User login
Dialysis Facilities: Web Site Updated
The Web site that helps patients compare dialysis facilities now has updated information on how well dialysis patients are treated for anemia and survival rates for each facility.
The Web site, www.medicare.gov/dialysis
The Web site that helps patients compare dialysis facilities now has updated information on how well dialysis patients are treated for anemia and survival rates for each facility.
The Web site, www.medicare.gov/dialysis
The Web site that helps patients compare dialysis facilities now has updated information on how well dialysis patients are treated for anemia and survival rates for each facility.
The Web site, www.medicare.gov/dialysis
Factors Tied to Chronic Kidney Disease Deaths
SAN DIEGO — The presence of an estimated glomerular filtration rate less than 60 mL/min per 1.73 m
“Don't just settle for measuring a patient's serum creatinine level. Know what the estimated GFR is,” lead study investigator Dr. David G. Warnock advised in an interview during a poster session at the annual meeting of the American Society of Nephrology.
“If it's less than 60 [mL/min per 1.73 m
Dr. Warnock and his colleagues evaluated the association between all-cause mortality and the three components of chronic kidney disease in 19,125 men and women who participated in the Reasons for Geographic and Racial Differences in Stroke (REGARDS) study, a population-based cohort investigation of incident stroke in whites and blacks aged 45 years and older in the United States.
In addition to the low glomerular filtration rate, albuminuria was defined as an albumin/creatinine ratio of 30 mg/g or greater and anemia was defined as a hemoglobin level less than 13.5 g/dL for men and less than 12.0 g/dL for women.
Study participants had single measurements of serum creatinine, urinary creatinine and albumin, and other baseline assessments. The researchers ascertained vital status based on telephone interviews every 6 months. Prevalent coronary heart disease included self-reported previous myocardial infarction, stroke, cardiovascular procedures, or evidence of previous myocardial infarction by electrocardiogram.
Of the 19,125 people in the cohort, 14,361 had no coronary heart disease or stroke over a mean follow-up of 3.6 years and 4,764 did. Study participants with prevalent coronary heart disease were slightly older than their unaffected counterparts (mean age of 67 years vs. 63 years, respectively).
Dr. Warnock, professor of medicine in the division of nephrology at the University of Alabama, Birmingham, reported that there were 650 deaths among study participants, evenly divided between those who had coronary heart disease or stroke and those who did not.
In both groups, significant hazard ratios for all-cause mortality were independently associated with an estimated glomerular filtration rate of less than 60 mL/min per 1.73 m
Dr. Warnock acknowledged certain limitations of the study, including the potential for ascertainment bias and the fact that only blacks and whites were enrolled, “so we can't say anything about Asians or Hispanics. There's no reason to suspect different results [in those populations], but we don't know.”
The study was supported by a grant from the National Institute of Neurological Disorders and Stroke and by a grant from Amgen to Dr. Warnock.
'Don't just settle for measuring a patient's serum creatinine level. Know what the estimated GFR is.'
Source DR. WARNOCK
SAN DIEGO — The presence of an estimated glomerular filtration rate less than 60 mL/min per 1.73 m
“Don't just settle for measuring a patient's serum creatinine level. Know what the estimated GFR is,” lead study investigator Dr. David G. Warnock advised in an interview during a poster session at the annual meeting of the American Society of Nephrology.
“If it's less than 60 [mL/min per 1.73 m
Dr. Warnock and his colleagues evaluated the association between all-cause mortality and the three components of chronic kidney disease in 19,125 men and women who participated in the Reasons for Geographic and Racial Differences in Stroke (REGARDS) study, a population-based cohort investigation of incident stroke in whites and blacks aged 45 years and older in the United States.
In addition to the low glomerular filtration rate, albuminuria was defined as an albumin/creatinine ratio of 30 mg/g or greater and anemia was defined as a hemoglobin level less than 13.5 g/dL for men and less than 12.0 g/dL for women.
Study participants had single measurements of serum creatinine, urinary creatinine and albumin, and other baseline assessments. The researchers ascertained vital status based on telephone interviews every 6 months. Prevalent coronary heart disease included self-reported previous myocardial infarction, stroke, cardiovascular procedures, or evidence of previous myocardial infarction by electrocardiogram.
Of the 19,125 people in the cohort, 14,361 had no coronary heart disease or stroke over a mean follow-up of 3.6 years and 4,764 did. Study participants with prevalent coronary heart disease were slightly older than their unaffected counterparts (mean age of 67 years vs. 63 years, respectively).
Dr. Warnock, professor of medicine in the division of nephrology at the University of Alabama, Birmingham, reported that there were 650 deaths among study participants, evenly divided between those who had coronary heart disease or stroke and those who did not.
In both groups, significant hazard ratios for all-cause mortality were independently associated with an estimated glomerular filtration rate of less than 60 mL/min per 1.73 m
Dr. Warnock acknowledged certain limitations of the study, including the potential for ascertainment bias and the fact that only blacks and whites were enrolled, “so we can't say anything about Asians or Hispanics. There's no reason to suspect different results [in those populations], but we don't know.”
The study was supported by a grant from the National Institute of Neurological Disorders and Stroke and by a grant from Amgen to Dr. Warnock.
'Don't just settle for measuring a patient's serum creatinine level. Know what the estimated GFR is.'
Source DR. WARNOCK
SAN DIEGO — The presence of an estimated glomerular filtration rate less than 60 mL/min per 1.73 m
“Don't just settle for measuring a patient's serum creatinine level. Know what the estimated GFR is,” lead study investigator Dr. David G. Warnock advised in an interview during a poster session at the annual meeting of the American Society of Nephrology.
“If it's less than 60 [mL/min per 1.73 m
Dr. Warnock and his colleagues evaluated the association between all-cause mortality and the three components of chronic kidney disease in 19,125 men and women who participated in the Reasons for Geographic and Racial Differences in Stroke (REGARDS) study, a population-based cohort investigation of incident stroke in whites and blacks aged 45 years and older in the United States.
In addition to the low glomerular filtration rate, albuminuria was defined as an albumin/creatinine ratio of 30 mg/g or greater and anemia was defined as a hemoglobin level less than 13.5 g/dL for men and less than 12.0 g/dL for women.
Study participants had single measurements of serum creatinine, urinary creatinine and albumin, and other baseline assessments. The researchers ascertained vital status based on telephone interviews every 6 months. Prevalent coronary heart disease included self-reported previous myocardial infarction, stroke, cardiovascular procedures, or evidence of previous myocardial infarction by electrocardiogram.
Of the 19,125 people in the cohort, 14,361 had no coronary heart disease or stroke over a mean follow-up of 3.6 years and 4,764 did. Study participants with prevalent coronary heart disease were slightly older than their unaffected counterparts (mean age of 67 years vs. 63 years, respectively).
Dr. Warnock, professor of medicine in the division of nephrology at the University of Alabama, Birmingham, reported that there were 650 deaths among study participants, evenly divided between those who had coronary heart disease or stroke and those who did not.
In both groups, significant hazard ratios for all-cause mortality were independently associated with an estimated glomerular filtration rate of less than 60 mL/min per 1.73 m
Dr. Warnock acknowledged certain limitations of the study, including the potential for ascertainment bias and the fact that only blacks and whites were enrolled, “so we can't say anything about Asians or Hispanics. There's no reason to suspect different results [in those populations], but we don't know.”
The study was supported by a grant from the National Institute of Neurological Disorders and Stroke and by a grant from Amgen to Dr. Warnock.
'Don't just settle for measuring a patient's serum creatinine level. Know what the estimated GFR is.'
Source DR. WARNOCK
Olmesartan May Help Prevent Microalbuminuria
SAN DIEGO — Olmesartan reduced the risk of microalbuminuria by 23% in normoalbuminuric patients with type 2 diabetes and at least one additional cardiovascular disease risk factor, results from a large European trial showed.
The angiotensin receptor blocker also yielded unprecedented blood pressure control for this population of patients.
Those are the first key findings from the Randomized Olmesartan and Diabetes Microalbuminuria Prevention (ROADMAP) study, which were unveiled during a press briefing at the annual meeting of the American Society of Nephrology.
“Despite all of our efforts, we still have problems effectively treating diabetic nephropathy,” said the study's steering committee chair, Dr. Hermann G. Haller of the department of nephrology at Hannover (Germany) Medical School. “The problem for prevention is that we have to diagnose and treat it early. Microalbuminuria is the first sign of the pathogenesis of diabetic nephropathy. It is also an important marker of early development of cardiovascular disease and can indicate microvascular disease.”
The primary end point of the study was the occurrence of microalbuminuria based on two or more positive morning spot urine measurements. Secondary end points were cardiovascular events, renal function, and microvascular morbidity.
With support from Daiichi Sankyo, which markets olmesartan, researchers in 19 countries enrolled 4,449 patients, aged 18-75 years, with well-controlled type 2 diabetes. All patients were normoalbuminuric (defined as a level of 25 mg/g or less for men and 35 mg/g or less for women) and had at lease one additional cardiovascular risk factor, such as high triglyceride levels or hypertension. None of the participants had received an ACE inhibitor or an angiotensin receptor blocker within 6 months of participation.
The patients were randomized to receive either 40 mg olmesartan per day or placebo (conventional antihypertensive treatment without blockade of the renin-angiotensin system). The urine albumin- creatinine ratio was determined every 6 months. Patients were followed for an average of 3.2 years.
At their discretion, study investigators could add calcium channel blockers, diuretics, or beta-blockers to the regimen to help patients achieve the target blood pressure goal of 130/80 mm Hg.
The patients' mean age was 58 years, mean duration of diabetes was 6 years, mean hemoglobin A1c level was 7.6%, and mean body mass index was 31 kg/m
Dr. Haller reported that nearly 80% of patients in the olmesartan group reached the target BP of 130/80 mm Hg at 42 months, compared with about 75% of patients in the placebo group. “The percentage of patients reaching the blood pressure goal was very high,” he said. “ROADMAP will need further analysis to find out what this high percentage of control actually means.”
Over the study period, microalbuminuria occurred in about 8% of the patients in the olmesartan group and 10% of the patients in the placebo group, a statistically significant difference (hazard ratio 0.77). This translated into a risk reduction of 23% for the olmesartan group, compared with the placebo group.
After 1 year, the first incidence of microalbuminuria occurred in about 3% of patients in both groups. For the remainder of the study, fewer patients in the olmesartan group experienced microalbuminuria, compared with patients in the placebo group. “The divergence after 1 year indicates that the specific effects of olmesartan are not due to early hemodynamic changes that would have happened in the first couple of months,” Dr. Haller said in an interview. “We think that olmesartan has a specific, perhaps structural effect on the kidney, either in the glomeruli or in the basal membrane, in the microcirculation.”
Dr. Haller disclosed that he has received honoraria and is a paid consultant for several pharmaceutical companies, including Daiichi Sankyo.
SAN DIEGO — Olmesartan reduced the risk of microalbuminuria by 23% in normoalbuminuric patients with type 2 diabetes and at least one additional cardiovascular disease risk factor, results from a large European trial showed.
The angiotensin receptor blocker also yielded unprecedented blood pressure control for this population of patients.
Those are the first key findings from the Randomized Olmesartan and Diabetes Microalbuminuria Prevention (ROADMAP) study, which were unveiled during a press briefing at the annual meeting of the American Society of Nephrology.
“Despite all of our efforts, we still have problems effectively treating diabetic nephropathy,” said the study's steering committee chair, Dr. Hermann G. Haller of the department of nephrology at Hannover (Germany) Medical School. “The problem for prevention is that we have to diagnose and treat it early. Microalbuminuria is the first sign of the pathogenesis of diabetic nephropathy. It is also an important marker of early development of cardiovascular disease and can indicate microvascular disease.”
The primary end point of the study was the occurrence of microalbuminuria based on two or more positive morning spot urine measurements. Secondary end points were cardiovascular events, renal function, and microvascular morbidity.
With support from Daiichi Sankyo, which markets olmesartan, researchers in 19 countries enrolled 4,449 patients, aged 18-75 years, with well-controlled type 2 diabetes. All patients were normoalbuminuric (defined as a level of 25 mg/g or less for men and 35 mg/g or less for women) and had at lease one additional cardiovascular risk factor, such as high triglyceride levels or hypertension. None of the participants had received an ACE inhibitor or an angiotensin receptor blocker within 6 months of participation.
The patients were randomized to receive either 40 mg olmesartan per day or placebo (conventional antihypertensive treatment without blockade of the renin-angiotensin system). The urine albumin- creatinine ratio was determined every 6 months. Patients were followed for an average of 3.2 years.
At their discretion, study investigators could add calcium channel blockers, diuretics, or beta-blockers to the regimen to help patients achieve the target blood pressure goal of 130/80 mm Hg.
The patients' mean age was 58 years, mean duration of diabetes was 6 years, mean hemoglobin A1c level was 7.6%, and mean body mass index was 31 kg/m
Dr. Haller reported that nearly 80% of patients in the olmesartan group reached the target BP of 130/80 mm Hg at 42 months, compared with about 75% of patients in the placebo group. “The percentage of patients reaching the blood pressure goal was very high,” he said. “ROADMAP will need further analysis to find out what this high percentage of control actually means.”
Over the study period, microalbuminuria occurred in about 8% of the patients in the olmesartan group and 10% of the patients in the placebo group, a statistically significant difference (hazard ratio 0.77). This translated into a risk reduction of 23% for the olmesartan group, compared with the placebo group.
After 1 year, the first incidence of microalbuminuria occurred in about 3% of patients in both groups. For the remainder of the study, fewer patients in the olmesartan group experienced microalbuminuria, compared with patients in the placebo group. “The divergence after 1 year indicates that the specific effects of olmesartan are not due to early hemodynamic changes that would have happened in the first couple of months,” Dr. Haller said in an interview. “We think that olmesartan has a specific, perhaps structural effect on the kidney, either in the glomeruli or in the basal membrane, in the microcirculation.”
Dr. Haller disclosed that he has received honoraria and is a paid consultant for several pharmaceutical companies, including Daiichi Sankyo.
SAN DIEGO — Olmesartan reduced the risk of microalbuminuria by 23% in normoalbuminuric patients with type 2 diabetes and at least one additional cardiovascular disease risk factor, results from a large European trial showed.
The angiotensin receptor blocker also yielded unprecedented blood pressure control for this population of patients.
Those are the first key findings from the Randomized Olmesartan and Diabetes Microalbuminuria Prevention (ROADMAP) study, which were unveiled during a press briefing at the annual meeting of the American Society of Nephrology.
“Despite all of our efforts, we still have problems effectively treating diabetic nephropathy,” said the study's steering committee chair, Dr. Hermann G. Haller of the department of nephrology at Hannover (Germany) Medical School. “The problem for prevention is that we have to diagnose and treat it early. Microalbuminuria is the first sign of the pathogenesis of diabetic nephropathy. It is also an important marker of early development of cardiovascular disease and can indicate microvascular disease.”
The primary end point of the study was the occurrence of microalbuminuria based on two or more positive morning spot urine measurements. Secondary end points were cardiovascular events, renal function, and microvascular morbidity.
With support from Daiichi Sankyo, which markets olmesartan, researchers in 19 countries enrolled 4,449 patients, aged 18-75 years, with well-controlled type 2 diabetes. All patients were normoalbuminuric (defined as a level of 25 mg/g or less for men and 35 mg/g or less for women) and had at lease one additional cardiovascular risk factor, such as high triglyceride levels or hypertension. None of the participants had received an ACE inhibitor or an angiotensin receptor blocker within 6 months of participation.
The patients were randomized to receive either 40 mg olmesartan per day or placebo (conventional antihypertensive treatment without blockade of the renin-angiotensin system). The urine albumin- creatinine ratio was determined every 6 months. Patients were followed for an average of 3.2 years.
At their discretion, study investigators could add calcium channel blockers, diuretics, or beta-blockers to the regimen to help patients achieve the target blood pressure goal of 130/80 mm Hg.
The patients' mean age was 58 years, mean duration of diabetes was 6 years, mean hemoglobin A1c level was 7.6%, and mean body mass index was 31 kg/m
Dr. Haller reported that nearly 80% of patients in the olmesartan group reached the target BP of 130/80 mm Hg at 42 months, compared with about 75% of patients in the placebo group. “The percentage of patients reaching the blood pressure goal was very high,” he said. “ROADMAP will need further analysis to find out what this high percentage of control actually means.”
Over the study period, microalbuminuria occurred in about 8% of the patients in the olmesartan group and 10% of the patients in the placebo group, a statistically significant difference (hazard ratio 0.77). This translated into a risk reduction of 23% for the olmesartan group, compared with the placebo group.
After 1 year, the first incidence of microalbuminuria occurred in about 3% of patients in both groups. For the remainder of the study, fewer patients in the olmesartan group experienced microalbuminuria, compared with patients in the placebo group. “The divergence after 1 year indicates that the specific effects of olmesartan are not due to early hemodynamic changes that would have happened in the first couple of months,” Dr. Haller said in an interview. “We think that olmesartan has a specific, perhaps structural effect on the kidney, either in the glomeruli or in the basal membrane, in the microcirculation.”
Dr. Haller disclosed that he has received honoraria and is a paid consultant for several pharmaceutical companies, including Daiichi Sankyo.
Managing diabetes in hemodialysis patients: Observations and recommendations
Although diabetes is the most common cause of end-stage renal disease (ESRD) worldwide, accounting for 44.2% of ESRD patients in the US Renal Data System in 2005,1 data are scarce on how diabetes should best be treated in patients in ESRD.
We do know that blood glucose levels need to be well controlled in these patients. Several observational studies and one nonrandomized interventional study2–10 showed that higher levels of hemoglobin A1c were associated with higher death rates in patients with diabetes and chronic kidney disease after adjusting for markers of inflammation and malnutrition.
However, ESRD significantly alters glycemic control, the results of hemoglobin A1c testing, and the excretion of antidiabetic medications. The various and opposing effects of ESRD and dialysis can make blood glucose levels fluctuate widely, placing patients at risk of hypoglycemia—and presenting a challenge for nephrologists and internists.
In this review, we summarize the available evidence and make practical recommendations for managing diabetes in patients on hemodialysis.
GLUCOSE LEVELS MAY FLUCTUATE WIDELY
In ESRD, both uremia and dialysis can complicate glycemic control by affecting the secretion, clearance, and peripheral tissue sensitivity of insulin.
Several factors, including uremic toxins, may increase insulin resistance in ESRD, leading to a blunted ability to suppress hepatic gluconeogenesis and regulate peripheral glucose utilization. In type 2 diabetes without kidney disease, insulin resistance leads to increased insulin secretion. This does not occur in ESRD because of concomitant metabolic acidosis, deficiency of 1,25 dihydroxyvitamin D, and secondary hyperparathyroidism.11,12 Hemodialysis further alters insulin secretion, clearance, and resistance as the result of periodic improvement in uremia, acidosis, and phosphate handling.
The dextrose concentration in the dialysate can also affect glucose control. In general, dialysates with lower dextrose concentrations are used and may be associated with hypoglycemia. Conversely, dialysates with higher dextrose concentrations are occasionally used in peritoneal dialysis to increase ultrafiltration, but this can lead to hyperglycemia.10,13
Thus, ESRD and hemodialysis exert opposing forces on insulin secretion, action, and metabolism, often creating unpredictable serum glucose values. For example, one would think that a patient who has insulin resistance would need more supplemental insulin; however, the reduced renal gluconeogenesis and insulin clearance seen in ESRD may result in variable net effects in different patients. In addition, ESRD and hemodialysis alter the pharmacokinetics of diabetic medications. Together, all of these factors contribute to wide fluctuations in glucose levels and increase the risk of hypoglycemic events.
HEMOGLOBIN A1c MAY BE FALSELY HIGH
Self-monitoring of blood glucose plus serial hemoglobin A1c measurements are the standard of care in diabetic patients without renal failure.
However, in diabetic patients with ESRD, elevated blood urea nitrogen causes formation of carbamylated hemoglobin, which is indistinguishable from glycosylated hemoglobin by electrical-charge-based assays and can cause the hemoglobin A1c measurement to be falsely elevated. Other factors such as the shorter red life span of red blood cells, iron deficiency, recent transfusion, and use of erythropoietin-stimulating agents may also cause underestimation of glucose control.14
Despite these limitations, the hemoglobin A1c level is considered a reasonable measure of glycemic control in ESRD. Glycated fructosamine and albumin are other measures of glycemic control with some advantages over hemoglobin A1c in dialysis patients. However, they are not readily available and can be affected by conditions that alter protein metabolism, including ESRD.15–18
Self-monitoring of blood glucose and continuous glucose monitoring systems provide real-time assessments of glycemic control, but both have limitations. Self-monitoring is subject to errors from poor technique, problems with the meters and strips, and lower sensitivity in measuring low blood glucose levels. Continuous monitoring is expensive and is less reliable at lower glucose concentrations, and thus it needs to be used in conjunction with other measures of glucose control. For these reasons, continuous glucose monitoring is not yet widely used.
The guidelines of the 2005 National Kidney Foundation Kidney Disease Outcomes Quality Initiative did not clearly establish a target hemoglobin A1c level for patients with diabetes and ESRD, but levels of 6% to 7% appear to be safe. The target fasting plasma glucose level should be lower than 140 mg/dL, and the target postprandial glucose level should be lower than 200 mg/dL.19
MOST ORAL DIABETES DRUGS ARE CONTRAINDICATED IN ESRD
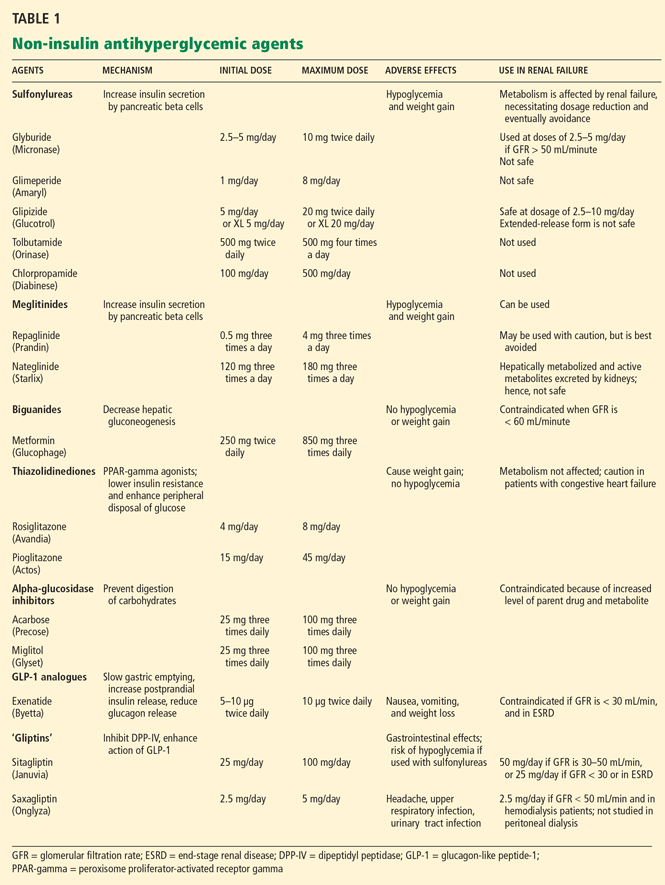
Sulfonylureas
Sulfonylureas reduce blood glucose by stimulating the pancreatic beta cells to increase insulin secretion.
Sulfonylureas have a wide volume of distribution and are highly protein-bound,20 but only the unbound drug exerts a clinical effect. Because of protein binding, dialysis cannot effectively clear elevated levels of sulfonylurea drugs. Furthermore, many ESRD patients take drugs such as salicylates, sulfonamides, vitamin K antagonists, beta-blockers, and fibric acid derivatives, which may displace sulfonylureas from albumin, thus increasing the risk of severe hypoglycemia.
The first-generation sulfonylureas—chlorpropamide (Diabinese), acetohexamide (Dymelor), tolbutamide (Orinase), and tolazamide (Tolinase)—are almost exclusively excreted by the kidney and are therefore contraindicated in ESRD.21 Second-generation agents include glipizide (Glucotrol), glimepiride (Amaryl), glyburide (Micronase), and gliclazide (not available in the United States). Although these drugs are metabolized in the liver, their active metabolites are excreted in the urine, and so they should be avoided in ESRD.22
The only sulfonylurea recommended in ESRD is glipizide, which is also metabolized in the liver but has inactive or weakly active metabolites excreted in the urine. The suggested dose of glipizide is 2.5 to 10 mg/day. In ESRD, sustained-release forms should be avoided because of concerns of hypoglycemia.23
Meglitinides
The meglitinides repaglinide (Prandin) and nateglinide (Starlix) are insulin secretagogues that stimulate pancreatic beta cells. Like the sulfonylureas, nateglinide is hepatically metabolized, with renal excretion of active metabolites. Repaglinide, in contrast, is almost completely converted to inactive metabolites in the liver, and less than 10% is excreted by the kidneys.24,25 The meglitinides still pose a risk of hypoglycemia, especially in ESRD, and hence are not recommended for patients on hemodialysis.24,25
Biguanides
Metformin (Glucophage) is a biguanide that reduces hepatic gluconeogenesis and glucose output. It is excreted essentially unchanged in the urine and is therefore contraindicated in patients with renal disease due to the risks of bioaccumulation and lactic acidosis.22
Thiazolidinediones
The thiazolidinediones rosiglitazone (Avandia) and pioglitazone (Actos) are highly potent, selective agonists that work by binding to and activating a nuclear transcription factor, specifically, peroxisome proliferator-activated receptor gamma (PPAR-gamma). These drugs do not bioaccumulate in renal failure and so do not need dosing adjustments.26
The main adverse effect of these agents is edema, especially when they are combined with insulin therapy. Because of this effect, a joint statement of the American Diabetes Association and the American Heart Association recommends avoiding thiazolidinediones in patients in New York Heart Association (NYHA) class III or IV heart failure.27 Furthermore, caution is required in patients in compensated heart failure (NYHA class I or II) or in those at risk of heart failure, such as patients with previous myocardial infarction or angina, hypertension, left ventricular hypertrophy, significant aortic or mitral valve disease, age greater than 70 years, or diabetes for more than 10 years.27
In summary, although ESRD and dialysis do not affect the metabolism of thiazolidinediones, these agents are not recommended in ESRD because of the associated risk of fluid accumulation and precipitation of heart failure.
Alpha-glucosidase inhibitors
The alpha-glucosidase inhibitors acarbose (Precose) and miglitol (Glyset) slow carbohydrate absorption from the intestine. The levels of these drugs and their active metabolites are higher in renal failure,22 and since data are scarce on the use of these drugs in ESRD, they are contraindicated in ESRD.
GLP-1 ANALOGUES AND ‘GLIPTINS,’ NEW CLASSES OF DRUGS
Glucagon-like peptide-1 (GLP-1) stimulates glucose-dependent insulin release from pancreatic beta cells and inhibits inappropriate postprandial glucagon release. It also slows gastric emptying and reduces food intake. Dipeptidyl peptidase IV (DPP-IV) is an active ubiquitous enzyme that deactivates a variety of bioactive peptides, including GLP-1.
Exenatide (Byetta) is a naturally occurring GLP-1 analogue that is resistant to degradation by DPP-IV and has a longer half-life. Given subcutaneously, exenatide undergoes minimal systemic metabolism and is excreted in the urine.
No dose adjustment is required if the glomerular filtration rate (GFR) is greater than 30 mL/min, but exenatide is contraindicated in patients undergoing hemodialysis or in patients who have a GFR less than 30 mL/min (Table 1).
Sitagliptin (Januvia) is a DPP-IV inhibitor, or “gliptin,” that can be used as initial pharmacologic therapy for type 2 diabetes, as a second agent in those who do not respond to a single agent such as a sulfonylurea,28 metformin,29–31 or a thiazolidinedione,32 and as an additional agent when dual therapy with metformin and a sulfonylurea does not provide adequate glycemic control.28 Sitagliptin is not extensively metabolized and is mainly excreted in the urine.
The usual dose of sitagliptin is 100 mg orally once daily, with reduction to 50 mg for patients with a GFR of 30 to 50 mL/min, and 25 mg for patients with a GFR less than 30 mL/min.33 Sitagliptin may be used at doses of 25 mg daily in ESRD, irrespective of dialysis timing (Table 1).
Other drugs of this class are being developed. Saxagliptin (Onglyza) was recently approved by the US Food and Drug Administration and can be used at a dosage of 2.5 mg daily after dialysis.
Sitagliptin has been associated with gastrointestinal adverse effects. Anaphylaxis, angioedema, and Steven-Johnson syndrome have been reported. The risk of hypoglycemia increases when sitagliptin is used with sulfonylureas.
ESRD REDUCES INSULIN CLEARANCE
In healthy nondiabetic people, the pancreatic beta cells secrete half of the daily insulin requirement (about 0.5 units/kg/day) at a steady basal rate independent of glucose levels. The other half is secreted in response to prandial glucose stimulation.
Secreted into the portal system, insulin passes through the liver, where about 75% is metabolized, with the remaining 25% metabolized by the kidneys. About 60% of the insulin in the arterial bed is filtered by the glomerulus, and 40% is actively secreted into the nephric tubules.34 Most of the insulin in the tubules is metabolized into amino acids, and only 1% of insulin is secreted intact.
For diabetic patients receiving exogenous insulin, renal metabolism plays a more significant role since there is no first-pass metabolism in the liver. As renal function starts to decline, insulin clearance does not change appreciably, due to compensatory peritubular insulin uptake.35 But once the GFR drops below 20 mL/min, the kidneys clear markedly less insulin, an effect compounded by a decrease in the hepatic metabolism of insulin that occurs in uremia.36 Thus, despite the increase in insulin resistance caused by renal failure, the net effect is a reduced requirement for exogenous insulin in ESRD.37
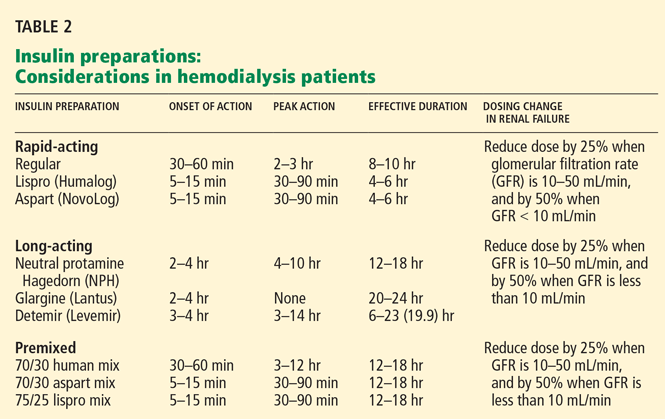
Aisenpreis et al38 showed that the pharmacokinetic profile of insulin lispro (Humalog), which has a short onset of action and a short duration of action, may not only facilitate the correction of hyperglycemia but may also decrease the risk of late hypoglycemic episodes, which is of increased relevance in hemodialysis patients.
On the basis of the available evidence,39,40 we recommend a long-acting insulin such as insulin glargine (Lantus) or NPH insulin for basal requirements, along with a rapid-acting insulin analogue such as lispro or insulin aspart (NovoLog) before meals two or three times daily.
When the GFR drops to between 10 and 50 mL/min, the total insulin dose should be reduced by 25%; once the filtration rate is below 10 mL/min, as in ESRD, the insulin dose should be decreased by 50% from the previous amount.41,42
The newer insulins such as glargine and lispro are more favorable than NPH and regular insulin, but they cost more, which can be an obstacle for some patients.
OBSERVATIONS AND RECOMMENDATIONS
After reviewing the available evidence for the use of diabetic therapy in ESRD, we offer the following observations and recommendations:
- Glycemic control and monitoring in ESRD are complex.
- Patients with ESRD are especially susceptible to hypoglycemia, so diabetic drug therapy requires special caution.
- ESRD patients need ongoing diabetes education, with an emphasis on how to recognize and treat hypoglycemia.
- Diabetic pharmacotherapy in ESRD should be individualized. The targets of therapy are a hemoglobin A1c value between 6% and 7%, a fasting blood glucose level less than 140 mg/dL, and a postprandial glucose level less than 200 mg/dL.
- Of the oral antidiabetic drugs available, glipizide, sitagliptin, and saxagliptin may be used in ESRD. Glipizide, starting with 2.5 mg daily, should be reserved for ESRD patients with a hemoglobin A1c value less than 8.5%.
- Thiazolidinediones may cause fluid overload and thus should be avoided in ESRD.
- We recommend a long-acting insulin (glargine or NPH) for basal requirements, along with rapid-acting insulin before meals two or three times daily.
- The newer basal insulin (glargine) and rapid-acting insulin analogues (lispro or aspart insulin) are more favorable than NPH and regular insulin, but their higher cost could be an issue.
- Some patients may prefer a premixed insulin combination for convenience of dosing. In that case, NPH plus lispro insulin may be better than NPH plus regular insulin.
- For ESRD patients with type 1 diabetes, insulin therapy should be started at 0.5 IU/kg, which is half the calculated dose in patients without renal failure.
- For ESRD patients with type 2 diabetes, insulin therapy should be started at a total daily dose of 0.25 IU/kg.
- Further adjustments to the regimen should be individualized based on self-monitored blood glucose patterns.
- We recommend consulting an endocrinologist with expertise in managing diabetes in ESRD.
- National Institute of Diabetes and Digestive and Kidney Diseases: United States Renal Data System: USRDS 2005 Annual Data Report. Bethesda, MD: National Institutes of Health, 2005.
- Wu MS, Yu CC, Yang CW, et al. Poor pre-dialysis glycaemic control is a predictor of mortality in type II diabetic patients on maintenance haemodialysis. Nephrol Dial Transplant 1997; 12:2105–2110.
- Morioka T, Emoto M, Tabata T, et al. Glycemic control is a predictor of survival for diabetic patients on hemodialysis. Diabetes Care 2001; 24:909–913.
- McMurray SD, Johnson G, Davis S, McDougall K. Diabetes education and care management significantly improve patient outcomes in the dialysis unit. Am J Kidney Dis 2002; 40:566–575.
- Oomichi T, Emoto M, Tabata T, et al. Impact of glycemic control on survival of diabetic patients on chronic regular hemodialysis: a 7-year observational study. Diabetes Care 2006; 29:1496–1500.
- Williams ME, Lacson E, Teng M, Ofsthun N, Lazarus JM. Hemodialyzed type I and type II diabetic patients in the US: characteristics, glycemic control, and survival. Kidney Int 2006; 70:1503–1509.
- Tzamaloukas AH, Yuan ZY, Murata GH, Avasthi PS, Oreopoulos DG. Clinical associations of glycemic control in diabetics on CAPD. Adv Perit Dial 1993; 9:291–294.
- Tzamaloukas AH, Murata GH, Zager PG, Eisenberg B, Avasthi PS. The relationship between glycemic control and morbidity and mortality for diabetics on dialysis. ASAIO J 1993; 39:880–885.
- Kalantar-Zadeh K, Kopple JD, Regidor DL, et al. A1C and survival in maintenance hemodialysis patients. Diabetes Care 2007; 30:1049–1055.
- Kovesdy C, Sharma K, Kalantar-Zadeh. Glycemic control in diabetic CKD patients: where do we stand? Am J Kidney Dis 2008; 52:766–777.
- Mak RH. Intravenous 1,25-dihydroxycholecalciferol corrects glucose intolerance in hemodialysis patients. Kidney Int 1992; 41:1049–1054.
- Hajjar SM, Fadda GZ, Thanakitcharu P, Smogorzewski M, Massry SG. Reduced activity of Na(+)-K+ ATPase of pancreatic islet cells in chronic renal failure: role of secondary hyperparathyroidism. J Am Soc Nephrol 1992; 2:1355–1359.
- Grodstein GP, Blumenkrantz MJ, Kopple JD, Moran JK, Coburn JW. Glucose absorption during continuous ambulatory peritoneal dialysis. Kidney Int 1981; 19:564–567.
- Joy MS, Cefali WT, Hogan SL, Nachman PH. Long-term glycemic control measurements in diabetic patients receiving hemodialysis. Am J Kidney Dis 2002; 39:297–307.
- Lamb E, Venton TR, Cattell WR, Dawnay A. Serum glycated albumin and fructosamine in renal dialysis patients. Nephron 1993; 64:82–88.
- Inaba M, Okuno S, Kumeda Y, et al; Osaka CKD Expert Research Group. Glycated albumin is a better glycemic indicator than glycated hemoglobin values in hemodialysis patients with diabetes: effect of anemia and erythropoietin injection. J Am Soc Nephrol 2007; 18:896–903.
- Constanti C, Simo JM, Joven J, Camps J. Serum fructosamine concentration in patients with nephrotic syndrome and with cirrhosis of the liver: the influence of hypoalbuminaemia and hypergammaglobulinaemia. Ann Clin Biochem 1992; 29:437–442.
- Ford HC, Lim WC, Crooke MJ. Hemoglobin A1 and serum fructosamine levels in hyperthyroidism. Clin Chim Acta 1987; 166:317–321.
- Mak RH. Impact of end-stage renal disease and dialysis on glycemic control. Semin Dial 2000; 13:4–8.
- Skillman TG, Feldman JM. The pharmacology of sulfonylureas. Am J Med 1981; 70:361–372.
- Krepinsky J, Ingram AJ, Clase CM. Prolonged sulfonylurea-induced hypoglycemia in diabetic patients with end-stage renal disease. Am J Kidney Dis 2000; 35:500–505.
- Snyder RW, Berns JS. Use of insulin and oral hypoglycemic medications in patients with diabetes mellitus and advanced kidney disease. Semin Dial 2004; 17:365–370.
- United Kingdom Prospective Diabetes Study (UKPDS) 13. Relative efficacy of randomly allocated diet, sulphonylureas, insulin, or metformin in patients with newly diagnosed non-insulin dependent diabetes followed for three years. BMJ 1995; 310:83–88.
- Inoue T, Shibahara N, Miyagawa K, et al. Pharmacokinetics of nateglinide and its metabolites in subjects with type 2 diabetes mellitus and renal failure. Clin Nephrol 2003; 60:90–95.
- Nagai T, Imamura M, Iizuka K, Mori M. Hypoglycemia due to nateglinide administration in diabetic patient with chronic renal failure. Diabetes Res Clin Pract 2003; 59:191–194.
- Thompson-Culkin K, Zussman B, Miller AK, Freed MI. Pharmacokinetics of rosiglitazone in patients with end-stage renal disease. J Int Med Res 2002; 30:391–399.
- Nesto RW, Bell D, Bonow RO, et al. Thiazolidinedione use, fluid retention, and congestive heart failure: a consensus statement from the American Heart Association and American Diabetes Association. Diabetes Care 2004; 27:256–263.
- Hermansen K, Kipnes M, Luo E, Fanurik D, Khatami H, Stein P; Sitagliptin Study 035 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor, sitagliptin, in patients with type 2 diabetes mellitus inadequately controlled on glimepiride alone or on glimepiride and metformin. Diabetes Obes Metab 2007; 9:733–745.
- Charbonnel B, Karasik A, Liu J, Wu M, Meininger G, et al; Sitagliptin Study 020 Group Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing metformin therapy in patients with type 2 diabetes inadequately controlled with metformin alone. Diabetes Care 2006; 29:2638–2643.
- Goldstein BJ, Feinglos MN, Lunceford JK, Johnson J, Williams-Herman DE; Sitagliptin 036 Study Group. Effect of initial combination therapy with sitagliptin, a dipeptidyl peptidase-4 inhibitor, and metformin on glycemic control in patients with type 2 diabetes. Diabetes Care 2007; 30:1979–1987.
- Nauck MA, Meininger G, Sheng D, Terranella L, Stein PP; Sitagliptin Study 024 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor, sitagliptin, compared with the sulfonylurea, glipizide, in patients with type 2 diabetes inadequately controlled on metformin alone: a randomized, double-blind, non-inferiority trial. Diabetes Obes Metab 2007; 9:194–205.
- Rosenstock J, Brazg R, Andryuk PJ, Lu K, Stein P; Sitagliptin Study 019 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing pioglitazone therapy in patients with type 2 diabetes: a 24-week, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Clin Ther 2006; 28:1556–1568.
- Bergman AJ, Cote J, Yi B, et al. Effect of renal insufficiency on the pharmacokinetics of sitagliptin, a dipeptidyl peptidase-4 inhibitor. Diabetes Care 2007; 30:1862–1864.
- Carone FA, Peterson DR. Hydrolysis and transport of small peptides by the proximal tubule. Am J Physiol 1980; 238:F151–F158.
- Rabkin R, Simon NM, Steiner S, Colwell JA. Effects of renal disease on renal uptake and excretion of insulin in man. N Engl J Med 1970; 282:182–187.
- Mak RH, DeFronzo RA. Glucose and insulin metabolism in uremia. Nephron 1992; 61:377–382.
- Biesenbach G, Raml A, Schmekal B, Eichbauer-Sturm G. Decreased insulin requirement in relation to GFR in nephropathic type 1 and insulin-treated type 2 diabetic patients. Diabet Med 2003; 20:642–645.
- Aisenpreis U, Pfützner A, Giehl M, Keller F, Jehle PM. Pharmacokinetics and pharmacodynamics of insulin Lispro compared with regular insulin in hemodialysis patients with diabetes mellitus. Nephrol Dial Transplant 1999; 14( suppl 4):5–6.
- Tunbridge FK, Newens A, Home PD, et al. A comparison of human ultralente- and lente-based twice-daily injection regimens. Diabet Med 1989; 6:496–501.
- Freeman SL, O’Brien PC, Rizza RA. Use of human ultralente as the basal insulin component in treatment of patients with IDDM. Diabetes Res Clin Pract 1991; 12:187–192.
- Charpentier G, Riveline JP, Varroud-Vial M. Management of drugs affecting blood glucose in diabetic patients with renal failure. Diabetes Metab 2000; 26( suppl 4):73–85.
- Aronoff GR, Berns JS, Brier ME, et al, eds. Drug Prescribing in Renal Failure: Dosing Guidelines for Adults, 4th ed. Philadelphia, PA: American College of Physicians, 1999.
Although diabetes is the most common cause of end-stage renal disease (ESRD) worldwide, accounting for 44.2% of ESRD patients in the US Renal Data System in 2005,1 data are scarce on how diabetes should best be treated in patients in ESRD.
We do know that blood glucose levels need to be well controlled in these patients. Several observational studies and one nonrandomized interventional study2–10 showed that higher levels of hemoglobin A1c were associated with higher death rates in patients with diabetes and chronic kidney disease after adjusting for markers of inflammation and malnutrition.
However, ESRD significantly alters glycemic control, the results of hemoglobin A1c testing, and the excretion of antidiabetic medications. The various and opposing effects of ESRD and dialysis can make blood glucose levels fluctuate widely, placing patients at risk of hypoglycemia—and presenting a challenge for nephrologists and internists.
In this review, we summarize the available evidence and make practical recommendations for managing diabetes in patients on hemodialysis.
GLUCOSE LEVELS MAY FLUCTUATE WIDELY
In ESRD, both uremia and dialysis can complicate glycemic control by affecting the secretion, clearance, and peripheral tissue sensitivity of insulin.
Several factors, including uremic toxins, may increase insulin resistance in ESRD, leading to a blunted ability to suppress hepatic gluconeogenesis and regulate peripheral glucose utilization. In type 2 diabetes without kidney disease, insulin resistance leads to increased insulin secretion. This does not occur in ESRD because of concomitant metabolic acidosis, deficiency of 1,25 dihydroxyvitamin D, and secondary hyperparathyroidism.11,12 Hemodialysis further alters insulin secretion, clearance, and resistance as the result of periodic improvement in uremia, acidosis, and phosphate handling.
The dextrose concentration in the dialysate can also affect glucose control. In general, dialysates with lower dextrose concentrations are used and may be associated with hypoglycemia. Conversely, dialysates with higher dextrose concentrations are occasionally used in peritoneal dialysis to increase ultrafiltration, but this can lead to hyperglycemia.10,13
Thus, ESRD and hemodialysis exert opposing forces on insulin secretion, action, and metabolism, often creating unpredictable serum glucose values. For example, one would think that a patient who has insulin resistance would need more supplemental insulin; however, the reduced renal gluconeogenesis and insulin clearance seen in ESRD may result in variable net effects in different patients. In addition, ESRD and hemodialysis alter the pharmacokinetics of diabetic medications. Together, all of these factors contribute to wide fluctuations in glucose levels and increase the risk of hypoglycemic events.
HEMOGLOBIN A1c MAY BE FALSELY HIGH
Self-monitoring of blood glucose plus serial hemoglobin A1c measurements are the standard of care in diabetic patients without renal failure.
However, in diabetic patients with ESRD, elevated blood urea nitrogen causes formation of carbamylated hemoglobin, which is indistinguishable from glycosylated hemoglobin by electrical-charge-based assays and can cause the hemoglobin A1c measurement to be falsely elevated. Other factors such as the shorter red life span of red blood cells, iron deficiency, recent transfusion, and use of erythropoietin-stimulating agents may also cause underestimation of glucose control.14
Despite these limitations, the hemoglobin A1c level is considered a reasonable measure of glycemic control in ESRD. Glycated fructosamine and albumin are other measures of glycemic control with some advantages over hemoglobin A1c in dialysis patients. However, they are not readily available and can be affected by conditions that alter protein metabolism, including ESRD.15–18
Self-monitoring of blood glucose and continuous glucose monitoring systems provide real-time assessments of glycemic control, but both have limitations. Self-monitoring is subject to errors from poor technique, problems with the meters and strips, and lower sensitivity in measuring low blood glucose levels. Continuous monitoring is expensive and is less reliable at lower glucose concentrations, and thus it needs to be used in conjunction with other measures of glucose control. For these reasons, continuous glucose monitoring is not yet widely used.
The guidelines of the 2005 National Kidney Foundation Kidney Disease Outcomes Quality Initiative did not clearly establish a target hemoglobin A1c level for patients with diabetes and ESRD, but levels of 6% to 7% appear to be safe. The target fasting plasma glucose level should be lower than 140 mg/dL, and the target postprandial glucose level should be lower than 200 mg/dL.19
MOST ORAL DIABETES DRUGS ARE CONTRAINDICATED IN ESRD
Sulfonylureas
Sulfonylureas reduce blood glucose by stimulating the pancreatic beta cells to increase insulin secretion.
Sulfonylureas have a wide volume of distribution and are highly protein-bound,20 but only the unbound drug exerts a clinical effect. Because of protein binding, dialysis cannot effectively clear elevated levels of sulfonylurea drugs. Furthermore, many ESRD patients take drugs such as salicylates, sulfonamides, vitamin K antagonists, beta-blockers, and fibric acid derivatives, which may displace sulfonylureas from albumin, thus increasing the risk of severe hypoglycemia.
The first-generation sulfonylureas—chlorpropamide (Diabinese), acetohexamide (Dymelor), tolbutamide (Orinase), and tolazamide (Tolinase)—are almost exclusively excreted by the kidney and are therefore contraindicated in ESRD.21 Second-generation agents include glipizide (Glucotrol), glimepiride (Amaryl), glyburide (Micronase), and gliclazide (not available in the United States). Although these drugs are metabolized in the liver, their active metabolites are excreted in the urine, and so they should be avoided in ESRD.22
The only sulfonylurea recommended in ESRD is glipizide, which is also metabolized in the liver but has inactive or weakly active metabolites excreted in the urine. The suggested dose of glipizide is 2.5 to 10 mg/day. In ESRD, sustained-release forms should be avoided because of concerns of hypoglycemia.23
Meglitinides
The meglitinides repaglinide (Prandin) and nateglinide (Starlix) are insulin secretagogues that stimulate pancreatic beta cells. Like the sulfonylureas, nateglinide is hepatically metabolized, with renal excretion of active metabolites. Repaglinide, in contrast, is almost completely converted to inactive metabolites in the liver, and less than 10% is excreted by the kidneys.24,25 The meglitinides still pose a risk of hypoglycemia, especially in ESRD, and hence are not recommended for patients on hemodialysis.24,25
Biguanides
Metformin (Glucophage) is a biguanide that reduces hepatic gluconeogenesis and glucose output. It is excreted essentially unchanged in the urine and is therefore contraindicated in patients with renal disease due to the risks of bioaccumulation and lactic acidosis.22
Thiazolidinediones
The thiazolidinediones rosiglitazone (Avandia) and pioglitazone (Actos) are highly potent, selective agonists that work by binding to and activating a nuclear transcription factor, specifically, peroxisome proliferator-activated receptor gamma (PPAR-gamma). These drugs do not bioaccumulate in renal failure and so do not need dosing adjustments.26
The main adverse effect of these agents is edema, especially when they are combined with insulin therapy. Because of this effect, a joint statement of the American Diabetes Association and the American Heart Association recommends avoiding thiazolidinediones in patients in New York Heart Association (NYHA) class III or IV heart failure.27 Furthermore, caution is required in patients in compensated heart failure (NYHA class I or II) or in those at risk of heart failure, such as patients with previous myocardial infarction or angina, hypertension, left ventricular hypertrophy, significant aortic or mitral valve disease, age greater than 70 years, or diabetes for more than 10 years.27
In summary, although ESRD and dialysis do not affect the metabolism of thiazolidinediones, these agents are not recommended in ESRD because of the associated risk of fluid accumulation and precipitation of heart failure.
Alpha-glucosidase inhibitors
The alpha-glucosidase inhibitors acarbose (Precose) and miglitol (Glyset) slow carbohydrate absorption from the intestine. The levels of these drugs and their active metabolites are higher in renal failure,22 and since data are scarce on the use of these drugs in ESRD, they are contraindicated in ESRD.
GLP-1 ANALOGUES AND ‘GLIPTINS,’ NEW CLASSES OF DRUGS
Glucagon-like peptide-1 (GLP-1) stimulates glucose-dependent insulin release from pancreatic beta cells and inhibits inappropriate postprandial glucagon release. It also slows gastric emptying and reduces food intake. Dipeptidyl peptidase IV (DPP-IV) is an active ubiquitous enzyme that deactivates a variety of bioactive peptides, including GLP-1.
Exenatide (Byetta) is a naturally occurring GLP-1 analogue that is resistant to degradation by DPP-IV and has a longer half-life. Given subcutaneously, exenatide undergoes minimal systemic metabolism and is excreted in the urine.
No dose adjustment is required if the glomerular filtration rate (GFR) is greater than 30 mL/min, but exenatide is contraindicated in patients undergoing hemodialysis or in patients who have a GFR less than 30 mL/min (Table 1).
Sitagliptin (Januvia) is a DPP-IV inhibitor, or “gliptin,” that can be used as initial pharmacologic therapy for type 2 diabetes, as a second agent in those who do not respond to a single agent such as a sulfonylurea,28 metformin,29–31 or a thiazolidinedione,32 and as an additional agent when dual therapy with metformin and a sulfonylurea does not provide adequate glycemic control.28 Sitagliptin is not extensively metabolized and is mainly excreted in the urine.
The usual dose of sitagliptin is 100 mg orally once daily, with reduction to 50 mg for patients with a GFR of 30 to 50 mL/min, and 25 mg for patients with a GFR less than 30 mL/min.33 Sitagliptin may be used at doses of 25 mg daily in ESRD, irrespective of dialysis timing (Table 1).
Other drugs of this class are being developed. Saxagliptin (Onglyza) was recently approved by the US Food and Drug Administration and can be used at a dosage of 2.5 mg daily after dialysis.
Sitagliptin has been associated with gastrointestinal adverse effects. Anaphylaxis, angioedema, and Steven-Johnson syndrome have been reported. The risk of hypoglycemia increases when sitagliptin is used with sulfonylureas.
ESRD REDUCES INSULIN CLEARANCE
In healthy nondiabetic people, the pancreatic beta cells secrete half of the daily insulin requirement (about 0.5 units/kg/day) at a steady basal rate independent of glucose levels. The other half is secreted in response to prandial glucose stimulation.
Secreted into the portal system, insulin passes through the liver, where about 75% is metabolized, with the remaining 25% metabolized by the kidneys. About 60% of the insulin in the arterial bed is filtered by the glomerulus, and 40% is actively secreted into the nephric tubules.34 Most of the insulin in the tubules is metabolized into amino acids, and only 1% of insulin is secreted intact.
For diabetic patients receiving exogenous insulin, renal metabolism plays a more significant role since there is no first-pass metabolism in the liver. As renal function starts to decline, insulin clearance does not change appreciably, due to compensatory peritubular insulin uptake.35 But once the GFR drops below 20 mL/min, the kidneys clear markedly less insulin, an effect compounded by a decrease in the hepatic metabolism of insulin that occurs in uremia.36 Thus, despite the increase in insulin resistance caused by renal failure, the net effect is a reduced requirement for exogenous insulin in ESRD.37
Aisenpreis et al38 showed that the pharmacokinetic profile of insulin lispro (Humalog), which has a short onset of action and a short duration of action, may not only facilitate the correction of hyperglycemia but may also decrease the risk of late hypoglycemic episodes, which is of increased relevance in hemodialysis patients.
On the basis of the available evidence,39,40 we recommend a long-acting insulin such as insulin glargine (Lantus) or NPH insulin for basal requirements, along with a rapid-acting insulin analogue such as lispro or insulin aspart (NovoLog) before meals two or three times daily.
When the GFR drops to between 10 and 50 mL/min, the total insulin dose should be reduced by 25%; once the filtration rate is below 10 mL/min, as in ESRD, the insulin dose should be decreased by 50% from the previous amount.41,42
The newer insulins such as glargine and lispro are more favorable than NPH and regular insulin, but they cost more, which can be an obstacle for some patients.
OBSERVATIONS AND RECOMMENDATIONS
After reviewing the available evidence for the use of diabetic therapy in ESRD, we offer the following observations and recommendations:
- Glycemic control and monitoring in ESRD are complex.
- Patients with ESRD are especially susceptible to hypoglycemia, so diabetic drug therapy requires special caution.
- ESRD patients need ongoing diabetes education, with an emphasis on how to recognize and treat hypoglycemia.
- Diabetic pharmacotherapy in ESRD should be individualized. The targets of therapy are a hemoglobin A1c value between 6% and 7%, a fasting blood glucose level less than 140 mg/dL, and a postprandial glucose level less than 200 mg/dL.
- Of the oral antidiabetic drugs available, glipizide, sitagliptin, and saxagliptin may be used in ESRD. Glipizide, starting with 2.5 mg daily, should be reserved for ESRD patients with a hemoglobin A1c value less than 8.5%.
- Thiazolidinediones may cause fluid overload and thus should be avoided in ESRD.
- We recommend a long-acting insulin (glargine or NPH) for basal requirements, along with rapid-acting insulin before meals two or three times daily.
- The newer basal insulin (glargine) and rapid-acting insulin analogues (lispro or aspart insulin) are more favorable than NPH and regular insulin, but their higher cost could be an issue.
- Some patients may prefer a premixed insulin combination for convenience of dosing. In that case, NPH plus lispro insulin may be better than NPH plus regular insulin.
- For ESRD patients with type 1 diabetes, insulin therapy should be started at 0.5 IU/kg, which is half the calculated dose in patients without renal failure.
- For ESRD patients with type 2 diabetes, insulin therapy should be started at a total daily dose of 0.25 IU/kg.
- Further adjustments to the regimen should be individualized based on self-monitored blood glucose patterns.
- We recommend consulting an endocrinologist with expertise in managing diabetes in ESRD.
Although diabetes is the most common cause of end-stage renal disease (ESRD) worldwide, accounting for 44.2% of ESRD patients in the US Renal Data System in 2005,1 data are scarce on how diabetes should best be treated in patients in ESRD.
We do know that blood glucose levels need to be well controlled in these patients. Several observational studies and one nonrandomized interventional study2–10 showed that higher levels of hemoglobin A1c were associated with higher death rates in patients with diabetes and chronic kidney disease after adjusting for markers of inflammation and malnutrition.
However, ESRD significantly alters glycemic control, the results of hemoglobin A1c testing, and the excretion of antidiabetic medications. The various and opposing effects of ESRD and dialysis can make blood glucose levels fluctuate widely, placing patients at risk of hypoglycemia—and presenting a challenge for nephrologists and internists.
In this review, we summarize the available evidence and make practical recommendations for managing diabetes in patients on hemodialysis.
GLUCOSE LEVELS MAY FLUCTUATE WIDELY
In ESRD, both uremia and dialysis can complicate glycemic control by affecting the secretion, clearance, and peripheral tissue sensitivity of insulin.
Several factors, including uremic toxins, may increase insulin resistance in ESRD, leading to a blunted ability to suppress hepatic gluconeogenesis and regulate peripheral glucose utilization. In type 2 diabetes without kidney disease, insulin resistance leads to increased insulin secretion. This does not occur in ESRD because of concomitant metabolic acidosis, deficiency of 1,25 dihydroxyvitamin D, and secondary hyperparathyroidism.11,12 Hemodialysis further alters insulin secretion, clearance, and resistance as the result of periodic improvement in uremia, acidosis, and phosphate handling.
The dextrose concentration in the dialysate can also affect glucose control. In general, dialysates with lower dextrose concentrations are used and may be associated with hypoglycemia. Conversely, dialysates with higher dextrose concentrations are occasionally used in peritoneal dialysis to increase ultrafiltration, but this can lead to hyperglycemia.10,13
Thus, ESRD and hemodialysis exert opposing forces on insulin secretion, action, and metabolism, often creating unpredictable serum glucose values. For example, one would think that a patient who has insulin resistance would need more supplemental insulin; however, the reduced renal gluconeogenesis and insulin clearance seen in ESRD may result in variable net effects in different patients. In addition, ESRD and hemodialysis alter the pharmacokinetics of diabetic medications. Together, all of these factors contribute to wide fluctuations in glucose levels and increase the risk of hypoglycemic events.
HEMOGLOBIN A1c MAY BE FALSELY HIGH
Self-monitoring of blood glucose plus serial hemoglobin A1c measurements are the standard of care in diabetic patients without renal failure.
However, in diabetic patients with ESRD, elevated blood urea nitrogen causes formation of carbamylated hemoglobin, which is indistinguishable from glycosylated hemoglobin by electrical-charge-based assays and can cause the hemoglobin A1c measurement to be falsely elevated. Other factors such as the shorter red life span of red blood cells, iron deficiency, recent transfusion, and use of erythropoietin-stimulating agents may also cause underestimation of glucose control.14
Despite these limitations, the hemoglobin A1c level is considered a reasonable measure of glycemic control in ESRD. Glycated fructosamine and albumin are other measures of glycemic control with some advantages over hemoglobin A1c in dialysis patients. However, they are not readily available and can be affected by conditions that alter protein metabolism, including ESRD.15–18
Self-monitoring of blood glucose and continuous glucose monitoring systems provide real-time assessments of glycemic control, but both have limitations. Self-monitoring is subject to errors from poor technique, problems with the meters and strips, and lower sensitivity in measuring low blood glucose levels. Continuous monitoring is expensive and is less reliable at lower glucose concentrations, and thus it needs to be used in conjunction with other measures of glucose control. For these reasons, continuous glucose monitoring is not yet widely used.
The guidelines of the 2005 National Kidney Foundation Kidney Disease Outcomes Quality Initiative did not clearly establish a target hemoglobin A1c level for patients with diabetes and ESRD, but levels of 6% to 7% appear to be safe. The target fasting plasma glucose level should be lower than 140 mg/dL, and the target postprandial glucose level should be lower than 200 mg/dL.19
MOST ORAL DIABETES DRUGS ARE CONTRAINDICATED IN ESRD
Sulfonylureas
Sulfonylureas reduce blood glucose by stimulating the pancreatic beta cells to increase insulin secretion.
Sulfonylureas have a wide volume of distribution and are highly protein-bound,20 but only the unbound drug exerts a clinical effect. Because of protein binding, dialysis cannot effectively clear elevated levels of sulfonylurea drugs. Furthermore, many ESRD patients take drugs such as salicylates, sulfonamides, vitamin K antagonists, beta-blockers, and fibric acid derivatives, which may displace sulfonylureas from albumin, thus increasing the risk of severe hypoglycemia.
The first-generation sulfonylureas—chlorpropamide (Diabinese), acetohexamide (Dymelor), tolbutamide (Orinase), and tolazamide (Tolinase)—are almost exclusively excreted by the kidney and are therefore contraindicated in ESRD.21 Second-generation agents include glipizide (Glucotrol), glimepiride (Amaryl), glyburide (Micronase), and gliclazide (not available in the United States). Although these drugs are metabolized in the liver, their active metabolites are excreted in the urine, and so they should be avoided in ESRD.22
The only sulfonylurea recommended in ESRD is glipizide, which is also metabolized in the liver but has inactive or weakly active metabolites excreted in the urine. The suggested dose of glipizide is 2.5 to 10 mg/day. In ESRD, sustained-release forms should be avoided because of concerns of hypoglycemia.23
Meglitinides
The meglitinides repaglinide (Prandin) and nateglinide (Starlix) are insulin secretagogues that stimulate pancreatic beta cells. Like the sulfonylureas, nateglinide is hepatically metabolized, with renal excretion of active metabolites. Repaglinide, in contrast, is almost completely converted to inactive metabolites in the liver, and less than 10% is excreted by the kidneys.24,25 The meglitinides still pose a risk of hypoglycemia, especially in ESRD, and hence are not recommended for patients on hemodialysis.24,25
Biguanides
Metformin (Glucophage) is a biguanide that reduces hepatic gluconeogenesis and glucose output. It is excreted essentially unchanged in the urine and is therefore contraindicated in patients with renal disease due to the risks of bioaccumulation and lactic acidosis.22
Thiazolidinediones
The thiazolidinediones rosiglitazone (Avandia) and pioglitazone (Actos) are highly potent, selective agonists that work by binding to and activating a nuclear transcription factor, specifically, peroxisome proliferator-activated receptor gamma (PPAR-gamma). These drugs do not bioaccumulate in renal failure and so do not need dosing adjustments.26
The main adverse effect of these agents is edema, especially when they are combined with insulin therapy. Because of this effect, a joint statement of the American Diabetes Association and the American Heart Association recommends avoiding thiazolidinediones in patients in New York Heart Association (NYHA) class III or IV heart failure.27 Furthermore, caution is required in patients in compensated heart failure (NYHA class I or II) or in those at risk of heart failure, such as patients with previous myocardial infarction or angina, hypertension, left ventricular hypertrophy, significant aortic or mitral valve disease, age greater than 70 years, or diabetes for more than 10 years.27
In summary, although ESRD and dialysis do not affect the metabolism of thiazolidinediones, these agents are not recommended in ESRD because of the associated risk of fluid accumulation and precipitation of heart failure.
Alpha-glucosidase inhibitors
The alpha-glucosidase inhibitors acarbose (Precose) and miglitol (Glyset) slow carbohydrate absorption from the intestine. The levels of these drugs and their active metabolites are higher in renal failure,22 and since data are scarce on the use of these drugs in ESRD, they are contraindicated in ESRD.
GLP-1 ANALOGUES AND ‘GLIPTINS,’ NEW CLASSES OF DRUGS
Glucagon-like peptide-1 (GLP-1) stimulates glucose-dependent insulin release from pancreatic beta cells and inhibits inappropriate postprandial glucagon release. It also slows gastric emptying and reduces food intake. Dipeptidyl peptidase IV (DPP-IV) is an active ubiquitous enzyme that deactivates a variety of bioactive peptides, including GLP-1.
Exenatide (Byetta) is a naturally occurring GLP-1 analogue that is resistant to degradation by DPP-IV and has a longer half-life. Given subcutaneously, exenatide undergoes minimal systemic metabolism and is excreted in the urine.
No dose adjustment is required if the glomerular filtration rate (GFR) is greater than 30 mL/min, but exenatide is contraindicated in patients undergoing hemodialysis or in patients who have a GFR less than 30 mL/min (Table 1).
Sitagliptin (Januvia) is a DPP-IV inhibitor, or “gliptin,” that can be used as initial pharmacologic therapy for type 2 diabetes, as a second agent in those who do not respond to a single agent such as a sulfonylurea,28 metformin,29–31 or a thiazolidinedione,32 and as an additional agent when dual therapy with metformin and a sulfonylurea does not provide adequate glycemic control.28 Sitagliptin is not extensively metabolized and is mainly excreted in the urine.
The usual dose of sitagliptin is 100 mg orally once daily, with reduction to 50 mg for patients with a GFR of 30 to 50 mL/min, and 25 mg for patients with a GFR less than 30 mL/min.33 Sitagliptin may be used at doses of 25 mg daily in ESRD, irrespective of dialysis timing (Table 1).
Other drugs of this class are being developed. Saxagliptin (Onglyza) was recently approved by the US Food and Drug Administration and can be used at a dosage of 2.5 mg daily after dialysis.
Sitagliptin has been associated with gastrointestinal adverse effects. Anaphylaxis, angioedema, and Steven-Johnson syndrome have been reported. The risk of hypoglycemia increases when sitagliptin is used with sulfonylureas.
ESRD REDUCES INSULIN CLEARANCE
In healthy nondiabetic people, the pancreatic beta cells secrete half of the daily insulin requirement (about 0.5 units/kg/day) at a steady basal rate independent of glucose levels. The other half is secreted in response to prandial glucose stimulation.
Secreted into the portal system, insulin passes through the liver, where about 75% is metabolized, with the remaining 25% metabolized by the kidneys. About 60% of the insulin in the arterial bed is filtered by the glomerulus, and 40% is actively secreted into the nephric tubules.34 Most of the insulin in the tubules is metabolized into amino acids, and only 1% of insulin is secreted intact.
For diabetic patients receiving exogenous insulin, renal metabolism plays a more significant role since there is no first-pass metabolism in the liver. As renal function starts to decline, insulin clearance does not change appreciably, due to compensatory peritubular insulin uptake.35 But once the GFR drops below 20 mL/min, the kidneys clear markedly less insulin, an effect compounded by a decrease in the hepatic metabolism of insulin that occurs in uremia.36 Thus, despite the increase in insulin resistance caused by renal failure, the net effect is a reduced requirement for exogenous insulin in ESRD.37
Aisenpreis et al38 showed that the pharmacokinetic profile of insulin lispro (Humalog), which has a short onset of action and a short duration of action, may not only facilitate the correction of hyperglycemia but may also decrease the risk of late hypoglycemic episodes, which is of increased relevance in hemodialysis patients.
On the basis of the available evidence,39,40 we recommend a long-acting insulin such as insulin glargine (Lantus) or NPH insulin for basal requirements, along with a rapid-acting insulin analogue such as lispro or insulin aspart (NovoLog) before meals two or three times daily.
When the GFR drops to between 10 and 50 mL/min, the total insulin dose should be reduced by 25%; once the filtration rate is below 10 mL/min, as in ESRD, the insulin dose should be decreased by 50% from the previous amount.41,42
The newer insulins such as glargine and lispro are more favorable than NPH and regular insulin, but they cost more, which can be an obstacle for some patients.
OBSERVATIONS AND RECOMMENDATIONS
After reviewing the available evidence for the use of diabetic therapy in ESRD, we offer the following observations and recommendations:
- Glycemic control and monitoring in ESRD are complex.
- Patients with ESRD are especially susceptible to hypoglycemia, so diabetic drug therapy requires special caution.
- ESRD patients need ongoing diabetes education, with an emphasis on how to recognize and treat hypoglycemia.
- Diabetic pharmacotherapy in ESRD should be individualized. The targets of therapy are a hemoglobin A1c value between 6% and 7%, a fasting blood glucose level less than 140 mg/dL, and a postprandial glucose level less than 200 mg/dL.
- Of the oral antidiabetic drugs available, glipizide, sitagliptin, and saxagliptin may be used in ESRD. Glipizide, starting with 2.5 mg daily, should be reserved for ESRD patients with a hemoglobin A1c value less than 8.5%.
- Thiazolidinediones may cause fluid overload and thus should be avoided in ESRD.
- We recommend a long-acting insulin (glargine or NPH) for basal requirements, along with rapid-acting insulin before meals two or three times daily.
- The newer basal insulin (glargine) and rapid-acting insulin analogues (lispro or aspart insulin) are more favorable than NPH and regular insulin, but their higher cost could be an issue.
- Some patients may prefer a premixed insulin combination for convenience of dosing. In that case, NPH plus lispro insulin may be better than NPH plus regular insulin.
- For ESRD patients with type 1 diabetes, insulin therapy should be started at 0.5 IU/kg, which is half the calculated dose in patients without renal failure.
- For ESRD patients with type 2 diabetes, insulin therapy should be started at a total daily dose of 0.25 IU/kg.
- Further adjustments to the regimen should be individualized based on self-monitored blood glucose patterns.
- We recommend consulting an endocrinologist with expertise in managing diabetes in ESRD.
- National Institute of Diabetes and Digestive and Kidney Diseases: United States Renal Data System: USRDS 2005 Annual Data Report. Bethesda, MD: National Institutes of Health, 2005.
- Wu MS, Yu CC, Yang CW, et al. Poor pre-dialysis glycaemic control is a predictor of mortality in type II diabetic patients on maintenance haemodialysis. Nephrol Dial Transplant 1997; 12:2105–2110.
- Morioka T, Emoto M, Tabata T, et al. Glycemic control is a predictor of survival for diabetic patients on hemodialysis. Diabetes Care 2001; 24:909–913.
- McMurray SD, Johnson G, Davis S, McDougall K. Diabetes education and care management significantly improve patient outcomes in the dialysis unit. Am J Kidney Dis 2002; 40:566–575.
- Oomichi T, Emoto M, Tabata T, et al. Impact of glycemic control on survival of diabetic patients on chronic regular hemodialysis: a 7-year observational study. Diabetes Care 2006; 29:1496–1500.
- Williams ME, Lacson E, Teng M, Ofsthun N, Lazarus JM. Hemodialyzed type I and type II diabetic patients in the US: characteristics, glycemic control, and survival. Kidney Int 2006; 70:1503–1509.
- Tzamaloukas AH, Yuan ZY, Murata GH, Avasthi PS, Oreopoulos DG. Clinical associations of glycemic control in diabetics on CAPD. Adv Perit Dial 1993; 9:291–294.
- Tzamaloukas AH, Murata GH, Zager PG, Eisenberg B, Avasthi PS. The relationship between glycemic control and morbidity and mortality for diabetics on dialysis. ASAIO J 1993; 39:880–885.
- Kalantar-Zadeh K, Kopple JD, Regidor DL, et al. A1C and survival in maintenance hemodialysis patients. Diabetes Care 2007; 30:1049–1055.
- Kovesdy C, Sharma K, Kalantar-Zadeh. Glycemic control in diabetic CKD patients: where do we stand? Am J Kidney Dis 2008; 52:766–777.
- Mak RH. Intravenous 1,25-dihydroxycholecalciferol corrects glucose intolerance in hemodialysis patients. Kidney Int 1992; 41:1049–1054.
- Hajjar SM, Fadda GZ, Thanakitcharu P, Smogorzewski M, Massry SG. Reduced activity of Na(+)-K+ ATPase of pancreatic islet cells in chronic renal failure: role of secondary hyperparathyroidism. J Am Soc Nephrol 1992; 2:1355–1359.
- Grodstein GP, Blumenkrantz MJ, Kopple JD, Moran JK, Coburn JW. Glucose absorption during continuous ambulatory peritoneal dialysis. Kidney Int 1981; 19:564–567.
- Joy MS, Cefali WT, Hogan SL, Nachman PH. Long-term glycemic control measurements in diabetic patients receiving hemodialysis. Am J Kidney Dis 2002; 39:297–307.
- Lamb E, Venton TR, Cattell WR, Dawnay A. Serum glycated albumin and fructosamine in renal dialysis patients. Nephron 1993; 64:82–88.
- Inaba M, Okuno S, Kumeda Y, et al; Osaka CKD Expert Research Group. Glycated albumin is a better glycemic indicator than glycated hemoglobin values in hemodialysis patients with diabetes: effect of anemia and erythropoietin injection. J Am Soc Nephrol 2007; 18:896–903.
- Constanti C, Simo JM, Joven J, Camps J. Serum fructosamine concentration in patients with nephrotic syndrome and with cirrhosis of the liver: the influence of hypoalbuminaemia and hypergammaglobulinaemia. Ann Clin Biochem 1992; 29:437–442.
- Ford HC, Lim WC, Crooke MJ. Hemoglobin A1 and serum fructosamine levels in hyperthyroidism. Clin Chim Acta 1987; 166:317–321.
- Mak RH. Impact of end-stage renal disease and dialysis on glycemic control. Semin Dial 2000; 13:4–8.
- Skillman TG, Feldman JM. The pharmacology of sulfonylureas. Am J Med 1981; 70:361–372.
- Krepinsky J, Ingram AJ, Clase CM. Prolonged sulfonylurea-induced hypoglycemia in diabetic patients with end-stage renal disease. Am J Kidney Dis 2000; 35:500–505.
- Snyder RW, Berns JS. Use of insulin and oral hypoglycemic medications in patients with diabetes mellitus and advanced kidney disease. Semin Dial 2004; 17:365–370.
- United Kingdom Prospective Diabetes Study (UKPDS) 13. Relative efficacy of randomly allocated diet, sulphonylureas, insulin, or metformin in patients with newly diagnosed non-insulin dependent diabetes followed for three years. BMJ 1995; 310:83–88.
- Inoue T, Shibahara N, Miyagawa K, et al. Pharmacokinetics of nateglinide and its metabolites in subjects with type 2 diabetes mellitus and renal failure. Clin Nephrol 2003; 60:90–95.
- Nagai T, Imamura M, Iizuka K, Mori M. Hypoglycemia due to nateglinide administration in diabetic patient with chronic renal failure. Diabetes Res Clin Pract 2003; 59:191–194.
- Thompson-Culkin K, Zussman B, Miller AK, Freed MI. Pharmacokinetics of rosiglitazone in patients with end-stage renal disease. J Int Med Res 2002; 30:391–399.
- Nesto RW, Bell D, Bonow RO, et al. Thiazolidinedione use, fluid retention, and congestive heart failure: a consensus statement from the American Heart Association and American Diabetes Association. Diabetes Care 2004; 27:256–263.
- Hermansen K, Kipnes M, Luo E, Fanurik D, Khatami H, Stein P; Sitagliptin Study 035 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor, sitagliptin, in patients with type 2 diabetes mellitus inadequately controlled on glimepiride alone or on glimepiride and metformin. Diabetes Obes Metab 2007; 9:733–745.
- Charbonnel B, Karasik A, Liu J, Wu M, Meininger G, et al; Sitagliptin Study 020 Group Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing metformin therapy in patients with type 2 diabetes inadequately controlled with metformin alone. Diabetes Care 2006; 29:2638–2643.
- Goldstein BJ, Feinglos MN, Lunceford JK, Johnson J, Williams-Herman DE; Sitagliptin 036 Study Group. Effect of initial combination therapy with sitagliptin, a dipeptidyl peptidase-4 inhibitor, and metformin on glycemic control in patients with type 2 diabetes. Diabetes Care 2007; 30:1979–1987.
- Nauck MA, Meininger G, Sheng D, Terranella L, Stein PP; Sitagliptin Study 024 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor, sitagliptin, compared with the sulfonylurea, glipizide, in patients with type 2 diabetes inadequately controlled on metformin alone: a randomized, double-blind, non-inferiority trial. Diabetes Obes Metab 2007; 9:194–205.
- Rosenstock J, Brazg R, Andryuk PJ, Lu K, Stein P; Sitagliptin Study 019 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing pioglitazone therapy in patients with type 2 diabetes: a 24-week, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Clin Ther 2006; 28:1556–1568.
- Bergman AJ, Cote J, Yi B, et al. Effect of renal insufficiency on the pharmacokinetics of sitagliptin, a dipeptidyl peptidase-4 inhibitor. Diabetes Care 2007; 30:1862–1864.
- Carone FA, Peterson DR. Hydrolysis and transport of small peptides by the proximal tubule. Am J Physiol 1980; 238:F151–F158.
- Rabkin R, Simon NM, Steiner S, Colwell JA. Effects of renal disease on renal uptake and excretion of insulin in man. N Engl J Med 1970; 282:182–187.
- Mak RH, DeFronzo RA. Glucose and insulin metabolism in uremia. Nephron 1992; 61:377–382.
- Biesenbach G, Raml A, Schmekal B, Eichbauer-Sturm G. Decreased insulin requirement in relation to GFR in nephropathic type 1 and insulin-treated type 2 diabetic patients. Diabet Med 2003; 20:642–645.
- Aisenpreis U, Pfützner A, Giehl M, Keller F, Jehle PM. Pharmacokinetics and pharmacodynamics of insulin Lispro compared with regular insulin in hemodialysis patients with diabetes mellitus. Nephrol Dial Transplant 1999; 14( suppl 4):5–6.
- Tunbridge FK, Newens A, Home PD, et al. A comparison of human ultralente- and lente-based twice-daily injection regimens. Diabet Med 1989; 6:496–501.
- Freeman SL, O’Brien PC, Rizza RA. Use of human ultralente as the basal insulin component in treatment of patients with IDDM. Diabetes Res Clin Pract 1991; 12:187–192.
- Charpentier G, Riveline JP, Varroud-Vial M. Management of drugs affecting blood glucose in diabetic patients with renal failure. Diabetes Metab 2000; 26( suppl 4):73–85.
- Aronoff GR, Berns JS, Brier ME, et al, eds. Drug Prescribing in Renal Failure: Dosing Guidelines for Adults, 4th ed. Philadelphia, PA: American College of Physicians, 1999.
- National Institute of Diabetes and Digestive and Kidney Diseases: United States Renal Data System: USRDS 2005 Annual Data Report. Bethesda, MD: National Institutes of Health, 2005.
- Wu MS, Yu CC, Yang CW, et al. Poor pre-dialysis glycaemic control is a predictor of mortality in type II diabetic patients on maintenance haemodialysis. Nephrol Dial Transplant 1997; 12:2105–2110.
- Morioka T, Emoto M, Tabata T, et al. Glycemic control is a predictor of survival for diabetic patients on hemodialysis. Diabetes Care 2001; 24:909–913.
- McMurray SD, Johnson G, Davis S, McDougall K. Diabetes education and care management significantly improve patient outcomes in the dialysis unit. Am J Kidney Dis 2002; 40:566–575.
- Oomichi T, Emoto M, Tabata T, et al. Impact of glycemic control on survival of diabetic patients on chronic regular hemodialysis: a 7-year observational study. Diabetes Care 2006; 29:1496–1500.
- Williams ME, Lacson E, Teng M, Ofsthun N, Lazarus JM. Hemodialyzed type I and type II diabetic patients in the US: characteristics, glycemic control, and survival. Kidney Int 2006; 70:1503–1509.
- Tzamaloukas AH, Yuan ZY, Murata GH, Avasthi PS, Oreopoulos DG. Clinical associations of glycemic control in diabetics on CAPD. Adv Perit Dial 1993; 9:291–294.
- Tzamaloukas AH, Murata GH, Zager PG, Eisenberg B, Avasthi PS. The relationship between glycemic control and morbidity and mortality for diabetics on dialysis. ASAIO J 1993; 39:880–885.
- Kalantar-Zadeh K, Kopple JD, Regidor DL, et al. A1C and survival in maintenance hemodialysis patients. Diabetes Care 2007; 30:1049–1055.
- Kovesdy C, Sharma K, Kalantar-Zadeh. Glycemic control in diabetic CKD patients: where do we stand? Am J Kidney Dis 2008; 52:766–777.
- Mak RH. Intravenous 1,25-dihydroxycholecalciferol corrects glucose intolerance in hemodialysis patients. Kidney Int 1992; 41:1049–1054.
- Hajjar SM, Fadda GZ, Thanakitcharu P, Smogorzewski M, Massry SG. Reduced activity of Na(+)-K+ ATPase of pancreatic islet cells in chronic renal failure: role of secondary hyperparathyroidism. J Am Soc Nephrol 1992; 2:1355–1359.
- Grodstein GP, Blumenkrantz MJ, Kopple JD, Moran JK, Coburn JW. Glucose absorption during continuous ambulatory peritoneal dialysis. Kidney Int 1981; 19:564–567.
- Joy MS, Cefali WT, Hogan SL, Nachman PH. Long-term glycemic control measurements in diabetic patients receiving hemodialysis. Am J Kidney Dis 2002; 39:297–307.
- Lamb E, Venton TR, Cattell WR, Dawnay A. Serum glycated albumin and fructosamine in renal dialysis patients. Nephron 1993; 64:82–88.
- Inaba M, Okuno S, Kumeda Y, et al; Osaka CKD Expert Research Group. Glycated albumin is a better glycemic indicator than glycated hemoglobin values in hemodialysis patients with diabetes: effect of anemia and erythropoietin injection. J Am Soc Nephrol 2007; 18:896–903.
- Constanti C, Simo JM, Joven J, Camps J. Serum fructosamine concentration in patients with nephrotic syndrome and with cirrhosis of the liver: the influence of hypoalbuminaemia and hypergammaglobulinaemia. Ann Clin Biochem 1992; 29:437–442.
- Ford HC, Lim WC, Crooke MJ. Hemoglobin A1 and serum fructosamine levels in hyperthyroidism. Clin Chim Acta 1987; 166:317–321.
- Mak RH. Impact of end-stage renal disease and dialysis on glycemic control. Semin Dial 2000; 13:4–8.
- Skillman TG, Feldman JM. The pharmacology of sulfonylureas. Am J Med 1981; 70:361–372.
- Krepinsky J, Ingram AJ, Clase CM. Prolonged sulfonylurea-induced hypoglycemia in diabetic patients with end-stage renal disease. Am J Kidney Dis 2000; 35:500–505.
- Snyder RW, Berns JS. Use of insulin and oral hypoglycemic medications in patients with diabetes mellitus and advanced kidney disease. Semin Dial 2004; 17:365–370.
- United Kingdom Prospective Diabetes Study (UKPDS) 13. Relative efficacy of randomly allocated diet, sulphonylureas, insulin, or metformin in patients with newly diagnosed non-insulin dependent diabetes followed for three years. BMJ 1995; 310:83–88.
- Inoue T, Shibahara N, Miyagawa K, et al. Pharmacokinetics of nateglinide and its metabolites in subjects with type 2 diabetes mellitus and renal failure. Clin Nephrol 2003; 60:90–95.
- Nagai T, Imamura M, Iizuka K, Mori M. Hypoglycemia due to nateglinide administration in diabetic patient with chronic renal failure. Diabetes Res Clin Pract 2003; 59:191–194.
- Thompson-Culkin K, Zussman B, Miller AK, Freed MI. Pharmacokinetics of rosiglitazone in patients with end-stage renal disease. J Int Med Res 2002; 30:391–399.
- Nesto RW, Bell D, Bonow RO, et al. Thiazolidinedione use, fluid retention, and congestive heart failure: a consensus statement from the American Heart Association and American Diabetes Association. Diabetes Care 2004; 27:256–263.
- Hermansen K, Kipnes M, Luo E, Fanurik D, Khatami H, Stein P; Sitagliptin Study 035 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor, sitagliptin, in patients with type 2 diabetes mellitus inadequately controlled on glimepiride alone or on glimepiride and metformin. Diabetes Obes Metab 2007; 9:733–745.
- Charbonnel B, Karasik A, Liu J, Wu M, Meininger G, et al; Sitagliptin Study 020 Group Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing metformin therapy in patients with type 2 diabetes inadequately controlled with metformin alone. Diabetes Care 2006; 29:2638–2643.
- Goldstein BJ, Feinglos MN, Lunceford JK, Johnson J, Williams-Herman DE; Sitagliptin 036 Study Group. Effect of initial combination therapy with sitagliptin, a dipeptidyl peptidase-4 inhibitor, and metformin on glycemic control in patients with type 2 diabetes. Diabetes Care 2007; 30:1979–1987.
- Nauck MA, Meininger G, Sheng D, Terranella L, Stein PP; Sitagliptin Study 024 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor, sitagliptin, compared with the sulfonylurea, glipizide, in patients with type 2 diabetes inadequately controlled on metformin alone: a randomized, double-blind, non-inferiority trial. Diabetes Obes Metab 2007; 9:194–205.
- Rosenstock J, Brazg R, Andryuk PJ, Lu K, Stein P; Sitagliptin Study 019 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing pioglitazone therapy in patients with type 2 diabetes: a 24-week, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Clin Ther 2006; 28:1556–1568.
- Bergman AJ, Cote J, Yi B, et al. Effect of renal insufficiency on the pharmacokinetics of sitagliptin, a dipeptidyl peptidase-4 inhibitor. Diabetes Care 2007; 30:1862–1864.
- Carone FA, Peterson DR. Hydrolysis and transport of small peptides by the proximal tubule. Am J Physiol 1980; 238:F151–F158.
- Rabkin R, Simon NM, Steiner S, Colwell JA. Effects of renal disease on renal uptake and excretion of insulin in man. N Engl J Med 1970; 282:182–187.
- Mak RH, DeFronzo RA. Glucose and insulin metabolism in uremia. Nephron 1992; 61:377–382.
- Biesenbach G, Raml A, Schmekal B, Eichbauer-Sturm G. Decreased insulin requirement in relation to GFR in nephropathic type 1 and insulin-treated type 2 diabetic patients. Diabet Med 2003; 20:642–645.
- Aisenpreis U, Pfützner A, Giehl M, Keller F, Jehle PM. Pharmacokinetics and pharmacodynamics of insulin Lispro compared with regular insulin in hemodialysis patients with diabetes mellitus. Nephrol Dial Transplant 1999; 14( suppl 4):5–6.
- Tunbridge FK, Newens A, Home PD, et al. A comparison of human ultralente- and lente-based twice-daily injection regimens. Diabet Med 1989; 6:496–501.
- Freeman SL, O’Brien PC, Rizza RA. Use of human ultralente as the basal insulin component in treatment of patients with IDDM. Diabetes Res Clin Pract 1991; 12:187–192.
- Charpentier G, Riveline JP, Varroud-Vial M. Management of drugs affecting blood glucose in diabetic patients with renal failure. Diabetes Metab 2000; 26( suppl 4):73–85.
- Aronoff GR, Berns JS, Brier ME, et al, eds. Drug Prescribing in Renal Failure: Dosing Guidelines for Adults, 4th ed. Philadelphia, PA: American College of Physicians, 1999.
KEY POINTS
- Blood glucose levels can fluctuate widely due to various and opposing effects of ESRD and dialysis.
- The hemoglobin A1c level can be falsely high in ESRD, but it is still a reasonable measure of glycemic control in this population.
- Most diabetes drugs are excreted at least in part by the kidney, so that patients in ESRD are at greater risk of hypoglycemia.
- Insulin is the cornerstone of treatment, since most oral diabetes drugs are contraindicated or not recommended in this population. Insulin doses should be lowered in those with low glomerular filtration rates.
Nephrolithiasis: Treatment, causes, and prevention
COMMON AND ON THE INCREASE
Nephrolithiasis is common, with a lifetime prevalence of 10% in men and 5% in women.1,2 Studies have shown that the prevalence is increasing in the United States. In the second National Health and Nutrition Examination Survey (1988–1994), the prevalence in adults ages 20 to 74 was greater than in the 1976–1980 survey (5.2% vs 3.2%).3 The increase was observed in whites but not in African Americans or Mexican Americans, was greater in men than in women, and was greater with age in each time period.
In addition, stones often recur, and each stone event can be associated with significant metabolic and intervention-related morbidity.
PRESENTATION: SEVERE COLIC
Most patients present with moderate to severe colic, caused by the stone entering the ureter. Stones in the proximal (upper) ureter cause pain in the flank or anterior upper abdomen. When the stone reaches the distal third of the ureter, pain is noted in the ipsilateral testicle or labia. A stone at the junction of the ureter and the bladder often causes dysuria, urgency, and frequency and may be mistaken for a lower urinary tract infection.
Less often, patients present with silent ureteral obstruction, unexplained persistent urinary infection, or painless hematuria. However, even in patients with symptoms, the absence of hematuria does not exclude urolithiasis. In a study of 397 patients presenting with acute symptomatic urolithiasis, 9% did not have hematuria.4
The differential diagnosis in a patient with symptoms suggesting renal colic includes:
- Musculoskeletal pain
- Herpes zoster
- Diverticulitis
- Duodenal ulcer
- Cholecystitis
- Pyelonephritis
- Renal infarct
- Renal hemorrhage
- Gynecologic disorders
- Ureteral obstruction from renal papillary necrosis with sloughed papillae, a blood clot, or a ureteral stricture.
HELICAL CT WITHOUT CONTRAST IS THE PREFERED IMAGING STUDY
The diagnosis can be confirmed by computed tomography (CT), renal ultrasonography, or intravenous pyelography.
Helical CT without contrast is the preferred imaging study in patients with suspected nephrolithiasis. It has several advantages over other imaging studies: it requires no radiocontrast material; it shows the distal ureters; it can detect radiolucent stones (ie, uric acid stones), radio-opaque stones, and stones as small as 1 to 2 mm; and it can detect hydronephrosis and intra-abdominal and renal disorders other than stones that could be causing the patient’s symptoms.
In a study in 100 consecutive patients presenting to an emergency department with flank pain, helical CT had a sensitivity of 98%, a specificity of 100%, a positive predictive value of 100%, and a negative predictive value of 97% for the diagnosis of ureteral stones.5 In a study of 1,000 consecutive patients with suspected stones, helical CT identified significant, additional, or alternative reasons for the patient’s symptoms in 10% of cases.6
Ultrasonography has the advantage of not using radiation, but it is less sensitive for detecting stones and can image only the kidney and the proximal ureter. A retrospective study in 123 patients found that, compared with helical CT as the gold standard, ultrasonography had a sensitivity of 24% and a specificity of 90%.7 Ultrasonography may also miss stones smaller than 3 mm in diameter.
Conventional radiography (kidney-ureter-bladder view) is inadequate for diagnosis as it may miss stones in the kidney or ureter (even small radio-opaque stones) and provides no information about possible obstruction.
Intravenous pyelography has few advantages in renal lithiasis, exposes the patient to the risk of radiocontrast infusion and contrast-mediated acute renal injury, and gives less information than noncontrast CT.
MEDICAL MANAGEMENT OF ACUTE STONE EVENTS
Most stones are smaller than 5 mm and readily pass without interventions such as lithotripsy, ureteroscopy, or percutaneous nephrolithotomy. (For more information on these interventions, see the review by Samplaski and colleagues in this issue of the Cleveland Clinic Journal of Medicine.8)
Even if the stone is as large as 1 cm, I would let the patient try to pass it spontaneously if it is in the distal ureter, and I would allow up to 4 weeks for this to happen.
For most patients, pain management is paramount. Randomized controlled trials suggest that parenteral nonsteroidal anti-inflammatory drugs (NSAIDs) are as effective as narcotics for controlling the pain of renal colic.9 Diclofenac (Voltaren) has been used in several studies.
To hasten stone passage, some recommend inducing high urine flow with oral intake of at least 2 to 3 L of fluids per 24 hours to ensure a urine output of at least 2 L per day.
Drugs may also help the stone to pass. A recent study in 210 patients with ureteral stones averaging 6 mm in diameter showed that tamsulosin (Flomax) increased the likelihood of spontaneous stone passage.10 A meta-analysis of 693 patients in nine randomized trials concluded that alpha-blockers and calcium channel blockers increased the likelihood of stone passage compared with no treatment.11 Borghi et al,12 in a randomized, double-blind study in 86 patients with unilateral ureteral stones, reported a higher rate of stone passage in patients treated with methylprednisolone (Medrol) 16 mg/day plus nifedipine (Procardia) 40 mg/day than in those given methylprednisolone alone.
PREVENTING RECURRENT STONES: PRINCIPLES AND SPECIFICS
Urinary stone disease recurs in 30% to 50% of patients within 5 years.1,13,14
In preventing recurrent stones, some principles apply to all patients and some are specific to the type of stone the patient had.
Stones form when the urine is supersaturated
Nephrolithiasis occurs when the concentration of stone-forming salts such as calcium oxalate, calcium phosphate, or uric acid is high. When the concentration is high enough to allow crystals to form or preformed crystals to grow, the urine is said to be supersaturated.
Several facors are the major determinants of whether the urine is supersaturated by different salts:
- Calcium oxalate—low urine volume and high concentrations of calcium and oxalate
- Calcium phosphate—a high urine calcium concentration and alkaline urine
- Uric acid—acidic urine
- Cystine—a high urinary cystine concentration and acidic urine.
Increasing daily fluid intake
Since the urinary concentration of stone-forming salts is strongly affected by the daily urine volume, it follows that increasing daily fluid intake is important in preventing recurrent stone disease.
In one study,15 199 patients with a first calcium stone were randomized to a program of high oral fluid intake or no intervention. Five years later, 12 (12%) of the 99 patients in the high-fluid intake group had had a second stone, compared with 27 (27%) in the untreated group (P = .008). Of interest, the baseline 24-hour urine volumes were significantly lower in patients with stones than in 101 normal controls (P = .001), suggesting that habitual low daily fluid intake is a risk factor for calcium stone disease.13
PREVENTING CALCIUM STONES
Most stones are composed of calcium oxalate or calcium phosphate. Calcium stone disease occurs most often in the 3rd to 5th decades of life.
Naturally occurring inhibitors of calcium crystal formation in the urine include citrate, nephrocalcin, uropontin, and magnesium. Of these, only citrate and magnesium levels are routinely measured; low levels of citrate are treated as a cause of calcium stone disease. It follows that the risk of calcium nephrolithiasis is the result of the interplay between the supersaturated state and the level of urinary inhibitors.16
Hypercalciuria and calcium oxalate stones
Calcium oxalate stones begin as crystals that form on the surface of the renal papillae over collections of suburothelial calcium phosphate particles called Randall plaque.17 The driving force for calcium oxalate overgrowth on plaque is calcium oxalate supersaturation, which is strongly linked to high urinary calcium excretion. The fraction of papillary surface covered by plaque in patients with idiopathic calcium oxalate stones correlates directly with the urine calcium level and inversely with urine volume and pH.18
Most patients with calcium oxalate stones have hypercalciuria (defined as 24-hour urinary calcium excretion > 300 mg in men, > 250 mg in women, or > 4 mg/kg in men or women).
Hypercalciuria can be idiopathic
Hypercalciuria can occur in primary hyperparathyroidism, sarcoidosis, vitamin D excess, corticosteroid treatment, renal tubular acidosis, hyperthyroidism, and malignant neoplasms. If none of these conditions is present, elevated urinary calcium excretion is considered idiopathic.
Some patients with idiopathic hypercalciuria have a strong family history of hypercalciuria and, likely, a genetic basis for the disease. This condition has been categorized by the presumed site of the primary abnormality:
Absorptive hypercalciuria. Most patients with idiopathic hypercalciuria absorb too much calcium from the intestine. In many of them, 1,25 dihydroxyvitamin D levels are slightly high and serum phosphorous levels are slightly low; the hypothesis is that they produce more 1,25 dihydroxyvitamin D or are more sensitive to it.19 However, Breslau et al20 showed that not all patients with idiopathic hypercalciuria have absorptive hypercalciuria mediated by 1,25 dihydroxyvitamin D, which suggests that the intestinal hyperabsorption of calcium has other mechanisms.
Resorptive hypercalciuria occurs if increased bone turnover leads to urinary loss of bone calcium.
Renal leak is due to a primary defect in renal tubular transport that causes loss of calcium into the urine and a secondary increase in intestinal calcium absorption or mobilization from bone.
This categorization is based on measuring fasting and 24-hour urine calcium, urinary calcium responses to a low-calcium diet, and responses to an oral calcium load.21 However, these studies are difficult to do and have been shown to have minimal clinical value.
To reduce calcium in the urine, limit sodium, give thiazides
Idiopathic hypercalciuria is worsened by a diet high in sodium22,23 and animal protein.24 Thiazide diuretics lower urinary calcium excretion and promote mineral retention.25 Therefore, treatment of idiopathic hypercalciuria consists of high fluid intake, dietary sodium restriction, and thiazide diuretics.
Calcium restriction is not advised
For several reasons, a calcium-restricted diet is not advised for patients with idiopathic hypercalciuria. 26 Dietary calcium restriction can put the patient into negative calcium balance. Further, it is thought that with less calcium to bind to dietary oxalate, more unbound oxalate can be absorbed in the colon and eventually excreted in the urine. This increase in urinary oxalate can be to the point of supersaturation, even though urinary calcium levels remain unchanged.25,27,28 This, in turn, increases the likelihood of stone formation.
Several studies showed that a higher intake of dietary calcium is actually associated with fewer calcium stone events in both men and women.25,27,28
Further, a study in 120 Italian patients with hypercalciuric calcium oxalate stones concluded that a diet that is normal in calcium, low in sodium, and low in animal protein was associated with a lower frequency of calcium stones than a low-calcium diet.29 Although both diets were associated with a reduction in urinary calcium concentrations, urinary oxalate excretion rose in those on a low-calcium diet and fell in those on a normal-calcium diet. The reduction in urinary oxalate excretion in patients on a normal-calcium diet was attributed to intestinal binding of dietary oxalate by dietary calcium, thus lessening the amount of free oxalate available for absorption. Although calcium oxalate excretion fell in both groups, it fell more in those on a normal calcium intake. Compared with those on a low-calcium diet, the patients on the normal-calcium, low-sodium, low-protein diet had a 50% lower risk of stones at 5 years.
Hyperparathyroidism
Primary hyperparathyroidism can cause hypercalciuria and nephrolithiasis. In one series,30 56 (4.9%) of 1,132 consecutive patients with nephrolithiasis had a confirmed diagnosis of hyperparathyroidism. Parathyroidectomy prevented subsequent stone disease in 48 patients.
However, only 17% to 24% of patients with primary hyperparathyroidism have urinary stones composed of calcium oxalate or calcium phosphate.31,32 In many studies, it was difficult to determine why a minority of these patients develop stones, but two studies shed some light on this.
Parks et al30 found that, compared with nephrolithiasis patients with idiopathic hypercalciuria, those with primary hyperparathyroidism have elevated serum calcium levels (but usually < 11.5 mg/dL), greater degrees of hypercalciuria (352 mg/day vs 252 mg/day, P < .001), and lower serum phosphate levels (2.45 vs 3.10 mg/dL, P < .001).
Odvina et al33 found, in a study of 131 patients with proven primary hyperparathyroidism, that 78 had nephrolithiasis and 53 did not. Those with stones excreted more calcium (343 mg/day) than those without stones (273 mg/day), had a higher urinary saturation of calcium oxalate and brushite, and excreted twice as much calcium following a 1-g oral calcium load.
These studies suggest that in patients with primary hyperparathyroidism, the risk of forming stones is related to the degree of hypercalciuria, and in particular to the increased intestinal absorption of dietary calcium.
Renal tubular acidosis
Features of distal renal tubular acidosis are systemic metabolic acidosis, alkaline urine, hypokalemia, hypercalciuria, hypocitraturia, and nephrolithiasis. The chronic metabolic acidosis results in loss of bone calcium, contributes to hypercalciuria, and is responsible for the hypocitraturia.34 Stone formation is the result of excessive urinary calcium excretion, the deficiency of the urinary crystal inhibitor citrate, and persistently alkaline urine.
Treatment with sodium bicarbonate or potassium citrate corrects the metabolic acidosis, reduces the loss of calcium from bone, corrects hypokalemia, and increases urinary citrate.
Too much uric acid in the urine
Elevated urinary uric acid excretion (> 800 mg/day in men, > 750 mg/day in women) is associated with formation of calcium oxalate stones35 and, in conjunction with low urine pH, with uric acid stones. An increase in dissolved uric acid salts induces heterogeneous calcium oxalate nucleation.36 In one randomized clinical trial,37 giving allopurinol (Zyloprim) lowered urinary uric acid excretion and was associated with a lower rate of calcium stone disease.
Too much oxalate in the urine
The 95th percentile for urinary oxalate excretion is 45 mg/day in women and 55 mg/day in men.38 Hyperoxaluria increases calcium oxalate supersaturation and contributes to calcium stone formation.
Normally, 90% of dietary oxalate binds to dietary calcium in the small intestine and passes into the stool as calcium oxalate; 10% of dietary oxalate remains free and is absorbed in the colon and subsequently excreted in the urine.
Hyperoxaluria may simply be a result of high dietary oxalate intake. However, increased enteric absorption of dietary oxalate can occur in those on a low-calcium diet (in which less calcium is available to bind to dietary oxalate, as described above) and may partially explain why a low-calcium diet has been associated with increased frequency of calcium stone disease.
Patients with enteric malabsorption of fat (eg, due to inflammatory bowel disease or intestinal bypass surgery for obesity) may also develop hyperoxaluria. This occurs because the excess enteric fat binds dietary calcium and allows free oxalate to be more readily absorbed in the colon.39
Rarely, hyperoxaluria is caused by one of several recessively inherited disorders of oxalate metabolism.40
The growing number of people with obesity has resulted in an upsurge in gastric bypass surgery. Although the current procedures do not pose the same metabolic risks as were noted in the 1970s when a different type of bypass was performed, the incidence of kidney stones does appear to be higher after these procedures. A recent analysis of 1,436 patients undergoing Roux-en-Y gastric bypass surgery found that 60 of them developed calcium stones afterward. Of these, 31 who underwent metabolic studies were found to have higher oxalate and lower citrate levels at 12 months of follow-up.41
Not enough citrate, a stone inhibitor
Hypocitraturia is defined as a daily urine citrate excretion less than 500 mg in women and 434 mg in men.42 As already mentioned, citrate plays an important role in inhibiting calcium crystal formation and preventing stone formation.
Urinary citrate excretion is mainly determined by tubular reabsorption, which is increased by acid loads and decreased by alkali loads.43 Low urine citrate levels are often seen in conditions that cause chronic metabolic acidosis, such as inflammatory bowel disease, intestinal malabsorption, and renal tubular acidosis—all of which are associated with increased occurrence of nephrolithiasis. However, in most nephrolithiasis patients with hypocitraturia, the cause is not apparent, and the mechanism of the hypocitraturia cannot be determined.44
In recent years, high-protein, low-carbohydrate diets have become popular for weight reduction, but they also have metabolic effects that increase the risk of stones.45 The metabolism of a diet high in animal protein produces more hydrogen ions that are buffered by bone, releasing calcium from bone and increasing urinary calcium excretion. These diets also cause intracellular acidosis, resulting in decreased urinary excretion of citrate. As a result of these effects, the stone-forming propensity of the urine is increased.
STRUVITE STONES MUST BE REMOVED
Struvite stones are the result of chronic upper urinary infection with urease-producing bacteria (Proteus sp, Haemophilus sp, Klebsiella sp, and Ureaplasma urealyticum).46,47 The hydrolysis of urea yields ammonium and hydroxyl ions and a persistently alkaline urine, and this scenario promotes the formation of stones composed of magnesium ammonium phosphate, ie, struvite.
Struvite stones, which are often branched (“staghorn” stones), occur more often in women and in patients who have chronic urinary obstruction or a neurologic disorder that impairs normal emptying of the bladder.
Treatment requires eradicating the infection with antibiotics and removing the bacteria-laden stones by one of several interventional techniques. Acetohydroxamic acid inhibits urease and has been used to treat struvite stone disease, but it has frequent and serious adverse effects.48
URIC ACID STONES FORM IN VERY ACIDIC URINE
Uric acid stones occur especially in patients with unusually low urine pH and hyperuricosuria. In some patients, this very low urine pH is the result of a defect in renal ammonia secretion, which results in less buffering of secreted hydrogen ions.49
The tendency to form uric acid stones is reported to be increasing in obese people with the metabolic syndrome. Some studies have shown that the defect in ammonia production by the kidney may be the result of insulin resistance.50
Urate stones are radiolucent but can be seen on ultrasonography and helical CT. On helical CT, they can be distinguished from calcium stones by their lower density.51
Since uric acid is much more soluble in an alkaline solution, both prevention and treatment should consist of alkalinization of urine to a pH of more than 6.0 with oral sodium bicarbonate or citrate solution and hydration. This treatment may actually dissolve uric acid stones. If hyperuricemia or hyperuricosuria is present, allopurinol can be prescribed.
CYSTINE STONES ALSO FORM IN ACIDIC URINE
Cystine stone disease occurs in people who have inherited an autosomally recessive gastrointestinal and renal tubular transport disorder of four amino acids, ie, cystine, ornithine, arginine, and lysine.52 Of these, cystine is the most insoluble in normally acidic urine and thus precipitates into stones. The onset is at a younger age than in calcium stone disease; the stones are radio-opaque.
Cystine solubility is about 243 mg/L in normal urine and rises with pH. Some patients can excrete as much as 1,000 mg per day.
Treatment53,54 consists of:
- Hydration, to achieve daily urine volumes of 3 to 3.5 L
- Alkalinization of the urine to a pH higher than 6.5 with potassium alkali (potassium citrate) or sodium bicarbonate
- Reduction of protein and sodium intake to reduce cystine excretion.
If these measures fail, D-penicillamine (Depen), tiopronin (Thiola), or captopril (Capoten)55–57 can be given to convert the cystine to a more soluble disulfide cysteine-drug complex. Captopril has only a modest effect at best and is usually given with another disulfide-complexing drug; it also has the disadvantage of producing hypotension. Adverse effects of D-penicillamine and tiopronin include abdominal pain, loss of taste, fever, proteinuria, and, in rare cases, nephrotic syndrome.
WORKUP AND MANAGEMENT OF NEPHROLITHIASIS

Anyone under age 20 with an initial stone deserves a more extensive evaluation, including screening for renal tubular acidosis, cystinuria, and hyperoxaluria. A more extensive workup is also warranted in patients with a history of chronic diarrhea, sarcoidosis, or a condition associated with renal tubular acidosis (eg, Sjögren syndrome), in patients with a family history of kidney stones, in patients with high-protein weight-loss diets, and in those undergoing gastric bypass surgery for obesity. In these high-risk patients, the evaluation should include 24-hour urine studies to measure calcium, oxalate, citrate, uric acid, creatinine, sodium, and volume.
Other diagnostic clues are often helpful in the decision to do a more comprehensive evaluation.
- Nephrocalcinosis on roentgenography suggests hyperparathyroidism, medullary sponge kidney, or renal tubular acidosis.
- Hypercalcemia that develops after treatment of hypercalciuria with a thiazide diuretic suggests latent hyperparathyroidism.
- A history of recurrent urinary tract infections or of anatomic abnormalities in the urinary tract should lead to an evaluation for struvite stone disease.
- Uric acid stones should be suspected in a patient with metabolic syndrome or a history of gout and are usually accompanied by a urine pH lower than 5.5.
- A urinalysis showing cystine crystals always indicates cystinuria, which should be confirmed by 24-hour urine cystine determination.
- A family history of renal stones is more common in idiopathic hypercalciuria, cystinuria, primary hyperoxaluria, and renal tubular acidosis.
- Johnson CM, Wilson DM, O’Fallon WM, Malek RS, Kurland LT. Renal stone epidemiology: a 25-year study in Rochester, Minnesota. Kidney Int 1979; 16:624–631.
- Hiatt RA, Dales LG, Friedman GD, Hunkeler EM. Frequency of urolithiasis in a prepaid medical care program. Am J Epidemiol 1982; 115:255–265.
- Stamatelou KK, Francis ME, Jones CA, Nyberg LM, Curhan GC. Time trends in reported prevalence of kidney stones in the United States: 1976–1994. Kidney Int 2003; 63:1817–1823.
- Li J, Kennedy D, Levine M, Kumar A, Mullen J. Absent hematuria and expensive computerized tomography: case characteristics of emergency urolithiasis. J Urol 2001; 165:782–784.
- Fielding JR, Steele G, Fox LA, Heller H, Loughlin KR. Spiral computerized tomography in the evaluation of acute flank pain: a replacement for excretory urography. J Urol 1997; 157:2071–2073.
- Katz DS, Scheer M, Lumerman JH, Mellinger BC, Stillman CA, Lane MJ. Alternative or additional diagnoses on unenhanced helical computed tomography for suspected renal colic: experience with 1,000 consecutive examinations. Urology 2000; 56:53–57.
- Fowler KAB, Locken JA, Duchesne JH, Williamson MR. US for detecting renal calculi with nonenhanced CT as a reference standard. Radiology 2002; 222:109–113.
- Samplaski MK, Irwin BH, Desai M. Less-invasive ways to remove stones from the kidneys and ureters. Cleve Clin J Med 2009; 76:592–598.
- Labrecque M, Dostaler LP, Rousselle R, Nguyen T, Poirier S. Efficacy of nonsteroidal anti-inflammatory drugs in the treatment of acute renal colic. A meta-analysis. Arch Intern Med 1994; 154:1381–1387.
- Dellabella M, Milanese G, Muzzonigro G. Randomized trial of the efficacy of tamsulosin, nifedipine, and phloroglucinol in medical expulsive therapy for distal ureteral calculi. J Urol 2005; 174:167–172.
- Hollingsworth JM, Togers MA, Kaufman SR, et al. Medical therapy to facilitate urinary stone passage: a meta-analysis. Lancet 2006; 368:1171–1179.
- Borghi L, Meschi T, Amato F, et al. Nifedipine and methylprednisolone in facilitating ureteral stone passage: a randomized, double blind, placebo-controlled study. J Urol 1994; 152:1095–1098.
- Williams RE. Long-term survey of 538 patients with upper urinary tract stone. Br J Urol 1963; 35:416–437.
- Coe FL, Keck J, Norton ER. The natural history of urolithiasis. JAMA 1977; 238:1519–1523.
- Borghi L, Meschi T, Amato F, Briganti A, Novarini A, Giannini A. Urinary volume, water, and recurrences in idiopathic calcium nephrolithiasis: a 5-year randomized prospective study. J Urol 1996; 155:839–843.
- Robertson WG, Peacock M, Marshall RW, Marshall DH, Nordin BE. Saturation-inhibition index as a measure of the risk of calcium oxalate stone formation in the urinary tract. N Engl J Med 1976; 294:249–252.
- Evan AP, Lingeman JE, Coe FL, et al. Randall’s plaque of patients with nephrolithiasis begins in basement membranes of thin loops of Henle. J Clin Invest 2003; 111:607–616.
- Kuo RL, Lingeman JE, Evan AP, et al. Urine calcium and volume predict coverage of renal papilla by Randall’s plaque. Kidney Int 2003; 64:2150–2154.
- Broadus AE, Horst RL, Lang R, Littledike ET, Rasmussen H. The importance of circulating 1,25-dihydroxyvitamin D in the pathogenesis of hypercalciuria and renal-stone formation in primary hyperparathyroidism. N Engl J Med 1980; 302:421–426.
- Breslau NA, Preminger GM, Adams BV, Otey J, Pak CY. Use of ketoconazole to probe the pathogenetic importance of 1,25-dihydroxyvitamin D in absorptive hypercalciuria. J Clin Endocrinol Metab 1992; 75:1446–1452.
- Levy FL, Adams-Huet B, Pak CY. Ambulatory evaluation of nephrolithiasis: an update of a 1980 protocol. Am J Med 1995; 98:50–59.
- Breslau NA, Sakhaee K, Pak CY. Impaired adaptation to saltinduced urinary calcium losses in postmenopausal osteoporosis. Trans Assoc Am Physicians 1985; 98:107–115.
- Burtis W, Gay L, Insogna K, Ellison A, Broadus A. Dietary hypercalciuria in patients with calcium oxalate kidney stones. Am J Clin Nutr 1994; 60:424–429.
- Hess B, Ackermann D, Essig M, Takkinen R, Jaeger P. Renal mass and serum calcitriol in male idiopathic calcium renal stone formers: role of protein intake. J Clin Endocrinol Metab 1995; 80:1916–1921.
- Coe FL, Parks JH, Bushinsky DA, Langman CB, Favus MJ. Chlorthalidone promotes mineral retention in patients with idiopathic hypercalciuria. Kidney Int 1988; 33:1140–1146.
- Pak CY, Britton F, Peterson R, et al. Ambulatory evaluation of nephrolithiasis. Classification, clinical presentation, and diagnostic criteria. Am J Med 1980; 69:19–30.
- Curhan GC, Willett WC, Rimm EB, Stampfer MJ. A prospective study of dietary calcium and other nutrients and the risk of symptomatic kidney stones. N Engl J Med 1993; 328:833–838.
- Curhan GC, Willett WC, Speizer FE, Spiegelman D, Stampfer MJ. Comparison of dietary calcium with supplemental calcium and other nutrients as factors affecting the risk for kidney stones in women. Ann Intern Med 1997; 126:497–504.
- Borghi L, Schianchi T, Meschi T, et al. Comparison of two diets for the prevention of recurrent stones in idiopathic hypercalciuria. N Engl J Med 2002; 346:77–84.
- Parks JH, Coe FL, Evan AP, Worcester EM. Clinical and laboratory characteristics of calcium stone-formers with and without primary hyperparathyroidism. Br J Urol 2008; 103:670–678.
- Silverberg SJ, Shane E, Jacobs TP, Siris E, Bilezikian JP. A 10-year prospective study of primary hyperparathyroidism with or without parathyroid surgery. N Engl J Med 1999; 341:1249–1255.
- Mollerup CL, Vestergaard P, Frokjaer VG, Mosekilde L, Christiansen P, Blichert-Toft M. Risk of renal stone events in primary hyperparathyroidism before and after parathyroid surgery: controlled retrospective follow up study. BMJ 2002; 325:807.
- Odvina CV, Sakhaee K, Heller HJ, et al. Biochemical characterization of primary hyperparathyroidism with and without kidney stones. Urol Res 2007; 35:123–128.
- Lemann J, Adams ND, Gray RW. Urinary calcium excretion in human beings. N Engl J Med 1979; 301:535–541.
- Coe FL. Treated and untreated recurrent calcium nephrolithiasis in patients with idiopathic hypercalciuria, hyperuricosuria, or no metabolic disorder. Ann Intern Med 1977; 87:404–410.
- Grover PK, Marshall VR, Ryall RL. Dissolved urate salts out calcium oxalate in undiluted human urine in vitro: implication for calcium oxalate stone genesis. Chem Biol 2003; 10:271–278.
- Ettinger B, Tang A, Citron JT, Livermore B, Williams T. Randomized trial of allopurinol in the prevention of calcium oxalate calculi. N Engl J Med 1986; 315:1386–1389.
- Coe FL, Parks JH. Pathogenesis and treatment of nephrolithiasis. In:The Kidney. Philadelphia: Lippincott Williams & Wilkins, 2000:1841–1867.
- Parks JH, Worcester EM, O’Connor RC, Coe FL. Urine stone risk factors in nephrolithiasis patients with and without bowel disease. Kidney Int 2003; 63:255–265.
- Danpure CJ, Rumsby G. Molecular aetiology of primary hyperoxaluria and its implications for clinical management. Expert Rev Mol Med 2004; 6:1–16.
- Sinha MK, Collazo-Clavell ML, Rule A, et al. Hyperoxaluric nephrolithiasis is a complication of Roux-en-Y gastric bypass surgery. Kidney Int 2007; 72:100–107.
- Parks JH, Coe FL. A urinary calcium-citrate index for the evaluation of nephrolithiasis. Kidney Int 1986; 30:85–90.
- Brennan S, Hering-Smith K, Hamm LL. Effect of pH on citrate reabsorption in the proximal convoluted tubule. Am J Physiol 1988; 255:F301–F306.
- Sakhaee K, Williams RH, Oh MS, et al. Alkali absorption and citrate excretion in calcium nephrolithiasis. J Bone Miner Res 1993; 8:789–794.
- Reddy ST, Wang CY, Sakhaee K, Brinkley L, Pak CY. Effect of low-carbohydrate, high-protein diets on acid-base balance, stone-forming propensity, and calcium metabolism. Am J Kidney Dis 2002; 40:265–274.
- Griffith DP. Struvite stones. Kidney Int 1978; 13:372–382.
- Jennis FS, Lavan JN, Neale FC, Posen S. Staghorn calculi of the kidney: clinical, bacteriological and biochemical features. Br J Urol 1970; 42:511–518.
- Griffith DP, Gibson JR, Clinton CW, Musher DM. Acetohydroxamic acid: clinical studies of a urease inhibitor in patients with staghorn renal calculi. J Urol 1978; 119:9–15.
- Kamel KS, Cheema-Dhadli S, Halperin ML. Studies on the pathophysiology of the low urine pH in patients with uric acid stones. Kidney Int 2002; 61:988–994.
- Abate N, Chandalia M, Cabo-Chan AV, Moe OW, Sakhaee K. The metabolic syndrome and uric acid nephrolithiasis: novel features of renal manifestation of insulin resistance. Kidney Int 2004; 65:386–392.
- Zarse CA, McAteer JA, Tann M, et al. Helical computed tomography accurately reports urinary stone composition using attenuation values: in vitro verification using high-resolution micro-computed tomography calibrated to fourier transform infrared microspectroscopy. Urology 2004; 63:828–833.
- Palacin M. The genetics of heteromeric amino acid transporters. Physiology (Bethesda) 2005; 20:112–124.
- Sakhaee K. Pathogenesis and medical management of cystinuria. Semin Nephrol 1996; 16:435–447.
- Shekarriz B, Stoller ML. Cystinuria and other noncalcareous calculi. Endocrinol Metab Clin North Am 2002; 31:951–977.
- Streem SB, Hall P. Effect of captopril on urinary cystine excretion in homozygous cystinuria. J Urol 1989; 142:1522–1524.
- Perazella MA, Buller GK. Successful treatment of cystinuria with captopril. Am J Kidney Dis 1993; 21:504–507.
- Sloand JA, Izzo JL. Captopril reduces urinary cystine excretion in cystinuria. Arch Intern Med 1987; 147:1409–1412.
COMMON AND ON THE INCREASE
Nephrolithiasis is common, with a lifetime prevalence of 10% in men and 5% in women.1,2 Studies have shown that the prevalence is increasing in the United States. In the second National Health and Nutrition Examination Survey (1988–1994), the prevalence in adults ages 20 to 74 was greater than in the 1976–1980 survey (5.2% vs 3.2%).3 The increase was observed in whites but not in African Americans or Mexican Americans, was greater in men than in women, and was greater with age in each time period.
In addition, stones often recur, and each stone event can be associated with significant metabolic and intervention-related morbidity.
PRESENTATION: SEVERE COLIC
Most patients present with moderate to severe colic, caused by the stone entering the ureter. Stones in the proximal (upper) ureter cause pain in the flank or anterior upper abdomen. When the stone reaches the distal third of the ureter, pain is noted in the ipsilateral testicle or labia. A stone at the junction of the ureter and the bladder often causes dysuria, urgency, and frequency and may be mistaken for a lower urinary tract infection.
Less often, patients present with silent ureteral obstruction, unexplained persistent urinary infection, or painless hematuria. However, even in patients with symptoms, the absence of hematuria does not exclude urolithiasis. In a study of 397 patients presenting with acute symptomatic urolithiasis, 9% did not have hematuria.4
The differential diagnosis in a patient with symptoms suggesting renal colic includes:
- Musculoskeletal pain
- Herpes zoster
- Diverticulitis
- Duodenal ulcer
- Cholecystitis
- Pyelonephritis
- Renal infarct
- Renal hemorrhage
- Gynecologic disorders
- Ureteral obstruction from renal papillary necrosis with sloughed papillae, a blood clot, or a ureteral stricture.
HELICAL CT WITHOUT CONTRAST IS THE PREFERED IMAGING STUDY
The diagnosis can be confirmed by computed tomography (CT), renal ultrasonography, or intravenous pyelography.
Helical CT without contrast is the preferred imaging study in patients with suspected nephrolithiasis. It has several advantages over other imaging studies: it requires no radiocontrast material; it shows the distal ureters; it can detect radiolucent stones (ie, uric acid stones), radio-opaque stones, and stones as small as 1 to 2 mm; and it can detect hydronephrosis and intra-abdominal and renal disorders other than stones that could be causing the patient’s symptoms.
In a study in 100 consecutive patients presenting to an emergency department with flank pain, helical CT had a sensitivity of 98%, a specificity of 100%, a positive predictive value of 100%, and a negative predictive value of 97% for the diagnosis of ureteral stones.5 In a study of 1,000 consecutive patients with suspected stones, helical CT identified significant, additional, or alternative reasons for the patient’s symptoms in 10% of cases.6
Ultrasonography has the advantage of not using radiation, but it is less sensitive for detecting stones and can image only the kidney and the proximal ureter. A retrospective study in 123 patients found that, compared with helical CT as the gold standard, ultrasonography had a sensitivity of 24% and a specificity of 90%.7 Ultrasonography may also miss stones smaller than 3 mm in diameter.
Conventional radiography (kidney-ureter-bladder view) is inadequate for diagnosis as it may miss stones in the kidney or ureter (even small radio-opaque stones) and provides no information about possible obstruction.
Intravenous pyelography has few advantages in renal lithiasis, exposes the patient to the risk of radiocontrast infusion and contrast-mediated acute renal injury, and gives less information than noncontrast CT.
MEDICAL MANAGEMENT OF ACUTE STONE EVENTS
Most stones are smaller than 5 mm and readily pass without interventions such as lithotripsy, ureteroscopy, or percutaneous nephrolithotomy. (For more information on these interventions, see the review by Samplaski and colleagues in this issue of the Cleveland Clinic Journal of Medicine.8)
Even if the stone is as large as 1 cm, I would let the patient try to pass it spontaneously if it is in the distal ureter, and I would allow up to 4 weeks for this to happen.
For most patients, pain management is paramount. Randomized controlled trials suggest that parenteral nonsteroidal anti-inflammatory drugs (NSAIDs) are as effective as narcotics for controlling the pain of renal colic.9 Diclofenac (Voltaren) has been used in several studies.
To hasten stone passage, some recommend inducing high urine flow with oral intake of at least 2 to 3 L of fluids per 24 hours to ensure a urine output of at least 2 L per day.
Drugs may also help the stone to pass. A recent study in 210 patients with ureteral stones averaging 6 mm in diameter showed that tamsulosin (Flomax) increased the likelihood of spontaneous stone passage.10 A meta-analysis of 693 patients in nine randomized trials concluded that alpha-blockers and calcium channel blockers increased the likelihood of stone passage compared with no treatment.11 Borghi et al,12 in a randomized, double-blind study in 86 patients with unilateral ureteral stones, reported a higher rate of stone passage in patients treated with methylprednisolone (Medrol) 16 mg/day plus nifedipine (Procardia) 40 mg/day than in those given methylprednisolone alone.
PREVENTING RECURRENT STONES: PRINCIPLES AND SPECIFICS
Urinary stone disease recurs in 30% to 50% of patients within 5 years.1,13,14
In preventing recurrent stones, some principles apply to all patients and some are specific to the type of stone the patient had.
Stones form when the urine is supersaturated
Nephrolithiasis occurs when the concentration of stone-forming salts such as calcium oxalate, calcium phosphate, or uric acid is high. When the concentration is high enough to allow crystals to form or preformed crystals to grow, the urine is said to be supersaturated.
Several facors are the major determinants of whether the urine is supersaturated by different salts:
- Calcium oxalate—low urine volume and high concentrations of calcium and oxalate
- Calcium phosphate—a high urine calcium concentration and alkaline urine
- Uric acid—acidic urine
- Cystine—a high urinary cystine concentration and acidic urine.
Increasing daily fluid intake
Since the urinary concentration of stone-forming salts is strongly affected by the daily urine volume, it follows that increasing daily fluid intake is important in preventing recurrent stone disease.
In one study,15 199 patients with a first calcium stone were randomized to a program of high oral fluid intake or no intervention. Five years later, 12 (12%) of the 99 patients in the high-fluid intake group had had a second stone, compared with 27 (27%) in the untreated group (P = .008). Of interest, the baseline 24-hour urine volumes were significantly lower in patients with stones than in 101 normal controls (P = .001), suggesting that habitual low daily fluid intake is a risk factor for calcium stone disease.13
PREVENTING CALCIUM STONES
Most stones are composed of calcium oxalate or calcium phosphate. Calcium stone disease occurs most often in the 3rd to 5th decades of life.
Naturally occurring inhibitors of calcium crystal formation in the urine include citrate, nephrocalcin, uropontin, and magnesium. Of these, only citrate and magnesium levels are routinely measured; low levels of citrate are treated as a cause of calcium stone disease. It follows that the risk of calcium nephrolithiasis is the result of the interplay between the supersaturated state and the level of urinary inhibitors.16
Hypercalciuria and calcium oxalate stones
Calcium oxalate stones begin as crystals that form on the surface of the renal papillae over collections of suburothelial calcium phosphate particles called Randall plaque.17 The driving force for calcium oxalate overgrowth on plaque is calcium oxalate supersaturation, which is strongly linked to high urinary calcium excretion. The fraction of papillary surface covered by plaque in patients with idiopathic calcium oxalate stones correlates directly with the urine calcium level and inversely with urine volume and pH.18
Most patients with calcium oxalate stones have hypercalciuria (defined as 24-hour urinary calcium excretion > 300 mg in men, > 250 mg in women, or > 4 mg/kg in men or women).
Hypercalciuria can be idiopathic
Hypercalciuria can occur in primary hyperparathyroidism, sarcoidosis, vitamin D excess, corticosteroid treatment, renal tubular acidosis, hyperthyroidism, and malignant neoplasms. If none of these conditions is present, elevated urinary calcium excretion is considered idiopathic.
Some patients with idiopathic hypercalciuria have a strong family history of hypercalciuria and, likely, a genetic basis for the disease. This condition has been categorized by the presumed site of the primary abnormality:
Absorptive hypercalciuria. Most patients with idiopathic hypercalciuria absorb too much calcium from the intestine. In many of them, 1,25 dihydroxyvitamin D levels are slightly high and serum phosphorous levels are slightly low; the hypothesis is that they produce more 1,25 dihydroxyvitamin D or are more sensitive to it.19 However, Breslau et al20 showed that not all patients with idiopathic hypercalciuria have absorptive hypercalciuria mediated by 1,25 dihydroxyvitamin D, which suggests that the intestinal hyperabsorption of calcium has other mechanisms.
Resorptive hypercalciuria occurs if increased bone turnover leads to urinary loss of bone calcium.
Renal leak is due to a primary defect in renal tubular transport that causes loss of calcium into the urine and a secondary increase in intestinal calcium absorption or mobilization from bone.
This categorization is based on measuring fasting and 24-hour urine calcium, urinary calcium responses to a low-calcium diet, and responses to an oral calcium load.21 However, these studies are difficult to do and have been shown to have minimal clinical value.
To reduce calcium in the urine, limit sodium, give thiazides
Idiopathic hypercalciuria is worsened by a diet high in sodium22,23 and animal protein.24 Thiazide diuretics lower urinary calcium excretion and promote mineral retention.25 Therefore, treatment of idiopathic hypercalciuria consists of high fluid intake, dietary sodium restriction, and thiazide diuretics.
Calcium restriction is not advised
For several reasons, a calcium-restricted diet is not advised for patients with idiopathic hypercalciuria. 26 Dietary calcium restriction can put the patient into negative calcium balance. Further, it is thought that with less calcium to bind to dietary oxalate, more unbound oxalate can be absorbed in the colon and eventually excreted in the urine. This increase in urinary oxalate can be to the point of supersaturation, even though urinary calcium levels remain unchanged.25,27,28 This, in turn, increases the likelihood of stone formation.
Several studies showed that a higher intake of dietary calcium is actually associated with fewer calcium stone events in both men and women.25,27,28
Further, a study in 120 Italian patients with hypercalciuric calcium oxalate stones concluded that a diet that is normal in calcium, low in sodium, and low in animal protein was associated with a lower frequency of calcium stones than a low-calcium diet.29 Although both diets were associated with a reduction in urinary calcium concentrations, urinary oxalate excretion rose in those on a low-calcium diet and fell in those on a normal-calcium diet. The reduction in urinary oxalate excretion in patients on a normal-calcium diet was attributed to intestinal binding of dietary oxalate by dietary calcium, thus lessening the amount of free oxalate available for absorption. Although calcium oxalate excretion fell in both groups, it fell more in those on a normal calcium intake. Compared with those on a low-calcium diet, the patients on the normal-calcium, low-sodium, low-protein diet had a 50% lower risk of stones at 5 years.
Hyperparathyroidism
Primary hyperparathyroidism can cause hypercalciuria and nephrolithiasis. In one series,30 56 (4.9%) of 1,132 consecutive patients with nephrolithiasis had a confirmed diagnosis of hyperparathyroidism. Parathyroidectomy prevented subsequent stone disease in 48 patients.
However, only 17% to 24% of patients with primary hyperparathyroidism have urinary stones composed of calcium oxalate or calcium phosphate.31,32 In many studies, it was difficult to determine why a minority of these patients develop stones, but two studies shed some light on this.
Parks et al30 found that, compared with nephrolithiasis patients with idiopathic hypercalciuria, those with primary hyperparathyroidism have elevated serum calcium levels (but usually < 11.5 mg/dL), greater degrees of hypercalciuria (352 mg/day vs 252 mg/day, P < .001), and lower serum phosphate levels (2.45 vs 3.10 mg/dL, P < .001).
Odvina et al33 found, in a study of 131 patients with proven primary hyperparathyroidism, that 78 had nephrolithiasis and 53 did not. Those with stones excreted more calcium (343 mg/day) than those without stones (273 mg/day), had a higher urinary saturation of calcium oxalate and brushite, and excreted twice as much calcium following a 1-g oral calcium load.
These studies suggest that in patients with primary hyperparathyroidism, the risk of forming stones is related to the degree of hypercalciuria, and in particular to the increased intestinal absorption of dietary calcium.
Renal tubular acidosis
Features of distal renal tubular acidosis are systemic metabolic acidosis, alkaline urine, hypokalemia, hypercalciuria, hypocitraturia, and nephrolithiasis. The chronic metabolic acidosis results in loss of bone calcium, contributes to hypercalciuria, and is responsible for the hypocitraturia.34 Stone formation is the result of excessive urinary calcium excretion, the deficiency of the urinary crystal inhibitor citrate, and persistently alkaline urine.
Treatment with sodium bicarbonate or potassium citrate corrects the metabolic acidosis, reduces the loss of calcium from bone, corrects hypokalemia, and increases urinary citrate.
Too much uric acid in the urine
Elevated urinary uric acid excretion (> 800 mg/day in men, > 750 mg/day in women) is associated with formation of calcium oxalate stones35 and, in conjunction with low urine pH, with uric acid stones. An increase in dissolved uric acid salts induces heterogeneous calcium oxalate nucleation.36 In one randomized clinical trial,37 giving allopurinol (Zyloprim) lowered urinary uric acid excretion and was associated with a lower rate of calcium stone disease.
Too much oxalate in the urine
The 95th percentile for urinary oxalate excretion is 45 mg/day in women and 55 mg/day in men.38 Hyperoxaluria increases calcium oxalate supersaturation and contributes to calcium stone formation.
Normally, 90% of dietary oxalate binds to dietary calcium in the small intestine and passes into the stool as calcium oxalate; 10% of dietary oxalate remains free and is absorbed in the colon and subsequently excreted in the urine.
Hyperoxaluria may simply be a result of high dietary oxalate intake. However, increased enteric absorption of dietary oxalate can occur in those on a low-calcium diet (in which less calcium is available to bind to dietary oxalate, as described above) and may partially explain why a low-calcium diet has been associated with increased frequency of calcium stone disease.
Patients with enteric malabsorption of fat (eg, due to inflammatory bowel disease or intestinal bypass surgery for obesity) may also develop hyperoxaluria. This occurs because the excess enteric fat binds dietary calcium and allows free oxalate to be more readily absorbed in the colon.39
Rarely, hyperoxaluria is caused by one of several recessively inherited disorders of oxalate metabolism.40
The growing number of people with obesity has resulted in an upsurge in gastric bypass surgery. Although the current procedures do not pose the same metabolic risks as were noted in the 1970s when a different type of bypass was performed, the incidence of kidney stones does appear to be higher after these procedures. A recent analysis of 1,436 patients undergoing Roux-en-Y gastric bypass surgery found that 60 of them developed calcium stones afterward. Of these, 31 who underwent metabolic studies were found to have higher oxalate and lower citrate levels at 12 months of follow-up.41
Not enough citrate, a stone inhibitor
Hypocitraturia is defined as a daily urine citrate excretion less than 500 mg in women and 434 mg in men.42 As already mentioned, citrate plays an important role in inhibiting calcium crystal formation and preventing stone formation.
Urinary citrate excretion is mainly determined by tubular reabsorption, which is increased by acid loads and decreased by alkali loads.43 Low urine citrate levels are often seen in conditions that cause chronic metabolic acidosis, such as inflammatory bowel disease, intestinal malabsorption, and renal tubular acidosis—all of which are associated with increased occurrence of nephrolithiasis. However, in most nephrolithiasis patients with hypocitraturia, the cause is not apparent, and the mechanism of the hypocitraturia cannot be determined.44
In recent years, high-protein, low-carbohydrate diets have become popular for weight reduction, but they also have metabolic effects that increase the risk of stones.45 The metabolism of a diet high in animal protein produces more hydrogen ions that are buffered by bone, releasing calcium from bone and increasing urinary calcium excretion. These diets also cause intracellular acidosis, resulting in decreased urinary excretion of citrate. As a result of these effects, the stone-forming propensity of the urine is increased.
STRUVITE STONES MUST BE REMOVED
Struvite stones are the result of chronic upper urinary infection with urease-producing bacteria (Proteus sp, Haemophilus sp, Klebsiella sp, and Ureaplasma urealyticum).46,47 The hydrolysis of urea yields ammonium and hydroxyl ions and a persistently alkaline urine, and this scenario promotes the formation of stones composed of magnesium ammonium phosphate, ie, struvite.
Struvite stones, which are often branched (“staghorn” stones), occur more often in women and in patients who have chronic urinary obstruction or a neurologic disorder that impairs normal emptying of the bladder.
Treatment requires eradicating the infection with antibiotics and removing the bacteria-laden stones by one of several interventional techniques. Acetohydroxamic acid inhibits urease and has been used to treat struvite stone disease, but it has frequent and serious adverse effects.48
URIC ACID STONES FORM IN VERY ACIDIC URINE
Uric acid stones occur especially in patients with unusually low urine pH and hyperuricosuria. In some patients, this very low urine pH is the result of a defect in renal ammonia secretion, which results in less buffering of secreted hydrogen ions.49
The tendency to form uric acid stones is reported to be increasing in obese people with the metabolic syndrome. Some studies have shown that the defect in ammonia production by the kidney may be the result of insulin resistance.50
Urate stones are radiolucent but can be seen on ultrasonography and helical CT. On helical CT, they can be distinguished from calcium stones by their lower density.51
Since uric acid is much more soluble in an alkaline solution, both prevention and treatment should consist of alkalinization of urine to a pH of more than 6.0 with oral sodium bicarbonate or citrate solution and hydration. This treatment may actually dissolve uric acid stones. If hyperuricemia or hyperuricosuria is present, allopurinol can be prescribed.
CYSTINE STONES ALSO FORM IN ACIDIC URINE
Cystine stone disease occurs in people who have inherited an autosomally recessive gastrointestinal and renal tubular transport disorder of four amino acids, ie, cystine, ornithine, arginine, and lysine.52 Of these, cystine is the most insoluble in normally acidic urine and thus precipitates into stones. The onset is at a younger age than in calcium stone disease; the stones are radio-opaque.
Cystine solubility is about 243 mg/L in normal urine and rises with pH. Some patients can excrete as much as 1,000 mg per day.
Treatment53,54 consists of:
- Hydration, to achieve daily urine volumes of 3 to 3.5 L
- Alkalinization of the urine to a pH higher than 6.5 with potassium alkali (potassium citrate) or sodium bicarbonate
- Reduction of protein and sodium intake to reduce cystine excretion.
If these measures fail, D-penicillamine (Depen), tiopronin (Thiola), or captopril (Capoten)55–57 can be given to convert the cystine to a more soluble disulfide cysteine-drug complex. Captopril has only a modest effect at best and is usually given with another disulfide-complexing drug; it also has the disadvantage of producing hypotension. Adverse effects of D-penicillamine and tiopronin include abdominal pain, loss of taste, fever, proteinuria, and, in rare cases, nephrotic syndrome.
WORKUP AND MANAGEMENT OF NEPHROLITHIASIS
Anyone under age 20 with an initial stone deserves a more extensive evaluation, including screening for renal tubular acidosis, cystinuria, and hyperoxaluria. A more extensive workup is also warranted in patients with a history of chronic diarrhea, sarcoidosis, or a condition associated with renal tubular acidosis (eg, Sjögren syndrome), in patients with a family history of kidney stones, in patients with high-protein weight-loss diets, and in those undergoing gastric bypass surgery for obesity. In these high-risk patients, the evaluation should include 24-hour urine studies to measure calcium, oxalate, citrate, uric acid, creatinine, sodium, and volume.
Other diagnostic clues are often helpful in the decision to do a more comprehensive evaluation.
- Nephrocalcinosis on roentgenography suggests hyperparathyroidism, medullary sponge kidney, or renal tubular acidosis.
- Hypercalcemia that develops after treatment of hypercalciuria with a thiazide diuretic suggests latent hyperparathyroidism.
- A history of recurrent urinary tract infections or of anatomic abnormalities in the urinary tract should lead to an evaluation for struvite stone disease.
- Uric acid stones should be suspected in a patient with metabolic syndrome or a history of gout and are usually accompanied by a urine pH lower than 5.5.
- A urinalysis showing cystine crystals always indicates cystinuria, which should be confirmed by 24-hour urine cystine determination.
- A family history of renal stones is more common in idiopathic hypercalciuria, cystinuria, primary hyperoxaluria, and renal tubular acidosis.
COMMON AND ON THE INCREASE
Nephrolithiasis is common, with a lifetime prevalence of 10% in men and 5% in women.1,2 Studies have shown that the prevalence is increasing in the United States. In the second National Health and Nutrition Examination Survey (1988–1994), the prevalence in adults ages 20 to 74 was greater than in the 1976–1980 survey (5.2% vs 3.2%).3 The increase was observed in whites but not in African Americans or Mexican Americans, was greater in men than in women, and was greater with age in each time period.
In addition, stones often recur, and each stone event can be associated with significant metabolic and intervention-related morbidity.
PRESENTATION: SEVERE COLIC
Most patients present with moderate to severe colic, caused by the stone entering the ureter. Stones in the proximal (upper) ureter cause pain in the flank or anterior upper abdomen. When the stone reaches the distal third of the ureter, pain is noted in the ipsilateral testicle or labia. A stone at the junction of the ureter and the bladder often causes dysuria, urgency, and frequency and may be mistaken for a lower urinary tract infection.
Less often, patients present with silent ureteral obstruction, unexplained persistent urinary infection, or painless hematuria. However, even in patients with symptoms, the absence of hematuria does not exclude urolithiasis. In a study of 397 patients presenting with acute symptomatic urolithiasis, 9% did not have hematuria.4
The differential diagnosis in a patient with symptoms suggesting renal colic includes:
- Musculoskeletal pain
- Herpes zoster
- Diverticulitis
- Duodenal ulcer
- Cholecystitis
- Pyelonephritis
- Renal infarct
- Renal hemorrhage
- Gynecologic disorders
- Ureteral obstruction from renal papillary necrosis with sloughed papillae, a blood clot, or a ureteral stricture.
HELICAL CT WITHOUT CONTRAST IS THE PREFERED IMAGING STUDY
The diagnosis can be confirmed by computed tomography (CT), renal ultrasonography, or intravenous pyelography.
Helical CT without contrast is the preferred imaging study in patients with suspected nephrolithiasis. It has several advantages over other imaging studies: it requires no radiocontrast material; it shows the distal ureters; it can detect radiolucent stones (ie, uric acid stones), radio-opaque stones, and stones as small as 1 to 2 mm; and it can detect hydronephrosis and intra-abdominal and renal disorders other than stones that could be causing the patient’s symptoms.
In a study in 100 consecutive patients presenting to an emergency department with flank pain, helical CT had a sensitivity of 98%, a specificity of 100%, a positive predictive value of 100%, and a negative predictive value of 97% for the diagnosis of ureteral stones.5 In a study of 1,000 consecutive patients with suspected stones, helical CT identified significant, additional, or alternative reasons for the patient’s symptoms in 10% of cases.6
Ultrasonography has the advantage of not using radiation, but it is less sensitive for detecting stones and can image only the kidney and the proximal ureter. A retrospective study in 123 patients found that, compared with helical CT as the gold standard, ultrasonography had a sensitivity of 24% and a specificity of 90%.7 Ultrasonography may also miss stones smaller than 3 mm in diameter.
Conventional radiography (kidney-ureter-bladder view) is inadequate for diagnosis as it may miss stones in the kidney or ureter (even small radio-opaque stones) and provides no information about possible obstruction.
Intravenous pyelography has few advantages in renal lithiasis, exposes the patient to the risk of radiocontrast infusion and contrast-mediated acute renal injury, and gives less information than noncontrast CT.
MEDICAL MANAGEMENT OF ACUTE STONE EVENTS
Most stones are smaller than 5 mm and readily pass without interventions such as lithotripsy, ureteroscopy, or percutaneous nephrolithotomy. (For more information on these interventions, see the review by Samplaski and colleagues in this issue of the Cleveland Clinic Journal of Medicine.8)
Even if the stone is as large as 1 cm, I would let the patient try to pass it spontaneously if it is in the distal ureter, and I would allow up to 4 weeks for this to happen.
For most patients, pain management is paramount. Randomized controlled trials suggest that parenteral nonsteroidal anti-inflammatory drugs (NSAIDs) are as effective as narcotics for controlling the pain of renal colic.9 Diclofenac (Voltaren) has been used in several studies.
To hasten stone passage, some recommend inducing high urine flow with oral intake of at least 2 to 3 L of fluids per 24 hours to ensure a urine output of at least 2 L per day.
Drugs may also help the stone to pass. A recent study in 210 patients with ureteral stones averaging 6 mm in diameter showed that tamsulosin (Flomax) increased the likelihood of spontaneous stone passage.10 A meta-analysis of 693 patients in nine randomized trials concluded that alpha-blockers and calcium channel blockers increased the likelihood of stone passage compared with no treatment.11 Borghi et al,12 in a randomized, double-blind study in 86 patients with unilateral ureteral stones, reported a higher rate of stone passage in patients treated with methylprednisolone (Medrol) 16 mg/day plus nifedipine (Procardia) 40 mg/day than in those given methylprednisolone alone.
PREVENTING RECURRENT STONES: PRINCIPLES AND SPECIFICS
Urinary stone disease recurs in 30% to 50% of patients within 5 years.1,13,14
In preventing recurrent stones, some principles apply to all patients and some are specific to the type of stone the patient had.
Stones form when the urine is supersaturated
Nephrolithiasis occurs when the concentration of stone-forming salts such as calcium oxalate, calcium phosphate, or uric acid is high. When the concentration is high enough to allow crystals to form or preformed crystals to grow, the urine is said to be supersaturated.
Several facors are the major determinants of whether the urine is supersaturated by different salts:
- Calcium oxalate—low urine volume and high concentrations of calcium and oxalate
- Calcium phosphate—a high urine calcium concentration and alkaline urine
- Uric acid—acidic urine
- Cystine—a high urinary cystine concentration and acidic urine.
Increasing daily fluid intake
Since the urinary concentration of stone-forming salts is strongly affected by the daily urine volume, it follows that increasing daily fluid intake is important in preventing recurrent stone disease.
In one study,15 199 patients with a first calcium stone were randomized to a program of high oral fluid intake or no intervention. Five years later, 12 (12%) of the 99 patients in the high-fluid intake group had had a second stone, compared with 27 (27%) in the untreated group (P = .008). Of interest, the baseline 24-hour urine volumes were significantly lower in patients with stones than in 101 normal controls (P = .001), suggesting that habitual low daily fluid intake is a risk factor for calcium stone disease.13
PREVENTING CALCIUM STONES
Most stones are composed of calcium oxalate or calcium phosphate. Calcium stone disease occurs most often in the 3rd to 5th decades of life.
Naturally occurring inhibitors of calcium crystal formation in the urine include citrate, nephrocalcin, uropontin, and magnesium. Of these, only citrate and magnesium levels are routinely measured; low levels of citrate are treated as a cause of calcium stone disease. It follows that the risk of calcium nephrolithiasis is the result of the interplay between the supersaturated state and the level of urinary inhibitors.16
Hypercalciuria and calcium oxalate stones
Calcium oxalate stones begin as crystals that form on the surface of the renal papillae over collections of suburothelial calcium phosphate particles called Randall plaque.17 The driving force for calcium oxalate overgrowth on plaque is calcium oxalate supersaturation, which is strongly linked to high urinary calcium excretion. The fraction of papillary surface covered by plaque in patients with idiopathic calcium oxalate stones correlates directly with the urine calcium level and inversely with urine volume and pH.18
Most patients with calcium oxalate stones have hypercalciuria (defined as 24-hour urinary calcium excretion > 300 mg in men, > 250 mg in women, or > 4 mg/kg in men or women).
Hypercalciuria can be idiopathic
Hypercalciuria can occur in primary hyperparathyroidism, sarcoidosis, vitamin D excess, corticosteroid treatment, renal tubular acidosis, hyperthyroidism, and malignant neoplasms. If none of these conditions is present, elevated urinary calcium excretion is considered idiopathic.
Some patients with idiopathic hypercalciuria have a strong family history of hypercalciuria and, likely, a genetic basis for the disease. This condition has been categorized by the presumed site of the primary abnormality:
Absorptive hypercalciuria. Most patients with idiopathic hypercalciuria absorb too much calcium from the intestine. In many of them, 1,25 dihydroxyvitamin D levels are slightly high and serum phosphorous levels are slightly low; the hypothesis is that they produce more 1,25 dihydroxyvitamin D or are more sensitive to it.19 However, Breslau et al20 showed that not all patients with idiopathic hypercalciuria have absorptive hypercalciuria mediated by 1,25 dihydroxyvitamin D, which suggests that the intestinal hyperabsorption of calcium has other mechanisms.
Resorptive hypercalciuria occurs if increased bone turnover leads to urinary loss of bone calcium.
Renal leak is due to a primary defect in renal tubular transport that causes loss of calcium into the urine and a secondary increase in intestinal calcium absorption or mobilization from bone.
This categorization is based on measuring fasting and 24-hour urine calcium, urinary calcium responses to a low-calcium diet, and responses to an oral calcium load.21 However, these studies are difficult to do and have been shown to have minimal clinical value.
To reduce calcium in the urine, limit sodium, give thiazides
Idiopathic hypercalciuria is worsened by a diet high in sodium22,23 and animal protein.24 Thiazide diuretics lower urinary calcium excretion and promote mineral retention.25 Therefore, treatment of idiopathic hypercalciuria consists of high fluid intake, dietary sodium restriction, and thiazide diuretics.
Calcium restriction is not advised
For several reasons, a calcium-restricted diet is not advised for patients with idiopathic hypercalciuria. 26 Dietary calcium restriction can put the patient into negative calcium balance. Further, it is thought that with less calcium to bind to dietary oxalate, more unbound oxalate can be absorbed in the colon and eventually excreted in the urine. This increase in urinary oxalate can be to the point of supersaturation, even though urinary calcium levels remain unchanged.25,27,28 This, in turn, increases the likelihood of stone formation.
Several studies showed that a higher intake of dietary calcium is actually associated with fewer calcium stone events in both men and women.25,27,28
Further, a study in 120 Italian patients with hypercalciuric calcium oxalate stones concluded that a diet that is normal in calcium, low in sodium, and low in animal protein was associated with a lower frequency of calcium stones than a low-calcium diet.29 Although both diets were associated with a reduction in urinary calcium concentrations, urinary oxalate excretion rose in those on a low-calcium diet and fell in those on a normal-calcium diet. The reduction in urinary oxalate excretion in patients on a normal-calcium diet was attributed to intestinal binding of dietary oxalate by dietary calcium, thus lessening the amount of free oxalate available for absorption. Although calcium oxalate excretion fell in both groups, it fell more in those on a normal calcium intake. Compared with those on a low-calcium diet, the patients on the normal-calcium, low-sodium, low-protein diet had a 50% lower risk of stones at 5 years.
Hyperparathyroidism
Primary hyperparathyroidism can cause hypercalciuria and nephrolithiasis. In one series,30 56 (4.9%) of 1,132 consecutive patients with nephrolithiasis had a confirmed diagnosis of hyperparathyroidism. Parathyroidectomy prevented subsequent stone disease in 48 patients.
However, only 17% to 24% of patients with primary hyperparathyroidism have urinary stones composed of calcium oxalate or calcium phosphate.31,32 In many studies, it was difficult to determine why a minority of these patients develop stones, but two studies shed some light on this.
Parks et al30 found that, compared with nephrolithiasis patients with idiopathic hypercalciuria, those with primary hyperparathyroidism have elevated serum calcium levels (but usually < 11.5 mg/dL), greater degrees of hypercalciuria (352 mg/day vs 252 mg/day, P < .001), and lower serum phosphate levels (2.45 vs 3.10 mg/dL, P < .001).
Odvina et al33 found, in a study of 131 patients with proven primary hyperparathyroidism, that 78 had nephrolithiasis and 53 did not. Those with stones excreted more calcium (343 mg/day) than those without stones (273 mg/day), had a higher urinary saturation of calcium oxalate and brushite, and excreted twice as much calcium following a 1-g oral calcium load.
These studies suggest that in patients with primary hyperparathyroidism, the risk of forming stones is related to the degree of hypercalciuria, and in particular to the increased intestinal absorption of dietary calcium.
Renal tubular acidosis
Features of distal renal tubular acidosis are systemic metabolic acidosis, alkaline urine, hypokalemia, hypercalciuria, hypocitraturia, and nephrolithiasis. The chronic metabolic acidosis results in loss of bone calcium, contributes to hypercalciuria, and is responsible for the hypocitraturia.34 Stone formation is the result of excessive urinary calcium excretion, the deficiency of the urinary crystal inhibitor citrate, and persistently alkaline urine.
Treatment with sodium bicarbonate or potassium citrate corrects the metabolic acidosis, reduces the loss of calcium from bone, corrects hypokalemia, and increases urinary citrate.
Too much uric acid in the urine
Elevated urinary uric acid excretion (> 800 mg/day in men, > 750 mg/day in women) is associated with formation of calcium oxalate stones35 and, in conjunction with low urine pH, with uric acid stones. An increase in dissolved uric acid salts induces heterogeneous calcium oxalate nucleation.36 In one randomized clinical trial,37 giving allopurinol (Zyloprim) lowered urinary uric acid excretion and was associated with a lower rate of calcium stone disease.
Too much oxalate in the urine
The 95th percentile for urinary oxalate excretion is 45 mg/day in women and 55 mg/day in men.38 Hyperoxaluria increases calcium oxalate supersaturation and contributes to calcium stone formation.
Normally, 90% of dietary oxalate binds to dietary calcium in the small intestine and passes into the stool as calcium oxalate; 10% of dietary oxalate remains free and is absorbed in the colon and subsequently excreted in the urine.
Hyperoxaluria may simply be a result of high dietary oxalate intake. However, increased enteric absorption of dietary oxalate can occur in those on a low-calcium diet (in which less calcium is available to bind to dietary oxalate, as described above) and may partially explain why a low-calcium diet has been associated with increased frequency of calcium stone disease.
Patients with enteric malabsorption of fat (eg, due to inflammatory bowel disease or intestinal bypass surgery for obesity) may also develop hyperoxaluria. This occurs because the excess enteric fat binds dietary calcium and allows free oxalate to be more readily absorbed in the colon.39
Rarely, hyperoxaluria is caused by one of several recessively inherited disorders of oxalate metabolism.40
The growing number of people with obesity has resulted in an upsurge in gastric bypass surgery. Although the current procedures do not pose the same metabolic risks as were noted in the 1970s when a different type of bypass was performed, the incidence of kidney stones does appear to be higher after these procedures. A recent analysis of 1,436 patients undergoing Roux-en-Y gastric bypass surgery found that 60 of them developed calcium stones afterward. Of these, 31 who underwent metabolic studies were found to have higher oxalate and lower citrate levels at 12 months of follow-up.41
Not enough citrate, a stone inhibitor
Hypocitraturia is defined as a daily urine citrate excretion less than 500 mg in women and 434 mg in men.42 As already mentioned, citrate plays an important role in inhibiting calcium crystal formation and preventing stone formation.
Urinary citrate excretion is mainly determined by tubular reabsorption, which is increased by acid loads and decreased by alkali loads.43 Low urine citrate levels are often seen in conditions that cause chronic metabolic acidosis, such as inflammatory bowel disease, intestinal malabsorption, and renal tubular acidosis—all of which are associated with increased occurrence of nephrolithiasis. However, in most nephrolithiasis patients with hypocitraturia, the cause is not apparent, and the mechanism of the hypocitraturia cannot be determined.44
In recent years, high-protein, low-carbohydrate diets have become popular for weight reduction, but they also have metabolic effects that increase the risk of stones.45 The metabolism of a diet high in animal protein produces more hydrogen ions that are buffered by bone, releasing calcium from bone and increasing urinary calcium excretion. These diets also cause intracellular acidosis, resulting in decreased urinary excretion of citrate. As a result of these effects, the stone-forming propensity of the urine is increased.
STRUVITE STONES MUST BE REMOVED
Struvite stones are the result of chronic upper urinary infection with urease-producing bacteria (Proteus sp, Haemophilus sp, Klebsiella sp, and Ureaplasma urealyticum).46,47 The hydrolysis of urea yields ammonium and hydroxyl ions and a persistently alkaline urine, and this scenario promotes the formation of stones composed of magnesium ammonium phosphate, ie, struvite.
Struvite stones, which are often branched (“staghorn” stones), occur more often in women and in patients who have chronic urinary obstruction or a neurologic disorder that impairs normal emptying of the bladder.
Treatment requires eradicating the infection with antibiotics and removing the bacteria-laden stones by one of several interventional techniques. Acetohydroxamic acid inhibits urease and has been used to treat struvite stone disease, but it has frequent and serious adverse effects.48
URIC ACID STONES FORM IN VERY ACIDIC URINE
Uric acid stones occur especially in patients with unusually low urine pH and hyperuricosuria. In some patients, this very low urine pH is the result of a defect in renal ammonia secretion, which results in less buffering of secreted hydrogen ions.49
The tendency to form uric acid stones is reported to be increasing in obese people with the metabolic syndrome. Some studies have shown that the defect in ammonia production by the kidney may be the result of insulin resistance.50
Urate stones are radiolucent but can be seen on ultrasonography and helical CT. On helical CT, they can be distinguished from calcium stones by their lower density.51
Since uric acid is much more soluble in an alkaline solution, both prevention and treatment should consist of alkalinization of urine to a pH of more than 6.0 with oral sodium bicarbonate or citrate solution and hydration. This treatment may actually dissolve uric acid stones. If hyperuricemia or hyperuricosuria is present, allopurinol can be prescribed.
CYSTINE STONES ALSO FORM IN ACIDIC URINE
Cystine stone disease occurs in people who have inherited an autosomally recessive gastrointestinal and renal tubular transport disorder of four amino acids, ie, cystine, ornithine, arginine, and lysine.52 Of these, cystine is the most insoluble in normally acidic urine and thus precipitates into stones. The onset is at a younger age than in calcium stone disease; the stones are radio-opaque.
Cystine solubility is about 243 mg/L in normal urine and rises with pH. Some patients can excrete as much as 1,000 mg per day.
Treatment53,54 consists of:
- Hydration, to achieve daily urine volumes of 3 to 3.5 L
- Alkalinization of the urine to a pH higher than 6.5 with potassium alkali (potassium citrate) or sodium bicarbonate
- Reduction of protein and sodium intake to reduce cystine excretion.
If these measures fail, D-penicillamine (Depen), tiopronin (Thiola), or captopril (Capoten)55–57 can be given to convert the cystine to a more soluble disulfide cysteine-drug complex. Captopril has only a modest effect at best and is usually given with another disulfide-complexing drug; it also has the disadvantage of producing hypotension. Adverse effects of D-penicillamine and tiopronin include abdominal pain, loss of taste, fever, proteinuria, and, in rare cases, nephrotic syndrome.
WORKUP AND MANAGEMENT OF NEPHROLITHIASIS
Anyone under age 20 with an initial stone deserves a more extensive evaluation, including screening for renal tubular acidosis, cystinuria, and hyperoxaluria. A more extensive workup is also warranted in patients with a history of chronic diarrhea, sarcoidosis, or a condition associated with renal tubular acidosis (eg, Sjögren syndrome), in patients with a family history of kidney stones, in patients with high-protein weight-loss diets, and in those undergoing gastric bypass surgery for obesity. In these high-risk patients, the evaluation should include 24-hour urine studies to measure calcium, oxalate, citrate, uric acid, creatinine, sodium, and volume.
Other diagnostic clues are often helpful in the decision to do a more comprehensive evaluation.
- Nephrocalcinosis on roentgenography suggests hyperparathyroidism, medullary sponge kidney, or renal tubular acidosis.
- Hypercalcemia that develops after treatment of hypercalciuria with a thiazide diuretic suggests latent hyperparathyroidism.
- A history of recurrent urinary tract infections or of anatomic abnormalities in the urinary tract should lead to an evaluation for struvite stone disease.
- Uric acid stones should be suspected in a patient with metabolic syndrome or a history of gout and are usually accompanied by a urine pH lower than 5.5.
- A urinalysis showing cystine crystals always indicates cystinuria, which should be confirmed by 24-hour urine cystine determination.
- A family history of renal stones is more common in idiopathic hypercalciuria, cystinuria, primary hyperoxaluria, and renal tubular acidosis.
- Johnson CM, Wilson DM, O’Fallon WM, Malek RS, Kurland LT. Renal stone epidemiology: a 25-year study in Rochester, Minnesota. Kidney Int 1979; 16:624–631.
- Hiatt RA, Dales LG, Friedman GD, Hunkeler EM. Frequency of urolithiasis in a prepaid medical care program. Am J Epidemiol 1982; 115:255–265.
- Stamatelou KK, Francis ME, Jones CA, Nyberg LM, Curhan GC. Time trends in reported prevalence of kidney stones in the United States: 1976–1994. Kidney Int 2003; 63:1817–1823.
- Li J, Kennedy D, Levine M, Kumar A, Mullen J. Absent hematuria and expensive computerized tomography: case characteristics of emergency urolithiasis. J Urol 2001; 165:782–784.
- Fielding JR, Steele G, Fox LA, Heller H, Loughlin KR. Spiral computerized tomography in the evaluation of acute flank pain: a replacement for excretory urography. J Urol 1997; 157:2071–2073.
- Katz DS, Scheer M, Lumerman JH, Mellinger BC, Stillman CA, Lane MJ. Alternative or additional diagnoses on unenhanced helical computed tomography for suspected renal colic: experience with 1,000 consecutive examinations. Urology 2000; 56:53–57.
- Fowler KAB, Locken JA, Duchesne JH, Williamson MR. US for detecting renal calculi with nonenhanced CT as a reference standard. Radiology 2002; 222:109–113.
- Samplaski MK, Irwin BH, Desai M. Less-invasive ways to remove stones from the kidneys and ureters. Cleve Clin J Med 2009; 76:592–598.
- Labrecque M, Dostaler LP, Rousselle R, Nguyen T, Poirier S. Efficacy of nonsteroidal anti-inflammatory drugs in the treatment of acute renal colic. A meta-analysis. Arch Intern Med 1994; 154:1381–1387.
- Dellabella M, Milanese G, Muzzonigro G. Randomized trial of the efficacy of tamsulosin, nifedipine, and phloroglucinol in medical expulsive therapy for distal ureteral calculi. J Urol 2005; 174:167–172.
- Hollingsworth JM, Togers MA, Kaufman SR, et al. Medical therapy to facilitate urinary stone passage: a meta-analysis. Lancet 2006; 368:1171–1179.
- Borghi L, Meschi T, Amato F, et al. Nifedipine and methylprednisolone in facilitating ureteral stone passage: a randomized, double blind, placebo-controlled study. J Urol 1994; 152:1095–1098.
- Williams RE. Long-term survey of 538 patients with upper urinary tract stone. Br J Urol 1963; 35:416–437.
- Coe FL, Keck J, Norton ER. The natural history of urolithiasis. JAMA 1977; 238:1519–1523.
- Borghi L, Meschi T, Amato F, Briganti A, Novarini A, Giannini A. Urinary volume, water, and recurrences in idiopathic calcium nephrolithiasis: a 5-year randomized prospective study. J Urol 1996; 155:839–843.
- Robertson WG, Peacock M, Marshall RW, Marshall DH, Nordin BE. Saturation-inhibition index as a measure of the risk of calcium oxalate stone formation in the urinary tract. N Engl J Med 1976; 294:249–252.
- Evan AP, Lingeman JE, Coe FL, et al. Randall’s plaque of patients with nephrolithiasis begins in basement membranes of thin loops of Henle. J Clin Invest 2003; 111:607–616.
- Kuo RL, Lingeman JE, Evan AP, et al. Urine calcium and volume predict coverage of renal papilla by Randall’s plaque. Kidney Int 2003; 64:2150–2154.
- Broadus AE, Horst RL, Lang R, Littledike ET, Rasmussen H. The importance of circulating 1,25-dihydroxyvitamin D in the pathogenesis of hypercalciuria and renal-stone formation in primary hyperparathyroidism. N Engl J Med 1980; 302:421–426.
- Breslau NA, Preminger GM, Adams BV, Otey J, Pak CY. Use of ketoconazole to probe the pathogenetic importance of 1,25-dihydroxyvitamin D in absorptive hypercalciuria. J Clin Endocrinol Metab 1992; 75:1446–1452.
- Levy FL, Adams-Huet B, Pak CY. Ambulatory evaluation of nephrolithiasis: an update of a 1980 protocol. Am J Med 1995; 98:50–59.
- Breslau NA, Sakhaee K, Pak CY. Impaired adaptation to saltinduced urinary calcium losses in postmenopausal osteoporosis. Trans Assoc Am Physicians 1985; 98:107–115.
- Burtis W, Gay L, Insogna K, Ellison A, Broadus A. Dietary hypercalciuria in patients with calcium oxalate kidney stones. Am J Clin Nutr 1994; 60:424–429.
- Hess B, Ackermann D, Essig M, Takkinen R, Jaeger P. Renal mass and serum calcitriol in male idiopathic calcium renal stone formers: role of protein intake. J Clin Endocrinol Metab 1995; 80:1916–1921.
- Coe FL, Parks JH, Bushinsky DA, Langman CB, Favus MJ. Chlorthalidone promotes mineral retention in patients with idiopathic hypercalciuria. Kidney Int 1988; 33:1140–1146.
- Pak CY, Britton F, Peterson R, et al. Ambulatory evaluation of nephrolithiasis. Classification, clinical presentation, and diagnostic criteria. Am J Med 1980; 69:19–30.
- Curhan GC, Willett WC, Rimm EB, Stampfer MJ. A prospective study of dietary calcium and other nutrients and the risk of symptomatic kidney stones. N Engl J Med 1993; 328:833–838.
- Curhan GC, Willett WC, Speizer FE, Spiegelman D, Stampfer MJ. Comparison of dietary calcium with supplemental calcium and other nutrients as factors affecting the risk for kidney stones in women. Ann Intern Med 1997; 126:497–504.
- Borghi L, Schianchi T, Meschi T, et al. Comparison of two diets for the prevention of recurrent stones in idiopathic hypercalciuria. N Engl J Med 2002; 346:77–84.
- Parks JH, Coe FL, Evan AP, Worcester EM. Clinical and laboratory characteristics of calcium stone-formers with and without primary hyperparathyroidism. Br J Urol 2008; 103:670–678.
- Silverberg SJ, Shane E, Jacobs TP, Siris E, Bilezikian JP. A 10-year prospective study of primary hyperparathyroidism with or without parathyroid surgery. N Engl J Med 1999; 341:1249–1255.
- Mollerup CL, Vestergaard P, Frokjaer VG, Mosekilde L, Christiansen P, Blichert-Toft M. Risk of renal stone events in primary hyperparathyroidism before and after parathyroid surgery: controlled retrospective follow up study. BMJ 2002; 325:807.
- Odvina CV, Sakhaee K, Heller HJ, et al. Biochemical characterization of primary hyperparathyroidism with and without kidney stones. Urol Res 2007; 35:123–128.
- Lemann J, Adams ND, Gray RW. Urinary calcium excretion in human beings. N Engl J Med 1979; 301:535–541.
- Coe FL. Treated and untreated recurrent calcium nephrolithiasis in patients with idiopathic hypercalciuria, hyperuricosuria, or no metabolic disorder. Ann Intern Med 1977; 87:404–410.
- Grover PK, Marshall VR, Ryall RL. Dissolved urate salts out calcium oxalate in undiluted human urine in vitro: implication for calcium oxalate stone genesis. Chem Biol 2003; 10:271–278.
- Ettinger B, Tang A, Citron JT, Livermore B, Williams T. Randomized trial of allopurinol in the prevention of calcium oxalate calculi. N Engl J Med 1986; 315:1386–1389.
- Coe FL, Parks JH. Pathogenesis and treatment of nephrolithiasis. In:The Kidney. Philadelphia: Lippincott Williams & Wilkins, 2000:1841–1867.
- Parks JH, Worcester EM, O’Connor RC, Coe FL. Urine stone risk factors in nephrolithiasis patients with and without bowel disease. Kidney Int 2003; 63:255–265.
- Danpure CJ, Rumsby G. Molecular aetiology of primary hyperoxaluria and its implications for clinical management. Expert Rev Mol Med 2004; 6:1–16.
- Sinha MK, Collazo-Clavell ML, Rule A, et al. Hyperoxaluric nephrolithiasis is a complication of Roux-en-Y gastric bypass surgery. Kidney Int 2007; 72:100–107.
- Parks JH, Coe FL. A urinary calcium-citrate index for the evaluation of nephrolithiasis. Kidney Int 1986; 30:85–90.
- Brennan S, Hering-Smith K, Hamm LL. Effect of pH on citrate reabsorption in the proximal convoluted tubule. Am J Physiol 1988; 255:F301–F306.
- Sakhaee K, Williams RH, Oh MS, et al. Alkali absorption and citrate excretion in calcium nephrolithiasis. J Bone Miner Res 1993; 8:789–794.
- Reddy ST, Wang CY, Sakhaee K, Brinkley L, Pak CY. Effect of low-carbohydrate, high-protein diets on acid-base balance, stone-forming propensity, and calcium metabolism. Am J Kidney Dis 2002; 40:265–274.
- Griffith DP. Struvite stones. Kidney Int 1978; 13:372–382.
- Jennis FS, Lavan JN, Neale FC, Posen S. Staghorn calculi of the kidney: clinical, bacteriological and biochemical features. Br J Urol 1970; 42:511–518.
- Griffith DP, Gibson JR, Clinton CW, Musher DM. Acetohydroxamic acid: clinical studies of a urease inhibitor in patients with staghorn renal calculi. J Urol 1978; 119:9–15.
- Kamel KS, Cheema-Dhadli S, Halperin ML. Studies on the pathophysiology of the low urine pH in patients with uric acid stones. Kidney Int 2002; 61:988–994.
- Abate N, Chandalia M, Cabo-Chan AV, Moe OW, Sakhaee K. The metabolic syndrome and uric acid nephrolithiasis: novel features of renal manifestation of insulin resistance. Kidney Int 2004; 65:386–392.
- Zarse CA, McAteer JA, Tann M, et al. Helical computed tomography accurately reports urinary stone composition using attenuation values: in vitro verification using high-resolution micro-computed tomography calibrated to fourier transform infrared microspectroscopy. Urology 2004; 63:828–833.
- Palacin M. The genetics of heteromeric amino acid transporters. Physiology (Bethesda) 2005; 20:112–124.
- Sakhaee K. Pathogenesis and medical management of cystinuria. Semin Nephrol 1996; 16:435–447.
- Shekarriz B, Stoller ML. Cystinuria and other noncalcareous calculi. Endocrinol Metab Clin North Am 2002; 31:951–977.
- Streem SB, Hall P. Effect of captopril on urinary cystine excretion in homozygous cystinuria. J Urol 1989; 142:1522–1524.
- Perazella MA, Buller GK. Successful treatment of cystinuria with captopril. Am J Kidney Dis 1993; 21:504–507.
- Sloand JA, Izzo JL. Captopril reduces urinary cystine excretion in cystinuria. Arch Intern Med 1987; 147:1409–1412.
- Johnson CM, Wilson DM, O’Fallon WM, Malek RS, Kurland LT. Renal stone epidemiology: a 25-year study in Rochester, Minnesota. Kidney Int 1979; 16:624–631.
- Hiatt RA, Dales LG, Friedman GD, Hunkeler EM. Frequency of urolithiasis in a prepaid medical care program. Am J Epidemiol 1982; 115:255–265.
- Stamatelou KK, Francis ME, Jones CA, Nyberg LM, Curhan GC. Time trends in reported prevalence of kidney stones in the United States: 1976–1994. Kidney Int 2003; 63:1817–1823.
- Li J, Kennedy D, Levine M, Kumar A, Mullen J. Absent hematuria and expensive computerized tomography: case characteristics of emergency urolithiasis. J Urol 2001; 165:782–784.
- Fielding JR, Steele G, Fox LA, Heller H, Loughlin KR. Spiral computerized tomography in the evaluation of acute flank pain: a replacement for excretory urography. J Urol 1997; 157:2071–2073.
- Katz DS, Scheer M, Lumerman JH, Mellinger BC, Stillman CA, Lane MJ. Alternative or additional diagnoses on unenhanced helical computed tomography for suspected renal colic: experience with 1,000 consecutive examinations. Urology 2000; 56:53–57.
- Fowler KAB, Locken JA, Duchesne JH, Williamson MR. US for detecting renal calculi with nonenhanced CT as a reference standard. Radiology 2002; 222:109–113.
- Samplaski MK, Irwin BH, Desai M. Less-invasive ways to remove stones from the kidneys and ureters. Cleve Clin J Med 2009; 76:592–598.
- Labrecque M, Dostaler LP, Rousselle R, Nguyen T, Poirier S. Efficacy of nonsteroidal anti-inflammatory drugs in the treatment of acute renal colic. A meta-analysis. Arch Intern Med 1994; 154:1381–1387.
- Dellabella M, Milanese G, Muzzonigro G. Randomized trial of the efficacy of tamsulosin, nifedipine, and phloroglucinol in medical expulsive therapy for distal ureteral calculi. J Urol 2005; 174:167–172.
- Hollingsworth JM, Togers MA, Kaufman SR, et al. Medical therapy to facilitate urinary stone passage: a meta-analysis. Lancet 2006; 368:1171–1179.
- Borghi L, Meschi T, Amato F, et al. Nifedipine and methylprednisolone in facilitating ureteral stone passage: a randomized, double blind, placebo-controlled study. J Urol 1994; 152:1095–1098.
- Williams RE. Long-term survey of 538 patients with upper urinary tract stone. Br J Urol 1963; 35:416–437.
- Coe FL, Keck J, Norton ER. The natural history of urolithiasis. JAMA 1977; 238:1519–1523.
- Borghi L, Meschi T, Amato F, Briganti A, Novarini A, Giannini A. Urinary volume, water, and recurrences in idiopathic calcium nephrolithiasis: a 5-year randomized prospective study. J Urol 1996; 155:839–843.
- Robertson WG, Peacock M, Marshall RW, Marshall DH, Nordin BE. Saturation-inhibition index as a measure of the risk of calcium oxalate stone formation in the urinary tract. N Engl J Med 1976; 294:249–252.
- Evan AP, Lingeman JE, Coe FL, et al. Randall’s plaque of patients with nephrolithiasis begins in basement membranes of thin loops of Henle. J Clin Invest 2003; 111:607–616.
- Kuo RL, Lingeman JE, Evan AP, et al. Urine calcium and volume predict coverage of renal papilla by Randall’s plaque. Kidney Int 2003; 64:2150–2154.
- Broadus AE, Horst RL, Lang R, Littledike ET, Rasmussen H. The importance of circulating 1,25-dihydroxyvitamin D in the pathogenesis of hypercalciuria and renal-stone formation in primary hyperparathyroidism. N Engl J Med 1980; 302:421–426.
- Breslau NA, Preminger GM, Adams BV, Otey J, Pak CY. Use of ketoconazole to probe the pathogenetic importance of 1,25-dihydroxyvitamin D in absorptive hypercalciuria. J Clin Endocrinol Metab 1992; 75:1446–1452.
- Levy FL, Adams-Huet B, Pak CY. Ambulatory evaluation of nephrolithiasis: an update of a 1980 protocol. Am J Med 1995; 98:50–59.
- Breslau NA, Sakhaee K, Pak CY. Impaired adaptation to saltinduced urinary calcium losses in postmenopausal osteoporosis. Trans Assoc Am Physicians 1985; 98:107–115.
- Burtis W, Gay L, Insogna K, Ellison A, Broadus A. Dietary hypercalciuria in patients with calcium oxalate kidney stones. Am J Clin Nutr 1994; 60:424–429.
- Hess B, Ackermann D, Essig M, Takkinen R, Jaeger P. Renal mass and serum calcitriol in male idiopathic calcium renal stone formers: role of protein intake. J Clin Endocrinol Metab 1995; 80:1916–1921.
- Coe FL, Parks JH, Bushinsky DA, Langman CB, Favus MJ. Chlorthalidone promotes mineral retention in patients with idiopathic hypercalciuria. Kidney Int 1988; 33:1140–1146.
- Pak CY, Britton F, Peterson R, et al. Ambulatory evaluation of nephrolithiasis. Classification, clinical presentation, and diagnostic criteria. Am J Med 1980; 69:19–30.
- Curhan GC, Willett WC, Rimm EB, Stampfer MJ. A prospective study of dietary calcium and other nutrients and the risk of symptomatic kidney stones. N Engl J Med 1993; 328:833–838.
- Curhan GC, Willett WC, Speizer FE, Spiegelman D, Stampfer MJ. Comparison of dietary calcium with supplemental calcium and other nutrients as factors affecting the risk for kidney stones in women. Ann Intern Med 1997; 126:497–504.
- Borghi L, Schianchi T, Meschi T, et al. Comparison of two diets for the prevention of recurrent stones in idiopathic hypercalciuria. N Engl J Med 2002; 346:77–84.
- Parks JH, Coe FL, Evan AP, Worcester EM. Clinical and laboratory characteristics of calcium stone-formers with and without primary hyperparathyroidism. Br J Urol 2008; 103:670–678.
- Silverberg SJ, Shane E, Jacobs TP, Siris E, Bilezikian JP. A 10-year prospective study of primary hyperparathyroidism with or without parathyroid surgery. N Engl J Med 1999; 341:1249–1255.
- Mollerup CL, Vestergaard P, Frokjaer VG, Mosekilde L, Christiansen P, Blichert-Toft M. Risk of renal stone events in primary hyperparathyroidism before and after parathyroid surgery: controlled retrospective follow up study. BMJ 2002; 325:807.
- Odvina CV, Sakhaee K, Heller HJ, et al. Biochemical characterization of primary hyperparathyroidism with and without kidney stones. Urol Res 2007; 35:123–128.
- Lemann J, Adams ND, Gray RW. Urinary calcium excretion in human beings. N Engl J Med 1979; 301:535–541.
- Coe FL. Treated and untreated recurrent calcium nephrolithiasis in patients with idiopathic hypercalciuria, hyperuricosuria, or no metabolic disorder. Ann Intern Med 1977; 87:404–410.
- Grover PK, Marshall VR, Ryall RL. Dissolved urate salts out calcium oxalate in undiluted human urine in vitro: implication for calcium oxalate stone genesis. Chem Biol 2003; 10:271–278.
- Ettinger B, Tang A, Citron JT, Livermore B, Williams T. Randomized trial of allopurinol in the prevention of calcium oxalate calculi. N Engl J Med 1986; 315:1386–1389.
- Coe FL, Parks JH. Pathogenesis and treatment of nephrolithiasis. In:The Kidney. Philadelphia: Lippincott Williams & Wilkins, 2000:1841–1867.
- Parks JH, Worcester EM, O’Connor RC, Coe FL. Urine stone risk factors in nephrolithiasis patients with and without bowel disease. Kidney Int 2003; 63:255–265.
- Danpure CJ, Rumsby G. Molecular aetiology of primary hyperoxaluria and its implications for clinical management. Expert Rev Mol Med 2004; 6:1–16.
- Sinha MK, Collazo-Clavell ML, Rule A, et al. Hyperoxaluric nephrolithiasis is a complication of Roux-en-Y gastric bypass surgery. Kidney Int 2007; 72:100–107.
- Parks JH, Coe FL. A urinary calcium-citrate index for the evaluation of nephrolithiasis. Kidney Int 1986; 30:85–90.
- Brennan S, Hering-Smith K, Hamm LL. Effect of pH on citrate reabsorption in the proximal convoluted tubule. Am J Physiol 1988; 255:F301–F306.
- Sakhaee K, Williams RH, Oh MS, et al. Alkali absorption and citrate excretion in calcium nephrolithiasis. J Bone Miner Res 1993; 8:789–794.
- Reddy ST, Wang CY, Sakhaee K, Brinkley L, Pak CY. Effect of low-carbohydrate, high-protein diets on acid-base balance, stone-forming propensity, and calcium metabolism. Am J Kidney Dis 2002; 40:265–274.
- Griffith DP. Struvite stones. Kidney Int 1978; 13:372–382.
- Jennis FS, Lavan JN, Neale FC, Posen S. Staghorn calculi of the kidney: clinical, bacteriological and biochemical features. Br J Urol 1970; 42:511–518.
- Griffith DP, Gibson JR, Clinton CW, Musher DM. Acetohydroxamic acid: clinical studies of a urease inhibitor in patients with staghorn renal calculi. J Urol 1978; 119:9–15.
- Kamel KS, Cheema-Dhadli S, Halperin ML. Studies on the pathophysiology of the low urine pH in patients with uric acid stones. Kidney Int 2002; 61:988–994.
- Abate N, Chandalia M, Cabo-Chan AV, Moe OW, Sakhaee K. The metabolic syndrome and uric acid nephrolithiasis: novel features of renal manifestation of insulin resistance. Kidney Int 2004; 65:386–392.
- Zarse CA, McAteer JA, Tann M, et al. Helical computed tomography accurately reports urinary stone composition using attenuation values: in vitro verification using high-resolution micro-computed tomography calibrated to fourier transform infrared microspectroscopy. Urology 2004; 63:828–833.
- Palacin M. The genetics of heteromeric amino acid transporters. Physiology (Bethesda) 2005; 20:112–124.
- Sakhaee K. Pathogenesis and medical management of cystinuria. Semin Nephrol 1996; 16:435–447.
- Shekarriz B, Stoller ML. Cystinuria and other noncalcareous calculi. Endocrinol Metab Clin North Am 2002; 31:951–977.
- Streem SB, Hall P. Effect of captopril on urinary cystine excretion in homozygous cystinuria. J Urol 1989; 142:1522–1524.
- Perazella MA, Buller GK. Successful treatment of cystinuria with captopril. Am J Kidney Dis 1993; 21:504–507.
- Sloand JA, Izzo JL. Captopril reduces urinary cystine excretion in cystinuria. Arch Intern Med 1987; 147:1409–1412.
KEY POINTS
- During an acute stone event, medical management focuses on pain control. Hydration and certain drugs may help the stone to pass.
- Most stones are composed of calcium oxalate or calcium phosphate. Less common are uric acid, magnesium ammonium phosphate, and cystine stones.
- To prevent stones from recurring, patients who have had any type of stone should maintain an adequate fluid intake to keep the urine dilute.
- Paradoxically, calcium restriction is not warranted for patients who have had calcium stones, and may even be harmful.
- Alkalinization of the urine may help prevent recurrent uric acid stones and cystine stones.
Less-invasive ways to remove stones from the kidneys and ureters
Very few patients undergo surgery for stones in the kidney or ureters anymore, now that less-invasive interventions are available, such as extracorporeal shock-wave lithotripsy, ureteroscopic stone removal, and percutaneous nephrolithotomy. Each of these options has advantages and disadvantages, depending on the characteristics of the stone or stones, such as size, number, location, and composition, as well as patient factors such as renal anatomy, body habitus, and comorbidities.
This article reviews the current interventional management of upper tract urolithiasis.
NOT ALL STONES NEED INTERVENTION

In a patient who has symptoms of urinary obstruction or sepsis, the decision to intervene is obvious. Stones that obstruct the flow of urine often cause symptoms due to distension of the ureter, the renal pelvis, or the renal capsule in a relatively predictable and characteristic pattern of pain originating in the flank and often radiating to the groin, testicle, or labia. And untreated struvite (“staghorn”) stones, a result of infection, can lead to life-threatening sepsis.
However, in patients with asymptomatic stones, the decision may not be clear-cut. Approximately 32% of patients with asymptomatic renal calculi go on to develop symptoms in the next 2.5 years, increasing to 49% at 5 years.3 Of the patients who develop symptoms, half will require a procedure to remove the stone, while half will pass the offending stone spontaneously.3
If even a small amount of stone is left in the kidney after surgery or other intervention, a large stone can form again, and ultimately, the function of that renal unit can decline. For this reason, most renal calculi should be treated or at least followed for signs of progression with serial imaging studies.
Today, although some patients are followed with kidney-ureter-bladder radiographic studies, most undergo computed tomography, which has the advantages of clearly delineating the stone location and size, the presence of small ureteral stones, and the presence and magnitude of hydronephrosis.
If the patient has no refractory symptoms related to obstruction and no signs of infection or of parenchymal damage, then observation with close follow-up is reasonable. However, infection with urinary tract obstruction, urosepsis, intractable pain or vomiting, acute kidney injury, obstruction in a solitary or transplanted kidney, or bilateral obstructing stones are all indications for urgent intervention.
Additionally, some patients who have asymptomatic stones should undergo evaluation and treatment because of their occupation. Examples are airline pilots and soldiers, in whom an episode of intractable renal colic could prove dangerous.
Stones in women
Women who are pregnant or of childbearing age and have an asymptomatic renal stone are not at any higher risk of stone growth and so should be treated the same as any other patient—except that ultrasonography should be used for imaging to minimize radiation exposure. Urine should be sent for culture. From 50% to 80% of these patients will pass their stones spontaneously with hydration and analgesia.4
If intervention is required, percutaneous nephrostomy and placement of ureteral stents can be done to expose the patient to the least possible amount of anesthesia or radiation.5
Ureteroscopic stone extraction in pregnant patients has also been shown not to cause pregnancy-related complications, and it entails minimal fluoroscopic exposure.6
Although lithotripsy has been used inadvertently in pregnant patients, its routine use in pregnant patients remains contraindicated.7
MEDICAL EXPULSIVE THERAPY
Conservative management, consisting of oral or intravenous hydration and analgesia, can be tried in patients with renal calculi whose condition is otherwise stable. Typically, intravenous hydration is given at a maintenance rate.8 Analgesia can be provided with both nonsteroidal anti-inflammatory drugs (NSAIDs) and narcotics, although NSAIDs, in particular ketorolac (Toradol), provide the best pain control.9
Calcium channel blockers and alphablockers inhibit ureteral spasms and promote the spontaneous passage of ureteral calculi.10 Compared with hydration alone, nifedipine (Procardia) has been shown to lead to an absolute increase of 9% in stone passage rates, and alpha-blockers have produced an absolute increase of 29%.11 These drugs can be given in conjunction with corticosteroids to reduce ureteral edema, which may contribute to stone retention in the ureter.12
As of this date, medical expulsive therapy is well established only for stones in the lower (distal) ureter. The applicability of this treatment for stones in the proximal ureter and kidney is still being investigated. In patients who have stones smaller than 1 cm in diameter and whose symptoms are under control, observation with medical expulsive therapy may well be appropriate. However, after 4 weeks, intervention is indicated, as the risk of complications and renal deterioration increase.
STONE SURGERY HAS BECOME RARE
Before the advent of lithotripsy and ureteroscopy (see below), most patients with symptomatic upper tract calculi underwent open surgical lithotomy. Many variations of pyelolithotomy and nephrolithotomy were performed, even bench surgery with autotransplantation (ie, removing the kidney, removing the stone, and then reimplanting the kidney). However, lithotripsy and ureteroscopic extraction have dramatically reduced the role of open stone surgery: it is currently done in only 0.3% to 0.7% of cases.13,14
Laparoscopic surgery for renal calculi is also rarely done. Although almost every type of stone procedure has been done laparoscopically,15–19 this approach is indicated only in situations in which lithotripsy or ureteroscopic treatment is expected to fail.
LESS-INVASIVE OPTIONS
Lithotripsy for small renal stones
Soon after it became available, lithotripsy became immensely popular because of its ability to break up stones without surgery. Ureteroscopic treatment has assumed a bigger role in recent years because it is more versatile, but lithotripsy remains the most common treatment for urolithiasis.
Advantages, uses. Lithotripsy is generally indicated for renal stones smaller than 2 cm,20 especially those not located in the calyx in the lower pole. It is most effective for stones in the renal pelvis (76% of patients become stone-free), and least effective for stones in the lower pole (59% stone-free).21 For this reason, for stones in the lower pole, only those smaller than 1 cm in diameter are treated with lithotripsy.
In the past, lithotripsy was also favored in patients who had stones in the proximal ureter, an area that was technically difficult to access with a ureteroscope. Recent advances in ureteroscope design have all but eliminated this difficulty.
Disadvantages. Lithotripsy can damage nearly any structure in the trajectory of the shock wave, causing bleeding, inflammation, or perforation. It can also cause disturbances in cardiac electrical signal transmission, leading to cardiac arrhythmias during treatment. Long-term concerns include a possible link between lithotripsy and the development of diabetes and hypertension.22 Lithotripsy is contraindicated in pregnancy and coagulopathic states and is less effective in morbidly obese patients.
Lithotripsy is more likely to fail if the skinto-stone distance is more than 10 mm, if the lower pole forms an acute angle with the ureter, or if the body mass index is greater than 30 kg/m2 (ie, if the patient is obese).23
Percutaneous nephrolithotomy for large or staghorn stones
Percutaneous nephrolithotomy is highly effective for renal calculi but is associated with more complications than lithotripsy or ureteroscopy. It involves inserting a needle through the skin into the renal collecting system and then dilating the tract to approximately 1 cm. Instruments are then inserted through this tract to break up and remove stones. In contrast to laparoscopy, no insufflation is used; the percutaneous tract provides direct access to the kidney for stone removal.
Advantages, uses. Outcomes of percutaneous nephrolithotomy are uniformly favorable across a wide spectrum of stone sizes, compositions, and locations.
Percutaneous nephrolithotomy is indicated in patients who have renal or ureteral stones larger than 2 cm or lower-pole stones larger than 1 cm (Figure 1).24,25
Staghorn stones, commonly associated with infection, lead to renal destruction with significant risk of morbidity and even death if left untreated.26 Because they must be completely removed, which is often difficult or impossible to do with ureteroscopy or lithotripsy, percutaneous nephrolithotomy is the first-line treatment.24
Disadvantages. Percutaneous nephrolithotomy is invasive and carries the associated risks of any major surgical procedure, including sepsis, perirenal hematoma or bleeding, and inadvertent injury to adjacent organs, including the pleurae, lungs, bowel, or spleen.
Ureteroscopy has improved
With improvements in design, stone treatment with flexible and semirigid ureteroscopy have become major options for urinary calculi, even those as far up as the kidney (Figure 1).
Advantages, uses. Ureteroscopy offers a low risk of complications (similar to that of lithotripsy), and stone-free rates approach those of percutaneous nephrolithotomy for small to moderate-sized renal stones.27,28 Outcomes are best for stones smaller than 1 cm, with residual fragments being seen with larger stones.
New flexible ureteroscopes that deflect up to 270° allow stones in the lower pole to be treated successfully.29 In conjunction with laser lithotripsy, ureteroscopy can be used to successfully treat hard stones (density > 1,000 Hounsfield units), stones in obese patients, and stones refractory to lithotripsy.
Rates of complications and second procedures are low, and, compared with lithotripsy, ureteroscopy takes less time to clear the stone.30 Ureteroscopy can also be used to treat stones in kidneys with complex anatomy, in which poor clearance of fragments may be a problem.28 It may also be used in coagulopathic, pregnant, or morbidly obese patients, in whom lithotripsy or percutaneous nephrolithotomy is less effective or contraindicated.
Disadvantages. Of note, ureteroscopy is a surgical skill, and better outcomes are obtained by surgeons with more experience.31
Complications of ureteroscopy include ureteral stricture, perforation, thermal injury, avulsion, intussusception, infection, or steinstrasse (obstruction with fragments of stones). In addition, after ureteroscopy, a temporary ureteral stent is often placed: the stent may cause discomfort and requires a minor adjunctive procedure for removal.
FACTORS THAT AFFECT THE CHOICE OF TREATMENT
Size and location of the stone
The most important predictors of spontaneous passage of ureteral stones are size and location. In general, small stones are more likely to pass spontaneously than large ones, and distal stones are more likely to pass than stones more proximal in the urinary tract.
Stones are typically classified as either ureteral (proximal, middle, or distal) or renal (pelvic or calyceal), depending on their location.
In the ureter. Most ureteral stones smaller than 5 mm in diameter pass spontaneously within 4 weeks of the onset of symptoms.25,32 In patients who have stones smaller than 1 cm, whose pain is controlled, and who show no evidence of sepsis or renal insufficiency, a period of observation is a reasonable option.11 Medications such as tamsulosin (Flomax) and nifedipine have been shown to reduce the need for analgesia and to reduce the time to stone passage.33,34
Lithotripsy and ureteroscopy are the two primary interventions for ureteral calculi.
Regardless of size, stones in the ureter can usually be removed by ureteroscopy. This may involve laser or pneumatic lithotripsy within the ureter or simple ureteroscopic basket retrieval of the intact stone. In situ lithotripsy is an option for proximal ureteral calculi and may be favored by patients who wish to avoid placement of a ureteral stent at the time of intervention. Percutaneous nephrolithotomy is reserved for large (> 2-cm) or impacted proximal ureteral stones, or for cases in which ureteroscopy has failed.35
For stones in the proximal ureter, no difference has been shown in stone passage rates between lithotripsy and ureteroscopy. For proximal stones smaller than 1 cm, lithotripsy has a higher stone-free rate, and for stones larger than 1 cm, ureteroscopy has been shown to have superior stone-free rates.11
For mid-ureteral and distal ureteral stones of all sizes, ureteroscopy has been shown to have superior stone-free rates, although the difference is statistically significant only for distal stones.11
In the kidney. Large renal stones (> 2 cm) or staghorn calculi within the renal collecting system are best treated with percutaneous nephrolithotomy, whereas renal stones smaller than 1 cm can usually be treated ureteroscopically or with lithotripsy.
Stones within the renal collecting system measuring between 1 and 2 cm in diameter can be treated with ureteroscopy, lithotripsy, percutaneous nephrolithotomy, or a combination, depending on the location and composition of the stone and the wishes of patient.
Stone composition
Cystine stones and calcium oxalate stones are hard, with a density greater than 1,000 Hounsfield units. Lithotripsy has a high failure rate with these types of stones.36
Uric acid stones are softer and do not show up well on x-ray imaging. While it is technically feasible to perform lithotripsy under ultrasonographic guidance, most practitioners prefer to use fluoroscopy to locate the stone. For this reason, patients with radiolucent stones (ie, uric acid stones) are also not good candidates for lithotripsy.
Struvite (staghorn) stones are by definition infected, with bacteria residing within the stone itself. Thus, it is imperative to remove all stone fragments during treatment to prevent sepsis and stone reformation. Over time, an untreated staghorn calculus will lead to failure of the renal unit.
Although lithotripsy, ureteroscopy, and percutaneous nephrolithotomy can all be used to treat staghorn calculi, percutaneous nephrolithotomy has the best stone-free rate (78%), and lithotripsy has the lowest (54%).24 Therefore, percutaneous nephrolithotomy is recommended as the first treatment for these stones, and if combination therapy is used, then percutaneous nephrolithotomy should be done last to ensure that the stone is completely removed.24 If lithotomy is to be used, drainage of the renal unit must be done in advance with either percutaneous nephrostomy or a ureteral stent, to ensure that all infected stone fragments will be flushed out.24
PREVENTING RECURRENCES
Metabolic abnormalities that increase the risk of urolithiasis can be identified and treated in up to 95% of patients who form recurrent stones.37 Most of these patients require simple dietary modifications, and just 15% require pharmacotherapy. (For more on this topic, see the review by Dr. Phillip Hall in this issue of the Journal.38) As urolithiasis is common and often recurrent, the appropriate interventive management, combined with dietary prophylaxis, should minimize patient morbidity and preserve renal function.
- Sierakowski R, Finlayson B, Landes RR, Finlayson CD, Sierakowski N. The frequency of urolithiasis in hospital discharge diagnoses in the United States. Invest Urol 1978; 15:438–441.
- Norlin A, Lindell B, Granberg PO, Lindvall N. Urolithiasis. A study of its frequency. Scand J Urol Nephrol 1976; 10:150–153.
- Glowacki LS, Beecroft ML, Cook RJ, Pahl D, Churchill DN. The natural history of asymptomatic urolithiasis. J Urol 1992; 147:319–321.
- Denstedt JD, Razvi H. Management of urinary calculi during pregnancy. J Urol 1992; 148:1072–1074.
- Swanson SK, Heilman RL, Eversman WG. Urinary tract stones in pregnancy. Surg Clin North Am 1995; 75:123–142.
- Watterson JD, Girvan AR, Beiko DT, et al. Ureteroscopy and holmium:YAG laser lithotripsy: an emerging definitive management strategy for symptomatic ureteral calculi in pregnancy. Urology 2002; 60:383–387.
- Frankenschmidt A, Sommerkamp H. Shock wave lithotripsy during pregnancy: a successful clinical experiment. J Urol 1998; 159:501–502.
- Springhart WP, Marquet CG, Sur RL, et al. Forced versus minimal intravenous hydration in the management of acute renal colic: a randomized trial. J Endourol 2006; 20:713–716.
- Holdgate A, Pollock T. Systematic review of the relative efficacy of nonsteroidal anti-inflammatory drugs and opioids in the treatment of acute renal colic. BMJ 2004; 328:1401.
- Hollingsworth JM, Rogers MA, Kaufman SR, et al. Medical therapy to facilitate urinary stone passage: a meta-analysis. Lancet 2006; 368:1171–1179.
- Preminger GM, Tiselius HG, Assimos DG, et al; American Urological Association Education and Research, Inc. 2007 Guideline for the management of ureteral calculi. Eur Urol 2007; 52:1610–1631.
- Pearle MS, Calhoun EA, Curhan GC; Urologic Diseases of America Project. Urologic diseases in America project: urolithiasis. J Urol 2005; 173:848–857.
- Matlaga BR, Assimos DG. Changing indications of open stone surgery. Urology 2002; 59:490–493.
- Paik ML, Wainstein MA, Spirnak JP, Hampel N, Resnick MI. Current indications for open stone surgery in the treatment of renal and ureteral calculi. J Urol 1998; 159:374–378.
- Raboy A, Ferzli GS, Ioffreda R, Albert PS. Laparoscopic ureterolithotomy. Urology 1992; 39:223–225.
- Winfield HN, Donovan JF, Godet AS, Clayman RV. Laparoscopic partial nephrectomy: initial case report for benign disease. J Endourol 1993; 7:521–526.
- Ruckle HC, Segura JW. Laparoscopic treatment of a stone-filled, caliceal diverticulum: a definitive, minimally invasive therapeutic option. J Urol 1994; 151:122–124.
- Deger S, Tuellmann M, Schoenberger B, Winkelmann B, Peters R, Loening SA. Laparoscopic anatrophic nephrolithotomy. Scand J Urol Nephrol 2004; 38:263–265.
- Harmon WJ, Kleer E, Segura JW. Laparoscopic pyelolithotomy for calculus removal in a pelvic kidney. J Urol 1996; 155:2019–2020.
- Abdel-Khalek M, Sheir KZ, Mokhtar AA, Eraky I, Kenawy M, Bazeed M. Prediction of success rate after extracorporeal shock-wave lithotripsy of renal stones—a multivariate analysis model. Scand J Urol Nephrol 2004; 38:161–167.
- Lingeman JE, Coury TA, Newman DM, et al. Comparison of results and morbidity of percutaneous nephrostolithotomy and extracorporeal shock wave lithotripsy. J Urol 1987; 138:485–490.
- Krambeck AE, Gettman MT, Rohlinger AL, Lohse CM, Patterson DE, Segura JW. Diabetes mellitus and hypertension associated with shock wave lithotripsy of renal and proximal ureteral stones at 19 years of followup. J Urol 2006; 175:1742–1747.
- Pareek G, Hedican SP, Lee FT, Nakada SY. Shock wave lithotripsy success determined by skin-to-stone distance on computed tomography. Urology 2005; 66:941–944.
- Preminger GM, Assimos DG, Lingeman JE, Nakada SY, Pearle MS, Wolf JS, Jr; AUA Nephrolithiasis Guideline Panel. Chapter 1: AUA guideline on management of staghorn calculi: diagnosis and treatment recommendations. J Urol 2005; 173:1991–2000.
- Grasso M, Conlin M, Bagley D. Retrograde ureteropyeloscopic treatment of 2 cm. or greater upper urinary tract and minor Staghorn calculi. J Urol 1998; 160:346–351.
- Blandy JP, Singh M. The case for a more aggressive approach to staghorn stones. J Urol 1976; 115:505–506.
- Fabrizio MD, Behari A, Bagley DH. Ureteroscopic management of intrarenal calculi. J Urol 1998; 159:1139–1143.
- Grasso M, Lang G, Loisides P, Bagley D, Taylor F. Endoscopic management of the symptomatic caliceal diverticular calculus. J Urol 1995; 153:1878–1881.
- Grasso M. Ureteropyeloscopic treatment of ureteral and intrarenal calculi. Urol Clin North Am 2000; 27:623–631.
- Peschel R, Janetschek G, Bartsch G. Extracorporeal shock wave lithotripsy versus ureteroscopy for distal ureteral calculi: a prospective randomized study. J Urol 1999; 162:1909–1912.
- Anagnostou T, Tolley D. Management of ureteric stones. Eur Urol 2004; 45:714–721.
- Segura JW, Preminger GM, Assimos DG, et al. Ureteral Stones Clinical Guidelines Panel summary report on the management of ureteral calculi. The American Urological Association. J Urol 1997; 158:1915–1921.
- Pearle MS. Nifedipine versus tamsulosin for the management of lower ureteral stones. Int Braz J Urol 2004; 30:337–338.
- Dellabella M, Milanese G, Muzzonigro G. Medical-expulsive therapy for distal ureterolithiasis: randomized prospective study on role of corticosteroids used in combination with tamsulosin-simplified treatment regimen and health-related quality of life. Urology 2005; 66:712–715.
- Albala DM, Assimos DG, Clayman RV, et al. Lower pole I: a prospective randomized trial of extracorporeal shock wave lithotripsy and percutaneous nephrostolithotomy for lower pole nephrolithiasis-initial results. J Urol 2001; 166:2072–2080.
- Pareek G, Armenakas NA, Fracchia JA. Hounsfield units on computerized tomography predict stone-free rates after extracorporeal shock wave lithotripsy. J Urol 2003; 169:1679–1681.
- Straub M, Hautmann RE. Developments in stone prevention. Curr Opin Urol 2005; 15:119–126.
- Hall PM. Kidney stones: formation, treatment, and prevention. Cleve Clin J Med 2009; 76:583–591.
Very few patients undergo surgery for stones in the kidney or ureters anymore, now that less-invasive interventions are available, such as extracorporeal shock-wave lithotripsy, ureteroscopic stone removal, and percutaneous nephrolithotomy. Each of these options has advantages and disadvantages, depending on the characteristics of the stone or stones, such as size, number, location, and composition, as well as patient factors such as renal anatomy, body habitus, and comorbidities.
This article reviews the current interventional management of upper tract urolithiasis.
NOT ALL STONES NEED INTERVENTION
In a patient who has symptoms of urinary obstruction or sepsis, the decision to intervene is obvious. Stones that obstruct the flow of urine often cause symptoms due to distension of the ureter, the renal pelvis, or the renal capsule in a relatively predictable and characteristic pattern of pain originating in the flank and often radiating to the groin, testicle, or labia. And untreated struvite (“staghorn”) stones, a result of infection, can lead to life-threatening sepsis.
However, in patients with asymptomatic stones, the decision may not be clear-cut. Approximately 32% of patients with asymptomatic renal calculi go on to develop symptoms in the next 2.5 years, increasing to 49% at 5 years.3 Of the patients who develop symptoms, half will require a procedure to remove the stone, while half will pass the offending stone spontaneously.3
If even a small amount of stone is left in the kidney after surgery or other intervention, a large stone can form again, and ultimately, the function of that renal unit can decline. For this reason, most renal calculi should be treated or at least followed for signs of progression with serial imaging studies.
Today, although some patients are followed with kidney-ureter-bladder radiographic studies, most undergo computed tomography, which has the advantages of clearly delineating the stone location and size, the presence of small ureteral stones, and the presence and magnitude of hydronephrosis.
If the patient has no refractory symptoms related to obstruction and no signs of infection or of parenchymal damage, then observation with close follow-up is reasonable. However, infection with urinary tract obstruction, urosepsis, intractable pain or vomiting, acute kidney injury, obstruction in a solitary or transplanted kidney, or bilateral obstructing stones are all indications for urgent intervention.
Additionally, some patients who have asymptomatic stones should undergo evaluation and treatment because of their occupation. Examples are airline pilots and soldiers, in whom an episode of intractable renal colic could prove dangerous.
Stones in women
Women who are pregnant or of childbearing age and have an asymptomatic renal stone are not at any higher risk of stone growth and so should be treated the same as any other patient—except that ultrasonography should be used for imaging to minimize radiation exposure. Urine should be sent for culture. From 50% to 80% of these patients will pass their stones spontaneously with hydration and analgesia.4
If intervention is required, percutaneous nephrostomy and placement of ureteral stents can be done to expose the patient to the least possible amount of anesthesia or radiation.5
Ureteroscopic stone extraction in pregnant patients has also been shown not to cause pregnancy-related complications, and it entails minimal fluoroscopic exposure.6
Although lithotripsy has been used inadvertently in pregnant patients, its routine use in pregnant patients remains contraindicated.7
MEDICAL EXPULSIVE THERAPY
Conservative management, consisting of oral or intravenous hydration and analgesia, can be tried in patients with renal calculi whose condition is otherwise stable. Typically, intravenous hydration is given at a maintenance rate.8 Analgesia can be provided with both nonsteroidal anti-inflammatory drugs (NSAIDs) and narcotics, although NSAIDs, in particular ketorolac (Toradol), provide the best pain control.9
Calcium channel blockers and alphablockers inhibit ureteral spasms and promote the spontaneous passage of ureteral calculi.10 Compared with hydration alone, nifedipine (Procardia) has been shown to lead to an absolute increase of 9% in stone passage rates, and alpha-blockers have produced an absolute increase of 29%.11 These drugs can be given in conjunction with corticosteroids to reduce ureteral edema, which may contribute to stone retention in the ureter.12
As of this date, medical expulsive therapy is well established only for stones in the lower (distal) ureter. The applicability of this treatment for stones in the proximal ureter and kidney is still being investigated. In patients who have stones smaller than 1 cm in diameter and whose symptoms are under control, observation with medical expulsive therapy may well be appropriate. However, after 4 weeks, intervention is indicated, as the risk of complications and renal deterioration increase.
STONE SURGERY HAS BECOME RARE
Before the advent of lithotripsy and ureteroscopy (see below), most patients with symptomatic upper tract calculi underwent open surgical lithotomy. Many variations of pyelolithotomy and nephrolithotomy were performed, even bench surgery with autotransplantation (ie, removing the kidney, removing the stone, and then reimplanting the kidney). However, lithotripsy and ureteroscopic extraction have dramatically reduced the role of open stone surgery: it is currently done in only 0.3% to 0.7% of cases.13,14
Laparoscopic surgery for renal calculi is also rarely done. Although almost every type of stone procedure has been done laparoscopically,15–19 this approach is indicated only in situations in which lithotripsy or ureteroscopic treatment is expected to fail.
LESS-INVASIVE OPTIONS
Lithotripsy for small renal stones
Soon after it became available, lithotripsy became immensely popular because of its ability to break up stones without surgery. Ureteroscopic treatment has assumed a bigger role in recent years because it is more versatile, but lithotripsy remains the most common treatment for urolithiasis.
Advantages, uses. Lithotripsy is generally indicated for renal stones smaller than 2 cm,20 especially those not located in the calyx in the lower pole. It is most effective for stones in the renal pelvis (76% of patients become stone-free), and least effective for stones in the lower pole (59% stone-free).21 For this reason, for stones in the lower pole, only those smaller than 1 cm in diameter are treated with lithotripsy.
In the past, lithotripsy was also favored in patients who had stones in the proximal ureter, an area that was technically difficult to access with a ureteroscope. Recent advances in ureteroscope design have all but eliminated this difficulty.
Disadvantages. Lithotripsy can damage nearly any structure in the trajectory of the shock wave, causing bleeding, inflammation, or perforation. It can also cause disturbances in cardiac electrical signal transmission, leading to cardiac arrhythmias during treatment. Long-term concerns include a possible link between lithotripsy and the development of diabetes and hypertension.22 Lithotripsy is contraindicated in pregnancy and coagulopathic states and is less effective in morbidly obese patients.
Lithotripsy is more likely to fail if the skinto-stone distance is more than 10 mm, if the lower pole forms an acute angle with the ureter, or if the body mass index is greater than 30 kg/m2 (ie, if the patient is obese).23
Percutaneous nephrolithotomy for large or staghorn stones
Percutaneous nephrolithotomy is highly effective for renal calculi but is associated with more complications than lithotripsy or ureteroscopy. It involves inserting a needle through the skin into the renal collecting system and then dilating the tract to approximately 1 cm. Instruments are then inserted through this tract to break up and remove stones. In contrast to laparoscopy, no insufflation is used; the percutaneous tract provides direct access to the kidney for stone removal.
Advantages, uses. Outcomes of percutaneous nephrolithotomy are uniformly favorable across a wide spectrum of stone sizes, compositions, and locations.
Percutaneous nephrolithotomy is indicated in patients who have renal or ureteral stones larger than 2 cm or lower-pole stones larger than 1 cm (Figure 1).24,25
Staghorn stones, commonly associated with infection, lead to renal destruction with significant risk of morbidity and even death if left untreated.26 Because they must be completely removed, which is often difficult or impossible to do with ureteroscopy or lithotripsy, percutaneous nephrolithotomy is the first-line treatment.24
Disadvantages. Percutaneous nephrolithotomy is invasive and carries the associated risks of any major surgical procedure, including sepsis, perirenal hematoma or bleeding, and inadvertent injury to adjacent organs, including the pleurae, lungs, bowel, or spleen.
Ureteroscopy has improved
With improvements in design, stone treatment with flexible and semirigid ureteroscopy have become major options for urinary calculi, even those as far up as the kidney (Figure 1).
Advantages, uses. Ureteroscopy offers a low risk of complications (similar to that of lithotripsy), and stone-free rates approach those of percutaneous nephrolithotomy for small to moderate-sized renal stones.27,28 Outcomes are best for stones smaller than 1 cm, with residual fragments being seen with larger stones.
New flexible ureteroscopes that deflect up to 270° allow stones in the lower pole to be treated successfully.29 In conjunction with laser lithotripsy, ureteroscopy can be used to successfully treat hard stones (density > 1,000 Hounsfield units), stones in obese patients, and stones refractory to lithotripsy.
Rates of complications and second procedures are low, and, compared with lithotripsy, ureteroscopy takes less time to clear the stone.30 Ureteroscopy can also be used to treat stones in kidneys with complex anatomy, in which poor clearance of fragments may be a problem.28 It may also be used in coagulopathic, pregnant, or morbidly obese patients, in whom lithotripsy or percutaneous nephrolithotomy is less effective or contraindicated.
Disadvantages. Of note, ureteroscopy is a surgical skill, and better outcomes are obtained by surgeons with more experience.31
Complications of ureteroscopy include ureteral stricture, perforation, thermal injury, avulsion, intussusception, infection, or steinstrasse (obstruction with fragments of stones). In addition, after ureteroscopy, a temporary ureteral stent is often placed: the stent may cause discomfort and requires a minor adjunctive procedure for removal.
FACTORS THAT AFFECT THE CHOICE OF TREATMENT
Size and location of the stone
The most important predictors of spontaneous passage of ureteral stones are size and location. In general, small stones are more likely to pass spontaneously than large ones, and distal stones are more likely to pass than stones more proximal in the urinary tract.
Stones are typically classified as either ureteral (proximal, middle, or distal) or renal (pelvic or calyceal), depending on their location.
In the ureter. Most ureteral stones smaller than 5 mm in diameter pass spontaneously within 4 weeks of the onset of symptoms.25,32 In patients who have stones smaller than 1 cm, whose pain is controlled, and who show no evidence of sepsis or renal insufficiency, a period of observation is a reasonable option.11 Medications such as tamsulosin (Flomax) and nifedipine have been shown to reduce the need for analgesia and to reduce the time to stone passage.33,34
Lithotripsy and ureteroscopy are the two primary interventions for ureteral calculi.
Regardless of size, stones in the ureter can usually be removed by ureteroscopy. This may involve laser or pneumatic lithotripsy within the ureter or simple ureteroscopic basket retrieval of the intact stone. In situ lithotripsy is an option for proximal ureteral calculi and may be favored by patients who wish to avoid placement of a ureteral stent at the time of intervention. Percutaneous nephrolithotomy is reserved for large (> 2-cm) or impacted proximal ureteral stones, or for cases in which ureteroscopy has failed.35
For stones in the proximal ureter, no difference has been shown in stone passage rates between lithotripsy and ureteroscopy. For proximal stones smaller than 1 cm, lithotripsy has a higher stone-free rate, and for stones larger than 1 cm, ureteroscopy has been shown to have superior stone-free rates.11
For mid-ureteral and distal ureteral stones of all sizes, ureteroscopy has been shown to have superior stone-free rates, although the difference is statistically significant only for distal stones.11
In the kidney. Large renal stones (> 2 cm) or staghorn calculi within the renal collecting system are best treated with percutaneous nephrolithotomy, whereas renal stones smaller than 1 cm can usually be treated ureteroscopically or with lithotripsy.
Stones within the renal collecting system measuring between 1 and 2 cm in diameter can be treated with ureteroscopy, lithotripsy, percutaneous nephrolithotomy, or a combination, depending on the location and composition of the stone and the wishes of patient.
Stone composition
Cystine stones and calcium oxalate stones are hard, with a density greater than 1,000 Hounsfield units. Lithotripsy has a high failure rate with these types of stones.36
Uric acid stones are softer and do not show up well on x-ray imaging. While it is technically feasible to perform lithotripsy under ultrasonographic guidance, most practitioners prefer to use fluoroscopy to locate the stone. For this reason, patients with radiolucent stones (ie, uric acid stones) are also not good candidates for lithotripsy.
Struvite (staghorn) stones are by definition infected, with bacteria residing within the stone itself. Thus, it is imperative to remove all stone fragments during treatment to prevent sepsis and stone reformation. Over time, an untreated staghorn calculus will lead to failure of the renal unit.
Although lithotripsy, ureteroscopy, and percutaneous nephrolithotomy can all be used to treat staghorn calculi, percutaneous nephrolithotomy has the best stone-free rate (78%), and lithotripsy has the lowest (54%).24 Therefore, percutaneous nephrolithotomy is recommended as the first treatment for these stones, and if combination therapy is used, then percutaneous nephrolithotomy should be done last to ensure that the stone is completely removed.24 If lithotomy is to be used, drainage of the renal unit must be done in advance with either percutaneous nephrostomy or a ureteral stent, to ensure that all infected stone fragments will be flushed out.24
PREVENTING RECURRENCES
Metabolic abnormalities that increase the risk of urolithiasis can be identified and treated in up to 95% of patients who form recurrent stones.37 Most of these patients require simple dietary modifications, and just 15% require pharmacotherapy. (For more on this topic, see the review by Dr. Phillip Hall in this issue of the Journal.38) As urolithiasis is common and often recurrent, the appropriate interventive management, combined with dietary prophylaxis, should minimize patient morbidity and preserve renal function.
Very few patients undergo surgery for stones in the kidney or ureters anymore, now that less-invasive interventions are available, such as extracorporeal shock-wave lithotripsy, ureteroscopic stone removal, and percutaneous nephrolithotomy. Each of these options has advantages and disadvantages, depending on the characteristics of the stone or stones, such as size, number, location, and composition, as well as patient factors such as renal anatomy, body habitus, and comorbidities.
This article reviews the current interventional management of upper tract urolithiasis.
NOT ALL STONES NEED INTERVENTION
In a patient who has symptoms of urinary obstruction or sepsis, the decision to intervene is obvious. Stones that obstruct the flow of urine often cause symptoms due to distension of the ureter, the renal pelvis, or the renal capsule in a relatively predictable and characteristic pattern of pain originating in the flank and often radiating to the groin, testicle, or labia. And untreated struvite (“staghorn”) stones, a result of infection, can lead to life-threatening sepsis.
However, in patients with asymptomatic stones, the decision may not be clear-cut. Approximately 32% of patients with asymptomatic renal calculi go on to develop symptoms in the next 2.5 years, increasing to 49% at 5 years.3 Of the patients who develop symptoms, half will require a procedure to remove the stone, while half will pass the offending stone spontaneously.3
If even a small amount of stone is left in the kidney after surgery or other intervention, a large stone can form again, and ultimately, the function of that renal unit can decline. For this reason, most renal calculi should be treated or at least followed for signs of progression with serial imaging studies.
Today, although some patients are followed with kidney-ureter-bladder radiographic studies, most undergo computed tomography, which has the advantages of clearly delineating the stone location and size, the presence of small ureteral stones, and the presence and magnitude of hydronephrosis.
If the patient has no refractory symptoms related to obstruction and no signs of infection or of parenchymal damage, then observation with close follow-up is reasonable. However, infection with urinary tract obstruction, urosepsis, intractable pain or vomiting, acute kidney injury, obstruction in a solitary or transplanted kidney, or bilateral obstructing stones are all indications for urgent intervention.
Additionally, some patients who have asymptomatic stones should undergo evaluation and treatment because of their occupation. Examples are airline pilots and soldiers, in whom an episode of intractable renal colic could prove dangerous.
Stones in women
Women who are pregnant or of childbearing age and have an asymptomatic renal stone are not at any higher risk of stone growth and so should be treated the same as any other patient—except that ultrasonography should be used for imaging to minimize radiation exposure. Urine should be sent for culture. From 50% to 80% of these patients will pass their stones spontaneously with hydration and analgesia.4
If intervention is required, percutaneous nephrostomy and placement of ureteral stents can be done to expose the patient to the least possible amount of anesthesia or radiation.5
Ureteroscopic stone extraction in pregnant patients has also been shown not to cause pregnancy-related complications, and it entails minimal fluoroscopic exposure.6
Although lithotripsy has been used inadvertently in pregnant patients, its routine use in pregnant patients remains contraindicated.7
MEDICAL EXPULSIVE THERAPY
Conservative management, consisting of oral or intravenous hydration and analgesia, can be tried in patients with renal calculi whose condition is otherwise stable. Typically, intravenous hydration is given at a maintenance rate.8 Analgesia can be provided with both nonsteroidal anti-inflammatory drugs (NSAIDs) and narcotics, although NSAIDs, in particular ketorolac (Toradol), provide the best pain control.9
Calcium channel blockers and alphablockers inhibit ureteral spasms and promote the spontaneous passage of ureteral calculi.10 Compared with hydration alone, nifedipine (Procardia) has been shown to lead to an absolute increase of 9% in stone passage rates, and alpha-blockers have produced an absolute increase of 29%.11 These drugs can be given in conjunction with corticosteroids to reduce ureteral edema, which may contribute to stone retention in the ureter.12
As of this date, medical expulsive therapy is well established only for stones in the lower (distal) ureter. The applicability of this treatment for stones in the proximal ureter and kidney is still being investigated. In patients who have stones smaller than 1 cm in diameter and whose symptoms are under control, observation with medical expulsive therapy may well be appropriate. However, after 4 weeks, intervention is indicated, as the risk of complications and renal deterioration increase.
STONE SURGERY HAS BECOME RARE
Before the advent of lithotripsy and ureteroscopy (see below), most patients with symptomatic upper tract calculi underwent open surgical lithotomy. Many variations of pyelolithotomy and nephrolithotomy were performed, even bench surgery with autotransplantation (ie, removing the kidney, removing the stone, and then reimplanting the kidney). However, lithotripsy and ureteroscopic extraction have dramatically reduced the role of open stone surgery: it is currently done in only 0.3% to 0.7% of cases.13,14
Laparoscopic surgery for renal calculi is also rarely done. Although almost every type of stone procedure has been done laparoscopically,15–19 this approach is indicated only in situations in which lithotripsy or ureteroscopic treatment is expected to fail.
LESS-INVASIVE OPTIONS
Lithotripsy for small renal stones
Soon after it became available, lithotripsy became immensely popular because of its ability to break up stones without surgery. Ureteroscopic treatment has assumed a bigger role in recent years because it is more versatile, but lithotripsy remains the most common treatment for urolithiasis.
Advantages, uses. Lithotripsy is generally indicated for renal stones smaller than 2 cm,20 especially those not located in the calyx in the lower pole. It is most effective for stones in the renal pelvis (76% of patients become stone-free), and least effective for stones in the lower pole (59% stone-free).21 For this reason, for stones in the lower pole, only those smaller than 1 cm in diameter are treated with lithotripsy.
In the past, lithotripsy was also favored in patients who had stones in the proximal ureter, an area that was technically difficult to access with a ureteroscope. Recent advances in ureteroscope design have all but eliminated this difficulty.
Disadvantages. Lithotripsy can damage nearly any structure in the trajectory of the shock wave, causing bleeding, inflammation, or perforation. It can also cause disturbances in cardiac electrical signal transmission, leading to cardiac arrhythmias during treatment. Long-term concerns include a possible link between lithotripsy and the development of diabetes and hypertension.22 Lithotripsy is contraindicated in pregnancy and coagulopathic states and is less effective in morbidly obese patients.
Lithotripsy is more likely to fail if the skinto-stone distance is more than 10 mm, if the lower pole forms an acute angle with the ureter, or if the body mass index is greater than 30 kg/m2 (ie, if the patient is obese).23
Percutaneous nephrolithotomy for large or staghorn stones
Percutaneous nephrolithotomy is highly effective for renal calculi but is associated with more complications than lithotripsy or ureteroscopy. It involves inserting a needle through the skin into the renal collecting system and then dilating the tract to approximately 1 cm. Instruments are then inserted through this tract to break up and remove stones. In contrast to laparoscopy, no insufflation is used; the percutaneous tract provides direct access to the kidney for stone removal.
Advantages, uses. Outcomes of percutaneous nephrolithotomy are uniformly favorable across a wide spectrum of stone sizes, compositions, and locations.
Percutaneous nephrolithotomy is indicated in patients who have renal or ureteral stones larger than 2 cm or lower-pole stones larger than 1 cm (Figure 1).24,25
Staghorn stones, commonly associated with infection, lead to renal destruction with significant risk of morbidity and even death if left untreated.26 Because they must be completely removed, which is often difficult or impossible to do with ureteroscopy or lithotripsy, percutaneous nephrolithotomy is the first-line treatment.24
Disadvantages. Percutaneous nephrolithotomy is invasive and carries the associated risks of any major surgical procedure, including sepsis, perirenal hematoma or bleeding, and inadvertent injury to adjacent organs, including the pleurae, lungs, bowel, or spleen.
Ureteroscopy has improved
With improvements in design, stone treatment with flexible and semirigid ureteroscopy have become major options for urinary calculi, even those as far up as the kidney (Figure 1).
Advantages, uses. Ureteroscopy offers a low risk of complications (similar to that of lithotripsy), and stone-free rates approach those of percutaneous nephrolithotomy for small to moderate-sized renal stones.27,28 Outcomes are best for stones smaller than 1 cm, with residual fragments being seen with larger stones.
New flexible ureteroscopes that deflect up to 270° allow stones in the lower pole to be treated successfully.29 In conjunction with laser lithotripsy, ureteroscopy can be used to successfully treat hard stones (density > 1,000 Hounsfield units), stones in obese patients, and stones refractory to lithotripsy.
Rates of complications and second procedures are low, and, compared with lithotripsy, ureteroscopy takes less time to clear the stone.30 Ureteroscopy can also be used to treat stones in kidneys with complex anatomy, in which poor clearance of fragments may be a problem.28 It may also be used in coagulopathic, pregnant, or morbidly obese patients, in whom lithotripsy or percutaneous nephrolithotomy is less effective or contraindicated.
Disadvantages. Of note, ureteroscopy is a surgical skill, and better outcomes are obtained by surgeons with more experience.31
Complications of ureteroscopy include ureteral stricture, perforation, thermal injury, avulsion, intussusception, infection, or steinstrasse (obstruction with fragments of stones). In addition, after ureteroscopy, a temporary ureteral stent is often placed: the stent may cause discomfort and requires a minor adjunctive procedure for removal.
FACTORS THAT AFFECT THE CHOICE OF TREATMENT
Size and location of the stone
The most important predictors of spontaneous passage of ureteral stones are size and location. In general, small stones are more likely to pass spontaneously than large ones, and distal stones are more likely to pass than stones more proximal in the urinary tract.
Stones are typically classified as either ureteral (proximal, middle, or distal) or renal (pelvic or calyceal), depending on their location.
In the ureter. Most ureteral stones smaller than 5 mm in diameter pass spontaneously within 4 weeks of the onset of symptoms.25,32 In patients who have stones smaller than 1 cm, whose pain is controlled, and who show no evidence of sepsis or renal insufficiency, a period of observation is a reasonable option.11 Medications such as tamsulosin (Flomax) and nifedipine have been shown to reduce the need for analgesia and to reduce the time to stone passage.33,34
Lithotripsy and ureteroscopy are the two primary interventions for ureteral calculi.
Regardless of size, stones in the ureter can usually be removed by ureteroscopy. This may involve laser or pneumatic lithotripsy within the ureter or simple ureteroscopic basket retrieval of the intact stone. In situ lithotripsy is an option for proximal ureteral calculi and may be favored by patients who wish to avoid placement of a ureteral stent at the time of intervention. Percutaneous nephrolithotomy is reserved for large (> 2-cm) or impacted proximal ureteral stones, or for cases in which ureteroscopy has failed.35
For stones in the proximal ureter, no difference has been shown in stone passage rates between lithotripsy and ureteroscopy. For proximal stones smaller than 1 cm, lithotripsy has a higher stone-free rate, and for stones larger than 1 cm, ureteroscopy has been shown to have superior stone-free rates.11
For mid-ureteral and distal ureteral stones of all sizes, ureteroscopy has been shown to have superior stone-free rates, although the difference is statistically significant only for distal stones.11
In the kidney. Large renal stones (> 2 cm) or staghorn calculi within the renal collecting system are best treated with percutaneous nephrolithotomy, whereas renal stones smaller than 1 cm can usually be treated ureteroscopically or with lithotripsy.
Stones within the renal collecting system measuring between 1 and 2 cm in diameter can be treated with ureteroscopy, lithotripsy, percutaneous nephrolithotomy, or a combination, depending on the location and composition of the stone and the wishes of patient.
Stone composition
Cystine stones and calcium oxalate stones are hard, with a density greater than 1,000 Hounsfield units. Lithotripsy has a high failure rate with these types of stones.36
Uric acid stones are softer and do not show up well on x-ray imaging. While it is technically feasible to perform lithotripsy under ultrasonographic guidance, most practitioners prefer to use fluoroscopy to locate the stone. For this reason, patients with radiolucent stones (ie, uric acid stones) are also not good candidates for lithotripsy.
Struvite (staghorn) stones are by definition infected, with bacteria residing within the stone itself. Thus, it is imperative to remove all stone fragments during treatment to prevent sepsis and stone reformation. Over time, an untreated staghorn calculus will lead to failure of the renal unit.
Although lithotripsy, ureteroscopy, and percutaneous nephrolithotomy can all be used to treat staghorn calculi, percutaneous nephrolithotomy has the best stone-free rate (78%), and lithotripsy has the lowest (54%).24 Therefore, percutaneous nephrolithotomy is recommended as the first treatment for these stones, and if combination therapy is used, then percutaneous nephrolithotomy should be done last to ensure that the stone is completely removed.24 If lithotomy is to be used, drainage of the renal unit must be done in advance with either percutaneous nephrostomy or a ureteral stent, to ensure that all infected stone fragments will be flushed out.24
PREVENTING RECURRENCES
Metabolic abnormalities that increase the risk of urolithiasis can be identified and treated in up to 95% of patients who form recurrent stones.37 Most of these patients require simple dietary modifications, and just 15% require pharmacotherapy. (For more on this topic, see the review by Dr. Phillip Hall in this issue of the Journal.38) As urolithiasis is common and often recurrent, the appropriate interventive management, combined with dietary prophylaxis, should minimize patient morbidity and preserve renal function.
- Sierakowski R, Finlayson B, Landes RR, Finlayson CD, Sierakowski N. The frequency of urolithiasis in hospital discharge diagnoses in the United States. Invest Urol 1978; 15:438–441.
- Norlin A, Lindell B, Granberg PO, Lindvall N. Urolithiasis. A study of its frequency. Scand J Urol Nephrol 1976; 10:150–153.
- Glowacki LS, Beecroft ML, Cook RJ, Pahl D, Churchill DN. The natural history of asymptomatic urolithiasis. J Urol 1992; 147:319–321.
- Denstedt JD, Razvi H. Management of urinary calculi during pregnancy. J Urol 1992; 148:1072–1074.
- Swanson SK, Heilman RL, Eversman WG. Urinary tract stones in pregnancy. Surg Clin North Am 1995; 75:123–142.
- Watterson JD, Girvan AR, Beiko DT, et al. Ureteroscopy and holmium:YAG laser lithotripsy: an emerging definitive management strategy for symptomatic ureteral calculi in pregnancy. Urology 2002; 60:383–387.
- Frankenschmidt A, Sommerkamp H. Shock wave lithotripsy during pregnancy: a successful clinical experiment. J Urol 1998; 159:501–502.
- Springhart WP, Marquet CG, Sur RL, et al. Forced versus minimal intravenous hydration in the management of acute renal colic: a randomized trial. J Endourol 2006; 20:713–716.
- Holdgate A, Pollock T. Systematic review of the relative efficacy of nonsteroidal anti-inflammatory drugs and opioids in the treatment of acute renal colic. BMJ 2004; 328:1401.
- Hollingsworth JM, Rogers MA, Kaufman SR, et al. Medical therapy to facilitate urinary stone passage: a meta-analysis. Lancet 2006; 368:1171–1179.
- Preminger GM, Tiselius HG, Assimos DG, et al; American Urological Association Education and Research, Inc. 2007 Guideline for the management of ureteral calculi. Eur Urol 2007; 52:1610–1631.
- Pearle MS, Calhoun EA, Curhan GC; Urologic Diseases of America Project. Urologic diseases in America project: urolithiasis. J Urol 2005; 173:848–857.
- Matlaga BR, Assimos DG. Changing indications of open stone surgery. Urology 2002; 59:490–493.
- Paik ML, Wainstein MA, Spirnak JP, Hampel N, Resnick MI. Current indications for open stone surgery in the treatment of renal and ureteral calculi. J Urol 1998; 159:374–378.
- Raboy A, Ferzli GS, Ioffreda R, Albert PS. Laparoscopic ureterolithotomy. Urology 1992; 39:223–225.
- Winfield HN, Donovan JF, Godet AS, Clayman RV. Laparoscopic partial nephrectomy: initial case report for benign disease. J Endourol 1993; 7:521–526.
- Ruckle HC, Segura JW. Laparoscopic treatment of a stone-filled, caliceal diverticulum: a definitive, minimally invasive therapeutic option. J Urol 1994; 151:122–124.
- Deger S, Tuellmann M, Schoenberger B, Winkelmann B, Peters R, Loening SA. Laparoscopic anatrophic nephrolithotomy. Scand J Urol Nephrol 2004; 38:263–265.
- Harmon WJ, Kleer E, Segura JW. Laparoscopic pyelolithotomy for calculus removal in a pelvic kidney. J Urol 1996; 155:2019–2020.
- Abdel-Khalek M, Sheir KZ, Mokhtar AA, Eraky I, Kenawy M, Bazeed M. Prediction of success rate after extracorporeal shock-wave lithotripsy of renal stones—a multivariate analysis model. Scand J Urol Nephrol 2004; 38:161–167.
- Lingeman JE, Coury TA, Newman DM, et al. Comparison of results and morbidity of percutaneous nephrostolithotomy and extracorporeal shock wave lithotripsy. J Urol 1987; 138:485–490.
- Krambeck AE, Gettman MT, Rohlinger AL, Lohse CM, Patterson DE, Segura JW. Diabetes mellitus and hypertension associated with shock wave lithotripsy of renal and proximal ureteral stones at 19 years of followup. J Urol 2006; 175:1742–1747.
- Pareek G, Hedican SP, Lee FT, Nakada SY. Shock wave lithotripsy success determined by skin-to-stone distance on computed tomography. Urology 2005; 66:941–944.
- Preminger GM, Assimos DG, Lingeman JE, Nakada SY, Pearle MS, Wolf JS, Jr; AUA Nephrolithiasis Guideline Panel. Chapter 1: AUA guideline on management of staghorn calculi: diagnosis and treatment recommendations. J Urol 2005; 173:1991–2000.
- Grasso M, Conlin M, Bagley D. Retrograde ureteropyeloscopic treatment of 2 cm. or greater upper urinary tract and minor Staghorn calculi. J Urol 1998; 160:346–351.
- Blandy JP, Singh M. The case for a more aggressive approach to staghorn stones. J Urol 1976; 115:505–506.
- Fabrizio MD, Behari A, Bagley DH. Ureteroscopic management of intrarenal calculi. J Urol 1998; 159:1139–1143.
- Grasso M, Lang G, Loisides P, Bagley D, Taylor F. Endoscopic management of the symptomatic caliceal diverticular calculus. J Urol 1995; 153:1878–1881.
- Grasso M. Ureteropyeloscopic treatment of ureteral and intrarenal calculi. Urol Clin North Am 2000; 27:623–631.
- Peschel R, Janetschek G, Bartsch G. Extracorporeal shock wave lithotripsy versus ureteroscopy for distal ureteral calculi: a prospective randomized study. J Urol 1999; 162:1909–1912.
- Anagnostou T, Tolley D. Management of ureteric stones. Eur Urol 2004; 45:714–721.
- Segura JW, Preminger GM, Assimos DG, et al. Ureteral Stones Clinical Guidelines Panel summary report on the management of ureteral calculi. The American Urological Association. J Urol 1997; 158:1915–1921.
- Pearle MS. Nifedipine versus tamsulosin for the management of lower ureteral stones. Int Braz J Urol 2004; 30:337–338.
- Dellabella M, Milanese G, Muzzonigro G. Medical-expulsive therapy for distal ureterolithiasis: randomized prospective study on role of corticosteroids used in combination with tamsulosin-simplified treatment regimen and health-related quality of life. Urology 2005; 66:712–715.
- Albala DM, Assimos DG, Clayman RV, et al. Lower pole I: a prospective randomized trial of extracorporeal shock wave lithotripsy and percutaneous nephrostolithotomy for lower pole nephrolithiasis-initial results. J Urol 2001; 166:2072–2080.
- Pareek G, Armenakas NA, Fracchia JA. Hounsfield units on computerized tomography predict stone-free rates after extracorporeal shock wave lithotripsy. J Urol 2003; 169:1679–1681.
- Straub M, Hautmann RE. Developments in stone prevention. Curr Opin Urol 2005; 15:119–126.
- Hall PM. Kidney stones: formation, treatment, and prevention. Cleve Clin J Med 2009; 76:583–591.
- Sierakowski R, Finlayson B, Landes RR, Finlayson CD, Sierakowski N. The frequency of urolithiasis in hospital discharge diagnoses in the United States. Invest Urol 1978; 15:438–441.
- Norlin A, Lindell B, Granberg PO, Lindvall N. Urolithiasis. A study of its frequency. Scand J Urol Nephrol 1976; 10:150–153.
- Glowacki LS, Beecroft ML, Cook RJ, Pahl D, Churchill DN. The natural history of asymptomatic urolithiasis. J Urol 1992; 147:319–321.
- Denstedt JD, Razvi H. Management of urinary calculi during pregnancy. J Urol 1992; 148:1072–1074.
- Swanson SK, Heilman RL, Eversman WG. Urinary tract stones in pregnancy. Surg Clin North Am 1995; 75:123–142.
- Watterson JD, Girvan AR, Beiko DT, et al. Ureteroscopy and holmium:YAG laser lithotripsy: an emerging definitive management strategy for symptomatic ureteral calculi in pregnancy. Urology 2002; 60:383–387.
- Frankenschmidt A, Sommerkamp H. Shock wave lithotripsy during pregnancy: a successful clinical experiment. J Urol 1998; 159:501–502.
- Springhart WP, Marquet CG, Sur RL, et al. Forced versus minimal intravenous hydration in the management of acute renal colic: a randomized trial. J Endourol 2006; 20:713–716.
- Holdgate A, Pollock T. Systematic review of the relative efficacy of nonsteroidal anti-inflammatory drugs and opioids in the treatment of acute renal colic. BMJ 2004; 328:1401.
- Hollingsworth JM, Rogers MA, Kaufman SR, et al. Medical therapy to facilitate urinary stone passage: a meta-analysis. Lancet 2006; 368:1171–1179.
- Preminger GM, Tiselius HG, Assimos DG, et al; American Urological Association Education and Research, Inc. 2007 Guideline for the management of ureteral calculi. Eur Urol 2007; 52:1610–1631.
- Pearle MS, Calhoun EA, Curhan GC; Urologic Diseases of America Project. Urologic diseases in America project: urolithiasis. J Urol 2005; 173:848–857.
- Matlaga BR, Assimos DG. Changing indications of open stone surgery. Urology 2002; 59:490–493.
- Paik ML, Wainstein MA, Spirnak JP, Hampel N, Resnick MI. Current indications for open stone surgery in the treatment of renal and ureteral calculi. J Urol 1998; 159:374–378.
- Raboy A, Ferzli GS, Ioffreda R, Albert PS. Laparoscopic ureterolithotomy. Urology 1992; 39:223–225.
- Winfield HN, Donovan JF, Godet AS, Clayman RV. Laparoscopic partial nephrectomy: initial case report for benign disease. J Endourol 1993; 7:521–526.
- Ruckle HC, Segura JW. Laparoscopic treatment of a stone-filled, caliceal diverticulum: a definitive, minimally invasive therapeutic option. J Urol 1994; 151:122–124.
- Deger S, Tuellmann M, Schoenberger B, Winkelmann B, Peters R, Loening SA. Laparoscopic anatrophic nephrolithotomy. Scand J Urol Nephrol 2004; 38:263–265.
- Harmon WJ, Kleer E, Segura JW. Laparoscopic pyelolithotomy for calculus removal in a pelvic kidney. J Urol 1996; 155:2019–2020.
- Abdel-Khalek M, Sheir KZ, Mokhtar AA, Eraky I, Kenawy M, Bazeed M. Prediction of success rate after extracorporeal shock-wave lithotripsy of renal stones—a multivariate analysis model. Scand J Urol Nephrol 2004; 38:161–167.
- Lingeman JE, Coury TA, Newman DM, et al. Comparison of results and morbidity of percutaneous nephrostolithotomy and extracorporeal shock wave lithotripsy. J Urol 1987; 138:485–490.
- Krambeck AE, Gettman MT, Rohlinger AL, Lohse CM, Patterson DE, Segura JW. Diabetes mellitus and hypertension associated with shock wave lithotripsy of renal and proximal ureteral stones at 19 years of followup. J Urol 2006; 175:1742–1747.
- Pareek G, Hedican SP, Lee FT, Nakada SY. Shock wave lithotripsy success determined by skin-to-stone distance on computed tomography. Urology 2005; 66:941–944.
- Preminger GM, Assimos DG, Lingeman JE, Nakada SY, Pearle MS, Wolf JS, Jr; AUA Nephrolithiasis Guideline Panel. Chapter 1: AUA guideline on management of staghorn calculi: diagnosis and treatment recommendations. J Urol 2005; 173:1991–2000.
- Grasso M, Conlin M, Bagley D. Retrograde ureteropyeloscopic treatment of 2 cm. or greater upper urinary tract and minor Staghorn calculi. J Urol 1998; 160:346–351.
- Blandy JP, Singh M. The case for a more aggressive approach to staghorn stones. J Urol 1976; 115:505–506.
- Fabrizio MD, Behari A, Bagley DH. Ureteroscopic management of intrarenal calculi. J Urol 1998; 159:1139–1143.
- Grasso M, Lang G, Loisides P, Bagley D, Taylor F. Endoscopic management of the symptomatic caliceal diverticular calculus. J Urol 1995; 153:1878–1881.
- Grasso M. Ureteropyeloscopic treatment of ureteral and intrarenal calculi. Urol Clin North Am 2000; 27:623–631.
- Peschel R, Janetschek G, Bartsch G. Extracorporeal shock wave lithotripsy versus ureteroscopy for distal ureteral calculi: a prospective randomized study. J Urol 1999; 162:1909–1912.
- Anagnostou T, Tolley D. Management of ureteric stones. Eur Urol 2004; 45:714–721.
- Segura JW, Preminger GM, Assimos DG, et al. Ureteral Stones Clinical Guidelines Panel summary report on the management of ureteral calculi. The American Urological Association. J Urol 1997; 158:1915–1921.
- Pearle MS. Nifedipine versus tamsulosin for the management of lower ureteral stones. Int Braz J Urol 2004; 30:337–338.
- Dellabella M, Milanese G, Muzzonigro G. Medical-expulsive therapy for distal ureterolithiasis: randomized prospective study on role of corticosteroids used in combination with tamsulosin-simplified treatment regimen and health-related quality of life. Urology 2005; 66:712–715.
- Albala DM, Assimos DG, Clayman RV, et al. Lower pole I: a prospective randomized trial of extracorporeal shock wave lithotripsy and percutaneous nephrostolithotomy for lower pole nephrolithiasis-initial results. J Urol 2001; 166:2072–2080.
- Pareek G, Armenakas NA, Fracchia JA. Hounsfield units on computerized tomography predict stone-free rates after extracorporeal shock wave lithotripsy. J Urol 2003; 169:1679–1681.
- Straub M, Hautmann RE. Developments in stone prevention. Curr Opin Urol 2005; 15:119–126.
- Hall PM. Kidney stones: formation, treatment, and prevention. Cleve Clin J Med 2009; 76:583–591.
KEY POINTS
- Stones that obstruct the flow of urine or that are associated with infection (ie, struvite or “staghorn” stones) should be removed promptly.
- For small stones in the distal ureter, medical therapy is an option: pain control, hydration, and control of ureteral spasms with calcium channel blockers and alpha-blockers help the patient pass the stone spontaneously.
- Extracorporeal shock-wave lithotripsy is the mostly commonly used option, but it is less effective for large stones and in obese patients.
- The ureteroscope can now be used to extract stones as high up as the kidney. Catheters that contain lasers and lithotripsy devices can break up large stones in situ for removal.
- Percutaneous nephrolithotomy is very effective for large stones in the kidney and is especially indicated for struvite stones.
Grand Rounds: Woman, 39, With Leg Weakness After Exercise Class
A 39-year-old woman presented to the emergency department (ED) with a chief complaint of muscle aches and pain. She stated that three days earlier, she had begun exercising in a 45-minute “spinning” class (ie, riding a stationary bicycle with a weighted front wheel). The patient had not engaged in any aerobic exercise for at least six months before the spinning class. She mentioned that much older participants in the class were outperforming her, but she did not feel the need to keep up with them.
After dismounting, the woman said, she experienced weakness in her legs and had great difficulty ambulating. She went home, took 400 mg of ibuprofen, and went to bed. She awoke with pain and swelling in both thighs and continued to take ibuprofen, in addition to applying a topical mentholated preparation to her thighs. She took an Epsom salts bath two days later.
On the morning of the third day after the spinning class, she voided black urine and presented to the ED.
The patient had no significant medical history. Surgical history was limited to removal of a ganglion cyst on her wrist. She denied any history of seizure disorder, thyroid disease, hepatitis, heart disease, or hyperlipidemia.
The patient had been taking ibuprofen as needed since the spinning class. She was taking no other medications. She denied any allergies to drugs or food.
The patient admitted to smoking one pack of cigarettes per week and to occasional alcohol consumption but denied use of illicit drugs. She was employed as an executive officer for a large business association.
On physical examination, the patient’s vital signs were blood pressure, 134/73 mm/Hg; pulse, 86 beats/min; and respirations, 16 breaths/min. She was afebrile, alert, and oriented. Her sclera were nonicteric. Her neck was supple with no anterior cervical lymphadenopathy. There was no thyroid enlargement, her lungs were clear to auscultation, and her heart sounds were regular. There was no peripheral edema, and dorsalis pedis pulses were present bilaterally. Her thighs appeared swollen but were not tender to palpation.
The patient’s history, combined with an extremely high level of serum creatine phosphokinase (CPK; ie, 123,800 U/L [reference range, 45 to 260 U/L1]), confirmed a diagnosis of rhabdomyolysis. She was admitted for close observation. The patient’s urinalysis revealed 2 to 5 red blood cells and 6 to 10 white blood cells per high-power field. Moderate occult blood was detected, with no casts or protein noted. A urine myoglobin test was not performed.
The patient underwent IV hydration with dextrose 5% in water and three ampules of sodium bicarbonate after being given a 2.0-L saline bolus. Ibuprofen was discontinued. IV hydration with bicarbonate solution was continued until the patient’s CPK level declined significantly. She underwent daily laboratory testing (see Table 1). Her renal function remained stable, and she was discharged on hospital day 7.
DISCUSSION
Rhabdomyolysis is a clinical condition defined as muscle necrosis resulting from the release of intracellular skeletal muscle components (including myoglobin, CPK, potassium, phosphorus, and aldolase) into the extracellular compartment.1-3 The condition was first described during the bombing of London in World War II, with high incidence of crush injuries, shock, and associated kidney damage.4 The preponderance of such injuries during a 1988 earthquake in Armenia led the International Society of Nephrology to form its Renal Disaster Relief Task Force, which has provided support at numerous other disaster scenes since then.5
Rhabdomyolysis has been identified with a variety of pathologic events: those that cause muscle trauma, those associated with muscle use or overuse, and other etiologies involving genetic, metabolic, infectious, or pharmaceutical factors.1 Many of the reported causes of rhabdomyolysis are listed in Table 2.1,2
For patients with muscle trauma, the etiology of rhabdomyolysis is clear, but for those with other disease states, diagnosis may be more elusive. Patients who present with rhabdomyolysis after excessive exercise, for example, may have underlying metabolic disorders that predispose them to exertional rhabdomyolysis, such as chronic hypokalemia resulting from primary hyperaldosteronism.1 Others may have a muscle enzyme deficiency, as in McArdle’s syndrome or carnitine deficiency.6
Alterations in blood chemistries can also contribute to development of rhabdomyolysis, even when more obvious etiologies for muscle necrosis are evident. Hypokalemia interferes with the vasodilation that normally occurs during exercise to increase muscle blood flow.1,7,8 Continued exercise can lead to muscle necrosis, raising a concern for athletes who take diuretics.1 Hypophosphatemia leads to a state of muscle necrosis; this is of particular concern for alcoholic patients who receive hyperalimentation without repletion of phosphates.9
Diagnosis
Patients with rhabdomyolysis usually present with myalgias, darkened urine (red, brown, or black), and a clinical scenario that corroborates the diagnosis (ie, history of trauma, excessive exercise, use of an offending medication).1 Some patients may have minimal to absent symptoms or symptoms that occur only after exercise.3
A careful history is key. While traumatic causes are obvious, it is important to ask a patient with rhabdomyolysis after exertion about previous history of excessive weakness during or immediately after exercise, excessive cramping, or discoloration of urine after exercise. The family history may point to a genetic abnormality. A thorough understanding of the patient’s use of medications, including OTC agents, is also important. Rhabdomyolysis has been reported in patients who use herbal remedies, including those taken to facilitate weight loss or to improve lipid profiles.10,11
For patients suspected of having rhabdomyolysis, a serum CPK level should be obtained; results exceeding normal values by five times confirm the diagnosis.3 Measurements for potassium, phosphorus, and calcium are also important to determine, as is renal function. A high level of serum aldolase (an enzyme that breaks down glucose in muscle tissue) can also support a diagnosis of rhabdomyolysis.1,12 Urinalysis and urine myoglobin testing are also warranted, although a negative urine myoglobin test result does not rule out rhabdomyolysis in the presence of an elevated CPK level. Myoglobin is cleared rapidly by the kidneys, whereas serum CPK levels change slowly.1
Any patient who presents with acute rhabdomyolysis and low to normal values for potassium or phosphate should be evaluated further for hypokalemia and hypophosphatemia as contributing or etiologic factors. Hypocalcemia may occur in the early course of rhabdomyolysis as calcium salt is deposited in muscle tissue. Patients recovering from rhabdomyolysis may experience rebound hypercalcemia as the damaged muscle releases the deposited calcium.7,13
In most cases of rhabdomyolysis, only laboratory values are needed to make the diagnosis and follow the course of the episode.1 However, when the etiology appears to involve metabolic deficiencies or genetic etiologies, it may become necessary to order additional diagnostic tests. These may include tests for thyroid function, a carnitine level to screen for glycogen storage diseases, and toxin screening (eg, for illicit drugs, such as cocaine).2,6
Treatment and Management
Effective treatment of rhabdomyolysis relies on recognizing the underlying disorder.1 For patients with muscle trauma (eg, crush injury) or muscle overuse, the mainstay of treatment is aggressive fluid resuscitation and prevention of acute injury to the kidneys.13 As for patients with an injury induced by a pharmaceutical agent or a toxin, removal of the offending agent is required, followed by hydration and prevention of renal damage. Supportive care during an infectious illness is also essential.14
Additionally, treatment must address the complications inherent with rhabdomyolysis.1 In addition to CPK, potassium, phosphorus, and myoglobin are also released from skeletal muscle tissue. Hyperkalemia can be fatal, and potassium levels must be monitored closely to avert this condition.7,8,13 During an episode of rhabdomyolysis, normal levels of both potassium and phosphorus should raise the clinician’s suspicion for underlying hypokalemia and hypophosphatemia—conditions that may have contributed to the episode of rhabdomyolysis. Hypocalcemia may also develop.13
Released myoglobin may cause acute kidney injury, as is the case in 33% to 50% of patients with rhabdomyolysis.3 In early studies, it was determined that alkalinizing the urine with IV isotonic bicarbonate might thwart onset of acute kidney injury.1,2,15 Time is critical, and even on the battlefield or at the scene of a recent disaster, most attempts at resuscitation are begun immediately. IV access may be problematic, but administration of oral bicarbonate solutions has also proven effective.15 Close follow-up of the serum urea and creatinine levels and measurement of the urine pH during alkalinization is warranted throughout the course of the episode.
Unfortunately, some patients respond poorly to these conservative measures, and the released myoglobin can cause renal tubular blockage and necrosis, resulting in acute kidney injury.1 Renal replacement therapy may be required.16 However, most episodes of dialysis-dependent acute renal injury do subside with time.
For patients with less elusive causes of rhabdomyolysis, treatment will hinge on a workup of the possible etiologies and follow-up treatment to target the apparent cause. For example, carnitine may be administered to patients with carnitine deficiency, and hypokalemic patients may be given potassium.1,6,7 These patients will also need counseling before they consider engaging in an exercise program.
Patient’s Outcome
The case patient presented with exertional rhabdomyolysis; improper hydration, severe deconditioning, and a relatively low serum potassium level may all have contributed to the muscle necrosis she experienced. She was given IV alkaline solutions and did not develop acute kidney injury. She was discharged from the hospital and at the time of this writing was awaiting outpatient follow-up.
It should be interesting to see whether the case patient experiences any further episodes of severe weakness after engaging in exercise. Her low-normal potassium level (reference range, 3.5 to 5.3 mmol/L17) warrants further follow-up, as does her mildly elevated thyroid-stimulating hormone level (reference range, 0.5 to 4.7 mcIU/mL17).
CONCLUSION
Patients with rhabdomyolysis may present with muscle aches, darkened urine, and/or weakness; an elevated CPK level confirms the diagnosis. Management is mainly conservative, with IV hydration accompanied by alkalinizing the urine and correcting any metabolic abnormalities, such as potassium deficiencies. For the few patients who experience severe acute kidney injury, renal replacement therapy may be necessary.
While most causes of rhabdomyolysis have obvious clinical scenarios, such as a crush injury, a search for muscle enzyme deficiencies, disorders of potassium homeostasis, and thyroid abnormalities is also warranted in patients who present with exertional rhabdomyolysis.
1. Huerta-Alardín AL, Varon J, Marik PE. Bench-to-bedside review: rhabdomyolysis—an overview for clinicians. Crit Care. 2005;9(2):158-169.
2. Warren JD, Blumbergs PC, Thompson PD. Rhabdomyolysis: a review. Muscle Nerve. 2002;25(3):332-347.
3. Lima RSA, da Silva GB Jr, Liborio AB, Daher ED. Acute kidney injury due to rhabdomyolysis. Saudi J Kidney Dis Transpl. 2008;19(5):721-729.
4. Bywaters EG, Beall D. Crush injuries with impairment of renal function [reprinted from BMJ, 1941]. J Am Soc Nephrol. 1998;9(2):322-332.
5. Vanholder R, Van Biesen W, Lameire N, Sever MS; International Society of Nephrology/Renal Disaster Relief Task Force. The role of the International Society of Nephrology/Renal Disaster Relief Task Force in the rescue of renal disaster victims. Contrib Nephrol. 2007;156:325-332.
6. Toledo R, López V, Martín G, et al. Rhabdomyolysis due to enzyme deficiency in muscles. Nefrología. 2009;29(1):77-80.
7. Agrawal S, Agrawal V, Taneja A. Hypokalemia causing rhabdomyolysis resulting in life-threatening hyperkalemia. Pediatr Nephrol. 2006;221(2): 289-291.
8. Knochel JP, Schlein EM. On the mechanism of rhabdomyolysis in potassium depletion. J Clin Invest. 1972:51(7):1750-1758.
9. Knochel JP. Hypophosphatemia and rhabdomyolysis. Am J Med. 1992;92(5):455-457.
10. Mansi IA, Huang J. Rhabdomyolysis in response to weight-loss herbal medicine. Am J Med Sci. 2004; 327(6): 356-357.
11. Heber D, Yip I, Ashley JM, et al. Cholesterol-lowering effects of a proprietary Chinese red-yeast-rice dietary supplement. Am J Clin Nutr. 1999;69(2): 231-236.
12. Hooda AK, Narula AS. Exertional rhabdomyolysis causing acute renal failure. Med J Armed Forces India. 2005;61(4):395-396.
13. Chatzizisis YS, Misirli G, Hatzitolios AI, Giannoglou GD. The syndrome of rhabdomyolysis: complications and treatment. Eur J Intern Med. 2008;19(8): 568-574.
14. Blanco JR, Zabalza M, Salcedo J, et al. Rhabdomyolysis of infectious and noninfectious causes. South Med J. 2002;95(5):542-544.
15. Ron D, Taitelman U, Michaelson M, et al. Prevention of acute renal failure in traumatic rhabdomyolysis. Arch Intern Med. 1984;144(2):277-280.
16. Soni SS, Nagarik AP, Adikey GK, Raman A. Using continuous renal replacement therapy to manage patients of shock and acute renal failure. J Emerg Trauma Shock. 2009;2(1):19-22.
17. Normal laboratory values. In: Beers MH, Berkow R, eds. The Merck Manual of Diagnosis and Therapy. 17th ed. Whitehouse Station, NJ: Merck Research Laboratories; 1999:2526-2546.
A 39-year-old woman presented to the emergency department (ED) with a chief complaint of muscle aches and pain. She stated that three days earlier, she had begun exercising in a 45-minute “spinning” class (ie, riding a stationary bicycle with a weighted front wheel). The patient had not engaged in any aerobic exercise for at least six months before the spinning class. She mentioned that much older participants in the class were outperforming her, but she did not feel the need to keep up with them.
After dismounting, the woman said, she experienced weakness in her legs and had great difficulty ambulating. She went home, took 400 mg of ibuprofen, and went to bed. She awoke with pain and swelling in both thighs and continued to take ibuprofen, in addition to applying a topical mentholated preparation to her thighs. She took an Epsom salts bath two days later.
On the morning of the third day after the spinning class, she voided black urine and presented to the ED.
The patient had no significant medical history. Surgical history was limited to removal of a ganglion cyst on her wrist. She denied any history of seizure disorder, thyroid disease, hepatitis, heart disease, or hyperlipidemia.
The patient had been taking ibuprofen as needed since the spinning class. She was taking no other medications. She denied any allergies to drugs or food.
The patient admitted to smoking one pack of cigarettes per week and to occasional alcohol consumption but denied use of illicit drugs. She was employed as an executive officer for a large business association.
On physical examination, the patient’s vital signs were blood pressure, 134/73 mm/Hg; pulse, 86 beats/min; and respirations, 16 breaths/min. She was afebrile, alert, and oriented. Her sclera were nonicteric. Her neck was supple with no anterior cervical lymphadenopathy. There was no thyroid enlargement, her lungs were clear to auscultation, and her heart sounds were regular. There was no peripheral edema, and dorsalis pedis pulses were present bilaterally. Her thighs appeared swollen but were not tender to palpation.
The patient’s history, combined with an extremely high level of serum creatine phosphokinase (CPK; ie, 123,800 U/L [reference range, 45 to 260 U/L1]), confirmed a diagnosis of rhabdomyolysis. She was admitted for close observation. The patient’s urinalysis revealed 2 to 5 red blood cells and 6 to 10 white blood cells per high-power field. Moderate occult blood was detected, with no casts or protein noted. A urine myoglobin test was not performed.
The patient underwent IV hydration with dextrose 5% in water and three ampules of sodium bicarbonate after being given a 2.0-L saline bolus. Ibuprofen was discontinued. IV hydration with bicarbonate solution was continued until the patient’s CPK level declined significantly. She underwent daily laboratory testing (see Table 1). Her renal function remained stable, and she was discharged on hospital day 7.
DISCUSSION
Rhabdomyolysis is a clinical condition defined as muscle necrosis resulting from the release of intracellular skeletal muscle components (including myoglobin, CPK, potassium, phosphorus, and aldolase) into the extracellular compartment.1-3 The condition was first described during the bombing of London in World War II, with high incidence of crush injuries, shock, and associated kidney damage.4 The preponderance of such injuries during a 1988 earthquake in Armenia led the International Society of Nephrology to form its Renal Disaster Relief Task Force, which has provided support at numerous other disaster scenes since then.5
Rhabdomyolysis has been identified with a variety of pathologic events: those that cause muscle trauma, those associated with muscle use or overuse, and other etiologies involving genetic, metabolic, infectious, or pharmaceutical factors.1 Many of the reported causes of rhabdomyolysis are listed in Table 2.1,2
For patients with muscle trauma, the etiology of rhabdomyolysis is clear, but for those with other disease states, diagnosis may be more elusive. Patients who present with rhabdomyolysis after excessive exercise, for example, may have underlying metabolic disorders that predispose them to exertional rhabdomyolysis, such as chronic hypokalemia resulting from primary hyperaldosteronism.1 Others may have a muscle enzyme deficiency, as in McArdle’s syndrome or carnitine deficiency.6
Alterations in blood chemistries can also contribute to development of rhabdomyolysis, even when more obvious etiologies for muscle necrosis are evident. Hypokalemia interferes with the vasodilation that normally occurs during exercise to increase muscle blood flow.1,7,8 Continued exercise can lead to muscle necrosis, raising a concern for athletes who take diuretics.1 Hypophosphatemia leads to a state of muscle necrosis; this is of particular concern for alcoholic patients who receive hyperalimentation without repletion of phosphates.9
Diagnosis
Patients with rhabdomyolysis usually present with myalgias, darkened urine (red, brown, or black), and a clinical scenario that corroborates the diagnosis (ie, history of trauma, excessive exercise, use of an offending medication).1 Some patients may have minimal to absent symptoms or symptoms that occur only after exercise.3
A careful history is key. While traumatic causes are obvious, it is important to ask a patient with rhabdomyolysis after exertion about previous history of excessive weakness during or immediately after exercise, excessive cramping, or discoloration of urine after exercise. The family history may point to a genetic abnormality. A thorough understanding of the patient’s use of medications, including OTC agents, is also important. Rhabdomyolysis has been reported in patients who use herbal remedies, including those taken to facilitate weight loss or to improve lipid profiles.10,11
For patients suspected of having rhabdomyolysis, a serum CPK level should be obtained; results exceeding normal values by five times confirm the diagnosis.3 Measurements for potassium, phosphorus, and calcium are also important to determine, as is renal function. A high level of serum aldolase (an enzyme that breaks down glucose in muscle tissue) can also support a diagnosis of rhabdomyolysis.1,12 Urinalysis and urine myoglobin testing are also warranted, although a negative urine myoglobin test result does not rule out rhabdomyolysis in the presence of an elevated CPK level. Myoglobin is cleared rapidly by the kidneys, whereas serum CPK levels change slowly.1
Any patient who presents with acute rhabdomyolysis and low to normal values for potassium or phosphate should be evaluated further for hypokalemia and hypophosphatemia as contributing or etiologic factors. Hypocalcemia may occur in the early course of rhabdomyolysis as calcium salt is deposited in muscle tissue. Patients recovering from rhabdomyolysis may experience rebound hypercalcemia as the damaged muscle releases the deposited calcium.7,13
In most cases of rhabdomyolysis, only laboratory values are needed to make the diagnosis and follow the course of the episode.1 However, when the etiology appears to involve metabolic deficiencies or genetic etiologies, it may become necessary to order additional diagnostic tests. These may include tests for thyroid function, a carnitine level to screen for glycogen storage diseases, and toxin screening (eg, for illicit drugs, such as cocaine).2,6
Treatment and Management
Effective treatment of rhabdomyolysis relies on recognizing the underlying disorder.1 For patients with muscle trauma (eg, crush injury) or muscle overuse, the mainstay of treatment is aggressive fluid resuscitation and prevention of acute injury to the kidneys.13 As for patients with an injury induced by a pharmaceutical agent or a toxin, removal of the offending agent is required, followed by hydration and prevention of renal damage. Supportive care during an infectious illness is also essential.14
Additionally, treatment must address the complications inherent with rhabdomyolysis.1 In addition to CPK, potassium, phosphorus, and myoglobin are also released from skeletal muscle tissue. Hyperkalemia can be fatal, and potassium levels must be monitored closely to avert this condition.7,8,13 During an episode of rhabdomyolysis, normal levels of both potassium and phosphorus should raise the clinician’s suspicion for underlying hypokalemia and hypophosphatemia—conditions that may have contributed to the episode of rhabdomyolysis. Hypocalcemia may also develop.13
Released myoglobin may cause acute kidney injury, as is the case in 33% to 50% of patients with rhabdomyolysis.3 In early studies, it was determined that alkalinizing the urine with IV isotonic bicarbonate might thwart onset of acute kidney injury.1,2,15 Time is critical, and even on the battlefield or at the scene of a recent disaster, most attempts at resuscitation are begun immediately. IV access may be problematic, but administration of oral bicarbonate solutions has also proven effective.15 Close follow-up of the serum urea and creatinine levels and measurement of the urine pH during alkalinization is warranted throughout the course of the episode.
Unfortunately, some patients respond poorly to these conservative measures, and the released myoglobin can cause renal tubular blockage and necrosis, resulting in acute kidney injury.1 Renal replacement therapy may be required.16 However, most episodes of dialysis-dependent acute renal injury do subside with time.
For patients with less elusive causes of rhabdomyolysis, treatment will hinge on a workup of the possible etiologies and follow-up treatment to target the apparent cause. For example, carnitine may be administered to patients with carnitine deficiency, and hypokalemic patients may be given potassium.1,6,7 These patients will also need counseling before they consider engaging in an exercise program.
Patient’s Outcome
The case patient presented with exertional rhabdomyolysis; improper hydration, severe deconditioning, and a relatively low serum potassium level may all have contributed to the muscle necrosis she experienced. She was given IV alkaline solutions and did not develop acute kidney injury. She was discharged from the hospital and at the time of this writing was awaiting outpatient follow-up.
It should be interesting to see whether the case patient experiences any further episodes of severe weakness after engaging in exercise. Her low-normal potassium level (reference range, 3.5 to 5.3 mmol/L17) warrants further follow-up, as does her mildly elevated thyroid-stimulating hormone level (reference range, 0.5 to 4.7 mcIU/mL17).
CONCLUSION
Patients with rhabdomyolysis may present with muscle aches, darkened urine, and/or weakness; an elevated CPK level confirms the diagnosis. Management is mainly conservative, with IV hydration accompanied by alkalinizing the urine and correcting any metabolic abnormalities, such as potassium deficiencies. For the few patients who experience severe acute kidney injury, renal replacement therapy may be necessary.
While most causes of rhabdomyolysis have obvious clinical scenarios, such as a crush injury, a search for muscle enzyme deficiencies, disorders of potassium homeostasis, and thyroid abnormalities is also warranted in patients who present with exertional rhabdomyolysis.
A 39-year-old woman presented to the emergency department (ED) with a chief complaint of muscle aches and pain. She stated that three days earlier, she had begun exercising in a 45-minute “spinning” class (ie, riding a stationary bicycle with a weighted front wheel). The patient had not engaged in any aerobic exercise for at least six months before the spinning class. She mentioned that much older participants in the class were outperforming her, but she did not feel the need to keep up with them.
After dismounting, the woman said, she experienced weakness in her legs and had great difficulty ambulating. She went home, took 400 mg of ibuprofen, and went to bed. She awoke with pain and swelling in both thighs and continued to take ibuprofen, in addition to applying a topical mentholated preparation to her thighs. She took an Epsom salts bath two days later.
On the morning of the third day after the spinning class, she voided black urine and presented to the ED.
The patient had no significant medical history. Surgical history was limited to removal of a ganglion cyst on her wrist. She denied any history of seizure disorder, thyroid disease, hepatitis, heart disease, or hyperlipidemia.
The patient had been taking ibuprofen as needed since the spinning class. She was taking no other medications. She denied any allergies to drugs or food.
The patient admitted to smoking one pack of cigarettes per week and to occasional alcohol consumption but denied use of illicit drugs. She was employed as an executive officer for a large business association.
On physical examination, the patient’s vital signs were blood pressure, 134/73 mm/Hg; pulse, 86 beats/min; and respirations, 16 breaths/min. She was afebrile, alert, and oriented. Her sclera were nonicteric. Her neck was supple with no anterior cervical lymphadenopathy. There was no thyroid enlargement, her lungs were clear to auscultation, and her heart sounds were regular. There was no peripheral edema, and dorsalis pedis pulses were present bilaterally. Her thighs appeared swollen but were not tender to palpation.
The patient’s history, combined with an extremely high level of serum creatine phosphokinase (CPK; ie, 123,800 U/L [reference range, 45 to 260 U/L1]), confirmed a diagnosis of rhabdomyolysis. She was admitted for close observation. The patient’s urinalysis revealed 2 to 5 red blood cells and 6 to 10 white blood cells per high-power field. Moderate occult blood was detected, with no casts or protein noted. A urine myoglobin test was not performed.
The patient underwent IV hydration with dextrose 5% in water and three ampules of sodium bicarbonate after being given a 2.0-L saline bolus. Ibuprofen was discontinued. IV hydration with bicarbonate solution was continued until the patient’s CPK level declined significantly. She underwent daily laboratory testing (see Table 1). Her renal function remained stable, and she was discharged on hospital day 7.
DISCUSSION
Rhabdomyolysis is a clinical condition defined as muscle necrosis resulting from the release of intracellular skeletal muscle components (including myoglobin, CPK, potassium, phosphorus, and aldolase) into the extracellular compartment.1-3 The condition was first described during the bombing of London in World War II, with high incidence of crush injuries, shock, and associated kidney damage.4 The preponderance of such injuries during a 1988 earthquake in Armenia led the International Society of Nephrology to form its Renal Disaster Relief Task Force, which has provided support at numerous other disaster scenes since then.5
Rhabdomyolysis has been identified with a variety of pathologic events: those that cause muscle trauma, those associated with muscle use or overuse, and other etiologies involving genetic, metabolic, infectious, or pharmaceutical factors.1 Many of the reported causes of rhabdomyolysis are listed in Table 2.1,2
For patients with muscle trauma, the etiology of rhabdomyolysis is clear, but for those with other disease states, diagnosis may be more elusive. Patients who present with rhabdomyolysis after excessive exercise, for example, may have underlying metabolic disorders that predispose them to exertional rhabdomyolysis, such as chronic hypokalemia resulting from primary hyperaldosteronism.1 Others may have a muscle enzyme deficiency, as in McArdle’s syndrome or carnitine deficiency.6
Alterations in blood chemistries can also contribute to development of rhabdomyolysis, even when more obvious etiologies for muscle necrosis are evident. Hypokalemia interferes with the vasodilation that normally occurs during exercise to increase muscle blood flow.1,7,8 Continued exercise can lead to muscle necrosis, raising a concern for athletes who take diuretics.1 Hypophosphatemia leads to a state of muscle necrosis; this is of particular concern for alcoholic patients who receive hyperalimentation without repletion of phosphates.9
Diagnosis
Patients with rhabdomyolysis usually present with myalgias, darkened urine (red, brown, or black), and a clinical scenario that corroborates the diagnosis (ie, history of trauma, excessive exercise, use of an offending medication).1 Some patients may have minimal to absent symptoms or symptoms that occur only after exercise.3
A careful history is key. While traumatic causes are obvious, it is important to ask a patient with rhabdomyolysis after exertion about previous history of excessive weakness during or immediately after exercise, excessive cramping, or discoloration of urine after exercise. The family history may point to a genetic abnormality. A thorough understanding of the patient’s use of medications, including OTC agents, is also important. Rhabdomyolysis has been reported in patients who use herbal remedies, including those taken to facilitate weight loss or to improve lipid profiles.10,11
For patients suspected of having rhabdomyolysis, a serum CPK level should be obtained; results exceeding normal values by five times confirm the diagnosis.3 Measurements for potassium, phosphorus, and calcium are also important to determine, as is renal function. A high level of serum aldolase (an enzyme that breaks down glucose in muscle tissue) can also support a diagnosis of rhabdomyolysis.1,12 Urinalysis and urine myoglobin testing are also warranted, although a negative urine myoglobin test result does not rule out rhabdomyolysis in the presence of an elevated CPK level. Myoglobin is cleared rapidly by the kidneys, whereas serum CPK levels change slowly.1
Any patient who presents with acute rhabdomyolysis and low to normal values for potassium or phosphate should be evaluated further for hypokalemia and hypophosphatemia as contributing or etiologic factors. Hypocalcemia may occur in the early course of rhabdomyolysis as calcium salt is deposited in muscle tissue. Patients recovering from rhabdomyolysis may experience rebound hypercalcemia as the damaged muscle releases the deposited calcium.7,13
In most cases of rhabdomyolysis, only laboratory values are needed to make the diagnosis and follow the course of the episode.1 However, when the etiology appears to involve metabolic deficiencies or genetic etiologies, it may become necessary to order additional diagnostic tests. These may include tests for thyroid function, a carnitine level to screen for glycogen storage diseases, and toxin screening (eg, for illicit drugs, such as cocaine).2,6
Treatment and Management
Effective treatment of rhabdomyolysis relies on recognizing the underlying disorder.1 For patients with muscle trauma (eg, crush injury) or muscle overuse, the mainstay of treatment is aggressive fluid resuscitation and prevention of acute injury to the kidneys.13 As for patients with an injury induced by a pharmaceutical agent or a toxin, removal of the offending agent is required, followed by hydration and prevention of renal damage. Supportive care during an infectious illness is also essential.14
Additionally, treatment must address the complications inherent with rhabdomyolysis.1 In addition to CPK, potassium, phosphorus, and myoglobin are also released from skeletal muscle tissue. Hyperkalemia can be fatal, and potassium levels must be monitored closely to avert this condition.7,8,13 During an episode of rhabdomyolysis, normal levels of both potassium and phosphorus should raise the clinician’s suspicion for underlying hypokalemia and hypophosphatemia—conditions that may have contributed to the episode of rhabdomyolysis. Hypocalcemia may also develop.13
Released myoglobin may cause acute kidney injury, as is the case in 33% to 50% of patients with rhabdomyolysis.3 In early studies, it was determined that alkalinizing the urine with IV isotonic bicarbonate might thwart onset of acute kidney injury.1,2,15 Time is critical, and even on the battlefield or at the scene of a recent disaster, most attempts at resuscitation are begun immediately. IV access may be problematic, but administration of oral bicarbonate solutions has also proven effective.15 Close follow-up of the serum urea and creatinine levels and measurement of the urine pH during alkalinization is warranted throughout the course of the episode.
Unfortunately, some patients respond poorly to these conservative measures, and the released myoglobin can cause renal tubular blockage and necrosis, resulting in acute kidney injury.1 Renal replacement therapy may be required.16 However, most episodes of dialysis-dependent acute renal injury do subside with time.
For patients with less elusive causes of rhabdomyolysis, treatment will hinge on a workup of the possible etiologies and follow-up treatment to target the apparent cause. For example, carnitine may be administered to patients with carnitine deficiency, and hypokalemic patients may be given potassium.1,6,7 These patients will also need counseling before they consider engaging in an exercise program.
Patient’s Outcome
The case patient presented with exertional rhabdomyolysis; improper hydration, severe deconditioning, and a relatively low serum potassium level may all have contributed to the muscle necrosis she experienced. She was given IV alkaline solutions and did not develop acute kidney injury. She was discharged from the hospital and at the time of this writing was awaiting outpatient follow-up.
It should be interesting to see whether the case patient experiences any further episodes of severe weakness after engaging in exercise. Her low-normal potassium level (reference range, 3.5 to 5.3 mmol/L17) warrants further follow-up, as does her mildly elevated thyroid-stimulating hormone level (reference range, 0.5 to 4.7 mcIU/mL17).
CONCLUSION
Patients with rhabdomyolysis may present with muscle aches, darkened urine, and/or weakness; an elevated CPK level confirms the diagnosis. Management is mainly conservative, with IV hydration accompanied by alkalinizing the urine and correcting any metabolic abnormalities, such as potassium deficiencies. For the few patients who experience severe acute kidney injury, renal replacement therapy may be necessary.
While most causes of rhabdomyolysis have obvious clinical scenarios, such as a crush injury, a search for muscle enzyme deficiencies, disorders of potassium homeostasis, and thyroid abnormalities is also warranted in patients who present with exertional rhabdomyolysis.
1. Huerta-Alardín AL, Varon J, Marik PE. Bench-to-bedside review: rhabdomyolysis—an overview for clinicians. Crit Care. 2005;9(2):158-169.
2. Warren JD, Blumbergs PC, Thompson PD. Rhabdomyolysis: a review. Muscle Nerve. 2002;25(3):332-347.
3. Lima RSA, da Silva GB Jr, Liborio AB, Daher ED. Acute kidney injury due to rhabdomyolysis. Saudi J Kidney Dis Transpl. 2008;19(5):721-729.
4. Bywaters EG, Beall D. Crush injuries with impairment of renal function [reprinted from BMJ, 1941]. J Am Soc Nephrol. 1998;9(2):322-332.
5. Vanholder R, Van Biesen W, Lameire N, Sever MS; International Society of Nephrology/Renal Disaster Relief Task Force. The role of the International Society of Nephrology/Renal Disaster Relief Task Force in the rescue of renal disaster victims. Contrib Nephrol. 2007;156:325-332.
6. Toledo R, López V, Martín G, et al. Rhabdomyolysis due to enzyme deficiency in muscles. Nefrología. 2009;29(1):77-80.
7. Agrawal S, Agrawal V, Taneja A. Hypokalemia causing rhabdomyolysis resulting in life-threatening hyperkalemia. Pediatr Nephrol. 2006;221(2): 289-291.
8. Knochel JP, Schlein EM. On the mechanism of rhabdomyolysis in potassium depletion. J Clin Invest. 1972:51(7):1750-1758.
9. Knochel JP. Hypophosphatemia and rhabdomyolysis. Am J Med. 1992;92(5):455-457.
10. Mansi IA, Huang J. Rhabdomyolysis in response to weight-loss herbal medicine. Am J Med Sci. 2004; 327(6): 356-357.
11. Heber D, Yip I, Ashley JM, et al. Cholesterol-lowering effects of a proprietary Chinese red-yeast-rice dietary supplement. Am J Clin Nutr. 1999;69(2): 231-236.
12. Hooda AK, Narula AS. Exertional rhabdomyolysis causing acute renal failure. Med J Armed Forces India. 2005;61(4):395-396.
13. Chatzizisis YS, Misirli G, Hatzitolios AI, Giannoglou GD. The syndrome of rhabdomyolysis: complications and treatment. Eur J Intern Med. 2008;19(8): 568-574.
14. Blanco JR, Zabalza M, Salcedo J, et al. Rhabdomyolysis of infectious and noninfectious causes. South Med J. 2002;95(5):542-544.
15. Ron D, Taitelman U, Michaelson M, et al. Prevention of acute renal failure in traumatic rhabdomyolysis. Arch Intern Med. 1984;144(2):277-280.
16. Soni SS, Nagarik AP, Adikey GK, Raman A. Using continuous renal replacement therapy to manage patients of shock and acute renal failure. J Emerg Trauma Shock. 2009;2(1):19-22.
17. Normal laboratory values. In: Beers MH, Berkow R, eds. The Merck Manual of Diagnosis and Therapy. 17th ed. Whitehouse Station, NJ: Merck Research Laboratories; 1999:2526-2546.
1. Huerta-Alardín AL, Varon J, Marik PE. Bench-to-bedside review: rhabdomyolysis—an overview for clinicians. Crit Care. 2005;9(2):158-169.
2. Warren JD, Blumbergs PC, Thompson PD. Rhabdomyolysis: a review. Muscle Nerve. 2002;25(3):332-347.
3. Lima RSA, da Silva GB Jr, Liborio AB, Daher ED. Acute kidney injury due to rhabdomyolysis. Saudi J Kidney Dis Transpl. 2008;19(5):721-729.
4. Bywaters EG, Beall D. Crush injuries with impairment of renal function [reprinted from BMJ, 1941]. J Am Soc Nephrol. 1998;9(2):322-332.
5. Vanholder R, Van Biesen W, Lameire N, Sever MS; International Society of Nephrology/Renal Disaster Relief Task Force. The role of the International Society of Nephrology/Renal Disaster Relief Task Force in the rescue of renal disaster victims. Contrib Nephrol. 2007;156:325-332.
6. Toledo R, López V, Martín G, et al. Rhabdomyolysis due to enzyme deficiency in muscles. Nefrología. 2009;29(1):77-80.
7. Agrawal S, Agrawal V, Taneja A. Hypokalemia causing rhabdomyolysis resulting in life-threatening hyperkalemia. Pediatr Nephrol. 2006;221(2): 289-291.
8. Knochel JP, Schlein EM. On the mechanism of rhabdomyolysis in potassium depletion. J Clin Invest. 1972:51(7):1750-1758.
9. Knochel JP. Hypophosphatemia and rhabdomyolysis. Am J Med. 1992;92(5):455-457.
10. Mansi IA, Huang J. Rhabdomyolysis in response to weight-loss herbal medicine. Am J Med Sci. 2004; 327(6): 356-357.
11. Heber D, Yip I, Ashley JM, et al. Cholesterol-lowering effects of a proprietary Chinese red-yeast-rice dietary supplement. Am J Clin Nutr. 1999;69(2): 231-236.
12. Hooda AK, Narula AS. Exertional rhabdomyolysis causing acute renal failure. Med J Armed Forces India. 2005;61(4):395-396.
13. Chatzizisis YS, Misirli G, Hatzitolios AI, Giannoglou GD. The syndrome of rhabdomyolysis: complications and treatment. Eur J Intern Med. 2008;19(8): 568-574.
14. Blanco JR, Zabalza M, Salcedo J, et al. Rhabdomyolysis of infectious and noninfectious causes. South Med J. 2002;95(5):542-544.
15. Ron D, Taitelman U, Michaelson M, et al. Prevention of acute renal failure in traumatic rhabdomyolysis. Arch Intern Med. 1984;144(2):277-280.
16. Soni SS, Nagarik AP, Adikey GK, Raman A. Using continuous renal replacement therapy to manage patients of shock and acute renal failure. J Emerg Trauma Shock. 2009;2(1):19-22.
17. Normal laboratory values. In: Beers MH, Berkow R, eds. The Merck Manual of Diagnosis and Therapy. 17th ed. Whitehouse Station, NJ: Merck Research Laboratories; 1999:2526-2546.
Nephrogenic Systemic Fibrosis
Recurrent pyelonephritis as a sign of ‘sponge kidney’
KEY FEATURES
Medullary sponge kidney causes extensive cystic dilation of medullary collecting tubules.1 It is usually an incidental finding in patients undergoing intravenous urography as part of the evaluation for infection, hematuria, or kidney stones.
The classic urographic appearance is linear striations with small brushes or “bouquets of flowers,” which represent the collection of contrast material in small papillary cysts.
Medullary sponge kidney has long been considered a congenital disorder, but the genetic defect has not yet been identified, and the pathogenesis is not yet known. It has only rarely been reported in children.2
It is usually asymptomatic, but complications may occur, including nephrocalcinosis or lithiasis, urinary tract infection, renal tubular acidosis, and impaired urine concentrating ability.
RISK OF LITHIASIS
Gambaro et al3 estimated that medullary sponge kidney is found in up to 20% of patients with urolithiasis, and that more than 70% of patients with medullary sponge kidney develop stones.
Cystic dilation of medullary collecting tubules (an anatomic abnormality) inevitably causes urinary stasis. This, combined with hypercalciuria and reduced excretion of urinary citrate and magnesium (metabolic abnormalities), contributes to lithiasis.4 Lithiasis in medullary sponge kidney is a well-known cause of urinary tract infection, and it tends to facilitate infective stone formation after episodes of urinary tract infection.5
The unique anatomic derangement of medullary sponge kidney contributes to the recurrence of pyelonephritis, in addition to the conventional risk factors—frequent sexual intercourse, avoidance of voiding because it is inconvenient, incomplete bladder emptying, ureteropelvic junction obstruction, ectopic ureter, and impaired immunity.6
DIAGNOSIS AND TREATMENT
Intravenous urography is the gold standard for the diagnosis of medullary sponge kidney; computed tomography and ultrasonography are generally limited in their ability to clearly show the tubular ectasia.7
Treatment includes antibiotics for acute pyelonephritis and thiazide diuretics and potassium citrate to prevent stone formation and renal tubular acidosis. Due to its silent course, medullary sponge kidney should be considered not only as a cause of nephrocalcinosis and nephrolithiasis, but also as a distinct entity complicating recurrent pyelonephritis.
- Gambaro G, Feltrin GP, Lupo A, Bonfante L, D'Angelo A, Antonello A. Medullary sponge kidney (Lenarduzzi-Cacchi-Ricci disease): a Padua Medical School discovery in the 1930s. Kidney Int 2006; 69:663–670.
- Kasap B, Soylu A, Oren O, Turkmen M, Kavukcu S. Medullary sponge kidney associated with distal renal tubular acidosis in a 5-year-old girl. Eur J Pediatr 2006; 165:648–651.
- Gambaro G, Fabris A, Puliatta D, Lupo A. Lithiasis in cystic kidney disease and malformations of the urinary tract. Urol Res 2006; 34:102–107.
- O'Neill M, Breslau NA, Pak CY. Metabolic evaluation of nephrolithiasis in patients with medullary sponge kidney. JAMA 1981; 245:1233–1236.
- Miano R, Germani S, Vespasiani G. Stones and urinary tract infections. Urol Int 2007; 79(suppl 1):32–36.
- Scholes D, Hooton TM, Roberts PL, Gupta K, Stapleton AE, Stamm WE. Risk factors associated with acute pyelonephritis in healthy women. Ann Intern Med 2005; 142:20–27.
- Maw AM, Megibow AJ, Grasso M, Goldfarb DS. Diagnosis of medullary sponge kidney by computed tomographic urography. Am J Kidney Dis 2007; 50:146–150.
KEY FEATURES
Medullary sponge kidney causes extensive cystic dilation of medullary collecting tubules.1 It is usually an incidental finding in patients undergoing intravenous urography as part of the evaluation for infection, hematuria, or kidney stones.
The classic urographic appearance is linear striations with small brushes or “bouquets of flowers,” which represent the collection of contrast material in small papillary cysts.
Medullary sponge kidney has long been considered a congenital disorder, but the genetic defect has not yet been identified, and the pathogenesis is not yet known. It has only rarely been reported in children.2
It is usually asymptomatic, but complications may occur, including nephrocalcinosis or lithiasis, urinary tract infection, renal tubular acidosis, and impaired urine concentrating ability.
RISK OF LITHIASIS
Gambaro et al3 estimated that medullary sponge kidney is found in up to 20% of patients with urolithiasis, and that more than 70% of patients with medullary sponge kidney develop stones.
Cystic dilation of medullary collecting tubules (an anatomic abnormality) inevitably causes urinary stasis. This, combined with hypercalciuria and reduced excretion of urinary citrate and magnesium (metabolic abnormalities), contributes to lithiasis.4 Lithiasis in medullary sponge kidney is a well-known cause of urinary tract infection, and it tends to facilitate infective stone formation after episodes of urinary tract infection.5
The unique anatomic derangement of medullary sponge kidney contributes to the recurrence of pyelonephritis, in addition to the conventional risk factors—frequent sexual intercourse, avoidance of voiding because it is inconvenient, incomplete bladder emptying, ureteropelvic junction obstruction, ectopic ureter, and impaired immunity.6
DIAGNOSIS AND TREATMENT
Intravenous urography is the gold standard for the diagnosis of medullary sponge kidney; computed tomography and ultrasonography are generally limited in their ability to clearly show the tubular ectasia.7
Treatment includes antibiotics for acute pyelonephritis and thiazide diuretics and potassium citrate to prevent stone formation and renal tubular acidosis. Due to its silent course, medullary sponge kidney should be considered not only as a cause of nephrocalcinosis and nephrolithiasis, but also as a distinct entity complicating recurrent pyelonephritis.
KEY FEATURES
Medullary sponge kidney causes extensive cystic dilation of medullary collecting tubules.1 It is usually an incidental finding in patients undergoing intravenous urography as part of the evaluation for infection, hematuria, or kidney stones.
The classic urographic appearance is linear striations with small brushes or “bouquets of flowers,” which represent the collection of contrast material in small papillary cysts.
Medullary sponge kidney has long been considered a congenital disorder, but the genetic defect has not yet been identified, and the pathogenesis is not yet known. It has only rarely been reported in children.2
It is usually asymptomatic, but complications may occur, including nephrocalcinosis or lithiasis, urinary tract infection, renal tubular acidosis, and impaired urine concentrating ability.
RISK OF LITHIASIS
Gambaro et al3 estimated that medullary sponge kidney is found in up to 20% of patients with urolithiasis, and that more than 70% of patients with medullary sponge kidney develop stones.
Cystic dilation of medullary collecting tubules (an anatomic abnormality) inevitably causes urinary stasis. This, combined with hypercalciuria and reduced excretion of urinary citrate and magnesium (metabolic abnormalities), contributes to lithiasis.4 Lithiasis in medullary sponge kidney is a well-known cause of urinary tract infection, and it tends to facilitate infective stone formation after episodes of urinary tract infection.5
The unique anatomic derangement of medullary sponge kidney contributes to the recurrence of pyelonephritis, in addition to the conventional risk factors—frequent sexual intercourse, avoidance of voiding because it is inconvenient, incomplete bladder emptying, ureteropelvic junction obstruction, ectopic ureter, and impaired immunity.6
DIAGNOSIS AND TREATMENT
Intravenous urography is the gold standard for the diagnosis of medullary sponge kidney; computed tomography and ultrasonography are generally limited in their ability to clearly show the tubular ectasia.7
Treatment includes antibiotics for acute pyelonephritis and thiazide diuretics and potassium citrate to prevent stone formation and renal tubular acidosis. Due to its silent course, medullary sponge kidney should be considered not only as a cause of nephrocalcinosis and nephrolithiasis, but also as a distinct entity complicating recurrent pyelonephritis.
- Gambaro G, Feltrin GP, Lupo A, Bonfante L, D'Angelo A, Antonello A. Medullary sponge kidney (Lenarduzzi-Cacchi-Ricci disease): a Padua Medical School discovery in the 1930s. Kidney Int 2006; 69:663–670.
- Kasap B, Soylu A, Oren O, Turkmen M, Kavukcu S. Medullary sponge kidney associated with distal renal tubular acidosis in a 5-year-old girl. Eur J Pediatr 2006; 165:648–651.
- Gambaro G, Fabris A, Puliatta D, Lupo A. Lithiasis in cystic kidney disease and malformations of the urinary tract. Urol Res 2006; 34:102–107.
- O'Neill M, Breslau NA, Pak CY. Metabolic evaluation of nephrolithiasis in patients with medullary sponge kidney. JAMA 1981; 245:1233–1236.
- Miano R, Germani S, Vespasiani G. Stones and urinary tract infections. Urol Int 2007; 79(suppl 1):32–36.
- Scholes D, Hooton TM, Roberts PL, Gupta K, Stapleton AE, Stamm WE. Risk factors associated with acute pyelonephritis in healthy women. Ann Intern Med 2005; 142:20–27.
- Maw AM, Megibow AJ, Grasso M, Goldfarb DS. Diagnosis of medullary sponge kidney by computed tomographic urography. Am J Kidney Dis 2007; 50:146–150.
- Gambaro G, Feltrin GP, Lupo A, Bonfante L, D'Angelo A, Antonello A. Medullary sponge kidney (Lenarduzzi-Cacchi-Ricci disease): a Padua Medical School discovery in the 1930s. Kidney Int 2006; 69:663–670.
- Kasap B, Soylu A, Oren O, Turkmen M, Kavukcu S. Medullary sponge kidney associated with distal renal tubular acidosis in a 5-year-old girl. Eur J Pediatr 2006; 165:648–651.
- Gambaro G, Fabris A, Puliatta D, Lupo A. Lithiasis in cystic kidney disease and malformations of the urinary tract. Urol Res 2006; 34:102–107.
- O'Neill M, Breslau NA, Pak CY. Metabolic evaluation of nephrolithiasis in patients with medullary sponge kidney. JAMA 1981; 245:1233–1236.
- Miano R, Germani S, Vespasiani G. Stones and urinary tract infections. Urol Int 2007; 79(suppl 1):32–36.
- Scholes D, Hooton TM, Roberts PL, Gupta K, Stapleton AE, Stamm WE. Risk factors associated with acute pyelonephritis in healthy women. Ann Intern Med 2005; 142:20–27.
- Maw AM, Megibow AJ, Grasso M, Goldfarb DS. Diagnosis of medullary sponge kidney by computed tomographic urography. Am J Kidney Dis 2007; 50:146–150.
{kind=link}
Does measuring natriuretic peptides have a role in patients with chronic kidney disease?
Yes, measuring the levels of certain natriuretic peptides can help diagnose decompensated heart failure and predict the risk of death and cardiac hospitalization in patients across a wide spectrum of renal function.
However, at this time, it is unclear whether routinely measuring natriuretic peptides will result in any change in the management of patients with chronic kidney disease. Additionally, using these peptides to monitor volume status in dialysis patients has not yet been deemed useful, although it may be complementary to echocardiography in evaluating cardiac risk in patients with end-stage renal disease.
A BRIEF REVIEW OF NATRIURETIC PEPTIDES
Natriuretic peptides include atrial natriuretic peptide, brain natriuretic peptide (BNP), C-type natriuretic peptide, and urodilantin.
BNP, which is homologous to atrial natriuretic peptide, is present in the brain and the heart. The circulating concentration of BNP is less than 20% of the atrial natriuretic peptide level in healthy people, but equals or exceeds that of atrial natriuretic peptide in patients with congestive heart failure.
BNP starts as a precursor protein. This is modified within the cell into a prohormone, proBNP, which is secreted from the left ventricle in response to myocardial wall stress. In the circulation, proBNP is cleaved into a biologically active C-terminal fragment—BNP—and a biologically inactive N-terminal fragment (NT-proBNP).1 NT-proBNP is primarily cleared by the kidney. BNP is cleared by receptor-mediated binding and removed by neutral endopeptidase, as well as by the kidney.
Both BNP and NT-proBNP have been investigated as diagnostic markers of suspected heart disease.
PEPTIDE LEVELS ARE HIGH IN CHRONIC KIDNEY DISEASE AND HEART FAILURE
An estimated 8.3 million people in the United States have stage 3, 4, or 5 chronic kidney disease,2 defined as an estimated glomerular filtration rate of less than 60 mL/min/1.73 m2. Approximately 50% of patients with heart failure have chronic kidney disease, and almost 60% of patients with chronic kidney disease have some abnormality in ventricular function.
A few years ago, researchers began investigating the benefits and limitations of using natriuretic peptides to diagnose cardiac dysfunction (left ventricular structural and functional abnormalities) in patients with chronic kidney disease.
One important study3 was conducted in almost 3,000 patients from the Dallas Heart Study who were between the ages of 30 and 65 years—a relatively young, mostly healthy population. The authors found that natriuretic peptide levels did not vary as long as the estimated glomerular filtration rate was within the normal range. However, when the estimated glomerular filtration rate dropped below a threshold of 90 mL/min/1.73 m2, the concentrations of both NT-proBNP and BNP increased exponentially. NT-proBNP levels rose more than BNP levels, as NT-proBNP is primarily cleared by the kidney.
More recent studies found that the high levels of NT-proBNP in patients with chronic kidney disease do not simply reflect the reduced clearance of this peptide; they also reflect compromised ventricular function.2,4 This relationship was supported by studies of the fractional renal excretion of NT-proBNP and BNP in several populations with and without renal impairment.5 Interestingly, fractional excretion of both peptides remained equivalent across a wide spectrum of renal function. Seemingly, cardiac disease drove the increase in values rather than the degree of renal impairment.
HIGH PEPTIDE LEVELS PREDICT DEATH, HOSPITALIZATION
Both BNP and NT-proBNP are strong predictors of death and cardiac hospitalization in kidney patients.1,4,6
In patients with end-stage renal disease, the risk of cardiovascular disease and death is significantly higher than that in the general population, and BNP has been found to be a valuable prognostic indicator of cardiac disease.7
Multiple studies showed that high levels of natriuretic peptides are associated with a higher risk of death in patients with acute coronary syndrome, independent of traditional cardiovascular risk factors such as electrocardiographic changes and levels of other biomarkers. However, these data were derived from patients with mild renal impairment.2
Apple et al8 compared the prognostic value of NT-proBNP with that of cardiac troponin T in hemodialysis patients who had no symptoms and found that NT-proBNP was more strongly associated with left ventricular systolic dysfunction and subsequent cardiovascular death.
PEPTIDE LEVELS ARE HIGHER IN ANEMIA
A significant number of patients with congestive heart failure have renal insufficiency and low hemoglobin levels, which may increase natriuretic peptide levels. It is unclear why anemia is associated with elevated levels of natriuretic peptides, even in the absence of clinical heart failure and independent of other cardiovascular risk factors.9 Nevertheless, anemia should be taken into consideration and treated effectively when evaluating patients with renal impairment and possible congestive heart failure.
PEPTIDES COMPLEMENT CARDIAC ECHO IN END-STAGE RENAL DISEASE
Numerous studies have found a close association between BNP and NT-proBNP levels and left ventricular mass and systolic function in patients with end-stage renal disease.10,11 Data from the Cardiovascular Risk Extended Evaluation in Dialysis Patients study12 suggest that BNP measurement can be reliably applied in end-stage renal disease to rule out systolic dysfunction and to detect left ventricular hypertrophy, but it has a very low negative predictive value for left ventricular hypertrophy in this patient population: someone with a normal BNP level can still have left ventricular hypertrophy.
In addition, volume status is harder to assess with BNP alone than with echocardiography, and an elevated BNP value is not very specific.13
In essence, both BNP and NT-proBNP can be used to complement echocardiography in evaluating cardiac risk in patients with end-stage renal disease. With additional data, it may be possible in the future to use them as substitutes for echocardiography when managing ventricular abnormalities in patients with end-stage renal disease.
USING SPECIFIC CUT POINTS IN RENAL DISEASE
When evaluating a patient with acute dyspnea and either chronic kidney disease or end-stage renal disease who is receiving dialysis, both BNP and NT-proBNP are affected similarly and necessitate a higher level of interpretation to diagnose decompensated heart failure. Currently, researchers disagree about specific cut points for natriuretic peptides. However, deFilippi and colleagues4 suggested the following cut points for NT-proBNP for diagnosing heart failure in patients of different ages with or without renal impairment:
- Younger than 50 years—450 ng/L
- Age 50 to 75 years—900 ng/L
- Older than 75 years—1,800 ng/L.
A BNP cutoff point of 225 pg/mL can be used for patients with an estimated glomerular filtration rate of less than 60 mL/min/1.73 m2, based on data from the Breathing Not Properly multinational study.14
There is no set cut-point for either BNP or NT-proBNP for predicting death and cardiac hospitalization in renal patients, but abnormally high levels should signal the need to optimize medical management and to monitor more closely.
- Austin WJ, Bhalla V, Hernandez-Arce I, et al. Correlation and prognostic utility of B-type natriuretic peptide and its amino-terminal fragment in patients with chronic kidney disease. Am J Clin Pathol 2006; 126:506–512.
- DeFilippi C, van Kimmenade RR, Pinto YM. Amino-terminal pro-B-type natriuretic peptide testing in renal disease. Am J Cardiol 2008; 101:82–88.
- Das SR, Abdullah SM, Leonard D, et al. Association between renal function and circulating levels of natriuretic peptides (from the Dallas Heart Study). Am J Cardiol 2008; 102:1394–1398.
- DeFilippi CR, Seliger SL, Maynard S, Christenson RH. Impact of renal disease on natriuretic peptide testing for diagnosing decompensated heart failure and predicting mortality. Clin Chem 2007; 53:1511–1519.
- Goetze JP, Jensen G, Møller S, Bendtsen F, Rehfeld JF, Henriksen JH. BNP and N-terminal proBNP are both extracted in the normal kidney. Eur J Clin Invest 2006; 36:8–15.
- Zoccali C. Biomarkers in chronic kidney disease: utility and issues towards better understanding. Curr Opin Nephrol Hypertens 2005; 14:532–537.
- Haapio M, Ronco C. BNP and a renal patient: emphasis on the unique characteristics of B-type natriuretic peptide in end-stage kidney disease. Contrib Nephrol 2008; 161:68–75.
- Apple FS, Murakami MM, Pearce LA, Herzog CA. Multibiomarker risk stratification of N-terminal pro-B-type natriuretic peptide, high-sensitivity C-reactive protein, and cardiac troponin T and I in end-stage renal disease for all-cause death. Clin Chem 2004: 50:2279–2285.
- Hogenhuis J, Voors AA, Jaarsma T, et al. Anemia and renal dysfunction are independently associated with BNP and NT-proBNP levels in patients with heart failure. Eur J Heart Fail 2007; 9:787–794.
- Madsen LH, Ladefoged S, Corell P, Schou M, Hildebrandt PR, Atar D. N-terminal pro brain natriuretic peptide predicts mortality in patients with end-stage renal disease on hemodialysis. Kidney Int 2007; 71:548–554.
- Wang AY, Lai KN. Use of cardiac biomarkers in end-stage renal disease. J Am Soc Nephrol 2008; 19:1643–1652.
- Mallamaci F, Zoccali C, Tripepi G, et al; on behalf of the CREED Investigators. Diagnostic potential of cardiac natriuretic peptides in dialysis patients. Kidney Int 2001; 59:1559–1566.
- Biasioli S, Zamperetti M, Borin D, Guidi G, De Fanti E, Schiavon R. Significance of plasma B-type natriuretic peptide in hemodialysis patients: blood sample timing and comorbidity burden. ASAIO J 2007; 53:587–591.
- McCullough PA, Duc P, Omland T, et al. B-type natriuretic peptide and renal function in the diagnosis of heart failure: an analysis from the Breathing Not Properly multinational study. Am J Kidney Dis 2003; 41:571–579.
Yes, measuring the levels of certain natriuretic peptides can help diagnose decompensated heart failure and predict the risk of death and cardiac hospitalization in patients across a wide spectrum of renal function.
However, at this time, it is unclear whether routinely measuring natriuretic peptides will result in any change in the management of patients with chronic kidney disease. Additionally, using these peptides to monitor volume status in dialysis patients has not yet been deemed useful, although it may be complementary to echocardiography in evaluating cardiac risk in patients with end-stage renal disease.
A BRIEF REVIEW OF NATRIURETIC PEPTIDES
Natriuretic peptides include atrial natriuretic peptide, brain natriuretic peptide (BNP), C-type natriuretic peptide, and urodilantin.
BNP, which is homologous to atrial natriuretic peptide, is present in the brain and the heart. The circulating concentration of BNP is less than 20% of the atrial natriuretic peptide level in healthy people, but equals or exceeds that of atrial natriuretic peptide in patients with congestive heart failure.
BNP starts as a precursor protein. This is modified within the cell into a prohormone, proBNP, which is secreted from the left ventricle in response to myocardial wall stress. In the circulation, proBNP is cleaved into a biologically active C-terminal fragment—BNP—and a biologically inactive N-terminal fragment (NT-proBNP).1 NT-proBNP is primarily cleared by the kidney. BNP is cleared by receptor-mediated binding and removed by neutral endopeptidase, as well as by the kidney.
Both BNP and NT-proBNP have been investigated as diagnostic markers of suspected heart disease.
PEPTIDE LEVELS ARE HIGH IN CHRONIC KIDNEY DISEASE AND HEART FAILURE
An estimated 8.3 million people in the United States have stage 3, 4, or 5 chronic kidney disease,2 defined as an estimated glomerular filtration rate of less than 60 mL/min/1.73 m2. Approximately 50% of patients with heart failure have chronic kidney disease, and almost 60% of patients with chronic kidney disease have some abnormality in ventricular function.
A few years ago, researchers began investigating the benefits and limitations of using natriuretic peptides to diagnose cardiac dysfunction (left ventricular structural and functional abnormalities) in patients with chronic kidney disease.
One important study3 was conducted in almost 3,000 patients from the Dallas Heart Study who were between the ages of 30 and 65 years—a relatively young, mostly healthy population. The authors found that natriuretic peptide levels did not vary as long as the estimated glomerular filtration rate was within the normal range. However, when the estimated glomerular filtration rate dropped below a threshold of 90 mL/min/1.73 m2, the concentrations of both NT-proBNP and BNP increased exponentially. NT-proBNP levels rose more than BNP levels, as NT-proBNP is primarily cleared by the kidney.
More recent studies found that the high levels of NT-proBNP in patients with chronic kidney disease do not simply reflect the reduced clearance of this peptide; they also reflect compromised ventricular function.2,4 This relationship was supported by studies of the fractional renal excretion of NT-proBNP and BNP in several populations with and without renal impairment.5 Interestingly, fractional excretion of both peptides remained equivalent across a wide spectrum of renal function. Seemingly, cardiac disease drove the increase in values rather than the degree of renal impairment.
HIGH PEPTIDE LEVELS PREDICT DEATH, HOSPITALIZATION
Both BNP and NT-proBNP are strong predictors of death and cardiac hospitalization in kidney patients.1,4,6
In patients with end-stage renal disease, the risk of cardiovascular disease and death is significantly higher than that in the general population, and BNP has been found to be a valuable prognostic indicator of cardiac disease.7
Multiple studies showed that high levels of natriuretic peptides are associated with a higher risk of death in patients with acute coronary syndrome, independent of traditional cardiovascular risk factors such as electrocardiographic changes and levels of other biomarkers. However, these data were derived from patients with mild renal impairment.2
Apple et al8 compared the prognostic value of NT-proBNP with that of cardiac troponin T in hemodialysis patients who had no symptoms and found that NT-proBNP was more strongly associated with left ventricular systolic dysfunction and subsequent cardiovascular death.
PEPTIDE LEVELS ARE HIGHER IN ANEMIA
A significant number of patients with congestive heart failure have renal insufficiency and low hemoglobin levels, which may increase natriuretic peptide levels. It is unclear why anemia is associated with elevated levels of natriuretic peptides, even in the absence of clinical heart failure and independent of other cardiovascular risk factors.9 Nevertheless, anemia should be taken into consideration and treated effectively when evaluating patients with renal impairment and possible congestive heart failure.
PEPTIDES COMPLEMENT CARDIAC ECHO IN END-STAGE RENAL DISEASE
Numerous studies have found a close association between BNP and NT-proBNP levels and left ventricular mass and systolic function in patients with end-stage renal disease.10,11 Data from the Cardiovascular Risk Extended Evaluation in Dialysis Patients study12 suggest that BNP measurement can be reliably applied in end-stage renal disease to rule out systolic dysfunction and to detect left ventricular hypertrophy, but it has a very low negative predictive value for left ventricular hypertrophy in this patient population: someone with a normal BNP level can still have left ventricular hypertrophy.
In addition, volume status is harder to assess with BNP alone than with echocardiography, and an elevated BNP value is not very specific.13
In essence, both BNP and NT-proBNP can be used to complement echocardiography in evaluating cardiac risk in patients with end-stage renal disease. With additional data, it may be possible in the future to use them as substitutes for echocardiography when managing ventricular abnormalities in patients with end-stage renal disease.
USING SPECIFIC CUT POINTS IN RENAL DISEASE
When evaluating a patient with acute dyspnea and either chronic kidney disease or end-stage renal disease who is receiving dialysis, both BNP and NT-proBNP are affected similarly and necessitate a higher level of interpretation to diagnose decompensated heart failure. Currently, researchers disagree about specific cut points for natriuretic peptides. However, deFilippi and colleagues4 suggested the following cut points for NT-proBNP for diagnosing heart failure in patients of different ages with or without renal impairment:
- Younger than 50 years—450 ng/L
- Age 50 to 75 years—900 ng/L
- Older than 75 years—1,800 ng/L.
A BNP cutoff point of 225 pg/mL can be used for patients with an estimated glomerular filtration rate of less than 60 mL/min/1.73 m2, based on data from the Breathing Not Properly multinational study.14
There is no set cut-point for either BNP or NT-proBNP for predicting death and cardiac hospitalization in renal patients, but abnormally high levels should signal the need to optimize medical management and to monitor more closely.
Yes, measuring the levels of certain natriuretic peptides can help diagnose decompensated heart failure and predict the risk of death and cardiac hospitalization in patients across a wide spectrum of renal function.
However, at this time, it is unclear whether routinely measuring natriuretic peptides will result in any change in the management of patients with chronic kidney disease. Additionally, using these peptides to monitor volume status in dialysis patients has not yet been deemed useful, although it may be complementary to echocardiography in evaluating cardiac risk in patients with end-stage renal disease.
A BRIEF REVIEW OF NATRIURETIC PEPTIDES
Natriuretic peptides include atrial natriuretic peptide, brain natriuretic peptide (BNP), C-type natriuretic peptide, and urodilantin.
BNP, which is homologous to atrial natriuretic peptide, is present in the brain and the heart. The circulating concentration of BNP is less than 20% of the atrial natriuretic peptide level in healthy people, but equals or exceeds that of atrial natriuretic peptide in patients with congestive heart failure.
BNP starts as a precursor protein. This is modified within the cell into a prohormone, proBNP, which is secreted from the left ventricle in response to myocardial wall stress. In the circulation, proBNP is cleaved into a biologically active C-terminal fragment—BNP—and a biologically inactive N-terminal fragment (NT-proBNP).1 NT-proBNP is primarily cleared by the kidney. BNP is cleared by receptor-mediated binding and removed by neutral endopeptidase, as well as by the kidney.
Both BNP and NT-proBNP have been investigated as diagnostic markers of suspected heart disease.
PEPTIDE LEVELS ARE HIGH IN CHRONIC KIDNEY DISEASE AND HEART FAILURE
An estimated 8.3 million people in the United States have stage 3, 4, or 5 chronic kidney disease,2 defined as an estimated glomerular filtration rate of less than 60 mL/min/1.73 m2. Approximately 50% of patients with heart failure have chronic kidney disease, and almost 60% of patients with chronic kidney disease have some abnormality in ventricular function.
A few years ago, researchers began investigating the benefits and limitations of using natriuretic peptides to diagnose cardiac dysfunction (left ventricular structural and functional abnormalities) in patients with chronic kidney disease.
One important study3 was conducted in almost 3,000 patients from the Dallas Heart Study who were between the ages of 30 and 65 years—a relatively young, mostly healthy population. The authors found that natriuretic peptide levels did not vary as long as the estimated glomerular filtration rate was within the normal range. However, when the estimated glomerular filtration rate dropped below a threshold of 90 mL/min/1.73 m2, the concentrations of both NT-proBNP and BNP increased exponentially. NT-proBNP levels rose more than BNP levels, as NT-proBNP is primarily cleared by the kidney.
More recent studies found that the high levels of NT-proBNP in patients with chronic kidney disease do not simply reflect the reduced clearance of this peptide; they also reflect compromised ventricular function.2,4 This relationship was supported by studies of the fractional renal excretion of NT-proBNP and BNP in several populations with and without renal impairment.5 Interestingly, fractional excretion of both peptides remained equivalent across a wide spectrum of renal function. Seemingly, cardiac disease drove the increase in values rather than the degree of renal impairment.
HIGH PEPTIDE LEVELS PREDICT DEATH, HOSPITALIZATION
Both BNP and NT-proBNP are strong predictors of death and cardiac hospitalization in kidney patients.1,4,6
In patients with end-stage renal disease, the risk of cardiovascular disease and death is significantly higher than that in the general population, and BNP has been found to be a valuable prognostic indicator of cardiac disease.7
Multiple studies showed that high levels of natriuretic peptides are associated with a higher risk of death in patients with acute coronary syndrome, independent of traditional cardiovascular risk factors such as electrocardiographic changes and levels of other biomarkers. However, these data were derived from patients with mild renal impairment.2
Apple et al8 compared the prognostic value of NT-proBNP with that of cardiac troponin T in hemodialysis patients who had no symptoms and found that NT-proBNP was more strongly associated with left ventricular systolic dysfunction and subsequent cardiovascular death.
PEPTIDE LEVELS ARE HIGHER IN ANEMIA
A significant number of patients with congestive heart failure have renal insufficiency and low hemoglobin levels, which may increase natriuretic peptide levels. It is unclear why anemia is associated with elevated levels of natriuretic peptides, even in the absence of clinical heart failure and independent of other cardiovascular risk factors.9 Nevertheless, anemia should be taken into consideration and treated effectively when evaluating patients with renal impairment and possible congestive heart failure.
PEPTIDES COMPLEMENT CARDIAC ECHO IN END-STAGE RENAL DISEASE
Numerous studies have found a close association between BNP and NT-proBNP levels and left ventricular mass and systolic function in patients with end-stage renal disease.10,11 Data from the Cardiovascular Risk Extended Evaluation in Dialysis Patients study12 suggest that BNP measurement can be reliably applied in end-stage renal disease to rule out systolic dysfunction and to detect left ventricular hypertrophy, but it has a very low negative predictive value for left ventricular hypertrophy in this patient population: someone with a normal BNP level can still have left ventricular hypertrophy.
In addition, volume status is harder to assess with BNP alone than with echocardiography, and an elevated BNP value is not very specific.13
In essence, both BNP and NT-proBNP can be used to complement echocardiography in evaluating cardiac risk in patients with end-stage renal disease. With additional data, it may be possible in the future to use them as substitutes for echocardiography when managing ventricular abnormalities in patients with end-stage renal disease.
USING SPECIFIC CUT POINTS IN RENAL DISEASE
When evaluating a patient with acute dyspnea and either chronic kidney disease or end-stage renal disease who is receiving dialysis, both BNP and NT-proBNP are affected similarly and necessitate a higher level of interpretation to diagnose decompensated heart failure. Currently, researchers disagree about specific cut points for natriuretic peptides. However, deFilippi and colleagues4 suggested the following cut points for NT-proBNP for diagnosing heart failure in patients of different ages with or without renal impairment:
- Younger than 50 years—450 ng/L
- Age 50 to 75 years—900 ng/L
- Older than 75 years—1,800 ng/L.
A BNP cutoff point of 225 pg/mL can be used for patients with an estimated glomerular filtration rate of less than 60 mL/min/1.73 m2, based on data from the Breathing Not Properly multinational study.14
There is no set cut-point for either BNP or NT-proBNP for predicting death and cardiac hospitalization in renal patients, but abnormally high levels should signal the need to optimize medical management and to monitor more closely.
- Austin WJ, Bhalla V, Hernandez-Arce I, et al. Correlation and prognostic utility of B-type natriuretic peptide and its amino-terminal fragment in patients with chronic kidney disease. Am J Clin Pathol 2006; 126:506–512.
- DeFilippi C, van Kimmenade RR, Pinto YM. Amino-terminal pro-B-type natriuretic peptide testing in renal disease. Am J Cardiol 2008; 101:82–88.
- Das SR, Abdullah SM, Leonard D, et al. Association between renal function and circulating levels of natriuretic peptides (from the Dallas Heart Study). Am J Cardiol 2008; 102:1394–1398.
- DeFilippi CR, Seliger SL, Maynard S, Christenson RH. Impact of renal disease on natriuretic peptide testing for diagnosing decompensated heart failure and predicting mortality. Clin Chem 2007; 53:1511–1519.
- Goetze JP, Jensen G, Møller S, Bendtsen F, Rehfeld JF, Henriksen JH. BNP and N-terminal proBNP are both extracted in the normal kidney. Eur J Clin Invest 2006; 36:8–15.
- Zoccali C. Biomarkers in chronic kidney disease: utility and issues towards better understanding. Curr Opin Nephrol Hypertens 2005; 14:532–537.
- Haapio M, Ronco C. BNP and a renal patient: emphasis on the unique characteristics of B-type natriuretic peptide in end-stage kidney disease. Contrib Nephrol 2008; 161:68–75.
- Apple FS, Murakami MM, Pearce LA, Herzog CA. Multibiomarker risk stratification of N-terminal pro-B-type natriuretic peptide, high-sensitivity C-reactive protein, and cardiac troponin T and I in end-stage renal disease for all-cause death. Clin Chem 2004: 50:2279–2285.
- Hogenhuis J, Voors AA, Jaarsma T, et al. Anemia and renal dysfunction are independently associated with BNP and NT-proBNP levels in patients with heart failure. Eur J Heart Fail 2007; 9:787–794.
- Madsen LH, Ladefoged S, Corell P, Schou M, Hildebrandt PR, Atar D. N-terminal pro brain natriuretic peptide predicts mortality in patients with end-stage renal disease on hemodialysis. Kidney Int 2007; 71:548–554.
- Wang AY, Lai KN. Use of cardiac biomarkers in end-stage renal disease. J Am Soc Nephrol 2008; 19:1643–1652.
- Mallamaci F, Zoccali C, Tripepi G, et al; on behalf of the CREED Investigators. Diagnostic potential of cardiac natriuretic peptides in dialysis patients. Kidney Int 2001; 59:1559–1566.
- Biasioli S, Zamperetti M, Borin D, Guidi G, De Fanti E, Schiavon R. Significance of plasma B-type natriuretic peptide in hemodialysis patients: blood sample timing and comorbidity burden. ASAIO J 2007; 53:587–591.
- McCullough PA, Duc P, Omland T, et al. B-type natriuretic peptide and renal function in the diagnosis of heart failure: an analysis from the Breathing Not Properly multinational study. Am J Kidney Dis 2003; 41:571–579.
- Austin WJ, Bhalla V, Hernandez-Arce I, et al. Correlation and prognostic utility of B-type natriuretic peptide and its amino-terminal fragment in patients with chronic kidney disease. Am J Clin Pathol 2006; 126:506–512.
- DeFilippi C, van Kimmenade RR, Pinto YM. Amino-terminal pro-B-type natriuretic peptide testing in renal disease. Am J Cardiol 2008; 101:82–88.
- Das SR, Abdullah SM, Leonard D, et al. Association between renal function and circulating levels of natriuretic peptides (from the Dallas Heart Study). Am J Cardiol 2008; 102:1394–1398.
- DeFilippi CR, Seliger SL, Maynard S, Christenson RH. Impact of renal disease on natriuretic peptide testing for diagnosing decompensated heart failure and predicting mortality. Clin Chem 2007; 53:1511–1519.
- Goetze JP, Jensen G, Møller S, Bendtsen F, Rehfeld JF, Henriksen JH. BNP and N-terminal proBNP are both extracted in the normal kidney. Eur J Clin Invest 2006; 36:8–15.
- Zoccali C. Biomarkers in chronic kidney disease: utility and issues towards better understanding. Curr Opin Nephrol Hypertens 2005; 14:532–537.
- Haapio M, Ronco C. BNP and a renal patient: emphasis on the unique characteristics of B-type natriuretic peptide in end-stage kidney disease. Contrib Nephrol 2008; 161:68–75.
- Apple FS, Murakami MM, Pearce LA, Herzog CA. Multibiomarker risk stratification of N-terminal pro-B-type natriuretic peptide, high-sensitivity C-reactive protein, and cardiac troponin T and I in end-stage renal disease for all-cause death. Clin Chem 2004: 50:2279–2285.
- Hogenhuis J, Voors AA, Jaarsma T, et al. Anemia and renal dysfunction are independently associated with BNP and NT-proBNP levels in patients with heart failure. Eur J Heart Fail 2007; 9:787–794.
- Madsen LH, Ladefoged S, Corell P, Schou M, Hildebrandt PR, Atar D. N-terminal pro brain natriuretic peptide predicts mortality in patients with end-stage renal disease on hemodialysis. Kidney Int 2007; 71:548–554.
- Wang AY, Lai KN. Use of cardiac biomarkers in end-stage renal disease. J Am Soc Nephrol 2008; 19:1643–1652.
- Mallamaci F, Zoccali C, Tripepi G, et al; on behalf of the CREED Investigators. Diagnostic potential of cardiac natriuretic peptides in dialysis patients. Kidney Int 2001; 59:1559–1566.
- Biasioli S, Zamperetti M, Borin D, Guidi G, De Fanti E, Schiavon R. Significance of plasma B-type natriuretic peptide in hemodialysis patients: blood sample timing and comorbidity burden. ASAIO J 2007; 53:587–591.
- McCullough PA, Duc P, Omland T, et al. B-type natriuretic peptide and renal function in the diagnosis of heart failure: an analysis from the Breathing Not Properly multinational study. Am J Kidney Dis 2003; 41:571–579.