User login
Comprehensive mechanical circulatory support guidelines issued
As a further sign of how much mechanical circulatory support for advanced heart failure has matured, the International Society of Heart and Lung Transplantation issued on Jan. 10 the first comprehensive guidelines for all phases of evaluating, implanting, and managing patients who receive left ventricular assist devices or related equipment.
"Traditionally management of patients with mechanical circulatory support [MCS] was very center specific, but because the number of treated patients has increased, and because patients now live with these devices for years, we reached a point where we needed best practices guidelines, an expert consensus on what is the best way to approach this treatment" said Dr. Salpy V. Pamboukian, a cardiologist and one of three cochairs of the guidelines-writing project.

"When MSC started, the role of the devices was as a bridge to heart transplantation, but the field has evolved over the past decade and now MCS for destination therapy has opened a new array of patients who could benefit from these devices," said Dr. Pamboukian, medical director of the MCS device program at the University of Alabama, Birmingham. "We hope these guidelines will serve as a springboard for further research into the long-term management of these patients," she said in an interview.
"As pumps improve and the number of patients with advanced heart failure increases more and more patients will receive a ventricular assist device [VAD], and heart transplant will grow less relevant. These guidelines are much more comprehensive [than anything previously published] and they represent the opinions of the physicians, surgeons, nurses, and other providers who care for these patients," said Dr. David S. Feldman, a cardiologist who is director of the heart failure, VAD, and cardiac transplantation program at the Minneapolis Heart Institute at Abbott Northwestern Hospital, and another cochair of the guidelines committee.
The guidelines, which took about 3 years to produce, came from a committee of 35 health care providers, with initial review by three independent experts followed by additional peer review and then a period of open comment from the society’s membership. The 146-page document consists of more than 250 individual recommendations presented in five sections: patient selection; risk management prior to surgery; intraoperative procedures and immediate postoperative management; in-patient management during the immediate postoperative period; and long-term outpatient management (J. Heart Lung Transplant. 2013;32:157-87).
The writing committee admitted up front in the paper that most of the recommendations are consensus opinions with no clear evidence base. "It’s a limitation," admitted Dr. Pamboukian, "but you need a common approach to patients. Even a busy center may put in 50 or 60 VADs a year. Hopefully, a result of the guidelines is that they will help centers get together and produce the critical mass of patients needed to conduct meaningful trials. It was time to get something on paper; the new guidelines are what we will now work off of." But despite an absence of evidence on which to base many recommendations, "I was pleasantly surprised that there was more consensus than controversy. There was more commonality in our approaches than differences," she added.
The most limited number of recommendations came from the third task force of the panel, which handled intraoperative procedures and immediate postoperative care. Though this section runs 17 pages and deals with topics such as anesthesia, implantation techniques, establishing hemostasis, performing concomitant procedures, methods for explantation, and management of postoperative hemodynamics and bleeding, it contains just three specific recommendations, all dealing with anesthesia. "There are essentially no studies that have looked at how to make things better in the surgical suite," explained Dr. Feldman.
"It’s very challenging to standardize a surgical procedure," added Dr. Pamboukian. "We tried to summarize useful practices, but consensus-based recommendations are difficult to do."
Another topic the guidelines finesse is patient selection. The field is currently trying to sort out the best stage of advanced heart failure for patients to receive mechanical circulatory support. "You’d be amazed at the disparity of who gets these devices now," Dr. Feldman said. In addition, the guideline writing committee decided to defer definitive choices until results are available from a large study starting later this year. The study, Evaluation of VAD Intervention Before Inotropic Therapy (REVIVE-IT), will examine the outcomes of patients with advanced New York Heart Association stage III heart failure who receive a VAD. "It didn’t seem appropriate to address this because of the trial," he added.
Both Dr. Pamboukian and Dr. Feldman agreed that the newly released guidelines will likely be in place for only a couple of years before a revision comes out, testament to the rapid changes in this field. Dr. Feldman cited new VADs from at least two manufacturers expected to enter first-in-man studies this year, and the continued snowballing of VAD implantation rates. The most recent 2012 numbers (through Sept. 30, 2012) from the Interagency Registry for Mechanically Assisted Circulatory Support (INTERMACS) showed nearly 2,000 VADS getting implanted into U.S. patients last year, the highest annual rate ever.
"Because the field is growing, a lot of new centers want to establish programs. We want this treatment to reach as many appropriate patients as possible, but we want it to grow responsibly. These guidelines help establish the best practices, and help ensure that patients get the best care wherever they go," Dr. Pamboukian said.
Dr. Pamboukian said that she had no disclosures. Dr. Feldman said that he has received research support from Terumo.
As a further sign of how much mechanical circulatory support for advanced heart failure has matured, the International Society of Heart and Lung Transplantation issued on Jan. 10 the first comprehensive guidelines for all phases of evaluating, implanting, and managing patients who receive left ventricular assist devices or related equipment.
"Traditionally management of patients with mechanical circulatory support [MCS] was very center specific, but because the number of treated patients has increased, and because patients now live with these devices for years, we reached a point where we needed best practices guidelines, an expert consensus on what is the best way to approach this treatment" said Dr. Salpy V. Pamboukian, a cardiologist and one of three cochairs of the guidelines-writing project.
"When MSC started, the role of the devices was as a bridge to heart transplantation, but the field has evolved over the past decade and now MCS for destination therapy has opened a new array of patients who could benefit from these devices," said Dr. Pamboukian, medical director of the MCS device program at the University of Alabama, Birmingham. "We hope these guidelines will serve as a springboard for further research into the long-term management of these patients," she said in an interview.
"As pumps improve and the number of patients with advanced heart failure increases more and more patients will receive a ventricular assist device [VAD], and heart transplant will grow less relevant. These guidelines are much more comprehensive [than anything previously published] and they represent the opinions of the physicians, surgeons, nurses, and other providers who care for these patients," said Dr. David S. Feldman, a cardiologist who is director of the heart failure, VAD, and cardiac transplantation program at the Minneapolis Heart Institute at Abbott Northwestern Hospital, and another cochair of the guidelines committee.
The guidelines, which took about 3 years to produce, came from a committee of 35 health care providers, with initial review by three independent experts followed by additional peer review and then a period of open comment from the society’s membership. The 146-page document consists of more than 250 individual recommendations presented in five sections: patient selection; risk management prior to surgery; intraoperative procedures and immediate postoperative management; in-patient management during the immediate postoperative period; and long-term outpatient management (J. Heart Lung Transplant. 2013;32:157-87).
The writing committee admitted up front in the paper that most of the recommendations are consensus opinions with no clear evidence base. "It’s a limitation," admitted Dr. Pamboukian, "but you need a common approach to patients. Even a busy center may put in 50 or 60 VADs a year. Hopefully, a result of the guidelines is that they will help centers get together and produce the critical mass of patients needed to conduct meaningful trials. It was time to get something on paper; the new guidelines are what we will now work off of." But despite an absence of evidence on which to base many recommendations, "I was pleasantly surprised that there was more consensus than controversy. There was more commonality in our approaches than differences," she added.
The most limited number of recommendations came from the third task force of the panel, which handled intraoperative procedures and immediate postoperative care. Though this section runs 17 pages and deals with topics such as anesthesia, implantation techniques, establishing hemostasis, performing concomitant procedures, methods for explantation, and management of postoperative hemodynamics and bleeding, it contains just three specific recommendations, all dealing with anesthesia. "There are essentially no studies that have looked at how to make things better in the surgical suite," explained Dr. Feldman.
"It’s very challenging to standardize a surgical procedure," added Dr. Pamboukian. "We tried to summarize useful practices, but consensus-based recommendations are difficult to do."
Another topic the guidelines finesse is patient selection. The field is currently trying to sort out the best stage of advanced heart failure for patients to receive mechanical circulatory support. "You’d be amazed at the disparity of who gets these devices now," Dr. Feldman said. In addition, the guideline writing committee decided to defer definitive choices until results are available from a large study starting later this year. The study, Evaluation of VAD Intervention Before Inotropic Therapy (REVIVE-IT), will examine the outcomes of patients with advanced New York Heart Association stage III heart failure who receive a VAD. "It didn’t seem appropriate to address this because of the trial," he added.
Both Dr. Pamboukian and Dr. Feldman agreed that the newly released guidelines will likely be in place for only a couple of years before a revision comes out, testament to the rapid changes in this field. Dr. Feldman cited new VADs from at least two manufacturers expected to enter first-in-man studies this year, and the continued snowballing of VAD implantation rates. The most recent 2012 numbers (through Sept. 30, 2012) from the Interagency Registry for Mechanically Assisted Circulatory Support (INTERMACS) showed nearly 2,000 VADS getting implanted into U.S. patients last year, the highest annual rate ever.
"Because the field is growing, a lot of new centers want to establish programs. We want this treatment to reach as many appropriate patients as possible, but we want it to grow responsibly. These guidelines help establish the best practices, and help ensure that patients get the best care wherever they go," Dr. Pamboukian said.
Dr. Pamboukian said that she had no disclosures. Dr. Feldman said that he has received research support from Terumo.
As a further sign of how much mechanical circulatory support for advanced heart failure has matured, the International Society of Heart and Lung Transplantation issued on Jan. 10 the first comprehensive guidelines for all phases of evaluating, implanting, and managing patients who receive left ventricular assist devices or related equipment.
"Traditionally management of patients with mechanical circulatory support [MCS] was very center specific, but because the number of treated patients has increased, and because patients now live with these devices for years, we reached a point where we needed best practices guidelines, an expert consensus on what is the best way to approach this treatment" said Dr. Salpy V. Pamboukian, a cardiologist and one of three cochairs of the guidelines-writing project.
"When MSC started, the role of the devices was as a bridge to heart transplantation, but the field has evolved over the past decade and now MCS for destination therapy has opened a new array of patients who could benefit from these devices," said Dr. Pamboukian, medical director of the MCS device program at the University of Alabama, Birmingham. "We hope these guidelines will serve as a springboard for further research into the long-term management of these patients," she said in an interview.
"As pumps improve and the number of patients with advanced heart failure increases more and more patients will receive a ventricular assist device [VAD], and heart transplant will grow less relevant. These guidelines are much more comprehensive [than anything previously published] and they represent the opinions of the physicians, surgeons, nurses, and other providers who care for these patients," said Dr. David S. Feldman, a cardiologist who is director of the heart failure, VAD, and cardiac transplantation program at the Minneapolis Heart Institute at Abbott Northwestern Hospital, and another cochair of the guidelines committee.
The guidelines, which took about 3 years to produce, came from a committee of 35 health care providers, with initial review by three independent experts followed by additional peer review and then a period of open comment from the society’s membership. The 146-page document consists of more than 250 individual recommendations presented in five sections: patient selection; risk management prior to surgery; intraoperative procedures and immediate postoperative management; in-patient management during the immediate postoperative period; and long-term outpatient management (J. Heart Lung Transplant. 2013;32:157-87).
The writing committee admitted up front in the paper that most of the recommendations are consensus opinions with no clear evidence base. "It’s a limitation," admitted Dr. Pamboukian, "but you need a common approach to patients. Even a busy center may put in 50 or 60 VADs a year. Hopefully, a result of the guidelines is that they will help centers get together and produce the critical mass of patients needed to conduct meaningful trials. It was time to get something on paper; the new guidelines are what we will now work off of." But despite an absence of evidence on which to base many recommendations, "I was pleasantly surprised that there was more consensus than controversy. There was more commonality in our approaches than differences," she added.
The most limited number of recommendations came from the third task force of the panel, which handled intraoperative procedures and immediate postoperative care. Though this section runs 17 pages and deals with topics such as anesthesia, implantation techniques, establishing hemostasis, performing concomitant procedures, methods for explantation, and management of postoperative hemodynamics and bleeding, it contains just three specific recommendations, all dealing with anesthesia. "There are essentially no studies that have looked at how to make things better in the surgical suite," explained Dr. Feldman.
"It’s very challenging to standardize a surgical procedure," added Dr. Pamboukian. "We tried to summarize useful practices, but consensus-based recommendations are difficult to do."
Another topic the guidelines finesse is patient selection. The field is currently trying to sort out the best stage of advanced heart failure for patients to receive mechanical circulatory support. "You’d be amazed at the disparity of who gets these devices now," Dr. Feldman said. In addition, the guideline writing committee decided to defer definitive choices until results are available from a large study starting later this year. The study, Evaluation of VAD Intervention Before Inotropic Therapy (REVIVE-IT), will examine the outcomes of patients with advanced New York Heart Association stage III heart failure who receive a VAD. "It didn’t seem appropriate to address this because of the trial," he added.
Both Dr. Pamboukian and Dr. Feldman agreed that the newly released guidelines will likely be in place for only a couple of years before a revision comes out, testament to the rapid changes in this field. Dr. Feldman cited new VADs from at least two manufacturers expected to enter first-in-man studies this year, and the continued snowballing of VAD implantation rates. The most recent 2012 numbers (through Sept. 30, 2012) from the Interagency Registry for Mechanically Assisted Circulatory Support (INTERMACS) showed nearly 2,000 VADS getting implanted into U.S. patients last year, the highest annual rate ever.
"Because the field is growing, a lot of new centers want to establish programs. We want this treatment to reach as many appropriate patients as possible, but we want it to grow responsibly. These guidelines help establish the best practices, and help ensure that patients get the best care wherever they go," Dr. Pamboukian said.
Dr. Pamboukian said that she had no disclosures. Dr. Feldman said that he has received research support from Terumo.
FROM THE JOURNAL OF HEART AND LUNG TRANSPLANTATION

FDA makes dabigatran contraindicated for mechanical valves
The FDA has declared the anticoagulant dabigatran contraindicated for patients with a mechanical heart valve, following anecdotal reports of blood clots forming on mechanical prosthetic valves and the abrupt stoppage of a phase II trial that had been testing dabigatran in this patient population.
"The U.S. Food and Drug Administration is informing health care professionals and the public that the blood thinner Pradaxa [dabigatran etexilate mesylate] should not be used to prevent stroke or blood clots in patients with mechanical heart valves," the agency said in a Drug Safety Communication.
"Health care professionals should promptly transition any patient with a mechanical heart valve who is taking Pradaxa to another medication," the agency added.
The statement also noted that the European-based, randomized phase II study to evaluate the safety and pharmacokinetics of oral dabigatran versus warfarin in patients after heart valve replacement (RE-ALIGN) (Am. Heart J. 2012;163:931-7) had been halted earlier in December by Boehringer Ingelheim, the company that markets Pradaxa, because the dabigatran-treated patients had shown an excess of strokes, myocardial infarctions, and blood clots that formed on the valves. Patients on dabigatran also had more episodes of bleeding after valve surgery than did patients on warfarin.
Although Boehringer Ingelheim and the researchers who ran the RE-ALIGN trial have not yet released the details of exactly what happened in the study, a report appeared in October from a group of cardiac surgeons at the Ottawa (Ont.) Heart Institute on their experience with two patients with mechanical heart valves who developed a thrombus on their valves and significant symptoms within 2 or 3 months of being switched from warfarin to dabigatran by their primary care physicians (J. Am. Coll. Cardiol. 2012;60:1710-1). These switches, which occurred even though dabigatran treatment in patients with mechanical prosthetic valves is an off-label use, probably represent the tip of the iceberg, said Dr. Munir Boodhwani, a cardiac surgeon at the Heart Institute and lead author of the two case reports.
"I suspect [this off-label use] is more common than we know. We see the problems, but we don’t know the denominator," he said in an interview. "In Ottawa, we routinely evaluate heart valve recipients every 6-12 months, and we have seen a few patients who had been switched from warfarin to dabigatran or another new oral anticoagulant. It has not been just one or two isolated cases. When we see these patients, we switch them back, and we send a message to their physician who made the switch that maybe this was not a good idea."
One of the cases he and his associates reported was a 51-year-old woman with a mechanical aortic valve who had been on warfarin for 8 years without complications and then was switched to 150 mg dabigatran twice daily by her general practitioner. Within 2 months, she developed crackles and a systolic murmur, and an echocardiogram revealed severe prosthetic aortic valve stenosis and a probable mass on the prosthesis. She arrived at the Heart Institute in cardiogenic shock and had cardiac arrest in the operating room. Surgery revealed an extensive thrombus on the valve, which was replaced. After surgery she had a complete recovery.
The second reported case was a 59-year-old woman with a mechanical mitral valve who had been on warfarin treatment without complications for about 4 years before being switched by her family physicians to 150 mg dabigatran twice daily. She developed progressive dyspnea, and an echocardiogram revealed a large thrombus on the valve. She underwent valve replacement and had an uneventful recovery.
Although phase III trial results showed dabigatran safe and effective for preventing blood clots and strokes in patients with nonvalvular atrial fibrillation, "atrial fibrillation is very different in a patient with a mechanical heart valve," Dr. Boodhwani said. "You cannot translate efficacy for one population to another. For atrial fibrillation, dosages of 110 mg b.i.d. and 150 mg b.i.d were effective [in the RE-LY trial; N. Engl. J. Med. 2009;361:1139-51], but in RE-ALIGN, dabigatran seems to have not been effective even at a dosage of 300 mg b.i.d. I think that Boehringer Ingelheim and the other companies that make the new anticoagulants need to go back to the drawing board and do more preclinical studies to determine what is a safe and effective dosage for anticoagulating patients with mechanical heart valves. It will likely need a higher dose, and then the question will be, What is the bleeding risk?"
The safety and efficacy results from nonvalvular atrial fibrillation patients in RE-LY are impossible to extrapolate to patients with mechanical valves, agreed Dr. Michael D. Ezekowitz, who was a coprincipal investigator for RE-LY but had no involvement in RE-ALIGN.
"It may be that a direct thrombin inhibitor [such as dabigatran] is not the drug of choice for preventing clots from forming on the surface of valves," Dr. Ezekowtiz said in an interview. "We have nearly 50 years of experience using warfarin for patients with mechanical heart valves, and that is clearly the drug of choice. This [RE-ALIGN] was probably a high-risk trial" in terms of trying to show that a new drug was at least as safe and effective as warfarin for heart valve patients.
Dr. Ezekowitz stressed that he has not been privy to any details of the RE-ALIGN results, but he suggested that it is hard to imagine that higher dabigatran dosages than the 300 mg b.i.d tested in RE-ALIGN could be used safely. "I was principal investigator for a phase II dabigatran study in atrial fibrillation, the PETRO [Prevention of Embolic and Thrombotic Events in Patients With Persistent AF] study (Am. J. Cardiol. 2007;100:1419-26). We tested up to 300 mg b.i.d., and in older atrial fibrillation patients this caused an excess of gastrointestinal bleeds and we decided to abandon that dosage in further testing. From a purely practical standpoint, I doubt whether a dabigatran dosage of more than 150 mg b.i.d would be tolerable," said Dr. Ezekowitz, professor of medicine at Jefferson Medical College in Philadelphia and director of atrial fibrillation research and education at the Cardiovascular Research Foundation in New York.
Some community physicians seem to have been mistakenly lured into prescribing dabigatran or other new anticoagulants to mechanical heart valve patients, perhaps because these physicians equated the newer drugs with better performance, Dr. Boodhwani said. But extrapolating the atrial fibrillation experience to heart-valve patients is premature, he warned.
"There is the potential for patients to die on these drugs. The off-label use can potentially be quite dangerous."
The FDA’s action and termination of the RE-ALIGN trial should send physicians a strong warning, he said. "This doesn’t close the door to future use of these drugs for mechanical valve patients, but physicians and industry need to be more cautious. The problems with warfarin need solutions, but only in a step-by-step way."
Dr. Boodhwani said he had no relevant financial disclosures. Dr. Ezekowitz said he has been a consultant to, has received honoraria as a lecturer on behalf of, and has received research funding from Boehringer Ingelheim, as well as from other drug companies that market antithrombotic drugs.
On Twitter @mitchelzoler
The FDA has declared the anticoagulant dabigatran contraindicated for patients with a mechanical heart valve, following anecdotal reports of blood clots forming on mechanical prosthetic valves and the abrupt stoppage of a phase II trial that had been testing dabigatran in this patient population.
"The U.S. Food and Drug Administration is informing health care professionals and the public that the blood thinner Pradaxa [dabigatran etexilate mesylate] should not be used to prevent stroke or blood clots in patients with mechanical heart valves," the agency said in a Drug Safety Communication.
"Health care professionals should promptly transition any patient with a mechanical heart valve who is taking Pradaxa to another medication," the agency added.
The statement also noted that the European-based, randomized phase II study to evaluate the safety and pharmacokinetics of oral dabigatran versus warfarin in patients after heart valve replacement (RE-ALIGN) (Am. Heart J. 2012;163:931-7) had been halted earlier in December by Boehringer Ingelheim, the company that markets Pradaxa, because the dabigatran-treated patients had shown an excess of strokes, myocardial infarctions, and blood clots that formed on the valves. Patients on dabigatran also had more episodes of bleeding after valve surgery than did patients on warfarin.
Although Boehringer Ingelheim and the researchers who ran the RE-ALIGN trial have not yet released the details of exactly what happened in the study, a report appeared in October from a group of cardiac surgeons at the Ottawa (Ont.) Heart Institute on their experience with two patients with mechanical heart valves who developed a thrombus on their valves and significant symptoms within 2 or 3 months of being switched from warfarin to dabigatran by their primary care physicians (J. Am. Coll. Cardiol. 2012;60:1710-1). These switches, which occurred even though dabigatran treatment in patients with mechanical prosthetic valves is an off-label use, probably represent the tip of the iceberg, said Dr. Munir Boodhwani, a cardiac surgeon at the Heart Institute and lead author of the two case reports.
"I suspect [this off-label use] is more common than we know. We see the problems, but we don’t know the denominator," he said in an interview. "In Ottawa, we routinely evaluate heart valve recipients every 6-12 months, and we have seen a few patients who had been switched from warfarin to dabigatran or another new oral anticoagulant. It has not been just one or two isolated cases. When we see these patients, we switch them back, and we send a message to their physician who made the switch that maybe this was not a good idea."
One of the cases he and his associates reported was a 51-year-old woman with a mechanical aortic valve who had been on warfarin for 8 years without complications and then was switched to 150 mg dabigatran twice daily by her general practitioner. Within 2 months, she developed crackles and a systolic murmur, and an echocardiogram revealed severe prosthetic aortic valve stenosis and a probable mass on the prosthesis. She arrived at the Heart Institute in cardiogenic shock and had cardiac arrest in the operating room. Surgery revealed an extensive thrombus on the valve, which was replaced. After surgery she had a complete recovery.
The second reported case was a 59-year-old woman with a mechanical mitral valve who had been on warfarin treatment without complications for about 4 years before being switched by her family physicians to 150 mg dabigatran twice daily. She developed progressive dyspnea, and an echocardiogram revealed a large thrombus on the valve. She underwent valve replacement and had an uneventful recovery.
Although phase III trial results showed dabigatran safe and effective for preventing blood clots and strokes in patients with nonvalvular atrial fibrillation, "atrial fibrillation is very different in a patient with a mechanical heart valve," Dr. Boodhwani said. "You cannot translate efficacy for one population to another. For atrial fibrillation, dosages of 110 mg b.i.d. and 150 mg b.i.d were effective [in the RE-LY trial; N. Engl. J. Med. 2009;361:1139-51], but in RE-ALIGN, dabigatran seems to have not been effective even at a dosage of 300 mg b.i.d. I think that Boehringer Ingelheim and the other companies that make the new anticoagulants need to go back to the drawing board and do more preclinical studies to determine what is a safe and effective dosage for anticoagulating patients with mechanical heart valves. It will likely need a higher dose, and then the question will be, What is the bleeding risk?"
The safety and efficacy results from nonvalvular atrial fibrillation patients in RE-LY are impossible to extrapolate to patients with mechanical valves, agreed Dr. Michael D. Ezekowitz, who was a coprincipal investigator for RE-LY but had no involvement in RE-ALIGN.
"It may be that a direct thrombin inhibitor [such as dabigatran] is not the drug of choice for preventing clots from forming on the surface of valves," Dr. Ezekowtiz said in an interview. "We have nearly 50 years of experience using warfarin for patients with mechanical heart valves, and that is clearly the drug of choice. This [RE-ALIGN] was probably a high-risk trial" in terms of trying to show that a new drug was at least as safe and effective as warfarin for heart valve patients.
Dr. Ezekowitz stressed that he has not been privy to any details of the RE-ALIGN results, but he suggested that it is hard to imagine that higher dabigatran dosages than the 300 mg b.i.d tested in RE-ALIGN could be used safely. "I was principal investigator for a phase II dabigatran study in atrial fibrillation, the PETRO [Prevention of Embolic and Thrombotic Events in Patients With Persistent AF] study (Am. J. Cardiol. 2007;100:1419-26). We tested up to 300 mg b.i.d., and in older atrial fibrillation patients this caused an excess of gastrointestinal bleeds and we decided to abandon that dosage in further testing. From a purely practical standpoint, I doubt whether a dabigatran dosage of more than 150 mg b.i.d would be tolerable," said Dr. Ezekowitz, professor of medicine at Jefferson Medical College in Philadelphia and director of atrial fibrillation research and education at the Cardiovascular Research Foundation in New York.
Some community physicians seem to have been mistakenly lured into prescribing dabigatran or other new anticoagulants to mechanical heart valve patients, perhaps because these physicians equated the newer drugs with better performance, Dr. Boodhwani said. But extrapolating the atrial fibrillation experience to heart-valve patients is premature, he warned.
"There is the potential for patients to die on these drugs. The off-label use can potentially be quite dangerous."
The FDA’s action and termination of the RE-ALIGN trial should send physicians a strong warning, he said. "This doesn’t close the door to future use of these drugs for mechanical valve patients, but physicians and industry need to be more cautious. The problems with warfarin need solutions, but only in a step-by-step way."
Dr. Boodhwani said he had no relevant financial disclosures. Dr. Ezekowitz said he has been a consultant to, has received honoraria as a lecturer on behalf of, and has received research funding from Boehringer Ingelheim, as well as from other drug companies that market antithrombotic drugs.
On Twitter @mitchelzoler
The FDA has declared the anticoagulant dabigatran contraindicated for patients with a mechanical heart valve, following anecdotal reports of blood clots forming on mechanical prosthetic valves and the abrupt stoppage of a phase II trial that had been testing dabigatran in this patient population.
"The U.S. Food and Drug Administration is informing health care professionals and the public that the blood thinner Pradaxa [dabigatran etexilate mesylate] should not be used to prevent stroke or blood clots in patients with mechanical heart valves," the agency said in a Drug Safety Communication.
"Health care professionals should promptly transition any patient with a mechanical heart valve who is taking Pradaxa to another medication," the agency added.
The statement also noted that the European-based, randomized phase II study to evaluate the safety and pharmacokinetics of oral dabigatran versus warfarin in patients after heart valve replacement (RE-ALIGN) (Am. Heart J. 2012;163:931-7) had been halted earlier in December by Boehringer Ingelheim, the company that markets Pradaxa, because the dabigatran-treated patients had shown an excess of strokes, myocardial infarctions, and blood clots that formed on the valves. Patients on dabigatran also had more episodes of bleeding after valve surgery than did patients on warfarin.
Although Boehringer Ingelheim and the researchers who ran the RE-ALIGN trial have not yet released the details of exactly what happened in the study, a report appeared in October from a group of cardiac surgeons at the Ottawa (Ont.) Heart Institute on their experience with two patients with mechanical heart valves who developed a thrombus on their valves and significant symptoms within 2 or 3 months of being switched from warfarin to dabigatran by their primary care physicians (J. Am. Coll. Cardiol. 2012;60:1710-1). These switches, which occurred even though dabigatran treatment in patients with mechanical prosthetic valves is an off-label use, probably represent the tip of the iceberg, said Dr. Munir Boodhwani, a cardiac surgeon at the Heart Institute and lead author of the two case reports.
"I suspect [this off-label use] is more common than we know. We see the problems, but we don’t know the denominator," he said in an interview. "In Ottawa, we routinely evaluate heart valve recipients every 6-12 months, and we have seen a few patients who had been switched from warfarin to dabigatran or another new oral anticoagulant. It has not been just one or two isolated cases. When we see these patients, we switch them back, and we send a message to their physician who made the switch that maybe this was not a good idea."
One of the cases he and his associates reported was a 51-year-old woman with a mechanical aortic valve who had been on warfarin for 8 years without complications and then was switched to 150 mg dabigatran twice daily by her general practitioner. Within 2 months, she developed crackles and a systolic murmur, and an echocardiogram revealed severe prosthetic aortic valve stenosis and a probable mass on the prosthesis. She arrived at the Heart Institute in cardiogenic shock and had cardiac arrest in the operating room. Surgery revealed an extensive thrombus on the valve, which was replaced. After surgery she had a complete recovery.
The second reported case was a 59-year-old woman with a mechanical mitral valve who had been on warfarin treatment without complications for about 4 years before being switched by her family physicians to 150 mg dabigatran twice daily. She developed progressive dyspnea, and an echocardiogram revealed a large thrombus on the valve. She underwent valve replacement and had an uneventful recovery.
Although phase III trial results showed dabigatran safe and effective for preventing blood clots and strokes in patients with nonvalvular atrial fibrillation, "atrial fibrillation is very different in a patient with a mechanical heart valve," Dr. Boodhwani said. "You cannot translate efficacy for one population to another. For atrial fibrillation, dosages of 110 mg b.i.d. and 150 mg b.i.d were effective [in the RE-LY trial; N. Engl. J. Med. 2009;361:1139-51], but in RE-ALIGN, dabigatran seems to have not been effective even at a dosage of 300 mg b.i.d. I think that Boehringer Ingelheim and the other companies that make the new anticoagulants need to go back to the drawing board and do more preclinical studies to determine what is a safe and effective dosage for anticoagulating patients with mechanical heart valves. It will likely need a higher dose, and then the question will be, What is the bleeding risk?"
The safety and efficacy results from nonvalvular atrial fibrillation patients in RE-LY are impossible to extrapolate to patients with mechanical valves, agreed Dr. Michael D. Ezekowitz, who was a coprincipal investigator for RE-LY but had no involvement in RE-ALIGN.
"It may be that a direct thrombin inhibitor [such as dabigatran] is not the drug of choice for preventing clots from forming on the surface of valves," Dr. Ezekowtiz said in an interview. "We have nearly 50 years of experience using warfarin for patients with mechanical heart valves, and that is clearly the drug of choice. This [RE-ALIGN] was probably a high-risk trial" in terms of trying to show that a new drug was at least as safe and effective as warfarin for heart valve patients.
Dr. Ezekowitz stressed that he has not been privy to any details of the RE-ALIGN results, but he suggested that it is hard to imagine that higher dabigatran dosages than the 300 mg b.i.d tested in RE-ALIGN could be used safely. "I was principal investigator for a phase II dabigatran study in atrial fibrillation, the PETRO [Prevention of Embolic and Thrombotic Events in Patients With Persistent AF] study (Am. J. Cardiol. 2007;100:1419-26). We tested up to 300 mg b.i.d., and in older atrial fibrillation patients this caused an excess of gastrointestinal bleeds and we decided to abandon that dosage in further testing. From a purely practical standpoint, I doubt whether a dabigatran dosage of more than 150 mg b.i.d would be tolerable," said Dr. Ezekowitz, professor of medicine at Jefferson Medical College in Philadelphia and director of atrial fibrillation research and education at the Cardiovascular Research Foundation in New York.
Some community physicians seem to have been mistakenly lured into prescribing dabigatran or other new anticoagulants to mechanical heart valve patients, perhaps because these physicians equated the newer drugs with better performance, Dr. Boodhwani said. But extrapolating the atrial fibrillation experience to heart-valve patients is premature, he warned.
"There is the potential for patients to die on these drugs. The off-label use can potentially be quite dangerous."
The FDA’s action and termination of the RE-ALIGN trial should send physicians a strong warning, he said. "This doesn’t close the door to future use of these drugs for mechanical valve patients, but physicians and industry need to be more cautious. The problems with warfarin need solutions, but only in a step-by-step way."
Dr. Boodhwani said he had no relevant financial disclosures. Dr. Ezekowitz said he has been a consultant to, has received honoraria as a lecturer on behalf of, and has received research funding from Boehringer Ingelheim, as well as from other drug companies that market antithrombotic drugs.
On Twitter @mitchelzoler
Progress Limited in Transcatheter Repair of Mitral Regurgitation
CHICAGO – Within narrow limits, transcatheter repair of mitral regurgitation can have outcomes meeting or even exceeding those of surgery.
"I believe that mitral regurgitation is mostly a surgical disease, but that interventional devices will, sooner or later, be able to manage some of the spectrum of mitral pathology," said Dr. Howard C. Herrmann at the Heart Valve Summit 2012. Dr. Herrmann is professor of medicine and director of interventional cardiology and cardiac catheterization at the University of Pennsylvania, Philadelphia.

"Transcatheter approaches are always going to be somewhat more limited than surgery," said Dr. Herrmann. The device that repairs a leaflet edge to edge is very different from the one that can be used to perform indirect annular dilatation via the coronary sinus, he noted.
Two devices relevant to those indications show promise, although neither is yet approved by the Food and Drug Administration (FDA), he said. These are Cardiac Dimensions’ Carillon for annular dilatation, and Abbott Vascular’s MitraClip for leaflet repair.
In the European TITAN clinical trial, 36 of 53 patients (68%) were implanted with the Carillon Mitral Contour System. The average baseline ejection fraction of all 53 patients was 28%, which Dr. Herrmann characterized as true functional mitral regurgitation (FMR). The prospective, nonrandomized, double-arm study compared results from the implanted group to results from a group without implants.
Implanted patients demonstrated significant reductions in FMR as assessed by multiple quantitative measures, including mean regurgitant volume, which decreased from 34.5 +/– 11.5 mL to 17.4 +/– 12.4 mL over 12 months (P less than .001) (Eur. J. Heart Fail. 2012;14:931-8).
These and other reductions correlated with symptomatic improvement, said Dr. Herrmann. However, the coronary sinus approach to percutaneous mitral annuloplasty is clearly limited by the variable anatomy between the coronary sinus and the posterior mitral annulus, as well as by the risk of coronary artery constriction, he said. Additionally, the long-term benefit of a surgical partial circumference ring is unknown (Circulation 2006;114:377-80).
The other device, the MitraClip, was evaluated in the EVEREST II trial for safety and efficacy in treatment of mitral valve regurgitation.
"Clearly, if you look at the primary effectiveness rate as defined by the trial, which is not a very high bar, surgery still beat the device," said Dr. Herrmann. However, he noted, the device clearly beat surgery in terms of morbidity and other safety end points.
"But when you look at it from an intent-to-treat analysis, those results are clearly inferior with MitraClip: At 2 years, 78.2% of percutaneous patients are free from mitral valve surgery, versus 96.1% of surgery patients (P less than .001).
"MitraClip should not and is not being used for low-risk patients with degenerative disease amenable to surgical repair. ... That said, in the patients who had good results with MitraClip, those results do appear to be fairly durable," he said. At 3 years, all-cause mortality was very similar between the two interventions. (J. Thorac. Cardiovasc. Surg. 2012;143:S60-3; J. Am. Coll. Cardiol. 2012;59:130-9).
Analysis of the high-surgical-risk cohort (n = 78) from EVEREST II revealed a 45% decrease in rehospitalizations (P = .02) at 12 months after intervention with MitraClip, said Dr. Herrmann. This finding led investigators to launch the COAPT trial, which has been approved by the FDA and is expected to start soon in the United States. Approximately 420 patients with significant FMR, thought to be at extremely high risk for surgery, will be randomized 1:1 to MitraClip (n = 210) or medical therapy (n = 210). The primary end point is a reduction in subsequent heart failure hospitalizations.
"This is truly a trial looking at palliative benefit, not mortality," he said.
He concluded by noting that several devices are under development, including others for leaflet repair, indirect annuloplasty, and direct annular or left ventricular remodeling; as well as direct LV shape-altering devices, transcatheter mitral valve replacement devices, and devices that mimic surgical ring annuloplasty.
The Heart Valve Summit was an educational program presented jointly by the American Association for Thoracic Surgery and the American College of Cardiology Foundation. Dr. Hermann has grant/research support from Abbott Vascular, Edwards Lifesciences, St. Jude Medical, Medtronic, and Gore, and has received consulting fees and honoraria from these and other device companies. His presentation was not sponsored.
CHICAGO – Within narrow limits, transcatheter repair of mitral regurgitation can have outcomes meeting or even exceeding those of surgery.
"I believe that mitral regurgitation is mostly a surgical disease, but that interventional devices will, sooner or later, be able to manage some of the spectrum of mitral pathology," said Dr. Howard C. Herrmann at the Heart Valve Summit 2012. Dr. Herrmann is professor of medicine and director of interventional cardiology and cardiac catheterization at the University of Pennsylvania, Philadelphia.
"Transcatheter approaches are always going to be somewhat more limited than surgery," said Dr. Herrmann. The device that repairs a leaflet edge to edge is very different from the one that can be used to perform indirect annular dilatation via the coronary sinus, he noted.
Two devices relevant to those indications show promise, although neither is yet approved by the Food and Drug Administration (FDA), he said. These are Cardiac Dimensions’ Carillon for annular dilatation, and Abbott Vascular’s MitraClip for leaflet repair.
In the European TITAN clinical trial, 36 of 53 patients (68%) were implanted with the Carillon Mitral Contour System. The average baseline ejection fraction of all 53 patients was 28%, which Dr. Herrmann characterized as true functional mitral regurgitation (FMR). The prospective, nonrandomized, double-arm study compared results from the implanted group to results from a group without implants.
Implanted patients demonstrated significant reductions in FMR as assessed by multiple quantitative measures, including mean regurgitant volume, which decreased from 34.5 +/– 11.5 mL to 17.4 +/– 12.4 mL over 12 months (P less than .001) (Eur. J. Heart Fail. 2012;14:931-8).
These and other reductions correlated with symptomatic improvement, said Dr. Herrmann. However, the coronary sinus approach to percutaneous mitral annuloplasty is clearly limited by the variable anatomy between the coronary sinus and the posterior mitral annulus, as well as by the risk of coronary artery constriction, he said. Additionally, the long-term benefit of a surgical partial circumference ring is unknown (Circulation 2006;114:377-80).
The other device, the MitraClip, was evaluated in the EVEREST II trial for safety and efficacy in treatment of mitral valve regurgitation.
"Clearly, if you look at the primary effectiveness rate as defined by the trial, which is not a very high bar, surgery still beat the device," said Dr. Herrmann. However, he noted, the device clearly beat surgery in terms of morbidity and other safety end points.
"But when you look at it from an intent-to-treat analysis, those results are clearly inferior with MitraClip: At 2 years, 78.2% of percutaneous patients are free from mitral valve surgery, versus 96.1% of surgery patients (P less than .001).
"MitraClip should not and is not being used for low-risk patients with degenerative disease amenable to surgical repair. ... That said, in the patients who had good results with MitraClip, those results do appear to be fairly durable," he said. At 3 years, all-cause mortality was very similar between the two interventions. (J. Thorac. Cardiovasc. Surg. 2012;143:S60-3; J. Am. Coll. Cardiol. 2012;59:130-9).
Analysis of the high-surgical-risk cohort (n = 78) from EVEREST II revealed a 45% decrease in rehospitalizations (P = .02) at 12 months after intervention with MitraClip, said Dr. Herrmann. This finding led investigators to launch the COAPT trial, which has been approved by the FDA and is expected to start soon in the United States. Approximately 420 patients with significant FMR, thought to be at extremely high risk for surgery, will be randomized 1:1 to MitraClip (n = 210) or medical therapy (n = 210). The primary end point is a reduction in subsequent heart failure hospitalizations.
"This is truly a trial looking at palliative benefit, not mortality," he said.
He concluded by noting that several devices are under development, including others for leaflet repair, indirect annuloplasty, and direct annular or left ventricular remodeling; as well as direct LV shape-altering devices, transcatheter mitral valve replacement devices, and devices that mimic surgical ring annuloplasty.
The Heart Valve Summit was an educational program presented jointly by the American Association for Thoracic Surgery and the American College of Cardiology Foundation. Dr. Hermann has grant/research support from Abbott Vascular, Edwards Lifesciences, St. Jude Medical, Medtronic, and Gore, and has received consulting fees and honoraria from these and other device companies. His presentation was not sponsored.
CHICAGO – Within narrow limits, transcatheter repair of mitral regurgitation can have outcomes meeting or even exceeding those of surgery.
"I believe that mitral regurgitation is mostly a surgical disease, but that interventional devices will, sooner or later, be able to manage some of the spectrum of mitral pathology," said Dr. Howard C. Herrmann at the Heart Valve Summit 2012. Dr. Herrmann is professor of medicine and director of interventional cardiology and cardiac catheterization at the University of Pennsylvania, Philadelphia.
"Transcatheter approaches are always going to be somewhat more limited than surgery," said Dr. Herrmann. The device that repairs a leaflet edge to edge is very different from the one that can be used to perform indirect annular dilatation via the coronary sinus, he noted.
Two devices relevant to those indications show promise, although neither is yet approved by the Food and Drug Administration (FDA), he said. These are Cardiac Dimensions’ Carillon for annular dilatation, and Abbott Vascular’s MitraClip for leaflet repair.
In the European TITAN clinical trial, 36 of 53 patients (68%) were implanted with the Carillon Mitral Contour System. The average baseline ejection fraction of all 53 patients was 28%, which Dr. Herrmann characterized as true functional mitral regurgitation (FMR). The prospective, nonrandomized, double-arm study compared results from the implanted group to results from a group without implants.
Implanted patients demonstrated significant reductions in FMR as assessed by multiple quantitative measures, including mean regurgitant volume, which decreased from 34.5 +/– 11.5 mL to 17.4 +/– 12.4 mL over 12 months (P less than .001) (Eur. J. Heart Fail. 2012;14:931-8).
These and other reductions correlated with symptomatic improvement, said Dr. Herrmann. However, the coronary sinus approach to percutaneous mitral annuloplasty is clearly limited by the variable anatomy between the coronary sinus and the posterior mitral annulus, as well as by the risk of coronary artery constriction, he said. Additionally, the long-term benefit of a surgical partial circumference ring is unknown (Circulation 2006;114:377-80).
The other device, the MitraClip, was evaluated in the EVEREST II trial for safety and efficacy in treatment of mitral valve regurgitation.
"Clearly, if you look at the primary effectiveness rate as defined by the trial, which is not a very high bar, surgery still beat the device," said Dr. Herrmann. However, he noted, the device clearly beat surgery in terms of morbidity and other safety end points.
"But when you look at it from an intent-to-treat analysis, those results are clearly inferior with MitraClip: At 2 years, 78.2% of percutaneous patients are free from mitral valve surgery, versus 96.1% of surgery patients (P less than .001).
"MitraClip should not and is not being used for low-risk patients with degenerative disease amenable to surgical repair. ... That said, in the patients who had good results with MitraClip, those results do appear to be fairly durable," he said. At 3 years, all-cause mortality was very similar between the two interventions. (J. Thorac. Cardiovasc. Surg. 2012;143:S60-3; J. Am. Coll. Cardiol. 2012;59:130-9).
Analysis of the high-surgical-risk cohort (n = 78) from EVEREST II revealed a 45% decrease in rehospitalizations (P = .02) at 12 months after intervention with MitraClip, said Dr. Herrmann. This finding led investigators to launch the COAPT trial, which has been approved by the FDA and is expected to start soon in the United States. Approximately 420 patients with significant FMR, thought to be at extremely high risk for surgery, will be randomized 1:1 to MitraClip (n = 210) or medical therapy (n = 210). The primary end point is a reduction in subsequent heart failure hospitalizations.
"This is truly a trial looking at palliative benefit, not mortality," he said.
He concluded by noting that several devices are under development, including others for leaflet repair, indirect annuloplasty, and direct annular or left ventricular remodeling; as well as direct LV shape-altering devices, transcatheter mitral valve replacement devices, and devices that mimic surgical ring annuloplasty.
The Heart Valve Summit was an educational program presented jointly by the American Association for Thoracic Surgery and the American College of Cardiology Foundation. Dr. Hermann has grant/research support from Abbott Vascular, Edwards Lifesciences, St. Jude Medical, Medtronic, and Gore, and has received consulting fees and honoraria from these and other device companies. His presentation was not sponsored.
EXPERT ANALYSIS FROM THE HEART VALVE SUMMIT 2012

FDA Approves Chest-Implanted LVAD
The HeartWare Ventricular Assist System, a small left ventricular assist device that is implanted in the chest instead of the abdomen, has been approved by the Food and Drug Administration as a bridge to heart transplant for end-stage heart failure patients.
The HeartWare Ventricular Assist System, manufactured by HeartWare, includes an implantable pump with an external driver and power source. The device is another treatment option for advanced heart failure patients, especially those who are smaller in size or can’t have an abdominal implant.
The approval of the continuous-flow pump device comes less than 3 years after the agency approved Thoratec’s HeartMate II left ventricular assist device (LVAD) for destination therapy. That device quickly replaced the previous generation of pulsatile-flow devices.
This is the first time that the FDA has approved a ventricular assist device using comparator data from a registry as a control, according to the agency.
The approval was based on data from the ADVANCE trial, which compared the outcomes of 137 patients implanted with the HeartWare System and of patients registered by the Interagency Registry for Mechanically Assisted Circulatory Support (INTERMACS). The registry has been in place since 2005, collecting information on patients who received an approved mechanical circulatory support device implant.
A total of 140 patients received the investigational pump, and 499 patients received a commercially available pump implanted contemporaneously. At 180 days, 90.7% of the investigational pump patients and 90.1% of the controls had survived, establishing the noninferiority of the investigational pump (P less than .001; 15% noninferiority margin). Infection, right heart failure, device replacement, stroke, kidney dysfunction, hemolysis, and arrhythmia rates for the HVAD were similar to those reported previously for the HeartMate II, according to Dr. Keith D. Aaronson and his colleagues in the study (Circulation. 2012;125:3191-3200).
Results of the ADVANCE trial showed that at 6 months, median 6-minute walk distance improved by 128.5 m, and "functional capacity and quality of life improved markedly, and the adverse event profile was favorable," according to the authors.
In a news release, FDA officials noted that "although rates of most key adverse events were comparable, the risk of stroke associated with the HeartWare LVAD necessitates patients and clinicians to discuss all treatment options before deciding to use the device."
"Well-designed registries in targeted product areas can enhance the public health and provide a cost-effective approach to clinical research for industry innovators," Christy Foreman, director of the Office of Device Evaluation in the FDA’s Center for Devices and Radiological Health, said in a statement. For HeartWare, registry data directly facilitated the development and availability of this new device. The registry is a joint effort involving the FDA; National Heart, Lung, and Blood Institute; Centers for Medicare and Medicaid Services (CMS); and clinicians, scientists, and industry.
The FDA approval of the bridge-to-transplant pump comes on the heels of a Medicare Advisory (MEDCAC) meeting, where heart societies and prominent heart failure experts discussed the state of VAD research. The panel stressed the importance of multidisciplinary heart teams, and said that there’s not enough data to show that the indications for VADs can be expanded to include lower-risk patients.

Medicare currently does not have an open National Coverage Determination (NCD) for VADs. Medicare currently reimburses VADs, under certain criteria, as a bridge-to-transplant and as destination therapy.
Dr. Sean Pinney, who spoke at the MEDCAC meeting on behalf of the Heart Failure Society of America, said that the society supported the NCD and did "not endorse any change in the current patient selection criteria which derive from prospective randomized trials." He added that there’s a need for more well-controlled clinical trials, including those that would examine "less sick" patients.
"We do not endorse expansion of destination therapy into this population in the absence of randomized clinical trials," said Dr. Pinney, associate professor of medicine at Mount Sinai Medical Center in New York.
The 140 U.S. centers that place LVADs are expected to implant nearly 3,000 devices this year. Nearly 4,600 patients have received an LVAD since 2010.
"This is a rapidly evolving field," said Dr. Mariell Jessup, president-elect of the American Heart Association and professor of medicine at the University of Pennsylvania Heart & Vascular Center in Philadelphia. "It’s only to be expected that CMS would open coverage determination. It’s a costly technology, and outcomes are a lot better than several years ago," she said.
Physicians quoted in this story reported no financial conflicts.
The HeartWare Ventricular Assist System, a small left ventricular assist device that is implanted in the chest instead of the abdomen, has been approved by the Food and Drug Administration as a bridge to heart transplant for end-stage heart failure patients.
The HeartWare Ventricular Assist System, manufactured by HeartWare, includes an implantable pump with an external driver and power source. The device is another treatment option for advanced heart failure patients, especially those who are smaller in size or can’t have an abdominal implant.
The approval of the continuous-flow pump device comes less than 3 years after the agency approved Thoratec’s HeartMate II left ventricular assist device (LVAD) for destination therapy. That device quickly replaced the previous generation of pulsatile-flow devices.
This is the first time that the FDA has approved a ventricular assist device using comparator data from a registry as a control, according to the agency.
The approval was based on data from the ADVANCE trial, which compared the outcomes of 137 patients implanted with the HeartWare System and of patients registered by the Interagency Registry for Mechanically Assisted Circulatory Support (INTERMACS). The registry has been in place since 2005, collecting information on patients who received an approved mechanical circulatory support device implant.
A total of 140 patients received the investigational pump, and 499 patients received a commercially available pump implanted contemporaneously. At 180 days, 90.7% of the investigational pump patients and 90.1% of the controls had survived, establishing the noninferiority of the investigational pump (P less than .001; 15% noninferiority margin). Infection, right heart failure, device replacement, stroke, kidney dysfunction, hemolysis, and arrhythmia rates for the HVAD were similar to those reported previously for the HeartMate II, according to Dr. Keith D. Aaronson and his colleagues in the study (Circulation. 2012;125:3191-3200).
Results of the ADVANCE trial showed that at 6 months, median 6-minute walk distance improved by 128.5 m, and "functional capacity and quality of life improved markedly, and the adverse event profile was favorable," according to the authors.
In a news release, FDA officials noted that "although rates of most key adverse events were comparable, the risk of stroke associated with the HeartWare LVAD necessitates patients and clinicians to discuss all treatment options before deciding to use the device."
"Well-designed registries in targeted product areas can enhance the public health and provide a cost-effective approach to clinical research for industry innovators," Christy Foreman, director of the Office of Device Evaluation in the FDA’s Center for Devices and Radiological Health, said in a statement. For HeartWare, registry data directly facilitated the development and availability of this new device. The registry is a joint effort involving the FDA; National Heart, Lung, and Blood Institute; Centers for Medicare and Medicaid Services (CMS); and clinicians, scientists, and industry.
The FDA approval of the bridge-to-transplant pump comes on the heels of a Medicare Advisory (MEDCAC) meeting, where heart societies and prominent heart failure experts discussed the state of VAD research. The panel stressed the importance of multidisciplinary heart teams, and said that there’s not enough data to show that the indications for VADs can be expanded to include lower-risk patients.
Medicare currently does not have an open National Coverage Determination (NCD) for VADs. Medicare currently reimburses VADs, under certain criteria, as a bridge-to-transplant and as destination therapy.
Dr. Sean Pinney, who spoke at the MEDCAC meeting on behalf of the Heart Failure Society of America, said that the society supported the NCD and did "not endorse any change in the current patient selection criteria which derive from prospective randomized trials." He added that there’s a need for more well-controlled clinical trials, including those that would examine "less sick" patients.
"We do not endorse expansion of destination therapy into this population in the absence of randomized clinical trials," said Dr. Pinney, associate professor of medicine at Mount Sinai Medical Center in New York.
The 140 U.S. centers that place LVADs are expected to implant nearly 3,000 devices this year. Nearly 4,600 patients have received an LVAD since 2010.
"This is a rapidly evolving field," said Dr. Mariell Jessup, president-elect of the American Heart Association and professor of medicine at the University of Pennsylvania Heart & Vascular Center in Philadelphia. "It’s only to be expected that CMS would open coverage determination. It’s a costly technology, and outcomes are a lot better than several years ago," she said.
Physicians quoted in this story reported no financial conflicts.
The HeartWare Ventricular Assist System, a small left ventricular assist device that is implanted in the chest instead of the abdomen, has been approved by the Food and Drug Administration as a bridge to heart transplant for end-stage heart failure patients.
The HeartWare Ventricular Assist System, manufactured by HeartWare, includes an implantable pump with an external driver and power source. The device is another treatment option for advanced heart failure patients, especially those who are smaller in size or can’t have an abdominal implant.
The approval of the continuous-flow pump device comes less than 3 years after the agency approved Thoratec’s HeartMate II left ventricular assist device (LVAD) for destination therapy. That device quickly replaced the previous generation of pulsatile-flow devices.
This is the first time that the FDA has approved a ventricular assist device using comparator data from a registry as a control, according to the agency.
The approval was based on data from the ADVANCE trial, which compared the outcomes of 137 patients implanted with the HeartWare System and of patients registered by the Interagency Registry for Mechanically Assisted Circulatory Support (INTERMACS). The registry has been in place since 2005, collecting information on patients who received an approved mechanical circulatory support device implant.
A total of 140 patients received the investigational pump, and 499 patients received a commercially available pump implanted contemporaneously. At 180 days, 90.7% of the investigational pump patients and 90.1% of the controls had survived, establishing the noninferiority of the investigational pump (P less than .001; 15% noninferiority margin). Infection, right heart failure, device replacement, stroke, kidney dysfunction, hemolysis, and arrhythmia rates for the HVAD were similar to those reported previously for the HeartMate II, according to Dr. Keith D. Aaronson and his colleagues in the study (Circulation. 2012;125:3191-3200).
Results of the ADVANCE trial showed that at 6 months, median 6-minute walk distance improved by 128.5 m, and "functional capacity and quality of life improved markedly, and the adverse event profile was favorable," according to the authors.
In a news release, FDA officials noted that "although rates of most key adverse events were comparable, the risk of stroke associated with the HeartWare LVAD necessitates patients and clinicians to discuss all treatment options before deciding to use the device."
"Well-designed registries in targeted product areas can enhance the public health and provide a cost-effective approach to clinical research for industry innovators," Christy Foreman, director of the Office of Device Evaluation in the FDA’s Center for Devices and Radiological Health, said in a statement. For HeartWare, registry data directly facilitated the development and availability of this new device. The registry is a joint effort involving the FDA; National Heart, Lung, and Blood Institute; Centers for Medicare and Medicaid Services (CMS); and clinicians, scientists, and industry.
The FDA approval of the bridge-to-transplant pump comes on the heels of a Medicare Advisory (MEDCAC) meeting, where heart societies and prominent heart failure experts discussed the state of VAD research. The panel stressed the importance of multidisciplinary heart teams, and said that there’s not enough data to show that the indications for VADs can be expanded to include lower-risk patients.
Medicare currently does not have an open National Coverage Determination (NCD) for VADs. Medicare currently reimburses VADs, under certain criteria, as a bridge-to-transplant and as destination therapy.
Dr. Sean Pinney, who spoke at the MEDCAC meeting on behalf of the Heart Failure Society of America, said that the society supported the NCD and did "not endorse any change in the current patient selection criteria which derive from prospective randomized trials." He added that there’s a need for more well-controlled clinical trials, including those that would examine "less sick" patients.
"We do not endorse expansion of destination therapy into this population in the absence of randomized clinical trials," said Dr. Pinney, associate professor of medicine at Mount Sinai Medical Center in New York.
The 140 U.S. centers that place LVADs are expected to implant nearly 3,000 devices this year. Nearly 4,600 patients have received an LVAD since 2010.
"This is a rapidly evolving field," said Dr. Mariell Jessup, president-elect of the American Heart Association and professor of medicine at the University of Pennsylvania Heart & Vascular Center in Philadelphia. "It’s only to be expected that CMS would open coverage determination. It’s a costly technology, and outcomes are a lot better than several years ago," she said.
Physicians quoted in this story reported no financial conflicts.
What's New? The Changing Landscape of Infective Endocarditis
CHICAGO -- Mitral valve endocarditis is a medical-surgical problem that demands a team approach, and the landscape of the disease has changed significantly for the worst in the recent past, according to a presentation at Heart Valve Summit 2012.
"If you’re lucky enough to have a valve infection with the Strep viridans microorganism, you’re likely to do well," said Dr. Patrick T. O’Gara, director of clinical cardiology at Brigham and Women’s Hospital, Boston.
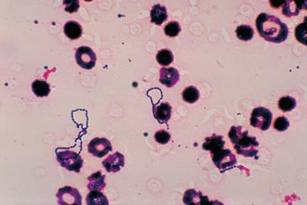
On the other hand, patients who’ve been infected with any other type of organism, particularly Staph aureus or its increasingly frequent relative methicillin-resistant Staph aureus, could be in real trouble.
"There’s no question that these organisms are smarter than we are and can lead to destruction of the valve and death of the patient, despite our best intentions," said Dr. O’Gara. He described the evolving epidemiology, natural history, and indications for surgery for this disease to this educational program of the American Association for Thoracic Surgery (AATS) and the American College of Cardiology Foundation (ACCF).
"Unfortunately, despite our best efforts, and worldwide, among centers with an interest in the care of patients with endocarditis, 6-month mortality rates still approach 25%," he said. This is close to the mortality rate of a type A aortic dissection, so it is by no means trivial. Early surgery is now performed on many who present with the disease in early stages, and perioperative mortality is 12%-20%.
It is a challenge to manage a patient who is asymptomatic with respect to heart failure but who has a large mobile vegetation involving the anterior mitral leaflet, said Dr. O’Gara. Vegetations in this location are the most prone to embolize.
"This is the common question now posed to consulting cardiologists: Does my patient require early surgery for prevention of embolic complications? We don’t really get asked too much any more: ‘Does my patient need early surgery because they’ve developed heart failure?’ I guess we’re much more confident in proceeding under those circumstances."
He discussed considerations for surgery, including the level of local surgical expertise. The intervention is complex and delicate. "This is not something for the faint of heart," said Dr. O’Gara.

In the early time frame following presentation, there is a relationship between the size of the vegetation and the risk of embolization.
The risk of stroke drops rapidly after initiation of antimicrobial therapy. Patients who develop stroke as a complication of endocarditis typically do so the day before, the day of, or the day after presentation, he said. Stroke risk continues to drop quickly in the first 2 weeks after initiation of antibiotics, and by week 5 it’s almost nil.
"If you’re thinking about intervention for prevention of stroke, it makes sense to do so in the first week, after identification of a patient at risk with a large mobile vegetation. It makes much less sense to do so in weeks 2 or 3."
The size of the vegetation alone may dictate mortality. A size of 1.5 cm or greater is associated with increased risk of death at 1 year in the setting of native valve endocarditis.
The good news is that event-free survival rates have been shown to be much better for those who undergo surgery in the first 7 days after presentation with high-risk native, left-sided endocarditis. In-hospital mortality as a function of early surgery appears to have declined over the course of time.
"So, in summary, I think for our management considerations, it’s early diagnosis, risk stratification, a heart team approach, consider early surgery, particularly if you have the operative expertise. The early risk of re-infection in implanted prosthetic material is very low," Dr. O’Gara said.
Dr. O’Gara disclosed ties with the Data Safety Monitoring Board and Lantheus Medical Imaging.
CHICAGO -- Mitral valve endocarditis is a medical-surgical problem that demands a team approach, and the landscape of the disease has changed significantly for the worst in the recent past, according to a presentation at Heart Valve Summit 2012.
"If you’re lucky enough to have a valve infection with the Strep viridans microorganism, you’re likely to do well," said Dr. Patrick T. O’Gara, director of clinical cardiology at Brigham and Women’s Hospital, Boston.
On the other hand, patients who’ve been infected with any other type of organism, particularly Staph aureus or its increasingly frequent relative methicillin-resistant Staph aureus, could be in real trouble.
"There’s no question that these organisms are smarter than we are and can lead to destruction of the valve and death of the patient, despite our best intentions," said Dr. O’Gara. He described the evolving epidemiology, natural history, and indications for surgery for this disease to this educational program of the American Association for Thoracic Surgery (AATS) and the American College of Cardiology Foundation (ACCF).
"Unfortunately, despite our best efforts, and worldwide, among centers with an interest in the care of patients with endocarditis, 6-month mortality rates still approach 25%," he said. This is close to the mortality rate of a type A aortic dissection, so it is by no means trivial. Early surgery is now performed on many who present with the disease in early stages, and perioperative mortality is 12%-20%.
It is a challenge to manage a patient who is asymptomatic with respect to heart failure but who has a large mobile vegetation involving the anterior mitral leaflet, said Dr. O’Gara. Vegetations in this location are the most prone to embolize.
"This is the common question now posed to consulting cardiologists: Does my patient require early surgery for prevention of embolic complications? We don’t really get asked too much any more: ‘Does my patient need early surgery because they’ve developed heart failure?’ I guess we’re much more confident in proceeding under those circumstances."
He discussed considerations for surgery, including the level of local surgical expertise. The intervention is complex and delicate. "This is not something for the faint of heart," said Dr. O’Gara.
In the early time frame following presentation, there is a relationship between the size of the vegetation and the risk of embolization.
The risk of stroke drops rapidly after initiation of antimicrobial therapy. Patients who develop stroke as a complication of endocarditis typically do so the day before, the day of, or the day after presentation, he said. Stroke risk continues to drop quickly in the first 2 weeks after initiation of antibiotics, and by week 5 it’s almost nil.
"If you’re thinking about intervention for prevention of stroke, it makes sense to do so in the first week, after identification of a patient at risk with a large mobile vegetation. It makes much less sense to do so in weeks 2 or 3."
The size of the vegetation alone may dictate mortality. A size of 1.5 cm or greater is associated with increased risk of death at 1 year in the setting of native valve endocarditis.
The good news is that event-free survival rates have been shown to be much better for those who undergo surgery in the first 7 days after presentation with high-risk native, left-sided endocarditis. In-hospital mortality as a function of early surgery appears to have declined over the course of time.
"So, in summary, I think for our management considerations, it’s early diagnosis, risk stratification, a heart team approach, consider early surgery, particularly if you have the operative expertise. The early risk of re-infection in implanted prosthetic material is very low," Dr. O’Gara said.
Dr. O’Gara disclosed ties with the Data Safety Monitoring Board and Lantheus Medical Imaging.
CHICAGO -- Mitral valve endocarditis is a medical-surgical problem that demands a team approach, and the landscape of the disease has changed significantly for the worst in the recent past, according to a presentation at Heart Valve Summit 2012.
"If you’re lucky enough to have a valve infection with the Strep viridans microorganism, you’re likely to do well," said Dr. Patrick T. O’Gara, director of clinical cardiology at Brigham and Women’s Hospital, Boston.
On the other hand, patients who’ve been infected with any other type of organism, particularly Staph aureus or its increasingly frequent relative methicillin-resistant Staph aureus, could be in real trouble.
"There’s no question that these organisms are smarter than we are and can lead to destruction of the valve and death of the patient, despite our best intentions," said Dr. O’Gara. He described the evolving epidemiology, natural history, and indications for surgery for this disease to this educational program of the American Association for Thoracic Surgery (AATS) and the American College of Cardiology Foundation (ACCF).
"Unfortunately, despite our best efforts, and worldwide, among centers with an interest in the care of patients with endocarditis, 6-month mortality rates still approach 25%," he said. This is close to the mortality rate of a type A aortic dissection, so it is by no means trivial. Early surgery is now performed on many who present with the disease in early stages, and perioperative mortality is 12%-20%.
It is a challenge to manage a patient who is asymptomatic with respect to heart failure but who has a large mobile vegetation involving the anterior mitral leaflet, said Dr. O’Gara. Vegetations in this location are the most prone to embolize.
"This is the common question now posed to consulting cardiologists: Does my patient require early surgery for prevention of embolic complications? We don’t really get asked too much any more: ‘Does my patient need early surgery because they’ve developed heart failure?’ I guess we’re much more confident in proceeding under those circumstances."
He discussed considerations for surgery, including the level of local surgical expertise. The intervention is complex and delicate. "This is not something for the faint of heart," said Dr. O’Gara.
In the early time frame following presentation, there is a relationship between the size of the vegetation and the risk of embolization.
The risk of stroke drops rapidly after initiation of antimicrobial therapy. Patients who develop stroke as a complication of endocarditis typically do so the day before, the day of, or the day after presentation, he said. Stroke risk continues to drop quickly in the first 2 weeks after initiation of antibiotics, and by week 5 it’s almost nil.
"If you’re thinking about intervention for prevention of stroke, it makes sense to do so in the first week, after identification of a patient at risk with a large mobile vegetation. It makes much less sense to do so in weeks 2 or 3."
The size of the vegetation alone may dictate mortality. A size of 1.5 cm or greater is associated with increased risk of death at 1 year in the setting of native valve endocarditis.
The good news is that event-free survival rates have been shown to be much better for those who undergo surgery in the first 7 days after presentation with high-risk native, left-sided endocarditis. In-hospital mortality as a function of early surgery appears to have declined over the course of time.
"So, in summary, I think for our management considerations, it’s early diagnosis, risk stratification, a heart team approach, consider early surgery, particularly if you have the operative expertise. The early risk of re-infection in implanted prosthetic material is very low," Dr. O’Gara said.
Dr. O’Gara disclosed ties with the Data Safety Monitoring Board and Lantheus Medical Imaging.
EXPERT ANALYSIS FROM HEART VALVE SUMMIT 2012
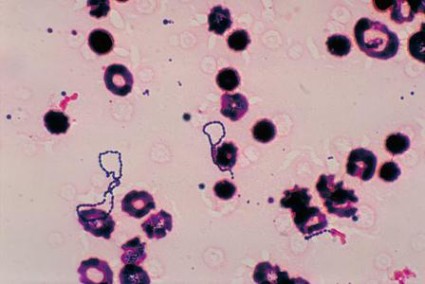
ADVANCE: TAVI Survival Remains High With CoreValve
Survival rates remained high 1 year after implantation with the transcatheter aortic CoreValve in the postmarket ADVANCE study, initial results show.
At 1 year, overall survival was 82.1% and cardiovascular survival 88.2%. This compares with survival rates of 87.4% and 91.7% at 6 months and 95.5% and 96.6% at 30 days, principal investigator Dr. Axel Linke reported at Transcatheter Cardiovascular Therapeutics 2012.
"I think they’re outstanding," he said in an interview. "If you put it into the perspective of the PARTNER A and B cohorts [in the pivotal trial in the Sapien transcatheter valve system], our mortality rate is, in absolute values, 8 to 13% lower."
One explanation is that the 1,015-patient, postmarket ADVANCE study sought out centers experienced with transcatheter aortic valve implantation (TAVI). The 44 centers in Western Europe, Asia, and South America were required to have performed at least 40 TAVI procedures, with some German centers having done as many as 500, to be certified by a TAVI proctor and to have a heart team in place.
"Clearly, our centers were out of the learning curve," remarked Dr. Linke of the University of Leipzig (Germany) Heart Center.
By comparison, some centers in the PARTNER trial of the Edwards Lifesciences Sapien valve contributed just six or seven patients and were selected based on their experience with general cardiologic intervention, he observed.
The 1-year survival rates in ADVANCE also surpass those from early registries, notably the French Aortic National CoreValve and Edwards Registry, where the initial experience with TAVI was associated with interventional mistakes, which were linked to early mortality, Dr. Linke said.
The CoreValve System has been implanted in more than 30,000 patients since its approval in the European Union in 2007, but is limited to investigational use in the United States and Japan.
No details were presented regarding mortality in various subgroups or complications such as stroke, paravalvular leaks or left bundle branch block (LBBB). A recent analysis raised concerns about LBBB, showing that one-third of 202 consecutive patients with no prior conduction disturbances developed new-onset LBBB after TAVI with a balloon-expandable valve (Sapien or Sapien XT). Although it resolved in 37.7% by hospital discharge and 57.3% at 6- to 12-month follow-up, patients with persistent LBBB had a significantly higher incidence of syncope and complete atrioventricular block requiring a permanent pacemaker (J. Am. Coll. Cardiol. 2012;60:1743-52 [doi:10.1016/j.jacc.2012.035]).
Dr. Linke said they will look at LBBB in more detailed analyses expected from ADVANCE in the coming weeks, but that there’s been no evidence of a problem with LBBB in earlier follow-up in ADVANCE or from other CoreValve users.
"Survival curves are absolutely identical up to the 6-month follow-up, so there’s no reason to believe they should be worse afterwards, although I can’t be exactly sure right now," he said, adding that in the publication, survival curves from patients with and without new-onset LBBB "started to diverge very early."
Quality of life data from ADVANCE, reported in a separate poster session at the meeting, showed significant benefits with the CoreValve, even among higher-risk patients.
Scores on the European Quality of Life–5 Dimensions (EQ-5D), which ranges from 0 (death) to 1 (perfect health), improved from 0.62 at baseline to 0.72 at 1 month, where it remained at 6 months, both highly significant differences from baseline.
On the Short Form Health Survey–12 (SF-12), scores at baseline, 1 month, and 6 months were 32.8, 39, and 39.7 for the physical component and 46.2, 48.5, and 50 for the mental component.
"Basically, whatever is gained, is gained very early from the baseline to the 1-month follow-up in the majority of the cases," said Dr. Linke.
The 322 higher-risk patients entering the study with a logistic EuroSCORE of more than 20 had significantly worse baseline health-related quality of life than did those with a EuroSCORE of 10 or less, but experienced significant improvements after TAVI on the EQ-5D at 1 month and on both components of the SF-12 at 6 months, all significant changes.
The access route used during TAVI had no impact on quality of life improvement at 6 months.
Dr. Linke reported serving as an adviser or consultant for Medtronic, which sponsored the study.
Survival rates remained high 1 year after implantation with the transcatheter aortic CoreValve in the postmarket ADVANCE study, initial results show.
At 1 year, overall survival was 82.1% and cardiovascular survival 88.2%. This compares with survival rates of 87.4% and 91.7% at 6 months and 95.5% and 96.6% at 30 days, principal investigator Dr. Axel Linke reported at Transcatheter Cardiovascular Therapeutics 2012.
"I think they’re outstanding," he said in an interview. "If you put it into the perspective of the PARTNER A and B cohorts [in the pivotal trial in the Sapien transcatheter valve system], our mortality rate is, in absolute values, 8 to 13% lower."
One explanation is that the 1,015-patient, postmarket ADVANCE study sought out centers experienced with transcatheter aortic valve implantation (TAVI). The 44 centers in Western Europe, Asia, and South America were required to have performed at least 40 TAVI procedures, with some German centers having done as many as 500, to be certified by a TAVI proctor and to have a heart team in place.
"Clearly, our centers were out of the learning curve," remarked Dr. Linke of the University of Leipzig (Germany) Heart Center.
By comparison, some centers in the PARTNER trial of the Edwards Lifesciences Sapien valve contributed just six or seven patients and were selected based on their experience with general cardiologic intervention, he observed.
The 1-year survival rates in ADVANCE also surpass those from early registries, notably the French Aortic National CoreValve and Edwards Registry, where the initial experience with TAVI was associated with interventional mistakes, which were linked to early mortality, Dr. Linke said.
The CoreValve System has been implanted in more than 30,000 patients since its approval in the European Union in 2007, but is limited to investigational use in the United States and Japan.
No details were presented regarding mortality in various subgroups or complications such as stroke, paravalvular leaks or left bundle branch block (LBBB). A recent analysis raised concerns about LBBB, showing that one-third of 202 consecutive patients with no prior conduction disturbances developed new-onset LBBB after TAVI with a balloon-expandable valve (Sapien or Sapien XT). Although it resolved in 37.7% by hospital discharge and 57.3% at 6- to 12-month follow-up, patients with persistent LBBB had a significantly higher incidence of syncope and complete atrioventricular block requiring a permanent pacemaker (J. Am. Coll. Cardiol. 2012;60:1743-52 [doi:10.1016/j.jacc.2012.035]).
Dr. Linke said they will look at LBBB in more detailed analyses expected from ADVANCE in the coming weeks, but that there’s been no evidence of a problem with LBBB in earlier follow-up in ADVANCE or from other CoreValve users.
"Survival curves are absolutely identical up to the 6-month follow-up, so there’s no reason to believe they should be worse afterwards, although I can’t be exactly sure right now," he said, adding that in the publication, survival curves from patients with and without new-onset LBBB "started to diverge very early."
Quality of life data from ADVANCE, reported in a separate poster session at the meeting, showed significant benefits with the CoreValve, even among higher-risk patients.
Scores on the European Quality of Life–5 Dimensions (EQ-5D), which ranges from 0 (death) to 1 (perfect health), improved from 0.62 at baseline to 0.72 at 1 month, where it remained at 6 months, both highly significant differences from baseline.
On the Short Form Health Survey–12 (SF-12), scores at baseline, 1 month, and 6 months were 32.8, 39, and 39.7 for the physical component and 46.2, 48.5, and 50 for the mental component.
"Basically, whatever is gained, is gained very early from the baseline to the 1-month follow-up in the majority of the cases," said Dr. Linke.
The 322 higher-risk patients entering the study with a logistic EuroSCORE of more than 20 had significantly worse baseline health-related quality of life than did those with a EuroSCORE of 10 or less, but experienced significant improvements after TAVI on the EQ-5D at 1 month and on both components of the SF-12 at 6 months, all significant changes.
The access route used during TAVI had no impact on quality of life improvement at 6 months.
Dr. Linke reported serving as an adviser or consultant for Medtronic, which sponsored the study.
Survival rates remained high 1 year after implantation with the transcatheter aortic CoreValve in the postmarket ADVANCE study, initial results show.
At 1 year, overall survival was 82.1% and cardiovascular survival 88.2%. This compares with survival rates of 87.4% and 91.7% at 6 months and 95.5% and 96.6% at 30 days, principal investigator Dr. Axel Linke reported at Transcatheter Cardiovascular Therapeutics 2012.
"I think they’re outstanding," he said in an interview. "If you put it into the perspective of the PARTNER A and B cohorts [in the pivotal trial in the Sapien transcatheter valve system], our mortality rate is, in absolute values, 8 to 13% lower."
One explanation is that the 1,015-patient, postmarket ADVANCE study sought out centers experienced with transcatheter aortic valve implantation (TAVI). The 44 centers in Western Europe, Asia, and South America were required to have performed at least 40 TAVI procedures, with some German centers having done as many as 500, to be certified by a TAVI proctor and to have a heart team in place.
"Clearly, our centers were out of the learning curve," remarked Dr. Linke of the University of Leipzig (Germany) Heart Center.
By comparison, some centers in the PARTNER trial of the Edwards Lifesciences Sapien valve contributed just six or seven patients and were selected based on their experience with general cardiologic intervention, he observed.
The 1-year survival rates in ADVANCE also surpass those from early registries, notably the French Aortic National CoreValve and Edwards Registry, where the initial experience with TAVI was associated with interventional mistakes, which were linked to early mortality, Dr. Linke said.
The CoreValve System has been implanted in more than 30,000 patients since its approval in the European Union in 2007, but is limited to investigational use in the United States and Japan.
No details were presented regarding mortality in various subgroups or complications such as stroke, paravalvular leaks or left bundle branch block (LBBB). A recent analysis raised concerns about LBBB, showing that one-third of 202 consecutive patients with no prior conduction disturbances developed new-onset LBBB after TAVI with a balloon-expandable valve (Sapien or Sapien XT). Although it resolved in 37.7% by hospital discharge and 57.3% at 6- to 12-month follow-up, patients with persistent LBBB had a significantly higher incidence of syncope and complete atrioventricular block requiring a permanent pacemaker (J. Am. Coll. Cardiol. 2012;60:1743-52 [doi:10.1016/j.jacc.2012.035]).
Dr. Linke said they will look at LBBB in more detailed analyses expected from ADVANCE in the coming weeks, but that there’s been no evidence of a problem with LBBB in earlier follow-up in ADVANCE or from other CoreValve users.
"Survival curves are absolutely identical up to the 6-month follow-up, so there’s no reason to believe they should be worse afterwards, although I can’t be exactly sure right now," he said, adding that in the publication, survival curves from patients with and without new-onset LBBB "started to diverge very early."
Quality of life data from ADVANCE, reported in a separate poster session at the meeting, showed significant benefits with the CoreValve, even among higher-risk patients.
Scores on the European Quality of Life–5 Dimensions (EQ-5D), which ranges from 0 (death) to 1 (perfect health), improved from 0.62 at baseline to 0.72 at 1 month, where it remained at 6 months, both highly significant differences from baseline.
On the Short Form Health Survey–12 (SF-12), scores at baseline, 1 month, and 6 months were 32.8, 39, and 39.7 for the physical component and 46.2, 48.5, and 50 for the mental component.
"Basically, whatever is gained, is gained very early from the baseline to the 1-month follow-up in the majority of the cases," said Dr. Linke.
The 322 higher-risk patients entering the study with a logistic EuroSCORE of more than 20 had significantly worse baseline health-related quality of life than did those with a EuroSCORE of 10 or less, but experienced significant improvements after TAVI on the EQ-5D at 1 month and on both components of the SF-12 at 6 months, all significant changes.
The access route used during TAVI had no impact on quality of life improvement at 6 months.
Dr. Linke reported serving as an adviser or consultant for Medtronic, which sponsored the study.
FROM TRANSCATHETER CARDIOVASCULAR THERAPEUTICS 2012
Major Finding: At 1 year, overall survival was 82.1% and cardiovascular survival 88.2%.
Data Source: International, postmarket phase IV study of 1,015 patients with severe aortic stenosis implanted with the transcatheter CoreValve.
Disclosures: Dr. Linke reported serving as an advisor or consultant for Medtronic, which sponsored the study.
Left-Atrial MAZE Ablation Compromises Atrial Function
LOS ANGELES – Full left-atrial ablation by the modified Cox MAZE procedure to treat atrial fibrillation led to significant reductions in left atrial function, based on a detailed assessment of 31 patients who underwent this treatment using bilateral bipolar radiofrequency.
The damaging effect of complete left-atrial ablation contrasted with the impact of a less extensive procedure, pulmonary vein isolation, which kept left-atrial function intact and even improved it by some parameters. Opinions split on the implications of these findings.
"Our advice is that left-atrial ablation should be restricted to those cases where pulmonary vein isolation will likely be insufficient" to restore and maintain sinus rhythm, Dr. Marieke G. Compier said as she presented the findings at the annual scientific sessions of the American Heart Association on Nov. 6.
But a cardiac surgeon who heard the results disagreed, contending that the top priority is performing the procedure that will result in durable prevention of atrial fibrillation (AF) recurrence.
"Recurrence of atrial fibrillation is a more important determinant of [successful] clinical outcomes than preserved atrial function," said Dr. Pierre Page, chief of cardiac surgery at the University of Montreal. "The data we have today show that left-atrial ablation is very beneficial," Dr. Page said in an interview.
Dr. Compier and her associates used echocardiography to assess left-atrial function in 31 patients who underwent a modified Cox-MAZE procedure for complete left-atrial ablation using bilateral bipolar radiofrequency, and 31 patients who underwent pulmonary-vein isolation (PVI) only. In the full ablation group, 25 patients had persistent AF and 6 had paroxysmal AF; in the PVI group, 25 patients had paroxysmal disease and 6 had persistent AF. All patients also underwent concurrent coronary artery bypass, valve surgery, or both.
The researchers assessed the efficacy of AF treatment using 24-hour ECG monitoring at 3, 6, and 12 months following surgery. They also used two-dimensional echocardiography to assess left atrial size and function at 3 months and 1 year after surgery.
After 1 year, 68% of the patients who had full ablation and 81% of those who underwent PVI were free of AF and completely off anti-arrhythmic drug treatment; the other patients in each group had AF recurrences. The different long-term success in maintaining sinus rhythm in the two groups probably stemmed from the unbalanced distribution of patients with paroxysmal and persistent AF, said Dr. Compier, a cardiologist in the Heart Center at Leiden (the Netherlands) University Medical Center. "I think this is why full ablation seemed less successful," she said.
After 1 year, echocardiographic examinations showed that patients treated with full ablation had statistically significant reductions in left atrial volume and strain, and 42% of patients had A-wave restoration. Compared with measurements made prior to surgery, strain rate fell by an average of about 50%, peak A-wave dropped by an average of about a third, and average left-atrial ejection fraction and filling fraction each dropped by about 20%. All of these changes were statistically significant, compared with baseline.
In contrast, patients who underwent PVI had no significant change in their strain rate or peak A wave, and their average left-atrial ejection fraction and filling fraction each rose by about 10% compared with baseline, statistically significant differences. A-wave restoration occurred in 87% of the PVI patients.
Stepwise regression analysis of baseline differences between the two study groups showed that the follow-up differences seen in left-atrial size and function were best explained by the different ablation treatments the two groups received, Dr. Compier said.
Dr. Compier said that she had no disclosures.
It comes as no surprise that complete left-atrial ablation during cardiac surgery produces impaired left-atrial function, although it is surprising to see how much damage occurs. But this finding is no reason to abandon atrial ablation and replace it with less extensive treatment with pulmonary-vein isolation unless the reduced left-atrial function is shown to have a clear impact on patient outcomes or survival. Based on what we know today, on balance, it’s more important to more thoroughly and reliably address our patients’ atrial arrhythmia than it is to preserve full atrial function. Substituting pulmonary-vein isolation for full ablation would increase the risk of atrial fibrillation recurrence.
The study done by Dr. Compier and her associates in Leiden is the first to document the functional impact of complete left-atrial ablation using the modified Cox-MAZE procedure in such a careful and systematic way using echocardiography. But anyone who is concerned about the impact of full ablation on atrial function must acknowledge that this treatment is also very beneficial to patients. The Leiden group clearly showed that full ablation is very detrimental to the atrium, but they did not associate these impairments with adverse clinical outcomes. As far as we know, atrial fibrillation is a more important determinant of clinical outcome than are changes in left-atrial function.

|
|
A major way in which full ablation differs from pulmonary vein isolation is the added isolation of the left-atrial appendage, and we know that contraction of the left atrium mostly depends on the left-atrial appendage. But isolation of the appendage also reduces the risk of stroke. Previously-reported findings from several studies have shown that pulmonary vein isolation is less effective at restoring sinus rhythm and preventing atrial fibrillation recurrence. Based on all the evidence collected so far, I will continue to preferentially use full ablation on most of my patients.
Dr. Pierre Page is chief of cardiac surgery at the University of Montreal. He had no disclosures. He made these comments in an interview.
It comes as no surprise that complete left-atrial ablation during cardiac surgery produces impaired left-atrial function, although it is surprising to see how much damage occurs. But this finding is no reason to abandon atrial ablation and replace it with less extensive treatment with pulmonary-vein isolation unless the reduced left-atrial function is shown to have a clear impact on patient outcomes or survival. Based on what we know today, on balance, it’s more important to more thoroughly and reliably address our patients’ atrial arrhythmia than it is to preserve full atrial function. Substituting pulmonary-vein isolation for full ablation would increase the risk of atrial fibrillation recurrence.
The study done by Dr. Compier and her associates in Leiden is the first to document the functional impact of complete left-atrial ablation using the modified Cox-MAZE procedure in such a careful and systematic way using echocardiography. But anyone who is concerned about the impact of full ablation on atrial function must acknowledge that this treatment is also very beneficial to patients. The Leiden group clearly showed that full ablation is very detrimental to the atrium, but they did not associate these impairments with adverse clinical outcomes. As far as we know, atrial fibrillation is a more important determinant of clinical outcome than are changes in left-atrial function.
|
|
|
A major way in which full ablation differs from pulmonary vein isolation is the added isolation of the left-atrial appendage, and we know that contraction of the left atrium mostly depends on the left-atrial appendage. But isolation of the appendage also reduces the risk of stroke. Previously-reported findings from several studies have shown that pulmonary vein isolation is less effective at restoring sinus rhythm and preventing atrial fibrillation recurrence. Based on all the evidence collected so far, I will continue to preferentially use full ablation on most of my patients.
Dr. Pierre Page is chief of cardiac surgery at the University of Montreal. He had no disclosures. He made these comments in an interview.
It comes as no surprise that complete left-atrial ablation during cardiac surgery produces impaired left-atrial function, although it is surprising to see how much damage occurs. But this finding is no reason to abandon atrial ablation and replace it with less extensive treatment with pulmonary-vein isolation unless the reduced left-atrial function is shown to have a clear impact on patient outcomes or survival. Based on what we know today, on balance, it’s more important to more thoroughly and reliably address our patients’ atrial arrhythmia than it is to preserve full atrial function. Substituting pulmonary-vein isolation for full ablation would increase the risk of atrial fibrillation recurrence.
The study done by Dr. Compier and her associates in Leiden is the first to document the functional impact of complete left-atrial ablation using the modified Cox-MAZE procedure in such a careful and systematic way using echocardiography. But anyone who is concerned about the impact of full ablation on atrial function must acknowledge that this treatment is also very beneficial to patients. The Leiden group clearly showed that full ablation is very detrimental to the atrium, but they did not associate these impairments with adverse clinical outcomes. As far as we know, atrial fibrillation is a more important determinant of clinical outcome than are changes in left-atrial function.
|
|
|
A major way in which full ablation differs from pulmonary vein isolation is the added isolation of the left-atrial appendage, and we know that contraction of the left atrium mostly depends on the left-atrial appendage. But isolation of the appendage also reduces the risk of stroke. Previously-reported findings from several studies have shown that pulmonary vein isolation is less effective at restoring sinus rhythm and preventing atrial fibrillation recurrence. Based on all the evidence collected so far, I will continue to preferentially use full ablation on most of my patients.
Dr. Pierre Page is chief of cardiac surgery at the University of Montreal. He had no disclosures. He made these comments in an interview.
LOS ANGELES – Full left-atrial ablation by the modified Cox MAZE procedure to treat atrial fibrillation led to significant reductions in left atrial function, based on a detailed assessment of 31 patients who underwent this treatment using bilateral bipolar radiofrequency.
The damaging effect of complete left-atrial ablation contrasted with the impact of a less extensive procedure, pulmonary vein isolation, which kept left-atrial function intact and even improved it by some parameters. Opinions split on the implications of these findings.
"Our advice is that left-atrial ablation should be restricted to those cases where pulmonary vein isolation will likely be insufficient" to restore and maintain sinus rhythm, Dr. Marieke G. Compier said as she presented the findings at the annual scientific sessions of the American Heart Association on Nov. 6.
But a cardiac surgeon who heard the results disagreed, contending that the top priority is performing the procedure that will result in durable prevention of atrial fibrillation (AF) recurrence.
"Recurrence of atrial fibrillation is a more important determinant of [successful] clinical outcomes than preserved atrial function," said Dr. Pierre Page, chief of cardiac surgery at the University of Montreal. "The data we have today show that left-atrial ablation is very beneficial," Dr. Page said in an interview.
Dr. Compier and her associates used echocardiography to assess left-atrial function in 31 patients who underwent a modified Cox-MAZE procedure for complete left-atrial ablation using bilateral bipolar radiofrequency, and 31 patients who underwent pulmonary-vein isolation (PVI) only. In the full ablation group, 25 patients had persistent AF and 6 had paroxysmal AF; in the PVI group, 25 patients had paroxysmal disease and 6 had persistent AF. All patients also underwent concurrent coronary artery bypass, valve surgery, or both.
The researchers assessed the efficacy of AF treatment using 24-hour ECG monitoring at 3, 6, and 12 months following surgery. They also used two-dimensional echocardiography to assess left atrial size and function at 3 months and 1 year after surgery.
After 1 year, 68% of the patients who had full ablation and 81% of those who underwent PVI were free of AF and completely off anti-arrhythmic drug treatment; the other patients in each group had AF recurrences. The different long-term success in maintaining sinus rhythm in the two groups probably stemmed from the unbalanced distribution of patients with paroxysmal and persistent AF, said Dr. Compier, a cardiologist in the Heart Center at Leiden (the Netherlands) University Medical Center. "I think this is why full ablation seemed less successful," she said.
After 1 year, echocardiographic examinations showed that patients treated with full ablation had statistically significant reductions in left atrial volume and strain, and 42% of patients had A-wave restoration. Compared with measurements made prior to surgery, strain rate fell by an average of about 50%, peak A-wave dropped by an average of about a third, and average left-atrial ejection fraction and filling fraction each dropped by about 20%. All of these changes were statistically significant, compared with baseline.
In contrast, patients who underwent PVI had no significant change in their strain rate or peak A wave, and their average left-atrial ejection fraction and filling fraction each rose by about 10% compared with baseline, statistically significant differences. A-wave restoration occurred in 87% of the PVI patients.
Stepwise regression analysis of baseline differences between the two study groups showed that the follow-up differences seen in left-atrial size and function were best explained by the different ablation treatments the two groups received, Dr. Compier said.
Dr. Compier said that she had no disclosures.
LOS ANGELES – Full left-atrial ablation by the modified Cox MAZE procedure to treat atrial fibrillation led to significant reductions in left atrial function, based on a detailed assessment of 31 patients who underwent this treatment using bilateral bipolar radiofrequency.
The damaging effect of complete left-atrial ablation contrasted with the impact of a less extensive procedure, pulmonary vein isolation, which kept left-atrial function intact and even improved it by some parameters. Opinions split on the implications of these findings.
"Our advice is that left-atrial ablation should be restricted to those cases where pulmonary vein isolation will likely be insufficient" to restore and maintain sinus rhythm, Dr. Marieke G. Compier said as she presented the findings at the annual scientific sessions of the American Heart Association on Nov. 6.
But a cardiac surgeon who heard the results disagreed, contending that the top priority is performing the procedure that will result in durable prevention of atrial fibrillation (AF) recurrence.
"Recurrence of atrial fibrillation is a more important determinant of [successful] clinical outcomes than preserved atrial function," said Dr. Pierre Page, chief of cardiac surgery at the University of Montreal. "The data we have today show that left-atrial ablation is very beneficial," Dr. Page said in an interview.
Dr. Compier and her associates used echocardiography to assess left-atrial function in 31 patients who underwent a modified Cox-MAZE procedure for complete left-atrial ablation using bilateral bipolar radiofrequency, and 31 patients who underwent pulmonary-vein isolation (PVI) only. In the full ablation group, 25 patients had persistent AF and 6 had paroxysmal AF; in the PVI group, 25 patients had paroxysmal disease and 6 had persistent AF. All patients also underwent concurrent coronary artery bypass, valve surgery, or both.
The researchers assessed the efficacy of AF treatment using 24-hour ECG monitoring at 3, 6, and 12 months following surgery. They also used two-dimensional echocardiography to assess left atrial size and function at 3 months and 1 year after surgery.
After 1 year, 68% of the patients who had full ablation and 81% of those who underwent PVI were free of AF and completely off anti-arrhythmic drug treatment; the other patients in each group had AF recurrences. The different long-term success in maintaining sinus rhythm in the two groups probably stemmed from the unbalanced distribution of patients with paroxysmal and persistent AF, said Dr. Compier, a cardiologist in the Heart Center at Leiden (the Netherlands) University Medical Center. "I think this is why full ablation seemed less successful," she said.
After 1 year, echocardiographic examinations showed that patients treated with full ablation had statistically significant reductions in left atrial volume and strain, and 42% of patients had A-wave restoration. Compared with measurements made prior to surgery, strain rate fell by an average of about 50%, peak A-wave dropped by an average of about a third, and average left-atrial ejection fraction and filling fraction each dropped by about 20%. All of these changes were statistically significant, compared with baseline.
In contrast, patients who underwent PVI had no significant change in their strain rate or peak A wave, and their average left-atrial ejection fraction and filling fraction each rose by about 10% compared with baseline, statistically significant differences. A-wave restoration occurred in 87% of the PVI patients.
Stepwise regression analysis of baseline differences between the two study groups showed that the follow-up differences seen in left-atrial size and function were best explained by the different ablation treatments the two groups received, Dr. Compier said.
Dr. Compier said that she had no disclosures.
AT THE ANNUAL SCIENTIFIC SESSIONS OF THE AMERICAN HEART ASSOCIATION
Major Finding: Full left-atrial ablation produced statistically significant impairments of left-atrial function, including an average 20% drop in ejection fraction.
Data Source: An echocardiographic assessment of left atrial size and function in 31 patients treated with surgical left-atrial ablation, and 31 treated by pulmonary vein isolation at one center.
Disclosures: Dr. Compier and Dr. Page said that they had no disclosures.

Perioperative Fish Oil Supplements Did Not Cut AF Risk
Perioperative supplementation with fish oil–derived fatty acids did not prevent postoperative atrial fibrillation in patients undergoing cardiac surgery, according to results from a large, randomized controlled trial.
Atrial fibrillation (AF) affects approximately one-third of patients who undergo cardiac surgery. Animal studies have suggested that the long-chain n-3-polyunsaturated fatty acids (PUFAs) found in fish oil have antiarrhythmic activity, and clinical trials have shown that habitual consumption of fish oil favorably affects physiological pathways that influence AF as well as overall cardiac mortality risk (J. Am. Coll. Cardiol. 2011;58:2047-67).
The new study, published online Nov. 5 in JAMA (doi:10.1001/jama.2012.28733) and presented simultaneously at the annual scientific sessions of the American Heart Association, demonstrated that fish oil supplementation beginning 2-5 days prior to cardiac surgery and continuing until hospital discharge did not reduce risk of postoperative atrial fibrillation.

For their research, Dr. Dariush Mozaffarian of the Harvard School of Public Health, Boston, and Dr. Roberto Marchioli of Consorzio Mario Negri Sud in Santa Maria Imbaro, Italy, and their colleagues randomized to fish oil capsules or placebo 1,516 patients who were scheduled for cardiac surgery at 28 centers in the United States, Italy, and Argentina.
Patients took 10 capsules, each 1-g capsule containing at least 840 mg of n-3-PUFAs, eicosapentaenoic acid (approximately 465 mg) plus docosahexaenoic acid (approximately 375 mg) as ethyl esters (Omacor) or matched placebo (olive oil) over 3-5 days before surgery, followed by two capsules per day until discharge or day 10 post surgery, whichever came first. The subjects’ average age was 64 years; 72% were men. Patients who took fish oil regularly were excluded from the study.
Centers were encouraged to use continuous electrocardiographic monitoring for at least 5 days post surgery. Twelve-lead ECGs were recommended daily and more frequently at the discretion of the treating physicians for symptoms or clinically suspected arrhythmia. Confirmatory rhythm strips or 12-lead ECGs were collected for all postoperative arrhythmias of at least 30 seconds’ duration.
AF episodes lasting 30 seconds or longer occurred after surgery in 233 (30.7%) controls and in 227 (30%) subjects given fish oil (odds ratio, 0.96 [95% confidence interval, 0.77-1.20]; P = .74). The two groups did not differ significantly in the number of AF episodes or the incidence of sustained AF episodes. Also, there was no significant difference in the risk of clinical bleeding, even though more than half of the study patients were also taking aspirin or other anticoagulants, the investigators reported.
Though the investigators hypothesized that they would see stronger efficacy of treatment among patients who consumed less than two servings a week of oily fish or those who had lower (<4%) plasma-PUFA levels at enrollment, no significant differences were found in those subgroups, they wrote.
Patients were identified and assigned to receive fish oil capsules for different durations (ranging from 2 to 5 days) prior to surgery, which could have resulted in the shorter durations being less effective, the investigators acknowledged. Subgroup analyses, however, did not detect significantly greater risk reduction associated with more days of fish oil before surgery.
The researchers noted that current best-practice guidelines for preventing postoperative AF were recommended to all participating surgical centers, "which could have reduced the influence of any additional therapy on risk of postoperative AF." Further, the supplement doses may have been too low to produce a benefit, even though phospholipid n-3-PUFA levels increased by an average of 40% by the time of surgery, "providing novel evidence that even short-term supplementation significantly influences circulating levels," they said.
Because of the known benefits of n-3-PUFAs on cardiovascular risk factors and physiologic pathways, "a more promising strategy may be long-term consumption to reduce the primary incidence of AF among ambulatory elderly adults with hypertension or other risk factors; such an approach should be tested in appropriately designed and powered clinical trials," the investigators wrote in their analysis.
The study was funded with grants from the National Institutes of Health, GlaxoSmithKline, Sigma Tau, and Pronova BioPharma, which also provided the capsules used in the study. None of the investigators declared conflicts of interest.
Perioperative supplementation with fish oil–derived fatty acids did not prevent postoperative atrial fibrillation in patients undergoing cardiac surgery, according to results from a large, randomized controlled trial.
Atrial fibrillation (AF) affects approximately one-third of patients who undergo cardiac surgery. Animal studies have suggested that the long-chain n-3-polyunsaturated fatty acids (PUFAs) found in fish oil have antiarrhythmic activity, and clinical trials have shown that habitual consumption of fish oil favorably affects physiological pathways that influence AF as well as overall cardiac mortality risk (J. Am. Coll. Cardiol. 2011;58:2047-67).
The new study, published online Nov. 5 in JAMA (doi:10.1001/jama.2012.28733) and presented simultaneously at the annual scientific sessions of the American Heart Association, demonstrated that fish oil supplementation beginning 2-5 days prior to cardiac surgery and continuing until hospital discharge did not reduce risk of postoperative atrial fibrillation.
For their research, Dr. Dariush Mozaffarian of the Harvard School of Public Health, Boston, and Dr. Roberto Marchioli of Consorzio Mario Negri Sud in Santa Maria Imbaro, Italy, and their colleagues randomized to fish oil capsules or placebo 1,516 patients who were scheduled for cardiac surgery at 28 centers in the United States, Italy, and Argentina.
Patients took 10 capsules, each 1-g capsule containing at least 840 mg of n-3-PUFAs, eicosapentaenoic acid (approximately 465 mg) plus docosahexaenoic acid (approximately 375 mg) as ethyl esters (Omacor) or matched placebo (olive oil) over 3-5 days before surgery, followed by two capsules per day until discharge or day 10 post surgery, whichever came first. The subjects’ average age was 64 years; 72% were men. Patients who took fish oil regularly were excluded from the study.
Centers were encouraged to use continuous electrocardiographic monitoring for at least 5 days post surgery. Twelve-lead ECGs were recommended daily and more frequently at the discretion of the treating physicians for symptoms or clinically suspected arrhythmia. Confirmatory rhythm strips or 12-lead ECGs were collected for all postoperative arrhythmias of at least 30 seconds’ duration.
AF episodes lasting 30 seconds or longer occurred after surgery in 233 (30.7%) controls and in 227 (30%) subjects given fish oil (odds ratio, 0.96 [95% confidence interval, 0.77-1.20]; P = .74). The two groups did not differ significantly in the number of AF episodes or the incidence of sustained AF episodes. Also, there was no significant difference in the risk of clinical bleeding, even though more than half of the study patients were also taking aspirin or other anticoagulants, the investigators reported.
Though the investigators hypothesized that they would see stronger efficacy of treatment among patients who consumed less than two servings a week of oily fish or those who had lower (<4%) plasma-PUFA levels at enrollment, no significant differences were found in those subgroups, they wrote.
Patients were identified and assigned to receive fish oil capsules for different durations (ranging from 2 to 5 days) prior to surgery, which could have resulted in the shorter durations being less effective, the investigators acknowledged. Subgroup analyses, however, did not detect significantly greater risk reduction associated with more days of fish oil before surgery.
The researchers noted that current best-practice guidelines for preventing postoperative AF were recommended to all participating surgical centers, "which could have reduced the influence of any additional therapy on risk of postoperative AF." Further, the supplement doses may have been too low to produce a benefit, even though phospholipid n-3-PUFA levels increased by an average of 40% by the time of surgery, "providing novel evidence that even short-term supplementation significantly influences circulating levels," they said.
Because of the known benefits of n-3-PUFAs on cardiovascular risk factors and physiologic pathways, "a more promising strategy may be long-term consumption to reduce the primary incidence of AF among ambulatory elderly adults with hypertension or other risk factors; such an approach should be tested in appropriately designed and powered clinical trials," the investigators wrote in their analysis.
The study was funded with grants from the National Institutes of Health, GlaxoSmithKline, Sigma Tau, and Pronova BioPharma, which also provided the capsules used in the study. None of the investigators declared conflicts of interest.
Perioperative supplementation with fish oil–derived fatty acids did not prevent postoperative atrial fibrillation in patients undergoing cardiac surgery, according to results from a large, randomized controlled trial.
Atrial fibrillation (AF) affects approximately one-third of patients who undergo cardiac surgery. Animal studies have suggested that the long-chain n-3-polyunsaturated fatty acids (PUFAs) found in fish oil have antiarrhythmic activity, and clinical trials have shown that habitual consumption of fish oil favorably affects physiological pathways that influence AF as well as overall cardiac mortality risk (J. Am. Coll. Cardiol. 2011;58:2047-67).
The new study, published online Nov. 5 in JAMA (doi:10.1001/jama.2012.28733) and presented simultaneously at the annual scientific sessions of the American Heart Association, demonstrated that fish oil supplementation beginning 2-5 days prior to cardiac surgery and continuing until hospital discharge did not reduce risk of postoperative atrial fibrillation.
For their research, Dr. Dariush Mozaffarian of the Harvard School of Public Health, Boston, and Dr. Roberto Marchioli of Consorzio Mario Negri Sud in Santa Maria Imbaro, Italy, and their colleagues randomized to fish oil capsules or placebo 1,516 patients who were scheduled for cardiac surgery at 28 centers in the United States, Italy, and Argentina.
Patients took 10 capsules, each 1-g capsule containing at least 840 mg of n-3-PUFAs, eicosapentaenoic acid (approximately 465 mg) plus docosahexaenoic acid (approximately 375 mg) as ethyl esters (Omacor) or matched placebo (olive oil) over 3-5 days before surgery, followed by two capsules per day until discharge or day 10 post surgery, whichever came first. The subjects’ average age was 64 years; 72% were men. Patients who took fish oil regularly were excluded from the study.
Centers were encouraged to use continuous electrocardiographic monitoring for at least 5 days post surgery. Twelve-lead ECGs were recommended daily and more frequently at the discretion of the treating physicians for symptoms or clinically suspected arrhythmia. Confirmatory rhythm strips or 12-lead ECGs were collected for all postoperative arrhythmias of at least 30 seconds’ duration.
AF episodes lasting 30 seconds or longer occurred after surgery in 233 (30.7%) controls and in 227 (30%) subjects given fish oil (odds ratio, 0.96 [95% confidence interval, 0.77-1.20]; P = .74). The two groups did not differ significantly in the number of AF episodes or the incidence of sustained AF episodes. Also, there was no significant difference in the risk of clinical bleeding, even though more than half of the study patients were also taking aspirin or other anticoagulants, the investigators reported.
Though the investigators hypothesized that they would see stronger efficacy of treatment among patients who consumed less than two servings a week of oily fish or those who had lower (<4%) plasma-PUFA levels at enrollment, no significant differences were found in those subgroups, they wrote.
Patients were identified and assigned to receive fish oil capsules for different durations (ranging from 2 to 5 days) prior to surgery, which could have resulted in the shorter durations being less effective, the investigators acknowledged. Subgroup analyses, however, did not detect significantly greater risk reduction associated with more days of fish oil before surgery.
The researchers noted that current best-practice guidelines for preventing postoperative AF were recommended to all participating surgical centers, "which could have reduced the influence of any additional therapy on risk of postoperative AF." Further, the supplement doses may have been too low to produce a benefit, even though phospholipid n-3-PUFA levels increased by an average of 40% by the time of surgery, "providing novel evidence that even short-term supplementation significantly influences circulating levels," they said.
Because of the known benefits of n-3-PUFAs on cardiovascular risk factors and physiologic pathways, "a more promising strategy may be long-term consumption to reduce the primary incidence of AF among ambulatory elderly adults with hypertension or other risk factors; such an approach should be tested in appropriately designed and powered clinical trials," the investigators wrote in their analysis.
The study was funded with grants from the National Institutes of Health, GlaxoSmithKline, Sigma Tau, and Pronova BioPharma, which also provided the capsules used in the study. None of the investigators declared conflicts of interest.
FROM JAMA
Major Finding: Atrial fibrillation lasting 30 seconds or longer occurred after cardiac surgery in 233 (30.7%) controls and 227 (30%) patients who started fish oil supplements 2-5 days before surgery (OR 0.96 [95% CI, 0.77-1.20]; P = .74).
Data Source: OPERA (Omega-3 Fatty Acids for Prevention of Post-operative Atrial Fibrillation) was a randomized, placebo-controlled trial enrolling more than 1,500 patients scheduled for cardiac surgery at 28 centers in three countries.
Disclosures: The study was funded with grants from the National Institutes of Health, GlaxoSmithKline, Sigma Tau, and Pronova BioPharma, which also provided the capsules used in the study. None of the investigators declared conflicts of interest.

FREEDOM: CABG Shows Excellent Cost Effectiveness
LOS ANGELES – Coronary artery bypass graft surgery not only provides better clinical outcomes than percutaneous coronary intervention in diabetic patients with multivessel disease, but it does so in a highly cost-effective manner, according to an economic analysis of the FREEDOM trial.
"The benefits are achieved at an overall cost that represents an attractive use of societal health care resources," Elizabeth A. Magnuson, Sc.D., said at the annual scientific sessions of the American Heart Association.

FREEDOM was a randomized international trial that compared the effectiveness of CABG with percutaneous coronary intervention (PCI) using drug-eluting stents in 1,900 diabetic patients with multivessel coronary artery disease who were candidates for both procedures.
The initial hospitalization for revascularization cost an average of $34,467 in the CABG group, $8,622 more than for PCI-treated patients. But during the next 5 years of follow-up, both repeat revascularizations and mortality were significantly more common in the PCI group.
Based on a conservative model of projected survival that assumed a gradual attenuation of CABG’s clinical benefits over time, bypass surgery was associated with an incremental cost-effectiveness ratio of $8,132 per quality-adjusted year of life (QALY) gained. That’s well below the figure of $50,000 per QALY widely accepted by health policy makers as defining the upper boundary of cost effectiveness, noted Dr. Magnuson, director of health economics and technology assessment at Saint Luke’s Mid-America Heart Institute, Kansas City, Mo.

"Even if we assume no further benefit beyond the trial period and we just captured the life-years lost due to the in-trial death events, we still get very favorable results for CABG, with an incremental cost-effectiveness ratio of roughly $27,000 per QALY," she said.
Discussant Dr. Mark A. Hlatky agreed with Dr. Magnuson that this was a very conservative analysis and said that the actual cost effectiveness of CABG in diabetes patients with multivessel disease might well be even more favorable than she projected.
"The cost-effectiveness results are quite good. This looks like a very economically attractive therapy for patients who have diabetes with multivessel disease," concluded Dr. Hlatky, professor of health research and policy and of cardiovascular medicine at Stanford (Calif.) University.
The study was funded by the National Heart, Lung, and Blood Institute. Dr. Magnuson and Dr. Hlatky reported having no relevant financial conflicts.
LOS ANGELES – Coronary artery bypass graft surgery not only provides better clinical outcomes than percutaneous coronary intervention in diabetic patients with multivessel disease, but it does so in a highly cost-effective manner, according to an economic analysis of the FREEDOM trial.
"The benefits are achieved at an overall cost that represents an attractive use of societal health care resources," Elizabeth A. Magnuson, Sc.D., said at the annual scientific sessions of the American Heart Association.
FREEDOM was a randomized international trial that compared the effectiveness of CABG with percutaneous coronary intervention (PCI) using drug-eluting stents in 1,900 diabetic patients with multivessel coronary artery disease who were candidates for both procedures.
The initial hospitalization for revascularization cost an average of $34,467 in the CABG group, $8,622 more than for PCI-treated patients. But during the next 5 years of follow-up, both repeat revascularizations and mortality were significantly more common in the PCI group.
Based on a conservative model of projected survival that assumed a gradual attenuation of CABG’s clinical benefits over time, bypass surgery was associated with an incremental cost-effectiveness ratio of $8,132 per quality-adjusted year of life (QALY) gained. That’s well below the figure of $50,000 per QALY widely accepted by health policy makers as defining the upper boundary of cost effectiveness, noted Dr. Magnuson, director of health economics and technology assessment at Saint Luke’s Mid-America Heart Institute, Kansas City, Mo.
"Even if we assume no further benefit beyond the trial period and we just captured the life-years lost due to the in-trial death events, we still get very favorable results for CABG, with an incremental cost-effectiveness ratio of roughly $27,000 per QALY," she said.
Discussant Dr. Mark A. Hlatky agreed with Dr. Magnuson that this was a very conservative analysis and said that the actual cost effectiveness of CABG in diabetes patients with multivessel disease might well be even more favorable than she projected.
"The cost-effectiveness results are quite good. This looks like a very economically attractive therapy for patients who have diabetes with multivessel disease," concluded Dr. Hlatky, professor of health research and policy and of cardiovascular medicine at Stanford (Calif.) University.
The study was funded by the National Heart, Lung, and Blood Institute. Dr. Magnuson and Dr. Hlatky reported having no relevant financial conflicts.
LOS ANGELES – Coronary artery bypass graft surgery not only provides better clinical outcomes than percutaneous coronary intervention in diabetic patients with multivessel disease, but it does so in a highly cost-effective manner, according to an economic analysis of the FREEDOM trial.
"The benefits are achieved at an overall cost that represents an attractive use of societal health care resources," Elizabeth A. Magnuson, Sc.D., said at the annual scientific sessions of the American Heart Association.
FREEDOM was a randomized international trial that compared the effectiveness of CABG with percutaneous coronary intervention (PCI) using drug-eluting stents in 1,900 diabetic patients with multivessel coronary artery disease who were candidates for both procedures.
The initial hospitalization for revascularization cost an average of $34,467 in the CABG group, $8,622 more than for PCI-treated patients. But during the next 5 years of follow-up, both repeat revascularizations and mortality were significantly more common in the PCI group.
Based on a conservative model of projected survival that assumed a gradual attenuation of CABG’s clinical benefits over time, bypass surgery was associated with an incremental cost-effectiveness ratio of $8,132 per quality-adjusted year of life (QALY) gained. That’s well below the figure of $50,000 per QALY widely accepted by health policy makers as defining the upper boundary of cost effectiveness, noted Dr. Magnuson, director of health economics and technology assessment at Saint Luke’s Mid-America Heart Institute, Kansas City, Mo.
"Even if we assume no further benefit beyond the trial period and we just captured the life-years lost due to the in-trial death events, we still get very favorable results for CABG, with an incremental cost-effectiveness ratio of roughly $27,000 per QALY," she said.
Discussant Dr. Mark A. Hlatky agreed with Dr. Magnuson that this was a very conservative analysis and said that the actual cost effectiveness of CABG in diabetes patients with multivessel disease might well be even more favorable than she projected.
"The cost-effectiveness results are quite good. This looks like a very economically attractive therapy for patients who have diabetes with multivessel disease," concluded Dr. Hlatky, professor of health research and policy and of cardiovascular medicine at Stanford (Calif.) University.
The study was funded by the National Heart, Lung, and Blood Institute. Dr. Magnuson and Dr. Hlatky reported having no relevant financial conflicts.
AT THE ANNUAL SCIENTIFIC SESSIONS OF THE AMERICAN HEART ASSOCIATION
Major Finding: Coronary artery bypass surgery in patients with diabetes and multivessel coronary artery disease had a highly favorable projected lifetime incremental cost-effectiveness ratio of $8,132 per quality-adjusted life year gained.
Data Source: Data are from a prespecified cost-effectiveness analysis from the randomized international FREEDOM trial comparing CABG and percutaneous coronary intervention using drug-eluting stents in 1,900 diabetic patients with multivessel coronary artery disease.
Disclosures: The study was funded by the National Heart, Lung, and Blood Institute. Dr. Magnuson and Dr. Hlatky reported having no relevant financial conflicts.

Lung-Volume Reduction Coils Boost Walk Distance
ATLANTA – The implantation of nitinol coils that grab and compress diseased lung tissue, thereby allowing for better functioning of healthy tissue, significantly improved quality of life, exercise capacity, and pulmonary lung function in a randomized controlled trial of patients with severe emphysema and hyperinflation.
Specifically, use of the investigational self-actuating, implantable devices in the RESET (Randomized Controlled Trial of RePneu Endobronchial Coils for the Treatment of Severe Emphysema with Hyperinflation) study was associated with a significantly improved mean St. George’s Respiratory Questionnaire score at 90 days after the final treatment in 23 patients who received active treatment, compared with 24 controls who received best medical care.
After adjustment for baseline variables, the between-group difference in the scores was 8.35 points in favor of the treatment group, Dr. Zaid Zoumot reported at the annual meeting of the American College of Chest Physicians.
The lung-volume reduction coils also were associated with significant improvements in mean 6-minute walk distance (mean between-group difference of 63.5 m in favor of the treatment group) and forced expiratory volume in 1 second (FEV1, mean between-group difference of 12% in favor of the treatment group), said Dr. Zoumot of Royal Brompton and Harefield Hospital Trust, London.
The between-group difference in change in mean residual volume did not reach statistical significance, despite a 0.64-L reduction in the treatment group compared with the control group, he noted.
RESET participants were adults with severe emphysema and hyperinflation with significant dyspnea and gas trapping who were screened at three participating centers in the United Kingdom. Those randomized to the treatment group initially underwent implantation of the coils in one lung, with treatment of the contralateral lung after 1 month if appropriate.
Treatment was generally safe and well tolerated; three patients in the treatment group had pneumothoraces, which were picked up on chest x-ray routinely performed 1 hour following the procedure and treated successfully, Dr. Zoumot said. No differences in adverse effects occurred between the groups after the first month of follow-up, including in exacerbations of chronic obstructive pulmonary disorder, he added.
"The safety profile was definitely acceptable, and in fact, the procedures were a lot safer than other endobronchial lung-volume reduction devices at this same stage of development, and certainly a lot safer than lung-volume reduction surgery, which has a quite high morbidity and mortality rate," he said.
The findings are encouraging given the limited therapeutic options for patients with severe emphysema with gas trapping and hyperinflation – particularly those with heterogeneous disease, he said.
Drug therapy is typically of little benefit in these patients, and although lung-volume reduction surgery and endobronchial valve treatment can be helpful in some patients, their use is precluded in many patients, including those with heterogeneous disease in the absence of collateral ventilation, he explained.
The RePneu lung-volume reduction coils, however, provide a minimally invasive mechanical approach to lung-volume reduction that is effective in both homogeneous and heterogeneous emphysema, with benefits unaffected by collateral ventilation, he said.
The coils, which are made entirely of nitinol – a highly biocompatible "shape memory" material used in numerous implantable devices – are deployed to the lung bronchoscopically using a proprietary delivery system. Initially, the coils are encased in a sheath to allow delivery in a straight configuration, but once they are in place they return to their original coil configuration, gathering and compressing the diseased tissue as they recoil.
The goal is to implant 10 coils per lobe in a fanlike distribution, Dr. Zoumot said. The procedure, which took about 45 minutes on average in this study, is typically performed under conscious sedation, he added.
Patients in the current study will be followed until 12 months after their final treatment, with results reported at both 6 and 12 months. A larger, multicenter randomized controlled trial with longer follow-up is also set to begin recruiting, Dr. Zoumot said.
This study was funded by PneumRx, the maker of the RePneu coils, and the study sites. Dr. Zoumot reported receiving grant funding and payment for travel expenses from PneumRx.
ATLANTA – The implantation of nitinol coils that grab and compress diseased lung tissue, thereby allowing for better functioning of healthy tissue, significantly improved quality of life, exercise capacity, and pulmonary lung function in a randomized controlled trial of patients with severe emphysema and hyperinflation.
Specifically, use of the investigational self-actuating, implantable devices in the RESET (Randomized Controlled Trial of RePneu Endobronchial Coils for the Treatment of Severe Emphysema with Hyperinflation) study was associated with a significantly improved mean St. George’s Respiratory Questionnaire score at 90 days after the final treatment in 23 patients who received active treatment, compared with 24 controls who received best medical care.
After adjustment for baseline variables, the between-group difference in the scores was 8.35 points in favor of the treatment group, Dr. Zaid Zoumot reported at the annual meeting of the American College of Chest Physicians.
The lung-volume reduction coils also were associated with significant improvements in mean 6-minute walk distance (mean between-group difference of 63.5 m in favor of the treatment group) and forced expiratory volume in 1 second (FEV1, mean between-group difference of 12% in favor of the treatment group), said Dr. Zoumot of Royal Brompton and Harefield Hospital Trust, London.
The between-group difference in change in mean residual volume did not reach statistical significance, despite a 0.64-L reduction in the treatment group compared with the control group, he noted.
RESET participants were adults with severe emphysema and hyperinflation with significant dyspnea and gas trapping who were screened at three participating centers in the United Kingdom. Those randomized to the treatment group initially underwent implantation of the coils in one lung, with treatment of the contralateral lung after 1 month if appropriate.
Treatment was generally safe and well tolerated; three patients in the treatment group had pneumothoraces, which were picked up on chest x-ray routinely performed 1 hour following the procedure and treated successfully, Dr. Zoumot said. No differences in adverse effects occurred between the groups after the first month of follow-up, including in exacerbations of chronic obstructive pulmonary disorder, he added.
"The safety profile was definitely acceptable, and in fact, the procedures were a lot safer than other endobronchial lung-volume reduction devices at this same stage of development, and certainly a lot safer than lung-volume reduction surgery, which has a quite high morbidity and mortality rate," he said.
The findings are encouraging given the limited therapeutic options for patients with severe emphysema with gas trapping and hyperinflation – particularly those with heterogeneous disease, he said.
Drug therapy is typically of little benefit in these patients, and although lung-volume reduction surgery and endobronchial valve treatment can be helpful in some patients, their use is precluded in many patients, including those with heterogeneous disease in the absence of collateral ventilation, he explained.
The RePneu lung-volume reduction coils, however, provide a minimally invasive mechanical approach to lung-volume reduction that is effective in both homogeneous and heterogeneous emphysema, with benefits unaffected by collateral ventilation, he said.
The coils, which are made entirely of nitinol – a highly biocompatible "shape memory" material used in numerous implantable devices – are deployed to the lung bronchoscopically using a proprietary delivery system. Initially, the coils are encased in a sheath to allow delivery in a straight configuration, but once they are in place they return to their original coil configuration, gathering and compressing the diseased tissue as they recoil.
The goal is to implant 10 coils per lobe in a fanlike distribution, Dr. Zoumot said. The procedure, which took about 45 minutes on average in this study, is typically performed under conscious sedation, he added.
Patients in the current study will be followed until 12 months after their final treatment, with results reported at both 6 and 12 months. A larger, multicenter randomized controlled trial with longer follow-up is also set to begin recruiting, Dr. Zoumot said.
This study was funded by PneumRx, the maker of the RePneu coils, and the study sites. Dr. Zoumot reported receiving grant funding and payment for travel expenses from PneumRx.
ATLANTA – The implantation of nitinol coils that grab and compress diseased lung tissue, thereby allowing for better functioning of healthy tissue, significantly improved quality of life, exercise capacity, and pulmonary lung function in a randomized controlled trial of patients with severe emphysema and hyperinflation.
Specifically, use of the investigational self-actuating, implantable devices in the RESET (Randomized Controlled Trial of RePneu Endobronchial Coils for the Treatment of Severe Emphysema with Hyperinflation) study was associated with a significantly improved mean St. George’s Respiratory Questionnaire score at 90 days after the final treatment in 23 patients who received active treatment, compared with 24 controls who received best medical care.
After adjustment for baseline variables, the between-group difference in the scores was 8.35 points in favor of the treatment group, Dr. Zaid Zoumot reported at the annual meeting of the American College of Chest Physicians.
The lung-volume reduction coils also were associated with significant improvements in mean 6-minute walk distance (mean between-group difference of 63.5 m in favor of the treatment group) and forced expiratory volume in 1 second (FEV1, mean between-group difference of 12% in favor of the treatment group), said Dr. Zoumot of Royal Brompton and Harefield Hospital Trust, London.
The between-group difference in change in mean residual volume did not reach statistical significance, despite a 0.64-L reduction in the treatment group compared with the control group, he noted.
RESET participants were adults with severe emphysema and hyperinflation with significant dyspnea and gas trapping who were screened at three participating centers in the United Kingdom. Those randomized to the treatment group initially underwent implantation of the coils in one lung, with treatment of the contralateral lung after 1 month if appropriate.
Treatment was generally safe and well tolerated; three patients in the treatment group had pneumothoraces, which were picked up on chest x-ray routinely performed 1 hour following the procedure and treated successfully, Dr. Zoumot said. No differences in adverse effects occurred between the groups after the first month of follow-up, including in exacerbations of chronic obstructive pulmonary disorder, he added.
"The safety profile was definitely acceptable, and in fact, the procedures were a lot safer than other endobronchial lung-volume reduction devices at this same stage of development, and certainly a lot safer than lung-volume reduction surgery, which has a quite high morbidity and mortality rate," he said.
The findings are encouraging given the limited therapeutic options for patients with severe emphysema with gas trapping and hyperinflation – particularly those with heterogeneous disease, he said.
Drug therapy is typically of little benefit in these patients, and although lung-volume reduction surgery and endobronchial valve treatment can be helpful in some patients, their use is precluded in many patients, including those with heterogeneous disease in the absence of collateral ventilation, he explained.
The RePneu lung-volume reduction coils, however, provide a minimally invasive mechanical approach to lung-volume reduction that is effective in both homogeneous and heterogeneous emphysema, with benefits unaffected by collateral ventilation, he said.
The coils, which are made entirely of nitinol – a highly biocompatible "shape memory" material used in numerous implantable devices – are deployed to the lung bronchoscopically using a proprietary delivery system. Initially, the coils are encased in a sheath to allow delivery in a straight configuration, but once they are in place they return to their original coil configuration, gathering and compressing the diseased tissue as they recoil.
The goal is to implant 10 coils per lobe in a fanlike distribution, Dr. Zoumot said. The procedure, which took about 45 minutes on average in this study, is typically performed under conscious sedation, he added.
Patients in the current study will be followed until 12 months after their final treatment, with results reported at both 6 and 12 months. A larger, multicenter randomized controlled trial with longer follow-up is also set to begin recruiting, Dr. Zoumot said.
This study was funded by PneumRx, the maker of the RePneu coils, and the study sites. Dr. Zoumot reported receiving grant funding and payment for travel expenses from PneumRx.
FROM THE ANNUAL MEETING OF THE AMERICAN COLLEGE OF CHEST PHYSICIANS
Major Finding: Implantation of lung-volume reduction coils was associated with a mean between-group difference of 63.5 m in 6-minute walk distance and a mean between-group difference of 12% in FEV1 in favor of the treatment group.
Data Source: This was a randomized controlled trial (RESET) of 23 patients who received active treatment and 24 controls who received best medical care.
Disclosures: This study was funded by PneumRx, the maker of the RePneu coils, and the study sites. Dr. Zoumot reported receiving grant funding and payment for travel expenses from PneumRx.
