User login
Prodrug infusion beats oral Parkinson’s disease therapy for motor symptoms
, according to a new study. The beneficial effects of these phosphate prodrugs of levodopa and carbidopa were most noticeable in the early morning, results of the phase 1B study showed.
As Parkinson’s disease progresses and dosing of oral levodopa/carbidopa (LD/CD) increases, its therapeutic window narrows, resulting in troublesome dyskinesia at peak drug levels and tremors and rigidity when levels fall.
“Foslevodopa/foscarbidopa shows lower ‘off’ time than oral levodopa/carbidopa, and this was statistically significant. Also, foslevodopa/foscarbidopa (fosL/fosC) showed more ‘on’ time without dyskinesia, compared with oral levodopa/carbidopa. This was also statistically significant,” lead author Sven Stodtmann, PhD, of AbbVie GmbH, Ludwigshafen, Germany, reported in his recorded presentation at the Movement Disorders Society’s 23rd International Congress of Parkinson’s Disease and Movement Disorder (Virtual) 2020.
Continuous infusion versus oral therapy
The analysis included 20 patients, and all data from these individuals were collected between 4:30 a.m. and 9:30 p.m.
Participants were 12 men and 8 women, aged 30-80 years, with advanced, idiopathic Parkinson’s disease responsive to levodopa but inadequately controlled on their current stable therapy, having a minimum of 2.5 off hours/day. Mean age was 61.3 plus or minus 10.5 years (range 35-77 years).
In this single-arm, open-label study, they received subcutaneous infusions of personalized therapeutic doses of fosL/fosC 24 hours/day for 28 days after a 10- to 30-day screening period during which they recorded LD/CD doses in a diary and had motor symptoms monitored using a wearable device.
Following the screening period, fosL/fosC doses were titrated over up to 5 days, with subsequent weekly study visits, for a total time on fosL/fosC of 28 days. Drug titration was aimed at maximizing functional on time and minimizing the number of off episodes while minimizing troublesome dyskinesia.
Continuous infusion of fosL/fosC performed better than oral LD/CD on all counts.
“The off time is much lower in the morning for people on foslevodopa/foscarbidopa [compared with oral LD/CD] because this is a 24-hour infusion product,” Dr. Stodtmann explained.
The effect was maintained over the course of the day with little fluctuation with fosL/fosC, off periods never exceeding about 25% between 4:30 a.m. and 9 p.m. For LD/CD, off periods were highest in the early morning and peaked at about 50% on a 3- to 4-hour cycle during the course of the day.
Increased on time without dyskinesia varied between about 60% and 80% during the day with fosL/fosC, showing the greatest difference between fosL/fosC and oral LD/CD in the early morning hours.
“On time with nontroublesome dyskinesia was lower for foscarbidopa/foslevodopa, compared to oral levodopa/carbidopa, but this was not statistically significant,” Dr. Stodtmann said. On time with troublesome dyskinesia followed the same pattern, again, not statistically significant.
Looking at the data another way, the investigators calculated the odds ratios of motor symptoms using fosL/fosC, compared with oral LD/CD. Use of fosL/fosC was associated with a 59% lower risk of being in the off state during the day, compared with oral LD/CD (odds ratio, 0.4; 95% confidence interval, 0.2-0.7; P < .01). Similarly, the probability of being in the on state without dyskinesia was much greater with fosL/fosC (OR, 2.75; 95% CI, 1.08-6.99; P < .05).
Encouraging, but more data needed
Indu Subramanian, MD, of the department of neurology at the University of California, Los Angeles, and director of the Parkinson’s Disease Research, Education, and Clinical Center at the West Los Angeles Veterans Affairs Hospital, commented that the field has been waiting to see data on fosL/fosC.
“It seems like it’s pretty reasonable in terms of what the goals were, which is to improve stability of Parkinson’s symptoms, to improve off time and give on time without troublesome dyskinesia,” she said. “So I think those [goals] have been met.”
Dr. Subramanian, who was not involved with the research, said she would have liked to have seen results concerning safety of this drug formulation, which the presentation lacked, “because historically, there have been issues with nodule formation and skin breakdown, things like that, due to the stability of the product in the subcutaneous form. … So, always to my understanding, there has been this search for things that are tolerated in the subcutaneous delivery.”
If this formulation proves safe and tolerable, Dr. Subramanian sees a potential place for it for some patients with advanced Parkinson’s disease.
“Certainly a subcutaneous formulation will be better than something that requires … deep brain surgery or even a pump insertion like Duopa [carbidopa/levodopa enteral suspension, AbbVie] or something like that,” she said. “I think [it] would be beneficial over something with the gut because the gut historically has been a problem to rely on in advanced Parkinson’s patients due to slower transit times, and the gut itself is affected with Parkinson’s disease.”
Dr. Stodtmann and all coauthors are employees of AbbVie, which was the sponsor of the study and was responsible for all aspects of it. Dr. Subramanian has given talks for Acadia Pharmaceuticals and Acorda Therapeutics in the past.
A version of this article originally appeared on Medscape.com.
, according to a new study. The beneficial effects of these phosphate prodrugs of levodopa and carbidopa were most noticeable in the early morning, results of the phase 1B study showed.
As Parkinson’s disease progresses and dosing of oral levodopa/carbidopa (LD/CD) increases, its therapeutic window narrows, resulting in troublesome dyskinesia at peak drug levels and tremors and rigidity when levels fall.
“Foslevodopa/foscarbidopa shows lower ‘off’ time than oral levodopa/carbidopa, and this was statistically significant. Also, foslevodopa/foscarbidopa (fosL/fosC) showed more ‘on’ time without dyskinesia, compared with oral levodopa/carbidopa. This was also statistically significant,” lead author Sven Stodtmann, PhD, of AbbVie GmbH, Ludwigshafen, Germany, reported in his recorded presentation at the Movement Disorders Society’s 23rd International Congress of Parkinson’s Disease and Movement Disorder (Virtual) 2020.
Continuous infusion versus oral therapy
The analysis included 20 patients, and all data from these individuals were collected between 4:30 a.m. and 9:30 p.m.
Participants were 12 men and 8 women, aged 30-80 years, with advanced, idiopathic Parkinson’s disease responsive to levodopa but inadequately controlled on their current stable therapy, having a minimum of 2.5 off hours/day. Mean age was 61.3 plus or minus 10.5 years (range 35-77 years).
In this single-arm, open-label study, they received subcutaneous infusions of personalized therapeutic doses of fosL/fosC 24 hours/day for 28 days after a 10- to 30-day screening period during which they recorded LD/CD doses in a diary and had motor symptoms monitored using a wearable device.
Following the screening period, fosL/fosC doses were titrated over up to 5 days, with subsequent weekly study visits, for a total time on fosL/fosC of 28 days. Drug titration was aimed at maximizing functional on time and minimizing the number of off episodes while minimizing troublesome dyskinesia.
Continuous infusion of fosL/fosC performed better than oral LD/CD on all counts.
“The off time is much lower in the morning for people on foslevodopa/foscarbidopa [compared with oral LD/CD] because this is a 24-hour infusion product,” Dr. Stodtmann explained.
The effect was maintained over the course of the day with little fluctuation with fosL/fosC, off periods never exceeding about 25% between 4:30 a.m. and 9 p.m. For LD/CD, off periods were highest in the early morning and peaked at about 50% on a 3- to 4-hour cycle during the course of the day.
Increased on time without dyskinesia varied between about 60% and 80% during the day with fosL/fosC, showing the greatest difference between fosL/fosC and oral LD/CD in the early morning hours.
“On time with nontroublesome dyskinesia was lower for foscarbidopa/foslevodopa, compared to oral levodopa/carbidopa, but this was not statistically significant,” Dr. Stodtmann said. On time with troublesome dyskinesia followed the same pattern, again, not statistically significant.
Looking at the data another way, the investigators calculated the odds ratios of motor symptoms using fosL/fosC, compared with oral LD/CD. Use of fosL/fosC was associated with a 59% lower risk of being in the off state during the day, compared with oral LD/CD (odds ratio, 0.4; 95% confidence interval, 0.2-0.7; P < .01). Similarly, the probability of being in the on state without dyskinesia was much greater with fosL/fosC (OR, 2.75; 95% CI, 1.08-6.99; P < .05).
Encouraging, but more data needed
Indu Subramanian, MD, of the department of neurology at the University of California, Los Angeles, and director of the Parkinson’s Disease Research, Education, and Clinical Center at the West Los Angeles Veterans Affairs Hospital, commented that the field has been waiting to see data on fosL/fosC.
“It seems like it’s pretty reasonable in terms of what the goals were, which is to improve stability of Parkinson’s symptoms, to improve off time and give on time without troublesome dyskinesia,” she said. “So I think those [goals] have been met.”
Dr. Subramanian, who was not involved with the research, said she would have liked to have seen results concerning safety of this drug formulation, which the presentation lacked, “because historically, there have been issues with nodule formation and skin breakdown, things like that, due to the stability of the product in the subcutaneous form. … So, always to my understanding, there has been this search for things that are tolerated in the subcutaneous delivery.”
If this formulation proves safe and tolerable, Dr. Subramanian sees a potential place for it for some patients with advanced Parkinson’s disease.
“Certainly a subcutaneous formulation will be better than something that requires … deep brain surgery or even a pump insertion like Duopa [carbidopa/levodopa enteral suspension, AbbVie] or something like that,” she said. “I think [it] would be beneficial over something with the gut because the gut historically has been a problem to rely on in advanced Parkinson’s patients due to slower transit times, and the gut itself is affected with Parkinson’s disease.”
Dr. Stodtmann and all coauthors are employees of AbbVie, which was the sponsor of the study and was responsible for all aspects of it. Dr. Subramanian has given talks for Acadia Pharmaceuticals and Acorda Therapeutics in the past.
A version of this article originally appeared on Medscape.com.
, according to a new study. The beneficial effects of these phosphate prodrugs of levodopa and carbidopa were most noticeable in the early morning, results of the phase 1B study showed.
As Parkinson’s disease progresses and dosing of oral levodopa/carbidopa (LD/CD) increases, its therapeutic window narrows, resulting in troublesome dyskinesia at peak drug levels and tremors and rigidity when levels fall.
“Foslevodopa/foscarbidopa shows lower ‘off’ time than oral levodopa/carbidopa, and this was statistically significant. Also, foslevodopa/foscarbidopa (fosL/fosC) showed more ‘on’ time without dyskinesia, compared with oral levodopa/carbidopa. This was also statistically significant,” lead author Sven Stodtmann, PhD, of AbbVie GmbH, Ludwigshafen, Germany, reported in his recorded presentation at the Movement Disorders Society’s 23rd International Congress of Parkinson’s Disease and Movement Disorder (Virtual) 2020.
Continuous infusion versus oral therapy
The analysis included 20 patients, and all data from these individuals were collected between 4:30 a.m. and 9:30 p.m.
Participants were 12 men and 8 women, aged 30-80 years, with advanced, idiopathic Parkinson’s disease responsive to levodopa but inadequately controlled on their current stable therapy, having a minimum of 2.5 off hours/day. Mean age was 61.3 plus or minus 10.5 years (range 35-77 years).
In this single-arm, open-label study, they received subcutaneous infusions of personalized therapeutic doses of fosL/fosC 24 hours/day for 28 days after a 10- to 30-day screening period during which they recorded LD/CD doses in a diary and had motor symptoms monitored using a wearable device.
Following the screening period, fosL/fosC doses were titrated over up to 5 days, with subsequent weekly study visits, for a total time on fosL/fosC of 28 days. Drug titration was aimed at maximizing functional on time and minimizing the number of off episodes while minimizing troublesome dyskinesia.
Continuous infusion of fosL/fosC performed better than oral LD/CD on all counts.
“The off time is much lower in the morning for people on foslevodopa/foscarbidopa [compared with oral LD/CD] because this is a 24-hour infusion product,” Dr. Stodtmann explained.
The effect was maintained over the course of the day with little fluctuation with fosL/fosC, off periods never exceeding about 25% between 4:30 a.m. and 9 p.m. For LD/CD, off periods were highest in the early morning and peaked at about 50% on a 3- to 4-hour cycle during the course of the day.
Increased on time without dyskinesia varied between about 60% and 80% during the day with fosL/fosC, showing the greatest difference between fosL/fosC and oral LD/CD in the early morning hours.
“On time with nontroublesome dyskinesia was lower for foscarbidopa/foslevodopa, compared to oral levodopa/carbidopa, but this was not statistically significant,” Dr. Stodtmann said. On time with troublesome dyskinesia followed the same pattern, again, not statistically significant.
Looking at the data another way, the investigators calculated the odds ratios of motor symptoms using fosL/fosC, compared with oral LD/CD. Use of fosL/fosC was associated with a 59% lower risk of being in the off state during the day, compared with oral LD/CD (odds ratio, 0.4; 95% confidence interval, 0.2-0.7; P < .01). Similarly, the probability of being in the on state without dyskinesia was much greater with fosL/fosC (OR, 2.75; 95% CI, 1.08-6.99; P < .05).
Encouraging, but more data needed
Indu Subramanian, MD, of the department of neurology at the University of California, Los Angeles, and director of the Parkinson’s Disease Research, Education, and Clinical Center at the West Los Angeles Veterans Affairs Hospital, commented that the field has been waiting to see data on fosL/fosC.
“It seems like it’s pretty reasonable in terms of what the goals were, which is to improve stability of Parkinson’s symptoms, to improve off time and give on time without troublesome dyskinesia,” she said. “So I think those [goals] have been met.”
Dr. Subramanian, who was not involved with the research, said she would have liked to have seen results concerning safety of this drug formulation, which the presentation lacked, “because historically, there have been issues with nodule formation and skin breakdown, things like that, due to the stability of the product in the subcutaneous form. … So, always to my understanding, there has been this search for things that are tolerated in the subcutaneous delivery.”
If this formulation proves safe and tolerable, Dr. Subramanian sees a potential place for it for some patients with advanced Parkinson’s disease.
“Certainly a subcutaneous formulation will be better than something that requires … deep brain surgery or even a pump insertion like Duopa [carbidopa/levodopa enteral suspension, AbbVie] or something like that,” she said. “I think [it] would be beneficial over something with the gut because the gut historically has been a problem to rely on in advanced Parkinson’s patients due to slower transit times, and the gut itself is affected with Parkinson’s disease.”
Dr. Stodtmann and all coauthors are employees of AbbVie, which was the sponsor of the study and was responsible for all aspects of it. Dr. Subramanian has given talks for Acadia Pharmaceuticals and Acorda Therapeutics in the past.
A version of this article originally appeared on Medscape.com.
Telemedicine feasible and reliable in Parkinson’s trial
, a 1-year, phase 3 clinical trial has shown. The trial was an add-on study involving a subset of subjects from the STEADY-PD III trial of isradipine in early Parkinson’s disease.
Although the trial was conducted before SARS-CoV-2 arrived on the scene, the findings have particular relevance for being able to conduct a variety of clinical trials in the face of COVID-19 and the need to limit in-person interactions.
The 40 participants used tablets to complete three remote, video-based assessments during 1 year, with each remote visit planned to be completed within 4 weeks of an in-person visit. It was easy to enroll patients, and they completed about 95% of planned visits, said neurologist Christopher Tarolli, MD, of the University of Rochester (N.Y.).
He presented the study findings at the Movement Disorder Society’s 23rd International Congress of Parkinson’s Disease and Movement Disorders (Virtual) 2020.
“The visits were clearly feasible, and we were able to do them [84%] within that 4-week time frame around the in-person visit,” he said. “The visits were also reasonably reliable, particularly so for what we call the nonmotor outcomes and the patient-reported outcomes.”
In-person versus remote assessment
For the remote visits, participants completed primarily the same battery of tests as the in-person visits. Responses on the Movement Disorder Society-Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) subscales demonstrated “that there was excellent correlation between patient-reported and nonmotor outcome measures and moderate correlation between in-person and remote-performed motor assessments,” Dr. Tarolli said.
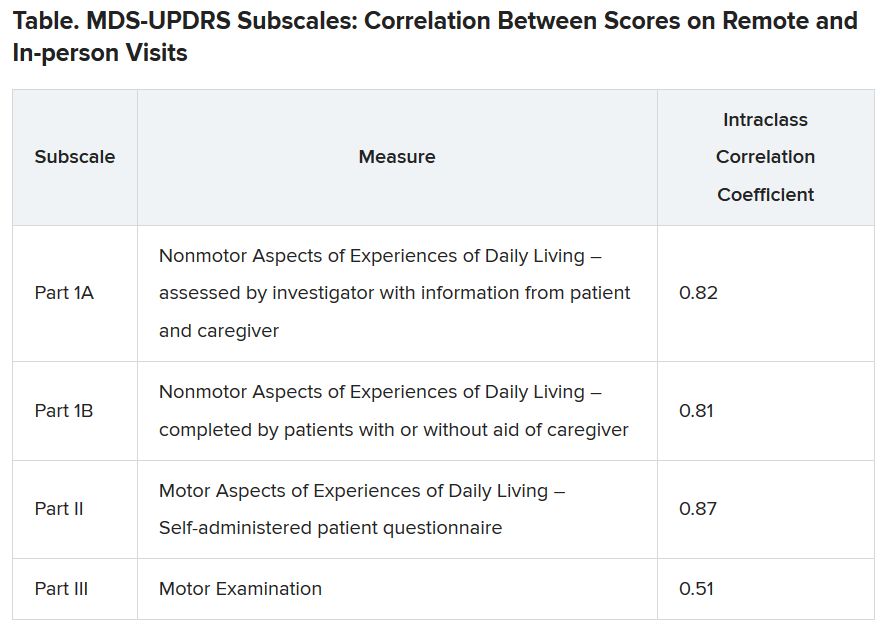
He explained that the study used modified motor assessments (MDS-UPDRS Part III) that excluded testing of rigidity and postural instability, which require hands-on testing by a trained examiner and thus are impossible to do remotely.
Additionally, the somewhat lower correlation on this subscale was probably the result of different investigators conducting in-person versus remote assessments, with a subset of in-person investigators who tended to rate participants more severely driving down the correlation. “I think if these methods were applied in future trials, the in-person and remote investigators would optimally be the same person,” Dr. Tarolli suggested.
Room for error?
Indu Subramanian, MD, of the department of neurology at the University of California, Los Angeles, and director of the Parkinson’s Disease Research, Education, and Clinical Center at the West Los Angeles Veterans Affairs Hospital, commented that “the reliability of UPDRS [part] III is where I would want to have, for sure, a little bit more of a deep dive. … possibly the same patient be rated by the same person.”
She also noted that doing remote and in-person assessments within 4 weeks of each other leaves a lot of room for variability. “You could see the same patient in the morning and then do UPDRS in the afternoon, and it can be totally different depending on when you meet the person,” she said.
Only so much testing can be done remotely. Nonetheless, she questioned whether it is really a valid UPDRS if rigidity and postural stability measures are eliminated. “[Is] this now a new modified UPDRS that we’re going to use that is as good as the old UPDRS moving forward, a home version of UPDRS or whatever we’re going to call it?”
Dr. Subramanian mentioned that patients have told her that UPDRS part III does not really measure what is most important to them, such as making pastries for their grandchildren rather than rapidly tapping their fingers.
“That speaks a little bit to the fact that we should have more patient-centered outcomes and things that patients can report. … things that are not going to require necessarily an in-person exam as maybe measures that really can be used moving forward in studies,” she suggested.
Patient satisfaction with remote visits
Greater than 90% of the patients were satisfied or very satisfied overall with the remote visits, including the convenience, comfort, and connection (using the devices and Internet connection), with “patients describing enjoying being able to do these visits from the comfort of their own home, not having to travel,” Dr. Tarolli said. Not having to drive in an ‘off’ state “was actually something that some participants identified as a safety benefit from this as well.”
There was also a time benefit to the patients and investigators. The average length of the remote visits was 54.3 minutes each versus 74 minutes of interaction for in-person visits, mainly a result of more efficient hand-offs between the neurologist and the study coordinator during the remote visits, plus being able to pause the remote visit to give a medication dose time to take effect.
For the patient, there was a large amount of time saved when travel time was considered – a total of 190.2 minutes on average for travel and testing for the in-person visits.
About three-quarters (76%) of the study patients said that remote visits would increase their likelihood of participating in future trials. However, that result may be skewed by the fact that these were already people willing to participate in a remote trial, so the generalizability of the result may be affected. Nonetheless, Dr. Tarolli said he thinks that, as technology gets better and older people become more comfortable with it, remote visits within Parkinson’s research studies may become more common.
One caveat he mentioned is that, with remote visits, the neurologist misses a chance to observe a patient’s whole body and construct a global impression of how he or she is moving. On the other hand, remote video gives the investigator the chance to see the living environment of the patient and suggest changes for safety, such as to reduce the risk of falling for a person with unsteadiness of gait living in a crowded house.
“It really allows us to make a more holistic assessment of how our patient is functioning outside the clinic, which I think we’ve traditionally had really no way of doing,” Dr. Tarolli said.
His final suggestion for anyone contemplating conducting studies with remote visits is to develop a team that is comfortable troubleshooting the technological aspects of those visits.
UCLA’s Dr. Subramanian lauded the University of Rochester team for their efforts in moving remote visits forward. “They’re at the cutting edge of these sorts of things,” she said. “So I’m assuming that they’ll come out with more things [for visits] to become better that are going to move this forward, which is exciting.”
Dr. Tarolli has disclosed no relevant financial relationships. Dr. Subramanian has given talks for Acorda Pharmaceuticals and Acadia Pharmaceuticals in the past. The study had only university, government, foundation, and other nonprofit support.
A version of this article originally appeared on Medscape.com.
, a 1-year, phase 3 clinical trial has shown. The trial was an add-on study involving a subset of subjects from the STEADY-PD III trial of isradipine in early Parkinson’s disease.
Although the trial was conducted before SARS-CoV-2 arrived on the scene, the findings have particular relevance for being able to conduct a variety of clinical trials in the face of COVID-19 and the need to limit in-person interactions.
The 40 participants used tablets to complete three remote, video-based assessments during 1 year, with each remote visit planned to be completed within 4 weeks of an in-person visit. It was easy to enroll patients, and they completed about 95% of planned visits, said neurologist Christopher Tarolli, MD, of the University of Rochester (N.Y.).
He presented the study findings at the Movement Disorder Society’s 23rd International Congress of Parkinson’s Disease and Movement Disorders (Virtual) 2020.
“The visits were clearly feasible, and we were able to do them [84%] within that 4-week time frame around the in-person visit,” he said. “The visits were also reasonably reliable, particularly so for what we call the nonmotor outcomes and the patient-reported outcomes.”
In-person versus remote assessment
For the remote visits, participants completed primarily the same battery of tests as the in-person visits. Responses on the Movement Disorder Society-Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) subscales demonstrated “that there was excellent correlation between patient-reported and nonmotor outcome measures and moderate correlation between in-person and remote-performed motor assessments,” Dr. Tarolli said.
He explained that the study used modified motor assessments (MDS-UPDRS Part III) that excluded testing of rigidity and postural instability, which require hands-on testing by a trained examiner and thus are impossible to do remotely.
Additionally, the somewhat lower correlation on this subscale was probably the result of different investigators conducting in-person versus remote assessments, with a subset of in-person investigators who tended to rate participants more severely driving down the correlation. “I think if these methods were applied in future trials, the in-person and remote investigators would optimally be the same person,” Dr. Tarolli suggested.
Room for error?
Indu Subramanian, MD, of the department of neurology at the University of California, Los Angeles, and director of the Parkinson’s Disease Research, Education, and Clinical Center at the West Los Angeles Veterans Affairs Hospital, commented that “the reliability of UPDRS [part] III is where I would want to have, for sure, a little bit more of a deep dive. … possibly the same patient be rated by the same person.”
She also noted that doing remote and in-person assessments within 4 weeks of each other leaves a lot of room for variability. “You could see the same patient in the morning and then do UPDRS in the afternoon, and it can be totally different depending on when you meet the person,” she said.
Only so much testing can be done remotely. Nonetheless, she questioned whether it is really a valid UPDRS if rigidity and postural stability measures are eliminated. “[Is] this now a new modified UPDRS that we’re going to use that is as good as the old UPDRS moving forward, a home version of UPDRS or whatever we’re going to call it?”
Dr. Subramanian mentioned that patients have told her that UPDRS part III does not really measure what is most important to them, such as making pastries for their grandchildren rather than rapidly tapping their fingers.
“That speaks a little bit to the fact that we should have more patient-centered outcomes and things that patients can report. … things that are not going to require necessarily an in-person exam as maybe measures that really can be used moving forward in studies,” she suggested.
Patient satisfaction with remote visits
Greater than 90% of the patients were satisfied or very satisfied overall with the remote visits, including the convenience, comfort, and connection (using the devices and Internet connection), with “patients describing enjoying being able to do these visits from the comfort of their own home, not having to travel,” Dr. Tarolli said. Not having to drive in an ‘off’ state “was actually something that some participants identified as a safety benefit from this as well.”
There was also a time benefit to the patients and investigators. The average length of the remote visits was 54.3 minutes each versus 74 minutes of interaction for in-person visits, mainly a result of more efficient hand-offs between the neurologist and the study coordinator during the remote visits, plus being able to pause the remote visit to give a medication dose time to take effect.
For the patient, there was a large amount of time saved when travel time was considered – a total of 190.2 minutes on average for travel and testing for the in-person visits.
About three-quarters (76%) of the study patients said that remote visits would increase their likelihood of participating in future trials. However, that result may be skewed by the fact that these were already people willing to participate in a remote trial, so the generalizability of the result may be affected. Nonetheless, Dr. Tarolli said he thinks that, as technology gets better and older people become more comfortable with it, remote visits within Parkinson’s research studies may become more common.
One caveat he mentioned is that, with remote visits, the neurologist misses a chance to observe a patient’s whole body and construct a global impression of how he or she is moving. On the other hand, remote video gives the investigator the chance to see the living environment of the patient and suggest changes for safety, such as to reduce the risk of falling for a person with unsteadiness of gait living in a crowded house.
“It really allows us to make a more holistic assessment of how our patient is functioning outside the clinic, which I think we’ve traditionally had really no way of doing,” Dr. Tarolli said.
His final suggestion for anyone contemplating conducting studies with remote visits is to develop a team that is comfortable troubleshooting the technological aspects of those visits.
UCLA’s Dr. Subramanian lauded the University of Rochester team for their efforts in moving remote visits forward. “They’re at the cutting edge of these sorts of things,” she said. “So I’m assuming that they’ll come out with more things [for visits] to become better that are going to move this forward, which is exciting.”
Dr. Tarolli has disclosed no relevant financial relationships. Dr. Subramanian has given talks for Acorda Pharmaceuticals and Acadia Pharmaceuticals in the past. The study had only university, government, foundation, and other nonprofit support.
A version of this article originally appeared on Medscape.com.
, a 1-year, phase 3 clinical trial has shown. The trial was an add-on study involving a subset of subjects from the STEADY-PD III trial of isradipine in early Parkinson’s disease.
Although the trial was conducted before SARS-CoV-2 arrived on the scene, the findings have particular relevance for being able to conduct a variety of clinical trials in the face of COVID-19 and the need to limit in-person interactions.
The 40 participants used tablets to complete three remote, video-based assessments during 1 year, with each remote visit planned to be completed within 4 weeks of an in-person visit. It was easy to enroll patients, and they completed about 95% of planned visits, said neurologist Christopher Tarolli, MD, of the University of Rochester (N.Y.).
He presented the study findings at the Movement Disorder Society’s 23rd International Congress of Parkinson’s Disease and Movement Disorders (Virtual) 2020.
“The visits were clearly feasible, and we were able to do them [84%] within that 4-week time frame around the in-person visit,” he said. “The visits were also reasonably reliable, particularly so for what we call the nonmotor outcomes and the patient-reported outcomes.”
In-person versus remote assessment
For the remote visits, participants completed primarily the same battery of tests as the in-person visits. Responses on the Movement Disorder Society-Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) subscales demonstrated “that there was excellent correlation between patient-reported and nonmotor outcome measures and moderate correlation between in-person and remote-performed motor assessments,” Dr. Tarolli said.
He explained that the study used modified motor assessments (MDS-UPDRS Part III) that excluded testing of rigidity and postural instability, which require hands-on testing by a trained examiner and thus are impossible to do remotely.
Additionally, the somewhat lower correlation on this subscale was probably the result of different investigators conducting in-person versus remote assessments, with a subset of in-person investigators who tended to rate participants more severely driving down the correlation. “I think if these methods were applied in future trials, the in-person and remote investigators would optimally be the same person,” Dr. Tarolli suggested.
Room for error?
Indu Subramanian, MD, of the department of neurology at the University of California, Los Angeles, and director of the Parkinson’s Disease Research, Education, and Clinical Center at the West Los Angeles Veterans Affairs Hospital, commented that “the reliability of UPDRS [part] III is where I would want to have, for sure, a little bit more of a deep dive. … possibly the same patient be rated by the same person.”
She also noted that doing remote and in-person assessments within 4 weeks of each other leaves a lot of room for variability. “You could see the same patient in the morning and then do UPDRS in the afternoon, and it can be totally different depending on when you meet the person,” she said.
Only so much testing can be done remotely. Nonetheless, she questioned whether it is really a valid UPDRS if rigidity and postural stability measures are eliminated. “[Is] this now a new modified UPDRS that we’re going to use that is as good as the old UPDRS moving forward, a home version of UPDRS or whatever we’re going to call it?”
Dr. Subramanian mentioned that patients have told her that UPDRS part III does not really measure what is most important to them, such as making pastries for their grandchildren rather than rapidly tapping their fingers.
“That speaks a little bit to the fact that we should have more patient-centered outcomes and things that patients can report. … things that are not going to require necessarily an in-person exam as maybe measures that really can be used moving forward in studies,” she suggested.
Patient satisfaction with remote visits
Greater than 90% of the patients were satisfied or very satisfied overall with the remote visits, including the convenience, comfort, and connection (using the devices and Internet connection), with “patients describing enjoying being able to do these visits from the comfort of their own home, not having to travel,” Dr. Tarolli said. Not having to drive in an ‘off’ state “was actually something that some participants identified as a safety benefit from this as well.”
There was also a time benefit to the patients and investigators. The average length of the remote visits was 54.3 minutes each versus 74 minutes of interaction for in-person visits, mainly a result of more efficient hand-offs between the neurologist and the study coordinator during the remote visits, plus being able to pause the remote visit to give a medication dose time to take effect.
For the patient, there was a large amount of time saved when travel time was considered – a total of 190.2 minutes on average for travel and testing for the in-person visits.
About three-quarters (76%) of the study patients said that remote visits would increase their likelihood of participating in future trials. However, that result may be skewed by the fact that these were already people willing to participate in a remote trial, so the generalizability of the result may be affected. Nonetheless, Dr. Tarolli said he thinks that, as technology gets better and older people become more comfortable with it, remote visits within Parkinson’s research studies may become more common.
One caveat he mentioned is that, with remote visits, the neurologist misses a chance to observe a patient’s whole body and construct a global impression of how he or she is moving. On the other hand, remote video gives the investigator the chance to see the living environment of the patient and suggest changes for safety, such as to reduce the risk of falling for a person with unsteadiness of gait living in a crowded house.
“It really allows us to make a more holistic assessment of how our patient is functioning outside the clinic, which I think we’ve traditionally had really no way of doing,” Dr. Tarolli said.
His final suggestion for anyone contemplating conducting studies with remote visits is to develop a team that is comfortable troubleshooting the technological aspects of those visits.
UCLA’s Dr. Subramanian lauded the University of Rochester team for their efforts in moving remote visits forward. “They’re at the cutting edge of these sorts of things,” she said. “So I’m assuming that they’ll come out with more things [for visits] to become better that are going to move this forward, which is exciting.”
Dr. Tarolli has disclosed no relevant financial relationships. Dr. Subramanian has given talks for Acorda Pharmaceuticals and Acadia Pharmaceuticals in the past. The study had only university, government, foundation, and other nonprofit support.
A version of this article originally appeared on Medscape.com.
The boy whose arm wouldn’t work
CASE Drooling, unsteady, and not himself
B, age 10, who is left handed and has autism spectrum disorder, is brought to the emergency department (ED) with a 1-day history of drooling, unsteady gait, and left wrist in sustained flexion. His parents report that for the past week, B has had cold symptoms, including rhinorrhea, a low-grade fever (100.0°F), and cough. Earlier in the day, he was seen at his pediatrician’s office, where he was diagnosed with an acute respiratory infection and started on amoxicillin, 500 mg twice daily for 7 days.
At baseline, B is nonverbal. He requires some assistance with his activities of daily living. He usually is able to walk without assistance and dress himself, but he is not toilet trained. His parents report that in the past day, he has had significant difficulties with tasks involving his left hand. Normally, B is able to feed himself “finger foods” but has been unable to do so today. His parents say that he has been unsteady on his feet, and has been “falling forward” when he tries to walk.
Two years ago, B was started on risperidone, 0.5 mg nightly, for behavioral aggression and self-mutilation. Over the next 12 months, the dosage was steadily increased to 1 mg twice daily, with good response. He has been taking his current dosage, 1 mg twice daily, for the past 12 months without adjustment. His parents report there have been no other medication changes, other than starting amoxicillin earlier that day.
As part of his initial ED evaluation, B is found to be mildly dehydrated, with an elevated sedimentation rate on urinalysis. His complete blood count (CBC) with differential is within normal limits. A comprehensive metabolic panel shows a slight increase in his creatinine level, indicating dehydration. B is administered IV fluid replacement because he is having difficulty drinking due to excessive drooling.
The ED physician is concerned that B may be experiencing an acute dystonic reaction from risperidone, so the team holds this medication, and gives B a one-time dose of IV diphenhydramine, 25 mg, for presumptive acute dystonic reaction. After several minutes, there is no improvement in the sustained flexion of his left wrist.
[polldaddy:10615848]
The authors’ observations
B presented with new-onset neurologic findings after a recently diagnosed upper respiratory viral illness. His symptoms appeared to be confined to his left upper extremity, specifically demonstrating left arm extension at the elbow with flexion of the left wrist. He also had new-onset unsteady gait with a stooped forward posture and required assistance with walking. Interestingly, despite B’s history of antipsychotic use, administering an anticholinergic agent did not lessen the dystonic posturing at his wrist and elbow.
EVALUATION Laboratory results reveal new clues
While in the ED, B undergoes MRI of the brain and spinal cord to rule out any mass lesions that could be impinging upon the motor pathways. Both brain and spinal cord imaging appear to be essentially normal, without evidence of impingement of the spinal nerves or lesions involving the brainstem or cerebellum.
Continue to: Due to concerns...
Due to concerns of possible airway obstruction, a CT scan of the neck is obtained to rule out any acute pathology, such as epiglottitis compromising his airway. The scan shows some inflammation and edema in the soft tissues that is thought to be secondary to his acute viral illness. B is able to maintain his airway and oxygenation, so intubation is not necessary.
A CPK test is ordered because there are concerns of sustained muscle contraction of B’s left wrist and elbow. The CPK level is 884 U/L (reference range 26 to 192 U/L). The elevation in CPK is consistent with prior laboratory findings of dehydration and indicating skeletal muscle breakdown from sustained muscle contraction. All other laboratory results, including a comprehensive metabolic panel, urine drug screen, and thyroid screening panel, are within normal limits.
[polldaddy:10615850]
EVALUATION No variation in facial expression
B is admitted to the general pediatrics service. Maintenance IV fluids are started due to concerns of dehydration and possible rhabdomyolysis due to his elevated CPK level. Risperidone is held throughout the hospital course due to concerns for an acute dystonic reaction. B is monitored for several days without clinical improvement and eventually discharged home with a diagnosis of inflammatory mononeuropathy due to viral infection. The patient is told to discontinue risperidone as part of discharge instructions.
Five days later, B returns to the hospital because there was no improvement in his left extremity or walking. His left elbow remains extended with left wrist in flexion. Psychiatry is consulted for further diagnostic clarity and evaluation.
On physical examination, B’s left arm remains unchanged. Despite discontinuing risperidone, there is evidence of cogwheel rigidity of the left wrist joint. Reflexes in the upper and lower extremities are 2+ and symmetrical bilaterally, suggesting intact upper and lower motor pathways. Babinski sign is absent bilaterally, which is a normal finding in B’s age group. B continues to have difficulty with ambulating and appears to “fall forward” while trying to walk with assistance. His parents also say that B is not laughing, smiling, or showing any variation in facial expression.
Continue to: Additional family history...
Additional family history is gathered from B’s parents for possible hereditary movement disorders such as Wilson’s disease. They report that no family members have developed involuntary movements or other neurologic syndromes. Additional considerations on the differential diagnosis for B include juvenile ALS or mononeuropathy involving the C5 and C6 nerve roots. B’s parents deny any recent shoulder trauma, and radiographic studies did not demonstrate any involvement of the nerve roots.
TREATMENT A trial of bromocriptine
At this point, B’s neurologic workup is essentially normal, and he is given a provisional diagnosis of antipsychotic-induced tardive dystonia vs tardive parkinsonism. Risperidone continues to be held, and B is monitored for clinical improvement. B is administered a one-time dose of diphenhydramine, 25 mg, for dystonia with no improvement in symptoms. He is then started on bromocriptine, 1.25 mg twice daily with meals, for parkinsonian symptoms secondary to antipsychotic medication use. After 1 day of treatment, B shows less sustained flexion of his left wrist. He is able to relax his left arm, shows improvements in ambulation, and requires less assistance. B continues to be observed closely and continues to improve toward his baseline.
At Day 4, he is discharged. B is able to walk mostly without assistance and demonstrates improvement in left wrist flexion. He is scheduled to see a movement disorders specialist a week after discharge. The initial diagnosis given by the movement disorder specialist is tardive dystonia.
The authors’ observations
Tardive dyskinesia is a well-known iatrogenic effect of antipsychotic medications that are commonly used to manage conditions such as schizophrenia or behavioral agitation associated with autism spectrum disorder. Symptoms of tardive dyskinesia typically emerge after 1 to 2 years of continuous exposure to dopamine receptor blocking agents (DRBAs). Tardive dyskinesia symptoms include involuntary, repetitive, purposeless movements of the tongue, jaw, lips, face, trunk, and upper and lower extremities, with significant functional impairment.1
Tardive syndromes refer to a diverse array of hyperkinetic, hypokinetic, and sensory movement disorders resulting from at least 3 months of continuous DRBA therapy.2 Tardive dyskinesia is perhaps the most well-known of the tardive syndromes, but is not the only one to consider when assessing for antipsychotic-induced movement disorders. A key feature differentiating a tardive syndrome is the persistence of the movement disorder after the DRBA is discontinued. In this case, B had been receiving a stable dose of risperidone for >1 year. He developed dystonic posturing of his left wrist and elbow that was both unresponsive to anticholinergic medication and persisted after risperidone was discontinued. The term “tardive” emphasizes the delay in development of abnormal involuntary movement symptoms after initiating antipsychotic medications.3 Table 12 shows a comparison of tardive dystonia vs an acute dystonic reaction.

Continue to: Other tardive syndromes include...
Other tardive syndromes include:
- tardive tics
- tardive parkinsonism
- tardive pain
- tardive myoclonus
- tardive akathisia
- tardive tremors.
The incidence of tardive syndromes increases 5% annually for the first 5 years of treatment. At 10 years of treatment, the annual incidence is thought to be 49%, and at 25 years of treatment, 68%.4 The predominant theory of the pathophysiology of tardive syndromes is that the chronic use of DRBAs causes a gradual hypersensitization of dopamine receptors.4 The diagnosis of a tardive syndrome is based on history of exposure to a DRBA as well as clinical observation of symptoms.
Compared with classic tardive dyskinesia, tardive dystonia is more common among younger patients. The mean age of onset of tardive dystonia is 40, and it typically affects young males.5 Typical posturing observed in cases of tardive dystonia include extension of the arms and flexion at the wrists.6 In contrast to cases of primary dystonia, tardive dystonia is typically associated with stereotypies, akathisia, or other movement disorders. Anticholinergic agents, such as
The American Psychiatric Association has issued guidelines on screening for involuntary movement syndromes by using the Abnormal Involuntary Movement Scale (AIMS).7 The current recommendations include assessment every 6 months for patients receiving first-generation antipsychotics, and every 12 months for those receiving second-generation antipsychotics.7 Prescribers should also carefully assess for any pre-existing involuntary movements before prescribing a DRBA.7
[polldaddy:10615855]
The authors’ observations
In 2013, the American Academy of Neurology (AAN) published guidelines on the treatment of tardive dyskinesia. According to these guidelines, at that time, the treatments with the most evidence supporting their use were clonazepam, ginkgo biloba,

Continue to: In 2017, valbenazine and deutetrabenazine...
In 2017, valbenazine and deutetrabenazine became the first FDA-approved treatments for tardive dyskinesia in adults. Both medications block the vesicular monoamine transporter 2 (VMAT2) system, which results in decreased synaptic dopamine and dopamine receptor stimulation. Both VMAT2 inhibitor medications have a category level A supporting their use for treating tardive dyskinesia.8-10
Currently, there are no published treatment guidelines on pharmacologic management of tardive dystonia. In B’s case, bromocriptine, a dopamine agonist, was used to counter the dopamine-blocking effects of risperidone on the nigrostriatal pathway and improve parkinsonian features of B’s presentation, including bradykinesia, stooped forward posture, and masked facies. Bromocriptine was found to be effective in alleviating parkinsonian features; however, to date there is no evidence demonstrating its effectiveness in countering delayed dystonic effects of DRBAs.
OUTCOME Improvement of dystonia symptoms
One week after discharge, B is seen for a follow-up visit. He continues taking bromocriptine, 1.25 mg twice daily, with meals after discharge. On examination, he has some evidence of tardive dystonia, including flexion of left wrist and posturing while ambulating. B’s parkinsonian features, including stooped forward posture, masked facies, and cogwheel rigidity of the left wrist muscle, have resolved. B is now able to walk on his own without unsteadiness. Bromocriptine is discontinued after 1 month, and his symptoms of dystonia continue to improve.
Two months after hospitalization, B is started on quetiapine, 25 mg twice daily, for behavioral aggression. Quetiapine is chosen because it has a lower dopamine receptor affinity compared with risperidone, and theoretically, quetiapine is associated with a lower risk of developing tardive symptoms. During the next 6 months, B is monitored closely for recurrence of tardive symptoms. Quetiapine is slowly titrated to 25 mg in the morning, and 50 mg at bedtime. His behavioral agitation improves significantly and he does not have a recurrence of tardive symptoms.
Bottom Line
Tardive dystonia is a possible iatrogenic adverse effect for patients receiving long-term dopamine receptor blocking agent (DRBA) therapy. Tardive syndromes encompass delayed-onset movement disorders caused by long-term blockade of the dopamine receptor by antipsychotic agents. Tardive dystonia can be contrasted from acute dystonic reaction based on the time course of development as well as by the persistence of symptoms after DRBAs are withheld.
Continue to: Related Resources
Related Resources
- American Academy of Neurology. Summary of evidence-based guideline for clinicians: treatment of tardive syndromes. https://www.aan.com/Guidelines/Home/GetGuidelineContent/613. Published 2013.
- Dystonia Medical Research Foundation. https://dystonia-foundation.org/.
Drug Brand Names
Amantadine • Gocovri, Symmetrel
Amoxicillin • Amoxil
Baclofen • Kemstro, Liroesal
Benztropine • Cogentin
Bromocriptine • Parlodel
Clonazepam • Klonopin
Deutetrabenazine • Austedo
Galantamine • Razadyne
Quetiapine • Seroquel
Risperidone • Risperdal
Tetrabenazine • Xenazine
Trihexyphenidyl • Artane, Tremin
Valbenazine • Ingrezza
1. Margolese HC, Chouinard G, Kolivakis TT, et al. Tardive dyskinesia in the era of typical and atypical antipsychotics. Part 1: pathophysiology and mechanisms of induction. Can J Psychiatr. 2005;50(9):541-547.
2. Truong D, Frei K. Setting the record straight: the nosology of tardive syndromes. Parkinsonism Relat Disord. 2019;59:146-150.
3. Cornett EM, Novitch M, Kaye AD, et al. Medication-induced tardive dyskinesia: a review and update. Ochsner J. 2017;17(2):162-174.
4. Schooler NR, Kane JM. Research diagnoses for tardive dyskinesia. Arch Gen Psychiatry. 1982;39(4):486-487.
5. Fahn S, Jankovic J, Hallett M. Principles and Practice of Movement Disorders. 2nd ed. Philadelphia, PA: Saunders; 2011:415-446.
6. Kang UJ, Burke RE, Fahn S. Natural history and treatment of tardive dystonia. Mov Disord. 1986;1(3):193-208.
7. Lehman AF, Lieberman JA, Dixon LB, et al. Practice guideline for the treatment of patients with schizophrenia, second edition. Am J Psychiatry. 2004;161(suppl 2):1-56.
8. Bhidayasiri R, Fahn S, Weiner WJ, et al, Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
9. Ingrezza [package insert]. San Diego, CA: Neurocrine Biosciences, Inc.; 2020.
10. Austedo [package insert]. North Wales, PA: Teva Pharmaceuticals; 2017.
CASE Drooling, unsteady, and not himself
B, age 10, who is left handed and has autism spectrum disorder, is brought to the emergency department (ED) with a 1-day history of drooling, unsteady gait, and left wrist in sustained flexion. His parents report that for the past week, B has had cold symptoms, including rhinorrhea, a low-grade fever (100.0°F), and cough. Earlier in the day, he was seen at his pediatrician’s office, where he was diagnosed with an acute respiratory infection and started on amoxicillin, 500 mg twice daily for 7 days.
At baseline, B is nonverbal. He requires some assistance with his activities of daily living. He usually is able to walk without assistance and dress himself, but he is not toilet trained. His parents report that in the past day, he has had significant difficulties with tasks involving his left hand. Normally, B is able to feed himself “finger foods” but has been unable to do so today. His parents say that he has been unsteady on his feet, and has been “falling forward” when he tries to walk.
Two years ago, B was started on risperidone, 0.5 mg nightly, for behavioral aggression and self-mutilation. Over the next 12 months, the dosage was steadily increased to 1 mg twice daily, with good response. He has been taking his current dosage, 1 mg twice daily, for the past 12 months without adjustment. His parents report there have been no other medication changes, other than starting amoxicillin earlier that day.
As part of his initial ED evaluation, B is found to be mildly dehydrated, with an elevated sedimentation rate on urinalysis. His complete blood count (CBC) with differential is within normal limits. A comprehensive metabolic panel shows a slight increase in his creatinine level, indicating dehydration. B is administered IV fluid replacement because he is having difficulty drinking due to excessive drooling.
The ED physician is concerned that B may be experiencing an acute dystonic reaction from risperidone, so the team holds this medication, and gives B a one-time dose of IV diphenhydramine, 25 mg, for presumptive acute dystonic reaction. After several minutes, there is no improvement in the sustained flexion of his left wrist.
[polldaddy:10615848]
The authors’ observations
B presented with new-onset neurologic findings after a recently diagnosed upper respiratory viral illness. His symptoms appeared to be confined to his left upper extremity, specifically demonstrating left arm extension at the elbow with flexion of the left wrist. He also had new-onset unsteady gait with a stooped forward posture and required assistance with walking. Interestingly, despite B’s history of antipsychotic use, administering an anticholinergic agent did not lessen the dystonic posturing at his wrist and elbow.
EVALUATION Laboratory results reveal new clues
While in the ED, B undergoes MRI of the brain and spinal cord to rule out any mass lesions that could be impinging upon the motor pathways. Both brain and spinal cord imaging appear to be essentially normal, without evidence of impingement of the spinal nerves or lesions involving the brainstem or cerebellum.
Continue to: Due to concerns...
Due to concerns of possible airway obstruction, a CT scan of the neck is obtained to rule out any acute pathology, such as epiglottitis compromising his airway. The scan shows some inflammation and edema in the soft tissues that is thought to be secondary to his acute viral illness. B is able to maintain his airway and oxygenation, so intubation is not necessary.
A CPK test is ordered because there are concerns of sustained muscle contraction of B’s left wrist and elbow. The CPK level is 884 U/L (reference range 26 to 192 U/L). The elevation in CPK is consistent with prior laboratory findings of dehydration and indicating skeletal muscle breakdown from sustained muscle contraction. All other laboratory results, including a comprehensive metabolic panel, urine drug screen, and thyroid screening panel, are within normal limits.
[polldaddy:10615850]
EVALUATION No variation in facial expression
B is admitted to the general pediatrics service. Maintenance IV fluids are started due to concerns of dehydration and possible rhabdomyolysis due to his elevated CPK level. Risperidone is held throughout the hospital course due to concerns for an acute dystonic reaction. B is monitored for several days without clinical improvement and eventually discharged home with a diagnosis of inflammatory mononeuropathy due to viral infection. The patient is told to discontinue risperidone as part of discharge instructions.
Five days later, B returns to the hospital because there was no improvement in his left extremity or walking. His left elbow remains extended with left wrist in flexion. Psychiatry is consulted for further diagnostic clarity and evaluation.
On physical examination, B’s left arm remains unchanged. Despite discontinuing risperidone, there is evidence of cogwheel rigidity of the left wrist joint. Reflexes in the upper and lower extremities are 2+ and symmetrical bilaterally, suggesting intact upper and lower motor pathways. Babinski sign is absent bilaterally, which is a normal finding in B’s age group. B continues to have difficulty with ambulating and appears to “fall forward” while trying to walk with assistance. His parents also say that B is not laughing, smiling, or showing any variation in facial expression.
Continue to: Additional family history...
Additional family history is gathered from B’s parents for possible hereditary movement disorders such as Wilson’s disease. They report that no family members have developed involuntary movements or other neurologic syndromes. Additional considerations on the differential diagnosis for B include juvenile ALS or mononeuropathy involving the C5 and C6 nerve roots. B’s parents deny any recent shoulder trauma, and radiographic studies did not demonstrate any involvement of the nerve roots.
TREATMENT A trial of bromocriptine
At this point, B’s neurologic workup is essentially normal, and he is given a provisional diagnosis of antipsychotic-induced tardive dystonia vs tardive parkinsonism. Risperidone continues to be held, and B is monitored for clinical improvement. B is administered a one-time dose of diphenhydramine, 25 mg, for dystonia with no improvement in symptoms. He is then started on bromocriptine, 1.25 mg twice daily with meals, for parkinsonian symptoms secondary to antipsychotic medication use. After 1 day of treatment, B shows less sustained flexion of his left wrist. He is able to relax his left arm, shows improvements in ambulation, and requires less assistance. B continues to be observed closely and continues to improve toward his baseline.
At Day 4, he is discharged. B is able to walk mostly without assistance and demonstrates improvement in left wrist flexion. He is scheduled to see a movement disorders specialist a week after discharge. The initial diagnosis given by the movement disorder specialist is tardive dystonia.
The authors’ observations
Tardive dyskinesia is a well-known iatrogenic effect of antipsychotic medications that are commonly used to manage conditions such as schizophrenia or behavioral agitation associated with autism spectrum disorder. Symptoms of tardive dyskinesia typically emerge after 1 to 2 years of continuous exposure to dopamine receptor blocking agents (DRBAs). Tardive dyskinesia symptoms include involuntary, repetitive, purposeless movements of the tongue, jaw, lips, face, trunk, and upper and lower extremities, with significant functional impairment.1
Tardive syndromes refer to a diverse array of hyperkinetic, hypokinetic, and sensory movement disorders resulting from at least 3 months of continuous DRBA therapy.2 Tardive dyskinesia is perhaps the most well-known of the tardive syndromes, but is not the only one to consider when assessing for antipsychotic-induced movement disorders. A key feature differentiating a tardive syndrome is the persistence of the movement disorder after the DRBA is discontinued. In this case, B had been receiving a stable dose of risperidone for >1 year. He developed dystonic posturing of his left wrist and elbow that was both unresponsive to anticholinergic medication and persisted after risperidone was discontinued. The term “tardive” emphasizes the delay in development of abnormal involuntary movement symptoms after initiating antipsychotic medications.3 Table 12 shows a comparison of tardive dystonia vs an acute dystonic reaction.
Continue to: Other tardive syndromes include...
Other tardive syndromes include:
- tardive tics
- tardive parkinsonism
- tardive pain
- tardive myoclonus
- tardive akathisia
- tardive tremors.
The incidence of tardive syndromes increases 5% annually for the first 5 years of treatment. At 10 years of treatment, the annual incidence is thought to be 49%, and at 25 years of treatment, 68%.4 The predominant theory of the pathophysiology of tardive syndromes is that the chronic use of DRBAs causes a gradual hypersensitization of dopamine receptors.4 The diagnosis of a tardive syndrome is based on history of exposure to a DRBA as well as clinical observation of symptoms.
Compared with classic tardive dyskinesia, tardive dystonia is more common among younger patients. The mean age of onset of tardive dystonia is 40, and it typically affects young males.5 Typical posturing observed in cases of tardive dystonia include extension of the arms and flexion at the wrists.6 In contrast to cases of primary dystonia, tardive dystonia is typically associated with stereotypies, akathisia, or other movement disorders. Anticholinergic agents, such as
The American Psychiatric Association has issued guidelines on screening for involuntary movement syndromes by using the Abnormal Involuntary Movement Scale (AIMS).7 The current recommendations include assessment every 6 months for patients receiving first-generation antipsychotics, and every 12 months for those receiving second-generation antipsychotics.7 Prescribers should also carefully assess for any pre-existing involuntary movements before prescribing a DRBA.7
[polldaddy:10615855]
The authors’ observations
In 2013, the American Academy of Neurology (AAN) published guidelines on the treatment of tardive dyskinesia. According to these guidelines, at that time, the treatments with the most evidence supporting their use were clonazepam, ginkgo biloba,
Continue to: In 2017, valbenazine and deutetrabenazine...
In 2017, valbenazine and deutetrabenazine became the first FDA-approved treatments for tardive dyskinesia in adults. Both medications block the vesicular monoamine transporter 2 (VMAT2) system, which results in decreased synaptic dopamine and dopamine receptor stimulation. Both VMAT2 inhibitor medications have a category level A supporting their use for treating tardive dyskinesia.8-10
Currently, there are no published treatment guidelines on pharmacologic management of tardive dystonia. In B’s case, bromocriptine, a dopamine agonist, was used to counter the dopamine-blocking effects of risperidone on the nigrostriatal pathway and improve parkinsonian features of B’s presentation, including bradykinesia, stooped forward posture, and masked facies. Bromocriptine was found to be effective in alleviating parkinsonian features; however, to date there is no evidence demonstrating its effectiveness in countering delayed dystonic effects of DRBAs.
OUTCOME Improvement of dystonia symptoms
One week after discharge, B is seen for a follow-up visit. He continues taking bromocriptine, 1.25 mg twice daily, with meals after discharge. On examination, he has some evidence of tardive dystonia, including flexion of left wrist and posturing while ambulating. B’s parkinsonian features, including stooped forward posture, masked facies, and cogwheel rigidity of the left wrist muscle, have resolved. B is now able to walk on his own without unsteadiness. Bromocriptine is discontinued after 1 month, and his symptoms of dystonia continue to improve.
Two months after hospitalization, B is started on quetiapine, 25 mg twice daily, for behavioral aggression. Quetiapine is chosen because it has a lower dopamine receptor affinity compared with risperidone, and theoretically, quetiapine is associated with a lower risk of developing tardive symptoms. During the next 6 months, B is monitored closely for recurrence of tardive symptoms. Quetiapine is slowly titrated to 25 mg in the morning, and 50 mg at bedtime. His behavioral agitation improves significantly and he does not have a recurrence of tardive symptoms.
Bottom Line
Tardive dystonia is a possible iatrogenic adverse effect for patients receiving long-term dopamine receptor blocking agent (DRBA) therapy. Tardive syndromes encompass delayed-onset movement disorders caused by long-term blockade of the dopamine receptor by antipsychotic agents. Tardive dystonia can be contrasted from acute dystonic reaction based on the time course of development as well as by the persistence of symptoms after DRBAs are withheld.
Continue to: Related Resources
Related Resources
- American Academy of Neurology. Summary of evidence-based guideline for clinicians: treatment of tardive syndromes. https://www.aan.com/Guidelines/Home/GetGuidelineContent/613. Published 2013.
- Dystonia Medical Research Foundation. https://dystonia-foundation.org/.
Drug Brand Names
Amantadine • Gocovri, Symmetrel
Amoxicillin • Amoxil
Baclofen • Kemstro, Liroesal
Benztropine • Cogentin
Bromocriptine • Parlodel
Clonazepam • Klonopin
Deutetrabenazine • Austedo
Galantamine • Razadyne
Quetiapine • Seroquel
Risperidone • Risperdal
Tetrabenazine • Xenazine
Trihexyphenidyl • Artane, Tremin
Valbenazine • Ingrezza
CASE Drooling, unsteady, and not himself
B, age 10, who is left handed and has autism spectrum disorder, is brought to the emergency department (ED) with a 1-day history of drooling, unsteady gait, and left wrist in sustained flexion. His parents report that for the past week, B has had cold symptoms, including rhinorrhea, a low-grade fever (100.0°F), and cough. Earlier in the day, he was seen at his pediatrician’s office, where he was diagnosed with an acute respiratory infection and started on amoxicillin, 500 mg twice daily for 7 days.
At baseline, B is nonverbal. He requires some assistance with his activities of daily living. He usually is able to walk without assistance and dress himself, but he is not toilet trained. His parents report that in the past day, he has had significant difficulties with tasks involving his left hand. Normally, B is able to feed himself “finger foods” but has been unable to do so today. His parents say that he has been unsteady on his feet, and has been “falling forward” when he tries to walk.
Two years ago, B was started on risperidone, 0.5 mg nightly, for behavioral aggression and self-mutilation. Over the next 12 months, the dosage was steadily increased to 1 mg twice daily, with good response. He has been taking his current dosage, 1 mg twice daily, for the past 12 months without adjustment. His parents report there have been no other medication changes, other than starting amoxicillin earlier that day.
As part of his initial ED evaluation, B is found to be mildly dehydrated, with an elevated sedimentation rate on urinalysis. His complete blood count (CBC) with differential is within normal limits. A comprehensive metabolic panel shows a slight increase in his creatinine level, indicating dehydration. B is administered IV fluid replacement because he is having difficulty drinking due to excessive drooling.
The ED physician is concerned that B may be experiencing an acute dystonic reaction from risperidone, so the team holds this medication, and gives B a one-time dose of IV diphenhydramine, 25 mg, for presumptive acute dystonic reaction. After several minutes, there is no improvement in the sustained flexion of his left wrist.
[polldaddy:10615848]
The authors’ observations
B presented with new-onset neurologic findings after a recently diagnosed upper respiratory viral illness. His symptoms appeared to be confined to his left upper extremity, specifically demonstrating left arm extension at the elbow with flexion of the left wrist. He also had new-onset unsteady gait with a stooped forward posture and required assistance with walking. Interestingly, despite B’s history of antipsychotic use, administering an anticholinergic agent did not lessen the dystonic posturing at his wrist and elbow.
EVALUATION Laboratory results reveal new clues
While in the ED, B undergoes MRI of the brain and spinal cord to rule out any mass lesions that could be impinging upon the motor pathways. Both brain and spinal cord imaging appear to be essentially normal, without evidence of impingement of the spinal nerves or lesions involving the brainstem or cerebellum.
Continue to: Due to concerns...
Due to concerns of possible airway obstruction, a CT scan of the neck is obtained to rule out any acute pathology, such as epiglottitis compromising his airway. The scan shows some inflammation and edema in the soft tissues that is thought to be secondary to his acute viral illness. B is able to maintain his airway and oxygenation, so intubation is not necessary.
A CPK test is ordered because there are concerns of sustained muscle contraction of B’s left wrist and elbow. The CPK level is 884 U/L (reference range 26 to 192 U/L). The elevation in CPK is consistent with prior laboratory findings of dehydration and indicating skeletal muscle breakdown from sustained muscle contraction. All other laboratory results, including a comprehensive metabolic panel, urine drug screen, and thyroid screening panel, are within normal limits.
[polldaddy:10615850]
EVALUATION No variation in facial expression
B is admitted to the general pediatrics service. Maintenance IV fluids are started due to concerns of dehydration and possible rhabdomyolysis due to his elevated CPK level. Risperidone is held throughout the hospital course due to concerns for an acute dystonic reaction. B is monitored for several days without clinical improvement and eventually discharged home with a diagnosis of inflammatory mononeuropathy due to viral infection. The patient is told to discontinue risperidone as part of discharge instructions.
Five days later, B returns to the hospital because there was no improvement in his left extremity or walking. His left elbow remains extended with left wrist in flexion. Psychiatry is consulted for further diagnostic clarity and evaluation.
On physical examination, B’s left arm remains unchanged. Despite discontinuing risperidone, there is evidence of cogwheel rigidity of the left wrist joint. Reflexes in the upper and lower extremities are 2+ and symmetrical bilaterally, suggesting intact upper and lower motor pathways. Babinski sign is absent bilaterally, which is a normal finding in B’s age group. B continues to have difficulty with ambulating and appears to “fall forward” while trying to walk with assistance. His parents also say that B is not laughing, smiling, or showing any variation in facial expression.
Continue to: Additional family history...
Additional family history is gathered from B’s parents for possible hereditary movement disorders such as Wilson’s disease. They report that no family members have developed involuntary movements or other neurologic syndromes. Additional considerations on the differential diagnosis for B include juvenile ALS or mononeuropathy involving the C5 and C6 nerve roots. B’s parents deny any recent shoulder trauma, and radiographic studies did not demonstrate any involvement of the nerve roots.
TREATMENT A trial of bromocriptine
At this point, B’s neurologic workup is essentially normal, and he is given a provisional diagnosis of antipsychotic-induced tardive dystonia vs tardive parkinsonism. Risperidone continues to be held, and B is monitored for clinical improvement. B is administered a one-time dose of diphenhydramine, 25 mg, for dystonia with no improvement in symptoms. He is then started on bromocriptine, 1.25 mg twice daily with meals, for parkinsonian symptoms secondary to antipsychotic medication use. After 1 day of treatment, B shows less sustained flexion of his left wrist. He is able to relax his left arm, shows improvements in ambulation, and requires less assistance. B continues to be observed closely and continues to improve toward his baseline.
At Day 4, he is discharged. B is able to walk mostly without assistance and demonstrates improvement in left wrist flexion. He is scheduled to see a movement disorders specialist a week after discharge. The initial diagnosis given by the movement disorder specialist is tardive dystonia.
The authors’ observations
Tardive dyskinesia is a well-known iatrogenic effect of antipsychotic medications that are commonly used to manage conditions such as schizophrenia or behavioral agitation associated with autism spectrum disorder. Symptoms of tardive dyskinesia typically emerge after 1 to 2 years of continuous exposure to dopamine receptor blocking agents (DRBAs). Tardive dyskinesia symptoms include involuntary, repetitive, purposeless movements of the tongue, jaw, lips, face, trunk, and upper and lower extremities, with significant functional impairment.1
Tardive syndromes refer to a diverse array of hyperkinetic, hypokinetic, and sensory movement disorders resulting from at least 3 months of continuous DRBA therapy.2 Tardive dyskinesia is perhaps the most well-known of the tardive syndromes, but is not the only one to consider when assessing for antipsychotic-induced movement disorders. A key feature differentiating a tardive syndrome is the persistence of the movement disorder after the DRBA is discontinued. In this case, B had been receiving a stable dose of risperidone for >1 year. He developed dystonic posturing of his left wrist and elbow that was both unresponsive to anticholinergic medication and persisted after risperidone was discontinued. The term “tardive” emphasizes the delay in development of abnormal involuntary movement symptoms after initiating antipsychotic medications.3 Table 12 shows a comparison of tardive dystonia vs an acute dystonic reaction.
Continue to: Other tardive syndromes include...
Other tardive syndromes include:
- tardive tics
- tardive parkinsonism
- tardive pain
- tardive myoclonus
- tardive akathisia
- tardive tremors.
The incidence of tardive syndromes increases 5% annually for the first 5 years of treatment. At 10 years of treatment, the annual incidence is thought to be 49%, and at 25 years of treatment, 68%.4 The predominant theory of the pathophysiology of tardive syndromes is that the chronic use of DRBAs causes a gradual hypersensitization of dopamine receptors.4 The diagnosis of a tardive syndrome is based on history of exposure to a DRBA as well as clinical observation of symptoms.
Compared with classic tardive dyskinesia, tardive dystonia is more common among younger patients. The mean age of onset of tardive dystonia is 40, and it typically affects young males.5 Typical posturing observed in cases of tardive dystonia include extension of the arms and flexion at the wrists.6 In contrast to cases of primary dystonia, tardive dystonia is typically associated with stereotypies, akathisia, or other movement disorders. Anticholinergic agents, such as
The American Psychiatric Association has issued guidelines on screening for involuntary movement syndromes by using the Abnormal Involuntary Movement Scale (AIMS).7 The current recommendations include assessment every 6 months for patients receiving first-generation antipsychotics, and every 12 months for those receiving second-generation antipsychotics.7 Prescribers should also carefully assess for any pre-existing involuntary movements before prescribing a DRBA.7
[polldaddy:10615855]
The authors’ observations
In 2013, the American Academy of Neurology (AAN) published guidelines on the treatment of tardive dyskinesia. According to these guidelines, at that time, the treatments with the most evidence supporting their use were clonazepam, ginkgo biloba,
Continue to: In 2017, valbenazine and deutetrabenazine...
In 2017, valbenazine and deutetrabenazine became the first FDA-approved treatments for tardive dyskinesia in adults. Both medications block the vesicular monoamine transporter 2 (VMAT2) system, which results in decreased synaptic dopamine and dopamine receptor stimulation. Both VMAT2 inhibitor medications have a category level A supporting their use for treating tardive dyskinesia.8-10
Currently, there are no published treatment guidelines on pharmacologic management of tardive dystonia. In B’s case, bromocriptine, a dopamine agonist, was used to counter the dopamine-blocking effects of risperidone on the nigrostriatal pathway and improve parkinsonian features of B’s presentation, including bradykinesia, stooped forward posture, and masked facies. Bromocriptine was found to be effective in alleviating parkinsonian features; however, to date there is no evidence demonstrating its effectiveness in countering delayed dystonic effects of DRBAs.
OUTCOME Improvement of dystonia symptoms
One week after discharge, B is seen for a follow-up visit. He continues taking bromocriptine, 1.25 mg twice daily, with meals after discharge. On examination, he has some evidence of tardive dystonia, including flexion of left wrist and posturing while ambulating. B’s parkinsonian features, including stooped forward posture, masked facies, and cogwheel rigidity of the left wrist muscle, have resolved. B is now able to walk on his own without unsteadiness. Bromocriptine is discontinued after 1 month, and his symptoms of dystonia continue to improve.
Two months after hospitalization, B is started on quetiapine, 25 mg twice daily, for behavioral aggression. Quetiapine is chosen because it has a lower dopamine receptor affinity compared with risperidone, and theoretically, quetiapine is associated with a lower risk of developing tardive symptoms. During the next 6 months, B is monitored closely for recurrence of tardive symptoms. Quetiapine is slowly titrated to 25 mg in the morning, and 50 mg at bedtime. His behavioral agitation improves significantly and he does not have a recurrence of tardive symptoms.
Bottom Line
Tardive dystonia is a possible iatrogenic adverse effect for patients receiving long-term dopamine receptor blocking agent (DRBA) therapy. Tardive syndromes encompass delayed-onset movement disorders caused by long-term blockade of the dopamine receptor by antipsychotic agents. Tardive dystonia can be contrasted from acute dystonic reaction based on the time course of development as well as by the persistence of symptoms after DRBAs are withheld.
Continue to: Related Resources
Related Resources
- American Academy of Neurology. Summary of evidence-based guideline for clinicians: treatment of tardive syndromes. https://www.aan.com/Guidelines/Home/GetGuidelineContent/613. Published 2013.
- Dystonia Medical Research Foundation. https://dystonia-foundation.org/.
Drug Brand Names
Amantadine • Gocovri, Symmetrel
Amoxicillin • Amoxil
Baclofen • Kemstro, Liroesal
Benztropine • Cogentin
Bromocriptine • Parlodel
Clonazepam • Klonopin
Deutetrabenazine • Austedo
Galantamine • Razadyne
Quetiapine • Seroquel
Risperidone • Risperdal
Tetrabenazine • Xenazine
Trihexyphenidyl • Artane, Tremin
Valbenazine • Ingrezza
1. Margolese HC, Chouinard G, Kolivakis TT, et al. Tardive dyskinesia in the era of typical and atypical antipsychotics. Part 1: pathophysiology and mechanisms of induction. Can J Psychiatr. 2005;50(9):541-547.
2. Truong D, Frei K. Setting the record straight: the nosology of tardive syndromes. Parkinsonism Relat Disord. 2019;59:146-150.
3. Cornett EM, Novitch M, Kaye AD, et al. Medication-induced tardive dyskinesia: a review and update. Ochsner J. 2017;17(2):162-174.
4. Schooler NR, Kane JM. Research diagnoses for tardive dyskinesia. Arch Gen Psychiatry. 1982;39(4):486-487.
5. Fahn S, Jankovic J, Hallett M. Principles and Practice of Movement Disorders. 2nd ed. Philadelphia, PA: Saunders; 2011:415-446.
6. Kang UJ, Burke RE, Fahn S. Natural history and treatment of tardive dystonia. Mov Disord. 1986;1(3):193-208.
7. Lehman AF, Lieberman JA, Dixon LB, et al. Practice guideline for the treatment of patients with schizophrenia, second edition. Am J Psychiatry. 2004;161(suppl 2):1-56.
8. Bhidayasiri R, Fahn S, Weiner WJ, et al, Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
9. Ingrezza [package insert]. San Diego, CA: Neurocrine Biosciences, Inc.; 2020.
10. Austedo [package insert]. North Wales, PA: Teva Pharmaceuticals; 2017.
1. Margolese HC, Chouinard G, Kolivakis TT, et al. Tardive dyskinesia in the era of typical and atypical antipsychotics. Part 1: pathophysiology and mechanisms of induction. Can J Psychiatr. 2005;50(9):541-547.
2. Truong D, Frei K. Setting the record straight: the nosology of tardive syndromes. Parkinsonism Relat Disord. 2019;59:146-150.
3. Cornett EM, Novitch M, Kaye AD, et al. Medication-induced tardive dyskinesia: a review and update. Ochsner J. 2017;17(2):162-174.
4. Schooler NR, Kane JM. Research diagnoses for tardive dyskinesia. Arch Gen Psychiatry. 1982;39(4):486-487.
5. Fahn S, Jankovic J, Hallett M. Principles and Practice of Movement Disorders. 2nd ed. Philadelphia, PA: Saunders; 2011:415-446.
6. Kang UJ, Burke RE, Fahn S. Natural history and treatment of tardive dystonia. Mov Disord. 1986;1(3):193-208.
7. Lehman AF, Lieberman JA, Dixon LB, et al. Practice guideline for the treatment of patients with schizophrenia, second edition. Am J Psychiatry. 2004;161(suppl 2):1-56.
8. Bhidayasiri R, Fahn S, Weiner WJ, et al, Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
9. Ingrezza [package insert]. San Diego, CA: Neurocrine Biosciences, Inc.; 2020.
10. Austedo [package insert]. North Wales, PA: Teva Pharmaceuticals; 2017.

Blood biomarker may predict Parkinson’s disease progression
Although the biomarker, neurofilament light chain (NfL), is not especially specific, it is the first blood-based biomarker for Parkinson’s disease.
Neurofilaments are components of the neural cytoskeleton, where they maintain structure along with other functions. Following axonal damage, NfL gets released into extracellular fluids. Previously, NfL has been detected in cerebrospinal fluid (CSF) in patients with multiple sclerosis and neurodegenerative dementias. NfL in the CSF can distinguish Parkinson’s disease (PD) from multiple system atrophy and progressive supranuclear palsy.
That’s useful, but a serum marker would open new doors. “An easily accessible biomarker that will serve as an indicator of diagnosis, disease state, and progression, as well as a marker of response to therapeutic intervention is needed. A biomarker will strengthen the ability to select patients for inclusion or stratification within clinical trials,” commented Okeanis Vaou, MD, director of the movement disorders program at St. Elizabeth’s Medical Center in Brighton, Mass. Dr. Vaou was not involved in the study, which was published Aug. 15 in Movement Disorders.
A potential biomarker?
To determine if serum NfL levels would correlate with CSF values and had potential as a biomarker, a large, multi-institutional team of researchers led by Brit Mollenhauer, MD, of the University Medical Center Goettingen (Germany), and Danielle Graham, MD, of Biogen, drew data from a prospective, longitudinal, single-center project called the De Novo Parkinson’s disease (DeNoPa) cohort.
The researchers analyzed data from 176 subjects, including drug-naive patients with newly diagnosed PD; age, sex, and education matched healthy controls; and patients who were initially diagnosed with Parkinson’s disease but had their diagnoses changed to a cognate or neurodegenerative disorder (OND). The researchers also drew 514 serum samples from the prospective longitudinal, observational, international multicenter study Parkinson’s Progression Marker Initiative (PPMI) cohort.
In the DeNoPa cohort, OND patients had the highest median CSF NfL levels at baseline (839 pg/mL) followed by PD patients (562 pg/mL) and healthy controls (494 pg/mL; P = .01). There was a strong correlation between CSF and serum NfL levels in a cross-sectional exploratory study with the PPMI cohort.
Age and sex covariates in the PPMI cohort explained 51% of NfL variability. After adjustment for age and sex, baseline median blood NfL levels were highest in the OND group (16.23 pg/mL), followed by the genetic PD group (13.36 pg/mL), prodromal participants (12.20 pg/mL), PD patients (11.73 pg/mL), unaffected mutation carriers (11.63 pg/mL), and healthy controls (11.05 pg/mL; F test P < .0001). Median serum NfL increased by 3.35% per year of age (P < .0001), and median serum NfL was 6.79% higher in women (P = .0002).
Doubling of adjusted serum NfL levels were associated with a median increase in the Movement Disorder Society Unified Parkinson’s Disease Rating Scale total score of 3.45 points (false-discovery rate–adjusted P = .0115), a median decrease in Symbol Digit Modality Test total score of 1.39 (FDR P = .026), a median decrease in Hopkins Verbal Learning Tests with discrimination recognition score of 0.3 (FDR P = .03), and a median decrease in Hopkins Verbal Learning Tests with retention score of 0.029 (FDR P = .04).
More specific markers needed
The findings are intriguing, said Dr Vaou, but “we need to acknowledge that increased NfL levels are not specific enough to Parkinson’s disease and reflect neuronal and axonal damage. Therefore, there is a need for more specific markers to support diagnostic accuracy, rate of progression, and ultimate prognosis. A serum NfL assay may be useful to clinicians evaluating patients with PD or OND diagnosis and mitigate the misdiagnosis of atypical PD. NfL may be particularly useful in differentiating PD from cognate disorders such as multiple system atrophy, progressive supranuclear palsy, and dementia with Lewy bodies.”
The current success is the result of large patient databases containing phenotypic data, imaging, and tests of tissue, blood, and cerebrospinal fluid, along with collaborations between advocacy groups, academia, and industry, according to Dr. Vaou. As that work continues, it could uncover more specific biomarkers “that will allow us not only to help with diagnosis and treatment but with disease progression, inclusion, recruitment and stratification in clinical studies, as well as (be an) indicator of response to therapeutic intervention of an investigational drug.”
The study was funded by the Michael J. Fox Foundation for Parkinson’s Research. Dr. Vaou had no relevant financial disclosures.
SOURCE: Mollenhauer B et al. Mov Disord. 2020 Aug 15. doi: 10.1002/mds.28206.
Although the biomarker, neurofilament light chain (NfL), is not especially specific, it is the first blood-based biomarker for Parkinson’s disease.
Neurofilaments are components of the neural cytoskeleton, where they maintain structure along with other functions. Following axonal damage, NfL gets released into extracellular fluids. Previously, NfL has been detected in cerebrospinal fluid (CSF) in patients with multiple sclerosis and neurodegenerative dementias. NfL in the CSF can distinguish Parkinson’s disease (PD) from multiple system atrophy and progressive supranuclear palsy.
That’s useful, but a serum marker would open new doors. “An easily accessible biomarker that will serve as an indicator of diagnosis, disease state, and progression, as well as a marker of response to therapeutic intervention is needed. A biomarker will strengthen the ability to select patients for inclusion or stratification within clinical trials,” commented Okeanis Vaou, MD, director of the movement disorders program at St. Elizabeth’s Medical Center in Brighton, Mass. Dr. Vaou was not involved in the study, which was published Aug. 15 in Movement Disorders.
A potential biomarker?
To determine if serum NfL levels would correlate with CSF values and had potential as a biomarker, a large, multi-institutional team of researchers led by Brit Mollenhauer, MD, of the University Medical Center Goettingen (Germany), and Danielle Graham, MD, of Biogen, drew data from a prospective, longitudinal, single-center project called the De Novo Parkinson’s disease (DeNoPa) cohort.
The researchers analyzed data from 176 subjects, including drug-naive patients with newly diagnosed PD; age, sex, and education matched healthy controls; and patients who were initially diagnosed with Parkinson’s disease but had their diagnoses changed to a cognate or neurodegenerative disorder (OND). The researchers also drew 514 serum samples from the prospective longitudinal, observational, international multicenter study Parkinson’s Progression Marker Initiative (PPMI) cohort.
In the DeNoPa cohort, OND patients had the highest median CSF NfL levels at baseline (839 pg/mL) followed by PD patients (562 pg/mL) and healthy controls (494 pg/mL; P = .01). There was a strong correlation between CSF and serum NfL levels in a cross-sectional exploratory study with the PPMI cohort.
Age and sex covariates in the PPMI cohort explained 51% of NfL variability. After adjustment for age and sex, baseline median blood NfL levels were highest in the OND group (16.23 pg/mL), followed by the genetic PD group (13.36 pg/mL), prodromal participants (12.20 pg/mL), PD patients (11.73 pg/mL), unaffected mutation carriers (11.63 pg/mL), and healthy controls (11.05 pg/mL; F test P < .0001). Median serum NfL increased by 3.35% per year of age (P < .0001), and median serum NfL was 6.79% higher in women (P = .0002).
Doubling of adjusted serum NfL levels were associated with a median increase in the Movement Disorder Society Unified Parkinson’s Disease Rating Scale total score of 3.45 points (false-discovery rate–adjusted P = .0115), a median decrease in Symbol Digit Modality Test total score of 1.39 (FDR P = .026), a median decrease in Hopkins Verbal Learning Tests with discrimination recognition score of 0.3 (FDR P = .03), and a median decrease in Hopkins Verbal Learning Tests with retention score of 0.029 (FDR P = .04).
More specific markers needed
The findings are intriguing, said Dr Vaou, but “we need to acknowledge that increased NfL levels are not specific enough to Parkinson’s disease and reflect neuronal and axonal damage. Therefore, there is a need for more specific markers to support diagnostic accuracy, rate of progression, and ultimate prognosis. A serum NfL assay may be useful to clinicians evaluating patients with PD or OND diagnosis and mitigate the misdiagnosis of atypical PD. NfL may be particularly useful in differentiating PD from cognate disorders such as multiple system atrophy, progressive supranuclear palsy, and dementia with Lewy bodies.”
The current success is the result of large patient databases containing phenotypic data, imaging, and tests of tissue, blood, and cerebrospinal fluid, along with collaborations between advocacy groups, academia, and industry, according to Dr. Vaou. As that work continues, it could uncover more specific biomarkers “that will allow us not only to help with diagnosis and treatment but with disease progression, inclusion, recruitment and stratification in clinical studies, as well as (be an) indicator of response to therapeutic intervention of an investigational drug.”
The study was funded by the Michael J. Fox Foundation for Parkinson’s Research. Dr. Vaou had no relevant financial disclosures.
SOURCE: Mollenhauer B et al. Mov Disord. 2020 Aug 15. doi: 10.1002/mds.28206.
Although the biomarker, neurofilament light chain (NfL), is not especially specific, it is the first blood-based biomarker for Parkinson’s disease.
Neurofilaments are components of the neural cytoskeleton, where they maintain structure along with other functions. Following axonal damage, NfL gets released into extracellular fluids. Previously, NfL has been detected in cerebrospinal fluid (CSF) in patients with multiple sclerosis and neurodegenerative dementias. NfL in the CSF can distinguish Parkinson’s disease (PD) from multiple system atrophy and progressive supranuclear palsy.
That’s useful, but a serum marker would open new doors. “An easily accessible biomarker that will serve as an indicator of diagnosis, disease state, and progression, as well as a marker of response to therapeutic intervention is needed. A biomarker will strengthen the ability to select patients for inclusion or stratification within clinical trials,” commented Okeanis Vaou, MD, director of the movement disorders program at St. Elizabeth’s Medical Center in Brighton, Mass. Dr. Vaou was not involved in the study, which was published Aug. 15 in Movement Disorders.
A potential biomarker?
To determine if serum NfL levels would correlate with CSF values and had potential as a biomarker, a large, multi-institutional team of researchers led by Brit Mollenhauer, MD, of the University Medical Center Goettingen (Germany), and Danielle Graham, MD, of Biogen, drew data from a prospective, longitudinal, single-center project called the De Novo Parkinson’s disease (DeNoPa) cohort.
The researchers analyzed data from 176 subjects, including drug-naive patients with newly diagnosed PD; age, sex, and education matched healthy controls; and patients who were initially diagnosed with Parkinson’s disease but had their diagnoses changed to a cognate or neurodegenerative disorder (OND). The researchers also drew 514 serum samples from the prospective longitudinal, observational, international multicenter study Parkinson’s Progression Marker Initiative (PPMI) cohort.
In the DeNoPa cohort, OND patients had the highest median CSF NfL levels at baseline (839 pg/mL) followed by PD patients (562 pg/mL) and healthy controls (494 pg/mL; P = .01). There was a strong correlation between CSF and serum NfL levels in a cross-sectional exploratory study with the PPMI cohort.
Age and sex covariates in the PPMI cohort explained 51% of NfL variability. After adjustment for age and sex, baseline median blood NfL levels were highest in the OND group (16.23 pg/mL), followed by the genetic PD group (13.36 pg/mL), prodromal participants (12.20 pg/mL), PD patients (11.73 pg/mL), unaffected mutation carriers (11.63 pg/mL), and healthy controls (11.05 pg/mL; F test P < .0001). Median serum NfL increased by 3.35% per year of age (P < .0001), and median serum NfL was 6.79% higher in women (P = .0002).
Doubling of adjusted serum NfL levels were associated with a median increase in the Movement Disorder Society Unified Parkinson’s Disease Rating Scale total score of 3.45 points (false-discovery rate–adjusted P = .0115), a median decrease in Symbol Digit Modality Test total score of 1.39 (FDR P = .026), a median decrease in Hopkins Verbal Learning Tests with discrimination recognition score of 0.3 (FDR P = .03), and a median decrease in Hopkins Verbal Learning Tests with retention score of 0.029 (FDR P = .04).
More specific markers needed
The findings are intriguing, said Dr Vaou, but “we need to acknowledge that increased NfL levels are not specific enough to Parkinson’s disease and reflect neuronal and axonal damage. Therefore, there is a need for more specific markers to support diagnostic accuracy, rate of progression, and ultimate prognosis. A serum NfL assay may be useful to clinicians evaluating patients with PD or OND diagnosis and mitigate the misdiagnosis of atypical PD. NfL may be particularly useful in differentiating PD from cognate disorders such as multiple system atrophy, progressive supranuclear palsy, and dementia with Lewy bodies.”
The current success is the result of large patient databases containing phenotypic data, imaging, and tests of tissue, blood, and cerebrospinal fluid, along with collaborations between advocacy groups, academia, and industry, according to Dr. Vaou. As that work continues, it could uncover more specific biomarkers “that will allow us not only to help with diagnosis and treatment but with disease progression, inclusion, recruitment and stratification in clinical studies, as well as (be an) indicator of response to therapeutic intervention of an investigational drug.”
The study was funded by the Michael J. Fox Foundation for Parkinson’s Research. Dr. Vaou had no relevant financial disclosures.
SOURCE: Mollenhauer B et al. Mov Disord. 2020 Aug 15. doi: 10.1002/mds.28206.
FROM MOVEMENT DISORDERS
Can DBS in early Parkinson’s disease reduce disease progression?
According to the investigators, a larger trial is needed to confirm these findings, which were published online ahead of print June 29 in Neurology.

Adverse events were similar between patients who underwent DBS and drug therapy and those who underwent drug therapy alone. This result is a preliminary indication of the safety of long-term DBS therapy, according to the researchers. Furthermore, patients who received DBS required a significantly lower levodopa equivalent daily dose (LEDD) and were less likely to need polypharmacy than were patients who received medical treatment alone.
“While we can be really excited about these findings, we can’t change our practice, what we recommend to patients, based on this [study],” said David Charles, MD, professor and vice chair of neurology at Vanderbilt University, Nashville, Tenn. “We have to do the next trial to get that class of evidence.”
An extension of a pilot trial
Previous research has indicated that treatment with DBS and optimal medical therapy provides benefits beyond those of medical therapy alone in patients with mid-stage or advanced Parkinson’s disease. Dr. Charles and colleagues conducted a randomized, single-blind pilot study to examine the safety and tolerability of STN DBS in 30 patients with early Parkinson’s disease. Eligible participants had Hoehn and Yahr stage II off medication, were between 50 and 75 years of age, had taken medication for 6 months to 4 years, and had no dyskinesia or other motor fluctuations.
Patients were randomly assigned in equal groups to optimal drug therapy plus STN DBS or to drug therapy alone. Investigators evaluated patients every 6 months for 2 years. The results suggested that STN DBS was safe and slowed the progression of rest tremor in this population.
Apart from research that included patients with advanced Parkinson’s disease, data relating to long-term follow-up of patients undergoing DBS for Parkinson’s disease have been limited. Prospective studies have found that DBS provides motor benefits in patients with advanced Parkinson’s disease after 5-10 years, but they have not included control groups of patients randomly assigned to medication alone. Understanding the durability of effect of DBS is particularly important in patients with early Parkinson’s disease, because they could be exposed to stimulation for a longer time than other patients.
DBS may slow progression of rest tremor
Dr. Charles and colleagues invited patients who completed their pilot study to participate in an observational follow-up study. All 29 patients who completed the pilot study consented to participate in the follow-up. The investigators conducted annual outpatient examinations at 3, 4, and 5 years after baseline. These examinations were similar to those conducted at baseline in the pilot trial. Patients’ scores on the Unified Parkinson’s Disease Rating Scale (UPDRS) Part III were obtained through blinded video assessment. Rigidity was not assessed. The investigators calculated patients’ levodopa equivalent daily dose (LEDD) and total electrical energy delivered (TEED). Adverse events were classified as mild, moderate, or severe.
Because of a problem with study funding, the investigators examined only eight patients in the optimal therapy group and nine patients in the DBS group at 3 years. The final analysis included 28 patients, because one patient was found not to have met inclusion criteria after the trial was completed.
At 5 years, participants’ mean age was 66.1 years. Participants had been taking medications for Parkinson’s disease for a mean duration of 7.2 years. No deaths occurred during the study. Four participants who had been assigned randomly to optimal drug therapy chose to receive STN DBS during the study. The investigators evaluated these participants in the treatment group to which they had been assigned at randomization using an intention-to-treat analysis that compared early STN DBS plus drug therapy with drug therapy alone.
Among patients with early DBS, the odds ratio (OR) of worse UPDRS III scores during 5 years was 0.42, compared with the medical therapy group. The difference in mean UPDRS III score between groups due to randomization was 3.70, which was a clinically important difference, according to the investigators.
In the early DBS group, the OR of worse rest tremor was 0.21, compared with the drug therapy group. The between-group difference in mean rest tremor score favored the DBS group. Excluding rest tremor from participants’ UPDRS III scores eliminated between-group differences in the odds of having worse motor symptoms and in the magnitude of difference of motor symptom score.
In the early DBS group, the OR of requiring a greater LEDD was 0.26, compared with the drug therapy group. The between group difference in mean LEDD significantly favored the DBS group. In addition, at 5 years, the proportion of patients requiring polypharmacy was 93% in the drug therapy group and 43% in the DBS group.
The investigators found no difference between groups in the prevalence of dyskinesia at baseline. At 5 years, the prevalence of dyskinesia was 50% in the drug therapy group and 21% in the DBS group. The difference was not statistically significant, however.
The study groups had similar adverse event profiles. Five adverse events during follow-up were related to surgery or the DBS device. The most common of the 13 study-related adverse events was nausea.
The study’s most significant finding is that “DBS implanted in early Parkinson’s disease decreases the risk of disease progression,” said Dr. Charles. No therapy, including DBS, has been proven to decrease this risk. “This is class II evidence. We have to get class I evidence before we change practice.”
Dr. Charles and colleagues have received Food and Drug Administration approval for a multicenter phase 3 trial to obtain this evidence. The new trial may extend findings regarding DBS in mid-stage and advanced Parkinson’s disease to early-stage Parkinson’s disease. That is, it may show that DBS plus drug therapy in early stage Parkinson’s disease is safe, efficacious, and superior to standard medical therapy alone. “But the reason to do the trial is to determine if it changes or slows the progression of the disease,” said Dr. Charles.
Effect on dyskinesia is unclear
“If a patient does go on to develop problems that need DBS management, and only a small fraction of patients with Parkinson’s disease evolve to this need, then this procedure can be performed at that time,” said Peter A. LeWitt, MD, Sastry Foundation Endowed Chair in Neurology at Wayne State University in Detroit.
“One confound of the study is that DBS provides symptomatic relief of dyskinesias if a patient has developed this problem after a few years of levodopa treatment,” Dr. LeWitt added. “To demonstrate that early use of DBS prevented the development of dyskinesias, the study design should have included a period of turning off the stimulators to determine whether the generation of dyskinesias was prevented, rather than merely suppressed by DBS, as any patient would experience.
“Finally, the goal of reducing use of levodopa dose medications or polypharmacy doesn’t justify subjecting a patient to a brain operation that is not without risks and great expense,” Dr. LeWitt continued. “The results of this underpowered study add to my opinion that the ‘premature’ use of DBS is not a good idea for the management of Parkinson’s disease.”
Medtronic, which manufactures the DBS device that the investigators used, provided part of the study’s funding. Vanderbilt University receives income for research or educational programs that Dr. Charles leads. Dr. LeWitt had no pertinent disclosures.
SOURCE: Hacker ML et al. Neurology. 2020 Jun 29. doi: 10.1212/WNL.0000000000009946.
According to the investigators, a larger trial is needed to confirm these findings, which were published online ahead of print June 29 in Neurology.
Adverse events were similar between patients who underwent DBS and drug therapy and those who underwent drug therapy alone. This result is a preliminary indication of the safety of long-term DBS therapy, according to the researchers. Furthermore, patients who received DBS required a significantly lower levodopa equivalent daily dose (LEDD) and were less likely to need polypharmacy than were patients who received medical treatment alone.
“While we can be really excited about these findings, we can’t change our practice, what we recommend to patients, based on this [study],” said David Charles, MD, professor and vice chair of neurology at Vanderbilt University, Nashville, Tenn. “We have to do the next trial to get that class of evidence.”
An extension of a pilot trial
Previous research has indicated that treatment with DBS and optimal medical therapy provides benefits beyond those of medical therapy alone in patients with mid-stage or advanced Parkinson’s disease. Dr. Charles and colleagues conducted a randomized, single-blind pilot study to examine the safety and tolerability of STN DBS in 30 patients with early Parkinson’s disease. Eligible participants had Hoehn and Yahr stage II off medication, were between 50 and 75 years of age, had taken medication for 6 months to 4 years, and had no dyskinesia or other motor fluctuations.
Patients were randomly assigned in equal groups to optimal drug therapy plus STN DBS or to drug therapy alone. Investigators evaluated patients every 6 months for 2 years. The results suggested that STN DBS was safe and slowed the progression of rest tremor in this population.
Apart from research that included patients with advanced Parkinson’s disease, data relating to long-term follow-up of patients undergoing DBS for Parkinson’s disease have been limited. Prospective studies have found that DBS provides motor benefits in patients with advanced Parkinson’s disease after 5-10 years, but they have not included control groups of patients randomly assigned to medication alone. Understanding the durability of effect of DBS is particularly important in patients with early Parkinson’s disease, because they could be exposed to stimulation for a longer time than other patients.
DBS may slow progression of rest tremor
Dr. Charles and colleagues invited patients who completed their pilot study to participate in an observational follow-up study. All 29 patients who completed the pilot study consented to participate in the follow-up. The investigators conducted annual outpatient examinations at 3, 4, and 5 years after baseline. These examinations were similar to those conducted at baseline in the pilot trial. Patients’ scores on the Unified Parkinson’s Disease Rating Scale (UPDRS) Part III were obtained through blinded video assessment. Rigidity was not assessed. The investigators calculated patients’ levodopa equivalent daily dose (LEDD) and total electrical energy delivered (TEED). Adverse events were classified as mild, moderate, or severe.
Because of a problem with study funding, the investigators examined only eight patients in the optimal therapy group and nine patients in the DBS group at 3 years. The final analysis included 28 patients, because one patient was found not to have met inclusion criteria after the trial was completed.
At 5 years, participants’ mean age was 66.1 years. Participants had been taking medications for Parkinson’s disease for a mean duration of 7.2 years. No deaths occurred during the study. Four participants who had been assigned randomly to optimal drug therapy chose to receive STN DBS during the study. The investigators evaluated these participants in the treatment group to which they had been assigned at randomization using an intention-to-treat analysis that compared early STN DBS plus drug therapy with drug therapy alone.
Among patients with early DBS, the odds ratio (OR) of worse UPDRS III scores during 5 years was 0.42, compared with the medical therapy group. The difference in mean UPDRS III score between groups due to randomization was 3.70, which was a clinically important difference, according to the investigators.
In the early DBS group, the OR of worse rest tremor was 0.21, compared with the drug therapy group. The between-group difference in mean rest tremor score favored the DBS group. Excluding rest tremor from participants’ UPDRS III scores eliminated between-group differences in the odds of having worse motor symptoms and in the magnitude of difference of motor symptom score.
In the early DBS group, the OR of requiring a greater LEDD was 0.26, compared with the drug therapy group. The between group difference in mean LEDD significantly favored the DBS group. In addition, at 5 years, the proportion of patients requiring polypharmacy was 93% in the drug therapy group and 43% in the DBS group.
The investigators found no difference between groups in the prevalence of dyskinesia at baseline. At 5 years, the prevalence of dyskinesia was 50% in the drug therapy group and 21% in the DBS group. The difference was not statistically significant, however.
The study groups had similar adverse event profiles. Five adverse events during follow-up were related to surgery or the DBS device. The most common of the 13 study-related adverse events was nausea.
The study’s most significant finding is that “DBS implanted in early Parkinson’s disease decreases the risk of disease progression,” said Dr. Charles. No therapy, including DBS, has been proven to decrease this risk. “This is class II evidence. We have to get class I evidence before we change practice.”
Dr. Charles and colleagues have received Food and Drug Administration approval for a multicenter phase 3 trial to obtain this evidence. The new trial may extend findings regarding DBS in mid-stage and advanced Parkinson’s disease to early-stage Parkinson’s disease. That is, it may show that DBS plus drug therapy in early stage Parkinson’s disease is safe, efficacious, and superior to standard medical therapy alone. “But the reason to do the trial is to determine if it changes or slows the progression of the disease,” said Dr. Charles.
Effect on dyskinesia is unclear
“If a patient does go on to develop problems that need DBS management, and only a small fraction of patients with Parkinson’s disease evolve to this need, then this procedure can be performed at that time,” said Peter A. LeWitt, MD, Sastry Foundation Endowed Chair in Neurology at Wayne State University in Detroit.
“One confound of the study is that DBS provides symptomatic relief of dyskinesias if a patient has developed this problem after a few years of levodopa treatment,” Dr. LeWitt added. “To demonstrate that early use of DBS prevented the development of dyskinesias, the study design should have included a period of turning off the stimulators to determine whether the generation of dyskinesias was prevented, rather than merely suppressed by DBS, as any patient would experience.
“Finally, the goal of reducing use of levodopa dose medications or polypharmacy doesn’t justify subjecting a patient to a brain operation that is not without risks and great expense,” Dr. LeWitt continued. “The results of this underpowered study add to my opinion that the ‘premature’ use of DBS is not a good idea for the management of Parkinson’s disease.”
Medtronic, which manufactures the DBS device that the investigators used, provided part of the study’s funding. Vanderbilt University receives income for research or educational programs that Dr. Charles leads. Dr. LeWitt had no pertinent disclosures.
SOURCE: Hacker ML et al. Neurology. 2020 Jun 29. doi: 10.1212/WNL.0000000000009946.
According to the investigators, a larger trial is needed to confirm these findings, which were published online ahead of print June 29 in Neurology.
Adverse events were similar between patients who underwent DBS and drug therapy and those who underwent drug therapy alone. This result is a preliminary indication of the safety of long-term DBS therapy, according to the researchers. Furthermore, patients who received DBS required a significantly lower levodopa equivalent daily dose (LEDD) and were less likely to need polypharmacy than were patients who received medical treatment alone.
“While we can be really excited about these findings, we can’t change our practice, what we recommend to patients, based on this [study],” said David Charles, MD, professor and vice chair of neurology at Vanderbilt University, Nashville, Tenn. “We have to do the next trial to get that class of evidence.”
An extension of a pilot trial
Previous research has indicated that treatment with DBS and optimal medical therapy provides benefits beyond those of medical therapy alone in patients with mid-stage or advanced Parkinson’s disease. Dr. Charles and colleagues conducted a randomized, single-blind pilot study to examine the safety and tolerability of STN DBS in 30 patients with early Parkinson’s disease. Eligible participants had Hoehn and Yahr stage II off medication, were between 50 and 75 years of age, had taken medication for 6 months to 4 years, and had no dyskinesia or other motor fluctuations.
Patients were randomly assigned in equal groups to optimal drug therapy plus STN DBS or to drug therapy alone. Investigators evaluated patients every 6 months for 2 years. The results suggested that STN DBS was safe and slowed the progression of rest tremor in this population.
Apart from research that included patients with advanced Parkinson’s disease, data relating to long-term follow-up of patients undergoing DBS for Parkinson’s disease have been limited. Prospective studies have found that DBS provides motor benefits in patients with advanced Parkinson’s disease after 5-10 years, but they have not included control groups of patients randomly assigned to medication alone. Understanding the durability of effect of DBS is particularly important in patients with early Parkinson’s disease, because they could be exposed to stimulation for a longer time than other patients.
DBS may slow progression of rest tremor
Dr. Charles and colleagues invited patients who completed their pilot study to participate in an observational follow-up study. All 29 patients who completed the pilot study consented to participate in the follow-up. The investigators conducted annual outpatient examinations at 3, 4, and 5 years after baseline. These examinations were similar to those conducted at baseline in the pilot trial. Patients’ scores on the Unified Parkinson’s Disease Rating Scale (UPDRS) Part III were obtained through blinded video assessment. Rigidity was not assessed. The investigators calculated patients’ levodopa equivalent daily dose (LEDD) and total electrical energy delivered (TEED). Adverse events were classified as mild, moderate, or severe.
Because of a problem with study funding, the investigators examined only eight patients in the optimal therapy group and nine patients in the DBS group at 3 years. The final analysis included 28 patients, because one patient was found not to have met inclusion criteria after the trial was completed.
At 5 years, participants’ mean age was 66.1 years. Participants had been taking medications for Parkinson’s disease for a mean duration of 7.2 years. No deaths occurred during the study. Four participants who had been assigned randomly to optimal drug therapy chose to receive STN DBS during the study. The investigators evaluated these participants in the treatment group to which they had been assigned at randomization using an intention-to-treat analysis that compared early STN DBS plus drug therapy with drug therapy alone.
Among patients with early DBS, the odds ratio (OR) of worse UPDRS III scores during 5 years was 0.42, compared with the medical therapy group. The difference in mean UPDRS III score between groups due to randomization was 3.70, which was a clinically important difference, according to the investigators.
In the early DBS group, the OR of worse rest tremor was 0.21, compared with the drug therapy group. The between-group difference in mean rest tremor score favored the DBS group. Excluding rest tremor from participants’ UPDRS III scores eliminated between-group differences in the odds of having worse motor symptoms and in the magnitude of difference of motor symptom score.
In the early DBS group, the OR of requiring a greater LEDD was 0.26, compared with the drug therapy group. The between group difference in mean LEDD significantly favored the DBS group. In addition, at 5 years, the proportion of patients requiring polypharmacy was 93% in the drug therapy group and 43% in the DBS group.
The investigators found no difference between groups in the prevalence of dyskinesia at baseline. At 5 years, the prevalence of dyskinesia was 50% in the drug therapy group and 21% in the DBS group. The difference was not statistically significant, however.
The study groups had similar adverse event profiles. Five adverse events during follow-up were related to surgery or the DBS device. The most common of the 13 study-related adverse events was nausea.
The study’s most significant finding is that “DBS implanted in early Parkinson’s disease decreases the risk of disease progression,” said Dr. Charles. No therapy, including DBS, has been proven to decrease this risk. “This is class II evidence. We have to get class I evidence before we change practice.”
Dr. Charles and colleagues have received Food and Drug Administration approval for a multicenter phase 3 trial to obtain this evidence. The new trial may extend findings regarding DBS in mid-stage and advanced Parkinson’s disease to early-stage Parkinson’s disease. That is, it may show that DBS plus drug therapy in early stage Parkinson’s disease is safe, efficacious, and superior to standard medical therapy alone. “But the reason to do the trial is to determine if it changes or slows the progression of the disease,” said Dr. Charles.
Effect on dyskinesia is unclear
“If a patient does go on to develop problems that need DBS management, and only a small fraction of patients with Parkinson’s disease evolve to this need, then this procedure can be performed at that time,” said Peter A. LeWitt, MD, Sastry Foundation Endowed Chair in Neurology at Wayne State University in Detroit.
“One confound of the study is that DBS provides symptomatic relief of dyskinesias if a patient has developed this problem after a few years of levodopa treatment,” Dr. LeWitt added. “To demonstrate that early use of DBS prevented the development of dyskinesias, the study design should have included a period of turning off the stimulators to determine whether the generation of dyskinesias was prevented, rather than merely suppressed by DBS, as any patient would experience.
“Finally, the goal of reducing use of levodopa dose medications or polypharmacy doesn’t justify subjecting a patient to a brain operation that is not without risks and great expense,” Dr. LeWitt continued. “The results of this underpowered study add to my opinion that the ‘premature’ use of DBS is not a good idea for the management of Parkinson’s disease.”
Medtronic, which manufactures the DBS device that the investigators used, provided part of the study’s funding. Vanderbilt University receives income for research or educational programs that Dr. Charles leads. Dr. LeWitt had no pertinent disclosures.
SOURCE: Hacker ML et al. Neurology. 2020 Jun 29. doi: 10.1212/WNL.0000000000009946.
FROM NEUROLOGY
Circadian rhythm changes linked to future Parkinson’s disease risk
a new study suggests. “We found that men with abnormal circadian rhythms had three times the risk of developing Parkinson’s disease over an 11-year follow-up period,” lead author, Yue Leng, MD, University of California, San Francisco, said in an interview.
“If confirmed to be a risk factor for Parkinson’s disease, then circadian rhythmicity could be a promising intervention target and will open new opportunities for the prevention and management of Parkinson’s disease,” the researchers concluded.
The study was published online in JAMA Neurology on June 15.
Circadian disruption is very common in neurodegenerative diseases such as Parkinson’s disease, but there isn’t much information on how it may predict the disease, Dr. Leng explained. “We wanted to see whether circadian abnormalities may predict Parkinson’s disease,” she said. “Parkinson’s disease has a long prodromal phase where brain changes have started to occur but no clinical symptoms have become evident. It would be useful to be able to identify these patients, and maybe changes in circadian rhythms may help us to do that,” she added.
For the study, the researchers analyzed data from 2,930 community-dwelling men aged 65 years or older (mean age, 76 years) who participated in the Osteoporotic Fractures in Men Study, in which they underwent comprehensive sleep and rest-activity rhythms assessment. “Patterns of rest and activity were measured with an actigraph device, which is worn on the wrist like a watch and captures movements which are translated into a rest-activity rhythm model – one of the most commonly used and evidence-based measures of circadian rhythm,” Dr. Leng said. Men were asked to wear the actigraphs continuously for a minimum of three 24-hour periods.
Results showed that 78 men (2.7%) developed Parkinson’s disease during the 11-year follow-up. After accounting for all covariates, the risk of Parkinson’s disease increased with decreasing circadian amplitude (strength of the rhythm) with an odds ratio of 1.77 per each decrease by one standard deviation; mesor (mean level of activity) with an odds ratio of 1.64; or robustness (how closely activity follows a 24-hour pattern) with an odds ratio of 1.54.
Those in the lowest quartile of amplitude, mesor, or robustness had approximately three times the risk of developing Parkinson’s disease compared with those in the highest quartile of amplitude. The association remained after further adjustment for nighttime sleep disturbances.
“It has previously been shown that daytime napping has been linked to risk of developing Parkinson’s disease. Now we have shown that abnormalities in the overall 24-hour circadian rest activity rhythm are also present in the prodromal phase of Parkinson’s disease, and this association was independent of several confounders, including nighttime sleep disturbances,” Dr. Leng said.
“This raises awareness of the importance of circadian rhythm in older individuals and changes in their 24-hour pattern of behavior could be an early signal of Parkinson’s disease,” she said.
“This study does not tell us whether these circadian changes are causal for Parkinson’s or not,” Dr. Leng noted.
Future studies are needed to explore underlying mechanisms and to determine whether circadian disruption itself might contribute to the development of Parkinson’s disease, the researchers said.
“If there is a causal link, then using techniques to improve circadian rhythm could help to prevent or slow the onset of Parkinson’s disease,” Dr. Leng suggested. There are many established therapies that act on circadian rhythm including bright light therapy, melatonin, and chronotherapy, she added.
Support for this study was provided by the National Institute on Aging (NIA); the National Institute of Arthritis and Musculoskeletal and Skin Diseases; the National Center for Advancing Translational Sciences; the National Heart, Lung, and Blood Institute; and the Weill Pilot Award. Dr. Leng reported grants from the NIA and the University of California, San Francisco, Weill Institute for Neurosciences during the conduct of the study; and grants from Global Brain Health Institute, the Alzheimer’s Association, and the Alzheimer’s Society outside the submitted work.
A version of this article originally appeared on Medscape.com.
a new study suggests. “We found that men with abnormal circadian rhythms had three times the risk of developing Parkinson’s disease over an 11-year follow-up period,” lead author, Yue Leng, MD, University of California, San Francisco, said in an interview.
“If confirmed to be a risk factor for Parkinson’s disease, then circadian rhythmicity could be a promising intervention target and will open new opportunities for the prevention and management of Parkinson’s disease,” the researchers concluded.
The study was published online in JAMA Neurology on June 15.
Circadian disruption is very common in neurodegenerative diseases such as Parkinson’s disease, but there isn’t much information on how it may predict the disease, Dr. Leng explained. “We wanted to see whether circadian abnormalities may predict Parkinson’s disease,” she said. “Parkinson’s disease has a long prodromal phase where brain changes have started to occur but no clinical symptoms have become evident. It would be useful to be able to identify these patients, and maybe changes in circadian rhythms may help us to do that,” she added.
For the study, the researchers analyzed data from 2,930 community-dwelling men aged 65 years or older (mean age, 76 years) who participated in the Osteoporotic Fractures in Men Study, in which they underwent comprehensive sleep and rest-activity rhythms assessment. “Patterns of rest and activity were measured with an actigraph device, which is worn on the wrist like a watch and captures movements which are translated into a rest-activity rhythm model – one of the most commonly used and evidence-based measures of circadian rhythm,” Dr. Leng said. Men were asked to wear the actigraphs continuously for a minimum of three 24-hour periods.
Results showed that 78 men (2.7%) developed Parkinson’s disease during the 11-year follow-up. After accounting for all covariates, the risk of Parkinson’s disease increased with decreasing circadian amplitude (strength of the rhythm) with an odds ratio of 1.77 per each decrease by one standard deviation; mesor (mean level of activity) with an odds ratio of 1.64; or robustness (how closely activity follows a 24-hour pattern) with an odds ratio of 1.54.
Those in the lowest quartile of amplitude, mesor, or robustness had approximately three times the risk of developing Parkinson’s disease compared with those in the highest quartile of amplitude. The association remained after further adjustment for nighttime sleep disturbances.
“It has previously been shown that daytime napping has been linked to risk of developing Parkinson’s disease. Now we have shown that abnormalities in the overall 24-hour circadian rest activity rhythm are also present in the prodromal phase of Parkinson’s disease, and this association was independent of several confounders, including nighttime sleep disturbances,” Dr. Leng said.
“This raises awareness of the importance of circadian rhythm in older individuals and changes in their 24-hour pattern of behavior could be an early signal of Parkinson’s disease,” she said.
“This study does not tell us whether these circadian changes are causal for Parkinson’s or not,” Dr. Leng noted.
Future studies are needed to explore underlying mechanisms and to determine whether circadian disruption itself might contribute to the development of Parkinson’s disease, the researchers said.
“If there is a causal link, then using techniques to improve circadian rhythm could help to prevent or slow the onset of Parkinson’s disease,” Dr. Leng suggested. There are many established therapies that act on circadian rhythm including bright light therapy, melatonin, and chronotherapy, she added.
Support for this study was provided by the National Institute on Aging (NIA); the National Institute of Arthritis and Musculoskeletal and Skin Diseases; the National Center for Advancing Translational Sciences; the National Heart, Lung, and Blood Institute; and the Weill Pilot Award. Dr. Leng reported grants from the NIA and the University of California, San Francisco, Weill Institute for Neurosciences during the conduct of the study; and grants from Global Brain Health Institute, the Alzheimer’s Association, and the Alzheimer’s Society outside the submitted work.
A version of this article originally appeared on Medscape.com.
a new study suggests. “We found that men with abnormal circadian rhythms had three times the risk of developing Parkinson’s disease over an 11-year follow-up period,” lead author, Yue Leng, MD, University of California, San Francisco, said in an interview.
“If confirmed to be a risk factor for Parkinson’s disease, then circadian rhythmicity could be a promising intervention target and will open new opportunities for the prevention and management of Parkinson’s disease,” the researchers concluded.
The study was published online in JAMA Neurology on June 15.
Circadian disruption is very common in neurodegenerative diseases such as Parkinson’s disease, but there isn’t much information on how it may predict the disease, Dr. Leng explained. “We wanted to see whether circadian abnormalities may predict Parkinson’s disease,” she said. “Parkinson’s disease has a long prodromal phase where brain changes have started to occur but no clinical symptoms have become evident. It would be useful to be able to identify these patients, and maybe changes in circadian rhythms may help us to do that,” she added.
For the study, the researchers analyzed data from 2,930 community-dwelling men aged 65 years or older (mean age, 76 years) who participated in the Osteoporotic Fractures in Men Study, in which they underwent comprehensive sleep and rest-activity rhythms assessment. “Patterns of rest and activity were measured with an actigraph device, which is worn on the wrist like a watch and captures movements which are translated into a rest-activity rhythm model – one of the most commonly used and evidence-based measures of circadian rhythm,” Dr. Leng said. Men were asked to wear the actigraphs continuously for a minimum of three 24-hour periods.
Results showed that 78 men (2.7%) developed Parkinson’s disease during the 11-year follow-up. After accounting for all covariates, the risk of Parkinson’s disease increased with decreasing circadian amplitude (strength of the rhythm) with an odds ratio of 1.77 per each decrease by one standard deviation; mesor (mean level of activity) with an odds ratio of 1.64; or robustness (how closely activity follows a 24-hour pattern) with an odds ratio of 1.54.
Those in the lowest quartile of amplitude, mesor, or robustness had approximately three times the risk of developing Parkinson’s disease compared with those in the highest quartile of amplitude. The association remained after further adjustment for nighttime sleep disturbances.
“It has previously been shown that daytime napping has been linked to risk of developing Parkinson’s disease. Now we have shown that abnormalities in the overall 24-hour circadian rest activity rhythm are also present in the prodromal phase of Parkinson’s disease, and this association was independent of several confounders, including nighttime sleep disturbances,” Dr. Leng said.
“This raises awareness of the importance of circadian rhythm in older individuals and changes in their 24-hour pattern of behavior could be an early signal of Parkinson’s disease,” she said.
“This study does not tell us whether these circadian changes are causal for Parkinson’s or not,” Dr. Leng noted.
Future studies are needed to explore underlying mechanisms and to determine whether circadian disruption itself might contribute to the development of Parkinson’s disease, the researchers said.
“If there is a causal link, then using techniques to improve circadian rhythm could help to prevent or slow the onset of Parkinson’s disease,” Dr. Leng suggested. There are many established therapies that act on circadian rhythm including bright light therapy, melatonin, and chronotherapy, she added.
Support for this study was provided by the National Institute on Aging (NIA); the National Institute of Arthritis and Musculoskeletal and Skin Diseases; the National Center for Advancing Translational Sciences; the National Heart, Lung, and Blood Institute; and the Weill Pilot Award. Dr. Leng reported grants from the NIA and the University of California, San Francisco, Weill Institute for Neurosciences during the conduct of the study; and grants from Global Brain Health Institute, the Alzheimer’s Association, and the Alzheimer’s Society outside the submitted work.
A version of this article originally appeared on Medscape.com.
FROM JAMA NEUROLOGY
Huntington’s disease biomarkers appear 24 years before clinical symptoms
, according to a study published in the June Lancet Neurology. The data come from the Huntington’s disease Young Adult Study (HD-YAS) conducted in the United Kingdom.
The genetic cause of Huntington’s disease provides a potential target for biomarker treatment, wrote joint first authors Rachael I. Scahill, PhD, and Paul Zeun, BMBS, of University College London and colleagues.
“A detailed characterization of the premanifest period in Huntington’s disease is crucial for disease staging, informing the optimum time to initiate treatments, and identifying biomarkers for future trials in people with premanifest Huntington’s disease (preHD),” they said.
Identifying biomarkers of pre-Huntington’s disease
For their study, the researchers recruited 64 young adults with presymptomatic Huntington’s disease (preHD) and 67 controls, with an average age of 29 years. Brain imaging was conducted between Aug. 2, 2017, and April 25, 2019. Individuals with preexisting measurable cognitive and psychiatric disorders were excluded.
The researchers found no significant evidence of cognitive or psychiatric impairment in the preHD group at 23.6 years from the predicted onset of symptoms. The preHD group showed smaller putamen volumes, compared with controls, but this difference had no apparent relation to the timing of symptom onset, the researchers said.
Brain imaging revealed elevations in the CSF mutant huntingtin, neurofilament light protein (NfL), YKL-40, and plasma NfL among individuals with preHD, compared with controls. Of these, CSF NfL showed the highest effect size of measures in the study and showed a significant increasing association with estimated years to the onset of clinical symptoms of HD carriers. Overall, 53% of individuals with preHD had CSF NfL values in the normal range, and 47% had elevated values, compared with controls.
“NfL is therefore a potential candidate to provide a measure of disease progression in early preHD and might eventually be used as a marker of response to treatment in future preventive trials,” the researchers said.
The study findings were limited by several factors including potential underpowering to detect associations with age and CAG gene segment repeats, the researchers noted.
However, “By identifying a cohort of individuals with preHD and no detectable functional impairment but who begin to exhibit subtle elevations in select biological measures of neurodegeneration, we have highlighted a crucial point early in the disease process,” they concluded.
“Intervening at this stage might offer the prospect of delaying or preventing further neurodegeneration while function is intact, giving gene carriers many more years of life without impairment,” they added.
What is the best window for treatment?
The study is “particularly important since the absence of any subclinical symptoms in preHD individuals far from onset shows that the abnormal developmental aspect of Huntington’s disease has no substantial effect on adults’ clinical pattern,” wrote Anne-Catherine Bachoud-Lévi, MD, of Université Paris Est, Créteil, France, in an accompanying comment.
“The most robust findings of [the study] are the sensitiveness of NfL, compared with mutant huntingtin in CSF of individuals with preHD, and that degenerative rather than developmental disorders are clinically relevant,” she said. However, potential limitations to the study include the exclusion absence of language and calculation as part of the cognitive assessments, she noted. “Ideally, more sensitive cognitive tasks including these domains should be designed for preHD participants.”
In addition, the risks versus benefits of any long-term treatment must be considered, Dr. Bachoud-Lévi noted.
“The best window for treatment should instead target the time when a detectable subclinical slope of cognitive performance allows for predicting disease onset within a few years,” she said. “Turning to machine learning methodology, such as that in oncology, might also permit combining the best window and the best disease-modifying therapy for individuals with preHD,” she added.
The study was supported by the Wellcome Trust, CHDI Foundation. The researchers had no financial conflicts to disclose. Dr. Bachoud-Lévi disclosed grants and personal fees from Roche, and grants from the French Ministry of Health and Direction de la Recherche Clinique.
SOURCES: Scahill RI et al. Lancet Neurol. 2020 June;19:502-12; Bachoud-Lévi A-C. Lancet Neurol. 2020 June;19:473-5.
, according to a study published in the June Lancet Neurology. The data come from the Huntington’s disease Young Adult Study (HD-YAS) conducted in the United Kingdom.
The genetic cause of Huntington’s disease provides a potential target for biomarker treatment, wrote joint first authors Rachael I. Scahill, PhD, and Paul Zeun, BMBS, of University College London and colleagues.
“A detailed characterization of the premanifest period in Huntington’s disease is crucial for disease staging, informing the optimum time to initiate treatments, and identifying biomarkers for future trials in people with premanifest Huntington’s disease (preHD),” they said.
Identifying biomarkers of pre-Huntington’s disease
For their study, the researchers recruited 64 young adults with presymptomatic Huntington’s disease (preHD) and 67 controls, with an average age of 29 years. Brain imaging was conducted between Aug. 2, 2017, and April 25, 2019. Individuals with preexisting measurable cognitive and psychiatric disorders were excluded.
The researchers found no significant evidence of cognitive or psychiatric impairment in the preHD group at 23.6 years from the predicted onset of symptoms. The preHD group showed smaller putamen volumes, compared with controls, but this difference had no apparent relation to the timing of symptom onset, the researchers said.
Brain imaging revealed elevations in the CSF mutant huntingtin, neurofilament light protein (NfL), YKL-40, and plasma NfL among individuals with preHD, compared with controls. Of these, CSF NfL showed the highest effect size of measures in the study and showed a significant increasing association with estimated years to the onset of clinical symptoms of HD carriers. Overall, 53% of individuals with preHD had CSF NfL values in the normal range, and 47% had elevated values, compared with controls.
“NfL is therefore a potential candidate to provide a measure of disease progression in early preHD and might eventually be used as a marker of response to treatment in future preventive trials,” the researchers said.
The study findings were limited by several factors including potential underpowering to detect associations with age and CAG gene segment repeats, the researchers noted.
However, “By identifying a cohort of individuals with preHD and no detectable functional impairment but who begin to exhibit subtle elevations in select biological measures of neurodegeneration, we have highlighted a crucial point early in the disease process,” they concluded.
“Intervening at this stage might offer the prospect of delaying or preventing further neurodegeneration while function is intact, giving gene carriers many more years of life without impairment,” they added.
What is the best window for treatment?
The study is “particularly important since the absence of any subclinical symptoms in preHD individuals far from onset shows that the abnormal developmental aspect of Huntington’s disease has no substantial effect on adults’ clinical pattern,” wrote Anne-Catherine Bachoud-Lévi, MD, of Université Paris Est, Créteil, France, in an accompanying comment.
“The most robust findings of [the study] are the sensitiveness of NfL, compared with mutant huntingtin in CSF of individuals with preHD, and that degenerative rather than developmental disorders are clinically relevant,” she said. However, potential limitations to the study include the exclusion absence of language and calculation as part of the cognitive assessments, she noted. “Ideally, more sensitive cognitive tasks including these domains should be designed for preHD participants.”
In addition, the risks versus benefits of any long-term treatment must be considered, Dr. Bachoud-Lévi noted.
“The best window for treatment should instead target the time when a detectable subclinical slope of cognitive performance allows for predicting disease onset within a few years,” she said. “Turning to machine learning methodology, such as that in oncology, might also permit combining the best window and the best disease-modifying therapy for individuals with preHD,” she added.
The study was supported by the Wellcome Trust, CHDI Foundation. The researchers had no financial conflicts to disclose. Dr. Bachoud-Lévi disclosed grants and personal fees from Roche, and grants from the French Ministry of Health and Direction de la Recherche Clinique.
SOURCES: Scahill RI et al. Lancet Neurol. 2020 June;19:502-12; Bachoud-Lévi A-C. Lancet Neurol. 2020 June;19:473-5.
, according to a study published in the June Lancet Neurology. The data come from the Huntington’s disease Young Adult Study (HD-YAS) conducted in the United Kingdom.
The genetic cause of Huntington’s disease provides a potential target for biomarker treatment, wrote joint first authors Rachael I. Scahill, PhD, and Paul Zeun, BMBS, of University College London and colleagues.
“A detailed characterization of the premanifest period in Huntington’s disease is crucial for disease staging, informing the optimum time to initiate treatments, and identifying biomarkers for future trials in people with premanifest Huntington’s disease (preHD),” they said.
Identifying biomarkers of pre-Huntington’s disease
For their study, the researchers recruited 64 young adults with presymptomatic Huntington’s disease (preHD) and 67 controls, with an average age of 29 years. Brain imaging was conducted between Aug. 2, 2017, and April 25, 2019. Individuals with preexisting measurable cognitive and psychiatric disorders were excluded.
The researchers found no significant evidence of cognitive or psychiatric impairment in the preHD group at 23.6 years from the predicted onset of symptoms. The preHD group showed smaller putamen volumes, compared with controls, but this difference had no apparent relation to the timing of symptom onset, the researchers said.
Brain imaging revealed elevations in the CSF mutant huntingtin, neurofilament light protein (NfL), YKL-40, and plasma NfL among individuals with preHD, compared with controls. Of these, CSF NfL showed the highest effect size of measures in the study and showed a significant increasing association with estimated years to the onset of clinical symptoms of HD carriers. Overall, 53% of individuals with preHD had CSF NfL values in the normal range, and 47% had elevated values, compared with controls.
“NfL is therefore a potential candidate to provide a measure of disease progression in early preHD and might eventually be used as a marker of response to treatment in future preventive trials,” the researchers said.
The study findings were limited by several factors including potential underpowering to detect associations with age and CAG gene segment repeats, the researchers noted.
However, “By identifying a cohort of individuals with preHD and no detectable functional impairment but who begin to exhibit subtle elevations in select biological measures of neurodegeneration, we have highlighted a crucial point early in the disease process,” they concluded.
“Intervening at this stage might offer the prospect of delaying or preventing further neurodegeneration while function is intact, giving gene carriers many more years of life without impairment,” they added.
What is the best window for treatment?
The study is “particularly important since the absence of any subclinical symptoms in preHD individuals far from onset shows that the abnormal developmental aspect of Huntington’s disease has no substantial effect on adults’ clinical pattern,” wrote Anne-Catherine Bachoud-Lévi, MD, of Université Paris Est, Créteil, France, in an accompanying comment.
“The most robust findings of [the study] are the sensitiveness of NfL, compared with mutant huntingtin in CSF of individuals with preHD, and that degenerative rather than developmental disorders are clinically relevant,” she said. However, potential limitations to the study include the exclusion absence of language and calculation as part of the cognitive assessments, she noted. “Ideally, more sensitive cognitive tasks including these domains should be designed for preHD participants.”
In addition, the risks versus benefits of any long-term treatment must be considered, Dr. Bachoud-Lévi noted.
“The best window for treatment should instead target the time when a detectable subclinical slope of cognitive performance allows for predicting disease onset within a few years,” she said. “Turning to machine learning methodology, such as that in oncology, might also permit combining the best window and the best disease-modifying therapy for individuals with preHD,” she added.
The study was supported by the Wellcome Trust, CHDI Foundation. The researchers had no financial conflicts to disclose. Dr. Bachoud-Lévi disclosed grants and personal fees from Roche, and grants from the French Ministry of Health and Direction de la Recherche Clinique.
SOURCES: Scahill RI et al. Lancet Neurol. 2020 June;19:502-12; Bachoud-Lévi A-C. Lancet Neurol. 2020 June;19:473-5.
FROM LANCET NEUROLOGY
Nilotinib is safe in moderate and advanced Parkinson’s disease
according to investigators. Nevertheless, other drugs that – like nilotinib – inhibit tyrosine kinase (c-Abl) may have a neuroprotective effect, they added. The study was presented online as part of the American Academy of Neurology’s 2020 Science Highlights.

Research using preclinical models of Parkinson’s disease has indicated that nilotinib offers neuroprotection. Tanya Simuni, MD, the Arthur C. Nielsen Jr., Research Professor of Parkinson’s Disease and Movement Disorders at Northwestern University in Chicago, and colleagues conducted a prospective study to evaluate the safety and tolerability of oral nilotinib in patients with moderate or advanced Parkinson’s disease. The investigators also sought to examine nilotinib’s symptomatic effect, as measured by the Movement Disorder Society–Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) part III. In addition, Dr. Simuni and colleagues analyzed the drug’s effect on progression of disability, as measured by various other Parkinson’s disease scales. The study’s exploratory outcomes included cognitive function, quality of life, pharmacokinetic profile, and a battery of serum and spinal fluid biomarkers.
The researchers conducted their randomized, double-blind, placebo-controlled, parallel-group study at 25 sites in the United States. They randomized 76 patients with Parkinson’s disease in approximately equal groups to a daily dose of placebo, 150 mg of nilotinib, or 300 mg of nilotinib. Safety visits occurred monthly. Patient assessments occurred at 3 months and at 6 months, which was the end of the treatment period. Patients presented off study medication at 1 month and 2 months after the end of the treatment period.
Treatment did not change dopamine levels
Baseline demographics and disease characteristics were balanced between groups. Mean age was about 66 years in the placebo group, 61 years in the 150-mg group, and 67 years in the 300-mg group. The proportion of male participants was 64% in the placebo group, 60% in the 150-mg group, and 81% in the 300-mg group. Disease duration was 9 years in the placebo group, approximately 9 years in the 150-mg group, and approximately 12 years in the 300-mg group. Mean MDS-UPDRS total on score was 46 in the placebo group, 47 in the 150-mg group, and 52 in the 300-mg group.
Tolerability was 84% in the placebo group, 76% in the in the 150-mg group, and 77% in the 300-mg group. The sole treatment-related serious adverse event, arrhythmia, occurred in one patient in the 300-mg group. The rate of any adverse event was 88% in the placebo group, 92% in the 150-mg group, and 88% in the 300-mg group. The rate of any serious adverse event was 8% in the placebo group and 4% in each nilotinib group.
From baseline to 1 month, MDS-UPDRS part III on scores decreased by 0.49 points in the placebo group, increased by 2.08 in the 150-mg group, and increased by 4.67 in the 300-mg group. Differences in other secondary measures (e.g., change in MDS-UPDRS part III on scores from baseline to 6 months and change in MDS-UPDRS part III off score from baseline to 6 months) were not statistically significant.
At 3 months, CSF levels of nilotinib were well below the threshold for c-Abl inhibition (approximately 11 ng/mL). The arithmetic mean levels were 0.91 ng/mL in the 150-mg group and 1.69 ng/mL in the 300-mg group. Nilotinib also failed to alter CSF levels of dopamine or its metabolites at 3 months. Dr. Simuni and colleagues did not see significant differences between treatment groups in the exploratory outcomes of cognitive function and quality of life.
“Nilotinib is not an optimal molecule to assess the therapeutic potential of c-Abl inhibition for Parkinson’s disease,” the investigators concluded.
Nilotinib may be an inappropriate candidate
The data “suggest that the hypothesis wasn’t tested, since the CSF and serum concentration of the drug were insufficient for enzyme inhibition,” said Peter LeWitt, MD, Sastry Foundation Endowed Chair in Neurology and professor of neurology at Wayne State University, Detroit. “A higher dose or a more CNS-penetrant drug would be needed for adequate testing of the hypothesis that c-Abl inhibition could provide disease modification.”
Nilotinib might not be an appropriate drug for this investigation, he continued. “There may be better choices among c-Abl inhibitors for penetration into the CNS, such as dasatinib, or for increased potency of effect, such as imatinib.”
Sun Pharma Advanced Research Company is conducting a clinical trial of KO706, another c-Abl inhibitor, added Dr. LeWitt, who is a researcher in that trial and an editorial adviser to Neurology Reviews. “The studies published recently in JAMA Neurology by Pagan et al. claiming target engagement with nilotinib in Parkinson’s disease patients need to be contrasted with the results of the current investigation. Disease modification with c-Abl inhibition continues to be a promising therapeutic avenue, but both positive and negative study results need careful reassessment and validation.”
The Michael J. Fox Foundation, the Cure Parkinson’s Trust, and Van Andel Research Institute funded the study. Novartis provided the study drug and placebo. The investigators reported no conflicts of interest.
SOURCE: Simuni T et al. AAN 2020. Abstract 43617.
according to investigators. Nevertheless, other drugs that – like nilotinib – inhibit tyrosine kinase (c-Abl) may have a neuroprotective effect, they added. The study was presented online as part of the American Academy of Neurology’s 2020 Science Highlights.
Research using preclinical models of Parkinson’s disease has indicated that nilotinib offers neuroprotection. Tanya Simuni, MD, the Arthur C. Nielsen Jr., Research Professor of Parkinson’s Disease and Movement Disorders at Northwestern University in Chicago, and colleagues conducted a prospective study to evaluate the safety and tolerability of oral nilotinib in patients with moderate or advanced Parkinson’s disease. The investigators also sought to examine nilotinib’s symptomatic effect, as measured by the Movement Disorder Society–Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) part III. In addition, Dr. Simuni and colleagues analyzed the drug’s effect on progression of disability, as measured by various other Parkinson’s disease scales. The study’s exploratory outcomes included cognitive function, quality of life, pharmacokinetic profile, and a battery of serum and spinal fluid biomarkers.
The researchers conducted their randomized, double-blind, placebo-controlled, parallel-group study at 25 sites in the United States. They randomized 76 patients with Parkinson’s disease in approximately equal groups to a daily dose of placebo, 150 mg of nilotinib, or 300 mg of nilotinib. Safety visits occurred monthly. Patient assessments occurred at 3 months and at 6 months, which was the end of the treatment period. Patients presented off study medication at 1 month and 2 months after the end of the treatment period.
Treatment did not change dopamine levels
Baseline demographics and disease characteristics were balanced between groups. Mean age was about 66 years in the placebo group, 61 years in the 150-mg group, and 67 years in the 300-mg group. The proportion of male participants was 64% in the placebo group, 60% in the 150-mg group, and 81% in the 300-mg group. Disease duration was 9 years in the placebo group, approximately 9 years in the 150-mg group, and approximately 12 years in the 300-mg group. Mean MDS-UPDRS total on score was 46 in the placebo group, 47 in the 150-mg group, and 52 in the 300-mg group.
Tolerability was 84% in the placebo group, 76% in the in the 150-mg group, and 77% in the 300-mg group. The sole treatment-related serious adverse event, arrhythmia, occurred in one patient in the 300-mg group. The rate of any adverse event was 88% in the placebo group, 92% in the 150-mg group, and 88% in the 300-mg group. The rate of any serious adverse event was 8% in the placebo group and 4% in each nilotinib group.
From baseline to 1 month, MDS-UPDRS part III on scores decreased by 0.49 points in the placebo group, increased by 2.08 in the 150-mg group, and increased by 4.67 in the 300-mg group. Differences in other secondary measures (e.g., change in MDS-UPDRS part III on scores from baseline to 6 months and change in MDS-UPDRS part III off score from baseline to 6 months) were not statistically significant.
At 3 months, CSF levels of nilotinib were well below the threshold for c-Abl inhibition (approximately 11 ng/mL). The arithmetic mean levels were 0.91 ng/mL in the 150-mg group and 1.69 ng/mL in the 300-mg group. Nilotinib also failed to alter CSF levels of dopamine or its metabolites at 3 months. Dr. Simuni and colleagues did not see significant differences between treatment groups in the exploratory outcomes of cognitive function and quality of life.
“Nilotinib is not an optimal molecule to assess the therapeutic potential of c-Abl inhibition for Parkinson’s disease,” the investigators concluded.
Nilotinib may be an inappropriate candidate
The data “suggest that the hypothesis wasn’t tested, since the CSF and serum concentration of the drug were insufficient for enzyme inhibition,” said Peter LeWitt, MD, Sastry Foundation Endowed Chair in Neurology and professor of neurology at Wayne State University, Detroit. “A higher dose or a more CNS-penetrant drug would be needed for adequate testing of the hypothesis that c-Abl inhibition could provide disease modification.”
Nilotinib might not be an appropriate drug for this investigation, he continued. “There may be better choices among c-Abl inhibitors for penetration into the CNS, such as dasatinib, or for increased potency of effect, such as imatinib.”
Sun Pharma Advanced Research Company is conducting a clinical trial of KO706, another c-Abl inhibitor, added Dr. LeWitt, who is a researcher in that trial and an editorial adviser to Neurology Reviews. “The studies published recently in JAMA Neurology by Pagan et al. claiming target engagement with nilotinib in Parkinson’s disease patients need to be contrasted with the results of the current investigation. Disease modification with c-Abl inhibition continues to be a promising therapeutic avenue, but both positive and negative study results need careful reassessment and validation.”
The Michael J. Fox Foundation, the Cure Parkinson’s Trust, and Van Andel Research Institute funded the study. Novartis provided the study drug and placebo. The investigators reported no conflicts of interest.
SOURCE: Simuni T et al. AAN 2020. Abstract 43617.
according to investigators. Nevertheless, other drugs that – like nilotinib – inhibit tyrosine kinase (c-Abl) may have a neuroprotective effect, they added. The study was presented online as part of the American Academy of Neurology’s 2020 Science Highlights.
Research using preclinical models of Parkinson’s disease has indicated that nilotinib offers neuroprotection. Tanya Simuni, MD, the Arthur C. Nielsen Jr., Research Professor of Parkinson’s Disease and Movement Disorders at Northwestern University in Chicago, and colleagues conducted a prospective study to evaluate the safety and tolerability of oral nilotinib in patients with moderate or advanced Parkinson’s disease. The investigators also sought to examine nilotinib’s symptomatic effect, as measured by the Movement Disorder Society–Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) part III. In addition, Dr. Simuni and colleagues analyzed the drug’s effect on progression of disability, as measured by various other Parkinson’s disease scales. The study’s exploratory outcomes included cognitive function, quality of life, pharmacokinetic profile, and a battery of serum and spinal fluid biomarkers.
The researchers conducted their randomized, double-blind, placebo-controlled, parallel-group study at 25 sites in the United States. They randomized 76 patients with Parkinson’s disease in approximately equal groups to a daily dose of placebo, 150 mg of nilotinib, or 300 mg of nilotinib. Safety visits occurred monthly. Patient assessments occurred at 3 months and at 6 months, which was the end of the treatment period. Patients presented off study medication at 1 month and 2 months after the end of the treatment period.
Treatment did not change dopamine levels
Baseline demographics and disease characteristics were balanced between groups. Mean age was about 66 years in the placebo group, 61 years in the 150-mg group, and 67 years in the 300-mg group. The proportion of male participants was 64% in the placebo group, 60% in the 150-mg group, and 81% in the 300-mg group. Disease duration was 9 years in the placebo group, approximately 9 years in the 150-mg group, and approximately 12 years in the 300-mg group. Mean MDS-UPDRS total on score was 46 in the placebo group, 47 in the 150-mg group, and 52 in the 300-mg group.
Tolerability was 84% in the placebo group, 76% in the in the 150-mg group, and 77% in the 300-mg group. The sole treatment-related serious adverse event, arrhythmia, occurred in one patient in the 300-mg group. The rate of any adverse event was 88% in the placebo group, 92% in the 150-mg group, and 88% in the 300-mg group. The rate of any serious adverse event was 8% in the placebo group and 4% in each nilotinib group.
From baseline to 1 month, MDS-UPDRS part III on scores decreased by 0.49 points in the placebo group, increased by 2.08 in the 150-mg group, and increased by 4.67 in the 300-mg group. Differences in other secondary measures (e.g., change in MDS-UPDRS part III on scores from baseline to 6 months and change in MDS-UPDRS part III off score from baseline to 6 months) were not statistically significant.
At 3 months, CSF levels of nilotinib were well below the threshold for c-Abl inhibition (approximately 11 ng/mL). The arithmetic mean levels were 0.91 ng/mL in the 150-mg group and 1.69 ng/mL in the 300-mg group. Nilotinib also failed to alter CSF levels of dopamine or its metabolites at 3 months. Dr. Simuni and colleagues did not see significant differences between treatment groups in the exploratory outcomes of cognitive function and quality of life.
“Nilotinib is not an optimal molecule to assess the therapeutic potential of c-Abl inhibition for Parkinson’s disease,” the investigators concluded.
Nilotinib may be an inappropriate candidate
The data “suggest that the hypothesis wasn’t tested, since the CSF and serum concentration of the drug were insufficient for enzyme inhibition,” said Peter LeWitt, MD, Sastry Foundation Endowed Chair in Neurology and professor of neurology at Wayne State University, Detroit. “A higher dose or a more CNS-penetrant drug would be needed for adequate testing of the hypothesis that c-Abl inhibition could provide disease modification.”
Nilotinib might not be an appropriate drug for this investigation, he continued. “There may be better choices among c-Abl inhibitors for penetration into the CNS, such as dasatinib, or for increased potency of effect, such as imatinib.”
Sun Pharma Advanced Research Company is conducting a clinical trial of KO706, another c-Abl inhibitor, added Dr. LeWitt, who is a researcher in that trial and an editorial adviser to Neurology Reviews. “The studies published recently in JAMA Neurology by Pagan et al. claiming target engagement with nilotinib in Parkinson’s disease patients need to be contrasted with the results of the current investigation. Disease modification with c-Abl inhibition continues to be a promising therapeutic avenue, but both positive and negative study results need careful reassessment and validation.”
The Michael J. Fox Foundation, the Cure Parkinson’s Trust, and Van Andel Research Institute funded the study. Novartis provided the study drug and placebo. The investigators reported no conflicts of interest.
SOURCE: Simuni T et al. AAN 2020. Abstract 43617.
FROM AAN 2020

FDA approves apomorphine sublingual film for ‘off’ episodes in Parkinson’s disease
, the manufacturer has announced. This marks the first approval for a sublingual therapy for this indication, which is defined as the re-emergence or worsening of Parkinson’s disease symptoms that have otherwise been controlled with standard care of levodopa/carbidopa, Sunovion reports. Almost 60% of patients with Parkinson’s disease experience off episodes.
The approval “affords healthcare providers with a needed option that can be added to their patients’ medication regimen to adequately address off episodes as their Parkinson’s disease progresses,” Stewart Factor, DO, professor of neurology and director of the Movement Disorders Program at Emory University School of Medicine, Atlanta, Georgia, said in a press release from the manufacturer.
“We know from our research and discussion with the Parkinson’s community that off episodes can significantly disrupt a patient’s daily life,” Todd Sherer, PhD, CEO of the Michael J. Fox Foundation for Parkinson’s Research, said in the same release. He added that the Fox Foundation “supported early clinical development of sublingual apomorphine.”
The treatment is expected to be available in US pharmacies in September.
Disruptive symptoms
Off episodes can include periods of tremor, slowed movement, and stiffness and occur during daytime hours.
“Several years after a person is diagnosed with [Parkinson’s disease] they may notice problems such as having trouble getting out of bed in the morning or having difficulty getting out of a chair, or that they feel frozen while trying to walk as the effect of their maintenance medication diminishes,” Dr. Factor noted.
Subcutaneous infusion of the dopamine agonist apomorphine previously has shown benefit in treating persistent motor fluctuations in patients with Parkinson’s disease.
Apomorphine hydrochloride sublingual film is a novel formulation of apomorphine. It dissolves under the tongue to help improve off episode symptoms as needed up to five times per day.
A phase 3 study of 109 patients that was published in December in Lancet Neurology showed that those who received the sublingual film therapy had a mean reduction of 11.1 points on the Movement Disorder Society Unified Parkinson’s Disease Rating Scale Part III 30 minutes after dosing at the 12-week assessment. This was a significant improvement in motor symptoms versus those who received placebo (mean difference, -7.6 points; P = .0002).
In addition, initial clinical improvement was found 15 minutes after dosing.
The most frequently reported treatment-emergent adverse events in the study population were oropharyngeal reactions, followed by nausea, somnolence, and dizziness.
Long-term safety?
“The availability of this new apomorphine sublingual formulation, along with an inhaled formulation under development, will broaden the treatment options for off periods,” Angelo Antonini, MD, PhD, from University of Padua, Italy, wrote in an accompanying editorial in The Lancet Neurology.
Although the results were encouraging, he noted some caution should be heeded.
Because of “the high rate of oropharyngeal adverse events, long-term safety needs to be monitored once the product is registered and available for chronic use in patients with Parkinson’s disease,” Dr. Antonini wrote.
Other safety information issued by the manufacturer includes a warning that patients who take the 5HT3 antagonists ondansetron, dolasetron, palonosetron, granisetron, or alosetron for nausea should not also use apomorphine hydrochloride sublingual film.
“People taking ondansetron together with apomorphine, the active ingredient in Kynmobi, have had very low blood pressure and lost consciousness or ‘blacked out,’ “ the warning notes.
It also should not be taken by individuals who are allergic to the ingredients in the medication, including sodium metabisulfite.
This article first appeared on Medscape.com.
, the manufacturer has announced. This marks the first approval for a sublingual therapy for this indication, which is defined as the re-emergence or worsening of Parkinson’s disease symptoms that have otherwise been controlled with standard care of levodopa/carbidopa, Sunovion reports. Almost 60% of patients with Parkinson’s disease experience off episodes.
The approval “affords healthcare providers with a needed option that can be added to their patients’ medication regimen to adequately address off episodes as their Parkinson’s disease progresses,” Stewart Factor, DO, professor of neurology and director of the Movement Disorders Program at Emory University School of Medicine, Atlanta, Georgia, said in a press release from the manufacturer.
“We know from our research and discussion with the Parkinson’s community that off episodes can significantly disrupt a patient’s daily life,” Todd Sherer, PhD, CEO of the Michael J. Fox Foundation for Parkinson’s Research, said in the same release. He added that the Fox Foundation “supported early clinical development of sublingual apomorphine.”
The treatment is expected to be available in US pharmacies in September.
Disruptive symptoms
Off episodes can include periods of tremor, slowed movement, and stiffness and occur during daytime hours.
“Several years after a person is diagnosed with [Parkinson’s disease] they may notice problems such as having trouble getting out of bed in the morning or having difficulty getting out of a chair, or that they feel frozen while trying to walk as the effect of their maintenance medication diminishes,” Dr. Factor noted.
Subcutaneous infusion of the dopamine agonist apomorphine previously has shown benefit in treating persistent motor fluctuations in patients with Parkinson’s disease.
Apomorphine hydrochloride sublingual film is a novel formulation of apomorphine. It dissolves under the tongue to help improve off episode symptoms as needed up to five times per day.
A phase 3 study of 109 patients that was published in December in Lancet Neurology showed that those who received the sublingual film therapy had a mean reduction of 11.1 points on the Movement Disorder Society Unified Parkinson’s Disease Rating Scale Part III 30 minutes after dosing at the 12-week assessment. This was a significant improvement in motor symptoms versus those who received placebo (mean difference, -7.6 points; P = .0002).
In addition, initial clinical improvement was found 15 minutes after dosing.
The most frequently reported treatment-emergent adverse events in the study population were oropharyngeal reactions, followed by nausea, somnolence, and dizziness.
Long-term safety?
“The availability of this new apomorphine sublingual formulation, along with an inhaled formulation under development, will broaden the treatment options for off periods,” Angelo Antonini, MD, PhD, from University of Padua, Italy, wrote in an accompanying editorial in The Lancet Neurology.
Although the results were encouraging, he noted some caution should be heeded.
Because of “the high rate of oropharyngeal adverse events, long-term safety needs to be monitored once the product is registered and available for chronic use in patients with Parkinson’s disease,” Dr. Antonini wrote.
Other safety information issued by the manufacturer includes a warning that patients who take the 5HT3 antagonists ondansetron, dolasetron, palonosetron, granisetron, or alosetron for nausea should not also use apomorphine hydrochloride sublingual film.
“People taking ondansetron together with apomorphine, the active ingredient in Kynmobi, have had very low blood pressure and lost consciousness or ‘blacked out,’ “ the warning notes.
It also should not be taken by individuals who are allergic to the ingredients in the medication, including sodium metabisulfite.
This article first appeared on Medscape.com.
, the manufacturer has announced. This marks the first approval for a sublingual therapy for this indication, which is defined as the re-emergence or worsening of Parkinson’s disease symptoms that have otherwise been controlled with standard care of levodopa/carbidopa, Sunovion reports. Almost 60% of patients with Parkinson’s disease experience off episodes.
The approval “affords healthcare providers with a needed option that can be added to their patients’ medication regimen to adequately address off episodes as their Parkinson’s disease progresses,” Stewart Factor, DO, professor of neurology and director of the Movement Disorders Program at Emory University School of Medicine, Atlanta, Georgia, said in a press release from the manufacturer.
“We know from our research and discussion with the Parkinson’s community that off episodes can significantly disrupt a patient’s daily life,” Todd Sherer, PhD, CEO of the Michael J. Fox Foundation for Parkinson’s Research, said in the same release. He added that the Fox Foundation “supported early clinical development of sublingual apomorphine.”
The treatment is expected to be available in US pharmacies in September.
Disruptive symptoms
Off episodes can include periods of tremor, slowed movement, and stiffness and occur during daytime hours.
“Several years after a person is diagnosed with [Parkinson’s disease] they may notice problems such as having trouble getting out of bed in the morning or having difficulty getting out of a chair, or that they feel frozen while trying to walk as the effect of their maintenance medication diminishes,” Dr. Factor noted.
Subcutaneous infusion of the dopamine agonist apomorphine previously has shown benefit in treating persistent motor fluctuations in patients with Parkinson’s disease.
Apomorphine hydrochloride sublingual film is a novel formulation of apomorphine. It dissolves under the tongue to help improve off episode symptoms as needed up to five times per day.
A phase 3 study of 109 patients that was published in December in Lancet Neurology showed that those who received the sublingual film therapy had a mean reduction of 11.1 points on the Movement Disorder Society Unified Parkinson’s Disease Rating Scale Part III 30 minutes after dosing at the 12-week assessment. This was a significant improvement in motor symptoms versus those who received placebo (mean difference, -7.6 points; P = .0002).
In addition, initial clinical improvement was found 15 minutes after dosing.
The most frequently reported treatment-emergent adverse events in the study population were oropharyngeal reactions, followed by nausea, somnolence, and dizziness.
Long-term safety?
“The availability of this new apomorphine sublingual formulation, along with an inhaled formulation under development, will broaden the treatment options for off periods,” Angelo Antonini, MD, PhD, from University of Padua, Italy, wrote in an accompanying editorial in The Lancet Neurology.
Although the results were encouraging, he noted some caution should be heeded.
Because of “the high rate of oropharyngeal adverse events, long-term safety needs to be monitored once the product is registered and available for chronic use in patients with Parkinson’s disease,” Dr. Antonini wrote.
Other safety information issued by the manufacturer includes a warning that patients who take the 5HT3 antagonists ondansetron, dolasetron, palonosetron, granisetron, or alosetron for nausea should not also use apomorphine hydrochloride sublingual film.
“People taking ondansetron together with apomorphine, the active ingredient in Kynmobi, have had very low blood pressure and lost consciousness or ‘blacked out,’ “ the warning notes.
It also should not be taken by individuals who are allergic to the ingredients in the medication, including sodium metabisulfite.
This article first appeared on Medscape.com.
GI symptoms in Parkinson’s disease correlate with less microbial diversity
according to research presented online as part of the 2020 American Academy of Neurology Science Highlights.
Jade E. Kenna, a PhD candidate and research assistant at the Perron Institute of Neurological and Translational Science in Perth, Australia, described findings from a multicenter assessment of 167 patients with Parkinson’s disease and 100 controls from movement disorders clinics in Australia. Participants completed the self-report Gastrointestinal Symptom Rating Scale (GSRS), which rates the frequency and severity of 15 GI symptoms. In addition, stool samples were analyzed using targeted sequencing to characterize gut microbiome composition.
Although Parkinson’s disease is recognized primarily as a motor disorder, GI dysfunction may be one of the first symptoms. “This is hypothesized to result from a change in microbiota towards an inflammatory, dysbiotic composition,” Ms. Kenna said. A limited number of studies have reported an association between altered microbiota composition, GI symptoms, and Parkinson’s disease, but not in Australian cohorts.
Total GSRS score was significantly higher in patients with Parkinson’s disease, compared with controls. Eight of the symptoms – heartburn, acid reflux, nausea or vomiting, borborygmus, increased flatus, decreased passage of stools, feeling of incomplete evacuation, and passing hard stools – were significantly increased in patients with Parkinson’s disease. GSRS symptoms can be categorized as upper, lower, general, hypoactive, or hyperactive, and patients with Parkinson’s disease had significantly increased ratings in the upper, lower, and hypoactive GI symptom domains.
“This is quite a novel finding as not only has this not been assessed in an Australian cohort of individuals before, but the majority of existing literature focuses on the presence of constipation only,” Ms. Kenna said. “The treatment and understanding of nonmotor symptoms of Parkinson’s disease, in particular GI symptoms, remain as one of the top unmet needs reported by patients with Parkinson’s disease themselves. Therefore, a better, more thorough understanding of these symptoms is clearly needed, and research into this area has such value in terms of improving current therapeutic approaches, management strategies, and patient education.”
Microbial analyses found that Firmicutes and Proteobacteria were significantly increased and Verrucomicrobia trended toward an increase in patients with Parkinson’s disease. Fusobacteria was increased in controls. “Proteobacteria and Verrucomicrobia are known to promote inflammation, which can lead to GI symptoms. Furthermore, Faecalibacterium and Ruminococcus, which are reduced in [Parkinson’s disease], can metabolize various substrates to produce [short-chain fatty acids] like butyrate, which are known to aid against intestinal barrier dysfunction and inflammation,” she said.
Individuals with Parkinson’s disease had significantly less microbial diversity. As Parkinson’s disease severity and GI symptom severity increased, microbiome diversity decreased, Ms. Kenna said. “As reduced diversity is associated with increased intestinal inflammation, this indicates that the altered microbiome we saw in [individuals with Parkinson’s disease] may be instigating the increase in incidence and severity of GI symptoms.”
Ms. Kenna reported that she had no disclosures.
SOURCE: Kenna JE. AAN 2020, Abstract S17.006.
according to research presented online as part of the 2020 American Academy of Neurology Science Highlights.
Jade E. Kenna, a PhD candidate and research assistant at the Perron Institute of Neurological and Translational Science in Perth, Australia, described findings from a multicenter assessment of 167 patients with Parkinson’s disease and 100 controls from movement disorders clinics in Australia. Participants completed the self-report Gastrointestinal Symptom Rating Scale (GSRS), which rates the frequency and severity of 15 GI symptoms. In addition, stool samples were analyzed using targeted sequencing to characterize gut microbiome composition.
Although Parkinson’s disease is recognized primarily as a motor disorder, GI dysfunction may be one of the first symptoms. “This is hypothesized to result from a change in microbiota towards an inflammatory, dysbiotic composition,” Ms. Kenna said. A limited number of studies have reported an association between altered microbiota composition, GI symptoms, and Parkinson’s disease, but not in Australian cohorts.
Total GSRS score was significantly higher in patients with Parkinson’s disease, compared with controls. Eight of the symptoms – heartburn, acid reflux, nausea or vomiting, borborygmus, increased flatus, decreased passage of stools, feeling of incomplete evacuation, and passing hard stools – were significantly increased in patients with Parkinson’s disease. GSRS symptoms can be categorized as upper, lower, general, hypoactive, or hyperactive, and patients with Parkinson’s disease had significantly increased ratings in the upper, lower, and hypoactive GI symptom domains.
“This is quite a novel finding as not only has this not been assessed in an Australian cohort of individuals before, but the majority of existing literature focuses on the presence of constipation only,” Ms. Kenna said. “The treatment and understanding of nonmotor symptoms of Parkinson’s disease, in particular GI symptoms, remain as one of the top unmet needs reported by patients with Parkinson’s disease themselves. Therefore, a better, more thorough understanding of these symptoms is clearly needed, and research into this area has such value in terms of improving current therapeutic approaches, management strategies, and patient education.”
Microbial analyses found that Firmicutes and Proteobacteria were significantly increased and Verrucomicrobia trended toward an increase in patients with Parkinson’s disease. Fusobacteria was increased in controls. “Proteobacteria and Verrucomicrobia are known to promote inflammation, which can lead to GI symptoms. Furthermore, Faecalibacterium and Ruminococcus, which are reduced in [Parkinson’s disease], can metabolize various substrates to produce [short-chain fatty acids] like butyrate, which are known to aid against intestinal barrier dysfunction and inflammation,” she said.
Individuals with Parkinson’s disease had significantly less microbial diversity. As Parkinson’s disease severity and GI symptom severity increased, microbiome diversity decreased, Ms. Kenna said. “As reduced diversity is associated with increased intestinal inflammation, this indicates that the altered microbiome we saw in [individuals with Parkinson’s disease] may be instigating the increase in incidence and severity of GI symptoms.”
Ms. Kenna reported that she had no disclosures.
SOURCE: Kenna JE. AAN 2020, Abstract S17.006.
according to research presented online as part of the 2020 American Academy of Neurology Science Highlights.
Jade E. Kenna, a PhD candidate and research assistant at the Perron Institute of Neurological and Translational Science in Perth, Australia, described findings from a multicenter assessment of 167 patients with Parkinson’s disease and 100 controls from movement disorders clinics in Australia. Participants completed the self-report Gastrointestinal Symptom Rating Scale (GSRS), which rates the frequency and severity of 15 GI symptoms. In addition, stool samples were analyzed using targeted sequencing to characterize gut microbiome composition.
Although Parkinson’s disease is recognized primarily as a motor disorder, GI dysfunction may be one of the first symptoms. “This is hypothesized to result from a change in microbiota towards an inflammatory, dysbiotic composition,” Ms. Kenna said. A limited number of studies have reported an association between altered microbiota composition, GI symptoms, and Parkinson’s disease, but not in Australian cohorts.
Total GSRS score was significantly higher in patients with Parkinson’s disease, compared with controls. Eight of the symptoms – heartburn, acid reflux, nausea or vomiting, borborygmus, increased flatus, decreased passage of stools, feeling of incomplete evacuation, and passing hard stools – were significantly increased in patients with Parkinson’s disease. GSRS symptoms can be categorized as upper, lower, general, hypoactive, or hyperactive, and patients with Parkinson’s disease had significantly increased ratings in the upper, lower, and hypoactive GI symptom domains.
“This is quite a novel finding as not only has this not been assessed in an Australian cohort of individuals before, but the majority of existing literature focuses on the presence of constipation only,” Ms. Kenna said. “The treatment and understanding of nonmotor symptoms of Parkinson’s disease, in particular GI symptoms, remain as one of the top unmet needs reported by patients with Parkinson’s disease themselves. Therefore, a better, more thorough understanding of these symptoms is clearly needed, and research into this area has such value in terms of improving current therapeutic approaches, management strategies, and patient education.”
Microbial analyses found that Firmicutes and Proteobacteria were significantly increased and Verrucomicrobia trended toward an increase in patients with Parkinson’s disease. Fusobacteria was increased in controls. “Proteobacteria and Verrucomicrobia are known to promote inflammation, which can lead to GI symptoms. Furthermore, Faecalibacterium and Ruminococcus, which are reduced in [Parkinson’s disease], can metabolize various substrates to produce [short-chain fatty acids] like butyrate, which are known to aid against intestinal barrier dysfunction and inflammation,” she said.
Individuals with Parkinson’s disease had significantly less microbial diversity. As Parkinson’s disease severity and GI symptom severity increased, microbiome diversity decreased, Ms. Kenna said. “As reduced diversity is associated with increased intestinal inflammation, this indicates that the altered microbiome we saw in [individuals with Parkinson’s disease] may be instigating the increase in incidence and severity of GI symptoms.”
Ms. Kenna reported that she had no disclosures.
SOURCE: Kenna JE. AAN 2020, Abstract S17.006.
FROM AAN 2020