User login
Allergic March

Oral immunotherapy desensitizes youth with peanut allergy
Peanut-allergic children and adolescents treated with a peanut-derived oral immunotherapy drug have shown significant improvements in response to a challenge dose of peanut protein, according to data presented at the annual meeting of the American College of Allergy, Asthma, and Immunology.
A phase 3 placebo-controlled study, published simultaneously Nov. 18 in the New England Journal of Medicine, randomized 551 individuals with peanut allergy to receive an escalating dose of AR101 – an investigational peanut-derived biologic oral immunotherapy drug – ranging from 0.5-300 mg daily, or placebo.
After 12 months, 67.2% of the 372 participants aged 4-17 years who received the immunotherapy drug were able to eat a dose of 600 mg or more of peanut protein with only mild symptoms, compared with 4% of the 124 participants aged 4-17 years in the placebo group.
The secondary endpoints were whether participants could tolerate either a 300 mg or 1,000 mg dose in the exit food challenge. For the 300 mg dose, 76.6% of the immunotherapy group and 8.1% of the placebo group were able to tolerate it, and for the 1,000 mg group, 50.3% of the immunotherapy group were able to tolerate it, compared with 2.4% of the placebo group.
During the exit food challenge, the severity of symptoms was significantly higher in the placebo group than in the treatment group. One-quarter of participants in the treatment group had at most moderate symptoms, compared with 59% in the placebo group. However, severe symptoms were experienced by 11% of the placebo group, compared with 5% of the treatment group.
One in 10 participants in the active group had to be treated with rescue epinephrine during the exit food challenge, compared with 53% of participants in the placebo group, and the number who required a second dose of rescue epinephrine was 1% and 15%, respectively.
“These data show that, in the context of a clinical trial, among participants 4-17 years of age, AR101 had immunomodulatory activity, raised the threshold dose of peanut exposure triggering the onset of clinically significant allergic symptoms (among participants having symptoms), during the double-blind, placebo-controlled exit food challenge, and attenuated the severity of those symptoms when they occurred,” wrote Brian P. Vickery, MD, of Emory University in Atlanta, and his coauthors.
The 55 participants aged 18-55 years were analyzed separately, and researchers found that for the 600 mg exit food test, the difference between the two groups did not reach statistical significance.
Apart from adverse events that occurred during the exit food challenge, the rate of adverse events was slightly higher in the treatment group compared to the placebo group (98.7% vs. 95.2%). The most common adverse events in the treatment arm were abdominal pain, vomiting, oral pruritis, and nausea.
The study was funded by Aimmune Therapeutics. Three authors were employees of or investigators for Aimmune Therapeutics and one also had a patent pending for oral immunotherapy for peanut allergy. Most authors declared funding, grants, consultancies, or other support from the pharmaceutical industry, including from some from Aimmune.
SOURCE: Vickery BP et al. N Engl J Med. 18 Nov 2018. doi: 10.1056/NEJMoa1812856.
Over the past decade, more case reports and small studies have suggested that the use of tiny and incrementally increasing amount of peanut could desensitize those who are allergic to peanuts. This study, which uses a product based on defatted peanut flour, has shown that by the end of the course of treatment, two-thirds of those treated could consume around four peanuts.
However, the treatment was associated with side effects, many participants needed treatment with epinephrine, and the study has not yet addressed concerns about the longer term side effects of sustained allergen consumption, such as eosinophilic esophagitis.
The question also still remains as to whether the allergen tolerance is long-lasting or whether it will need to be maintained with regular exposure.
Michael R. Perkin, PhD, is affiliated with the Population Health Research Unit at St George’s, University of London. These comments are taken from an accompanying editorial (N Engl J Med. 18 Nov 2018. doi: 10.1056/NEJMe1813314). No conflicts of interest were declared.
Over the past decade, more case reports and small studies have suggested that the use of tiny and incrementally increasing amount of peanut could desensitize those who are allergic to peanuts. This study, which uses a product based on defatted peanut flour, has shown that by the end of the course of treatment, two-thirds of those treated could consume around four peanuts.
However, the treatment was associated with side effects, many participants needed treatment with epinephrine, and the study has not yet addressed concerns about the longer term side effects of sustained allergen consumption, such as eosinophilic esophagitis.
The question also still remains as to whether the allergen tolerance is long-lasting or whether it will need to be maintained with regular exposure.
Michael R. Perkin, PhD, is affiliated with the Population Health Research Unit at St George’s, University of London. These comments are taken from an accompanying editorial (N Engl J Med. 18 Nov 2018. doi: 10.1056/NEJMe1813314). No conflicts of interest were declared.
Over the past decade, more case reports and small studies have suggested that the use of tiny and incrementally increasing amount of peanut could desensitize those who are allergic to peanuts. This study, which uses a product based on defatted peanut flour, has shown that by the end of the course of treatment, two-thirds of those treated could consume around four peanuts.
However, the treatment was associated with side effects, many participants needed treatment with epinephrine, and the study has not yet addressed concerns about the longer term side effects of sustained allergen consumption, such as eosinophilic esophagitis.
The question also still remains as to whether the allergen tolerance is long-lasting or whether it will need to be maintained with regular exposure.
Michael R. Perkin, PhD, is affiliated with the Population Health Research Unit at St George’s, University of London. These comments are taken from an accompanying editorial (N Engl J Med. 18 Nov 2018. doi: 10.1056/NEJMe1813314). No conflicts of interest were declared.
Peanut-allergic children and adolescents treated with a peanut-derived oral immunotherapy drug have shown significant improvements in response to a challenge dose of peanut protein, according to data presented at the annual meeting of the American College of Allergy, Asthma, and Immunology.
A phase 3 placebo-controlled study, published simultaneously Nov. 18 in the New England Journal of Medicine, randomized 551 individuals with peanut allergy to receive an escalating dose of AR101 – an investigational peanut-derived biologic oral immunotherapy drug – ranging from 0.5-300 mg daily, or placebo.
After 12 months, 67.2% of the 372 participants aged 4-17 years who received the immunotherapy drug were able to eat a dose of 600 mg or more of peanut protein with only mild symptoms, compared with 4% of the 124 participants aged 4-17 years in the placebo group.
The secondary endpoints were whether participants could tolerate either a 300 mg or 1,000 mg dose in the exit food challenge. For the 300 mg dose, 76.6% of the immunotherapy group and 8.1% of the placebo group were able to tolerate it, and for the 1,000 mg group, 50.3% of the immunotherapy group were able to tolerate it, compared with 2.4% of the placebo group.
During the exit food challenge, the severity of symptoms was significantly higher in the placebo group than in the treatment group. One-quarter of participants in the treatment group had at most moderate symptoms, compared with 59% in the placebo group. However, severe symptoms were experienced by 11% of the placebo group, compared with 5% of the treatment group.
One in 10 participants in the active group had to be treated with rescue epinephrine during the exit food challenge, compared with 53% of participants in the placebo group, and the number who required a second dose of rescue epinephrine was 1% and 15%, respectively.
“These data show that, in the context of a clinical trial, among participants 4-17 years of age, AR101 had immunomodulatory activity, raised the threshold dose of peanut exposure triggering the onset of clinically significant allergic symptoms (among participants having symptoms), during the double-blind, placebo-controlled exit food challenge, and attenuated the severity of those symptoms when they occurred,” wrote Brian P. Vickery, MD, of Emory University in Atlanta, and his coauthors.
The 55 participants aged 18-55 years were analyzed separately, and researchers found that for the 600 mg exit food test, the difference between the two groups did not reach statistical significance.
Apart from adverse events that occurred during the exit food challenge, the rate of adverse events was slightly higher in the treatment group compared to the placebo group (98.7% vs. 95.2%). The most common adverse events in the treatment arm were abdominal pain, vomiting, oral pruritis, and nausea.
The study was funded by Aimmune Therapeutics. Three authors were employees of or investigators for Aimmune Therapeutics and one also had a patent pending for oral immunotherapy for peanut allergy. Most authors declared funding, grants, consultancies, or other support from the pharmaceutical industry, including from some from Aimmune.
SOURCE: Vickery BP et al. N Engl J Med. 18 Nov 2018. doi: 10.1056/NEJMoa1812856.
Peanut-allergic children and adolescents treated with a peanut-derived oral immunotherapy drug have shown significant improvements in response to a challenge dose of peanut protein, according to data presented at the annual meeting of the American College of Allergy, Asthma, and Immunology.
A phase 3 placebo-controlled study, published simultaneously Nov. 18 in the New England Journal of Medicine, randomized 551 individuals with peanut allergy to receive an escalating dose of AR101 – an investigational peanut-derived biologic oral immunotherapy drug – ranging from 0.5-300 mg daily, or placebo.
After 12 months, 67.2% of the 372 participants aged 4-17 years who received the immunotherapy drug were able to eat a dose of 600 mg or more of peanut protein with only mild symptoms, compared with 4% of the 124 participants aged 4-17 years in the placebo group.
The secondary endpoints were whether participants could tolerate either a 300 mg or 1,000 mg dose in the exit food challenge. For the 300 mg dose, 76.6% of the immunotherapy group and 8.1% of the placebo group were able to tolerate it, and for the 1,000 mg group, 50.3% of the immunotherapy group were able to tolerate it, compared with 2.4% of the placebo group.
During the exit food challenge, the severity of symptoms was significantly higher in the placebo group than in the treatment group. One-quarter of participants in the treatment group had at most moderate symptoms, compared with 59% in the placebo group. However, severe symptoms were experienced by 11% of the placebo group, compared with 5% of the treatment group.
One in 10 participants in the active group had to be treated with rescue epinephrine during the exit food challenge, compared with 53% of participants in the placebo group, and the number who required a second dose of rescue epinephrine was 1% and 15%, respectively.
“These data show that, in the context of a clinical trial, among participants 4-17 years of age, AR101 had immunomodulatory activity, raised the threshold dose of peanut exposure triggering the onset of clinically significant allergic symptoms (among participants having symptoms), during the double-blind, placebo-controlled exit food challenge, and attenuated the severity of those symptoms when they occurred,” wrote Brian P. Vickery, MD, of Emory University in Atlanta, and his coauthors.
The 55 participants aged 18-55 years were analyzed separately, and researchers found that for the 600 mg exit food test, the difference between the two groups did not reach statistical significance.
Apart from adverse events that occurred during the exit food challenge, the rate of adverse events was slightly higher in the treatment group compared to the placebo group (98.7% vs. 95.2%). The most common adverse events in the treatment arm were abdominal pain, vomiting, oral pruritis, and nausea.
The study was funded by Aimmune Therapeutics. Three authors were employees of or investigators for Aimmune Therapeutics and one also had a patent pending for oral immunotherapy for peanut allergy. Most authors declared funding, grants, consultancies, or other support from the pharmaceutical industry, including from some from Aimmune.
SOURCE: Vickery BP et al. N Engl J Med. 18 Nov 2018. doi: 10.1056/NEJMoa1812856.
FROM ACAAI 2018
Key clinical point: Oral peanut immunotherapy can improve tolerance in patients aged 4-17 with peanut allergy.
Major finding: Among patients treated with oral peanut immunotherapy, 67.2% were able to tolerate 600 mg of peanut protein, compared with 4% of the placebo group.
Study details: A randomized, placebo-controlled phase 3 study in 551 individuals with peanut allergy.
Disclosures: The study was funded by Aimmune Therapeutics. Three authors were employees of or investigators for Aimmune Therapeutics, and one also had a patent pending for oral immunotherapy for peanut allergy. Most authors declared funding, grants, consultancies, or other support from the pharmaceutical industry, including from some from Aimmune.
Source: Vickery BP et al. N Engl J Med. 18 Nov 2018. doi: 10.1056/NEJMoa1812856.
Small fibers, large impact

The details about an individual’s search for information tell us a lot about healthcare concerns and uncertainty across the medical universe. For nearly a decade, one of the most “clicked on” papers we have published in the Journal has been a review of small fiber neuropathy—a clinical entity with a prevalence of perhaps 1 in 1,000 to 2,000 people and, to my knowledge, no associated walkathons or arm bracelets. Yet it certainly piques the interest of clinicians from many specialties far broader than neurology. In this issue of the Journal, Dr. Jinny Tavee updates her 2009 review and provides us with a clinical overview of the disorder and the opportunity to assess how much further we need to more fully understand its management and associated comorbid conditions.
The wide interest in this disorder plugs into our current seeming epidemic of patients with chronic pain. It seems that almost half of my new patients have issues related to chronic pain that are not directly explained by active inflammation or anatomic damage. Many of these patients have diffuse body pains with associated fatigue and sleep disorders and are diagnosed with fibromyalgia. But others describe pain with a burning and tingling quality that seems of neurologic origin, yet their neurologic examination is normal. A few describe a predominantly distal symmetric stocking-and-glove distribution, but most do not. In some patients these pains are spatially random and evanescent, which to me are usually the hardest to fathom. Nerve conduction studies, when performed, are unrevealing.
A number of systemic autoimmune disorders, as discussed by Dr. Tavee in her article, are suggested to have an association with these symptoms. Given the chronicity and the frustrating nature of the symptoms, it is no surprise that a panoply of immune serologies are frequently ordered. Invariably, since serologies (eg, ANA, SSA, SSB, rheumatoid factor) are not specific for any single entity, some will return as positive. The strength of many of these associations is weak; even when the clinical diagnosis of lupus, for example, is definite, treatment of the underlying disease does not necessarily improve the dysesthetic pain. In an alternative scenario, the small fiber neuropathy is ascribed to a systemic autoimmune disorder that has been diagnosed because an autoantibody has been detected, but this rarely helps the patient and may in fact worsen symptoms by increasing anxiety and concern over having a systemic disease such as Sjögren syndrome or lupus (both of which sound terrible when reviewed on the Internet).
Some patients describe autonomic symptoms. Given the biologic basis that has been defined for this entity, it is no surprise that some patients have marked symptoms of decreased exocrine gland function, gastrointestinal dysmotility, and orthostasis. These symptoms may not be recognized unless specifically sought out when interviewing the patient.
Given the chronicity and sometimes the vagaries of symptoms, it is often comforting for patients to get an actual diagnosis. Dr. Tavee notes the relative simplicity of diagnostic procedures. But determining the clinical implications of the results may not be straightforward, and devising a fully and uniformly effective therapeutic approach eludes us still. As she points out, a multidisciplinary approach to therapy and diagnosis can be quite helpful.
The details about an individual’s search for information tell us a lot about healthcare concerns and uncertainty across the medical universe. For nearly a decade, one of the most “clicked on” papers we have published in the Journal has been a review of small fiber neuropathy—a clinical entity with a prevalence of perhaps 1 in 1,000 to 2,000 people and, to my knowledge, no associated walkathons or arm bracelets. Yet it certainly piques the interest of clinicians from many specialties far broader than neurology. In this issue of the Journal, Dr. Jinny Tavee updates her 2009 review and provides us with a clinical overview of the disorder and the opportunity to assess how much further we need to more fully understand its management and associated comorbid conditions.
The wide interest in this disorder plugs into our current seeming epidemic of patients with chronic pain. It seems that almost half of my new patients have issues related to chronic pain that are not directly explained by active inflammation or anatomic damage. Many of these patients have diffuse body pains with associated fatigue and sleep disorders and are diagnosed with fibromyalgia. But others describe pain with a burning and tingling quality that seems of neurologic origin, yet their neurologic examination is normal. A few describe a predominantly distal symmetric stocking-and-glove distribution, but most do not. In some patients these pains are spatially random and evanescent, which to me are usually the hardest to fathom. Nerve conduction studies, when performed, are unrevealing.
A number of systemic autoimmune disorders, as discussed by Dr. Tavee in her article, are suggested to have an association with these symptoms. Given the chronicity and the frustrating nature of the symptoms, it is no surprise that a panoply of immune serologies are frequently ordered. Invariably, since serologies (eg, ANA, SSA, SSB, rheumatoid factor) are not specific for any single entity, some will return as positive. The strength of many of these associations is weak; even when the clinical diagnosis of lupus, for example, is definite, treatment of the underlying disease does not necessarily improve the dysesthetic pain. In an alternative scenario, the small fiber neuropathy is ascribed to a systemic autoimmune disorder that has been diagnosed because an autoantibody has been detected, but this rarely helps the patient and may in fact worsen symptoms by increasing anxiety and concern over having a systemic disease such as Sjögren syndrome or lupus (both of which sound terrible when reviewed on the Internet).
Some patients describe autonomic symptoms. Given the biologic basis that has been defined for this entity, it is no surprise that some patients have marked symptoms of decreased exocrine gland function, gastrointestinal dysmotility, and orthostasis. These symptoms may not be recognized unless specifically sought out when interviewing the patient.
Given the chronicity and sometimes the vagaries of symptoms, it is often comforting for patients to get an actual diagnosis. Dr. Tavee notes the relative simplicity of diagnostic procedures. But determining the clinical implications of the results may not be straightforward, and devising a fully and uniformly effective therapeutic approach eludes us still. As she points out, a multidisciplinary approach to therapy and diagnosis can be quite helpful.
The details about an individual’s search for information tell us a lot about healthcare concerns and uncertainty across the medical universe. For nearly a decade, one of the most “clicked on” papers we have published in the Journal has been a review of small fiber neuropathy—a clinical entity with a prevalence of perhaps 1 in 1,000 to 2,000 people and, to my knowledge, no associated walkathons or arm bracelets. Yet it certainly piques the interest of clinicians from many specialties far broader than neurology. In this issue of the Journal, Dr. Jinny Tavee updates her 2009 review and provides us with a clinical overview of the disorder and the opportunity to assess how much further we need to more fully understand its management and associated comorbid conditions.
The wide interest in this disorder plugs into our current seeming epidemic of patients with chronic pain. It seems that almost half of my new patients have issues related to chronic pain that are not directly explained by active inflammation or anatomic damage. Many of these patients have diffuse body pains with associated fatigue and sleep disorders and are diagnosed with fibromyalgia. But others describe pain with a burning and tingling quality that seems of neurologic origin, yet their neurologic examination is normal. A few describe a predominantly distal symmetric stocking-and-glove distribution, but most do not. In some patients these pains are spatially random and evanescent, which to me are usually the hardest to fathom. Nerve conduction studies, when performed, are unrevealing.
A number of systemic autoimmune disorders, as discussed by Dr. Tavee in her article, are suggested to have an association with these symptoms. Given the chronicity and the frustrating nature of the symptoms, it is no surprise that a panoply of immune serologies are frequently ordered. Invariably, since serologies (eg, ANA, SSA, SSB, rheumatoid factor) are not specific for any single entity, some will return as positive. The strength of many of these associations is weak; even when the clinical diagnosis of lupus, for example, is definite, treatment of the underlying disease does not necessarily improve the dysesthetic pain. In an alternative scenario, the small fiber neuropathy is ascribed to a systemic autoimmune disorder that has been diagnosed because an autoantibody has been detected, but this rarely helps the patient and may in fact worsen symptoms by increasing anxiety and concern over having a systemic disease such as Sjögren syndrome or lupus (both of which sound terrible when reviewed on the Internet).
Some patients describe autonomic symptoms. Given the biologic basis that has been defined for this entity, it is no surprise that some patients have marked symptoms of decreased exocrine gland function, gastrointestinal dysmotility, and orthostasis. These symptoms may not be recognized unless specifically sought out when interviewing the patient.
Given the chronicity and sometimes the vagaries of symptoms, it is often comforting for patients to get an actual diagnosis. Dr. Tavee notes the relative simplicity of diagnostic procedures. But determining the clinical implications of the results may not be straightforward, and devising a fully and uniformly effective therapeutic approach eludes us still. As she points out, a multidisciplinary approach to therapy and diagnosis can be quite helpful.
Screen sooner and more often for those with family history of CRC
WASHINGTON – The number of first- and second-degree relatives with colorectal cancer can increase an individual’s risk for CRC, which could require screening to be done more frequently.
“There have been multiple guidelines reported as to what should be done for these individuals,” Harminder Singh, MD, of the University of Manitoba and his associates stated. “However, for the most part, they have not systematically analyzed the data.” He went on to say that, “more importantly, there’s been no recent AGA [American Gastroenterological Association] or Canadian Association of Gastroenterology statement, which led the development of this guideline.”
To address this issue, Dr. Singh and his colleagues conducted a systematic review of 10 literature searches to answer the following five questions concerning colorectal risk and screening practices: What is the effect of a family history of CRC on an individual’s risk of CRC? What is the effect of a family history of adenoma on an individual’s risk of CRC? At what age should CRC screening begin? Which screening tests are optimal? What are the optimal testing intervals for people with a family history of CRC or adenoma?
These questions were developed via an iterative online platform and then further developed and voted on by a team of specialists. GRADE (Grading of Recommendation Assessment, Development and Evaluation) was used to assess the quality of evidence to support these questions.
Similarly, individuals with two or more first-degree relatives with CRC had a two- to fourfold increased risk of developing CRC, compared with the general population. The review also found that, of the 20 recommendation statements from the review panel, there was consensus about 19 of them.
Colorectal cancer screening is recommended for all individuals with a family history of CRC or documented adenoma. Similarly, colonoscopy is recommended as the preferred test for individuals at the highest risk– those with one or more affected first-degree relatives. Fecal immunochemistry tests are considered a viable alternative except in patients with two or more first-degree relatives.
If a patient is considered to have an elevated risk of CRC because of family history, then screening should begin when they are aged 10 years younger than when that first-degree relative was diagnosed, and a 5-year screening interval should be followed after that.
Dr. Singh pointed out that the age of the affected first-degree relative should be considered when weighing an individual’s related risk of developing CRC. For example, having an first-degree relative who is diagnosed after the age of 75 is not likely to elevate an individual’s risk of developing CRC. Individuals with one or more second-degree relatives with CRC or nonadvanced adenoma do not appear to have an elevated risk of developing CRC and should be screened according to average-risk guidelines.
Dr. Singh reported receiving funding for from Merck Canada.
SOURCE: Leddin D. Gastroenterology. 2018 Jun. doi: 10.1016/S0016-5085(18)31083-7.
WASHINGTON – The number of first- and second-degree relatives with colorectal cancer can increase an individual’s risk for CRC, which could require screening to be done more frequently.
“There have been multiple guidelines reported as to what should be done for these individuals,” Harminder Singh, MD, of the University of Manitoba and his associates stated. “However, for the most part, they have not systematically analyzed the data.” He went on to say that, “more importantly, there’s been no recent AGA [American Gastroenterological Association] or Canadian Association of Gastroenterology statement, which led the development of this guideline.”
To address this issue, Dr. Singh and his colleagues conducted a systematic review of 10 literature searches to answer the following five questions concerning colorectal risk and screening practices: What is the effect of a family history of CRC on an individual’s risk of CRC? What is the effect of a family history of adenoma on an individual’s risk of CRC? At what age should CRC screening begin? Which screening tests are optimal? What are the optimal testing intervals for people with a family history of CRC or adenoma?
These questions were developed via an iterative online platform and then further developed and voted on by a team of specialists. GRADE (Grading of Recommendation Assessment, Development and Evaluation) was used to assess the quality of evidence to support these questions.
Similarly, individuals with two or more first-degree relatives with CRC had a two- to fourfold increased risk of developing CRC, compared with the general population. The review also found that, of the 20 recommendation statements from the review panel, there was consensus about 19 of them.
Colorectal cancer screening is recommended for all individuals with a family history of CRC or documented adenoma. Similarly, colonoscopy is recommended as the preferred test for individuals at the highest risk– those with one or more affected first-degree relatives. Fecal immunochemistry tests are considered a viable alternative except in patients with two or more first-degree relatives.
If a patient is considered to have an elevated risk of CRC because of family history, then screening should begin when they are aged 10 years younger than when that first-degree relative was diagnosed, and a 5-year screening interval should be followed after that.
Dr. Singh pointed out that the age of the affected first-degree relative should be considered when weighing an individual’s related risk of developing CRC. For example, having an first-degree relative who is diagnosed after the age of 75 is not likely to elevate an individual’s risk of developing CRC. Individuals with one or more second-degree relatives with CRC or nonadvanced adenoma do not appear to have an elevated risk of developing CRC and should be screened according to average-risk guidelines.
Dr. Singh reported receiving funding for from Merck Canada.
SOURCE: Leddin D. Gastroenterology. 2018 Jun. doi: 10.1016/S0016-5085(18)31083-7.
WASHINGTON – The number of first- and second-degree relatives with colorectal cancer can increase an individual’s risk for CRC, which could require screening to be done more frequently.
“There have been multiple guidelines reported as to what should be done for these individuals,” Harminder Singh, MD, of the University of Manitoba and his associates stated. “However, for the most part, they have not systematically analyzed the data.” He went on to say that, “more importantly, there’s been no recent AGA [American Gastroenterological Association] or Canadian Association of Gastroenterology statement, which led the development of this guideline.”
To address this issue, Dr. Singh and his colleagues conducted a systematic review of 10 literature searches to answer the following five questions concerning colorectal risk and screening practices: What is the effect of a family history of CRC on an individual’s risk of CRC? What is the effect of a family history of adenoma on an individual’s risk of CRC? At what age should CRC screening begin? Which screening tests are optimal? What are the optimal testing intervals for people with a family history of CRC or adenoma?
These questions were developed via an iterative online platform and then further developed and voted on by a team of specialists. GRADE (Grading of Recommendation Assessment, Development and Evaluation) was used to assess the quality of evidence to support these questions.
Similarly, individuals with two or more first-degree relatives with CRC had a two- to fourfold increased risk of developing CRC, compared with the general population. The review also found that, of the 20 recommendation statements from the review panel, there was consensus about 19 of them.
Colorectal cancer screening is recommended for all individuals with a family history of CRC or documented adenoma. Similarly, colonoscopy is recommended as the preferred test for individuals at the highest risk– those with one or more affected first-degree relatives. Fecal immunochemistry tests are considered a viable alternative except in patients with two or more first-degree relatives.
If a patient is considered to have an elevated risk of CRC because of family history, then screening should begin when they are aged 10 years younger than when that first-degree relative was diagnosed, and a 5-year screening interval should be followed after that.
Dr. Singh pointed out that the age of the affected first-degree relative should be considered when weighing an individual’s related risk of developing CRC. For example, having an first-degree relative who is diagnosed after the age of 75 is not likely to elevate an individual’s risk of developing CRC. Individuals with one or more second-degree relatives with CRC or nonadvanced adenoma do not appear to have an elevated risk of developing CRC and should be screened according to average-risk guidelines.
Dr. Singh reported receiving funding for from Merck Canada.
SOURCE: Leddin D. Gastroenterology. 2018 Jun. doi: 10.1016/S0016-5085(18)31083-7.
REPORTING FROM DDW 2018
Key clinical point: Patients with 1 or more first-degree relative with CRC should be screened more often.
Major finding: Patients with one or more first-degree relatives with CRC or adenoma had a twofold greater risk of developing CRC, compared with those without a family history of these diseases.
Study details: A systematic review of 10 literature searches assessing risk of CRC in those with a family history of CRC.
Disclosures: Dr. Singh has received funding from Merck Canada.
Source: Leddin D. Gastroenterology. 2018 Jun. doi: 10.1016/S0016-5085(18)31083-7.
Neuropathic Pain in MS
Q) How do I assess for and treat neuropathic pain in MS?
In multiple sclerosis (MS), pain is a common symptom; patients may experience varying forms during their disease course. One type is neuropathic pain, which is initiated or caused by a demyelinating lesion in the central nervous system.1 It may occur spontaneously or be evoked, and it can be intermittent or steady. Given the nature of the disease course in MS, it is important to complete a pain assessment at each visit.
A patient experiencing neuropathic pain is likely to report abnormal sensations or hypersensitivity in the affected area. It is often combined with or adjacent to areas of sensory deficit.1 This includes altered sensations such as pins and needles, numbness, crawling, or burning. The most common MS-related neuropathic pain conditions are ongoing dysaesthetic extremity pain and paroxysmal pain, such as trigeminal neuralgia and Lhermitte phenomenon.1-3
Assessment. When assessing the history of neuropathic pain, it is beneficial to remember that abnormal sensory findings should be neuroanatomically aligned with a lesion site. The mnemonic OPQRST is a helpful reminder to ask about
Onset
Provoking/palliating factors
Quality of the sensation
If it radiates
Severity of the pain (using a scale of 0-10 can be helpful)
Time when the pain occurs.
These probing questions will aid diagnosis and uncover clues on areas to pay special attention to during the examination. For example, when a patient reports numbness of both feet, the clinician might suspect a lesion in the spinal cord and then can try to determine the level during the sensory exam.
Screening tools that capture the patient experience, such as the modified version of the Brief Pain Inventory (BPI), can assist in diagnosis as well as measure the impact of treatment.4
A physical assessment for neuropathic pain includes a full neurologic evaluation of motor, sensory, and autonomic systems to identify all signs of neurologic dysfunction. Attention should be paid to the possible types of negative sensory symptoms (eg, sensory loss) and positive findings (eg, paresthesia). When completing the sensory exam, the clinician can gauge pain by using a sharp object such as a toothpick. Tactile sense can be assessed with a piece of cotton, and temperature can be tested with warm and cold objects. A tuning fork can identify vibration sense. Body sensory maps, on which the clinician draws the sensory disturbance on schematic charts, can provide valuable information.
Diagnostic tests, such as MRI, can also assist in confirming the lesion of the somatosensory nervous system that explains the pain.
Continue to: Treatment
Treatment. Many patients who experience neuropathic pain require a multidisciplinary approach.5 Support from colleagues in rehabilitation can help the patient identify alternative approaches to functioning that avoid triggering or exacerbating the pain. Equipment can also maximize independence and improve quality of life. For example, a soft neck collar is often used to prevent the forward movement that triggers pain in Lhermitte phenomenon.6
When prescribing pain medication, it is important to understand that neuropathic pain is inadequately relieved or not relieved at all with conventional analgesics, such as NSAIDs, or opioid analgesics (eg, morphine).2,3
Dysesthesias are most frequently treated with medications that are categorized as antiseizure, such as gabapentin and pregabalin. Carbamazepam and phenytoin are used as secondline therapy. Sometimes, anti-anxiety medication (eg, duloxetine hydrochloride and clonazepam or tricyclic antidepressants, including amitriptyline or nortriptyline) can be helpful.7 When treating paroxysmal symptoms such as trigeminal neuralgia, antiseizure medications can be effective. Carbamazepine is often the firstline of treatment. As a secondline, oxcarbazepine, lamotrigine, and/or baclofen may be used. In some cases, a referral to neurosurgery for a procedure to reduce pressure on the trigeminal nerve is required.5,8
It is also important to treat any additional symptoms that the pain may be causing, such as depression or social isolation. Referral for counseling as well as integrative health and wellness services can support the patient through a difficult time.5 —RS
Rachael Stacom, MS, ANP-BC, MSCN
Independence Care System, New York, NY
1. Zagon IS, Mclaughlin PJ. Multiple Sclerosis: Perspectives in Treatment and Pathogenesis. Brisbane, Australia: Codon Publications. 2017.
2. O’Connor AB, Schwid SR, Hermann DN, et al. Pain associated with multiple sclerosis: systematic review and proposed classification. Pain. 2008;137(1):96-111.
3. Truini A, Galeotti F, Cruccu G. Treating pain in multiple sclerosis. Expert Opin Pharmacother. 2011;12(15):2355-2368.
4. Osborne TL, Raichle KA, Jensen MP, et al. The reliability and validity of pain interference measures in persons with multiple sclerosis. J Pain Symptom Manage. 2006;32(3):217-229.
5. Sullivan AB, Scheman J, Lopresti A, Prayor-Patterson H. Interdisciplinary treatment of patients with multiple sclerosis and chronic pain. Int J MS Care. 2012;14(4):216-220.
6. MS Australia. Pain and multiple sclerosis (MS). www.msaustralia.org.au/publications/pain-and-multiple-sclerosis-ms. Accessed May 15, 2018.
7. Maloni H; National Multiple Sclerosis Society. Clinical bulletin: pain in multiple sclerosis. www.nationalmssociety.org/NationalMSSociety/media/MSNationalFiles/Brochures/Clinical_Bulletin_Pain-in-MS.pdf. Accessed May 15, 2018.
8. Multiple Sclerosis Association of America (MSAA). (H. Maloni, Ed.) The Motivator Winter/Spring. Retrieved from https://mymsaa.org/publications/motivator/winter-spring13/cover-story/pain. Accessed May 15, 2018.
Clinician Reviews in partnership with

MS Consult is edited by Colleen J. Harris, MN, NP, MSCN, Nurse Practitioner/Manager of the Multiple Sclerosis Clinic at Foothills Medical Centre in Calgary, Alberta, Canada, and Bryan Walker, MHS, PA-C, who is in the Department of Neurology, Division of MS and Neuroimmunology, at Duke University Medical Center in Durham, North Carolina. This month's response was authored by Rachel Stacom, MS, ANP-BC, MSCN, who is Senior Vice President, Care Management, at Independence Care System, New York, NY.
Clinician Reviews in partnership with
MS Consult is edited by Colleen J. Harris, MN, NP, MSCN, Nurse Practitioner/Manager of the Multiple Sclerosis Clinic at Foothills Medical Centre in Calgary, Alberta, Canada, and Bryan Walker, MHS, PA-C, who is in the Department of Neurology, Division of MS and Neuroimmunology, at Duke University Medical Center in Durham, North Carolina. This month's response was authored by Rachel Stacom, MS, ANP-BC, MSCN, who is Senior Vice President, Care Management, at Independence Care System, New York, NY.
Clinician Reviews in partnership with
MS Consult is edited by Colleen J. Harris, MN, NP, MSCN, Nurse Practitioner/Manager of the Multiple Sclerosis Clinic at Foothills Medical Centre in Calgary, Alberta, Canada, and Bryan Walker, MHS, PA-C, who is in the Department of Neurology, Division of MS and Neuroimmunology, at Duke University Medical Center in Durham, North Carolina. This month's response was authored by Rachel Stacom, MS, ANP-BC, MSCN, who is Senior Vice President, Care Management, at Independence Care System, New York, NY.
Q) How do I assess for and treat neuropathic pain in MS?
In multiple sclerosis (MS), pain is a common symptom; patients may experience varying forms during their disease course. One type is neuropathic pain, which is initiated or caused by a demyelinating lesion in the central nervous system.1 It may occur spontaneously or be evoked, and it can be intermittent or steady. Given the nature of the disease course in MS, it is important to complete a pain assessment at each visit.
A patient experiencing neuropathic pain is likely to report abnormal sensations or hypersensitivity in the affected area. It is often combined with or adjacent to areas of sensory deficit.1 This includes altered sensations such as pins and needles, numbness, crawling, or burning. The most common MS-related neuropathic pain conditions are ongoing dysaesthetic extremity pain and paroxysmal pain, such as trigeminal neuralgia and Lhermitte phenomenon.1-3
Assessment. When assessing the history of neuropathic pain, it is beneficial to remember that abnormal sensory findings should be neuroanatomically aligned with a lesion site. The mnemonic OPQRST is a helpful reminder to ask about
Onset
Provoking/palliating factors
Quality of the sensation
If it radiates
Severity of the pain (using a scale of 0-10 can be helpful)
Time when the pain occurs.
These probing questions will aid diagnosis and uncover clues on areas to pay special attention to during the examination. For example, when a patient reports numbness of both feet, the clinician might suspect a lesion in the spinal cord and then can try to determine the level during the sensory exam.
Screening tools that capture the patient experience, such as the modified version of the Brief Pain Inventory (BPI), can assist in diagnosis as well as measure the impact of treatment.4
A physical assessment for neuropathic pain includes a full neurologic evaluation of motor, sensory, and autonomic systems to identify all signs of neurologic dysfunction. Attention should be paid to the possible types of negative sensory symptoms (eg, sensory loss) and positive findings (eg, paresthesia). When completing the sensory exam, the clinician can gauge pain by using a sharp object such as a toothpick. Tactile sense can be assessed with a piece of cotton, and temperature can be tested with warm and cold objects. A tuning fork can identify vibration sense. Body sensory maps, on which the clinician draws the sensory disturbance on schematic charts, can provide valuable information.
Diagnostic tests, such as MRI, can also assist in confirming the lesion of the somatosensory nervous system that explains the pain.
Continue to: Treatment
Treatment. Many patients who experience neuropathic pain require a multidisciplinary approach.5 Support from colleagues in rehabilitation can help the patient identify alternative approaches to functioning that avoid triggering or exacerbating the pain. Equipment can also maximize independence and improve quality of life. For example, a soft neck collar is often used to prevent the forward movement that triggers pain in Lhermitte phenomenon.6
When prescribing pain medication, it is important to understand that neuropathic pain is inadequately relieved or not relieved at all with conventional analgesics, such as NSAIDs, or opioid analgesics (eg, morphine).2,3
Dysesthesias are most frequently treated with medications that are categorized as antiseizure, such as gabapentin and pregabalin. Carbamazepam and phenytoin are used as secondline therapy. Sometimes, anti-anxiety medication (eg, duloxetine hydrochloride and clonazepam or tricyclic antidepressants, including amitriptyline or nortriptyline) can be helpful.7 When treating paroxysmal symptoms such as trigeminal neuralgia, antiseizure medications can be effective. Carbamazepine is often the firstline of treatment. As a secondline, oxcarbazepine, lamotrigine, and/or baclofen may be used. In some cases, a referral to neurosurgery for a procedure to reduce pressure on the trigeminal nerve is required.5,8
It is also important to treat any additional symptoms that the pain may be causing, such as depression or social isolation. Referral for counseling as well as integrative health and wellness services can support the patient through a difficult time.5 —RS
Rachael Stacom, MS, ANP-BC, MSCN
Independence Care System, New York, NY
Q) How do I assess for and treat neuropathic pain in MS?
In multiple sclerosis (MS), pain is a common symptom; patients may experience varying forms during their disease course. One type is neuropathic pain, which is initiated or caused by a demyelinating lesion in the central nervous system.1 It may occur spontaneously or be evoked, and it can be intermittent or steady. Given the nature of the disease course in MS, it is important to complete a pain assessment at each visit.
A patient experiencing neuropathic pain is likely to report abnormal sensations or hypersensitivity in the affected area. It is often combined with or adjacent to areas of sensory deficit.1 This includes altered sensations such as pins and needles, numbness, crawling, or burning. The most common MS-related neuropathic pain conditions are ongoing dysaesthetic extremity pain and paroxysmal pain, such as trigeminal neuralgia and Lhermitte phenomenon.1-3
Assessment. When assessing the history of neuropathic pain, it is beneficial to remember that abnormal sensory findings should be neuroanatomically aligned with a lesion site. The mnemonic OPQRST is a helpful reminder to ask about
Onset
Provoking/palliating factors
Quality of the sensation
If it radiates
Severity of the pain (using a scale of 0-10 can be helpful)
Time when the pain occurs.
These probing questions will aid diagnosis and uncover clues on areas to pay special attention to during the examination. For example, when a patient reports numbness of both feet, the clinician might suspect a lesion in the spinal cord and then can try to determine the level during the sensory exam.
Screening tools that capture the patient experience, such as the modified version of the Brief Pain Inventory (BPI), can assist in diagnosis as well as measure the impact of treatment.4
A physical assessment for neuropathic pain includes a full neurologic evaluation of motor, sensory, and autonomic systems to identify all signs of neurologic dysfunction. Attention should be paid to the possible types of negative sensory symptoms (eg, sensory loss) and positive findings (eg, paresthesia). When completing the sensory exam, the clinician can gauge pain by using a sharp object such as a toothpick. Tactile sense can be assessed with a piece of cotton, and temperature can be tested with warm and cold objects. A tuning fork can identify vibration sense. Body sensory maps, on which the clinician draws the sensory disturbance on schematic charts, can provide valuable information.
Diagnostic tests, such as MRI, can also assist in confirming the lesion of the somatosensory nervous system that explains the pain.
Continue to: Treatment
Treatment. Many patients who experience neuropathic pain require a multidisciplinary approach.5 Support from colleagues in rehabilitation can help the patient identify alternative approaches to functioning that avoid triggering or exacerbating the pain. Equipment can also maximize independence and improve quality of life. For example, a soft neck collar is often used to prevent the forward movement that triggers pain in Lhermitte phenomenon.6
When prescribing pain medication, it is important to understand that neuropathic pain is inadequately relieved or not relieved at all with conventional analgesics, such as NSAIDs, or opioid analgesics (eg, morphine).2,3
Dysesthesias are most frequently treated with medications that are categorized as antiseizure, such as gabapentin and pregabalin. Carbamazepam and phenytoin are used as secondline therapy. Sometimes, anti-anxiety medication (eg, duloxetine hydrochloride and clonazepam or tricyclic antidepressants, including amitriptyline or nortriptyline) can be helpful.7 When treating paroxysmal symptoms such as trigeminal neuralgia, antiseizure medications can be effective. Carbamazepine is often the firstline of treatment. As a secondline, oxcarbazepine, lamotrigine, and/or baclofen may be used. In some cases, a referral to neurosurgery for a procedure to reduce pressure on the trigeminal nerve is required.5,8
It is also important to treat any additional symptoms that the pain may be causing, such as depression or social isolation. Referral for counseling as well as integrative health and wellness services can support the patient through a difficult time.5 —RS
Rachael Stacom, MS, ANP-BC, MSCN
Independence Care System, New York, NY
1. Zagon IS, Mclaughlin PJ. Multiple Sclerosis: Perspectives in Treatment and Pathogenesis. Brisbane, Australia: Codon Publications. 2017.
2. O’Connor AB, Schwid SR, Hermann DN, et al. Pain associated with multiple sclerosis: systematic review and proposed classification. Pain. 2008;137(1):96-111.
3. Truini A, Galeotti F, Cruccu G. Treating pain in multiple sclerosis. Expert Opin Pharmacother. 2011;12(15):2355-2368.
4. Osborne TL, Raichle KA, Jensen MP, et al. The reliability and validity of pain interference measures in persons with multiple sclerosis. J Pain Symptom Manage. 2006;32(3):217-229.
5. Sullivan AB, Scheman J, Lopresti A, Prayor-Patterson H. Interdisciplinary treatment of patients with multiple sclerosis and chronic pain. Int J MS Care. 2012;14(4):216-220.
6. MS Australia. Pain and multiple sclerosis (MS). www.msaustralia.org.au/publications/pain-and-multiple-sclerosis-ms. Accessed May 15, 2018.
7. Maloni H; National Multiple Sclerosis Society. Clinical bulletin: pain in multiple sclerosis. www.nationalmssociety.org/NationalMSSociety/media/MSNationalFiles/Brochures/Clinical_Bulletin_Pain-in-MS.pdf. Accessed May 15, 2018.
8. Multiple Sclerosis Association of America (MSAA). (H. Maloni, Ed.) The Motivator Winter/Spring. Retrieved from https://mymsaa.org/publications/motivator/winter-spring13/cover-story/pain. Accessed May 15, 2018.
1. Zagon IS, Mclaughlin PJ. Multiple Sclerosis: Perspectives in Treatment and Pathogenesis. Brisbane, Australia: Codon Publications. 2017.
2. O’Connor AB, Schwid SR, Hermann DN, et al. Pain associated with multiple sclerosis: systematic review and proposed classification. Pain. 2008;137(1):96-111.
3. Truini A, Galeotti F, Cruccu G. Treating pain in multiple sclerosis. Expert Opin Pharmacother. 2011;12(15):2355-2368.
4. Osborne TL, Raichle KA, Jensen MP, et al. The reliability and validity of pain interference measures in persons with multiple sclerosis. J Pain Symptom Manage. 2006;32(3):217-229.
5. Sullivan AB, Scheman J, Lopresti A, Prayor-Patterson H. Interdisciplinary treatment of patients with multiple sclerosis and chronic pain. Int J MS Care. 2012;14(4):216-220.
6. MS Australia. Pain and multiple sclerosis (MS). www.msaustralia.org.au/publications/pain-and-multiple-sclerosis-ms. Accessed May 15, 2018.
7. Maloni H; National Multiple Sclerosis Society. Clinical bulletin: pain in multiple sclerosis. www.nationalmssociety.org/NationalMSSociety/media/MSNationalFiles/Brochures/Clinical_Bulletin_Pain-in-MS.pdf. Accessed May 15, 2018.
8. Multiple Sclerosis Association of America (MSAA). (H. Maloni, Ed.) The Motivator Winter/Spring. Retrieved from https://mymsaa.org/publications/motivator/winter-spring13/cover-story/pain. Accessed May 15, 2018.

Hydroxychloroquine: An old drug with new relevance
A 29-year-old African American woman presents with a photosensitive malar rash, fatigue, morning stiffness, and swelling in her hands. She is found to have elevated antinuclear antibody at a titer of 1:320. A complete blood cell count demonstrates leukopenia and thrombocytopenia. Results of renal function testing and urinalysis are within normal limits. She has no other medical problems and no history of blood clots or pregnancy loss.
Her arthritis and rash suggest systemic lupus erythematosus. She is counseled to avoid sun exposure, and treatment with hydroxychloroquine is considered.
WHAT IS HYDROXYCHLOROQUINE?
Hydroxychloroquine was developed to treat malaria but was later found to have immunomodulatory properties. It is now approved by the US Food and Drug Administration for treatment of discoid lupus, systemic lupus erythematosus, and rheumatoid arthritis. It is also approved to treat malaria; however, of the several malarial parasites, only Plasmodium falciparum can still be cured by hydroxychloroquine, and growing resistance limits the geographic locations where this drug can be used effectively.1,2
HISTORICAL BACKGROUND
Antimalarial drugs were discovered shortly before World War II. Their production was industrialized during the war because malaria was a leading cause of disease among soldiers, especially those deployed to the South Pacific.3
Atabrine (quinacrine), the first antimalarial widely used, had numerous side effects including yellowing of the skin. Aggressive research efforts to develop an alternative led to field testing of one of its derivative compounds, chloroquine, by the US Army in 1943. Continued chemical modification would create hydroxychloroquine, introduced in 1955.
A serendipitous consequence of the mass use of antimalarials during World War II was the discovery that they could be used to treat inflammatory arthritis and lupus. Eight years after the war ended, Shee4 reported that chloroquine had a beneficial effect on lupus and rheumatoid arthritis in US soldiers. Hydroxychloroquine is now the most commonly prescribed antimalarial for treatment of autoimmune disease.
HOW HYDROXYCHLOROQUINE WORKS
The primary mechanism by which hydroxychloroquine modulates systemic lupus erythematosus is by suppressing activation of Toll-like receptors, which exist on the surface of endosomes and play a significant role in the innate immune response and in autoimmune disease. Their activation is necessary for the expression of interferon-regulated genes and production of tumor necrosis factor alpha, which are key in the cell-mediated inflammatory response.
Antimalarial drugs such as hydroxychloroquine prevent Toll-like receptor activation by binding directly to nucleic acids in the activation pathway.5 In vitro studies show that blocking this pathway blunts the body’s primary cell-mediated inflammatory response; in vivo studies show that use of hydroxychloroquine is strongly correlated with a reduction in interferon alpha levels.6 The powerful effect of hydroxychloroquine on the cell-mediated pattern of inflammation found in lupus is consistent with this theory.
It was previously hypothesized that the immune-modulating effects of hydroxychloroquine were associated with a more general dysregulation of cellular lysosomes through inhibition of proteolysis or changes in cellular pH.7 This theory has since been displaced by the more specific and elegant mechanism described above.5
HOW WELL DOES IT WORK?
Benefit in systemic lupus erythematosus
Hydroxychloroquine has consistently demonstrated significant and multifaceted benefit in patients with systemic lupus erythematosus.
A systematic review of 95 articles8 concluded that this drug decreases lupus flares and decreases mortality rates in lupus patients by at least 50%, with a high level of evidence. Beneficial effects that had a moderate level of evidence were an increase in bone mineral density, fewer thrombotic events, and fewer cases of irreversible organ damage.
The preventive effect of hydroxychloroquine on thrombosis in lupus patients has been consistently demonstrated and is one of the key reasons the drug is considered a cornerstone of therapy in this disease.9 A nested case-control study of patients with lupus and thromboembolism demonstrated an odds ratio of 0.31 and relative risk reduction of 68% for those using antimalarials.10
Benefit in antiphospholipid antibody syndrome
Hydroxychloroquine prevents thrombosis in other diseases as well. For example, it has been shown to reduce the incidence of thrombotic events in patients with primary antiphospholipid syndrome.
In a retrospective cohort study in 114 patients with this disease, hydroxychloroquine significantly reduced the incidence of arterial thrombotic events over 10 years of follow-up (recurrence incidence 0 in those treated with hydroxychloroquine vs 1.14% in those not treated).11 The study also tracked levels of antiphospholipid antibodies and reported that hydroxychloroquine significantly reduced the levels of antibodies to cardiolipin and beta-2 glycoprotein 1, both implicated in the pathology of thrombosis.11
In vitro studies have also demonstrated that hydroxychloroquine can modulate a dysregulated inflammatory system to reduce thrombosis. For example, it has been shown that hydroxychloroquine can reverse platelet activation by antiphospholipid antibodies, prevent linking of antibody complexes to cell membranes, and promote proper membrane protein expression, thereby reducing the thrombotic qualities of antiphospholipid antibodies and even improving clearance times of antiphospholipid-related thrombi.12
Benefit in rheumatoid arthritis
Though there is less evidence, hydroxychloroquine has also shown benefit in rheumatoid arthritis, where it can be used by itself in mild disease or as part of combination therapy with active arthritis. Compared with biologic therapy in patients with early aggressive rheumatoid arthritis, triple therapy with methotrexate, sulfasalazine, and hydroxychloroquine was nearly as effective in terms of quality of life, and it cost only one-third as much, saving $20,000 per year of therapy per patient.13
Hydroxychloroquine has also been compared directly with chloroquine, its closest relation, in a large study incorporating patients with rheumatoid arthritis and patients with systemic lupus erythematosus. Patients using chloroquine experienced significantly more side effects, though it did prove marginally more effective.14
No benefit shown in Sjögren syndrome
Unfortunately, despite widespread use, hydroxychloroquine has not demonstrated positive clinical effects when used to treat primary Sjögren syndrome. Most notably, a 2014 randomized controlled trial of hydroxychloroquine vs placebo in 120 Sjögren patients found no significant improvement in primary symptoms of dryness, pain, or fatigue after 6 months of therapy.15
Metabolic benefits
Unexpectedly, hydroxychloroquine is associated with multiple metabolic benefits including improved lipid profiles and lower blood glucose levels. These findings, in addition to a reduced incidence of thrombosis, were initially reported in the Baltimore Lupus Cohort in 1996.16 Specifically, longitudinal evaluation of a cohort of lupus patients showed that hydroxychloroquine use was associated with a 7.6% reduction in total cholesterol and a 13.7% reduction in low-density lipoprotein cholesterol (LDL-C) over 3 months of therapy.17
Similar findings, including a reduction in LDL-C and an increase in high-density lipoprotein cholesterol, were strongly associated with the addition of hydroxychloroquine to methotrexate or to methotrexate and etanercept in a large cohort of rheumatoid arthritis patients followed over 2 years of therapy.18
In nondiabetic women with systemic lupus erythematosus or rheumatoid arthritis, average blood glucose was significantly lower in those taking hydroxychloroquine than in nonusers. The incidence of insulin resistance was also lower, but the difference was not statistically significant.19
Some have suggested that hydroxychloroquine may prevent diabetes mellitus. In a retrospective case series, compared with rheumatoid arthritis patients not taking the drug, patients treated with hydroxychloroquine for more than 4 years had a 25% lower risk of developing diabetes mellitus.20
In view of these metabolic benefits, especially regarding lipid regulation, and the above described antithrombotic properties of hydroxychloroquine, some researchers have recently hypothesized that hydroxychloroquine may be of benefit in patients with coronary artery disease.21 They suggested that the inflammatory contribution to the mechanism of coronary artery disease could be lessened by hydroxychloroquine even in patients without lupus erythematosus or rheumatoid arthritis.
PHARMACOLOGIC PROPERTIES
Understanding the pharmacologic properties of hydroxychloroquine is key to using it appropriately in clinical practice.
The half-life of elimination of hydroxychloroquine is 40 to 50 days, with half of the drug excreted renally in a concentration-dependent fashion.22,23 The drug reaches 95% of its steady-state concentration by about 6 months of therapy. Shorter durations of therapy do not provide adequate time for the drug to achieve steady-state concentration and may not allow patients and providers time to see its full clinical results. Therefore, its manufacturers recommend a 6-month trial of therapy to adequately determine if the drug improves symptoms.1
The oral bioavailability of hydroxychloroquine is about 75%, but pharmacokinetics vary among individuals.22,23 It has been suggested that this variability affects the efficacy of hydroxychloroquine. In a study of 300 patients with cutaneous lupus erythematosus, those whose treatment failed had significantly lower blood concentrations of hydroxychloroquine, while those who achieved complete remission had significantly higher concentrations.24
Another study found that titrating doses to target therapeutic blood concentrations can reduce disease activity in cutaneous lupus erythematosus.25 Measuring the blood concentration of hydroxychloroquine is not common in clinical practice but may have a role in select patients in whom initial therapy using a standard dosing regimen does not produce the desired results.
HOW SAFE IS HYDROXYCHLOROQUINE?
Hydroxychloroquine has numerous adverse effects, necessitating vigilance on the part of the prescriber. Most commonly reported are retinopathy, hyperpigmentation, myopathy, and skin reactions.1
Retinopathy
Retinopathy’s irreversibility—the threat of permanent vision loss—and its substantial prevalence in patients with a large drug exposure history, have marked retinopathy as the most concerning potential toxicity. The risk of ocular toxicity increases with the cumulative hydroxychloroquine dose. The prevalence of retinopathy in those using the drug less than 10 years is less than 2%; in contrast, the prevalence in patients with more than 20 years of exposure is reported to be as high as 20%.26
The American Academy of Ophthalmology has long stated that retinopathy is a significant risk of hydroxychloroquine therapy and that patients taking hydroxychloroquine should therefore undergo routine retinal and visual field screening by an ophthalmologist.

Currently, initial screening followed by yearly screening beginning 5 years thereafter is recommended for patients at low risk of toxicity (Table 1).27 Patients determined by an ophthalmologist to be at higher risk of retinopathy should be screened yearly. As identified by the American Academy of Ophthalmology, major risk factors for retinopathy include duration of use, concomitant tamoxifen exposure, significant renal disease, and preexisting retinal and macular disease.26,28
Recommendations for hydroxychloroquine dosing and screening were recently revised, for 2 reasons. Initially, its manufacturers recommended that hydroxychloroquine dosage be no higher than 6.5 mg/kg of ideal body weight to prevent retinopathy.1,29,30 However, it has recently been demonstrated that real body weight is a better predictor of risk of retinopathy than ideal body weight when dosing hydroxychloroquine, perhaps because of the increasing variance of real body weight in our patient population.26

Further, an atypical pattern of retinopathy called pericentral retinopathy is more common in Asians. A study of about 200 patients with a history of hydroxychloroquine retinopathy, including 36 Asian patients, found that the pericentral pattern occurred in half the Asian patients but only 2% of the white patients.31 The mechanism for this finding is unclear, but because pericentral retinopathy spares the macula, it can be missed using standard screening methods. Therefore, the American Academy of Ophthalmology now recommends that the dose limit be reduced from 6.5 mg/kg of ideal body weight to no more than 5.0 mg/kg of real body weight (Table 2).28
It is also recommended that screening methods such as automated visual fields and optical coherence tomography extend their fields beyond the macula in Asian patients to ensure that pericentral retinopathy is not missed.28
Optical coherence tomography is a particularly useful tool in the ocular evaluation of patients taking hydroxychloroquine. It can detect subtle changes such as thinning of the foveal photoreceptor outer segment, thickening of the retinal pigment epithelium, and loss of the macular ganglion cell–inner plexiform layer before there are visible signs of retinopathy and before symptoms arise.32
Currently, these guidelines are underutilized in clinical practice. Physician adherence to ophthalmologic guidelines is reported at about 50%.33 This statistic is jarring, given the potential for permanent loss of vision in those with hydroxychloroquine-mediated retinopathy, and demonstrates the importance of reinforcing proper understanding of the use of hydroxychloroquine in clinical practice.
Other adverse effects
Cutaneous hyperpigmentation can occur with hydroxychloroquine use (Figure 1). The hyperpigmentation appears to be due to local bruising following deposition of iron in the soft tissue.

While the pigmentation may persist permanently and cause an undesirable cosmetic effect, it has not been associated with other adverse outcomes.
Myopathy is a rare adverse effect. In one case series, 3 of 214 patients treated with hydroxychloroquine developed hydroxychloroquine-induced myopathy.35 Over the duration of their therapy, this was equivalent to an incidence of 1 case of myopathy in 100 patient-years of therapy. Myopathy improves with discontinuation of therapy, though it can persist for weeks, likely because of hydroxychloroquine’s prolonged elimination half-life.
Cardiomyopathy, specifically neurocardiomyopathy, is also an extremely rare adverse effect of hydroxychloroquine use. The mechanism is believed to be associated with the effect of hydroxychloroquine on lysosomal action, leading to an acquired lysosomal storage disorder with the typical cardiac hypertrophy and conduction abnormalities associated with this family of diseases.36
Acute generalized exanthematous pustulosis is another rare complication of hydroxychloroquine therapy. The appearance of the reaction is similar to that of pustular psoriasis, with pustules overlying flaking and scaling skin. It usually resolves within 2 weeks after cessation of hydroxychloroquine therapy. In a select few cases, the reaction persists or waxes and wanes over a period of weeks to months, and longer durations of recovery are thought to be due to hydroxychloroquine’s long half-life, as in hydroxychloroquine-induced myopathy.37
In view of this rare reaction, manufacturers of hydroxychloroquine recommend caution when using the drug in patients with psoriasis.1
Hematologic abnormalities. In very rare cases, hydroxychloroquine is associated with hematologic abnormalities including agranulocytosis, anemia, aplastic anemia, leukopenia, and thrombocytopenia.1
While no specific guidelines exist, caution is warranted when using hydroxychloroquine in patients with porphyria. Additionally, hydroxychloroquine and other antimalarials including primaquine have been associated with hemolysis in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency. The risk of hemolysis is generally considered low except at high hydroxychloroquine doses in patients with severe G6PD deficiency.38

For the above reasons, manufacturers recommended baseline and routine blood counts, and some providers screen patients for G6PD deficiency when prescribing hydroxychloroquine (Table 3).
PREGNANCY
Hydroxychloroquine is in pregnancy category C. Information is limited, and in view of the risks, the manufacturer says that it should be avoided in pregnancy.1 Nevertheless, it is generally considered safe during pregnancy, and its benefits may make it acceptable to continue in a patient who becomes pregnant, in spite of the possible risks.
We favor continuing hydroxychloroquine. This drug has been associated with improved maternal and fetal outcomes in lupus patients. Its use during pregnancy has not been associated with congenital malformations. The adverse visual effects of long-term hydroxychloroquine use, namely retinopathy, have never been reported in children as a consequence of exposure in utero.
In addition, hydroxychloroquine is transmitted only in minute quantities in breast milk.39 In pregnant women with systemic lupus erythematosus, hydroxychloroquine was associated with a lower risk of adverse outcomes, including preterm delivery and intrauterine growth restriction.40 However, hydroxychloroquine is far more toxic when ingested directly by infants than in adults.1
Maternal outcomes are also improved with the use of hydroxychloroquine. Stopping hydroxychloroquine during pregnancy in women with systemic lupus erythematosus is associated with significantly higher disease activity—fully twice as high as in those who continue hydroxychloroquine.41 These study results were corroborated in a small randomized trial in which pregnant women with lupus on placebo had significantly higher lupus disease activity scores than those pregnant women who were given hydroxychloroquine.42 The women taking hydroxychloroquine experienced no severe lupus flares for the duration of their pregnancies.
These findings suggest not only that hydroxychloroquine is safe in pregnancy, but also that it should be continued in lupus patients during pregnancy to prevent disease flares and adverse fetal outcomes.
AREAS OF UNCERTAINTY
Benefit in preclinical lupus?
Hydroxychloroquine has a consistently profound effect on outcomes in systemic lupus erythematosus. These findings, in addition to the more widespread use of antibody screening, have led to suggestions that hydroxychloroquine could be of benefit even before systemic lupus erythematosus is diagnosed.
A study in US military personnel found that patients taking hydroxychloroquine experienced a significantly longer lag time between first reported clinical symptoms of lupus and official diagnosis compared with matched controls who also went on to develop the disease, averaging 1.08 vs 0.29 years to disease classification.43 Those who used hydroxychloroquine also had lower rates of autoantibody accumulation. Therefore, hydroxychloroquine could be of benefit in carefully selected candidates at high risk of developing systemic lupus erythematosus.
The beneficial effects of hydroxychloroquine on patients with lupus and rheumatoid arthritis, in terms of primary measures of disease activity and secondary outcomes, were discovered fortuitously and were not the original intended targets of the drug. Because of its versatility, there are numerous other disease states in which hydroxychloroquine has shown a degree of benefit or has shown a potential for benefit.
Antiviral activity?
It has been suggested that antimalarial drugs could serve as adjunctive therapies against filoviruses such as Marburg and Ebola. There is a small body of in vitro and in vivo evidence that hydroxychloroquine could temper severe systemic inflammatory responses to filoviruses both through dysregulation of lysosomes and lysosomal pH (filoviruses have a pH-dependent mechanism of action) and through decreased production of tumor necrosis factor alpha and interferons. Heavy burdens of interferons and tumor necrosis factor alpha are associated with increased mortality rates in those infected with filoviruses.44
Antineoplastic activity?
Hydroxychloroquine has undergone in vitro testing as an adjunct to cancer therapies. There are several mechanisms by which it is theorized that hydroxychloroquine could target malignant cells, including inhibition of multidrug resistance pumps or autophagy, improvement of chemotherapy cell penetration, potentiation of presentation of major histocompatibility complexes, or even intercalation directly into DNA.45,46 However, it can also impair natural anticancer immunity and may allow cancer cells better nutrient supply through vascular effects.
In vitro studies have shown tumoricidal effects in lymphoma and melanoma, and inhibition of growth in lung, colon, breast, cervix, larynx, liver, and prostate cancers. In vivo studies have shown that hydroxychloroquine in high doses can prolong survival in glioblastoma.45
Unfortunately, all of these theorized or observed effects are dose-dependent and likely require doses that exceed currently recommended maximums.
Negative chronotropic effect?
Hydroxychloroquine has been found to decrease the resting heart rate in a cumulative dose-dependent fashion.47 Further, hydroxychloroquine has been known to increase digoxin levels, and the medications should not be used in combination.1
Whether the decrease in resting heart rate is associated with harm or benefit and whether the effect is significant enough to be considered when implementing therapy remain unanswered and deserve further investigation, as does the primary use of hydroxychloroquine for beneficial lipid and glucose reduction in patients who are otherwise healthy.
CASE CONCLUSION
The patient described at the beginning of this article was provided with information on the risks and benefits of hydroxychloroquine for treatment of her arthritis and rash suggestive of mild systemic lupus, and she opted to begin therapy. Her baseline eye screening was within normal limits. Based on her weight of 62 kg, she was started on 300 mg of hydroxychloroquine daily.
She had no significant adverse effects from the medication and reported slow improvement in her rash and joint complaints over the next 2 months. She remained on hydroxychloroquine over the next year without adverse effects or new evidence of autoimmune disease.
- Sanofi-Aventis. Product monograph: Plaquenil. http://products.sanofi.ca/en/plaquenil.pdf. Accessed May 2, 2018.
- Centers for Disease Control and Prevention (CDC). Malaria information and prophylaxis, by country. www.cdc.gov/malaria/travelers/country_table/a.html. Accessed May 2, 2018.
- Wallace DJ. The history of antimalarials. Lupus 1996; 5(suppl 1):S2–S3. pmid:8803902
- Shee JC. Lupus erythematosus treated with chloroquine. Lancet 1953; 265(6778):201–202. pmid:13070595
- Kuznik A, Bencina M, Svajger U, Jeras M, Rozman B, Jerala R. Mechanism of endosomal TLR inhibition by antimalarial drugs and imidazoquinolines. J Immunol 2011; 186:4794–4804. doi:10.4049/jimmunol.1000702
- Willis R, Seif AM, McGwin G Jr, et al. Effect of hydroxychloroquine treatment on pro-inflammatory cytokines and disease activity in SLE patients: data from LUMINA, a multiethnic US cohort. Lupus 2012; 21(8):830–835. doi:10.1177/0961203312437270
- Fox R. Anti-malarial drugs: possible mechanisms of action in autoimmune disease and prospects for drug development. Lupus 1996; 5(suppl 1):S4–S10. pmid:8803903
- Ruiz-Irastorza G, Ramos-Casals M, Brito-Zeron P, Khamashta MA. Clinical efficacy and side effects of antimalarials in systemic lupus erythematosus: a systematic review. Ann Rheum Dis 2010; 69(1):20–28. doi:10.1136/ard.2008.101766
- Lam NC, Ghetu MV, Bieniek ML. Systemic lupus erythematosus: primary care approach to diagnosis and management. Am Fam Physician 2016; 94(4):284–294. pmid:27548593
- Jung H, Bobba R, Su J, et al. The protective effect of antimalarial drugs on thrombovascular events in systemic lupus erythematosus. Arthritis Rheum 2010; 62(3):863–868. doi:10.1002/art.27289
- Nuri E, Taraborelli M, Andreoli L, et al. Long-term use of hydroxychloroquine reduces antiphospholipid antibodies levels in patients with primary antiphospholipid syndrome. Immunol Res 2017; 65(1):17–24. doi:10.1007/s12026-016-8812-z
- Erkan D, Aguiar CL, Andrade D, et al. 14th International Congress on Antiphospholipid Antibodies: task force report on antiphospholipid syndrome treatment trends. Autoimmun Rev 2014; 13(6):685–696. doi:10.1016/j.autrev.2014.01.053
- Jalal H, O’Dell JR, Bridges SL Jr, et al. Cost-effectiveness of triple therapy versus etanercept plus methotrexate in early aggressive rheumatoid arthritis. Arthritis Care Res (Hoboken) 2016; 68(12):1751–1757. doi:10.1002/acr.22895
- Avina-Zubieta JA, Galindo-Rodriguez G, Newman S, Suarez-Almazor ME, Russell AS. Long-term effectiveness of antimalarial drugs in rheumatic diseases. Ann Rheum Dis 1998; 57(10):582–587. pmid:9893568
- Gottenberg JE, Ravaud P, Puechal X, et al. Effects of hydroxychloroquine on symptomatic improvement in primary Sjogren syndrome. JAMA 2014; 312(3):249–258. doi:10.1001/jama.2014.7682
- Petri M. Hydroxychloroquine use in the Baltimore Lupus Cohort: effects on lipids, glucose and thrombosis. Lupus 1996; 5(suppl 1):S16–S22. pmid:8803905
- Cairoli E, Rebella M, Danese N, Garra V, Borba EF. Hydroxychloroquine reduces low-density lipoprotein cholesterol levels in systemic lupus erythematosus: a longitudinal evaluation of the lipid-lowering effect. Lupus 2012; 21(11):1178–1182. doi:10.1177/0961203312450084
- Charles-Schoeman C, Wang X, Lee YY, et al. Association of triple therapy with improvement in cholesterol profiles over two-year followup in the treatment of early aggressive rheumatoid arthritis trial. Arthritis Rheumatol 2016; 68(3):577–586. doi:10.1002/art.39502
- Penn SK, Kao AH, Schott LL, et al. Hydroxychloroquine and glycemia in women with rheumatoid arthritis and systemic lupus erythematosus. J Rheumatol 2010; 37(6):1136–1142. doi:10.3899/jrheum.090994
- Wasko MC, Hubert HB, Lingala VB, et al. Hydroxychloroquine and risk of diabetes in patients with rheumatoid arthritis. JAMA 2007; 298(2):187–193. doi:10.1001/jama.298.2.187
- Sun L, Liu M, Li R, et al. Hydroxychloroquine, a promising choice for coronary artery disease? Med Hypotheses 2016; 93:5–7. doi:10.1016/j.mehy.2016.04.045
- Tett SE, Cutler DJ, Day RO, Brown KF. Bioavailability of hydroxychloroquine tablets in healthy volunteers. Br J Clin Pharmacol 1989; 27(6):771–779. pmid:2757893
- Furst DE. Pharmacokinetics of hydroxychloroquine and chloroquine during treatment of rheumatic diseases. Lupus 1996; 5(suppl 1):S11–S15. pmid:8803904
- Frances C, Cosnes A, Duhaut P, et al. Low blood concentration of hydroxychloroquine in patients with refractory cutaneous lupus erythematosus. Arch Dermatol 2012; 148(4):479–484. doi:10.1001/archdermatol.2011.2558
- Chasset F, Arnaud L, Costedoat-Chalumeau N, Zahr N, Bessis D, Francès C. The effect of increasing the dose of hydroxychloroquine in patients with refractory cutaneous lupus erythematosus: an open-label prospective pilot study. J Am Acad Dermatol 2016; 74(4):693–699.e3. doi:10.1016/j.jaad.2015.09.064
- Melles RB, Marmor MF. The risk of toxic retinopathy in patients on long-term hydroxychloroquine therapy. JAMA Ophthalmol 2014; 132(12):1453–1460. doi:10.1001/jamaophthalmol.2014.3459
- Committee on Rheumatologic Care. American College of Rheumatology position statement. Screening for hydroxychloroquine retinopathy. www.rheumatology.org/Portals/0/Files/Screening-for-Hydroxychloroquine-Retinopathy-Position-Statement.pdf. Accessed April 2, 2018.
- Marmor MF, Kellner U, Lai TY, Melles RB, Mieler WF; American Academy of Ophthalmology. Recommendations on screening for chloroquine and hydroxychloroquine retinopathy (2016 revision). Ophthalmology 2016; 123(6):1386–1394. doi:10.1016/j.ophtha.2016.01.058
- Mackenzie AH. Antimalarial drugs for rheumatoid arthritis. Am J Med 1983; 75(6A):48–58. pmid:6362406
- Mackenzie AH. Dose refinements in long-term therapy of rheumatoid arthritis with antimalarials. Am J Med 1983; 75(1A):40–45. pmid:6869410
- Melles RB, Marmor MF. Pericentral retinopathy and racial differences in hydroxychloroquine toxicity. Ophthalmology 2015; 122(1):110–116. doi:10.1016/j.ophtha.2014.07.018
- Uslu H, Gurler B, Yildirim A, et al. Effect of hydroxychloroquine on the retinal layers: a quantitative evaluation with spectral-domain optical coherence tomography. J Ophthalmol 2016; 2016:8643174. doi:10.1155/2016/8643174
- Au A, Parikh V, Modi YS, Ehlers JP, Schachat AP, Singh RP. Hydroxychloroquine screening practice patterns within a large multispecialty ophthalmic practice. Am J Ophthalmol 2015; 160(3):561–568.e2. doi:10.1016/j.ajo.2015.06.009
- Jallouli M, Frances C, Plette JC, et al; Plaquenil Lupus Systemic Study Group. Hydroxychloroquine-induced pigmentation in patients with systemic lupus erythematosus. JAMA Dermatol 2013; 149(8):935–940. doi:10.1001/jamadermatol.2013.709
- Avina-Zubieta JA, Johnson ES, Suarez-Almazor ME, Russell AS. Incidence of myopathy in patients treated with antimalarials: a report of three cases and review of the literature. Br J Rheumatol 1995; 34(2):166–170. pmid:7704464
- Yogasundaram H, Putko BN, Tien J, et al. Hydroxychloroquine-induced cardiomyopathy: case report, pathophysiology, diagnosis, and treatment. Can J Cardiol 2014; 30:1706–1715. doi:10.1016/j.cjca.2014.08.016
- Pearson KC, Morrell DS, Runge SR, Jolly P. Prolonged pustular eruption from hydroxychloroquine: an unusual case of acute generalized exanthematous pustulosis. Cutis 2016; 97(3):212–216. pmid:27023083
- Youngster I, Arcavi L, Schechmaster R, et al. Medications and glucose-6-phosphate dehydrogenase deficiency: an evidence-based review. Drug Saf 2010; 33(9):713–726. doi:10.2165/11536520-000000000-00000
- Ostensen M, Khamashta M, Lockshin M, et al. Anti-inflammatory and immunosuppressive drugs and reproduction. Arthritis Res Ther 2006; 8(3):209. doi:10.1186/ar1957
- Leroux M, Desveaux C, Parcevaux M, et al. Impact of hydroxychloroquine on preterm delivery and intrauterine growth restriction in pregnant women with systemic lupus erythematosus: a descriptive cohort study. Lupus 2015; 24(13):1384–1391. doi:10.1177/0961203315591027
- Clowse MEB, Magder L, Witter F, Petri M. Hydroxychloroquine in lupus pregnancy. Arthritis Rheum 2006; 54(11):3640–3647. doi:10.1002/art.22159
- Levy RA, Vilela VS, Cataldo MJ, et al. Hydroxychloroquine in lupus pregnancy: double-blind and placebo-controlled study. Lupus 2001; 10(6):401–404. doi:10.1191/096120301678646137
- James JA, Kim-Howard XR, Bruner BF, et al. Hydroxychloroquine sulfate treatment is associated with later onset of systemic lupus erythematosus. Lupus 2007; 16(6):401–409. doi:10.1177/0961203307078579
- Akpovwa H. Chloroquine could be used for the treatment of filoviral infections and other viral infections that emerge or emerged from viruses requiring an acidic pH for infectivity. Cell Biochem Funct 2016; 34(4):191–196. doi:10.1002/cbf.3182
- Pascolo S. Time to use a dose of chloroquine as an adjuvant to anti-cancer chemotherapies. Eur J Pharmacol 2016; 771:139–144. doi:10.1016/j.ejphar.2015.12.017
- Furlong HC, Wessels JM, Guerra MT, Stämpfli MR, Foster WG. Hydroxychloroquine attenuates cigarette smoke induced autophagic signaling in the mouse ovary. Reprod Toxicol 2016; 61:105–113. doi:10.1016/j.reprotox.2016.03.044
- Cairoli E, Danese N, Teliz M, et al. Cumulative dose of hydroxychloroquine is associated with a decrease of resting heart rate in patients with systemic lupus erythematosus: a pilot study. Lupus 2015; 24(11):1204–1209. doi:10.1177/0961203315580870
A 29-year-old African American woman presents with a photosensitive malar rash, fatigue, morning stiffness, and swelling in her hands. She is found to have elevated antinuclear antibody at a titer of 1:320. A complete blood cell count demonstrates leukopenia and thrombocytopenia. Results of renal function testing and urinalysis are within normal limits. She has no other medical problems and no history of blood clots or pregnancy loss.
Her arthritis and rash suggest systemic lupus erythematosus. She is counseled to avoid sun exposure, and treatment with hydroxychloroquine is considered.
WHAT IS HYDROXYCHLOROQUINE?
Hydroxychloroquine was developed to treat malaria but was later found to have immunomodulatory properties. It is now approved by the US Food and Drug Administration for treatment of discoid lupus, systemic lupus erythematosus, and rheumatoid arthritis. It is also approved to treat malaria; however, of the several malarial parasites, only Plasmodium falciparum can still be cured by hydroxychloroquine, and growing resistance limits the geographic locations where this drug can be used effectively.1,2
HISTORICAL BACKGROUND
Antimalarial drugs were discovered shortly before World War II. Their production was industrialized during the war because malaria was a leading cause of disease among soldiers, especially those deployed to the South Pacific.3
Atabrine (quinacrine), the first antimalarial widely used, had numerous side effects including yellowing of the skin. Aggressive research efforts to develop an alternative led to field testing of one of its derivative compounds, chloroquine, by the US Army in 1943. Continued chemical modification would create hydroxychloroquine, introduced in 1955.
A serendipitous consequence of the mass use of antimalarials during World War II was the discovery that they could be used to treat inflammatory arthritis and lupus. Eight years after the war ended, Shee4 reported that chloroquine had a beneficial effect on lupus and rheumatoid arthritis in US soldiers. Hydroxychloroquine is now the most commonly prescribed antimalarial for treatment of autoimmune disease.
HOW HYDROXYCHLOROQUINE WORKS
The primary mechanism by which hydroxychloroquine modulates systemic lupus erythematosus is by suppressing activation of Toll-like receptors, which exist on the surface of endosomes and play a significant role in the innate immune response and in autoimmune disease. Their activation is necessary for the expression of interferon-regulated genes and production of tumor necrosis factor alpha, which are key in the cell-mediated inflammatory response.
Antimalarial drugs such as hydroxychloroquine prevent Toll-like receptor activation by binding directly to nucleic acids in the activation pathway.5 In vitro studies show that blocking this pathway blunts the body’s primary cell-mediated inflammatory response; in vivo studies show that use of hydroxychloroquine is strongly correlated with a reduction in interferon alpha levels.6 The powerful effect of hydroxychloroquine on the cell-mediated pattern of inflammation found in lupus is consistent with this theory.
It was previously hypothesized that the immune-modulating effects of hydroxychloroquine were associated with a more general dysregulation of cellular lysosomes through inhibition of proteolysis or changes in cellular pH.7 This theory has since been displaced by the more specific and elegant mechanism described above.5
HOW WELL DOES IT WORK?
Benefit in systemic lupus erythematosus
Hydroxychloroquine has consistently demonstrated significant and multifaceted benefit in patients with systemic lupus erythematosus.
A systematic review of 95 articles8 concluded that this drug decreases lupus flares and decreases mortality rates in lupus patients by at least 50%, with a high level of evidence. Beneficial effects that had a moderate level of evidence were an increase in bone mineral density, fewer thrombotic events, and fewer cases of irreversible organ damage.
The preventive effect of hydroxychloroquine on thrombosis in lupus patients has been consistently demonstrated and is one of the key reasons the drug is considered a cornerstone of therapy in this disease.9 A nested case-control study of patients with lupus and thromboembolism demonstrated an odds ratio of 0.31 and relative risk reduction of 68% for those using antimalarials.10
Benefit in antiphospholipid antibody syndrome
Hydroxychloroquine prevents thrombosis in other diseases as well. For example, it has been shown to reduce the incidence of thrombotic events in patients with primary antiphospholipid syndrome.
In a retrospective cohort study in 114 patients with this disease, hydroxychloroquine significantly reduced the incidence of arterial thrombotic events over 10 years of follow-up (recurrence incidence 0 in those treated with hydroxychloroquine vs 1.14% in those not treated).11 The study also tracked levels of antiphospholipid antibodies and reported that hydroxychloroquine significantly reduced the levels of antibodies to cardiolipin and beta-2 glycoprotein 1, both implicated in the pathology of thrombosis.11
In vitro studies have also demonstrated that hydroxychloroquine can modulate a dysregulated inflammatory system to reduce thrombosis. For example, it has been shown that hydroxychloroquine can reverse platelet activation by antiphospholipid antibodies, prevent linking of antibody complexes to cell membranes, and promote proper membrane protein expression, thereby reducing the thrombotic qualities of antiphospholipid antibodies and even improving clearance times of antiphospholipid-related thrombi.12
Benefit in rheumatoid arthritis
Though there is less evidence, hydroxychloroquine has also shown benefit in rheumatoid arthritis, where it can be used by itself in mild disease or as part of combination therapy with active arthritis. Compared with biologic therapy in patients with early aggressive rheumatoid arthritis, triple therapy with methotrexate, sulfasalazine, and hydroxychloroquine was nearly as effective in terms of quality of life, and it cost only one-third as much, saving $20,000 per year of therapy per patient.13
Hydroxychloroquine has also been compared directly with chloroquine, its closest relation, in a large study incorporating patients with rheumatoid arthritis and patients with systemic lupus erythematosus. Patients using chloroquine experienced significantly more side effects, though it did prove marginally more effective.14
No benefit shown in Sjögren syndrome
Unfortunately, despite widespread use, hydroxychloroquine has not demonstrated positive clinical effects when used to treat primary Sjögren syndrome. Most notably, a 2014 randomized controlled trial of hydroxychloroquine vs placebo in 120 Sjögren patients found no significant improvement in primary symptoms of dryness, pain, or fatigue after 6 months of therapy.15
Metabolic benefits
Unexpectedly, hydroxychloroquine is associated with multiple metabolic benefits including improved lipid profiles and lower blood glucose levels. These findings, in addition to a reduced incidence of thrombosis, were initially reported in the Baltimore Lupus Cohort in 1996.16 Specifically, longitudinal evaluation of a cohort of lupus patients showed that hydroxychloroquine use was associated with a 7.6% reduction in total cholesterol and a 13.7% reduction in low-density lipoprotein cholesterol (LDL-C) over 3 months of therapy.17
Similar findings, including a reduction in LDL-C and an increase in high-density lipoprotein cholesterol, were strongly associated with the addition of hydroxychloroquine to methotrexate or to methotrexate and etanercept in a large cohort of rheumatoid arthritis patients followed over 2 years of therapy.18
In nondiabetic women with systemic lupus erythematosus or rheumatoid arthritis, average blood glucose was significantly lower in those taking hydroxychloroquine than in nonusers. The incidence of insulin resistance was also lower, but the difference was not statistically significant.19
Some have suggested that hydroxychloroquine may prevent diabetes mellitus. In a retrospective case series, compared with rheumatoid arthritis patients not taking the drug, patients treated with hydroxychloroquine for more than 4 years had a 25% lower risk of developing diabetes mellitus.20
In view of these metabolic benefits, especially regarding lipid regulation, and the above described antithrombotic properties of hydroxychloroquine, some researchers have recently hypothesized that hydroxychloroquine may be of benefit in patients with coronary artery disease.21 They suggested that the inflammatory contribution to the mechanism of coronary artery disease could be lessened by hydroxychloroquine even in patients without lupus erythematosus or rheumatoid arthritis.
PHARMACOLOGIC PROPERTIES
Understanding the pharmacologic properties of hydroxychloroquine is key to using it appropriately in clinical practice.
The half-life of elimination of hydroxychloroquine is 40 to 50 days, with half of the drug excreted renally in a concentration-dependent fashion.22,23 The drug reaches 95% of its steady-state concentration by about 6 months of therapy. Shorter durations of therapy do not provide adequate time for the drug to achieve steady-state concentration and may not allow patients and providers time to see its full clinical results. Therefore, its manufacturers recommend a 6-month trial of therapy to adequately determine if the drug improves symptoms.1
The oral bioavailability of hydroxychloroquine is about 75%, but pharmacokinetics vary among individuals.22,23 It has been suggested that this variability affects the efficacy of hydroxychloroquine. In a study of 300 patients with cutaneous lupus erythematosus, those whose treatment failed had significantly lower blood concentrations of hydroxychloroquine, while those who achieved complete remission had significantly higher concentrations.24
Another study found that titrating doses to target therapeutic blood concentrations can reduce disease activity in cutaneous lupus erythematosus.25 Measuring the blood concentration of hydroxychloroquine is not common in clinical practice but may have a role in select patients in whom initial therapy using a standard dosing regimen does not produce the desired results.
HOW SAFE IS HYDROXYCHLOROQUINE?
Hydroxychloroquine has numerous adverse effects, necessitating vigilance on the part of the prescriber. Most commonly reported are retinopathy, hyperpigmentation, myopathy, and skin reactions.1
Retinopathy
Retinopathy’s irreversibility—the threat of permanent vision loss—and its substantial prevalence in patients with a large drug exposure history, have marked retinopathy as the most concerning potential toxicity. The risk of ocular toxicity increases with the cumulative hydroxychloroquine dose. The prevalence of retinopathy in those using the drug less than 10 years is less than 2%; in contrast, the prevalence in patients with more than 20 years of exposure is reported to be as high as 20%.26
The American Academy of Ophthalmology has long stated that retinopathy is a significant risk of hydroxychloroquine therapy and that patients taking hydroxychloroquine should therefore undergo routine retinal and visual field screening by an ophthalmologist.
Currently, initial screening followed by yearly screening beginning 5 years thereafter is recommended for patients at low risk of toxicity (Table 1).27 Patients determined by an ophthalmologist to be at higher risk of retinopathy should be screened yearly. As identified by the American Academy of Ophthalmology, major risk factors for retinopathy include duration of use, concomitant tamoxifen exposure, significant renal disease, and preexisting retinal and macular disease.26,28
Recommendations for hydroxychloroquine dosing and screening were recently revised, for 2 reasons. Initially, its manufacturers recommended that hydroxychloroquine dosage be no higher than 6.5 mg/kg of ideal body weight to prevent retinopathy.1,29,30 However, it has recently been demonstrated that real body weight is a better predictor of risk of retinopathy than ideal body weight when dosing hydroxychloroquine, perhaps because of the increasing variance of real body weight in our patient population.26
Further, an atypical pattern of retinopathy called pericentral retinopathy is more common in Asians. A study of about 200 patients with a history of hydroxychloroquine retinopathy, including 36 Asian patients, found that the pericentral pattern occurred in half the Asian patients but only 2% of the white patients.31 The mechanism for this finding is unclear, but because pericentral retinopathy spares the macula, it can be missed using standard screening methods. Therefore, the American Academy of Ophthalmology now recommends that the dose limit be reduced from 6.5 mg/kg of ideal body weight to no more than 5.0 mg/kg of real body weight (Table 2).28
It is also recommended that screening methods such as automated visual fields and optical coherence tomography extend their fields beyond the macula in Asian patients to ensure that pericentral retinopathy is not missed.28
Optical coherence tomography is a particularly useful tool in the ocular evaluation of patients taking hydroxychloroquine. It can detect subtle changes such as thinning of the foveal photoreceptor outer segment, thickening of the retinal pigment epithelium, and loss of the macular ganglion cell–inner plexiform layer before there are visible signs of retinopathy and before symptoms arise.32
Currently, these guidelines are underutilized in clinical practice. Physician adherence to ophthalmologic guidelines is reported at about 50%.33 This statistic is jarring, given the potential for permanent loss of vision in those with hydroxychloroquine-mediated retinopathy, and demonstrates the importance of reinforcing proper understanding of the use of hydroxychloroquine in clinical practice.
Other adverse effects
Cutaneous hyperpigmentation can occur with hydroxychloroquine use (Figure 1). The hyperpigmentation appears to be due to local bruising following deposition of iron in the soft tissue.
While the pigmentation may persist permanently and cause an undesirable cosmetic effect, it has not been associated with other adverse outcomes.
Myopathy is a rare adverse effect. In one case series, 3 of 214 patients treated with hydroxychloroquine developed hydroxychloroquine-induced myopathy.35 Over the duration of their therapy, this was equivalent to an incidence of 1 case of myopathy in 100 patient-years of therapy. Myopathy improves with discontinuation of therapy, though it can persist for weeks, likely because of hydroxychloroquine’s prolonged elimination half-life.
Cardiomyopathy, specifically neurocardiomyopathy, is also an extremely rare adverse effect of hydroxychloroquine use. The mechanism is believed to be associated with the effect of hydroxychloroquine on lysosomal action, leading to an acquired lysosomal storage disorder with the typical cardiac hypertrophy and conduction abnormalities associated with this family of diseases.36
Acute generalized exanthematous pustulosis is another rare complication of hydroxychloroquine therapy. The appearance of the reaction is similar to that of pustular psoriasis, with pustules overlying flaking and scaling skin. It usually resolves within 2 weeks after cessation of hydroxychloroquine therapy. In a select few cases, the reaction persists or waxes and wanes over a period of weeks to months, and longer durations of recovery are thought to be due to hydroxychloroquine’s long half-life, as in hydroxychloroquine-induced myopathy.37
In view of this rare reaction, manufacturers of hydroxychloroquine recommend caution when using the drug in patients with psoriasis.1
Hematologic abnormalities. In very rare cases, hydroxychloroquine is associated with hematologic abnormalities including agranulocytosis, anemia, aplastic anemia, leukopenia, and thrombocytopenia.1
While no specific guidelines exist, caution is warranted when using hydroxychloroquine in patients with porphyria. Additionally, hydroxychloroquine and other antimalarials including primaquine have been associated with hemolysis in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency. The risk of hemolysis is generally considered low except at high hydroxychloroquine doses in patients with severe G6PD deficiency.38
For the above reasons, manufacturers recommended baseline and routine blood counts, and some providers screen patients for G6PD deficiency when prescribing hydroxychloroquine (Table 3).
PREGNANCY
Hydroxychloroquine is in pregnancy category C. Information is limited, and in view of the risks, the manufacturer says that it should be avoided in pregnancy.1 Nevertheless, it is generally considered safe during pregnancy, and its benefits may make it acceptable to continue in a patient who becomes pregnant, in spite of the possible risks.
We favor continuing hydroxychloroquine. This drug has been associated with improved maternal and fetal outcomes in lupus patients. Its use during pregnancy has not been associated with congenital malformations. The adverse visual effects of long-term hydroxychloroquine use, namely retinopathy, have never been reported in children as a consequence of exposure in utero.
In addition, hydroxychloroquine is transmitted only in minute quantities in breast milk.39 In pregnant women with systemic lupus erythematosus, hydroxychloroquine was associated with a lower risk of adverse outcomes, including preterm delivery and intrauterine growth restriction.40 However, hydroxychloroquine is far more toxic when ingested directly by infants than in adults.1
Maternal outcomes are also improved with the use of hydroxychloroquine. Stopping hydroxychloroquine during pregnancy in women with systemic lupus erythematosus is associated with significantly higher disease activity—fully twice as high as in those who continue hydroxychloroquine.41 These study results were corroborated in a small randomized trial in which pregnant women with lupus on placebo had significantly higher lupus disease activity scores than those pregnant women who were given hydroxychloroquine.42 The women taking hydroxychloroquine experienced no severe lupus flares for the duration of their pregnancies.
These findings suggest not only that hydroxychloroquine is safe in pregnancy, but also that it should be continued in lupus patients during pregnancy to prevent disease flares and adverse fetal outcomes.
AREAS OF UNCERTAINTY
Benefit in preclinical lupus?
Hydroxychloroquine has a consistently profound effect on outcomes in systemic lupus erythematosus. These findings, in addition to the more widespread use of antibody screening, have led to suggestions that hydroxychloroquine could be of benefit even before systemic lupus erythematosus is diagnosed.
A study in US military personnel found that patients taking hydroxychloroquine experienced a significantly longer lag time between first reported clinical symptoms of lupus and official diagnosis compared with matched controls who also went on to develop the disease, averaging 1.08 vs 0.29 years to disease classification.43 Those who used hydroxychloroquine also had lower rates of autoantibody accumulation. Therefore, hydroxychloroquine could be of benefit in carefully selected candidates at high risk of developing systemic lupus erythematosus.
The beneficial effects of hydroxychloroquine on patients with lupus and rheumatoid arthritis, in terms of primary measures of disease activity and secondary outcomes, were discovered fortuitously and were not the original intended targets of the drug. Because of its versatility, there are numerous other disease states in which hydroxychloroquine has shown a degree of benefit or has shown a potential for benefit.
Antiviral activity?
It has been suggested that antimalarial drugs could serve as adjunctive therapies against filoviruses such as Marburg and Ebola. There is a small body of in vitro and in vivo evidence that hydroxychloroquine could temper severe systemic inflammatory responses to filoviruses both through dysregulation of lysosomes and lysosomal pH (filoviruses have a pH-dependent mechanism of action) and through decreased production of tumor necrosis factor alpha and interferons. Heavy burdens of interferons and tumor necrosis factor alpha are associated with increased mortality rates in those infected with filoviruses.44
Antineoplastic activity?
Hydroxychloroquine has undergone in vitro testing as an adjunct to cancer therapies. There are several mechanisms by which it is theorized that hydroxychloroquine could target malignant cells, including inhibition of multidrug resistance pumps or autophagy, improvement of chemotherapy cell penetration, potentiation of presentation of major histocompatibility complexes, or even intercalation directly into DNA.45,46 However, it can also impair natural anticancer immunity and may allow cancer cells better nutrient supply through vascular effects.
In vitro studies have shown tumoricidal effects in lymphoma and melanoma, and inhibition of growth in lung, colon, breast, cervix, larynx, liver, and prostate cancers. In vivo studies have shown that hydroxychloroquine in high doses can prolong survival in glioblastoma.45
Unfortunately, all of these theorized or observed effects are dose-dependent and likely require doses that exceed currently recommended maximums.
Negative chronotropic effect?
Hydroxychloroquine has been found to decrease the resting heart rate in a cumulative dose-dependent fashion.47 Further, hydroxychloroquine has been known to increase digoxin levels, and the medications should not be used in combination.1
Whether the decrease in resting heart rate is associated with harm or benefit and whether the effect is significant enough to be considered when implementing therapy remain unanswered and deserve further investigation, as does the primary use of hydroxychloroquine for beneficial lipid and glucose reduction in patients who are otherwise healthy.
CASE CONCLUSION
The patient described at the beginning of this article was provided with information on the risks and benefits of hydroxychloroquine for treatment of her arthritis and rash suggestive of mild systemic lupus, and she opted to begin therapy. Her baseline eye screening was within normal limits. Based on her weight of 62 kg, she was started on 300 mg of hydroxychloroquine daily.
She had no significant adverse effects from the medication and reported slow improvement in her rash and joint complaints over the next 2 months. She remained on hydroxychloroquine over the next year without adverse effects or new evidence of autoimmune disease.
A 29-year-old African American woman presents with a photosensitive malar rash, fatigue, morning stiffness, and swelling in her hands. She is found to have elevated antinuclear antibody at a titer of 1:320. A complete blood cell count demonstrates leukopenia and thrombocytopenia. Results of renal function testing and urinalysis are within normal limits. She has no other medical problems and no history of blood clots or pregnancy loss.
Her arthritis and rash suggest systemic lupus erythematosus. She is counseled to avoid sun exposure, and treatment with hydroxychloroquine is considered.
WHAT IS HYDROXYCHLOROQUINE?
Hydroxychloroquine was developed to treat malaria but was later found to have immunomodulatory properties. It is now approved by the US Food and Drug Administration for treatment of discoid lupus, systemic lupus erythematosus, and rheumatoid arthritis. It is also approved to treat malaria; however, of the several malarial parasites, only Plasmodium falciparum can still be cured by hydroxychloroquine, and growing resistance limits the geographic locations where this drug can be used effectively.1,2
HISTORICAL BACKGROUND
Antimalarial drugs were discovered shortly before World War II. Their production was industrialized during the war because malaria was a leading cause of disease among soldiers, especially those deployed to the South Pacific.3
Atabrine (quinacrine), the first antimalarial widely used, had numerous side effects including yellowing of the skin. Aggressive research efforts to develop an alternative led to field testing of one of its derivative compounds, chloroquine, by the US Army in 1943. Continued chemical modification would create hydroxychloroquine, introduced in 1955.
A serendipitous consequence of the mass use of antimalarials during World War II was the discovery that they could be used to treat inflammatory arthritis and lupus. Eight years after the war ended, Shee4 reported that chloroquine had a beneficial effect on lupus and rheumatoid arthritis in US soldiers. Hydroxychloroquine is now the most commonly prescribed antimalarial for treatment of autoimmune disease.
HOW HYDROXYCHLOROQUINE WORKS
The primary mechanism by which hydroxychloroquine modulates systemic lupus erythematosus is by suppressing activation of Toll-like receptors, which exist on the surface of endosomes and play a significant role in the innate immune response and in autoimmune disease. Their activation is necessary for the expression of interferon-regulated genes and production of tumor necrosis factor alpha, which are key in the cell-mediated inflammatory response.
Antimalarial drugs such as hydroxychloroquine prevent Toll-like receptor activation by binding directly to nucleic acids in the activation pathway.5 In vitro studies show that blocking this pathway blunts the body’s primary cell-mediated inflammatory response; in vivo studies show that use of hydroxychloroquine is strongly correlated with a reduction in interferon alpha levels.6 The powerful effect of hydroxychloroquine on the cell-mediated pattern of inflammation found in lupus is consistent with this theory.
It was previously hypothesized that the immune-modulating effects of hydroxychloroquine were associated with a more general dysregulation of cellular lysosomes through inhibition of proteolysis or changes in cellular pH.7 This theory has since been displaced by the more specific and elegant mechanism described above.5
HOW WELL DOES IT WORK?
Benefit in systemic lupus erythematosus
Hydroxychloroquine has consistently demonstrated significant and multifaceted benefit in patients with systemic lupus erythematosus.
A systematic review of 95 articles8 concluded that this drug decreases lupus flares and decreases mortality rates in lupus patients by at least 50%, with a high level of evidence. Beneficial effects that had a moderate level of evidence were an increase in bone mineral density, fewer thrombotic events, and fewer cases of irreversible organ damage.
The preventive effect of hydroxychloroquine on thrombosis in lupus patients has been consistently demonstrated and is one of the key reasons the drug is considered a cornerstone of therapy in this disease.9 A nested case-control study of patients with lupus and thromboembolism demonstrated an odds ratio of 0.31 and relative risk reduction of 68% for those using antimalarials.10
Benefit in antiphospholipid antibody syndrome
Hydroxychloroquine prevents thrombosis in other diseases as well. For example, it has been shown to reduce the incidence of thrombotic events in patients with primary antiphospholipid syndrome.
In a retrospective cohort study in 114 patients with this disease, hydroxychloroquine significantly reduced the incidence of arterial thrombotic events over 10 years of follow-up (recurrence incidence 0 in those treated with hydroxychloroquine vs 1.14% in those not treated).11 The study also tracked levels of antiphospholipid antibodies and reported that hydroxychloroquine significantly reduced the levels of antibodies to cardiolipin and beta-2 glycoprotein 1, both implicated in the pathology of thrombosis.11
In vitro studies have also demonstrated that hydroxychloroquine can modulate a dysregulated inflammatory system to reduce thrombosis. For example, it has been shown that hydroxychloroquine can reverse platelet activation by antiphospholipid antibodies, prevent linking of antibody complexes to cell membranes, and promote proper membrane protein expression, thereby reducing the thrombotic qualities of antiphospholipid antibodies and even improving clearance times of antiphospholipid-related thrombi.12
Benefit in rheumatoid arthritis
Though there is less evidence, hydroxychloroquine has also shown benefit in rheumatoid arthritis, where it can be used by itself in mild disease or as part of combination therapy with active arthritis. Compared with biologic therapy in patients with early aggressive rheumatoid arthritis, triple therapy with methotrexate, sulfasalazine, and hydroxychloroquine was nearly as effective in terms of quality of life, and it cost only one-third as much, saving $20,000 per year of therapy per patient.13
Hydroxychloroquine has also been compared directly with chloroquine, its closest relation, in a large study incorporating patients with rheumatoid arthritis and patients with systemic lupus erythematosus. Patients using chloroquine experienced significantly more side effects, though it did prove marginally more effective.14
No benefit shown in Sjögren syndrome
Unfortunately, despite widespread use, hydroxychloroquine has not demonstrated positive clinical effects when used to treat primary Sjögren syndrome. Most notably, a 2014 randomized controlled trial of hydroxychloroquine vs placebo in 120 Sjögren patients found no significant improvement in primary symptoms of dryness, pain, or fatigue after 6 months of therapy.15
Metabolic benefits
Unexpectedly, hydroxychloroquine is associated with multiple metabolic benefits including improved lipid profiles and lower blood glucose levels. These findings, in addition to a reduced incidence of thrombosis, were initially reported in the Baltimore Lupus Cohort in 1996.16 Specifically, longitudinal evaluation of a cohort of lupus patients showed that hydroxychloroquine use was associated with a 7.6% reduction in total cholesterol and a 13.7% reduction in low-density lipoprotein cholesterol (LDL-C) over 3 months of therapy.17
Similar findings, including a reduction in LDL-C and an increase in high-density lipoprotein cholesterol, were strongly associated with the addition of hydroxychloroquine to methotrexate or to methotrexate and etanercept in a large cohort of rheumatoid arthritis patients followed over 2 years of therapy.18
In nondiabetic women with systemic lupus erythematosus or rheumatoid arthritis, average blood glucose was significantly lower in those taking hydroxychloroquine than in nonusers. The incidence of insulin resistance was also lower, but the difference was not statistically significant.19
Some have suggested that hydroxychloroquine may prevent diabetes mellitus. In a retrospective case series, compared with rheumatoid arthritis patients not taking the drug, patients treated with hydroxychloroquine for more than 4 years had a 25% lower risk of developing diabetes mellitus.20
In view of these metabolic benefits, especially regarding lipid regulation, and the above described antithrombotic properties of hydroxychloroquine, some researchers have recently hypothesized that hydroxychloroquine may be of benefit in patients with coronary artery disease.21 They suggested that the inflammatory contribution to the mechanism of coronary artery disease could be lessened by hydroxychloroquine even in patients without lupus erythematosus or rheumatoid arthritis.
PHARMACOLOGIC PROPERTIES
Understanding the pharmacologic properties of hydroxychloroquine is key to using it appropriately in clinical practice.
The half-life of elimination of hydroxychloroquine is 40 to 50 days, with half of the drug excreted renally in a concentration-dependent fashion.22,23 The drug reaches 95% of its steady-state concentration by about 6 months of therapy. Shorter durations of therapy do not provide adequate time for the drug to achieve steady-state concentration and may not allow patients and providers time to see its full clinical results. Therefore, its manufacturers recommend a 6-month trial of therapy to adequately determine if the drug improves symptoms.1
The oral bioavailability of hydroxychloroquine is about 75%, but pharmacokinetics vary among individuals.22,23 It has been suggested that this variability affects the efficacy of hydroxychloroquine. In a study of 300 patients with cutaneous lupus erythematosus, those whose treatment failed had significantly lower blood concentrations of hydroxychloroquine, while those who achieved complete remission had significantly higher concentrations.24
Another study found that titrating doses to target therapeutic blood concentrations can reduce disease activity in cutaneous lupus erythematosus.25 Measuring the blood concentration of hydroxychloroquine is not common in clinical practice but may have a role in select patients in whom initial therapy using a standard dosing regimen does not produce the desired results.
HOW SAFE IS HYDROXYCHLOROQUINE?
Hydroxychloroquine has numerous adverse effects, necessitating vigilance on the part of the prescriber. Most commonly reported are retinopathy, hyperpigmentation, myopathy, and skin reactions.1
Retinopathy
Retinopathy’s irreversibility—the threat of permanent vision loss—and its substantial prevalence in patients with a large drug exposure history, have marked retinopathy as the most concerning potential toxicity. The risk of ocular toxicity increases with the cumulative hydroxychloroquine dose. The prevalence of retinopathy in those using the drug less than 10 years is less than 2%; in contrast, the prevalence in patients with more than 20 years of exposure is reported to be as high as 20%.26
The American Academy of Ophthalmology has long stated that retinopathy is a significant risk of hydroxychloroquine therapy and that patients taking hydroxychloroquine should therefore undergo routine retinal and visual field screening by an ophthalmologist.
Currently, initial screening followed by yearly screening beginning 5 years thereafter is recommended for patients at low risk of toxicity (Table 1).27 Patients determined by an ophthalmologist to be at higher risk of retinopathy should be screened yearly. As identified by the American Academy of Ophthalmology, major risk factors for retinopathy include duration of use, concomitant tamoxifen exposure, significant renal disease, and preexisting retinal and macular disease.26,28
Recommendations for hydroxychloroquine dosing and screening were recently revised, for 2 reasons. Initially, its manufacturers recommended that hydroxychloroquine dosage be no higher than 6.5 mg/kg of ideal body weight to prevent retinopathy.1,29,30 However, it has recently been demonstrated that real body weight is a better predictor of risk of retinopathy than ideal body weight when dosing hydroxychloroquine, perhaps because of the increasing variance of real body weight in our patient population.26
Further, an atypical pattern of retinopathy called pericentral retinopathy is more common in Asians. A study of about 200 patients with a history of hydroxychloroquine retinopathy, including 36 Asian patients, found that the pericentral pattern occurred in half the Asian patients but only 2% of the white patients.31 The mechanism for this finding is unclear, but because pericentral retinopathy spares the macula, it can be missed using standard screening methods. Therefore, the American Academy of Ophthalmology now recommends that the dose limit be reduced from 6.5 mg/kg of ideal body weight to no more than 5.0 mg/kg of real body weight (Table 2).28
It is also recommended that screening methods such as automated visual fields and optical coherence tomography extend their fields beyond the macula in Asian patients to ensure that pericentral retinopathy is not missed.28
Optical coherence tomography is a particularly useful tool in the ocular evaluation of patients taking hydroxychloroquine. It can detect subtle changes such as thinning of the foveal photoreceptor outer segment, thickening of the retinal pigment epithelium, and loss of the macular ganglion cell–inner plexiform layer before there are visible signs of retinopathy and before symptoms arise.32
Currently, these guidelines are underutilized in clinical practice. Physician adherence to ophthalmologic guidelines is reported at about 50%.33 This statistic is jarring, given the potential for permanent loss of vision in those with hydroxychloroquine-mediated retinopathy, and demonstrates the importance of reinforcing proper understanding of the use of hydroxychloroquine in clinical practice.
Other adverse effects
Cutaneous hyperpigmentation can occur with hydroxychloroquine use (Figure 1). The hyperpigmentation appears to be due to local bruising following deposition of iron in the soft tissue.
While the pigmentation may persist permanently and cause an undesirable cosmetic effect, it has not been associated with other adverse outcomes.
Myopathy is a rare adverse effect. In one case series, 3 of 214 patients treated with hydroxychloroquine developed hydroxychloroquine-induced myopathy.35 Over the duration of their therapy, this was equivalent to an incidence of 1 case of myopathy in 100 patient-years of therapy. Myopathy improves with discontinuation of therapy, though it can persist for weeks, likely because of hydroxychloroquine’s prolonged elimination half-life.
Cardiomyopathy, specifically neurocardiomyopathy, is also an extremely rare adverse effect of hydroxychloroquine use. The mechanism is believed to be associated with the effect of hydroxychloroquine on lysosomal action, leading to an acquired lysosomal storage disorder with the typical cardiac hypertrophy and conduction abnormalities associated with this family of diseases.36
Acute generalized exanthematous pustulosis is another rare complication of hydroxychloroquine therapy. The appearance of the reaction is similar to that of pustular psoriasis, with pustules overlying flaking and scaling skin. It usually resolves within 2 weeks after cessation of hydroxychloroquine therapy. In a select few cases, the reaction persists or waxes and wanes over a period of weeks to months, and longer durations of recovery are thought to be due to hydroxychloroquine’s long half-life, as in hydroxychloroquine-induced myopathy.37
In view of this rare reaction, manufacturers of hydroxychloroquine recommend caution when using the drug in patients with psoriasis.1
Hematologic abnormalities. In very rare cases, hydroxychloroquine is associated with hematologic abnormalities including agranulocytosis, anemia, aplastic anemia, leukopenia, and thrombocytopenia.1
While no specific guidelines exist, caution is warranted when using hydroxychloroquine in patients with porphyria. Additionally, hydroxychloroquine and other antimalarials including primaquine have been associated with hemolysis in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency. The risk of hemolysis is generally considered low except at high hydroxychloroquine doses in patients with severe G6PD deficiency.38
For the above reasons, manufacturers recommended baseline and routine blood counts, and some providers screen patients for G6PD deficiency when prescribing hydroxychloroquine (Table 3).
PREGNANCY
Hydroxychloroquine is in pregnancy category C. Information is limited, and in view of the risks, the manufacturer says that it should be avoided in pregnancy.1 Nevertheless, it is generally considered safe during pregnancy, and its benefits may make it acceptable to continue in a patient who becomes pregnant, in spite of the possible risks.
We favor continuing hydroxychloroquine. This drug has been associated with improved maternal and fetal outcomes in lupus patients. Its use during pregnancy has not been associated with congenital malformations. The adverse visual effects of long-term hydroxychloroquine use, namely retinopathy, have never been reported in children as a consequence of exposure in utero.
In addition, hydroxychloroquine is transmitted only in minute quantities in breast milk.39 In pregnant women with systemic lupus erythematosus, hydroxychloroquine was associated with a lower risk of adverse outcomes, including preterm delivery and intrauterine growth restriction.40 However, hydroxychloroquine is far more toxic when ingested directly by infants than in adults.1
Maternal outcomes are also improved with the use of hydroxychloroquine. Stopping hydroxychloroquine during pregnancy in women with systemic lupus erythematosus is associated with significantly higher disease activity—fully twice as high as in those who continue hydroxychloroquine.41 These study results were corroborated in a small randomized trial in which pregnant women with lupus on placebo had significantly higher lupus disease activity scores than those pregnant women who were given hydroxychloroquine.42 The women taking hydroxychloroquine experienced no severe lupus flares for the duration of their pregnancies.
These findings suggest not only that hydroxychloroquine is safe in pregnancy, but also that it should be continued in lupus patients during pregnancy to prevent disease flares and adverse fetal outcomes.
AREAS OF UNCERTAINTY
Benefit in preclinical lupus?
Hydroxychloroquine has a consistently profound effect on outcomes in systemic lupus erythematosus. These findings, in addition to the more widespread use of antibody screening, have led to suggestions that hydroxychloroquine could be of benefit even before systemic lupus erythematosus is diagnosed.
A study in US military personnel found that patients taking hydroxychloroquine experienced a significantly longer lag time between first reported clinical symptoms of lupus and official diagnosis compared with matched controls who also went on to develop the disease, averaging 1.08 vs 0.29 years to disease classification.43 Those who used hydroxychloroquine also had lower rates of autoantibody accumulation. Therefore, hydroxychloroquine could be of benefit in carefully selected candidates at high risk of developing systemic lupus erythematosus.
The beneficial effects of hydroxychloroquine on patients with lupus and rheumatoid arthritis, in terms of primary measures of disease activity and secondary outcomes, were discovered fortuitously and were not the original intended targets of the drug. Because of its versatility, there are numerous other disease states in which hydroxychloroquine has shown a degree of benefit or has shown a potential for benefit.
Antiviral activity?
It has been suggested that antimalarial drugs could serve as adjunctive therapies against filoviruses such as Marburg and Ebola. There is a small body of in vitro and in vivo evidence that hydroxychloroquine could temper severe systemic inflammatory responses to filoviruses both through dysregulation of lysosomes and lysosomal pH (filoviruses have a pH-dependent mechanism of action) and through decreased production of tumor necrosis factor alpha and interferons. Heavy burdens of interferons and tumor necrosis factor alpha are associated with increased mortality rates in those infected with filoviruses.44
Antineoplastic activity?
Hydroxychloroquine has undergone in vitro testing as an adjunct to cancer therapies. There are several mechanisms by which it is theorized that hydroxychloroquine could target malignant cells, including inhibition of multidrug resistance pumps or autophagy, improvement of chemotherapy cell penetration, potentiation of presentation of major histocompatibility complexes, or even intercalation directly into DNA.45,46 However, it can also impair natural anticancer immunity and may allow cancer cells better nutrient supply through vascular effects.
In vitro studies have shown tumoricidal effects in lymphoma and melanoma, and inhibition of growth in lung, colon, breast, cervix, larynx, liver, and prostate cancers. In vivo studies have shown that hydroxychloroquine in high doses can prolong survival in glioblastoma.45
Unfortunately, all of these theorized or observed effects are dose-dependent and likely require doses that exceed currently recommended maximums.
Negative chronotropic effect?
Hydroxychloroquine has been found to decrease the resting heart rate in a cumulative dose-dependent fashion.47 Further, hydroxychloroquine has been known to increase digoxin levels, and the medications should not be used in combination.1
Whether the decrease in resting heart rate is associated with harm or benefit and whether the effect is significant enough to be considered when implementing therapy remain unanswered and deserve further investigation, as does the primary use of hydroxychloroquine for beneficial lipid and glucose reduction in patients who are otherwise healthy.
CASE CONCLUSION
The patient described at the beginning of this article was provided with information on the risks and benefits of hydroxychloroquine for treatment of her arthritis and rash suggestive of mild systemic lupus, and she opted to begin therapy. Her baseline eye screening was within normal limits. Based on her weight of 62 kg, she was started on 300 mg of hydroxychloroquine daily.
She had no significant adverse effects from the medication and reported slow improvement in her rash and joint complaints over the next 2 months. She remained on hydroxychloroquine over the next year without adverse effects or new evidence of autoimmune disease.
- Sanofi-Aventis. Product monograph: Plaquenil. http://products.sanofi.ca/en/plaquenil.pdf. Accessed May 2, 2018.
- Centers for Disease Control and Prevention (CDC). Malaria information and prophylaxis, by country. www.cdc.gov/malaria/travelers/country_table/a.html. Accessed May 2, 2018.
- Wallace DJ. The history of antimalarials. Lupus 1996; 5(suppl 1):S2–S3. pmid:8803902
- Shee JC. Lupus erythematosus treated with chloroquine. Lancet 1953; 265(6778):201–202. pmid:13070595
- Kuznik A, Bencina M, Svajger U, Jeras M, Rozman B, Jerala R. Mechanism of endosomal TLR inhibition by antimalarial drugs and imidazoquinolines. J Immunol 2011; 186:4794–4804. doi:10.4049/jimmunol.1000702
- Willis R, Seif AM, McGwin G Jr, et al. Effect of hydroxychloroquine treatment on pro-inflammatory cytokines and disease activity in SLE patients: data from LUMINA, a multiethnic US cohort. Lupus 2012; 21(8):830–835. doi:10.1177/0961203312437270
- Fox R. Anti-malarial drugs: possible mechanisms of action in autoimmune disease and prospects for drug development. Lupus 1996; 5(suppl 1):S4–S10. pmid:8803903
- Ruiz-Irastorza G, Ramos-Casals M, Brito-Zeron P, Khamashta MA. Clinical efficacy and side effects of antimalarials in systemic lupus erythematosus: a systematic review. Ann Rheum Dis 2010; 69(1):20–28. doi:10.1136/ard.2008.101766
- Lam NC, Ghetu MV, Bieniek ML. Systemic lupus erythematosus: primary care approach to diagnosis and management. Am Fam Physician 2016; 94(4):284–294. pmid:27548593
- Jung H, Bobba R, Su J, et al. The protective effect of antimalarial drugs on thrombovascular events in systemic lupus erythematosus. Arthritis Rheum 2010; 62(3):863–868. doi:10.1002/art.27289
- Nuri E, Taraborelli M, Andreoli L, et al. Long-term use of hydroxychloroquine reduces antiphospholipid antibodies levels in patients with primary antiphospholipid syndrome. Immunol Res 2017; 65(1):17–24. doi:10.1007/s12026-016-8812-z
- Erkan D, Aguiar CL, Andrade D, et al. 14th International Congress on Antiphospholipid Antibodies: task force report on antiphospholipid syndrome treatment trends. Autoimmun Rev 2014; 13(6):685–696. doi:10.1016/j.autrev.2014.01.053
- Jalal H, O’Dell JR, Bridges SL Jr, et al. Cost-effectiveness of triple therapy versus etanercept plus methotrexate in early aggressive rheumatoid arthritis. Arthritis Care Res (Hoboken) 2016; 68(12):1751–1757. doi:10.1002/acr.22895
- Avina-Zubieta JA, Galindo-Rodriguez G, Newman S, Suarez-Almazor ME, Russell AS. Long-term effectiveness of antimalarial drugs in rheumatic diseases. Ann Rheum Dis 1998; 57(10):582–587. pmid:9893568
- Gottenberg JE, Ravaud P, Puechal X, et al. Effects of hydroxychloroquine on symptomatic improvement in primary Sjogren syndrome. JAMA 2014; 312(3):249–258. doi:10.1001/jama.2014.7682
- Petri M. Hydroxychloroquine use in the Baltimore Lupus Cohort: effects on lipids, glucose and thrombosis. Lupus 1996; 5(suppl 1):S16–S22. pmid:8803905
- Cairoli E, Rebella M, Danese N, Garra V, Borba EF. Hydroxychloroquine reduces low-density lipoprotein cholesterol levels in systemic lupus erythematosus: a longitudinal evaluation of the lipid-lowering effect. Lupus 2012; 21(11):1178–1182. doi:10.1177/0961203312450084
- Charles-Schoeman C, Wang X, Lee YY, et al. Association of triple therapy with improvement in cholesterol profiles over two-year followup in the treatment of early aggressive rheumatoid arthritis trial. Arthritis Rheumatol 2016; 68(3):577–586. doi:10.1002/art.39502
- Penn SK, Kao AH, Schott LL, et al. Hydroxychloroquine and glycemia in women with rheumatoid arthritis and systemic lupus erythematosus. J Rheumatol 2010; 37(6):1136–1142. doi:10.3899/jrheum.090994
- Wasko MC, Hubert HB, Lingala VB, et al. Hydroxychloroquine and risk of diabetes in patients with rheumatoid arthritis. JAMA 2007; 298(2):187–193. doi:10.1001/jama.298.2.187
- Sun L, Liu M, Li R, et al. Hydroxychloroquine, a promising choice for coronary artery disease? Med Hypotheses 2016; 93:5–7. doi:10.1016/j.mehy.2016.04.045
- Tett SE, Cutler DJ, Day RO, Brown KF. Bioavailability of hydroxychloroquine tablets in healthy volunteers. Br J Clin Pharmacol 1989; 27(6):771–779. pmid:2757893
- Furst DE. Pharmacokinetics of hydroxychloroquine and chloroquine during treatment of rheumatic diseases. Lupus 1996; 5(suppl 1):S11–S15. pmid:8803904
- Frances C, Cosnes A, Duhaut P, et al. Low blood concentration of hydroxychloroquine in patients with refractory cutaneous lupus erythematosus. Arch Dermatol 2012; 148(4):479–484. doi:10.1001/archdermatol.2011.2558
- Chasset F, Arnaud L, Costedoat-Chalumeau N, Zahr N, Bessis D, Francès C. The effect of increasing the dose of hydroxychloroquine in patients with refractory cutaneous lupus erythematosus: an open-label prospective pilot study. J Am Acad Dermatol 2016; 74(4):693–699.e3. doi:10.1016/j.jaad.2015.09.064
- Melles RB, Marmor MF. The risk of toxic retinopathy in patients on long-term hydroxychloroquine therapy. JAMA Ophthalmol 2014; 132(12):1453–1460. doi:10.1001/jamaophthalmol.2014.3459
- Committee on Rheumatologic Care. American College of Rheumatology position statement. Screening for hydroxychloroquine retinopathy. www.rheumatology.org/Portals/0/Files/Screening-for-Hydroxychloroquine-Retinopathy-Position-Statement.pdf. Accessed April 2, 2018.
- Marmor MF, Kellner U, Lai TY, Melles RB, Mieler WF; American Academy of Ophthalmology. Recommendations on screening for chloroquine and hydroxychloroquine retinopathy (2016 revision). Ophthalmology 2016; 123(6):1386–1394. doi:10.1016/j.ophtha.2016.01.058
- Mackenzie AH. Antimalarial drugs for rheumatoid arthritis. Am J Med 1983; 75(6A):48–58. pmid:6362406
- Mackenzie AH. Dose refinements in long-term therapy of rheumatoid arthritis with antimalarials. Am J Med 1983; 75(1A):40–45. pmid:6869410
- Melles RB, Marmor MF. Pericentral retinopathy and racial differences in hydroxychloroquine toxicity. Ophthalmology 2015; 122(1):110–116. doi:10.1016/j.ophtha.2014.07.018
- Uslu H, Gurler B, Yildirim A, et al. Effect of hydroxychloroquine on the retinal layers: a quantitative evaluation with spectral-domain optical coherence tomography. J Ophthalmol 2016; 2016:8643174. doi:10.1155/2016/8643174
- Au A, Parikh V, Modi YS, Ehlers JP, Schachat AP, Singh RP. Hydroxychloroquine screening practice patterns within a large multispecialty ophthalmic practice. Am J Ophthalmol 2015; 160(3):561–568.e2. doi:10.1016/j.ajo.2015.06.009
- Jallouli M, Frances C, Plette JC, et al; Plaquenil Lupus Systemic Study Group. Hydroxychloroquine-induced pigmentation in patients with systemic lupus erythematosus. JAMA Dermatol 2013; 149(8):935–940. doi:10.1001/jamadermatol.2013.709
- Avina-Zubieta JA, Johnson ES, Suarez-Almazor ME, Russell AS. Incidence of myopathy in patients treated with antimalarials: a report of three cases and review of the literature. Br J Rheumatol 1995; 34(2):166–170. pmid:7704464
- Yogasundaram H, Putko BN, Tien J, et al. Hydroxychloroquine-induced cardiomyopathy: case report, pathophysiology, diagnosis, and treatment. Can J Cardiol 2014; 30:1706–1715. doi:10.1016/j.cjca.2014.08.016
- Pearson KC, Morrell DS, Runge SR, Jolly P. Prolonged pustular eruption from hydroxychloroquine: an unusual case of acute generalized exanthematous pustulosis. Cutis 2016; 97(3):212–216. pmid:27023083
- Youngster I, Arcavi L, Schechmaster R, et al. Medications and glucose-6-phosphate dehydrogenase deficiency: an evidence-based review. Drug Saf 2010; 33(9):713–726. doi:10.2165/11536520-000000000-00000
- Ostensen M, Khamashta M, Lockshin M, et al. Anti-inflammatory and immunosuppressive drugs and reproduction. Arthritis Res Ther 2006; 8(3):209. doi:10.1186/ar1957
- Leroux M, Desveaux C, Parcevaux M, et al. Impact of hydroxychloroquine on preterm delivery and intrauterine growth restriction in pregnant women with systemic lupus erythematosus: a descriptive cohort study. Lupus 2015; 24(13):1384–1391. doi:10.1177/0961203315591027
- Clowse MEB, Magder L, Witter F, Petri M. Hydroxychloroquine in lupus pregnancy. Arthritis Rheum 2006; 54(11):3640–3647. doi:10.1002/art.22159
- Levy RA, Vilela VS, Cataldo MJ, et al. Hydroxychloroquine in lupus pregnancy: double-blind and placebo-controlled study. Lupus 2001; 10(6):401–404. doi:10.1191/096120301678646137
- James JA, Kim-Howard XR, Bruner BF, et al. Hydroxychloroquine sulfate treatment is associated with later onset of systemic lupus erythematosus. Lupus 2007; 16(6):401–409. doi:10.1177/0961203307078579
- Akpovwa H. Chloroquine could be used for the treatment of filoviral infections and other viral infections that emerge or emerged from viruses requiring an acidic pH for infectivity. Cell Biochem Funct 2016; 34(4):191–196. doi:10.1002/cbf.3182
- Pascolo S. Time to use a dose of chloroquine as an adjuvant to anti-cancer chemotherapies. Eur J Pharmacol 2016; 771:139–144. doi:10.1016/j.ejphar.2015.12.017
- Furlong HC, Wessels JM, Guerra MT, Stämpfli MR, Foster WG. Hydroxychloroquine attenuates cigarette smoke induced autophagic signaling in the mouse ovary. Reprod Toxicol 2016; 61:105–113. doi:10.1016/j.reprotox.2016.03.044
- Cairoli E, Danese N, Teliz M, et al. Cumulative dose of hydroxychloroquine is associated with a decrease of resting heart rate in patients with systemic lupus erythematosus: a pilot study. Lupus 2015; 24(11):1204–1209. doi:10.1177/0961203315580870
- Sanofi-Aventis. Product monograph: Plaquenil. http://products.sanofi.ca/en/plaquenil.pdf. Accessed May 2, 2018.
- Centers for Disease Control and Prevention (CDC). Malaria information and prophylaxis, by country. www.cdc.gov/malaria/travelers/country_table/a.html. Accessed May 2, 2018.
- Wallace DJ. The history of antimalarials. Lupus 1996; 5(suppl 1):S2–S3. pmid:8803902
- Shee JC. Lupus erythematosus treated with chloroquine. Lancet 1953; 265(6778):201–202. pmid:13070595
- Kuznik A, Bencina M, Svajger U, Jeras M, Rozman B, Jerala R. Mechanism of endosomal TLR inhibition by antimalarial drugs and imidazoquinolines. J Immunol 2011; 186:4794–4804. doi:10.4049/jimmunol.1000702
- Willis R, Seif AM, McGwin G Jr, et al. Effect of hydroxychloroquine treatment on pro-inflammatory cytokines and disease activity in SLE patients: data from LUMINA, a multiethnic US cohort. Lupus 2012; 21(8):830–835. doi:10.1177/0961203312437270
- Fox R. Anti-malarial drugs: possible mechanisms of action in autoimmune disease and prospects for drug development. Lupus 1996; 5(suppl 1):S4–S10. pmid:8803903
- Ruiz-Irastorza G, Ramos-Casals M, Brito-Zeron P, Khamashta MA. Clinical efficacy and side effects of antimalarials in systemic lupus erythematosus: a systematic review. Ann Rheum Dis 2010; 69(1):20–28. doi:10.1136/ard.2008.101766
- Lam NC, Ghetu MV, Bieniek ML. Systemic lupus erythematosus: primary care approach to diagnosis and management. Am Fam Physician 2016; 94(4):284–294. pmid:27548593
- Jung H, Bobba R, Su J, et al. The protective effect of antimalarial drugs on thrombovascular events in systemic lupus erythematosus. Arthritis Rheum 2010; 62(3):863–868. doi:10.1002/art.27289
- Nuri E, Taraborelli M, Andreoli L, et al. Long-term use of hydroxychloroquine reduces antiphospholipid antibodies levels in patients with primary antiphospholipid syndrome. Immunol Res 2017; 65(1):17–24. doi:10.1007/s12026-016-8812-z
- Erkan D, Aguiar CL, Andrade D, et al. 14th International Congress on Antiphospholipid Antibodies: task force report on antiphospholipid syndrome treatment trends. Autoimmun Rev 2014; 13(6):685–696. doi:10.1016/j.autrev.2014.01.053
- Jalal H, O’Dell JR, Bridges SL Jr, et al. Cost-effectiveness of triple therapy versus etanercept plus methotrexate in early aggressive rheumatoid arthritis. Arthritis Care Res (Hoboken) 2016; 68(12):1751–1757. doi:10.1002/acr.22895
- Avina-Zubieta JA, Galindo-Rodriguez G, Newman S, Suarez-Almazor ME, Russell AS. Long-term effectiveness of antimalarial drugs in rheumatic diseases. Ann Rheum Dis 1998; 57(10):582–587. pmid:9893568
- Gottenberg JE, Ravaud P, Puechal X, et al. Effects of hydroxychloroquine on symptomatic improvement in primary Sjogren syndrome. JAMA 2014; 312(3):249–258. doi:10.1001/jama.2014.7682
- Petri M. Hydroxychloroquine use in the Baltimore Lupus Cohort: effects on lipids, glucose and thrombosis. Lupus 1996; 5(suppl 1):S16–S22. pmid:8803905
- Cairoli E, Rebella M, Danese N, Garra V, Borba EF. Hydroxychloroquine reduces low-density lipoprotein cholesterol levels in systemic lupus erythematosus: a longitudinal evaluation of the lipid-lowering effect. Lupus 2012; 21(11):1178–1182. doi:10.1177/0961203312450084
- Charles-Schoeman C, Wang X, Lee YY, et al. Association of triple therapy with improvement in cholesterol profiles over two-year followup in the treatment of early aggressive rheumatoid arthritis trial. Arthritis Rheumatol 2016; 68(3):577–586. doi:10.1002/art.39502
- Penn SK, Kao AH, Schott LL, et al. Hydroxychloroquine and glycemia in women with rheumatoid arthritis and systemic lupus erythematosus. J Rheumatol 2010; 37(6):1136–1142. doi:10.3899/jrheum.090994
- Wasko MC, Hubert HB, Lingala VB, et al. Hydroxychloroquine and risk of diabetes in patients with rheumatoid arthritis. JAMA 2007; 298(2):187–193. doi:10.1001/jama.298.2.187
- Sun L, Liu M, Li R, et al. Hydroxychloroquine, a promising choice for coronary artery disease? Med Hypotheses 2016; 93:5–7. doi:10.1016/j.mehy.2016.04.045
- Tett SE, Cutler DJ, Day RO, Brown KF. Bioavailability of hydroxychloroquine tablets in healthy volunteers. Br J Clin Pharmacol 1989; 27(6):771–779. pmid:2757893
- Furst DE. Pharmacokinetics of hydroxychloroquine and chloroquine during treatment of rheumatic diseases. Lupus 1996; 5(suppl 1):S11–S15. pmid:8803904
- Frances C, Cosnes A, Duhaut P, et al. Low blood concentration of hydroxychloroquine in patients with refractory cutaneous lupus erythematosus. Arch Dermatol 2012; 148(4):479–484. doi:10.1001/archdermatol.2011.2558
- Chasset F, Arnaud L, Costedoat-Chalumeau N, Zahr N, Bessis D, Francès C. The effect of increasing the dose of hydroxychloroquine in patients with refractory cutaneous lupus erythematosus: an open-label prospective pilot study. J Am Acad Dermatol 2016; 74(4):693–699.e3. doi:10.1016/j.jaad.2015.09.064
- Melles RB, Marmor MF. The risk of toxic retinopathy in patients on long-term hydroxychloroquine therapy. JAMA Ophthalmol 2014; 132(12):1453–1460. doi:10.1001/jamaophthalmol.2014.3459
- Committee on Rheumatologic Care. American College of Rheumatology position statement. Screening for hydroxychloroquine retinopathy. www.rheumatology.org/Portals/0/Files/Screening-for-Hydroxychloroquine-Retinopathy-Position-Statement.pdf. Accessed April 2, 2018.
- Marmor MF, Kellner U, Lai TY, Melles RB, Mieler WF; American Academy of Ophthalmology. Recommendations on screening for chloroquine and hydroxychloroquine retinopathy (2016 revision). Ophthalmology 2016; 123(6):1386–1394. doi:10.1016/j.ophtha.2016.01.058
- Mackenzie AH. Antimalarial drugs for rheumatoid arthritis. Am J Med 1983; 75(6A):48–58. pmid:6362406
- Mackenzie AH. Dose refinements in long-term therapy of rheumatoid arthritis with antimalarials. Am J Med 1983; 75(1A):40–45. pmid:6869410
- Melles RB, Marmor MF. Pericentral retinopathy and racial differences in hydroxychloroquine toxicity. Ophthalmology 2015; 122(1):110–116. doi:10.1016/j.ophtha.2014.07.018
- Uslu H, Gurler B, Yildirim A, et al. Effect of hydroxychloroquine on the retinal layers: a quantitative evaluation with spectral-domain optical coherence tomography. J Ophthalmol 2016; 2016:8643174. doi:10.1155/2016/8643174
- Au A, Parikh V, Modi YS, Ehlers JP, Schachat AP, Singh RP. Hydroxychloroquine screening practice patterns within a large multispecialty ophthalmic practice. Am J Ophthalmol 2015; 160(3):561–568.e2. doi:10.1016/j.ajo.2015.06.009
- Jallouli M, Frances C, Plette JC, et al; Plaquenil Lupus Systemic Study Group. Hydroxychloroquine-induced pigmentation in patients with systemic lupus erythematosus. JAMA Dermatol 2013; 149(8):935–940. doi:10.1001/jamadermatol.2013.709
- Avina-Zubieta JA, Johnson ES, Suarez-Almazor ME, Russell AS. Incidence of myopathy in patients treated with antimalarials: a report of three cases and review of the literature. Br J Rheumatol 1995; 34(2):166–170. pmid:7704464
- Yogasundaram H, Putko BN, Tien J, et al. Hydroxychloroquine-induced cardiomyopathy: case report, pathophysiology, diagnosis, and treatment. Can J Cardiol 2014; 30:1706–1715. doi:10.1016/j.cjca.2014.08.016
- Pearson KC, Morrell DS, Runge SR, Jolly P. Prolonged pustular eruption from hydroxychloroquine: an unusual case of acute generalized exanthematous pustulosis. Cutis 2016; 97(3):212–216. pmid:27023083
- Youngster I, Arcavi L, Schechmaster R, et al. Medications and glucose-6-phosphate dehydrogenase deficiency: an evidence-based review. Drug Saf 2010; 33(9):713–726. doi:10.2165/11536520-000000000-00000
- Ostensen M, Khamashta M, Lockshin M, et al. Anti-inflammatory and immunosuppressive drugs and reproduction. Arthritis Res Ther 2006; 8(3):209. doi:10.1186/ar1957
- Leroux M, Desveaux C, Parcevaux M, et al. Impact of hydroxychloroquine on preterm delivery and intrauterine growth restriction in pregnant women with systemic lupus erythematosus: a descriptive cohort study. Lupus 2015; 24(13):1384–1391. doi:10.1177/0961203315591027
- Clowse MEB, Magder L, Witter F, Petri M. Hydroxychloroquine in lupus pregnancy. Arthritis Rheum 2006; 54(11):3640–3647. doi:10.1002/art.22159
- Levy RA, Vilela VS, Cataldo MJ, et al. Hydroxychloroquine in lupus pregnancy: double-blind and placebo-controlled study. Lupus 2001; 10(6):401–404. doi:10.1191/096120301678646137
- James JA, Kim-Howard XR, Bruner BF, et al. Hydroxychloroquine sulfate treatment is associated with later onset of systemic lupus erythematosus. Lupus 2007; 16(6):401–409. doi:10.1177/0961203307078579
- Akpovwa H. Chloroquine could be used for the treatment of filoviral infections and other viral infections that emerge or emerged from viruses requiring an acidic pH for infectivity. Cell Biochem Funct 2016; 34(4):191–196. doi:10.1002/cbf.3182
- Pascolo S. Time to use a dose of chloroquine as an adjuvant to anti-cancer chemotherapies. Eur J Pharmacol 2016; 771:139–144. doi:10.1016/j.ejphar.2015.12.017
- Furlong HC, Wessels JM, Guerra MT, Stämpfli MR, Foster WG. Hydroxychloroquine attenuates cigarette smoke induced autophagic signaling in the mouse ovary. Reprod Toxicol 2016; 61:105–113. doi:10.1016/j.reprotox.2016.03.044
- Cairoli E, Danese N, Teliz M, et al. Cumulative dose of hydroxychloroquine is associated with a decrease of resting heart rate in patients with systemic lupus erythematosus: a pilot study. Lupus 2015; 24(11):1204–1209. doi:10.1177/0961203315580870
KEY POINTS
- Hydroxychloroquine acts by suppressing Toll-like receptors to trigger important immunomodulatory effects.
- Hydroxychloroquine is a well-established and effective therapy for systemic and cutaneous lupus and other autoimmune diseases.
- Patients with systemic lupus erythematosus treated with hydroxychloroquine have lower mortality rates and a lower risk of lupus nephritis.
- Retinal toxicity is the most serious potential complication of hydroxychloroquine therapy. Adherence to current ophthalmologic screening recommendations and proper dosing protocols lowers this risk.

The Evidence for Herbal and Botanical Remedies
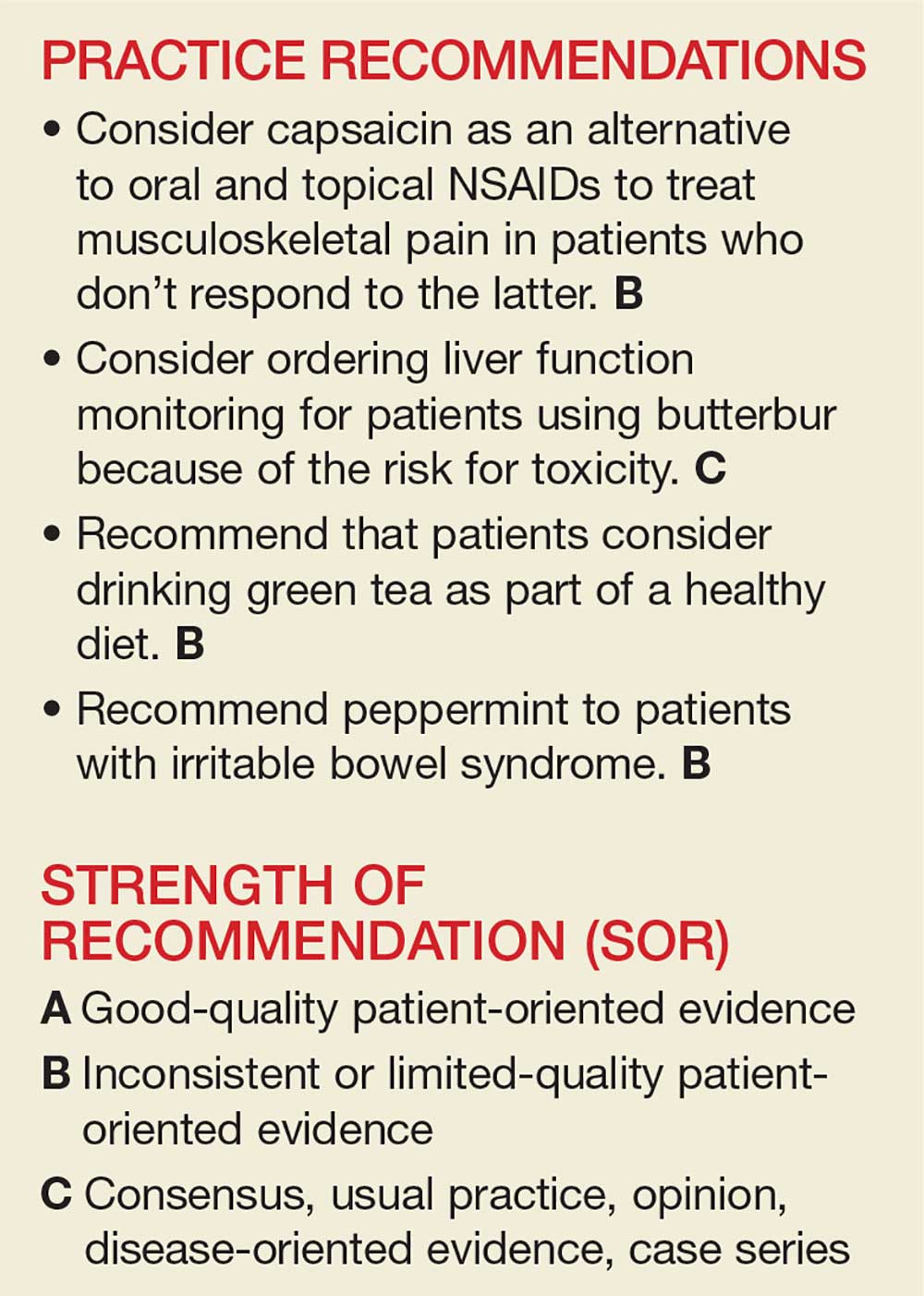
The National Center for Complementary and Integrative Health, a division of the National Institutes of Medicine, estimates that 38% of American adults use complementary and alternative medicine (including 17.7% who say they use “natural products”).1 Despite the popularity of these products, many providers remain skeptical—and for good reason. Enthusiasts may offer dramatic anecdotes to “prove” their supplements’ worth, but little scientific support is available for most herbal remedies. There are, however, exceptions—capsaicin, butterbur, green tea, and peppermint—as this review of the medical literature reveals.
Worth noting as you consider this—or any—review of herbals is that while there is limited scientific evidence to establish the safety and efficacy of most herbal products, they are nonetheless freely sold without FDA approval because, under current regulations, they are considered dietary supplements. That legal designation means companies can manufacture, sell, and market herbs without first demonstrating safety and efficacy, as is required for pharmaceutical drugs. Because herbal medications do not require the same testing through the large randomized controlled trials (RCTs) required for pharmaceuticals, evidence is often based on smaller RCTs and other studies of lower overall quality. Despite these limitations, we believe it’s worth keeping an open mind about the value of evidence-based herbal and botanical treatments.
CAPSAICIN
Capsaicin, an active compound in chili peppers, provokes a burning sensation but also has a long history of use in pain treatment.2 Qutenza, an FDA-approved, chemically synthesized 8% capsaicin patch, is identical to the naturally occurring molecule.2 Topical capsaicin exerts its therapeutic effect by rapidly depleting substance P, thus reducing the transmission of pain from C fibers to higher neurologic centers in the area of administration.3
Meta-analyses and systematic reviews have shown capsaicin is effective for various painful conditions, including peripheral diabetic neuropathy, osteoarthritis (OA), low back pain (LBP), and postherpetic neuralgia (PHN).
Peripheral neuropathy. A Cochrane review of six randomized, double-blind, placebo-controlled studies of at least six weeks’ duration using topical 8% capsaicin to treat PHN and HIV-associated neuropathy concluded that high-concentration topical capsaicin provided more relief in patients with high pain levels than control patients who received a subtherapeutic (0.04%) capsaicin cream. Number-needed-to-treat values were between 8 and 12. Local adverse events were common, but not consistently reported enough to calculate a number needed to harm.4
OA. In randomized trials, capsaicin provided mild-to-moderate efficacy for patients with hand and knee OA, when compared with placebo.5-7 A systematic review of capsaicin for all osteoarthritic conditions noted that there was consistent evidence that capsaicin gel was effective for OA.8 However, a 2013 Cochrane review of only knee OA noted that capsicum extract did not provide significant clinical improvement for pain or function and resulted in a significant number of adverse events.9
LBP. Based on a 2014 Cochrane review of three trials (755 subjects) of moderate quality, capsicum frutescens cream or plaster appeared more efficacious than placebo in people with chronic LBP.10 Based on current (low-quality) evidence in one trial, however, it’s not clear whether topical capsicum cream is more beneficial for acute LBP than placebo.10
PHN. Topical capsaicin is an FDA-approved treatment for PHN. A review and cost-effectiveness analysis demonstrated that 8% capsaicin had significantly higher effectiveness rates than the oral agents (tricyclic antidepressants, duloxetine, gabapentin, pregabalin) used to treat PHN.11 The cost of the capsaicin patch was similar to a topical lidocaine patch and oral products for PHN.11 A meta-analysis of seven RCTs indicated that 8% topical capsaicin was superior to the low-dose capsaicin patch for relieving pain associated with PHN.12
Continue to: Adverse effects
Adverse effects
Very few toxic effects have been reported during a half-century of capsaicin use. Those that have been reported are mainly limited to mild local reactions.2 The most common adverse effect of topical capsaicin is local irritation (burning, stinging, and erythema), which was reported in approximately 40% of patients.6 Nevertheless, more than 90% of the subjects in clinical studies were able to complete the studies, and pain rapidly resolved after patch removal.2 Washing with soap and water may help prevent the compound from spreading to other parts of the body unintentionally.
The safety of the patch has been demonstrated with repeated dosing every three months for up to one year. However, the long-term risks of chronic capsaicin use and its effect on epidermal innervation are uncertain.5
The bottom line
Capsaicin appears to be an effective treatment for neuropathy and chronic LBP. It is FDA approved for the treatment of PHN. It may also benefit patients with OA and acute LBP. Serious adverse effects are uncommon with topical use. Common adverse effects include burning pain and irritation in the area of application, which can be intense and cause discontinuation.2
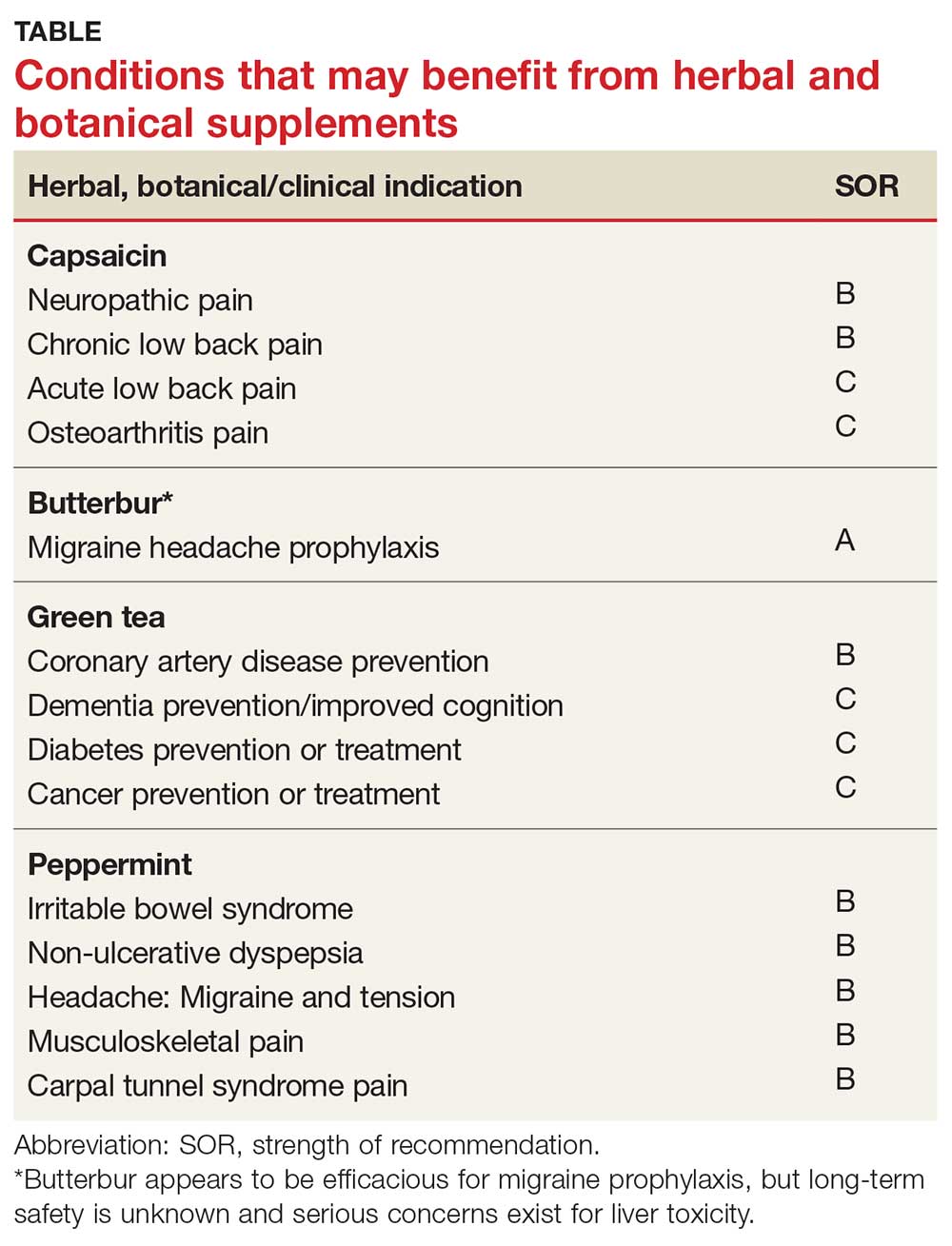
Continue to: BUTTERBUR
BUTTERBUR
Petasites hybridus, also known as butterbur, is a member of the daisy family, Asteraceae, and is a perennial plant found throughout Europe and Asia.13 It was used as a remedy for ulcers, wounds, and inflammation in ancient Greece. Its calcium channel–blocking effects may counteract vasoconstriction and play a role in preventing hyperexcitation of neurons.14 Sesquiterpenes, the pharmacologically active compounds in butterbur, have strong anti-inflammatory and vasodilatory effects through lipoxygenase and leukotriene inhibition.14
Migraine headache. Butterbur appears to be effective in migraine prophylaxis. Several studies have shown butterbur to significantly reduce the number of migraine attacks per month when compared with placebo. In a small, randomized, placebo-controlled, parallel-group study on the efficacy and tolerability of a special butterbur root extract (Petadolex) for the prevention of migraine, response rate was 45% in the butterbur group vs 15% in the placebo group. Butterbur was well tolerated.15 Similar results were found in another RCT in which butterbur 75 mg bid significantly reduced migraine frequency by 48%, compared with 26% for the placebo group.16 Butterbur was well tolerated in this study, too, and no serious adverse events occurred. Findings suggest that 75 mg bid may be a good option for migraine prevention, given the agent’s safety profile.
Petadolex may also be a good option in pediatric migraine. A 2005 study in children and adolescents found that 77% of patients experienced a reduction in attacks by at least 50% with butterbur. Patients were treated with 50 mg to 150 mg over four months.17
In their 2012 guidelines for migraine prevention, the American Academy of Neurology (AAN) and American Headache Society gave butterbur a Level A recommendation, concluding that butterbur should be offered to patients with migraine to reduce the frequency and severity of migraine attacks.18 However, the AAN changed its position in 2015, redacting the recommendation due to serious safety concerns.19
Allergic rhinitis. Although the data are not convincing, some studies have shown that butterbur may be beneficial for the treatment of allergic rhinitis.20,21
Continue to: Adverse effects
Adverse effects
While the butterbur plant itself contains pyrrolizidine alkaloids (PA), which are hepatotoxic and carcinogenic, extracts of butterbur root that are almost completely free from these alkaloids are available. Patients who choose to use butterbur should be advised to use only products that are certified and labeled PA free.
Petadolex, the medication used in migraine studies, was initially approved by the German health regulatory authority, but approval was later withdrawn due to concerns about liver toxicity.22 In 2012, the United Kingdom’s Medicines and Health Care Products Regulatory Agency withdrew all butterbur products from the market due to associated cases of liver toxicity.22 Butterbur products are still available in the US market, and the risks and benefits should be discussed with all patients considering this treatment. Liver function monitoring is recommended for all patients using butterbur.22
The herb can also cause dyspepsia, headache, itchy eyes, gastrointestinal symptoms, asthma, fatigue, and drowsiness. Additionally, people who are allergic to ragweed and daisies may have allergic reactions to butterbur. Eructation (belching) occurred in 7% of patients in a pediatric study.17
The bottom line
Butterbur appears to be efficacious for migraine prophylaxis, but long-term safety is unknown and serious concerns exist for liver toxicity.
Continue to: GREEN TEA
GREEN TEA
Most tea leaves come from the Camellia sinensis bush, but green and black tea are processed differently to produce different end products.23 It is estimated that green tea accounts for approximately a quarter of all tea consumption and is most commonly consumed in Asian countries.23 The health-promoting effects of green tea are mainly attributed to its polyphenol content.24 Of the many types of tea, green tea has the highest concentration of polyphenols, including catechins, which are powerful antioxidants.23,24 Green tea has been used in traditional Chinese and Indian medicine to control bleeding, improve digestion, and promote overall health.23
Dementia. Green tea polyphenols may enhance cognition and may protect against the development of dementia. In-vitro studies have shown that green tea reduces hydrogen peroxide and ß-amyloid peptides, which are significant in the development of Alzheimer’s disease.25 A 12-subject double-blind study found green tea increased working memory and had an impact on frontoparietal brain connections.26 Furthermore, a cohort study with 13,645 Japanese participants over a five-year period found that frequent green tea consumption (> 5 cups per day) was associated with a lower risk for dementia.27 Additional studies are needed, but green tea may be useful in the treatment or prevention of dementia in the future.
Coronary artery disease. In one study, green tea plasma and urinary concentrations were associated with plasma biomarkers of cardiovascular disease and diabetes.28 In one review, the consumption of green tea was associated with a statistically significant reduction in LDL cholesterol.29 Furthermore, a 2015 systematic review and meta-analysis of prospective observational studies concluded that increased tea consumption (of any type) is associated with a reduced risk for coronary heart disease, cardiac death, stroke, and total mortality.30
Cancer. Many studies have shown that green tea may reduce the risk for cancer, although epidemiologic evidence is inconsistent. Studies have shown that cancer rates tend to be lower in those who consume higher levels of green tea.31,32 Whether this can be attributed solely to green tea remains debatable. Several other studies have shown that polyphenols in green tea can inhibit the growth of cancer cells, but the exact mechanism by which tea interacts with cancerous cells is unknown.23
Several population-based studies have been performed, mostly in Japan, which showed green tea consumption reduced the risk for cancer. Fewer prostate cancer cases have been reported in men who consume green tea.33 While studies have been performed to determine whether green tea has effects on pancreatic, esophageal, ovarian, breast, bladder, and colorectal cancer, the evidence remains inadequate.32
Diabetes. Green tea has been shown in several studies to have a beneficial effect on diabetes. A retrospective Japanese cohort study showed that those who consumed green tea were one-third less likely to develop type 2 diabetes.34 A 10-year study from Taiwan found lower body fat and smaller waist circumference in those who consumed green tea regularly.35 A 2014 meta-analysis and systematic review of tea (any type) consumption and the risk for diabetes concluded that three or more cups of tea per day was associated with a lower risk for diabetes.36 Another meta-analysis of 17 RCTs focused on green tea concluded that green tea improves glucose control and A1C values.37
Continue to: Adverse effects
Adverse effects
There have been concerns about potential hepatotoxicity induced by green tea intake.38 However, a systematic review of 34 RCTs on liver-related adverse events from green tea showed only a slight elevation in liver function tests; no serious liver-related adverse events were reported.38 This review suggested that liver-related adverse events after intake of green tea extracts are rare.38
Consuming green tea in the diet may lower the risk for adverse effects since the concentration consumed is generally much lower than that found in extracts.
Contraindications to drinking green tea are few. Individuals with caffeine sensitivities could experience insomnia, anxiety, irritability, or upset stomach. Additionally, patients who are taking anticoagulation drugs, such as warfarin, should avoid green tea due to its vitamin K content, which can counter the effects of warfarin. Pregnant or breastfeeding women, those with heart problems or high blood pressure, kidney or liver problems, stomach ulcers, or anxiety disorders should use caution with green tea consumption.
The bottom line
Green tea consumption in the diet appears to be safe and may have beneficial effects on weight, dementia, and risk for diabetes, cancer, and cardiovascular disease. Patients may want to consider drinking green tea as part of a healthy diet, in combination with exercise.
Continue to: PEPPERMINT
PEPPERMINT
Mentha piperita, also known as peppermint, is a hybrid between water mint and spearmint. It is found throughout Europe and North America and is commonly used in tea and toothpaste and as a flavoring for gum. Menthol and methyl salicylate are the main active ingredients in peppermint, and peppermint has calcium channel–blocker effects.39 Menthol has been shown to help regulate cold and pain sensation through the TRPM8 receptor.40 The peppermint herb is used both orally and topically, and has been studied in the treatment of multiple conditions.
Irritable bowel syndrome (IBS). It appears that peppermint inhibits spontaneous peristaltic activity, which reduces gastric emptying, decreases basal tone in the gastrointestinal tract, and slows down peristalsis in the gut.39
The American College of Gastroenterology guidelines currently note that there is moderate-quality evidence for peppermint oil in the treatment of IBS.41 A Cochrane review concluded that peppermint appears to be beneficial for IBS-related symptoms and pain.42 In a systematic review of nine studies from 2014, peppermint oil was found to be more effective than placebo for IBS symptoms such as pain, bloating, gas, and diarrhea.43 The review also indicated that peppermint oil is safe, with heartburn being the most common complaint.43 A 2016 study also found that triple-coated microspheres containing peppermint oil reduced the frequency and intensity of IBS symptoms.44
Non-ulcer dyspepsia. In combination with caraway oil, peppermint oil can be used to reduce symptoms of non-ulcer dyspepsia.45,46 A multicenter, randomized, placebo-controlled, double-blind study found that 43.3% of subjects improved with a peppermint-caraway oil combination after eight weeks, compared with 3.5% receiving placebo.46
Barium enema–related colonic spasm. Peppermint can relax the lower esophageal sphincter, and it has been shown to be useful as an antispasmodic agent for barium enema–related colonic spasm.47,48
Itching/skin irritation. Peppermint, when applied topically, has been used to calm pruritus and relieve irritation and inflammation. It has a soothing and cooling effect on the skin. At least one study found it to be effective in the treatment of pruritus gravidarum, although the study population consisted of only 96 subjects.49
Migraine headache. Initial small trials suggest that menthol is likely beneficial for migraine headaches. A pilot trial of 25 patients treated with topical menthol 6% gel for an acute migraine attack showed a significant improvement in headache intensity two hours after gel application.50 In a randomized, triple-blind, placebo-controlled, crossover study of 35 patients, a menthol 10% solution was shown to be more efficacious as abortive treatment of migraine headaches than placebo.51
Tension headache. In a randomized, placebo-controlled, double-blind crossover study, topical peppermint oil showed a significant clinical reduction in tension headache pain.52 Another small, randomized, double-blind trial showed that tiger balm (containing menthol as the main ingredient) also produced statistically significant improvement in tension headache discomfort compared with placebo.53
Continue to: Musculoskeletal pain
Musculoskeletal pain. A small study comparing topical menthol to ice for muscle soreness noted decreased perceived discomfort with menthol.54 Menthol has also been shown to reduce pain in patients with knee OA.55
Carpal tunnel syndrome (CTS). A triple-blind RCT concluded that topical menthol acutely reduced pain intensity in slaughterhouse workers with CTS, and it should be considered as an effective nonsystemic alternative to regular analgesics in the workplace management of chronic and neuropathic pain.56
Adverse effects
Peppermint appears to be safe for most adults when used in small doses, and serious adverse effects are rare.43,57 While peppermint tea appears to be safe in moderate-to-large amounts, people allergic to plants in the peppermint family (eg, mint, thyme, sage, rosemary, marjoram, basil, lavender) may experience allergic reactions with swelling, wheals, or erythema. Peppermint may also cause heartburn due to relaxation of the cardiac sphincter.
Other symptoms may include nausea, vomiting, flushing, and headache.58 The herb may also be both hepatotoxic and nephrotoxic at extremely high doses.59 Other considerations for women are that it can trigger menstruation and should be avoided during pregnancy. Due to uncertain efficacy in this population, peppermint oil should not be used on the face of infants, young children, or pregnant women.58,59
The bottom line
Peppermint appears to be safe and well tolerated. It is useful in alleviating IBS symptoms and may be effective in the treatment of non-ulcerative dyspepsia, musculoskeletal pain, headache, and CTS.54,55
1. National Center for Complementary and Integrative Health. The Use of Complementary and Alternative Medicine in the United States. https://nccih.nih.gov/research/statistics/2007/camsurvey_fs1.htm. Accessed April 19, 2018.
2. Wallace M, Pappagallo M. Qutenza: a capsaicin 8% patch for the management of postherpetic neuralgia. Expert Rev Neurother. 2011;11:15-27.
3. Rains C, Bryson HM. Topical capsaicin. A review of its pharmacological properties and therapeutic potential in post-herpetic neuralgia, diabetic neuropathy and osteoarthritis. Drugs Aging. 1995;7:317-328.
4. Derry S, Sven-Rice A, Cole P, et al. Topical capsaicin (high concentration) for chronic neuropathic pain in adults. Cochrane Database Syst Rev. 2013;(2):CD007393.
5. Mason L, Moore RA, Derry S, et al. Systematic review of topical capsaicin for the treatment of chronic pain. BMJ. 2004;328:991.
6. Deal CL, Schnitzer TJ, Lipstein E, et al. Treatment of arthritis with topical capsaicin: a double-blind trial. Clin Ther. 1991; 13:383.
7. McCarthy GM, McCarty DJ. Effect of topical capsaicin in the therapy of painful osteoarthritis of the hands. J Rheumatol. 1992;19:604.
8. De Silva V, El-Metwally A, Ernst E, et al; Arthritis Research UK Working Group on Complementary and Alternative Medicines. Evidence for the efficacy of complementary and alternative medicines in the management of osteoarthritis: a systematic review. Rheumatology (Oxford). 2011;50:911-920.
9. Cameron M, Chrubasik S. Topical herbal therapies for treating osteoarthritis. Cochrane Database Syst Rev. 2013;(5): CD010538.
10. Oltean H, Robbins C, van Tulder MW, et al. Herbal medicine for low-back pain. Cochrane Database Syst Rev. 2014;(12): CD004504.
11. Armstrong EP, Malone DC, McCarberg B, et al. Cost-effectiveness analysis of a new 8% capsaicin patch compared to existing therapies for postherpetic neuralgia. Curr Med Res Opin. 2011;27:939-950.
12. Mou J, Paillard F, Turnbull B, et al. Efficacy of Qutenza (capsaicin) 8% patch for neuropathic pain: a meta-analysis of the Qutenza Clinical Trials Database. Pain. 2013;154:1632-1639.
13. Sun-Edelstein C, Mauskop A. Alternative headache treatments: nutraceuticals, behavioral and physical treatments. Headache. 2011;51:469-483.
14. D’Andrea G, Cevoli S, Cologno D. Herbal therapy in migraine. Neurol Sci. 2014;35(suppl 1):135-140.
15. Diener HC, Rahlfs VW, Danesch U. The first placebo-controlled trial of a special butterbur root extract for the prevention of migraine: reanalysis of efficacy criteria. Eur Neurol. 2004;51:89-97.
16. Lipton RB, Göbel H, Einhäupl KM, et al. Petasites hybridus root (butterbur) is an effective preventive treatment for migraine. Neurology. 2004;63:2240-2244.
17. Pothmann R, Danesch U. Migraine prevention in children and adolescents: results of an open study with a special butterbur root extract. Headache. 2005;45:196-203.
18. Holland S, Silberstein SD, Freitag F, et al; Quality Standards Subcommittee of the American Academy of Neurology and the American Headache Society. Evidence-based guideline update: NSAIDs and other complementary treatments for episodic migraine prevention in adults: report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Headache Society. Neurology. 2012;78:1346-1353.
19. American Academy of Neurology. Evidence-based guideline update: NSAIDs and other complementary treatments for episodic migraine prevention in adults: [RETIRED]. http://n.neurology.org/content/78/17/1346. Accessed April 29, 2018.
20. Man LX. Complementary and alternative medicine for allergic rhinitis. Curr Opin Otolaryngol Head Neck Surg. 2009;17:226-231.
21. Guo R, Pittler MH, Ernst E. Herbal medicines for the treatment of allergic rhinitis: a systematic review. Ann Allergy Asthma Immunol. 2007;99:483-495.
22. Daniel O, Mauskop A. Nutraceuticals in acute and prophylactic treatment of migraine. Curr Treat Options Neurol. 2016; 18:14.
23. Chacko SM, Thambi PT, Kuttan R, et al. Beneficial effects of green tea: a literature review. Chin Med. 2010;6:13.
24. Naghma K, Hasan M. Tea polyphenols for health promotion. Life Sci. 2007;81:519-533.
25. Okello EJ, McDougall GJ, Kumar S, et al. In vitro protective effects of colon-available extract of Camellia sinensis (tea) against hydrogen peroxide and beta-amyloid (Aβ((1-42))) induced cytotoxicity in differentiated PC12 cells. Phytomedicine. 2011;18:691-696.
26. Schmidt A, Hammann F, Wölnerhanssen B, et al. Green tea extract enhances parieto-frontal connectivity during working memory processing. Psychopharmacology (Berl). 2014;231: 3879-3888.
27. Tomata Y, Sugiyama K, Kaiho Y, et al. Green tea consumption and the risk of incident dementia in elderly Japanese: The Ohsaki Cohort 2006 Study. Am J Geriatr Psychiatry. 2016;24: 881-889.
28. Takechi R, Alfonso H, Hiramatsu N, et al. Elevated plasma and urinary concentrations of green tea catechins associated with improved plasma lipid profile in healthy Japanese women. Nutr Res. 2016;36:220-226.
29. Kim A, Chiu A, Barone MK, et al. Green tea catechins decrease total and low-density lipoprotein cholesterol: a systematic review and meta-analysis. J Am Diet Assoc. 2011; 111:1720-1729.
30. Zhang C, Qin YY, Wei X, et al. Tea consumption and risk of cardiovascular outcomes and total mortality: a systematic review and meta-analysis of prospective observational studies. Eur J Epidemiol. 2015;30:103-113.
31. Imai K, Suga K, Nakachi K. Cancer-preventive effects of drinking green tea among a Japanese population. Prev Med. 1997;26:769-775.
32. Yuan JM. Cancer prevention by green tea: evidence from epidemiologic studies. Am J Clin Nutr. 2013;98(6 suppl): 1676S-1681S.
33. Kurahashi N, Sasazuki S, Iwasaki M, et al. Green tea consumption and prostate cancer risk in Japanese men: a prospective study. Am J Epidemiol. 2008;167:71-77.
34. Iso H, Date C, Wakai K, et al. The relationship between green tea and total caffeine intake and risk for self-reported type 2 diabetes among Japanese adults. Ann Intern Med. 2006; 144:554-562.
35. Kim HM, Kim J. The effects of green tea on obesity and type 2 diabetes. Diab Metab J. 2013;37:173-175.
36. Yang J, Mao Q, Xu H, et al. Tea consumption and risk of type 2 diabetes mellitus: a systematic review and meta-analysis update. BMJ Open. 2014;4:e005632.
37. Liu K, Zhou R, Wang B, et al. Effect of green tea on glucose control and insulin sensitivity: a meta-analysis of 17 randomized controlled trials. Am J Clin Nutr. 2013;98:340-348.
38. Isomura T, Suzuki S, Origasa H, et al. Liver-related safety assessment of green tea extracts in humans: a systematic review of randomized controlled trials. Eur J Clin Nutr. 2016;70:1340.
39. Tillisch K. Complementary and alternative medicine for gastrointestinal disorders. Clin Med (Lond). 2007;7:224-227.
40. Knowlton WM, McKemy DD. TRPM8: from cold to cancer, peppermint to pain. Curr Pharm Biotechnol. 2011;12:68-77.
41. Ford AC, Moayyedi P, Lacy BE, et al. Task Force on the Management of Functional Bowel Disorders. American College of Gastroenterology monograph on the management of irritable bowel syndrome and chronic idiopathic constipation. Am J Gastroenterol. 2014;109(suppl 1):S2-S26.
42. Ruepert L, Quartero AO, de Wit NJ, et al. Bulking agents, antispasmodics and antidepressants for the treatment of irritable bowel syndrome. Cochrane Database Syst Rev. 2011;(8):CD003460.
43. Khanna R, MacDonald JK, Levesque BG. Peppermint oil for the treatment of irritable bowel syndrome: a systematic review and meta-analysis. J Clin Gastroenterol. 2014;48:505-512.
44. Cash BD, Epstein MS, Shah SM. A novel delivery system of peppermint oil is an effective therapy for irritable bowel syndrome symptoms. Dig Dis Sci. 2016;61:560-571.
45. Holtmann G, Haag S, Adam B, et al. Effects of a fixed combination of peppermint oil and caraway oil on symptoms and quality of life in patients suffering from functional dyspepsia. Phytomedicine. 2003;10(suppl 4):56-57.
46. Madisch A, Heydenreich CJ, Wieland V, et al. Treatment of functional dyspepsia with a fixed peppermint oil and caraway oil combination preparation as compared to cisapride. A multicenter, reference-controlled double-blind equivalence study. Arzneimittelforschung. 1999;49:925-932.
47. Asao T, Kuwano H, Ide M, et al. Spasmolytic effect of peppermint oil in barium during double-contrast barium enema compared with Buscopan. Clin Radiol. 2003;58:301-305.
48. Sparks MJ, O’Sullivan P, Herrington AA, et al. Does peppermint oil relieve spasm during barium enema? Br J Radiol. 1995;68:841-843.
49. Akhavan Amjadi M, Mojab F, Kamranpour SB. The effect of peppermint oil on symptomatic treatment of pruritus in pregnant women. Iran J Pharm Res. 2012;11:1073-1077.
50. St Cyr A, Chen A, Bradley KC, et al. Efficacy and tolerability of STOPAIN for a migraine attack. Front Neurol. 2015;6:11.
51. Borhani Haghighi A, Motazedian S, Rezaii R, et al. Cutaneous application of menthol 10% solution as an abortive treatment of migraine without aura: a randomised, double-blind, placebo-controlled, crossed-over study. Int J Clin Pract. 2010; 64:451-456.
52. Gobel H, Fresenius J, Heinze A, et al. Effectiveness of oleum menthae piperitae and paracetamol in therapy of headache of the tension type [German]. Nervenarzt. 1996;67:672-681.
53. Schattner P, Randerson D. Tiger Balm as a treatment of tension headache. A clinical trial in general practice. Aust Fam Physician. 1996;25:216-220.
54. Johar P, Grover V, Topp R, et al. A comparison of topical menthol to ice on pain, evoked tetanic and voluntary force during delayed onset muscle soreness. Int J Sports Phys Ther. 2012;7:314-322.
55. Topp R, Brosky JA Jr, Pieschel D. The effect of either topical menthol or a placebo on functioning and knee pain among patients with knee OA. J Geriatr Phys Ther. 2013;36:92-99.
56. Sundstrup E, Jakobsen MD, Brandt M, et al. Acute effect of topical menthol on chronic pain in slaughterhouse workers with carpal tunnel syndrome: triple-blind, randomized placebo-controlled trial. Rehabil Res Pract. 2014;2014:310913.
57. Nair B. Final report on the safety assessment of mentha piperita (peppermint) oil, mentha piperita (peppermint) leaf extract, mentha piperita (peppermint) leaf, and mentha piperita (peppermint) leaf water. Int J Toxicol. 2001;20(suppl 3):61-73.
58. Klingler B, Chadhary S. Peppermint oil. Am Fam Physician. 2007;75:1027-1030.
59. Nath SS, Pandey C, Roy D. A near fatal case of high dose peppermint oil ingestion—lessons learnt. Indian J Anaesth. 2012;56:582-584.
The National Center for Complementary and Integrative Health, a division of the National Institutes of Medicine, estimates that 38% of American adults use complementary and alternative medicine (including 17.7% who say they use “natural products”).1 Despite the popularity of these products, many providers remain skeptical—and for good reason. Enthusiasts may offer dramatic anecdotes to “prove” their supplements’ worth, but little scientific support is available for most herbal remedies. There are, however, exceptions—capsaicin, butterbur, green tea, and peppermint—as this review of the medical literature reveals.
Worth noting as you consider this—or any—review of herbals is that while there is limited scientific evidence to establish the safety and efficacy of most herbal products, they are nonetheless freely sold without FDA approval because, under current regulations, they are considered dietary supplements. That legal designation means companies can manufacture, sell, and market herbs without first demonstrating safety and efficacy, as is required for pharmaceutical drugs. Because herbal medications do not require the same testing through the large randomized controlled trials (RCTs) required for pharmaceuticals, evidence is often based on smaller RCTs and other studies of lower overall quality. Despite these limitations, we believe it’s worth keeping an open mind about the value of evidence-based herbal and botanical treatments.
CAPSAICIN
Capsaicin, an active compound in chili peppers, provokes a burning sensation but also has a long history of use in pain treatment.2 Qutenza, an FDA-approved, chemically synthesized 8% capsaicin patch, is identical to the naturally occurring molecule.2 Topical capsaicin exerts its therapeutic effect by rapidly depleting substance P, thus reducing the transmission of pain from C fibers to higher neurologic centers in the area of administration.3
Meta-analyses and systematic reviews have shown capsaicin is effective for various painful conditions, including peripheral diabetic neuropathy, osteoarthritis (OA), low back pain (LBP), and postherpetic neuralgia (PHN).
Peripheral neuropathy. A Cochrane review of six randomized, double-blind, placebo-controlled studies of at least six weeks’ duration using topical 8% capsaicin to treat PHN and HIV-associated neuropathy concluded that high-concentration topical capsaicin provided more relief in patients with high pain levels than control patients who received a subtherapeutic (0.04%) capsaicin cream. Number-needed-to-treat values were between 8 and 12. Local adverse events were common, but not consistently reported enough to calculate a number needed to harm.4
OA. In randomized trials, capsaicin provided mild-to-moderate efficacy for patients with hand and knee OA, when compared with placebo.5-7 A systematic review of capsaicin for all osteoarthritic conditions noted that there was consistent evidence that capsaicin gel was effective for OA.8 However, a 2013 Cochrane review of only knee OA noted that capsicum extract did not provide significant clinical improvement for pain or function and resulted in a significant number of adverse events.9
LBP. Based on a 2014 Cochrane review of three trials (755 subjects) of moderate quality, capsicum frutescens cream or plaster appeared more efficacious than placebo in people with chronic LBP.10 Based on current (low-quality) evidence in one trial, however, it’s not clear whether topical capsicum cream is more beneficial for acute LBP than placebo.10
PHN. Topical capsaicin is an FDA-approved treatment for PHN. A review and cost-effectiveness analysis demonstrated that 8% capsaicin had significantly higher effectiveness rates than the oral agents (tricyclic antidepressants, duloxetine, gabapentin, pregabalin) used to treat PHN.11 The cost of the capsaicin patch was similar to a topical lidocaine patch and oral products for PHN.11 A meta-analysis of seven RCTs indicated that 8% topical capsaicin was superior to the low-dose capsaicin patch for relieving pain associated with PHN.12
Continue to: Adverse effects
Adverse effects
Very few toxic effects have been reported during a half-century of capsaicin use. Those that have been reported are mainly limited to mild local reactions.2 The most common adverse effect of topical capsaicin is local irritation (burning, stinging, and erythema), which was reported in approximately 40% of patients.6 Nevertheless, more than 90% of the subjects in clinical studies were able to complete the studies, and pain rapidly resolved after patch removal.2 Washing with soap and water may help prevent the compound from spreading to other parts of the body unintentionally.
The safety of the patch has been demonstrated with repeated dosing every three months for up to one year. However, the long-term risks of chronic capsaicin use and its effect on epidermal innervation are uncertain.5
The bottom line
Capsaicin appears to be an effective treatment for neuropathy and chronic LBP. It is FDA approved for the treatment of PHN. It may also benefit patients with OA and acute LBP. Serious adverse effects are uncommon with topical use. Common adverse effects include burning pain and irritation in the area of application, which can be intense and cause discontinuation.2
Continue to: BUTTERBUR
BUTTERBUR
Petasites hybridus, also known as butterbur, is a member of the daisy family, Asteraceae, and is a perennial plant found throughout Europe and Asia.13 It was used as a remedy for ulcers, wounds, and inflammation in ancient Greece. Its calcium channel–blocking effects may counteract vasoconstriction and play a role in preventing hyperexcitation of neurons.14 Sesquiterpenes, the pharmacologically active compounds in butterbur, have strong anti-inflammatory and vasodilatory effects through lipoxygenase and leukotriene inhibition.14
Migraine headache. Butterbur appears to be effective in migraine prophylaxis. Several studies have shown butterbur to significantly reduce the number of migraine attacks per month when compared with placebo. In a small, randomized, placebo-controlled, parallel-group study on the efficacy and tolerability of a special butterbur root extract (Petadolex) for the prevention of migraine, response rate was 45% in the butterbur group vs 15% in the placebo group. Butterbur was well tolerated.15 Similar results were found in another RCT in which butterbur 75 mg bid significantly reduced migraine frequency by 48%, compared with 26% for the placebo group.16 Butterbur was well tolerated in this study, too, and no serious adverse events occurred. Findings suggest that 75 mg bid may be a good option for migraine prevention, given the agent’s safety profile.
Petadolex may also be a good option in pediatric migraine. A 2005 study in children and adolescents found that 77% of patients experienced a reduction in attacks by at least 50% with butterbur. Patients were treated with 50 mg to 150 mg over four months.17
In their 2012 guidelines for migraine prevention, the American Academy of Neurology (AAN) and American Headache Society gave butterbur a Level A recommendation, concluding that butterbur should be offered to patients with migraine to reduce the frequency and severity of migraine attacks.18 However, the AAN changed its position in 2015, redacting the recommendation due to serious safety concerns.19
Allergic rhinitis. Although the data are not convincing, some studies have shown that butterbur may be beneficial for the treatment of allergic rhinitis.20,21
Continue to: Adverse effects
Adverse effects
While the butterbur plant itself contains pyrrolizidine alkaloids (PA), which are hepatotoxic and carcinogenic, extracts of butterbur root that are almost completely free from these alkaloids are available. Patients who choose to use butterbur should be advised to use only products that are certified and labeled PA free.
Petadolex, the medication used in migraine studies, was initially approved by the German health regulatory authority, but approval was later withdrawn due to concerns about liver toxicity.22 In 2012, the United Kingdom’s Medicines and Health Care Products Regulatory Agency withdrew all butterbur products from the market due to associated cases of liver toxicity.22 Butterbur products are still available in the US market, and the risks and benefits should be discussed with all patients considering this treatment. Liver function monitoring is recommended for all patients using butterbur.22
The herb can also cause dyspepsia, headache, itchy eyes, gastrointestinal symptoms, asthma, fatigue, and drowsiness. Additionally, people who are allergic to ragweed and daisies may have allergic reactions to butterbur. Eructation (belching) occurred in 7% of patients in a pediatric study.17
The bottom line
Butterbur appears to be efficacious for migraine prophylaxis, but long-term safety is unknown and serious concerns exist for liver toxicity.
Continue to: GREEN TEA
GREEN TEA
Most tea leaves come from the Camellia sinensis bush, but green and black tea are processed differently to produce different end products.23 It is estimated that green tea accounts for approximately a quarter of all tea consumption and is most commonly consumed in Asian countries.23 The health-promoting effects of green tea are mainly attributed to its polyphenol content.24 Of the many types of tea, green tea has the highest concentration of polyphenols, including catechins, which are powerful antioxidants.23,24 Green tea has been used in traditional Chinese and Indian medicine to control bleeding, improve digestion, and promote overall health.23
Dementia. Green tea polyphenols may enhance cognition and may protect against the development of dementia. In-vitro studies have shown that green tea reduces hydrogen peroxide and ß-amyloid peptides, which are significant in the development of Alzheimer’s disease.25 A 12-subject double-blind study found green tea increased working memory and had an impact on frontoparietal brain connections.26 Furthermore, a cohort study with 13,645 Japanese participants over a five-year period found that frequent green tea consumption (> 5 cups per day) was associated with a lower risk for dementia.27 Additional studies are needed, but green tea may be useful in the treatment or prevention of dementia in the future.
Coronary artery disease. In one study, green tea plasma and urinary concentrations were associated with plasma biomarkers of cardiovascular disease and diabetes.28 In one review, the consumption of green tea was associated with a statistically significant reduction in LDL cholesterol.29 Furthermore, a 2015 systematic review and meta-analysis of prospective observational studies concluded that increased tea consumption (of any type) is associated with a reduced risk for coronary heart disease, cardiac death, stroke, and total mortality.30
Cancer. Many studies have shown that green tea may reduce the risk for cancer, although epidemiologic evidence is inconsistent. Studies have shown that cancer rates tend to be lower in those who consume higher levels of green tea.31,32 Whether this can be attributed solely to green tea remains debatable. Several other studies have shown that polyphenols in green tea can inhibit the growth of cancer cells, but the exact mechanism by which tea interacts with cancerous cells is unknown.23
Several population-based studies have been performed, mostly in Japan, which showed green tea consumption reduced the risk for cancer. Fewer prostate cancer cases have been reported in men who consume green tea.33 While studies have been performed to determine whether green tea has effects on pancreatic, esophageal, ovarian, breast, bladder, and colorectal cancer, the evidence remains inadequate.32
Diabetes. Green tea has been shown in several studies to have a beneficial effect on diabetes. A retrospective Japanese cohort study showed that those who consumed green tea were one-third less likely to develop type 2 diabetes.34 A 10-year study from Taiwan found lower body fat and smaller waist circumference in those who consumed green tea regularly.35 A 2014 meta-analysis and systematic review of tea (any type) consumption and the risk for diabetes concluded that three or more cups of tea per day was associated with a lower risk for diabetes.36 Another meta-analysis of 17 RCTs focused on green tea concluded that green tea improves glucose control and A1C values.37
Continue to: Adverse effects
Adverse effects
There have been concerns about potential hepatotoxicity induced by green tea intake.38 However, a systematic review of 34 RCTs on liver-related adverse events from green tea showed only a slight elevation in liver function tests; no serious liver-related adverse events were reported.38 This review suggested that liver-related adverse events after intake of green tea extracts are rare.38
Consuming green tea in the diet may lower the risk for adverse effects since the concentration consumed is generally much lower than that found in extracts.
Contraindications to drinking green tea are few. Individuals with caffeine sensitivities could experience insomnia, anxiety, irritability, or upset stomach. Additionally, patients who are taking anticoagulation drugs, such as warfarin, should avoid green tea due to its vitamin K content, which can counter the effects of warfarin. Pregnant or breastfeeding women, those with heart problems or high blood pressure, kidney or liver problems, stomach ulcers, or anxiety disorders should use caution with green tea consumption.
The bottom line
Green tea consumption in the diet appears to be safe and may have beneficial effects on weight, dementia, and risk for diabetes, cancer, and cardiovascular disease. Patients may want to consider drinking green tea as part of a healthy diet, in combination with exercise.
Continue to: PEPPERMINT
PEPPERMINT
Mentha piperita, also known as peppermint, is a hybrid between water mint and spearmint. It is found throughout Europe and North America and is commonly used in tea and toothpaste and as a flavoring for gum. Menthol and methyl salicylate are the main active ingredients in peppermint, and peppermint has calcium channel–blocker effects.39 Menthol has been shown to help regulate cold and pain sensation through the TRPM8 receptor.40 The peppermint herb is used both orally and topically, and has been studied in the treatment of multiple conditions.
Irritable bowel syndrome (IBS). It appears that peppermint inhibits spontaneous peristaltic activity, which reduces gastric emptying, decreases basal tone in the gastrointestinal tract, and slows down peristalsis in the gut.39
The American College of Gastroenterology guidelines currently note that there is moderate-quality evidence for peppermint oil in the treatment of IBS.41 A Cochrane review concluded that peppermint appears to be beneficial for IBS-related symptoms and pain.42 In a systematic review of nine studies from 2014, peppermint oil was found to be more effective than placebo for IBS symptoms such as pain, bloating, gas, and diarrhea.43 The review also indicated that peppermint oil is safe, with heartburn being the most common complaint.43 A 2016 study also found that triple-coated microspheres containing peppermint oil reduced the frequency and intensity of IBS symptoms.44
Non-ulcer dyspepsia. In combination with caraway oil, peppermint oil can be used to reduce symptoms of non-ulcer dyspepsia.45,46 A multicenter, randomized, placebo-controlled, double-blind study found that 43.3% of subjects improved with a peppermint-caraway oil combination after eight weeks, compared with 3.5% receiving placebo.46
Barium enema–related colonic spasm. Peppermint can relax the lower esophageal sphincter, and it has been shown to be useful as an antispasmodic agent for barium enema–related colonic spasm.47,48
Itching/skin irritation. Peppermint, when applied topically, has been used to calm pruritus and relieve irritation and inflammation. It has a soothing and cooling effect on the skin. At least one study found it to be effective in the treatment of pruritus gravidarum, although the study population consisted of only 96 subjects.49
Migraine headache. Initial small trials suggest that menthol is likely beneficial for migraine headaches. A pilot trial of 25 patients treated with topical menthol 6% gel for an acute migraine attack showed a significant improvement in headache intensity two hours after gel application.50 In a randomized, triple-blind, placebo-controlled, crossover study of 35 patients, a menthol 10% solution was shown to be more efficacious as abortive treatment of migraine headaches than placebo.51
Tension headache. In a randomized, placebo-controlled, double-blind crossover study, topical peppermint oil showed a significant clinical reduction in tension headache pain.52 Another small, randomized, double-blind trial showed that tiger balm (containing menthol as the main ingredient) also produced statistically significant improvement in tension headache discomfort compared with placebo.53
Continue to: Musculoskeletal pain
Musculoskeletal pain. A small study comparing topical menthol to ice for muscle soreness noted decreased perceived discomfort with menthol.54 Menthol has also been shown to reduce pain in patients with knee OA.55
Carpal tunnel syndrome (CTS). A triple-blind RCT concluded that topical menthol acutely reduced pain intensity in slaughterhouse workers with CTS, and it should be considered as an effective nonsystemic alternative to regular analgesics in the workplace management of chronic and neuropathic pain.56
Adverse effects
Peppermint appears to be safe for most adults when used in small doses, and serious adverse effects are rare.43,57 While peppermint tea appears to be safe in moderate-to-large amounts, people allergic to plants in the peppermint family (eg, mint, thyme, sage, rosemary, marjoram, basil, lavender) may experience allergic reactions with swelling, wheals, or erythema. Peppermint may also cause heartburn due to relaxation of the cardiac sphincter.
Other symptoms may include nausea, vomiting, flushing, and headache.58 The herb may also be both hepatotoxic and nephrotoxic at extremely high doses.59 Other considerations for women are that it can trigger menstruation and should be avoided during pregnancy. Due to uncertain efficacy in this population, peppermint oil should not be used on the face of infants, young children, or pregnant women.58,59
The bottom line
Peppermint appears to be safe and well tolerated. It is useful in alleviating IBS symptoms and may be effective in the treatment of non-ulcerative dyspepsia, musculoskeletal pain, headache, and CTS.54,55
The National Center for Complementary and Integrative Health, a division of the National Institutes of Medicine, estimates that 38% of American adults use complementary and alternative medicine (including 17.7% who say they use “natural products”).1 Despite the popularity of these products, many providers remain skeptical—and for good reason. Enthusiasts may offer dramatic anecdotes to “prove” their supplements’ worth, but little scientific support is available for most herbal remedies. There are, however, exceptions—capsaicin, butterbur, green tea, and peppermint—as this review of the medical literature reveals.
Worth noting as you consider this—or any—review of herbals is that while there is limited scientific evidence to establish the safety and efficacy of most herbal products, they are nonetheless freely sold without FDA approval because, under current regulations, they are considered dietary supplements. That legal designation means companies can manufacture, sell, and market herbs without first demonstrating safety and efficacy, as is required for pharmaceutical drugs. Because herbal medications do not require the same testing through the large randomized controlled trials (RCTs) required for pharmaceuticals, evidence is often based on smaller RCTs and other studies of lower overall quality. Despite these limitations, we believe it’s worth keeping an open mind about the value of evidence-based herbal and botanical treatments.
CAPSAICIN
Capsaicin, an active compound in chili peppers, provokes a burning sensation but also has a long history of use in pain treatment.2 Qutenza, an FDA-approved, chemically synthesized 8% capsaicin patch, is identical to the naturally occurring molecule.2 Topical capsaicin exerts its therapeutic effect by rapidly depleting substance P, thus reducing the transmission of pain from C fibers to higher neurologic centers in the area of administration.3
Meta-analyses and systematic reviews have shown capsaicin is effective for various painful conditions, including peripheral diabetic neuropathy, osteoarthritis (OA), low back pain (LBP), and postherpetic neuralgia (PHN).
Peripheral neuropathy. A Cochrane review of six randomized, double-blind, placebo-controlled studies of at least six weeks’ duration using topical 8% capsaicin to treat PHN and HIV-associated neuropathy concluded that high-concentration topical capsaicin provided more relief in patients with high pain levels than control patients who received a subtherapeutic (0.04%) capsaicin cream. Number-needed-to-treat values were between 8 and 12. Local adverse events were common, but not consistently reported enough to calculate a number needed to harm.4
OA. In randomized trials, capsaicin provided mild-to-moderate efficacy for patients with hand and knee OA, when compared with placebo.5-7 A systematic review of capsaicin for all osteoarthritic conditions noted that there was consistent evidence that capsaicin gel was effective for OA.8 However, a 2013 Cochrane review of only knee OA noted that capsicum extract did not provide significant clinical improvement for pain or function and resulted in a significant number of adverse events.9
LBP. Based on a 2014 Cochrane review of three trials (755 subjects) of moderate quality, capsicum frutescens cream or plaster appeared more efficacious than placebo in people with chronic LBP.10 Based on current (low-quality) evidence in one trial, however, it’s not clear whether topical capsicum cream is more beneficial for acute LBP than placebo.10
PHN. Topical capsaicin is an FDA-approved treatment for PHN. A review and cost-effectiveness analysis demonstrated that 8% capsaicin had significantly higher effectiveness rates than the oral agents (tricyclic antidepressants, duloxetine, gabapentin, pregabalin) used to treat PHN.11 The cost of the capsaicin patch was similar to a topical lidocaine patch and oral products for PHN.11 A meta-analysis of seven RCTs indicated that 8% topical capsaicin was superior to the low-dose capsaicin patch for relieving pain associated with PHN.12
Continue to: Adverse effects
Adverse effects
Very few toxic effects have been reported during a half-century of capsaicin use. Those that have been reported are mainly limited to mild local reactions.2 The most common adverse effect of topical capsaicin is local irritation (burning, stinging, and erythema), which was reported in approximately 40% of patients.6 Nevertheless, more than 90% of the subjects in clinical studies were able to complete the studies, and pain rapidly resolved after patch removal.2 Washing with soap and water may help prevent the compound from spreading to other parts of the body unintentionally.
The safety of the patch has been demonstrated with repeated dosing every three months for up to one year. However, the long-term risks of chronic capsaicin use and its effect on epidermal innervation are uncertain.5
The bottom line
Capsaicin appears to be an effective treatment for neuropathy and chronic LBP. It is FDA approved for the treatment of PHN. It may also benefit patients with OA and acute LBP. Serious adverse effects are uncommon with topical use. Common adverse effects include burning pain and irritation in the area of application, which can be intense and cause discontinuation.2
Continue to: BUTTERBUR
BUTTERBUR
Petasites hybridus, also known as butterbur, is a member of the daisy family, Asteraceae, and is a perennial plant found throughout Europe and Asia.13 It was used as a remedy for ulcers, wounds, and inflammation in ancient Greece. Its calcium channel–blocking effects may counteract vasoconstriction and play a role in preventing hyperexcitation of neurons.14 Sesquiterpenes, the pharmacologically active compounds in butterbur, have strong anti-inflammatory and vasodilatory effects through lipoxygenase and leukotriene inhibition.14
Migraine headache. Butterbur appears to be effective in migraine prophylaxis. Several studies have shown butterbur to significantly reduce the number of migraine attacks per month when compared with placebo. In a small, randomized, placebo-controlled, parallel-group study on the efficacy and tolerability of a special butterbur root extract (Petadolex) for the prevention of migraine, response rate was 45% in the butterbur group vs 15% in the placebo group. Butterbur was well tolerated.15 Similar results were found in another RCT in which butterbur 75 mg bid significantly reduced migraine frequency by 48%, compared with 26% for the placebo group.16 Butterbur was well tolerated in this study, too, and no serious adverse events occurred. Findings suggest that 75 mg bid may be a good option for migraine prevention, given the agent’s safety profile.
Petadolex may also be a good option in pediatric migraine. A 2005 study in children and adolescents found that 77% of patients experienced a reduction in attacks by at least 50% with butterbur. Patients were treated with 50 mg to 150 mg over four months.17
In their 2012 guidelines for migraine prevention, the American Academy of Neurology (AAN) and American Headache Society gave butterbur a Level A recommendation, concluding that butterbur should be offered to patients with migraine to reduce the frequency and severity of migraine attacks.18 However, the AAN changed its position in 2015, redacting the recommendation due to serious safety concerns.19
Allergic rhinitis. Although the data are not convincing, some studies have shown that butterbur may be beneficial for the treatment of allergic rhinitis.20,21
Continue to: Adverse effects
Adverse effects
While the butterbur plant itself contains pyrrolizidine alkaloids (PA), which are hepatotoxic and carcinogenic, extracts of butterbur root that are almost completely free from these alkaloids are available. Patients who choose to use butterbur should be advised to use only products that are certified and labeled PA free.
Petadolex, the medication used in migraine studies, was initially approved by the German health regulatory authority, but approval was later withdrawn due to concerns about liver toxicity.22 In 2012, the United Kingdom’s Medicines and Health Care Products Regulatory Agency withdrew all butterbur products from the market due to associated cases of liver toxicity.22 Butterbur products are still available in the US market, and the risks and benefits should be discussed with all patients considering this treatment. Liver function monitoring is recommended for all patients using butterbur.22
The herb can also cause dyspepsia, headache, itchy eyes, gastrointestinal symptoms, asthma, fatigue, and drowsiness. Additionally, people who are allergic to ragweed and daisies may have allergic reactions to butterbur. Eructation (belching) occurred in 7% of patients in a pediatric study.17
The bottom line
Butterbur appears to be efficacious for migraine prophylaxis, but long-term safety is unknown and serious concerns exist for liver toxicity.
Continue to: GREEN TEA
GREEN TEA
Most tea leaves come from the Camellia sinensis bush, but green and black tea are processed differently to produce different end products.23 It is estimated that green tea accounts for approximately a quarter of all tea consumption and is most commonly consumed in Asian countries.23 The health-promoting effects of green tea are mainly attributed to its polyphenol content.24 Of the many types of tea, green tea has the highest concentration of polyphenols, including catechins, which are powerful antioxidants.23,24 Green tea has been used in traditional Chinese and Indian medicine to control bleeding, improve digestion, and promote overall health.23
Dementia. Green tea polyphenols may enhance cognition and may protect against the development of dementia. In-vitro studies have shown that green tea reduces hydrogen peroxide and ß-amyloid peptides, which are significant in the development of Alzheimer’s disease.25 A 12-subject double-blind study found green tea increased working memory and had an impact on frontoparietal brain connections.26 Furthermore, a cohort study with 13,645 Japanese participants over a five-year period found that frequent green tea consumption (> 5 cups per day) was associated with a lower risk for dementia.27 Additional studies are needed, but green tea may be useful in the treatment or prevention of dementia in the future.
Coronary artery disease. In one study, green tea plasma and urinary concentrations were associated with plasma biomarkers of cardiovascular disease and diabetes.28 In one review, the consumption of green tea was associated with a statistically significant reduction in LDL cholesterol.29 Furthermore, a 2015 systematic review and meta-analysis of prospective observational studies concluded that increased tea consumption (of any type) is associated with a reduced risk for coronary heart disease, cardiac death, stroke, and total mortality.30
Cancer. Many studies have shown that green tea may reduce the risk for cancer, although epidemiologic evidence is inconsistent. Studies have shown that cancer rates tend to be lower in those who consume higher levels of green tea.31,32 Whether this can be attributed solely to green tea remains debatable. Several other studies have shown that polyphenols in green tea can inhibit the growth of cancer cells, but the exact mechanism by which tea interacts with cancerous cells is unknown.23
Several population-based studies have been performed, mostly in Japan, which showed green tea consumption reduced the risk for cancer. Fewer prostate cancer cases have been reported in men who consume green tea.33 While studies have been performed to determine whether green tea has effects on pancreatic, esophageal, ovarian, breast, bladder, and colorectal cancer, the evidence remains inadequate.32
Diabetes. Green tea has been shown in several studies to have a beneficial effect on diabetes. A retrospective Japanese cohort study showed that those who consumed green tea were one-third less likely to develop type 2 diabetes.34 A 10-year study from Taiwan found lower body fat and smaller waist circumference in those who consumed green tea regularly.35 A 2014 meta-analysis and systematic review of tea (any type) consumption and the risk for diabetes concluded that three or more cups of tea per day was associated with a lower risk for diabetes.36 Another meta-analysis of 17 RCTs focused on green tea concluded that green tea improves glucose control and A1C values.37
Continue to: Adverse effects
Adverse effects
There have been concerns about potential hepatotoxicity induced by green tea intake.38 However, a systematic review of 34 RCTs on liver-related adverse events from green tea showed only a slight elevation in liver function tests; no serious liver-related adverse events were reported.38 This review suggested that liver-related adverse events after intake of green tea extracts are rare.38
Consuming green tea in the diet may lower the risk for adverse effects since the concentration consumed is generally much lower than that found in extracts.
Contraindications to drinking green tea are few. Individuals with caffeine sensitivities could experience insomnia, anxiety, irritability, or upset stomach. Additionally, patients who are taking anticoagulation drugs, such as warfarin, should avoid green tea due to its vitamin K content, which can counter the effects of warfarin. Pregnant or breastfeeding women, those with heart problems or high blood pressure, kidney or liver problems, stomach ulcers, or anxiety disorders should use caution with green tea consumption.
The bottom line
Green tea consumption in the diet appears to be safe and may have beneficial effects on weight, dementia, and risk for diabetes, cancer, and cardiovascular disease. Patients may want to consider drinking green tea as part of a healthy diet, in combination with exercise.
Continue to: PEPPERMINT
PEPPERMINT
Mentha piperita, also known as peppermint, is a hybrid between water mint and spearmint. It is found throughout Europe and North America and is commonly used in tea and toothpaste and as a flavoring for gum. Menthol and methyl salicylate are the main active ingredients in peppermint, and peppermint has calcium channel–blocker effects.39 Menthol has been shown to help regulate cold and pain sensation through the TRPM8 receptor.40 The peppermint herb is used both orally and topically, and has been studied in the treatment of multiple conditions.
Irritable bowel syndrome (IBS). It appears that peppermint inhibits spontaneous peristaltic activity, which reduces gastric emptying, decreases basal tone in the gastrointestinal tract, and slows down peristalsis in the gut.39
The American College of Gastroenterology guidelines currently note that there is moderate-quality evidence for peppermint oil in the treatment of IBS.41 A Cochrane review concluded that peppermint appears to be beneficial for IBS-related symptoms and pain.42 In a systematic review of nine studies from 2014, peppermint oil was found to be more effective than placebo for IBS symptoms such as pain, bloating, gas, and diarrhea.43 The review also indicated that peppermint oil is safe, with heartburn being the most common complaint.43 A 2016 study also found that triple-coated microspheres containing peppermint oil reduced the frequency and intensity of IBS symptoms.44
Non-ulcer dyspepsia. In combination with caraway oil, peppermint oil can be used to reduce symptoms of non-ulcer dyspepsia.45,46 A multicenter, randomized, placebo-controlled, double-blind study found that 43.3% of subjects improved with a peppermint-caraway oil combination after eight weeks, compared with 3.5% receiving placebo.46
Barium enema–related colonic spasm. Peppermint can relax the lower esophageal sphincter, and it has been shown to be useful as an antispasmodic agent for barium enema–related colonic spasm.47,48
Itching/skin irritation. Peppermint, when applied topically, has been used to calm pruritus and relieve irritation and inflammation. It has a soothing and cooling effect on the skin. At least one study found it to be effective in the treatment of pruritus gravidarum, although the study population consisted of only 96 subjects.49
Migraine headache. Initial small trials suggest that menthol is likely beneficial for migraine headaches. A pilot trial of 25 patients treated with topical menthol 6% gel for an acute migraine attack showed a significant improvement in headache intensity two hours after gel application.50 In a randomized, triple-blind, placebo-controlled, crossover study of 35 patients, a menthol 10% solution was shown to be more efficacious as abortive treatment of migraine headaches than placebo.51
Tension headache. In a randomized, placebo-controlled, double-blind crossover study, topical peppermint oil showed a significant clinical reduction in tension headache pain.52 Another small, randomized, double-blind trial showed that tiger balm (containing menthol as the main ingredient) also produced statistically significant improvement in tension headache discomfort compared with placebo.53
Continue to: Musculoskeletal pain
Musculoskeletal pain. A small study comparing topical menthol to ice for muscle soreness noted decreased perceived discomfort with menthol.54 Menthol has also been shown to reduce pain in patients with knee OA.55
Carpal tunnel syndrome (CTS). A triple-blind RCT concluded that topical menthol acutely reduced pain intensity in slaughterhouse workers with CTS, and it should be considered as an effective nonsystemic alternative to regular analgesics in the workplace management of chronic and neuropathic pain.56
Adverse effects
Peppermint appears to be safe for most adults when used in small doses, and serious adverse effects are rare.43,57 While peppermint tea appears to be safe in moderate-to-large amounts, people allergic to plants in the peppermint family (eg, mint, thyme, sage, rosemary, marjoram, basil, lavender) may experience allergic reactions with swelling, wheals, or erythema. Peppermint may also cause heartburn due to relaxation of the cardiac sphincter.
Other symptoms may include nausea, vomiting, flushing, and headache.58 The herb may also be both hepatotoxic and nephrotoxic at extremely high doses.59 Other considerations for women are that it can trigger menstruation and should be avoided during pregnancy. Due to uncertain efficacy in this population, peppermint oil should not be used on the face of infants, young children, or pregnant women.58,59
The bottom line
Peppermint appears to be safe and well tolerated. It is useful in alleviating IBS symptoms and may be effective in the treatment of non-ulcerative dyspepsia, musculoskeletal pain, headache, and CTS.54,55
1. National Center for Complementary and Integrative Health. The Use of Complementary and Alternative Medicine in the United States. https://nccih.nih.gov/research/statistics/2007/camsurvey_fs1.htm. Accessed April 19, 2018.
2. Wallace M, Pappagallo M. Qutenza: a capsaicin 8% patch for the management of postherpetic neuralgia. Expert Rev Neurother. 2011;11:15-27.
3. Rains C, Bryson HM. Topical capsaicin. A review of its pharmacological properties and therapeutic potential in post-herpetic neuralgia, diabetic neuropathy and osteoarthritis. Drugs Aging. 1995;7:317-328.
4. Derry S, Sven-Rice A, Cole P, et al. Topical capsaicin (high concentration) for chronic neuropathic pain in adults. Cochrane Database Syst Rev. 2013;(2):CD007393.
5. Mason L, Moore RA, Derry S, et al. Systematic review of topical capsaicin for the treatment of chronic pain. BMJ. 2004;328:991.
6. Deal CL, Schnitzer TJ, Lipstein E, et al. Treatment of arthritis with topical capsaicin: a double-blind trial. Clin Ther. 1991; 13:383.
7. McCarthy GM, McCarty DJ. Effect of topical capsaicin in the therapy of painful osteoarthritis of the hands. J Rheumatol. 1992;19:604.
8. De Silva V, El-Metwally A, Ernst E, et al; Arthritis Research UK Working Group on Complementary and Alternative Medicines. Evidence for the efficacy of complementary and alternative medicines in the management of osteoarthritis: a systematic review. Rheumatology (Oxford). 2011;50:911-920.
9. Cameron M, Chrubasik S. Topical herbal therapies for treating osteoarthritis. Cochrane Database Syst Rev. 2013;(5): CD010538.
10. Oltean H, Robbins C, van Tulder MW, et al. Herbal medicine for low-back pain. Cochrane Database Syst Rev. 2014;(12): CD004504.
11. Armstrong EP, Malone DC, McCarberg B, et al. Cost-effectiveness analysis of a new 8% capsaicin patch compared to existing therapies for postherpetic neuralgia. Curr Med Res Opin. 2011;27:939-950.
12. Mou J, Paillard F, Turnbull B, et al. Efficacy of Qutenza (capsaicin) 8% patch for neuropathic pain: a meta-analysis of the Qutenza Clinical Trials Database. Pain. 2013;154:1632-1639.
13. Sun-Edelstein C, Mauskop A. Alternative headache treatments: nutraceuticals, behavioral and physical treatments. Headache. 2011;51:469-483.
14. D’Andrea G, Cevoli S, Cologno D. Herbal therapy in migraine. Neurol Sci. 2014;35(suppl 1):135-140.
15. Diener HC, Rahlfs VW, Danesch U. The first placebo-controlled trial of a special butterbur root extract for the prevention of migraine: reanalysis of efficacy criteria. Eur Neurol. 2004;51:89-97.
16. Lipton RB, Göbel H, Einhäupl KM, et al. Petasites hybridus root (butterbur) is an effective preventive treatment for migraine. Neurology. 2004;63:2240-2244.
17. Pothmann R, Danesch U. Migraine prevention in children and adolescents: results of an open study with a special butterbur root extract. Headache. 2005;45:196-203.
18. Holland S, Silberstein SD, Freitag F, et al; Quality Standards Subcommittee of the American Academy of Neurology and the American Headache Society. Evidence-based guideline update: NSAIDs and other complementary treatments for episodic migraine prevention in adults: report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Headache Society. Neurology. 2012;78:1346-1353.
19. American Academy of Neurology. Evidence-based guideline update: NSAIDs and other complementary treatments for episodic migraine prevention in adults: [RETIRED]. http://n.neurology.org/content/78/17/1346. Accessed April 29, 2018.
20. Man LX. Complementary and alternative medicine for allergic rhinitis. Curr Opin Otolaryngol Head Neck Surg. 2009;17:226-231.
21. Guo R, Pittler MH, Ernst E. Herbal medicines for the treatment of allergic rhinitis: a systematic review. Ann Allergy Asthma Immunol. 2007;99:483-495.
22. Daniel O, Mauskop A. Nutraceuticals in acute and prophylactic treatment of migraine. Curr Treat Options Neurol. 2016; 18:14.
23. Chacko SM, Thambi PT, Kuttan R, et al. Beneficial effects of green tea: a literature review. Chin Med. 2010;6:13.
24. Naghma K, Hasan M. Tea polyphenols for health promotion. Life Sci. 2007;81:519-533.
25. Okello EJ, McDougall GJ, Kumar S, et al. In vitro protective effects of colon-available extract of Camellia sinensis (tea) against hydrogen peroxide and beta-amyloid (Aβ((1-42))) induced cytotoxicity in differentiated PC12 cells. Phytomedicine. 2011;18:691-696.
26. Schmidt A, Hammann F, Wölnerhanssen B, et al. Green tea extract enhances parieto-frontal connectivity during working memory processing. Psychopharmacology (Berl). 2014;231: 3879-3888.
27. Tomata Y, Sugiyama K, Kaiho Y, et al. Green tea consumption and the risk of incident dementia in elderly Japanese: The Ohsaki Cohort 2006 Study. Am J Geriatr Psychiatry. 2016;24: 881-889.
28. Takechi R, Alfonso H, Hiramatsu N, et al. Elevated plasma and urinary concentrations of green tea catechins associated with improved plasma lipid profile in healthy Japanese women. Nutr Res. 2016;36:220-226.
29. Kim A, Chiu A, Barone MK, et al. Green tea catechins decrease total and low-density lipoprotein cholesterol: a systematic review and meta-analysis. J Am Diet Assoc. 2011; 111:1720-1729.
30. Zhang C, Qin YY, Wei X, et al. Tea consumption and risk of cardiovascular outcomes and total mortality: a systematic review and meta-analysis of prospective observational studies. Eur J Epidemiol. 2015;30:103-113.
31. Imai K, Suga K, Nakachi K. Cancer-preventive effects of drinking green tea among a Japanese population. Prev Med. 1997;26:769-775.
32. Yuan JM. Cancer prevention by green tea: evidence from epidemiologic studies. Am J Clin Nutr. 2013;98(6 suppl): 1676S-1681S.
33. Kurahashi N, Sasazuki S, Iwasaki M, et al. Green tea consumption and prostate cancer risk in Japanese men: a prospective study. Am J Epidemiol. 2008;167:71-77.
34. Iso H, Date C, Wakai K, et al. The relationship between green tea and total caffeine intake and risk for self-reported type 2 diabetes among Japanese adults. Ann Intern Med. 2006; 144:554-562.
35. Kim HM, Kim J. The effects of green tea on obesity and type 2 diabetes. Diab Metab J. 2013;37:173-175.
36. Yang J, Mao Q, Xu H, et al. Tea consumption and risk of type 2 diabetes mellitus: a systematic review and meta-analysis update. BMJ Open. 2014;4:e005632.
37. Liu K, Zhou R, Wang B, et al. Effect of green tea on glucose control and insulin sensitivity: a meta-analysis of 17 randomized controlled trials. Am J Clin Nutr. 2013;98:340-348.
38. Isomura T, Suzuki S, Origasa H, et al. Liver-related safety assessment of green tea extracts in humans: a systematic review of randomized controlled trials. Eur J Clin Nutr. 2016;70:1340.
39. Tillisch K. Complementary and alternative medicine for gastrointestinal disorders. Clin Med (Lond). 2007;7:224-227.
40. Knowlton WM, McKemy DD. TRPM8: from cold to cancer, peppermint to pain. Curr Pharm Biotechnol. 2011;12:68-77.
41. Ford AC, Moayyedi P, Lacy BE, et al. Task Force on the Management of Functional Bowel Disorders. American College of Gastroenterology monograph on the management of irritable bowel syndrome and chronic idiopathic constipation. Am J Gastroenterol. 2014;109(suppl 1):S2-S26.
42. Ruepert L, Quartero AO, de Wit NJ, et al. Bulking agents, antispasmodics and antidepressants for the treatment of irritable bowel syndrome. Cochrane Database Syst Rev. 2011;(8):CD003460.
43. Khanna R, MacDonald JK, Levesque BG. Peppermint oil for the treatment of irritable bowel syndrome: a systematic review and meta-analysis. J Clin Gastroenterol. 2014;48:505-512.
44. Cash BD, Epstein MS, Shah SM. A novel delivery system of peppermint oil is an effective therapy for irritable bowel syndrome symptoms. Dig Dis Sci. 2016;61:560-571.
45. Holtmann G, Haag S, Adam B, et al. Effects of a fixed combination of peppermint oil and caraway oil on symptoms and quality of life in patients suffering from functional dyspepsia. Phytomedicine. 2003;10(suppl 4):56-57.
46. Madisch A, Heydenreich CJ, Wieland V, et al. Treatment of functional dyspepsia with a fixed peppermint oil and caraway oil combination preparation as compared to cisapride. A multicenter, reference-controlled double-blind equivalence study. Arzneimittelforschung. 1999;49:925-932.
47. Asao T, Kuwano H, Ide M, et al. Spasmolytic effect of peppermint oil in barium during double-contrast barium enema compared with Buscopan. Clin Radiol. 2003;58:301-305.
48. Sparks MJ, O’Sullivan P, Herrington AA, et al. Does peppermint oil relieve spasm during barium enema? Br J Radiol. 1995;68:841-843.
49. Akhavan Amjadi M, Mojab F, Kamranpour SB. The effect of peppermint oil on symptomatic treatment of pruritus in pregnant women. Iran J Pharm Res. 2012;11:1073-1077.
50. St Cyr A, Chen A, Bradley KC, et al. Efficacy and tolerability of STOPAIN for a migraine attack. Front Neurol. 2015;6:11.
51. Borhani Haghighi A, Motazedian S, Rezaii R, et al. Cutaneous application of menthol 10% solution as an abortive treatment of migraine without aura: a randomised, double-blind, placebo-controlled, crossed-over study. Int J Clin Pract. 2010; 64:451-456.
52. Gobel H, Fresenius J, Heinze A, et al. Effectiveness of oleum menthae piperitae and paracetamol in therapy of headache of the tension type [German]. Nervenarzt. 1996;67:672-681.
53. Schattner P, Randerson D. Tiger Balm as a treatment of tension headache. A clinical trial in general practice. Aust Fam Physician. 1996;25:216-220.
54. Johar P, Grover V, Topp R, et al. A comparison of topical menthol to ice on pain, evoked tetanic and voluntary force during delayed onset muscle soreness. Int J Sports Phys Ther. 2012;7:314-322.
55. Topp R, Brosky JA Jr, Pieschel D. The effect of either topical menthol or a placebo on functioning and knee pain among patients with knee OA. J Geriatr Phys Ther. 2013;36:92-99.
56. Sundstrup E, Jakobsen MD, Brandt M, et al. Acute effect of topical menthol on chronic pain in slaughterhouse workers with carpal tunnel syndrome: triple-blind, randomized placebo-controlled trial. Rehabil Res Pract. 2014;2014:310913.
57. Nair B. Final report on the safety assessment of mentha piperita (peppermint) oil, mentha piperita (peppermint) leaf extract, mentha piperita (peppermint) leaf, and mentha piperita (peppermint) leaf water. Int J Toxicol. 2001;20(suppl 3):61-73.
58. Klingler B, Chadhary S. Peppermint oil. Am Fam Physician. 2007;75:1027-1030.
59. Nath SS, Pandey C, Roy D. A near fatal case of high dose peppermint oil ingestion—lessons learnt. Indian J Anaesth. 2012;56:582-584.
1. National Center for Complementary and Integrative Health. The Use of Complementary and Alternative Medicine in the United States. https://nccih.nih.gov/research/statistics/2007/camsurvey_fs1.htm. Accessed April 19, 2018.
2. Wallace M, Pappagallo M. Qutenza: a capsaicin 8% patch for the management of postherpetic neuralgia. Expert Rev Neurother. 2011;11:15-27.
3. Rains C, Bryson HM. Topical capsaicin. A review of its pharmacological properties and therapeutic potential in post-herpetic neuralgia, diabetic neuropathy and osteoarthritis. Drugs Aging. 1995;7:317-328.
4. Derry S, Sven-Rice A, Cole P, et al. Topical capsaicin (high concentration) for chronic neuropathic pain in adults. Cochrane Database Syst Rev. 2013;(2):CD007393.
5. Mason L, Moore RA, Derry S, et al. Systematic review of topical capsaicin for the treatment of chronic pain. BMJ. 2004;328:991.
6. Deal CL, Schnitzer TJ, Lipstein E, et al. Treatment of arthritis with topical capsaicin: a double-blind trial. Clin Ther. 1991; 13:383.
7. McCarthy GM, McCarty DJ. Effect of topical capsaicin in the therapy of painful osteoarthritis of the hands. J Rheumatol. 1992;19:604.
8. De Silva V, El-Metwally A, Ernst E, et al; Arthritis Research UK Working Group on Complementary and Alternative Medicines. Evidence for the efficacy of complementary and alternative medicines in the management of osteoarthritis: a systematic review. Rheumatology (Oxford). 2011;50:911-920.
9. Cameron M, Chrubasik S. Topical herbal therapies for treating osteoarthritis. Cochrane Database Syst Rev. 2013;(5): CD010538.
10. Oltean H, Robbins C, van Tulder MW, et al. Herbal medicine for low-back pain. Cochrane Database Syst Rev. 2014;(12): CD004504.
11. Armstrong EP, Malone DC, McCarberg B, et al. Cost-effectiveness analysis of a new 8% capsaicin patch compared to existing therapies for postherpetic neuralgia. Curr Med Res Opin. 2011;27:939-950.
12. Mou J, Paillard F, Turnbull B, et al. Efficacy of Qutenza (capsaicin) 8% patch for neuropathic pain: a meta-analysis of the Qutenza Clinical Trials Database. Pain. 2013;154:1632-1639.
13. Sun-Edelstein C, Mauskop A. Alternative headache treatments: nutraceuticals, behavioral and physical treatments. Headache. 2011;51:469-483.
14. D’Andrea G, Cevoli S, Cologno D. Herbal therapy in migraine. Neurol Sci. 2014;35(suppl 1):135-140.
15. Diener HC, Rahlfs VW, Danesch U. The first placebo-controlled trial of a special butterbur root extract for the prevention of migraine: reanalysis of efficacy criteria. Eur Neurol. 2004;51:89-97.
16. Lipton RB, Göbel H, Einhäupl KM, et al. Petasites hybridus root (butterbur) is an effective preventive treatment for migraine. Neurology. 2004;63:2240-2244.
17. Pothmann R, Danesch U. Migraine prevention in children and adolescents: results of an open study with a special butterbur root extract. Headache. 2005;45:196-203.
18. Holland S, Silberstein SD, Freitag F, et al; Quality Standards Subcommittee of the American Academy of Neurology and the American Headache Society. Evidence-based guideline update: NSAIDs and other complementary treatments for episodic migraine prevention in adults: report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Headache Society. Neurology. 2012;78:1346-1353.
19. American Academy of Neurology. Evidence-based guideline update: NSAIDs and other complementary treatments for episodic migraine prevention in adults: [RETIRED]. http://n.neurology.org/content/78/17/1346. Accessed April 29, 2018.
20. Man LX. Complementary and alternative medicine for allergic rhinitis. Curr Opin Otolaryngol Head Neck Surg. 2009;17:226-231.
21. Guo R, Pittler MH, Ernst E. Herbal medicines for the treatment of allergic rhinitis: a systematic review. Ann Allergy Asthma Immunol. 2007;99:483-495.
22. Daniel O, Mauskop A. Nutraceuticals in acute and prophylactic treatment of migraine. Curr Treat Options Neurol. 2016; 18:14.
23. Chacko SM, Thambi PT, Kuttan R, et al. Beneficial effects of green tea: a literature review. Chin Med. 2010;6:13.
24. Naghma K, Hasan M. Tea polyphenols for health promotion. Life Sci. 2007;81:519-533.
25. Okello EJ, McDougall GJ, Kumar S, et al. In vitro protective effects of colon-available extract of Camellia sinensis (tea) against hydrogen peroxide and beta-amyloid (Aβ((1-42))) induced cytotoxicity in differentiated PC12 cells. Phytomedicine. 2011;18:691-696.
26. Schmidt A, Hammann F, Wölnerhanssen B, et al. Green tea extract enhances parieto-frontal connectivity during working memory processing. Psychopharmacology (Berl). 2014;231: 3879-3888.
27. Tomata Y, Sugiyama K, Kaiho Y, et al. Green tea consumption and the risk of incident dementia in elderly Japanese: The Ohsaki Cohort 2006 Study. Am J Geriatr Psychiatry. 2016;24: 881-889.
28. Takechi R, Alfonso H, Hiramatsu N, et al. Elevated plasma and urinary concentrations of green tea catechins associated with improved plasma lipid profile in healthy Japanese women. Nutr Res. 2016;36:220-226.
29. Kim A, Chiu A, Barone MK, et al. Green tea catechins decrease total and low-density lipoprotein cholesterol: a systematic review and meta-analysis. J Am Diet Assoc. 2011; 111:1720-1729.
30. Zhang C, Qin YY, Wei X, et al. Tea consumption and risk of cardiovascular outcomes and total mortality: a systematic review and meta-analysis of prospective observational studies. Eur J Epidemiol. 2015;30:103-113.
31. Imai K, Suga K, Nakachi K. Cancer-preventive effects of drinking green tea among a Japanese population. Prev Med. 1997;26:769-775.
32. Yuan JM. Cancer prevention by green tea: evidence from epidemiologic studies. Am J Clin Nutr. 2013;98(6 suppl): 1676S-1681S.
33. Kurahashi N, Sasazuki S, Iwasaki M, et al. Green tea consumption and prostate cancer risk in Japanese men: a prospective study. Am J Epidemiol. 2008;167:71-77.
34. Iso H, Date C, Wakai K, et al. The relationship between green tea and total caffeine intake and risk for self-reported type 2 diabetes among Japanese adults. Ann Intern Med. 2006; 144:554-562.
35. Kim HM, Kim J. The effects of green tea on obesity and type 2 diabetes. Diab Metab J. 2013;37:173-175.
36. Yang J, Mao Q, Xu H, et al. Tea consumption and risk of type 2 diabetes mellitus: a systematic review and meta-analysis update. BMJ Open. 2014;4:e005632.
37. Liu K, Zhou R, Wang B, et al. Effect of green tea on glucose control and insulin sensitivity: a meta-analysis of 17 randomized controlled trials. Am J Clin Nutr. 2013;98:340-348.
38. Isomura T, Suzuki S, Origasa H, et al. Liver-related safety assessment of green tea extracts in humans: a systematic review of randomized controlled trials. Eur J Clin Nutr. 2016;70:1340.
39. Tillisch K. Complementary and alternative medicine for gastrointestinal disorders. Clin Med (Lond). 2007;7:224-227.
40. Knowlton WM, McKemy DD. TRPM8: from cold to cancer, peppermint to pain. Curr Pharm Biotechnol. 2011;12:68-77.
41. Ford AC, Moayyedi P, Lacy BE, et al. Task Force on the Management of Functional Bowel Disorders. American College of Gastroenterology monograph on the management of irritable bowel syndrome and chronic idiopathic constipation. Am J Gastroenterol. 2014;109(suppl 1):S2-S26.
42. Ruepert L, Quartero AO, de Wit NJ, et al. Bulking agents, antispasmodics and antidepressants for the treatment of irritable bowel syndrome. Cochrane Database Syst Rev. 2011;(8):CD003460.
43. Khanna R, MacDonald JK, Levesque BG. Peppermint oil for the treatment of irritable bowel syndrome: a systematic review and meta-analysis. J Clin Gastroenterol. 2014;48:505-512.
44. Cash BD, Epstein MS, Shah SM. A novel delivery system of peppermint oil is an effective therapy for irritable bowel syndrome symptoms. Dig Dis Sci. 2016;61:560-571.
45. Holtmann G, Haag S, Adam B, et al. Effects of a fixed combination of peppermint oil and caraway oil on symptoms and quality of life in patients suffering from functional dyspepsia. Phytomedicine. 2003;10(suppl 4):56-57.
46. Madisch A, Heydenreich CJ, Wieland V, et al. Treatment of functional dyspepsia with a fixed peppermint oil and caraway oil combination preparation as compared to cisapride. A multicenter, reference-controlled double-blind equivalence study. Arzneimittelforschung. 1999;49:925-932.
47. Asao T, Kuwano H, Ide M, et al. Spasmolytic effect of peppermint oil in barium during double-contrast barium enema compared with Buscopan. Clin Radiol. 2003;58:301-305.
48. Sparks MJ, O’Sullivan P, Herrington AA, et al. Does peppermint oil relieve spasm during barium enema? Br J Radiol. 1995;68:841-843.
49. Akhavan Amjadi M, Mojab F, Kamranpour SB. The effect of peppermint oil on symptomatic treatment of pruritus in pregnant women. Iran J Pharm Res. 2012;11:1073-1077.
50. St Cyr A, Chen A, Bradley KC, et al. Efficacy and tolerability of STOPAIN for a migraine attack. Front Neurol. 2015;6:11.
51. Borhani Haghighi A, Motazedian S, Rezaii R, et al. Cutaneous application of menthol 10% solution as an abortive treatment of migraine without aura: a randomised, double-blind, placebo-controlled, crossed-over study. Int J Clin Pract. 2010; 64:451-456.
52. Gobel H, Fresenius J, Heinze A, et al. Effectiveness of oleum menthae piperitae and paracetamol in therapy of headache of the tension type [German]. Nervenarzt. 1996;67:672-681.
53. Schattner P, Randerson D. Tiger Balm as a treatment of tension headache. A clinical trial in general practice. Aust Fam Physician. 1996;25:216-220.
54. Johar P, Grover V, Topp R, et al. A comparison of topical menthol to ice on pain, evoked tetanic and voluntary force during delayed onset muscle soreness. Int J Sports Phys Ther. 2012;7:314-322.
55. Topp R, Brosky JA Jr, Pieschel D. The effect of either topical menthol or a placebo on functioning and knee pain among patients with knee OA. J Geriatr Phys Ther. 2013;36:92-99.
56. Sundstrup E, Jakobsen MD, Brandt M, et al. Acute effect of topical menthol on chronic pain in slaughterhouse workers with carpal tunnel syndrome: triple-blind, randomized placebo-controlled trial. Rehabil Res Pract. 2014;2014:310913.
57. Nair B. Final report on the safety assessment of mentha piperita (peppermint) oil, mentha piperita (peppermint) leaf extract, mentha piperita (peppermint) leaf, and mentha piperita (peppermint) leaf water. Int J Toxicol. 2001;20(suppl 3):61-73.
58. Klingler B, Chadhary S. Peppermint oil. Am Fam Physician. 2007;75:1027-1030.
59. Nath SS, Pandey C, Roy D. A near fatal case of high dose peppermint oil ingestion—lessons learnt. Indian J Anaesth. 2012;56:582-584.

Hyperkeratotic fissured plaques on both hands: Mechanic’s hands
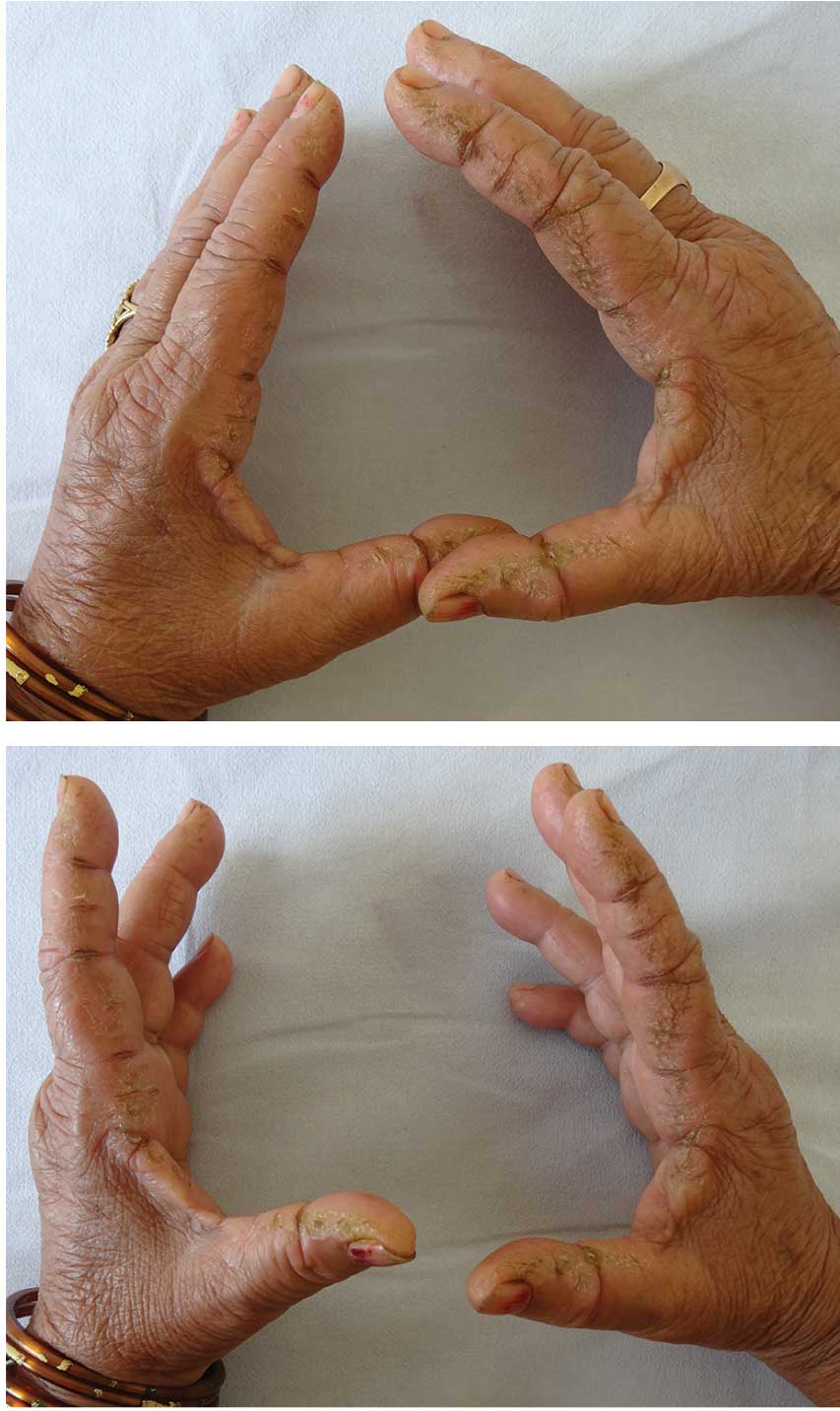
Nail fold capillaroscopy did not reveal telangiectasia or ragged cuticles. Further examination of the skin showed confluent macular violaceous erythema on the eyelids (suggestive of the heliotrope sign), V area of the neck, upper arms, and back.
She also had a low-grade intermittent fever for the past 2 months, as well as difficulty in getting up from a squatting position and combing her hair, dyspnea on exertion, blue discoloration of the fingers on exposure to cold, and intermittent pain, stiffness, and swelling in the small joints of both hands that was worse in the morning and seemed to be relieved by activity. She had no history of dysphagia or nasal regurgitation of food. Strength against resistance was reduced in both arms and knee extensors. A diagnosis of dermatomyositis with “mechanic’s hands” was considered.
Laboratory testing was negative for antinuclear antibodies and showed elevated creatine kinase and positive anti-Jo-1 antibodies. High-resolution computed tomography of the chest showed evidence of interstitial lung disease. Features were consistent with antisynthetase syndrome and dermatomyositis. An age-appropriate malignancy screen was normal.
The patient was started on oral prednisolone 60 mg, hydroxychloroquine 300 mg, and azathioprine 100 mg. For her hands, topical clobetasol propionate 0.05% with 3% salicyclic acid and emollients were advised. Her muscle weakness improved considerably after 2 months, but the photosensitivity and mechanic’s hands improved only minimally.
ANTISYNTHETASE SYNDROME
Antisynthetase syndrome is a subset of idiopathic inflammatory myopathies characterized by fever, Raynaud phenomenon, arthritis, myositis, interstitial lung disease, and mechanic’s hands. It is associated with myositis-specific antibodies directed against aminoacyl-tRNA synthetases, of which anti-Jo-1 is the most common. Other antibodies including anti-PL-7 and anti-PL-12 may be present, whereas antinuclear antibodies may be negative.
Mechanic’s hands is seen in about 30% of patients with antisynthetase syndrome and is an important physical sign, as its presence in a patient with myositis and arthritis prompts an evaluation to exclude interstitial lung disease. Its onset later in the disease course may herald the flare-up of interstitial lung disease.1 A similar hyperkeratosis may also affect the feet, and the importance of a careful cutaneous examination of the hands and feet should be stressed in patients presenting with polymyositis and dermatomyositis.
In contrast, hyperkeratotic eczema of the hand is usually pruritic, and involvement of the tips and palmar aspects of the fingers and palms is characteristic.2,3 Vesicles (pompholyx) and coarse pitting of the nails may also be seen in eczema. Other features that help rule out eczema are the development of these features over a short period of time, asymptomatic nature, presence of systemic symptoms, and involvement of only the lateral margins of the index fingers, with no involvement of the palmar aspects and other fingers.
Degenerative collagenous plaque, closely resembling mechanic’s hands, is common in elderly people with photodamaged skin. It is asymptomatic, is not associated with systemic illness, and features clumping and thickening of elastic fibers on histopathology.
Association with cancer risk
Though antisynthetase syndrome was not previously considered to be associated with an increased risk of malignancy, a retrospective review of 124 patients with antisynthetase syndrome recently showed a malignancy risk of 6.5%.4 Overall, the data regarding the association of malignancy and antisynthetase syndrome are conflicting, and this needs further study. Therefore, an age-appropriate malignancy screen is recommended.4–6 Also, the presence of malignancy and interstitial lung disease is associated with a poor prognosis in these patients.
Treatment
Glucocorticoids are the mainstay of treatment, and azathioprine and methotrexate are important steroid-sparing agents.5,7 Use of methotrexate warrants caution in patients with interstitial lung disease, since methotrexate itself can cause pulmonary fibrosis.
In our patient, prednisolone was slowly tapered to 20 mg/day, and hydroxychloroquine and azathioprine were continued at 300 mg/day and 100 mg/day, respectively. Topical treatment for mechanic’s hands was continued, with only minimal improvement.
- Bartoloni E, Gonzalez-Gay MA, Scire C, et al. Clinical follow-up predictors of disease pattern change in anti-Jo1 positive anti-synthetase syndrome: results from a multicenter, international and retrospective study. Autoimmun Rev 2017; 16(3):253–257. doi:10.1016/j.autrev.2017.01.008
- Bachmeyer C, Tillie-Leblond I, Lacert A, Cadranel J, Aractingi S. “Mechanic's hands”: a misleading cutaneous sign of the antisynthetase syndrome. Br J Dermatol 2007; 156(1):192–194. doi:10.1111/j.1365-2133.2006.07593.x
- Mii S, Kobayashi R, Nakano T, et al. A histopathologic study of mechanic's hands associated with dermatomyositis: a report of five cases. Int J Dermatol 2009; 48(11):1177–1182. doi:10.1111/j.1365-4632.2009.04164.x
- Shi J, Li S, Yang H, et al. Clinical profiles and prognosis of patients with distinct antisynthetase autoantibodies. J Rheumatol 2017; 44(7):1051–1057. doi:10.3899/jrheum.161480
- Chatterjee S, Prayson R, Farver C. Antisynthetase syndrome: not just an inflammatory myopathy. Cleve Clin J Med 2013; 80(10):655–666. doi:10.3949/ccjm.80a.12171
- Boleto G, Perotin JM, Eschard JP, Salmon JH. Squamous cell carcinoma of the lung associated with anti-Jo1 antisynthetase syndrome: a case report and review of the literature. Rheumatol Int 2017; 37(7):1203–1206. doi:10.1007/s00296-017-3728-z
- Mirrakhimov AE. Antisynthetase syndrome: a review of etiopathogenesis, diagnosis and management. Curr Med Chem 2015; 22(16):1963–1975. doi:10.2174/0929867322666150514094935
Nail fold capillaroscopy did not reveal telangiectasia or ragged cuticles. Further examination of the skin showed confluent macular violaceous erythema on the eyelids (suggestive of the heliotrope sign), V area of the neck, upper arms, and back.
She also had a low-grade intermittent fever for the past 2 months, as well as difficulty in getting up from a squatting position and combing her hair, dyspnea on exertion, blue discoloration of the fingers on exposure to cold, and intermittent pain, stiffness, and swelling in the small joints of both hands that was worse in the morning and seemed to be relieved by activity. She had no history of dysphagia or nasal regurgitation of food. Strength against resistance was reduced in both arms and knee extensors. A diagnosis of dermatomyositis with “mechanic’s hands” was considered.
Laboratory testing was negative for antinuclear antibodies and showed elevated creatine kinase and positive anti-Jo-1 antibodies. High-resolution computed tomography of the chest showed evidence of interstitial lung disease. Features were consistent with antisynthetase syndrome and dermatomyositis. An age-appropriate malignancy screen was normal.
The patient was started on oral prednisolone 60 mg, hydroxychloroquine 300 mg, and azathioprine 100 mg. For her hands, topical clobetasol propionate 0.05% with 3% salicyclic acid and emollients were advised. Her muscle weakness improved considerably after 2 months, but the photosensitivity and mechanic’s hands improved only minimally.
ANTISYNTHETASE SYNDROME
Antisynthetase syndrome is a subset of idiopathic inflammatory myopathies characterized by fever, Raynaud phenomenon, arthritis, myositis, interstitial lung disease, and mechanic’s hands. It is associated with myositis-specific antibodies directed against aminoacyl-tRNA synthetases, of which anti-Jo-1 is the most common. Other antibodies including anti-PL-7 and anti-PL-12 may be present, whereas antinuclear antibodies may be negative.
Mechanic’s hands is seen in about 30% of patients with antisynthetase syndrome and is an important physical sign, as its presence in a patient with myositis and arthritis prompts an evaluation to exclude interstitial lung disease. Its onset later in the disease course may herald the flare-up of interstitial lung disease.1 A similar hyperkeratosis may also affect the feet, and the importance of a careful cutaneous examination of the hands and feet should be stressed in patients presenting with polymyositis and dermatomyositis.
In contrast, hyperkeratotic eczema of the hand is usually pruritic, and involvement of the tips and palmar aspects of the fingers and palms is characteristic.2,3 Vesicles (pompholyx) and coarse pitting of the nails may also be seen in eczema. Other features that help rule out eczema are the development of these features over a short period of time, asymptomatic nature, presence of systemic symptoms, and involvement of only the lateral margins of the index fingers, with no involvement of the palmar aspects and other fingers.
Degenerative collagenous plaque, closely resembling mechanic’s hands, is common in elderly people with photodamaged skin. It is asymptomatic, is not associated with systemic illness, and features clumping and thickening of elastic fibers on histopathology.
Association with cancer risk
Though antisynthetase syndrome was not previously considered to be associated with an increased risk of malignancy, a retrospective review of 124 patients with antisynthetase syndrome recently showed a malignancy risk of 6.5%.4 Overall, the data regarding the association of malignancy and antisynthetase syndrome are conflicting, and this needs further study. Therefore, an age-appropriate malignancy screen is recommended.4–6 Also, the presence of malignancy and interstitial lung disease is associated with a poor prognosis in these patients.
Treatment
Glucocorticoids are the mainstay of treatment, and azathioprine and methotrexate are important steroid-sparing agents.5,7 Use of methotrexate warrants caution in patients with interstitial lung disease, since methotrexate itself can cause pulmonary fibrosis.
In our patient, prednisolone was slowly tapered to 20 mg/day, and hydroxychloroquine and azathioprine were continued at 300 mg/day and 100 mg/day, respectively. Topical treatment for mechanic’s hands was continued, with only minimal improvement.
Nail fold capillaroscopy did not reveal telangiectasia or ragged cuticles. Further examination of the skin showed confluent macular violaceous erythema on the eyelids (suggestive of the heliotrope sign), V area of the neck, upper arms, and back.
She also had a low-grade intermittent fever for the past 2 months, as well as difficulty in getting up from a squatting position and combing her hair, dyspnea on exertion, blue discoloration of the fingers on exposure to cold, and intermittent pain, stiffness, and swelling in the small joints of both hands that was worse in the morning and seemed to be relieved by activity. She had no history of dysphagia or nasal regurgitation of food. Strength against resistance was reduced in both arms and knee extensors. A diagnosis of dermatomyositis with “mechanic’s hands” was considered.
Laboratory testing was negative for antinuclear antibodies and showed elevated creatine kinase and positive anti-Jo-1 antibodies. High-resolution computed tomography of the chest showed evidence of interstitial lung disease. Features were consistent with antisynthetase syndrome and dermatomyositis. An age-appropriate malignancy screen was normal.
The patient was started on oral prednisolone 60 mg, hydroxychloroquine 300 mg, and azathioprine 100 mg. For her hands, topical clobetasol propionate 0.05% with 3% salicyclic acid and emollients were advised. Her muscle weakness improved considerably after 2 months, but the photosensitivity and mechanic’s hands improved only minimally.
ANTISYNTHETASE SYNDROME
Antisynthetase syndrome is a subset of idiopathic inflammatory myopathies characterized by fever, Raynaud phenomenon, arthritis, myositis, interstitial lung disease, and mechanic’s hands. It is associated with myositis-specific antibodies directed against aminoacyl-tRNA synthetases, of which anti-Jo-1 is the most common. Other antibodies including anti-PL-7 and anti-PL-12 may be present, whereas antinuclear antibodies may be negative.
Mechanic’s hands is seen in about 30% of patients with antisynthetase syndrome and is an important physical sign, as its presence in a patient with myositis and arthritis prompts an evaluation to exclude interstitial lung disease. Its onset later in the disease course may herald the flare-up of interstitial lung disease.1 A similar hyperkeratosis may also affect the feet, and the importance of a careful cutaneous examination of the hands and feet should be stressed in patients presenting with polymyositis and dermatomyositis.
In contrast, hyperkeratotic eczema of the hand is usually pruritic, and involvement of the tips and palmar aspects of the fingers and palms is characteristic.2,3 Vesicles (pompholyx) and coarse pitting of the nails may also be seen in eczema. Other features that help rule out eczema are the development of these features over a short period of time, asymptomatic nature, presence of systemic symptoms, and involvement of only the lateral margins of the index fingers, with no involvement of the palmar aspects and other fingers.
Degenerative collagenous plaque, closely resembling mechanic’s hands, is common in elderly people with photodamaged skin. It is asymptomatic, is not associated with systemic illness, and features clumping and thickening of elastic fibers on histopathology.
Association with cancer risk
Though antisynthetase syndrome was not previously considered to be associated with an increased risk of malignancy, a retrospective review of 124 patients with antisynthetase syndrome recently showed a malignancy risk of 6.5%.4 Overall, the data regarding the association of malignancy and antisynthetase syndrome are conflicting, and this needs further study. Therefore, an age-appropriate malignancy screen is recommended.4–6 Also, the presence of malignancy and interstitial lung disease is associated with a poor prognosis in these patients.
Treatment
Glucocorticoids are the mainstay of treatment, and azathioprine and methotrexate are important steroid-sparing agents.5,7 Use of methotrexate warrants caution in patients with interstitial lung disease, since methotrexate itself can cause pulmonary fibrosis.
In our patient, prednisolone was slowly tapered to 20 mg/day, and hydroxychloroquine and azathioprine were continued at 300 mg/day and 100 mg/day, respectively. Topical treatment for mechanic’s hands was continued, with only minimal improvement.
- Bartoloni E, Gonzalez-Gay MA, Scire C, et al. Clinical follow-up predictors of disease pattern change in anti-Jo1 positive anti-synthetase syndrome: results from a multicenter, international and retrospective study. Autoimmun Rev 2017; 16(3):253–257. doi:10.1016/j.autrev.2017.01.008
- Bachmeyer C, Tillie-Leblond I, Lacert A, Cadranel J, Aractingi S. “Mechanic's hands”: a misleading cutaneous sign of the antisynthetase syndrome. Br J Dermatol 2007; 156(1):192–194. doi:10.1111/j.1365-2133.2006.07593.x
- Mii S, Kobayashi R, Nakano T, et al. A histopathologic study of mechanic's hands associated with dermatomyositis: a report of five cases. Int J Dermatol 2009; 48(11):1177–1182. doi:10.1111/j.1365-4632.2009.04164.x
- Shi J, Li S, Yang H, et al. Clinical profiles and prognosis of patients with distinct antisynthetase autoantibodies. J Rheumatol 2017; 44(7):1051–1057. doi:10.3899/jrheum.161480
- Chatterjee S, Prayson R, Farver C. Antisynthetase syndrome: not just an inflammatory myopathy. Cleve Clin J Med 2013; 80(10):655–666. doi:10.3949/ccjm.80a.12171
- Boleto G, Perotin JM, Eschard JP, Salmon JH. Squamous cell carcinoma of the lung associated with anti-Jo1 antisynthetase syndrome: a case report and review of the literature. Rheumatol Int 2017; 37(7):1203–1206. doi:10.1007/s00296-017-3728-z
- Mirrakhimov AE. Antisynthetase syndrome: a review of etiopathogenesis, diagnosis and management. Curr Med Chem 2015; 22(16):1963–1975. doi:10.2174/0929867322666150514094935
- Bartoloni E, Gonzalez-Gay MA, Scire C, et al. Clinical follow-up predictors of disease pattern change in anti-Jo1 positive anti-synthetase syndrome: results from a multicenter, international and retrospective study. Autoimmun Rev 2017; 16(3):253–257. doi:10.1016/j.autrev.2017.01.008
- Bachmeyer C, Tillie-Leblond I, Lacert A, Cadranel J, Aractingi S. “Mechanic's hands”: a misleading cutaneous sign of the antisynthetase syndrome. Br J Dermatol 2007; 156(1):192–194. doi:10.1111/j.1365-2133.2006.07593.x
- Mii S, Kobayashi R, Nakano T, et al. A histopathologic study of mechanic's hands associated with dermatomyositis: a report of five cases. Int J Dermatol 2009; 48(11):1177–1182. doi:10.1111/j.1365-4632.2009.04164.x
- Shi J, Li S, Yang H, et al. Clinical profiles and prognosis of patients with distinct antisynthetase autoantibodies. J Rheumatol 2017; 44(7):1051–1057. doi:10.3899/jrheum.161480
- Chatterjee S, Prayson R, Farver C. Antisynthetase syndrome: not just an inflammatory myopathy. Cleve Clin J Med 2013; 80(10):655–666. doi:10.3949/ccjm.80a.12171
- Boleto G, Perotin JM, Eschard JP, Salmon JH. Squamous cell carcinoma of the lung associated with anti-Jo1 antisynthetase syndrome: a case report and review of the literature. Rheumatol Int 2017; 37(7):1203–1206. doi:10.1007/s00296-017-3728-z
- Mirrakhimov AE. Antisynthetase syndrome: a review of etiopathogenesis, diagnosis and management. Curr Med Chem 2015; 22(16):1963–1975. doi:10.2174/0929867322666150514094935
Olopatadine/mometasone combo is safe and effective
ORLANDO – Twice-daily treatment with a combination of olopatadine and mometasone showed significant clinical benefit and demonstrated safety for patients with seasonal allergic rhinitis, according to the results of a phase 3 trial.
The combination, known as GSP301, is a fixed-dose nasal spray containing olopatadine, a Food and Drug Adminstration–approved antihistamine, and mometasone, an FDA-approved corticosteroid.

The combination therapy demonstrated statistically significant improvement in scores, compared with those associated with placebo (P less than .001) and olopatadine alone (P = .003). It also showed benefit when compared with mometasone alone (P = .059), Dr. Hampel reported at the joint congress of the American Academy of Allergy, Asthma, and Immunology and the World Allergy Organization.
“You expect this outcome,” Dr. Hampel said in an interview. “Olopatadine is a good drug, mometasone is a good drug, so you’d expect the combination to be good, and it was.”
Dr. Hampel and his colleagues also analyzed changes in baseline scores for patients in the olopatadine group and the mometasone group and compared those changes with those seen in the placebo group. Patients in the mometasone group experienced statistically significant improvement, compared with those in the placebo group (P = .004), and patients in the olopatadine group also appeared to experienced benefit (P = .076).
Adverse events were similar across all treatment groups, although a slightly higher percentage of patients in the GSP301 group (12.9%) and olopatadine groups (12.5%) experienced adverse events, Dr. Hampel said.
Glenmark Pharmaceuticals sponsored the study. Dr. Hampel reported funding from Glenmark Pharmaceuticals and other pharmaceutical companies
SOURCE: Hampel F et al. AAAAI/WAO Joint Congress, Abstract 546.
ORLANDO – Twice-daily treatment with a combination of olopatadine and mometasone showed significant clinical benefit and demonstrated safety for patients with seasonal allergic rhinitis, according to the results of a phase 3 trial.
The combination, known as GSP301, is a fixed-dose nasal spray containing olopatadine, a Food and Drug Adminstration–approved antihistamine, and mometasone, an FDA-approved corticosteroid.
The combination therapy demonstrated statistically significant improvement in scores, compared with those associated with placebo (P less than .001) and olopatadine alone (P = .003). It also showed benefit when compared with mometasone alone (P = .059), Dr. Hampel reported at the joint congress of the American Academy of Allergy, Asthma, and Immunology and the World Allergy Organization.
“You expect this outcome,” Dr. Hampel said in an interview. “Olopatadine is a good drug, mometasone is a good drug, so you’d expect the combination to be good, and it was.”
Dr. Hampel and his colleagues also analyzed changes in baseline scores for patients in the olopatadine group and the mometasone group and compared those changes with those seen in the placebo group. Patients in the mometasone group experienced statistically significant improvement, compared with those in the placebo group (P = .004), and patients in the olopatadine group also appeared to experienced benefit (P = .076).
Adverse events were similar across all treatment groups, although a slightly higher percentage of patients in the GSP301 group (12.9%) and olopatadine groups (12.5%) experienced adverse events, Dr. Hampel said.
Glenmark Pharmaceuticals sponsored the study. Dr. Hampel reported funding from Glenmark Pharmaceuticals and other pharmaceutical companies
SOURCE: Hampel F et al. AAAAI/WAO Joint Congress, Abstract 546.
ORLANDO – Twice-daily treatment with a combination of olopatadine and mometasone showed significant clinical benefit and demonstrated safety for patients with seasonal allergic rhinitis, according to the results of a phase 3 trial.
The combination, known as GSP301, is a fixed-dose nasal spray containing olopatadine, a Food and Drug Adminstration–approved antihistamine, and mometasone, an FDA-approved corticosteroid.
The combination therapy demonstrated statistically significant improvement in scores, compared with those associated with placebo (P less than .001) and olopatadine alone (P = .003). It also showed benefit when compared with mometasone alone (P = .059), Dr. Hampel reported at the joint congress of the American Academy of Allergy, Asthma, and Immunology and the World Allergy Organization.
“You expect this outcome,” Dr. Hampel said in an interview. “Olopatadine is a good drug, mometasone is a good drug, so you’d expect the combination to be good, and it was.”
Dr. Hampel and his colleagues also analyzed changes in baseline scores for patients in the olopatadine group and the mometasone group and compared those changes with those seen in the placebo group. Patients in the mometasone group experienced statistically significant improvement, compared with those in the placebo group (P = .004), and patients in the olopatadine group also appeared to experienced benefit (P = .076).
Adverse events were similar across all treatment groups, although a slightly higher percentage of patients in the GSP301 group (12.9%) and olopatadine groups (12.5%) experienced adverse events, Dr. Hampel said.
Glenmark Pharmaceuticals sponsored the study. Dr. Hampel reported funding from Glenmark Pharmaceuticals and other pharmaceutical companies
SOURCE: Hampel F et al. AAAAI/WAO Joint Congress, Abstract 546.
REPORTING FROM AAAAI/WAO JOINT CONGRESS
Key clinical point: Twice-daily combination therapy improved reflective total nasal symptom scores better than either component alone.
Major finding: Combination therapy significantly improved total nasal symptom scores (P less than .001).
Study details: Phase 3, double-blind, randomized, parallel-group study of 1,180 patients aged 12 years and older with seasonal allergic rhinitis.
Disclosures: Glenmark Pharmaceuticals sponsored the study. Dr. Hampel reported funding from Glenmark Pharmaceuticals and other pharmaceutical companies.
Source: Hampel F et al. AAAAI/WAO Joint Congress, Abstract 546.
The ACA and Multiple Sclerosis
Q) How has the Affordable Care Act affected people living with multiple sclerosis—an Americans with Disabilities Act recognized disease?
The Affordable Care Act (ACA) has been a source of controversy since it became law in 2010. Perhaps some of the tension surrounding it stems from misunderstanding; however, it is clear that individual experiences and/or perceptions flavor the ongoing debate. Rather than perpetuate the contention, we’d simply like to outline some of the ways in which patients with multiple sclerosis (MS) have benefited from the ACA—and what we must do to ensure continued quality and affordability of care in the event of changes to the law.
Living with MS in the United States is costly. According to the National Multiple Sclerosis Society, average annual costs—both direct and indirect (ie, lost wages)—are about $69,000. Health care costs account for more than half of this total (about $39,000). Total costs for all people in the US living with MS are estimated at $28 billion per year.1
In 2016, according to the US Census Bureau, almost 13% of Americans lived below the federal poverty level, and 6% of Americans reported “deep poverty”—defined as household income below 50% of the poverty threshold for that year.2 It has been reported that while at least 90% of people living with MS are insured, 70% are struggling to pay for health care. In fact, 30% put off seeking care because of costs; one consequence is delay in filling prescriptions.3
The burden of expense for our MS patients is considerable. Here’s how the ACA has impacted our patients by attempting to minimize the devastating cost.
Guaranteed Health Insurance Coverage for Pre-existing Conditions. When the ACA became law in March 2010, there were three main goals: making affordable health insurance available to more people, expanding the Medicaid program to cover all adults with income below 138% of the federal poverty level, and supporting innovative medical care delivery methods to lower the cost of health care.4
Following the ACA’s full implementation in 2014, private health insurance companies were prevented from refusing coverage to those with pre-existing conditions, such as MS. This was a game changer, since patients, regardless of their MS diagnosis, were now guaranteed individual insurance. Furthermore, they could not be charged increased premiums based on their prior medical history.5
Preventive Services Covered Without Cost-sharing. Under the ACA, health plans generally must provide preventive services, such as those rated A or B by the US Preventive Services Task Force. This includes routine immunizations for both adults and children, which represents a cost savings to patients living with MS. Another advantage is that women, including those living with MS, have access to sexually transmitted infection screenings, breastfeeding support and supplies, domestic violence screening, and contraceptives.6
Improved Coverage Through Medicare. The ACA mandated improvement in coverage with Medicare Part D benefits. In addition to the preventive care benefits noted above, which apply to Medicare recipients as well, the ACA reduced federal payments to Medicare Advantage plans over time and provided bonus payments to plans with high quality ratings.7
Further changes in Medicare spending included the creation of a 15-person, by-appointment board (known as the Independent Payment Advisory Board) tasked with identifying ways to “modify benefits, eligibility, premiums, or taxes,” which will hopefully continue to optimize the cost of care for patients living with MS and utilizing Medicare.7
Cost Savings With Medicaid Expansion. Medicaid expansion was enacted to keep patients with a costly illness, such as MS, from financial destitution because of their condition. As of January 2018, 32 states and the District of Columbia have seen expansion of their programs.8 In those states, people with a household income below 138% of the poverty level (less than $27,000 for a family of three) can now qualify for Medicaid. States that have not expanded coverage include Idaho, Wyoming, Utah, South Dakota, Nebraska, Kansas, Oklahoma, Texas, Missouri, Wisconsin, Tennessee, Mississippi, Alabama, Georgia, Virginia, North Carolina, South Carolina, and Florida.8 The expansion of Medicaid helps MS patients by shrinking the ever-present gap that still prevents some from qualifying for the additional financial assistance they need due to their chronic illness.
One thing we have learned is that MS patients may not realize they have access to some of these services—particularly preventive care—or they may hesitate to obtain services due to a lack of clarity on whether they are covered. Health care providers can remind patients that they may qualify for “unrealized services,” which could provide value and optimize general preventive care. MS patients with Medicare and Medicaid, for example, may not know that they have access to colorectal cancer screenings via a waived deductible.6
Since last year, there has been vigorous discussion about repealing, replacing, or otherwise amending the ACA. While a political discussion is beyond the bounds of this column, we do need to be aware of how changes to the ACA would affect patients with MS.
Optimizing wellness and prevention and providing access to care to patients with a costly disease, such as MS, is important. In addition to ensuring ongoing access to affordable services, we need to do more to improve mental health access and reduce the cost of needed medications. We also need to close the insurance gap in all 50 states. Continued dialogue will be necessary to help government leaders understand the cost impact of MS (and other diseases), in order to keep our country moving in a positive direction that optimizes wellness and health care reform. —ALD
Amy L. Dix, MPAS, PA-C, MSCS
Department of Neurology at Kansas City Multiple Sclerosis Center in Overland Park, Kansas
1. National Multiple Sclerosis Society. Health Policy Fact Sheet #2: Financial burdens for people with MS, their families, and society. www.nationalmssociety.org/NationalMSSociety/media/MSNationalFiles/Documents/Health-Policy-Fact-Sheet-2-Costs.pdf. Accessed February 8, 2018.
2. Center for Poverty Research, University of California—Davis. What is the current poverty rate in the United States? https://poverty.ucdavis.edu/faq/what-current-poverty-rate-united-states. Accessed February 8, 2018.
3. Iezzoni LI, Ngo L. Health, disability, and life insurance experiences of working-age persons with multiple sclerosis. Mult Scler. 2007;13(4):534-546.
4. Centers for Medicare & Medicaid Services. Affordable Care Act (ACA). HealthCare.gov. www.healthcare.gov/glossary/affordable-care-act. Accessed February 8, 2018.
5. US Department of Health and Human Services. About the ACA: pre-existing conditions. www.hhs.gov/healthcare/about-the-aca/pre-existing-conditions/index.html. Accessed February 8, 2018.
6. Tolbert J. The coverage provisions in the Affordable Care Act: an update. Kaiser Family Foundation. www.kff.org/report-section/the-coverage-provisions-in-the-affordable-care-act-an-update-health-insurance-market-reforms. Accessed February 8, 2018.
7. Kaiser Family Foundation. Focus on health reform: summary of key changes to Medicare in 2010 health reform law. https://kaiserfamilyfoundation.files.wordpress.com/2013/01/7948-02.pdf. Accessed February 8, 2018.
8. Families USA. A 50-state look at Medicaid expansion. http://familiesusa.org/product/50-state-look-medicaid-expansion. Accessed February 8, 2018.
Clinician Reviews in partnership with
MS Consult is edited by Colleen J. Harris, MN, NP, MSCN, Nurse Practitioner/Manager of the Multiple Sclerosis Clinic at Foothills Medical Centre in Calgary, Alberta, Canada, and Bryan Walker, MHS, PA-C, who is in the Department of Neurology, Division of MS and Neuroimmunology, at Duke University Medical Center in Durham, North Carolina. This month's responses were authored by Christen Kutz, PhD, PA-C, who practices at Colorado Springs Neurological Associates, and Amy L. Dix, MPAS, PA-C, MSCS, who practices in the Department of Neurology at Kansas City Multiple Sclerosis Center in Overland Park, Kansas.
Clinician Reviews in partnership with
MS Consult is edited by Colleen J. Harris, MN, NP, MSCN, Nurse Practitioner/Manager of the Multiple Sclerosis Clinic at Foothills Medical Centre in Calgary, Alberta, Canada, and Bryan Walker, MHS, PA-C, who is in the Department of Neurology, Division of MS and Neuroimmunology, at Duke University Medical Center in Durham, North Carolina. This month's responses were authored by Christen Kutz, PhD, PA-C, who practices at Colorado Springs Neurological Associates, and Amy L. Dix, MPAS, PA-C, MSCS, who practices in the Department of Neurology at Kansas City Multiple Sclerosis Center in Overland Park, Kansas.
Clinician Reviews in partnership with
MS Consult is edited by Colleen J. Harris, MN, NP, MSCN, Nurse Practitioner/Manager of the Multiple Sclerosis Clinic at Foothills Medical Centre in Calgary, Alberta, Canada, and Bryan Walker, MHS, PA-C, who is in the Department of Neurology, Division of MS and Neuroimmunology, at Duke University Medical Center in Durham, North Carolina. This month's responses were authored by Christen Kutz, PhD, PA-C, who practices at Colorado Springs Neurological Associates, and Amy L. Dix, MPAS, PA-C, MSCS, who practices in the Department of Neurology at Kansas City Multiple Sclerosis Center in Overland Park, Kansas.
Q) How has the Affordable Care Act affected people living with multiple sclerosis—an Americans with Disabilities Act recognized disease?
The Affordable Care Act (ACA) has been a source of controversy since it became law in 2010. Perhaps some of the tension surrounding it stems from misunderstanding; however, it is clear that individual experiences and/or perceptions flavor the ongoing debate. Rather than perpetuate the contention, we’d simply like to outline some of the ways in which patients with multiple sclerosis (MS) have benefited from the ACA—and what we must do to ensure continued quality and affordability of care in the event of changes to the law.
Living with MS in the United States is costly. According to the National Multiple Sclerosis Society, average annual costs—both direct and indirect (ie, lost wages)—are about $69,000. Health care costs account for more than half of this total (about $39,000). Total costs for all people in the US living with MS are estimated at $28 billion per year.1
In 2016, according to the US Census Bureau, almost 13% of Americans lived below the federal poverty level, and 6% of Americans reported “deep poverty”—defined as household income below 50% of the poverty threshold for that year.2 It has been reported that while at least 90% of people living with MS are insured, 70% are struggling to pay for health care. In fact, 30% put off seeking care because of costs; one consequence is delay in filling prescriptions.3
The burden of expense for our MS patients is considerable. Here’s how the ACA has impacted our patients by attempting to minimize the devastating cost.
Guaranteed Health Insurance Coverage for Pre-existing Conditions. When the ACA became law in March 2010, there were three main goals: making affordable health insurance available to more people, expanding the Medicaid program to cover all adults with income below 138% of the federal poverty level, and supporting innovative medical care delivery methods to lower the cost of health care.4
Following the ACA’s full implementation in 2014, private health insurance companies were prevented from refusing coverage to those with pre-existing conditions, such as MS. This was a game changer, since patients, regardless of their MS diagnosis, were now guaranteed individual insurance. Furthermore, they could not be charged increased premiums based on their prior medical history.5
Preventive Services Covered Without Cost-sharing. Under the ACA, health plans generally must provide preventive services, such as those rated A or B by the US Preventive Services Task Force. This includes routine immunizations for both adults and children, which represents a cost savings to patients living with MS. Another advantage is that women, including those living with MS, have access to sexually transmitted infection screenings, breastfeeding support and supplies, domestic violence screening, and contraceptives.6
Improved Coverage Through Medicare. The ACA mandated improvement in coverage with Medicare Part D benefits. In addition to the preventive care benefits noted above, which apply to Medicare recipients as well, the ACA reduced federal payments to Medicare Advantage plans over time and provided bonus payments to plans with high quality ratings.7
Further changes in Medicare spending included the creation of a 15-person, by-appointment board (known as the Independent Payment Advisory Board) tasked with identifying ways to “modify benefits, eligibility, premiums, or taxes,” which will hopefully continue to optimize the cost of care for patients living with MS and utilizing Medicare.7
Cost Savings With Medicaid Expansion. Medicaid expansion was enacted to keep patients with a costly illness, such as MS, from financial destitution because of their condition. As of January 2018, 32 states and the District of Columbia have seen expansion of their programs.8 In those states, people with a household income below 138% of the poverty level (less than $27,000 for a family of three) can now qualify for Medicaid. States that have not expanded coverage include Idaho, Wyoming, Utah, South Dakota, Nebraska, Kansas, Oklahoma, Texas, Missouri, Wisconsin, Tennessee, Mississippi, Alabama, Georgia, Virginia, North Carolina, South Carolina, and Florida.8 The expansion of Medicaid helps MS patients by shrinking the ever-present gap that still prevents some from qualifying for the additional financial assistance they need due to their chronic illness.
One thing we have learned is that MS patients may not realize they have access to some of these services—particularly preventive care—or they may hesitate to obtain services due to a lack of clarity on whether they are covered. Health care providers can remind patients that they may qualify for “unrealized services,” which could provide value and optimize general preventive care. MS patients with Medicare and Medicaid, for example, may not know that they have access to colorectal cancer screenings via a waived deductible.6
Since last year, there has been vigorous discussion about repealing, replacing, or otherwise amending the ACA. While a political discussion is beyond the bounds of this column, we do need to be aware of how changes to the ACA would affect patients with MS.
Optimizing wellness and prevention and providing access to care to patients with a costly disease, such as MS, is important. In addition to ensuring ongoing access to affordable services, we need to do more to improve mental health access and reduce the cost of needed medications. We also need to close the insurance gap in all 50 states. Continued dialogue will be necessary to help government leaders understand the cost impact of MS (and other diseases), in order to keep our country moving in a positive direction that optimizes wellness and health care reform. —ALD
Amy L. Dix, MPAS, PA-C, MSCS
Department of Neurology at Kansas City Multiple Sclerosis Center in Overland Park, Kansas
Q) How has the Affordable Care Act affected people living with multiple sclerosis—an Americans with Disabilities Act recognized disease?
The Affordable Care Act (ACA) has been a source of controversy since it became law in 2010. Perhaps some of the tension surrounding it stems from misunderstanding; however, it is clear that individual experiences and/or perceptions flavor the ongoing debate. Rather than perpetuate the contention, we’d simply like to outline some of the ways in which patients with multiple sclerosis (MS) have benefited from the ACA—and what we must do to ensure continued quality and affordability of care in the event of changes to the law.
Living with MS in the United States is costly. According to the National Multiple Sclerosis Society, average annual costs—both direct and indirect (ie, lost wages)—are about $69,000. Health care costs account for more than half of this total (about $39,000). Total costs for all people in the US living with MS are estimated at $28 billion per year.1
In 2016, according to the US Census Bureau, almost 13% of Americans lived below the federal poverty level, and 6% of Americans reported “deep poverty”—defined as household income below 50% of the poverty threshold for that year.2 It has been reported that while at least 90% of people living with MS are insured, 70% are struggling to pay for health care. In fact, 30% put off seeking care because of costs; one consequence is delay in filling prescriptions.3
The burden of expense for our MS patients is considerable. Here’s how the ACA has impacted our patients by attempting to minimize the devastating cost.
Guaranteed Health Insurance Coverage for Pre-existing Conditions. When the ACA became law in March 2010, there were three main goals: making affordable health insurance available to more people, expanding the Medicaid program to cover all adults with income below 138% of the federal poverty level, and supporting innovative medical care delivery methods to lower the cost of health care.4
Following the ACA’s full implementation in 2014, private health insurance companies were prevented from refusing coverage to those with pre-existing conditions, such as MS. This was a game changer, since patients, regardless of their MS diagnosis, were now guaranteed individual insurance. Furthermore, they could not be charged increased premiums based on their prior medical history.5
Preventive Services Covered Without Cost-sharing. Under the ACA, health plans generally must provide preventive services, such as those rated A or B by the US Preventive Services Task Force. This includes routine immunizations for both adults and children, which represents a cost savings to patients living with MS. Another advantage is that women, including those living with MS, have access to sexually transmitted infection screenings, breastfeeding support and supplies, domestic violence screening, and contraceptives.6
Improved Coverage Through Medicare. The ACA mandated improvement in coverage with Medicare Part D benefits. In addition to the preventive care benefits noted above, which apply to Medicare recipients as well, the ACA reduced federal payments to Medicare Advantage plans over time and provided bonus payments to plans with high quality ratings.7
Further changes in Medicare spending included the creation of a 15-person, by-appointment board (known as the Independent Payment Advisory Board) tasked with identifying ways to “modify benefits, eligibility, premiums, or taxes,” which will hopefully continue to optimize the cost of care for patients living with MS and utilizing Medicare.7
Cost Savings With Medicaid Expansion. Medicaid expansion was enacted to keep patients with a costly illness, such as MS, from financial destitution because of their condition. As of January 2018, 32 states and the District of Columbia have seen expansion of their programs.8 In those states, people with a household income below 138% of the poverty level (less than $27,000 for a family of three) can now qualify for Medicaid. States that have not expanded coverage include Idaho, Wyoming, Utah, South Dakota, Nebraska, Kansas, Oklahoma, Texas, Missouri, Wisconsin, Tennessee, Mississippi, Alabama, Georgia, Virginia, North Carolina, South Carolina, and Florida.8 The expansion of Medicaid helps MS patients by shrinking the ever-present gap that still prevents some from qualifying for the additional financial assistance they need due to their chronic illness.
One thing we have learned is that MS patients may not realize they have access to some of these services—particularly preventive care—or they may hesitate to obtain services due to a lack of clarity on whether they are covered. Health care providers can remind patients that they may qualify for “unrealized services,” which could provide value and optimize general preventive care. MS patients with Medicare and Medicaid, for example, may not know that they have access to colorectal cancer screenings via a waived deductible.6
Since last year, there has been vigorous discussion about repealing, replacing, or otherwise amending the ACA. While a political discussion is beyond the bounds of this column, we do need to be aware of how changes to the ACA would affect patients with MS.
Optimizing wellness and prevention and providing access to care to patients with a costly disease, such as MS, is important. In addition to ensuring ongoing access to affordable services, we need to do more to improve mental health access and reduce the cost of needed medications. We also need to close the insurance gap in all 50 states. Continued dialogue will be necessary to help government leaders understand the cost impact of MS (and other diseases), in order to keep our country moving in a positive direction that optimizes wellness and health care reform. —ALD
Amy L. Dix, MPAS, PA-C, MSCS
Department of Neurology at Kansas City Multiple Sclerosis Center in Overland Park, Kansas
1. National Multiple Sclerosis Society. Health Policy Fact Sheet #2: Financial burdens for people with MS, their families, and society. www.nationalmssociety.org/NationalMSSociety/media/MSNationalFiles/Documents/Health-Policy-Fact-Sheet-2-Costs.pdf. Accessed February 8, 2018.
2. Center for Poverty Research, University of California—Davis. What is the current poverty rate in the United States? https://poverty.ucdavis.edu/faq/what-current-poverty-rate-united-states. Accessed February 8, 2018.
3. Iezzoni LI, Ngo L. Health, disability, and life insurance experiences of working-age persons with multiple sclerosis. Mult Scler. 2007;13(4):534-546.
4. Centers for Medicare & Medicaid Services. Affordable Care Act (ACA). HealthCare.gov. www.healthcare.gov/glossary/affordable-care-act. Accessed February 8, 2018.
5. US Department of Health and Human Services. About the ACA: pre-existing conditions. www.hhs.gov/healthcare/about-the-aca/pre-existing-conditions/index.html. Accessed February 8, 2018.
6. Tolbert J. The coverage provisions in the Affordable Care Act: an update. Kaiser Family Foundation. www.kff.org/report-section/the-coverage-provisions-in-the-affordable-care-act-an-update-health-insurance-market-reforms. Accessed February 8, 2018.
7. Kaiser Family Foundation. Focus on health reform: summary of key changes to Medicare in 2010 health reform law. https://kaiserfamilyfoundation.files.wordpress.com/2013/01/7948-02.pdf. Accessed February 8, 2018.
8. Families USA. A 50-state look at Medicaid expansion. http://familiesusa.org/product/50-state-look-medicaid-expansion. Accessed February 8, 2018.
1. National Multiple Sclerosis Society. Health Policy Fact Sheet #2: Financial burdens for people with MS, their families, and society. www.nationalmssociety.org/NationalMSSociety/media/MSNationalFiles/Documents/Health-Policy-Fact-Sheet-2-Costs.pdf. Accessed February 8, 2018.
2. Center for Poverty Research, University of California—Davis. What is the current poverty rate in the United States? https://poverty.ucdavis.edu/faq/what-current-poverty-rate-united-states. Accessed February 8, 2018.
3. Iezzoni LI, Ngo L. Health, disability, and life insurance experiences of working-age persons with multiple sclerosis. Mult Scler. 2007;13(4):534-546.
4. Centers for Medicare & Medicaid Services. Affordable Care Act (ACA). HealthCare.gov. www.healthcare.gov/glossary/affordable-care-act. Accessed February 8, 2018.
5. US Department of Health and Human Services. About the ACA: pre-existing conditions. www.hhs.gov/healthcare/about-the-aca/pre-existing-conditions/index.html. Accessed February 8, 2018.
6. Tolbert J. The coverage provisions in the Affordable Care Act: an update. Kaiser Family Foundation. www.kff.org/report-section/the-coverage-provisions-in-the-affordable-care-act-an-update-health-insurance-market-reforms. Accessed February 8, 2018.
7. Kaiser Family Foundation. Focus on health reform: summary of key changes to Medicare in 2010 health reform law. https://kaiserfamilyfoundation.files.wordpress.com/2013/01/7948-02.pdf. Accessed February 8, 2018.
8. Families USA. A 50-state look at Medicaid expansion. http://familiesusa.org/product/50-state-look-medicaid-expansion. Accessed February 8, 2018.