User login
GLOW: Ibrutinib+venetoclax shines in first line for CLL/SLL
For older, unfit patients with chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), first-line treatment with the all-oral combination of ibrutinib (Imbruvica) and venetoclax (Venclexta) was associated with superior progression-free survival, compared with chlorambucil and obinutuzumab (Gazyva), results of the phase 3 GLOW trial showed.
Among 211 patients with CLL/SLL who were 65 or older, or younger patients with a high comorbidity burden, the median progression-free survival (PFS) after 27.7 months of follow-up was not reached for patients treated with a fixed duration combination of ibrutinib plus venetoclax (I+V), compared with 21 months for patients who received chlorambucil plus obinutuzumab (Clb+O), reported Arnon P. Kater, MD, PhD, from Amsterdam University Medical Centers.
The oral combination was also associated with a higher rate of undetectable minimal residual disease (MRD) 3 months after the end of treatment, at 51.9% vs. 17.1% with Clb+O, he said in a late-breaking abstract presented during the European Hematology Association annual meeting (Abstract LB1902).
“Overall, the results from GLOW support a positive clinical profile of I+V as an all-oral, once-daily, fixed-duration regimen in older patients with newly diagnosed CLL,” he said.
A low bar
But the bar for success in the GLOW trial may have been set low, with the older combination of chlorambucil plus obinutuzumab used as the comparator, rather than a more contemporary regimen, said Clive S. Zent , MD, from the Wilmot Cancer Institute at the University of Rochester (New York) Medical Center, who was not involved in the study.
“Really, nobody in this country that I’m aware of, certainly not in academic medicine, would be using chlorambucil and obinutuzumab or rituximab for standard of care,” Dr. Zent said in an interview.
“This is like comparing a mule cart to a Tesla,” he said.
Apart from possibly providing a rationale for using ibrutinib and venetoclax in this population, the GLOW results do not add much beyond that already in the CAPTIVE MRD trial, he added.
“What we’re really interested in, is ibrutinib and venetoclax better than ibrutinib alone or venetoclax alone? And that has not been asked or answered yet,” he said.
Dr. Zent acknowledged that the combination works very well and is well tolerated, and the idea of fixed duration therapy is attractive, as are the long-term outcomes for many patients following cessation of therapy.
“But remember, if you take ibrutinib, you’ve got a 90% chance of going into remission, and an over 80% chance of being in remission 5 years later, so that’s pretty good as well,” he said.
Paolo P. Ghia, MD, from Univerista Vita-Salute San Raffaele in Milan, who has studied fixed-duration I+V in younger patients with CLL in the CAPTIVATE trial agreed that “in general, there is very little role for chemoimmunotherapy in CLL.”
But Dr. Ghia, who was not involved in the GLOW study, said in an interview that the results add to the growing body of evidence of the efficacy and safety of ibrutinib -venetoclax in a wide range of patients.
“Overall between the two studies [CAPTIVATE and GLOW] we have now over 400 patients who have been treated with the combination, and the message is rather similar in the two studies: you have a high frequency of undetectable MRD in peripheral blood and in the bone marrow, with a high concordance between the two tissues, and in particular we have a durability of the response,” he said.
GLOW details
Dr. Kater noted that ibrutinib and venetoclax have distinct and complementary modes of action, with ibrutinib mobilizing CLL cells out of their “protective lymphoid niches” and inhibiting their proliferation, as well as accelerating apoptosis by sensitizing cells to inhibition by the anti–B-cell lymphoma 2 (BCL-2) agent venetoclax. The combination leads to high levels of MRD negative by eliminating subpopulations of resting and dividing CLL cells.
The GLOW investigators enrolled 211 patients who were 65 or older, or were younger than 65 with a cumulative illness rating scale (CIRS) score of greater than 6, or creatinine clearance rate of less than 70 mL/min, and no known deletion 17p (del17p) or TP53 mutation.
The patients all had Eastern Cooperative Oncology Group performance status scores of 0-2.
After stratification by immunoglobulin heavy chain variable (IGHV) region genes and presence of deletion 11q (del11q), the patients were randomly assigned to either a three cycle run-in with ibrutinib 420 mg daily followed by ibrutinib plus venetoclax ramped up from 20 to 400 mg, or to chlorambucil 0.5 mg/kg on days and 15 for six cycles, and obinutuzumab 1,000 mg on days 1,2, 8, and 15 of cycle 1, and day 1 of cycles 2-6.
About one-third of patients in each study arm were 75 or older. Baseline characteristics were generally similar between the arms, except for a higher frequency of CIRS scores above 6 in the I+V arm, and a higher frequency of elevated lactate dehydrogenase in the Clb+O arm.
Superior PFS
As noted, the primary endpoint of PFS as assessed by independent review committee (IRC) after 27.7 months of follow-up had not been reached in I+V arm, compared with 21 months in the Clb+O arm, translating into a hazard ratio or progression with I+V of 0.21 (P < .0001). Investigator-assessed PFS was similar, with an HR of 2.07 (P < .0001).
PFS was superior with I+V across all subgroups, including age, baseline performance status. CIRS total score, Rai stage, bulky disease, elevated LDH at baseline, IGHV mutated or unmutated, and presence or absence of del(11q).
IRC-assessed combined complete response (CR) or CR with incomplete recovery of blood counts (CRi) rates were 38.7% with I+V vs. 11.4% with Clb+O (P < .0001).
Responses were also more durable with the oral combination, with 90% of patients having a sustained IRC-assessed response at 24 months, compared with 41% in the chemoimmunotherapy arm.
Rates of undetectable MRD by next-generation sequencing 3 months after the end of treatment were also significantly higher with I+V in both bone marrow (51.9% vs. 17.1%, respectively, P < .0001) and peripheral blood (54.7% vs. 39%, P = .0259).
One year post treatment 49% of patients assigned to I+V had undetectable MRD in peripheral blood, compared with 12% of patients assigned to Clb+O).
Safety
In all, 11 patients assigned to I+V discontinued treatment because of adverse events, compared with 2 in the Clb+O arm. Two patients in the I+V arm (1.9%) discontinued ibrutinib because of atrial fibrillation (AF). Serious adverse events in 5% or more of patients that were more frequent with I+V include infections (12.3% vs. 8.6% and AF (6.6% vs. o%). The tumor lysis syndrome (TLS) was not seen in the I+V arm, but occurred in 5.7% of patients in the Clb+O arm.
There were a total of 11 deaths in the I+V arm and 12 in the Clb+O arm during treatment or follow-up.
Causes of death were generally similar between the arms, with infections and cardiac events being the most common causes, Dr. Kater said.
Of the four deaths that occurred during ibrutinib lead-in, one was due to infection, one to metastatic carcinoma, and two due to cardiac disorders. Of the three that occurred in the I+V arm during treatment, two were from sudden death, and one from a nervous system disorder. Four patients in this arm died during follow-up, two from infections, one from sudden death, and one from progressive disease with Richter transformation.
In the Clb+O arm, one patient died during treatment from an infection and one died from hepatobiliary disease. Of the 10 that died during follow-up, 6 died from infections/infestations, 2 from cardiac disorders, and 1 each from nervous system and respiratory/thoracic/mediastinal disorder.
The study was supported by Janssen Research & Development. Dr. Kater disclosed advisory board activity, research committee, and steering committee participation for Janssen, and similar relationships with others. Dr. Zent disclosed research funding to the University of Rochester from AstraZenca/Acerta and TG Therpeutics. Dr. Ghia disclosed consultancy, honoraria, travel expenses, and research funding from Janssen and others.
For older, unfit patients with chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), first-line treatment with the all-oral combination of ibrutinib (Imbruvica) and venetoclax (Venclexta) was associated with superior progression-free survival, compared with chlorambucil and obinutuzumab (Gazyva), results of the phase 3 GLOW trial showed.
Among 211 patients with CLL/SLL who were 65 or older, or younger patients with a high comorbidity burden, the median progression-free survival (PFS) after 27.7 months of follow-up was not reached for patients treated with a fixed duration combination of ibrutinib plus venetoclax (I+V), compared with 21 months for patients who received chlorambucil plus obinutuzumab (Clb+O), reported Arnon P. Kater, MD, PhD, from Amsterdam University Medical Centers.
The oral combination was also associated with a higher rate of undetectable minimal residual disease (MRD) 3 months after the end of treatment, at 51.9% vs. 17.1% with Clb+O, he said in a late-breaking abstract presented during the European Hematology Association annual meeting (Abstract LB1902).
“Overall, the results from GLOW support a positive clinical profile of I+V as an all-oral, once-daily, fixed-duration regimen in older patients with newly diagnosed CLL,” he said.
A low bar
But the bar for success in the GLOW trial may have been set low, with the older combination of chlorambucil plus obinutuzumab used as the comparator, rather than a more contemporary regimen, said Clive S. Zent , MD, from the Wilmot Cancer Institute at the University of Rochester (New York) Medical Center, who was not involved in the study.
“Really, nobody in this country that I’m aware of, certainly not in academic medicine, would be using chlorambucil and obinutuzumab or rituximab for standard of care,” Dr. Zent said in an interview.
“This is like comparing a mule cart to a Tesla,” he said.
Apart from possibly providing a rationale for using ibrutinib and venetoclax in this population, the GLOW results do not add much beyond that already in the CAPTIVE MRD trial, he added.
“What we’re really interested in, is ibrutinib and venetoclax better than ibrutinib alone or venetoclax alone? And that has not been asked or answered yet,” he said.
Dr. Zent acknowledged that the combination works very well and is well tolerated, and the idea of fixed duration therapy is attractive, as are the long-term outcomes for many patients following cessation of therapy.
“But remember, if you take ibrutinib, you’ve got a 90% chance of going into remission, and an over 80% chance of being in remission 5 years later, so that’s pretty good as well,” he said.
Paolo P. Ghia, MD, from Univerista Vita-Salute San Raffaele in Milan, who has studied fixed-duration I+V in younger patients with CLL in the CAPTIVATE trial agreed that “in general, there is very little role for chemoimmunotherapy in CLL.”
But Dr. Ghia, who was not involved in the GLOW study, said in an interview that the results add to the growing body of evidence of the efficacy and safety of ibrutinib -venetoclax in a wide range of patients.
“Overall between the two studies [CAPTIVATE and GLOW] we have now over 400 patients who have been treated with the combination, and the message is rather similar in the two studies: you have a high frequency of undetectable MRD in peripheral blood and in the bone marrow, with a high concordance between the two tissues, and in particular we have a durability of the response,” he said.
GLOW details
Dr. Kater noted that ibrutinib and venetoclax have distinct and complementary modes of action, with ibrutinib mobilizing CLL cells out of their “protective lymphoid niches” and inhibiting their proliferation, as well as accelerating apoptosis by sensitizing cells to inhibition by the anti–B-cell lymphoma 2 (BCL-2) agent venetoclax. The combination leads to high levels of MRD negative by eliminating subpopulations of resting and dividing CLL cells.
The GLOW investigators enrolled 211 patients who were 65 or older, or were younger than 65 with a cumulative illness rating scale (CIRS) score of greater than 6, or creatinine clearance rate of less than 70 mL/min, and no known deletion 17p (del17p) or TP53 mutation.
The patients all had Eastern Cooperative Oncology Group performance status scores of 0-2.
After stratification by immunoglobulin heavy chain variable (IGHV) region genes and presence of deletion 11q (del11q), the patients were randomly assigned to either a three cycle run-in with ibrutinib 420 mg daily followed by ibrutinib plus venetoclax ramped up from 20 to 400 mg, or to chlorambucil 0.5 mg/kg on days and 15 for six cycles, and obinutuzumab 1,000 mg on days 1,2, 8, and 15 of cycle 1, and day 1 of cycles 2-6.
About one-third of patients in each study arm were 75 or older. Baseline characteristics were generally similar between the arms, except for a higher frequency of CIRS scores above 6 in the I+V arm, and a higher frequency of elevated lactate dehydrogenase in the Clb+O arm.
Superior PFS
As noted, the primary endpoint of PFS as assessed by independent review committee (IRC) after 27.7 months of follow-up had not been reached in I+V arm, compared with 21 months in the Clb+O arm, translating into a hazard ratio or progression with I+V of 0.21 (P < .0001). Investigator-assessed PFS was similar, with an HR of 2.07 (P < .0001).
PFS was superior with I+V across all subgroups, including age, baseline performance status. CIRS total score, Rai stage, bulky disease, elevated LDH at baseline, IGHV mutated or unmutated, and presence or absence of del(11q).
IRC-assessed combined complete response (CR) or CR with incomplete recovery of blood counts (CRi) rates were 38.7% with I+V vs. 11.4% with Clb+O (P < .0001).
Responses were also more durable with the oral combination, with 90% of patients having a sustained IRC-assessed response at 24 months, compared with 41% in the chemoimmunotherapy arm.
Rates of undetectable MRD by next-generation sequencing 3 months after the end of treatment were also significantly higher with I+V in both bone marrow (51.9% vs. 17.1%, respectively, P < .0001) and peripheral blood (54.7% vs. 39%, P = .0259).
One year post treatment 49% of patients assigned to I+V had undetectable MRD in peripheral blood, compared with 12% of patients assigned to Clb+O).
Safety
In all, 11 patients assigned to I+V discontinued treatment because of adverse events, compared with 2 in the Clb+O arm. Two patients in the I+V arm (1.9%) discontinued ibrutinib because of atrial fibrillation (AF). Serious adverse events in 5% or more of patients that were more frequent with I+V include infections (12.3% vs. 8.6% and AF (6.6% vs. o%). The tumor lysis syndrome (TLS) was not seen in the I+V arm, but occurred in 5.7% of patients in the Clb+O arm.
There were a total of 11 deaths in the I+V arm and 12 in the Clb+O arm during treatment or follow-up.
Causes of death were generally similar between the arms, with infections and cardiac events being the most common causes, Dr. Kater said.
Of the four deaths that occurred during ibrutinib lead-in, one was due to infection, one to metastatic carcinoma, and two due to cardiac disorders. Of the three that occurred in the I+V arm during treatment, two were from sudden death, and one from a nervous system disorder. Four patients in this arm died during follow-up, two from infections, one from sudden death, and one from progressive disease with Richter transformation.
In the Clb+O arm, one patient died during treatment from an infection and one died from hepatobiliary disease. Of the 10 that died during follow-up, 6 died from infections/infestations, 2 from cardiac disorders, and 1 each from nervous system and respiratory/thoracic/mediastinal disorder.
The study was supported by Janssen Research & Development. Dr. Kater disclosed advisory board activity, research committee, and steering committee participation for Janssen, and similar relationships with others. Dr. Zent disclosed research funding to the University of Rochester from AstraZenca/Acerta and TG Therpeutics. Dr. Ghia disclosed consultancy, honoraria, travel expenses, and research funding from Janssen and others.
For older, unfit patients with chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), first-line treatment with the all-oral combination of ibrutinib (Imbruvica) and venetoclax (Venclexta) was associated with superior progression-free survival, compared with chlorambucil and obinutuzumab (Gazyva), results of the phase 3 GLOW trial showed.
Among 211 patients with CLL/SLL who were 65 or older, or younger patients with a high comorbidity burden, the median progression-free survival (PFS) after 27.7 months of follow-up was not reached for patients treated with a fixed duration combination of ibrutinib plus venetoclax (I+V), compared with 21 months for patients who received chlorambucil plus obinutuzumab (Clb+O), reported Arnon P. Kater, MD, PhD, from Amsterdam University Medical Centers.
The oral combination was also associated with a higher rate of undetectable minimal residual disease (MRD) 3 months after the end of treatment, at 51.9% vs. 17.1% with Clb+O, he said in a late-breaking abstract presented during the European Hematology Association annual meeting (Abstract LB1902).
“Overall, the results from GLOW support a positive clinical profile of I+V as an all-oral, once-daily, fixed-duration regimen in older patients with newly diagnosed CLL,” he said.
A low bar
But the bar for success in the GLOW trial may have been set low, with the older combination of chlorambucil plus obinutuzumab used as the comparator, rather than a more contemporary regimen, said Clive S. Zent , MD, from the Wilmot Cancer Institute at the University of Rochester (New York) Medical Center, who was not involved in the study.
“Really, nobody in this country that I’m aware of, certainly not in academic medicine, would be using chlorambucil and obinutuzumab or rituximab for standard of care,” Dr. Zent said in an interview.
“This is like comparing a mule cart to a Tesla,” he said.
Apart from possibly providing a rationale for using ibrutinib and venetoclax in this population, the GLOW results do not add much beyond that already in the CAPTIVE MRD trial, he added.
“What we’re really interested in, is ibrutinib and venetoclax better than ibrutinib alone or venetoclax alone? And that has not been asked or answered yet,” he said.
Dr. Zent acknowledged that the combination works very well and is well tolerated, and the idea of fixed duration therapy is attractive, as are the long-term outcomes for many patients following cessation of therapy.
“But remember, if you take ibrutinib, you’ve got a 90% chance of going into remission, and an over 80% chance of being in remission 5 years later, so that’s pretty good as well,” he said.
Paolo P. Ghia, MD, from Univerista Vita-Salute San Raffaele in Milan, who has studied fixed-duration I+V in younger patients with CLL in the CAPTIVATE trial agreed that “in general, there is very little role for chemoimmunotherapy in CLL.”
But Dr. Ghia, who was not involved in the GLOW study, said in an interview that the results add to the growing body of evidence of the efficacy and safety of ibrutinib -venetoclax in a wide range of patients.
“Overall between the two studies [CAPTIVATE and GLOW] we have now over 400 patients who have been treated with the combination, and the message is rather similar in the two studies: you have a high frequency of undetectable MRD in peripheral blood and in the bone marrow, with a high concordance between the two tissues, and in particular we have a durability of the response,” he said.
GLOW details
Dr. Kater noted that ibrutinib and venetoclax have distinct and complementary modes of action, with ibrutinib mobilizing CLL cells out of their “protective lymphoid niches” and inhibiting their proliferation, as well as accelerating apoptosis by sensitizing cells to inhibition by the anti–B-cell lymphoma 2 (BCL-2) agent venetoclax. The combination leads to high levels of MRD negative by eliminating subpopulations of resting and dividing CLL cells.
The GLOW investigators enrolled 211 patients who were 65 or older, or were younger than 65 with a cumulative illness rating scale (CIRS) score of greater than 6, or creatinine clearance rate of less than 70 mL/min, and no known deletion 17p (del17p) or TP53 mutation.
The patients all had Eastern Cooperative Oncology Group performance status scores of 0-2.
After stratification by immunoglobulin heavy chain variable (IGHV) region genes and presence of deletion 11q (del11q), the patients were randomly assigned to either a three cycle run-in with ibrutinib 420 mg daily followed by ibrutinib plus venetoclax ramped up from 20 to 400 mg, or to chlorambucil 0.5 mg/kg on days and 15 for six cycles, and obinutuzumab 1,000 mg on days 1,2, 8, and 15 of cycle 1, and day 1 of cycles 2-6.
About one-third of patients in each study arm were 75 or older. Baseline characteristics were generally similar between the arms, except for a higher frequency of CIRS scores above 6 in the I+V arm, and a higher frequency of elevated lactate dehydrogenase in the Clb+O arm.
Superior PFS
As noted, the primary endpoint of PFS as assessed by independent review committee (IRC) after 27.7 months of follow-up had not been reached in I+V arm, compared with 21 months in the Clb+O arm, translating into a hazard ratio or progression with I+V of 0.21 (P < .0001). Investigator-assessed PFS was similar, with an HR of 2.07 (P < .0001).
PFS was superior with I+V across all subgroups, including age, baseline performance status. CIRS total score, Rai stage, bulky disease, elevated LDH at baseline, IGHV mutated or unmutated, and presence or absence of del(11q).
IRC-assessed combined complete response (CR) or CR with incomplete recovery of blood counts (CRi) rates were 38.7% with I+V vs. 11.4% with Clb+O (P < .0001).
Responses were also more durable with the oral combination, with 90% of patients having a sustained IRC-assessed response at 24 months, compared with 41% in the chemoimmunotherapy arm.
Rates of undetectable MRD by next-generation sequencing 3 months after the end of treatment were also significantly higher with I+V in both bone marrow (51.9% vs. 17.1%, respectively, P < .0001) and peripheral blood (54.7% vs. 39%, P = .0259).
One year post treatment 49% of patients assigned to I+V had undetectable MRD in peripheral blood, compared with 12% of patients assigned to Clb+O).
Safety
In all, 11 patients assigned to I+V discontinued treatment because of adverse events, compared with 2 in the Clb+O arm. Two patients in the I+V arm (1.9%) discontinued ibrutinib because of atrial fibrillation (AF). Serious adverse events in 5% or more of patients that were more frequent with I+V include infections (12.3% vs. 8.6% and AF (6.6% vs. o%). The tumor lysis syndrome (TLS) was not seen in the I+V arm, but occurred in 5.7% of patients in the Clb+O arm.
There were a total of 11 deaths in the I+V arm and 12 in the Clb+O arm during treatment or follow-up.
Causes of death were generally similar between the arms, with infections and cardiac events being the most common causes, Dr. Kater said.
Of the four deaths that occurred during ibrutinib lead-in, one was due to infection, one to metastatic carcinoma, and two due to cardiac disorders. Of the three that occurred in the I+V arm during treatment, two were from sudden death, and one from a nervous system disorder. Four patients in this arm died during follow-up, two from infections, one from sudden death, and one from progressive disease with Richter transformation.
In the Clb+O arm, one patient died during treatment from an infection and one died from hepatobiliary disease. Of the 10 that died during follow-up, 6 died from infections/infestations, 2 from cardiac disorders, and 1 each from nervous system and respiratory/thoracic/mediastinal disorder.
The study was supported by Janssen Research & Development. Dr. Kater disclosed advisory board activity, research committee, and steering committee participation for Janssen, and similar relationships with others. Dr. Zent disclosed research funding to the University of Rochester from AstraZenca/Acerta and TG Therpeutics. Dr. Ghia disclosed consultancy, honoraria, travel expenses, and research funding from Janssen and others.
FROM EHA 2021
Choosing the right R-CHOP dosage for elderly patients with DLBCL
Physicians often face the choice of whether to treat elderly patients with diffuse large B-cell lymphoma (DLBCL) with a full or reduced dose intensity (DI) of R-CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisolone + rituximab), according to Edward J. Bataillard of the Imperial College Healthcare National Health Service Trust, London, and colleagues.
To address this issue, the researchers conducted a systematic review assessing the impact of R-CHOP DI on DLBCL survival outcomes, according to Preferred Reporting Items for Systematic Reviews and Meta-Analyses for Protocols (PRISMA-P) guidelines. They found that greater than 80 years of age is an important cutoff for treating patients with a reduced R-CHOP dosage, according to their results, published in Blood Advances (2021;5[9]:2426-37).
Cutoff at 80 years of age
Their final review comprised 13 studies including 5,188 patients. Overall, the lower DI (intended or relative) was associated with inferior survival in seven of nine studies reporting crude survival analyses. In addition, most studies and those larger studies of higher quality showed poorer outcomes associated with reduced R-CHOP DI.
However, in subgroups of patients aged 80 years or more, survival was not consistently affected by the use of lower dosage R-CHOP, according to the researchers.
“We found evidence of improved survival with higher RDIs (up to R-CHOP-21) in those aged < 80 years, but the literature to date does not support full-dose intensity in those 80 years [or older],” they stated.
However, the researchers concluded that: “In the absence of improved options beyond R-CHOP in DLBCL over the past 20 years, prospective studies of DI are warranted, despite the recognized challenges involved.”
Two of the authors reported being previously employed by Roche. A third served as a consultant and adviser and received honoraria from Roche and other pharmaceutical companies. Several authors reported disclosures related to multiple other pharmaceutical companies.
Physicians often face the choice of whether to treat elderly patients with diffuse large B-cell lymphoma (DLBCL) with a full or reduced dose intensity (DI) of R-CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisolone + rituximab), according to Edward J. Bataillard of the Imperial College Healthcare National Health Service Trust, London, and colleagues.
To address this issue, the researchers conducted a systematic review assessing the impact of R-CHOP DI on DLBCL survival outcomes, according to Preferred Reporting Items for Systematic Reviews and Meta-Analyses for Protocols (PRISMA-P) guidelines. They found that greater than 80 years of age is an important cutoff for treating patients with a reduced R-CHOP dosage, according to their results, published in Blood Advances (2021;5[9]:2426-37).
Cutoff at 80 years of age
Their final review comprised 13 studies including 5,188 patients. Overall, the lower DI (intended or relative) was associated with inferior survival in seven of nine studies reporting crude survival analyses. In addition, most studies and those larger studies of higher quality showed poorer outcomes associated with reduced R-CHOP DI.
However, in subgroups of patients aged 80 years or more, survival was not consistently affected by the use of lower dosage R-CHOP, according to the researchers.
“We found evidence of improved survival with higher RDIs (up to R-CHOP-21) in those aged < 80 years, but the literature to date does not support full-dose intensity in those 80 years [or older],” they stated.
However, the researchers concluded that: “In the absence of improved options beyond R-CHOP in DLBCL over the past 20 years, prospective studies of DI are warranted, despite the recognized challenges involved.”
Two of the authors reported being previously employed by Roche. A third served as a consultant and adviser and received honoraria from Roche and other pharmaceutical companies. Several authors reported disclosures related to multiple other pharmaceutical companies.
Physicians often face the choice of whether to treat elderly patients with diffuse large B-cell lymphoma (DLBCL) with a full or reduced dose intensity (DI) of R-CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisolone + rituximab), according to Edward J. Bataillard of the Imperial College Healthcare National Health Service Trust, London, and colleagues.
To address this issue, the researchers conducted a systematic review assessing the impact of R-CHOP DI on DLBCL survival outcomes, according to Preferred Reporting Items for Systematic Reviews and Meta-Analyses for Protocols (PRISMA-P) guidelines. They found that greater than 80 years of age is an important cutoff for treating patients with a reduced R-CHOP dosage, according to their results, published in Blood Advances (2021;5[9]:2426-37).
Cutoff at 80 years of age
Their final review comprised 13 studies including 5,188 patients. Overall, the lower DI (intended or relative) was associated with inferior survival in seven of nine studies reporting crude survival analyses. In addition, most studies and those larger studies of higher quality showed poorer outcomes associated with reduced R-CHOP DI.
However, in subgroups of patients aged 80 years or more, survival was not consistently affected by the use of lower dosage R-CHOP, according to the researchers.
“We found evidence of improved survival with higher RDIs (up to R-CHOP-21) in those aged < 80 years, but the literature to date does not support full-dose intensity in those 80 years [or older],” they stated.
However, the researchers concluded that: “In the absence of improved options beyond R-CHOP in DLBCL over the past 20 years, prospective studies of DI are warranted, despite the recognized challenges involved.”
Two of the authors reported being previously employed by Roche. A third served as a consultant and adviser and received honoraria from Roche and other pharmaceutical companies. Several authors reported disclosures related to multiple other pharmaceutical companies.
FROM BLOOD ADVANCES
Patients with CLL have significantly reduced response to COVID-19 vaccine
Patients with chronic lymphocytic leukemia (CLL) have increased risk for severe COVID-19 disease as well as mortality.
Such patients are likely to have compromised immune systems, making them respond poorly to vaccines, as has been seen in studies involving pneumococcal, hepatitis B, and influenza A and B vaccination.
In order to determine if vaccination against COVID-19 disease will be effective among these patients, researchers performed a study to determine the efficacy of a single COVID-19 vaccine in patients with CLL. They found that the response rate of patients with CLL to vaccination was significantly lower than that of healthy controls, according to the study published in Blood Advances.
Study details
The study (NCT04746092) assessed the humoral immune responses to BNT162b2 mRNA COVID-19 (Pfizer) vaccination in adult patients with CLL and compared responses with those obtained in age-matched healthy controls. Patients received two vaccine doses, 21 days apart, and antibody titers were measured 2-3 weeks after administration of the second dose, according to Yair Herishanu, MD, of the Tel-Aviv Sourasky Medical Center, Tel Aviv University, and colleagues.
Troubling results
The researchers found an antibody-mediated response to the BNT162b2 mRNA COVID-19 vaccine in only 66 of 167 (39.5%) of all patients with CLL. The response rate of 52 of these responding patients with CLL to the vaccine was significantly lower than that occurring in 52 age- and sex-matched healthy controls (52% vs. 100%, respectively; adjusted odds ratio, 0.010; 95% confidence interval, 0.001-0.162; P < .001).
Among the patients with CLL, the response rate was highest in those who obtained clinical remission after treatment (79.2%), followed by 55.2% in treatment-naive patients, and it was only 16% in patients under treatment at the time of vaccination.
In patients treated with either BTK inhibitors or venetoclax with and without anti-CD20 antibody, response rates were low (16.0% and 13.6%, respectively). In particular, none of the patients exposed to anti-CD20 antibodies less than 12 months prior to vaccination responded, according to the researchers.
Multivariate analysis showed that the independent predictors of a vaccine response were age (65 years or younger; odds ratio, 3.17; P = .025), sex (women; OR, 3.66; P = .006), lack of active therapy (including treatment naive and previously treated patients; OR 6.59; P < .001), IgG levels 550 mg/dL or greater (OR, 3.70; P = .037), and IgM levels 40mg/dL or greater (OR, 2.92; P = .017).
Within a median follow-up period of 75 days since the first vaccine dose, none of the CLL patients developed COVID-19 infection, the researchers reported.
“Vaccinated patients with CLL should continue to adhere to masking, social distancing, and vaccination of their close contacts should be strongly recommended. Serological tests after the second injection of the COVID-19 vaccine can provide valuable information to the individual patient and perhaps, may be integrated in future clinical decisions,” the researchers concluded.
The study was sponsored by the Tel-Aviv Sourasky Medical Center. The authors reported that they had no conflicts of interest.
Patients with chronic lymphocytic leukemia (CLL) have increased risk for severe COVID-19 disease as well as mortality.
Such patients are likely to have compromised immune systems, making them respond poorly to vaccines, as has been seen in studies involving pneumococcal, hepatitis B, and influenza A and B vaccination.
In order to determine if vaccination against COVID-19 disease will be effective among these patients, researchers performed a study to determine the efficacy of a single COVID-19 vaccine in patients with CLL. They found that the response rate of patients with CLL to vaccination was significantly lower than that of healthy controls, according to the study published in Blood Advances.
Study details
The study (NCT04746092) assessed the humoral immune responses to BNT162b2 mRNA COVID-19 (Pfizer) vaccination in adult patients with CLL and compared responses with those obtained in age-matched healthy controls. Patients received two vaccine doses, 21 days apart, and antibody titers were measured 2-3 weeks after administration of the second dose, according to Yair Herishanu, MD, of the Tel-Aviv Sourasky Medical Center, Tel Aviv University, and colleagues.
Troubling results
The researchers found an antibody-mediated response to the BNT162b2 mRNA COVID-19 vaccine in only 66 of 167 (39.5%) of all patients with CLL. The response rate of 52 of these responding patients with CLL to the vaccine was significantly lower than that occurring in 52 age- and sex-matched healthy controls (52% vs. 100%, respectively; adjusted odds ratio, 0.010; 95% confidence interval, 0.001-0.162; P < .001).
Among the patients with CLL, the response rate was highest in those who obtained clinical remission after treatment (79.2%), followed by 55.2% in treatment-naive patients, and it was only 16% in patients under treatment at the time of vaccination.
In patients treated with either BTK inhibitors or venetoclax with and without anti-CD20 antibody, response rates were low (16.0% and 13.6%, respectively). In particular, none of the patients exposed to anti-CD20 antibodies less than 12 months prior to vaccination responded, according to the researchers.
Multivariate analysis showed that the independent predictors of a vaccine response were age (65 years or younger; odds ratio, 3.17; P = .025), sex (women; OR, 3.66; P = .006), lack of active therapy (including treatment naive and previously treated patients; OR 6.59; P < .001), IgG levels 550 mg/dL or greater (OR, 3.70; P = .037), and IgM levels 40mg/dL or greater (OR, 2.92; P = .017).
Within a median follow-up period of 75 days since the first vaccine dose, none of the CLL patients developed COVID-19 infection, the researchers reported.
“Vaccinated patients with CLL should continue to adhere to masking, social distancing, and vaccination of their close contacts should be strongly recommended. Serological tests after the second injection of the COVID-19 vaccine can provide valuable information to the individual patient and perhaps, may be integrated in future clinical decisions,” the researchers concluded.
The study was sponsored by the Tel-Aviv Sourasky Medical Center. The authors reported that they had no conflicts of interest.
Patients with chronic lymphocytic leukemia (CLL) have increased risk for severe COVID-19 disease as well as mortality.
Such patients are likely to have compromised immune systems, making them respond poorly to vaccines, as has been seen in studies involving pneumococcal, hepatitis B, and influenza A and B vaccination.
In order to determine if vaccination against COVID-19 disease will be effective among these patients, researchers performed a study to determine the efficacy of a single COVID-19 vaccine in patients with CLL. They found that the response rate of patients with CLL to vaccination was significantly lower than that of healthy controls, according to the study published in Blood Advances.
Study details
The study (NCT04746092) assessed the humoral immune responses to BNT162b2 mRNA COVID-19 (Pfizer) vaccination in adult patients with CLL and compared responses with those obtained in age-matched healthy controls. Patients received two vaccine doses, 21 days apart, and antibody titers were measured 2-3 weeks after administration of the second dose, according to Yair Herishanu, MD, of the Tel-Aviv Sourasky Medical Center, Tel Aviv University, and colleagues.
Troubling results
The researchers found an antibody-mediated response to the BNT162b2 mRNA COVID-19 vaccine in only 66 of 167 (39.5%) of all patients with CLL. The response rate of 52 of these responding patients with CLL to the vaccine was significantly lower than that occurring in 52 age- and sex-matched healthy controls (52% vs. 100%, respectively; adjusted odds ratio, 0.010; 95% confidence interval, 0.001-0.162; P < .001).
Among the patients with CLL, the response rate was highest in those who obtained clinical remission after treatment (79.2%), followed by 55.2% in treatment-naive patients, and it was only 16% in patients under treatment at the time of vaccination.
In patients treated with either BTK inhibitors or venetoclax with and without anti-CD20 antibody, response rates were low (16.0% and 13.6%, respectively). In particular, none of the patients exposed to anti-CD20 antibodies less than 12 months prior to vaccination responded, according to the researchers.
Multivariate analysis showed that the independent predictors of a vaccine response were age (65 years or younger; odds ratio, 3.17; P = .025), sex (women; OR, 3.66; P = .006), lack of active therapy (including treatment naive and previously treated patients; OR 6.59; P < .001), IgG levels 550 mg/dL or greater (OR, 3.70; P = .037), and IgM levels 40mg/dL or greater (OR, 2.92; P = .017).
Within a median follow-up period of 75 days since the first vaccine dose, none of the CLL patients developed COVID-19 infection, the researchers reported.
“Vaccinated patients with CLL should continue to adhere to masking, social distancing, and vaccination of their close contacts should be strongly recommended. Serological tests after the second injection of the COVID-19 vaccine can provide valuable information to the individual patient and perhaps, may be integrated in future clinical decisions,” the researchers concluded.
The study was sponsored by the Tel-Aviv Sourasky Medical Center. The authors reported that they had no conflicts of interest.
FROM BLOOD ADVANCES
GENUINE improvements: Ublituximab plus ibrutinib for CLL
Chronic lymphocytic leukemia (CLL) is a clinically heterogeneous disease associated with several known genetic abnormalities, including 17p deletion (del[17p]), 11q deletion (del[11q]), and TP53 gene mutations, which are adverse prognostic markers among patients treated with chemoimmunotherapy.
The Bruton tyrosine kinase (BTK) inhibitor ibrutinib is approved for patients with untreated, relapsed, or refractory disease, including those with del(17p). Clinicians will soon have the chance to pair it with ublituximab, a next-generation, glycoengineered, type I, anti-CD20 monoclonal antibody that binds to a unique epitope on CD20, differentiating it from rituximab, ofatumumab, and obinutuzumab. Results from the phase 3 GENUINE trial, which were recently published in The Lancet Haematology, showed that ublituximab plus ibrutinib was superior to ibrutinib alone for patients with relapsed or refractory high-risk CLL.
This news organization spoke with Jennifer R. Brown, MD, PhD, director of the CLL Center and institute physician at the Dana-Farber Cancer Institute and professor of medicine at Harvard Medical School, both in Boston, about the GENUINE trial and its potential impact on treatment choices going forward.
What type of patients were treated in the GENUINE trial?
Dr. Brown: This is a trial among relapsed/refractory CLL patients with 17p or 11q deletion or TP53 mutation. Patients aged 18 years or older with CLL who warranted treatment, as defined by International Workshop on CLL criteria, were eligible if they had previously received at least two cycles of at least one standard treatment regimen, had an Eastern Cooperative Oncology Group performance status of 2 or lower, and had high-risk cytogenetics, defined as the presence of at least one of del(17p), del(11q), or TP53 mutation confirmed by a central laboratory with fluorescence in situ hybridization and/or next-generation sequencing.
What were the main outcomes of the trial?
Originally, the GENUINE trial had coprimary endpoints of progression-free survival (PFS) and overall response rate. Because of slow accrual, it was amended to have one primary endpoint of independent review committee (IRC)–assessed ORR.
IRC-assessed ORR was improved from 65% to 83% with the addition of ublituximab. PFS also improved significantly in the ublituximab group, with an even greater improvement when the analysis was limited to those with del(17p) or TP53 aberrancy, but this outcome was limited by the reduced sample size of the study as well as the relatively short PFS of the ibrutinib arm.
After a median follow-up of 41.6 months, the median IRC-assessed PFS in all treated patients was not reached in the ublituximab plus ibrutinib group after 15 PFS events but was 35.9 months in the ibrutinib group after 25 PFS events (hazard ratio, 0.46; 95% confidence interval, 0.24-0.87; P = .016).
Undetectable minimal residual disease was also seen in 42% of the combination arm, compared with 6% of the ibrutinib arm.
What types of adverse events were found in the trial?
The researchers found mostly mild and known side effects of ibrutinib. More atrial fibrillation and neutropenia were seen in the antibody group, but this was not marked.
Most adverse events were of grade 1 or 2. The most common grade 3 and 4 adverse events were neutropenia (11 [19%] patients in the ublituximab plus ibrutinib group and 7 [12%] in the ibrutinib group), anemia (5 [8%] and 5 [9%], respectively), and diarrhea (6 [10%] and 3 [5%], respectively).
What about serious adverse events?
Hospitalization from infection was seen, as expected. There were two cardiac arrests and an unexplained death, across both arms, which was concerning, given the known association of ibrutinib with ventricular arrhythmia and sudden death. There were also several hemorrhages, including one fatal one, which was again consistent with the known side effects of ibrutinib.
Are there treatments comparable with ublituximab plus ibrutinib that clinicians should perhaps first consider using?
In terms of other anti-CD20 antibodies, we have two randomized trials that have failed to show a benefit from adding rituximab to ibrutinib.
Obinutuzumab, like ublituximab, is also a next-generation glycoengineered antibody, and it is reasonably likely that it might lead to similar results. However, the only data we have on ibrutinib with obinutuzumab are from a single arm in a more heterogeneous, lower-risk patient population, and it is unlikely that a randomized comparison will ever be done.
On the basis of these trial results, how would you use the combination of ublituximab and ibrutinib for your patients?
I would consider the addition of ublituximab to a BTK inhibitor in high-risk patients (once ublituximab is approved). I already usually use a next-generation BTK inhibitor rather than ibrutinib.
Are there any other implications of the GENUINE trial?
I think this trial underscores the importance of studying genetic subgroups of patients separately. In this case, that was done in high-risk patients, but this observation likely also applies to low-risk patients.
Most trials to date have enrolled unselected patient populations, often without stratification, and their results therefore tend to obscure the outcomes in both the very high risk (as studied here) and in the low risk (patients with immunoglobulin heavy chain variable region gene mutations).
Dr. Brown has served as a consultant for AbbVie, Acerta/AstraZeneca, Beigene, Bristol-Myers Squibb/Juno/Celgene, Catapult, Genentech/Roche, Janssen, MEI Pharma, Morphosys, and Novartis, and has received research funding from Gilead, Loxo/Lilly, TG Therapeutics, Verastem/SecuraBio.
A version of this article first appeared on Medscape.com.
Chronic lymphocytic leukemia (CLL) is a clinically heterogeneous disease associated with several known genetic abnormalities, including 17p deletion (del[17p]), 11q deletion (del[11q]), and TP53 gene mutations, which are adverse prognostic markers among patients treated with chemoimmunotherapy.
The Bruton tyrosine kinase (BTK) inhibitor ibrutinib is approved for patients with untreated, relapsed, or refractory disease, including those with del(17p). Clinicians will soon have the chance to pair it with ublituximab, a next-generation, glycoengineered, type I, anti-CD20 monoclonal antibody that binds to a unique epitope on CD20, differentiating it from rituximab, ofatumumab, and obinutuzumab. Results from the phase 3 GENUINE trial, which were recently published in The Lancet Haematology, showed that ublituximab plus ibrutinib was superior to ibrutinib alone for patients with relapsed or refractory high-risk CLL.
This news organization spoke with Jennifer R. Brown, MD, PhD, director of the CLL Center and institute physician at the Dana-Farber Cancer Institute and professor of medicine at Harvard Medical School, both in Boston, about the GENUINE trial and its potential impact on treatment choices going forward.
What type of patients were treated in the GENUINE trial?
Dr. Brown: This is a trial among relapsed/refractory CLL patients with 17p or 11q deletion or TP53 mutation. Patients aged 18 years or older with CLL who warranted treatment, as defined by International Workshop on CLL criteria, were eligible if they had previously received at least two cycles of at least one standard treatment regimen, had an Eastern Cooperative Oncology Group performance status of 2 or lower, and had high-risk cytogenetics, defined as the presence of at least one of del(17p), del(11q), or TP53 mutation confirmed by a central laboratory with fluorescence in situ hybridization and/or next-generation sequencing.
What were the main outcomes of the trial?
Originally, the GENUINE trial had coprimary endpoints of progression-free survival (PFS) and overall response rate. Because of slow accrual, it was amended to have one primary endpoint of independent review committee (IRC)–assessed ORR.
IRC-assessed ORR was improved from 65% to 83% with the addition of ublituximab. PFS also improved significantly in the ublituximab group, with an even greater improvement when the analysis was limited to those with del(17p) or TP53 aberrancy, but this outcome was limited by the reduced sample size of the study as well as the relatively short PFS of the ibrutinib arm.
After a median follow-up of 41.6 months, the median IRC-assessed PFS in all treated patients was not reached in the ublituximab plus ibrutinib group after 15 PFS events but was 35.9 months in the ibrutinib group after 25 PFS events (hazard ratio, 0.46; 95% confidence interval, 0.24-0.87; P = .016).
Undetectable minimal residual disease was also seen in 42% of the combination arm, compared with 6% of the ibrutinib arm.
What types of adverse events were found in the trial?
The researchers found mostly mild and known side effects of ibrutinib. More atrial fibrillation and neutropenia were seen in the antibody group, but this was not marked.
Most adverse events were of grade 1 or 2. The most common grade 3 and 4 adverse events were neutropenia (11 [19%] patients in the ublituximab plus ibrutinib group and 7 [12%] in the ibrutinib group), anemia (5 [8%] and 5 [9%], respectively), and diarrhea (6 [10%] and 3 [5%], respectively).
What about serious adverse events?
Hospitalization from infection was seen, as expected. There were two cardiac arrests and an unexplained death, across both arms, which was concerning, given the known association of ibrutinib with ventricular arrhythmia and sudden death. There were also several hemorrhages, including one fatal one, which was again consistent with the known side effects of ibrutinib.
Are there treatments comparable with ublituximab plus ibrutinib that clinicians should perhaps first consider using?
In terms of other anti-CD20 antibodies, we have two randomized trials that have failed to show a benefit from adding rituximab to ibrutinib.
Obinutuzumab, like ublituximab, is also a next-generation glycoengineered antibody, and it is reasonably likely that it might lead to similar results. However, the only data we have on ibrutinib with obinutuzumab are from a single arm in a more heterogeneous, lower-risk patient population, and it is unlikely that a randomized comparison will ever be done.
On the basis of these trial results, how would you use the combination of ublituximab and ibrutinib for your patients?
I would consider the addition of ublituximab to a BTK inhibitor in high-risk patients (once ublituximab is approved). I already usually use a next-generation BTK inhibitor rather than ibrutinib.
Are there any other implications of the GENUINE trial?
I think this trial underscores the importance of studying genetic subgroups of patients separately. In this case, that was done in high-risk patients, but this observation likely also applies to low-risk patients.
Most trials to date have enrolled unselected patient populations, often without stratification, and their results therefore tend to obscure the outcomes in both the very high risk (as studied here) and in the low risk (patients with immunoglobulin heavy chain variable region gene mutations).
Dr. Brown has served as a consultant for AbbVie, Acerta/AstraZeneca, Beigene, Bristol-Myers Squibb/Juno/Celgene, Catapult, Genentech/Roche, Janssen, MEI Pharma, Morphosys, and Novartis, and has received research funding from Gilead, Loxo/Lilly, TG Therapeutics, Verastem/SecuraBio.
A version of this article first appeared on Medscape.com.
Chronic lymphocytic leukemia (CLL) is a clinically heterogeneous disease associated with several known genetic abnormalities, including 17p deletion (del[17p]), 11q deletion (del[11q]), and TP53 gene mutations, which are adverse prognostic markers among patients treated with chemoimmunotherapy.
The Bruton tyrosine kinase (BTK) inhibitor ibrutinib is approved for patients with untreated, relapsed, or refractory disease, including those with del(17p). Clinicians will soon have the chance to pair it with ublituximab, a next-generation, glycoengineered, type I, anti-CD20 monoclonal antibody that binds to a unique epitope on CD20, differentiating it from rituximab, ofatumumab, and obinutuzumab. Results from the phase 3 GENUINE trial, which were recently published in The Lancet Haematology, showed that ublituximab plus ibrutinib was superior to ibrutinib alone for patients with relapsed or refractory high-risk CLL.
This news organization spoke with Jennifer R. Brown, MD, PhD, director of the CLL Center and institute physician at the Dana-Farber Cancer Institute and professor of medicine at Harvard Medical School, both in Boston, about the GENUINE trial and its potential impact on treatment choices going forward.
What type of patients were treated in the GENUINE trial?
Dr. Brown: This is a trial among relapsed/refractory CLL patients with 17p or 11q deletion or TP53 mutation. Patients aged 18 years or older with CLL who warranted treatment, as defined by International Workshop on CLL criteria, were eligible if they had previously received at least two cycles of at least one standard treatment regimen, had an Eastern Cooperative Oncology Group performance status of 2 or lower, and had high-risk cytogenetics, defined as the presence of at least one of del(17p), del(11q), or TP53 mutation confirmed by a central laboratory with fluorescence in situ hybridization and/or next-generation sequencing.
What were the main outcomes of the trial?
Originally, the GENUINE trial had coprimary endpoints of progression-free survival (PFS) and overall response rate. Because of slow accrual, it was amended to have one primary endpoint of independent review committee (IRC)–assessed ORR.
IRC-assessed ORR was improved from 65% to 83% with the addition of ublituximab. PFS also improved significantly in the ublituximab group, with an even greater improvement when the analysis was limited to those with del(17p) or TP53 aberrancy, but this outcome was limited by the reduced sample size of the study as well as the relatively short PFS of the ibrutinib arm.
After a median follow-up of 41.6 months, the median IRC-assessed PFS in all treated patients was not reached in the ublituximab plus ibrutinib group after 15 PFS events but was 35.9 months in the ibrutinib group after 25 PFS events (hazard ratio, 0.46; 95% confidence interval, 0.24-0.87; P = .016).
Undetectable minimal residual disease was also seen in 42% of the combination arm, compared with 6% of the ibrutinib arm.
What types of adverse events were found in the trial?
The researchers found mostly mild and known side effects of ibrutinib. More atrial fibrillation and neutropenia were seen in the antibody group, but this was not marked.
Most adverse events were of grade 1 or 2. The most common grade 3 and 4 adverse events were neutropenia (11 [19%] patients in the ublituximab plus ibrutinib group and 7 [12%] in the ibrutinib group), anemia (5 [8%] and 5 [9%], respectively), and diarrhea (6 [10%] and 3 [5%], respectively).
What about serious adverse events?
Hospitalization from infection was seen, as expected. There were two cardiac arrests and an unexplained death, across both arms, which was concerning, given the known association of ibrutinib with ventricular arrhythmia and sudden death. There were also several hemorrhages, including one fatal one, which was again consistent with the known side effects of ibrutinib.
Are there treatments comparable with ublituximab plus ibrutinib that clinicians should perhaps first consider using?
In terms of other anti-CD20 antibodies, we have two randomized trials that have failed to show a benefit from adding rituximab to ibrutinib.
Obinutuzumab, like ublituximab, is also a next-generation glycoengineered antibody, and it is reasonably likely that it might lead to similar results. However, the only data we have on ibrutinib with obinutuzumab are from a single arm in a more heterogeneous, lower-risk patient population, and it is unlikely that a randomized comparison will ever be done.
On the basis of these trial results, how would you use the combination of ublituximab and ibrutinib for your patients?
I would consider the addition of ublituximab to a BTK inhibitor in high-risk patients (once ublituximab is approved). I already usually use a next-generation BTK inhibitor rather than ibrutinib.
Are there any other implications of the GENUINE trial?
I think this trial underscores the importance of studying genetic subgroups of patients separately. In this case, that was done in high-risk patients, but this observation likely also applies to low-risk patients.
Most trials to date have enrolled unselected patient populations, often without stratification, and their results therefore tend to obscure the outcomes in both the very high risk (as studied here) and in the low risk (patients with immunoglobulin heavy chain variable region gene mutations).
Dr. Brown has served as a consultant for AbbVie, Acerta/AstraZeneca, Beigene, Bristol-Myers Squibb/Juno/Celgene, Catapult, Genentech/Roche, Janssen, MEI Pharma, Morphosys, and Novartis, and has received research funding from Gilead, Loxo/Lilly, TG Therapeutics, Verastem/SecuraBio.
A version of this article first appeared on Medscape.com.
CLL patients: Diagnostic difficulties, treatment confusion with COVID-19
Chronic lymphocytic leukemia (CLL) patients present significant problems with regard to COVID-19 disease, according to a literature review by Yousef Roosta, MD, of Urmia (Iran) University of Medical Sciences, and colleagues.
Diagnostic interaction between CLL and COVID-19 provides a major challenge. CLL patients have a lower rate of anti–SARS-CoV-2 IgG development, and evidence shows worse therapeutic outcomes in these patients, according to study published in Leukemia Research Reports.
The researchers assessed 20 retrieved articles, 11 of which examined patients with CLL and with concomitant COVID-19; and 9 articles were designed as prospective or retrospective case series of such patients. The studies were assessed qualitatively by the QUADAS-2 tool.
Troubling results
Although the overall prevalence of CLL and COVID-19 concurrence was low, at 0.6% (95% confidence interval 0.5%-0.7%) according to the meta-analysis, the results showed some special challenges in the diagnosis and care of these patients.
Diagnostic difficulties are a unique problem. Lymphopenia is common in patients with COVID-19, while lymphocytosis may be considered a transient or even rare finding. The interplay between the two diseases is sometimes very misleading for specialists, and in patients with lymphocytosis, the diagnosis of CLL may be completely ignored, according to the researchers. They added that when performing a diagnostic approach for concurrent COVID-19 and CLL, due to differences in the amount and type of immune response, “relying on serological testing, and especially the evaluation of the anti–SARS-CoV-2 IgG levels may not be beneficial,” they indicated.
In addition, studies showed unacceptable therapeutic outcome in patients with concurrent CLL and COVID-19, with mortality ranging from 33% to 41.7%, showing a need to revise current treatment protocols, according to the authors. In one study, 85.7% of surviving patients showed a considerable decrease in functional class and significant fatigue, with such a poor prognosis occurring more commonly in the elderly.
With regard to treatment, “it is quite obvious that despite the use of current standard protocols, the prognosis of these patients will be much worse than the prognosis of CLL patients with no evidence of COVID-19. Even in the first-line treatment protocol for these patients, there is no agreement in combination therapy with selected CLL drugs along with management protocols of COVID-19 patients,” the researchers stated.
“[The] different hematological behaviors of two diseases might mimic the detection of COVID-19 in the CLL state and vise versa. Also, due to the low level of immune response against SARS-CoV-2 in CLL patients, both scheduled immunological-based diagnosis and treatment may fail,” the researchers added.
The authors reported that they had no disclosures.
Chronic lymphocytic leukemia (CLL) patients present significant problems with regard to COVID-19 disease, according to a literature review by Yousef Roosta, MD, of Urmia (Iran) University of Medical Sciences, and colleagues.
Diagnostic interaction between CLL and COVID-19 provides a major challenge. CLL patients have a lower rate of anti–SARS-CoV-2 IgG development, and evidence shows worse therapeutic outcomes in these patients, according to study published in Leukemia Research Reports.
The researchers assessed 20 retrieved articles, 11 of which examined patients with CLL and with concomitant COVID-19; and 9 articles were designed as prospective or retrospective case series of such patients. The studies were assessed qualitatively by the QUADAS-2 tool.
Troubling results
Although the overall prevalence of CLL and COVID-19 concurrence was low, at 0.6% (95% confidence interval 0.5%-0.7%) according to the meta-analysis, the results showed some special challenges in the diagnosis and care of these patients.
Diagnostic difficulties are a unique problem. Lymphopenia is common in patients with COVID-19, while lymphocytosis may be considered a transient or even rare finding. The interplay between the two diseases is sometimes very misleading for specialists, and in patients with lymphocytosis, the diagnosis of CLL may be completely ignored, according to the researchers. They added that when performing a diagnostic approach for concurrent COVID-19 and CLL, due to differences in the amount and type of immune response, “relying on serological testing, and especially the evaluation of the anti–SARS-CoV-2 IgG levels may not be beneficial,” they indicated.
In addition, studies showed unacceptable therapeutic outcome in patients with concurrent CLL and COVID-19, with mortality ranging from 33% to 41.7%, showing a need to revise current treatment protocols, according to the authors. In one study, 85.7% of surviving patients showed a considerable decrease in functional class and significant fatigue, with such a poor prognosis occurring more commonly in the elderly.
With regard to treatment, “it is quite obvious that despite the use of current standard protocols, the prognosis of these patients will be much worse than the prognosis of CLL patients with no evidence of COVID-19. Even in the first-line treatment protocol for these patients, there is no agreement in combination therapy with selected CLL drugs along with management protocols of COVID-19 patients,” the researchers stated.
“[The] different hematological behaviors of two diseases might mimic the detection of COVID-19 in the CLL state and vise versa. Also, due to the low level of immune response against SARS-CoV-2 in CLL patients, both scheduled immunological-based diagnosis and treatment may fail,” the researchers added.
The authors reported that they had no disclosures.
Chronic lymphocytic leukemia (CLL) patients present significant problems with regard to COVID-19 disease, according to a literature review by Yousef Roosta, MD, of Urmia (Iran) University of Medical Sciences, and colleagues.
Diagnostic interaction between CLL and COVID-19 provides a major challenge. CLL patients have a lower rate of anti–SARS-CoV-2 IgG development, and evidence shows worse therapeutic outcomes in these patients, according to study published in Leukemia Research Reports.
The researchers assessed 20 retrieved articles, 11 of which examined patients with CLL and with concomitant COVID-19; and 9 articles were designed as prospective or retrospective case series of such patients. The studies were assessed qualitatively by the QUADAS-2 tool.
Troubling results
Although the overall prevalence of CLL and COVID-19 concurrence was low, at 0.6% (95% confidence interval 0.5%-0.7%) according to the meta-analysis, the results showed some special challenges in the diagnosis and care of these patients.
Diagnostic difficulties are a unique problem. Lymphopenia is common in patients with COVID-19, while lymphocytosis may be considered a transient or even rare finding. The interplay between the two diseases is sometimes very misleading for specialists, and in patients with lymphocytosis, the diagnosis of CLL may be completely ignored, according to the researchers. They added that when performing a diagnostic approach for concurrent COVID-19 and CLL, due to differences in the amount and type of immune response, “relying on serological testing, and especially the evaluation of the anti–SARS-CoV-2 IgG levels may not be beneficial,” they indicated.
In addition, studies showed unacceptable therapeutic outcome in patients with concurrent CLL and COVID-19, with mortality ranging from 33% to 41.7%, showing a need to revise current treatment protocols, according to the authors. In one study, 85.7% of surviving patients showed a considerable decrease in functional class and significant fatigue, with such a poor prognosis occurring more commonly in the elderly.
With regard to treatment, “it is quite obvious that despite the use of current standard protocols, the prognosis of these patients will be much worse than the prognosis of CLL patients with no evidence of COVID-19. Even in the first-line treatment protocol for these patients, there is no agreement in combination therapy with selected CLL drugs along with management protocols of COVID-19 patients,” the researchers stated.
“[The] different hematological behaviors of two diseases might mimic the detection of COVID-19 in the CLL state and vise versa. Also, due to the low level of immune response against SARS-CoV-2 in CLL patients, both scheduled immunological-based diagnosis and treatment may fail,” the researchers added.
The authors reported that they had no disclosures.
FROM LEUKEMIA RESEARCH REPORTS
COVID-19 vaccine failure in patients with blood cancers
COVID vaccines do not work well for patients with hematologic malignancies, new data suggest.
A small study involving 67 such patients shows that nearly half did not produce antibodies and were therefore still at risk of contracting COVID-19, even though they had all received both doses of one of the new mRNA COVID vaccines (Moderna or Pfizer).
“[This] is in stark contrast with the results of phase 1 mRNA vaccine immunogenicity trials, in which robust antibody responses were seen in essentially 100% of participants,” said the authors, led by Mounzer Agha, MD, director of the Mario Lemieux Center for Blood Cancers at the University of Pittsburgh Medical Center’s Hillman Cancer Center.
“Clinicians caring for patients with hematological malignancies and other immunocompromising conditions should be aware of the possibility of COVID-19 vaccine failure,” they emphasized.
“It’s critically important for these patients to be aware of their continued risk [for SARS-CoV-2 infection] and to seek prompt medical attention if they have COVID-19 symptoms, even after vaccination,” Dr. Agha said in a statement.
The study was published online on April 9 as preprint in medRxiv and has not yet undergone peer review.
Antibody responses
The authors analyzed responses in a group of 67 patients who had a hematologic malignancy, including chronic lymphocytic leukemia (CLL), lymphoma, and multiple myeloma. Approximately 45% of the patients were receiving therapy for their cancer at the time of vaccination; the rest were under observation.
All patients received two doses of an mRNA COVID vaccine and so were considered to be fully vaccinated.
Antibody responses for these fully vaccinated patients were then analyzed. The median duration between receipt of the second dose of the vaccine and the antibody test was 23 days.
“In total ... 46.3% ... had a negative antibody result after vaccination and were therefore considered to be vaccine nonresponders,” the authors reported.
The worst responses occurred in patients with CLL, of whom only 23% produced measurable antibodies to either vaccine, although approximately 70% of these patients were not receiving any form of cancer therapy at the time of vaccination.
Older patients were more likely not to have a response to either vaccine compared with younger patients, the investigators added.
In contrast, gender, immunoglobulin G levels, the number of days between the second dose and the measurement of antibodies, and status of cancer therapy did not differ among patients who had a response to the vaccines and those who did not.
“Our findings underscore the importance of adherence to nonpharmaceutical interventions to prevent COVID-19 in hematological malignancy patients,” the authors wrote. This is particularly important, given the fact that among patients with hematologic malignancies who become infected with SARS-CoV-2, the mortality rate is in excess of 30%.
Moreover, among such patients, viral shedding may be prolonged, often lasting several months. As such, “these patients should be advised to wear masks and observe social distancing regardless of vaccination status,” the investigators advised.
As of March 2021, guidance from the Centers for Disease Control and Prevention has allowed gatherings of unmasked people who have been vaccinated and of those at low risk for COVID-19 who have not yet been vaccinated. “As we see more national guidance allowing for unmasked gatherings among vaccinated people, clinicians should counsel their immunocompromised patients about the possibility that COVID-19 vaccines may not fully protect them against SARS-CoV-2,” coauthor Ghady Haidar, MD, assistant professor of medicine at the University of Pittsburgh, said in a statement.
“Our results show that the odds of the vaccine producing an antibody response in people with hematologic malignancies are the equivalent of a coin flip,” he said.
The authors have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
COVID vaccines do not work well for patients with hematologic malignancies, new data suggest.
A small study involving 67 such patients shows that nearly half did not produce antibodies and were therefore still at risk of contracting COVID-19, even though they had all received both doses of one of the new mRNA COVID vaccines (Moderna or Pfizer).
“[This] is in stark contrast with the results of phase 1 mRNA vaccine immunogenicity trials, in which robust antibody responses were seen in essentially 100% of participants,” said the authors, led by Mounzer Agha, MD, director of the Mario Lemieux Center for Blood Cancers at the University of Pittsburgh Medical Center’s Hillman Cancer Center.
“Clinicians caring for patients with hematological malignancies and other immunocompromising conditions should be aware of the possibility of COVID-19 vaccine failure,” they emphasized.
“It’s critically important for these patients to be aware of their continued risk [for SARS-CoV-2 infection] and to seek prompt medical attention if they have COVID-19 symptoms, even after vaccination,” Dr. Agha said in a statement.
The study was published online on April 9 as preprint in medRxiv and has not yet undergone peer review.
Antibody responses
The authors analyzed responses in a group of 67 patients who had a hematologic malignancy, including chronic lymphocytic leukemia (CLL), lymphoma, and multiple myeloma. Approximately 45% of the patients were receiving therapy for their cancer at the time of vaccination; the rest were under observation.
All patients received two doses of an mRNA COVID vaccine and so were considered to be fully vaccinated.
Antibody responses for these fully vaccinated patients were then analyzed. The median duration between receipt of the second dose of the vaccine and the antibody test was 23 days.
“In total ... 46.3% ... had a negative antibody result after vaccination and were therefore considered to be vaccine nonresponders,” the authors reported.
The worst responses occurred in patients with CLL, of whom only 23% produced measurable antibodies to either vaccine, although approximately 70% of these patients were not receiving any form of cancer therapy at the time of vaccination.
Older patients were more likely not to have a response to either vaccine compared with younger patients, the investigators added.
In contrast, gender, immunoglobulin G levels, the number of days between the second dose and the measurement of antibodies, and status of cancer therapy did not differ among patients who had a response to the vaccines and those who did not.
“Our findings underscore the importance of adherence to nonpharmaceutical interventions to prevent COVID-19 in hematological malignancy patients,” the authors wrote. This is particularly important, given the fact that among patients with hematologic malignancies who become infected with SARS-CoV-2, the mortality rate is in excess of 30%.
Moreover, among such patients, viral shedding may be prolonged, often lasting several months. As such, “these patients should be advised to wear masks and observe social distancing regardless of vaccination status,” the investigators advised.
As of March 2021, guidance from the Centers for Disease Control and Prevention has allowed gatherings of unmasked people who have been vaccinated and of those at low risk for COVID-19 who have not yet been vaccinated. “As we see more national guidance allowing for unmasked gatherings among vaccinated people, clinicians should counsel their immunocompromised patients about the possibility that COVID-19 vaccines may not fully protect them against SARS-CoV-2,” coauthor Ghady Haidar, MD, assistant professor of medicine at the University of Pittsburgh, said in a statement.
“Our results show that the odds of the vaccine producing an antibody response in people with hematologic malignancies are the equivalent of a coin flip,” he said.
The authors have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
COVID vaccines do not work well for patients with hematologic malignancies, new data suggest.
A small study involving 67 such patients shows that nearly half did not produce antibodies and were therefore still at risk of contracting COVID-19, even though they had all received both doses of one of the new mRNA COVID vaccines (Moderna or Pfizer).
“[This] is in stark contrast with the results of phase 1 mRNA vaccine immunogenicity trials, in which robust antibody responses were seen in essentially 100% of participants,” said the authors, led by Mounzer Agha, MD, director of the Mario Lemieux Center for Blood Cancers at the University of Pittsburgh Medical Center’s Hillman Cancer Center.
“Clinicians caring for patients with hematological malignancies and other immunocompromising conditions should be aware of the possibility of COVID-19 vaccine failure,” they emphasized.
“It’s critically important for these patients to be aware of their continued risk [for SARS-CoV-2 infection] and to seek prompt medical attention if they have COVID-19 symptoms, even after vaccination,” Dr. Agha said in a statement.
The study was published online on April 9 as preprint in medRxiv and has not yet undergone peer review.
Antibody responses
The authors analyzed responses in a group of 67 patients who had a hematologic malignancy, including chronic lymphocytic leukemia (CLL), lymphoma, and multiple myeloma. Approximately 45% of the patients were receiving therapy for their cancer at the time of vaccination; the rest were under observation.
All patients received two doses of an mRNA COVID vaccine and so were considered to be fully vaccinated.
Antibody responses for these fully vaccinated patients were then analyzed. The median duration between receipt of the second dose of the vaccine and the antibody test was 23 days.
“In total ... 46.3% ... had a negative antibody result after vaccination and were therefore considered to be vaccine nonresponders,” the authors reported.
The worst responses occurred in patients with CLL, of whom only 23% produced measurable antibodies to either vaccine, although approximately 70% of these patients were not receiving any form of cancer therapy at the time of vaccination.
Older patients were more likely not to have a response to either vaccine compared with younger patients, the investigators added.
In contrast, gender, immunoglobulin G levels, the number of days between the second dose and the measurement of antibodies, and status of cancer therapy did not differ among patients who had a response to the vaccines and those who did not.
“Our findings underscore the importance of adherence to nonpharmaceutical interventions to prevent COVID-19 in hematological malignancy patients,” the authors wrote. This is particularly important, given the fact that among patients with hematologic malignancies who become infected with SARS-CoV-2, the mortality rate is in excess of 30%.
Moreover, among such patients, viral shedding may be prolonged, often lasting several months. As such, “these patients should be advised to wear masks and observe social distancing regardless of vaccination status,” the investigators advised.
As of March 2021, guidance from the Centers for Disease Control and Prevention has allowed gatherings of unmasked people who have been vaccinated and of those at low risk for COVID-19 who have not yet been vaccinated. “As we see more national guidance allowing for unmasked gatherings among vaccinated people, clinicians should counsel their immunocompromised patients about the possibility that COVID-19 vaccines may not fully protect them against SARS-CoV-2,” coauthor Ghady Haidar, MD, assistant professor of medicine at the University of Pittsburgh, said in a statement.
“Our results show that the odds of the vaccine producing an antibody response in people with hematologic malignancies are the equivalent of a coin flip,” he said.
The authors have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Steroids can be stopped in some older multiple myeloma patients
For select older patients, it is safe to switch to a lower dose of lenalidomide maintenance therapy and discontinue dexamethasone after 9 months. The regimen is safe and yields outcomes similar to those of standard, continuous lenalidomide/dexamethasone (Rd), according to new findings.
At a median follow-up of 37 months, event-free survival was 10.4 months in the experimental arm in which dexamethasone therapy was stopped (Rd-R) versus 6.9 months for standard therapy. The tailored approach also resulted in fewer adverse effects.
The authors noted that there was no difference in progression-free survival (PFS) and overall survival between the two groups.
“These results may be useful for the treatment of myeloma patients, since approximately one-third of patients not eligible for stem cell transplantation are intermediate fit, the population in our study,” said lead author Alessandra Larocca, MD, PhD, from the department of hematology-oncology of the University Hospital Città della Salute e della Scienza, Torino, Italy.
She said in an interview that they expect that these findings “may help to optimize the treatment of less-fit elderly patients by reducing the occurrence of adverse events and thus improving outcomes and preserving quality of life of these patients.”
This approach is a viable option for clinicians to consider for some patient subgroups. “This steroid-sparing approach can also be used in other combinations,” she said. “Ongoing trials are now evaluating steroid sparing in combination with monoclonal antibodies or the role of frailty-guided treatment.”
The study was published March 19, 2021, in Blood.
Curtailing steroids
Myeloma patients aged 75 years or older or who have comorbidities and functional impairments are an understudied population. They are more susceptible to adverse events that may negatively affect the duration of treatment and outcomes. Steroids are “scarcely tolerated” in the long term, even among younger patients, and “whether sparing dexamethasone is as effective as prolonged steroid exposure remains an open issue,” the authors wrote. There are still no clear data on the advantage of continuous steroid treatment as opposed to fixed-duration treatment for newly diagnosed patients.
In 2010, a study compared high-dose with low-dose dexamethasone. As expected, the rate of adverse events was lower among patients who received the low-dose steroid, but quite unexpectedly, deaths with high-dose dexamethasone were significantly higher than with low-dose dexamethasone.
The 1-year overall survival was 96% among patients who received the low dose of dexamethasone versus 87% with the standard high dose.
S. Vincent Rajkumar, MD, of the Mayo Clinic, Rochester, Minn., who was the lead author of the 2010 study, spoke with this new organization about the current study. “This is an important and practice-changing study,” he said. “We have already changed our practice and recommendations based on this study.”
He explained that, for transplant-ineligible patients, instead of initial therapy with bortezomib-lenalidomide-dexamethasone followed by Rd, they use lenalidomide alone without steroids.
“After 9 months of initial therapy, I now recommend we stop dexamethasone unless we are having problems controlling the myeloma, such as progressive disease,” Dr. Rajkumar said. “I congratulate the authors on a study that will improve the quality of life for our patients.”
Improved event-free survival
In this study, Dr. Larocca and colleagues investigated the efficacy and feasibility of a dose- and schedule-adjusted Rd regimen that was followed by maintenance Rd-R 10 mg/d and compared the regimen with continuous Rd in elderly, intermediate-fit patients who were newly diagnosed with multiple myeloma.
The primary endpoint was event-free survival, defined as progression/death from any cause, lenalidomide discontinuation, and any hematologic grade 4 or nonhematologic grade 3-4 adverse events.
The cohort consisted of 199 patients who were randomly assigned to receive either Rd-R (n = 101) or continuous Rd (n = 98). The median age was 75 years in the Rd-R arm and 76 years in the Rd arm; 52% of patients in the Rd-R group and 43% in the Rd group were classified as being intermediate fit not for age but for geriatric impairments.
With a median follow-up of 37 months, event-free survival was 10.4 months in the Rd-R arm versus 6.9 months in the Rd arm (hazard ratio, 0.70; P = .02). This benefit was maintained beyond nine cycles (median: 19.8 vs. 10.6 months for Rd-R vs. Rd; HR, 0.55; P = .03)
The median PFS was 20.2 months with Rd-R and 18.3 months with Rd (HR, 0.78; P = .16). The median overall survival was not reached. The 3-year overall survival was 74% with Rd-R and 63% with continuous Rd (HR, 0.62; P = .06). Among patients remaining on therapy after nine cycles, no difference in median PFS was observed between the two groups (24.3 vs. 18.7 months; HR, 0.73; P = .19).
Best response was similar for both groups, with an overall response rate of 78% versus 68% (P = .15). The very good partial response rate was 51% in the Rd-R arm versus 39% in the continuous Rd arm (P = .09).
Toxicities were similar between the two groups. Hematologic adverse events of at least grade 3 were reported in 26% of Rd-R patients versus 20% of Rd patients (P = .40). In both groups, the most frequent grade ≥3 hematologic toxicity was neutropenia (21% vs 18%). The most frequent grade ≥3 toxicities were nonhematologic. They occurred in 33% of Rd-R patients and 43% of Rd patients (P = .15). The most frequent nonhematologic toxicities were infections (10% vs. 12%), constitutional (3% vs. 12%), dermatologic (7% vs. 3%), and central nervous toxicities (2% vs. 6%).
The study was sponsored by Fondazione EMN Italy Onlus. Dr. Larocca has received honoraria from Amgen, Bristol-Myers Squibb, Celgene, Janssen, and GlaxoSmithKline, and has served on the advisory boards for Bristol-Myers Squibb, Celgene, Janssen, and Takeda. Several coauthors also have disclosed relationships with industry. Dr. Rajkumar disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
For select older patients, it is safe to switch to a lower dose of lenalidomide maintenance therapy and discontinue dexamethasone after 9 months. The regimen is safe and yields outcomes similar to those of standard, continuous lenalidomide/dexamethasone (Rd), according to new findings.
At a median follow-up of 37 months, event-free survival was 10.4 months in the experimental arm in which dexamethasone therapy was stopped (Rd-R) versus 6.9 months for standard therapy. The tailored approach also resulted in fewer adverse effects.
The authors noted that there was no difference in progression-free survival (PFS) and overall survival between the two groups.
“These results may be useful for the treatment of myeloma patients, since approximately one-third of patients not eligible for stem cell transplantation are intermediate fit, the population in our study,” said lead author Alessandra Larocca, MD, PhD, from the department of hematology-oncology of the University Hospital Città della Salute e della Scienza, Torino, Italy.
She said in an interview that they expect that these findings “may help to optimize the treatment of less-fit elderly patients by reducing the occurrence of adverse events and thus improving outcomes and preserving quality of life of these patients.”
This approach is a viable option for clinicians to consider for some patient subgroups. “This steroid-sparing approach can also be used in other combinations,” she said. “Ongoing trials are now evaluating steroid sparing in combination with monoclonal antibodies or the role of frailty-guided treatment.”
The study was published March 19, 2021, in Blood.
Curtailing steroids
Myeloma patients aged 75 years or older or who have comorbidities and functional impairments are an understudied population. They are more susceptible to adverse events that may negatively affect the duration of treatment and outcomes. Steroids are “scarcely tolerated” in the long term, even among younger patients, and “whether sparing dexamethasone is as effective as prolonged steroid exposure remains an open issue,” the authors wrote. There are still no clear data on the advantage of continuous steroid treatment as opposed to fixed-duration treatment for newly diagnosed patients.
In 2010, a study compared high-dose with low-dose dexamethasone. As expected, the rate of adverse events was lower among patients who received the low-dose steroid, but quite unexpectedly, deaths with high-dose dexamethasone were significantly higher than with low-dose dexamethasone.
The 1-year overall survival was 96% among patients who received the low dose of dexamethasone versus 87% with the standard high dose.
S. Vincent Rajkumar, MD, of the Mayo Clinic, Rochester, Minn., who was the lead author of the 2010 study, spoke with this new organization about the current study. “This is an important and practice-changing study,” he said. “We have already changed our practice and recommendations based on this study.”
He explained that, for transplant-ineligible patients, instead of initial therapy with bortezomib-lenalidomide-dexamethasone followed by Rd, they use lenalidomide alone without steroids.
“After 9 months of initial therapy, I now recommend we stop dexamethasone unless we are having problems controlling the myeloma, such as progressive disease,” Dr. Rajkumar said. “I congratulate the authors on a study that will improve the quality of life for our patients.”
Improved event-free survival
In this study, Dr. Larocca and colleagues investigated the efficacy and feasibility of a dose- and schedule-adjusted Rd regimen that was followed by maintenance Rd-R 10 mg/d and compared the regimen with continuous Rd in elderly, intermediate-fit patients who were newly diagnosed with multiple myeloma.
The primary endpoint was event-free survival, defined as progression/death from any cause, lenalidomide discontinuation, and any hematologic grade 4 or nonhematologic grade 3-4 adverse events.
The cohort consisted of 199 patients who were randomly assigned to receive either Rd-R (n = 101) or continuous Rd (n = 98). The median age was 75 years in the Rd-R arm and 76 years in the Rd arm; 52% of patients in the Rd-R group and 43% in the Rd group were classified as being intermediate fit not for age but for geriatric impairments.
With a median follow-up of 37 months, event-free survival was 10.4 months in the Rd-R arm versus 6.9 months in the Rd arm (hazard ratio, 0.70; P = .02). This benefit was maintained beyond nine cycles (median: 19.8 vs. 10.6 months for Rd-R vs. Rd; HR, 0.55; P = .03)
The median PFS was 20.2 months with Rd-R and 18.3 months with Rd (HR, 0.78; P = .16). The median overall survival was not reached. The 3-year overall survival was 74% with Rd-R and 63% with continuous Rd (HR, 0.62; P = .06). Among patients remaining on therapy after nine cycles, no difference in median PFS was observed between the two groups (24.3 vs. 18.7 months; HR, 0.73; P = .19).
Best response was similar for both groups, with an overall response rate of 78% versus 68% (P = .15). The very good partial response rate was 51% in the Rd-R arm versus 39% in the continuous Rd arm (P = .09).
Toxicities were similar between the two groups. Hematologic adverse events of at least grade 3 were reported in 26% of Rd-R patients versus 20% of Rd patients (P = .40). In both groups, the most frequent grade ≥3 hematologic toxicity was neutropenia (21% vs 18%). The most frequent grade ≥3 toxicities were nonhematologic. They occurred in 33% of Rd-R patients and 43% of Rd patients (P = .15). The most frequent nonhematologic toxicities were infections (10% vs. 12%), constitutional (3% vs. 12%), dermatologic (7% vs. 3%), and central nervous toxicities (2% vs. 6%).
The study was sponsored by Fondazione EMN Italy Onlus. Dr. Larocca has received honoraria from Amgen, Bristol-Myers Squibb, Celgene, Janssen, and GlaxoSmithKline, and has served on the advisory boards for Bristol-Myers Squibb, Celgene, Janssen, and Takeda. Several coauthors also have disclosed relationships with industry. Dr. Rajkumar disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
For select older patients, it is safe to switch to a lower dose of lenalidomide maintenance therapy and discontinue dexamethasone after 9 months. The regimen is safe and yields outcomes similar to those of standard, continuous lenalidomide/dexamethasone (Rd), according to new findings.
At a median follow-up of 37 months, event-free survival was 10.4 months in the experimental arm in which dexamethasone therapy was stopped (Rd-R) versus 6.9 months for standard therapy. The tailored approach also resulted in fewer adverse effects.
The authors noted that there was no difference in progression-free survival (PFS) and overall survival between the two groups.
“These results may be useful for the treatment of myeloma patients, since approximately one-third of patients not eligible for stem cell transplantation are intermediate fit, the population in our study,” said lead author Alessandra Larocca, MD, PhD, from the department of hematology-oncology of the University Hospital Città della Salute e della Scienza, Torino, Italy.
She said in an interview that they expect that these findings “may help to optimize the treatment of less-fit elderly patients by reducing the occurrence of adverse events and thus improving outcomes and preserving quality of life of these patients.”
This approach is a viable option for clinicians to consider for some patient subgroups. “This steroid-sparing approach can also be used in other combinations,” she said. “Ongoing trials are now evaluating steroid sparing in combination with monoclonal antibodies or the role of frailty-guided treatment.”
The study was published March 19, 2021, in Blood.
Curtailing steroids
Myeloma patients aged 75 years or older or who have comorbidities and functional impairments are an understudied population. They are more susceptible to adverse events that may negatively affect the duration of treatment and outcomes. Steroids are “scarcely tolerated” in the long term, even among younger patients, and “whether sparing dexamethasone is as effective as prolonged steroid exposure remains an open issue,” the authors wrote. There are still no clear data on the advantage of continuous steroid treatment as opposed to fixed-duration treatment for newly diagnosed patients.
In 2010, a study compared high-dose with low-dose dexamethasone. As expected, the rate of adverse events was lower among patients who received the low-dose steroid, but quite unexpectedly, deaths with high-dose dexamethasone were significantly higher than with low-dose dexamethasone.
The 1-year overall survival was 96% among patients who received the low dose of dexamethasone versus 87% with the standard high dose.
S. Vincent Rajkumar, MD, of the Mayo Clinic, Rochester, Minn., who was the lead author of the 2010 study, spoke with this new organization about the current study. “This is an important and practice-changing study,” he said. “We have already changed our practice and recommendations based on this study.”
He explained that, for transplant-ineligible patients, instead of initial therapy with bortezomib-lenalidomide-dexamethasone followed by Rd, they use lenalidomide alone without steroids.
“After 9 months of initial therapy, I now recommend we stop dexamethasone unless we are having problems controlling the myeloma, such as progressive disease,” Dr. Rajkumar said. “I congratulate the authors on a study that will improve the quality of life for our patients.”
Improved event-free survival
In this study, Dr. Larocca and colleagues investigated the efficacy and feasibility of a dose- and schedule-adjusted Rd regimen that was followed by maintenance Rd-R 10 mg/d and compared the regimen with continuous Rd in elderly, intermediate-fit patients who were newly diagnosed with multiple myeloma.
The primary endpoint was event-free survival, defined as progression/death from any cause, lenalidomide discontinuation, and any hematologic grade 4 or nonhematologic grade 3-4 adverse events.
The cohort consisted of 199 patients who were randomly assigned to receive either Rd-R (n = 101) or continuous Rd (n = 98). The median age was 75 years in the Rd-R arm and 76 years in the Rd arm; 52% of patients in the Rd-R group and 43% in the Rd group were classified as being intermediate fit not for age but for geriatric impairments.
With a median follow-up of 37 months, event-free survival was 10.4 months in the Rd-R arm versus 6.9 months in the Rd arm (hazard ratio, 0.70; P = .02). This benefit was maintained beyond nine cycles (median: 19.8 vs. 10.6 months for Rd-R vs. Rd; HR, 0.55; P = .03)
The median PFS was 20.2 months with Rd-R and 18.3 months with Rd (HR, 0.78; P = .16). The median overall survival was not reached. The 3-year overall survival was 74% with Rd-R and 63% with continuous Rd (HR, 0.62; P = .06). Among patients remaining on therapy after nine cycles, no difference in median PFS was observed between the two groups (24.3 vs. 18.7 months; HR, 0.73; P = .19).
Best response was similar for both groups, with an overall response rate of 78% versus 68% (P = .15). The very good partial response rate was 51% in the Rd-R arm versus 39% in the continuous Rd arm (P = .09).
Toxicities were similar between the two groups. Hematologic adverse events of at least grade 3 were reported in 26% of Rd-R patients versus 20% of Rd patients (P = .40). In both groups, the most frequent grade ≥3 hematologic toxicity was neutropenia (21% vs 18%). The most frequent grade ≥3 toxicities were nonhematologic. They occurred in 33% of Rd-R patients and 43% of Rd patients (P = .15). The most frequent nonhematologic toxicities were infections (10% vs. 12%), constitutional (3% vs. 12%), dermatologic (7% vs. 3%), and central nervous toxicities (2% vs. 6%).
The study was sponsored by Fondazione EMN Italy Onlus. Dr. Larocca has received honoraria from Amgen, Bristol-Myers Squibb, Celgene, Janssen, and GlaxoSmithKline, and has served on the advisory boards for Bristol-Myers Squibb, Celgene, Janssen, and Takeda. Several coauthors also have disclosed relationships with industry. Dr. Rajkumar disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
First CAR T-cell therapy for multiple myeloma: Abecma
Chimeric antigen receptor (CAR) T-cell therapy, described as a “living drug,” is now available for patients with relapsed/refractory multiple myeloma who have been treated with four or more prior lines of therapy.
The Food and Drug Administration said these patients represent an “unmet medical need” when it granted approval for the new product – idecabtagene vicleucel (ide-cel; Abecma), developed by bluebird bio and Bristol-Myers Squibb.
Ide-cel is the first CAR T-cell therapy to gain approval for use in multiple myeloma. It is also the first CAR T-cell therapy to target B-cell maturation antigen.
Previously approved CAR T-cell products target CD19 and have been approved for use in certain types of leukemia and lymphoma.
All the CAR T-cell therapies are customized treatments that are created specifically for each individual patient from their own blood. The patient’s own T cells are removed from the blood, are genetically modified and expanded, and are then infused back into the patient. These modified T cells then seek out and destroy blood cancer cells, and they continue to do so long term.
In some patients, this has led to eradication of disease that had previously progressed with every other treatment that had been tried – results that have been described as “absolutely remarkable” and “one-shot therapy that looks to be curative.”
However, this cell therapy comes with serious adverse effects, including neurologic toxicity and cytokine release syndrome (CRS), which can be life threatening. For this reason, all these products have a risk evaluation and mitigation strategy, and the use of CAR T-cell therapies is limited to designated centers.
In addition, these CAR T-cells products are phenomenally expensive; hospitals have reported heavy financial losses with their use, and patients have turned to crowdfunding to pay for these therapies.
‘Phenomenal’ results in MM
The FDA noted that approval of ide-cel for multiple myeloma is based on data from a multicenter study that involved 127 patients with relapsed/refractory disease who had received at least three prior lines of treatment.
The results from this trial were published Feb. 25 in the New England Journal of Medicine.
An expert not involved in the trial described the results as “phenomenal.”
Krina Patel, MD, an associate professor in the department of lymphoma/myeloma at the University of Texas MD Anderson Cancer Center, Houston, said that “the response rate of 73% in a patient population with a median of six lines of therapy, and with one-third of those patients achieving a deep response of complete response or better, is phenomenal.
“We are very excited as a myeloma community for this study of idecabtagene vicleucel for relapsed/refractory patients,” Dr. Patel told this news organization at the time.
The lead investigator of the study, Nikhil Munshi, MD, of Dana-Farber Cancer Institute, Boston, commented:
Both experts highlighted the poor prognosis for patients with relapsed/refractory disease. Recent decades have seen a flurry of new agents for myeloma, and there are now three main classes of agents: immunomodulatory agents, proteasome inhibitors, and anti-CD38 antibodies.
Nevertheless, in some patients, the disease continues to progress. For patients for whom treatments with all three classes of drugs have failed, the median progression-free survival is 3-4 months, and the median overall survival is 9 months.
In contrast, the results reported in the NEJM article showed that overall median progression-free survival was 8.8 months, but it was more than double that (20.2 months) for patients who achieved a complete or stringent complete response.
Estimated median overall survival was 19.4 months, and the overall survival was 78% at 12 months. The authors note that overall survival data are not yet mature.
The patients who were enrolled in the CAR T-cell trial had undergone many previous treatments. They had undergone a median of six prior drug therapies (range, 3-16), and most of the patients (120, 94%) had also undergone autologous hematopoietic stem cell transplant.
In addition, the majority of patients (84%) had disease that was triple refractory (to an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 antibody), 60% had disease that was penta-exposed (to bortezomib, carfilzomib, lenalidomide, pomalidomide, and daratumumab), and 26% had disease that was penta-refractory.
In the NEJM article, the authors report that about a third of patients had a complete response to CAR T-cell therapy.
At a median follow-up of 13.3 months, 94 of 128 patients (73%) showed a response to therapy (P < .001); 42 (33%) showed a complete or stringent complete response; and 67 patients (52%) showed a “very good partial response or better,” they write.
In the FDA announcement of the product approval, the figures for complete response were slightly lower. “Of those studied, 28% of patients showed complete response – or disappearance of all signs of multiple myeloma – to Abecma, and 65% of this group remained in complete response to the treatment for at least 12 months,” the agency noted.
The FDA also noted that treatment with Abecma can cause severe side effects. The label carries a boxed warning regarding CRS, hemophagocytic lymphohistiocytosis/macrophage activation syndrome, neurologic toxicity, and prolonged cytopenia, all of which can be fatal or life threatening.
The most common side effects of Abecma are CRS, infections, fatigue, musculoskeletal pain, and a weakened immune system. Side effects from treatment usually appear within the first 1-2 weeks after treatment, but some side effects may occur later.
The agency also noted that, to further evaluate the long-term safety of the drug, it is requiring the manufacturer to conduct a postmarketing observational study.
“The FDA remains committed to advancing novel treatment options for areas of unmet patient need,” said Peter Marks, MD, PhD, director of the FDA’s Center for Biologics Evaluation and Research.
“While there is no cure for multiple myeloma, the long-term outlook can vary based on the individual’s age and the stage of the condition at the time of diagnosis. Today’s approval provides a new treatment option for patients who have this uncommon type of cancer.”
A version of this article first appeared on Medscape.com.
Chimeric antigen receptor (CAR) T-cell therapy, described as a “living drug,” is now available for patients with relapsed/refractory multiple myeloma who have been treated with four or more prior lines of therapy.
The Food and Drug Administration said these patients represent an “unmet medical need” when it granted approval for the new product – idecabtagene vicleucel (ide-cel; Abecma), developed by bluebird bio and Bristol-Myers Squibb.
Ide-cel is the first CAR T-cell therapy to gain approval for use in multiple myeloma. It is also the first CAR T-cell therapy to target B-cell maturation antigen.
Previously approved CAR T-cell products target CD19 and have been approved for use in certain types of leukemia and lymphoma.
All the CAR T-cell therapies are customized treatments that are created specifically for each individual patient from their own blood. The patient’s own T cells are removed from the blood, are genetically modified and expanded, and are then infused back into the patient. These modified T cells then seek out and destroy blood cancer cells, and they continue to do so long term.
In some patients, this has led to eradication of disease that had previously progressed with every other treatment that had been tried – results that have been described as “absolutely remarkable” and “one-shot therapy that looks to be curative.”
However, this cell therapy comes with serious adverse effects, including neurologic toxicity and cytokine release syndrome (CRS), which can be life threatening. For this reason, all these products have a risk evaluation and mitigation strategy, and the use of CAR T-cell therapies is limited to designated centers.
In addition, these CAR T-cells products are phenomenally expensive; hospitals have reported heavy financial losses with their use, and patients have turned to crowdfunding to pay for these therapies.
‘Phenomenal’ results in MM
The FDA noted that approval of ide-cel for multiple myeloma is based on data from a multicenter study that involved 127 patients with relapsed/refractory disease who had received at least three prior lines of treatment.
The results from this trial were published Feb. 25 in the New England Journal of Medicine.
An expert not involved in the trial described the results as “phenomenal.”
Krina Patel, MD, an associate professor in the department of lymphoma/myeloma at the University of Texas MD Anderson Cancer Center, Houston, said that “the response rate of 73% in a patient population with a median of six lines of therapy, and with one-third of those patients achieving a deep response of complete response or better, is phenomenal.
“We are very excited as a myeloma community for this study of idecabtagene vicleucel for relapsed/refractory patients,” Dr. Patel told this news organization at the time.
The lead investigator of the study, Nikhil Munshi, MD, of Dana-Farber Cancer Institute, Boston, commented:
Both experts highlighted the poor prognosis for patients with relapsed/refractory disease. Recent decades have seen a flurry of new agents for myeloma, and there are now three main classes of agents: immunomodulatory agents, proteasome inhibitors, and anti-CD38 antibodies.
Nevertheless, in some patients, the disease continues to progress. For patients for whom treatments with all three classes of drugs have failed, the median progression-free survival is 3-4 months, and the median overall survival is 9 months.
In contrast, the results reported in the NEJM article showed that overall median progression-free survival was 8.8 months, but it was more than double that (20.2 months) for patients who achieved a complete or stringent complete response.
Estimated median overall survival was 19.4 months, and the overall survival was 78% at 12 months. The authors note that overall survival data are not yet mature.
The patients who were enrolled in the CAR T-cell trial had undergone many previous treatments. They had undergone a median of six prior drug therapies (range, 3-16), and most of the patients (120, 94%) had also undergone autologous hematopoietic stem cell transplant.
In addition, the majority of patients (84%) had disease that was triple refractory (to an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 antibody), 60% had disease that was penta-exposed (to bortezomib, carfilzomib, lenalidomide, pomalidomide, and daratumumab), and 26% had disease that was penta-refractory.
In the NEJM article, the authors report that about a third of patients had a complete response to CAR T-cell therapy.
At a median follow-up of 13.3 months, 94 of 128 patients (73%) showed a response to therapy (P < .001); 42 (33%) showed a complete or stringent complete response; and 67 patients (52%) showed a “very good partial response or better,” they write.
In the FDA announcement of the product approval, the figures for complete response were slightly lower. “Of those studied, 28% of patients showed complete response – or disappearance of all signs of multiple myeloma – to Abecma, and 65% of this group remained in complete response to the treatment for at least 12 months,” the agency noted.
The FDA also noted that treatment with Abecma can cause severe side effects. The label carries a boxed warning regarding CRS, hemophagocytic lymphohistiocytosis/macrophage activation syndrome, neurologic toxicity, and prolonged cytopenia, all of which can be fatal or life threatening.
The most common side effects of Abecma are CRS, infections, fatigue, musculoskeletal pain, and a weakened immune system. Side effects from treatment usually appear within the first 1-2 weeks after treatment, but some side effects may occur later.
The agency also noted that, to further evaluate the long-term safety of the drug, it is requiring the manufacturer to conduct a postmarketing observational study.
“The FDA remains committed to advancing novel treatment options for areas of unmet patient need,” said Peter Marks, MD, PhD, director of the FDA’s Center for Biologics Evaluation and Research.
“While there is no cure for multiple myeloma, the long-term outlook can vary based on the individual’s age and the stage of the condition at the time of diagnosis. Today’s approval provides a new treatment option for patients who have this uncommon type of cancer.”
A version of this article first appeared on Medscape.com.
Chimeric antigen receptor (CAR) T-cell therapy, described as a “living drug,” is now available for patients with relapsed/refractory multiple myeloma who have been treated with four or more prior lines of therapy.
The Food and Drug Administration said these patients represent an “unmet medical need” when it granted approval for the new product – idecabtagene vicleucel (ide-cel; Abecma), developed by bluebird bio and Bristol-Myers Squibb.
Ide-cel is the first CAR T-cell therapy to gain approval for use in multiple myeloma. It is also the first CAR T-cell therapy to target B-cell maturation antigen.
Previously approved CAR T-cell products target CD19 and have been approved for use in certain types of leukemia and lymphoma.
All the CAR T-cell therapies are customized treatments that are created specifically for each individual patient from their own blood. The patient’s own T cells are removed from the blood, are genetically modified and expanded, and are then infused back into the patient. These modified T cells then seek out and destroy blood cancer cells, and they continue to do so long term.
In some patients, this has led to eradication of disease that had previously progressed with every other treatment that had been tried – results that have been described as “absolutely remarkable” and “one-shot therapy that looks to be curative.”
However, this cell therapy comes with serious adverse effects, including neurologic toxicity and cytokine release syndrome (CRS), which can be life threatening. For this reason, all these products have a risk evaluation and mitigation strategy, and the use of CAR T-cell therapies is limited to designated centers.
In addition, these CAR T-cells products are phenomenally expensive; hospitals have reported heavy financial losses with their use, and patients have turned to crowdfunding to pay for these therapies.
‘Phenomenal’ results in MM
The FDA noted that approval of ide-cel for multiple myeloma is based on data from a multicenter study that involved 127 patients with relapsed/refractory disease who had received at least three prior lines of treatment.
The results from this trial were published Feb. 25 in the New England Journal of Medicine.
An expert not involved in the trial described the results as “phenomenal.”
Krina Patel, MD, an associate professor in the department of lymphoma/myeloma at the University of Texas MD Anderson Cancer Center, Houston, said that “the response rate of 73% in a patient population with a median of six lines of therapy, and with one-third of those patients achieving a deep response of complete response or better, is phenomenal.
“We are very excited as a myeloma community for this study of idecabtagene vicleucel for relapsed/refractory patients,” Dr. Patel told this news organization at the time.
The lead investigator of the study, Nikhil Munshi, MD, of Dana-Farber Cancer Institute, Boston, commented:
Both experts highlighted the poor prognosis for patients with relapsed/refractory disease. Recent decades have seen a flurry of new agents for myeloma, and there are now three main classes of agents: immunomodulatory agents, proteasome inhibitors, and anti-CD38 antibodies.
Nevertheless, in some patients, the disease continues to progress. For patients for whom treatments with all three classes of drugs have failed, the median progression-free survival is 3-4 months, and the median overall survival is 9 months.
In contrast, the results reported in the NEJM article showed that overall median progression-free survival was 8.8 months, but it was more than double that (20.2 months) for patients who achieved a complete or stringent complete response.
Estimated median overall survival was 19.4 months, and the overall survival was 78% at 12 months. The authors note that overall survival data are not yet mature.
The patients who were enrolled in the CAR T-cell trial had undergone many previous treatments. They had undergone a median of six prior drug therapies (range, 3-16), and most of the patients (120, 94%) had also undergone autologous hematopoietic stem cell transplant.
In addition, the majority of patients (84%) had disease that was triple refractory (to an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 antibody), 60% had disease that was penta-exposed (to bortezomib, carfilzomib, lenalidomide, pomalidomide, and daratumumab), and 26% had disease that was penta-refractory.
In the NEJM article, the authors report that about a third of patients had a complete response to CAR T-cell therapy.
At a median follow-up of 13.3 months, 94 of 128 patients (73%) showed a response to therapy (P < .001); 42 (33%) showed a complete or stringent complete response; and 67 patients (52%) showed a “very good partial response or better,” they write.
In the FDA announcement of the product approval, the figures for complete response were slightly lower. “Of those studied, 28% of patients showed complete response – or disappearance of all signs of multiple myeloma – to Abecma, and 65% of this group remained in complete response to the treatment for at least 12 months,” the agency noted.
The FDA also noted that treatment with Abecma can cause severe side effects. The label carries a boxed warning regarding CRS, hemophagocytic lymphohistiocytosis/macrophage activation syndrome, neurologic toxicity, and prolonged cytopenia, all of which can be fatal or life threatening.
The most common side effects of Abecma are CRS, infections, fatigue, musculoskeletal pain, and a weakened immune system. Side effects from treatment usually appear within the first 1-2 weeks after treatment, but some side effects may occur later.
The agency also noted that, to further evaluate the long-term safety of the drug, it is requiring the manufacturer to conduct a postmarketing observational study.
“The FDA remains committed to advancing novel treatment options for areas of unmet patient need,” said Peter Marks, MD, PhD, director of the FDA’s Center for Biologics Evaluation and Research.
“While there is no cure for multiple myeloma, the long-term outlook can vary based on the individual’s age and the stage of the condition at the time of diagnosis. Today’s approval provides a new treatment option for patients who have this uncommon type of cancer.”
A version of this article first appeared on Medscape.com.
Concern over response to COVID-19 in patients with blood cancers
Patients with cancer, particularly those with solid tumors, mounted an immune response to COVID-19 similar to that seen in people without cancer, but among patients with hematologic cancers, immune responses were less pronounced and were highly variable, typically taking longer to clear the virus.
The findings come from a small U.K. study published online Jan. 4 in Cancer Cell as a fast-track preprint article.
The findings may have implications for vaccinating against COVID-19, said the researchers, led by Sheeba Irshad, MD, PhD, a Cancer Research UK clinician scientist based at King’s College London.
“Our study provides some confidence and reassurance to care providers that many of our patients with solid cancers will mount a good immune response against the virus, develop antibodies that last, and hopefully resume their cancer treatment as soon as possible,” Dr. Irshad said in a statement.
“These conclusions imply that many patients, despite being on immunosuppressive therapies, will respond satisfactorily to COVID-19 vaccines,” she added.
Although “the data would suggest that solid cancer patients are likely to mount an efficient immune response to the vaccine ... the same cannot be said for hematological cancers, especially those with B-cell malignancies,” Dr. Irshad said in an interview.
“They may be susceptible to persistent infection despite developing antibodies, so the next stage of our study will focus on monitoring their response to the vaccines.
“At present, the best way to protect them alongside vaccinating them may be to vaccinate all their health care providers and carers to achieve herd immunity and continue to respect the public health measures put in place,” such as wearing a mask, practicing social distancing, and testing asymptomatic persons, she commented.
Study details
This study, known as the SARS-CoV-2 for Cancer Patients study, involved 76 patients with cancer; 41 of these patients had COVID-19, and 35 served as non-COVID cancer control patients.
Peripheral blood was collected from all patients; multiple samples were taken every 2-4 days where possible.
The COVID-19 and control groups were matched for age, body mass index, and tumor type, and both groups included patients with solid and hematologic cancers.
The groups were also comparable in terms of the proportion of patients with stage IV disease, those who received palliative as opposed to radical treatment, and patients who were treated within 4 weeks of recruitment to the study.
The results showed that 24.4% of cancer patients who were exposed to COVID-19 remained asymptomatic, 21.9% had mild disease, 31.7% had moderate disease, and 21.9% had severe disease.
Patients with hematologic cancers were more likely to experience dyspnea than those with solid tumors, and 39% received corticosteroid/antiviral therapies that specifically targeted COVID-19 infection.
The median duration of virus shedding was 39 days across the whole cohort. It was notably longer among patients with hematologic cancers, at a median of 55 days versus 29 days for patients with solid tumors.
Of 46 patients who survived beyond 30 days and for whom complete data were available, the team found that those with moderate or severe COVID-19 were more likely to be diagnosed with progressive cancer at their next assessment in comparison with those who were asymptomatic with COVID-19 or with control patients.
Solid-cancer patients with moderate to severe COVID-19 had sustained lymphopenia and increased neutrophil-to-lymphocyte ratios up to days 40-49 of the infection, whereas among those with mild infection, clinical blood parameters were typically in the normal range.
Although overall blood profiles of patients with hematologic cancers were similar to those of patients with solid cancers, the trajectories between mild and moderate/severe COVID-19 overlapped, and there was a large degree of heterogeneity between patients.
The team also reports that among patients with solid tumors, all parameters returned to values that were close to baseline 4-6 weeks after the patients tested negative for COVID-19 on nasopharyngeal swabbing; by contrast, many of the patients with hematologic cancers experienced ongoing immune dysregulation.
Further analysis revealed differences in immune signatures between patients with solid cancers who had active SARS-CoV-2 infection and noninfected control patients. The former showed, for example, interleukin-8, IL-6, and IL-10, IP-10 enrichment.
In contrast, there were few differences between infected and noninfected hematologic cancer patients.
Across both cohorts, approximately 75% of patients had detectable antibodies against COVID-19. Antibodies were sustained for up to 78 days after exposure to the virus.
However, patients with solid tumors showed earlier seroconversion than those with hematologic cancers. The latter had more varied responses to infection, displaying three distinct phenotypes: failure to mount an antibody response, with prolonged viral shedding, even beyond day 50 after the first positive swab; an antibody response but failure to clear the virus; and an antibody response and successful clearing of the virus.
The team noted that overall patients with hematologic cancers showed a mild response to COVID-19 in the active/early phases of the disease and that the response grew stronger over time, similar to the immune changes typically seen with chronic infections.
This was particularly the case for patients with cancers that affect B cells.
The team acknowledged that there are several limitations to the study, including its small sample size and lack of statistical power to detect differences between, for example, different treatment modalities.
“An important question which remains unanswered is if a ‘reinforced’ immune system following immunotherapy results in an under-/overactivation of the immune response” to COVID-19, the investigators commented. They note that one such patient had a good response.
The SOAP study is sponsored by King’s College London and Guy’s and St. Thomas’ Foundation NHS Trust. It is funded from grants from the KCL Charity funds, MRC, Cancer Research UK, program grants from Breast Cancer Now at King’s College London and by grants to the Breast Cancer Now Toby Robin’s Research Center at the Institute of Cancer Research, London, and the Wellcome Trust Investigator Award, and is supported by the Cancer Research UK Cancer Immunotherapy Accelerator and the UK COVID-Immunology-Consortium. The authors have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Patients with cancer, particularly those with solid tumors, mounted an immune response to COVID-19 similar to that seen in people without cancer, but among patients with hematologic cancers, immune responses were less pronounced and were highly variable, typically taking longer to clear the virus.
The findings come from a small U.K. study published online Jan. 4 in Cancer Cell as a fast-track preprint article.
The findings may have implications for vaccinating against COVID-19, said the researchers, led by Sheeba Irshad, MD, PhD, a Cancer Research UK clinician scientist based at King’s College London.
“Our study provides some confidence and reassurance to care providers that many of our patients with solid cancers will mount a good immune response against the virus, develop antibodies that last, and hopefully resume their cancer treatment as soon as possible,” Dr. Irshad said in a statement.
“These conclusions imply that many patients, despite being on immunosuppressive therapies, will respond satisfactorily to COVID-19 vaccines,” she added.
Although “the data would suggest that solid cancer patients are likely to mount an efficient immune response to the vaccine ... the same cannot be said for hematological cancers, especially those with B-cell malignancies,” Dr. Irshad said in an interview.
“They may be susceptible to persistent infection despite developing antibodies, so the next stage of our study will focus on monitoring their response to the vaccines.
“At present, the best way to protect them alongside vaccinating them may be to vaccinate all their health care providers and carers to achieve herd immunity and continue to respect the public health measures put in place,” such as wearing a mask, practicing social distancing, and testing asymptomatic persons, she commented.
Study details
This study, known as the SARS-CoV-2 for Cancer Patients study, involved 76 patients with cancer; 41 of these patients had COVID-19, and 35 served as non-COVID cancer control patients.
Peripheral blood was collected from all patients; multiple samples were taken every 2-4 days where possible.
The COVID-19 and control groups were matched for age, body mass index, and tumor type, and both groups included patients with solid and hematologic cancers.
The groups were also comparable in terms of the proportion of patients with stage IV disease, those who received palliative as opposed to radical treatment, and patients who were treated within 4 weeks of recruitment to the study.
The results showed that 24.4% of cancer patients who were exposed to COVID-19 remained asymptomatic, 21.9% had mild disease, 31.7% had moderate disease, and 21.9% had severe disease.
Patients with hematologic cancers were more likely to experience dyspnea than those with solid tumors, and 39% received corticosteroid/antiviral therapies that specifically targeted COVID-19 infection.
The median duration of virus shedding was 39 days across the whole cohort. It was notably longer among patients with hematologic cancers, at a median of 55 days versus 29 days for patients with solid tumors.
Of 46 patients who survived beyond 30 days and for whom complete data were available, the team found that those with moderate or severe COVID-19 were more likely to be diagnosed with progressive cancer at their next assessment in comparison with those who were asymptomatic with COVID-19 or with control patients.
Solid-cancer patients with moderate to severe COVID-19 had sustained lymphopenia and increased neutrophil-to-lymphocyte ratios up to days 40-49 of the infection, whereas among those with mild infection, clinical blood parameters were typically in the normal range.
Although overall blood profiles of patients with hematologic cancers were similar to those of patients with solid cancers, the trajectories between mild and moderate/severe COVID-19 overlapped, and there was a large degree of heterogeneity between patients.
The team also reports that among patients with solid tumors, all parameters returned to values that were close to baseline 4-6 weeks after the patients tested negative for COVID-19 on nasopharyngeal swabbing; by contrast, many of the patients with hematologic cancers experienced ongoing immune dysregulation.
Further analysis revealed differences in immune signatures between patients with solid cancers who had active SARS-CoV-2 infection and noninfected control patients. The former showed, for example, interleukin-8, IL-6, and IL-10, IP-10 enrichment.
In contrast, there were few differences between infected and noninfected hematologic cancer patients.
Across both cohorts, approximately 75% of patients had detectable antibodies against COVID-19. Antibodies were sustained for up to 78 days after exposure to the virus.
However, patients with solid tumors showed earlier seroconversion than those with hematologic cancers. The latter had more varied responses to infection, displaying three distinct phenotypes: failure to mount an antibody response, with prolonged viral shedding, even beyond day 50 after the first positive swab; an antibody response but failure to clear the virus; and an antibody response and successful clearing of the virus.
The team noted that overall patients with hematologic cancers showed a mild response to COVID-19 in the active/early phases of the disease and that the response grew stronger over time, similar to the immune changes typically seen with chronic infections.
This was particularly the case for patients with cancers that affect B cells.
The team acknowledged that there are several limitations to the study, including its small sample size and lack of statistical power to detect differences between, for example, different treatment modalities.
“An important question which remains unanswered is if a ‘reinforced’ immune system following immunotherapy results in an under-/overactivation of the immune response” to COVID-19, the investigators commented. They note that one such patient had a good response.
The SOAP study is sponsored by King’s College London and Guy’s and St. Thomas’ Foundation NHS Trust. It is funded from grants from the KCL Charity funds, MRC, Cancer Research UK, program grants from Breast Cancer Now at King’s College London and by grants to the Breast Cancer Now Toby Robin’s Research Center at the Institute of Cancer Research, London, and the Wellcome Trust Investigator Award, and is supported by the Cancer Research UK Cancer Immunotherapy Accelerator and the UK COVID-Immunology-Consortium. The authors have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Patients with cancer, particularly those with solid tumors, mounted an immune response to COVID-19 similar to that seen in people without cancer, but among patients with hematologic cancers, immune responses were less pronounced and were highly variable, typically taking longer to clear the virus.
The findings come from a small U.K. study published online Jan. 4 in Cancer Cell as a fast-track preprint article.
The findings may have implications for vaccinating against COVID-19, said the researchers, led by Sheeba Irshad, MD, PhD, a Cancer Research UK clinician scientist based at King’s College London.
“Our study provides some confidence and reassurance to care providers that many of our patients with solid cancers will mount a good immune response against the virus, develop antibodies that last, and hopefully resume their cancer treatment as soon as possible,” Dr. Irshad said in a statement.
“These conclusions imply that many patients, despite being on immunosuppressive therapies, will respond satisfactorily to COVID-19 vaccines,” she added.
Although “the data would suggest that solid cancer patients are likely to mount an efficient immune response to the vaccine ... the same cannot be said for hematological cancers, especially those with B-cell malignancies,” Dr. Irshad said in an interview.
“They may be susceptible to persistent infection despite developing antibodies, so the next stage of our study will focus on monitoring their response to the vaccines.
“At present, the best way to protect them alongside vaccinating them may be to vaccinate all their health care providers and carers to achieve herd immunity and continue to respect the public health measures put in place,” such as wearing a mask, practicing social distancing, and testing asymptomatic persons, she commented.
Study details
This study, known as the SARS-CoV-2 for Cancer Patients study, involved 76 patients with cancer; 41 of these patients had COVID-19, and 35 served as non-COVID cancer control patients.
Peripheral blood was collected from all patients; multiple samples were taken every 2-4 days where possible.
The COVID-19 and control groups were matched for age, body mass index, and tumor type, and both groups included patients with solid and hematologic cancers.
The groups were also comparable in terms of the proportion of patients with stage IV disease, those who received palliative as opposed to radical treatment, and patients who were treated within 4 weeks of recruitment to the study.
The results showed that 24.4% of cancer patients who were exposed to COVID-19 remained asymptomatic, 21.9% had mild disease, 31.7% had moderate disease, and 21.9% had severe disease.
Patients with hematologic cancers were more likely to experience dyspnea than those with solid tumors, and 39% received corticosteroid/antiviral therapies that specifically targeted COVID-19 infection.
The median duration of virus shedding was 39 days across the whole cohort. It was notably longer among patients with hematologic cancers, at a median of 55 days versus 29 days for patients with solid tumors.
Of 46 patients who survived beyond 30 days and for whom complete data were available, the team found that those with moderate or severe COVID-19 were more likely to be diagnosed with progressive cancer at their next assessment in comparison with those who were asymptomatic with COVID-19 or with control patients.
Solid-cancer patients with moderate to severe COVID-19 had sustained lymphopenia and increased neutrophil-to-lymphocyte ratios up to days 40-49 of the infection, whereas among those with mild infection, clinical blood parameters were typically in the normal range.
Although overall blood profiles of patients with hematologic cancers were similar to those of patients with solid cancers, the trajectories between mild and moderate/severe COVID-19 overlapped, and there was a large degree of heterogeneity between patients.
The team also reports that among patients with solid tumors, all parameters returned to values that were close to baseline 4-6 weeks after the patients tested negative for COVID-19 on nasopharyngeal swabbing; by contrast, many of the patients with hematologic cancers experienced ongoing immune dysregulation.
Further analysis revealed differences in immune signatures between patients with solid cancers who had active SARS-CoV-2 infection and noninfected control patients. The former showed, for example, interleukin-8, IL-6, and IL-10, IP-10 enrichment.
In contrast, there were few differences between infected and noninfected hematologic cancer patients.
Across both cohorts, approximately 75% of patients had detectable antibodies against COVID-19. Antibodies were sustained for up to 78 days after exposure to the virus.
However, patients with solid tumors showed earlier seroconversion than those with hematologic cancers. The latter had more varied responses to infection, displaying three distinct phenotypes: failure to mount an antibody response, with prolonged viral shedding, even beyond day 50 after the first positive swab; an antibody response but failure to clear the virus; and an antibody response and successful clearing of the virus.
The team noted that overall patients with hematologic cancers showed a mild response to COVID-19 in the active/early phases of the disease and that the response grew stronger over time, similar to the immune changes typically seen with chronic infections.
This was particularly the case for patients with cancers that affect B cells.
The team acknowledged that there are several limitations to the study, including its small sample size and lack of statistical power to detect differences between, for example, different treatment modalities.
“An important question which remains unanswered is if a ‘reinforced’ immune system following immunotherapy results in an under-/overactivation of the immune response” to COVID-19, the investigators commented. They note that one such patient had a good response.
The SOAP study is sponsored by King’s College London and Guy’s and St. Thomas’ Foundation NHS Trust. It is funded from grants from the KCL Charity funds, MRC, Cancer Research UK, program grants from Breast Cancer Now at King’s College London and by grants to the Breast Cancer Now Toby Robin’s Research Center at the Institute of Cancer Research, London, and the Wellcome Trust Investigator Award, and is supported by the Cancer Research UK Cancer Immunotherapy Accelerator and the UK COVID-Immunology-Consortium. The authors have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Sequential Targeted Treatment for a Geriatric Patient with Acute Myeloid Leukemia with Concurrent FLT3-TKD and IDH1 Mutations
Nearly 20,000 patients are diagnosed with acute myeloid leukemia (AML) in the US annually.1 Despite the use of aggressive chemotherapeutic agents, the prognosis remains poor, with a mean 5-year survival of 28.3%.2 Fortunately, with the refinement of next-generation sequencing (NGS) hematology panels and development of systemic targeted therapies, the treatment landscape for eligible patients has improved, both in frontline and relapsed or refractory (R/R) patients.
Specifically, investigations into alterations within the FMS-like tyrosine kinase (FLT3) and isocitrate dehydrogenase (IDH) genes have led to the discovery of a number of targeted treatments. Midostaurin is US Food and Drug Administration (FDA)-approved for use in combination with induction chemotherapy for patients with internal tandem duplication of the FLT3 (FLT3-ITD) gene or mutations within the tyrosine kinase domain (FLT3-TKD).3 Ivosidenib is indicated for frontline treatment for those who are poor candidates for induction chemotherapy, and R/R patients who have an R132H mutation in IDH1.4,5 Enasidenib is FDA-approved for R/R patients with R140Q, R172S, and R172K mutations in IDH2.6
The optimal treatment for patients with AML with ≥ 2 clinically actionable mutations has not been established. In this article we describe a geriatric patient who initially was diagnosed with AML with concurrent FLT3-TKD and IDH1 mutations and received targeted, sequential management. We detail changes in disease phenotype and mutational status by repeating an NGS hematology panel and cytogenetic studies after each stage of therapy. Lastly, we discuss the clonal evolution apparent within leukemic cells with use of ≥ 1 or more targeted agents.
Case Presentation
A 68-year-old man presented to the Emergency Department at The Durham Veterans Affairs Medical Center in North Carolina with fatigue and light-headedness. Because of his symptoms and pancytopenia, a bone marrow aspiration and trephine biopsy were performed, which showed 57% myeloblasts, 12% promyelocytes/myelocytes, and 2% metamyelocytes in 20 to 30% cellular bone marrow. Flow cytometry confirmed a blast population consistent with AML. A LeukoVantage (Quest Diagnostics) hematologic NGS panel revealed the presence of FLT3-TKD, IDH1, RUNX1, BCOR-E1477, and SF3B1 mutations (Table). Initial fluorescence in situ hybridization (FISH) results showed a normal pattern of hybridization with no translocations. His disease was deemed to be intermediate-high risk because of the presence of FLT3-TKD and RUNX1 mutations, despite the normal cytogenetic profile and absence of additional clinical features.
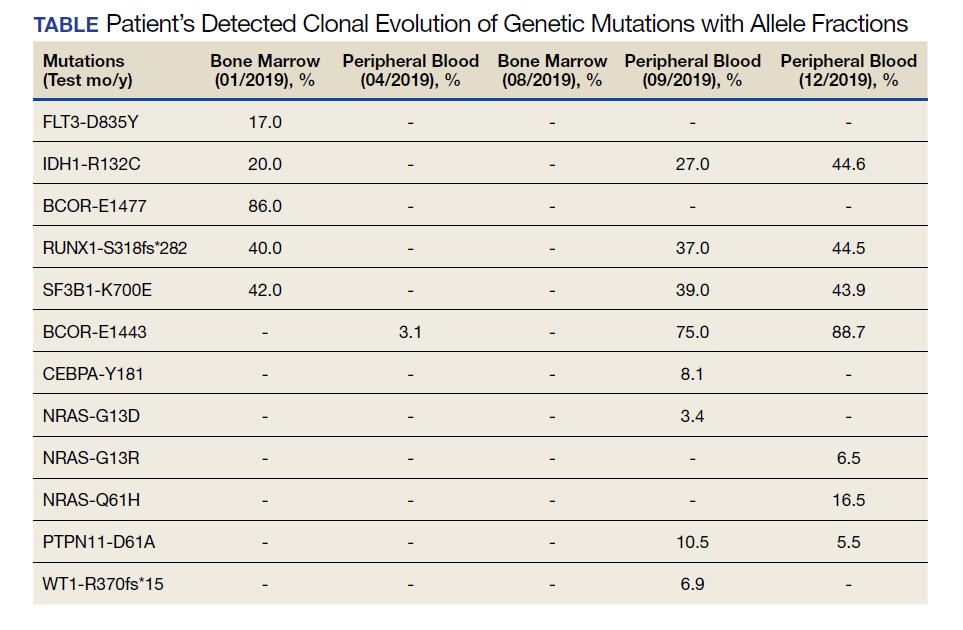
Induction chemotherapy was started with idarubicin, 12 mg/m2, on days 1 to 3 and cytarabine, 200 mg/m2, on days 1 to 7. Because of the presence of a FLT3-TKD mutation, midostaurin was planned for days 8 to 21. After induction chemotherapy, a bone marrow biopsy on day 14 revealed an acellular marrow with no observed myeloblasts. A bone marrow biopsy conducted before initiating consolidation therapy, revealed 30% cellularity with morphologic remission. However, flow cytometry found 5% myeloblasts expressing CD34, CD117, CD13, CD38, and HLA-DR, consistent with measurable residual disease. He received 2 cycles of consolidation therapy with high-dose cytarabine combined with midostaurin. After the patient's second cycle of consolidation, he continued to experience transfusion-dependent cytopenias. Another bone marrow evaluation demonstrated 10% cellularity with nearly all cells appearing to be myeloblasts. A repeat LeukoVantage NGS panel demonstrated undetectable FLT3-TKD mutation and persistent IDH1-R123C mutation. FISH studies revealed a complex karyotype with monosomy of chromosomes 5 and 7 and trisomy of chromosome 8.
We discussed with the patient and his family the options available, which included initiating targeted therapy for his IDH1 mutation, administering hypomethylation therapy with or without venetoclax, or pursuing palliative measures. We collectively decided to pursue therapy with single-agent oral ivosidenib, 500 mg daily. After 1 month of treatment, our patient developed worsening fatigue. His white blood cell count had increased to > 43 k/cm2, raising concern for differentiation syndrome.
A review of the peripheral smear showed a wide-spectrum of maturing granulocytes, with a large percentage of blasts. Peripheral flow cytometry confirmed a blast population of 15%. After a short period of symptom improvement with steroids, the patient developed worsening confusion. Brain imaging identified 2 subdural hemorrhages. Because of a significant peripheral blast population and the development of these hemorrhages, palliative measures were pursued, and the patient was discharged to an inpatient hospice facility. A final NGS panel performed from peripheral blood detected mutations in IDH1, RUNX1, PTPN11, NRAS, BCOR-E1443, and SF3B1 genes.
Discussion
To our knowledge, this is the first reported case of a patient who sequentially received targeted treatments directed against both FLT3 and IDH1 mutations. Initial management with midostaurin and cytarabine resulted in sustained remission of his FLT3-TKD mutation. However, despite receiving prompt standard of care with combination induction chemotherapy and targeted therapy, the patient experienced unfavorable clonal evolution based upon his molecular and cytogenetic testing. Addition of ivosidenib as a second targeting agent for his IDH1 mutation did not achieve a second remission.
Clonal evolution is a well-described phenomenon in hematology. Indolent conditions, such as clonal hematopoiesis of intermediate potential, or malignancies, such as myelodysplastic syndromes and myeloproliferative neoplasms, could transform into acute leukemia through the accumulation of driver mutations and/or cytogenetic abnormalities. Clonal evolution often is viewed as the culprit in patients with AML whose disease relapses after remission with initial chemotherapy.7-10 With the increasing availability of commercial NGS panels designed to assess mutations among patients experiencing hematologic malignancies, patterns of relapse, and, models of clonal evolution could be observed closely in patients with AML.
We were able to monitor molecular changes within our patient’s predominant clonal populations by repeating peripheral comprehensive NGS panels after lines of targeted therapies. The repeated sequencing revealed that clones with FLT3-TKD mutations responded to midostaurin with first-line chemotherapy whereas it was unclear whether clones with IDH1 mutation responded to ivosidenib. Development of complex cytogenetic findings along with the clonal expansion of BCOR mutation-harboring cells likely contributed to our patient’s acutely worsening condition. Several studies have found that the presence of a BCOR mutation in adults with AML leads to lower overall survival and relapse-free survival.11,12 As of now, there are no treatments specifically targeting BCOR mutations.
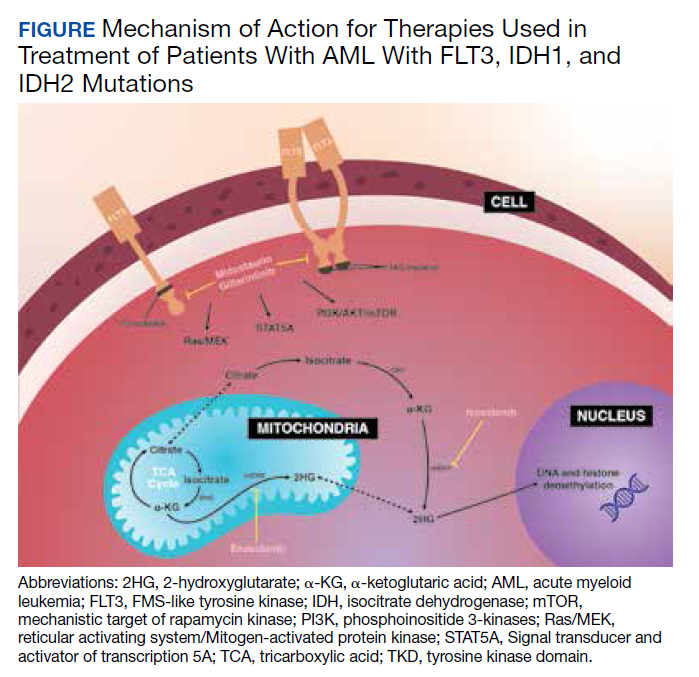
Although there are novel targeting agents with proven efficacy for both FLT3 and IDH1 mutations (Figure), it is difficult to determine which pathogenic mutation drives disease onset. No evidence suggests that these drugs could be administered in tandem. At the present time, interest is directed towards targeting all AML subclones simultaneously, which could reduce the likelihood of evolution among founder clones.7,10,13 In their comparison between molecular profiles and outcomes of patients with AML, Papaemmanuil and colleagues observed that > 80% of patients with AML harbor ≥ 2 driver mutations concurrently.14 Moreover, FLT3-ITD and IDH1 mutations tend to co-occur in approximately 9 to 27% of AML cases.15-18 Available targeted agents for AML are relatively new and hematologists’ familiarity with these drugs is continuing to grow. As the number of novel agents increases, investigations directed toward assessing the safety profile and efficacy of combining targeted agents will be beneficial for patients with AML with ≥ 1 driver mutation.
Conclusions
For our patient with AML, sequential targeted management of FLT3-TKD and IDH1 mutations was not beneficial. Higher-risk disease features, such as the development of a complex karyotype, likely contributed to our patient’s poor response to second-line ivosidenib. The sequential NGS malignant hematology panels allowed us to closely monitor changes to the molecular structure of our patient’s AML after each line of targeted therapy. Future investigations of combining targeted agents for patients with AML with concurrent actionable mutations would provide insight into outcomes of treating multiple clonal populations simultaneously.
1. De Kouchkovsky I, Abdul-Hay M. Acute myeloid leukemia: a comprehensive review and 2016 update. Blood Cancer J. 2016;6(7):e441. doi:10.1038/bcj.2016.50.
2. National Cancer Institute. Cancer Stat Facts: Leukemia — acute myeloid leukemia (AML). Accessed November 4, 2020. https://seer.cancer.gov/statfacts/html/amyl.html
3. Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med. 2017;377(5):454-464. doi:10.1056/NEJMoa1614359.
4. DiNardo CD, Stein EM, de Botton S, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med. 2018;378(25):2386-2398. doi:10.1056/NEJMoa1716984.
5. Roboz, GJ, DiNardo, CD, Stein, EM, et al. Ivosidenib induces deep durable remissions in patients with newly diagnosed IDH1-mutant acute myeloid leukemia. Blood. 2019;135(7), 463-471. doi: 10.1182/blood.2019002140
6. Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722-731. doi:10.1182/blood-2017-04-779405.
7. Jan M, Majeti R. Clonal evolution of acute leukemia genomes. Oncogene. 2013;32(2):135-140. doi:10.1038/onc.2012.48.
8. Grove CS, Vassiliou GS. Acute myeloid leukaemia: a paradigm for the clonal evolution of cancer? Dis Model Mech. 2014;7(8):941-951. doi:10.1242/dmm.015974.
9. Anderson K, Lutz C, van Delft FW, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. 2011;469(7330):356-561. doi: 10.1038/nature09650.
10. Ding L, Ley TJ, Larson DE, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481(7382):506-510. doi:10.1038/nature10738.
11. Terada K, Yamaguchi H, Ueki T, et al. Usefulness of BCOR gene mutation as a prognostic factor in acute myeloid leukemia with intermediate cytogenetic prognosis. Genes Chromosomes Cancer. 2018;57(8):401-408. doi:10.1002/gcc.22542.
12. Grossmann V, Tiacci E, Holmes AB, et al. Whole-exome sequencing identifies somatic mutations of BCOR in acute myeloid leukemia with normal karyotype. Blood. 2011;118(23):6153-6163. doi:10.1182/blood-2011-07-365320.
13. Parkin B, Ouillette P, Li Y, et al. Clonal evolution and devolution after chemotherapy in adult acute myelogenous leukemia. Blood. 2013;121(2):369-377. doi:10.1182/blood-2012-04-427039.
14. Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209-2221. doi:10.1056/NEJMoa1516192.
15. DiNardo CD, Ravandi F, Agresta S, et al. Characteristics, clinical outcome, and prognostic significance of IDH mutations in AML. Am J Hematol. 2015;90(8):732-736. doi:10.1002/ajh.24072.
16. Rakheja D, Konoplev S, Medeiros LJ, Chen W. IDH mutations in acute myeloid leukemia. Hum Pathol. 2012;43 (10):1541-1551. doi:10.1016/j.humpath.2012.05.003.
17. Lai C, Doucette K, Norsworthy K. Recent drug approvals for acute myeloid leukemia. J H Oncol. 2019;12(1):100. doi:10.1186/s13045-019-0774-x.
18. Boddu P, Takahashi K, Pemmaraju N, et al. Influence of IDH on FLT3-ITD status in newly diagnosed AML. Leukemia. 2017;31(11):2526-2529. doi:10.1038/leu.2017.244.
Nearly 20,000 patients are diagnosed with acute myeloid leukemia (AML) in the US annually.1 Despite the use of aggressive chemotherapeutic agents, the prognosis remains poor, with a mean 5-year survival of 28.3%.2 Fortunately, with the refinement of next-generation sequencing (NGS) hematology panels and development of systemic targeted therapies, the treatment landscape for eligible patients has improved, both in frontline and relapsed or refractory (R/R) patients.
Specifically, investigations into alterations within the FMS-like tyrosine kinase (FLT3) and isocitrate dehydrogenase (IDH) genes have led to the discovery of a number of targeted treatments. Midostaurin is US Food and Drug Administration (FDA)-approved for use in combination with induction chemotherapy for patients with internal tandem duplication of the FLT3 (FLT3-ITD) gene or mutations within the tyrosine kinase domain (FLT3-TKD).3 Ivosidenib is indicated for frontline treatment for those who are poor candidates for induction chemotherapy, and R/R patients who have an R132H mutation in IDH1.4,5 Enasidenib is FDA-approved for R/R patients with R140Q, R172S, and R172K mutations in IDH2.6
The optimal treatment for patients with AML with ≥ 2 clinically actionable mutations has not been established. In this article we describe a geriatric patient who initially was diagnosed with AML with concurrent FLT3-TKD and IDH1 mutations and received targeted, sequential management. We detail changes in disease phenotype and mutational status by repeating an NGS hematology panel and cytogenetic studies after each stage of therapy. Lastly, we discuss the clonal evolution apparent within leukemic cells with use of ≥ 1 or more targeted agents.
Case Presentation
A 68-year-old man presented to the Emergency Department at The Durham Veterans Affairs Medical Center in North Carolina with fatigue and light-headedness. Because of his symptoms and pancytopenia, a bone marrow aspiration and trephine biopsy were performed, which showed 57% myeloblasts, 12% promyelocytes/myelocytes, and 2% metamyelocytes in 20 to 30% cellular bone marrow. Flow cytometry confirmed a blast population consistent with AML. A LeukoVantage (Quest Diagnostics) hematologic NGS panel revealed the presence of FLT3-TKD, IDH1, RUNX1, BCOR-E1477, and SF3B1 mutations (Table). Initial fluorescence in situ hybridization (FISH) results showed a normal pattern of hybridization with no translocations. His disease was deemed to be intermediate-high risk because of the presence of FLT3-TKD and RUNX1 mutations, despite the normal cytogenetic profile and absence of additional clinical features.
Induction chemotherapy was started with idarubicin, 12 mg/m2, on days 1 to 3 and cytarabine, 200 mg/m2, on days 1 to 7. Because of the presence of a FLT3-TKD mutation, midostaurin was planned for days 8 to 21. After induction chemotherapy, a bone marrow biopsy on day 14 revealed an acellular marrow with no observed myeloblasts. A bone marrow biopsy conducted before initiating consolidation therapy, revealed 30% cellularity with morphologic remission. However, flow cytometry found 5% myeloblasts expressing CD34, CD117, CD13, CD38, and HLA-DR, consistent with measurable residual disease. He received 2 cycles of consolidation therapy with high-dose cytarabine combined with midostaurin. After the patient's second cycle of consolidation, he continued to experience transfusion-dependent cytopenias. Another bone marrow evaluation demonstrated 10% cellularity with nearly all cells appearing to be myeloblasts. A repeat LeukoVantage NGS panel demonstrated undetectable FLT3-TKD mutation and persistent IDH1-R123C mutation. FISH studies revealed a complex karyotype with monosomy of chromosomes 5 and 7 and trisomy of chromosome 8.
We discussed with the patient and his family the options available, which included initiating targeted therapy for his IDH1 mutation, administering hypomethylation therapy with or without venetoclax, or pursuing palliative measures. We collectively decided to pursue therapy with single-agent oral ivosidenib, 500 mg daily. After 1 month of treatment, our patient developed worsening fatigue. His white blood cell count had increased to > 43 k/cm2, raising concern for differentiation syndrome.
A review of the peripheral smear showed a wide-spectrum of maturing granulocytes, with a large percentage of blasts. Peripheral flow cytometry confirmed a blast population of 15%. After a short period of symptom improvement with steroids, the patient developed worsening confusion. Brain imaging identified 2 subdural hemorrhages. Because of a significant peripheral blast population and the development of these hemorrhages, palliative measures were pursued, and the patient was discharged to an inpatient hospice facility. A final NGS panel performed from peripheral blood detected mutations in IDH1, RUNX1, PTPN11, NRAS, BCOR-E1443, and SF3B1 genes.
Discussion
To our knowledge, this is the first reported case of a patient who sequentially received targeted treatments directed against both FLT3 and IDH1 mutations. Initial management with midostaurin and cytarabine resulted in sustained remission of his FLT3-TKD mutation. However, despite receiving prompt standard of care with combination induction chemotherapy and targeted therapy, the patient experienced unfavorable clonal evolution based upon his molecular and cytogenetic testing. Addition of ivosidenib as a second targeting agent for his IDH1 mutation did not achieve a second remission.
Clonal evolution is a well-described phenomenon in hematology. Indolent conditions, such as clonal hematopoiesis of intermediate potential, or malignancies, such as myelodysplastic syndromes and myeloproliferative neoplasms, could transform into acute leukemia through the accumulation of driver mutations and/or cytogenetic abnormalities. Clonal evolution often is viewed as the culprit in patients with AML whose disease relapses after remission with initial chemotherapy.7-10 With the increasing availability of commercial NGS panels designed to assess mutations among patients experiencing hematologic malignancies, patterns of relapse, and, models of clonal evolution could be observed closely in patients with AML.
We were able to monitor molecular changes within our patient’s predominant clonal populations by repeating peripheral comprehensive NGS panels after lines of targeted therapies. The repeated sequencing revealed that clones with FLT3-TKD mutations responded to midostaurin with first-line chemotherapy whereas it was unclear whether clones with IDH1 mutation responded to ivosidenib. Development of complex cytogenetic findings along with the clonal expansion of BCOR mutation-harboring cells likely contributed to our patient’s acutely worsening condition. Several studies have found that the presence of a BCOR mutation in adults with AML leads to lower overall survival and relapse-free survival.11,12 As of now, there are no treatments specifically targeting BCOR mutations.
Although there are novel targeting agents with proven efficacy for both FLT3 and IDH1 mutations (Figure), it is difficult to determine which pathogenic mutation drives disease onset. No evidence suggests that these drugs could be administered in tandem. At the present time, interest is directed towards targeting all AML subclones simultaneously, which could reduce the likelihood of evolution among founder clones.7,10,13 In their comparison between molecular profiles and outcomes of patients with AML, Papaemmanuil and colleagues observed that > 80% of patients with AML harbor ≥ 2 driver mutations concurrently.14 Moreover, FLT3-ITD and IDH1 mutations tend to co-occur in approximately 9 to 27% of AML cases.15-18 Available targeted agents for AML are relatively new and hematologists’ familiarity with these drugs is continuing to grow. As the number of novel agents increases, investigations directed toward assessing the safety profile and efficacy of combining targeted agents will be beneficial for patients with AML with ≥ 1 driver mutation.
Conclusions
For our patient with AML, sequential targeted management of FLT3-TKD and IDH1 mutations was not beneficial. Higher-risk disease features, such as the development of a complex karyotype, likely contributed to our patient’s poor response to second-line ivosidenib. The sequential NGS malignant hematology panels allowed us to closely monitor changes to the molecular structure of our patient’s AML after each line of targeted therapy. Future investigations of combining targeted agents for patients with AML with concurrent actionable mutations would provide insight into outcomes of treating multiple clonal populations simultaneously.
Nearly 20,000 patients are diagnosed with acute myeloid leukemia (AML) in the US annually.1 Despite the use of aggressive chemotherapeutic agents, the prognosis remains poor, with a mean 5-year survival of 28.3%.2 Fortunately, with the refinement of next-generation sequencing (NGS) hematology panels and development of systemic targeted therapies, the treatment landscape for eligible patients has improved, both in frontline and relapsed or refractory (R/R) patients.
Specifically, investigations into alterations within the FMS-like tyrosine kinase (FLT3) and isocitrate dehydrogenase (IDH) genes have led to the discovery of a number of targeted treatments. Midostaurin is US Food and Drug Administration (FDA)-approved for use in combination with induction chemotherapy for patients with internal tandem duplication of the FLT3 (FLT3-ITD) gene or mutations within the tyrosine kinase domain (FLT3-TKD).3 Ivosidenib is indicated for frontline treatment for those who are poor candidates for induction chemotherapy, and R/R patients who have an R132H mutation in IDH1.4,5 Enasidenib is FDA-approved for R/R patients with R140Q, R172S, and R172K mutations in IDH2.6
The optimal treatment for patients with AML with ≥ 2 clinically actionable mutations has not been established. In this article we describe a geriatric patient who initially was diagnosed with AML with concurrent FLT3-TKD and IDH1 mutations and received targeted, sequential management. We detail changes in disease phenotype and mutational status by repeating an NGS hematology panel and cytogenetic studies after each stage of therapy. Lastly, we discuss the clonal evolution apparent within leukemic cells with use of ≥ 1 or more targeted agents.
Case Presentation
A 68-year-old man presented to the Emergency Department at The Durham Veterans Affairs Medical Center in North Carolina with fatigue and light-headedness. Because of his symptoms and pancytopenia, a bone marrow aspiration and trephine biopsy were performed, which showed 57% myeloblasts, 12% promyelocytes/myelocytes, and 2% metamyelocytes in 20 to 30% cellular bone marrow. Flow cytometry confirmed a blast population consistent with AML. A LeukoVantage (Quest Diagnostics) hematologic NGS panel revealed the presence of FLT3-TKD, IDH1, RUNX1, BCOR-E1477, and SF3B1 mutations (Table). Initial fluorescence in situ hybridization (FISH) results showed a normal pattern of hybridization with no translocations. His disease was deemed to be intermediate-high risk because of the presence of FLT3-TKD and RUNX1 mutations, despite the normal cytogenetic profile and absence of additional clinical features.
Induction chemotherapy was started with idarubicin, 12 mg/m2, on days 1 to 3 and cytarabine, 200 mg/m2, on days 1 to 7. Because of the presence of a FLT3-TKD mutation, midostaurin was planned for days 8 to 21. After induction chemotherapy, a bone marrow biopsy on day 14 revealed an acellular marrow with no observed myeloblasts. A bone marrow biopsy conducted before initiating consolidation therapy, revealed 30% cellularity with morphologic remission. However, flow cytometry found 5% myeloblasts expressing CD34, CD117, CD13, CD38, and HLA-DR, consistent with measurable residual disease. He received 2 cycles of consolidation therapy with high-dose cytarabine combined with midostaurin. After the patient's second cycle of consolidation, he continued to experience transfusion-dependent cytopenias. Another bone marrow evaluation demonstrated 10% cellularity with nearly all cells appearing to be myeloblasts. A repeat LeukoVantage NGS panel demonstrated undetectable FLT3-TKD mutation and persistent IDH1-R123C mutation. FISH studies revealed a complex karyotype with monosomy of chromosomes 5 and 7 and trisomy of chromosome 8.
We discussed with the patient and his family the options available, which included initiating targeted therapy for his IDH1 mutation, administering hypomethylation therapy with or without venetoclax, or pursuing palliative measures. We collectively decided to pursue therapy with single-agent oral ivosidenib, 500 mg daily. After 1 month of treatment, our patient developed worsening fatigue. His white blood cell count had increased to > 43 k/cm2, raising concern for differentiation syndrome.
A review of the peripheral smear showed a wide-spectrum of maturing granulocytes, with a large percentage of blasts. Peripheral flow cytometry confirmed a blast population of 15%. After a short period of symptom improvement with steroids, the patient developed worsening confusion. Brain imaging identified 2 subdural hemorrhages. Because of a significant peripheral blast population and the development of these hemorrhages, palliative measures were pursued, and the patient was discharged to an inpatient hospice facility. A final NGS panel performed from peripheral blood detected mutations in IDH1, RUNX1, PTPN11, NRAS, BCOR-E1443, and SF3B1 genes.
Discussion
To our knowledge, this is the first reported case of a patient who sequentially received targeted treatments directed against both FLT3 and IDH1 mutations. Initial management with midostaurin and cytarabine resulted in sustained remission of his FLT3-TKD mutation. However, despite receiving prompt standard of care with combination induction chemotherapy and targeted therapy, the patient experienced unfavorable clonal evolution based upon his molecular and cytogenetic testing. Addition of ivosidenib as a second targeting agent for his IDH1 mutation did not achieve a second remission.
Clonal evolution is a well-described phenomenon in hematology. Indolent conditions, such as clonal hematopoiesis of intermediate potential, or malignancies, such as myelodysplastic syndromes and myeloproliferative neoplasms, could transform into acute leukemia through the accumulation of driver mutations and/or cytogenetic abnormalities. Clonal evolution often is viewed as the culprit in patients with AML whose disease relapses after remission with initial chemotherapy.7-10 With the increasing availability of commercial NGS panels designed to assess mutations among patients experiencing hematologic malignancies, patterns of relapse, and, models of clonal evolution could be observed closely in patients with AML.
We were able to monitor molecular changes within our patient’s predominant clonal populations by repeating peripheral comprehensive NGS panels after lines of targeted therapies. The repeated sequencing revealed that clones with FLT3-TKD mutations responded to midostaurin with first-line chemotherapy whereas it was unclear whether clones with IDH1 mutation responded to ivosidenib. Development of complex cytogenetic findings along with the clonal expansion of BCOR mutation-harboring cells likely contributed to our patient’s acutely worsening condition. Several studies have found that the presence of a BCOR mutation in adults with AML leads to lower overall survival and relapse-free survival.11,12 As of now, there are no treatments specifically targeting BCOR mutations.
Although there are novel targeting agents with proven efficacy for both FLT3 and IDH1 mutations (Figure), it is difficult to determine which pathogenic mutation drives disease onset. No evidence suggests that these drugs could be administered in tandem. At the present time, interest is directed towards targeting all AML subclones simultaneously, which could reduce the likelihood of evolution among founder clones.7,10,13 In their comparison between molecular profiles and outcomes of patients with AML, Papaemmanuil and colleagues observed that > 80% of patients with AML harbor ≥ 2 driver mutations concurrently.14 Moreover, FLT3-ITD and IDH1 mutations tend to co-occur in approximately 9 to 27% of AML cases.15-18 Available targeted agents for AML are relatively new and hematologists’ familiarity with these drugs is continuing to grow. As the number of novel agents increases, investigations directed toward assessing the safety profile and efficacy of combining targeted agents will be beneficial for patients with AML with ≥ 1 driver mutation.
Conclusions
For our patient with AML, sequential targeted management of FLT3-TKD and IDH1 mutations was not beneficial. Higher-risk disease features, such as the development of a complex karyotype, likely contributed to our patient’s poor response to second-line ivosidenib. The sequential NGS malignant hematology panels allowed us to closely monitor changes to the molecular structure of our patient’s AML after each line of targeted therapy. Future investigations of combining targeted agents for patients with AML with concurrent actionable mutations would provide insight into outcomes of treating multiple clonal populations simultaneously.
1. De Kouchkovsky I, Abdul-Hay M. Acute myeloid leukemia: a comprehensive review and 2016 update. Blood Cancer J. 2016;6(7):e441. doi:10.1038/bcj.2016.50.
2. National Cancer Institute. Cancer Stat Facts: Leukemia — acute myeloid leukemia (AML). Accessed November 4, 2020. https://seer.cancer.gov/statfacts/html/amyl.html
3. Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med. 2017;377(5):454-464. doi:10.1056/NEJMoa1614359.
4. DiNardo CD, Stein EM, de Botton S, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med. 2018;378(25):2386-2398. doi:10.1056/NEJMoa1716984.
5. Roboz, GJ, DiNardo, CD, Stein, EM, et al. Ivosidenib induces deep durable remissions in patients with newly diagnosed IDH1-mutant acute myeloid leukemia. Blood. 2019;135(7), 463-471. doi: 10.1182/blood.2019002140
6. Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722-731. doi:10.1182/blood-2017-04-779405.
7. Jan M, Majeti R. Clonal evolution of acute leukemia genomes. Oncogene. 2013;32(2):135-140. doi:10.1038/onc.2012.48.
8. Grove CS, Vassiliou GS. Acute myeloid leukaemia: a paradigm for the clonal evolution of cancer? Dis Model Mech. 2014;7(8):941-951. doi:10.1242/dmm.015974.
9. Anderson K, Lutz C, van Delft FW, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. 2011;469(7330):356-561. doi: 10.1038/nature09650.
10. Ding L, Ley TJ, Larson DE, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481(7382):506-510. doi:10.1038/nature10738.
11. Terada K, Yamaguchi H, Ueki T, et al. Usefulness of BCOR gene mutation as a prognostic factor in acute myeloid leukemia with intermediate cytogenetic prognosis. Genes Chromosomes Cancer. 2018;57(8):401-408. doi:10.1002/gcc.22542.
12. Grossmann V, Tiacci E, Holmes AB, et al. Whole-exome sequencing identifies somatic mutations of BCOR in acute myeloid leukemia with normal karyotype. Blood. 2011;118(23):6153-6163. doi:10.1182/blood-2011-07-365320.
13. Parkin B, Ouillette P, Li Y, et al. Clonal evolution and devolution after chemotherapy in adult acute myelogenous leukemia. Blood. 2013;121(2):369-377. doi:10.1182/blood-2012-04-427039.
14. Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209-2221. doi:10.1056/NEJMoa1516192.
15. DiNardo CD, Ravandi F, Agresta S, et al. Characteristics, clinical outcome, and prognostic significance of IDH mutations in AML. Am J Hematol. 2015;90(8):732-736. doi:10.1002/ajh.24072.
16. Rakheja D, Konoplev S, Medeiros LJ, Chen W. IDH mutations in acute myeloid leukemia. Hum Pathol. 2012;43 (10):1541-1551. doi:10.1016/j.humpath.2012.05.003.
17. Lai C, Doucette K, Norsworthy K. Recent drug approvals for acute myeloid leukemia. J H Oncol. 2019;12(1):100. doi:10.1186/s13045-019-0774-x.
18. Boddu P, Takahashi K, Pemmaraju N, et al. Influence of IDH on FLT3-ITD status in newly diagnosed AML. Leukemia. 2017;31(11):2526-2529. doi:10.1038/leu.2017.244.
1. De Kouchkovsky I, Abdul-Hay M. Acute myeloid leukemia: a comprehensive review and 2016 update. Blood Cancer J. 2016;6(7):e441. doi:10.1038/bcj.2016.50.
2. National Cancer Institute. Cancer Stat Facts: Leukemia — acute myeloid leukemia (AML). Accessed November 4, 2020. https://seer.cancer.gov/statfacts/html/amyl.html
3. Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med. 2017;377(5):454-464. doi:10.1056/NEJMoa1614359.
4. DiNardo CD, Stein EM, de Botton S, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med. 2018;378(25):2386-2398. doi:10.1056/NEJMoa1716984.
5. Roboz, GJ, DiNardo, CD, Stein, EM, et al. Ivosidenib induces deep durable remissions in patients with newly diagnosed IDH1-mutant acute myeloid leukemia. Blood. 2019;135(7), 463-471. doi: 10.1182/blood.2019002140
6. Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722-731. doi:10.1182/blood-2017-04-779405.
7. Jan M, Majeti R. Clonal evolution of acute leukemia genomes. Oncogene. 2013;32(2):135-140. doi:10.1038/onc.2012.48.
8. Grove CS, Vassiliou GS. Acute myeloid leukaemia: a paradigm for the clonal evolution of cancer? Dis Model Mech. 2014;7(8):941-951. doi:10.1242/dmm.015974.
9. Anderson K, Lutz C, van Delft FW, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. 2011;469(7330):356-561. doi: 10.1038/nature09650.
10. Ding L, Ley TJ, Larson DE, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481(7382):506-510. doi:10.1038/nature10738.
11. Terada K, Yamaguchi H, Ueki T, et al. Usefulness of BCOR gene mutation as a prognostic factor in acute myeloid leukemia with intermediate cytogenetic prognosis. Genes Chromosomes Cancer. 2018;57(8):401-408. doi:10.1002/gcc.22542.
12. Grossmann V, Tiacci E, Holmes AB, et al. Whole-exome sequencing identifies somatic mutations of BCOR in acute myeloid leukemia with normal karyotype. Blood. 2011;118(23):6153-6163. doi:10.1182/blood-2011-07-365320.
13. Parkin B, Ouillette P, Li Y, et al. Clonal evolution and devolution after chemotherapy in adult acute myelogenous leukemia. Blood. 2013;121(2):369-377. doi:10.1182/blood-2012-04-427039.
14. Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209-2221. doi:10.1056/NEJMoa1516192.
15. DiNardo CD, Ravandi F, Agresta S, et al. Characteristics, clinical outcome, and prognostic significance of IDH mutations in AML. Am J Hematol. 2015;90(8):732-736. doi:10.1002/ajh.24072.
16. Rakheja D, Konoplev S, Medeiros LJ, Chen W. IDH mutations in acute myeloid leukemia. Hum Pathol. 2012;43 (10):1541-1551. doi:10.1016/j.humpath.2012.05.003.
17. Lai C, Doucette K, Norsworthy K. Recent drug approvals for acute myeloid leukemia. J H Oncol. 2019;12(1):100. doi:10.1186/s13045-019-0774-x.
18. Boddu P, Takahashi K, Pemmaraju N, et al. Influence of IDH on FLT3-ITD status in newly diagnosed AML. Leukemia. 2017;31(11):2526-2529. doi:10.1038/leu.2017.244.