User login
Frequent BCCs linked to blood cancers

New research suggests people who develop frequent cases of basal cell carcinoma (BCC) have an increased risk of leukemias, lymphomas, and other cancers.
“We discovered that people who develop 6 or more basal cell carcinomas during a 10-year period are about 3 times more likely than the general population to develop other, unrelated cancers,” said Kavita Sarin, MD, PhD, of Stanford University School of Medicine in California.
“We’re hopeful that this finding could be a way to identify people at an increased risk for a life-threatening malignancy before those cancers develop.”
Dr Sarin and her colleagues reported their findings in JCI Insight.
Stanford cohort
The researchers first studied 61 patients treated at Stanford Health Care for unusually frequent BCCs—an average of 11 per patient over a 10-year period. The team investigated whether these patients may have mutations in 29 genes that code for DNA damage repair proteins.
“We found that about 20% of the people with frequent basal cell carcinomas have a mutation in one of the genes responsible for repairing DNA damage, versus about 3% of the general population,” Dr Sarin said. “That’s shockingly high.”
Specifically, there were 12 BCC patients (19.7%) who had 13 pathogenic mutations in 12 genes—APC, BARD1, BRCA1, BRCA2, CDH1, CHEK2, MLH1, MSH2, MSH6, MUTYH, NBN, and PALB2. And 3.0% of non-Finnish European subjects in the Exome Aggregation Consortium had pathogenic mutations in these 12 genes.
Furthermore, 21 of the 61 BCC patients (64.4%) had a history of additional cancers. This included 5 hematologic malignancies (leukemia/lymphoma), 5 invasive melanomas, and 2 breast, 2 colon, and 5 prostate cancers.
When the researchers compared the cancer prevalence in these patients to the Surveillance, Epidemiology, and End Results-estimated prevalence of cancer in the 60- to 69-year-old population of European descent, the BCC cohort had an increased risk of any cancer—a relative risk (RR) of 3.5 (P<0.001).
The RR was 3.5 for leukemia and lymphoma (P=0.004), 11.9 for invasive melanoma (P<0.001), 4.5 for colon cancer (P=0.030), 5.6 for breast cancer (P=0.009), and 4.7 for prostate cancer (P<0.001).
Insurance cohort
To confirm the findings in the Stanford cohort, the researchers applied a similar analysis to a large medical insurance claims database, Truven MarketScan.
The database contained 111,562 patients with 1 case of BCC, 13,264 patients with 6 or more BCCs, and 2920 patients with 12 or more BCCs. Truven patients with no history of BCC served as controls.
The researchers adjusted for age and sex and found that patients with 1 BCC, 6 or more BCCs, and 12 or more BCCs had an increased risk of any cancer compared to controls.
The odds ratio (OR) for any cancer was 1.61 for patients with 1 BCC, 3.12 for those with 6 or more BCCs, and 4.15 for patients with 12 or more BCCs.
The OR for Hodgkin lymphoma was 2.27 for patients with 1 BCC, 8.94 for patients with 6 or more BCCs, and 15.41 for patients with 12 or more BCCs.
The OR for non-Hodgkin lymphoma was 1.40 for patients with 1 BCC, 2.59 for patients with 6 or more BCCs, and 3.10 for patients with 12 or more BCCs.
The OR for leukemia was 1.76 for patients with 1 BCC, 3.23 for patients with 6 or more BCCs, and 5.78 for patients with 12 or more BCCs.
The researchers pointed out that, the more BCCs an individual had, the more likely that person was to have had other cancers as well.
“I was surprised to see such a strong correlation, but it’s also very gratifying,” Dr Sarin said. “Now, we can ask patients with repeated basal cell carcinomas whether they have family members with other types of cancers and perhaps suggest that they consider genetic testing and increased screening.”
The researchers are continuing to enroll Stanford patients in their study to learn whether particular mutations in genes responsible for repairing DNA damage are linked to the development of specific malignancies. The team would also like to conduct a similar study in patients with frequent melanomas.
The current study was supported by the Dermatology Foundation, the Stanford Society of Physician Scholars, the American Skin Association, and Pellepharm Inc.
New research suggests people who develop frequent cases of basal cell carcinoma (BCC) have an increased risk of leukemias, lymphomas, and other cancers.
“We discovered that people who develop 6 or more basal cell carcinomas during a 10-year period are about 3 times more likely than the general population to develop other, unrelated cancers,” said Kavita Sarin, MD, PhD, of Stanford University School of Medicine in California.
“We’re hopeful that this finding could be a way to identify people at an increased risk for a life-threatening malignancy before those cancers develop.”
Dr Sarin and her colleagues reported their findings in JCI Insight.
Stanford cohort
The researchers first studied 61 patients treated at Stanford Health Care for unusually frequent BCCs—an average of 11 per patient over a 10-year period. The team investigated whether these patients may have mutations in 29 genes that code for DNA damage repair proteins.
“We found that about 20% of the people with frequent basal cell carcinomas have a mutation in one of the genes responsible for repairing DNA damage, versus about 3% of the general population,” Dr Sarin said. “That’s shockingly high.”
Specifically, there were 12 BCC patients (19.7%) who had 13 pathogenic mutations in 12 genes—APC, BARD1, BRCA1, BRCA2, CDH1, CHEK2, MLH1, MSH2, MSH6, MUTYH, NBN, and PALB2. And 3.0% of non-Finnish European subjects in the Exome Aggregation Consortium had pathogenic mutations in these 12 genes.
Furthermore, 21 of the 61 BCC patients (64.4%) had a history of additional cancers. This included 5 hematologic malignancies (leukemia/lymphoma), 5 invasive melanomas, and 2 breast, 2 colon, and 5 prostate cancers.
When the researchers compared the cancer prevalence in these patients to the Surveillance, Epidemiology, and End Results-estimated prevalence of cancer in the 60- to 69-year-old population of European descent, the BCC cohort had an increased risk of any cancer—a relative risk (RR) of 3.5 (P<0.001).
The RR was 3.5 for leukemia and lymphoma (P=0.004), 11.9 for invasive melanoma (P<0.001), 4.5 for colon cancer (P=0.030), 5.6 for breast cancer (P=0.009), and 4.7 for prostate cancer (P<0.001).
Insurance cohort
To confirm the findings in the Stanford cohort, the researchers applied a similar analysis to a large medical insurance claims database, Truven MarketScan.
The database contained 111,562 patients with 1 case of BCC, 13,264 patients with 6 or more BCCs, and 2920 patients with 12 or more BCCs. Truven patients with no history of BCC served as controls.
The researchers adjusted for age and sex and found that patients with 1 BCC, 6 or more BCCs, and 12 or more BCCs had an increased risk of any cancer compared to controls.
The odds ratio (OR) for any cancer was 1.61 for patients with 1 BCC, 3.12 for those with 6 or more BCCs, and 4.15 for patients with 12 or more BCCs.
The OR for Hodgkin lymphoma was 2.27 for patients with 1 BCC, 8.94 for patients with 6 or more BCCs, and 15.41 for patients with 12 or more BCCs.
The OR for non-Hodgkin lymphoma was 1.40 for patients with 1 BCC, 2.59 for patients with 6 or more BCCs, and 3.10 for patients with 12 or more BCCs.
The OR for leukemia was 1.76 for patients with 1 BCC, 3.23 for patients with 6 or more BCCs, and 5.78 for patients with 12 or more BCCs.
The researchers pointed out that, the more BCCs an individual had, the more likely that person was to have had other cancers as well.
“I was surprised to see such a strong correlation, but it’s also very gratifying,” Dr Sarin said. “Now, we can ask patients with repeated basal cell carcinomas whether they have family members with other types of cancers and perhaps suggest that they consider genetic testing and increased screening.”
The researchers are continuing to enroll Stanford patients in their study to learn whether particular mutations in genes responsible for repairing DNA damage are linked to the development of specific malignancies. The team would also like to conduct a similar study in patients with frequent melanomas.
The current study was supported by the Dermatology Foundation, the Stanford Society of Physician Scholars, the American Skin Association, and Pellepharm Inc.
New research suggests people who develop frequent cases of basal cell carcinoma (BCC) have an increased risk of leukemias, lymphomas, and other cancers.
“We discovered that people who develop 6 or more basal cell carcinomas during a 10-year period are about 3 times more likely than the general population to develop other, unrelated cancers,” said Kavita Sarin, MD, PhD, of Stanford University School of Medicine in California.
“We’re hopeful that this finding could be a way to identify people at an increased risk for a life-threatening malignancy before those cancers develop.”
Dr Sarin and her colleagues reported their findings in JCI Insight.
Stanford cohort
The researchers first studied 61 patients treated at Stanford Health Care for unusually frequent BCCs—an average of 11 per patient over a 10-year period. The team investigated whether these patients may have mutations in 29 genes that code for DNA damage repair proteins.
“We found that about 20% of the people with frequent basal cell carcinomas have a mutation in one of the genes responsible for repairing DNA damage, versus about 3% of the general population,” Dr Sarin said. “That’s shockingly high.”
Specifically, there were 12 BCC patients (19.7%) who had 13 pathogenic mutations in 12 genes—APC, BARD1, BRCA1, BRCA2, CDH1, CHEK2, MLH1, MSH2, MSH6, MUTYH, NBN, and PALB2. And 3.0% of non-Finnish European subjects in the Exome Aggregation Consortium had pathogenic mutations in these 12 genes.
Furthermore, 21 of the 61 BCC patients (64.4%) had a history of additional cancers. This included 5 hematologic malignancies (leukemia/lymphoma), 5 invasive melanomas, and 2 breast, 2 colon, and 5 prostate cancers.
When the researchers compared the cancer prevalence in these patients to the Surveillance, Epidemiology, and End Results-estimated prevalence of cancer in the 60- to 69-year-old population of European descent, the BCC cohort had an increased risk of any cancer—a relative risk (RR) of 3.5 (P<0.001).
The RR was 3.5 for leukemia and lymphoma (P=0.004), 11.9 for invasive melanoma (P<0.001), 4.5 for colon cancer (P=0.030), 5.6 for breast cancer (P=0.009), and 4.7 for prostate cancer (P<0.001).
Insurance cohort
To confirm the findings in the Stanford cohort, the researchers applied a similar analysis to a large medical insurance claims database, Truven MarketScan.
The database contained 111,562 patients with 1 case of BCC, 13,264 patients with 6 or more BCCs, and 2920 patients with 12 or more BCCs. Truven patients with no history of BCC served as controls.
The researchers adjusted for age and sex and found that patients with 1 BCC, 6 or more BCCs, and 12 or more BCCs had an increased risk of any cancer compared to controls.
The odds ratio (OR) for any cancer was 1.61 for patients with 1 BCC, 3.12 for those with 6 or more BCCs, and 4.15 for patients with 12 or more BCCs.
The OR for Hodgkin lymphoma was 2.27 for patients with 1 BCC, 8.94 for patients with 6 or more BCCs, and 15.41 for patients with 12 or more BCCs.
The OR for non-Hodgkin lymphoma was 1.40 for patients with 1 BCC, 2.59 for patients with 6 or more BCCs, and 3.10 for patients with 12 or more BCCs.
The OR for leukemia was 1.76 for patients with 1 BCC, 3.23 for patients with 6 or more BCCs, and 5.78 for patients with 12 or more BCCs.
The researchers pointed out that, the more BCCs an individual had, the more likely that person was to have had other cancers as well.
“I was surprised to see such a strong correlation, but it’s also very gratifying,” Dr Sarin said. “Now, we can ask patients with repeated basal cell carcinomas whether they have family members with other types of cancers and perhaps suggest that they consider genetic testing and increased screening.”
The researchers are continuing to enroll Stanford patients in their study to learn whether particular mutations in genes responsible for repairing DNA damage are linked to the development of specific malignancies. The team would also like to conduct a similar study in patients with frequent melanomas.
The current study was supported by the Dermatology Foundation, the Stanford Society of Physician Scholars, the American Skin Association, and Pellepharm Inc.
New chronic lymphocytic leukemia guidelines from the UK
Fludarabine, cyclophosphamide, and rituximab are recommended as initial therapy for patients with chronic lymphocytic leukemia who do not have TP53 disruption, according to new guidelines from the British Society for Haematology.
The guidelines update the 2012 recommendations on chronic lymphocytic leukemia (CLL) to include “significant” developments in treatment. They were published in the British Journal of Haematology.
Anna H. Schuh, MD, of the department of oncology at the University of Oxford (England), and her coauthors noted that, while these guidelines apply to treatments available outside clinical trials, wherever possible patients with CLL should be treated within the clinical trial setting.
While recommending fludarabine, cyclophosphamide, and rituximab as first-line therapy, the guideline authors acknowledged that the combination of bendamustine and rituximab is an acceptable alternative for patients who could not take the triple therapy because of comorbidities such as advanced age, renal impairment, or issues with marrow capacity.
Similarly, less-fit patients could also be considered for chlorambucil-obinutuzumab or chlorambucil-ofatumumab combinations.
All patients diagnosed with CLL should be tested for TP53 deletions and mutations before each line of therapy, the guideline committee recommended. TP53 disruption makes chemoimmunotherapy ineffective because of either a deletion of chromosome 17p or a mutation in the TP53 gene. However, there is compelling evidence for the efficacy of ibrutinib in these patients, or idelalisib and rituximab for those with cardiac disease or receiving vitamin K antagonists.
With respect to maintenance therapy, the guidelines noted that this was not routinely recommended in CLL as “it is unclear to what extent the progression-free survival benefit is offset by long-term toxicity.”
Patients who are refractory to chemoimmunotherapy, who have relapsed, or who cannot be retreated with chemoimmunotherapy should be treated with idelalisib with rituximab or ibrutinib monotherapy, the guidelines suggested.
“Deciding whether ibrutinib or idelalisib with rituximab is most appropriate for an individual patient depends on a range of factors, including toxicity profile and convenience of delivery,” the authors wrote. However, they noted that the value of adding bendamustine to either option was unclear as research had not shown significant, associated gains in median progression-free survival.
Allogeneic stem cell transplantation should be considered as a treatment option for patients who have either failed chemotherapy, have a TP53 disruption and have not responded to B-cell receptor signaling pathway inhibitors such as ibrutinib, or have a Richter transformation.
The guidelines also addressed the issue of autoimmune cytopenias, which occur in 5%-10% of patients with CLL and can actually precede the diagnosis of CLL in about 9% of cases.
In patients where autoimmune cytopenia is the dominant clinical feature, they should be treated with corticosteroids, intravenous immunoglobulin, or rituximab. However, for patients where the cytopenia is triggered by CLL therapy, the guidelines recommended halting treatment and beginning immunosuppression.
The guideline development was supported by the British Society for Haematology. The UK CLL Forum is a registered charity that receives funding from a number of pharmaceutical companies.
SOURCE: Schuh AH et al. Br J Haematol. 2018 Jul 15. doi: 10.1111/bjh.15460.
Fludarabine, cyclophosphamide, and rituximab are recommended as initial therapy for patients with chronic lymphocytic leukemia who do not have TP53 disruption, according to new guidelines from the British Society for Haematology.
The guidelines update the 2012 recommendations on chronic lymphocytic leukemia (CLL) to include “significant” developments in treatment. They were published in the British Journal of Haematology.
Anna H. Schuh, MD, of the department of oncology at the University of Oxford (England), and her coauthors noted that, while these guidelines apply to treatments available outside clinical trials, wherever possible patients with CLL should be treated within the clinical trial setting.
While recommending fludarabine, cyclophosphamide, and rituximab as first-line therapy, the guideline authors acknowledged that the combination of bendamustine and rituximab is an acceptable alternative for patients who could not take the triple therapy because of comorbidities such as advanced age, renal impairment, or issues with marrow capacity.
Similarly, less-fit patients could also be considered for chlorambucil-obinutuzumab or chlorambucil-ofatumumab combinations.
All patients diagnosed with CLL should be tested for TP53 deletions and mutations before each line of therapy, the guideline committee recommended. TP53 disruption makes chemoimmunotherapy ineffective because of either a deletion of chromosome 17p or a mutation in the TP53 gene. However, there is compelling evidence for the efficacy of ibrutinib in these patients, or idelalisib and rituximab for those with cardiac disease or receiving vitamin K antagonists.
With respect to maintenance therapy, the guidelines noted that this was not routinely recommended in CLL as “it is unclear to what extent the progression-free survival benefit is offset by long-term toxicity.”
Patients who are refractory to chemoimmunotherapy, who have relapsed, or who cannot be retreated with chemoimmunotherapy should be treated with idelalisib with rituximab or ibrutinib monotherapy, the guidelines suggested.
“Deciding whether ibrutinib or idelalisib with rituximab is most appropriate for an individual patient depends on a range of factors, including toxicity profile and convenience of delivery,” the authors wrote. However, they noted that the value of adding bendamustine to either option was unclear as research had not shown significant, associated gains in median progression-free survival.
Allogeneic stem cell transplantation should be considered as a treatment option for patients who have either failed chemotherapy, have a TP53 disruption and have not responded to B-cell receptor signaling pathway inhibitors such as ibrutinib, or have a Richter transformation.
The guidelines also addressed the issue of autoimmune cytopenias, which occur in 5%-10% of patients with CLL and can actually precede the diagnosis of CLL in about 9% of cases.
In patients where autoimmune cytopenia is the dominant clinical feature, they should be treated with corticosteroids, intravenous immunoglobulin, or rituximab. However, for patients where the cytopenia is triggered by CLL therapy, the guidelines recommended halting treatment and beginning immunosuppression.
The guideline development was supported by the British Society for Haematology. The UK CLL Forum is a registered charity that receives funding from a number of pharmaceutical companies.
SOURCE: Schuh AH et al. Br J Haematol. 2018 Jul 15. doi: 10.1111/bjh.15460.
Fludarabine, cyclophosphamide, and rituximab are recommended as initial therapy for patients with chronic lymphocytic leukemia who do not have TP53 disruption, according to new guidelines from the British Society for Haematology.
The guidelines update the 2012 recommendations on chronic lymphocytic leukemia (CLL) to include “significant” developments in treatment. They were published in the British Journal of Haematology.
Anna H. Schuh, MD, of the department of oncology at the University of Oxford (England), and her coauthors noted that, while these guidelines apply to treatments available outside clinical trials, wherever possible patients with CLL should be treated within the clinical trial setting.
While recommending fludarabine, cyclophosphamide, and rituximab as first-line therapy, the guideline authors acknowledged that the combination of bendamustine and rituximab is an acceptable alternative for patients who could not take the triple therapy because of comorbidities such as advanced age, renal impairment, or issues with marrow capacity.
Similarly, less-fit patients could also be considered for chlorambucil-obinutuzumab or chlorambucil-ofatumumab combinations.
All patients diagnosed with CLL should be tested for TP53 deletions and mutations before each line of therapy, the guideline committee recommended. TP53 disruption makes chemoimmunotherapy ineffective because of either a deletion of chromosome 17p or a mutation in the TP53 gene. However, there is compelling evidence for the efficacy of ibrutinib in these patients, or idelalisib and rituximab for those with cardiac disease or receiving vitamin K antagonists.
With respect to maintenance therapy, the guidelines noted that this was not routinely recommended in CLL as “it is unclear to what extent the progression-free survival benefit is offset by long-term toxicity.”
Patients who are refractory to chemoimmunotherapy, who have relapsed, or who cannot be retreated with chemoimmunotherapy should be treated with idelalisib with rituximab or ibrutinib monotherapy, the guidelines suggested.
“Deciding whether ibrutinib or idelalisib with rituximab is most appropriate for an individual patient depends on a range of factors, including toxicity profile and convenience of delivery,” the authors wrote. However, they noted that the value of adding bendamustine to either option was unclear as research had not shown significant, associated gains in median progression-free survival.
Allogeneic stem cell transplantation should be considered as a treatment option for patients who have either failed chemotherapy, have a TP53 disruption and have not responded to B-cell receptor signaling pathway inhibitors such as ibrutinib, or have a Richter transformation.
The guidelines also addressed the issue of autoimmune cytopenias, which occur in 5%-10% of patients with CLL and can actually precede the diagnosis of CLL in about 9% of cases.
In patients where autoimmune cytopenia is the dominant clinical feature, they should be treated with corticosteroids, intravenous immunoglobulin, or rituximab. However, for patients where the cytopenia is triggered by CLL therapy, the guidelines recommended halting treatment and beginning immunosuppression.
The guideline development was supported by the British Society for Haematology. The UK CLL Forum is a registered charity that receives funding from a number of pharmaceutical companies.
SOURCE: Schuh AH et al. Br J Haematol. 2018 Jul 15. doi: 10.1111/bjh.15460.
FROM THE BRITISH JOURNAL OF HAEMATOLOGY
Key clinical point:
Major finding: All patients diagnosed with CLL should be tested for TP53 disruption.
Study details: A guideline developed by the British Society for Haematology offering recommendations for CLL treatment outside clinical trials.
Disclosures: The guideline development was supported by the British Society for Haematology. The UK CLL Forum is a registered charity that receives funding from a number of pharmaceutical companies.
Source: Schuh AH et al. Br J Haematol. 2018 Jul 15. doi: 10.1111/bjh.15460.
Drug could be repurposed for T-ALL
Venetoclax might improve the treatment of certain patients with T-cell acute lymphoblastic leukemia (T-ALL), according to preclinical research published in Leukemia.
Researchers found that a ribosomal defect—the R98S mutation in ribosomal protein L10 (RPL10 R98S)—causes overexpression of BCL-2 in T-ALL.
The BCL-2 inhibitor venetoclax induced apoptosis of RPL10 R98S T-ALL cells and inhibited leukemia progression in mouse models of RPL10 R98S T-ALL.
The researchers therefore believe venetoclax could be used, in combination with other drugs, to treat T-ALL patients with RPL10 R98S.
“In the past couple of years, it has become clear that ribosome defects play a role in different types of cancer,” said study author Kim De Keersmaecker, PhD, of KU Leuven in Leuven, Belgium.
“In the case of a ribosome defect, the cells still produce proteins, but the balance between their quantities is slightly off, which leads to cancer.”
Dr De Keersmaecker and her colleagues noted that RPL10 R98S affects 8% of pediatric patients with T-ALL.
With this study, the researchers found that RPL10 R98S mutant cells were more resilient than wild-type (WT) cells. In overgrowth condition, Ba/F3 RPL10 R98S mutant cells “displayed a clear survival benefit” over RPL10 WT cells.
Likewise, RPL10 R98S Jurkat cells exhibited a survival benefit over WT Jurkat cells in overgrowth condition. And RPL10 R98S Jurkat cells were more resistant to treatment with doxorubicin.
Dr De Keersmaecker and her colleagues said the increased survival they observed in RPL10 R98S mutant cells is associated with enhanced BCL-2 expression. So the team decided to test a BCL-2 inhibitor in RPL10 R98S leukemic cells.
In vitro, venetoclax induced slightly more apoptosis in Jurkat RPL10 R98S cells than WT Jurkat cells. In vivo, venetoclax induced apoptosis in RPL10 R98S T-ALL cells but not WT T-ALL cells.
The researchers also found that venetoclax could re-sensitize RPL10 R98S cells to doxorubicin.
Finally, the team injected RPL10 WT and R98S samples from pediatric T-ALL patients into mice and treated the animals with DMSO or venetoclax (50 mg/kg) once a week.
Venetoclax had very little effect on the RPL10 WT mice. Percentages of human CD45 T-ALL cells in the peripheral blood were similar whether mice received DMSO or venetoclax.
However, in the RPL10 R98S mice, those that received DMSO experienced disease progression, while there were no signs of leukemia progression in the peripheral blood of mice that received venetoclax.
The splenomegaly observed in DMSO-treated mice was “almost completely suppressed” in mice that received venetoclax, according to the researchers.
The team also said they observed a 30% to 50% suppression of human CD45 leukemia cell engraftment in the bone marrow and the presence of 30% to 40% mouse CD45 cells in mice treated with venetoclax.
On the other hand, mice treated with DMSO had more than 95% human leukemia infiltration in the bone marrow and no mouse CD45-expressing cells.
Dr De Keersmaecker and her colleagues said these results suggest RPL10 R98S pediatric T-ALL is sensitive to BCL-2 targeted therapies such as venetoclax. However, venetoclax alone would not be sufficient to treat this type of T-ALL.
“Patients with leukemia often get a drug cocktail, while our study only tested the BCL-2 inhibitor,” Dr De Keersmaecker said. “That’s why our follow-up study will focus on a cocktail of this BCL-2 inhibitor and other drugs. For patients with the ribosome defect analyzed in our study, this avenue is definitely worth examining in greater detail.”
Venetoclax might improve the treatment of certain patients with T-cell acute lymphoblastic leukemia (T-ALL), according to preclinical research published in Leukemia.
Researchers found that a ribosomal defect—the R98S mutation in ribosomal protein L10 (RPL10 R98S)—causes overexpression of BCL-2 in T-ALL.
The BCL-2 inhibitor venetoclax induced apoptosis of RPL10 R98S T-ALL cells and inhibited leukemia progression in mouse models of RPL10 R98S T-ALL.
The researchers therefore believe venetoclax could be used, in combination with other drugs, to treat T-ALL patients with RPL10 R98S.
“In the past couple of years, it has become clear that ribosome defects play a role in different types of cancer,” said study author Kim De Keersmaecker, PhD, of KU Leuven in Leuven, Belgium.
“In the case of a ribosome defect, the cells still produce proteins, but the balance between their quantities is slightly off, which leads to cancer.”
Dr De Keersmaecker and her colleagues noted that RPL10 R98S affects 8% of pediatric patients with T-ALL.
With this study, the researchers found that RPL10 R98S mutant cells were more resilient than wild-type (WT) cells. In overgrowth condition, Ba/F3 RPL10 R98S mutant cells “displayed a clear survival benefit” over RPL10 WT cells.
Likewise, RPL10 R98S Jurkat cells exhibited a survival benefit over WT Jurkat cells in overgrowth condition. And RPL10 R98S Jurkat cells were more resistant to treatment with doxorubicin.
Dr De Keersmaecker and her colleagues said the increased survival they observed in RPL10 R98S mutant cells is associated with enhanced BCL-2 expression. So the team decided to test a BCL-2 inhibitor in RPL10 R98S leukemic cells.
In vitro, venetoclax induced slightly more apoptosis in Jurkat RPL10 R98S cells than WT Jurkat cells. In vivo, venetoclax induced apoptosis in RPL10 R98S T-ALL cells but not WT T-ALL cells.
The researchers also found that venetoclax could re-sensitize RPL10 R98S cells to doxorubicin.
Finally, the team injected RPL10 WT and R98S samples from pediatric T-ALL patients into mice and treated the animals with DMSO or venetoclax (50 mg/kg) once a week.
Venetoclax had very little effect on the RPL10 WT mice. Percentages of human CD45 T-ALL cells in the peripheral blood were similar whether mice received DMSO or venetoclax.
However, in the RPL10 R98S mice, those that received DMSO experienced disease progression, while there were no signs of leukemia progression in the peripheral blood of mice that received venetoclax.
The splenomegaly observed in DMSO-treated mice was “almost completely suppressed” in mice that received venetoclax, according to the researchers.
The team also said they observed a 30% to 50% suppression of human CD45 leukemia cell engraftment in the bone marrow and the presence of 30% to 40% mouse CD45 cells in mice treated with venetoclax.
On the other hand, mice treated with DMSO had more than 95% human leukemia infiltration in the bone marrow and no mouse CD45-expressing cells.
Dr De Keersmaecker and her colleagues said these results suggest RPL10 R98S pediatric T-ALL is sensitive to BCL-2 targeted therapies such as venetoclax. However, venetoclax alone would not be sufficient to treat this type of T-ALL.
“Patients with leukemia often get a drug cocktail, while our study only tested the BCL-2 inhibitor,” Dr De Keersmaecker said. “That’s why our follow-up study will focus on a cocktail of this BCL-2 inhibitor and other drugs. For patients with the ribosome defect analyzed in our study, this avenue is definitely worth examining in greater detail.”
Venetoclax might improve the treatment of certain patients with T-cell acute lymphoblastic leukemia (T-ALL), according to preclinical research published in Leukemia.
Researchers found that a ribosomal defect—the R98S mutation in ribosomal protein L10 (RPL10 R98S)—causes overexpression of BCL-2 in T-ALL.
The BCL-2 inhibitor venetoclax induced apoptosis of RPL10 R98S T-ALL cells and inhibited leukemia progression in mouse models of RPL10 R98S T-ALL.
The researchers therefore believe venetoclax could be used, in combination with other drugs, to treat T-ALL patients with RPL10 R98S.
“In the past couple of years, it has become clear that ribosome defects play a role in different types of cancer,” said study author Kim De Keersmaecker, PhD, of KU Leuven in Leuven, Belgium.
“In the case of a ribosome defect, the cells still produce proteins, but the balance between their quantities is slightly off, which leads to cancer.”
Dr De Keersmaecker and her colleagues noted that RPL10 R98S affects 8% of pediatric patients with T-ALL.
With this study, the researchers found that RPL10 R98S mutant cells were more resilient than wild-type (WT) cells. In overgrowth condition, Ba/F3 RPL10 R98S mutant cells “displayed a clear survival benefit” over RPL10 WT cells.
Likewise, RPL10 R98S Jurkat cells exhibited a survival benefit over WT Jurkat cells in overgrowth condition. And RPL10 R98S Jurkat cells were more resistant to treatment with doxorubicin.
Dr De Keersmaecker and her colleagues said the increased survival they observed in RPL10 R98S mutant cells is associated with enhanced BCL-2 expression. So the team decided to test a BCL-2 inhibitor in RPL10 R98S leukemic cells.
In vitro, venetoclax induced slightly more apoptosis in Jurkat RPL10 R98S cells than WT Jurkat cells. In vivo, venetoclax induced apoptosis in RPL10 R98S T-ALL cells but not WT T-ALL cells.
The researchers also found that venetoclax could re-sensitize RPL10 R98S cells to doxorubicin.
Finally, the team injected RPL10 WT and R98S samples from pediatric T-ALL patients into mice and treated the animals with DMSO or venetoclax (50 mg/kg) once a week.
Venetoclax had very little effect on the RPL10 WT mice. Percentages of human CD45 T-ALL cells in the peripheral blood were similar whether mice received DMSO or venetoclax.
However, in the RPL10 R98S mice, those that received DMSO experienced disease progression, while there were no signs of leukemia progression in the peripheral blood of mice that received venetoclax.
The splenomegaly observed in DMSO-treated mice was “almost completely suppressed” in mice that received venetoclax, according to the researchers.
The team also said they observed a 30% to 50% suppression of human CD45 leukemia cell engraftment in the bone marrow and the presence of 30% to 40% mouse CD45 cells in mice treated with venetoclax.
On the other hand, mice treated with DMSO had more than 95% human leukemia infiltration in the bone marrow and no mouse CD45-expressing cells.
Dr De Keersmaecker and her colleagues said these results suggest RPL10 R98S pediatric T-ALL is sensitive to BCL-2 targeted therapies such as venetoclax. However, venetoclax alone would not be sufficient to treat this type of T-ALL.
“Patients with leukemia often get a drug cocktail, while our study only tested the BCL-2 inhibitor,” Dr De Keersmaecker said. “That’s why our follow-up study will focus on a cocktail of this BCL-2 inhibitor and other drugs. For patients with the ribosome defect analyzed in our study, this avenue is definitely worth examining in greater detail.”
Treatment guidelines for CAR T-cell therapy

Researchers have developed treatment guidelines for pediatric patients receiving chimeric antigen receptor (CAR) T-cell therapy.
The guidelines include recommendations for patient selection and consent, treatment details, and advice on managing cytokine release syndrome (CRS) and other adverse events associated with CAR T-cell therapy.
The guidelines were published in Nature Reviews Clinical Oncology.
“CAR T-cell therapy has been associated with remarkable response rates for children and young adults with ALL [acute lymphoblastic leukemia], yet this innovative form of cellular immunotherapy has resulted in unique and severe toxicities which can lead to rapid cardiorespiratory and/or neurological deterioration,” said guidelines author Kris Mahadeo, MD, of The University of Texas MD Anderson Cancer Center in Houston.
“This novel therapy requires the medical vigilance of a diverse multi-disciplinary team and associated clinical infrastructure to ensure optimal patient outcomes.”
Pediatric patient selection and consent
The guidelines state that providers of CAR T-cell therapies should adhere to product information labels and guidance from risk evaluation and mitigation strategy programs (level of evidence: IV, grade: D).
In addition, patient selection should be based on the indications approved by the US Food and Drug Administration and criteria used in pivotal studies. However, this can change as new information becomes available (level of evidence: IV, grade: D).
Informed consent should include descriptions of the risks and benefits associated with leukapheresis, lymphodepletion, CRS, CAR T-cell-related encephalopathy syndrome (CRES), bridging chemotherapy, intensive care support, and anti-IL-6 therapy (level of evidence: IIA, grade: B).
Providers should obtain child assent when appropriate and may benefit from incorporating child life and psychological services in assent discussions (level of evidence: IV, grade: D).
Treatment specifics
The guidelines recommend cyclophosphamide–fludarabine regimens for lymphodepletion, although exceptions can be considered in cases of hemorrhagic cystitis and/or resistance to a prior cyclophosphamide-based regimen (level of evidence: IIA, grade: B).
Providers should consider inpatient admission for a minimum of 3 to 7 days after receipt of tisagenlecleucel. This was based on the experience in pediatric and young adult patients with CD19+ relapsed and/or refractory B-cell acute lymphoblastic leukemia (level of evidence: IIA, grade: B).
Patients should be closely monitored for hypotension, hypocalcemia, and catheter-related pain during leukapheresis (level of evidence: IIA, grade: B).
For patients receiving tocilizumab, those weighing <30 kg should receive 12 mg/kg, and those weighing ≥30 kg should receive 8 mg/kg (level of evidence: IIA, grade: B).
Adverse events
The guidelines say parent and/or caregiver concerns should be addressed as these individuals may be best equipped to recognize early signs or symptoms of CRS (level of evidence: III, grade: C).
When CAR T-cell therapy is administered in an outpatient setting, there should be a low threshold for patient admission upon the development of signs or symptoms suggestive of CRS and/or CRES (level of evidence: IIA, grade: B).
CRS grading should be performed at least once every 12 hours (level of evidence: IIA, grade: B). Detailed information on grading is provided in the guidelines.
Providers should suspect CRS if any of the following signs/symptoms are present within the first 2 weeks of CAR T-cell infusion:
- Fever ≥38 °C
- Hypotension
- Hypoxia with an arterial oxygen saturation of <90% on room air
- Evidence of organ toxicity as determined by the most recent CTCAE grading system and considerations detailed in the guidelines (level of evidence: IIA, grade: C).
The guidelines also recommend “high vigilance” for sinus tachycardia as an early sign of CRS (level of evidence: IIA, grade: B) as well as application of the PALICC (Pediatric Acute Lung Injury Consensus Conference) at-risk P-ARDS (pediatric acute respiratory distress syndrome) criteria for the CRS grading of hypoxia (level of evidence: IIA, grade: B).
Hemophagocytic lymphohistiocytosis and/or macrophage-activation syndrome can be treated with anti-IL-6 therapy and corticosteroids. However, refractory cases may require systemic and/or intrathecal therapy or use of the IL-1 receptor antagonist anakinra (level of evidence: IIA, grade: C).
The guidelines recommend that delirium screening be performed at least twice per 24-hour period among admitted patients and at least daily among outpatients during the high-risk periods for CRES (level of evidence: IIA, grade: C). Delirium screening should be performed with the CAPD (Cornell Assessment of Pediatric Delirium) tool or CARTOX-10 (CAR T-Cell Therapy-Associated Toxicity 10-point assessment scale) for patients age 12 and older who have sufficient cognitive abilities.
Acute kidney injury in children can be graded according to the CTCAE (Common Terminology Criteria for Adverse Events) using pRIFLE (Pediatric Risk, Injury, Failure, Loss, End-Stage Renal Disease) and KDIGO (Kidney Disease: Improving Global Outcomes) definitions of oliguria (level of evidence: IIA, grade: B).
Other considerations
The guidelines “strongly encourage” consideration of quality-adjusted life-years gained for pediatric patients who might achieve long-term remission from CAR T-cell therapy and encourage efforts to reduce the cost of care (level of evidence: IV, grade: D).
The guidelines also recommend that CAR T-cell programs seek FACT IEC (Foundation for the Accreditation of Cellular Therapy for Immune Effector Cells) accreditation to ensure adherence to quality standards (level of evidence: IV, grade: D).
Finally, the guidelines suggest the possibility of a prospective collaboration with intensive-care registries, which could allow accurate data entry of cell therapy variables into the CIBMTR registry with concurrent entry of intensive-care variables into an appropriate registry by pediatric critical care teams (level of evidence: IV, grade: D).
Researchers have developed treatment guidelines for pediatric patients receiving chimeric antigen receptor (CAR) T-cell therapy.
The guidelines include recommendations for patient selection and consent, treatment details, and advice on managing cytokine release syndrome (CRS) and other adverse events associated with CAR T-cell therapy.
The guidelines were published in Nature Reviews Clinical Oncology.
“CAR T-cell therapy has been associated with remarkable response rates for children and young adults with ALL [acute lymphoblastic leukemia], yet this innovative form of cellular immunotherapy has resulted in unique and severe toxicities which can lead to rapid cardiorespiratory and/or neurological deterioration,” said guidelines author Kris Mahadeo, MD, of The University of Texas MD Anderson Cancer Center in Houston.
“This novel therapy requires the medical vigilance of a diverse multi-disciplinary team and associated clinical infrastructure to ensure optimal patient outcomes.”
Pediatric patient selection and consent
The guidelines state that providers of CAR T-cell therapies should adhere to product information labels and guidance from risk evaluation and mitigation strategy programs (level of evidence: IV, grade: D).
In addition, patient selection should be based on the indications approved by the US Food and Drug Administration and criteria used in pivotal studies. However, this can change as new information becomes available (level of evidence: IV, grade: D).
Informed consent should include descriptions of the risks and benefits associated with leukapheresis, lymphodepletion, CRS, CAR T-cell-related encephalopathy syndrome (CRES), bridging chemotherapy, intensive care support, and anti-IL-6 therapy (level of evidence: IIA, grade: B).
Providers should obtain child assent when appropriate and may benefit from incorporating child life and psychological services in assent discussions (level of evidence: IV, grade: D).
Treatment specifics
The guidelines recommend cyclophosphamide–fludarabine regimens for lymphodepletion, although exceptions can be considered in cases of hemorrhagic cystitis and/or resistance to a prior cyclophosphamide-based regimen (level of evidence: IIA, grade: B).
Providers should consider inpatient admission for a minimum of 3 to 7 days after receipt of tisagenlecleucel. This was based on the experience in pediatric and young adult patients with CD19+ relapsed and/or refractory B-cell acute lymphoblastic leukemia (level of evidence: IIA, grade: B).
Patients should be closely monitored for hypotension, hypocalcemia, and catheter-related pain during leukapheresis (level of evidence: IIA, grade: B).
For patients receiving tocilizumab, those weighing <30 kg should receive 12 mg/kg, and those weighing ≥30 kg should receive 8 mg/kg (level of evidence: IIA, grade: B).
Adverse events
The guidelines say parent and/or caregiver concerns should be addressed as these individuals may be best equipped to recognize early signs or symptoms of CRS (level of evidence: III, grade: C).
When CAR T-cell therapy is administered in an outpatient setting, there should be a low threshold for patient admission upon the development of signs or symptoms suggestive of CRS and/or CRES (level of evidence: IIA, grade: B).
CRS grading should be performed at least once every 12 hours (level of evidence: IIA, grade: B). Detailed information on grading is provided in the guidelines.
Providers should suspect CRS if any of the following signs/symptoms are present within the first 2 weeks of CAR T-cell infusion:
- Fever ≥38 °C
- Hypotension
- Hypoxia with an arterial oxygen saturation of <90% on room air
- Evidence of organ toxicity as determined by the most recent CTCAE grading system and considerations detailed in the guidelines (level of evidence: IIA, grade: C).
The guidelines also recommend “high vigilance” for sinus tachycardia as an early sign of CRS (level of evidence: IIA, grade: B) as well as application of the PALICC (Pediatric Acute Lung Injury Consensus Conference) at-risk P-ARDS (pediatric acute respiratory distress syndrome) criteria for the CRS grading of hypoxia (level of evidence: IIA, grade: B).
Hemophagocytic lymphohistiocytosis and/or macrophage-activation syndrome can be treated with anti-IL-6 therapy and corticosteroids. However, refractory cases may require systemic and/or intrathecal therapy or use of the IL-1 receptor antagonist anakinra (level of evidence: IIA, grade: C).
The guidelines recommend that delirium screening be performed at least twice per 24-hour period among admitted patients and at least daily among outpatients during the high-risk periods for CRES (level of evidence: IIA, grade: C). Delirium screening should be performed with the CAPD (Cornell Assessment of Pediatric Delirium) tool or CARTOX-10 (CAR T-Cell Therapy-Associated Toxicity 10-point assessment scale) for patients age 12 and older who have sufficient cognitive abilities.
Acute kidney injury in children can be graded according to the CTCAE (Common Terminology Criteria for Adverse Events) using pRIFLE (Pediatric Risk, Injury, Failure, Loss, End-Stage Renal Disease) and KDIGO (Kidney Disease: Improving Global Outcomes) definitions of oliguria (level of evidence: IIA, grade: B).
Other considerations
The guidelines “strongly encourage” consideration of quality-adjusted life-years gained for pediatric patients who might achieve long-term remission from CAR T-cell therapy and encourage efforts to reduce the cost of care (level of evidence: IV, grade: D).
The guidelines also recommend that CAR T-cell programs seek FACT IEC (Foundation for the Accreditation of Cellular Therapy for Immune Effector Cells) accreditation to ensure adherence to quality standards (level of evidence: IV, grade: D).
Finally, the guidelines suggest the possibility of a prospective collaboration with intensive-care registries, which could allow accurate data entry of cell therapy variables into the CIBMTR registry with concurrent entry of intensive-care variables into an appropriate registry by pediatric critical care teams (level of evidence: IV, grade: D).
Researchers have developed treatment guidelines for pediatric patients receiving chimeric antigen receptor (CAR) T-cell therapy.
The guidelines include recommendations for patient selection and consent, treatment details, and advice on managing cytokine release syndrome (CRS) and other adverse events associated with CAR T-cell therapy.
The guidelines were published in Nature Reviews Clinical Oncology.
“CAR T-cell therapy has been associated with remarkable response rates for children and young adults with ALL [acute lymphoblastic leukemia], yet this innovative form of cellular immunotherapy has resulted in unique and severe toxicities which can lead to rapid cardiorespiratory and/or neurological deterioration,” said guidelines author Kris Mahadeo, MD, of The University of Texas MD Anderson Cancer Center in Houston.
“This novel therapy requires the medical vigilance of a diverse multi-disciplinary team and associated clinical infrastructure to ensure optimal patient outcomes.”
Pediatric patient selection and consent
The guidelines state that providers of CAR T-cell therapies should adhere to product information labels and guidance from risk evaluation and mitigation strategy programs (level of evidence: IV, grade: D).
In addition, patient selection should be based on the indications approved by the US Food and Drug Administration and criteria used in pivotal studies. However, this can change as new information becomes available (level of evidence: IV, grade: D).
Informed consent should include descriptions of the risks and benefits associated with leukapheresis, lymphodepletion, CRS, CAR T-cell-related encephalopathy syndrome (CRES), bridging chemotherapy, intensive care support, and anti-IL-6 therapy (level of evidence: IIA, grade: B).
Providers should obtain child assent when appropriate and may benefit from incorporating child life and psychological services in assent discussions (level of evidence: IV, grade: D).
Treatment specifics
The guidelines recommend cyclophosphamide–fludarabine regimens for lymphodepletion, although exceptions can be considered in cases of hemorrhagic cystitis and/or resistance to a prior cyclophosphamide-based regimen (level of evidence: IIA, grade: B).
Providers should consider inpatient admission for a minimum of 3 to 7 days after receipt of tisagenlecleucel. This was based on the experience in pediatric and young adult patients with CD19+ relapsed and/or refractory B-cell acute lymphoblastic leukemia (level of evidence: IIA, grade: B).
Patients should be closely monitored for hypotension, hypocalcemia, and catheter-related pain during leukapheresis (level of evidence: IIA, grade: B).
For patients receiving tocilizumab, those weighing <30 kg should receive 12 mg/kg, and those weighing ≥30 kg should receive 8 mg/kg (level of evidence: IIA, grade: B).
Adverse events
The guidelines say parent and/or caregiver concerns should be addressed as these individuals may be best equipped to recognize early signs or symptoms of CRS (level of evidence: III, grade: C).
When CAR T-cell therapy is administered in an outpatient setting, there should be a low threshold for patient admission upon the development of signs or symptoms suggestive of CRS and/or CRES (level of evidence: IIA, grade: B).
CRS grading should be performed at least once every 12 hours (level of evidence: IIA, grade: B). Detailed information on grading is provided in the guidelines.
Providers should suspect CRS if any of the following signs/symptoms are present within the first 2 weeks of CAR T-cell infusion:
- Fever ≥38 °C
- Hypotension
- Hypoxia with an arterial oxygen saturation of <90% on room air
- Evidence of organ toxicity as determined by the most recent CTCAE grading system and considerations detailed in the guidelines (level of evidence: IIA, grade: C).
The guidelines also recommend “high vigilance” for sinus tachycardia as an early sign of CRS (level of evidence: IIA, grade: B) as well as application of the PALICC (Pediatric Acute Lung Injury Consensus Conference) at-risk P-ARDS (pediatric acute respiratory distress syndrome) criteria for the CRS grading of hypoxia (level of evidence: IIA, grade: B).
Hemophagocytic lymphohistiocytosis and/or macrophage-activation syndrome can be treated with anti-IL-6 therapy and corticosteroids. However, refractory cases may require systemic and/or intrathecal therapy or use of the IL-1 receptor antagonist anakinra (level of evidence: IIA, grade: C).
The guidelines recommend that delirium screening be performed at least twice per 24-hour period among admitted patients and at least daily among outpatients during the high-risk periods for CRES (level of evidence: IIA, grade: C). Delirium screening should be performed with the CAPD (Cornell Assessment of Pediatric Delirium) tool or CARTOX-10 (CAR T-Cell Therapy-Associated Toxicity 10-point assessment scale) for patients age 12 and older who have sufficient cognitive abilities.
Acute kidney injury in children can be graded according to the CTCAE (Common Terminology Criteria for Adverse Events) using pRIFLE (Pediatric Risk, Injury, Failure, Loss, End-Stage Renal Disease) and KDIGO (Kidney Disease: Improving Global Outcomes) definitions of oliguria (level of evidence: IIA, grade: B).
Other considerations
The guidelines “strongly encourage” consideration of quality-adjusted life-years gained for pediatric patients who might achieve long-term remission from CAR T-cell therapy and encourage efforts to reduce the cost of care (level of evidence: IV, grade: D).
The guidelines also recommend that CAR T-cell programs seek FACT IEC (Foundation for the Accreditation of Cellular Therapy for Immune Effector Cells) accreditation to ensure adherence to quality standards (level of evidence: IV, grade: D).
Finally, the guidelines suggest the possibility of a prospective collaboration with intensive-care registries, which could allow accurate data entry of cell therapy variables into the CIBMTR registry with concurrent entry of intensive-care variables into an appropriate registry by pediatric critical care teams (level of evidence: IV, grade: D).
Orphan designation recommended for PCM-075

The European Medicines Agency’s Committee for Orphan Medicinal Products (COMP) has recommended that PCM-075 receive orphan drug designation as a treatment for acute myeloid leukemia (AML).
PCM-075 is an oral adenosine triphosphate competitive inhibitor of the serine/threonine Polo-like kinase 1 (PLK1) enzyme, which is overexpressed in hematologic and solid tumor malignancies.
The COMP’s recommendation for PCM-075 is expected to be adopted by the European Commission at the end of this month.
Orphan drug designation in Europe is available to companies developing products intended to treat a life-threatening or chronically debilitating condition that affects fewer than 5 in 10,000 people in the European Union (EU).
The designation allows for financial and regulatory incentives that include 10 years of marketing exclusivity in the EU after product approval, eligibility for conditional marketing authorization, protocol assistance from the European Medicines Agency at reduced fees during the product development phase, and direct access to centralized marketing authorization in the EU.
PCM-075 research
PCM-075 only targets the PLK1 isoform (not PLK2 or PLK3) and has a 24-hour drug half-life with reversible, on-target hematologic toxicities, according to Trovagene, Inc., the company developing PCM-075.
Trovagene believes that PCM-075’s reversible, on-target activity, combined with an improved dose/scheduling protocol, could mean that PCM-075 will improve upon long-term outcomes observed in previous studies with a PLK inhibitor in AML.
This includes a phase 2 study in which AML patients who received a PLK inhibitor plus low-dose cytarabine (LDAC) had a higher response rate than patients who received LDAC alone—31% and 13.3%, respectively.
Trovagene said preclinical studies have shown that PCM-075 synergizes with more than 10 drugs used to treat hematologic and solid tumor malignancies. This includes FLT3 and HDAC inhibitors, taxanes, and cytotoxins.
Trovagene is now conducting a phase 1b/2 trial of PCM-075 in combination with standard care (LDAC or decitabine) in patients with AML (NCT03303339).
The company has already completed a phase 1 dose-escalation study of PCM-075 in patients with advanced metastatic solid tumor malignancies. Results from this study were published in Investigational New Drugs.
The European Medicines Agency’s Committee for Orphan Medicinal Products (COMP) has recommended that PCM-075 receive orphan drug designation as a treatment for acute myeloid leukemia (AML).
PCM-075 is an oral adenosine triphosphate competitive inhibitor of the serine/threonine Polo-like kinase 1 (PLK1) enzyme, which is overexpressed in hematologic and solid tumor malignancies.
The COMP’s recommendation for PCM-075 is expected to be adopted by the European Commission at the end of this month.
Orphan drug designation in Europe is available to companies developing products intended to treat a life-threatening or chronically debilitating condition that affects fewer than 5 in 10,000 people in the European Union (EU).
The designation allows for financial and regulatory incentives that include 10 years of marketing exclusivity in the EU after product approval, eligibility for conditional marketing authorization, protocol assistance from the European Medicines Agency at reduced fees during the product development phase, and direct access to centralized marketing authorization in the EU.
PCM-075 research
PCM-075 only targets the PLK1 isoform (not PLK2 or PLK3) and has a 24-hour drug half-life with reversible, on-target hematologic toxicities, according to Trovagene, Inc., the company developing PCM-075.
Trovagene believes that PCM-075’s reversible, on-target activity, combined with an improved dose/scheduling protocol, could mean that PCM-075 will improve upon long-term outcomes observed in previous studies with a PLK inhibitor in AML.
This includes a phase 2 study in which AML patients who received a PLK inhibitor plus low-dose cytarabine (LDAC) had a higher response rate than patients who received LDAC alone—31% and 13.3%, respectively.
Trovagene said preclinical studies have shown that PCM-075 synergizes with more than 10 drugs used to treat hematologic and solid tumor malignancies. This includes FLT3 and HDAC inhibitors, taxanes, and cytotoxins.
Trovagene is now conducting a phase 1b/2 trial of PCM-075 in combination with standard care (LDAC or decitabine) in patients with AML (NCT03303339).
The company has already completed a phase 1 dose-escalation study of PCM-075 in patients with advanced metastatic solid tumor malignancies. Results from this study were published in Investigational New Drugs.
The European Medicines Agency’s Committee for Orphan Medicinal Products (COMP) has recommended that PCM-075 receive orphan drug designation as a treatment for acute myeloid leukemia (AML).
PCM-075 is an oral adenosine triphosphate competitive inhibitor of the serine/threonine Polo-like kinase 1 (PLK1) enzyme, which is overexpressed in hematologic and solid tumor malignancies.
The COMP’s recommendation for PCM-075 is expected to be adopted by the European Commission at the end of this month.
Orphan drug designation in Europe is available to companies developing products intended to treat a life-threatening or chronically debilitating condition that affects fewer than 5 in 10,000 people in the European Union (EU).
The designation allows for financial and regulatory incentives that include 10 years of marketing exclusivity in the EU after product approval, eligibility for conditional marketing authorization, protocol assistance from the European Medicines Agency at reduced fees during the product development phase, and direct access to centralized marketing authorization in the EU.
PCM-075 research
PCM-075 only targets the PLK1 isoform (not PLK2 or PLK3) and has a 24-hour drug half-life with reversible, on-target hematologic toxicities, according to Trovagene, Inc., the company developing PCM-075.
Trovagene believes that PCM-075’s reversible, on-target activity, combined with an improved dose/scheduling protocol, could mean that PCM-075 will improve upon long-term outcomes observed in previous studies with a PLK inhibitor in AML.
This includes a phase 2 study in which AML patients who received a PLK inhibitor plus low-dose cytarabine (LDAC) had a higher response rate than patients who received LDAC alone—31% and 13.3%, respectively.
Trovagene said preclinical studies have shown that PCM-075 synergizes with more than 10 drugs used to treat hematologic and solid tumor malignancies. This includes FLT3 and HDAC inhibitors, taxanes, and cytotoxins.
Trovagene is now conducting a phase 1b/2 trial of PCM-075 in combination with standard care (LDAC or decitabine) in patients with AML (NCT03303339).
The company has already completed a phase 1 dose-escalation study of PCM-075 in patients with advanced metastatic solid tumor malignancies. Results from this study were published in Investigational New Drugs.
Method may enable eradication of LSCs in AML
Disrupting mitophagy may be a “promising strategy” for eliminating leukemia stem cells (LSCs) in acute myeloid leukemia (AML), according to researchers.
The team found that AML LSCs depend on mitophagy to maintain their “stemness,” but targeting the central metabolic stress regulator AMPK or the mitochondrial dynamics regulator FIS1 can disrupt mitophagy and impair LSC function.
Craig T. Jordan, PhD, of the University of Colorado in Aurora, and his colleagues reported these findings in Cell Stem Cell.
The researchers said in vitro experiments showed that LSCs have elevated levels of FIS1 and “distinct mitochondrial morphology.”
When the team inhibited FIS1 in the AML cell line MOLM-13 and primary AML cells, they observed disruption of mitochondrial dynamics. Experiments in mouse models indicated that FIS1 is required for LSC self-renewal.
Specifically, the researchers said they found that depletion of FIS1 hinders mitophagy and leads to inactivation of GSK3, myeloid differentiation, cell-cycle arrest, and loss of LSC function.
Dr Jordan and his colleagues also found that AMPK is an upstream regulator of FIS1, and targeting AMPK produces similar effects as targeting FIS1—namely, disrupting mitophagy and impairing LSC self-renewal.
The researchers said their findings suggest that mitochondrial stress generated from oncogenic transformation may activate AMPK signaling in LSCs. And the AMPK signaling drives FIS1-mediated mitophagy, which eliminates stressed mitochondria and allows LSCs to thrive.
However, when AMPK or FIS1 is inhibited, the damaged mitochondria are not eliminated. This leads to “GSK3 inhibition and other unknown events” that prompt differentiation and hinder LSC function.
“Leukemia stem cells require AMPK for their survival, but normal hematopoietic cells can do without it,” Dr Jordan noted. “The reason this study is so important is that, so far, nobody’s come up with a good way to kill leukemia stem cells while sparing normal blood-forming cells. If we can translate this concept to patients, the potential for improved therapy is very exciting.”
Disrupting mitophagy may be a “promising strategy” for eliminating leukemia stem cells (LSCs) in acute myeloid leukemia (AML), according to researchers.
The team found that AML LSCs depend on mitophagy to maintain their “stemness,” but targeting the central metabolic stress regulator AMPK or the mitochondrial dynamics regulator FIS1 can disrupt mitophagy and impair LSC function.
Craig T. Jordan, PhD, of the University of Colorado in Aurora, and his colleagues reported these findings in Cell Stem Cell.
The researchers said in vitro experiments showed that LSCs have elevated levels of FIS1 and “distinct mitochondrial morphology.”
When the team inhibited FIS1 in the AML cell line MOLM-13 and primary AML cells, they observed disruption of mitochondrial dynamics. Experiments in mouse models indicated that FIS1 is required for LSC self-renewal.
Specifically, the researchers said they found that depletion of FIS1 hinders mitophagy and leads to inactivation of GSK3, myeloid differentiation, cell-cycle arrest, and loss of LSC function.
Dr Jordan and his colleagues also found that AMPK is an upstream regulator of FIS1, and targeting AMPK produces similar effects as targeting FIS1—namely, disrupting mitophagy and impairing LSC self-renewal.
The researchers said their findings suggest that mitochondrial stress generated from oncogenic transformation may activate AMPK signaling in LSCs. And the AMPK signaling drives FIS1-mediated mitophagy, which eliminates stressed mitochondria and allows LSCs to thrive.
However, when AMPK or FIS1 is inhibited, the damaged mitochondria are not eliminated. This leads to “GSK3 inhibition and other unknown events” that prompt differentiation and hinder LSC function.
“Leukemia stem cells require AMPK for their survival, but normal hematopoietic cells can do without it,” Dr Jordan noted. “The reason this study is so important is that, so far, nobody’s come up with a good way to kill leukemia stem cells while sparing normal blood-forming cells. If we can translate this concept to patients, the potential for improved therapy is very exciting.”
Disrupting mitophagy may be a “promising strategy” for eliminating leukemia stem cells (LSCs) in acute myeloid leukemia (AML), according to researchers.
The team found that AML LSCs depend on mitophagy to maintain their “stemness,” but targeting the central metabolic stress regulator AMPK or the mitochondrial dynamics regulator FIS1 can disrupt mitophagy and impair LSC function.
Craig T. Jordan, PhD, of the University of Colorado in Aurora, and his colleagues reported these findings in Cell Stem Cell.
The researchers said in vitro experiments showed that LSCs have elevated levels of FIS1 and “distinct mitochondrial morphology.”
When the team inhibited FIS1 in the AML cell line MOLM-13 and primary AML cells, they observed disruption of mitochondrial dynamics. Experiments in mouse models indicated that FIS1 is required for LSC self-renewal.
Specifically, the researchers said they found that depletion of FIS1 hinders mitophagy and leads to inactivation of GSK3, myeloid differentiation, cell-cycle arrest, and loss of LSC function.
Dr Jordan and his colleagues also found that AMPK is an upstream regulator of FIS1, and targeting AMPK produces similar effects as targeting FIS1—namely, disrupting mitophagy and impairing LSC self-renewal.
The researchers said their findings suggest that mitochondrial stress generated from oncogenic transformation may activate AMPK signaling in LSCs. And the AMPK signaling drives FIS1-mediated mitophagy, which eliminates stressed mitochondria and allows LSCs to thrive.
However, when AMPK or FIS1 is inhibited, the damaged mitochondria are not eliminated. This leads to “GSK3 inhibition and other unknown events” that prompt differentiation and hinder LSC function.
“Leukemia stem cells require AMPK for their survival, but normal hematopoietic cells can do without it,” Dr Jordan noted. “The reason this study is so important is that, so far, nobody’s come up with a good way to kill leukemia stem cells while sparing normal blood-forming cells. If we can translate this concept to patients, the potential for improved therapy is very exciting.”
Vadastuximab talirine gives big boost to AML remission
For elderly patients with CD33-positive acute myeloid leukemia (AML), vadastuximab talirine in combination with a hypomethylating agent (HMA) improves remission rates, compared with HMA therapy alone, according to a phase 1 trial.
More than half of the patients treated with combination therapy achieved deep remission, defined as a negative-flow cytometry test for minimal residual disease. Despite these promising results, hematologic toxicity concerns may limit future trials.
“Outcomes for patients with acute myeloid leukemia (AML) remain poor, particularly in older patients,” wrote Amir T. Fathi, MD, of the division of hematology and oncology at Massachusetts General Hospital Cancer Center, Boston, and his coauthors.
Many elderly patients currently receive hypomethylating agents HMAs as a form of low-intensity therapy, but associated remission rates are low. “The development of novel, well-tolerated therapies to enhance the efficacy of HMAs could meaningfully improve the standard of care for older patients with AML,” the investigators wrote in Blood. Vadastuximab talirine is a novel antibody therapy that targets CD33; preclinical data suggested that it could be an effective combination with HMA therapy.
The phase 1 trial involved 53 patients with newly diagnosed, CD33-positive AML and a median age of 75 years. Patients were naive to HMA therapy but could have previously received other low-intensity treatments. HMA therapy was administered first; either azacitidine (75 mg/m2 subcutaneous IV for 7 days) or decitabine (20 mg/m2 IV for 5 days), according to institutional standards. On the last day of HMA therapy, vadastuximab talirine (10 mcg/kg IV) was given. This protocol was repeated in 28-day cycles for up to four cycles. Patients who tolerated the combination and showed a clinical response were eligible to continue therapy.
The composite remission rate (CRc: complete remission and complete remission with incomplete blood count recovery) with combination therapy was 70%. Historically, HMA monotherapies have much lower composite remission rates (decitabine, 17.8%; azacytidine, 27.8%). Of all patients achieving remission, 51% tested negative by flow cytometry for minimal residual disease. Median overall survival was 11.3 months and median relapse-free survival was 7.7 months.
“Nevertheless, the increased response rate with the addition of vadastuximab talirine to HMAs was also associated with increased toxicity when compared to single-agent HMA therapy – indicative of the greater degree of myelosuppression,” the researchers wrote. The most common grade 3 or higher adverse events were thrombocytopenia (57%), febrile neutropenia (49%), anemia (45%), neutropenia (42%), and fatigue (15%).
The investigators stated that “the overall safety profile was similar for patients treated with vadastuximab talirine in combination with azacitidine versus decitabine (with the exception of incidence of febrile neutropenia).”
Following the encouraging results of this phase 1 trial, the CASCADE phase 3 trial was launched to again compare this combination with HMA monotherapy; however, the trial was halted early because of deaths in the combination arm. The investigators cited the need for stricter protocols to ensure safety during future trials.
“With such guidance and precaution, promising combinations for AML, a disease affecting predominantly older and more frail patients, may be more effectively studied so as to enhance our current suboptimal therapeutic options,” they wrote.
Seattle Genetics provided study funding and author compensation.
SOURCE: Fathi AT et al. Blood. 2018 Jul 25. doi: 10.1182/blood-2018-03-841171.
For elderly patients with CD33-positive acute myeloid leukemia (AML), vadastuximab talirine in combination with a hypomethylating agent (HMA) improves remission rates, compared with HMA therapy alone, according to a phase 1 trial.
More than half of the patients treated with combination therapy achieved deep remission, defined as a negative-flow cytometry test for minimal residual disease. Despite these promising results, hematologic toxicity concerns may limit future trials.
“Outcomes for patients with acute myeloid leukemia (AML) remain poor, particularly in older patients,” wrote Amir T. Fathi, MD, of the division of hematology and oncology at Massachusetts General Hospital Cancer Center, Boston, and his coauthors.
Many elderly patients currently receive hypomethylating agents HMAs as a form of low-intensity therapy, but associated remission rates are low. “The development of novel, well-tolerated therapies to enhance the efficacy of HMAs could meaningfully improve the standard of care for older patients with AML,” the investigators wrote in Blood. Vadastuximab talirine is a novel antibody therapy that targets CD33; preclinical data suggested that it could be an effective combination with HMA therapy.
The phase 1 trial involved 53 patients with newly diagnosed, CD33-positive AML and a median age of 75 years. Patients were naive to HMA therapy but could have previously received other low-intensity treatments. HMA therapy was administered first; either azacitidine (75 mg/m2 subcutaneous IV for 7 days) or decitabine (20 mg/m2 IV for 5 days), according to institutional standards. On the last day of HMA therapy, vadastuximab talirine (10 mcg/kg IV) was given. This protocol was repeated in 28-day cycles for up to four cycles. Patients who tolerated the combination and showed a clinical response were eligible to continue therapy.
The composite remission rate (CRc: complete remission and complete remission with incomplete blood count recovery) with combination therapy was 70%. Historically, HMA monotherapies have much lower composite remission rates (decitabine, 17.8%; azacytidine, 27.8%). Of all patients achieving remission, 51% tested negative by flow cytometry for minimal residual disease. Median overall survival was 11.3 months and median relapse-free survival was 7.7 months.
“Nevertheless, the increased response rate with the addition of vadastuximab talirine to HMAs was also associated with increased toxicity when compared to single-agent HMA therapy – indicative of the greater degree of myelosuppression,” the researchers wrote. The most common grade 3 or higher adverse events were thrombocytopenia (57%), febrile neutropenia (49%), anemia (45%), neutropenia (42%), and fatigue (15%).
The investigators stated that “the overall safety profile was similar for patients treated with vadastuximab talirine in combination with azacitidine versus decitabine (with the exception of incidence of febrile neutropenia).”
Following the encouraging results of this phase 1 trial, the CASCADE phase 3 trial was launched to again compare this combination with HMA monotherapy; however, the trial was halted early because of deaths in the combination arm. The investigators cited the need for stricter protocols to ensure safety during future trials.
“With such guidance and precaution, promising combinations for AML, a disease affecting predominantly older and more frail patients, may be more effectively studied so as to enhance our current suboptimal therapeutic options,” they wrote.
Seattle Genetics provided study funding and author compensation.
SOURCE: Fathi AT et al. Blood. 2018 Jul 25. doi: 10.1182/blood-2018-03-841171.
For elderly patients with CD33-positive acute myeloid leukemia (AML), vadastuximab talirine in combination with a hypomethylating agent (HMA) improves remission rates, compared with HMA therapy alone, according to a phase 1 trial.
More than half of the patients treated with combination therapy achieved deep remission, defined as a negative-flow cytometry test for minimal residual disease. Despite these promising results, hematologic toxicity concerns may limit future trials.
“Outcomes for patients with acute myeloid leukemia (AML) remain poor, particularly in older patients,” wrote Amir T. Fathi, MD, of the division of hematology and oncology at Massachusetts General Hospital Cancer Center, Boston, and his coauthors.
Many elderly patients currently receive hypomethylating agents HMAs as a form of low-intensity therapy, but associated remission rates are low. “The development of novel, well-tolerated therapies to enhance the efficacy of HMAs could meaningfully improve the standard of care for older patients with AML,” the investigators wrote in Blood. Vadastuximab talirine is a novel antibody therapy that targets CD33; preclinical data suggested that it could be an effective combination with HMA therapy.
The phase 1 trial involved 53 patients with newly diagnosed, CD33-positive AML and a median age of 75 years. Patients were naive to HMA therapy but could have previously received other low-intensity treatments. HMA therapy was administered first; either azacitidine (75 mg/m2 subcutaneous IV for 7 days) or decitabine (20 mg/m2 IV for 5 days), according to institutional standards. On the last day of HMA therapy, vadastuximab talirine (10 mcg/kg IV) was given. This protocol was repeated in 28-day cycles for up to four cycles. Patients who tolerated the combination and showed a clinical response were eligible to continue therapy.
The composite remission rate (CRc: complete remission and complete remission with incomplete blood count recovery) with combination therapy was 70%. Historically, HMA monotherapies have much lower composite remission rates (decitabine, 17.8%; azacytidine, 27.8%). Of all patients achieving remission, 51% tested negative by flow cytometry for minimal residual disease. Median overall survival was 11.3 months and median relapse-free survival was 7.7 months.
“Nevertheless, the increased response rate with the addition of vadastuximab talirine to HMAs was also associated with increased toxicity when compared to single-agent HMA therapy – indicative of the greater degree of myelosuppression,” the researchers wrote. The most common grade 3 or higher adverse events were thrombocytopenia (57%), febrile neutropenia (49%), anemia (45%), neutropenia (42%), and fatigue (15%).
The investigators stated that “the overall safety profile was similar for patients treated with vadastuximab talirine in combination with azacitidine versus decitabine (with the exception of incidence of febrile neutropenia).”
Following the encouraging results of this phase 1 trial, the CASCADE phase 3 trial was launched to again compare this combination with HMA monotherapy; however, the trial was halted early because of deaths in the combination arm. The investigators cited the need for stricter protocols to ensure safety during future trials.
“With such guidance and precaution, promising combinations for AML, a disease affecting predominantly older and more frail patients, may be more effectively studied so as to enhance our current suboptimal therapeutic options,” they wrote.
Seattle Genetics provided study funding and author compensation.
SOURCE: Fathi AT et al. Blood. 2018 Jul 25. doi: 10.1182/blood-2018-03-841171.
FROM BLOOD
Key clinical point:
Major finding: The composite remission rate in patients treated with vadastuximab talirine and HMA therapy was 70%, compared with 17.8%-27.8% for patients treated with HMA therapy alone historically.
Study details: A prospective, phase 1 trial involving 53 elderly patients with CD33-positive AML at 14 treatment centers.
Disclosures: Seattle Genetics provided study funding and author compensation.
Source: Fathi AT et al. Blood. 2018 Jul 25. doi: 10.1182/blood-2018-03-841171.
Adult CCSs report financial hardships

Health-related financial hardship is common among adult survivors of childhood cancer, according to a study published in the Journal of the National Cancer Institute.
Researchers analyzed more than 2800 long-term childhood cancer survivors (CCSs) and found that 65% had financial challenges related to their cancer diagnosis.
“These findings suggest primary care doctors and oncologists should routinely screen childhood cancer survivors for possible financial hardship,” said I-Chan Huang, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
Specifically, Dr Huang recommends that healthcare providers routinely ask CCSs if they are unable to purchase medications, ever skip appointments for economic reasons, or worry about how to pay their medical bills.
For this study, Dr Huang and his colleagues analyzed data from 2811 CCSs. The subjects had a mean age of 31.8 (range, 18 to 65) and were a mean of 23.6 years from cancer diagnosis. Most (57.8%) had been diagnosed with hematologic malignancies, 32.0% with solid tumors, and 10.1% with central nervous system malignancies.
All subjects had been treated at St. Jude and enrolled in the St. Jude LIFE study. Participants return to St. Jude periodically for several days of clinical and functional assessments. Data for this study were collected during the CCSs’ first St. Jude LIFE evaluations.
Assessing hardship
The researchers measured 3 types of financial hardship—material, psychological, and coping/behavioral.
About 1 in 5 CCSs (22.4%) reported material financial hardship. In other words, their cancer had an impact on their financial situation.
More than half of CCSs (51.1%) reported psychological hardship—concern about their ability to pay for medical expenses.
And 33% of CCSs reported coping/behavioral hardship—an inability to see a doctor or go to the hospital due to finances.
Roughly 65% of CCSs reported at least 1 type of financial hardship.
All 3 types of hardship were significantly associated with somatization (all P<0.001), anxiety (all P<0.001), depression (all P<0.001), suicidal thoughts (all P<0.05), and difficulty in retirement planning (all P<0.001).
Furthermore, CCSs who reported financial hardship had significantly lower health-related quality of life (P<0.001 for all 3 domains), sensation abnormality (all P<0.001), pulmonary symptoms (all P<0.05), and cardiac symptoms (all P<0.05).
Predicting hardship
Intensive cancer treatment, chronic health conditions, second cancers, age at the time of study evaluation, education level, and annual household income were all significantly associated with a greater risk of financial hardship.
CCSs age 40 and older had an increased risk of psychological and coping/behavioral hardship (P<0.001 for both domains).
CCSs with an annual household income of less than $40,000 had an increased risk of material, psychological, and coping/behavioral hardship, compared to CCSs with an income of $80,000 or more (P<0.001 for all domains).
CCSs who did not obtain a high school diploma had an increased risk of material (P<0.001), psychological (P<0.01), and coping/behavioral hardship (P<0.001) compared to college graduates.
CCSs who received cancer treatments associated with a high-risk disease burden (vs low-risk) had an increased risk of material (P=0.01) and psychological (P=0.004) hardship.
Health conditions associated with material financial hardship included grade 2-4 myocardial infarction (P<0.001), peripheral neuropathy (P<0.001), subsequent neoplasm (P<0.001), seizure (P=0.007), reproductive disorders (P=0.01), stroke (P=0.02), amputation (P=0.02), upper gastrointestinal disease (P=0.04), and hearing loss (P=0.05).
Grade 2-4 myocardial infarction and reproductive disorders were significantly associated with psychological financial hardship (P=0.02 for both).
“Severe late effects that emerge early in life and disrupt education and training opportunities are a double hit for survivors,” Dr Huang said. “These health problems decrease the survivors’ earning mobility and financial security later in life. The phenomenon leaves them at risk for poor health and psychological outcomes compared to healthier survivors.”
Health-related financial hardship is common among adult survivors of childhood cancer, according to a study published in the Journal of the National Cancer Institute.
Researchers analyzed more than 2800 long-term childhood cancer survivors (CCSs) and found that 65% had financial challenges related to their cancer diagnosis.
“These findings suggest primary care doctors and oncologists should routinely screen childhood cancer survivors for possible financial hardship,” said I-Chan Huang, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
Specifically, Dr Huang recommends that healthcare providers routinely ask CCSs if they are unable to purchase medications, ever skip appointments for economic reasons, or worry about how to pay their medical bills.
For this study, Dr Huang and his colleagues analyzed data from 2811 CCSs. The subjects had a mean age of 31.8 (range, 18 to 65) and were a mean of 23.6 years from cancer diagnosis. Most (57.8%) had been diagnosed with hematologic malignancies, 32.0% with solid tumors, and 10.1% with central nervous system malignancies.
All subjects had been treated at St. Jude and enrolled in the St. Jude LIFE study. Participants return to St. Jude periodically for several days of clinical and functional assessments. Data for this study were collected during the CCSs’ first St. Jude LIFE evaluations.
Assessing hardship
The researchers measured 3 types of financial hardship—material, psychological, and coping/behavioral.
About 1 in 5 CCSs (22.4%) reported material financial hardship. In other words, their cancer had an impact on their financial situation.
More than half of CCSs (51.1%) reported psychological hardship—concern about their ability to pay for medical expenses.
And 33% of CCSs reported coping/behavioral hardship—an inability to see a doctor or go to the hospital due to finances.
Roughly 65% of CCSs reported at least 1 type of financial hardship.
All 3 types of hardship were significantly associated with somatization (all P<0.001), anxiety (all P<0.001), depression (all P<0.001), suicidal thoughts (all P<0.05), and difficulty in retirement planning (all P<0.001).
Furthermore, CCSs who reported financial hardship had significantly lower health-related quality of life (P<0.001 for all 3 domains), sensation abnormality (all P<0.001), pulmonary symptoms (all P<0.05), and cardiac symptoms (all P<0.05).
Predicting hardship
Intensive cancer treatment, chronic health conditions, second cancers, age at the time of study evaluation, education level, and annual household income were all significantly associated with a greater risk of financial hardship.
CCSs age 40 and older had an increased risk of psychological and coping/behavioral hardship (P<0.001 for both domains).
CCSs with an annual household income of less than $40,000 had an increased risk of material, psychological, and coping/behavioral hardship, compared to CCSs with an income of $80,000 or more (P<0.001 for all domains).
CCSs who did not obtain a high school diploma had an increased risk of material (P<0.001), psychological (P<0.01), and coping/behavioral hardship (P<0.001) compared to college graduates.
CCSs who received cancer treatments associated with a high-risk disease burden (vs low-risk) had an increased risk of material (P=0.01) and psychological (P=0.004) hardship.
Health conditions associated with material financial hardship included grade 2-4 myocardial infarction (P<0.001), peripheral neuropathy (P<0.001), subsequent neoplasm (P<0.001), seizure (P=0.007), reproductive disorders (P=0.01), stroke (P=0.02), amputation (P=0.02), upper gastrointestinal disease (P=0.04), and hearing loss (P=0.05).
Grade 2-4 myocardial infarction and reproductive disorders were significantly associated with psychological financial hardship (P=0.02 for both).
“Severe late effects that emerge early in life and disrupt education and training opportunities are a double hit for survivors,” Dr Huang said. “These health problems decrease the survivors’ earning mobility and financial security later in life. The phenomenon leaves them at risk for poor health and psychological outcomes compared to healthier survivors.”
Health-related financial hardship is common among adult survivors of childhood cancer, according to a study published in the Journal of the National Cancer Institute.
Researchers analyzed more than 2800 long-term childhood cancer survivors (CCSs) and found that 65% had financial challenges related to their cancer diagnosis.
“These findings suggest primary care doctors and oncologists should routinely screen childhood cancer survivors for possible financial hardship,” said I-Chan Huang, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
Specifically, Dr Huang recommends that healthcare providers routinely ask CCSs if they are unable to purchase medications, ever skip appointments for economic reasons, or worry about how to pay their medical bills.
For this study, Dr Huang and his colleagues analyzed data from 2811 CCSs. The subjects had a mean age of 31.8 (range, 18 to 65) and were a mean of 23.6 years from cancer diagnosis. Most (57.8%) had been diagnosed with hematologic malignancies, 32.0% with solid tumors, and 10.1% with central nervous system malignancies.
All subjects had been treated at St. Jude and enrolled in the St. Jude LIFE study. Participants return to St. Jude periodically for several days of clinical and functional assessments. Data for this study were collected during the CCSs’ first St. Jude LIFE evaluations.
Assessing hardship
The researchers measured 3 types of financial hardship—material, psychological, and coping/behavioral.
About 1 in 5 CCSs (22.4%) reported material financial hardship. In other words, their cancer had an impact on their financial situation.
More than half of CCSs (51.1%) reported psychological hardship—concern about their ability to pay for medical expenses.
And 33% of CCSs reported coping/behavioral hardship—an inability to see a doctor or go to the hospital due to finances.
Roughly 65% of CCSs reported at least 1 type of financial hardship.
All 3 types of hardship were significantly associated with somatization (all P<0.001), anxiety (all P<0.001), depression (all P<0.001), suicidal thoughts (all P<0.05), and difficulty in retirement planning (all P<0.001).
Furthermore, CCSs who reported financial hardship had significantly lower health-related quality of life (P<0.001 for all 3 domains), sensation abnormality (all P<0.001), pulmonary symptoms (all P<0.05), and cardiac symptoms (all P<0.05).
Predicting hardship
Intensive cancer treatment, chronic health conditions, second cancers, age at the time of study evaluation, education level, and annual household income were all significantly associated with a greater risk of financial hardship.
CCSs age 40 and older had an increased risk of psychological and coping/behavioral hardship (P<0.001 for both domains).
CCSs with an annual household income of less than $40,000 had an increased risk of material, psychological, and coping/behavioral hardship, compared to CCSs with an income of $80,000 or more (P<0.001 for all domains).
CCSs who did not obtain a high school diploma had an increased risk of material (P<0.001), psychological (P<0.01), and coping/behavioral hardship (P<0.001) compared to college graduates.
CCSs who received cancer treatments associated with a high-risk disease burden (vs low-risk) had an increased risk of material (P=0.01) and psychological (P=0.004) hardship.
Health conditions associated with material financial hardship included grade 2-4 myocardial infarction (P<0.001), peripheral neuropathy (P<0.001), subsequent neoplasm (P<0.001), seizure (P=0.007), reproductive disorders (P=0.01), stroke (P=0.02), amputation (P=0.02), upper gastrointestinal disease (P=0.04), and hearing loss (P=0.05).
Grade 2-4 myocardial infarction and reproductive disorders were significantly associated with psychological financial hardship (P=0.02 for both).
“Severe late effects that emerge early in life and disrupt education and training opportunities are a double hit for survivors,” Dr Huang said. “These health problems decrease the survivors’ earning mobility and financial security later in life. The phenomenon leaves them at risk for poor health and psychological outcomes compared to healthier survivors.”
Inhibitor receives breakthrough designation for AML

The US Food and Drug Administration (FDA) has granted breakthrough therapy designation to quizartinib, an investigational FLT3 inhibitor, for the treatment of adults with relapsed/refractory FLT3-ITD acute myeloid leukemia (AML).
The FDA granted quizartinib breakthrough designation based on results from the phase 3 QuANTUM-R study, which were presented at the 23rd Congress of the European Hematology Association in June.
QuANTUM-R enrolled adults with FLT3-ITD AML (at least 3% FLT3-ITD allelic ratio) who had refractory disease or had relapsed within 6 months of their first complete remission.
Patients were randomized to receive once-daily treatment with quizartinib (n=245) or a salvage chemotherapy regimen (n=122)—low-dose cytarabine (LoDAC, n=29); combination mitoxantrone, etoposide, and cytarabine (MEC, n=40); or combination fludarabine, cytarabine, and idarubicin (FLAG-IDA, n=53).
Responders could proceed to hematopoietic stem cell transplant (HSCT), and those in the quizartinib arm could resume quizartinib after HSCT. Thirty-two percent of quizartinib-treated patients and 12% of the chemotherapy group went on to HSCT.
The median follow-up was 23.5 months. The efficacy results include all randomized patients.
The overall response rate was 69% in the quizartinib arm and 30% in the chemotherapy arm. The complete response (CR) rate was 4% and 1%, respectively; the rate of CR with incomplete platelet recovery was 4% and 0%, respectively; and the rate of CR with incomplete hematologic recovery was 40% and 26%, respectively.
The median overall survival was 6.2 months in the quizartinib arm and 4.7 months in the chemotherapy arm (hazard ratio=0.76, P=0.0177). The 1-year overall survival rate was 27% and 20%, respectively.
The median event-free survival was 6.0 weeks in the quizartinib arm and 3.7 weeks in the chemotherapy arm (hazard ratio=0.90, P=0.1071).
The safety results include only patients who received their assigned treatment—241 patients who received quizartinib and 94 who received salvage chemotherapy (22 on LoDAC, 25 on MEC, and 47 on FLAG-IDA).
Grade 3 or higher hematologic treatment-emergent adverse events occurring in at least 5% of patients (in the quizartinib and chemotherapy groups, respectively) included thrombocytopenia (35% and 34%), anemia (30% and 29%), neutropenia (32% and 25%), febrile neutropenia (31% and 21%), and leukopenia (17% and 16%).
Grade 3 or higher nonhematologic treatment-emergent adverse events occurring in at least 5% of patients (in the quizartinib and chemotherapy groups, respectively) included fatigue (8% and 1%), hypokalemia (12% and 9%), sepsis/septic shock (16% and 18%), dyspnea (5% for both), hypophosphatemia (5% for both), and pneumonia (12% and 9%).
Three percent of patients in the quizartinib arm had grade 3 QTcF prolongation, and 2 patients discontinued quizartinib due to QTcF prolongation.
About breakthrough designation
Breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
The designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
Other designations for quizartinib
In addition to breakthrough therapy designation, quizartinib has fast track and orphan drug designations from the FDA.
The FDA’s fast track development program is designed to expedite clinical development and submission of applications for products with the potential to treat serious or life-threatening conditions and address unmet medical needs.
Fast track designation facilitates frequent interactions with the FDA review team, including meetings to discuss the product’s development plan and written communications about issues such as trial design and use of biomarkers.
Products that receive fast track designation may be eligible for accelerated approval and priority review if relevant criteria are met. Such products may also be eligible for rolling review, which allows a developer to submit individual sections of a product’s application for review as they are ready, rather than waiting until all sections are complete.
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
Orphan designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation to quizartinib, an investigational FLT3 inhibitor, for the treatment of adults with relapsed/refractory FLT3-ITD acute myeloid leukemia (AML).
The FDA granted quizartinib breakthrough designation based on results from the phase 3 QuANTUM-R study, which were presented at the 23rd Congress of the European Hematology Association in June.
QuANTUM-R enrolled adults with FLT3-ITD AML (at least 3% FLT3-ITD allelic ratio) who had refractory disease or had relapsed within 6 months of their first complete remission.
Patients were randomized to receive once-daily treatment with quizartinib (n=245) or a salvage chemotherapy regimen (n=122)—low-dose cytarabine (LoDAC, n=29); combination mitoxantrone, etoposide, and cytarabine (MEC, n=40); or combination fludarabine, cytarabine, and idarubicin (FLAG-IDA, n=53).
Responders could proceed to hematopoietic stem cell transplant (HSCT), and those in the quizartinib arm could resume quizartinib after HSCT. Thirty-two percent of quizartinib-treated patients and 12% of the chemotherapy group went on to HSCT.
The median follow-up was 23.5 months. The efficacy results include all randomized patients.
The overall response rate was 69% in the quizartinib arm and 30% in the chemotherapy arm. The complete response (CR) rate was 4% and 1%, respectively; the rate of CR with incomplete platelet recovery was 4% and 0%, respectively; and the rate of CR with incomplete hematologic recovery was 40% and 26%, respectively.
The median overall survival was 6.2 months in the quizartinib arm and 4.7 months in the chemotherapy arm (hazard ratio=0.76, P=0.0177). The 1-year overall survival rate was 27% and 20%, respectively.
The median event-free survival was 6.0 weeks in the quizartinib arm and 3.7 weeks in the chemotherapy arm (hazard ratio=0.90, P=0.1071).
The safety results include only patients who received their assigned treatment—241 patients who received quizartinib and 94 who received salvage chemotherapy (22 on LoDAC, 25 on MEC, and 47 on FLAG-IDA).
Grade 3 or higher hematologic treatment-emergent adverse events occurring in at least 5% of patients (in the quizartinib and chemotherapy groups, respectively) included thrombocytopenia (35% and 34%), anemia (30% and 29%), neutropenia (32% and 25%), febrile neutropenia (31% and 21%), and leukopenia (17% and 16%).
Grade 3 or higher nonhematologic treatment-emergent adverse events occurring in at least 5% of patients (in the quizartinib and chemotherapy groups, respectively) included fatigue (8% and 1%), hypokalemia (12% and 9%), sepsis/septic shock (16% and 18%), dyspnea (5% for both), hypophosphatemia (5% for both), and pneumonia (12% and 9%).
Three percent of patients in the quizartinib arm had grade 3 QTcF prolongation, and 2 patients discontinued quizartinib due to QTcF prolongation.
About breakthrough designation
Breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
The designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
Other designations for quizartinib
In addition to breakthrough therapy designation, quizartinib has fast track and orphan drug designations from the FDA.
The FDA’s fast track development program is designed to expedite clinical development and submission of applications for products with the potential to treat serious or life-threatening conditions and address unmet medical needs.
Fast track designation facilitates frequent interactions with the FDA review team, including meetings to discuss the product’s development plan and written communications about issues such as trial design and use of biomarkers.
Products that receive fast track designation may be eligible for accelerated approval and priority review if relevant criteria are met. Such products may also be eligible for rolling review, which allows a developer to submit individual sections of a product’s application for review as they are ready, rather than waiting until all sections are complete.
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
Orphan designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation to quizartinib, an investigational FLT3 inhibitor, for the treatment of adults with relapsed/refractory FLT3-ITD acute myeloid leukemia (AML).
The FDA granted quizartinib breakthrough designation based on results from the phase 3 QuANTUM-R study, which were presented at the 23rd Congress of the European Hematology Association in June.
QuANTUM-R enrolled adults with FLT3-ITD AML (at least 3% FLT3-ITD allelic ratio) who had refractory disease or had relapsed within 6 months of their first complete remission.
Patients were randomized to receive once-daily treatment with quizartinib (n=245) or a salvage chemotherapy regimen (n=122)—low-dose cytarabine (LoDAC, n=29); combination mitoxantrone, etoposide, and cytarabine (MEC, n=40); or combination fludarabine, cytarabine, and idarubicin (FLAG-IDA, n=53).
Responders could proceed to hematopoietic stem cell transplant (HSCT), and those in the quizartinib arm could resume quizartinib after HSCT. Thirty-two percent of quizartinib-treated patients and 12% of the chemotherapy group went on to HSCT.
The median follow-up was 23.5 months. The efficacy results include all randomized patients.
The overall response rate was 69% in the quizartinib arm and 30% in the chemotherapy arm. The complete response (CR) rate was 4% and 1%, respectively; the rate of CR with incomplete platelet recovery was 4% and 0%, respectively; and the rate of CR with incomplete hematologic recovery was 40% and 26%, respectively.
The median overall survival was 6.2 months in the quizartinib arm and 4.7 months in the chemotherapy arm (hazard ratio=0.76, P=0.0177). The 1-year overall survival rate was 27% and 20%, respectively.
The median event-free survival was 6.0 weeks in the quizartinib arm and 3.7 weeks in the chemotherapy arm (hazard ratio=0.90, P=0.1071).
The safety results include only patients who received their assigned treatment—241 patients who received quizartinib and 94 who received salvage chemotherapy (22 on LoDAC, 25 on MEC, and 47 on FLAG-IDA).
Grade 3 or higher hematologic treatment-emergent adverse events occurring in at least 5% of patients (in the quizartinib and chemotherapy groups, respectively) included thrombocytopenia (35% and 34%), anemia (30% and 29%), neutropenia (32% and 25%), febrile neutropenia (31% and 21%), and leukopenia (17% and 16%).
Grade 3 or higher nonhematologic treatment-emergent adverse events occurring in at least 5% of patients (in the quizartinib and chemotherapy groups, respectively) included fatigue (8% and 1%), hypokalemia (12% and 9%), sepsis/septic shock (16% and 18%), dyspnea (5% for both), hypophosphatemia (5% for both), and pneumonia (12% and 9%).
Three percent of patients in the quizartinib arm had grade 3 QTcF prolongation, and 2 patients discontinued quizartinib due to QTcF prolongation.
About breakthrough designation
Breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
The designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
Other designations for quizartinib
In addition to breakthrough therapy designation, quizartinib has fast track and orphan drug designations from the FDA.
The FDA’s fast track development program is designed to expedite clinical development and submission of applications for products with the potential to treat serious or life-threatening conditions and address unmet medical needs.
Fast track designation facilitates frequent interactions with the FDA review team, including meetings to discuss the product’s development plan and written communications about issues such as trial design and use of biomarkers.
Products that receive fast track designation may be eligible for accelerated approval and priority review if relevant criteria are met. Such products may also be eligible for rolling review, which allows a developer to submit individual sections of a product’s application for review as they are ready, rather than waiting until all sections are complete.
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
Orphan designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
Acute Myeloid Leukemia
Introduction
Acute myeloid leukemia (AML) comprises a heterogeneous group of disorders characterized by proliferation of clonal, abnormally differentiated hematopoietic progenitor cells of myeloid lineage that infiltrate the bone marrow, blood, and other tissues.1 In most cases, AML is rapidly fatal if left untreated. Over the past 2 decades, our understanding of the underlying disease biology responsible for the development of AML has improved substantially. We have learned that biological differences drive the various clinical, cytogenetic, and molecular subentities of AML; distinguishing among these subentities helps to identify optimal therapies, while offering improved clinical outcomes for select groups. After years of stagnation in therapeutic advances, 4 new drugs for treating AML were approved by the US Food and Drug Administration (FDA) in 2017. In this article, we review key features of AML diagnosis and management in the context of 2 case presentations.
Epidemiology and Risk Factors
An estimated 21,380 new cases of AML were diagnosed in the United States in 2017, constituting roughly 1.3% of all new cases of cancer.2 Approximately 10,590 patients died of AML in 2017. The median age of patients at the time of diagnosis is 68 years, and the incidence is approximately 4.2 per 100,000 persons per year. The 5-year survival for AML has steadily risen from a meager 6.3% in 1975 to 17.3% in 1995 and 28.1% in 2009.2 The cure rates for AML vary drastically with age. Long-term survival is achieved in approximately 35% to 40% of adults who present at age 60 years or younger, but only 5% to 15% of those older than 60 years at presentation will achieve long-term survival.3
Most cases of AML occur in the absence of any known risk factors. High-dose radiation exposure, chronic benzene exposure, chronic tobacco smoking, and certain chemotherapeutics are known to increase the risk for AML.4 Inconsistent correlations have also been made between exposure to organic solvents, petroleum products, radon, pesticides, and herbicides and the development of AML.4 Obesity may also increase AML risk.4
Two distinct subcategories of therapy-related AML (t-AML) are known. Patients who have been exposed to alkylating chemotherapeutics (eg, melphalan, cyclophosphamide, and nitrogen mustard) can develop t-AML with chromosomal 5 and/or 7 abnormalities after a latency period of approximately 4 to 8 years.5 In contrast, patients exposed to topoisomerase II inhibitors (notably etoposide) develop AML with abnormalities of 11q23 (leading to MLL gene rearrangement) or 21q22 (RUNX1) after a latency period of about 1 to 3 years.6 AML can also arise out of other myeloid disorders such as myelodysplastic syndrome and myeloproliferative neoplasms, and other bone marrow failure syndromes such as aplastic anemia.4 Various inherited or congenital conditions such as Down syndrome, Bloom syndrome, Fanconi anemia, neurofibromatosis 1, and dyskeratosis congenita can also predispose to the development of AML. A more detailed listing of conditions associated with AML can be found elsewhere.4
Molecular Landscape
The first cancer genome sequence was reported in an AML patient in 2008.7 Since then, various elegantly conducted studies have expanded our understanding of the molecular abnormalities in AML. The Cancer Genome Atlas Research Network analyzed the genomes of 200 cases of de novo AML in adults.8 Only 13 mutations were found on average, much fewer than the number of mutations in most adult cancers. Twenty-three genes were commonly mutated, and another 237 were mutated in 2 or more cases. Essentially, all cases had at least 1 nonsynonymous mutation in 1 of 9 categories of genes: transcription-factor fusions (18%), the gene encoding nucleophosmin (NPM1) (27%), tumor-suppressor genes (16%), DNA-methylation–related genes (44%), signaling genes (59%), chromatin-modifying genes (30%), myeloid transcription-factor genes (22%), spliceosome-complex genes (14%), and cohesin-complex genes (13%).
In another study, samples from 1540 patients from 3 prospective trials of intensive chemotherapy were analyzed to understand how genetic diversity defines the pathophysiology of AML.9 The study authors identified 5234 driver mutations from 76 genes or genomic regions, with 2 or more drivers identified in 86% of the samples. Eleven classes of mutational events, each with distinct diagnostic features and clinical outcomes, were identified. Acting as an internal positive control in this analysis, previously recognized mutational and cytogenetic groups emerged as distinct entities, including the groups with biallelic CEBPA mutations, mutations in NPM1, MLL fusions, and the cytogenetic entities t(6;9), inv(3), t(8;21), t(15;17), and inv(16). Three additional categories emerged as distinct entities: AML with mutations in genes encoding chromatin, RNA splicing regulators, or both (18% of patients); AML with TP53 mutations, chromosomal aneuploidies, or both (13%); and, provisionally, AML with IDH2R172 mutations (1%). An additional level of complexity was also revealed within the subgroup of patients with NPM1 mutations, where gene–gene interactions identified co-mutational events associated with both favorable or adverse prognosis.
Further supporting this molecular classification of AML, a study that performed targeted mutational analysis of 194 patients with defined secondary AML (s-AML) or t-AML and 105 unselected AML patients found that the presence of mutations in SRSF2, SF3B1, U2AF1, ZRSR2, ASXL1, EZH2, BCOR, or STAG2 (all members of the chromatin or RNA splicing families) was highly specific for the diagnosis of s-AML.10 These findings are particularly clinically useful in those without a known history of antecedent hematologic disorder. These mutations defining the AML ontogeny were found to occur early in leukemogenesis, persist in clonal remissions, and predict worse clinical outcomes. Mutations in genes involved in regulation of DNA modification and of chromatin state (commonly DNMT3A, ASXL1, and TET2) have also been shown to be present in preleukemic stem or progenitor cells and to occur early in leukemogenesis.3 Unsurprisingly, some of these same mutations, including those in epigenetic regulators (DNMT3A, ASXL1, and TET2) and less frequently in splicing factor genes (SF3B1, SRSF2), have been associated with clonal hematopoietic expansion in elderly, seemingly healthy adults, a condition termed clonal hematopoiesis of indeterminate potential (CHIP).3,11,12 The presence of CHIP is associated with increased risk of hematologic neoplasms and all-cause mortality, the latter being possibly driven by a near doubling in the risk of coronary heart disease in humans and by accelerated atherosclerosis in a mouse model.11,13,14
Clinical Presentation and Work-up
Case Patient 1
A 57-year-old woman with a history of hypertension presents to the emergency department with complaints of productive cough and fevers for the previous 3 days. Examination reveals conjunctival pallor, gingival hyperplasia, and decreased breath sounds at the posterior right lung field. Investigations reveal a white blood cell (WBC) count of 51,000/µL with 15% blasts, a hemoglobin of 7.8 g/dL, and a platelet count of 56 × 103/µL. Peripheral blood smear is notable for large myeloblasts with occasional Auer rods. Chest radiograph shows a consolidation in the right lower lobe.
Case Patient 2
A 69-year-old man presents to his primary care physician for evaluation of worsening fatigue for the previous 4 months. Ten years prior to presentation, he had received 6 cycles of RCHOP (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone) as treatment for diffuse large B-cell lymphoma. Conjunctival pallor, patches of purpura over the extremities, and mucosal petechiae are noted on examination. Laboratory analyisis reveals a WBC count of 2400/µL with 12% blasts, hemoglobin of 9.0 g/dL, and platelet count of 10 × 103/µL. Peripheral smear shows dysplastic myeloid cells and blasts.
Clinical Features
Patients with AML typically present with features secondary to proliferation of blasts (ie, findings of bone marrow failure and end organ damage).4,5 Fatigue, pallor, dizziness, dyspnea, and headaches occur secondary to anemia. Easy and prolonged bruising, petechiae, epistaxis, gingival bleeding, and conjunctival hemorrhages result from thrombocytopenia. Bleeding from other sites such as the central nervous system and gastrointestinal tract occurs but is uncommon. Patients may also present with infections resulting from unrecognized neutropenia. Constitutional symptoms including anorexia, fevers, and weight loss are frequently reported, while organomegaly (hepatomegaly and/or splenomegaly) is seen in about a quarter of patients.4 Infiltration of blasts into almost every organ has been noted, a condition known as myeloid (or granulocytic) sarcoma.15 This condition is more commonly found in patients with blastic, monoblastic, or myelomonocytic variants of AML, and is known as isolated myeloid sarcoma if no concurrent marrow or blood involvement is identified. In the absence of induction chemotherapy, systemic involvement occurs in a matter of weeks to months following such presentation.16
Laboratory analysis will usually demonstrate derangements in peripheral blood cell lines. At least half of patients have a total WBC count less than 5000/µL, a platelet count less than 50 × 103/µL, or both at the time of diagnosis.4,17 Approximately 10% of patients present with hyperleukocytosis and a WBC count greater than 100,000/µL, which can be associated with leukostasis.5 Additionally, spontaneous electrolyte derangement consistent with tumor lysis syndrome and coagulation abnormalities found in disseminated intravascular coagulation may be noted, even before initiation of therapy.
Work-Up of Suspected AML
Bone marrow biopsy and aspirate, along with touch preparations of the core biopsy sample, are crucial in the workup of suspected AML. At least 200 WBCs on blood smears and 500 nucleated cells on spiculated marrow smears should be counted.3 Reactivity with specific histochemical stains (myeloperoxidase, Sudan black B, or naphthyl AS-D-chloroacetate), presence of Auer rods, and reactivity to monoclonal antibodies against epitopes present on myeloblasts (eg, CD13, CD33, CD117) help distinguish myeloblasts from lymphoblasts.4 Flow cytometric analysis helps in confirming myeloid lineage; blasts generally express CD34 and HLA-DR, markers of immature hematopoietic precursors, and dim CD45 (common leukocyte antigen). One or more lymphoid antigens may be aberrantly expressed as well. Of note, in about 2% to 3% of acute leukemia cases, immunohistochemistry and/or flow cytometry findings demonstrate immature cells with features of both myeloid and lymphoid lineages (biphenotypic) or different populations of myeloid and lymphoid leukemia cells (bilineal). These leukemias are termed mixed-phenotype acute leukemia and are typically treated with either AML or acute lymphoblastic leukemia regimens.18
Cytogenetics, as assessed through conventional karyotype and fluorescence in situ hybridization (FISH), constitutes an essential part of the work-up. Eight balanced translocations and inversions and their variants are included in the World Health Organization (WHO) category “AML with recurrent genetic abnormalities,” while 9 balanced rearrangements and multiple unbalanced abnormalities in the presence of a blast count ≥ 20% are sufficient to establish the diagnosis of “AML with myelodysplasia-related changes.”3,19 Various other gene rearrangements thought to represent disease-initiating events are recognized as well, but these rearrangements do not yet formally define WHO disease categories.3 FISH can help detect RUNX1-RUNX1T1, CBFB-MYH11, KMT2A (MLL), and MECOM (EVI1) gene fusions, as well as chromosomal changes like 5q, 7q, or 17p, especially when fewer than 20 metaphases are assessable (due to failure of culture) by conventional cytogenetic methods.3
As certain molecular markers help with disease prognosis and the selection of personalized therapies, testing for these markers is recommended as part of a complete work-up of AML. The current standard of care is to test for nucleophosmin (NPM1), fms-like tyrosine kinase 3 (FLT3), and CEBPA mutations in all newly diagnosed patients.1RUNX1 mutation analysis should also be considered as its presence defines a provisional WHO subcategory.19 In the case of FLT3, the analysis should include both internal tandem duplications (FLT3-ITD, associated with worse prognosis especially at high allelic ratio) and tyrosine-kinase domain mutations (FLT3-TKD; D835 and I836), especially now that FLT3 inhibitors are regularly used.20 Most academic centers now routinely use next-generation sequencing–based panels to assess multiple mutations. 
Diagnosis and Classification
A marrow or blood blast (myeloblasts, monoblasts, megakaryoblasts, or promonocytes [considered blast equivalents]) count of ≥ 20% is required for AML diagnosis.3,19 The presence of t(15;17), t(8;21), inv(16), or t(16;16), however, is considered diagnostic of AML irrespective of blast count.3,19 The previously used French-American-British (FAB) classification scheme has been replaced by the WHO classification (Table 2), which takes into account the morphologic, cytogenetic, genetic, and clinical features of the leukemia. 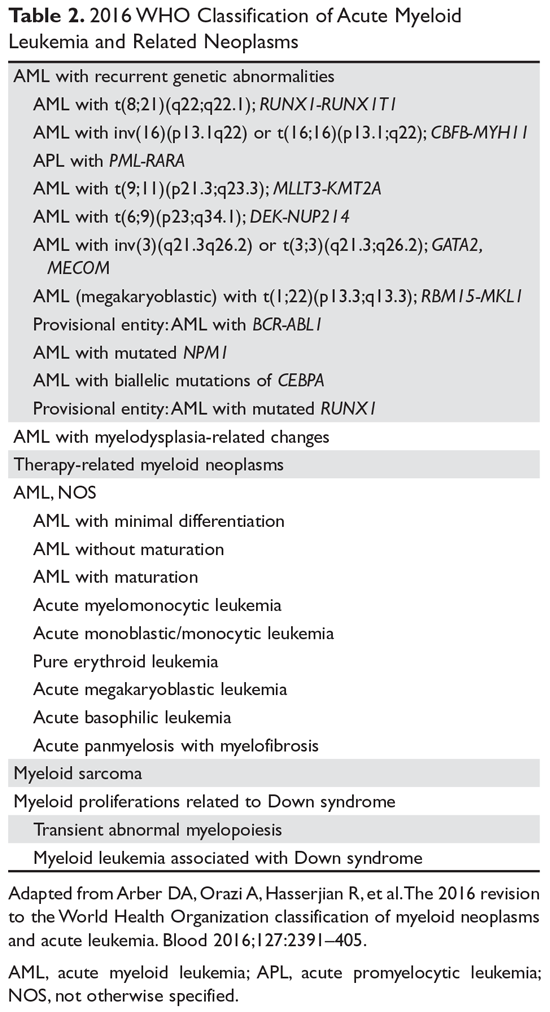
The category “AML with myelodysplasia-related changes” includes AML that has evolved out of an antecedent myelodysplastic syndrome, has ≥ 50% dysplasia in 2 or more lineages, or has myelodysplasia-related cytogenetic changes (eg, –5/del(5q), –7/del(7q), ≥ 3 cytogenetic abnormalities).19 “Therapy-related myeloid neoplasm,” or therapy-related AML, is diagnosed when the patient has previously received cytotoxic agents or ionizing radiation.19
Cases which do not meet the criteria for 1 of the previously mentioned categories are currently classified as “AML, not otherwise specified.” Further subclassification is pursued as per the older FAB scheme; however, no additional prognostic information is obtained in doing so.3,19 Myeloid sarcoma is strictly not a subcategory of AML. Rather, it is an extramedullary mass of myeloid blasts that effaces the normal tissue architecture.16 Rarely, myeloid sarcoma can be present without systemic disease involvement; it is important to note that management of such cases is identical to management of overt AML.16
Finally, myeloid proliferations related to Down syndrome include 2 entities seen in children with Down syndrome.19 Transient abnormal myelopoiesis, seen in 10% to 30% of newborns with Down syndrome, presents with circulating blasts that resolve in a couple of months. Myeloid leukemia associated with Down syndrome is AML that occurs usually in the first 3 years of life and persists if not treated.19
Case 1 Continued
The presence of 15% blasts in the peripheral blood is concerning for, but not diagnostic of, AML. On the other hand, the presence of Auer rods is virtually pathognomonic for AML. Gingival hyperplasia in this patient may be reflective of extramedullary disease. Cytogenetics from the peripheral blood and marrow aspirate show inv(16) in 20 of 20 cells. Molecular panel is notable for mutation in c-KIT. As such, the patient is diagnosed with core-binding factor AML, which per the ELN classification is considered a favorable-risk AML. The presence of c-KIT mutation, however, confers a relatively worse outcome.
Case 2 Continued
Presence of pancytopenia in a patient who previously received cytotoxic chemotherapy is highly concerning for therapy-related myeloid neoplasm. The presence of 12% blasts in the peripheral blood does not meet the criteria for diagnosis of AML. However, marrow specimens show 40% blasts, thus meeting the criteria for an AML diagnosis. Additionally, cytogenetics are notable for the presence of monosomy 7, while a next-generation sequencing panel shows a mutation in TP53. Put together, this patient meets the criteria for therapy-related AML which is an adverse-risk AML according to the ELN classification.
Management
The 2 most significant factors that must be considered when selecting AML therapies are the patient’s suitability for intensive chemotherapy and the biological characteristics of the AML. The former is a nuanced decision that incorporates age, performance status, and existing comorbidities. Treatment-related mortality calculators can guide physicians when making therapy decisions, especially in older patients (≥ 65 years). Retrospective evidence from various studies suggests that older, medically fit patients may derive clinically comparable benefits from intensive and less intensive induction therapies.25–27 The biological characteristics of the leukemia can be suggested by morphologic findings, cytogenetics, and molecular information, in addition to a history of antecedent myeloid neoplasms. Recently, an AML composite model incorporating an augmented Hematopoietic Cell Transplantation–specific Comorbidity Index (HCT-CI) score, age, and cytogenetic/molecular risks was shown to improve treatment decision-making about AML; this model potentially could be used to guide patient stratification in clinical trials as well.28 The overall treatment model of AML is largely unchanged otherwise. It is generally divided into induction, consolidation, and maintenance therapies.
Induction Therapy
In patients who can tolerate intensive therapies, the role of anthracycline- and cytarabine-based treatment is well established. However, the choice of specific anthracycline is not well established. One study concluded that idarubicin and mitoxantrone led to better outcomes as compared to daunorubicin, while another showed no difference between these agents.29,30 A pooled study of AML trials conducted in patients aged 50 years and older showed that while idarubicin led to a higher complete remission rate (69% versus 61%), the overall survival (OS) did not differ significantly.31 As for dosing, daunorubicin given at 45 mg/m2 daily for 3 days has been shown to have lower complete remission rates and higher relapse rates than a dose of 90 mg/m2 daily for 3 days in younger patients.32–34 However, it is not clear whether the 90 mg/m2 dose is superior to the frequently used dose of 60 mg/m2.35 A French study has shown comparable rates of complete remission, relapse, and OS between the 60 mg/m2 and 90 mg/m2 doses in patients with intermediate or unfavorable cytogenetics.36
If idarubicin is used, a dose of 12 mg/m2 for 3 days is considered the standard. In patients aged 50 to 70 years, there were no statistically significant differences in rates of relapse or OS between daunorubicin 80 mg/m2 for 3 days versus idarubicin 12 mg/m2 for 3 days versus idarubicin 12 mg/m2 for 4 days.37 As for cytarabine, the bulk of the evidence indicates that a dose of 1000 mg/m2 or higher should not be used.38 As such, the typical induction chemotherapy regimen of choice is 3 days of anthracycline (daunorubicin or idarubicin) and 7 days of cytarabine (100–200 mg/m2 continuous infusion), also known as the 7+3 regimen, which was first pioneered in the 1970s. In a recent phase 3 trial, 309 patients aged 60 to 75 years with high-risk AML (AML with myelodysplasia-related changes or t-AML) were randomly assigned to either the 7+3 regimen or CPX-351 (ie, nano-liposomal encapsulation of cytarabine and daunorubicin in a 5:1 molar ratio).39 A higher composite complete response rate (47.7% versus 33.3%; P = 0.016) and improved survival (9.56 months versus 5.95 months; hazard ratio [HR] 0.69, P = 0.005) were seen with CPX-351, leading to its approval by the FDA in patients with high-risk AML.
The 7+3 regimen has served as a backbone onto which other drugs have been added in clinical trials—the majority without any clinical benefits—for patients who can tolerate intensive therapy. In this context, the role of 2 therapies recently approved by the FDA must be discussed. In the RATIFY trial, 717 patients aged 18 to 59 years with AML and a FLT3 mutation were randomly assigned to receive standard chemotherapy (induction and consolidation therapy) plus either midostaurin or placebo; those who were in remission after consolidation therapy received either midostaurin or placebo in the maintenance phase.40 The primary endpoint was met as midostaurin improved OS (HR 0.78, P = 0.009). The benefit of midostaurin was consistent across all FLT3 subtypes and mutant allele burdens, regardless of whether patients proceeded to allogeneic stem cell transplant (allo-SCT). Based on the results of RATIFY, midostaurin was approved by the FDA for treatment of AML patients who are positive for the FLT3 mutation. Whether more potent and selective FLT3 inhibitors like gilteritinib, quizartinib, or crenolanib improve the outcomes is currently under investigation in various clinical trials.20
The development of gemtuzumab ozogamicin (GO) has been more complicated. GO, an antibody-drug conjugate comprised of a CD33-directed humanized monoclonal antibody linked covalently to the cytotoxic agent calicheamicin, binds CD33 present on the surface of myeloid leukemic blasts and immature normal cells of myelomonocytic lineage.41 The drug first received an accelerated approval in 2000 as monotherapy (2 doses of 9 mg/m2 14 days apart) for the treatment of patients 60 years of age and older with CD33-positive AML in first relapse based on the results of 3 open-label multicenter trials.41,42 However, a confirmatory S0106 trial in which GO 6 mg/m2 was added on day 4 in newly diagnosed AML patients was terminated early when an interim analysis showed an increased rate of death in induction (6% versus 1%) and lack of improvement in complete response, disease-free survival, or OS with the addition of GO.43 This study led to the withdrawal of GO from the US market in 2010. However, 2 randomized trials that studied GO using a different dose and schedule suggested that the addition of GO to intensive chemotherapy improved survival outcomes in patients with favorable and intermediate-risk cytogenetics.44,45 The results of the multicenter, open-label phase 3 ALFA-0701 trial, which randomly assigned 271 patients aged 50 to 70 years with newly diagnosed AML to daunorubicin and cytarabine alone or in combination with GO (3 mg/m2 on days 1, 4, and 7 during induction and day 1 of 2 consolidation courses), showed a statistically significant improvement in event-free survival (17.3 months versus 9.5 months; HR 0.56 [95% confidence interval 0.42 to 0.76]).45 Again, the survival benefits were more pronounced in patients with favorable or intermediate-risk cytogenetics than in those with unfavorable cytogenetics. The results of this trial led to the re-approval of GO in newly diagnosed AML patients.
For patients who cannot tolerate intensive therapies, the 2 main therapeutic options are low-dose cytarabine (LDAC) and the hypomethylating agents (HMA) azacitidine and decitabine. A phase 3 trial of decitabine versus mostly LDAC (or best supportive care, BSC) demonstrated favorable survival with decitabine (7.7 months versus 5.0 months).46 In the AZA-AML-001 trial, azacitidine improved median survival (10.4 months versus 6.5 months) in comparison to the control arm (LDAC, 7+3, BSC).47 Emerging data has also suggested that HMAs may be particularly active in patients with unfavorable-risk AML, a group for which LDAC has been shown to be especially useless.48 As such, HMA therapies are generally preferred over LDAC in practice. Finally, it is pertinent to note that GO can also be used as monotherapy based on the results of the open-label phase 3 AML-19 study in which GO demonstrated a survival advantage over BSC (4.9 months versus 3.6 months, P = 0.005).49
Postremission or Consolidation Therapy
There is no standard consolidation therapy for AML at present. In general, for patients who received HMA in the induction phase, the same HMA should be continued indefinitely until disease progression or allo-SCT.3 For those who received intensive chemotherapy in the induction phase, the consensus is to use cytarabine-based consolidation therapies. Cytarabine given as a single agent in high-doses has generally led to similar outcomes as multiagent chemotherapy.50 In this regard, cytarabine regimens, with or without anthracycline, at 3000 mg/m2 have similar efficacy as an intermediate dose of 1000 mg/m2.38 A total of 2 to 4 cycles of post-remission therapy is considered standard.3 Intensified post-remission chemotherapy has not been associated with consistent benefit in older AML patients or those with poor-risk disease. In recent years, measurable residual disease (MRD) assessment has emerged as a potentially useful tool in risk stratification and treatment planning, with various studies suggesting that MRD status in complete remission is one of the most important prognostic factors.51 Prospective studies confirming the significance of MRD as a marker for therapy selection are awaited. Finally, maintenance chemotherapy is not part of standard AML treatment.3
Role of Stem Cell Transplant
AML is the most common indication for allo-SCT. The availability of alternative donor strategies, which include mismatched, unrelated, haplo-identical, and cord blood donor sources, and the development of non-myeloablative and reduced-intensity conditioning (RIC) regimens (which take advantage of graft-versus-leukemia effect while decreasing cytotoxicity from myeloablative regimens) have expanded the possibility of allo-SCT to most patients under the age of 75 years.3 The decision to perform transplant is now largely based upon assessment of the risk (nonrelapse mortality) to benefit (reduction in risk of relapse) ratio, as determined by both disease-related features (cytogenetics, molecular profile) and clinical characteristics of the donor (type, availability, match) and the recipient (comorbidities, performance status).3 In a meta-analysis of 24 prospective trials involving more than 6000 AML patients in first complete remission, allo-SCT was associated with a significant survival benefit in patients with intermediate- and poor-risk AML but not in patients with good-risk AML.52 In line with this, good-risk AML patients are generally not recommended for transplant in first complete remission. For patients with normal karyotype who were said to have de novo AML (historically an intermediate-risk AML group), superior OS was demonstrated with transplant over intensive chemotherapy in those patients with either FLT3-ITD mutations or those with the molecular profile characterized by negativity for mutations in NPM1/CEBPA/FLT3.53 For patients with primary refractory disease and high-risk AML, transplant is probably the only curative option.
The choice of conditioning regimen is guided by several factors, including the subtype of AML, disease status, donor-recipient genetic disparity, graft source, comorbidities in the recipient (ie, tolerability for intensive conditioning regimen), as well as the reliance on graft-versus-leukemia effect as compared to cytotoxic effect of the regimen. The BMT CTN 0901 trial, which randomly assigned 218 patients aged 18 to 65 years to RIC (typically fludarabine/busulfan) or myeloablative regimens, showed an advantage for myeloablative regimens.54 The trial demonstrated a lower risk of relapse (13.5% versus 48.3%, P < 0.01) and higher rates of relapse-free survival (67.7% versus 47.3%, P < 0.01) and OS (67.7% versus. 77.4%, P = 0.07) at 18 months despite higher treatment-related mortality (15.8% versus 4.4%, P = 0.02) and a higher rate of grade 2 to 4 acute graft-versus-host disease (44.7% versus 31.6%, P = 0.024). At present, a RIC regimen is generally recommended for older patients or those with a higher comorbidity burden, while the myeloablative regimen is recommended for younger, fit patients.
Relapsed/Refractory Disease
The treatment of relapsed and refractory AML constitutes a major challenge, with OS estimated around 10% at 3 years.55 Currently, there is no standard salvage therapy in this setting, thus underscoring the need for clinical trials. For younger, fitter patients, the typical approach is to use intensive chemotherapy to achieve a second complete remission followed by a stem cell transplant. In younger patients, a second complete remission is achievable in about 55% of patients, although this rate is lower (~20%–30%) in more unselected patients.56,57 About two thirds of those who achieve complete remission may be able to proceed to transplant.57 For older patients where transplant is not possible, the goal is to use less intensive therapies that help with palliation. HMAs (azacitidine, decitabine) are used and have complete remission rates of 16% to 21% and median survival of 6 to 9 months in older patients.3 LDAC is another option in this setting. The recent approval of GO in this setting has further expanded the options. This approval was based on the outcomes of the phase 2 single-arm MyloFrance-1 study in which single-agent GO administered at 3 mg/m2 on days 1, 4, and 7 led to complete remission in 15 of 57 patients.58
With greater elucidation of the molecular characteristics of AML, the emergence of more effective targeted therapies is possible. Enasidenib, an inhibitor of mutant isocitrate dehydrogenase 2 (IDH2) protein that promotes differentiation of leukemic myeloblasts, recently received regulatory approval based on a single-arm trial. The overall response rate in this study was 38.5%, including a composite complete remission rate of 26.6% at a dose of 100 mg daily.59 IDH differentiation syndrome, akin to the differentiation syndrome seen in acute promyelocytic leukemia, occurred in approximately 12% of the patients, with the most frequent manifestations being dyspnea, fever, pulmonary infiltrates, and hypoxia.60
Survival of patients who relapse following transplant is particularly poor. A recent Center for International Blood and Marrow Transplant Research study found a 3-year OS ranging from a dismal 4% for those who present with early relapses (within 1 to 6 months) post-transplant to a more modest 38% for those who relapsed ≥ 3 years after their first transplant.61 The German Cooperative Transplant Study Group have suggested that azacitidine or chemotherapy followed by donor-lymphocyte infusions might improve responses over chemotherapy alone.62 Ipilimumab-based CTLA-4 blockade was reported to produce responses in a small cohort of patients, which was particularly notable in patients presenting with extramedullary manifestations of relapse.63 In patients who are otherwise fit but have a florid relapse, a second transplant can sometimes be sought, but the value of a different donor for second transplant is unclear.3
Case 1 Conclusion
Given his relatively young age, suitability for intensive therapy, and the presence of a core- binding factor abnormality, the patient is treated with an induction regimen containing daunorubicin, cytarabine, and GO (7+3 + GO). He achieves complete remission. This is followed by consolidation chemotherapy with high-dose cytarabine and GO. Allo-SCT is reserved for later should the AML relapse. Note that dasatinib, a c-KIT inhibitor, can be added to the treatment regimens as per the results of the CALGB 10801 protocol.64 Also, autologous SCT, instead of allo-SCT, can be considered in rare situations with relapsed core-binding factor AML (especially with inv(16) AML, younger patients, longer time in complete remission prior to relapse, and use of GO).
Case 2 Conclusion
The patient is deemed suitable for intensive chemotherapy. As such, CPX-351 is given in induction and consolidation and complete remission is achieved. Because he has adverse-risk AML, an allo-SCT is planned, but the patient relapses before it can be performed. Following 3 courses of decitabine therapy, the patient achieves complete remission once again but declines transplant. He maintains remission for an additional 4 months but then the leukemia progresses. Clinical trials are recommended to the patient, but he decides to pursue hospice care.
Conclusion
AML is the most common acute leukemia in adults. As defined currently, AML represents a group of related but distinct myeloid disorders that are characterized by various chromosomal, genetic, and epigenetic alterations. Early diagnosis and treatment can help prevent the emergence or manage the detrimental effects of its various complications such as leukostasis and tumor lysis syndrome. Improvements in supportive care, incremental treatment advances, and the wide adoption of allo-SCT for less than favorable cases have significantly improved survival of AML patients since the initial design of combinatorial (7+3) induction chemotherapy, particularly in patients presenting at a younger age. HMAs and the emergence of targeted therapies like FLT-3 and IDH2 inhibitors have added to our therapeutic armamentarium. Despite these advances, long-term survival rates in AML patients continue to be only approximately 40% to 50%. Older patients (particularly those over age 65 at the time of diagnosis), those with relapsed disease, and those with AML with certain unfavorable genetic abnormalities continue to have dismal outcomes. The design of newer targeted therapies, epigenetic agents, and immunotherapies will hopefully address this unmet need.
1. Dohner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med 2015;373:1136–52.
2. National Cancer Institute. Surveillance, Epidemiology, and End Results (SEER) Program. Cancer Stat Facts. Leukemia: Acute Myeloid Leukemia (AML). 2018;2018.
3. Dohner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017;129:424–47.
4. Liesveld JL, Lichtman MA. Acute myelogenous leukemia. In: Kaushansky K, Lichtman MA, Prchal JT, et al, eds. New York: Williams Hematology. 9th ed. New York: McGraw-Hill Education; 2015.
5. Randhawa JK, Khoury J, Ravandi-Kashani F. Adult acute myeloid leukemia. In: Kantarjian HM, Wolff RA, eds. The MD Anderson Manual of Medical Oncology. 3rd ed. New York: McGraw-Hill Medical; 2016.
6. Armstrong SA, Staunton JE, Silverman LB, et al. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet 2002;30:41–7.
7. Graubert TA, Mardis ER. Genomics of acute myeloid leukemia. Cancer J 2011;17:487–91.
8. Cancer Genome Atlas Research Network, Ley TJ, Miller C, Ding L, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 2013;368:2059–74.
9. Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med 2016;374:2209–21.
10. Lindsley RC, Mar BG, Mazzola E, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 2015;125:1367–76.
11. Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014;371:2488–98.
12. Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015;126:9–16.
13. Jaiswal S, Natarajan P, Silver AJ, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med 2017;377:111–21.
14. Genovese G, Kahler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014;371:2477–87.
15. Pileri S, Ascani S, Cox M, et al. Myeloid sarcoma: clinico-pathologic, phenotypic and cytogenetic analysis of 92 adult patients. Leukemia 2007;21:340–50.
16. Vachhani P, Bose P. Isolated gastric myeloid sarcoma: a case report and review of the literature. Case Rep Hematol 2014;2014:541807.
17. Rowe JM. Clinical and laboratory features of the myeloid and lymphocytic leukemias. Am J Med Technol 1983;49:103–9.
18. Wolach O, Stone RM. Mixed-phenotype acute leukemia: current challenges in diagnosis and therapy. Curr Opin Hematol 2017;24:139–45.
19. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016;127:2391–405.
20. Assi R, Ravandi F. FLT3 inhibitors in acute myeloid leukemia: Choosing the best when the optimal does not exist. Am J Hematol 2018;93:553–63.
21. Patel JP, Gonen M, Figueroa ME, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med 2012;366:1079–89.
22. Ley TJ, Ding L, Walter MJ, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med 2010;363:2424–33.
23. Dores GM, Devesa SS, Curtis RE, et al. Acute leukemia incidence and patient survival among children and adults in the United States, 2001-2007. Blood 2012;119:34–43.
24. Cairoli R, Beghini A, Grillo G, et al. Prognostic impact of c-KIT mutations in core binding factor leukemias: an Italian retrospective study. Blood 2006;107:3463–8.
25. Sorror ML, Storer BE, Elsawy M, et al. Intensive versus non-intensive induction therapy for patients (Pts) with newly diagnosed acute myeloid leukemia (AML) using two different novel prognostic models [abstract]. Blood 2016;128(22):216.
26. Quintás-Cardama A, Ravandi F, Liu-Dumlao T, et al. Epigenetic therapy is associated with similar survival compared with intensive chemotherapy in older patients with newly diagnosed acute myeloid leukemia. Blood 2012;120;4840-5.
27. Gupta N, Miller A, Gandhi Set al. Comparison of epigenetic versus standard induction chemotherapy for newly diagnosed acute myeloid leukemia patients ≥60 years old.Am J Hematol 2015;90:639-46.
28. Sorror ML, Storer BE, Fathi AT, et al. Development and validation of a novel acute myeloid leukemia-composite model to estimate risks of mortality. JAMA Oncol 2017;3:1675–82.
29. Rowe JM, Neuberg D, Friedenberg W, et al. A phase 3 study of three induction regimens and of priming with GM-CSF in older adults with acute myeloid leukemia: a trial by the Eastern Cooperative Oncology Group. Blood 2004;103:479–85.
30. Mandelli F, Vignetti M, Suciu S, et al. Daunorubicin versus mitoxantrone versus idarubicin as induction and consolidation chemotherapy for adults with acute myeloid leukemia: the EORTC and GIMEMA Groups Study AML-10. J Clin Oncol 2009;27:5397–403.
31. Gardin C, Chevret S, Pautas C, et al. Superior long-term outcome with idarubicin compared with high-dose daunorubicin in patients with acute myeloid leukemia age 50 years and older. J Clin Oncol 2013;31:321–7.
32. Fernandez HF, Sun Z, Yao X, et al. Anthracycline dose intensification in acute myeloid leukemia. N Engl J Med 2009;361:1249–59.
33. Lee JH, Joo YD, Kim H, et al. A randomized trial comparing standard versus high-dose daunorubicin induction in patients with acute myeloid leukemia. Blood 2011;118:3832–41.
34. Lowenberg B, Ossenkoppele GJ, van Putten W, et al. High-dose daunorubicin in older patients with acute myeloid leukemia. N Engl J Med 2009;361:1235–48.
35. Burnett AK, Russell NH, Hills RK, et al. A randomized comparison of daunorubicin 90 mg/m2 vs 60 mg/m2 in AML induction: results from the UK NCRI AML17 trial in 1206 patients. Blood 2015;125:3878–85.
36. Devillier R, Bertoli S, Prebet T, et al. Comparison of 60 or 90 mg/m(2) of daunorubicin in induction therapy for acute myeloid leukemia with intermediate or unfavorable cytogenetics. Am J Hematol 2015;90:E29–30.
37. Pautas C, Merabet F, Thomas X, et al. Randomized study of intensified anthracycline doses for induction and recombinant interleukin-2 for maintenance in patients with acute myeloid leukemia age 50 to 70 years: results of the ALFA-9801 study. J Clin Oncol 2010;28:808–14.
38. Lowenberg B. Sense and nonsense of high-dose cytarabine for acute myeloid leukemia. Blood 2013;121:26–8.
39. Lancet JE, Uy GL, Cortes JE, et al. Final results of a phase III randomized trial of CPX-351 versus 7 + 3 in older patients with newly diagnosed high risk (secondary) AML [abstract]. J Clin Oncol 2016;34(15_suppl):7000-7000.
40. Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med 2017;377:454–64.
41. Jen EY, Ko CW, Lee JE, et al. FDA approval: Gemtuzumab ozogamicin for the treatment of adults with newly-diagnosed CD33-positive acute myeloid leukemia. Clin Cancer Res 2018; doi: 10.1158/1078-0432. CCR-17-3179.
42. Sievers EL, Larson RA, Stadtmauer EA, et al. Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukemia in first relapse. J Clin Oncol 2001;19:3244–54.
43. Petersdorf SH, Kopecky KJ, Slovak M, et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 2013;121:4854–60.
44. Burnett AK, Russell NH, Hills RK, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy improves survival in older patients with acute myeloid leukemia. J Clin Oncol 2012;30:3924–31.
45. Castaigne S, Pautas C, Terre C, et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet 2012;379:1508–16.
46. Kantarjian HM, Thomas XG, Dmoszynska A, et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J Clin Oncol 2012;30:2670–7.
47. Dombret H, Seymour JF, Butrym A, et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood 2015;126:291–9.
48. Welch JS, Petti AA, Miller CA, et al. TP53 and decitabine in acute myeloid leukemia and myelodysplastic syndromes. N Engl J Med 2016;375:2023–36.
49. Amadori S, Suciu S, Selleslag D, et al. Gemtuzumab ozogamicin versus best supportive care in older patients with newly diagnosed acute myeloid leukemia unsuitable for intensive chemotherapy: results of the randomized phase III EORTC-GIMEMA AML-19 trial. J Clin Oncol 2016;34:972–9.
50. Miyawaki S, Ohtake S, Fujisawa S, et al. A randomized comparison of 4 courses of standard-dose multiagent chemotherapy versus 3 courses of high-dose cytarabine alone in postremission therapy for acute myeloid leukemia in adults: the JALSG AML201 Study. Blood 2011;117:2366–72.
51. Schuurhuis GJ, Heuser M, Freeman S, et al. Minimal/measurable residual disease in AML: a consensus document from the European LeukemiaNet MRD Working Party. Blood 2018;131:1275–91.
52. Koreth J, Schlenk R, Kopecky KJ, et al. Allogeneic stem cell transplantation for acute myeloid leukemia in first complete remission: systematic review and meta-analysis of prospective clinical trials. JAMA 2009;301:2349–61.
53. Schlenk RF, Dohner K, Krauter J, et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med 2008;358:1909–18.
54. Pasquini MC, Logan B, Wu J, et al. Results of a phase III randomized, multi-center study of allogeneic stem cell transplantation after high versus reduced intensity conditioning in patients with myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML): Blood and Marrow Transplant Clinical Trials Network (BMT CTN) 0901. Blood 2015;126:LBA–8.
55. Bose P, Vachhani P, Cortes JE. Treatment of relapsed/refractory acute myeloid leukemia. Curr Treat Options Oncol 2017;18:17,017-0456-2.
56. Burnett AK, Goldstone A, Hills RK, et al. Curability of patients with acute myeloid leukemia who did not undergo transplantation in first remission. J Clin Oncol 2013;31:1293–301.
57. Ravandi F, Ritchie EK, Sayar H, et al. Vosaroxin plus cytarabine versus placebo plus cytarabine in patients with first relapsed or refractory acute myeloid leukaemia (VALOR): a randomised, controlled, double-blind, multinational, phase 3 study. Lancet Oncol 2015;16:1025–36.
58. Taksin AL, Legrand O, Raffoux E, et al. High efficacy and safety profile of fractionated doses of Mylotarg as induction therapy in patients with relapsed acute myeloblastic leukemia: a prospective study of the alfa group. Leukemia 2007;21:66–71.
59. Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017;130:722–31.
60. Fathi AT, DiNardo CD, Kline I, et al. Differentiation syndrome associated with enasidenib, a selective inhibitor of mutant isocitrate dehydrogenase 2: analysis of a phase 1/2 study. JAMA Oncol 2018;doi: 10.1001/jamaoncol.2017.4695.
61. Bejanyan N, Weisdorf DJ, Logan BR, et al. Survival of patients with acute myeloid leukemia relapsing after allogeneic hematopoietic cell transplantation: a center for international blood and marrow transplant research study. Biol Blood Marrow Transplant 2015;21:454–9.
62. Schroeder T, Rachlis E, Bug G, et al. Treatment of acute myeloid leukemia or myelodysplastic syndrome relapse after allogeneic stem cell transplantation with azacitidine and donor lymphocyte infusions--a retrospective multicenter analysis from the German Cooperative Transplant Study Group. Biol Blood Marrow Transplant 2015;21:653–60.
63. Davids MS, Kim HT, Bachireddy P, et al. Ipilimumab for patients with relapse after allogeneic transplantation. N Engl J Med 2016;375:143–53.
64. Marcucci G, Geyer S, Zhao W, et al. Adding KIT inhibitor dasatinib (DAS) to chemotherapy overcomes the negative impact of KIT mutation/over-expression in core binding factor (CBF) acute myeloid leukemia (AML): results from CALGB 10801 (Alliance) [abstract]. Blood 2014;124:8.
Introduction
Acute myeloid leukemia (AML) comprises a heterogeneous group of disorders characterized by proliferation of clonal, abnormally differentiated hematopoietic progenitor cells of myeloid lineage that infiltrate the bone marrow, blood, and other tissues.1 In most cases, AML is rapidly fatal if left untreated. Over the past 2 decades, our understanding of the underlying disease biology responsible for the development of AML has improved substantially. We have learned that biological differences drive the various clinical, cytogenetic, and molecular subentities of AML; distinguishing among these subentities helps to identify optimal therapies, while offering improved clinical outcomes for select groups. After years of stagnation in therapeutic advances, 4 new drugs for treating AML were approved by the US Food and Drug Administration (FDA) in 2017. In this article, we review key features of AML diagnosis and management in the context of 2 case presentations.
Epidemiology and Risk Factors
An estimated 21,380 new cases of AML were diagnosed in the United States in 2017, constituting roughly 1.3% of all new cases of cancer.2 Approximately 10,590 patients died of AML in 2017. The median age of patients at the time of diagnosis is 68 years, and the incidence is approximately 4.2 per 100,000 persons per year. The 5-year survival for AML has steadily risen from a meager 6.3% in 1975 to 17.3% in 1995 and 28.1% in 2009.2 The cure rates for AML vary drastically with age. Long-term survival is achieved in approximately 35% to 40% of adults who present at age 60 years or younger, but only 5% to 15% of those older than 60 years at presentation will achieve long-term survival.3
Most cases of AML occur in the absence of any known risk factors. High-dose radiation exposure, chronic benzene exposure, chronic tobacco smoking, and certain chemotherapeutics are known to increase the risk for AML.4 Inconsistent correlations have also been made between exposure to organic solvents, petroleum products, radon, pesticides, and herbicides and the development of AML.4 Obesity may also increase AML risk.4
Two distinct subcategories of therapy-related AML (t-AML) are known. Patients who have been exposed to alkylating chemotherapeutics (eg, melphalan, cyclophosphamide, and nitrogen mustard) can develop t-AML with chromosomal 5 and/or 7 abnormalities after a latency period of approximately 4 to 8 years.5 In contrast, patients exposed to topoisomerase II inhibitors (notably etoposide) develop AML with abnormalities of 11q23 (leading to MLL gene rearrangement) or 21q22 (RUNX1) after a latency period of about 1 to 3 years.6 AML can also arise out of other myeloid disorders such as myelodysplastic syndrome and myeloproliferative neoplasms, and other bone marrow failure syndromes such as aplastic anemia.4 Various inherited or congenital conditions such as Down syndrome, Bloom syndrome, Fanconi anemia, neurofibromatosis 1, and dyskeratosis congenita can also predispose to the development of AML. A more detailed listing of conditions associated with AML can be found elsewhere.4
Molecular Landscape
The first cancer genome sequence was reported in an AML patient in 2008.7 Since then, various elegantly conducted studies have expanded our understanding of the molecular abnormalities in AML. The Cancer Genome Atlas Research Network analyzed the genomes of 200 cases of de novo AML in adults.8 Only 13 mutations were found on average, much fewer than the number of mutations in most adult cancers. Twenty-three genes were commonly mutated, and another 237 were mutated in 2 or more cases. Essentially, all cases had at least 1 nonsynonymous mutation in 1 of 9 categories of genes: transcription-factor fusions (18%), the gene encoding nucleophosmin (NPM1) (27%), tumor-suppressor genes (16%), DNA-methylation–related genes (44%), signaling genes (59%), chromatin-modifying genes (30%), myeloid transcription-factor genes (22%), spliceosome-complex genes (14%), and cohesin-complex genes (13%).
In another study, samples from 1540 patients from 3 prospective trials of intensive chemotherapy were analyzed to understand how genetic diversity defines the pathophysiology of AML.9 The study authors identified 5234 driver mutations from 76 genes or genomic regions, with 2 or more drivers identified in 86% of the samples. Eleven classes of mutational events, each with distinct diagnostic features and clinical outcomes, were identified. Acting as an internal positive control in this analysis, previously recognized mutational and cytogenetic groups emerged as distinct entities, including the groups with biallelic CEBPA mutations, mutations in NPM1, MLL fusions, and the cytogenetic entities t(6;9), inv(3), t(8;21), t(15;17), and inv(16). Three additional categories emerged as distinct entities: AML with mutations in genes encoding chromatin, RNA splicing regulators, or both (18% of patients); AML with TP53 mutations, chromosomal aneuploidies, or both (13%); and, provisionally, AML with IDH2R172 mutations (1%). An additional level of complexity was also revealed within the subgroup of patients with NPM1 mutations, where gene–gene interactions identified co-mutational events associated with both favorable or adverse prognosis.
Further supporting this molecular classification of AML, a study that performed targeted mutational analysis of 194 patients with defined secondary AML (s-AML) or t-AML and 105 unselected AML patients found that the presence of mutations in SRSF2, SF3B1, U2AF1, ZRSR2, ASXL1, EZH2, BCOR, or STAG2 (all members of the chromatin or RNA splicing families) was highly specific for the diagnosis of s-AML.10 These findings are particularly clinically useful in those without a known history of antecedent hematologic disorder. These mutations defining the AML ontogeny were found to occur early in leukemogenesis, persist in clonal remissions, and predict worse clinical outcomes. Mutations in genes involved in regulation of DNA modification and of chromatin state (commonly DNMT3A, ASXL1, and TET2) have also been shown to be present in preleukemic stem or progenitor cells and to occur early in leukemogenesis.3 Unsurprisingly, some of these same mutations, including those in epigenetic regulators (DNMT3A, ASXL1, and TET2) and less frequently in splicing factor genes (SF3B1, SRSF2), have been associated with clonal hematopoietic expansion in elderly, seemingly healthy adults, a condition termed clonal hematopoiesis of indeterminate potential (CHIP).3,11,12 The presence of CHIP is associated with increased risk of hematologic neoplasms and all-cause mortality, the latter being possibly driven by a near doubling in the risk of coronary heart disease in humans and by accelerated atherosclerosis in a mouse model.11,13,14
Clinical Presentation and Work-up
Case Patient 1
A 57-year-old woman with a history of hypertension presents to the emergency department with complaints of productive cough and fevers for the previous 3 days. Examination reveals conjunctival pallor, gingival hyperplasia, and decreased breath sounds at the posterior right lung field. Investigations reveal a white blood cell (WBC) count of 51,000/µL with 15% blasts, a hemoglobin of 7.8 g/dL, and a platelet count of 56 × 103/µL. Peripheral blood smear is notable for large myeloblasts with occasional Auer rods. Chest radiograph shows a consolidation in the right lower lobe.
Case Patient 2
A 69-year-old man presents to his primary care physician for evaluation of worsening fatigue for the previous 4 months. Ten years prior to presentation, he had received 6 cycles of RCHOP (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone) as treatment for diffuse large B-cell lymphoma. Conjunctival pallor, patches of purpura over the extremities, and mucosal petechiae are noted on examination. Laboratory analyisis reveals a WBC count of 2400/µL with 12% blasts, hemoglobin of 9.0 g/dL, and platelet count of 10 × 103/µL. Peripheral smear shows dysplastic myeloid cells and blasts.
Clinical Features
Patients with AML typically present with features secondary to proliferation of blasts (ie, findings of bone marrow failure and end organ damage).4,5 Fatigue, pallor, dizziness, dyspnea, and headaches occur secondary to anemia. Easy and prolonged bruising, petechiae, epistaxis, gingival bleeding, and conjunctival hemorrhages result from thrombocytopenia. Bleeding from other sites such as the central nervous system and gastrointestinal tract occurs but is uncommon. Patients may also present with infections resulting from unrecognized neutropenia. Constitutional symptoms including anorexia, fevers, and weight loss are frequently reported, while organomegaly (hepatomegaly and/or splenomegaly) is seen in about a quarter of patients.4 Infiltration of blasts into almost every organ has been noted, a condition known as myeloid (or granulocytic) sarcoma.15 This condition is more commonly found in patients with blastic, monoblastic, or myelomonocytic variants of AML, and is known as isolated myeloid sarcoma if no concurrent marrow or blood involvement is identified. In the absence of induction chemotherapy, systemic involvement occurs in a matter of weeks to months following such presentation.16
Laboratory analysis will usually demonstrate derangements in peripheral blood cell lines. At least half of patients have a total WBC count less than 5000/µL, a platelet count less than 50 × 103/µL, or both at the time of diagnosis.4,17 Approximately 10% of patients present with hyperleukocytosis and a WBC count greater than 100,000/µL, which can be associated with leukostasis.5 Additionally, spontaneous electrolyte derangement consistent with tumor lysis syndrome and coagulation abnormalities found in disseminated intravascular coagulation may be noted, even before initiation of therapy.
Work-Up of Suspected AML
Bone marrow biopsy and aspirate, along with touch preparations of the core biopsy sample, are crucial in the workup of suspected AML. At least 200 WBCs on blood smears and 500 nucleated cells on spiculated marrow smears should be counted.3 Reactivity with specific histochemical stains (myeloperoxidase, Sudan black B, or naphthyl AS-D-chloroacetate), presence of Auer rods, and reactivity to monoclonal antibodies against epitopes present on myeloblasts (eg, CD13, CD33, CD117) help distinguish myeloblasts from lymphoblasts.4 Flow cytometric analysis helps in confirming myeloid lineage; blasts generally express CD34 and HLA-DR, markers of immature hematopoietic precursors, and dim CD45 (common leukocyte antigen). One or more lymphoid antigens may be aberrantly expressed as well. Of note, in about 2% to 3% of acute leukemia cases, immunohistochemistry and/or flow cytometry findings demonstrate immature cells with features of both myeloid and lymphoid lineages (biphenotypic) or different populations of myeloid and lymphoid leukemia cells (bilineal). These leukemias are termed mixed-phenotype acute leukemia and are typically treated with either AML or acute lymphoblastic leukemia regimens.18
Cytogenetics, as assessed through conventional karyotype and fluorescence in situ hybridization (FISH), constitutes an essential part of the work-up. Eight balanced translocations and inversions and their variants are included in the World Health Organization (WHO) category “AML with recurrent genetic abnormalities,” while 9 balanced rearrangements and multiple unbalanced abnormalities in the presence of a blast count ≥ 20% are sufficient to establish the diagnosis of “AML with myelodysplasia-related changes.”3,19 Various other gene rearrangements thought to represent disease-initiating events are recognized as well, but these rearrangements do not yet formally define WHO disease categories.3 FISH can help detect RUNX1-RUNX1T1, CBFB-MYH11, KMT2A (MLL), and MECOM (EVI1) gene fusions, as well as chromosomal changes like 5q, 7q, or 17p, especially when fewer than 20 metaphases are assessable (due to failure of culture) by conventional cytogenetic methods.3
As certain molecular markers help with disease prognosis and the selection of personalized therapies, testing for these markers is recommended as part of a complete work-up of AML. The current standard of care is to test for nucleophosmin (NPM1), fms-like tyrosine kinase 3 (FLT3), and CEBPA mutations in all newly diagnosed patients.1RUNX1 mutation analysis should also be considered as its presence defines a provisional WHO subcategory.19 In the case of FLT3, the analysis should include both internal tandem duplications (FLT3-ITD, associated with worse prognosis especially at high allelic ratio) and tyrosine-kinase domain mutations (FLT3-TKD; D835 and I836), especially now that FLT3 inhibitors are regularly used.20 Most academic centers now routinely use next-generation sequencing–based panels to assess multiple mutations.
Diagnosis and Classification
A marrow or blood blast (myeloblasts, monoblasts, megakaryoblasts, or promonocytes [considered blast equivalents]) count of ≥ 20% is required for AML diagnosis.3,19 The presence of t(15;17), t(8;21), inv(16), or t(16;16), however, is considered diagnostic of AML irrespective of blast count.3,19 The previously used French-American-British (FAB) classification scheme has been replaced by the WHO classification (Table 2), which takes into account the morphologic, cytogenetic, genetic, and clinical features of the leukemia.
The category “AML with myelodysplasia-related changes” includes AML that has evolved out of an antecedent myelodysplastic syndrome, has ≥ 50% dysplasia in 2 or more lineages, or has myelodysplasia-related cytogenetic changes (eg, –5/del(5q), –7/del(7q), ≥ 3 cytogenetic abnormalities).19 “Therapy-related myeloid neoplasm,” or therapy-related AML, is diagnosed when the patient has previously received cytotoxic agents or ionizing radiation.19
Cases which do not meet the criteria for 1 of the previously mentioned categories are currently classified as “AML, not otherwise specified.” Further subclassification is pursued as per the older FAB scheme; however, no additional prognostic information is obtained in doing so.3,19 Myeloid sarcoma is strictly not a subcategory of AML. Rather, it is an extramedullary mass of myeloid blasts that effaces the normal tissue architecture.16 Rarely, myeloid sarcoma can be present without systemic disease involvement; it is important to note that management of such cases is identical to management of overt AML.16
Finally, myeloid proliferations related to Down syndrome include 2 entities seen in children with Down syndrome.19 Transient abnormal myelopoiesis, seen in 10% to 30% of newborns with Down syndrome, presents with circulating blasts that resolve in a couple of months. Myeloid leukemia associated with Down syndrome is AML that occurs usually in the first 3 years of life and persists if not treated.19
Case 1 Continued
The presence of 15% blasts in the peripheral blood is concerning for, but not diagnostic of, AML. On the other hand, the presence of Auer rods is virtually pathognomonic for AML. Gingival hyperplasia in this patient may be reflective of extramedullary disease. Cytogenetics from the peripheral blood and marrow aspirate show inv(16) in 20 of 20 cells. Molecular panel is notable for mutation in c-KIT. As such, the patient is diagnosed with core-binding factor AML, which per the ELN classification is considered a favorable-risk AML. The presence of c-KIT mutation, however, confers a relatively worse outcome.
Case 2 Continued
Presence of pancytopenia in a patient who previously received cytotoxic chemotherapy is highly concerning for therapy-related myeloid neoplasm. The presence of 12% blasts in the peripheral blood does not meet the criteria for diagnosis of AML. However, marrow specimens show 40% blasts, thus meeting the criteria for an AML diagnosis. Additionally, cytogenetics are notable for the presence of monosomy 7, while a next-generation sequencing panel shows a mutation in TP53. Put together, this patient meets the criteria for therapy-related AML which is an adverse-risk AML according to the ELN classification.
Management
The 2 most significant factors that must be considered when selecting AML therapies are the patient’s suitability for intensive chemotherapy and the biological characteristics of the AML. The former is a nuanced decision that incorporates age, performance status, and existing comorbidities. Treatment-related mortality calculators can guide physicians when making therapy decisions, especially in older patients (≥ 65 years). Retrospective evidence from various studies suggests that older, medically fit patients may derive clinically comparable benefits from intensive and less intensive induction therapies.25–27 The biological characteristics of the leukemia can be suggested by morphologic findings, cytogenetics, and molecular information, in addition to a history of antecedent myeloid neoplasms. Recently, an AML composite model incorporating an augmented Hematopoietic Cell Transplantation–specific Comorbidity Index (HCT-CI) score, age, and cytogenetic/molecular risks was shown to improve treatment decision-making about AML; this model potentially could be used to guide patient stratification in clinical trials as well.28 The overall treatment model of AML is largely unchanged otherwise. It is generally divided into induction, consolidation, and maintenance therapies.
Induction Therapy
In patients who can tolerate intensive therapies, the role of anthracycline- and cytarabine-based treatment is well established. However, the choice of specific anthracycline is not well established. One study concluded that idarubicin and mitoxantrone led to better outcomes as compared to daunorubicin, while another showed no difference between these agents.29,30 A pooled study of AML trials conducted in patients aged 50 years and older showed that while idarubicin led to a higher complete remission rate (69% versus 61%), the overall survival (OS) did not differ significantly.31 As for dosing, daunorubicin given at 45 mg/m2 daily for 3 days has been shown to have lower complete remission rates and higher relapse rates than a dose of 90 mg/m2 daily for 3 days in younger patients.32–34 However, it is not clear whether the 90 mg/m2 dose is superior to the frequently used dose of 60 mg/m2.35 A French study has shown comparable rates of complete remission, relapse, and OS between the 60 mg/m2 and 90 mg/m2 doses in patients with intermediate or unfavorable cytogenetics.36
If idarubicin is used, a dose of 12 mg/m2 for 3 days is considered the standard. In patients aged 50 to 70 years, there were no statistically significant differences in rates of relapse or OS between daunorubicin 80 mg/m2 for 3 days versus idarubicin 12 mg/m2 for 3 days versus idarubicin 12 mg/m2 for 4 days.37 As for cytarabine, the bulk of the evidence indicates that a dose of 1000 mg/m2 or higher should not be used.38 As such, the typical induction chemotherapy regimen of choice is 3 days of anthracycline (daunorubicin or idarubicin) and 7 days of cytarabine (100–200 mg/m2 continuous infusion), also known as the 7+3 regimen, which was first pioneered in the 1970s. In a recent phase 3 trial, 309 patients aged 60 to 75 years with high-risk AML (AML with myelodysplasia-related changes or t-AML) were randomly assigned to either the 7+3 regimen or CPX-351 (ie, nano-liposomal encapsulation of cytarabine and daunorubicin in a 5:1 molar ratio).39 A higher composite complete response rate (47.7% versus 33.3%; P = 0.016) and improved survival (9.56 months versus 5.95 months; hazard ratio [HR] 0.69, P = 0.005) were seen with CPX-351, leading to its approval by the FDA in patients with high-risk AML.
The 7+3 regimen has served as a backbone onto which other drugs have been added in clinical trials—the majority without any clinical benefits—for patients who can tolerate intensive therapy. In this context, the role of 2 therapies recently approved by the FDA must be discussed. In the RATIFY trial, 717 patients aged 18 to 59 years with AML and a FLT3 mutation were randomly assigned to receive standard chemotherapy (induction and consolidation therapy) plus either midostaurin or placebo; those who were in remission after consolidation therapy received either midostaurin or placebo in the maintenance phase.40 The primary endpoint was met as midostaurin improved OS (HR 0.78, P = 0.009). The benefit of midostaurin was consistent across all FLT3 subtypes and mutant allele burdens, regardless of whether patients proceeded to allogeneic stem cell transplant (allo-SCT). Based on the results of RATIFY, midostaurin was approved by the FDA for treatment of AML patients who are positive for the FLT3 mutation. Whether more potent and selective FLT3 inhibitors like gilteritinib, quizartinib, or crenolanib improve the outcomes is currently under investigation in various clinical trials.20
The development of gemtuzumab ozogamicin (GO) has been more complicated. GO, an antibody-drug conjugate comprised of a CD33-directed humanized monoclonal antibody linked covalently to the cytotoxic agent calicheamicin, binds CD33 present on the surface of myeloid leukemic blasts and immature normal cells of myelomonocytic lineage.41 The drug first received an accelerated approval in 2000 as monotherapy (2 doses of 9 mg/m2 14 days apart) for the treatment of patients 60 years of age and older with CD33-positive AML in first relapse based on the results of 3 open-label multicenter trials.41,42 However, a confirmatory S0106 trial in which GO 6 mg/m2 was added on day 4 in newly diagnosed AML patients was terminated early when an interim analysis showed an increased rate of death in induction (6% versus 1%) and lack of improvement in complete response, disease-free survival, or OS with the addition of GO.43 This study led to the withdrawal of GO from the US market in 2010. However, 2 randomized trials that studied GO using a different dose and schedule suggested that the addition of GO to intensive chemotherapy improved survival outcomes in patients with favorable and intermediate-risk cytogenetics.44,45 The results of the multicenter, open-label phase 3 ALFA-0701 trial, which randomly assigned 271 patients aged 50 to 70 years with newly diagnosed AML to daunorubicin and cytarabine alone or in combination with GO (3 mg/m2 on days 1, 4, and 7 during induction and day 1 of 2 consolidation courses), showed a statistically significant improvement in event-free survival (17.3 months versus 9.5 months; HR 0.56 [95% confidence interval 0.42 to 0.76]).45 Again, the survival benefits were more pronounced in patients with favorable or intermediate-risk cytogenetics than in those with unfavorable cytogenetics. The results of this trial led to the re-approval of GO in newly diagnosed AML patients.
For patients who cannot tolerate intensive therapies, the 2 main therapeutic options are low-dose cytarabine (LDAC) and the hypomethylating agents (HMA) azacitidine and decitabine. A phase 3 trial of decitabine versus mostly LDAC (or best supportive care, BSC) demonstrated favorable survival with decitabine (7.7 months versus 5.0 months).46 In the AZA-AML-001 trial, azacitidine improved median survival (10.4 months versus 6.5 months) in comparison to the control arm (LDAC, 7+3, BSC).47 Emerging data has also suggested that HMAs may be particularly active in patients with unfavorable-risk AML, a group for which LDAC has been shown to be especially useless.48 As such, HMA therapies are generally preferred over LDAC in practice. Finally, it is pertinent to note that GO can also be used as monotherapy based on the results of the open-label phase 3 AML-19 study in which GO demonstrated a survival advantage over BSC (4.9 months versus 3.6 months, P = 0.005).49
Postremission or Consolidation Therapy
There is no standard consolidation therapy for AML at present. In general, for patients who received HMA in the induction phase, the same HMA should be continued indefinitely until disease progression or allo-SCT.3 For those who received intensive chemotherapy in the induction phase, the consensus is to use cytarabine-based consolidation therapies. Cytarabine given as a single agent in high-doses has generally led to similar outcomes as multiagent chemotherapy.50 In this regard, cytarabine regimens, with or without anthracycline, at 3000 mg/m2 have similar efficacy as an intermediate dose of 1000 mg/m2.38 A total of 2 to 4 cycles of post-remission therapy is considered standard.3 Intensified post-remission chemotherapy has not been associated with consistent benefit in older AML patients or those with poor-risk disease. In recent years, measurable residual disease (MRD) assessment has emerged as a potentially useful tool in risk stratification and treatment planning, with various studies suggesting that MRD status in complete remission is one of the most important prognostic factors.51 Prospective studies confirming the significance of MRD as a marker for therapy selection are awaited. Finally, maintenance chemotherapy is not part of standard AML treatment.3
Role of Stem Cell Transplant
AML is the most common indication for allo-SCT. The availability of alternative donor strategies, which include mismatched, unrelated, haplo-identical, and cord blood donor sources, and the development of non-myeloablative and reduced-intensity conditioning (RIC) regimens (which take advantage of graft-versus-leukemia effect while decreasing cytotoxicity from myeloablative regimens) have expanded the possibility of allo-SCT to most patients under the age of 75 years.3 The decision to perform transplant is now largely based upon assessment of the risk (nonrelapse mortality) to benefit (reduction in risk of relapse) ratio, as determined by both disease-related features (cytogenetics, molecular profile) and clinical characteristics of the donor (type, availability, match) and the recipient (comorbidities, performance status).3 In a meta-analysis of 24 prospective trials involving more than 6000 AML patients in first complete remission, allo-SCT was associated with a significant survival benefit in patients with intermediate- and poor-risk AML but not in patients with good-risk AML.52 In line with this, good-risk AML patients are generally not recommended for transplant in first complete remission. For patients with normal karyotype who were said to have de novo AML (historically an intermediate-risk AML group), superior OS was demonstrated with transplant over intensive chemotherapy in those patients with either FLT3-ITD mutations or those with the molecular profile characterized by negativity for mutations in NPM1/CEBPA/FLT3.53 For patients with primary refractory disease and high-risk AML, transplant is probably the only curative option.
The choice of conditioning regimen is guided by several factors, including the subtype of AML, disease status, donor-recipient genetic disparity, graft source, comorbidities in the recipient (ie, tolerability for intensive conditioning regimen), as well as the reliance on graft-versus-leukemia effect as compared to cytotoxic effect of the regimen. The BMT CTN 0901 trial, which randomly assigned 218 patients aged 18 to 65 years to RIC (typically fludarabine/busulfan) or myeloablative regimens, showed an advantage for myeloablative regimens.54 The trial demonstrated a lower risk of relapse (13.5% versus 48.3%, P < 0.01) and higher rates of relapse-free survival (67.7% versus 47.3%, P < 0.01) and OS (67.7% versus. 77.4%, P = 0.07) at 18 months despite higher treatment-related mortality (15.8% versus 4.4%, P = 0.02) and a higher rate of grade 2 to 4 acute graft-versus-host disease (44.7% versus 31.6%, P = 0.024). At present, a RIC regimen is generally recommended for older patients or those with a higher comorbidity burden, while the myeloablative regimen is recommended for younger, fit patients.
Relapsed/Refractory Disease
The treatment of relapsed and refractory AML constitutes a major challenge, with OS estimated around 10% at 3 years.55 Currently, there is no standard salvage therapy in this setting, thus underscoring the need for clinical trials. For younger, fitter patients, the typical approach is to use intensive chemotherapy to achieve a second complete remission followed by a stem cell transplant. In younger patients, a second complete remission is achievable in about 55% of patients, although this rate is lower (~20%–30%) in more unselected patients.56,57 About two thirds of those who achieve complete remission may be able to proceed to transplant.57 For older patients where transplant is not possible, the goal is to use less intensive therapies that help with palliation. HMAs (azacitidine, decitabine) are used and have complete remission rates of 16% to 21% and median survival of 6 to 9 months in older patients.3 LDAC is another option in this setting. The recent approval of GO in this setting has further expanded the options. This approval was based on the outcomes of the phase 2 single-arm MyloFrance-1 study in which single-agent GO administered at 3 mg/m2 on days 1, 4, and 7 led to complete remission in 15 of 57 patients.58
With greater elucidation of the molecular characteristics of AML, the emergence of more effective targeted therapies is possible. Enasidenib, an inhibitor of mutant isocitrate dehydrogenase 2 (IDH2) protein that promotes differentiation of leukemic myeloblasts, recently received regulatory approval based on a single-arm trial. The overall response rate in this study was 38.5%, including a composite complete remission rate of 26.6% at a dose of 100 mg daily.59 IDH differentiation syndrome, akin to the differentiation syndrome seen in acute promyelocytic leukemia, occurred in approximately 12% of the patients, with the most frequent manifestations being dyspnea, fever, pulmonary infiltrates, and hypoxia.60
Survival of patients who relapse following transplant is particularly poor. A recent Center for International Blood and Marrow Transplant Research study found a 3-year OS ranging from a dismal 4% for those who present with early relapses (within 1 to 6 months) post-transplant to a more modest 38% for those who relapsed ≥ 3 years after their first transplant.61 The German Cooperative Transplant Study Group have suggested that azacitidine or chemotherapy followed by donor-lymphocyte infusions might improve responses over chemotherapy alone.62 Ipilimumab-based CTLA-4 blockade was reported to produce responses in a small cohort of patients, which was particularly notable in patients presenting with extramedullary manifestations of relapse.63 In patients who are otherwise fit but have a florid relapse, a second transplant can sometimes be sought, but the value of a different donor for second transplant is unclear.3
Case 1 Conclusion
Given his relatively young age, suitability for intensive therapy, and the presence of a core- binding factor abnormality, the patient is treated with an induction regimen containing daunorubicin, cytarabine, and GO (7+3 + GO). He achieves complete remission. This is followed by consolidation chemotherapy with high-dose cytarabine and GO. Allo-SCT is reserved for later should the AML relapse. Note that dasatinib, a c-KIT inhibitor, can be added to the treatment regimens as per the results of the CALGB 10801 protocol.64 Also, autologous SCT, instead of allo-SCT, can be considered in rare situations with relapsed core-binding factor AML (especially with inv(16) AML, younger patients, longer time in complete remission prior to relapse, and use of GO).
Case 2 Conclusion
The patient is deemed suitable for intensive chemotherapy. As such, CPX-351 is given in induction and consolidation and complete remission is achieved. Because he has adverse-risk AML, an allo-SCT is planned, but the patient relapses before it can be performed. Following 3 courses of decitabine therapy, the patient achieves complete remission once again but declines transplant. He maintains remission for an additional 4 months but then the leukemia progresses. Clinical trials are recommended to the patient, but he decides to pursue hospice care.
Conclusion
AML is the most common acute leukemia in adults. As defined currently, AML represents a group of related but distinct myeloid disorders that are characterized by various chromosomal, genetic, and epigenetic alterations. Early diagnosis and treatment can help prevent the emergence or manage the detrimental effects of its various complications such as leukostasis and tumor lysis syndrome. Improvements in supportive care, incremental treatment advances, and the wide adoption of allo-SCT for less than favorable cases have significantly improved survival of AML patients since the initial design of combinatorial (7+3) induction chemotherapy, particularly in patients presenting at a younger age. HMAs and the emergence of targeted therapies like FLT-3 and IDH2 inhibitors have added to our therapeutic armamentarium. Despite these advances, long-term survival rates in AML patients continue to be only approximately 40% to 50%. Older patients (particularly those over age 65 at the time of diagnosis), those with relapsed disease, and those with AML with certain unfavorable genetic abnormalities continue to have dismal outcomes. The design of newer targeted therapies, epigenetic agents, and immunotherapies will hopefully address this unmet need.
Introduction
Acute myeloid leukemia (AML) comprises a heterogeneous group of disorders characterized by proliferation of clonal, abnormally differentiated hematopoietic progenitor cells of myeloid lineage that infiltrate the bone marrow, blood, and other tissues.1 In most cases, AML is rapidly fatal if left untreated. Over the past 2 decades, our understanding of the underlying disease biology responsible for the development of AML has improved substantially. We have learned that biological differences drive the various clinical, cytogenetic, and molecular subentities of AML; distinguishing among these subentities helps to identify optimal therapies, while offering improved clinical outcomes for select groups. After years of stagnation in therapeutic advances, 4 new drugs for treating AML were approved by the US Food and Drug Administration (FDA) in 2017. In this article, we review key features of AML diagnosis and management in the context of 2 case presentations.
Epidemiology and Risk Factors
An estimated 21,380 new cases of AML were diagnosed in the United States in 2017, constituting roughly 1.3% of all new cases of cancer.2 Approximately 10,590 patients died of AML in 2017. The median age of patients at the time of diagnosis is 68 years, and the incidence is approximately 4.2 per 100,000 persons per year. The 5-year survival for AML has steadily risen from a meager 6.3% in 1975 to 17.3% in 1995 and 28.1% in 2009.2 The cure rates for AML vary drastically with age. Long-term survival is achieved in approximately 35% to 40% of adults who present at age 60 years or younger, but only 5% to 15% of those older than 60 years at presentation will achieve long-term survival.3
Most cases of AML occur in the absence of any known risk factors. High-dose radiation exposure, chronic benzene exposure, chronic tobacco smoking, and certain chemotherapeutics are known to increase the risk for AML.4 Inconsistent correlations have also been made between exposure to organic solvents, petroleum products, radon, pesticides, and herbicides and the development of AML.4 Obesity may also increase AML risk.4
Two distinct subcategories of therapy-related AML (t-AML) are known. Patients who have been exposed to alkylating chemotherapeutics (eg, melphalan, cyclophosphamide, and nitrogen mustard) can develop t-AML with chromosomal 5 and/or 7 abnormalities after a latency period of approximately 4 to 8 years.5 In contrast, patients exposed to topoisomerase II inhibitors (notably etoposide) develop AML with abnormalities of 11q23 (leading to MLL gene rearrangement) or 21q22 (RUNX1) after a latency period of about 1 to 3 years.6 AML can also arise out of other myeloid disorders such as myelodysplastic syndrome and myeloproliferative neoplasms, and other bone marrow failure syndromes such as aplastic anemia.4 Various inherited or congenital conditions such as Down syndrome, Bloom syndrome, Fanconi anemia, neurofibromatosis 1, and dyskeratosis congenita can also predispose to the development of AML. A more detailed listing of conditions associated with AML can be found elsewhere.4
Molecular Landscape
The first cancer genome sequence was reported in an AML patient in 2008.7 Since then, various elegantly conducted studies have expanded our understanding of the molecular abnormalities in AML. The Cancer Genome Atlas Research Network analyzed the genomes of 200 cases of de novo AML in adults.8 Only 13 mutations were found on average, much fewer than the number of mutations in most adult cancers. Twenty-three genes were commonly mutated, and another 237 were mutated in 2 or more cases. Essentially, all cases had at least 1 nonsynonymous mutation in 1 of 9 categories of genes: transcription-factor fusions (18%), the gene encoding nucleophosmin (NPM1) (27%), tumor-suppressor genes (16%), DNA-methylation–related genes (44%), signaling genes (59%), chromatin-modifying genes (30%), myeloid transcription-factor genes (22%), spliceosome-complex genes (14%), and cohesin-complex genes (13%).
In another study, samples from 1540 patients from 3 prospective trials of intensive chemotherapy were analyzed to understand how genetic diversity defines the pathophysiology of AML.9 The study authors identified 5234 driver mutations from 76 genes or genomic regions, with 2 or more drivers identified in 86% of the samples. Eleven classes of mutational events, each with distinct diagnostic features and clinical outcomes, were identified. Acting as an internal positive control in this analysis, previously recognized mutational and cytogenetic groups emerged as distinct entities, including the groups with biallelic CEBPA mutations, mutations in NPM1, MLL fusions, and the cytogenetic entities t(6;9), inv(3), t(8;21), t(15;17), and inv(16). Three additional categories emerged as distinct entities: AML with mutations in genes encoding chromatin, RNA splicing regulators, or both (18% of patients); AML with TP53 mutations, chromosomal aneuploidies, or both (13%); and, provisionally, AML with IDH2R172 mutations (1%). An additional level of complexity was also revealed within the subgroup of patients with NPM1 mutations, where gene–gene interactions identified co-mutational events associated with both favorable or adverse prognosis.
Further supporting this molecular classification of AML, a study that performed targeted mutational analysis of 194 patients with defined secondary AML (s-AML) or t-AML and 105 unselected AML patients found that the presence of mutations in SRSF2, SF3B1, U2AF1, ZRSR2, ASXL1, EZH2, BCOR, or STAG2 (all members of the chromatin or RNA splicing families) was highly specific for the diagnosis of s-AML.10 These findings are particularly clinically useful in those without a known history of antecedent hematologic disorder. These mutations defining the AML ontogeny were found to occur early in leukemogenesis, persist in clonal remissions, and predict worse clinical outcomes. Mutations in genes involved in regulation of DNA modification and of chromatin state (commonly DNMT3A, ASXL1, and TET2) have also been shown to be present in preleukemic stem or progenitor cells and to occur early in leukemogenesis.3 Unsurprisingly, some of these same mutations, including those in epigenetic regulators (DNMT3A, ASXL1, and TET2) and less frequently in splicing factor genes (SF3B1, SRSF2), have been associated with clonal hematopoietic expansion in elderly, seemingly healthy adults, a condition termed clonal hematopoiesis of indeterminate potential (CHIP).3,11,12 The presence of CHIP is associated with increased risk of hematologic neoplasms and all-cause mortality, the latter being possibly driven by a near doubling in the risk of coronary heart disease in humans and by accelerated atherosclerosis in a mouse model.11,13,14
Clinical Presentation and Work-up
Case Patient 1
A 57-year-old woman with a history of hypertension presents to the emergency department with complaints of productive cough and fevers for the previous 3 days. Examination reveals conjunctival pallor, gingival hyperplasia, and decreased breath sounds at the posterior right lung field. Investigations reveal a white blood cell (WBC) count of 51,000/µL with 15% blasts, a hemoglobin of 7.8 g/dL, and a platelet count of 56 × 103/µL. Peripheral blood smear is notable for large myeloblasts with occasional Auer rods. Chest radiograph shows a consolidation in the right lower lobe.
Case Patient 2
A 69-year-old man presents to his primary care physician for evaluation of worsening fatigue for the previous 4 months. Ten years prior to presentation, he had received 6 cycles of RCHOP (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone) as treatment for diffuse large B-cell lymphoma. Conjunctival pallor, patches of purpura over the extremities, and mucosal petechiae are noted on examination. Laboratory analyisis reveals a WBC count of 2400/µL with 12% blasts, hemoglobin of 9.0 g/dL, and platelet count of 10 × 103/µL. Peripheral smear shows dysplastic myeloid cells and blasts.
Clinical Features
Patients with AML typically present with features secondary to proliferation of blasts (ie, findings of bone marrow failure and end organ damage).4,5 Fatigue, pallor, dizziness, dyspnea, and headaches occur secondary to anemia. Easy and prolonged bruising, petechiae, epistaxis, gingival bleeding, and conjunctival hemorrhages result from thrombocytopenia. Bleeding from other sites such as the central nervous system and gastrointestinal tract occurs but is uncommon. Patients may also present with infections resulting from unrecognized neutropenia. Constitutional symptoms including anorexia, fevers, and weight loss are frequently reported, while organomegaly (hepatomegaly and/or splenomegaly) is seen in about a quarter of patients.4 Infiltration of blasts into almost every organ has been noted, a condition known as myeloid (or granulocytic) sarcoma.15 This condition is more commonly found in patients with blastic, monoblastic, or myelomonocytic variants of AML, and is known as isolated myeloid sarcoma if no concurrent marrow or blood involvement is identified. In the absence of induction chemotherapy, systemic involvement occurs in a matter of weeks to months following such presentation.16
Laboratory analysis will usually demonstrate derangements in peripheral blood cell lines. At least half of patients have a total WBC count less than 5000/µL, a platelet count less than 50 × 103/µL, or both at the time of diagnosis.4,17 Approximately 10% of patients present with hyperleukocytosis and a WBC count greater than 100,000/µL, which can be associated with leukostasis.5 Additionally, spontaneous electrolyte derangement consistent with tumor lysis syndrome and coagulation abnormalities found in disseminated intravascular coagulation may be noted, even before initiation of therapy.
Work-Up of Suspected AML
Bone marrow biopsy and aspirate, along with touch preparations of the core biopsy sample, are crucial in the workup of suspected AML. At least 200 WBCs on blood smears and 500 nucleated cells on spiculated marrow smears should be counted.3 Reactivity with specific histochemical stains (myeloperoxidase, Sudan black B, or naphthyl AS-D-chloroacetate), presence of Auer rods, and reactivity to monoclonal antibodies against epitopes present on myeloblasts (eg, CD13, CD33, CD117) help distinguish myeloblasts from lymphoblasts.4 Flow cytometric analysis helps in confirming myeloid lineage; blasts generally express CD34 and HLA-DR, markers of immature hematopoietic precursors, and dim CD45 (common leukocyte antigen). One or more lymphoid antigens may be aberrantly expressed as well. Of note, in about 2% to 3% of acute leukemia cases, immunohistochemistry and/or flow cytometry findings demonstrate immature cells with features of both myeloid and lymphoid lineages (biphenotypic) or different populations of myeloid and lymphoid leukemia cells (bilineal). These leukemias are termed mixed-phenotype acute leukemia and are typically treated with either AML or acute lymphoblastic leukemia regimens.18
Cytogenetics, as assessed through conventional karyotype and fluorescence in situ hybridization (FISH), constitutes an essential part of the work-up. Eight balanced translocations and inversions and their variants are included in the World Health Organization (WHO) category “AML with recurrent genetic abnormalities,” while 9 balanced rearrangements and multiple unbalanced abnormalities in the presence of a blast count ≥ 20% are sufficient to establish the diagnosis of “AML with myelodysplasia-related changes.”3,19 Various other gene rearrangements thought to represent disease-initiating events are recognized as well, but these rearrangements do not yet formally define WHO disease categories.3 FISH can help detect RUNX1-RUNX1T1, CBFB-MYH11, KMT2A (MLL), and MECOM (EVI1) gene fusions, as well as chromosomal changes like 5q, 7q, or 17p, especially when fewer than 20 metaphases are assessable (due to failure of culture) by conventional cytogenetic methods.3
As certain molecular markers help with disease prognosis and the selection of personalized therapies, testing for these markers is recommended as part of a complete work-up of AML. The current standard of care is to test for nucleophosmin (NPM1), fms-like tyrosine kinase 3 (FLT3), and CEBPA mutations in all newly diagnosed patients.1RUNX1 mutation analysis should also be considered as its presence defines a provisional WHO subcategory.19 In the case of FLT3, the analysis should include both internal tandem duplications (FLT3-ITD, associated with worse prognosis especially at high allelic ratio) and tyrosine-kinase domain mutations (FLT3-TKD; D835 and I836), especially now that FLT3 inhibitors are regularly used.20 Most academic centers now routinely use next-generation sequencing–based panels to assess multiple mutations.
Diagnosis and Classification
A marrow or blood blast (myeloblasts, monoblasts, megakaryoblasts, or promonocytes [considered blast equivalents]) count of ≥ 20% is required for AML diagnosis.3,19 The presence of t(15;17), t(8;21), inv(16), or t(16;16), however, is considered diagnostic of AML irrespective of blast count.3,19 The previously used French-American-British (FAB) classification scheme has been replaced by the WHO classification (Table 2), which takes into account the morphologic, cytogenetic, genetic, and clinical features of the leukemia.
The category “AML with myelodysplasia-related changes” includes AML that has evolved out of an antecedent myelodysplastic syndrome, has ≥ 50% dysplasia in 2 or more lineages, or has myelodysplasia-related cytogenetic changes (eg, –5/del(5q), –7/del(7q), ≥ 3 cytogenetic abnormalities).19 “Therapy-related myeloid neoplasm,” or therapy-related AML, is diagnosed when the patient has previously received cytotoxic agents or ionizing radiation.19
Cases which do not meet the criteria for 1 of the previously mentioned categories are currently classified as “AML, not otherwise specified.” Further subclassification is pursued as per the older FAB scheme; however, no additional prognostic information is obtained in doing so.3,19 Myeloid sarcoma is strictly not a subcategory of AML. Rather, it is an extramedullary mass of myeloid blasts that effaces the normal tissue architecture.16 Rarely, myeloid sarcoma can be present without systemic disease involvement; it is important to note that management of such cases is identical to management of overt AML.16
Finally, myeloid proliferations related to Down syndrome include 2 entities seen in children with Down syndrome.19 Transient abnormal myelopoiesis, seen in 10% to 30% of newborns with Down syndrome, presents with circulating blasts that resolve in a couple of months. Myeloid leukemia associated with Down syndrome is AML that occurs usually in the first 3 years of life and persists if not treated.19
Case 1 Continued
The presence of 15% blasts in the peripheral blood is concerning for, but not diagnostic of, AML. On the other hand, the presence of Auer rods is virtually pathognomonic for AML. Gingival hyperplasia in this patient may be reflective of extramedullary disease. Cytogenetics from the peripheral blood and marrow aspirate show inv(16) in 20 of 20 cells. Molecular panel is notable for mutation in c-KIT. As such, the patient is diagnosed with core-binding factor AML, which per the ELN classification is considered a favorable-risk AML. The presence of c-KIT mutation, however, confers a relatively worse outcome.
Case 2 Continued
Presence of pancytopenia in a patient who previously received cytotoxic chemotherapy is highly concerning for therapy-related myeloid neoplasm. The presence of 12% blasts in the peripheral blood does not meet the criteria for diagnosis of AML. However, marrow specimens show 40% blasts, thus meeting the criteria for an AML diagnosis. Additionally, cytogenetics are notable for the presence of monosomy 7, while a next-generation sequencing panel shows a mutation in TP53. Put together, this patient meets the criteria for therapy-related AML which is an adverse-risk AML according to the ELN classification.
Management
The 2 most significant factors that must be considered when selecting AML therapies are the patient’s suitability for intensive chemotherapy and the biological characteristics of the AML. The former is a nuanced decision that incorporates age, performance status, and existing comorbidities. Treatment-related mortality calculators can guide physicians when making therapy decisions, especially in older patients (≥ 65 years). Retrospective evidence from various studies suggests that older, medically fit patients may derive clinically comparable benefits from intensive and less intensive induction therapies.25–27 The biological characteristics of the leukemia can be suggested by morphologic findings, cytogenetics, and molecular information, in addition to a history of antecedent myeloid neoplasms. Recently, an AML composite model incorporating an augmented Hematopoietic Cell Transplantation–specific Comorbidity Index (HCT-CI) score, age, and cytogenetic/molecular risks was shown to improve treatment decision-making about AML; this model potentially could be used to guide patient stratification in clinical trials as well.28 The overall treatment model of AML is largely unchanged otherwise. It is generally divided into induction, consolidation, and maintenance therapies.
Induction Therapy
In patients who can tolerate intensive therapies, the role of anthracycline- and cytarabine-based treatment is well established. However, the choice of specific anthracycline is not well established. One study concluded that idarubicin and mitoxantrone led to better outcomes as compared to daunorubicin, while another showed no difference between these agents.29,30 A pooled study of AML trials conducted in patients aged 50 years and older showed that while idarubicin led to a higher complete remission rate (69% versus 61%), the overall survival (OS) did not differ significantly.31 As for dosing, daunorubicin given at 45 mg/m2 daily for 3 days has been shown to have lower complete remission rates and higher relapse rates than a dose of 90 mg/m2 daily for 3 days in younger patients.32–34 However, it is not clear whether the 90 mg/m2 dose is superior to the frequently used dose of 60 mg/m2.35 A French study has shown comparable rates of complete remission, relapse, and OS between the 60 mg/m2 and 90 mg/m2 doses in patients with intermediate or unfavorable cytogenetics.36
If idarubicin is used, a dose of 12 mg/m2 for 3 days is considered the standard. In patients aged 50 to 70 years, there were no statistically significant differences in rates of relapse or OS between daunorubicin 80 mg/m2 for 3 days versus idarubicin 12 mg/m2 for 3 days versus idarubicin 12 mg/m2 for 4 days.37 As for cytarabine, the bulk of the evidence indicates that a dose of 1000 mg/m2 or higher should not be used.38 As such, the typical induction chemotherapy regimen of choice is 3 days of anthracycline (daunorubicin or idarubicin) and 7 days of cytarabine (100–200 mg/m2 continuous infusion), also known as the 7+3 regimen, which was first pioneered in the 1970s. In a recent phase 3 trial, 309 patients aged 60 to 75 years with high-risk AML (AML with myelodysplasia-related changes or t-AML) were randomly assigned to either the 7+3 regimen or CPX-351 (ie, nano-liposomal encapsulation of cytarabine and daunorubicin in a 5:1 molar ratio).39 A higher composite complete response rate (47.7% versus 33.3%; P = 0.016) and improved survival (9.56 months versus 5.95 months; hazard ratio [HR] 0.69, P = 0.005) were seen with CPX-351, leading to its approval by the FDA in patients with high-risk AML.
The 7+3 regimen has served as a backbone onto which other drugs have been added in clinical trials—the majority without any clinical benefits—for patients who can tolerate intensive therapy. In this context, the role of 2 therapies recently approved by the FDA must be discussed. In the RATIFY trial, 717 patients aged 18 to 59 years with AML and a FLT3 mutation were randomly assigned to receive standard chemotherapy (induction and consolidation therapy) plus either midostaurin or placebo; those who were in remission after consolidation therapy received either midostaurin or placebo in the maintenance phase.40 The primary endpoint was met as midostaurin improved OS (HR 0.78, P = 0.009). The benefit of midostaurin was consistent across all FLT3 subtypes and mutant allele burdens, regardless of whether patients proceeded to allogeneic stem cell transplant (allo-SCT). Based on the results of RATIFY, midostaurin was approved by the FDA for treatment of AML patients who are positive for the FLT3 mutation. Whether more potent and selective FLT3 inhibitors like gilteritinib, quizartinib, or crenolanib improve the outcomes is currently under investigation in various clinical trials.20
The development of gemtuzumab ozogamicin (GO) has been more complicated. GO, an antibody-drug conjugate comprised of a CD33-directed humanized monoclonal antibody linked covalently to the cytotoxic agent calicheamicin, binds CD33 present on the surface of myeloid leukemic blasts and immature normal cells of myelomonocytic lineage.41 The drug first received an accelerated approval in 2000 as monotherapy (2 doses of 9 mg/m2 14 days apart) for the treatment of patients 60 years of age and older with CD33-positive AML in first relapse based on the results of 3 open-label multicenter trials.41,42 However, a confirmatory S0106 trial in which GO 6 mg/m2 was added on day 4 in newly diagnosed AML patients was terminated early when an interim analysis showed an increased rate of death in induction (6% versus 1%) and lack of improvement in complete response, disease-free survival, or OS with the addition of GO.43 This study led to the withdrawal of GO from the US market in 2010. However, 2 randomized trials that studied GO using a different dose and schedule suggested that the addition of GO to intensive chemotherapy improved survival outcomes in patients with favorable and intermediate-risk cytogenetics.44,45 The results of the multicenter, open-label phase 3 ALFA-0701 trial, which randomly assigned 271 patients aged 50 to 70 years with newly diagnosed AML to daunorubicin and cytarabine alone or in combination with GO (3 mg/m2 on days 1, 4, and 7 during induction and day 1 of 2 consolidation courses), showed a statistically significant improvement in event-free survival (17.3 months versus 9.5 months; HR 0.56 [95% confidence interval 0.42 to 0.76]).45 Again, the survival benefits were more pronounced in patients with favorable or intermediate-risk cytogenetics than in those with unfavorable cytogenetics. The results of this trial led to the re-approval of GO in newly diagnosed AML patients.
For patients who cannot tolerate intensive therapies, the 2 main therapeutic options are low-dose cytarabine (LDAC) and the hypomethylating agents (HMA) azacitidine and decitabine. A phase 3 trial of decitabine versus mostly LDAC (or best supportive care, BSC) demonstrated favorable survival with decitabine (7.7 months versus 5.0 months).46 In the AZA-AML-001 trial, azacitidine improved median survival (10.4 months versus 6.5 months) in comparison to the control arm (LDAC, 7+3, BSC).47 Emerging data has also suggested that HMAs may be particularly active in patients with unfavorable-risk AML, a group for which LDAC has been shown to be especially useless.48 As such, HMA therapies are generally preferred over LDAC in practice. Finally, it is pertinent to note that GO can also be used as monotherapy based on the results of the open-label phase 3 AML-19 study in which GO demonstrated a survival advantage over BSC (4.9 months versus 3.6 months, P = 0.005).49
Postremission or Consolidation Therapy
There is no standard consolidation therapy for AML at present. In general, for patients who received HMA in the induction phase, the same HMA should be continued indefinitely until disease progression or allo-SCT.3 For those who received intensive chemotherapy in the induction phase, the consensus is to use cytarabine-based consolidation therapies. Cytarabine given as a single agent in high-doses has generally led to similar outcomes as multiagent chemotherapy.50 In this regard, cytarabine regimens, with or without anthracycline, at 3000 mg/m2 have similar efficacy as an intermediate dose of 1000 mg/m2.38 A total of 2 to 4 cycles of post-remission therapy is considered standard.3 Intensified post-remission chemotherapy has not been associated with consistent benefit in older AML patients or those with poor-risk disease. In recent years, measurable residual disease (MRD) assessment has emerged as a potentially useful tool in risk stratification and treatment planning, with various studies suggesting that MRD status in complete remission is one of the most important prognostic factors.51 Prospective studies confirming the significance of MRD as a marker for therapy selection are awaited. Finally, maintenance chemotherapy is not part of standard AML treatment.3
Role of Stem Cell Transplant
AML is the most common indication for allo-SCT. The availability of alternative donor strategies, which include mismatched, unrelated, haplo-identical, and cord blood donor sources, and the development of non-myeloablative and reduced-intensity conditioning (RIC) regimens (which take advantage of graft-versus-leukemia effect while decreasing cytotoxicity from myeloablative regimens) have expanded the possibility of allo-SCT to most patients under the age of 75 years.3 The decision to perform transplant is now largely based upon assessment of the risk (nonrelapse mortality) to benefit (reduction in risk of relapse) ratio, as determined by both disease-related features (cytogenetics, molecular profile) and clinical characteristics of the donor (type, availability, match) and the recipient (comorbidities, performance status).3 In a meta-analysis of 24 prospective trials involving more than 6000 AML patients in first complete remission, allo-SCT was associated with a significant survival benefit in patients with intermediate- and poor-risk AML but not in patients with good-risk AML.52 In line with this, good-risk AML patients are generally not recommended for transplant in first complete remission. For patients with normal karyotype who were said to have de novo AML (historically an intermediate-risk AML group), superior OS was demonstrated with transplant over intensive chemotherapy in those patients with either FLT3-ITD mutations or those with the molecular profile characterized by negativity for mutations in NPM1/CEBPA/FLT3.53 For patients with primary refractory disease and high-risk AML, transplant is probably the only curative option.
The choice of conditioning regimen is guided by several factors, including the subtype of AML, disease status, donor-recipient genetic disparity, graft source, comorbidities in the recipient (ie, tolerability for intensive conditioning regimen), as well as the reliance on graft-versus-leukemia effect as compared to cytotoxic effect of the regimen. The BMT CTN 0901 trial, which randomly assigned 218 patients aged 18 to 65 years to RIC (typically fludarabine/busulfan) or myeloablative regimens, showed an advantage for myeloablative regimens.54 The trial demonstrated a lower risk of relapse (13.5% versus 48.3%, P < 0.01) and higher rates of relapse-free survival (67.7% versus 47.3%, P < 0.01) and OS (67.7% versus. 77.4%, P = 0.07) at 18 months despite higher treatment-related mortality (15.8% versus 4.4%, P = 0.02) and a higher rate of grade 2 to 4 acute graft-versus-host disease (44.7% versus 31.6%, P = 0.024). At present, a RIC regimen is generally recommended for older patients or those with a higher comorbidity burden, while the myeloablative regimen is recommended for younger, fit patients.
Relapsed/Refractory Disease
The treatment of relapsed and refractory AML constitutes a major challenge, with OS estimated around 10% at 3 years.55 Currently, there is no standard salvage therapy in this setting, thus underscoring the need for clinical trials. For younger, fitter patients, the typical approach is to use intensive chemotherapy to achieve a second complete remission followed by a stem cell transplant. In younger patients, a second complete remission is achievable in about 55% of patients, although this rate is lower (~20%–30%) in more unselected patients.56,57 About two thirds of those who achieve complete remission may be able to proceed to transplant.57 For older patients where transplant is not possible, the goal is to use less intensive therapies that help with palliation. HMAs (azacitidine, decitabine) are used and have complete remission rates of 16% to 21% and median survival of 6 to 9 months in older patients.3 LDAC is another option in this setting. The recent approval of GO in this setting has further expanded the options. This approval was based on the outcomes of the phase 2 single-arm MyloFrance-1 study in which single-agent GO administered at 3 mg/m2 on days 1, 4, and 7 led to complete remission in 15 of 57 patients.58
With greater elucidation of the molecular characteristics of AML, the emergence of more effective targeted therapies is possible. Enasidenib, an inhibitor of mutant isocitrate dehydrogenase 2 (IDH2) protein that promotes differentiation of leukemic myeloblasts, recently received regulatory approval based on a single-arm trial. The overall response rate in this study was 38.5%, including a composite complete remission rate of 26.6% at a dose of 100 mg daily.59 IDH differentiation syndrome, akin to the differentiation syndrome seen in acute promyelocytic leukemia, occurred in approximately 12% of the patients, with the most frequent manifestations being dyspnea, fever, pulmonary infiltrates, and hypoxia.60
Survival of patients who relapse following transplant is particularly poor. A recent Center for International Blood and Marrow Transplant Research study found a 3-year OS ranging from a dismal 4% for those who present with early relapses (within 1 to 6 months) post-transplant to a more modest 38% for those who relapsed ≥ 3 years after their first transplant.61 The German Cooperative Transplant Study Group have suggested that azacitidine or chemotherapy followed by donor-lymphocyte infusions might improve responses over chemotherapy alone.62 Ipilimumab-based CTLA-4 blockade was reported to produce responses in a small cohort of patients, which was particularly notable in patients presenting with extramedullary manifestations of relapse.63 In patients who are otherwise fit but have a florid relapse, a second transplant can sometimes be sought, but the value of a different donor for second transplant is unclear.3
Case 1 Conclusion
Given his relatively young age, suitability for intensive therapy, and the presence of a core- binding factor abnormality, the patient is treated with an induction regimen containing daunorubicin, cytarabine, and GO (7+3 + GO). He achieves complete remission. This is followed by consolidation chemotherapy with high-dose cytarabine and GO. Allo-SCT is reserved for later should the AML relapse. Note that dasatinib, a c-KIT inhibitor, can be added to the treatment regimens as per the results of the CALGB 10801 protocol.64 Also, autologous SCT, instead of allo-SCT, can be considered in rare situations with relapsed core-binding factor AML (especially with inv(16) AML, younger patients, longer time in complete remission prior to relapse, and use of GO).
Case 2 Conclusion
The patient is deemed suitable for intensive chemotherapy. As such, CPX-351 is given in induction and consolidation and complete remission is achieved. Because he has adverse-risk AML, an allo-SCT is planned, but the patient relapses before it can be performed. Following 3 courses of decitabine therapy, the patient achieves complete remission once again but declines transplant. He maintains remission for an additional 4 months but then the leukemia progresses. Clinical trials are recommended to the patient, but he decides to pursue hospice care.
Conclusion
AML is the most common acute leukemia in adults. As defined currently, AML represents a group of related but distinct myeloid disorders that are characterized by various chromosomal, genetic, and epigenetic alterations. Early diagnosis and treatment can help prevent the emergence or manage the detrimental effects of its various complications such as leukostasis and tumor lysis syndrome. Improvements in supportive care, incremental treatment advances, and the wide adoption of allo-SCT for less than favorable cases have significantly improved survival of AML patients since the initial design of combinatorial (7+3) induction chemotherapy, particularly in patients presenting at a younger age. HMAs and the emergence of targeted therapies like FLT-3 and IDH2 inhibitors have added to our therapeutic armamentarium. Despite these advances, long-term survival rates in AML patients continue to be only approximately 40% to 50%. Older patients (particularly those over age 65 at the time of diagnosis), those with relapsed disease, and those with AML with certain unfavorable genetic abnormalities continue to have dismal outcomes. The design of newer targeted therapies, epigenetic agents, and immunotherapies will hopefully address this unmet need.
1. Dohner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med 2015;373:1136–52.
2. National Cancer Institute. Surveillance, Epidemiology, and End Results (SEER) Program. Cancer Stat Facts. Leukemia: Acute Myeloid Leukemia (AML). 2018;2018.
3. Dohner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017;129:424–47.
4. Liesveld JL, Lichtman MA. Acute myelogenous leukemia. In: Kaushansky K, Lichtman MA, Prchal JT, et al, eds. New York: Williams Hematology. 9th ed. New York: McGraw-Hill Education; 2015.
5. Randhawa JK, Khoury J, Ravandi-Kashani F. Adult acute myeloid leukemia. In: Kantarjian HM, Wolff RA, eds. The MD Anderson Manual of Medical Oncology. 3rd ed. New York: McGraw-Hill Medical; 2016.
6. Armstrong SA, Staunton JE, Silverman LB, et al. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet 2002;30:41–7.
7. Graubert TA, Mardis ER. Genomics of acute myeloid leukemia. Cancer J 2011;17:487–91.
8. Cancer Genome Atlas Research Network, Ley TJ, Miller C, Ding L, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 2013;368:2059–74.
9. Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med 2016;374:2209–21.
10. Lindsley RC, Mar BG, Mazzola E, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 2015;125:1367–76.
11. Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014;371:2488–98.
12. Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015;126:9–16.
13. Jaiswal S, Natarajan P, Silver AJ, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med 2017;377:111–21.
14. Genovese G, Kahler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014;371:2477–87.
15. Pileri S, Ascani S, Cox M, et al. Myeloid sarcoma: clinico-pathologic, phenotypic and cytogenetic analysis of 92 adult patients. Leukemia 2007;21:340–50.
16. Vachhani P, Bose P. Isolated gastric myeloid sarcoma: a case report and review of the literature. Case Rep Hematol 2014;2014:541807.
17. Rowe JM. Clinical and laboratory features of the myeloid and lymphocytic leukemias. Am J Med Technol 1983;49:103–9.
18. Wolach O, Stone RM. Mixed-phenotype acute leukemia: current challenges in diagnosis and therapy. Curr Opin Hematol 2017;24:139–45.
19. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016;127:2391–405.
20. Assi R, Ravandi F. FLT3 inhibitors in acute myeloid leukemia: Choosing the best when the optimal does not exist. Am J Hematol 2018;93:553–63.
21. Patel JP, Gonen M, Figueroa ME, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med 2012;366:1079–89.
22. Ley TJ, Ding L, Walter MJ, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med 2010;363:2424–33.
23. Dores GM, Devesa SS, Curtis RE, et al. Acute leukemia incidence and patient survival among children and adults in the United States, 2001-2007. Blood 2012;119:34–43.
24. Cairoli R, Beghini A, Grillo G, et al. Prognostic impact of c-KIT mutations in core binding factor leukemias: an Italian retrospective study. Blood 2006;107:3463–8.
25. Sorror ML, Storer BE, Elsawy M, et al. Intensive versus non-intensive induction therapy for patients (Pts) with newly diagnosed acute myeloid leukemia (AML) using two different novel prognostic models [abstract]. Blood 2016;128(22):216.
26. Quintás-Cardama A, Ravandi F, Liu-Dumlao T, et al. Epigenetic therapy is associated with similar survival compared with intensive chemotherapy in older patients with newly diagnosed acute myeloid leukemia. Blood 2012;120;4840-5.
27. Gupta N, Miller A, Gandhi Set al. Comparison of epigenetic versus standard induction chemotherapy for newly diagnosed acute myeloid leukemia patients ≥60 years old.Am J Hematol 2015;90:639-46.
28. Sorror ML, Storer BE, Fathi AT, et al. Development and validation of a novel acute myeloid leukemia-composite model to estimate risks of mortality. JAMA Oncol 2017;3:1675–82.
29. Rowe JM, Neuberg D, Friedenberg W, et al. A phase 3 study of three induction regimens and of priming with GM-CSF in older adults with acute myeloid leukemia: a trial by the Eastern Cooperative Oncology Group. Blood 2004;103:479–85.
30. Mandelli F, Vignetti M, Suciu S, et al. Daunorubicin versus mitoxantrone versus idarubicin as induction and consolidation chemotherapy for adults with acute myeloid leukemia: the EORTC and GIMEMA Groups Study AML-10. J Clin Oncol 2009;27:5397–403.
31. Gardin C, Chevret S, Pautas C, et al. Superior long-term outcome with idarubicin compared with high-dose daunorubicin in patients with acute myeloid leukemia age 50 years and older. J Clin Oncol 2013;31:321–7.
32. Fernandez HF, Sun Z, Yao X, et al. Anthracycline dose intensification in acute myeloid leukemia. N Engl J Med 2009;361:1249–59.
33. Lee JH, Joo YD, Kim H, et al. A randomized trial comparing standard versus high-dose daunorubicin induction in patients with acute myeloid leukemia. Blood 2011;118:3832–41.
34. Lowenberg B, Ossenkoppele GJ, van Putten W, et al. High-dose daunorubicin in older patients with acute myeloid leukemia. N Engl J Med 2009;361:1235–48.
35. Burnett AK, Russell NH, Hills RK, et al. A randomized comparison of daunorubicin 90 mg/m2 vs 60 mg/m2 in AML induction: results from the UK NCRI AML17 trial in 1206 patients. Blood 2015;125:3878–85.
36. Devillier R, Bertoli S, Prebet T, et al. Comparison of 60 or 90 mg/m(2) of daunorubicin in induction therapy for acute myeloid leukemia with intermediate or unfavorable cytogenetics. Am J Hematol 2015;90:E29–30.
37. Pautas C, Merabet F, Thomas X, et al. Randomized study of intensified anthracycline doses for induction and recombinant interleukin-2 for maintenance in patients with acute myeloid leukemia age 50 to 70 years: results of the ALFA-9801 study. J Clin Oncol 2010;28:808–14.
38. Lowenberg B. Sense and nonsense of high-dose cytarabine for acute myeloid leukemia. Blood 2013;121:26–8.
39. Lancet JE, Uy GL, Cortes JE, et al. Final results of a phase III randomized trial of CPX-351 versus 7 + 3 in older patients with newly diagnosed high risk (secondary) AML [abstract]. J Clin Oncol 2016;34(15_suppl):7000-7000.
40. Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med 2017;377:454–64.
41. Jen EY, Ko CW, Lee JE, et al. FDA approval: Gemtuzumab ozogamicin for the treatment of adults with newly-diagnosed CD33-positive acute myeloid leukemia. Clin Cancer Res 2018; doi: 10.1158/1078-0432. CCR-17-3179.
42. Sievers EL, Larson RA, Stadtmauer EA, et al. Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukemia in first relapse. J Clin Oncol 2001;19:3244–54.
43. Petersdorf SH, Kopecky KJ, Slovak M, et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 2013;121:4854–60.
44. Burnett AK, Russell NH, Hills RK, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy improves survival in older patients with acute myeloid leukemia. J Clin Oncol 2012;30:3924–31.
45. Castaigne S, Pautas C, Terre C, et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet 2012;379:1508–16.
46. Kantarjian HM, Thomas XG, Dmoszynska A, et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J Clin Oncol 2012;30:2670–7.
47. Dombret H, Seymour JF, Butrym A, et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood 2015;126:291–9.
48. Welch JS, Petti AA, Miller CA, et al. TP53 and decitabine in acute myeloid leukemia and myelodysplastic syndromes. N Engl J Med 2016;375:2023–36.
49. Amadori S, Suciu S, Selleslag D, et al. Gemtuzumab ozogamicin versus best supportive care in older patients with newly diagnosed acute myeloid leukemia unsuitable for intensive chemotherapy: results of the randomized phase III EORTC-GIMEMA AML-19 trial. J Clin Oncol 2016;34:972–9.
50. Miyawaki S, Ohtake S, Fujisawa S, et al. A randomized comparison of 4 courses of standard-dose multiagent chemotherapy versus 3 courses of high-dose cytarabine alone in postremission therapy for acute myeloid leukemia in adults: the JALSG AML201 Study. Blood 2011;117:2366–72.
51. Schuurhuis GJ, Heuser M, Freeman S, et al. Minimal/measurable residual disease in AML: a consensus document from the European LeukemiaNet MRD Working Party. Blood 2018;131:1275–91.
52. Koreth J, Schlenk R, Kopecky KJ, et al. Allogeneic stem cell transplantation for acute myeloid leukemia in first complete remission: systematic review and meta-analysis of prospective clinical trials. JAMA 2009;301:2349–61.
53. Schlenk RF, Dohner K, Krauter J, et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med 2008;358:1909–18.
54. Pasquini MC, Logan B, Wu J, et al. Results of a phase III randomized, multi-center study of allogeneic stem cell transplantation after high versus reduced intensity conditioning in patients with myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML): Blood and Marrow Transplant Clinical Trials Network (BMT CTN) 0901. Blood 2015;126:LBA–8.
55. Bose P, Vachhani P, Cortes JE. Treatment of relapsed/refractory acute myeloid leukemia. Curr Treat Options Oncol 2017;18:17,017-0456-2.
56. Burnett AK, Goldstone A, Hills RK, et al. Curability of patients with acute myeloid leukemia who did not undergo transplantation in first remission. J Clin Oncol 2013;31:1293–301.
57. Ravandi F, Ritchie EK, Sayar H, et al. Vosaroxin plus cytarabine versus placebo plus cytarabine in patients with first relapsed or refractory acute myeloid leukaemia (VALOR): a randomised, controlled, double-blind, multinational, phase 3 study. Lancet Oncol 2015;16:1025–36.
58. Taksin AL, Legrand O, Raffoux E, et al. High efficacy and safety profile of fractionated doses of Mylotarg as induction therapy in patients with relapsed acute myeloblastic leukemia: a prospective study of the alfa group. Leukemia 2007;21:66–71.
59. Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017;130:722–31.
60. Fathi AT, DiNardo CD, Kline I, et al. Differentiation syndrome associated with enasidenib, a selective inhibitor of mutant isocitrate dehydrogenase 2: analysis of a phase 1/2 study. JAMA Oncol 2018;doi: 10.1001/jamaoncol.2017.4695.
61. Bejanyan N, Weisdorf DJ, Logan BR, et al. Survival of patients with acute myeloid leukemia relapsing after allogeneic hematopoietic cell transplantation: a center for international blood and marrow transplant research study. Biol Blood Marrow Transplant 2015;21:454–9.
62. Schroeder T, Rachlis E, Bug G, et al. Treatment of acute myeloid leukemia or myelodysplastic syndrome relapse after allogeneic stem cell transplantation with azacitidine and donor lymphocyte infusions--a retrospective multicenter analysis from the German Cooperative Transplant Study Group. Biol Blood Marrow Transplant 2015;21:653–60.
63. Davids MS, Kim HT, Bachireddy P, et al. Ipilimumab for patients with relapse after allogeneic transplantation. N Engl J Med 2016;375:143–53.
64. Marcucci G, Geyer S, Zhao W, et al. Adding KIT inhibitor dasatinib (DAS) to chemotherapy overcomes the negative impact of KIT mutation/over-expression in core binding factor (CBF) acute myeloid leukemia (AML): results from CALGB 10801 (Alliance) [abstract]. Blood 2014;124:8.
1. Dohner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med 2015;373:1136–52.
2. National Cancer Institute. Surveillance, Epidemiology, and End Results (SEER) Program. Cancer Stat Facts. Leukemia: Acute Myeloid Leukemia (AML). 2018;2018.
3. Dohner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017;129:424–47.
4. Liesveld JL, Lichtman MA. Acute myelogenous leukemia. In: Kaushansky K, Lichtman MA, Prchal JT, et al, eds. New York: Williams Hematology. 9th ed. New York: McGraw-Hill Education; 2015.
5. Randhawa JK, Khoury J, Ravandi-Kashani F. Adult acute myeloid leukemia. In: Kantarjian HM, Wolff RA, eds. The MD Anderson Manual of Medical Oncology. 3rd ed. New York: McGraw-Hill Medical; 2016.
6. Armstrong SA, Staunton JE, Silverman LB, et al. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet 2002;30:41–7.
7. Graubert TA, Mardis ER. Genomics of acute myeloid leukemia. Cancer J 2011;17:487–91.
8. Cancer Genome Atlas Research Network, Ley TJ, Miller C, Ding L, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 2013;368:2059–74.
9. Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med 2016;374:2209–21.
10. Lindsley RC, Mar BG, Mazzola E, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 2015;125:1367–76.
11. Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014;371:2488–98.
12. Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015;126:9–16.
13. Jaiswal S, Natarajan P, Silver AJ, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med 2017;377:111–21.
14. Genovese G, Kahler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014;371:2477–87.
15. Pileri S, Ascani S, Cox M, et al. Myeloid sarcoma: clinico-pathologic, phenotypic and cytogenetic analysis of 92 adult patients. Leukemia 2007;21:340–50.
16. Vachhani P, Bose P. Isolated gastric myeloid sarcoma: a case report and review of the literature. Case Rep Hematol 2014;2014:541807.
17. Rowe JM. Clinical and laboratory features of the myeloid and lymphocytic leukemias. Am J Med Technol 1983;49:103–9.
18. Wolach O, Stone RM. Mixed-phenotype acute leukemia: current challenges in diagnosis and therapy. Curr Opin Hematol 2017;24:139–45.
19. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016;127:2391–405.
20. Assi R, Ravandi F. FLT3 inhibitors in acute myeloid leukemia: Choosing the best when the optimal does not exist. Am J Hematol 2018;93:553–63.
21. Patel JP, Gonen M, Figueroa ME, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med 2012;366:1079–89.
22. Ley TJ, Ding L, Walter MJ, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med 2010;363:2424–33.
23. Dores GM, Devesa SS, Curtis RE, et al. Acute leukemia incidence and patient survival among children and adults in the United States, 2001-2007. Blood 2012;119:34–43.
24. Cairoli R, Beghini A, Grillo G, et al. Prognostic impact of c-KIT mutations in core binding factor leukemias: an Italian retrospective study. Blood 2006;107:3463–8.
25. Sorror ML, Storer BE, Elsawy M, et al. Intensive versus non-intensive induction therapy for patients (Pts) with newly diagnosed acute myeloid leukemia (AML) using two different novel prognostic models [abstract]. Blood 2016;128(22):216.
26. Quintás-Cardama A, Ravandi F, Liu-Dumlao T, et al. Epigenetic therapy is associated with similar survival compared with intensive chemotherapy in older patients with newly diagnosed acute myeloid leukemia. Blood 2012;120;4840-5.
27. Gupta N, Miller A, Gandhi Set al. Comparison of epigenetic versus standard induction chemotherapy for newly diagnosed acute myeloid leukemia patients ≥60 years old.Am J Hematol 2015;90:639-46.
28. Sorror ML, Storer BE, Fathi AT, et al. Development and validation of a novel acute myeloid leukemia-composite model to estimate risks of mortality. JAMA Oncol 2017;3:1675–82.
29. Rowe JM, Neuberg D, Friedenberg W, et al. A phase 3 study of three induction regimens and of priming with GM-CSF in older adults with acute myeloid leukemia: a trial by the Eastern Cooperative Oncology Group. Blood 2004;103:479–85.
30. Mandelli F, Vignetti M, Suciu S, et al. Daunorubicin versus mitoxantrone versus idarubicin as induction and consolidation chemotherapy for adults with acute myeloid leukemia: the EORTC and GIMEMA Groups Study AML-10. J Clin Oncol 2009;27:5397–403.
31. Gardin C, Chevret S, Pautas C, et al. Superior long-term outcome with idarubicin compared with high-dose daunorubicin in patients with acute myeloid leukemia age 50 years and older. J Clin Oncol 2013;31:321–7.
32. Fernandez HF, Sun Z, Yao X, et al. Anthracycline dose intensification in acute myeloid leukemia. N Engl J Med 2009;361:1249–59.
33. Lee JH, Joo YD, Kim H, et al. A randomized trial comparing standard versus high-dose daunorubicin induction in patients with acute myeloid leukemia. Blood 2011;118:3832–41.
34. Lowenberg B, Ossenkoppele GJ, van Putten W, et al. High-dose daunorubicin in older patients with acute myeloid leukemia. N Engl J Med 2009;361:1235–48.
35. Burnett AK, Russell NH, Hills RK, et al. A randomized comparison of daunorubicin 90 mg/m2 vs 60 mg/m2 in AML induction: results from the UK NCRI AML17 trial in 1206 patients. Blood 2015;125:3878–85.
36. Devillier R, Bertoli S, Prebet T, et al. Comparison of 60 or 90 mg/m(2) of daunorubicin in induction therapy for acute myeloid leukemia with intermediate or unfavorable cytogenetics. Am J Hematol 2015;90:E29–30.
37. Pautas C, Merabet F, Thomas X, et al. Randomized study of intensified anthracycline doses for induction and recombinant interleukin-2 for maintenance in patients with acute myeloid leukemia age 50 to 70 years: results of the ALFA-9801 study. J Clin Oncol 2010;28:808–14.
38. Lowenberg B. Sense and nonsense of high-dose cytarabine for acute myeloid leukemia. Blood 2013;121:26–8.
39. Lancet JE, Uy GL, Cortes JE, et al. Final results of a phase III randomized trial of CPX-351 versus 7 + 3 in older patients with newly diagnosed high risk (secondary) AML [abstract]. J Clin Oncol 2016;34(15_suppl):7000-7000.
40. Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med 2017;377:454–64.
41. Jen EY, Ko CW, Lee JE, et al. FDA approval: Gemtuzumab ozogamicin for the treatment of adults with newly-diagnosed CD33-positive acute myeloid leukemia. Clin Cancer Res 2018; doi: 10.1158/1078-0432. CCR-17-3179.
42. Sievers EL, Larson RA, Stadtmauer EA, et al. Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukemia in first relapse. J Clin Oncol 2001;19:3244–54.
43. Petersdorf SH, Kopecky KJ, Slovak M, et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 2013;121:4854–60.
44. Burnett AK, Russell NH, Hills RK, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy improves survival in older patients with acute myeloid leukemia. J Clin Oncol 2012;30:3924–31.
45. Castaigne S, Pautas C, Terre C, et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet 2012;379:1508–16.
46. Kantarjian HM, Thomas XG, Dmoszynska A, et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J Clin Oncol 2012;30:2670–7.
47. Dombret H, Seymour JF, Butrym A, et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood 2015;126:291–9.
48. Welch JS, Petti AA, Miller CA, et al. TP53 and decitabine in acute myeloid leukemia and myelodysplastic syndromes. N Engl J Med 2016;375:2023–36.
49. Amadori S, Suciu S, Selleslag D, et al. Gemtuzumab ozogamicin versus best supportive care in older patients with newly diagnosed acute myeloid leukemia unsuitable for intensive chemotherapy: results of the randomized phase III EORTC-GIMEMA AML-19 trial. J Clin Oncol 2016;34:972–9.
50. Miyawaki S, Ohtake S, Fujisawa S, et al. A randomized comparison of 4 courses of standard-dose multiagent chemotherapy versus 3 courses of high-dose cytarabine alone in postremission therapy for acute myeloid leukemia in adults: the JALSG AML201 Study. Blood 2011;117:2366–72.
51. Schuurhuis GJ, Heuser M, Freeman S, et al. Minimal/measurable residual disease in AML: a consensus document from the European LeukemiaNet MRD Working Party. Blood 2018;131:1275–91.
52. Koreth J, Schlenk R, Kopecky KJ, et al. Allogeneic stem cell transplantation for acute myeloid leukemia in first complete remission: systematic review and meta-analysis of prospective clinical trials. JAMA 2009;301:2349–61.
53. Schlenk RF, Dohner K, Krauter J, et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med 2008;358:1909–18.
54. Pasquini MC, Logan B, Wu J, et al. Results of a phase III randomized, multi-center study of allogeneic stem cell transplantation after high versus reduced intensity conditioning in patients with myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML): Blood and Marrow Transplant Clinical Trials Network (BMT CTN) 0901. Blood 2015;126:LBA–8.
55. Bose P, Vachhani P, Cortes JE. Treatment of relapsed/refractory acute myeloid leukemia. Curr Treat Options Oncol 2017;18:17,017-0456-2.
56. Burnett AK, Goldstone A, Hills RK, et al. Curability of patients with acute myeloid leukemia who did not undergo transplantation in first remission. J Clin Oncol 2013;31:1293–301.
57. Ravandi F, Ritchie EK, Sayar H, et al. Vosaroxin plus cytarabine versus placebo plus cytarabine in patients with first relapsed or refractory acute myeloid leukaemia (VALOR): a randomised, controlled, double-blind, multinational, phase 3 study. Lancet Oncol 2015;16:1025–36.
58. Taksin AL, Legrand O, Raffoux E, et al. High efficacy and safety profile of fractionated doses of Mylotarg as induction therapy in patients with relapsed acute myeloblastic leukemia: a prospective study of the alfa group. Leukemia 2007;21:66–71.
59. Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017;130:722–31.
60. Fathi AT, DiNardo CD, Kline I, et al. Differentiation syndrome associated with enasidenib, a selective inhibitor of mutant isocitrate dehydrogenase 2: analysis of a phase 1/2 study. JAMA Oncol 2018;doi: 10.1001/jamaoncol.2017.4695.
61. Bejanyan N, Weisdorf DJ, Logan BR, et al. Survival of patients with acute myeloid leukemia relapsing after allogeneic hematopoietic cell transplantation: a center for international blood and marrow transplant research study. Biol Blood Marrow Transplant 2015;21:454–9.
62. Schroeder T, Rachlis E, Bug G, et al. Treatment of acute myeloid leukemia or myelodysplastic syndrome relapse after allogeneic stem cell transplantation with azacitidine and donor lymphocyte infusions--a retrospective multicenter analysis from the German Cooperative Transplant Study Group. Biol Blood Marrow Transplant 2015;21:653–60.
63. Davids MS, Kim HT, Bachireddy P, et al. Ipilimumab for patients with relapse after allogeneic transplantation. N Engl J Med 2016;375:143–53.
64. Marcucci G, Geyer S, Zhao W, et al. Adding KIT inhibitor dasatinib (DAS) to chemotherapy overcomes the negative impact of KIT mutation/over-expression in core binding factor (CBF) acute myeloid leukemia (AML): results from CALGB 10801 (Alliance) [abstract]. Blood 2014;124:8.