User login
Study points to need to improve identification of culprit in SJS/TEN cases
demonstrated.
“Prompt identification and discontinuation of a culprit drug is critical to improving patient outcomes and preventing recurrence,” researchers led by Sherrie J. Divito, MD, PhD, of the department of dermatology at Brigham and Women’s Hospital, Boston, wrote in the study, which was published online in JAMA Dermatology. “Identification is difficult because there is no laboratory test or other criterion standard (in the absence of rechallenge) to identify a culprit drug, and patients take on average six medications at the time of their reaction. Consequently, many patients may be labeled as allergic to multiple agents.”
Although failing to identify the culprit drug can have severe consequences, they added, “overlabeling” (labeling a patient as allergic to a drug or drugs that they can tolerate safely) “is not insignificant.” As a result of overlabeling, “the patient may receive a less efficacious, more toxic, and/or more expensive agent than necessary, and in some cases may be left without treatment for their underlying disease.”
To evaluate the outcomes of patients’ allergy lists, current approaches to identify culprit drugs such as the Algorithm for Drug Causality for Epidermal Necrolysis (ALDEN), which was published in 2010, and potential methods of improving culprit drug identification, the researchers performed a retrospective cohort study of 48 patients at Brigham and Women’s Hospital and Massachusetts General Hospital, with clinically and histologically confirmed cases of SJS/TEN between January 2000 and July 2018. Of the 48 patients, 26 had SJS/TEN overlap and 22 had TEN. Their median age at diagnosis was 40 years; 60.4% were female; and 52.1% were white, 12.5% were Black, 10.4% were Hispanic, and 8.3% were Asian. They took a median of 6.5 drugs in the 3 months prior to disease onset.
The researchers observed that all patients had at least one drug labeled as an allergy. A single culprit drug was labeled in 17 cases, but physicians communicated certainty in only 7 of those cases. Among all 48 patients, 104 drugs were labeled as allergies.
To identify a culprit drug, physicians appeared to mainly rely on two factors: drug notoriety and timing of exposure, compared with the onset of SJS/TEN. “Identifying high-risk medications seemed heuristic, with one or more drugs in question noted in the record as a common culprit without reference to published or vetted data regarding risk,” the researchers wrote. “Regarding timing, drug charts when present in medical records were incomplete, as they focused predominantly on high-notoriety drugs.”
In other findings, ALDEN scoring was discordant with physician-labeled lists in 28 cases. It labeled an additional 9 drugs missed by physicians and scored 43 drugs labeled as allergens by physicians as “unlikely.” The researchers also reported that 20 cases could have potentially benefited from human leukocyte antigen testing.
Their results “underscore the need for a laboratory test to identify culprit drugs,” but without such a test, “a systematic unbiased approach, such as ALDEN or the RegiSCAR database, with possibly HLA testing, should be considered to ensure the true culprit drug is not missed and exonerate as many nonculprit drugs as possible,” Dr. Divito and colleagues concluded.
They acknowledged certain limitations of the analysis, including its retrospective design and that many cases predated research advances in the topic area that took place during the 18-year study period.
Karl Saardi, MD, director of the inpatient dermatology service at George Washington University Hospital, Washington, who was asked to comment on the study, said that the findings “are consistent with clinical practice in that drug causality is usually determined by ‘gestalt’ rather than objective tools like the ALDEN algorithm.”
“The main limitation is the small size, which suggests the study sites are low-volume centers for SJS/TEN. The fact that the ALDEN score wasn’t developed until 2010 means that all the cases included prior to 2010 would not have applied the ALDEN algorithm, so I think the metric about how infrequently ALDEN was applied is not very meaningful.”
Still, he said that he was “surprised” by the number of medications that were added as allergies based on clinical impression, “and I’m glad this article does cast some light on the issue. In my experience, beta-lactam antibiotics are often – incorrectly – deemed to be the cause of SJS/TEN when further review of the patient’s medication history clearly shows they have tolerated these drugs multiple times in the past.”
Since 2000, he added, “our understanding of SJS/TEN has grown substantially including the identification of MIRM [mycoplasma-induced rash and mucositis]/RIME [reactive infections mucocutaneous eruptions] and GBFDE [generalized bullous fixed drug eruption] as mimickers.”
Christine Ko, MD, professor of dermatology and pathology at Yale University, New Haven, Conn., who was also asked to comment on the study, agreed that a limitation of the study is that it partially preceded development of the unbiased approaches to determining the cause of a medication reaction, such as the ALDEN system. “A strength of this study is the examination of heuristics in dermatology and how they relate to patient safety,” she added.
The study was funded by grants from the National Institutes of Health, a Dermatology Foundation Diversity Research Supplement Award, and by the German Research Foundation. Dr. Divito reported receiving personal fees from Adaptimmune and MEI Pharma and a provisional patent issued from Brigham and Women’s Hospital outside the submitted work. Neither Dr. Saardi nor Dr. Ko reported having relevant disclosures.
demonstrated.
“Prompt identification and discontinuation of a culprit drug is critical to improving patient outcomes and preventing recurrence,” researchers led by Sherrie J. Divito, MD, PhD, of the department of dermatology at Brigham and Women’s Hospital, Boston, wrote in the study, which was published online in JAMA Dermatology. “Identification is difficult because there is no laboratory test or other criterion standard (in the absence of rechallenge) to identify a culprit drug, and patients take on average six medications at the time of their reaction. Consequently, many patients may be labeled as allergic to multiple agents.”
Although failing to identify the culprit drug can have severe consequences, they added, “overlabeling” (labeling a patient as allergic to a drug or drugs that they can tolerate safely) “is not insignificant.” As a result of overlabeling, “the patient may receive a less efficacious, more toxic, and/or more expensive agent than necessary, and in some cases may be left without treatment for their underlying disease.”
To evaluate the outcomes of patients’ allergy lists, current approaches to identify culprit drugs such as the Algorithm for Drug Causality for Epidermal Necrolysis (ALDEN), which was published in 2010, and potential methods of improving culprit drug identification, the researchers performed a retrospective cohort study of 48 patients at Brigham and Women’s Hospital and Massachusetts General Hospital, with clinically and histologically confirmed cases of SJS/TEN between January 2000 and July 2018. Of the 48 patients, 26 had SJS/TEN overlap and 22 had TEN. Their median age at diagnosis was 40 years; 60.4% were female; and 52.1% were white, 12.5% were Black, 10.4% were Hispanic, and 8.3% were Asian. They took a median of 6.5 drugs in the 3 months prior to disease onset.
The researchers observed that all patients had at least one drug labeled as an allergy. A single culprit drug was labeled in 17 cases, but physicians communicated certainty in only 7 of those cases. Among all 48 patients, 104 drugs were labeled as allergies.
To identify a culprit drug, physicians appeared to mainly rely on two factors: drug notoriety and timing of exposure, compared with the onset of SJS/TEN. “Identifying high-risk medications seemed heuristic, with one or more drugs in question noted in the record as a common culprit without reference to published or vetted data regarding risk,” the researchers wrote. “Regarding timing, drug charts when present in medical records were incomplete, as they focused predominantly on high-notoriety drugs.”
In other findings, ALDEN scoring was discordant with physician-labeled lists in 28 cases. It labeled an additional 9 drugs missed by physicians and scored 43 drugs labeled as allergens by physicians as “unlikely.” The researchers also reported that 20 cases could have potentially benefited from human leukocyte antigen testing.
Their results “underscore the need for a laboratory test to identify culprit drugs,” but without such a test, “a systematic unbiased approach, such as ALDEN or the RegiSCAR database, with possibly HLA testing, should be considered to ensure the true culprit drug is not missed and exonerate as many nonculprit drugs as possible,” Dr. Divito and colleagues concluded.
They acknowledged certain limitations of the analysis, including its retrospective design and that many cases predated research advances in the topic area that took place during the 18-year study period.
Karl Saardi, MD, director of the inpatient dermatology service at George Washington University Hospital, Washington, who was asked to comment on the study, said that the findings “are consistent with clinical practice in that drug causality is usually determined by ‘gestalt’ rather than objective tools like the ALDEN algorithm.”
“The main limitation is the small size, which suggests the study sites are low-volume centers for SJS/TEN. The fact that the ALDEN score wasn’t developed until 2010 means that all the cases included prior to 2010 would not have applied the ALDEN algorithm, so I think the metric about how infrequently ALDEN was applied is not very meaningful.”
Still, he said that he was “surprised” by the number of medications that were added as allergies based on clinical impression, “and I’m glad this article does cast some light on the issue. In my experience, beta-lactam antibiotics are often – incorrectly – deemed to be the cause of SJS/TEN when further review of the patient’s medication history clearly shows they have tolerated these drugs multiple times in the past.”
Since 2000, he added, “our understanding of SJS/TEN has grown substantially including the identification of MIRM [mycoplasma-induced rash and mucositis]/RIME [reactive infections mucocutaneous eruptions] and GBFDE [generalized bullous fixed drug eruption] as mimickers.”
Christine Ko, MD, professor of dermatology and pathology at Yale University, New Haven, Conn., who was also asked to comment on the study, agreed that a limitation of the study is that it partially preceded development of the unbiased approaches to determining the cause of a medication reaction, such as the ALDEN system. “A strength of this study is the examination of heuristics in dermatology and how they relate to patient safety,” she added.
The study was funded by grants from the National Institutes of Health, a Dermatology Foundation Diversity Research Supplement Award, and by the German Research Foundation. Dr. Divito reported receiving personal fees from Adaptimmune and MEI Pharma and a provisional patent issued from Brigham and Women’s Hospital outside the submitted work. Neither Dr. Saardi nor Dr. Ko reported having relevant disclosures.
demonstrated.
“Prompt identification and discontinuation of a culprit drug is critical to improving patient outcomes and preventing recurrence,” researchers led by Sherrie J. Divito, MD, PhD, of the department of dermatology at Brigham and Women’s Hospital, Boston, wrote in the study, which was published online in JAMA Dermatology. “Identification is difficult because there is no laboratory test or other criterion standard (in the absence of rechallenge) to identify a culprit drug, and patients take on average six medications at the time of their reaction. Consequently, many patients may be labeled as allergic to multiple agents.”
Although failing to identify the culprit drug can have severe consequences, they added, “overlabeling” (labeling a patient as allergic to a drug or drugs that they can tolerate safely) “is not insignificant.” As a result of overlabeling, “the patient may receive a less efficacious, more toxic, and/or more expensive agent than necessary, and in some cases may be left without treatment for their underlying disease.”
To evaluate the outcomes of patients’ allergy lists, current approaches to identify culprit drugs such as the Algorithm for Drug Causality for Epidermal Necrolysis (ALDEN), which was published in 2010, and potential methods of improving culprit drug identification, the researchers performed a retrospective cohort study of 48 patients at Brigham and Women’s Hospital and Massachusetts General Hospital, with clinically and histologically confirmed cases of SJS/TEN between January 2000 and July 2018. Of the 48 patients, 26 had SJS/TEN overlap and 22 had TEN. Their median age at diagnosis was 40 years; 60.4% were female; and 52.1% were white, 12.5% were Black, 10.4% were Hispanic, and 8.3% were Asian. They took a median of 6.5 drugs in the 3 months prior to disease onset.
The researchers observed that all patients had at least one drug labeled as an allergy. A single culprit drug was labeled in 17 cases, but physicians communicated certainty in only 7 of those cases. Among all 48 patients, 104 drugs were labeled as allergies.
To identify a culprit drug, physicians appeared to mainly rely on two factors: drug notoriety and timing of exposure, compared with the onset of SJS/TEN. “Identifying high-risk medications seemed heuristic, with one or more drugs in question noted in the record as a common culprit without reference to published or vetted data regarding risk,” the researchers wrote. “Regarding timing, drug charts when present in medical records were incomplete, as they focused predominantly on high-notoriety drugs.”
In other findings, ALDEN scoring was discordant with physician-labeled lists in 28 cases. It labeled an additional 9 drugs missed by physicians and scored 43 drugs labeled as allergens by physicians as “unlikely.” The researchers also reported that 20 cases could have potentially benefited from human leukocyte antigen testing.
Their results “underscore the need for a laboratory test to identify culprit drugs,” but without such a test, “a systematic unbiased approach, such as ALDEN or the RegiSCAR database, with possibly HLA testing, should be considered to ensure the true culprit drug is not missed and exonerate as many nonculprit drugs as possible,” Dr. Divito and colleagues concluded.
They acknowledged certain limitations of the analysis, including its retrospective design and that many cases predated research advances in the topic area that took place during the 18-year study period.
Karl Saardi, MD, director of the inpatient dermatology service at George Washington University Hospital, Washington, who was asked to comment on the study, said that the findings “are consistent with clinical practice in that drug causality is usually determined by ‘gestalt’ rather than objective tools like the ALDEN algorithm.”
“The main limitation is the small size, which suggests the study sites are low-volume centers for SJS/TEN. The fact that the ALDEN score wasn’t developed until 2010 means that all the cases included prior to 2010 would not have applied the ALDEN algorithm, so I think the metric about how infrequently ALDEN was applied is not very meaningful.”
Still, he said that he was “surprised” by the number of medications that were added as allergies based on clinical impression, “and I’m glad this article does cast some light on the issue. In my experience, beta-lactam antibiotics are often – incorrectly – deemed to be the cause of SJS/TEN when further review of the patient’s medication history clearly shows they have tolerated these drugs multiple times in the past.”
Since 2000, he added, “our understanding of SJS/TEN has grown substantially including the identification of MIRM [mycoplasma-induced rash and mucositis]/RIME [reactive infections mucocutaneous eruptions] and GBFDE [generalized bullous fixed drug eruption] as mimickers.”
Christine Ko, MD, professor of dermatology and pathology at Yale University, New Haven, Conn., who was also asked to comment on the study, agreed that a limitation of the study is that it partially preceded development of the unbiased approaches to determining the cause of a medication reaction, such as the ALDEN system. “A strength of this study is the examination of heuristics in dermatology and how they relate to patient safety,” she added.
The study was funded by grants from the National Institutes of Health, a Dermatology Foundation Diversity Research Supplement Award, and by the German Research Foundation. Dr. Divito reported receiving personal fees from Adaptimmune and MEI Pharma and a provisional patent issued from Brigham and Women’s Hospital outside the submitted work. Neither Dr. Saardi nor Dr. Ko reported having relevant disclosures.
FROM JAMA DERMATOLOGY
FDA passes on olorofim despite critical need for antifungals
The U.S. Food and Drug Administration is declining to approve the investigational antifungal olorofim and is asking for more data, according to a news release from the manufacturer, F2G.
Olorofim, (formerly known as F901318) is the first in the orotomide class of antifungals to be evaluated clinically for the treatment of invasive mold infections. Its maker, F2G, is a biotech company based in Manchester, England, that focuses on developing drugs for rare fungal diseases.
The company says it remains optimistic and will address the FDA’s requirements and continue to seek approval.
The FDA’s denial comes as fungal infections are becoming increasingly common and resistant to treatment. There are only four antifungal classes currently available, and there are few new candidates in the pipeline. No new classes of antifungals have been developed in 2 decades.
David Andes, MD, chief of the division of infectious diseases at the University of Wisconsin–Madison, told this news organization he shares the hope that the company can meet the requirements to gain approval.
“Some of the early results were really exciting,” he said. “People are enthusiastic about the compound because it has a novel mechanism of action, and it is active against a group of fungi that we have limited to no options for.”
Early results ‘exciting’
Dr. Andes said several physicians have been able to prescribe olorofim under the compassionate use program “and have witnessed success.”
Olorofim is the first antifungal agent to be granted breakthrough therapy designation, which the FDA granted in November 2019 for the treatment of invasive mold infections for patients with limited or no treatment options, including patients with refractory aspergillosis or those who are intolerant of currently available therapy. It is also indicated for infections due to Lomentospora prolificans, Scedosporium, and Scopulariopsis species.
Olorofim received a second breakthrough therapy designation in October 2020. The second designation was granted for treatment of central nervous system coccidioidomycosis that is refractory or for cases that cannot be treated with standard-of-care therapy.
It is very difficult for patients to be approved to receive compassionate use medicines, Dr. Andes pointed out. “I’d like to have access sooner rather than later,” he added.
Dr. Andes says the drugs are expensive and are time consuming to produce. And with antifungals, it is difficult to demonstrate safety in comparison with other antimicrobial agents because “it’s hard to hurt a fungus without having toxicity with human cells.”
Complete response letter issued
F2G received a complete response letter from the FDA regarding its new drug application for olorofim, according to the news release issued by the company. “While F2G is disappointed with this outcome, we remain optimistic about olorofim’s potential to address an unmet need for patients with invasive fungal infections who have exhausted their treatment alternatives,” Francesco Maria Lavino, chief executive officer, said in the release. “We are assessing the details of the Complete Response Letter, and we plan to meet with the FDA to discuss it further.”
Dr. Andes says few other antifungals have made it as far as olorofim in clinical trials.
Lance B. Price, PhD, codirector of the Antibiotic Resistance Action Center at George Washington University in Washington, told this news organization that despite the lack of antifungals in the pipeline, “We can’t allow our desperation to override the checkpoints that ensure that antifungals are safe to use in people.”
In the meantime, he said, it is important to preserve the utility of current antifungals by avoiding overusing them in medicine and agriculture.
“Sadly,” he said, “a drug called ipflufenoquin, which works by a similar mode of action as olorofim, has already been approved by the U.S. Environmental Protection Agency for use in plant agriculture. This could weaken the effectiveness of olorofim for treating things like Aspergillus infections even before the drug has been approved for use in humans.”
Plant drug undermining olorofim efficacy in humans
“While I’m sure this makes financial sense for the makers of ipflufenoquin, it borders on insanity from a public health perspective,” Dr. Price said.
Meanwhile, the global threat of fungal infections grows. The World Health Organization has launched its first-ever list of health-threatening fungi. Authors of a WHO report that contains the list write, “The invasive forms of these fungal infections often affect severely ill patients and those with significant underlying immune system–related conditions.”
F2G will continue to expand olorofim’s clinical trial program, according to the company’s statement. Along with its partner, Shionogi, it is enrolling patients with proven or probable invasive aspergillosis in a global phase 3 trial (OASIS), which will compare outcomes after treatment with olorofim in comparison with amphotericin B liposome (AmBisome) followed by standard of care.
A version of this article first appeared on Medscape.com.
The U.S. Food and Drug Administration is declining to approve the investigational antifungal olorofim and is asking for more data, according to a news release from the manufacturer, F2G.
Olorofim, (formerly known as F901318) is the first in the orotomide class of antifungals to be evaluated clinically for the treatment of invasive mold infections. Its maker, F2G, is a biotech company based in Manchester, England, that focuses on developing drugs for rare fungal diseases.
The company says it remains optimistic and will address the FDA’s requirements and continue to seek approval.
The FDA’s denial comes as fungal infections are becoming increasingly common and resistant to treatment. There are only four antifungal classes currently available, and there are few new candidates in the pipeline. No new classes of antifungals have been developed in 2 decades.
David Andes, MD, chief of the division of infectious diseases at the University of Wisconsin–Madison, told this news organization he shares the hope that the company can meet the requirements to gain approval.
“Some of the early results were really exciting,” he said. “People are enthusiastic about the compound because it has a novel mechanism of action, and it is active against a group of fungi that we have limited to no options for.”
Early results ‘exciting’
Dr. Andes said several physicians have been able to prescribe olorofim under the compassionate use program “and have witnessed success.”
Olorofim is the first antifungal agent to be granted breakthrough therapy designation, which the FDA granted in November 2019 for the treatment of invasive mold infections for patients with limited or no treatment options, including patients with refractory aspergillosis or those who are intolerant of currently available therapy. It is also indicated for infections due to Lomentospora prolificans, Scedosporium, and Scopulariopsis species.
Olorofim received a second breakthrough therapy designation in October 2020. The second designation was granted for treatment of central nervous system coccidioidomycosis that is refractory or for cases that cannot be treated with standard-of-care therapy.
It is very difficult for patients to be approved to receive compassionate use medicines, Dr. Andes pointed out. “I’d like to have access sooner rather than later,” he added.
Dr. Andes says the drugs are expensive and are time consuming to produce. And with antifungals, it is difficult to demonstrate safety in comparison with other antimicrobial agents because “it’s hard to hurt a fungus without having toxicity with human cells.”
Complete response letter issued
F2G received a complete response letter from the FDA regarding its new drug application for olorofim, according to the news release issued by the company. “While F2G is disappointed with this outcome, we remain optimistic about olorofim’s potential to address an unmet need for patients with invasive fungal infections who have exhausted their treatment alternatives,” Francesco Maria Lavino, chief executive officer, said in the release. “We are assessing the details of the Complete Response Letter, and we plan to meet with the FDA to discuss it further.”
Dr. Andes says few other antifungals have made it as far as olorofim in clinical trials.
Lance B. Price, PhD, codirector of the Antibiotic Resistance Action Center at George Washington University in Washington, told this news organization that despite the lack of antifungals in the pipeline, “We can’t allow our desperation to override the checkpoints that ensure that antifungals are safe to use in people.”
In the meantime, he said, it is important to preserve the utility of current antifungals by avoiding overusing them in medicine and agriculture.
“Sadly,” he said, “a drug called ipflufenoquin, which works by a similar mode of action as olorofim, has already been approved by the U.S. Environmental Protection Agency for use in plant agriculture. This could weaken the effectiveness of olorofim for treating things like Aspergillus infections even before the drug has been approved for use in humans.”
Plant drug undermining olorofim efficacy in humans
“While I’m sure this makes financial sense for the makers of ipflufenoquin, it borders on insanity from a public health perspective,” Dr. Price said.
Meanwhile, the global threat of fungal infections grows. The World Health Organization has launched its first-ever list of health-threatening fungi. Authors of a WHO report that contains the list write, “The invasive forms of these fungal infections often affect severely ill patients and those with significant underlying immune system–related conditions.”
F2G will continue to expand olorofim’s clinical trial program, according to the company’s statement. Along with its partner, Shionogi, it is enrolling patients with proven or probable invasive aspergillosis in a global phase 3 trial (OASIS), which will compare outcomes after treatment with olorofim in comparison with amphotericin B liposome (AmBisome) followed by standard of care.
A version of this article first appeared on Medscape.com.
The U.S. Food and Drug Administration is declining to approve the investigational antifungal olorofim and is asking for more data, according to a news release from the manufacturer, F2G.
Olorofim, (formerly known as F901318) is the first in the orotomide class of antifungals to be evaluated clinically for the treatment of invasive mold infections. Its maker, F2G, is a biotech company based in Manchester, England, that focuses on developing drugs for rare fungal diseases.
The company says it remains optimistic and will address the FDA’s requirements and continue to seek approval.
The FDA’s denial comes as fungal infections are becoming increasingly common and resistant to treatment. There are only four antifungal classes currently available, and there are few new candidates in the pipeline. No new classes of antifungals have been developed in 2 decades.
David Andes, MD, chief of the division of infectious diseases at the University of Wisconsin–Madison, told this news organization he shares the hope that the company can meet the requirements to gain approval.
“Some of the early results were really exciting,” he said. “People are enthusiastic about the compound because it has a novel mechanism of action, and it is active against a group of fungi that we have limited to no options for.”
Early results ‘exciting’
Dr. Andes said several physicians have been able to prescribe olorofim under the compassionate use program “and have witnessed success.”
Olorofim is the first antifungal agent to be granted breakthrough therapy designation, which the FDA granted in November 2019 for the treatment of invasive mold infections for patients with limited or no treatment options, including patients with refractory aspergillosis or those who are intolerant of currently available therapy. It is also indicated for infections due to Lomentospora prolificans, Scedosporium, and Scopulariopsis species.
Olorofim received a second breakthrough therapy designation in October 2020. The second designation was granted for treatment of central nervous system coccidioidomycosis that is refractory or for cases that cannot be treated with standard-of-care therapy.
It is very difficult for patients to be approved to receive compassionate use medicines, Dr. Andes pointed out. “I’d like to have access sooner rather than later,” he added.
Dr. Andes says the drugs are expensive and are time consuming to produce. And with antifungals, it is difficult to demonstrate safety in comparison with other antimicrobial agents because “it’s hard to hurt a fungus without having toxicity with human cells.”
Complete response letter issued
F2G received a complete response letter from the FDA regarding its new drug application for olorofim, according to the news release issued by the company. “While F2G is disappointed with this outcome, we remain optimistic about olorofim’s potential to address an unmet need for patients with invasive fungal infections who have exhausted their treatment alternatives,” Francesco Maria Lavino, chief executive officer, said in the release. “We are assessing the details of the Complete Response Letter, and we plan to meet with the FDA to discuss it further.”
Dr. Andes says few other antifungals have made it as far as olorofim in clinical trials.
Lance B. Price, PhD, codirector of the Antibiotic Resistance Action Center at George Washington University in Washington, told this news organization that despite the lack of antifungals in the pipeline, “We can’t allow our desperation to override the checkpoints that ensure that antifungals are safe to use in people.”
In the meantime, he said, it is important to preserve the utility of current antifungals by avoiding overusing them in medicine and agriculture.
“Sadly,” he said, “a drug called ipflufenoquin, which works by a similar mode of action as olorofim, has already been approved by the U.S. Environmental Protection Agency for use in plant agriculture. This could weaken the effectiveness of olorofim for treating things like Aspergillus infections even before the drug has been approved for use in humans.”
Plant drug undermining olorofim efficacy in humans
“While I’m sure this makes financial sense for the makers of ipflufenoquin, it borders on insanity from a public health perspective,” Dr. Price said.
Meanwhile, the global threat of fungal infections grows. The World Health Organization has launched its first-ever list of health-threatening fungi. Authors of a WHO report that contains the list write, “The invasive forms of these fungal infections often affect severely ill patients and those with significant underlying immune system–related conditions.”
F2G will continue to expand olorofim’s clinical trial program, according to the company’s statement. Along with its partner, Shionogi, it is enrolling patients with proven or probable invasive aspergillosis in a global phase 3 trial (OASIS), which will compare outcomes after treatment with olorofim in comparison with amphotericin B liposome (AmBisome) followed by standard of care.
A version of this article first appeared on Medscape.com.
A 63-year-old male presented for evaluation of worsening genital lesions and associated swelling
.1 Clinically, ENV presents as verrucous, hyperkeratotic, cobblestone-like patches, plaques, and nodules with associated nonpitting edema of the affected body area.1 Secondary bacterial infections are common and often worsen the clinical course. The etiology of ENV involves chronic lymphatic obstruction and venous insufficiency, with additional risk factors including obesity, chronic lymphedema, bacterial infection, surgery or trauma, neoplasia, radiation, congestive heart failure, or scleroderma.2,3 While most commonly presenting on the lower extremities, cases have been reported involving the abdomen, sacrum, ears, buttocks, and penoscrotal area.1,2

Regardless of location, the pathogenesis of ENV remains the same. Chronic lymphatic obstruction results in accumulation and lymphostasis of protein-rich dermal fluid, which subsequently precipitates fibroblast proliferation and activation, suppression of the local immune response and development of recurrent lymphangitis, chronic inflammation, and potential secondary bacterial infection.2,4
There is no standard of care for the treatment and management of ENV and recurrence is common. Interventions often involve those used for chronic lymphedema – including leg elevation, compression stockings or devices, skin hygiene, and lymphatic pumping.2,3 Medical management with topical and oral retinoids has been reported, as well as emphasis on weight loss and infection control.1,4 Surgical intervention is often reserved for refractory cases that fail to respond to more conservative management, or severe presentations resulting in extensive functional and aesthetic impairment. Less commonly reported treatment modalities include lymphaticovenular anastomosis and ablative carbon dioxide laser use, although this latter intervention demonstrated minimal improvement in this patient.5,6
Penoscrotal ENV is a rare form of ENV affecting the genital region of males, often resulting in significant disfigurement, functional impairment, and psychosocial distress. Penoscrotal elephantiasis can be idiopathic, due to filarial infections, scleroinflammatory stricture of the urethra, Chlamydia trachomatis infection, and lymphostasis secondary to chronic inflammatory conditions such as streptococcal infections, radiotherapy, surgery, chronic venous stasis, or Kaposi sarcoma.7
In addition, hidradenitis suppurativa (HS) has been documented multiple times in the literature in association with the development of ENV, detailing lymphatic scarring secondary to chronic inguinal HS as the main pathogenic factor.8,9
Surgery is the mainstay of treatment for penoscrotal ENV, which not only improves functionality and cosmesis, but also aids in prevention of rare malignant sequelae, such as lymphangiosarcoma.10 Such interventions can involve lymphangioplasty to aid in lymphatic drainage or excision of the mass and subcutaneous tissue with full-thickness skin grafting for reconstruction.7 Collaboration between urology, plastic surgery, and dermatology is often essential to obtain adequate care with satisfactory outcomes and minimal recurrence for patients with this uncommon condition.
This case and photo were submitted by Marlee Hill, a medical student at the University of Oklahoma, Oklahoma City; and Michael Franzetti, MD, and Jeffrey McBride, MD, department of dermatology, University of Oklahoma Health Sciences Center. The column was edited by Donna Bilu Martin, MD.
Dr. Donna Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to d[email protected].
References
1. Hadian Y et al. Dermatol Online J. 2019 Dec 15;25(12):13030/qt6rn1s8ff.
2. Judge N and Kilic A. J Dermatol Case Rep. 2016 Nov 13;10(2):32-4.
3. Dean SM et al. J Am Acad Dermatol. 2011 Jun;64(6):1104-10.
4. Sisto K and Khachemoune A. Am J Clin Dermatol. 2008;9(3):141-6.
5. Motegi S et al. Dermatology. 2007;215(2):147-51.
6. Robinson CG et al. J Cutan Med Surg. 2018;22(6):611-3.
7. Koualla S et al. Ann Chir Plast Esthet. 2023 Apr 10;S0294-1260(23)00035-3.
8. Lelonek E et al. Acta Derm Venereol. 2021 Feb 11;101(2):adv00389.
9. Good LM et al. J Am Acad Dermatol. 2011 May;64(5):993-4.
10. Cerri A et al. Eur J Dermatol. 1998 Oct-Nov;8(7):511-4.
.1 Clinically, ENV presents as verrucous, hyperkeratotic, cobblestone-like patches, plaques, and nodules with associated nonpitting edema of the affected body area.1 Secondary bacterial infections are common and often worsen the clinical course. The etiology of ENV involves chronic lymphatic obstruction and venous insufficiency, with additional risk factors including obesity, chronic lymphedema, bacterial infection, surgery or trauma, neoplasia, radiation, congestive heart failure, or scleroderma.2,3 While most commonly presenting on the lower extremities, cases have been reported involving the abdomen, sacrum, ears, buttocks, and penoscrotal area.1,2
Regardless of location, the pathogenesis of ENV remains the same. Chronic lymphatic obstruction results in accumulation and lymphostasis of protein-rich dermal fluid, which subsequently precipitates fibroblast proliferation and activation, suppression of the local immune response and development of recurrent lymphangitis, chronic inflammation, and potential secondary bacterial infection.2,4
There is no standard of care for the treatment and management of ENV and recurrence is common. Interventions often involve those used for chronic lymphedema – including leg elevation, compression stockings or devices, skin hygiene, and lymphatic pumping.2,3 Medical management with topical and oral retinoids has been reported, as well as emphasis on weight loss and infection control.1,4 Surgical intervention is often reserved for refractory cases that fail to respond to more conservative management, or severe presentations resulting in extensive functional and aesthetic impairment. Less commonly reported treatment modalities include lymphaticovenular anastomosis and ablative carbon dioxide laser use, although this latter intervention demonstrated minimal improvement in this patient.5,6
Penoscrotal ENV is a rare form of ENV affecting the genital region of males, often resulting in significant disfigurement, functional impairment, and psychosocial distress. Penoscrotal elephantiasis can be idiopathic, due to filarial infections, scleroinflammatory stricture of the urethra, Chlamydia trachomatis infection, and lymphostasis secondary to chronic inflammatory conditions such as streptococcal infections, radiotherapy, surgery, chronic venous stasis, or Kaposi sarcoma.7
In addition, hidradenitis suppurativa (HS) has been documented multiple times in the literature in association with the development of ENV, detailing lymphatic scarring secondary to chronic inguinal HS as the main pathogenic factor.8,9
Surgery is the mainstay of treatment for penoscrotal ENV, which not only improves functionality and cosmesis, but also aids in prevention of rare malignant sequelae, such as lymphangiosarcoma.10 Such interventions can involve lymphangioplasty to aid in lymphatic drainage or excision of the mass and subcutaneous tissue with full-thickness skin grafting for reconstruction.7 Collaboration between urology, plastic surgery, and dermatology is often essential to obtain adequate care with satisfactory outcomes and minimal recurrence for patients with this uncommon condition.
This case and photo were submitted by Marlee Hill, a medical student at the University of Oklahoma, Oklahoma City; and Michael Franzetti, MD, and Jeffrey McBride, MD, department of dermatology, University of Oklahoma Health Sciences Center. The column was edited by Donna Bilu Martin, MD.
Dr. Donna Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to d[email protected].
References
1. Hadian Y et al. Dermatol Online J. 2019 Dec 15;25(12):13030/qt6rn1s8ff.
2. Judge N and Kilic A. J Dermatol Case Rep. 2016 Nov 13;10(2):32-4.
3. Dean SM et al. J Am Acad Dermatol. 2011 Jun;64(6):1104-10.
4. Sisto K and Khachemoune A. Am J Clin Dermatol. 2008;9(3):141-6.
5. Motegi S et al. Dermatology. 2007;215(2):147-51.
6. Robinson CG et al. J Cutan Med Surg. 2018;22(6):611-3.
7. Koualla S et al. Ann Chir Plast Esthet. 2023 Apr 10;S0294-1260(23)00035-3.
8. Lelonek E et al. Acta Derm Venereol. 2021 Feb 11;101(2):adv00389.
9. Good LM et al. J Am Acad Dermatol. 2011 May;64(5):993-4.
10. Cerri A et al. Eur J Dermatol. 1998 Oct-Nov;8(7):511-4.
.1 Clinically, ENV presents as verrucous, hyperkeratotic, cobblestone-like patches, plaques, and nodules with associated nonpitting edema of the affected body area.1 Secondary bacterial infections are common and often worsen the clinical course. The etiology of ENV involves chronic lymphatic obstruction and venous insufficiency, with additional risk factors including obesity, chronic lymphedema, bacterial infection, surgery or trauma, neoplasia, radiation, congestive heart failure, or scleroderma.2,3 While most commonly presenting on the lower extremities, cases have been reported involving the abdomen, sacrum, ears, buttocks, and penoscrotal area.1,2
Regardless of location, the pathogenesis of ENV remains the same. Chronic lymphatic obstruction results in accumulation and lymphostasis of protein-rich dermal fluid, which subsequently precipitates fibroblast proliferation and activation, suppression of the local immune response and development of recurrent lymphangitis, chronic inflammation, and potential secondary bacterial infection.2,4
There is no standard of care for the treatment and management of ENV and recurrence is common. Interventions often involve those used for chronic lymphedema – including leg elevation, compression stockings or devices, skin hygiene, and lymphatic pumping.2,3 Medical management with topical and oral retinoids has been reported, as well as emphasis on weight loss and infection control.1,4 Surgical intervention is often reserved for refractory cases that fail to respond to more conservative management, or severe presentations resulting in extensive functional and aesthetic impairment. Less commonly reported treatment modalities include lymphaticovenular anastomosis and ablative carbon dioxide laser use, although this latter intervention demonstrated minimal improvement in this patient.5,6
Penoscrotal ENV is a rare form of ENV affecting the genital region of males, often resulting in significant disfigurement, functional impairment, and psychosocial distress. Penoscrotal elephantiasis can be idiopathic, due to filarial infections, scleroinflammatory stricture of the urethra, Chlamydia trachomatis infection, and lymphostasis secondary to chronic inflammatory conditions such as streptococcal infections, radiotherapy, surgery, chronic venous stasis, or Kaposi sarcoma.7
In addition, hidradenitis suppurativa (HS) has been documented multiple times in the literature in association with the development of ENV, detailing lymphatic scarring secondary to chronic inguinal HS as the main pathogenic factor.8,9
Surgery is the mainstay of treatment for penoscrotal ENV, which not only improves functionality and cosmesis, but also aids in prevention of rare malignant sequelae, such as lymphangiosarcoma.10 Such interventions can involve lymphangioplasty to aid in lymphatic drainage or excision of the mass and subcutaneous tissue with full-thickness skin grafting for reconstruction.7 Collaboration between urology, plastic surgery, and dermatology is often essential to obtain adequate care with satisfactory outcomes and minimal recurrence for patients with this uncommon condition.
This case and photo were submitted by Marlee Hill, a medical student at the University of Oklahoma, Oklahoma City; and Michael Franzetti, MD, and Jeffrey McBride, MD, department of dermatology, University of Oklahoma Health Sciences Center. The column was edited by Donna Bilu Martin, MD.
Dr. Donna Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to d[email protected].
References
1. Hadian Y et al. Dermatol Online J. 2019 Dec 15;25(12):13030/qt6rn1s8ff.
2. Judge N and Kilic A. J Dermatol Case Rep. 2016 Nov 13;10(2):32-4.
3. Dean SM et al. J Am Acad Dermatol. 2011 Jun;64(6):1104-10.
4. Sisto K and Khachemoune A. Am J Clin Dermatol. 2008;9(3):141-6.
5. Motegi S et al. Dermatology. 2007;215(2):147-51.
6. Robinson CG et al. J Cutan Med Surg. 2018;22(6):611-3.
7. Koualla S et al. Ann Chir Plast Esthet. 2023 Apr 10;S0294-1260(23)00035-3.
8. Lelonek E et al. Acta Derm Venereol. 2021 Feb 11;101(2):adv00389.
9. Good LM et al. J Am Acad Dermatol. 2011 May;64(5):993-4.
10. Cerri A et al. Eur J Dermatol. 1998 Oct-Nov;8(7):511-4.

Report eyes complications from microwave energy devices for hyperhidrosis
database showed.

While microwave energy devices (MEDs) are used to treat hyperhidrosis, the largest MED clinical trial included only 101 patients, Samantha Jo Albucker and Shari Lipner, MD, PhD, wrote in a research letter reporting the results.
For the study, published online in the Journal of the American Academy of Dermatology, Ms. Albucker, a student at Tulane University, New Orleans, and Dr. Lipner, associate professor of clinical dermatology at Weill Cornell Medicine, New York, searched the MAUDE database between Feb. 28, 2013, and Dec. 29, 2022, for adverse events (AEs) involving MEDs for hyperhidrosis treatment. Of the 502 medical device reports identified over the study period, the axilla was the most frequent injury site in 50.4% of cases. The three most common complications were infections (45.4%); neurological symptoms including neuropathy, nerve damage, and numbness (21.7%); and burns/ulcerations/erosions (19.1%).
In other findings, 2.4% of patients required hospitalization, most often because of infection (83.3%), followed by burn and coma (8.3% each). The average symptom onset was 2 months postprocedure, and the most common treatment was antibiotics in 62.2% of cases, followed by incision and drainage/aspiration in 21.7% of cases.
A codiagnosis of hidradenitis suppurativa (HS) was reported in 5.4% of all medical device reports. The researchers noted that in a published randomized clinical trial of eight HS patients undergoing MED treatment to assess the effect on HS symptoms, the treatment showed no clinical advantage. In addition, they referred to two case reports describing new-onset HS after MED treatment for hyperhidrosis.
“Therefore, we recommend questioning patients about HS history and examining for HS clinical findings before performing MED for hyperhidrosis,” they wrote, adding that the data, “taken together, suggests that avoidance of MED treatment of hyperhidrosis in HS patients is prudent and alternative treatments may be prescribed.”
The researchers acknowledged certain limitations of their analysis, including uncompleted medical device reports, patient reporting, and unverified causes of adverse events. “Large multicenter studies are needed to corroborate our results,” they concluded.

Adam Friedman, MD, professor and chair of dermatology at George Washington University, Washington, who was asked to comment on the study, said that primary idiopathic hyperhidrosis is a common medical condition that is often overlooked as a legitimate concern, and causes a quality-of-life burden. “Even with the striking numbers in the millions, there are limited treatment options available for axillary let alone other forms of primary hyperhidrosis,” said Dr. Friedman, who was not involved with the study.
“Therefore, for the short treatment list we have, it is important to have some predictive power with respect to clinical impact to provide realistic expectations as well as potential adverse events to ensure best practices and meaningful patient guidance. In this research letter, our colleagues highlight complications that can ensue from microwave therapy for hyperhidrosis and the frequency of said adverse events. Knowing these data is half the battle, and I for one would not have assumed infection was number one on the list of adverse events.”
Ms. Albucker had no relevant conflicts of interest to disclose. Dr. Lipner disclosed that she has served as a consultant for Ortho Dermatologics, Hoth Therapeutics, BelleTorus Corporation, and Moberg Pharmaceuticals.
Dr. Friedman disclosed that he is a consultant and/or advisory board member for Medscape/SanovaWorks, Oakstone Institute, L’Oréal, La Roche Posay, Galderma, Aveeno, Ortho Dermatologic, Microcures, Pfizer, Novartis, Lilly, Hoth Therapeutics, Zylo Therapeutics, BMS, Vial, Janssen, Novocure, Dermavant, Regeneron/Sanofi, and Incyte. He has also received grants from Pfizer, the Dermatology Foundation, Lilly, Janssen, Incyte, and Galderma.
database showed.
While microwave energy devices (MEDs) are used to treat hyperhidrosis, the largest MED clinical trial included only 101 patients, Samantha Jo Albucker and Shari Lipner, MD, PhD, wrote in a research letter reporting the results.
For the study, published online in the Journal of the American Academy of Dermatology, Ms. Albucker, a student at Tulane University, New Orleans, and Dr. Lipner, associate professor of clinical dermatology at Weill Cornell Medicine, New York, searched the MAUDE database between Feb. 28, 2013, and Dec. 29, 2022, for adverse events (AEs) involving MEDs for hyperhidrosis treatment. Of the 502 medical device reports identified over the study period, the axilla was the most frequent injury site in 50.4% of cases. The three most common complications were infections (45.4%); neurological symptoms including neuropathy, nerve damage, and numbness (21.7%); and burns/ulcerations/erosions (19.1%).
In other findings, 2.4% of patients required hospitalization, most often because of infection (83.3%), followed by burn and coma (8.3% each). The average symptom onset was 2 months postprocedure, and the most common treatment was antibiotics in 62.2% of cases, followed by incision and drainage/aspiration in 21.7% of cases.
A codiagnosis of hidradenitis suppurativa (HS) was reported in 5.4% of all medical device reports. The researchers noted that in a published randomized clinical trial of eight HS patients undergoing MED treatment to assess the effect on HS symptoms, the treatment showed no clinical advantage. In addition, they referred to two case reports describing new-onset HS after MED treatment for hyperhidrosis.
“Therefore, we recommend questioning patients about HS history and examining for HS clinical findings before performing MED for hyperhidrosis,” they wrote, adding that the data, “taken together, suggests that avoidance of MED treatment of hyperhidrosis in HS patients is prudent and alternative treatments may be prescribed.”
The researchers acknowledged certain limitations of their analysis, including uncompleted medical device reports, patient reporting, and unverified causes of adverse events. “Large multicenter studies are needed to corroborate our results,” they concluded.
Adam Friedman, MD, professor and chair of dermatology at George Washington University, Washington, who was asked to comment on the study, said that primary idiopathic hyperhidrosis is a common medical condition that is often overlooked as a legitimate concern, and causes a quality-of-life burden. “Even with the striking numbers in the millions, there are limited treatment options available for axillary let alone other forms of primary hyperhidrosis,” said Dr. Friedman, who was not involved with the study.
“Therefore, for the short treatment list we have, it is important to have some predictive power with respect to clinical impact to provide realistic expectations as well as potential adverse events to ensure best practices and meaningful patient guidance. In this research letter, our colleagues highlight complications that can ensue from microwave therapy for hyperhidrosis and the frequency of said adverse events. Knowing these data is half the battle, and I for one would not have assumed infection was number one on the list of adverse events.”
Ms. Albucker had no relevant conflicts of interest to disclose. Dr. Lipner disclosed that she has served as a consultant for Ortho Dermatologics, Hoth Therapeutics, BelleTorus Corporation, and Moberg Pharmaceuticals.
Dr. Friedman disclosed that he is a consultant and/or advisory board member for Medscape/SanovaWorks, Oakstone Institute, L’Oréal, La Roche Posay, Galderma, Aveeno, Ortho Dermatologic, Microcures, Pfizer, Novartis, Lilly, Hoth Therapeutics, Zylo Therapeutics, BMS, Vial, Janssen, Novocure, Dermavant, Regeneron/Sanofi, and Incyte. He has also received grants from Pfizer, the Dermatology Foundation, Lilly, Janssen, Incyte, and Galderma.
database showed.
While microwave energy devices (MEDs) are used to treat hyperhidrosis, the largest MED clinical trial included only 101 patients, Samantha Jo Albucker and Shari Lipner, MD, PhD, wrote in a research letter reporting the results.
For the study, published online in the Journal of the American Academy of Dermatology, Ms. Albucker, a student at Tulane University, New Orleans, and Dr. Lipner, associate professor of clinical dermatology at Weill Cornell Medicine, New York, searched the MAUDE database between Feb. 28, 2013, and Dec. 29, 2022, for adverse events (AEs) involving MEDs for hyperhidrosis treatment. Of the 502 medical device reports identified over the study period, the axilla was the most frequent injury site in 50.4% of cases. The three most common complications were infections (45.4%); neurological symptoms including neuropathy, nerve damage, and numbness (21.7%); and burns/ulcerations/erosions (19.1%).
In other findings, 2.4% of patients required hospitalization, most often because of infection (83.3%), followed by burn and coma (8.3% each). The average symptom onset was 2 months postprocedure, and the most common treatment was antibiotics in 62.2% of cases, followed by incision and drainage/aspiration in 21.7% of cases.
A codiagnosis of hidradenitis suppurativa (HS) was reported in 5.4% of all medical device reports. The researchers noted that in a published randomized clinical trial of eight HS patients undergoing MED treatment to assess the effect on HS symptoms, the treatment showed no clinical advantage. In addition, they referred to two case reports describing new-onset HS after MED treatment for hyperhidrosis.
“Therefore, we recommend questioning patients about HS history and examining for HS clinical findings before performing MED for hyperhidrosis,” they wrote, adding that the data, “taken together, suggests that avoidance of MED treatment of hyperhidrosis in HS patients is prudent and alternative treatments may be prescribed.”
The researchers acknowledged certain limitations of their analysis, including uncompleted medical device reports, patient reporting, and unverified causes of adverse events. “Large multicenter studies are needed to corroborate our results,” they concluded.
Adam Friedman, MD, professor and chair of dermatology at George Washington University, Washington, who was asked to comment on the study, said that primary idiopathic hyperhidrosis is a common medical condition that is often overlooked as a legitimate concern, and causes a quality-of-life burden. “Even with the striking numbers in the millions, there are limited treatment options available for axillary let alone other forms of primary hyperhidrosis,” said Dr. Friedman, who was not involved with the study.
“Therefore, for the short treatment list we have, it is important to have some predictive power with respect to clinical impact to provide realistic expectations as well as potential adverse events to ensure best practices and meaningful patient guidance. In this research letter, our colleagues highlight complications that can ensue from microwave therapy for hyperhidrosis and the frequency of said adverse events. Knowing these data is half the battle, and I for one would not have assumed infection was number one on the list of adverse events.”
Ms. Albucker had no relevant conflicts of interest to disclose. Dr. Lipner disclosed that she has served as a consultant for Ortho Dermatologics, Hoth Therapeutics, BelleTorus Corporation, and Moberg Pharmaceuticals.
Dr. Friedman disclosed that he is a consultant and/or advisory board member for Medscape/SanovaWorks, Oakstone Institute, L’Oréal, La Roche Posay, Galderma, Aveeno, Ortho Dermatologic, Microcures, Pfizer, Novartis, Lilly, Hoth Therapeutics, Zylo Therapeutics, BMS, Vial, Janssen, Novocure, Dermavant, Regeneron/Sanofi, and Incyte. He has also received grants from Pfizer, the Dermatology Foundation, Lilly, Janssen, Incyte, and Galderma.
FROM THE JOURNAL OF THE AMERICAN ACADEMY OF DERMATOLOGY
IL-17 inhibitor approved in Europe for hidradenitis suppurativa
The biologic is the first interleukin-17A (IL-17A) inhibitor to be approved for the treatment of moderate to severe HS. The manufacturer, Novartis, expects a regulatory decision from the U.S. Food and Drug Administration later this year, according to a company press release announcing the approval.
The European approval is based on the results from the phase 3 SUNSHINE and SUNRISE trials, which evaluated the efficacy, safety, and tolerability of the drug. The multicenter, randomized, placebo-controlled, double-blind trials enrolled a total of more than 1,000 adults with moderate to severe HS.
Patients were randomly assigned either to receive subcutaneous secukinumab 300 mg every 2 weeks or 4 weeks or to receive placebo. The treatment was effective at improving the symptoms of HS when given every 2 weeks, according to results recently published in The Lancet.
The primary outcome measure for both trials was HS clinical response – defined as a decrease in abscess and inflammatory nodule count by 50% or more with no increase in the number of abscesses or draining fistulae, compared with baseline.
In the studies, 42% and 45% of patients treated with secukinumab every 2 weeks in the SUNRISE and SUNSHINE trials, respectively, had a clinical response at 16 weeks, compared with 31% and 34% among those who received placebo, which were statistically significant differences. A significant clinical response was seen at week 4 in the SUNSHINE trial and in week 2 in the SUNRISE trial. In both trials, clinical efficacy was sustained to the end of the trial, at week 52.
Headaches were the most common side effect. They affected approximately 1 in 10 patients in both trials.
HS, also called acne inversa, is a chronic skin condition that causes painful lesions. The condition affects 1%- 2% of the U.S. population, according to the nonprofit Hidradenitis Suppurativa Foundation. It also disproportionately affects young adults, women, and Black patients.
In Europe, about 200,000 people live with moderate to severe stages of the condition, according to the Novartis press release.
Secukinumab inhibits IL-17A, a cytokine involved in the inflammation of psoriatic arthritis, plaque psoriasis, ankylosing spondylitis, and nonradiographic axial spondylarthritis. It has been approved for the treatment of those conditions, as well as for the treatment of juvenile idiopathic arthritis and enthesitis-related arthritis in the United States and the European Union.
The only other approved biologic therapy for HS is the tumor necrosis factor inhibitor adalimumab.
Novartis is investigating the potential application of secukinumab for the treatment of lupus nephritis and giant cell arteritis, as well as polymyalgia rheumatica and rotator cuff tendinopathy, according to the company press release.
The study published in The Lancet was funded by Novartis.
A version of this article first appeared on Medscape.com.
The biologic is the first interleukin-17A (IL-17A) inhibitor to be approved for the treatment of moderate to severe HS. The manufacturer, Novartis, expects a regulatory decision from the U.S. Food and Drug Administration later this year, according to a company press release announcing the approval.
The European approval is based on the results from the phase 3 SUNSHINE and SUNRISE trials, which evaluated the efficacy, safety, and tolerability of the drug. The multicenter, randomized, placebo-controlled, double-blind trials enrolled a total of more than 1,000 adults with moderate to severe HS.
Patients were randomly assigned either to receive subcutaneous secukinumab 300 mg every 2 weeks or 4 weeks or to receive placebo. The treatment was effective at improving the symptoms of HS when given every 2 weeks, according to results recently published in The Lancet.
The primary outcome measure for both trials was HS clinical response – defined as a decrease in abscess and inflammatory nodule count by 50% or more with no increase in the number of abscesses or draining fistulae, compared with baseline.
In the studies, 42% and 45% of patients treated with secukinumab every 2 weeks in the SUNRISE and SUNSHINE trials, respectively, had a clinical response at 16 weeks, compared with 31% and 34% among those who received placebo, which were statistically significant differences. A significant clinical response was seen at week 4 in the SUNSHINE trial and in week 2 in the SUNRISE trial. In both trials, clinical efficacy was sustained to the end of the trial, at week 52.
Headaches were the most common side effect. They affected approximately 1 in 10 patients in both trials.
HS, also called acne inversa, is a chronic skin condition that causes painful lesions. The condition affects 1%- 2% of the U.S. population, according to the nonprofit Hidradenitis Suppurativa Foundation. It also disproportionately affects young adults, women, and Black patients.
In Europe, about 200,000 people live with moderate to severe stages of the condition, according to the Novartis press release.
Secukinumab inhibits IL-17A, a cytokine involved in the inflammation of psoriatic arthritis, plaque psoriasis, ankylosing spondylitis, and nonradiographic axial spondylarthritis. It has been approved for the treatment of those conditions, as well as for the treatment of juvenile idiopathic arthritis and enthesitis-related arthritis in the United States and the European Union.
The only other approved biologic therapy for HS is the tumor necrosis factor inhibitor adalimumab.
Novartis is investigating the potential application of secukinumab for the treatment of lupus nephritis and giant cell arteritis, as well as polymyalgia rheumatica and rotator cuff tendinopathy, according to the company press release.
The study published in The Lancet was funded by Novartis.
A version of this article first appeared on Medscape.com.
The biologic is the first interleukin-17A (IL-17A) inhibitor to be approved for the treatment of moderate to severe HS. The manufacturer, Novartis, expects a regulatory decision from the U.S. Food and Drug Administration later this year, according to a company press release announcing the approval.
The European approval is based on the results from the phase 3 SUNSHINE and SUNRISE trials, which evaluated the efficacy, safety, and tolerability of the drug. The multicenter, randomized, placebo-controlled, double-blind trials enrolled a total of more than 1,000 adults with moderate to severe HS.
Patients were randomly assigned either to receive subcutaneous secukinumab 300 mg every 2 weeks or 4 weeks or to receive placebo. The treatment was effective at improving the symptoms of HS when given every 2 weeks, according to results recently published in The Lancet.
The primary outcome measure for both trials was HS clinical response – defined as a decrease in abscess and inflammatory nodule count by 50% or more with no increase in the number of abscesses or draining fistulae, compared with baseline.
In the studies, 42% and 45% of patients treated with secukinumab every 2 weeks in the SUNRISE and SUNSHINE trials, respectively, had a clinical response at 16 weeks, compared with 31% and 34% among those who received placebo, which were statistically significant differences. A significant clinical response was seen at week 4 in the SUNSHINE trial and in week 2 in the SUNRISE trial. In both trials, clinical efficacy was sustained to the end of the trial, at week 52.
Headaches were the most common side effect. They affected approximately 1 in 10 patients in both trials.
HS, also called acne inversa, is a chronic skin condition that causes painful lesions. The condition affects 1%- 2% of the U.S. population, according to the nonprofit Hidradenitis Suppurativa Foundation. It also disproportionately affects young adults, women, and Black patients.
In Europe, about 200,000 people live with moderate to severe stages of the condition, according to the Novartis press release.
Secukinumab inhibits IL-17A, a cytokine involved in the inflammation of psoriatic arthritis, plaque psoriasis, ankylosing spondylitis, and nonradiographic axial spondylarthritis. It has been approved for the treatment of those conditions, as well as for the treatment of juvenile idiopathic arthritis and enthesitis-related arthritis in the United States and the European Union.
The only other approved biologic therapy for HS is the tumor necrosis factor inhibitor adalimumab.
Novartis is investigating the potential application of secukinumab for the treatment of lupus nephritis and giant cell arteritis, as well as polymyalgia rheumatica and rotator cuff tendinopathy, according to the company press release.
The study published in The Lancet was funded by Novartis.
A version of this article first appeared on Medscape.com.
Scientists discover variants, therapy for disabling pansclerotic morphea
A team of especially in patients who have not responded to other interventions.
DPM was first reported in 1923, and while a genetic cause has been suspected, it had not been identified until now. The disease is the most severe form of deep morphea, which affects individuals with juvenile localized scleroderma. Patients, generally children under age 14, experience rapid sclerosis of all layers of the skin, fascia, muscle, and bone. DPM is also deadly: Most patients do not live more than 10 years after diagnosis, as they contract squamous cell carcinoma, restrictive pulmonary disease, sepsis, and gangrene.
In the study, published in the New England Journal of Medicine, the researchers discovered that people with DPM have an overactive version of the protein STAT4, which regulates inflammation and wound healing. The scientists studied four patients from three unrelated families with an autosomal dominant pattern of inheritance of DPM.
“Researchers previously thought that this disorder was caused by the immune system attacking the skin,” Sarah Blackstone, a predoctoral fellow in the inflammatory disease section at the National Human Genome Research Institute and co–first author of the study, said in a statement from the National Institutes of Health describing the results. “However, we found that this is an oversimplification, and that both skin and the immune system play an active role in disabling pansclerotic morphea,” added Ms. Blackstone, also a medical student at the University of South Dakota, Sioux Falls.
The overactive STAT4 protein creates a positive feedback loop of inflammation and impaired wound-healing. By targeting JAK, the researchers were able to stop the feedback and patients’ wounds dramatically improved. After 18 months of treatment with oral ruxolitinib, one patient had discontinued all other medications, and had complete resolution of a chest rash, substantial clearing on the arms and legs, and global clinical improvement.
The authors said that oral systemic JAK inhibitor therapy is preferred over topical therapy. Their research also suggested that anti–interleukin-6 monoclonal antibodies – such as tocilizumab, approved for indications that include rheumatoid arthritis and systemic sclerosis–associated interstitial lung disease, “may be an alternative therapy or may be useful in combination with JAK inhibitors in patients with DPM,” the authors wrote.
Most current DPM therapies – including methotrexate, mycophenolate mofetil, and ultraviolet A light therapy – have been ineffective, and some have severe side effects.
“The findings of this study open doors for JAK inhibitors to be a potential treatment for other inflammatory skin disorders or disorders related to tissue scarring, whether it is scarring of the lungs, liver or bone marrow,” Dan Kastner, MD, PhD, an NIH distinguished investigator, head of the NHGRI’s inflammatory disease section, and a senior author of the paper, said in the NIH statement.
“We hope to continue studying other molecules in this pathway and how they are altered in patients with disabling pansclerotic morphea and related conditions to find clues to understanding a broader array of more common diseases,” Lori Broderick, MD, PhD, a senior author of the paper and an associate professor at University of California, San Diego, said in the statement.
The study was led by researchers at NHGRI in collaboration with researchers from UCSD and the University of Pittsburgh. Researchers from the National Institute of Arthritis and Musculoskeletal and Skin Diseases and the National Institute of Allergy and Infectious Diseases also participated.
The study was supported by grants from the American Academy of Allergy, Asthma, and Immunology Foundation; the Ludwig Institute for Cancer Research; the University of California, San Diego, department of pediatrics; and the Novo Nordisk Foundation. Additional support and grants were given by the Deutsche Forschungsgemeinschaft, various institutes at the NIH, the California Institute for Regenerative Medicine, the Hydrocephalus Association, the Scleroderma Research Foundation, the Biowulf High-Performance Computing Cluster of the Center for Information Technology, the Undiagnosed Diseases Program of the Common Fund of the Office of the Director of the NIH, and the NIH Clinical Center.
A team of especially in patients who have not responded to other interventions.
DPM was first reported in 1923, and while a genetic cause has been suspected, it had not been identified until now. The disease is the most severe form of deep morphea, which affects individuals with juvenile localized scleroderma. Patients, generally children under age 14, experience rapid sclerosis of all layers of the skin, fascia, muscle, and bone. DPM is also deadly: Most patients do not live more than 10 years after diagnosis, as they contract squamous cell carcinoma, restrictive pulmonary disease, sepsis, and gangrene.
In the study, published in the New England Journal of Medicine, the researchers discovered that people with DPM have an overactive version of the protein STAT4, which regulates inflammation and wound healing. The scientists studied four patients from three unrelated families with an autosomal dominant pattern of inheritance of DPM.
“Researchers previously thought that this disorder was caused by the immune system attacking the skin,” Sarah Blackstone, a predoctoral fellow in the inflammatory disease section at the National Human Genome Research Institute and co–first author of the study, said in a statement from the National Institutes of Health describing the results. “However, we found that this is an oversimplification, and that both skin and the immune system play an active role in disabling pansclerotic morphea,” added Ms. Blackstone, also a medical student at the University of South Dakota, Sioux Falls.
The overactive STAT4 protein creates a positive feedback loop of inflammation and impaired wound-healing. By targeting JAK, the researchers were able to stop the feedback and patients’ wounds dramatically improved. After 18 months of treatment with oral ruxolitinib, one patient had discontinued all other medications, and had complete resolution of a chest rash, substantial clearing on the arms and legs, and global clinical improvement.
The authors said that oral systemic JAK inhibitor therapy is preferred over topical therapy. Their research also suggested that anti–interleukin-6 monoclonal antibodies – such as tocilizumab, approved for indications that include rheumatoid arthritis and systemic sclerosis–associated interstitial lung disease, “may be an alternative therapy or may be useful in combination with JAK inhibitors in patients with DPM,” the authors wrote.
Most current DPM therapies – including methotrexate, mycophenolate mofetil, and ultraviolet A light therapy – have been ineffective, and some have severe side effects.
“The findings of this study open doors for JAK inhibitors to be a potential treatment for other inflammatory skin disorders or disorders related to tissue scarring, whether it is scarring of the lungs, liver or bone marrow,” Dan Kastner, MD, PhD, an NIH distinguished investigator, head of the NHGRI’s inflammatory disease section, and a senior author of the paper, said in the NIH statement.
“We hope to continue studying other molecules in this pathway and how they are altered in patients with disabling pansclerotic morphea and related conditions to find clues to understanding a broader array of more common diseases,” Lori Broderick, MD, PhD, a senior author of the paper and an associate professor at University of California, San Diego, said in the statement.
The study was led by researchers at NHGRI in collaboration with researchers from UCSD and the University of Pittsburgh. Researchers from the National Institute of Arthritis and Musculoskeletal and Skin Diseases and the National Institute of Allergy and Infectious Diseases also participated.
The study was supported by grants from the American Academy of Allergy, Asthma, and Immunology Foundation; the Ludwig Institute for Cancer Research; the University of California, San Diego, department of pediatrics; and the Novo Nordisk Foundation. Additional support and grants were given by the Deutsche Forschungsgemeinschaft, various institutes at the NIH, the California Institute for Regenerative Medicine, the Hydrocephalus Association, the Scleroderma Research Foundation, the Biowulf High-Performance Computing Cluster of the Center for Information Technology, the Undiagnosed Diseases Program of the Common Fund of the Office of the Director of the NIH, and the NIH Clinical Center.
A team of especially in patients who have not responded to other interventions.
DPM was first reported in 1923, and while a genetic cause has been suspected, it had not been identified until now. The disease is the most severe form of deep morphea, which affects individuals with juvenile localized scleroderma. Patients, generally children under age 14, experience rapid sclerosis of all layers of the skin, fascia, muscle, and bone. DPM is also deadly: Most patients do not live more than 10 years after diagnosis, as they contract squamous cell carcinoma, restrictive pulmonary disease, sepsis, and gangrene.
In the study, published in the New England Journal of Medicine, the researchers discovered that people with DPM have an overactive version of the protein STAT4, which regulates inflammation and wound healing. The scientists studied four patients from three unrelated families with an autosomal dominant pattern of inheritance of DPM.
“Researchers previously thought that this disorder was caused by the immune system attacking the skin,” Sarah Blackstone, a predoctoral fellow in the inflammatory disease section at the National Human Genome Research Institute and co–first author of the study, said in a statement from the National Institutes of Health describing the results. “However, we found that this is an oversimplification, and that both skin and the immune system play an active role in disabling pansclerotic morphea,” added Ms. Blackstone, also a medical student at the University of South Dakota, Sioux Falls.
The overactive STAT4 protein creates a positive feedback loop of inflammation and impaired wound-healing. By targeting JAK, the researchers were able to stop the feedback and patients’ wounds dramatically improved. After 18 months of treatment with oral ruxolitinib, one patient had discontinued all other medications, and had complete resolution of a chest rash, substantial clearing on the arms and legs, and global clinical improvement.
The authors said that oral systemic JAK inhibitor therapy is preferred over topical therapy. Their research also suggested that anti–interleukin-6 monoclonal antibodies – such as tocilizumab, approved for indications that include rheumatoid arthritis and systemic sclerosis–associated interstitial lung disease, “may be an alternative therapy or may be useful in combination with JAK inhibitors in patients with DPM,” the authors wrote.
Most current DPM therapies – including methotrexate, mycophenolate mofetil, and ultraviolet A light therapy – have been ineffective, and some have severe side effects.
“The findings of this study open doors for JAK inhibitors to be a potential treatment for other inflammatory skin disorders or disorders related to tissue scarring, whether it is scarring of the lungs, liver or bone marrow,” Dan Kastner, MD, PhD, an NIH distinguished investigator, head of the NHGRI’s inflammatory disease section, and a senior author of the paper, said in the NIH statement.
“We hope to continue studying other molecules in this pathway and how they are altered in patients with disabling pansclerotic morphea and related conditions to find clues to understanding a broader array of more common diseases,” Lori Broderick, MD, PhD, a senior author of the paper and an associate professor at University of California, San Diego, said in the statement.
The study was led by researchers at NHGRI in collaboration with researchers from UCSD and the University of Pittsburgh. Researchers from the National Institute of Arthritis and Musculoskeletal and Skin Diseases and the National Institute of Allergy and Infectious Diseases also participated.
The study was supported by grants from the American Academy of Allergy, Asthma, and Immunology Foundation; the Ludwig Institute for Cancer Research; the University of California, San Diego, department of pediatrics; and the Novo Nordisk Foundation. Additional support and grants were given by the Deutsche Forschungsgemeinschaft, various institutes at the NIH, the California Institute for Regenerative Medicine, the Hydrocephalus Association, the Scleroderma Research Foundation, the Biowulf High-Performance Computing Cluster of the Center for Information Technology, the Undiagnosed Diseases Program of the Common Fund of the Office of the Director of the NIH, and the NIH Clinical Center.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Beta-blocker gel shows promise for diabetic foot ulcers
say Indian researchers.
Esmolol is a short-acting beta-adrenergic receptor blocker that is currently approved by the Food and Drug Administration for cardiac indications such as short-term use for supraventricular tachycardia.
As a gel, esmolol hydrochloride is administered topically to stimulate wound healing via mechanisms such as the migration of keratinocytes, fibroblasts, and endothelial cells into wound tissue.
The current trial enrolled patients with type 1 or 2 diabetes, finding that, among 140 assessed, target ulcer closure within 12 weeks was more than twice as likely in those assigned esmolol gel plus standard of care than those given standard of care alone.
The impact of adding esmolol gel to standard of care was even greater in patients with a body mass index (BMI) over 25 kg/m2 and in those who weighed more than 80 kg (176 lb).
“The use of esmolol in the treatment of diabetic foot ulcers in addition to standard of care may be an important addition to the endeavor of healing diabetic foot ulcers,” wrote Ashu Rastogi, MD, DM, department of endocrinology, Post Graduate Institute of Medical Education and Research, Chandigarh, India, and colleagues, in their article recently published in JAMA Network Open.
Dr. Rastogi first presented the findings at the 2022 annual meeting of the European Association for the Study of Diabetes. The results were well received, with one clinician describing them as “astounding.”
However, Andrew Boulton, MD, PhD, said in an interview that, although the final published data are “interesting,” they “need further confirmation” because “there are one or two unusual features” about the study. Dr. Boulton is a professor of medicine, division of diabetes, endocrinology & gastroenterology, at the University of Manchester (England).
He highlighted that the study was of “basically neuropathic ulcers, many of which were plantar and should be able to heal without any specific additional therapy.”
In addition, the inclusion criteria state that the ulcers could be below the malleoli or 5 cm above them, which Dr. Boulton explained is “very unusual and would therefore include some atypical and not truly diabetic ‘foot’ ulcers.”
And Frances Game, MBBCh, department of diabetes and endocrinology, University Hospitals of Derby (England) and Burton NHS Foundation Trust, added that there are questions about the study methodology.
She said in an interview that although it is a “fascinating study,” the main comparison group did not receive vehicle, or placebo, gel in addition to standard of care. “How were they blinded [to treatment]?”
The “biggest problem” with the study, however, is that the primary outcome was reported as a per-protocol endpoint, not as a standard intention-to-treat analysis, which allowed the researchers to exclude patients whose ulcers increased in size by over 30% on two consecutive visits.
“That kind of makes [esmolol gel] look better than it is because they’ve taken out the ones who got worse,” Dr. Game noted. However, the findings, while not conclusive, do warrant further study of esmolol gel.
The authors noted that diabetic foot ulcers are a severe complication of diabetes, with a prevalence of 1.3%-12.0% across various countries, And the complication contributes to patient morbidity and mortality, with a 5-year mortality that is substantially higher than that of many cancers.
Moreover, “even with the best therapy,” such as advanced moist wound therapy, bioengineered tissue or skin substitutes, peptides, growth factors, electric stimulation, and negative-pressure wound therapy, just 30% of wounds linked to diabetes heal and recurrence is as high as 70%.
Against this backdrop, topical esmolol 14% gel was shown in a phase 1/2 study to be associated with ulcer area reduction and earlier wound closure versus standard of care plus a control vehicle gel.
The current phase 3, randomized, controlled trial involved individuals aged 18-75 years with type 1 or type 2 diabetes and noninfected diabetic foot ulcers classified as grade 1A and 1C on the University of Texas Wound Classification System, which had been open for at least 6 weeks and had an area of 2-25 cm2.
Patients from 27 tertiary care centers across India were enrolled in 2018-2020. They were randomized in a 3:3:1 ratio to one of three groups: esmolol 14% gel plus standard of care, standard of care only, or vehicle plus standard of care.
The study lasted 25 weeks and included a 1-week screening phase, during which all patients received standard of care, a 12-week treatment phase, and a 12-week follow-up phase. The latter included a closure confirmation period of 4 weeks and an observation period of 8 weeks.
Patients were assessed once a week during the treatment phase, and then at weeks 14, 16, 20, and 24.
In all, 176 patients were enrolled. Participants were a mean age of 56.4 years and 69.3% were men. Average hemoglobin A1c was 8.6%. Mean diabetic foot ulcer area was 4.7 cm2 and the average ulcer duration was 49.8 weeks.
The primary outcome was the proportion of patients who achieved target ulcer closure during the 12-week treatment phase and was assessed in 140 patients.
Overall, 60.3% of patients treated with esmolol gel plus standard of care achieved target ulcer closure versus 41.7% of those in the standard of care alone group (odds ratio, 2.13; P = .03).
The secondary outcome was the proportion of patients with target ulcer closure by the study end and was assessed in 120 patients.
In total, 77.2% of patients in the esmolol gel plus standard of care group met the secondary endpoint, compared with 55.6% of those receiving standard of care alone (OR, 1.72; P = .01).
Further analysis suggested the benefit seen with esmolol gel plus standard of care was greater in patients with a weight greater than 80 kg versus standard of care alone (OR, 4.04; P = .04), and in those with a BMI greater than 25 (OR, 2.72; P = .03).
Treatment-emergent adverse events were reported by 33 (18.8%) participants, with 12 events deemed serious. “However, none of the serious adverse events were considered as drug-related by the investigators,” concluded the researchers.
The study was partly funded by NovaLead Pharma and the Biotechnology Industry Research Assistance Council, New Delhi, set up by the Department of Biotechnology, Government of India. Dr. Rastogi reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
say Indian researchers.
Esmolol is a short-acting beta-adrenergic receptor blocker that is currently approved by the Food and Drug Administration for cardiac indications such as short-term use for supraventricular tachycardia.
As a gel, esmolol hydrochloride is administered topically to stimulate wound healing via mechanisms such as the migration of keratinocytes, fibroblasts, and endothelial cells into wound tissue.
The current trial enrolled patients with type 1 or 2 diabetes, finding that, among 140 assessed, target ulcer closure within 12 weeks was more than twice as likely in those assigned esmolol gel plus standard of care than those given standard of care alone.
The impact of adding esmolol gel to standard of care was even greater in patients with a body mass index (BMI) over 25 kg/m2 and in those who weighed more than 80 kg (176 lb).
“The use of esmolol in the treatment of diabetic foot ulcers in addition to standard of care may be an important addition to the endeavor of healing diabetic foot ulcers,” wrote Ashu Rastogi, MD, DM, department of endocrinology, Post Graduate Institute of Medical Education and Research, Chandigarh, India, and colleagues, in their article recently published in JAMA Network Open.
Dr. Rastogi first presented the findings at the 2022 annual meeting of the European Association for the Study of Diabetes. The results were well received, with one clinician describing them as “astounding.”
However, Andrew Boulton, MD, PhD, said in an interview that, although the final published data are “interesting,” they “need further confirmation” because “there are one or two unusual features” about the study. Dr. Boulton is a professor of medicine, division of diabetes, endocrinology & gastroenterology, at the University of Manchester (England).
He highlighted that the study was of “basically neuropathic ulcers, many of which were plantar and should be able to heal without any specific additional therapy.”
In addition, the inclusion criteria state that the ulcers could be below the malleoli or 5 cm above them, which Dr. Boulton explained is “very unusual and would therefore include some atypical and not truly diabetic ‘foot’ ulcers.”
And Frances Game, MBBCh, department of diabetes and endocrinology, University Hospitals of Derby (England) and Burton NHS Foundation Trust, added that there are questions about the study methodology.
She said in an interview that although it is a “fascinating study,” the main comparison group did not receive vehicle, or placebo, gel in addition to standard of care. “How were they blinded [to treatment]?”
The “biggest problem” with the study, however, is that the primary outcome was reported as a per-protocol endpoint, not as a standard intention-to-treat analysis, which allowed the researchers to exclude patients whose ulcers increased in size by over 30% on two consecutive visits.
“That kind of makes [esmolol gel] look better than it is because they’ve taken out the ones who got worse,” Dr. Game noted. However, the findings, while not conclusive, do warrant further study of esmolol gel.
The authors noted that diabetic foot ulcers are a severe complication of diabetes, with a prevalence of 1.3%-12.0% across various countries, And the complication contributes to patient morbidity and mortality, with a 5-year mortality that is substantially higher than that of many cancers.
Moreover, “even with the best therapy,” such as advanced moist wound therapy, bioengineered tissue or skin substitutes, peptides, growth factors, electric stimulation, and negative-pressure wound therapy, just 30% of wounds linked to diabetes heal and recurrence is as high as 70%.
Against this backdrop, topical esmolol 14% gel was shown in a phase 1/2 study to be associated with ulcer area reduction and earlier wound closure versus standard of care plus a control vehicle gel.
The current phase 3, randomized, controlled trial involved individuals aged 18-75 years with type 1 or type 2 diabetes and noninfected diabetic foot ulcers classified as grade 1A and 1C on the University of Texas Wound Classification System, which had been open for at least 6 weeks and had an area of 2-25 cm2.
Patients from 27 tertiary care centers across India were enrolled in 2018-2020. They were randomized in a 3:3:1 ratio to one of three groups: esmolol 14% gel plus standard of care, standard of care only, or vehicle plus standard of care.
The study lasted 25 weeks and included a 1-week screening phase, during which all patients received standard of care, a 12-week treatment phase, and a 12-week follow-up phase. The latter included a closure confirmation period of 4 weeks and an observation period of 8 weeks.
Patients were assessed once a week during the treatment phase, and then at weeks 14, 16, 20, and 24.
In all, 176 patients were enrolled. Participants were a mean age of 56.4 years and 69.3% were men. Average hemoglobin A1c was 8.6%. Mean diabetic foot ulcer area was 4.7 cm2 and the average ulcer duration was 49.8 weeks.
The primary outcome was the proportion of patients who achieved target ulcer closure during the 12-week treatment phase and was assessed in 140 patients.
Overall, 60.3% of patients treated with esmolol gel plus standard of care achieved target ulcer closure versus 41.7% of those in the standard of care alone group (odds ratio, 2.13; P = .03).
The secondary outcome was the proportion of patients with target ulcer closure by the study end and was assessed in 120 patients.
In total, 77.2% of patients in the esmolol gel plus standard of care group met the secondary endpoint, compared with 55.6% of those receiving standard of care alone (OR, 1.72; P = .01).
Further analysis suggested the benefit seen with esmolol gel plus standard of care was greater in patients with a weight greater than 80 kg versus standard of care alone (OR, 4.04; P = .04), and in those with a BMI greater than 25 (OR, 2.72; P = .03).
Treatment-emergent adverse events were reported by 33 (18.8%) participants, with 12 events deemed serious. “However, none of the serious adverse events were considered as drug-related by the investigators,” concluded the researchers.
The study was partly funded by NovaLead Pharma and the Biotechnology Industry Research Assistance Council, New Delhi, set up by the Department of Biotechnology, Government of India. Dr. Rastogi reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
say Indian researchers.
Esmolol is a short-acting beta-adrenergic receptor blocker that is currently approved by the Food and Drug Administration for cardiac indications such as short-term use for supraventricular tachycardia.
As a gel, esmolol hydrochloride is administered topically to stimulate wound healing via mechanisms such as the migration of keratinocytes, fibroblasts, and endothelial cells into wound tissue.
The current trial enrolled patients with type 1 or 2 diabetes, finding that, among 140 assessed, target ulcer closure within 12 weeks was more than twice as likely in those assigned esmolol gel plus standard of care than those given standard of care alone.
The impact of adding esmolol gel to standard of care was even greater in patients with a body mass index (BMI) over 25 kg/m2 and in those who weighed more than 80 kg (176 lb).
“The use of esmolol in the treatment of diabetic foot ulcers in addition to standard of care may be an important addition to the endeavor of healing diabetic foot ulcers,” wrote Ashu Rastogi, MD, DM, department of endocrinology, Post Graduate Institute of Medical Education and Research, Chandigarh, India, and colleagues, in their article recently published in JAMA Network Open.
Dr. Rastogi first presented the findings at the 2022 annual meeting of the European Association for the Study of Diabetes. The results were well received, with one clinician describing them as “astounding.”
However, Andrew Boulton, MD, PhD, said in an interview that, although the final published data are “interesting,” they “need further confirmation” because “there are one or two unusual features” about the study. Dr. Boulton is a professor of medicine, division of diabetes, endocrinology & gastroenterology, at the University of Manchester (England).
He highlighted that the study was of “basically neuropathic ulcers, many of which were plantar and should be able to heal without any specific additional therapy.”
In addition, the inclusion criteria state that the ulcers could be below the malleoli or 5 cm above them, which Dr. Boulton explained is “very unusual and would therefore include some atypical and not truly diabetic ‘foot’ ulcers.”
And Frances Game, MBBCh, department of diabetes and endocrinology, University Hospitals of Derby (England) and Burton NHS Foundation Trust, added that there are questions about the study methodology.
She said in an interview that although it is a “fascinating study,” the main comparison group did not receive vehicle, or placebo, gel in addition to standard of care. “How were they blinded [to treatment]?”
The “biggest problem” with the study, however, is that the primary outcome was reported as a per-protocol endpoint, not as a standard intention-to-treat analysis, which allowed the researchers to exclude patients whose ulcers increased in size by over 30% on two consecutive visits.
“That kind of makes [esmolol gel] look better than it is because they’ve taken out the ones who got worse,” Dr. Game noted. However, the findings, while not conclusive, do warrant further study of esmolol gel.
The authors noted that diabetic foot ulcers are a severe complication of diabetes, with a prevalence of 1.3%-12.0% across various countries, And the complication contributes to patient morbidity and mortality, with a 5-year mortality that is substantially higher than that of many cancers.
Moreover, “even with the best therapy,” such as advanced moist wound therapy, bioengineered tissue or skin substitutes, peptides, growth factors, electric stimulation, and negative-pressure wound therapy, just 30% of wounds linked to diabetes heal and recurrence is as high as 70%.
Against this backdrop, topical esmolol 14% gel was shown in a phase 1/2 study to be associated with ulcer area reduction and earlier wound closure versus standard of care plus a control vehicle gel.
The current phase 3, randomized, controlled trial involved individuals aged 18-75 years with type 1 or type 2 diabetes and noninfected diabetic foot ulcers classified as grade 1A and 1C on the University of Texas Wound Classification System, which had been open for at least 6 weeks and had an area of 2-25 cm2.
Patients from 27 tertiary care centers across India were enrolled in 2018-2020. They were randomized in a 3:3:1 ratio to one of three groups: esmolol 14% gel plus standard of care, standard of care only, or vehicle plus standard of care.
The study lasted 25 weeks and included a 1-week screening phase, during which all patients received standard of care, a 12-week treatment phase, and a 12-week follow-up phase. The latter included a closure confirmation period of 4 weeks and an observation period of 8 weeks.
Patients were assessed once a week during the treatment phase, and then at weeks 14, 16, 20, and 24.
In all, 176 patients were enrolled. Participants were a mean age of 56.4 years and 69.3% were men. Average hemoglobin A1c was 8.6%. Mean diabetic foot ulcer area was 4.7 cm2 and the average ulcer duration was 49.8 weeks.
The primary outcome was the proportion of patients who achieved target ulcer closure during the 12-week treatment phase and was assessed in 140 patients.
Overall, 60.3% of patients treated with esmolol gel plus standard of care achieved target ulcer closure versus 41.7% of those in the standard of care alone group (odds ratio, 2.13; P = .03).
The secondary outcome was the proportion of patients with target ulcer closure by the study end and was assessed in 120 patients.
In total, 77.2% of patients in the esmolol gel plus standard of care group met the secondary endpoint, compared with 55.6% of those receiving standard of care alone (OR, 1.72; P = .01).
Further analysis suggested the benefit seen with esmolol gel plus standard of care was greater in patients with a weight greater than 80 kg versus standard of care alone (OR, 4.04; P = .04), and in those with a BMI greater than 25 (OR, 2.72; P = .03).
Treatment-emergent adverse events were reported by 33 (18.8%) participants, with 12 events deemed serious. “However, none of the serious adverse events were considered as drug-related by the investigators,” concluded the researchers.
The study was partly funded by NovaLead Pharma and the Biotechnology Industry Research Assistance Council, New Delhi, set up by the Department of Biotechnology, Government of India. Dr. Rastogi reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM JAMA NETWORK OPEN
FDA OKs first-ever topical gene therapy, for rare skin disease
(DEB), a rare skin disease.

The therapy, Vyjuvek (beremagene geperpavec, formerly known as B-VEC), uses a nonreplicating herpes simplex virus type 1 (HSV-1) vector to deliver the COL7A1 gene directly to skin cells, restoring the COL7 protein fibrils that stabilize skin structure.
Vyjuvek, developed by Krystal Biotech, is designed to be used repetitively to heal a single wound or on more than one wound. In the pivotal study, the gene therapy, delivered in a topical gel, was administered once a week for 6 months.
The FDA gave Vyjuvek priority review, approving the therapy just 9 months after Krystal filed its application for approval. Vyjuvek is also an orphan drug, giving it potentially 7 years of marketing exclusivity.
“Vyjuvek is the first FDA-approved gene therapy treatment for DEB, a rare and serious genetic skin disorder,” said Peter Marks, MD, PhD, director of the FDA’s Center for Biologics Evaluation and Research, in the FDA’s statement announcing the approval.
“With the FDA approval of Vyjuvek, the DEB population has reached a monumental milestone in the treatment of this horrible disorder,” said Brett Kopelan, executive director of debra of America, a national advocacy group for people with DEB, in a statement issued by Krystal Biotech. “Our hopes have now been realized for a safe and effective treatment for one of the most devastating symptoms of the disorder,” said Mr. Kopelan.
“This is a devastating disease,” said M. Peter Marinkovich, MD, primary investigator of Krystal’s pivotal GEM-3 trial and director of the Blistering Disease Clinic at Stanford Health Care, in the statement issued by Krystal. “Until now, doctors and nurses had no way to stop blisters and wounds from developing on dystrophic EB patient skin, and all we could do was to give them bandages and helplessly watch as new blisters formed,” said Dr. Marinkovich, who is also associate professor of dermatology at Stanford (Calif.) University School of Medicine.
“Because it’s safe and easy to apply directly to wounds, it doesn’t require a lot of supporting technology or specialized expertise, making Vyjuvek highly accessible even to patients who live far away from specialized centers,” he said.
Paras P. Vakharia, MD, PharmD, assistant professor of dermatology at Northwestern University, Chicago, said that Vyjuvek is an important advance for DEB. “This to me would be a treatment that I would consider for almost all patients,” Dr. Vakharia said in an interview.
Dr. Vakharia, who was not involved with trials of Vyjuvek, said he had concerns about whether patients might develop antibodies to either HSV or C7 but that the greater initial worry is if Vyjuvek will be affordable and accessible. “I envision that it will be a costly medication,” he said.
Mr. Kopelan, the patient advocate, said that his organization “will continue to work closely with Krystal to assure patients have ready access to Vyjuvek.”
Krystal did not respond before press time to a request for comment on pricing.
Dystrophic epidermolysis bullosa affects 1 to 3 people per million in the United States. It is caused by mutations in the COL7A1 gene, which encodes the alpha-1 chain of collagen type VII (C7) protein. C7 forms the anchoring fibrils that attach the epidermis to the underlying dermal connective tissue. COL71A mutations that lead to defective, decreased, or absent C7 can make the skin so fragile that it tears with the slightest touch.
DEB usually presents at birth and is divided into two major types depending on the inheritance pattern: recessive dystrophic epidermolysis bullosa and dominant dystrophic epidermolysis bullosa.
People with the dominant form typically have mild cases with blistering primarily on the hands, feet, knees, and elbows. The recessive form can be painful and debilitating, causing widespread blistering. Depending on the severity, it can lead to nonhealing wounds, fusing of fingers and toes, anemia, weak bones, and esophageal and genitourinary strictures. Squamous cell cancers are a frequent cause of death.
Vyjuvek is mixed into an inactive gel and is evenly applied to a wound once a week by a health care professional, according to the FDA.
The agency recommends the following precautions for patients and caregivers:
- Avoid direct contact with treated wounds and dressings of treated wounds for 24 hours following application. Clean any area that is accidentally exposed.
- Wash hands and wear protective gloves when changing wound dressings.
- Disinfect the bandages used for the first dressing with a viricidal agent, such as 70% isopropyl alcohol, 6% hydrogen peroxide, or less than 0.4% ammonium chloride, and dispose of them in a separate, sealed plastic bag in household waste. Subsequent used dressings and cleaning materials should be disposed of in sealed plastic bags in household waste.
FDA approval of Vyjuvek was based on a randomized, double-blinded, placebo-controlled, 31-patient, phase 3 trial published in the New England Journal of Medicine, which showed that 71% of wounds treated with the gene therapy had complete healing at 3 months, compared with 20% of those treated with placebo (95% confidence interval, 29-73; P < .001). At 6 months, 67% of wounds treated with the gene therapy had complete healing, compared with 22% of wounds treated with placebo (95% CI, 24-68; P = .002).
Almost two-thirds of the patients had at least one adverse event. Most were mild or moderate. The most common events were pruritus, chills, and squamous cell carcinoma, reported in three patients each. SCC cases occurred at wound sites that had not been exposed to Vyjuvek or placebo. Serious adverse events, which were unrelated to the treatment, occurred in three patients and included diarrhea, anemia, and cellulitis.
Krystal will also be seeking marketing approval for Vyjuvek in the European Union, likely in 2024, said the company. In September, the European Medicines Agency’s Pediatric Committee issued a positive opinion on the gene therapy and Krystal’s plan to test B-VEC in children.
Dr. Marinkovich and Dr. Vakharia have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
(DEB), a rare skin disease.
The therapy, Vyjuvek (beremagene geperpavec, formerly known as B-VEC), uses a nonreplicating herpes simplex virus type 1 (HSV-1) vector to deliver the COL7A1 gene directly to skin cells, restoring the COL7 protein fibrils that stabilize skin structure.
Vyjuvek, developed by Krystal Biotech, is designed to be used repetitively to heal a single wound or on more than one wound. In the pivotal study, the gene therapy, delivered in a topical gel, was administered once a week for 6 months.
The FDA gave Vyjuvek priority review, approving the therapy just 9 months after Krystal filed its application for approval. Vyjuvek is also an orphan drug, giving it potentially 7 years of marketing exclusivity.
“Vyjuvek is the first FDA-approved gene therapy treatment for DEB, a rare and serious genetic skin disorder,” said Peter Marks, MD, PhD, director of the FDA’s Center for Biologics Evaluation and Research, in the FDA’s statement announcing the approval.
“With the FDA approval of Vyjuvek, the DEB population has reached a monumental milestone in the treatment of this horrible disorder,” said Brett Kopelan, executive director of debra of America, a national advocacy group for people with DEB, in a statement issued by Krystal Biotech. “Our hopes have now been realized for a safe and effective treatment for one of the most devastating symptoms of the disorder,” said Mr. Kopelan.
“This is a devastating disease,” said M. Peter Marinkovich, MD, primary investigator of Krystal’s pivotal GEM-3 trial and director of the Blistering Disease Clinic at Stanford Health Care, in the statement issued by Krystal. “Until now, doctors and nurses had no way to stop blisters and wounds from developing on dystrophic EB patient skin, and all we could do was to give them bandages and helplessly watch as new blisters formed,” said Dr. Marinkovich, who is also associate professor of dermatology at Stanford (Calif.) University School of Medicine.
“Because it’s safe and easy to apply directly to wounds, it doesn’t require a lot of supporting technology or specialized expertise, making Vyjuvek highly accessible even to patients who live far away from specialized centers,” he said.
Paras P. Vakharia, MD, PharmD, assistant professor of dermatology at Northwestern University, Chicago, said that Vyjuvek is an important advance for DEB. “This to me would be a treatment that I would consider for almost all patients,” Dr. Vakharia said in an interview.
Dr. Vakharia, who was not involved with trials of Vyjuvek, said he had concerns about whether patients might develop antibodies to either HSV or C7 but that the greater initial worry is if Vyjuvek will be affordable and accessible. “I envision that it will be a costly medication,” he said.
Mr. Kopelan, the patient advocate, said that his organization “will continue to work closely with Krystal to assure patients have ready access to Vyjuvek.”
Krystal did not respond before press time to a request for comment on pricing.
Dystrophic epidermolysis bullosa affects 1 to 3 people per million in the United States. It is caused by mutations in the COL7A1 gene, which encodes the alpha-1 chain of collagen type VII (C7) protein. C7 forms the anchoring fibrils that attach the epidermis to the underlying dermal connective tissue. COL71A mutations that lead to defective, decreased, or absent C7 can make the skin so fragile that it tears with the slightest touch.
DEB usually presents at birth and is divided into two major types depending on the inheritance pattern: recessive dystrophic epidermolysis bullosa and dominant dystrophic epidermolysis bullosa.
People with the dominant form typically have mild cases with blistering primarily on the hands, feet, knees, and elbows. The recessive form can be painful and debilitating, causing widespread blistering. Depending on the severity, it can lead to nonhealing wounds, fusing of fingers and toes, anemia, weak bones, and esophageal and genitourinary strictures. Squamous cell cancers are a frequent cause of death.
Vyjuvek is mixed into an inactive gel and is evenly applied to a wound once a week by a health care professional, according to the FDA.
The agency recommends the following precautions for patients and caregivers:
- Avoid direct contact with treated wounds and dressings of treated wounds for 24 hours following application. Clean any area that is accidentally exposed.
- Wash hands and wear protective gloves when changing wound dressings.
- Disinfect the bandages used for the first dressing with a viricidal agent, such as 70% isopropyl alcohol, 6% hydrogen peroxide, or less than 0.4% ammonium chloride, and dispose of them in a separate, sealed plastic bag in household waste. Subsequent used dressings and cleaning materials should be disposed of in sealed plastic bags in household waste.
FDA approval of Vyjuvek was based on a randomized, double-blinded, placebo-controlled, 31-patient, phase 3 trial published in the New England Journal of Medicine, which showed that 71% of wounds treated with the gene therapy had complete healing at 3 months, compared with 20% of those treated with placebo (95% confidence interval, 29-73; P < .001). At 6 months, 67% of wounds treated with the gene therapy had complete healing, compared with 22% of wounds treated with placebo (95% CI, 24-68; P = .002).
Almost two-thirds of the patients had at least one adverse event. Most were mild or moderate. The most common events were pruritus, chills, and squamous cell carcinoma, reported in three patients each. SCC cases occurred at wound sites that had not been exposed to Vyjuvek or placebo. Serious adverse events, which were unrelated to the treatment, occurred in three patients and included diarrhea, anemia, and cellulitis.
Krystal will also be seeking marketing approval for Vyjuvek in the European Union, likely in 2024, said the company. In September, the European Medicines Agency’s Pediatric Committee issued a positive opinion on the gene therapy and Krystal’s plan to test B-VEC in children.
Dr. Marinkovich and Dr. Vakharia have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
(DEB), a rare skin disease.
The therapy, Vyjuvek (beremagene geperpavec, formerly known as B-VEC), uses a nonreplicating herpes simplex virus type 1 (HSV-1) vector to deliver the COL7A1 gene directly to skin cells, restoring the COL7 protein fibrils that stabilize skin structure.
Vyjuvek, developed by Krystal Biotech, is designed to be used repetitively to heal a single wound or on more than one wound. In the pivotal study, the gene therapy, delivered in a topical gel, was administered once a week for 6 months.
The FDA gave Vyjuvek priority review, approving the therapy just 9 months after Krystal filed its application for approval. Vyjuvek is also an orphan drug, giving it potentially 7 years of marketing exclusivity.
“Vyjuvek is the first FDA-approved gene therapy treatment for DEB, a rare and serious genetic skin disorder,” said Peter Marks, MD, PhD, director of the FDA’s Center for Biologics Evaluation and Research, in the FDA’s statement announcing the approval.
“With the FDA approval of Vyjuvek, the DEB population has reached a monumental milestone in the treatment of this horrible disorder,” said Brett Kopelan, executive director of debra of America, a national advocacy group for people with DEB, in a statement issued by Krystal Biotech. “Our hopes have now been realized for a safe and effective treatment for one of the most devastating symptoms of the disorder,” said Mr. Kopelan.
“This is a devastating disease,” said M. Peter Marinkovich, MD, primary investigator of Krystal’s pivotal GEM-3 trial and director of the Blistering Disease Clinic at Stanford Health Care, in the statement issued by Krystal. “Until now, doctors and nurses had no way to stop blisters and wounds from developing on dystrophic EB patient skin, and all we could do was to give them bandages and helplessly watch as new blisters formed,” said Dr. Marinkovich, who is also associate professor of dermatology at Stanford (Calif.) University School of Medicine.
“Because it’s safe and easy to apply directly to wounds, it doesn’t require a lot of supporting technology or specialized expertise, making Vyjuvek highly accessible even to patients who live far away from specialized centers,” he said.
Paras P. Vakharia, MD, PharmD, assistant professor of dermatology at Northwestern University, Chicago, said that Vyjuvek is an important advance for DEB. “This to me would be a treatment that I would consider for almost all patients,” Dr. Vakharia said in an interview.
Dr. Vakharia, who was not involved with trials of Vyjuvek, said he had concerns about whether patients might develop antibodies to either HSV or C7 but that the greater initial worry is if Vyjuvek will be affordable and accessible. “I envision that it will be a costly medication,” he said.
Mr. Kopelan, the patient advocate, said that his organization “will continue to work closely with Krystal to assure patients have ready access to Vyjuvek.”
Krystal did not respond before press time to a request for comment on pricing.
Dystrophic epidermolysis bullosa affects 1 to 3 people per million in the United States. It is caused by mutations in the COL7A1 gene, which encodes the alpha-1 chain of collagen type VII (C7) protein. C7 forms the anchoring fibrils that attach the epidermis to the underlying dermal connective tissue. COL71A mutations that lead to defective, decreased, or absent C7 can make the skin so fragile that it tears with the slightest touch.
DEB usually presents at birth and is divided into two major types depending on the inheritance pattern: recessive dystrophic epidermolysis bullosa and dominant dystrophic epidermolysis bullosa.
People with the dominant form typically have mild cases with blistering primarily on the hands, feet, knees, and elbows. The recessive form can be painful and debilitating, causing widespread blistering. Depending on the severity, it can lead to nonhealing wounds, fusing of fingers and toes, anemia, weak bones, and esophageal and genitourinary strictures. Squamous cell cancers are a frequent cause of death.
Vyjuvek is mixed into an inactive gel and is evenly applied to a wound once a week by a health care professional, according to the FDA.
The agency recommends the following precautions for patients and caregivers:
- Avoid direct contact with treated wounds and dressings of treated wounds for 24 hours following application. Clean any area that is accidentally exposed.
- Wash hands and wear protective gloves when changing wound dressings.
- Disinfect the bandages used for the first dressing with a viricidal agent, such as 70% isopropyl alcohol, 6% hydrogen peroxide, or less than 0.4% ammonium chloride, and dispose of them in a separate, sealed plastic bag in household waste. Subsequent used dressings and cleaning materials should be disposed of in sealed plastic bags in household waste.
FDA approval of Vyjuvek was based on a randomized, double-blinded, placebo-controlled, 31-patient, phase 3 trial published in the New England Journal of Medicine, which showed that 71% of wounds treated with the gene therapy had complete healing at 3 months, compared with 20% of those treated with placebo (95% confidence interval, 29-73; P < .001). At 6 months, 67% of wounds treated with the gene therapy had complete healing, compared with 22% of wounds treated with placebo (95% CI, 24-68; P = .002).
Almost two-thirds of the patients had at least one adverse event. Most were mild or moderate. The most common events were pruritus, chills, and squamous cell carcinoma, reported in three patients each. SCC cases occurred at wound sites that had not been exposed to Vyjuvek or placebo. Serious adverse events, which were unrelated to the treatment, occurred in three patients and included diarrhea, anemia, and cellulitis.
Krystal will also be seeking marketing approval for Vyjuvek in the European Union, likely in 2024, said the company. In September, the European Medicines Agency’s Pediatric Committee issued a positive opinion on the gene therapy and Krystal’s plan to test B-VEC in children.
Dr. Marinkovich and Dr. Vakharia have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
A healthy 36-year-old female presented with 4 days of itchy lesions on the right upper extremity
Additionally, Orthopox DNA by PCR and Monkeypox (mpox) virus DNA by PCR were detected. Herpes simplex virus and bacterial viral cultures were negative. Valacyclovir was started at the time of presentation and the patient’s lesions resolved without sequelae.
Mpox is a zoonotic double-stranded DNA virus that is part of the Orthopoxvirus family, including the West African and Central African variants. This disease presents similarly to smallpox, so most mpox research was conducted around the time smallpox was eradicated. It was not until 1970, when the disease was isolated from a patient with suspected smallpox in the Democratic Republic of the Congo (DRC), that human mpox was considered a distinct disease. An epidemic outbreak in the United States occurred in 2003 related to infected prairie dogs, and travel-related outbreaks have been more recently reported up until May 2022, in which mpox was reported in nonendemic areas including North America, Europe, and Australia. Most cases in this outbreak occurred in men who have sex with men (MSM), but this is not always the case, and mpox is not necessarily considered a sexually transmitted infection. Mpox presents similarly to smallpox and VZV, so using laboratory tests is important in diagnosing and tracking this disease.
Although it is not easily transmitted, the disease can spread through bodily secretions both directly and indirectly. Mpox typically begins with a prodrome that includes fever, headache, myalgia, and fatigue. This is followed by lymphadenopathy that precedes and coincides with rash development. The lymph nodes are firm, tender, may be painful, and are a defining factor in presentation that differs from smallpox and varicella. The rash typically starts on the face, then presents on the body in a centrifugal distribution. However, cases related to sexual transmission present with anogenital lesions. The lesions are characterized by a progression from maculopapular to vesiculopustular, and can vary widely in quantity.
Notably, individuals are contagious from the onset of the prodrome until the lesions have scabbed over and fallen off. The eruptive nature of the later lesions poses a threat of secondary infection, and is often accompanied by a second febrile period that signifies deterioration of the patient’s condition. Other signs of secondary infection are variable and include pulmonary symptoms, vomiting, diarrhea, ocular infections, and in rare cases, encephalitis. These sequelae are more common in unvaccinated and immunocompromised individuals. Long-term complications of mpox include pitted scarring from cutaneous lesions with children being more susceptible to severe disease. The mortality rate for the disease is very low. (As of May 10, 2023, there have been 30,395 mpox cases reported in the United States, and 42 deaths, according to the Centers for Disease Control and Prevention.)
There are a variety of diagnostic tests that can aid in mpox identification, but they are most strongly supported when combined with clinical and epidemiological data. The best, least invasive method includes collection of lesion exudate or crust on a swab, and viral DNA is best preserved by keeping the specimen in a cool, dry, and dark environment. PCR is considered the standard, and electron microscopy and immunohistochemistry are valid tests, but all modalities require sophisticated technicians with the proper laboratory equipment. This is limiting because many cases present in underserved areas that lack the facilities for proper, real-time analysis. Antigen and antibody-based tests can be used, but cross-reactivity of other orthopoxviridae limits confirmation of mpox infection. Vaccination status, history and location must be considered.
Vaccination is the chief form of prevention for mpox, although it is not considered entirely protective. Smallpox vaccination provides protection, but widespread administration of the vaccine is no longer practiced, and an estimated 70% of the global population is no longer vaccinated. Vaccination is recommended for anyone at risk of exposure, but as this is a live, attenuated vaccine, the immune status of the patient is important to keep in mind. Tecovirimat and other antiviral medications including cidofovir and brincidofovir may be considered in severe cases.
This case is unique as our patient, who had no known risk factors for mpox, presented with mpox and VZV, simultaneously. Although clinical presentation and epidemiological patterns between these diseases differ, there have been a limited number of cases of coinfection reported in the literature, mainly in the DRC where mpox is endemic. Diagnosis must be made by separate laboratory tests and there are differences in presentation between independent and coinfection for these viruses. Notably, patients with mpox/VZV coinfection may be less likely to present with lesions on the face, thorax, arms, palms, and soles than those with only mpox but experience a higher lesion burden than those afflicted by only VZV. Coinfection may be related to reactivation of dormant VZV, or increased susceptibility to secondary infection when infected with one virus.
This case and photo were submitted by Lucas Shapiro, BS, of the Dr. Kiran C. Patel College of Osteopathic Medicine at Nova Southeastern University, Fort Lauderdale, Fla., and Donna Bilu Martin, MD.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
1. Macneil A et al. Clin Infect Dis. 2009 Jan 1;48(1):e6-8.
2. Di Gennaro F et al. Microorganisms. 2022 Aug 12;10(8):1633.
3. Hughes CM et al. Am J Trop Med Hyg. 2020 Dec 7;104(2):604-11.
Additionally, Orthopox DNA by PCR and Monkeypox (mpox) virus DNA by PCR were detected. Herpes simplex virus and bacterial viral cultures were negative. Valacyclovir was started at the time of presentation and the patient’s lesions resolved without sequelae.
Mpox is a zoonotic double-stranded DNA virus that is part of the Orthopoxvirus family, including the West African and Central African variants. This disease presents similarly to smallpox, so most mpox research was conducted around the time smallpox was eradicated. It was not until 1970, when the disease was isolated from a patient with suspected smallpox in the Democratic Republic of the Congo (DRC), that human mpox was considered a distinct disease. An epidemic outbreak in the United States occurred in 2003 related to infected prairie dogs, and travel-related outbreaks have been more recently reported up until May 2022, in which mpox was reported in nonendemic areas including North America, Europe, and Australia. Most cases in this outbreak occurred in men who have sex with men (MSM), but this is not always the case, and mpox is not necessarily considered a sexually transmitted infection. Mpox presents similarly to smallpox and VZV, so using laboratory tests is important in diagnosing and tracking this disease.
Although it is not easily transmitted, the disease can spread through bodily secretions both directly and indirectly. Mpox typically begins with a prodrome that includes fever, headache, myalgia, and fatigue. This is followed by lymphadenopathy that precedes and coincides with rash development. The lymph nodes are firm, tender, may be painful, and are a defining factor in presentation that differs from smallpox and varicella. The rash typically starts on the face, then presents on the body in a centrifugal distribution. However, cases related to sexual transmission present with anogenital lesions. The lesions are characterized by a progression from maculopapular to vesiculopustular, and can vary widely in quantity.
Notably, individuals are contagious from the onset of the prodrome until the lesions have scabbed over and fallen off. The eruptive nature of the later lesions poses a threat of secondary infection, and is often accompanied by a second febrile period that signifies deterioration of the patient’s condition. Other signs of secondary infection are variable and include pulmonary symptoms, vomiting, diarrhea, ocular infections, and in rare cases, encephalitis. These sequelae are more common in unvaccinated and immunocompromised individuals. Long-term complications of mpox include pitted scarring from cutaneous lesions with children being more susceptible to severe disease. The mortality rate for the disease is very low. (As of May 10, 2023, there have been 30,395 mpox cases reported in the United States, and 42 deaths, according to the Centers for Disease Control and Prevention.)
There are a variety of diagnostic tests that can aid in mpox identification, but they are most strongly supported when combined with clinical and epidemiological data. The best, least invasive method includes collection of lesion exudate or crust on a swab, and viral DNA is best preserved by keeping the specimen in a cool, dry, and dark environment. PCR is considered the standard, and electron microscopy and immunohistochemistry are valid tests, but all modalities require sophisticated technicians with the proper laboratory equipment. This is limiting because many cases present in underserved areas that lack the facilities for proper, real-time analysis. Antigen and antibody-based tests can be used, but cross-reactivity of other orthopoxviridae limits confirmation of mpox infection. Vaccination status, history and location must be considered.
Vaccination is the chief form of prevention for mpox, although it is not considered entirely protective. Smallpox vaccination provides protection, but widespread administration of the vaccine is no longer practiced, and an estimated 70% of the global population is no longer vaccinated. Vaccination is recommended for anyone at risk of exposure, but as this is a live, attenuated vaccine, the immune status of the patient is important to keep in mind. Tecovirimat and other antiviral medications including cidofovir and brincidofovir may be considered in severe cases.
This case is unique as our patient, who had no known risk factors for mpox, presented with mpox and VZV, simultaneously. Although clinical presentation and epidemiological patterns between these diseases differ, there have been a limited number of cases of coinfection reported in the literature, mainly in the DRC where mpox is endemic. Diagnosis must be made by separate laboratory tests and there are differences in presentation between independent and coinfection for these viruses. Notably, patients with mpox/VZV coinfection may be less likely to present with lesions on the face, thorax, arms, palms, and soles than those with only mpox but experience a higher lesion burden than those afflicted by only VZV. Coinfection may be related to reactivation of dormant VZV, or increased susceptibility to secondary infection when infected with one virus.
This case and photo were submitted by Lucas Shapiro, BS, of the Dr. Kiran C. Patel College of Osteopathic Medicine at Nova Southeastern University, Fort Lauderdale, Fla., and Donna Bilu Martin, MD.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
1. Macneil A et al. Clin Infect Dis. 2009 Jan 1;48(1):e6-8.
2. Di Gennaro F et al. Microorganisms. 2022 Aug 12;10(8):1633.
3. Hughes CM et al. Am J Trop Med Hyg. 2020 Dec 7;104(2):604-11.
Additionally, Orthopox DNA by PCR and Monkeypox (mpox) virus DNA by PCR were detected. Herpes simplex virus and bacterial viral cultures were negative. Valacyclovir was started at the time of presentation and the patient’s lesions resolved without sequelae.
Mpox is a zoonotic double-stranded DNA virus that is part of the Orthopoxvirus family, including the West African and Central African variants. This disease presents similarly to smallpox, so most mpox research was conducted around the time smallpox was eradicated. It was not until 1970, when the disease was isolated from a patient with suspected smallpox in the Democratic Republic of the Congo (DRC), that human mpox was considered a distinct disease. An epidemic outbreak in the United States occurred in 2003 related to infected prairie dogs, and travel-related outbreaks have been more recently reported up until May 2022, in which mpox was reported in nonendemic areas including North America, Europe, and Australia. Most cases in this outbreak occurred in men who have sex with men (MSM), but this is not always the case, and mpox is not necessarily considered a sexually transmitted infection. Mpox presents similarly to smallpox and VZV, so using laboratory tests is important in diagnosing and tracking this disease.
Although it is not easily transmitted, the disease can spread through bodily secretions both directly and indirectly. Mpox typically begins with a prodrome that includes fever, headache, myalgia, and fatigue. This is followed by lymphadenopathy that precedes and coincides with rash development. The lymph nodes are firm, tender, may be painful, and are a defining factor in presentation that differs from smallpox and varicella. The rash typically starts on the face, then presents on the body in a centrifugal distribution. However, cases related to sexual transmission present with anogenital lesions. The lesions are characterized by a progression from maculopapular to vesiculopustular, and can vary widely in quantity.
Notably, individuals are contagious from the onset of the prodrome until the lesions have scabbed over and fallen off. The eruptive nature of the later lesions poses a threat of secondary infection, and is often accompanied by a second febrile period that signifies deterioration of the patient’s condition. Other signs of secondary infection are variable and include pulmonary symptoms, vomiting, diarrhea, ocular infections, and in rare cases, encephalitis. These sequelae are more common in unvaccinated and immunocompromised individuals. Long-term complications of mpox include pitted scarring from cutaneous lesions with children being more susceptible to severe disease. The mortality rate for the disease is very low. (As of May 10, 2023, there have been 30,395 mpox cases reported in the United States, and 42 deaths, according to the Centers for Disease Control and Prevention.)
There are a variety of diagnostic tests that can aid in mpox identification, but they are most strongly supported when combined with clinical and epidemiological data. The best, least invasive method includes collection of lesion exudate or crust on a swab, and viral DNA is best preserved by keeping the specimen in a cool, dry, and dark environment. PCR is considered the standard, and electron microscopy and immunohistochemistry are valid tests, but all modalities require sophisticated technicians with the proper laboratory equipment. This is limiting because many cases present in underserved areas that lack the facilities for proper, real-time analysis. Antigen and antibody-based tests can be used, but cross-reactivity of other orthopoxviridae limits confirmation of mpox infection. Vaccination status, history and location must be considered.
Vaccination is the chief form of prevention for mpox, although it is not considered entirely protective. Smallpox vaccination provides protection, but widespread administration of the vaccine is no longer practiced, and an estimated 70% of the global population is no longer vaccinated. Vaccination is recommended for anyone at risk of exposure, but as this is a live, attenuated vaccine, the immune status of the patient is important to keep in mind. Tecovirimat and other antiviral medications including cidofovir and brincidofovir may be considered in severe cases.
This case is unique as our patient, who had no known risk factors for mpox, presented with mpox and VZV, simultaneously. Although clinical presentation and epidemiological patterns between these diseases differ, there have been a limited number of cases of coinfection reported in the literature, mainly in the DRC where mpox is endemic. Diagnosis must be made by separate laboratory tests and there are differences in presentation between independent and coinfection for these viruses. Notably, patients with mpox/VZV coinfection may be less likely to present with lesions on the face, thorax, arms, palms, and soles than those with only mpox but experience a higher lesion burden than those afflicted by only VZV. Coinfection may be related to reactivation of dormant VZV, or increased susceptibility to secondary infection when infected with one virus.
This case and photo were submitted by Lucas Shapiro, BS, of the Dr. Kiran C. Patel College of Osteopathic Medicine at Nova Southeastern University, Fort Lauderdale, Fla., and Donna Bilu Martin, MD.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
1. Macneil A et al. Clin Infect Dis. 2009 Jan 1;48(1):e6-8.
2. Di Gennaro F et al. Microorganisms. 2022 Aug 12;10(8):1633.
3. Hughes CM et al. Am J Trop Med Hyg. 2020 Dec 7;104(2):604-11.
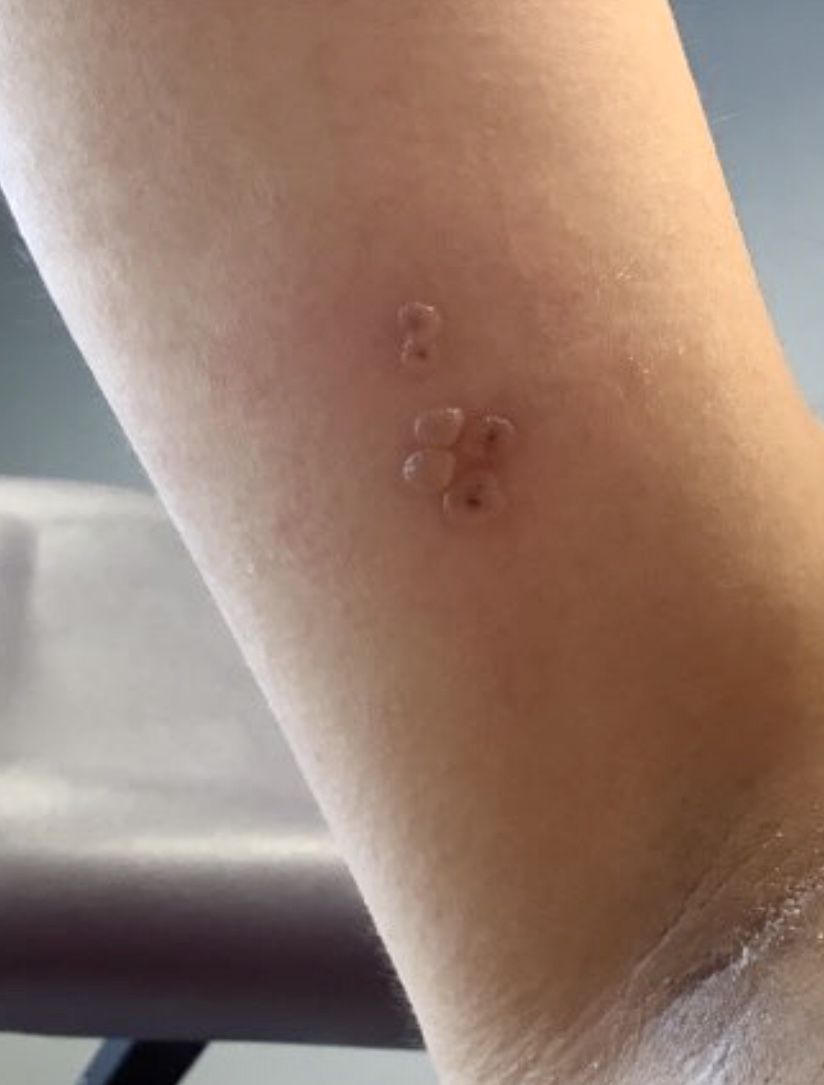
Two phase 3 trials show benefits of dupilumab for prurigo nodularis
The results, which were published online in Nature Medicine, were the basis for the FDA approval of dupilumab (Dupixent) for adults with PN in September 2022, the first treatment approved for treating PN in the United States.
“These positive studies support the involvement of type 2 cytokines in driving PN disease pathogenesis and the targeting of the [interleukin]-4/IL-13 axis as a novel therapeutic paradigm for patients with PN,” wrote the researchers, who were led by principal investigator Gil Yosipovitch, MD, professor of dermatology at the University of Miami, Fla. Dupilumab, an IL-4 receptor alpha antagonist, blocks the shared receptor component (IL-4R alpha) for IL-4 and IL-13.
For the two phase 3 trials, which were called LIBERTY-PN PRIME and PRIME2 and were sponsored by Sanofi and Regeneron Pharmaceuticals, researchers randomized adults with PN with 20 or more nodules and severe itch uncontrolled with topical therapies 1:1 to 300 mg dupilumab or placebo subcutaneously every 2 weeks for 24 weeks. The primary endpoint was pruritus improvement, which was measured by the proportion of patients with a 4-point or greater reduction in Worst Itch Numeric Rating Scale (WI-NRS) from baseline at week 24 (PRIME) or week 12 (PRIME2). Key secondary endpoints included a reduction in the number of nodules to 5 or fewer at week 24.
PRIME and PRIME2 enrolled 151 and 160 patients, respectively. In PRIME, 60% of patients in the dupilumab arm achieved a 4-point or greater reduction in the WI-NRS at week 24, compared with 18.4% of patients in the placebo arm (P < .001). In PRIME2, 37.2% of patients in the dupilumab arm achieved a 4-point or greater reduction in the WI-NRS at week 12, compared with 22% of patients in the placebo arm (P = .022).
The researchers also reported that, from an initial baseline of 20 to greater than 100 nodules, 32.0% of dupilumab-treated patients in PRIME and 25.6% in PRIME2 showed a reduction to 5 nodules or fewer, which corresponded to a response of “clear” or “almost clear” skin at week 12, compared with 11.8% and 12.2% of placebo-treated patients, respectively. This treatment effect on skin lesions continued to improve after week 12, with 48% of dupilumab-treated patients in PRIME and 44.9% in PRIME2 having five nodules or fewer at week 24, compared with 18.4% and 15.9% of placebo-treated patients, respectively. Safety was consistent with the known dupilumab safety profile.
“Validation is the first success of this paper,” said Adam Friedman, MD, professor and chair of dermatology at George Washington University, Washington, who was asked to comment on the study. “While both the safety and efficacy of dupilumab in these two phase 3 programs is the meat of the matter, nuanced highlights for me include the rigid nature of the exclusion criteria to ensure a study population that truly has PN as a stand-alone disease, rather than a secondary finding as we once believed to be the entire story. I think it’s important for us to recognize that it’s not one or the other, rather there is both ‘primary’ prurigo nodularis, and then there is secondary prurigo nodularis associated with something else [a wide range of underlying medical conditions], just like we divide primary and secondary hyperhidrosis.”
Dr. Yosipovitch reported having competing interests with several pharmaceutical companies, including Regeneron and Sanofi. Dr. Friedman disclosed that he is a consultant to and a speaker for Regeneron.
The results, which were published online in Nature Medicine, were the basis for the FDA approval of dupilumab (Dupixent) for adults with PN in September 2022, the first treatment approved for treating PN in the United States.
“These positive studies support the involvement of type 2 cytokines in driving PN disease pathogenesis and the targeting of the [interleukin]-4/IL-13 axis as a novel therapeutic paradigm for patients with PN,” wrote the researchers, who were led by principal investigator Gil Yosipovitch, MD, professor of dermatology at the University of Miami, Fla. Dupilumab, an IL-4 receptor alpha antagonist, blocks the shared receptor component (IL-4R alpha) for IL-4 and IL-13.
For the two phase 3 trials, which were called LIBERTY-PN PRIME and PRIME2 and were sponsored by Sanofi and Regeneron Pharmaceuticals, researchers randomized adults with PN with 20 or more nodules and severe itch uncontrolled with topical therapies 1:1 to 300 mg dupilumab or placebo subcutaneously every 2 weeks for 24 weeks. The primary endpoint was pruritus improvement, which was measured by the proportion of patients with a 4-point or greater reduction in Worst Itch Numeric Rating Scale (WI-NRS) from baseline at week 24 (PRIME) or week 12 (PRIME2). Key secondary endpoints included a reduction in the number of nodules to 5 or fewer at week 24.
PRIME and PRIME2 enrolled 151 and 160 patients, respectively. In PRIME, 60% of patients in the dupilumab arm achieved a 4-point or greater reduction in the WI-NRS at week 24, compared with 18.4% of patients in the placebo arm (P < .001). In PRIME2, 37.2% of patients in the dupilumab arm achieved a 4-point or greater reduction in the WI-NRS at week 12, compared with 22% of patients in the placebo arm (P = .022).
The researchers also reported that, from an initial baseline of 20 to greater than 100 nodules, 32.0% of dupilumab-treated patients in PRIME and 25.6% in PRIME2 showed a reduction to 5 nodules or fewer, which corresponded to a response of “clear” or “almost clear” skin at week 12, compared with 11.8% and 12.2% of placebo-treated patients, respectively. This treatment effect on skin lesions continued to improve after week 12, with 48% of dupilumab-treated patients in PRIME and 44.9% in PRIME2 having five nodules or fewer at week 24, compared with 18.4% and 15.9% of placebo-treated patients, respectively. Safety was consistent with the known dupilumab safety profile.
“Validation is the first success of this paper,” said Adam Friedman, MD, professor and chair of dermatology at George Washington University, Washington, who was asked to comment on the study. “While both the safety and efficacy of dupilumab in these two phase 3 programs is the meat of the matter, nuanced highlights for me include the rigid nature of the exclusion criteria to ensure a study population that truly has PN as a stand-alone disease, rather than a secondary finding as we once believed to be the entire story. I think it’s important for us to recognize that it’s not one or the other, rather there is both ‘primary’ prurigo nodularis, and then there is secondary prurigo nodularis associated with something else [a wide range of underlying medical conditions], just like we divide primary and secondary hyperhidrosis.”
Dr. Yosipovitch reported having competing interests with several pharmaceutical companies, including Regeneron and Sanofi. Dr. Friedman disclosed that he is a consultant to and a speaker for Regeneron.
The results, which were published online in Nature Medicine, were the basis for the FDA approval of dupilumab (Dupixent) for adults with PN in September 2022, the first treatment approved for treating PN in the United States.
“These positive studies support the involvement of type 2 cytokines in driving PN disease pathogenesis and the targeting of the [interleukin]-4/IL-13 axis as a novel therapeutic paradigm for patients with PN,” wrote the researchers, who were led by principal investigator Gil Yosipovitch, MD, professor of dermatology at the University of Miami, Fla. Dupilumab, an IL-4 receptor alpha antagonist, blocks the shared receptor component (IL-4R alpha) for IL-4 and IL-13.
For the two phase 3 trials, which were called LIBERTY-PN PRIME and PRIME2 and were sponsored by Sanofi and Regeneron Pharmaceuticals, researchers randomized adults with PN with 20 or more nodules and severe itch uncontrolled with topical therapies 1:1 to 300 mg dupilumab or placebo subcutaneously every 2 weeks for 24 weeks. The primary endpoint was pruritus improvement, which was measured by the proportion of patients with a 4-point or greater reduction in Worst Itch Numeric Rating Scale (WI-NRS) from baseline at week 24 (PRIME) or week 12 (PRIME2). Key secondary endpoints included a reduction in the number of nodules to 5 or fewer at week 24.
PRIME and PRIME2 enrolled 151 and 160 patients, respectively. In PRIME, 60% of patients in the dupilumab arm achieved a 4-point or greater reduction in the WI-NRS at week 24, compared with 18.4% of patients in the placebo arm (P < .001). In PRIME2, 37.2% of patients in the dupilumab arm achieved a 4-point or greater reduction in the WI-NRS at week 12, compared with 22% of patients in the placebo arm (P = .022).
The researchers also reported that, from an initial baseline of 20 to greater than 100 nodules, 32.0% of dupilumab-treated patients in PRIME and 25.6% in PRIME2 showed a reduction to 5 nodules or fewer, which corresponded to a response of “clear” or “almost clear” skin at week 12, compared with 11.8% and 12.2% of placebo-treated patients, respectively. This treatment effect on skin lesions continued to improve after week 12, with 48% of dupilumab-treated patients in PRIME and 44.9% in PRIME2 having five nodules or fewer at week 24, compared with 18.4% and 15.9% of placebo-treated patients, respectively. Safety was consistent with the known dupilumab safety profile.
“Validation is the first success of this paper,” said Adam Friedman, MD, professor and chair of dermatology at George Washington University, Washington, who was asked to comment on the study. “While both the safety and efficacy of dupilumab in these two phase 3 programs is the meat of the matter, nuanced highlights for me include the rigid nature of the exclusion criteria to ensure a study population that truly has PN as a stand-alone disease, rather than a secondary finding as we once believed to be the entire story. I think it’s important for us to recognize that it’s not one or the other, rather there is both ‘primary’ prurigo nodularis, and then there is secondary prurigo nodularis associated with something else [a wide range of underlying medical conditions], just like we divide primary and secondary hyperhidrosis.”
Dr. Yosipovitch reported having competing interests with several pharmaceutical companies, including Regeneron and Sanofi. Dr. Friedman disclosed that he is a consultant to and a speaker for Regeneron.
FROM NATURE MEDICINE