User login
R2-CHOP doesn’t improve survival in DLBCL
LUGANO, Switzerland – Adding the immunomodulator lenalidomide (Revlimid) to standard chemotherapy for patients with newly diagnosed ABC-type diffuse large B-cell lymphoma (DLBCL) – the so-called R2-CHOP regimen – did not significantly improve either progression-free or overall survival, compared with R-CHOP alone, investigators in the phase 3 ROBUST trial found.

Among 570 patients with activated B-cell (ABC) type DLBCL followed for a median of 27.1 months, median progression-free survival (PFS) – the primary endpoint – had not been reached either for patients randomized to R-CHOP (rituximab, cyclophosphamide, vincristine, doxorubicin and prednisone) plus lenalidomide (R2-CHOP) or R-CHOP plus placebo.
The 1-year and 2-year PFS rate with R2-CHOP was 77%, compared with 75% for R-CHOP, and 2-year PFS rates were 67% and 64%, respectively, and neither comparison was statistically significant reported Umberto Vitolo, MD, from the Citta della Salute e della Scienzia Hospital and University in Turin, Italy.“The future direction is that promising preclinical data with next-generation immunomodulatory agents will be evaluated in future DLBCL clinical trials,” he said at the International Conference on Malignant Lymphoma.
The ROBUST trial is the latest in a long line of studies that failed to show improvement in outcomes with the addition of a novel agent to R-CHOP.
The rationale for adding lenalidomide to R-CHOP came from in-vitro studies showing antiproliferative and immunomodulatory action of lenalidomide against DLBCL, as well as two proof-of-concept clinical studies (REAL07 and MC078E) indicating efficacy against non–germinal center–like B (GCB) type DLBCL, Dr. Vitolo said.
In the ROBUST trial, investigators across 257 global sites enrolled 570 patients with ABC-type DLBCL, stratified them by International Prognostic Index (IPI) score (2 vs. 3 or greater), bulky disease (less than 7 cm vs. 7 cm or more), and age (younger than 65 years vs. 65 years and older) and randomly assigned them to receive R-CHOP with either oral lenalidomide 15 mg or placebo daily on days 1-14 of each 21-day cycle for six cycles.
All patients were required to have neutropenia prophylaxis according to local practice, with either a granulocyte- or granulocyte-macrophage colony-stimulating factor.
The efficacy analysis was by intention-to-treat and included 285 patients in each arm.
The investigators found no significant difference in the primary endpoint of PFS. Overall response rates (ORR) and complete response (CR) rates were high in both arms. The ORR was 91% in each arm, and the CR rate was 69% for R2-CHOP and 65% for R-CHOP.
Event-free survival (EFS) – a composite of first disease progression, death, or relapse after CR or start of second-line therapy – also did not differ significantly between the groups. The 1-year and 2-year EFS rates were 68% vs. 71% and 59% vs. 61%, respectively. The median EFS was not reached in either arm.
Similarly, overall survival did not differ between the groups. At 48 months of follow-up, 57 patients in the R2-CHOP arm and 62 patients in the R-CHOP arm had died. Respective 1- and 2-year overall survival rates were 91% vs. 90%, and 79% vs. 80%.
In the safety analysis, which included 283 patients in the R2-CHOP arm and 284 in the placebo/R-CHOP arm, there were no new safety signals observed. In all, 78% of patients in the lenalidomide arm and 71% in the placebo arm had at least one grade 3 or greater adverse events. The most common adverse events were hematologic, including neutropenia, febrile neutropenia, anemia, thrombocytopenia, and leukopenia.
The ROBUST study was funded by Celgene. Dr. Vitolo reported consulting and speaker’s bureau fees and research funding from the company.
SOURCE: Vitolo U et al. 15-ICML, Abstract 005.
LUGANO, Switzerland – Adding the immunomodulator lenalidomide (Revlimid) to standard chemotherapy for patients with newly diagnosed ABC-type diffuse large B-cell lymphoma (DLBCL) – the so-called R2-CHOP regimen – did not significantly improve either progression-free or overall survival, compared with R-CHOP alone, investigators in the phase 3 ROBUST trial found.
Among 570 patients with activated B-cell (ABC) type DLBCL followed for a median of 27.1 months, median progression-free survival (PFS) – the primary endpoint – had not been reached either for patients randomized to R-CHOP (rituximab, cyclophosphamide, vincristine, doxorubicin and prednisone) plus lenalidomide (R2-CHOP) or R-CHOP plus placebo.
The 1-year and 2-year PFS rate with R2-CHOP was 77%, compared with 75% for R-CHOP, and 2-year PFS rates were 67% and 64%, respectively, and neither comparison was statistically significant reported Umberto Vitolo, MD, from the Citta della Salute e della Scienzia Hospital and University in Turin, Italy.“The future direction is that promising preclinical data with next-generation immunomodulatory agents will be evaluated in future DLBCL clinical trials,” he said at the International Conference on Malignant Lymphoma.
The ROBUST trial is the latest in a long line of studies that failed to show improvement in outcomes with the addition of a novel agent to R-CHOP.
The rationale for adding lenalidomide to R-CHOP came from in-vitro studies showing antiproliferative and immunomodulatory action of lenalidomide against DLBCL, as well as two proof-of-concept clinical studies (REAL07 and MC078E) indicating efficacy against non–germinal center–like B (GCB) type DLBCL, Dr. Vitolo said.
In the ROBUST trial, investigators across 257 global sites enrolled 570 patients with ABC-type DLBCL, stratified them by International Prognostic Index (IPI) score (2 vs. 3 or greater), bulky disease (less than 7 cm vs. 7 cm or more), and age (younger than 65 years vs. 65 years and older) and randomly assigned them to receive R-CHOP with either oral lenalidomide 15 mg or placebo daily on days 1-14 of each 21-day cycle for six cycles.
All patients were required to have neutropenia prophylaxis according to local practice, with either a granulocyte- or granulocyte-macrophage colony-stimulating factor.
The efficacy analysis was by intention-to-treat and included 285 patients in each arm.
The investigators found no significant difference in the primary endpoint of PFS. Overall response rates (ORR) and complete response (CR) rates were high in both arms. The ORR was 91% in each arm, and the CR rate was 69% for R2-CHOP and 65% for R-CHOP.
Event-free survival (EFS) – a composite of first disease progression, death, or relapse after CR or start of second-line therapy – also did not differ significantly between the groups. The 1-year and 2-year EFS rates were 68% vs. 71% and 59% vs. 61%, respectively. The median EFS was not reached in either arm.
Similarly, overall survival did not differ between the groups. At 48 months of follow-up, 57 patients in the R2-CHOP arm and 62 patients in the R-CHOP arm had died. Respective 1- and 2-year overall survival rates were 91% vs. 90%, and 79% vs. 80%.
In the safety analysis, which included 283 patients in the R2-CHOP arm and 284 in the placebo/R-CHOP arm, there were no new safety signals observed. In all, 78% of patients in the lenalidomide arm and 71% in the placebo arm had at least one grade 3 or greater adverse events. The most common adverse events were hematologic, including neutropenia, febrile neutropenia, anemia, thrombocytopenia, and leukopenia.
The ROBUST study was funded by Celgene. Dr. Vitolo reported consulting and speaker’s bureau fees and research funding from the company.
SOURCE: Vitolo U et al. 15-ICML, Abstract 005.
LUGANO, Switzerland – Adding the immunomodulator lenalidomide (Revlimid) to standard chemotherapy for patients with newly diagnosed ABC-type diffuse large B-cell lymphoma (DLBCL) – the so-called R2-CHOP regimen – did not significantly improve either progression-free or overall survival, compared with R-CHOP alone, investigators in the phase 3 ROBUST trial found.
Among 570 patients with activated B-cell (ABC) type DLBCL followed for a median of 27.1 months, median progression-free survival (PFS) – the primary endpoint – had not been reached either for patients randomized to R-CHOP (rituximab, cyclophosphamide, vincristine, doxorubicin and prednisone) plus lenalidomide (R2-CHOP) or R-CHOP plus placebo.
The 1-year and 2-year PFS rate with R2-CHOP was 77%, compared with 75% for R-CHOP, and 2-year PFS rates were 67% and 64%, respectively, and neither comparison was statistically significant reported Umberto Vitolo, MD, from the Citta della Salute e della Scienzia Hospital and University in Turin, Italy.“The future direction is that promising preclinical data with next-generation immunomodulatory agents will be evaluated in future DLBCL clinical trials,” he said at the International Conference on Malignant Lymphoma.
The ROBUST trial is the latest in a long line of studies that failed to show improvement in outcomes with the addition of a novel agent to R-CHOP.
The rationale for adding lenalidomide to R-CHOP came from in-vitro studies showing antiproliferative and immunomodulatory action of lenalidomide against DLBCL, as well as two proof-of-concept clinical studies (REAL07 and MC078E) indicating efficacy against non–germinal center–like B (GCB) type DLBCL, Dr. Vitolo said.
In the ROBUST trial, investigators across 257 global sites enrolled 570 patients with ABC-type DLBCL, stratified them by International Prognostic Index (IPI) score (2 vs. 3 or greater), bulky disease (less than 7 cm vs. 7 cm or more), and age (younger than 65 years vs. 65 years and older) and randomly assigned them to receive R-CHOP with either oral lenalidomide 15 mg or placebo daily on days 1-14 of each 21-day cycle for six cycles.
All patients were required to have neutropenia prophylaxis according to local practice, with either a granulocyte- or granulocyte-macrophage colony-stimulating factor.
The efficacy analysis was by intention-to-treat and included 285 patients in each arm.
The investigators found no significant difference in the primary endpoint of PFS. Overall response rates (ORR) and complete response (CR) rates were high in both arms. The ORR was 91% in each arm, and the CR rate was 69% for R2-CHOP and 65% for R-CHOP.
Event-free survival (EFS) – a composite of first disease progression, death, or relapse after CR or start of second-line therapy – also did not differ significantly between the groups. The 1-year and 2-year EFS rates were 68% vs. 71% and 59% vs. 61%, respectively. The median EFS was not reached in either arm.
Similarly, overall survival did not differ between the groups. At 48 months of follow-up, 57 patients in the R2-CHOP arm and 62 patients in the R-CHOP arm had died. Respective 1- and 2-year overall survival rates were 91% vs. 90%, and 79% vs. 80%.
In the safety analysis, which included 283 patients in the R2-CHOP arm and 284 in the placebo/R-CHOP arm, there were no new safety signals observed. In all, 78% of patients in the lenalidomide arm and 71% in the placebo arm had at least one grade 3 or greater adverse events. The most common adverse events were hematologic, including neutropenia, febrile neutropenia, anemia, thrombocytopenia, and leukopenia.
The ROBUST study was funded by Celgene. Dr. Vitolo reported consulting and speaker’s bureau fees and research funding from the company.
SOURCE: Vitolo U et al. 15-ICML, Abstract 005.
REPORTING FROM 15-ICML
No reduction in PE risk with vena cava filters after severe injury
MELBOURNE – Use of a prophylactic vena cava filter to trap blood clots in severely injured patients does not appear to reduce the risk of pulmonary embolism or death, according to data presented at the International Society on Thrombosis and Haemostasis congress.
The researchers reported the outcomes of a multicenter, controlled trial in which 240 severely injured patients with a contraindication to anticoagulants were randomized to receive a vena cava filter within 72 hours of admission, or no filter. The findings were published simultaneously in the New England Journal of Medicine.
The study showed no significant differences between the filter and no-filter groups in the primary outcome of a composite of symptomatic pulmonary embolism or death from any cause at 90 days after enrollment (13.9% vs. 14.4% respectively, P = .98).
In a prespecified subgroup analysis, researchers examined patients who survived 7 days after injury and did not receive prophylactic anticoagulation in those 7 days. Among this group of patients, none of those who received the vena cava filter experienced a symptomatic pulmonary embolism between day 8 and day 90, but five patients (14.7%) in the no-filter group did.
Filters were left in place for a median duration of 27 days (11-90 days). Among the 122 patients who received a filter – which included two patients in the control group – researchers found trapped thrombi in the filter in six patients.
Transfusion requirements, and the incidence of major and nonmajor bleeding and leg deep vein thrombosis, were similar between the filter and no-filter groups. Seven patients in the filter group (5.7%) required more than one attempt to remove the filter, and in one patient the filter had to be removed surgically.
Kwok M. Ho, PhD, of the department of intensive care medicine at Royal Perth Hospital, Australia, and coauthors wrote that while vena cava filters are widely used in trauma centers to prevent pulmonary embolism in patients at high risk of bleeding, there are conflicting recommendations regarding their use, and most studies so far have been observational.
“Given the cost and risks associated with a vena cava filter, our data suggest that there is no urgency to insert the filter in patients who can be treated with prophylactic anticoagulation within 7 days after injury,” they wrote. “Unnecessary insertion of a vena cava filter has the potential to cause harm.”
However, they noted that patients with multiple, large intracranial hematomas were particularly at risk from bleeding with anticoagulant therapy, and therefore may benefit from the use of a vena cava filter.
The Medical Research Foundation of Royal Perth Hospital and the Western Australian Department of Health funded the study. Dr. Ho reported funding from the Western Australian Department of Health and the Raine Medical Research Foundation to conduct the study, as well as serving as an adviser to Medtronic and Cardinal Health.
SOURCE: Ho KM et al. N Engl J Med. 2019 Jul 7. doi: 10.156/NEJMoa1806515.
MELBOURNE – Use of a prophylactic vena cava filter to trap blood clots in severely injured patients does not appear to reduce the risk of pulmonary embolism or death, according to data presented at the International Society on Thrombosis and Haemostasis congress.
The researchers reported the outcomes of a multicenter, controlled trial in which 240 severely injured patients with a contraindication to anticoagulants were randomized to receive a vena cava filter within 72 hours of admission, or no filter. The findings were published simultaneously in the New England Journal of Medicine.
The study showed no significant differences between the filter and no-filter groups in the primary outcome of a composite of symptomatic pulmonary embolism or death from any cause at 90 days after enrollment (13.9% vs. 14.4% respectively, P = .98).
In a prespecified subgroup analysis, researchers examined patients who survived 7 days after injury and did not receive prophylactic anticoagulation in those 7 days. Among this group of patients, none of those who received the vena cava filter experienced a symptomatic pulmonary embolism between day 8 and day 90, but five patients (14.7%) in the no-filter group did.
Filters were left in place for a median duration of 27 days (11-90 days). Among the 122 patients who received a filter – which included two patients in the control group – researchers found trapped thrombi in the filter in six patients.
Transfusion requirements, and the incidence of major and nonmajor bleeding and leg deep vein thrombosis, were similar between the filter and no-filter groups. Seven patients in the filter group (5.7%) required more than one attempt to remove the filter, and in one patient the filter had to be removed surgically.
Kwok M. Ho, PhD, of the department of intensive care medicine at Royal Perth Hospital, Australia, and coauthors wrote that while vena cava filters are widely used in trauma centers to prevent pulmonary embolism in patients at high risk of bleeding, there are conflicting recommendations regarding their use, and most studies so far have been observational.
“Given the cost and risks associated with a vena cava filter, our data suggest that there is no urgency to insert the filter in patients who can be treated with prophylactic anticoagulation within 7 days after injury,” they wrote. “Unnecessary insertion of a vena cava filter has the potential to cause harm.”
However, they noted that patients with multiple, large intracranial hematomas were particularly at risk from bleeding with anticoagulant therapy, and therefore may benefit from the use of a vena cava filter.
The Medical Research Foundation of Royal Perth Hospital and the Western Australian Department of Health funded the study. Dr. Ho reported funding from the Western Australian Department of Health and the Raine Medical Research Foundation to conduct the study, as well as serving as an adviser to Medtronic and Cardinal Health.
SOURCE: Ho KM et al. N Engl J Med. 2019 Jul 7. doi: 10.156/NEJMoa1806515.
MELBOURNE – Use of a prophylactic vena cava filter to trap blood clots in severely injured patients does not appear to reduce the risk of pulmonary embolism or death, according to data presented at the International Society on Thrombosis and Haemostasis congress.
The researchers reported the outcomes of a multicenter, controlled trial in which 240 severely injured patients with a contraindication to anticoagulants were randomized to receive a vena cava filter within 72 hours of admission, or no filter. The findings were published simultaneously in the New England Journal of Medicine.
The study showed no significant differences between the filter and no-filter groups in the primary outcome of a composite of symptomatic pulmonary embolism or death from any cause at 90 days after enrollment (13.9% vs. 14.4% respectively, P = .98).
In a prespecified subgroup analysis, researchers examined patients who survived 7 days after injury and did not receive prophylactic anticoagulation in those 7 days. Among this group of patients, none of those who received the vena cava filter experienced a symptomatic pulmonary embolism between day 8 and day 90, but five patients (14.7%) in the no-filter group did.
Filters were left in place for a median duration of 27 days (11-90 days). Among the 122 patients who received a filter – which included two patients in the control group – researchers found trapped thrombi in the filter in six patients.
Transfusion requirements, and the incidence of major and nonmajor bleeding and leg deep vein thrombosis, were similar between the filter and no-filter groups. Seven patients in the filter group (5.7%) required more than one attempt to remove the filter, and in one patient the filter had to be removed surgically.
Kwok M. Ho, PhD, of the department of intensive care medicine at Royal Perth Hospital, Australia, and coauthors wrote that while vena cava filters are widely used in trauma centers to prevent pulmonary embolism in patients at high risk of bleeding, there are conflicting recommendations regarding their use, and most studies so far have been observational.
“Given the cost and risks associated with a vena cava filter, our data suggest that there is no urgency to insert the filter in patients who can be treated with prophylactic anticoagulation within 7 days after injury,” they wrote. “Unnecessary insertion of a vena cava filter has the potential to cause harm.”
However, they noted that patients with multiple, large intracranial hematomas were particularly at risk from bleeding with anticoagulant therapy, and therefore may benefit from the use of a vena cava filter.
The Medical Research Foundation of Royal Perth Hospital and the Western Australian Department of Health funded the study. Dr. Ho reported funding from the Western Australian Department of Health and the Raine Medical Research Foundation to conduct the study, as well as serving as an adviser to Medtronic and Cardinal Health.
SOURCE: Ho KM et al. N Engl J Med. 2019 Jul 7. doi: 10.156/NEJMoa1806515.
REPORTING FROM 2019 ISTH CONGRESS
CNS-directed therapy appears more effective for synDLBCL
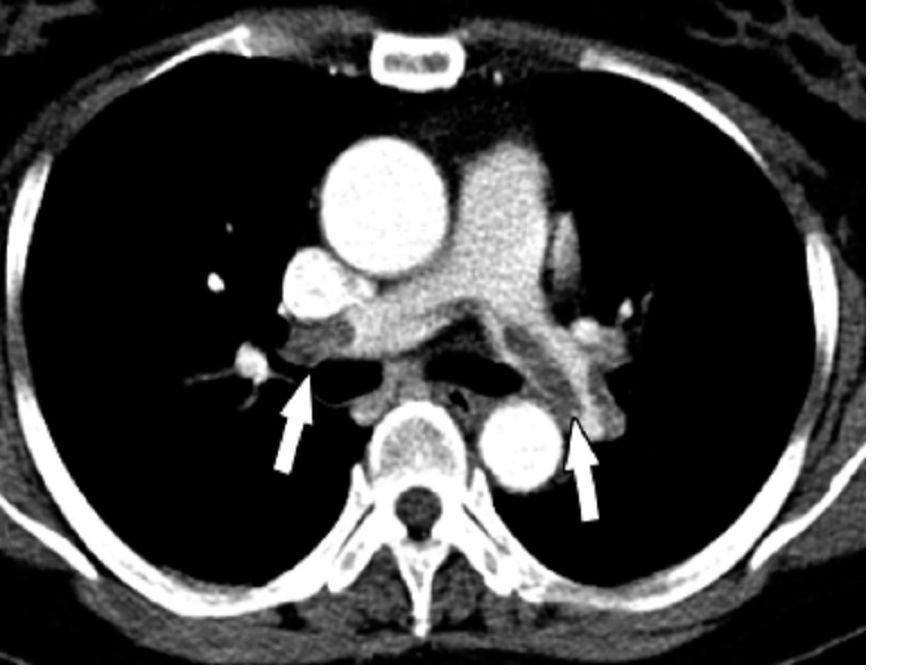
Controlling CNS disease is “paramount” in treating diffuse large B-cell lymphoma with synchronous CNS and systemic disease (synDLBCL), according to researchers.
In a retrospective study, the CNS was the most common site of relapse in patients with synDLBCL, and patients had better outcomes when they received CNS-directed therapy.
The 2-year progression-free survival rate was 50% in patients who received CNS-intensive therapy and 31% in those who received CNS-conservative therapy. The 2-year overall survival rate was 54% and 44%, respectively.
Dr. Joel C. Wight, of Austin Health in Heidelberg, Australia, and colleagues conducted this study and recounted their findings in the British Journal of Haematology.
The researchers retrospectively analyzed 80 patients with synDLBCL treated at 10 centers in Australia and the United Kingdom. Patients had DLBCL not otherwise specified (n = 67); high-grade B-cell lymphoma, including double-hit lymphoma (n = 12); or T-cell histiocyte-rich DLBCL (n = 1).
At baseline, all patients were treatment-naive, they had a median age of 64 years (range, 18-87 years), and 68% were male. Seventy percent of patients had high-risk disease according to the CNS International Prognostic Index (IPI), and 96% had non-CNS extranodal disease. The median number of extranodal sites outside the CNS was 2 (range, 0 to more than 10).
Patients were divided into those who received CNS-intensive therapy (n = 38) and those given CNS-conservative therapy (n = 42). The CNS-conservative group was significantly older (P less than .001), significantly more likely to have high-risk disease according to the National Comprehensive Cancer Network IPI (P = .009) or CNS IPI (P = .01) and significantly more likely to have leptomeningeal disease only (P less than .001).
Treatment
CNS-intensive therapy was defined as any established multiagent IV chemotherapy regimen with two or more CNS-penetrating drugs and cytarabine, with or without intrathecal chemotherapy and/or radiotherapy.
CNS-conservative therapy was defined as one or fewer IV CNS-penetrating chemotherapy agents in induction, with or without intrathecal chemotherapy and/or radiotherapy.
Systemic induction in the CNS-intensive group consisted of R-HyperCVAD (rituximab, hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone alternating with IV methotrexate and cytarabine) in 66% of patients and R-CODOX-M/IVAC (rituximab, hyperfractionated cyclophosphamide, vincristine, doxorubicin, methotrexate/ifosfamide, etoposide, cytarabine) in 24% of patients.
The most common systemic induction regimens in the CNS-conservative group were R-CHOP (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisolone) or CHOP-like regimens, given to 83% of patients.
CNS-directed IV therapy was given to 100% of the CNS-intensive group and 60% of the CNS-conservative group. This consisted of IV methotrexate plus cytarabine (97%) or MATRix (methotrexate, cytarabine, and thiotepa; 3%) in the CNS-intensive group and high-dose methotrexate in the conservative group.
Intrathecal chemotherapy was given to 97% of the CNS-intensive group and 60% of the CNS-conservative group. CNS-directed radiation was given to 32% and 19%, respectively.
Thirteen patients in the CNS-intensive group and one in the CNS-conservative group underwent autologous transplant as consolidation.
Outcomes
Dose reductions were more frequent in the CNS-conservative group than in the CNS-intensive group, at 48% and 18% (P = .009), as was early cessation of chemotherapy, at 52% and 18% (P = .002). Rates of treatment-related mortality were similar, at 13% in the CNS-intensive group and 12% in the CNS-conservative group.
At the end of induction, the complete response rate was 69% in the CNS-intensive group and 51% in the CNS-conservative group (P = .16). Primary refractory disease was observed in 19% and 38% of patients, respectively (P = .07).
The CNS was the most common site of relapse or progression (n = 28). CNS progression or relapse occurred in 25% of the CNS-intensive group and 49% of the CNS-conservative group (P = .03).
The 2-year progression-free survival rate was 50% for the CNS-intensive group and 31% for the CNS-conservative group (P = .006). The 2-year overall survival rate was 54% and 44%, respectively (P = .037).
When patients were matched for induction outcomes, consolidative transplant did not improve survival.
“The most significant factor affecting survival was the ability to control the CNS disease, which was improved by the addition of IV cytarabine to [high-dose methotrexate],” the researchers wrote.
“Whilst the younger age and more intensive systemic treatment of the CNS-intensive group may have contributed to the improved survival, it is clear that CNS disease control was substantially improved by the addition of cytarabine with lower rates of CNS relapse/progression observed.”
The researchers noted that “adequate control of the CNS disease is paramount and is best achieved by intensive CNS-directed induction.”
There was no formal funding for this study, and the researchers did not provide financial disclosures.
SOURCE: Wight JC et al. Br J Haematol. 2019 Jun 24. doi: 10.1111/bjh.16064.
Controlling CNS disease is “paramount” in treating diffuse large B-cell lymphoma with synchronous CNS and systemic disease (synDLBCL), according to researchers.
In a retrospective study, the CNS was the most common site of relapse in patients with synDLBCL, and patients had better outcomes when they received CNS-directed therapy.
The 2-year progression-free survival rate was 50% in patients who received CNS-intensive therapy and 31% in those who received CNS-conservative therapy. The 2-year overall survival rate was 54% and 44%, respectively.
Dr. Joel C. Wight, of Austin Health in Heidelberg, Australia, and colleagues conducted this study and recounted their findings in the British Journal of Haematology.
The researchers retrospectively analyzed 80 patients with synDLBCL treated at 10 centers in Australia and the United Kingdom. Patients had DLBCL not otherwise specified (n = 67); high-grade B-cell lymphoma, including double-hit lymphoma (n = 12); or T-cell histiocyte-rich DLBCL (n = 1).
At baseline, all patients were treatment-naive, they had a median age of 64 years (range, 18-87 years), and 68% were male. Seventy percent of patients had high-risk disease according to the CNS International Prognostic Index (IPI), and 96% had non-CNS extranodal disease. The median number of extranodal sites outside the CNS was 2 (range, 0 to more than 10).
Patients were divided into those who received CNS-intensive therapy (n = 38) and those given CNS-conservative therapy (n = 42). The CNS-conservative group was significantly older (P less than .001), significantly more likely to have high-risk disease according to the National Comprehensive Cancer Network IPI (P = .009) or CNS IPI (P = .01) and significantly more likely to have leptomeningeal disease only (P less than .001).
Treatment
CNS-intensive therapy was defined as any established multiagent IV chemotherapy regimen with two or more CNS-penetrating drugs and cytarabine, with or without intrathecal chemotherapy and/or radiotherapy.
CNS-conservative therapy was defined as one or fewer IV CNS-penetrating chemotherapy agents in induction, with or without intrathecal chemotherapy and/or radiotherapy.
Systemic induction in the CNS-intensive group consisted of R-HyperCVAD (rituximab, hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone alternating with IV methotrexate and cytarabine) in 66% of patients and R-CODOX-M/IVAC (rituximab, hyperfractionated cyclophosphamide, vincristine, doxorubicin, methotrexate/ifosfamide, etoposide, cytarabine) in 24% of patients.
The most common systemic induction regimens in the CNS-conservative group were R-CHOP (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisolone) or CHOP-like regimens, given to 83% of patients.
CNS-directed IV therapy was given to 100% of the CNS-intensive group and 60% of the CNS-conservative group. This consisted of IV methotrexate plus cytarabine (97%) or MATRix (methotrexate, cytarabine, and thiotepa; 3%) in the CNS-intensive group and high-dose methotrexate in the conservative group.
Intrathecal chemotherapy was given to 97% of the CNS-intensive group and 60% of the CNS-conservative group. CNS-directed radiation was given to 32% and 19%, respectively.
Thirteen patients in the CNS-intensive group and one in the CNS-conservative group underwent autologous transplant as consolidation.
Outcomes
Dose reductions were more frequent in the CNS-conservative group than in the CNS-intensive group, at 48% and 18% (P = .009), as was early cessation of chemotherapy, at 52% and 18% (P = .002). Rates of treatment-related mortality were similar, at 13% in the CNS-intensive group and 12% in the CNS-conservative group.
At the end of induction, the complete response rate was 69% in the CNS-intensive group and 51% in the CNS-conservative group (P = .16). Primary refractory disease was observed in 19% and 38% of patients, respectively (P = .07).
The CNS was the most common site of relapse or progression (n = 28). CNS progression or relapse occurred in 25% of the CNS-intensive group and 49% of the CNS-conservative group (P = .03).
The 2-year progression-free survival rate was 50% for the CNS-intensive group and 31% for the CNS-conservative group (P = .006). The 2-year overall survival rate was 54% and 44%, respectively (P = .037).
When patients were matched for induction outcomes, consolidative transplant did not improve survival.
“The most significant factor affecting survival was the ability to control the CNS disease, which was improved by the addition of IV cytarabine to [high-dose methotrexate],” the researchers wrote.
“Whilst the younger age and more intensive systemic treatment of the CNS-intensive group may have contributed to the improved survival, it is clear that CNS disease control was substantially improved by the addition of cytarabine with lower rates of CNS relapse/progression observed.”
The researchers noted that “adequate control of the CNS disease is paramount and is best achieved by intensive CNS-directed induction.”
There was no formal funding for this study, and the researchers did not provide financial disclosures.
SOURCE: Wight JC et al. Br J Haematol. 2019 Jun 24. doi: 10.1111/bjh.16064.
Controlling CNS disease is “paramount” in treating diffuse large B-cell lymphoma with synchronous CNS and systemic disease (synDLBCL), according to researchers.
In a retrospective study, the CNS was the most common site of relapse in patients with synDLBCL, and patients had better outcomes when they received CNS-directed therapy.
The 2-year progression-free survival rate was 50% in patients who received CNS-intensive therapy and 31% in those who received CNS-conservative therapy. The 2-year overall survival rate was 54% and 44%, respectively.
Dr. Joel C. Wight, of Austin Health in Heidelberg, Australia, and colleagues conducted this study and recounted their findings in the British Journal of Haematology.
The researchers retrospectively analyzed 80 patients with synDLBCL treated at 10 centers in Australia and the United Kingdom. Patients had DLBCL not otherwise specified (n = 67); high-grade B-cell lymphoma, including double-hit lymphoma (n = 12); or T-cell histiocyte-rich DLBCL (n = 1).
At baseline, all patients were treatment-naive, they had a median age of 64 years (range, 18-87 years), and 68% were male. Seventy percent of patients had high-risk disease according to the CNS International Prognostic Index (IPI), and 96% had non-CNS extranodal disease. The median number of extranodal sites outside the CNS was 2 (range, 0 to more than 10).
Patients were divided into those who received CNS-intensive therapy (n = 38) and those given CNS-conservative therapy (n = 42). The CNS-conservative group was significantly older (P less than .001), significantly more likely to have high-risk disease according to the National Comprehensive Cancer Network IPI (P = .009) or CNS IPI (P = .01) and significantly more likely to have leptomeningeal disease only (P less than .001).
Treatment
CNS-intensive therapy was defined as any established multiagent IV chemotherapy regimen with two or more CNS-penetrating drugs and cytarabine, with or without intrathecal chemotherapy and/or radiotherapy.
CNS-conservative therapy was defined as one or fewer IV CNS-penetrating chemotherapy agents in induction, with or without intrathecal chemotherapy and/or radiotherapy.
Systemic induction in the CNS-intensive group consisted of R-HyperCVAD (rituximab, hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone alternating with IV methotrexate and cytarabine) in 66% of patients and R-CODOX-M/IVAC (rituximab, hyperfractionated cyclophosphamide, vincristine, doxorubicin, methotrexate/ifosfamide, etoposide, cytarabine) in 24% of patients.
The most common systemic induction regimens in the CNS-conservative group were R-CHOP (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisolone) or CHOP-like regimens, given to 83% of patients.
CNS-directed IV therapy was given to 100% of the CNS-intensive group and 60% of the CNS-conservative group. This consisted of IV methotrexate plus cytarabine (97%) or MATRix (methotrexate, cytarabine, and thiotepa; 3%) in the CNS-intensive group and high-dose methotrexate in the conservative group.
Intrathecal chemotherapy was given to 97% of the CNS-intensive group and 60% of the CNS-conservative group. CNS-directed radiation was given to 32% and 19%, respectively.
Thirteen patients in the CNS-intensive group and one in the CNS-conservative group underwent autologous transplant as consolidation.
Outcomes
Dose reductions were more frequent in the CNS-conservative group than in the CNS-intensive group, at 48% and 18% (P = .009), as was early cessation of chemotherapy, at 52% and 18% (P = .002). Rates of treatment-related mortality were similar, at 13% in the CNS-intensive group and 12% in the CNS-conservative group.
At the end of induction, the complete response rate was 69% in the CNS-intensive group and 51% in the CNS-conservative group (P = .16). Primary refractory disease was observed in 19% and 38% of patients, respectively (P = .07).
The CNS was the most common site of relapse or progression (n = 28). CNS progression or relapse occurred in 25% of the CNS-intensive group and 49% of the CNS-conservative group (P = .03).
The 2-year progression-free survival rate was 50% for the CNS-intensive group and 31% for the CNS-conservative group (P = .006). The 2-year overall survival rate was 54% and 44%, respectively (P = .037).
When patients were matched for induction outcomes, consolidative transplant did not improve survival.
“The most significant factor affecting survival was the ability to control the CNS disease, which was improved by the addition of IV cytarabine to [high-dose methotrexate],” the researchers wrote.
“Whilst the younger age and more intensive systemic treatment of the CNS-intensive group may have contributed to the improved survival, it is clear that CNS disease control was substantially improved by the addition of cytarabine with lower rates of CNS relapse/progression observed.”
The researchers noted that “adequate control of the CNS disease is paramount and is best achieved by intensive CNS-directed induction.”
There was no formal funding for this study, and the researchers did not provide financial disclosures.
SOURCE: Wight JC et al. Br J Haematol. 2019 Jun 24. doi: 10.1111/bjh.16064.
FROM BRITISH JOURNAL OF HAEMATOLOGY
FDA approves Xpovio for relapsed/refractory multiple myeloma

The oral therapy was approved for patients who have received at least four prior therapies and whose disease is resistant to several other forms of treatment, including at least two proteasome inhibitors, at least two immunomodulatory agents, and an anti-CD38 monoclonal antibody, according to the FDA.
The approval provides a “treatment option for patients with multiple myeloma with no (other) available therapy,” said Richard Pazdur, MD, director of the FDA Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA Center for Drug Evaluation and Research.
The approval was based on a study that included 83 patients with RRMM who had an overall response rate of 25.3% to Xpovio in combination with dexamethasone.
“The median time to first response was 4 weeks, with a range of 1-10 weeks. The median duration of response was 3.8 months. The efficacy evaluation was supported by additional information from an ongoing, randomized trial in patients with multiple myeloma,” according to the statement.
Common side effects seen in patients taking Xpovio in combination with dexamethasone include leukopenia, neutropenia, thrombocytopenia, and anemia. Patients also reported vomiting, nausea, fatigue, diarrhea, fever, decreased appetite and weight, constipation, upper respiratory tract infections, and hyponatremia.
Patients taking Xpovio should be monitored for low blood counts, platelets, and sodium levels, and should avoid other medications that may cause dizziness or confusion. Patients’ hydration status, blood counts, and other medications should be optimized to avoid dizziness or confusion. Females of reproductive age and males with a female partner of reproductive potential must use effective contraception during treatment with Xpovio. Women who are pregnant or breastfeeding should not take Xpovio.
Xpovio must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks.
Xpovio in combination with dexamethasone was granted accelerated approval, and further clinical trials are required to verify and describe the drug’s clinical benefit.
The FDA granted the approval of Xpovio to Karyopharm Therapeutics.
The oral therapy was approved for patients who have received at least four prior therapies and whose disease is resistant to several other forms of treatment, including at least two proteasome inhibitors, at least two immunomodulatory agents, and an anti-CD38 monoclonal antibody, according to the FDA.
The approval provides a “treatment option for patients with multiple myeloma with no (other) available therapy,” said Richard Pazdur, MD, director of the FDA Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA Center for Drug Evaluation and Research.
The approval was based on a study that included 83 patients with RRMM who had an overall response rate of 25.3% to Xpovio in combination with dexamethasone.
“The median time to first response was 4 weeks, with a range of 1-10 weeks. The median duration of response was 3.8 months. The efficacy evaluation was supported by additional information from an ongoing, randomized trial in patients with multiple myeloma,” according to the statement.
Common side effects seen in patients taking Xpovio in combination with dexamethasone include leukopenia, neutropenia, thrombocytopenia, and anemia. Patients also reported vomiting, nausea, fatigue, diarrhea, fever, decreased appetite and weight, constipation, upper respiratory tract infections, and hyponatremia.
Patients taking Xpovio should be monitored for low blood counts, platelets, and sodium levels, and should avoid other medications that may cause dizziness or confusion. Patients’ hydration status, blood counts, and other medications should be optimized to avoid dizziness or confusion. Females of reproductive age and males with a female partner of reproductive potential must use effective contraception during treatment with Xpovio. Women who are pregnant or breastfeeding should not take Xpovio.
Xpovio must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks.
Xpovio in combination with dexamethasone was granted accelerated approval, and further clinical trials are required to verify and describe the drug’s clinical benefit.
The FDA granted the approval of Xpovio to Karyopharm Therapeutics.
The oral therapy was approved for patients who have received at least four prior therapies and whose disease is resistant to several other forms of treatment, including at least two proteasome inhibitors, at least two immunomodulatory agents, and an anti-CD38 monoclonal antibody, according to the FDA.
The approval provides a “treatment option for patients with multiple myeloma with no (other) available therapy,” said Richard Pazdur, MD, director of the FDA Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA Center for Drug Evaluation and Research.
The approval was based on a study that included 83 patients with RRMM who had an overall response rate of 25.3% to Xpovio in combination with dexamethasone.
“The median time to first response was 4 weeks, with a range of 1-10 weeks. The median duration of response was 3.8 months. The efficacy evaluation was supported by additional information from an ongoing, randomized trial in patients with multiple myeloma,” according to the statement.
Common side effects seen in patients taking Xpovio in combination with dexamethasone include leukopenia, neutropenia, thrombocytopenia, and anemia. Patients also reported vomiting, nausea, fatigue, diarrhea, fever, decreased appetite and weight, constipation, upper respiratory tract infections, and hyponatremia.
Patients taking Xpovio should be monitored for low blood counts, platelets, and sodium levels, and should avoid other medications that may cause dizziness or confusion. Patients’ hydration status, blood counts, and other medications should be optimized to avoid dizziness or confusion. Females of reproductive age and males with a female partner of reproductive potential must use effective contraception during treatment with Xpovio. Women who are pregnant or breastfeeding should not take Xpovio.
Xpovio must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks.
Xpovio in combination with dexamethasone was granted accelerated approval, and further clinical trials are required to verify and describe the drug’s clinical benefit.
The FDA granted the approval of Xpovio to Karyopharm Therapeutics.
Cancer deaths cost over $94 billion in lost earnings in 2015
Cancer led to 492,000 deaths for Americans aged 16-84 years in 2015, and those deaths cost $94.4 billion in lost earnings that year, according to a study published in JAMA Oncology.
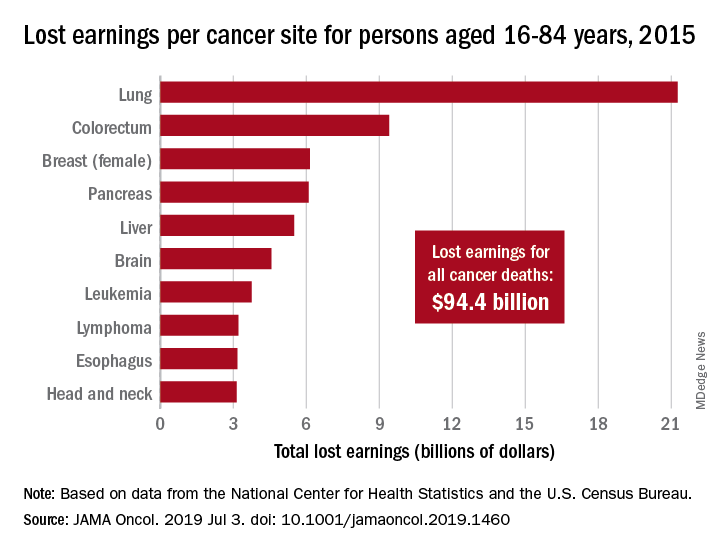
Cancer also took more than 8.7 million years of life from those individuals, with lung cancer being the most costly in terms of both lost earnings and years of life lost, said Farhad Islami, MD, PhD, and associates at the American Cancer Society, Atlanta.
“Person-years of life lost and lost earnings were high for many cancers associated with modifiable risk factors and effective screening and treatment, suggesting that a substantial proportion of the mortality burden is potentially avoidable,” they wrote, adding that “implementation of comprehensive cancer prevention interventions and equitable access to high-quality care across all states could reduce the burden of cancer and associated geographic and other differences in the country.”
In 2015, lung cancer took more than 2.2 million years of life and $21.3 billion in earnings from Americans aged 16-84 years. Colorectal cancer was next with 766,000 years of life lost and $9.4 billion in lost earnings, followed by female breast cancer with losses of 746,000 years of life and 6.2 billion in earnings, Dr. Islami and associated reported.
For all cancers, the cost in lost earnings per death was almost $192,000, with the highest costs coming from cancers of the brain and nervous system ($315,000) and cervix ($311,000). On that basis, lung cancer cost was lower than average at $159,000 per death, they noted.
At the state level, lost-earnings rates were lowest in Utah ($19.6 million per 100,000 persons) and highest in Kentucky ($35.3 million per 100,000). “If all states had Utah’s lost earnings rate in 2015, lost earnings in the U.S. would have been reduced by 29.3%, or $27.7 billion, and life years lost nationwide in 2015 would be reduced by 2.4 million,” Dr. Islami and his associates said in a written statement.
Data for the study were obtained from the National Center for Health Statistics (mortality) and the U.S. Census Bureau (earnings). The study was supported by the Intramural Research Department of the American Cancer Society, and all of the investigators are employees of the society.
SOURCE: Islami F et al. JAMA Oncol. 2019 Jul 3. doi: 10.1001/jamaoncol.2019.1460.
Cancer led to 492,000 deaths for Americans aged 16-84 years in 2015, and those deaths cost $94.4 billion in lost earnings that year, according to a study published in JAMA Oncology.
Cancer also took more than 8.7 million years of life from those individuals, with lung cancer being the most costly in terms of both lost earnings and years of life lost, said Farhad Islami, MD, PhD, and associates at the American Cancer Society, Atlanta.
“Person-years of life lost and lost earnings were high for many cancers associated with modifiable risk factors and effective screening and treatment, suggesting that a substantial proportion of the mortality burden is potentially avoidable,” they wrote, adding that “implementation of comprehensive cancer prevention interventions and equitable access to high-quality care across all states could reduce the burden of cancer and associated geographic and other differences in the country.”
In 2015, lung cancer took more than 2.2 million years of life and $21.3 billion in earnings from Americans aged 16-84 years. Colorectal cancer was next with 766,000 years of life lost and $9.4 billion in lost earnings, followed by female breast cancer with losses of 746,000 years of life and 6.2 billion in earnings, Dr. Islami and associated reported.
For all cancers, the cost in lost earnings per death was almost $192,000, with the highest costs coming from cancers of the brain and nervous system ($315,000) and cervix ($311,000). On that basis, lung cancer cost was lower than average at $159,000 per death, they noted.
At the state level, lost-earnings rates were lowest in Utah ($19.6 million per 100,000 persons) and highest in Kentucky ($35.3 million per 100,000). “If all states had Utah’s lost earnings rate in 2015, lost earnings in the U.S. would have been reduced by 29.3%, or $27.7 billion, and life years lost nationwide in 2015 would be reduced by 2.4 million,” Dr. Islami and his associates said in a written statement.
Data for the study were obtained from the National Center for Health Statistics (mortality) and the U.S. Census Bureau (earnings). The study was supported by the Intramural Research Department of the American Cancer Society, and all of the investigators are employees of the society.
SOURCE: Islami F et al. JAMA Oncol. 2019 Jul 3. doi: 10.1001/jamaoncol.2019.1460.
Cancer led to 492,000 deaths for Americans aged 16-84 years in 2015, and those deaths cost $94.4 billion in lost earnings that year, according to a study published in JAMA Oncology.
Cancer also took more than 8.7 million years of life from those individuals, with lung cancer being the most costly in terms of both lost earnings and years of life lost, said Farhad Islami, MD, PhD, and associates at the American Cancer Society, Atlanta.
“Person-years of life lost and lost earnings were high for many cancers associated with modifiable risk factors and effective screening and treatment, suggesting that a substantial proportion of the mortality burden is potentially avoidable,” they wrote, adding that “implementation of comprehensive cancer prevention interventions and equitable access to high-quality care across all states could reduce the burden of cancer and associated geographic and other differences in the country.”
In 2015, lung cancer took more than 2.2 million years of life and $21.3 billion in earnings from Americans aged 16-84 years. Colorectal cancer was next with 766,000 years of life lost and $9.4 billion in lost earnings, followed by female breast cancer with losses of 746,000 years of life and 6.2 billion in earnings, Dr. Islami and associated reported.
For all cancers, the cost in lost earnings per death was almost $192,000, with the highest costs coming from cancers of the brain and nervous system ($315,000) and cervix ($311,000). On that basis, lung cancer cost was lower than average at $159,000 per death, they noted.
At the state level, lost-earnings rates were lowest in Utah ($19.6 million per 100,000 persons) and highest in Kentucky ($35.3 million per 100,000). “If all states had Utah’s lost earnings rate in 2015, lost earnings in the U.S. would have been reduced by 29.3%, or $27.7 billion, and life years lost nationwide in 2015 would be reduced by 2.4 million,” Dr. Islami and his associates said in a written statement.
Data for the study were obtained from the National Center for Health Statistics (mortality) and the U.S. Census Bureau (earnings). The study was supported by the Intramural Research Department of the American Cancer Society, and all of the investigators are employees of the society.
SOURCE: Islami F et al. JAMA Oncol. 2019 Jul 3. doi: 10.1001/jamaoncol.2019.1460.
FROM JAMA ONCOLOGY
AGA Clinical Practice Update: Coagulation in cirrhosis
Cirrhosis can involve “precarious” changes in hemostatic pathways that tip the scales toward either bleeding or hypercoagulation, experts wrote in an American Gastroenterological Association Clinical Practice Update.
Based on current evidence, clinicians should not routinely correct thrombocytopenia and coagulopathy in patients with cirrhosis prior to low-risk procedures, such as therapeutic paracentesis, thoracentesis, and routine upper endoscopy for variceal ligation, Jacqueline G. O’Leary, MD, of Dallas VA Medical Center and her three coreviewers wrote in Gastroenterology.
To optimize clot formation prior to high-risk procedures, and in patients with active bleeding, a platelet count above 50,000 per mcL is still recommended. However, it may be more meaningful to couple that platelet target with a fibrinogen level above 120 mg/dL rather than rely on the international normalized ratio (INR), the experts wrote. Not only does INR vary significantly depending on which thromboplastin is used in the test, but “correcting” INR with a fresh frozen plasma infusion does not affect thrombin production and worsens portal hypertension. Using cryoprecipitate to replenish fibrinogen has less impact on portal hypertension. “Global tests of clot formation, such as rotational thromboelastometry (ROTEM), thromboelastography (TEG), sonorheometry, and thrombin generation may eventually have a role in the evaluation of clotting in patients with cirrhosis but currently lack validated target levels,” the experts wrote.
They advised clinicians to limit the use of blood products (such as fresh frozen plasma and pooled platelet transfusions) because of cost and the risk of exacerbated portal hypertension, infection, and immunologic complications. For severe anemia and uremia, red blood cell transfusion (250 mL) can be considered. Platelet-rich plasma from one donor is less immunologically risky than a pooled platelet transfusion. Thrombopoietin agonists are “a good alternative” to platelet transfusion but require about 10 days for response. Alternative prothrombotic therapies include oral thrombopoietin receptor agonists (avatrombopag and lusutrombopag) to boost platelet count before an invasive procedure, antifibrinolytic therapy (aminocaproic acid and tranexamic acid) for persistent bleeding from mucosal oozing or puncture wounds. Desmopressin should only be considered for patients with comorbid renal failure.
For anticoagulation, the practice update recommends considering systemic heparin infusion for cirrhotic patients with symptomatic deep venous thrombosis (DVT) or portal vein thrombosis (PVT). However, the anti–factor Xa assay will not reliably monitor response if patients have low liver-derived antithrombin III (heparin cofactor). “With newly diagnosed PVT, the decision to intervene with directed therapy rests on the extent of the thrombosis, presence or absence of attributable symptoms, and the risk of bleeding and falls,” the experts stated.
Six-month follow-up imaging is recommended to assess anticoagulation efficacy. More frequent imaging can be considered for PVT patients considered at high risk for therapeutic anticoagulation. If clots do not fully resolve after 6 months of treatment, options including extending therapy with the same agent, switching to a different anticoagulant class, or receiving transjugular intrahepatic portosystemic shunt (TIPS). “The role for TIPS in PVT is evolving and may address complications like portal hypertensive bleeding, medically refractory clot, and the need for repeated banding after variceal bleeding,” the experts noted.
Prophylaxis of DVT is recommended for all hospitalized patients with cirrhosis. Vitamin K antagonists and direct-acting oral anticoagulants (dabigatran, apixaban, rivaroxaban, and edoxaban) are alternatives to heparin for anticoagulation of cirrhotic patients with either PVT and DVT, the experts wrote. However, DOACs are not recommended for most Child-Pugh B patients or for any Child-Pugh C patients.
No funding sources or conflicts of interest were reported.
SOURCE: O’Leary JG et al. Gastroenterology. 2019. doi: 10.1053/j.gastro.2019.03.070.
Cirrhosis can involve “precarious” changes in hemostatic pathways that tip the scales toward either bleeding or hypercoagulation, experts wrote in an American Gastroenterological Association Clinical Practice Update.
Based on current evidence, clinicians should not routinely correct thrombocytopenia and coagulopathy in patients with cirrhosis prior to low-risk procedures, such as therapeutic paracentesis, thoracentesis, and routine upper endoscopy for variceal ligation, Jacqueline G. O’Leary, MD, of Dallas VA Medical Center and her three coreviewers wrote in Gastroenterology.
To optimize clot formation prior to high-risk procedures, and in patients with active bleeding, a platelet count above 50,000 per mcL is still recommended. However, it may be more meaningful to couple that platelet target with a fibrinogen level above 120 mg/dL rather than rely on the international normalized ratio (INR), the experts wrote. Not only does INR vary significantly depending on which thromboplastin is used in the test, but “correcting” INR with a fresh frozen plasma infusion does not affect thrombin production and worsens portal hypertension. Using cryoprecipitate to replenish fibrinogen has less impact on portal hypertension. “Global tests of clot formation, such as rotational thromboelastometry (ROTEM), thromboelastography (TEG), sonorheometry, and thrombin generation may eventually have a role in the evaluation of clotting in patients with cirrhosis but currently lack validated target levels,” the experts wrote.
They advised clinicians to limit the use of blood products (such as fresh frozen plasma and pooled platelet transfusions) because of cost and the risk of exacerbated portal hypertension, infection, and immunologic complications. For severe anemia and uremia, red blood cell transfusion (250 mL) can be considered. Platelet-rich plasma from one donor is less immunologically risky than a pooled platelet transfusion. Thrombopoietin agonists are “a good alternative” to platelet transfusion but require about 10 days for response. Alternative prothrombotic therapies include oral thrombopoietin receptor agonists (avatrombopag and lusutrombopag) to boost platelet count before an invasive procedure, antifibrinolytic therapy (aminocaproic acid and tranexamic acid) for persistent bleeding from mucosal oozing or puncture wounds. Desmopressin should only be considered for patients with comorbid renal failure.
For anticoagulation, the practice update recommends considering systemic heparin infusion for cirrhotic patients with symptomatic deep venous thrombosis (DVT) or portal vein thrombosis (PVT). However, the anti–factor Xa assay will not reliably monitor response if patients have low liver-derived antithrombin III (heparin cofactor). “With newly diagnosed PVT, the decision to intervene with directed therapy rests on the extent of the thrombosis, presence or absence of attributable symptoms, and the risk of bleeding and falls,” the experts stated.
Six-month follow-up imaging is recommended to assess anticoagulation efficacy. More frequent imaging can be considered for PVT patients considered at high risk for therapeutic anticoagulation. If clots do not fully resolve after 6 months of treatment, options including extending therapy with the same agent, switching to a different anticoagulant class, or receiving transjugular intrahepatic portosystemic shunt (TIPS). “The role for TIPS in PVT is evolving and may address complications like portal hypertensive bleeding, medically refractory clot, and the need for repeated banding after variceal bleeding,” the experts noted.
Prophylaxis of DVT is recommended for all hospitalized patients with cirrhosis. Vitamin K antagonists and direct-acting oral anticoagulants (dabigatran, apixaban, rivaroxaban, and edoxaban) are alternatives to heparin for anticoagulation of cirrhotic patients with either PVT and DVT, the experts wrote. However, DOACs are not recommended for most Child-Pugh B patients or for any Child-Pugh C patients.
No funding sources or conflicts of interest were reported.
SOURCE: O’Leary JG et al. Gastroenterology. 2019. doi: 10.1053/j.gastro.2019.03.070.
Cirrhosis can involve “precarious” changes in hemostatic pathways that tip the scales toward either bleeding or hypercoagulation, experts wrote in an American Gastroenterological Association Clinical Practice Update.
Based on current evidence, clinicians should not routinely correct thrombocytopenia and coagulopathy in patients with cirrhosis prior to low-risk procedures, such as therapeutic paracentesis, thoracentesis, and routine upper endoscopy for variceal ligation, Jacqueline G. O’Leary, MD, of Dallas VA Medical Center and her three coreviewers wrote in Gastroenterology.
To optimize clot formation prior to high-risk procedures, and in patients with active bleeding, a platelet count above 50,000 per mcL is still recommended. However, it may be more meaningful to couple that platelet target with a fibrinogen level above 120 mg/dL rather than rely on the international normalized ratio (INR), the experts wrote. Not only does INR vary significantly depending on which thromboplastin is used in the test, but “correcting” INR with a fresh frozen plasma infusion does not affect thrombin production and worsens portal hypertension. Using cryoprecipitate to replenish fibrinogen has less impact on portal hypertension. “Global tests of clot formation, such as rotational thromboelastometry (ROTEM), thromboelastography (TEG), sonorheometry, and thrombin generation may eventually have a role in the evaluation of clotting in patients with cirrhosis but currently lack validated target levels,” the experts wrote.
They advised clinicians to limit the use of blood products (such as fresh frozen plasma and pooled platelet transfusions) because of cost and the risk of exacerbated portal hypertension, infection, and immunologic complications. For severe anemia and uremia, red blood cell transfusion (250 mL) can be considered. Platelet-rich plasma from one donor is less immunologically risky than a pooled platelet transfusion. Thrombopoietin agonists are “a good alternative” to platelet transfusion but require about 10 days for response. Alternative prothrombotic therapies include oral thrombopoietin receptor agonists (avatrombopag and lusutrombopag) to boost platelet count before an invasive procedure, antifibrinolytic therapy (aminocaproic acid and tranexamic acid) for persistent bleeding from mucosal oozing or puncture wounds. Desmopressin should only be considered for patients with comorbid renal failure.
For anticoagulation, the practice update recommends considering systemic heparin infusion for cirrhotic patients with symptomatic deep venous thrombosis (DVT) or portal vein thrombosis (PVT). However, the anti–factor Xa assay will not reliably monitor response if patients have low liver-derived antithrombin III (heparin cofactor). “With newly diagnosed PVT, the decision to intervene with directed therapy rests on the extent of the thrombosis, presence or absence of attributable symptoms, and the risk of bleeding and falls,” the experts stated.
Six-month follow-up imaging is recommended to assess anticoagulation efficacy. More frequent imaging can be considered for PVT patients considered at high risk for therapeutic anticoagulation. If clots do not fully resolve after 6 months of treatment, options including extending therapy with the same agent, switching to a different anticoagulant class, or receiving transjugular intrahepatic portosystemic shunt (TIPS). “The role for TIPS in PVT is evolving and may address complications like portal hypertensive bleeding, medically refractory clot, and the need for repeated banding after variceal bleeding,” the experts noted.
Prophylaxis of DVT is recommended for all hospitalized patients with cirrhosis. Vitamin K antagonists and direct-acting oral anticoagulants (dabigatran, apixaban, rivaroxaban, and edoxaban) are alternatives to heparin for anticoagulation of cirrhotic patients with either PVT and DVT, the experts wrote. However, DOACs are not recommended for most Child-Pugh B patients or for any Child-Pugh C patients.
No funding sources or conflicts of interest were reported.
SOURCE: O’Leary JG et al. Gastroenterology. 2019. doi: 10.1053/j.gastro.2019.03.070.
FROM GASTROENTEROLOGY
Study: Why urban sickle cell patients quit hydroxyurea
FORT LAUDERDALE, Fla. – A study of sickle cell patients at a clinic in the Bronx found that upwards of 75% of them get a prescription for hydroxyurea to improve hemoglobin levels, but that one-third have discontinued use for various reasons, according to results reported at the 13th annual Foundation for Sickle Cell Disease Research symposium here.
“The results identify variability in reported side effects and reasons for discontinuation, and highlight the importance of clear communication between providers and patients to discuss the benefits and challenges of hydroxyurea,” said Caterina Minniti, MD, professor of clinical medicine and pediatrics at Einstein College of Medicine and director of the Sickle Cell Center for Adults at Montefiore Hospital, Bronx, N.Y. The study analyzed self-reporting surveys completed by 224 adult outpatients in the Montefiore sickle cell clinic, and then verified the data in the electronic medical record, Dr. Minniti said. She noted, “Our population is unique in the Bronx in that we have a high percentage of Hispanic patients.” They comprised 24.1% of the study population.
“We found that 77.2% of the patients have ever been prescribed hydroxyurea,” she said. “That was really great.” Also, 91% of those with severe genotypes of SCD had been prescribed the drug; 68% of them were still taking hydroxyurea at the time of the survey, she said. Among patients with the mild genotype, 42.1% had been prescribed hydroxyurea and half were still on it when they completed their surveys.
When the survey evaluated how long patients had been taking the drug, she said, “That’s where I start to get concerned.” About half – 48.6% – had taken the drug for one to five years, “which is a very short period of time,” Dr. Minniti said. Another 15% were on hydroxyurea for less than a year, 23% for 5 to 10 years and 19% for 10 years or more.
The study drilled down into reasons why patients discontinued the drug. Side effects were cited by 24.6% (n=15). They include fatigue, hair loss, and GI upset. Other reasons include perceived ineffectiveness (16.4%, n=10); physician direction (14.8%, n=9), and reproductive health and ulcer formation (each at 8.2%, n=5).
“Many patients perceive ineffectiveness of hydroxyurea in the short term, but the benefits of hydoxyurea stem from chronic use over the long term,” Dr. Minniti said. She noted that some patients discontinued the drug for legitimate medical indications, “such as pregnancy and breast feeding, but were not restarted afterward.”
Dr. Minniti disclosed relationships with Novartis, Global Blood Therapeutics, Teutona, Bluebird Bio, GBT and Bayer.
SOURCE: Minniti C, et al. Abstract no. JSCDH-D-19-00058. Foundation for Sickle Cell Disease Research Symposium; Fort Lauderdale, Fla.; June 9, 2019.
FORT LAUDERDALE, Fla. – A study of sickle cell patients at a clinic in the Bronx found that upwards of 75% of them get a prescription for hydroxyurea to improve hemoglobin levels, but that one-third have discontinued use for various reasons, according to results reported at the 13th annual Foundation for Sickle Cell Disease Research symposium here.
“The results identify variability in reported side effects and reasons for discontinuation, and highlight the importance of clear communication between providers and patients to discuss the benefits and challenges of hydroxyurea,” said Caterina Minniti, MD, professor of clinical medicine and pediatrics at Einstein College of Medicine and director of the Sickle Cell Center for Adults at Montefiore Hospital, Bronx, N.Y. The study analyzed self-reporting surveys completed by 224 adult outpatients in the Montefiore sickle cell clinic, and then verified the data in the electronic medical record, Dr. Minniti said. She noted, “Our population is unique in the Bronx in that we have a high percentage of Hispanic patients.” They comprised 24.1% of the study population.
“We found that 77.2% of the patients have ever been prescribed hydroxyurea,” she said. “That was really great.” Also, 91% of those with severe genotypes of SCD had been prescribed the drug; 68% of them were still taking hydroxyurea at the time of the survey, she said. Among patients with the mild genotype, 42.1% had been prescribed hydroxyurea and half were still on it when they completed their surveys.
When the survey evaluated how long patients had been taking the drug, she said, “That’s where I start to get concerned.” About half – 48.6% – had taken the drug for one to five years, “which is a very short period of time,” Dr. Minniti said. Another 15% were on hydroxyurea for less than a year, 23% for 5 to 10 years and 19% for 10 years or more.
The study drilled down into reasons why patients discontinued the drug. Side effects were cited by 24.6% (n=15). They include fatigue, hair loss, and GI upset. Other reasons include perceived ineffectiveness (16.4%, n=10); physician direction (14.8%, n=9), and reproductive health and ulcer formation (each at 8.2%, n=5).
“Many patients perceive ineffectiveness of hydroxyurea in the short term, but the benefits of hydoxyurea stem from chronic use over the long term,” Dr. Minniti said. She noted that some patients discontinued the drug for legitimate medical indications, “such as pregnancy and breast feeding, but were not restarted afterward.”
Dr. Minniti disclosed relationships with Novartis, Global Blood Therapeutics, Teutona, Bluebird Bio, GBT and Bayer.
SOURCE: Minniti C, et al. Abstract no. JSCDH-D-19-00058. Foundation for Sickle Cell Disease Research Symposium; Fort Lauderdale, Fla.; June 9, 2019.
FORT LAUDERDALE, Fla. – A study of sickle cell patients at a clinic in the Bronx found that upwards of 75% of them get a prescription for hydroxyurea to improve hemoglobin levels, but that one-third have discontinued use for various reasons, according to results reported at the 13th annual Foundation for Sickle Cell Disease Research symposium here.
“The results identify variability in reported side effects and reasons for discontinuation, and highlight the importance of clear communication between providers and patients to discuss the benefits and challenges of hydroxyurea,” said Caterina Minniti, MD, professor of clinical medicine and pediatrics at Einstein College of Medicine and director of the Sickle Cell Center for Adults at Montefiore Hospital, Bronx, N.Y. The study analyzed self-reporting surveys completed by 224 adult outpatients in the Montefiore sickle cell clinic, and then verified the data in the electronic medical record, Dr. Minniti said. She noted, “Our population is unique in the Bronx in that we have a high percentage of Hispanic patients.” They comprised 24.1% of the study population.
“We found that 77.2% of the patients have ever been prescribed hydroxyurea,” she said. “That was really great.” Also, 91% of those with severe genotypes of SCD had been prescribed the drug; 68% of them were still taking hydroxyurea at the time of the survey, she said. Among patients with the mild genotype, 42.1% had been prescribed hydroxyurea and half were still on it when they completed their surveys.
When the survey evaluated how long patients had been taking the drug, she said, “That’s where I start to get concerned.” About half – 48.6% – had taken the drug for one to five years, “which is a very short period of time,” Dr. Minniti said. Another 15% were on hydroxyurea for less than a year, 23% for 5 to 10 years and 19% for 10 years or more.
The study drilled down into reasons why patients discontinued the drug. Side effects were cited by 24.6% (n=15). They include fatigue, hair loss, and GI upset. Other reasons include perceived ineffectiveness (16.4%, n=10); physician direction (14.8%, n=9), and reproductive health and ulcer formation (each at 8.2%, n=5).
“Many patients perceive ineffectiveness of hydroxyurea in the short term, but the benefits of hydoxyurea stem from chronic use over the long term,” Dr. Minniti said. She noted that some patients discontinued the drug for legitimate medical indications, “such as pregnancy and breast feeding, but were not restarted afterward.”
Dr. Minniti disclosed relationships with Novartis, Global Blood Therapeutics, Teutona, Bluebird Bio, GBT and Bayer.
SOURCE: Minniti C, et al. Abstract no. JSCDH-D-19-00058. Foundation for Sickle Cell Disease Research Symposium; Fort Lauderdale, Fla.; June 9, 2019.
REPORTING FROM THE ANNUAL SICKLE CELL DISEASE RESEARCH AND EDUCATIONAL SYMPOSIUM
Femoral head decompression relieves SCD hip pain
FORT LAUDERDALE, FLA. – Hip joint pain and deterioration can be a painful and disabling outcome for patients with sickle cell disease, but femoral head core decompression with the addition of bone marrow aspirate concentrate decreases their pain and may help avoid or delay hip replacement, according to results of a pilot study presented at the annual meeting of the Foundation for Sickle Cell Disease Research.
Eric Fornari, MD, of the Children’s Hospital at Montefiore in Bronx, N.Y., reported on results of core decompression (CD) in 35 hips of 26 sickle cell patients; 17 underwent CD only and 18 had CD with injection of bone marrow aspirate concentrate (CD+BMAC). The average patient age was 24.3 years, with a range from 9.7-50.7 years.
“Compared to patients treated with CD alone, patients treated with CD+BMAC complained of significantly less pain and had significant improvement in their functional scores and patient-related outcomes at short-term follow-up,” Dr. Fornari said.
Among the CD+BMAC patients, pain scores declined two points on average, from 6 preoperatively to 4 postoperatively, he said. This was clinically significant, compared with the CD-only group, Dr. Fornari said.
Patients in the CD+BMAC group also reported consistently superior hip outcome and modified Harris hip scores. With either treatment, more than 90% of patients were pain-free and walked independently at their most recent follow-up, he said.
The objective of CD is to relieve pressure within the head of the femur, stimulate vascularity and target the avascular necrosis (AVN) lesion within the head that is visible on imaging. To get the bone marrow aspirate concentrate, Dr. Fornari extracts 120 cc of bone marrow from the iliac crest, then concentrates it to 12 cc. The same instrument is used to tap into the femoral head and inject the bone marrow aspirate concentrate. The study looked at clinical and radiographic outcomes of treated patients.
Average follow-up for the entire study population was 3.6 years, but that varied widely between the two groups (CD-only at almost 6 years, CD+BMAC at 1.4 years) because CD+BMAC has only been done for the last 3 years, Dr. Fornari said.
Progression to total hip arthroplasty (THA) was similar between both groups: 5 of 17 patients (29%) for CD-only vs. 4 of 18 patients (22%) for CD+BMAC (P = .711).
“When you look at progression, there were a number of hips that got CD or CD+BMAC and were better postoperatively; they went from a Ficat score of stage II to a stage I, or stage III to stage II,” he said.
X-rays were not always a reliable marker of outcome after either CD procedure, Dr. Fornari noted. “I’ve seen patients who’ve had terrible looking X-rays who have no pain, and patients who have totally normal X-rays that are completely debilitated,” he said. “We have to start asking ourselves, ‘What is the marker of success?’ because when we do this patients are feeling better.”
Multivariate analysis was used to identify factors predictive of progression to THA after the procedure, Dr. Fornari said. “Age of diagnosis, age of surgery, female gender, and lower hydroxyurea dose at surgery were predictive of advancing disease, whereas a higher dose of hydroxyurea was predictive against advancement,” he said.
The average age of patients who had no THA after either procedure was 21 years, compared with 33.9 years for those who had THA (P = .003). Average hydroxyurea dose at surgery was 24.7 mg/kg in the no-THA group vs. 12.5 mg/kg in those who had THA (P = .005).
Notably, there were no readmissions, fractures, deep vein thromboses, pulmonary embolisms or infarctions after CD, Dr. Fornari said. Transfusions were required in two CD-only and three CD+BMAC patients. Hospitalization rates for vaso-occlusive crisis were similar between groups (P = .103).
Dr. Fornari said the challenge is to identify suitable patients for these procedures. “These are complicated patients and you don’t want to put them through the process of having surgery, putting them on crutches and restricted weight bearing, if they’re not going to get better,” he said. “This procedure done minimally invasively is not the end all and be all, but we have to figure out who are the right patients for it. Patient selection is key.”
Finding those patients starts with a rigorous history and physical exam, he said. Physicians should have a “low threshold” for MRI in these patients because that will reveal findings, such as pre-collapse disease and characteristic of AVN lesions, that may appear normal on X-ray. Patient education is also important. “To think that an injection into the top of the hip is going to solve all their problems is a little naive, so you have to have an honest conversation with the patient,” he said.
Dr. Fornari reported having no financial disclosures.
SOURCE: Fornari ED et al. FSCDR 2019, Abstract JSCDH-D-19-00004.
FORT LAUDERDALE, FLA. – Hip joint pain and deterioration can be a painful and disabling outcome for patients with sickle cell disease, but femoral head core decompression with the addition of bone marrow aspirate concentrate decreases their pain and may help avoid or delay hip replacement, according to results of a pilot study presented at the annual meeting of the Foundation for Sickle Cell Disease Research.
Eric Fornari, MD, of the Children’s Hospital at Montefiore in Bronx, N.Y., reported on results of core decompression (CD) in 35 hips of 26 sickle cell patients; 17 underwent CD only and 18 had CD with injection of bone marrow aspirate concentrate (CD+BMAC). The average patient age was 24.3 years, with a range from 9.7-50.7 years.
“Compared to patients treated with CD alone, patients treated with CD+BMAC complained of significantly less pain and had significant improvement in their functional scores and patient-related outcomes at short-term follow-up,” Dr. Fornari said.
Among the CD+BMAC patients, pain scores declined two points on average, from 6 preoperatively to 4 postoperatively, he said. This was clinically significant, compared with the CD-only group, Dr. Fornari said.
Patients in the CD+BMAC group also reported consistently superior hip outcome and modified Harris hip scores. With either treatment, more than 90% of patients were pain-free and walked independently at their most recent follow-up, he said.
The objective of CD is to relieve pressure within the head of the femur, stimulate vascularity and target the avascular necrosis (AVN) lesion within the head that is visible on imaging. To get the bone marrow aspirate concentrate, Dr. Fornari extracts 120 cc of bone marrow from the iliac crest, then concentrates it to 12 cc. The same instrument is used to tap into the femoral head and inject the bone marrow aspirate concentrate. The study looked at clinical and radiographic outcomes of treated patients.
Average follow-up for the entire study population was 3.6 years, but that varied widely between the two groups (CD-only at almost 6 years, CD+BMAC at 1.4 years) because CD+BMAC has only been done for the last 3 years, Dr. Fornari said.
Progression to total hip arthroplasty (THA) was similar between both groups: 5 of 17 patients (29%) for CD-only vs. 4 of 18 patients (22%) for CD+BMAC (P = .711).
“When you look at progression, there were a number of hips that got CD or CD+BMAC and were better postoperatively; they went from a Ficat score of stage II to a stage I, or stage III to stage II,” he said.
X-rays were not always a reliable marker of outcome after either CD procedure, Dr. Fornari noted. “I’ve seen patients who’ve had terrible looking X-rays who have no pain, and patients who have totally normal X-rays that are completely debilitated,” he said. “We have to start asking ourselves, ‘What is the marker of success?’ because when we do this patients are feeling better.”
Multivariate analysis was used to identify factors predictive of progression to THA after the procedure, Dr. Fornari said. “Age of diagnosis, age of surgery, female gender, and lower hydroxyurea dose at surgery were predictive of advancing disease, whereas a higher dose of hydroxyurea was predictive against advancement,” he said.
The average age of patients who had no THA after either procedure was 21 years, compared with 33.9 years for those who had THA (P = .003). Average hydroxyurea dose at surgery was 24.7 mg/kg in the no-THA group vs. 12.5 mg/kg in those who had THA (P = .005).
Notably, there were no readmissions, fractures, deep vein thromboses, pulmonary embolisms or infarctions after CD, Dr. Fornari said. Transfusions were required in two CD-only and three CD+BMAC patients. Hospitalization rates for vaso-occlusive crisis were similar between groups (P = .103).
Dr. Fornari said the challenge is to identify suitable patients for these procedures. “These are complicated patients and you don’t want to put them through the process of having surgery, putting them on crutches and restricted weight bearing, if they’re not going to get better,” he said. “This procedure done minimally invasively is not the end all and be all, but we have to figure out who are the right patients for it. Patient selection is key.”
Finding those patients starts with a rigorous history and physical exam, he said. Physicians should have a “low threshold” for MRI in these patients because that will reveal findings, such as pre-collapse disease and characteristic of AVN lesions, that may appear normal on X-ray. Patient education is also important. “To think that an injection into the top of the hip is going to solve all their problems is a little naive, so you have to have an honest conversation with the patient,” he said.
Dr. Fornari reported having no financial disclosures.
SOURCE: Fornari ED et al. FSCDR 2019, Abstract JSCDH-D-19-00004.
FORT LAUDERDALE, FLA. – Hip joint pain and deterioration can be a painful and disabling outcome for patients with sickle cell disease, but femoral head core decompression with the addition of bone marrow aspirate concentrate decreases their pain and may help avoid or delay hip replacement, according to results of a pilot study presented at the annual meeting of the Foundation for Sickle Cell Disease Research.
Eric Fornari, MD, of the Children’s Hospital at Montefiore in Bronx, N.Y., reported on results of core decompression (CD) in 35 hips of 26 sickle cell patients; 17 underwent CD only and 18 had CD with injection of bone marrow aspirate concentrate (CD+BMAC). The average patient age was 24.3 years, with a range from 9.7-50.7 years.
“Compared to patients treated with CD alone, patients treated with CD+BMAC complained of significantly less pain and had significant improvement in their functional scores and patient-related outcomes at short-term follow-up,” Dr. Fornari said.
Among the CD+BMAC patients, pain scores declined two points on average, from 6 preoperatively to 4 postoperatively, he said. This was clinically significant, compared with the CD-only group, Dr. Fornari said.
Patients in the CD+BMAC group also reported consistently superior hip outcome and modified Harris hip scores. With either treatment, more than 90% of patients were pain-free and walked independently at their most recent follow-up, he said.
The objective of CD is to relieve pressure within the head of the femur, stimulate vascularity and target the avascular necrosis (AVN) lesion within the head that is visible on imaging. To get the bone marrow aspirate concentrate, Dr. Fornari extracts 120 cc of bone marrow from the iliac crest, then concentrates it to 12 cc. The same instrument is used to tap into the femoral head and inject the bone marrow aspirate concentrate. The study looked at clinical and radiographic outcomes of treated patients.
Average follow-up for the entire study population was 3.6 years, but that varied widely between the two groups (CD-only at almost 6 years, CD+BMAC at 1.4 years) because CD+BMAC has only been done for the last 3 years, Dr. Fornari said.
Progression to total hip arthroplasty (THA) was similar between both groups: 5 of 17 patients (29%) for CD-only vs. 4 of 18 patients (22%) for CD+BMAC (P = .711).
“When you look at progression, there were a number of hips that got CD or CD+BMAC and were better postoperatively; they went from a Ficat score of stage II to a stage I, or stage III to stage II,” he said.
X-rays were not always a reliable marker of outcome after either CD procedure, Dr. Fornari noted. “I’ve seen patients who’ve had terrible looking X-rays who have no pain, and patients who have totally normal X-rays that are completely debilitated,” he said. “We have to start asking ourselves, ‘What is the marker of success?’ because when we do this patients are feeling better.”
Multivariate analysis was used to identify factors predictive of progression to THA after the procedure, Dr. Fornari said. “Age of diagnosis, age of surgery, female gender, and lower hydroxyurea dose at surgery were predictive of advancing disease, whereas a higher dose of hydroxyurea was predictive against advancement,” he said.
The average age of patients who had no THA after either procedure was 21 years, compared with 33.9 years for those who had THA (P = .003). Average hydroxyurea dose at surgery was 24.7 mg/kg in the no-THA group vs. 12.5 mg/kg in those who had THA (P = .005).
Notably, there were no readmissions, fractures, deep vein thromboses, pulmonary embolisms or infarctions after CD, Dr. Fornari said. Transfusions were required in two CD-only and three CD+BMAC patients. Hospitalization rates for vaso-occlusive crisis were similar between groups (P = .103).
Dr. Fornari said the challenge is to identify suitable patients for these procedures. “These are complicated patients and you don’t want to put them through the process of having surgery, putting them on crutches and restricted weight bearing, if they’re not going to get better,” he said. “This procedure done minimally invasively is not the end all and be all, but we have to figure out who are the right patients for it. Patient selection is key.”
Finding those patients starts with a rigorous history and physical exam, he said. Physicians should have a “low threshold” for MRI in these patients because that will reveal findings, such as pre-collapse disease and characteristic of AVN lesions, that may appear normal on X-ray. Patient education is also important. “To think that an injection into the top of the hip is going to solve all their problems is a little naive, so you have to have an honest conversation with the patient,” he said.
Dr. Fornari reported having no financial disclosures.
SOURCE: Fornari ED et al. FSCDR 2019, Abstract JSCDH-D-19-00004.
REPORTING FROM FSCDR 2019
Orthopedic complications in sickle cell require prompt action
FORT LAUDERDALE, FLA. – Orthopedic crises are common in patients with sickle cell disease, ranging from osteonecrosis to bone infarction, and physicians who manage these patients should know how to recognize these crises and not hesitate to consult an orthopedic surgeon early on, according to one expert at the annual meeting of the Foundation for Sickle Cell Disease Research.

“Sickle cell is a common entity in orthopedic surgery, so you shouldn’t hesitate in the hospital or outpatient settings to call for an orthopedic surgeon when you’re dealing with acute pain crises, medullary infarcts, and osteonecrosis,” said Mark W. Bridges, MD, an orthopedic surgeon with Orthopaedic Associates in Southern Florida.
Dr. Bridges noted that the femoral head is the most common location for osteonecrosis, one of the four major orthopedic manifestations of sickle cell disease that he reviewed. The others are septic arthritis, osteomyelitis, and bone infarction.
“Bone infarction is more common than osteomyelitis, and gadolinium-enhanced MRI can help to differentiate the two,” he said.
Osteonecrosis occurs when ischemic cells die, weakening the subchondral bone. Besides the femoral head, osteonecrosis commonly affects the humeral head of the shoulder and the femoral condyles of the knee. Dr. Bridges reviewed the five stages of the Ficat and Arlet classification of osteonecrosis:
- 0 – no pain, normal x-rays.
- I – pain, normal x-rays but abnormal MRI.
- II – pain, abnormal x-ray (sclerosis without collapse).
- III – pain (subchondral collapse without joint degeneration).
- IV – pain (arthritic changes with subchondral collapse).
For osteonecrosis of the shoulder, Dr. Bridges said four surgical options exist: core decompression for stages I and II; humeral head resurfacing for stages II and III; and hemiarthroplasty or total shoulder replacement for stages III and IV.
“No medical therapies are known to slow the progression,” he said.
Total joint replacement can be inevitable in these patients when total collapse of the joint occurs, but Dr. Bridges added a word of caution. “Overall when it comes down to replacing joints, there are more complications in patients that have [sickle cell disease],” he said. “Normally the complication rate is about 1%; that typically goes up to about 10% in SCD patients, but when you have a patient with end-stage disease – shoulder collapse or hip collapse – you have to do something.”
Septic arthritis is an infection within the joint space, most commonly the hip, and it affects 5% of children and 0.3% of adults with sickle cell disease (Clin Orthop Relat Res. 2010;468:1676-81).
“This is very similar to a vaso-occlusive crisis,” Dr. Bridges said.
MRI with gadolinium can help guide treatment, and blood cultures and joint aspiration can identify the infectious microbe. Staphylococcus aureus is the most common, Dr. Bridges said. Treatment consists of IV antibiotics, irrigation, and debridement.
Osteomyelitis is an infection within the bone with symptoms similar to those of septic arthritis, although osteomyelitis patients are typically sicker, he said. MRI with gadolinium is indicated in patients who don’t respond to IV fluid, oxygenation, and nonsteroidal anti-inflammatory drugs. “Try to treat them like they have vaso-occlusive crisis,” he said. Blood cultures usually suffice in these patients; bone aspiration is rarely needed, Dr. Bridges said.
The most common organisms are Staphylococcus aureus and Salmonella, and sickle cell disease patients can have infections in more than one location, Dr. Bridges noted.
“If IV antibiotics don’t work, then these patients need surgical debridement,” he added.
Adults are prone to a higher rate of complications than are children, including joint stiffness, osteonecrosis, pathologic fracture, and chronic osteomyelitis.
Ischemic marrow from vaso-occlusion can result in bone infarction. With its severe pain, swelling, erythema, and loss of motion, bone infarction can appear similar to osteomyelitis and septic arthritis, although a high-grade fever is uncommon in bone infarction. Unlike osteomyelitis, gadolinium on MRI does not enhance in bone infarction.
Treatment “consists of supportive management with pain medications, hydration, and antibiotics until osteomyelitis is ruled out,” Dr. Bridges said.
When a patient with one of these orthopedic conditions needs surgery, there are three considerations: preoperative transfusions to achieve hemoglobin level of 10 mg/dL for major procedures; no transfusions for arthroscopy and small closed reductions; and postoperative oxygenation and hydration to prevent a vaso-occlusive event and acute chest syndrome, he said.
Dr. Bridges reported having no conflicts of interest.
FORT LAUDERDALE, FLA. – Orthopedic crises are common in patients with sickle cell disease, ranging from osteonecrosis to bone infarction, and physicians who manage these patients should know how to recognize these crises and not hesitate to consult an orthopedic surgeon early on, according to one expert at the annual meeting of the Foundation for Sickle Cell Disease Research.
“Sickle cell is a common entity in orthopedic surgery, so you shouldn’t hesitate in the hospital or outpatient settings to call for an orthopedic surgeon when you’re dealing with acute pain crises, medullary infarcts, and osteonecrosis,” said Mark W. Bridges, MD, an orthopedic surgeon with Orthopaedic Associates in Southern Florida.
Dr. Bridges noted that the femoral head is the most common location for osteonecrosis, one of the four major orthopedic manifestations of sickle cell disease that he reviewed. The others are septic arthritis, osteomyelitis, and bone infarction.
“Bone infarction is more common than osteomyelitis, and gadolinium-enhanced MRI can help to differentiate the two,” he said.
Osteonecrosis occurs when ischemic cells die, weakening the subchondral bone. Besides the femoral head, osteonecrosis commonly affects the humeral head of the shoulder and the femoral condyles of the knee. Dr. Bridges reviewed the five stages of the Ficat and Arlet classification of osteonecrosis:
- 0 – no pain, normal x-rays.
- I – pain, normal x-rays but abnormal MRI.
- II – pain, abnormal x-ray (sclerosis without collapse).
- III – pain (subchondral collapse without joint degeneration).
- IV – pain (arthritic changes with subchondral collapse).
For osteonecrosis of the shoulder, Dr. Bridges said four surgical options exist: core decompression for stages I and II; humeral head resurfacing for stages II and III; and hemiarthroplasty or total shoulder replacement for stages III and IV.
“No medical therapies are known to slow the progression,” he said.
Total joint replacement can be inevitable in these patients when total collapse of the joint occurs, but Dr. Bridges added a word of caution. “Overall when it comes down to replacing joints, there are more complications in patients that have [sickle cell disease],” he said. “Normally the complication rate is about 1%; that typically goes up to about 10% in SCD patients, but when you have a patient with end-stage disease – shoulder collapse or hip collapse – you have to do something.”
Septic arthritis is an infection within the joint space, most commonly the hip, and it affects 5% of children and 0.3% of adults with sickle cell disease (Clin Orthop Relat Res. 2010;468:1676-81).
“This is very similar to a vaso-occlusive crisis,” Dr. Bridges said.
MRI with gadolinium can help guide treatment, and blood cultures and joint aspiration can identify the infectious microbe. Staphylococcus aureus is the most common, Dr. Bridges said. Treatment consists of IV antibiotics, irrigation, and debridement.
Osteomyelitis is an infection within the bone with symptoms similar to those of septic arthritis, although osteomyelitis patients are typically sicker, he said. MRI with gadolinium is indicated in patients who don’t respond to IV fluid, oxygenation, and nonsteroidal anti-inflammatory drugs. “Try to treat them like they have vaso-occlusive crisis,” he said. Blood cultures usually suffice in these patients; bone aspiration is rarely needed, Dr. Bridges said.
The most common organisms are Staphylococcus aureus and Salmonella, and sickle cell disease patients can have infections in more than one location, Dr. Bridges noted.
“If IV antibiotics don’t work, then these patients need surgical debridement,” he added.
Adults are prone to a higher rate of complications than are children, including joint stiffness, osteonecrosis, pathologic fracture, and chronic osteomyelitis.
Ischemic marrow from vaso-occlusion can result in bone infarction. With its severe pain, swelling, erythema, and loss of motion, bone infarction can appear similar to osteomyelitis and septic arthritis, although a high-grade fever is uncommon in bone infarction. Unlike osteomyelitis, gadolinium on MRI does not enhance in bone infarction.
Treatment “consists of supportive management with pain medications, hydration, and antibiotics until osteomyelitis is ruled out,” Dr. Bridges said.
When a patient with one of these orthopedic conditions needs surgery, there are three considerations: preoperative transfusions to achieve hemoglobin level of 10 mg/dL for major procedures; no transfusions for arthroscopy and small closed reductions; and postoperative oxygenation and hydration to prevent a vaso-occlusive event and acute chest syndrome, he said.
Dr. Bridges reported having no conflicts of interest.
FORT LAUDERDALE, FLA. – Orthopedic crises are common in patients with sickle cell disease, ranging from osteonecrosis to bone infarction, and physicians who manage these patients should know how to recognize these crises and not hesitate to consult an orthopedic surgeon early on, according to one expert at the annual meeting of the Foundation for Sickle Cell Disease Research.
“Sickle cell is a common entity in orthopedic surgery, so you shouldn’t hesitate in the hospital or outpatient settings to call for an orthopedic surgeon when you’re dealing with acute pain crises, medullary infarcts, and osteonecrosis,” said Mark W. Bridges, MD, an orthopedic surgeon with Orthopaedic Associates in Southern Florida.
Dr. Bridges noted that the femoral head is the most common location for osteonecrosis, one of the four major orthopedic manifestations of sickle cell disease that he reviewed. The others are septic arthritis, osteomyelitis, and bone infarction.
“Bone infarction is more common than osteomyelitis, and gadolinium-enhanced MRI can help to differentiate the two,” he said.
Osteonecrosis occurs when ischemic cells die, weakening the subchondral bone. Besides the femoral head, osteonecrosis commonly affects the humeral head of the shoulder and the femoral condyles of the knee. Dr. Bridges reviewed the five stages of the Ficat and Arlet classification of osteonecrosis:
- 0 – no pain, normal x-rays.
- I – pain, normal x-rays but abnormal MRI.
- II – pain, abnormal x-ray (sclerosis without collapse).
- III – pain (subchondral collapse without joint degeneration).
- IV – pain (arthritic changes with subchondral collapse).
For osteonecrosis of the shoulder, Dr. Bridges said four surgical options exist: core decompression for stages I and II; humeral head resurfacing for stages II and III; and hemiarthroplasty or total shoulder replacement for stages III and IV.
“No medical therapies are known to slow the progression,” he said.
Total joint replacement can be inevitable in these patients when total collapse of the joint occurs, but Dr. Bridges added a word of caution. “Overall when it comes down to replacing joints, there are more complications in patients that have [sickle cell disease],” he said. “Normally the complication rate is about 1%; that typically goes up to about 10% in SCD patients, but when you have a patient with end-stage disease – shoulder collapse or hip collapse – you have to do something.”
Septic arthritis is an infection within the joint space, most commonly the hip, and it affects 5% of children and 0.3% of adults with sickle cell disease (Clin Orthop Relat Res. 2010;468:1676-81).
“This is very similar to a vaso-occlusive crisis,” Dr. Bridges said.
MRI with gadolinium can help guide treatment, and blood cultures and joint aspiration can identify the infectious microbe. Staphylococcus aureus is the most common, Dr. Bridges said. Treatment consists of IV antibiotics, irrigation, and debridement.
Osteomyelitis is an infection within the bone with symptoms similar to those of septic arthritis, although osteomyelitis patients are typically sicker, he said. MRI with gadolinium is indicated in patients who don’t respond to IV fluid, oxygenation, and nonsteroidal anti-inflammatory drugs. “Try to treat them like they have vaso-occlusive crisis,” he said. Blood cultures usually suffice in these patients; bone aspiration is rarely needed, Dr. Bridges said.
The most common organisms are Staphylococcus aureus and Salmonella, and sickle cell disease patients can have infections in more than one location, Dr. Bridges noted.
“If IV antibiotics don’t work, then these patients need surgical debridement,” he added.
Adults are prone to a higher rate of complications than are children, including joint stiffness, osteonecrosis, pathologic fracture, and chronic osteomyelitis.
Ischemic marrow from vaso-occlusion can result in bone infarction. With its severe pain, swelling, erythema, and loss of motion, bone infarction can appear similar to osteomyelitis and septic arthritis, although a high-grade fever is uncommon in bone infarction. Unlike osteomyelitis, gadolinium on MRI does not enhance in bone infarction.
Treatment “consists of supportive management with pain medications, hydration, and antibiotics until osteomyelitis is ruled out,” Dr. Bridges said.
When a patient with one of these orthopedic conditions needs surgery, there are three considerations: preoperative transfusions to achieve hemoglobin level of 10 mg/dL for major procedures; no transfusions for arthroscopy and small closed reductions; and postoperative oxygenation and hydration to prevent a vaso-occlusive event and acute chest syndrome, he said.
Dr. Bridges reported having no conflicts of interest.
EXPERT ANALYSIS FROM FSCDR 2019
Cell count ratios appear to predict thromboembolism in lymphoma
AMSTERDAM – When predicting the risk of thromboembolism in lymphoma patients receiving chemotherapy, clinicians can rely on a routine diagnostic tool: complete blood count, investigators reported.

A recent study found that high neutrophil to lymphocyte (NLR) and platelet to lymphocyte (PLR) ratios were prognostic for thromboembolism in this setting, reported lead author Vladimir Otasevic, MD, of the Clinical Centre of Serbia in Belgrade.
“Because of the presence of a broad spectrum of risk factors [in patients with lymphoma undergoing chemotherapy], some authors have published risk-assessment models for prediction of thromboembolism,” Dr. Otasevic said during a presentation at the annual congress of the European Hematology Association. While the underlying pathophysiology that precedes thromboembolism is complex, Dr. Otasevic suggested that risk prediction may not have to be, noting that NLR and PLR were recently proposed as risk biomarkers.
To test the utility of these potential biomarkers, Dr. Otasevic and his colleagues retrospectively analyzed data from 484 patients with non-Hodgkin and Hodgkin lymphoma who had undergone at least one cycle of chemotherapy at the Clinic for Hematology, Clinical Centre of Serbia. Patients were followed for venous and arterial thromboembolic events from the time of diagnosis to 3 months beyond their final cycle of chemotherapy. NLR and PLR ratios were calculated from complete blood count. Thromboembolism was diagnosed by radiography, clinical exam, and laboratory evaluation, with probable diagnoses reviewed by an internist and radiologist.
The median patient age was 53 years with a range from 18 to 89 years. Most patients were recently diagnosed with advanced disease (21.1% stage III and 42.5% stage IV). Half of the population had high-grade non-Hodgkin lymphoma (50.0%) and slightly more than a quarter had low-grade non-Hodgkin lymphoma (28.3%). Low-grade Hodgkin lymphoma was less common (17.4%) and followed distantly by other forms (4.3%).
Thirty-five patients (7.2%) developed thromboembolic events; of these, 30 had venous thromboembolism (6.2%), 6 had arterial thromboembolism (1.2%), and 1 had both. Patients who experienced thromboembolic events had significantly higher NLR and PLR than patients without thromboembolism, and both ratios were significantly associated with one another.
A positive NLR, defined as a ratio of 3.1 or more, was associated with a relative risk of 4.1 for thromboembolism (P less than .001), while a positive PLR, defined as a ratio of 10 or more, was associated with a relative risk of 2.9 (P = .008). Using a multivariate model, a positive NLR was associated with an even higher relative risk (RR = 4.5; P less than .001).
“NLR and PLR demonstrated significant powerfulness in prediction of future risk of [thromboembolism] in lymphoma patients,” the investigators concluded. “Simplicity, effectiveness, modesty, and practicability qualify these new tools for routine [thromboembolism] prognostic assessment.”
Dr. Otasevic said that he and his colleagues have plans to build on these findings with further analysis involving progression-free and overall survival.
The investigators reported no disclosures.
SOURCE: Otasevic V et al. EHA Congress, Abstract S1645.
AMSTERDAM – When predicting the risk of thromboembolism in lymphoma patients receiving chemotherapy, clinicians can rely on a routine diagnostic tool: complete blood count, investigators reported.
A recent study found that high neutrophil to lymphocyte (NLR) and platelet to lymphocyte (PLR) ratios were prognostic for thromboembolism in this setting, reported lead author Vladimir Otasevic, MD, of the Clinical Centre of Serbia in Belgrade.
“Because of the presence of a broad spectrum of risk factors [in patients with lymphoma undergoing chemotherapy], some authors have published risk-assessment models for prediction of thromboembolism,” Dr. Otasevic said during a presentation at the annual congress of the European Hematology Association. While the underlying pathophysiology that precedes thromboembolism is complex, Dr. Otasevic suggested that risk prediction may not have to be, noting that NLR and PLR were recently proposed as risk biomarkers.
To test the utility of these potential biomarkers, Dr. Otasevic and his colleagues retrospectively analyzed data from 484 patients with non-Hodgkin and Hodgkin lymphoma who had undergone at least one cycle of chemotherapy at the Clinic for Hematology, Clinical Centre of Serbia. Patients were followed for venous and arterial thromboembolic events from the time of diagnosis to 3 months beyond their final cycle of chemotherapy. NLR and PLR ratios were calculated from complete blood count. Thromboembolism was diagnosed by radiography, clinical exam, and laboratory evaluation, with probable diagnoses reviewed by an internist and radiologist.
The median patient age was 53 years with a range from 18 to 89 years. Most patients were recently diagnosed with advanced disease (21.1% stage III and 42.5% stage IV). Half of the population had high-grade non-Hodgkin lymphoma (50.0%) and slightly more than a quarter had low-grade non-Hodgkin lymphoma (28.3%). Low-grade Hodgkin lymphoma was less common (17.4%) and followed distantly by other forms (4.3%).
Thirty-five patients (7.2%) developed thromboembolic events; of these, 30 had venous thromboembolism (6.2%), 6 had arterial thromboembolism (1.2%), and 1 had both. Patients who experienced thromboembolic events had significantly higher NLR and PLR than patients without thromboembolism, and both ratios were significantly associated with one another.
A positive NLR, defined as a ratio of 3.1 or more, was associated with a relative risk of 4.1 for thromboembolism (P less than .001), while a positive PLR, defined as a ratio of 10 or more, was associated with a relative risk of 2.9 (P = .008). Using a multivariate model, a positive NLR was associated with an even higher relative risk (RR = 4.5; P less than .001).
“NLR and PLR demonstrated significant powerfulness in prediction of future risk of [thromboembolism] in lymphoma patients,” the investigators concluded. “Simplicity, effectiveness, modesty, and practicability qualify these new tools for routine [thromboembolism] prognostic assessment.”
Dr. Otasevic said that he and his colleagues have plans to build on these findings with further analysis involving progression-free and overall survival.
The investigators reported no disclosures.
SOURCE: Otasevic V et al. EHA Congress, Abstract S1645.
AMSTERDAM – When predicting the risk of thromboembolism in lymphoma patients receiving chemotherapy, clinicians can rely on a routine diagnostic tool: complete blood count, investigators reported.
A recent study found that high neutrophil to lymphocyte (NLR) and platelet to lymphocyte (PLR) ratios were prognostic for thromboembolism in this setting, reported lead author Vladimir Otasevic, MD, of the Clinical Centre of Serbia in Belgrade.
“Because of the presence of a broad spectrum of risk factors [in patients with lymphoma undergoing chemotherapy], some authors have published risk-assessment models for prediction of thromboembolism,” Dr. Otasevic said during a presentation at the annual congress of the European Hematology Association. While the underlying pathophysiology that precedes thromboembolism is complex, Dr. Otasevic suggested that risk prediction may not have to be, noting that NLR and PLR were recently proposed as risk biomarkers.
To test the utility of these potential biomarkers, Dr. Otasevic and his colleagues retrospectively analyzed data from 484 patients with non-Hodgkin and Hodgkin lymphoma who had undergone at least one cycle of chemotherapy at the Clinic for Hematology, Clinical Centre of Serbia. Patients were followed for venous and arterial thromboembolic events from the time of diagnosis to 3 months beyond their final cycle of chemotherapy. NLR and PLR ratios were calculated from complete blood count. Thromboembolism was diagnosed by radiography, clinical exam, and laboratory evaluation, with probable diagnoses reviewed by an internist and radiologist.
The median patient age was 53 years with a range from 18 to 89 years. Most patients were recently diagnosed with advanced disease (21.1% stage III and 42.5% stage IV). Half of the population had high-grade non-Hodgkin lymphoma (50.0%) and slightly more than a quarter had low-grade non-Hodgkin lymphoma (28.3%). Low-grade Hodgkin lymphoma was less common (17.4%) and followed distantly by other forms (4.3%).
Thirty-five patients (7.2%) developed thromboembolic events; of these, 30 had venous thromboembolism (6.2%), 6 had arterial thromboembolism (1.2%), and 1 had both. Patients who experienced thromboembolic events had significantly higher NLR and PLR than patients without thromboembolism, and both ratios were significantly associated with one another.
A positive NLR, defined as a ratio of 3.1 or more, was associated with a relative risk of 4.1 for thromboembolism (P less than .001), while a positive PLR, defined as a ratio of 10 or more, was associated with a relative risk of 2.9 (P = .008). Using a multivariate model, a positive NLR was associated with an even higher relative risk (RR = 4.5; P less than .001).
“NLR and PLR demonstrated significant powerfulness in prediction of future risk of [thromboembolism] in lymphoma patients,” the investigators concluded. “Simplicity, effectiveness, modesty, and practicability qualify these new tools for routine [thromboembolism] prognostic assessment.”
Dr. Otasevic said that he and his colleagues have plans to build on these findings with further analysis involving progression-free and overall survival.
The investigators reported no disclosures.
SOURCE: Otasevic V et al. EHA Congress, Abstract S1645.
REPORTING FROM EHA CONGRESS