User login
Dataset could reveal better therapies for AML

Researchers have released a dataset detailing the molecular makeup of tumor cells from more than 500 patients with acute myeloid leukemia (AML).
The team discovered mutations not previously observed in AML and found associations between mutations and responses to certain therapies.
For instance, AML cases with FLT3, NPM1, and DNMT3A mutations proved sensitive to the BTK inhibitor ibrutinib.
The researchers described their findings in Nature.
The team also made their dataset available via Vizome, an online data viewer. Other researchers can use Vizome to find out which targeted therapies might be most effective against specific subsets of AML cells.
“People can get online, search our database, and very quickly get answers to ‘Is this a good drug?’ or ‘Is there a patient population my drug can work in?’” said study author Brian Druker, MD, of Oregon Health & Science University (OHSU) in Portland, Oregon.
Newly identified mutations
For this study, part of the Beat AML initiative, Dr. Druker and his colleagues performed whole-exome and RNA sequencing on 672 samples from 562 AML patients.
The team identified mutations in 11 genes that were called in 1% or more of patients in this dataset but had not been observed in previous AML sequencing studies. The genes were:
- CUB and Sushi multiple domains 2 (CSMD2)
- NAC alpha domain containing (NACAD)
- Teneurin transmembrane protein 2 (TENM2)
- Aggrecan (ACAN)
- ADAM metallopeptidase with thrombospondin type 1 motif 7 (ADAMTS7)
- Immunoglobulin-like and fibronectin type III domain containing 1 (IGFN1)
- Neurobeachin-like 2 (NBEAL2)
- Poly(U) binding splicing factor 60 (PUF60)
- Zinc-finger protein 687 (ZNF687)
- Cadherin EGF LAG sevenpass G-type receptor 2 (CELSR2)
- Glutamate ionotropic receptor NMDA type subunit 2B (GRIN2B).
Testing therapies
The researchers also assessed how AML cells from 409 of the patient samples responded to each of 122 targeted therapies.
The team found that mutations in TP53, ASXL1, NRAS, and KRAS caused “a broad pattern of drug resistance.”
However, cases with TP53 mutations were sensitive to elesclomol (a drug that targets cancer cell metabolism), cases with ASXL1 mutations were sensitive to the HDAC inhibitor panobinostat, and cases with KRAS/NRAS mutations were sensitive to MAPK inhibitors (with NRAS-mutated cases demonstrating greater sensitivity).
The researchers also found that IDH2 mutations “conferred sensitivity to a broad spectrum of drugs,” but IDH1 mutations were associated with resistance to most drugs.
As previously mentioned, the researchers found a significant association between mutations in FLT3, NPM1, and DNMT3A and sensitivity to ibrutinib. However, the team found that cases with DNMT3A mutations alone or mutations in DNMT3A and FLT3 were not significantly different from cases with wild-type genes.
On the other hand, cases with FLT3-ITD alone or any combination with a mutation in NPM1 (including mutations in all three genes) were significantly more sensitive to ibrutinib than cases with wild-type genes.
Cases with FLT3-ITD and mutations in NPM1 were sensitive to another kinase inhibitor, entospletinib, as well.
The researchers also found that mutations in both BCOR and RUNX1 correlated with increased sensitivity to four JAK inhibitors—momelotinib, ruxolitinib, tofacitinib, and JAK inhibitor I.
However, cases with BCOR mutations alone or mutations in BCOR and DNMT3A or SRSF2 showed no difference in sensitivity to the JAK inhibitors from cases with wild-type genes.
Next steps
“We’re just starting to scratch the surface of what we can do when we analyze the data,” Dr. Druker said. “The real power comes when you start to integrate all that data. You can analyze what drug worked and why it worked.”
In fact, the researchers are already developing and initiating clinical trials to test hypotheses generated by this study.
“You can start to sense some momentum building with new, better therapeutics for AML patients, and, hopefully, this dataset will help fuel that momentum even further,” said study author Jeff Tyner, PhD, of the OHSU School of Medicine.
“We want to parlay this information into clinical trials as much as we can, and we also want the broader community to use this dataset to accelerate their own work.”
Funding for the current study was provided by grants from The Leukemia & Lymphoma Society, the National Cancer Institute, the National Library of Medicine, and other groups.
Researchers have released a dataset detailing the molecular makeup of tumor cells from more than 500 patients with acute myeloid leukemia (AML).
The team discovered mutations not previously observed in AML and found associations between mutations and responses to certain therapies.
For instance, AML cases with FLT3, NPM1, and DNMT3A mutations proved sensitive to the BTK inhibitor ibrutinib.
The researchers described their findings in Nature.
The team also made their dataset available via Vizome, an online data viewer. Other researchers can use Vizome to find out which targeted therapies might be most effective against specific subsets of AML cells.
“People can get online, search our database, and very quickly get answers to ‘Is this a good drug?’ or ‘Is there a patient population my drug can work in?’” said study author Brian Druker, MD, of Oregon Health & Science University (OHSU) in Portland, Oregon.
Newly identified mutations
For this study, part of the Beat AML initiative, Dr. Druker and his colleagues performed whole-exome and RNA sequencing on 672 samples from 562 AML patients.
The team identified mutations in 11 genes that were called in 1% or more of patients in this dataset but had not been observed in previous AML sequencing studies. The genes were:
- CUB and Sushi multiple domains 2 (CSMD2)
- NAC alpha domain containing (NACAD)
- Teneurin transmembrane protein 2 (TENM2)
- Aggrecan (ACAN)
- ADAM metallopeptidase with thrombospondin type 1 motif 7 (ADAMTS7)
- Immunoglobulin-like and fibronectin type III domain containing 1 (IGFN1)
- Neurobeachin-like 2 (NBEAL2)
- Poly(U) binding splicing factor 60 (PUF60)
- Zinc-finger protein 687 (ZNF687)
- Cadherin EGF LAG sevenpass G-type receptor 2 (CELSR2)
- Glutamate ionotropic receptor NMDA type subunit 2B (GRIN2B).
Testing therapies
The researchers also assessed how AML cells from 409 of the patient samples responded to each of 122 targeted therapies.
The team found that mutations in TP53, ASXL1, NRAS, and KRAS caused “a broad pattern of drug resistance.”
However, cases with TP53 mutations were sensitive to elesclomol (a drug that targets cancer cell metabolism), cases with ASXL1 mutations were sensitive to the HDAC inhibitor panobinostat, and cases with KRAS/NRAS mutations were sensitive to MAPK inhibitors (with NRAS-mutated cases demonstrating greater sensitivity).
The researchers also found that IDH2 mutations “conferred sensitivity to a broad spectrum of drugs,” but IDH1 mutations were associated with resistance to most drugs.
As previously mentioned, the researchers found a significant association between mutations in FLT3, NPM1, and DNMT3A and sensitivity to ibrutinib. However, the team found that cases with DNMT3A mutations alone or mutations in DNMT3A and FLT3 were not significantly different from cases with wild-type genes.
On the other hand, cases with FLT3-ITD alone or any combination with a mutation in NPM1 (including mutations in all three genes) were significantly more sensitive to ibrutinib than cases with wild-type genes.
Cases with FLT3-ITD and mutations in NPM1 were sensitive to another kinase inhibitor, entospletinib, as well.
The researchers also found that mutations in both BCOR and RUNX1 correlated with increased sensitivity to four JAK inhibitors—momelotinib, ruxolitinib, tofacitinib, and JAK inhibitor I.
However, cases with BCOR mutations alone or mutations in BCOR and DNMT3A or SRSF2 showed no difference in sensitivity to the JAK inhibitors from cases with wild-type genes.
Next steps
“We’re just starting to scratch the surface of what we can do when we analyze the data,” Dr. Druker said. “The real power comes when you start to integrate all that data. You can analyze what drug worked and why it worked.”
In fact, the researchers are already developing and initiating clinical trials to test hypotheses generated by this study.
“You can start to sense some momentum building with new, better therapeutics for AML patients, and, hopefully, this dataset will help fuel that momentum even further,” said study author Jeff Tyner, PhD, of the OHSU School of Medicine.
“We want to parlay this information into clinical trials as much as we can, and we also want the broader community to use this dataset to accelerate their own work.”
Funding for the current study was provided by grants from The Leukemia & Lymphoma Society, the National Cancer Institute, the National Library of Medicine, and other groups.
Researchers have released a dataset detailing the molecular makeup of tumor cells from more than 500 patients with acute myeloid leukemia (AML).
The team discovered mutations not previously observed in AML and found associations between mutations and responses to certain therapies.
For instance, AML cases with FLT3, NPM1, and DNMT3A mutations proved sensitive to the BTK inhibitor ibrutinib.
The researchers described their findings in Nature.
The team also made their dataset available via Vizome, an online data viewer. Other researchers can use Vizome to find out which targeted therapies might be most effective against specific subsets of AML cells.
“People can get online, search our database, and very quickly get answers to ‘Is this a good drug?’ or ‘Is there a patient population my drug can work in?’” said study author Brian Druker, MD, of Oregon Health & Science University (OHSU) in Portland, Oregon.
Newly identified mutations
For this study, part of the Beat AML initiative, Dr. Druker and his colleagues performed whole-exome and RNA sequencing on 672 samples from 562 AML patients.
The team identified mutations in 11 genes that were called in 1% or more of patients in this dataset but had not been observed in previous AML sequencing studies. The genes were:
- CUB and Sushi multiple domains 2 (CSMD2)
- NAC alpha domain containing (NACAD)
- Teneurin transmembrane protein 2 (TENM2)
- Aggrecan (ACAN)
- ADAM metallopeptidase with thrombospondin type 1 motif 7 (ADAMTS7)
- Immunoglobulin-like and fibronectin type III domain containing 1 (IGFN1)
- Neurobeachin-like 2 (NBEAL2)
- Poly(U) binding splicing factor 60 (PUF60)
- Zinc-finger protein 687 (ZNF687)
- Cadherin EGF LAG sevenpass G-type receptor 2 (CELSR2)
- Glutamate ionotropic receptor NMDA type subunit 2B (GRIN2B).
Testing therapies
The researchers also assessed how AML cells from 409 of the patient samples responded to each of 122 targeted therapies.
The team found that mutations in TP53, ASXL1, NRAS, and KRAS caused “a broad pattern of drug resistance.”
However, cases with TP53 mutations were sensitive to elesclomol (a drug that targets cancer cell metabolism), cases with ASXL1 mutations were sensitive to the HDAC inhibitor panobinostat, and cases with KRAS/NRAS mutations were sensitive to MAPK inhibitors (with NRAS-mutated cases demonstrating greater sensitivity).
The researchers also found that IDH2 mutations “conferred sensitivity to a broad spectrum of drugs,” but IDH1 mutations were associated with resistance to most drugs.
As previously mentioned, the researchers found a significant association between mutations in FLT3, NPM1, and DNMT3A and sensitivity to ibrutinib. However, the team found that cases with DNMT3A mutations alone or mutations in DNMT3A and FLT3 were not significantly different from cases with wild-type genes.
On the other hand, cases with FLT3-ITD alone or any combination with a mutation in NPM1 (including mutations in all three genes) were significantly more sensitive to ibrutinib than cases with wild-type genes.
Cases with FLT3-ITD and mutations in NPM1 were sensitive to another kinase inhibitor, entospletinib, as well.
The researchers also found that mutations in both BCOR and RUNX1 correlated with increased sensitivity to four JAK inhibitors—momelotinib, ruxolitinib, tofacitinib, and JAK inhibitor I.
However, cases with BCOR mutations alone or mutations in BCOR and DNMT3A or SRSF2 showed no difference in sensitivity to the JAK inhibitors from cases with wild-type genes.
Next steps
“We’re just starting to scratch the surface of what we can do when we analyze the data,” Dr. Druker said. “The real power comes when you start to integrate all that data. You can analyze what drug worked and why it worked.”
In fact, the researchers are already developing and initiating clinical trials to test hypotheses generated by this study.
“You can start to sense some momentum building with new, better therapeutics for AML patients, and, hopefully, this dataset will help fuel that momentum even further,” said study author Jeff Tyner, PhD, of the OHSU School of Medicine.
“We want to parlay this information into clinical trials as much as we can, and we also want the broader community to use this dataset to accelerate their own work.”
Funding for the current study was provided by grants from The Leukemia & Lymphoma Society, the National Cancer Institute, the National Library of Medicine, and other groups.
Researchers consider R/R ALL drugs in the first-line setting
CHICAGO – Novel antibodies are improving outcomes in relapsed and refractory acute lymphoblastic leukemia (ALL), and the hope is that they will also show benefit in the up-front treatment setting and thereby improve overall outcomes, according to Anjali Advani, MD.
“It has been a really exciting time in ALL because several drugs have now been FDA approved: blinatumomab, inotuzumab, and now – for patients who are less than 26 years of age – we actually have CAR [chimeric antigen receptor] T cells that have been approved,” Dr. Advani, a hematologist and director of the inpatient leukemia program at the Cleveland Clinic said at the American Society of Hematology Meeting on Hematologic Malignancies.
At the time of relapse, however, the only known cure is allogeneic bone marrow transplant. That may change as more data regarding CAR T cells become available, but the typical goal at this time is to get patients into remission and then to transplant, she said.
Blinatumomab
“Blinatumomab is a very interesting antibody,” Dr. Advani said, explaining that it is a bispecific, T cell–engaging antibody with an anti-CD3 arm that engages the T cell and an anti-CD19 antibody that engages the B lymphoblast.
“Basically this drug then acts as a bridge between the lymphoblast and the T cell to lead to proliferation of the cytotoxic T cell and apoptosis of the lymphoblast,” she said. “It’s interesting because it’s an antibody but it actually works through the immune system through the T cells.”
The largest study to date of blinatumomab in the relapsed/refractory ALL setting showed a 43% complete remission (CR) or CR with partial hematological recovery of peripheral blood counts (CRi) in 189 treated patients with Philadelphia chromosome–negative ALL. It also demonstrated and a 39% rate of salvage status 2 or higher, she said, noting that the response was impressive given that about 30% of participants had a prior transplant (Lancet. 2015 Jan 1;16[1]:57-66).
Of the responders, 40% went on to allogeneic transplant. This was a “fairly impressive” rate given the 30% prior-transplant rate, Dr. Advani said.
“There also was a high minimal residual disease response in those patients achieving CR,” she said, adding that the only significant predictor of response was bone marrow blast count; patients with 50% or more blasts in the bone marrow had a reduced likelihood of responding to blinatumomab.
The agent was approved by the Food and Drug Administration in December 2014 based on these phase 2 findings.
Adverse events mainly included toxicities that are expected in leukemia patients; the most frequent were febrile neutropenia, neutropenia, and anemia. Two patients developed cytokine release syndrome, and about half of the blinatumomab-treated patients experienced neurological events, although the majority of those were grade 1 or 2 and were easily manageable, she noted.
Blinatumomab was further evaluated in the phase 3 TOWER study (NCT02013167), which compared it with standard-of-care chemotherapy regimens. This study showed much higher response rates with blinatumomab than with the chemotherapy regimens (CR with full, partial, or incomplete hematologic recovery, 44% vs. 25%, respectively), Dr. Advani said (N Engl J Med. 2017 Mar 2;376[9]:836-47).
“The main things to remember [are that blinatumomab is] generally very well tolerated and it has been shown to be superior over standard chemotherapy,” she said. “I think it’s a very good drug to use as a bridge to transplant.”
One setting where blinatumomab perhaps should not be used is in patients with central nervous system disease, she noted.
“There is some concern, at least theoretically, that if you have to use concurrent intrathecal chemo along with blinatumomab, there could be some neurotoxicity,” Dr. Advani said, adding that there are no clear data in that setting because patients with CNS disease were not included in the trials.
Patients with high tumor burden may also be poor candidates for blinatumomab because they tend to have lower response rates.
“That doesn’t mean you can’t use it, but you have to kind of think about what the best option would be,” she said.
Additionally, patients treated with CAR T-cell therapy may develop CD19 loss or CD19-negative disease, and blinatumomab should be avoided in these patients.
“The nice thing ... is you don’t have to worry about veno-occlusive disease [VOD] in patients who are proceeding to transplant,” she said, explaining that no increased risk of VOD was seen in these trials.
Inotuzumab
Inotuzumab, which was approved in 2017, differs from blinatumomab in that it is an anti-CD22-calicheamicin conjugate; however, it also showed high response rates in the initial phase 2 trial in relapsed/refractory ALL. The overall response rate was 57%, with 18% achieving a complete response and 63% achieving complete molecular remission.
Of 49 treated patients, 22 patients proceeded to allogeneic transplant, and 5 of those developed VOD.
“Interestingly, four out of five of these patients had received a clofarabine-based preparative regimen, and this likely explains why there was a higher risk of VOD in this study,” she said, noting that the VOD risk has been lower in subsequent studies of inotuzumab.
The international INO-VATE ALL study (NCT01564784) that led to FDA approval was similar in design to the TOWER study in that it compared inotuzumab with standard chemotherapy regimens, and response rates were clearly higher (81% vs. 33%) with inotuzumab (N Engl J Med. 2016 Aug 25;375[8]:740-53).
The VOD risk in the INO-VATE trial was 11%, and it seemed to be higher in those who received dual alkylator–conditioning regimens, which are commonly used in Europe.
Longer-term outcomes after transplant in INO-VATE participants show that median survival has not been reached.
“It’s encouraging that with longer follow-up these patients actually look like they’re doing well,” Dr. Advani said, adding that inotuzumab is a good treatment option for relapsed patients with high disease burden or with CNS disease.
The continuous hookup required for this treatment may be problematic for some younger and older patients, but it is generally not an issue, she noted.
It is important, though, to give as few cycles prior to transplant as possible and to “really think about the preparative regimen to decrease the risk of VOD.”
CAR T-cell therapy
As for CAR T-cell therapy in the relapsed/refractory ALL setting, tisagenlecleucel was approved in 2017 for those up to age 25 years with B-cell precursor ALL that is refractory or in second or later relapse.
Approval was based on a single-arm trial of 63 patients with relapsed or refractory pediatric precursor B-cell ALL, including 35 patients who had prior transplant. The confirmed overall remission rate was 82%, with a 63% CR rate and 19% CRi rate.
“This is a very exciting area,” Dr. Advani said. “There are multiple trials being done in adults with ALL to really look at the older subgroup of patients.”
Overall outcomes
“These treatments we have now really seem to be effective in the relapse setting, but the problem is that once patients relapse and then go to transplant, their overall survival is still poor,” Dr. Advani said. “So the question is how can we improve the up-front treatment of patients so that hopefully they don’t relapse, and hopefully we also can send a smaller number of patients to transplant.”
Two trials seek to address this, she said.
The A041501 study (NCT03150693) is comparing C10403 chemotherapy with C10403 induction followed by two cycles of inotuzumab before continuing with chemotherapy in adults under age 40 years with previously untreated B ALL.
The primary objective is improved 3-year event-free survival, she said, adding that minimal residual disease (MRD) testing will be used and that CD20-positive patients will receive rituximab, as is now standard.
The phase 3 E1910 study (NCT02003222) is evaluating up-front blinatumomab in patients aged 30-70 years with newly diagnosed BCR-ABL–negative B-lineage ALL. This trial was complicated by the recent approval of blinatumomab for MRD-positive disease, which rendered randomization of MRD-positive patients unethical. MRD-negative patients will be randomized, however.
“The hope is that, by incorporating blinatumomab up front, this will again improve outcomes for patients,” she said.
Dr. Advani reported consultancy for Pfizer; research funding from Genzyme, Novartis, Pfizer, and Sigma Tau; and honoraria from Genzyme, Pfizer, and Sigma Tau. She is also on the speakers bureau for Sigma Tau.
CHICAGO – Novel antibodies are improving outcomes in relapsed and refractory acute lymphoblastic leukemia (ALL), and the hope is that they will also show benefit in the up-front treatment setting and thereby improve overall outcomes, according to Anjali Advani, MD.
“It has been a really exciting time in ALL because several drugs have now been FDA approved: blinatumomab, inotuzumab, and now – for patients who are less than 26 years of age – we actually have CAR [chimeric antigen receptor] T cells that have been approved,” Dr. Advani, a hematologist and director of the inpatient leukemia program at the Cleveland Clinic said at the American Society of Hematology Meeting on Hematologic Malignancies.
At the time of relapse, however, the only known cure is allogeneic bone marrow transplant. That may change as more data regarding CAR T cells become available, but the typical goal at this time is to get patients into remission and then to transplant, she said.
Blinatumomab
“Blinatumomab is a very interesting antibody,” Dr. Advani said, explaining that it is a bispecific, T cell–engaging antibody with an anti-CD3 arm that engages the T cell and an anti-CD19 antibody that engages the B lymphoblast.
“Basically this drug then acts as a bridge between the lymphoblast and the T cell to lead to proliferation of the cytotoxic T cell and apoptosis of the lymphoblast,” she said. “It’s interesting because it’s an antibody but it actually works through the immune system through the T cells.”
The largest study to date of blinatumomab in the relapsed/refractory ALL setting showed a 43% complete remission (CR) or CR with partial hematological recovery of peripheral blood counts (CRi) in 189 treated patients with Philadelphia chromosome–negative ALL. It also demonstrated and a 39% rate of salvage status 2 or higher, she said, noting that the response was impressive given that about 30% of participants had a prior transplant (Lancet. 2015 Jan 1;16[1]:57-66).
Of the responders, 40% went on to allogeneic transplant. This was a “fairly impressive” rate given the 30% prior-transplant rate, Dr. Advani said.
“There also was a high minimal residual disease response in those patients achieving CR,” she said, adding that the only significant predictor of response was bone marrow blast count; patients with 50% or more blasts in the bone marrow had a reduced likelihood of responding to blinatumomab.
The agent was approved by the Food and Drug Administration in December 2014 based on these phase 2 findings.
Adverse events mainly included toxicities that are expected in leukemia patients; the most frequent were febrile neutropenia, neutropenia, and anemia. Two patients developed cytokine release syndrome, and about half of the blinatumomab-treated patients experienced neurological events, although the majority of those were grade 1 or 2 and were easily manageable, she noted.
Blinatumomab was further evaluated in the phase 3 TOWER study (NCT02013167), which compared it with standard-of-care chemotherapy regimens. This study showed much higher response rates with blinatumomab than with the chemotherapy regimens (CR with full, partial, or incomplete hematologic recovery, 44% vs. 25%, respectively), Dr. Advani said (N Engl J Med. 2017 Mar 2;376[9]:836-47).
“The main things to remember [are that blinatumomab is] generally very well tolerated and it has been shown to be superior over standard chemotherapy,” she said. “I think it’s a very good drug to use as a bridge to transplant.”
One setting where blinatumomab perhaps should not be used is in patients with central nervous system disease, she noted.
“There is some concern, at least theoretically, that if you have to use concurrent intrathecal chemo along with blinatumomab, there could be some neurotoxicity,” Dr. Advani said, adding that there are no clear data in that setting because patients with CNS disease were not included in the trials.
Patients with high tumor burden may also be poor candidates for blinatumomab because they tend to have lower response rates.
“That doesn’t mean you can’t use it, but you have to kind of think about what the best option would be,” she said.
Additionally, patients treated with CAR T-cell therapy may develop CD19 loss or CD19-negative disease, and blinatumomab should be avoided in these patients.
“The nice thing ... is you don’t have to worry about veno-occlusive disease [VOD] in patients who are proceeding to transplant,” she said, explaining that no increased risk of VOD was seen in these trials.
Inotuzumab
Inotuzumab, which was approved in 2017, differs from blinatumomab in that it is an anti-CD22-calicheamicin conjugate; however, it also showed high response rates in the initial phase 2 trial in relapsed/refractory ALL. The overall response rate was 57%, with 18% achieving a complete response and 63% achieving complete molecular remission.
Of 49 treated patients, 22 patients proceeded to allogeneic transplant, and 5 of those developed VOD.
“Interestingly, four out of five of these patients had received a clofarabine-based preparative regimen, and this likely explains why there was a higher risk of VOD in this study,” she said, noting that the VOD risk has been lower in subsequent studies of inotuzumab.
The international INO-VATE ALL study (NCT01564784) that led to FDA approval was similar in design to the TOWER study in that it compared inotuzumab with standard chemotherapy regimens, and response rates were clearly higher (81% vs. 33%) with inotuzumab (N Engl J Med. 2016 Aug 25;375[8]:740-53).
The VOD risk in the INO-VATE trial was 11%, and it seemed to be higher in those who received dual alkylator–conditioning regimens, which are commonly used in Europe.
Longer-term outcomes after transplant in INO-VATE participants show that median survival has not been reached.
“It’s encouraging that with longer follow-up these patients actually look like they’re doing well,” Dr. Advani said, adding that inotuzumab is a good treatment option for relapsed patients with high disease burden or with CNS disease.
The continuous hookup required for this treatment may be problematic for some younger and older patients, but it is generally not an issue, she noted.
It is important, though, to give as few cycles prior to transplant as possible and to “really think about the preparative regimen to decrease the risk of VOD.”
CAR T-cell therapy
As for CAR T-cell therapy in the relapsed/refractory ALL setting, tisagenlecleucel was approved in 2017 for those up to age 25 years with B-cell precursor ALL that is refractory or in second or later relapse.
Approval was based on a single-arm trial of 63 patients with relapsed or refractory pediatric precursor B-cell ALL, including 35 patients who had prior transplant. The confirmed overall remission rate was 82%, with a 63% CR rate and 19% CRi rate.
“This is a very exciting area,” Dr. Advani said. “There are multiple trials being done in adults with ALL to really look at the older subgroup of patients.”
Overall outcomes
“These treatments we have now really seem to be effective in the relapse setting, but the problem is that once patients relapse and then go to transplant, their overall survival is still poor,” Dr. Advani said. “So the question is how can we improve the up-front treatment of patients so that hopefully they don’t relapse, and hopefully we also can send a smaller number of patients to transplant.”
Two trials seek to address this, she said.
The A041501 study (NCT03150693) is comparing C10403 chemotherapy with C10403 induction followed by two cycles of inotuzumab before continuing with chemotherapy in adults under age 40 years with previously untreated B ALL.
The primary objective is improved 3-year event-free survival, she said, adding that minimal residual disease (MRD) testing will be used and that CD20-positive patients will receive rituximab, as is now standard.
The phase 3 E1910 study (NCT02003222) is evaluating up-front blinatumomab in patients aged 30-70 years with newly diagnosed BCR-ABL–negative B-lineage ALL. This trial was complicated by the recent approval of blinatumomab for MRD-positive disease, which rendered randomization of MRD-positive patients unethical. MRD-negative patients will be randomized, however.
“The hope is that, by incorporating blinatumomab up front, this will again improve outcomes for patients,” she said.
Dr. Advani reported consultancy for Pfizer; research funding from Genzyme, Novartis, Pfizer, and Sigma Tau; and honoraria from Genzyme, Pfizer, and Sigma Tau. She is also on the speakers bureau for Sigma Tau.
CHICAGO – Novel antibodies are improving outcomes in relapsed and refractory acute lymphoblastic leukemia (ALL), and the hope is that they will also show benefit in the up-front treatment setting and thereby improve overall outcomes, according to Anjali Advani, MD.
“It has been a really exciting time in ALL because several drugs have now been FDA approved: blinatumomab, inotuzumab, and now – for patients who are less than 26 years of age – we actually have CAR [chimeric antigen receptor] T cells that have been approved,” Dr. Advani, a hematologist and director of the inpatient leukemia program at the Cleveland Clinic said at the American Society of Hematology Meeting on Hematologic Malignancies.
At the time of relapse, however, the only known cure is allogeneic bone marrow transplant. That may change as more data regarding CAR T cells become available, but the typical goal at this time is to get patients into remission and then to transplant, she said.
Blinatumomab
“Blinatumomab is a very interesting antibody,” Dr. Advani said, explaining that it is a bispecific, T cell–engaging antibody with an anti-CD3 arm that engages the T cell and an anti-CD19 antibody that engages the B lymphoblast.
“Basically this drug then acts as a bridge between the lymphoblast and the T cell to lead to proliferation of the cytotoxic T cell and apoptosis of the lymphoblast,” she said. “It’s interesting because it’s an antibody but it actually works through the immune system through the T cells.”
The largest study to date of blinatumomab in the relapsed/refractory ALL setting showed a 43% complete remission (CR) or CR with partial hematological recovery of peripheral blood counts (CRi) in 189 treated patients with Philadelphia chromosome–negative ALL. It also demonstrated and a 39% rate of salvage status 2 or higher, she said, noting that the response was impressive given that about 30% of participants had a prior transplant (Lancet. 2015 Jan 1;16[1]:57-66).
Of the responders, 40% went on to allogeneic transplant. This was a “fairly impressive” rate given the 30% prior-transplant rate, Dr. Advani said.
“There also was a high minimal residual disease response in those patients achieving CR,” she said, adding that the only significant predictor of response was bone marrow blast count; patients with 50% or more blasts in the bone marrow had a reduced likelihood of responding to blinatumomab.
The agent was approved by the Food and Drug Administration in December 2014 based on these phase 2 findings.
Adverse events mainly included toxicities that are expected in leukemia patients; the most frequent were febrile neutropenia, neutropenia, and anemia. Two patients developed cytokine release syndrome, and about half of the blinatumomab-treated patients experienced neurological events, although the majority of those were grade 1 or 2 and were easily manageable, she noted.
Blinatumomab was further evaluated in the phase 3 TOWER study (NCT02013167), which compared it with standard-of-care chemotherapy regimens. This study showed much higher response rates with blinatumomab than with the chemotherapy regimens (CR with full, partial, or incomplete hematologic recovery, 44% vs. 25%, respectively), Dr. Advani said (N Engl J Med. 2017 Mar 2;376[9]:836-47).
“The main things to remember [are that blinatumomab is] generally very well tolerated and it has been shown to be superior over standard chemotherapy,” she said. “I think it’s a very good drug to use as a bridge to transplant.”
One setting where blinatumomab perhaps should not be used is in patients with central nervous system disease, she noted.
“There is some concern, at least theoretically, that if you have to use concurrent intrathecal chemo along with blinatumomab, there could be some neurotoxicity,” Dr. Advani said, adding that there are no clear data in that setting because patients with CNS disease were not included in the trials.
Patients with high tumor burden may also be poor candidates for blinatumomab because they tend to have lower response rates.
“That doesn’t mean you can’t use it, but you have to kind of think about what the best option would be,” she said.
Additionally, patients treated with CAR T-cell therapy may develop CD19 loss or CD19-negative disease, and blinatumomab should be avoided in these patients.
“The nice thing ... is you don’t have to worry about veno-occlusive disease [VOD] in patients who are proceeding to transplant,” she said, explaining that no increased risk of VOD was seen in these trials.
Inotuzumab
Inotuzumab, which was approved in 2017, differs from blinatumomab in that it is an anti-CD22-calicheamicin conjugate; however, it also showed high response rates in the initial phase 2 trial in relapsed/refractory ALL. The overall response rate was 57%, with 18% achieving a complete response and 63% achieving complete molecular remission.
Of 49 treated patients, 22 patients proceeded to allogeneic transplant, and 5 of those developed VOD.
“Interestingly, four out of five of these patients had received a clofarabine-based preparative regimen, and this likely explains why there was a higher risk of VOD in this study,” she said, noting that the VOD risk has been lower in subsequent studies of inotuzumab.
The international INO-VATE ALL study (NCT01564784) that led to FDA approval was similar in design to the TOWER study in that it compared inotuzumab with standard chemotherapy regimens, and response rates were clearly higher (81% vs. 33%) with inotuzumab (N Engl J Med. 2016 Aug 25;375[8]:740-53).
The VOD risk in the INO-VATE trial was 11%, and it seemed to be higher in those who received dual alkylator–conditioning regimens, which are commonly used in Europe.
Longer-term outcomes after transplant in INO-VATE participants show that median survival has not been reached.
“It’s encouraging that with longer follow-up these patients actually look like they’re doing well,” Dr. Advani said, adding that inotuzumab is a good treatment option for relapsed patients with high disease burden or with CNS disease.
The continuous hookup required for this treatment may be problematic for some younger and older patients, but it is generally not an issue, she noted.
It is important, though, to give as few cycles prior to transplant as possible and to “really think about the preparative regimen to decrease the risk of VOD.”
CAR T-cell therapy
As for CAR T-cell therapy in the relapsed/refractory ALL setting, tisagenlecleucel was approved in 2017 for those up to age 25 years with B-cell precursor ALL that is refractory or in second or later relapse.
Approval was based on a single-arm trial of 63 patients with relapsed or refractory pediatric precursor B-cell ALL, including 35 patients who had prior transplant. The confirmed overall remission rate was 82%, with a 63% CR rate and 19% CRi rate.
“This is a very exciting area,” Dr. Advani said. “There are multiple trials being done in adults with ALL to really look at the older subgroup of patients.”
Overall outcomes
“These treatments we have now really seem to be effective in the relapse setting, but the problem is that once patients relapse and then go to transplant, their overall survival is still poor,” Dr. Advani said. “So the question is how can we improve the up-front treatment of patients so that hopefully they don’t relapse, and hopefully we also can send a smaller number of patients to transplant.”
Two trials seek to address this, she said.
The A041501 study (NCT03150693) is comparing C10403 chemotherapy with C10403 induction followed by two cycles of inotuzumab before continuing with chemotherapy in adults under age 40 years with previously untreated B ALL.
The primary objective is improved 3-year event-free survival, she said, adding that minimal residual disease (MRD) testing will be used and that CD20-positive patients will receive rituximab, as is now standard.
The phase 3 E1910 study (NCT02003222) is evaluating up-front blinatumomab in patients aged 30-70 years with newly diagnosed BCR-ABL–negative B-lineage ALL. This trial was complicated by the recent approval of blinatumomab for MRD-positive disease, which rendered randomization of MRD-positive patients unethical. MRD-negative patients will be randomized, however.
“The hope is that, by incorporating blinatumomab up front, this will again improve outcomes for patients,” she said.
Dr. Advani reported consultancy for Pfizer; research funding from Genzyme, Novartis, Pfizer, and Sigma Tau; and honoraria from Genzyme, Pfizer, and Sigma Tau. She is also on the speakers bureau for Sigma Tau.
EXPERT ANALYSIS FROM MHM 2018
Ibrutinib plus obinutuzumab gets priority review in CLL/SLL
The Food and Drug Administration has granted priority review to an anti-CD20, chemotherapy-free combination – ibrutinib plus obinutuzumab – for the frontline treatment of chronic lymphocytic leukemia or small lymphocytic lymphoma (CLL/SLL).

The agency will review the combination in previously untreated adults.
Ibrutinib (Imbruvica) is already approved as a single agent for adults with CLL/SLL for all lines of therapy and in combination with bendamustine and rituximab. Obinutuzumab (Gazyva) has been approved for patients with previously untreated CLL, in combination with chlorambucil.
The current application, which is sponsored by Janssen and Pharmacyclics, is based on results from the phase 3 iLLUMINATE trial. Preliminary results announced by Janssen and Pharmacyclics showed that ibrutinib plus obinutuzumab had statistically significant better progression-free survival, compared with chlorambucil plus obinutuzumab, as assessed by an independent review committee.
Complete results from the trial will be presented at an upcoming medical meeting, according to the sponsors.
The Food and Drug Administration has granted priority review to an anti-CD20, chemotherapy-free combination – ibrutinib plus obinutuzumab – for the frontline treatment of chronic lymphocytic leukemia or small lymphocytic lymphoma (CLL/SLL).
The agency will review the combination in previously untreated adults.
Ibrutinib (Imbruvica) is already approved as a single agent for adults with CLL/SLL for all lines of therapy and in combination with bendamustine and rituximab. Obinutuzumab (Gazyva) has been approved for patients with previously untreated CLL, in combination with chlorambucil.
The current application, which is sponsored by Janssen and Pharmacyclics, is based on results from the phase 3 iLLUMINATE trial. Preliminary results announced by Janssen and Pharmacyclics showed that ibrutinib plus obinutuzumab had statistically significant better progression-free survival, compared with chlorambucil plus obinutuzumab, as assessed by an independent review committee.
Complete results from the trial will be presented at an upcoming medical meeting, according to the sponsors.
The Food and Drug Administration has granted priority review to an anti-CD20, chemotherapy-free combination – ibrutinib plus obinutuzumab – for the frontline treatment of chronic lymphocytic leukemia or small lymphocytic lymphoma (CLL/SLL).
The agency will review the combination in previously untreated adults.
Ibrutinib (Imbruvica) is already approved as a single agent for adults with CLL/SLL for all lines of therapy and in combination with bendamustine and rituximab. Obinutuzumab (Gazyva) has been approved for patients with previously untreated CLL, in combination with chlorambucil.
The current application, which is sponsored by Janssen and Pharmacyclics, is based on results from the phase 3 iLLUMINATE trial. Preliminary results announced by Janssen and Pharmacyclics showed that ibrutinib plus obinutuzumab had statistically significant better progression-free survival, compared with chlorambucil plus obinutuzumab, as assessed by an independent review committee.
Complete results from the trial will be presented at an upcoming medical meeting, according to the sponsors.
FDA offers guidance on MRD assessment in blood cancer trials
The of patients with hematologic malignancies.

The FDA said it developed the document to assist drug sponsors who are planning to use minimal residual disease (MRD) as a biomarker in clinical trials conducted under an investigational new drug application or to support FDA approval of products intended to treat hematologic malignancies.
“As a result of important workshops where we’ve heard from stakeholders and an analysis of marketing applications showing inconsistent quality of MRD data, the FDA identified a need to provide sponsors with guidance on the use of MRD as a biomarker in regulatory submissions,” FDA Commissioner Scott Gottlieb, MD, said in a statement.
The guidance explains how MRD might be used in clinical trials, highlights considerations for MRD assessment that are specific to certain hematologic malignancies, and lists requirements for regulatory submissions that utilize MRD.
MRD could potentially be used as a biomarker in clinical trials – specifically as a diagnostic, prognostic, predictive, efficacy-response, or monitoring biomarker, according to the draft guidance. Additionally, MRD could be used as a surrogate endpoint or “to select patients at high risk or to enrich the trial population.”
The draft guidance also provides specific considerations for MRD assessment in individual hematologic malignancies, including acute lymphoblastic leukemia, acute myeloid leukemia, acute promyelocytic leukemia, chronic lymphocytic leukemia, chronic myeloid leukemia, and multiple myeloma.
The full document is available on the FDA website.
The of patients with hematologic malignancies.
The FDA said it developed the document to assist drug sponsors who are planning to use minimal residual disease (MRD) as a biomarker in clinical trials conducted under an investigational new drug application or to support FDA approval of products intended to treat hematologic malignancies.
“As a result of important workshops where we’ve heard from stakeholders and an analysis of marketing applications showing inconsistent quality of MRD data, the FDA identified a need to provide sponsors with guidance on the use of MRD as a biomarker in regulatory submissions,” FDA Commissioner Scott Gottlieb, MD, said in a statement.
The guidance explains how MRD might be used in clinical trials, highlights considerations for MRD assessment that are specific to certain hematologic malignancies, and lists requirements for regulatory submissions that utilize MRD.
MRD could potentially be used as a biomarker in clinical trials – specifically as a diagnostic, prognostic, predictive, efficacy-response, or monitoring biomarker, according to the draft guidance. Additionally, MRD could be used as a surrogate endpoint or “to select patients at high risk or to enrich the trial population.”
The draft guidance also provides specific considerations for MRD assessment in individual hematologic malignancies, including acute lymphoblastic leukemia, acute myeloid leukemia, acute promyelocytic leukemia, chronic lymphocytic leukemia, chronic myeloid leukemia, and multiple myeloma.
The full document is available on the FDA website.
The of patients with hematologic malignancies.
The FDA said it developed the document to assist drug sponsors who are planning to use minimal residual disease (MRD) as a biomarker in clinical trials conducted under an investigational new drug application or to support FDA approval of products intended to treat hematologic malignancies.
“As a result of important workshops where we’ve heard from stakeholders and an analysis of marketing applications showing inconsistent quality of MRD data, the FDA identified a need to provide sponsors with guidance on the use of MRD as a biomarker in regulatory submissions,” FDA Commissioner Scott Gottlieb, MD, said in a statement.
The guidance explains how MRD might be used in clinical trials, highlights considerations for MRD assessment that are specific to certain hematologic malignancies, and lists requirements for regulatory submissions that utilize MRD.
MRD could potentially be used as a biomarker in clinical trials – specifically as a diagnostic, prognostic, predictive, efficacy-response, or monitoring biomarker, according to the draft guidance. Additionally, MRD could be used as a surrogate endpoint or “to select patients at high risk or to enrich the trial population.”
The draft guidance also provides specific considerations for MRD assessment in individual hematologic malignancies, including acute lymphoblastic leukemia, acute myeloid leukemia, acute promyelocytic leukemia, chronic lymphocytic leukemia, chronic myeloid leukemia, and multiple myeloma.
The full document is available on the FDA website.
FDA issues draft guidance on MRD

The U.S. Food and Drug Administration (FDA) has issued a draft guidance on the use of minimal residual disease (MRD) assessment in trials of patients with hematologic malignancies.
The FDA said it developed this guidance to assist sponsors who are planning to use MRD as a biomarker in clinical trials conducted under an investigational new drug application or to support FDA approval of products intended to treat hematologic malignancies.
“As a result of important workshops where we’ve heard from stakeholders and an analysis of marketing applications showing inconsistent quality of MRD data, the FDA identified a need to provide sponsors with guidance on the use of MRD as a biomarker in regulatory submissions,” said FDA Commissioner Scott Gottlieb, MD.
The guidance explains how MRD might be used in clinical trials, highlights considerations for MRD assessment that are specific to certain hematologic malignancies, and lists requirements for regulatory submissions that utilize MRD.
The full document, “Hematologic Malignancies: Regulatory Considerations for Use of Minimal Residual Disease in Development of Drug and Biological Products for Treatment,” is available for download from the FDA website.
How MRD can be used
The guidance notes that MRD could potentially be used as a biomarker in clinical trials, specifically, as a diagnostic, prognostic, predictive, efficacy-response, or monitoring biomarker.
MRD could also be used as a surrogate endpoint, and there are two mechanisms for obtaining FDA feedback on the use of a novel surrogate endpoint to support approval of a product:
- The drug development tool qualification process
- Discussions with the specific Center for Drug Evaluation and Research or Center for Biologics Evaluation and Research review division.
Furthermore, a sponsor can use MRD “to select patients at high risk or to enrich the trial population,” according to the guidance.
Disease specifics
The guidance also details specific considerations for MRD assessment in individual hematologic malignancies. For example:
- In acute lymphoblastic leukemia, a patient with an MRD level of 0.1% or more in first or second complete remission has a high risk of relapse.
- In trials of acute myeloid leukemia, the sponsor should provide data showing that the marker selected to assess MRD “reflects the leukemia and not underlying clonal hematopoiesis.”
- Patients with low-risk acute promyelocytic leukemia who achieve MRD negativity after arsenic/tretinoin-based therapy are generally considered cured.
- In chronic lymphocytic leukemia, MRD can be assessed in the peripheral blood or bone marrow, but the sample source should remain the same throughout a trial.
- In chronic myeloid leukemia, MRD can be used to select and monitor patients who are eligible to discontinue treatment with tyrosine kinase inhibitors.
- In multiple myeloma, imaging techniques may be combined with MRD assessment of the bone marrow to assess patient response to treatment.
Types of technology
The guidance lists the four general technologies used for MRD assessment in hematologic malignancies:
- Multiparametric flow cytometry
- Next-generation sequencing
- Quantitative reverse transcription polymerase chain reaction of specific gene fusions
- Allele-specific oligonucleotide polymerase chain reaction.
The FDA said it does not have a preference as to which technology is used in a trial. However, the sponsor must pre-specify the technology used and should utilize the same technology throughout a trial.
The FDA also said it “does not foresee the need for co-development of an MRD assay with a drug product.” However, the assay must be analytically valid for results important to the trial, and MRD assessment must be a clinically valid biomarker in the context in which it’s used.
If the MRD assay used is not FDA-cleared or -approved, additional information about the assay must be provided to the FDA.
The U.S. Food and Drug Administration (FDA) has issued a draft guidance on the use of minimal residual disease (MRD) assessment in trials of patients with hematologic malignancies.
The FDA said it developed this guidance to assist sponsors who are planning to use MRD as a biomarker in clinical trials conducted under an investigational new drug application or to support FDA approval of products intended to treat hematologic malignancies.
“As a result of important workshops where we’ve heard from stakeholders and an analysis of marketing applications showing inconsistent quality of MRD data, the FDA identified a need to provide sponsors with guidance on the use of MRD as a biomarker in regulatory submissions,” said FDA Commissioner Scott Gottlieb, MD.
The guidance explains how MRD might be used in clinical trials, highlights considerations for MRD assessment that are specific to certain hematologic malignancies, and lists requirements for regulatory submissions that utilize MRD.
The full document, “Hematologic Malignancies: Regulatory Considerations for Use of Minimal Residual Disease in Development of Drug and Biological Products for Treatment,” is available for download from the FDA website.
How MRD can be used
The guidance notes that MRD could potentially be used as a biomarker in clinical trials, specifically, as a diagnostic, prognostic, predictive, efficacy-response, or monitoring biomarker.
MRD could also be used as a surrogate endpoint, and there are two mechanisms for obtaining FDA feedback on the use of a novel surrogate endpoint to support approval of a product:
- The drug development tool qualification process
- Discussions with the specific Center for Drug Evaluation and Research or Center for Biologics Evaluation and Research review division.
Furthermore, a sponsor can use MRD “to select patients at high risk or to enrich the trial population,” according to the guidance.
Disease specifics
The guidance also details specific considerations for MRD assessment in individual hematologic malignancies. For example:
- In acute lymphoblastic leukemia, a patient with an MRD level of 0.1% or more in first or second complete remission has a high risk of relapse.
- In trials of acute myeloid leukemia, the sponsor should provide data showing that the marker selected to assess MRD “reflects the leukemia and not underlying clonal hematopoiesis.”
- Patients with low-risk acute promyelocytic leukemia who achieve MRD negativity after arsenic/tretinoin-based therapy are generally considered cured.
- In chronic lymphocytic leukemia, MRD can be assessed in the peripheral blood or bone marrow, but the sample source should remain the same throughout a trial.
- In chronic myeloid leukemia, MRD can be used to select and monitor patients who are eligible to discontinue treatment with tyrosine kinase inhibitors.
- In multiple myeloma, imaging techniques may be combined with MRD assessment of the bone marrow to assess patient response to treatment.
Types of technology
The guidance lists the four general technologies used for MRD assessment in hematologic malignancies:
- Multiparametric flow cytometry
- Next-generation sequencing
- Quantitative reverse transcription polymerase chain reaction of specific gene fusions
- Allele-specific oligonucleotide polymerase chain reaction.
The FDA said it does not have a preference as to which technology is used in a trial. However, the sponsor must pre-specify the technology used and should utilize the same technology throughout a trial.
The FDA also said it “does not foresee the need for co-development of an MRD assay with a drug product.” However, the assay must be analytically valid for results important to the trial, and MRD assessment must be a clinically valid biomarker in the context in which it’s used.
If the MRD assay used is not FDA-cleared or -approved, additional information about the assay must be provided to the FDA.
The U.S. Food and Drug Administration (FDA) has issued a draft guidance on the use of minimal residual disease (MRD) assessment in trials of patients with hematologic malignancies.
The FDA said it developed this guidance to assist sponsors who are planning to use MRD as a biomarker in clinical trials conducted under an investigational new drug application or to support FDA approval of products intended to treat hematologic malignancies.
“As a result of important workshops where we’ve heard from stakeholders and an analysis of marketing applications showing inconsistent quality of MRD data, the FDA identified a need to provide sponsors with guidance on the use of MRD as a biomarker in regulatory submissions,” said FDA Commissioner Scott Gottlieb, MD.
The guidance explains how MRD might be used in clinical trials, highlights considerations for MRD assessment that are specific to certain hematologic malignancies, and lists requirements for regulatory submissions that utilize MRD.
The full document, “Hematologic Malignancies: Regulatory Considerations for Use of Minimal Residual Disease in Development of Drug and Biological Products for Treatment,” is available for download from the FDA website.
How MRD can be used
The guidance notes that MRD could potentially be used as a biomarker in clinical trials, specifically, as a diagnostic, prognostic, predictive, efficacy-response, or monitoring biomarker.
MRD could also be used as a surrogate endpoint, and there are two mechanisms for obtaining FDA feedback on the use of a novel surrogate endpoint to support approval of a product:
- The drug development tool qualification process
- Discussions with the specific Center for Drug Evaluation and Research or Center for Biologics Evaluation and Research review division.
Furthermore, a sponsor can use MRD “to select patients at high risk or to enrich the trial population,” according to the guidance.
Disease specifics
The guidance also details specific considerations for MRD assessment in individual hematologic malignancies. For example:
- In acute lymphoblastic leukemia, a patient with an MRD level of 0.1% or more in first or second complete remission has a high risk of relapse.
- In trials of acute myeloid leukemia, the sponsor should provide data showing that the marker selected to assess MRD “reflects the leukemia and not underlying clonal hematopoiesis.”
- Patients with low-risk acute promyelocytic leukemia who achieve MRD negativity after arsenic/tretinoin-based therapy are generally considered cured.
- In chronic lymphocytic leukemia, MRD can be assessed in the peripheral blood or bone marrow, but the sample source should remain the same throughout a trial.
- In chronic myeloid leukemia, MRD can be used to select and monitor patients who are eligible to discontinue treatment with tyrosine kinase inhibitors.
- In multiple myeloma, imaging techniques may be combined with MRD assessment of the bone marrow to assess patient response to treatment.
Types of technology
The guidance lists the four general technologies used for MRD assessment in hematologic malignancies:
- Multiparametric flow cytometry
- Next-generation sequencing
- Quantitative reverse transcription polymerase chain reaction of specific gene fusions
- Allele-specific oligonucleotide polymerase chain reaction.
The FDA said it does not have a preference as to which technology is used in a trial. However, the sponsor must pre-specify the technology used and should utilize the same technology throughout a trial.
The FDA also said it “does not foresee the need for co-development of an MRD assay with a drug product.” However, the assay must be analytically valid for results important to the trial, and MRD assessment must be a clinically valid biomarker in the context in which it’s used.
If the MRD assay used is not FDA-cleared or -approved, additional information about the assay must be provided to the FDA.
Optimizing use of TKIs in chronic leukemia
DUBROVNIK, CROATIA – Long-term efficacy and toxicity should inform decisions about tyrosine kinase inhibitors (TKIs) in chronic myeloid leukemia (CML), according to one expert.

Studies have indicated that long-term survival rates are similar whether CML patients receive frontline treatment with imatinib or second-generation TKIs. But the newer TKIs pose a higher risk of uncommon toxicities, Hagop M. Kantarjian, MD, said during the keynote presentation at Leukemia and Lymphoma, a meeting jointly sponsored by the University of Texas MD Anderson Cancer Center and the School of Medicine at the University of Zagreb, Croatia.
Dr. Kantarjian, a professor at MD Anderson Cancer Center in Houston, said most CML patients should receive daily treatment with TKIs – even if they are in complete cytogenetic response or 100% Philadelphia chromosome positive – because they will live longer.
Frontline treatment options for CML that are approved by the Food and Drug Administration include imatinib, dasatinib, nilotinib, and bosutinib.
Dr. Kantarjian noted that dasatinib and nilotinib bested imatinib in early analyses from clinical trials, but all three TKIs produced similar rates of overall survival (OS) and progression-free survival (PFS) at extended follow-up.
Dasatinib and imatinib produced similar rates of 5-year OS and PFS in the DASISION trial (J Clin Oncol. 2016 Jul 10;34[20]:2333-40).
In ENESTnd, 5-year OS and PFS rates were similar with nilotinib and imatinib (Leukemia. 2016 May;30[5]:1044-54).
However, the higher incidence of uncommon toxicities with the newer TKIs must be taken into account, Dr. Kantarjian said.
Choosing a TKI
Dr. Kantarjian recommends frontline imatinib for older patients (aged 65-70) and those who are low risk based on their Sokal score.
Second-generation TKIs should be given up front to patients who are at higher risk by Sokal and for “very young patients in whom early treatment discontinuation is important,” he said.
“In accelerated or blast phase, I always use the second-generation TKIs,” he said. “If there’s no binding mutation, I prefer dasatinib. I think it’s the most potent of them. If there are toxicities with dasatinib, bosutinib is equivalent in efficacy, so they are interchangeable.”
A TKI should not be discarded unless there is loss of complete cytogenetic response – not major molecular response – at the maximum tolerated adjusted dose that does not cause grade 3-4 toxicities or chronic grade 2 toxicities, Dr. Kantarjian added.
“We have to remember that we can go down on the dosages of, for example, imatinib, down to 200 mg a day, dasatinib as low as 20 mg a day, nilotinib as low as 150 mg twice a day or even 200 mg daily, and bosutinib down to 200 mg daily,” he said. “So if we have a patient who’s responding with side effects, we should not abandon the particular TKI, we should try to manipulate the dose schedule if they are having a good response.”
Dr. Kantarjian noted that pleural effusion is a toxicity of particular concern with dasatinib, but lowering the dose to 50 mg daily results in similar efficacy and significantly less toxicity than 100 mg daily. For patients over the age of 70, a 20-mg dose can be used.
Vaso-occlusive and vasospastic reactions are increasingly observed in patients treated with nilotinib. For that reason, Dr. Kantarjian said he prefers to forgo up-front nilotinib, particularly in patients who have cardiovascular or neurotoxic problems.
“The incidence of vaso-occlusive and vasospastic reactions is now close to 10%-15% at about 10 years with nilotinib,” Dr. Kantarjian said. “So it is not a trivial toxicity.”
For patients with vaso-occlusive/vasospastic reactions, “bosutinib is probably the safest drug,” Dr. Kantarjian said.
For second- or third-line therapy, patients can receive ponatinib or a second-generation TKI (dasatinib, nilotinib, or bosutinib), as well as omacetaxine or allogeneic stem cell transplant.
“If you disregard toxicities, I think ponatinib is the most powerful TKI, and I think that’s because we are using it at a higher dose that produces so many toxicities,” Dr. Kantarjian said.
Ponatinib is not used up front because of these toxicities, particularly pancreatitis, skin rashes, vaso-occlusive disorders, and hypertension, he added.
Dr. Kantarjian suggests giving ponatinib at 30 mg daily in patients with T315I mutation and those without guiding mutations who are resistant to second-generation TKIs.
Discontinuing a TKI
Dr. Kantarjian said patients can discontinue TKI therapy if they:
- Are low- or intermediate-risk by Sokal.
- Have quantifiable BCR-ABL transcripts.
- Are in chronic phase.
- Achieved an optimal response to their first TKI.
- Have been on TKI therapy for more than 8 years.
- Achieved a complete molecular response.
- Have had a molecular response for more than 2-3 years.
- Are available for monitoring every other month for the first 2 years.
Dr. Kantarjian did not report any conflicts of interest at the meeting. However, he has previously reported relationships with Novartis, Bristol-Myers Squibb, Pfizer, and Ariad Pharmaceuticals.
The Leukemia and Lymphoma meeting is organized by Jonathan Wood & Association, which is owned by the parent company of this news organization.
DUBROVNIK, CROATIA – Long-term efficacy and toxicity should inform decisions about tyrosine kinase inhibitors (TKIs) in chronic myeloid leukemia (CML), according to one expert.
Studies have indicated that long-term survival rates are similar whether CML patients receive frontline treatment with imatinib or second-generation TKIs. But the newer TKIs pose a higher risk of uncommon toxicities, Hagop M. Kantarjian, MD, said during the keynote presentation at Leukemia and Lymphoma, a meeting jointly sponsored by the University of Texas MD Anderson Cancer Center and the School of Medicine at the University of Zagreb, Croatia.
Dr. Kantarjian, a professor at MD Anderson Cancer Center in Houston, said most CML patients should receive daily treatment with TKIs – even if they are in complete cytogenetic response or 100% Philadelphia chromosome positive – because they will live longer.
Frontline treatment options for CML that are approved by the Food and Drug Administration include imatinib, dasatinib, nilotinib, and bosutinib.
Dr. Kantarjian noted that dasatinib and nilotinib bested imatinib in early analyses from clinical trials, but all three TKIs produced similar rates of overall survival (OS) and progression-free survival (PFS) at extended follow-up.
Dasatinib and imatinib produced similar rates of 5-year OS and PFS in the DASISION trial (J Clin Oncol. 2016 Jul 10;34[20]:2333-40).
In ENESTnd, 5-year OS and PFS rates were similar with nilotinib and imatinib (Leukemia. 2016 May;30[5]:1044-54).
However, the higher incidence of uncommon toxicities with the newer TKIs must be taken into account, Dr. Kantarjian said.
Choosing a TKI
Dr. Kantarjian recommends frontline imatinib for older patients (aged 65-70) and those who are low risk based on their Sokal score.
Second-generation TKIs should be given up front to patients who are at higher risk by Sokal and for “very young patients in whom early treatment discontinuation is important,” he said.
“In accelerated or blast phase, I always use the second-generation TKIs,” he said. “If there’s no binding mutation, I prefer dasatinib. I think it’s the most potent of them. If there are toxicities with dasatinib, bosutinib is equivalent in efficacy, so they are interchangeable.”
A TKI should not be discarded unless there is loss of complete cytogenetic response – not major molecular response – at the maximum tolerated adjusted dose that does not cause grade 3-4 toxicities or chronic grade 2 toxicities, Dr. Kantarjian added.
“We have to remember that we can go down on the dosages of, for example, imatinib, down to 200 mg a day, dasatinib as low as 20 mg a day, nilotinib as low as 150 mg twice a day or even 200 mg daily, and bosutinib down to 200 mg daily,” he said. “So if we have a patient who’s responding with side effects, we should not abandon the particular TKI, we should try to manipulate the dose schedule if they are having a good response.”
Dr. Kantarjian noted that pleural effusion is a toxicity of particular concern with dasatinib, but lowering the dose to 50 mg daily results in similar efficacy and significantly less toxicity than 100 mg daily. For patients over the age of 70, a 20-mg dose can be used.
Vaso-occlusive and vasospastic reactions are increasingly observed in patients treated with nilotinib. For that reason, Dr. Kantarjian said he prefers to forgo up-front nilotinib, particularly in patients who have cardiovascular or neurotoxic problems.
“The incidence of vaso-occlusive and vasospastic reactions is now close to 10%-15% at about 10 years with nilotinib,” Dr. Kantarjian said. “So it is not a trivial toxicity.”
For patients with vaso-occlusive/vasospastic reactions, “bosutinib is probably the safest drug,” Dr. Kantarjian said.
For second- or third-line therapy, patients can receive ponatinib or a second-generation TKI (dasatinib, nilotinib, or bosutinib), as well as omacetaxine or allogeneic stem cell transplant.
“If you disregard toxicities, I think ponatinib is the most powerful TKI, and I think that’s because we are using it at a higher dose that produces so many toxicities,” Dr. Kantarjian said.
Ponatinib is not used up front because of these toxicities, particularly pancreatitis, skin rashes, vaso-occlusive disorders, and hypertension, he added.
Dr. Kantarjian suggests giving ponatinib at 30 mg daily in patients with T315I mutation and those without guiding mutations who are resistant to second-generation TKIs.
Discontinuing a TKI
Dr. Kantarjian said patients can discontinue TKI therapy if they:
- Are low- or intermediate-risk by Sokal.
- Have quantifiable BCR-ABL transcripts.
- Are in chronic phase.
- Achieved an optimal response to their first TKI.
- Have been on TKI therapy for more than 8 years.
- Achieved a complete molecular response.
- Have had a molecular response for more than 2-3 years.
- Are available for monitoring every other month for the first 2 years.
Dr. Kantarjian did not report any conflicts of interest at the meeting. However, he has previously reported relationships with Novartis, Bristol-Myers Squibb, Pfizer, and Ariad Pharmaceuticals.
The Leukemia and Lymphoma meeting is organized by Jonathan Wood & Association, which is owned by the parent company of this news organization.
DUBROVNIK, CROATIA – Long-term efficacy and toxicity should inform decisions about tyrosine kinase inhibitors (TKIs) in chronic myeloid leukemia (CML), according to one expert.
Studies have indicated that long-term survival rates are similar whether CML patients receive frontline treatment with imatinib or second-generation TKIs. But the newer TKIs pose a higher risk of uncommon toxicities, Hagop M. Kantarjian, MD, said during the keynote presentation at Leukemia and Lymphoma, a meeting jointly sponsored by the University of Texas MD Anderson Cancer Center and the School of Medicine at the University of Zagreb, Croatia.
Dr. Kantarjian, a professor at MD Anderson Cancer Center in Houston, said most CML patients should receive daily treatment with TKIs – even if they are in complete cytogenetic response or 100% Philadelphia chromosome positive – because they will live longer.
Frontline treatment options for CML that are approved by the Food and Drug Administration include imatinib, dasatinib, nilotinib, and bosutinib.
Dr. Kantarjian noted that dasatinib and nilotinib bested imatinib in early analyses from clinical trials, but all three TKIs produced similar rates of overall survival (OS) and progression-free survival (PFS) at extended follow-up.
Dasatinib and imatinib produced similar rates of 5-year OS and PFS in the DASISION trial (J Clin Oncol. 2016 Jul 10;34[20]:2333-40).
In ENESTnd, 5-year OS and PFS rates were similar with nilotinib and imatinib (Leukemia. 2016 May;30[5]:1044-54).
However, the higher incidence of uncommon toxicities with the newer TKIs must be taken into account, Dr. Kantarjian said.
Choosing a TKI
Dr. Kantarjian recommends frontline imatinib for older patients (aged 65-70) and those who are low risk based on their Sokal score.
Second-generation TKIs should be given up front to patients who are at higher risk by Sokal and for “very young patients in whom early treatment discontinuation is important,” he said.
“In accelerated or blast phase, I always use the second-generation TKIs,” he said. “If there’s no binding mutation, I prefer dasatinib. I think it’s the most potent of them. If there are toxicities with dasatinib, bosutinib is equivalent in efficacy, so they are interchangeable.”
A TKI should not be discarded unless there is loss of complete cytogenetic response – not major molecular response – at the maximum tolerated adjusted dose that does not cause grade 3-4 toxicities or chronic grade 2 toxicities, Dr. Kantarjian added.
“We have to remember that we can go down on the dosages of, for example, imatinib, down to 200 mg a day, dasatinib as low as 20 mg a day, nilotinib as low as 150 mg twice a day or even 200 mg daily, and bosutinib down to 200 mg daily,” he said. “So if we have a patient who’s responding with side effects, we should not abandon the particular TKI, we should try to manipulate the dose schedule if they are having a good response.”
Dr. Kantarjian noted that pleural effusion is a toxicity of particular concern with dasatinib, but lowering the dose to 50 mg daily results in similar efficacy and significantly less toxicity than 100 mg daily. For patients over the age of 70, a 20-mg dose can be used.
Vaso-occlusive and vasospastic reactions are increasingly observed in patients treated with nilotinib. For that reason, Dr. Kantarjian said he prefers to forgo up-front nilotinib, particularly in patients who have cardiovascular or neurotoxic problems.
“The incidence of vaso-occlusive and vasospastic reactions is now close to 10%-15% at about 10 years with nilotinib,” Dr. Kantarjian said. “So it is not a trivial toxicity.”
For patients with vaso-occlusive/vasospastic reactions, “bosutinib is probably the safest drug,” Dr. Kantarjian said.
For second- or third-line therapy, patients can receive ponatinib or a second-generation TKI (dasatinib, nilotinib, or bosutinib), as well as omacetaxine or allogeneic stem cell transplant.
“If you disregard toxicities, I think ponatinib is the most powerful TKI, and I think that’s because we are using it at a higher dose that produces so many toxicities,” Dr. Kantarjian said.
Ponatinib is not used up front because of these toxicities, particularly pancreatitis, skin rashes, vaso-occlusive disorders, and hypertension, he added.
Dr. Kantarjian suggests giving ponatinib at 30 mg daily in patients with T315I mutation and those without guiding mutations who are resistant to second-generation TKIs.
Discontinuing a TKI
Dr. Kantarjian said patients can discontinue TKI therapy if they:
- Are low- or intermediate-risk by Sokal.
- Have quantifiable BCR-ABL transcripts.
- Are in chronic phase.
- Achieved an optimal response to their first TKI.
- Have been on TKI therapy for more than 8 years.
- Achieved a complete molecular response.
- Have had a molecular response for more than 2-3 years.
- Are available for monitoring every other month for the first 2 years.
Dr. Kantarjian did not report any conflicts of interest at the meeting. However, he has previously reported relationships with Novartis, Bristol-Myers Squibb, Pfizer, and Ariad Pharmaceuticals.
The Leukemia and Lymphoma meeting is organized by Jonathan Wood & Association, which is owned by the parent company of this news organization.
REPORTING FROM LEUKEMIA AND LYMPHOMA 2018
The challenges of diagnosing CMML
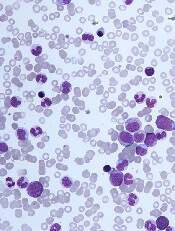
DUBROVNIK, CROATIA—Diagnosing chronic myelomonocytic leukemia (CMML) remains a challenge in 2018, according to a presentation at Leukemia and Lymphoma: Europe and the USA, Linking Knowledge and Practice.
Even with updated World Health Organization (WHO) criteria, karyotyping, and genetic analyses, it can be difficult to distinguish CMML from other conditions, according to Nadira Duraković, MD, PhD, of the University Hospital Zagreb in Croatia.
However, Dr. Duraković said there are characteristics that differentiate CMML from myelodysplastic syndromes (MDS), myeloproliferative neoplasms (MPNs), and atypical chronic myeloid leukemia (CML).
Furthermore, studies have suggested that monocyte subset distribution analysis can be useful for diagnosing CMML.
Dr. Duraković began her presentation with an overview of the 2016 WHO classification of CMML (Blood 2016 127:2391-2405).
According to the WHO, patients have CMML if:
- They have persistent peripheral blood monocytosis (1×109/L) with monocytes accounting for 10% of the white blood cell count
- They do not meet WHO criteria for BCR-ABL1-positive CML, primary myelofibrosis, polycythemia vera, or essential thrombocythemia
- There is no evidence of PCM1-JAK2 or PDGFRA, PDGFRB, or FGFR1 rearrangement
- They have fewer than 20% blasts in the blood and bone marrow
- They have dysplasia in one or more myeloid lineages
- If myelodysplasia is absent or minimal, an acquired clonal cytogenetic or molecular genetic abnormality must be present.
Alternatively, if patients have monocytosis that has persisted for at least 3 months, and all other causes of monocytosis have been excluded, “you can say that your patient has CMML,” Dr. Duraković said.
Other causes of monocytosis include infections, malignancies, medications, inflammatory conditions, and other conditions such as pregnancy.
However, Dr. Duraković pointed out that the cause of monocytosis cannot always be determined, and, in some cases, CMML patients may not meet the WHO criteria.
“[T]here are cases where there just aren’t enough monocytes to fulfill the WHO criteria,” Dr. Duraković said. “You can have a patient with peripheral blood cytopenia and monocytosis who does not have 1,000 monocytes. Patients can have progressive dysplasia, can have splenomegaly, be really sick, but fail to meet WHO criteria.”
Distinguishing CMML from other conditions
“Differentiating CMML from myelodysplastic syndromes can be tough,” Dr. Duraković said. “There are dysplastic features that are present in CMML . . . but, in CMML, they are more subtle, and they are more difficult to appreciate than in myelodysplastic syndromes.”
The ratio of myeloid to erythroid cells is elevated in CMML, and patients may have atypical monocytes (paramyeloid cells) that are unique to CMML.
Dr. Duraković noted that megakaryocyte dysplasia in CMML can be characterized by “myeloproliferative megakaryocytes,” which are large cells that cluster and have hyperlobulated nuclei, or “MDS megakaryocytes,” which are small, solitary cells with hypolobulated nuclei.
She went on to explain that “MPN phenotype” CMML is characterized by leukocytosis, monocytosis, hepatomegaly, splenomegaly, and clinical features of myeloproliferation (fatigue, night sweats, bone pain, weight loss, etc.).
Thirty percent of cases are associated with splenomegaly, and 30% of patients can have an increase in bone marrow reticulin fibrosis.
Dr. Duraković also noted that a prior MPN diagnosis excludes CMML. The presence of common MPN mutations, such as JAK2, CALR, or MPL, suggests a patient has an MPN with monocytosis rather than CMML.
Patients who have unclassified MPNs or MDS, rather than CMML, either do not have 1,000 monocytes or the monocytes do not represent more than 10% of the differential, Dr. Duraković said.
She also noted that it can be difficult to differentiate CMML from atypical CML.
“Atypical CML is characterized by profound dysgranulopoiesis, absence of the BCR-ABL1 fusion gene, and neutrophilia,” Dr. Duraković explained. “Those patients [commonly] have monocytosis, but, here, that 10% rule is valuable because their monocytes comprise less than 10% of the entire white blood cell count.”
Karyotyping, genotyping, and immunophenotyping
“There is no disease-defining karyotype abnormality [in CMML],” Dr. Duraković noted.
She said 30% of patients have abnormal karyotype, and the most common abnormality is trisomy 8. Unlike in patients with MDS, del(5q) and monosomal karyotypes are infrequent in patients with CMML.
Similarly, there are no “disease-defining” mutations or genetic changes in CMML, although CMML is genetically distinct from MDS, Dr. Duraković said.
For instance, SRSF2 encodes a component of the spliceosome that is mutated in almost half of CMML patients and less than 10% of MDS patients. Likewise, ASLX1 and TET2 are “much more frequently involved” in CMML than in MDS, Dr. Duraković said.
In a 2012 study of 275 CMML patients, researchers found that 93% of patients had at least one somatic mutation in nine recurrently mutated genes—SRFS2, ASXL1, CBL, EZH2, JAK2V617F, KRAS, NRAS, RUNX1, and TET2 (Blood 2012 120:3080-3088).
However, Dr. Duraković noted that these mutations are found in other disorders as well, so this information may not be helpful in differentiating CMML from other disorders.
A 2015 study revealed a technique that does appear useful for identifying CMML—monocyte subset distribution analysis (Blood 2015 125(23): 3618–3626).
For this analysis, monocytes are divided into the following categories:
- Classical/MO1 (CD14bright/CD16−)
- Intermediate/MO2 (CD14bright/CD16+)
- Non-classical/MO3 (CD14dim/CD16+).
The researchers found that CMML patients had an increase in the fraction of classical monocytes (with a cutoff value of 94.0%), as compared to healthy control subjects, patients with another hematologic disorder, and patients with reactive monocytosis.
A 2018 study confirmed that monocyte subset distribution analysis could differentiate CMML from other hematologic disorders, with the exception of atypical CML (Am J Clin Pathol 2018 150(4):293-302).
This study also suggested that a decreased percentage of non-classical monocytes was more sensitive than an increased percentage of classical monocytes.
Despite the differences between these studies, “monocyte subset distribution analysis is showing promise as a method of identifying hard-to-identify CMML patients with ease and affordability,” Dr. Duraković said.
She added that the technique can be implemented in clinical practice using the HematoflowTM solution (Cytometry B Clin Cytom 2018 94(5):658-661).
Dr. Duraković did not report any conflicts of interest.
DUBROVNIK, CROATIA—Diagnosing chronic myelomonocytic leukemia (CMML) remains a challenge in 2018, according to a presentation at Leukemia and Lymphoma: Europe and the USA, Linking Knowledge and Practice.
Even with updated World Health Organization (WHO) criteria, karyotyping, and genetic analyses, it can be difficult to distinguish CMML from other conditions, according to Nadira Duraković, MD, PhD, of the University Hospital Zagreb in Croatia.
However, Dr. Duraković said there are characteristics that differentiate CMML from myelodysplastic syndromes (MDS), myeloproliferative neoplasms (MPNs), and atypical chronic myeloid leukemia (CML).
Furthermore, studies have suggested that monocyte subset distribution analysis can be useful for diagnosing CMML.
Dr. Duraković began her presentation with an overview of the 2016 WHO classification of CMML (Blood 2016 127:2391-2405).
According to the WHO, patients have CMML if:
- They have persistent peripheral blood monocytosis (1×109/L) with monocytes accounting for 10% of the white blood cell count
- They do not meet WHO criteria for BCR-ABL1-positive CML, primary myelofibrosis, polycythemia vera, or essential thrombocythemia
- There is no evidence of PCM1-JAK2 or PDGFRA, PDGFRB, or FGFR1 rearrangement
- They have fewer than 20% blasts in the blood and bone marrow
- They have dysplasia in one or more myeloid lineages
- If myelodysplasia is absent or minimal, an acquired clonal cytogenetic or molecular genetic abnormality must be present.
Alternatively, if patients have monocytosis that has persisted for at least 3 months, and all other causes of monocytosis have been excluded, “you can say that your patient has CMML,” Dr. Duraković said.
Other causes of monocytosis include infections, malignancies, medications, inflammatory conditions, and other conditions such as pregnancy.
However, Dr. Duraković pointed out that the cause of monocytosis cannot always be determined, and, in some cases, CMML patients may not meet the WHO criteria.
“[T]here are cases where there just aren’t enough monocytes to fulfill the WHO criteria,” Dr. Duraković said. “You can have a patient with peripheral blood cytopenia and monocytosis who does not have 1,000 monocytes. Patients can have progressive dysplasia, can have splenomegaly, be really sick, but fail to meet WHO criteria.”
Distinguishing CMML from other conditions
“Differentiating CMML from myelodysplastic syndromes can be tough,” Dr. Duraković said. “There are dysplastic features that are present in CMML . . . but, in CMML, they are more subtle, and they are more difficult to appreciate than in myelodysplastic syndromes.”
The ratio of myeloid to erythroid cells is elevated in CMML, and patients may have atypical monocytes (paramyeloid cells) that are unique to CMML.
Dr. Duraković noted that megakaryocyte dysplasia in CMML can be characterized by “myeloproliferative megakaryocytes,” which are large cells that cluster and have hyperlobulated nuclei, or “MDS megakaryocytes,” which are small, solitary cells with hypolobulated nuclei.
She went on to explain that “MPN phenotype” CMML is characterized by leukocytosis, monocytosis, hepatomegaly, splenomegaly, and clinical features of myeloproliferation (fatigue, night sweats, bone pain, weight loss, etc.).
Thirty percent of cases are associated with splenomegaly, and 30% of patients can have an increase in bone marrow reticulin fibrosis.
Dr. Duraković also noted that a prior MPN diagnosis excludes CMML. The presence of common MPN mutations, such as JAK2, CALR, or MPL, suggests a patient has an MPN with monocytosis rather than CMML.
Patients who have unclassified MPNs or MDS, rather than CMML, either do not have 1,000 monocytes or the monocytes do not represent more than 10% of the differential, Dr. Duraković said.
She also noted that it can be difficult to differentiate CMML from atypical CML.
“Atypical CML is characterized by profound dysgranulopoiesis, absence of the BCR-ABL1 fusion gene, and neutrophilia,” Dr. Duraković explained. “Those patients [commonly] have monocytosis, but, here, that 10% rule is valuable because their monocytes comprise less than 10% of the entire white blood cell count.”
Karyotyping, genotyping, and immunophenotyping
“There is no disease-defining karyotype abnormality [in CMML],” Dr. Duraković noted.
She said 30% of patients have abnormal karyotype, and the most common abnormality is trisomy 8. Unlike in patients with MDS, del(5q) and monosomal karyotypes are infrequent in patients with CMML.
Similarly, there are no “disease-defining” mutations or genetic changes in CMML, although CMML is genetically distinct from MDS, Dr. Duraković said.
For instance, SRSF2 encodes a component of the spliceosome that is mutated in almost half of CMML patients and less than 10% of MDS patients. Likewise, ASLX1 and TET2 are “much more frequently involved” in CMML than in MDS, Dr. Duraković said.
In a 2012 study of 275 CMML patients, researchers found that 93% of patients had at least one somatic mutation in nine recurrently mutated genes—SRFS2, ASXL1, CBL, EZH2, JAK2V617F, KRAS, NRAS, RUNX1, and TET2 (Blood 2012 120:3080-3088).
However, Dr. Duraković noted that these mutations are found in other disorders as well, so this information may not be helpful in differentiating CMML from other disorders.
A 2015 study revealed a technique that does appear useful for identifying CMML—monocyte subset distribution analysis (Blood 2015 125(23): 3618–3626).
For this analysis, monocytes are divided into the following categories:
- Classical/MO1 (CD14bright/CD16−)
- Intermediate/MO2 (CD14bright/CD16+)
- Non-classical/MO3 (CD14dim/CD16+).
The researchers found that CMML patients had an increase in the fraction of classical monocytes (with a cutoff value of 94.0%), as compared to healthy control subjects, patients with another hematologic disorder, and patients with reactive monocytosis.
A 2018 study confirmed that monocyte subset distribution analysis could differentiate CMML from other hematologic disorders, with the exception of atypical CML (Am J Clin Pathol 2018 150(4):293-302).
This study also suggested that a decreased percentage of non-classical monocytes was more sensitive than an increased percentage of classical monocytes.
Despite the differences between these studies, “monocyte subset distribution analysis is showing promise as a method of identifying hard-to-identify CMML patients with ease and affordability,” Dr. Duraković said.
She added that the technique can be implemented in clinical practice using the HematoflowTM solution (Cytometry B Clin Cytom 2018 94(5):658-661).
Dr. Duraković did not report any conflicts of interest.
DUBROVNIK, CROATIA—Diagnosing chronic myelomonocytic leukemia (CMML) remains a challenge in 2018, according to a presentation at Leukemia and Lymphoma: Europe and the USA, Linking Knowledge and Practice.
Even with updated World Health Organization (WHO) criteria, karyotyping, and genetic analyses, it can be difficult to distinguish CMML from other conditions, according to Nadira Duraković, MD, PhD, of the University Hospital Zagreb in Croatia.
However, Dr. Duraković said there are characteristics that differentiate CMML from myelodysplastic syndromes (MDS), myeloproliferative neoplasms (MPNs), and atypical chronic myeloid leukemia (CML).
Furthermore, studies have suggested that monocyte subset distribution analysis can be useful for diagnosing CMML.
Dr. Duraković began her presentation with an overview of the 2016 WHO classification of CMML (Blood 2016 127:2391-2405).
According to the WHO, patients have CMML if:
- They have persistent peripheral blood monocytosis (1×109/L) with monocytes accounting for 10% of the white blood cell count
- They do not meet WHO criteria for BCR-ABL1-positive CML, primary myelofibrosis, polycythemia vera, or essential thrombocythemia
- There is no evidence of PCM1-JAK2 or PDGFRA, PDGFRB, or FGFR1 rearrangement
- They have fewer than 20% blasts in the blood and bone marrow
- They have dysplasia in one or more myeloid lineages
- If myelodysplasia is absent or minimal, an acquired clonal cytogenetic or molecular genetic abnormality must be present.
Alternatively, if patients have monocytosis that has persisted for at least 3 months, and all other causes of monocytosis have been excluded, “you can say that your patient has CMML,” Dr. Duraković said.
Other causes of monocytosis include infections, malignancies, medications, inflammatory conditions, and other conditions such as pregnancy.
However, Dr. Duraković pointed out that the cause of monocytosis cannot always be determined, and, in some cases, CMML patients may not meet the WHO criteria.
“[T]here are cases where there just aren’t enough monocytes to fulfill the WHO criteria,” Dr. Duraković said. “You can have a patient with peripheral blood cytopenia and monocytosis who does not have 1,000 monocytes. Patients can have progressive dysplasia, can have splenomegaly, be really sick, but fail to meet WHO criteria.”
Distinguishing CMML from other conditions
“Differentiating CMML from myelodysplastic syndromes can be tough,” Dr. Duraković said. “There are dysplastic features that are present in CMML . . . but, in CMML, they are more subtle, and they are more difficult to appreciate than in myelodysplastic syndromes.”
The ratio of myeloid to erythroid cells is elevated in CMML, and patients may have atypical monocytes (paramyeloid cells) that are unique to CMML.
Dr. Duraković noted that megakaryocyte dysplasia in CMML can be characterized by “myeloproliferative megakaryocytes,” which are large cells that cluster and have hyperlobulated nuclei, or “MDS megakaryocytes,” which are small, solitary cells with hypolobulated nuclei.
She went on to explain that “MPN phenotype” CMML is characterized by leukocytosis, monocytosis, hepatomegaly, splenomegaly, and clinical features of myeloproliferation (fatigue, night sweats, bone pain, weight loss, etc.).
Thirty percent of cases are associated with splenomegaly, and 30% of patients can have an increase in bone marrow reticulin fibrosis.
Dr. Duraković also noted that a prior MPN diagnosis excludes CMML. The presence of common MPN mutations, such as JAK2, CALR, or MPL, suggests a patient has an MPN with monocytosis rather than CMML.
Patients who have unclassified MPNs or MDS, rather than CMML, either do not have 1,000 monocytes or the monocytes do not represent more than 10% of the differential, Dr. Duraković said.
She also noted that it can be difficult to differentiate CMML from atypical CML.
“Atypical CML is characterized by profound dysgranulopoiesis, absence of the BCR-ABL1 fusion gene, and neutrophilia,” Dr. Duraković explained. “Those patients [commonly] have monocytosis, but, here, that 10% rule is valuable because their monocytes comprise less than 10% of the entire white blood cell count.”
Karyotyping, genotyping, and immunophenotyping
“There is no disease-defining karyotype abnormality [in CMML],” Dr. Duraković noted.
She said 30% of patients have abnormal karyotype, and the most common abnormality is trisomy 8. Unlike in patients with MDS, del(5q) and monosomal karyotypes are infrequent in patients with CMML.
Similarly, there are no “disease-defining” mutations or genetic changes in CMML, although CMML is genetically distinct from MDS, Dr. Duraković said.
For instance, SRSF2 encodes a component of the spliceosome that is mutated in almost half of CMML patients and less than 10% of MDS patients. Likewise, ASLX1 and TET2 are “much more frequently involved” in CMML than in MDS, Dr. Duraković said.
In a 2012 study of 275 CMML patients, researchers found that 93% of patients had at least one somatic mutation in nine recurrently mutated genes—SRFS2, ASXL1, CBL, EZH2, JAK2V617F, KRAS, NRAS, RUNX1, and TET2 (Blood 2012 120:3080-3088).
However, Dr. Duraković noted that these mutations are found in other disorders as well, so this information may not be helpful in differentiating CMML from other disorders.
A 2015 study revealed a technique that does appear useful for identifying CMML—monocyte subset distribution analysis (Blood 2015 125(23): 3618–3626).
For this analysis, monocytes are divided into the following categories:
- Classical/MO1 (CD14bright/CD16−)
- Intermediate/MO2 (CD14bright/CD16+)
- Non-classical/MO3 (CD14dim/CD16+).
The researchers found that CMML patients had an increase in the fraction of classical monocytes (with a cutoff value of 94.0%), as compared to healthy control subjects, patients with another hematologic disorder, and patients with reactive monocytosis.
A 2018 study confirmed that monocyte subset distribution analysis could differentiate CMML from other hematologic disorders, with the exception of atypical CML (Am J Clin Pathol 2018 150(4):293-302).
This study also suggested that a decreased percentage of non-classical monocytes was more sensitive than an increased percentage of classical monocytes.
Despite the differences between these studies, “monocyte subset distribution analysis is showing promise as a method of identifying hard-to-identify CMML patients with ease and affordability,” Dr. Duraković said.
She added that the technique can be implemented in clinical practice using the HematoflowTM solution (Cytometry B Clin Cytom 2018 94(5):658-661).
Dr. Duraković did not report any conflicts of interest.
Bacteremic sepsis in ALL tied to neurocognitive dysfunction

Bacteremic sepsis during acute lymphoblastic leukemia (ALL) treatment may contribute to neurocognitive dysfunction later in life, results of a cohort study suggest.
Pediatric ALL survivors who had sepsis while on treatment performed worse on measures of intelligence, attention, executive function, and processing speed than survivors with no sepsis history, according to study results.
Links between sepsis and impaired neurocognitive function found in this study have “practice-changing implications” for cancer survivors, investigators reported in JAMA Pediatrics.
“Prevention of infection, early recognition and appropriate management of sepsis, and preemptive neurocognitive interventions should be prioritized, because these might prevent or ameliorate neurologic damage,” said Joshua Wolf, MBBS, of St. Jude Children’s Research Hospital, Memphis, and the coauthors of the report.
The study included 212 children who, at a median age of 5 years, had received risk-adapted chemotherapy for ALL with no hematopoietic cell transplant or cranial irradiation.
Sixteen of the patients (7.5%) had a history of bacteremic sepsis during ALL therapy, according to retrospectively obtained data.
As a part of the study, all patients participated in neurocognitive testing, which was done at a median of 7.7 years after diagnosis.
Patients with a history of bacteremic sepsis performed poorly on multiple measures of neurocognitive function, as compared with all other patients, according to results of analyses that were adjusted for multiple potentially confounding factors, such as age, race, and leukemia risk category.
Although not all neurocognitive measures were significantly different between groups, survivors with a sepsis history performed worse on evaluations of spatial planning (difference, 0.78; 95% CI, 0.57-1.00), verbal fluency (0.38; 95% CI, 0.14-0.62), and attention (0.63; 95% CI, 0.30-0.95), among other measures.
This is believed to be the first published study looking at potential links between sepsis during ALL treatment and long-term neurocognitive dysfunction, investigators said. However, similar observations have been made in other patient populations, they added.
Exactly how sepsis might lead to neurocognitive deficits remains unclear.
“In the population of children with cancer, these mechanisms might be augmented by increased blood-brain barrier permeability to neurotoxic chemotherapy drugs,” the investigators said in their report.
Further study is needed to look at potential brain injury mechanisms and to validate the current findings in other ALL patient cohorts, they concluded.
The study was supported by the National Institute of Mental Health, the National Cancer Institute, and the American Lebanese Syrian Associated Charities. The researchers reported having no conflicts of interest.
Bacteremic sepsis during acute lymphoblastic leukemia (ALL) treatment may contribute to neurocognitive dysfunction later in life, results of a cohort study suggest.
Pediatric ALL survivors who had sepsis while on treatment performed worse on measures of intelligence, attention, executive function, and processing speed than survivors with no sepsis history, according to study results.
Links between sepsis and impaired neurocognitive function found in this study have “practice-changing implications” for cancer survivors, investigators reported in JAMA Pediatrics.
“Prevention of infection, early recognition and appropriate management of sepsis, and preemptive neurocognitive interventions should be prioritized, because these might prevent or ameliorate neurologic damage,” said Joshua Wolf, MBBS, of St. Jude Children’s Research Hospital, Memphis, and the coauthors of the report.
The study included 212 children who, at a median age of 5 years, had received risk-adapted chemotherapy for ALL with no hematopoietic cell transplant or cranial irradiation.
Sixteen of the patients (7.5%) had a history of bacteremic sepsis during ALL therapy, according to retrospectively obtained data.
As a part of the study, all patients participated in neurocognitive testing, which was done at a median of 7.7 years after diagnosis.
Patients with a history of bacteremic sepsis performed poorly on multiple measures of neurocognitive function, as compared with all other patients, according to results of analyses that were adjusted for multiple potentially confounding factors, such as age, race, and leukemia risk category.
Although not all neurocognitive measures were significantly different between groups, survivors with a sepsis history performed worse on evaluations of spatial planning (difference, 0.78; 95% CI, 0.57-1.00), verbal fluency (0.38; 95% CI, 0.14-0.62), and attention (0.63; 95% CI, 0.30-0.95), among other measures.
This is believed to be the first published study looking at potential links between sepsis during ALL treatment and long-term neurocognitive dysfunction, investigators said. However, similar observations have been made in other patient populations, they added.
Exactly how sepsis might lead to neurocognitive deficits remains unclear.
“In the population of children with cancer, these mechanisms might be augmented by increased blood-brain barrier permeability to neurotoxic chemotherapy drugs,” the investigators said in their report.
Further study is needed to look at potential brain injury mechanisms and to validate the current findings in other ALL patient cohorts, they concluded.
The study was supported by the National Institute of Mental Health, the National Cancer Institute, and the American Lebanese Syrian Associated Charities. The researchers reported having no conflicts of interest.
Bacteremic sepsis during acute lymphoblastic leukemia (ALL) treatment may contribute to neurocognitive dysfunction later in life, results of a cohort study suggest.
Pediatric ALL survivors who had sepsis while on treatment performed worse on measures of intelligence, attention, executive function, and processing speed than survivors with no sepsis history, according to study results.
Links between sepsis and impaired neurocognitive function found in this study have “practice-changing implications” for cancer survivors, investigators reported in JAMA Pediatrics.
“Prevention of infection, early recognition and appropriate management of sepsis, and preemptive neurocognitive interventions should be prioritized, because these might prevent or ameliorate neurologic damage,” said Joshua Wolf, MBBS, of St. Jude Children’s Research Hospital, Memphis, and the coauthors of the report.
The study included 212 children who, at a median age of 5 years, had received risk-adapted chemotherapy for ALL with no hematopoietic cell transplant or cranial irradiation.
Sixteen of the patients (7.5%) had a history of bacteremic sepsis during ALL therapy, according to retrospectively obtained data.
As a part of the study, all patients participated in neurocognitive testing, which was done at a median of 7.7 years after diagnosis.
Patients with a history of bacteremic sepsis performed poorly on multiple measures of neurocognitive function, as compared with all other patients, according to results of analyses that were adjusted for multiple potentially confounding factors, such as age, race, and leukemia risk category.
Although not all neurocognitive measures were significantly different between groups, survivors with a sepsis history performed worse on evaluations of spatial planning (difference, 0.78; 95% CI, 0.57-1.00), verbal fluency (0.38; 95% CI, 0.14-0.62), and attention (0.63; 95% CI, 0.30-0.95), among other measures.
This is believed to be the first published study looking at potential links between sepsis during ALL treatment and long-term neurocognitive dysfunction, investigators said. However, similar observations have been made in other patient populations, they added.
Exactly how sepsis might lead to neurocognitive deficits remains unclear.
“In the population of children with cancer, these mechanisms might be augmented by increased blood-brain barrier permeability to neurotoxic chemotherapy drugs,” the investigators said in their report.
Further study is needed to look at potential brain injury mechanisms and to validate the current findings in other ALL patient cohorts, they concluded.
The study was supported by the National Institute of Mental Health, the National Cancer Institute, and the American Lebanese Syrian Associated Charities. The researchers reported having no conflicts of interest.
Optimizing use of TKIs in CML

DUBROVNIK, CROATIA—Long-term efficacy and toxicity should inform decisions about tyrosine kinase inhibitors (TKIs) in chronic myeloid leukemia (CML), according to the keynote presenter at Leukemia and Lymphoma: Europe and the USA, Linking Knowledge and Practice.
Studies have indicated that long-term survival rates are similar whether CML patients receive frontline treatment with imatinib or second-generation TKIs.
However, the newer TKIs pose a higher risk of uncommon toxicities, said Hagop Kantarjian, MD, a professor at MD Anderson Cancer Center in Houston, Texas, who gave the meeting’s keynote speech.
Dr. Kantarjian said most CML patients should receive daily treatment with TKIs—even if they are in complete cytogenetic response or 100% Ph-positive—because they will live longer.
Frontline treatment options for CML that are approved by the U.S. Food and Drug Administration include imatinib, dasatinib, nilotinib, and bosutinib.
Dr. Kantarjian noted that dasatinib and nilotinib bested imatinib in early analyses from clinical trials, but all three TKIs produced similar rates of overall survival (OS) and progression-free survival (PFS) at extended follow-up.
Dasatinib and imatinib produced similar rates of 5-year OS and PFS in the DASISION trial.1 In ENESTnd, 5-year OS and PFS rates were similar with nilotinib and imatinib.2
However, Dr. Kantarjian said the higher incidence of uncommon toxicities with the newer TKIs must be taken into account.
Choosing a TKI
Dr. Kantarjian recommends frontline imatinib for older patients (≥ 65 to 70) and those who are low-risk according to Sokal score.
Second-generation TKIs should be given upfront to patients who are higher-risk by Sokal and for “very young patients in whom early treatment discontinuation is important,” according to Dr. Kantarjian.
“In accelerated or blast phase, I always use the second-generation TKIs,” he said. “If there’s no binding mutation, I prefer dasatinib. I think it’s the most potent of them. If there are toxicities with dasatinib, bosutinib is equivalent in efficacy, so they are interchangeable.”
Dr. Kantarjian also said a TKI should not be discarded unless there is loss of complete cytogenetic response (not major molecular response) at the maximum tolerated adjusted dose that does not cause grade 3-4 toxicities or chronic grade 2 toxicities.
“[W]e have to remember that we can go down on the dosages of, for example, imatinib down to 200 mg a day, dasatinib as low as 20 mg a day, nilotinib as low as 150 mg twice a day or even 200 mg daily, and bosutinib down to 200 mg daily,” Dr. Kantarjian said.
“So if we have a patient who’s responding with side effects, we should not abandon the particular TKI, we should try to manipulate the dose schedule if they are having a good response.”
Dr. Kantarjian noted that pleural effusion is a toxicity of particular concern with dasatinib, but lowering the dose to 50 mg daily results in similar efficacy and significantly less toxicity than 100 mg daily. For patients over the age of 70, a 20 mg dose can be used.
Dr. Kantarjian said vaso-occlusive and vasospastic reactions are increasingly observed in patients treated with nilotinib. Therefore, he prefers to forgo upfront nilotinib, particularly in patients who have cardiovascular or neurotoxic problems.
“The incidence of vaso-occlusive and vasospastic reactions is now close to 10% to 15% at about 10 years with nilotinib,” Dr. Kantarjian said. “So it is not a trivial toxicity.”
For patients with vaso-occlusive/vasospastic reactions, “bosutinib is probably the safest drug,” Dr. Kantarjian said.
For second- or third-line therapy, patients can receive ponatinib or a second-generation TKI (dasatinib, nilotinib, or bosutinib) as well as omacetaxine or allogeneic stem cell transplant.
“If you disregard toxicities, I think ponatinib is the most powerful TKI, and I think that’s because we are using it at a higher dose that produces so many toxicities,” Dr. Kantarjian said.
He added that the reason ponatinib is not used upfront is because of these toxicities, particularly pancreatitis, skin rashes, vaso-occlusive disorders, and hypertension.
Dr. Kantarjian suggests giving ponatinib at 30 mg daily in patients with T315I mutation and those without guiding mutations who are resistant to second-generation TKIs.
When to discontinue TKIs
Dr. Kantarjian said patients can discontinue TKI therapy if they:
- Are low- or intermediate-risk by Sokal
- Have quantifiable BCR-ABL transcripts—B2A2, B3A2 (e13a2 or e14a2)
- Are in chronic phase
- Achieved an optimal response to their first TKI
- Have been on TKI therapy for more than 8 years
- Achieved a complete molecular response (MR4.5)
- Have had a molecular response for more than 2 to 3 years
- Are available for monitoring every other month for the first 2 years.
Dr. Kantarjian did not report any conflicts of interest at the meeting. However, he has previously reported relationships with Novartis (makers of imatinib and nilotinib), Bristol-Myers Squibb (makers of dasatinib), Pfizer (makers of bosutinib), and Ariad Pharmaceuticals (makers of ponatinib, now owned by Takeda Pharmaceutical Company Limited).
1. Cortes JE et al. J Clin Oncol. 2016 Jul 10; 34(20): 2333–2340. doi: 10.1200/JCO.2015.64.8899
2. Hochhaus A et al. Leukemia. 2016 May; 30(5): 1044–1054. doi: 10.1038/leu.2016.5
DUBROVNIK, CROATIA—Long-term efficacy and toxicity should inform decisions about tyrosine kinase inhibitors (TKIs) in chronic myeloid leukemia (CML), according to the keynote presenter at Leukemia and Lymphoma: Europe and the USA, Linking Knowledge and Practice.
Studies have indicated that long-term survival rates are similar whether CML patients receive frontline treatment with imatinib or second-generation TKIs.
However, the newer TKIs pose a higher risk of uncommon toxicities, said Hagop Kantarjian, MD, a professor at MD Anderson Cancer Center in Houston, Texas, who gave the meeting’s keynote speech.
Dr. Kantarjian said most CML patients should receive daily treatment with TKIs—even if they are in complete cytogenetic response or 100% Ph-positive—because they will live longer.
Frontline treatment options for CML that are approved by the U.S. Food and Drug Administration include imatinib, dasatinib, nilotinib, and bosutinib.
Dr. Kantarjian noted that dasatinib and nilotinib bested imatinib in early analyses from clinical trials, but all three TKIs produced similar rates of overall survival (OS) and progression-free survival (PFS) at extended follow-up.
Dasatinib and imatinib produced similar rates of 5-year OS and PFS in the DASISION trial.1 In ENESTnd, 5-year OS and PFS rates were similar with nilotinib and imatinib.2
However, Dr. Kantarjian said the higher incidence of uncommon toxicities with the newer TKIs must be taken into account.
Choosing a TKI
Dr. Kantarjian recommends frontline imatinib for older patients (≥ 65 to 70) and those who are low-risk according to Sokal score.
Second-generation TKIs should be given upfront to patients who are higher-risk by Sokal and for “very young patients in whom early treatment discontinuation is important,” according to Dr. Kantarjian.
“In accelerated or blast phase, I always use the second-generation TKIs,” he said. “If there’s no binding mutation, I prefer dasatinib. I think it’s the most potent of them. If there are toxicities with dasatinib, bosutinib is equivalent in efficacy, so they are interchangeable.”
Dr. Kantarjian also said a TKI should not be discarded unless there is loss of complete cytogenetic response (not major molecular response) at the maximum tolerated adjusted dose that does not cause grade 3-4 toxicities or chronic grade 2 toxicities.
“[W]e have to remember that we can go down on the dosages of, for example, imatinib down to 200 mg a day, dasatinib as low as 20 mg a day, nilotinib as low as 150 mg twice a day or even 200 mg daily, and bosutinib down to 200 mg daily,” Dr. Kantarjian said.
“So if we have a patient who’s responding with side effects, we should not abandon the particular TKI, we should try to manipulate the dose schedule if they are having a good response.”
Dr. Kantarjian noted that pleural effusion is a toxicity of particular concern with dasatinib, but lowering the dose to 50 mg daily results in similar efficacy and significantly less toxicity than 100 mg daily. For patients over the age of 70, a 20 mg dose can be used.
Dr. Kantarjian said vaso-occlusive and vasospastic reactions are increasingly observed in patients treated with nilotinib. Therefore, he prefers to forgo upfront nilotinib, particularly in patients who have cardiovascular or neurotoxic problems.
“The incidence of vaso-occlusive and vasospastic reactions is now close to 10% to 15% at about 10 years with nilotinib,” Dr. Kantarjian said. “So it is not a trivial toxicity.”
For patients with vaso-occlusive/vasospastic reactions, “bosutinib is probably the safest drug,” Dr. Kantarjian said.
For second- or third-line therapy, patients can receive ponatinib or a second-generation TKI (dasatinib, nilotinib, or bosutinib) as well as omacetaxine or allogeneic stem cell transplant.
“If you disregard toxicities, I think ponatinib is the most powerful TKI, and I think that’s because we are using it at a higher dose that produces so many toxicities,” Dr. Kantarjian said.
He added that the reason ponatinib is not used upfront is because of these toxicities, particularly pancreatitis, skin rashes, vaso-occlusive disorders, and hypertension.
Dr. Kantarjian suggests giving ponatinib at 30 mg daily in patients with T315I mutation and those without guiding mutations who are resistant to second-generation TKIs.
When to discontinue TKIs
Dr. Kantarjian said patients can discontinue TKI therapy if they:
- Are low- or intermediate-risk by Sokal
- Have quantifiable BCR-ABL transcripts—B2A2, B3A2 (e13a2 or e14a2)
- Are in chronic phase
- Achieved an optimal response to their first TKI
- Have been on TKI therapy for more than 8 years
- Achieved a complete molecular response (MR4.5)
- Have had a molecular response for more than 2 to 3 years
- Are available for monitoring every other month for the first 2 years.
Dr. Kantarjian did not report any conflicts of interest at the meeting. However, he has previously reported relationships with Novartis (makers of imatinib and nilotinib), Bristol-Myers Squibb (makers of dasatinib), Pfizer (makers of bosutinib), and Ariad Pharmaceuticals (makers of ponatinib, now owned by Takeda Pharmaceutical Company Limited).
1. Cortes JE et al. J Clin Oncol. 2016 Jul 10; 34(20): 2333–2340. doi: 10.1200/JCO.2015.64.8899
2. Hochhaus A et al. Leukemia. 2016 May; 30(5): 1044–1054. doi: 10.1038/leu.2016.5
DUBROVNIK, CROATIA—Long-term efficacy and toxicity should inform decisions about tyrosine kinase inhibitors (TKIs) in chronic myeloid leukemia (CML), according to the keynote presenter at Leukemia and Lymphoma: Europe and the USA, Linking Knowledge and Practice.
Studies have indicated that long-term survival rates are similar whether CML patients receive frontline treatment with imatinib or second-generation TKIs.
However, the newer TKIs pose a higher risk of uncommon toxicities, said Hagop Kantarjian, MD, a professor at MD Anderson Cancer Center in Houston, Texas, who gave the meeting’s keynote speech.
Dr. Kantarjian said most CML patients should receive daily treatment with TKIs—even if they are in complete cytogenetic response or 100% Ph-positive—because they will live longer.
Frontline treatment options for CML that are approved by the U.S. Food and Drug Administration include imatinib, dasatinib, nilotinib, and bosutinib.
Dr. Kantarjian noted that dasatinib and nilotinib bested imatinib in early analyses from clinical trials, but all three TKIs produced similar rates of overall survival (OS) and progression-free survival (PFS) at extended follow-up.
Dasatinib and imatinib produced similar rates of 5-year OS and PFS in the DASISION trial.1 In ENESTnd, 5-year OS and PFS rates were similar with nilotinib and imatinib.2
However, Dr. Kantarjian said the higher incidence of uncommon toxicities with the newer TKIs must be taken into account.
Choosing a TKI
Dr. Kantarjian recommends frontline imatinib for older patients (≥ 65 to 70) and those who are low-risk according to Sokal score.
Second-generation TKIs should be given upfront to patients who are higher-risk by Sokal and for “very young patients in whom early treatment discontinuation is important,” according to Dr. Kantarjian.
“In accelerated or blast phase, I always use the second-generation TKIs,” he said. “If there’s no binding mutation, I prefer dasatinib. I think it’s the most potent of them. If there are toxicities with dasatinib, bosutinib is equivalent in efficacy, so they are interchangeable.”
Dr. Kantarjian also said a TKI should not be discarded unless there is loss of complete cytogenetic response (not major molecular response) at the maximum tolerated adjusted dose that does not cause grade 3-4 toxicities or chronic grade 2 toxicities.
“[W]e have to remember that we can go down on the dosages of, for example, imatinib down to 200 mg a day, dasatinib as low as 20 mg a day, nilotinib as low as 150 mg twice a day or even 200 mg daily, and bosutinib down to 200 mg daily,” Dr. Kantarjian said.
“So if we have a patient who’s responding with side effects, we should not abandon the particular TKI, we should try to manipulate the dose schedule if they are having a good response.”
Dr. Kantarjian noted that pleural effusion is a toxicity of particular concern with dasatinib, but lowering the dose to 50 mg daily results in similar efficacy and significantly less toxicity than 100 mg daily. For patients over the age of 70, a 20 mg dose can be used.
Dr. Kantarjian said vaso-occlusive and vasospastic reactions are increasingly observed in patients treated with nilotinib. Therefore, he prefers to forgo upfront nilotinib, particularly in patients who have cardiovascular or neurotoxic problems.
“The incidence of vaso-occlusive and vasospastic reactions is now close to 10% to 15% at about 10 years with nilotinib,” Dr. Kantarjian said. “So it is not a trivial toxicity.”
For patients with vaso-occlusive/vasospastic reactions, “bosutinib is probably the safest drug,” Dr. Kantarjian said.
For second- or third-line therapy, patients can receive ponatinib or a second-generation TKI (dasatinib, nilotinib, or bosutinib) as well as omacetaxine or allogeneic stem cell transplant.
“If you disregard toxicities, I think ponatinib is the most powerful TKI, and I think that’s because we are using it at a higher dose that produces so many toxicities,” Dr. Kantarjian said.
He added that the reason ponatinib is not used upfront is because of these toxicities, particularly pancreatitis, skin rashes, vaso-occlusive disorders, and hypertension.
Dr. Kantarjian suggests giving ponatinib at 30 mg daily in patients with T315I mutation and those without guiding mutations who are resistant to second-generation TKIs.
When to discontinue TKIs
Dr. Kantarjian said patients can discontinue TKI therapy if they:
- Are low- or intermediate-risk by Sokal
- Have quantifiable BCR-ABL transcripts—B2A2, B3A2 (e13a2 or e14a2)
- Are in chronic phase
- Achieved an optimal response to their first TKI
- Have been on TKI therapy for more than 8 years
- Achieved a complete molecular response (MR4.5)
- Have had a molecular response for more than 2 to 3 years
- Are available for monitoring every other month for the first 2 years.
Dr. Kantarjian did not report any conflicts of interest at the meeting. However, he has previously reported relationships with Novartis (makers of imatinib and nilotinib), Bristol-Myers Squibb (makers of dasatinib), Pfizer (makers of bosutinib), and Ariad Pharmaceuticals (makers of ponatinib, now owned by Takeda Pharmaceutical Company Limited).
1. Cortes JE et al. J Clin Oncol. 2016 Jul 10; 34(20): 2333–2340. doi: 10.1200/JCO.2015.64.8899
2. Hochhaus A et al. Leukemia. 2016 May; 30(5): 1044–1054. doi: 10.1038/leu.2016.5
Genomic profiling predicts outcomes in patients with MPN
Genomic characteristics of patients with myeloproliferative neoplasms (MPN) can predict clinical outcomes, a recent study found.

Eight genomic subgroups of MPN were recognized, each with distinct clinical features, including event-free survival, risk of leukemic transformation, and blood counts, according to Jacob Grinfeld, MD, of the Wellcome-MRC Cambridge (England) Stem Cell Institute and Cambridge Institute for Medical Research and his colleagues.
“Current classification schemes distinguish among the subtypes of myeloproliferative neoplasms according to clinical and laboratory features, but uncertainty clouds where and how to draw dividing lines among them,” the investigators wrote in the New England Journal of Medicine. “In blood cancers, a progressive shift is under way, from clinical and morphologic classification schemes to those that are based on genomics.”
MPNs are often driven by mutations in CALR, MPL, or JAK2 genes, but classification is not confined to just three genomic types; many patients have additional driver mutations throughout a variety of cancer genes, and it is these additional mutations that are responsible for the wide range of disease phenotypes and clinical outcomes.
This study included 2,035 patients with MPNs, including essential thrombocythemia, polycythemia vera, myelofibrosis, and other MPN diagnoses. The investigators performed targeted sequencing for the full coding sequence of 69 genes and genomewide copy-number information in 1,887 patients. Another 148 patients underwent whole-exome sequencing.
By sequencing coding exons from 69 myeloid cancer genes, the investigators were able to survey the diversity of mutations across a population of patients with MPNs and identify mutation-associated clinical outcomes.
The results showed that slightly less than half (45%) of the patients had a solitary abnormality in CALR, MPL, or JAK2, while the remaining patients had additional driver mutations. In some instances, additional mutations were numerous, particularly in older patients with advanced disease. In at least five cases, 33 genes had driver mutations.
Further analysis identified eight genomic subgroups that could predict clinical outcomes based on shared chromosomal abnormalities and mutations. For example, one subgroup included patients with TP53 mutations; these individuals had a “dismal prognosis” and were 15.5 times more likely to transform to acute myeloid leukemia (AML), compared with the JAK2-heterozygous subgroup (P less than .001).
Because prognosis is “a key determinant of the treatment of patients with MPNs,” genomic subgrouping may one day guide clinical decision making, the investigators concluded.
To further this cause, the investigators have made available an online calculator of individualized patient outcomes, which can be accessed at https://cancer.sanger.ac.uk/mpn-multistage/.
The study was funded by the Wellcome Trust, the National Institute for Health Research Cambridge Biomedical Research Centre, Cancer Research UK, and others. Some study authors reported fees from Celgene, Novartis, Gilead, Shire, and others outside of the study.
SOURCE: Grinfeld J et al. N Engl J Med. 2018;379:1416-30.
Genomic characteristics of patients with myeloproliferative neoplasms (MPN) can predict clinical outcomes, a recent study found.
Eight genomic subgroups of MPN were recognized, each with distinct clinical features, including event-free survival, risk of leukemic transformation, and blood counts, according to Jacob Grinfeld, MD, of the Wellcome-MRC Cambridge (England) Stem Cell Institute and Cambridge Institute for Medical Research and his colleagues.
“Current classification schemes distinguish among the subtypes of myeloproliferative neoplasms according to clinical and laboratory features, but uncertainty clouds where and how to draw dividing lines among them,” the investigators wrote in the New England Journal of Medicine. “In blood cancers, a progressive shift is under way, from clinical and morphologic classification schemes to those that are based on genomics.”
MPNs are often driven by mutations in CALR, MPL, or JAK2 genes, but classification is not confined to just three genomic types; many patients have additional driver mutations throughout a variety of cancer genes, and it is these additional mutations that are responsible for the wide range of disease phenotypes and clinical outcomes.
This study included 2,035 patients with MPNs, including essential thrombocythemia, polycythemia vera, myelofibrosis, and other MPN diagnoses. The investigators performed targeted sequencing for the full coding sequence of 69 genes and genomewide copy-number information in 1,887 patients. Another 148 patients underwent whole-exome sequencing.
By sequencing coding exons from 69 myeloid cancer genes, the investigators were able to survey the diversity of mutations across a population of patients with MPNs and identify mutation-associated clinical outcomes.
The results showed that slightly less than half (45%) of the patients had a solitary abnormality in CALR, MPL, or JAK2, while the remaining patients had additional driver mutations. In some instances, additional mutations were numerous, particularly in older patients with advanced disease. In at least five cases, 33 genes had driver mutations.
Further analysis identified eight genomic subgroups that could predict clinical outcomes based on shared chromosomal abnormalities and mutations. For example, one subgroup included patients with TP53 mutations; these individuals had a “dismal prognosis” and were 15.5 times more likely to transform to acute myeloid leukemia (AML), compared with the JAK2-heterozygous subgroup (P less than .001).
Because prognosis is “a key determinant of the treatment of patients with MPNs,” genomic subgrouping may one day guide clinical decision making, the investigators concluded.
To further this cause, the investigators have made available an online calculator of individualized patient outcomes, which can be accessed at https://cancer.sanger.ac.uk/mpn-multistage/.
The study was funded by the Wellcome Trust, the National Institute for Health Research Cambridge Biomedical Research Centre, Cancer Research UK, and others. Some study authors reported fees from Celgene, Novartis, Gilead, Shire, and others outside of the study.
SOURCE: Grinfeld J et al. N Engl J Med. 2018;379:1416-30.
Genomic characteristics of patients with myeloproliferative neoplasms (MPN) can predict clinical outcomes, a recent study found.
Eight genomic subgroups of MPN were recognized, each with distinct clinical features, including event-free survival, risk of leukemic transformation, and blood counts, according to Jacob Grinfeld, MD, of the Wellcome-MRC Cambridge (England) Stem Cell Institute and Cambridge Institute for Medical Research and his colleagues.
“Current classification schemes distinguish among the subtypes of myeloproliferative neoplasms according to clinical and laboratory features, but uncertainty clouds where and how to draw dividing lines among them,” the investigators wrote in the New England Journal of Medicine. “In blood cancers, a progressive shift is under way, from clinical and morphologic classification schemes to those that are based on genomics.”
MPNs are often driven by mutations in CALR, MPL, or JAK2 genes, but classification is not confined to just three genomic types; many patients have additional driver mutations throughout a variety of cancer genes, and it is these additional mutations that are responsible for the wide range of disease phenotypes and clinical outcomes.
This study included 2,035 patients with MPNs, including essential thrombocythemia, polycythemia vera, myelofibrosis, and other MPN diagnoses. The investigators performed targeted sequencing for the full coding sequence of 69 genes and genomewide copy-number information in 1,887 patients. Another 148 patients underwent whole-exome sequencing.
By sequencing coding exons from 69 myeloid cancer genes, the investigators were able to survey the diversity of mutations across a population of patients with MPNs and identify mutation-associated clinical outcomes.
The results showed that slightly less than half (45%) of the patients had a solitary abnormality in CALR, MPL, or JAK2, while the remaining patients had additional driver mutations. In some instances, additional mutations were numerous, particularly in older patients with advanced disease. In at least five cases, 33 genes had driver mutations.
Further analysis identified eight genomic subgroups that could predict clinical outcomes based on shared chromosomal abnormalities and mutations. For example, one subgroup included patients with TP53 mutations; these individuals had a “dismal prognosis” and were 15.5 times more likely to transform to acute myeloid leukemia (AML), compared with the JAK2-heterozygous subgroup (P less than .001).
Because prognosis is “a key determinant of the treatment of patients with MPNs,” genomic subgrouping may one day guide clinical decision making, the investigators concluded.
To further this cause, the investigators have made available an online calculator of individualized patient outcomes, which can be accessed at https://cancer.sanger.ac.uk/mpn-multistage/.
The study was funded by the Wellcome Trust, the National Institute for Health Research Cambridge Biomedical Research Centre, Cancer Research UK, and others. Some study authors reported fees from Celgene, Novartis, Gilead, Shire, and others outside of the study.
SOURCE: Grinfeld J et al. N Engl J Med. 2018;379:1416-30.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point:
Major finding: Eight genomic subgroups of MPN were recognized, each with distinct clinical features, including event-free survival, risk of leukemic transformation, and blood counts.
Study details: A gene sequencing study involving 2,035 patients with MPN.
Disclosures: The study was funded by the Wellcome Trust, the National Institute for Health Research Cambridge Biomedical Research Centre, Cancer Research UK, and others. Some study authors reported fees from Celgene, Novartis, Gilead, Shire, and others outside of the study.
Source: Grinfeld J et al. N Engl J Med. 2018;379:1416-30.

{kind=link}