User login
Study reveals lack of sexual aids for cancer survivors

ORLANDO—A new study suggests many US cancer centers do not have therapeutic aids for patients who experience sexual dysfunction after cancer treatment.
Of 25 cancer centers polled, 80% said they had no sexual aids available on site for men, and 64% said they had no such aids for women.
Sharon Bober, PhD, of the Dana-Farber Cancer Institute in Boston, Massachusetts, and her colleagues presented this research at the 2018 Cancer Survivorship Symposium (abstract 134*).
“[P]roviding sexual aids is one step toward treating sexual health like any other aspect of survivorship care,” Dr Bober said.
“It should be no different than providing wigs and head coverings to women who have lost their hair due to chemotherapy. It’s important to give patients the message that regaining sexual health is a perfectly valid and life-affirming aspect of regaining overall quality of life.”
Dr Bober and her colleagues conducted this study to determine the availability of sexual aids at 25 National Cancer Institute-designated cancer centers.
The researchers called these centers posing as a spouse, adult child, or sibling of a patient. The team made separate calls to ask about sexual aids for women and those for men.
Women’s sexual aids
Twenty-four percent of cancer centers (n=6) said they had sexual aids for women, 64% (n=16) did not, and 12% of centers were unreachable (n=3).
The most common aids were personal lubrication, vaginal moisturizer, and vaginal dilators—all of which were available at 5 centers.
Three centers had vibrators, 2 had books/pamphlets, 2 had pelvic floor exercisers, and 2 had product lists.
Men’s sexual aids
Twelve percent of cancer centers (n=3) said they had sexual aids for men, 80% (n=20) did not, and 8% (n=2) were unreachable.
Two centers said they had personal lubrication available for men, 2 had penile support rings, 1 had vacuum erection devices, and 1 had books/pamphlets.
Next steps
Now, Dr Bober and her colleagues hope to query the other 44 National Cancer Institute-designated cancer centers to see what products they are selling and perhaps conduct patient surveys to find out what types of resources are most useful for cancer survivors.
“What we really need to do is go to the centers that are successfully providing sexual health products and find out how they promote and provide resources to their patients,” Dr Bober said.
“We can’t keep the conversation at the 10,000-foot level. We need to talk concretely about how to partner with providers to make sexual health resources, including sexual health aids, available so cancer survivors can get the help that they need.”
*Information presented differs from the abstract.
ORLANDO—A new study suggests many US cancer centers do not have therapeutic aids for patients who experience sexual dysfunction after cancer treatment.
Of 25 cancer centers polled, 80% said they had no sexual aids available on site for men, and 64% said they had no such aids for women.
Sharon Bober, PhD, of the Dana-Farber Cancer Institute in Boston, Massachusetts, and her colleagues presented this research at the 2018 Cancer Survivorship Symposium (abstract 134*).
“[P]roviding sexual aids is one step toward treating sexual health like any other aspect of survivorship care,” Dr Bober said.
“It should be no different than providing wigs and head coverings to women who have lost their hair due to chemotherapy. It’s important to give patients the message that regaining sexual health is a perfectly valid and life-affirming aspect of regaining overall quality of life.”
Dr Bober and her colleagues conducted this study to determine the availability of sexual aids at 25 National Cancer Institute-designated cancer centers.
The researchers called these centers posing as a spouse, adult child, or sibling of a patient. The team made separate calls to ask about sexual aids for women and those for men.
Women’s sexual aids
Twenty-four percent of cancer centers (n=6) said they had sexual aids for women, 64% (n=16) did not, and 12% of centers were unreachable (n=3).
The most common aids were personal lubrication, vaginal moisturizer, and vaginal dilators—all of which were available at 5 centers.
Three centers had vibrators, 2 had books/pamphlets, 2 had pelvic floor exercisers, and 2 had product lists.
Men’s sexual aids
Twelve percent of cancer centers (n=3) said they had sexual aids for men, 80% (n=20) did not, and 8% (n=2) were unreachable.
Two centers said they had personal lubrication available for men, 2 had penile support rings, 1 had vacuum erection devices, and 1 had books/pamphlets.
Next steps
Now, Dr Bober and her colleagues hope to query the other 44 National Cancer Institute-designated cancer centers to see what products they are selling and perhaps conduct patient surveys to find out what types of resources are most useful for cancer survivors.
“What we really need to do is go to the centers that are successfully providing sexual health products and find out how they promote and provide resources to their patients,” Dr Bober said.
“We can’t keep the conversation at the 10,000-foot level. We need to talk concretely about how to partner with providers to make sexual health resources, including sexual health aids, available so cancer survivors can get the help that they need.”
*Information presented differs from the abstract.
ORLANDO—A new study suggests many US cancer centers do not have therapeutic aids for patients who experience sexual dysfunction after cancer treatment.
Of 25 cancer centers polled, 80% said they had no sexual aids available on site for men, and 64% said they had no such aids for women.
Sharon Bober, PhD, of the Dana-Farber Cancer Institute in Boston, Massachusetts, and her colleagues presented this research at the 2018 Cancer Survivorship Symposium (abstract 134*).
“[P]roviding sexual aids is one step toward treating sexual health like any other aspect of survivorship care,” Dr Bober said.
“It should be no different than providing wigs and head coverings to women who have lost their hair due to chemotherapy. It’s important to give patients the message that regaining sexual health is a perfectly valid and life-affirming aspect of regaining overall quality of life.”
Dr Bober and her colleagues conducted this study to determine the availability of sexual aids at 25 National Cancer Institute-designated cancer centers.
The researchers called these centers posing as a spouse, adult child, or sibling of a patient. The team made separate calls to ask about sexual aids for women and those for men.
Women’s sexual aids
Twenty-four percent of cancer centers (n=6) said they had sexual aids for women, 64% (n=16) did not, and 12% of centers were unreachable (n=3).
The most common aids were personal lubrication, vaginal moisturizer, and vaginal dilators—all of which were available at 5 centers.
Three centers had vibrators, 2 had books/pamphlets, 2 had pelvic floor exercisers, and 2 had product lists.
Men’s sexual aids
Twelve percent of cancer centers (n=3) said they had sexual aids for men, 80% (n=20) did not, and 8% (n=2) were unreachable.
Two centers said they had personal lubrication available for men, 2 had penile support rings, 1 had vacuum erection devices, and 1 had books/pamphlets.
Next steps
Now, Dr Bober and her colleagues hope to query the other 44 National Cancer Institute-designated cancer centers to see what products they are selling and perhaps conduct patient surveys to find out what types of resources are most useful for cancer survivors.
“What we really need to do is go to the centers that are successfully providing sexual health products and find out how they promote and provide resources to their patients,” Dr Bober said.
“We can’t keep the conversation at the 10,000-foot level. We need to talk concretely about how to partner with providers to make sexual health resources, including sexual health aids, available so cancer survivors can get the help that they need.”
*Information presented differs from the abstract.
FDA grants ivosidenib NDA priority review

The US Food and Drug Administration (FDA) has accepted for priority review the new drug application (NDA) for ivosidenib, a targeted inhibitor of mutant IDH1.
With this NDA, Agios Pharmaceuticals, Inc., is seeking approval for ivosidenib (formerly AG-120) to treat patients with relapsed or refractory acute myeloid leukemia (AML) with an IDH1 mutation.
The FDA expects to make a decision on the NDA by August 21, 2018.
The agency aims to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
Phase 1 data
The priority review for the ivosidenib NDA is based on results from AG120-C-001, a phase 1 trial of patients with advanced hematologic malignancies and an IDH1 mutation. Data from this study were presented at the 2017 ASH Annual Meeting (abstract 725).
This ongoing trial includes a dose-escalation phase and 4 expansion arms. Ivosidenib doses ranged from 200 mg to 1200 mg in the dose-escalation phase. Patients in the dose-expansion arms received a 500 mg daily dose of the drug.
Arm 1 includes IDH1-mutant-positive AML patients who relapsed after bone marrow transplant, were in second or later relapse, were refractory to initial induction or re-induction treatment, or who relapsed within a year of initial treatment, excluding patients with favorable-risk status.
Arms 2, 3 and 4 were not included in the primary efficacy analysis.
The primary analysis set consists of 125 relapsed/refractory AML patients—92 from arm 1 of the expansion and 33 patients from the dose-escalation who met the eligibility criteria for arm 1 and received ivosidenib at 500 mg once daily.
The median age of these patients was 67 (range, 18-87), and the median number of prior regimens they received was 2 (range, 1-6).
The primary endpoint for these patients is the rate of complete response (CR) and CR with partial hematologic recovery (CRh), which was 30.4%. The CR rate was 21.6% (27/125), and the CRh rate was 8.8% (11/125).
The overall response rate was 41.6% (52/125). The median duration of response was 6.5 months for all patients, 9.3 months for those who achieved a CR, and 8.2 months for those who had a CR/CRh.
At the time of the data cut-off, the median overall survival was 8.8 months. The median overall survival was not reached for patients who achieved a CR/CRh, was 9.3 months for non-CR/CRh responders, and was 3.9 months for non-responders.
There were a few adverse events of interest. Eight percent of patients reported grade 3 or higher leukocytosis, which was managed with hydroxyurea, and none of the cases were fatal.
Eight percent of patients reported grade 3 QT prolongation. Ivosidenib was reduced in 1 patient and held in 5 patients (for any grade of QT prolongation). There were no grade 4 or 5 cases of QT prolongation.
Finally, 9.6% of patients reported IDH-differentiation syndrome, which was managed with corticosteroids and diuretics. None of the cases were grade 4 or 5.
Companion diagnostic
Abbott has submitted a premarket approval application to the FDA for an IDH1 assay to be used on the Abbott m2000 RealTime System, an automated sample preparation and batch analyzer system for nucleic acid amplification and detection.
In 2014, Abbott and Agios entered into an exclusive agreement under which Abbott is responsible for the development and commercialization of a RealTime PCR assay for detection of the IDH1 mutation in bone marrow and blood. The Abbott assay is intended to serve as a companion diagnostic for ivosidenib.
The US Food and Drug Administration (FDA) has accepted for priority review the new drug application (NDA) for ivosidenib, a targeted inhibitor of mutant IDH1.
With this NDA, Agios Pharmaceuticals, Inc., is seeking approval for ivosidenib (formerly AG-120) to treat patients with relapsed or refractory acute myeloid leukemia (AML) with an IDH1 mutation.
The FDA expects to make a decision on the NDA by August 21, 2018.
The agency aims to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
Phase 1 data
The priority review for the ivosidenib NDA is based on results from AG120-C-001, a phase 1 trial of patients with advanced hematologic malignancies and an IDH1 mutation. Data from this study were presented at the 2017 ASH Annual Meeting (abstract 725).
This ongoing trial includes a dose-escalation phase and 4 expansion arms. Ivosidenib doses ranged from 200 mg to 1200 mg in the dose-escalation phase. Patients in the dose-expansion arms received a 500 mg daily dose of the drug.
Arm 1 includes IDH1-mutant-positive AML patients who relapsed after bone marrow transplant, were in second or later relapse, were refractory to initial induction or re-induction treatment, or who relapsed within a year of initial treatment, excluding patients with favorable-risk status.
Arms 2, 3 and 4 were not included in the primary efficacy analysis.
The primary analysis set consists of 125 relapsed/refractory AML patients—92 from arm 1 of the expansion and 33 patients from the dose-escalation who met the eligibility criteria for arm 1 and received ivosidenib at 500 mg once daily.
The median age of these patients was 67 (range, 18-87), and the median number of prior regimens they received was 2 (range, 1-6).
The primary endpoint for these patients is the rate of complete response (CR) and CR with partial hematologic recovery (CRh), which was 30.4%. The CR rate was 21.6% (27/125), and the CRh rate was 8.8% (11/125).
The overall response rate was 41.6% (52/125). The median duration of response was 6.5 months for all patients, 9.3 months for those who achieved a CR, and 8.2 months for those who had a CR/CRh.
At the time of the data cut-off, the median overall survival was 8.8 months. The median overall survival was not reached for patients who achieved a CR/CRh, was 9.3 months for non-CR/CRh responders, and was 3.9 months for non-responders.
There were a few adverse events of interest. Eight percent of patients reported grade 3 or higher leukocytosis, which was managed with hydroxyurea, and none of the cases were fatal.
Eight percent of patients reported grade 3 QT prolongation. Ivosidenib was reduced in 1 patient and held in 5 patients (for any grade of QT prolongation). There were no grade 4 or 5 cases of QT prolongation.
Finally, 9.6% of patients reported IDH-differentiation syndrome, which was managed with corticosteroids and diuretics. None of the cases were grade 4 or 5.
Companion diagnostic
Abbott has submitted a premarket approval application to the FDA for an IDH1 assay to be used on the Abbott m2000 RealTime System, an automated sample preparation and batch analyzer system for nucleic acid amplification and detection.
In 2014, Abbott and Agios entered into an exclusive agreement under which Abbott is responsible for the development and commercialization of a RealTime PCR assay for detection of the IDH1 mutation in bone marrow and blood. The Abbott assay is intended to serve as a companion diagnostic for ivosidenib.
The US Food and Drug Administration (FDA) has accepted for priority review the new drug application (NDA) for ivosidenib, a targeted inhibitor of mutant IDH1.
With this NDA, Agios Pharmaceuticals, Inc., is seeking approval for ivosidenib (formerly AG-120) to treat patients with relapsed or refractory acute myeloid leukemia (AML) with an IDH1 mutation.
The FDA expects to make a decision on the NDA by August 21, 2018.
The agency aims to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
Phase 1 data
The priority review for the ivosidenib NDA is based on results from AG120-C-001, a phase 1 trial of patients with advanced hematologic malignancies and an IDH1 mutation. Data from this study were presented at the 2017 ASH Annual Meeting (abstract 725).
This ongoing trial includes a dose-escalation phase and 4 expansion arms. Ivosidenib doses ranged from 200 mg to 1200 mg in the dose-escalation phase. Patients in the dose-expansion arms received a 500 mg daily dose of the drug.
Arm 1 includes IDH1-mutant-positive AML patients who relapsed after bone marrow transplant, were in second or later relapse, were refractory to initial induction or re-induction treatment, or who relapsed within a year of initial treatment, excluding patients with favorable-risk status.
Arms 2, 3 and 4 were not included in the primary efficacy analysis.
The primary analysis set consists of 125 relapsed/refractory AML patients—92 from arm 1 of the expansion and 33 patients from the dose-escalation who met the eligibility criteria for arm 1 and received ivosidenib at 500 mg once daily.
The median age of these patients was 67 (range, 18-87), and the median number of prior regimens they received was 2 (range, 1-6).
The primary endpoint for these patients is the rate of complete response (CR) and CR with partial hematologic recovery (CRh), which was 30.4%. The CR rate was 21.6% (27/125), and the CRh rate was 8.8% (11/125).
The overall response rate was 41.6% (52/125). The median duration of response was 6.5 months for all patients, 9.3 months for those who achieved a CR, and 8.2 months for those who had a CR/CRh.
At the time of the data cut-off, the median overall survival was 8.8 months. The median overall survival was not reached for patients who achieved a CR/CRh, was 9.3 months for non-CR/CRh responders, and was 3.9 months for non-responders.
There were a few adverse events of interest. Eight percent of patients reported grade 3 or higher leukocytosis, which was managed with hydroxyurea, and none of the cases were fatal.
Eight percent of patients reported grade 3 QT prolongation. Ivosidenib was reduced in 1 patient and held in 5 patients (for any grade of QT prolongation). There were no grade 4 or 5 cases of QT prolongation.
Finally, 9.6% of patients reported IDH-differentiation syndrome, which was managed with corticosteroids and diuretics. None of the cases were grade 4 or 5.
Companion diagnostic
Abbott has submitted a premarket approval application to the FDA for an IDH1 assay to be used on the Abbott m2000 RealTime System, an automated sample preparation and batch analyzer system for nucleic acid amplification and detection.
In 2014, Abbott and Agios entered into an exclusive agreement under which Abbott is responsible for the development and commercialization of a RealTime PCR assay for detection of the IDH1 mutation in bone marrow and blood. The Abbott assay is intended to serve as a companion diagnostic for ivosidenib.
NK-cell therapy in resistant MDS, AML

Results of a phase 1/2 trial suggest treatment with haploidentical natural killer (NK) cells can be effective against relapsed/refractory myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML).
NK-cell therapy elicited responses in 6 of the 16 patients studied and provided a bridge to transplant for 5 patients.
Three responders were still alive at more than 3 years of follow-up.
There were 4 grade 3 adverse events (AEs) and 2 grade 5 AEs considered possibly or probably related to NK-cell therapy.
Investigators reported these results in Clinical Cancer Research.
The trial enrolled 16 patients. Eight had MDS/AML, 3 had de novo AML, and 5 had high-risk MDS, including refractory anemia with excess blasts (RAEB) type 1 progressing toward type 2, RAEB-2, and chronic myelomonocytic leukemia type 2.
The patients’ median age was 64 (range, 40-70), and they had received a median of 3 prior therapies (range, 1-6). Six patients had received an allogeneic hematopoietic stem cell transplant (HSCT).
For this study, all patients received fludarabine, cyclophosphamide, and total lymphoid irradiation prior to receiving haploidentical NK cells.
The median follow-up was 8 months for all patients and 28 months for responders.
Efficacy
Six patients responded to treatment. One patient with de novo AML had a complete response (CR). Two high-risk MDS patients had a marrow CR (mCR), as did 2 MDS/AML patients. One MDS/AML patient had a partial response (PR).
Two patients had stable disease (SD)—1 with MDS and 1 with MDS/AML. One patient with de novo AML had a morphologic leukemia-free state after NK-cell therapy.
Five patients proceeded to HSCT—3 in mCR, 1 in PR, and 1 with SD.
Three patients were still alive at last follow-up—1 with MDS who achieved an mCR and went on to HSCT, 1 with MDS/AML who achieved an mCR and went on to HSCT, and 1 with MDS/AML who achieved an mCR and went on to receive chemotherapy and donor lymphocyte infusion.
One survivor has more than 5 years of follow-up (the MDS patient), and the other 2 have more than 3 years of follow-up.
“Our study shows that patients with MDS, AML, and MDS/AML can be treated with NK cell-based immunotherapy and that the therapy can be highly efficacious,” said study author Hans-Gustaf Ljunggren, MD, PhD, of Karolinska Institutet in Stockholm, Sweden.
Safety
The most common AEs of any grade considered possibly or probably related to NK-cell therapy were chills (n=13) and nausea (n=4).
Two patients had cytokine release syndrome (CRS) likely associated with hemophagocytic lymphohistiocytosis (HLH).
Each of the following potentially related AEs were reported once: headache, vomiting, encephalitis infection, sinus tachycardia, bone pain, pain in extremity, and maculopapular rash.
There were 4 grade 3 AEs—CRS/HLH (n=1), chills (n=1), and nausea (n=2)—but no grade 4 AEs.
There were 2 grade 5 AEs—CRS/HLH and encephalitis infection. These occurred in a single patient who died with HLH, human herpes virus-6 encephalitis, and AML relapse.
Two investigators involved in this study serve on the scientific advisory board of Fate Therapeutics. Dr Ljunggren serves on the scientific advisory board of CellProtect, Nordic Pharmaceuticals, and HOPE Bio-Sciences. He is also on the board of directors of Vycellix and is a collaborator with Fate Therapeutics.
Results of a phase 1/2 trial suggest treatment with haploidentical natural killer (NK) cells can be effective against relapsed/refractory myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML).
NK-cell therapy elicited responses in 6 of the 16 patients studied and provided a bridge to transplant for 5 patients.
Three responders were still alive at more than 3 years of follow-up.
There were 4 grade 3 adverse events (AEs) and 2 grade 5 AEs considered possibly or probably related to NK-cell therapy.
Investigators reported these results in Clinical Cancer Research.
The trial enrolled 16 patients. Eight had MDS/AML, 3 had de novo AML, and 5 had high-risk MDS, including refractory anemia with excess blasts (RAEB) type 1 progressing toward type 2, RAEB-2, and chronic myelomonocytic leukemia type 2.
The patients’ median age was 64 (range, 40-70), and they had received a median of 3 prior therapies (range, 1-6). Six patients had received an allogeneic hematopoietic stem cell transplant (HSCT).
For this study, all patients received fludarabine, cyclophosphamide, and total lymphoid irradiation prior to receiving haploidentical NK cells.
The median follow-up was 8 months for all patients and 28 months for responders.
Efficacy
Six patients responded to treatment. One patient with de novo AML had a complete response (CR). Two high-risk MDS patients had a marrow CR (mCR), as did 2 MDS/AML patients. One MDS/AML patient had a partial response (PR).
Two patients had stable disease (SD)—1 with MDS and 1 with MDS/AML. One patient with de novo AML had a morphologic leukemia-free state after NK-cell therapy.
Five patients proceeded to HSCT—3 in mCR, 1 in PR, and 1 with SD.
Three patients were still alive at last follow-up—1 with MDS who achieved an mCR and went on to HSCT, 1 with MDS/AML who achieved an mCR and went on to HSCT, and 1 with MDS/AML who achieved an mCR and went on to receive chemotherapy and donor lymphocyte infusion.
One survivor has more than 5 years of follow-up (the MDS patient), and the other 2 have more than 3 years of follow-up.
“Our study shows that patients with MDS, AML, and MDS/AML can be treated with NK cell-based immunotherapy and that the therapy can be highly efficacious,” said study author Hans-Gustaf Ljunggren, MD, PhD, of Karolinska Institutet in Stockholm, Sweden.
Safety
The most common AEs of any grade considered possibly or probably related to NK-cell therapy were chills (n=13) and nausea (n=4).
Two patients had cytokine release syndrome (CRS) likely associated with hemophagocytic lymphohistiocytosis (HLH).
Each of the following potentially related AEs were reported once: headache, vomiting, encephalitis infection, sinus tachycardia, bone pain, pain in extremity, and maculopapular rash.
There were 4 grade 3 AEs—CRS/HLH (n=1), chills (n=1), and nausea (n=2)—but no grade 4 AEs.
There were 2 grade 5 AEs—CRS/HLH and encephalitis infection. These occurred in a single patient who died with HLH, human herpes virus-6 encephalitis, and AML relapse.
Two investigators involved in this study serve on the scientific advisory board of Fate Therapeutics. Dr Ljunggren serves on the scientific advisory board of CellProtect, Nordic Pharmaceuticals, and HOPE Bio-Sciences. He is also on the board of directors of Vycellix and is a collaborator with Fate Therapeutics.
Results of a phase 1/2 trial suggest treatment with haploidentical natural killer (NK) cells can be effective against relapsed/refractory myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML).
NK-cell therapy elicited responses in 6 of the 16 patients studied and provided a bridge to transplant for 5 patients.
Three responders were still alive at more than 3 years of follow-up.
There were 4 grade 3 adverse events (AEs) and 2 grade 5 AEs considered possibly or probably related to NK-cell therapy.
Investigators reported these results in Clinical Cancer Research.
The trial enrolled 16 patients. Eight had MDS/AML, 3 had de novo AML, and 5 had high-risk MDS, including refractory anemia with excess blasts (RAEB) type 1 progressing toward type 2, RAEB-2, and chronic myelomonocytic leukemia type 2.
The patients’ median age was 64 (range, 40-70), and they had received a median of 3 prior therapies (range, 1-6). Six patients had received an allogeneic hematopoietic stem cell transplant (HSCT).
For this study, all patients received fludarabine, cyclophosphamide, and total lymphoid irradiation prior to receiving haploidentical NK cells.
The median follow-up was 8 months for all patients and 28 months for responders.
Efficacy
Six patients responded to treatment. One patient with de novo AML had a complete response (CR). Two high-risk MDS patients had a marrow CR (mCR), as did 2 MDS/AML patients. One MDS/AML patient had a partial response (PR).
Two patients had stable disease (SD)—1 with MDS and 1 with MDS/AML. One patient with de novo AML had a morphologic leukemia-free state after NK-cell therapy.
Five patients proceeded to HSCT—3 in mCR, 1 in PR, and 1 with SD.
Three patients were still alive at last follow-up—1 with MDS who achieved an mCR and went on to HSCT, 1 with MDS/AML who achieved an mCR and went on to HSCT, and 1 with MDS/AML who achieved an mCR and went on to receive chemotherapy and donor lymphocyte infusion.
One survivor has more than 5 years of follow-up (the MDS patient), and the other 2 have more than 3 years of follow-up.
“Our study shows that patients with MDS, AML, and MDS/AML can be treated with NK cell-based immunotherapy and that the therapy can be highly efficacious,” said study author Hans-Gustaf Ljunggren, MD, PhD, of Karolinska Institutet in Stockholm, Sweden.
Safety
The most common AEs of any grade considered possibly or probably related to NK-cell therapy were chills (n=13) and nausea (n=4).
Two patients had cytokine release syndrome (CRS) likely associated with hemophagocytic lymphohistiocytosis (HLH).
Each of the following potentially related AEs were reported once: headache, vomiting, encephalitis infection, sinus tachycardia, bone pain, pain in extremity, and maculopapular rash.
There were 4 grade 3 AEs—CRS/HLH (n=1), chills (n=1), and nausea (n=2)—but no grade 4 AEs.
There were 2 grade 5 AEs—CRS/HLH and encephalitis infection. These occurred in a single patient who died with HLH, human herpes virus-6 encephalitis, and AML relapse.
Two investigators involved in this study serve on the scientific advisory board of Fate Therapeutics. Dr Ljunggren serves on the scientific advisory board of CellProtect, Nordic Pharmaceuticals, and HOPE Bio-Sciences. He is also on the board of directors of Vycellix and is a collaborator with Fate Therapeutics.
Azacitidine now available in China

Azacitidine for injection (Vidaza®) is now available in China.
The nucleoside metabolic inhibitor was approved in China to treat patients with intermediate-2/high-risk myelodysplastic syndromes (MDS), acute myeloid leukemia (AML) with 20% to 30% bone marrow blasts, and chronic myelomonocytic leukemia (CMML).
Azacitidine for injection is marketed in China by BeiGene Ltd. under an exclusive license from Celgene Corporation.
“Vidaza is the only approved hypomethylating agent shown to prolong survival for patients with MDS and the first new treatment for MDS patients approved in China since 2009,” said John V. Oyler, founder, chief executive officer, and chairman of BeiGene.
“We are excited to announce that the first prescription was made in January 2018. From now on, Chinese patients can benefit from Vidaza in hospitals around China.”
Azacitidine was evaluated in a global phase 3 trial of patients with intermediate-2- and high-risk MDS, CMML, or AML (AZA-001). Results from this trial were published in The Lancet Oncology in 2009.
Patients were randomized to receive azacitidine plus best supportive care (BSC, n=179) or conventional care regimens plus BSC (105 to BSC alone, 49 to low-dose cytarabine, and 25 to chemotherapy with cytarabine and anthracycline).
Azacitidine was given subcutaneously at a dose of 75 mg/m2 daily for 7 consecutive days every 28 days until disease progression, relapse after response, or unacceptable toxicity.
The median overall survival was 24.5 months with azacitidine, compared to 15 months for patients treated with conventional care regimens.
There was a higher hematologic response rate in the azacitidine arm than the conventional care arm—29% and 12%, respectively.
In the azacitidine group, 45% of patients who were dependent on red blood cell transfusions at baseline became transfusion independent, compared with 11% in the conventional care group.
Forty-six percent of patients in the azacitidine arm and 63% in the conventional care arm died.
Grade 3/4 hematologic toxicity (in the azacitidine and conventional care arms, respectively) included neutropenia (91% and 76%), thrombocytopenia (85% and 80%), and anemia (57% and 68%). ![]()
Azacitidine for injection (Vidaza®) is now available in China.
The nucleoside metabolic inhibitor was approved in China to treat patients with intermediate-2/high-risk myelodysplastic syndromes (MDS), acute myeloid leukemia (AML) with 20% to 30% bone marrow blasts, and chronic myelomonocytic leukemia (CMML).
Azacitidine for injection is marketed in China by BeiGene Ltd. under an exclusive license from Celgene Corporation.
“Vidaza is the only approved hypomethylating agent shown to prolong survival for patients with MDS and the first new treatment for MDS patients approved in China since 2009,” said John V. Oyler, founder, chief executive officer, and chairman of BeiGene.
“We are excited to announce that the first prescription was made in January 2018. From now on, Chinese patients can benefit from Vidaza in hospitals around China.”
Azacitidine was evaluated in a global phase 3 trial of patients with intermediate-2- and high-risk MDS, CMML, or AML (AZA-001). Results from this trial were published in The Lancet Oncology in 2009.
Patients were randomized to receive azacitidine plus best supportive care (BSC, n=179) or conventional care regimens plus BSC (105 to BSC alone, 49 to low-dose cytarabine, and 25 to chemotherapy with cytarabine and anthracycline).
Azacitidine was given subcutaneously at a dose of 75 mg/m2 daily for 7 consecutive days every 28 days until disease progression, relapse after response, or unacceptable toxicity.
The median overall survival was 24.5 months with azacitidine, compared to 15 months for patients treated with conventional care regimens.
There was a higher hematologic response rate in the azacitidine arm than the conventional care arm—29% and 12%, respectively.
In the azacitidine group, 45% of patients who were dependent on red blood cell transfusions at baseline became transfusion independent, compared with 11% in the conventional care group.
Forty-six percent of patients in the azacitidine arm and 63% in the conventional care arm died.
Grade 3/4 hematologic toxicity (in the azacitidine and conventional care arms, respectively) included neutropenia (91% and 76%), thrombocytopenia (85% and 80%), and anemia (57% and 68%). ![]()
Azacitidine for injection (Vidaza®) is now available in China.
The nucleoside metabolic inhibitor was approved in China to treat patients with intermediate-2/high-risk myelodysplastic syndromes (MDS), acute myeloid leukemia (AML) with 20% to 30% bone marrow blasts, and chronic myelomonocytic leukemia (CMML).
Azacitidine for injection is marketed in China by BeiGene Ltd. under an exclusive license from Celgene Corporation.
“Vidaza is the only approved hypomethylating agent shown to prolong survival for patients with MDS and the first new treatment for MDS patients approved in China since 2009,” said John V. Oyler, founder, chief executive officer, and chairman of BeiGene.
“We are excited to announce that the first prescription was made in January 2018. From now on, Chinese patients can benefit from Vidaza in hospitals around China.”
Azacitidine was evaluated in a global phase 3 trial of patients with intermediate-2- and high-risk MDS, CMML, or AML (AZA-001). Results from this trial were published in The Lancet Oncology in 2009.
Patients were randomized to receive azacitidine plus best supportive care (BSC, n=179) or conventional care regimens plus BSC (105 to BSC alone, 49 to low-dose cytarabine, and 25 to chemotherapy with cytarabine and anthracycline).
Azacitidine was given subcutaneously at a dose of 75 mg/m2 daily for 7 consecutive days every 28 days until disease progression, relapse after response, or unacceptable toxicity.
The median overall survival was 24.5 months with azacitidine, compared to 15 months for patients treated with conventional care regimens.
There was a higher hematologic response rate in the azacitidine arm than the conventional care arm—29% and 12%, respectively.
In the azacitidine group, 45% of patients who were dependent on red blood cell transfusions at baseline became transfusion independent, compared with 11% in the conventional care group.
Forty-six percent of patients in the azacitidine arm and 63% in the conventional care arm died.
Grade 3/4 hematologic toxicity (in the azacitidine and conventional care arms, respectively) included neutropenia (91% and 76%), thrombocytopenia (85% and 80%), and anemia (57% and 68%). ![]()
Disease burden impacts survival, toxicity after CAR T-cell therapy

Adults with relapsed or refractory acute lymphoblastic leukemia (ALL) have better outcomes if they have a low disease burden when receiving chimeric antigen receptor (CAR) T-cell therapy, according to research published in NEJM.
The final analysis of a phase 1 trial showed that patients with a low disease burden at baseline had superior event-free survival (EFS) and overall survival (OS) after therapy, compared to patients with a high disease burden.
Patients with a low disease burden also had a lower rate of cytokine release syndrome (CRS) and neurotoxic events.
“This is the longest follow-up study of people with ALL treated with CAR therapy,” said study author Jae Park, MD, of Memorial Sloan Kettering Cancer Center in New York, New York.
“With the long follow-up, we were able to demonstrate, for the first time, that patients with a lower disease burden benefited the most from CAR therapy, with significantly improved survival and reduced toxicity.”
The study included 53 adults with ALL who had a median age of 44 (range, 23-74).
They were heavily pretreated, with 68% receiving CAR T-cell therapy as a third or later salvage treatment. Thirty-six percent of patients had received an allogeneic transplant, and 23% had primary refractory disease.
In this trial, patients received a single infusion of 19-28z CAR T cells after conditioning chemotherapy. The maximum follow-up time was 5.5 years, with a median follow-up of 29 months.
In all, 83% of patients achieved a complete response. The median EFS was 6.1 months, and the median OS was 12.9 months.
The median EFS was significantly longer for patients with a low disease burden (<5% bone marrow blasts) compared to a high disease burden (≥5% bone marrow blasts or extramedullary disease)—10.6 months and 5.6 months, respectively (P=0.01).
The same was true for the median OS, which was 20.1 months in patients with a low disease burden and 12.4 months in those with a high disease burden (P=0.02).
For the entire study population, the rate of CRS was 85%, and 26% of patients had severe CRS. One patient died of severe CRS and multi-organ failure before the researchers began modifying the dose of CAR T cells according to the pretreatment disease burden.
Severe CRS occurred in 41% of patients with a high disease burden and 5% of those with a low disease burden.
In the entire study population, 2% of patients had grade 2 neurotoxic effects, 36% had grade 3, 6% had grade 4, and none had grade 5. The rate of severe neurotoxicity was 42%.
Neurotoxic effects occurred in 59% of patients with a high disease burden and 14% of those with a low disease burden.
“Among all of the clinical and disease factors we examined, pretreatment disease burden was the strongest predictor of long-term outcome after CAR therapy,” Dr Park said. “Our data supports the incorporation of CAR therapy in an earlier treatment setting in ALL, when the disease volume is small, so as to achieve the greatest long-term efficacy and lowest toxicity.”
This work was supported by Juno Therapeutics, the National Institutes of Health, the Carson Family Charitable Trust, the Emerald Foundation, the Mr. and Mrs. Goodwyn Commonwealth Fund, the Terry Fox Run for Cancer Research organized by the Canadian Association of New York, Kate’s Team, William Laurence and Blanche Hughes Foundation, the Center for Experimental Therapeutics at Memorial Sloan Kettering, and the Lake Road Foundation. ![]()
Adults with relapsed or refractory acute lymphoblastic leukemia (ALL) have better outcomes if they have a low disease burden when receiving chimeric antigen receptor (CAR) T-cell therapy, according to research published in NEJM.
The final analysis of a phase 1 trial showed that patients with a low disease burden at baseline had superior event-free survival (EFS) and overall survival (OS) after therapy, compared to patients with a high disease burden.
Patients with a low disease burden also had a lower rate of cytokine release syndrome (CRS) and neurotoxic events.
“This is the longest follow-up study of people with ALL treated with CAR therapy,” said study author Jae Park, MD, of Memorial Sloan Kettering Cancer Center in New York, New York.
“With the long follow-up, we were able to demonstrate, for the first time, that patients with a lower disease burden benefited the most from CAR therapy, with significantly improved survival and reduced toxicity.”
The study included 53 adults with ALL who had a median age of 44 (range, 23-74).
They were heavily pretreated, with 68% receiving CAR T-cell therapy as a third or later salvage treatment. Thirty-six percent of patients had received an allogeneic transplant, and 23% had primary refractory disease.
In this trial, patients received a single infusion of 19-28z CAR T cells after conditioning chemotherapy. The maximum follow-up time was 5.5 years, with a median follow-up of 29 months.
In all, 83% of patients achieved a complete response. The median EFS was 6.1 months, and the median OS was 12.9 months.
The median EFS was significantly longer for patients with a low disease burden (<5% bone marrow blasts) compared to a high disease burden (≥5% bone marrow blasts or extramedullary disease)—10.6 months and 5.6 months, respectively (P=0.01).
The same was true for the median OS, which was 20.1 months in patients with a low disease burden and 12.4 months in those with a high disease burden (P=0.02).
For the entire study population, the rate of CRS was 85%, and 26% of patients had severe CRS. One patient died of severe CRS and multi-organ failure before the researchers began modifying the dose of CAR T cells according to the pretreatment disease burden.
Severe CRS occurred in 41% of patients with a high disease burden and 5% of those with a low disease burden.
In the entire study population, 2% of patients had grade 2 neurotoxic effects, 36% had grade 3, 6% had grade 4, and none had grade 5. The rate of severe neurotoxicity was 42%.
Neurotoxic effects occurred in 59% of patients with a high disease burden and 14% of those with a low disease burden.
“Among all of the clinical and disease factors we examined, pretreatment disease burden was the strongest predictor of long-term outcome after CAR therapy,” Dr Park said. “Our data supports the incorporation of CAR therapy in an earlier treatment setting in ALL, when the disease volume is small, so as to achieve the greatest long-term efficacy and lowest toxicity.”
This work was supported by Juno Therapeutics, the National Institutes of Health, the Carson Family Charitable Trust, the Emerald Foundation, the Mr. and Mrs. Goodwyn Commonwealth Fund, the Terry Fox Run for Cancer Research organized by the Canadian Association of New York, Kate’s Team, William Laurence and Blanche Hughes Foundation, the Center for Experimental Therapeutics at Memorial Sloan Kettering, and the Lake Road Foundation. ![]()
Adults with relapsed or refractory acute lymphoblastic leukemia (ALL) have better outcomes if they have a low disease burden when receiving chimeric antigen receptor (CAR) T-cell therapy, according to research published in NEJM.
The final analysis of a phase 1 trial showed that patients with a low disease burden at baseline had superior event-free survival (EFS) and overall survival (OS) after therapy, compared to patients with a high disease burden.
Patients with a low disease burden also had a lower rate of cytokine release syndrome (CRS) and neurotoxic events.
“This is the longest follow-up study of people with ALL treated with CAR therapy,” said study author Jae Park, MD, of Memorial Sloan Kettering Cancer Center in New York, New York.
“With the long follow-up, we were able to demonstrate, for the first time, that patients with a lower disease burden benefited the most from CAR therapy, with significantly improved survival and reduced toxicity.”
The study included 53 adults with ALL who had a median age of 44 (range, 23-74).
They were heavily pretreated, with 68% receiving CAR T-cell therapy as a third or later salvage treatment. Thirty-six percent of patients had received an allogeneic transplant, and 23% had primary refractory disease.
In this trial, patients received a single infusion of 19-28z CAR T cells after conditioning chemotherapy. The maximum follow-up time was 5.5 years, with a median follow-up of 29 months.
In all, 83% of patients achieved a complete response. The median EFS was 6.1 months, and the median OS was 12.9 months.
The median EFS was significantly longer for patients with a low disease burden (<5% bone marrow blasts) compared to a high disease burden (≥5% bone marrow blasts or extramedullary disease)—10.6 months and 5.6 months, respectively (P=0.01).
The same was true for the median OS, which was 20.1 months in patients with a low disease burden and 12.4 months in those with a high disease burden (P=0.02).
For the entire study population, the rate of CRS was 85%, and 26% of patients had severe CRS. One patient died of severe CRS and multi-organ failure before the researchers began modifying the dose of CAR T cells according to the pretreatment disease burden.
Severe CRS occurred in 41% of patients with a high disease burden and 5% of those with a low disease burden.
In the entire study population, 2% of patients had grade 2 neurotoxic effects, 36% had grade 3, 6% had grade 4, and none had grade 5. The rate of severe neurotoxicity was 42%.
Neurotoxic effects occurred in 59% of patients with a high disease burden and 14% of those with a low disease burden.
“Among all of the clinical and disease factors we examined, pretreatment disease burden was the strongest predictor of long-term outcome after CAR therapy,” Dr Park said. “Our data supports the incorporation of CAR therapy in an earlier treatment setting in ALL, when the disease volume is small, so as to achieve the greatest long-term efficacy and lowest toxicity.”
This work was supported by Juno Therapeutics, the National Institutes of Health, the Carson Family Charitable Trust, the Emerald Foundation, the Mr. and Mrs. Goodwyn Commonwealth Fund, the Terry Fox Run for Cancer Research organized by the Canadian Association of New York, Kate’s Team, William Laurence and Blanche Hughes Foundation, the Center for Experimental Therapeutics at Memorial Sloan Kettering, and the Lake Road Foundation. ![]()
Polycythemia Vera and Essential Thrombocythemia: Current Management
Introduction
Polycythemia vera (PV) and essential thrombocythemia (ET), along with primary myelofibrosis (PMF), belong to the group of Philadelphia-negative myeloproliferative neoplasms (MPN). All these malignancies arise from the clonal proliferation of an aberrant hematopoietic stem cell, but are characterized by distinct clinical phenotypes.1,2 Although the clinical course of PV and ET is indolent, it can be complicated by thrombohemorrhagic episodes and/or evolution into myelofibrosis and/or acute myeloid leukemia (AML).3 Since vascular events are the most frequent life-threatening complications of PV and ET, therapeutic strategies are aimed at reducing this risk. Treatment may also help control other disease-associated symptoms.4 No therapy has been shown to prevent evolution of PV or ET into myelofibrosis or AML. The discovery of the Janus kinase 2 (JAK2)/V617F mutation in most patients with PV and over half of those with ET (and PMF)5,6 has opened new avenues of research and led to the development of targeted therapies, such as the JAK1/2 inhibitor ruxolitinib, for patients with MPN.7,8
Epidemiology
PV and ET are typically diagnosed in the fifth to seventh decade of life.9 Although these disorders are generally associated with a long clinical course, survival of patients with PV or ET may be shorter than that of the general population.10–13 Estimating the incidence and prevalence of MPN is a challenge because most patients remain asymptomatic for long periods of time and do not seek medical attention.13 The annual incidence rates of PV and ET are estimated at 0.01 to 2.61 and 0.21 to 2.53 per 100,000, respectively. PV occurs slightly more frequently in males, whereas ET has a predilection for females.14 Given the long course and low mortality associated with these disorders, the prevalence of PV and ET are significantly higher than the respective incidence: up to 47 and 57 per 100,000, respectively.15–17
Molecular Pathogenesis
In 2005 researchers discovered a gain-of-function mutation of the JAK2 gene in nearly all patients with PV and more than half of those with ET and PMF.5,6,18,19 JAK2 is a non-receptor tyrosine kinase that plays a central role in normal hematopoiesis. Substitution of a valine for a phenylalanine at codon 617 (ie, V617F) leads to its constitutive activation and signaling through the JAK-STAT pathway.5,6,18,19 More rarely (and exclusively in patients with PV), JAK2 mutations involve exon 12.20–22 The vast majority of JAK2-negative ET patients harbor mutations in either the myeloproliferative leukemia (MPL) gene, which encodes the thrombopoietin receptor,23–25 or the calreticulin (CALR) gene,26,27 which encodes for a chaperone protein that plays a role in cellular proliferation, differentiation, and apoptosis.28 Both the MPL and CALR mutations ultimately result in the constitutive activation of the JAK-STAT pathway. Thus, JAK2, MPL, and CALR alterations are collectively referred to as driver mutations. Moreover, because these mutations affect the same oncogenic pathway (ie, JAK-STAT), they are almost always mutually exclusive in a given patient. Patients with ET (or myelofibrosis) who are wild-type for JAK2, MPL, and CALR are referred to as having “triple-negative” disease. Many recurrent non-driver mutations are also found in patients with MPN that are not exclusive of each other (ie, patients may have many at the same time), and involve for example ten-eleven translocation-2 (TET2), additional sex combs like 1 (ASXL1), enhancer of zeste homolog 2 (EZH2), isocitrate dehydrogenase 1 and isocitrate dehydrogenase 2 (IDH1/2), and DNA methyltransferase 3A (DNMT3A) genes, among others.29 The biologic and prognostic significance of these non-driver alterations remain to be fully defined in ET and PV.
Diagnosis and Risk Assessment
Case Presentations
Patient A is a 68-year-old man with a history of gouty arthritis who presents with a 6-month history of recurrent headaches and itching that increases after a hot shower. Over the past 2 months, he has also noticed worsening fatigue and redness of his face. He is a nonsmoker. Physical exam reveals erythromelalgia (ie, erythema, edema, and warmth) of the upper and lower extremities, scattered scratch marks, and splenomegaly 4 cm below the costal margin. Complete blood count (CBC) shows a white blood cell (WBC) count of 8100/µL, hemoglobin 194 g/L, and platelets 582 × 103/µL. Serum erythropoietin level is decreased at 2 mU/mL. Peripheral blood testing reveals a JAK2V617F mutation.
Patient B is a 51-year-old woman with a history of severe depression treated with sertraline and hypertension controlled with lisinopril and amlodipine who presents to her primary care physician for her “50-year-old physical.” She denies symptoms and is a nonsmoker. Physical exam is unrevealing. CBC shows a WBC count of 7400/µL (normal differential), hemoglobin 135 g/L, and platelets 1282 × 103/µL. A bone marrow biopsy shows normal cellularity with clusters of large, hyperlobulated megakaryocytes. Reverse transcriptase-polymerase chain reaction fails to reveal a BCR-ABL fusion product. The patient is diagnosed with ET.
Diagnostic Criteria
Diagnostic criteria for PV and ET according to the World Health Organization (WHO) classification30 are summarized in Table 1. Criteria for the diagnosis of prefibrotic myelofibrosis are included as well since this entity was formally recognized as separate from ET and part of the PMF spectrum in the 2016 WHO classification of myeloid tumors.30 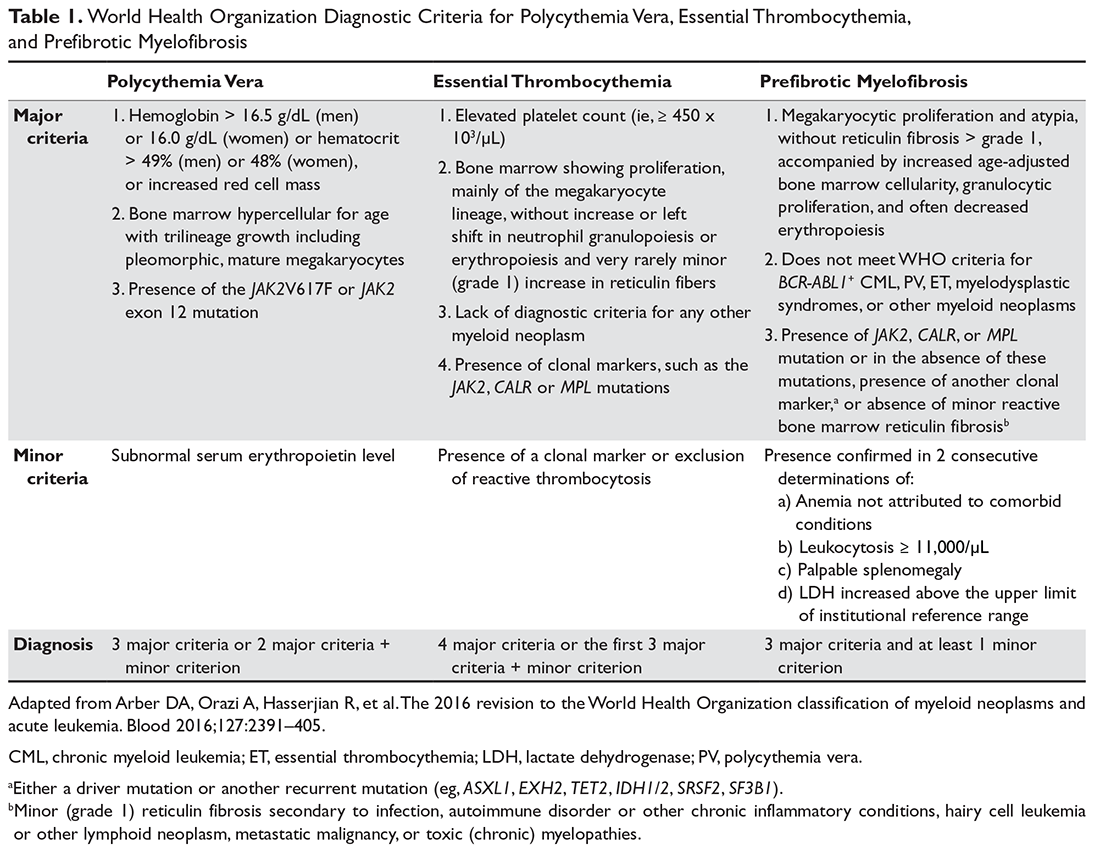
Risk Stratification
Thrombohemorrhagic events, evolution into myelofibrosis, and leukemic transformation are the most serious complications in the course of PV or ET. Only thrombohemorrhagic events are, at least partially, preventable. Arterial or venous thrombotic complications are observed at rates of 1.8 to 10.9 per 100 patient-years in PV (arterial thrombosis being more common than venous) and 0.74 to 7.7 per 100 patient-years in ET, depending on the risk group35 and the presence of other factors (see below).
Thrombosis Risk Stratification in PV
The risk stratification of patients with PV is based on 2 factors: age ≥ 60 years and prior history of thrombosis. If either is present the patient is assigned to the high-risk category, whereas if none is present the patient is considered at low risk.36 In addition, high hematocrit37 and high WBC,38 but not thrombocytosis, have been associated with the development of vascular complications. In one study, the risk of new arterial thrombosis was increased by the presence of leukoerythroblastosis, hypertension, and prior arterial thrombosis, while karyotypic abnormalities and prior venous thrombosis were predictors of new venous thrombosis.39 Another emerging risk factor for thrombosis in patients with PV is high JAK2 allele burden (ie, the normal-to-mutated gene product ratio), although the evidence supporting this conclusion is equivocal.40
Thrombosis Risk Stratification in ET
Traditionally, in ET patients, thrombotic risk was assessed using the same 2 factors (age ≥ 60 years and prior history of thrombosis), separating patients into low- and high-risk groups. However, the prognostication of ET patients has been refined recently with the identification of new relevant factors. In particular, the impact of JAK2 mutations on thrombotic risk has been thoroughly studied. Clinically, the presence of JAK2V617F is associated with older age, higher hemoglobin and hematocrit, lower platelet counts, more frequent need for cytoreductive treatment, and greater tendency to evolve into PV (a rare event).41,42 Many,41,43–46 but not all,47–51 studies suggested a correlation between JAK2 mutation and risk of both arterial and venous thrombosis. Although infrequent, a JAK2V617F homozygous state (ie, the mutation is present in both alleles) might confer an even higher thrombotic risk.52 Moreover, the impact of the JAK2 mutation on vascular events persists over time,53 particularly in patients with high or unstable mutation burden.54 Based on JAK2V617F’s influence on the thrombotic risk of ET patients, a new prognostic score was proposed, the International Prognostic Score for ET (IPSET)-thrombosis (Table 2). The revised version of this model is currently endorsed by the National Comprehensive Cancer Network and divides patients into 4 risk groups: high, intermediate, low, and very low. Treatment recommendations vary according to the risk group (as described below).55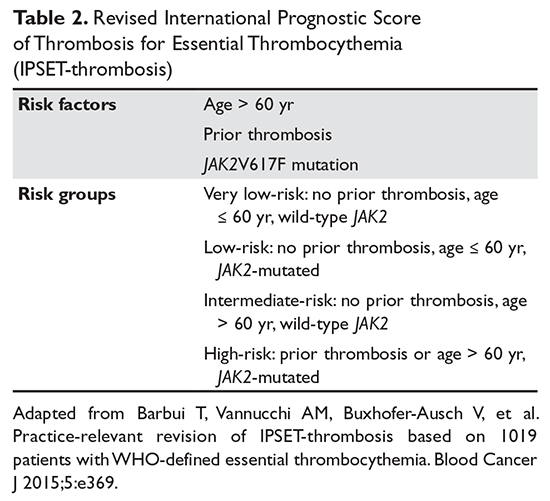
Other thrombotic risk factors have been identified, but deemed not significant enough to be included in the model. Cardiovascular risk factors (hypercholesterolemia, hypertension, smoking, diabetes mellitus) can increase the risk of vascular events,56–59 as can splenomegaly60 and baseline or persistent leukocytosis.61–63 Thrombocytosis has been correlated with thrombotic risk in some studies,64–68 whereas others did not support this conclusion and/or suggested a lower rate of thrombosis and, in some cases, increased risk of bleeding in ET patients with platelet counts greater than 1000 × 103/µL (due to acquired von Willebrand syndrome).56,61,63,68,69
CALR mutations tend to occur in younger males with lower hemoglobin and WBC count, higher platelet count, and greater marrow megakaryocytic predominance as compared to JAK2 mutations.26,27,70–72 The associated incidence of thrombosis was less than 10% at 15 years in patients with CALR mutations, lower than the incidence reported for ET patients with JAK2V617F mutations.73 The presence of the mutation per se does not appear to affect the thrombotic risk.74–76 Information on the thrombotic risk associated with MPL mutations or a triple-negative state is scarce. In both instances, however, the risk appears to be lower than with the JAK2 mutation.73,77–79
Venous thromboembolism in patients with PV or ET may occur at unusual sites, such as the splanchnic or cerebral venous systems.80 Risk factors for unusual venous thromboembolism include younger age,81 female gender (especially with concomitant use of oral contraceptive pills),82 and splenomegaly/splenectomy.83JAK2 mutation has also been associated with thrombosis at unusual sites. However, the prevalence of MPN or JAK2V617F in patients presenting with splanchnic venous thromboembolism has varied.80 In addition, MPN may be occult (ie, no clinical or laboratory abnormalities) in around 15% of patients.84 Screening for JAK2V617F and underlying MPN is recommended in patients presenting with isolated unexplained splanchnic venous thromboembolism. Treatment entails long-term anticoagulation therapy. JAK2V617F screening in patients with nonsplanchnic venous thromboembolism is not recommended, as its prevalence in this group is low (< 3%).85,86
Treatment
Cases Continued
Patient A is diagnosed with PV based on the presence of 2 major criteria (elevated hemoglobin and presence of the JAK2V617F mutation) and 1 minor criterion (low erythropoietin level). Given his age, he belongs to the high-risk disease category. He is now seeking advice regarding the management of his newly diagnosed PV.
Patient B presents to the emergency department with right lower extremity swelling and is found to have deep femoral thrombosis extending to the iliac vein. Five days after being discharged from the emergency department, she presents for follow-up. She is taking warfarin compliantly and her INR is within therapeutic range. The patient now has high-risk ET and would like to know more about thrombosis in her condition and how to best manage her risk.
Risk-Adapted Therapy
Low-Risk PV
All patients with PV should receive counseling to mitigate cardiovascular risk factors, including smoking cessation, lifestyle modifications, and lipid-lowering therapy, as indicated. Furthermore, all PV patients should receive acetylsalicylic acid (ASA) to decrease their risk for thrombosis and control vasomotor symptoms.55,87 Aspirin 81 to 100 mg daily is the preferred regimen because it provides adequate antithrombotic effect without the associated bleeding risk of higher-dose aspirin.88 Low-risk PV patients should also receive periodic phlebotomies to reduce and maintain their hematocrit below 45%. This recommendation is based on the results of the Cytoreductive Therapy in Polycythemia Vera (CYTO PV) randomized controlled trial. In the CYTO PV study, patients receiving more intense therapy to maintain the hematocrit below 45% had a lower incidence of cardiovascular-related deaths or major thrombotic events than those with hematocrit goals of 45% to 50% (2.7% versus 9.8%).89 Cytoreduction is an option for low-risk patients who do not tolerate phlebotomy or require frequent phlebotomy, or who have disease-related bleeding, severe symptoms, symptomatic splenomegaly, or progressive leukocytosis.38
High-Risk PV
Patients older than 60 years and/or with a history of thrombosis should be considered for cytoreductive therapy in addition to the above measures. Front-line cytoreductive therapies include hydroxyurea or interferon (IFN)- alfa.87 Hydroxyurea is a potent ribonucleotide reductase inhibitor that interferes with DNA repair and is the treatment of choice for most high-risk patients with PV.90 In a small trial hydroxyurea reduced the risk of thrombosis compared with historical controls treated with phlebotomy alone.91 Hydroxyurea is generally well tolerated; common side effects include cytopenias, nail changes, and mucosal and/or skin ulcers. Although never formally proven to be leukemogenic, this agent should be used with caution in younger patients.87 Indeed, in the original study, the rates of transformation were 5.9% and 1.5% for patients receiving hydroxyurea and phlebotomy alone,92 respectively, although an independent role for hydroxyurea in leukemic transformation was not supported in the much larger European Collaboration on Low-dose Aspirin in Polycythemia Vera (ECLAP) study.93 About 70% of patients will have a sustained response to hydroxyurea,94 while the remaining patients become resistant to or intolerant of the drug. Resistant individuals have a higher risk of progression to acute leukemia and death.95
IFN alfa is a pleiotropic antitumor agent that has found application in many types of malignancies96 and is sometimes employed as treatment for patients with newly diagnosed high-risk PV. Early studies showed responses in up to 100% of cases,97,98 albeit at the expense of a high discontinuation rate due to adverse events, such as flu-like symptoms, fatigue, and neuropsychiatric manifestations.99 A newer formulation of the drug obtained by adding a polyethylene glycol (PEG) moiety to the native IFN alfa molecule (PEG-IFN alfa) was shown to have a longer half-life, greater stability, less immunogenicity, and, potentially, better tolerability.100 Pilot phase 2 trials of PEG-IFN alfa-2a demonstrated its remarkable activity, with symptomatic and hematologic responses seen in the majority of patients (which, in some cases, persisted beyond discontinuation), and reasonable tolerability, with long-term discontinuation rates of around 20% to 30%.101–103 In some patients JAK2V617F became undetectable over time.104 Results of 2 ongoing trials, MDP-RC111 (single-arm study, PEG-IFN alfa-2a in high-risk PV or ET [NCT01259817]) and MPD-RC112 (randomized controlled trial, PEG-IFN alfa-2a versus hydroxyurea in the same population [NCT01258856]), will shed light on the role of PEG-IFN alfa in the management of patients with high-risk PV or ET. In 2 phase 2 studies of PEG-IFN alfa-2b, complete responses were seen in 70% to 100% of patients and discontinuation occurred in around a third of cases.105,106 A new, longer-acting formulation of PEG-IFN alfa-2a (peg-proline INF alfa-2b, AOP2014) is also undergoing clinical development.107,108
The approach to treatment of PV based on thrombotic risk level is illustrated in Figure 1. 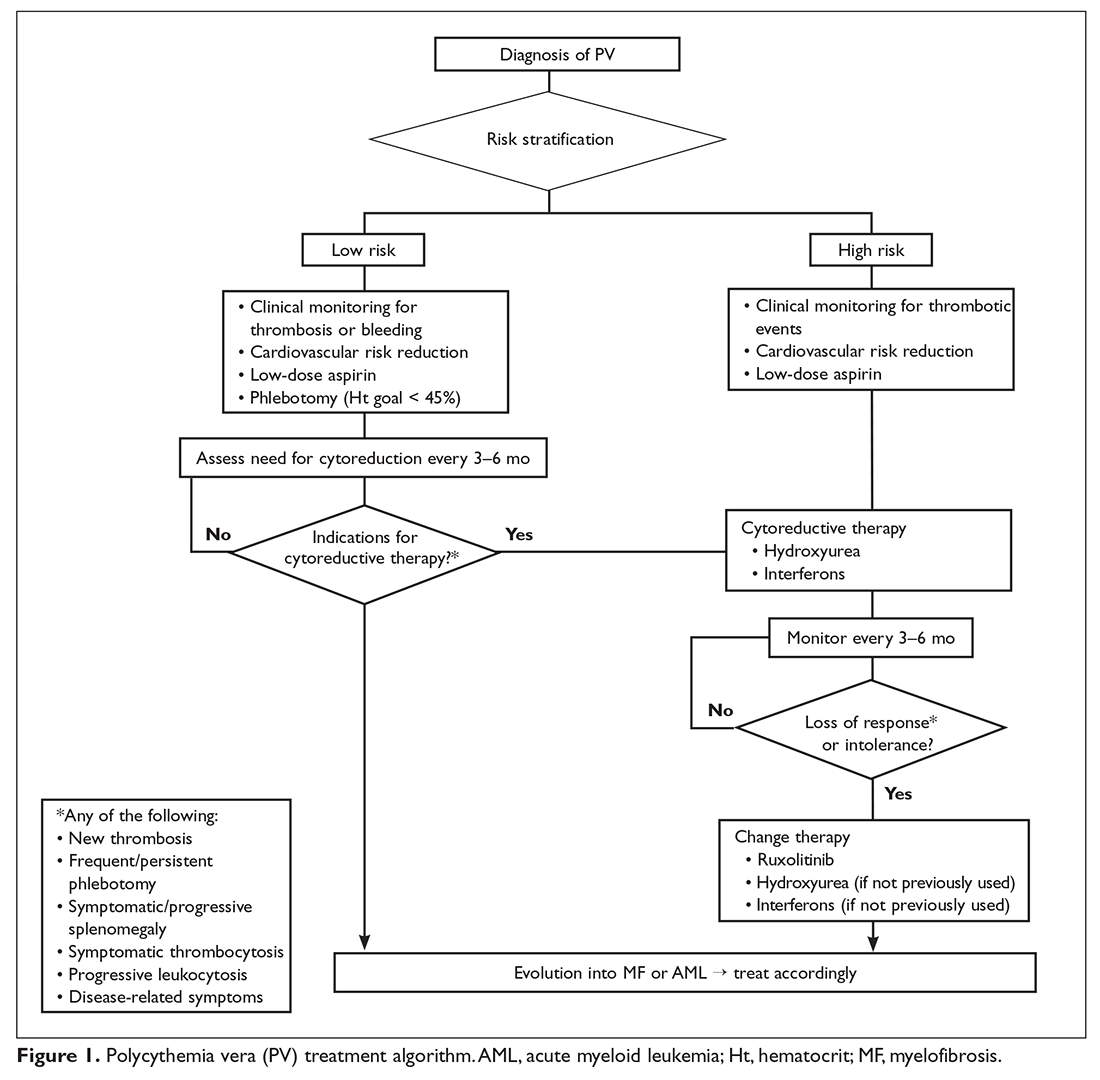
Very Low- and Low-Risk ET
Like patients with PV, individuals with ET should undergo rigorous cardiovascular risk management and generally receive ASA to decrease their thrombotic risk and improve symptom control. Antiplatelet therapy may not be warranted in patients with documented
Intermediate-Risk ET
This category includes patients older than 60 years but without thrombosis or JAK2 mutations. These individuals would have been considered high risk (and thus candidates for cytoreductive therapy) according to the traditional risk stratification. Guidelines currently recommend ASA as the sole therapy for these patients, while reserving cytoreduction for those who experience thrombosis (ie, become high-risk) or have uncontrolled vasomotor or general symptoms, symptomatic splenomegaly, symptomatic thrombocytosis, or progressive leukocytosis.
High-Risk ET
For patients with ET in need of cytoreductive therapy (ie, those with prior thrombosis or older than 60 years with a JAK2V617F mutation), first-line options include hydroxyurea, IFN, and anagrelide. Hydroxyurea remains the treatment of choice in the majority of patients.110 In a seminal study, 114 patients with ET were randomly assigned to either observation or hydroxyurea treatment with the goal of maintaining the platelet count below 600 × 103/µL. At a median follow-up of 27 months, patients in the hydroxyurea group had a lower thrombosis rate (3.6% versus 24%, P = 0.003) and longer thrombosis-free survival, regardless of the use of antiplatelet drugs.64
Anagrelide, a selective inhibitor of megakaryocytic differentiation and proliferation, was compared with hydroxyurea in patients with ET in 2 randomized trials. In the first (N = 809), the group receiving anagrelide had a higher risk of arterial thrombosis, major bleeding, and fibrotic evolution, but lower incidence of venous thrombosis. Hydroxyurea was better tolerated, mainly due to anagrelide-related cardiovascular adverse events.111 As a result of this study, hydroxyurea is often preferred to anagrelide as front-line therapy for patients with newly diagnosed high-risk ET. In the second, more recent study (N = 259), however, the 2 agents proved equivalent in terms of major or minor arterial or venous thrombosis, as well as discontinuation rate.112 The discrepancy between the 2 trials may be partly explained by the different ET diagnostic criteria used, with the latter only enrolling patients with WHO-defined true ET, while the former utilized Polycythemia Vera Study Group-ET diagnostic criteria that included patients with increases in other blood counts or varying degrees of marrow fibrosis.
Interferons were studied in ET in parallel with PV. PEG-IFN alfa-2a proved effective in patients with ET, with responses observed in 80% of patients.103 PEG-IFN alfa-2b produced similar results, with responses in 70% to 90% of patients in small studies and discontinuation observed in 20% to 38% of cases.105,106,113 Because the very long-term leukemogenic potential of hydroxyurea has remained somewhat uncertain, anagrelide or IFN might be preferable choices in younger patients.
The approach to treatment of ET based on thrombotic risk level is illustrated in Figure 2.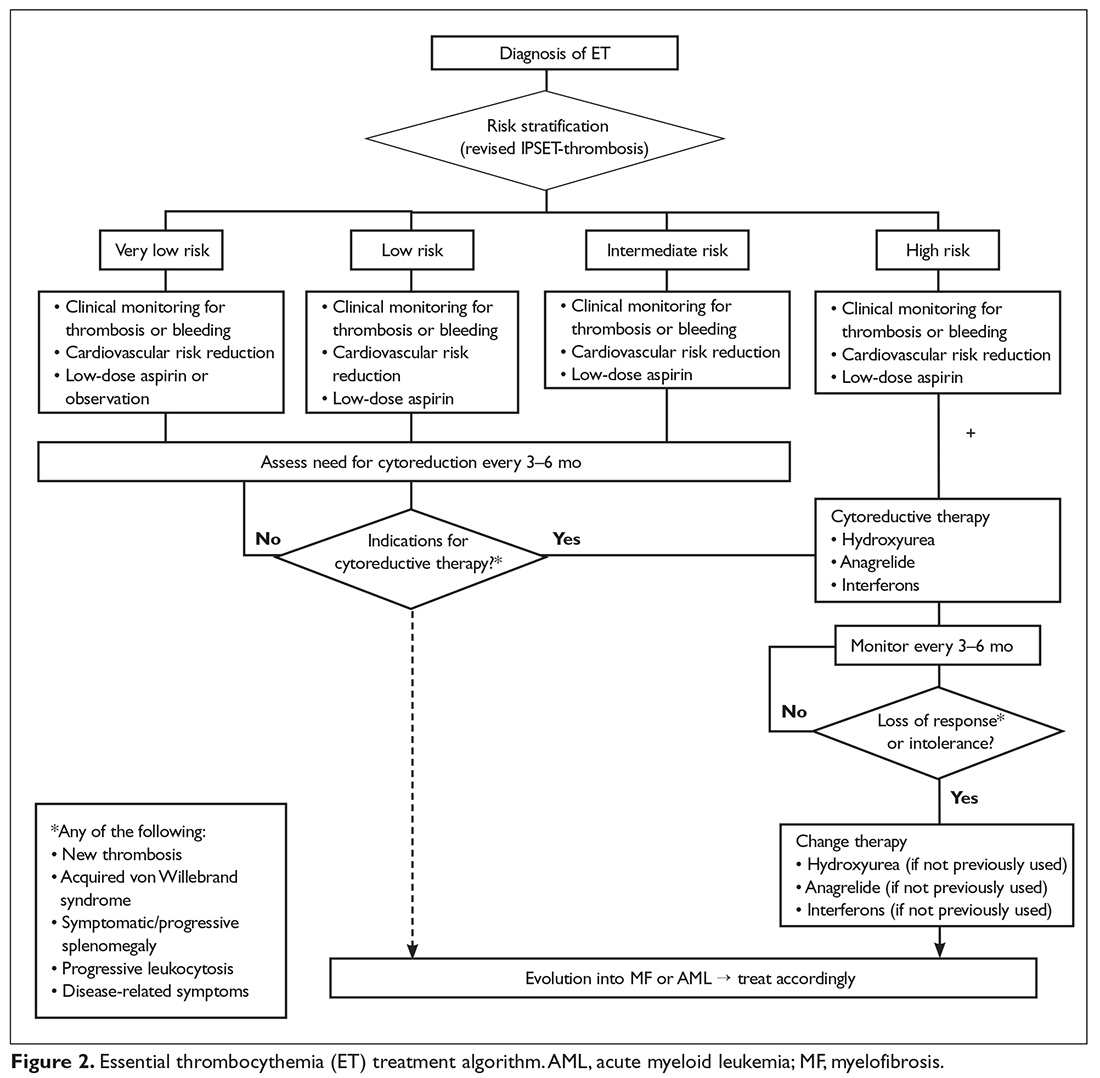
Assessing Response to Therapy
For both patients with PV and ET the endpoint of treatment set forth for clinical trials has been the achievement of a clinicohematologic response. However, studies have failed to show a correlation between response and reduction of the thrombohemorrhagic risk.114 Therefore, proposed clinical trial response criteria were revised to include absence of hemorrhagic or thrombotic events as part of the definition of response (Table 3).94 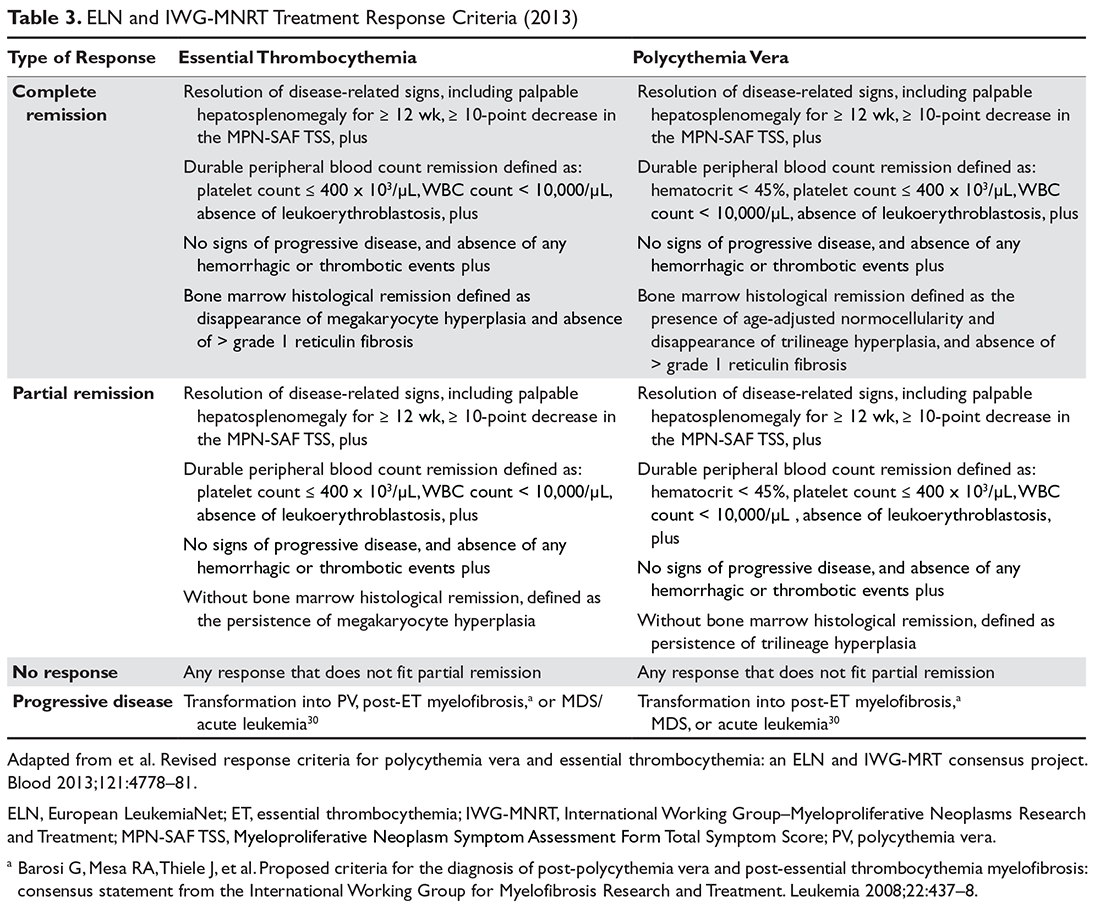
Cases Continued
Patient A was initially treated with phlebotomies and his blood counts were subsequently controlled with hydroxyurea, which he took uninterruptedly at an average dose of 2.5 g daily. He also took ASA daily throughout. Now, 18 months after the start of therapy, he presents with a complaint of fatigue for the past 3 months, which more recently has been associated with recurrent itching. A repeat CBC shows a WBC count of 17,200/µL, hemoglobin 181 g/L, and platelets 940 × 103/µL.
Patient B presents for scheduled follow-up. She has had no further thrombotic episodes. However, she spontaneously discontinued hydroxyurea 1 month ago because of worsening mouth ulcers that impaired her ability to eat even small meals. She seeks recommendations for further treatment options.
Approach to Patients Refractory to or Intolerant of First-Line Therapy
According to the European LeukemiaNet recommendations, an inadequate response to hydroxyurea in patients with PV (or myelofibrosis) is defined as a need for phlebotomy to maintain hematocrit below < 45%, platelet count > 400 × 103/µL, and a WBC count > 10,000/µL, or failure to reduce splenomegaly > 10 cm by > 50% at a dose of ≥ 2 g/day or maximum tolerated dose. Historically, treatment options for patients with PV or ET who failed first-line therapy (most commonly hydroxyurea) have included alkylating agents, such as busulfan, chlorambucil, or pipobroman, and phosphorus (P)-32. However, the use of these drugs is limited by the associated risk of leukemic transformation.93,115,116 The use of IFN (or anagrelide for ET) is often considered in patients previously treated with hydroxyurea, and vice versa.
Ruxolitinib is a JAK1 and JAK2 inhibitor currently approved for the treatment of PV patients refractory to or intolerant of hydroxyurea.7 Following promising results of a phase 2 trial,117 ruxolitinib 10 mg twice daily was compared with best available therapy in the pivotal RESPONSE trial (N = 222). Ruxolitinib proved superior in achieving hematocrit control, reduction of spleen volume, and improvement of symptoms. Grade 3-4 hematologic toxicity was infrequent and similar in the 2 arms.118 In addition, longer follow-up of that study suggested a lower rate of thrombotic events in patients receiving ruxolitinib (1.8 versus 8.2 per 100 patient-years).119 In a similarly designed randomized phase 3 study in PV patients without splenomegaly (RESPONSE-2), more patients in the ruxolitinib arm had hematocrit reduction without an increase in toxicity. Based on the results of the above studies, ruxolitinib can be considered a standard of care for second-line therapy in this post-hydroxyurea patient population.120
Ruxolitinib is also being tested in patients with high-risk ET who have become resistant to, or were intolerant of hydroxyurea, but currently has no approved indication in this setting.121,122 Common side effects of ruxolitinib include cytopenias (especially anemia), increased risk of infections, hyperlipidemia, and increased risk of non-melanoma skin cancer.
Novel Agents
Novel agents that have been studied in patients with PV and ET are histone deacetylase inhibitors, murine double minute 2 (MDM2, or HDM2 for their human counterpart) inhibitors (which restore the function of p53), Bcl-2 homology domain 3 mimetics such as navitoclax and venetoclax, and, for patients with ET, the telomerase inhibitor imetelstat.123
Disease Evolution
Cases Continued
Patient A’s PV has been well controlled with PEG-IFN alfa-2a 90 μg subcutaneously weekly. However, he now presents with a complaint of worsening fatigue and early satiety. On exam the patient appears ill and splenomegaly is appreciated 12 cm below the costal margin. CBC shows a WBC count of 2600/µL, hemoglobin 73 g/L, and platelets 122 × 103/µL. Peripheral blood smear reveals leukoerythroblastosis and dacrocytosis. CBC 6 months ago was normal. A bone marrow biopsy is consistent with myelofibrosis.
After discontinuing hydroxyurea, patient B’s ET has been well controlled with anagrelide. However, for the past 4 weeks she has complained of severe fatigue and easy bruising. Physical exam reveals a pale, ill-appearing woman with scattered bruises. CBC shows a WBC count of 14,600/µL with 44% myeloblasts, hemoglobin 73 g/L, and platelets 22 × 103/µL. CBC 6 months ago was normal. A bone marrow biopsy is consistent with leukemic transformation of ET.
Post-PV/Post-ET Myelofibrosis
Diagnostic criteria for post-PV and post-ET myelofibrosis are outlined in Table 4. 
Leukemic Transformation
The presence of more than 20% blasts in peripheral blood or bone marrow in a patient with MPN defines leukemic transformation. This occurs in up to 5% to 10% of patients and may or may not be preceded by a myelofibrosis phase.126 In cases of extramedullary transformation, a lower percentage of blasts can be seen in the bone marrow compared to the peripheral blood. The pathogenesis of leukemic transformation has remained elusive, but it is believed to be associated with genetic instability, which facilitates the acquisition of additional mutations, including those of TET2, ASXL1, EZH2 and DNMT3, IDH1/2, and TP53.127
Clinical risk factors for leukemic transformation include advanced age, karyotypic abnormalities, prior therapy with alkylating agents or P-32, splenectomy, increased peripheral blood or bone marrow blasts, leukocytosis, anemia, thrombocytopenia, and cytogenetic abnormalities. Hydroxyurea, interferon, and ruxolitinib have not been shown to have leukemogenic potential thus far. Prognosis of leukemic transformation is uniformly poor and patient survival rarely exceeds 6 months.
There is no standard of care for leukemic transformation of MPN (MPN-LT). Treatment options range from low-intensity regimens to more aggressive AML-type induction chemotherapy. No strategy appears clearly superior to others.128 Hematopoietic stem cell transplantation is the only therapy that provides clinically meaningful benefit to patients,129 but it is applicable only to a minority of patients with chemosensitive disease and good performance status.130 Notable experimental approaches to MPN–LT include hypomethylating agents, such as decitabine131 or azacitidine,132 with or without ruxolitinib.133-135
Conclusion
PV and ET are rare, chronic myeloid disorders. Patients typically experience a long clinical course and enjoy near-normal quality of life if properly managed. The 2 most important life-limiting complications of PV and ET are thrombohemorrhagic events and myelofibrosis/AML transformation. Vascular events are at least in part preventable with counseling on risk factors, phlebotomy (for patients with PV), antiplatelet therapy, and cytoreduction with hydroxyurea, IFNs, or anagrelide (for patients with ET). In addition, ruxolitinib was recently approved for PV patients after hydroxyurea failure. PV/ET transformation in myelofibrosis or AML is part of the natural history of the disease and no therapy has been shown to prevent it. Treatment follows recommendations set forth for PMF and AML, but results are generally poorer and novel strategies are needed to improve patients’ outcomes.
1. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon: IARC; 2008.
2. Alvarez-Larran A, Pereira A, Arellano-Rodrigo E, et al. Cytoreduction plus low-dose aspirin versus cytoreduction alone as primary prophylaxis of thrombosis in patients with high-risk essential thrombocythaemia: an observational study. Br J Haematol 2013;161:865–71.
3. Tefferi A. Polycythemia vera and essential thrombocythemia: 2013 update on diagnosis, risk-stratification, and management. Am J Hematol 2013;88:507–16.
4. Michiels JJ, Berneman Z, Schroyens W, et al. Platelet-mediated erythromelalgic, cerebral, ocular and coronary microvascular ischemic and thrombotic manifestations in patients with essential thrombocythemia and polycythemia vera: a distinct aspirin-responsive and coumadin-resistant arterial thrombophilia. Platelets 2006;17:528–44.
5. James C, Ugo V, Couedic J-PL, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005;434:1144–8.
6. Kralovics R, Passamonti F, Buser A, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders.N Engl J Med 2005;352:1779–90.
7. Verstovsek S, Mesa R, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med 2012;366:799–807.
8. Harrison C, Kiladjian J, Al-Ali H, et al. JAK Inhibition with ruxolitinib versus best available therapy for myelofibrosis.N Engl J Med 2012;366:787–98.
9. Berglund S, Zettervall O. Incidence of polycythemia vera in a defined population. Eur J Haematol 1992;48:20–6.
10. Rozman C, Giralt M, Feliu E, et al. Life expectancy of patients with chronic nonleukemic myeloproliferative disorders. Cancer 1991;67:2658–63.
11. Passamonti F, Rumi E, Pungolino E, et al. Life expectancy and prognostic factors for survival in patients with polycythemia vera and essential thrombocythemia. Am J Med 2004;117:755–61.
12. Hultcrantz M, Kristinsson SY, Andersson TM, et al. Patterns of survival among patients with myeloproliferative neoplasms diagnosed in Sweden from 1973 to 2008: a population-based study. J Clin Oncol 2012;30:2995–3001.
13. Wolanskyj AP, Schwager SM, et al. Essential thrombocythemia beyond the first decade: life expectancy, long-term complication rates, and prognostic factors. Mayo Clin Proc 2006;81:159–66.
14. Srour SA, Devesa SS, Morton LM, et al. Incidence and patient survival of myeloproliferative neoplasms and myelodysplastic/myeloproliferative neoplasms in the United States, 2001-12. Br J Haematol 2016;174:382–96.
15. Titmarsh GJ, Duncombe AS, McMullin MF, et al. How common are myeloproliferative neoplasms? A systematic review and meta-analysis. Am J Hematol 2014;89:581–7.
16. Moulard O, Mehta J, Fryzek J, et al. Epidemiology of myelofibrosis, essential thrombocythemia, and polycythemia vera in the European Union. Eur J Haematol 2014;92:289–97.
17. Stein BL, Gotlib J, Arcasoy M, et al. Historical views, conventional approaches, and evolving management strategies for myeloproliferative neoplasms. J Natl Compr Canc Netw 2015;13:424–34.
18. Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005;7:387–97.
19. Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005;365:1054–61.
20. Scott LM, Tong W, Levine RL, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med 2007;356:459–68.
21. Pardanani A, Lasho TL, Finke C, et al. Prevalence and clinicopathologic correlates of JAK2 exon 12 mutations in JAK2V617F-negative polycythemia vera. Leukemia 2007;21:1960–3.
22. Passamonti F, Elena C, Schnittger S, et al. Molecular and clinical features of the myeloproliferative neoplasm associated with JAK2 exon 12 mutations. Blood 2011;117:2813–6.
23. Abe M, Suzuki K, Inagaki O, et al. A novel MPL point mutation resulting in thrombopoietin-independent activation. Leukemia 2002;16:1500–6.
24. Pikman Y, Lee BH, Mercher T, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med 2006;3:e270.
25. Pardanani AD, Levine RL, Lasho T, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood 2006;108:3472–6.
26. Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med 2013;369:2391–405.
27. Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med 2013;369:2379–90.
28. Wang WA, Groenendyk J, Michalak M. Calreticulin signaling in health and disease. Int J Biochem Cell Biol 2012;44:842–6.
29. Saeidi K. Myeloproliferative neoplasms: Current molecular biology and genetics. Crit Rev Oncol Hematol 2016;98:375–89.
30. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016;127:2391–405.
31. Mesa RA, Niblack J, Wadleigh M, et al. The burden of fatigue and quality of life in myeloproliferative disorders (MPDs): an international Internet-based survey of 1179 MPD patients. Cancer 2007;109:68–76.
32. Marchioli R, Finazzi G, Landolfi R, et al. Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol 2005;23:2224–32.
33. Scherber R, Dueck AC, Johansson P, et al. The Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF): international prospective validation and reliability trial in 402 patients. Blood 2011;118:401–8.
34. Emanuel RM, Dueck AC, Geyer HL, et al. Myeloproliferative neoplasm (MPN) symptom assessment form total symptom score: prospective international assessment of an abbreviated symptom burden scoring system among patients with MPNs. J Clin Oncol 2012;30:4098–103.
35. Casini A, Fontana P, Lecompte TP. Thrombotic complications of myeloproliferative neoplasms: risk assessment and risk-guided management. J Thromb Haemost 2013;11:1215–27.
36. Barbui T, Finazzi G, Falanga A. Myeloproliferative neoplasms and thrombosis. Blood 2013;122:2176-–84.
37. Pearson TC, Wetherley-Mein G. Vascular occlusive episodes and venous haematocrit in primary myeloproliferative polychytemia. Lancet 1978;2:1219–22.
38. Landolfi R, Di Gennaro L, Barbui T, et al. Leukocytosis as a major thrombotic risk factor in patients with polycythemia vera. Blood 2007;109:2446–52.
39. Tefferi A, Rumi E, Finazzi G, et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia 2013;27:1874–81.
40. Barbui T, Falanga A. Molecular biomarkers of thrombosis in myeloproliferative neoplasms. Thromb Res 2016;140 Suppl 1:S71–75.
41. Campbell PJ, Scott LM, Buck G, et al. Definition of subtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2 V617F mutation status: a prospective study. Lancet 2005;366:1945–53.
42. Tefferi A, Guglielmelli P, Larson DR, et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 2014;124:2507–13.
43. Qin Y, Wang X, Zhao C, et al. The impact of JAK2V617F mutation on different types of thrombosis risk in patients with essential thrombocythemia: a meta-analysis. Int J Hematol 2015;102:170–80.
44. Ziakas PD. Effect of JAK2 V617F on thrombotic risk in patients with essential thrombocythemia: measuring the uncertain. Haematologica 2008;93:1412–4.
45. Lussana F, Dentali F, Abbate R, et al. Screening for thrombophilia and antithrombotic prophylaxis in pregnancy: Guidelines of the Italian Society for Haemostasis and Thrombosis (SISET). Thromb Res 2009;124:e19–25.
46. Dahabreh IJ, Zoi K, Giannouli S, et al. Is JAK2 V617F mutation more than a diagnostic index? A meta-analysis of clinical outcomes in essential thrombocythemia. Leuk Res 2009;33:67–73.
47. Cho YU, Chi HS, Lee EH, et al. Comparison of clinicopathologic findings according to JAK2 V617F mutation in patients with essential thrombocythemia. Int J Hematol 2009;89:39–44.
48. Wolanskyj AP, Lasho TL, Schwager SM, et al. JAK2 mutation in essential thrombocythaemia: clinical associations and long-term prognostic relevance. Br J Haematol 2005;131:208–13.
49. Antonioli E, Guglielmelli P, Pancrazzi A, et al. Clinical implications of the JAK2 V617F mutation in essential thrombocythemia. Leukemia 2005;19:1847–9.
50. Palandri F, Catani L, Testoni N, et al. Long-term follow-up of 386 consecutive patients with essential thrombocythemia: safety of cytoreductive therapy. Am J Hematol 2009;84:215–20.
51. Chim CS, Sim JP, Chan CC, et al. Impact of JAK2V617F mutation on thrombosis and myeloid transformation in essential thrombocythemia: a multivariate analysis by Cox regression in 141 patients. Hematology 2010;15:187–92.
52. Vannucchi AM, Antonioli E, Guglielmelli P, et al. Clinical profile of homozygous JAK2 617V>F mutation in patients with polycythemia vera or essential thrombocythemia. Blood 2007;110:840–6.
53. Carobbio A, Finazzi G, Antonioli E, et al. JAK2V617F allele burden and thrombosis: a direct comparison in essential thrombocythemia and polycythemia vera. Exp Hematol 2009;37:1016–21.
54. Alvarez-Larran A, Bellosillo B, Pereira A, et al. JAK2V617F monitoring in polycythemia vera and essential thrombocythemia: clinical usefulness for predicting myelofibrotic transformation and thrombotic events. Am J Hematol 2014;89:517–23.
55. Barbui T, Vannucchi AM, Buxhofer-Ausch V, et al. Practice-relevant revision of IPSET-thrombosis based on 1019 patients with WHO-defined essential thrombocythemia. Blood Cancer J 2015;5:e369.
56. Carobbio A, Thiele J, Passamonti F, et al. Risk factors for arterial and venous thrombosis in WHO-defined essential thrombocythemia: an international study of 891 patients. Blood 2011;117:5857–9.
57. Alvarez-Larran A, Cervantes F, Bellosillo B, et al. Essential thrombocythemia in young individuals: frequency and risk factors for vascular events and evolution to myelofibrosis in 126 patients. Leukemia 2007;21:1218–23.
58. Jantunen R, Juvonen E, Ikkala E, et al. The predictive value of vascular risk factors and gender for the development of thrombotic complications in essential thrombocythemia. Ann Hematol 2001;80:74–8.
59. Besses C, Cervantes F, Pereira A, et al. Major vascular complications in essential thrombocythemia: a study of the predictive factors in a series of 148 patients. Leukemia 1999;13:150–4.
60. Haider M, Gangat N, Hanson C, Tefferi A. Splenomegaly and thrombosis risk in essential thrombocythemia: the mayo clinic experience. Am J Hematol 2016;91:E296–297.
61. Carobbio A, Finazzi G, Antonioli E, et al. Thrombocytosis and leukocytosis interaction in vascular complications of essential thrombocythemia. Blood 2008;112:3135–7.
62. Palandri F, Polverelli N, Catani L, et al. Impact of leukocytosis on thrombotic risk and survival in 532 patients with essential thrombocythemia: a retrospective study. Ann Hematol 2011;90:933–8.
63. Campbell PJ, MacLean C, Beer PA, et al. Correlation of blood counts with vascular complications in essential thrombocythemia: analysis of the prospective PT1 cohort. Blood 2012;120:1409–11.
64. Cortelazzo S, Finazzi G, Ruggeri M, et al. Hydroxyurea for patients with essential thrombocythemia and a high risk of thrombosis. N Engl J Med 1995;332:1132–6.
65. van Genderen PJ, Mulder PG, Waleboer M, et al. Prevention and treatment of thrombotic complications in essential thrombocythaemia: efficacy and safety of aspirin. Br J Haematol 1997;97:179–84.
66. Storen EC, Tefferi A. Long-term use of anagrelide in young patients with essential thrombocythemia. Blood 2001;97:863–6.
67. De Stefano V, Za T, Rossi E, et al. Recurrent thrombosis in patients with polycythemia vera and essential thrombocythemia: incidence, risk factors, and effect of treatments. Haematologica 2008;93:372–80.
68. Alvarez-Larran A, Cervantes F, Pereira A, et al. Observation versus antiplatelet therapy as primary prophylaxis for thrombosis in low-risk essential thrombocythemia. Blood 2010;116:1205–10.
69. Palandri F, Polverelli N, Catani L, et al. Bleeding in essential thrombocythaemia: a retrospective analysis on 565 patients. Br J Haematol 2012;156:281–4.
70. Rotunno G, Mannarelli C, Guglielmelli P, et al. Impact of calreticulin mutations on clinical and hematological phenotype and outcome in essential thrombocythemia. Blood 2014;123:1552–5.
71. Tefferi A, Wassie EA, Lasho TL, et al. Calreticulin mutations and long-term survival in essential thrombocythemia. Leukemia 2014;28:2300–3.
72. Rumi E, Pietra D, Ferretti V, et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood 2014;123:1544–51.
73. Palandri F, Latagliata R, Polverelli N, et al. Mutations and long-term outcome of 217 young patients with essential thrombocythemia or early primary myelofibrosis. Leukemia 2015;29:1344–9.
74. Fu R, Xuan M, Zhou Y, et al. Analysis of calreticulin mutations in Chinese patients with essential thrombocythemia: clinical implications in diagnosis, prognosis and treatment. Leukemia 2014;28:1912–4.
75. Tefferi A, Wassie EA, Guglielmelli P, et al. Type 1 versus Type 2 calreticulin mutations in essential thrombocythemia: a collaborative study of 1027 patients. Am J Hematol 2014;89:E121–4.
76. Pietra D, Rumi E, Ferretti VV, et al. Differential clinical effects of different mutation subtypes in CALR-mutant myeloproliferative neoplasms. Leukemia 2016;30:431–8.
77. Rumi E, Pietra D, Guglielmelli P, et al. Acquired copy-neutral loss of heterozygosity of chromosome 1p as a molecular event associated with marrow fibrosis in MPL-mutated myeloproliferative neoplasms. Blood 2013;121:4388–95.
78. Beer PA, Campbell PJ, Scott LM, et al. MPL mutations in myeloproliferative disorders: analysis of the PT-1 cohort. Blood 2008;112:141–9.
79. Gangat N, Wassie EA, Lasho TL, et al. Mutations and thrombosis in essential thrombocythemia: prognostic interaction with age and thrombosis history. Eur J Haematol 2015;94:31–6.
80. Sekhar M, McVinnie K, Burroughs AK. Splanchnic vein thrombosis in myeloproliferative neoplasms. Br J Haematol 2013;162:730–47.
81. Stein BL, Saraf S, Sobol U, et al. Age-related differences in disease characteristics and clinical outcomes in polycythemia vera. Leuk Lymph 2013;54:1989–95.
82. Landolfi R, Di Gennaro L, Nicolazzi MA, et al. Polycythemia vera: gender-related phenotypic differences. Intern Emerg Med 2012;7:509–15.
83. Winslow ER, Brunt LM, Drebin JA, et al. Portal vein thrombosis after splenectomy. Am J Surg 2002;184:631–6.
84. Smalberg JH, Arends LR, Valla DC, et al. Myeloproliferative neoplasms in Budd-Chiari syndrome and portal vein thrombosis: a meta-analysis. Blood 2012;120:4921–8.
85. Dentali F, Squizzato A, Brivio L, et al. JAK2V617F mutation for the early diagnosis of Ph- myeloproliferative neoplasms in patients with venous thromboembolism: a meta-analysis. Blood 2009;113:5617–23.
86. Pardanani A, Lasho TL, Hussein K, et al. JAK2V617F mutation screening as part of the hypercoagulable work-up in the absence of splanchnic venous thrombosis or overt myeloproliferative neoplasm: assessment of value in a series of 664 consecutive patients. Mayo Clin Proc 2008;83:457–9.
87. Barbui T, Barosi G, Birgegard G, et al. Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European LeukemiaNet. J Clin Oncol 2011;29:761–70.
88. Landolfi R, Marchioli R, Kutti J, et al. Efficacy and safety of low-dose aspirin in polycythemia vera. N Engl J Med 2004;350:114–24.
89. Marchioli R, Finazzi G, Specchia G, et al. Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med 2013;368:22–33.
90. Kiladjian JJ, Chevret S, Dosquet C, et al. Treatment of polycythemia vera with hydroxyurea and pipobroman: final results of a randomized trial initiated in 1980. J Clin Oncol 2011;29:3907–13.
91. Kaplan ME, Mack K, Goldberg JD, et al. Long-term management of polycythemia vera with hydroxyurea: a progress report. Semin Hematol 1986;23:167–71.
92. Fruchtman SM, Mack K, Kaplan ME, et al. From efficacy to safety: a Polycythemia Vera Study group report on hydroxyurea in patients with polycythemia vera. Semin Hematol 1997;34:17–23.
93. Finazzi G, Caruso V, Marchioli R, et al. Acute leukemia in polycythemia vera: an analysis of 1638 patients enrolled in a prospective observational study. Blood 2005;105:2664–70.
94. Barosi G, Mesa R, Finazzi G, et al. Revised response criteria for polycythemia vera and essential thrombocythemia: an ELN and IWG-MRT consensus project. Blood 2013;121:4778–81.
95. Alvarez-Larran A, Pereira A, Cervantes F, et al. Assessment and prognostic value of the European LeukemiaNet criteria for clinicohematologic response, resistance, and intolerance to hydroxyurea in polycythemia vera. Blood 2012;119:1363–9.
96. Stein BL, Tiu RV. Biological rationale and clinical use of interferon in the classical BCR-ABL-negative myeloproliferative neoplasms. J Interferon Cytokine Res 2013;33:145–53.
97. Ludwig H, Cortelezzi A, Van Camp BG, et al. Treatment with recombinant interferon-alpha-2C: multiple myeloma and thrombocythaemia in myeloproliferative diseases. Oncology 1985;42 Suppl 1:19–25.
98. Silver RT. Long-term effects of the treatment of polycythemia vera with recombinant interferon-alpha. Cancer 2006;107:451–8.
99. Kiladjian JJ, Mesa RA, Hoffman R. The renaissance of interferon therapy for the treatment of myeloid malignancies. Blood 2011;117:4706–15.
100. Veronese FM, Mero A. The impact of PEGylation on biological therapies. BioDrugs 2008;22:315–29.
101. Kiladjian JJ, Cassinat B, Chevret S, et al. Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood 2008;112:3065–72.
102. Turlure P, Cambier N, Roussel M, et al. Complete hematological, molecular and histological remissions without cytoreductive treatment lasting after pegylated-interferon {alpha}-2a (peg-IFN{alpha}-2a) therapy in polycythemia vera (PV): long term results of a phase 2 trial [abstract]. Blood 2011;118(21). Abstract 280.
103. Quintas-Cardama A, Kantarjian H, Manshouri T, et al. Pegylated interferon alfa-2a yields high rates of hematologic and molecular response in patients with advanced essential thrombocythemia and polycythemia vera. J Clin Oncol 2009;27:5418–24.
104. Quintas-Cardama A, Abdel-Wahab O, Manshouri T, et al. Molecular analysis of patients with polycythemia vera or essential thrombocythemia receiving pegylated interferon a-2a. Blood 2013;122:893–901.
105. Samuelsson J, Hasselbalch H, Bruserud O, et al. A phase II trial of pegylated interferon alpha-2b therapy for polycythemia vera and essential thrombocythemia: feasibility, clinical and biologic effects, and impact on quality of life. Cancer 2006;106:2397–405.
106. Jabbour E, Kantarjian H, Cortes J, et al. PEG-IFN-alpha-2b therapy in BCR-ABL-negative myeloproliferative disorders: final result of a phase 2 study. Cancer 2007;110:2012–18.
107. Them NC, Bagienski K, Berg T, et al. Molecular responses and chromosomal aberrations in patients with polycythemia vera treated with peg-proline-interferon alpha-2b. Am J Hematol 2015;90:288–94.
108. Gisslinger H, Klade C, Georgiev P, et al. Final results from PROUD-PV a randomized controlled phase 3 trial comparing ropeginterferon alfa-2b to hydroxyurea in polycythemia vera patients [abstract]. Blood 2016;128(suppl 22). Abstract 475.
109. van Genderen PJ, van Vliet HH, Prins FJ, et al. Excessive prolongation of the bleeding time by aspirin in essential thrombocythemia is related to a decrease of large von Willebrand factor multimers in plasma. Ann Hematol 1997;75:215–20.
110. Cortelazzo S, Finazzi G, Ruggeri M, et al. Hydroxyurea for patients with essential thrombocythemia and a high risk of thrombosis. N Engl J Med 1995;332:1132–7.
111. Harrison CN, Campbell PJ, Buck G, et al. Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. N Engl J Med 2005;353:33–45.
112. Gisslinger H, Gotic M, Holowiecki J, et al. Anagrelide compared with hydroxyurea in WHO-classified essential thrombocythemia: the ANAHYDRET Study, a randomized controlled trial. Blood 2013;121:1720–8.
113. Alvarado Y, Cortes J, Verstovsek S, et al. Pilot study of pegylated interferon-alpha 2b in patients with essential thrombocythemia. Cancer Chemother Pharmacol 2003;51:81–6.
114. Barosi G, Tefferi A, Barbui T, ad hoc committee ‘Definition of clinically relevant outcomes for contemporarily clinical trials in Ph-neg M. Do current response criteria in classical Ph-negative myeloproliferative neoplasms capture benefit for patients? Leukemia 2012;26:1148–9.
115. Bjorkholm M, Derolf AR, Hultcrantz M, et al. Treatment-related risk factors for transformation to acute myeloid leukemia and myelodysplastic syndromes in myeloproliferative neoplasms. J Clin Oncol 2011;29:2410–5.
116. Alvarez-Larran A, Martinez-Aviles L, Hernandez-Boluda JC, et al. Busulfan in patients with polycythemia vera or essential thrombocythemia refractory or intolerant to hydroxyurea. Ann Hematol 2014;93:2037–43.
117. Verstovsek S, Passamonti F, Rambaldi A, et al. A phase 2 study of ruxolitinib, an oral JAK1 and JAK2 Inhibitor, in patients with advanced polycythemia vera who are refractory or intolerant to hydroxyurea. Cancer 2014;120:513–20.
118. Vannucchi AM, Kiladjian JJ, Griesshammer M, et al. Ruxolitinib in polycythemia vera resistant to or intolerant of hydroxyurea. N Engl J Med 2015; 372:426–35.
119. Verstovsek S, Vannucchi AM, Griesshammer M, et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80-week follow-up from the RESPONSE trial. Haematologica 2016;101:821–9.
120. Passamonti F, Griesshammer M, Palandri F, et al. Ruxolitinib for the treatment of inadequately controlled polycythaemia vera without splenomegaly (RESPONSE-2): a randomised, open-label, phase 3b study. Lancet Oncol 2017;18:88–99.
121. Verstovsek S, Passamonti F, Rambaldi A, et al. Long-term results from a phase II open-label study of ruxolitinib in patients with essential thrombocythemia refractory to or intolerant of hydroxyurea [abstract]. Blood 2014;124. Abstract 1847.
122. Harrison CN, Mead AJ, Panchal A, et al. Ruxolitinib versus best available therapy for ET intolerant or resistant to hydroxycarbamide in a randomized trial. Blood 2017 Aug 9. pii: blood-2017-05-785790 .
123. Bose P, Verstovsek S. Drug development pipeline for myeloproliferative neoplasms: potential future impact on guidelines and management. J Natl Compr Canc Netw 2016;14:1613–24.
124. Cerquozzi S, Teffieri A. Blast transformation and fibrotic progression in polycythemia vera and essential thrombocythemia: a literature review of incidence and risk factors. Blood Cancer J 2015;Nov 13;5:e366.
125. Passamonti F, Rumi E, Caramella M, et al. A dynamic prognostic model to predict survival in post-polycythemia vera myelofibrosis. Blood 2008;111:3383–7.
126. Mesa RA, Verstovsek S, Cervantes F, et al. Primary myelofibrosis (PMF), post polycythemia vera myelofibrosis (post-PV MF), post essential thrombocythemia myelofibrosis (post-ET MF), blast phase PMF (PMF-BP): Consensus on terminology by the international working group for myelofibrosis research and treatment (IWG-MRT). Leuk Res 2007;31:737–40.
127. Rampal R, Mascarenhas J. Pathogenesis and management of acute myeloid leukemia that has evolved from a myeloproliferative neoplasm. Curr Opin Hematol 2014;21:65–71.
128. Chihara D, Kantarjian HM, Newberry KJ, et al. Survival outcome of patients with acute myeloid leukemia transformed from myeloproliferative neoplasms [abstract]. Blood 2016;128. Abstract 1940.
129. Tam CS, Nussenzveig RM, Popat U, et al. The natural history and treatment outcome of blast phase BCR-ABL- myeloproliferative neoplasms. Blood 2008;112:1628–37.
130. Kundranda MN, Tibes R, Mesa RA. Transformation of a chronic myeloproliferative neoplasm to acute myelogenous leukemia: does anything work? Curr Hematol Malig Rep 2012;7:78–86.
131. Badar T, Kantarjian HM, Ravandi F, et al. Therapeutic benefit of decitabine, a hypomethylating agent, in patients with high-risk primary myelofibrosis and myeloproliferative neoplasm in accelerated or blastic/acute myeloid leukemia phase. Leuk Res 2015;39:950–6.
132. Thepot S, Itzykson R, Seegers V, et al. Treatment of progression of Philadelphia-negative myeloproliferative neoplasms to myelodysplastic syndrome or acute myeloid leukemia by azacitidine: a report on 54 cases on the behalf of the Groupe Francophone des Myelodysplasies (GFM). Blood 2010;116:3735–42.
133. Pemmaraju N, Kantarjian H, Kadia T, et al. A phase I/II study of the Janus kinase (JAK)1 and 2 inhibitor ruxolitinib in patients with relapsed or refractory acute myeloid leukemia. Clin Lymphoma Myeloma Leuk 2015;15:171–6.
134. Rampal RK, Mascarenhas JO, Kosiorek HE, et al. Safety and efficacy of combined ruxolitinib and decitabine in patients with blast-phase MPN and post-MPN AML: results of a phase I study (Myeloproliferative Disorders Research Consortium 109 trial) [abstract]. Blood 2016;128. Abstract 1124.
135. Bose P, Verstovsek S, Gasior Y, et al. Phase I/II study of ruxolitinib (RUX) with decitabine (DAC) in patients with post-myeloproliferative neoplasm acute myeloid leukemia (post-MPN AML): phase I results [abstract]. Blood 2016;128. Abstract 4262.
Introduction
Polycythemia vera (PV) and essential thrombocythemia (ET), along with primary myelofibrosis (PMF), belong to the group of Philadelphia-negative myeloproliferative neoplasms (MPN). All these malignancies arise from the clonal proliferation of an aberrant hematopoietic stem cell, but are characterized by distinct clinical phenotypes.1,2 Although the clinical course of PV and ET is indolent, it can be complicated by thrombohemorrhagic episodes and/or evolution into myelofibrosis and/or acute myeloid leukemia (AML).3 Since vascular events are the most frequent life-threatening complications of PV and ET, therapeutic strategies are aimed at reducing this risk. Treatment may also help control other disease-associated symptoms.4 No therapy has been shown to prevent evolution of PV or ET into myelofibrosis or AML. The discovery of the Janus kinase 2 (JAK2)/V617F mutation in most patients with PV and over half of those with ET (and PMF)5,6 has opened new avenues of research and led to the development of targeted therapies, such as the JAK1/2 inhibitor ruxolitinib, for patients with MPN.7,8
Epidemiology
PV and ET are typically diagnosed in the fifth to seventh decade of life.9 Although these disorders are generally associated with a long clinical course, survival of patients with PV or ET may be shorter than that of the general population.10–13 Estimating the incidence and prevalence of MPN is a challenge because most patients remain asymptomatic for long periods of time and do not seek medical attention.13 The annual incidence rates of PV and ET are estimated at 0.01 to 2.61 and 0.21 to 2.53 per 100,000, respectively. PV occurs slightly more frequently in males, whereas ET has a predilection for females.14 Given the long course and low mortality associated with these disorders, the prevalence of PV and ET are significantly higher than the respective incidence: up to 47 and 57 per 100,000, respectively.15–17
Molecular Pathogenesis
In 2005 researchers discovered a gain-of-function mutation of the JAK2 gene in nearly all patients with PV and more than half of those with ET and PMF.5,6,18,19 JAK2 is a non-receptor tyrosine kinase that plays a central role in normal hematopoiesis. Substitution of a valine for a phenylalanine at codon 617 (ie, V617F) leads to its constitutive activation and signaling through the JAK-STAT pathway.5,6,18,19 More rarely (and exclusively in patients with PV), JAK2 mutations involve exon 12.20–22 The vast majority of JAK2-negative ET patients harbor mutations in either the myeloproliferative leukemia (MPL) gene, which encodes the thrombopoietin receptor,23–25 or the calreticulin (CALR) gene,26,27 which encodes for a chaperone protein that plays a role in cellular proliferation, differentiation, and apoptosis.28 Both the MPL and CALR mutations ultimately result in the constitutive activation of the JAK-STAT pathway. Thus, JAK2, MPL, and CALR alterations are collectively referred to as driver mutations. Moreover, because these mutations affect the same oncogenic pathway (ie, JAK-STAT), they are almost always mutually exclusive in a given patient. Patients with ET (or myelofibrosis) who are wild-type for JAK2, MPL, and CALR are referred to as having “triple-negative” disease. Many recurrent non-driver mutations are also found in patients with MPN that are not exclusive of each other (ie, patients may have many at the same time), and involve for example ten-eleven translocation-2 (TET2), additional sex combs like 1 (ASXL1), enhancer of zeste homolog 2 (EZH2), isocitrate dehydrogenase 1 and isocitrate dehydrogenase 2 (IDH1/2), and DNA methyltransferase 3A (DNMT3A) genes, among others.29 The biologic and prognostic significance of these non-driver alterations remain to be fully defined in ET and PV.
Diagnosis and Risk Assessment
Case Presentations
Patient A is a 68-year-old man with a history of gouty arthritis who presents with a 6-month history of recurrent headaches and itching that increases after a hot shower. Over the past 2 months, he has also noticed worsening fatigue and redness of his face. He is a nonsmoker. Physical exam reveals erythromelalgia (ie, erythema, edema, and warmth) of the upper and lower extremities, scattered scratch marks, and splenomegaly 4 cm below the costal margin. Complete blood count (CBC) shows a white blood cell (WBC) count of 8100/µL, hemoglobin 194 g/L, and platelets 582 × 103/µL. Serum erythropoietin level is decreased at 2 mU/mL. Peripheral blood testing reveals a JAK2V617F mutation.
Patient B is a 51-year-old woman with a history of severe depression treated with sertraline and hypertension controlled with lisinopril and amlodipine who presents to her primary care physician for her “50-year-old physical.” She denies symptoms and is a nonsmoker. Physical exam is unrevealing. CBC shows a WBC count of 7400/µL (normal differential), hemoglobin 135 g/L, and platelets 1282 × 103/µL. A bone marrow biopsy shows normal cellularity with clusters of large, hyperlobulated megakaryocytes. Reverse transcriptase-polymerase chain reaction fails to reveal a BCR-ABL fusion product. The patient is diagnosed with ET.
Diagnostic Criteria
Diagnostic criteria for PV and ET according to the World Health Organization (WHO) classification30 are summarized in Table 1. Criteria for the diagnosis of prefibrotic myelofibrosis are included as well since this entity was formally recognized as separate from ET and part of the PMF spectrum in the 2016 WHO classification of myeloid tumors.30
Risk Stratification
Thrombohemorrhagic events, evolution into myelofibrosis, and leukemic transformation are the most serious complications in the course of PV or ET. Only thrombohemorrhagic events are, at least partially, preventable. Arterial or venous thrombotic complications are observed at rates of 1.8 to 10.9 per 100 patient-years in PV (arterial thrombosis being more common than venous) and 0.74 to 7.7 per 100 patient-years in ET, depending on the risk group35 and the presence of other factors (see below).
Thrombosis Risk Stratification in PV
The risk stratification of patients with PV is based on 2 factors: age ≥ 60 years and prior history of thrombosis. If either is present the patient is assigned to the high-risk category, whereas if none is present the patient is considered at low risk.36 In addition, high hematocrit37 and high WBC,38 but not thrombocytosis, have been associated with the development of vascular complications. In one study, the risk of new arterial thrombosis was increased by the presence of leukoerythroblastosis, hypertension, and prior arterial thrombosis, while karyotypic abnormalities and prior venous thrombosis were predictors of new venous thrombosis.39 Another emerging risk factor for thrombosis in patients with PV is high JAK2 allele burden (ie, the normal-to-mutated gene product ratio), although the evidence supporting this conclusion is equivocal.40
Thrombosis Risk Stratification in ET
Traditionally, in ET patients, thrombotic risk was assessed using the same 2 factors (age ≥ 60 years and prior history of thrombosis), separating patients into low- and high-risk groups. However, the prognostication of ET patients has been refined recently with the identification of new relevant factors. In particular, the impact of JAK2 mutations on thrombotic risk has been thoroughly studied. Clinically, the presence of JAK2V617F is associated with older age, higher hemoglobin and hematocrit, lower platelet counts, more frequent need for cytoreductive treatment, and greater tendency to evolve into PV (a rare event).41,42 Many,41,43–46 but not all,47–51 studies suggested a correlation between JAK2 mutation and risk of both arterial and venous thrombosis. Although infrequent, a JAK2V617F homozygous state (ie, the mutation is present in both alleles) might confer an even higher thrombotic risk.52 Moreover, the impact of the JAK2 mutation on vascular events persists over time,53 particularly in patients with high or unstable mutation burden.54 Based on JAK2V617F’s influence on the thrombotic risk of ET patients, a new prognostic score was proposed, the International Prognostic Score for ET (IPSET)-thrombosis (Table 2). The revised version of this model is currently endorsed by the National Comprehensive Cancer Network and divides patients into 4 risk groups: high, intermediate, low, and very low. Treatment recommendations vary according to the risk group (as described below).55
Other thrombotic risk factors have been identified, but deemed not significant enough to be included in the model. Cardiovascular risk factors (hypercholesterolemia, hypertension, smoking, diabetes mellitus) can increase the risk of vascular events,56–59 as can splenomegaly60 and baseline or persistent leukocytosis.61–63 Thrombocytosis has been correlated with thrombotic risk in some studies,64–68 whereas others did not support this conclusion and/or suggested a lower rate of thrombosis and, in some cases, increased risk of bleeding in ET patients with platelet counts greater than 1000 × 103/µL (due to acquired von Willebrand syndrome).56,61,63,68,69
CALR mutations tend to occur in younger males with lower hemoglobin and WBC count, higher platelet count, and greater marrow megakaryocytic predominance as compared to JAK2 mutations.26,27,70–72 The associated incidence of thrombosis was less than 10% at 15 years in patients with CALR mutations, lower than the incidence reported for ET patients with JAK2V617F mutations.73 The presence of the mutation per se does not appear to affect the thrombotic risk.74–76 Information on the thrombotic risk associated with MPL mutations or a triple-negative state is scarce. In both instances, however, the risk appears to be lower than with the JAK2 mutation.73,77–79
Venous thromboembolism in patients with PV or ET may occur at unusual sites, such as the splanchnic or cerebral venous systems.80 Risk factors for unusual venous thromboembolism include younger age,81 female gender (especially with concomitant use of oral contraceptive pills),82 and splenomegaly/splenectomy.83JAK2 mutation has also been associated with thrombosis at unusual sites. However, the prevalence of MPN or JAK2V617F in patients presenting with splanchnic venous thromboembolism has varied.80 In addition, MPN may be occult (ie, no clinical or laboratory abnormalities) in around 15% of patients.84 Screening for JAK2V617F and underlying MPN is recommended in patients presenting with isolated unexplained splanchnic venous thromboembolism. Treatment entails long-term anticoagulation therapy. JAK2V617F screening in patients with nonsplanchnic venous thromboembolism is not recommended, as its prevalence in this group is low (< 3%).85,86
Treatment
Cases Continued
Patient A is diagnosed with PV based on the presence of 2 major criteria (elevated hemoglobin and presence of the JAK2V617F mutation) and 1 minor criterion (low erythropoietin level). Given his age, he belongs to the high-risk disease category. He is now seeking advice regarding the management of his newly diagnosed PV.
Patient B presents to the emergency department with right lower extremity swelling and is found to have deep femoral thrombosis extending to the iliac vein. Five days after being discharged from the emergency department, she presents for follow-up. She is taking warfarin compliantly and her INR is within therapeutic range. The patient now has high-risk ET and would like to know more about thrombosis in her condition and how to best manage her risk.
Risk-Adapted Therapy
Low-Risk PV
All patients with PV should receive counseling to mitigate cardiovascular risk factors, including smoking cessation, lifestyle modifications, and lipid-lowering therapy, as indicated. Furthermore, all PV patients should receive acetylsalicylic acid (ASA) to decrease their risk for thrombosis and control vasomotor symptoms.55,87 Aspirin 81 to 100 mg daily is the preferred regimen because it provides adequate antithrombotic effect without the associated bleeding risk of higher-dose aspirin.88 Low-risk PV patients should also receive periodic phlebotomies to reduce and maintain their hematocrit below 45%. This recommendation is based on the results of the Cytoreductive Therapy in Polycythemia Vera (CYTO PV) randomized controlled trial. In the CYTO PV study, patients receiving more intense therapy to maintain the hematocrit below 45% had a lower incidence of cardiovascular-related deaths or major thrombotic events than those with hematocrit goals of 45% to 50% (2.7% versus 9.8%).89 Cytoreduction is an option for low-risk patients who do not tolerate phlebotomy or require frequent phlebotomy, or who have disease-related bleeding, severe symptoms, symptomatic splenomegaly, or progressive leukocytosis.38
High-Risk PV
Patients older than 60 years and/or with a history of thrombosis should be considered for cytoreductive therapy in addition to the above measures. Front-line cytoreductive therapies include hydroxyurea or interferon (IFN)- alfa.87 Hydroxyurea is a potent ribonucleotide reductase inhibitor that interferes with DNA repair and is the treatment of choice for most high-risk patients with PV.90 In a small trial hydroxyurea reduced the risk of thrombosis compared with historical controls treated with phlebotomy alone.91 Hydroxyurea is generally well tolerated; common side effects include cytopenias, nail changes, and mucosal and/or skin ulcers. Although never formally proven to be leukemogenic, this agent should be used with caution in younger patients.87 Indeed, in the original study, the rates of transformation were 5.9% and 1.5% for patients receiving hydroxyurea and phlebotomy alone,92 respectively, although an independent role for hydroxyurea in leukemic transformation was not supported in the much larger European Collaboration on Low-dose Aspirin in Polycythemia Vera (ECLAP) study.93 About 70% of patients will have a sustained response to hydroxyurea,94 while the remaining patients become resistant to or intolerant of the drug. Resistant individuals have a higher risk of progression to acute leukemia and death.95
IFN alfa is a pleiotropic antitumor agent that has found application in many types of malignancies96 and is sometimes employed as treatment for patients with newly diagnosed high-risk PV. Early studies showed responses in up to 100% of cases,97,98 albeit at the expense of a high discontinuation rate due to adverse events, such as flu-like symptoms, fatigue, and neuropsychiatric manifestations.99 A newer formulation of the drug obtained by adding a polyethylene glycol (PEG) moiety to the native IFN alfa molecule (PEG-IFN alfa) was shown to have a longer half-life, greater stability, less immunogenicity, and, potentially, better tolerability.100 Pilot phase 2 trials of PEG-IFN alfa-2a demonstrated its remarkable activity, with symptomatic and hematologic responses seen in the majority of patients (which, in some cases, persisted beyond discontinuation), and reasonable tolerability, with long-term discontinuation rates of around 20% to 30%.101–103 In some patients JAK2V617F became undetectable over time.104 Results of 2 ongoing trials, MDP-RC111 (single-arm study, PEG-IFN alfa-2a in high-risk PV or ET [NCT01259817]) and MPD-RC112 (randomized controlled trial, PEG-IFN alfa-2a versus hydroxyurea in the same population [NCT01258856]), will shed light on the role of PEG-IFN alfa in the management of patients with high-risk PV or ET. In 2 phase 2 studies of PEG-IFN alfa-2b, complete responses were seen in 70% to 100% of patients and discontinuation occurred in around a third of cases.105,106 A new, longer-acting formulation of PEG-IFN alfa-2a (peg-proline INF alfa-2b, AOP2014) is also undergoing clinical development.107,108
The approach to treatment of PV based on thrombotic risk level is illustrated in Figure 1.
Very Low- and Low-Risk ET
Like patients with PV, individuals with ET should undergo rigorous cardiovascular risk management and generally receive ASA to decrease their thrombotic risk and improve symptom control. Antiplatelet therapy may not be warranted in patients with documented
Intermediate-Risk ET
This category includes patients older than 60 years but without thrombosis or JAK2 mutations. These individuals would have been considered high risk (and thus candidates for cytoreductive therapy) according to the traditional risk stratification. Guidelines currently recommend ASA as the sole therapy for these patients, while reserving cytoreduction for those who experience thrombosis (ie, become high-risk) or have uncontrolled vasomotor or general symptoms, symptomatic splenomegaly, symptomatic thrombocytosis, or progressive leukocytosis.
High-Risk ET
For patients with ET in need of cytoreductive therapy (ie, those with prior thrombosis or older than 60 years with a JAK2V617F mutation), first-line options include hydroxyurea, IFN, and anagrelide. Hydroxyurea remains the treatment of choice in the majority of patients.110 In a seminal study, 114 patients with ET were randomly assigned to either observation or hydroxyurea treatment with the goal of maintaining the platelet count below 600 × 103/µL. At a median follow-up of 27 months, patients in the hydroxyurea group had a lower thrombosis rate (3.6% versus 24%, P = 0.003) and longer thrombosis-free survival, regardless of the use of antiplatelet drugs.64
Anagrelide, a selective inhibitor of megakaryocytic differentiation and proliferation, was compared with hydroxyurea in patients with ET in 2 randomized trials. In the first (N = 809), the group receiving anagrelide had a higher risk of arterial thrombosis, major bleeding, and fibrotic evolution, but lower incidence of venous thrombosis. Hydroxyurea was better tolerated, mainly due to anagrelide-related cardiovascular adverse events.111 As a result of this study, hydroxyurea is often preferred to anagrelide as front-line therapy for patients with newly diagnosed high-risk ET. In the second, more recent study (N = 259), however, the 2 agents proved equivalent in terms of major or minor arterial or venous thrombosis, as well as discontinuation rate.112 The discrepancy between the 2 trials may be partly explained by the different ET diagnostic criteria used, with the latter only enrolling patients with WHO-defined true ET, while the former utilized Polycythemia Vera Study Group-ET diagnostic criteria that included patients with increases in other blood counts or varying degrees of marrow fibrosis.
Interferons were studied in ET in parallel with PV. PEG-IFN alfa-2a proved effective in patients with ET, with responses observed in 80% of patients.103 PEG-IFN alfa-2b produced similar results, with responses in 70% to 90% of patients in small studies and discontinuation observed in 20% to 38% of cases.105,106,113 Because the very long-term leukemogenic potential of hydroxyurea has remained somewhat uncertain, anagrelide or IFN might be preferable choices in younger patients.
The approach to treatment of ET based on thrombotic risk level is illustrated in Figure 2.
Assessing Response to Therapy
For both patients with PV and ET the endpoint of treatment set forth for clinical trials has been the achievement of a clinicohematologic response. However, studies have failed to show a correlation between response and reduction of the thrombohemorrhagic risk.114 Therefore, proposed clinical trial response criteria were revised to include absence of hemorrhagic or thrombotic events as part of the definition of response (Table 3).94
Cases Continued
Patient A was initially treated with phlebotomies and his blood counts were subsequently controlled with hydroxyurea, which he took uninterruptedly at an average dose of 2.5 g daily. He also took ASA daily throughout. Now, 18 months after the start of therapy, he presents with a complaint of fatigue for the past 3 months, which more recently has been associated with recurrent itching. A repeat CBC shows a WBC count of 17,200/µL, hemoglobin 181 g/L, and platelets 940 × 103/µL.
Patient B presents for scheduled follow-up. She has had no further thrombotic episodes. However, she spontaneously discontinued hydroxyurea 1 month ago because of worsening mouth ulcers that impaired her ability to eat even small meals. She seeks recommendations for further treatment options.
Approach to Patients Refractory to or Intolerant of First-Line Therapy
According to the European LeukemiaNet recommendations, an inadequate response to hydroxyurea in patients with PV (or myelofibrosis) is defined as a need for phlebotomy to maintain hematocrit below < 45%, platelet count > 400 × 103/µL, and a WBC count > 10,000/µL, or failure to reduce splenomegaly > 10 cm by > 50% at a dose of ≥ 2 g/day or maximum tolerated dose. Historically, treatment options for patients with PV or ET who failed first-line therapy (most commonly hydroxyurea) have included alkylating agents, such as busulfan, chlorambucil, or pipobroman, and phosphorus (P)-32. However, the use of these drugs is limited by the associated risk of leukemic transformation.93,115,116 The use of IFN (or anagrelide for ET) is often considered in patients previously treated with hydroxyurea, and vice versa.
Ruxolitinib is a JAK1 and JAK2 inhibitor currently approved for the treatment of PV patients refractory to or intolerant of hydroxyurea.7 Following promising results of a phase 2 trial,117 ruxolitinib 10 mg twice daily was compared with best available therapy in the pivotal RESPONSE trial (N = 222). Ruxolitinib proved superior in achieving hematocrit control, reduction of spleen volume, and improvement of symptoms. Grade 3-4 hematologic toxicity was infrequent and similar in the 2 arms.118 In addition, longer follow-up of that study suggested a lower rate of thrombotic events in patients receiving ruxolitinib (1.8 versus 8.2 per 100 patient-years).119 In a similarly designed randomized phase 3 study in PV patients without splenomegaly (RESPONSE-2), more patients in the ruxolitinib arm had hematocrit reduction without an increase in toxicity. Based on the results of the above studies, ruxolitinib can be considered a standard of care for second-line therapy in this post-hydroxyurea patient population.120
Ruxolitinib is also being tested in patients with high-risk ET who have become resistant to, or were intolerant of hydroxyurea, but currently has no approved indication in this setting.121,122 Common side effects of ruxolitinib include cytopenias (especially anemia), increased risk of infections, hyperlipidemia, and increased risk of non-melanoma skin cancer.
Novel Agents
Novel agents that have been studied in patients with PV and ET are histone deacetylase inhibitors, murine double minute 2 (MDM2, or HDM2 for their human counterpart) inhibitors (which restore the function of p53), Bcl-2 homology domain 3 mimetics such as navitoclax and venetoclax, and, for patients with ET, the telomerase inhibitor imetelstat.123
Disease Evolution
Cases Continued
Patient A’s PV has been well controlled with PEG-IFN alfa-2a 90 μg subcutaneously weekly. However, he now presents with a complaint of worsening fatigue and early satiety. On exam the patient appears ill and splenomegaly is appreciated 12 cm below the costal margin. CBC shows a WBC count of 2600/µL, hemoglobin 73 g/L, and platelets 122 × 103/µL. Peripheral blood smear reveals leukoerythroblastosis and dacrocytosis. CBC 6 months ago was normal. A bone marrow biopsy is consistent with myelofibrosis.
After discontinuing hydroxyurea, patient B’s ET has been well controlled with anagrelide. However, for the past 4 weeks she has complained of severe fatigue and easy bruising. Physical exam reveals a pale, ill-appearing woman with scattered bruises. CBC shows a WBC count of 14,600/µL with 44% myeloblasts, hemoglobin 73 g/L, and platelets 22 × 103/µL. CBC 6 months ago was normal. A bone marrow biopsy is consistent with leukemic transformation of ET.
Post-PV/Post-ET Myelofibrosis
Diagnostic criteria for post-PV and post-ET myelofibrosis are outlined in Table 4.
Leukemic Transformation
The presence of more than 20% blasts in peripheral blood or bone marrow in a patient with MPN defines leukemic transformation. This occurs in up to 5% to 10% of patients and may or may not be preceded by a myelofibrosis phase.126 In cases of extramedullary transformation, a lower percentage of blasts can be seen in the bone marrow compared to the peripheral blood. The pathogenesis of leukemic transformation has remained elusive, but it is believed to be associated with genetic instability, which facilitates the acquisition of additional mutations, including those of TET2, ASXL1, EZH2 and DNMT3, IDH1/2, and TP53.127
Clinical risk factors for leukemic transformation include advanced age, karyotypic abnormalities, prior therapy with alkylating agents or P-32, splenectomy, increased peripheral blood or bone marrow blasts, leukocytosis, anemia, thrombocytopenia, and cytogenetic abnormalities. Hydroxyurea, interferon, and ruxolitinib have not been shown to have leukemogenic potential thus far. Prognosis of leukemic transformation is uniformly poor and patient survival rarely exceeds 6 months.
There is no standard of care for leukemic transformation of MPN (MPN-LT). Treatment options range from low-intensity regimens to more aggressive AML-type induction chemotherapy. No strategy appears clearly superior to others.128 Hematopoietic stem cell transplantation is the only therapy that provides clinically meaningful benefit to patients,129 but it is applicable only to a minority of patients with chemosensitive disease and good performance status.130 Notable experimental approaches to MPN–LT include hypomethylating agents, such as decitabine131 or azacitidine,132 with or without ruxolitinib.133-135
Conclusion
PV and ET are rare, chronic myeloid disorders. Patients typically experience a long clinical course and enjoy near-normal quality of life if properly managed. The 2 most important life-limiting complications of PV and ET are thrombohemorrhagic events and myelofibrosis/AML transformation. Vascular events are at least in part preventable with counseling on risk factors, phlebotomy (for patients with PV), antiplatelet therapy, and cytoreduction with hydroxyurea, IFNs, or anagrelide (for patients with ET). In addition, ruxolitinib was recently approved for PV patients after hydroxyurea failure. PV/ET transformation in myelofibrosis or AML is part of the natural history of the disease and no therapy has been shown to prevent it. Treatment follows recommendations set forth for PMF and AML, but results are generally poorer and novel strategies are needed to improve patients’ outcomes.
Introduction
Polycythemia vera (PV) and essential thrombocythemia (ET), along with primary myelofibrosis (PMF), belong to the group of Philadelphia-negative myeloproliferative neoplasms (MPN). All these malignancies arise from the clonal proliferation of an aberrant hematopoietic stem cell, but are characterized by distinct clinical phenotypes.1,2 Although the clinical course of PV and ET is indolent, it can be complicated by thrombohemorrhagic episodes and/or evolution into myelofibrosis and/or acute myeloid leukemia (AML).3 Since vascular events are the most frequent life-threatening complications of PV and ET, therapeutic strategies are aimed at reducing this risk. Treatment may also help control other disease-associated symptoms.4 No therapy has been shown to prevent evolution of PV or ET into myelofibrosis or AML. The discovery of the Janus kinase 2 (JAK2)/V617F mutation in most patients with PV and over half of those with ET (and PMF)5,6 has opened new avenues of research and led to the development of targeted therapies, such as the JAK1/2 inhibitor ruxolitinib, for patients with MPN.7,8
Epidemiology
PV and ET are typically diagnosed in the fifth to seventh decade of life.9 Although these disorders are generally associated with a long clinical course, survival of patients with PV or ET may be shorter than that of the general population.10–13 Estimating the incidence and prevalence of MPN is a challenge because most patients remain asymptomatic for long periods of time and do not seek medical attention.13 The annual incidence rates of PV and ET are estimated at 0.01 to 2.61 and 0.21 to 2.53 per 100,000, respectively. PV occurs slightly more frequently in males, whereas ET has a predilection for females.14 Given the long course and low mortality associated with these disorders, the prevalence of PV and ET are significantly higher than the respective incidence: up to 47 and 57 per 100,000, respectively.15–17
Molecular Pathogenesis
In 2005 researchers discovered a gain-of-function mutation of the JAK2 gene in nearly all patients with PV and more than half of those with ET and PMF.5,6,18,19 JAK2 is a non-receptor tyrosine kinase that plays a central role in normal hematopoiesis. Substitution of a valine for a phenylalanine at codon 617 (ie, V617F) leads to its constitutive activation and signaling through the JAK-STAT pathway.5,6,18,19 More rarely (and exclusively in patients with PV), JAK2 mutations involve exon 12.20–22 The vast majority of JAK2-negative ET patients harbor mutations in either the myeloproliferative leukemia (MPL) gene, which encodes the thrombopoietin receptor,23–25 or the calreticulin (CALR) gene,26,27 which encodes for a chaperone protein that plays a role in cellular proliferation, differentiation, and apoptosis.28 Both the MPL and CALR mutations ultimately result in the constitutive activation of the JAK-STAT pathway. Thus, JAK2, MPL, and CALR alterations are collectively referred to as driver mutations. Moreover, because these mutations affect the same oncogenic pathway (ie, JAK-STAT), they are almost always mutually exclusive in a given patient. Patients with ET (or myelofibrosis) who are wild-type for JAK2, MPL, and CALR are referred to as having “triple-negative” disease. Many recurrent non-driver mutations are also found in patients with MPN that are not exclusive of each other (ie, patients may have many at the same time), and involve for example ten-eleven translocation-2 (TET2), additional sex combs like 1 (ASXL1), enhancer of zeste homolog 2 (EZH2), isocitrate dehydrogenase 1 and isocitrate dehydrogenase 2 (IDH1/2), and DNA methyltransferase 3A (DNMT3A) genes, among others.29 The biologic and prognostic significance of these non-driver alterations remain to be fully defined in ET and PV.
Diagnosis and Risk Assessment
Case Presentations
Patient A is a 68-year-old man with a history of gouty arthritis who presents with a 6-month history of recurrent headaches and itching that increases after a hot shower. Over the past 2 months, he has also noticed worsening fatigue and redness of his face. He is a nonsmoker. Physical exam reveals erythromelalgia (ie, erythema, edema, and warmth) of the upper and lower extremities, scattered scratch marks, and splenomegaly 4 cm below the costal margin. Complete blood count (CBC) shows a white blood cell (WBC) count of 8100/µL, hemoglobin 194 g/L, and platelets 582 × 103/µL. Serum erythropoietin level is decreased at 2 mU/mL. Peripheral blood testing reveals a JAK2V617F mutation.
Patient B is a 51-year-old woman with a history of severe depression treated with sertraline and hypertension controlled with lisinopril and amlodipine who presents to her primary care physician for her “50-year-old physical.” She denies symptoms and is a nonsmoker. Physical exam is unrevealing. CBC shows a WBC count of 7400/µL (normal differential), hemoglobin 135 g/L, and platelets 1282 × 103/µL. A bone marrow biopsy shows normal cellularity with clusters of large, hyperlobulated megakaryocytes. Reverse transcriptase-polymerase chain reaction fails to reveal a BCR-ABL fusion product. The patient is diagnosed with ET.
Diagnostic Criteria
Diagnostic criteria for PV and ET according to the World Health Organization (WHO) classification30 are summarized in Table 1. Criteria for the diagnosis of prefibrotic myelofibrosis are included as well since this entity was formally recognized as separate from ET and part of the PMF spectrum in the 2016 WHO classification of myeloid tumors.30
Risk Stratification
Thrombohemorrhagic events, evolution into myelofibrosis, and leukemic transformation are the most serious complications in the course of PV or ET. Only thrombohemorrhagic events are, at least partially, preventable. Arterial or venous thrombotic complications are observed at rates of 1.8 to 10.9 per 100 patient-years in PV (arterial thrombosis being more common than venous) and 0.74 to 7.7 per 100 patient-years in ET, depending on the risk group35 and the presence of other factors (see below).
Thrombosis Risk Stratification in PV
The risk stratification of patients with PV is based on 2 factors: age ≥ 60 years and prior history of thrombosis. If either is present the patient is assigned to the high-risk category, whereas if none is present the patient is considered at low risk.36 In addition, high hematocrit37 and high WBC,38 but not thrombocytosis, have been associated with the development of vascular complications. In one study, the risk of new arterial thrombosis was increased by the presence of leukoerythroblastosis, hypertension, and prior arterial thrombosis, while karyotypic abnormalities and prior venous thrombosis were predictors of new venous thrombosis.39 Another emerging risk factor for thrombosis in patients with PV is high JAK2 allele burden (ie, the normal-to-mutated gene product ratio), although the evidence supporting this conclusion is equivocal.40
Thrombosis Risk Stratification in ET
Traditionally, in ET patients, thrombotic risk was assessed using the same 2 factors (age ≥ 60 years and prior history of thrombosis), separating patients into low- and high-risk groups. However, the prognostication of ET patients has been refined recently with the identification of new relevant factors. In particular, the impact of JAK2 mutations on thrombotic risk has been thoroughly studied. Clinically, the presence of JAK2V617F is associated with older age, higher hemoglobin and hematocrit, lower platelet counts, more frequent need for cytoreductive treatment, and greater tendency to evolve into PV (a rare event).41,42 Many,41,43–46 but not all,47–51 studies suggested a correlation between JAK2 mutation and risk of both arterial and venous thrombosis. Although infrequent, a JAK2V617F homozygous state (ie, the mutation is present in both alleles) might confer an even higher thrombotic risk.52 Moreover, the impact of the JAK2 mutation on vascular events persists over time,53 particularly in patients with high or unstable mutation burden.54 Based on JAK2V617F’s influence on the thrombotic risk of ET patients, a new prognostic score was proposed, the International Prognostic Score for ET (IPSET)-thrombosis (Table 2). The revised version of this model is currently endorsed by the National Comprehensive Cancer Network and divides patients into 4 risk groups: high, intermediate, low, and very low. Treatment recommendations vary according to the risk group (as described below).55
Other thrombotic risk factors have been identified, but deemed not significant enough to be included in the model. Cardiovascular risk factors (hypercholesterolemia, hypertension, smoking, diabetes mellitus) can increase the risk of vascular events,56–59 as can splenomegaly60 and baseline or persistent leukocytosis.61–63 Thrombocytosis has been correlated with thrombotic risk in some studies,64–68 whereas others did not support this conclusion and/or suggested a lower rate of thrombosis and, in some cases, increased risk of bleeding in ET patients with platelet counts greater than 1000 × 103/µL (due to acquired von Willebrand syndrome).56,61,63,68,69
CALR mutations tend to occur in younger males with lower hemoglobin and WBC count, higher platelet count, and greater marrow megakaryocytic predominance as compared to JAK2 mutations.26,27,70–72 The associated incidence of thrombosis was less than 10% at 15 years in patients with CALR mutations, lower than the incidence reported for ET patients with JAK2V617F mutations.73 The presence of the mutation per se does not appear to affect the thrombotic risk.74–76 Information on the thrombotic risk associated with MPL mutations or a triple-negative state is scarce. In both instances, however, the risk appears to be lower than with the JAK2 mutation.73,77–79
Venous thromboembolism in patients with PV or ET may occur at unusual sites, such as the splanchnic or cerebral venous systems.80 Risk factors for unusual venous thromboembolism include younger age,81 female gender (especially with concomitant use of oral contraceptive pills),82 and splenomegaly/splenectomy.83JAK2 mutation has also been associated with thrombosis at unusual sites. However, the prevalence of MPN or JAK2V617F in patients presenting with splanchnic venous thromboembolism has varied.80 In addition, MPN may be occult (ie, no clinical or laboratory abnormalities) in around 15% of patients.84 Screening for JAK2V617F and underlying MPN is recommended in patients presenting with isolated unexplained splanchnic venous thromboembolism. Treatment entails long-term anticoagulation therapy. JAK2V617F screening in patients with nonsplanchnic venous thromboembolism is not recommended, as its prevalence in this group is low (< 3%).85,86
Treatment
Cases Continued
Patient A is diagnosed with PV based on the presence of 2 major criteria (elevated hemoglobin and presence of the JAK2V617F mutation) and 1 minor criterion (low erythropoietin level). Given his age, he belongs to the high-risk disease category. He is now seeking advice regarding the management of his newly diagnosed PV.
Patient B presents to the emergency department with right lower extremity swelling and is found to have deep femoral thrombosis extending to the iliac vein. Five days after being discharged from the emergency department, she presents for follow-up. She is taking warfarin compliantly and her INR is within therapeutic range. The patient now has high-risk ET and would like to know more about thrombosis in her condition and how to best manage her risk.
Risk-Adapted Therapy
Low-Risk PV
All patients with PV should receive counseling to mitigate cardiovascular risk factors, including smoking cessation, lifestyle modifications, and lipid-lowering therapy, as indicated. Furthermore, all PV patients should receive acetylsalicylic acid (ASA) to decrease their risk for thrombosis and control vasomotor symptoms.55,87 Aspirin 81 to 100 mg daily is the preferred regimen because it provides adequate antithrombotic effect without the associated bleeding risk of higher-dose aspirin.88 Low-risk PV patients should also receive periodic phlebotomies to reduce and maintain their hematocrit below 45%. This recommendation is based on the results of the Cytoreductive Therapy in Polycythemia Vera (CYTO PV) randomized controlled trial. In the CYTO PV study, patients receiving more intense therapy to maintain the hematocrit below 45% had a lower incidence of cardiovascular-related deaths or major thrombotic events than those with hematocrit goals of 45% to 50% (2.7% versus 9.8%).89 Cytoreduction is an option for low-risk patients who do not tolerate phlebotomy or require frequent phlebotomy, or who have disease-related bleeding, severe symptoms, symptomatic splenomegaly, or progressive leukocytosis.38
High-Risk PV
Patients older than 60 years and/or with a history of thrombosis should be considered for cytoreductive therapy in addition to the above measures. Front-line cytoreductive therapies include hydroxyurea or interferon (IFN)- alfa.87 Hydroxyurea is a potent ribonucleotide reductase inhibitor that interferes with DNA repair and is the treatment of choice for most high-risk patients with PV.90 In a small trial hydroxyurea reduced the risk of thrombosis compared with historical controls treated with phlebotomy alone.91 Hydroxyurea is generally well tolerated; common side effects include cytopenias, nail changes, and mucosal and/or skin ulcers. Although never formally proven to be leukemogenic, this agent should be used with caution in younger patients.87 Indeed, in the original study, the rates of transformation were 5.9% and 1.5% for patients receiving hydroxyurea and phlebotomy alone,92 respectively, although an independent role for hydroxyurea in leukemic transformation was not supported in the much larger European Collaboration on Low-dose Aspirin in Polycythemia Vera (ECLAP) study.93 About 70% of patients will have a sustained response to hydroxyurea,94 while the remaining patients become resistant to or intolerant of the drug. Resistant individuals have a higher risk of progression to acute leukemia and death.95
IFN alfa is a pleiotropic antitumor agent that has found application in many types of malignancies96 and is sometimes employed as treatment for patients with newly diagnosed high-risk PV. Early studies showed responses in up to 100% of cases,97,98 albeit at the expense of a high discontinuation rate due to adverse events, such as flu-like symptoms, fatigue, and neuropsychiatric manifestations.99 A newer formulation of the drug obtained by adding a polyethylene glycol (PEG) moiety to the native IFN alfa molecule (PEG-IFN alfa) was shown to have a longer half-life, greater stability, less immunogenicity, and, potentially, better tolerability.100 Pilot phase 2 trials of PEG-IFN alfa-2a demonstrated its remarkable activity, with symptomatic and hematologic responses seen in the majority of patients (which, in some cases, persisted beyond discontinuation), and reasonable tolerability, with long-term discontinuation rates of around 20% to 30%.101–103 In some patients JAK2V617F became undetectable over time.104 Results of 2 ongoing trials, MDP-RC111 (single-arm study, PEG-IFN alfa-2a in high-risk PV or ET [NCT01259817]) and MPD-RC112 (randomized controlled trial, PEG-IFN alfa-2a versus hydroxyurea in the same population [NCT01258856]), will shed light on the role of PEG-IFN alfa in the management of patients with high-risk PV or ET. In 2 phase 2 studies of PEG-IFN alfa-2b, complete responses were seen in 70% to 100% of patients and discontinuation occurred in around a third of cases.105,106 A new, longer-acting formulation of PEG-IFN alfa-2a (peg-proline INF alfa-2b, AOP2014) is also undergoing clinical development.107,108
The approach to treatment of PV based on thrombotic risk level is illustrated in Figure 1.
Very Low- and Low-Risk ET
Like patients with PV, individuals with ET should undergo rigorous cardiovascular risk management and generally receive ASA to decrease their thrombotic risk and improve symptom control. Antiplatelet therapy may not be warranted in patients with documented
Intermediate-Risk ET
This category includes patients older than 60 years but without thrombosis or JAK2 mutations. These individuals would have been considered high risk (and thus candidates for cytoreductive therapy) according to the traditional risk stratification. Guidelines currently recommend ASA as the sole therapy for these patients, while reserving cytoreduction for those who experience thrombosis (ie, become high-risk) or have uncontrolled vasomotor or general symptoms, symptomatic splenomegaly, symptomatic thrombocytosis, or progressive leukocytosis.
High-Risk ET
For patients with ET in need of cytoreductive therapy (ie, those with prior thrombosis or older than 60 years with a JAK2V617F mutation), first-line options include hydroxyurea, IFN, and anagrelide. Hydroxyurea remains the treatment of choice in the majority of patients.110 In a seminal study, 114 patients with ET were randomly assigned to either observation or hydroxyurea treatment with the goal of maintaining the platelet count below 600 × 103/µL. At a median follow-up of 27 months, patients in the hydroxyurea group had a lower thrombosis rate (3.6% versus 24%, P = 0.003) and longer thrombosis-free survival, regardless of the use of antiplatelet drugs.64
Anagrelide, a selective inhibitor of megakaryocytic differentiation and proliferation, was compared with hydroxyurea in patients with ET in 2 randomized trials. In the first (N = 809), the group receiving anagrelide had a higher risk of arterial thrombosis, major bleeding, and fibrotic evolution, but lower incidence of venous thrombosis. Hydroxyurea was better tolerated, mainly due to anagrelide-related cardiovascular adverse events.111 As a result of this study, hydroxyurea is often preferred to anagrelide as front-line therapy for patients with newly diagnosed high-risk ET. In the second, more recent study (N = 259), however, the 2 agents proved equivalent in terms of major or minor arterial or venous thrombosis, as well as discontinuation rate.112 The discrepancy between the 2 trials may be partly explained by the different ET diagnostic criteria used, with the latter only enrolling patients with WHO-defined true ET, while the former utilized Polycythemia Vera Study Group-ET diagnostic criteria that included patients with increases in other blood counts or varying degrees of marrow fibrosis.
Interferons were studied in ET in parallel with PV. PEG-IFN alfa-2a proved effective in patients with ET, with responses observed in 80% of patients.103 PEG-IFN alfa-2b produced similar results, with responses in 70% to 90% of patients in small studies and discontinuation observed in 20% to 38% of cases.105,106,113 Because the very long-term leukemogenic potential of hydroxyurea has remained somewhat uncertain, anagrelide or IFN might be preferable choices in younger patients.
The approach to treatment of ET based on thrombotic risk level is illustrated in Figure 2.
Assessing Response to Therapy
For both patients with PV and ET the endpoint of treatment set forth for clinical trials has been the achievement of a clinicohematologic response. However, studies have failed to show a correlation between response and reduction of the thrombohemorrhagic risk.114 Therefore, proposed clinical trial response criteria were revised to include absence of hemorrhagic or thrombotic events as part of the definition of response (Table 3).94
Cases Continued
Patient A was initially treated with phlebotomies and his blood counts were subsequently controlled with hydroxyurea, which he took uninterruptedly at an average dose of 2.5 g daily. He also took ASA daily throughout. Now, 18 months after the start of therapy, he presents with a complaint of fatigue for the past 3 months, which more recently has been associated with recurrent itching. A repeat CBC shows a WBC count of 17,200/µL, hemoglobin 181 g/L, and platelets 940 × 103/µL.
Patient B presents for scheduled follow-up. She has had no further thrombotic episodes. However, she spontaneously discontinued hydroxyurea 1 month ago because of worsening mouth ulcers that impaired her ability to eat even small meals. She seeks recommendations for further treatment options.
Approach to Patients Refractory to or Intolerant of First-Line Therapy
According to the European LeukemiaNet recommendations, an inadequate response to hydroxyurea in patients with PV (or myelofibrosis) is defined as a need for phlebotomy to maintain hematocrit below < 45%, platelet count > 400 × 103/µL, and a WBC count > 10,000/µL, or failure to reduce splenomegaly > 10 cm by > 50% at a dose of ≥ 2 g/day or maximum tolerated dose. Historically, treatment options for patients with PV or ET who failed first-line therapy (most commonly hydroxyurea) have included alkylating agents, such as busulfan, chlorambucil, or pipobroman, and phosphorus (P)-32. However, the use of these drugs is limited by the associated risk of leukemic transformation.93,115,116 The use of IFN (or anagrelide for ET) is often considered in patients previously treated with hydroxyurea, and vice versa.
Ruxolitinib is a JAK1 and JAK2 inhibitor currently approved for the treatment of PV patients refractory to or intolerant of hydroxyurea.7 Following promising results of a phase 2 trial,117 ruxolitinib 10 mg twice daily was compared with best available therapy in the pivotal RESPONSE trial (N = 222). Ruxolitinib proved superior in achieving hematocrit control, reduction of spleen volume, and improvement of symptoms. Grade 3-4 hematologic toxicity was infrequent and similar in the 2 arms.118 In addition, longer follow-up of that study suggested a lower rate of thrombotic events in patients receiving ruxolitinib (1.8 versus 8.2 per 100 patient-years).119 In a similarly designed randomized phase 3 study in PV patients without splenomegaly (RESPONSE-2), more patients in the ruxolitinib arm had hematocrit reduction without an increase in toxicity. Based on the results of the above studies, ruxolitinib can be considered a standard of care for second-line therapy in this post-hydroxyurea patient population.120
Ruxolitinib is also being tested in patients with high-risk ET who have become resistant to, or were intolerant of hydroxyurea, but currently has no approved indication in this setting.121,122 Common side effects of ruxolitinib include cytopenias (especially anemia), increased risk of infections, hyperlipidemia, and increased risk of non-melanoma skin cancer.
Novel Agents
Novel agents that have been studied in patients with PV and ET are histone deacetylase inhibitors, murine double minute 2 (MDM2, or HDM2 for their human counterpart) inhibitors (which restore the function of p53), Bcl-2 homology domain 3 mimetics such as navitoclax and venetoclax, and, for patients with ET, the telomerase inhibitor imetelstat.123
Disease Evolution
Cases Continued
Patient A’s PV has been well controlled with PEG-IFN alfa-2a 90 μg subcutaneously weekly. However, he now presents with a complaint of worsening fatigue and early satiety. On exam the patient appears ill and splenomegaly is appreciated 12 cm below the costal margin. CBC shows a WBC count of 2600/µL, hemoglobin 73 g/L, and platelets 122 × 103/µL. Peripheral blood smear reveals leukoerythroblastosis and dacrocytosis. CBC 6 months ago was normal. A bone marrow biopsy is consistent with myelofibrosis.
After discontinuing hydroxyurea, patient B’s ET has been well controlled with anagrelide. However, for the past 4 weeks she has complained of severe fatigue and easy bruising. Physical exam reveals a pale, ill-appearing woman with scattered bruises. CBC shows a WBC count of 14,600/µL with 44% myeloblasts, hemoglobin 73 g/L, and platelets 22 × 103/µL. CBC 6 months ago was normal. A bone marrow biopsy is consistent with leukemic transformation of ET.
Post-PV/Post-ET Myelofibrosis
Diagnostic criteria for post-PV and post-ET myelofibrosis are outlined in Table 4.
Leukemic Transformation
The presence of more than 20% blasts in peripheral blood or bone marrow in a patient with MPN defines leukemic transformation. This occurs in up to 5% to 10% of patients and may or may not be preceded by a myelofibrosis phase.126 In cases of extramedullary transformation, a lower percentage of blasts can be seen in the bone marrow compared to the peripheral blood. The pathogenesis of leukemic transformation has remained elusive, but it is believed to be associated with genetic instability, which facilitates the acquisition of additional mutations, including those of TET2, ASXL1, EZH2 and DNMT3, IDH1/2, and TP53.127
Clinical risk factors for leukemic transformation include advanced age, karyotypic abnormalities, prior therapy with alkylating agents or P-32, splenectomy, increased peripheral blood or bone marrow blasts, leukocytosis, anemia, thrombocytopenia, and cytogenetic abnormalities. Hydroxyurea, interferon, and ruxolitinib have not been shown to have leukemogenic potential thus far. Prognosis of leukemic transformation is uniformly poor and patient survival rarely exceeds 6 months.
There is no standard of care for leukemic transformation of MPN (MPN-LT). Treatment options range from low-intensity regimens to more aggressive AML-type induction chemotherapy. No strategy appears clearly superior to others.128 Hematopoietic stem cell transplantation is the only therapy that provides clinically meaningful benefit to patients,129 but it is applicable only to a minority of patients with chemosensitive disease and good performance status.130 Notable experimental approaches to MPN–LT include hypomethylating agents, such as decitabine131 or azacitidine,132 with or without ruxolitinib.133-135
Conclusion
PV and ET are rare, chronic myeloid disorders. Patients typically experience a long clinical course and enjoy near-normal quality of life if properly managed. The 2 most important life-limiting complications of PV and ET are thrombohemorrhagic events and myelofibrosis/AML transformation. Vascular events are at least in part preventable with counseling on risk factors, phlebotomy (for patients with PV), antiplatelet therapy, and cytoreduction with hydroxyurea, IFNs, or anagrelide (for patients with ET). In addition, ruxolitinib was recently approved for PV patients after hydroxyurea failure. PV/ET transformation in myelofibrosis or AML is part of the natural history of the disease and no therapy has been shown to prevent it. Treatment follows recommendations set forth for PMF and AML, but results are generally poorer and novel strategies are needed to improve patients’ outcomes.
1. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon: IARC; 2008.
2. Alvarez-Larran A, Pereira A, Arellano-Rodrigo E, et al. Cytoreduction plus low-dose aspirin versus cytoreduction alone as primary prophylaxis of thrombosis in patients with high-risk essential thrombocythaemia: an observational study. Br J Haematol 2013;161:865–71.
3. Tefferi A. Polycythemia vera and essential thrombocythemia: 2013 update on diagnosis, risk-stratification, and management. Am J Hematol 2013;88:507–16.
4. Michiels JJ, Berneman Z, Schroyens W, et al. Platelet-mediated erythromelalgic, cerebral, ocular and coronary microvascular ischemic and thrombotic manifestations in patients with essential thrombocythemia and polycythemia vera: a distinct aspirin-responsive and coumadin-resistant arterial thrombophilia. Platelets 2006;17:528–44.
5. James C, Ugo V, Couedic J-PL, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005;434:1144–8.
6. Kralovics R, Passamonti F, Buser A, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders.N Engl J Med 2005;352:1779–90.
7. Verstovsek S, Mesa R, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med 2012;366:799–807.
8. Harrison C, Kiladjian J, Al-Ali H, et al. JAK Inhibition with ruxolitinib versus best available therapy for myelofibrosis.N Engl J Med 2012;366:787–98.
9. Berglund S, Zettervall O. Incidence of polycythemia vera in a defined population. Eur J Haematol 1992;48:20–6.
10. Rozman C, Giralt M, Feliu E, et al. Life expectancy of patients with chronic nonleukemic myeloproliferative disorders. Cancer 1991;67:2658–63.
11. Passamonti F, Rumi E, Pungolino E, et al. Life expectancy and prognostic factors for survival in patients with polycythemia vera and essential thrombocythemia. Am J Med 2004;117:755–61.
12. Hultcrantz M, Kristinsson SY, Andersson TM, et al. Patterns of survival among patients with myeloproliferative neoplasms diagnosed in Sweden from 1973 to 2008: a population-based study. J Clin Oncol 2012;30:2995–3001.
13. Wolanskyj AP, Schwager SM, et al. Essential thrombocythemia beyond the first decade: life expectancy, long-term complication rates, and prognostic factors. Mayo Clin Proc 2006;81:159–66.
14. Srour SA, Devesa SS, Morton LM, et al. Incidence and patient survival of myeloproliferative neoplasms and myelodysplastic/myeloproliferative neoplasms in the United States, 2001-12. Br J Haematol 2016;174:382–96.
15. Titmarsh GJ, Duncombe AS, McMullin MF, et al. How common are myeloproliferative neoplasms? A systematic review and meta-analysis. Am J Hematol 2014;89:581–7.
16. Moulard O, Mehta J, Fryzek J, et al. Epidemiology of myelofibrosis, essential thrombocythemia, and polycythemia vera in the European Union. Eur J Haematol 2014;92:289–97.
17. Stein BL, Gotlib J, Arcasoy M, et al. Historical views, conventional approaches, and evolving management strategies for myeloproliferative neoplasms. J Natl Compr Canc Netw 2015;13:424–34.
18. Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005;7:387–97.
19. Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005;365:1054–61.
20. Scott LM, Tong W, Levine RL, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med 2007;356:459–68.
21. Pardanani A, Lasho TL, Finke C, et al. Prevalence and clinicopathologic correlates of JAK2 exon 12 mutations in JAK2V617F-negative polycythemia vera. Leukemia 2007;21:1960–3.
22. Passamonti F, Elena C, Schnittger S, et al. Molecular and clinical features of the myeloproliferative neoplasm associated with JAK2 exon 12 mutations. Blood 2011;117:2813–6.
23. Abe M, Suzuki K, Inagaki O, et al. A novel MPL point mutation resulting in thrombopoietin-independent activation. Leukemia 2002;16:1500–6.
24. Pikman Y, Lee BH, Mercher T, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med 2006;3:e270.
25. Pardanani AD, Levine RL, Lasho T, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood 2006;108:3472–6.
26. Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med 2013;369:2391–405.
27. Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med 2013;369:2379–90.
28. Wang WA, Groenendyk J, Michalak M. Calreticulin signaling in health and disease. Int J Biochem Cell Biol 2012;44:842–6.
29. Saeidi K. Myeloproliferative neoplasms: Current molecular biology and genetics. Crit Rev Oncol Hematol 2016;98:375–89.
30. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016;127:2391–405.
31. Mesa RA, Niblack J, Wadleigh M, et al. The burden of fatigue and quality of life in myeloproliferative disorders (MPDs): an international Internet-based survey of 1179 MPD patients. Cancer 2007;109:68–76.
32. Marchioli R, Finazzi G, Landolfi R, et al. Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol 2005;23:2224–32.
33. Scherber R, Dueck AC, Johansson P, et al. The Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF): international prospective validation and reliability trial in 402 patients. Blood 2011;118:401–8.
34. Emanuel RM, Dueck AC, Geyer HL, et al. Myeloproliferative neoplasm (MPN) symptom assessment form total symptom score: prospective international assessment of an abbreviated symptom burden scoring system among patients with MPNs. J Clin Oncol 2012;30:4098–103.
35. Casini A, Fontana P, Lecompte TP. Thrombotic complications of myeloproliferative neoplasms: risk assessment and risk-guided management. J Thromb Haemost 2013;11:1215–27.
36. Barbui T, Finazzi G, Falanga A. Myeloproliferative neoplasms and thrombosis. Blood 2013;122:2176-–84.
37. Pearson TC, Wetherley-Mein G. Vascular occlusive episodes and venous haematocrit in primary myeloproliferative polychytemia. Lancet 1978;2:1219–22.
38. Landolfi R, Di Gennaro L, Barbui T, et al. Leukocytosis as a major thrombotic risk factor in patients with polycythemia vera. Blood 2007;109:2446–52.
39. Tefferi A, Rumi E, Finazzi G, et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia 2013;27:1874–81.
40. Barbui T, Falanga A. Molecular biomarkers of thrombosis in myeloproliferative neoplasms. Thromb Res 2016;140 Suppl 1:S71–75.
41. Campbell PJ, Scott LM, Buck G, et al. Definition of subtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2 V617F mutation status: a prospective study. Lancet 2005;366:1945–53.
42. Tefferi A, Guglielmelli P, Larson DR, et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 2014;124:2507–13.
43. Qin Y, Wang X, Zhao C, et al. The impact of JAK2V617F mutation on different types of thrombosis risk in patients with essential thrombocythemia: a meta-analysis. Int J Hematol 2015;102:170–80.
44. Ziakas PD. Effect of JAK2 V617F on thrombotic risk in patients with essential thrombocythemia: measuring the uncertain. Haematologica 2008;93:1412–4.
45. Lussana F, Dentali F, Abbate R, et al. Screening for thrombophilia and antithrombotic prophylaxis in pregnancy: Guidelines of the Italian Society for Haemostasis and Thrombosis (SISET). Thromb Res 2009;124:e19–25.
46. Dahabreh IJ, Zoi K, Giannouli S, et al. Is JAK2 V617F mutation more than a diagnostic index? A meta-analysis of clinical outcomes in essential thrombocythemia. Leuk Res 2009;33:67–73.
47. Cho YU, Chi HS, Lee EH, et al. Comparison of clinicopathologic findings according to JAK2 V617F mutation in patients with essential thrombocythemia. Int J Hematol 2009;89:39–44.
48. Wolanskyj AP, Lasho TL, Schwager SM, et al. JAK2 mutation in essential thrombocythaemia: clinical associations and long-term prognostic relevance. Br J Haematol 2005;131:208–13.
49. Antonioli E, Guglielmelli P, Pancrazzi A, et al. Clinical implications of the JAK2 V617F mutation in essential thrombocythemia. Leukemia 2005;19:1847–9.
50. Palandri F, Catani L, Testoni N, et al. Long-term follow-up of 386 consecutive patients with essential thrombocythemia: safety of cytoreductive therapy. Am J Hematol 2009;84:215–20.
51. Chim CS, Sim JP, Chan CC, et al. Impact of JAK2V617F mutation on thrombosis and myeloid transformation in essential thrombocythemia: a multivariate analysis by Cox regression in 141 patients. Hematology 2010;15:187–92.
52. Vannucchi AM, Antonioli E, Guglielmelli P, et al. Clinical profile of homozygous JAK2 617V>F mutation in patients with polycythemia vera or essential thrombocythemia. Blood 2007;110:840–6.
53. Carobbio A, Finazzi G, Antonioli E, et al. JAK2V617F allele burden and thrombosis: a direct comparison in essential thrombocythemia and polycythemia vera. Exp Hematol 2009;37:1016–21.
54. Alvarez-Larran A, Bellosillo B, Pereira A, et al. JAK2V617F monitoring in polycythemia vera and essential thrombocythemia: clinical usefulness for predicting myelofibrotic transformation and thrombotic events. Am J Hematol 2014;89:517–23.
55. Barbui T, Vannucchi AM, Buxhofer-Ausch V, et al. Practice-relevant revision of IPSET-thrombosis based on 1019 patients with WHO-defined essential thrombocythemia. Blood Cancer J 2015;5:e369.
56. Carobbio A, Thiele J, Passamonti F, et al. Risk factors for arterial and venous thrombosis in WHO-defined essential thrombocythemia: an international study of 891 patients. Blood 2011;117:5857–9.
57. Alvarez-Larran A, Cervantes F, Bellosillo B, et al. Essential thrombocythemia in young individuals: frequency and risk factors for vascular events and evolution to myelofibrosis in 126 patients. Leukemia 2007;21:1218–23.
58. Jantunen R, Juvonen E, Ikkala E, et al. The predictive value of vascular risk factors and gender for the development of thrombotic complications in essential thrombocythemia. Ann Hematol 2001;80:74–8.
59. Besses C, Cervantes F, Pereira A, et al. Major vascular complications in essential thrombocythemia: a study of the predictive factors in a series of 148 patients. Leukemia 1999;13:150–4.
60. Haider M, Gangat N, Hanson C, Tefferi A. Splenomegaly and thrombosis risk in essential thrombocythemia: the mayo clinic experience. Am J Hematol 2016;91:E296–297.
61. Carobbio A, Finazzi G, Antonioli E, et al. Thrombocytosis and leukocytosis interaction in vascular complications of essential thrombocythemia. Blood 2008;112:3135–7.
62. Palandri F, Polverelli N, Catani L, et al. Impact of leukocytosis on thrombotic risk and survival in 532 patients with essential thrombocythemia: a retrospective study. Ann Hematol 2011;90:933–8.
63. Campbell PJ, MacLean C, Beer PA, et al. Correlation of blood counts with vascular complications in essential thrombocythemia: analysis of the prospective PT1 cohort. Blood 2012;120:1409–11.
64. Cortelazzo S, Finazzi G, Ruggeri M, et al. Hydroxyurea for patients with essential thrombocythemia and a high risk of thrombosis. N Engl J Med 1995;332:1132–6.
65. van Genderen PJ, Mulder PG, Waleboer M, et al. Prevention and treatment of thrombotic complications in essential thrombocythaemia: efficacy and safety of aspirin. Br J Haematol 1997;97:179–84.
66. Storen EC, Tefferi A. Long-term use of anagrelide in young patients with essential thrombocythemia. Blood 2001;97:863–6.
67. De Stefano V, Za T, Rossi E, et al. Recurrent thrombosis in patients with polycythemia vera and essential thrombocythemia: incidence, risk factors, and effect of treatments. Haematologica 2008;93:372–80.
68. Alvarez-Larran A, Cervantes F, Pereira A, et al. Observation versus antiplatelet therapy as primary prophylaxis for thrombosis in low-risk essential thrombocythemia. Blood 2010;116:1205–10.
69. Palandri F, Polverelli N, Catani L, et al. Bleeding in essential thrombocythaemia: a retrospective analysis on 565 patients. Br J Haematol 2012;156:281–4.
70. Rotunno G, Mannarelli C, Guglielmelli P, et al. Impact of calreticulin mutations on clinical and hematological phenotype and outcome in essential thrombocythemia. Blood 2014;123:1552–5.
71. Tefferi A, Wassie EA, Lasho TL, et al. Calreticulin mutations and long-term survival in essential thrombocythemia. Leukemia 2014;28:2300–3.
72. Rumi E, Pietra D, Ferretti V, et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood 2014;123:1544–51.
73. Palandri F, Latagliata R, Polverelli N, et al. Mutations and long-term outcome of 217 young patients with essential thrombocythemia or early primary myelofibrosis. Leukemia 2015;29:1344–9.
74. Fu R, Xuan M, Zhou Y, et al. Analysis of calreticulin mutations in Chinese patients with essential thrombocythemia: clinical implications in diagnosis, prognosis and treatment. Leukemia 2014;28:1912–4.
75. Tefferi A, Wassie EA, Guglielmelli P, et al. Type 1 versus Type 2 calreticulin mutations in essential thrombocythemia: a collaborative study of 1027 patients. Am J Hematol 2014;89:E121–4.
76. Pietra D, Rumi E, Ferretti VV, et al. Differential clinical effects of different mutation subtypes in CALR-mutant myeloproliferative neoplasms. Leukemia 2016;30:431–8.
77. Rumi E, Pietra D, Guglielmelli P, et al. Acquired copy-neutral loss of heterozygosity of chromosome 1p as a molecular event associated with marrow fibrosis in MPL-mutated myeloproliferative neoplasms. Blood 2013;121:4388–95.
78. Beer PA, Campbell PJ, Scott LM, et al. MPL mutations in myeloproliferative disorders: analysis of the PT-1 cohort. Blood 2008;112:141–9.
79. Gangat N, Wassie EA, Lasho TL, et al. Mutations and thrombosis in essential thrombocythemia: prognostic interaction with age and thrombosis history. Eur J Haematol 2015;94:31–6.
80. Sekhar M, McVinnie K, Burroughs AK. Splanchnic vein thrombosis in myeloproliferative neoplasms. Br J Haematol 2013;162:730–47.
81. Stein BL, Saraf S, Sobol U, et al. Age-related differences in disease characteristics and clinical outcomes in polycythemia vera. Leuk Lymph 2013;54:1989–95.
82. Landolfi R, Di Gennaro L, Nicolazzi MA, et al. Polycythemia vera: gender-related phenotypic differences. Intern Emerg Med 2012;7:509–15.
83. Winslow ER, Brunt LM, Drebin JA, et al. Portal vein thrombosis after splenectomy. Am J Surg 2002;184:631–6.
84. Smalberg JH, Arends LR, Valla DC, et al. Myeloproliferative neoplasms in Budd-Chiari syndrome and portal vein thrombosis: a meta-analysis. Blood 2012;120:4921–8.
85. Dentali F, Squizzato A, Brivio L, et al. JAK2V617F mutation for the early diagnosis of Ph- myeloproliferative neoplasms in patients with venous thromboembolism: a meta-analysis. Blood 2009;113:5617–23.
86. Pardanani A, Lasho TL, Hussein K, et al. JAK2V617F mutation screening as part of the hypercoagulable work-up in the absence of splanchnic venous thrombosis or overt myeloproliferative neoplasm: assessment of value in a series of 664 consecutive patients. Mayo Clin Proc 2008;83:457–9.
87. Barbui T, Barosi G, Birgegard G, et al. Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European LeukemiaNet. J Clin Oncol 2011;29:761–70.
88. Landolfi R, Marchioli R, Kutti J, et al. Efficacy and safety of low-dose aspirin in polycythemia vera. N Engl J Med 2004;350:114–24.
89. Marchioli R, Finazzi G, Specchia G, et al. Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med 2013;368:22–33.
90. Kiladjian JJ, Chevret S, Dosquet C, et al. Treatment of polycythemia vera with hydroxyurea and pipobroman: final results of a randomized trial initiated in 1980. J Clin Oncol 2011;29:3907–13.
91. Kaplan ME, Mack K, Goldberg JD, et al. Long-term management of polycythemia vera with hydroxyurea: a progress report. Semin Hematol 1986;23:167–71.
92. Fruchtman SM, Mack K, Kaplan ME, et al. From efficacy to safety: a Polycythemia Vera Study group report on hydroxyurea in patients with polycythemia vera. Semin Hematol 1997;34:17–23.
93. Finazzi G, Caruso V, Marchioli R, et al. Acute leukemia in polycythemia vera: an analysis of 1638 patients enrolled in a prospective observational study. Blood 2005;105:2664–70.
94. Barosi G, Mesa R, Finazzi G, et al. Revised response criteria for polycythemia vera and essential thrombocythemia: an ELN and IWG-MRT consensus project. Blood 2013;121:4778–81.
95. Alvarez-Larran A, Pereira A, Cervantes F, et al. Assessment and prognostic value of the European LeukemiaNet criteria for clinicohematologic response, resistance, and intolerance to hydroxyurea in polycythemia vera. Blood 2012;119:1363–9.
96. Stein BL, Tiu RV. Biological rationale and clinical use of interferon in the classical BCR-ABL-negative myeloproliferative neoplasms. J Interferon Cytokine Res 2013;33:145–53.
97. Ludwig H, Cortelezzi A, Van Camp BG, et al. Treatment with recombinant interferon-alpha-2C: multiple myeloma and thrombocythaemia in myeloproliferative diseases. Oncology 1985;42 Suppl 1:19–25.
98. Silver RT. Long-term effects of the treatment of polycythemia vera with recombinant interferon-alpha. Cancer 2006;107:451–8.
99. Kiladjian JJ, Mesa RA, Hoffman R. The renaissance of interferon therapy for the treatment of myeloid malignancies. Blood 2011;117:4706–15.
100. Veronese FM, Mero A. The impact of PEGylation on biological therapies. BioDrugs 2008;22:315–29.
101. Kiladjian JJ, Cassinat B, Chevret S, et al. Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood 2008;112:3065–72.
102. Turlure P, Cambier N, Roussel M, et al. Complete hematological, molecular and histological remissions without cytoreductive treatment lasting after pegylated-interferon {alpha}-2a (peg-IFN{alpha}-2a) therapy in polycythemia vera (PV): long term results of a phase 2 trial [abstract]. Blood 2011;118(21). Abstract 280.
103. Quintas-Cardama A, Kantarjian H, Manshouri T, et al. Pegylated interferon alfa-2a yields high rates of hematologic and molecular response in patients with advanced essential thrombocythemia and polycythemia vera. J Clin Oncol 2009;27:5418–24.
104. Quintas-Cardama A, Abdel-Wahab O, Manshouri T, et al. Molecular analysis of patients with polycythemia vera or essential thrombocythemia receiving pegylated interferon a-2a. Blood 2013;122:893–901.
105. Samuelsson J, Hasselbalch H, Bruserud O, et al. A phase II trial of pegylated interferon alpha-2b therapy for polycythemia vera and essential thrombocythemia: feasibility, clinical and biologic effects, and impact on quality of life. Cancer 2006;106:2397–405.
106. Jabbour E, Kantarjian H, Cortes J, et al. PEG-IFN-alpha-2b therapy in BCR-ABL-negative myeloproliferative disorders: final result of a phase 2 study. Cancer 2007;110:2012–18.
107. Them NC, Bagienski K, Berg T, et al. Molecular responses and chromosomal aberrations in patients with polycythemia vera treated with peg-proline-interferon alpha-2b. Am J Hematol 2015;90:288–94.
108. Gisslinger H, Klade C, Georgiev P, et al. Final results from PROUD-PV a randomized controlled phase 3 trial comparing ropeginterferon alfa-2b to hydroxyurea in polycythemia vera patients [abstract]. Blood 2016;128(suppl 22). Abstract 475.
109. van Genderen PJ, van Vliet HH, Prins FJ, et al. Excessive prolongation of the bleeding time by aspirin in essential thrombocythemia is related to a decrease of large von Willebrand factor multimers in plasma. Ann Hematol 1997;75:215–20.
110. Cortelazzo S, Finazzi G, Ruggeri M, et al. Hydroxyurea for patients with essential thrombocythemia and a high risk of thrombosis. N Engl J Med 1995;332:1132–7.
111. Harrison CN, Campbell PJ, Buck G, et al. Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. N Engl J Med 2005;353:33–45.
112. Gisslinger H, Gotic M, Holowiecki J, et al. Anagrelide compared with hydroxyurea in WHO-classified essential thrombocythemia: the ANAHYDRET Study, a randomized controlled trial. Blood 2013;121:1720–8.
113. Alvarado Y, Cortes J, Verstovsek S, et al. Pilot study of pegylated interferon-alpha 2b in patients with essential thrombocythemia. Cancer Chemother Pharmacol 2003;51:81–6.
114. Barosi G, Tefferi A, Barbui T, ad hoc committee ‘Definition of clinically relevant outcomes for contemporarily clinical trials in Ph-neg M. Do current response criteria in classical Ph-negative myeloproliferative neoplasms capture benefit for patients? Leukemia 2012;26:1148–9.
115. Bjorkholm M, Derolf AR, Hultcrantz M, et al. Treatment-related risk factors for transformation to acute myeloid leukemia and myelodysplastic syndromes in myeloproliferative neoplasms. J Clin Oncol 2011;29:2410–5.
116. Alvarez-Larran A, Martinez-Aviles L, Hernandez-Boluda JC, et al. Busulfan in patients with polycythemia vera or essential thrombocythemia refractory or intolerant to hydroxyurea. Ann Hematol 2014;93:2037–43.
117. Verstovsek S, Passamonti F, Rambaldi A, et al. A phase 2 study of ruxolitinib, an oral JAK1 and JAK2 Inhibitor, in patients with advanced polycythemia vera who are refractory or intolerant to hydroxyurea. Cancer 2014;120:513–20.
118. Vannucchi AM, Kiladjian JJ, Griesshammer M, et al. Ruxolitinib in polycythemia vera resistant to or intolerant of hydroxyurea. N Engl J Med 2015; 372:426–35.
119. Verstovsek S, Vannucchi AM, Griesshammer M, et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80-week follow-up from the RESPONSE trial. Haematologica 2016;101:821–9.
120. Passamonti F, Griesshammer M, Palandri F, et al. Ruxolitinib for the treatment of inadequately controlled polycythaemia vera without splenomegaly (RESPONSE-2): a randomised, open-label, phase 3b study. Lancet Oncol 2017;18:88–99.
121. Verstovsek S, Passamonti F, Rambaldi A, et al. Long-term results from a phase II open-label study of ruxolitinib in patients with essential thrombocythemia refractory to or intolerant of hydroxyurea [abstract]. Blood 2014;124. Abstract 1847.
122. Harrison CN, Mead AJ, Panchal A, et al. Ruxolitinib versus best available therapy for ET intolerant or resistant to hydroxycarbamide in a randomized trial. Blood 2017 Aug 9. pii: blood-2017-05-785790 .
123. Bose P, Verstovsek S. Drug development pipeline for myeloproliferative neoplasms: potential future impact on guidelines and management. J Natl Compr Canc Netw 2016;14:1613–24.
124. Cerquozzi S, Teffieri A. Blast transformation and fibrotic progression in polycythemia vera and essential thrombocythemia: a literature review of incidence and risk factors. Blood Cancer J 2015;Nov 13;5:e366.
125. Passamonti F, Rumi E, Caramella M, et al. A dynamic prognostic model to predict survival in post-polycythemia vera myelofibrosis. Blood 2008;111:3383–7.
126. Mesa RA, Verstovsek S, Cervantes F, et al. Primary myelofibrosis (PMF), post polycythemia vera myelofibrosis (post-PV MF), post essential thrombocythemia myelofibrosis (post-ET MF), blast phase PMF (PMF-BP): Consensus on terminology by the international working group for myelofibrosis research and treatment (IWG-MRT). Leuk Res 2007;31:737–40.
127. Rampal R, Mascarenhas J. Pathogenesis and management of acute myeloid leukemia that has evolved from a myeloproliferative neoplasm. Curr Opin Hematol 2014;21:65–71.
128. Chihara D, Kantarjian HM, Newberry KJ, et al. Survival outcome of patients with acute myeloid leukemia transformed from myeloproliferative neoplasms [abstract]. Blood 2016;128. Abstract 1940.
129. Tam CS, Nussenzveig RM, Popat U, et al. The natural history and treatment outcome of blast phase BCR-ABL- myeloproliferative neoplasms. Blood 2008;112:1628–37.
130. Kundranda MN, Tibes R, Mesa RA. Transformation of a chronic myeloproliferative neoplasm to acute myelogenous leukemia: does anything work? Curr Hematol Malig Rep 2012;7:78–86.
131. Badar T, Kantarjian HM, Ravandi F, et al. Therapeutic benefit of decitabine, a hypomethylating agent, in patients with high-risk primary myelofibrosis and myeloproliferative neoplasm in accelerated or blastic/acute myeloid leukemia phase. Leuk Res 2015;39:950–6.
132. Thepot S, Itzykson R, Seegers V, et al. Treatment of progression of Philadelphia-negative myeloproliferative neoplasms to myelodysplastic syndrome or acute myeloid leukemia by azacitidine: a report on 54 cases on the behalf of the Groupe Francophone des Myelodysplasies (GFM). Blood 2010;116:3735–42.
133. Pemmaraju N, Kantarjian H, Kadia T, et al. A phase I/II study of the Janus kinase (JAK)1 and 2 inhibitor ruxolitinib in patients with relapsed or refractory acute myeloid leukemia. Clin Lymphoma Myeloma Leuk 2015;15:171–6.
134. Rampal RK, Mascarenhas JO, Kosiorek HE, et al. Safety and efficacy of combined ruxolitinib and decitabine in patients with blast-phase MPN and post-MPN AML: results of a phase I study (Myeloproliferative Disorders Research Consortium 109 trial) [abstract]. Blood 2016;128. Abstract 1124.
135. Bose P, Verstovsek S, Gasior Y, et al. Phase I/II study of ruxolitinib (RUX) with decitabine (DAC) in patients with post-myeloproliferative neoplasm acute myeloid leukemia (post-MPN AML): phase I results [abstract]. Blood 2016;128. Abstract 4262.
1. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon: IARC; 2008.
2. Alvarez-Larran A, Pereira A, Arellano-Rodrigo E, et al. Cytoreduction plus low-dose aspirin versus cytoreduction alone as primary prophylaxis of thrombosis in patients with high-risk essential thrombocythaemia: an observational study. Br J Haematol 2013;161:865–71.
3. Tefferi A. Polycythemia vera and essential thrombocythemia: 2013 update on diagnosis, risk-stratification, and management. Am J Hematol 2013;88:507–16.
4. Michiels JJ, Berneman Z, Schroyens W, et al. Platelet-mediated erythromelalgic, cerebral, ocular and coronary microvascular ischemic and thrombotic manifestations in patients with essential thrombocythemia and polycythemia vera: a distinct aspirin-responsive and coumadin-resistant arterial thrombophilia. Platelets 2006;17:528–44.
5. James C, Ugo V, Couedic J-PL, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005;434:1144–8.
6. Kralovics R, Passamonti F, Buser A, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders.N Engl J Med 2005;352:1779–90.
7. Verstovsek S, Mesa R, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med 2012;366:799–807.
8. Harrison C, Kiladjian J, Al-Ali H, et al. JAK Inhibition with ruxolitinib versus best available therapy for myelofibrosis.N Engl J Med 2012;366:787–98.
9. Berglund S, Zettervall O. Incidence of polycythemia vera in a defined population. Eur J Haematol 1992;48:20–6.
10. Rozman C, Giralt M, Feliu E, et al. Life expectancy of patients with chronic nonleukemic myeloproliferative disorders. Cancer 1991;67:2658–63.
11. Passamonti F, Rumi E, Pungolino E, et al. Life expectancy and prognostic factors for survival in patients with polycythemia vera and essential thrombocythemia. Am J Med 2004;117:755–61.
12. Hultcrantz M, Kristinsson SY, Andersson TM, et al. Patterns of survival among patients with myeloproliferative neoplasms diagnosed in Sweden from 1973 to 2008: a population-based study. J Clin Oncol 2012;30:2995–3001.
13. Wolanskyj AP, Schwager SM, et al. Essential thrombocythemia beyond the first decade: life expectancy, long-term complication rates, and prognostic factors. Mayo Clin Proc 2006;81:159–66.
14. Srour SA, Devesa SS, Morton LM, et al. Incidence and patient survival of myeloproliferative neoplasms and myelodysplastic/myeloproliferative neoplasms in the United States, 2001-12. Br J Haematol 2016;174:382–96.
15. Titmarsh GJ, Duncombe AS, McMullin MF, et al. How common are myeloproliferative neoplasms? A systematic review and meta-analysis. Am J Hematol 2014;89:581–7.
16. Moulard O, Mehta J, Fryzek J, et al. Epidemiology of myelofibrosis, essential thrombocythemia, and polycythemia vera in the European Union. Eur J Haematol 2014;92:289–97.
17. Stein BL, Gotlib J, Arcasoy M, et al. Historical views, conventional approaches, and evolving management strategies for myeloproliferative neoplasms. J Natl Compr Canc Netw 2015;13:424–34.
18. Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005;7:387–97.
19. Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005;365:1054–61.
20. Scott LM, Tong W, Levine RL, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med 2007;356:459–68.
21. Pardanani A, Lasho TL, Finke C, et al. Prevalence and clinicopathologic correlates of JAK2 exon 12 mutations in JAK2V617F-negative polycythemia vera. Leukemia 2007;21:1960–3.
22. Passamonti F, Elena C, Schnittger S, et al. Molecular and clinical features of the myeloproliferative neoplasm associated with JAK2 exon 12 mutations. Blood 2011;117:2813–6.
23. Abe M, Suzuki K, Inagaki O, et al. A novel MPL point mutation resulting in thrombopoietin-independent activation. Leukemia 2002;16:1500–6.
24. Pikman Y, Lee BH, Mercher T, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med 2006;3:e270.
25. Pardanani AD, Levine RL, Lasho T, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood 2006;108:3472–6.
26. Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med 2013;369:2391–405.
27. Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med 2013;369:2379–90.
28. Wang WA, Groenendyk J, Michalak M. Calreticulin signaling in health and disease. Int J Biochem Cell Biol 2012;44:842–6.
29. Saeidi K. Myeloproliferative neoplasms: Current molecular biology and genetics. Crit Rev Oncol Hematol 2016;98:375–89.
30. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016;127:2391–405.
31. Mesa RA, Niblack J, Wadleigh M, et al. The burden of fatigue and quality of life in myeloproliferative disorders (MPDs): an international Internet-based survey of 1179 MPD patients. Cancer 2007;109:68–76.
32. Marchioli R, Finazzi G, Landolfi R, et al. Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol 2005;23:2224–32.
33. Scherber R, Dueck AC, Johansson P, et al. The Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF): international prospective validation and reliability trial in 402 patients. Blood 2011;118:401–8.
34. Emanuel RM, Dueck AC, Geyer HL, et al. Myeloproliferative neoplasm (MPN) symptom assessment form total symptom score: prospective international assessment of an abbreviated symptom burden scoring system among patients with MPNs. J Clin Oncol 2012;30:4098–103.
35. Casini A, Fontana P, Lecompte TP. Thrombotic complications of myeloproliferative neoplasms: risk assessment and risk-guided management. J Thromb Haemost 2013;11:1215–27.
36. Barbui T, Finazzi G, Falanga A. Myeloproliferative neoplasms and thrombosis. Blood 2013;122:2176-–84.
37. Pearson TC, Wetherley-Mein G. Vascular occlusive episodes and venous haematocrit in primary myeloproliferative polychytemia. Lancet 1978;2:1219–22.
38. Landolfi R, Di Gennaro L, Barbui T, et al. Leukocytosis as a major thrombotic risk factor in patients with polycythemia vera. Blood 2007;109:2446–52.
39. Tefferi A, Rumi E, Finazzi G, et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia 2013;27:1874–81.
40. Barbui T, Falanga A. Molecular biomarkers of thrombosis in myeloproliferative neoplasms. Thromb Res 2016;140 Suppl 1:S71–75.
41. Campbell PJ, Scott LM, Buck G, et al. Definition of subtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2 V617F mutation status: a prospective study. Lancet 2005;366:1945–53.
42. Tefferi A, Guglielmelli P, Larson DR, et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 2014;124:2507–13.
43. Qin Y, Wang X, Zhao C, et al. The impact of JAK2V617F mutation on different types of thrombosis risk in patients with essential thrombocythemia: a meta-analysis. Int J Hematol 2015;102:170–80.
44. Ziakas PD. Effect of JAK2 V617F on thrombotic risk in patients with essential thrombocythemia: measuring the uncertain. Haematologica 2008;93:1412–4.
45. Lussana F, Dentali F, Abbate R, et al. Screening for thrombophilia and antithrombotic prophylaxis in pregnancy: Guidelines of the Italian Society for Haemostasis and Thrombosis (SISET). Thromb Res 2009;124:e19–25.
46. Dahabreh IJ, Zoi K, Giannouli S, et al. Is JAK2 V617F mutation more than a diagnostic index? A meta-analysis of clinical outcomes in essential thrombocythemia. Leuk Res 2009;33:67–73.
47. Cho YU, Chi HS, Lee EH, et al. Comparison of clinicopathologic findings according to JAK2 V617F mutation in patients with essential thrombocythemia. Int J Hematol 2009;89:39–44.
48. Wolanskyj AP, Lasho TL, Schwager SM, et al. JAK2 mutation in essential thrombocythaemia: clinical associations and long-term prognostic relevance. Br J Haematol 2005;131:208–13.
49. Antonioli E, Guglielmelli P, Pancrazzi A, et al. Clinical implications of the JAK2 V617F mutation in essential thrombocythemia. Leukemia 2005;19:1847–9.
50. Palandri F, Catani L, Testoni N, et al. Long-term follow-up of 386 consecutive patients with essential thrombocythemia: safety of cytoreductive therapy. Am J Hematol 2009;84:215–20.
51. Chim CS, Sim JP, Chan CC, et al. Impact of JAK2V617F mutation on thrombosis and myeloid transformation in essential thrombocythemia: a multivariate analysis by Cox regression in 141 patients. Hematology 2010;15:187–92.
52. Vannucchi AM, Antonioli E, Guglielmelli P, et al. Clinical profile of homozygous JAK2 617V>F mutation in patients with polycythemia vera or essential thrombocythemia. Blood 2007;110:840–6.
53. Carobbio A, Finazzi G, Antonioli E, et al. JAK2V617F allele burden and thrombosis: a direct comparison in essential thrombocythemia and polycythemia vera. Exp Hematol 2009;37:1016–21.
54. Alvarez-Larran A, Bellosillo B, Pereira A, et al. JAK2V617F monitoring in polycythemia vera and essential thrombocythemia: clinical usefulness for predicting myelofibrotic transformation and thrombotic events. Am J Hematol 2014;89:517–23.
55. Barbui T, Vannucchi AM, Buxhofer-Ausch V, et al. Practice-relevant revision of IPSET-thrombosis based on 1019 patients with WHO-defined essential thrombocythemia. Blood Cancer J 2015;5:e369.
56. Carobbio A, Thiele J, Passamonti F, et al. Risk factors for arterial and venous thrombosis in WHO-defined essential thrombocythemia: an international study of 891 patients. Blood 2011;117:5857–9.
57. Alvarez-Larran A, Cervantes F, Bellosillo B, et al. Essential thrombocythemia in young individuals: frequency and risk factors for vascular events and evolution to myelofibrosis in 126 patients. Leukemia 2007;21:1218–23.
58. Jantunen R, Juvonen E, Ikkala E, et al. The predictive value of vascular risk factors and gender for the development of thrombotic complications in essential thrombocythemia. Ann Hematol 2001;80:74–8.
59. Besses C, Cervantes F, Pereira A, et al. Major vascular complications in essential thrombocythemia: a study of the predictive factors in a series of 148 patients. Leukemia 1999;13:150–4.
60. Haider M, Gangat N, Hanson C, Tefferi A. Splenomegaly and thrombosis risk in essential thrombocythemia: the mayo clinic experience. Am J Hematol 2016;91:E296–297.
61. Carobbio A, Finazzi G, Antonioli E, et al. Thrombocytosis and leukocytosis interaction in vascular complications of essential thrombocythemia. Blood 2008;112:3135–7.
62. Palandri F, Polverelli N, Catani L, et al. Impact of leukocytosis on thrombotic risk and survival in 532 patients with essential thrombocythemia: a retrospective study. Ann Hematol 2011;90:933–8.
63. Campbell PJ, MacLean C, Beer PA, et al. Correlation of blood counts with vascular complications in essential thrombocythemia: analysis of the prospective PT1 cohort. Blood 2012;120:1409–11.
64. Cortelazzo S, Finazzi G, Ruggeri M, et al. Hydroxyurea for patients with essential thrombocythemia and a high risk of thrombosis. N Engl J Med 1995;332:1132–6.
65. van Genderen PJ, Mulder PG, Waleboer M, et al. Prevention and treatment of thrombotic complications in essential thrombocythaemia: efficacy and safety of aspirin. Br J Haematol 1997;97:179–84.
66. Storen EC, Tefferi A. Long-term use of anagrelide in young patients with essential thrombocythemia. Blood 2001;97:863–6.
67. De Stefano V, Za T, Rossi E, et al. Recurrent thrombosis in patients with polycythemia vera and essential thrombocythemia: incidence, risk factors, and effect of treatments. Haematologica 2008;93:372–80.
68. Alvarez-Larran A, Cervantes F, Pereira A, et al. Observation versus antiplatelet therapy as primary prophylaxis for thrombosis in low-risk essential thrombocythemia. Blood 2010;116:1205–10.
69. Palandri F, Polverelli N, Catani L, et al. Bleeding in essential thrombocythaemia: a retrospective analysis on 565 patients. Br J Haematol 2012;156:281–4.
70. Rotunno G, Mannarelli C, Guglielmelli P, et al. Impact of calreticulin mutations on clinical and hematological phenotype and outcome in essential thrombocythemia. Blood 2014;123:1552–5.
71. Tefferi A, Wassie EA, Lasho TL, et al. Calreticulin mutations and long-term survival in essential thrombocythemia. Leukemia 2014;28:2300–3.
72. Rumi E, Pietra D, Ferretti V, et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood 2014;123:1544–51.
73. Palandri F, Latagliata R, Polverelli N, et al. Mutations and long-term outcome of 217 young patients with essential thrombocythemia or early primary myelofibrosis. Leukemia 2015;29:1344–9.
74. Fu R, Xuan M, Zhou Y, et al. Analysis of calreticulin mutations in Chinese patients with essential thrombocythemia: clinical implications in diagnosis, prognosis and treatment. Leukemia 2014;28:1912–4.
75. Tefferi A, Wassie EA, Guglielmelli P, et al. Type 1 versus Type 2 calreticulin mutations in essential thrombocythemia: a collaborative study of 1027 patients. Am J Hematol 2014;89:E121–4.
76. Pietra D, Rumi E, Ferretti VV, et al. Differential clinical effects of different mutation subtypes in CALR-mutant myeloproliferative neoplasms. Leukemia 2016;30:431–8.
77. Rumi E, Pietra D, Guglielmelli P, et al. Acquired copy-neutral loss of heterozygosity of chromosome 1p as a molecular event associated with marrow fibrosis in MPL-mutated myeloproliferative neoplasms. Blood 2013;121:4388–95.
78. Beer PA, Campbell PJ, Scott LM, et al. MPL mutations in myeloproliferative disorders: analysis of the PT-1 cohort. Blood 2008;112:141–9.
79. Gangat N, Wassie EA, Lasho TL, et al. Mutations and thrombosis in essential thrombocythemia: prognostic interaction with age and thrombosis history. Eur J Haematol 2015;94:31–6.
80. Sekhar M, McVinnie K, Burroughs AK. Splanchnic vein thrombosis in myeloproliferative neoplasms. Br J Haematol 2013;162:730–47.
81. Stein BL, Saraf S, Sobol U, et al. Age-related differences in disease characteristics and clinical outcomes in polycythemia vera. Leuk Lymph 2013;54:1989–95.
82. Landolfi R, Di Gennaro L, Nicolazzi MA, et al. Polycythemia vera: gender-related phenotypic differences. Intern Emerg Med 2012;7:509–15.
83. Winslow ER, Brunt LM, Drebin JA, et al. Portal vein thrombosis after splenectomy. Am J Surg 2002;184:631–6.
84. Smalberg JH, Arends LR, Valla DC, et al. Myeloproliferative neoplasms in Budd-Chiari syndrome and portal vein thrombosis: a meta-analysis. Blood 2012;120:4921–8.
85. Dentali F, Squizzato A, Brivio L, et al. JAK2V617F mutation for the early diagnosis of Ph- myeloproliferative neoplasms in patients with venous thromboembolism: a meta-analysis. Blood 2009;113:5617–23.
86. Pardanani A, Lasho TL, Hussein K, et al. JAK2V617F mutation screening as part of the hypercoagulable work-up in the absence of splanchnic venous thrombosis or overt myeloproliferative neoplasm: assessment of value in a series of 664 consecutive patients. Mayo Clin Proc 2008;83:457–9.
87. Barbui T, Barosi G, Birgegard G, et al. Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European LeukemiaNet. J Clin Oncol 2011;29:761–70.
88. Landolfi R, Marchioli R, Kutti J, et al. Efficacy and safety of low-dose aspirin in polycythemia vera. N Engl J Med 2004;350:114–24.
89. Marchioli R, Finazzi G, Specchia G, et al. Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med 2013;368:22–33.
90. Kiladjian JJ, Chevret S, Dosquet C, et al. Treatment of polycythemia vera with hydroxyurea and pipobroman: final results of a randomized trial initiated in 1980. J Clin Oncol 2011;29:3907–13.
91. Kaplan ME, Mack K, Goldberg JD, et al. Long-term management of polycythemia vera with hydroxyurea: a progress report. Semin Hematol 1986;23:167–71.
92. Fruchtman SM, Mack K, Kaplan ME, et al. From efficacy to safety: a Polycythemia Vera Study group report on hydroxyurea in patients with polycythemia vera. Semin Hematol 1997;34:17–23.
93. Finazzi G, Caruso V, Marchioli R, et al. Acute leukemia in polycythemia vera: an analysis of 1638 patients enrolled in a prospective observational study. Blood 2005;105:2664–70.
94. Barosi G, Mesa R, Finazzi G, et al. Revised response criteria for polycythemia vera and essential thrombocythemia: an ELN and IWG-MRT consensus project. Blood 2013;121:4778–81.
95. Alvarez-Larran A, Pereira A, Cervantes F, et al. Assessment and prognostic value of the European LeukemiaNet criteria for clinicohematologic response, resistance, and intolerance to hydroxyurea in polycythemia vera. Blood 2012;119:1363–9.
96. Stein BL, Tiu RV. Biological rationale and clinical use of interferon in the classical BCR-ABL-negative myeloproliferative neoplasms. J Interferon Cytokine Res 2013;33:145–53.
97. Ludwig H, Cortelezzi A, Van Camp BG, et al. Treatment with recombinant interferon-alpha-2C: multiple myeloma and thrombocythaemia in myeloproliferative diseases. Oncology 1985;42 Suppl 1:19–25.
98. Silver RT. Long-term effects of the treatment of polycythemia vera with recombinant interferon-alpha. Cancer 2006;107:451–8.
99. Kiladjian JJ, Mesa RA, Hoffman R. The renaissance of interferon therapy for the treatment of myeloid malignancies. Blood 2011;117:4706–15.
100. Veronese FM, Mero A. The impact of PEGylation on biological therapies. BioDrugs 2008;22:315–29.
101. Kiladjian JJ, Cassinat B, Chevret S, et al. Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood 2008;112:3065–72.
102. Turlure P, Cambier N, Roussel M, et al. Complete hematological, molecular and histological remissions without cytoreductive treatment lasting after pegylated-interferon {alpha}-2a (peg-IFN{alpha}-2a) therapy in polycythemia vera (PV): long term results of a phase 2 trial [abstract]. Blood 2011;118(21). Abstract 280.
103. Quintas-Cardama A, Kantarjian H, Manshouri T, et al. Pegylated interferon alfa-2a yields high rates of hematologic and molecular response in patients with advanced essential thrombocythemia and polycythemia vera. J Clin Oncol 2009;27:5418–24.
104. Quintas-Cardama A, Abdel-Wahab O, Manshouri T, et al. Molecular analysis of patients with polycythemia vera or essential thrombocythemia receiving pegylated interferon a-2a. Blood 2013;122:893–901.
105. Samuelsson J, Hasselbalch H, Bruserud O, et al. A phase II trial of pegylated interferon alpha-2b therapy for polycythemia vera and essential thrombocythemia: feasibility, clinical and biologic effects, and impact on quality of life. Cancer 2006;106:2397–405.
106. Jabbour E, Kantarjian H, Cortes J, et al. PEG-IFN-alpha-2b therapy in BCR-ABL-negative myeloproliferative disorders: final result of a phase 2 study. Cancer 2007;110:2012–18.
107. Them NC, Bagienski K, Berg T, et al. Molecular responses and chromosomal aberrations in patients with polycythemia vera treated with peg-proline-interferon alpha-2b. Am J Hematol 2015;90:288–94.
108. Gisslinger H, Klade C, Georgiev P, et al. Final results from PROUD-PV a randomized controlled phase 3 trial comparing ropeginterferon alfa-2b to hydroxyurea in polycythemia vera patients [abstract]. Blood 2016;128(suppl 22). Abstract 475.
109. van Genderen PJ, van Vliet HH, Prins FJ, et al. Excessive prolongation of the bleeding time by aspirin in essential thrombocythemia is related to a decrease of large von Willebrand factor multimers in plasma. Ann Hematol 1997;75:215–20.
110. Cortelazzo S, Finazzi G, Ruggeri M, et al. Hydroxyurea for patients with essential thrombocythemia and a high risk of thrombosis. N Engl J Med 1995;332:1132–7.
111. Harrison CN, Campbell PJ, Buck G, et al. Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. N Engl J Med 2005;353:33–45.
112. Gisslinger H, Gotic M, Holowiecki J, et al. Anagrelide compared with hydroxyurea in WHO-classified essential thrombocythemia: the ANAHYDRET Study, a randomized controlled trial. Blood 2013;121:1720–8.
113. Alvarado Y, Cortes J, Verstovsek S, et al. Pilot study of pegylated interferon-alpha 2b in patients with essential thrombocythemia. Cancer Chemother Pharmacol 2003;51:81–6.
114. Barosi G, Tefferi A, Barbui T, ad hoc committee ‘Definition of clinically relevant outcomes for contemporarily clinical trials in Ph-neg M. Do current response criteria in classical Ph-negative myeloproliferative neoplasms capture benefit for patients? Leukemia 2012;26:1148–9.
115. Bjorkholm M, Derolf AR, Hultcrantz M, et al. Treatment-related risk factors for transformation to acute myeloid leukemia and myelodysplastic syndromes in myeloproliferative neoplasms. J Clin Oncol 2011;29:2410–5.
116. Alvarez-Larran A, Martinez-Aviles L, Hernandez-Boluda JC, et al. Busulfan in patients with polycythemia vera or essential thrombocythemia refractory or intolerant to hydroxyurea. Ann Hematol 2014;93:2037–43.
117. Verstovsek S, Passamonti F, Rambaldi A, et al. A phase 2 study of ruxolitinib, an oral JAK1 and JAK2 Inhibitor, in patients with advanced polycythemia vera who are refractory or intolerant to hydroxyurea. Cancer 2014;120:513–20.
118. Vannucchi AM, Kiladjian JJ, Griesshammer M, et al. Ruxolitinib in polycythemia vera resistant to or intolerant of hydroxyurea. N Engl J Med 2015; 372:426–35.
119. Verstovsek S, Vannucchi AM, Griesshammer M, et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80-week follow-up from the RESPONSE trial. Haematologica 2016;101:821–9.
120. Passamonti F, Griesshammer M, Palandri F, et al. Ruxolitinib for the treatment of inadequately controlled polycythaemia vera without splenomegaly (RESPONSE-2): a randomised, open-label, phase 3b study. Lancet Oncol 2017;18:88–99.
121. Verstovsek S, Passamonti F, Rambaldi A, et al. Long-term results from a phase II open-label study of ruxolitinib in patients with essential thrombocythemia refractory to or intolerant of hydroxyurea [abstract]. Blood 2014;124. Abstract 1847.
122. Harrison CN, Mead AJ, Panchal A, et al. Ruxolitinib versus best available therapy for ET intolerant or resistant to hydroxycarbamide in a randomized trial. Blood 2017 Aug 9. pii: blood-2017-05-785790 .
123. Bose P, Verstovsek S. Drug development pipeline for myeloproliferative neoplasms: potential future impact on guidelines and management. J Natl Compr Canc Netw 2016;14:1613–24.
124. Cerquozzi S, Teffieri A. Blast transformation and fibrotic progression in polycythemia vera and essential thrombocythemia: a literature review of incidence and risk factors. Blood Cancer J 2015;Nov 13;5:e366.
125. Passamonti F, Rumi E, Caramella M, et al. A dynamic prognostic model to predict survival in post-polycythemia vera myelofibrosis. Blood 2008;111:3383–7.
126. Mesa RA, Verstovsek S, Cervantes F, et al. Primary myelofibrosis (PMF), post polycythemia vera myelofibrosis (post-PV MF), post essential thrombocythemia myelofibrosis (post-ET MF), blast phase PMF (PMF-BP): Consensus on terminology by the international working group for myelofibrosis research and treatment (IWG-MRT). Leuk Res 2007;31:737–40.
127. Rampal R, Mascarenhas J. Pathogenesis and management of acute myeloid leukemia that has evolved from a myeloproliferative neoplasm. Curr Opin Hematol 2014;21:65–71.
128. Chihara D, Kantarjian HM, Newberry KJ, et al. Survival outcome of patients with acute myeloid leukemia transformed from myeloproliferative neoplasms [abstract]. Blood 2016;128. Abstract 1940.
129. Tam CS, Nussenzveig RM, Popat U, et al. The natural history and treatment outcome of blast phase BCR-ABL- myeloproliferative neoplasms. Blood 2008;112:1628–37.
130. Kundranda MN, Tibes R, Mesa RA. Transformation of a chronic myeloproliferative neoplasm to acute myelogenous leukemia: does anything work? Curr Hematol Malig Rep 2012;7:78–86.
131. Badar T, Kantarjian HM, Ravandi F, et al. Therapeutic benefit of decitabine, a hypomethylating agent, in patients with high-risk primary myelofibrosis and myeloproliferative neoplasm in accelerated or blastic/acute myeloid leukemia phase. Leuk Res 2015;39:950–6.
132. Thepot S, Itzykson R, Seegers V, et al. Treatment of progression of Philadelphia-negative myeloproliferative neoplasms to myelodysplastic syndrome or acute myeloid leukemia by azacitidine: a report on 54 cases on the behalf of the Groupe Francophone des Myelodysplasies (GFM). Blood 2010;116:3735–42.
133. Pemmaraju N, Kantarjian H, Kadia T, et al. A phase I/II study of the Janus kinase (JAK)1 and 2 inhibitor ruxolitinib in patients with relapsed or refractory acute myeloid leukemia. Clin Lymphoma Myeloma Leuk 2015;15:171–6.
134. Rampal RK, Mascarenhas JO, Kosiorek HE, et al. Safety and efficacy of combined ruxolitinib and decitabine in patients with blast-phase MPN and post-MPN AML: results of a phase I study (Myeloproliferative Disorders Research Consortium 109 trial) [abstract]. Blood 2016;128. Abstract 1124.
135. Bose P, Verstovsek S, Gasior Y, et al. Phase I/II study of ruxolitinib (RUX) with decitabine (DAC) in patients with post-myeloproliferative neoplasm acute myeloid leukemia (post-MPN AML): phase I results [abstract]. Blood 2016;128. Abstract 4262.
CLL Index proves accurate in predicting survival, time to treat
An international prognostic index for patients with chronic lymphocytic leukemia (CLL), known as CLL-IPI, was predictive of time from diagnosis to first treatment (TTFT) and 5-year median overall survival in patients across different risk categories treated with chemoimmunotherapy, according to a systematic review and meta-analysis published in Blood (2018 Jan 18;131[3]:365-8).
But limited data were available for patients treated with targeted therapies that are likely to have a profound effect on overall survival; thereby restricting the use of CLL-IPI in current clinical practice.
Novel therapies such as ibrutinib, idelalisib, and venetoclax have changed the treatment landscape for CLL, Stefano Molica, MD, of Azienda Ospedaliera Pugliese-Ciaccio, Catanzaro, Italy, and his colleagues wrote. “Because observation remains the standard of care for asymptomatic early-stage patients, the introduction of these agents does not impact the utility of the CLL-IPI for predicting time from diagnosis to first treatment, but it likely has a profound impact on the survival of patients of all risk categories once treatment is indicated.”
The CLL-IPI tool, first published in 2016 to predict clinical outcomes in CLL patients, combines five parameters: age, clinical stage, TP53 status [normal vs. del(17p) and/or TP53 mutation], immunoglobulin heavy chain–variable mutational status, and serum b2-microglobulin. The prognostic tool was validated across several studies conducted in different countries with diverse practice settings, including academic hospitals, national population-based cohorts, and clinical trials.
The researchers conducted a systematic review and meta-analysis to understand the utility of CLL-IPI tool in predicting OS and TTFT across each risk category of CLL patients.
They included nine studies with 7,843 patients to assess the impact of the CLL-IPI on overall survival. The patient distribution into the CLL-IPI risk categories was low risk (median 45.9%), intermediate risk (median 30%), high risk (median 16.5%), and very high risk (median 3.6%).
The researchers relied on 11 series comprising 7,383 patients to assess 5-year survival probability, which was 92% for low risk, 81% for intermediate risk, 60% for high risk, and 34% for very high risk. They used seven studies comprising 5,206 patients to assess TTFT and found that the probability of remaining treatment free at 5 years was 82% in the low-risk group, 45% in the intermediate-risk group, 30% in the high-risk group, and 16% in the very-high-risk group.
Although a significant step toward harmonizing international prognostication for CLL, additional studies validating the utility of the CLL-IPI for predicting OS in patients treated with targeted therapy are needed, they wrote.
The researchers reported having no financial disclosures.
An international prognostic index for patients with chronic lymphocytic leukemia (CLL), known as CLL-IPI, was predictive of time from diagnosis to first treatment (TTFT) and 5-year median overall survival in patients across different risk categories treated with chemoimmunotherapy, according to a systematic review and meta-analysis published in Blood (2018 Jan 18;131[3]:365-8).
But limited data were available for patients treated with targeted therapies that are likely to have a profound effect on overall survival; thereby restricting the use of CLL-IPI in current clinical practice.
Novel therapies such as ibrutinib, idelalisib, and venetoclax have changed the treatment landscape for CLL, Stefano Molica, MD, of Azienda Ospedaliera Pugliese-Ciaccio, Catanzaro, Italy, and his colleagues wrote. “Because observation remains the standard of care for asymptomatic early-stage patients, the introduction of these agents does not impact the utility of the CLL-IPI for predicting time from diagnosis to first treatment, but it likely has a profound impact on the survival of patients of all risk categories once treatment is indicated.”
The CLL-IPI tool, first published in 2016 to predict clinical outcomes in CLL patients, combines five parameters: age, clinical stage, TP53 status [normal vs. del(17p) and/or TP53 mutation], immunoglobulin heavy chain–variable mutational status, and serum b2-microglobulin. The prognostic tool was validated across several studies conducted in different countries with diverse practice settings, including academic hospitals, national population-based cohorts, and clinical trials.
The researchers conducted a systematic review and meta-analysis to understand the utility of CLL-IPI tool in predicting OS and TTFT across each risk category of CLL patients.
They included nine studies with 7,843 patients to assess the impact of the CLL-IPI on overall survival. The patient distribution into the CLL-IPI risk categories was low risk (median 45.9%), intermediate risk (median 30%), high risk (median 16.5%), and very high risk (median 3.6%).
The researchers relied on 11 series comprising 7,383 patients to assess 5-year survival probability, which was 92% for low risk, 81% for intermediate risk, 60% for high risk, and 34% for very high risk. They used seven studies comprising 5,206 patients to assess TTFT and found that the probability of remaining treatment free at 5 years was 82% in the low-risk group, 45% in the intermediate-risk group, 30% in the high-risk group, and 16% in the very-high-risk group.
Although a significant step toward harmonizing international prognostication for CLL, additional studies validating the utility of the CLL-IPI for predicting OS in patients treated with targeted therapy are needed, they wrote.
The researchers reported having no financial disclosures.
An international prognostic index for patients with chronic lymphocytic leukemia (CLL), known as CLL-IPI, was predictive of time from diagnosis to first treatment (TTFT) and 5-year median overall survival in patients across different risk categories treated with chemoimmunotherapy, according to a systematic review and meta-analysis published in Blood (2018 Jan 18;131[3]:365-8).
But limited data were available for patients treated with targeted therapies that are likely to have a profound effect on overall survival; thereby restricting the use of CLL-IPI in current clinical practice.
Novel therapies such as ibrutinib, idelalisib, and venetoclax have changed the treatment landscape for CLL, Stefano Molica, MD, of Azienda Ospedaliera Pugliese-Ciaccio, Catanzaro, Italy, and his colleagues wrote. “Because observation remains the standard of care for asymptomatic early-stage patients, the introduction of these agents does not impact the utility of the CLL-IPI for predicting time from diagnosis to first treatment, but it likely has a profound impact on the survival of patients of all risk categories once treatment is indicated.”
The CLL-IPI tool, first published in 2016 to predict clinical outcomes in CLL patients, combines five parameters: age, clinical stage, TP53 status [normal vs. del(17p) and/or TP53 mutation], immunoglobulin heavy chain–variable mutational status, and serum b2-microglobulin. The prognostic tool was validated across several studies conducted in different countries with diverse practice settings, including academic hospitals, national population-based cohorts, and clinical trials.
The researchers conducted a systematic review and meta-analysis to understand the utility of CLL-IPI tool in predicting OS and TTFT across each risk category of CLL patients.
They included nine studies with 7,843 patients to assess the impact of the CLL-IPI on overall survival. The patient distribution into the CLL-IPI risk categories was low risk (median 45.9%), intermediate risk (median 30%), high risk (median 16.5%), and very high risk (median 3.6%).
The researchers relied on 11 series comprising 7,383 patients to assess 5-year survival probability, which was 92% for low risk, 81% for intermediate risk, 60% for high risk, and 34% for very high risk. They used seven studies comprising 5,206 patients to assess TTFT and found that the probability of remaining treatment free at 5 years was 82% in the low-risk group, 45% in the intermediate-risk group, 30% in the high-risk group, and 16% in the very-high-risk group.
Although a significant step toward harmonizing international prognostication for CLL, additional studies validating the utility of the CLL-IPI for predicting OS in patients treated with targeted therapy are needed, they wrote.
The researchers reported having no financial disclosures.
FROM BLOOD
AML immune profiles correlate with relapse-free survival
SAN FRANCISCO – Immune-enriched acute myeloid leukemias might be amenable to immunotherapy that is tailored to the bone marrow tumor microenvironment, according to findings from a pan-cancer analysis of bone marrow samples.
The analysis, performed with 3D biology technology and an RNA pan-cancer immune profiling panel to characterize bone marrow specimens from 46 children and 28 adults with acute myeloid leukemia (AML), identified heterogeneous immune profiles that correlated with relapse-free survival (RFS) and overall survival (OS), Jayakumar Vadakekolathu, PhD, reported at the ASCO-SITC Clinical Immuno-Oncology Symposium.
The specimens, including 63 from nonpromyelocytic de novo AML, 7 from AML in children with complete remission, 3 from adults with secondary AML, and 1 from an adult with treatment-related AML, were analyzed with the nCounter system from NanoString Technologies, and were visualized via digital spatial profiling, said Dr. Vadakekolathu of Nottingham Trent University in England.

The investigators identified two distinct immune gene expression profiles (GEPs) that were largely age-differentiated: Cluster A (myeloid-enriched specimens) included 26 children and 8 adults, and cluster B (myeloid-depleted specimens) included 9 children and 18 adults. These GEPs predicted clinical outcome; relapse free survival was 2.2 months in cluster A versus 18.3 months in cluster B (hazard ratio, 2.58) and overall survival was 6.3 months in cluster A, compared with 22.4 months in cluster B (HR, 2.39), Dr. Vadakekolathu reported.
The findings could have implications for the development of new treatment strategies, he said, noting that AML is a highly heterogeneous disease in terms of genetics, clinical manifestations, and outcome.
“Prognosis is determined by cytogenetic and molecular abnormalities, as well as by response to chemotherapy. De novo AML is cured in roughly 70% of children, 35%-40% of adults, and 5%-15% of elderly patients,” he said. Some patients with AML fail to respond to induction chemotherapy, and others eventually relapse despite the lack of adverse risk factors, he added.
The general therapeutic strategy in patients with AML has not changed substantially in more than 30 years, he said.
“High degrees of molecular complexity in AML present a considerable challenge in clinical implementation, and there is an urgent need to discover better biomarkers to identify high-risk patients before starting chemotherapy, which would enable testing of investigational therapeutic strategies in clinical trials,” he said.
In an effort to identify immune gene signatures across the spectrum of AML genotypes and to correlate transcriptomic and proteomic profiles with patient outcomes, he and his colleagues used a pediatric cohort from Children’s Hospital of Philadelphia (median age at diagnosis of 10 years), and an adult cohort from the Technical University of Dresden, Germany (median age at diagnosis, 55.5 years). Bone marrow samples were collected and analyzed at diagnosis.
Hierarchical clustering identified the two distinct clusters. The immune-enriched cluster A had heightened expression of T cells, natural killer cells, and cytotoxic cells, and also expressed CD8A, IFNG, FOXP3, the cell chemoattractants CXCL9 and CXCL10, and inhibitory molecules including IDO1 and the immune checkpoints LAG3, CTLA4, and PD-L1. The immune-depleted cluster B overexpressed genes associated with mast cell functions and CD8 T-cell exhaustion, and showed low expression of T-cell and B-cell genes.
Further analysis of 10 of the inflamed samples from patients with newly diagnosed AML was performed with digital spatial profiling (DSP) to visualize in situ leukemia–immune system interactions, Dr. Vadakekolathu said.
Surface antigens CD123 and CD3 were used as a visualization marker for leukemia cells and to identify bone marrow-infiltrating T cells, respectively. Protein quantification showed a higher concentration of CD3 counts in T-cell-rich versus T-cell-poor areas, and protein expression profiles showed strong correlations with various immunologically relevant molecules. The co-localization of CD8 T cells with FoxP3 Treg cells and PD-L1- and VISTA-expressing cell types evident on DSP represents an immune landscape consistent with the establishment of adaptive immune resistance mechanisms of immune escape, he noted.
The findings suggest immune enriched AMLs might be amenable to combination immunotherapies tailored to the bone marrow tumor microenvironment, such as IDO1 inhibitors and checkpoint blockade, Dr. Vadakekolathu said. “Immune gene expression profiles of AML might support rapid prediction of patient outcomes, discovery of novel immune biomarkers and therapeutic targets, and development of integrated patient stratifications,” he said.
This study was supported by grants from the Roger Counter Foundation and the Qatar National Research Fund. Dr. Vadakekolathu reported having no disclosures. Some authors reported employment or other financial relationships with NanoString Technologies.
SOURCE: Rutella S et al. ASCO-SITC Abstract 50
SAN FRANCISCO – Immune-enriched acute myeloid leukemias might be amenable to immunotherapy that is tailored to the bone marrow tumor microenvironment, according to findings from a pan-cancer analysis of bone marrow samples.
The analysis, performed with 3D biology technology and an RNA pan-cancer immune profiling panel to characterize bone marrow specimens from 46 children and 28 adults with acute myeloid leukemia (AML), identified heterogeneous immune profiles that correlated with relapse-free survival (RFS) and overall survival (OS), Jayakumar Vadakekolathu, PhD, reported at the ASCO-SITC Clinical Immuno-Oncology Symposium.
The specimens, including 63 from nonpromyelocytic de novo AML, 7 from AML in children with complete remission, 3 from adults with secondary AML, and 1 from an adult with treatment-related AML, were analyzed with the nCounter system from NanoString Technologies, and were visualized via digital spatial profiling, said Dr. Vadakekolathu of Nottingham Trent University in England.
The investigators identified two distinct immune gene expression profiles (GEPs) that were largely age-differentiated: Cluster A (myeloid-enriched specimens) included 26 children and 8 adults, and cluster B (myeloid-depleted specimens) included 9 children and 18 adults. These GEPs predicted clinical outcome; relapse free survival was 2.2 months in cluster A versus 18.3 months in cluster B (hazard ratio, 2.58) and overall survival was 6.3 months in cluster A, compared with 22.4 months in cluster B (HR, 2.39), Dr. Vadakekolathu reported.
The findings could have implications for the development of new treatment strategies, he said, noting that AML is a highly heterogeneous disease in terms of genetics, clinical manifestations, and outcome.
“Prognosis is determined by cytogenetic and molecular abnormalities, as well as by response to chemotherapy. De novo AML is cured in roughly 70% of children, 35%-40% of adults, and 5%-15% of elderly patients,” he said. Some patients with AML fail to respond to induction chemotherapy, and others eventually relapse despite the lack of adverse risk factors, he added.
The general therapeutic strategy in patients with AML has not changed substantially in more than 30 years, he said.
“High degrees of molecular complexity in AML present a considerable challenge in clinical implementation, and there is an urgent need to discover better biomarkers to identify high-risk patients before starting chemotherapy, which would enable testing of investigational therapeutic strategies in clinical trials,” he said.
In an effort to identify immune gene signatures across the spectrum of AML genotypes and to correlate transcriptomic and proteomic profiles with patient outcomes, he and his colleagues used a pediatric cohort from Children’s Hospital of Philadelphia (median age at diagnosis of 10 years), and an adult cohort from the Technical University of Dresden, Germany (median age at diagnosis, 55.5 years). Bone marrow samples were collected and analyzed at diagnosis.
Hierarchical clustering identified the two distinct clusters. The immune-enriched cluster A had heightened expression of T cells, natural killer cells, and cytotoxic cells, and also expressed CD8A, IFNG, FOXP3, the cell chemoattractants CXCL9 and CXCL10, and inhibitory molecules including IDO1 and the immune checkpoints LAG3, CTLA4, and PD-L1. The immune-depleted cluster B overexpressed genes associated with mast cell functions and CD8 T-cell exhaustion, and showed low expression of T-cell and B-cell genes.
Further analysis of 10 of the inflamed samples from patients with newly diagnosed AML was performed with digital spatial profiling (DSP) to visualize in situ leukemia–immune system interactions, Dr. Vadakekolathu said.
Surface antigens CD123 and CD3 were used as a visualization marker for leukemia cells and to identify bone marrow-infiltrating T cells, respectively. Protein quantification showed a higher concentration of CD3 counts in T-cell-rich versus T-cell-poor areas, and protein expression profiles showed strong correlations with various immunologically relevant molecules. The co-localization of CD8 T cells with FoxP3 Treg cells and PD-L1- and VISTA-expressing cell types evident on DSP represents an immune landscape consistent with the establishment of adaptive immune resistance mechanisms of immune escape, he noted.
The findings suggest immune enriched AMLs might be amenable to combination immunotherapies tailored to the bone marrow tumor microenvironment, such as IDO1 inhibitors and checkpoint blockade, Dr. Vadakekolathu said. “Immune gene expression profiles of AML might support rapid prediction of patient outcomes, discovery of novel immune biomarkers and therapeutic targets, and development of integrated patient stratifications,” he said.
This study was supported by grants from the Roger Counter Foundation and the Qatar National Research Fund. Dr. Vadakekolathu reported having no disclosures. Some authors reported employment or other financial relationships with NanoString Technologies.
SOURCE: Rutella S et al. ASCO-SITC Abstract 50
SAN FRANCISCO – Immune-enriched acute myeloid leukemias might be amenable to immunotherapy that is tailored to the bone marrow tumor microenvironment, according to findings from a pan-cancer analysis of bone marrow samples.
The analysis, performed with 3D biology technology and an RNA pan-cancer immune profiling panel to characterize bone marrow specimens from 46 children and 28 adults with acute myeloid leukemia (AML), identified heterogeneous immune profiles that correlated with relapse-free survival (RFS) and overall survival (OS), Jayakumar Vadakekolathu, PhD, reported at the ASCO-SITC Clinical Immuno-Oncology Symposium.
The specimens, including 63 from nonpromyelocytic de novo AML, 7 from AML in children with complete remission, 3 from adults with secondary AML, and 1 from an adult with treatment-related AML, were analyzed with the nCounter system from NanoString Technologies, and were visualized via digital spatial profiling, said Dr. Vadakekolathu of Nottingham Trent University in England.
The investigators identified two distinct immune gene expression profiles (GEPs) that were largely age-differentiated: Cluster A (myeloid-enriched specimens) included 26 children and 8 adults, and cluster B (myeloid-depleted specimens) included 9 children and 18 adults. These GEPs predicted clinical outcome; relapse free survival was 2.2 months in cluster A versus 18.3 months in cluster B (hazard ratio, 2.58) and overall survival was 6.3 months in cluster A, compared with 22.4 months in cluster B (HR, 2.39), Dr. Vadakekolathu reported.
The findings could have implications for the development of new treatment strategies, he said, noting that AML is a highly heterogeneous disease in terms of genetics, clinical manifestations, and outcome.
“Prognosis is determined by cytogenetic and molecular abnormalities, as well as by response to chemotherapy. De novo AML is cured in roughly 70% of children, 35%-40% of adults, and 5%-15% of elderly patients,” he said. Some patients with AML fail to respond to induction chemotherapy, and others eventually relapse despite the lack of adverse risk factors, he added.
The general therapeutic strategy in patients with AML has not changed substantially in more than 30 years, he said.
“High degrees of molecular complexity in AML present a considerable challenge in clinical implementation, and there is an urgent need to discover better biomarkers to identify high-risk patients before starting chemotherapy, which would enable testing of investigational therapeutic strategies in clinical trials,” he said.
In an effort to identify immune gene signatures across the spectrum of AML genotypes and to correlate transcriptomic and proteomic profiles with patient outcomes, he and his colleagues used a pediatric cohort from Children’s Hospital of Philadelphia (median age at diagnosis of 10 years), and an adult cohort from the Technical University of Dresden, Germany (median age at diagnosis, 55.5 years). Bone marrow samples were collected and analyzed at diagnosis.
Hierarchical clustering identified the two distinct clusters. The immune-enriched cluster A had heightened expression of T cells, natural killer cells, and cytotoxic cells, and also expressed CD8A, IFNG, FOXP3, the cell chemoattractants CXCL9 and CXCL10, and inhibitory molecules including IDO1 and the immune checkpoints LAG3, CTLA4, and PD-L1. The immune-depleted cluster B overexpressed genes associated with mast cell functions and CD8 T-cell exhaustion, and showed low expression of T-cell and B-cell genes.
Further analysis of 10 of the inflamed samples from patients with newly diagnosed AML was performed with digital spatial profiling (DSP) to visualize in situ leukemia–immune system interactions, Dr. Vadakekolathu said.
Surface antigens CD123 and CD3 were used as a visualization marker for leukemia cells and to identify bone marrow-infiltrating T cells, respectively. Protein quantification showed a higher concentration of CD3 counts in T-cell-rich versus T-cell-poor areas, and protein expression profiles showed strong correlations with various immunologically relevant molecules. The co-localization of CD8 T cells with FoxP3 Treg cells and PD-L1- and VISTA-expressing cell types evident on DSP represents an immune landscape consistent with the establishment of adaptive immune resistance mechanisms of immune escape, he noted.
The findings suggest immune enriched AMLs might be amenable to combination immunotherapies tailored to the bone marrow tumor microenvironment, such as IDO1 inhibitors and checkpoint blockade, Dr. Vadakekolathu said. “Immune gene expression profiles of AML might support rapid prediction of patient outcomes, discovery of novel immune biomarkers and therapeutic targets, and development of integrated patient stratifications,” he said.
This study was supported by grants from the Roger Counter Foundation and the Qatar National Research Fund. Dr. Vadakekolathu reported having no disclosures. Some authors reported employment or other financial relationships with NanoString Technologies.
SOURCE: Rutella S et al. ASCO-SITC Abstract 50
REPORTING FROM THE CLINICAL IMMUNO-ONCOLOGY SYMPOSIUM
Key clinical point:
Major finding: Relapse-free survival was 2.2 months in a cluster of myeloid-enriched specimens, compared with 18.3 months in a cluster of myeloid-depleted specimens (hazard ratio, 2.58).
Study details: A pan-cancer analysis of bone marrow specimens from 74 patients.
Disclosures: This study was supported by grants from the Roger Counter Foundation and the Qatar National Research Fund. Dr. Vadakekolathu reported having no disclosures. Some authors reported employment and other financial relationships with NanoString Technologies.
Source: Rutella S et al. ASCO-SITC Abstract 50.
Socioeconomic deprivation tied to survival in ALL

Socioeconomic deprivation may decrease survival in adults with acute lymphoblastic leukemia (ALL), according to research published in BMC Cancer.
Researchers found that ALL patients living in more deprived areas of England had a 16% to 21% greater risk of dying than patients living in the least deprived areas.
The researchers also observed a 33% higher risk of mortality in patients who were treated at hospitals that manage few ALL patients.
“The findings are likely to have significant implications for the organization of NHS [National Health Service] services for the treatment of adults with this rare but serious condition,” said study author Ravi Maheswaran, MD, of the University of Sheffield in Sheffield, UK.
To conduct this study, Dr Maheswaran and Nick Morley, MBBS, of Royal Hallamshire Hospital in Sheffield, analyzed anonymized NHS data on hospital admissions.
The researchers identified 2921 adults (age 18 and older) who were diagnosed with ALL from 2001 to 2012 and assessed follow-up data on survival rates up to 2013.
There were 1870 deaths during follow-up, and the 5-year survival rate was 32%.
As expected, survival decreased with age but increased over time. The mortality hazard ratio (HR) was 1.38 for patients ages 30 to 39, 3.72 for patients ages 60 to 69, and 9.02 for patients age 80 and older.
The HR was 0.98 for patients diagnosed from 2005 to 2008 and 0.70 for patients diagnosed from 2009 to 2012, compared to 1.00 for patients diagnosed from 2001 to 2004.
Patients living in areas of socioeconomic deprivation had a greater risk of death, but it did not seem to matter whether the patients lived in rural or urban areas.
The mortality HR was 1.16 for patients living in the most deprived areas and 1.21 for patients in intermediate areas (with the least deprived areas as the reference, 1.00). The HR was 1.00 for both rural and urban areas.
The risk of death was higher for patients treated at hospitals with low volumes of adults with ALL. The mortality HR was 1.33 for low-volume hospitals, which were defined as hospitals with 15 or fewer ALL patients admitted over a 3-year time period for which data were available.
“These results, although concerning, are from a single study, and further work is needed to confirm our findings,” Dr Maheswaran said.
“If the association between high deprivation and poorer survival is confirmed, more investigation will be needed to understand why adults with this type of leukemia living in deprived areas have poorer survival and what can be done to address this inequality.”
“Confirmation that hospitals treating few patients with this rare condition have worse outcomes would mean that the NHS should seriously consider if treatment services for adults with acute lymphoblastic leukemia should mainly be provided by specialist centers in order to improve survival.” ![]()
Socioeconomic deprivation may decrease survival in adults with acute lymphoblastic leukemia (ALL), according to research published in BMC Cancer.
Researchers found that ALL patients living in more deprived areas of England had a 16% to 21% greater risk of dying than patients living in the least deprived areas.
The researchers also observed a 33% higher risk of mortality in patients who were treated at hospitals that manage few ALL patients.
“The findings are likely to have significant implications for the organization of NHS [National Health Service] services for the treatment of adults with this rare but serious condition,” said study author Ravi Maheswaran, MD, of the University of Sheffield in Sheffield, UK.
To conduct this study, Dr Maheswaran and Nick Morley, MBBS, of Royal Hallamshire Hospital in Sheffield, analyzed anonymized NHS data on hospital admissions.
The researchers identified 2921 adults (age 18 and older) who were diagnosed with ALL from 2001 to 2012 and assessed follow-up data on survival rates up to 2013.
There were 1870 deaths during follow-up, and the 5-year survival rate was 32%.
As expected, survival decreased with age but increased over time. The mortality hazard ratio (HR) was 1.38 for patients ages 30 to 39, 3.72 for patients ages 60 to 69, and 9.02 for patients age 80 and older.
The HR was 0.98 for patients diagnosed from 2005 to 2008 and 0.70 for patients diagnosed from 2009 to 2012, compared to 1.00 for patients diagnosed from 2001 to 2004.
Patients living in areas of socioeconomic deprivation had a greater risk of death, but it did not seem to matter whether the patients lived in rural or urban areas.
The mortality HR was 1.16 for patients living in the most deprived areas and 1.21 for patients in intermediate areas (with the least deprived areas as the reference, 1.00). The HR was 1.00 for both rural and urban areas.
The risk of death was higher for patients treated at hospitals with low volumes of adults with ALL. The mortality HR was 1.33 for low-volume hospitals, which were defined as hospitals with 15 or fewer ALL patients admitted over a 3-year time period for which data were available.
“These results, although concerning, are from a single study, and further work is needed to confirm our findings,” Dr Maheswaran said.
“If the association between high deprivation and poorer survival is confirmed, more investigation will be needed to understand why adults with this type of leukemia living in deprived areas have poorer survival and what can be done to address this inequality.”
“Confirmation that hospitals treating few patients with this rare condition have worse outcomes would mean that the NHS should seriously consider if treatment services for adults with acute lymphoblastic leukemia should mainly be provided by specialist centers in order to improve survival.” ![]()
Socioeconomic deprivation may decrease survival in adults with acute lymphoblastic leukemia (ALL), according to research published in BMC Cancer.
Researchers found that ALL patients living in more deprived areas of England had a 16% to 21% greater risk of dying than patients living in the least deprived areas.
The researchers also observed a 33% higher risk of mortality in patients who were treated at hospitals that manage few ALL patients.
“The findings are likely to have significant implications for the organization of NHS [National Health Service] services for the treatment of adults with this rare but serious condition,” said study author Ravi Maheswaran, MD, of the University of Sheffield in Sheffield, UK.
To conduct this study, Dr Maheswaran and Nick Morley, MBBS, of Royal Hallamshire Hospital in Sheffield, analyzed anonymized NHS data on hospital admissions.
The researchers identified 2921 adults (age 18 and older) who were diagnosed with ALL from 2001 to 2012 and assessed follow-up data on survival rates up to 2013.
There were 1870 deaths during follow-up, and the 5-year survival rate was 32%.
As expected, survival decreased with age but increased over time. The mortality hazard ratio (HR) was 1.38 for patients ages 30 to 39, 3.72 for patients ages 60 to 69, and 9.02 for patients age 80 and older.
The HR was 0.98 for patients diagnosed from 2005 to 2008 and 0.70 for patients diagnosed from 2009 to 2012, compared to 1.00 for patients diagnosed from 2001 to 2004.
Patients living in areas of socioeconomic deprivation had a greater risk of death, but it did not seem to matter whether the patients lived in rural or urban areas.
The mortality HR was 1.16 for patients living in the most deprived areas and 1.21 for patients in intermediate areas (with the least deprived areas as the reference, 1.00). The HR was 1.00 for both rural and urban areas.
The risk of death was higher for patients treated at hospitals with low volumes of adults with ALL. The mortality HR was 1.33 for low-volume hospitals, which were defined as hospitals with 15 or fewer ALL patients admitted over a 3-year time period for which data were available.
“These results, although concerning, are from a single study, and further work is needed to confirm our findings,” Dr Maheswaran said.
“If the association between high deprivation and poorer survival is confirmed, more investigation will be needed to understand why adults with this type of leukemia living in deprived areas have poorer survival and what can be done to address this inequality.”
“Confirmation that hospitals treating few patients with this rare condition have worse outcomes would mean that the NHS should seriously consider if treatment services for adults with acute lymphoblastic leukemia should mainly be provided by specialist centers in order to improve survival.” ![]()
CAR T-cell therapy produces durable CRs in ALL

Updated results from the phase 2 ELIANA study have shown that tisagenlecleucel can produce durable complete responses (CRs) in children and young adults with relapsed/refractory acute lymphoblastic leukemia (ALL).
Sixty percent of patients who received the chimeric antigen receptor (CAR) T-cell therapy achieved a CR, and 21% had a CR with incomplete hematologic recovery (CRi).
The median duration of CR/CRi was not reached at a median follow-up of 13.1 months.
The most common treatment-related adverse event (AE) was cytokine release syndrome (CRS), occurring in 77% of patients.
Researchers reported these results in NEJM. The study was sponsored by Novartis.
“This expanded, global study of CAR T-cell therapy gives us further evidence of how remarkable this treatment can be for our young patients in whom all other treatments failed,” said study author Shannon L. Maude, MD, PhD, of Children’s Hospital of Philadelphia in Pennsylvania.
“Our data show not only can we can achieve longer-term durable remissions and longer-term survival for our patients but that these personalized, cancer-fighting cells can remain in the body for months or even years, effectively doing their job.”
The trial included 75 patients who received tisagenlecleucel. At enrollment, the patients’ median age was 11 (range, 3 to 23).
Patients had received a median of 3 prior therapies (range, 1 to 8), and they had a median marrow blast percentage of 74% (range, 5 to 99).
All patients received a single infusion of tisagenlecleucel. Most (n=72) received lymphodepleting chemotherapy prior to the CAR T cells.
Results
The median duration of follow-up was 13.1 months.
The study’s primary endpoint was overall remission rate, which was defined as the rate of a best overall response of either CR or CRi within 3 months. The overall remission rate was 81% (61/75), with 60% of patients (n=45) achieving a CR and 21% (n=16) achieving a CRi.
All patients whose best response was CR/CRi were negative for minimal residual disease. The median duration of response was not met.
The researchers said tisagenlecleucel persisted in the blood for as long as 20 months.
The relapse-free survival rate among patients with a CR/CRi was 80% at 6 months and 59% at 12 months.
Seventeen patients who had achieved a CR relapsed before receiving subsequent treatment. Three patients went on to subsequent therapy before relapse but ultimately relapsed.
Relapse was also reported in 2 patients who had been classified as non-responders because they did not maintain a response for at least 28 days.
Eight patients underwent allogeneic hematopoietic stem cell transplant while in remission, and all 8 were alive when the manuscript for this study was submitted. Four patients had not relapsed, and the other 4 had unknown disease status.
At 6 months, the event-free survival rate was 73%, and the overall survival rate was 90%. At 12 months, the rates were 50% and 76%, respectively.
All patients experienced at least 1 AE, and 95% had AEs thought to be related to tisagenlecleucel. Grade 3/4 AEs occurred in 88% of patients. In 73% of patients, these AEs were thought to be related to treatment.
AEs of special interest included CRS (77%), neurologic events (40%), infections (43%), febrile neutropenia (35%), cytopenias not resolved by day 28 (37%), and tumor lysis syndrome (4%).
The median duration of CRS was 8 days (range, 1-36). Forty-seven patients were admitted to the intensive care unit to receive treatment for CRS, with a median stay of 7 days (range, 1-34).
“One of our more challenging questions—‘Can we manage the serious side effects of CAR T-cell therapy?’—was asked and answered in this global study,” said author Stephan A. Grupp, MD, PhD, of Children’s Hospital of Philadelphia.
“Some of our patients get very sick, but we showed that most toxic effects can be short-lived and reversible, with the potential for our patients to achieve durable complete remissions. That’s a pretty amazing turnaround for the high-risk child who, up until now, had little chance of surviving.” ![]()
Updated results from the phase 2 ELIANA study have shown that tisagenlecleucel can produce durable complete responses (CRs) in children and young adults with relapsed/refractory acute lymphoblastic leukemia (ALL).
Sixty percent of patients who received the chimeric antigen receptor (CAR) T-cell therapy achieved a CR, and 21% had a CR with incomplete hematologic recovery (CRi).
The median duration of CR/CRi was not reached at a median follow-up of 13.1 months.
The most common treatment-related adverse event (AE) was cytokine release syndrome (CRS), occurring in 77% of patients.
Researchers reported these results in NEJM. The study was sponsored by Novartis.
“This expanded, global study of CAR T-cell therapy gives us further evidence of how remarkable this treatment can be for our young patients in whom all other treatments failed,” said study author Shannon L. Maude, MD, PhD, of Children’s Hospital of Philadelphia in Pennsylvania.
“Our data show not only can we can achieve longer-term durable remissions and longer-term survival for our patients but that these personalized, cancer-fighting cells can remain in the body for months or even years, effectively doing their job.”
The trial included 75 patients who received tisagenlecleucel. At enrollment, the patients’ median age was 11 (range, 3 to 23).
Patients had received a median of 3 prior therapies (range, 1 to 8), and they had a median marrow blast percentage of 74% (range, 5 to 99).
All patients received a single infusion of tisagenlecleucel. Most (n=72) received lymphodepleting chemotherapy prior to the CAR T cells.
Results
The median duration of follow-up was 13.1 months.
The study’s primary endpoint was overall remission rate, which was defined as the rate of a best overall response of either CR or CRi within 3 months. The overall remission rate was 81% (61/75), with 60% of patients (n=45) achieving a CR and 21% (n=16) achieving a CRi.
All patients whose best response was CR/CRi were negative for minimal residual disease. The median duration of response was not met.
The researchers said tisagenlecleucel persisted in the blood for as long as 20 months.
The relapse-free survival rate among patients with a CR/CRi was 80% at 6 months and 59% at 12 months.
Seventeen patients who had achieved a CR relapsed before receiving subsequent treatment. Three patients went on to subsequent therapy before relapse but ultimately relapsed.
Relapse was also reported in 2 patients who had been classified as non-responders because they did not maintain a response for at least 28 days.
Eight patients underwent allogeneic hematopoietic stem cell transplant while in remission, and all 8 were alive when the manuscript for this study was submitted. Four patients had not relapsed, and the other 4 had unknown disease status.
At 6 months, the event-free survival rate was 73%, and the overall survival rate was 90%. At 12 months, the rates were 50% and 76%, respectively.
All patients experienced at least 1 AE, and 95% had AEs thought to be related to tisagenlecleucel. Grade 3/4 AEs occurred in 88% of patients. In 73% of patients, these AEs were thought to be related to treatment.
AEs of special interest included CRS (77%), neurologic events (40%), infections (43%), febrile neutropenia (35%), cytopenias not resolved by day 28 (37%), and tumor lysis syndrome (4%).
The median duration of CRS was 8 days (range, 1-36). Forty-seven patients were admitted to the intensive care unit to receive treatment for CRS, with a median stay of 7 days (range, 1-34).
“One of our more challenging questions—‘Can we manage the serious side effects of CAR T-cell therapy?’—was asked and answered in this global study,” said author Stephan A. Grupp, MD, PhD, of Children’s Hospital of Philadelphia.
“Some of our patients get very sick, but we showed that most toxic effects can be short-lived and reversible, with the potential for our patients to achieve durable complete remissions. That’s a pretty amazing turnaround for the high-risk child who, up until now, had little chance of surviving.” ![]()
Updated results from the phase 2 ELIANA study have shown that tisagenlecleucel can produce durable complete responses (CRs) in children and young adults with relapsed/refractory acute lymphoblastic leukemia (ALL).
Sixty percent of patients who received the chimeric antigen receptor (CAR) T-cell therapy achieved a CR, and 21% had a CR with incomplete hematologic recovery (CRi).
The median duration of CR/CRi was not reached at a median follow-up of 13.1 months.
The most common treatment-related adverse event (AE) was cytokine release syndrome (CRS), occurring in 77% of patients.
Researchers reported these results in NEJM. The study was sponsored by Novartis.
“This expanded, global study of CAR T-cell therapy gives us further evidence of how remarkable this treatment can be for our young patients in whom all other treatments failed,” said study author Shannon L. Maude, MD, PhD, of Children’s Hospital of Philadelphia in Pennsylvania.
“Our data show not only can we can achieve longer-term durable remissions and longer-term survival for our patients but that these personalized, cancer-fighting cells can remain in the body for months or even years, effectively doing their job.”
The trial included 75 patients who received tisagenlecleucel. At enrollment, the patients’ median age was 11 (range, 3 to 23).
Patients had received a median of 3 prior therapies (range, 1 to 8), and they had a median marrow blast percentage of 74% (range, 5 to 99).
All patients received a single infusion of tisagenlecleucel. Most (n=72) received lymphodepleting chemotherapy prior to the CAR T cells.
Results
The median duration of follow-up was 13.1 months.
The study’s primary endpoint was overall remission rate, which was defined as the rate of a best overall response of either CR or CRi within 3 months. The overall remission rate was 81% (61/75), with 60% of patients (n=45) achieving a CR and 21% (n=16) achieving a CRi.
All patients whose best response was CR/CRi were negative for minimal residual disease. The median duration of response was not met.
The researchers said tisagenlecleucel persisted in the blood for as long as 20 months.
The relapse-free survival rate among patients with a CR/CRi was 80% at 6 months and 59% at 12 months.
Seventeen patients who had achieved a CR relapsed before receiving subsequent treatment. Three patients went on to subsequent therapy before relapse but ultimately relapsed.
Relapse was also reported in 2 patients who had been classified as non-responders because they did not maintain a response for at least 28 days.
Eight patients underwent allogeneic hematopoietic stem cell transplant while in remission, and all 8 were alive when the manuscript for this study was submitted. Four patients had not relapsed, and the other 4 had unknown disease status.
At 6 months, the event-free survival rate was 73%, and the overall survival rate was 90%. At 12 months, the rates were 50% and 76%, respectively.
All patients experienced at least 1 AE, and 95% had AEs thought to be related to tisagenlecleucel. Grade 3/4 AEs occurred in 88% of patients. In 73% of patients, these AEs were thought to be related to treatment.
AEs of special interest included CRS (77%), neurologic events (40%), infections (43%), febrile neutropenia (35%), cytopenias not resolved by day 28 (37%), and tumor lysis syndrome (4%).
The median duration of CRS was 8 days (range, 1-36). Forty-seven patients were admitted to the intensive care unit to receive treatment for CRS, with a median stay of 7 days (range, 1-34).
“One of our more challenging questions—‘Can we manage the serious side effects of CAR T-cell therapy?’—was asked and answered in this global study,” said author Stephan A. Grupp, MD, PhD, of Children’s Hospital of Philadelphia.
“Some of our patients get very sick, but we showed that most toxic effects can be short-lived and reversible, with the potential for our patients to achieve durable complete remissions. That’s a pretty amazing turnaround for the high-risk child who, up until now, had little chance of surviving.” ![]()