User login
CDK8 inhibitor can fight AML, though it’s unclear how

DUBROVNIK, CROATIA—The CDK8 inhibitor SEL120 has demonstrated preclinical activity against acute myeloid leukemia (AML), but the agent’s mechanism of action is still unclear.
Researchers found that several AML cell lines were “highly sensitive” to SEL120, and the inhibitor was active in primary patient samples.
SEL120 also reduced tumor growth in mouse models of AML and demonstrated synergy with venetoclax.
The researchers believe SEL120 works by affecting the maintenance of AML cells and leukemic stem cells (LSCs), inducing differentiation and, sometimes, apoptosis. However, the mechanism is not well defined.
Eliza Majewska, PhD, of Selvita S.A. in Krakow, Poland, discussed research with SEL120 at Leukemia and Lymphoma: Europe and the USA, Linking Knowledge and Practice.
Dr. Majewska explained that CDK8 is a transcriptional kinase working in the context of the Mediator complex, and previous research1 indicated that CDK8 drives oncogenic transcription in AML.
In a prior study2, researchers found that SEL120 inhibits CDK8 activity in AML cells with high levels of STAT phosphorylation.
Dr. Majewska said the MV4-11 cell line responds particularly well to SEL120, and other sensitive cell lines include SKNO-1, Oci-AML5, GDM-1, KG-1, MOLM-16, and Oci-AML3.
“The fact that STAT signaling was upregulated in those cell lines that were very sensitive to SEL120 gave us the hint that perhaps we are looking at a mechanism of action of the compound that has something to do with leukemic stem cells,” Dr. Majewska said.
In fact, she and her colleagues found that cell lines sensitive to SEL120 had upregulation of genes linked to LSCs and high levels of CD34 surface expression.
Experiments in CD34+ TEX cells showed that SEL120 specifically depletes CD34+ cells, leads to downregulation of stemness-related genes, and induces myeloid differentiation.
After 6 days of treatment with SEL120, TEX cells showed decreased expression of the LSC-linked genes MEIS1 and LILRB2, enrichment of gene sets downregulated in LSCs and linked to differentiation, and increased expression of differentiation markers and immune response genes.
SEL120 also demonstrated antileukemic activity in vivo. The researchers tested SEL120 in a CD34+ model of AML (KG-1) and a FLT3-ITD model of AML (MV4-11).
In both models, SEL120 induced “significant tumor regression” of about 80%. In some cases, the researchers observed apoptosis.
Toxicities observed in the mice included weight loss and upregulation of inflammation.
The researchers also found that SEL120 was synergistic with venetoclax. In fact, the combination of these drugs resulted in “almost complete remission cures” in the MV4-11 model, according to Dr. Majewska.
Finally, she and her colleagues discovered that SEL120 was active against primary patient cells. Samples from 3 of 4 AML patients had a significant reduction in cell numbers after 7 days of treatment with SEL120. For one patient, there were no viable cells on day 7.
Dr. Majewska said a phase 1 trial of SEL120 is planned for 2019 or 2020, and SEL120’s mechanism of action is still under investigation.
“The mechanism of action . . . is, in our mind, at least in some cases, linked to the fact that CDK8 functions within the context of the Mediator complex, which contributes to gene expression related to leukemic stem cells,” Dr. Majewska said.
“And when we inhibit this specific transcription, of course, the Mediator complex still works because this is just one of the components of the complex. However, the function that it has is suddenly very different, and it’s actually linked to lack of maintenance of leukemic stem cells, resulting in differentiation [and], in some cases, the induction of apoptosis, but we do not fully understand the mechanism of this induction.”
Dr. Majewska works for Selvita, the company developing SEL120. This research was funded by Selvita, the Leukemia & Lymphoma Society, and the National Centre for Research and Development.
1. Pelish HE et al. Nature. 2015 Oct 8;526(7572):273-276. doi: 10.1038/nature14904
2. Rzymski T et al. Oncotarget. 2017 May 16;8(20):33779-33795. doi: 10.18632/oncotarget.16810.
DUBROVNIK, CROATIA—The CDK8 inhibitor SEL120 has demonstrated preclinical activity against acute myeloid leukemia (AML), but the agent’s mechanism of action is still unclear.
Researchers found that several AML cell lines were “highly sensitive” to SEL120, and the inhibitor was active in primary patient samples.
SEL120 also reduced tumor growth in mouse models of AML and demonstrated synergy with venetoclax.
The researchers believe SEL120 works by affecting the maintenance of AML cells and leukemic stem cells (LSCs), inducing differentiation and, sometimes, apoptosis. However, the mechanism is not well defined.
Eliza Majewska, PhD, of Selvita S.A. in Krakow, Poland, discussed research with SEL120 at Leukemia and Lymphoma: Europe and the USA, Linking Knowledge and Practice.
Dr. Majewska explained that CDK8 is a transcriptional kinase working in the context of the Mediator complex, and previous research1 indicated that CDK8 drives oncogenic transcription in AML.
In a prior study2, researchers found that SEL120 inhibits CDK8 activity in AML cells with high levels of STAT phosphorylation.
Dr. Majewska said the MV4-11 cell line responds particularly well to SEL120, and other sensitive cell lines include SKNO-1, Oci-AML5, GDM-1, KG-1, MOLM-16, and Oci-AML3.
“The fact that STAT signaling was upregulated in those cell lines that were very sensitive to SEL120 gave us the hint that perhaps we are looking at a mechanism of action of the compound that has something to do with leukemic stem cells,” Dr. Majewska said.
In fact, she and her colleagues found that cell lines sensitive to SEL120 had upregulation of genes linked to LSCs and high levels of CD34 surface expression.
Experiments in CD34+ TEX cells showed that SEL120 specifically depletes CD34+ cells, leads to downregulation of stemness-related genes, and induces myeloid differentiation.
After 6 days of treatment with SEL120, TEX cells showed decreased expression of the LSC-linked genes MEIS1 and LILRB2, enrichment of gene sets downregulated in LSCs and linked to differentiation, and increased expression of differentiation markers and immune response genes.
SEL120 also demonstrated antileukemic activity in vivo. The researchers tested SEL120 in a CD34+ model of AML (KG-1) and a FLT3-ITD model of AML (MV4-11).
In both models, SEL120 induced “significant tumor regression” of about 80%. In some cases, the researchers observed apoptosis.
Toxicities observed in the mice included weight loss and upregulation of inflammation.
The researchers also found that SEL120 was synergistic with venetoclax. In fact, the combination of these drugs resulted in “almost complete remission cures” in the MV4-11 model, according to Dr. Majewska.
Finally, she and her colleagues discovered that SEL120 was active against primary patient cells. Samples from 3 of 4 AML patients had a significant reduction in cell numbers after 7 days of treatment with SEL120. For one patient, there were no viable cells on day 7.
Dr. Majewska said a phase 1 trial of SEL120 is planned for 2019 or 2020, and SEL120’s mechanism of action is still under investigation.
“The mechanism of action . . . is, in our mind, at least in some cases, linked to the fact that CDK8 functions within the context of the Mediator complex, which contributes to gene expression related to leukemic stem cells,” Dr. Majewska said.
“And when we inhibit this specific transcription, of course, the Mediator complex still works because this is just one of the components of the complex. However, the function that it has is suddenly very different, and it’s actually linked to lack of maintenance of leukemic stem cells, resulting in differentiation [and], in some cases, the induction of apoptosis, but we do not fully understand the mechanism of this induction.”
Dr. Majewska works for Selvita, the company developing SEL120. This research was funded by Selvita, the Leukemia & Lymphoma Society, and the National Centre for Research and Development.
1. Pelish HE et al. Nature. 2015 Oct 8;526(7572):273-276. doi: 10.1038/nature14904
2. Rzymski T et al. Oncotarget. 2017 May 16;8(20):33779-33795. doi: 10.18632/oncotarget.16810.
DUBROVNIK, CROATIA—The CDK8 inhibitor SEL120 has demonstrated preclinical activity against acute myeloid leukemia (AML), but the agent’s mechanism of action is still unclear.
Researchers found that several AML cell lines were “highly sensitive” to SEL120, and the inhibitor was active in primary patient samples.
SEL120 also reduced tumor growth in mouse models of AML and demonstrated synergy with venetoclax.
The researchers believe SEL120 works by affecting the maintenance of AML cells and leukemic stem cells (LSCs), inducing differentiation and, sometimes, apoptosis. However, the mechanism is not well defined.
Eliza Majewska, PhD, of Selvita S.A. in Krakow, Poland, discussed research with SEL120 at Leukemia and Lymphoma: Europe and the USA, Linking Knowledge and Practice.
Dr. Majewska explained that CDK8 is a transcriptional kinase working in the context of the Mediator complex, and previous research1 indicated that CDK8 drives oncogenic transcription in AML.
In a prior study2, researchers found that SEL120 inhibits CDK8 activity in AML cells with high levels of STAT phosphorylation.
Dr. Majewska said the MV4-11 cell line responds particularly well to SEL120, and other sensitive cell lines include SKNO-1, Oci-AML5, GDM-1, KG-1, MOLM-16, and Oci-AML3.
“The fact that STAT signaling was upregulated in those cell lines that were very sensitive to SEL120 gave us the hint that perhaps we are looking at a mechanism of action of the compound that has something to do with leukemic stem cells,” Dr. Majewska said.
In fact, she and her colleagues found that cell lines sensitive to SEL120 had upregulation of genes linked to LSCs and high levels of CD34 surface expression.
Experiments in CD34+ TEX cells showed that SEL120 specifically depletes CD34+ cells, leads to downregulation of stemness-related genes, and induces myeloid differentiation.
After 6 days of treatment with SEL120, TEX cells showed decreased expression of the LSC-linked genes MEIS1 and LILRB2, enrichment of gene sets downregulated in LSCs and linked to differentiation, and increased expression of differentiation markers and immune response genes.
SEL120 also demonstrated antileukemic activity in vivo. The researchers tested SEL120 in a CD34+ model of AML (KG-1) and a FLT3-ITD model of AML (MV4-11).
In both models, SEL120 induced “significant tumor regression” of about 80%. In some cases, the researchers observed apoptosis.
Toxicities observed in the mice included weight loss and upregulation of inflammation.
The researchers also found that SEL120 was synergistic with venetoclax. In fact, the combination of these drugs resulted in “almost complete remission cures” in the MV4-11 model, according to Dr. Majewska.
Finally, she and her colleagues discovered that SEL120 was active against primary patient cells. Samples from 3 of 4 AML patients had a significant reduction in cell numbers after 7 days of treatment with SEL120. For one patient, there were no viable cells on day 7.
Dr. Majewska said a phase 1 trial of SEL120 is planned for 2019 or 2020, and SEL120’s mechanism of action is still under investigation.
“The mechanism of action . . . is, in our mind, at least in some cases, linked to the fact that CDK8 functions within the context of the Mediator complex, which contributes to gene expression related to leukemic stem cells,” Dr. Majewska said.
“And when we inhibit this specific transcription, of course, the Mediator complex still works because this is just one of the components of the complex. However, the function that it has is suddenly very different, and it’s actually linked to lack of maintenance of leukemic stem cells, resulting in differentiation [and], in some cases, the induction of apoptosis, but we do not fully understand the mechanism of this induction.”
Dr. Majewska works for Selvita, the company developing SEL120. This research was funded by Selvita, the Leukemia & Lymphoma Society, and the National Centre for Research and Development.
1. Pelish HE et al. Nature. 2015 Oct 8;526(7572):273-276. doi: 10.1038/nature14904
2. Rzymski T et al. Oncotarget. 2017 May 16;8(20):33779-33795. doi: 10.18632/oncotarget.16810.
Phase 1 NHL, ALL trials placed on clinical hold

Update: On October 12, 2018, Affimed N.V. received a notification from the U.S. Food and Drug Administration (FDA) saying the agency concurred with Affimed’s decision and formally placed the investigational new drug application for AFM11 on full clinical hold. Affimed said it will comply with the FDA and other global health authorities’ requests for information to resolve the clinical hold.
Affimed N.V. has placed trials of AFM11 on clinical hold and notified the global health authorities of its decision.
AFM11 is a CD19/CD3-targeting T-cell engager being evaluated in two phase 1 trials—one in patients with relapsed or refractory, CD19-positive B-cell non-Hodgkin lymphoma (NHL) and one in adults with relapsed or refractory B-precursor acute lymphoblastic leukemia (ALL).
Affimed initiated the clinical hold on these trials after serious adverse events occurred in three patients treated with AFM11.
This included a death in the ALL study and two life-threatening events in the NHL study.
The serious adverse events occurred in patients enrolled in the highest dose cohorts of each study.
A total of 33 patients have been treated in the two studies (NCT02848911 and NCT02106091), and preliminary signs of clinical activity have been observed in several patients.
Affimed said it will be working closely with the global health authorities, safety monitoring committees, and the studies’ clinical investigators to review the adverse events, assess all the data, and determine next steps for the AFM11 program.
Affimed intends to provide an update on AFM11 upon completing the evaluation.
Update: On October 12, 2018, Affimed N.V. received a notification from the U.S. Food and Drug Administration (FDA) saying the agency concurred with Affimed’s decision and formally placed the investigational new drug application for AFM11 on full clinical hold. Affimed said it will comply with the FDA and other global health authorities’ requests for information to resolve the clinical hold.
Affimed N.V. has placed trials of AFM11 on clinical hold and notified the global health authorities of its decision.
AFM11 is a CD19/CD3-targeting T-cell engager being evaluated in two phase 1 trials—one in patients with relapsed or refractory, CD19-positive B-cell non-Hodgkin lymphoma (NHL) and one in adults with relapsed or refractory B-precursor acute lymphoblastic leukemia (ALL).
Affimed initiated the clinical hold on these trials after serious adverse events occurred in three patients treated with AFM11.
This included a death in the ALL study and two life-threatening events in the NHL study.
The serious adverse events occurred in patients enrolled in the highest dose cohorts of each study.
A total of 33 patients have been treated in the two studies (NCT02848911 and NCT02106091), and preliminary signs of clinical activity have been observed in several patients.
Affimed said it will be working closely with the global health authorities, safety monitoring committees, and the studies’ clinical investigators to review the adverse events, assess all the data, and determine next steps for the AFM11 program.
Affimed intends to provide an update on AFM11 upon completing the evaluation.
Update: On October 12, 2018, Affimed N.V. received a notification from the U.S. Food and Drug Administration (FDA) saying the agency concurred with Affimed’s decision and formally placed the investigational new drug application for AFM11 on full clinical hold. Affimed said it will comply with the FDA and other global health authorities’ requests for information to resolve the clinical hold.
Affimed N.V. has placed trials of AFM11 on clinical hold and notified the global health authorities of its decision.
AFM11 is a CD19/CD3-targeting T-cell engager being evaluated in two phase 1 trials—one in patients with relapsed or refractory, CD19-positive B-cell non-Hodgkin lymphoma (NHL) and one in adults with relapsed or refractory B-precursor acute lymphoblastic leukemia (ALL).
Affimed initiated the clinical hold on these trials after serious adverse events occurred in three patients treated with AFM11.
This included a death in the ALL study and two life-threatening events in the NHL study.
The serious adverse events occurred in patients enrolled in the highest dose cohorts of each study.
A total of 33 patients have been treated in the two studies (NCT02848911 and NCT02106091), and preliminary signs of clinical activity have been observed in several patients.
Affimed said it will be working closely with the global health authorities, safety monitoring committees, and the studies’ clinical investigators to review the adverse events, assess all the data, and determine next steps for the AFM11 program.
Affimed intends to provide an update on AFM11 upon completing the evaluation.
Weighing the costs of CAR T-cell therapy

The cost-effectiveness of tisagenlecleucel (Kymriah) depends on long-term clinical outcomes, which are presently unknown, according to investigators.
If the long-term outcomes are more modest than clinical trials suggest, then payers may be unwilling to cover the costly therapy, reported John K. Lin, MD, of Stanford University, and his colleagues.
Lowering the price or setting up an outcomes-based pricing structure may be necessary to get insurers to cover the therapy.
Tisagenlecleucel is an anti-CD19 chimeric antigen receptor (CAR) T-cell therapy that was approved by the U.S. Food and Drug Administration in August 2017 for relapsed or refractory pediatric B-cell acute lymphoblastic leukemia (ALL).
In 2018, the FDA expanded the indication for tisagenlecleucel to include adults with relapsed or refractory large B-cell lymphoma, though outcomes from lymphoma trials are not analyzed in the current study.
At a wholesale acquisition cost of $475,000 per infusion, it is the most expensive existing oncology therapy to date, and can be accompanied by expensive, potentially fatal adverse effects.
However, clinical trials suggest that tisagenlecleucel can offer years of relapse-free remission, thereby allowing patients to forgo other expensive therapies such as hematopoietic stem cell transplantation (HSCT).
“Although tisagenlecleucel-induced remission rates are promising, compared with those of established therapies (greater than 80% vs. less than 50%), only short-term follow-up data currently exist,” the investigators wrote in the Journal of Clinical Oncology.
“Given the high cost and broad applicability in other malignancies of tisagenlecleucel, a pressing question for policy makers, payers, patients, and clinicians is whether the cost of therapy represents reasonable value.”
The study used a Markov model to assess various long-term clinical outcome rates and cost thresholds of tisagenlecleucel. The lifetime cost of therapy was assessed and compared with costs of existing therapies.
The results showed that a 5-year relapse free survival rate of 40% would make the present cost ($475,000) of tisagenlecleucel economically reasonable. In this scenario, the increased life expectancy would be 12.1 years and would result in an additional 5.07 quality-adjusted life years (QALY) gained at a cost of $61,000 per QALY, compared with blinatumomab.
But if long-term outcomes are less favorable, tisagenlecleucel becomes much less cost effective. A 5-year relapse-free survival rate of 20% would drop increased life expectancy to 3.8 years, resulting in 1.80 QALYs gained and raising the cost to $151,000 per QALY.
“Our results suggest that at tisagenlecleucel’s current price and payment structure, its economic value is uncertain,” the investigators wrote.
They suggested a price drop to $200,000 or $350,000, which would allow the drug to remain cost effective even in a worse-case scenario, in which patients relapse and tisagenlecleucel is a bridge to transplant.
Another option is to move to outcomes-based pricing. Making payment conditional on 7 months of remission would make the treatment cost effective, according to the analysis.
“Price reductions of tisagenlecleucel or payment only for longer-term remissions would favorably influence cost-effectiveness, even if long-term clinical outcomes are modest,” the investigators wrote.
The study was funded by a Veterans Affairs Office of Academic Affiliations advanced fellowship in health service and research development, and a National Center for Advancing Translational Science Clinical and Translational Science Award.
One of the study coauthors reported consulting and research funding from Novartis.
The cost-effectiveness of tisagenlecleucel (Kymriah) depends on long-term clinical outcomes, which are presently unknown, according to investigators.
If the long-term outcomes are more modest than clinical trials suggest, then payers may be unwilling to cover the costly therapy, reported John K. Lin, MD, of Stanford University, and his colleagues.
Lowering the price or setting up an outcomes-based pricing structure may be necessary to get insurers to cover the therapy.
Tisagenlecleucel is an anti-CD19 chimeric antigen receptor (CAR) T-cell therapy that was approved by the U.S. Food and Drug Administration in August 2017 for relapsed or refractory pediatric B-cell acute lymphoblastic leukemia (ALL).
In 2018, the FDA expanded the indication for tisagenlecleucel to include adults with relapsed or refractory large B-cell lymphoma, though outcomes from lymphoma trials are not analyzed in the current study.
At a wholesale acquisition cost of $475,000 per infusion, it is the most expensive existing oncology therapy to date, and can be accompanied by expensive, potentially fatal adverse effects.
However, clinical trials suggest that tisagenlecleucel can offer years of relapse-free remission, thereby allowing patients to forgo other expensive therapies such as hematopoietic stem cell transplantation (HSCT).
“Although tisagenlecleucel-induced remission rates are promising, compared with those of established therapies (greater than 80% vs. less than 50%), only short-term follow-up data currently exist,” the investigators wrote in the Journal of Clinical Oncology.
“Given the high cost and broad applicability in other malignancies of tisagenlecleucel, a pressing question for policy makers, payers, patients, and clinicians is whether the cost of therapy represents reasonable value.”
The study used a Markov model to assess various long-term clinical outcome rates and cost thresholds of tisagenlecleucel. The lifetime cost of therapy was assessed and compared with costs of existing therapies.
The results showed that a 5-year relapse free survival rate of 40% would make the present cost ($475,000) of tisagenlecleucel economically reasonable. In this scenario, the increased life expectancy would be 12.1 years and would result in an additional 5.07 quality-adjusted life years (QALY) gained at a cost of $61,000 per QALY, compared with blinatumomab.
But if long-term outcomes are less favorable, tisagenlecleucel becomes much less cost effective. A 5-year relapse-free survival rate of 20% would drop increased life expectancy to 3.8 years, resulting in 1.80 QALYs gained and raising the cost to $151,000 per QALY.
“Our results suggest that at tisagenlecleucel’s current price and payment structure, its economic value is uncertain,” the investigators wrote.
They suggested a price drop to $200,000 or $350,000, which would allow the drug to remain cost effective even in a worse-case scenario, in which patients relapse and tisagenlecleucel is a bridge to transplant.
Another option is to move to outcomes-based pricing. Making payment conditional on 7 months of remission would make the treatment cost effective, according to the analysis.
“Price reductions of tisagenlecleucel or payment only for longer-term remissions would favorably influence cost-effectiveness, even if long-term clinical outcomes are modest,” the investigators wrote.
The study was funded by a Veterans Affairs Office of Academic Affiliations advanced fellowship in health service and research development, and a National Center for Advancing Translational Science Clinical and Translational Science Award.
One of the study coauthors reported consulting and research funding from Novartis.
The cost-effectiveness of tisagenlecleucel (Kymriah) depends on long-term clinical outcomes, which are presently unknown, according to investigators.
If the long-term outcomes are more modest than clinical trials suggest, then payers may be unwilling to cover the costly therapy, reported John K. Lin, MD, of Stanford University, and his colleagues.
Lowering the price or setting up an outcomes-based pricing structure may be necessary to get insurers to cover the therapy.
Tisagenlecleucel is an anti-CD19 chimeric antigen receptor (CAR) T-cell therapy that was approved by the U.S. Food and Drug Administration in August 2017 for relapsed or refractory pediatric B-cell acute lymphoblastic leukemia (ALL).
In 2018, the FDA expanded the indication for tisagenlecleucel to include adults with relapsed or refractory large B-cell lymphoma, though outcomes from lymphoma trials are not analyzed in the current study.
At a wholesale acquisition cost of $475,000 per infusion, it is the most expensive existing oncology therapy to date, and can be accompanied by expensive, potentially fatal adverse effects.
However, clinical trials suggest that tisagenlecleucel can offer years of relapse-free remission, thereby allowing patients to forgo other expensive therapies such as hematopoietic stem cell transplantation (HSCT).
“Although tisagenlecleucel-induced remission rates are promising, compared with those of established therapies (greater than 80% vs. less than 50%), only short-term follow-up data currently exist,” the investigators wrote in the Journal of Clinical Oncology.
“Given the high cost and broad applicability in other malignancies of tisagenlecleucel, a pressing question for policy makers, payers, patients, and clinicians is whether the cost of therapy represents reasonable value.”
The study used a Markov model to assess various long-term clinical outcome rates and cost thresholds of tisagenlecleucel. The lifetime cost of therapy was assessed and compared with costs of existing therapies.
The results showed that a 5-year relapse free survival rate of 40% would make the present cost ($475,000) of tisagenlecleucel economically reasonable. In this scenario, the increased life expectancy would be 12.1 years and would result in an additional 5.07 quality-adjusted life years (QALY) gained at a cost of $61,000 per QALY, compared with blinatumomab.
But if long-term outcomes are less favorable, tisagenlecleucel becomes much less cost effective. A 5-year relapse-free survival rate of 20% would drop increased life expectancy to 3.8 years, resulting in 1.80 QALYs gained and raising the cost to $151,000 per QALY.
“Our results suggest that at tisagenlecleucel’s current price and payment structure, its economic value is uncertain,” the investigators wrote.
They suggested a price drop to $200,000 or $350,000, which would allow the drug to remain cost effective even in a worse-case scenario, in which patients relapse and tisagenlecleucel is a bridge to transplant.
Another option is to move to outcomes-based pricing. Making payment conditional on 7 months of remission would make the treatment cost effective, according to the analysis.
“Price reductions of tisagenlecleucel or payment only for longer-term remissions would favorably influence cost-effectiveness, even if long-term clinical outcomes are modest,” the investigators wrote.
The study was funded by a Veterans Affairs Office of Academic Affiliations advanced fellowship in health service and research development, and a National Center for Advancing Translational Science Clinical and Translational Science Award.
One of the study coauthors reported consulting and research funding from Novartis.
Some mutation testing can be useful at CLL diagnosis
CHICAGO – A number of mutation tests – including immunoglobulin heavy chain gene (IgVH), fluorescence in situ hybridization (FISH), and TP53 – provide useful prognostic information at the time of chronic lymphocytic leukemia (CLL) diagnosis, according to Paul M. Barr, MD.
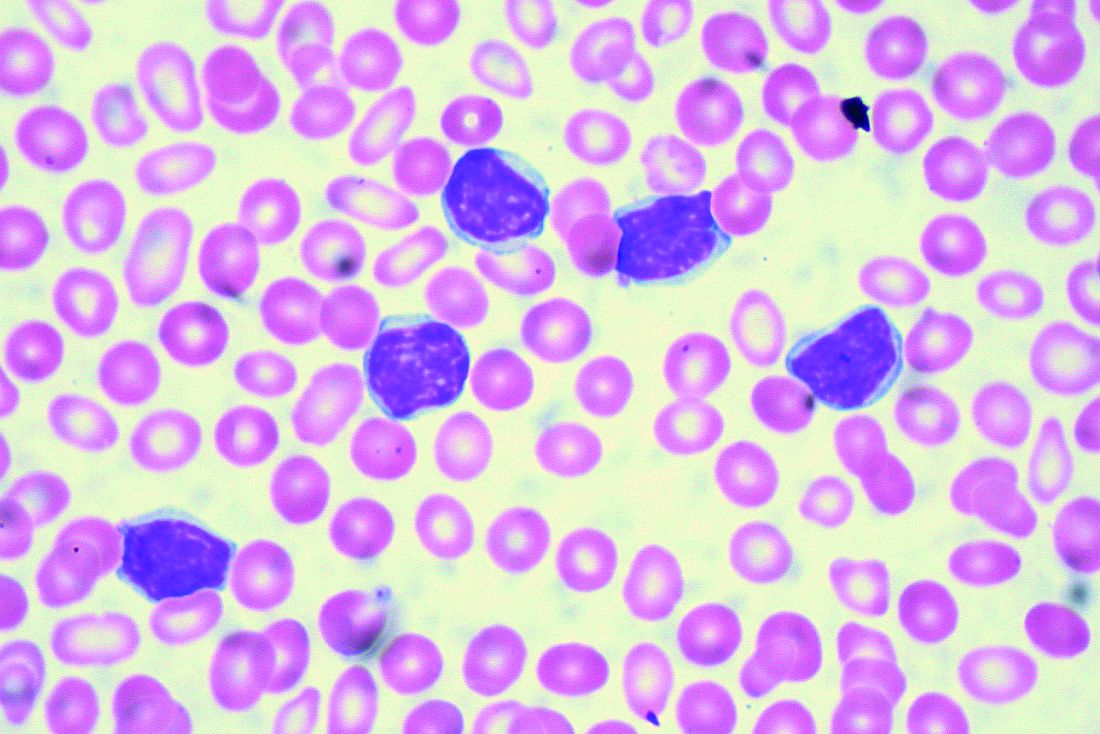
“It’s understood that IgVH mutation status is certainly prognostic,” Dr. Barr, associate professor of hematology/oncology at the University of Rochester (N.Y.), said during a presentation at the American Society of Hematology Meeting on Hematologic Malignancies.
The B-cell receptor of the CLL cells uses IgVH genes that may or may not have undergone somatic mutations, with unmutated being defined as 98% or more sequence homology to germline.
“This is indicative of stronger signaling through the B-cell receptor and, as we all know, predicts for an inferior prognosis,” he explained, citing a study that demonstrated superior survival rates with mutated IgVH genes (Blood. 1999;94[6]:1840-7).
“It’s also well understood and accepted that we should perform a FISH panel; we should look for interphase cytogenetics based on FISH in our patients,” Dr. Barr said. “Having said that, we, as medical oncologists, do not do a very good job of using this testing appropriately. Patterns of care studies have suggested that a significant number of patients don’t get FISH testing at diagnosis or before first-line therapy.”
In fact, a typical interphase FISH panel identifies cytogenetic lesions, including del(17p), del(11q), del(13q), and trisomy 12 in more than 80% of CLL cases, with del(13q) being the most common.
Another marker that can be assessed in CLL patients and has maintained prognostic value across multiple analyses is serum beta-2 microglobulin, Dr. Barr noted.
TP53 sequencing is valuable as well and has been associated with outcomes similar to those seen in patients with del(17p), he said, citing data from a study that found similarly poor outcomes with TP53 mutations or deletions and del(17p), even when minor subclones are identified using next-generation sequencing (Blood. 2014;123:2139-47).
“One of the primary reasons for this is that the two aberrations go together. Most often, if you have del(17p) you’re also going to find a TP53 mutation, but in about 30% of patients or so, only one allele is affected, so this is why it’s still important to test for TP53 mutations when you’re looking for a 17p deletion,” he said.
Numerous other recurrent mutations in CLL have been associated with poor overall survival and/or progression-free survival, including SF3B1, ATM, NOTCH1, POT1, BIRC3, and NFKBIE.
“The gut instinct is that maybe we should start testing for all of these mutations now, but I would caution everybody that we still need further validation before we can apply these to the majority of patients,” Dr. Barr said. “We still don’t know exactly what to do with all of these mutations – when and how often we should test for them, if the novel agents are truly better – so while, again, they can predict for inferior outcomes, I would say these are not yet standard of care to be tested in all patients.”
It is likely, though, that new prognostic systems will evolve as more is learned about how to use these molecular aberrations. Attempts are already being made to incorporate novel mutations into a prognostic system. Dr. Barr pointed to a report that looked at the integration of mutations and cytogenetic lesions to improve the accuracy of survival prediction in CLL (Blood. 2013;121:1403-12).
“But this still requires prospective testing, especially in patients getting the novel agents,” he said.
Conventional karyotyping also has potential, though a limited role in this setting, he said, noting that it can be reliably performed with stimulation of CLL cells.
“We also know additional aberrations are prognostic and that a complex karyotype predicts for a very poor outcome,” he said. The International Workshop on CLL (iwCLL) guidelines, which were recently updated for the first time in a decade, state that further validation is needed.
“I think it’s potentially useful in a very young patient you are considering taking to transplant, but again, I agree with the stance that this is not something that should be performed in every patient across the board,” he said.
The tests currently recommended by iwCLL before CLL treatment include IgVH mutation status; FISH for del(13q), del(11q), del(17p), and trisomy 12 in peripheral blood lymphocytes; and TP53.
“Some folks... don’t check a lot of these markers at diagnosis, but wait for patients to require therapy, and that’s a reasonable way to practice,” Dr. Barr said, noting, however, that he prefers knowing patients’ risk up front – especially for those patients he will see just once before they are “managed closer to home for the majority of their course.
“But if you [wait], then knowing what to repeat later is important,” he added. Namely, the FISH and TP53 tests are worth repeating as patients can acquire additional molecular aberrations over time.
Dr. Barr reported serving as a consultant for Pharmacyclics, AbbVie, Celgene, Gilead Sciences, Infinity Pharmaceuticals, Novartis, and Seattle Genetics. He also reported receiving research funding from Pharmacyclics and AbbVie.
CHICAGO – A number of mutation tests – including immunoglobulin heavy chain gene (IgVH), fluorescence in situ hybridization (FISH), and TP53 – provide useful prognostic information at the time of chronic lymphocytic leukemia (CLL) diagnosis, according to Paul M. Barr, MD.
“It’s understood that IgVH mutation status is certainly prognostic,” Dr. Barr, associate professor of hematology/oncology at the University of Rochester (N.Y.), said during a presentation at the American Society of Hematology Meeting on Hematologic Malignancies.
The B-cell receptor of the CLL cells uses IgVH genes that may or may not have undergone somatic mutations, with unmutated being defined as 98% or more sequence homology to germline.
“This is indicative of stronger signaling through the B-cell receptor and, as we all know, predicts for an inferior prognosis,” he explained, citing a study that demonstrated superior survival rates with mutated IgVH genes (Blood. 1999;94[6]:1840-7).
“It’s also well understood and accepted that we should perform a FISH panel; we should look for interphase cytogenetics based on FISH in our patients,” Dr. Barr said. “Having said that, we, as medical oncologists, do not do a very good job of using this testing appropriately. Patterns of care studies have suggested that a significant number of patients don’t get FISH testing at diagnosis or before first-line therapy.”
In fact, a typical interphase FISH panel identifies cytogenetic lesions, including del(17p), del(11q), del(13q), and trisomy 12 in more than 80% of CLL cases, with del(13q) being the most common.
Another marker that can be assessed in CLL patients and has maintained prognostic value across multiple analyses is serum beta-2 microglobulin, Dr. Barr noted.
TP53 sequencing is valuable as well and has been associated with outcomes similar to those seen in patients with del(17p), he said, citing data from a study that found similarly poor outcomes with TP53 mutations or deletions and del(17p), even when minor subclones are identified using next-generation sequencing (Blood. 2014;123:2139-47).
“One of the primary reasons for this is that the two aberrations go together. Most often, if you have del(17p) you’re also going to find a TP53 mutation, but in about 30% of patients or so, only one allele is affected, so this is why it’s still important to test for TP53 mutations when you’re looking for a 17p deletion,” he said.
Numerous other recurrent mutations in CLL have been associated with poor overall survival and/or progression-free survival, including SF3B1, ATM, NOTCH1, POT1, BIRC3, and NFKBIE.
“The gut instinct is that maybe we should start testing for all of these mutations now, but I would caution everybody that we still need further validation before we can apply these to the majority of patients,” Dr. Barr said. “We still don’t know exactly what to do with all of these mutations – when and how often we should test for them, if the novel agents are truly better – so while, again, they can predict for inferior outcomes, I would say these are not yet standard of care to be tested in all patients.”
It is likely, though, that new prognostic systems will evolve as more is learned about how to use these molecular aberrations. Attempts are already being made to incorporate novel mutations into a prognostic system. Dr. Barr pointed to a report that looked at the integration of mutations and cytogenetic lesions to improve the accuracy of survival prediction in CLL (Blood. 2013;121:1403-12).
“But this still requires prospective testing, especially in patients getting the novel agents,” he said.
Conventional karyotyping also has potential, though a limited role in this setting, he said, noting that it can be reliably performed with stimulation of CLL cells.
“We also know additional aberrations are prognostic and that a complex karyotype predicts for a very poor outcome,” he said. The International Workshop on CLL (iwCLL) guidelines, which were recently updated for the first time in a decade, state that further validation is needed.
“I think it’s potentially useful in a very young patient you are considering taking to transplant, but again, I agree with the stance that this is not something that should be performed in every patient across the board,” he said.
The tests currently recommended by iwCLL before CLL treatment include IgVH mutation status; FISH for del(13q), del(11q), del(17p), and trisomy 12 in peripheral blood lymphocytes; and TP53.
“Some folks... don’t check a lot of these markers at diagnosis, but wait for patients to require therapy, and that’s a reasonable way to practice,” Dr. Barr said, noting, however, that he prefers knowing patients’ risk up front – especially for those patients he will see just once before they are “managed closer to home for the majority of their course.
“But if you [wait], then knowing what to repeat later is important,” he added. Namely, the FISH and TP53 tests are worth repeating as patients can acquire additional molecular aberrations over time.
Dr. Barr reported serving as a consultant for Pharmacyclics, AbbVie, Celgene, Gilead Sciences, Infinity Pharmaceuticals, Novartis, and Seattle Genetics. He also reported receiving research funding from Pharmacyclics and AbbVie.
CHICAGO – A number of mutation tests – including immunoglobulin heavy chain gene (IgVH), fluorescence in situ hybridization (FISH), and TP53 – provide useful prognostic information at the time of chronic lymphocytic leukemia (CLL) diagnosis, according to Paul M. Barr, MD.
“It’s understood that IgVH mutation status is certainly prognostic,” Dr. Barr, associate professor of hematology/oncology at the University of Rochester (N.Y.), said during a presentation at the American Society of Hematology Meeting on Hematologic Malignancies.
The B-cell receptor of the CLL cells uses IgVH genes that may or may not have undergone somatic mutations, with unmutated being defined as 98% or more sequence homology to germline.
“This is indicative of stronger signaling through the B-cell receptor and, as we all know, predicts for an inferior prognosis,” he explained, citing a study that demonstrated superior survival rates with mutated IgVH genes (Blood. 1999;94[6]:1840-7).
“It’s also well understood and accepted that we should perform a FISH panel; we should look for interphase cytogenetics based on FISH in our patients,” Dr. Barr said. “Having said that, we, as medical oncologists, do not do a very good job of using this testing appropriately. Patterns of care studies have suggested that a significant number of patients don’t get FISH testing at diagnosis or before first-line therapy.”
In fact, a typical interphase FISH panel identifies cytogenetic lesions, including del(17p), del(11q), del(13q), and trisomy 12 in more than 80% of CLL cases, with del(13q) being the most common.
Another marker that can be assessed in CLL patients and has maintained prognostic value across multiple analyses is serum beta-2 microglobulin, Dr. Barr noted.
TP53 sequencing is valuable as well and has been associated with outcomes similar to those seen in patients with del(17p), he said, citing data from a study that found similarly poor outcomes with TP53 mutations or deletions and del(17p), even when minor subclones are identified using next-generation sequencing (Blood. 2014;123:2139-47).
“One of the primary reasons for this is that the two aberrations go together. Most often, if you have del(17p) you’re also going to find a TP53 mutation, but in about 30% of patients or so, only one allele is affected, so this is why it’s still important to test for TP53 mutations when you’re looking for a 17p deletion,” he said.
Numerous other recurrent mutations in CLL have been associated with poor overall survival and/or progression-free survival, including SF3B1, ATM, NOTCH1, POT1, BIRC3, and NFKBIE.
“The gut instinct is that maybe we should start testing for all of these mutations now, but I would caution everybody that we still need further validation before we can apply these to the majority of patients,” Dr. Barr said. “We still don’t know exactly what to do with all of these mutations – when and how often we should test for them, if the novel agents are truly better – so while, again, they can predict for inferior outcomes, I would say these are not yet standard of care to be tested in all patients.”
It is likely, though, that new prognostic systems will evolve as more is learned about how to use these molecular aberrations. Attempts are already being made to incorporate novel mutations into a prognostic system. Dr. Barr pointed to a report that looked at the integration of mutations and cytogenetic lesions to improve the accuracy of survival prediction in CLL (Blood. 2013;121:1403-12).
“But this still requires prospective testing, especially in patients getting the novel agents,” he said.
Conventional karyotyping also has potential, though a limited role in this setting, he said, noting that it can be reliably performed with stimulation of CLL cells.
“We also know additional aberrations are prognostic and that a complex karyotype predicts for a very poor outcome,” he said. The International Workshop on CLL (iwCLL) guidelines, which were recently updated for the first time in a decade, state that further validation is needed.
“I think it’s potentially useful in a very young patient you are considering taking to transplant, but again, I agree with the stance that this is not something that should be performed in every patient across the board,” he said.
The tests currently recommended by iwCLL before CLL treatment include IgVH mutation status; FISH for del(13q), del(11q), del(17p), and trisomy 12 in peripheral blood lymphocytes; and TP53.
“Some folks... don’t check a lot of these markers at diagnosis, but wait for patients to require therapy, and that’s a reasonable way to practice,” Dr. Barr said, noting, however, that he prefers knowing patients’ risk up front – especially for those patients he will see just once before they are “managed closer to home for the majority of their course.
“But if you [wait], then knowing what to repeat later is important,” he added. Namely, the FISH and TP53 tests are worth repeating as patients can acquire additional molecular aberrations over time.
Dr. Barr reported serving as a consultant for Pharmacyclics, AbbVie, Celgene, Gilead Sciences, Infinity Pharmaceuticals, Novartis, and Seattle Genetics. He also reported receiving research funding from Pharmacyclics and AbbVie.
EXPERT ANALYSIS FROM MHM 2018
Novel options for treating hairy cell leukemia
NEW YORK – Ibrutinib, and now moxetumomab pasudotox, are two novel therapies that can be tried in patients with previously treated hairy cell leukemia, although data and experience with them are so far limited in this rare disease, experts said during a panel discussion at the National Comprehensive Cancer Network Hematologic Malignancies Annual Congress.
Since there are so few patients, data on the BTK inhibitor ibrutinib in hairy cell leukemia is largely “anecdotal,” said Andrew D. Zelenetz, MD, PhD, of Memorial Sloan Kettering Cancer Center in New York.
The anti-CD22 monoclonal antibody moxetumomab pasudotox – approved for hairy cell leukemia in September – isn’t yet on the formulary at Memorial Sloan Kettering, Dr. Zelenetz added in a panel discussion of treatment options for a patient previously treated with purine analogueues and vemurafenib.
Between the two agents, moxetumomab pasudotox has more robust data in this disease, said John N. Allan, MD, of Weill Cornell Medicine, New York.
“I think if you can get access to the drug, that’s probably the best answer,” Dr. Allan said in the case discussion.
Hairy cell leukemia is an indolent B-cell lymphoma that makes up just 2% of all lymphoid leukemias, according to NCCN guidelines.
It is a chronic disease that requires long-term management, according to Dr. Allan.
First-line treatment is usually a purine analogue, either cladribine or pentostatin, and multiple treatments are possible as long as responses of greater than 2 years are achieved, he told attendees at the NCCN conference.
For relapses more than 2 years after first-line treatment, patients can be retreated with the same purine analogue, with or without rituximab, or can be switched to the alternative purine analogue, he said.
Vemurafenib, the BRAF inhibitor, is “surprisingly” effective in 90% of classic hairy cell leukemia patients with the BRAF V600E mutation, Dr. Allan added, though only about 40% of patients achieve complete response.
In discussing therapy options for a hairy cell leukemia patient previously treated with purine analogues and vemurafenib, Dr. Allan noted that the data behind ibrutinib includes case reports and early clinical investigations.
Several phase 1 studies with small numbers of patients show response rates “in the 50% range,” he said.
“This is an option,” he said. “It’s in the guidelines, and it’s something to consider.”
Moxetumomab pasudotox was recently approved for intravenous use in adults with relapsed or refractory hairy cell leukemia who have had at least two previous systemic treatments, including a purine nucleoside analogue. The CD22-directed cytotoxin is the first of its kind for treating patients with hairy cell leukemia, according to the Food and Drug Administration.
In a single-arm, open-label clinical trial including 80 patients with hairy cell leukemia who had previous treatment in line with that indication, 75% had a partial or complete response, of whom 30% had a durable complete response (CR), defined as maintaining hematologic remission for at least 180 days following CR.
Following the FDA’s approval of moxetumomab pasudotox, the NCCN updated its hairy cell leukemia clinical practice guidelines to include the drug as a category 2A recommendation for relapsed/refractory treatment. Other category 2A options in that setting include ibrutinib, vemurafenib with or without rituximab, or a clinical trial.
Along with that, NCCN guideline authors added a full page on special considerations for use of moxetumomab pasudotox. That includes advice on monitoring for capillary leak syndrome and hemolytic uremic syndrome, along with guidance on capillary leak syndrome grading and management by grade.
Dr. Zelenetz reported financial disclosures related to Adaptive Biotechnology, Amgen, AstraZeneca, Celgene, Genentech, Gilead, Hoffman La Roche, MEI Pharma, MorphoSys AG, Novartis, Pfizer, Pharmacyclics, Roche, and Verastem Oncology. Dr. Allan reported disclosures related to AbbVie, Acerta Pharma, Genentech, Pharmacyclics, Sunesis, and Verastem Oncology.
NEW YORK – Ibrutinib, and now moxetumomab pasudotox, are two novel therapies that can be tried in patients with previously treated hairy cell leukemia, although data and experience with them are so far limited in this rare disease, experts said during a panel discussion at the National Comprehensive Cancer Network Hematologic Malignancies Annual Congress.
Since there are so few patients, data on the BTK inhibitor ibrutinib in hairy cell leukemia is largely “anecdotal,” said Andrew D. Zelenetz, MD, PhD, of Memorial Sloan Kettering Cancer Center in New York.
The anti-CD22 monoclonal antibody moxetumomab pasudotox – approved for hairy cell leukemia in September – isn’t yet on the formulary at Memorial Sloan Kettering, Dr. Zelenetz added in a panel discussion of treatment options for a patient previously treated with purine analogueues and vemurafenib.
Between the two agents, moxetumomab pasudotox has more robust data in this disease, said John N. Allan, MD, of Weill Cornell Medicine, New York.
“I think if you can get access to the drug, that’s probably the best answer,” Dr. Allan said in the case discussion.
Hairy cell leukemia is an indolent B-cell lymphoma that makes up just 2% of all lymphoid leukemias, according to NCCN guidelines.
It is a chronic disease that requires long-term management, according to Dr. Allan.
First-line treatment is usually a purine analogue, either cladribine or pentostatin, and multiple treatments are possible as long as responses of greater than 2 years are achieved, he told attendees at the NCCN conference.
For relapses more than 2 years after first-line treatment, patients can be retreated with the same purine analogue, with or without rituximab, or can be switched to the alternative purine analogue, he said.
Vemurafenib, the BRAF inhibitor, is “surprisingly” effective in 90% of classic hairy cell leukemia patients with the BRAF V600E mutation, Dr. Allan added, though only about 40% of patients achieve complete response.
In discussing therapy options for a hairy cell leukemia patient previously treated with purine analogues and vemurafenib, Dr. Allan noted that the data behind ibrutinib includes case reports and early clinical investigations.
Several phase 1 studies with small numbers of patients show response rates “in the 50% range,” he said.
“This is an option,” he said. “It’s in the guidelines, and it’s something to consider.”
Moxetumomab pasudotox was recently approved for intravenous use in adults with relapsed or refractory hairy cell leukemia who have had at least two previous systemic treatments, including a purine nucleoside analogue. The CD22-directed cytotoxin is the first of its kind for treating patients with hairy cell leukemia, according to the Food and Drug Administration.
In a single-arm, open-label clinical trial including 80 patients with hairy cell leukemia who had previous treatment in line with that indication, 75% had a partial or complete response, of whom 30% had a durable complete response (CR), defined as maintaining hematologic remission for at least 180 days following CR.
Following the FDA’s approval of moxetumomab pasudotox, the NCCN updated its hairy cell leukemia clinical practice guidelines to include the drug as a category 2A recommendation for relapsed/refractory treatment. Other category 2A options in that setting include ibrutinib, vemurafenib with or without rituximab, or a clinical trial.
Along with that, NCCN guideline authors added a full page on special considerations for use of moxetumomab pasudotox. That includes advice on monitoring for capillary leak syndrome and hemolytic uremic syndrome, along with guidance on capillary leak syndrome grading and management by grade.
Dr. Zelenetz reported financial disclosures related to Adaptive Biotechnology, Amgen, AstraZeneca, Celgene, Genentech, Gilead, Hoffman La Roche, MEI Pharma, MorphoSys AG, Novartis, Pfizer, Pharmacyclics, Roche, and Verastem Oncology. Dr. Allan reported disclosures related to AbbVie, Acerta Pharma, Genentech, Pharmacyclics, Sunesis, and Verastem Oncology.
NEW YORK – Ibrutinib, and now moxetumomab pasudotox, are two novel therapies that can be tried in patients with previously treated hairy cell leukemia, although data and experience with them are so far limited in this rare disease, experts said during a panel discussion at the National Comprehensive Cancer Network Hematologic Malignancies Annual Congress.
Since there are so few patients, data on the BTK inhibitor ibrutinib in hairy cell leukemia is largely “anecdotal,” said Andrew D. Zelenetz, MD, PhD, of Memorial Sloan Kettering Cancer Center in New York.
The anti-CD22 monoclonal antibody moxetumomab pasudotox – approved for hairy cell leukemia in September – isn’t yet on the formulary at Memorial Sloan Kettering, Dr. Zelenetz added in a panel discussion of treatment options for a patient previously treated with purine analogueues and vemurafenib.
Between the two agents, moxetumomab pasudotox has more robust data in this disease, said John N. Allan, MD, of Weill Cornell Medicine, New York.
“I think if you can get access to the drug, that’s probably the best answer,” Dr. Allan said in the case discussion.
Hairy cell leukemia is an indolent B-cell lymphoma that makes up just 2% of all lymphoid leukemias, according to NCCN guidelines.
It is a chronic disease that requires long-term management, according to Dr. Allan.
First-line treatment is usually a purine analogue, either cladribine or pentostatin, and multiple treatments are possible as long as responses of greater than 2 years are achieved, he told attendees at the NCCN conference.
For relapses more than 2 years after first-line treatment, patients can be retreated with the same purine analogue, with or without rituximab, or can be switched to the alternative purine analogue, he said.
Vemurafenib, the BRAF inhibitor, is “surprisingly” effective in 90% of classic hairy cell leukemia patients with the BRAF V600E mutation, Dr. Allan added, though only about 40% of patients achieve complete response.
In discussing therapy options for a hairy cell leukemia patient previously treated with purine analogues and vemurafenib, Dr. Allan noted that the data behind ibrutinib includes case reports and early clinical investigations.
Several phase 1 studies with small numbers of patients show response rates “in the 50% range,” he said.
“This is an option,” he said. “It’s in the guidelines, and it’s something to consider.”
Moxetumomab pasudotox was recently approved for intravenous use in adults with relapsed or refractory hairy cell leukemia who have had at least two previous systemic treatments, including a purine nucleoside analogue. The CD22-directed cytotoxin is the first of its kind for treating patients with hairy cell leukemia, according to the Food and Drug Administration.
In a single-arm, open-label clinical trial including 80 patients with hairy cell leukemia who had previous treatment in line with that indication, 75% had a partial or complete response, of whom 30% had a durable complete response (CR), defined as maintaining hematologic remission for at least 180 days following CR.
Following the FDA’s approval of moxetumomab pasudotox, the NCCN updated its hairy cell leukemia clinical practice guidelines to include the drug as a category 2A recommendation for relapsed/refractory treatment. Other category 2A options in that setting include ibrutinib, vemurafenib with or without rituximab, or a clinical trial.
Along with that, NCCN guideline authors added a full page on special considerations for use of moxetumomab pasudotox. That includes advice on monitoring for capillary leak syndrome and hemolytic uremic syndrome, along with guidance on capillary leak syndrome grading and management by grade.
Dr. Zelenetz reported financial disclosures related to Adaptive Biotechnology, Amgen, AstraZeneca, Celgene, Genentech, Gilead, Hoffman La Roche, MEI Pharma, MorphoSys AG, Novartis, Pfizer, Pharmacyclics, Roche, and Verastem Oncology. Dr. Allan reported disclosures related to AbbVie, Acerta Pharma, Genentech, Pharmacyclics, Sunesis, and Verastem Oncology.
EXPERT ANALYSIS FROM NCCN HEMATOLOGIC MALIGNANCIES
Single leukemic cell can contaminate CAR T-cell product
Investigators report that a single leukemic cell unintentionally engineered into the chimeric antigen receptor (CAR) T-cell product can mask it from recognition and confer resistance to CAR T-cell therapy.
They described the case of a 20-year-old male who received the anti-CD19 CAR tisagenlecleucel (Kymriah) and relapsed at day 252 after the infusion.
The transduction of a leukemic cell during manufacture of the CAR T-cell product “is a rare event,” they wrote, and indicated that “this is the only case out of 369 patients reported worldwide at the time of publication.”
Lead author Marco Ruella, MD, of the University of Pennsylvania, and colleagues described the case in a Brief Communication published in Nature Medicine.
"In this case,” Dr. Ruella said, “we found that 100 percent of relapsed leukemic cells carried the CAR that we use to genetically modify T cells."
The patient had B-cell acute lymphoblastic leukemia (B-ALL) and relapsed three times after chemotherapy and a cord blood transplant before enrolling in the phase 1 trial of CTL019 (NCT 01626495).
The investigators reported that the infused CAR cells “displayed the typical pattern of in vivo engraftment and expansion.” At day 28 after the infusion, the patient was in complete remission.
But by day 252, he experienced a second expansion of CAR cells that did not correspond to the re-expansion of CAR+ T cells.
At day 261, the patient relapsed with more than 90% CD10+CD19- leukemic cells in the bone marrow and circulating blasts. The cells were CAR-transduced B-cell leukemia (CARB) cells.
The CARB cells continued to expand, and the patient died of progressive leukemia.
The investigators tracked the origin of the CARB cells by analyzing the relapsed CAR19+ cells using next-generation sequencing.
They hypothesized that the CAR19+ leukemia relapse occurred through lentiviral transduction during the manufacturing process, since they detected no replication-competent lentivirus when testing the patient’s peripheral blood at numerous time points after CTL019 infusion.
Further analysis confirmed the CARB cells were a byproduct made during CTL019 cell manufacturing.
To confirm that the leukemia relapse originated from a single clone, the investigators expanded in mice blast cells detected in the patient at month 9. Nine of 71 cells analyzed were positive for vector-host junctions. This confirmed that the relapsed cells originated from a single blast clone.
The investigators also excluded other possible reasons for the loss of CD19, including mutations, splicing variants, and structural alteration of the B-cell receptor complex.
They found that expression of the CAR in cis on B-ALL blasts masked the CAR target epitope.
The investigators concluded that their results “provide a direct confirmation of the cancer stem cell hypothesis in humans, given that clonal analysis indicated that the relapse and subsequent death of the patient were attributed to the progeny of a single leukemic blast cell with extensive replicative capacity, both in culture and in vivo.”
They called for improved manufacturing technologies that can eliminate contamination by residual tumor cells from engineered T cells.
Interestingly, this case developed not long after a case that showed essentially the opposite situation—a patient with chronic lymphocytic leukemia went into remission because of a single CAR T cell that reproduced and fought off the disease.
Investigators report that a single leukemic cell unintentionally engineered into the chimeric antigen receptor (CAR) T-cell product can mask it from recognition and confer resistance to CAR T-cell therapy.
They described the case of a 20-year-old male who received the anti-CD19 CAR tisagenlecleucel (Kymriah) and relapsed at day 252 after the infusion.
The transduction of a leukemic cell during manufacture of the CAR T-cell product “is a rare event,” they wrote, and indicated that “this is the only case out of 369 patients reported worldwide at the time of publication.”
Lead author Marco Ruella, MD, of the University of Pennsylvania, and colleagues described the case in a Brief Communication published in Nature Medicine.
"In this case,” Dr. Ruella said, “we found that 100 percent of relapsed leukemic cells carried the CAR that we use to genetically modify T cells."
The patient had B-cell acute lymphoblastic leukemia (B-ALL) and relapsed three times after chemotherapy and a cord blood transplant before enrolling in the phase 1 trial of CTL019 (NCT 01626495).
The investigators reported that the infused CAR cells “displayed the typical pattern of in vivo engraftment and expansion.” At day 28 after the infusion, the patient was in complete remission.
But by day 252, he experienced a second expansion of CAR cells that did not correspond to the re-expansion of CAR+ T cells.
At day 261, the patient relapsed with more than 90% CD10+CD19- leukemic cells in the bone marrow and circulating blasts. The cells were CAR-transduced B-cell leukemia (CARB) cells.
The CARB cells continued to expand, and the patient died of progressive leukemia.
The investigators tracked the origin of the CARB cells by analyzing the relapsed CAR19+ cells using next-generation sequencing.
They hypothesized that the CAR19+ leukemia relapse occurred through lentiviral transduction during the manufacturing process, since they detected no replication-competent lentivirus when testing the patient’s peripheral blood at numerous time points after CTL019 infusion.
Further analysis confirmed the CARB cells were a byproduct made during CTL019 cell manufacturing.
To confirm that the leukemia relapse originated from a single clone, the investigators expanded in mice blast cells detected in the patient at month 9. Nine of 71 cells analyzed were positive for vector-host junctions. This confirmed that the relapsed cells originated from a single blast clone.
The investigators also excluded other possible reasons for the loss of CD19, including mutations, splicing variants, and structural alteration of the B-cell receptor complex.
They found that expression of the CAR in cis on B-ALL blasts masked the CAR target epitope.
The investigators concluded that their results “provide a direct confirmation of the cancer stem cell hypothesis in humans, given that clonal analysis indicated that the relapse and subsequent death of the patient were attributed to the progeny of a single leukemic blast cell with extensive replicative capacity, both in culture and in vivo.”
They called for improved manufacturing technologies that can eliminate contamination by residual tumor cells from engineered T cells.
Interestingly, this case developed not long after a case that showed essentially the opposite situation—a patient with chronic lymphocytic leukemia went into remission because of a single CAR T cell that reproduced and fought off the disease.
Investigators report that a single leukemic cell unintentionally engineered into the chimeric antigen receptor (CAR) T-cell product can mask it from recognition and confer resistance to CAR T-cell therapy.
They described the case of a 20-year-old male who received the anti-CD19 CAR tisagenlecleucel (Kymriah) and relapsed at day 252 after the infusion.
The transduction of a leukemic cell during manufacture of the CAR T-cell product “is a rare event,” they wrote, and indicated that “this is the only case out of 369 patients reported worldwide at the time of publication.”
Lead author Marco Ruella, MD, of the University of Pennsylvania, and colleagues described the case in a Brief Communication published in Nature Medicine.
"In this case,” Dr. Ruella said, “we found that 100 percent of relapsed leukemic cells carried the CAR that we use to genetically modify T cells."
The patient had B-cell acute lymphoblastic leukemia (B-ALL) and relapsed three times after chemotherapy and a cord blood transplant before enrolling in the phase 1 trial of CTL019 (NCT 01626495).
The investigators reported that the infused CAR cells “displayed the typical pattern of in vivo engraftment and expansion.” At day 28 after the infusion, the patient was in complete remission.
But by day 252, he experienced a second expansion of CAR cells that did not correspond to the re-expansion of CAR+ T cells.
At day 261, the patient relapsed with more than 90% CD10+CD19- leukemic cells in the bone marrow and circulating blasts. The cells were CAR-transduced B-cell leukemia (CARB) cells.
The CARB cells continued to expand, and the patient died of progressive leukemia.
The investigators tracked the origin of the CARB cells by analyzing the relapsed CAR19+ cells using next-generation sequencing.
They hypothesized that the CAR19+ leukemia relapse occurred through lentiviral transduction during the manufacturing process, since they detected no replication-competent lentivirus when testing the patient’s peripheral blood at numerous time points after CTL019 infusion.
Further analysis confirmed the CARB cells were a byproduct made during CTL019 cell manufacturing.
To confirm that the leukemia relapse originated from a single clone, the investigators expanded in mice blast cells detected in the patient at month 9. Nine of 71 cells analyzed were positive for vector-host junctions. This confirmed that the relapsed cells originated from a single blast clone.
The investigators also excluded other possible reasons for the loss of CD19, including mutations, splicing variants, and structural alteration of the B-cell receptor complex.
They found that expression of the CAR in cis on B-ALL blasts masked the CAR target epitope.
The investigators concluded that their results “provide a direct confirmation of the cancer stem cell hypothesis in humans, given that clonal analysis indicated that the relapse and subsequent death of the patient were attributed to the progeny of a single leukemic blast cell with extensive replicative capacity, both in culture and in vivo.”
They called for improved manufacturing technologies that can eliminate contamination by residual tumor cells from engineered T cells.
Interestingly, this case developed not long after a case that showed essentially the opposite situation—a patient with chronic lymphocytic leukemia went into remission because of a single CAR T cell that reproduced and fought off the disease.
Venetoclax promising in unfit elderly AML patients
New York – For unfit elderly patients with acute myeloid leukemia (AML), venetoclax may be one of the most promising potential options that is emerging, according to an expert in the field.
The oral B-cell lymphoma 2 (BCL-2) inhibitor is the treatment that some in the AML community are “most excited about” for this population, Eunice S. Wang, MD, of Roswell Park Comprehensive Cancer Center in Buffalo, N.Y., said at the National Comprehensive Cancer Network (NCCN) Annual Congress: Hematologic Malignancies.
Venetoclax is currently approved by the Food and Drug Administration for previously treated chronic lymphocytic leukemia (CLL), alone or in combination with rituximab. It has been granted four Breakthrough Therapy designations from the FDA in AML. In July 2018, AbbVie submitted a Supplemental New Drug Application to the FDA for its use in combination with a hypomethylating agent (HMA) or in combination with low-dose cytarabine (LDAC) for the treatment of newly diagnosed AML patients who are ineligible for intensive chemotherapy.
“This agent doesn’t work on its own but had worked in the refractory setting and can be a great option upfront,” Dr. Wang said.
About half the patients were alive at 1 year following treatment with venetoclax plus low-dose chemotherapy, whether that was LDAC or an HMA, she said, commenting on recently reported results.
In a phase 1b trial, venetoclax was evaluated in combination with either azacitidine or decitabine. In recently reported preliminary results that included 57 patients aged 65 years or older who were ineligible for standard induction therapy, the combination was well tolerated and had promising activity (Lancet Oncol. 2018 Feb;19[2]:216-28).
Overall, 35 patients (61%) had complete remission (CR) or complete remission with incomplete marrow recovery (CRi).
In another report on this same trial, which included 33 patients from a single participating center who received venetoclax and azacytidine, the overall response rate was 91%, including 19 (58%) with CR and 9 (27%) with CRi (Blood. 2017 Dec;130 [Suppl 1]:181).
A separate phase 1/2 trial examined venetoclax plus LDAC in treatment-naive elderly patients unfit for intensive chemotherapy. In the 1-year outcomes that have been reported, the observed CR/CRi rate was 62%, median overall survival was an “encouraging” 11.4 months, and the observed 12-month overall survival was 46% in 61 patients treated at a venetoclax dose of 600 mg (Blood. 2017 Dec;130 [Suppl 1]:890).
In those 61 patients, treatment-related grade 3/4 adverse events included thrombocytopenia in 59%, neutropenia in 46%, febrile neutropenia in 36%, anemia in 28%, decreased white blood cell count in 26%, and one case of tumor lysis syndrome, according to the report.
Based on those findings in a cohort of patients with poor risk features, venetoclax 600 mg plus LDAC was carried forward to be evaluated in an ongoing phase 3 study, investigators noted at the time.
Dr. Wang reported financial relationships with AbbVie, Amgen, ImmunoGen, Incyte, Novartis, and Otsuka.
New York – For unfit elderly patients with acute myeloid leukemia (AML), venetoclax may be one of the most promising potential options that is emerging, according to an expert in the field.
The oral B-cell lymphoma 2 (BCL-2) inhibitor is the treatment that some in the AML community are “most excited about” for this population, Eunice S. Wang, MD, of Roswell Park Comprehensive Cancer Center in Buffalo, N.Y., said at the National Comprehensive Cancer Network (NCCN) Annual Congress: Hematologic Malignancies.
Venetoclax is currently approved by the Food and Drug Administration for previously treated chronic lymphocytic leukemia (CLL), alone or in combination with rituximab. It has been granted four Breakthrough Therapy designations from the FDA in AML. In July 2018, AbbVie submitted a Supplemental New Drug Application to the FDA for its use in combination with a hypomethylating agent (HMA) or in combination with low-dose cytarabine (LDAC) for the treatment of newly diagnosed AML patients who are ineligible for intensive chemotherapy.
“This agent doesn’t work on its own but had worked in the refractory setting and can be a great option upfront,” Dr. Wang said.
About half the patients were alive at 1 year following treatment with venetoclax plus low-dose chemotherapy, whether that was LDAC or an HMA, she said, commenting on recently reported results.
In a phase 1b trial, venetoclax was evaluated in combination with either azacitidine or decitabine. In recently reported preliminary results that included 57 patients aged 65 years or older who were ineligible for standard induction therapy, the combination was well tolerated and had promising activity (Lancet Oncol. 2018 Feb;19[2]:216-28).
Overall, 35 patients (61%) had complete remission (CR) or complete remission with incomplete marrow recovery (CRi).
In another report on this same trial, which included 33 patients from a single participating center who received venetoclax and azacytidine, the overall response rate was 91%, including 19 (58%) with CR and 9 (27%) with CRi (Blood. 2017 Dec;130 [Suppl 1]:181).
A separate phase 1/2 trial examined venetoclax plus LDAC in treatment-naive elderly patients unfit for intensive chemotherapy. In the 1-year outcomes that have been reported, the observed CR/CRi rate was 62%, median overall survival was an “encouraging” 11.4 months, and the observed 12-month overall survival was 46% in 61 patients treated at a venetoclax dose of 600 mg (Blood. 2017 Dec;130 [Suppl 1]:890).
In those 61 patients, treatment-related grade 3/4 adverse events included thrombocytopenia in 59%, neutropenia in 46%, febrile neutropenia in 36%, anemia in 28%, decreased white blood cell count in 26%, and one case of tumor lysis syndrome, according to the report.
Based on those findings in a cohort of patients with poor risk features, venetoclax 600 mg plus LDAC was carried forward to be evaluated in an ongoing phase 3 study, investigators noted at the time.
Dr. Wang reported financial relationships with AbbVie, Amgen, ImmunoGen, Incyte, Novartis, and Otsuka.
New York – For unfit elderly patients with acute myeloid leukemia (AML), venetoclax may be one of the most promising potential options that is emerging, according to an expert in the field.
The oral B-cell lymphoma 2 (BCL-2) inhibitor is the treatment that some in the AML community are “most excited about” for this population, Eunice S. Wang, MD, of Roswell Park Comprehensive Cancer Center in Buffalo, N.Y., said at the National Comprehensive Cancer Network (NCCN) Annual Congress: Hematologic Malignancies.
Venetoclax is currently approved by the Food and Drug Administration for previously treated chronic lymphocytic leukemia (CLL), alone or in combination with rituximab. It has been granted four Breakthrough Therapy designations from the FDA in AML. In July 2018, AbbVie submitted a Supplemental New Drug Application to the FDA for its use in combination with a hypomethylating agent (HMA) or in combination with low-dose cytarabine (LDAC) for the treatment of newly diagnosed AML patients who are ineligible for intensive chemotherapy.
“This agent doesn’t work on its own but had worked in the refractory setting and can be a great option upfront,” Dr. Wang said.
About half the patients were alive at 1 year following treatment with venetoclax plus low-dose chemotherapy, whether that was LDAC or an HMA, she said, commenting on recently reported results.
In a phase 1b trial, venetoclax was evaluated in combination with either azacitidine or decitabine. In recently reported preliminary results that included 57 patients aged 65 years or older who were ineligible for standard induction therapy, the combination was well tolerated and had promising activity (Lancet Oncol. 2018 Feb;19[2]:216-28).
Overall, 35 patients (61%) had complete remission (CR) or complete remission with incomplete marrow recovery (CRi).
In another report on this same trial, which included 33 patients from a single participating center who received venetoclax and azacytidine, the overall response rate was 91%, including 19 (58%) with CR and 9 (27%) with CRi (Blood. 2017 Dec;130 [Suppl 1]:181).
A separate phase 1/2 trial examined venetoclax plus LDAC in treatment-naive elderly patients unfit for intensive chemotherapy. In the 1-year outcomes that have been reported, the observed CR/CRi rate was 62%, median overall survival was an “encouraging” 11.4 months, and the observed 12-month overall survival was 46% in 61 patients treated at a venetoclax dose of 600 mg (Blood. 2017 Dec;130 [Suppl 1]:890).
In those 61 patients, treatment-related grade 3/4 adverse events included thrombocytopenia in 59%, neutropenia in 46%, febrile neutropenia in 36%, anemia in 28%, decreased white blood cell count in 26%, and one case of tumor lysis syndrome, according to the report.
Based on those findings in a cohort of patients with poor risk features, venetoclax 600 mg plus LDAC was carried forward to be evaluated in an ongoing phase 3 study, investigators noted at the time.
Dr. Wang reported financial relationships with AbbVie, Amgen, ImmunoGen, Incyte, Novartis, and Otsuka.
EXPERT ANALYSIS FROM THE NCCN ANNUAL CONGRESS: HEMATOLOGIC MALIGNANCIES
First reported case of induced resistance to tisagenlecleucel
Unintentional transduction of a single leukemic B cell appears to have induced resistance to CTL019 (tisagenlecleucel, Kymriah) therapy, a recent case study suggests.
A total of 9 months after receiving a seemingly successful CD19-targeted chimeric antigen receptor (CAR) T-cell (CTL019; tisagenlecleucel) infusion, a 20-year-old man with B-cell acute lymphoblastic leukemia (B-ALL) had a frank relapse, with more than 90% bone marrow infiltration of CAR-transduced B-cell leukemia cells. Further investigation showed that the CAR gene had unintentionally been added to a solitary leukemic B cell during the CAR T-cell manufacturing process, reported Marco Ruella, MD, of the University of Pennsylvania, Philadelphia.
“The transduction of a single leukemic cell with an anti-CD19 CAR lentivirus during CTL019 manufacturing is sufficient to mediate resistance through masking of the CD19 epitope. This is a rare event, as this is the only case out of 369 patients reported worldwide at the time of publication. ... These findings illustrate the need for improved manufacturing technologies that can purge residual contaminating tumor cells from engineered T cells,” the authors wrote in Nature Medicine.
The findings also confirm the cancer stem cell hypothesis in humans, “given that clonal analysis indicated that the relapse and subsequent death of the patient were attributed to the progeny of a single leukemic blast cell with extensive replicative capacity, both in culture and in vivo,” they wrote.
Initially, “the infused CTL019 cells displayed the typical pattern of in vivo engraftment and expansion by CAR19-specific flow cytometry, followed by decline to an undetectable level in the peripheral blood” of the affected patient, the authors wrote. “The expansion and contraction phases and long-term persistence of CAR T cells were confirmed via qPCR using CAR-specific primers.” The patient was in complete remission at day 28.
However, they added, routine peripheral blood monitoring with quantitative polymerase chain reaction for CAR-specific sequences identified “the emergence of a second expansion phase of CAR cells starting at day 252, which did not correlate with re-expansion of CAR + T cells by flow cytometry.” Frank relapse soon followed.
Analysis confirmed “that the lack of detection of CD19 by flow cytometry was due to CAR19 binding in cis to CD19 on the surface of leukemic blasts, thus masking the epitope from detection by standard flow cytometry,” the authors wrote.
Study funding was provided by Bristol-Myers Squibb, Novartis, the National Institutes of Health, and others. Dr. Ruella and several of his colleagues work under a research collaboration involving the University of Pennsylvania and the Novartis Institutes of Biomedical Research and are inventors of intellectual property licensed by the University of Pennsylvania to Novartis.
SOURCE: Ruella M et al. Nat Med. 2018 Oct 1. doi: 10.1038/s41591-018-0201-9.
Unintentional transduction of a single leukemic B cell appears to have induced resistance to CTL019 (tisagenlecleucel, Kymriah) therapy, a recent case study suggests.
A total of 9 months after receiving a seemingly successful CD19-targeted chimeric antigen receptor (CAR) T-cell (CTL019; tisagenlecleucel) infusion, a 20-year-old man with B-cell acute lymphoblastic leukemia (B-ALL) had a frank relapse, with more than 90% bone marrow infiltration of CAR-transduced B-cell leukemia cells. Further investigation showed that the CAR gene had unintentionally been added to a solitary leukemic B cell during the CAR T-cell manufacturing process, reported Marco Ruella, MD, of the University of Pennsylvania, Philadelphia.
“The transduction of a single leukemic cell with an anti-CD19 CAR lentivirus during CTL019 manufacturing is sufficient to mediate resistance through masking of the CD19 epitope. This is a rare event, as this is the only case out of 369 patients reported worldwide at the time of publication. ... These findings illustrate the need for improved manufacturing technologies that can purge residual contaminating tumor cells from engineered T cells,” the authors wrote in Nature Medicine.
The findings also confirm the cancer stem cell hypothesis in humans, “given that clonal analysis indicated that the relapse and subsequent death of the patient were attributed to the progeny of a single leukemic blast cell with extensive replicative capacity, both in culture and in vivo,” they wrote.
Initially, “the infused CTL019 cells displayed the typical pattern of in vivo engraftment and expansion by CAR19-specific flow cytometry, followed by decline to an undetectable level in the peripheral blood” of the affected patient, the authors wrote. “The expansion and contraction phases and long-term persistence of CAR T cells were confirmed via qPCR using CAR-specific primers.” The patient was in complete remission at day 28.
However, they added, routine peripheral blood monitoring with quantitative polymerase chain reaction for CAR-specific sequences identified “the emergence of a second expansion phase of CAR cells starting at day 252, which did not correlate with re-expansion of CAR + T cells by flow cytometry.” Frank relapse soon followed.
Analysis confirmed “that the lack of detection of CD19 by flow cytometry was due to CAR19 binding in cis to CD19 on the surface of leukemic blasts, thus masking the epitope from detection by standard flow cytometry,” the authors wrote.
Study funding was provided by Bristol-Myers Squibb, Novartis, the National Institutes of Health, and others. Dr. Ruella and several of his colleagues work under a research collaboration involving the University of Pennsylvania and the Novartis Institutes of Biomedical Research and are inventors of intellectual property licensed by the University of Pennsylvania to Novartis.
SOURCE: Ruella M et al. Nat Med. 2018 Oct 1. doi: 10.1038/s41591-018-0201-9.
Unintentional transduction of a single leukemic B cell appears to have induced resistance to CTL019 (tisagenlecleucel, Kymriah) therapy, a recent case study suggests.
A total of 9 months after receiving a seemingly successful CD19-targeted chimeric antigen receptor (CAR) T-cell (CTL019; tisagenlecleucel) infusion, a 20-year-old man with B-cell acute lymphoblastic leukemia (B-ALL) had a frank relapse, with more than 90% bone marrow infiltration of CAR-transduced B-cell leukemia cells. Further investigation showed that the CAR gene had unintentionally been added to a solitary leukemic B cell during the CAR T-cell manufacturing process, reported Marco Ruella, MD, of the University of Pennsylvania, Philadelphia.
“The transduction of a single leukemic cell with an anti-CD19 CAR lentivirus during CTL019 manufacturing is sufficient to mediate resistance through masking of the CD19 epitope. This is a rare event, as this is the only case out of 369 patients reported worldwide at the time of publication. ... These findings illustrate the need for improved manufacturing technologies that can purge residual contaminating tumor cells from engineered T cells,” the authors wrote in Nature Medicine.
The findings also confirm the cancer stem cell hypothesis in humans, “given that clonal analysis indicated that the relapse and subsequent death of the patient were attributed to the progeny of a single leukemic blast cell with extensive replicative capacity, both in culture and in vivo,” they wrote.
Initially, “the infused CTL019 cells displayed the typical pattern of in vivo engraftment and expansion by CAR19-specific flow cytometry, followed by decline to an undetectable level in the peripheral blood” of the affected patient, the authors wrote. “The expansion and contraction phases and long-term persistence of CAR T cells were confirmed via qPCR using CAR-specific primers.” The patient was in complete remission at day 28.
However, they added, routine peripheral blood monitoring with quantitative polymerase chain reaction for CAR-specific sequences identified “the emergence of a second expansion phase of CAR cells starting at day 252, which did not correlate with re-expansion of CAR + T cells by flow cytometry.” Frank relapse soon followed.
Analysis confirmed “that the lack of detection of CD19 by flow cytometry was due to CAR19 binding in cis to CD19 on the surface of leukemic blasts, thus masking the epitope from detection by standard flow cytometry,” the authors wrote.
Study funding was provided by Bristol-Myers Squibb, Novartis, the National Institutes of Health, and others. Dr. Ruella and several of his colleagues work under a research collaboration involving the University of Pennsylvania and the Novartis Institutes of Biomedical Research and are inventors of intellectual property licensed by the University of Pennsylvania to Novartis.
SOURCE: Ruella M et al. Nat Med. 2018 Oct 1. doi: 10.1038/s41591-018-0201-9.
FROM NATURE MEDICINE
Key clinical point: Unintentional transduction of a single leukemic B cell induced resistance to CTL019 (tisagenlecleucel) therapy.
Major finding: A patient with B-cell acute lymphoblastic leukemia (B-ALL) had frank relapse 9 months after a CTL019 infusion, with more than 90% bone marrow infiltration of chimeric antigen receptor–transduced B-cell leukemia cells.
Study details: A case study of a 20-year-old male with B-ALL undergoing CTL019 therapy.
Disclosures: Study funding was provided by Bristol-Myers Squibb, Novartis, the National Institutes of Health, and others. Dr. Ruella and several of his colleagues work under a research collaboration involving the University of Pennsylvania and the Novartis Institutes of Biomedical Research and are inventors of intellectual property licensed by the University of Pennsylvania to Novartis.
Source: Ruella M et al. Nat Med. 2018 Oct 1. doi: 10.1038/s41591-018-0201-9.
FDA authorizes ClonoSEQ to detect MRD in ALL, myeloma
, the U.S. Food and Drug Administration announced. Marketing authorization of the ClonoSEQ assay was granted to Adaptive Biotechnologies.

The ClonoSEQ assay is an in vitro diagnostic test that uses multiplex polymerase chain reaction and next-generation sequencing to identify and quantify certain gene sequences in DNA extracted from the bone marrow from patients with ALL or multiple myeloma. This is a single-site assay collected by the patient’s provider and sent to Adaptive Biotechnologies for evaluation.
The ClonoSEQ assay is capable of detecting minimal residual disease at levels below 1 in 1 million cells. Currently, providers test for MRD using flow cytometry assays or polymerase chain reaction–based assays. Those methods are usually capable of measuring MRD down to 1 in 10,000 or 1 in 100,000 cells.
“Determining whether a patient has residual cancer cells remaining after treatment provides information on how well a patient has responded to therapy and how long remission may last. Having a highly sensitive test available to measure minimal residual disease in ALL or multiple myeloma patients can help providers manage their patients’ care,” FDA Commissioner Scott Gottlieb, MD, said in a press release.
Along with this authorization, the FDA is establishing criteria, called special controls, which clarify the agency’s expectations in assuring the accuracy, reliability, and effectiveness of tests intended to be used as an aid to measure MRD to assess the change in burden of disease during and after treatment. These special controls, when met along with general controls, provide a reasonable assurance of safety and effectiveness for these tests, the agency said in the release. This action also creates a new regulatory classification, which means that subsequent devices of the same type with the same intended use may go through the FDA’s 510(k) process, whereby devices can obtain marketing authorization by demonstrating substantial equivalence to a previously approved device.
“The FDA is applying novel regulatory approaches to make sure that these rapidly evolving [next-generation sequencing] tests are accurate and reliable. At the same time, we’re seeing more and more laboratory-developed tests seek marketing authorization from the FDA,” he said, adding that the agency has put forward a plan to modernize the regulatory framework for all in vitro clinical tests.
The FDA evaluated data to demonstrate clinical validity from a retrospective analysis of samples obtained from three previously conducted clinical studies including 273 patients with ALL, an ongoing study of 323 patients with multiple myeloma, and a study of 706 patients with multiple myeloma, according to the FDA release.
For patients with ALL, the ClonoSEQ assay was used to assess MRD at various disease burden thresholds to show that the MRD level correlated with event-free survival – the length of time, after treatment, that the patient remains free of certain complications or events. Patients whose ClonoSEQ assay result was MRD negative had longer event-free survival, while patients with higher MRD assay results had lower event-free survival. Similar patterns of results were seen for progression-free and disease-free survival in patients with multiple myeloma.
, the U.S. Food and Drug Administration announced. Marketing authorization of the ClonoSEQ assay was granted to Adaptive Biotechnologies.
The ClonoSEQ assay is an in vitro diagnostic test that uses multiplex polymerase chain reaction and next-generation sequencing to identify and quantify certain gene sequences in DNA extracted from the bone marrow from patients with ALL or multiple myeloma. This is a single-site assay collected by the patient’s provider and sent to Adaptive Biotechnologies for evaluation.
The ClonoSEQ assay is capable of detecting minimal residual disease at levels below 1 in 1 million cells. Currently, providers test for MRD using flow cytometry assays or polymerase chain reaction–based assays. Those methods are usually capable of measuring MRD down to 1 in 10,000 or 1 in 100,000 cells.
“Determining whether a patient has residual cancer cells remaining after treatment provides information on how well a patient has responded to therapy and how long remission may last. Having a highly sensitive test available to measure minimal residual disease in ALL or multiple myeloma patients can help providers manage their patients’ care,” FDA Commissioner Scott Gottlieb, MD, said in a press release.
Along with this authorization, the FDA is establishing criteria, called special controls, which clarify the agency’s expectations in assuring the accuracy, reliability, and effectiveness of tests intended to be used as an aid to measure MRD to assess the change in burden of disease during and after treatment. These special controls, when met along with general controls, provide a reasonable assurance of safety and effectiveness for these tests, the agency said in the release. This action also creates a new regulatory classification, which means that subsequent devices of the same type with the same intended use may go through the FDA’s 510(k) process, whereby devices can obtain marketing authorization by demonstrating substantial equivalence to a previously approved device.
“The FDA is applying novel regulatory approaches to make sure that these rapidly evolving [next-generation sequencing] tests are accurate and reliable. At the same time, we’re seeing more and more laboratory-developed tests seek marketing authorization from the FDA,” he said, adding that the agency has put forward a plan to modernize the regulatory framework for all in vitro clinical tests.
The FDA evaluated data to demonstrate clinical validity from a retrospective analysis of samples obtained from three previously conducted clinical studies including 273 patients with ALL, an ongoing study of 323 patients with multiple myeloma, and a study of 706 patients with multiple myeloma, according to the FDA release.
For patients with ALL, the ClonoSEQ assay was used to assess MRD at various disease burden thresholds to show that the MRD level correlated with event-free survival – the length of time, after treatment, that the patient remains free of certain complications or events. Patients whose ClonoSEQ assay result was MRD negative had longer event-free survival, while patients with higher MRD assay results had lower event-free survival. Similar patterns of results were seen for progression-free and disease-free survival in patients with multiple myeloma.
, the U.S. Food and Drug Administration announced. Marketing authorization of the ClonoSEQ assay was granted to Adaptive Biotechnologies.
The ClonoSEQ assay is an in vitro diagnostic test that uses multiplex polymerase chain reaction and next-generation sequencing to identify and quantify certain gene sequences in DNA extracted from the bone marrow from patients with ALL or multiple myeloma. This is a single-site assay collected by the patient’s provider and sent to Adaptive Biotechnologies for evaluation.
The ClonoSEQ assay is capable of detecting minimal residual disease at levels below 1 in 1 million cells. Currently, providers test for MRD using flow cytometry assays or polymerase chain reaction–based assays. Those methods are usually capable of measuring MRD down to 1 in 10,000 or 1 in 100,000 cells.
“Determining whether a patient has residual cancer cells remaining after treatment provides information on how well a patient has responded to therapy and how long remission may last. Having a highly sensitive test available to measure minimal residual disease in ALL or multiple myeloma patients can help providers manage their patients’ care,” FDA Commissioner Scott Gottlieb, MD, said in a press release.
Along with this authorization, the FDA is establishing criteria, called special controls, which clarify the agency’s expectations in assuring the accuracy, reliability, and effectiveness of tests intended to be used as an aid to measure MRD to assess the change in burden of disease during and after treatment. These special controls, when met along with general controls, provide a reasonable assurance of safety and effectiveness for these tests, the agency said in the release. This action also creates a new regulatory classification, which means that subsequent devices of the same type with the same intended use may go through the FDA’s 510(k) process, whereby devices can obtain marketing authorization by demonstrating substantial equivalence to a previously approved device.
“The FDA is applying novel regulatory approaches to make sure that these rapidly evolving [next-generation sequencing] tests are accurate and reliable. At the same time, we’re seeing more and more laboratory-developed tests seek marketing authorization from the FDA,” he said, adding that the agency has put forward a plan to modernize the regulatory framework for all in vitro clinical tests.
The FDA evaluated data to demonstrate clinical validity from a retrospective analysis of samples obtained from three previously conducted clinical studies including 273 patients with ALL, an ongoing study of 323 patients with multiple myeloma, and a study of 706 patients with multiple myeloma, according to the FDA release.
For patients with ALL, the ClonoSEQ assay was used to assess MRD at various disease burden thresholds to show that the MRD level correlated with event-free survival – the length of time, after treatment, that the patient remains free of certain complications or events. Patients whose ClonoSEQ assay result was MRD negative had longer event-free survival, while patients with higher MRD assay results had lower event-free survival. Similar patterns of results were seen for progression-free and disease-free survival in patients with multiple myeloma.
First NGS assay approved for MRD detection in ALL or MM

The U.S. Food and Drug Administration has authorized the first next-generation sequencing (NGS)-based assay to be marketed for minimal residual disease (MRD) testing in patients with acute lymphoblastic leukemia (ALL) or multiple myeloma (MM).
The assay, called clonoSEQ®, uses both polymerase chain reaction (PCR) and NGS to identify and quantify gene sequences in DNA from patients’ bone marrow.
ClonoSEQ Assay can detect MRD levels below 1 in 1 million cells. By comparison flow cytometry assays or PCR-based assays are capable of measuring MRD down to 1 in 10,000 or 1 in 100,000 cells.
The clonoSEQ Assay is marketed by Adaptive Biotechnologies.
The FDA based its authorization on data from three clinical studies, one with 273 ALL patients, an ongoing study of 323 MM patients, and another MM trial with 706 patients.
Validation in ALL
As described in the clonoSEQ Assay Technical Information, a subset of 273 patients originally enrolled in the Children’s Oncology Group AALL0232 (NCT00075725) and AALL0331 (NCT00103285) studies had left-over bone marrow specimens to evaluate the performance of the clonoSEQ Assay.
MRD as determined by MRD negativity at less than 10-4 predicted improved event-free survival (EFS) irrespective of age. MRD-positive patients had a 2.74 higher event risk compared to MRD-negative patients.
Similar findings between MRD negativity and EFS in pediatric ALL using an earlier version of the assay were published in Blood.
Validation in MM
The ongoing phase 3 DFCI Study 10-106 (NCT01208662) is comparing conventional treatment with lenalidomide, bortezomib and dexamethasone to high-dose treatment with stem cell transplant as initial management of MM patients less than 65 years.
According to clonoSEQ’s technical information, bone marrow samples from 323 of the 720 patients originally enrolled were available and evaluable for MRD assessment.
ClonoSEQ measurements demonstrated that MRD status at a threshold of 10-5 significantly predicts progression-free survival (PFS) in all patients (P=0.027).
And samples from 75 patients who had achieved complete remission (CR) showed a modest association with disease-free survival (DFS) and lower MRD levels (P=0.064).
In the phase 3 ALCYONE trial, investigators randomly assigned 706 treatment-naïve MM patients ineligible for hematopoietic stem cell transplant to bortezomib, melphalan, and prednisone with or without daratumumab.
MRD assessments were made using the clonoSEQ Assay at screening, at confirmation of CR or stringent CR, and at intervals after patients achieved a CR.
Patients who did not achieve CR were considered MRD positive. The threshold for the MRD analysis was 10-5.
Investigators found that patients who were MRD negative by the clonoSEQ Assay had longer PFS compared to MRD-positive patients, regardless of treatment group.
For additional information on the clonoSEQ Assay consult the Technical Information available online.
The U.S. Food and Drug Administration has authorized the first next-generation sequencing (NGS)-based assay to be marketed for minimal residual disease (MRD) testing in patients with acute lymphoblastic leukemia (ALL) or multiple myeloma (MM).
The assay, called clonoSEQ®, uses both polymerase chain reaction (PCR) and NGS to identify and quantify gene sequences in DNA from patients’ bone marrow.
ClonoSEQ Assay can detect MRD levels below 1 in 1 million cells. By comparison flow cytometry assays or PCR-based assays are capable of measuring MRD down to 1 in 10,000 or 1 in 100,000 cells.
The clonoSEQ Assay is marketed by Adaptive Biotechnologies.
The FDA based its authorization on data from three clinical studies, one with 273 ALL patients, an ongoing study of 323 MM patients, and another MM trial with 706 patients.
Validation in ALL
As described in the clonoSEQ Assay Technical Information, a subset of 273 patients originally enrolled in the Children’s Oncology Group AALL0232 (NCT00075725) and AALL0331 (NCT00103285) studies had left-over bone marrow specimens to evaluate the performance of the clonoSEQ Assay.
MRD as determined by MRD negativity at less than 10-4 predicted improved event-free survival (EFS) irrespective of age. MRD-positive patients had a 2.74 higher event risk compared to MRD-negative patients.
Similar findings between MRD negativity and EFS in pediatric ALL using an earlier version of the assay were published in Blood.
Validation in MM
The ongoing phase 3 DFCI Study 10-106 (NCT01208662) is comparing conventional treatment with lenalidomide, bortezomib and dexamethasone to high-dose treatment with stem cell transplant as initial management of MM patients less than 65 years.
According to clonoSEQ’s technical information, bone marrow samples from 323 of the 720 patients originally enrolled were available and evaluable for MRD assessment.
ClonoSEQ measurements demonstrated that MRD status at a threshold of 10-5 significantly predicts progression-free survival (PFS) in all patients (P=0.027).
And samples from 75 patients who had achieved complete remission (CR) showed a modest association with disease-free survival (DFS) and lower MRD levels (P=0.064).
In the phase 3 ALCYONE trial, investigators randomly assigned 706 treatment-naïve MM patients ineligible for hematopoietic stem cell transplant to bortezomib, melphalan, and prednisone with or without daratumumab.
MRD assessments were made using the clonoSEQ Assay at screening, at confirmation of CR or stringent CR, and at intervals after patients achieved a CR.
Patients who did not achieve CR were considered MRD positive. The threshold for the MRD analysis was 10-5.
Investigators found that patients who were MRD negative by the clonoSEQ Assay had longer PFS compared to MRD-positive patients, regardless of treatment group.
For additional information on the clonoSEQ Assay consult the Technical Information available online.
The U.S. Food and Drug Administration has authorized the first next-generation sequencing (NGS)-based assay to be marketed for minimal residual disease (MRD) testing in patients with acute lymphoblastic leukemia (ALL) or multiple myeloma (MM).
The assay, called clonoSEQ®, uses both polymerase chain reaction (PCR) and NGS to identify and quantify gene sequences in DNA from patients’ bone marrow.
ClonoSEQ Assay can detect MRD levels below 1 in 1 million cells. By comparison flow cytometry assays or PCR-based assays are capable of measuring MRD down to 1 in 10,000 or 1 in 100,000 cells.
The clonoSEQ Assay is marketed by Adaptive Biotechnologies.
The FDA based its authorization on data from three clinical studies, one with 273 ALL patients, an ongoing study of 323 MM patients, and another MM trial with 706 patients.
Validation in ALL
As described in the clonoSEQ Assay Technical Information, a subset of 273 patients originally enrolled in the Children’s Oncology Group AALL0232 (NCT00075725) and AALL0331 (NCT00103285) studies had left-over bone marrow specimens to evaluate the performance of the clonoSEQ Assay.
MRD as determined by MRD negativity at less than 10-4 predicted improved event-free survival (EFS) irrespective of age. MRD-positive patients had a 2.74 higher event risk compared to MRD-negative patients.
Similar findings between MRD negativity and EFS in pediatric ALL using an earlier version of the assay were published in Blood.
Validation in MM
The ongoing phase 3 DFCI Study 10-106 (NCT01208662) is comparing conventional treatment with lenalidomide, bortezomib and dexamethasone to high-dose treatment with stem cell transplant as initial management of MM patients less than 65 years.
According to clonoSEQ’s technical information, bone marrow samples from 323 of the 720 patients originally enrolled were available and evaluable for MRD assessment.
ClonoSEQ measurements demonstrated that MRD status at a threshold of 10-5 significantly predicts progression-free survival (PFS) in all patients (P=0.027).
And samples from 75 patients who had achieved complete remission (CR) showed a modest association with disease-free survival (DFS) and lower MRD levels (P=0.064).
In the phase 3 ALCYONE trial, investigators randomly assigned 706 treatment-naïve MM patients ineligible for hematopoietic stem cell transplant to bortezomib, melphalan, and prednisone with or without daratumumab.
MRD assessments were made using the clonoSEQ Assay at screening, at confirmation of CR or stringent CR, and at intervals after patients achieved a CR.
Patients who did not achieve CR were considered MRD positive. The threshold for the MRD analysis was 10-5.
Investigators found that patients who were MRD negative by the clonoSEQ Assay had longer PFS compared to MRD-positive patients, regardless of treatment group.
For additional information on the clonoSEQ Assay consult the Technical Information available online.