User login
Monitoring for Infection in MS Patients
Q) How do you monitor for infection in patients with multiple sclerosis who take disease-modifying therapies?
The answer to this question is “it depends”—on several factors, including current and previous use of disease-modifying therapies (DMTs), concomitant medications, comorbidities, vaccination history, presence of John Cunningham virus (JCV) antibodies (in the case of natalizumab use), and prior or current use of immunosuppressive therapies.
There are many FDA-approved DMTs for multiple sclerosis (MS). Each has a different rate of infection occurring in clinical trials and varying requirements and/or recommendations for safety monitoring. The package inserts for each DMT offer some guidance for clinicians.
Injectable therapies. For two interferon therapies—interferon ß-1b SC and interferon ß-1a—it is recommended to order a complete blood count (CBC), blood chemistry, and liver function tests (LFTs) at baseline, then again at one, three, and six months, and then at clinician discretion thereafter.1,2 For peginterferon ß-1a, ordering a CBC, basic chemistry, and LFTs, at the clinician’s discretion, is advised.3 The package insert for interferon ß-1a IM does not offer specific recommendations for routine safety monitoring.4
The package insert for glatiramer acetate offers no recommendations for routine safety monitoring.5
In patients for whom two or more DMTs have failed to work, the monoclonal antibody daclizumab may be indicated. Compared to placebo and active comparator, this drug was associated with a higher risk for infection in clinical trials. The most commonly observed types were upper respiratory, urinary tract, and viral infections. There are no recommendations for CBC monitoring with daclizumab, but monthly LFTs are required due to increased risk for hepatic injury.6
Oral DMTs. Patients taking fingolimod, teriflunomide, and dimethyl fumarate have increased risk for infection; as a result, there are more safety monitoring recommendations for these medications.7-9
Prior to starting therapy with fingolimod, baseline CBC, blood chemistries, and varicella antibody testing should be done. During therapy, routine CBC testing and LFTs are advised at the clinician’s discretion or if the patient exhibits signs and symptoms of infection (see Table). In clinical trials, fingolimod use was interrupted if the lymphocyte count was sustained at < 200. In rare cases, progressive multifocal leukoencephalopathy (PML) has occurred—so the patient’s age, JCV antibody status, prior use of immunosuppressant therapy, and length of fingolimod treatment should be taken into consideration.7

Patients starting teriflunomide should have baseline LFTs and CBC and tuberculosis (TB) testing (either skin or serum), with subsequent monthly LFTs for the first six months on treatment. Some patients may experience neutropenia, thrombocytopenia, and lymphopenia. As a result, patients may have an increased risk for infection. Safety monitoring is at the clinician’s discretion.8
For patients initiating dimethyl fumarate, a baseline CBC is recommended, to be repeated every six to 12 months thereafter, and/or as clinically indicated. Since lymphopenia may occur, consider interruption of dimethyl fumarate in patients with lymphocyte counts < 0.5 persisting for more than six months. Rare cases of PML have also occurred; at the first suggestive sign or symptom, dimethyl fumarate should be withheld and appropriate diagnostic testing should be completed.9
Infusion therapies. There are four infusion therapies available for MS treatment. Mitoxantrone, though not commonly used, is still available for relapsing and secondary progressive forms of MS. Common infections seen in clinical trials include upper respiratory, urinary tract, and sinus infections. A CBC, including platelets, should be obtained prior to each course of mitoxantrone and again if signs and symptoms of infection develop.10
Natalizumab is an integrin receptor antagonist administered in monthly IV infusions. Patients receiving natalizumab may have increased risk for urinary tract infections, lower respiratory infections, gastroenteritis, vaginitis, and herpes infections. These risks should be monitored at the clinician’s discretion. There have been several cases of PML associated with natalizumab; risk factors include duration of therapy, prior use of immunosuppressants, and presence of JCV antibodies.11
Alemtuzumab is a CD52-directed monoclonal antibody indicated in patients with relapsing forms of MS who have had an inadequate response to at least two DMTs. In clinical trials, subjects had a higher risk for nasopharyngitis, urinary tract infections, upper respiratory infections, sinusitis, herpetic infections, influenza, and bronchitis. Due to the increased risk for infection and secondary autoimmunities, patients are required to have monthly CBC testing, LFTs, and urinalysis for up to 48 months after their last infusion.12
Lastly, ocrelizumab is a CD20-directed cytolytic antibody for the treatment of relapsing and progressive forms of MS. In clinical trials, there was a higher incidence of upper and lower respiratory infections, skin infections, and herpes-related infections. Prior to initiating ocrelizumab, hepatitis B virus screening should be completed. There are no specific recommendations for routine monitoring during therapy, although providers should monitor patients clinically for any signs and symptoms of infection.13
A word of caution: The common signs and symptoms of infection are listed in the Table. If these symptoms are present in your patient, consider ordering diagnostic testing to evaluate for infection.
Symptoms of PML include progressive unilateral weakness, clumsiness of limbs, disturbance of vision, and changes in thinking, memory, and orientation leading to confusion and personality changes. At the first sign or symptom suggestive of PML, the DMT should be discontinued and diagnostic testing performed.
Providers may contact the manufacturer directly for further guidance on DMT surveillance and treatment protocols. —CK
Christen Kutz, PhD, PA-C
Colorado Springs Neurological Associates
1. Betaseron (interferon [b]-1b) [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals; 1993.
2. Rebif (interferon [b]-1a) [package insert]. Rockland, MA: EMD Serono; revised 2015.
3. Plegridy (peginterferon [b]-1a) [package insert]. Cambridge, MA: Biogen Idec; 2013.
4. Avonex (interferon [b] -1a) [package insert]. Cambridge, MA: Biogen Inc.; 1996.
5. Copaxone (glatiramer acetate) [package insert]. Overland Park, KS: Teva Neuroscience; revised 2016.
6. Zinbryta (daclizumab) [package insert]. Cambridge, MA: Biogen Idec; 2016.
7. Gilenya (fingolimod) [package insert]. Hanover, NJ: Novartis; revised 2016.
8. Aubagio (teriflunomide) [package insert]. Cambridge, MA: Genzyme Corporation; revised 2016.
9. Tecfidera (dimethyl fumarate) [package insert]. Cambridge, MA: Biogen Idec; revised 2017.
10. Novantrone (mitoxantrone) [package insert]. Rockland, MA: EMD Serono; 2008.
11. Tysabri (natalizumab) [package insert]. Cambridge, MA: Biogen Idec; revised 2017.
12. Lemtrada (alemtuzumab) [package insert]. Cambridge, MA: Genzyme Corporation; revised 2017.
13. Ocrevus (ocrelizumab) [package insert]. San Francisco, CA: Genentech; 2017.
Clinician Reviews in partnership with
MS Consult is edited by Colleen J. Harris, MN, NP, MSCN, Nurse Practitioner/Manager of the Multiple Sclerosis Clinic at Foothills Medical Centre in Calgary, Alberta, Canada, and Bryan Walker, MHS, PA-C, who is in the Department of Neurology, Division of MS and Neuroimmunology, at Duke University Medical Center in Durham, North Carolina. This month's responses were authored by Christen Kutz, PhD, PA-C, who practices at Colorado Springs Neurological Associates, and Amy L. Dix, MPAS, PA-C, MSCS, who practices in the Department of Neurology at Kansas City Multiple Sclerosis Center in Overland Park, Kansas.
Clinician Reviews in partnership with
MS Consult is edited by Colleen J. Harris, MN, NP, MSCN, Nurse Practitioner/Manager of the Multiple Sclerosis Clinic at Foothills Medical Centre in Calgary, Alberta, Canada, and Bryan Walker, MHS, PA-C, who is in the Department of Neurology, Division of MS and Neuroimmunology, at Duke University Medical Center in Durham, North Carolina. This month's responses were authored by Christen Kutz, PhD, PA-C, who practices at Colorado Springs Neurological Associates, and Amy L. Dix, MPAS, PA-C, MSCS, who practices in the Department of Neurology at Kansas City Multiple Sclerosis Center in Overland Park, Kansas.
Clinician Reviews in partnership with
MS Consult is edited by Colleen J. Harris, MN, NP, MSCN, Nurse Practitioner/Manager of the Multiple Sclerosis Clinic at Foothills Medical Centre in Calgary, Alberta, Canada, and Bryan Walker, MHS, PA-C, who is in the Department of Neurology, Division of MS and Neuroimmunology, at Duke University Medical Center in Durham, North Carolina. This month's responses were authored by Christen Kutz, PhD, PA-C, who practices at Colorado Springs Neurological Associates, and Amy L. Dix, MPAS, PA-C, MSCS, who practices in the Department of Neurology at Kansas City Multiple Sclerosis Center in Overland Park, Kansas.
Q) How do you monitor for infection in patients with multiple sclerosis who take disease-modifying therapies?
The answer to this question is “it depends”—on several factors, including current and previous use of disease-modifying therapies (DMTs), concomitant medications, comorbidities, vaccination history, presence of John Cunningham virus (JCV) antibodies (in the case of natalizumab use), and prior or current use of immunosuppressive therapies.
There are many FDA-approved DMTs for multiple sclerosis (MS). Each has a different rate of infection occurring in clinical trials and varying requirements and/or recommendations for safety monitoring. The package inserts for each DMT offer some guidance for clinicians.
Injectable therapies. For two interferon therapies—interferon ß-1b SC and interferon ß-1a—it is recommended to order a complete blood count (CBC), blood chemistry, and liver function tests (LFTs) at baseline, then again at one, three, and six months, and then at clinician discretion thereafter.1,2 For peginterferon ß-1a, ordering a CBC, basic chemistry, and LFTs, at the clinician’s discretion, is advised.3 The package insert for interferon ß-1a IM does not offer specific recommendations for routine safety monitoring.4
The package insert for glatiramer acetate offers no recommendations for routine safety monitoring.5
In patients for whom two or more DMTs have failed to work, the monoclonal antibody daclizumab may be indicated. Compared to placebo and active comparator, this drug was associated with a higher risk for infection in clinical trials. The most commonly observed types were upper respiratory, urinary tract, and viral infections. There are no recommendations for CBC monitoring with daclizumab, but monthly LFTs are required due to increased risk for hepatic injury.6
Oral DMTs. Patients taking fingolimod, teriflunomide, and dimethyl fumarate have increased risk for infection; as a result, there are more safety monitoring recommendations for these medications.7-9
Prior to starting therapy with fingolimod, baseline CBC, blood chemistries, and varicella antibody testing should be done. During therapy, routine CBC testing and LFTs are advised at the clinician’s discretion or if the patient exhibits signs and symptoms of infection (see Table). In clinical trials, fingolimod use was interrupted if the lymphocyte count was sustained at < 200. In rare cases, progressive multifocal leukoencephalopathy (PML) has occurred—so the patient’s age, JCV antibody status, prior use of immunosuppressant therapy, and length of fingolimod treatment should be taken into consideration.7
Patients starting teriflunomide should have baseline LFTs and CBC and tuberculosis (TB) testing (either skin or serum), with subsequent monthly LFTs for the first six months on treatment. Some patients may experience neutropenia, thrombocytopenia, and lymphopenia. As a result, patients may have an increased risk for infection. Safety monitoring is at the clinician’s discretion.8
For patients initiating dimethyl fumarate, a baseline CBC is recommended, to be repeated every six to 12 months thereafter, and/or as clinically indicated. Since lymphopenia may occur, consider interruption of dimethyl fumarate in patients with lymphocyte counts < 0.5 persisting for more than six months. Rare cases of PML have also occurred; at the first suggestive sign or symptom, dimethyl fumarate should be withheld and appropriate diagnostic testing should be completed.9
Infusion therapies. There are four infusion therapies available for MS treatment. Mitoxantrone, though not commonly used, is still available for relapsing and secondary progressive forms of MS. Common infections seen in clinical trials include upper respiratory, urinary tract, and sinus infections. A CBC, including platelets, should be obtained prior to each course of mitoxantrone and again if signs and symptoms of infection develop.10
Natalizumab is an integrin receptor antagonist administered in monthly IV infusions. Patients receiving natalizumab may have increased risk for urinary tract infections, lower respiratory infections, gastroenteritis, vaginitis, and herpes infections. These risks should be monitored at the clinician’s discretion. There have been several cases of PML associated with natalizumab; risk factors include duration of therapy, prior use of immunosuppressants, and presence of JCV antibodies.11
Alemtuzumab is a CD52-directed monoclonal antibody indicated in patients with relapsing forms of MS who have had an inadequate response to at least two DMTs. In clinical trials, subjects had a higher risk for nasopharyngitis, urinary tract infections, upper respiratory infections, sinusitis, herpetic infections, influenza, and bronchitis. Due to the increased risk for infection and secondary autoimmunities, patients are required to have monthly CBC testing, LFTs, and urinalysis for up to 48 months after their last infusion.12
Lastly, ocrelizumab is a CD20-directed cytolytic antibody for the treatment of relapsing and progressive forms of MS. In clinical trials, there was a higher incidence of upper and lower respiratory infections, skin infections, and herpes-related infections. Prior to initiating ocrelizumab, hepatitis B virus screening should be completed. There are no specific recommendations for routine monitoring during therapy, although providers should monitor patients clinically for any signs and symptoms of infection.13
A word of caution: The common signs and symptoms of infection are listed in the Table. If these symptoms are present in your patient, consider ordering diagnostic testing to evaluate for infection.
Symptoms of PML include progressive unilateral weakness, clumsiness of limbs, disturbance of vision, and changes in thinking, memory, and orientation leading to confusion and personality changes. At the first sign or symptom suggestive of PML, the DMT should be discontinued and diagnostic testing performed.
Providers may contact the manufacturer directly for further guidance on DMT surveillance and treatment protocols. —CK
Christen Kutz, PhD, PA-C
Colorado Springs Neurological Associates
Q) How do you monitor for infection in patients with multiple sclerosis who take disease-modifying therapies?
The answer to this question is “it depends”—on several factors, including current and previous use of disease-modifying therapies (DMTs), concomitant medications, comorbidities, vaccination history, presence of John Cunningham virus (JCV) antibodies (in the case of natalizumab use), and prior or current use of immunosuppressive therapies.
There are many FDA-approved DMTs for multiple sclerosis (MS). Each has a different rate of infection occurring in clinical trials and varying requirements and/or recommendations for safety monitoring. The package inserts for each DMT offer some guidance for clinicians.
Injectable therapies. For two interferon therapies—interferon ß-1b SC and interferon ß-1a—it is recommended to order a complete blood count (CBC), blood chemistry, and liver function tests (LFTs) at baseline, then again at one, three, and six months, and then at clinician discretion thereafter.1,2 For peginterferon ß-1a, ordering a CBC, basic chemistry, and LFTs, at the clinician’s discretion, is advised.3 The package insert for interferon ß-1a IM does not offer specific recommendations for routine safety monitoring.4
The package insert for glatiramer acetate offers no recommendations for routine safety monitoring.5
In patients for whom two or more DMTs have failed to work, the monoclonal antibody daclizumab may be indicated. Compared to placebo and active comparator, this drug was associated with a higher risk for infection in clinical trials. The most commonly observed types were upper respiratory, urinary tract, and viral infections. There are no recommendations for CBC monitoring with daclizumab, but monthly LFTs are required due to increased risk for hepatic injury.6
Oral DMTs. Patients taking fingolimod, teriflunomide, and dimethyl fumarate have increased risk for infection; as a result, there are more safety monitoring recommendations for these medications.7-9
Prior to starting therapy with fingolimod, baseline CBC, blood chemistries, and varicella antibody testing should be done. During therapy, routine CBC testing and LFTs are advised at the clinician’s discretion or if the patient exhibits signs and symptoms of infection (see Table). In clinical trials, fingolimod use was interrupted if the lymphocyte count was sustained at < 200. In rare cases, progressive multifocal leukoencephalopathy (PML) has occurred—so the patient’s age, JCV antibody status, prior use of immunosuppressant therapy, and length of fingolimod treatment should be taken into consideration.7
Patients starting teriflunomide should have baseline LFTs and CBC and tuberculosis (TB) testing (either skin or serum), with subsequent monthly LFTs for the first six months on treatment. Some patients may experience neutropenia, thrombocytopenia, and lymphopenia. As a result, patients may have an increased risk for infection. Safety monitoring is at the clinician’s discretion.8
For patients initiating dimethyl fumarate, a baseline CBC is recommended, to be repeated every six to 12 months thereafter, and/or as clinically indicated. Since lymphopenia may occur, consider interruption of dimethyl fumarate in patients with lymphocyte counts < 0.5 persisting for more than six months. Rare cases of PML have also occurred; at the first suggestive sign or symptom, dimethyl fumarate should be withheld and appropriate diagnostic testing should be completed.9
Infusion therapies. There are four infusion therapies available for MS treatment. Mitoxantrone, though not commonly used, is still available for relapsing and secondary progressive forms of MS. Common infections seen in clinical trials include upper respiratory, urinary tract, and sinus infections. A CBC, including platelets, should be obtained prior to each course of mitoxantrone and again if signs and symptoms of infection develop.10
Natalizumab is an integrin receptor antagonist administered in monthly IV infusions. Patients receiving natalizumab may have increased risk for urinary tract infections, lower respiratory infections, gastroenteritis, vaginitis, and herpes infections. These risks should be monitored at the clinician’s discretion. There have been several cases of PML associated with natalizumab; risk factors include duration of therapy, prior use of immunosuppressants, and presence of JCV antibodies.11
Alemtuzumab is a CD52-directed monoclonal antibody indicated in patients with relapsing forms of MS who have had an inadequate response to at least two DMTs. In clinical trials, subjects had a higher risk for nasopharyngitis, urinary tract infections, upper respiratory infections, sinusitis, herpetic infections, influenza, and bronchitis. Due to the increased risk for infection and secondary autoimmunities, patients are required to have monthly CBC testing, LFTs, and urinalysis for up to 48 months after their last infusion.12
Lastly, ocrelizumab is a CD20-directed cytolytic antibody for the treatment of relapsing and progressive forms of MS. In clinical trials, there was a higher incidence of upper and lower respiratory infections, skin infections, and herpes-related infections. Prior to initiating ocrelizumab, hepatitis B virus screening should be completed. There are no specific recommendations for routine monitoring during therapy, although providers should monitor patients clinically for any signs and symptoms of infection.13
A word of caution: The common signs and symptoms of infection are listed in the Table. If these symptoms are present in your patient, consider ordering diagnostic testing to evaluate for infection.
Symptoms of PML include progressive unilateral weakness, clumsiness of limbs, disturbance of vision, and changes in thinking, memory, and orientation leading to confusion and personality changes. At the first sign or symptom suggestive of PML, the DMT should be discontinued and diagnostic testing performed.
Providers may contact the manufacturer directly for further guidance on DMT surveillance and treatment protocols. —CK
Christen Kutz, PhD, PA-C
Colorado Springs Neurological Associates
1. Betaseron (interferon [b]-1b) [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals; 1993.
2. Rebif (interferon [b]-1a) [package insert]. Rockland, MA: EMD Serono; revised 2015.
3. Plegridy (peginterferon [b]-1a) [package insert]. Cambridge, MA: Biogen Idec; 2013.
4. Avonex (interferon [b] -1a) [package insert]. Cambridge, MA: Biogen Inc.; 1996.
5. Copaxone (glatiramer acetate) [package insert]. Overland Park, KS: Teva Neuroscience; revised 2016.
6. Zinbryta (daclizumab) [package insert]. Cambridge, MA: Biogen Idec; 2016.
7. Gilenya (fingolimod) [package insert]. Hanover, NJ: Novartis; revised 2016.
8. Aubagio (teriflunomide) [package insert]. Cambridge, MA: Genzyme Corporation; revised 2016.
9. Tecfidera (dimethyl fumarate) [package insert]. Cambridge, MA: Biogen Idec; revised 2017.
10. Novantrone (mitoxantrone) [package insert]. Rockland, MA: EMD Serono; 2008.
11. Tysabri (natalizumab) [package insert]. Cambridge, MA: Biogen Idec; revised 2017.
12. Lemtrada (alemtuzumab) [package insert]. Cambridge, MA: Genzyme Corporation; revised 2017.
13. Ocrevus (ocrelizumab) [package insert]. San Francisco, CA: Genentech; 2017.
1. Betaseron (interferon [b]-1b) [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals; 1993.
2. Rebif (interferon [b]-1a) [package insert]. Rockland, MA: EMD Serono; revised 2015.
3. Plegridy (peginterferon [b]-1a) [package insert]. Cambridge, MA: Biogen Idec; 2013.
4. Avonex (interferon [b] -1a) [package insert]. Cambridge, MA: Biogen Inc.; 1996.
5. Copaxone (glatiramer acetate) [package insert]. Overland Park, KS: Teva Neuroscience; revised 2016.
6. Zinbryta (daclizumab) [package insert]. Cambridge, MA: Biogen Idec; 2016.
7. Gilenya (fingolimod) [package insert]. Hanover, NJ: Novartis; revised 2016.
8. Aubagio (teriflunomide) [package insert]. Cambridge, MA: Genzyme Corporation; revised 2016.
9. Tecfidera (dimethyl fumarate) [package insert]. Cambridge, MA: Biogen Idec; revised 2017.
10. Novantrone (mitoxantrone) [package insert]. Rockland, MA: EMD Serono; 2008.
11. Tysabri (natalizumab) [package insert]. Cambridge, MA: Biogen Idec; revised 2017.
12. Lemtrada (alemtuzumab) [package insert]. Cambridge, MA: Genzyme Corporation; revised 2017.
13. Ocrevus (ocrelizumab) [package insert]. San Francisco, CA: Genentech; 2017.
Macrophage activation syndrome’s impact in childhood SLE felt mostly early
Nearly 10% of children with systemic lupus erythematosus (SLE) developed macrophage activation syndrome (MAS) at some point during a mean follow-up time of more than 3 years at one center, and most were concomitantly diagnosed with the syndrome.
Although the investigators from the University of Toronto reported significantly higher mortality among patients with MAS, most cases were successfully treated with corticosteroids, and no relapses were observed during follow-up.
MAS was first identified in patients with juvenile idiopathic arthritis and is most well known as a complication of that broadly named disease, but data on outcomes and disease course in SLE patients are limited, first author Roberto Ezequiel Borgia, MD, and his colleagues wrote in their report in Arthritis & Rheumatology.
The researchers identified 403 children with SLE seen at the Hospital for Sick Children in Toronto during 2002-2012. Overall, 38 patients (9%) had MAS; of those patients, 68% received a MAS diagnosis within 7 days of the SLE diagnosis – termed “concomitant” diagnosis – while another 29% received a MAS diagnosis within 180 days of their SLE diagnosis.
The researchers explained that “since there are no validated nor universally accepted diagnostic criteria for MAS in SLE, the definition of MAS was based on the treating pediatric rheumatologist’s expert opinion at the time of the initial presentation.” The most common presenting feature of MAS was fever (100%), followed by generalized lymphadenopathy (24%), hepatomegaly (18%), CNS dysfunction secondary to MAS (18%), hemorrhage (13%), and splenomegaly (10%).
The average age of the children at diagnosis was nearly 14 years, and 79% were female. The average follow-up was 3.5 years. There were no significant differences in the demographic features of children with and without MAS nor were there any in variables used to assess lupus outcomes, which included immunosuppressive drug use, average daily corticosteroid dose (18.3 mg/day with MAS vs. 18.6 mg/day without MAS), and the number of pediatric ICU visits (incidence rate ratio for MAS vs. non-MAS, 1.60 [95% CI, 0.74-3.18]).
Mortality was significantly higher in children with MAS, compared with those without MAS (5.3% vs. 0.3%; P = .02), although the overall number of deaths in the cohort was small (n = 3). Apart from the “acute illness which was associated with 2 deaths secondary to MAS,” the investigators said that they “did not find any significant differences in the number of deaths or damage accrual between the cohorts, including overall SLICC [Systemic Lupus International Collaborating Clinics] damage score or any specific damage feature within the score.”
The study findings were limited by several factors including the lack of validated MAS criteria for children with SLE and a lack of follow-up data on the patients beyond 18 years of age, the researchers said.
The results suggest that MAS remains a life-threatening complication in children with SLE and should be considered an important cause of mortality for them, but “if the initial presentation does not result in death, the long-term outcome seem[s] to be comparable to those without MAS,” the investigators wrote.
The researchers had no financial conflicts to disclose.
SOURCE: Borgia R et al. Arthritis Rheumatol. 2018 Jan 17. doi: 10.1002/art.40417
As we learn more about the role of macrophage activation syndrome (MAS), a secondary form of hemophagocytic lymphohistiocytosis in rheumatic diseases, it has become clear that patients may develop this syndrome in a variety of settings. The most common presentation of MAS is in association with systemic onset juvenile idiopathic arthritis, but is has been described in other forms of childhood rheumatic diseases, including other types of juvenile idiopathic arthritis, lupus, mixed connective tissue disease, Kawasaki disease, and sarcoidosis. Study of secondary MAS has led to suggested diagnostic criteria; however, those criteria are very similar to the presentation of adult and childhood systemic lupus with cytopenias, hepatitis, and coagulopathy.

The work by Borgia et al. encourages us to look for evidence of MAS in our lupus patients as it allows us to identify patients at risk for poor outcomes and to provide interventions to reduce those risks.
Marisa S. Klein-Gitelman, MD , is a professor of pediatrics at Northwestern University, Chicago, and is a pediatric rheumatologist at the Ann & Robert H. Lurie Children’s Hospital of Chicago. She has no relevant disclosures.
As we learn more about the role of macrophage activation syndrome (MAS), a secondary form of hemophagocytic lymphohistiocytosis in rheumatic diseases, it has become clear that patients may develop this syndrome in a variety of settings. The most common presentation of MAS is in association with systemic onset juvenile idiopathic arthritis, but is has been described in other forms of childhood rheumatic diseases, including other types of juvenile idiopathic arthritis, lupus, mixed connective tissue disease, Kawasaki disease, and sarcoidosis. Study of secondary MAS has led to suggested diagnostic criteria; however, those criteria are very similar to the presentation of adult and childhood systemic lupus with cytopenias, hepatitis, and coagulopathy.
The work by Borgia et al. encourages us to look for evidence of MAS in our lupus patients as it allows us to identify patients at risk for poor outcomes and to provide interventions to reduce those risks.
Marisa S. Klein-Gitelman, MD , is a professor of pediatrics at Northwestern University, Chicago, and is a pediatric rheumatologist at the Ann & Robert H. Lurie Children’s Hospital of Chicago. She has no relevant disclosures.
As we learn more about the role of macrophage activation syndrome (MAS), a secondary form of hemophagocytic lymphohistiocytosis in rheumatic diseases, it has become clear that patients may develop this syndrome in a variety of settings. The most common presentation of MAS is in association with systemic onset juvenile idiopathic arthritis, but is has been described in other forms of childhood rheumatic diseases, including other types of juvenile idiopathic arthritis, lupus, mixed connective tissue disease, Kawasaki disease, and sarcoidosis. Study of secondary MAS has led to suggested diagnostic criteria; however, those criteria are very similar to the presentation of adult and childhood systemic lupus with cytopenias, hepatitis, and coagulopathy.
The work by Borgia et al. encourages us to look for evidence of MAS in our lupus patients as it allows us to identify patients at risk for poor outcomes and to provide interventions to reduce those risks.
Marisa S. Klein-Gitelman, MD , is a professor of pediatrics at Northwestern University, Chicago, and is a pediatric rheumatologist at the Ann & Robert H. Lurie Children’s Hospital of Chicago. She has no relevant disclosures.
Nearly 10% of children with systemic lupus erythematosus (SLE) developed macrophage activation syndrome (MAS) at some point during a mean follow-up time of more than 3 years at one center, and most were concomitantly diagnosed with the syndrome.
Although the investigators from the University of Toronto reported significantly higher mortality among patients with MAS, most cases were successfully treated with corticosteroids, and no relapses were observed during follow-up.
MAS was first identified in patients with juvenile idiopathic arthritis and is most well known as a complication of that broadly named disease, but data on outcomes and disease course in SLE patients are limited, first author Roberto Ezequiel Borgia, MD, and his colleagues wrote in their report in Arthritis & Rheumatology.
The researchers identified 403 children with SLE seen at the Hospital for Sick Children in Toronto during 2002-2012. Overall, 38 patients (9%) had MAS; of those patients, 68% received a MAS diagnosis within 7 days of the SLE diagnosis – termed “concomitant” diagnosis – while another 29% received a MAS diagnosis within 180 days of their SLE diagnosis.
The researchers explained that “since there are no validated nor universally accepted diagnostic criteria for MAS in SLE, the definition of MAS was based on the treating pediatric rheumatologist’s expert opinion at the time of the initial presentation.” The most common presenting feature of MAS was fever (100%), followed by generalized lymphadenopathy (24%), hepatomegaly (18%), CNS dysfunction secondary to MAS (18%), hemorrhage (13%), and splenomegaly (10%).
The average age of the children at diagnosis was nearly 14 years, and 79% were female. The average follow-up was 3.5 years. There were no significant differences in the demographic features of children with and without MAS nor were there any in variables used to assess lupus outcomes, which included immunosuppressive drug use, average daily corticosteroid dose (18.3 mg/day with MAS vs. 18.6 mg/day without MAS), and the number of pediatric ICU visits (incidence rate ratio for MAS vs. non-MAS, 1.60 [95% CI, 0.74-3.18]).
Mortality was significantly higher in children with MAS, compared with those without MAS (5.3% vs. 0.3%; P = .02), although the overall number of deaths in the cohort was small (n = 3). Apart from the “acute illness which was associated with 2 deaths secondary to MAS,” the investigators said that they “did not find any significant differences in the number of deaths or damage accrual between the cohorts, including overall SLICC [Systemic Lupus International Collaborating Clinics] damage score or any specific damage feature within the score.”
The study findings were limited by several factors including the lack of validated MAS criteria for children with SLE and a lack of follow-up data on the patients beyond 18 years of age, the researchers said.
The results suggest that MAS remains a life-threatening complication in children with SLE and should be considered an important cause of mortality for them, but “if the initial presentation does not result in death, the long-term outcome seem[s] to be comparable to those without MAS,” the investigators wrote.
The researchers had no financial conflicts to disclose.
SOURCE: Borgia R et al. Arthritis Rheumatol. 2018 Jan 17. doi: 10.1002/art.40417
Nearly 10% of children with systemic lupus erythematosus (SLE) developed macrophage activation syndrome (MAS) at some point during a mean follow-up time of more than 3 years at one center, and most were concomitantly diagnosed with the syndrome.
Although the investigators from the University of Toronto reported significantly higher mortality among patients with MAS, most cases were successfully treated with corticosteroids, and no relapses were observed during follow-up.
MAS was first identified in patients with juvenile idiopathic arthritis and is most well known as a complication of that broadly named disease, but data on outcomes and disease course in SLE patients are limited, first author Roberto Ezequiel Borgia, MD, and his colleagues wrote in their report in Arthritis & Rheumatology.
The researchers identified 403 children with SLE seen at the Hospital for Sick Children in Toronto during 2002-2012. Overall, 38 patients (9%) had MAS; of those patients, 68% received a MAS diagnosis within 7 days of the SLE diagnosis – termed “concomitant” diagnosis – while another 29% received a MAS diagnosis within 180 days of their SLE diagnosis.
The researchers explained that “since there are no validated nor universally accepted diagnostic criteria for MAS in SLE, the definition of MAS was based on the treating pediatric rheumatologist’s expert opinion at the time of the initial presentation.” The most common presenting feature of MAS was fever (100%), followed by generalized lymphadenopathy (24%), hepatomegaly (18%), CNS dysfunction secondary to MAS (18%), hemorrhage (13%), and splenomegaly (10%).
The average age of the children at diagnosis was nearly 14 years, and 79% were female. The average follow-up was 3.5 years. There were no significant differences in the demographic features of children with and without MAS nor were there any in variables used to assess lupus outcomes, which included immunosuppressive drug use, average daily corticosteroid dose (18.3 mg/day with MAS vs. 18.6 mg/day without MAS), and the number of pediatric ICU visits (incidence rate ratio for MAS vs. non-MAS, 1.60 [95% CI, 0.74-3.18]).
Mortality was significantly higher in children with MAS, compared with those without MAS (5.3% vs. 0.3%; P = .02), although the overall number of deaths in the cohort was small (n = 3). Apart from the “acute illness which was associated with 2 deaths secondary to MAS,” the investigators said that they “did not find any significant differences in the number of deaths or damage accrual between the cohorts, including overall SLICC [Systemic Lupus International Collaborating Clinics] damage score or any specific damage feature within the score.”
The study findings were limited by several factors including the lack of validated MAS criteria for children with SLE and a lack of follow-up data on the patients beyond 18 years of age, the researchers said.
The results suggest that MAS remains a life-threatening complication in children with SLE and should be considered an important cause of mortality for them, but “if the initial presentation does not result in death, the long-term outcome seem[s] to be comparable to those without MAS,” the investigators wrote.
The researchers had no financial conflicts to disclose.
SOURCE: Borgia R et al. Arthritis Rheumatol. 2018 Jan 17. doi: 10.1002/art.40417
FROM ARTHRITIS & RHEUMATOLOGY
Key clinical point: Nearly 10% of children with SLE developed MAS at some point during a mean follow-up time of more than 3 years, but many outcomes were the same in patients with and without MAS.
Major finding: Mortality was 5.3% in children with MAS, compared with 0.3% in those without MAS (P = .02), over a 3.5-year follow-up period.
Study details: The data come from 403 children with SLE seen at a single center during 2002-2012.
Disclosures: The researchers had no financial conflicts to disclose.
Source: Borgia R et al. Arthritis Rheumatol. 2018 Jan 17. doi: 10.1002/art.40417.

JAK inhibitors look good for severe alopecia areata treatment
said Lucy Yichu Liu, MD, and Brett Andrew King, MD, of Yale University, New Haven, Conn.
Standard medical therapies for alopecia areata – usually topical or injected corticosteroids and allergic contact sensitization – are not very effective for severe disease, particularly alopecia totalis and alopecia universalis. The Janus kinase (JAK) pathway recently has been suggested as a target for treatment.
Dr. Liu and Dr. King reviewed several studies, including a retrospective cohort study of 13 patients aged 12-17 years, in which 7 patients had 100% hair loss and 6 had 20%-70% scalp hair loss. The adolescents were treated with the JAK1/3 inhibitor tofacitinib citrate 5 mg twice daily for 2-16 months (median, 5 months). That led to 93% median improvement in Severity of Alopecia Tool (SALT) score (range, 1%-100%) from baseline. Nine patients experienced hair regrowth. There were mild adverse effects, such as upper respiratory infections and headaches.
In a retrospective cohort study of 90 adults taking tofacitinib at a dosage of 5-10 mg twice daily for 4 months or longer with or without prednisone (300 mg once monthly for three doses), patients were divided into those who were more or less likely to respond based on duration of disease. Of 65 patients with alopecia totalis, or alopecia universalis that had lasted 10 years or less, or alopecia areata, 77% had some hair regrowth; 58% had more than 50% improvement from baseline, and 20% achieved full regrowth of hair, Dr. Liu and Dr. King reported in the Journal of Investigative Dermatology Symposium Proceedings.
“Given the finding in adults that complete scalp hair loss for more than 10 years is less likely to respond to treatment, there may be merit to pursuing treatment, even if only intermittently, in adolescents or even younger patients with stable, severe alopecia areata, to prevent irreversible hair loss in the future,” they wrote.
A patient with alopecia universalis achieved partial scalp hair regrowth and complete eyebrow regrowth with compounded ruxolitinib, a topical JAK inhibitor, according to a 2016 case report. Dr. Liu and Dr. King reported that clinical trials with topical JAK inhibitors, including topical tofacitinib and topical ruxolitinib, currently are ongoing.
SOURCE: Liu LY et al. J Investig Dermatol Symp Proc. 2018 Jan. doi: 10.1016/j.jisp.2017.10.003.
said Lucy Yichu Liu, MD, and Brett Andrew King, MD, of Yale University, New Haven, Conn.
Standard medical therapies for alopecia areata – usually topical or injected corticosteroids and allergic contact sensitization – are not very effective for severe disease, particularly alopecia totalis and alopecia universalis. The Janus kinase (JAK) pathway recently has been suggested as a target for treatment.
Dr. Liu and Dr. King reviewed several studies, including a retrospective cohort study of 13 patients aged 12-17 years, in which 7 patients had 100% hair loss and 6 had 20%-70% scalp hair loss. The adolescents were treated with the JAK1/3 inhibitor tofacitinib citrate 5 mg twice daily for 2-16 months (median, 5 months). That led to 93% median improvement in Severity of Alopecia Tool (SALT) score (range, 1%-100%) from baseline. Nine patients experienced hair regrowth. There were mild adverse effects, such as upper respiratory infections and headaches.
In a retrospective cohort study of 90 adults taking tofacitinib at a dosage of 5-10 mg twice daily for 4 months or longer with or without prednisone (300 mg once monthly for three doses), patients were divided into those who were more or less likely to respond based on duration of disease. Of 65 patients with alopecia totalis, or alopecia universalis that had lasted 10 years or less, or alopecia areata, 77% had some hair regrowth; 58% had more than 50% improvement from baseline, and 20% achieved full regrowth of hair, Dr. Liu and Dr. King reported in the Journal of Investigative Dermatology Symposium Proceedings.
“Given the finding in adults that complete scalp hair loss for more than 10 years is less likely to respond to treatment, there may be merit to pursuing treatment, even if only intermittently, in adolescents or even younger patients with stable, severe alopecia areata, to prevent irreversible hair loss in the future,” they wrote.
A patient with alopecia universalis achieved partial scalp hair regrowth and complete eyebrow regrowth with compounded ruxolitinib, a topical JAK inhibitor, according to a 2016 case report. Dr. Liu and Dr. King reported that clinical trials with topical JAK inhibitors, including topical tofacitinib and topical ruxolitinib, currently are ongoing.
SOURCE: Liu LY et al. J Investig Dermatol Symp Proc. 2018 Jan. doi: 10.1016/j.jisp.2017.10.003.
said Lucy Yichu Liu, MD, and Brett Andrew King, MD, of Yale University, New Haven, Conn.
Standard medical therapies for alopecia areata – usually topical or injected corticosteroids and allergic contact sensitization – are not very effective for severe disease, particularly alopecia totalis and alopecia universalis. The Janus kinase (JAK) pathway recently has been suggested as a target for treatment.
Dr. Liu and Dr. King reviewed several studies, including a retrospective cohort study of 13 patients aged 12-17 years, in which 7 patients had 100% hair loss and 6 had 20%-70% scalp hair loss. The adolescents were treated with the JAK1/3 inhibitor tofacitinib citrate 5 mg twice daily for 2-16 months (median, 5 months). That led to 93% median improvement in Severity of Alopecia Tool (SALT) score (range, 1%-100%) from baseline. Nine patients experienced hair regrowth. There were mild adverse effects, such as upper respiratory infections and headaches.
In a retrospective cohort study of 90 adults taking tofacitinib at a dosage of 5-10 mg twice daily for 4 months or longer with or without prednisone (300 mg once monthly for three doses), patients were divided into those who were more or less likely to respond based on duration of disease. Of 65 patients with alopecia totalis, or alopecia universalis that had lasted 10 years or less, or alopecia areata, 77% had some hair regrowth; 58% had more than 50% improvement from baseline, and 20% achieved full regrowth of hair, Dr. Liu and Dr. King reported in the Journal of Investigative Dermatology Symposium Proceedings.
“Given the finding in adults that complete scalp hair loss for more than 10 years is less likely to respond to treatment, there may be merit to pursuing treatment, even if only intermittently, in adolescents or even younger patients with stable, severe alopecia areata, to prevent irreversible hair loss in the future,” they wrote.
A patient with alopecia universalis achieved partial scalp hair regrowth and complete eyebrow regrowth with compounded ruxolitinib, a topical JAK inhibitor, according to a 2016 case report. Dr. Liu and Dr. King reported that clinical trials with topical JAK inhibitors, including topical tofacitinib and topical ruxolitinib, currently are ongoing.
SOURCE: Liu LY et al. J Investig Dermatol Symp Proc. 2018 Jan. doi: 10.1016/j.jisp.2017.10.003.
FROM JOURNAL OF INVESTIGATIVE DERMATOLOGY SYMPOSIUM PROCEEDINGS
Birth cohort affected 2015-2016 flu vaccine effectiveness
The influenza vaccine introduced in 2009 showed reduced effectiveness during the 2015-2016 influenza season, but only in adults born between 1958 and 1979, according to an analysis published online in the Journal of Infectious Diseases.
Using the Influenza Vaccine Effectiveness Network, researchers analyzed data from 2,115 patients with medically attended acute respiratory illness who tested positive for A(H1N1)pdm09 influenza virus, and 14,696 patients who tested negative for the influenza virus, from 2010-2011 to 2015-2016 (excluding the 2014-2015 influenza season).
Overall, 48% of the influenza virus–negative patients and 28% of the virus-positive patients had received at least one dose of the seasonal inactivated influenza vaccine more than 2 weeks before they fell ill.
However, the vaccine, which was based on the A/California/07/2009 strain of the A(H1N1)pdm09 virus, was only 47% effective during the 2015-2016 season, compared with 61% effectiveness during the 2010-2011 season through to the 2013-2014 season.
When researchers looked at vaccine effectiveness by birth cohort, they found that one particular cohort – individuals born between 1958 and 1979 – showed a significantly reduced vaccine effectiveness (22%) during the 2015-2016 season. By comparison, vaccine effectiveness in this cohort was 61% during the 2010-2013 seasons, and 56% during the 2013-2014 season.
When this birth cohort was excluded from analysis of the 2015-2016 season, the overall vaccine effectiveness for that season was 61%.
While the vaccine was based on an early reference strain of A(H1N1)pdm09, the virus itself later acquired mutations in the hemagglutinin gene, leading to the emergence of new genetic clades, including 6B, which dominated in the 2013-2014 influenza season, and 6B.1, which dominated in 2015-2016.
“Limited serologic data suggest that some adults born during 1958-1979 (age range in 2015-2016, 36-57 years) have decreased antibody titers against A(H1N1)pdm09 group 6B and 6B.1 viruses,” wrote Brendan Flannery, PhD, from the Centers for Disease Control and Prevention, and his coauthors.
They suggested that individuals in this cohort may have been immunologically primed with A/USSR/90/1977-like viruses, which were the first group of A(H1N1) viruses that this cohort would have been exposed to. A(H1N1) strains didn’t circulate between 1958 and 1977. Vaccination with A(H1N1)pdm09 viruses may have induced antibodies against shared antigenic components found on early versions of A(H1N1)pdm09.
If these shared antigenic epitopes were then altered in the later 6B and 6B.1 viruses, that might account for decreased antibody titers in this age group.
“Replacement of the A/California/07/2009(H1N1)pdm09 vaccine reference strain with A/Michigan/45/2015 (group 6B.1) should lead to improved [vaccine effectiveness] against circulating A(H1N1)pdm09 viruses,” the investigators noted.
The study was supported by the Centers for Disease Control and Prevention, the National Institutes of Health, and the National Center for Advancing Translational Sciences. Eight authors declared funding, grants, and consultancies with the pharmaceutical industry, with five also declaring funding from the CDC.
SOURCE: Flannery B et al. J Infect Dis. 2018 Jan 18. doi: 10.1093/infdis/jix634.
This study proposes that influenza virus strains encountered early in life focus the immune response to later infection or vaccination on shared epitopes between the early and later strains. Supporting this hypothesis is evidence from other studies showing that 60% of the serological response to inactivated influenza vaccines is the result of boosting pre-existing antibodies, rather than the creation of new, vaccine-induced antibodies.
However there are also some flaws to this argument, and we should be careful to avoid confirmation bias. For example, the reduction in effectiveness of vaccines against A(H1N1) has been observed in North America, where this study is located, but to a lesser extent in studies conducted in other regions. Reductions in vaccine effectiveness have also been observed in other birth cohorts and during other influenza seasons.
That aside, accumulating evidence suggests that the vaccine strain be updated from A/California/7/2009 to A/Michigan/45/2015 (a clade 6B.1 strain) for the 2016-2017 influenza seasons.
Allen C. Cheng, PhD, is from the School of Public Health and Preventive Medicine at Monash University, Melbourne, and Kanta Subbarao, MBBS, is from the World Health Organization Collaborating Centre for Reference and Research on Influenza and the Peter Doherty Institute for Infection and Immunity, Australia. These comments are taken from an accompanying editorial (J Infect Dis. 2018, Jan 18. doi: 10.1093/infdis/jix635). The authors declared support from the Australian Department of Health and the Australian National Health and Medical Research Council. No conflicts of interest were declared.
This study proposes that influenza virus strains encountered early in life focus the immune response to later infection or vaccination on shared epitopes between the early and later strains. Supporting this hypothesis is evidence from other studies showing that 60% of the serological response to inactivated influenza vaccines is the result of boosting pre-existing antibodies, rather than the creation of new, vaccine-induced antibodies.
However there are also some flaws to this argument, and we should be careful to avoid confirmation bias. For example, the reduction in effectiveness of vaccines against A(H1N1) has been observed in North America, where this study is located, but to a lesser extent in studies conducted in other regions. Reductions in vaccine effectiveness have also been observed in other birth cohorts and during other influenza seasons.
That aside, accumulating evidence suggests that the vaccine strain be updated from A/California/7/2009 to A/Michigan/45/2015 (a clade 6B.1 strain) for the 2016-2017 influenza seasons.
Allen C. Cheng, PhD, is from the School of Public Health and Preventive Medicine at Monash University, Melbourne, and Kanta Subbarao, MBBS, is from the World Health Organization Collaborating Centre for Reference and Research on Influenza and the Peter Doherty Institute for Infection and Immunity, Australia. These comments are taken from an accompanying editorial (J Infect Dis. 2018, Jan 18. doi: 10.1093/infdis/jix635). The authors declared support from the Australian Department of Health and the Australian National Health and Medical Research Council. No conflicts of interest were declared.
This study proposes that influenza virus strains encountered early in life focus the immune response to later infection or vaccination on shared epitopes between the early and later strains. Supporting this hypothesis is evidence from other studies showing that 60% of the serological response to inactivated influenza vaccines is the result of boosting pre-existing antibodies, rather than the creation of new, vaccine-induced antibodies.
However there are also some flaws to this argument, and we should be careful to avoid confirmation bias. For example, the reduction in effectiveness of vaccines against A(H1N1) has been observed in North America, where this study is located, but to a lesser extent in studies conducted in other regions. Reductions in vaccine effectiveness have also been observed in other birth cohorts and during other influenza seasons.
That aside, accumulating evidence suggests that the vaccine strain be updated from A/California/7/2009 to A/Michigan/45/2015 (a clade 6B.1 strain) for the 2016-2017 influenza seasons.
Allen C. Cheng, PhD, is from the School of Public Health and Preventive Medicine at Monash University, Melbourne, and Kanta Subbarao, MBBS, is from the World Health Organization Collaborating Centre for Reference and Research on Influenza and the Peter Doherty Institute for Infection and Immunity, Australia. These comments are taken from an accompanying editorial (J Infect Dis. 2018, Jan 18. doi: 10.1093/infdis/jix635). The authors declared support from the Australian Department of Health and the Australian National Health and Medical Research Council. No conflicts of interest were declared.
The influenza vaccine introduced in 2009 showed reduced effectiveness during the 2015-2016 influenza season, but only in adults born between 1958 and 1979, according to an analysis published online in the Journal of Infectious Diseases.
Using the Influenza Vaccine Effectiveness Network, researchers analyzed data from 2,115 patients with medically attended acute respiratory illness who tested positive for A(H1N1)pdm09 influenza virus, and 14,696 patients who tested negative for the influenza virus, from 2010-2011 to 2015-2016 (excluding the 2014-2015 influenza season).
Overall, 48% of the influenza virus–negative patients and 28% of the virus-positive patients had received at least one dose of the seasonal inactivated influenza vaccine more than 2 weeks before they fell ill.
However, the vaccine, which was based on the A/California/07/2009 strain of the A(H1N1)pdm09 virus, was only 47% effective during the 2015-2016 season, compared with 61% effectiveness during the 2010-2011 season through to the 2013-2014 season.
When researchers looked at vaccine effectiveness by birth cohort, they found that one particular cohort – individuals born between 1958 and 1979 – showed a significantly reduced vaccine effectiveness (22%) during the 2015-2016 season. By comparison, vaccine effectiveness in this cohort was 61% during the 2010-2013 seasons, and 56% during the 2013-2014 season.
When this birth cohort was excluded from analysis of the 2015-2016 season, the overall vaccine effectiveness for that season was 61%.
While the vaccine was based on an early reference strain of A(H1N1)pdm09, the virus itself later acquired mutations in the hemagglutinin gene, leading to the emergence of new genetic clades, including 6B, which dominated in the 2013-2014 influenza season, and 6B.1, which dominated in 2015-2016.
“Limited serologic data suggest that some adults born during 1958-1979 (age range in 2015-2016, 36-57 years) have decreased antibody titers against A(H1N1)pdm09 group 6B and 6B.1 viruses,” wrote Brendan Flannery, PhD, from the Centers for Disease Control and Prevention, and his coauthors.
They suggested that individuals in this cohort may have been immunologically primed with A/USSR/90/1977-like viruses, which were the first group of A(H1N1) viruses that this cohort would have been exposed to. A(H1N1) strains didn’t circulate between 1958 and 1977. Vaccination with A(H1N1)pdm09 viruses may have induced antibodies against shared antigenic components found on early versions of A(H1N1)pdm09.
If these shared antigenic epitopes were then altered in the later 6B and 6B.1 viruses, that might account for decreased antibody titers in this age group.
“Replacement of the A/California/07/2009(H1N1)pdm09 vaccine reference strain with A/Michigan/45/2015 (group 6B.1) should lead to improved [vaccine effectiveness] against circulating A(H1N1)pdm09 viruses,” the investigators noted.
The study was supported by the Centers for Disease Control and Prevention, the National Institutes of Health, and the National Center for Advancing Translational Sciences. Eight authors declared funding, grants, and consultancies with the pharmaceutical industry, with five also declaring funding from the CDC.
SOURCE: Flannery B et al. J Infect Dis. 2018 Jan 18. doi: 10.1093/infdis/jix634.
The influenza vaccine introduced in 2009 showed reduced effectiveness during the 2015-2016 influenza season, but only in adults born between 1958 and 1979, according to an analysis published online in the Journal of Infectious Diseases.
Using the Influenza Vaccine Effectiveness Network, researchers analyzed data from 2,115 patients with medically attended acute respiratory illness who tested positive for A(H1N1)pdm09 influenza virus, and 14,696 patients who tested negative for the influenza virus, from 2010-2011 to 2015-2016 (excluding the 2014-2015 influenza season).
Overall, 48% of the influenza virus–negative patients and 28% of the virus-positive patients had received at least one dose of the seasonal inactivated influenza vaccine more than 2 weeks before they fell ill.
However, the vaccine, which was based on the A/California/07/2009 strain of the A(H1N1)pdm09 virus, was only 47% effective during the 2015-2016 season, compared with 61% effectiveness during the 2010-2011 season through to the 2013-2014 season.
When researchers looked at vaccine effectiveness by birth cohort, they found that one particular cohort – individuals born between 1958 and 1979 – showed a significantly reduced vaccine effectiveness (22%) during the 2015-2016 season. By comparison, vaccine effectiveness in this cohort was 61% during the 2010-2013 seasons, and 56% during the 2013-2014 season.
When this birth cohort was excluded from analysis of the 2015-2016 season, the overall vaccine effectiveness for that season was 61%.
While the vaccine was based on an early reference strain of A(H1N1)pdm09, the virus itself later acquired mutations in the hemagglutinin gene, leading to the emergence of new genetic clades, including 6B, which dominated in the 2013-2014 influenza season, and 6B.1, which dominated in 2015-2016.
“Limited serologic data suggest that some adults born during 1958-1979 (age range in 2015-2016, 36-57 years) have decreased antibody titers against A(H1N1)pdm09 group 6B and 6B.1 viruses,” wrote Brendan Flannery, PhD, from the Centers for Disease Control and Prevention, and his coauthors.
They suggested that individuals in this cohort may have been immunologically primed with A/USSR/90/1977-like viruses, which were the first group of A(H1N1) viruses that this cohort would have been exposed to. A(H1N1) strains didn’t circulate between 1958 and 1977. Vaccination with A(H1N1)pdm09 viruses may have induced antibodies against shared antigenic components found on early versions of A(H1N1)pdm09.
If these shared antigenic epitopes were then altered in the later 6B and 6B.1 viruses, that might account for decreased antibody titers in this age group.
“Replacement of the A/California/07/2009(H1N1)pdm09 vaccine reference strain with A/Michigan/45/2015 (group 6B.1) should lead to improved [vaccine effectiveness] against circulating A(H1N1)pdm09 viruses,” the investigators noted.
The study was supported by the Centers for Disease Control and Prevention, the National Institutes of Health, and the National Center for Advancing Translational Sciences. Eight authors declared funding, grants, and consultancies with the pharmaceutical industry, with five also declaring funding from the CDC.
SOURCE: Flannery B et al. J Infect Dis. 2018 Jan 18. doi: 10.1093/infdis/jix634.
FROM THE JOURNAL OF INFECTIOUS DISEASES
Key clinical point:
Major finding: The influenza vaccine effectiveness during the 2015-2016 season was just 22% in individuals born between 1958 and 1979.
Data source: A retrospective case-control study of 2,115 patients who tested positive for A(H1N1)pdm09 influenza virus, and 14,696 negative controls.
Disclosures: The study was supported by the Centers for Disease Control and Prevention, the National Institutes of Health, and the National Center for Advancing Translational Sciences. Eight authors declared funding, grants, and consultancies with the pharmaceutical industry, with five also declaring funding from the CDC.
Source: Flannery B et al. J Infect Dis. 2018 Jan 18. doi: 10.1093/infdis/jix634.
Dental Health: What It Means in Kidney Disease
Q) I teach nephrology at a local PA program, and they want us to integrate dental care into each module. What’s the connection between the two?
Dental health is frequently overlooked in the medical realm, as many clinicians feel that dental issues are out of our purview. Hematuria worries us, but bleeding gums and other signs of periodontal disease are often ignored. Surprisingly, many patients don’t seem to mind when their gums bleed every time they brush; they believe that this is normal, when really, it’s not.
Growing evidence supports associations between dental health and multiple medical issues—chronic kidney disease (CKD) among them. Periodontal disease is one of several inflammatory diseases caused by an interaction between gram-negative periodontal bacterial species and the immune system. It manifests with sore, red, bleeding gums and can lead to tooth loss if left untreated.

Chronic inflammation in the gums is a good indicator of inflammation elsewhere in the body. In and of itself, periodontitis can set off an inflammatory cascade in the body. Poor dentition can also lead to poor nutrition, which then causes a feedback loop, leading to even more inflammation.
Patients with periodontal disease have higher levels of C-reactive protein and a higher erythrocyte sedimentation rate than those without the disease.1 And a recent study by Zhang et al showed that periodontal disease increased risk for all-cause mortality in patients with CKD.2
The high cost of CKD from both a financial and personal view makes any intervention worth exploring, as the risk factors are difficult to modify and the CKD population is growing worldwide. We, as medical providers, should reiterate what our dental colleagues have been saying for years: Encourage patients with CKD to practice good dental hygiene by brushing twice a day and flossing daily, in an attempt to improve their overall outcomes.
LCDR Julie Taylor, PA-C
United States Public Health Service, Boston
1. Zhang J, Jiang H, Sun M, Chen J. Association between periodontal disease and mortality in people with CKD: a meta-analysis of cohort studies. BMC Nephrol. 2017;18(1):269.
2. Chen YT, Shin CJ, Ou SM, et al; Taiwan Geriatric Kidney Disease (TGKD) Research Group. Periodontal disease and risks of kidney function decline and mortality in older people: a community-based cohort study. Am J Kidney Dis. 2015; 66(2):223-230.
Clinician Reviews in partnership with
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation's Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a semi-retired PA who works with the American Academy of Nephrology PAs and is a past chair of the NKF-CAP. This month's responses were authored by Nicole DeFeo McCormick, DNP, MBA, NP-C, CCTC, who is an Assistant Professor in the School of Medicine at the University of Colorado, and LCDR Julie Cure, PA-C, who is with the United States Public Health Service in Boston.
Clinician Reviews in partnership with
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation's Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a semi-retired PA who works with the American Academy of Nephrology PAs and is a past chair of the NKF-CAP. This month's responses were authored by Nicole DeFeo McCormick, DNP, MBA, NP-C, CCTC, who is an Assistant Professor in the School of Medicine at the University of Colorado, and LCDR Julie Cure, PA-C, who is with the United States Public Health Service in Boston.
Clinician Reviews in partnership with
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation's Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a semi-retired PA who works with the American Academy of Nephrology PAs and is a past chair of the NKF-CAP. This month's responses were authored by Nicole DeFeo McCormick, DNP, MBA, NP-C, CCTC, who is an Assistant Professor in the School of Medicine at the University of Colorado, and LCDR Julie Cure, PA-C, who is with the United States Public Health Service in Boston.
Q) I teach nephrology at a local PA program, and they want us to integrate dental care into each module. What’s the connection between the two?
Dental health is frequently overlooked in the medical realm, as many clinicians feel that dental issues are out of our purview. Hematuria worries us, but bleeding gums and other signs of periodontal disease are often ignored. Surprisingly, many patients don’t seem to mind when their gums bleed every time they brush; they believe that this is normal, when really, it’s not.
Growing evidence supports associations between dental health and multiple medical issues—chronic kidney disease (CKD) among them. Periodontal disease is one of several inflammatory diseases caused by an interaction between gram-negative periodontal bacterial species and the immune system. It manifests with sore, red, bleeding gums and can lead to tooth loss if left untreated.
Chronic inflammation in the gums is a good indicator of inflammation elsewhere in the body. In and of itself, periodontitis can set off an inflammatory cascade in the body. Poor dentition can also lead to poor nutrition, which then causes a feedback loop, leading to even more inflammation.
Patients with periodontal disease have higher levels of C-reactive protein and a higher erythrocyte sedimentation rate than those without the disease.1 And a recent study by Zhang et al showed that periodontal disease increased risk for all-cause mortality in patients with CKD.2
The high cost of CKD from both a financial and personal view makes any intervention worth exploring, as the risk factors are difficult to modify and the CKD population is growing worldwide. We, as medical providers, should reiterate what our dental colleagues have been saying for years: Encourage patients with CKD to practice good dental hygiene by brushing twice a day and flossing daily, in an attempt to improve their overall outcomes.
LCDR Julie Taylor, PA-C
United States Public Health Service, Boston
Q) I teach nephrology at a local PA program, and they want us to integrate dental care into each module. What’s the connection between the two?
Dental health is frequently overlooked in the medical realm, as many clinicians feel that dental issues are out of our purview. Hematuria worries us, but bleeding gums and other signs of periodontal disease are often ignored. Surprisingly, many patients don’t seem to mind when their gums bleed every time they brush; they believe that this is normal, when really, it’s not.
Growing evidence supports associations between dental health and multiple medical issues—chronic kidney disease (CKD) among them. Periodontal disease is one of several inflammatory diseases caused by an interaction between gram-negative periodontal bacterial species and the immune system. It manifests with sore, red, bleeding gums and can lead to tooth loss if left untreated.
Chronic inflammation in the gums is a good indicator of inflammation elsewhere in the body. In and of itself, periodontitis can set off an inflammatory cascade in the body. Poor dentition can also lead to poor nutrition, which then causes a feedback loop, leading to even more inflammation.
Patients with periodontal disease have higher levels of C-reactive protein and a higher erythrocyte sedimentation rate than those without the disease.1 And a recent study by Zhang et al showed that periodontal disease increased risk for all-cause mortality in patients with CKD.2
The high cost of CKD from both a financial and personal view makes any intervention worth exploring, as the risk factors are difficult to modify and the CKD population is growing worldwide. We, as medical providers, should reiterate what our dental colleagues have been saying for years: Encourage patients with CKD to practice good dental hygiene by brushing twice a day and flossing daily, in an attempt to improve their overall outcomes.
LCDR Julie Taylor, PA-C
United States Public Health Service, Boston
1. Zhang J, Jiang H, Sun M, Chen J. Association between periodontal disease and mortality in people with CKD: a meta-analysis of cohort studies. BMC Nephrol. 2017;18(1):269.
2. Chen YT, Shin CJ, Ou SM, et al; Taiwan Geriatric Kidney Disease (TGKD) Research Group. Periodontal disease and risks of kidney function decline and mortality in older people: a community-based cohort study. Am J Kidney Dis. 2015; 66(2):223-230.
1. Zhang J, Jiang H, Sun M, Chen J. Association between periodontal disease and mortality in people with CKD: a meta-analysis of cohort studies. BMC Nephrol. 2017;18(1):269.
2. Chen YT, Shin CJ, Ou SM, et al; Taiwan Geriatric Kidney Disease (TGKD) Research Group. Periodontal disease and risks of kidney function decline and mortality in older people: a community-based cohort study. Am J Kidney Dis. 2015; 66(2):223-230.

For Patients With CKD, Don’t Wait—Vaccinate!
Q) What can I tell my kidney patients to increase acceptance of the influenza and pneumonia vaccines during cold and flu season?
The CDC recommends that everyone ages 6 months and older receive an annual flu vaccination, unless contraindicated.1 Additionally, administration of either the 13-valent pneumococcal conjugate vaccine (PCV13) or the 23-valent pneumococcal polysaccharide vaccine (PPSV23) is recommended for all adults ages 65 and older and for younger adults (ages 19 to 64) with diabetes, chronic kidney disease (CKD), chronic heart disease, and/or solid organ transplant.1 Despite these recommendations, patients often decline vaccination. What they may not realize is that CKD increases their risk for infection.
In a cohort of more than 1 million Swedish patients, researchers found that any stage of CKD increased risk for community-acquired infection and that the risk for lower respiratory tract infection increased as glomerular filtration rate declined.2 Patients on hemodialysis have an increased risk for pneumonia and an incidence of pneumonia-related mortality that is up to 16 times higher than that of the general population.3 Pneumonia also increases the risk for cardiovascular events among all patients with CKD, regardless of stage.4
So, can vaccines reduce these risks in our kidney patients? McGrath and colleagues found that patients with end-stage renal disease (ESRD) who were vaccinated against the flu had lower mortality rates than those who were not vaccinated—even when the vaccine was poorly matched to the circulating virus strain.5 Additional research has demonstrated that for patients with any stage of CKD, including those on dialysis, the flu vaccine is safe and effective, and its protection may be durable over time.6
For pneumonia vaccines, antibody response in patients with CKD may be suboptimal; however, Medicare data have demonstrated that patients with ESRD who are vaccinated against pneumonia have lower rates of all-cause and cardiovascular mortality than unvaccinated patients do.5 Given their increased vulnerability to vaccine-preventable respiratory illnesses, it is imperative that our kidney patients receive both the flu and pneumonia vaccines.
Nicole DeFeo McCormick, DNP, MBA, NP-C, CCTC
Assistant Professor
School of Medicine at the University of Colorado
1. CDC. Recommended immunization schedule for adults aged 19 years or older, United States, 2017. www.cdc.gov/vaccines/schedules/hcp/index.html. Accessed November 22, 2017.
2. Xu H, Gasparini A, Ishigami J, et al. eGFR and the risk of community-acquired infections. Clin J Am Soc Nephrol. 2017; 12(9):1399-1408.
3. Sarnak MJ, Jaber BL. Pulmonary infectious mortality among patients with end-stage renal disease. Chest. 2001;120(6): 1883-1887.
4. Mathew R, Mason D, Kennedy JS. Vaccination issues in patients with chronic kidney disease. Expert Rev Vaccines. 2014;13(2):285-298.
5. McGrath LJ, Kshirsagar AV, Cole SR, et al. Evaluating influenza vaccine effectiveness among hemodialysis patients using a natural experiment. Arch Intern Med. 2012;172(7): 548-554.
6. Janus N, Vacher L, Karie S, et al. Vaccination and chronic kidney disease. Nephrol Dial Transplant. 2008;23(3):800-807.
Clinician Reviews in partnership with
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation's Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a semi-retired PA who works with the American Academy of Nephrology PAs and is a past chair of the NKF-CAP. This month's responses were authored by Nicole DeFeo McCormick, DNP, MBA, NP-C, CCTC, who is an Assistant Professor in the School of Medicine at the University of Colorado, and LCDR Julie Taylor, PA-C, who is with the United States Public Health Service in Boston.
Clinician Reviews in partnership with
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation's Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a semi-retired PA who works with the American Academy of Nephrology PAs and is a past chair of the NKF-CAP. This month's responses were authored by Nicole DeFeo McCormick, DNP, MBA, NP-C, CCTC, who is an Assistant Professor in the School of Medicine at the University of Colorado, and LCDR Julie Taylor, PA-C, who is with the United States Public Health Service in Boston.
Clinician Reviews in partnership with
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation's Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a semi-retired PA who works with the American Academy of Nephrology PAs and is a past chair of the NKF-CAP. This month's responses were authored by Nicole DeFeo McCormick, DNP, MBA, NP-C, CCTC, who is an Assistant Professor in the School of Medicine at the University of Colorado, and LCDR Julie Taylor, PA-C, who is with the United States Public Health Service in Boston.
Q) What can I tell my kidney patients to increase acceptance of the influenza and pneumonia vaccines during cold and flu season?
The CDC recommends that everyone ages 6 months and older receive an annual flu vaccination, unless contraindicated.1 Additionally, administration of either the 13-valent pneumococcal conjugate vaccine (PCV13) or the 23-valent pneumococcal polysaccharide vaccine (PPSV23) is recommended for all adults ages 65 and older and for younger adults (ages 19 to 64) with diabetes, chronic kidney disease (CKD), chronic heart disease, and/or solid organ transplant.1 Despite these recommendations, patients often decline vaccination. What they may not realize is that CKD increases their risk for infection.
In a cohort of more than 1 million Swedish patients, researchers found that any stage of CKD increased risk for community-acquired infection and that the risk for lower respiratory tract infection increased as glomerular filtration rate declined.2 Patients on hemodialysis have an increased risk for pneumonia and an incidence of pneumonia-related mortality that is up to 16 times higher than that of the general population.3 Pneumonia also increases the risk for cardiovascular events among all patients with CKD, regardless of stage.4
So, can vaccines reduce these risks in our kidney patients? McGrath and colleagues found that patients with end-stage renal disease (ESRD) who were vaccinated against the flu had lower mortality rates than those who were not vaccinated—even when the vaccine was poorly matched to the circulating virus strain.5 Additional research has demonstrated that for patients with any stage of CKD, including those on dialysis, the flu vaccine is safe and effective, and its protection may be durable over time.6
For pneumonia vaccines, antibody response in patients with CKD may be suboptimal; however, Medicare data have demonstrated that patients with ESRD who are vaccinated against pneumonia have lower rates of all-cause and cardiovascular mortality than unvaccinated patients do.5 Given their increased vulnerability to vaccine-preventable respiratory illnesses, it is imperative that our kidney patients receive both the flu and pneumonia vaccines.
Nicole DeFeo McCormick, DNP, MBA, NP-C, CCTC
Assistant Professor
School of Medicine at the University of Colorado
Q) What can I tell my kidney patients to increase acceptance of the influenza and pneumonia vaccines during cold and flu season?
The CDC recommends that everyone ages 6 months and older receive an annual flu vaccination, unless contraindicated.1 Additionally, administration of either the 13-valent pneumococcal conjugate vaccine (PCV13) or the 23-valent pneumococcal polysaccharide vaccine (PPSV23) is recommended for all adults ages 65 and older and for younger adults (ages 19 to 64) with diabetes, chronic kidney disease (CKD), chronic heart disease, and/or solid organ transplant.1 Despite these recommendations, patients often decline vaccination. What they may not realize is that CKD increases their risk for infection.
In a cohort of more than 1 million Swedish patients, researchers found that any stage of CKD increased risk for community-acquired infection and that the risk for lower respiratory tract infection increased as glomerular filtration rate declined.2 Patients on hemodialysis have an increased risk for pneumonia and an incidence of pneumonia-related mortality that is up to 16 times higher than that of the general population.3 Pneumonia also increases the risk for cardiovascular events among all patients with CKD, regardless of stage.4
So, can vaccines reduce these risks in our kidney patients? McGrath and colleagues found that patients with end-stage renal disease (ESRD) who were vaccinated against the flu had lower mortality rates than those who were not vaccinated—even when the vaccine was poorly matched to the circulating virus strain.5 Additional research has demonstrated that for patients with any stage of CKD, including those on dialysis, the flu vaccine is safe and effective, and its protection may be durable over time.6
For pneumonia vaccines, antibody response in patients with CKD may be suboptimal; however, Medicare data have demonstrated that patients with ESRD who are vaccinated against pneumonia have lower rates of all-cause and cardiovascular mortality than unvaccinated patients do.5 Given their increased vulnerability to vaccine-preventable respiratory illnesses, it is imperative that our kidney patients receive both the flu and pneumonia vaccines.
Nicole DeFeo McCormick, DNP, MBA, NP-C, CCTC
Assistant Professor
School of Medicine at the University of Colorado
1. CDC. Recommended immunization schedule for adults aged 19 years or older, United States, 2017. www.cdc.gov/vaccines/schedules/hcp/index.html. Accessed November 22, 2017.
2. Xu H, Gasparini A, Ishigami J, et al. eGFR and the risk of community-acquired infections. Clin J Am Soc Nephrol. 2017; 12(9):1399-1408.
3. Sarnak MJ, Jaber BL. Pulmonary infectious mortality among patients with end-stage renal disease. Chest. 2001;120(6): 1883-1887.
4. Mathew R, Mason D, Kennedy JS. Vaccination issues in patients with chronic kidney disease. Expert Rev Vaccines. 2014;13(2):285-298.
5. McGrath LJ, Kshirsagar AV, Cole SR, et al. Evaluating influenza vaccine effectiveness among hemodialysis patients using a natural experiment. Arch Intern Med. 2012;172(7): 548-554.
6. Janus N, Vacher L, Karie S, et al. Vaccination and chronic kidney disease. Nephrol Dial Transplant. 2008;23(3):800-807.
1. CDC. Recommended immunization schedule for adults aged 19 years or older, United States, 2017. www.cdc.gov/vaccines/schedules/hcp/index.html. Accessed November 22, 2017.
2. Xu H, Gasparini A, Ishigami J, et al. eGFR and the risk of community-acquired infections. Clin J Am Soc Nephrol. 2017; 12(9):1399-1408.
3. Sarnak MJ, Jaber BL. Pulmonary infectious mortality among patients with end-stage renal disease. Chest. 2001;120(6): 1883-1887.
4. Mathew R, Mason D, Kennedy JS. Vaccination issues in patients with chronic kidney disease. Expert Rev Vaccines. 2014;13(2):285-298.
5. McGrath LJ, Kshirsagar AV, Cole SR, et al. Evaluating influenza vaccine effectiveness among hemodialysis patients using a natural experiment. Arch Intern Med. 2012;172(7): 548-554.
6. Janus N, Vacher L, Karie S, et al. Vaccination and chronic kidney disease. Nephrol Dial Transplant. 2008;23(3):800-807.
Dysmorphic red blood cell formation
A 23-year-old woman presented with hematuria. Her blood pressure was normal, and she had no rash, joint pain, or other symptoms. Urinalysis was positive for proteinuria and hematuria, and urinary sediment analysis showed dysmorphic red blood cells (RBCs) and red cell casts, leading to a diagnosis of glomerulonephritis. She had proteinuria of 1.2 g/24 hours. Laboratory tests for systemic diseases were negative. Renal biopsy study revealed stage III immunoglobulin A (IgA) nephropathy.
GLOMERULAR HEMATURIA
Glomerular hematuria may represent an immune-mediated injury to the glomerular capillary wall, but it can also be present in noninflammatory glomerulopathies.1
The type of dysmorphic RBCs (crenated or misshapen cells, acanthocytes) may be of diagnostic importance. In particular, dysmorphic red cells alone may be predictive of only renal bleeding, while acanthocytes (ring-shaped RBCs with vesicle-shaped protrusions best seen on phase-contrast microscopy) appear to be most predictive of glomerular disease.2 For example, in 1 study,3 the presence of acanthocytes comprising at least 5% of excreted RBCs had a sensitivity of 52% for glomerular disease and a specificity of 98%.3
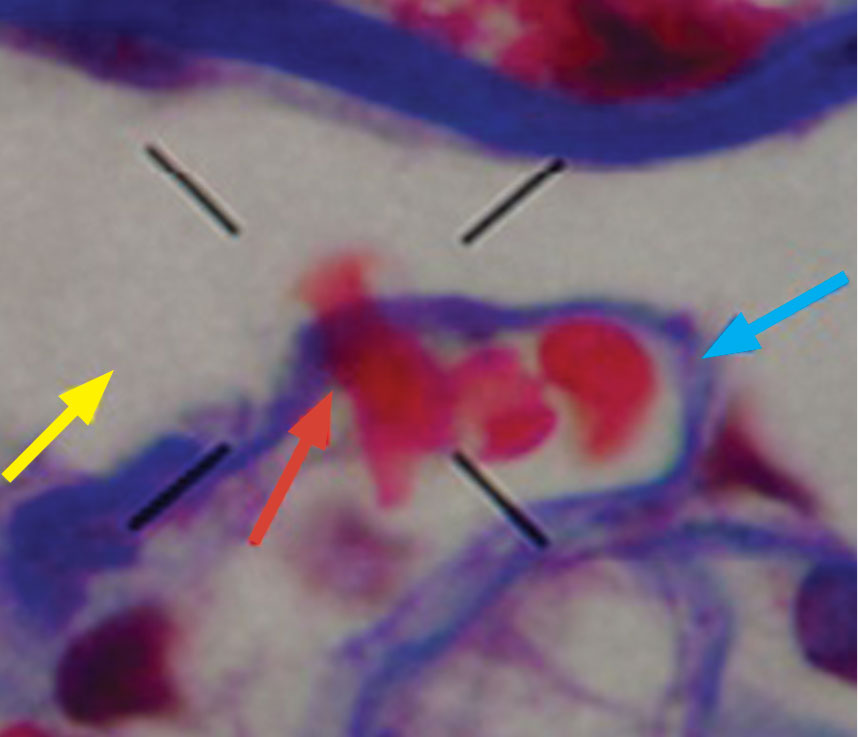
.")
- Collar JE, Ladva S, Cairns TD, Cattell V. Red cell traverse through thin glomerular basement membranes. Kidney Int 2001; 59:2069–2072.
- Fogazzi GB, Ponticelli C, Ritz E. The Urinary Sediment: An Integrated View. 2nd ed. Oxford: Oxford University Press; 1999:30.
- Köhler H, Wandel E, Brunck B. Acanthocyturia—a characteristic marker for glomerular bleeding. Kidney Int 1991; 40:115–120.
- Fogazzi GB. The Urinary Sediment: An Integrated View. 3rd ed. France: Elsevier; 2010.
- Briner VA, Reinhart WH. In vitro production of ‘glomerular red cells’: role of pH and osmolality. Nephron 1990; 56:13–18.
- Schramek P, Moritsch A, Haschkowitz H, Binder BR, Maier M. In vitro generation of dysmorphic erythrocytes. Kidney Int 1989; 36:72–77.
- Pollock C, Liu PL, Györy AZ, et al. Dysmorphism of urinary red blood cells—value in diagnosis. Kidney Int 1989; 36:1045–1049.
- Shichiri M, Hosoda K, Nishio Y, et al. Red-cell-volume distribution curves in diagnosis of glomerular and non-glomerular haematuria. Lancet 1988; 1:908–911.
A 23-year-old woman presented with hematuria. Her blood pressure was normal, and she had no rash, joint pain, or other symptoms. Urinalysis was positive for proteinuria and hematuria, and urinary sediment analysis showed dysmorphic red blood cells (RBCs) and red cell casts, leading to a diagnosis of glomerulonephritis. She had proteinuria of 1.2 g/24 hours. Laboratory tests for systemic diseases were negative. Renal biopsy study revealed stage III immunoglobulin A (IgA) nephropathy.
GLOMERULAR HEMATURIA
Glomerular hematuria may represent an immune-mediated injury to the glomerular capillary wall, but it can also be present in noninflammatory glomerulopathies.1
The type of dysmorphic RBCs (crenated or misshapen cells, acanthocytes) may be of diagnostic importance. In particular, dysmorphic red cells alone may be predictive of only renal bleeding, while acanthocytes (ring-shaped RBCs with vesicle-shaped protrusions best seen on phase-contrast microscopy) appear to be most predictive of glomerular disease.2 For example, in 1 study,3 the presence of acanthocytes comprising at least 5% of excreted RBCs had a sensitivity of 52% for glomerular disease and a specificity of 98%.3
A 23-year-old woman presented with hematuria. Her blood pressure was normal, and she had no rash, joint pain, or other symptoms. Urinalysis was positive for proteinuria and hematuria, and urinary sediment analysis showed dysmorphic red blood cells (RBCs) and red cell casts, leading to a diagnosis of glomerulonephritis. She had proteinuria of 1.2 g/24 hours. Laboratory tests for systemic diseases were negative. Renal biopsy study revealed stage III immunoglobulin A (IgA) nephropathy.
GLOMERULAR HEMATURIA
Glomerular hematuria may represent an immune-mediated injury to the glomerular capillary wall, but it can also be present in noninflammatory glomerulopathies.1
The type of dysmorphic RBCs (crenated or misshapen cells, acanthocytes) may be of diagnostic importance. In particular, dysmorphic red cells alone may be predictive of only renal bleeding, while acanthocytes (ring-shaped RBCs with vesicle-shaped protrusions best seen on phase-contrast microscopy) appear to be most predictive of glomerular disease.2 For example, in 1 study,3 the presence of acanthocytes comprising at least 5% of excreted RBCs had a sensitivity of 52% for glomerular disease and a specificity of 98%.3
- Collar JE, Ladva S, Cairns TD, Cattell V. Red cell traverse through thin glomerular basement membranes. Kidney Int 2001; 59:2069–2072.
- Fogazzi GB, Ponticelli C, Ritz E. The Urinary Sediment: An Integrated View. 2nd ed. Oxford: Oxford University Press; 1999:30.
- Köhler H, Wandel E, Brunck B. Acanthocyturia—a characteristic marker for glomerular bleeding. Kidney Int 1991; 40:115–120.
- Fogazzi GB. The Urinary Sediment: An Integrated View. 3rd ed. France: Elsevier; 2010.
- Briner VA, Reinhart WH. In vitro production of ‘glomerular red cells’: role of pH and osmolality. Nephron 1990; 56:13–18.
- Schramek P, Moritsch A, Haschkowitz H, Binder BR, Maier M. In vitro generation of dysmorphic erythrocytes. Kidney Int 1989; 36:72–77.
- Pollock C, Liu PL, Györy AZ, et al. Dysmorphism of urinary red blood cells—value in diagnosis. Kidney Int 1989; 36:1045–1049.
- Shichiri M, Hosoda K, Nishio Y, et al. Red-cell-volume distribution curves in diagnosis of glomerular and non-glomerular haematuria. Lancet 1988; 1:908–911.
- Collar JE, Ladva S, Cairns TD, Cattell V. Red cell traverse through thin glomerular basement membranes. Kidney Int 2001; 59:2069–2072.
- Fogazzi GB, Ponticelli C, Ritz E. The Urinary Sediment: An Integrated View. 2nd ed. Oxford: Oxford University Press; 1999:30.
- Köhler H, Wandel E, Brunck B. Acanthocyturia—a characteristic marker for glomerular bleeding. Kidney Int 1991; 40:115–120.
- Fogazzi GB. The Urinary Sediment: An Integrated View. 3rd ed. France: Elsevier; 2010.
- Briner VA, Reinhart WH. In vitro production of ‘glomerular red cells’: role of pH and osmolality. Nephron 1990; 56:13–18.
- Schramek P, Moritsch A, Haschkowitz H, Binder BR, Maier M. In vitro generation of dysmorphic erythrocytes. Kidney Int 1989; 36:72–77.
- Pollock C, Liu PL, Györy AZ, et al. Dysmorphism of urinary red blood cells—value in diagnosis. Kidney Int 1989; 36:1045–1049.
- Shichiri M, Hosoda K, Nishio Y, et al. Red-cell-volume distribution curves in diagnosis of glomerular and non-glomerular haematuria. Lancet 1988; 1:908–911.
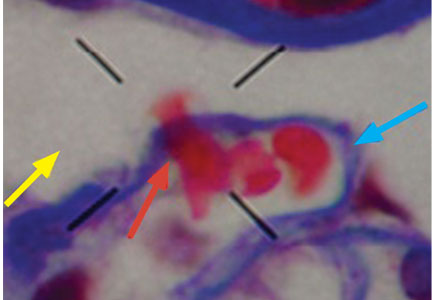
Chronic Urticaria: It’s More Than Just Antihistamines!
CE/CME No: CR-1801
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Differentiate between acute and chronic urticaria.
• List common history questions required for the diagnosis of chronic urticaria.
• Explain a stepwise plan for treatment of chronic urticaria.
• Describe serologic testing that should be ordered for chronic urticaria.
• Demonstrate knowledge of when to refer patients to a specialist for alternative treatment options.
FACULTY
Randy D. Danielsen is Professor and Dean of the Arizona School of Health Sciences, and Director of the Center for the Future of the Health Professions at A.T. Still University in Mesa, Arizona. Gabriel Ortiz practices at Breathe America El Paso in Texas and is a former AAPA liaison to the American Academy of Allergy, Asthma & Immunology (AAAAI) and National Institutes of Health/National Asthma Education and Prevention Program—Coordinating Committee. Susan Symington has practiced in allergy, asthma, and immunology for more than 10 years. She is the current AAPA liaison to theAAAAI and is President-Elect of the AAPA-Allergy, Asthma, and Immunology subspecialty organization.
The authors have no financial relationships to disclose.
![]()
ACCREDITATION STATEMENT
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid through December 31, 2018.
Article begins on next page >>
The discomfort caused by an urticarial rash, along with its unpredictable course, can interfere with a patient’s sleep and work/school. Adding to the frustration of patients and providers alike, an underlying cause is seldom identified. But a stepwise treatment approach can bring relief to all.
Urticaria, often referred to as hives, is a common cutaneous disorder with a lifetime incidence between 15% and 25%.1 Urticaria is characterized by recurring pruritic wheals that arise due to allergic and nonallergic reactions to internal and external agents. The name urticaria comes from the Latin word for “nettle,” urtica, derived from the Latin word uro, meaning “to burn.”2
Urticaria can be debilitating for patients, who may complain of a burning sensation. It can last for years in some and reduces quality of life for many. Recently, more successful treatments for urticaria have emerged that can provide tremendous relief.
It is important to understand some of the ways to diagnose and treat patients in a primary care setting and also to know when referral is appropriate. This article will discuss the diagnosis, treatment, and referral process for patients with chronic urticaria.
PATHOPHYSIOLOGY
Hives most commonly arise from an immunologic reaction in the superficial skin layers that results in the release of histamine, which causes swelling, itching, and erythema. The mast cell is the major effector cell in the pathophysiology of urticaria.3 In immunologic urticaria, the antigen binds to immunoglobulin (Ig) E on the mast cell surface, causing degranulation and release of histamine, which accounts for the wheals and itching associated with the condition. Histamine binds to H1 and H2 receptors in the skin to cause arteriolar dilation, venous constriction, and increased capillary permeability, accounting for the accompanying swelling.3 Not all urticaria is mediated by IgE; it can result from systemic disease processes in the body that are immune related but not related to IgE. An example would be autoimmune urticaria.
Urticaria commonly occurs with angioedema, which is marked by a greater degree of swelling and results from mast cell activation in the deeper dermis and subcutaneous tissue. Either condition can occur independently, however. Angioedema typically affects the lips, tongue, face, pharynx, and bilateral extremities; rarely, it affects the gastrointestinal tract. Angioedema may be hereditary, but its nonhereditary causes can be similar to those of urticaria.3 For example, a patient could be severely allergic to cat dander and, when exposed to this allergic trigger, develop swelling of the lips, facial edema, and flushing.
FORMS OF URTICARIA
Urticaria can be broadly divided based on the duration of illness: less than six weeks is termed acute urticaria, and continuous or intermittent presence for six weeks or more, chronic urticaria.4
Acute urticaria may occur in any age group but is most often seen in children.1 Acute urticaria and angioedema frequently resolve within a few days, without an identified cause. An inciting cause can be identified in only about 15% to 20% of cases; the most common cause is viral infection, followed by foods, drugs, insect stings, transfusion reactions, and, rarely, contactants and inhalants (see Table 1).1,5 Acute urticaria that is not associated with angioedema or respiratory distress is usually self-limited. The condition typically resolves before extensive evaluation, including testing for possible allergic triggers, can be done. The associated skin lesions are often self-limited or can be controlled symptomatically with antihistamines and avoidance of known possible triggers.1
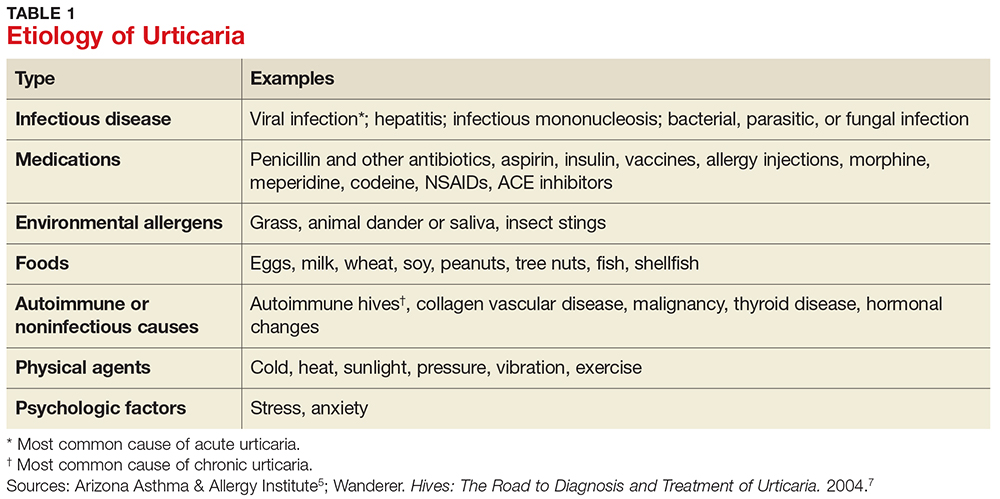
Chronic urticaria, sometimes called chronic idiopathic urticaria, is more common in adults, occurs on most days of the week, and, as noted, persists for more than six weeks with no identifiable triggers.6 It affects about 0.5% to 1% of the population (lifetime prevalence).3 Approximately 45% of patients with chronic urticaria have accompanying episodes of angioedema, and 30% to 50% have an autoimmune process involving autoantibodies against the thyroid, IgE, or the high-affinity IgE receptor (FcR1).3 The diagnosis is based primarily on clinical history and presentation; this will guide the determination of what types of diagnostic testing are necessary.
Chronic urticaria requires an extensive, but not indiscriminate, evaluation with history, physical examination, allergy testing, and laboratory testing for immune system, liver, kidney, thyroid, and collagen vascular diseases.3 Unfortunately, an identifiable cause of chronic urticaria is found in only 10% to 20% of patients; most cases are idiopathic.3,7
Several forms of chronic urticaria can be precipitated by physical stimuli, such as exercise, generalized heat, or sweating (cholinergic urticaria); localized heat (localized heat urticaria); low temperatures (cold urticaria); sun exposure (solar urticaria); water (aquagenic urticaria); and vibration.1 In another form (pressure urticaria), pressure on the skin increases histamine release, leading to the development of wheals and itching; this form is also called dermatographism, which means “write on skin” (see Figure 1). These types of urticaria should be evaluated and treated by a board-certified allergist, as there are special evaluations that can confirm the diagnosis.
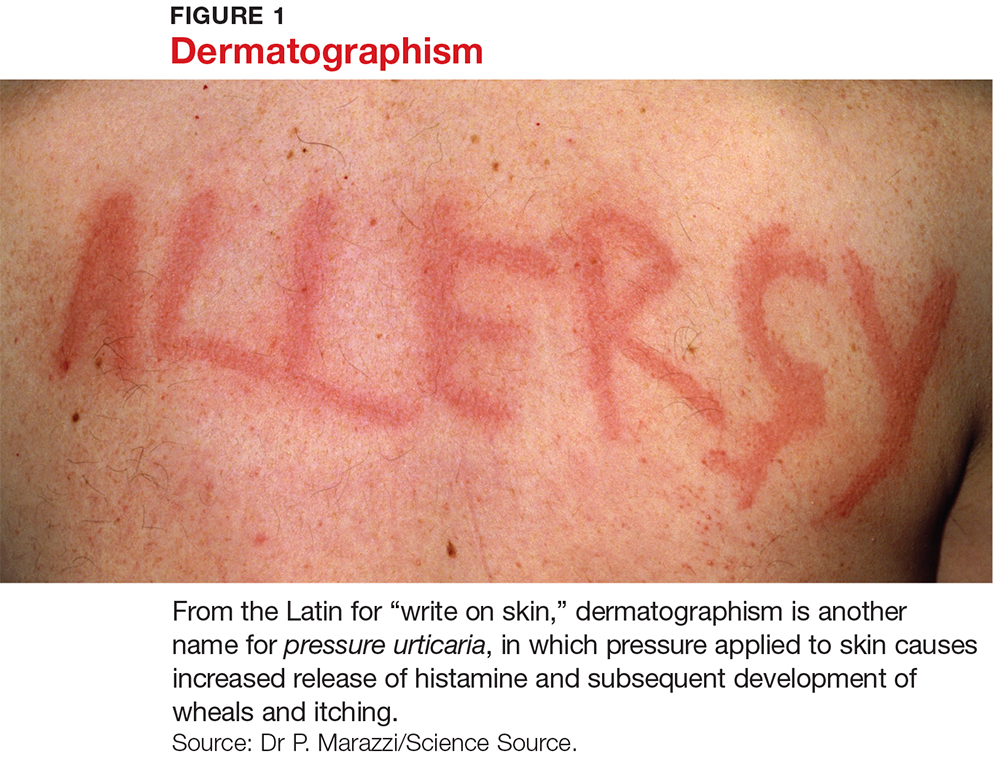
CLINICAL FEATURES
The main feature of urticaria is raised skin lesions that appear pale to pink to erythematous and most commonly are intensely pruritic (see Figure 2). These lesions range from a few millimeters to several centimeters in size and may coalesce.
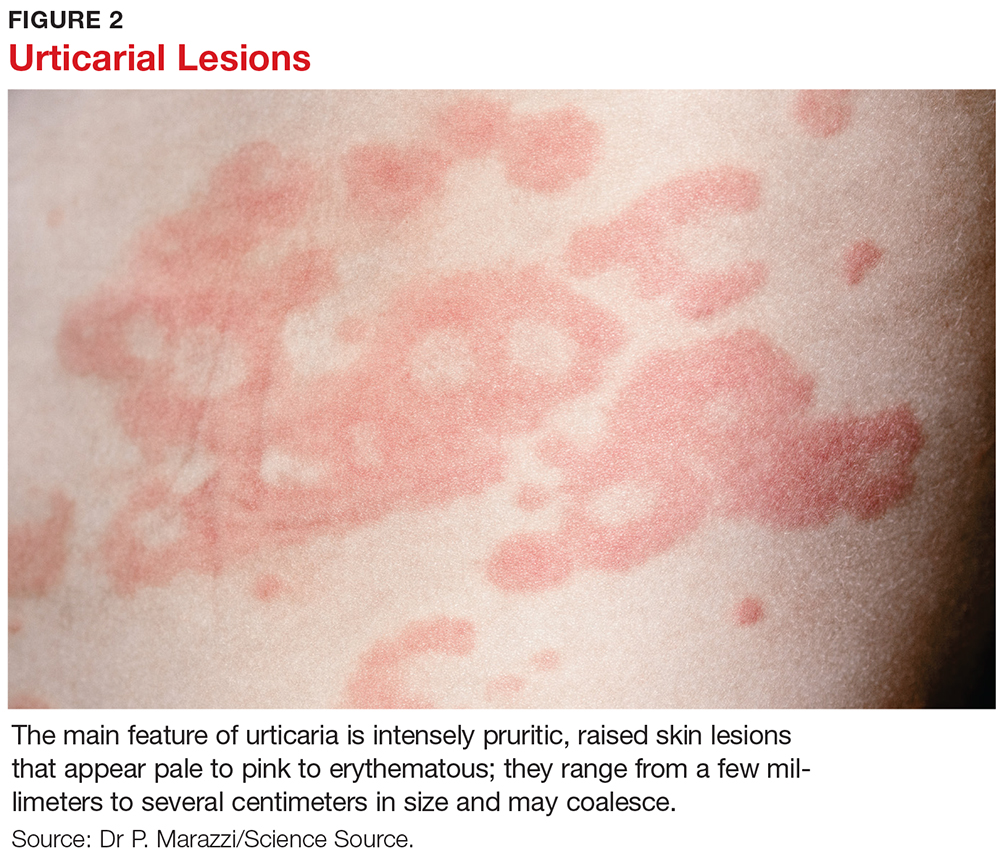
Characteristically, evanescent old lesions resolve, and new ones develop over 24 hours, usually without scarring. Scratching generally worsens dermatographism, with new urticaria produced over the scratched area. Any area of the body may be involved.
The lesions of early urticaria may vary in size and blanch when pressure is applied. An individual hive may last minutes or up to 24 hours and may reoccur intermittently on various sites on the body for an unspecified period of time.1,6
DIFFERENTIAL DIAGNOSIS
Other dermatologic conditions may be mistaken for chronic urticaria. Common rashes that may mimic it include anaphylaxis, atopic dermatitis, medication allergy or fixed drug eruption, ACE inhibitor–related angioedema, mastocytosis, contact dermatitis, autoimmune thyroid disease, bullous pemphigoid, and dermatitis herpetiformis.
Patients should be encouraged to bring pictures of the rash to the office visit, since the rash may have waned at the time of the visit and diagnosis based on the patient’s description alone can be challenging. Most rashes in the differential can be identified or eliminated through a careful history and complete physical exam. When necessary, serologic testing and skin punch biopsies can elucidate and confirm the diagnosis.
EVALUATION
History and physical examination
The medical history is the most important part of the evaluation of a patient with urticaria. The information that should be elicited and documented during the history is shown in Table 2.
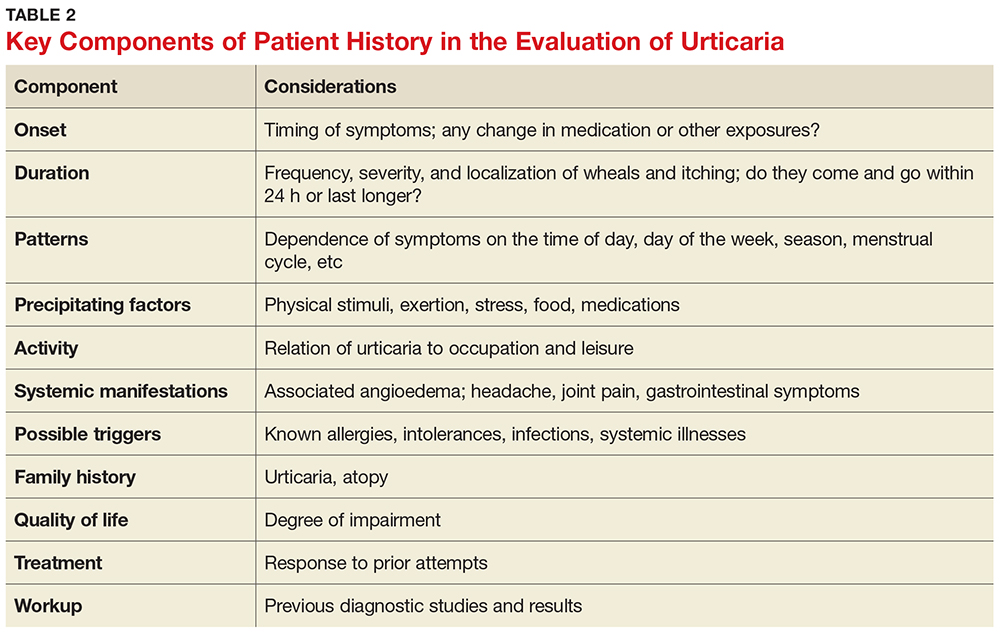
A general comprehensive physical exam should be undertaken and the findings carefully documented. As noted, it can be helpful for patients to bring in pictures of the rash if the lesions wax and wane. It is also important to assess whether the urticarial lesions blanch when palpated, since this is a characteristic feature of acute and chronic urticarial lesions (but not of those with an autoimmune, cholinergic, or vasculitic cause). Thus, blanching of the wheal is a key finding on physical exam to discriminate between possible causes.8 Lesions pigmented with purpuric areas that scar or last longer than 24 hours suggest urticarial vasculitis; other features that distinguish urticarial vasculitis from chronic urticaria are listed in Table 3.2
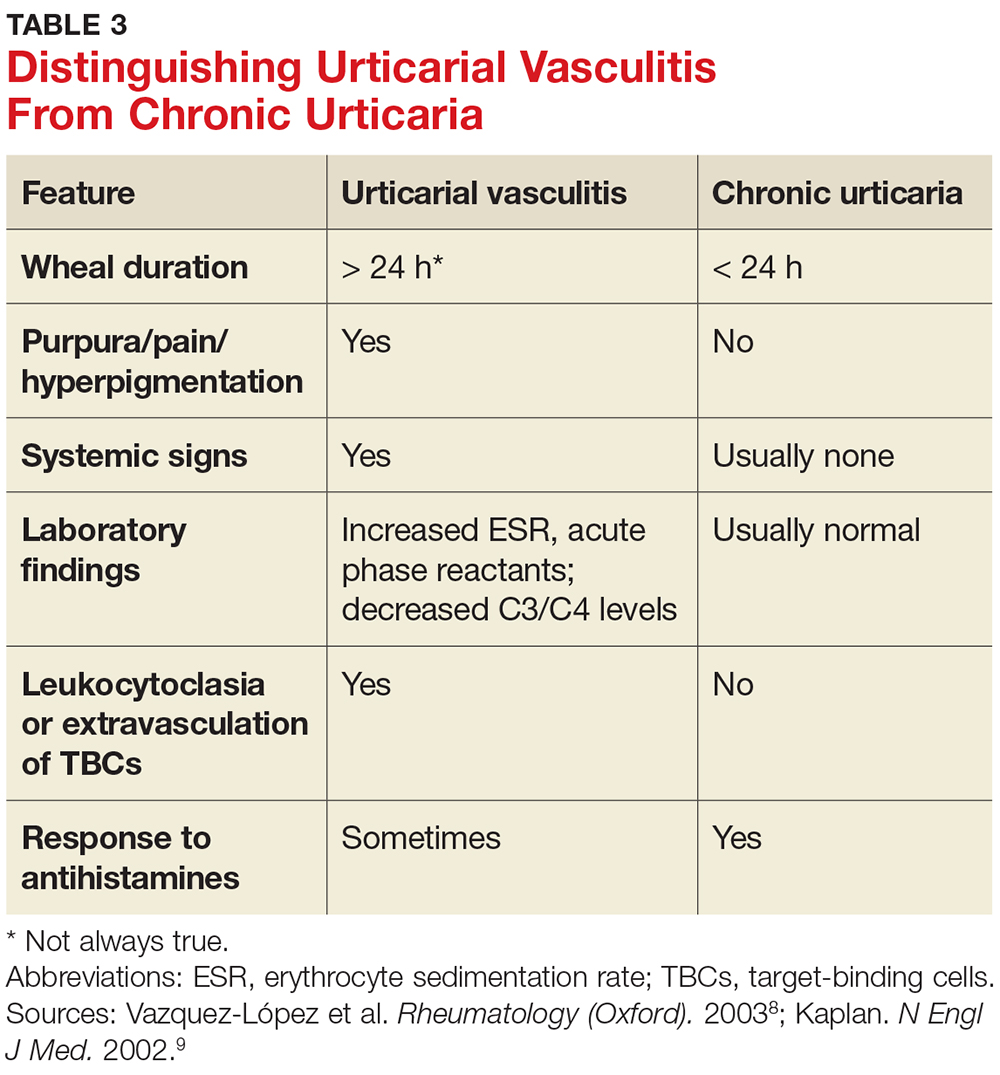
Laboratory evaluation
Although there is no consensus regarding appropriate laboratory testing, the following tests should be considered for patients with chronic urticaria after completion of a thorough history and physical exam: complete blood count (CBC) with differential; erythrocyte sedimentation rate (ESR) and/or C-reactive protein (CRP); chemistry panel and hepatic panel; and thyroid-stimulating hormone, antimicrosomal antibodies, and antithyroglobulin antibodies measurements.7
While the CBC is usually within normal limits, if eosinophilia is present, a workup for an atopic disorder or parasitic infection should be considered. If the ESR/CRP results are positive, consider ordering a larger antinuclear antibody (ANA) panel. Note: The utility of performing these tests routinely for chronic urticaria patients is unclear, as studies have demonstrated that results are usually normal. But it is important to order the appropriate tests to help you rule in or out a likely diagnosis.
Additional testing may be indicated by non-IgE or possible autoimmune findings on the history and/or physical exam. This can include a functional autoantibody assay (for autoantibodies to the high-affinity IgE receptor [FcR1]); complement analysis (eg, C3, C4, CH50), especially when concerned about hereditary angioedema; stool analysis for ova and parasites; Helicobacter pylori workup (there is limited experimental evidence to recommend this, however); hepatitis B and C workup; chest radiograph and/or other imaging studies; ANA panel; rheumatoid factor; cryoglobulin levels; skin biopsy; and urinalysis.7
Local urticaria can occur following contact with allergens via an IgE-mediated mechanism. If an allergen is suspected as a possible trigger, serologic testing to assess allergen-specific IgE levels that may be contributing to the urticaria can be performed in a primary care setting. The specific IgE levels most commonly assessed are for the endemic outdoor aeroallergens (eg, pets [cat, dog], dust mites); measurement of food-specific IgE levels can be ordered if a specific allergy is a concern. Allergy skin prick testing for immediate hypersensitivity and a physical challenge test are usually performed in an allergy office by board-certified allergists.
Skin biopsy should be done on all lesions concerning for urticarial vasculitis (see Table 4).2 Biopsy is also important if the hives are painful rather than pruritic, as this may suggest a different cause. The clinician should consider more detailed lab testing and skin biopsy if urticaria does not respond to therapy as anticipated. Also, specific lab testing may be required screening for certain planned medical therapies (eg, glucose-6-phosphate dehydrogenase enzyme deficiency screening before dapsone or hydroxychloroquine therapy).3
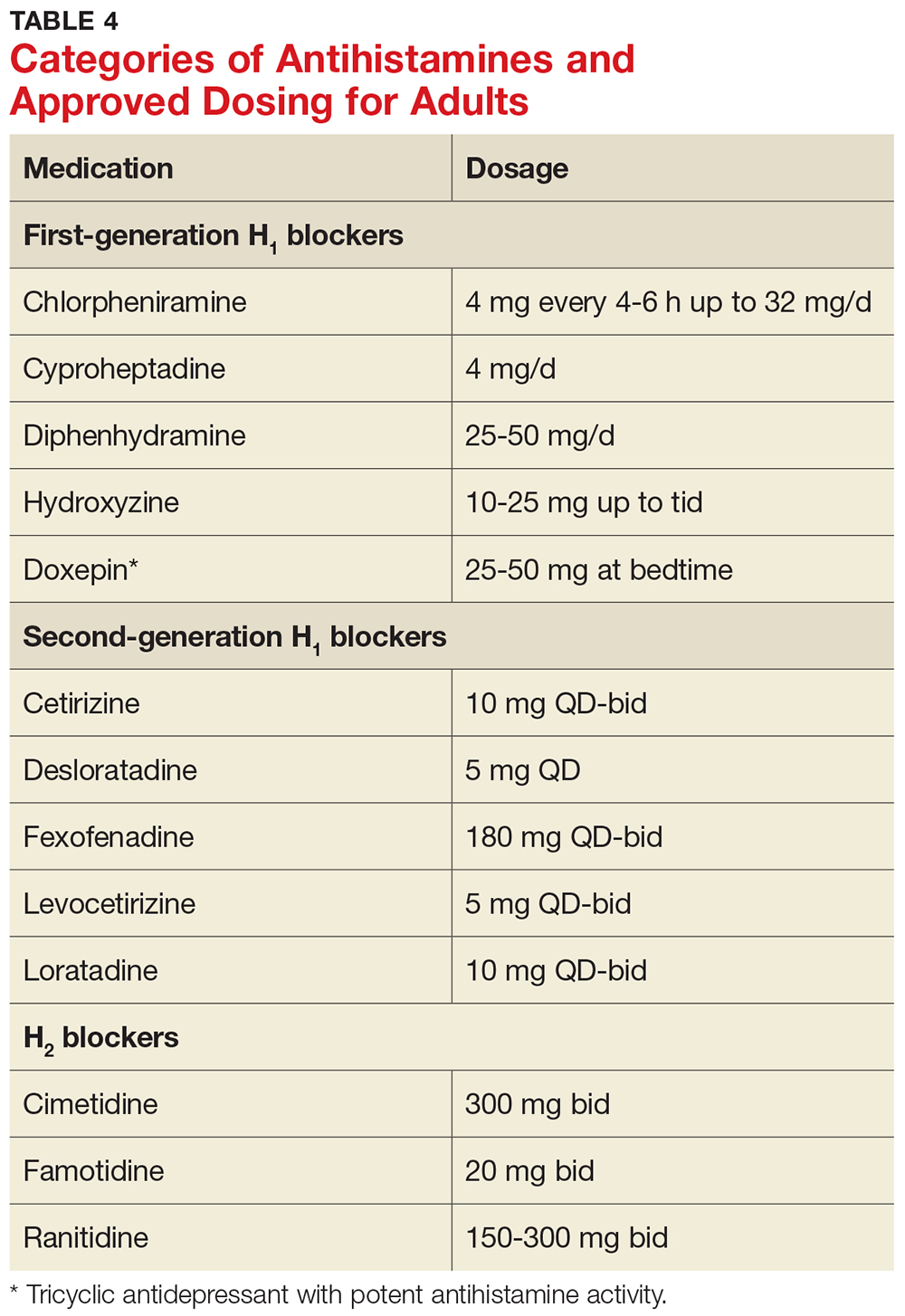
MANAGEMENT
Nonpharmacologic therapy
Treatment of the underlying cause, if identified, may be helpful and should be considered. For example, if a thyroid disorder is found on serologic testing, correcting the disorder may resolve the urticaria.9 Similarly, if a complement deficiency consistent with hereditary angioedema is detected, there are medications to correct it, which can be life-saving.3 Medications for treating hereditary angioedema are best prescribed in an allergy practice.
If triggers are discovered, the patient must be made aware of them and advised to avoid them as much as possible; however, total avoidance can be very difficult. Other common potentiating factors—such as alcohol overuse, excessive tiredness, emotional stress, hyperthermia, and use of aspirin and NSAIDs—should be avoided.10 These factors can worsen what is already triggering the urticaria and make it more difficult to treat; an example would be a patient who develops urticaria from a new household dog and is taking anti-inflammatory drugs for arthritis symptoms.
Topical agents rarely result in any improvement, and their use is therefore discouraged. In fact, high-potency corticosteroids may cause dermal atrophy.11 Also, dietary changes are not indicated for most patients with chronic urticaria, because undiscovered allergy to food or food additives is not likely to be responsible.4
Antihistamines
Antihistamines are the most commonly used pharmacologic treatment for chronic urticaria (see Table 4). H2-receptor blockers, taken in combination with first- and second-generation H1-receptor blockers, have been reported to be more efficacious than H1 antihistamines alone for the treatment of chronic urticaria.6 This added efficacy may be related to pharmacologic interactions and increased blood levels achieved with first-generation antihistamines. Increased doses of second-generation antihistamines—as high as four times the standard dose—are advocated by the 2014 Joint Task Force on Practice Parameters (JTFPP) for the diagnosis and management of acute and chronic urticaria.4
A stepwise approach to treatment is imperative. The JTFPP guidelines (available at www.allergyparameters.org) are summarized below.
Step 1: Administer a second-generation antihistamine at the standard therapeutic dose (see Table 4) and avoid triggers, NSAIDs, and other exacerbating factors.
If symptom control is not achieved in one to two weeks, move on to
Step 2: Increase therapy by one or more of the following methods: increase the dose of the second-generation antihistamine used in Step 1 (up to 4x the standard dose); add another second-generation antihistamine to the regimen; add an H2 blocker (ranitidine, famotidine, cimetidine); and/or add a leukotriene-receptor antagonist (montelukast 10 mg/d).
If these measures do not result in adequate symptom control, it’s time for
Step 3: Gradually increase the dose of H1 antihistamine(s) and discontinue any medications added in Step 2 that did not appear beneficial. Add a first-generation antihistamine (hydroxyzine, doxepin, cyproheptadine), which should be taken at bedtime due to risk for sedation.12
If symptoms are not controlled by Step 3 measures, or if the patient is unable to tolerate an increased dose of first-generation antihistamines, the urticaria is considered refractory. At this point, the clinician should consider referral to an allergy specialist for
Step 4: Add an alternative medication, such as cyclosporine (an anti-inflammatory, immunosuppressive agent) or omalizumab (a monoclonal antibody that selectively binds to IgE).
It should be noted that while the recent FDA approval of omalizumab for treatment of chronic urticaria has been life-changing for many patients, the product label does carry a black box warning about anaphylaxis. Because special monitoring is needed (and prior authorization will likely be required by the patient’s insurer), omalizumab is best prescribed in an allergy office.
It is not uncommon for patients with chronic urticaria to require multiple medications to control their symptoms. Once controlled, they will require maintenance and reevaluation on a regular basis.13
When to refer
Clinicians must know when to refer a patient with chronic urticaria to an allergist/immunologist. Referral is indicated when an underlying disorder is suspected, when symptoms are not controlled with Steps 1 to 3 of the management guidelines, or when the patient requires repeated or prolonged treatment with glucocorticoids.
Unfortunately, out of frustration on both the provider and the patient side, glucocorticoids may be started, after determining that that is “all that works” for the patient. There appears to be a limited role for glucocorticoids, so they should be avoided unless absolutely necessary (ie, if there is no response to antihistamines).
If signs and symptoms suggest urticarial vasculitis, it is prudent to consider referral to a specialist in rheumatology. Urticarial vasculitis requires a special skin punch biopsy to confirm the diagnosis.8 The biopsy procedure may be performed by a primary care provider; if the clinician is not comfortable doing so, referral to an appropriate dermatology provider is indicated.
PATIENT EDUCATION/REASSURANCE
Effective patient education is critical, because patients often experience considerable distress as the symptoms of chronic urticaria wax and wane unpredictably. It is not uncommon for patients with this condition to complain of symptoms that interfere with work, school, and sleep. Reassurance can help to alleviate frustration and anxiety. Patients should understand that the symptoms of chronic urticaria can be successfully managed in the majority of patients, and that chronic idiopathic urticaria is rarely permanent, with about 50% of patients experiencing remission within one year.6
CONCLUSION
The diagnosis of chronic urticaria is based primarily on the presentation, clinical history, and laboratory workup. Management of this chronic and uncomfortable condition requires the identification and exclusion of possible triggers, followed by effective patient education/counseling and a personalized management plan. By knowing when to suspect chronic urticaria, being familiar with the approach to evaluation and initial treatment, and knowing when referral to a specialist is indicated, primary care providers can help their patients find a path to relief.
1. Riedl MA, Ortiz G, Casillas AM. A primary care guide to managing chronic urticaria. JAAPA. 2003;16:WEB.
2. Grieve M. Nettles. http://botanical.com/botanical/mgmh/n/nettle03.html. Accessed December 19, 2017.
3. Powell RJ, Du Toit GL, Siddique N, et al; British Society for Allergy and Clinical Immunology. BSACI guidelines for the management of chronic urticaria and angioedema. Clin Exp Allergy. 2007;37(5):631-650.
4. Bernstein JA, Lang DM, Khan DA, et al. The diagnosis and management of acute and chronic urticaria: 2014 update. J Allergy Clin Immunol. 2014;133(5):1270-1277.
5. Arizona Asthma & Allergy Institute. Possible causes of hives. www.azsneeze.com/hives. Accessed December 19, 2017.
6. Kozel MM, Mekkes JR, Bossuyt PM, Bos JD. Natural course of physical and chronic urticaria and angioedema in 220 patients. J Am Acad Dermatol. 2001;45(3):387-391.
7. Wanderer AA. Hives: The Road to Diagnosis and Treatment of Urticaria. Bozeman, MT: Anson Publishing; 2004.
8. Vazquez-López F, Maldonado-Seral C, Soler-Sánchez T, et al. Surface microscopy for discriminating between common urticaria and urticarial vasculitis. Rheumatology (Oxford). 2003;42(9):1079-1082.
9. Kaplan AP. Chronic urticaria and angioedema. N Engl J Med. 2002;346(3):175-179.
10. Yadav S, Bajaj AK. Management of difficult urticaria. Indian J Dermatol. 2009;54(3):275-279.
11. Ellingsen AR, Thestrup-Pedersen K. Treatment of chronic idiopathic urticaria with topical steroids. An open trial. Acta Derm Venereol. 1996;76(1):43-44.
12. Goldsobel AB, Rohr AS, Siegel SC, et al. Efficacy of doxepin in the treatment of chronic idiopathic urticaria. J Allergy Clin Immunol. 1986;78(5 pt 1):867-873.
13. Ferrer M, Bartra J, Gimenez-Arnau A, et al. Management of urticaria: not too complicated, not too simple. Clin Exp Allergy. 2015;45(4):731-743.
CE/CME No: CR-1801
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Differentiate between acute and chronic urticaria.
• List common history questions required for the diagnosis of chronic urticaria.
• Explain a stepwise plan for treatment of chronic urticaria.
• Describe serologic testing that should be ordered for chronic urticaria.
• Demonstrate knowledge of when to refer patients to a specialist for alternative treatment options.
FACULTY
Randy D. Danielsen is Professor and Dean of the Arizona School of Health Sciences, and Director of the Center for the Future of the Health Professions at A.T. Still University in Mesa, Arizona. Gabriel Ortiz practices at Breathe America El Paso in Texas and is a former AAPA liaison to the American Academy of Allergy, Asthma & Immunology (AAAAI) and National Institutes of Health/National Asthma Education and Prevention Program—Coordinating Committee. Susan Symington has practiced in allergy, asthma, and immunology for more than 10 years. She is the current AAPA liaison to theAAAAI and is President-Elect of the AAPA-Allergy, Asthma, and Immunology subspecialty organization.
The authors have no financial relationships to disclose.
![]()
ACCREDITATION STATEMENT
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid through December 31, 2018.
Article begins on next page >>
The discomfort caused by an urticarial rash, along with its unpredictable course, can interfere with a patient’s sleep and work/school. Adding to the frustration of patients and providers alike, an underlying cause is seldom identified. But a stepwise treatment approach can bring relief to all.
Urticaria, often referred to as hives, is a common cutaneous disorder with a lifetime incidence between 15% and 25%.1 Urticaria is characterized by recurring pruritic wheals that arise due to allergic and nonallergic reactions to internal and external agents. The name urticaria comes from the Latin word for “nettle,” urtica, derived from the Latin word uro, meaning “to burn.”2
Urticaria can be debilitating for patients, who may complain of a burning sensation. It can last for years in some and reduces quality of life for many. Recently, more successful treatments for urticaria have emerged that can provide tremendous relief.
It is important to understand some of the ways to diagnose and treat patients in a primary care setting and also to know when referral is appropriate. This article will discuss the diagnosis, treatment, and referral process for patients with chronic urticaria.
PATHOPHYSIOLOGY
Hives most commonly arise from an immunologic reaction in the superficial skin layers that results in the release of histamine, which causes swelling, itching, and erythema. The mast cell is the major effector cell in the pathophysiology of urticaria.3 In immunologic urticaria, the antigen binds to immunoglobulin (Ig) E on the mast cell surface, causing degranulation and release of histamine, which accounts for the wheals and itching associated with the condition. Histamine binds to H1 and H2 receptors in the skin to cause arteriolar dilation, venous constriction, and increased capillary permeability, accounting for the accompanying swelling.3 Not all urticaria is mediated by IgE; it can result from systemic disease processes in the body that are immune related but not related to IgE. An example would be autoimmune urticaria.
Urticaria commonly occurs with angioedema, which is marked by a greater degree of swelling and results from mast cell activation in the deeper dermis and subcutaneous tissue. Either condition can occur independently, however. Angioedema typically affects the lips, tongue, face, pharynx, and bilateral extremities; rarely, it affects the gastrointestinal tract. Angioedema may be hereditary, but its nonhereditary causes can be similar to those of urticaria.3 For example, a patient could be severely allergic to cat dander and, when exposed to this allergic trigger, develop swelling of the lips, facial edema, and flushing.
FORMS OF URTICARIA
Urticaria can be broadly divided based on the duration of illness: less than six weeks is termed acute urticaria, and continuous or intermittent presence for six weeks or more, chronic urticaria.4
Acute urticaria may occur in any age group but is most often seen in children.1 Acute urticaria and angioedema frequently resolve within a few days, without an identified cause. An inciting cause can be identified in only about 15% to 20% of cases; the most common cause is viral infection, followed by foods, drugs, insect stings, transfusion reactions, and, rarely, contactants and inhalants (see Table 1).1,5 Acute urticaria that is not associated with angioedema or respiratory distress is usually self-limited. The condition typically resolves before extensive evaluation, including testing for possible allergic triggers, can be done. The associated skin lesions are often self-limited or can be controlled symptomatically with antihistamines and avoidance of known possible triggers.1
Chronic urticaria, sometimes called chronic idiopathic urticaria, is more common in adults, occurs on most days of the week, and, as noted, persists for more than six weeks with no identifiable triggers.6 It affects about 0.5% to 1% of the population (lifetime prevalence).3 Approximately 45% of patients with chronic urticaria have accompanying episodes of angioedema, and 30% to 50% have an autoimmune process involving autoantibodies against the thyroid, IgE, or the high-affinity IgE receptor (FcR1).3 The diagnosis is based primarily on clinical history and presentation; this will guide the determination of what types of diagnostic testing are necessary.
Chronic urticaria requires an extensive, but not indiscriminate, evaluation with history, physical examination, allergy testing, and laboratory testing for immune system, liver, kidney, thyroid, and collagen vascular diseases.3 Unfortunately, an identifiable cause of chronic urticaria is found in only 10% to 20% of patients; most cases are idiopathic.3,7
Several forms of chronic urticaria can be precipitated by physical stimuli, such as exercise, generalized heat, or sweating (cholinergic urticaria); localized heat (localized heat urticaria); low temperatures (cold urticaria); sun exposure (solar urticaria); water (aquagenic urticaria); and vibration.1 In another form (pressure urticaria), pressure on the skin increases histamine release, leading to the development of wheals and itching; this form is also called dermatographism, which means “write on skin” (see Figure 1). These types of urticaria should be evaluated and treated by a board-certified allergist, as there are special evaluations that can confirm the diagnosis.
CLINICAL FEATURES
The main feature of urticaria is raised skin lesions that appear pale to pink to erythematous and most commonly are intensely pruritic (see Figure 2). These lesions range from a few millimeters to several centimeters in size and may coalesce.
Characteristically, evanescent old lesions resolve, and new ones develop over 24 hours, usually without scarring. Scratching generally worsens dermatographism, with new urticaria produced over the scratched area. Any area of the body may be involved.
The lesions of early urticaria may vary in size and blanch when pressure is applied. An individual hive may last minutes or up to 24 hours and may reoccur intermittently on various sites on the body for an unspecified period of time.1,6
DIFFERENTIAL DIAGNOSIS
Other dermatologic conditions may be mistaken for chronic urticaria. Common rashes that may mimic it include anaphylaxis, atopic dermatitis, medication allergy or fixed drug eruption, ACE inhibitor–related angioedema, mastocytosis, contact dermatitis, autoimmune thyroid disease, bullous pemphigoid, and dermatitis herpetiformis.
Patients should be encouraged to bring pictures of the rash to the office visit, since the rash may have waned at the time of the visit and diagnosis based on the patient’s description alone can be challenging. Most rashes in the differential can be identified or eliminated through a careful history and complete physical exam. When necessary, serologic testing and skin punch biopsies can elucidate and confirm the diagnosis.
EVALUATION
History and physical examination
The medical history is the most important part of the evaluation of a patient with urticaria. The information that should be elicited and documented during the history is shown in Table 2.
A general comprehensive physical exam should be undertaken and the findings carefully documented. As noted, it can be helpful for patients to bring in pictures of the rash if the lesions wax and wane. It is also important to assess whether the urticarial lesions blanch when palpated, since this is a characteristic feature of acute and chronic urticarial lesions (but not of those with an autoimmune, cholinergic, or vasculitic cause). Thus, blanching of the wheal is a key finding on physical exam to discriminate between possible causes.8 Lesions pigmented with purpuric areas that scar or last longer than 24 hours suggest urticarial vasculitis; other features that distinguish urticarial vasculitis from chronic urticaria are listed in Table 3.2
Laboratory evaluation
Although there is no consensus regarding appropriate laboratory testing, the following tests should be considered for patients with chronic urticaria after completion of a thorough history and physical exam: complete blood count (CBC) with differential; erythrocyte sedimentation rate (ESR) and/or C-reactive protein (CRP); chemistry panel and hepatic panel; and thyroid-stimulating hormone, antimicrosomal antibodies, and antithyroglobulin antibodies measurements.7
While the CBC is usually within normal limits, if eosinophilia is present, a workup for an atopic disorder or parasitic infection should be considered. If the ESR/CRP results are positive, consider ordering a larger antinuclear antibody (ANA) panel. Note: The utility of performing these tests routinely for chronic urticaria patients is unclear, as studies have demonstrated that results are usually normal. But it is important to order the appropriate tests to help you rule in or out a likely diagnosis.
Additional testing may be indicated by non-IgE or possible autoimmune findings on the history and/or physical exam. This can include a functional autoantibody assay (for autoantibodies to the high-affinity IgE receptor [FcR1]); complement analysis (eg, C3, C4, CH50), especially when concerned about hereditary angioedema; stool analysis for ova and parasites; Helicobacter pylori workup (there is limited experimental evidence to recommend this, however); hepatitis B and C workup; chest radiograph and/or other imaging studies; ANA panel; rheumatoid factor; cryoglobulin levels; skin biopsy; and urinalysis.7
Local urticaria can occur following contact with allergens via an IgE-mediated mechanism. If an allergen is suspected as a possible trigger, serologic testing to assess allergen-specific IgE levels that may be contributing to the urticaria can be performed in a primary care setting. The specific IgE levels most commonly assessed are for the endemic outdoor aeroallergens (eg, pets [cat, dog], dust mites); measurement of food-specific IgE levels can be ordered if a specific allergy is a concern. Allergy skin prick testing for immediate hypersensitivity and a physical challenge test are usually performed in an allergy office by board-certified allergists.
Skin biopsy should be done on all lesions concerning for urticarial vasculitis (see Table 4).2 Biopsy is also important if the hives are painful rather than pruritic, as this may suggest a different cause. The clinician should consider more detailed lab testing and skin biopsy if urticaria does not respond to therapy as anticipated. Also, specific lab testing may be required screening for certain planned medical therapies (eg, glucose-6-phosphate dehydrogenase enzyme deficiency screening before dapsone or hydroxychloroquine therapy).3
MANAGEMENT
Nonpharmacologic therapy
Treatment of the underlying cause, if identified, may be helpful and should be considered. For example, if a thyroid disorder is found on serologic testing, correcting the disorder may resolve the urticaria.9 Similarly, if a complement deficiency consistent with hereditary angioedema is detected, there are medications to correct it, which can be life-saving.3 Medications for treating hereditary angioedema are best prescribed in an allergy practice.
If triggers are discovered, the patient must be made aware of them and advised to avoid them as much as possible; however, total avoidance can be very difficult. Other common potentiating factors—such as alcohol overuse, excessive tiredness, emotional stress, hyperthermia, and use of aspirin and NSAIDs—should be avoided.10 These factors can worsen what is already triggering the urticaria and make it more difficult to treat; an example would be a patient who develops urticaria from a new household dog and is taking anti-inflammatory drugs for arthritis symptoms.
Topical agents rarely result in any improvement, and their use is therefore discouraged. In fact, high-potency corticosteroids may cause dermal atrophy.11 Also, dietary changes are not indicated for most patients with chronic urticaria, because undiscovered allergy to food or food additives is not likely to be responsible.4
Antihistamines
Antihistamines are the most commonly used pharmacologic treatment for chronic urticaria (see Table 4). H2-receptor blockers, taken in combination with first- and second-generation H1-receptor blockers, have been reported to be more efficacious than H1 antihistamines alone for the treatment of chronic urticaria.6 This added efficacy may be related to pharmacologic interactions and increased blood levels achieved with first-generation antihistamines. Increased doses of second-generation antihistamines—as high as four times the standard dose—are advocated by the 2014 Joint Task Force on Practice Parameters (JTFPP) for the diagnosis and management of acute and chronic urticaria.4
A stepwise approach to treatment is imperative. The JTFPP guidelines (available at www.allergyparameters.org) are summarized below.
Step 1: Administer a second-generation antihistamine at the standard therapeutic dose (see Table 4) and avoid triggers, NSAIDs, and other exacerbating factors.
If symptom control is not achieved in one to two weeks, move on to
Step 2: Increase therapy by one or more of the following methods: increase the dose of the second-generation antihistamine used in Step 1 (up to 4x the standard dose); add another second-generation antihistamine to the regimen; add an H2 blocker (ranitidine, famotidine, cimetidine); and/or add a leukotriene-receptor antagonist (montelukast 10 mg/d).
If these measures do not result in adequate symptom control, it’s time for
Step 3: Gradually increase the dose of H1 antihistamine(s) and discontinue any medications added in Step 2 that did not appear beneficial. Add a first-generation antihistamine (hydroxyzine, doxepin, cyproheptadine), which should be taken at bedtime due to risk for sedation.12
If symptoms are not controlled by Step 3 measures, or if the patient is unable to tolerate an increased dose of first-generation antihistamines, the urticaria is considered refractory. At this point, the clinician should consider referral to an allergy specialist for
Step 4: Add an alternative medication, such as cyclosporine (an anti-inflammatory, immunosuppressive agent) or omalizumab (a monoclonal antibody that selectively binds to IgE).
It should be noted that while the recent FDA approval of omalizumab for treatment of chronic urticaria has been life-changing for many patients, the product label does carry a black box warning about anaphylaxis. Because special monitoring is needed (and prior authorization will likely be required by the patient’s insurer), omalizumab is best prescribed in an allergy office.
It is not uncommon for patients with chronic urticaria to require multiple medications to control their symptoms. Once controlled, they will require maintenance and reevaluation on a regular basis.13
When to refer
Clinicians must know when to refer a patient with chronic urticaria to an allergist/immunologist. Referral is indicated when an underlying disorder is suspected, when symptoms are not controlled with Steps 1 to 3 of the management guidelines, or when the patient requires repeated or prolonged treatment with glucocorticoids.
Unfortunately, out of frustration on both the provider and the patient side, glucocorticoids may be started, after determining that that is “all that works” for the patient. There appears to be a limited role for glucocorticoids, so they should be avoided unless absolutely necessary (ie, if there is no response to antihistamines).
If signs and symptoms suggest urticarial vasculitis, it is prudent to consider referral to a specialist in rheumatology. Urticarial vasculitis requires a special skin punch biopsy to confirm the diagnosis.8 The biopsy procedure may be performed by a primary care provider; if the clinician is not comfortable doing so, referral to an appropriate dermatology provider is indicated.
PATIENT EDUCATION/REASSURANCE
Effective patient education is critical, because patients often experience considerable distress as the symptoms of chronic urticaria wax and wane unpredictably. It is not uncommon for patients with this condition to complain of symptoms that interfere with work, school, and sleep. Reassurance can help to alleviate frustration and anxiety. Patients should understand that the symptoms of chronic urticaria can be successfully managed in the majority of patients, and that chronic idiopathic urticaria is rarely permanent, with about 50% of patients experiencing remission within one year.6
CONCLUSION
The diagnosis of chronic urticaria is based primarily on the presentation, clinical history, and laboratory workup. Management of this chronic and uncomfortable condition requires the identification and exclusion of possible triggers, followed by effective patient education/counseling and a personalized management plan. By knowing when to suspect chronic urticaria, being familiar with the approach to evaluation and initial treatment, and knowing when referral to a specialist is indicated, primary care providers can help their patients find a path to relief.
CE/CME No: CR-1801
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Differentiate between acute and chronic urticaria.
• List common history questions required for the diagnosis of chronic urticaria.
• Explain a stepwise plan for treatment of chronic urticaria.
• Describe serologic testing that should be ordered for chronic urticaria.
• Demonstrate knowledge of when to refer patients to a specialist for alternative treatment options.
FACULTY
Randy D. Danielsen is Professor and Dean of the Arizona School of Health Sciences, and Director of the Center for the Future of the Health Professions at A.T. Still University in Mesa, Arizona. Gabriel Ortiz practices at Breathe America El Paso in Texas and is a former AAPA liaison to the American Academy of Allergy, Asthma & Immunology (AAAAI) and National Institutes of Health/National Asthma Education and Prevention Program—Coordinating Committee. Susan Symington has practiced in allergy, asthma, and immunology for more than 10 years. She is the current AAPA liaison to theAAAAI and is President-Elect of the AAPA-Allergy, Asthma, and Immunology subspecialty organization.
The authors have no financial relationships to disclose.
![]()
ACCREDITATION STATEMENT
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid through December 31, 2018.
Article begins on next page >>
The discomfort caused by an urticarial rash, along with its unpredictable course, can interfere with a patient’s sleep and work/school. Adding to the frustration of patients and providers alike, an underlying cause is seldom identified. But a stepwise treatment approach can bring relief to all.
Urticaria, often referred to as hives, is a common cutaneous disorder with a lifetime incidence between 15% and 25%.1 Urticaria is characterized by recurring pruritic wheals that arise due to allergic and nonallergic reactions to internal and external agents. The name urticaria comes from the Latin word for “nettle,” urtica, derived from the Latin word uro, meaning “to burn.”2
Urticaria can be debilitating for patients, who may complain of a burning sensation. It can last for years in some and reduces quality of life for many. Recently, more successful treatments for urticaria have emerged that can provide tremendous relief.
It is important to understand some of the ways to diagnose and treat patients in a primary care setting and also to know when referral is appropriate. This article will discuss the diagnosis, treatment, and referral process for patients with chronic urticaria.
PATHOPHYSIOLOGY
Hives most commonly arise from an immunologic reaction in the superficial skin layers that results in the release of histamine, which causes swelling, itching, and erythema. The mast cell is the major effector cell in the pathophysiology of urticaria.3 In immunologic urticaria, the antigen binds to immunoglobulin (Ig) E on the mast cell surface, causing degranulation and release of histamine, which accounts for the wheals and itching associated with the condition. Histamine binds to H1 and H2 receptors in the skin to cause arteriolar dilation, venous constriction, and increased capillary permeability, accounting for the accompanying swelling.3 Not all urticaria is mediated by IgE; it can result from systemic disease processes in the body that are immune related but not related to IgE. An example would be autoimmune urticaria.
Urticaria commonly occurs with angioedema, which is marked by a greater degree of swelling and results from mast cell activation in the deeper dermis and subcutaneous tissue. Either condition can occur independently, however. Angioedema typically affects the lips, tongue, face, pharynx, and bilateral extremities; rarely, it affects the gastrointestinal tract. Angioedema may be hereditary, but its nonhereditary causes can be similar to those of urticaria.3 For example, a patient could be severely allergic to cat dander and, when exposed to this allergic trigger, develop swelling of the lips, facial edema, and flushing.
FORMS OF URTICARIA
Urticaria can be broadly divided based on the duration of illness: less than six weeks is termed acute urticaria, and continuous or intermittent presence for six weeks or more, chronic urticaria.4
Acute urticaria may occur in any age group but is most often seen in children.1 Acute urticaria and angioedema frequently resolve within a few days, without an identified cause. An inciting cause can be identified in only about 15% to 20% of cases; the most common cause is viral infection, followed by foods, drugs, insect stings, transfusion reactions, and, rarely, contactants and inhalants (see Table 1).1,5 Acute urticaria that is not associated with angioedema or respiratory distress is usually self-limited. The condition typically resolves before extensive evaluation, including testing for possible allergic triggers, can be done. The associated skin lesions are often self-limited or can be controlled symptomatically with antihistamines and avoidance of known possible triggers.1
Chronic urticaria, sometimes called chronic idiopathic urticaria, is more common in adults, occurs on most days of the week, and, as noted, persists for more than six weeks with no identifiable triggers.6 It affects about 0.5% to 1% of the population (lifetime prevalence).3 Approximately 45% of patients with chronic urticaria have accompanying episodes of angioedema, and 30% to 50% have an autoimmune process involving autoantibodies against the thyroid, IgE, or the high-affinity IgE receptor (FcR1).3 The diagnosis is based primarily on clinical history and presentation; this will guide the determination of what types of diagnostic testing are necessary.
Chronic urticaria requires an extensive, but not indiscriminate, evaluation with history, physical examination, allergy testing, and laboratory testing for immune system, liver, kidney, thyroid, and collagen vascular diseases.3 Unfortunately, an identifiable cause of chronic urticaria is found in only 10% to 20% of patients; most cases are idiopathic.3,7
Several forms of chronic urticaria can be precipitated by physical stimuli, such as exercise, generalized heat, or sweating (cholinergic urticaria); localized heat (localized heat urticaria); low temperatures (cold urticaria); sun exposure (solar urticaria); water (aquagenic urticaria); and vibration.1 In another form (pressure urticaria), pressure on the skin increases histamine release, leading to the development of wheals and itching; this form is also called dermatographism, which means “write on skin” (see Figure 1). These types of urticaria should be evaluated and treated by a board-certified allergist, as there are special evaluations that can confirm the diagnosis.
CLINICAL FEATURES
The main feature of urticaria is raised skin lesions that appear pale to pink to erythematous and most commonly are intensely pruritic (see Figure 2). These lesions range from a few millimeters to several centimeters in size and may coalesce.
Characteristically, evanescent old lesions resolve, and new ones develop over 24 hours, usually without scarring. Scratching generally worsens dermatographism, with new urticaria produced over the scratched area. Any area of the body may be involved.
The lesions of early urticaria may vary in size and blanch when pressure is applied. An individual hive may last minutes or up to 24 hours and may reoccur intermittently on various sites on the body for an unspecified period of time.1,6
DIFFERENTIAL DIAGNOSIS
Other dermatologic conditions may be mistaken for chronic urticaria. Common rashes that may mimic it include anaphylaxis, atopic dermatitis, medication allergy or fixed drug eruption, ACE inhibitor–related angioedema, mastocytosis, contact dermatitis, autoimmune thyroid disease, bullous pemphigoid, and dermatitis herpetiformis.
Patients should be encouraged to bring pictures of the rash to the office visit, since the rash may have waned at the time of the visit and diagnosis based on the patient’s description alone can be challenging. Most rashes in the differential can be identified or eliminated through a careful history and complete physical exam. When necessary, serologic testing and skin punch biopsies can elucidate and confirm the diagnosis.
EVALUATION
History and physical examination
The medical history is the most important part of the evaluation of a patient with urticaria. The information that should be elicited and documented during the history is shown in Table 2.
A general comprehensive physical exam should be undertaken and the findings carefully documented. As noted, it can be helpful for patients to bring in pictures of the rash if the lesions wax and wane. It is also important to assess whether the urticarial lesions blanch when palpated, since this is a characteristic feature of acute and chronic urticarial lesions (but not of those with an autoimmune, cholinergic, or vasculitic cause). Thus, blanching of the wheal is a key finding on physical exam to discriminate between possible causes.8 Lesions pigmented with purpuric areas that scar or last longer than 24 hours suggest urticarial vasculitis; other features that distinguish urticarial vasculitis from chronic urticaria are listed in Table 3.2
Laboratory evaluation
Although there is no consensus regarding appropriate laboratory testing, the following tests should be considered for patients with chronic urticaria after completion of a thorough history and physical exam: complete blood count (CBC) with differential; erythrocyte sedimentation rate (ESR) and/or C-reactive protein (CRP); chemistry panel and hepatic panel; and thyroid-stimulating hormone, antimicrosomal antibodies, and antithyroglobulin antibodies measurements.7
While the CBC is usually within normal limits, if eosinophilia is present, a workup for an atopic disorder or parasitic infection should be considered. If the ESR/CRP results are positive, consider ordering a larger antinuclear antibody (ANA) panel. Note: The utility of performing these tests routinely for chronic urticaria patients is unclear, as studies have demonstrated that results are usually normal. But it is important to order the appropriate tests to help you rule in or out a likely diagnosis.
Additional testing may be indicated by non-IgE or possible autoimmune findings on the history and/or physical exam. This can include a functional autoantibody assay (for autoantibodies to the high-affinity IgE receptor [FcR1]); complement analysis (eg, C3, C4, CH50), especially when concerned about hereditary angioedema; stool analysis for ova and parasites; Helicobacter pylori workup (there is limited experimental evidence to recommend this, however); hepatitis B and C workup; chest radiograph and/or other imaging studies; ANA panel; rheumatoid factor; cryoglobulin levels; skin biopsy; and urinalysis.7
Local urticaria can occur following contact with allergens via an IgE-mediated mechanism. If an allergen is suspected as a possible trigger, serologic testing to assess allergen-specific IgE levels that may be contributing to the urticaria can be performed in a primary care setting. The specific IgE levels most commonly assessed are for the endemic outdoor aeroallergens (eg, pets [cat, dog], dust mites); measurement of food-specific IgE levels can be ordered if a specific allergy is a concern. Allergy skin prick testing for immediate hypersensitivity and a physical challenge test are usually performed in an allergy office by board-certified allergists.
Skin biopsy should be done on all lesions concerning for urticarial vasculitis (see Table 4).2 Biopsy is also important if the hives are painful rather than pruritic, as this may suggest a different cause. The clinician should consider more detailed lab testing and skin biopsy if urticaria does not respond to therapy as anticipated. Also, specific lab testing may be required screening for certain planned medical therapies (eg, glucose-6-phosphate dehydrogenase enzyme deficiency screening before dapsone or hydroxychloroquine therapy).3
MANAGEMENT
Nonpharmacologic therapy
Treatment of the underlying cause, if identified, may be helpful and should be considered. For example, if a thyroid disorder is found on serologic testing, correcting the disorder may resolve the urticaria.9 Similarly, if a complement deficiency consistent with hereditary angioedema is detected, there are medications to correct it, which can be life-saving.3 Medications for treating hereditary angioedema are best prescribed in an allergy practice.
If triggers are discovered, the patient must be made aware of them and advised to avoid them as much as possible; however, total avoidance can be very difficult. Other common potentiating factors—such as alcohol overuse, excessive tiredness, emotional stress, hyperthermia, and use of aspirin and NSAIDs—should be avoided.10 These factors can worsen what is already triggering the urticaria and make it more difficult to treat; an example would be a patient who develops urticaria from a new household dog and is taking anti-inflammatory drugs for arthritis symptoms.
Topical agents rarely result in any improvement, and their use is therefore discouraged. In fact, high-potency corticosteroids may cause dermal atrophy.11 Also, dietary changes are not indicated for most patients with chronic urticaria, because undiscovered allergy to food or food additives is not likely to be responsible.4
Antihistamines
Antihistamines are the most commonly used pharmacologic treatment for chronic urticaria (see Table 4). H2-receptor blockers, taken in combination with first- and second-generation H1-receptor blockers, have been reported to be more efficacious than H1 antihistamines alone for the treatment of chronic urticaria.6 This added efficacy may be related to pharmacologic interactions and increased blood levels achieved with first-generation antihistamines. Increased doses of second-generation antihistamines—as high as four times the standard dose—are advocated by the 2014 Joint Task Force on Practice Parameters (JTFPP) for the diagnosis and management of acute and chronic urticaria.4
A stepwise approach to treatment is imperative. The JTFPP guidelines (available at www.allergyparameters.org) are summarized below.
Step 1: Administer a second-generation antihistamine at the standard therapeutic dose (see Table 4) and avoid triggers, NSAIDs, and other exacerbating factors.
If symptom control is not achieved in one to two weeks, move on to
Step 2: Increase therapy by one or more of the following methods: increase the dose of the second-generation antihistamine used in Step 1 (up to 4x the standard dose); add another second-generation antihistamine to the regimen; add an H2 blocker (ranitidine, famotidine, cimetidine); and/or add a leukotriene-receptor antagonist (montelukast 10 mg/d).
If these measures do not result in adequate symptom control, it’s time for
Step 3: Gradually increase the dose of H1 antihistamine(s) and discontinue any medications added in Step 2 that did not appear beneficial. Add a first-generation antihistamine (hydroxyzine, doxepin, cyproheptadine), which should be taken at bedtime due to risk for sedation.12
If symptoms are not controlled by Step 3 measures, or if the patient is unable to tolerate an increased dose of first-generation antihistamines, the urticaria is considered refractory. At this point, the clinician should consider referral to an allergy specialist for
Step 4: Add an alternative medication, such as cyclosporine (an anti-inflammatory, immunosuppressive agent) or omalizumab (a monoclonal antibody that selectively binds to IgE).
It should be noted that while the recent FDA approval of omalizumab for treatment of chronic urticaria has been life-changing for many patients, the product label does carry a black box warning about anaphylaxis. Because special monitoring is needed (and prior authorization will likely be required by the patient’s insurer), omalizumab is best prescribed in an allergy office.
It is not uncommon for patients with chronic urticaria to require multiple medications to control their symptoms. Once controlled, they will require maintenance and reevaluation on a regular basis.13
When to refer
Clinicians must know when to refer a patient with chronic urticaria to an allergist/immunologist. Referral is indicated when an underlying disorder is suspected, when symptoms are not controlled with Steps 1 to 3 of the management guidelines, or when the patient requires repeated or prolonged treatment with glucocorticoids.
Unfortunately, out of frustration on both the provider and the patient side, glucocorticoids may be started, after determining that that is “all that works” for the patient. There appears to be a limited role for glucocorticoids, so they should be avoided unless absolutely necessary (ie, if there is no response to antihistamines).
If signs and symptoms suggest urticarial vasculitis, it is prudent to consider referral to a specialist in rheumatology. Urticarial vasculitis requires a special skin punch biopsy to confirm the diagnosis.8 The biopsy procedure may be performed by a primary care provider; if the clinician is not comfortable doing so, referral to an appropriate dermatology provider is indicated.
PATIENT EDUCATION/REASSURANCE
Effective patient education is critical, because patients often experience considerable distress as the symptoms of chronic urticaria wax and wane unpredictably. It is not uncommon for patients with this condition to complain of symptoms that interfere with work, school, and sleep. Reassurance can help to alleviate frustration and anxiety. Patients should understand that the symptoms of chronic urticaria can be successfully managed in the majority of patients, and that chronic idiopathic urticaria is rarely permanent, with about 50% of patients experiencing remission within one year.6
CONCLUSION
The diagnosis of chronic urticaria is based primarily on the presentation, clinical history, and laboratory workup. Management of this chronic and uncomfortable condition requires the identification and exclusion of possible triggers, followed by effective patient education/counseling and a personalized management plan. By knowing when to suspect chronic urticaria, being familiar with the approach to evaluation and initial treatment, and knowing when referral to a specialist is indicated, primary care providers can help their patients find a path to relief.
1. Riedl MA, Ortiz G, Casillas AM. A primary care guide to managing chronic urticaria. JAAPA. 2003;16:WEB.
2. Grieve M. Nettles. http://botanical.com/botanical/mgmh/n/nettle03.html. Accessed December 19, 2017.
3. Powell RJ, Du Toit GL, Siddique N, et al; British Society for Allergy and Clinical Immunology. BSACI guidelines for the management of chronic urticaria and angioedema. Clin Exp Allergy. 2007;37(5):631-650.
4. Bernstein JA, Lang DM, Khan DA, et al. The diagnosis and management of acute and chronic urticaria: 2014 update. J Allergy Clin Immunol. 2014;133(5):1270-1277.
5. Arizona Asthma & Allergy Institute. Possible causes of hives. www.azsneeze.com/hives. Accessed December 19, 2017.
6. Kozel MM, Mekkes JR, Bossuyt PM, Bos JD. Natural course of physical and chronic urticaria and angioedema in 220 patients. J Am Acad Dermatol. 2001;45(3):387-391.
7. Wanderer AA. Hives: The Road to Diagnosis and Treatment of Urticaria. Bozeman, MT: Anson Publishing; 2004.
8. Vazquez-López F, Maldonado-Seral C, Soler-Sánchez T, et al. Surface microscopy for discriminating between common urticaria and urticarial vasculitis. Rheumatology (Oxford). 2003;42(9):1079-1082.
9. Kaplan AP. Chronic urticaria and angioedema. N Engl J Med. 2002;346(3):175-179.
10. Yadav S, Bajaj AK. Management of difficult urticaria. Indian J Dermatol. 2009;54(3):275-279.
11. Ellingsen AR, Thestrup-Pedersen K. Treatment of chronic idiopathic urticaria with topical steroids. An open trial. Acta Derm Venereol. 1996;76(1):43-44.
12. Goldsobel AB, Rohr AS, Siegel SC, et al. Efficacy of doxepin in the treatment of chronic idiopathic urticaria. J Allergy Clin Immunol. 1986;78(5 pt 1):867-873.
13. Ferrer M, Bartra J, Gimenez-Arnau A, et al. Management of urticaria: not too complicated, not too simple. Clin Exp Allergy. 2015;45(4):731-743.
1. Riedl MA, Ortiz G, Casillas AM. A primary care guide to managing chronic urticaria. JAAPA. 2003;16:WEB.
2. Grieve M. Nettles. http://botanical.com/botanical/mgmh/n/nettle03.html. Accessed December 19, 2017.
3. Powell RJ, Du Toit GL, Siddique N, et al; British Society for Allergy and Clinical Immunology. BSACI guidelines for the management of chronic urticaria and angioedema. Clin Exp Allergy. 2007;37(5):631-650.
4. Bernstein JA, Lang DM, Khan DA, et al. The diagnosis and management of acute and chronic urticaria: 2014 update. J Allergy Clin Immunol. 2014;133(5):1270-1277.
5. Arizona Asthma & Allergy Institute. Possible causes of hives. www.azsneeze.com/hives. Accessed December 19, 2017.
6. Kozel MM, Mekkes JR, Bossuyt PM, Bos JD. Natural course of physical and chronic urticaria and angioedema in 220 patients. J Am Acad Dermatol. 2001;45(3):387-391.
7. Wanderer AA. Hives: The Road to Diagnosis and Treatment of Urticaria. Bozeman, MT: Anson Publishing; 2004.
8. Vazquez-López F, Maldonado-Seral C, Soler-Sánchez T, et al. Surface microscopy for discriminating between common urticaria and urticarial vasculitis. Rheumatology (Oxford). 2003;42(9):1079-1082.
9. Kaplan AP. Chronic urticaria and angioedema. N Engl J Med. 2002;346(3):175-179.
10. Yadav S, Bajaj AK. Management of difficult urticaria. Indian J Dermatol. 2009;54(3):275-279.
11. Ellingsen AR, Thestrup-Pedersen K. Treatment of chronic idiopathic urticaria with topical steroids. An open trial. Acta Derm Venereol. 1996;76(1):43-44.
12. Goldsobel AB, Rohr AS, Siegel SC, et al. Efficacy of doxepin in the treatment of chronic idiopathic urticaria. J Allergy Clin Immunol. 1986;78(5 pt 1):867-873.
13. Ferrer M, Bartra J, Gimenez-Arnau A, et al. Management of urticaria: not too complicated, not too simple. Clin Exp Allergy. 2015;45(4):731-743.
Diagnosing & Treating Neuromyelitis Optica Spectrum Disorder
Q) How do you know if a neurologic symptom is due to a relapse of neuromyelitis optica spectrum disorder? And how should a confirmed relapse be treated?
Neuromyelitis optica spectrum disorder (NMOSD) is a severe, relapsing autoimmune disease of the central nervous system (CNS) that targets the optic nerves and spinal cord, leading to blindness and paralysis.1,2 Whereas multiple sclerosis (MS) is characterized by demyelination, NMOSD is associated with astrocytic damage and tissue necrosis.3 Because longitudinally extensive inflammatory lesions are typical with NMOSD, permanent CNS damage is common with each relapse.4
Health care providers first need to determine whether a patient with NMOSD who presents with new or worsening symptoms is having a relapse. A relapse is caused by a breach of the blood-brain barrier by the peripheral immune system, which leads to inflammation and damage to the CNS.5 This causes neurologic symptoms that depend on the anatomic location. Once damage has occurred, symptoms may result either from a new relapse in the same location as a previous inflammatory event or from a pseudorelapse.6
Pseudorelapses are triggered by a systemic metabolic imbalance; they exacerbate symptoms from previous CNS damage. Differentiating between a true relapse and a pseudorelapse can be a diagnostic challenge for even the most seasoned of health care providers. Kessler et al retrospectively examined which clinical factors can distinguish relapses from pseudorelapses.6 Their findings suggest that while clinical examination alone may be effective in events involving vision loss, MRI may be necessary when signs and symptoms are attributable to a spinal cord lesion.
In fact, they found that the degree of clinical worsening in patients with spinal cord symptoms caused by a pseudorelapse was similar to that of a true relapse. The most common causes of pseudorelapse included infection, dysautonomia, metabolic abnormalities, and changes to medication regimens. Interestingly, the presence of infection did not rule out a relapse, as patients experiencing relapses were equally likely as those with pseudorelapse to have a urinary tract infection. The authors concluded, based on their data, that an MRI is warranted to verify a relapse in patients who experience worsening of symptoms localized to the spinal cord but is not necessary to rule out a pseudorelapse of optic neu
In contrast to MS, a progressive phase is not believed to be associated with NMOSD.7 Instead, accrual of disability occurs with each relapse. The majority of patients with NMOSD do not return to baseline following an untreated relapse, making it especially important that patients receive adequate acute treatment to mitigate the damage.8
Currently, there are no medications approved by the FDA for the acute or preventive treatment of NMOSD. However, off-label use of immunotherapies, including rituximab, mycophenolate mofetil, azathioprine, prednisone, methotrexate, tocilizumab, and mitoxantrone, have been studied for relapse prevention.2 In addition, there are three ongoing phase III trials investigating eculizumab (C5 complement inhibitor), inebilizumab (CD19 monoclonal antibody), and SA237 (IL6R blocker); results from these studies could potentially widen the landscape of immunotherapy use in NMOSD.2
Less investigation into appropriate acute treatment of new relapses has been conducted, however, leaving clinicians and patients uncertain about how to manage a new inflammatory event. Traditionally, firstline treatment for acute NMOSD relapses has been the same as for MS relapses—high-dose methylprednisolone. However, due to the severity of NMOSD relapses and the relative lack of response to steroids alone, methylprednisolone is commonly followed by plasma exchange (PLEX).2
Most data to guide clinical decision-making suggest that patients with NMOSD relapses recover better when PLEX is added to steroid treatment. Abboud et al found that 65% of patients who received both PLEX and methylprednisolone recovered to their prerelapse baseline, compared to 35% of those who received methylprednisolone alone.9 These findings were supported by a larger retrospective investigation by Kleiter et al, which found improved recovery with treatment escalation in their cohort.8 These data support the recommendation to use PLEX as an adjunct therapy in acute relapses—particularly in relapses with severe presentations.

Because diagnosis and treatment of relapses involve many factors, ranging from accrual of disability, long-term immunotherapy decisions, and medical costs, diligence in provider decision-making is essential when caring for patients with NMOSD. -MAM
Maureen A. Mealy, BSN, MSCN
Neuromyelitis Optica Research Program Manager, Senior Research Nurse of the Transverse Myelitis & Multiple Sclerosis Centers, PhD candidate at Johns Hopkins School of Nursing in Baltimore
1. Wingerchuk DM, Hogancamp WF, O’Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic’s syndrome). Neurology. 1999;53(5):1107-1114.
2. Kessler RA, Mealy MA, Levy M. Treatment of neuromyelitis optica spectrum disorder: acute, preventive, and symptomatic. Curr Treat Options Neurol. 2016;18(1):2.
3. Popescu BF, Lucchinetti CF. Immunopathology: autoimmune glial diseases and differentiation from multiple sclerosis. Handb Clin Neurol. 2016;133:95-106.
4. Jarius S, Ruprecht K, Wildemann B, et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: a multicentre study of 175 patients. J Neuroinflammation. 2012;9:14.
5. Orman G, Wang KY, Pekcevik Y, et al. Enhancing brain lesions during acute optic neuritis and/or longitudinally extensive transverse myelitis may portend a higher relapse rate in neuromyelitis optica spectrum disorders. Am J Neuroradiol. 2017;38(5):949-953.
6. Kessler RA, Mealy MA, Levy M. Early indicators of relapses vs pseudorelapses in neuromyelitis optica spectrum disorder. Neurol Neuroimmunol Neuroinflamm. 2016;3(5):e269.
7. Wingerchuk DM, Pittock SJ, Lucchinetti CF, et al. A secondary progressive clinical course is uncommon in neuromyelitis optica. Neurology. 2007;68(8):603-605.
8. Kleiter I, Gahlen A, Borisow N, et al. Neuromyelitis optica: evaluation of 871 attacks and 1,153 treatment courses. Ann Neurol. 2016;79(2):206-216.
9. Abboud H, Petrak A, Mealy M, et al. Treatment of acute relapses in neuromyelitis optica: steroids alone versus steroids plus plasma exchange. Mult Scler. 2016;22(2):185-192.
Clinician Reviews in partnership with
MS Consult is edited by Colleen J. Harris, MN, NP, MSCN, Nurse Practitioner/Manager of the Multiple Sclerosis Clinic at Foothills Medical Centre in Calgary, Alberta, Canada, and Bryan Walker, MHS, PA-C, who is in the Department of Neurology, Division of MS and Neuroimmunology, at Duke University Medical Center in Durham, North Carolina. This month's responses were authored by Denise R. Bruen, MSN, APRN-BC, MSCN, who is with the University of Virginia in Charlottesville, and Maureen A. Mealy, BSN, MSCN, who is Neuromyelitis Optica Clinical Research Program Manager, Senior Research Nurse of the Transverse Myelitis & Multiple Sclerosis Centers, and PhD candidate at Johns Hopkins School of Nursing in Baltimore.
Clinician Reviews in partnership with
MS Consult is edited by Colleen J. Harris, MN, NP, MSCN, Nurse Practitioner/Manager of the Multiple Sclerosis Clinic at Foothills Medical Centre in Calgary, Alberta, Canada, and Bryan Walker, MHS, PA-C, who is in the Department of Neurology, Division of MS and Neuroimmunology, at Duke University Medical Center in Durham, North Carolina. This month's responses were authored by Denise R. Bruen, MSN, APRN-BC, MSCN, who is with the University of Virginia in Charlottesville, and Maureen A. Mealy, BSN, MSCN, who is Neuromyelitis Optica Clinical Research Program Manager, Senior Research Nurse of the Transverse Myelitis & Multiple Sclerosis Centers, and PhD candidate at Johns Hopkins School of Nursing in Baltimore.
Clinician Reviews in partnership with
MS Consult is edited by Colleen J. Harris, MN, NP, MSCN, Nurse Practitioner/Manager of the Multiple Sclerosis Clinic at Foothills Medical Centre in Calgary, Alberta, Canada, and Bryan Walker, MHS, PA-C, who is in the Department of Neurology, Division of MS and Neuroimmunology, at Duke University Medical Center in Durham, North Carolina. This month's responses were authored by Denise R. Bruen, MSN, APRN-BC, MSCN, who is with the University of Virginia in Charlottesville, and Maureen A. Mealy, BSN, MSCN, who is Neuromyelitis Optica Clinical Research Program Manager, Senior Research Nurse of the Transverse Myelitis & Multiple Sclerosis Centers, and PhD candidate at Johns Hopkins School of Nursing in Baltimore.
Q) How do you know if a neurologic symptom is due to a relapse of neuromyelitis optica spectrum disorder? And how should a confirmed relapse be treated?
Neuromyelitis optica spectrum disorder (NMOSD) is a severe, relapsing autoimmune disease of the central nervous system (CNS) that targets the optic nerves and spinal cord, leading to blindness and paralysis.1,2 Whereas multiple sclerosis (MS) is characterized by demyelination, NMOSD is associated with astrocytic damage and tissue necrosis.3 Because longitudinally extensive inflammatory lesions are typical with NMOSD, permanent CNS damage is common with each relapse.4
Health care providers first need to determine whether a patient with NMOSD who presents with new or worsening symptoms is having a relapse. A relapse is caused by a breach of the blood-brain barrier by the peripheral immune system, which leads to inflammation and damage to the CNS.5 This causes neurologic symptoms that depend on the anatomic location. Once damage has occurred, symptoms may result either from a new relapse in the same location as a previous inflammatory event or from a pseudorelapse.6
Pseudorelapses are triggered by a systemic metabolic imbalance; they exacerbate symptoms from previous CNS damage. Differentiating between a true relapse and a pseudorelapse can be a diagnostic challenge for even the most seasoned of health care providers. Kessler et al retrospectively examined which clinical factors can distinguish relapses from pseudorelapses.6 Their findings suggest that while clinical examination alone may be effective in events involving vision loss, MRI may be necessary when signs and symptoms are attributable to a spinal cord lesion.
In fact, they found that the degree of clinical worsening in patients with spinal cord symptoms caused by a pseudorelapse was similar to that of a true relapse. The most common causes of pseudorelapse included infection, dysautonomia, metabolic abnormalities, and changes to medication regimens. Interestingly, the presence of infection did not rule out a relapse, as patients experiencing relapses were equally likely as those with pseudorelapse to have a urinary tract infection. The authors concluded, based on their data, that an MRI is warranted to verify a relapse in patients who experience worsening of symptoms localized to the spinal cord but is not necessary to rule out a pseudorelapse of optic neu
In contrast to MS, a progressive phase is not believed to be associated with NMOSD.7 Instead, accrual of disability occurs with each relapse. The majority of patients with NMOSD do not return to baseline following an untreated relapse, making it especially important that patients receive adequate acute treatment to mitigate the damage.8
Currently, there are no medications approved by the FDA for the acute or preventive treatment of NMOSD. However, off-label use of immunotherapies, including rituximab, mycophenolate mofetil, azathioprine, prednisone, methotrexate, tocilizumab, and mitoxantrone, have been studied for relapse prevention.2 In addition, there are three ongoing phase III trials investigating eculizumab (C5 complement inhibitor), inebilizumab (CD19 monoclonal antibody), and SA237 (IL6R blocker); results from these studies could potentially widen the landscape of immunotherapy use in NMOSD.2
Less investigation into appropriate acute treatment of new relapses has been conducted, however, leaving clinicians and patients uncertain about how to manage a new inflammatory event. Traditionally, firstline treatment for acute NMOSD relapses has been the same as for MS relapses—high-dose methylprednisolone. However, due to the severity of NMOSD relapses and the relative lack of response to steroids alone, methylprednisolone is commonly followed by plasma exchange (PLEX).2
Most data to guide clinical decision-making suggest that patients with NMOSD relapses recover better when PLEX is added to steroid treatment. Abboud et al found that 65% of patients who received both PLEX and methylprednisolone recovered to their prerelapse baseline, compared to 35% of those who received methylprednisolone alone.9 These findings were supported by a larger retrospective investigation by Kleiter et al, which found improved recovery with treatment escalation in their cohort.8 These data support the recommendation to use PLEX as an adjunct therapy in acute relapses—particularly in relapses with severe presentations.
Because diagnosis and treatment of relapses involve many factors, ranging from accrual of disability, long-term immunotherapy decisions, and medical costs, diligence in provider decision-making is essential when caring for patients with NMOSD. -MAM
Maureen A. Mealy, BSN, MSCN
Neuromyelitis Optica Research Program Manager, Senior Research Nurse of the Transverse Myelitis & Multiple Sclerosis Centers, PhD candidate at Johns Hopkins School of Nursing in Baltimore
Q) How do you know if a neurologic symptom is due to a relapse of neuromyelitis optica spectrum disorder? And how should a confirmed relapse be treated?
Neuromyelitis optica spectrum disorder (NMOSD) is a severe, relapsing autoimmune disease of the central nervous system (CNS) that targets the optic nerves and spinal cord, leading to blindness and paralysis.1,2 Whereas multiple sclerosis (MS) is characterized by demyelination, NMOSD is associated with astrocytic damage and tissue necrosis.3 Because longitudinally extensive inflammatory lesions are typical with NMOSD, permanent CNS damage is common with each relapse.4
Health care providers first need to determine whether a patient with NMOSD who presents with new or worsening symptoms is having a relapse. A relapse is caused by a breach of the blood-brain barrier by the peripheral immune system, which leads to inflammation and damage to the CNS.5 This causes neurologic symptoms that depend on the anatomic location. Once damage has occurred, symptoms may result either from a new relapse in the same location as a previous inflammatory event or from a pseudorelapse.6
Pseudorelapses are triggered by a systemic metabolic imbalance; they exacerbate symptoms from previous CNS damage. Differentiating between a true relapse and a pseudorelapse can be a diagnostic challenge for even the most seasoned of health care providers. Kessler et al retrospectively examined which clinical factors can distinguish relapses from pseudorelapses.6 Their findings suggest that while clinical examination alone may be effective in events involving vision loss, MRI may be necessary when signs and symptoms are attributable to a spinal cord lesion.
In fact, they found that the degree of clinical worsening in patients with spinal cord symptoms caused by a pseudorelapse was similar to that of a true relapse. The most common causes of pseudorelapse included infection, dysautonomia, metabolic abnormalities, and changes to medication regimens. Interestingly, the presence of infection did not rule out a relapse, as patients experiencing relapses were equally likely as those with pseudorelapse to have a urinary tract infection. The authors concluded, based on their data, that an MRI is warranted to verify a relapse in patients who experience worsening of symptoms localized to the spinal cord but is not necessary to rule out a pseudorelapse of optic neu
In contrast to MS, a progressive phase is not believed to be associated with NMOSD.7 Instead, accrual of disability occurs with each relapse. The majority of patients with NMOSD do not return to baseline following an untreated relapse, making it especially important that patients receive adequate acute treatment to mitigate the damage.8
Currently, there are no medications approved by the FDA for the acute or preventive treatment of NMOSD. However, off-label use of immunotherapies, including rituximab, mycophenolate mofetil, azathioprine, prednisone, methotrexate, tocilizumab, and mitoxantrone, have been studied for relapse prevention.2 In addition, there are three ongoing phase III trials investigating eculizumab (C5 complement inhibitor), inebilizumab (CD19 monoclonal antibody), and SA237 (IL6R blocker); results from these studies could potentially widen the landscape of immunotherapy use in NMOSD.2
Less investigation into appropriate acute treatment of new relapses has been conducted, however, leaving clinicians and patients uncertain about how to manage a new inflammatory event. Traditionally, firstline treatment for acute NMOSD relapses has been the same as for MS relapses—high-dose methylprednisolone. However, due to the severity of NMOSD relapses and the relative lack of response to steroids alone, methylprednisolone is commonly followed by plasma exchange (PLEX).2
Most data to guide clinical decision-making suggest that patients with NMOSD relapses recover better when PLEX is added to steroid treatment. Abboud et al found that 65% of patients who received both PLEX and methylprednisolone recovered to their prerelapse baseline, compared to 35% of those who received methylprednisolone alone.9 These findings were supported by a larger retrospective investigation by Kleiter et al, which found improved recovery with treatment escalation in their cohort.8 These data support the recommendation to use PLEX as an adjunct therapy in acute relapses—particularly in relapses with severe presentations.
Because diagnosis and treatment of relapses involve many factors, ranging from accrual of disability, long-term immunotherapy decisions, and medical costs, diligence in provider decision-making is essential when caring for patients with NMOSD. -MAM
Maureen A. Mealy, BSN, MSCN
Neuromyelitis Optica Research Program Manager, Senior Research Nurse of the Transverse Myelitis & Multiple Sclerosis Centers, PhD candidate at Johns Hopkins School of Nursing in Baltimore
1. Wingerchuk DM, Hogancamp WF, O’Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic’s syndrome). Neurology. 1999;53(5):1107-1114.
2. Kessler RA, Mealy MA, Levy M. Treatment of neuromyelitis optica spectrum disorder: acute, preventive, and symptomatic. Curr Treat Options Neurol. 2016;18(1):2.
3. Popescu BF, Lucchinetti CF. Immunopathology: autoimmune glial diseases and differentiation from multiple sclerosis. Handb Clin Neurol. 2016;133:95-106.
4. Jarius S, Ruprecht K, Wildemann B, et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: a multicentre study of 175 patients. J Neuroinflammation. 2012;9:14.
5. Orman G, Wang KY, Pekcevik Y, et al. Enhancing brain lesions during acute optic neuritis and/or longitudinally extensive transverse myelitis may portend a higher relapse rate in neuromyelitis optica spectrum disorders. Am J Neuroradiol. 2017;38(5):949-953.
6. Kessler RA, Mealy MA, Levy M. Early indicators of relapses vs pseudorelapses in neuromyelitis optica spectrum disorder. Neurol Neuroimmunol Neuroinflamm. 2016;3(5):e269.
7. Wingerchuk DM, Pittock SJ, Lucchinetti CF, et al. A secondary progressive clinical course is uncommon in neuromyelitis optica. Neurology. 2007;68(8):603-605.
8. Kleiter I, Gahlen A, Borisow N, et al. Neuromyelitis optica: evaluation of 871 attacks and 1,153 treatment courses. Ann Neurol. 2016;79(2):206-216.
9. Abboud H, Petrak A, Mealy M, et al. Treatment of acute relapses in neuromyelitis optica: steroids alone versus steroids plus plasma exchange. Mult Scler. 2016;22(2):185-192.
1. Wingerchuk DM, Hogancamp WF, O’Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic’s syndrome). Neurology. 1999;53(5):1107-1114.
2. Kessler RA, Mealy MA, Levy M. Treatment of neuromyelitis optica spectrum disorder: acute, preventive, and symptomatic. Curr Treat Options Neurol. 2016;18(1):2.
3. Popescu BF, Lucchinetti CF. Immunopathology: autoimmune glial diseases and differentiation from multiple sclerosis. Handb Clin Neurol. 2016;133:95-106.
4. Jarius S, Ruprecht K, Wildemann B, et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: a multicentre study of 175 patients. J Neuroinflammation. 2012;9:14.
5. Orman G, Wang KY, Pekcevik Y, et al. Enhancing brain lesions during acute optic neuritis and/or longitudinally extensive transverse myelitis may portend a higher relapse rate in neuromyelitis optica spectrum disorders. Am J Neuroradiol. 2017;38(5):949-953.
6. Kessler RA, Mealy MA, Levy M. Early indicators of relapses vs pseudorelapses in neuromyelitis optica spectrum disorder. Neurol Neuroimmunol Neuroinflamm. 2016;3(5):e269.
7. Wingerchuk DM, Pittock SJ, Lucchinetti CF, et al. A secondary progressive clinical course is uncommon in neuromyelitis optica. Neurology. 2007;68(8):603-605.
8. Kleiter I, Gahlen A, Borisow N, et al. Neuromyelitis optica: evaluation of 871 attacks and 1,153 treatment courses. Ann Neurol. 2016;79(2):206-216.
9. Abboud H, Petrak A, Mealy M, et al. Treatment of acute relapses in neuromyelitis optica: steroids alone versus steroids plus plasma exchange. Mult Scler. 2016;22(2):185-192.

Bladder Complications in MS
Q) My patient has multiple sclerosis and complains of feeling weaker, but denies urinary symptoms. Why have I been told to check for urinary tract infection and not just administer steroids?
Bladder complications are extremely common in patients living with multiple sclerosis (MS), occurring in around 80% of this population.1 These complications—which include urinary urgency, failure to fully empty the bladder, incontinence, and difficulty getting to a toilet in time—can increase risk for urinary tract infection (UTI). And because many patients with MS also have sensory problems (eg, neurogenic bladder), they do not always present with the hallmark UTI symptoms of burning or pain with urination.
Often, presenting symptoms include generalized weakness, increased spasticity, or intensified neurologic issues. These can lead patients to believe they are having a relapse, when in fact, a UTI is causing a pseudoexacerbation of their baseline neurologic issues. In addition, frequent nocturia can disrupt sleep and further contribute to MS-related fatigue. Patients may self-induce dehydration by limiting their daytime fluid intake in an effort to avoid bathroom visits.1
In partnership with urology colleagues, you can help mitigate bladder complications in patients with MS; this can entail use of medication or interventions such as in-and-out or straight catheterization, timed voids, Botox, or pelvic floor physical therapy. Behavior modifications—ie, minimizing caffeine intake, limiting alcohol consumption, and stopping fluids early in the evening—can also be beneficial.1,2
Before initiating bladder medication, it is important to review potential adverse effects with the patient. It’s also crucial to ensure that patients are fully emptying their bladders before starting anticholinergic medications, as these can worsen retention.
Which treatment should you choose? Insurance companies tend to prefer generic, older-generation anticholinergics, but bear in mind that these can cause or contribute to cognitive issues (which many patients with MS already have).3 Another medication, such as mirabegron, may be preferable; it’s less likely than anticholinergics to cause dry mouth, which may help with compliance. Also, be aware that anticholinergics can cause blurred vision, which might lead patients to believe they are having optic neuritis or another MS-related visual change.4
That said, it is possible for patients to have a relapse and a UTI simultaneously. Due to potential adverse effects, it is essential to balance the risks and benefits of steroid therapy. Steroids could worsen an untreated infection and may not be appropriate for the patient’s symptoms or chief complaint.
Addressing bladder symptoms can not only help prevent UTIs but can also improve skin integrity, sleep quality, independence, and overall quality of life. A thorough exam and history-taking can alleviate secondary and tertiary urinary complications, as well as avoid unnecessary use of corticosteroids. -DRB
Denise R. Bruen, MSN, APRN-BC, MSCN
University of Virgina, Charlottesville
1. Sheehan J. Coping with MS bladder dysfunction. www.everydayhealth.com/multiple-sclerosis/symptoms/coping-with-bladder-dysfunction/. Accessed November 18, 2017.
2. Mayo Clinic. Bladder control: medications for urinary problems. www.mayoclinic.org/diseases-conditions/urinary-incontinence/in-depth/bladder-control-problems/art-20044220. Accessed November 18, 2017.
3. Staskin DR, Zoltan E. Anticholinergics and central nervous system effects: are we confused? Rev Urol. 2007;9(4):191-196.
4. Geller EJ, Crane AK, Wells EC, et al. Effect of anticholinergic use for the treatment of overactive bladder on cognitive function in post-menopausal women. Clin Drug Investig. 2012;32(10):697-705.
Clinician Reviews in partnership with
MS Consult is edited by Colleen J. Harris, MN, NP, MSCN, Nurse Practitioner/Manager of the Multiple Sclerosis Clinic at Foothills Medical Centre in Calgary, Alberta, Canada, and Bryan Walker, MHS, PA-C, who is in the Department of Neurology, Division of MS and Neuroimmunology, at Duke University Medical Center in Durham, North Carolina. This month's responses were authored by Denise R. Bruen, MSN, APRN-BC, MSCN, who is with the University of Virginia in Charlottesville, and Maureen A. Mealy, BSN, MSCN, who is Neuromyelitis Optica Clinical Research Program Manager, Senior Research Nurse of the Transverse Myelitis & Multiple Sclerosis Centers, and PhD candidate at Johns Hopkins School of Nursing in Baltimore.
Clinician Reviews in partnership with
MS Consult is edited by Colleen J. Harris, MN, NP, MSCN, Nurse Practitioner/Manager of the Multiple Sclerosis Clinic at Foothills Medical Centre in Calgary, Alberta, Canada, and Bryan Walker, MHS, PA-C, who is in the Department of Neurology, Division of MS and Neuroimmunology, at Duke University Medical Center in Durham, North Carolina. This month's responses were authored by Denise R. Bruen, MSN, APRN-BC, MSCN, who is with the University of Virginia in Charlottesville, and Maureen A. Mealy, BSN, MSCN, who is Neuromyelitis Optica Clinical Research Program Manager, Senior Research Nurse of the Transverse Myelitis & Multiple Sclerosis Centers, and PhD candidate at Johns Hopkins School of Nursing in Baltimore.
Clinician Reviews in partnership with
MS Consult is edited by Colleen J. Harris, MN, NP, MSCN, Nurse Practitioner/Manager of the Multiple Sclerosis Clinic at Foothills Medical Centre in Calgary, Alberta, Canada, and Bryan Walker, MHS, PA-C, who is in the Department of Neurology, Division of MS and Neuroimmunology, at Duke University Medical Center in Durham, North Carolina. This month's responses were authored by Denise R. Bruen, MSN, APRN-BC, MSCN, who is with the University of Virginia in Charlottesville, and Maureen A. Mealy, BSN, MSCN, who is Neuromyelitis Optica Clinical Research Program Manager, Senior Research Nurse of the Transverse Myelitis & Multiple Sclerosis Centers, and PhD candidate at Johns Hopkins School of Nursing in Baltimore.
Q) My patient has multiple sclerosis and complains of feeling weaker, but denies urinary symptoms. Why have I been told to check for urinary tract infection and not just administer steroids?
Bladder complications are extremely common in patients living with multiple sclerosis (MS), occurring in around 80% of this population.1 These complications—which include urinary urgency, failure to fully empty the bladder, incontinence, and difficulty getting to a toilet in time—can increase risk for urinary tract infection (UTI). And because many patients with MS also have sensory problems (eg, neurogenic bladder), they do not always present with the hallmark UTI symptoms of burning or pain with urination.
Often, presenting symptoms include generalized weakness, increased spasticity, or intensified neurologic issues. These can lead patients to believe they are having a relapse, when in fact, a UTI is causing a pseudoexacerbation of their baseline neurologic issues. In addition, frequent nocturia can disrupt sleep and further contribute to MS-related fatigue. Patients may self-induce dehydration by limiting their daytime fluid intake in an effort to avoid bathroom visits.1
In partnership with urology colleagues, you can help mitigate bladder complications in patients with MS; this can entail use of medication or interventions such as in-and-out or straight catheterization, timed voids, Botox, or pelvic floor physical therapy. Behavior modifications—ie, minimizing caffeine intake, limiting alcohol consumption, and stopping fluids early in the evening—can also be beneficial.1,2
Before initiating bladder medication, it is important to review potential adverse effects with the patient. It’s also crucial to ensure that patients are fully emptying their bladders before starting anticholinergic medications, as these can worsen retention.
Which treatment should you choose? Insurance companies tend to prefer generic, older-generation anticholinergics, but bear in mind that these can cause or contribute to cognitive issues (which many patients with MS already have).3 Another medication, such as mirabegron, may be preferable; it’s less likely than anticholinergics to cause dry mouth, which may help with compliance. Also, be aware that anticholinergics can cause blurred vision, which might lead patients to believe they are having optic neuritis or another MS-related visual change.4
That said, it is possible for patients to have a relapse and a UTI simultaneously. Due to potential adverse effects, it is essential to balance the risks and benefits of steroid therapy. Steroids could worsen an untreated infection and may not be appropriate for the patient’s symptoms or chief complaint.
Addressing bladder symptoms can not only help prevent UTIs but can also improve skin integrity, sleep quality, independence, and overall quality of life. A thorough exam and history-taking can alleviate secondary and tertiary urinary complications, as well as avoid unnecessary use of corticosteroids. -DRB
Denise R. Bruen, MSN, APRN-BC, MSCN
University of Virgina, Charlottesville
Q) My patient has multiple sclerosis and complains of feeling weaker, but denies urinary symptoms. Why have I been told to check for urinary tract infection and not just administer steroids?
Bladder complications are extremely common in patients living with multiple sclerosis (MS), occurring in around 80% of this population.1 These complications—which include urinary urgency, failure to fully empty the bladder, incontinence, and difficulty getting to a toilet in time—can increase risk for urinary tract infection (UTI). And because many patients with MS also have sensory problems (eg, neurogenic bladder), they do not always present with the hallmark UTI symptoms of burning or pain with urination.
Often, presenting symptoms include generalized weakness, increased spasticity, or intensified neurologic issues. These can lead patients to believe they are having a relapse, when in fact, a UTI is causing a pseudoexacerbation of their baseline neurologic issues. In addition, frequent nocturia can disrupt sleep and further contribute to MS-related fatigue. Patients may self-induce dehydration by limiting their daytime fluid intake in an effort to avoid bathroom visits.1
In partnership with urology colleagues, you can help mitigate bladder complications in patients with MS; this can entail use of medication or interventions such as in-and-out or straight catheterization, timed voids, Botox, or pelvic floor physical therapy. Behavior modifications—ie, minimizing caffeine intake, limiting alcohol consumption, and stopping fluids early in the evening—can also be beneficial.1,2
Before initiating bladder medication, it is important to review potential adverse effects with the patient. It’s also crucial to ensure that patients are fully emptying their bladders before starting anticholinergic medications, as these can worsen retention.
Which treatment should you choose? Insurance companies tend to prefer generic, older-generation anticholinergics, but bear in mind that these can cause or contribute to cognitive issues (which many patients with MS already have).3 Another medication, such as mirabegron, may be preferable; it’s less likely than anticholinergics to cause dry mouth, which may help with compliance. Also, be aware that anticholinergics can cause blurred vision, which might lead patients to believe they are having optic neuritis or another MS-related visual change.4
That said, it is possible for patients to have a relapse and a UTI simultaneously. Due to potential adverse effects, it is essential to balance the risks and benefits of steroid therapy. Steroids could worsen an untreated infection and may not be appropriate for the patient’s symptoms or chief complaint.
Addressing bladder symptoms can not only help prevent UTIs but can also improve skin integrity, sleep quality, independence, and overall quality of life. A thorough exam and history-taking can alleviate secondary and tertiary urinary complications, as well as avoid unnecessary use of corticosteroids. -DRB
Denise R. Bruen, MSN, APRN-BC, MSCN
University of Virgina, Charlottesville
1. Sheehan J. Coping with MS bladder dysfunction. www.everydayhealth.com/multiple-sclerosis/symptoms/coping-with-bladder-dysfunction/. Accessed November 18, 2017.
2. Mayo Clinic. Bladder control: medications for urinary problems. www.mayoclinic.org/diseases-conditions/urinary-incontinence/in-depth/bladder-control-problems/art-20044220. Accessed November 18, 2017.
3. Staskin DR, Zoltan E. Anticholinergics and central nervous system effects: are we confused? Rev Urol. 2007;9(4):191-196.
4. Geller EJ, Crane AK, Wells EC, et al. Effect of anticholinergic use for the treatment of overactive bladder on cognitive function in post-menopausal women. Clin Drug Investig. 2012;32(10):697-705.
1. Sheehan J. Coping with MS bladder dysfunction. www.everydayhealth.com/multiple-sclerosis/symptoms/coping-with-bladder-dysfunction/. Accessed November 18, 2017.
2. Mayo Clinic. Bladder control: medications for urinary problems. www.mayoclinic.org/diseases-conditions/urinary-incontinence/in-depth/bladder-control-problems/art-20044220. Accessed November 18, 2017.
3. Staskin DR, Zoltan E. Anticholinergics and central nervous system effects: are we confused? Rev Urol. 2007;9(4):191-196.
4. Geller EJ, Crane AK, Wells EC, et al. Effect of anticholinergic use for the treatment of overactive bladder on cognitive function in post-menopausal women. Clin Drug Investig. 2012;32(10):697-705.