancer involving 4 lymph nodes. She underwent left radical mastectomy, chemotherapy, radiation therapy, and breast reconstruction. At time of trial, the cancer had invaded her vertebrae, was Stage IV, and most likely incurable.
PATIENT'S CLAIM: Although the surgeon admittedly did not possess the qualifications required under the Mammography Quality Standards Act, he interpreted about 5,000 mammograms per year in his office. In this case, he failed to detect a small breast tumor in May 2009. He also failed to perform testing when the patient reported nipple discharge. A more timely diagnosis of breast cancer at Stage I would have provided a 90% chance of long-term survival.
DEFENDANTS' DEFENSE: The defense held the radiologist fully liable because the surgeon was not a qualified interpreter of mammography, therefore relying on the radiologist’s interpretation. The radiologist was legally responsible for the missed diagnosis.
VERDICT: A $15M New York verdict was reached, finding the breast surgeon 75% at fault and the radiologist 25%. The radiologist settled before the trial (the jury was not informed of this). The breast surgeon was responsible for $11.25M. The defense indicated intent to appeal.
Alleged failure to evacuate uterus after cesarean delivery
A 37-year-old woman underwent cesarean delivery (CD) performed by 2 ObGyns. After delivery, she began to hemorrhage and the uterus became atonic. Hysterectomy was performed but the bleeding did not stop. The ObGyns called in 3 other ObGyns. During exploratory laparotomy, the bleeding was halted.
PATIENT'S CLAIM: She and her husband had hoped to have more children but the hysterectomy precluded that. She sued all 5 ObGyns, alleging that the delivering ObGyns failed to properly perform the CD and that each physician failed to properly perform the laparotomy, causing a large scar. The claim was discontinued against the 3 surgical ObGyns; trial addressed the 2 delivering ObGyns.
The patient’s expert ObGyn remarked that the hemorrhage was caused by a small placental remnant that remained in the uterus as a result of inadequate evacuation following delivery. The presence of the remnant was indicated by the uterine atony and should have prompted immediate investigation. The physicians’ notes did not document exploration of the uterus prior to closure.
PHYSICIAN'S DEFENSE: The defense’s expert contended that atony would not be a result of a small remnant of placenta. The patient’s uterus was properly evacuated, the hemorrhage was an unforeseeable complication, and the ObGyns properly addressed the hemorrhage.
VERDICT: A New York defense verdict was returned.
Alleged bowel injury during hysterectomy
Two days after a woman underwent a hysterectomy performed by her ObGyn, she went to the emergency department with increasing pain. Her ObGyn admitted her to the hospital. A general surgeon performed an exploratory laparotomy the next day that revealed an abscess; a 1-cm perforation of the patient’s bowel was surgically repaired. The patient had a difficult recovery. She developed pneumonia and respiratory failure. She underwent multiple repair surgeries for recurrent abscesses and fistulas because the wound was slow to heal.
PATIENT'S CLAIM: The ObGyn’s surgical technique was negligent. He injured the bowel when inserting a trocar and did not identify the injury in a timely manner. The expert witness commented that such an injury can sometimes be a surgical complication, but not in this case: the ObGyn rushed the procedure because he had another patient waiting for CD at another hospital.
PHYSICIAN'S DEFENSE: The ObGyn denied negligence and contended that the trocar used in surgery was too blunt to have caused a perforation. It would have been obvious to the ObGyn during surgery if a perforation had occurred. The perforation developed days after surgery within an abscess.
VERDICT: A Mississippi defense verdict was returned.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Delayed diagnosis of breast cancer: $15M award
A woman in her mid-50s had been seen by a breast surgeon for 16 years for regular mammograms and sonograms. In May 2009, the breast surgeon misinterpreted a mammogram as negative, as did a radiologist who re-read the mammogram weeks later. In December 2010, the patient returned to the breast surgeon with nipple discharge. No further testing was conducted. In October 2011, the patient was found to have Stage IIIA breast cancer involving 4 lymph nodes. She underwent left radical mastectomy, chemotherapy, radiation therapy, and breast reconstruction. At time of trial, the cancer had invaded her vertebrae, was Stage IV, and most likely incurable.
PATIENT'S CLAIM: Although the surgeon admittedly did not possess the qualifications required under the Mammography Quality Standards Act, he interpreted about 5,000 mammograms per year in his office. In this case, he failed to detect a small breast tumor in May 2009. He also failed to perform testing when the patient reported nipple discharge. A more timely diagnosis of breast cancer at Stage I would have provided a 90% chance of long-term survival.
DEFENDANTS' DEFENSE: The defense held the radiologist fully liable because the surgeon was not a qualified interpreter of mammography, therefore relying on the radiologist’s interpretation. The radiologist was legally responsible for the missed diagnosis.
VERDICT: A $15M New York verdict was reached, finding the breast surgeon 75% at fault and the radiologist 25%. The radiologist settled before the trial (the jury was not informed of this). The breast surgeon was responsible for $11.25M. The defense indicated intent to appeal.
Alleged failure to evacuate uterus after cesarean delivery
A 37-year-old woman underwent cesarean delivery (CD) performed by 2 ObGyns. After delivery, she began to hemorrhage and the uterus became atonic. Hysterectomy was performed but the bleeding did not stop. The ObGyns called in 3 other ObGyns. During exploratory laparotomy, the bleeding was halted.
PATIENT'S CLAIM: She and her husband had hoped to have more children but the hysterectomy precluded that. She sued all 5 ObGyns, alleging that the delivering ObGyns failed to properly perform the CD and that each physician failed to properly perform the laparotomy, causing a large scar. The claim was discontinued against the 3 surgical ObGyns; trial addressed the 2 delivering ObGyns.
The patient’s expert ObGyn remarked that the hemorrhage was caused by a small placental remnant that remained in the uterus as a result of inadequate evacuation following delivery. The presence of the remnant was indicated by the uterine atony and should have prompted immediate investigation. The physicians’ notes did not document exploration of the uterus prior to closure.
PHYSICIAN'S DEFENSE: The defense’s expert contended that atony would not be a result of a small remnant of placenta. The patient’s uterus was properly evacuated, the hemorrhage was an unforeseeable complication, and the ObGyns properly addressed the hemorrhage.
VERDICT: A New York defense verdict was returned.
Alleged bowel injury during hysterectomy
Two days after a woman underwent a hysterectomy performed by her ObGyn, she went to the emergency department with increasing pain. Her ObGyn admitted her to the hospital. A general surgeon performed an exploratory laparotomy the next day that revealed an abscess; a 1-cm perforation of the patient’s bowel was surgically repaired. The patient had a difficult recovery. She developed pneumonia and respiratory failure. She underwent multiple repair surgeries for recurrent abscesses and fistulas because the wound was slow to heal.
PATIENT'S CLAIM: The ObGyn’s surgical technique was negligent. He injured the bowel when inserting a trocar and did not identify the injury in a timely manner. The expert witness commented that such an injury can sometimes be a surgical complication, but not in this case: the ObGyn rushed the procedure because he had another patient waiting for CD at another hospital.
PHYSICIAN'S DEFENSE: The ObGyn denied negligence and contended that the trocar used in surgery was too blunt to have caused a perforation. It would have been obvious to the ObGyn during surgery if a perforation had occurred. The perforation developed days after surgery within an abscess.
VERDICT: A Mississippi defense verdict was returned.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Inherited mutations drive 12% of Nigerian breast cancer
About one in eight Nigerian women with breast cancer has an inherited mutation of the BRCA1, BRCA2, PALB2, or TP53 gene.
A new analysis of the Nigerian Breast Cancer Study confirmed that these inherited mutations drive about 12% of the country’s breast cancer cases. The findings could pave the way for the first large-scale national breast cancer gene screening program, wrote Olufunmilayo I. Olopade, MD, and her colleagues. The report is in the Journal of Clinical Oncology.
“We suggest that genomic sequencing to identify women at extremely high risk of breast cancer could be a highly innovative approach to tailored risk management and life-saving interventions,” wrote Dr. Olopade, director of the Center for Clinical Cancer Genetics at the University of Chicago, and her colleagues. “Nigeria now has data to prioritize the integration of genetic testing into its cancer control plan. Women with an extremely high risk of breast cancer because of mutations in these genes can be identified inexpensively and unambiguously and offered interventions to reduce cancer risk.”
And, since about half of the sisters and daughters of affected women will carry the same mutation, such a screening program could reach far beyond every index patient identified, the investigators noted.
“If these women at very high risk can be identified either through their relatives with breast cancer or in the general population, resources can be focused particularly on their behalf. For as-yet unaffected women at high genetic risk, these resources would be intensive surveillance for early detection of breast cancer and, after childbearing is completed, the possibility of preventive salpingo-oophorectomy. Integrated population screening for cancer for all women is the goal, but focused outreach to women at extremely high risk represents an especially efficient use of resources and an attainable evidence-based global health approach.”
The Nigerian Breast Cancer Study enrolled 1,136 women with invasive breast cancer from 1998 to 2014. These were compared with 997 women without cancer, matched from the same communities. Genetic sequencing searched for mutations in both known and breast cancer genes.
Cases and controls were a mean of 47 years old; only 6% of cases reported a family history of breast cancer. Of 577 patients with information on tumor stage, 86% (497) were diagnosed at stage III (241) or IV (256).
Among the cases, 167 (14.7%) carried a mutation in a breast cancer risk gene, compared with 1.8% of controls. BRCA1 was the most common mutation, occurring in 7% of patients; these women were 23 times more likely to develop breast cancer than were those without the gene (odds ratio, 23.4). BRCA2 was the next most common, occurring in 4% of cases and conferring a nearly 11-fold increased risk (OR, 10.76). PALB2 occurred in 11 cases (1%) and no controls, and TP53 in four cases (0.4%).
Women with the BRCA1 mutation were diagnosed at a significantly younger age than were other patients (42.6 vs. 47.9 years), as were carriers of the TP53 mutation (32.8 vs. 47.6 years).
Ten other genes (ATM, BARD1, BRIP1, CHEK1, CHEK2, GEN1, NBN, RAD51C, RAD51D, and XRCC2) carried a mutation in at least one patient each. “When limited to mutations in the four high-risk genes, 11%-12% of cases in this study carried a loss-of-function variant.”
Dr. Olopade had no financial disclosures.
SOURCE: Olopade et al. J Clin Oncol. 2018 Aug 21. doi: 10.1200/JCO.2018.78.3977.
The findings of the Nigerian Breast Cancer Study make a case for large-scale breast cancer gene screening. But even in a wealthy country with good infrastructure, such a program would be dauntingly complex, Ophira Ginsburg, MD, and Paul Brennan, PhD, wrote in an accompanying editorial.
“Given the estimated 40,983 women in Nigeria younger than age 65 years who will be newly diagnosed with breast cancer in 2030, the estimated mutation carrier frequency for a high-risk gene of 11%-12% translates to approximately 5,000 women with breast cancer each year who might benefit directly from tailored risk-reducing strategies. Moreover, 50% of these women’s sisters and daughters would also stand to benefit,” they wrote.
However, 32 million women would need to be screened to find the 220,000 with one of the mutations – a task that is “clearly beyond the scope of most countries.
“Furthermore, women with pathogenic variants would require intensive follow-up and intervention strategies to reduce their risk of developing breast, ovarian/fallopian tube, and potentially other cancers depending on the gene involved. Importantly, this approach would not address the larger problem of the high breast cancer mortality among the vast majority of women without a pathogenic variant but who make up approximately 85% of the breast cancer burden.”
The World Health Organization recognizes this challenge; the agency doesn’t even recommend mammogram-based population screening unless there is a basic, reliable infrastructure including electricity, quality-assurance measures, referral and recall mechanisms, and monitoring and evaluation frameworks. But WHO does suggest some core elements to guide a country’s comprehensive cancer management strategy, including:
• Considering the whole continuum from prevention to palliation.
• Providing a sustainable strategic plan on the basis of the country’s cancer burden, risk factor prevalence, and the resources available to implement the plan.
• Developing an evidence-based approach generated by population-based cancer registries.
“As many countries improve their cancer systems, investing in human resources, infrastructure, monitoring, and evaluation, it is timely to consider how to evaluate readiness to undertake a population-level cancer genetics intervention and consider the core elements that should be in place to make a substantive effect on cancer mortality.”
Dr. Ginsburg is with the Perlmutter Cancer Center of New York University. Dr. Brennan is with the International Agency for Research on Cancer, Lyon, France.
The findings of the Nigerian Breast Cancer Study make a case for large-scale breast cancer gene screening. But even in a wealthy country with good infrastructure, such a program would be dauntingly complex, Ophira Ginsburg, MD, and Paul Brennan, PhD, wrote in an accompanying editorial.
“Given the estimated 40,983 women in Nigeria younger than age 65 years who will be newly diagnosed with breast cancer in 2030, the estimated mutation carrier frequency for a high-risk gene of 11%-12% translates to approximately 5,000 women with breast cancer each year who might benefit directly from tailored risk-reducing strategies. Moreover, 50% of these women’s sisters and daughters would also stand to benefit,” they wrote.
However, 32 million women would need to be screened to find the 220,000 with one of the mutations – a task that is “clearly beyond the scope of most countries.
“Furthermore, women with pathogenic variants would require intensive follow-up and intervention strategies to reduce their risk of developing breast, ovarian/fallopian tube, and potentially other cancers depending on the gene involved. Importantly, this approach would not address the larger problem of the high breast cancer mortality among the vast majority of women without a pathogenic variant but who make up approximately 85% of the breast cancer burden.”
The World Health Organization recognizes this challenge; the agency doesn’t even recommend mammogram-based population screening unless there is a basic, reliable infrastructure including electricity, quality-assurance measures, referral and recall mechanisms, and monitoring and evaluation frameworks. But WHO does suggest some core elements to guide a country’s comprehensive cancer management strategy, including:
• Considering the whole continuum from prevention to palliation.
• Providing a sustainable strategic plan on the basis of the country’s cancer burden, risk factor prevalence, and the resources available to implement the plan.
• Developing an evidence-based approach generated by population-based cancer registries.
“As many countries improve their cancer systems, investing in human resources, infrastructure, monitoring, and evaluation, it is timely to consider how to evaluate readiness to undertake a population-level cancer genetics intervention and consider the core elements that should be in place to make a substantive effect on cancer mortality.”
Dr. Ginsburg is with the Perlmutter Cancer Center of New York University. Dr. Brennan is with the International Agency for Research on Cancer, Lyon, France.
The findings of the Nigerian Breast Cancer Study make a case for large-scale breast cancer gene screening. But even in a wealthy country with good infrastructure, such a program would be dauntingly complex, Ophira Ginsburg, MD, and Paul Brennan, PhD, wrote in an accompanying editorial.
“Given the estimated 40,983 women in Nigeria younger than age 65 years who will be newly diagnosed with breast cancer in 2030, the estimated mutation carrier frequency for a high-risk gene of 11%-12% translates to approximately 5,000 women with breast cancer each year who might benefit directly from tailored risk-reducing strategies. Moreover, 50% of these women’s sisters and daughters would also stand to benefit,” they wrote.
However, 32 million women would need to be screened to find the 220,000 with one of the mutations – a task that is “clearly beyond the scope of most countries.
“Furthermore, women with pathogenic variants would require intensive follow-up and intervention strategies to reduce their risk of developing breast, ovarian/fallopian tube, and potentially other cancers depending on the gene involved. Importantly, this approach would not address the larger problem of the high breast cancer mortality among the vast majority of women without a pathogenic variant but who make up approximately 85% of the breast cancer burden.”
The World Health Organization recognizes this challenge; the agency doesn’t even recommend mammogram-based population screening unless there is a basic, reliable infrastructure including electricity, quality-assurance measures, referral and recall mechanisms, and monitoring and evaluation frameworks. But WHO does suggest some core elements to guide a country’s comprehensive cancer management strategy, including:
• Considering the whole continuum from prevention to palliation.
• Providing a sustainable strategic plan on the basis of the country’s cancer burden, risk factor prevalence, and the resources available to implement the plan.
• Developing an evidence-based approach generated by population-based cancer registries.
“As many countries improve their cancer systems, investing in human resources, infrastructure, monitoring, and evaluation, it is timely to consider how to evaluate readiness to undertake a population-level cancer genetics intervention and consider the core elements that should be in place to make a substantive effect on cancer mortality.”
Dr. Ginsburg is with the Perlmutter Cancer Center of New York University. Dr. Brennan is with the International Agency for Research on Cancer, Lyon, France.
About one in eight Nigerian women with breast cancer has an inherited mutation of the BRCA1, BRCA2, PALB2, or TP53 gene.
A new analysis of the Nigerian Breast Cancer Study confirmed that these inherited mutations drive about 12% of the country’s breast cancer cases. The findings could pave the way for the first large-scale national breast cancer gene screening program, wrote Olufunmilayo I. Olopade, MD, and her colleagues. The report is in the Journal of Clinical Oncology.
“We suggest that genomic sequencing to identify women at extremely high risk of breast cancer could be a highly innovative approach to tailored risk management and life-saving interventions,” wrote Dr. Olopade, director of the Center for Clinical Cancer Genetics at the University of Chicago, and her colleagues. “Nigeria now has data to prioritize the integration of genetic testing into its cancer control plan. Women with an extremely high risk of breast cancer because of mutations in these genes can be identified inexpensively and unambiguously and offered interventions to reduce cancer risk.”
And, since about half of the sisters and daughters of affected women will carry the same mutation, such a screening program could reach far beyond every index patient identified, the investigators noted.
“If these women at very high risk can be identified either through their relatives with breast cancer or in the general population, resources can be focused particularly on their behalf. For as-yet unaffected women at high genetic risk, these resources would be intensive surveillance for early detection of breast cancer and, after childbearing is completed, the possibility of preventive salpingo-oophorectomy. Integrated population screening for cancer for all women is the goal, but focused outreach to women at extremely high risk represents an especially efficient use of resources and an attainable evidence-based global health approach.”
The Nigerian Breast Cancer Study enrolled 1,136 women with invasive breast cancer from 1998 to 2014. These were compared with 997 women without cancer, matched from the same communities. Genetic sequencing searched for mutations in both known and breast cancer genes.
Cases and controls were a mean of 47 years old; only 6% of cases reported a family history of breast cancer. Of 577 patients with information on tumor stage, 86% (497) were diagnosed at stage III (241) or IV (256).
Among the cases, 167 (14.7%) carried a mutation in a breast cancer risk gene, compared with 1.8% of controls. BRCA1 was the most common mutation, occurring in 7% of patients; these women were 23 times more likely to develop breast cancer than were those without the gene (odds ratio, 23.4). BRCA2 was the next most common, occurring in 4% of cases and conferring a nearly 11-fold increased risk (OR, 10.76). PALB2 occurred in 11 cases (1%) and no controls, and TP53 in four cases (0.4%).
Women with the BRCA1 mutation were diagnosed at a significantly younger age than were other patients (42.6 vs. 47.9 years), as were carriers of the TP53 mutation (32.8 vs. 47.6 years).
Ten other genes (ATM, BARD1, BRIP1, CHEK1, CHEK2, GEN1, NBN, RAD51C, RAD51D, and XRCC2) carried a mutation in at least one patient each. “When limited to mutations in the four high-risk genes, 11%-12% of cases in this study carried a loss-of-function variant.”
Dr. Olopade had no financial disclosures.
SOURCE: Olopade et al. J Clin Oncol. 2018 Aug 21. doi: 10.1200/JCO.2018.78.3977.
About one in eight Nigerian women with breast cancer has an inherited mutation of the BRCA1, BRCA2, PALB2, or TP53 gene.
A new analysis of the Nigerian Breast Cancer Study confirmed that these inherited mutations drive about 12% of the country’s breast cancer cases. The findings could pave the way for the first large-scale national breast cancer gene screening program, wrote Olufunmilayo I. Olopade, MD, and her colleagues. The report is in the Journal of Clinical Oncology.
“We suggest that genomic sequencing to identify women at extremely high risk of breast cancer could be a highly innovative approach to tailored risk management and life-saving interventions,” wrote Dr. Olopade, director of the Center for Clinical Cancer Genetics at the University of Chicago, and her colleagues. “Nigeria now has data to prioritize the integration of genetic testing into its cancer control plan. Women with an extremely high risk of breast cancer because of mutations in these genes can be identified inexpensively and unambiguously and offered interventions to reduce cancer risk.”
And, since about half of the sisters and daughters of affected women will carry the same mutation, such a screening program could reach far beyond every index patient identified, the investigators noted.
“If these women at very high risk can be identified either through their relatives with breast cancer or in the general population, resources can be focused particularly on their behalf. For as-yet unaffected women at high genetic risk, these resources would be intensive surveillance for early detection of breast cancer and, after childbearing is completed, the possibility of preventive salpingo-oophorectomy. Integrated population screening for cancer for all women is the goal, but focused outreach to women at extremely high risk represents an especially efficient use of resources and an attainable evidence-based global health approach.”
The Nigerian Breast Cancer Study enrolled 1,136 women with invasive breast cancer from 1998 to 2014. These were compared with 997 women without cancer, matched from the same communities. Genetic sequencing searched for mutations in both known and breast cancer genes.
Cases and controls were a mean of 47 years old; only 6% of cases reported a family history of breast cancer. Of 577 patients with information on tumor stage, 86% (497) were diagnosed at stage III (241) or IV (256).
Among the cases, 167 (14.7%) carried a mutation in a breast cancer risk gene, compared with 1.8% of controls. BRCA1 was the most common mutation, occurring in 7% of patients; these women were 23 times more likely to develop breast cancer than were those without the gene (odds ratio, 23.4). BRCA2 was the next most common, occurring in 4% of cases and conferring a nearly 11-fold increased risk (OR, 10.76). PALB2 occurred in 11 cases (1%) and no controls, and TP53 in four cases (0.4%).
Women with the BRCA1 mutation were diagnosed at a significantly younger age than were other patients (42.6 vs. 47.9 years), as were carriers of the TP53 mutation (32.8 vs. 47.6 years).
Ten other genes (ATM, BARD1, BRIP1, CHEK1, CHEK2, GEN1, NBN, RAD51C, RAD51D, and XRCC2) carried a mutation in at least one patient each. “When limited to mutations in the four high-risk genes, 11%-12% of cases in this study carried a loss-of-function variant.”
Dr. Olopade had no financial disclosures.
SOURCE: Olopade et al. J Clin Oncol. 2018 Aug 21. doi: 10.1200/JCO.2018.78.3977.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: Loss-of-function mutations in four breast cancer risk genes account for much of the disease among Nigerian women with the disease.
Major finding: Inherited mutations of the BRCA1, BRCA2, PALB2, or TP53 gene account for 12% of breast cancer in Nigerian women.
Study details: The Nigerian Breast Cancer Study comprised 1,136 women with invasive breast cancer and 997 controls.
Disclosures: Dr. Olopade had no financial disclosures. The study was largely funded by the National Institutes of Health and the Susan G Komen Foundation.
Source: Olopade et al. J Clin Oncol. 2018 Aug 21. doi: 10.1200/JCO.2018.78.3977.
Neratinib extends adjuvant treatment of patients with HER2-positive breast cancer
The small-molecule tyrosine kinase inhibitor neratinib is now approved for the extended adjuvant treatment of patients with early-stage HER2 [human epidermal growth factor receptor]-positive breast cancer following postoperative trastuzumab. Trastuzumab is a HER2-targeted monoclonal antibody that has become standard of care in combination with chemotherapy for the treatment of this patient population in which it significantly improves survival. However, disease recurrence will occur in about a quarter of trastuzumab-treated patients owing to the development of resistance.
Neratinib may help overcome trastuzumab resistance thanks to its potent inhibition of the downstream phosphorylation of HER2 and other members of the HER family. Its approval was based on the phase 3 ExteNET trial, in which extended adjuvant treatment with neratinib was compared with placebo among 2,840 patients who remained disease free after 1 year of adjuvant trastuzumab.1
The ExteNET trial was performed at 495 centers in Europe, Asia, Australia, New Zealand, and South America. Patients aged 18 years or older (≥20 years in Japan), with stage 1-3 HER2-positive breast cancer, who completed neoadjuvant and adjuvant trastuzumab therapy up to 1 year before randomization were eligible. Patients also had an Eastern Cooperative Oncology Group Performance Status of 0 or 1 (range, 0-5; 0, fully active, and 5, dead), normal organ function, and a left ventricular ejection fraction within normal institutional range. Patients with clinically significant cardiac, gastrointesintal or psychiatric comorbidities and those who were not able to swallow oral medication were excluded from the study.
Patients randomly received oral neratinib 240 mg per day or matching placebo, and randomization was stratified according to HR status (positive or negative), nodal status (0, 1-3, or ≥4) and trastuzumab-adjuvant regimen (sequentially or concurrently with chemotherapy).
The primary outcome was invasive disease-free survival (iDFS). The 2-year iDFS rate was 93.9% for neratinib, compared with 91.6% for placebo (hazard ratio [HR], 0.66; P < .008). Recently, a 5-year analysis of the ExteNET trial showed that after a median follow-up of 5.2 years, the iDFS rates were 90.2% vs 87.7% (HR, 0.73; P = .0083).2
Adverse events
The most common adverse event (AE) was diarrhea, in 95% of patients, 40% of whom had grade 3 diarrhea, leading to dose reduction in 26% of patients and discontinuation in 16.8% of patients. Serious AEs occurred in 7% of patients in the neratinib and 6% of those in the placebo arms. In the 5-year analysis, there was no evidence of increased risk of long-term toxicity or adverse consequences of neratinib-associated diarrhea. Furthermore, the ongoing, open-label phase 2 CONTROL trial suggests that the occurrence and severity of neratinib-associated diarrhea can be effectively controlled with antidiarrheal prophylaxis, with drugs such as loperamide.3
At the January 2017 cut-off, 137 patients treated with neratinib (240 mg/day) for 1 year had also received treatment with loperamide monotherapy, 64 patients had received loperamide and budesonide, and 10 patients had received loperamide and colestipol. The safety data from the loperamide monotherapy arm were compared with the safety data from the ExteNET trial, which was based in a similar population of patients who did not receive antidiarrheal prophylaxis. The incidence of all-grade diarrhea was 77% vs 95%, respectively, for those who received antidiarrheal prophylaxis in the CONTROL trial compared with those in the ExteNET trial who did not, and the repective rates of grade 3 diarrhea were 31% and 40%. The rate of dose reductions and holds owing to diarrhea were also lower among those who received antidiarrheal prophylaxis, but the rate of discontinuation due to diarrhea was higher in the loperamide-treated cohort.
Warnings and precautions
Neratinib is marketed as Nerlynx by Puma Biotechnology Inc. The prescribing information describes warnings and precautions relating to diarrhea, hepatotoxicity, and embryofetal toxicity. Patients should be monitored for diarrhea and treated with antidiarrheals as needed. Severe diarrhea with dehydration should be treated with fluids and electrolytes as needed, treatment should be interrupted and resumed at a reduced dose. For grade 3/4 diarrhea or diarrhea with complicating features (eg, dehydration, fever, neutropenia), stool cultures should be performed to rule out infectious causes.
Total bilirubin, aspartate and alanine aminotransferase, and alkaline phosphatase levels should be measured before starting treatment, every 3 months during treatment, or as clinically indicated. Neratinib can cause fetal harm, so pregnant women should be advised of the risk to the fetus and patients of reproductive potential should be counseled on the need for effective contraception during treatment and for at least 1 month after the last dose.4
1. Chan A, Delaloge S, Holmes FA, et al. Neratinib after trastuzumab-based adjuvant therapy in patients with HER2-positive breast cancer (ExteNET): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2016;17: 367-377.
2. Martin M, Holmes FA, Ejlertsen B, et al. Neratinib after trastuzumab- based adjuvant therapy in HER2-positive breast cancer (ExteNET): a 5-year analysis of a randomised, double-blind, placebo- controlled, phase 3 trial. Lancet Oncol. 2017;18(12):1688-1700.
3. Ibrahim E, Tripathy D, Wilkinson M, et al. E£ects of adding budesonide or colestipol to loperamide prophylaxis on neratinib-associated diarrhea in patients (pts) with HER2+ early-stage breast cancer (EBC): The CONTROL trial. Cancer Res. 2017; 77(13 supplement): Abstract CT128.
4. Nerlynx (neratinib) tablets, for oral use. Prescribing information. Puma Biotechnology Inc. https://nerlynx.com/pdf/full-prescribinginformation. pdf. Revised July 2017. Accessed November 20th, 2017.
The small-molecule tyrosine kinase inhibitor neratinib is now approved for the extended adjuvant treatment of patients with early-stage HER2 [human epidermal growth factor receptor]-positive breast cancer following postoperative trastuzumab. Trastuzumab is a HER2-targeted monoclonal antibody that has become standard of care in combination with chemotherapy for the treatment of this patient population in which it significantly improves survival. However, disease recurrence will occur in about a quarter of trastuzumab-treated patients owing to the development of resistance.
Neratinib may help overcome trastuzumab resistance thanks to its potent inhibition of the downstream phosphorylation of HER2 and other members of the HER family. Its approval was based on the phase 3 ExteNET trial, in which extended adjuvant treatment with neratinib was compared with placebo among 2,840 patients who remained disease free after 1 year of adjuvant trastuzumab.1
The ExteNET trial was performed at 495 centers in Europe, Asia, Australia, New Zealand, and South America. Patients aged 18 years or older (≥20 years in Japan), with stage 1-3 HER2-positive breast cancer, who completed neoadjuvant and adjuvant trastuzumab therapy up to 1 year before randomization were eligible. Patients also had an Eastern Cooperative Oncology Group Performance Status of 0 or 1 (range, 0-5; 0, fully active, and 5, dead), normal organ function, and a left ventricular ejection fraction within normal institutional range. Patients with clinically significant cardiac, gastrointesintal or psychiatric comorbidities and those who were not able to swallow oral medication were excluded from the study.
Patients randomly received oral neratinib 240 mg per day or matching placebo, and randomization was stratified according to HR status (positive or negative), nodal status (0, 1-3, or ≥4) and trastuzumab-adjuvant regimen (sequentially or concurrently with chemotherapy).
The primary outcome was invasive disease-free survival (iDFS). The 2-year iDFS rate was 93.9% for neratinib, compared with 91.6% for placebo (hazard ratio [HR], 0.66; P < .008). Recently, a 5-year analysis of the ExteNET trial showed that after a median follow-up of 5.2 years, the iDFS rates were 90.2% vs 87.7% (HR, 0.73; P = .0083).2
Adverse events
The most common adverse event (AE) was diarrhea, in 95% of patients, 40% of whom had grade 3 diarrhea, leading to dose reduction in 26% of patients and discontinuation in 16.8% of patients. Serious AEs occurred in 7% of patients in the neratinib and 6% of those in the placebo arms. In the 5-year analysis, there was no evidence of increased risk of long-term toxicity or adverse consequences of neratinib-associated diarrhea. Furthermore, the ongoing, open-label phase 2 CONTROL trial suggests that the occurrence and severity of neratinib-associated diarrhea can be effectively controlled with antidiarrheal prophylaxis, with drugs such as loperamide.3
At the January 2017 cut-off, 137 patients treated with neratinib (240 mg/day) for 1 year had also received treatment with loperamide monotherapy, 64 patients had received loperamide and budesonide, and 10 patients had received loperamide and colestipol. The safety data from the loperamide monotherapy arm were compared with the safety data from the ExteNET trial, which was based in a similar population of patients who did not receive antidiarrheal prophylaxis. The incidence of all-grade diarrhea was 77% vs 95%, respectively, for those who received antidiarrheal prophylaxis in the CONTROL trial compared with those in the ExteNET trial who did not, and the repective rates of grade 3 diarrhea were 31% and 40%. The rate of dose reductions and holds owing to diarrhea were also lower among those who received antidiarrheal prophylaxis, but the rate of discontinuation due to diarrhea was higher in the loperamide-treated cohort.
Warnings and precautions
Neratinib is marketed as Nerlynx by Puma Biotechnology Inc. The prescribing information describes warnings and precautions relating to diarrhea, hepatotoxicity, and embryofetal toxicity. Patients should be monitored for diarrhea and treated with antidiarrheals as needed. Severe diarrhea with dehydration should be treated with fluids and electrolytes as needed, treatment should be interrupted and resumed at a reduced dose. For grade 3/4 diarrhea or diarrhea with complicating features (eg, dehydration, fever, neutropenia), stool cultures should be performed to rule out infectious causes.
Total bilirubin, aspartate and alanine aminotransferase, and alkaline phosphatase levels should be measured before starting treatment, every 3 months during treatment, or as clinically indicated. Neratinib can cause fetal harm, so pregnant women should be advised of the risk to the fetus and patients of reproductive potential should be counseled on the need for effective contraception during treatment and for at least 1 month after the last dose.4
The small-molecule tyrosine kinase inhibitor neratinib is now approved for the extended adjuvant treatment of patients with early-stage HER2 [human epidermal growth factor receptor]-positive breast cancer following postoperative trastuzumab. Trastuzumab is a HER2-targeted monoclonal antibody that has become standard of care in combination with chemotherapy for the treatment of this patient population in which it significantly improves survival. However, disease recurrence will occur in about a quarter of trastuzumab-treated patients owing to the development of resistance.
Neratinib may help overcome trastuzumab resistance thanks to its potent inhibition of the downstream phosphorylation of HER2 and other members of the HER family. Its approval was based on the phase 3 ExteNET trial, in which extended adjuvant treatment with neratinib was compared with placebo among 2,840 patients who remained disease free after 1 year of adjuvant trastuzumab.1
The ExteNET trial was performed at 495 centers in Europe, Asia, Australia, New Zealand, and South America. Patients aged 18 years or older (≥20 years in Japan), with stage 1-3 HER2-positive breast cancer, who completed neoadjuvant and adjuvant trastuzumab therapy up to 1 year before randomization were eligible. Patients also had an Eastern Cooperative Oncology Group Performance Status of 0 or 1 (range, 0-5; 0, fully active, and 5, dead), normal organ function, and a left ventricular ejection fraction within normal institutional range. Patients with clinically significant cardiac, gastrointesintal or psychiatric comorbidities and those who were not able to swallow oral medication were excluded from the study.
Patients randomly received oral neratinib 240 mg per day or matching placebo, and randomization was stratified according to HR status (positive or negative), nodal status (0, 1-3, or ≥4) and trastuzumab-adjuvant regimen (sequentially or concurrently with chemotherapy).
The primary outcome was invasive disease-free survival (iDFS). The 2-year iDFS rate was 93.9% for neratinib, compared with 91.6% for placebo (hazard ratio [HR], 0.66; P < .008). Recently, a 5-year analysis of the ExteNET trial showed that after a median follow-up of 5.2 years, the iDFS rates were 90.2% vs 87.7% (HR, 0.73; P = .0083).2
Adverse events
The most common adverse event (AE) was diarrhea, in 95% of patients, 40% of whom had grade 3 diarrhea, leading to dose reduction in 26% of patients and discontinuation in 16.8% of patients. Serious AEs occurred in 7% of patients in the neratinib and 6% of those in the placebo arms. In the 5-year analysis, there was no evidence of increased risk of long-term toxicity or adverse consequences of neratinib-associated diarrhea. Furthermore, the ongoing, open-label phase 2 CONTROL trial suggests that the occurrence and severity of neratinib-associated diarrhea can be effectively controlled with antidiarrheal prophylaxis, with drugs such as loperamide.3
At the January 2017 cut-off, 137 patients treated with neratinib (240 mg/day) for 1 year had also received treatment with loperamide monotherapy, 64 patients had received loperamide and budesonide, and 10 patients had received loperamide and colestipol. The safety data from the loperamide monotherapy arm were compared with the safety data from the ExteNET trial, which was based in a similar population of patients who did not receive antidiarrheal prophylaxis. The incidence of all-grade diarrhea was 77% vs 95%, respectively, for those who received antidiarrheal prophylaxis in the CONTROL trial compared with those in the ExteNET trial who did not, and the repective rates of grade 3 diarrhea were 31% and 40%. The rate of dose reductions and holds owing to diarrhea were also lower among those who received antidiarrheal prophylaxis, but the rate of discontinuation due to diarrhea was higher in the loperamide-treated cohort.
Warnings and precautions
Neratinib is marketed as Nerlynx by Puma Biotechnology Inc. The prescribing information describes warnings and precautions relating to diarrhea, hepatotoxicity, and embryofetal toxicity. Patients should be monitored for diarrhea and treated with antidiarrheals as needed. Severe diarrhea with dehydration should be treated with fluids and electrolytes as needed, treatment should be interrupted and resumed at a reduced dose. For grade 3/4 diarrhea or diarrhea with complicating features (eg, dehydration, fever, neutropenia), stool cultures should be performed to rule out infectious causes.
Total bilirubin, aspartate and alanine aminotransferase, and alkaline phosphatase levels should be measured before starting treatment, every 3 months during treatment, or as clinically indicated. Neratinib can cause fetal harm, so pregnant women should be advised of the risk to the fetus and patients of reproductive potential should be counseled on the need for effective contraception during treatment and for at least 1 month after the last dose.4
1. Chan A, Delaloge S, Holmes FA, et al. Neratinib after trastuzumab-based adjuvant therapy in patients with HER2-positive breast cancer (ExteNET): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2016;17: 367-377.
2. Martin M, Holmes FA, Ejlertsen B, et al. Neratinib after trastuzumab- based adjuvant therapy in HER2-positive breast cancer (ExteNET): a 5-year analysis of a randomised, double-blind, placebo- controlled, phase 3 trial. Lancet Oncol. 2017;18(12):1688-1700.
3. Ibrahim E, Tripathy D, Wilkinson M, et al. E£ects of adding budesonide or colestipol to loperamide prophylaxis on neratinib-associated diarrhea in patients (pts) with HER2+ early-stage breast cancer (EBC): The CONTROL trial. Cancer Res. 2017; 77(13 supplement): Abstract CT128.
4. Nerlynx (neratinib) tablets, for oral use. Prescribing information. Puma Biotechnology Inc. https://nerlynx.com/pdf/full-prescribinginformation. pdf. Revised July 2017. Accessed November 20th, 2017.
1. Chan A, Delaloge S, Holmes FA, et al. Neratinib after trastuzumab-based adjuvant therapy in patients with HER2-positive breast cancer (ExteNET): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2016;17: 367-377.
2. Martin M, Holmes FA, Ejlertsen B, et al. Neratinib after trastuzumab- based adjuvant therapy in HER2-positive breast cancer (ExteNET): a 5-year analysis of a randomised, double-blind, placebo- controlled, phase 3 trial. Lancet Oncol. 2017;18(12):1688-1700.
3. Ibrahim E, Tripathy D, Wilkinson M, et al. E£ects of adding budesonide or colestipol to loperamide prophylaxis on neratinib-associated diarrhea in patients (pts) with HER2+ early-stage breast cancer (EBC): The CONTROL trial. Cancer Res. 2017; 77(13 supplement): Abstract CT128.
4. Nerlynx (neratinib) tablets, for oral use. Prescribing information. Puma Biotechnology Inc. https://nerlynx.com/pdf/full-prescribinginformation. pdf. Revised July 2017. Accessed November 20th, 2017.
Meeting the potential of immunotherapy: new targets provide rational combinations
The relationship between the immune system and tumors is complex and dynamic, and for immunotherapy to reach its full potential it will likely need to attack on multiple fronts. Here, we discuss some of the latest and most promising developments in the immuno-oncology field designed to build on the successes and address limitations.
The anti-tumor immune response
Cancer is a disease of genomic instability, whereby genetic alterations ranging from a single nucleotide to the whole chromosome level frequently occur. Although cancers derive from a patient’s own tissues, these genetic differences can mark the cancer cell as non-self, triggering an immune response to eliminate these cells.
The first hints of this anti-tumor immunity date back more than a century and a half and sparked the concept of mobilizing the immune system to treat patients.1-3 Although early pioneers achieved little progress in this regard, their efforts provided invaluable insights into the complex and dynamic relationship between a tumor and the immune system that are now translating into real clinical successes.
We now understand that the immune system has a dual role in both restraining and promoting cancer development and have translated this understanding into the theory of cancer immunoediting. Immunoediting has three stages: elimination, wherein the tumor is seemingly destroyed by the innate and adaptive immune response; equilibrium, in which cancer cells that were able to escape elimination are selected for growth; and escape, whereby these resistant cancer cells overwhelm the immune system and develop into a symptomatic lesion.4,5
Immuno-oncologists have also described the cancer immunity cycle to capture the steps that are required for an effective anti-tumor immune response and defects in this cycle form the basis of the most common mechanisms used by cancer cells to subvert the anti-tumor immune response. Much like the cancer hallmarks did for molecularly targeted cancer drugs, the cancer immunity cycle serves as the intellectual framework for cancer immunotherapy.6,7
Exploiting nature’s weapon of mass destruction
Initially, attempts at immunotherapy focused on boosting the immune response using adjuvants and cytokines. The characterization of subtle differences between tumor cells and normal cells led to the development of vaccines and cell-based therapies that exploited these tumor-associated antigens (TAAs).1-6
Despite the approval of a therapeutic vaccine, sipuleucel-T, in 2010 for the treatment of metastatic prostate cancer, in general the success of vaccines has been limited. Marketing authorization for sipuleucel-T was recently withdrawn in Europe, and although it is still available in the United States, it is not widely used because of issues with production and administration. Other vaccines, such as GVAX, which looked particularly promising in early-stage clinical trials, failed to show clinical efficacy in subsequent testing.8,9
Cell-based therapies, such as adoptive cellular therapy (ACT), in which immune cells are removed from the host, primed to attack cancer cells, and then reinfused back into the patient, have focused on T cells because they are the major effectors of the adaptive immune response. Clinical success with the most common approach, tumor-infiltrating lymphocyte (TIL)
Two key techniques have been developed (Figure 1). T-cell receptor (TCR) therapy involves genetically modifying the receptor on the surface of T cells that is responsible for recognizing antigens bound to major histocompatibility complex (MHC) molecules on the surface of antigen-presenting cells (APCs). The TCR can be altered to recognize a specific TAA or modified to improve its antigen recognition and binding capabilities. This type of therapy is limited by the fact that the TCRs need to be genetically matched to the patient’s immune type.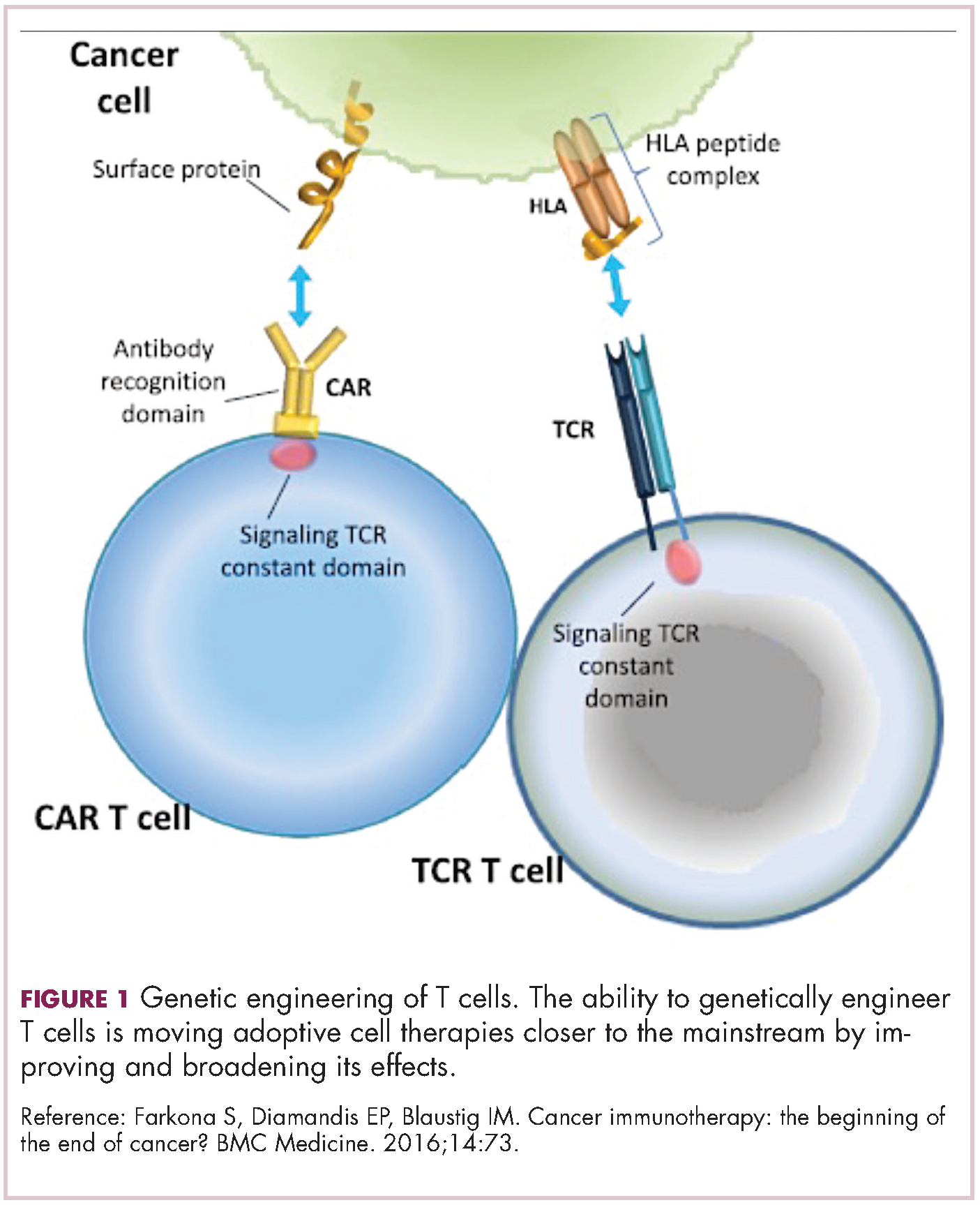
Releasing the brakes
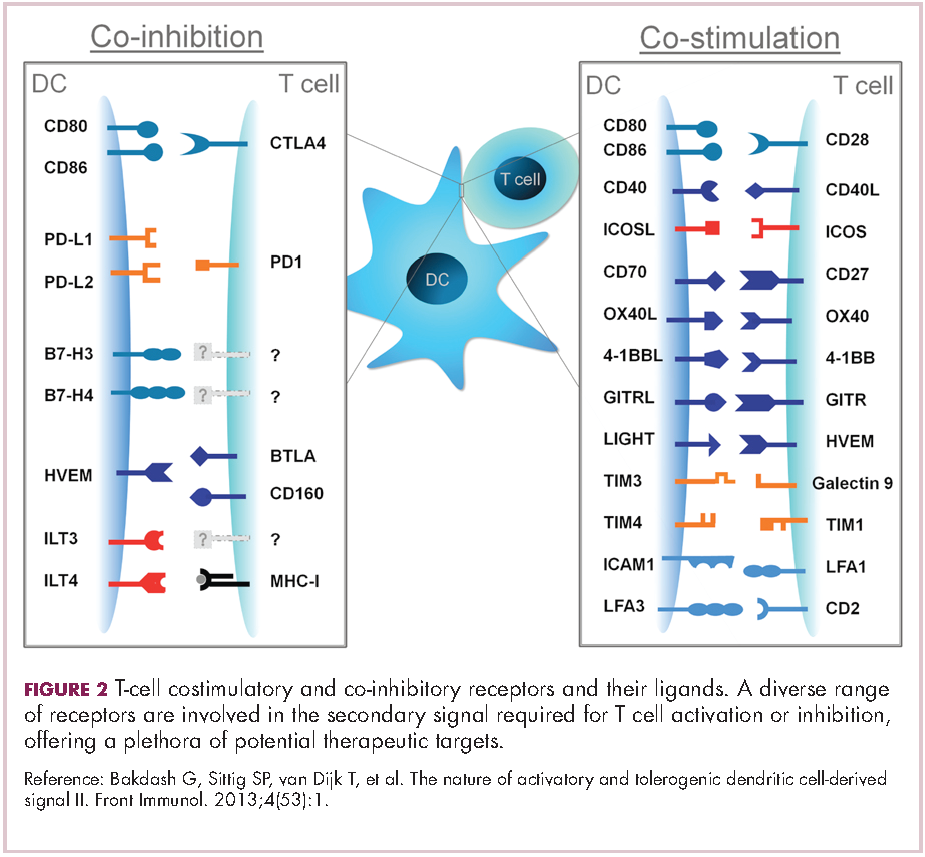
To ensure that it is only activated at the appropriate time and not in response to the antigens expressed on the surface of the host’s own tissues or harmless materials, the immune system has developed numerous mechanisms for immunological tolerance. Cancer cells are able to exploit these mechanisms to allow them to evade the anti-tumor immune response. One of the main ways in which they do this is by manipulating the signaling pathways involved in T-cell activation, which play a vital role in tolerance.12
To become fully activated, T cells require a primary signal generated by an interaction between the TCR and the antigen-MHC complex on the surface of an APC, followed by secondary costimulatory signals generated by a range of different receptors present on the T-cell surface binding to their ligands on the APC.
If the second signal is inhibitory rather than stimulatory, then the T cell is deactivated instead of becoming activated. Two key coinhibitory receptors are programmed cell death 1 (PD-1) and cytotoxic T-lymphocyte antigen 4 (CTLA-4) and tumor cells are able to overcome the anti-tumor immune response in part by expressing the ligands that bind these receptors to dampen the activity of tumor-infiltrating T cells and induce tolerance.13
The development of inhibitors of CTLA-4 and PD-1 and their respective ligands has driven some of the most dramatic successes with cancer immunotherapy, particularly with PD-1-targeting drugs which have fewer side effects. Targeting of this pathway has resulted in durable responses, revolutionizing the treatment of metastatic melanoma, with recently published long-term survival data for pembrolizumab showing that 40% of patients were alive 3 years after initiating treatment and, in a separate study, 34% of nivolumab-treated patients were still alive after 5 years.14,15 More recently, PD-1 inhibitors have been slowly expanding into a range of other cancer types and 4 immune checkpoint inhibitors are now approved by the United States Food and Drug Administration (FDA): ipilimumab (Yervoy), nivolumab (Opdivo), pembrolizumab (Keytruda) and atezolizumab (Tecentriq).
Six years on from the first approval in this drug class and an extensive network of coinhibitory receptors has been uncovered – so-called immune checkpoints – many of which are now also serving as therapeutic targets (Table, Figure 2).16 Lymphocyte activation gene 3 (LAG-3) is a member of the immunoglobulin superfamily of receptors that is expressed on a number of different types of immune cell. In addition to negatively regulating cytotoxic T-cell activation like PD-1 and CTLA-4, it is also thought to regulate the immunosuppressive functions of regulatory T cells and the maturation and activation of dendritic cells. T-cell immunoglobulin and mucin domain-containing 3 (TIM-3) is found on the surface of helper and cytotoxic T cells and regulates T-cell inhibition as well as macrophage activation. Inhibitors of both proteins have been developed that are being evaluated in phase 1 or 2 clinical trials in a variety of tumor types.17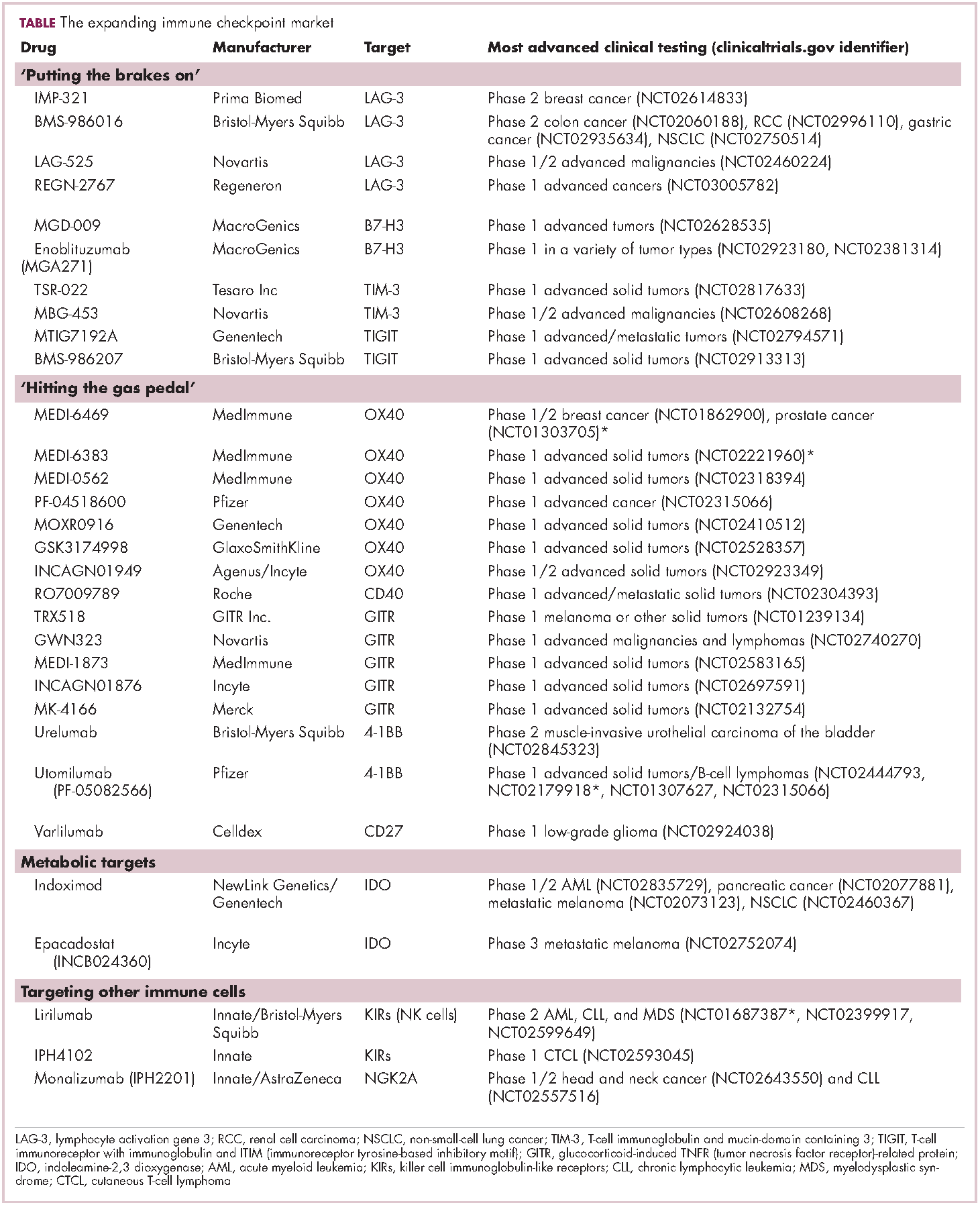
Indeed, although T cells have commanded the most attention, there is growing appreciation of the potential for targeting other types of immune cell that play a role in the anti-tumor immune response or in fostering an immunosuppressive microenvironment. NK cells have been a particular focus, since they represent the body’s first line of immune defense and they appear to have analogous inhibitory and activating receptors expressed on their surface that regulate their cytotoxic activity.
The best-defined NK cell receptors are the killer cell immunoglobulin-like receptors (KIRs) that bind to the MHC class I proteins found on the surface of all cells that distinguish them as ‘self’ or ‘non-self’. KIRs can be either activating or inhibitory, depending upon their structure and the ligands to which they bind.19 To date, 2 antibodies targeting inhibitory KIRs have been developed. Though there has been some disappointment with these drugs, most recently a phase 2 trial of lirilumab in elderly patients with acute myeloid leukemia, which missed its primary endpoint, they continue to be evaluated in clinical trials.20
The inhibitory immune checkpoint field has also expanded to include molecules that regulate T-cell activity in other ways. Most prominently, this includes enzymes like indoleamine-2,3 dioxygenase (IDO), which is involved in the metabolism of the essential amino acid tryptophan. IDO-induced depletion of tryptophan and generation of tryptophan metabolites is toxic to cytotoxic T cells, and IDO is also thought to directly activate regulatory T cells, thus the net effect of IDO is immunosuppression. Two IDO inhibitors are currently being developed.21
Stepping on the gas
Despite their unprecedented success, immune checkpoint inhibitors are not effective in all patients or in all tumor types. Their efficacy is limited in large part by the requirement for a pre-existing anti-tumor immune response. If there are no T cells within the tumor microenvironment then releasing the brakes on the immune system won’t help.
More recently, researchers have returned to the idea of stimulating an anti-tumor immune response, this time by targeting the other side of the immune checkpoint coin, the costimulatory molecules. These drugs could prove more effective as they aren’t reliant on a pre-existing anti-tumor immune response. A number of agonist antibodies designed to target these receptors have now been developed and are undergoing clinical evaluation.22
Furthest along in development are those targeting OX40, a costimulatory molecule that is upregulated on the surface of T cells once they have been fully activated by the TCR signal and an initial costimulatory signal. OX40 is thought to be involved in a more long-term immune response and in the formation of a memory response. A mouse monoclonal antibody had a potent immune-stimulating effect accompanied by the regression of at least 1 metastatic lesion in 30% of patients treated in a phase 1 clinical trial, but was limited by the generation of anti-mouse antibodies. 7 OX40 agonists are now in clinical development, 6 fully human monoclonal antibodies and 1 OX40 ligand-Fc fusion protein, MEDI-6383.23
Combinations are key
Many researchers are now reaching the conclusion that combination therapy is likely to be key in expanding the scope of immunotherapy into currently unresponsive patient populations. Investigating rational combinations is already becoming a burgeoning area of the immuno-oncology field, with a variety of different strategies being tested.
Now the question becomes what are the optimal combinations and the timing and sequencing of combination therapy is likely to be a paramount consideration. Developing combinations that have distinct mechanisms of action or target multiple steps in the cancer immunity cycle offers the greatest potential for therapeutic synergy since this is most likely to address potential mechanisms of resistance by blocking other paths to immune evasion for cancer cells (Figure 3).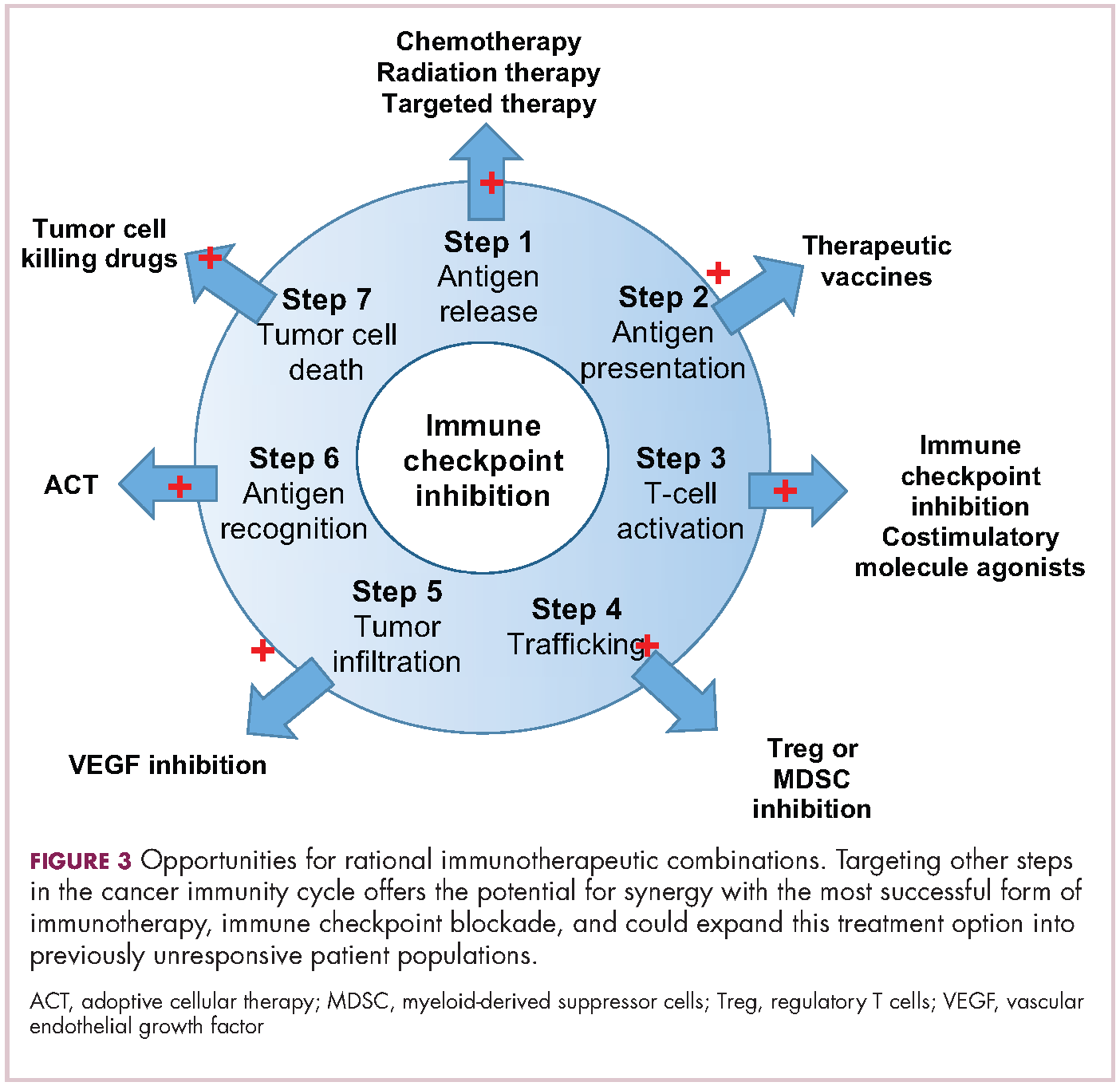
Given the expanding network of immune-checkpoint inhibitors and agonists, the focal point of combination therapy has been combining immune checkpoint-targeting drugs with different mechanisms of action, including those that would simultaneously release the brakes and step on the gas pedal. The vast majority of ongoing clinical trials of approved checkpoint inhibitors and the drugs in development listed in the table are combination trials.
These efforts yielded the first FDA-approved combination immunotherapy regimen in 2015; nivolumab and ipilimumab for the treatment of metastatic melanoma. Approval was based on the demonstration of improved ORR, prolonged response duration, and improved progression-free survival among 142 patients treated with the combination, compared to either drug alone.24
The results of a phase 1/2 trial evaluating the combination of a 4-1BB receptor agonist urelumab with nivolumab in hematologic malignancies and solid tumors found the combination to be safe and particularly effective in patients with advanced/metastatic melanoma, with an ORR of 50%.25 Nivolumab was also combined with the CD27 agonist varlilumab in a phase 1/2 clinical trial of patients with solid tumors, for which data was also recently released. Among 46 patients enrolled, primarily those with colorectal and ovarian cancer the combination had an acceptable safety profile and favorable changes in intratumoral immune biomarkers were observed. The phase 2 portion of the trial is ongoing.26
Meanwhile, Incyte’s IDO inhibitor epacadostat has recently been making waves in combination with pembrolizumab in patients with advanced solid tumors. It demonstrated particularly promising clinical activity in patients with metastatic melanoma, with an overall response rate (ORR) of 57%, including 2 complete responses (CRs), prompting initiation of a phase 3 trial of this combination (NCT02752074).27
1. Adams JL, Smothers J, Srinivasan R, et al. Big opportunities for small molecules in immuno-oncology. Nat Rev Drug Disc. 2015;14:603-622.
2. D’Errico G, Machado HL, Sainz Jr B. A current perspective on cancer immune therapy: step-by-step approach to constructing the magic bullet. Clin Trans Med. 2017;6:3.
3. Farkona S, Diamandis EP, Blaustig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14:73.
4. Meiliana A, Dewi NM, Wijaya A. Cancer immunotherapy: a review. Indones Biomed J. 2016;8(1):1-20.
5. Smyth MJ, Ngiow SF, Ribas A, et al. Combination cancer immunotherapies tailored to the tumor microenvironment. Nat Rev Clin Oncol. 2016;13:143-158.
6. de Charette M, Marabelle A, Houot R. Turning tumor cells into antigen presenting cells: The next step to improve cancer immunotherapy? Eur J Cancer 2016;68:134-147.
7. Chen DS and Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013;39:1-10.
8. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011;480:480-489.
9. Le DT, Wang-Gillam A, Picozzi V Jr, et al. A phase 2, randomized trial of GVAX Pancreas and CRS-207 immunotherapy versus GVAX alone in patients with metastatic pancreatic adenocarcinoma: Updated results. Presented at: the ASCO Gastrointestinal Cancers Symposium; January 16-18, 2014; San Francisco, CA. Abstract 177.
10. Sharpe M and Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
11. Perica K, Varela JC, Oelke M, et al. Adoptive T Cell Immunotherapy for Cancer. Ram Mai Med J. 2015;6(1):e0004.
12. Xing Y and Hogquist KA. T-Cell Tolerance: Central and Peripheral. Cold Spring Harb Perspect Biol. 2012;4:a006957.
13. Buchbinder EI and Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol. 2016;39(1):98-106.
14. Robert C, Ribas A, Hamid O, et al. 3-year overall survival for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. J Clin Oncol. 2016(suppl;abstr 9503).
15. Hodi SF, Kluger HM, Sznol M, et al. Durable, long-term survival in previously treated patients with advanced melanoma who received nivolumab monotherapy in a phase I trial. Presented at the 2016 AACR Annual Meeting; April 16-20; New Orleans, LA. Abstract CT001.
16. Bakdash G, Sittig SP, van Dijk T, et al. The nature of activatory and tolerogenic dendritic cell-derived signal II. Front Immunol. 2013;4(53):1-18.
17. Sheridan C. Immuno-oncology moves beyond PD-1. Nat Biotechnol. 2015;33(7):673-675.
18. Blake SJ, Dougall WC, Miles JJ, et al. Molecular pathways: targeting CD96 and TIGIT for cancer immunotherapy. Clin Cancer Res. 2016;22(21):5183-5188.
19. Carotta S. Targeting NK cells for anticancer immunotherapy: clinical and preclinical approaches. Front Immunol. 2016;7:152.
20. Innate Pharma Web site. Innate Pharma Announces Top-Line Results from EFFIKIR Trial Evaluating the Efficacy of Lirilumab as a Single Agent in Elderly Patients with Acute Myeloid Leukemia. http://www.innate-pharma.com/en/news-events/press-releases/innate-pharma-announces-top-line-results-effikir-trial-evaluating-efficacy-lirilumab-single-agent-elderly-patients-acute-myeloid-leukemia. Last updated February 6, 2017. Accessed online February 22, 2017.
21. Sheridan C. IDO inhibitors move center stage in immuno-oncology. Nat Biotechnol. 2015;33(4):321-322.
22. Sanmamed MF, Pastor F, Rodriguez A, et al. Agonists of co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin Oncol. 2015;42(4):640-655.
23. Linch SN, McNamara MJ, Redmond WL. OX40 agonists and combination immunotherapy: putting the pedal to the metal. Front Oncol. 2015;5:34.
24. U.S. Food and Drug Administration Web site. Nivolumab in combination with ipilimumab. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm465274.htm. Last updated October 1, 2015. Accessed online February 22, 2017.
25. Massarelli E. Clinical safety and efficacy assessment of the CD137 agonist urelumab alone and in combination with nivolumab in patients with hematologic and solid tumor malignancies. Presented at the 31st Annual Meeting of the Society for the Immunotherapy of Cancer; November 9-13, 2016; National Harbor, MD. Abstract 239.
26. Sanborn RE, Pishvain MJ, Callahan MK, et al. Phase I results from the combination of an immune-activating anti-CD27 antibody (varlilumab) in combination with PD-1 blockade (nivolumab): activation across multiple immune pathways without untoward immune-related adverse events. Clin Cancer Res. 2016;76(14):suppl. Abstract CT023.
27. Gangadhar T, Hamid O, Smith D.C, et al. Epacadostat plus pembrolizumab in patients with advanced melanoma and select solid tumors: updated phase 1 results from ECHO-202/KEYNOTE-037. Ann Oncol. 2016;27(6):379-400.
The relationship between the immune system and tumors is complex and dynamic, and for immunotherapy to reach its full potential it will likely need to attack on multiple fronts. Here, we discuss some of the latest and most promising developments in the immuno-oncology field designed to build on the successes and address limitations.
The anti-tumor immune response
Cancer is a disease of genomic instability, whereby genetic alterations ranging from a single nucleotide to the whole chromosome level frequently occur. Although cancers derive from a patient’s own tissues, these genetic differences can mark the cancer cell as non-self, triggering an immune response to eliminate these cells.
The first hints of this anti-tumor immunity date back more than a century and a half and sparked the concept of mobilizing the immune system to treat patients.1-3 Although early pioneers achieved little progress in this regard, their efforts provided invaluable insights into the complex and dynamic relationship between a tumor and the immune system that are now translating into real clinical successes.
We now understand that the immune system has a dual role in both restraining and promoting cancer development and have translated this understanding into the theory of cancer immunoediting. Immunoediting has three stages: elimination, wherein the tumor is seemingly destroyed by the innate and adaptive immune response; equilibrium, in which cancer cells that were able to escape elimination are selected for growth; and escape, whereby these resistant cancer cells overwhelm the immune system and develop into a symptomatic lesion.4,5
Immuno-oncologists have also described the cancer immunity cycle to capture the steps that are required for an effective anti-tumor immune response and defects in this cycle form the basis of the most common mechanisms used by cancer cells to subvert the anti-tumor immune response. Much like the cancer hallmarks did for molecularly targeted cancer drugs, the cancer immunity cycle serves as the intellectual framework for cancer immunotherapy.6,7
Exploiting nature’s weapon of mass destruction
Initially, attempts at immunotherapy focused on boosting the immune response using adjuvants and cytokines. The characterization of subtle differences between tumor cells and normal cells led to the development of vaccines and cell-based therapies that exploited these tumor-associated antigens (TAAs).1-6
Despite the approval of a therapeutic vaccine, sipuleucel-T, in 2010 for the treatment of metastatic prostate cancer, in general the success of vaccines has been limited. Marketing authorization for sipuleucel-T was recently withdrawn in Europe, and although it is still available in the United States, it is not widely used because of issues with production and administration. Other vaccines, such as GVAX, which looked particularly promising in early-stage clinical trials, failed to show clinical efficacy in subsequent testing.8,9
Cell-based therapies, such as adoptive cellular therapy (ACT), in which immune cells are removed from the host, primed to attack cancer cells, and then reinfused back into the patient, have focused on T cells because they are the major effectors of the adaptive immune response. Clinical success with the most common approach, tumor-infiltrating lymphocyte (TIL)
Two key techniques have been developed (Figure 1). T-cell receptor (TCR) therapy involves genetically modifying the receptor on the surface of T cells that is responsible for recognizing antigens bound to major histocompatibility complex (MHC) molecules on the surface of antigen-presenting cells (APCs). The TCR can be altered to recognize a specific TAA or modified to improve its antigen recognition and binding capabilities. This type of therapy is limited by the fact that the TCRs need to be genetically matched to the patient’s immune type.
Releasing the brakes
To ensure that it is only activated at the appropriate time and not in response to the antigens expressed on the surface of the host’s own tissues or harmless materials, the immune system has developed numerous mechanisms for immunological tolerance. Cancer cells are able to exploit these mechanisms to allow them to evade the anti-tumor immune response. One of the main ways in which they do this is by manipulating the signaling pathways involved in T-cell activation, which play a vital role in tolerance.12
To become fully activated, T cells require a primary signal generated by an interaction between the TCR and the antigen-MHC complex on the surface of an APC, followed by secondary costimulatory signals generated by a range of different receptors present on the T-cell surface binding to their ligands on the APC.
If the second signal is inhibitory rather than stimulatory, then the T cell is deactivated instead of becoming activated. Two key coinhibitory receptors are programmed cell death 1 (PD-1) and cytotoxic T-lymphocyte antigen 4 (CTLA-4) and tumor cells are able to overcome the anti-tumor immune response in part by expressing the ligands that bind these receptors to dampen the activity of tumor-infiltrating T cells and induce tolerance.13
The development of inhibitors of CTLA-4 and PD-1 and their respective ligands has driven some of the most dramatic successes with cancer immunotherapy, particularly with PD-1-targeting drugs which have fewer side effects. Targeting of this pathway has resulted in durable responses, revolutionizing the treatment of metastatic melanoma, with recently published long-term survival data for pembrolizumab showing that 40% of patients were alive 3 years after initiating treatment and, in a separate study, 34% of nivolumab-treated patients were still alive after 5 years.14,15 More recently, PD-1 inhibitors have been slowly expanding into a range of other cancer types and 4 immune checkpoint inhibitors are now approved by the United States Food and Drug Administration (FDA): ipilimumab (Yervoy), nivolumab (Opdivo), pembrolizumab (Keytruda) and atezolizumab (Tecentriq).
Six years on from the first approval in this drug class and an extensive network of coinhibitory receptors has been uncovered – so-called immune checkpoints – many of which are now also serving as therapeutic targets (Table, Figure 2).16 Lymphocyte activation gene 3 (LAG-3) is a member of the immunoglobulin superfamily of receptors that is expressed on a number of different types of immune cell. In addition to negatively regulating cytotoxic T-cell activation like PD-1 and CTLA-4, it is also thought to regulate the immunosuppressive functions of regulatory T cells and the maturation and activation of dendritic cells. T-cell immunoglobulin and mucin domain-containing 3 (TIM-3) is found on the surface of helper and cytotoxic T cells and regulates T-cell inhibition as well as macrophage activation. Inhibitors of both proteins have been developed that are being evaluated in phase 1 or 2 clinical trials in a variety of tumor types.17
Indeed, although T cells have commanded the most attention, there is growing appreciation of the potential for targeting other types of immune cell that play a role in the anti-tumor immune response or in fostering an immunosuppressive microenvironment. NK cells have been a particular focus, since they represent the body’s first line of immune defense and they appear to have analogous inhibitory and activating receptors expressed on their surface that regulate their cytotoxic activity.
The best-defined NK cell receptors are the killer cell immunoglobulin-like receptors (KIRs) that bind to the MHC class I proteins found on the surface of all cells that distinguish them as ‘self’ or ‘non-self’. KIRs can be either activating or inhibitory, depending upon their structure and the ligands to which they bind.19 To date, 2 antibodies targeting inhibitory KIRs have been developed. Though there has been some disappointment with these drugs, most recently a phase 2 trial of lirilumab in elderly patients with acute myeloid leukemia, which missed its primary endpoint, they continue to be evaluated in clinical trials.20
The inhibitory immune checkpoint field has also expanded to include molecules that regulate T-cell activity in other ways. Most prominently, this includes enzymes like indoleamine-2,3 dioxygenase (IDO), which is involved in the metabolism of the essential amino acid tryptophan. IDO-induced depletion of tryptophan and generation of tryptophan metabolites is toxic to cytotoxic T cells, and IDO is also thought to directly activate regulatory T cells, thus the net effect of IDO is immunosuppression. Two IDO inhibitors are currently being developed.21
Stepping on the gas
Despite their unprecedented success, immune checkpoint inhibitors are not effective in all patients or in all tumor types. Their efficacy is limited in large part by the requirement for a pre-existing anti-tumor immune response. If there are no T cells within the tumor microenvironment then releasing the brakes on the immune system won’t help.
More recently, researchers have returned to the idea of stimulating an anti-tumor immune response, this time by targeting the other side of the immune checkpoint coin, the costimulatory molecules. These drugs could prove more effective as they aren’t reliant on a pre-existing anti-tumor immune response. A number of agonist antibodies designed to target these receptors have now been developed and are undergoing clinical evaluation.22
Furthest along in development are those targeting OX40, a costimulatory molecule that is upregulated on the surface of T cells once they have been fully activated by the TCR signal and an initial costimulatory signal. OX40 is thought to be involved in a more long-term immune response and in the formation of a memory response. A mouse monoclonal antibody had a potent immune-stimulating effect accompanied by the regression of at least 1 metastatic lesion in 30% of patients treated in a phase 1 clinical trial, but was limited by the generation of anti-mouse antibodies. 7 OX40 agonists are now in clinical development, 6 fully human monoclonal antibodies and 1 OX40 ligand-Fc fusion protein, MEDI-6383.23
Combinations are key
Many researchers are now reaching the conclusion that combination therapy is likely to be key in expanding the scope of immunotherapy into currently unresponsive patient populations. Investigating rational combinations is already becoming a burgeoning area of the immuno-oncology field, with a variety of different strategies being tested.
Now the question becomes what are the optimal combinations and the timing and sequencing of combination therapy is likely to be a paramount consideration. Developing combinations that have distinct mechanisms of action or target multiple steps in the cancer immunity cycle offers the greatest potential for therapeutic synergy since this is most likely to address potential mechanisms of resistance by blocking other paths to immune evasion for cancer cells (Figure 3).
Given the expanding network of immune-checkpoint inhibitors and agonists, the focal point of combination therapy has been combining immune checkpoint-targeting drugs with different mechanisms of action, including those that would simultaneously release the brakes and step on the gas pedal. The vast majority of ongoing clinical trials of approved checkpoint inhibitors and the drugs in development listed in the table are combination trials.
These efforts yielded the first FDA-approved combination immunotherapy regimen in 2015; nivolumab and ipilimumab for the treatment of metastatic melanoma. Approval was based on the demonstration of improved ORR, prolonged response duration, and improved progression-free survival among 142 patients treated with the combination, compared to either drug alone.24
The results of a phase 1/2 trial evaluating the combination of a 4-1BB receptor agonist urelumab with nivolumab in hematologic malignancies and solid tumors found the combination to be safe and particularly effective in patients with advanced/metastatic melanoma, with an ORR of 50%.25 Nivolumab was also combined with the CD27 agonist varlilumab in a phase 1/2 clinical trial of patients with solid tumors, for which data was also recently released. Among 46 patients enrolled, primarily those with colorectal and ovarian cancer the combination had an acceptable safety profile and favorable changes in intratumoral immune biomarkers were observed. The phase 2 portion of the trial is ongoing.26
Meanwhile, Incyte’s IDO inhibitor epacadostat has recently been making waves in combination with pembrolizumab in patients with advanced solid tumors. It demonstrated particularly promising clinical activity in patients with metastatic melanoma, with an overall response rate (ORR) of 57%, including 2 complete responses (CRs), prompting initiation of a phase 3 trial of this combination (NCT02752074).27
The relationship between the immune system and tumors is complex and dynamic, and for immunotherapy to reach its full potential it will likely need to attack on multiple fronts. Here, we discuss some of the latest and most promising developments in the immuno-oncology field designed to build on the successes and address limitations.
The anti-tumor immune response
Cancer is a disease of genomic instability, whereby genetic alterations ranging from a single nucleotide to the whole chromosome level frequently occur. Although cancers derive from a patient’s own tissues, these genetic differences can mark the cancer cell as non-self, triggering an immune response to eliminate these cells.
The first hints of this anti-tumor immunity date back more than a century and a half and sparked the concept of mobilizing the immune system to treat patients.1-3 Although early pioneers achieved little progress in this regard, their efforts provided invaluable insights into the complex and dynamic relationship between a tumor and the immune system that are now translating into real clinical successes.
We now understand that the immune system has a dual role in both restraining and promoting cancer development and have translated this understanding into the theory of cancer immunoediting. Immunoediting has three stages: elimination, wherein the tumor is seemingly destroyed by the innate and adaptive immune response; equilibrium, in which cancer cells that were able to escape elimination are selected for growth; and escape, whereby these resistant cancer cells overwhelm the immune system and develop into a symptomatic lesion.4,5
Immuno-oncologists have also described the cancer immunity cycle to capture the steps that are required for an effective anti-tumor immune response and defects in this cycle form the basis of the most common mechanisms used by cancer cells to subvert the anti-tumor immune response. Much like the cancer hallmarks did for molecularly targeted cancer drugs, the cancer immunity cycle serves as the intellectual framework for cancer immunotherapy.6,7
Exploiting nature’s weapon of mass destruction
Initially, attempts at immunotherapy focused on boosting the immune response using adjuvants and cytokines. The characterization of subtle differences between tumor cells and normal cells led to the development of vaccines and cell-based therapies that exploited these tumor-associated antigens (TAAs).1-6
Despite the approval of a therapeutic vaccine, sipuleucel-T, in 2010 for the treatment of metastatic prostate cancer, in general the success of vaccines has been limited. Marketing authorization for sipuleucel-T was recently withdrawn in Europe, and although it is still available in the United States, it is not widely used because of issues with production and administration. Other vaccines, such as GVAX, which looked particularly promising in early-stage clinical trials, failed to show clinical efficacy in subsequent testing.8,9
Cell-based therapies, such as adoptive cellular therapy (ACT), in which immune cells are removed from the host, primed to attack cancer cells, and then reinfused back into the patient, have focused on T cells because they are the major effectors of the adaptive immune response. Clinical success with the most common approach, tumor-infiltrating lymphocyte (TIL)
Two key techniques have been developed (Figure 1). T-cell receptor (TCR) therapy involves genetically modifying the receptor on the surface of T cells that is responsible for recognizing antigens bound to major histocompatibility complex (MHC) molecules on the surface of antigen-presenting cells (APCs). The TCR can be altered to recognize a specific TAA or modified to improve its antigen recognition and binding capabilities. This type of therapy is limited by the fact that the TCRs need to be genetically matched to the patient’s immune type.
Releasing the brakes
To ensure that it is only activated at the appropriate time and not in response to the antigens expressed on the surface of the host’s own tissues or harmless materials, the immune system has developed numerous mechanisms for immunological tolerance. Cancer cells are able to exploit these mechanisms to allow them to evade the anti-tumor immune response. One of the main ways in which they do this is by manipulating the signaling pathways involved in T-cell activation, which play a vital role in tolerance.12
To become fully activated, T cells require a primary signal generated by an interaction between the TCR and the antigen-MHC complex on the surface of an APC, followed by secondary costimulatory signals generated by a range of different receptors present on the T-cell surface binding to their ligands on the APC.
If the second signal is inhibitory rather than stimulatory, then the T cell is deactivated instead of becoming activated. Two key coinhibitory receptors are programmed cell death 1 (PD-1) and cytotoxic T-lymphocyte antigen 4 (CTLA-4) and tumor cells are able to overcome the anti-tumor immune response in part by expressing the ligands that bind these receptors to dampen the activity of tumor-infiltrating T cells and induce tolerance.13
The development of inhibitors of CTLA-4 and PD-1 and their respective ligands has driven some of the most dramatic successes with cancer immunotherapy, particularly with PD-1-targeting drugs which have fewer side effects. Targeting of this pathway has resulted in durable responses, revolutionizing the treatment of metastatic melanoma, with recently published long-term survival data for pembrolizumab showing that 40% of patients were alive 3 years after initiating treatment and, in a separate study, 34% of nivolumab-treated patients were still alive after 5 years.14,15 More recently, PD-1 inhibitors have been slowly expanding into a range of other cancer types and 4 immune checkpoint inhibitors are now approved by the United States Food and Drug Administration (FDA): ipilimumab (Yervoy), nivolumab (Opdivo), pembrolizumab (Keytruda) and atezolizumab (Tecentriq).
Six years on from the first approval in this drug class and an extensive network of coinhibitory receptors has been uncovered – so-called immune checkpoints – many of which are now also serving as therapeutic targets (Table, Figure 2).16 Lymphocyte activation gene 3 (LAG-3) is a member of the immunoglobulin superfamily of receptors that is expressed on a number of different types of immune cell. In addition to negatively regulating cytotoxic T-cell activation like PD-1 and CTLA-4, it is also thought to regulate the immunosuppressive functions of regulatory T cells and the maturation and activation of dendritic cells. T-cell immunoglobulin and mucin domain-containing 3 (TIM-3) is found on the surface of helper and cytotoxic T cells and regulates T-cell inhibition as well as macrophage activation. Inhibitors of both proteins have been developed that are being evaluated in phase 1 or 2 clinical trials in a variety of tumor types.17
Indeed, although T cells have commanded the most attention, there is growing appreciation of the potential for targeting other types of immune cell that play a role in the anti-tumor immune response or in fostering an immunosuppressive microenvironment. NK cells have been a particular focus, since they represent the body’s first line of immune defense and they appear to have analogous inhibitory and activating receptors expressed on their surface that regulate their cytotoxic activity.
The best-defined NK cell receptors are the killer cell immunoglobulin-like receptors (KIRs) that bind to the MHC class I proteins found on the surface of all cells that distinguish them as ‘self’ or ‘non-self’. KIRs can be either activating or inhibitory, depending upon their structure and the ligands to which they bind.19 To date, 2 antibodies targeting inhibitory KIRs have been developed. Though there has been some disappointment with these drugs, most recently a phase 2 trial of lirilumab in elderly patients with acute myeloid leukemia, which missed its primary endpoint, they continue to be evaluated in clinical trials.20
The inhibitory immune checkpoint field has also expanded to include molecules that regulate T-cell activity in other ways. Most prominently, this includes enzymes like indoleamine-2,3 dioxygenase (IDO), which is involved in the metabolism of the essential amino acid tryptophan. IDO-induced depletion of tryptophan and generation of tryptophan metabolites is toxic to cytotoxic T cells, and IDO is also thought to directly activate regulatory T cells, thus the net effect of IDO is immunosuppression. Two IDO inhibitors are currently being developed.21
Stepping on the gas
Despite their unprecedented success, immune checkpoint inhibitors are not effective in all patients or in all tumor types. Their efficacy is limited in large part by the requirement for a pre-existing anti-tumor immune response. If there are no T cells within the tumor microenvironment then releasing the brakes on the immune system won’t help.
More recently, researchers have returned to the idea of stimulating an anti-tumor immune response, this time by targeting the other side of the immune checkpoint coin, the costimulatory molecules. These drugs could prove more effective as they aren’t reliant on a pre-existing anti-tumor immune response. A number of agonist antibodies designed to target these receptors have now been developed and are undergoing clinical evaluation.22
Furthest along in development are those targeting OX40, a costimulatory molecule that is upregulated on the surface of T cells once they have been fully activated by the TCR signal and an initial costimulatory signal. OX40 is thought to be involved in a more long-term immune response and in the formation of a memory response. A mouse monoclonal antibody had a potent immune-stimulating effect accompanied by the regression of at least 1 metastatic lesion in 30% of patients treated in a phase 1 clinical trial, but was limited by the generation of anti-mouse antibodies. 7 OX40 agonists are now in clinical development, 6 fully human monoclonal antibodies and 1 OX40 ligand-Fc fusion protein, MEDI-6383.23
Combinations are key
Many researchers are now reaching the conclusion that combination therapy is likely to be key in expanding the scope of immunotherapy into currently unresponsive patient populations. Investigating rational combinations is already becoming a burgeoning area of the immuno-oncology field, with a variety of different strategies being tested.
Now the question becomes what are the optimal combinations and the timing and sequencing of combination therapy is likely to be a paramount consideration. Developing combinations that have distinct mechanisms of action or target multiple steps in the cancer immunity cycle offers the greatest potential for therapeutic synergy since this is most likely to address potential mechanisms of resistance by blocking other paths to immune evasion for cancer cells (Figure 3).
Given the expanding network of immune-checkpoint inhibitors and agonists, the focal point of combination therapy has been combining immune checkpoint-targeting drugs with different mechanisms of action, including those that would simultaneously release the brakes and step on the gas pedal. The vast majority of ongoing clinical trials of approved checkpoint inhibitors and the drugs in development listed in the table are combination trials.
These efforts yielded the first FDA-approved combination immunotherapy regimen in 2015; nivolumab and ipilimumab for the treatment of metastatic melanoma. Approval was based on the demonstration of improved ORR, prolonged response duration, and improved progression-free survival among 142 patients treated with the combination, compared to either drug alone.24
The results of a phase 1/2 trial evaluating the combination of a 4-1BB receptor agonist urelumab with nivolumab in hematologic malignancies and solid tumors found the combination to be safe and particularly effective in patients with advanced/metastatic melanoma, with an ORR of 50%.25 Nivolumab was also combined with the CD27 agonist varlilumab in a phase 1/2 clinical trial of patients with solid tumors, for which data was also recently released. Among 46 patients enrolled, primarily those with colorectal and ovarian cancer the combination had an acceptable safety profile and favorable changes in intratumoral immune biomarkers were observed. The phase 2 portion of the trial is ongoing.26
Meanwhile, Incyte’s IDO inhibitor epacadostat has recently been making waves in combination with pembrolizumab in patients with advanced solid tumors. It demonstrated particularly promising clinical activity in patients with metastatic melanoma, with an overall response rate (ORR) of 57%, including 2 complete responses (CRs), prompting initiation of a phase 3 trial of this combination (NCT02752074).27
1. Adams JL, Smothers J, Srinivasan R, et al. Big opportunities for small molecules in immuno-oncology. Nat Rev Drug Disc. 2015;14:603-622.
2. D’Errico G, Machado HL, Sainz Jr B. A current perspective on cancer immune therapy: step-by-step approach to constructing the magic bullet. Clin Trans Med. 2017;6:3.
3. Farkona S, Diamandis EP, Blaustig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14:73.
4. Meiliana A, Dewi NM, Wijaya A. Cancer immunotherapy: a review. Indones Biomed J. 2016;8(1):1-20.
5. Smyth MJ, Ngiow SF, Ribas A, et al. Combination cancer immunotherapies tailored to the tumor microenvironment. Nat Rev Clin Oncol. 2016;13:143-158.
6. de Charette M, Marabelle A, Houot R. Turning tumor cells into antigen presenting cells: The next step to improve cancer immunotherapy? Eur J Cancer 2016;68:134-147.
7. Chen DS and Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013;39:1-10.
8. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011;480:480-489.
9. Le DT, Wang-Gillam A, Picozzi V Jr, et al. A phase 2, randomized trial of GVAX Pancreas and CRS-207 immunotherapy versus GVAX alone in patients with metastatic pancreatic adenocarcinoma: Updated results. Presented at: the ASCO Gastrointestinal Cancers Symposium; January 16-18, 2014; San Francisco, CA. Abstract 177.
10. Sharpe M and Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
11. Perica K, Varela JC, Oelke M, et al. Adoptive T Cell Immunotherapy for Cancer. Ram Mai Med J. 2015;6(1):e0004.
12. Xing Y and Hogquist KA. T-Cell Tolerance: Central and Peripheral. Cold Spring Harb Perspect Biol. 2012;4:a006957.
13. Buchbinder EI and Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol. 2016;39(1):98-106.
14. Robert C, Ribas A, Hamid O, et al. 3-year overall survival for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. J Clin Oncol. 2016(suppl;abstr 9503).
15. Hodi SF, Kluger HM, Sznol M, et al. Durable, long-term survival in previously treated patients with advanced melanoma who received nivolumab monotherapy in a phase I trial. Presented at the 2016 AACR Annual Meeting; April 16-20; New Orleans, LA. Abstract CT001.
16. Bakdash G, Sittig SP, van Dijk T, et al. The nature of activatory and tolerogenic dendritic cell-derived signal II. Front Immunol. 2013;4(53):1-18.
17. Sheridan C. Immuno-oncology moves beyond PD-1. Nat Biotechnol. 2015;33(7):673-675.
18. Blake SJ, Dougall WC, Miles JJ, et al. Molecular pathways: targeting CD96 and TIGIT for cancer immunotherapy. Clin Cancer Res. 2016;22(21):5183-5188.
19. Carotta S. Targeting NK cells for anticancer immunotherapy: clinical and preclinical approaches. Front Immunol. 2016;7:152.
20. Innate Pharma Web site. Innate Pharma Announces Top-Line Results from EFFIKIR Trial Evaluating the Efficacy of Lirilumab as a Single Agent in Elderly Patients with Acute Myeloid Leukemia. http://www.innate-pharma.com/en/news-events/press-releases/innate-pharma-announces-top-line-results-effikir-trial-evaluating-efficacy-lirilumab-single-agent-elderly-patients-acute-myeloid-leukemia. Last updated February 6, 2017. Accessed online February 22, 2017.
21. Sheridan C. IDO inhibitors move center stage in immuno-oncology. Nat Biotechnol. 2015;33(4):321-322.
22. Sanmamed MF, Pastor F, Rodriguez A, et al. Agonists of co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin Oncol. 2015;42(4):640-655.
23. Linch SN, McNamara MJ, Redmond WL. OX40 agonists and combination immunotherapy: putting the pedal to the metal. Front Oncol. 2015;5:34.
24. U.S. Food and Drug Administration Web site. Nivolumab in combination with ipilimumab. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm465274.htm. Last updated October 1, 2015. Accessed online February 22, 2017.
25. Massarelli E. Clinical safety and efficacy assessment of the CD137 agonist urelumab alone and in combination with nivolumab in patients with hematologic and solid tumor malignancies. Presented at the 31st Annual Meeting of the Society for the Immunotherapy of Cancer; November 9-13, 2016; National Harbor, MD. Abstract 239.
26. Sanborn RE, Pishvain MJ, Callahan MK, et al. Phase I results from the combination of an immune-activating anti-CD27 antibody (varlilumab) in combination with PD-1 blockade (nivolumab): activation across multiple immune pathways without untoward immune-related adverse events. Clin Cancer Res. 2016;76(14):suppl. Abstract CT023.
27. Gangadhar T, Hamid O, Smith D.C, et al. Epacadostat plus pembrolizumab in patients with advanced melanoma and select solid tumors: updated phase 1 results from ECHO-202/KEYNOTE-037. Ann Oncol. 2016;27(6):379-400.
1. Adams JL, Smothers J, Srinivasan R, et al. Big opportunities for small molecules in immuno-oncology. Nat Rev Drug Disc. 2015;14:603-622.
2. D’Errico G, Machado HL, Sainz Jr B. A current perspective on cancer immune therapy: step-by-step approach to constructing the magic bullet. Clin Trans Med. 2017;6:3.
3. Farkona S, Diamandis EP, Blaustig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14:73.
4. Meiliana A, Dewi NM, Wijaya A. Cancer immunotherapy: a review. Indones Biomed J. 2016;8(1):1-20.
5. Smyth MJ, Ngiow SF, Ribas A, et al. Combination cancer immunotherapies tailored to the tumor microenvironment. Nat Rev Clin Oncol. 2016;13:143-158.
6. de Charette M, Marabelle A, Houot R. Turning tumor cells into antigen presenting cells: The next step to improve cancer immunotherapy? Eur J Cancer 2016;68:134-147.
7. Chen DS and Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013;39:1-10.
8. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011;480:480-489.
9. Le DT, Wang-Gillam A, Picozzi V Jr, et al. A phase 2, randomized trial of GVAX Pancreas and CRS-207 immunotherapy versus GVAX alone in patients with metastatic pancreatic adenocarcinoma: Updated results. Presented at: the ASCO Gastrointestinal Cancers Symposium; January 16-18, 2014; San Francisco, CA. Abstract 177.
10. Sharpe M and Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
11. Perica K, Varela JC, Oelke M, et al. Adoptive T Cell Immunotherapy for Cancer. Ram Mai Med J. 2015;6(1):e0004.
12. Xing Y and Hogquist KA. T-Cell Tolerance: Central and Peripheral. Cold Spring Harb Perspect Biol. 2012;4:a006957.
13. Buchbinder EI and Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol. 2016;39(1):98-106.
14. Robert C, Ribas A, Hamid O, et al. 3-year overall survival for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. J Clin Oncol. 2016(suppl;abstr 9503).
15. Hodi SF, Kluger HM, Sznol M, et al. Durable, long-term survival in previously treated patients with advanced melanoma who received nivolumab monotherapy in a phase I trial. Presented at the 2016 AACR Annual Meeting; April 16-20; New Orleans, LA. Abstract CT001.
16. Bakdash G, Sittig SP, van Dijk T, et al. The nature of activatory and tolerogenic dendritic cell-derived signal II. Front Immunol. 2013;4(53):1-18.
17. Sheridan C. Immuno-oncology moves beyond PD-1. Nat Biotechnol. 2015;33(7):673-675.
18. Blake SJ, Dougall WC, Miles JJ, et al. Molecular pathways: targeting CD96 and TIGIT for cancer immunotherapy. Clin Cancer Res. 2016;22(21):5183-5188.
19. Carotta S. Targeting NK cells for anticancer immunotherapy: clinical and preclinical approaches. Front Immunol. 2016;7:152.
20. Innate Pharma Web site. Innate Pharma Announces Top-Line Results from EFFIKIR Trial Evaluating the Efficacy of Lirilumab as a Single Agent in Elderly Patients with Acute Myeloid Leukemia. http://www.innate-pharma.com/en/news-events/press-releases/innate-pharma-announces-top-line-results-effikir-trial-evaluating-efficacy-lirilumab-single-agent-elderly-patients-acute-myeloid-leukemia. Last updated February 6, 2017. Accessed online February 22, 2017.
21. Sheridan C. IDO inhibitors move center stage in immuno-oncology. Nat Biotechnol. 2015;33(4):321-322.
22. Sanmamed MF, Pastor F, Rodriguez A, et al. Agonists of co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin Oncol. 2015;42(4):640-655.
23. Linch SN, McNamara MJ, Redmond WL. OX40 agonists and combination immunotherapy: putting the pedal to the metal. Front Oncol. 2015;5:34.
24. U.S. Food and Drug Administration Web site. Nivolumab in combination with ipilimumab. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm465274.htm. Last updated October 1, 2015. Accessed online February 22, 2017.
25. Massarelli E. Clinical safety and efficacy assessment of the CD137 agonist urelumab alone and in combination with nivolumab in patients with hematologic and solid tumor malignancies. Presented at the 31st Annual Meeting of the Society for the Immunotherapy of Cancer; November 9-13, 2016; National Harbor, MD. Abstract 239.
26. Sanborn RE, Pishvain MJ, Callahan MK, et al. Phase I results from the combination of an immune-activating anti-CD27 antibody (varlilumab) in combination with PD-1 blockade (nivolumab): activation across multiple immune pathways without untoward immune-related adverse events. Clin Cancer Res. 2016;76(14):suppl. Abstract CT023.
27. Gangadhar T, Hamid O, Smith D.C, et al. Epacadostat plus pembrolizumab in patients with advanced melanoma and select solid tumors: updated phase 1 results from ECHO-202/KEYNOTE-037. Ann Oncol. 2016;27(6):379-400.
Variants in five genes signal TNBC risk
Women with germline pathogenic variants in five genes are at high risk for developing triple-negative breast cancer, and have a greater than 20% lifetime risk for breast cancer in general, results of a large study suggest.
Multigene testing of nearly 11,000 women with triple-negative breast cancer (TNBC; lacking estrogen, progesterone, and human epidermal growth factor receptors) showed that germline pathogenic variants in BARD1, BRCA1, BRCA2, PALB2, and RAD51D were associated with risk for clinical TNBC ranging from nearly sixfold to more than 16-fold higher than that of women without the genetic variants, reported Fergus J. Couch, PhD, of the Mayo Clinic, Rochester, Minn., and his colleagues.
“The results suggest that all TNBC patients should undergo multigene panel testing, regardless of age at diagnosis or family history of cancer, for improved cancer risk assessment and because of the ongoing development of targeted therapeutic approaches for TNBC patients with mutations in predisposition genes,” they wrote. Their report is in the Journal of the National Cancer Institute.
Although National Comprehensive Cancer Network guidelines recommend testing for the cancer predisposition genes BRCA1 and BRCA2 in women with a TNBC diagnosis at age 60 or younger or those with a family history of breast and/or ovarian cancer, the picture is less clear regarding genetic predisposition to TNBC, the authors noted.
“[R]ecommendations for testing of other genes are not fully established because the risks of TNBC associated with mutations in cancer predisposition genes have not been established. Thus, a better understanding of gene-specific risks for TNBC is needed to identify the genes that should be tested in the setting of TNBC,” they wrote.
The investigators looked for associations between deleterious mutations in cancer predisposition genes and TNBC among 8,753 patients with TNBC testing with a 21-gene assay, and among 2,148 women tested for 17 genes in studies conducted by the Triple Negative Breast Cancer Consortium.
They found that among white women, germline pathogenic variants were associated with the following odds ratios (OR) for TNBC (all P values less than .0001):
- BRCA2 = 5.42
- BARD1 = 5.92
- RAD51D = 6.97
- PALB2 = 14.41
- BRCA1 = 16.27
Although there were insufficient data on the risks for African American women, an exploratory analysis showed that risks for TNBC associated with specific pathogenic variants were similar to those for white women, the authors said.
The pathogenic variants were detected in 12% of all patients in the study.
“Continued study of gene-specific risks for breast cancer subtypes may lead to tailored medical management recommendations for PV [pathogenic variant] carriers. Consistent with this hypothesis, initial studies evaluating intensified screening in high-risk women have suggested that a decrease in mortality from TNBC can be achieved,” they wrote.
The study was supported in part by the National Institutes of Health and the Breast Cancer Research Foundation, and was sponsored by Ambry Genetics Inc. The authors reported having no conflicts of interest.
SOURCE: Shimelis H et al. J Natl Cancer Inst. 2018 Aug 7. doi: 10.1093/jnci/djy106.
Women with germline pathogenic variants in five genes are at high risk for developing triple-negative breast cancer, and have a greater than 20% lifetime risk for breast cancer in general, results of a large study suggest.
Multigene testing of nearly 11,000 women with triple-negative breast cancer (TNBC; lacking estrogen, progesterone, and human epidermal growth factor receptors) showed that germline pathogenic variants in BARD1, BRCA1, BRCA2, PALB2, and RAD51D were associated with risk for clinical TNBC ranging from nearly sixfold to more than 16-fold higher than that of women without the genetic variants, reported Fergus J. Couch, PhD, of the Mayo Clinic, Rochester, Minn., and his colleagues.
“The results suggest that all TNBC patients should undergo multigene panel testing, regardless of age at diagnosis or family history of cancer, for improved cancer risk assessment and because of the ongoing development of targeted therapeutic approaches for TNBC patients with mutations in predisposition genes,” they wrote. Their report is in the Journal of the National Cancer Institute.
Although National Comprehensive Cancer Network guidelines recommend testing for the cancer predisposition genes BRCA1 and BRCA2 in women with a TNBC diagnosis at age 60 or younger or those with a family history of breast and/or ovarian cancer, the picture is less clear regarding genetic predisposition to TNBC, the authors noted.
“[R]ecommendations for testing of other genes are not fully established because the risks of TNBC associated with mutations in cancer predisposition genes have not been established. Thus, a better understanding of gene-specific risks for TNBC is needed to identify the genes that should be tested in the setting of TNBC,” they wrote.
The investigators looked for associations between deleterious mutations in cancer predisposition genes and TNBC among 8,753 patients with TNBC testing with a 21-gene assay, and among 2,148 women tested for 17 genes in studies conducted by the Triple Negative Breast Cancer Consortium.
They found that among white women, germline pathogenic variants were associated with the following odds ratios (OR) for TNBC (all P values less than .0001):
- BRCA2 = 5.42
- BARD1 = 5.92
- RAD51D = 6.97
- PALB2 = 14.41
- BRCA1 = 16.27
Although there were insufficient data on the risks for African American women, an exploratory analysis showed that risks for TNBC associated with specific pathogenic variants were similar to those for white women, the authors said.
The pathogenic variants were detected in 12% of all patients in the study.
“Continued study of gene-specific risks for breast cancer subtypes may lead to tailored medical management recommendations for PV [pathogenic variant] carriers. Consistent with this hypothesis, initial studies evaluating intensified screening in high-risk women have suggested that a decrease in mortality from TNBC can be achieved,” they wrote.
The study was supported in part by the National Institutes of Health and the Breast Cancer Research Foundation, and was sponsored by Ambry Genetics Inc. The authors reported having no conflicts of interest.
SOURCE: Shimelis H et al. J Natl Cancer Inst. 2018 Aug 7. doi: 10.1093/jnci/djy106.
Women with germline pathogenic variants in five genes are at high risk for developing triple-negative breast cancer, and have a greater than 20% lifetime risk for breast cancer in general, results of a large study suggest.
Multigene testing of nearly 11,000 women with triple-negative breast cancer (TNBC; lacking estrogen, progesterone, and human epidermal growth factor receptors) showed that germline pathogenic variants in BARD1, BRCA1, BRCA2, PALB2, and RAD51D were associated with risk for clinical TNBC ranging from nearly sixfold to more than 16-fold higher than that of women without the genetic variants, reported Fergus J. Couch, PhD, of the Mayo Clinic, Rochester, Minn., and his colleagues.
“The results suggest that all TNBC patients should undergo multigene panel testing, regardless of age at diagnosis or family history of cancer, for improved cancer risk assessment and because of the ongoing development of targeted therapeutic approaches for TNBC patients with mutations in predisposition genes,” they wrote. Their report is in the Journal of the National Cancer Institute.
Although National Comprehensive Cancer Network guidelines recommend testing for the cancer predisposition genes BRCA1 and BRCA2 in women with a TNBC diagnosis at age 60 or younger or those with a family history of breast and/or ovarian cancer, the picture is less clear regarding genetic predisposition to TNBC, the authors noted.
“[R]ecommendations for testing of other genes are not fully established because the risks of TNBC associated with mutations in cancer predisposition genes have not been established. Thus, a better understanding of gene-specific risks for TNBC is needed to identify the genes that should be tested in the setting of TNBC,” they wrote.
The investigators looked for associations between deleterious mutations in cancer predisposition genes and TNBC among 8,753 patients with TNBC testing with a 21-gene assay, and among 2,148 women tested for 17 genes in studies conducted by the Triple Negative Breast Cancer Consortium.
They found that among white women, germline pathogenic variants were associated with the following odds ratios (OR) for TNBC (all P values less than .0001):
- BRCA2 = 5.42
- BARD1 = 5.92
- RAD51D = 6.97
- PALB2 = 14.41
- BRCA1 = 16.27
Although there were insufficient data on the risks for African American women, an exploratory analysis showed that risks for TNBC associated with specific pathogenic variants were similar to those for white women, the authors said.
The pathogenic variants were detected in 12% of all patients in the study.
“Continued study of gene-specific risks for breast cancer subtypes may lead to tailored medical management recommendations for PV [pathogenic variant] carriers. Consistent with this hypothesis, initial studies evaluating intensified screening in high-risk women have suggested that a decrease in mortality from TNBC can be achieved,” they wrote.
The study was supported in part by the National Institutes of Health and the Breast Cancer Research Foundation, and was sponsored by Ambry Genetics Inc. The authors reported having no conflicts of interest.
SOURCE: Shimelis H et al. J Natl Cancer Inst. 2018 Aug 7. doi: 10.1093/jnci/djy106.
FROM JOURNAL OF THE NATIONAL CANCER INSTITUTE
Key clinical point: Pathogenic variants in five cancer predisposition genes are associated with significantly increased risk for triple negative breast cancer (TNBC).
Major finding: Pathogenic variants in BRCA1 were associated with a more than 16-fold risk for TNBC.
Study details: Retrospective review of multigene assay testing in 10,901 women with TNBC.
Disclosures: The study was supported in part by the National Institutes of Health and the Breast Cancer Research Foundation, and was sponsored by Ambry Genetics Inc. The authors reported having no conflicts of interest.
Source: Shimelis H et al. J Natl Cancer Inst. 2018 Aug 7. doi: 10.1093/jnci/djy106.
RT linked with better survival in DCIS
Treatment with lumpectomy and radiotherapy was associated with a significant reduction in breast cancer mortality in patients with ductal carcinoma in situ (DCIS), compared with a lumpectomy alone or a mastectomy alone, investigators reported in JAMA Oncology.
Among women who received adjuvant radiation, there was an associated 23% reduced risk of dying of breast cancer. This extrapolated to a cumulative mortality of 2.33% for those treated with lumpectomy alone and 1.74% for women treated with lumpectomy and radiotherapy at 15 years (adjusted hazard ratio, 0.77; 95% confidence interval, 0.67-0.88; P less than .001).
“Although the clinical benefit is small, it is intriguing that radiotherapy has this effect, which appears to be attributable to systemic activity rather than local control,” wrote Vasily Giannakeas, MPH, of the Women’s College Research Institute, Toronto, and colleagues.
Emerging evidence suggests that adding radiotherapy to breast conserving surgery can reduce the risk of breast cancer mortality among women with DCIS and lower the risk of local recurrence. Because of the low rate of mortality associated with DCIS, the authors noted that it has been difficult to investigate deaths related to DCIS. The association of adjuvant radiotherapy with breast cancer survival in this population has also not yet been clearly established.
To determine the extent to which radiotherapy is associated with reduced risk of breast cancer mortality in patients treated for DCIS and identify patient subgroups who might derive the most benefit from radiotherapy, the authors conducted a historical cohort study using the Surveillance, Epidemiology, and End Results database. A total of 140,366 women diagnosed with first primary DCIS between 1998 and 2014 were identified, and three separate comparisons were made using 1:1 matching: lumpectomy with radiation versus lumpectomy alone, lumpectomy alone versus mastectomy, and lumpectomy with radiation therapy versus mastectomy.
A total of 35,070 women (25.0%) were treated with lumpectomy alone, 65,301 (46.5%) were treated with lumpectomy and radiotherapy, and 39,995 (28.5%) were treated with mastectomy.
The overall cumulative mortality for the entire cohort from breast cancer at 15 years was 2.03%. The actuarial 15-year mortality rate for the mastectomy group (2.26%) was similar to those who had lumpectomy without radiotherapy (2.33%).
The adjusted HR for death for mastectomy versus lumpectomy alone (based on 20,832 propensity-matched pairs) was 0.91 (95% CI, 0.78-1.05). The adjusted hazard ratios for death were 0.77 (95% CI, 0.67-0.88) for lumpectomy and radiotherapy versus lumpectomy alone (29,465 propensity-matched pairs), 0.91 (95% CI, 0.78-1.05) for mastectomy alone versus lumpectomy alone (20,832 propensity-matched pairs), and 0.75 (95% CI, 0.65-0.87) for lumpectomy and radiotherapy versus mastectomy (29,865 propensity-matched pairs).
When looking at subgroups and the effect of radiotherapy on mortality, the authors found the following: The HR was 0.59 (95% CI, 0.43-0.80) for patients aged younger than 50 years and 0.86 (95% CI, 0.73- 1.01) for those aged 50 years and older; it was 0.67 (95% CI, 0.51-0.87) for patients with ER-positive cancers, 0.50 (95% CI, 0.32-0.78) for ER-negative cancers, and 0.93 (95% CI, 0.77-1.13) for those with unknown ER status.
“How exactly radiotherapy affects survival is an important question that should be explored in future studies,” the authors concluded.
There was no outside funding source reported. Mr. Giannakeas is supported by the Canadian Institutes of Health Research Frederick Banting and Charles Best Doctoral Research Award.
SOURCE: Giannakeas V et al. JAMA Network Open. 2018 Aug 10. doi:10.1001/jamanetworkopen.2018.1100.
In an accompanying editorial, Mira Goldberg, MD, and Timothy J. Whelan, BM, BCh, of the department of oncology at McMaster University, Hamilton, Ont., noted that the primary goal of using adjuvant radiotherapy in patients with ductal carcinoma in situ (DCIS) is to reduce the risk of local recurrence of DCIS or of invasive breast cancer.
Despite widespread screening with mammography, along with improvements in technology so as to detect even smaller lesions, “there is increased concern about the overdiagnosis of DCIS,” they wrote. Results from recent studies generally suggest that patients with good prognostic factors and who have a low risk of local recurrence at 10 years (10%) are unlikely to gain any major benefit from being treated with radiotherapy.
They also pointed out that there is growing interest in the use of molecular markers as a means to help detect patients who are at a lower risk of recurrence and thus who may not benefit from radiotherapy.
“The results of the study by Giannakeas and colleagues are reassuring,” the editorialists wrote, as it demonstrated that the risk of breast cancer mortality in patients with DCIS is very low, and the potential absolute benefit of radiotherapy is also quite small. (The number of patients that need to be treated to prevent a breast cancer death was 370.) These data continue to support a strategy for low-risk DCIS of omitting radiotherapy after lumpectomy. This is especially pertinent when “one considers the negative effects of treatment: the cost and inconvenience of 5-6 weeks of daily treatments, acute adverse effects such as breast pain and fatigue, and potential long-term toxic effects of cardiac disease and second cancers.”
The editorialists also highlighted the authors’ speculation that there could be additional systemic effects of radiotherapy, possibly resulting from an elicited immune response or radiation scatter to distant tissues. While this hypothesis is theoretically possible, their results could also be explained by confounding factors, such as a higher use of endocrine therapy in patients who received adjuvant radiotherapy.
Dr. Whelan has received research support from Genomic Health. This editorial accompanied the article by Giannakeas et al. (JAMA Network Open. 2018;1[4]e181102). No other disclosures were reported.
In an accompanying editorial, Mira Goldberg, MD, and Timothy J. Whelan, BM, BCh, of the department of oncology at McMaster University, Hamilton, Ont., noted that the primary goal of using adjuvant radiotherapy in patients with ductal carcinoma in situ (DCIS) is to reduce the risk of local recurrence of DCIS or of invasive breast cancer.
Despite widespread screening with mammography, along with improvements in technology so as to detect even smaller lesions, “there is increased concern about the overdiagnosis of DCIS,” they wrote. Results from recent studies generally suggest that patients with good prognostic factors and who have a low risk of local recurrence at 10 years (10%) are unlikely to gain any major benefit from being treated with radiotherapy.
They also pointed out that there is growing interest in the use of molecular markers as a means to help detect patients who are at a lower risk of recurrence and thus who may not benefit from radiotherapy.
“The results of the study by Giannakeas and colleagues are reassuring,” the editorialists wrote, as it demonstrated that the risk of breast cancer mortality in patients with DCIS is very low, and the potential absolute benefit of radiotherapy is also quite small. (The number of patients that need to be treated to prevent a breast cancer death was 370.) These data continue to support a strategy for low-risk DCIS of omitting radiotherapy after lumpectomy. This is especially pertinent when “one considers the negative effects of treatment: the cost and inconvenience of 5-6 weeks of daily treatments, acute adverse effects such as breast pain and fatigue, and potential long-term toxic effects of cardiac disease and second cancers.”
The editorialists also highlighted the authors’ speculation that there could be additional systemic effects of radiotherapy, possibly resulting from an elicited immune response or radiation scatter to distant tissues. While this hypothesis is theoretically possible, their results could also be explained by confounding factors, such as a higher use of endocrine therapy in patients who received adjuvant radiotherapy.
Dr. Whelan has received research support from Genomic Health. This editorial accompanied the article by Giannakeas et al. (JAMA Network Open. 2018;1[4]e181102). No other disclosures were reported.
In an accompanying editorial, Mira Goldberg, MD, and Timothy J. Whelan, BM, BCh, of the department of oncology at McMaster University, Hamilton, Ont., noted that the primary goal of using adjuvant radiotherapy in patients with ductal carcinoma in situ (DCIS) is to reduce the risk of local recurrence of DCIS or of invasive breast cancer.
Despite widespread screening with mammography, along with improvements in technology so as to detect even smaller lesions, “there is increased concern about the overdiagnosis of DCIS,” they wrote. Results from recent studies generally suggest that patients with good prognostic factors and who have a low risk of local recurrence at 10 years (10%) are unlikely to gain any major benefit from being treated with radiotherapy.
They also pointed out that there is growing interest in the use of molecular markers as a means to help detect patients who are at a lower risk of recurrence and thus who may not benefit from radiotherapy.
“The results of the study by Giannakeas and colleagues are reassuring,” the editorialists wrote, as it demonstrated that the risk of breast cancer mortality in patients with DCIS is very low, and the potential absolute benefit of radiotherapy is also quite small. (The number of patients that need to be treated to prevent a breast cancer death was 370.) These data continue to support a strategy for low-risk DCIS of omitting radiotherapy after lumpectomy. This is especially pertinent when “one considers the negative effects of treatment: the cost and inconvenience of 5-6 weeks of daily treatments, acute adverse effects such as breast pain and fatigue, and potential long-term toxic effects of cardiac disease and second cancers.”
The editorialists also highlighted the authors’ speculation that there could be additional systemic effects of radiotherapy, possibly resulting from an elicited immune response or radiation scatter to distant tissues. While this hypothesis is theoretically possible, their results could also be explained by confounding factors, such as a higher use of endocrine therapy in patients who received adjuvant radiotherapy.
Dr. Whelan has received research support from Genomic Health. This editorial accompanied the article by Giannakeas et al. (JAMA Network Open. 2018;1[4]e181102). No other disclosures were reported.
Treatment with lumpectomy and radiotherapy was associated with a significant reduction in breast cancer mortality in patients with ductal carcinoma in situ (DCIS), compared with a lumpectomy alone or a mastectomy alone, investigators reported in JAMA Oncology.
Among women who received adjuvant radiation, there was an associated 23% reduced risk of dying of breast cancer. This extrapolated to a cumulative mortality of 2.33% for those treated with lumpectomy alone and 1.74% for women treated with lumpectomy and radiotherapy at 15 years (adjusted hazard ratio, 0.77; 95% confidence interval, 0.67-0.88; P less than .001).
“Although the clinical benefit is small, it is intriguing that radiotherapy has this effect, which appears to be attributable to systemic activity rather than local control,” wrote Vasily Giannakeas, MPH, of the Women’s College Research Institute, Toronto, and colleagues.
Emerging evidence suggests that adding radiotherapy to breast conserving surgery can reduce the risk of breast cancer mortality among women with DCIS and lower the risk of local recurrence. Because of the low rate of mortality associated with DCIS, the authors noted that it has been difficult to investigate deaths related to DCIS. The association of adjuvant radiotherapy with breast cancer survival in this population has also not yet been clearly established.
To determine the extent to which radiotherapy is associated with reduced risk of breast cancer mortality in patients treated for DCIS and identify patient subgroups who might derive the most benefit from radiotherapy, the authors conducted a historical cohort study using the Surveillance, Epidemiology, and End Results database. A total of 140,366 women diagnosed with first primary DCIS between 1998 and 2014 were identified, and three separate comparisons were made using 1:1 matching: lumpectomy with radiation versus lumpectomy alone, lumpectomy alone versus mastectomy, and lumpectomy with radiation therapy versus mastectomy.
A total of 35,070 women (25.0%) were treated with lumpectomy alone, 65,301 (46.5%) were treated with lumpectomy and radiotherapy, and 39,995 (28.5%) were treated with mastectomy.
The overall cumulative mortality for the entire cohort from breast cancer at 15 years was 2.03%. The actuarial 15-year mortality rate for the mastectomy group (2.26%) was similar to those who had lumpectomy without radiotherapy (2.33%).
The adjusted HR for death for mastectomy versus lumpectomy alone (based on 20,832 propensity-matched pairs) was 0.91 (95% CI, 0.78-1.05). The adjusted hazard ratios for death were 0.77 (95% CI, 0.67-0.88) for lumpectomy and radiotherapy versus lumpectomy alone (29,465 propensity-matched pairs), 0.91 (95% CI, 0.78-1.05) for mastectomy alone versus lumpectomy alone (20,832 propensity-matched pairs), and 0.75 (95% CI, 0.65-0.87) for lumpectomy and radiotherapy versus mastectomy (29,865 propensity-matched pairs).
When looking at subgroups and the effect of radiotherapy on mortality, the authors found the following: The HR was 0.59 (95% CI, 0.43-0.80) for patients aged younger than 50 years and 0.86 (95% CI, 0.73- 1.01) for those aged 50 years and older; it was 0.67 (95% CI, 0.51-0.87) for patients with ER-positive cancers, 0.50 (95% CI, 0.32-0.78) for ER-negative cancers, and 0.93 (95% CI, 0.77-1.13) for those with unknown ER status.
“How exactly radiotherapy affects survival is an important question that should be explored in future studies,” the authors concluded.
There was no outside funding source reported. Mr. Giannakeas is supported by the Canadian Institutes of Health Research Frederick Banting and Charles Best Doctoral Research Award.
SOURCE: Giannakeas V et al. JAMA Network Open. 2018 Aug 10. doi:10.1001/jamanetworkopen.2018.1100.
Treatment with lumpectomy and radiotherapy was associated with a significant reduction in breast cancer mortality in patients with ductal carcinoma in situ (DCIS), compared with a lumpectomy alone or a mastectomy alone, investigators reported in JAMA Oncology.
Among women who received adjuvant radiation, there was an associated 23% reduced risk of dying of breast cancer. This extrapolated to a cumulative mortality of 2.33% for those treated with lumpectomy alone and 1.74% for women treated with lumpectomy and radiotherapy at 15 years (adjusted hazard ratio, 0.77; 95% confidence interval, 0.67-0.88; P less than .001).
“Although the clinical benefit is small, it is intriguing that radiotherapy has this effect, which appears to be attributable to systemic activity rather than local control,” wrote Vasily Giannakeas, MPH, of the Women’s College Research Institute, Toronto, and colleagues.
Emerging evidence suggests that adding radiotherapy to breast conserving surgery can reduce the risk of breast cancer mortality among women with DCIS and lower the risk of local recurrence. Because of the low rate of mortality associated with DCIS, the authors noted that it has been difficult to investigate deaths related to DCIS. The association of adjuvant radiotherapy with breast cancer survival in this population has also not yet been clearly established.
To determine the extent to which radiotherapy is associated with reduced risk of breast cancer mortality in patients treated for DCIS and identify patient subgroups who might derive the most benefit from radiotherapy, the authors conducted a historical cohort study using the Surveillance, Epidemiology, and End Results database. A total of 140,366 women diagnosed with first primary DCIS between 1998 and 2014 were identified, and three separate comparisons were made using 1:1 matching: lumpectomy with radiation versus lumpectomy alone, lumpectomy alone versus mastectomy, and lumpectomy with radiation therapy versus mastectomy.
A total of 35,070 women (25.0%) were treated with lumpectomy alone, 65,301 (46.5%) were treated with lumpectomy and radiotherapy, and 39,995 (28.5%) were treated with mastectomy.
The overall cumulative mortality for the entire cohort from breast cancer at 15 years was 2.03%. The actuarial 15-year mortality rate for the mastectomy group (2.26%) was similar to those who had lumpectomy without radiotherapy (2.33%).
The adjusted HR for death for mastectomy versus lumpectomy alone (based on 20,832 propensity-matched pairs) was 0.91 (95% CI, 0.78-1.05). The adjusted hazard ratios for death were 0.77 (95% CI, 0.67-0.88) for lumpectomy and radiotherapy versus lumpectomy alone (29,465 propensity-matched pairs), 0.91 (95% CI, 0.78-1.05) for mastectomy alone versus lumpectomy alone (20,832 propensity-matched pairs), and 0.75 (95% CI, 0.65-0.87) for lumpectomy and radiotherapy versus mastectomy (29,865 propensity-matched pairs).
When looking at subgroups and the effect of radiotherapy on mortality, the authors found the following: The HR was 0.59 (95% CI, 0.43-0.80) for patients aged younger than 50 years and 0.86 (95% CI, 0.73- 1.01) for those aged 50 years and older; it was 0.67 (95% CI, 0.51-0.87) for patients with ER-positive cancers, 0.50 (95% CI, 0.32-0.78) for ER-negative cancers, and 0.93 (95% CI, 0.77-1.13) for those with unknown ER status.
“How exactly radiotherapy affects survival is an important question that should be explored in future studies,” the authors concluded.
There was no outside funding source reported. Mr. Giannakeas is supported by the Canadian Institutes of Health Research Frederick Banting and Charles Best Doctoral Research Award.
SOURCE: Giannakeas V et al. JAMA Network Open. 2018 Aug 10. doi:10.1001/jamanetworkopen.2018.1100.
FROM JAMA ONCOLOGY
Key clinical point: Lumpectomy and adjuvant radiotherapy together was superior to lumpectomy or mastectomy alone.
Major finding: The 15-year breast cancer mortality rate was 2.33% for lumpectomy alone, 1.74% for lumpectomy and radiation, and 2.26% for mastectomy.
Study details: A historical cohort study using Surveillance, Epidemiology, and End Results data that included 140,366 women diagnosed with first primary ductal carcinoma in situ.
Disclosures: There was no outside funding source reported. Mr. Giannakeas is supported by the Canadian Institutes of Health Research Frederick Banting and Charles Best Doctoral Research Award. No other disclosures were reported.
Source: Giannakeas V et al. JAMA Network Open. 2018 Aug 10. doi: 10.1001/jamanetworkopen.2018.1100.
Herceptin linked to doubling of HF risk in women with breast cancer
Adding more evidence to an ongoing debate, a large new study suggests that patients with breast cancer who take trastuzumab (Herceptin) may face double the adjusted risk of developing heart failure, with older women at highest risk.
The study also found that most patients who took trastuzumab didn’t receive recommended cardiac screening.
The researchers said their findings are unique because they tracked both younger and older patients. “By examining the rates of both cardiac monitoring and cardiotoxicity, we could begin to address the controversial issue of whether cardiac monitoring is warranted in young breast cancer patients,” wrote Mariana Chavez-MacGregor, MD, MSC, of the University of Texas MD Anderson Cancer Center, and her associates. The report was published in JACC: Cardiovascular Imaging.
While Trastuzumab has boosted breast cancer survival rates for patients with HER2-positive tumors, it’s also raised concerns about cardiotoxicity that could be an indicator of subsequent congestive heart failure (Cochrane Database Syst Rev. 2014 Jun 12;(6):CD006242).
According to the new study, the risk of trastuzumab risk is linked to damage to cardiac myocytes that can cause reversible cardiotoxicity.
The prescribing information for trastuzumab advises patients to undergo cardiac monitoring before treatment with trastuzumab and every 3 months during treatment. Recommendations by medical organizations have varied.
Now, as a 2016 report put it, it’s “increasingly unclear” whether frequent routine monitoring is appropriate for all patients (J Clin Oncol. 2016 Apr 1;34[10]:1030-3).
For the current study, Dr. Chavez-MacGregor and her associates identified 16,456 adult women in the United States who were diagnosed with nonmetastatic invasive breast cancer from 2009 to 2014. Researchers tracked the group, with a median age of 56, through as late as 2015.
The women were treated with chemotherapy within 6 months of diagnosis, and 4,325 received trastuzumab.
Of all the subjects, 692 patients (4.2%) developed heart failure following chemotherapy. The rate among patients treated with trastuzumab was higher, at 8.3%, compared with 2.7% for those not treated with trastuzumab (P less than .001).
The researchers also looked at anthracycline users and found that they were slightly more likely to develop HF (4.6% vs. 4.0% among nonusers, P = .048).
Increased age boosted the risk of HF in the trastuzumab-treated patients, and the risk was highest in those treated with both anthracyclines and trastuzumab. Other factors linked to more risk were comorbidities, hypertension, and valve disease.
After adjusting for confounders, the researchers estimated that those treated with trastuzumab were 2.01 times more likely to develop HF (HR, 2.01; 95% confidence interval, 1.72-2.36), and those who took anthracycline were 1.53 times more likely (HR, 1.53; 95% CI, 1.30-1.80)
The researchers also examined medical records for evidence that subjects underwent cardiac screening at least once every 4 months, not 3 months, as the prescribing information recommends. The study team chose to focus on 4-month intervals “to compensate for differences in scheduling, resources, or levels of accessibility to medical care.”
Medical records suggest that 73.5% of patients who took trastuzumab underwent cardiac screening at the beginning of therapy, but only 46.2% continued to do so at least every 4 months.
An adjusted model linked more screening to the use of anthracyclines and taxanes, radiation treatment, and living in the Northeast vs. the West.
“HF was more frequently identified among patients undergoing recommended cardiac monitoring (10.4% compared with 6.5%, respectively; P less than.001), suggesting that, as more patients are screened, more patients are likely to be found having HF,” the researchers reported.
However, they added that “our sensitivity analysis using inpatient claims allowed us to determine that the HF identified using cardiac monitoring was not severe enough to require hospitalization and was likely asymptomatic. The clinical implications of the diagnosis of asymptomatic HF are hard to determine and are beyond the scope of this study.”
The researchers also noted that the findings suggest that screening has become more common in recent years.
“The number of cancer survivors is expected to increase over time, and we will continue to see patients develop treatment-related cardiotoxicity,” the researchers wrote. “Thus, more research, evidence-based guidelines, and tools for prediction of cancer treatment–related cardiotoxicity are needed.”
The National Cancer Institute and Cancer Prevention and Research Institute of Texas funded the study. Two study authors reported grant funding from the Susan G. Komen Breast Cancer Foundation, and one reports consulting for Pfizer and Roche. The other authors reported no disclosures.
SOURCE: Chavez-MacGregor M et al. JACC: Cardiovasc Imaging. 2018 Aug;11[8]1084-93.
While trastuzumab clearly benefits patients with HER2-positive breast cancer at various stages of progression, concerns about heart failure persist. Studies have suggested that the drug doesn’t boost the risk of late cardiac events, but it’s not clear if this is due to mandated screening in these trials. The new study provides more evidence that adherence to screening guidelines is limited, and recent trials offer evidence that the general cardiac risk may be overblown. Future studies could be designed to offer insight into the wisdom of adjusting screening regimens based on stratification of risk. A meta-analysis could also be helpful, and the upcoming results of the SAFE-HEART study will provide information about the safety of anti-HER2 antibody therapy in patients with low but asymptomatic left-ventricular ejection fraction.
These comments are excerpted from a commentary by Chau T. Dang, MD, of Memorial Sloan Kettering Cancer Center, New York, and her associates (JACC: Cardiovasc Imaging. 2018 Aug;11[8]:1094-7). Most of the commentary authors report various disclosures.
While trastuzumab clearly benefits patients with HER2-positive breast cancer at various stages of progression, concerns about heart failure persist. Studies have suggested that the drug doesn’t boost the risk of late cardiac events, but it’s not clear if this is due to mandated screening in these trials. The new study provides more evidence that adherence to screening guidelines is limited, and recent trials offer evidence that the general cardiac risk may be overblown. Future studies could be designed to offer insight into the wisdom of adjusting screening regimens based on stratification of risk. A meta-analysis could also be helpful, and the upcoming results of the SAFE-HEART study will provide information about the safety of anti-HER2 antibody therapy in patients with low but asymptomatic left-ventricular ejection fraction.
These comments are excerpted from a commentary by Chau T. Dang, MD, of Memorial Sloan Kettering Cancer Center, New York, and her associates (JACC: Cardiovasc Imaging. 2018 Aug;11[8]:1094-7). Most of the commentary authors report various disclosures.
While trastuzumab clearly benefits patients with HER2-positive breast cancer at various stages of progression, concerns about heart failure persist. Studies have suggested that the drug doesn’t boost the risk of late cardiac events, but it’s not clear if this is due to mandated screening in these trials. The new study provides more evidence that adherence to screening guidelines is limited, and recent trials offer evidence that the general cardiac risk may be overblown. Future studies could be designed to offer insight into the wisdom of adjusting screening regimens based on stratification of risk. A meta-analysis could also be helpful, and the upcoming results of the SAFE-HEART study will provide information about the safety of anti-HER2 antibody therapy in patients with low but asymptomatic left-ventricular ejection fraction.
These comments are excerpted from a commentary by Chau T. Dang, MD, of Memorial Sloan Kettering Cancer Center, New York, and her associates (JACC: Cardiovasc Imaging. 2018 Aug;11[8]:1094-7). Most of the commentary authors report various disclosures.
Adding more evidence to an ongoing debate, a large new study suggests that patients with breast cancer who take trastuzumab (Herceptin) may face double the adjusted risk of developing heart failure, with older women at highest risk.
The study also found that most patients who took trastuzumab didn’t receive recommended cardiac screening.
The researchers said their findings are unique because they tracked both younger and older patients. “By examining the rates of both cardiac monitoring and cardiotoxicity, we could begin to address the controversial issue of whether cardiac monitoring is warranted in young breast cancer patients,” wrote Mariana Chavez-MacGregor, MD, MSC, of the University of Texas MD Anderson Cancer Center, and her associates. The report was published in JACC: Cardiovascular Imaging.
While Trastuzumab has boosted breast cancer survival rates for patients with HER2-positive tumors, it’s also raised concerns about cardiotoxicity that could be an indicator of subsequent congestive heart failure (Cochrane Database Syst Rev. 2014 Jun 12;(6):CD006242).
According to the new study, the risk of trastuzumab risk is linked to damage to cardiac myocytes that can cause reversible cardiotoxicity.
The prescribing information for trastuzumab advises patients to undergo cardiac monitoring before treatment with trastuzumab and every 3 months during treatment. Recommendations by medical organizations have varied.
Now, as a 2016 report put it, it’s “increasingly unclear” whether frequent routine monitoring is appropriate for all patients (J Clin Oncol. 2016 Apr 1;34[10]:1030-3).
For the current study, Dr. Chavez-MacGregor and her associates identified 16,456 adult women in the United States who were diagnosed with nonmetastatic invasive breast cancer from 2009 to 2014. Researchers tracked the group, with a median age of 56, through as late as 2015.
The women were treated with chemotherapy within 6 months of diagnosis, and 4,325 received trastuzumab.
Of all the subjects, 692 patients (4.2%) developed heart failure following chemotherapy. The rate among patients treated with trastuzumab was higher, at 8.3%, compared with 2.7% for those not treated with trastuzumab (P less than .001).
The researchers also looked at anthracycline users and found that they were slightly more likely to develop HF (4.6% vs. 4.0% among nonusers, P = .048).
Increased age boosted the risk of HF in the trastuzumab-treated patients, and the risk was highest in those treated with both anthracyclines and trastuzumab. Other factors linked to more risk were comorbidities, hypertension, and valve disease.
After adjusting for confounders, the researchers estimated that those treated with trastuzumab were 2.01 times more likely to develop HF (HR, 2.01; 95% confidence interval, 1.72-2.36), and those who took anthracycline were 1.53 times more likely (HR, 1.53; 95% CI, 1.30-1.80)
The researchers also examined medical records for evidence that subjects underwent cardiac screening at least once every 4 months, not 3 months, as the prescribing information recommends. The study team chose to focus on 4-month intervals “to compensate for differences in scheduling, resources, or levels of accessibility to medical care.”
Medical records suggest that 73.5% of patients who took trastuzumab underwent cardiac screening at the beginning of therapy, but only 46.2% continued to do so at least every 4 months.
An adjusted model linked more screening to the use of anthracyclines and taxanes, radiation treatment, and living in the Northeast vs. the West.
“HF was more frequently identified among patients undergoing recommended cardiac monitoring (10.4% compared with 6.5%, respectively; P less than.001), suggesting that, as more patients are screened, more patients are likely to be found having HF,” the researchers reported.
However, they added that “our sensitivity analysis using inpatient claims allowed us to determine that the HF identified using cardiac monitoring was not severe enough to require hospitalization and was likely asymptomatic. The clinical implications of the diagnosis of asymptomatic HF are hard to determine and are beyond the scope of this study.”
The researchers also noted that the findings suggest that screening has become more common in recent years.
“The number of cancer survivors is expected to increase over time, and we will continue to see patients develop treatment-related cardiotoxicity,” the researchers wrote. “Thus, more research, evidence-based guidelines, and tools for prediction of cancer treatment–related cardiotoxicity are needed.”
The National Cancer Institute and Cancer Prevention and Research Institute of Texas funded the study. Two study authors reported grant funding from the Susan G. Komen Breast Cancer Foundation, and one reports consulting for Pfizer and Roche. The other authors reported no disclosures.
SOURCE: Chavez-MacGregor M et al. JACC: Cardiovasc Imaging. 2018 Aug;11[8]1084-93.
Adding more evidence to an ongoing debate, a large new study suggests that patients with breast cancer who take trastuzumab (Herceptin) may face double the adjusted risk of developing heart failure, with older women at highest risk.
The study also found that most patients who took trastuzumab didn’t receive recommended cardiac screening.
The researchers said their findings are unique because they tracked both younger and older patients. “By examining the rates of both cardiac monitoring and cardiotoxicity, we could begin to address the controversial issue of whether cardiac monitoring is warranted in young breast cancer patients,” wrote Mariana Chavez-MacGregor, MD, MSC, of the University of Texas MD Anderson Cancer Center, and her associates. The report was published in JACC: Cardiovascular Imaging.
While Trastuzumab has boosted breast cancer survival rates for patients with HER2-positive tumors, it’s also raised concerns about cardiotoxicity that could be an indicator of subsequent congestive heart failure (Cochrane Database Syst Rev. 2014 Jun 12;(6):CD006242).
According to the new study, the risk of trastuzumab risk is linked to damage to cardiac myocytes that can cause reversible cardiotoxicity.
The prescribing information for trastuzumab advises patients to undergo cardiac monitoring before treatment with trastuzumab and every 3 months during treatment. Recommendations by medical organizations have varied.
Now, as a 2016 report put it, it’s “increasingly unclear” whether frequent routine monitoring is appropriate for all patients (J Clin Oncol. 2016 Apr 1;34[10]:1030-3).
For the current study, Dr. Chavez-MacGregor and her associates identified 16,456 adult women in the United States who were diagnosed with nonmetastatic invasive breast cancer from 2009 to 2014. Researchers tracked the group, with a median age of 56, through as late as 2015.
The women were treated with chemotherapy within 6 months of diagnosis, and 4,325 received trastuzumab.
Of all the subjects, 692 patients (4.2%) developed heart failure following chemotherapy. The rate among patients treated with trastuzumab was higher, at 8.3%, compared with 2.7% for those not treated with trastuzumab (P less than .001).
The researchers also looked at anthracycline users and found that they were slightly more likely to develop HF (4.6% vs. 4.0% among nonusers, P = .048).
Increased age boosted the risk of HF in the trastuzumab-treated patients, and the risk was highest in those treated with both anthracyclines and trastuzumab. Other factors linked to more risk were comorbidities, hypertension, and valve disease.
After adjusting for confounders, the researchers estimated that those treated with trastuzumab were 2.01 times more likely to develop HF (HR, 2.01; 95% confidence interval, 1.72-2.36), and those who took anthracycline were 1.53 times more likely (HR, 1.53; 95% CI, 1.30-1.80)
The researchers also examined medical records for evidence that subjects underwent cardiac screening at least once every 4 months, not 3 months, as the prescribing information recommends. The study team chose to focus on 4-month intervals “to compensate for differences in scheduling, resources, or levels of accessibility to medical care.”
Medical records suggest that 73.5% of patients who took trastuzumab underwent cardiac screening at the beginning of therapy, but only 46.2% continued to do so at least every 4 months.
An adjusted model linked more screening to the use of anthracyclines and taxanes, radiation treatment, and living in the Northeast vs. the West.
“HF was more frequently identified among patients undergoing recommended cardiac monitoring (10.4% compared with 6.5%, respectively; P less than.001), suggesting that, as more patients are screened, more patients are likely to be found having HF,” the researchers reported.
However, they added that “our sensitivity analysis using inpatient claims allowed us to determine that the HF identified using cardiac monitoring was not severe enough to require hospitalization and was likely asymptomatic. The clinical implications of the diagnosis of asymptomatic HF are hard to determine and are beyond the scope of this study.”
The researchers also noted that the findings suggest that screening has become more common in recent years.
“The number of cancer survivors is expected to increase over time, and we will continue to see patients develop treatment-related cardiotoxicity,” the researchers wrote. “Thus, more research, evidence-based guidelines, and tools for prediction of cancer treatment–related cardiotoxicity are needed.”
The National Cancer Institute and Cancer Prevention and Research Institute of Texas funded the study. Two study authors reported grant funding from the Susan G. Komen Breast Cancer Foundation, and one reports consulting for Pfizer and Roche. The other authors reported no disclosures.
SOURCE: Chavez-MacGregor M et al. JACC: Cardiovasc Imaging. 2018 Aug;11[8]1084-93.
FROM JACC: CARDIOVASCULAR IMAGING
Key clinical point: Women with breast cancer who take trastuzumab (Herceptin) may face double the adjusted risk of heart failure, but most aren’t screened frequently.
Major finding: Patients who took trastuzumab were 2.01 times more likely to develop HF (HR, 95% CI, 1.72-2.36) than were those who didn’t. Of all patients who took the drug, fewer than half received recommended frequency of screening.
Study details: Analysis of 16,456 U.S. adult women with nonmetastatic breast cancer diagnosed from 2009 to 2014 and tracked through 2015. Of those, 4.2% developed HF.
Disclosures: The National Cancer Institute and Cancer Prevention and Research Institute of Texas funded the study. Two study authors report grant funding from the Susan G. Komen Breast Cancer Foundation, and one reports consulting for Pfizer and Roche. The other authors report no disclosures.
Source: Chavez-MacGregor M et al. JACC: Cardiovasc Imaging 2018 Aug;11[8]1084-93.
Breast Implant Rupture After Radiation
The rupture rate for breast implants is about 10% at 10 years after insertion. That means women aged ≥ 70 years have a greater risk of rupture. For women who had breast augmentation or reconstruction before the advent of fifth-generation implants, there are no specific recommendations regarding follow-up and very little guidance in the literature about management for those who have had implants after radiation, say clinicians from Mayo Clinic.
They report on a 74-year-old patient who was treated for breast cancer in 1987 and 1988. She underwent lumpectomy, adjuvant unilateral radiation, a right simple mastectomy, left modified radical mastectomy, and implant-based reconstruction. Nearly 30 years later, she felt an asymmetry in 1 breast. Magnetic resonance imaging and ultrasound revealed that both implants had ruptured.
It is well known, the clinicians say, that complications of postmastectomy radiotherapy include capsular contracture, infection, and loss of prosthesis in implant-based reconstruction. Studies have shown that fibrosis, a hallmark of chronic radiation therapy, can show up even several years after radiotherapy—underscoring the importance of long-term follow-up for these patients. Moreover, the fact that the consequences of silicone on irradiated mastectomy flaps is unknown posed a further challenge.
While the cause of their patient’s implant rupture is unknown, the clinicians say it is “very likely” that delayed-onset fibrosis and capsular contracture secondary to radiation played a role. Such complications, though rare, should be kept in mind, the clinicians advise, when evaluating patients who had radiation and implants.
Source:
Molinar VE, Sabbagh MD, Manrique OJ. BMJ Case Rep. 2018; pii: bcr-2018-224578.
doi: 10.1136/bcr-2018-224578.
The rupture rate for breast implants is about 10% at 10 years after insertion. That means women aged ≥ 70 years have a greater risk of rupture. For women who had breast augmentation or reconstruction before the advent of fifth-generation implants, there are no specific recommendations regarding follow-up and very little guidance in the literature about management for those who have had implants after radiation, say clinicians from Mayo Clinic.
They report on a 74-year-old patient who was treated for breast cancer in 1987 and 1988. She underwent lumpectomy, adjuvant unilateral radiation, a right simple mastectomy, left modified radical mastectomy, and implant-based reconstruction. Nearly 30 years later, she felt an asymmetry in 1 breast. Magnetic resonance imaging and ultrasound revealed that both implants had ruptured.
It is well known, the clinicians say, that complications of postmastectomy radiotherapy include capsular contracture, infection, and loss of prosthesis in implant-based reconstruction. Studies have shown that fibrosis, a hallmark of chronic radiation therapy, can show up even several years after radiotherapy—underscoring the importance of long-term follow-up for these patients. Moreover, the fact that the consequences of silicone on irradiated mastectomy flaps is unknown posed a further challenge.
While the cause of their patient’s implant rupture is unknown, the clinicians say it is “very likely” that delayed-onset fibrosis and capsular contracture secondary to radiation played a role. Such complications, though rare, should be kept in mind, the clinicians advise, when evaluating patients who had radiation and implants.
Source:
Molinar VE, Sabbagh MD, Manrique OJ. BMJ Case Rep. 2018; pii: bcr-2018-224578.
doi: 10.1136/bcr-2018-224578.
The rupture rate for breast implants is about 10% at 10 years after insertion. That means women aged ≥ 70 years have a greater risk of rupture. For women who had breast augmentation or reconstruction before the advent of fifth-generation implants, there are no specific recommendations regarding follow-up and very little guidance in the literature about management for those who have had implants after radiation, say clinicians from Mayo Clinic.
They report on a 74-year-old patient who was treated for breast cancer in 1987 and 1988. She underwent lumpectomy, adjuvant unilateral radiation, a right simple mastectomy, left modified radical mastectomy, and implant-based reconstruction. Nearly 30 years later, she felt an asymmetry in 1 breast. Magnetic resonance imaging and ultrasound revealed that both implants had ruptured.
It is well known, the clinicians say, that complications of postmastectomy radiotherapy include capsular contracture, infection, and loss of prosthesis in implant-based reconstruction. Studies have shown that fibrosis, a hallmark of chronic radiation therapy, can show up even several years after radiotherapy—underscoring the importance of long-term follow-up for these patients. Moreover, the fact that the consequences of silicone on irradiated mastectomy flaps is unknown posed a further challenge.
While the cause of their patient’s implant rupture is unknown, the clinicians say it is “very likely” that delayed-onset fibrosis and capsular contracture secondary to radiation played a role. Such complications, though rare, should be kept in mind, the clinicians advise, when evaluating patients who had radiation and implants.
Source:
Molinar VE, Sabbagh MD, Manrique OJ. BMJ Case Rep. 2018; pii: bcr-2018-224578.
doi: 10.1136/bcr-2018-224578.
CTCs linked to late recurrence in HER2–, HR+ breast cancer
Circulating tumor cells could be used to stratify patients with hormone receptor (HR)–positive, HER2-negative breast cancer for late recurrence risk, results of a secondary analysis of a randomized clinical trial suggest.
Risk of late clinical recurrence was about 13-fold higher among HR-positive patients with a positive circulating tumor cell (CTC) assay result, according to results of the study, published in JAMA Oncology.
“This prospectively conducted study offers a high level of evidence supporting the association between a positive CTC assay result and risk of clinical recurrence,” said Joseph A. Sparano, MD, of Albert Einstein College of Medicine, New York, and his coauthors.
The present study is the first to show that this CTC assay may play a role in determining late clinical recurrence after local and systemic adjuvant therapy, according to the investigators.
The study is a secondary analysis of E5103, a phase 3 trial of adjuvant doxorubicin and cyclophosphamide followed by paclitaxel with bevacizumab in patients with HER2-negative stage II-III breast cancer. Investigators included a total of 547 patients who had no clinical evidence of recurrence between 4.5 and 7.5 years of registration in that trial.
Positive CTC assay results occurred in 26 of those patients (4.8%), they found.
At a median follow-up of 2.6 years, 24 patients had a clinical recurrence, including 23 HR-positive patients and just 1 HR-negative patient. Accordingly, the investigators focused most of their further analysis on the HR-positive subset.
A total of 7 of 23 patients with HR-positive disease (30.4%) had a positive CTC assay result.
A positive CTC result in HR-positive patients was associated with a 13.1-fold increased risk of recurrence, multivariate analysis showed.
Higher CTC burden appeared to be associated with a numerically higher recurrence risk in HR-positive patients, the investigators found. They saw recurrences in 16 of 335 patients with a CTC count of 0 cells per 7.5 mL blood (4.8%), compared with 2 of 12 patients with 1 cell per 7.5 mL blood (16.7%), and 5 of 6 patients with 2 or more cells per 7.5 mL (83.3%).
Taken together, these results provided proof of concept to support additional investigations of the CTC assay and other blood-based biomarker tests in the setting of late clinical recurrence in HR-positive patients, the researchers said.
They acknowledged several limitations of this study: It was small, it had relatively short follow-up, and it did not evaluate the CTC assay in the context of other assays.
“Notwithstanding proof of concept, further evaluation is required to confirm the clinical validity and determine the clinical utility of performing the CTC assay in this context,” Dr. Sparano and his coauthors wrote.
Late recurrences, or those that occur more than 5 years after diagnosis, account for about half of all recurrences among HR-positive receptive breast cancers, Dr. Sparano and his colleagues said.
The researchers had no conflicts of interest to report. The study was supported by grants from the National Cancer Institute, National Institutes of Health, Breast Cancer Research Foundation, and Susan G. Komen Foundation.
SOURCE: Sparano J et al. JAMA Oncol. 2018 Jul 26. doi: 10.1001/jamaoncol.2018.2574.
Circulating tumor cells could be used to stratify patients with hormone receptor (HR)–positive, HER2-negative breast cancer for late recurrence risk, results of a secondary analysis of a randomized clinical trial suggest.
Risk of late clinical recurrence was about 13-fold higher among HR-positive patients with a positive circulating tumor cell (CTC) assay result, according to results of the study, published in JAMA Oncology.
“This prospectively conducted study offers a high level of evidence supporting the association between a positive CTC assay result and risk of clinical recurrence,” said Joseph A. Sparano, MD, of Albert Einstein College of Medicine, New York, and his coauthors.
The present study is the first to show that this CTC assay may play a role in determining late clinical recurrence after local and systemic adjuvant therapy, according to the investigators.
The study is a secondary analysis of E5103, a phase 3 trial of adjuvant doxorubicin and cyclophosphamide followed by paclitaxel with bevacizumab in patients with HER2-negative stage II-III breast cancer. Investigators included a total of 547 patients who had no clinical evidence of recurrence between 4.5 and 7.5 years of registration in that trial.
Positive CTC assay results occurred in 26 of those patients (4.8%), they found.
At a median follow-up of 2.6 years, 24 patients had a clinical recurrence, including 23 HR-positive patients and just 1 HR-negative patient. Accordingly, the investigators focused most of their further analysis on the HR-positive subset.
A total of 7 of 23 patients with HR-positive disease (30.4%) had a positive CTC assay result.
A positive CTC result in HR-positive patients was associated with a 13.1-fold increased risk of recurrence, multivariate analysis showed.
Higher CTC burden appeared to be associated with a numerically higher recurrence risk in HR-positive patients, the investigators found. They saw recurrences in 16 of 335 patients with a CTC count of 0 cells per 7.5 mL blood (4.8%), compared with 2 of 12 patients with 1 cell per 7.5 mL blood (16.7%), and 5 of 6 patients with 2 or more cells per 7.5 mL (83.3%).
Taken together, these results provided proof of concept to support additional investigations of the CTC assay and other blood-based biomarker tests in the setting of late clinical recurrence in HR-positive patients, the researchers said.
They acknowledged several limitations of this study: It was small, it had relatively short follow-up, and it did not evaluate the CTC assay in the context of other assays.
“Notwithstanding proof of concept, further evaluation is required to confirm the clinical validity and determine the clinical utility of performing the CTC assay in this context,” Dr. Sparano and his coauthors wrote.
Late recurrences, or those that occur more than 5 years after diagnosis, account for about half of all recurrences among HR-positive receptive breast cancers, Dr. Sparano and his colleagues said.
The researchers had no conflicts of interest to report. The study was supported by grants from the National Cancer Institute, National Institutes of Health, Breast Cancer Research Foundation, and Susan G. Komen Foundation.
SOURCE: Sparano J et al. JAMA Oncol. 2018 Jul 26. doi: 10.1001/jamaoncol.2018.2574.
Circulating tumor cells could be used to stratify patients with hormone receptor (HR)–positive, HER2-negative breast cancer for late recurrence risk, results of a secondary analysis of a randomized clinical trial suggest.
Risk of late clinical recurrence was about 13-fold higher among HR-positive patients with a positive circulating tumor cell (CTC) assay result, according to results of the study, published in JAMA Oncology.
“This prospectively conducted study offers a high level of evidence supporting the association between a positive CTC assay result and risk of clinical recurrence,” said Joseph A. Sparano, MD, of Albert Einstein College of Medicine, New York, and his coauthors.
The present study is the first to show that this CTC assay may play a role in determining late clinical recurrence after local and systemic adjuvant therapy, according to the investigators.
The study is a secondary analysis of E5103, a phase 3 trial of adjuvant doxorubicin and cyclophosphamide followed by paclitaxel with bevacizumab in patients with HER2-negative stage II-III breast cancer. Investigators included a total of 547 patients who had no clinical evidence of recurrence between 4.5 and 7.5 years of registration in that trial.
Positive CTC assay results occurred in 26 of those patients (4.8%), they found.
At a median follow-up of 2.6 years, 24 patients had a clinical recurrence, including 23 HR-positive patients and just 1 HR-negative patient. Accordingly, the investigators focused most of their further analysis on the HR-positive subset.
A total of 7 of 23 patients with HR-positive disease (30.4%) had a positive CTC assay result.
A positive CTC result in HR-positive patients was associated with a 13.1-fold increased risk of recurrence, multivariate analysis showed.
Higher CTC burden appeared to be associated with a numerically higher recurrence risk in HR-positive patients, the investigators found. They saw recurrences in 16 of 335 patients with a CTC count of 0 cells per 7.5 mL blood (4.8%), compared with 2 of 12 patients with 1 cell per 7.5 mL blood (16.7%), and 5 of 6 patients with 2 or more cells per 7.5 mL (83.3%).
Taken together, these results provided proof of concept to support additional investigations of the CTC assay and other blood-based biomarker tests in the setting of late clinical recurrence in HR-positive patients, the researchers said.
They acknowledged several limitations of this study: It was small, it had relatively short follow-up, and it did not evaluate the CTC assay in the context of other assays.
“Notwithstanding proof of concept, further evaluation is required to confirm the clinical validity and determine the clinical utility of performing the CTC assay in this context,” Dr. Sparano and his coauthors wrote.
Late recurrences, or those that occur more than 5 years after diagnosis, account for about half of all recurrences among HR-positive receptive breast cancers, Dr. Sparano and his colleagues said.
The researchers had no conflicts of interest to report. The study was supported by grants from the National Cancer Institute, National Institutes of Health, Breast Cancer Research Foundation, and Susan G. Komen Foundation.
SOURCE: Sparano J et al. JAMA Oncol. 2018 Jul 26. doi: 10.1001/jamaoncol.2018.2574.
FROM JAMA ONCOLOGY
Key clinical point: Circulating tumor cells (CTC) may help to evaluate late recurrence risk in patients with HER2-negative breast cancer.
Major finding: A positive CTC result was associated with a 13.1-fold increased risk of recurrence in hormone receptor–positive patients.
Study details: Secondary analysis of a randomized clinical trial including 547 patients with HER2-negative stage II-III breast cancer.
Disclosures: The study was supported by grants from the National Cancer Institute, National Institutes of Health, Breast Cancer Research Foundation, and Susan G. Komen Foundation. The authors reported no conflicts of interest.
Source: Sparano J et al. JAMA Oncol. 2018 Jul 26. doi: 10.1001/jamaoncol.2018.2574.
Lower CTC count IDs indolent MBC disease subset
CHICAGO – A circulating tumor cell (CTC) count less than 5 per 7.5 mL of blood in patients with metastatic breast cancer indicates an indolent disease subset, according to a pooled analysis of individual patient data from two large cohorts.
The findings, which were independent of molecular subtype, disease location, or line of treatment, have important implications for CTC-based staging of metastatic breast cancer (MBC), which in turn could guide treatment decision making and drug development, Andrew A. Davis, MD, of Northwestern University, Chicago, and his colleagues reported in a poster at the annual meeting of the American Society of Clinical Oncology.
In 1,944 patients from the European Pooled Analysis Consortium (EPAC) and 492 from MD Anderson Cancer Center, CTC counts of 5 per 7.5mL or greater were associated with worse outcomes overall (hazard ratio, 2.43), the investigators said.
Median overall survival (OS) among all patients with CTC counts less than 5, who were considered to have stage IV indolent disease (stage IVindolent), was 36.3 months, and OS among those with de novo disease and CDC counts less than 5 was greater than 5.5 years, they said, noting that the survival benefit persisted across all disease subtypes.
For example, median OS in patients with stage IVindolent vs. stage IVaggressive (those with CTC counts of 5 or greater ) was 44.0 vs. 17.3 months in patients with hormone receptor–positive disease, 23.8 vs. 9.0 months in triple negative breast cancer patients, and 36.7 vs. 20.4 months in patients with HER2+ disease, respectively, they explained.They also noted that stage IVindolent disease could discriminate a less aggressive cohort both for patients with and without prior treatment; the hazard ratios were 0.40 and 0.42 favoring indolent disease for both first-line treatment and treatment beyond the first line, respectively.
In early-stage breast cancer, diagnostic tools have been incorporated into practice to help identify patients who will benefit from conservative vs. aggressive therapy, and the current findings suggest that CTC counts could be used in that manner for staging MBC.
“We propose a CTC-based staging system for MBC based on indolent and aggressive disease to incorporate into the American Joint Committee on Cancer [tumor node metastasis] staging classification,” they wrote, adding that prospective studies of single-agent, cost-effective treatments for stage IVindolent disease in the first-line setting are needed.
This study was supported by the Lynn Sage Breast Cancer Research OncoSET Program at Robert H. Lurie Cancer Center. Dr. Davis reported having no disclosures.
SOURCE: Davis A et al., ASCO 2018 Poster 1019.
CHICAGO – A circulating tumor cell (CTC) count less than 5 per 7.5 mL of blood in patients with metastatic breast cancer indicates an indolent disease subset, according to a pooled analysis of individual patient data from two large cohorts.
The findings, which were independent of molecular subtype, disease location, or line of treatment, have important implications for CTC-based staging of metastatic breast cancer (MBC), which in turn could guide treatment decision making and drug development, Andrew A. Davis, MD, of Northwestern University, Chicago, and his colleagues reported in a poster at the annual meeting of the American Society of Clinical Oncology.
In 1,944 patients from the European Pooled Analysis Consortium (EPAC) and 492 from MD Anderson Cancer Center, CTC counts of 5 per 7.5mL or greater were associated with worse outcomes overall (hazard ratio, 2.43), the investigators said.
Median overall survival (OS) among all patients with CTC counts less than 5, who were considered to have stage IV indolent disease (stage IVindolent), was 36.3 months, and OS among those with de novo disease and CDC counts less than 5 was greater than 5.5 years, they said, noting that the survival benefit persisted across all disease subtypes.
For example, median OS in patients with stage IVindolent vs. stage IVaggressive (those with CTC counts of 5 or greater ) was 44.0 vs. 17.3 months in patients with hormone receptor–positive disease, 23.8 vs. 9.0 months in triple negative breast cancer patients, and 36.7 vs. 20.4 months in patients with HER2+ disease, respectively, they explained.They also noted that stage IVindolent disease could discriminate a less aggressive cohort both for patients with and without prior treatment; the hazard ratios were 0.40 and 0.42 favoring indolent disease for both first-line treatment and treatment beyond the first line, respectively.
In early-stage breast cancer, diagnostic tools have been incorporated into practice to help identify patients who will benefit from conservative vs. aggressive therapy, and the current findings suggest that CTC counts could be used in that manner for staging MBC.
“We propose a CTC-based staging system for MBC based on indolent and aggressive disease to incorporate into the American Joint Committee on Cancer [tumor node metastasis] staging classification,” they wrote, adding that prospective studies of single-agent, cost-effective treatments for stage IVindolent disease in the first-line setting are needed.
This study was supported by the Lynn Sage Breast Cancer Research OncoSET Program at Robert H. Lurie Cancer Center. Dr. Davis reported having no disclosures.
SOURCE: Davis A et al., ASCO 2018 Poster 1019.
CHICAGO – A circulating tumor cell (CTC) count less than 5 per 7.5 mL of blood in patients with metastatic breast cancer indicates an indolent disease subset, according to a pooled analysis of individual patient data from two large cohorts.
The findings, which were independent of molecular subtype, disease location, or line of treatment, have important implications for CTC-based staging of metastatic breast cancer (MBC), which in turn could guide treatment decision making and drug development, Andrew A. Davis, MD, of Northwestern University, Chicago, and his colleagues reported in a poster at the annual meeting of the American Society of Clinical Oncology.
In 1,944 patients from the European Pooled Analysis Consortium (EPAC) and 492 from MD Anderson Cancer Center, CTC counts of 5 per 7.5mL or greater were associated with worse outcomes overall (hazard ratio, 2.43), the investigators said.
Median overall survival (OS) among all patients with CTC counts less than 5, who were considered to have stage IV indolent disease (stage IVindolent), was 36.3 months, and OS among those with de novo disease and CDC counts less than 5 was greater than 5.5 years, they said, noting that the survival benefit persisted across all disease subtypes.
For example, median OS in patients with stage IVindolent vs. stage IVaggressive (those with CTC counts of 5 or greater ) was 44.0 vs. 17.3 months in patients with hormone receptor–positive disease, 23.8 vs. 9.0 months in triple negative breast cancer patients, and 36.7 vs. 20.4 months in patients with HER2+ disease, respectively, they explained.They also noted that stage IVindolent disease could discriminate a less aggressive cohort both for patients with and without prior treatment; the hazard ratios were 0.40 and 0.42 favoring indolent disease for both first-line treatment and treatment beyond the first line, respectively.
In early-stage breast cancer, diagnostic tools have been incorporated into practice to help identify patients who will benefit from conservative vs. aggressive therapy, and the current findings suggest that CTC counts could be used in that manner for staging MBC.
“We propose a CTC-based staging system for MBC based on indolent and aggressive disease to incorporate into the American Joint Committee on Cancer [tumor node metastasis] staging classification,” they wrote, adding that prospective studies of single-agent, cost-effective treatments for stage IVindolent disease in the first-line setting are needed.
This study was supported by the Lynn Sage Breast Cancer Research OncoSET Program at Robert H. Lurie Cancer Center. Dr. Davis reported having no disclosures.
SOURCE: Davis A et al., ASCO 2018 Poster 1019.
REPORTING FROM ASCO 2018
Key clinical point: A CTC count less than 5 per 7.5 mL of blood in patients with MBC indicates an indolent disease subset.
Major finding: Median OS for stage IVindolent vs. stage IVaggressive disease was 4.0 vs. 17.3 months in HER2-negative patients, 23.8 vs. 9.0 months in TNBC patients, and 36.7 vs. 20.4 months in HER2-positive disease.
Study details: A pooled analysis of data from two cohort studies including 2,436 patients.
Disclosures: This study was supported by the Lynn Sage Breast Cancer Research OncoSET Program at Robert H. Lurie Cancer Center. Dr. Davis reported having no disclosures.
Source: Davis A et al. ASCO 2018 Poster 1019.