User login
Huntington’s research returns to Latin America, as scientists tread with care
BARRANQUILLA, COLOMBIA – “We don’t like to call them brigades. That sounds militant,” said neuropsychologist Johan Acosta-López, PhD.
Dr. Acosta-López, the head of cognitive neurosciences at Simón Bolivar University in this city on Colombia’s Atlantic coast, was among five Colombian clinicians – neurologists, psychiatrists, and neuropsychologists – stuffed into a car on their way to a conference hotel in July 2018.

The following day they would be joined by clinicians and researchers from North America, other Latin American countries, and Europe for a first-of-its kind meeting on Huntington’s disease (HD) in the region, sponsored by Factor-H, an HD charity working in Latin America.
Once the talks wrapped up, the researchers – clinicians and basic scientists – were invited to see patients at a hospital in a town an hour inland with a large concentration of HD families, most of them extremely poor. For some, the Factor-H–sponsored “brigade” would be their first hands-on experience with HD patients in a developing country.
There was some debate in the car about what to call such events: brigades, “integrated health days,” or clinics. Around here – where HD abounded but patients were weary of researchers – terminology mattered.
“We’ve had so many investigators arrive in this area – foreigners and Colombians – telling people ‘we’ve got this huge, great project that you’ll benefit from.’ And they take blood samples and never return,” Dr. Acosta-López said.

Even as a local investigator, Dr. Acosta-López has faced challenges getting a new study off the ground. Dr. Acosta-López and his colleagues are working under a grant from the Colombian government to recruit 241 presymptomatic subjects with confirmed genetic markers for HD, and evaluate them for cognitive and neurologic changes preceding disease onset.
It’s a cross-sectional study, and such studies are usually funded for a year. But the investigators knew it would take much more than a year to recruit patients here, and planned their study for 3 years. As of July, the team had been engaging with the community for 6 months but still didn’t have a single blood sample.
“We’ve had to convince everyone that this time is different,” he said, “and that means focusing on the social aspect” – setting up a legal-assistance program through the university to help families claim health benefits and a job-training program sponsored by local businesses.
It’s unusual for researchers to find themselves playing such extensive roles in coordinating social and economic support for their subjects. But with HD, it’s happening across Latin America, where researchers speak frequently of a “debt” owed to HD families in this region.
Huntington’s disease is a neurodegenerative disease caused by a genetic mutation in the huntingtin (HTT) gene, changing the normal protein it expresses in the body to a toxic form that damages cortical and basal ganglia neurons. It affects between 0.5 to 1 in 10,000 people worldwide, with higher prevalence in the United States, Europe, and Australia.
HD is inherited in an autosomal dominant pattern; a child of a parent with the mutation has a 50% chance of developing the disease. Patients develop cognitive symptoms that progress to dementia, along with the debilitating involuntary, dancelike movements that gave the disease the name by which it was formerly known: Huntington’s chorea.
In the 1980s and 1990s, several generations of Latin American HD families provided data that allowed for some of the greatest research advances in the disease – and they may represent a large share of the world’s HD cases. Yet, they continue to live in extreme poverty and have benefited little from the findings of the past 3 decades.
Without recognizing this and working to improve the families’ well-being, the researchers at the conference said it’s unlikely that promising therapies in the pipeline will ever reach the populations that need them the most.
Discovery in Venezuela

Some 8 hours by car from Barranquilla sits Lake Maracaibo, Venezuela, home to the largest known clusters of HD patients worldwide. The disease is believed to have come to the shores of Lake Maracaibo with a lone European immigrant – a Spanish sailor, many claim – at the end of the 18th century. Cases were first described in the 1950s by a young Venezuelan physician named Américo Negrette, MD.
Dr. Negrette’s findings were ignored by health officials in Venezuela and went unnoticed in the international research community until 1972, when a student of Dr. Negrette’s presented at an HD conference in Ohio. There he drew the attention of the American neuropsychologist Nancy S. Wexler, PhD. Dr. Wexler’s own mother had died of HD, and her father Milton, a noted psychoanalyst, founded the first research foundation dedicated to the disease.

While prevalence of HD in North America, Australia, and Europe is about 1 in 10,000, the region around Lake Maracaibo saw 70 times that rate at the time, thanks to high birth rates, geographic isolation, and extensive intermarriage within a handful of families. The families comprised mostly poor fishermen who lived in makeshift homes in towns ringing the lake.
In 1979, Dr. Wexler, with funding from the U.S. National Institutes of Health, began making annual research visits to Lake Maracaibo, and in 1983, the research group she coordinated, using data from blood and tissue samples donated by affected families, identified the location of the huntingtin gene on chromosome 4 (Nature. 1983 Nov 17;306[5940]:234-8). A decade later, the researchers isolated the mutant version of the gene and found it to be a triplet (CAG) expansion mutation, with more CAG repetitions associated with earlier age at disease onset (Cell. 1993 Mar 26;72[6]:971-83). Dr. Wexler and her colleagues’ findings led to the first genetic tests for HD.
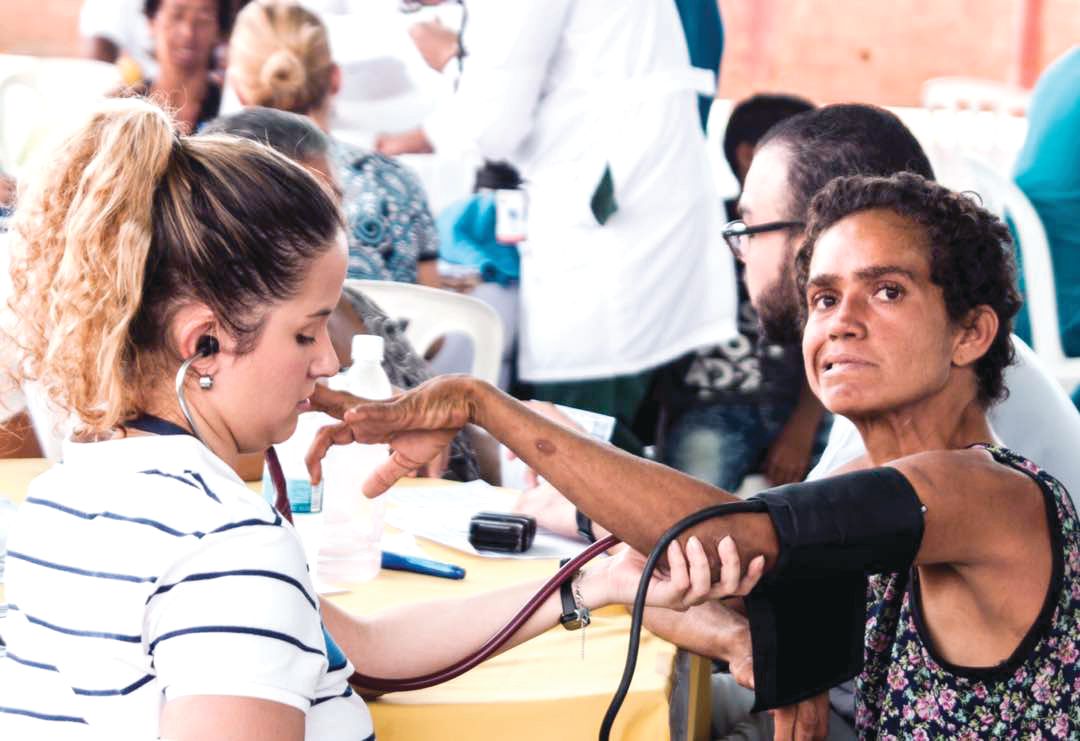
Nationalist policies in Venezuela ended Dr. Wexler and her colleagues’ annual visits to Lake Maracaibo in 2002, along with the food, clothing, and medicines that the group routinely distributed to the families when they came.
Over 23 years, the researchers obtained data from some 18,000 individuals, but the families did not benefit in any durable way from the research. Local investigators with whom Dr. Wexler’s group collaborated lacked the resources and training to continue independently.
Access to medications is limited in Venezuela, and there is no institutional support for hundreds of HD patients living in extreme poverty, many of them descendants of the patients who contributed to the research and generation of these samples. The families’ biological material was sent to labs abroad, where investigators continue to derive findings from it today. Though genetic testing was performed on thousands at risk for the disease, few received access to their results through genetic counseling. A hospice established by Dr. Wexler’s foundation limped along until 2014, when it was finally shuttered.
Rebuilding bridges
A handful of families from the Lake Maracaibo towns attended the conference in Barranquilla. Their travel costs were picked up by Factor-H, which sponsored the event.
Ignacio Muñoz-Sanjuán, PhD, Factor-H’s founder and president, knew the families personally. He’s visited them regularly for years. In 2017, Dr. Muñoz-Sanjuán, a molecular biologist known affectionately in the HD community as “Nacho,” invited several to Rome for a meeting with Pope Francis, as part of an effort to raise awareness of HD and to request support from the Catholic Church for the Latin American families.

Humanitarian work is relatively new to Dr. Muñoz-Sanjuán, who’s spent his career in drug development. In addition to his unpaid work with Factor-H, he is vice president of biology with the CHDI Foundation, a Los Angeles–based nonprofit that funds drug research in HD. CHDI is reported to have about $100 million in annual funding – about triple the NIH budget in recent years for HD research. Its major donors are a group of investors who for years have remained anonymous and do not publicly discuss their philanthropy.
The Spanish-born Dr. Muñoz-Sanjuán had little direct experience with HD populations in Latin America until a few years ago, he said.
At a CHDI meeting in Brazil, he said, “I was talking with physicians and patient advocates from Latin America, telling them they had to be willing to be involved, that these communities with high prevalence had a lot to offer science,” Dr. Muñoz-Sanjuán said in an interview. “I was told that it was me who needed to understand the conditions in which HD patients lived. It completely put me on the spot.”
HD tends to strike during the most productive years of a person’s life, from the late 30s onward, keeping them from working and obliging family members to stop working to care for them. In a poor community, it can condemn a family to a state of extreme poverty for generations. Tetrabenazine (Xenazine), a medication to quiet chorea symptoms, is costly enough that many patients must do without it. Ensuring adequate calorie intake is difficult in HD patients, whose constant movements cause them to lose weight.
Dr. Muñoz-Sanjuán traveled to Colombia, Venezuela, and Brazil, meeting HD families and doctors like neurologist Gustavo Barrios, MD, of Hospital Occidente de Kennedy in Bogotá, Colombia. In a talk at the Barranquilla conference, Dr. Barrios related the experience of his first visit to El Dificil, a community in northern Colombia where some large HD families are forced to survive on the equivalent of $5 a day. “I had to confront not only the fact that these families were living with a terrible disease but in conditions of extreme deprivation,” he said. “My life as a doctor changed that day.”
Dr. Muñoz-Sanjuán helped form a Latin American HD network to involve clinicians like Dr. Barrios who worked with HD clusters, most of them poorly studied. “These are all neglected communities that share similar features,” Dr. Muñoz-Sanjuán said.
On Colombia’s Caribbean coast, for example, HD had been documented since the early 1990s, but genotyping was not performed until recently. Prevalence data are “virtually nonexistent” in Colombia, said Sonia Moreno, PhD, a neuropsychologist at the University of Antioquia in Medellin. In a pilot study presented this year at the CHDI Foundation’s Enroll-HD Congress, Dr. Moreno and her colleagues mined Colombian public health data for likely HD cases, and argued for the creation of a national registry.
In 2012, Dr. Muñoz-Sanjuán founded Factor-H with the aim of improving living conditions for Latin American communities with HD.

Factor-H does not receive funds from CHDI Foundation and instead relies on donations from individuals and companies; its annual budget is less than $200,000. But through contracts with local nongovernmental organizations, it has sponsored health clinics and ongoing food assistance, delivered shipments of medicines and clothing, and started a sponsorship program for young people in HD families, whose studies often are interrupted caring for sick parents. It hopes to build permanent support centers in Colombia and Venezuela where HD families can get their food and medical needs met.
“The traditional thinking in the HD research community is that we’re helping people by doing the legwork to make medicine – and that’s not necessarily enough. You need a more holistic approach,” Dr. Muñoz-Sanjuán said.
Lennie Pineda, MSc, who recently retired as a geneticist with the University of Zulia in Maracaibo, Venezuela, said that Dr. Muñoz-Sanjuán was viewed skeptically when he first visited, in part because of his biomedical research background.
Ms. Pineda, who worked with the region’s HD families her whole career, has been wary of past research efforts in Venezuela. In 2010, she published a paper critical of Dr. Wexler’s and his colleagues’ approach (Revista Redbioética/UNESCO. 2010;1[2]:50-61), particularly regarding issues of informed consent.
“I was very cold to Nacho,” she laughed. “We all looked at him suspiciously.”
Ms. Pineda said Dr. Muñoz-Sanjuán won her over with his interest and creativity in finding concrete ways improve the lives of families in the Lake Maracaibo towns.
In a talk at the conference, Edison Soto, a young man from San Luis, a town on Lake Maracaibo that is a key cluster of HD, said Dr. Muñoz-Sanjuán’s visits had reawakened hope among the families there. “For years, no one thought about us, and because of the situation in the country it’s been hard, really hard,” he said.
“Nacho’s smart,” Ms. Pineda said. “He’s not coming to build a research cohort, he’s coming with genuine intention to help. But if one day conditions are adequate to support investigation, and the people here are well informed and volunteer for a study with full consent, well, all the better,” she said.
Dr. Muñoz-Sanjuán acknowledged that his humanitarian work could be perceived as preparing the ground for future clinical trials.
“I’m not doing anything research oriented with Latin America,” he said. “I would never approach these communities and recommend they take part in a study or give samples, unless their conditions change significantly. But the idea of cross-contamination is a problem I might need to fix. There may come a day where I need to depersonalize Factor-H from me.”
A research platform, a novel agent
Though HD research in Latin America remains rife with challenges, a number of investigators at the conference talked optimistically about planned and ongoing HD studies in Latin America.
The biggest of these is ENROLL-HD, a long-term global observational study of families with HD that uses a standardized approach to data collection. The platform, launched in 2013, aims to enroll 20,000 participants for yearly (or more frequent) assessment. Data from ENROLL-HD will support a diverse range of studies on everything from biomarkers to genetic modifiers to quality of life measures in HD.
ENROLL-HD has opened study sites in Argentina, Chile, and Colombia, and plans to launch a site near Lima, Peru, that is home to an HD cluster. Venezuela is considered out of reach, at least for now.
In Barranquilla, Claudia Perandones, MD, PhD, a genetics researcher in Argentina who manages ENROLL-HD for Latin America and is a cofounder of Factor-H, explained why the kind of clusters seen in Latin America are so valuable scientifically.
The extended family groups share a disease haplotype, eat the same foods, and live in similar environments, Dr. Perandones noted. Because not all the variation in HD can be explained by the number of CAG repeats a patient has, having a large sample with a common haplotype would help researchers pinpoint other environmental and genetic factors that can modify the onset or progress of the disease.
Another key goal of ENROLL-HD, investigators say, is to speed recruitment into clinical trials as they arise. And for the first time in history, potentially game-changing therapies are being developed specifically for HD.
For the past 5 years the Swiss pharmaceutical giant Roche has worked with a smaller biotech firm, Ionis Pharmaceuticals, on an agent called RG6042, which was known until recently as IONIS-HTTRx. CHDI was extensively involved in the agent’s preclinical development, contributing some $10 million to get it off the ground.
RG6042 is an antisense oligonucleotide, delivered by spinal injection, which works by interrupting an mRNA signaling pathway to suppress production of mutant HTT (mHTT) protein in the brain. Antisense oligonucleotides, sometimes called gene silencing therapies, are a new and promising approach in neurodegenerative diseases. Two have received FDA approval to treat spinal muscular atrophy and Duchenne muscular dystrophy.
In April 2018, Roche announced positive results from phase 1/2a study in 46 HD patients in Europe and North America. Patients in that 13-week study saw significant (up to 60%) dose-dependent reductions of the mHTT in their cerebrospinal fluid; a post hoc analysis also found some evidence of functional improvement (Neurology. 2018;90[15 Supplement]:CT.002).
These encouraging findings led to Roche’s announcement of a global phase 3 randomized, controlled trial that is scheduled to begin enrolling in 2019. Roche hopes to randomize 660 patients with mild HD across 15 countries for the 2-year trial, called GENERATION-HD1.
Sites in Latin America are expected to include Argentina, Chile, and Colombia.
At the Barranquilla meeting, Daniel Ciriano, MD, Roche’s Argentina-based medical director for Latin America, extolled the company’s commitment to ethics and social welfare in the region. In recent years, Roche has increased its humanitarian commitments across Latin America, including helping rebuild a Chilean village after an earthquake and offering free breast cancer and kidney disease treatments.
RG6042 is only one of a number of promising approaches to HD. Other therapies in the pipeline include gene silencing delivered by viral vectors instead of repeated spinal injections, an oral drug that interrupts mHTT production, immunotherapies, and even CRISPR gene–editing techniques.
Little was said at the conference, however, about how Latin American HD communities might be able to afford RG6042 or any other therapy that emerges from the pipeline.
Dr. Muñoz-Sanjuán called the issue “a theme for future discussion.”
“This is an area that has to be handled carefully and not one we are heavily invested in yet, although it’s very important,” he said.
On the ground
Several of the European and North American scientists who presented in Barranquilla took pains to express their concern with the well-being of HD patients in Latin America and to demonstrate goodwill toward the local researchers and clinicians.
Hilal A. Lashuel, PhD, a molecular biologist working on the structure and behavior of the HTT protein, said his participation in the Factor-H event at the Vatican the year before had awakened him to “the real human part of HD,” and changed the way he does science.
Normally, Dr. Lashuel said, “we do research disconnected from the realities of the diseases we work with.”
“We need to not just to do research but [to ensure] that research is done right,” he said, which means also focusing on improving patients’ standard of living.
The room broke out in applause when Dr. Lashuel announced new internships for investigators from developing countries. He also presented a parting video from his research team at the École Polytechnique Fédérale de Lausanne (Switzerland), complete with music and affectionate messages in Spanish.
Pharmacologist Elena Cattaneo, PhD, a stem cell researcher long active in the HD community, and also a senator in Italy’s parliament, delivered a similarly warm, carefully choreographed video message from her laboratory at the University of Milan.
Just days later in the town of Juan de Acosta, an hour inland of Barranquilla, the same researchers sat down with patients and families who crowded the waiting room of the town’s only hospital, as the sun beat in through the windows and as mule carts, stray dogs, and buses passed by on the main drag outside.
The event had been titled a “brigade” after all, but the HD families did not seem to mind – and indeed so many showed up that a sign had to be placed on the door saying that no one who arrived after noon could be seen. Consults were not limited to HD-related matters, so families could be seen for any complaint.
HD was first documented in this town in the early 1990s, but much remains to be understood about the size of the cluster, the haplotype, and its relation to other clusters in Colombia or Venezuela. The families here share a handful of last names and likely share a common ancestor. In the early 19th century, the Barranquilla region was flooded with European migrants who reached the city by ship. (HD clusters in Latin America tend to be concentrated in coastal regions, possibly because of migration patterns.)
The waiting room of the hospital was loud with chatter. Small children played as their relatives waited for consults. Some showed the characteristic restless movements and emaciated bodies of people with advanced HD.
The foreign scientists were barred from taking any patient data out of the hospital or asking for samples. Even picture taking was prohibited. Instead they performed genetic counseling and neuropsychological tests; they sorted out differential diagnoses and advised on medications. Visiting Colombian and Venezuelan physicians did the same, while their assistants met with families in the waiting room, taking medical histories and sketching out basic genealogies.
Some of the foreign researchers reported fruitful interactions with patients, while others seemed perplexed by what they’d experienced. Alba di Pardo, PhD, a genetic epidemiologist at the Istituto Neurologico Mediterraneo Pozzilli (Italy), said she’d spent the morning doing genetic counseling with families and going over genealogies to assess risk. Yet, despite the fact that anyone with an HD parent has a 50% chance of developing the disease, some family members acted uninterested, she said.
Dr. di Pardo’s colleague at the Istituto, biologist Vittorio Maglione, PhD, reported having a similar experience. As he was counseling a young woman about her risk for HD, she scrolled indifferently through Facebook posts on her phone, he said.
On some level, Dr. Maglione said, he could understand patients’ reluctance to engage noting that, while there were many potential HD therapies to try, any new treatment paradigm for HD was many years away from a place like this – and potentially very costly. Dr. Maglione – along with Dr. di Pardo – is researching the SP1 axis, a sphingolipid pathway implicated in neurodegenerative such diseases as HD and which has potential as a drug target (Trends Pharmacol Sci. 2018;39[5]:468-80).
Psychologist Pedro Puentes Rozo of Simón Bolivar University, who is working with Dr. Acosta-López on the local cohort study of presymptomatic HD patients, said that, for most of the families in the clinic that day, any seeming indifference probably masked deeper fears. People already were well aware of their risk. “They’ve known about it forever, said Dr. Puentes Rozo, who has been working with this HD population for a decade. “But this is a catastrophic illness and can generate a lot of anxiety.”
Dr. Puentes Rozo said the group’s planned study, unlike studies in the past, would be conducted under strict “international ethical norms and standards.” Subjects would receive ongoing psychological support, and the researchers were working to establish a genetic counseling center so that people who want to know their status “can be prepared,” he said, and plan for their lives and families.
By fall 2018, the cohort study was underway. The group had sponsored several more hospital brigades – or “integrated health days” as they preferred to call them, at the hospital in Juan de Acosta, giving them a chance to work face to face with families.
They drew no blood during the clinics, as investigators in the past had done. Instead, they explained the study to patients, performed the initial screenings, and invited them to designated study appointments at the university. Legal assistance was up and running, and the jobs program would start in 2019.
Enrollment was climbing. And the group was steadily accumulating data.
BARRANQUILLA, COLOMBIA – “We don’t like to call them brigades. That sounds militant,” said neuropsychologist Johan Acosta-López, PhD.
Dr. Acosta-López, the head of cognitive neurosciences at Simón Bolivar University in this city on Colombia’s Atlantic coast, was among five Colombian clinicians – neurologists, psychiatrists, and neuropsychologists – stuffed into a car on their way to a conference hotel in July 2018.
The following day they would be joined by clinicians and researchers from North America, other Latin American countries, and Europe for a first-of-its kind meeting on Huntington’s disease (HD) in the region, sponsored by Factor-H, an HD charity working in Latin America.
Once the talks wrapped up, the researchers – clinicians and basic scientists – were invited to see patients at a hospital in a town an hour inland with a large concentration of HD families, most of them extremely poor. For some, the Factor-H–sponsored “brigade” would be their first hands-on experience with HD patients in a developing country.
There was some debate in the car about what to call such events: brigades, “integrated health days,” or clinics. Around here – where HD abounded but patients were weary of researchers – terminology mattered.
“We’ve had so many investigators arrive in this area – foreigners and Colombians – telling people ‘we’ve got this huge, great project that you’ll benefit from.’ And they take blood samples and never return,” Dr. Acosta-López said.
Even as a local investigator, Dr. Acosta-López has faced challenges getting a new study off the ground. Dr. Acosta-López and his colleagues are working under a grant from the Colombian government to recruit 241 presymptomatic subjects with confirmed genetic markers for HD, and evaluate them for cognitive and neurologic changes preceding disease onset.
It’s a cross-sectional study, and such studies are usually funded for a year. But the investigators knew it would take much more than a year to recruit patients here, and planned their study for 3 years. As of July, the team had been engaging with the community for 6 months but still didn’t have a single blood sample.
“We’ve had to convince everyone that this time is different,” he said, “and that means focusing on the social aspect” – setting up a legal-assistance program through the university to help families claim health benefits and a job-training program sponsored by local businesses.
It’s unusual for researchers to find themselves playing such extensive roles in coordinating social and economic support for their subjects. But with HD, it’s happening across Latin America, where researchers speak frequently of a “debt” owed to HD families in this region.
Huntington’s disease is a neurodegenerative disease caused by a genetic mutation in the huntingtin (HTT) gene, changing the normal protein it expresses in the body to a toxic form that damages cortical and basal ganglia neurons. It affects between 0.5 to 1 in 10,000 people worldwide, with higher prevalence in the United States, Europe, and Australia.
HD is inherited in an autosomal dominant pattern; a child of a parent with the mutation has a 50% chance of developing the disease. Patients develop cognitive symptoms that progress to dementia, along with the debilitating involuntary, dancelike movements that gave the disease the name by which it was formerly known: Huntington’s chorea.
In the 1980s and 1990s, several generations of Latin American HD families provided data that allowed for some of the greatest research advances in the disease – and they may represent a large share of the world’s HD cases. Yet, they continue to live in extreme poverty and have benefited little from the findings of the past 3 decades.
Without recognizing this and working to improve the families’ well-being, the researchers at the conference said it’s unlikely that promising therapies in the pipeline will ever reach the populations that need them the most.
Discovery in Venezuela
Some 8 hours by car from Barranquilla sits Lake Maracaibo, Venezuela, home to the largest known clusters of HD patients worldwide. The disease is believed to have come to the shores of Lake Maracaibo with a lone European immigrant – a Spanish sailor, many claim – at the end of the 18th century. Cases were first described in the 1950s by a young Venezuelan physician named Américo Negrette, MD.
Dr. Negrette’s findings were ignored by health officials in Venezuela and went unnoticed in the international research community until 1972, when a student of Dr. Negrette’s presented at an HD conference in Ohio. There he drew the attention of the American neuropsychologist Nancy S. Wexler, PhD. Dr. Wexler’s own mother had died of HD, and her father Milton, a noted psychoanalyst, founded the first research foundation dedicated to the disease.
While prevalence of HD in North America, Australia, and Europe is about 1 in 10,000, the region around Lake Maracaibo saw 70 times that rate at the time, thanks to high birth rates, geographic isolation, and extensive intermarriage within a handful of families. The families comprised mostly poor fishermen who lived in makeshift homes in towns ringing the lake.
In 1979, Dr. Wexler, with funding from the U.S. National Institutes of Health, began making annual research visits to Lake Maracaibo, and in 1983, the research group she coordinated, using data from blood and tissue samples donated by affected families, identified the location of the huntingtin gene on chromosome 4 (Nature. 1983 Nov 17;306[5940]:234-8). A decade later, the researchers isolated the mutant version of the gene and found it to be a triplet (CAG) expansion mutation, with more CAG repetitions associated with earlier age at disease onset (Cell. 1993 Mar 26;72[6]:971-83). Dr. Wexler and her colleagues’ findings led to the first genetic tests for HD.
Nationalist policies in Venezuela ended Dr. Wexler and her colleagues’ annual visits to Lake Maracaibo in 2002, along with the food, clothing, and medicines that the group routinely distributed to the families when they came.
Over 23 years, the researchers obtained data from some 18,000 individuals, but the families did not benefit in any durable way from the research. Local investigators with whom Dr. Wexler’s group collaborated lacked the resources and training to continue independently.
Access to medications is limited in Venezuela, and there is no institutional support for hundreds of HD patients living in extreme poverty, many of them descendants of the patients who contributed to the research and generation of these samples. The families’ biological material was sent to labs abroad, where investigators continue to derive findings from it today. Though genetic testing was performed on thousands at risk for the disease, few received access to their results through genetic counseling. A hospice established by Dr. Wexler’s foundation limped along until 2014, when it was finally shuttered.
Rebuilding bridges
A handful of families from the Lake Maracaibo towns attended the conference in Barranquilla. Their travel costs were picked up by Factor-H, which sponsored the event.
Ignacio Muñoz-Sanjuán, PhD, Factor-H’s founder and president, knew the families personally. He’s visited them regularly for years. In 2017, Dr. Muñoz-Sanjuán, a molecular biologist known affectionately in the HD community as “Nacho,” invited several to Rome for a meeting with Pope Francis, as part of an effort to raise awareness of HD and to request support from the Catholic Church for the Latin American families.
Humanitarian work is relatively new to Dr. Muñoz-Sanjuán, who’s spent his career in drug development. In addition to his unpaid work with Factor-H, he is vice president of biology with the CHDI Foundation, a Los Angeles–based nonprofit that funds drug research in HD. CHDI is reported to have about $100 million in annual funding – about triple the NIH budget in recent years for HD research. Its major donors are a group of investors who for years have remained anonymous and do not publicly discuss their philanthropy.
The Spanish-born Dr. Muñoz-Sanjuán had little direct experience with HD populations in Latin America until a few years ago, he said.
At a CHDI meeting in Brazil, he said, “I was talking with physicians and patient advocates from Latin America, telling them they had to be willing to be involved, that these communities with high prevalence had a lot to offer science,” Dr. Muñoz-Sanjuán said in an interview. “I was told that it was me who needed to understand the conditions in which HD patients lived. It completely put me on the spot.”
HD tends to strike during the most productive years of a person’s life, from the late 30s onward, keeping them from working and obliging family members to stop working to care for them. In a poor community, it can condemn a family to a state of extreme poverty for generations. Tetrabenazine (Xenazine), a medication to quiet chorea symptoms, is costly enough that many patients must do without it. Ensuring adequate calorie intake is difficult in HD patients, whose constant movements cause them to lose weight.
Dr. Muñoz-Sanjuán traveled to Colombia, Venezuela, and Brazil, meeting HD families and doctors like neurologist Gustavo Barrios, MD, of Hospital Occidente de Kennedy in Bogotá, Colombia. In a talk at the Barranquilla conference, Dr. Barrios related the experience of his first visit to El Dificil, a community in northern Colombia where some large HD families are forced to survive on the equivalent of $5 a day. “I had to confront not only the fact that these families were living with a terrible disease but in conditions of extreme deprivation,” he said. “My life as a doctor changed that day.”
Dr. Muñoz-Sanjuán helped form a Latin American HD network to involve clinicians like Dr. Barrios who worked with HD clusters, most of them poorly studied. “These are all neglected communities that share similar features,” Dr. Muñoz-Sanjuán said.
On Colombia’s Caribbean coast, for example, HD had been documented since the early 1990s, but genotyping was not performed until recently. Prevalence data are “virtually nonexistent” in Colombia, said Sonia Moreno, PhD, a neuropsychologist at the University of Antioquia in Medellin. In a pilot study presented this year at the CHDI Foundation’s Enroll-HD Congress, Dr. Moreno and her colleagues mined Colombian public health data for likely HD cases, and argued for the creation of a national registry.
In 2012, Dr. Muñoz-Sanjuán founded Factor-H with the aim of improving living conditions for Latin American communities with HD.
Factor-H does not receive funds from CHDI Foundation and instead relies on donations from individuals and companies; its annual budget is less than $200,000. But through contracts with local nongovernmental organizations, it has sponsored health clinics and ongoing food assistance, delivered shipments of medicines and clothing, and started a sponsorship program for young people in HD families, whose studies often are interrupted caring for sick parents. It hopes to build permanent support centers in Colombia and Venezuela where HD families can get their food and medical needs met.
“The traditional thinking in the HD research community is that we’re helping people by doing the legwork to make medicine – and that’s not necessarily enough. You need a more holistic approach,” Dr. Muñoz-Sanjuán said.
Lennie Pineda, MSc, who recently retired as a geneticist with the University of Zulia in Maracaibo, Venezuela, said that Dr. Muñoz-Sanjuán was viewed skeptically when he first visited, in part because of his biomedical research background.
Ms. Pineda, who worked with the region’s HD families her whole career, has been wary of past research efforts in Venezuela. In 2010, she published a paper critical of Dr. Wexler’s and his colleagues’ approach (Revista Redbioética/UNESCO. 2010;1[2]:50-61), particularly regarding issues of informed consent.
“I was very cold to Nacho,” she laughed. “We all looked at him suspiciously.”
Ms. Pineda said Dr. Muñoz-Sanjuán won her over with his interest and creativity in finding concrete ways improve the lives of families in the Lake Maracaibo towns.
In a talk at the conference, Edison Soto, a young man from San Luis, a town on Lake Maracaibo that is a key cluster of HD, said Dr. Muñoz-Sanjuán’s visits had reawakened hope among the families there. “For years, no one thought about us, and because of the situation in the country it’s been hard, really hard,” he said.
“Nacho’s smart,” Ms. Pineda said. “He’s not coming to build a research cohort, he’s coming with genuine intention to help. But if one day conditions are adequate to support investigation, and the people here are well informed and volunteer for a study with full consent, well, all the better,” she said.
Dr. Muñoz-Sanjuán acknowledged that his humanitarian work could be perceived as preparing the ground for future clinical trials.
“I’m not doing anything research oriented with Latin America,” he said. “I would never approach these communities and recommend they take part in a study or give samples, unless their conditions change significantly. But the idea of cross-contamination is a problem I might need to fix. There may come a day where I need to depersonalize Factor-H from me.”
A research platform, a novel agent
Though HD research in Latin America remains rife with challenges, a number of investigators at the conference talked optimistically about planned and ongoing HD studies in Latin America.
The biggest of these is ENROLL-HD, a long-term global observational study of families with HD that uses a standardized approach to data collection. The platform, launched in 2013, aims to enroll 20,000 participants for yearly (or more frequent) assessment. Data from ENROLL-HD will support a diverse range of studies on everything from biomarkers to genetic modifiers to quality of life measures in HD.
ENROLL-HD has opened study sites in Argentina, Chile, and Colombia, and plans to launch a site near Lima, Peru, that is home to an HD cluster. Venezuela is considered out of reach, at least for now.
In Barranquilla, Claudia Perandones, MD, PhD, a genetics researcher in Argentina who manages ENROLL-HD for Latin America and is a cofounder of Factor-H, explained why the kind of clusters seen in Latin America are so valuable scientifically.
The extended family groups share a disease haplotype, eat the same foods, and live in similar environments, Dr. Perandones noted. Because not all the variation in HD can be explained by the number of CAG repeats a patient has, having a large sample with a common haplotype would help researchers pinpoint other environmental and genetic factors that can modify the onset or progress of the disease.
Another key goal of ENROLL-HD, investigators say, is to speed recruitment into clinical trials as they arise. And for the first time in history, potentially game-changing therapies are being developed specifically for HD.
For the past 5 years the Swiss pharmaceutical giant Roche has worked with a smaller biotech firm, Ionis Pharmaceuticals, on an agent called RG6042, which was known until recently as IONIS-HTTRx. CHDI was extensively involved in the agent’s preclinical development, contributing some $10 million to get it off the ground.
RG6042 is an antisense oligonucleotide, delivered by spinal injection, which works by interrupting an mRNA signaling pathway to suppress production of mutant HTT (mHTT) protein in the brain. Antisense oligonucleotides, sometimes called gene silencing therapies, are a new and promising approach in neurodegenerative diseases. Two have received FDA approval to treat spinal muscular atrophy and Duchenne muscular dystrophy.
In April 2018, Roche announced positive results from phase 1/2a study in 46 HD patients in Europe and North America. Patients in that 13-week study saw significant (up to 60%) dose-dependent reductions of the mHTT in their cerebrospinal fluid; a post hoc analysis also found some evidence of functional improvement (Neurology. 2018;90[15 Supplement]:CT.002).
These encouraging findings led to Roche’s announcement of a global phase 3 randomized, controlled trial that is scheduled to begin enrolling in 2019. Roche hopes to randomize 660 patients with mild HD across 15 countries for the 2-year trial, called GENERATION-HD1.
Sites in Latin America are expected to include Argentina, Chile, and Colombia.
At the Barranquilla meeting, Daniel Ciriano, MD, Roche’s Argentina-based medical director for Latin America, extolled the company’s commitment to ethics and social welfare in the region. In recent years, Roche has increased its humanitarian commitments across Latin America, including helping rebuild a Chilean village after an earthquake and offering free breast cancer and kidney disease treatments.
RG6042 is only one of a number of promising approaches to HD. Other therapies in the pipeline include gene silencing delivered by viral vectors instead of repeated spinal injections, an oral drug that interrupts mHTT production, immunotherapies, and even CRISPR gene–editing techniques.
Little was said at the conference, however, about how Latin American HD communities might be able to afford RG6042 or any other therapy that emerges from the pipeline.
Dr. Muñoz-Sanjuán called the issue “a theme for future discussion.”
“This is an area that has to be handled carefully and not one we are heavily invested in yet, although it’s very important,” he said.
On the ground
Several of the European and North American scientists who presented in Barranquilla took pains to express their concern with the well-being of HD patients in Latin America and to demonstrate goodwill toward the local researchers and clinicians.
Hilal A. Lashuel, PhD, a molecular biologist working on the structure and behavior of the HTT protein, said his participation in the Factor-H event at the Vatican the year before had awakened him to “the real human part of HD,” and changed the way he does science.
Normally, Dr. Lashuel said, “we do research disconnected from the realities of the diseases we work with.”
“We need to not just to do research but [to ensure] that research is done right,” he said, which means also focusing on improving patients’ standard of living.
The room broke out in applause when Dr. Lashuel announced new internships for investigators from developing countries. He also presented a parting video from his research team at the École Polytechnique Fédérale de Lausanne (Switzerland), complete with music and affectionate messages in Spanish.
Pharmacologist Elena Cattaneo, PhD, a stem cell researcher long active in the HD community, and also a senator in Italy’s parliament, delivered a similarly warm, carefully choreographed video message from her laboratory at the University of Milan.
Just days later in the town of Juan de Acosta, an hour inland of Barranquilla, the same researchers sat down with patients and families who crowded the waiting room of the town’s only hospital, as the sun beat in through the windows and as mule carts, stray dogs, and buses passed by on the main drag outside.
The event had been titled a “brigade” after all, but the HD families did not seem to mind – and indeed so many showed up that a sign had to be placed on the door saying that no one who arrived after noon could be seen. Consults were not limited to HD-related matters, so families could be seen for any complaint.
HD was first documented in this town in the early 1990s, but much remains to be understood about the size of the cluster, the haplotype, and its relation to other clusters in Colombia or Venezuela. The families here share a handful of last names and likely share a common ancestor. In the early 19th century, the Barranquilla region was flooded with European migrants who reached the city by ship. (HD clusters in Latin America tend to be concentrated in coastal regions, possibly because of migration patterns.)
The waiting room of the hospital was loud with chatter. Small children played as their relatives waited for consults. Some showed the characteristic restless movements and emaciated bodies of people with advanced HD.
The foreign scientists were barred from taking any patient data out of the hospital or asking for samples. Even picture taking was prohibited. Instead they performed genetic counseling and neuropsychological tests; they sorted out differential diagnoses and advised on medications. Visiting Colombian and Venezuelan physicians did the same, while their assistants met with families in the waiting room, taking medical histories and sketching out basic genealogies.
Some of the foreign researchers reported fruitful interactions with patients, while others seemed perplexed by what they’d experienced. Alba di Pardo, PhD, a genetic epidemiologist at the Istituto Neurologico Mediterraneo Pozzilli (Italy), said she’d spent the morning doing genetic counseling with families and going over genealogies to assess risk. Yet, despite the fact that anyone with an HD parent has a 50% chance of developing the disease, some family members acted uninterested, she said.
Dr. di Pardo’s colleague at the Istituto, biologist Vittorio Maglione, PhD, reported having a similar experience. As he was counseling a young woman about her risk for HD, she scrolled indifferently through Facebook posts on her phone, he said.
On some level, Dr. Maglione said, he could understand patients’ reluctance to engage noting that, while there were many potential HD therapies to try, any new treatment paradigm for HD was many years away from a place like this – and potentially very costly. Dr. Maglione – along with Dr. di Pardo – is researching the SP1 axis, a sphingolipid pathway implicated in neurodegenerative such diseases as HD and which has potential as a drug target (Trends Pharmacol Sci. 2018;39[5]:468-80).
Psychologist Pedro Puentes Rozo of Simón Bolivar University, who is working with Dr. Acosta-López on the local cohort study of presymptomatic HD patients, said that, for most of the families in the clinic that day, any seeming indifference probably masked deeper fears. People already were well aware of their risk. “They’ve known about it forever, said Dr. Puentes Rozo, who has been working with this HD population for a decade. “But this is a catastrophic illness and can generate a lot of anxiety.”
Dr. Puentes Rozo said the group’s planned study, unlike studies in the past, would be conducted under strict “international ethical norms and standards.” Subjects would receive ongoing psychological support, and the researchers were working to establish a genetic counseling center so that people who want to know their status “can be prepared,” he said, and plan for their lives and families.
By fall 2018, the cohort study was underway. The group had sponsored several more hospital brigades – or “integrated health days” as they preferred to call them, at the hospital in Juan de Acosta, giving them a chance to work face to face with families.
They drew no blood during the clinics, as investigators in the past had done. Instead, they explained the study to patients, performed the initial screenings, and invited them to designated study appointments at the university. Legal assistance was up and running, and the jobs program would start in 2019.
Enrollment was climbing. And the group was steadily accumulating data.
BARRANQUILLA, COLOMBIA – “We don’t like to call them brigades. That sounds militant,” said neuropsychologist Johan Acosta-López, PhD.
Dr. Acosta-López, the head of cognitive neurosciences at Simón Bolivar University in this city on Colombia’s Atlantic coast, was among five Colombian clinicians – neurologists, psychiatrists, and neuropsychologists – stuffed into a car on their way to a conference hotel in July 2018.
The following day they would be joined by clinicians and researchers from North America, other Latin American countries, and Europe for a first-of-its kind meeting on Huntington’s disease (HD) in the region, sponsored by Factor-H, an HD charity working in Latin America.
Once the talks wrapped up, the researchers – clinicians and basic scientists – were invited to see patients at a hospital in a town an hour inland with a large concentration of HD families, most of them extremely poor. For some, the Factor-H–sponsored “brigade” would be their first hands-on experience with HD patients in a developing country.
There was some debate in the car about what to call such events: brigades, “integrated health days,” or clinics. Around here – where HD abounded but patients were weary of researchers – terminology mattered.
“We’ve had so many investigators arrive in this area – foreigners and Colombians – telling people ‘we’ve got this huge, great project that you’ll benefit from.’ And they take blood samples and never return,” Dr. Acosta-López said.
Even as a local investigator, Dr. Acosta-López has faced challenges getting a new study off the ground. Dr. Acosta-López and his colleagues are working under a grant from the Colombian government to recruit 241 presymptomatic subjects with confirmed genetic markers for HD, and evaluate them for cognitive and neurologic changes preceding disease onset.
It’s a cross-sectional study, and such studies are usually funded for a year. But the investigators knew it would take much more than a year to recruit patients here, and planned their study for 3 years. As of July, the team had been engaging with the community for 6 months but still didn’t have a single blood sample.
“We’ve had to convince everyone that this time is different,” he said, “and that means focusing on the social aspect” – setting up a legal-assistance program through the university to help families claim health benefits and a job-training program sponsored by local businesses.
It’s unusual for researchers to find themselves playing such extensive roles in coordinating social and economic support for their subjects. But with HD, it’s happening across Latin America, where researchers speak frequently of a “debt” owed to HD families in this region.
Huntington’s disease is a neurodegenerative disease caused by a genetic mutation in the huntingtin (HTT) gene, changing the normal protein it expresses in the body to a toxic form that damages cortical and basal ganglia neurons. It affects between 0.5 to 1 in 10,000 people worldwide, with higher prevalence in the United States, Europe, and Australia.
HD is inherited in an autosomal dominant pattern; a child of a parent with the mutation has a 50% chance of developing the disease. Patients develop cognitive symptoms that progress to dementia, along with the debilitating involuntary, dancelike movements that gave the disease the name by which it was formerly known: Huntington’s chorea.
In the 1980s and 1990s, several generations of Latin American HD families provided data that allowed for some of the greatest research advances in the disease – and they may represent a large share of the world’s HD cases. Yet, they continue to live in extreme poverty and have benefited little from the findings of the past 3 decades.
Without recognizing this and working to improve the families’ well-being, the researchers at the conference said it’s unlikely that promising therapies in the pipeline will ever reach the populations that need them the most.
Discovery in Venezuela
Some 8 hours by car from Barranquilla sits Lake Maracaibo, Venezuela, home to the largest known clusters of HD patients worldwide. The disease is believed to have come to the shores of Lake Maracaibo with a lone European immigrant – a Spanish sailor, many claim – at the end of the 18th century. Cases were first described in the 1950s by a young Venezuelan physician named Américo Negrette, MD.
Dr. Negrette’s findings were ignored by health officials in Venezuela and went unnoticed in the international research community until 1972, when a student of Dr. Negrette’s presented at an HD conference in Ohio. There he drew the attention of the American neuropsychologist Nancy S. Wexler, PhD. Dr. Wexler’s own mother had died of HD, and her father Milton, a noted psychoanalyst, founded the first research foundation dedicated to the disease.
While prevalence of HD in North America, Australia, and Europe is about 1 in 10,000, the region around Lake Maracaibo saw 70 times that rate at the time, thanks to high birth rates, geographic isolation, and extensive intermarriage within a handful of families. The families comprised mostly poor fishermen who lived in makeshift homes in towns ringing the lake.
In 1979, Dr. Wexler, with funding from the U.S. National Institutes of Health, began making annual research visits to Lake Maracaibo, and in 1983, the research group she coordinated, using data from blood and tissue samples donated by affected families, identified the location of the huntingtin gene on chromosome 4 (Nature. 1983 Nov 17;306[5940]:234-8). A decade later, the researchers isolated the mutant version of the gene and found it to be a triplet (CAG) expansion mutation, with more CAG repetitions associated with earlier age at disease onset (Cell. 1993 Mar 26;72[6]:971-83). Dr. Wexler and her colleagues’ findings led to the first genetic tests for HD.
Nationalist policies in Venezuela ended Dr. Wexler and her colleagues’ annual visits to Lake Maracaibo in 2002, along with the food, clothing, and medicines that the group routinely distributed to the families when they came.
Over 23 years, the researchers obtained data from some 18,000 individuals, but the families did not benefit in any durable way from the research. Local investigators with whom Dr. Wexler’s group collaborated lacked the resources and training to continue independently.
Access to medications is limited in Venezuela, and there is no institutional support for hundreds of HD patients living in extreme poverty, many of them descendants of the patients who contributed to the research and generation of these samples. The families’ biological material was sent to labs abroad, where investigators continue to derive findings from it today. Though genetic testing was performed on thousands at risk for the disease, few received access to their results through genetic counseling. A hospice established by Dr. Wexler’s foundation limped along until 2014, when it was finally shuttered.
Rebuilding bridges
A handful of families from the Lake Maracaibo towns attended the conference in Barranquilla. Their travel costs were picked up by Factor-H, which sponsored the event.
Ignacio Muñoz-Sanjuán, PhD, Factor-H’s founder and president, knew the families personally. He’s visited them regularly for years. In 2017, Dr. Muñoz-Sanjuán, a molecular biologist known affectionately in the HD community as “Nacho,” invited several to Rome for a meeting with Pope Francis, as part of an effort to raise awareness of HD and to request support from the Catholic Church for the Latin American families.
Humanitarian work is relatively new to Dr. Muñoz-Sanjuán, who’s spent his career in drug development. In addition to his unpaid work with Factor-H, he is vice president of biology with the CHDI Foundation, a Los Angeles–based nonprofit that funds drug research in HD. CHDI is reported to have about $100 million in annual funding – about triple the NIH budget in recent years for HD research. Its major donors are a group of investors who for years have remained anonymous and do not publicly discuss their philanthropy.
The Spanish-born Dr. Muñoz-Sanjuán had little direct experience with HD populations in Latin America until a few years ago, he said.
At a CHDI meeting in Brazil, he said, “I was talking with physicians and patient advocates from Latin America, telling them they had to be willing to be involved, that these communities with high prevalence had a lot to offer science,” Dr. Muñoz-Sanjuán said in an interview. “I was told that it was me who needed to understand the conditions in which HD patients lived. It completely put me on the spot.”
HD tends to strike during the most productive years of a person’s life, from the late 30s onward, keeping them from working and obliging family members to stop working to care for them. In a poor community, it can condemn a family to a state of extreme poverty for generations. Tetrabenazine (Xenazine), a medication to quiet chorea symptoms, is costly enough that many patients must do without it. Ensuring adequate calorie intake is difficult in HD patients, whose constant movements cause them to lose weight.
Dr. Muñoz-Sanjuán traveled to Colombia, Venezuela, and Brazil, meeting HD families and doctors like neurologist Gustavo Barrios, MD, of Hospital Occidente de Kennedy in Bogotá, Colombia. In a talk at the Barranquilla conference, Dr. Barrios related the experience of his first visit to El Dificil, a community in northern Colombia where some large HD families are forced to survive on the equivalent of $5 a day. “I had to confront not only the fact that these families were living with a terrible disease but in conditions of extreme deprivation,” he said. “My life as a doctor changed that day.”
Dr. Muñoz-Sanjuán helped form a Latin American HD network to involve clinicians like Dr. Barrios who worked with HD clusters, most of them poorly studied. “These are all neglected communities that share similar features,” Dr. Muñoz-Sanjuán said.
On Colombia’s Caribbean coast, for example, HD had been documented since the early 1990s, but genotyping was not performed until recently. Prevalence data are “virtually nonexistent” in Colombia, said Sonia Moreno, PhD, a neuropsychologist at the University of Antioquia in Medellin. In a pilot study presented this year at the CHDI Foundation’s Enroll-HD Congress, Dr. Moreno and her colleagues mined Colombian public health data for likely HD cases, and argued for the creation of a national registry.
In 2012, Dr. Muñoz-Sanjuán founded Factor-H with the aim of improving living conditions for Latin American communities with HD.
Factor-H does not receive funds from CHDI Foundation and instead relies on donations from individuals and companies; its annual budget is less than $200,000. But through contracts with local nongovernmental organizations, it has sponsored health clinics and ongoing food assistance, delivered shipments of medicines and clothing, and started a sponsorship program for young people in HD families, whose studies often are interrupted caring for sick parents. It hopes to build permanent support centers in Colombia and Venezuela where HD families can get their food and medical needs met.
“The traditional thinking in the HD research community is that we’re helping people by doing the legwork to make medicine – and that’s not necessarily enough. You need a more holistic approach,” Dr. Muñoz-Sanjuán said.
Lennie Pineda, MSc, who recently retired as a geneticist with the University of Zulia in Maracaibo, Venezuela, said that Dr. Muñoz-Sanjuán was viewed skeptically when he first visited, in part because of his biomedical research background.
Ms. Pineda, who worked with the region’s HD families her whole career, has been wary of past research efforts in Venezuela. In 2010, she published a paper critical of Dr. Wexler’s and his colleagues’ approach (Revista Redbioética/UNESCO. 2010;1[2]:50-61), particularly regarding issues of informed consent.
“I was very cold to Nacho,” she laughed. “We all looked at him suspiciously.”
Ms. Pineda said Dr. Muñoz-Sanjuán won her over with his interest and creativity in finding concrete ways improve the lives of families in the Lake Maracaibo towns.
In a talk at the conference, Edison Soto, a young man from San Luis, a town on Lake Maracaibo that is a key cluster of HD, said Dr. Muñoz-Sanjuán’s visits had reawakened hope among the families there. “For years, no one thought about us, and because of the situation in the country it’s been hard, really hard,” he said.
“Nacho’s smart,” Ms. Pineda said. “He’s not coming to build a research cohort, he’s coming with genuine intention to help. But if one day conditions are adequate to support investigation, and the people here are well informed and volunteer for a study with full consent, well, all the better,” she said.
Dr. Muñoz-Sanjuán acknowledged that his humanitarian work could be perceived as preparing the ground for future clinical trials.
“I’m not doing anything research oriented with Latin America,” he said. “I would never approach these communities and recommend they take part in a study or give samples, unless their conditions change significantly. But the idea of cross-contamination is a problem I might need to fix. There may come a day where I need to depersonalize Factor-H from me.”
A research platform, a novel agent
Though HD research in Latin America remains rife with challenges, a number of investigators at the conference talked optimistically about planned and ongoing HD studies in Latin America.
The biggest of these is ENROLL-HD, a long-term global observational study of families with HD that uses a standardized approach to data collection. The platform, launched in 2013, aims to enroll 20,000 participants for yearly (or more frequent) assessment. Data from ENROLL-HD will support a diverse range of studies on everything from biomarkers to genetic modifiers to quality of life measures in HD.
ENROLL-HD has opened study sites in Argentina, Chile, and Colombia, and plans to launch a site near Lima, Peru, that is home to an HD cluster. Venezuela is considered out of reach, at least for now.
In Barranquilla, Claudia Perandones, MD, PhD, a genetics researcher in Argentina who manages ENROLL-HD for Latin America and is a cofounder of Factor-H, explained why the kind of clusters seen in Latin America are so valuable scientifically.
The extended family groups share a disease haplotype, eat the same foods, and live in similar environments, Dr. Perandones noted. Because not all the variation in HD can be explained by the number of CAG repeats a patient has, having a large sample with a common haplotype would help researchers pinpoint other environmental and genetic factors that can modify the onset or progress of the disease.
Another key goal of ENROLL-HD, investigators say, is to speed recruitment into clinical trials as they arise. And for the first time in history, potentially game-changing therapies are being developed specifically for HD.
For the past 5 years the Swiss pharmaceutical giant Roche has worked with a smaller biotech firm, Ionis Pharmaceuticals, on an agent called RG6042, which was known until recently as IONIS-HTTRx. CHDI was extensively involved in the agent’s preclinical development, contributing some $10 million to get it off the ground.
RG6042 is an antisense oligonucleotide, delivered by spinal injection, which works by interrupting an mRNA signaling pathway to suppress production of mutant HTT (mHTT) protein in the brain. Antisense oligonucleotides, sometimes called gene silencing therapies, are a new and promising approach in neurodegenerative diseases. Two have received FDA approval to treat spinal muscular atrophy and Duchenne muscular dystrophy.
In April 2018, Roche announced positive results from phase 1/2a study in 46 HD patients in Europe and North America. Patients in that 13-week study saw significant (up to 60%) dose-dependent reductions of the mHTT in their cerebrospinal fluid; a post hoc analysis also found some evidence of functional improvement (Neurology. 2018;90[15 Supplement]:CT.002).
These encouraging findings led to Roche’s announcement of a global phase 3 randomized, controlled trial that is scheduled to begin enrolling in 2019. Roche hopes to randomize 660 patients with mild HD across 15 countries for the 2-year trial, called GENERATION-HD1.
Sites in Latin America are expected to include Argentina, Chile, and Colombia.
At the Barranquilla meeting, Daniel Ciriano, MD, Roche’s Argentina-based medical director for Latin America, extolled the company’s commitment to ethics and social welfare in the region. In recent years, Roche has increased its humanitarian commitments across Latin America, including helping rebuild a Chilean village after an earthquake and offering free breast cancer and kidney disease treatments.
RG6042 is only one of a number of promising approaches to HD. Other therapies in the pipeline include gene silencing delivered by viral vectors instead of repeated spinal injections, an oral drug that interrupts mHTT production, immunotherapies, and even CRISPR gene–editing techniques.
Little was said at the conference, however, about how Latin American HD communities might be able to afford RG6042 or any other therapy that emerges from the pipeline.
Dr. Muñoz-Sanjuán called the issue “a theme for future discussion.”
“This is an area that has to be handled carefully and not one we are heavily invested in yet, although it’s very important,” he said.
On the ground
Several of the European and North American scientists who presented in Barranquilla took pains to express their concern with the well-being of HD patients in Latin America and to demonstrate goodwill toward the local researchers and clinicians.
Hilal A. Lashuel, PhD, a molecular biologist working on the structure and behavior of the HTT protein, said his participation in the Factor-H event at the Vatican the year before had awakened him to “the real human part of HD,” and changed the way he does science.
Normally, Dr. Lashuel said, “we do research disconnected from the realities of the diseases we work with.”
“We need to not just to do research but [to ensure] that research is done right,” he said, which means also focusing on improving patients’ standard of living.
The room broke out in applause when Dr. Lashuel announced new internships for investigators from developing countries. He also presented a parting video from his research team at the École Polytechnique Fédérale de Lausanne (Switzerland), complete with music and affectionate messages in Spanish.
Pharmacologist Elena Cattaneo, PhD, a stem cell researcher long active in the HD community, and also a senator in Italy’s parliament, delivered a similarly warm, carefully choreographed video message from her laboratory at the University of Milan.
Just days later in the town of Juan de Acosta, an hour inland of Barranquilla, the same researchers sat down with patients and families who crowded the waiting room of the town’s only hospital, as the sun beat in through the windows and as mule carts, stray dogs, and buses passed by on the main drag outside.
The event had been titled a “brigade” after all, but the HD families did not seem to mind – and indeed so many showed up that a sign had to be placed on the door saying that no one who arrived after noon could be seen. Consults were not limited to HD-related matters, so families could be seen for any complaint.
HD was first documented in this town in the early 1990s, but much remains to be understood about the size of the cluster, the haplotype, and its relation to other clusters in Colombia or Venezuela. The families here share a handful of last names and likely share a common ancestor. In the early 19th century, the Barranquilla region was flooded with European migrants who reached the city by ship. (HD clusters in Latin America tend to be concentrated in coastal regions, possibly because of migration patterns.)
The waiting room of the hospital was loud with chatter. Small children played as their relatives waited for consults. Some showed the characteristic restless movements and emaciated bodies of people with advanced HD.
The foreign scientists were barred from taking any patient data out of the hospital or asking for samples. Even picture taking was prohibited. Instead they performed genetic counseling and neuropsychological tests; they sorted out differential diagnoses and advised on medications. Visiting Colombian and Venezuelan physicians did the same, while their assistants met with families in the waiting room, taking medical histories and sketching out basic genealogies.
Some of the foreign researchers reported fruitful interactions with patients, while others seemed perplexed by what they’d experienced. Alba di Pardo, PhD, a genetic epidemiologist at the Istituto Neurologico Mediterraneo Pozzilli (Italy), said she’d spent the morning doing genetic counseling with families and going over genealogies to assess risk. Yet, despite the fact that anyone with an HD parent has a 50% chance of developing the disease, some family members acted uninterested, she said.
Dr. di Pardo’s colleague at the Istituto, biologist Vittorio Maglione, PhD, reported having a similar experience. As he was counseling a young woman about her risk for HD, she scrolled indifferently through Facebook posts on her phone, he said.
On some level, Dr. Maglione said, he could understand patients’ reluctance to engage noting that, while there were many potential HD therapies to try, any new treatment paradigm for HD was many years away from a place like this – and potentially very costly. Dr. Maglione – along with Dr. di Pardo – is researching the SP1 axis, a sphingolipid pathway implicated in neurodegenerative such diseases as HD and which has potential as a drug target (Trends Pharmacol Sci. 2018;39[5]:468-80).
Psychologist Pedro Puentes Rozo of Simón Bolivar University, who is working with Dr. Acosta-López on the local cohort study of presymptomatic HD patients, said that, for most of the families in the clinic that day, any seeming indifference probably masked deeper fears. People already were well aware of their risk. “They’ve known about it forever, said Dr. Puentes Rozo, who has been working with this HD population for a decade. “But this is a catastrophic illness and can generate a lot of anxiety.”
Dr. Puentes Rozo said the group’s planned study, unlike studies in the past, would be conducted under strict “international ethical norms and standards.” Subjects would receive ongoing psychological support, and the researchers were working to establish a genetic counseling center so that people who want to know their status “can be prepared,” he said, and plan for their lives and families.
By fall 2018, the cohort study was underway. The group had sponsored several more hospital brigades – or “integrated health days” as they preferred to call them, at the hospital in Juan de Acosta, giving them a chance to work face to face with families.
They drew no blood during the clinics, as investigators in the past had done. Instead, they explained the study to patients, performed the initial screenings, and invited them to designated study appointments at the university. Legal assistance was up and running, and the jobs program would start in 2019.
Enrollment was climbing. And the group was steadily accumulating data.
New brain circuitry found with Parkinson’s disease gene therapy
A gene therapy for Parkinson’s disease, focusing on the subthalamic nucleus, appears to lead to the formation of unique brain circuitry that correlates with clinical improvement.
In a paper published online Nov. 28 in Science Translational Medicine, researchers describe the findings of a metabolic imaging study to explore the mechanism underlying benefits seen in a phase 2, blinded, sham-controlled clinical trial of the gene therapy.
The therapy in question used an adeno-associated viral vector to deliver the gene for glutamic acid decarboxylase into the subthalamic nucleus – a region of the brain known to be overactivated in Parkinson’s disease – which was intended to have an inhibitory effect on the neurons in that region.
Martin Niethammer, MD, PhD, of the Center for Neurosciences at The Feinstein Institute for Medical Research in New York, and his coauthors used 18F-fluorodeoxyglucose positron emission tomography at baseline, 6, and 12 months in 15 gene-therapy patients and 21 sham-treated patients, which revealed the development of new brain circuits in patients treated with the gene therapy.
The circuits, which researchers called the glutamic acid decarboxylase-related pattern, or GADRP, presented with increased metabolism in the premotor region – which also extended into the adjacent motor cortex – and in the supramarginal gyrus. There was also decreased metabolic activity in the caudate, anterior putamen, and adjacent globus pallidus; the ventral anterior and medial dorsal thalamic nuclei; and in the inferior frontal gyrus.
All 15 patients who received the gene therapy showed significant trends in GADRP expression after the treatment, compared with patients who underwent the sham procedure. Furthermore, these correlated significantly with improved clinical outcomes.
The imaging also revealed increased connectivity between regions in the GADRP space among patients who received the gene therapy, with researchers noting five new intrahemispheric node-to-node connections in these patients that were not seen in the sham procedure group.
These included connection linking the left caudate nucleus to the left superior frontal node, the right superior frontal node to the right supramarginal gyrus, and linking the left anterior putamen and globus pallidus with the ipsilateral thalamic node.
The authors also found that overall connectivity in the network rose to “abnormal” levels in the 12 months after gene therapy, while no similar increases were seen in the sham group.
Given that deep brain stimulation for Parkinson’s disease also targets the subthalamic nucleus, researchers looked at changes to the GADRP network in these patients, compared with those who received sham therapy and those who received gene therapy.
They saw that changes in GADRP expression were significantly different between the gene therapy-treated patients and those treated with deep brain stimulation and sham surgery. However, the differences between deep brain stimulation and sham surgery were not significant.
“The current study indicates that customized networks can be characterized using functional imaging data acquired in randomized, controlled phase 2 clinical trials and, if validated, could be used as quantitative outcome measures in more definitive, later-stage clinical trials,” the authors wrote.
The study was supported by Neurologix. Two authors were consultants and stockholders of MeiraGTx.
SOURCE: Niethammer N et al. Sci Transl Med. 2018 Nov 28. doi: 10.1126/scitranslmed.aau0713.
A gene therapy for Parkinson’s disease, focusing on the subthalamic nucleus, appears to lead to the formation of unique brain circuitry that correlates with clinical improvement.
In a paper published online Nov. 28 in Science Translational Medicine, researchers describe the findings of a metabolic imaging study to explore the mechanism underlying benefits seen in a phase 2, blinded, sham-controlled clinical trial of the gene therapy.
The therapy in question used an adeno-associated viral vector to deliver the gene for glutamic acid decarboxylase into the subthalamic nucleus – a region of the brain known to be overactivated in Parkinson’s disease – which was intended to have an inhibitory effect on the neurons in that region.
Martin Niethammer, MD, PhD, of the Center for Neurosciences at The Feinstein Institute for Medical Research in New York, and his coauthors used 18F-fluorodeoxyglucose positron emission tomography at baseline, 6, and 12 months in 15 gene-therapy patients and 21 sham-treated patients, which revealed the development of new brain circuits in patients treated with the gene therapy.
The circuits, which researchers called the glutamic acid decarboxylase-related pattern, or GADRP, presented with increased metabolism in the premotor region – which also extended into the adjacent motor cortex – and in the supramarginal gyrus. There was also decreased metabolic activity in the caudate, anterior putamen, and adjacent globus pallidus; the ventral anterior and medial dorsal thalamic nuclei; and in the inferior frontal gyrus.
All 15 patients who received the gene therapy showed significant trends in GADRP expression after the treatment, compared with patients who underwent the sham procedure. Furthermore, these correlated significantly with improved clinical outcomes.
The imaging also revealed increased connectivity between regions in the GADRP space among patients who received the gene therapy, with researchers noting five new intrahemispheric node-to-node connections in these patients that were not seen in the sham procedure group.
These included connection linking the left caudate nucleus to the left superior frontal node, the right superior frontal node to the right supramarginal gyrus, and linking the left anterior putamen and globus pallidus with the ipsilateral thalamic node.
The authors also found that overall connectivity in the network rose to “abnormal” levels in the 12 months after gene therapy, while no similar increases were seen in the sham group.
Given that deep brain stimulation for Parkinson’s disease also targets the subthalamic nucleus, researchers looked at changes to the GADRP network in these patients, compared with those who received sham therapy and those who received gene therapy.
They saw that changes in GADRP expression were significantly different between the gene therapy-treated patients and those treated with deep brain stimulation and sham surgery. However, the differences between deep brain stimulation and sham surgery were not significant.
“The current study indicates that customized networks can be characterized using functional imaging data acquired in randomized, controlled phase 2 clinical trials and, if validated, could be used as quantitative outcome measures in more definitive, later-stage clinical trials,” the authors wrote.
The study was supported by Neurologix. Two authors were consultants and stockholders of MeiraGTx.
SOURCE: Niethammer N et al. Sci Transl Med. 2018 Nov 28. doi: 10.1126/scitranslmed.aau0713.
A gene therapy for Parkinson’s disease, focusing on the subthalamic nucleus, appears to lead to the formation of unique brain circuitry that correlates with clinical improvement.
In a paper published online Nov. 28 in Science Translational Medicine, researchers describe the findings of a metabolic imaging study to explore the mechanism underlying benefits seen in a phase 2, blinded, sham-controlled clinical trial of the gene therapy.
The therapy in question used an adeno-associated viral vector to deliver the gene for glutamic acid decarboxylase into the subthalamic nucleus – a region of the brain known to be overactivated in Parkinson’s disease – which was intended to have an inhibitory effect on the neurons in that region.
Martin Niethammer, MD, PhD, of the Center for Neurosciences at The Feinstein Institute for Medical Research in New York, and his coauthors used 18F-fluorodeoxyglucose positron emission tomography at baseline, 6, and 12 months in 15 gene-therapy patients and 21 sham-treated patients, which revealed the development of new brain circuits in patients treated with the gene therapy.
The circuits, which researchers called the glutamic acid decarboxylase-related pattern, or GADRP, presented with increased metabolism in the premotor region – which also extended into the adjacent motor cortex – and in the supramarginal gyrus. There was also decreased metabolic activity in the caudate, anterior putamen, and adjacent globus pallidus; the ventral anterior and medial dorsal thalamic nuclei; and in the inferior frontal gyrus.
All 15 patients who received the gene therapy showed significant trends in GADRP expression after the treatment, compared with patients who underwent the sham procedure. Furthermore, these correlated significantly with improved clinical outcomes.
The imaging also revealed increased connectivity between regions in the GADRP space among patients who received the gene therapy, with researchers noting five new intrahemispheric node-to-node connections in these patients that were not seen in the sham procedure group.
These included connection linking the left caudate nucleus to the left superior frontal node, the right superior frontal node to the right supramarginal gyrus, and linking the left anterior putamen and globus pallidus with the ipsilateral thalamic node.
The authors also found that overall connectivity in the network rose to “abnormal” levels in the 12 months after gene therapy, while no similar increases were seen in the sham group.
Given that deep brain stimulation for Parkinson’s disease also targets the subthalamic nucleus, researchers looked at changes to the GADRP network in these patients, compared with those who received sham therapy and those who received gene therapy.
They saw that changes in GADRP expression were significantly different between the gene therapy-treated patients and those treated with deep brain stimulation and sham surgery. However, the differences between deep brain stimulation and sham surgery were not significant.
“The current study indicates that customized networks can be characterized using functional imaging data acquired in randomized, controlled phase 2 clinical trials and, if validated, could be used as quantitative outcome measures in more definitive, later-stage clinical trials,” the authors wrote.
The study was supported by Neurologix. Two authors were consultants and stockholders of MeiraGTx.
SOURCE: Niethammer N et al. Sci Transl Med. 2018 Nov 28. doi: 10.1126/scitranslmed.aau0713.
FROM SCIENCE TRANSLATIONAL MEDICINE
Key clinical point:
Major finding: Gene therapy for Parkinson’s disease was associated with increased brain connectivity.
Study details: A phase 2, blinded, sham-controlled study of 36 patients with Parkinson’s disease.
Disclosures: The study was supported by Neurologix. Two authors were consultants and stockholders of MeiraGTx.
Source: Niethammer N et al. Sci Transl Med. 2018 Nov 28. doi: 10.1126/scitranslmed.aau0713.
Home-Based Therapy May Benefit Young Children With Motor Stereotypies
Teaching parents to deliver behavioral therapy via an instructional DVD may be an effective treatment approach for patients 5 and older.
CHICAGO—Home-based, parent-administered behavioral therapy supplemented by telephone contact with a therapist is effective in reducing complex motor stereotypies in children as young as 5, according to a study presented at the 47th Annual Meeting of the Child Neurology Society. “We recommend this combined approach for children ages 5 and older,” said Harvey S. Singer, MD, Professor of Neurology at Johns Hopkins School of Medicine in Baltimore, and colleagues.

Rhythmic Movements
Motor stereotypies are repetitive, rhythmic, fixed movements that last for seconds or minutes, stop with distraction, and are thought to arise from alterations within habitual motor pathways in the brain. Complex motor stereotypies typically involve the upper extremities (eg, hand flapping and finger wiggling) and begin in early childhood. Pharmacologic therapy has not been effective, but behavioral therapy has benefited patients, Dr. Singer and colleagues said. In one study of 54 children ages 7 to 14, home-based, parent-administered behavioral therapy using an instructional DVD significantly reduced stereotypies versus baseline.
To evaluate the effectiveness of a home-based, parent-provided therapy accompanied by scheduled telephone calls with a therapist in patients ages 5 to 7 with primary complex motor stereotypies, Dr. Singer and colleagues conducted a study with 38 children (24 boys; mean age, 6).
Patients had confirmed complex motor stereotypies with onset before age 3. Patients reported no premonitory urge and had temporary suspension of movement by an external stimulus or distraction. The researchers did not exclude patients due to inattentiveness, hyperactivity, impulsivity, or obsessive-compulsive behaviors. They did exclude patients with autism spectrum disorder, evidence of intellectual disability, seizures or a known neurologic disorder, or motor or vocal tics. Primary outcome measures included the Stereotypy Severity Scale (SSS) Motor and Impairment scores and the Stereotypy Linear Analog Scale (SLAS). Secondary outcomes included Patient Global Impression of Improvement (PGI-I).
Behavioral Therapy
The investigators sent participants a 44-minute instructional DVD about complex motor stereotypies with instructions provided by a behavioral psychologist. Parents were instructed to implement awareness training in the first week and to collect data and reinforce suppression starting in Week 2. Awareness training aims to make the child aware of his or her movements by using videos showing the activity and by the practice of voluntarily starting and stopping the movement.
Fourteen of the 38 participants were lost to follow-up (ie, they completed no post-DVD receipt assessments). The intent-to-treat group included 24 participants (15 boys) who completed at least one phone call with the study psychologist after receiving the DVD. There was no difference in stereotypy severity among those who were lost to follow-up and those in the intent-to-treat group. Those lost to follow-up had higher scores on ADHD ratings, however, which “suggests those with greater ADHD symptomatology at baseline were more likely to drop out,” the researchers said.
Compared with baseline measures, participants in the intent-to-treat group had significant reductions in SSS Motor and SSS Impairment scores at the last available assessment, and outcome measures progressively improved during the study. On the PGI-I, 56.5% of participants were very much or much improved, 30% were improved, and 13% reported no change.
All primary outcome measures significantly decreased. SSS Motor scores decreased by 23% as measured by telephone and 30% as measured online, SSS Impairment scores decreased by 31% measured by telephone and 32% measured online, and SLAS decreased by 54% as measured online. “The similarity between beneficial outcomes on the SSS Motor and SSS Impairment scales suggest a positive impact on both the movements themselves and their functional impact,” Dr. Singer and colleagues said.
Teaching parents to deliver behavioral therapy via an instructional DVD may be an effective treatment approach for patients 5 and older.
Teaching parents to deliver behavioral therapy via an instructional DVD may be an effective treatment approach for patients 5 and older.
CHICAGO—Home-based, parent-administered behavioral therapy supplemented by telephone contact with a therapist is effective in reducing complex motor stereotypies in children as young as 5, according to a study presented at the 47th Annual Meeting of the Child Neurology Society. “We recommend this combined approach for children ages 5 and older,” said Harvey S. Singer, MD, Professor of Neurology at Johns Hopkins School of Medicine in Baltimore, and colleagues.
Rhythmic Movements
Motor stereotypies are repetitive, rhythmic, fixed movements that last for seconds or minutes, stop with distraction, and are thought to arise from alterations within habitual motor pathways in the brain. Complex motor stereotypies typically involve the upper extremities (eg, hand flapping and finger wiggling) and begin in early childhood. Pharmacologic therapy has not been effective, but behavioral therapy has benefited patients, Dr. Singer and colleagues said. In one study of 54 children ages 7 to 14, home-based, parent-administered behavioral therapy using an instructional DVD significantly reduced stereotypies versus baseline.
To evaluate the effectiveness of a home-based, parent-provided therapy accompanied by scheduled telephone calls with a therapist in patients ages 5 to 7 with primary complex motor stereotypies, Dr. Singer and colleagues conducted a study with 38 children (24 boys; mean age, 6).
Patients had confirmed complex motor stereotypies with onset before age 3. Patients reported no premonitory urge and had temporary suspension of movement by an external stimulus or distraction. The researchers did not exclude patients due to inattentiveness, hyperactivity, impulsivity, or obsessive-compulsive behaviors. They did exclude patients with autism spectrum disorder, evidence of intellectual disability, seizures or a known neurologic disorder, or motor or vocal tics. Primary outcome measures included the Stereotypy Severity Scale (SSS) Motor and Impairment scores and the Stereotypy Linear Analog Scale (SLAS). Secondary outcomes included Patient Global Impression of Improvement (PGI-I).
Behavioral Therapy
The investigators sent participants a 44-minute instructional DVD about complex motor stereotypies with instructions provided by a behavioral psychologist. Parents were instructed to implement awareness training in the first week and to collect data and reinforce suppression starting in Week 2. Awareness training aims to make the child aware of his or her movements by using videos showing the activity and by the practice of voluntarily starting and stopping the movement.
Fourteen of the 38 participants were lost to follow-up (ie, they completed no post-DVD receipt assessments). The intent-to-treat group included 24 participants (15 boys) who completed at least one phone call with the study psychologist after receiving the DVD. There was no difference in stereotypy severity among those who were lost to follow-up and those in the intent-to-treat group. Those lost to follow-up had higher scores on ADHD ratings, however, which “suggests those with greater ADHD symptomatology at baseline were more likely to drop out,” the researchers said.
Compared with baseline measures, participants in the intent-to-treat group had significant reductions in SSS Motor and SSS Impairment scores at the last available assessment, and outcome measures progressively improved during the study. On the PGI-I, 56.5% of participants were very much or much improved, 30% were improved, and 13% reported no change.
All primary outcome measures significantly decreased. SSS Motor scores decreased by 23% as measured by telephone and 30% as measured online, SSS Impairment scores decreased by 31% measured by telephone and 32% measured online, and SLAS decreased by 54% as measured online. “The similarity between beneficial outcomes on the SSS Motor and SSS Impairment scales suggest a positive impact on both the movements themselves and their functional impact,” Dr. Singer and colleagues said.
CHICAGO—Home-based, parent-administered behavioral therapy supplemented by telephone contact with a therapist is effective in reducing complex motor stereotypies in children as young as 5, according to a study presented at the 47th Annual Meeting of the Child Neurology Society. “We recommend this combined approach for children ages 5 and older,” said Harvey S. Singer, MD, Professor of Neurology at Johns Hopkins School of Medicine in Baltimore, and colleagues.
Rhythmic Movements
Motor stereotypies are repetitive, rhythmic, fixed movements that last for seconds or minutes, stop with distraction, and are thought to arise from alterations within habitual motor pathways in the brain. Complex motor stereotypies typically involve the upper extremities (eg, hand flapping and finger wiggling) and begin in early childhood. Pharmacologic therapy has not been effective, but behavioral therapy has benefited patients, Dr. Singer and colleagues said. In one study of 54 children ages 7 to 14, home-based, parent-administered behavioral therapy using an instructional DVD significantly reduced stereotypies versus baseline.
To evaluate the effectiveness of a home-based, parent-provided therapy accompanied by scheduled telephone calls with a therapist in patients ages 5 to 7 with primary complex motor stereotypies, Dr. Singer and colleagues conducted a study with 38 children (24 boys; mean age, 6).
Patients had confirmed complex motor stereotypies with onset before age 3. Patients reported no premonitory urge and had temporary suspension of movement by an external stimulus or distraction. The researchers did not exclude patients due to inattentiveness, hyperactivity, impulsivity, or obsessive-compulsive behaviors. They did exclude patients with autism spectrum disorder, evidence of intellectual disability, seizures or a known neurologic disorder, or motor or vocal tics. Primary outcome measures included the Stereotypy Severity Scale (SSS) Motor and Impairment scores and the Stereotypy Linear Analog Scale (SLAS). Secondary outcomes included Patient Global Impression of Improvement (PGI-I).
Behavioral Therapy
The investigators sent participants a 44-minute instructional DVD about complex motor stereotypies with instructions provided by a behavioral psychologist. Parents were instructed to implement awareness training in the first week and to collect data and reinforce suppression starting in Week 2. Awareness training aims to make the child aware of his or her movements by using videos showing the activity and by the practice of voluntarily starting and stopping the movement.
Fourteen of the 38 participants were lost to follow-up (ie, they completed no post-DVD receipt assessments). The intent-to-treat group included 24 participants (15 boys) who completed at least one phone call with the study psychologist after receiving the DVD. There was no difference in stereotypy severity among those who were lost to follow-up and those in the intent-to-treat group. Those lost to follow-up had higher scores on ADHD ratings, however, which “suggests those with greater ADHD symptomatology at baseline were more likely to drop out,” the researchers said.
Compared with baseline measures, participants in the intent-to-treat group had significant reductions in SSS Motor and SSS Impairment scores at the last available assessment, and outcome measures progressively improved during the study. On the PGI-I, 56.5% of participants were very much or much improved, 30% were improved, and 13% reported no change.
All primary outcome measures significantly decreased. SSS Motor scores decreased by 23% as measured by telephone and 30% as measured online, SSS Impairment scores decreased by 31% measured by telephone and 32% measured online, and SLAS decreased by 54% as measured online. “The similarity between beneficial outcomes on the SSS Motor and SSS Impairment scales suggest a positive impact on both the movements themselves and their functional impact,” Dr. Singer and colleagues said.
Robin Williams’ widow recounts ‘terror’ of late husband’s Lewy body dementia
ATLANTA –

“With our medical team’s care, for the next 10 months we chased symptoms, but they were so elusive,” Mr. Williams’ widow, Susan Schneider Williams, said during a keynote address at the annual meeting of the American Neurological Association. “One hallmark of LBD is that symptoms appear and disappear randomly. The game whack-a-mole comes to mind. As soon as you think you are about to figure out a symptom, it disappears, and another one pops up.”
Mr. Williams’ medical team included one general physician, one neurologist, one motor specialist, two psychiatrists, one hypnotherapist, one physical trainer, and assorted alternative specialists. “We had been celebrating our second wedding anniversary when Robin started having gut discomfort,” Ms. Williams recalled. “He was tested for diverticulitis [but] the results came back negative. The pain eventually subsided but what was alarming was Robin’s reaction to it. He had a sudden and sustained spike in fear and anxiety unlike anything I’d seen before. By that point, we’d been by each other’s side long enough that I knew his normal baseline moods, fears, and anxieties. This was totally out of character, and I wondered privately: ‘Is my husband a hypochondriac?’ What I know now is that he was exhibiting a notable hallmark of LBD: new onset anxiety, sustained.” Lewy body disease is characterized by more than 40 symptoms, she continued, “and Robin experienced nearly all of them. He was particularly debilitated by fear, anxiety, delusions, paranoia, and as I came to find out later, hallucinations.”
The medical team continued running all sorts of tests, but everything kept came back negative, except for a very high cortisol count. By the late spring of 2014, however, Mr. Williams was diagnosed with Parkinson’s disease. “I was relieved to find out we finally had an answer, but I could tell, Robin was not buying it,” said Ms. Williams, who is a California-based fine artist, author, and brain health advocate. “The motor specialist said it was early and mild and that he’d be feeling better once he adjusted to the medications, [that] he had another 10 good years.”
In an attempt to treat the Parkinson’s and what was assumed to be depression, his care plan involved adjusting Parkinson’s medications, combined with an antidepressant. His physician also recommended a visit to the Dan Anderson Renewal Center in Minnesota, “for enhanced 12-step work to augment his sobriety,” Ms. Williams said. “The hope was this might help with fear and anxiety. Robin was clean and sober for 8 continuous years when he passed. I watched how he gained spiritually in so many ways from all the work he’d been doing, but his brain biology was going in the exact opposite direction. He tried desperately to join the parts of his heart, mind, and spirit, but his brain was pulling him apart. I felt like I was watching my husband disintegrate before my eyes, and there was nothing anyone could do about it. There came a day when we were getting ready to go to one of our dear friend’s birthday party. I came and saw Robin as he lay on our bed, imprisoned by fear and anxiety. Through tears, he pleaded, ‘I just want to reboot my brain!’ I promised him, ‘I know, honey. I swear we’re going to get to the bottom of this.’ ”
The couple was about a week away from choosing which neurocognitive testing facility to go to for further evaluation when Mr. Williams took his own life in his Paradise Cay, Calif., home on Aug. 11, 2014. “Robin was exhausted from the terror coming from his brain,” Ms. Williams said. “He took [his own life] before it could take any more of him.”
About 3 months later, the underlying cause of death was revealed: diffuse Lewy body dementia, “one of the worst cases they’d ever seen,” she said. “Because Robin’s disease pathway was extreme and unfolded the way it did, it highlights quite strikingly this disease spectrum. He had a perfusion of Lewy bodies, the essential underlying shared biology between Parkinson’s and Lewy body disease, scattered throughout his entire brain and brain stem.” She added that her husband’s prior history of depression from earlier in life “added to the challenge of getting a proper diagnosis. That single symptom of depression was being treated as its own illness, rather than part of the larger neurocognitive disease. It seems that one of the biggest challenges to getting an accurate diagnosis is that LBD symptoms have tremendous crossover with normal human psychology and behavior, mood, cognition and sleep issues. All of us experience fear, stress, anxiety, paranoia, trouble sleeping, mild depression, and other issues from time to time. We would hardly be human if we didn’t. The challenge of LBD is seeing the giant constellation that it is, rather than just a few of its stars.”
In early 2016, Ms. Williams received the “Commitment to Cures Award” from American Brain Foundation, honoring work she’s done raising awareness for Lewy body disease since her husband’s death. “The day I accepted that award and told our story to a room full of neurologists, my path was forever changed,” she said. “The ABF’s mission of connecting donors to researchers and curing brain disease was an alignment with my mission and hope.” She currently serves as vice chair of the ABF’s board of directors.
“From my own research and from the myriad of letters and information that has come to me, I have distilled what I think are the top three overlooked ideas in this disease space,” Ms. Williams said. “1. Diagnosis: The norm seems to be misdiagnosis, switched diagnosis, or no diagnosis at all. 2. Symptoms: They are being treated independently, apart from the neurological disorder. 3. Suicides: If more autopsies were done, more suicides would be attributed to this disease.”
She concluded her address by reflecting on the impact of her husband’s death has had in bringing an international spotlight to LBD. “When I meet individuals who have lost someone they loved to LBD, I see the pain in their eyes, but I hear the determination in their voice as they chart their own course toward making a difference,” Ms. Williams said. “I have been blessed to learn over and over again that I am not alone. I believe that Robin’s death in this battle against these diseases holds a profound purpose. There was tremendous power in what he suffered, and I saw that power up close. I’m here doing all that I can to see that power transformed into something good.”
ATLANTA –
“With our medical team’s care, for the next 10 months we chased symptoms, but they were so elusive,” Mr. Williams’ widow, Susan Schneider Williams, said during a keynote address at the annual meeting of the American Neurological Association. “One hallmark of LBD is that symptoms appear and disappear randomly. The game whack-a-mole comes to mind. As soon as you think you are about to figure out a symptom, it disappears, and another one pops up.”
Mr. Williams’ medical team included one general physician, one neurologist, one motor specialist, two psychiatrists, one hypnotherapist, one physical trainer, and assorted alternative specialists. “We had been celebrating our second wedding anniversary when Robin started having gut discomfort,” Ms. Williams recalled. “He was tested for diverticulitis [but] the results came back negative. The pain eventually subsided but what was alarming was Robin’s reaction to it. He had a sudden and sustained spike in fear and anxiety unlike anything I’d seen before. By that point, we’d been by each other’s side long enough that I knew his normal baseline moods, fears, and anxieties. This was totally out of character, and I wondered privately: ‘Is my husband a hypochondriac?’ What I know now is that he was exhibiting a notable hallmark of LBD: new onset anxiety, sustained.” Lewy body disease is characterized by more than 40 symptoms, she continued, “and Robin experienced nearly all of them. He was particularly debilitated by fear, anxiety, delusions, paranoia, and as I came to find out later, hallucinations.”
The medical team continued running all sorts of tests, but everything kept came back negative, except for a very high cortisol count. By the late spring of 2014, however, Mr. Williams was diagnosed with Parkinson’s disease. “I was relieved to find out we finally had an answer, but I could tell, Robin was not buying it,” said Ms. Williams, who is a California-based fine artist, author, and brain health advocate. “The motor specialist said it was early and mild and that he’d be feeling better once he adjusted to the medications, [that] he had another 10 good years.”
In an attempt to treat the Parkinson’s and what was assumed to be depression, his care plan involved adjusting Parkinson’s medications, combined with an antidepressant. His physician also recommended a visit to the Dan Anderson Renewal Center in Minnesota, “for enhanced 12-step work to augment his sobriety,” Ms. Williams said. “The hope was this might help with fear and anxiety. Robin was clean and sober for 8 continuous years when he passed. I watched how he gained spiritually in so many ways from all the work he’d been doing, but his brain biology was going in the exact opposite direction. He tried desperately to join the parts of his heart, mind, and spirit, but his brain was pulling him apart. I felt like I was watching my husband disintegrate before my eyes, and there was nothing anyone could do about it. There came a day when we were getting ready to go to one of our dear friend’s birthday party. I came and saw Robin as he lay on our bed, imprisoned by fear and anxiety. Through tears, he pleaded, ‘I just want to reboot my brain!’ I promised him, ‘I know, honey. I swear we’re going to get to the bottom of this.’ ”
The couple was about a week away from choosing which neurocognitive testing facility to go to for further evaluation when Mr. Williams took his own life in his Paradise Cay, Calif., home on Aug. 11, 2014. “Robin was exhausted from the terror coming from his brain,” Ms. Williams said. “He took [his own life] before it could take any more of him.”
About 3 months later, the underlying cause of death was revealed: diffuse Lewy body dementia, “one of the worst cases they’d ever seen,” she said. “Because Robin’s disease pathway was extreme and unfolded the way it did, it highlights quite strikingly this disease spectrum. He had a perfusion of Lewy bodies, the essential underlying shared biology between Parkinson’s and Lewy body disease, scattered throughout his entire brain and brain stem.” She added that her husband’s prior history of depression from earlier in life “added to the challenge of getting a proper diagnosis. That single symptom of depression was being treated as its own illness, rather than part of the larger neurocognitive disease. It seems that one of the biggest challenges to getting an accurate diagnosis is that LBD symptoms have tremendous crossover with normal human psychology and behavior, mood, cognition and sleep issues. All of us experience fear, stress, anxiety, paranoia, trouble sleeping, mild depression, and other issues from time to time. We would hardly be human if we didn’t. The challenge of LBD is seeing the giant constellation that it is, rather than just a few of its stars.”
In early 2016, Ms. Williams received the “Commitment to Cures Award” from American Brain Foundation, honoring work she’s done raising awareness for Lewy body disease since her husband’s death. “The day I accepted that award and told our story to a room full of neurologists, my path was forever changed,” she said. “The ABF’s mission of connecting donors to researchers and curing brain disease was an alignment with my mission and hope.” She currently serves as vice chair of the ABF’s board of directors.
“From my own research and from the myriad of letters and information that has come to me, I have distilled what I think are the top three overlooked ideas in this disease space,” Ms. Williams said. “1. Diagnosis: The norm seems to be misdiagnosis, switched diagnosis, or no diagnosis at all. 2. Symptoms: They are being treated independently, apart from the neurological disorder. 3. Suicides: If more autopsies were done, more suicides would be attributed to this disease.”
She concluded her address by reflecting on the impact of her husband’s death has had in bringing an international spotlight to LBD. “When I meet individuals who have lost someone they loved to LBD, I see the pain in their eyes, but I hear the determination in their voice as they chart their own course toward making a difference,” Ms. Williams said. “I have been blessed to learn over and over again that I am not alone. I believe that Robin’s death in this battle against these diseases holds a profound purpose. There was tremendous power in what he suffered, and I saw that power up close. I’m here doing all that I can to see that power transformed into something good.”
ATLANTA –
“With our medical team’s care, for the next 10 months we chased symptoms, but they were so elusive,” Mr. Williams’ widow, Susan Schneider Williams, said during a keynote address at the annual meeting of the American Neurological Association. “One hallmark of LBD is that symptoms appear and disappear randomly. The game whack-a-mole comes to mind. As soon as you think you are about to figure out a symptom, it disappears, and another one pops up.”
Mr. Williams’ medical team included one general physician, one neurologist, one motor specialist, two psychiatrists, one hypnotherapist, one physical trainer, and assorted alternative specialists. “We had been celebrating our second wedding anniversary when Robin started having gut discomfort,” Ms. Williams recalled. “He was tested for diverticulitis [but] the results came back negative. The pain eventually subsided but what was alarming was Robin’s reaction to it. He had a sudden and sustained spike in fear and anxiety unlike anything I’d seen before. By that point, we’d been by each other’s side long enough that I knew his normal baseline moods, fears, and anxieties. This was totally out of character, and I wondered privately: ‘Is my husband a hypochondriac?’ What I know now is that he was exhibiting a notable hallmark of LBD: new onset anxiety, sustained.” Lewy body disease is characterized by more than 40 symptoms, she continued, “and Robin experienced nearly all of them. He was particularly debilitated by fear, anxiety, delusions, paranoia, and as I came to find out later, hallucinations.”
The medical team continued running all sorts of tests, but everything kept came back negative, except for a very high cortisol count. By the late spring of 2014, however, Mr. Williams was diagnosed with Parkinson’s disease. “I was relieved to find out we finally had an answer, but I could tell, Robin was not buying it,” said Ms. Williams, who is a California-based fine artist, author, and brain health advocate. “The motor specialist said it was early and mild and that he’d be feeling better once he adjusted to the medications, [that] he had another 10 good years.”
In an attempt to treat the Parkinson’s and what was assumed to be depression, his care plan involved adjusting Parkinson’s medications, combined with an antidepressant. His physician also recommended a visit to the Dan Anderson Renewal Center in Minnesota, “for enhanced 12-step work to augment his sobriety,” Ms. Williams said. “The hope was this might help with fear and anxiety. Robin was clean and sober for 8 continuous years when he passed. I watched how he gained spiritually in so many ways from all the work he’d been doing, but his brain biology was going in the exact opposite direction. He tried desperately to join the parts of his heart, mind, and spirit, but his brain was pulling him apart. I felt like I was watching my husband disintegrate before my eyes, and there was nothing anyone could do about it. There came a day when we were getting ready to go to one of our dear friend’s birthday party. I came and saw Robin as he lay on our bed, imprisoned by fear and anxiety. Through tears, he pleaded, ‘I just want to reboot my brain!’ I promised him, ‘I know, honey. I swear we’re going to get to the bottom of this.’ ”
The couple was about a week away from choosing which neurocognitive testing facility to go to for further evaluation when Mr. Williams took his own life in his Paradise Cay, Calif., home on Aug. 11, 2014. “Robin was exhausted from the terror coming from his brain,” Ms. Williams said. “He took [his own life] before it could take any more of him.”
About 3 months later, the underlying cause of death was revealed: diffuse Lewy body dementia, “one of the worst cases they’d ever seen,” she said. “Because Robin’s disease pathway was extreme and unfolded the way it did, it highlights quite strikingly this disease spectrum. He had a perfusion of Lewy bodies, the essential underlying shared biology between Parkinson’s and Lewy body disease, scattered throughout his entire brain and brain stem.” She added that her husband’s prior history of depression from earlier in life “added to the challenge of getting a proper diagnosis. That single symptom of depression was being treated as its own illness, rather than part of the larger neurocognitive disease. It seems that one of the biggest challenges to getting an accurate diagnosis is that LBD symptoms have tremendous crossover with normal human psychology and behavior, mood, cognition and sleep issues. All of us experience fear, stress, anxiety, paranoia, trouble sleeping, mild depression, and other issues from time to time. We would hardly be human if we didn’t. The challenge of LBD is seeing the giant constellation that it is, rather than just a few of its stars.”
In early 2016, Ms. Williams received the “Commitment to Cures Award” from American Brain Foundation, honoring work she’s done raising awareness for Lewy body disease since her husband’s death. “The day I accepted that award and told our story to a room full of neurologists, my path was forever changed,” she said. “The ABF’s mission of connecting donors to researchers and curing brain disease was an alignment with my mission and hope.” She currently serves as vice chair of the ABF’s board of directors.
“From my own research and from the myriad of letters and information that has come to me, I have distilled what I think are the top three overlooked ideas in this disease space,” Ms. Williams said. “1. Diagnosis: The norm seems to be misdiagnosis, switched diagnosis, or no diagnosis at all. 2. Symptoms: They are being treated independently, apart from the neurological disorder. 3. Suicides: If more autopsies were done, more suicides would be attributed to this disease.”
She concluded her address by reflecting on the impact of her husband’s death has had in bringing an international spotlight to LBD. “When I meet individuals who have lost someone they loved to LBD, I see the pain in their eyes, but I hear the determination in their voice as they chart their own course toward making a difference,” Ms. Williams said. “I have been blessed to learn over and over again that I am not alone. I believe that Robin’s death in this battle against these diseases holds a profound purpose. There was tremendous power in what he suffered, and I saw that power up close. I’m here doing all that I can to see that power transformed into something good.”
REPORTING FROM ANA 2018
Propolis plus L-dopa buffers PD symptoms in rat model
NEW YORK – A bee-derived supplement known to have anti-inflammatory properties showed promise in potentiating the effect of levodopa in a rodent model of Parkinson’s disease, with biochemical and behavioral improvements seen that exceeded high-dose levodopa alone.
Levodopa (L-dopa) has long been a keystone in treatment for individuals with Parkinson’s disease (PD). However, its effectiveness wanes with time and long-term use is associated with significant undesirable side effects, including dyskinesias.
A dopamine precursor, L-dopa, once converted to dopamine, replaces the dopamine no longer made by the substantia nigra. Increasingly, neuroinflammation and oxidative stress are felt to play a role in the natural history of PD, said Azza Ali, PhD, who presented the findings at a poster session of the International Conference on Parkinson’s Disease and Movement Disorders. Whether mitigating these effects can alter the disease course for individuals with PD has not been well investigated, she added.
Dr. Ali and her research group at Al-Azhar University, Cairo, Egypt, where she heads the department of pharmacology and toxicology, are investigating several nutritional and supplement strategies to dampen inflammation. Among the substances she and her colleagues are investigating is propolis, a plant-derived product produced by bees and distinct from honey or beeswax.
Propolis has been shown to have antioxidant properties in addition to other reported health benefits.
Dr. Ali and her colleagues used a rodent model for PD: Rats were dosed with rotenone, which is known to induce a histologically verifiable parkinsonian syndrome. The investigators used six groups of rats; one group was the normal control, while the others all had rotenone-induced parkinsonism.
Of the remaining groups, one rotenone-dosed group was given neither L-dopa nor propolis. Two groups were treated with oral L-dopa alone, at 10 or 25 mg/kg per day. Another group was given an oral dose of 300 mg/kg per day of propolis, and the last group received propolis at that dose while also receiving the lower dose of L-dopa.
All groups were subject to behavioral assessments including a swim test, an open field test, a maze test, and other tasks designed to assess motor function and other aspects of behavior. Additionally, Dr. Ali and her collaborators assessed a variety of biochemical parameters that looked at oxidative stress and neuroinflammation in all rodent groups.
For most parameters, the rotenone-dosed rats that showed results most like normal controls were those who received both low-dose L-dopa and propolis. In particular, the rats receiving both L-dopa and propolis outperformed the other rotenone groups in the behavioral open field test and a grid test, where their performance neared the control group.
Levels of interleukin-1 beta fell for all treated rodents, but fell the most for those treated with the combination. Tissue dopamine levels were also lower for the rats who received combination treatment, and acetylcholinesterase and malondialdehyde levels also fell to near normal for rats receiving the combination treatment, but not for the other groups.
“Propolis is efficient in protection from PD development and represents a suitable adjuvant therapy, which can be translated to serious reduction of the long-term therapy side effects of the mainstay drug L-dopa,” wrote Dr. Ali and her coauthors. “Consequently, propolis could be recommended as a disease-modifying therapy of PD as well as a promising adjunct therapy with L-dopa especially when given early in the treatment course.”
NEW YORK – A bee-derived supplement known to have anti-inflammatory properties showed promise in potentiating the effect of levodopa in a rodent model of Parkinson’s disease, with biochemical and behavioral improvements seen that exceeded high-dose levodopa alone.
Levodopa (L-dopa) has long been a keystone in treatment for individuals with Parkinson’s disease (PD). However, its effectiveness wanes with time and long-term use is associated with significant undesirable side effects, including dyskinesias.
A dopamine precursor, L-dopa, once converted to dopamine, replaces the dopamine no longer made by the substantia nigra. Increasingly, neuroinflammation and oxidative stress are felt to play a role in the natural history of PD, said Azza Ali, PhD, who presented the findings at a poster session of the International Conference on Parkinson’s Disease and Movement Disorders. Whether mitigating these effects can alter the disease course for individuals with PD has not been well investigated, she added.
Dr. Ali and her research group at Al-Azhar University, Cairo, Egypt, where she heads the department of pharmacology and toxicology, are investigating several nutritional and supplement strategies to dampen inflammation. Among the substances she and her colleagues are investigating is propolis, a plant-derived product produced by bees and distinct from honey or beeswax.
Propolis has been shown to have antioxidant properties in addition to other reported health benefits.
Dr. Ali and her colleagues used a rodent model for PD: Rats were dosed with rotenone, which is known to induce a histologically verifiable parkinsonian syndrome. The investigators used six groups of rats; one group was the normal control, while the others all had rotenone-induced parkinsonism.
Of the remaining groups, one rotenone-dosed group was given neither L-dopa nor propolis. Two groups were treated with oral L-dopa alone, at 10 or 25 mg/kg per day. Another group was given an oral dose of 300 mg/kg per day of propolis, and the last group received propolis at that dose while also receiving the lower dose of L-dopa.
All groups were subject to behavioral assessments including a swim test, an open field test, a maze test, and other tasks designed to assess motor function and other aspects of behavior. Additionally, Dr. Ali and her collaborators assessed a variety of biochemical parameters that looked at oxidative stress and neuroinflammation in all rodent groups.
For most parameters, the rotenone-dosed rats that showed results most like normal controls were those who received both low-dose L-dopa and propolis. In particular, the rats receiving both L-dopa and propolis outperformed the other rotenone groups in the behavioral open field test and a grid test, where their performance neared the control group.
Levels of interleukin-1 beta fell for all treated rodents, but fell the most for those treated with the combination. Tissue dopamine levels were also lower for the rats who received combination treatment, and acetylcholinesterase and malondialdehyde levels also fell to near normal for rats receiving the combination treatment, but not for the other groups.
“Propolis is efficient in protection from PD development and represents a suitable adjuvant therapy, which can be translated to serious reduction of the long-term therapy side effects of the mainstay drug L-dopa,” wrote Dr. Ali and her coauthors. “Consequently, propolis could be recommended as a disease-modifying therapy of PD as well as a promising adjunct therapy with L-dopa especially when given early in the treatment course.”
NEW YORK – A bee-derived supplement known to have anti-inflammatory properties showed promise in potentiating the effect of levodopa in a rodent model of Parkinson’s disease, with biochemical and behavioral improvements seen that exceeded high-dose levodopa alone.
Levodopa (L-dopa) has long been a keystone in treatment for individuals with Parkinson’s disease (PD). However, its effectiveness wanes with time and long-term use is associated with significant undesirable side effects, including dyskinesias.
A dopamine precursor, L-dopa, once converted to dopamine, replaces the dopamine no longer made by the substantia nigra. Increasingly, neuroinflammation and oxidative stress are felt to play a role in the natural history of PD, said Azza Ali, PhD, who presented the findings at a poster session of the International Conference on Parkinson’s Disease and Movement Disorders. Whether mitigating these effects can alter the disease course for individuals with PD has not been well investigated, she added.
Dr. Ali and her research group at Al-Azhar University, Cairo, Egypt, where she heads the department of pharmacology and toxicology, are investigating several nutritional and supplement strategies to dampen inflammation. Among the substances she and her colleagues are investigating is propolis, a plant-derived product produced by bees and distinct from honey or beeswax.
Propolis has been shown to have antioxidant properties in addition to other reported health benefits.
Dr. Ali and her colleagues used a rodent model for PD: Rats were dosed with rotenone, which is known to induce a histologically verifiable parkinsonian syndrome. The investigators used six groups of rats; one group was the normal control, while the others all had rotenone-induced parkinsonism.
Of the remaining groups, one rotenone-dosed group was given neither L-dopa nor propolis. Two groups were treated with oral L-dopa alone, at 10 or 25 mg/kg per day. Another group was given an oral dose of 300 mg/kg per day of propolis, and the last group received propolis at that dose while also receiving the lower dose of L-dopa.
All groups were subject to behavioral assessments including a swim test, an open field test, a maze test, and other tasks designed to assess motor function and other aspects of behavior. Additionally, Dr. Ali and her collaborators assessed a variety of biochemical parameters that looked at oxidative stress and neuroinflammation in all rodent groups.
For most parameters, the rotenone-dosed rats that showed results most like normal controls were those who received both low-dose L-dopa and propolis. In particular, the rats receiving both L-dopa and propolis outperformed the other rotenone groups in the behavioral open field test and a grid test, where their performance neared the control group.
Levels of interleukin-1 beta fell for all treated rodents, but fell the most for those treated with the combination. Tissue dopamine levels were also lower for the rats who received combination treatment, and acetylcholinesterase and malondialdehyde levels also fell to near normal for rats receiving the combination treatment, but not for the other groups.
“Propolis is efficient in protection from PD development and represents a suitable adjuvant therapy, which can be translated to serious reduction of the long-term therapy side effects of the mainstay drug L-dopa,” wrote Dr. Ali and her coauthors. “Consequently, propolis could be recommended as a disease-modifying therapy of PD as well as a promising adjunct therapy with L-dopa especially when given early in the treatment course.”
REPORTING FROM ICPDMD 2018
Combination supplements show neuroprotective potential in Parkinson’s
NEW YORK – Preclinical studies have shown the natural supplements vinpocetine and pomegranate, along with vitamin B complex and vitamin E, may have some effect individually in providing neuroprotection in Parkinson’s disease, but may have a more profound effect when used in combination, a researcher from Egypt reported at the International Conference on Parkinson’s Disease and Movement Disorders.

“We need to carry out a clinical trial to ensure that this multiple direct strategy can provide protection in different stages of neurodegenerative disease – during induction and even during the progression of the disease,” said Azza Ali, PhD, of Al-Azhar University, Cairo. She presented a poster of her research in an unspecified number of rats with manganese-induced Parkinsonian symptoms. The goal of the study was to compare the oral supplements to each other and to evaluate their impact in combinations. Excessive levels of manganese have been associated with movement disorders similar to Parkinson’s disease.
The research involved histologic studies to evaluate the impact of the supplements in the brains, and evaluated biochemical, neuroinflammatory, apoptotic, and oxidative markers. Behavioral tests evaluated cognition, memory, and motor skills.
Histological studies of manganese-induced brains exhibited nuclear pyknosis – clumping of chromosomes, excessive chromatic aberrations, and shrinkage of the nucleus – in the neurons of the cerebral cortex as well as in some areas of the hippocampus, although no alteration was seen in the subiculum, Dr. Ali reported. The stria showed multiple plaque formations with nuclear pyknosis and degeneration in some neurons.
All the studied treatments improved motor, memory, and cognitive decline induced by manganese, with pomegranate and vinpocetine yielding the best results, Dr. Ali said. However, a combination of treatments showed more pronounced improvements in some biochemical markers, as well as the neuroinflammatory, apoptotic, and oxidative markers. “They have a high antioxidant and antiapoptotic effect,” Dr. Ali said. Histopathologic studies confirmed those results, she noted.
Pomegranate (150 mg/kg) had a somewhat positive effect in the subiculum and fascia dentate areas of the hippocampus, although the stria appeared similar to manganese-induced brains. With vinpocetine (20 mg/kg), neurons in the cerebral cortex showed intact histological structure with some degeneration and nuclear pyknosis in the subiculum. There was no alteration in the neurons of the fascia dentate, hilus, and stria of the hippocampus.
Histologic studies of induced brains after treatment with vitamin-B complex (8.5 mg/kg) showed nuclear pyknosis and degeneration in the neurons of the cerebral cortex and hippocampus, including the subiculum. The stria also showed multiple focal eosinophilic plaques with nuclear pyknosis and degeneration in some neurons. Vitamin E (100 mg/kg) resulted in intact neurons in the cerebral cortex but not in the hippocampus.
Histopathologic studies of brains that received combination treatment showed no alteration in the neurons of the cerebral cortex or in the subiculum and fascia dentate of the hippocampus, although a few neurons in the stria showed nuclear pyknosis and degeneration, Dr. Ali said.
She had no financial relationships to disclose.
NEW YORK – Preclinical studies have shown the natural supplements vinpocetine and pomegranate, along with vitamin B complex and vitamin E, may have some effect individually in providing neuroprotection in Parkinson’s disease, but may have a more profound effect when used in combination, a researcher from Egypt reported at the International Conference on Parkinson’s Disease and Movement Disorders.
“We need to carry out a clinical trial to ensure that this multiple direct strategy can provide protection in different stages of neurodegenerative disease – during induction and even during the progression of the disease,” said Azza Ali, PhD, of Al-Azhar University, Cairo. She presented a poster of her research in an unspecified number of rats with manganese-induced Parkinsonian symptoms. The goal of the study was to compare the oral supplements to each other and to evaluate their impact in combinations. Excessive levels of manganese have been associated with movement disorders similar to Parkinson’s disease.
The research involved histologic studies to evaluate the impact of the supplements in the brains, and evaluated biochemical, neuroinflammatory, apoptotic, and oxidative markers. Behavioral tests evaluated cognition, memory, and motor skills.
Histological studies of manganese-induced brains exhibited nuclear pyknosis – clumping of chromosomes, excessive chromatic aberrations, and shrinkage of the nucleus – in the neurons of the cerebral cortex as well as in some areas of the hippocampus, although no alteration was seen in the subiculum, Dr. Ali reported. The stria showed multiple plaque formations with nuclear pyknosis and degeneration in some neurons.
All the studied treatments improved motor, memory, and cognitive decline induced by manganese, with pomegranate and vinpocetine yielding the best results, Dr. Ali said. However, a combination of treatments showed more pronounced improvements in some biochemical markers, as well as the neuroinflammatory, apoptotic, and oxidative markers. “They have a high antioxidant and antiapoptotic effect,” Dr. Ali said. Histopathologic studies confirmed those results, she noted.
Pomegranate (150 mg/kg) had a somewhat positive effect in the subiculum and fascia dentate areas of the hippocampus, although the stria appeared similar to manganese-induced brains. With vinpocetine (20 mg/kg), neurons in the cerebral cortex showed intact histological structure with some degeneration and nuclear pyknosis in the subiculum. There was no alteration in the neurons of the fascia dentate, hilus, and stria of the hippocampus.
Histologic studies of induced brains after treatment with vitamin-B complex (8.5 mg/kg) showed nuclear pyknosis and degeneration in the neurons of the cerebral cortex and hippocampus, including the subiculum. The stria also showed multiple focal eosinophilic plaques with nuclear pyknosis and degeneration in some neurons. Vitamin E (100 mg/kg) resulted in intact neurons in the cerebral cortex but not in the hippocampus.
Histopathologic studies of brains that received combination treatment showed no alteration in the neurons of the cerebral cortex or in the subiculum and fascia dentate of the hippocampus, although a few neurons in the stria showed nuclear pyknosis and degeneration, Dr. Ali said.
She had no financial relationships to disclose.
NEW YORK – Preclinical studies have shown the natural supplements vinpocetine and pomegranate, along with vitamin B complex and vitamin E, may have some effect individually in providing neuroprotection in Parkinson’s disease, but may have a more profound effect when used in combination, a researcher from Egypt reported at the International Conference on Parkinson’s Disease and Movement Disorders.
“We need to carry out a clinical trial to ensure that this multiple direct strategy can provide protection in different stages of neurodegenerative disease – during induction and even during the progression of the disease,” said Azza Ali, PhD, of Al-Azhar University, Cairo. She presented a poster of her research in an unspecified number of rats with manganese-induced Parkinsonian symptoms. The goal of the study was to compare the oral supplements to each other and to evaluate their impact in combinations. Excessive levels of manganese have been associated with movement disorders similar to Parkinson’s disease.
The research involved histologic studies to evaluate the impact of the supplements in the brains, and evaluated biochemical, neuroinflammatory, apoptotic, and oxidative markers. Behavioral tests evaluated cognition, memory, and motor skills.
Histological studies of manganese-induced brains exhibited nuclear pyknosis – clumping of chromosomes, excessive chromatic aberrations, and shrinkage of the nucleus – in the neurons of the cerebral cortex as well as in some areas of the hippocampus, although no alteration was seen in the subiculum, Dr. Ali reported. The stria showed multiple plaque formations with nuclear pyknosis and degeneration in some neurons.
All the studied treatments improved motor, memory, and cognitive decline induced by manganese, with pomegranate and vinpocetine yielding the best results, Dr. Ali said. However, a combination of treatments showed more pronounced improvements in some biochemical markers, as well as the neuroinflammatory, apoptotic, and oxidative markers. “They have a high antioxidant and antiapoptotic effect,” Dr. Ali said. Histopathologic studies confirmed those results, she noted.
Pomegranate (150 mg/kg) had a somewhat positive effect in the subiculum and fascia dentate areas of the hippocampus, although the stria appeared similar to manganese-induced brains. With vinpocetine (20 mg/kg), neurons in the cerebral cortex showed intact histological structure with some degeneration and nuclear pyknosis in the subiculum. There was no alteration in the neurons of the fascia dentate, hilus, and stria of the hippocampus.
Histologic studies of induced brains after treatment with vitamin-B complex (8.5 mg/kg) showed nuclear pyknosis and degeneration in the neurons of the cerebral cortex and hippocampus, including the subiculum. The stria also showed multiple focal eosinophilic plaques with nuclear pyknosis and degeneration in some neurons. Vitamin E (100 mg/kg) resulted in intact neurons in the cerebral cortex but not in the hippocampus.
Histopathologic studies of brains that received combination treatment showed no alteration in the neurons of the cerebral cortex or in the subiculum and fascia dentate of the hippocampus, although a few neurons in the stria showed nuclear pyknosis and degeneration, Dr. Ali said.
She had no financial relationships to disclose.
REPORTING FROM ICPDMD 2018
Appendix linked to Parkinson’s disease in series of unexpected findings
Appendectomy has been associated with a reduced risk of Parkinson’s disease (PD), which supports the potential for a reservoir of aggregated alpha-synuclein in the appendix to affect risk of the condition, according to new epidemiologic and translational evidence from two data sets that promotes a new and emerging theory for PD etiology.
When placed into the context of other recent studies, these epidemiologic data “point to the appendix as a site of origin for Parkinson’s and provide a path forward for devising new treatment strategies,” reported senior author Viviane Labrie, PhD, of the Van Andel Research Institute (VARI) in Grand Rapids, Mich.
The epidemiologic data was the most recent step in a series of findings summarized in a newly published paper in Science Translational Medicine. As the researchers explained, it is relevant to a separate body of evidence that alpha-synuclein, a protein that serves as the hallmark of PD when it appears in Lewy bodies, can be isolated in the nerve fibers and nerve cells of the appendix.
“We have shown that alpha-synuclein proteins, including the truncated forms observed in Lewy bodies, are abundant in the appendix,” reported first author Bryan A. Killinger, PhD, also at VARI, in a press teleconference. He said this finding is likely to explain the reduced risk of PD from appendectomy.
In the largest of the epidemiologic studies, the effect of appendectomy on subsequent risk of PD was evaluated through the health records from more than 1.6 million individuals in Sweden. The incidence of PD was found to be 19.3% lower among 551,647 patients who had an appendectomy, compared with controls.
In addition, the data showed that when PD did occur after appendectomy, it was delayed on average by 3.6 years. It is notable that appendectomy was not associated with protection from PD in patients with a familial link to PD, a group they said comprises less than 10% of cases.
In patients with PD, nonmotor symptoms often include GI tract dysfunction, which can, in some cases, be part of a prodromal presentation that precedes the onset of classical PD symptoms by several years, the authors reported. However, the new research upends previous conceptions of disease. The demonstration of abundant alpha-synuclein in the appendix coupled with the protective effect of appendectomy, suggests that PD may originate in the GI tract and then spread to the central nervous system (CNS) rather than the other way around.
“The vermiform appendix was once considered to be an unnecessary organ. Although there is now good evidence that the appendix plays a major role in the regulation of the immune system, including the regulation of gut bacteria, our work suggests it is also mediates risk of Parkinson’s,” Dr. Labrie said in the teleconference.
In the paper, numerous pieces of the puzzle are brought together to suggest that alpha-synuclein in the appendix is linked to alpha-synuclein in the CNS. Many of the findings along this investigative pathway were described as surprising. For example, immunohistochemistry studies revealed high amounts of alpha-synuclein in nearly every sample of appendiceal tissue examined, including normal and inflamed tissue, tissue from individuals with PD and those without, and tissues from young and old individuals.
“The normal tissue, as well as appendiceal tissue from PD patients, contained high levels of alpha-synuclein in the truncated forms analogous to those seen in Lewy body pathology,” Dr. Killinger said. Based on these and other findings, he believes that alpha-synuclein in the appendix forms a reservoir for seeding the aggregates involved in the pathology of PD, although he acknowledged that it is not yet clear how the proteins in the appendix find their way to the brain.
From these data, it appears that most individuals with an intact appendix have alpha-synuclein in the nerve fibers, but Dr. Labrie pointed out that the only about 1% of the population develops PD. She speculated that there is “some confluence of events,” such as an environmental trigger altering the GI microbiome, that mediates ultimate risk of PD, but she noted that these events may take place decades before signs and symptoms of PD develop. The data appear to be a substantial reorientation in understanding PD.
“We have shown that the appendix is a hub for the accumulation of clumped forms of alpha-synuclein proteins, which are implicated in Parkinson’s,” Dr. Killinger said. “This knowledge will be invaluable as we explore new prevention and treatment strategies.”
The research was funded by a variety of governmental and private grants to individual authors. Dr. Killinger and Dr. Labrie report no financial relationships relevant to this study.
SOURCE: Killinger BA et al. Sci Transl Med. 2018;10:eaar5380.
Appendectomy has been associated with a reduced risk of Parkinson’s disease (PD), which supports the potential for a reservoir of aggregated alpha-synuclein in the appendix to affect risk of the condition, according to new epidemiologic and translational evidence from two data sets that promotes a new and emerging theory for PD etiology.
When placed into the context of other recent studies, these epidemiologic data “point to the appendix as a site of origin for Parkinson’s and provide a path forward for devising new treatment strategies,” reported senior author Viviane Labrie, PhD, of the Van Andel Research Institute (VARI) in Grand Rapids, Mich.
The epidemiologic data was the most recent step in a series of findings summarized in a newly published paper in Science Translational Medicine. As the researchers explained, it is relevant to a separate body of evidence that alpha-synuclein, a protein that serves as the hallmark of PD when it appears in Lewy bodies, can be isolated in the nerve fibers and nerve cells of the appendix.
“We have shown that alpha-synuclein proteins, including the truncated forms observed in Lewy bodies, are abundant in the appendix,” reported first author Bryan A. Killinger, PhD, also at VARI, in a press teleconference. He said this finding is likely to explain the reduced risk of PD from appendectomy.
In the largest of the epidemiologic studies, the effect of appendectomy on subsequent risk of PD was evaluated through the health records from more than 1.6 million individuals in Sweden. The incidence of PD was found to be 19.3% lower among 551,647 patients who had an appendectomy, compared with controls.
In addition, the data showed that when PD did occur after appendectomy, it was delayed on average by 3.6 years. It is notable that appendectomy was not associated with protection from PD in patients with a familial link to PD, a group they said comprises less than 10% of cases.
In patients with PD, nonmotor symptoms often include GI tract dysfunction, which can, in some cases, be part of a prodromal presentation that precedes the onset of classical PD symptoms by several years, the authors reported. However, the new research upends previous conceptions of disease. The demonstration of abundant alpha-synuclein in the appendix coupled with the protective effect of appendectomy, suggests that PD may originate in the GI tract and then spread to the central nervous system (CNS) rather than the other way around.
“The vermiform appendix was once considered to be an unnecessary organ. Although there is now good evidence that the appendix plays a major role in the regulation of the immune system, including the regulation of gut bacteria, our work suggests it is also mediates risk of Parkinson’s,” Dr. Labrie said in the teleconference.
In the paper, numerous pieces of the puzzle are brought together to suggest that alpha-synuclein in the appendix is linked to alpha-synuclein in the CNS. Many of the findings along this investigative pathway were described as surprising. For example, immunohistochemistry studies revealed high amounts of alpha-synuclein in nearly every sample of appendiceal tissue examined, including normal and inflamed tissue, tissue from individuals with PD and those without, and tissues from young and old individuals.
“The normal tissue, as well as appendiceal tissue from PD patients, contained high levels of alpha-synuclein in the truncated forms analogous to those seen in Lewy body pathology,” Dr. Killinger said. Based on these and other findings, he believes that alpha-synuclein in the appendix forms a reservoir for seeding the aggregates involved in the pathology of PD, although he acknowledged that it is not yet clear how the proteins in the appendix find their way to the brain.
From these data, it appears that most individuals with an intact appendix have alpha-synuclein in the nerve fibers, but Dr. Labrie pointed out that the only about 1% of the population develops PD. She speculated that there is “some confluence of events,” such as an environmental trigger altering the GI microbiome, that mediates ultimate risk of PD, but she noted that these events may take place decades before signs and symptoms of PD develop. The data appear to be a substantial reorientation in understanding PD.
“We have shown that the appendix is a hub for the accumulation of clumped forms of alpha-synuclein proteins, which are implicated in Parkinson’s,” Dr. Killinger said. “This knowledge will be invaluable as we explore new prevention and treatment strategies.”
The research was funded by a variety of governmental and private grants to individual authors. Dr. Killinger and Dr. Labrie report no financial relationships relevant to this study.
SOURCE: Killinger BA et al. Sci Transl Med. 2018;10:eaar5380.
Appendectomy has been associated with a reduced risk of Parkinson’s disease (PD), which supports the potential for a reservoir of aggregated alpha-synuclein in the appendix to affect risk of the condition, according to new epidemiologic and translational evidence from two data sets that promotes a new and emerging theory for PD etiology.
When placed into the context of other recent studies, these epidemiologic data “point to the appendix as a site of origin for Parkinson’s and provide a path forward for devising new treatment strategies,” reported senior author Viviane Labrie, PhD, of the Van Andel Research Institute (VARI) in Grand Rapids, Mich.
The epidemiologic data was the most recent step in a series of findings summarized in a newly published paper in Science Translational Medicine. As the researchers explained, it is relevant to a separate body of evidence that alpha-synuclein, a protein that serves as the hallmark of PD when it appears in Lewy bodies, can be isolated in the nerve fibers and nerve cells of the appendix.
“We have shown that alpha-synuclein proteins, including the truncated forms observed in Lewy bodies, are abundant in the appendix,” reported first author Bryan A. Killinger, PhD, also at VARI, in a press teleconference. He said this finding is likely to explain the reduced risk of PD from appendectomy.
In the largest of the epidemiologic studies, the effect of appendectomy on subsequent risk of PD was evaluated through the health records from more than 1.6 million individuals in Sweden. The incidence of PD was found to be 19.3% lower among 551,647 patients who had an appendectomy, compared with controls.
In addition, the data showed that when PD did occur after appendectomy, it was delayed on average by 3.6 years. It is notable that appendectomy was not associated with protection from PD in patients with a familial link to PD, a group they said comprises less than 10% of cases.
In patients with PD, nonmotor symptoms often include GI tract dysfunction, which can, in some cases, be part of a prodromal presentation that precedes the onset of classical PD symptoms by several years, the authors reported. However, the new research upends previous conceptions of disease. The demonstration of abundant alpha-synuclein in the appendix coupled with the protective effect of appendectomy, suggests that PD may originate in the GI tract and then spread to the central nervous system (CNS) rather than the other way around.
“The vermiform appendix was once considered to be an unnecessary organ. Although there is now good evidence that the appendix plays a major role in the regulation of the immune system, including the regulation of gut bacteria, our work suggests it is also mediates risk of Parkinson’s,” Dr. Labrie said in the teleconference.
In the paper, numerous pieces of the puzzle are brought together to suggest that alpha-synuclein in the appendix is linked to alpha-synuclein in the CNS. Many of the findings along this investigative pathway were described as surprising. For example, immunohistochemistry studies revealed high amounts of alpha-synuclein in nearly every sample of appendiceal tissue examined, including normal and inflamed tissue, tissue from individuals with PD and those without, and tissues from young and old individuals.
“The normal tissue, as well as appendiceal tissue from PD patients, contained high levels of alpha-synuclein in the truncated forms analogous to those seen in Lewy body pathology,” Dr. Killinger said. Based on these and other findings, he believes that alpha-synuclein in the appendix forms a reservoir for seeding the aggregates involved in the pathology of PD, although he acknowledged that it is not yet clear how the proteins in the appendix find their way to the brain.
From these data, it appears that most individuals with an intact appendix have alpha-synuclein in the nerve fibers, but Dr. Labrie pointed out that the only about 1% of the population develops PD. She speculated that there is “some confluence of events,” such as an environmental trigger altering the GI microbiome, that mediates ultimate risk of PD, but she noted that these events may take place decades before signs and symptoms of PD develop. The data appear to be a substantial reorientation in understanding PD.
“We have shown that the appendix is a hub for the accumulation of clumped forms of alpha-synuclein proteins, which are implicated in Parkinson’s,” Dr. Killinger said. “This knowledge will be invaluable as we explore new prevention and treatment strategies.”
The research was funded by a variety of governmental and private grants to individual authors. Dr. Killinger and Dr. Labrie report no financial relationships relevant to this study.
SOURCE: Killinger BA et al. Sci Transl Med. 2018;10:eaar5380.
FROM SCIENCE TRANSLATIONAL MEDICINE
Key clinical point:
Major finding: A 19.3% reduction in risk of PD from appendectomy may relate to alpha-synuclein in the appendix.
Study details: Series of related epidemiologic and translational studies.
Disclosures: The research was funded by a variety of governmental and private grants to individual authors. Dr. Killinger and Dr. Labrie report no financial relationships relevant to this study.
Source: Killinger BA et al. Sci Transl Med. 2018;10:eaar5380.
Management of Lewy body dementia remains complex
ATLANTA – In the not-so-distant past, neurologists viewed dementia with Lewy bodies as a disorder primarily of the brain, but it turned out to be far more complex than that.

At the annual meeting of the American Neurological Association, Bradley F. Boeve, MD, described dementia with Lewy bodies (DLB) as a systemic neurologic disorder affecting the brain, including brain stem, spinal cord, and peripheral nervous system, especially the autonomic nervous system. “This leads to the complex array of clinical manifestations, which are quite different from patient to patient cross-sectionally and longitudinally,” said Dr. Boeve, the Little Family Foundation Professor of Lewy Body Dementia in the department of neurology at the Mayo Clinic, Rochester, Minn.
, he said. The four core clinical features are Parkinsonism unrelated to medications; recurrent, fully-formed visual hallucinations; fluctuations in cognition and/or arousal; and rapid eye movement (REM) sleep behavior disorder. “This is the most predictive of all four features,” Dr. Boeve said. He described REM sleep behavior disorder as a parasomnia manifested by the tendency to repeatedly “act out one’s dreams.” The dreams tend to contain a chasing/attacking theme, and behaviors mirror dream content. Injuries to the patient and bed partner can occur.
Typically, patients will present with REM sleep behavior disorder in their 50s and 60s, and sometimes in their 30s and 40s, “decades before cognitive changes begin,” he said. “This is usually followed by Parkinsonism and visual hallucinations. That’s the prototypical DLB [case], but there are many examples where this is not followed. Prominent neuropsychiatric features can also begin before any cognitive changes.”
Neuropsychological features of DLB often include impairment of executive functions and visuospatial functions. “Early in the course of Alzheimer’s disease, typically performance on memory measures – especially delayed recall – are down and the other measures are borderline or mildly impaired,” Dr. Boeve noted. “By contrast, in DLB, attention, executive function, and visuospatial measures are down, but memory is often pretty good. What’s remarkable is that in the office setting, when you take a history the person often says, ‘I’m very forgetful,’ yet in the testing environment people tend to rise to the occasion pretty well.”
Imaging isn’t always helpful in establishing a diagnosis of DLB. MRI scans, for example, “can look pretty normal, including the hippocampi,” he said. “This is really the norm in DLB and it seems to be a disconnect. The person can have significant symptoms yet their MRI scan can be pretty normal.”
In Alzheimer’s disease, 18F-fluorodeoxyglucose-PET (FDG-PET) shows temporal, parietal, and frontal hypometabolism, sparing of the sensory-motor strip and sparing of the primary occipital cortex, while in DLB, FDG-PET shows marked deficits in the occipital regions with relative sparing of the frontal and temporal lobes. Another key neuroimaging sign of DLB is the posterior cingulate island sign, which is characterized by sparing of the posterior cingulate cortex relative to the precuneus plus cuneus on FDG-PET.
In 2017, the Dementia with Lewy Bodies Consortium published updated recommendations on the diagnosis and management of the disease (Neurology. 2017;89[1]:88-100). In its consensus report, the consortium defines probable DLB as dementia plus two or more clinical features or one core clinical feature plus one or more indicative biomarker. These biomarkers include reduced dopamine transport uptake in basal ganglia by SPECT or PET; abnormal (low uptake) meta-iodobenzylguanidine (MIBG) myocardial scintigraphy, and/or polysomnographic confirmation of REM sleep without atonia.
“Neuropathologically, limbic with or without neocortical Lewy bodies and Lewy neurites are the defining characteristics of pathologically-proven DLB,” added Dr. Boeve, a member of the DLB consortium. “The classic DLB phenotype can occur in limbic-predominant DLB. Lewy bodies in the neocortex are not necessary to cause a dementia syndrome.”
He characterized management of DLB as “very complicated. Consider symptoms as they relate to cognitive impairment, neuropsychiatric features, motor features, sleep disorders, and autonomic dysfunction.” He often asks the patient/family to prioritize the three most troublesome issues they seek to change, and develops a plan based on their input.
There is no Food and Drug Administration–approved medication for DLB, but the standard of care is an acetylcholinesterase inhibitor such as donepezil. “There is evidence that memantine can provide a modest benefit,” Dr. Boeve said. “Hypersomnia is quite prominent in DLB and worthy of assessing and treating.” Clinicians must weigh the pros and cons of pharmacotherapy with each patient. “For example, in the atypical neuroleptic class [of drugs], there may be a benefit to the hallucinations and delusions in DLB but hypersomnia can worsen,” he said. “Selecting agents is challenging but worth the effort.”
Survival is lower and more rapid with DLB, compared with Alzheimer’s. Most people pass away from primary DLB-related features or failure to thrive. The second most common is pneumonia or aspiration. Median survival was 4 years after diagnosis in one study, and end-of life discussions occurred in less than half of all patients. “This is a frustrating reminder that we as clinicians are not very good at discussing important topics such as end-of-life care with patients and their families,” Dr. Boeve said. Resources that he recommends for education and support include the Lewy Body Dementia Association and The Lewy Body Society.
At the 2016 Alzheimer’s Disease-Related Dementias Summit, clinicians formed a list of DLB research priorities (Neurology 2017;89[23]:2381-91). Among them were recommendations to “initiate clinical trials in diverse populations using therapies that address symptoms that have the greatest effect on patient function and caregiver burden” and “identify novel common and rare genetic variants, epigenetic changes, and environmental influences that affect the risk for and clinical features of” the disease.
Meanwhile, several research protocols are under way, including the development of a DLB module by the U.S. Alzheimer’s Research Disease Centers and a number of DLB-focused projects from the National Institute of Neurological Disorders and Stroke (NINDS) Parkinson’s Disease Biomarkers Program. In addition, the Lewy Body Dementia Association Research Centers of Excellence program is focused on optimizing clinical care and setting up the infrastructure for clinical trials, while the North American Prodromal Synucleinopathy Consortium is conducting longitudinal studies in those with REM sleep behavior disorder.
Dr. Boeve disclosed that he has been an investigator for clinical trials sponsored by GE Healthcare, Axovant, and Biogen. He is a member of the scientific advisory board for the Tau Consortium and has received research support from the National Institute on Aging, the NINDS, the Mangurian Foundation, and the Little Family Foundation.
ATLANTA – In the not-so-distant past, neurologists viewed dementia with Lewy bodies as a disorder primarily of the brain, but it turned out to be far more complex than that.
At the annual meeting of the American Neurological Association, Bradley F. Boeve, MD, described dementia with Lewy bodies (DLB) as a systemic neurologic disorder affecting the brain, including brain stem, spinal cord, and peripheral nervous system, especially the autonomic nervous system. “This leads to the complex array of clinical manifestations, which are quite different from patient to patient cross-sectionally and longitudinally,” said Dr. Boeve, the Little Family Foundation Professor of Lewy Body Dementia in the department of neurology at the Mayo Clinic, Rochester, Minn.
, he said. The four core clinical features are Parkinsonism unrelated to medications; recurrent, fully-formed visual hallucinations; fluctuations in cognition and/or arousal; and rapid eye movement (REM) sleep behavior disorder. “This is the most predictive of all four features,” Dr. Boeve said. He described REM sleep behavior disorder as a parasomnia manifested by the tendency to repeatedly “act out one’s dreams.” The dreams tend to contain a chasing/attacking theme, and behaviors mirror dream content. Injuries to the patient and bed partner can occur.
Typically, patients will present with REM sleep behavior disorder in their 50s and 60s, and sometimes in their 30s and 40s, “decades before cognitive changes begin,” he said. “This is usually followed by Parkinsonism and visual hallucinations. That’s the prototypical DLB [case], but there are many examples where this is not followed. Prominent neuropsychiatric features can also begin before any cognitive changes.”
Neuropsychological features of DLB often include impairment of executive functions and visuospatial functions. “Early in the course of Alzheimer’s disease, typically performance on memory measures – especially delayed recall – are down and the other measures are borderline or mildly impaired,” Dr. Boeve noted. “By contrast, in DLB, attention, executive function, and visuospatial measures are down, but memory is often pretty good. What’s remarkable is that in the office setting, when you take a history the person often says, ‘I’m very forgetful,’ yet in the testing environment people tend to rise to the occasion pretty well.”
Imaging isn’t always helpful in establishing a diagnosis of DLB. MRI scans, for example, “can look pretty normal, including the hippocampi,” he said. “This is really the norm in DLB and it seems to be a disconnect. The person can have significant symptoms yet their MRI scan can be pretty normal.”
In Alzheimer’s disease, 18F-fluorodeoxyglucose-PET (FDG-PET) shows temporal, parietal, and frontal hypometabolism, sparing of the sensory-motor strip and sparing of the primary occipital cortex, while in DLB, FDG-PET shows marked deficits in the occipital regions with relative sparing of the frontal and temporal lobes. Another key neuroimaging sign of DLB is the posterior cingulate island sign, which is characterized by sparing of the posterior cingulate cortex relative to the precuneus plus cuneus on FDG-PET.
In 2017, the Dementia with Lewy Bodies Consortium published updated recommendations on the diagnosis and management of the disease (Neurology. 2017;89[1]:88-100). In its consensus report, the consortium defines probable DLB as dementia plus two or more clinical features or one core clinical feature plus one or more indicative biomarker. These biomarkers include reduced dopamine transport uptake in basal ganglia by SPECT or PET; abnormal (low uptake) meta-iodobenzylguanidine (MIBG) myocardial scintigraphy, and/or polysomnographic confirmation of REM sleep without atonia.
“Neuropathologically, limbic with or without neocortical Lewy bodies and Lewy neurites are the defining characteristics of pathologically-proven DLB,” added Dr. Boeve, a member of the DLB consortium. “The classic DLB phenotype can occur in limbic-predominant DLB. Lewy bodies in the neocortex are not necessary to cause a dementia syndrome.”
He characterized management of DLB as “very complicated. Consider symptoms as they relate to cognitive impairment, neuropsychiatric features, motor features, sleep disorders, and autonomic dysfunction.” He often asks the patient/family to prioritize the three most troublesome issues they seek to change, and develops a plan based on their input.
There is no Food and Drug Administration–approved medication for DLB, but the standard of care is an acetylcholinesterase inhibitor such as donepezil. “There is evidence that memantine can provide a modest benefit,” Dr. Boeve said. “Hypersomnia is quite prominent in DLB and worthy of assessing and treating.” Clinicians must weigh the pros and cons of pharmacotherapy with each patient. “For example, in the atypical neuroleptic class [of drugs], there may be a benefit to the hallucinations and delusions in DLB but hypersomnia can worsen,” he said. “Selecting agents is challenging but worth the effort.”
Survival is lower and more rapid with DLB, compared with Alzheimer’s. Most people pass away from primary DLB-related features or failure to thrive. The second most common is pneumonia or aspiration. Median survival was 4 years after diagnosis in one study, and end-of life discussions occurred in less than half of all patients. “This is a frustrating reminder that we as clinicians are not very good at discussing important topics such as end-of-life care with patients and their families,” Dr. Boeve said. Resources that he recommends for education and support include the Lewy Body Dementia Association and The Lewy Body Society.
At the 2016 Alzheimer’s Disease-Related Dementias Summit, clinicians formed a list of DLB research priorities (Neurology 2017;89[23]:2381-91). Among them were recommendations to “initiate clinical trials in diverse populations using therapies that address symptoms that have the greatest effect on patient function and caregiver burden” and “identify novel common and rare genetic variants, epigenetic changes, and environmental influences that affect the risk for and clinical features of” the disease.
Meanwhile, several research protocols are under way, including the development of a DLB module by the U.S. Alzheimer’s Research Disease Centers and a number of DLB-focused projects from the National Institute of Neurological Disorders and Stroke (NINDS) Parkinson’s Disease Biomarkers Program. In addition, the Lewy Body Dementia Association Research Centers of Excellence program is focused on optimizing clinical care and setting up the infrastructure for clinical trials, while the North American Prodromal Synucleinopathy Consortium is conducting longitudinal studies in those with REM sleep behavior disorder.
Dr. Boeve disclosed that he has been an investigator for clinical trials sponsored by GE Healthcare, Axovant, and Biogen. He is a member of the scientific advisory board for the Tau Consortium and has received research support from the National Institute on Aging, the NINDS, the Mangurian Foundation, and the Little Family Foundation.
ATLANTA – In the not-so-distant past, neurologists viewed dementia with Lewy bodies as a disorder primarily of the brain, but it turned out to be far more complex than that.
At the annual meeting of the American Neurological Association, Bradley F. Boeve, MD, described dementia with Lewy bodies (DLB) as a systemic neurologic disorder affecting the brain, including brain stem, spinal cord, and peripheral nervous system, especially the autonomic nervous system. “This leads to the complex array of clinical manifestations, which are quite different from patient to patient cross-sectionally and longitudinally,” said Dr. Boeve, the Little Family Foundation Professor of Lewy Body Dementia in the department of neurology at the Mayo Clinic, Rochester, Minn.
, he said. The four core clinical features are Parkinsonism unrelated to medications; recurrent, fully-formed visual hallucinations; fluctuations in cognition and/or arousal; and rapid eye movement (REM) sleep behavior disorder. “This is the most predictive of all four features,” Dr. Boeve said. He described REM sleep behavior disorder as a parasomnia manifested by the tendency to repeatedly “act out one’s dreams.” The dreams tend to contain a chasing/attacking theme, and behaviors mirror dream content. Injuries to the patient and bed partner can occur.
Typically, patients will present with REM sleep behavior disorder in their 50s and 60s, and sometimes in their 30s and 40s, “decades before cognitive changes begin,” he said. “This is usually followed by Parkinsonism and visual hallucinations. That’s the prototypical DLB [case], but there are many examples where this is not followed. Prominent neuropsychiatric features can also begin before any cognitive changes.”
Neuropsychological features of DLB often include impairment of executive functions and visuospatial functions. “Early in the course of Alzheimer’s disease, typically performance on memory measures – especially delayed recall – are down and the other measures are borderline or mildly impaired,” Dr. Boeve noted. “By contrast, in DLB, attention, executive function, and visuospatial measures are down, but memory is often pretty good. What’s remarkable is that in the office setting, when you take a history the person often says, ‘I’m very forgetful,’ yet in the testing environment people tend to rise to the occasion pretty well.”
Imaging isn’t always helpful in establishing a diagnosis of DLB. MRI scans, for example, “can look pretty normal, including the hippocampi,” he said. “This is really the norm in DLB and it seems to be a disconnect. The person can have significant symptoms yet their MRI scan can be pretty normal.”
In Alzheimer’s disease, 18F-fluorodeoxyglucose-PET (FDG-PET) shows temporal, parietal, and frontal hypometabolism, sparing of the sensory-motor strip and sparing of the primary occipital cortex, while in DLB, FDG-PET shows marked deficits in the occipital regions with relative sparing of the frontal and temporal lobes. Another key neuroimaging sign of DLB is the posterior cingulate island sign, which is characterized by sparing of the posterior cingulate cortex relative to the precuneus plus cuneus on FDG-PET.
In 2017, the Dementia with Lewy Bodies Consortium published updated recommendations on the diagnosis and management of the disease (Neurology. 2017;89[1]:88-100). In its consensus report, the consortium defines probable DLB as dementia plus two or more clinical features or one core clinical feature plus one or more indicative biomarker. These biomarkers include reduced dopamine transport uptake in basal ganglia by SPECT or PET; abnormal (low uptake) meta-iodobenzylguanidine (MIBG) myocardial scintigraphy, and/or polysomnographic confirmation of REM sleep without atonia.
“Neuropathologically, limbic with or without neocortical Lewy bodies and Lewy neurites are the defining characteristics of pathologically-proven DLB,” added Dr. Boeve, a member of the DLB consortium. “The classic DLB phenotype can occur in limbic-predominant DLB. Lewy bodies in the neocortex are not necessary to cause a dementia syndrome.”
He characterized management of DLB as “very complicated. Consider symptoms as they relate to cognitive impairment, neuropsychiatric features, motor features, sleep disorders, and autonomic dysfunction.” He often asks the patient/family to prioritize the three most troublesome issues they seek to change, and develops a plan based on their input.
There is no Food and Drug Administration–approved medication for DLB, but the standard of care is an acetylcholinesterase inhibitor such as donepezil. “There is evidence that memantine can provide a modest benefit,” Dr. Boeve said. “Hypersomnia is quite prominent in DLB and worthy of assessing and treating.” Clinicians must weigh the pros and cons of pharmacotherapy with each patient. “For example, in the atypical neuroleptic class [of drugs], there may be a benefit to the hallucinations and delusions in DLB but hypersomnia can worsen,” he said. “Selecting agents is challenging but worth the effort.”
Survival is lower and more rapid with DLB, compared with Alzheimer’s. Most people pass away from primary DLB-related features or failure to thrive. The second most common is pneumonia or aspiration. Median survival was 4 years after diagnosis in one study, and end-of life discussions occurred in less than half of all patients. “This is a frustrating reminder that we as clinicians are not very good at discussing important topics such as end-of-life care with patients and their families,” Dr. Boeve said. Resources that he recommends for education and support include the Lewy Body Dementia Association and The Lewy Body Society.
At the 2016 Alzheimer’s Disease-Related Dementias Summit, clinicians formed a list of DLB research priorities (Neurology 2017;89[23]:2381-91). Among them were recommendations to “initiate clinical trials in diverse populations using therapies that address symptoms that have the greatest effect on patient function and caregiver burden” and “identify novel common and rare genetic variants, epigenetic changes, and environmental influences that affect the risk for and clinical features of” the disease.
Meanwhile, several research protocols are under way, including the development of a DLB module by the U.S. Alzheimer’s Research Disease Centers and a number of DLB-focused projects from the National Institute of Neurological Disorders and Stroke (NINDS) Parkinson’s Disease Biomarkers Program. In addition, the Lewy Body Dementia Association Research Centers of Excellence program is focused on optimizing clinical care and setting up the infrastructure for clinical trials, while the North American Prodromal Synucleinopathy Consortium is conducting longitudinal studies in those with REM sleep behavior disorder.
Dr. Boeve disclosed that he has been an investigator for clinical trials sponsored by GE Healthcare, Axovant, and Biogen. He is a member of the scientific advisory board for the Tau Consortium and has received research support from the National Institute on Aging, the NINDS, the Mangurian Foundation, and the Little Family Foundation.
EXPERT ANALYSIS FROM ANA 2018
Parkinson’s prevalence varies significantly from state to state
ATLANTA – The prevalence of Parkinson’s disease and associated health care spending on the condition vary significantly from state to state, an analysis of Medicare data showed.

“There is a big variation in not only the prevalence of Parkinson’s disease but also in spending and in health care utilization” among Medicare beneficiaries diagnosed with the condition, lead study author Michelle E. Fullard, MD, said in an interview at the annual meeting of the American Neurological Association. “As neurologists, we should be aware of this. We can use this information to identify and target areas in which Parkinson’s patients may have increased need and require more resources. It can also inform planning at the state and federal levels.”
Dr. Fullard, formerly of the department of neurology at the University of Pennsylvania, Philadelphia, and her colleagues evaluated data from Medicare Beneficiary Summary and Medicare Carrier Files for 27,538,023 individuals aged 65 years and older who were continuously enrolled in Medicare parts A and B during 2014. They calculated state-level differences in Parkinson’s disease prevalence, demographic and eligibility characteristics, costs, and health care use, including number of emergency room visits, number of outpatient clinic visits, and inpatient hospitalizations. The researchers used reimbursement data to calculate the mean out-of-pocket and Medicare cost per individual in each state, and compared direct costs and health service utilization for individuals with and without Parkinson’s disease.
Of all Medicare beneficiaries studied, 392,214 (1.42%) had a diagnosis of Parkinson’s disease. Nearly half (46%) were women and 26% were aged 85 years and older. States with the highest prevalence of Parkinson’s disease included New York (1,720/100,000), Illinois (1,566/100,000), Connecticut (1,560/100,000), Florida (1,551/100,000), Pennsylvania (1,549/100,000), Rhode Island (1,543/100,000), New Jersey (1,541/100,000), Texas (1,522/100,000), California (1,520/100,000) and Louisiana (1,519/100,000). Minnesota had the lowest prevalence (803/100,000).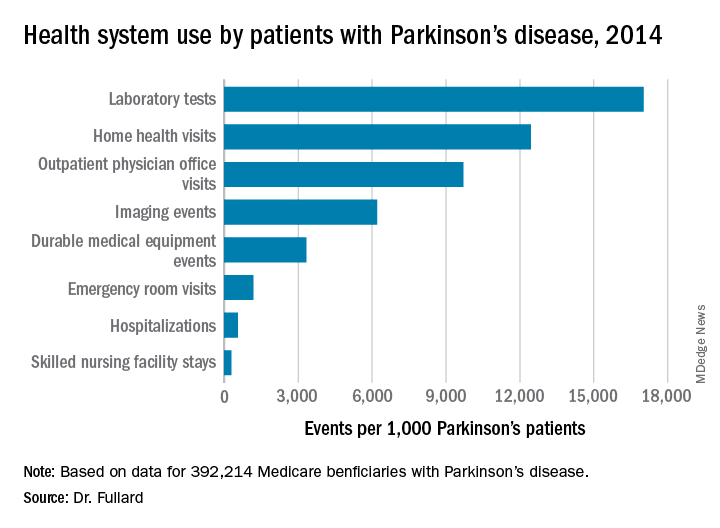
Among the national sample of patients with Parkinson’s disease, there were 219,049 hospitalizations (which represented 558/1,000 Parkinson’s patients), 37,839 readmissions (172/1,000 hospitalizations), 9,740,609 outpatient physician office visits (9,700/1,000 patients), 34,159 hospice stays (87/1,000 patients), 113,027 skilled nursing facility stays (288/1,000 patients), 466,160 emergency room visits (1,188/1,000 patients, 39% of which resulted in hospital admission). In addition, there were 1,308,934 durable medical equipment events (3,337/1,000 patients), 6,676,119 laboratory tests (17,021/1,000 patients), 2,435,654 imaging events (6,210/1,000 patients), and 4,879,538 home health visits (12,441/1,000 patients). The costliest services were inpatient care ($2.1 billion), skilled nursing facility care ($1.4 billion), prescription drugs used by those with prescription coverage ($974.8 million), hospital outpatient care ($881 million), and home health care ($776.5 million).
“States with a higher prevalence of Parkinson’s disease may have a larger proportion of high-risk factor patient groups, a higher concentration of providers who recognize and document Parkinson’s disease, increased public awareness of symptoms, or increased health care–seeking behaviors among people living in the state,” the researchers wrote in their abstract. “Among our top Parkinson’s disease prevalence states, Florida and New York also rank high in terms of absolute number of Medicare beneficiaries and have large supplies of health care providers.”
They also noted that Medicare beneficiaries with Parkinson’s had increased use of health care and spending, compared with their counterparts without the disease. “This was true across all sectors of care (inpatient, outpatient, skilled nursing, and ancillary services) and is in line with data demonstrating that PD, its complications, and the shift away from comorbid disease care and prevention that occurs after a Parkinson’s disease diagnosis drive health care spending and utilization among these individuals,” they wrote.
The study was supported by the Parkinson’s Foundation. Dr. Fullard, who now holds a faculty position at the University of Colorado, Aurora, reported having no financial disclosures.
SOURCE: Ann Neurol. 2018;84[S22]:S89-90, Abstract S215.
ATLANTA – The prevalence of Parkinson’s disease and associated health care spending on the condition vary significantly from state to state, an analysis of Medicare data showed.
“There is a big variation in not only the prevalence of Parkinson’s disease but also in spending and in health care utilization” among Medicare beneficiaries diagnosed with the condition, lead study author Michelle E. Fullard, MD, said in an interview at the annual meeting of the American Neurological Association. “As neurologists, we should be aware of this. We can use this information to identify and target areas in which Parkinson’s patients may have increased need and require more resources. It can also inform planning at the state and federal levels.”
Dr. Fullard, formerly of the department of neurology at the University of Pennsylvania, Philadelphia, and her colleagues evaluated data from Medicare Beneficiary Summary and Medicare Carrier Files for 27,538,023 individuals aged 65 years and older who were continuously enrolled in Medicare parts A and B during 2014. They calculated state-level differences in Parkinson’s disease prevalence, demographic and eligibility characteristics, costs, and health care use, including number of emergency room visits, number of outpatient clinic visits, and inpatient hospitalizations. The researchers used reimbursement data to calculate the mean out-of-pocket and Medicare cost per individual in each state, and compared direct costs and health service utilization for individuals with and without Parkinson’s disease.
Of all Medicare beneficiaries studied, 392,214 (1.42%) had a diagnosis of Parkinson’s disease. Nearly half (46%) were women and 26% were aged 85 years and older. States with the highest prevalence of Parkinson’s disease included New York (1,720/100,000), Illinois (1,566/100,000), Connecticut (1,560/100,000), Florida (1,551/100,000), Pennsylvania (1,549/100,000), Rhode Island (1,543/100,000), New Jersey (1,541/100,000), Texas (1,522/100,000), California (1,520/100,000) and Louisiana (1,519/100,000). Minnesota had the lowest prevalence (803/100,000).
Among the national sample of patients with Parkinson’s disease, there were 219,049 hospitalizations (which represented 558/1,000 Parkinson’s patients), 37,839 readmissions (172/1,000 hospitalizations), 9,740,609 outpatient physician office visits (9,700/1,000 patients), 34,159 hospice stays (87/1,000 patients), 113,027 skilled nursing facility stays (288/1,000 patients), 466,160 emergency room visits (1,188/1,000 patients, 39% of which resulted in hospital admission). In addition, there were 1,308,934 durable medical equipment events (3,337/1,000 patients), 6,676,119 laboratory tests (17,021/1,000 patients), 2,435,654 imaging events (6,210/1,000 patients), and 4,879,538 home health visits (12,441/1,000 patients). The costliest services were inpatient care ($2.1 billion), skilled nursing facility care ($1.4 billion), prescription drugs used by those with prescription coverage ($974.8 million), hospital outpatient care ($881 million), and home health care ($776.5 million).
“States with a higher prevalence of Parkinson’s disease may have a larger proportion of high-risk factor patient groups, a higher concentration of providers who recognize and document Parkinson’s disease, increased public awareness of symptoms, or increased health care–seeking behaviors among people living in the state,” the researchers wrote in their abstract. “Among our top Parkinson’s disease prevalence states, Florida and New York also rank high in terms of absolute number of Medicare beneficiaries and have large supplies of health care providers.”
They also noted that Medicare beneficiaries with Parkinson’s had increased use of health care and spending, compared with their counterparts without the disease. “This was true across all sectors of care (inpatient, outpatient, skilled nursing, and ancillary services) and is in line with data demonstrating that PD, its complications, and the shift away from comorbid disease care and prevention that occurs after a Parkinson’s disease diagnosis drive health care spending and utilization among these individuals,” they wrote.
The study was supported by the Parkinson’s Foundation. Dr. Fullard, who now holds a faculty position at the University of Colorado, Aurora, reported having no financial disclosures.
SOURCE: Ann Neurol. 2018;84[S22]:S89-90, Abstract S215.
ATLANTA – The prevalence of Parkinson’s disease and associated health care spending on the condition vary significantly from state to state, an analysis of Medicare data showed.
“There is a big variation in not only the prevalence of Parkinson’s disease but also in spending and in health care utilization” among Medicare beneficiaries diagnosed with the condition, lead study author Michelle E. Fullard, MD, said in an interview at the annual meeting of the American Neurological Association. “As neurologists, we should be aware of this. We can use this information to identify and target areas in which Parkinson’s patients may have increased need and require more resources. It can also inform planning at the state and federal levels.”
Dr. Fullard, formerly of the department of neurology at the University of Pennsylvania, Philadelphia, and her colleagues evaluated data from Medicare Beneficiary Summary and Medicare Carrier Files for 27,538,023 individuals aged 65 years and older who were continuously enrolled in Medicare parts A and B during 2014. They calculated state-level differences in Parkinson’s disease prevalence, demographic and eligibility characteristics, costs, and health care use, including number of emergency room visits, number of outpatient clinic visits, and inpatient hospitalizations. The researchers used reimbursement data to calculate the mean out-of-pocket and Medicare cost per individual in each state, and compared direct costs and health service utilization for individuals with and without Parkinson’s disease.
Of all Medicare beneficiaries studied, 392,214 (1.42%) had a diagnosis of Parkinson’s disease. Nearly half (46%) were women and 26% were aged 85 years and older. States with the highest prevalence of Parkinson’s disease included New York (1,720/100,000), Illinois (1,566/100,000), Connecticut (1,560/100,000), Florida (1,551/100,000), Pennsylvania (1,549/100,000), Rhode Island (1,543/100,000), New Jersey (1,541/100,000), Texas (1,522/100,000), California (1,520/100,000) and Louisiana (1,519/100,000). Minnesota had the lowest prevalence (803/100,000).
Among the national sample of patients with Parkinson’s disease, there were 219,049 hospitalizations (which represented 558/1,000 Parkinson’s patients), 37,839 readmissions (172/1,000 hospitalizations), 9,740,609 outpatient physician office visits (9,700/1,000 patients), 34,159 hospice stays (87/1,000 patients), 113,027 skilled nursing facility stays (288/1,000 patients), 466,160 emergency room visits (1,188/1,000 patients, 39% of which resulted in hospital admission). In addition, there were 1,308,934 durable medical equipment events (3,337/1,000 patients), 6,676,119 laboratory tests (17,021/1,000 patients), 2,435,654 imaging events (6,210/1,000 patients), and 4,879,538 home health visits (12,441/1,000 patients). The costliest services were inpatient care ($2.1 billion), skilled nursing facility care ($1.4 billion), prescription drugs used by those with prescription coverage ($974.8 million), hospital outpatient care ($881 million), and home health care ($776.5 million).
“States with a higher prevalence of Parkinson’s disease may have a larger proportion of high-risk factor patient groups, a higher concentration of providers who recognize and document Parkinson’s disease, increased public awareness of symptoms, or increased health care–seeking behaviors among people living in the state,” the researchers wrote in their abstract. “Among our top Parkinson’s disease prevalence states, Florida and New York also rank high in terms of absolute number of Medicare beneficiaries and have large supplies of health care providers.”
They also noted that Medicare beneficiaries with Parkinson’s had increased use of health care and spending, compared with their counterparts without the disease. “This was true across all sectors of care (inpatient, outpatient, skilled nursing, and ancillary services) and is in line with data demonstrating that PD, its complications, and the shift away from comorbid disease care and prevention that occurs after a Parkinson’s disease diagnosis drive health care spending and utilization among these individuals,” they wrote.
The study was supported by the Parkinson’s Foundation. Dr. Fullard, who now holds a faculty position at the University of Colorado, Aurora, reported having no financial disclosures.
SOURCE: Ann Neurol. 2018;84[S22]:S89-90, Abstract S215.
REPORTING FROM ANA 2018
Key clinical point:
Major finding: States with the highest prevalence of Parkinson’s disease included New York (1,720/100,000), Illinois (1,566/100,000), and Connecticut (1,560/100,000), while Minnesota had the lowest prevalence (803/100,000).
Study details: An analysis of 392,214 Medicare beneficiaries who carried a diagnosis of Parkinson’s disease.
Disclosures: The study was supported by the Parkinson’s Foundation. Dr. Fullard reported having no financial disclosures.
Source: Ann Neurol. 2018;84[S22]:S89-90. Abstract S215.
Can boxing training improve Parkinson reaction times?
NEW YORK – A small pilot study has shown that patients with Parkinson’s disease who participated in the Rock Steady Boxing non-contact training program may have faster reaction times than PD patients who did not participate in the program, according to a poster presented at the International Conference on Parkinson’s Disease and Movement Disorders.

“The novelty of this is that it shows how Rock Steady Boxing and exercise programs that use sequences and the learning of sequences could possibly help slow the decline, or maintain a level of functioning longer, in Parkinson’s disease,” said Christopher McLeod, a second-year medical student at New York Institute of Technology (NYIT) College of Osteopathic Medicine, Old Westbury, N.Y.
Rock Steady Boxing is a non-contact program tailored to Parkinson’s patients founded in 2006 by Scott Newman, an Indiana lawyer who was diagnosed with early onset Parkinson’s at age 40. The regimen involves intense one-on-one training centered around boxing. Rock Steady Boxing offers classes from coast to coast in the United States and in 13 other countries. Mr. McLeod is a volunteer at the NYIT chapter of Rock Steady Boxing in Old Westbury, N.Y.
Mr. McLeod studied 28 PD patients – 14 who had been taking Rock Steady Boxing classes at NYIT for at least 6 months and 14 controls. The goal of the study was to evaluate if the Rock Steady Boxing participants showed any improvement in procedural motor learning. His coauthor was Adena Leder, DO, a faculty neurologist and movement disorder specialist at NYIT,
“What’s new about this research is the procedural memory component and the Rock Steady Boxing program is just more of the vessel, so to speak,” Mr. McLeod said. “This is a pilot study. We wanted to see if Rock Steady Boxing would show benefits in these patients. There are some trends in my research that [indicate] it would; it did not have statistical significance, but we did see trend lines.”
The researchers used a modified Serial Reaction Time Test (SRTT) composed of seven blocks of 10 stimuli each with 30-second breaks between blocks. Blocks consisted of a random familiarization block, four learning blocks repeating the same sequence of stimuli, a transfer block of random stimuli, and a posttransfer block presenting the same sequence of stimuli from the four learning blocks.
They assessed procedural learning by comparing the reduction in response time over the four identical learning blocks as well as by comparing changes in response time when the subjects were subsequently exposed to the random transfer block.
Experienced boxers demonstrated faster reaction time over the four learning blocks, ranging from 795.32 vs. 906.89 ms in the first learning block to 674.79 vs. 787.32 ms in the fourth learning block (P = .19). In the random sequence transfer block, controls showed a 93.5-ms decrease in median reaction time vs. a 27.3-ms increase in reaction time of experienced boxers. One possible explanation the investigators noted is that the controls simply got better at reading the stimuli over time without actually learning the repeated sequence.
Mr. McLeod noted that a typical Rock Steady Boxing session starts with a warmup and stretch, then learning the boxing stance with the nondominant foot back, shoulders over the body and the head over the feet. The boxing moves involve sequences of different punching combinations — jab, jab, cross; left, left, right; jab, cross, hook. Then the class divides into separate circuits for boxing and exercise. The boxing circuit involves punching the speed bag – the small, air-filled, pear-shaped bag attached to a hook at eye level – as well as heavy bag and partner-held focus mitts, all with the aim of reinforcing the learned sequences. The exercise circuit focuses on muscle training and exercise with the goal of improving balance and gait.
“The boxing sequences help not only with cognitive ability but motor control,” Mr. McLeod said. “The program also helps with some of the nonmotor aspects of Parkinson’s disease. Depression is almost synonymous with Parkinson’s disease; this brings people together and builds camaraderie.”
Mr. McLeod said he hopes the research continues. “I’m hoping that this can be a jumping-off point for research going forward with procedural memory, Parkinson’s, and Rock Steady Boxing or programs like it,” he said. Future research should involve more subjects, measure improvement within same subjects who participate in the program, and account for variables such as age and gender.
Mr. McLeod and Dr. Leder reported having no relevant financial disclosures.
NEW YORK – A small pilot study has shown that patients with Parkinson’s disease who participated in the Rock Steady Boxing non-contact training program may have faster reaction times than PD patients who did not participate in the program, according to a poster presented at the International Conference on Parkinson’s Disease and Movement Disorders.
“The novelty of this is that it shows how Rock Steady Boxing and exercise programs that use sequences and the learning of sequences could possibly help slow the decline, or maintain a level of functioning longer, in Parkinson’s disease,” said Christopher McLeod, a second-year medical student at New York Institute of Technology (NYIT) College of Osteopathic Medicine, Old Westbury, N.Y.
Rock Steady Boxing is a non-contact program tailored to Parkinson’s patients founded in 2006 by Scott Newman, an Indiana lawyer who was diagnosed with early onset Parkinson’s at age 40. The regimen involves intense one-on-one training centered around boxing. Rock Steady Boxing offers classes from coast to coast in the United States and in 13 other countries. Mr. McLeod is a volunteer at the NYIT chapter of Rock Steady Boxing in Old Westbury, N.Y.
Mr. McLeod studied 28 PD patients – 14 who had been taking Rock Steady Boxing classes at NYIT for at least 6 months and 14 controls. The goal of the study was to evaluate if the Rock Steady Boxing participants showed any improvement in procedural motor learning. His coauthor was Adena Leder, DO, a faculty neurologist and movement disorder specialist at NYIT,
“What’s new about this research is the procedural memory component and the Rock Steady Boxing program is just more of the vessel, so to speak,” Mr. McLeod said. “This is a pilot study. We wanted to see if Rock Steady Boxing would show benefits in these patients. There are some trends in my research that [indicate] it would; it did not have statistical significance, but we did see trend lines.”
The researchers used a modified Serial Reaction Time Test (SRTT) composed of seven blocks of 10 stimuli each with 30-second breaks between blocks. Blocks consisted of a random familiarization block, four learning blocks repeating the same sequence of stimuli, a transfer block of random stimuli, and a posttransfer block presenting the same sequence of stimuli from the four learning blocks.
They assessed procedural learning by comparing the reduction in response time over the four identical learning blocks as well as by comparing changes in response time when the subjects were subsequently exposed to the random transfer block.
Experienced boxers demonstrated faster reaction time over the four learning blocks, ranging from 795.32 vs. 906.89 ms in the first learning block to 674.79 vs. 787.32 ms in the fourth learning block (P = .19). In the random sequence transfer block, controls showed a 93.5-ms decrease in median reaction time vs. a 27.3-ms increase in reaction time of experienced boxers. One possible explanation the investigators noted is that the controls simply got better at reading the stimuli over time without actually learning the repeated sequence.
Mr. McLeod noted that a typical Rock Steady Boxing session starts with a warmup and stretch, then learning the boxing stance with the nondominant foot back, shoulders over the body and the head over the feet. The boxing moves involve sequences of different punching combinations — jab, jab, cross; left, left, right; jab, cross, hook. Then the class divides into separate circuits for boxing and exercise. The boxing circuit involves punching the speed bag – the small, air-filled, pear-shaped bag attached to a hook at eye level – as well as heavy bag and partner-held focus mitts, all with the aim of reinforcing the learned sequences. The exercise circuit focuses on muscle training and exercise with the goal of improving balance and gait.
“The boxing sequences help not only with cognitive ability but motor control,” Mr. McLeod said. “The program also helps with some of the nonmotor aspects of Parkinson’s disease. Depression is almost synonymous with Parkinson’s disease; this brings people together and builds camaraderie.”
Mr. McLeod said he hopes the research continues. “I’m hoping that this can be a jumping-off point for research going forward with procedural memory, Parkinson’s, and Rock Steady Boxing or programs like it,” he said. Future research should involve more subjects, measure improvement within same subjects who participate in the program, and account for variables such as age and gender.
Mr. McLeod and Dr. Leder reported having no relevant financial disclosures.
NEW YORK – A small pilot study has shown that patients with Parkinson’s disease who participated in the Rock Steady Boxing non-contact training program may have faster reaction times than PD patients who did not participate in the program, according to a poster presented at the International Conference on Parkinson’s Disease and Movement Disorders.
“The novelty of this is that it shows how Rock Steady Boxing and exercise programs that use sequences and the learning of sequences could possibly help slow the decline, or maintain a level of functioning longer, in Parkinson’s disease,” said Christopher McLeod, a second-year medical student at New York Institute of Technology (NYIT) College of Osteopathic Medicine, Old Westbury, N.Y.
Rock Steady Boxing is a non-contact program tailored to Parkinson’s patients founded in 2006 by Scott Newman, an Indiana lawyer who was diagnosed with early onset Parkinson’s at age 40. The regimen involves intense one-on-one training centered around boxing. Rock Steady Boxing offers classes from coast to coast in the United States and in 13 other countries. Mr. McLeod is a volunteer at the NYIT chapter of Rock Steady Boxing in Old Westbury, N.Y.
Mr. McLeod studied 28 PD patients – 14 who had been taking Rock Steady Boxing classes at NYIT for at least 6 months and 14 controls. The goal of the study was to evaluate if the Rock Steady Boxing participants showed any improvement in procedural motor learning. His coauthor was Adena Leder, DO, a faculty neurologist and movement disorder specialist at NYIT,
“What’s new about this research is the procedural memory component and the Rock Steady Boxing program is just more of the vessel, so to speak,” Mr. McLeod said. “This is a pilot study. We wanted to see if Rock Steady Boxing would show benefits in these patients. There are some trends in my research that [indicate] it would; it did not have statistical significance, but we did see trend lines.”
The researchers used a modified Serial Reaction Time Test (SRTT) composed of seven blocks of 10 stimuli each with 30-second breaks between blocks. Blocks consisted of a random familiarization block, four learning blocks repeating the same sequence of stimuli, a transfer block of random stimuli, and a posttransfer block presenting the same sequence of stimuli from the four learning blocks.
They assessed procedural learning by comparing the reduction in response time over the four identical learning blocks as well as by comparing changes in response time when the subjects were subsequently exposed to the random transfer block.
Experienced boxers demonstrated faster reaction time over the four learning blocks, ranging from 795.32 vs. 906.89 ms in the first learning block to 674.79 vs. 787.32 ms in the fourth learning block (P = .19). In the random sequence transfer block, controls showed a 93.5-ms decrease in median reaction time vs. a 27.3-ms increase in reaction time of experienced boxers. One possible explanation the investigators noted is that the controls simply got better at reading the stimuli over time without actually learning the repeated sequence.
Mr. McLeod noted that a typical Rock Steady Boxing session starts with a warmup and stretch, then learning the boxing stance with the nondominant foot back, shoulders over the body and the head over the feet. The boxing moves involve sequences of different punching combinations — jab, jab, cross; left, left, right; jab, cross, hook. Then the class divides into separate circuits for boxing and exercise. The boxing circuit involves punching the speed bag – the small, air-filled, pear-shaped bag attached to a hook at eye level – as well as heavy bag and partner-held focus mitts, all with the aim of reinforcing the learned sequences. The exercise circuit focuses on muscle training and exercise with the goal of improving balance and gait.
“The boxing sequences help not only with cognitive ability but motor control,” Mr. McLeod said. “The program also helps with some of the nonmotor aspects of Parkinson’s disease. Depression is almost synonymous with Parkinson’s disease; this brings people together and builds camaraderie.”
Mr. McLeod said he hopes the research continues. “I’m hoping that this can be a jumping-off point for research going forward with procedural memory, Parkinson’s, and Rock Steady Boxing or programs like it,” he said. Future research should involve more subjects, measure improvement within same subjects who participate in the program, and account for variables such as age and gender.
Mr. McLeod and Dr. Leder reported having no relevant financial disclosures.
REPORTING FROM ICPDMD 2018
Key clinical point: Exercise programs may help improve procedural learning in individuals with Parkinson’s disease.
Major finding: Rock Steady Boxing experienced boxers demonstrated reaction times ranging from 795.32 vs. 906.89 ms to 674.79 vs. 787.32 ms across four test blocks.
Study details: Pilot study of 14 Parkinson’s patients who participated in Rock Steady Boxing vs. 14 controls.
Disclosures: Mr. McLeod reported no relevant financial disclosures.