User login
<p>For ClinicalEdge use only</p>
FDA Grants Full Approval to Encorafenib in Metastatic CRC
FDA Grants Full Approval to Encorafenib in Metastatic CRC
The FDA has granted traditional approval to encorafenib (Braftovi, Pfizer) in combination with cetuximab (Erbitux, Eli Lilly) and fluorouracil-based chemotherapy for treatment of adults with metastatic colorectal cancer with a BRAF V600E mutation, as detected by an FDA-authorized test.
Encorafenib received accelerated approval for use with cetuximab plus mFOLFOX6 in this patient population in 2024, based on results from the BREAKWATER trial showing improved objective response rates. The conversion to full approval is supported by progression-free and overall survival outcomes.
As reported previously by Medscape Medical News, the combination of encorafenib/cetuximab/mFOLFOX6 doubled median overall survival compared with standard chemotherapy with or without bevacizumab. At a median follow-up of 22 months, overall survival was 30 months with the encorafenib regimen vs 15 months with standard chemotherapy (hazard ratio [HR], 0.49; P < .0001).
At median follow up of 16.8 months, median progression-free survival was 12.8 in the encorafenib group vs 7.1 months in the standard chemotherapy group (HR, 0.53; P < .0001).
The survival results are “unprecedented” and “practice changing” for these patients, who historically have a poor prognosis, lead investigator Elena Élez, MD, PhD, of Vall d’Hebron University Hospital in Barcelona, Spain, said in presenting the findings at the American Society of Clinical Oncology (ASCO) 2025 annual meeting.
The results were simultaneously published in The New England Journal of Medicine.
Speaking at the ASCO meeting, study discussant Andrea Sartore-Bianchi, MD, of the University of Milan, Italy, called the results “striking” and said the encorafenib combination should be considered the first-line standard of care.
As for safety, the rate of treatment-related grade 3/4 adverse events in the trial was 76% with encorafenib vs 59% with standard chemotherapy. Patients receiving encorafenib also had higher rates of anemia, arthralgia, rash, and pyrexia, but there was no substantial increase in treatment discontinuation.
The recommended encorafenib dose is 300 mg (four 75 mg capsules) once daily, in combination with cetuximab and mFOLFOX6 or in combination with cetuximab and FOLFIRI until disease progression or unacceptable toxicity, the FDA said in its approval announcement.
Prescribing information includes warnings and precautions for new primary malignancies (cutaneous and noncutaneous), tumor promotion in BRAF-wild-type tumors, cardiomyopathy, hepatotoxicity, hemorrhage, uveitis, QT prolongation, and embryo-fetal toxicity.
A version of this article first appeared on Medscape.com.
The FDA has granted traditional approval to encorafenib (Braftovi, Pfizer) in combination with cetuximab (Erbitux, Eli Lilly) and fluorouracil-based chemotherapy for treatment of adults with metastatic colorectal cancer with a BRAF V600E mutation, as detected by an FDA-authorized test.
Encorafenib received accelerated approval for use with cetuximab plus mFOLFOX6 in this patient population in 2024, based on results from the BREAKWATER trial showing improved objective response rates. The conversion to full approval is supported by progression-free and overall survival outcomes.
As reported previously by Medscape Medical News, the combination of encorafenib/cetuximab/mFOLFOX6 doubled median overall survival compared with standard chemotherapy with or without bevacizumab. At a median follow-up of 22 months, overall survival was 30 months with the encorafenib regimen vs 15 months with standard chemotherapy (hazard ratio [HR], 0.49; P < .0001).
At median follow up of 16.8 months, median progression-free survival was 12.8 in the encorafenib group vs 7.1 months in the standard chemotherapy group (HR, 0.53; P < .0001).
The survival results are “unprecedented” and “practice changing” for these patients, who historically have a poor prognosis, lead investigator Elena Élez, MD, PhD, of Vall d’Hebron University Hospital in Barcelona, Spain, said in presenting the findings at the American Society of Clinical Oncology (ASCO) 2025 annual meeting.
The results were simultaneously published in The New England Journal of Medicine.
Speaking at the ASCO meeting, study discussant Andrea Sartore-Bianchi, MD, of the University of Milan, Italy, called the results “striking” and said the encorafenib combination should be considered the first-line standard of care.
As for safety, the rate of treatment-related grade 3/4 adverse events in the trial was 76% with encorafenib vs 59% with standard chemotherapy. Patients receiving encorafenib also had higher rates of anemia, arthralgia, rash, and pyrexia, but there was no substantial increase in treatment discontinuation.
The recommended encorafenib dose is 300 mg (four 75 mg capsules) once daily, in combination with cetuximab and mFOLFOX6 or in combination with cetuximab and FOLFIRI until disease progression or unacceptable toxicity, the FDA said in its approval announcement.
Prescribing information includes warnings and precautions for new primary malignancies (cutaneous and noncutaneous), tumor promotion in BRAF-wild-type tumors, cardiomyopathy, hepatotoxicity, hemorrhage, uveitis, QT prolongation, and embryo-fetal toxicity.
A version of this article first appeared on Medscape.com.
The FDA has granted traditional approval to encorafenib (Braftovi, Pfizer) in combination with cetuximab (Erbitux, Eli Lilly) and fluorouracil-based chemotherapy for treatment of adults with metastatic colorectal cancer with a BRAF V600E mutation, as detected by an FDA-authorized test.
Encorafenib received accelerated approval for use with cetuximab plus mFOLFOX6 in this patient population in 2024, based on results from the BREAKWATER trial showing improved objective response rates. The conversion to full approval is supported by progression-free and overall survival outcomes.
As reported previously by Medscape Medical News, the combination of encorafenib/cetuximab/mFOLFOX6 doubled median overall survival compared with standard chemotherapy with or without bevacizumab. At a median follow-up of 22 months, overall survival was 30 months with the encorafenib regimen vs 15 months with standard chemotherapy (hazard ratio [HR], 0.49; P < .0001).
At median follow up of 16.8 months, median progression-free survival was 12.8 in the encorafenib group vs 7.1 months in the standard chemotherapy group (HR, 0.53; P < .0001).
The survival results are “unprecedented” and “practice changing” for these patients, who historically have a poor prognosis, lead investigator Elena Élez, MD, PhD, of Vall d’Hebron University Hospital in Barcelona, Spain, said in presenting the findings at the American Society of Clinical Oncology (ASCO) 2025 annual meeting.
The results were simultaneously published in The New England Journal of Medicine.
Speaking at the ASCO meeting, study discussant Andrea Sartore-Bianchi, MD, of the University of Milan, Italy, called the results “striking” and said the encorafenib combination should be considered the first-line standard of care.
As for safety, the rate of treatment-related grade 3/4 adverse events in the trial was 76% with encorafenib vs 59% with standard chemotherapy. Patients receiving encorafenib also had higher rates of anemia, arthralgia, rash, and pyrexia, but there was no substantial increase in treatment discontinuation.
The recommended encorafenib dose is 300 mg (four 75 mg capsules) once daily, in combination with cetuximab and mFOLFOX6 or in combination with cetuximab and FOLFIRI until disease progression or unacceptable toxicity, the FDA said in its approval announcement.
Prescribing information includes warnings and precautions for new primary malignancies (cutaneous and noncutaneous), tumor promotion in BRAF-wild-type tumors, cardiomyopathy, hepatotoxicity, hemorrhage, uveitis, QT prolongation, and embryo-fetal toxicity.
A version of this article first appeared on Medscape.com.
FDA Grants Full Approval to Encorafenib in Metastatic CRC
FDA Grants Full Approval to Encorafenib in Metastatic CRC
FDA OKs Subcutaneous Mosunetuzumab for Follicular Lymphoma
The FDA has granted accelerated approval for a subcutaneous (SC) formulation of mosunetuzumab (Lunsumio VELO, Roche) for the treatment of certain adults with relapsed or refractory follicular lymphoma.
Specifically, the CD20 × CD3 bispecific antibody — which was initially approved as an intravenous (IV) formulation and was the first of its kind approved for relapsed or refractory follicular lymphoma after at least 2 prior lines of therapy — is now approved for SC administration in the same setting, according to a Roche press release.
SC delivery reduces treatment time to about 1 minute compared with the 2–4 hours required with IV infusion. Like the IV formulation, the SC version can be administered in the outpatient setting and is a fixed-duration treatment given for a defined period, Roche noted, adding that “[b]y contrast, treat-to-progression treatment options are designed to be given to patients indefinitely until disease progression or until treatment can no longer be tolerated.”
Full approval, which may be contingent on verification of benefit in a confirmatory trial, was based on findings from the phase 1/2 G029781 study of both IV and SC formulations in patients with relapsed or refractory non–Hodgkin lymphoma, including follicular lymphoma.
The objective response rate and complete response rate with SC formulation were 75% and 59%, respectively. The median duration of response was 22.4 months.
Adverse reactions occurring in at least 20% of patients were injection site reactions, fatigue, rash, cytokine release syndrome (CRS), SARS–CoV–2 infection, musculoskeletal pain, and diarrhea. CRS occurred in 30% of patients. Most of those events were low-grade, and all resolved after a median of 2 days.
“This approval is a significant step in broadening access to effective treatments for people living with follicular lymphoma,” stated Ian Flinn, MD, PhD, of Tennessee Oncology and OneOncology. “With its manageable cytokine release syndrome profile and reduced administration time, Lunsumio VELO enables oncologists to deliver advanced care in community practice settings.”
A version of this article first appeared on Medscape.com.
The FDA has granted accelerated approval for a subcutaneous (SC) formulation of mosunetuzumab (Lunsumio VELO, Roche) for the treatment of certain adults with relapsed or refractory follicular lymphoma.
Specifically, the CD20 × CD3 bispecific antibody — which was initially approved as an intravenous (IV) formulation and was the first of its kind approved for relapsed or refractory follicular lymphoma after at least 2 prior lines of therapy — is now approved for SC administration in the same setting, according to a Roche press release.
SC delivery reduces treatment time to about 1 minute compared with the 2–4 hours required with IV infusion. Like the IV formulation, the SC version can be administered in the outpatient setting and is a fixed-duration treatment given for a defined period, Roche noted, adding that “[b]y contrast, treat-to-progression treatment options are designed to be given to patients indefinitely until disease progression or until treatment can no longer be tolerated.”
Full approval, which may be contingent on verification of benefit in a confirmatory trial, was based on findings from the phase 1/2 G029781 study of both IV and SC formulations in patients with relapsed or refractory non–Hodgkin lymphoma, including follicular lymphoma.
The objective response rate and complete response rate with SC formulation were 75% and 59%, respectively. The median duration of response was 22.4 months.
Adverse reactions occurring in at least 20% of patients were injection site reactions, fatigue, rash, cytokine release syndrome (CRS), SARS–CoV–2 infection, musculoskeletal pain, and diarrhea. CRS occurred in 30% of patients. Most of those events were low-grade, and all resolved after a median of 2 days.
“This approval is a significant step in broadening access to effective treatments for people living with follicular lymphoma,” stated Ian Flinn, MD, PhD, of Tennessee Oncology and OneOncology. “With its manageable cytokine release syndrome profile and reduced administration time, Lunsumio VELO enables oncologists to deliver advanced care in community practice settings.”
A version of this article first appeared on Medscape.com.
The FDA has granted accelerated approval for a subcutaneous (SC) formulation of mosunetuzumab (Lunsumio VELO, Roche) for the treatment of certain adults with relapsed or refractory follicular lymphoma.
Specifically, the CD20 × CD3 bispecific antibody — which was initially approved as an intravenous (IV) formulation and was the first of its kind approved for relapsed or refractory follicular lymphoma after at least 2 prior lines of therapy — is now approved for SC administration in the same setting, according to a Roche press release.
SC delivery reduces treatment time to about 1 minute compared with the 2–4 hours required with IV infusion. Like the IV formulation, the SC version can be administered in the outpatient setting and is a fixed-duration treatment given for a defined period, Roche noted, adding that “[b]y contrast, treat-to-progression treatment options are designed to be given to patients indefinitely until disease progression or until treatment can no longer be tolerated.”
Full approval, which may be contingent on verification of benefit in a confirmatory trial, was based on findings from the phase 1/2 G029781 study of both IV and SC formulations in patients with relapsed or refractory non–Hodgkin lymphoma, including follicular lymphoma.
The objective response rate and complete response rate with SC formulation were 75% and 59%, respectively. The median duration of response was 22.4 months.
Adverse reactions occurring in at least 20% of patients were injection site reactions, fatigue, rash, cytokine release syndrome (CRS), SARS–CoV–2 infection, musculoskeletal pain, and diarrhea. CRS occurred in 30% of patients. Most of those events were low-grade, and all resolved after a median of 2 days.
“This approval is a significant step in broadening access to effective treatments for people living with follicular lymphoma,” stated Ian Flinn, MD, PhD, of Tennessee Oncology and OneOncology. “With its manageable cytokine release syndrome profile and reduced administration time, Lunsumio VELO enables oncologists to deliver advanced care in community practice settings.”
A version of this article first appeared on Medscape.com.
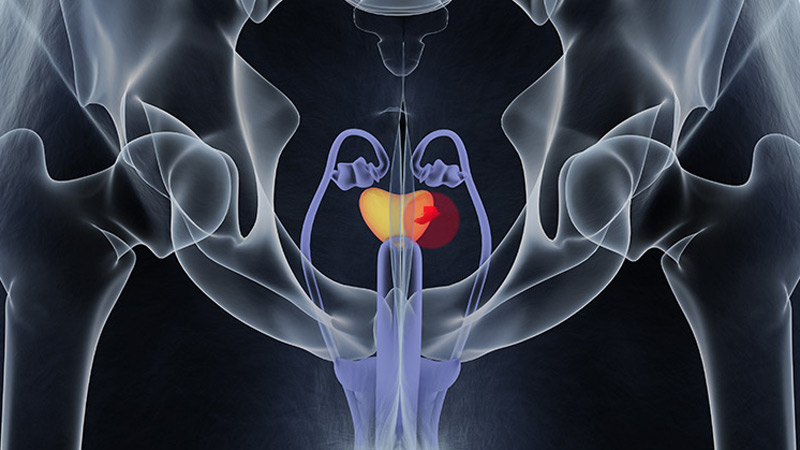
FDA OKs Blood-Based Test to Help Diagnose Prostate Cancer
FDA OKs Blood-Based Test to Help Diagnose Prostate Cancer
The FDA has granted Cleveland Diagnostics' IsoPSA test premarket approval (PMA) to help detect prostate cancer in men aged ≥ 50 years with elevated PSA levels.
IsoPSA is a blood assay that detects variations of the PSA protein that signal a higher likelihood of high-grade tumors. It is one of several biomarker tests included in the National Comprehensive Cancer Network's guidelines on early detection of prostate cancer.
Cleveland Diagnostics noted that 75% of prostate biopsies are negative for high-grade disease. IsoPSA and similar tests aim to help identify men who need a biopsy while allowing others avoid an unnecessary procedure.
IsoPSA has been available since 2020 under the FDA's Laboratory-Developed Test rubric, meaning that blood samples had to be shipped for analysis to Cleveland Diagnostics' lab. With the PMA, testing can now be done at CLIA-certified labs across the country.
The company expects the approval should increase access to IsoPSA and reduce turnaround time. "We remain focused on executing our commercial strategy and expanding access to IsoPSA," company President and CEO Arnon Chait, PhD, said in a press release.
The approval was based, in part, on a prospective validation study of 888 men scheduled for prostate biopsy. IsoPSA demonstrated an AUC of 0.783 for high-grade tumors, with a sensitivity of 90.2% and a specificity of 45.5%. In a real-world clinical utility study with 900 patients, IsoPSA testing led to a 55% decrease in biopsy recommendations.
The test is covered by Medicare and a growing number of commercial payers, Cleveland Diagnostics said.
M. Alexander Otto is a physician assistant with a master's degree in medical science and a journalism degree from Newhouse. He is an award-winning medical journalist who worked for several major news outlets before joining Medscape Medical News. Alex is also an MIT Knight Science Journalism Fellow. Email: [email protected].
A version of this article first appeared on Medscape.com.
The FDA has granted Cleveland Diagnostics' IsoPSA test premarket approval (PMA) to help detect prostate cancer in men aged ≥ 50 years with elevated PSA levels.
IsoPSA is a blood assay that detects variations of the PSA protein that signal a higher likelihood of high-grade tumors. It is one of several biomarker tests included in the National Comprehensive Cancer Network's guidelines on early detection of prostate cancer.
Cleveland Diagnostics noted that 75% of prostate biopsies are negative for high-grade disease. IsoPSA and similar tests aim to help identify men who need a biopsy while allowing others avoid an unnecessary procedure.
IsoPSA has been available since 2020 under the FDA's Laboratory-Developed Test rubric, meaning that blood samples had to be shipped for analysis to Cleveland Diagnostics' lab. With the PMA, testing can now be done at CLIA-certified labs across the country.
The company expects the approval should increase access to IsoPSA and reduce turnaround time. "We remain focused on executing our commercial strategy and expanding access to IsoPSA," company President and CEO Arnon Chait, PhD, said in a press release.
The approval was based, in part, on a prospective validation study of 888 men scheduled for prostate biopsy. IsoPSA demonstrated an AUC of 0.783 for high-grade tumors, with a sensitivity of 90.2% and a specificity of 45.5%. In a real-world clinical utility study with 900 patients, IsoPSA testing led to a 55% decrease in biopsy recommendations.
The test is covered by Medicare and a growing number of commercial payers, Cleveland Diagnostics said.
M. Alexander Otto is a physician assistant with a master's degree in medical science and a journalism degree from Newhouse. He is an award-winning medical journalist who worked for several major news outlets before joining Medscape Medical News. Alex is also an MIT Knight Science Journalism Fellow. Email: [email protected].
A version of this article first appeared on Medscape.com.
The FDA has granted Cleveland Diagnostics' IsoPSA test premarket approval (PMA) to help detect prostate cancer in men aged ≥ 50 years with elevated PSA levels.
IsoPSA is a blood assay that detects variations of the PSA protein that signal a higher likelihood of high-grade tumors. It is one of several biomarker tests included in the National Comprehensive Cancer Network's guidelines on early detection of prostate cancer.
Cleveland Diagnostics noted that 75% of prostate biopsies are negative for high-grade disease. IsoPSA and similar tests aim to help identify men who need a biopsy while allowing others avoid an unnecessary procedure.
IsoPSA has been available since 2020 under the FDA's Laboratory-Developed Test rubric, meaning that blood samples had to be shipped for analysis to Cleveland Diagnostics' lab. With the PMA, testing can now be done at CLIA-certified labs across the country.
The company expects the approval should increase access to IsoPSA and reduce turnaround time. "We remain focused on executing our commercial strategy and expanding access to IsoPSA," company President and CEO Arnon Chait, PhD, said in a press release.
The approval was based, in part, on a prospective validation study of 888 men scheduled for prostate biopsy. IsoPSA demonstrated an AUC of 0.783 for high-grade tumors, with a sensitivity of 90.2% and a specificity of 45.5%. In a real-world clinical utility study with 900 patients, IsoPSA testing led to a 55% decrease in biopsy recommendations.
The test is covered by Medicare and a growing number of commercial payers, Cleveland Diagnostics said.
M. Alexander Otto is a physician assistant with a master's degree in medical science and a journalism degree from Newhouse. He is an award-winning medical journalist who worked for several major news outlets before joining Medscape Medical News. Alex is also an MIT Knight Science Journalism Fellow. Email: [email protected].
A version of this article first appeared on Medscape.com.
FDA OKs Blood-Based Test to Help Diagnose Prostate Cancer
FDA OKs Blood-Based Test to Help Diagnose Prostate Cancer
Mitomycin approved for low-grade upper tract urothelial cancer
pyelocalyceal

“This is the first approval specifically for patients with low-grade [upper tract urothelial cancer] and provides an option for some patients who may otherwise require a nephroureterectomy,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, said in a statement.
“Due to substantial treatment challenges associated with the complex anatomy of the upper urinary tract, many patients need to be treated with radical surgery – usually complete removal of the affected kidney, ureter, and bladder cuff," Dr. Pazdur added. "Jelmyto gives patients, for the first time, an alternative treatment option for low-grade [upper tract urothelial cancer].”
The FDA’s approval of mitomycin is based on results from the phase 3 OLYMPUS trial (NCT02793128). This ongoing, single-arm trial enrolled 71 patients with treatment-naive or recurrent low-grade noninvasive upper tract urothelial cancer with at least one measurable papillary tumor located above the ureteropelvic junction. Patients with larger tumors were allowed to have prior tumor debulking.
The patients received mitomycin weekly for 6 weeks at the recommended dose of 4 mg/mL, instilled via ureteral catheter or nephrostomy tube, with the total instillation volume based on volumetric measurements using pyelography, not exceeding 15 mL (60 mg mitomycin).
Patients who achieved a complete response at 3 months could receive monthly instillations up to a maximum of 11 additional instillations.
At 3 months, 41 patients (58%) achieved a complete response (CR). At 12 months after CR determination, 19 patients were still in CR, and 7 patients had documented recurrences. The median duration of CR was not reached.
The most common adverse events (occurring in at least 20% of patients) were ureteric obstruction, flank pain, urinary tract infection, hematuria, renal dysfunction, fatigue, nausea, abdominal pain, dysuria, and vomiting. Ureteric obstruction occurred in 58% of patients, and 88% of those patients required ureteral stent placement.
In all, 23% of patients discontinued mitomycin due to adverse events, and 34% had dose interruptions due to adverse events.
The approval of mitomycin was granted to UroGen Pharma. The FDA granted the application priority review, fast track designation, and breakthrough therapy designation.
The full prescribing information for mitomycin is available for download from the FDA website.
pyelocalyceal
“This is the first approval specifically for patients with low-grade [upper tract urothelial cancer] and provides an option for some patients who may otherwise require a nephroureterectomy,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, said in a statement.
“Due to substantial treatment challenges associated with the complex anatomy of the upper urinary tract, many patients need to be treated with radical surgery – usually complete removal of the affected kidney, ureter, and bladder cuff," Dr. Pazdur added. "Jelmyto gives patients, for the first time, an alternative treatment option for low-grade [upper tract urothelial cancer].”
The FDA’s approval of mitomycin is based on results from the phase 3 OLYMPUS trial (NCT02793128). This ongoing, single-arm trial enrolled 71 patients with treatment-naive or recurrent low-grade noninvasive upper tract urothelial cancer with at least one measurable papillary tumor located above the ureteropelvic junction. Patients with larger tumors were allowed to have prior tumor debulking.
The patients received mitomycin weekly for 6 weeks at the recommended dose of 4 mg/mL, instilled via ureteral catheter or nephrostomy tube, with the total instillation volume based on volumetric measurements using pyelography, not exceeding 15 mL (60 mg mitomycin).
Patients who achieved a complete response at 3 months could receive monthly instillations up to a maximum of 11 additional instillations.
At 3 months, 41 patients (58%) achieved a complete response (CR). At 12 months after CR determination, 19 patients were still in CR, and 7 patients had documented recurrences. The median duration of CR was not reached.
The most common adverse events (occurring in at least 20% of patients) were ureteric obstruction, flank pain, urinary tract infection, hematuria, renal dysfunction, fatigue, nausea, abdominal pain, dysuria, and vomiting. Ureteric obstruction occurred in 58% of patients, and 88% of those patients required ureteral stent placement.
In all, 23% of patients discontinued mitomycin due to adverse events, and 34% had dose interruptions due to adverse events.
The approval of mitomycin was granted to UroGen Pharma. The FDA granted the application priority review, fast track designation, and breakthrough therapy designation.
The full prescribing information for mitomycin is available for download from the FDA website.
pyelocalyceal
“This is the first approval specifically for patients with low-grade [upper tract urothelial cancer] and provides an option for some patients who may otherwise require a nephroureterectomy,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, said in a statement.
“Due to substantial treatment challenges associated with the complex anatomy of the upper urinary tract, many patients need to be treated with radical surgery – usually complete removal of the affected kidney, ureter, and bladder cuff," Dr. Pazdur added. "Jelmyto gives patients, for the first time, an alternative treatment option for low-grade [upper tract urothelial cancer].”
The FDA’s approval of mitomycin is based on results from the phase 3 OLYMPUS trial (NCT02793128). This ongoing, single-arm trial enrolled 71 patients with treatment-naive or recurrent low-grade noninvasive upper tract urothelial cancer with at least one measurable papillary tumor located above the ureteropelvic junction. Patients with larger tumors were allowed to have prior tumor debulking.
The patients received mitomycin weekly for 6 weeks at the recommended dose of 4 mg/mL, instilled via ureteral catheter or nephrostomy tube, with the total instillation volume based on volumetric measurements using pyelography, not exceeding 15 mL (60 mg mitomycin).
Patients who achieved a complete response at 3 months could receive monthly instillations up to a maximum of 11 additional instillations.
At 3 months, 41 patients (58%) achieved a complete response (CR). At 12 months after CR determination, 19 patients were still in CR, and 7 patients had documented recurrences. The median duration of CR was not reached.
The most common adverse events (occurring in at least 20% of patients) were ureteric obstruction, flank pain, urinary tract infection, hematuria, renal dysfunction, fatigue, nausea, abdominal pain, dysuria, and vomiting. Ureteric obstruction occurred in 58% of patients, and 88% of those patients required ureteral stent placement.
In all, 23% of patients discontinued mitomycin due to adverse events, and 34% had dose interruptions due to adverse events.
The approval of mitomycin was granted to UroGen Pharma. The FDA granted the application priority review, fast track designation, and breakthrough therapy designation.
The full prescribing information for mitomycin is available for download from the FDA website.
FROM FDA
FDA approves new drug for relapsed/refractory multiple myeloma
The U.S. Food and Drug Administration today approved isatuximab (Sarclisa, Sanofi) in combination with pomalidomide (Revlimid, Celgene) and dexamethasone for the treatment of adult patients with multiple myeloma who have received two or more prior therapies including lenalidomide and a proteasome inhibitor.

Isatuximab is an anti-CD38 monoclonal antibody administered by intravenous infusion that works by helping the immune system attack multiple myeloma cancer cells.
“While there is no cure for multiple myeloma, Sarclisa is now another CD38-directed treatment option added to the list of FDA-approved treatments of patients with multiple myeloma who have progressive disease after previous therapies,” said Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research.
“In the clinical trial, there was a 40% reduction in the risk of disease progression or death with this therapy,” he added.
The new approval is based on results from ICARIA-MM, an open-label, randomized phase 3 clinical trial of isatuximab among 307 patients in this setting.
In the trial, at a median follow-up of 11.6 months, median progression-free survival was 11.5 months in the isatuximab-pomalidomide-dexamethasone group versus 6.5 months in the pomalidomide-dexamethasone group (hazard ratio, 0.60; P = .001), as reported last year. Overall response rates were 60.4% for the triplet-treated group versus 35.3% for the doublet-treated group.
The most common side effects for isatuximab included neutropenia, infusion-related reactions, pneumonia, upper respiratory tract infection, diarrhea, anemia, lymphopenia, and thrombocytopenia.
Deaths because of treatment-related adverse events were reported for one patient (less than 1%) in the isatuximab-pomalidomide-dexamethasone group (sepsis) and two patients (1%) in the pomalidomide-dexamethasone group (pneumonia and urinary tract infection).
The drug can also cause serious side effects, including IV infusion-related reactions. In the case of a grade 3 or higher reaction, the drug should be permanently discontinued and health care professionals should institute appropriate medical management.
The FDA notes there have been higher incidences of second primary malignancies observed in a controlled clinical trial of patients with multiple myeloma receiving the drug.
The FDA also highlighted that laboratory test interference may be caused by isatuximab and that blood banks should be informed that patients are receiving the drug. Isatuximab may interfere with, for example, antibody screening for patients who need a blood transfusion. Isatuximab may also interfere with the assays used to monitor M-protein, which may impact the determination of complete response.
This article originally appeared on Medscape.com.
The U.S. Food and Drug Administration today approved isatuximab (Sarclisa, Sanofi) in combination with pomalidomide (Revlimid, Celgene) and dexamethasone for the treatment of adult patients with multiple myeloma who have received two or more prior therapies including lenalidomide and a proteasome inhibitor.
Isatuximab is an anti-CD38 monoclonal antibody administered by intravenous infusion that works by helping the immune system attack multiple myeloma cancer cells.
“While there is no cure for multiple myeloma, Sarclisa is now another CD38-directed treatment option added to the list of FDA-approved treatments of patients with multiple myeloma who have progressive disease after previous therapies,” said Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research.
“In the clinical trial, there was a 40% reduction in the risk of disease progression or death with this therapy,” he added.
The new approval is based on results from ICARIA-MM, an open-label, randomized phase 3 clinical trial of isatuximab among 307 patients in this setting.
In the trial, at a median follow-up of 11.6 months, median progression-free survival was 11.5 months in the isatuximab-pomalidomide-dexamethasone group versus 6.5 months in the pomalidomide-dexamethasone group (hazard ratio, 0.60; P = .001), as reported last year. Overall response rates were 60.4% for the triplet-treated group versus 35.3% for the doublet-treated group.
The most common side effects for isatuximab included neutropenia, infusion-related reactions, pneumonia, upper respiratory tract infection, diarrhea, anemia, lymphopenia, and thrombocytopenia.
Deaths because of treatment-related adverse events were reported for one patient (less than 1%) in the isatuximab-pomalidomide-dexamethasone group (sepsis) and two patients (1%) in the pomalidomide-dexamethasone group (pneumonia and urinary tract infection).
The drug can also cause serious side effects, including IV infusion-related reactions. In the case of a grade 3 or higher reaction, the drug should be permanently discontinued and health care professionals should institute appropriate medical management.
The FDA notes there have been higher incidences of second primary malignancies observed in a controlled clinical trial of patients with multiple myeloma receiving the drug.
The FDA also highlighted that laboratory test interference may be caused by isatuximab and that blood banks should be informed that patients are receiving the drug. Isatuximab may interfere with, for example, antibody screening for patients who need a blood transfusion. Isatuximab may also interfere with the assays used to monitor M-protein, which may impact the determination of complete response.
This article originally appeared on Medscape.com.
The U.S. Food and Drug Administration today approved isatuximab (Sarclisa, Sanofi) in combination with pomalidomide (Revlimid, Celgene) and dexamethasone for the treatment of adult patients with multiple myeloma who have received two or more prior therapies including lenalidomide and a proteasome inhibitor.
Isatuximab is an anti-CD38 monoclonal antibody administered by intravenous infusion that works by helping the immune system attack multiple myeloma cancer cells.
“While there is no cure for multiple myeloma, Sarclisa is now another CD38-directed treatment option added to the list of FDA-approved treatments of patients with multiple myeloma who have progressive disease after previous therapies,” said Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research.
“In the clinical trial, there was a 40% reduction in the risk of disease progression or death with this therapy,” he added.
The new approval is based on results from ICARIA-MM, an open-label, randomized phase 3 clinical trial of isatuximab among 307 patients in this setting.
In the trial, at a median follow-up of 11.6 months, median progression-free survival was 11.5 months in the isatuximab-pomalidomide-dexamethasone group versus 6.5 months in the pomalidomide-dexamethasone group (hazard ratio, 0.60; P = .001), as reported last year. Overall response rates were 60.4% for the triplet-treated group versus 35.3% for the doublet-treated group.
The most common side effects for isatuximab included neutropenia, infusion-related reactions, pneumonia, upper respiratory tract infection, diarrhea, anemia, lymphopenia, and thrombocytopenia.
Deaths because of treatment-related adverse events were reported for one patient (less than 1%) in the isatuximab-pomalidomide-dexamethasone group (sepsis) and two patients (1%) in the pomalidomide-dexamethasone group (pneumonia and urinary tract infection).
The drug can also cause serious side effects, including IV infusion-related reactions. In the case of a grade 3 or higher reaction, the drug should be permanently discontinued and health care professionals should institute appropriate medical management.
The FDA notes there have been higher incidences of second primary malignancies observed in a controlled clinical trial of patients with multiple myeloma receiving the drug.
The FDA also highlighted that laboratory test interference may be caused by isatuximab and that blood banks should be informed that patients are receiving the drug. Isatuximab may interfere with, for example, antibody screening for patients who need a blood transfusion. Isatuximab may also interfere with the assays used to monitor M-protein, which may impact the determination of complete response.
This article originally appeared on Medscape.com.
FDA approves neratinib in combination for metastatic HER2-positive breast cancer
The Food and Drug Administration has approved neratinib (NERLYNX) in combination with capecitabine for use in adults with advanced or metastatic HER2-positive breast cancer who have received at least two prior anti-HER2 based regimens in the metastatic setting.
The recommended dose for neratinib in this population is 240 mg once daily with food on days 1-21 of a 21-day cycle. Neratinib should be given with capecitabine at 750 mg/m2 twice daily on days 1-14 until progression or unacceptable toxicity.
The full prescribing information for neratinib is available from the FDA website.
The FDA’s new approval of neratinib is based on results from the NALA trial (NCT01808573). The trial enrolled 621 patients with metastatic HER2-positive breast cancer who had received at least two prior anti-HER2 based regimens in the metastatic setting.
The patients were randomized to neratinib plus capecitabine or lapatinib plus capecitabine and received treatment until progression or unacceptable toxicity.
The objective response rate was 32.8% in the neratinib arm and 26.7% in the lapatinib arm. The median duration of response was 8.5 months and 5.6 months, respectively.
The median progression-free survival was 5.6 months in the neratinib arm and 5.5 months in the lapatinib arm (hazard ratio 0.76; P = .0059). The median overall survival was 21 months and 18.7 months, respectively (HR 0.88; P = .2086).
The most common grade 3/4 adverse events in the neratinib arm were diarrhea, nausea, vomiting, fatigue, and decreased appetite.
The Food and Drug Administration has approved neratinib (NERLYNX) in combination with capecitabine for use in adults with advanced or metastatic HER2-positive breast cancer who have received at least two prior anti-HER2 based regimens in the metastatic setting.
The recommended dose for neratinib in this population is 240 mg once daily with food on days 1-21 of a 21-day cycle. Neratinib should be given with capecitabine at 750 mg/m2 twice daily on days 1-14 until progression or unacceptable toxicity.
The full prescribing information for neratinib is available from the FDA website.
The FDA’s new approval of neratinib is based on results from the NALA trial (NCT01808573). The trial enrolled 621 patients with metastatic HER2-positive breast cancer who had received at least two prior anti-HER2 based regimens in the metastatic setting.
The patients were randomized to neratinib plus capecitabine or lapatinib plus capecitabine and received treatment until progression or unacceptable toxicity.
The objective response rate was 32.8% in the neratinib arm and 26.7% in the lapatinib arm. The median duration of response was 8.5 months and 5.6 months, respectively.
The median progression-free survival was 5.6 months in the neratinib arm and 5.5 months in the lapatinib arm (hazard ratio 0.76; P = .0059). The median overall survival was 21 months and 18.7 months, respectively (HR 0.88; P = .2086).
The most common grade 3/4 adverse events in the neratinib arm were diarrhea, nausea, vomiting, fatigue, and decreased appetite.
The Food and Drug Administration has approved neratinib (NERLYNX) in combination with capecitabine for use in adults with advanced or metastatic HER2-positive breast cancer who have received at least two prior anti-HER2 based regimens in the metastatic setting.
The recommended dose for neratinib in this population is 240 mg once daily with food on days 1-21 of a 21-day cycle. Neratinib should be given with capecitabine at 750 mg/m2 twice daily on days 1-14 until progression or unacceptable toxicity.
The full prescribing information for neratinib is available from the FDA website.
The FDA’s new approval of neratinib is based on results from the NALA trial (NCT01808573). The trial enrolled 621 patients with metastatic HER2-positive breast cancer who had received at least two prior anti-HER2 based regimens in the metastatic setting.
The patients were randomized to neratinib plus capecitabine or lapatinib plus capecitabine and received treatment until progression or unacceptable toxicity.
The objective response rate was 32.8% in the neratinib arm and 26.7% in the lapatinib arm. The median duration of response was 8.5 months and 5.6 months, respectively.
The median progression-free survival was 5.6 months in the neratinib arm and 5.5 months in the lapatinib arm (hazard ratio 0.76; P = .0059). The median overall survival was 21 months and 18.7 months, respectively (HR 0.88; P = .2086).
The most common grade 3/4 adverse events in the neratinib arm were diarrhea, nausea, vomiting, fatigue, and decreased appetite.
Pharmacogenomics testing: What the FDA says

Mr. R, age 30, is referred to you by his primary care physician, who diagnosed him with depression approximately 2 years ago. When he was first diagnosed, Mr. R was prescribed
Mr. R says that based on his primary care physician’s recommendation, he had undergone pharmacogenomics testing to help guide therapy. He presents the results to you, and you notice that he has the cytochrome P450 (CYP) 2C19 *2/*3 genotype and a CYP2D6*4/*5 genotype. Both are associated with a poor metabolism phenotype. Should you use these findings to determine which medication Mr. R should be treated with next?
While the field of pharmacogenomics is not new, within the last few years this science has begun to transition into clinical practice. A recent meta-analysis found support for using pharmacogenomics testing results in clinical practice.1 This study included more than 1,700 patients who took part in 5 controlled trials that randomized participants to either pharmacogenetics-guided or unguided (ie, standard) treatment. Each participant was assessed using the Hamilton Depression Rating Scale-17 (HDRS-17) a minimum of 3 times over a minimum of 8 weeks.1 While the exact inclusion and exclusion criteria for each trial differed, they all defined remission of depression as achieving an HDRS-17 score ≤7. Overall, the authors concluded that based on the random-effects pooled risk ratio, there was a significant association between pharmacogenetics-guided prescribing and remission (relative risk = 1.71, 95% confidence interval [CI], 1.17 to 2.48; P = .005). The results of this meta-analysis are controversial, however, because all 5 studies were industry-funded, and interpretation of the testing results was based on proprietary algorithms.
Experts in the field and professional societies, such as the International Society of Psychiatric Genetics (ISPG), have issued policy statements on genetic testing within psychiatry.2,3 While the ISPG did not necessarily endorse use of pharmacogenomics in practice, they recommended that clinicians follow good medical practice and stay current on changes to drug labeling and adverse event reports.3 The ISPG also noted that useful but not exhaustive lists of pharmacogenetic tests are maintained by the Clinical Pharmacogenetics Implementation Consortium (CPIC) and the US FDA.3
Laboratory developed vs direct-to-consumer tests
In a previous Savvy Psychopharmacology article,4 we had discussed the role of CPIC, but not the role of the FDA. This issue is key because there is a lack of clarity regarding pharmacogenomics tests and whether they are considered Class II devices by the FDA, which would require their review and approval. Until recently, the FDA was fairly quiet regarding pharmacogenomics tests because most of these tests were considered laboratory developed tests, which were regulated under the Clinic Laboratory Improvements Amendments program. The critical distinction of a laboratory developed test is that it is developed and performed in a single laboratory and is offered to patients only when prescribed by a clinician. Due to this distinction, laboratory developed pharmacogenomics tests did not need FDA 510(k) clearance, which is a premarket submission common for medical devices.
Direct-to-consumer pharmacogenomics tests are different in that the FDA has classified these platforms as medical devices; however, they are reviewed by the FDA only if they are being used for moderate- to high-risk medical purposes, or if the results of the testing may have a higher impact on medical care. As part of its review, the FDA examines test accuracy and reliably measures to determine if the measurement is predictive of a certain state of health and supported by what the company claims about the test and how well it works. Additionally, the FDA examines the company-provided descriptive information to ensure that consumers can easily understand it without the help of a clinician.5
Conflicting FDA statements
Recently the FDA issued 2 statements—one a policy statement and the other a safety communication—about laboratory developed tests and direct-to-consumer tests. The statements appear to contradict themselves, despite focusing on using pharmacogenomics testing in practice.
Continue to: The FDA's first statement
The FDA’s first statement. On October 31, 2018, the FDA released a policy statement that they had “permitted marketing, with special controls,” of the Personal Genome Service Pharmacogenetic Reports test through 23andMe (a direct-to-consumer genetic testing company) for 33 different variants within specific pharmacogenomic genes (CYP2C19, CYP2C9, CYP3A5, UGT1A1, DPYD, TPMT, SLC01B1, and CYP2D6) that may impact drug metabolism or response.6 As part of its review of this Personal Genome Service Pharmacogenetic Reports test, the FDA found that the company-provided data showed that the test is accurate and can correctly identify the 33 specific genetic variants. The FDA review also showed that the testing results were reproducible, and the test instructions and reports could be understood by consumers.
While the specific reports related to this testing are not yet available within 23andMe, this approval allows for greater oversight by the FDA with regard to the pharmacogenomics information provided through this company’s Personal Genome Service Pharmacogenetic Reports test. The FDA noted that this approval was only for adults age >185 and that consumers “should not use the test results to stop or change any medication.”6 Further, the FDA stated that the results of the direct-to-consumer test should be confirmed with independent pharmacogenomics testing before making any medical decision. Unfortunately, the FDA did not offer guidance on what would be an appropriate independent pharmacogenomics test, but it did provide a link to a list of FDA-approved nucleic acid–based tests, on which 23andMe’s Personal Genome Service Pharmacogenetic Reports test is included.7
The FDA’s second statement. On November 1, 2018, the FDA issued a separate safety communication that cautioned clinicians and patients that most of the current commercially available testing platforms for pharmacogenomics have not been FDA-reviewed, meaning that they may lack clinical evidence supporting their use.8 Further, the FDA safety communication stated, “Changing drug treatment based on the results from such a genetic test could lead to inappropriate treatment decisions and potentially serious health consequences for the patient.”8
Taken together, these FDA statements appear to support pharmacogenomics testing with approval of the 23andMe’s Personal Genome Service Pharmacogenetic Reports test but warn that the testing results should not be used to make treatment decisions, and that they should be verified. However, the FDA does not offer any guidance on what an appropriate testing platform would be
What the FDA advises
The FDA has provided some guidance to clinicians and patients regarding next steps for patients who are interested in having pharmacogenomics testing or who have already undergone testing. The FDA’s first point is that both clinicians and patients need to be aware that pharmacogenomics testing is not FDA-reviewed, that patients should discuss the results of their testing with their clinicians, and that they should not stop their medication based on the results of the testing. Additionally, the FDA recommends that clinicians and patients should be aware that any claims made by the testing companies regarding the specific effect of a medication may not be supported by evidence. Furthermore, the FDA strongly recommends that clinicians consult the FDA-approved drug label, or the label of the FDA-cleared or FDA-approved genetic test, for information regarding how genetic information should be used in making treatment decisions. The FDA recommends reviewing the Warning section, as well as the Indications and Usage, Dosage and Administration, or Use in Specific Populations sections of the FDA-approved drug labeling.
Continue to: Unfortunately, this information...
Unfortunately, this information might be difficult to locate due to the lack of consistency regarding where it is placed in the FDA-approved drug labeling. The Pharmacogenomics Knowledgebase (https://www.pharmgkb.org/) can help clinicians quickly identify information regarding medications, their metabolic pathways, CPIC dosing guidelines, and the FDA-approved drug labeling information.9 By searching for specific medications within the Pharmacogenomic Knowledge Base, information regarding the FDA-approved drug labeling can be easily found, which is important because currently >120 medications contain pharmacogenomics information in their FDA-approved drug labeling.10
CASE CONTINUED
Overall Mr. R’s pharmacogenomics testing results indicate that he has 2 genotypes that are associated with poor metabolism phenotypes and could result in reduced metabolism of medications that are metabolized by these CYP enzymes, leading to higher blood levels and an increased risk of adverse effects. The Table11 lists pharmacogenomics information from the FDA-approved drug labeling and from the Pharmacogenomics Knowledgebase for both the medications Mr. R has previously been prescribed and for several potential medications to consider.

It would be prudent to first discuss with Mr. R the FDA’s recent policy statement and safety communication. While you could recommend that he pursue additional pharmacogenomics testing, it is unclear which specific laboratory is available to conduct this confirmatory analysis.
Because Mr. R has had unsuccessful trials of several medications that primarily fall in the selective serotonin reuptake inhibitors class, it might be time to consider a medication from a different class. A quick review of the FDA-approved drug labeling for
Related Resources
- Gammal RS, Gardner KN, Burghardt KJ. Where to find guidance on using pharmacogenomics in psychiatric practice. Current Psychiatry. 2016;15(9):93-94.
- Clinical Pharmacogenomics Implementation Consortium. What is CPIC? https://www.pharmgkb.org/page/cpic.
Drug Brand Names
Bupropion • Wellbutrin, Zyban
Citalopram • Celexa
Paroxetine • Paxil
Sertraline • Zoloft
Venlafaxine • Effexor
1. Bousman CA, Arandjelovic K, Mancuso SG, et al. Pharmacogenetic tests and depressive symptom remission: a meta-analysis of randomized controlled trials. Pharmacogenomics. 2019;20(1):37-47.
2. Zubenko GS, Sommer BR, Cohen BM. Pharmacogenetics in psychiatry: a companion, rather than competitor, to protocol-based care-reply. JAMA Psychiatry. 2018;75(10):1090-1091.
3. International Society for Psychiatric Genetics. Genetic testing statement: genetic testing and psychiatric disorders: a statement from the International Society of Psychiatric Genetics. https://ispg.net/genetic-testing-statement/. Revised January 26, 2017. Accessed January 1, 2019.
4. Ellingrod VL, Ward KM. Using pharmacogenetics guidelines when prescribing: what’s available. Current Psychiatry. 2018;17(1):43-46.
5. U.S. Food and Drug Administration. Medical devices: direct-to-consumer tests. https://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/InVitroDiagnostics/ucm624726.htm. Published November 1, 2018. Accessed January 1, 2019.
6. U.S. Food and Drug Administration. FDA news releases: FDA authorizes first direct-to consumer test for detecting variants that may be associated with medication metabolism. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm624753.htm. Published October 31, 2018. Accessed January 1, 2019.
7. U.S. Food and Drug Administration. Medical devices: nucleic acid based tests. https://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/InVitroDiagnostics/ucm330711.htm. Published February 5, 2019. Accessed March 1, 2019.
8. U.S. Food and Drug Administration. Medical devices. The FDA warns against the use of many genetic tests with unapproved claims to predict patient response to specific medications: FDA Safety Communications. https://www.fda.gov/MedicalDevices/Safety/AlertsandNotices/ucm624725.htm. Published November 1, 2018. Accessed January 1, 2019.
9. Whirl-Carrillo EM, McDonagh JM, Hebert L, et al. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther. 2012;92(4):414-417.
10. U.S. Food and Drug Administration. Drugs. Table of pharmacogenomic biomarkers in drug labeling. https://www.fda.gov/Drugs/ScienceResearch/ucm572698.htm. Published August 3, 2018. Accessed January 1, 2019.
11. U.S. Food and Drug Administration. Drugs@FDA: FDA approved drug products. https://www.accessdata.fda.gov/scripts/cder/daf. Accessed March 4, 2019.
Mr. R, age 30, is referred to you by his primary care physician, who diagnosed him with depression approximately 2 years ago. When he was first diagnosed, Mr. R was prescribed
Mr. R says that based on his primary care physician’s recommendation, he had undergone pharmacogenomics testing to help guide therapy. He presents the results to you, and you notice that he has the cytochrome P450 (CYP) 2C19 *2/*3 genotype and a CYP2D6*4/*5 genotype. Both are associated with a poor metabolism phenotype. Should you use these findings to determine which medication Mr. R should be treated with next?
While the field of pharmacogenomics is not new, within the last few years this science has begun to transition into clinical practice. A recent meta-analysis found support for using pharmacogenomics testing results in clinical practice.1 This study included more than 1,700 patients who took part in 5 controlled trials that randomized participants to either pharmacogenetics-guided or unguided (ie, standard) treatment. Each participant was assessed using the Hamilton Depression Rating Scale-17 (HDRS-17) a minimum of 3 times over a minimum of 8 weeks.1 While the exact inclusion and exclusion criteria for each trial differed, they all defined remission of depression as achieving an HDRS-17 score ≤7. Overall, the authors concluded that based on the random-effects pooled risk ratio, there was a significant association between pharmacogenetics-guided prescribing and remission (relative risk = 1.71, 95% confidence interval [CI], 1.17 to 2.48; P = .005). The results of this meta-analysis are controversial, however, because all 5 studies were industry-funded, and interpretation of the testing results was based on proprietary algorithms.
Experts in the field and professional societies, such as the International Society of Psychiatric Genetics (ISPG), have issued policy statements on genetic testing within psychiatry.2,3 While the ISPG did not necessarily endorse use of pharmacogenomics in practice, they recommended that clinicians follow good medical practice and stay current on changes to drug labeling and adverse event reports.3 The ISPG also noted that useful but not exhaustive lists of pharmacogenetic tests are maintained by the Clinical Pharmacogenetics Implementation Consortium (CPIC) and the US FDA.3
Laboratory developed vs direct-to-consumer tests
In a previous Savvy Psychopharmacology article,4 we had discussed the role of CPIC, but not the role of the FDA. This issue is key because there is a lack of clarity regarding pharmacogenomics tests and whether they are considered Class II devices by the FDA, which would require their review and approval. Until recently, the FDA was fairly quiet regarding pharmacogenomics tests because most of these tests were considered laboratory developed tests, which were regulated under the Clinic Laboratory Improvements Amendments program. The critical distinction of a laboratory developed test is that it is developed and performed in a single laboratory and is offered to patients only when prescribed by a clinician. Due to this distinction, laboratory developed pharmacogenomics tests did not need FDA 510(k) clearance, which is a premarket submission common for medical devices.
Direct-to-consumer pharmacogenomics tests are different in that the FDA has classified these platforms as medical devices; however, they are reviewed by the FDA only if they are being used for moderate- to high-risk medical purposes, or if the results of the testing may have a higher impact on medical care. As part of its review, the FDA examines test accuracy and reliably measures to determine if the measurement is predictive of a certain state of health and supported by what the company claims about the test and how well it works. Additionally, the FDA examines the company-provided descriptive information to ensure that consumers can easily understand it without the help of a clinician.5
Conflicting FDA statements
Recently the FDA issued 2 statements—one a policy statement and the other a safety communication—about laboratory developed tests and direct-to-consumer tests. The statements appear to contradict themselves, despite focusing on using pharmacogenomics testing in practice.
Continue to: The FDA's first statement
The FDA’s first statement. On October 31, 2018, the FDA released a policy statement that they had “permitted marketing, with special controls,” of the Personal Genome Service Pharmacogenetic Reports test through 23andMe (a direct-to-consumer genetic testing company) for 33 different variants within specific pharmacogenomic genes (CYP2C19, CYP2C9, CYP3A5, UGT1A1, DPYD, TPMT, SLC01B1, and CYP2D6) that may impact drug metabolism or response.6 As part of its review of this Personal Genome Service Pharmacogenetic Reports test, the FDA found that the company-provided data showed that the test is accurate and can correctly identify the 33 specific genetic variants. The FDA review also showed that the testing results were reproducible, and the test instructions and reports could be understood by consumers.
While the specific reports related to this testing are not yet available within 23andMe, this approval allows for greater oversight by the FDA with regard to the pharmacogenomics information provided through this company’s Personal Genome Service Pharmacogenetic Reports test. The FDA noted that this approval was only for adults age >185 and that consumers “should not use the test results to stop or change any medication.”6 Further, the FDA stated that the results of the direct-to-consumer test should be confirmed with independent pharmacogenomics testing before making any medical decision. Unfortunately, the FDA did not offer guidance on what would be an appropriate independent pharmacogenomics test, but it did provide a link to a list of FDA-approved nucleic acid–based tests, on which 23andMe’s Personal Genome Service Pharmacogenetic Reports test is included.7
The FDA’s second statement. On November 1, 2018, the FDA issued a separate safety communication that cautioned clinicians and patients that most of the current commercially available testing platforms for pharmacogenomics have not been FDA-reviewed, meaning that they may lack clinical evidence supporting their use.8 Further, the FDA safety communication stated, “Changing drug treatment based on the results from such a genetic test could lead to inappropriate treatment decisions and potentially serious health consequences for the patient.”8
Taken together, these FDA statements appear to support pharmacogenomics testing with approval of the 23andMe’s Personal Genome Service Pharmacogenetic Reports test but warn that the testing results should not be used to make treatment decisions, and that they should be verified. However, the FDA does not offer any guidance on what an appropriate testing platform would be
What the FDA advises
The FDA has provided some guidance to clinicians and patients regarding next steps for patients who are interested in having pharmacogenomics testing or who have already undergone testing. The FDA’s first point is that both clinicians and patients need to be aware that pharmacogenomics testing is not FDA-reviewed, that patients should discuss the results of their testing with their clinicians, and that they should not stop their medication based on the results of the testing. Additionally, the FDA recommends that clinicians and patients should be aware that any claims made by the testing companies regarding the specific effect of a medication may not be supported by evidence. Furthermore, the FDA strongly recommends that clinicians consult the FDA-approved drug label, or the label of the FDA-cleared or FDA-approved genetic test, for information regarding how genetic information should be used in making treatment decisions. The FDA recommends reviewing the Warning section, as well as the Indications and Usage, Dosage and Administration, or Use in Specific Populations sections of the FDA-approved drug labeling.
Continue to: Unfortunately, this information...
Unfortunately, this information might be difficult to locate due to the lack of consistency regarding where it is placed in the FDA-approved drug labeling. The Pharmacogenomics Knowledgebase (https://www.pharmgkb.org/) can help clinicians quickly identify information regarding medications, their metabolic pathways, CPIC dosing guidelines, and the FDA-approved drug labeling information.9 By searching for specific medications within the Pharmacogenomic Knowledge Base, information regarding the FDA-approved drug labeling can be easily found, which is important because currently >120 medications contain pharmacogenomics information in their FDA-approved drug labeling.10
CASE CONTINUED
Overall Mr. R’s pharmacogenomics testing results indicate that he has 2 genotypes that are associated with poor metabolism phenotypes and could result in reduced metabolism of medications that are metabolized by these CYP enzymes, leading to higher blood levels and an increased risk of adverse effects. The Table11 lists pharmacogenomics information from the FDA-approved drug labeling and from the Pharmacogenomics Knowledgebase for both the medications Mr. R has previously been prescribed and for several potential medications to consider.
It would be prudent to first discuss with Mr. R the FDA’s recent policy statement and safety communication. While you could recommend that he pursue additional pharmacogenomics testing, it is unclear which specific laboratory is available to conduct this confirmatory analysis.
Because Mr. R has had unsuccessful trials of several medications that primarily fall in the selective serotonin reuptake inhibitors class, it might be time to consider a medication from a different class. A quick review of the FDA-approved drug labeling for
Related Resources
- Gammal RS, Gardner KN, Burghardt KJ. Where to find guidance on using pharmacogenomics in psychiatric practice. Current Psychiatry. 2016;15(9):93-94.
- Clinical Pharmacogenomics Implementation Consortium. What is CPIC? https://www.pharmgkb.org/page/cpic.
Drug Brand Names
Bupropion • Wellbutrin, Zyban
Citalopram • Celexa
Paroxetine • Paxil
Sertraline • Zoloft
Venlafaxine • Effexor
Mr. R, age 30, is referred to you by his primary care physician, who diagnosed him with depression approximately 2 years ago. When he was first diagnosed, Mr. R was prescribed
Mr. R says that based on his primary care physician’s recommendation, he had undergone pharmacogenomics testing to help guide therapy. He presents the results to you, and you notice that he has the cytochrome P450 (CYP) 2C19 *2/*3 genotype and a CYP2D6*4/*5 genotype. Both are associated with a poor metabolism phenotype. Should you use these findings to determine which medication Mr. R should be treated with next?
While the field of pharmacogenomics is not new, within the last few years this science has begun to transition into clinical practice. A recent meta-analysis found support for using pharmacogenomics testing results in clinical practice.1 This study included more than 1,700 patients who took part in 5 controlled trials that randomized participants to either pharmacogenetics-guided or unguided (ie, standard) treatment. Each participant was assessed using the Hamilton Depression Rating Scale-17 (HDRS-17) a minimum of 3 times over a minimum of 8 weeks.1 While the exact inclusion and exclusion criteria for each trial differed, they all defined remission of depression as achieving an HDRS-17 score ≤7. Overall, the authors concluded that based on the random-effects pooled risk ratio, there was a significant association between pharmacogenetics-guided prescribing and remission (relative risk = 1.71, 95% confidence interval [CI], 1.17 to 2.48; P = .005). The results of this meta-analysis are controversial, however, because all 5 studies were industry-funded, and interpretation of the testing results was based on proprietary algorithms.
Experts in the field and professional societies, such as the International Society of Psychiatric Genetics (ISPG), have issued policy statements on genetic testing within psychiatry.2,3 While the ISPG did not necessarily endorse use of pharmacogenomics in practice, they recommended that clinicians follow good medical practice and stay current on changes to drug labeling and adverse event reports.3 The ISPG also noted that useful but not exhaustive lists of pharmacogenetic tests are maintained by the Clinical Pharmacogenetics Implementation Consortium (CPIC) and the US FDA.3
Laboratory developed vs direct-to-consumer tests
In a previous Savvy Psychopharmacology article,4 we had discussed the role of CPIC, but not the role of the FDA. This issue is key because there is a lack of clarity regarding pharmacogenomics tests and whether they are considered Class II devices by the FDA, which would require their review and approval. Until recently, the FDA was fairly quiet regarding pharmacogenomics tests because most of these tests were considered laboratory developed tests, which were regulated under the Clinic Laboratory Improvements Amendments program. The critical distinction of a laboratory developed test is that it is developed and performed in a single laboratory and is offered to patients only when prescribed by a clinician. Due to this distinction, laboratory developed pharmacogenomics tests did not need FDA 510(k) clearance, which is a premarket submission common for medical devices.
Direct-to-consumer pharmacogenomics tests are different in that the FDA has classified these platforms as medical devices; however, they are reviewed by the FDA only if they are being used for moderate- to high-risk medical purposes, or if the results of the testing may have a higher impact on medical care. As part of its review, the FDA examines test accuracy and reliably measures to determine if the measurement is predictive of a certain state of health and supported by what the company claims about the test and how well it works. Additionally, the FDA examines the company-provided descriptive information to ensure that consumers can easily understand it without the help of a clinician.5
Conflicting FDA statements
Recently the FDA issued 2 statements—one a policy statement and the other a safety communication—about laboratory developed tests and direct-to-consumer tests. The statements appear to contradict themselves, despite focusing on using pharmacogenomics testing in practice.
Continue to: The FDA's first statement
The FDA’s first statement. On October 31, 2018, the FDA released a policy statement that they had “permitted marketing, with special controls,” of the Personal Genome Service Pharmacogenetic Reports test through 23andMe (a direct-to-consumer genetic testing company) for 33 different variants within specific pharmacogenomic genes (CYP2C19, CYP2C9, CYP3A5, UGT1A1, DPYD, TPMT, SLC01B1, and CYP2D6) that may impact drug metabolism or response.6 As part of its review of this Personal Genome Service Pharmacogenetic Reports test, the FDA found that the company-provided data showed that the test is accurate and can correctly identify the 33 specific genetic variants. The FDA review also showed that the testing results were reproducible, and the test instructions and reports could be understood by consumers.
While the specific reports related to this testing are not yet available within 23andMe, this approval allows for greater oversight by the FDA with regard to the pharmacogenomics information provided through this company’s Personal Genome Service Pharmacogenetic Reports test. The FDA noted that this approval was only for adults age >185 and that consumers “should not use the test results to stop or change any medication.”6 Further, the FDA stated that the results of the direct-to-consumer test should be confirmed with independent pharmacogenomics testing before making any medical decision. Unfortunately, the FDA did not offer guidance on what would be an appropriate independent pharmacogenomics test, but it did provide a link to a list of FDA-approved nucleic acid–based tests, on which 23andMe’s Personal Genome Service Pharmacogenetic Reports test is included.7
The FDA’s second statement. On November 1, 2018, the FDA issued a separate safety communication that cautioned clinicians and patients that most of the current commercially available testing platforms for pharmacogenomics have not been FDA-reviewed, meaning that they may lack clinical evidence supporting their use.8 Further, the FDA safety communication stated, “Changing drug treatment based on the results from such a genetic test could lead to inappropriate treatment decisions and potentially serious health consequences for the patient.”8
Taken together, these FDA statements appear to support pharmacogenomics testing with approval of the 23andMe’s Personal Genome Service Pharmacogenetic Reports test but warn that the testing results should not be used to make treatment decisions, and that they should be verified. However, the FDA does not offer any guidance on what an appropriate testing platform would be
What the FDA advises
The FDA has provided some guidance to clinicians and patients regarding next steps for patients who are interested in having pharmacogenomics testing or who have already undergone testing. The FDA’s first point is that both clinicians and patients need to be aware that pharmacogenomics testing is not FDA-reviewed, that patients should discuss the results of their testing with their clinicians, and that they should not stop their medication based on the results of the testing. Additionally, the FDA recommends that clinicians and patients should be aware that any claims made by the testing companies regarding the specific effect of a medication may not be supported by evidence. Furthermore, the FDA strongly recommends that clinicians consult the FDA-approved drug label, or the label of the FDA-cleared or FDA-approved genetic test, for information regarding how genetic information should be used in making treatment decisions. The FDA recommends reviewing the Warning section, as well as the Indications and Usage, Dosage and Administration, or Use in Specific Populations sections of the FDA-approved drug labeling.
Continue to: Unfortunately, this information...
Unfortunately, this information might be difficult to locate due to the lack of consistency regarding where it is placed in the FDA-approved drug labeling. The Pharmacogenomics Knowledgebase (https://www.pharmgkb.org/) can help clinicians quickly identify information regarding medications, their metabolic pathways, CPIC dosing guidelines, and the FDA-approved drug labeling information.9 By searching for specific medications within the Pharmacogenomic Knowledge Base, information regarding the FDA-approved drug labeling can be easily found, which is important because currently >120 medications contain pharmacogenomics information in their FDA-approved drug labeling.10
CASE CONTINUED
Overall Mr. R’s pharmacogenomics testing results indicate that he has 2 genotypes that are associated with poor metabolism phenotypes and could result in reduced metabolism of medications that are metabolized by these CYP enzymes, leading to higher blood levels and an increased risk of adverse effects. The Table11 lists pharmacogenomics information from the FDA-approved drug labeling and from the Pharmacogenomics Knowledgebase for both the medications Mr. R has previously been prescribed and for several potential medications to consider.
It would be prudent to first discuss with Mr. R the FDA’s recent policy statement and safety communication. While you could recommend that he pursue additional pharmacogenomics testing, it is unclear which specific laboratory is available to conduct this confirmatory analysis.
Because Mr. R has had unsuccessful trials of several medications that primarily fall in the selective serotonin reuptake inhibitors class, it might be time to consider a medication from a different class. A quick review of the FDA-approved drug labeling for
Related Resources
- Gammal RS, Gardner KN, Burghardt KJ. Where to find guidance on using pharmacogenomics in psychiatric practice. Current Psychiatry. 2016;15(9):93-94.
- Clinical Pharmacogenomics Implementation Consortium. What is CPIC? https://www.pharmgkb.org/page/cpic.
Drug Brand Names
Bupropion • Wellbutrin, Zyban
Citalopram • Celexa
Paroxetine • Paxil
Sertraline • Zoloft
Venlafaxine • Effexor
1. Bousman CA, Arandjelovic K, Mancuso SG, et al. Pharmacogenetic tests and depressive symptom remission: a meta-analysis of randomized controlled trials. Pharmacogenomics. 2019;20(1):37-47.
2. Zubenko GS, Sommer BR, Cohen BM. Pharmacogenetics in psychiatry: a companion, rather than competitor, to protocol-based care-reply. JAMA Psychiatry. 2018;75(10):1090-1091.
3. International Society for Psychiatric Genetics. Genetic testing statement: genetic testing and psychiatric disorders: a statement from the International Society of Psychiatric Genetics. https://ispg.net/genetic-testing-statement/. Revised January 26, 2017. Accessed January 1, 2019.
4. Ellingrod VL, Ward KM. Using pharmacogenetics guidelines when prescribing: what’s available. Current Psychiatry. 2018;17(1):43-46.
5. U.S. Food and Drug Administration. Medical devices: direct-to-consumer tests. https://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/InVitroDiagnostics/ucm624726.htm. Published November 1, 2018. Accessed January 1, 2019.
6. U.S. Food and Drug Administration. FDA news releases: FDA authorizes first direct-to consumer test for detecting variants that may be associated with medication metabolism. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm624753.htm. Published October 31, 2018. Accessed January 1, 2019.
7. U.S. Food and Drug Administration. Medical devices: nucleic acid based tests. https://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/InVitroDiagnostics/ucm330711.htm. Published February 5, 2019. Accessed March 1, 2019.
8. U.S. Food and Drug Administration. Medical devices. The FDA warns against the use of many genetic tests with unapproved claims to predict patient response to specific medications: FDA Safety Communications. https://www.fda.gov/MedicalDevices/Safety/AlertsandNotices/ucm624725.htm. Published November 1, 2018. Accessed January 1, 2019.
9. Whirl-Carrillo EM, McDonagh JM, Hebert L, et al. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther. 2012;92(4):414-417.
10. U.S. Food and Drug Administration. Drugs. Table of pharmacogenomic biomarkers in drug labeling. https://www.fda.gov/Drugs/ScienceResearch/ucm572698.htm. Published August 3, 2018. Accessed January 1, 2019.
11. U.S. Food and Drug Administration. Drugs@FDA: FDA approved drug products. https://www.accessdata.fda.gov/scripts/cder/daf. Accessed March 4, 2019.
1. Bousman CA, Arandjelovic K, Mancuso SG, et al. Pharmacogenetic tests and depressive symptom remission: a meta-analysis of randomized controlled trials. Pharmacogenomics. 2019;20(1):37-47.
2. Zubenko GS, Sommer BR, Cohen BM. Pharmacogenetics in psychiatry: a companion, rather than competitor, to protocol-based care-reply. JAMA Psychiatry. 2018;75(10):1090-1091.
3. International Society for Psychiatric Genetics. Genetic testing statement: genetic testing and psychiatric disorders: a statement from the International Society of Psychiatric Genetics. https://ispg.net/genetic-testing-statement/. Revised January 26, 2017. Accessed January 1, 2019.
4. Ellingrod VL, Ward KM. Using pharmacogenetics guidelines when prescribing: what’s available. Current Psychiatry. 2018;17(1):43-46.
5. U.S. Food and Drug Administration. Medical devices: direct-to-consumer tests. https://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/InVitroDiagnostics/ucm624726.htm. Published November 1, 2018. Accessed January 1, 2019.
6. U.S. Food and Drug Administration. FDA news releases: FDA authorizes first direct-to consumer test for detecting variants that may be associated with medication metabolism. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm624753.htm. Published October 31, 2018. Accessed January 1, 2019.
7. U.S. Food and Drug Administration. Medical devices: nucleic acid based tests. https://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/InVitroDiagnostics/ucm330711.htm. Published February 5, 2019. Accessed March 1, 2019.
8. U.S. Food and Drug Administration. Medical devices. The FDA warns against the use of many genetic tests with unapproved claims to predict patient response to specific medications: FDA Safety Communications. https://www.fda.gov/MedicalDevices/Safety/AlertsandNotices/ucm624725.htm. Published November 1, 2018. Accessed January 1, 2019.
9. Whirl-Carrillo EM, McDonagh JM, Hebert L, et al. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther. 2012;92(4):414-417.
10. U.S. Food and Drug Administration. Drugs. Table of pharmacogenomic biomarkers in drug labeling. https://www.fda.gov/Drugs/ScienceResearch/ucm572698.htm. Published August 3, 2018. Accessed January 1, 2019.
11. U.S. Food and Drug Administration. Drugs@FDA: FDA approved drug products. https://www.accessdata.fda.gov/scripts/cder/daf. Accessed March 4, 2019.

FDA Boxed Warnings Updates: October 2018
The FDA’s MedWatch program safety labeling changes for boxed warnings are compiled quarterly for drugs and therapeutic biologics where important changes have been made to the safety information. These and other label changes are searchable in the Drug Safety Labeling Changes (SLC) database, where data are available to the public in downloadable and searchable formats. Boxed warnings are ordinarily used to highlight either adverse reactions so serious in proportion to the potential benefi t from the drug that it is essential that it be considered in assessing the risks and benefi ts of using the drug; or serious adverse reactions that can be prevented/reduced in frequency or severity by appropriate use of the drug; or FDA approved the drug with restrictions to ensure safe use because FDA concluded that the drug can be safely used only if distribution or use is restricted. For complete FDA Drug Safety Labeling changes, please visit http://www.accessdata.fda.gov/scripts/cder/safetylabelingchanges.
DESOGEN (DESOGESTREL AND ETHINYL ESTRADIOL TABLETS
- Edited boxed warning, June 2018
WARNING: CIGARETTE SMOKING AND SERIOUS CARDIOVASCULAR EVENTS
Cigarette smoking increases the risk of serious cardiovascular events from combination oral contraceptive (COC) use. This risk increases with age, particularly in women over 35 years of age, and with the number of cigarettes smoked. For this reason, COCs are contraindicated in women who are over 35 years of age, and smoke.
TRIZIVIR (ABACAVIR, LAMIVUDINE, AND ZIDOVUDINE TABLETS)
- Edited boxed warning, April 2018
Lactic Acidosis and Severe Hepatomegaly with Steatosis: Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogues, including abacavir, lamivudine, and zidovudine (components of TRIZIVIR). Discontinue TRIZIVIR if clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity occur [see Warnings and Precautions (5.4)].
ERBITUX (CETUXIMAB)
- Edited boxed warning, June 2018
WARNING: INFUSION REACTIONS and CARDIOPULMONARY ARREST
Infusion Reactions: ERBITUX can cause serious and fatal infusion reactions [see Warnings and Precautions (5.1), Adverse Reactions (6)]. Immediately interrupt and permanently discontinue ERBITUX for serious infusion reactions [see Dosage and Administration (2.4)].
Cardiopulmonary Arrest: Cardiopulmonary arrest or sudden death occurred in patients with squamous cell carcinoma of the head and neck receiving ERBITUX with radiation therapy or a cetuximab product with platinum-based therapy and fluorouracil. Monitor serum electrolytes, including serum magnesium, potassium, and calcium, during and after ERBITUX administration [see Warnings and Precautions (5.2, 5.6)].
AUSTEDO (DEUTETRABENAZINE)
- Edited boxed warning, June 2018
WARNING: DEPRESSION AND SUICIDALITY IN PATIENTS WITH HUNTINGTON’S DISEASE
AUSTEDO can increase the risk of depression …
EXJADE (DEFERASIROX)
- Edited boxed warning, May 2018
Renal Failure: EXJADE can cause acute renal failure and death, particularly in patients with comorbidities and those who are in the advanced stages of their hematologic disorders. Evaluate baseline renal function prior to starting or increasing Exjade dosing in all patients. Exjade is contraindicated in adult and pediatric patients with eGFR less than 40 mL/min/1.73 m2. Measure serum creatinine in duplicate prior to initiation of therapy. Monitor renal function at least monthly. For patients with baseline renal impairment or increased risk of acute renal failure, monitor renal function weekly for the first month, then at least monthly. Reduce the starting dose in patients with pre-existing renal disease. During therapy, increase the frequency of monitoring and modify the dose for patients with an increased risk of renal impairment, including use of concomitant nephrotoxic drugs, and pediatric patients with volume depletion or overchelation.
TUXARIN ER (CODEINE PHOSPHATE AND CHLORPHENIRAMINE MALEATE)
- Edited boxed warning, June 2018
WARNING: ADDICTION, ABUSE, AND MISUSE; LIFE-THREATENING RESPIRATORY DEPRESSION; ACCIDENTAL INGESTION; ULTRA-RAPID METABOLISM OF CODEINE AND OTHER RISK FACTORS FOR LIFE-THREATENING RESPIRATORY DEPRESSION IN CHILDREN; MEDICATION ERRORS; INTERACTIONS WITH DRUGS AFFECTING CYTOCHROME P450 ISOENZYMES; CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS; NEONATAL OPIOID WITHDRAWAL SYNDROME.
Addiction, Abuse, and Misuse: TUXARIN ER exposes patients and other users to the risks of opioid addiction, abuse, and misuse,
which can lead to overdose and death. Reserve TUXARIN ER for use in adult patients for whom the benefits of cough suppression
are expected to outweigh the risks, and in whom an adequate assessment of the etiology of the cough has been made. Assess each
patient’s risk prior to prescribing TUXARIN ER, prescribe TUXARIN ER for the shortest duration that is consistent with individual patient
treatment goals, monitor all patients regularly for the development of addition or abuse, and refill only after reevaluation of the need for continued treatment. [see Warnings and Precautions (5.1)]
Life-Threatening Respiratory Depression: Serious, life-threatening, or fatal respiratory depression may occur with use of TUXARIN ER.
Monitor for respiratory depression, especially during initiation of TUXARIN ER therapy or when used in patients at higher risk [see Warnings and Precautions (5.2)].
Accidental Ingestion: Accidental ingestion of even one dose of TUXARIN ER, especially by children, can result in a fatal overdose of codeine [see Warnings and Precautions (5.2)].
Ultra-Rapid Metabolism of Codeine and Other Risk Factors for Life-Threatening Respiratory Depression in Children: Life threatening
respiratory depression and death have occurred in children who received codeine. Most of the reported cases occurred following tonsillectomy and/or adenoidectomy, and many of the children had evidence of being an ultra-rapid metabolizer of codeine due to a
CYP2D6 polymorphism. [see Warnings and Precautions (5.3)]. TUXARIN ER is contraindicated in children younger than 12 years of age and
in children younger than 18 years of age following tonsillectomy and/or adenoidectomy [see Contraindications (4)]. Avoid the use of TUXARIN ER in adolescents 12 to 18 years of age who have other risk factors that may increase their sensitivity to the respiratory depressant effects of codeine. [see Warnings and Precautions (5.1)].
Interactions with Drugs Affecting Cytochrome P450 Isoenzymes: The effects of concomitant use or discontinuation of cytochrome P450 3A4 inducers, 3A4 inhibitors, or 2D6 inhibitors with codeine are complex, requiring careful consideration of the effects on the parent drug, codeine, and the active metabolite, morphine. Avoid the use of TUXARIN ER in patients who are taking a CYP3A4 inhibitor, CYP3A4 inducer, or 2D6 inhibitor [see Warnings and Precautions (5.8), Drug Interactions (7.1,7.2, 7.4)]
Risks from Concomitant Use with Benzodiazepines, CNS Depressants: Concomitant use of opioids with benzodiazepines or other central nervous system (CNS) depressants, including alcohol, may result in profound sedation, respiratory depression, coma, and death. Avoid use of TUXARIN ER in patients taking benzodiazepines, other CNS depressants, or alcohol. [see Warning and Precautions (5.9) Drug Interactions (7.5)].
Neonatal Opioid Withdrawal Syndrome: TUXARIN ER is not recommended for use in pregnant women [see Use in Specific Populations
(8.1)]. Prolonged use of TUXARIN ER during pregnancy can result in neonatal opioid withdrawal syndrome, which may be life-threatening if not recognized and treated, and requires management according to protocols developed by neonatology experts. If TUXARIN ER is used for a prolonged period in a pregnant woman, advise the patient of the risk of neonatal opioid withdrawal syndrome and ensure that appropriate treatment will be available [see Warnings and Precautions (5.15)].
PROGRAF (TACROLIMUS)
- Edited boxed warning, May 2018
Increased risk for developing serious infections and malignancies with PROGRAF or other immunosuppressants that may lead to hospitalization or death [(see Warnings and Precautions 5.1,5.2)].
SAMSCA (TOLVAPTAN)
- Edited boxed warning, April 2018
Addition of the following:
WARNING: NOT FOR USE FOR AUTOSOMAL DOMINANT POLYCYSTIC KIDNEY DISEASE (ADPKD)
Because of the risk of hepatoxicity, tolvaptan should not be used for ADPKD outside of the FDA-approved REMS
GLUCOPHAGE/GLUCOPHAGE XR (METFORMIN HYDROCHLORIDE)
- Edited boxed warning, May 2018
PLR conversion; Lactic Acidosis warning is highlighted as boxed warning.
The FDA’s MedWatch program safety labeling changes for boxed warnings are compiled quarterly for drugs and therapeutic biologics where important changes have been made to the safety information. These and other label changes are searchable in the Drug Safety Labeling Changes (SLC) database, where data are available to the public in downloadable and searchable formats. Boxed warnings are ordinarily used to highlight either adverse reactions so serious in proportion to the potential benefi t from the drug that it is essential that it be considered in assessing the risks and benefi ts of using the drug; or serious adverse reactions that can be prevented/reduced in frequency or severity by appropriate use of the drug; or FDA approved the drug with restrictions to ensure safe use because FDA concluded that the drug can be safely used only if distribution or use is restricted. For complete FDA Drug Safety Labeling changes, please visit http://www.accessdata.fda.gov/scripts/cder/safetylabelingchanges.
DESOGEN (DESOGESTREL AND ETHINYL ESTRADIOL TABLETS
- Edited boxed warning, June 2018
WARNING: CIGARETTE SMOKING AND SERIOUS CARDIOVASCULAR EVENTS
Cigarette smoking increases the risk of serious cardiovascular events from combination oral contraceptive (COC) use. This risk increases with age, particularly in women over 35 years of age, and with the number of cigarettes smoked. For this reason, COCs are contraindicated in women who are over 35 years of age, and smoke.
TRIZIVIR (ABACAVIR, LAMIVUDINE, AND ZIDOVUDINE TABLETS)
- Edited boxed warning, April 2018
Lactic Acidosis and Severe Hepatomegaly with Steatosis: Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogues, including abacavir, lamivudine, and zidovudine (components of TRIZIVIR). Discontinue TRIZIVIR if clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity occur [see Warnings and Precautions (5.4)].
ERBITUX (CETUXIMAB)
- Edited boxed warning, June 2018
WARNING: INFUSION REACTIONS and CARDIOPULMONARY ARREST
Infusion Reactions: ERBITUX can cause serious and fatal infusion reactions [see Warnings and Precautions (5.1), Adverse Reactions (6)]. Immediately interrupt and permanently discontinue ERBITUX for serious infusion reactions [see Dosage and Administration (2.4)].
Cardiopulmonary Arrest: Cardiopulmonary arrest or sudden death occurred in patients with squamous cell carcinoma of the head and neck receiving ERBITUX with radiation therapy or a cetuximab product with platinum-based therapy and fluorouracil. Monitor serum electrolytes, including serum magnesium, potassium, and calcium, during and after ERBITUX administration [see Warnings and Precautions (5.2, 5.6)].
AUSTEDO (DEUTETRABENAZINE)
- Edited boxed warning, June 2018
WARNING: DEPRESSION AND SUICIDALITY IN PATIENTS WITH HUNTINGTON’S DISEASE
AUSTEDO can increase the risk of depression …
EXJADE (DEFERASIROX)
- Edited boxed warning, May 2018
Renal Failure: EXJADE can cause acute renal failure and death, particularly in patients with comorbidities and those who are in the advanced stages of their hematologic disorders. Evaluate baseline renal function prior to starting or increasing Exjade dosing in all patients. Exjade is contraindicated in adult and pediatric patients with eGFR less than 40 mL/min/1.73 m2. Measure serum creatinine in duplicate prior to initiation of therapy. Monitor renal function at least monthly. For patients with baseline renal impairment or increased risk of acute renal failure, monitor renal function weekly for the first month, then at least monthly. Reduce the starting dose in patients with pre-existing renal disease. During therapy, increase the frequency of monitoring and modify the dose for patients with an increased risk of renal impairment, including use of concomitant nephrotoxic drugs, and pediatric patients with volume depletion or overchelation.
TUXARIN ER (CODEINE PHOSPHATE AND CHLORPHENIRAMINE MALEATE)
- Edited boxed warning, June 2018
WARNING: ADDICTION, ABUSE, AND MISUSE; LIFE-THREATENING RESPIRATORY DEPRESSION; ACCIDENTAL INGESTION; ULTRA-RAPID METABOLISM OF CODEINE AND OTHER RISK FACTORS FOR LIFE-THREATENING RESPIRATORY DEPRESSION IN CHILDREN; MEDICATION ERRORS; INTERACTIONS WITH DRUGS AFFECTING CYTOCHROME P450 ISOENZYMES; CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS; NEONATAL OPIOID WITHDRAWAL SYNDROME.
Addiction, Abuse, and Misuse: TUXARIN ER exposes patients and other users to the risks of opioid addiction, abuse, and misuse,
which can lead to overdose and death. Reserve TUXARIN ER for use in adult patients for whom the benefits of cough suppression
are expected to outweigh the risks, and in whom an adequate assessment of the etiology of the cough has been made. Assess each
patient’s risk prior to prescribing TUXARIN ER, prescribe TUXARIN ER for the shortest duration that is consistent with individual patient
treatment goals, monitor all patients regularly for the development of addition or abuse, and refill only after reevaluation of the need for continued treatment. [see Warnings and Precautions (5.1)]
Life-Threatening Respiratory Depression: Serious, life-threatening, or fatal respiratory depression may occur with use of TUXARIN ER.
Monitor for respiratory depression, especially during initiation of TUXARIN ER therapy or when used in patients at higher risk [see Warnings and Precautions (5.2)].
Accidental Ingestion: Accidental ingestion of even one dose of TUXARIN ER, especially by children, can result in a fatal overdose of codeine [see Warnings and Precautions (5.2)].
Ultra-Rapid Metabolism of Codeine and Other Risk Factors for Life-Threatening Respiratory Depression in Children: Life threatening
respiratory depression and death have occurred in children who received codeine. Most of the reported cases occurred following tonsillectomy and/or adenoidectomy, and many of the children had evidence of being an ultra-rapid metabolizer of codeine due to a
CYP2D6 polymorphism. [see Warnings and Precautions (5.3)]. TUXARIN ER is contraindicated in children younger than 12 years of age and
in children younger than 18 years of age following tonsillectomy and/or adenoidectomy [see Contraindications (4)]. Avoid the use of TUXARIN ER in adolescents 12 to 18 years of age who have other risk factors that may increase their sensitivity to the respiratory depressant effects of codeine. [see Warnings and Precautions (5.1)].
Interactions with Drugs Affecting Cytochrome P450 Isoenzymes: The effects of concomitant use or discontinuation of cytochrome P450 3A4 inducers, 3A4 inhibitors, or 2D6 inhibitors with codeine are complex, requiring careful consideration of the effects on the parent drug, codeine, and the active metabolite, morphine. Avoid the use of TUXARIN ER in patients who are taking a CYP3A4 inhibitor, CYP3A4 inducer, or 2D6 inhibitor [see Warnings and Precautions (5.8), Drug Interactions (7.1,7.2, 7.4)]
Risks from Concomitant Use with Benzodiazepines, CNS Depressants: Concomitant use of opioids with benzodiazepines or other central nervous system (CNS) depressants, including alcohol, may result in profound sedation, respiratory depression, coma, and death. Avoid use of TUXARIN ER in patients taking benzodiazepines, other CNS depressants, or alcohol. [see Warning and Precautions (5.9) Drug Interactions (7.5)].
Neonatal Opioid Withdrawal Syndrome: TUXARIN ER is not recommended for use in pregnant women [see Use in Specific Populations
(8.1)]. Prolonged use of TUXARIN ER during pregnancy can result in neonatal opioid withdrawal syndrome, which may be life-threatening if not recognized and treated, and requires management according to protocols developed by neonatology experts. If TUXARIN ER is used for a prolonged period in a pregnant woman, advise the patient of the risk of neonatal opioid withdrawal syndrome and ensure that appropriate treatment will be available [see Warnings and Precautions (5.15)].
PROGRAF (TACROLIMUS)
- Edited boxed warning, May 2018
Increased risk for developing serious infections and malignancies with PROGRAF or other immunosuppressants that may lead to hospitalization or death [(see Warnings and Precautions 5.1,5.2)].
SAMSCA (TOLVAPTAN)
- Edited boxed warning, April 2018
Addition of the following:
WARNING: NOT FOR USE FOR AUTOSOMAL DOMINANT POLYCYSTIC KIDNEY DISEASE (ADPKD)
Because of the risk of hepatoxicity, tolvaptan should not be used for ADPKD outside of the FDA-approved REMS
GLUCOPHAGE/GLUCOPHAGE XR (METFORMIN HYDROCHLORIDE)
- Edited boxed warning, May 2018
PLR conversion; Lactic Acidosis warning is highlighted as boxed warning.
The FDA’s MedWatch program safety labeling changes for boxed warnings are compiled quarterly for drugs and therapeutic biologics where important changes have been made to the safety information. These and other label changes are searchable in the Drug Safety Labeling Changes (SLC) database, where data are available to the public in downloadable and searchable formats. Boxed warnings are ordinarily used to highlight either adverse reactions so serious in proportion to the potential benefi t from the drug that it is essential that it be considered in assessing the risks and benefi ts of using the drug; or serious adverse reactions that can be prevented/reduced in frequency or severity by appropriate use of the drug; or FDA approved the drug with restrictions to ensure safe use because FDA concluded that the drug can be safely used only if distribution or use is restricted. For complete FDA Drug Safety Labeling changes, please visit http://www.accessdata.fda.gov/scripts/cder/safetylabelingchanges.
DESOGEN (DESOGESTREL AND ETHINYL ESTRADIOL TABLETS
- Edited boxed warning, June 2018
WARNING: CIGARETTE SMOKING AND SERIOUS CARDIOVASCULAR EVENTS
Cigarette smoking increases the risk of serious cardiovascular events from combination oral contraceptive (COC) use. This risk increases with age, particularly in women over 35 years of age, and with the number of cigarettes smoked. For this reason, COCs are contraindicated in women who are over 35 years of age, and smoke.
TRIZIVIR (ABACAVIR, LAMIVUDINE, AND ZIDOVUDINE TABLETS)
- Edited boxed warning, April 2018
Lactic Acidosis and Severe Hepatomegaly with Steatosis: Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogues, including abacavir, lamivudine, and zidovudine (components of TRIZIVIR). Discontinue TRIZIVIR if clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity occur [see Warnings and Precautions (5.4)].
ERBITUX (CETUXIMAB)
- Edited boxed warning, June 2018
WARNING: INFUSION REACTIONS and CARDIOPULMONARY ARREST
Infusion Reactions: ERBITUX can cause serious and fatal infusion reactions [see Warnings and Precautions (5.1), Adverse Reactions (6)]. Immediately interrupt and permanently discontinue ERBITUX for serious infusion reactions [see Dosage and Administration (2.4)].
Cardiopulmonary Arrest: Cardiopulmonary arrest or sudden death occurred in patients with squamous cell carcinoma of the head and neck receiving ERBITUX with radiation therapy or a cetuximab product with platinum-based therapy and fluorouracil. Monitor serum electrolytes, including serum magnesium, potassium, and calcium, during and after ERBITUX administration [see Warnings and Precautions (5.2, 5.6)].
AUSTEDO (DEUTETRABENAZINE)
- Edited boxed warning, June 2018
WARNING: DEPRESSION AND SUICIDALITY IN PATIENTS WITH HUNTINGTON’S DISEASE
AUSTEDO can increase the risk of depression …
EXJADE (DEFERASIROX)
- Edited boxed warning, May 2018
Renal Failure: EXJADE can cause acute renal failure and death, particularly in patients with comorbidities and those who are in the advanced stages of their hematologic disorders. Evaluate baseline renal function prior to starting or increasing Exjade dosing in all patients. Exjade is contraindicated in adult and pediatric patients with eGFR less than 40 mL/min/1.73 m2. Measure serum creatinine in duplicate prior to initiation of therapy. Monitor renal function at least monthly. For patients with baseline renal impairment or increased risk of acute renal failure, monitor renal function weekly for the first month, then at least monthly. Reduce the starting dose in patients with pre-existing renal disease. During therapy, increase the frequency of monitoring and modify the dose for patients with an increased risk of renal impairment, including use of concomitant nephrotoxic drugs, and pediatric patients with volume depletion or overchelation.
TUXARIN ER (CODEINE PHOSPHATE AND CHLORPHENIRAMINE MALEATE)
- Edited boxed warning, June 2018
WARNING: ADDICTION, ABUSE, AND MISUSE; LIFE-THREATENING RESPIRATORY DEPRESSION; ACCIDENTAL INGESTION; ULTRA-RAPID METABOLISM OF CODEINE AND OTHER RISK FACTORS FOR LIFE-THREATENING RESPIRATORY DEPRESSION IN CHILDREN; MEDICATION ERRORS; INTERACTIONS WITH DRUGS AFFECTING CYTOCHROME P450 ISOENZYMES; CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS; NEONATAL OPIOID WITHDRAWAL SYNDROME.
Addiction, Abuse, and Misuse: TUXARIN ER exposes patients and other users to the risks of opioid addiction, abuse, and misuse,
which can lead to overdose and death. Reserve TUXARIN ER for use in adult patients for whom the benefits of cough suppression
are expected to outweigh the risks, and in whom an adequate assessment of the etiology of the cough has been made. Assess each
patient’s risk prior to prescribing TUXARIN ER, prescribe TUXARIN ER for the shortest duration that is consistent with individual patient
treatment goals, monitor all patients regularly for the development of addition or abuse, and refill only after reevaluation of the need for continued treatment. [see Warnings and Precautions (5.1)]
Life-Threatening Respiratory Depression: Serious, life-threatening, or fatal respiratory depression may occur with use of TUXARIN ER.
Monitor for respiratory depression, especially during initiation of TUXARIN ER therapy or when used in patients at higher risk [see Warnings and Precautions (5.2)].
Accidental Ingestion: Accidental ingestion of even one dose of TUXARIN ER, especially by children, can result in a fatal overdose of codeine [see Warnings and Precautions (5.2)].
Ultra-Rapid Metabolism of Codeine and Other Risk Factors for Life-Threatening Respiratory Depression in Children: Life threatening
respiratory depression and death have occurred in children who received codeine. Most of the reported cases occurred following tonsillectomy and/or adenoidectomy, and many of the children had evidence of being an ultra-rapid metabolizer of codeine due to a
CYP2D6 polymorphism. [see Warnings and Precautions (5.3)]. TUXARIN ER is contraindicated in children younger than 12 years of age and
in children younger than 18 years of age following tonsillectomy and/or adenoidectomy [see Contraindications (4)]. Avoid the use of TUXARIN ER in adolescents 12 to 18 years of age who have other risk factors that may increase their sensitivity to the respiratory depressant effects of codeine. [see Warnings and Precautions (5.1)].
Interactions with Drugs Affecting Cytochrome P450 Isoenzymes: The effects of concomitant use or discontinuation of cytochrome P450 3A4 inducers, 3A4 inhibitors, or 2D6 inhibitors with codeine are complex, requiring careful consideration of the effects on the parent drug, codeine, and the active metabolite, morphine. Avoid the use of TUXARIN ER in patients who are taking a CYP3A4 inhibitor, CYP3A4 inducer, or 2D6 inhibitor [see Warnings and Precautions (5.8), Drug Interactions (7.1,7.2, 7.4)]
Risks from Concomitant Use with Benzodiazepines, CNS Depressants: Concomitant use of opioids with benzodiazepines or other central nervous system (CNS) depressants, including alcohol, may result in profound sedation, respiratory depression, coma, and death. Avoid use of TUXARIN ER in patients taking benzodiazepines, other CNS depressants, or alcohol. [see Warning and Precautions (5.9) Drug Interactions (7.5)].
Neonatal Opioid Withdrawal Syndrome: TUXARIN ER is not recommended for use in pregnant women [see Use in Specific Populations
(8.1)]. Prolonged use of TUXARIN ER during pregnancy can result in neonatal opioid withdrawal syndrome, which may be life-threatening if not recognized and treated, and requires management according to protocols developed by neonatology experts. If TUXARIN ER is used for a prolonged period in a pregnant woman, advise the patient of the risk of neonatal opioid withdrawal syndrome and ensure that appropriate treatment will be available [see Warnings and Precautions (5.15)].
PROGRAF (TACROLIMUS)
- Edited boxed warning, May 2018
Increased risk for developing serious infections and malignancies with PROGRAF or other immunosuppressants that may lead to hospitalization or death [(see Warnings and Precautions 5.1,5.2)].
SAMSCA (TOLVAPTAN)
- Edited boxed warning, April 2018
Addition of the following:
WARNING: NOT FOR USE FOR AUTOSOMAL DOMINANT POLYCYSTIC KIDNEY DISEASE (ADPKD)
Because of the risk of hepatoxicity, tolvaptan should not be used for ADPKD outside of the FDA-approved REMS
GLUCOPHAGE/GLUCOPHAGE XR (METFORMIN HYDROCHLORIDE)
- Edited boxed warning, May 2018
PLR conversion; Lactic Acidosis warning is highlighted as boxed warning.
Do Erythropoiesis-Stimulating Agents Have a Risk Evaluation and Mitigation Strategy? (FULL)
Epoetin alfa and darbepoetin alfa are erythropoiesis-stimulating agents (ESAs), approved for the treatment of anemia (low red blood cells [RBCs]) resulting from chronic kidney disease, chemotherapy, and certain treatments for HIV. These ESAs also are used to reduce the number of blood transfusions during and after certain major surgeries. Erythropoiesis-stimulating agents work like the human protein erythropoietin, which stimulates bone marrow to make RBCs. Epoetin alfa (marketed as Procrit and Epogen) and darbepoetin alfa (marketed as Aranesp) are manufactured by Amgen, Inc. (Thousand Oaks, CA).
In 1989 epoetin alfa was approved for the treatment of anemia associated with chronic renal failure, including patients on dialysis and patients not on dialysis, and in 1993 for the treatment of anemia due to the effects of concomitant myelosuppressive chemotherapy. Epoetin alfa also is indicated for anemia due to zidovudine in patients with HIV and reduction of RBC transfusions during certain surgeries.
Darbepoetin alfa was approved in 2001 for the treatment of anemia associated with chronic renal failure, including patients on dialysis and patients not on dialysis, and in 2006 for the treatment of anemia due to the effects of concomitant myelosuppressive chemotherapy.
Risk Evaluation and Mitigation Strategies
Both epoetin alfa and darbepoetin alfa increase the risk of death, myocardial infarction, stroke, venous thromboembolism, and thrombosis of vascular access and tumor progression or recurrence. Epoetin alfa also can lead to an increase in adverse cardiovascular events, hypertension, seizures, and severe anemia.
In 2008, the FDA determined that Risk Evaluation and Mitigation Strategies (REMS) were necessary for ESAs (darbopoetin alfa and epoetin alfa), to ensure that the benefits for use as treatment for anemia associated with myelosuppressive chemotherapy outweigh the risk of shortened overall survival (OS) and/or the increased risk of tumor progression or recurrence in patients with cancer. The REMS was approved in 2010.
Under the ESA REMS program, referred to as the ESA APPRISE Oncology Program, health care providers (HCPs) that prescribed and/or dispensed darbopoetin alfa to patients with cancer and hospitals that dispensed darbopoetin alfa to patients with cancer were required to enroll and become certified in the ESA REMS. The ESA REMS also required the completion of a Patient and Healthcare Provider Acknowledgement Form for each patient with cancer before the new ESA treatment course to ensure patients were counseled about the benefits and risks of these products.
In April 2017, the FDA determined that the ESA REMS that was limited to the use of epoetin alfa and darbopoetin alfa to treat patients with anemia due to associated myelosuppressive chemotherapy was no longer necessary; the benefits of ESAs outweighed the risks of shortened OS and/or increased risk of tumor progression or recurrence in patients with cancer. 1 The FDA recognized the burden that some REMS can place on HCPs and patients. The agency has authority to modify or remove the REMS to minimize the burden on the health care delivery system of complying with the strategy.
Data
The FDA discontinued the REMS based on an evaluation of the results of the REMS Assessments submitted by Amgen and additional FDA analyses to understand the impact of the various regulatory and other actions on the use of ESAs. The REMS Assessment showed the following:
- The results from surveyed prescribers demonstrated acceptable knowledge of the product risks of decreased survival and/or the increased risk of tumor progression or recurrence and the need to counsel patients about these risks; and
- The drug utilization data indicated appropriate prescribing of ESAs consistent with the intended use as a treatment alternative to RBC transfusion for anemia associated with myelosuppressive chemotherapy.
The FDA also conducted an evaluation of the impact of multiple actions, including the ESA REMS, on the use of the ESAs using sponsor-submitted data from outpatient oncology practices between 2006 and 2014. During 2004 to 2009, the FDA took multiple regulatory actions, including labeling changes. In 2007, the Center for Medicare and Medicaid Services (CMS) made a National Coverage Determination (NCD) to limit coverage of ESAs for nonrenal disease indications. These actions coincided with the following:
- A decrease in the proportion of patients receiving chemotherapy using ESAs;
- An increase in the proportion of patients receiving chemotherapy who initiate ESAs at a hemoglobin level < 10 g/dL; and
- An increase in the proportion of patients who initiate ESAs at a dosage consistent with product prescribing information.
Full implementation of the ESA REMS in 2011 had minimal impact on trends in these 3 ESA utilization metrics beyond the changes observed after the CMS coverage determination and multiple other FDA regulatory actions.
This information led the FDA to conclude that it was no longer necessary to require the certification of prescribers and hospitals that prescribe and/or dispense ESAs to patients with cancer in order to ensure that the benefits outweigh the risks.
The FDA has released the REMS requirements for the epoetin alfa and darbopoetin alfa ESA products, and the risks can be communicated by the current product prescribing information. The appropriate use of ESAs is supported by the CMS NCD, the American Society of Clinical Oncology, and American Society of Hematology clinical guidelines, which are evidence-based guidelines intended to provide a basis for the standard of care in clinical oncology.
Education
While the REMS is no longer necessary to ensure the benefits outweigh the risks, the serious risks of shortened OS and/or increased risk of tumor progression or recurrence associated with these drugs remain. The boxed warning language remains as follows: ESAs INCREASE THE RISK OF DEATH, MYOCARDIAL INFARCTION, STROKE, VENOUS THROMBOEMBOLISM, THROMBOSIS OF VASCULAR ACCESS AND TUMOR PROGRESSION OR RECURRENCE. Health care providers are encouraged to discuss the risks and benefits of using ESAs with each patient before initiating use.
Click here to read the digital edition.
1. U.S. Food & Drug Administration. Information on erythropoiesis-stimulating agents (ESA) epoetin alfa (marketed as Procrit, Epogen), darbepoetin alfa (marketed as Aranesp). https://www.fda.gov/Drugs/DrugSafety/ucm109375.htm. Updated April 13, 2017. Accessed July 13, 2017.
Epoetin alfa and darbepoetin alfa are erythropoiesis-stimulating agents (ESAs), approved for the treatment of anemia (low red blood cells [RBCs]) resulting from chronic kidney disease, chemotherapy, and certain treatments for HIV. These ESAs also are used to reduce the number of blood transfusions during and after certain major surgeries. Erythropoiesis-stimulating agents work like the human protein erythropoietin, which stimulates bone marrow to make RBCs. Epoetin alfa (marketed as Procrit and Epogen) and darbepoetin alfa (marketed as Aranesp) are manufactured by Amgen, Inc. (Thousand Oaks, CA).
In 1989 epoetin alfa was approved for the treatment of anemia associated with chronic renal failure, including patients on dialysis and patients not on dialysis, and in 1993 for the treatment of anemia due to the effects of concomitant myelosuppressive chemotherapy. Epoetin alfa also is indicated for anemia due to zidovudine in patients with HIV and reduction of RBC transfusions during certain surgeries.
Darbepoetin alfa was approved in 2001 for the treatment of anemia associated with chronic renal failure, including patients on dialysis and patients not on dialysis, and in 2006 for the treatment of anemia due to the effects of concomitant myelosuppressive chemotherapy.
Risk Evaluation and Mitigation Strategies
Both epoetin alfa and darbepoetin alfa increase the risk of death, myocardial infarction, stroke, venous thromboembolism, and thrombosis of vascular access and tumor progression or recurrence. Epoetin alfa also can lead to an increase in adverse cardiovascular events, hypertension, seizures, and severe anemia.
In 2008, the FDA determined that Risk Evaluation and Mitigation Strategies (REMS) were necessary for ESAs (darbopoetin alfa and epoetin alfa), to ensure that the benefits for use as treatment for anemia associated with myelosuppressive chemotherapy outweigh the risk of shortened overall survival (OS) and/or the increased risk of tumor progression or recurrence in patients with cancer. The REMS was approved in 2010.
Under the ESA REMS program, referred to as the ESA APPRISE Oncology Program, health care providers (HCPs) that prescribed and/or dispensed darbopoetin alfa to patients with cancer and hospitals that dispensed darbopoetin alfa to patients with cancer were required to enroll and become certified in the ESA REMS. The ESA REMS also required the completion of a Patient and Healthcare Provider Acknowledgement Form for each patient with cancer before the new ESA treatment course to ensure patients were counseled about the benefits and risks of these products.
In April 2017, the FDA determined that the ESA REMS that was limited to the use of epoetin alfa and darbopoetin alfa to treat patients with anemia due to associated myelosuppressive chemotherapy was no longer necessary; the benefits of ESAs outweighed the risks of shortened OS and/or increased risk of tumor progression or recurrence in patients with cancer. 1 The FDA recognized the burden that some REMS can place on HCPs and patients. The agency has authority to modify or remove the REMS to minimize the burden on the health care delivery system of complying with the strategy.
Data
The FDA discontinued the REMS based on an evaluation of the results of the REMS Assessments submitted by Amgen and additional FDA analyses to understand the impact of the various regulatory and other actions on the use of ESAs. The REMS Assessment showed the following:
- The results from surveyed prescribers demonstrated acceptable knowledge of the product risks of decreased survival and/or the increased risk of tumor progression or recurrence and the need to counsel patients about these risks; and
- The drug utilization data indicated appropriate prescribing of ESAs consistent with the intended use as a treatment alternative to RBC transfusion for anemia associated with myelosuppressive chemotherapy.
The FDA also conducted an evaluation of the impact of multiple actions, including the ESA REMS, on the use of the ESAs using sponsor-submitted data from outpatient oncology practices between 2006 and 2014. During 2004 to 2009, the FDA took multiple regulatory actions, including labeling changes. In 2007, the Center for Medicare and Medicaid Services (CMS) made a National Coverage Determination (NCD) to limit coverage of ESAs for nonrenal disease indications. These actions coincided with the following:
- A decrease in the proportion of patients receiving chemotherapy using ESAs;
- An increase in the proportion of patients receiving chemotherapy who initiate ESAs at a hemoglobin level < 10 g/dL; and
- An increase in the proportion of patients who initiate ESAs at a dosage consistent with product prescribing information.
Full implementation of the ESA REMS in 2011 had minimal impact on trends in these 3 ESA utilization metrics beyond the changes observed after the CMS coverage determination and multiple other FDA regulatory actions.
This information led the FDA to conclude that it was no longer necessary to require the certification of prescribers and hospitals that prescribe and/or dispense ESAs to patients with cancer in order to ensure that the benefits outweigh the risks.
The FDA has released the REMS requirements for the epoetin alfa and darbopoetin alfa ESA products, and the risks can be communicated by the current product prescribing information. The appropriate use of ESAs is supported by the CMS NCD, the American Society of Clinical Oncology, and American Society of Hematology clinical guidelines, which are evidence-based guidelines intended to provide a basis for the standard of care in clinical oncology.
Education
While the REMS is no longer necessary to ensure the benefits outweigh the risks, the serious risks of shortened OS and/or increased risk of tumor progression or recurrence associated with these drugs remain. The boxed warning language remains as follows: ESAs INCREASE THE RISK OF DEATH, MYOCARDIAL INFARCTION, STROKE, VENOUS THROMBOEMBOLISM, THROMBOSIS OF VASCULAR ACCESS AND TUMOR PROGRESSION OR RECURRENCE. Health care providers are encouraged to discuss the risks and benefits of using ESAs with each patient before initiating use.
Click here to read the digital edition.
Epoetin alfa and darbepoetin alfa are erythropoiesis-stimulating agents (ESAs), approved for the treatment of anemia (low red blood cells [RBCs]) resulting from chronic kidney disease, chemotherapy, and certain treatments for HIV. These ESAs also are used to reduce the number of blood transfusions during and after certain major surgeries. Erythropoiesis-stimulating agents work like the human protein erythropoietin, which stimulates bone marrow to make RBCs. Epoetin alfa (marketed as Procrit and Epogen) and darbepoetin alfa (marketed as Aranesp) are manufactured by Amgen, Inc. (Thousand Oaks, CA).
In 1989 epoetin alfa was approved for the treatment of anemia associated with chronic renal failure, including patients on dialysis and patients not on dialysis, and in 1993 for the treatment of anemia due to the effects of concomitant myelosuppressive chemotherapy. Epoetin alfa also is indicated for anemia due to zidovudine in patients with HIV and reduction of RBC transfusions during certain surgeries.
Darbepoetin alfa was approved in 2001 for the treatment of anemia associated with chronic renal failure, including patients on dialysis and patients not on dialysis, and in 2006 for the treatment of anemia due to the effects of concomitant myelosuppressive chemotherapy.
Risk Evaluation and Mitigation Strategies
Both epoetin alfa and darbepoetin alfa increase the risk of death, myocardial infarction, stroke, venous thromboembolism, and thrombosis of vascular access and tumor progression or recurrence. Epoetin alfa also can lead to an increase in adverse cardiovascular events, hypertension, seizures, and severe anemia.
In 2008, the FDA determined that Risk Evaluation and Mitigation Strategies (REMS) were necessary for ESAs (darbopoetin alfa and epoetin alfa), to ensure that the benefits for use as treatment for anemia associated with myelosuppressive chemotherapy outweigh the risk of shortened overall survival (OS) and/or the increased risk of tumor progression or recurrence in patients with cancer. The REMS was approved in 2010.
Under the ESA REMS program, referred to as the ESA APPRISE Oncology Program, health care providers (HCPs) that prescribed and/or dispensed darbopoetin alfa to patients with cancer and hospitals that dispensed darbopoetin alfa to patients with cancer were required to enroll and become certified in the ESA REMS. The ESA REMS also required the completion of a Patient and Healthcare Provider Acknowledgement Form for each patient with cancer before the new ESA treatment course to ensure patients were counseled about the benefits and risks of these products.
In April 2017, the FDA determined that the ESA REMS that was limited to the use of epoetin alfa and darbopoetin alfa to treat patients with anemia due to associated myelosuppressive chemotherapy was no longer necessary; the benefits of ESAs outweighed the risks of shortened OS and/or increased risk of tumor progression or recurrence in patients with cancer. 1 The FDA recognized the burden that some REMS can place on HCPs and patients. The agency has authority to modify or remove the REMS to minimize the burden on the health care delivery system of complying with the strategy.
Data
The FDA discontinued the REMS based on an evaluation of the results of the REMS Assessments submitted by Amgen and additional FDA analyses to understand the impact of the various regulatory and other actions on the use of ESAs. The REMS Assessment showed the following:
- The results from surveyed prescribers demonstrated acceptable knowledge of the product risks of decreased survival and/or the increased risk of tumor progression or recurrence and the need to counsel patients about these risks; and
- The drug utilization data indicated appropriate prescribing of ESAs consistent with the intended use as a treatment alternative to RBC transfusion for anemia associated with myelosuppressive chemotherapy.
The FDA also conducted an evaluation of the impact of multiple actions, including the ESA REMS, on the use of the ESAs using sponsor-submitted data from outpatient oncology practices between 2006 and 2014. During 2004 to 2009, the FDA took multiple regulatory actions, including labeling changes. In 2007, the Center for Medicare and Medicaid Services (CMS) made a National Coverage Determination (NCD) to limit coverage of ESAs for nonrenal disease indications. These actions coincided with the following:
- A decrease in the proportion of patients receiving chemotherapy using ESAs;
- An increase in the proportion of patients receiving chemotherapy who initiate ESAs at a hemoglobin level < 10 g/dL; and
- An increase in the proportion of patients who initiate ESAs at a dosage consistent with product prescribing information.
Full implementation of the ESA REMS in 2011 had minimal impact on trends in these 3 ESA utilization metrics beyond the changes observed after the CMS coverage determination and multiple other FDA regulatory actions.
This information led the FDA to conclude that it was no longer necessary to require the certification of prescribers and hospitals that prescribe and/or dispense ESAs to patients with cancer in order to ensure that the benefits outweigh the risks.
The FDA has released the REMS requirements for the epoetin alfa and darbopoetin alfa ESA products, and the risks can be communicated by the current product prescribing information. The appropriate use of ESAs is supported by the CMS NCD, the American Society of Clinical Oncology, and American Society of Hematology clinical guidelines, which are evidence-based guidelines intended to provide a basis for the standard of care in clinical oncology.
Education
While the REMS is no longer necessary to ensure the benefits outweigh the risks, the serious risks of shortened OS and/or increased risk of tumor progression or recurrence associated with these drugs remain. The boxed warning language remains as follows: ESAs INCREASE THE RISK OF DEATH, MYOCARDIAL INFARCTION, STROKE, VENOUS THROMBOEMBOLISM, THROMBOSIS OF VASCULAR ACCESS AND TUMOR PROGRESSION OR RECURRENCE. Health care providers are encouraged to discuss the risks and benefits of using ESAs with each patient before initiating use.
Click here to read the digital edition.
1. U.S. Food & Drug Administration. Information on erythropoiesis-stimulating agents (ESA) epoetin alfa (marketed as Procrit, Epogen), darbepoetin alfa (marketed as Aranesp). https://www.fda.gov/Drugs/DrugSafety/ucm109375.htm. Updated April 13, 2017. Accessed July 13, 2017.
1. U.S. Food & Drug Administration. Information on erythropoiesis-stimulating agents (ESA) epoetin alfa (marketed as Procrit, Epogen), darbepoetin alfa (marketed as Aranesp). https://www.fda.gov/Drugs/DrugSafety/ucm109375.htm. Updated April 13, 2017. Accessed July 13, 2017.
FDA Boxed Warning Updates: June 2018
The FDA’s MedWatch program safety labeling changes for boxed warnings are compiled quarterly for drugs and therapeutic biologics where important changes have been made to the safety information. These and other label changes are searchable in the Drug Safety Labeling Changes (SLC) database, where data are available to the public in downloadable and searchable formats. Boxed warnings are ordinarily used to highlight either adverse reactions so serious in proportion to the potential benefi t from the drug that it is essential that it be considered in assessing the risks and benefi ts of using the drug; or serious adverse reactions that can be prevented/reduced in frequency or severity by appropriate use of the drug; or FDA approved the drug with restrictions to ensure safe use because FDA concluded that the drug can be safely used only if distribution or use is restricted. For complete FDA Drug Safety Labeling changes, please visit http://www.accessdata.fda.gov/scripts/cder/safetylabelingchanges.
VIDEX AND VIDEX EC (DIDANOSINE):
- Edited boxed warning, January 2018
WARNING: PANCREATITIS, LACTIC ACIDOSIS and HEPATOMEGALY with STEATOSIS
Coadministration of VIDEX or VIDEX EC and stavudine is contraindicated because of increased risk of serious and/or life-threatening events. Suspend treatment if clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity occurs.
ZYDELIG (IDELALISIB):
- Edited boxed warning, January 2018
WARNING: FATAL AND SERIOUS TOXICITIES: HEPATIC, SEVERE DIARRHEA, COLITIS, PNEUMONITIS, INFECTIONS, and INTESTINAL PERFORATION
Fatal and/or serious hepatotoxicity occurred in 16% to 18% of Zydelig-treated patients…
Fatal and/or serious and severe diarrhea or colitis occurred in 14% to 20% of Zydelig-treated patients…
Fatal and/or serious infections occurred in 21% to 48% of Zydelig-treated patients…
AQUAMEPHYTON (PHYTONADIONE):
- Edited boxed warning, March 2018
WARNING: HYPERSENSITIVITY REACTIONS WITH INTRAVENOUS AND INTRAMUSCULAR USE
Fatal hypersensitivity reactions, including anaphylaxis, have occurred during and immediately after INTRAVENOUS and INTRAMUSCULAR injection of Aqua-MEPHYTON. Reactions have occurred despite dilution to avoid rapid infusion and upon first dose. Avoid the intravenous and intramuscular routes of administration unless the subcutaneous route is not feasible and the serious risk is justified.
FERAHEME (FERUMOXYTOL):
- Edited boxed warning, February 2018
WARNING: RISK FOR SERIOUS HYPERSENSITIVITY/ANAPHYLAXIS REACTIONS
Fatal and serious hypersensitivity reactions including anaphylaxis have occurred in patients receiving feraheme. Initial symptoms may
include hypotension, syncope, unresponsiveness, cardiac/cardiorespiratory arrest.
Only administer feraheme as an intravenous infusion over at least 15 minutes and only when personnel and therapies are immediately available for the treatment of anaphylaxis and other hypersensitivity reactions.
METHADONE HYDROCHLORIDE, METHADOSE (METHADONE HYDROCHLORIDE):
- Edited boxed warning, February 2018
RISKS FROM CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS
Concomitant use with benzodiazepines or other central nervous system (CNS) depressants, including alcohol, is a risk factor for respiratory depression and death.
Reserve concomitant prescribing of benzodiazepines or other CNS depressants for use in patients for whom alternatives to benzodiazepines or other CNS depressants are inadequate.
Follow patients for signs and symptoms of respiratory depression and sedation. If the patient is visibly sedated, evaluate the cause of sedation, and consider delaying or omitting daily methadone dosing.
DOLOPHINE HYDROCHLORIDE (METHADONE HYDROCHLORIDE):
- Edited boxed warning, February 2018
RISKS FROM CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS
Concomitant use of opioids with benzodiazepines or other central nervous system (CNS) depressants, including alcohol, may result in profound sedation, respiratory depression, coma, and death.
Reserve concomitant prescribing of DOLOPHINE Tablets and benzodiazepines or other CNS depressants for use in patients for whom alternatives to benzodiazepines or other CNS depressants are inadequate.
Limit dosages and durations to the minimum required for patients being treated for pain.
Follow patients for signs and symptoms of respiratory depression and sedation. If the patient is visibly sedated, evaluate the cause of sedation, and consider delaying or omitting the daily methadone dose.
PARNATE (TRANYLCYPROMINE SULFATE):
- Edited boxed warning, January 2018
WARNING: SUICIDAL THOUGHTS AND BEHAVIORS AND HYPERTENSIVE CRISIS WITH SIGNIFICANT TYRAMINE USE
SUICIDAL THOUGHTS AND BEHAVIORS
Antidepressants increased the risk of suicidal thoughts and behaviors in pediatric and young adult patients in short-term studies. Closely monitor all antidepressant-treated patients for clinical worsening, and for emergence of suicidal thoughts and behaviors. PARNATE is not approved for use in pediatric patients.
HYPERTENSIVE CRISIS WITH SIGNIFICANT TYRAMINE USE
Excessive consumption of foods or beverages with significant tyramine content or the use of certain drugs with PARNATE or after PARNATE discontinuation can precipitate hypertensive crisis. Monitor blood pressure and allow for medication-free intervals between administration of PARNATE and interacting drugs. Instruct patients to avoid ingestion of foods and beverages with high tyramine content.
OCALIVA (OBETICHOLIC ACID):
- New boxed warning/Newly added section, February 2018
WARNING: HEPATIC DECOMPENSATION AND FAILURE IN INCORRECTLY DOSED PBC PATIENTS WITH CHILD-PUGH CLASS B OR C OR DECOMPENSATED CIRRHOSIS
In postmarketing reports, hepatic decompensation and failure, in some cases fatal, have been reported in patients with primary biliary cholangitis (PBC) with decompensated cirrhosis or Child-Pugh Class B or C hepatic impairment when OCALIVA was dosed more frequently than recommended.
The recommended starting dosage of OCALIVA is 5 mg once weekly for patients with Child-Pugh Class B or C hepatic impairment or a prior decompensation event.
The FDA’s MedWatch program safety labeling changes for boxed warnings are compiled quarterly for drugs and therapeutic biologics where important changes have been made to the safety information. These and other label changes are searchable in the Drug Safety Labeling Changes (SLC) database, where data are available to the public in downloadable and searchable formats. Boxed warnings are ordinarily used to highlight either adverse reactions so serious in proportion to the potential benefi t from the drug that it is essential that it be considered in assessing the risks and benefi ts of using the drug; or serious adverse reactions that can be prevented/reduced in frequency or severity by appropriate use of the drug; or FDA approved the drug with restrictions to ensure safe use because FDA concluded that the drug can be safely used only if distribution or use is restricted. For complete FDA Drug Safety Labeling changes, please visit http://www.accessdata.fda.gov/scripts/cder/safetylabelingchanges.
VIDEX AND VIDEX EC (DIDANOSINE):
- Edited boxed warning, January 2018
WARNING: PANCREATITIS, LACTIC ACIDOSIS and HEPATOMEGALY with STEATOSIS
Coadministration of VIDEX or VIDEX EC and stavudine is contraindicated because of increased risk of serious and/or life-threatening events. Suspend treatment if clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity occurs.
ZYDELIG (IDELALISIB):
- Edited boxed warning, January 2018
WARNING: FATAL AND SERIOUS TOXICITIES: HEPATIC, SEVERE DIARRHEA, COLITIS, PNEUMONITIS, INFECTIONS, and INTESTINAL PERFORATION
Fatal and/or serious hepatotoxicity occurred in 16% to 18% of Zydelig-treated patients…
Fatal and/or serious and severe diarrhea or colitis occurred in 14% to 20% of Zydelig-treated patients…
Fatal and/or serious infections occurred in 21% to 48% of Zydelig-treated patients…
AQUAMEPHYTON (PHYTONADIONE):
- Edited boxed warning, March 2018
WARNING: HYPERSENSITIVITY REACTIONS WITH INTRAVENOUS AND INTRAMUSCULAR USE
Fatal hypersensitivity reactions, including anaphylaxis, have occurred during and immediately after INTRAVENOUS and INTRAMUSCULAR injection of Aqua-MEPHYTON. Reactions have occurred despite dilution to avoid rapid infusion and upon first dose. Avoid the intravenous and intramuscular routes of administration unless the subcutaneous route is not feasible and the serious risk is justified.
FERAHEME (FERUMOXYTOL):
- Edited boxed warning, February 2018
WARNING: RISK FOR SERIOUS HYPERSENSITIVITY/ANAPHYLAXIS REACTIONS
Fatal and serious hypersensitivity reactions including anaphylaxis have occurred in patients receiving feraheme. Initial symptoms may
include hypotension, syncope, unresponsiveness, cardiac/cardiorespiratory arrest.
Only administer feraheme as an intravenous infusion over at least 15 minutes and only when personnel and therapies are immediately available for the treatment of anaphylaxis and other hypersensitivity reactions.
METHADONE HYDROCHLORIDE, METHADOSE (METHADONE HYDROCHLORIDE):
- Edited boxed warning, February 2018
RISKS FROM CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS
Concomitant use with benzodiazepines or other central nervous system (CNS) depressants, including alcohol, is a risk factor for respiratory depression and death.
Reserve concomitant prescribing of benzodiazepines or other CNS depressants for use in patients for whom alternatives to benzodiazepines or other CNS depressants are inadequate.
Follow patients for signs and symptoms of respiratory depression and sedation. If the patient is visibly sedated, evaluate the cause of sedation, and consider delaying or omitting daily methadone dosing.
DOLOPHINE HYDROCHLORIDE (METHADONE HYDROCHLORIDE):
- Edited boxed warning, February 2018
RISKS FROM CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS
Concomitant use of opioids with benzodiazepines or other central nervous system (CNS) depressants, including alcohol, may result in profound sedation, respiratory depression, coma, and death.
Reserve concomitant prescribing of DOLOPHINE Tablets and benzodiazepines or other CNS depressants for use in patients for whom alternatives to benzodiazepines or other CNS depressants are inadequate.
Limit dosages and durations to the minimum required for patients being treated for pain.
Follow patients for signs and symptoms of respiratory depression and sedation. If the patient is visibly sedated, evaluate the cause of sedation, and consider delaying or omitting the daily methadone dose.
PARNATE (TRANYLCYPROMINE SULFATE):
- Edited boxed warning, January 2018
WARNING: SUICIDAL THOUGHTS AND BEHAVIORS AND HYPERTENSIVE CRISIS WITH SIGNIFICANT TYRAMINE USE
SUICIDAL THOUGHTS AND BEHAVIORS
Antidepressants increased the risk of suicidal thoughts and behaviors in pediatric and young adult patients in short-term studies. Closely monitor all antidepressant-treated patients for clinical worsening, and for emergence of suicidal thoughts and behaviors. PARNATE is not approved for use in pediatric patients.
HYPERTENSIVE CRISIS WITH SIGNIFICANT TYRAMINE USE
Excessive consumption of foods or beverages with significant tyramine content or the use of certain drugs with PARNATE or after PARNATE discontinuation can precipitate hypertensive crisis. Monitor blood pressure and allow for medication-free intervals between administration of PARNATE and interacting drugs. Instruct patients to avoid ingestion of foods and beverages with high tyramine content.
OCALIVA (OBETICHOLIC ACID):
- New boxed warning/Newly added section, February 2018
WARNING: HEPATIC DECOMPENSATION AND FAILURE IN INCORRECTLY DOSED PBC PATIENTS WITH CHILD-PUGH CLASS B OR C OR DECOMPENSATED CIRRHOSIS
In postmarketing reports, hepatic decompensation and failure, in some cases fatal, have been reported in patients with primary biliary cholangitis (PBC) with decompensated cirrhosis or Child-Pugh Class B or C hepatic impairment when OCALIVA was dosed more frequently than recommended.
The recommended starting dosage of OCALIVA is 5 mg once weekly for patients with Child-Pugh Class B or C hepatic impairment or a prior decompensation event.
The FDA’s MedWatch program safety labeling changes for boxed warnings are compiled quarterly for drugs and therapeutic biologics where important changes have been made to the safety information. These and other label changes are searchable in the Drug Safety Labeling Changes (SLC) database, where data are available to the public in downloadable and searchable formats. Boxed warnings are ordinarily used to highlight either adverse reactions so serious in proportion to the potential benefi t from the drug that it is essential that it be considered in assessing the risks and benefi ts of using the drug; or serious adverse reactions that can be prevented/reduced in frequency or severity by appropriate use of the drug; or FDA approved the drug with restrictions to ensure safe use because FDA concluded that the drug can be safely used only if distribution or use is restricted. For complete FDA Drug Safety Labeling changes, please visit http://www.accessdata.fda.gov/scripts/cder/safetylabelingchanges.
VIDEX AND VIDEX EC (DIDANOSINE):
- Edited boxed warning, January 2018
WARNING: PANCREATITIS, LACTIC ACIDOSIS and HEPATOMEGALY with STEATOSIS
Coadministration of VIDEX or VIDEX EC and stavudine is contraindicated because of increased risk of serious and/or life-threatening events. Suspend treatment if clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity occurs.
ZYDELIG (IDELALISIB):
- Edited boxed warning, January 2018
WARNING: FATAL AND SERIOUS TOXICITIES: HEPATIC, SEVERE DIARRHEA, COLITIS, PNEUMONITIS, INFECTIONS, and INTESTINAL PERFORATION
Fatal and/or serious hepatotoxicity occurred in 16% to 18% of Zydelig-treated patients…
Fatal and/or serious and severe diarrhea or colitis occurred in 14% to 20% of Zydelig-treated patients…
Fatal and/or serious infections occurred in 21% to 48% of Zydelig-treated patients…
AQUAMEPHYTON (PHYTONADIONE):
- Edited boxed warning, March 2018
WARNING: HYPERSENSITIVITY REACTIONS WITH INTRAVENOUS AND INTRAMUSCULAR USE
Fatal hypersensitivity reactions, including anaphylaxis, have occurred during and immediately after INTRAVENOUS and INTRAMUSCULAR injection of Aqua-MEPHYTON. Reactions have occurred despite dilution to avoid rapid infusion and upon first dose. Avoid the intravenous and intramuscular routes of administration unless the subcutaneous route is not feasible and the serious risk is justified.
FERAHEME (FERUMOXYTOL):
- Edited boxed warning, February 2018
WARNING: RISK FOR SERIOUS HYPERSENSITIVITY/ANAPHYLAXIS REACTIONS
Fatal and serious hypersensitivity reactions including anaphylaxis have occurred in patients receiving feraheme. Initial symptoms may
include hypotension, syncope, unresponsiveness, cardiac/cardiorespiratory arrest.
Only administer feraheme as an intravenous infusion over at least 15 minutes and only when personnel and therapies are immediately available for the treatment of anaphylaxis and other hypersensitivity reactions.
METHADONE HYDROCHLORIDE, METHADOSE (METHADONE HYDROCHLORIDE):
- Edited boxed warning, February 2018
RISKS FROM CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS
Concomitant use with benzodiazepines or other central nervous system (CNS) depressants, including alcohol, is a risk factor for respiratory depression and death.
Reserve concomitant prescribing of benzodiazepines or other CNS depressants for use in patients for whom alternatives to benzodiazepines or other CNS depressants are inadequate.
Follow patients for signs and symptoms of respiratory depression and sedation. If the patient is visibly sedated, evaluate the cause of sedation, and consider delaying or omitting daily methadone dosing.
DOLOPHINE HYDROCHLORIDE (METHADONE HYDROCHLORIDE):
- Edited boxed warning, February 2018
RISKS FROM CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS
Concomitant use of opioids with benzodiazepines or other central nervous system (CNS) depressants, including alcohol, may result in profound sedation, respiratory depression, coma, and death.
Reserve concomitant prescribing of DOLOPHINE Tablets and benzodiazepines or other CNS depressants for use in patients for whom alternatives to benzodiazepines or other CNS depressants are inadequate.
Limit dosages and durations to the minimum required for patients being treated for pain.
Follow patients for signs and symptoms of respiratory depression and sedation. If the patient is visibly sedated, evaluate the cause of sedation, and consider delaying or omitting the daily methadone dose.
PARNATE (TRANYLCYPROMINE SULFATE):
- Edited boxed warning, January 2018
WARNING: SUICIDAL THOUGHTS AND BEHAVIORS AND HYPERTENSIVE CRISIS WITH SIGNIFICANT TYRAMINE USE
SUICIDAL THOUGHTS AND BEHAVIORS
Antidepressants increased the risk of suicidal thoughts and behaviors in pediatric and young adult patients in short-term studies. Closely monitor all antidepressant-treated patients for clinical worsening, and for emergence of suicidal thoughts and behaviors. PARNATE is not approved for use in pediatric patients.
HYPERTENSIVE CRISIS WITH SIGNIFICANT TYRAMINE USE
Excessive consumption of foods or beverages with significant tyramine content or the use of certain drugs with PARNATE or after PARNATE discontinuation can precipitate hypertensive crisis. Monitor blood pressure and allow for medication-free intervals between administration of PARNATE and interacting drugs. Instruct patients to avoid ingestion of foods and beverages with high tyramine content.
OCALIVA (OBETICHOLIC ACID):
- New boxed warning/Newly added section, February 2018
WARNING: HEPATIC DECOMPENSATION AND FAILURE IN INCORRECTLY DOSED PBC PATIENTS WITH CHILD-PUGH CLASS B OR C OR DECOMPENSATED CIRRHOSIS
In postmarketing reports, hepatic decompensation and failure, in some cases fatal, have been reported in patients with primary biliary cholangitis (PBC) with decompensated cirrhosis or Child-Pugh Class B or C hepatic impairment when OCALIVA was dosed more frequently than recommended.
The recommended starting dosage of OCALIVA is 5 mg once weekly for patients with Child-Pugh Class B or C hepatic impairment or a prior decompensation event.