User login
Calcified cysts in the upper abdomen
Q: Which is the most plausible diagnosis?
- Nephrolithiasis
- Hydatid cyst
- Polycystic kidney and liver disease
- Metastatic calcification
- Intra-abdominal abscesses
- Cystadenoma
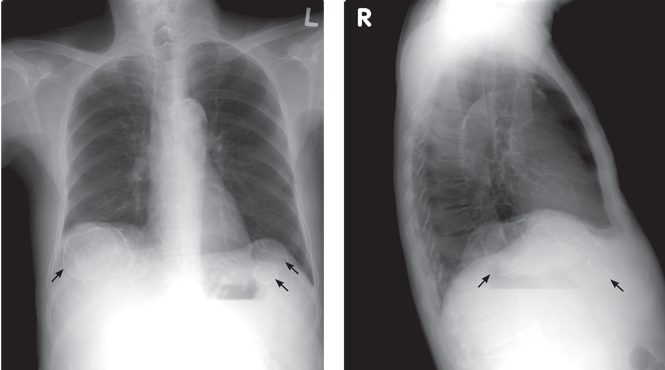
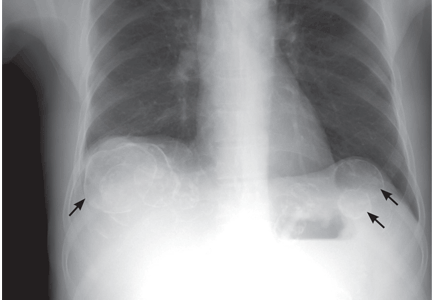
A: This patient has polycystic kidney disease. He first came to our hospital 21 years ago because of upper gastrointestinal bleeding, and he was found to have multiple cystic lesions. He was diagnosed as having polycystic kidney disease with multiple hepatic cysts.
About 15 years ago, he had several episodes of gross hematuria with right flank pain, which were attributed to bleeding from the cysts. Because his renal function was deteriorating, he started regular hemodialysis at that time. He underwent parathyroidectomy for secondary hyperparathyroidism 2 years ago.
The differential diagnosis of calcified cystic lesions in the upper abdomen includes infectious diseases (eg, chronic intra-abdominal abscess, echinococcosis, cysticercosis), neoplasms (eg, cystadenomas, cystadenocarcinomas), organized hematomas, liver cysts, renal cysts, gallstones, renal stones, calcified lymph nodes, and vessel calcification.
Autosomal dominant polycystic kidney disease is the most common inherited renal disease.1 Typically, patients with this disease have a positive family history, although about 5% to 10% do not.1,3 (Our patient did not.)
The diagnosis is suggested by family history and imaging, with typical findings including large kidneys and extensive cysts scattered throughout both kidneys.1–3 The history, epidemiologic data, and the character of the cystic lesions help in making the final diagnosis.
In patients at risk, the presence of at least two renal cysts (unilateral or bilateral) at age 15 to 30, of at least two cysts in each kidney at age 30 to 59, and of at least four cysts in each kidney at age 60 or older is regarded as sufficient to establish the diagnosis.1–3
Approximately 80% of patients with autosomal dominant polycystic kidney disease also have hepatic cysts.1,3 Cyst calcifications occur in about 25% of patients, either as a residual effect of intracystic hemorrhage or as a consequence of secondary hyperparathyroidism.4,5
- Braun WE. Autosomal dominant polycystic kidney disease: emerging concepts of pathogenesis and new treatments. Cleve Clin J Med 2009; 76:97–104.
- Torres VE, Bennett WM. Diagnosis of and screening for autosomal dominant polycystic kidney disease. In:Rose BD, editor: UpToDate. Waltham, MA: UpToDate, 2009.
- Grantham JJ. Clinical practice. Autosomal dominant polycystic kidney disease. N Engl J Med 2008; 359:1477–1485.
- Coffin B, Hadengue A, Degos F, Benhamou JP. Calcified hepatic and renal cysts in adult dominant polycystic kidney disease. Dig Dis Sci 1990; 35:1172–1175.
- Levine E, Grantham JJ. Calcified renal stones and cyst calcifications in autosomal dominant polycystic kidney disease: clinical and CT study in 84 patients. AJR Am J Roentgenol 1992; 159:77–81.
Q: Which is the most plausible diagnosis?
- Nephrolithiasis
- Hydatid cyst
- Polycystic kidney and liver disease
- Metastatic calcification
- Intra-abdominal abscesses
- Cystadenoma
A: This patient has polycystic kidney disease. He first came to our hospital 21 years ago because of upper gastrointestinal bleeding, and he was found to have multiple cystic lesions. He was diagnosed as having polycystic kidney disease with multiple hepatic cysts.
About 15 years ago, he had several episodes of gross hematuria with right flank pain, which were attributed to bleeding from the cysts. Because his renal function was deteriorating, he started regular hemodialysis at that time. He underwent parathyroidectomy for secondary hyperparathyroidism 2 years ago.
The differential diagnosis of calcified cystic lesions in the upper abdomen includes infectious diseases (eg, chronic intra-abdominal abscess, echinococcosis, cysticercosis), neoplasms (eg, cystadenomas, cystadenocarcinomas), organized hematomas, liver cysts, renal cysts, gallstones, renal stones, calcified lymph nodes, and vessel calcification.
Autosomal dominant polycystic kidney disease is the most common inherited renal disease.1 Typically, patients with this disease have a positive family history, although about 5% to 10% do not.1,3 (Our patient did not.)
The diagnosis is suggested by family history and imaging, with typical findings including large kidneys and extensive cysts scattered throughout both kidneys.1–3 The history, epidemiologic data, and the character of the cystic lesions help in making the final diagnosis.
In patients at risk, the presence of at least two renal cysts (unilateral or bilateral) at age 15 to 30, of at least two cysts in each kidney at age 30 to 59, and of at least four cysts in each kidney at age 60 or older is regarded as sufficient to establish the diagnosis.1–3
Approximately 80% of patients with autosomal dominant polycystic kidney disease also have hepatic cysts.1,3 Cyst calcifications occur in about 25% of patients, either as a residual effect of intracystic hemorrhage or as a consequence of secondary hyperparathyroidism.4,5
Q: Which is the most plausible diagnosis?
- Nephrolithiasis
- Hydatid cyst
- Polycystic kidney and liver disease
- Metastatic calcification
- Intra-abdominal abscesses
- Cystadenoma
A: This patient has polycystic kidney disease. He first came to our hospital 21 years ago because of upper gastrointestinal bleeding, and he was found to have multiple cystic lesions. He was diagnosed as having polycystic kidney disease with multiple hepatic cysts.
About 15 years ago, he had several episodes of gross hematuria with right flank pain, which were attributed to bleeding from the cysts. Because his renal function was deteriorating, he started regular hemodialysis at that time. He underwent parathyroidectomy for secondary hyperparathyroidism 2 years ago.
The differential diagnosis of calcified cystic lesions in the upper abdomen includes infectious diseases (eg, chronic intra-abdominal abscess, echinococcosis, cysticercosis), neoplasms (eg, cystadenomas, cystadenocarcinomas), organized hematomas, liver cysts, renal cysts, gallstones, renal stones, calcified lymph nodes, and vessel calcification.
Autosomal dominant polycystic kidney disease is the most common inherited renal disease.1 Typically, patients with this disease have a positive family history, although about 5% to 10% do not.1,3 (Our patient did not.)
The diagnosis is suggested by family history and imaging, with typical findings including large kidneys and extensive cysts scattered throughout both kidneys.1–3 The history, epidemiologic data, and the character of the cystic lesions help in making the final diagnosis.
In patients at risk, the presence of at least two renal cysts (unilateral or bilateral) at age 15 to 30, of at least two cysts in each kidney at age 30 to 59, and of at least four cysts in each kidney at age 60 or older is regarded as sufficient to establish the diagnosis.1–3
Approximately 80% of patients with autosomal dominant polycystic kidney disease also have hepatic cysts.1,3 Cyst calcifications occur in about 25% of patients, either as a residual effect of intracystic hemorrhage or as a consequence of secondary hyperparathyroidism.4,5
- Braun WE. Autosomal dominant polycystic kidney disease: emerging concepts of pathogenesis and new treatments. Cleve Clin J Med 2009; 76:97–104.
- Torres VE, Bennett WM. Diagnosis of and screening for autosomal dominant polycystic kidney disease. In:Rose BD, editor: UpToDate. Waltham, MA: UpToDate, 2009.
- Grantham JJ. Clinical practice. Autosomal dominant polycystic kidney disease. N Engl J Med 2008; 359:1477–1485.
- Coffin B, Hadengue A, Degos F, Benhamou JP. Calcified hepatic and renal cysts in adult dominant polycystic kidney disease. Dig Dis Sci 1990; 35:1172–1175.
- Levine E, Grantham JJ. Calcified renal stones and cyst calcifications in autosomal dominant polycystic kidney disease: clinical and CT study in 84 patients. AJR Am J Roentgenol 1992; 159:77–81.
- Braun WE. Autosomal dominant polycystic kidney disease: emerging concepts of pathogenesis and new treatments. Cleve Clin J Med 2009; 76:97–104.
- Torres VE, Bennett WM. Diagnosis of and screening for autosomal dominant polycystic kidney disease. In:Rose BD, editor: UpToDate. Waltham, MA: UpToDate, 2009.
- Grantham JJ. Clinical practice. Autosomal dominant polycystic kidney disease. N Engl J Med 2008; 359:1477–1485.
- Coffin B, Hadengue A, Degos F, Benhamou JP. Calcified hepatic and renal cysts in adult dominant polycystic kidney disease. Dig Dis Sci 1990; 35:1172–1175.
- Levine E, Grantham JJ. Calcified renal stones and cyst calcifications in autosomal dominant polycystic kidney disease: clinical and CT study in 84 patients. AJR Am J Roentgenol 1992; 159:77–81.
Inadequate follow-up ends in kidney transplant … Teenager dies of undiagnosed pneumonia … more
Inadequate follow-up ends in a kidney transplant
SMALL AMOUNTS OF PROTEIN AND BLOOD appeared in urine samples obtained during routine screenings of a 34-year-old man by his primary care physician. The doctor never told the patient about the proteinuria and reassured him that the presence of blood was normal for some adults and nothing to worry about.
The physician requested a urology consult on 1 occasion, but no cause was found for the blood and protein in the urine. After a further workup, the primary care physician concluded that it was benign. The urologist maintained that it wasn’t his job to do a workup for kidney disease or proteinuria; a kidney specialist would normally do such a work-up.
The blood and protein in the patient’s urine increased during subsequent years. The primary care physician didn’t order additional testing or consult a kidney specialist.
At a routine physical exam 5 years after the initial finding of proteinuria and hematuria, the patient’s blood and urine screening tests were grossly abnormal; he had anemia and kidney failure and needed immediate hospitalization. The primary care physician didn’t tell the patient about the abnormal test results because he didn’t see them—a lapse he blamed on a system error and office staff.
Several weeks after his latest doctor visit, the patient became acutely ill. His kidneys stopped functioning, and he went into hypertensive crisis. He was hospitalized and IgA nephropathy was diagnosed. His kidneys never recovered. The patient was placed on hemodialysis and received a kidney transplant 6 months later.
PLAINTIFF’S CLAIM Although IgA nephropathy has no known cause or cure, it can be treated with diet modification, lifestyle change, blood pressure control, and medication. With proper diagnosis and treatment, the patient would have retained kidney function for another 2½ years or more.
DOCTORS’ DEFENSE Earlier diagnosis would have prolonged kidney function for only about 6 months.
VERDICT $400,000 Massachusetts settlement.
COMMENT Blaming a bad outcome on “a system error and office staff ” is unlikely to be a winning defense in a court of law.
Teenager dies of undiagnosed pneumonia
A 16-YEAR-OLD GIRL was taken to the emergency room with diarrhea, fever, a nonproductive cough, chest pain, and rhinorrhea. The pediatrician and nurse who examined her found no abnormalities of the lungs, respiration, or oxygenation. A viral syndrome and/or infection of the upper respiratory tract was diagnosed. The girl was discharged with instructions to see her primary physician and return to the ER if her condition worsened.
The patient saw her pediatrician 3 days later after becoming increasingly weak. The pediatrician noted abnormalities in her respiration. He diagnosed a virus but prescribed antibiotics, and told the girl to return if her condition became worse. The girl didn’t return and died 3 days later. Her death was attributed to pneumonia.
PLAINTIFF’S CLAIM The pediatrician and nurse in the ER should have diagnosed pneumonia. The differential diagnosis in the ER should have included pneumonia, and the patient shouldn’t have been released until pneumonia had been ruled out. The patient’s pediatrician should have given IV antibiotics and ordered a chest radiograph and white blood cell count.
DOCTORS’ DEFENSE The patient’s symptoms were characteristic of a viral infection and not typical of a bacterial infection. The pneumonia originated after the patient was last seen and was an aggressive form.
VERDICT $3.9 million New York verdict reduced to $500,000 under a high/low agreement.
COMMENT Our worst nightmare: treating a patient appropriately by withholding antibiotics (in the case of the emergency room staff ) followed by a catastrophic outcome. This case is a great example of why we practice defensive medicine and what’s wrong with our tort system.
Serious symptoms and history fail to prompt stroke workup
A MAN WITH DIABETES AND HYPERTENSION went to his primary care physician’s office complaining of right-sided headache, dizziness, some weakness and tingling on his left side, and difficulty picking up his left foot. The 56-year-old patient was seen by a nurse practitioner. The nurse consulted the physician twice during the visit, but the physician didn’t examine the patient personally.
An electrocardiogram was performed. The nurse found no neurologic indications of a transient ischemic attack. The patient was sent home with prescriptions for aspirin and atenolol and instructions to return in a week.
The patient’s condition deteriorated, and he went to the emergency department, where he was treated for a stroke. The symptoms progressed, however, leading to significant physical and cognitive disabilities.
PLAINTIFF’S CLAIM The physician and nurse practitioner failed to appreciate the patient’s risk of a stroke and recognize that his symptoms suggested a serious neurologic event. Immediate referral to an ED for a stroke work-up and treatment would have prevented progression of the stroke and the resulting disabilities. The physician should have evaluated the patient personally. The patient had not received proper treatment for hypertension, diabetes, and high cholesterol for many years before the stroke.
THE DEFENSE The treatment given was proper; earlier admission wouldn’t have made a difference.
VERDICT $750,000 Massachusetts settlement.
COMMENT Supervision of midlevel employees carries its own risks. When in doubt, see the patient!
Inadequate follow-up ends in a kidney transplant
SMALL AMOUNTS OF PROTEIN AND BLOOD appeared in urine samples obtained during routine screenings of a 34-year-old man by his primary care physician. The doctor never told the patient about the proteinuria and reassured him that the presence of blood was normal for some adults and nothing to worry about.
The physician requested a urology consult on 1 occasion, but no cause was found for the blood and protein in the urine. After a further workup, the primary care physician concluded that it was benign. The urologist maintained that it wasn’t his job to do a workup for kidney disease or proteinuria; a kidney specialist would normally do such a work-up.
The blood and protein in the patient’s urine increased during subsequent years. The primary care physician didn’t order additional testing or consult a kidney specialist.
At a routine physical exam 5 years after the initial finding of proteinuria and hematuria, the patient’s blood and urine screening tests were grossly abnormal; he had anemia and kidney failure and needed immediate hospitalization. The primary care physician didn’t tell the patient about the abnormal test results because he didn’t see them—a lapse he blamed on a system error and office staff.
Several weeks after his latest doctor visit, the patient became acutely ill. His kidneys stopped functioning, and he went into hypertensive crisis. He was hospitalized and IgA nephropathy was diagnosed. His kidneys never recovered. The patient was placed on hemodialysis and received a kidney transplant 6 months later.
PLAINTIFF’S CLAIM Although IgA nephropathy has no known cause or cure, it can be treated with diet modification, lifestyle change, blood pressure control, and medication. With proper diagnosis and treatment, the patient would have retained kidney function for another 2½ years or more.
DOCTORS’ DEFENSE Earlier diagnosis would have prolonged kidney function for only about 6 months.
VERDICT $400,000 Massachusetts settlement.
COMMENT Blaming a bad outcome on “a system error and office staff ” is unlikely to be a winning defense in a court of law.
Teenager dies of undiagnosed pneumonia
A 16-YEAR-OLD GIRL was taken to the emergency room with diarrhea, fever, a nonproductive cough, chest pain, and rhinorrhea. The pediatrician and nurse who examined her found no abnormalities of the lungs, respiration, or oxygenation. A viral syndrome and/or infection of the upper respiratory tract was diagnosed. The girl was discharged with instructions to see her primary physician and return to the ER if her condition worsened.
The patient saw her pediatrician 3 days later after becoming increasingly weak. The pediatrician noted abnormalities in her respiration. He diagnosed a virus but prescribed antibiotics, and told the girl to return if her condition became worse. The girl didn’t return and died 3 days later. Her death was attributed to pneumonia.
PLAINTIFF’S CLAIM The pediatrician and nurse in the ER should have diagnosed pneumonia. The differential diagnosis in the ER should have included pneumonia, and the patient shouldn’t have been released until pneumonia had been ruled out. The patient’s pediatrician should have given IV antibiotics and ordered a chest radiograph and white blood cell count.
DOCTORS’ DEFENSE The patient’s symptoms were characteristic of a viral infection and not typical of a bacterial infection. The pneumonia originated after the patient was last seen and was an aggressive form.
VERDICT $3.9 million New York verdict reduced to $500,000 under a high/low agreement.
COMMENT Our worst nightmare: treating a patient appropriately by withholding antibiotics (in the case of the emergency room staff ) followed by a catastrophic outcome. This case is a great example of why we practice defensive medicine and what’s wrong with our tort system.
Serious symptoms and history fail to prompt stroke workup
A MAN WITH DIABETES AND HYPERTENSION went to his primary care physician’s office complaining of right-sided headache, dizziness, some weakness and tingling on his left side, and difficulty picking up his left foot. The 56-year-old patient was seen by a nurse practitioner. The nurse consulted the physician twice during the visit, but the physician didn’t examine the patient personally.
An electrocardiogram was performed. The nurse found no neurologic indications of a transient ischemic attack. The patient was sent home with prescriptions for aspirin and atenolol and instructions to return in a week.
The patient’s condition deteriorated, and he went to the emergency department, where he was treated for a stroke. The symptoms progressed, however, leading to significant physical and cognitive disabilities.
PLAINTIFF’S CLAIM The physician and nurse practitioner failed to appreciate the patient’s risk of a stroke and recognize that his symptoms suggested a serious neurologic event. Immediate referral to an ED for a stroke work-up and treatment would have prevented progression of the stroke and the resulting disabilities. The physician should have evaluated the patient personally. The patient had not received proper treatment for hypertension, diabetes, and high cholesterol for many years before the stroke.
THE DEFENSE The treatment given was proper; earlier admission wouldn’t have made a difference.
VERDICT $750,000 Massachusetts settlement.
COMMENT Supervision of midlevel employees carries its own risks. When in doubt, see the patient!
Inadequate follow-up ends in a kidney transplant
SMALL AMOUNTS OF PROTEIN AND BLOOD appeared in urine samples obtained during routine screenings of a 34-year-old man by his primary care physician. The doctor never told the patient about the proteinuria and reassured him that the presence of blood was normal for some adults and nothing to worry about.
The physician requested a urology consult on 1 occasion, but no cause was found for the blood and protein in the urine. After a further workup, the primary care physician concluded that it was benign. The urologist maintained that it wasn’t his job to do a workup for kidney disease or proteinuria; a kidney specialist would normally do such a work-up.
The blood and protein in the patient’s urine increased during subsequent years. The primary care physician didn’t order additional testing or consult a kidney specialist.
At a routine physical exam 5 years after the initial finding of proteinuria and hematuria, the patient’s blood and urine screening tests were grossly abnormal; he had anemia and kidney failure and needed immediate hospitalization. The primary care physician didn’t tell the patient about the abnormal test results because he didn’t see them—a lapse he blamed on a system error and office staff.
Several weeks after his latest doctor visit, the patient became acutely ill. His kidneys stopped functioning, and he went into hypertensive crisis. He was hospitalized and IgA nephropathy was diagnosed. His kidneys never recovered. The patient was placed on hemodialysis and received a kidney transplant 6 months later.
PLAINTIFF’S CLAIM Although IgA nephropathy has no known cause or cure, it can be treated with diet modification, lifestyle change, blood pressure control, and medication. With proper diagnosis and treatment, the patient would have retained kidney function for another 2½ years or more.
DOCTORS’ DEFENSE Earlier diagnosis would have prolonged kidney function for only about 6 months.
VERDICT $400,000 Massachusetts settlement.
COMMENT Blaming a bad outcome on “a system error and office staff ” is unlikely to be a winning defense in a court of law.
Teenager dies of undiagnosed pneumonia
A 16-YEAR-OLD GIRL was taken to the emergency room with diarrhea, fever, a nonproductive cough, chest pain, and rhinorrhea. The pediatrician and nurse who examined her found no abnormalities of the lungs, respiration, or oxygenation. A viral syndrome and/or infection of the upper respiratory tract was diagnosed. The girl was discharged with instructions to see her primary physician and return to the ER if her condition worsened.
The patient saw her pediatrician 3 days later after becoming increasingly weak. The pediatrician noted abnormalities in her respiration. He diagnosed a virus but prescribed antibiotics, and told the girl to return if her condition became worse. The girl didn’t return and died 3 days later. Her death was attributed to pneumonia.
PLAINTIFF’S CLAIM The pediatrician and nurse in the ER should have diagnosed pneumonia. The differential diagnosis in the ER should have included pneumonia, and the patient shouldn’t have been released until pneumonia had been ruled out. The patient’s pediatrician should have given IV antibiotics and ordered a chest radiograph and white blood cell count.
DOCTORS’ DEFENSE The patient’s symptoms were characteristic of a viral infection and not typical of a bacterial infection. The pneumonia originated after the patient was last seen and was an aggressive form.
VERDICT $3.9 million New York verdict reduced to $500,000 under a high/low agreement.
COMMENT Our worst nightmare: treating a patient appropriately by withholding antibiotics (in the case of the emergency room staff ) followed by a catastrophic outcome. This case is a great example of why we practice defensive medicine and what’s wrong with our tort system.
Serious symptoms and history fail to prompt stroke workup
A MAN WITH DIABETES AND HYPERTENSION went to his primary care physician’s office complaining of right-sided headache, dizziness, some weakness and tingling on his left side, and difficulty picking up his left foot. The 56-year-old patient was seen by a nurse practitioner. The nurse consulted the physician twice during the visit, but the physician didn’t examine the patient personally.
An electrocardiogram was performed. The nurse found no neurologic indications of a transient ischemic attack. The patient was sent home with prescriptions for aspirin and atenolol and instructions to return in a week.
The patient’s condition deteriorated, and he went to the emergency department, where he was treated for a stroke. The symptoms progressed, however, leading to significant physical and cognitive disabilities.
PLAINTIFF’S CLAIM The physician and nurse practitioner failed to appreciate the patient’s risk of a stroke and recognize that his symptoms suggested a serious neurologic event. Immediate referral to an ED for a stroke work-up and treatment would have prevented progression of the stroke and the resulting disabilities. The physician should have evaluated the patient personally. The patient had not received proper treatment for hypertension, diabetes, and high cholesterol for many years before the stroke.
THE DEFENSE The treatment given was proper; earlier admission wouldn’t have made a difference.
VERDICT $750,000 Massachusetts settlement.
COMMENT Supervision of midlevel employees carries its own risks. When in doubt, see the patient!
Anemia and chronic kidney disease: What’s the connection?
• Evaluate for chronic kidney disease (CKD) anemia when a patient has a serum creatinine ≥2 mg/dL and hemoglobin <12 g/dL (adult males and postmenopausal females) or <11 g/dL (premenopausal females). A
• Before you treat CKD anemia, correct any underlying iron deficiency. A
• Start anemia therapy with erythropoietin-stimulating agents when hemoglobin is ≤10 g/dL, and maintain target hemoglobin levels between 11 and 12 g/dL, in accordance with National Kidney Foundation guidelines. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE Mary J, a 65-year-old woman with stage 3 chronic kidney disease (CKD), is in your office for a follow-up appointment. Over the past 6 months, she has noticed a decrease in her energy level. On her routine blood work, you see that her hemoglobin has been slowly declining over the past year. It is now 9 g/dL and her estimated glomerular filtration rate (GFR) is 40 mL/min.
How would you evaluate Mary’s anemia, and would you suspect that it was related to her CKD?
Most physicians are aware that CKD—which affects approximately 10% of the US population1—has a deleterious effect on cardiovascular disease, but many fail to recognize the impact it has on the hematopoietic system. Managing the anemia that accompanies CKD in patients like Mary requires a finely tuned diagnostic approach and treatment strategy. This article will help toward that end.
Anemia of CKD: A common problem
Anemia of CKD is one of the first signs of kidney dysfunction, yet it often goes undetected because of its insidious onset. Anemia develops gradually as kidney function declines and the GFR drops to 70 mL/min in male patients and 50 mL/min in females.2 Epidemiologic data indicate that two-thirds of patients in the early stages of kidney failure are also anemic, with a hemoglobin level of less than 11 g/dL, yet only one-third of these patients have ever received erythropoietin-stimulating agents (ESAs) to treat their anemia.1 The National Kidney Foundation Kidney Disease Outcomes Quality Initiative (NKF KDOQI) guidelines recommend that the evaluation of anemia of CKD begin in patients with a serum creatinine ≥2 mg/dL when the hemoglobin is <12 g/dL in adult males and postmenopausal females and <11 g/dL in premenopausal females.3
How kidney failure leads to anemia
Patients like Mary develop anemia of CKD because failing kidneys produce less erythropoietin (EPO) than the body requires for the production of red blood cells. EPO is an endogenous hormone produced by peritubular fibroblasts in the renal cortex.4 Most of this hormone (90%) is produced in the kidney, with the remainder manufactured by hepatocytes.
Erythropoiesis is stimulated by blood loss, decreased oxygen tension, and an increase in oxygen affinity, which leads to an increase in EPO production via upregulation of the EPO gene. In healthy individuals, detection of hypoxia by the kidney can result in a 1000-fold increase in EPO production.5 Patients with CKD don’t have that kind of robust response, and their EPO levels remain normal or below normal even when challenged by lack of oxygen. Anemia in CKD can also be caused by nutritional deficiencies, decreased red blood cell survival because of uremic toxins, oxidative stress, inflammation, and the use of angiotensin-converting enzyme (ACE) inhibitors.
Chronic anemia, CKD, and CV disease: A deadly triad
The leading cause of death in patients with CKD is cardiovascular disease. Patients with cardiorenal anemia syndrome develop a self-perpetuating triad that increases the risk of death when all 3 conditions are present. Anemic patients double their relative risk of death when CKD is present and triple their risk if they have anemia, CKD, and cardiovascular disease.6
Epidemiologic studies suggest an association among anemia, left ventricular hypertrophy (LVH), mortality, and cardiovascular outcomes. One study evaluated 2423 stage 3 and 4 CKD patients with anemia, defined as hemoglobin <13 g/dL in males and <12 g/dL in females. The results showed an increase in composite outcomes of myocardial infarction, stroke, and death.7 A prospective study evaluating 246 people with stages 2 to 4 CKD reported anemia to be an independent risk factor for the development of LVH.8 The stages of CKD are shown in the TABLE.
Suspected mechanisms of cardiovascular disease progression due to chronic anemia include tissue hypoxia, free radical formation, endothelial dysfunction, and vascular damage. Compensatory neurohumeral adaptations result in an increased sympathetic response and upregulation of the reninangiotensin-aldosterone system.9
TABLE
Stages of chronic kidney disease
| Stage | Description | GFR (mL/min/1.73 m2) |
|---|---|---|
| 1 | Kidney damage with normal or increased GFR | ≥90 |
| 2 | Kidney damage with mildly decreased GFR | 60-89 |
| 3 | Moderately decreased GFR | 30-59 |
| 4 | Severely decreased GFR | 15-29 |
| 5 | Kidney failure | <15 or dialysis |
| GFR, glomerular filtration rate. | ||
| Source: KDOQI Clinical Practice Guideline and Clinical Practice Recommendations for anemia in chronic kidney disease: 2007 update of hemoglobin target. Am J Kidney Dis. 2007.3 | ||
Anemia of CKD: A diagnosis of exclusion
Because anemia can have many causes, other possibilities must be ruled out before a diagnosis of CKD anemia can be made. Testing should be tailored to each individual situation, determined by a thorough history and physical. Steps in the diagnosis are shown in the FLOW CHART. A basic work-up should include complete blood count with differential, iron studies (ferritin, serum Fe, and total iron binding capacity), reticulocyte count, and a guaiac test. Other blood tests, such as thyroid-stimulating hormone (TSH), B12, and folate levels, and a hemolysis panel (lactate dehydrogenase, haptoglobin), should be obtained if the history suggests these disorders. A peripheral blood smear showing normocytic red blood cells with a normochromic pattern would favor the diagnosis of anemia of CKD.
FLOW CHART
A step-by-step guide to CKD anemia diagnosis and treatment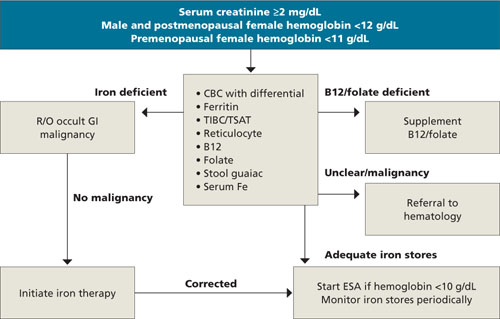
CBC, complete blood count; CKD, chronic kidney disease; ESA, erythropoietin-stimulating agents; R/O, rule out; TIBC/TSAT, total iron-binding capacity/transferrin saturation.
A look at the iron connection
Many patients with CKD anemia have iron deficiency and are unable to produce adequate numbers of red blood cells. Iron deficiency can have many causes: not enough iron-rich food in the diet, chronic bleeding, malabsorption, or an occult gastrointestinal malignancy. Once iron deficiency anemia is diagnosed, a colonoscopy is warranted to rule out occult malignancy. Ferritin, a protein found mostly in macrophages and hepatocytes, stores iron and serves as a marker for total iron stores. Using stored iron requires transferrin, a transporting protein, to shuttle iron from the reticuloendothelial system and gut to the bone marrow. CKD is a pro-inflammatory state that results in a limited ability to use iron stores. For this reason, patients with CKD require higher levels of iron.
Absolute iron deficiency. Iron deficiency in CKD patients with serum ferritin <100 ng/mL and transferrin saturation (TSAT) <20% is characterized as absolute iron deficiency. The TSAT represents the percent of iron bound to transferrin and is a good indicator of the body’s functional capacity to use stored iron.
Relative iron deficiency and iron block. Patients who do not respond to ESA therapy even though they have adequate iron stores are said to have a functional or relative iron deficiency. Iron block is a condition that results in anemia from a chronic inflammatory state such as infection, autoimmune disorders, or malignancies. It resolves once the inflammatory process abates. Both conditions have similar anemia profiles, with a serum ferritin >100 ng/mL and a TSAT <20%. Differentiating between these conditions requires dynamic testing using serial iron studies and observing responses to ESAs and iron supplementation.
Options for correcting iron deficiency
After a thorough history and physical with appropriate screening, you find that Mary has an iron deficiency that must be corrected before her anemia can be treated effectively. Treatment for iron deficiency is usually initiated with oral therapy, at the recommended dose of 200 mg oral elemental iron a day in 3 divided doses.
If the oral therapy does not correct iron deficiency within 3 months, or a patient cannot tolerate the constipation that is often a side effect of this therapy, IV iron administration can be considered. Because CKD patients do not have the ongoing iron losses seen in patients with end-stage renal disease (ESRD), a conservative approach using a single IV dose followed by repeat testing is warranted. The goal is to achieve ferritin levels >100 ng/dL and TSAT >20%. A number of products for IV iron administration are available. The most widely used are iron dextran (INFeD), ferric gluconate (Ferrlecit), and iron sucrose (Venofer).
Iron stores are replenished? Time to treat the anemia
When ferritin levels and TSAT show that iron deficiency has been corrected, ESA treatment for anemia can begin. Two major brands of ESAs currently in use in the United States are a recombinant human erythropoietin (rHuEPO) known as epoetin alfa (Procrit, Epogen), and darbepoetin alpha (Aranesp). Both medications are effective and can be given intravenously or subcutaneously. Subcutaneous darbepoetin alpha has a longer half-life compared with epoetin alpha (70 vs 24 hours), so dosing intervals can be longer.10,11 ESAs should not be started in patients with uncontrolled hypertension until the blood pressure is controlled, or in patients with an active malignancy unless the treatment is directly supervised by an oncologist.
Aim for complete anemia resolution? That’s controversial
Treatment of CKD anemia with ESAs is widely practiced, but controversy over whether it is beneficial to aim for complete resolution of anemia is ongoing. The CREATE (Cardiovascular Risk Reduction by Early Anemia Treatment) and CHOIR (Correction of Hemoglobin and Outcomes in Renal Insufficiency) trials published in 2006 failed to resolve the issue.12,13
In the CREATE trial, patients targeted to achieve normal hemoglobin levels did no better in avoiding cardiovascular events than patients targeted for lower levels. The CHOIR trial was stopped early because of an increased trend toward death and hospitalization for congestive heart failure in the group with therapy targeted to achieve normal hemoglobin levels.
The recently published TREAT (Trial to Reduce Cardiovascular Events with Aranesp Therapy) study of patients with type 2 diabetes and CKD showed no reduction in all-cause mortality, cardiovascular morbidity, or ESRD in patients receiving Aranesp targeted to achieve a hemoglobin level of approximately 13 g/dL, compared with placebo.14 The study did demonstrate, however, that patients receiving Aranesp were about twice as likely to have a stroke than the placebo subjects (101 vs 53)—which might lead clinicians to ponder whether the gains, if any, were worth the risk.
Revised labeling. Late last year, the US Food and Drug Administration approved a label change for Procrit and Aranesp, warning that patients with renal failure “experienced greater risks for death and serious cardiovascular events when administered ESAs to target higher vs lower hemoglobin levels” and advising physicians to “individualize dosing to achieve and maintain hemoglobin levels within the range of 10 to 12 g/dL.”10,11 The 2007 NKF KDOQI guidelines suggest maintaining a hemoglobin level between 11 and 12 g/dL and have not incorporated the results of the TREAT trial.
Some patients don’t respond to ESAs
Inadequate response to ESAs is most commonly caused by underdosing or inadequate iron stores. NKF KDOQI guidelines recommend checking TSAT and ferritin prior to initiating therapy and monitoring these levels every 3 months.3 True nonresponders are individuals with good iron stores who are unable to achieve target hemoglobin within 4 to 6 months despite receiving subcutaneous epoetin 300 IU/kg per week. Inadequate response to ESAs can be caused by ongoing occult blood loss, infection, inflammation, nutritional deficiencies, hemolysis, hemoglobinemias, aluminum toxicity, anti-EPO antibody, hyperparathyroidism, multiple myeloma, and bone marrow dysfunction.10,11 If patients do not respond to ESA therapy, the NKF KDOQI guidelines recommend referral to a nephrologist or hematologist.3
How did Mary fare?
Mary did well taking oral iron supplementation. Once her iron deficiency was corrected, you were able to begin treating her anemia. After appropriate titration of her ESA, she was able to maintain a hemoglobin level between 11 and 12 g/dL 4 months into therapy. On a follow-up visit, she had no side effects from the medication and reported an increase in her energy level.
CORRESPONDENCE
Jonathan Taliercio, DO, Cleveland Clinic, Department of Nephrology and Hypertension, 9500 Euclid Avenue, Cleveland, OH 44195; [email protected]
1. United States Renal Data System, USRDS. 2009 Annual Data Report. Atlas of Chronic Kidney Disease in the United States. Bethesda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; 2009.
2. Hsu CJ, McCulloch CE, Curhan GC. Epidemiology of anemia associated with chronic renal insufficiency among adults in the United States: results from the Third National Health and Nutrition Examination Survey. J Am Soc Nephrol. 2002;13:504-510.
3. KDOQI Clinical Practice Guideline and Clinical Practice Recommendations for anemia in chronic kidney disease: 2007 update of hemoglobin target. Am J Kidney Dis. 2007;50:471-530.
4. Donnelly S. Why is erythropoietin made in the kidney? The kidney functions as a critmeter. Am J Kidney Dis. 2001;38:415-425.
5. Ebert B, Franklin H. Regulation of the erythropoietin gene. Blood. 1999;94:1864-1877.
6. Silverberg D, Wexler D, Blum M, et al. The cardio-renal anaemia syndrome: does it exist? Nephrol Dial Transplant. 2003;18(suppl 8):viii 7-viii 12.
7. Weiner D, Tighiouart H, Vlagopoulos P, et al. Effects of anemia and left ventricular hypertrophy on cardiovascular disease in patients with chronic kidney disease. J Am Soc Nephrol. 2005;16:1803-1810.
8. Levin A, Thompson C, Ethier J, et al. Left ventricular mass index increase in early renal disease: impact of decline in hemoglobin. Am J Kidney Dis. 1999;34:125-134.
9. Rao M, Pereira B. Optimal anemia management reduces cardiovascular morbidity, mortality, and costs in chronic kidney disease. Kidney Int. 2005;68:1432-1438.
10. Amgen. Aranesp (Darbepoetin Alpha) package insert. Available at www.aranesp.com/professional/crf/full_prescribing_info/pi.jsp. Accessed November 16, 2009.
11. Amgen. Procrit (Epoetin Alpha) package insert. Available at www.procrit.com/sites/default/files/shared/OBI/PI/ProcritBooklet.pdf#page=1. Accessed November 16, 2009.
12. Drueke T, Locatelli F, Clyne N, et al. Normalization of hemoglobin level in patients with CKD and anemia. N Engl J Med. 2006;355:2071-2084.
13. Singh A, Szczech L, Tang K. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med. 2006;355:2085-2098.
14. Pfeffer MA, Burdmann EA, Chen CY, et al. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med. 2009;361:2019-2032.
• Evaluate for chronic kidney disease (CKD) anemia when a patient has a serum creatinine ≥2 mg/dL and hemoglobin <12 g/dL (adult males and postmenopausal females) or <11 g/dL (premenopausal females). A
• Before you treat CKD anemia, correct any underlying iron deficiency. A
• Start anemia therapy with erythropoietin-stimulating agents when hemoglobin is ≤10 g/dL, and maintain target hemoglobin levels between 11 and 12 g/dL, in accordance with National Kidney Foundation guidelines. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE Mary J, a 65-year-old woman with stage 3 chronic kidney disease (CKD), is in your office for a follow-up appointment. Over the past 6 months, she has noticed a decrease in her energy level. On her routine blood work, you see that her hemoglobin has been slowly declining over the past year. It is now 9 g/dL and her estimated glomerular filtration rate (GFR) is 40 mL/min.
How would you evaluate Mary’s anemia, and would you suspect that it was related to her CKD?
Most physicians are aware that CKD—which affects approximately 10% of the US population1—has a deleterious effect on cardiovascular disease, but many fail to recognize the impact it has on the hematopoietic system. Managing the anemia that accompanies CKD in patients like Mary requires a finely tuned diagnostic approach and treatment strategy. This article will help toward that end.
Anemia of CKD: A common problem
Anemia of CKD is one of the first signs of kidney dysfunction, yet it often goes undetected because of its insidious onset. Anemia develops gradually as kidney function declines and the GFR drops to 70 mL/min in male patients and 50 mL/min in females.2 Epidemiologic data indicate that two-thirds of patients in the early stages of kidney failure are also anemic, with a hemoglobin level of less than 11 g/dL, yet only one-third of these patients have ever received erythropoietin-stimulating agents (ESAs) to treat their anemia.1 The National Kidney Foundation Kidney Disease Outcomes Quality Initiative (NKF KDOQI) guidelines recommend that the evaluation of anemia of CKD begin in patients with a serum creatinine ≥2 mg/dL when the hemoglobin is <12 g/dL in adult males and postmenopausal females and <11 g/dL in premenopausal females.3
How kidney failure leads to anemia
Patients like Mary develop anemia of CKD because failing kidneys produce less erythropoietin (EPO) than the body requires for the production of red blood cells. EPO is an endogenous hormone produced by peritubular fibroblasts in the renal cortex.4 Most of this hormone (90%) is produced in the kidney, with the remainder manufactured by hepatocytes.
Erythropoiesis is stimulated by blood loss, decreased oxygen tension, and an increase in oxygen affinity, which leads to an increase in EPO production via upregulation of the EPO gene. In healthy individuals, detection of hypoxia by the kidney can result in a 1000-fold increase in EPO production.5 Patients with CKD don’t have that kind of robust response, and their EPO levels remain normal or below normal even when challenged by lack of oxygen. Anemia in CKD can also be caused by nutritional deficiencies, decreased red blood cell survival because of uremic toxins, oxidative stress, inflammation, and the use of angiotensin-converting enzyme (ACE) inhibitors.
Chronic anemia, CKD, and CV disease: A deadly triad
The leading cause of death in patients with CKD is cardiovascular disease. Patients with cardiorenal anemia syndrome develop a self-perpetuating triad that increases the risk of death when all 3 conditions are present. Anemic patients double their relative risk of death when CKD is present and triple their risk if they have anemia, CKD, and cardiovascular disease.6
Epidemiologic studies suggest an association among anemia, left ventricular hypertrophy (LVH), mortality, and cardiovascular outcomes. One study evaluated 2423 stage 3 and 4 CKD patients with anemia, defined as hemoglobin <13 g/dL in males and <12 g/dL in females. The results showed an increase in composite outcomes of myocardial infarction, stroke, and death.7 A prospective study evaluating 246 people with stages 2 to 4 CKD reported anemia to be an independent risk factor for the development of LVH.8 The stages of CKD are shown in the TABLE.
Suspected mechanisms of cardiovascular disease progression due to chronic anemia include tissue hypoxia, free radical formation, endothelial dysfunction, and vascular damage. Compensatory neurohumeral adaptations result in an increased sympathetic response and upregulation of the reninangiotensin-aldosterone system.9
TABLE
Stages of chronic kidney disease
| Stage | Description | GFR (mL/min/1.73 m2) |
|---|---|---|
| 1 | Kidney damage with normal or increased GFR | ≥90 |
| 2 | Kidney damage with mildly decreased GFR | 60-89 |
| 3 | Moderately decreased GFR | 30-59 |
| 4 | Severely decreased GFR | 15-29 |
| 5 | Kidney failure | <15 or dialysis |
| GFR, glomerular filtration rate. | ||
| Source: KDOQI Clinical Practice Guideline and Clinical Practice Recommendations for anemia in chronic kidney disease: 2007 update of hemoglobin target. Am J Kidney Dis. 2007.3 | ||
Anemia of CKD: A diagnosis of exclusion
Because anemia can have many causes, other possibilities must be ruled out before a diagnosis of CKD anemia can be made. Testing should be tailored to each individual situation, determined by a thorough history and physical. Steps in the diagnosis are shown in the FLOW CHART. A basic work-up should include complete blood count with differential, iron studies (ferritin, serum Fe, and total iron binding capacity), reticulocyte count, and a guaiac test. Other blood tests, such as thyroid-stimulating hormone (TSH), B12, and folate levels, and a hemolysis panel (lactate dehydrogenase, haptoglobin), should be obtained if the history suggests these disorders. A peripheral blood smear showing normocytic red blood cells with a normochromic pattern would favor the diagnosis of anemia of CKD.
FLOW CHART
A step-by-step guide to CKD anemia diagnosis and treatment
CBC, complete blood count; CKD, chronic kidney disease; ESA, erythropoietin-stimulating agents; R/O, rule out; TIBC/TSAT, total iron-binding capacity/transferrin saturation.
A look at the iron connection
Many patients with CKD anemia have iron deficiency and are unable to produce adequate numbers of red blood cells. Iron deficiency can have many causes: not enough iron-rich food in the diet, chronic bleeding, malabsorption, or an occult gastrointestinal malignancy. Once iron deficiency anemia is diagnosed, a colonoscopy is warranted to rule out occult malignancy. Ferritin, a protein found mostly in macrophages and hepatocytes, stores iron and serves as a marker for total iron stores. Using stored iron requires transferrin, a transporting protein, to shuttle iron from the reticuloendothelial system and gut to the bone marrow. CKD is a pro-inflammatory state that results in a limited ability to use iron stores. For this reason, patients with CKD require higher levels of iron.
Absolute iron deficiency. Iron deficiency in CKD patients with serum ferritin <100 ng/mL and transferrin saturation (TSAT) <20% is characterized as absolute iron deficiency. The TSAT represents the percent of iron bound to transferrin and is a good indicator of the body’s functional capacity to use stored iron.
Relative iron deficiency and iron block. Patients who do not respond to ESA therapy even though they have adequate iron stores are said to have a functional or relative iron deficiency. Iron block is a condition that results in anemia from a chronic inflammatory state such as infection, autoimmune disorders, or malignancies. It resolves once the inflammatory process abates. Both conditions have similar anemia profiles, with a serum ferritin >100 ng/mL and a TSAT <20%. Differentiating between these conditions requires dynamic testing using serial iron studies and observing responses to ESAs and iron supplementation.
Options for correcting iron deficiency
After a thorough history and physical with appropriate screening, you find that Mary has an iron deficiency that must be corrected before her anemia can be treated effectively. Treatment for iron deficiency is usually initiated with oral therapy, at the recommended dose of 200 mg oral elemental iron a day in 3 divided doses.
If the oral therapy does not correct iron deficiency within 3 months, or a patient cannot tolerate the constipation that is often a side effect of this therapy, IV iron administration can be considered. Because CKD patients do not have the ongoing iron losses seen in patients with end-stage renal disease (ESRD), a conservative approach using a single IV dose followed by repeat testing is warranted. The goal is to achieve ferritin levels >100 ng/dL and TSAT >20%. A number of products for IV iron administration are available. The most widely used are iron dextran (INFeD), ferric gluconate (Ferrlecit), and iron sucrose (Venofer).
Iron stores are replenished? Time to treat the anemia
When ferritin levels and TSAT show that iron deficiency has been corrected, ESA treatment for anemia can begin. Two major brands of ESAs currently in use in the United States are a recombinant human erythropoietin (rHuEPO) known as epoetin alfa (Procrit, Epogen), and darbepoetin alpha (Aranesp). Both medications are effective and can be given intravenously or subcutaneously. Subcutaneous darbepoetin alpha has a longer half-life compared with epoetin alpha (70 vs 24 hours), so dosing intervals can be longer.10,11 ESAs should not be started in patients with uncontrolled hypertension until the blood pressure is controlled, or in patients with an active malignancy unless the treatment is directly supervised by an oncologist.
Aim for complete anemia resolution? That’s controversial
Treatment of CKD anemia with ESAs is widely practiced, but controversy over whether it is beneficial to aim for complete resolution of anemia is ongoing. The CREATE (Cardiovascular Risk Reduction by Early Anemia Treatment) and CHOIR (Correction of Hemoglobin and Outcomes in Renal Insufficiency) trials published in 2006 failed to resolve the issue.12,13
In the CREATE trial, patients targeted to achieve normal hemoglobin levels did no better in avoiding cardiovascular events than patients targeted for lower levels. The CHOIR trial was stopped early because of an increased trend toward death and hospitalization for congestive heart failure in the group with therapy targeted to achieve normal hemoglobin levels.
The recently published TREAT (Trial to Reduce Cardiovascular Events with Aranesp Therapy) study of patients with type 2 diabetes and CKD showed no reduction in all-cause mortality, cardiovascular morbidity, or ESRD in patients receiving Aranesp targeted to achieve a hemoglobin level of approximately 13 g/dL, compared with placebo.14 The study did demonstrate, however, that patients receiving Aranesp were about twice as likely to have a stroke than the placebo subjects (101 vs 53)—which might lead clinicians to ponder whether the gains, if any, were worth the risk.
Revised labeling. Late last year, the US Food and Drug Administration approved a label change for Procrit and Aranesp, warning that patients with renal failure “experienced greater risks for death and serious cardiovascular events when administered ESAs to target higher vs lower hemoglobin levels” and advising physicians to “individualize dosing to achieve and maintain hemoglobin levels within the range of 10 to 12 g/dL.”10,11 The 2007 NKF KDOQI guidelines suggest maintaining a hemoglobin level between 11 and 12 g/dL and have not incorporated the results of the TREAT trial.
Some patients don’t respond to ESAs
Inadequate response to ESAs is most commonly caused by underdosing or inadequate iron stores. NKF KDOQI guidelines recommend checking TSAT and ferritin prior to initiating therapy and monitoring these levels every 3 months.3 True nonresponders are individuals with good iron stores who are unable to achieve target hemoglobin within 4 to 6 months despite receiving subcutaneous epoetin 300 IU/kg per week. Inadequate response to ESAs can be caused by ongoing occult blood loss, infection, inflammation, nutritional deficiencies, hemolysis, hemoglobinemias, aluminum toxicity, anti-EPO antibody, hyperparathyroidism, multiple myeloma, and bone marrow dysfunction.10,11 If patients do not respond to ESA therapy, the NKF KDOQI guidelines recommend referral to a nephrologist or hematologist.3
How did Mary fare?
Mary did well taking oral iron supplementation. Once her iron deficiency was corrected, you were able to begin treating her anemia. After appropriate titration of her ESA, she was able to maintain a hemoglobin level between 11 and 12 g/dL 4 months into therapy. On a follow-up visit, she had no side effects from the medication and reported an increase in her energy level.
CORRESPONDENCE
Jonathan Taliercio, DO, Cleveland Clinic, Department of Nephrology and Hypertension, 9500 Euclid Avenue, Cleveland, OH 44195; [email protected]
• Evaluate for chronic kidney disease (CKD) anemia when a patient has a serum creatinine ≥2 mg/dL and hemoglobin <12 g/dL (adult males and postmenopausal females) or <11 g/dL (premenopausal females). A
• Before you treat CKD anemia, correct any underlying iron deficiency. A
• Start anemia therapy with erythropoietin-stimulating agents when hemoglobin is ≤10 g/dL, and maintain target hemoglobin levels between 11 and 12 g/dL, in accordance with National Kidney Foundation guidelines. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE Mary J, a 65-year-old woman with stage 3 chronic kidney disease (CKD), is in your office for a follow-up appointment. Over the past 6 months, she has noticed a decrease in her energy level. On her routine blood work, you see that her hemoglobin has been slowly declining over the past year. It is now 9 g/dL and her estimated glomerular filtration rate (GFR) is 40 mL/min.
How would you evaluate Mary’s anemia, and would you suspect that it was related to her CKD?
Most physicians are aware that CKD—which affects approximately 10% of the US population1—has a deleterious effect on cardiovascular disease, but many fail to recognize the impact it has on the hematopoietic system. Managing the anemia that accompanies CKD in patients like Mary requires a finely tuned diagnostic approach and treatment strategy. This article will help toward that end.
Anemia of CKD: A common problem
Anemia of CKD is one of the first signs of kidney dysfunction, yet it often goes undetected because of its insidious onset. Anemia develops gradually as kidney function declines and the GFR drops to 70 mL/min in male patients and 50 mL/min in females.2 Epidemiologic data indicate that two-thirds of patients in the early stages of kidney failure are also anemic, with a hemoglobin level of less than 11 g/dL, yet only one-third of these patients have ever received erythropoietin-stimulating agents (ESAs) to treat their anemia.1 The National Kidney Foundation Kidney Disease Outcomes Quality Initiative (NKF KDOQI) guidelines recommend that the evaluation of anemia of CKD begin in patients with a serum creatinine ≥2 mg/dL when the hemoglobin is <12 g/dL in adult males and postmenopausal females and <11 g/dL in premenopausal females.3
How kidney failure leads to anemia
Patients like Mary develop anemia of CKD because failing kidneys produce less erythropoietin (EPO) than the body requires for the production of red blood cells. EPO is an endogenous hormone produced by peritubular fibroblasts in the renal cortex.4 Most of this hormone (90%) is produced in the kidney, with the remainder manufactured by hepatocytes.
Erythropoiesis is stimulated by blood loss, decreased oxygen tension, and an increase in oxygen affinity, which leads to an increase in EPO production via upregulation of the EPO gene. In healthy individuals, detection of hypoxia by the kidney can result in a 1000-fold increase in EPO production.5 Patients with CKD don’t have that kind of robust response, and their EPO levels remain normal or below normal even when challenged by lack of oxygen. Anemia in CKD can also be caused by nutritional deficiencies, decreased red blood cell survival because of uremic toxins, oxidative stress, inflammation, and the use of angiotensin-converting enzyme (ACE) inhibitors.
Chronic anemia, CKD, and CV disease: A deadly triad
The leading cause of death in patients with CKD is cardiovascular disease. Patients with cardiorenal anemia syndrome develop a self-perpetuating triad that increases the risk of death when all 3 conditions are present. Anemic patients double their relative risk of death when CKD is present and triple their risk if they have anemia, CKD, and cardiovascular disease.6
Epidemiologic studies suggest an association among anemia, left ventricular hypertrophy (LVH), mortality, and cardiovascular outcomes. One study evaluated 2423 stage 3 and 4 CKD patients with anemia, defined as hemoglobin <13 g/dL in males and <12 g/dL in females. The results showed an increase in composite outcomes of myocardial infarction, stroke, and death.7 A prospective study evaluating 246 people with stages 2 to 4 CKD reported anemia to be an independent risk factor for the development of LVH.8 The stages of CKD are shown in the TABLE.
Suspected mechanisms of cardiovascular disease progression due to chronic anemia include tissue hypoxia, free radical formation, endothelial dysfunction, and vascular damage. Compensatory neurohumeral adaptations result in an increased sympathetic response and upregulation of the reninangiotensin-aldosterone system.9
TABLE
Stages of chronic kidney disease
| Stage | Description | GFR (mL/min/1.73 m2) |
|---|---|---|
| 1 | Kidney damage with normal or increased GFR | ≥90 |
| 2 | Kidney damage with mildly decreased GFR | 60-89 |
| 3 | Moderately decreased GFR | 30-59 |
| 4 | Severely decreased GFR | 15-29 |
| 5 | Kidney failure | <15 or dialysis |
| GFR, glomerular filtration rate. | ||
| Source: KDOQI Clinical Practice Guideline and Clinical Practice Recommendations for anemia in chronic kidney disease: 2007 update of hemoglobin target. Am J Kidney Dis. 2007.3 | ||
Anemia of CKD: A diagnosis of exclusion
Because anemia can have many causes, other possibilities must be ruled out before a diagnosis of CKD anemia can be made. Testing should be tailored to each individual situation, determined by a thorough history and physical. Steps in the diagnosis are shown in the FLOW CHART. A basic work-up should include complete blood count with differential, iron studies (ferritin, serum Fe, and total iron binding capacity), reticulocyte count, and a guaiac test. Other blood tests, such as thyroid-stimulating hormone (TSH), B12, and folate levels, and a hemolysis panel (lactate dehydrogenase, haptoglobin), should be obtained if the history suggests these disorders. A peripheral blood smear showing normocytic red blood cells with a normochromic pattern would favor the diagnosis of anemia of CKD.
FLOW CHART
A step-by-step guide to CKD anemia diagnosis and treatment
CBC, complete blood count; CKD, chronic kidney disease; ESA, erythropoietin-stimulating agents; R/O, rule out; TIBC/TSAT, total iron-binding capacity/transferrin saturation.
A look at the iron connection
Many patients with CKD anemia have iron deficiency and are unable to produce adequate numbers of red blood cells. Iron deficiency can have many causes: not enough iron-rich food in the diet, chronic bleeding, malabsorption, or an occult gastrointestinal malignancy. Once iron deficiency anemia is diagnosed, a colonoscopy is warranted to rule out occult malignancy. Ferritin, a protein found mostly in macrophages and hepatocytes, stores iron and serves as a marker for total iron stores. Using stored iron requires transferrin, a transporting protein, to shuttle iron from the reticuloendothelial system and gut to the bone marrow. CKD is a pro-inflammatory state that results in a limited ability to use iron stores. For this reason, patients with CKD require higher levels of iron.
Absolute iron deficiency. Iron deficiency in CKD patients with serum ferritin <100 ng/mL and transferrin saturation (TSAT) <20% is characterized as absolute iron deficiency. The TSAT represents the percent of iron bound to transferrin and is a good indicator of the body’s functional capacity to use stored iron.
Relative iron deficiency and iron block. Patients who do not respond to ESA therapy even though they have adequate iron stores are said to have a functional or relative iron deficiency. Iron block is a condition that results in anemia from a chronic inflammatory state such as infection, autoimmune disorders, or malignancies. It resolves once the inflammatory process abates. Both conditions have similar anemia profiles, with a serum ferritin >100 ng/mL and a TSAT <20%. Differentiating between these conditions requires dynamic testing using serial iron studies and observing responses to ESAs and iron supplementation.
Options for correcting iron deficiency
After a thorough history and physical with appropriate screening, you find that Mary has an iron deficiency that must be corrected before her anemia can be treated effectively. Treatment for iron deficiency is usually initiated with oral therapy, at the recommended dose of 200 mg oral elemental iron a day in 3 divided doses.
If the oral therapy does not correct iron deficiency within 3 months, or a patient cannot tolerate the constipation that is often a side effect of this therapy, IV iron administration can be considered. Because CKD patients do not have the ongoing iron losses seen in patients with end-stage renal disease (ESRD), a conservative approach using a single IV dose followed by repeat testing is warranted. The goal is to achieve ferritin levels >100 ng/dL and TSAT >20%. A number of products for IV iron administration are available. The most widely used are iron dextran (INFeD), ferric gluconate (Ferrlecit), and iron sucrose (Venofer).
Iron stores are replenished? Time to treat the anemia
When ferritin levels and TSAT show that iron deficiency has been corrected, ESA treatment for anemia can begin. Two major brands of ESAs currently in use in the United States are a recombinant human erythropoietin (rHuEPO) known as epoetin alfa (Procrit, Epogen), and darbepoetin alpha (Aranesp). Both medications are effective and can be given intravenously or subcutaneously. Subcutaneous darbepoetin alpha has a longer half-life compared with epoetin alpha (70 vs 24 hours), so dosing intervals can be longer.10,11 ESAs should not be started in patients with uncontrolled hypertension until the blood pressure is controlled, or in patients with an active malignancy unless the treatment is directly supervised by an oncologist.
Aim for complete anemia resolution? That’s controversial
Treatment of CKD anemia with ESAs is widely practiced, but controversy over whether it is beneficial to aim for complete resolution of anemia is ongoing. The CREATE (Cardiovascular Risk Reduction by Early Anemia Treatment) and CHOIR (Correction of Hemoglobin and Outcomes in Renal Insufficiency) trials published in 2006 failed to resolve the issue.12,13
In the CREATE trial, patients targeted to achieve normal hemoglobin levels did no better in avoiding cardiovascular events than patients targeted for lower levels. The CHOIR trial was stopped early because of an increased trend toward death and hospitalization for congestive heart failure in the group with therapy targeted to achieve normal hemoglobin levels.
The recently published TREAT (Trial to Reduce Cardiovascular Events with Aranesp Therapy) study of patients with type 2 diabetes and CKD showed no reduction in all-cause mortality, cardiovascular morbidity, or ESRD in patients receiving Aranesp targeted to achieve a hemoglobin level of approximately 13 g/dL, compared with placebo.14 The study did demonstrate, however, that patients receiving Aranesp were about twice as likely to have a stroke than the placebo subjects (101 vs 53)—which might lead clinicians to ponder whether the gains, if any, were worth the risk.
Revised labeling. Late last year, the US Food and Drug Administration approved a label change for Procrit and Aranesp, warning that patients with renal failure “experienced greater risks for death and serious cardiovascular events when administered ESAs to target higher vs lower hemoglobin levels” and advising physicians to “individualize dosing to achieve and maintain hemoglobin levels within the range of 10 to 12 g/dL.”10,11 The 2007 NKF KDOQI guidelines suggest maintaining a hemoglobin level between 11 and 12 g/dL and have not incorporated the results of the TREAT trial.
Some patients don’t respond to ESAs
Inadequate response to ESAs is most commonly caused by underdosing or inadequate iron stores. NKF KDOQI guidelines recommend checking TSAT and ferritin prior to initiating therapy and monitoring these levels every 3 months.3 True nonresponders are individuals with good iron stores who are unable to achieve target hemoglobin within 4 to 6 months despite receiving subcutaneous epoetin 300 IU/kg per week. Inadequate response to ESAs can be caused by ongoing occult blood loss, infection, inflammation, nutritional deficiencies, hemolysis, hemoglobinemias, aluminum toxicity, anti-EPO antibody, hyperparathyroidism, multiple myeloma, and bone marrow dysfunction.10,11 If patients do not respond to ESA therapy, the NKF KDOQI guidelines recommend referral to a nephrologist or hematologist.3
How did Mary fare?
Mary did well taking oral iron supplementation. Once her iron deficiency was corrected, you were able to begin treating her anemia. After appropriate titration of her ESA, she was able to maintain a hemoglobin level between 11 and 12 g/dL 4 months into therapy. On a follow-up visit, she had no side effects from the medication and reported an increase in her energy level.
CORRESPONDENCE
Jonathan Taliercio, DO, Cleveland Clinic, Department of Nephrology and Hypertension, 9500 Euclid Avenue, Cleveland, OH 44195; [email protected]
1. United States Renal Data System, USRDS. 2009 Annual Data Report. Atlas of Chronic Kidney Disease in the United States. Bethesda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; 2009.
2. Hsu CJ, McCulloch CE, Curhan GC. Epidemiology of anemia associated with chronic renal insufficiency among adults in the United States: results from the Third National Health and Nutrition Examination Survey. J Am Soc Nephrol. 2002;13:504-510.
3. KDOQI Clinical Practice Guideline and Clinical Practice Recommendations for anemia in chronic kidney disease: 2007 update of hemoglobin target. Am J Kidney Dis. 2007;50:471-530.
4. Donnelly S. Why is erythropoietin made in the kidney? The kidney functions as a critmeter. Am J Kidney Dis. 2001;38:415-425.
5. Ebert B, Franklin H. Regulation of the erythropoietin gene. Blood. 1999;94:1864-1877.
6. Silverberg D, Wexler D, Blum M, et al. The cardio-renal anaemia syndrome: does it exist? Nephrol Dial Transplant. 2003;18(suppl 8):viii 7-viii 12.
7. Weiner D, Tighiouart H, Vlagopoulos P, et al. Effects of anemia and left ventricular hypertrophy on cardiovascular disease in patients with chronic kidney disease. J Am Soc Nephrol. 2005;16:1803-1810.
8. Levin A, Thompson C, Ethier J, et al. Left ventricular mass index increase in early renal disease: impact of decline in hemoglobin. Am J Kidney Dis. 1999;34:125-134.
9. Rao M, Pereira B. Optimal anemia management reduces cardiovascular morbidity, mortality, and costs in chronic kidney disease. Kidney Int. 2005;68:1432-1438.
10. Amgen. Aranesp (Darbepoetin Alpha) package insert. Available at www.aranesp.com/professional/crf/full_prescribing_info/pi.jsp. Accessed November 16, 2009.
11. Amgen. Procrit (Epoetin Alpha) package insert. Available at www.procrit.com/sites/default/files/shared/OBI/PI/ProcritBooklet.pdf#page=1. Accessed November 16, 2009.
12. Drueke T, Locatelli F, Clyne N, et al. Normalization of hemoglobin level in patients with CKD and anemia. N Engl J Med. 2006;355:2071-2084.
13. Singh A, Szczech L, Tang K. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med. 2006;355:2085-2098.
14. Pfeffer MA, Burdmann EA, Chen CY, et al. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med. 2009;361:2019-2032.
1. United States Renal Data System, USRDS. 2009 Annual Data Report. Atlas of Chronic Kidney Disease in the United States. Bethesda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; 2009.
2. Hsu CJ, McCulloch CE, Curhan GC. Epidemiology of anemia associated with chronic renal insufficiency among adults in the United States: results from the Third National Health and Nutrition Examination Survey. J Am Soc Nephrol. 2002;13:504-510.
3. KDOQI Clinical Practice Guideline and Clinical Practice Recommendations for anemia in chronic kidney disease: 2007 update of hemoglobin target. Am J Kidney Dis. 2007;50:471-530.
4. Donnelly S. Why is erythropoietin made in the kidney? The kidney functions as a critmeter. Am J Kidney Dis. 2001;38:415-425.
5. Ebert B, Franklin H. Regulation of the erythropoietin gene. Blood. 1999;94:1864-1877.
6. Silverberg D, Wexler D, Blum M, et al. The cardio-renal anaemia syndrome: does it exist? Nephrol Dial Transplant. 2003;18(suppl 8):viii 7-viii 12.
7. Weiner D, Tighiouart H, Vlagopoulos P, et al. Effects of anemia and left ventricular hypertrophy on cardiovascular disease in patients with chronic kidney disease. J Am Soc Nephrol. 2005;16:1803-1810.
8. Levin A, Thompson C, Ethier J, et al. Left ventricular mass index increase in early renal disease: impact of decline in hemoglobin. Am J Kidney Dis. 1999;34:125-134.
9. Rao M, Pereira B. Optimal anemia management reduces cardiovascular morbidity, mortality, and costs in chronic kidney disease. Kidney Int. 2005;68:1432-1438.
10. Amgen. Aranesp (Darbepoetin Alpha) package insert. Available at www.aranesp.com/professional/crf/full_prescribing_info/pi.jsp. Accessed November 16, 2009.
11. Amgen. Procrit (Epoetin Alpha) package insert. Available at www.procrit.com/sites/default/files/shared/OBI/PI/ProcritBooklet.pdf#page=1. Accessed November 16, 2009.
12. Drueke T, Locatelli F, Clyne N, et al. Normalization of hemoglobin level in patients with CKD and anemia. N Engl J Med. 2006;355:2071-2084.
13. Singh A, Szczech L, Tang K. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med. 2006;355:2085-2098.
14. Pfeffer MA, Burdmann EA, Chen CY, et al. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med. 2009;361:2019-2032.
Low bone density is not always bisphosphonate deficiency

A few common conditions dramatically underscore the potential difficulties in distinguishing the hyperosteolytic biology of osteoporosis from disorders of bone hypoproduction or defective mineralization. Patients with a severe or complicated gastrointestinal malabsorptive state such as a history of gastric bypass surgery are included in this group. Management of transplantation patients may also be challenging.
But perhaps the most complicated metabolic bone patients to manage are those with severe chronic kidney disease. In this issue of the Journal, Dr. Paul Miller and, in an accompanying commentary, Dr. Maria Coco discuss the problems, some potential bone-protective strategies, and some of the controversies faced by clinicians treating bone disease in patients with chronic kidney disease.
While patients with chronic kidney disease who have low T scores are often comanaged by nephrologists and specialists in metabolic bone disease, the discussion of the pathophysiologic pathways resulting in reduced bone density is germane to many of us. A documented low T score does not equal osteoporosis and thus should not lead us to automatically prescribe a bisphosphonate.
Clues to the presence of a disease associated with secondary osteoporosis or osteomalacia should be sought in any patient with a low T score. Some of these clues are adenopathy on examination, a personal or striking family history of nephrolithiasis, unexplained anemia, thyroid disease, a low anion gap, an unexplained change in blood pressure, a particularly alkaline urine, frequent loose stools, and disturbances of phosphate or calcium.
The era of ignoring osteoporosis is fortunately coming to a close. But we should not cavalierly go where the generation of internists before us could not go—to our prescription pads. Low bone density is not a one-size-fits-all disorder. We need to carefully consider other diagnostic and therapeutic options before assuming that low bone density is due to osteoporosis in every patient. These two articles should stimulate serious thought about possible alternative diagnoses to the now frequently diagnosed “osteoporosis.”
A few common conditions dramatically underscore the potential difficulties in distinguishing the hyperosteolytic biology of osteoporosis from disorders of bone hypoproduction or defective mineralization. Patients with a severe or complicated gastrointestinal malabsorptive state such as a history of gastric bypass surgery are included in this group. Management of transplantation patients may also be challenging.
But perhaps the most complicated metabolic bone patients to manage are those with severe chronic kidney disease. In this issue of the Journal, Dr. Paul Miller and, in an accompanying commentary, Dr. Maria Coco discuss the problems, some potential bone-protective strategies, and some of the controversies faced by clinicians treating bone disease in patients with chronic kidney disease.
While patients with chronic kidney disease who have low T scores are often comanaged by nephrologists and specialists in metabolic bone disease, the discussion of the pathophysiologic pathways resulting in reduced bone density is germane to many of us. A documented low T score does not equal osteoporosis and thus should not lead us to automatically prescribe a bisphosphonate.
Clues to the presence of a disease associated with secondary osteoporosis or osteomalacia should be sought in any patient with a low T score. Some of these clues are adenopathy on examination, a personal or striking family history of nephrolithiasis, unexplained anemia, thyroid disease, a low anion gap, an unexplained change in blood pressure, a particularly alkaline urine, frequent loose stools, and disturbances of phosphate or calcium.
The era of ignoring osteoporosis is fortunately coming to a close. But we should not cavalierly go where the generation of internists before us could not go—to our prescription pads. Low bone density is not a one-size-fits-all disorder. We need to carefully consider other diagnostic and therapeutic options before assuming that low bone density is due to osteoporosis in every patient. These two articles should stimulate serious thought about possible alternative diagnoses to the now frequently diagnosed “osteoporosis.”
A few common conditions dramatically underscore the potential difficulties in distinguishing the hyperosteolytic biology of osteoporosis from disorders of bone hypoproduction or defective mineralization. Patients with a severe or complicated gastrointestinal malabsorptive state such as a history of gastric bypass surgery are included in this group. Management of transplantation patients may also be challenging.
But perhaps the most complicated metabolic bone patients to manage are those with severe chronic kidney disease. In this issue of the Journal, Dr. Paul Miller and, in an accompanying commentary, Dr. Maria Coco discuss the problems, some potential bone-protective strategies, and some of the controversies faced by clinicians treating bone disease in patients with chronic kidney disease.
While patients with chronic kidney disease who have low T scores are often comanaged by nephrologists and specialists in metabolic bone disease, the discussion of the pathophysiologic pathways resulting in reduced bone density is germane to many of us. A documented low T score does not equal osteoporosis and thus should not lead us to automatically prescribe a bisphosphonate.
Clues to the presence of a disease associated with secondary osteoporosis or osteomalacia should be sought in any patient with a low T score. Some of these clues are adenopathy on examination, a personal or striking family history of nephrolithiasis, unexplained anemia, thyroid disease, a low anion gap, an unexplained change in blood pressure, a particularly alkaline urine, frequent loose stools, and disturbances of phosphate or calcium.
The era of ignoring osteoporosis is fortunately coming to a close. But we should not cavalierly go where the generation of internists before us could not go—to our prescription pads. Low bone density is not a one-size-fits-all disorder. We need to carefully consider other diagnostic and therapeutic options before assuming that low bone density is due to osteoporosis in every patient. These two articles should stimulate serious thought about possible alternative diagnoses to the now frequently diagnosed “osteoporosis.”
Fragility fractures in chronic kidney disease: An opinion-based approach

But even in chronic kidney disease, many fractures are due to postmenopausal or agerelated osteoporosis, and estrogen-deficiency osteoporosis is the most common cause of fragility fractures overall.1–3 Osteoporosis can be diagnosed only after other causes of skeletal fragility have been ruled out. And how to diagnose and treat osteoporosis in the most severe stage of kidney disease is a matter of opinion, as we have almost no data to guide us.
Nevertheless, in the pages that follow, I will outline my admittedly opinion-based approach to diagnosing and managing the causes of fragility fractures in patients with chronic kidney disease.
T SCORES DO NOT DISTINGUISH THE CAUSES OF FRAGILITY
The most common cause of fragility fractures is osteoporosis due to estrogen deficiency.1–3 But since many other medical conditions can lead to osteoporosis, simple diagnostic criteria have been difficult to find.
Before 1994, the diagnosis of osteoporosis was made on the basis of low-trauma fractures.4 Now, we use the World Health Organization criteria,5 based on bone mineral density T scores:
- Normal—a T score of −1.0 standard deviations or higher
- Osteopenia—a T score of less than −1.0 but higher than −2.5
- Osteoporosis—a T score of −2.5 or less
- Severe osteoporosis—a T score of −2.5 or less with a fragility fracture.
However, fractures can also be due to metabolic bone diseases that are not osteoporosis, including renal bone diseases.6–7 While a low T score or a fracture provides a working diagnosis of osteoporosis, it does not help distinguish the different types of osteoporosis and nonosteoporotic metabolic bone diseases. For example, osteomalacia and osteogenesis imperfecta can also cause fragility fractures and can be associated with low bone density. Using these criteria to define osteoporosis is even more problematic in patients with chronic kidney disease.
FIVE STAGES OF CHRONIC KIDNEY DISEASE
The National Kidney Foundation8 classifies the severity of chronic kidney disease on the basis of the glomerular filtration rate (GFR), as measured by 24-hour urine for creatinine clearance, or as estimated by the Cockcroft-Gault equation or, preferably, the Modification of Diet in Renal Disease (MDRD) equation (calculators are available at www.kidney.org/professionals/kdoqi/gfr_calculator.cfm):
- Stage 1—GFR 90 mL/minute/1.73 m2 or higher
- Stage 2—GFR 60 to 89
- Stage 3—GFR 30 to 59
- Stage 4—GFR 15 to 29
- Stage 5—GFR lower than 15, or if the patient is on dialysis. (Another stage, called 5D, was added to the list to denote patients on dialysis, since the metabolic derangements in bone and systemic biology may differ between patients on dialysis vs those not on dialysis.)
This staging system is relevant to the discussion of bone fragility that follows.
CHRONIC KIDNEY DISEASE IS COMMON IN THE ELDERLY
The third National Health and Nutrition Examination Survey9 found that, at least as estimated by the Cockcroft-Gault equation, the GFR declines with age, so that by the age of 70 at least 20% of the US population has stage 4 or 5 chronic kidney disease.
Although the Cockcroft-Gault and MDRD equations may yield lower GFR values in the general population than one would get by measuring creatinine, inulin, or iothalamate clearance,10,11 the point is that both osteoporosis and chronic kidney disease are common.
THE GAMUT OF RENAL OSTEODYSTROPHY
In kidney failure (stage 5 chronic kidney disease), all forms of renal osteodystrophy may be associated with fragility fractures. Renal osteodystrophy can be defined by quantitative bone histomorphometry.12,13 The systemic conditions that may be associated with the bone disease and systemic vascular disease (chronic kidney disease–mineral and bone disorder) are characterized by one or more of the following14:
- Abnormalities of calcium, phosphorus, parathyroid hormone, or vitamin D metabolism
- Abnormalities in bone turnover, mineralization, volume, linear growth, or strength
- Vascular or other soft-tissue calcification.
The National Kidney Foundation14 classifies renal osteodystrophy on the basis of:
- Turnover—high, normal, or low
- Mineralization—normal or abnormal
- Volume—high, normal, or low.
Although this system helps us understand these diseases better, it does not provide a working diagnosis of osteoporosis.14
WHAT IS OSTEOPOROSIS?
In an attempt to define osteoporosis by a pathophysiologic mechanism, the National Institutes of Health15 have held two consensus conferences and have stated that “osteoporosis is a skeletal disorder characterized by impairment in bone strength predisposing a person to an increased risk of fracture. Bone strength primarily reflects the integration of bone density and bone quality.”15 However, the consensus statement also does not provide a working diagnosis of osteoporosis—one that clinicians can apply to management decisions, and one that is also accepted by the US International Classification of Disease codes for reimbursement purposes.
The 1994 World Health Organization criteria offer the most pragmatic operational definition of osteoporosis, and they can be applied in both men and women, as well as in younger patients with medical conditions associated with increased risk of low-trauma fracture.5,16 Although the main purpose of these criteria was to advise international health authorities of the potential future economic impact of osteoporosis, the T score also became the pragmatic diagnostic threshold for defining normal, osteopenia, and osteoporosis in clinical practice.
The T score also calls attention to an important observation: of people who have fractures and subsequently undergo bone densitometry, more are found to have osteopenia than osteoporosis. The reasons are that there are more people with osteopenia than osteoporosis,17,18 and many other factors independent of low bone mineral density contribute to bone strength.19,20
How is osteoporosis diagnosed in stage 1–3 chronic kidney disease?
In patients with chronic kidney disease who develop fragility fractures, the reasonable question is: Is the cause of the fracture osteoporosis or some other metabolic bone disease associated with chronic kidney disease?
The National Kidney Foundation guidelines14 say that the diagnosis of osteoporosis can be established in patients with stage 1, 2, or 3 chronic kidney disease on the basis of either of the World Health Organization criteria, ie, a T score of −2.5 or lower or fragility fractures, as in the postmenopausal population, as long as there are no biochemical abnormalities that suggest chronic kidney disease–mineral and bone disorder.
How is osteoporosis diagnosed in stage 4 or 5 chronic kidney disease?
The answer is neither straightforward nor clearly defined in severe (stage 4 or 5) chronic kidney disease.
In stage 5 and especially in patients on dialysis, the derangements in bone and mineral metabolism become serious enough to impair bone strength and increase the risk of lowtrauma fractures. The risk of hip fracture in stage 5 may be four times higher than in agematched controls.21–24
Adynamic, severe hyperparathyroid bone disease as well as osteomalacia can be associated with a higher risk of fragility fractures than in aged-matched controls in population studies of postmenopausal women or elderly men. These are bone fragility conditions that are not osteoporosis but that can mimic osteoporosis by the World Health Organization criteria.
Thus, when a patient in stage 5 has severe fragility fractures that by themselves may be life-threatening, it is reasonable to ask if the drugs that reduce the risk of fractures in many other osteoporotic conditions (postmenopausal, steroid-induced, elderly male osteoporosis, after solid organ transplantation) can also be used in patients with advanced chronic kidney disease.
The diagnosis of osteoporosis in these patients has no universally accepted criteria. The diagnosis is best suggested by excluding other forms of renal osteodystrophy by quantitative histomorphometry or by attempting to classify the form of renal osteodystrophy by noninvasive means of assessing bone turnover, mineralization, and volume. However, we lack clinical tools to make these distinctions in individual patients.
While many promising radiologic techniques that examine bone microarchitecture offer hope of being able to define turnover, mineralization, and volume noninvasively in severe chronic kidney disease, they are investigational and unproven at this time in discriminating between renal osteodystrophy and osteoporosis.6,25–27 As we increase our understanding of the relationships between turnover, mineralization, volume, and bone strength, these noninvasive imaging technologies may become the means to correlate turnover, mineralization, and volume to bone strength and open up an entirely new way to classify skeletal strength.
In the meantime, the clinician is left with quantitative bone histomorphometry (which requires biopsy) and biochemical markers of bone turnover to characterize the bone disease that may be responsible for low-trauma fractures in stage 5 chronic kidney disease. The clinician should first use biochemical markers before bone biopsy to distinguish the form of renal osteodystrophy, as this distinction may be able to prevent unnecessary biopsy.
Biochemical markers of bone metabolism
In chronic kidney disease, the bone biochemical tests that nephrologists usually assess during the course of declining renal function are the serum levels of:
- Phosphorus
- Parathyroid hormone
- Calcium
- Other electrolytes
- Total alkaline phosphatase or bone-specific alkaline phosphatase
- 1,25 dihydroxyvitamin D.
In postmenopausal osteoporosis, the biochemical markers of bone turnover that are measured to reflect baseline levels of bone turnover or change in bone turnover in response to drug therapy are:
- The serum or urine collagen cross-links N-telopeptide (NTx) and C-telopeptide (CTx), markers of bone resorption
- Bone-specific alkaline phosphatase (an osteoblast activity marker)
- Serum osteocalcin, a bone formation marker
- Propeptide type 1 collagen (P1NP), a marker of osteoblast activity, highly correlated with bone formation
- 25-hydroxyvitamin D levels.
Biochemical markers of bone turnover cannot be used to diagnose osteoporosis. They can, however, provide clinical guidance as to whether a patient has high or low bone turnover and whether therapy is affecting bone turnover.28–36 Although these markers have value in making these distinctions in groups of patients, they are less sensitive and specific for classifying an individual patient’s bone turnover status.
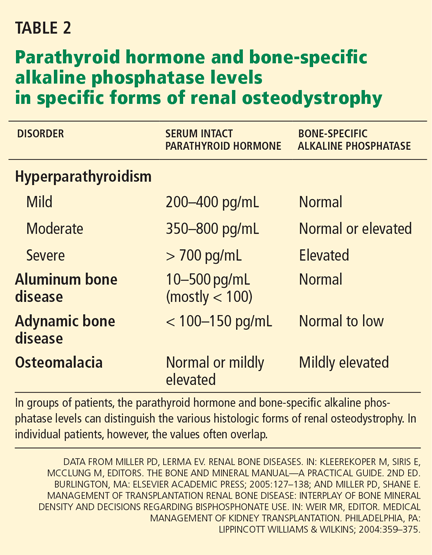
Bone-specific alkaline phosphatase, parathyroid hormone, and adynamic bone disease
If a patient’s bone-specific alkaline phosphatase level is elevated, adynamic bone disease is highly unlikely. Assuming that other causes of this elevated level (eg, Paget disease of bone, metastatic cancer) have already been excluded, the elevated level could represent either osteomalacia or hyperparathyroid bone disease.
However, a “normal” bone-specific alkaline phosphatase level does not exclude adynamic bone disease, whereas a low level is more often associated with low bone turnover.
An elevated parathyroid hormone level does not exclude adynamic renal bone disease, but a low level (< 150 pg/mL) suggests a lowbone-turnover state. A level six times or more greater than the upper limit of normal is far more likely to be associated with high bone turnover.
Thus, in clinical practice, patients with stage 4 or 5 chronic kidney disease who have elevated bone-specific alkaline phosphatase or very high parathyroid hormone values do not have adynamic bone disease. Furthermore, once other causes of these aberrant biochemical abnormalities have been defined, then “high-bone-turnover osteoporosis” may be a consideration. Certainly, in my opinion, if bone turnover markers suggest low bone turnover, bone biopsy is necessary before starting an antiresorptive agent.35
Quantitative bone histomorphometry
Double tetracycline-labeled quantitative histomorphometry is still the only accepted way to measure turnover, mineralization, and volume in clinical practice.43–45 A committee of the American Society for Bone and Mineral Research has developed histomorphometric criteria for distinguishing among the different types of metabolic bone diseases (osteomalacia, adynamic bone disease, hyperparathyroid bone disease).12 These criteria can be used to distinguish among the various metabolic bone diseases that accompany stage 5 chronic kidney disease, including adynamic bone disease.43,46–48
For patients in stage 5 who have had a fragility fracture, adynamic bone disease should be excluded before the off-label use of an osteoporosis drug that reduces bone turnover, such as a bisphosphonate, calcitonin, estrogen, a selective estrogen receptor modulator, or denosumab (anti-RANK ligand antibody). While there is no evidence, for example, that starting a bisphosphonate in a patient who already has adynamic bone disease is detrimental to either bone strength or systemic vascular calcification (which may be linked to low bone turnover),49 it seems unreasonable to do so until solid prospective data clarify the harm or benefit.50 Preliminary experimental and clinical data suggest that bisphosphonates may even reduce progression of extraosseous calcification and inhibit the development of atherosclerosis.50
Hence, quantitative bone histomorphometry can discriminate among the various forms of renal osteodystrophy. If a distinct form of renal osteodystrophy is not present in a patient with stage 4 or 5 chronic kidney disease who has had a fracture and who, on biopsy, has a low trabecular bone volume, the patient probably has osteoporosis by exclusion.
TREATING OSTEOPOROSIS IN STAGE 1–3 CHRONIC KIDNEY DISEASE
As previously mentioned, patients who have fragility fractures in stage 1, 2, or 3 chronic kidney disease are more likely to have osteoporosis than renal osteodystrophy as the cause of their impaired bone strength. Although several articles have described a higher risk of fragility fractures in patients with age-related reduction in renal function than in agematched patients with normal renal function, the specific metabolic bone disease other than osteoporosis accounting for this bone fragility has not been defined.6
Hence, patients with osteoporosis who are in stage 1, 2, or 3 chronic kidney disease and do not have a known biochemical abnormality that might suggest some form of renal osteodystrophy can and should be considered for treatment with approved drugs that reduce the risk of fractures in postmenopausal, male, or glucocorticoid-induced osteoporosis.51–53 In clinical trials, these agents were shown to be effective in patients with serum creatinine concentrations as high as 2.0 mg/dL or a GFR as low as 30 mL/min, as estimated by the Cockcroft-Gault equation.
While all of the approved agents show evidence of reducing the risk of vertebral fractures, patients at higher risk of fractures or those who have already suffered a nonvertebral fracture are more often considered candidates for treatment with a bisphosphonate or teriparatide (Forteo), both of which have shown evidence of reducing the risk of all fractures.
Bisphosphonates in stage 1–3 chronic kidney disease
There is prospective evidence that patients with an age-related reduction in GFR down to 30 mL/min benefit from oral and intravenous bisphosphonates, since all of the clinical trials that led to the approval of bisphosphonates included patients with GFRs as low as this.54–57 Bisphosphonates seem to have an excellent safety profile as measured by renal adverse events in patients with a GFR of 30 mL/min or greater.52–59
From the intravenous bisphosphonate studies, it appears that ibandronate (Boniva) at the approved dose of 3 mg intravenously every 3 months and zoledronic acid (Reclast) 5 mg once a year given over 15 minutes are safe in patients with a GFR greater than 30 or 35 mL/min.
However, the safety of these drugs might not be the same in patients with preexisting renal parenchymal disease (eg, in diabetes) or in patients using other agents that could affect renal function (eg, nonsteroidal antiinflammatory drugs). Therefore, caution is still needed when deciding to use intravenous bisphosphonates in specific higher-risk renal subpopulations.
In the clinical trials of zoledronic acid, a substantial proportion of patients had diabetes, and no difference was seen in adverse renal effects between diabetic and nondiabetic patients. Also, GFRs declined equally between the treated and placebo groups over time and were no different at the end of 3 years.55 However, in patients in whom serum creatinine was measured 9 to 11 days after the infusion of zoledronic acid, there was a small but statistically significant transient increase in serum creatinine concentration (0.5–2.0 mg/dL above baseline) after the second annual infusion. 58 The serum creatinine concentrations returned to their baseline values in all of these patients before the next annual infusion.
It is important that infusions of zoledronic acid be given no faster than over 15 minutes. More rapid infusion has been associated with acute renal failure, suggesting that the tubular damage that mimics acute tubular necrosis is related to the maximal concentration and not to the area under the curve. I infuse zoledronic acid over 30 minutes in patients with normal renal function or in those with stage 1, 2, or 3 chronic kidney disease.
Teriparatide
Teriparatide’s approval trial did not require baseline measurements of GFR, but patients were enrolled only if their baseline serum creatinine concentrations were less than 2.0 mg/dL.60 In a post hoc analysis, a small subset of patients had GFRs as low as 30 mL/min as estimated by the Cockcroft-Gault equation. In these patients, teriparatide 20 or 40 μg/day had an anabolic effect as measured by increases in osteoblast activity markers and bone mineral density, similar to that seen in patients with higher estimated GFRs and without any adverse renal effects.61
There are no data on using teriparatide in stage 4 or 5 chronic kidney disease, and I emphasize that in all of the clinical trials of teriparatide, all patients, even those with estimated GFRs as low as 30 mL/min, had normal baseline serum intact parathyroid hormone levels. It is possible that the bone biologic response could differ between patients with chronic kidney disease who have an elevated as compared with a normal serum parathyroid hormone level. This issue should be investigated.
TREATING OSTEOPOROSIS IN STAGE 4 OR 5 CHRONIC KIDNEY DISEASE
Treatment decisions are more difficult in patients with stage 4 and especially stage 5 chronic kidney disease who have had fragility fractures. This is even the case when the clinician has determined to the best of his or her ability that the patient has osteoporosis rather than renal osteodystrophy.
There are no prospective data showing any of the approved drugs to be effective in treating osteoporosis in patients whose GFRs are lower than 30 mL/min. However, a post hoc analysis of pooled data from nine clinical trials62 found that risedronate (Actonel) 5 mg/day reduced the incidence of new vertebral fractures. Another post hoc analysis, from the Fracture Intervention Trial,63 found that alendronate (Fosamax) 5 mg/day for the first 2 years and 10 mg/day for the 3rd year reduced the incidence of all clinical fractures. In neither of these post hoc analyses did the drug affect the serum creatinine concentration. The patients—postmenopausal women—had GFRs as low as 15 mL/min as estimated by the Cockcroft-Gault equation. Similar post hoc data have been published on raloxifene (Evista).64
There are no data on the efficacy (reduction in fracture risk) or safety of any bisphosphonate in patients with GFRs lower than 15 mL/min (stage 5 chronic kidney disease). Nevertheless, the question often arises when fragility fractures occur in this population. Here, only opinion and controversy exist, and we fervently await good science and randomized prospective data.
How to manage renal bone disease after transplantation is a distinctly separate issue in which bisphosphonate use may be even more controversial than in end-stage renal disease.65,66
In my opinion, patients without fractures with stage 5 chronic kidney disease should not be given bisphosphonates or teriparatide offlabel. Treating only on the basis of low bone mineral density and other risk factors seems to be associated with greater risk than benefit.
In stage 5 patients suffering fragility fractures, a bisphosphonate may be considered, but only after renal osteodystrophy has been thoroughly ruled out, which most often requires a bone biopsy.43,67,68 In skilled hands, transiliac bone biopsy is a safe procedure with little morbidity.
If osteoporosis appears to be the cause of the fracture, and if one chooses to use a bisphosphonate and the patient gives his or her informed consent, then I would give half the usual dose and restrict the therapy to no more than 3 years. The reason for halving the dose is based on the known pharmacokinetics of bisphosphonates in people with normal renal function: 50% of a given dose goes to bone and 50% is excreted by the kidney. Furthermore, the dialyzability of bisphosphonates has not been well studied. Limiting the treatment to 3 years is based on the unknown but probably greater bone retention of bisphosphonates when excretion is impaired.
I must emphasize that these approaches are not based on any evidence of efficacy, but rather are considered in extreme cases of often-recurrent fragility fractures in which the fractures per se pose a great risk of morbidity and death. These approaches should be clearly discussed with the patient, undertaken by specialists knowledgeable in complex metabolic bone disease management, and initiated only after the skeletal fragility disorder is well characterized.
SUMMING UP
No consensus exists on the criteria for diagnosing osteoporosis in stage 4 or 5 chronic kidney disease.
In higher-risk patients in stage 1, 2, or 3 chronic kidney disease who have osteoporosis, it appears that any drug approved for osteoporosis can be used, eg, a bisphosphonate, teriparatide, or both.
Considerations for management are far more complex in stage 4 or 5 because of the increased prevalence of other metabolic bone diseases and renal osteodystrophy, and because the World Health Organization criteria cannot be used to diagnose osteoporosis. In stage 5, the differential diagnosis requires careful analysis of a broad range of biochemical markers of bone turnover and, at times, quantitative bone histomorphometry, especially if one is considering using a bisphosphonate. It is unknown if bisphosphonates, by reducing bone turnover in a preexisting low-bone-turnover state, would help or harm bone or would lead to less or more cardiovascular disease. These questions must be addressed by better science and prospective data.
In the future, newer noninvasive radiologic tools to measure microstructure and mineralization of bone promise to help us better understand osteoporosis and renal osteodystrophy in a noninvasive manner.
In clinical practice, at the current time and with current limited knowledge, treatment of osteoporosis in stage 4 or 5 chronic kidney disease is opinion-based. Nevertheless, in very specific clinical cases of severe fragility fractures that, by themselves, may cause disability and death, bisphosphonates should be considered by experts in bone metabolism and, as with any off-label application, after careful informed discussions with the patient.
- Melton LJ. Epidemiology worldwide. Endocrinol Metab Clin North Am 2003; 32:1–13.
- Burge R, Dawson-Hughes B, Solomon DH, Wong JB, King A, Tosteson A. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005–2025. J Bone Miner Res 2007; 22:465–475.
- Barrett-Connor E, Siris ES, Wehren LE, et al. Osteoporosis and fracture risk in women of different ethnic groups. J Bone Miner Res 2005; 20:185–194.
- Assessment of fracture risk and its application to screening for postmenopausal osteoporosis. Report of a WHO Study Group. World Health Organ Tech Rep Ser 1994; 843:1–129.
- Miller PD, Bonnick SL. Clinical application of bone densitometry. In:Favus MJ, editor. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 4th ed. American Society for Bone and Mineral Research. Philadelphia, PA: Lippincott Williams & Wilkins; 1999.
- Nickolas TL, Leonard MB, Shane E. Chronic kidney disease and bone fracture: a growing concern. Kidney Int 2008; 74:721–731.
- Gal-Moscovici A, Sprague SM. Osteoporosis and chronic kidney disease. Semin Dial 2007; 20:423–430.
- National Kidney Foundation. Clinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am J Kidney Dis 2003; 42(suppl 3):S1–S201.
- Coresh J, Astor BC, Greene T, Eknoyan G, Levey AS. Prevalence of chronic kidney disease and decreased kidney function in the adult US population: Third National Health and Nutrition Examination Survey. Am J Kidney Dis 2003; 41:1–12.
- Bennett WM. Reporting eGFR. Clin J Am Soc Nephrol 2008; 3:1561–1562.
- Glassock RJ, Winearls C. Screening for CKD with eGFR: doubts and dangers. Clin J Am Soc Nephrol 2008; 3:1563–1568.
- Parfitt AM, Drezner M, Glorieux F, et al. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res 1987; 2:595–610.
- Andress DL, Sherrard DJ. The osteodystrophy of chronic renal failure. In:Schrier RW, editor: Diseases of the Kidney and Urinary Tract. Philadelphia, PA: Lippincott Williams & Wilkins; 2007:2431–2453.
- Moe S, Drueke T, Cunningham J, et al. Definition, evaluation, and classification of renal osteodystrophy: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int 2006; 69:1945–1953.
- NIH Consensus Development Panel Osteoporosis Prevention Diagnosis and Therapy. Osteoporosis prevention, diagnosis, and therapy. JAMA 2001; 285:785–795.
- Baim S, Binkley N, Bilezikian JP, et al. Official positions of the International Society for Clinical Densitometry and executive summary of the 2007 ISCD Position Development Conference. J Clin Densitom 2008; 11:75–91.
- Siris E, Miller P, Barrett-Connor E, et al. Identification and fracture outcomes of undiagnosed low bone mineral density in postmenopausal women: results from the National Osteoporosis Risk Assessment (NORA). JAMA 2001; 286:2815–2822.
- Schuit SCE, Oei HH, Witteman JC, et al. Fracture incidence and association with bone mineral density in elderly men and women: the Rotterdam Study. Bone 2004; 34:195–202.
- Bouxsein ML. Non-invasive measurements of bone strength: promise and peril. J Musculoskelet Neuronal Interact 2004; 4:404–405.
- Seeman E. Bone quality: the material and structural basis of bone strength. J Bone Miner Metab 2008; 26:1–8.
- Alem AM, Sherrard DJ, Gillen DL, et al. Increased risk of hip fracture among patients with end-stage renal disease. Kidney Int 2000; 58:396–399.
- Ball AM, Gillen DL, Sherrard D, et al. Risk of hip fracture among dialysis and renal transplant recipients. JAMA 2002; 288:3014–3018.
- Jadoul M, Albert JM, Akiba T, et al. Incidence and risk factors for hip or other bone fractures among hemodialysis patients in the Dialysis Outcomes and Practice Patterns Study. Kidney Int 2006; 70:1358–1366.
- Stehman-Breen CO, Sherrard DJ, Alem AM, et al. Risk factors for hip fracture among patients with end-stage renal disease. Kidney Int 2000; 58:2200–2205.
- Wehrli FW, Leonard MB, Saha PK, Gomberg BR. Quantitative highresolution magnetic resonance imaging reveals structural implications of renal osteodystrophy on trabecular and cortical bone. J Magn Reson Imaging 2004; 20:83–89.
- Genant HK, Lang TF, Engelke K, et al. Advances in the noninvasive assessment of bone density, quality, and structure. Calcif Tissue Int 1996; 59(suppl 1):S10–S15.
- Roschger P, Paschalis EP, Fratzl P, Klaushofer K. Bone mineralization density distribution in health and disease. Bone 2008; 42:456–466.
- Miller PD. Bone density and markers of bone turnover in predicting fracture risk and how changes in these measures predict fracture risk reduction. Curr Osteoporos Rep 2005; 3:103–110.
- Miller PD, Hochberg MC, Wehren LE, Ross PD, Wasnich RD. How useful are measures of BMD and bone turnover? Curr Med Res Opin 2005; 21:545–554.
- Chavassieux PM, Delmas PD. Bone remodeling: biochemical markers or bone biopsy? J Bone Miner Res 2006; 21:178–179.
- Garnero P. Biomarkers for osteoporosis management: utility in diagnosis, fracture risk prediction and therapy monitoring. Mol Diagn Ther 2008; 12:157–170.
- Hochberg M, Greenspan S, Wasnich R, Miller P, Thompson D, Ross P. Changes in bone density and turnover explain the reductions in incidence of nonvertebral fractures that occur during treatment with antiresorptive agents. J Clin Endocrinol Metab 2002; 87:1586–1592.
- Bouxsein ML, Delmas PD. Considerations for development of surrogate endpoints for antifracture efficacy of new treatments in osteoporosis: a perspective. J Bone Miner Res 2008; 23:1155–1167.
- Chen P, Satterwhite JH, Licatta AA, et al. Early changes in biochemical markers of bone formation predict BMD response to teriparatide in postmenopausal women with osteoporosis. J Bone Miner Res 2005; 20:962–970.
- Miller PD, Delmas PD, Lindsay R, et al. Early responsiveness of women with osteoporosis to teriparatide after therapy with alendronate or risedronate. J Clin Endocrinol Metab 2008; 93:3785–3793.
- Baim S, Miller PD. Assessing the clinical utility of serum CTX in postmenopausal osteoporosis and its use in predicting risk of osteonecrosis of the jaw. J Bone Miner Res 2009; 24:561–574.
- Miller PD, Lerma EV. Renal bone diseases. In:Kleerekoper M, Siris E, McClung M, editors. The Bone and Mineral Manual—A Practical Guide. 2nd ed. Burlington, MA: Elsevier Academic Press; 2005:127–138.
- Miller PD, Shane E. Management of transplantation renal bone disease: Interplay of bone mineral density and decisions regarding bisphosphonate use. In:Weir MR, editor. Medical Management of Kidney Transplantation. Philadelphia, PA: Lippincott Williams & Wilkins; 2004:359–375.
- Levin A, Bakris GL, Molitch M, et al. Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: results of the study to evaluate early kidney disease. Kidney int 2007; 71:31–38.
- Vassalotti JA, Uribarri J, Chen SC, et al. Trends in mineral metabolism: Kidney Early Evaluation Program (KEEP) and the National Health and Nutrition Examination Survey (NHANES) 1999–2004. Am J Kidney Dis 2008; 51(suppl 2):S56–S68.
- Meier C, Seibel MJ, Kraenzlin ME. Use of bone turnover markers in the real world: are we there yet? J Bone Miner Res 2009; 24:386–388.
- Lehmann G, Ott U, Kaemmerer D, Schuetze J, Wolf G. Bone histomorphometry and biochemical markers of bone turnover in patients with chronic kidney disease stages 3–5. Clin Nephrol 2008; 70:296–305.
- Miller PD. The role of bone biopsy in patients with chronic renal failure. Clin J Am Soc Nephrol 2008; 3(suppl 3):S140–S150.
- Frost HM. Tetracycline-based histological analysis of bone remodeling. Calcif Tissue Res 1969; 3:211–237.
- Hitt O, Jaworski ZF, Shimizu AG, Frost HM. Tissue-level bone formation rates in chronic renal failure, measured by means of tetracycline bone labeling. Can J Physiol Pharmacol 1970; 48:824–828.
- Coen G. Adynamic bone disease: an update and overview. J Nephrol 2005; 18:117–122.
- Parfitt AM. Renal bone disease: a new conceptual framework for the interpretation of bone histomorphometry. Curr Opin Nephrol Hypertens 2003; 12:387–403.
- Brandenburg VM, Floege J. Adynamic bone diseaseùbone and beyond. NDT Plus 2008; 3:135–147. doi:10.1093/ndtplus/sfn040.
- Hruska KA, Saab G, Mathew S, Lund R. Renal osteodystrophy, phosphate homeostasis, and vascular calcification. Semin Dial 2007; 20:309–315.
- Toussaint ND, Elder GJ, Kerr PG. Bisphosphonates in chronic kidney disease; balancing potential benefits and adverse effects on bone and soft tissue. Clin J Am Soc Nephrol 2009; 4:221–233.
- Miller PD. Is there a role for bisphosphonates in chronic kidney disease? Semin Dial 2007; 20:186–190.
- Miller PD. Bisphosphonates: pharmacology and use in the treatment of osteoporosis. In:Marcus R, Feldman D, Nelson DA, Rosen CJ, editors. Osteoporosis. 3rd ed. Boston, MA: Elsevier Academic Press; 2008:1725–1736.
- Russell RG, Watts NB, Ebetino FH, Rogers MJ. Mechanisms of action of bisphosphonates: similarities and differences and their potential influence on clinical efficacy. Osteoporosis Int 2008; 19:733–759.
- Eisman JA, Civitelli R, Adami S, et al. Efficacy and tolerability of intravenous ibandronate injections in postmenopausal osteoporosis: 2-year results from the DIVA study. J Rheumatol 2008; 35:488–497.
- Black DM, Delmas PD, Eastell RR, et al. Once-yearly zoledronic acid for treatment of postmenopausal osteoporosis. N Engl J Med 2007; 356:1809–1822.
- Lewiecki EM, Miller PD. Renal safety of intravenous bisphosphonates in the treatment of osteoporosis. Expert Opin Drug Saf 2007; 6:663–672.
- Miller PD. Anti-resorptives in the management of osteoporosis. Best Pract Res Clin Endocrinol Metab 2008; 22:849–868.
- Perazella MA, Markowitz GS. Bisphosphonate nephrotoxicity. Kidney Int 2008; 74:1385–1393.
- Boonen S, Sellmeyer DE, Lippuner K, et al. Renal safety of annual zoledronic acid infusions in osteoporotic postmenopausal women. Kidney Int 2008; 74:641–648.
- Neer RM, Arnaud CD, Zanchetta JR, et al. Effect of parathyroid hormone (1–34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med 2001; 344:1434–1441.
- Miller PD, Schwartz EN, Chen P, Misurski DA, Krege JH. Teriparatide in postmenopausal women with osteoporosis and mild or moderate renal impairment. Osteoporosis Int 2007; 18:59–68.
- Miller PD, Roux C, Boonen S, Barton I, Dunlap L, Burgio D. Safety and efficacy of risedronate in patients with age-related reduced renal function as estimated by the Cockcroft and Gault method: a pooled analysis of nine clinical trials. J Bone Miner Res 2005; 20:2105–2115.
- Jamal SA, Bauer DC, Ensrud KE, et al. Alendronate treatment in women with normal to severely impaired renal function: an analysis of the fracture intervention trial. J Bone Miner Res 2007; 22:503–508.
- Ishani A, Blackwell T, Jamal SA, Cummings SR, Ensrud KE; MORE Investigators. The effect of raloxifene treatment in postmenopausal women with CKD. J Am Soc Nephrol 2008; 19:1430–1438.
- Coco M, Glicklich D, Faugere MC, et al. Prevention of bone loss in renal transplant recipients: a prospective, randomized trial of intravenous pamidronate. J Am Soc Nephrol 2003; 14:2669–2676.
- Palmer SC, McGregor DO, Strippoli GF. Interventions for preventing bone disease in kidney transplant recipients. Cochrane Database Syst Rev 2007;CD005015.
- Ferreira MA. Diagnosis of renal osteodystrophy: when and how to use biochemical markers and non-invasive methods; when bone biopsy is needed. Nephrol Dial Transplant 2000; 15(suppl 5):8–14.
- Trueba D, Sawaya BP, Mawad H, Malluche HH. Bone biopsy: indications, techniques, and complications. Semin Dial 2003; 16:341–345.
But even in chronic kidney disease, many fractures are due to postmenopausal or agerelated osteoporosis, and estrogen-deficiency osteoporosis is the most common cause of fragility fractures overall.1–3 Osteoporosis can be diagnosed only after other causes of skeletal fragility have been ruled out. And how to diagnose and treat osteoporosis in the most severe stage of kidney disease is a matter of opinion, as we have almost no data to guide us.
Nevertheless, in the pages that follow, I will outline my admittedly opinion-based approach to diagnosing and managing the causes of fragility fractures in patients with chronic kidney disease.
T SCORES DO NOT DISTINGUISH THE CAUSES OF FRAGILITY
The most common cause of fragility fractures is osteoporosis due to estrogen deficiency.1–3 But since many other medical conditions can lead to osteoporosis, simple diagnostic criteria have been difficult to find.
Before 1994, the diagnosis of osteoporosis was made on the basis of low-trauma fractures.4 Now, we use the World Health Organization criteria,5 based on bone mineral density T scores:
- Normal—a T score of −1.0 standard deviations or higher
- Osteopenia—a T score of less than −1.0 but higher than −2.5
- Osteoporosis—a T score of −2.5 or less
- Severe osteoporosis—a T score of −2.5 or less with a fragility fracture.
However, fractures can also be due to metabolic bone diseases that are not osteoporosis, including renal bone diseases.6–7 While a low T score or a fracture provides a working diagnosis of osteoporosis, it does not help distinguish the different types of osteoporosis and nonosteoporotic metabolic bone diseases. For example, osteomalacia and osteogenesis imperfecta can also cause fragility fractures and can be associated with low bone density. Using these criteria to define osteoporosis is even more problematic in patients with chronic kidney disease.
FIVE STAGES OF CHRONIC KIDNEY DISEASE
The National Kidney Foundation8 classifies the severity of chronic kidney disease on the basis of the glomerular filtration rate (GFR), as measured by 24-hour urine for creatinine clearance, or as estimated by the Cockcroft-Gault equation or, preferably, the Modification of Diet in Renal Disease (MDRD) equation (calculators are available at www.kidney.org/professionals/kdoqi/gfr_calculator.cfm):
- Stage 1—GFR 90 mL/minute/1.73 m2 or higher
- Stage 2—GFR 60 to 89
- Stage 3—GFR 30 to 59
- Stage 4—GFR 15 to 29
- Stage 5—GFR lower than 15, or if the patient is on dialysis. (Another stage, called 5D, was added to the list to denote patients on dialysis, since the metabolic derangements in bone and systemic biology may differ between patients on dialysis vs those not on dialysis.)
This staging system is relevant to the discussion of bone fragility that follows.
CHRONIC KIDNEY DISEASE IS COMMON IN THE ELDERLY
The third National Health and Nutrition Examination Survey9 found that, at least as estimated by the Cockcroft-Gault equation, the GFR declines with age, so that by the age of 70 at least 20% of the US population has stage 4 or 5 chronic kidney disease.
Although the Cockcroft-Gault and MDRD equations may yield lower GFR values in the general population than one would get by measuring creatinine, inulin, or iothalamate clearance,10,11 the point is that both osteoporosis and chronic kidney disease are common.
THE GAMUT OF RENAL OSTEODYSTROPHY
In kidney failure (stage 5 chronic kidney disease), all forms of renal osteodystrophy may be associated with fragility fractures. Renal osteodystrophy can be defined by quantitative bone histomorphometry.12,13 The systemic conditions that may be associated with the bone disease and systemic vascular disease (chronic kidney disease–mineral and bone disorder) are characterized by one or more of the following14:
- Abnormalities of calcium, phosphorus, parathyroid hormone, or vitamin D metabolism
- Abnormalities in bone turnover, mineralization, volume, linear growth, or strength
- Vascular or other soft-tissue calcification.
The National Kidney Foundation14 classifies renal osteodystrophy on the basis of:
- Turnover—high, normal, or low
- Mineralization—normal or abnormal
- Volume—high, normal, or low.
Although this system helps us understand these diseases better, it does not provide a working diagnosis of osteoporosis.14
WHAT IS OSTEOPOROSIS?
In an attempt to define osteoporosis by a pathophysiologic mechanism, the National Institutes of Health15 have held two consensus conferences and have stated that “osteoporosis is a skeletal disorder characterized by impairment in bone strength predisposing a person to an increased risk of fracture. Bone strength primarily reflects the integration of bone density and bone quality.”15 However, the consensus statement also does not provide a working diagnosis of osteoporosis—one that clinicians can apply to management decisions, and one that is also accepted by the US International Classification of Disease codes for reimbursement purposes.
The 1994 World Health Organization criteria offer the most pragmatic operational definition of osteoporosis, and they can be applied in both men and women, as well as in younger patients with medical conditions associated with increased risk of low-trauma fracture.5,16 Although the main purpose of these criteria was to advise international health authorities of the potential future economic impact of osteoporosis, the T score also became the pragmatic diagnostic threshold for defining normal, osteopenia, and osteoporosis in clinical practice.
The T score also calls attention to an important observation: of people who have fractures and subsequently undergo bone densitometry, more are found to have osteopenia than osteoporosis. The reasons are that there are more people with osteopenia than osteoporosis,17,18 and many other factors independent of low bone mineral density contribute to bone strength.19,20
How is osteoporosis diagnosed in stage 1–3 chronic kidney disease?
In patients with chronic kidney disease who develop fragility fractures, the reasonable question is: Is the cause of the fracture osteoporosis or some other metabolic bone disease associated with chronic kidney disease?
The National Kidney Foundation guidelines14 say that the diagnosis of osteoporosis can be established in patients with stage 1, 2, or 3 chronic kidney disease on the basis of either of the World Health Organization criteria, ie, a T score of −2.5 or lower or fragility fractures, as in the postmenopausal population, as long as there are no biochemical abnormalities that suggest chronic kidney disease–mineral and bone disorder.
How is osteoporosis diagnosed in stage 4 or 5 chronic kidney disease?
The answer is neither straightforward nor clearly defined in severe (stage 4 or 5) chronic kidney disease.
In stage 5 and especially in patients on dialysis, the derangements in bone and mineral metabolism become serious enough to impair bone strength and increase the risk of lowtrauma fractures. The risk of hip fracture in stage 5 may be four times higher than in agematched controls.21–24
Adynamic, severe hyperparathyroid bone disease as well as osteomalacia can be associated with a higher risk of fragility fractures than in aged-matched controls in population studies of postmenopausal women or elderly men. These are bone fragility conditions that are not osteoporosis but that can mimic osteoporosis by the World Health Organization criteria.
Thus, when a patient in stage 5 has severe fragility fractures that by themselves may be life-threatening, it is reasonable to ask if the drugs that reduce the risk of fractures in many other osteoporotic conditions (postmenopausal, steroid-induced, elderly male osteoporosis, after solid organ transplantation) can also be used in patients with advanced chronic kidney disease.
The diagnosis of osteoporosis in these patients has no universally accepted criteria. The diagnosis is best suggested by excluding other forms of renal osteodystrophy by quantitative histomorphometry or by attempting to classify the form of renal osteodystrophy by noninvasive means of assessing bone turnover, mineralization, and volume. However, we lack clinical tools to make these distinctions in individual patients.
While many promising radiologic techniques that examine bone microarchitecture offer hope of being able to define turnover, mineralization, and volume noninvasively in severe chronic kidney disease, they are investigational and unproven at this time in discriminating between renal osteodystrophy and osteoporosis.6,25–27 As we increase our understanding of the relationships between turnover, mineralization, volume, and bone strength, these noninvasive imaging technologies may become the means to correlate turnover, mineralization, and volume to bone strength and open up an entirely new way to classify skeletal strength.
In the meantime, the clinician is left with quantitative bone histomorphometry (which requires biopsy) and biochemical markers of bone turnover to characterize the bone disease that may be responsible for low-trauma fractures in stage 5 chronic kidney disease. The clinician should first use biochemical markers before bone biopsy to distinguish the form of renal osteodystrophy, as this distinction may be able to prevent unnecessary biopsy.
Biochemical markers of bone metabolism
In chronic kidney disease, the bone biochemical tests that nephrologists usually assess during the course of declining renal function are the serum levels of:
- Phosphorus
- Parathyroid hormone
- Calcium
- Other electrolytes
- Total alkaline phosphatase or bone-specific alkaline phosphatase
- 1,25 dihydroxyvitamin D.
In postmenopausal osteoporosis, the biochemical markers of bone turnover that are measured to reflect baseline levels of bone turnover or change in bone turnover in response to drug therapy are:
- The serum or urine collagen cross-links N-telopeptide (NTx) and C-telopeptide (CTx), markers of bone resorption
- Bone-specific alkaline phosphatase (an osteoblast activity marker)
- Serum osteocalcin, a bone formation marker
- Propeptide type 1 collagen (P1NP), a marker of osteoblast activity, highly correlated with bone formation
- 25-hydroxyvitamin D levels.
Biochemical markers of bone turnover cannot be used to diagnose osteoporosis. They can, however, provide clinical guidance as to whether a patient has high or low bone turnover and whether therapy is affecting bone turnover.28–36 Although these markers have value in making these distinctions in groups of patients, they are less sensitive and specific for classifying an individual patient’s bone turnover status.
Bone-specific alkaline phosphatase, parathyroid hormone, and adynamic bone disease
If a patient’s bone-specific alkaline phosphatase level is elevated, adynamic bone disease is highly unlikely. Assuming that other causes of this elevated level (eg, Paget disease of bone, metastatic cancer) have already been excluded, the elevated level could represent either osteomalacia or hyperparathyroid bone disease.
However, a “normal” bone-specific alkaline phosphatase level does not exclude adynamic bone disease, whereas a low level is more often associated with low bone turnover.
An elevated parathyroid hormone level does not exclude adynamic renal bone disease, but a low level (< 150 pg/mL) suggests a lowbone-turnover state. A level six times or more greater than the upper limit of normal is far more likely to be associated with high bone turnover.
Thus, in clinical practice, patients with stage 4 or 5 chronic kidney disease who have elevated bone-specific alkaline phosphatase or very high parathyroid hormone values do not have adynamic bone disease. Furthermore, once other causes of these aberrant biochemical abnormalities have been defined, then “high-bone-turnover osteoporosis” may be a consideration. Certainly, in my opinion, if bone turnover markers suggest low bone turnover, bone biopsy is necessary before starting an antiresorptive agent.35
Quantitative bone histomorphometry
Double tetracycline-labeled quantitative histomorphometry is still the only accepted way to measure turnover, mineralization, and volume in clinical practice.43–45 A committee of the American Society for Bone and Mineral Research has developed histomorphometric criteria for distinguishing among the different types of metabolic bone diseases (osteomalacia, adynamic bone disease, hyperparathyroid bone disease).12 These criteria can be used to distinguish among the various metabolic bone diseases that accompany stage 5 chronic kidney disease, including adynamic bone disease.43,46–48
For patients in stage 5 who have had a fragility fracture, adynamic bone disease should be excluded before the off-label use of an osteoporosis drug that reduces bone turnover, such as a bisphosphonate, calcitonin, estrogen, a selective estrogen receptor modulator, or denosumab (anti-RANK ligand antibody). While there is no evidence, for example, that starting a bisphosphonate in a patient who already has adynamic bone disease is detrimental to either bone strength or systemic vascular calcification (which may be linked to low bone turnover),49 it seems unreasonable to do so until solid prospective data clarify the harm or benefit.50 Preliminary experimental and clinical data suggest that bisphosphonates may even reduce progression of extraosseous calcification and inhibit the development of atherosclerosis.50
Hence, quantitative bone histomorphometry can discriminate among the various forms of renal osteodystrophy. If a distinct form of renal osteodystrophy is not present in a patient with stage 4 or 5 chronic kidney disease who has had a fracture and who, on biopsy, has a low trabecular bone volume, the patient probably has osteoporosis by exclusion.
TREATING OSTEOPOROSIS IN STAGE 1–3 CHRONIC KIDNEY DISEASE
As previously mentioned, patients who have fragility fractures in stage 1, 2, or 3 chronic kidney disease are more likely to have osteoporosis than renal osteodystrophy as the cause of their impaired bone strength. Although several articles have described a higher risk of fragility fractures in patients with age-related reduction in renal function than in agematched patients with normal renal function, the specific metabolic bone disease other than osteoporosis accounting for this bone fragility has not been defined.6
Hence, patients with osteoporosis who are in stage 1, 2, or 3 chronic kidney disease and do not have a known biochemical abnormality that might suggest some form of renal osteodystrophy can and should be considered for treatment with approved drugs that reduce the risk of fractures in postmenopausal, male, or glucocorticoid-induced osteoporosis.51–53 In clinical trials, these agents were shown to be effective in patients with serum creatinine concentrations as high as 2.0 mg/dL or a GFR as low as 30 mL/min, as estimated by the Cockcroft-Gault equation.
While all of the approved agents show evidence of reducing the risk of vertebral fractures, patients at higher risk of fractures or those who have already suffered a nonvertebral fracture are more often considered candidates for treatment with a bisphosphonate or teriparatide (Forteo), both of which have shown evidence of reducing the risk of all fractures.
Bisphosphonates in stage 1–3 chronic kidney disease
There is prospective evidence that patients with an age-related reduction in GFR down to 30 mL/min benefit from oral and intravenous bisphosphonates, since all of the clinical trials that led to the approval of bisphosphonates included patients with GFRs as low as this.54–57 Bisphosphonates seem to have an excellent safety profile as measured by renal adverse events in patients with a GFR of 30 mL/min or greater.52–59
From the intravenous bisphosphonate studies, it appears that ibandronate (Boniva) at the approved dose of 3 mg intravenously every 3 months and zoledronic acid (Reclast) 5 mg once a year given over 15 minutes are safe in patients with a GFR greater than 30 or 35 mL/min.
However, the safety of these drugs might not be the same in patients with preexisting renal parenchymal disease (eg, in diabetes) or in patients using other agents that could affect renal function (eg, nonsteroidal antiinflammatory drugs). Therefore, caution is still needed when deciding to use intravenous bisphosphonates in specific higher-risk renal subpopulations.
In the clinical trials of zoledronic acid, a substantial proportion of patients had diabetes, and no difference was seen in adverse renal effects between diabetic and nondiabetic patients. Also, GFRs declined equally between the treated and placebo groups over time and were no different at the end of 3 years.55 However, in patients in whom serum creatinine was measured 9 to 11 days after the infusion of zoledronic acid, there was a small but statistically significant transient increase in serum creatinine concentration (0.5–2.0 mg/dL above baseline) after the second annual infusion. 58 The serum creatinine concentrations returned to their baseline values in all of these patients before the next annual infusion.
It is important that infusions of zoledronic acid be given no faster than over 15 minutes. More rapid infusion has been associated with acute renal failure, suggesting that the tubular damage that mimics acute tubular necrosis is related to the maximal concentration and not to the area under the curve. I infuse zoledronic acid over 30 minutes in patients with normal renal function or in those with stage 1, 2, or 3 chronic kidney disease.
Teriparatide
Teriparatide’s approval trial did not require baseline measurements of GFR, but patients were enrolled only if their baseline serum creatinine concentrations were less than 2.0 mg/dL.60 In a post hoc analysis, a small subset of patients had GFRs as low as 30 mL/min as estimated by the Cockcroft-Gault equation. In these patients, teriparatide 20 or 40 μg/day had an anabolic effect as measured by increases in osteoblast activity markers and bone mineral density, similar to that seen in patients with higher estimated GFRs and without any adverse renal effects.61
There are no data on using teriparatide in stage 4 or 5 chronic kidney disease, and I emphasize that in all of the clinical trials of teriparatide, all patients, even those with estimated GFRs as low as 30 mL/min, had normal baseline serum intact parathyroid hormone levels. It is possible that the bone biologic response could differ between patients with chronic kidney disease who have an elevated as compared with a normal serum parathyroid hormone level. This issue should be investigated.
TREATING OSTEOPOROSIS IN STAGE 4 OR 5 CHRONIC KIDNEY DISEASE
Treatment decisions are more difficult in patients with stage 4 and especially stage 5 chronic kidney disease who have had fragility fractures. This is even the case when the clinician has determined to the best of his or her ability that the patient has osteoporosis rather than renal osteodystrophy.
There are no prospective data showing any of the approved drugs to be effective in treating osteoporosis in patients whose GFRs are lower than 30 mL/min. However, a post hoc analysis of pooled data from nine clinical trials62 found that risedronate (Actonel) 5 mg/day reduced the incidence of new vertebral fractures. Another post hoc analysis, from the Fracture Intervention Trial,63 found that alendronate (Fosamax) 5 mg/day for the first 2 years and 10 mg/day for the 3rd year reduced the incidence of all clinical fractures. In neither of these post hoc analyses did the drug affect the serum creatinine concentration. The patients—postmenopausal women—had GFRs as low as 15 mL/min as estimated by the Cockcroft-Gault equation. Similar post hoc data have been published on raloxifene (Evista).64
There are no data on the efficacy (reduction in fracture risk) or safety of any bisphosphonate in patients with GFRs lower than 15 mL/min (stage 5 chronic kidney disease). Nevertheless, the question often arises when fragility fractures occur in this population. Here, only opinion and controversy exist, and we fervently await good science and randomized prospective data.
How to manage renal bone disease after transplantation is a distinctly separate issue in which bisphosphonate use may be even more controversial than in end-stage renal disease.65,66
In my opinion, patients without fractures with stage 5 chronic kidney disease should not be given bisphosphonates or teriparatide offlabel. Treating only on the basis of low bone mineral density and other risk factors seems to be associated with greater risk than benefit.
In stage 5 patients suffering fragility fractures, a bisphosphonate may be considered, but only after renal osteodystrophy has been thoroughly ruled out, which most often requires a bone biopsy.43,67,68 In skilled hands, transiliac bone biopsy is a safe procedure with little morbidity.
If osteoporosis appears to be the cause of the fracture, and if one chooses to use a bisphosphonate and the patient gives his or her informed consent, then I would give half the usual dose and restrict the therapy to no more than 3 years. The reason for halving the dose is based on the known pharmacokinetics of bisphosphonates in people with normal renal function: 50% of a given dose goes to bone and 50% is excreted by the kidney. Furthermore, the dialyzability of bisphosphonates has not been well studied. Limiting the treatment to 3 years is based on the unknown but probably greater bone retention of bisphosphonates when excretion is impaired.
I must emphasize that these approaches are not based on any evidence of efficacy, but rather are considered in extreme cases of often-recurrent fragility fractures in which the fractures per se pose a great risk of morbidity and death. These approaches should be clearly discussed with the patient, undertaken by specialists knowledgeable in complex metabolic bone disease management, and initiated only after the skeletal fragility disorder is well characterized.
SUMMING UP
No consensus exists on the criteria for diagnosing osteoporosis in stage 4 or 5 chronic kidney disease.
In higher-risk patients in stage 1, 2, or 3 chronic kidney disease who have osteoporosis, it appears that any drug approved for osteoporosis can be used, eg, a bisphosphonate, teriparatide, or both.
Considerations for management are far more complex in stage 4 or 5 because of the increased prevalence of other metabolic bone diseases and renal osteodystrophy, and because the World Health Organization criteria cannot be used to diagnose osteoporosis. In stage 5, the differential diagnosis requires careful analysis of a broad range of biochemical markers of bone turnover and, at times, quantitative bone histomorphometry, especially if one is considering using a bisphosphonate. It is unknown if bisphosphonates, by reducing bone turnover in a preexisting low-bone-turnover state, would help or harm bone or would lead to less or more cardiovascular disease. These questions must be addressed by better science and prospective data.
In the future, newer noninvasive radiologic tools to measure microstructure and mineralization of bone promise to help us better understand osteoporosis and renal osteodystrophy in a noninvasive manner.
In clinical practice, at the current time and with current limited knowledge, treatment of osteoporosis in stage 4 or 5 chronic kidney disease is opinion-based. Nevertheless, in very specific clinical cases of severe fragility fractures that, by themselves, may cause disability and death, bisphosphonates should be considered by experts in bone metabolism and, as with any off-label application, after careful informed discussions with the patient.
But even in chronic kidney disease, many fractures are due to postmenopausal or agerelated osteoporosis, and estrogen-deficiency osteoporosis is the most common cause of fragility fractures overall.1–3 Osteoporosis can be diagnosed only after other causes of skeletal fragility have been ruled out. And how to diagnose and treat osteoporosis in the most severe stage of kidney disease is a matter of opinion, as we have almost no data to guide us.
Nevertheless, in the pages that follow, I will outline my admittedly opinion-based approach to diagnosing and managing the causes of fragility fractures in patients with chronic kidney disease.
T SCORES DO NOT DISTINGUISH THE CAUSES OF FRAGILITY
The most common cause of fragility fractures is osteoporosis due to estrogen deficiency.1–3 But since many other medical conditions can lead to osteoporosis, simple diagnostic criteria have been difficult to find.
Before 1994, the diagnosis of osteoporosis was made on the basis of low-trauma fractures.4 Now, we use the World Health Organization criteria,5 based on bone mineral density T scores:
- Normal—a T score of −1.0 standard deviations or higher
- Osteopenia—a T score of less than −1.0 but higher than −2.5
- Osteoporosis—a T score of −2.5 or less
- Severe osteoporosis—a T score of −2.5 or less with a fragility fracture.
However, fractures can also be due to metabolic bone diseases that are not osteoporosis, including renal bone diseases.6–7 While a low T score or a fracture provides a working diagnosis of osteoporosis, it does not help distinguish the different types of osteoporosis and nonosteoporotic metabolic bone diseases. For example, osteomalacia and osteogenesis imperfecta can also cause fragility fractures and can be associated with low bone density. Using these criteria to define osteoporosis is even more problematic in patients with chronic kidney disease.
FIVE STAGES OF CHRONIC KIDNEY DISEASE
The National Kidney Foundation8 classifies the severity of chronic kidney disease on the basis of the glomerular filtration rate (GFR), as measured by 24-hour urine for creatinine clearance, or as estimated by the Cockcroft-Gault equation or, preferably, the Modification of Diet in Renal Disease (MDRD) equation (calculators are available at www.kidney.org/professionals/kdoqi/gfr_calculator.cfm):
- Stage 1—GFR 90 mL/minute/1.73 m2 or higher
- Stage 2—GFR 60 to 89
- Stage 3—GFR 30 to 59
- Stage 4—GFR 15 to 29
- Stage 5—GFR lower than 15, or if the patient is on dialysis. (Another stage, called 5D, was added to the list to denote patients on dialysis, since the metabolic derangements in bone and systemic biology may differ between patients on dialysis vs those not on dialysis.)
This staging system is relevant to the discussion of bone fragility that follows.
CHRONIC KIDNEY DISEASE IS COMMON IN THE ELDERLY
The third National Health and Nutrition Examination Survey9 found that, at least as estimated by the Cockcroft-Gault equation, the GFR declines with age, so that by the age of 70 at least 20% of the US population has stage 4 or 5 chronic kidney disease.
Although the Cockcroft-Gault and MDRD equations may yield lower GFR values in the general population than one would get by measuring creatinine, inulin, or iothalamate clearance,10,11 the point is that both osteoporosis and chronic kidney disease are common.
THE GAMUT OF RENAL OSTEODYSTROPHY
In kidney failure (stage 5 chronic kidney disease), all forms of renal osteodystrophy may be associated with fragility fractures. Renal osteodystrophy can be defined by quantitative bone histomorphometry.12,13 The systemic conditions that may be associated with the bone disease and systemic vascular disease (chronic kidney disease–mineral and bone disorder) are characterized by one or more of the following14:
- Abnormalities of calcium, phosphorus, parathyroid hormone, or vitamin D metabolism
- Abnormalities in bone turnover, mineralization, volume, linear growth, or strength
- Vascular or other soft-tissue calcification.
The National Kidney Foundation14 classifies renal osteodystrophy on the basis of:
- Turnover—high, normal, or low
- Mineralization—normal or abnormal
- Volume—high, normal, or low.
Although this system helps us understand these diseases better, it does not provide a working diagnosis of osteoporosis.14
WHAT IS OSTEOPOROSIS?
In an attempt to define osteoporosis by a pathophysiologic mechanism, the National Institutes of Health15 have held two consensus conferences and have stated that “osteoporosis is a skeletal disorder characterized by impairment in bone strength predisposing a person to an increased risk of fracture. Bone strength primarily reflects the integration of bone density and bone quality.”15 However, the consensus statement also does not provide a working diagnosis of osteoporosis—one that clinicians can apply to management decisions, and one that is also accepted by the US International Classification of Disease codes for reimbursement purposes.
The 1994 World Health Organization criteria offer the most pragmatic operational definition of osteoporosis, and they can be applied in both men and women, as well as in younger patients with medical conditions associated with increased risk of low-trauma fracture.5,16 Although the main purpose of these criteria was to advise international health authorities of the potential future economic impact of osteoporosis, the T score also became the pragmatic diagnostic threshold for defining normal, osteopenia, and osteoporosis in clinical practice.
The T score also calls attention to an important observation: of people who have fractures and subsequently undergo bone densitometry, more are found to have osteopenia than osteoporosis. The reasons are that there are more people with osteopenia than osteoporosis,17,18 and many other factors independent of low bone mineral density contribute to bone strength.19,20
How is osteoporosis diagnosed in stage 1–3 chronic kidney disease?
In patients with chronic kidney disease who develop fragility fractures, the reasonable question is: Is the cause of the fracture osteoporosis or some other metabolic bone disease associated with chronic kidney disease?
The National Kidney Foundation guidelines14 say that the diagnosis of osteoporosis can be established in patients with stage 1, 2, or 3 chronic kidney disease on the basis of either of the World Health Organization criteria, ie, a T score of −2.5 or lower or fragility fractures, as in the postmenopausal population, as long as there are no biochemical abnormalities that suggest chronic kidney disease–mineral and bone disorder.
How is osteoporosis diagnosed in stage 4 or 5 chronic kidney disease?
The answer is neither straightforward nor clearly defined in severe (stage 4 or 5) chronic kidney disease.
In stage 5 and especially in patients on dialysis, the derangements in bone and mineral metabolism become serious enough to impair bone strength and increase the risk of lowtrauma fractures. The risk of hip fracture in stage 5 may be four times higher than in agematched controls.21–24
Adynamic, severe hyperparathyroid bone disease as well as osteomalacia can be associated with a higher risk of fragility fractures than in aged-matched controls in population studies of postmenopausal women or elderly men. These are bone fragility conditions that are not osteoporosis but that can mimic osteoporosis by the World Health Organization criteria.
Thus, when a patient in stage 5 has severe fragility fractures that by themselves may be life-threatening, it is reasonable to ask if the drugs that reduce the risk of fractures in many other osteoporotic conditions (postmenopausal, steroid-induced, elderly male osteoporosis, after solid organ transplantation) can also be used in patients with advanced chronic kidney disease.
The diagnosis of osteoporosis in these patients has no universally accepted criteria. The diagnosis is best suggested by excluding other forms of renal osteodystrophy by quantitative histomorphometry or by attempting to classify the form of renal osteodystrophy by noninvasive means of assessing bone turnover, mineralization, and volume. However, we lack clinical tools to make these distinctions in individual patients.
While many promising radiologic techniques that examine bone microarchitecture offer hope of being able to define turnover, mineralization, and volume noninvasively in severe chronic kidney disease, they are investigational and unproven at this time in discriminating between renal osteodystrophy and osteoporosis.6,25–27 As we increase our understanding of the relationships between turnover, mineralization, volume, and bone strength, these noninvasive imaging technologies may become the means to correlate turnover, mineralization, and volume to bone strength and open up an entirely new way to classify skeletal strength.
In the meantime, the clinician is left with quantitative bone histomorphometry (which requires biopsy) and biochemical markers of bone turnover to characterize the bone disease that may be responsible for low-trauma fractures in stage 5 chronic kidney disease. The clinician should first use biochemical markers before bone biopsy to distinguish the form of renal osteodystrophy, as this distinction may be able to prevent unnecessary biopsy.
Biochemical markers of bone metabolism
In chronic kidney disease, the bone biochemical tests that nephrologists usually assess during the course of declining renal function are the serum levels of:
- Phosphorus
- Parathyroid hormone
- Calcium
- Other electrolytes
- Total alkaline phosphatase or bone-specific alkaline phosphatase
- 1,25 dihydroxyvitamin D.
In postmenopausal osteoporosis, the biochemical markers of bone turnover that are measured to reflect baseline levels of bone turnover or change in bone turnover in response to drug therapy are:
- The serum or urine collagen cross-links N-telopeptide (NTx) and C-telopeptide (CTx), markers of bone resorption
- Bone-specific alkaline phosphatase (an osteoblast activity marker)
- Serum osteocalcin, a bone formation marker
- Propeptide type 1 collagen (P1NP), a marker of osteoblast activity, highly correlated with bone formation
- 25-hydroxyvitamin D levels.
Biochemical markers of bone turnover cannot be used to diagnose osteoporosis. They can, however, provide clinical guidance as to whether a patient has high or low bone turnover and whether therapy is affecting bone turnover.28–36 Although these markers have value in making these distinctions in groups of patients, they are less sensitive and specific for classifying an individual patient’s bone turnover status.
Bone-specific alkaline phosphatase, parathyroid hormone, and adynamic bone disease
If a patient’s bone-specific alkaline phosphatase level is elevated, adynamic bone disease is highly unlikely. Assuming that other causes of this elevated level (eg, Paget disease of bone, metastatic cancer) have already been excluded, the elevated level could represent either osteomalacia or hyperparathyroid bone disease.
However, a “normal” bone-specific alkaline phosphatase level does not exclude adynamic bone disease, whereas a low level is more often associated with low bone turnover.
An elevated parathyroid hormone level does not exclude adynamic renal bone disease, but a low level (< 150 pg/mL) suggests a lowbone-turnover state. A level six times or more greater than the upper limit of normal is far more likely to be associated with high bone turnover.
Thus, in clinical practice, patients with stage 4 or 5 chronic kidney disease who have elevated bone-specific alkaline phosphatase or very high parathyroid hormone values do not have adynamic bone disease. Furthermore, once other causes of these aberrant biochemical abnormalities have been defined, then “high-bone-turnover osteoporosis” may be a consideration. Certainly, in my opinion, if bone turnover markers suggest low bone turnover, bone biopsy is necessary before starting an antiresorptive agent.35
Quantitative bone histomorphometry
Double tetracycline-labeled quantitative histomorphometry is still the only accepted way to measure turnover, mineralization, and volume in clinical practice.43–45 A committee of the American Society for Bone and Mineral Research has developed histomorphometric criteria for distinguishing among the different types of metabolic bone diseases (osteomalacia, adynamic bone disease, hyperparathyroid bone disease).12 These criteria can be used to distinguish among the various metabolic bone diseases that accompany stage 5 chronic kidney disease, including adynamic bone disease.43,46–48
For patients in stage 5 who have had a fragility fracture, adynamic bone disease should be excluded before the off-label use of an osteoporosis drug that reduces bone turnover, such as a bisphosphonate, calcitonin, estrogen, a selective estrogen receptor modulator, or denosumab (anti-RANK ligand antibody). While there is no evidence, for example, that starting a bisphosphonate in a patient who already has adynamic bone disease is detrimental to either bone strength or systemic vascular calcification (which may be linked to low bone turnover),49 it seems unreasonable to do so until solid prospective data clarify the harm or benefit.50 Preliminary experimental and clinical data suggest that bisphosphonates may even reduce progression of extraosseous calcification and inhibit the development of atherosclerosis.50
Hence, quantitative bone histomorphometry can discriminate among the various forms of renal osteodystrophy. If a distinct form of renal osteodystrophy is not present in a patient with stage 4 or 5 chronic kidney disease who has had a fracture and who, on biopsy, has a low trabecular bone volume, the patient probably has osteoporosis by exclusion.
TREATING OSTEOPOROSIS IN STAGE 1–3 CHRONIC KIDNEY DISEASE
As previously mentioned, patients who have fragility fractures in stage 1, 2, or 3 chronic kidney disease are more likely to have osteoporosis than renal osteodystrophy as the cause of their impaired bone strength. Although several articles have described a higher risk of fragility fractures in patients with age-related reduction in renal function than in agematched patients with normal renal function, the specific metabolic bone disease other than osteoporosis accounting for this bone fragility has not been defined.6
Hence, patients with osteoporosis who are in stage 1, 2, or 3 chronic kidney disease and do not have a known biochemical abnormality that might suggest some form of renal osteodystrophy can and should be considered for treatment with approved drugs that reduce the risk of fractures in postmenopausal, male, or glucocorticoid-induced osteoporosis.51–53 In clinical trials, these agents were shown to be effective in patients with serum creatinine concentrations as high as 2.0 mg/dL or a GFR as low as 30 mL/min, as estimated by the Cockcroft-Gault equation.
While all of the approved agents show evidence of reducing the risk of vertebral fractures, patients at higher risk of fractures or those who have already suffered a nonvertebral fracture are more often considered candidates for treatment with a bisphosphonate or teriparatide (Forteo), both of which have shown evidence of reducing the risk of all fractures.
Bisphosphonates in stage 1–3 chronic kidney disease
There is prospective evidence that patients with an age-related reduction in GFR down to 30 mL/min benefit from oral and intravenous bisphosphonates, since all of the clinical trials that led to the approval of bisphosphonates included patients with GFRs as low as this.54–57 Bisphosphonates seem to have an excellent safety profile as measured by renal adverse events in patients with a GFR of 30 mL/min or greater.52–59
From the intravenous bisphosphonate studies, it appears that ibandronate (Boniva) at the approved dose of 3 mg intravenously every 3 months and zoledronic acid (Reclast) 5 mg once a year given over 15 minutes are safe in patients with a GFR greater than 30 or 35 mL/min.
However, the safety of these drugs might not be the same in patients with preexisting renal parenchymal disease (eg, in diabetes) or in patients using other agents that could affect renal function (eg, nonsteroidal antiinflammatory drugs). Therefore, caution is still needed when deciding to use intravenous bisphosphonates in specific higher-risk renal subpopulations.
In the clinical trials of zoledronic acid, a substantial proportion of patients had diabetes, and no difference was seen in adverse renal effects between diabetic and nondiabetic patients. Also, GFRs declined equally between the treated and placebo groups over time and were no different at the end of 3 years.55 However, in patients in whom serum creatinine was measured 9 to 11 days after the infusion of zoledronic acid, there was a small but statistically significant transient increase in serum creatinine concentration (0.5–2.0 mg/dL above baseline) after the second annual infusion. 58 The serum creatinine concentrations returned to their baseline values in all of these patients before the next annual infusion.
It is important that infusions of zoledronic acid be given no faster than over 15 minutes. More rapid infusion has been associated with acute renal failure, suggesting that the tubular damage that mimics acute tubular necrosis is related to the maximal concentration and not to the area under the curve. I infuse zoledronic acid over 30 minutes in patients with normal renal function or in those with stage 1, 2, or 3 chronic kidney disease.
Teriparatide
Teriparatide’s approval trial did not require baseline measurements of GFR, but patients were enrolled only if their baseline serum creatinine concentrations were less than 2.0 mg/dL.60 In a post hoc analysis, a small subset of patients had GFRs as low as 30 mL/min as estimated by the Cockcroft-Gault equation. In these patients, teriparatide 20 or 40 μg/day had an anabolic effect as measured by increases in osteoblast activity markers and bone mineral density, similar to that seen in patients with higher estimated GFRs and without any adverse renal effects.61
There are no data on using teriparatide in stage 4 or 5 chronic kidney disease, and I emphasize that in all of the clinical trials of teriparatide, all patients, even those with estimated GFRs as low as 30 mL/min, had normal baseline serum intact parathyroid hormone levels. It is possible that the bone biologic response could differ between patients with chronic kidney disease who have an elevated as compared with a normal serum parathyroid hormone level. This issue should be investigated.
TREATING OSTEOPOROSIS IN STAGE 4 OR 5 CHRONIC KIDNEY DISEASE
Treatment decisions are more difficult in patients with stage 4 and especially stage 5 chronic kidney disease who have had fragility fractures. This is even the case when the clinician has determined to the best of his or her ability that the patient has osteoporosis rather than renal osteodystrophy.
There are no prospective data showing any of the approved drugs to be effective in treating osteoporosis in patients whose GFRs are lower than 30 mL/min. However, a post hoc analysis of pooled data from nine clinical trials62 found that risedronate (Actonel) 5 mg/day reduced the incidence of new vertebral fractures. Another post hoc analysis, from the Fracture Intervention Trial,63 found that alendronate (Fosamax) 5 mg/day for the first 2 years and 10 mg/day for the 3rd year reduced the incidence of all clinical fractures. In neither of these post hoc analyses did the drug affect the serum creatinine concentration. The patients—postmenopausal women—had GFRs as low as 15 mL/min as estimated by the Cockcroft-Gault equation. Similar post hoc data have been published on raloxifene (Evista).64
There are no data on the efficacy (reduction in fracture risk) or safety of any bisphosphonate in patients with GFRs lower than 15 mL/min (stage 5 chronic kidney disease). Nevertheless, the question often arises when fragility fractures occur in this population. Here, only opinion and controversy exist, and we fervently await good science and randomized prospective data.
How to manage renal bone disease after transplantation is a distinctly separate issue in which bisphosphonate use may be even more controversial than in end-stage renal disease.65,66
In my opinion, patients without fractures with stage 5 chronic kidney disease should not be given bisphosphonates or teriparatide offlabel. Treating only on the basis of low bone mineral density and other risk factors seems to be associated with greater risk than benefit.
In stage 5 patients suffering fragility fractures, a bisphosphonate may be considered, but only after renal osteodystrophy has been thoroughly ruled out, which most often requires a bone biopsy.43,67,68 In skilled hands, transiliac bone biopsy is a safe procedure with little morbidity.
If osteoporosis appears to be the cause of the fracture, and if one chooses to use a bisphosphonate and the patient gives his or her informed consent, then I would give half the usual dose and restrict the therapy to no more than 3 years. The reason for halving the dose is based on the known pharmacokinetics of bisphosphonates in people with normal renal function: 50% of a given dose goes to bone and 50% is excreted by the kidney. Furthermore, the dialyzability of bisphosphonates has not been well studied. Limiting the treatment to 3 years is based on the unknown but probably greater bone retention of bisphosphonates when excretion is impaired.
I must emphasize that these approaches are not based on any evidence of efficacy, but rather are considered in extreme cases of often-recurrent fragility fractures in which the fractures per se pose a great risk of morbidity and death. These approaches should be clearly discussed with the patient, undertaken by specialists knowledgeable in complex metabolic bone disease management, and initiated only after the skeletal fragility disorder is well characterized.
SUMMING UP
No consensus exists on the criteria for diagnosing osteoporosis in stage 4 or 5 chronic kidney disease.
In higher-risk patients in stage 1, 2, or 3 chronic kidney disease who have osteoporosis, it appears that any drug approved for osteoporosis can be used, eg, a bisphosphonate, teriparatide, or both.
Considerations for management are far more complex in stage 4 or 5 because of the increased prevalence of other metabolic bone diseases and renal osteodystrophy, and because the World Health Organization criteria cannot be used to diagnose osteoporosis. In stage 5, the differential diagnosis requires careful analysis of a broad range of biochemical markers of bone turnover and, at times, quantitative bone histomorphometry, especially if one is considering using a bisphosphonate. It is unknown if bisphosphonates, by reducing bone turnover in a preexisting low-bone-turnover state, would help or harm bone or would lead to less or more cardiovascular disease. These questions must be addressed by better science and prospective data.
In the future, newer noninvasive radiologic tools to measure microstructure and mineralization of bone promise to help us better understand osteoporosis and renal osteodystrophy in a noninvasive manner.
In clinical practice, at the current time and with current limited knowledge, treatment of osteoporosis in stage 4 or 5 chronic kidney disease is opinion-based. Nevertheless, in very specific clinical cases of severe fragility fractures that, by themselves, may cause disability and death, bisphosphonates should be considered by experts in bone metabolism and, as with any off-label application, after careful informed discussions with the patient.
- Melton LJ. Epidemiology worldwide. Endocrinol Metab Clin North Am 2003; 32:1–13.
- Burge R, Dawson-Hughes B, Solomon DH, Wong JB, King A, Tosteson A. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005–2025. J Bone Miner Res 2007; 22:465–475.
- Barrett-Connor E, Siris ES, Wehren LE, et al. Osteoporosis and fracture risk in women of different ethnic groups. J Bone Miner Res 2005; 20:185–194.
- Assessment of fracture risk and its application to screening for postmenopausal osteoporosis. Report of a WHO Study Group. World Health Organ Tech Rep Ser 1994; 843:1–129.
- Miller PD, Bonnick SL. Clinical application of bone densitometry. In:Favus MJ, editor. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 4th ed. American Society for Bone and Mineral Research. Philadelphia, PA: Lippincott Williams & Wilkins; 1999.
- Nickolas TL, Leonard MB, Shane E. Chronic kidney disease and bone fracture: a growing concern. Kidney Int 2008; 74:721–731.
- Gal-Moscovici A, Sprague SM. Osteoporosis and chronic kidney disease. Semin Dial 2007; 20:423–430.
- National Kidney Foundation. Clinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am J Kidney Dis 2003; 42(suppl 3):S1–S201.
- Coresh J, Astor BC, Greene T, Eknoyan G, Levey AS. Prevalence of chronic kidney disease and decreased kidney function in the adult US population: Third National Health and Nutrition Examination Survey. Am J Kidney Dis 2003; 41:1–12.
- Bennett WM. Reporting eGFR. Clin J Am Soc Nephrol 2008; 3:1561–1562.
- Glassock RJ, Winearls C. Screening for CKD with eGFR: doubts and dangers. Clin J Am Soc Nephrol 2008; 3:1563–1568.
- Parfitt AM, Drezner M, Glorieux F, et al. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res 1987; 2:595–610.
- Andress DL, Sherrard DJ. The osteodystrophy of chronic renal failure. In:Schrier RW, editor: Diseases of the Kidney and Urinary Tract. Philadelphia, PA: Lippincott Williams & Wilkins; 2007:2431–2453.
- Moe S, Drueke T, Cunningham J, et al. Definition, evaluation, and classification of renal osteodystrophy: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int 2006; 69:1945–1953.
- NIH Consensus Development Panel Osteoporosis Prevention Diagnosis and Therapy. Osteoporosis prevention, diagnosis, and therapy. JAMA 2001; 285:785–795.
- Baim S, Binkley N, Bilezikian JP, et al. Official positions of the International Society for Clinical Densitometry and executive summary of the 2007 ISCD Position Development Conference. J Clin Densitom 2008; 11:75–91.
- Siris E, Miller P, Barrett-Connor E, et al. Identification and fracture outcomes of undiagnosed low bone mineral density in postmenopausal women: results from the National Osteoporosis Risk Assessment (NORA). JAMA 2001; 286:2815–2822.
- Schuit SCE, Oei HH, Witteman JC, et al. Fracture incidence and association with bone mineral density in elderly men and women: the Rotterdam Study. Bone 2004; 34:195–202.
- Bouxsein ML. Non-invasive measurements of bone strength: promise and peril. J Musculoskelet Neuronal Interact 2004; 4:404–405.
- Seeman E. Bone quality: the material and structural basis of bone strength. J Bone Miner Metab 2008; 26:1–8.
- Alem AM, Sherrard DJ, Gillen DL, et al. Increased risk of hip fracture among patients with end-stage renal disease. Kidney Int 2000; 58:396–399.
- Ball AM, Gillen DL, Sherrard D, et al. Risk of hip fracture among dialysis and renal transplant recipients. JAMA 2002; 288:3014–3018.
- Jadoul M, Albert JM, Akiba T, et al. Incidence and risk factors for hip or other bone fractures among hemodialysis patients in the Dialysis Outcomes and Practice Patterns Study. Kidney Int 2006; 70:1358–1366.
- Stehman-Breen CO, Sherrard DJ, Alem AM, et al. Risk factors for hip fracture among patients with end-stage renal disease. Kidney Int 2000; 58:2200–2205.
- Wehrli FW, Leonard MB, Saha PK, Gomberg BR. Quantitative highresolution magnetic resonance imaging reveals structural implications of renal osteodystrophy on trabecular and cortical bone. J Magn Reson Imaging 2004; 20:83–89.
- Genant HK, Lang TF, Engelke K, et al. Advances in the noninvasive assessment of bone density, quality, and structure. Calcif Tissue Int 1996; 59(suppl 1):S10–S15.
- Roschger P, Paschalis EP, Fratzl P, Klaushofer K. Bone mineralization density distribution in health and disease. Bone 2008; 42:456–466.
- Miller PD. Bone density and markers of bone turnover in predicting fracture risk and how changes in these measures predict fracture risk reduction. Curr Osteoporos Rep 2005; 3:103–110.
- Miller PD, Hochberg MC, Wehren LE, Ross PD, Wasnich RD. How useful are measures of BMD and bone turnover? Curr Med Res Opin 2005; 21:545–554.
- Chavassieux PM, Delmas PD. Bone remodeling: biochemical markers or bone biopsy? J Bone Miner Res 2006; 21:178–179.
- Garnero P. Biomarkers for osteoporosis management: utility in diagnosis, fracture risk prediction and therapy monitoring. Mol Diagn Ther 2008; 12:157–170.
- Hochberg M, Greenspan S, Wasnich R, Miller P, Thompson D, Ross P. Changes in bone density and turnover explain the reductions in incidence of nonvertebral fractures that occur during treatment with antiresorptive agents. J Clin Endocrinol Metab 2002; 87:1586–1592.
- Bouxsein ML, Delmas PD. Considerations for development of surrogate endpoints for antifracture efficacy of new treatments in osteoporosis: a perspective. J Bone Miner Res 2008; 23:1155–1167.
- Chen P, Satterwhite JH, Licatta AA, et al. Early changes in biochemical markers of bone formation predict BMD response to teriparatide in postmenopausal women with osteoporosis. J Bone Miner Res 2005; 20:962–970.
- Miller PD, Delmas PD, Lindsay R, et al. Early responsiveness of women with osteoporosis to teriparatide after therapy with alendronate or risedronate. J Clin Endocrinol Metab 2008; 93:3785–3793.
- Baim S, Miller PD. Assessing the clinical utility of serum CTX in postmenopausal osteoporosis and its use in predicting risk of osteonecrosis of the jaw. J Bone Miner Res 2009; 24:561–574.
- Miller PD, Lerma EV. Renal bone diseases. In:Kleerekoper M, Siris E, McClung M, editors. The Bone and Mineral Manual—A Practical Guide. 2nd ed. Burlington, MA: Elsevier Academic Press; 2005:127–138.
- Miller PD, Shane E. Management of transplantation renal bone disease: Interplay of bone mineral density and decisions regarding bisphosphonate use. In:Weir MR, editor. Medical Management of Kidney Transplantation. Philadelphia, PA: Lippincott Williams & Wilkins; 2004:359–375.
- Levin A, Bakris GL, Molitch M, et al. Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: results of the study to evaluate early kidney disease. Kidney int 2007; 71:31–38.
- Vassalotti JA, Uribarri J, Chen SC, et al. Trends in mineral metabolism: Kidney Early Evaluation Program (KEEP) and the National Health and Nutrition Examination Survey (NHANES) 1999–2004. Am J Kidney Dis 2008; 51(suppl 2):S56–S68.
- Meier C, Seibel MJ, Kraenzlin ME. Use of bone turnover markers in the real world: are we there yet? J Bone Miner Res 2009; 24:386–388.
- Lehmann G, Ott U, Kaemmerer D, Schuetze J, Wolf G. Bone histomorphometry and biochemical markers of bone turnover in patients with chronic kidney disease stages 3–5. Clin Nephrol 2008; 70:296–305.
- Miller PD. The role of bone biopsy in patients with chronic renal failure. Clin J Am Soc Nephrol 2008; 3(suppl 3):S140–S150.
- Frost HM. Tetracycline-based histological analysis of bone remodeling. Calcif Tissue Res 1969; 3:211–237.
- Hitt O, Jaworski ZF, Shimizu AG, Frost HM. Tissue-level bone formation rates in chronic renal failure, measured by means of tetracycline bone labeling. Can J Physiol Pharmacol 1970; 48:824–828.
- Coen G. Adynamic bone disease: an update and overview. J Nephrol 2005; 18:117–122.
- Parfitt AM. Renal bone disease: a new conceptual framework for the interpretation of bone histomorphometry. Curr Opin Nephrol Hypertens 2003; 12:387–403.
- Brandenburg VM, Floege J. Adynamic bone diseaseùbone and beyond. NDT Plus 2008; 3:135–147. doi:10.1093/ndtplus/sfn040.
- Hruska KA, Saab G, Mathew S, Lund R. Renal osteodystrophy, phosphate homeostasis, and vascular calcification. Semin Dial 2007; 20:309–315.
- Toussaint ND, Elder GJ, Kerr PG. Bisphosphonates in chronic kidney disease; balancing potential benefits and adverse effects on bone and soft tissue. Clin J Am Soc Nephrol 2009; 4:221–233.
- Miller PD. Is there a role for bisphosphonates in chronic kidney disease? Semin Dial 2007; 20:186–190.
- Miller PD. Bisphosphonates: pharmacology and use in the treatment of osteoporosis. In:Marcus R, Feldman D, Nelson DA, Rosen CJ, editors. Osteoporosis. 3rd ed. Boston, MA: Elsevier Academic Press; 2008:1725–1736.
- Russell RG, Watts NB, Ebetino FH, Rogers MJ. Mechanisms of action of bisphosphonates: similarities and differences and their potential influence on clinical efficacy. Osteoporosis Int 2008; 19:733–759.
- Eisman JA, Civitelli R, Adami S, et al. Efficacy and tolerability of intravenous ibandronate injections in postmenopausal osteoporosis: 2-year results from the DIVA study. J Rheumatol 2008; 35:488–497.
- Black DM, Delmas PD, Eastell RR, et al. Once-yearly zoledronic acid for treatment of postmenopausal osteoporosis. N Engl J Med 2007; 356:1809–1822.
- Lewiecki EM, Miller PD. Renal safety of intravenous bisphosphonates in the treatment of osteoporosis. Expert Opin Drug Saf 2007; 6:663–672.
- Miller PD. Anti-resorptives in the management of osteoporosis. Best Pract Res Clin Endocrinol Metab 2008; 22:849–868.
- Perazella MA, Markowitz GS. Bisphosphonate nephrotoxicity. Kidney Int 2008; 74:1385–1393.
- Boonen S, Sellmeyer DE, Lippuner K, et al. Renal safety of annual zoledronic acid infusions in osteoporotic postmenopausal women. Kidney Int 2008; 74:641–648.
- Neer RM, Arnaud CD, Zanchetta JR, et al. Effect of parathyroid hormone (1–34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med 2001; 344:1434–1441.
- Miller PD, Schwartz EN, Chen P, Misurski DA, Krege JH. Teriparatide in postmenopausal women with osteoporosis and mild or moderate renal impairment. Osteoporosis Int 2007; 18:59–68.
- Miller PD, Roux C, Boonen S, Barton I, Dunlap L, Burgio D. Safety and efficacy of risedronate in patients with age-related reduced renal function as estimated by the Cockcroft and Gault method: a pooled analysis of nine clinical trials. J Bone Miner Res 2005; 20:2105–2115.
- Jamal SA, Bauer DC, Ensrud KE, et al. Alendronate treatment in women with normal to severely impaired renal function: an analysis of the fracture intervention trial. J Bone Miner Res 2007; 22:503–508.
- Ishani A, Blackwell T, Jamal SA, Cummings SR, Ensrud KE; MORE Investigators. The effect of raloxifene treatment in postmenopausal women with CKD. J Am Soc Nephrol 2008; 19:1430–1438.
- Coco M, Glicklich D, Faugere MC, et al. Prevention of bone loss in renal transplant recipients: a prospective, randomized trial of intravenous pamidronate. J Am Soc Nephrol 2003; 14:2669–2676.
- Palmer SC, McGregor DO, Strippoli GF. Interventions for preventing bone disease in kidney transplant recipients. Cochrane Database Syst Rev 2007;CD005015.
- Ferreira MA. Diagnosis of renal osteodystrophy: when and how to use biochemical markers and non-invasive methods; when bone biopsy is needed. Nephrol Dial Transplant 2000; 15(suppl 5):8–14.
- Trueba D, Sawaya BP, Mawad H, Malluche HH. Bone biopsy: indications, techniques, and complications. Semin Dial 2003; 16:341–345.
- Melton LJ. Epidemiology worldwide. Endocrinol Metab Clin North Am 2003; 32:1–13.
- Burge R, Dawson-Hughes B, Solomon DH, Wong JB, King A, Tosteson A. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005–2025. J Bone Miner Res 2007; 22:465–475.
- Barrett-Connor E, Siris ES, Wehren LE, et al. Osteoporosis and fracture risk in women of different ethnic groups. J Bone Miner Res 2005; 20:185–194.
- Assessment of fracture risk and its application to screening for postmenopausal osteoporosis. Report of a WHO Study Group. World Health Organ Tech Rep Ser 1994; 843:1–129.
- Miller PD, Bonnick SL. Clinical application of bone densitometry. In:Favus MJ, editor. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 4th ed. American Society for Bone and Mineral Research. Philadelphia, PA: Lippincott Williams & Wilkins; 1999.
- Nickolas TL, Leonard MB, Shane E. Chronic kidney disease and bone fracture: a growing concern. Kidney Int 2008; 74:721–731.
- Gal-Moscovici A, Sprague SM. Osteoporosis and chronic kidney disease. Semin Dial 2007; 20:423–430.
- National Kidney Foundation. Clinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am J Kidney Dis 2003; 42(suppl 3):S1–S201.
- Coresh J, Astor BC, Greene T, Eknoyan G, Levey AS. Prevalence of chronic kidney disease and decreased kidney function in the adult US population: Third National Health and Nutrition Examination Survey. Am J Kidney Dis 2003; 41:1–12.
- Bennett WM. Reporting eGFR. Clin J Am Soc Nephrol 2008; 3:1561–1562.
- Glassock RJ, Winearls C. Screening for CKD with eGFR: doubts and dangers. Clin J Am Soc Nephrol 2008; 3:1563–1568.
- Parfitt AM, Drezner M, Glorieux F, et al. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res 1987; 2:595–610.
- Andress DL, Sherrard DJ. The osteodystrophy of chronic renal failure. In:Schrier RW, editor: Diseases of the Kidney and Urinary Tract. Philadelphia, PA: Lippincott Williams & Wilkins; 2007:2431–2453.
- Moe S, Drueke T, Cunningham J, et al. Definition, evaluation, and classification of renal osteodystrophy: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int 2006; 69:1945–1953.
- NIH Consensus Development Panel Osteoporosis Prevention Diagnosis and Therapy. Osteoporosis prevention, diagnosis, and therapy. JAMA 2001; 285:785–795.
- Baim S, Binkley N, Bilezikian JP, et al. Official positions of the International Society for Clinical Densitometry and executive summary of the 2007 ISCD Position Development Conference. J Clin Densitom 2008; 11:75–91.
- Siris E, Miller P, Barrett-Connor E, et al. Identification and fracture outcomes of undiagnosed low bone mineral density in postmenopausal women: results from the National Osteoporosis Risk Assessment (NORA). JAMA 2001; 286:2815–2822.
- Schuit SCE, Oei HH, Witteman JC, et al. Fracture incidence and association with bone mineral density in elderly men and women: the Rotterdam Study. Bone 2004; 34:195–202.
- Bouxsein ML. Non-invasive measurements of bone strength: promise and peril. J Musculoskelet Neuronal Interact 2004; 4:404–405.
- Seeman E. Bone quality: the material and structural basis of bone strength. J Bone Miner Metab 2008; 26:1–8.
- Alem AM, Sherrard DJ, Gillen DL, et al. Increased risk of hip fracture among patients with end-stage renal disease. Kidney Int 2000; 58:396–399.
- Ball AM, Gillen DL, Sherrard D, et al. Risk of hip fracture among dialysis and renal transplant recipients. JAMA 2002; 288:3014–3018.
- Jadoul M, Albert JM, Akiba T, et al. Incidence and risk factors for hip or other bone fractures among hemodialysis patients in the Dialysis Outcomes and Practice Patterns Study. Kidney Int 2006; 70:1358–1366.
- Stehman-Breen CO, Sherrard DJ, Alem AM, et al. Risk factors for hip fracture among patients with end-stage renal disease. Kidney Int 2000; 58:2200–2205.
- Wehrli FW, Leonard MB, Saha PK, Gomberg BR. Quantitative highresolution magnetic resonance imaging reveals structural implications of renal osteodystrophy on trabecular and cortical bone. J Magn Reson Imaging 2004; 20:83–89.
- Genant HK, Lang TF, Engelke K, et al. Advances in the noninvasive assessment of bone density, quality, and structure. Calcif Tissue Int 1996; 59(suppl 1):S10–S15.
- Roschger P, Paschalis EP, Fratzl P, Klaushofer K. Bone mineralization density distribution in health and disease. Bone 2008; 42:456–466.
- Miller PD. Bone density and markers of bone turnover in predicting fracture risk and how changes in these measures predict fracture risk reduction. Curr Osteoporos Rep 2005; 3:103–110.
- Miller PD, Hochberg MC, Wehren LE, Ross PD, Wasnich RD. How useful are measures of BMD and bone turnover? Curr Med Res Opin 2005; 21:545–554.
- Chavassieux PM, Delmas PD. Bone remodeling: biochemical markers or bone biopsy? J Bone Miner Res 2006; 21:178–179.
- Garnero P. Biomarkers for osteoporosis management: utility in diagnosis, fracture risk prediction and therapy monitoring. Mol Diagn Ther 2008; 12:157–170.
- Hochberg M, Greenspan S, Wasnich R, Miller P, Thompson D, Ross P. Changes in bone density and turnover explain the reductions in incidence of nonvertebral fractures that occur during treatment with antiresorptive agents. J Clin Endocrinol Metab 2002; 87:1586–1592.
- Bouxsein ML, Delmas PD. Considerations for development of surrogate endpoints for antifracture efficacy of new treatments in osteoporosis: a perspective. J Bone Miner Res 2008; 23:1155–1167.
- Chen P, Satterwhite JH, Licatta AA, et al. Early changes in biochemical markers of bone formation predict BMD response to teriparatide in postmenopausal women with osteoporosis. J Bone Miner Res 2005; 20:962–970.
- Miller PD, Delmas PD, Lindsay R, et al. Early responsiveness of women with osteoporosis to teriparatide after therapy with alendronate or risedronate. J Clin Endocrinol Metab 2008; 93:3785–3793.
- Baim S, Miller PD. Assessing the clinical utility of serum CTX in postmenopausal osteoporosis and its use in predicting risk of osteonecrosis of the jaw. J Bone Miner Res 2009; 24:561–574.
- Miller PD, Lerma EV. Renal bone diseases. In:Kleerekoper M, Siris E, McClung M, editors. The Bone and Mineral Manual—A Practical Guide. 2nd ed. Burlington, MA: Elsevier Academic Press; 2005:127–138.
- Miller PD, Shane E. Management of transplantation renal bone disease: Interplay of bone mineral density and decisions regarding bisphosphonate use. In:Weir MR, editor. Medical Management of Kidney Transplantation. Philadelphia, PA: Lippincott Williams & Wilkins; 2004:359–375.
- Levin A, Bakris GL, Molitch M, et al. Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: results of the study to evaluate early kidney disease. Kidney int 2007; 71:31–38.
- Vassalotti JA, Uribarri J, Chen SC, et al. Trends in mineral metabolism: Kidney Early Evaluation Program (KEEP) and the National Health and Nutrition Examination Survey (NHANES) 1999–2004. Am J Kidney Dis 2008; 51(suppl 2):S56–S68.
- Meier C, Seibel MJ, Kraenzlin ME. Use of bone turnover markers in the real world: are we there yet? J Bone Miner Res 2009; 24:386–388.
- Lehmann G, Ott U, Kaemmerer D, Schuetze J, Wolf G. Bone histomorphometry and biochemical markers of bone turnover in patients with chronic kidney disease stages 3–5. Clin Nephrol 2008; 70:296–305.
- Miller PD. The role of bone biopsy in patients with chronic renal failure. Clin J Am Soc Nephrol 2008; 3(suppl 3):S140–S150.
- Frost HM. Tetracycline-based histological analysis of bone remodeling. Calcif Tissue Res 1969; 3:211–237.
- Hitt O, Jaworski ZF, Shimizu AG, Frost HM. Tissue-level bone formation rates in chronic renal failure, measured by means of tetracycline bone labeling. Can J Physiol Pharmacol 1970; 48:824–828.
- Coen G. Adynamic bone disease: an update and overview. J Nephrol 2005; 18:117–122.
- Parfitt AM. Renal bone disease: a new conceptual framework for the interpretation of bone histomorphometry. Curr Opin Nephrol Hypertens 2003; 12:387–403.
- Brandenburg VM, Floege J. Adynamic bone diseaseùbone and beyond. NDT Plus 2008; 3:135–147. doi:10.1093/ndtplus/sfn040.
- Hruska KA, Saab G, Mathew S, Lund R. Renal osteodystrophy, phosphate homeostasis, and vascular calcification. Semin Dial 2007; 20:309–315.
- Toussaint ND, Elder GJ, Kerr PG. Bisphosphonates in chronic kidney disease; balancing potential benefits and adverse effects on bone and soft tissue. Clin J Am Soc Nephrol 2009; 4:221–233.
- Miller PD. Is there a role for bisphosphonates in chronic kidney disease? Semin Dial 2007; 20:186–190.
- Miller PD. Bisphosphonates: pharmacology and use in the treatment of osteoporosis. In:Marcus R, Feldman D, Nelson DA, Rosen CJ, editors. Osteoporosis. 3rd ed. Boston, MA: Elsevier Academic Press; 2008:1725–1736.
- Russell RG, Watts NB, Ebetino FH, Rogers MJ. Mechanisms of action of bisphosphonates: similarities and differences and their potential influence on clinical efficacy. Osteoporosis Int 2008; 19:733–759.
- Eisman JA, Civitelli R, Adami S, et al. Efficacy and tolerability of intravenous ibandronate injections in postmenopausal osteoporosis: 2-year results from the DIVA study. J Rheumatol 2008; 35:488–497.
- Black DM, Delmas PD, Eastell RR, et al. Once-yearly zoledronic acid for treatment of postmenopausal osteoporosis. N Engl J Med 2007; 356:1809–1822.
- Lewiecki EM, Miller PD. Renal safety of intravenous bisphosphonates in the treatment of osteoporosis. Expert Opin Drug Saf 2007; 6:663–672.
- Miller PD. Anti-resorptives in the management of osteoporosis. Best Pract Res Clin Endocrinol Metab 2008; 22:849–868.
- Perazella MA, Markowitz GS. Bisphosphonate nephrotoxicity. Kidney Int 2008; 74:1385–1393.
- Boonen S, Sellmeyer DE, Lippuner K, et al. Renal safety of annual zoledronic acid infusions in osteoporotic postmenopausal women. Kidney Int 2008; 74:641–648.
- Neer RM, Arnaud CD, Zanchetta JR, et al. Effect of parathyroid hormone (1–34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med 2001; 344:1434–1441.
- Miller PD, Schwartz EN, Chen P, Misurski DA, Krege JH. Teriparatide in postmenopausal women with osteoporosis and mild or moderate renal impairment. Osteoporosis Int 2007; 18:59–68.
- Miller PD, Roux C, Boonen S, Barton I, Dunlap L, Burgio D. Safety and efficacy of risedronate in patients with age-related reduced renal function as estimated by the Cockcroft and Gault method: a pooled analysis of nine clinical trials. J Bone Miner Res 2005; 20:2105–2115.
- Jamal SA, Bauer DC, Ensrud KE, et al. Alendronate treatment in women with normal to severely impaired renal function: an analysis of the fracture intervention trial. J Bone Miner Res 2007; 22:503–508.
- Ishani A, Blackwell T, Jamal SA, Cummings SR, Ensrud KE; MORE Investigators. The effect of raloxifene treatment in postmenopausal women with CKD. J Am Soc Nephrol 2008; 19:1430–1438.
- Coco M, Glicklich D, Faugere MC, et al. Prevention of bone loss in renal transplant recipients: a prospective, randomized trial of intravenous pamidronate. J Am Soc Nephrol 2003; 14:2669–2676.
- Palmer SC, McGregor DO, Strippoli GF. Interventions for preventing bone disease in kidney transplant recipients. Cochrane Database Syst Rev 2007;CD005015.
- Ferreira MA. Diagnosis of renal osteodystrophy: when and how to use biochemical markers and non-invasive methods; when bone biopsy is needed. Nephrol Dial Transplant 2000; 15(suppl 5):8–14.
- Trueba D, Sawaya BP, Mawad H, Malluche HH. Bone biopsy: indications, techniques, and complications. Semin Dial 2003; 16:341–345.
KEY POINTS
- If the patient’s glomerular filtration rate (GFR) is at least 30 mL/min/1.73 m2 and if no biochemical test results suggest renal osteodystrophy, osteoporosis can be diagnosed if the T score is less than −2.5 or if the patient has had a fragility fracture. These criteria can also probably be applied, though with less certainty, if the patient’s GFR is as low as 15.
- If the patient’s GFR is less than 15 or if he or she is on dialysis, biochemical profiling often cannot distinguish among the heterogeneous forms of renal bone disease. In some cases of severe chronic kidney disease with fractures, bone biopsy is needed to rule out renal osteodystrophy and to diagnose osteoporosis by exclusion.
- In the author’s opinion, in patients with severe chronic kidney disease and fractures who have “osteoporosis” by exclusion, off-label use of bisphosphonates is an option, but only after very careful consideration.
Treating the renal patient who has a fracture: Opinion vs evidence
Managing bone health in patients with chronic kidney disease presents unique challenges. While the common end point—a fracture—is comparable to that in patients with osteoporosis, the underlying metabolic conditions differ from patient to patient with chronic kidney disease and may be dramatically different from those in patients who have osteoporosis without chronic kidney disease.
Renal osteodystrophy is not osteoporosis
Renal osteodystrophy is not osteoporosis. While osteoporosis in people without kidney disease is defined clinically on the basis of bone mineral density (measured by bone densitometry), renal osteodystrophy is a histologic diagnosis made on bone biopsy: it is a continuum between frankly low-turnoverbone disease—encompassing adynamic bone disease and osteomalacia—and frankly highturnover-bone disease, with severe secondary hyperparathyroid bone disease and osteitis fibrosa. Histologically, there may or may not be low trabecular bone volume or loss of connectivity typical of the bone loss in osteoporosis.
Patients at both ends of the spectrum of bone turnover in renal osteodystrophy may have the same bone mineral density on densitometry. Low bone mineral density may reflect inadequate mineralization (seen in osteomalacia and adynamic bone disease) or increased peritrabecular fibrosis (seen in secondary hyperparathyroid bone disease). High bone mineral density readings may capture extraosseous calcifications, which are very common in chronic kidney disease.
Renal osteodystrophy is part of the syndrome called chronic kidney disease-mineral and bone disease,1 which is not limited to bone fractures but may also affect vascular health. Abnormal calcium deposits in vascular tissue—consistent with calciphylaxis and associated with increased morbidity and mortality rates in chronic kidney disease—may occur with low bone turnover.
The diagnosis of osteoporosis in the general population is based on clinical evidence: the measured bone mineral density is compared with normalized scores. Histologically, the bone of the osteoporotic patient shows osteopenia with increased bone turnover and a shift toward increased bone resorption, resulting in loss of connectivity of the trabeculae, as well as decreased trabecular volume. These conditions are common in advanced age and in certain pathologic states (eg, steroid therapy, metastatic bone disease, Paget disease of bone).
It is well accepted that the risk of fracture in osteoporosis increases as measured bone mineral density decreases. Conversely, increasing bone mineral density has been correlated with fewer fractures. The clinician is often guided by biomarkers of bone metabolism such as urinary N-terminal cross-linked telopeptides of collagen (NTx) in diagnosing and treating bone breakdown.
Can bisphosphonates be used in chronic kidney disease?
Bisphosphonates are antiresorptive agents that bind to the hydroxyapatite of bone. They poison the osteoclast (the bone-resorbing cell), causing its death and thereby halting the resorption of bone. Osteoblasts—the boneforming cells—are presumably not affected, and the bone continues to make osteoid, which is subsequently mineralized. Bone turnover is dramatically decreased. The net effect is increased bone density in people with osteoporosis. The half-life of these agents is years.
In the general population, bisphosphonate therapy has been associated with decreased risk of fragility bone fractures. However, the long-term effects are not yet known. Indeed, jaw necrosis—possibly due to low bone turnover—is being reported with increasing frequency.2 Fractures associated with low bone turnover in patients without chronic kidney disease treated with bisphosphonates longterm are now being reported.3,4
In an article in this issue of the Journal,5 the author advocates the use of bisphosphonate therapy in patients with chronic kidney disease who have low bone mineral density. However, treating patients who have chronic kidney disease on the basis of low bone mineral density with bone-suppressing agents may further depress bone turnover and lead to more extraosseous calcifications as the turnedoff bone is unable to accept serum calcium.6
Further, it is unclear how long “long-term” would be in a patient with advanced chronic kidney disease: Would the half-life of the bisphosphonates be tremendously increased, leading to adverse events sooner? Would adynamic bone disease promptly develop, leading to rampant jaw necrosis and bone fractures? Would vascular calcification flourish?
Bone biomarkers are hard to interpret in chronic kidney disease
In chronic kidney disease, the interpretation of biomarkers of bone metabolism is notoriously unreliable. The usual chemistry values associated with clinical osteoporosis in the general population—ie, elevated levels of urinary NTx, serum C-terminal cross-linked telopeptides of collagen (CTx), osteocalcin, and bone-specific alkaline phosphatase—are not valid in patients with chronic kidney disease, for obvious reasons: with declining renal function, the various markers accumulate in the serum. Urinary NTx does not apply in patients with advanced chronic kidney disease or end-stage renal disease.
How should renal osteodystrophy be treated?
Nephrologists currently focus therapy on reducing hyperphosphatemia (associated with increased morbidity across all stages of chronic kidney disease), replenishing vitamin D as much as possible without causing hyperphosphatemia and hypercalcemia, and suppressing parathyroid hormone secretion.
However, there is not enough evidence on what the goal should be with respect to parathyroid hormone in patients with chronic kidney disease who are not on dialysis. Although in the recent past many believed that parathyroid hormone goals should be 150 to 300 pg/mL in dialysis patients, the latest guidelines suggest that perhaps this goal is too narrow and may lead to more adynamic bone disease. Similarly, there is no consensus on the use of synthetic parathyroid hormone analogues.
Bisphosphonate therapy, particularly with pamidronate (Aredia) and zolendronic acid (Reclast), has been associated with adverse renal effects even in patients without chronic kidney disease. There are no prospective studies of the effects of these agents in patients with depressed renal function.
The patient with chronic kidney disease who has a fracture remains a unique problem for the nephrologist, primary care physician, and subspecialist. Efforts should be concentrated on preventing and treating metabolic bone disease in its entire spectrum, with rational, prospective studies, and should not depend on anecdotal reports. Opinions abound, without adequate evidence to back them up.
- Moe S, Drüeke T, Cunningham J, et al; Kidney Disease: Improving Global Outcomes (KDIGO). Definition, evaluation, and classification of renal osteodystrophy: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int 2006; 69:1945–1953.
- Rustemeyer J, Bremerich A. Bisphosphonate-associated osteonecrosis of the jaw: what do we currently know? A survey of knowledge given in the recent literature. Clin Oral Investig 2009; Epub ahead of print.
- Armamento-Villareal R, Napoli N, Diemer K, et al. Bone turnover in bone biopsies of patients with low-energy cortical fractures receiving bisphosphonates: a case series. Calcif Tissue Int 2009; 85:37–44.
- Ali T, Jay RH. Spontaneous femoral shaft fracture after long-term alendronate. Age Ageing 2009; Epub ahead of print.
- Miller PD. Fragility fractures in chronic kidney disease: an opinionbased approach. Cleve Clin J Med 2009; 76:713–721.
- Toussaint ND, Elder GJ, Kerr PG. Bisphosphonates in chronic kidney disease; balancing potential benefits and adverse effects on bone and soft tissue. Clin J Am Soc Nephrol 2009; 4:221–233.
Managing bone health in patients with chronic kidney disease presents unique challenges. While the common end point—a fracture—is comparable to that in patients with osteoporosis, the underlying metabolic conditions differ from patient to patient with chronic kidney disease and may be dramatically different from those in patients who have osteoporosis without chronic kidney disease.
Renal osteodystrophy is not osteoporosis
Renal osteodystrophy is not osteoporosis. While osteoporosis in people without kidney disease is defined clinically on the basis of bone mineral density (measured by bone densitometry), renal osteodystrophy is a histologic diagnosis made on bone biopsy: it is a continuum between frankly low-turnoverbone disease—encompassing adynamic bone disease and osteomalacia—and frankly highturnover-bone disease, with severe secondary hyperparathyroid bone disease and osteitis fibrosa. Histologically, there may or may not be low trabecular bone volume or loss of connectivity typical of the bone loss in osteoporosis.
Patients at both ends of the spectrum of bone turnover in renal osteodystrophy may have the same bone mineral density on densitometry. Low bone mineral density may reflect inadequate mineralization (seen in osteomalacia and adynamic bone disease) or increased peritrabecular fibrosis (seen in secondary hyperparathyroid bone disease). High bone mineral density readings may capture extraosseous calcifications, which are very common in chronic kidney disease.
Renal osteodystrophy is part of the syndrome called chronic kidney disease-mineral and bone disease,1 which is not limited to bone fractures but may also affect vascular health. Abnormal calcium deposits in vascular tissue—consistent with calciphylaxis and associated with increased morbidity and mortality rates in chronic kidney disease—may occur with low bone turnover.
The diagnosis of osteoporosis in the general population is based on clinical evidence: the measured bone mineral density is compared with normalized scores. Histologically, the bone of the osteoporotic patient shows osteopenia with increased bone turnover and a shift toward increased bone resorption, resulting in loss of connectivity of the trabeculae, as well as decreased trabecular volume. These conditions are common in advanced age and in certain pathologic states (eg, steroid therapy, metastatic bone disease, Paget disease of bone).
It is well accepted that the risk of fracture in osteoporosis increases as measured bone mineral density decreases. Conversely, increasing bone mineral density has been correlated with fewer fractures. The clinician is often guided by biomarkers of bone metabolism such as urinary N-terminal cross-linked telopeptides of collagen (NTx) in diagnosing and treating bone breakdown.
Can bisphosphonates be used in chronic kidney disease?
Bisphosphonates are antiresorptive agents that bind to the hydroxyapatite of bone. They poison the osteoclast (the bone-resorbing cell), causing its death and thereby halting the resorption of bone. Osteoblasts—the boneforming cells—are presumably not affected, and the bone continues to make osteoid, which is subsequently mineralized. Bone turnover is dramatically decreased. The net effect is increased bone density in people with osteoporosis. The half-life of these agents is years.
In the general population, bisphosphonate therapy has been associated with decreased risk of fragility bone fractures. However, the long-term effects are not yet known. Indeed, jaw necrosis—possibly due to low bone turnover—is being reported with increasing frequency.2 Fractures associated with low bone turnover in patients without chronic kidney disease treated with bisphosphonates longterm are now being reported.3,4
In an article in this issue of the Journal,5 the author advocates the use of bisphosphonate therapy in patients with chronic kidney disease who have low bone mineral density. However, treating patients who have chronic kidney disease on the basis of low bone mineral density with bone-suppressing agents may further depress bone turnover and lead to more extraosseous calcifications as the turnedoff bone is unable to accept serum calcium.6
Further, it is unclear how long “long-term” would be in a patient with advanced chronic kidney disease: Would the half-life of the bisphosphonates be tremendously increased, leading to adverse events sooner? Would adynamic bone disease promptly develop, leading to rampant jaw necrosis and bone fractures? Would vascular calcification flourish?
Bone biomarkers are hard to interpret in chronic kidney disease
In chronic kidney disease, the interpretation of biomarkers of bone metabolism is notoriously unreliable. The usual chemistry values associated with clinical osteoporosis in the general population—ie, elevated levels of urinary NTx, serum C-terminal cross-linked telopeptides of collagen (CTx), osteocalcin, and bone-specific alkaline phosphatase—are not valid in patients with chronic kidney disease, for obvious reasons: with declining renal function, the various markers accumulate in the serum. Urinary NTx does not apply in patients with advanced chronic kidney disease or end-stage renal disease.
How should renal osteodystrophy be treated?
Nephrologists currently focus therapy on reducing hyperphosphatemia (associated with increased morbidity across all stages of chronic kidney disease), replenishing vitamin D as much as possible without causing hyperphosphatemia and hypercalcemia, and suppressing parathyroid hormone secretion.
However, there is not enough evidence on what the goal should be with respect to parathyroid hormone in patients with chronic kidney disease who are not on dialysis. Although in the recent past many believed that parathyroid hormone goals should be 150 to 300 pg/mL in dialysis patients, the latest guidelines suggest that perhaps this goal is too narrow and may lead to more adynamic bone disease. Similarly, there is no consensus on the use of synthetic parathyroid hormone analogues.
Bisphosphonate therapy, particularly with pamidronate (Aredia) and zolendronic acid (Reclast), has been associated with adverse renal effects even in patients without chronic kidney disease. There are no prospective studies of the effects of these agents in patients with depressed renal function.
The patient with chronic kidney disease who has a fracture remains a unique problem for the nephrologist, primary care physician, and subspecialist. Efforts should be concentrated on preventing and treating metabolic bone disease in its entire spectrum, with rational, prospective studies, and should not depend on anecdotal reports. Opinions abound, without adequate evidence to back them up.
Managing bone health in patients with chronic kidney disease presents unique challenges. While the common end point—a fracture—is comparable to that in patients with osteoporosis, the underlying metabolic conditions differ from patient to patient with chronic kidney disease and may be dramatically different from those in patients who have osteoporosis without chronic kidney disease.
Renal osteodystrophy is not osteoporosis
Renal osteodystrophy is not osteoporosis. While osteoporosis in people without kidney disease is defined clinically on the basis of bone mineral density (measured by bone densitometry), renal osteodystrophy is a histologic diagnosis made on bone biopsy: it is a continuum between frankly low-turnoverbone disease—encompassing adynamic bone disease and osteomalacia—and frankly highturnover-bone disease, with severe secondary hyperparathyroid bone disease and osteitis fibrosa. Histologically, there may or may not be low trabecular bone volume or loss of connectivity typical of the bone loss in osteoporosis.
Patients at both ends of the spectrum of bone turnover in renal osteodystrophy may have the same bone mineral density on densitometry. Low bone mineral density may reflect inadequate mineralization (seen in osteomalacia and adynamic bone disease) or increased peritrabecular fibrosis (seen in secondary hyperparathyroid bone disease). High bone mineral density readings may capture extraosseous calcifications, which are very common in chronic kidney disease.
Renal osteodystrophy is part of the syndrome called chronic kidney disease-mineral and bone disease,1 which is not limited to bone fractures but may also affect vascular health. Abnormal calcium deposits in vascular tissue—consistent with calciphylaxis and associated with increased morbidity and mortality rates in chronic kidney disease—may occur with low bone turnover.
The diagnosis of osteoporosis in the general population is based on clinical evidence: the measured bone mineral density is compared with normalized scores. Histologically, the bone of the osteoporotic patient shows osteopenia with increased bone turnover and a shift toward increased bone resorption, resulting in loss of connectivity of the trabeculae, as well as decreased trabecular volume. These conditions are common in advanced age and in certain pathologic states (eg, steroid therapy, metastatic bone disease, Paget disease of bone).
It is well accepted that the risk of fracture in osteoporosis increases as measured bone mineral density decreases. Conversely, increasing bone mineral density has been correlated with fewer fractures. The clinician is often guided by biomarkers of bone metabolism such as urinary N-terminal cross-linked telopeptides of collagen (NTx) in diagnosing and treating bone breakdown.
Can bisphosphonates be used in chronic kidney disease?
Bisphosphonates are antiresorptive agents that bind to the hydroxyapatite of bone. They poison the osteoclast (the bone-resorbing cell), causing its death and thereby halting the resorption of bone. Osteoblasts—the boneforming cells—are presumably not affected, and the bone continues to make osteoid, which is subsequently mineralized. Bone turnover is dramatically decreased. The net effect is increased bone density in people with osteoporosis. The half-life of these agents is years.
In the general population, bisphosphonate therapy has been associated with decreased risk of fragility bone fractures. However, the long-term effects are not yet known. Indeed, jaw necrosis—possibly due to low bone turnover—is being reported with increasing frequency.2 Fractures associated with low bone turnover in patients without chronic kidney disease treated with bisphosphonates longterm are now being reported.3,4
In an article in this issue of the Journal,5 the author advocates the use of bisphosphonate therapy in patients with chronic kidney disease who have low bone mineral density. However, treating patients who have chronic kidney disease on the basis of low bone mineral density with bone-suppressing agents may further depress bone turnover and lead to more extraosseous calcifications as the turnedoff bone is unable to accept serum calcium.6
Further, it is unclear how long “long-term” would be in a patient with advanced chronic kidney disease: Would the half-life of the bisphosphonates be tremendously increased, leading to adverse events sooner? Would adynamic bone disease promptly develop, leading to rampant jaw necrosis and bone fractures? Would vascular calcification flourish?
Bone biomarkers are hard to interpret in chronic kidney disease
In chronic kidney disease, the interpretation of biomarkers of bone metabolism is notoriously unreliable. The usual chemistry values associated with clinical osteoporosis in the general population—ie, elevated levels of urinary NTx, serum C-terminal cross-linked telopeptides of collagen (CTx), osteocalcin, and bone-specific alkaline phosphatase—are not valid in patients with chronic kidney disease, for obvious reasons: with declining renal function, the various markers accumulate in the serum. Urinary NTx does not apply in patients with advanced chronic kidney disease or end-stage renal disease.
How should renal osteodystrophy be treated?
Nephrologists currently focus therapy on reducing hyperphosphatemia (associated with increased morbidity across all stages of chronic kidney disease), replenishing vitamin D as much as possible without causing hyperphosphatemia and hypercalcemia, and suppressing parathyroid hormone secretion.
However, there is not enough evidence on what the goal should be with respect to parathyroid hormone in patients with chronic kidney disease who are not on dialysis. Although in the recent past many believed that parathyroid hormone goals should be 150 to 300 pg/mL in dialysis patients, the latest guidelines suggest that perhaps this goal is too narrow and may lead to more adynamic bone disease. Similarly, there is no consensus on the use of synthetic parathyroid hormone analogues.
Bisphosphonate therapy, particularly with pamidronate (Aredia) and zolendronic acid (Reclast), has been associated with adverse renal effects even in patients without chronic kidney disease. There are no prospective studies of the effects of these agents in patients with depressed renal function.
The patient with chronic kidney disease who has a fracture remains a unique problem for the nephrologist, primary care physician, and subspecialist. Efforts should be concentrated on preventing and treating metabolic bone disease in its entire spectrum, with rational, prospective studies, and should not depend on anecdotal reports. Opinions abound, without adequate evidence to back them up.
- Moe S, Drüeke T, Cunningham J, et al; Kidney Disease: Improving Global Outcomes (KDIGO). Definition, evaluation, and classification of renal osteodystrophy: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int 2006; 69:1945–1953.
- Rustemeyer J, Bremerich A. Bisphosphonate-associated osteonecrosis of the jaw: what do we currently know? A survey of knowledge given in the recent literature. Clin Oral Investig 2009; Epub ahead of print.
- Armamento-Villareal R, Napoli N, Diemer K, et al. Bone turnover in bone biopsies of patients with low-energy cortical fractures receiving bisphosphonates: a case series. Calcif Tissue Int 2009; 85:37–44.
- Ali T, Jay RH. Spontaneous femoral shaft fracture after long-term alendronate. Age Ageing 2009; Epub ahead of print.
- Miller PD. Fragility fractures in chronic kidney disease: an opinionbased approach. Cleve Clin J Med 2009; 76:713–721.
- Toussaint ND, Elder GJ, Kerr PG. Bisphosphonates in chronic kidney disease; balancing potential benefits and adverse effects on bone and soft tissue. Clin J Am Soc Nephrol 2009; 4:221–233.
- Moe S, Drüeke T, Cunningham J, et al; Kidney Disease: Improving Global Outcomes (KDIGO). Definition, evaluation, and classification of renal osteodystrophy: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int 2006; 69:1945–1953.
- Rustemeyer J, Bremerich A. Bisphosphonate-associated osteonecrosis of the jaw: what do we currently know? A survey of knowledge given in the recent literature. Clin Oral Investig 2009; Epub ahead of print.
- Armamento-Villareal R, Napoli N, Diemer K, et al. Bone turnover in bone biopsies of patients with low-energy cortical fractures receiving bisphosphonates: a case series. Calcif Tissue Int 2009; 85:37–44.
- Ali T, Jay RH. Spontaneous femoral shaft fracture after long-term alendronate. Age Ageing 2009; Epub ahead of print.
- Miller PD. Fragility fractures in chronic kidney disease: an opinionbased approach. Cleve Clin J Med 2009; 76:713–721.
- Toussaint ND, Elder GJ, Kerr PG. Bisphosphonates in chronic kidney disease; balancing potential benefits and adverse effects on bone and soft tissue. Clin J Am Soc Nephrol 2009; 4:221–233.
Nighttime Voiding Similar With OAB, Insomnia
HOLLYWOOD, FLA. — The nighttime void volumes of people with an overactive bladder or primary insomnia were each significantly lower than controls, in an unexpected finding from a small pilot study.
“This is the first study to evaluate sleep and bladder diaries of insomniacs, people with overactive bladders, and controls,” said Dr. Cindy L. Amundsen, a urogynecologist at Duke University in Durham, N.C.
The objective was to compare the nighttime bladder symptoms between 10 people with overactive bladder (OAB), 10 people with insomnia, and 5 control patients with neither condition.
Some patients said “they've been told for years they had an overactive bladder, but [they] acted more like insomniacs and vice versa,” she said at the annual meeting of the American Urogynecologic Society.
All participants completed 7-day sleep diaries that included the number of awakenings and the duration of sleep. They also filled out 3-day bladder diaries with timing and volume of all voids.
Dr. Amundsen and her colleagues had expected to find notably lower nighttime urinary volumes in the participants with detrusor overactivity because of a greater voiding frequency. However, they recorded lower average nighttime void volumes in both the cohort with OAB (211 mL) and with insomnia (294 mL), compared with controls (460 mL). The study was funded by grants from Astellas Pharma and GlaxoSmithKline. Dr. Amundsen and her associates reported no relevant disclosures.
The mean daytime voided volume was statistically lower in the OAB patients (171 mL), compared with the insomnia patients (246 mL) and controls (291 mL).
The OAB patients had a significantly greater number of voids in 24 hours despite comparable total urine output per day. The total mean number of voids was 11.5 in the OAB group, 6.4 in the insomnia group, and 6.1 in controls. The total number of nocturia episodes likewise was significantly higher in the OAB group at 2.9, compared with 0.4 in insomniacs and 0.3 in controls.
All participants rated their perceived urgency on a 1-5 scale in the bladder diaries. The mean degree of urgency was 2.6 in the OAB group, 1.5 in the insomnia group, and 1.6 for the controls.
There was no significant difference in age or body mass index between the groups. Eligibility criteria for the OAB cohort included 3 or more months of symptoms, eight or more voids in a typical 24 hours, and two or more nocturia episodes per night. Those in the primary insomnia group reported awakening two or more times per night.
In addition, participants with OAB tended to void more quickly after awakening at night. “Polysomnogram and cystogram information was not presented here. But insomniacs spend more time being awake before they void compared to OAB patients. They are up 6-10 minutes, [and voiding is] almost an afterthought, whereas OAB patients void within 2 minutes in general,” she said.
In related data presented as a poster at the meeting, she and her colleagues found mean total sleep time was 423 minutes for the same 10 patients in the OAB group, 295 minutes for the insomniacs, and 444 minutes for controls.
Nocturia caused all the awakenings in the OAB group. In contrast to insomniacs, the participants with OAB were able to fall back to sleep faster—the mean time awake after sleep onset was 39 minutes in this group, compared with 103 minutes in insomniacs and 14 minutes in controls. These differences were statistically significant, and confirmed the sleep-disrupting effects of nocturia in the OAB group, the researchers noted.
HOLLYWOOD, FLA. — The nighttime void volumes of people with an overactive bladder or primary insomnia were each significantly lower than controls, in an unexpected finding from a small pilot study.
“This is the first study to evaluate sleep and bladder diaries of insomniacs, people with overactive bladders, and controls,” said Dr. Cindy L. Amundsen, a urogynecologist at Duke University in Durham, N.C.
The objective was to compare the nighttime bladder symptoms between 10 people with overactive bladder (OAB), 10 people with insomnia, and 5 control patients with neither condition.
Some patients said “they've been told for years they had an overactive bladder, but [they] acted more like insomniacs and vice versa,” she said at the annual meeting of the American Urogynecologic Society.
All participants completed 7-day sleep diaries that included the number of awakenings and the duration of sleep. They also filled out 3-day bladder diaries with timing and volume of all voids.
Dr. Amundsen and her colleagues had expected to find notably lower nighttime urinary volumes in the participants with detrusor overactivity because of a greater voiding frequency. However, they recorded lower average nighttime void volumes in both the cohort with OAB (211 mL) and with insomnia (294 mL), compared with controls (460 mL). The study was funded by grants from Astellas Pharma and GlaxoSmithKline. Dr. Amundsen and her associates reported no relevant disclosures.
The mean daytime voided volume was statistically lower in the OAB patients (171 mL), compared with the insomnia patients (246 mL) and controls (291 mL).
The OAB patients had a significantly greater number of voids in 24 hours despite comparable total urine output per day. The total mean number of voids was 11.5 in the OAB group, 6.4 in the insomnia group, and 6.1 in controls. The total number of nocturia episodes likewise was significantly higher in the OAB group at 2.9, compared with 0.4 in insomniacs and 0.3 in controls.
All participants rated their perceived urgency on a 1-5 scale in the bladder diaries. The mean degree of urgency was 2.6 in the OAB group, 1.5 in the insomnia group, and 1.6 for the controls.
There was no significant difference in age or body mass index between the groups. Eligibility criteria for the OAB cohort included 3 or more months of symptoms, eight or more voids in a typical 24 hours, and two or more nocturia episodes per night. Those in the primary insomnia group reported awakening two or more times per night.
In addition, participants with OAB tended to void more quickly after awakening at night. “Polysomnogram and cystogram information was not presented here. But insomniacs spend more time being awake before they void compared to OAB patients. They are up 6-10 minutes, [and voiding is] almost an afterthought, whereas OAB patients void within 2 minutes in general,” she said.
In related data presented as a poster at the meeting, she and her colleagues found mean total sleep time was 423 minutes for the same 10 patients in the OAB group, 295 minutes for the insomniacs, and 444 minutes for controls.
Nocturia caused all the awakenings in the OAB group. In contrast to insomniacs, the participants with OAB were able to fall back to sleep faster—the mean time awake after sleep onset was 39 minutes in this group, compared with 103 minutes in insomniacs and 14 minutes in controls. These differences were statistically significant, and confirmed the sleep-disrupting effects of nocturia in the OAB group, the researchers noted.
HOLLYWOOD, FLA. — The nighttime void volumes of people with an overactive bladder or primary insomnia were each significantly lower than controls, in an unexpected finding from a small pilot study.
“This is the first study to evaluate sleep and bladder diaries of insomniacs, people with overactive bladders, and controls,” said Dr. Cindy L. Amundsen, a urogynecologist at Duke University in Durham, N.C.
The objective was to compare the nighttime bladder symptoms between 10 people with overactive bladder (OAB), 10 people with insomnia, and 5 control patients with neither condition.
Some patients said “they've been told for years they had an overactive bladder, but [they] acted more like insomniacs and vice versa,” she said at the annual meeting of the American Urogynecologic Society.
All participants completed 7-day sleep diaries that included the number of awakenings and the duration of sleep. They also filled out 3-day bladder diaries with timing and volume of all voids.
Dr. Amundsen and her colleagues had expected to find notably lower nighttime urinary volumes in the participants with detrusor overactivity because of a greater voiding frequency. However, they recorded lower average nighttime void volumes in both the cohort with OAB (211 mL) and with insomnia (294 mL), compared with controls (460 mL). The study was funded by grants from Astellas Pharma and GlaxoSmithKline. Dr. Amundsen and her associates reported no relevant disclosures.
The mean daytime voided volume was statistically lower in the OAB patients (171 mL), compared with the insomnia patients (246 mL) and controls (291 mL).
The OAB patients had a significantly greater number of voids in 24 hours despite comparable total urine output per day. The total mean number of voids was 11.5 in the OAB group, 6.4 in the insomnia group, and 6.1 in controls. The total number of nocturia episodes likewise was significantly higher in the OAB group at 2.9, compared with 0.4 in insomniacs and 0.3 in controls.
All participants rated their perceived urgency on a 1-5 scale in the bladder diaries. The mean degree of urgency was 2.6 in the OAB group, 1.5 in the insomnia group, and 1.6 for the controls.
There was no significant difference in age or body mass index between the groups. Eligibility criteria for the OAB cohort included 3 or more months of symptoms, eight or more voids in a typical 24 hours, and two or more nocturia episodes per night. Those in the primary insomnia group reported awakening two or more times per night.
In addition, participants with OAB tended to void more quickly after awakening at night. “Polysomnogram and cystogram information was not presented here. But insomniacs spend more time being awake before they void compared to OAB patients. They are up 6-10 minutes, [and voiding is] almost an afterthought, whereas OAB patients void within 2 minutes in general,” she said.
In related data presented as a poster at the meeting, she and her colleagues found mean total sleep time was 423 minutes for the same 10 patients in the OAB group, 295 minutes for the insomniacs, and 444 minutes for controls.
Nocturia caused all the awakenings in the OAB group. In contrast to insomniacs, the participants with OAB were able to fall back to sleep faster—the mean time awake after sleep onset was 39 minutes in this group, compared with 103 minutes in insomniacs and 14 minutes in controls. These differences were statistically significant, and confirmed the sleep-disrupting effects of nocturia in the OAB group, the researchers noted.
Hormone Tx Boosts Radiotherapy in Prostate Ca
CHICAGO — The addition of hormone therapy to radiation improves overall survival in men with locally confined prostate cancer, but the benefit appears to be concentrated in intermediate-risk patients, according to initial results from the largest prostate cancer treatment trial to date.
The overall survival rate among the 1,979 men in the Radiation Therapy Oncology Group (RTOG) 9408 trial was 62% with combined therapy and 57% with radiotherapy alone at 8 years, Dr. Christopher U. Jones reported in a late-breaking abstract at the annual meeting of the American Society for Therapeutic Radiology and Oncology.
Intermediate-risk men who received 4 months of androgen suppression plus radiotherapy had the most pronounced benefit, with an overall survival rate of 72% vs. 66% for their radiation-only controls. The overall survival rate was 66% and 58%, respectively, in high-risk men and 76% compared with 73% in low-risk men.
RTOG 9408 is “a landmark, practice-changing study,.” said Dr. Matthew R. Smith, of the department of hematology/oncology, Massachusetts General Hospital, Boston. It provides “the first compelling evidence of a survival benefit for short-term androgen deprivation therapy in this intermediate-risk subgroup” treated with conventional radiation, he said. The number needed to treat was 17.
The results of RTOG 9408 had been eagerly anticipated because androgen-deprivation therapy has been widely adopted in men with localized disease, including those at low risk, despite the lack of a compelling survival benefit and emerging evidence of treatment-related morbidity including decreased bone mineral density, greater risk for clinical fractures, and increased triglycerides and insulin sensitivity, Dr. Smith said.
“RTOG 9408 definitively establishes that there is no benefit for androgen deprivation therapy in patients with low-risk disease,” he said. “Cancer control rates are outstanding with both conventional and high-dose radiation therapy in this low-risk group. Unquestionably this is a setting where less is more.”
The trial does not answer whether androgen deprivation is necessary in patients with intermediate-risk disease treated with more modern high-dose radiation techniques, he added. This question will be addressed in other trials, including the recently opened RTOG 0815 trial.
Patients in the current trial were enrolled from October 1994 to April 2001, and randomized to hormones plus radiotherapy (987 patients) or radiotherapy alone (992). All received 66.6 Gy of radiation, a dose slightly lower than that currently used with newer techniques such as intensity-modulated radiation therapy. Androgen deprivation therapy was administered for 2 months before and 2 months during radiation.
At baseline, patients had T1b-T2b adenocarcinoma of the prostate and a prostate-specific antigen (PSA) level of 20 or less. Median age was 71 years.
The low-risk group included 685 patients with a Gleason score of 6 or less, a PSA of 10 or less and no T2b disease. The 1,068 intermediate-risk patients had a Gleason score of 7 or a Gleason of 6 or less and either a PSA of 10-20 or T2b disease. The 226 high-risk patients had a Gleason score of 8-10.
The addition of short-course hormones to radiation did not increase the risk of death from intercurrent disease, said Dr. Jones, a radiation oncologist in Sacramento, Calif.
The actuarial 10-year death rate from intercurrent disease, excluding deaths from prostate cancer, was 35% in the combination arm and 37% in the radiation-only arm.
Dr. Jones reported no conflicts of interest.
The study was supported by grants from the National Cancer Institute in Bethesda, Md.
CHICAGO — The addition of hormone therapy to radiation improves overall survival in men with locally confined prostate cancer, but the benefit appears to be concentrated in intermediate-risk patients, according to initial results from the largest prostate cancer treatment trial to date.
The overall survival rate among the 1,979 men in the Radiation Therapy Oncology Group (RTOG) 9408 trial was 62% with combined therapy and 57% with radiotherapy alone at 8 years, Dr. Christopher U. Jones reported in a late-breaking abstract at the annual meeting of the American Society for Therapeutic Radiology and Oncology.
Intermediate-risk men who received 4 months of androgen suppression plus radiotherapy had the most pronounced benefit, with an overall survival rate of 72% vs. 66% for their radiation-only controls. The overall survival rate was 66% and 58%, respectively, in high-risk men and 76% compared with 73% in low-risk men.
RTOG 9408 is “a landmark, practice-changing study,.” said Dr. Matthew R. Smith, of the department of hematology/oncology, Massachusetts General Hospital, Boston. It provides “the first compelling evidence of a survival benefit for short-term androgen deprivation therapy in this intermediate-risk subgroup” treated with conventional radiation, he said. The number needed to treat was 17.
The results of RTOG 9408 had been eagerly anticipated because androgen-deprivation therapy has been widely adopted in men with localized disease, including those at low risk, despite the lack of a compelling survival benefit and emerging evidence of treatment-related morbidity including decreased bone mineral density, greater risk for clinical fractures, and increased triglycerides and insulin sensitivity, Dr. Smith said.
“RTOG 9408 definitively establishes that there is no benefit for androgen deprivation therapy in patients with low-risk disease,” he said. “Cancer control rates are outstanding with both conventional and high-dose radiation therapy in this low-risk group. Unquestionably this is a setting where less is more.”
The trial does not answer whether androgen deprivation is necessary in patients with intermediate-risk disease treated with more modern high-dose radiation techniques, he added. This question will be addressed in other trials, including the recently opened RTOG 0815 trial.
Patients in the current trial were enrolled from October 1994 to April 2001, and randomized to hormones plus radiotherapy (987 patients) or radiotherapy alone (992). All received 66.6 Gy of radiation, a dose slightly lower than that currently used with newer techniques such as intensity-modulated radiation therapy. Androgen deprivation therapy was administered for 2 months before and 2 months during radiation.
At baseline, patients had T1b-T2b adenocarcinoma of the prostate and a prostate-specific antigen (PSA) level of 20 or less. Median age was 71 years.
The low-risk group included 685 patients with a Gleason score of 6 or less, a PSA of 10 or less and no T2b disease. The 1,068 intermediate-risk patients had a Gleason score of 7 or a Gleason of 6 or less and either a PSA of 10-20 or T2b disease. The 226 high-risk patients had a Gleason score of 8-10.
The addition of short-course hormones to radiation did not increase the risk of death from intercurrent disease, said Dr. Jones, a radiation oncologist in Sacramento, Calif.
The actuarial 10-year death rate from intercurrent disease, excluding deaths from prostate cancer, was 35% in the combination arm and 37% in the radiation-only arm.
Dr. Jones reported no conflicts of interest.
The study was supported by grants from the National Cancer Institute in Bethesda, Md.
CHICAGO — The addition of hormone therapy to radiation improves overall survival in men with locally confined prostate cancer, but the benefit appears to be concentrated in intermediate-risk patients, according to initial results from the largest prostate cancer treatment trial to date.
The overall survival rate among the 1,979 men in the Radiation Therapy Oncology Group (RTOG) 9408 trial was 62% with combined therapy and 57% with radiotherapy alone at 8 years, Dr. Christopher U. Jones reported in a late-breaking abstract at the annual meeting of the American Society for Therapeutic Radiology and Oncology.
Intermediate-risk men who received 4 months of androgen suppression plus radiotherapy had the most pronounced benefit, with an overall survival rate of 72% vs. 66% for their radiation-only controls. The overall survival rate was 66% and 58%, respectively, in high-risk men and 76% compared with 73% in low-risk men.
RTOG 9408 is “a landmark, practice-changing study,.” said Dr. Matthew R. Smith, of the department of hematology/oncology, Massachusetts General Hospital, Boston. It provides “the first compelling evidence of a survival benefit for short-term androgen deprivation therapy in this intermediate-risk subgroup” treated with conventional radiation, he said. The number needed to treat was 17.
The results of RTOG 9408 had been eagerly anticipated because androgen-deprivation therapy has been widely adopted in men with localized disease, including those at low risk, despite the lack of a compelling survival benefit and emerging evidence of treatment-related morbidity including decreased bone mineral density, greater risk for clinical fractures, and increased triglycerides and insulin sensitivity, Dr. Smith said.
“RTOG 9408 definitively establishes that there is no benefit for androgen deprivation therapy in patients with low-risk disease,” he said. “Cancer control rates are outstanding with both conventional and high-dose radiation therapy in this low-risk group. Unquestionably this is a setting where less is more.”
The trial does not answer whether androgen deprivation is necessary in patients with intermediate-risk disease treated with more modern high-dose radiation techniques, he added. This question will be addressed in other trials, including the recently opened RTOG 0815 trial.
Patients in the current trial were enrolled from October 1994 to April 2001, and randomized to hormones plus radiotherapy (987 patients) or radiotherapy alone (992). All received 66.6 Gy of radiation, a dose slightly lower than that currently used with newer techniques such as intensity-modulated radiation therapy. Androgen deprivation therapy was administered for 2 months before and 2 months during radiation.
At baseline, patients had T1b-T2b adenocarcinoma of the prostate and a prostate-specific antigen (PSA) level of 20 or less. Median age was 71 years.
The low-risk group included 685 patients with a Gleason score of 6 or less, a PSA of 10 or less and no T2b disease. The 1,068 intermediate-risk patients had a Gleason score of 7 or a Gleason of 6 or less and either a PSA of 10-20 or T2b disease. The 226 high-risk patients had a Gleason score of 8-10.
The addition of short-course hormones to radiation did not increase the risk of death from intercurrent disease, said Dr. Jones, a radiation oncologist in Sacramento, Calif.
The actuarial 10-year death rate from intercurrent disease, excluding deaths from prostate cancer, was 35% in the combination arm and 37% in the radiation-only arm.
Dr. Jones reported no conflicts of interest.
The study was supported by grants from the National Cancer Institute in Bethesda, Md.
Short-Course Radiotherapy Limits T3 Prostate Ca
CHICAGO — Delivery of a lower total radiation dose in fewer but more intense fractions improved control of high-risk prostate cancer without increasing toxicity in a multicenter, phase III trial of 168 men.
After a median follow-up of 3 years, the freedom from biochemical failure rate was 87% with hypofractionated radiotherapy vs. 79% with conventional radiation.
The difference in this co-primary end point was significant (P = .035).
In a multivariate Cox analysis, hypofractionated radiation therapy reduced the risk of biochemical failure by roughly 70% (hazard ratio 0.35), said lead author Dr. Giorgio Arcangeli, a radiation oncologist at the Regina Elena National Cancer Institute in Rome. Metastasis-free survival was similar in both groups.
Men in the hypofractionated arm received 62 Gy of radiation in 20 fractions of 3.1 Gy over 5 weeks, compared with 80 Gy of radiation in 40 fractions of 2 Gy over 8 weeks in the conventional arm.
New data suggest that prostate cancer may have a unique biology that makes it more sensitive than other tumors and normal tissue to higher daily doses of radiation, potentially allowing clinicians to complete radiation treatment in a shorter time.
Dr. Arcangeli acknowledged that longer follow-up is required to definitively validate this treatment strategy, but suggested there are important up-front benefits for patients.
“It offers convenience to patients by halving the number of visits to radiotherapy departments, an important benefit for these patients, who are typically an older, less mobile group,” Dr. Arcangeli said.
The investigators hypothesized that the two treatment schedules would be equally effective because they have the same biological equivalent dose and same tumor control probability, but that late complications would be reduced with hypofractionation.
So far, no significant differences between the two groups have been observed in the other primary end point of late side effects in urinary and bowel function, Dr. Arcangeli said at the annual meeting of the American Society for Radiation Oncology, where the findings were presented.
Three-year rates of grade 2 or higher toxicity were 15% in the hypofractionation arm and 17% in the conventional arm for gastrointestinal side effects, and 11% vs. 15% for genitourinary toxicity.
The severity of toxicity scores did not differ between groups, but acute toxicity in the hypofractionation arm developed and ended earlier than in the conventional arm, he said.
Men were eligible for the study if they had a prostate-specific antigen (PSA) level of more than 20 ng/mL, a Gleason score of 7, T3 or higher disease, or at least two of the follow characteristics: Gleason score of 7, PSA level of 11-20 ng/mL, and T2c disease.
Overall, 83 men received hypofractionated and 85 men conventional fractionated schedules of 3-D conformal radiotherapy to the prostate and seminal vesicles, beginning 2 months after initiation of a 9-month course of total androgen blockade.
They had no distant metastases, previous pelvic irradiation, or previous prostate surgery other than transurethral resection of the prostate. Their median age was 75 years.
Studies are in progress to test the benefits of even shorter treatment schedules, Dr. Arcangeli commented in a statement.
The authors reported no conflicts of interest related to their study.
CHICAGO — Delivery of a lower total radiation dose in fewer but more intense fractions improved control of high-risk prostate cancer without increasing toxicity in a multicenter, phase III trial of 168 men.
After a median follow-up of 3 years, the freedom from biochemical failure rate was 87% with hypofractionated radiotherapy vs. 79% with conventional radiation.
The difference in this co-primary end point was significant (P = .035).
In a multivariate Cox analysis, hypofractionated radiation therapy reduced the risk of biochemical failure by roughly 70% (hazard ratio 0.35), said lead author Dr. Giorgio Arcangeli, a radiation oncologist at the Regina Elena National Cancer Institute in Rome. Metastasis-free survival was similar in both groups.
Men in the hypofractionated arm received 62 Gy of radiation in 20 fractions of 3.1 Gy over 5 weeks, compared with 80 Gy of radiation in 40 fractions of 2 Gy over 8 weeks in the conventional arm.
New data suggest that prostate cancer may have a unique biology that makes it more sensitive than other tumors and normal tissue to higher daily doses of radiation, potentially allowing clinicians to complete radiation treatment in a shorter time.
Dr. Arcangeli acknowledged that longer follow-up is required to definitively validate this treatment strategy, but suggested there are important up-front benefits for patients.
“It offers convenience to patients by halving the number of visits to radiotherapy departments, an important benefit for these patients, who are typically an older, less mobile group,” Dr. Arcangeli said.
The investigators hypothesized that the two treatment schedules would be equally effective because they have the same biological equivalent dose and same tumor control probability, but that late complications would be reduced with hypofractionation.
So far, no significant differences between the two groups have been observed in the other primary end point of late side effects in urinary and bowel function, Dr. Arcangeli said at the annual meeting of the American Society for Radiation Oncology, where the findings were presented.
Three-year rates of grade 2 or higher toxicity were 15% in the hypofractionation arm and 17% in the conventional arm for gastrointestinal side effects, and 11% vs. 15% for genitourinary toxicity.
The severity of toxicity scores did not differ between groups, but acute toxicity in the hypofractionation arm developed and ended earlier than in the conventional arm, he said.
Men were eligible for the study if they had a prostate-specific antigen (PSA) level of more than 20 ng/mL, a Gleason score of 7, T3 or higher disease, or at least two of the follow characteristics: Gleason score of 7, PSA level of 11-20 ng/mL, and T2c disease.
Overall, 83 men received hypofractionated and 85 men conventional fractionated schedules of 3-D conformal radiotherapy to the prostate and seminal vesicles, beginning 2 months after initiation of a 9-month course of total androgen blockade.
They had no distant metastases, previous pelvic irradiation, or previous prostate surgery other than transurethral resection of the prostate. Their median age was 75 years.
Studies are in progress to test the benefits of even shorter treatment schedules, Dr. Arcangeli commented in a statement.
The authors reported no conflicts of interest related to their study.
CHICAGO — Delivery of a lower total radiation dose in fewer but more intense fractions improved control of high-risk prostate cancer without increasing toxicity in a multicenter, phase III trial of 168 men.
After a median follow-up of 3 years, the freedom from biochemical failure rate was 87% with hypofractionated radiotherapy vs. 79% with conventional radiation.
The difference in this co-primary end point was significant (P = .035).
In a multivariate Cox analysis, hypofractionated radiation therapy reduced the risk of biochemical failure by roughly 70% (hazard ratio 0.35), said lead author Dr. Giorgio Arcangeli, a radiation oncologist at the Regina Elena National Cancer Institute in Rome. Metastasis-free survival was similar in both groups.
Men in the hypofractionated arm received 62 Gy of radiation in 20 fractions of 3.1 Gy over 5 weeks, compared with 80 Gy of radiation in 40 fractions of 2 Gy over 8 weeks in the conventional arm.
New data suggest that prostate cancer may have a unique biology that makes it more sensitive than other tumors and normal tissue to higher daily doses of radiation, potentially allowing clinicians to complete radiation treatment in a shorter time.
Dr. Arcangeli acknowledged that longer follow-up is required to definitively validate this treatment strategy, but suggested there are important up-front benefits for patients.
“It offers convenience to patients by halving the number of visits to radiotherapy departments, an important benefit for these patients, who are typically an older, less mobile group,” Dr. Arcangeli said.
The investigators hypothesized that the two treatment schedules would be equally effective because they have the same biological equivalent dose and same tumor control probability, but that late complications would be reduced with hypofractionation.
So far, no significant differences between the two groups have been observed in the other primary end point of late side effects in urinary and bowel function, Dr. Arcangeli said at the annual meeting of the American Society for Radiation Oncology, where the findings were presented.
Three-year rates of grade 2 or higher toxicity were 15% in the hypofractionation arm and 17% in the conventional arm for gastrointestinal side effects, and 11% vs. 15% for genitourinary toxicity.
The severity of toxicity scores did not differ between groups, but acute toxicity in the hypofractionation arm developed and ended earlier than in the conventional arm, he said.
Men were eligible for the study if they had a prostate-specific antigen (PSA) level of more than 20 ng/mL, a Gleason score of 7, T3 or higher disease, or at least two of the follow characteristics: Gleason score of 7, PSA level of 11-20 ng/mL, and T2c disease.
Overall, 83 men received hypofractionated and 85 men conventional fractionated schedules of 3-D conformal radiotherapy to the prostate and seminal vesicles, beginning 2 months after initiation of a 9-month course of total androgen blockade.
They had no distant metastases, previous pelvic irradiation, or previous prostate surgery other than transurethral resection of the prostate. Their median age was 75 years.
Studies are in progress to test the benefits of even shorter treatment schedules, Dr. Arcangeli commented in a statement.
The authors reported no conflicts of interest related to their study.
Anticoagulant May Control Localized Prostate Ca
CHICAGO — Along with its known cardiovascular benefits, anticoagulation therapy may improve biochemical control of localized prostate cancer treated with radiotherapy.
In a retrospective study of 662 patients, the biochemical control rate at 48 months was significantly better (at 91%) in men taking warfarin, clopidogrel, and/or aspirin, compared with 78% in men not taking blood-thinning therapy. Distant metastases were also significantly reduced in the anticoagulant group, compared with the nonanticoagulant group (1% vs. 5%).
The overall survival rates were 92% and 90%, respectively, which did not reach statistical significance, Dr. Kevin S. Choe and his colleagues reported in a poster at the annual meeting of the American Society for Radiation Oncology.
Previous clinical trials have produced limited and inconsistent data in metastatic prostate disease, although epidemiologic studies have shown that men on anticoagulants develop prostate cancer less frequently. There is also substantial evidence from preclinical models suggesting that anticoagulants may influence multiple tumor processes including tumor growth, angiogenesis, and the metastatic pathway, Dr. Choe of the University of Chicago said at a press briefing.
“According to our data, we think that the most plausible path [by which an anticoagulant influences prostate cancer patients] … is by limiting metastases, because we see the biggest effect among patients who have very aggressive types of prostate cancer that tend to spread,” he said.
In subgroup analysis, the improvement in biochemical control was statistically significant only for patients with high-risk disease as defined by National Comprehensive Cancer Network criteria.
The 4-year, freedom-from-biochemical-failure rate using the Phoenix definition (prostate-specific antigen greater than nadir plus 2 ng/mL) was 82.4% in high-risk men on anticoagulants vs. 57.6% in high-risk controls. The biochemical failure rate for patients both on and off anticoagulants was 92.5% vs. 83% in intermediate-risk men and 95% vs. 90.5% in low-risk men.
In multivariate analysis, anticoagulant use was independently associated with improved biochemical control, lowering the risk of biochemical failure by almost half (hazard ratio, 0.54). The type of anticoagulant did not significantly influence biochemical failure rates, nor was the combination of two agents better than a single agent.
The current study grew out of another study in the same cohort by Dr. Choe and his colleagues showing that warfarin and clopidogrel use during external-beam radiotherapy substantially increasesd the risk of grade 3 or higher rectal bleeding (Int. J. Radiat. Oncol. Biol. Phys. 2009 May 20 [doi:10.1016/j.ijrobp.2009.02.026]).
Although aspirin and other less potent blood-thinning agents such as enoxaparin may lessen the risk of this bleeding toxicity, Dr. Choe balked at recommending anticoagulation for all prostate cancer patients.
“In patients already taking anticoagulants for cardiovascular risks, there may be additional benefits in prostate cancer,” he said, adding that if an anticoagulant were ever to be recommended, “it would need to be planned out very carefully” and will require larger prospective studies to determine whether the benefit is worth the risk.
The median dosage used by patients at consult or during follow-up was warfarin 5 mg/day and clopidogrel 75 mg/day. Aspirin dosage was not recorded. All patients were treated with external-beam radiotherapy, permanent seed implant, or both. No patients underwent surgery. Their median age was 69 years, and median initial PSA was 8.4 ng/mL.
Dr. Choe plans to conduct a prospective database analysis of prostate cancer patients who had surgery instead of radiotherapy to test the hypothesis that the benefit results from an effect on the cancer itself and not an interaction between the anticoagulants and radiotherapy.
The investigators reported no study sponsorship or conflicts of interest.
CHICAGO — Along with its known cardiovascular benefits, anticoagulation therapy may improve biochemical control of localized prostate cancer treated with radiotherapy.
In a retrospective study of 662 patients, the biochemical control rate at 48 months was significantly better (at 91%) in men taking warfarin, clopidogrel, and/or aspirin, compared with 78% in men not taking blood-thinning therapy. Distant metastases were also significantly reduced in the anticoagulant group, compared with the nonanticoagulant group (1% vs. 5%).
The overall survival rates were 92% and 90%, respectively, which did not reach statistical significance, Dr. Kevin S. Choe and his colleagues reported in a poster at the annual meeting of the American Society for Radiation Oncology.
Previous clinical trials have produced limited and inconsistent data in metastatic prostate disease, although epidemiologic studies have shown that men on anticoagulants develop prostate cancer less frequently. There is also substantial evidence from preclinical models suggesting that anticoagulants may influence multiple tumor processes including tumor growth, angiogenesis, and the metastatic pathway, Dr. Choe of the University of Chicago said at a press briefing.
“According to our data, we think that the most plausible path [by which an anticoagulant influences prostate cancer patients] … is by limiting metastases, because we see the biggest effect among patients who have very aggressive types of prostate cancer that tend to spread,” he said.
In subgroup analysis, the improvement in biochemical control was statistically significant only for patients with high-risk disease as defined by National Comprehensive Cancer Network criteria.
The 4-year, freedom-from-biochemical-failure rate using the Phoenix definition (prostate-specific antigen greater than nadir plus 2 ng/mL) was 82.4% in high-risk men on anticoagulants vs. 57.6% in high-risk controls. The biochemical failure rate for patients both on and off anticoagulants was 92.5% vs. 83% in intermediate-risk men and 95% vs. 90.5% in low-risk men.
In multivariate analysis, anticoagulant use was independently associated with improved biochemical control, lowering the risk of biochemical failure by almost half (hazard ratio, 0.54). The type of anticoagulant did not significantly influence biochemical failure rates, nor was the combination of two agents better than a single agent.
The current study grew out of another study in the same cohort by Dr. Choe and his colleagues showing that warfarin and clopidogrel use during external-beam radiotherapy substantially increasesd the risk of grade 3 or higher rectal bleeding (Int. J. Radiat. Oncol. Biol. Phys. 2009 May 20 [doi:10.1016/j.ijrobp.2009.02.026]).
Although aspirin and other less potent blood-thinning agents such as enoxaparin may lessen the risk of this bleeding toxicity, Dr. Choe balked at recommending anticoagulation for all prostate cancer patients.
“In patients already taking anticoagulants for cardiovascular risks, there may be additional benefits in prostate cancer,” he said, adding that if an anticoagulant were ever to be recommended, “it would need to be planned out very carefully” and will require larger prospective studies to determine whether the benefit is worth the risk.
The median dosage used by patients at consult or during follow-up was warfarin 5 mg/day and clopidogrel 75 mg/day. Aspirin dosage was not recorded. All patients were treated with external-beam radiotherapy, permanent seed implant, or both. No patients underwent surgery. Their median age was 69 years, and median initial PSA was 8.4 ng/mL.
Dr. Choe plans to conduct a prospective database analysis of prostate cancer patients who had surgery instead of radiotherapy to test the hypothesis that the benefit results from an effect on the cancer itself and not an interaction between the anticoagulants and radiotherapy.
The investigators reported no study sponsorship or conflicts of interest.
CHICAGO — Along with its known cardiovascular benefits, anticoagulation therapy may improve biochemical control of localized prostate cancer treated with radiotherapy.
In a retrospective study of 662 patients, the biochemical control rate at 48 months was significantly better (at 91%) in men taking warfarin, clopidogrel, and/or aspirin, compared with 78% in men not taking blood-thinning therapy. Distant metastases were also significantly reduced in the anticoagulant group, compared with the nonanticoagulant group (1% vs. 5%).
The overall survival rates were 92% and 90%, respectively, which did not reach statistical significance, Dr. Kevin S. Choe and his colleagues reported in a poster at the annual meeting of the American Society for Radiation Oncology.
Previous clinical trials have produced limited and inconsistent data in metastatic prostate disease, although epidemiologic studies have shown that men on anticoagulants develop prostate cancer less frequently. There is also substantial evidence from preclinical models suggesting that anticoagulants may influence multiple tumor processes including tumor growth, angiogenesis, and the metastatic pathway, Dr. Choe of the University of Chicago said at a press briefing.
“According to our data, we think that the most plausible path [by which an anticoagulant influences prostate cancer patients] … is by limiting metastases, because we see the biggest effect among patients who have very aggressive types of prostate cancer that tend to spread,” he said.
In subgroup analysis, the improvement in biochemical control was statistically significant only for patients with high-risk disease as defined by National Comprehensive Cancer Network criteria.
The 4-year, freedom-from-biochemical-failure rate using the Phoenix definition (prostate-specific antigen greater than nadir plus 2 ng/mL) was 82.4% in high-risk men on anticoagulants vs. 57.6% in high-risk controls. The biochemical failure rate for patients both on and off anticoagulants was 92.5% vs. 83% in intermediate-risk men and 95% vs. 90.5% in low-risk men.
In multivariate analysis, anticoagulant use was independently associated with improved biochemical control, lowering the risk of biochemical failure by almost half (hazard ratio, 0.54). The type of anticoagulant did not significantly influence biochemical failure rates, nor was the combination of two agents better than a single agent.
The current study grew out of another study in the same cohort by Dr. Choe and his colleagues showing that warfarin and clopidogrel use during external-beam radiotherapy substantially increasesd the risk of grade 3 or higher rectal bleeding (Int. J. Radiat. Oncol. Biol. Phys. 2009 May 20 [doi:10.1016/j.ijrobp.2009.02.026]).
Although aspirin and other less potent blood-thinning agents such as enoxaparin may lessen the risk of this bleeding toxicity, Dr. Choe balked at recommending anticoagulation for all prostate cancer patients.
“In patients already taking anticoagulants for cardiovascular risks, there may be additional benefits in prostate cancer,” he said, adding that if an anticoagulant were ever to be recommended, “it would need to be planned out very carefully” and will require larger prospective studies to determine whether the benefit is worth the risk.
The median dosage used by patients at consult or during follow-up was warfarin 5 mg/day and clopidogrel 75 mg/day. Aspirin dosage was not recorded. All patients were treated with external-beam radiotherapy, permanent seed implant, or both. No patients underwent surgery. Their median age was 69 years, and median initial PSA was 8.4 ng/mL.
Dr. Choe plans to conduct a prospective database analysis of prostate cancer patients who had surgery instead of radiotherapy to test the hypothesis that the benefit results from an effect on the cancer itself and not an interaction between the anticoagulants and radiotherapy.
The investigators reported no study sponsorship or conflicts of interest.