User login
FDA Approves Tadalafil for Benign Prostatic Hyperplasia
The erectile dysfunction drug tadalafil has been approved for treatment of the signs and symptoms of benign prostatic hyperplasia, the Food and Drug Administration announced on Oct. 6.
Tadalafil, the phosphodiesterase-5 (PDE5) inhibitor marketed as Cialis by Eli Lilly, was also approved for treating BPH and erectile dysfunction (ED), when they occur simultaneously, according to the FDA statement. The agency first approved tadalafil for treating erectile dysfunction in 2003.
Men with BPH have an enlarged prostate, which can cause symptoms ranging from difficulty urinating and a weak urine stream to a sudden urge to urinate and more frequent urination.
In two studies of men with BPH, those treated with 5 mg/day of tadalafil experienced statistically significant improvements in symptoms, as indicated by reductions in the total International Prostate Symptom Score (IPSS), when compared with the score in men who received a placebo.
Similarly, in a placebo-controlled study of men with both ED and BPH, those treated with 5 mg/day of tadalafil had improvements in symptoms of both conditions, with the ED improvement measured by the erectile function domain score of the International Index of Erectile Function.
The FDA noted that tadalafil is contraindicated in patients taking nitrates, such as nitroglycerin, because it has been shown to potentiate the hypotensive effects of nitrates. In addition, combining tadalafil with alpha-blockers for treating BPH "is not recommended because the combination has not been adequately studied for the treatment of BPH, and there is a risk of lowering blood pressure," the statement said.
Tadalafil is the first PDE5 inhibitor to be approved for BPH. The eight drugs previously approved for treating BPH symptoms are the 5-alpha reductase inhibitors finasteride (Proscar) and dutasteride (Avodart); alpha-blockers terazosin (Hytrin), doxazosin (Cardura), tamsulosin (Flomax), alfuzosin (Uroxatral), and silodosin (Rapaflo); and the combination of dutasteride plus tamsulosin (Jalyn).
The erectile dysfunction drug tadalafil has been approved for treatment of the signs and symptoms of benign prostatic hyperplasia, the Food and Drug Administration announced on Oct. 6.
Tadalafil, the phosphodiesterase-5 (PDE5) inhibitor marketed as Cialis by Eli Lilly, was also approved for treating BPH and erectile dysfunction (ED), when they occur simultaneously, according to the FDA statement. The agency first approved tadalafil for treating erectile dysfunction in 2003.
Men with BPH have an enlarged prostate, which can cause symptoms ranging from difficulty urinating and a weak urine stream to a sudden urge to urinate and more frequent urination.
In two studies of men with BPH, those treated with 5 mg/day of tadalafil experienced statistically significant improvements in symptoms, as indicated by reductions in the total International Prostate Symptom Score (IPSS), when compared with the score in men who received a placebo.
Similarly, in a placebo-controlled study of men with both ED and BPH, those treated with 5 mg/day of tadalafil had improvements in symptoms of both conditions, with the ED improvement measured by the erectile function domain score of the International Index of Erectile Function.
The FDA noted that tadalafil is contraindicated in patients taking nitrates, such as nitroglycerin, because it has been shown to potentiate the hypotensive effects of nitrates. In addition, combining tadalafil with alpha-blockers for treating BPH "is not recommended because the combination has not been adequately studied for the treatment of BPH, and there is a risk of lowering blood pressure," the statement said.
Tadalafil is the first PDE5 inhibitor to be approved for BPH. The eight drugs previously approved for treating BPH symptoms are the 5-alpha reductase inhibitors finasteride (Proscar) and dutasteride (Avodart); alpha-blockers terazosin (Hytrin), doxazosin (Cardura), tamsulosin (Flomax), alfuzosin (Uroxatral), and silodosin (Rapaflo); and the combination of dutasteride plus tamsulosin (Jalyn).
The erectile dysfunction drug tadalafil has been approved for treatment of the signs and symptoms of benign prostatic hyperplasia, the Food and Drug Administration announced on Oct. 6.
Tadalafil, the phosphodiesterase-5 (PDE5) inhibitor marketed as Cialis by Eli Lilly, was also approved for treating BPH and erectile dysfunction (ED), when they occur simultaneously, according to the FDA statement. The agency first approved tadalafil for treating erectile dysfunction in 2003.
Men with BPH have an enlarged prostate, which can cause symptoms ranging from difficulty urinating and a weak urine stream to a sudden urge to urinate and more frequent urination.
In two studies of men with BPH, those treated with 5 mg/day of tadalafil experienced statistically significant improvements in symptoms, as indicated by reductions in the total International Prostate Symptom Score (IPSS), when compared with the score in men who received a placebo.
Similarly, in a placebo-controlled study of men with both ED and BPH, those treated with 5 mg/day of tadalafil had improvements in symptoms of both conditions, with the ED improvement measured by the erectile function domain score of the International Index of Erectile Function.
The FDA noted that tadalafil is contraindicated in patients taking nitrates, such as nitroglycerin, because it has been shown to potentiate the hypotensive effects of nitrates. In addition, combining tadalafil with alpha-blockers for treating BPH "is not recommended because the combination has not been adequately studied for the treatment of BPH, and there is a risk of lowering blood pressure," the statement said.
Tadalafil is the first PDE5 inhibitor to be approved for BPH. The eight drugs previously approved for treating BPH symptoms are the 5-alpha reductase inhibitors finasteride (Proscar) and dutasteride (Avodart); alpha-blockers terazosin (Hytrin), doxazosin (Cardura), tamsulosin (Flomax), alfuzosin (Uroxatral), and silodosin (Rapaflo); and the combination of dutasteride plus tamsulosin (Jalyn).
Necrotic skin lesions after hemodialysis
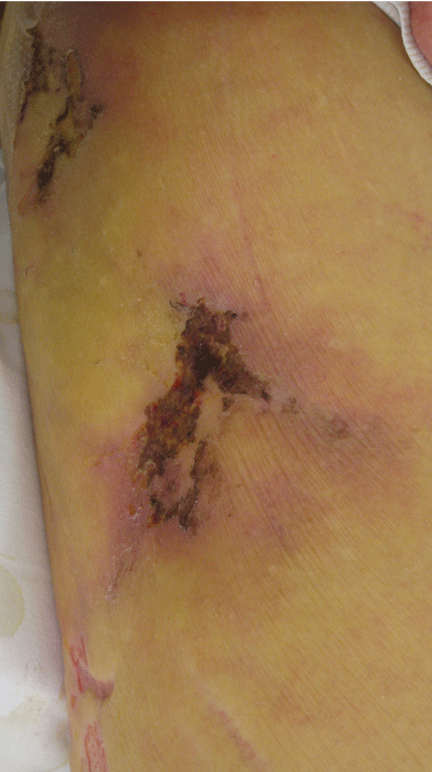
Current laboratory values:
- Serum calcium concentration 7.8 mg/dL (reference range 8.5–10.5)
- Phosphorus 6.4 mg/dL (2.5–4.5)
- Corrected calcium-phosphorus product 55
- Parathyroid hormone 275 pg/mL (10–60)
- 25-hydroxyvitamin D 7.4 ng/mL (31–80).
Q: Given the patient’s history, which of the following does her skin lesion likely represent?
- Necrotizing fasciitis
- Calciphylaxis
- Disseminated intravascular coagulation
- Anticoagulant-induced skin necrosis
A: Calciphylaxis, or calcific uremic arteriolopathy, is the most likely. It is rare in people with normal renal function, and still rare but somewhat less so in end-stage renal disease patients undergoing chronic hemodialysis.
WHAT CAUSED IT IN OUR PATIENT?
The cause of calciphylaxis is unknown. Theories have focused on protein C and parathyroid hormone. Putative precipitating factors include acute tubular necrosis, albumin infusion with paracentesis, deficiency of protein C or S, hyperparathyroidism, hyperphosphatemia, hypercalcemia, vitamin D supplementation, steroids, trauma, and warfarin use.
Our patient had a history of hypothyroidism, ulcerative colitis, and end-stage liver disease due to primary sclerosing cholangitis, but no previous history of renal disease.
At the time of her acute renal failure, her calcium-phosphorus level was 55, parathyroid hormone level 274 pg/mL (normal 10–60), and protein C level 26% (normal 76%–147%). At the time the skin lesions were discovered, her protein C level had dropped to 14%; her parathyroid level had returned to normal.
Her home medications included furosemide (Lasix), levothyroxine (Synthroid), mesalamine (Pentasa), azathioprine (Imuran), ursodiol (Actigall), spironolactone (Aldactone), and omeprazole (Prilosec).
NONHEALING LESIONS
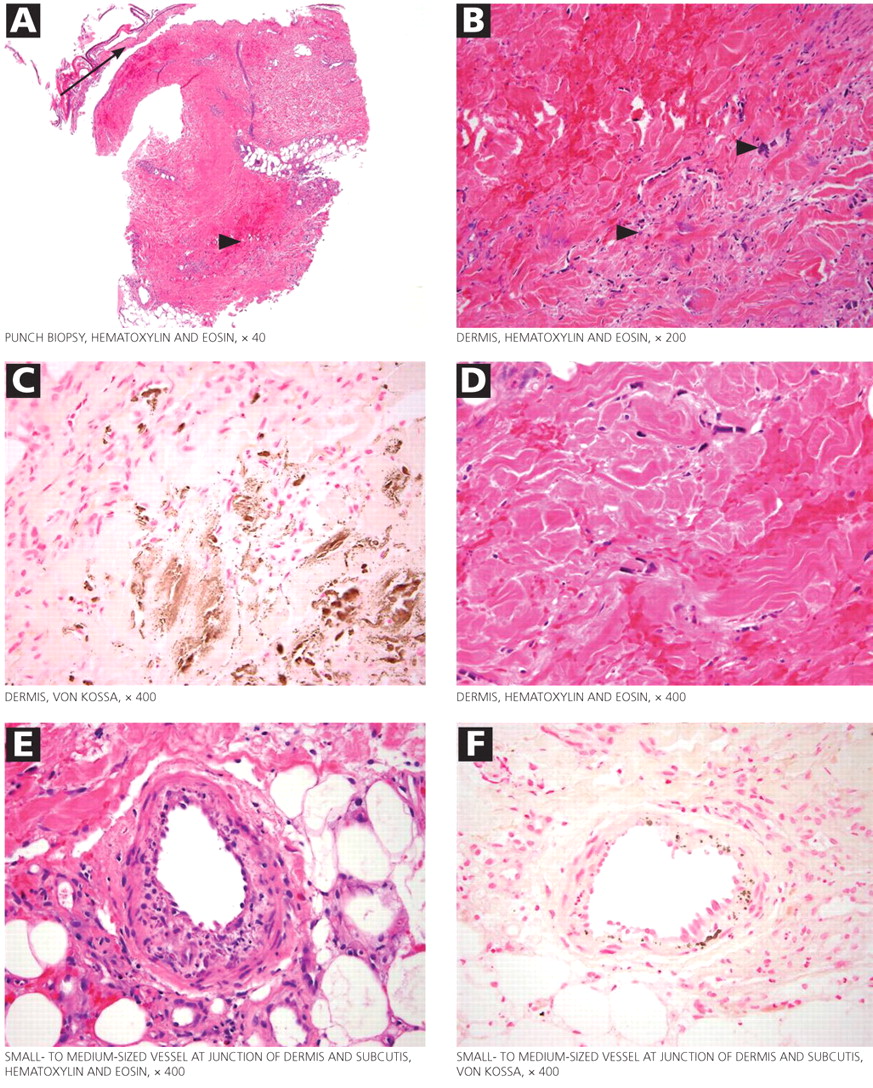
The skin lesions of calciphylaxis usually occur in areas of increased adipose tissue. The lesions may not manifest until several weeks after the initial insult (ie, the elevated calcium-phosphate level). Skin biopsy is recommended if a necrotic skin lesion is identified in a patient with an elevated calcium-phosphate level or in a patient with risk factors for renal, liver, or parathyroid disease.
PROGNOSIS IS POOR
Treatment is supportive. Intensive wound care (with surgical evaluation for skin grafting), hyperbaric oxygen, and possibly tissue plasminogen activator (if there is evidence of a hypercoagulable state and occlusive vasculopathy) may be the most beneficial. Identifying the underlying cause and regulating the calcium-phosphorus product level with diet, phosphate binders, bisphosphonates, and sodium thiosulfate are also important in wound healing. Cinacalcet (Sensipar) and parathyroidectomy should be considered in cases of secondary hyperparathyroidism.
Calciphylaxis is important to recognize early in its course and may require a multidisciplinary approach to treatment. Its prognosis is poor, with death rates ranging from 40% to 60%.
Our patient developed recurrent hepatorenal syndrome and sepsis and eventually died of septic shock.
- Daudén E, Oñate MJ. Calciphylaxis. Dermatol Clin 2008; 26:557–568.
- Pliquett RU, Schwock J, Paschke R, Achenbach H. Calciphylaxis in chronic, non-dialysis-dependent renal disease. BMC Nephrol 2003; 4:8.
- Nigwekar SU, Wolf M, Sterns RH, Hix JK. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol 2008; 3:1139–1143.
Suggested Reading
- Rogers NM, Coates PT. Calcific uraemic arteriolopathy:an update. Curr Opin Nephrol Hypertens 2008; 17:629–634.
- Weenig RH, Sewell LD, Davis MD, McCarthy JT, Pittelkow MR. Calciphylaxis: natural history, risk factor analysis, and outcome. J Am Acad Dermatol 2007; 56:569–579.
Current laboratory values:
- Serum calcium concentration 7.8 mg/dL (reference range 8.5–10.5)
- Phosphorus 6.4 mg/dL (2.5–4.5)
- Corrected calcium-phosphorus product 55
- Parathyroid hormone 275 pg/mL (10–60)
- 25-hydroxyvitamin D 7.4 ng/mL (31–80).
Q: Given the patient’s history, which of the following does her skin lesion likely represent?
- Necrotizing fasciitis
- Calciphylaxis
- Disseminated intravascular coagulation
- Anticoagulant-induced skin necrosis
A: Calciphylaxis, or calcific uremic arteriolopathy, is the most likely. It is rare in people with normal renal function, and still rare but somewhat less so in end-stage renal disease patients undergoing chronic hemodialysis.
WHAT CAUSED IT IN OUR PATIENT?
The cause of calciphylaxis is unknown. Theories have focused on protein C and parathyroid hormone. Putative precipitating factors include acute tubular necrosis, albumin infusion with paracentesis, deficiency of protein C or S, hyperparathyroidism, hyperphosphatemia, hypercalcemia, vitamin D supplementation, steroids, trauma, and warfarin use.
Our patient had a history of hypothyroidism, ulcerative colitis, and end-stage liver disease due to primary sclerosing cholangitis, but no previous history of renal disease.
At the time of her acute renal failure, her calcium-phosphorus level was 55, parathyroid hormone level 274 pg/mL (normal 10–60), and protein C level 26% (normal 76%–147%). At the time the skin lesions were discovered, her protein C level had dropped to 14%; her parathyroid level had returned to normal.
Her home medications included furosemide (Lasix), levothyroxine (Synthroid), mesalamine (Pentasa), azathioprine (Imuran), ursodiol (Actigall), spironolactone (Aldactone), and omeprazole (Prilosec).
NONHEALING LESIONS
The skin lesions of calciphylaxis usually occur in areas of increased adipose tissue. The lesions may not manifest until several weeks after the initial insult (ie, the elevated calcium-phosphate level). Skin biopsy is recommended if a necrotic skin lesion is identified in a patient with an elevated calcium-phosphate level or in a patient with risk factors for renal, liver, or parathyroid disease.
PROGNOSIS IS POOR
Treatment is supportive. Intensive wound care (with surgical evaluation for skin grafting), hyperbaric oxygen, and possibly tissue plasminogen activator (if there is evidence of a hypercoagulable state and occlusive vasculopathy) may be the most beneficial. Identifying the underlying cause and regulating the calcium-phosphorus product level with diet, phosphate binders, bisphosphonates, and sodium thiosulfate are also important in wound healing. Cinacalcet (Sensipar) and parathyroidectomy should be considered in cases of secondary hyperparathyroidism.
Calciphylaxis is important to recognize early in its course and may require a multidisciplinary approach to treatment. Its prognosis is poor, with death rates ranging from 40% to 60%.
Our patient developed recurrent hepatorenal syndrome and sepsis and eventually died of septic shock.
Current laboratory values:
- Serum calcium concentration 7.8 mg/dL (reference range 8.5–10.5)
- Phosphorus 6.4 mg/dL (2.5–4.5)
- Corrected calcium-phosphorus product 55
- Parathyroid hormone 275 pg/mL (10–60)
- 25-hydroxyvitamin D 7.4 ng/mL (31–80).
Q: Given the patient’s history, which of the following does her skin lesion likely represent?
- Necrotizing fasciitis
- Calciphylaxis
- Disseminated intravascular coagulation
- Anticoagulant-induced skin necrosis
A: Calciphylaxis, or calcific uremic arteriolopathy, is the most likely. It is rare in people with normal renal function, and still rare but somewhat less so in end-stage renal disease patients undergoing chronic hemodialysis.
WHAT CAUSED IT IN OUR PATIENT?
The cause of calciphylaxis is unknown. Theories have focused on protein C and parathyroid hormone. Putative precipitating factors include acute tubular necrosis, albumin infusion with paracentesis, deficiency of protein C or S, hyperparathyroidism, hyperphosphatemia, hypercalcemia, vitamin D supplementation, steroids, trauma, and warfarin use.
Our patient had a history of hypothyroidism, ulcerative colitis, and end-stage liver disease due to primary sclerosing cholangitis, but no previous history of renal disease.
At the time of her acute renal failure, her calcium-phosphorus level was 55, parathyroid hormone level 274 pg/mL (normal 10–60), and protein C level 26% (normal 76%–147%). At the time the skin lesions were discovered, her protein C level had dropped to 14%; her parathyroid level had returned to normal.
Her home medications included furosemide (Lasix), levothyroxine (Synthroid), mesalamine (Pentasa), azathioprine (Imuran), ursodiol (Actigall), spironolactone (Aldactone), and omeprazole (Prilosec).
NONHEALING LESIONS
The skin lesions of calciphylaxis usually occur in areas of increased adipose tissue. The lesions may not manifest until several weeks after the initial insult (ie, the elevated calcium-phosphate level). Skin biopsy is recommended if a necrotic skin lesion is identified in a patient with an elevated calcium-phosphate level or in a patient with risk factors for renal, liver, or parathyroid disease.
PROGNOSIS IS POOR
Treatment is supportive. Intensive wound care (with surgical evaluation for skin grafting), hyperbaric oxygen, and possibly tissue plasminogen activator (if there is evidence of a hypercoagulable state and occlusive vasculopathy) may be the most beneficial. Identifying the underlying cause and regulating the calcium-phosphorus product level with diet, phosphate binders, bisphosphonates, and sodium thiosulfate are also important in wound healing. Cinacalcet (Sensipar) and parathyroidectomy should be considered in cases of secondary hyperparathyroidism.
Calciphylaxis is important to recognize early in its course and may require a multidisciplinary approach to treatment. Its prognosis is poor, with death rates ranging from 40% to 60%.
Our patient developed recurrent hepatorenal syndrome and sepsis and eventually died of septic shock.
- Daudén E, Oñate MJ. Calciphylaxis. Dermatol Clin 2008; 26:557–568.
- Pliquett RU, Schwock J, Paschke R, Achenbach H. Calciphylaxis in chronic, non-dialysis-dependent renal disease. BMC Nephrol 2003; 4:8.
- Nigwekar SU, Wolf M, Sterns RH, Hix JK. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol 2008; 3:1139–1143.
Suggested Reading
- Rogers NM, Coates PT. Calcific uraemic arteriolopathy:an update. Curr Opin Nephrol Hypertens 2008; 17:629–634.
- Weenig RH, Sewell LD, Davis MD, McCarthy JT, Pittelkow MR. Calciphylaxis: natural history, risk factor analysis, and outcome. J Am Acad Dermatol 2007; 56:569–579.
- Daudén E, Oñate MJ. Calciphylaxis. Dermatol Clin 2008; 26:557–568.
- Pliquett RU, Schwock J, Paschke R, Achenbach H. Calciphylaxis in chronic, non-dialysis-dependent renal disease. BMC Nephrol 2003; 4:8.
- Nigwekar SU, Wolf M, Sterns RH, Hix JK. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol 2008; 3:1139–1143.
Suggested Reading
- Rogers NM, Coates PT. Calcific uraemic arteriolopathy:an update. Curr Opin Nephrol Hypertens 2008; 17:629–634.
- Weenig RH, Sewell LD, Davis MD, McCarthy JT, Pittelkow MR. Calciphylaxis: natural history, risk factor analysis, and outcome. J Am Acad Dermatol 2007; 56:569–579.
Kidney Transplantation: Who is Eligible?
For many years, the medical community speculated about the possibility of organ transplantation. The first successful transplant of any kind involving humans was a corneal transplant in 1905.1
It wasn’t until 1954 that the first successful organ transplant, a kidney transplant between identical twins, occurred.2 Several new concepts emerged: organ rejection plays a major role in the failure or success of a transplant; and donors and recipients must be matched based on blood group.
Today, about 169,000 people in the US live with a donated kidney. Each year, some 10,500 cadaveric organs are transplanted, and 6,400 donors are living donors.3 The National Kidney Foundation’s recent 10-year initiative, End the Wait!,4 seeks to close the gap between the more than 50,000 people on the transplant waiting list3 and the number of available donor organs.
Since many patients live for years with their transplanted organs, the primary care clinician is likely to see transplant recipients in a family practice or internal medicine setting. While each patient has unique needs, there are commonalities among them.
Renal Consult welcomes any additional comments or questions regarding care of the renal patient. Please address them to [email protected].
Jane S. Davis, CRNP, DNP
Q: I have a 70-year-old male patient who is losing kidney function. He asked me about transplantation, but I really don’t know whether he is eligible to get on the list. Who is eligible? Is there an age limit? Are patients with chronic illnesses (hepatitis B, hepatitis C, HIV) eligible? How long is the list? Where can I find these answers?
There are no specific guidelines regarding eligibility or age restrictions for kidney transplantation in the United States. Most transplant centers look at patients older than 65 a little more carefully than younger patients—they have to be in good health apart from their renal disease. Some centers will not transplant patients older than 70, while others transplant patients who are 80 or older.15 The best thing to do is to refer the patient to the local center or call and find out. Again, the Organ Procurement and Transplantation Network,7 which lists transplant centers and contact information, can be accessed at optn.transplant.hrsa.gov/mem bers/search.asp
Chronic illnesses are not automatic rule-outs for the most part. Very few centers transplant HIV-positive patients, but this does occur, especially in major cities with a large population of persons with HIV (eg, Washington, DC; San Francisco, New York City, Cincinnati). An infectious disease specialist must follow these patients after transplantation and adjust their HAART (highly active antiretroviral therapy) medications to compensate for both the decreased renal function and anti-rejection medications. Hepatitis B and C patients are often accepted as long as liver biopsy shows no cirrhosis and the viral load is low or manageable. If the patient is found to have cirrhosis or decompensation, a combined liver-kidney transplant can be planned, although the success rate of this procedure is low.16,17
Patients with certain types of hepatitis C may be eligible to receive a kidney from a donor with hepatitis C18 in order to shorten the wait time and make use of a kidney that cannot be transplanted into a person not infected with hepatitis C.
Transplant waiting lists vary by region across the country. There is a centralized electronic list managed by UNOS, on which eligible recipients are placed once they have been approved by the transplant center, following the medical work-up and acceptance by the transplant committee at each center. This is referred to as “being listed” or “on the list.” Patients begin to accrue waiting time as soon as they are added, and this list is precise to the second! There is a list for each blood type, with its own set of waiting times in each region.20 Average waiting times, by blood type (ie, ABO), can be searched at www.ustransplant.org/Calcula tors/KidneyWaitTime.aspx
When a donor organ becomes available and has been evaluated by the procurement team, the donor’s information is entered into the system and the computer generates a list of eligible candidates, based on a variety of factors. This is called a “match-run.”
Waiting time is the most important factor, but consideration is given to patients younger than 18, those who have previously donated an organ, and those with high antibody levels (ie, panel-reactive antibodies, or PRAs). Patients in the latter group may find it more difficult to locate a compatible donor, as these patients have been sensitized as a result of prior transplantation, pregnancy, or blood transfusions. It is very rare for a patient to be a perfect match (0 mismatch), but should the right organ become available, the matched patient receives priority consideration.
Besides the ABO match, human leukocyte antigen (HLA) matching of six main HLA antibodies is done. Within each of these six antibodies (HLA-A, B, C, DP, DR, DQ), subgroup matching is also done because some HLA subgroups are more highly correlated with rejection than others.21,22 A more complete explanation of organ matching and allocation can be found on the “Transplant Living” Web site: www.transplantliving.org/beforethetransplant/allocation/matchingorgans.aspx.
Patients should be encouraged to access “Transplant Living” (www.transplantliving.org) and UNOS for information and links. Additional information about transplantation, eligibility, performance statistics, policies, procedures, and other questions and answers, for both clinicians and patients, can be found on the Organ Procurement and Transplantation Network Web site (optn.transplant.hrsa.gov).
Annette Needham, MSN, ARNP, NP-C, CNN-NP, CCTC, Florida Hospital Transplant Center, Orlando
References
1. Armitage WJ, Tullo AB, Larkin DFP. The first successful full-thickness corneal transplant: a commentary on Eduard Zirm’s landmark paper of 1906. Br J Ophthalmol. 2006;90(10):1222-1223.
2. Kidney transplantation: past, present, and future. www.stanford.edu/dept/HPS/transplant/html/history.html. Accessed September 16, 2011.
3. United States Renal Data System. Atlas. www .usrds.org/atlas.htm. Accessed September 16, 2011.
4. National Kidney Foundation. End the wait! www.kidney.org/news/end_the_wait/index.cfm. Accessed September 16, 2011.
5. National Kidney Foundation. Kidney transplant (2011). www.kidney.org/atoz/content/kidneytransnewlease.cfm. Accessed September 16, 2011.
6. United States Renal Data Systems. Presentations and posters (2000-2011). www.usrds.org/presentations.htm. Accessed September 16, 2011.
7. Organ Procurement and Transplantation Network. Members: member directory. optn.transplant.hrsa.gov/members/search.asp. Accessed September 16, 2011.
8. Jensen P, Møller B, Hansen S. Skin cancer in kidney and heart transplant recipients and different long-term immunosuppressive therapy regimens. J Am Acad Dermatol. 2000;42(2 pt 1):307.
9. Wong G, Chapman JR, Craig JC. Cancer screening in renal transplant recipients: what is the evidence? Clin J Am Soc Nephrol. 2008;3 suppl 2:S87-S100.
10. Parker A, Bowles K, Bradley JA, et al; Haemato-oncology subgroup of the British Committee for Standards in Haematology and the British Transplantation Society. Diagnosis of post-transplant lymphoproliferative disorder in solid organ transplant patients. Br J Haematol. 2010;149(5):675-692.
11. Transplant Living. After the transplant (2011). www.transplantliving.org/afterthetransplant/default.aspx. Accessed September 16, 2011.
12. United Network for Organ Sharing. www.unos.org. Accessed September 16, 2011.
13. Kidney Disease Improving Global Outcomes. Managing your adult patients who have a kidney transplant (2010). www.kidney.org/professionals/tools/pdf/02-50-4079_ABB_ManagingTransRecip Bk_PC.pdf. Accessed September 16, 2011.
14. Abbud-Filho M, Adams P, Alberu J, et al. A report of the Lisbon Conference on the care of the kidney transplant recipient. Transplantation. 2007; (Suppl 8):83:1-22.
15. Heldal K, Hartmann A, Leivestad T, et al. Risk variables associated with the outcome of kidney recipients >70 years of age in the new millennium. Nephrol Dial Transplant. 2011;26(8):2706-2711.
16. Chava SP, Singh B, Stangou A, et al. Simultaneous combined liver and kidney transplantation: a single center experience. Clin Transplant. 2010; 24(3):E62-E68.
17. Ruiz R, Kunitake H, Wilkinson AH, et al. Long-term analysis of combined liver and kidney transplantation at a single center. Arch Surg. 2006;141 (8):735-741.
18. Veroux P, Veroux M, Puliatti C, et al. Kidney transplantation from hepatitis C virus-positive donors into hepatitis C virus-positive recipients: a safe way to expand the donor pool? Transplant Proc. 2005;37(6):2571-2573.
19. United States Renal Data Systems, Annual Data Reports. National Kidney and Urologic Disease Information Clearinghouse. Figure 6ii. Transplant (kidney only) wait list and wait times. www.usrds.org/2010/pdf/v2_07.pdf. Accessed September 16, 2011.
20. Arbor Research Collaborative for Health. Kidney waiting time calculator. www.ustransplant.org/Calculators/KidneyWaitTime.aspx. Accessed September 16, 2011.
21. Karakayali FY, Ozdemir H, Kivrakdal S, et al. Recurrent glomerular diseases after renal transplantation. Transplant Proc. 2006;38(2):470-472.
22. Nojima M, Ichikawa Y, Ihara H, et al. Significant effect of HLA-DRB1 matching on acute rejection of kidney transplants within 3 months. Transplant Proc. 2001;33(1-2):1182-1184.
For many years, the medical community speculated about the possibility of organ transplantation. The first successful transplant of any kind involving humans was a corneal transplant in 1905.1
It wasn’t until 1954 that the first successful organ transplant, a kidney transplant between identical twins, occurred.2 Several new concepts emerged: organ rejection plays a major role in the failure or success of a transplant; and donors and recipients must be matched based on blood group.
Today, about 169,000 people in the US live with a donated kidney. Each year, some 10,500 cadaveric organs are transplanted, and 6,400 donors are living donors.3 The National Kidney Foundation’s recent 10-year initiative, End the Wait!,4 seeks to close the gap between the more than 50,000 people on the transplant waiting list3 and the number of available donor organs.
Since many patients live for years with their transplanted organs, the primary care clinician is likely to see transplant recipients in a family practice or internal medicine setting. While each patient has unique needs, there are commonalities among them.
Renal Consult welcomes any additional comments or questions regarding care of the renal patient. Please address them to [email protected].
Jane S. Davis, CRNP, DNP
Q: I have a 70-year-old male patient who is losing kidney function. He asked me about transplantation, but I really don’t know whether he is eligible to get on the list. Who is eligible? Is there an age limit? Are patients with chronic illnesses (hepatitis B, hepatitis C, HIV) eligible? How long is the list? Where can I find these answers?
There are no specific guidelines regarding eligibility or age restrictions for kidney transplantation in the United States. Most transplant centers look at patients older than 65 a little more carefully than younger patients—they have to be in good health apart from their renal disease. Some centers will not transplant patients older than 70, while others transplant patients who are 80 or older.15 The best thing to do is to refer the patient to the local center or call and find out. Again, the Organ Procurement and Transplantation Network,7 which lists transplant centers and contact information, can be accessed at optn.transplant.hrsa.gov/mem bers/search.asp
Chronic illnesses are not automatic rule-outs for the most part. Very few centers transplant HIV-positive patients, but this does occur, especially in major cities with a large population of persons with HIV (eg, Washington, DC; San Francisco, New York City, Cincinnati). An infectious disease specialist must follow these patients after transplantation and adjust their HAART (highly active antiretroviral therapy) medications to compensate for both the decreased renal function and anti-rejection medications. Hepatitis B and C patients are often accepted as long as liver biopsy shows no cirrhosis and the viral load is low or manageable. If the patient is found to have cirrhosis or decompensation, a combined liver-kidney transplant can be planned, although the success rate of this procedure is low.16,17
Patients with certain types of hepatitis C may be eligible to receive a kidney from a donor with hepatitis C18 in order to shorten the wait time and make use of a kidney that cannot be transplanted into a person not infected with hepatitis C.
Transplant waiting lists vary by region across the country. There is a centralized electronic list managed by UNOS, on which eligible recipients are placed once they have been approved by the transplant center, following the medical work-up and acceptance by the transplant committee at each center. This is referred to as “being listed” or “on the list.” Patients begin to accrue waiting time as soon as they are added, and this list is precise to the second! There is a list for each blood type, with its own set of waiting times in each region.20 Average waiting times, by blood type (ie, ABO), can be searched at www.ustransplant.org/Calcula tors/KidneyWaitTime.aspx
When a donor organ becomes available and has been evaluated by the procurement team, the donor’s information is entered into the system and the computer generates a list of eligible candidates, based on a variety of factors. This is called a “match-run.”
Waiting time is the most important factor, but consideration is given to patients younger than 18, those who have previously donated an organ, and those with high antibody levels (ie, panel-reactive antibodies, or PRAs). Patients in the latter group may find it more difficult to locate a compatible donor, as these patients have been sensitized as a result of prior transplantation, pregnancy, or blood transfusions. It is very rare for a patient to be a perfect match (0 mismatch), but should the right organ become available, the matched patient receives priority consideration.
Besides the ABO match, human leukocyte antigen (HLA) matching of six main HLA antibodies is done. Within each of these six antibodies (HLA-A, B, C, DP, DR, DQ), subgroup matching is also done because some HLA subgroups are more highly correlated with rejection than others.21,22 A more complete explanation of organ matching and allocation can be found on the “Transplant Living” Web site: www.transplantliving.org/beforethetransplant/allocation/matchingorgans.aspx.
Patients should be encouraged to access “Transplant Living” (www.transplantliving.org) and UNOS for information and links. Additional information about transplantation, eligibility, performance statistics, policies, procedures, and other questions and answers, for both clinicians and patients, can be found on the Organ Procurement and Transplantation Network Web site (optn.transplant.hrsa.gov).
Annette Needham, MSN, ARNP, NP-C, CNN-NP, CCTC, Florida Hospital Transplant Center, Orlando
References
1. Armitage WJ, Tullo AB, Larkin DFP. The first successful full-thickness corneal transplant: a commentary on Eduard Zirm’s landmark paper of 1906. Br J Ophthalmol. 2006;90(10):1222-1223.
2. Kidney transplantation: past, present, and future. www.stanford.edu/dept/HPS/transplant/html/history.html. Accessed September 16, 2011.
3. United States Renal Data System. Atlas. www .usrds.org/atlas.htm. Accessed September 16, 2011.
4. National Kidney Foundation. End the wait! www.kidney.org/news/end_the_wait/index.cfm. Accessed September 16, 2011.
5. National Kidney Foundation. Kidney transplant (2011). www.kidney.org/atoz/content/kidneytransnewlease.cfm. Accessed September 16, 2011.
6. United States Renal Data Systems. Presentations and posters (2000-2011). www.usrds.org/presentations.htm. Accessed September 16, 2011.
7. Organ Procurement and Transplantation Network. Members: member directory. optn.transplant.hrsa.gov/members/search.asp. Accessed September 16, 2011.
8. Jensen P, Møller B, Hansen S. Skin cancer in kidney and heart transplant recipients and different long-term immunosuppressive therapy regimens. J Am Acad Dermatol. 2000;42(2 pt 1):307.
9. Wong G, Chapman JR, Craig JC. Cancer screening in renal transplant recipients: what is the evidence? Clin J Am Soc Nephrol. 2008;3 suppl 2:S87-S100.
10. Parker A, Bowles K, Bradley JA, et al; Haemato-oncology subgroup of the British Committee for Standards in Haematology and the British Transplantation Society. Diagnosis of post-transplant lymphoproliferative disorder in solid organ transplant patients. Br J Haematol. 2010;149(5):675-692.
11. Transplant Living. After the transplant (2011). www.transplantliving.org/afterthetransplant/default.aspx. Accessed September 16, 2011.
12. United Network for Organ Sharing. www.unos.org. Accessed September 16, 2011.
13. Kidney Disease Improving Global Outcomes. Managing your adult patients who have a kidney transplant (2010). www.kidney.org/professionals/tools/pdf/02-50-4079_ABB_ManagingTransRecip Bk_PC.pdf. Accessed September 16, 2011.
14. Abbud-Filho M, Adams P, Alberu J, et al. A report of the Lisbon Conference on the care of the kidney transplant recipient. Transplantation. 2007; (Suppl 8):83:1-22.
15. Heldal K, Hartmann A, Leivestad T, et al. Risk variables associated with the outcome of kidney recipients >70 years of age in the new millennium. Nephrol Dial Transplant. 2011;26(8):2706-2711.
16. Chava SP, Singh B, Stangou A, et al. Simultaneous combined liver and kidney transplantation: a single center experience. Clin Transplant. 2010; 24(3):E62-E68.
17. Ruiz R, Kunitake H, Wilkinson AH, et al. Long-term analysis of combined liver and kidney transplantation at a single center. Arch Surg. 2006;141 (8):735-741.
18. Veroux P, Veroux M, Puliatti C, et al. Kidney transplantation from hepatitis C virus-positive donors into hepatitis C virus-positive recipients: a safe way to expand the donor pool? Transplant Proc. 2005;37(6):2571-2573.
19. United States Renal Data Systems, Annual Data Reports. National Kidney and Urologic Disease Information Clearinghouse. Figure 6ii. Transplant (kidney only) wait list and wait times. www.usrds.org/2010/pdf/v2_07.pdf. Accessed September 16, 2011.
20. Arbor Research Collaborative for Health. Kidney waiting time calculator. www.ustransplant.org/Calculators/KidneyWaitTime.aspx. Accessed September 16, 2011.
21. Karakayali FY, Ozdemir H, Kivrakdal S, et al. Recurrent glomerular diseases after renal transplantation. Transplant Proc. 2006;38(2):470-472.
22. Nojima M, Ichikawa Y, Ihara H, et al. Significant effect of HLA-DRB1 matching on acute rejection of kidney transplants within 3 months. Transplant Proc. 2001;33(1-2):1182-1184.
For many years, the medical community speculated about the possibility of organ transplantation. The first successful transplant of any kind involving humans was a corneal transplant in 1905.1
It wasn’t until 1954 that the first successful organ transplant, a kidney transplant between identical twins, occurred.2 Several new concepts emerged: organ rejection plays a major role in the failure or success of a transplant; and donors and recipients must be matched based on blood group.
Today, about 169,000 people in the US live with a donated kidney. Each year, some 10,500 cadaveric organs are transplanted, and 6,400 donors are living donors.3 The National Kidney Foundation’s recent 10-year initiative, End the Wait!,4 seeks to close the gap between the more than 50,000 people on the transplant waiting list3 and the number of available donor organs.
Since many patients live for years with their transplanted organs, the primary care clinician is likely to see transplant recipients in a family practice or internal medicine setting. While each patient has unique needs, there are commonalities among them.
Renal Consult welcomes any additional comments or questions regarding care of the renal patient. Please address them to [email protected].
Jane S. Davis, CRNP, DNP
Q: I have a 70-year-old male patient who is losing kidney function. He asked me about transplantation, but I really don’t know whether he is eligible to get on the list. Who is eligible? Is there an age limit? Are patients with chronic illnesses (hepatitis B, hepatitis C, HIV) eligible? How long is the list? Where can I find these answers?
There are no specific guidelines regarding eligibility or age restrictions for kidney transplantation in the United States. Most transplant centers look at patients older than 65 a little more carefully than younger patients—they have to be in good health apart from their renal disease. Some centers will not transplant patients older than 70, while others transplant patients who are 80 or older.15 The best thing to do is to refer the patient to the local center or call and find out. Again, the Organ Procurement and Transplantation Network,7 which lists transplant centers and contact information, can be accessed at optn.transplant.hrsa.gov/mem bers/search.asp
Chronic illnesses are not automatic rule-outs for the most part. Very few centers transplant HIV-positive patients, but this does occur, especially in major cities with a large population of persons with HIV (eg, Washington, DC; San Francisco, New York City, Cincinnati). An infectious disease specialist must follow these patients after transplantation and adjust their HAART (highly active antiretroviral therapy) medications to compensate for both the decreased renal function and anti-rejection medications. Hepatitis B and C patients are often accepted as long as liver biopsy shows no cirrhosis and the viral load is low or manageable. If the patient is found to have cirrhosis or decompensation, a combined liver-kidney transplant can be planned, although the success rate of this procedure is low.16,17
Patients with certain types of hepatitis C may be eligible to receive a kidney from a donor with hepatitis C18 in order to shorten the wait time and make use of a kidney that cannot be transplanted into a person not infected with hepatitis C.
Transplant waiting lists vary by region across the country. There is a centralized electronic list managed by UNOS, on which eligible recipients are placed once they have been approved by the transplant center, following the medical work-up and acceptance by the transplant committee at each center. This is referred to as “being listed” or “on the list.” Patients begin to accrue waiting time as soon as they are added, and this list is precise to the second! There is a list for each blood type, with its own set of waiting times in each region.20 Average waiting times, by blood type (ie, ABO), can be searched at www.ustransplant.org/Calcula tors/KidneyWaitTime.aspx
When a donor organ becomes available and has been evaluated by the procurement team, the donor’s information is entered into the system and the computer generates a list of eligible candidates, based on a variety of factors. This is called a “match-run.”
Waiting time is the most important factor, but consideration is given to patients younger than 18, those who have previously donated an organ, and those with high antibody levels (ie, panel-reactive antibodies, or PRAs). Patients in the latter group may find it more difficult to locate a compatible donor, as these patients have been sensitized as a result of prior transplantation, pregnancy, or blood transfusions. It is very rare for a patient to be a perfect match (0 mismatch), but should the right organ become available, the matched patient receives priority consideration.
Besides the ABO match, human leukocyte antigen (HLA) matching of six main HLA antibodies is done. Within each of these six antibodies (HLA-A, B, C, DP, DR, DQ), subgroup matching is also done because some HLA subgroups are more highly correlated with rejection than others.21,22 A more complete explanation of organ matching and allocation can be found on the “Transplant Living” Web site: www.transplantliving.org/beforethetransplant/allocation/matchingorgans.aspx.
Patients should be encouraged to access “Transplant Living” (www.transplantliving.org) and UNOS for information and links. Additional information about transplantation, eligibility, performance statistics, policies, procedures, and other questions and answers, for both clinicians and patients, can be found on the Organ Procurement and Transplantation Network Web site (optn.transplant.hrsa.gov).
Annette Needham, MSN, ARNP, NP-C, CNN-NP, CCTC, Florida Hospital Transplant Center, Orlando
References
1. Armitage WJ, Tullo AB, Larkin DFP. The first successful full-thickness corneal transplant: a commentary on Eduard Zirm’s landmark paper of 1906. Br J Ophthalmol. 2006;90(10):1222-1223.
2. Kidney transplantation: past, present, and future. www.stanford.edu/dept/HPS/transplant/html/history.html. Accessed September 16, 2011.
3. United States Renal Data System. Atlas. www .usrds.org/atlas.htm. Accessed September 16, 2011.
4. National Kidney Foundation. End the wait! www.kidney.org/news/end_the_wait/index.cfm. Accessed September 16, 2011.
5. National Kidney Foundation. Kidney transplant (2011). www.kidney.org/atoz/content/kidneytransnewlease.cfm. Accessed September 16, 2011.
6. United States Renal Data Systems. Presentations and posters (2000-2011). www.usrds.org/presentations.htm. Accessed September 16, 2011.
7. Organ Procurement and Transplantation Network. Members: member directory. optn.transplant.hrsa.gov/members/search.asp. Accessed September 16, 2011.
8. Jensen P, Møller B, Hansen S. Skin cancer in kidney and heart transplant recipients and different long-term immunosuppressive therapy regimens. J Am Acad Dermatol. 2000;42(2 pt 1):307.
9. Wong G, Chapman JR, Craig JC. Cancer screening in renal transplant recipients: what is the evidence? Clin J Am Soc Nephrol. 2008;3 suppl 2:S87-S100.
10. Parker A, Bowles K, Bradley JA, et al; Haemato-oncology subgroup of the British Committee for Standards in Haematology and the British Transplantation Society. Diagnosis of post-transplant lymphoproliferative disorder in solid organ transplant patients. Br J Haematol. 2010;149(5):675-692.
11. Transplant Living. After the transplant (2011). www.transplantliving.org/afterthetransplant/default.aspx. Accessed September 16, 2011.
12. United Network for Organ Sharing. www.unos.org. Accessed September 16, 2011.
13. Kidney Disease Improving Global Outcomes. Managing your adult patients who have a kidney transplant (2010). www.kidney.org/professionals/tools/pdf/02-50-4079_ABB_ManagingTransRecip Bk_PC.pdf. Accessed September 16, 2011.
14. Abbud-Filho M, Adams P, Alberu J, et al. A report of the Lisbon Conference on the care of the kidney transplant recipient. Transplantation. 2007; (Suppl 8):83:1-22.
15. Heldal K, Hartmann A, Leivestad T, et al. Risk variables associated with the outcome of kidney recipients >70 years of age in the new millennium. Nephrol Dial Transplant. 2011;26(8):2706-2711.
16. Chava SP, Singh B, Stangou A, et al. Simultaneous combined liver and kidney transplantation: a single center experience. Clin Transplant. 2010; 24(3):E62-E68.
17. Ruiz R, Kunitake H, Wilkinson AH, et al. Long-term analysis of combined liver and kidney transplantation at a single center. Arch Surg. 2006;141 (8):735-741.
18. Veroux P, Veroux M, Puliatti C, et al. Kidney transplantation from hepatitis C virus-positive donors into hepatitis C virus-positive recipients: a safe way to expand the donor pool? Transplant Proc. 2005;37(6):2571-2573.
19. United States Renal Data Systems, Annual Data Reports. National Kidney and Urologic Disease Information Clearinghouse. Figure 6ii. Transplant (kidney only) wait list and wait times. www.usrds.org/2010/pdf/v2_07.pdf. Accessed September 16, 2011.
20. Arbor Research Collaborative for Health. Kidney waiting time calculator. www.ustransplant.org/Calculators/KidneyWaitTime.aspx. Accessed September 16, 2011.
21. Karakayali FY, Ozdemir H, Kivrakdal S, et al. Recurrent glomerular diseases after renal transplantation. Transplant Proc. 2006;38(2):470-472.
22. Nojima M, Ichikawa Y, Ihara H, et al. Significant effect of HLA-DRB1 matching on acute rejection of kidney transplants within 3 months. Transplant Proc. 2001;33(1-2):1182-1184.
Kidney Transplantation: Posttransplant Preventive Care
For many years, the medical community speculated about the possibility of organ transplantation. The first successful transplant of any kind involving humans was a corneal transplant in 1905.1
It wasn’t until 1954 that the first successful organ transplant, a kidney transplant between identical twins, occurred.2 Several new concepts emerged: organ rejection plays a major role in the failure or success of a transplant; and donors and recipients must be matched based on blood group.
Today, about 169,000 people in the US live with a donated kidney. Each year, some 10,500 cadaveric organs are transplanted, and 6,400 donors are living donors.3 The National Kidney Foundation’s recent 10-year initiative, End the Wait!,4 seeks to close the gap between the more than 50,000 people on the transplant waiting list3 and the number of available donor organs.
Since many patients live for years with their transplanted organs, the primary care clinician is likely to see transplant recipients in a family practice or internal medicine setting. While each patient has unique needs, there are commonalities among them.
Renal Consult welcomes any additional comments or questions regarding care of the renal patient. Please address them to [email protected].
Jane S. Davis, CRNP, DNP
Q: I am in primary care and have a kidney transplant patient that I see annually for her Pap test and pelvic exam. Is there anything in particular that I am supposed to look for? I feel out of my comfort zone.
As with most people, preventive care is vital and posttransplant patients are no different. However, there are a few “special circumstances” to keep in mind.
Besides ascertaining that posttransplant patients are taking their medications every day, determine whether they have recently had a generic substituted for their regular anti-rejection meds. Many transplant medications have generic equivalents now; while we want changes made only with the approval of a transplant center, it is legal for a pharmacy to substitute a generic without notifying the transplant nephrologist. We have seen rejection, toxicities, or changes in creatinine levels due to substitution of generics—or even substitution from one generic equivalent to another. These medications have a small effective window and have to be closely monitored whenever different manufacturers are used.
In addition, some patients will stop taking their immunosuppressive drug, either because they “feel better” and don’t believe they need it anymore, or because they can no longer afford it. Medicare will only pay for 36 months of these medications, and patients often halve the dose or stop taking the medication altogether when the cost becomes too high.5
There is a very useful Web site on transplant medications from the United States Renal Data System.6 The site, which also offers a wealth of information on chronic kidney disease (CKD), is www.usrds.org/presentations.htm
Dosing for any medication is based on the patient’s glomerular filtration rate (GFR). Your transplant patients have been taught their baseline creatinine level, but some do forget. Even after transplant (whether of a kidney, a pancreas, a liver, lungs, or a heart), the immunosuppressive medications will affect the GFR, and the patient is a CKD patient.
If a patient’s creatinine level is 1.9 mg/dL (normal range, 0.6 to 1.2), but it has varied between 1.8 and 2.0 ever since the transplant and they are not having any other issues, this is “normal” for them and no cause for alarm. On the other hand, if the creatinine level is 1.9 mg/dL and the patient reports that it is always 1.2, they need immediate referral. If the patient is new to the area, you can find a local transplant center on the Organ Procurement and Transplantation Network directory7: optn.transplant.hrsa.gov/mem bers/search.asp
Screening for infections and malignancies is another important aspect of posttransplant care. I advise all patients to see a dermatologist at least once annually, as the risk for skin cancer is increased sevenfold in a transplant patient, compared with the general population.8 Annual Pap test, pelvic exam, and mammogram are important for female posttransplant patients, as is annual prostate-specific antigen testing for male posttransplant patients older than 45 with a life expectancy of at least 10 years.9
During the physical exam, the clinician should always check for lymphadenopathy or any other “lumps and bumps,” as posttransplant lymphoproliferative disorder is also a risk associated with long-term immunosuppression.10 A wonderful online resource for patients and providers, “Transplant Living,”11 has an excellent section on posttransplant care: www.transplantliving.org/af terthetransplant/default.aspx. This Web site is managed by the United Network of Organ Sharing12 (UNOS; www.unos.org), the organization that manages organ transplantation and donation under contract with the federal government.
Routine vaccinations are recommended—especially pneumococcal vaccine and an annual flu shot. Diphtheria-pertussis-tetanus, hepatitis A, hepatitis B, inactivated polio, and typhoid are also acceptable vaccines for a transplant patient. Vaccines that are contraindicated after transplantation include varicella, bacillus Calmette-Guérin, smallpox, intranasal influenza, live oral typhoid, measles, mumps, rubella, oral polio, live Japanese B encephalitis, and yellow fever.13,14
References
1. Armitage WJ, Tullo AB, Larkin DFP. The first successful full-thickness corneal transplant: a commentary on Eduard Zirm’s landmark paper of 1906. Br J Ophthalmol. 2006;90(10):1222-1223.
2. Kidney transplantation: past, present, and future. www.stanford.edu/dept/HPS/transplant/html/history.html. Accessed September 16, 2011.
3. United States Renal Data System. Atlas. www .usrds.org/atlas.htm. Accessed September 16, 2011.
4. National Kidney Foundation. End the wait! www.kidney.org/news/end_the_wait/index.cfm. Accessed September 16, 2011.
5. National Kidney Foundation. Kidney transplant (2011). www.kidney.org/atoz/content/kidneytransnewlease.cfm. Accessed September 16, 2011.
6. United States Renal Data Systems. Presentations and posters (2000-2011). www.usrds.org/presentations.htm. Accessed September 16, 2011.
7. Organ Procurement and Transplantation Network. Members: member directory. optn.transplant.hrsa.gov/members/search.asp. Accessed September 16, 2011.
8. Jensen P, Møller B, Hansen S. Skin cancer in kidney and heart transplant recipients and different long-term immunosuppressive therapy regimens. J Am Acad Dermatol. 2000;42(2 pt 1):307.
9. Wong G, Chapman JR, Craig JC. Cancer screening in renal transplant recipients: what is the evidence? Clin J Am Soc Nephrol. 2008;3 suppl 2:S87-S100.
10. Parker A, Bowles K, Bradley JA, et al; Haemato-oncology subgroup of the British Committee for Standards in Haematology and the British Transplantation Society. Diagnosis of post-transplant lymphoproliferative disorder in solid organ transplant patients. Br J Haematol. 2010;149(5):675-692.
11. Transplant Living. After the transplant (2011). www.transplantliving.org/afterthetransplant/default.aspx. Accessed September 16, 2011.
12. United Network for Organ Sharing. www.unos.org. Accessed September 16, 2011.
13. Kidney Disease Improving Global Outcomes. Managing your adult patients who have a kidney transplant (2010). www.kidney.org/professionals/tools/pdf/02-50-4079_ABB_ManagingTransRecip Bk_PC.pdf. Accessed September 16, 2011.
14. Abbud-Filho M, Adams P, Alberu J, et al. A report of the Lisbon Conference on the care of the kidney transplant recipient. Transplantation. 2007; (Suppl 8):83:1-22.
15. Heldal K, Hartmann A, Leivestad T, et al. Risk variables associated with the outcome of kidney recipients >70 years of age in the new millennium. Nephrol Dial Transplant. 2011;26(8):2706-2711.
16. Chava SP, Singh B, Stangou A, et al. Simultaneous combined liver and kidney transplantation: a single center experience. Clin Transplant. 2010; 24(3):E62-E68.
17. Ruiz R, Kunitake H, Wilkinson AH, et al. Long-term analysis of combined liver and kidney transplantation at a single center. Arch Surg. 2006;141 (8):735-741.
18. Veroux P, Veroux M, Puliatti C, et al. Kidney transplantation from hepatitis C virus-positive donors into hepatitis C virus-positive recipients: a safe way to expand the donor pool? Transplant Proc. 2005;37(6):2571-2573.
19. United States Renal Data Systems, Annual Data Reports. National Kidney and Urologic Disease Information Clearinghouse. Figure 6ii. Transplant (kidney only) wait list and wait times. www.usrds.org/2010/pdf/v2_07.pdf. Accessed September 16, 2011.
20. Arbor Research Collaborative for Health. Kidney waiting time calculator. www.ustransplant.org/Calculators/KidneyWaitTime.aspx. Accessed September 16, 2011.
21. Karakayali FY, Ozdemir H, Kivrakdal S, et al. Recurrent glomerular diseases after renal transplantation. Transplant Proc. 2006;38(2):470-472.
22. Nojima M, Ichikawa Y, Ihara H, et al. Significant effect of HLA-DRB1 matching on acute rejection of kidney transplants within 3 months. Transplant Proc. 2001;33(1-2):1182-1184.
For many years, the medical community speculated about the possibility of organ transplantation. The first successful transplant of any kind involving humans was a corneal transplant in 1905.1
It wasn’t until 1954 that the first successful organ transplant, a kidney transplant between identical twins, occurred.2 Several new concepts emerged: organ rejection plays a major role in the failure or success of a transplant; and donors and recipients must be matched based on blood group.
Today, about 169,000 people in the US live with a donated kidney. Each year, some 10,500 cadaveric organs are transplanted, and 6,400 donors are living donors.3 The National Kidney Foundation’s recent 10-year initiative, End the Wait!,4 seeks to close the gap between the more than 50,000 people on the transplant waiting list3 and the number of available donor organs.
Since many patients live for years with their transplanted organs, the primary care clinician is likely to see transplant recipients in a family practice or internal medicine setting. While each patient has unique needs, there are commonalities among them.
Renal Consult welcomes any additional comments or questions regarding care of the renal patient. Please address them to [email protected].
Jane S. Davis, CRNP, DNP
Q: I am in primary care and have a kidney transplant patient that I see annually for her Pap test and pelvic exam. Is there anything in particular that I am supposed to look for? I feel out of my comfort zone.
As with most people, preventive care is vital and posttransplant patients are no different. However, there are a few “special circumstances” to keep in mind.
Besides ascertaining that posttransplant patients are taking their medications every day, determine whether they have recently had a generic substituted for their regular anti-rejection meds. Many transplant medications have generic equivalents now; while we want changes made only with the approval of a transplant center, it is legal for a pharmacy to substitute a generic without notifying the transplant nephrologist. We have seen rejection, toxicities, or changes in creatinine levels due to substitution of generics—or even substitution from one generic equivalent to another. These medications have a small effective window and have to be closely monitored whenever different manufacturers are used.
In addition, some patients will stop taking their immunosuppressive drug, either because they “feel better” and don’t believe they need it anymore, or because they can no longer afford it. Medicare will only pay for 36 months of these medications, and patients often halve the dose or stop taking the medication altogether when the cost becomes too high.5
There is a very useful Web site on transplant medications from the United States Renal Data System.6 The site, which also offers a wealth of information on chronic kidney disease (CKD), is www.usrds.org/presentations.htm
Dosing for any medication is based on the patient’s glomerular filtration rate (GFR). Your transplant patients have been taught their baseline creatinine level, but some do forget. Even after transplant (whether of a kidney, a pancreas, a liver, lungs, or a heart), the immunosuppressive medications will affect the GFR, and the patient is a CKD patient.
If a patient’s creatinine level is 1.9 mg/dL (normal range, 0.6 to 1.2), but it has varied between 1.8 and 2.0 ever since the transplant and they are not having any other issues, this is “normal” for them and no cause for alarm. On the other hand, if the creatinine level is 1.9 mg/dL and the patient reports that it is always 1.2, they need immediate referral. If the patient is new to the area, you can find a local transplant center on the Organ Procurement and Transplantation Network directory7: optn.transplant.hrsa.gov/mem bers/search.asp
Screening for infections and malignancies is another important aspect of posttransplant care. I advise all patients to see a dermatologist at least once annually, as the risk for skin cancer is increased sevenfold in a transplant patient, compared with the general population.8 Annual Pap test, pelvic exam, and mammogram are important for female posttransplant patients, as is annual prostate-specific antigen testing for male posttransplant patients older than 45 with a life expectancy of at least 10 years.9
During the physical exam, the clinician should always check for lymphadenopathy or any other “lumps and bumps,” as posttransplant lymphoproliferative disorder is also a risk associated with long-term immunosuppression.10 A wonderful online resource for patients and providers, “Transplant Living,”11 has an excellent section on posttransplant care: www.transplantliving.org/af terthetransplant/default.aspx. This Web site is managed by the United Network of Organ Sharing12 (UNOS; www.unos.org), the organization that manages organ transplantation and donation under contract with the federal government.
Routine vaccinations are recommended—especially pneumococcal vaccine and an annual flu shot. Diphtheria-pertussis-tetanus, hepatitis A, hepatitis B, inactivated polio, and typhoid are also acceptable vaccines for a transplant patient. Vaccines that are contraindicated after transplantation include varicella, bacillus Calmette-Guérin, smallpox, intranasal influenza, live oral typhoid, measles, mumps, rubella, oral polio, live Japanese B encephalitis, and yellow fever.13,14
References
1. Armitage WJ, Tullo AB, Larkin DFP. The first successful full-thickness corneal transplant: a commentary on Eduard Zirm’s landmark paper of 1906. Br J Ophthalmol. 2006;90(10):1222-1223.
2. Kidney transplantation: past, present, and future. www.stanford.edu/dept/HPS/transplant/html/history.html. Accessed September 16, 2011.
3. United States Renal Data System. Atlas. www .usrds.org/atlas.htm. Accessed September 16, 2011.
4. National Kidney Foundation. End the wait! www.kidney.org/news/end_the_wait/index.cfm. Accessed September 16, 2011.
5. National Kidney Foundation. Kidney transplant (2011). www.kidney.org/atoz/content/kidneytransnewlease.cfm. Accessed September 16, 2011.
6. United States Renal Data Systems. Presentations and posters (2000-2011). www.usrds.org/presentations.htm. Accessed September 16, 2011.
7. Organ Procurement and Transplantation Network. Members: member directory. optn.transplant.hrsa.gov/members/search.asp. Accessed September 16, 2011.
8. Jensen P, Møller B, Hansen S. Skin cancer in kidney and heart transplant recipients and different long-term immunosuppressive therapy regimens. J Am Acad Dermatol. 2000;42(2 pt 1):307.
9. Wong G, Chapman JR, Craig JC. Cancer screening in renal transplant recipients: what is the evidence? Clin J Am Soc Nephrol. 2008;3 suppl 2:S87-S100.
10. Parker A, Bowles K, Bradley JA, et al; Haemato-oncology subgroup of the British Committee for Standards in Haematology and the British Transplantation Society. Diagnosis of post-transplant lymphoproliferative disorder in solid organ transplant patients. Br J Haematol. 2010;149(5):675-692.
11. Transplant Living. After the transplant (2011). www.transplantliving.org/afterthetransplant/default.aspx. Accessed September 16, 2011.
12. United Network for Organ Sharing. www.unos.org. Accessed September 16, 2011.
13. Kidney Disease Improving Global Outcomes. Managing your adult patients who have a kidney transplant (2010). www.kidney.org/professionals/tools/pdf/02-50-4079_ABB_ManagingTransRecip Bk_PC.pdf. Accessed September 16, 2011.
14. Abbud-Filho M, Adams P, Alberu J, et al. A report of the Lisbon Conference on the care of the kidney transplant recipient. Transplantation. 2007; (Suppl 8):83:1-22.
15. Heldal K, Hartmann A, Leivestad T, et al. Risk variables associated with the outcome of kidney recipients >70 years of age in the new millennium. Nephrol Dial Transplant. 2011;26(8):2706-2711.
16. Chava SP, Singh B, Stangou A, et al. Simultaneous combined liver and kidney transplantation: a single center experience. Clin Transplant. 2010; 24(3):E62-E68.
17. Ruiz R, Kunitake H, Wilkinson AH, et al. Long-term analysis of combined liver and kidney transplantation at a single center. Arch Surg. 2006;141 (8):735-741.
18. Veroux P, Veroux M, Puliatti C, et al. Kidney transplantation from hepatitis C virus-positive donors into hepatitis C virus-positive recipients: a safe way to expand the donor pool? Transplant Proc. 2005;37(6):2571-2573.
19. United States Renal Data Systems, Annual Data Reports. National Kidney and Urologic Disease Information Clearinghouse. Figure 6ii. Transplant (kidney only) wait list and wait times. www.usrds.org/2010/pdf/v2_07.pdf. Accessed September 16, 2011.
20. Arbor Research Collaborative for Health. Kidney waiting time calculator. www.ustransplant.org/Calculators/KidneyWaitTime.aspx. Accessed September 16, 2011.
21. Karakayali FY, Ozdemir H, Kivrakdal S, et al. Recurrent glomerular diseases after renal transplantation. Transplant Proc. 2006;38(2):470-472.
22. Nojima M, Ichikawa Y, Ihara H, et al. Significant effect of HLA-DRB1 matching on acute rejection of kidney transplants within 3 months. Transplant Proc. 2001;33(1-2):1182-1184.
For many years, the medical community speculated about the possibility of organ transplantation. The first successful transplant of any kind involving humans was a corneal transplant in 1905.1
It wasn’t until 1954 that the first successful organ transplant, a kidney transplant between identical twins, occurred.2 Several new concepts emerged: organ rejection plays a major role in the failure or success of a transplant; and donors and recipients must be matched based on blood group.
Today, about 169,000 people in the US live with a donated kidney. Each year, some 10,500 cadaveric organs are transplanted, and 6,400 donors are living donors.3 The National Kidney Foundation’s recent 10-year initiative, End the Wait!,4 seeks to close the gap between the more than 50,000 people on the transplant waiting list3 and the number of available donor organs.
Since many patients live for years with their transplanted organs, the primary care clinician is likely to see transplant recipients in a family practice or internal medicine setting. While each patient has unique needs, there are commonalities among them.
Renal Consult welcomes any additional comments or questions regarding care of the renal patient. Please address them to [email protected].
Jane S. Davis, CRNP, DNP
Q: I am in primary care and have a kidney transplant patient that I see annually for her Pap test and pelvic exam. Is there anything in particular that I am supposed to look for? I feel out of my comfort zone.
As with most people, preventive care is vital and posttransplant patients are no different. However, there are a few “special circumstances” to keep in mind.
Besides ascertaining that posttransplant patients are taking their medications every day, determine whether they have recently had a generic substituted for their regular anti-rejection meds. Many transplant medications have generic equivalents now; while we want changes made only with the approval of a transplant center, it is legal for a pharmacy to substitute a generic without notifying the transplant nephrologist. We have seen rejection, toxicities, or changes in creatinine levels due to substitution of generics—or even substitution from one generic equivalent to another. These medications have a small effective window and have to be closely monitored whenever different manufacturers are used.
In addition, some patients will stop taking their immunosuppressive drug, either because they “feel better” and don’t believe they need it anymore, or because they can no longer afford it. Medicare will only pay for 36 months of these medications, and patients often halve the dose or stop taking the medication altogether when the cost becomes too high.5
There is a very useful Web site on transplant medications from the United States Renal Data System.6 The site, which also offers a wealth of information on chronic kidney disease (CKD), is www.usrds.org/presentations.htm
Dosing for any medication is based on the patient’s glomerular filtration rate (GFR). Your transplant patients have been taught their baseline creatinine level, but some do forget. Even after transplant (whether of a kidney, a pancreas, a liver, lungs, or a heart), the immunosuppressive medications will affect the GFR, and the patient is a CKD patient.
If a patient’s creatinine level is 1.9 mg/dL (normal range, 0.6 to 1.2), but it has varied between 1.8 and 2.0 ever since the transplant and they are not having any other issues, this is “normal” for them and no cause for alarm. On the other hand, if the creatinine level is 1.9 mg/dL and the patient reports that it is always 1.2, they need immediate referral. If the patient is new to the area, you can find a local transplant center on the Organ Procurement and Transplantation Network directory7: optn.transplant.hrsa.gov/mem bers/search.asp
Screening for infections and malignancies is another important aspect of posttransplant care. I advise all patients to see a dermatologist at least once annually, as the risk for skin cancer is increased sevenfold in a transplant patient, compared with the general population.8 Annual Pap test, pelvic exam, and mammogram are important for female posttransplant patients, as is annual prostate-specific antigen testing for male posttransplant patients older than 45 with a life expectancy of at least 10 years.9
During the physical exam, the clinician should always check for lymphadenopathy or any other “lumps and bumps,” as posttransplant lymphoproliferative disorder is also a risk associated with long-term immunosuppression.10 A wonderful online resource for patients and providers, “Transplant Living,”11 has an excellent section on posttransplant care: www.transplantliving.org/af terthetransplant/default.aspx. This Web site is managed by the United Network of Organ Sharing12 (UNOS; www.unos.org), the organization that manages organ transplantation and donation under contract with the federal government.
Routine vaccinations are recommended—especially pneumococcal vaccine and an annual flu shot. Diphtheria-pertussis-tetanus, hepatitis A, hepatitis B, inactivated polio, and typhoid are also acceptable vaccines for a transplant patient. Vaccines that are contraindicated after transplantation include varicella, bacillus Calmette-Guérin, smallpox, intranasal influenza, live oral typhoid, measles, mumps, rubella, oral polio, live Japanese B encephalitis, and yellow fever.13,14
References
1. Armitage WJ, Tullo AB, Larkin DFP. The first successful full-thickness corneal transplant: a commentary on Eduard Zirm’s landmark paper of 1906. Br J Ophthalmol. 2006;90(10):1222-1223.
2. Kidney transplantation: past, present, and future. www.stanford.edu/dept/HPS/transplant/html/history.html. Accessed September 16, 2011.
3. United States Renal Data System. Atlas. www .usrds.org/atlas.htm. Accessed September 16, 2011.
4. National Kidney Foundation. End the wait! www.kidney.org/news/end_the_wait/index.cfm. Accessed September 16, 2011.
5. National Kidney Foundation. Kidney transplant (2011). www.kidney.org/atoz/content/kidneytransnewlease.cfm. Accessed September 16, 2011.
6. United States Renal Data Systems. Presentations and posters (2000-2011). www.usrds.org/presentations.htm. Accessed September 16, 2011.
7. Organ Procurement and Transplantation Network. Members: member directory. optn.transplant.hrsa.gov/members/search.asp. Accessed September 16, 2011.
8. Jensen P, Møller B, Hansen S. Skin cancer in kidney and heart transplant recipients and different long-term immunosuppressive therapy regimens. J Am Acad Dermatol. 2000;42(2 pt 1):307.
9. Wong G, Chapman JR, Craig JC. Cancer screening in renal transplant recipients: what is the evidence? Clin J Am Soc Nephrol. 2008;3 suppl 2:S87-S100.
10. Parker A, Bowles K, Bradley JA, et al; Haemato-oncology subgroup of the British Committee for Standards in Haematology and the British Transplantation Society. Diagnosis of post-transplant lymphoproliferative disorder in solid organ transplant patients. Br J Haematol. 2010;149(5):675-692.
11. Transplant Living. After the transplant (2011). www.transplantliving.org/afterthetransplant/default.aspx. Accessed September 16, 2011.
12. United Network for Organ Sharing. www.unos.org. Accessed September 16, 2011.
13. Kidney Disease Improving Global Outcomes. Managing your adult patients who have a kidney transplant (2010). www.kidney.org/professionals/tools/pdf/02-50-4079_ABB_ManagingTransRecip Bk_PC.pdf. Accessed September 16, 2011.
14. Abbud-Filho M, Adams P, Alberu J, et al. A report of the Lisbon Conference on the care of the kidney transplant recipient. Transplantation. 2007; (Suppl 8):83:1-22.
15. Heldal K, Hartmann A, Leivestad T, et al. Risk variables associated with the outcome of kidney recipients >70 years of age in the new millennium. Nephrol Dial Transplant. 2011;26(8):2706-2711.
16. Chava SP, Singh B, Stangou A, et al. Simultaneous combined liver and kidney transplantation: a single center experience. Clin Transplant. 2010; 24(3):E62-E68.
17. Ruiz R, Kunitake H, Wilkinson AH, et al. Long-term analysis of combined liver and kidney transplantation at a single center. Arch Surg. 2006;141 (8):735-741.
18. Veroux P, Veroux M, Puliatti C, et al. Kidney transplantation from hepatitis C virus-positive donors into hepatitis C virus-positive recipients: a safe way to expand the donor pool? Transplant Proc. 2005;37(6):2571-2573.
19. United States Renal Data Systems, Annual Data Reports. National Kidney and Urologic Disease Information Clearinghouse. Figure 6ii. Transplant (kidney only) wait list and wait times. www.usrds.org/2010/pdf/v2_07.pdf. Accessed September 16, 2011.
20. Arbor Research Collaborative for Health. Kidney waiting time calculator. www.ustransplant.org/Calculators/KidneyWaitTime.aspx. Accessed September 16, 2011.
21. Karakayali FY, Ozdemir H, Kivrakdal S, et al. Recurrent glomerular diseases after renal transplantation. Transplant Proc. 2006;38(2):470-472.
22. Nojima M, Ichikawa Y, Ihara H, et al. Significant effect of HLA-DRB1 matching on acute rejection of kidney transplants within 3 months. Transplant Proc. 2001;33(1-2):1182-1184.
Are serum uric acid levels always elevated in acute gout?
NO. Many patients with acute gout (11%-49%) have normal serum uric acid (SUA) levels (strength of recommendation [SOR]: A, prospective cohort studies). Patients taking allopurinol are significantly more likely to have normal uric acid levels during acute gout attacks (SOR: B, extrapolated from prospective cohorts).
Evidence summary
Six studies have evaluated SUA levels in patients with acute gout. Despite variations in diagnostic approach (clinical criteria vs synovial crystal analysis) and definitions of normal SUA (based on laboratory methods and sex), all 6 studies found normal levels in 11% to 49% of patients with acute gout (TABLE 1).
TABLE 1
Serum uric acid and acute gout: The evidence
| Type of cohort (n) | LOE* | Setting | Method of diagnosis | % with normal serum uric acid |
|---|---|---|---|---|
| Prospective1 (28) | 1b | Veterans Administration rheumatology clinic | Crystal positivity | 11% |
| Prospective2 (38) | 1b | Multiple settings (eg, inpatient, clinic, ED) | Clinical criteria or crystal positivity | 43% |
| Retrospective3 (226) | 2b | Hospitalized patients | Clinical criteria or crystal positivity | 12% |
| Retrospective4 (339) | 2b | Multiple settings | Crystal positivity | 32% |
| Retrospective5 (41) | 2b | Rheumatology clinic | Clinical criteria | 49% |
| Retrospective6 (69) | 2b | Multiple settings | Clinical criteria | 33%† |
| ED, emergency department; LOE, level of evidence. *1b, prospective cohort study with good follow-up (>80%); 2b, retrospective cohort study or prospective study with poor follow-up. †Not necessarily during acute gout. | ||||
Elevated SUA can be an indicator of gout—or not
A prospective cohort study of 82 patients at a Veterans Administration rheumatology clinic found elevated SUA to be the most sensitive indicator among various clinical criteria for diagnosing acute gout. However, 3 (11%) of the 28 patients who had crystal-proven gout also had a normal SUA.1
A second prospective cohort study that evaluated 38 patients during 42 episodes of acute gout in various clinical settings reported a normal SUA in 43% of patients diagnosed on clinical grounds or by joint aspiration.2
Some patients become hyperuricemic after diagnosis
The largest retrospective cohort study evaluated 226 Korean inpatients with acute gout diagnosed either by synovial crystals or American College of Rheumatology (ACR) criteria (TABLE 2). It found that 12% (27) had a normal SUA at diagnosis. Interestingly, 81% became hyperuricemic some time after diagnosis.3
TABLE 2
American College of Rheumatology criteria for classifying acute gouty arthritis
|
| Source: Wallace SL et al. Arthritis Rheum. 1977.7 |
What is a normal SUA value?
Another study reviewed SUA levels in a cohort derived from 2 large prospective RCTs of etoricoxib in patients diagnosed with acute gout by crystal analysis. The proportion of patients with a normal SUA varied substantially according to the definition of a normal value: 32% were normal using a value of 0.48 mmol/L; 11% had normal SUA levels when 0.36 mmol/L was used as the cutoff.4
A secondary analysis evaluated the effect of allopurinol on SUA. The proportion of patients on allopurinol with a normal SUA level compared with patients not taking allopurinol was 49% vs 29% using the higher normal cutoff value, and 29% vs 11% using the lower normal value (P<.001).4
Two studies find many gout patients with a normal SUA
A Japanese retrospective cohort study using ACR criteria found that nearly half of patients diagnosed with acute gout had a normal SUA level.5 A 1967 retrospective examination of Framingham Heart Study data found that one-third of patients clinically diagnosed with gout had a normal level. Some of the patients hadn’t been diagnosed at the time their SUA was measured, however.6
Recommendations
The ACR’s 1977 criteria for diagnosing gout include hyperuricemia as one potential indicator.7 The European League Against Rheumatism advises that normal SUA levels may accompany crystal-proven gout because uric acid either acts as a negative acute-phase reactant or increases in renal excretion during acute episodes. They conclude that SUA has “limited diagnostic value,” especially during acute gout.8
1. Malik A, Schumacher HR, Dinnella JE, et al. Clinical diagnostic criteria for gout: comparison with the gold standard of synovial fluid crystal analysis. J Clin Rheumatol. 2009;15:22-24.
2. Logan JA, Morrison E, McGill PE. Serum uric acid in acute gout. Ann Rheum Dis. 1997;56:696-697.
3. Park YB, Park YS, Lee SC, et al. Clinical analysis of gouty patients with normouricaemia at diagnosis. Ann Rheum Dis. 2003;62:90-92.
4. Schlesinger N, Norquist JM, Watson DJ. Serum urate during acute gout. J Rheumatol. 2009;36:1287-1289.
5. Urano W, Yamanaka H, Tsutani H, et al. The inflammatory process in the mechanism of decreased serum uric acid concentrations during acute gouty arthritis. J Rheumatol. 2002;29:1950-1953.
6. Hall AP, Barry PE, Dawber TR, et al. Epidemiology of gout and hyperuricemia. A long-term population study. Am J Med. 1967;42:27-37.
7. Wallace SL, Robinson H, Masi AT, et al. Preliminary criteria for the classification of the acute arthritis of primary gout. Arthritis Rheum. 1977;20:895-900.
8. Zhang W, Doherty M, Pascual E, et al. EULAR evidence based recommendations for gout. Part I: diagnosis. Report of a task force of the Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann Rheum Dis. 2006;65:1301-1311.
NO. Many patients with acute gout (11%-49%) have normal serum uric acid (SUA) levels (strength of recommendation [SOR]: A, prospective cohort studies). Patients taking allopurinol are significantly more likely to have normal uric acid levels during acute gout attacks (SOR: B, extrapolated from prospective cohorts).
Evidence summary
Six studies have evaluated SUA levels in patients with acute gout. Despite variations in diagnostic approach (clinical criteria vs synovial crystal analysis) and definitions of normal SUA (based on laboratory methods and sex), all 6 studies found normal levels in 11% to 49% of patients with acute gout (TABLE 1).
TABLE 1
Serum uric acid and acute gout: The evidence
| Type of cohort (n) | LOE* | Setting | Method of diagnosis | % with normal serum uric acid |
|---|---|---|---|---|
| Prospective1 (28) | 1b | Veterans Administration rheumatology clinic | Crystal positivity | 11% |
| Prospective2 (38) | 1b | Multiple settings (eg, inpatient, clinic, ED) | Clinical criteria or crystal positivity | 43% |
| Retrospective3 (226) | 2b | Hospitalized patients | Clinical criteria or crystal positivity | 12% |
| Retrospective4 (339) | 2b | Multiple settings | Crystal positivity | 32% |
| Retrospective5 (41) | 2b | Rheumatology clinic | Clinical criteria | 49% |
| Retrospective6 (69) | 2b | Multiple settings | Clinical criteria | 33%† |
| ED, emergency department; LOE, level of evidence. *1b, prospective cohort study with good follow-up (>80%); 2b, retrospective cohort study or prospective study with poor follow-up. †Not necessarily during acute gout. | ||||
Elevated SUA can be an indicator of gout—or not
A prospective cohort study of 82 patients at a Veterans Administration rheumatology clinic found elevated SUA to be the most sensitive indicator among various clinical criteria for diagnosing acute gout. However, 3 (11%) of the 28 patients who had crystal-proven gout also had a normal SUA.1
A second prospective cohort study that evaluated 38 patients during 42 episodes of acute gout in various clinical settings reported a normal SUA in 43% of patients diagnosed on clinical grounds or by joint aspiration.2
Some patients become hyperuricemic after diagnosis
The largest retrospective cohort study evaluated 226 Korean inpatients with acute gout diagnosed either by synovial crystals or American College of Rheumatology (ACR) criteria (TABLE 2). It found that 12% (27) had a normal SUA at diagnosis. Interestingly, 81% became hyperuricemic some time after diagnosis.3
TABLE 2
American College of Rheumatology criteria for classifying acute gouty arthritis
|
| Source: Wallace SL et al. Arthritis Rheum. 1977.7 |
What is a normal SUA value?
Another study reviewed SUA levels in a cohort derived from 2 large prospective RCTs of etoricoxib in patients diagnosed with acute gout by crystal analysis. The proportion of patients with a normal SUA varied substantially according to the definition of a normal value: 32% were normal using a value of 0.48 mmol/L; 11% had normal SUA levels when 0.36 mmol/L was used as the cutoff.4
A secondary analysis evaluated the effect of allopurinol on SUA. The proportion of patients on allopurinol with a normal SUA level compared with patients not taking allopurinol was 49% vs 29% using the higher normal cutoff value, and 29% vs 11% using the lower normal value (P<.001).4
Two studies find many gout patients with a normal SUA
A Japanese retrospective cohort study using ACR criteria found that nearly half of patients diagnosed with acute gout had a normal SUA level.5 A 1967 retrospective examination of Framingham Heart Study data found that one-third of patients clinically diagnosed with gout had a normal level. Some of the patients hadn’t been diagnosed at the time their SUA was measured, however.6
Recommendations
The ACR’s 1977 criteria for diagnosing gout include hyperuricemia as one potential indicator.7 The European League Against Rheumatism advises that normal SUA levels may accompany crystal-proven gout because uric acid either acts as a negative acute-phase reactant or increases in renal excretion during acute episodes. They conclude that SUA has “limited diagnostic value,” especially during acute gout.8
NO. Many patients with acute gout (11%-49%) have normal serum uric acid (SUA) levels (strength of recommendation [SOR]: A, prospective cohort studies). Patients taking allopurinol are significantly more likely to have normal uric acid levels during acute gout attacks (SOR: B, extrapolated from prospective cohorts).
Evidence summary
Six studies have evaluated SUA levels in patients with acute gout. Despite variations in diagnostic approach (clinical criteria vs synovial crystal analysis) and definitions of normal SUA (based on laboratory methods and sex), all 6 studies found normal levels in 11% to 49% of patients with acute gout (TABLE 1).
TABLE 1
Serum uric acid and acute gout: The evidence
| Type of cohort (n) | LOE* | Setting | Method of diagnosis | % with normal serum uric acid |
|---|---|---|---|---|
| Prospective1 (28) | 1b | Veterans Administration rheumatology clinic | Crystal positivity | 11% |
| Prospective2 (38) | 1b | Multiple settings (eg, inpatient, clinic, ED) | Clinical criteria or crystal positivity | 43% |
| Retrospective3 (226) | 2b | Hospitalized patients | Clinical criteria or crystal positivity | 12% |
| Retrospective4 (339) | 2b | Multiple settings | Crystal positivity | 32% |
| Retrospective5 (41) | 2b | Rheumatology clinic | Clinical criteria | 49% |
| Retrospective6 (69) | 2b | Multiple settings | Clinical criteria | 33%† |
| ED, emergency department; LOE, level of evidence. *1b, prospective cohort study with good follow-up (>80%); 2b, retrospective cohort study or prospective study with poor follow-up. †Not necessarily during acute gout. | ||||
Elevated SUA can be an indicator of gout—or not
A prospective cohort study of 82 patients at a Veterans Administration rheumatology clinic found elevated SUA to be the most sensitive indicator among various clinical criteria for diagnosing acute gout. However, 3 (11%) of the 28 patients who had crystal-proven gout also had a normal SUA.1
A second prospective cohort study that evaluated 38 patients during 42 episodes of acute gout in various clinical settings reported a normal SUA in 43% of patients diagnosed on clinical grounds or by joint aspiration.2
Some patients become hyperuricemic after diagnosis
The largest retrospective cohort study evaluated 226 Korean inpatients with acute gout diagnosed either by synovial crystals or American College of Rheumatology (ACR) criteria (TABLE 2). It found that 12% (27) had a normal SUA at diagnosis. Interestingly, 81% became hyperuricemic some time after diagnosis.3
TABLE 2
American College of Rheumatology criteria for classifying acute gouty arthritis
|
| Source: Wallace SL et al. Arthritis Rheum. 1977.7 |
What is a normal SUA value?
Another study reviewed SUA levels in a cohort derived from 2 large prospective RCTs of etoricoxib in patients diagnosed with acute gout by crystal analysis. The proportion of patients with a normal SUA varied substantially according to the definition of a normal value: 32% were normal using a value of 0.48 mmol/L; 11% had normal SUA levels when 0.36 mmol/L was used as the cutoff.4
A secondary analysis evaluated the effect of allopurinol on SUA. The proportion of patients on allopurinol with a normal SUA level compared with patients not taking allopurinol was 49% vs 29% using the higher normal cutoff value, and 29% vs 11% using the lower normal value (P<.001).4
Two studies find many gout patients with a normal SUA
A Japanese retrospective cohort study using ACR criteria found that nearly half of patients diagnosed with acute gout had a normal SUA level.5 A 1967 retrospective examination of Framingham Heart Study data found that one-third of patients clinically diagnosed with gout had a normal level. Some of the patients hadn’t been diagnosed at the time their SUA was measured, however.6
Recommendations
The ACR’s 1977 criteria for diagnosing gout include hyperuricemia as one potential indicator.7 The European League Against Rheumatism advises that normal SUA levels may accompany crystal-proven gout because uric acid either acts as a negative acute-phase reactant or increases in renal excretion during acute episodes. They conclude that SUA has “limited diagnostic value,” especially during acute gout.8
1. Malik A, Schumacher HR, Dinnella JE, et al. Clinical diagnostic criteria for gout: comparison with the gold standard of synovial fluid crystal analysis. J Clin Rheumatol. 2009;15:22-24.
2. Logan JA, Morrison E, McGill PE. Serum uric acid in acute gout. Ann Rheum Dis. 1997;56:696-697.
3. Park YB, Park YS, Lee SC, et al. Clinical analysis of gouty patients with normouricaemia at diagnosis. Ann Rheum Dis. 2003;62:90-92.
4. Schlesinger N, Norquist JM, Watson DJ. Serum urate during acute gout. J Rheumatol. 2009;36:1287-1289.
5. Urano W, Yamanaka H, Tsutani H, et al. The inflammatory process in the mechanism of decreased serum uric acid concentrations during acute gouty arthritis. J Rheumatol. 2002;29:1950-1953.
6. Hall AP, Barry PE, Dawber TR, et al. Epidemiology of gout and hyperuricemia. A long-term population study. Am J Med. 1967;42:27-37.
7. Wallace SL, Robinson H, Masi AT, et al. Preliminary criteria for the classification of the acute arthritis of primary gout. Arthritis Rheum. 1977;20:895-900.
8. Zhang W, Doherty M, Pascual E, et al. EULAR evidence based recommendations for gout. Part I: diagnosis. Report of a task force of the Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann Rheum Dis. 2006;65:1301-1311.
1. Malik A, Schumacher HR, Dinnella JE, et al. Clinical diagnostic criteria for gout: comparison with the gold standard of synovial fluid crystal analysis. J Clin Rheumatol. 2009;15:22-24.
2. Logan JA, Morrison E, McGill PE. Serum uric acid in acute gout. Ann Rheum Dis. 1997;56:696-697.
3. Park YB, Park YS, Lee SC, et al. Clinical analysis of gouty patients with normouricaemia at diagnosis. Ann Rheum Dis. 2003;62:90-92.
4. Schlesinger N, Norquist JM, Watson DJ. Serum urate during acute gout. J Rheumatol. 2009;36:1287-1289.
5. Urano W, Yamanaka H, Tsutani H, et al. The inflammatory process in the mechanism of decreased serum uric acid concentrations during acute gouty arthritis. J Rheumatol. 2002;29:1950-1953.
6. Hall AP, Barry PE, Dawber TR, et al. Epidemiology of gout and hyperuricemia. A long-term population study. Am J Med. 1967;42:27-37.
7. Wallace SL, Robinson H, Masi AT, et al. Preliminary criteria for the classification of the acute arthritis of primary gout. Arthritis Rheum. 1977;20:895-900.
8. Zhang W, Doherty M, Pascual E, et al. EULAR evidence based recommendations for gout. Part I: diagnosis. Report of a task force of the Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann Rheum Dis. 2006;65:1301-1311.
Evidence-based answers from the Family Physicians Inquiries Network
BPH: Saw Palmetto No Better Than Placebo
Even at doses three times higher than usual, saw palmetto extract was no better than placebo at improving lower urinary tract symptoms attributed to benign prostatic hyperplasia in a study in the Sept. 28 issue of JAMA.
Extracts from the fruit of the saw palmetto dwarf palm tree are the most widely used plant extracts in the United States and Europe for urinary symptoms. They are purported to have anti-androgenic, anti-inflammatory, and antiproliferative effects, but none of these properties have been proved.
A meta-analysis from approximately 10 years ago showed that, compared with placebo, saw palmetto significantly reduced nocturia and improved peak uroflow, as well as being rated by study subjects as more beneficial. But more recent clinical trials and reviews of the literature have had more negative findings.
"We conducted a randomized clinical trial to determine if a standard daily dose of saw palmetto extract increased to a double and then a triple daily dose over 72 weeks would improve lower urinary tract symptoms attributed to benign prostatic hypertrophy," said Dr. Michael J. Barry of Massachusetts General Hospital, Boston, and his associates in the Complementary and Alternative Medicine for Urological Symptoms (CAMUS) study group.
This study design allowed an adequate duration of treatment – 24 weeks at each dose level – to assess outcomes, they noted.
The double-blind trial included 369 men aged 45 years and older who had a peak uroflow rate of at least 4 mL per second and a score of 8-24 on the American Urological Association Symptom Index (AUASI) at baseline. They were treated at 11 sites across North America with a standard dose (320 mg/day) of gelcaps containing saw palmetto extract or placebo, which was escalated to 640 mg/day at 24 weeks and 960 mg/day at 48 weeks.
The primary outcome measure was the change in AUASI score at 72 weeks. This decreased only slightly, and to nearly the same degree, in both groups: The reduction was 2.20 points with saw palmetto and 2.99 points with placebo, the investigators said (JAMA 2011;306:1344-51).
The result was the same in a per-protocol analysis of the 151 subjects who received saw palmetto and the 155 who received placebo for the entire duration of the study.
Similarly, the proportion of men who achieved a minimal (3-point) decrease in AUASI score over time was 42.6% with saw palmetto and 44.2% with placebo, slightly favoring placebo. A dose-response analysis showed that saw palmetto was no better than placebo at any dose level.
Further analyses also showed that the active treatment was no better than placebo for a wide range of secondary outcomes including change in BPH Index scores and change in measures of nocturia, peak uroflow, postvoiding residual volume, and incontinence.
The trial also did not reveal any subgroup of patients, such as men with higher PSA levels or men with lower peak uroflow, who showed "a clinically important differential response" to saw palmetto, compared with placebo.
At the conclusion of the study, two measures of patient satisfaction with treatment did not differ between the two groups. Both men who received saw palmetto and men who received placebo rated their symptoms as "between ‘a little better’ and ‘about the same.’ "
This study was supported by the National Institutes of Health, the National Institute of Diabetes and Digestive and Kidney Diseases, the National Center for Complementary and Alternative Medicine, and the NIH Office of Dietary Supplements. Saw palmetto extract and matching placebo gelcaps were donated by Rottapharm/Madaus, Cologne, Germany. This study was conducted under an Investigational New Drug Application from the Food and Drug Administration. Dr. Barry’s associates reported ties to numerous industry sources.
Even at doses three times higher than usual, saw palmetto extract was no better than placebo at improving lower urinary tract symptoms attributed to benign prostatic hyperplasia in a study in the Sept. 28 issue of JAMA.
Extracts from the fruit of the saw palmetto dwarf palm tree are the most widely used plant extracts in the United States and Europe for urinary symptoms. They are purported to have anti-androgenic, anti-inflammatory, and antiproliferative effects, but none of these properties have been proved.
A meta-analysis from approximately 10 years ago showed that, compared with placebo, saw palmetto significantly reduced nocturia and improved peak uroflow, as well as being rated by study subjects as more beneficial. But more recent clinical trials and reviews of the literature have had more negative findings.
"We conducted a randomized clinical trial to determine if a standard daily dose of saw palmetto extract increased to a double and then a triple daily dose over 72 weeks would improve lower urinary tract symptoms attributed to benign prostatic hypertrophy," said Dr. Michael J. Barry of Massachusetts General Hospital, Boston, and his associates in the Complementary and Alternative Medicine for Urological Symptoms (CAMUS) study group.
This study design allowed an adequate duration of treatment – 24 weeks at each dose level – to assess outcomes, they noted.
The double-blind trial included 369 men aged 45 years and older who had a peak uroflow rate of at least 4 mL per second and a score of 8-24 on the American Urological Association Symptom Index (AUASI) at baseline. They were treated at 11 sites across North America with a standard dose (320 mg/day) of gelcaps containing saw palmetto extract or placebo, which was escalated to 640 mg/day at 24 weeks and 960 mg/day at 48 weeks.
The primary outcome measure was the change in AUASI score at 72 weeks. This decreased only slightly, and to nearly the same degree, in both groups: The reduction was 2.20 points with saw palmetto and 2.99 points with placebo, the investigators said (JAMA 2011;306:1344-51).
The result was the same in a per-protocol analysis of the 151 subjects who received saw palmetto and the 155 who received placebo for the entire duration of the study.
Similarly, the proportion of men who achieved a minimal (3-point) decrease in AUASI score over time was 42.6% with saw palmetto and 44.2% with placebo, slightly favoring placebo. A dose-response analysis showed that saw palmetto was no better than placebo at any dose level.
Further analyses also showed that the active treatment was no better than placebo for a wide range of secondary outcomes including change in BPH Index scores and change in measures of nocturia, peak uroflow, postvoiding residual volume, and incontinence.
The trial also did not reveal any subgroup of patients, such as men with higher PSA levels or men with lower peak uroflow, who showed "a clinically important differential response" to saw palmetto, compared with placebo.
At the conclusion of the study, two measures of patient satisfaction with treatment did not differ between the two groups. Both men who received saw palmetto and men who received placebo rated their symptoms as "between ‘a little better’ and ‘about the same.’ "
This study was supported by the National Institutes of Health, the National Institute of Diabetes and Digestive and Kidney Diseases, the National Center for Complementary and Alternative Medicine, and the NIH Office of Dietary Supplements. Saw palmetto extract and matching placebo gelcaps were donated by Rottapharm/Madaus, Cologne, Germany. This study was conducted under an Investigational New Drug Application from the Food and Drug Administration. Dr. Barry’s associates reported ties to numerous industry sources.
Even at doses three times higher than usual, saw palmetto extract was no better than placebo at improving lower urinary tract symptoms attributed to benign prostatic hyperplasia in a study in the Sept. 28 issue of JAMA.
Extracts from the fruit of the saw palmetto dwarf palm tree are the most widely used plant extracts in the United States and Europe for urinary symptoms. They are purported to have anti-androgenic, anti-inflammatory, and antiproliferative effects, but none of these properties have been proved.
A meta-analysis from approximately 10 years ago showed that, compared with placebo, saw palmetto significantly reduced nocturia and improved peak uroflow, as well as being rated by study subjects as more beneficial. But more recent clinical trials and reviews of the literature have had more negative findings.
"We conducted a randomized clinical trial to determine if a standard daily dose of saw palmetto extract increased to a double and then a triple daily dose over 72 weeks would improve lower urinary tract symptoms attributed to benign prostatic hypertrophy," said Dr. Michael J. Barry of Massachusetts General Hospital, Boston, and his associates in the Complementary and Alternative Medicine for Urological Symptoms (CAMUS) study group.
This study design allowed an adequate duration of treatment – 24 weeks at each dose level – to assess outcomes, they noted.
The double-blind trial included 369 men aged 45 years and older who had a peak uroflow rate of at least 4 mL per second and a score of 8-24 on the American Urological Association Symptom Index (AUASI) at baseline. They were treated at 11 sites across North America with a standard dose (320 mg/day) of gelcaps containing saw palmetto extract or placebo, which was escalated to 640 mg/day at 24 weeks and 960 mg/day at 48 weeks.
The primary outcome measure was the change in AUASI score at 72 weeks. This decreased only slightly, and to nearly the same degree, in both groups: The reduction was 2.20 points with saw palmetto and 2.99 points with placebo, the investigators said (JAMA 2011;306:1344-51).
The result was the same in a per-protocol analysis of the 151 subjects who received saw palmetto and the 155 who received placebo for the entire duration of the study.
Similarly, the proportion of men who achieved a minimal (3-point) decrease in AUASI score over time was 42.6% with saw palmetto and 44.2% with placebo, slightly favoring placebo. A dose-response analysis showed that saw palmetto was no better than placebo at any dose level.
Further analyses also showed that the active treatment was no better than placebo for a wide range of secondary outcomes including change in BPH Index scores and change in measures of nocturia, peak uroflow, postvoiding residual volume, and incontinence.
The trial also did not reveal any subgroup of patients, such as men with higher PSA levels or men with lower peak uroflow, who showed "a clinically important differential response" to saw palmetto, compared with placebo.
At the conclusion of the study, two measures of patient satisfaction with treatment did not differ between the two groups. Both men who received saw palmetto and men who received placebo rated their symptoms as "between ‘a little better’ and ‘about the same.’ "
This study was supported by the National Institutes of Health, the National Institute of Diabetes and Digestive and Kidney Diseases, the National Center for Complementary and Alternative Medicine, and the NIH Office of Dietary Supplements. Saw palmetto extract and matching placebo gelcaps were donated by Rottapharm/Madaus, Cologne, Germany. This study was conducted under an Investigational New Drug Application from the Food and Drug Administration. Dr. Barry’s associates reported ties to numerous industry sources.
FROM JAMA
Major Finding: After 72 weeks in which the standard dose of saw palmetto was doubled and then tripled, AUASI score improved by only 2.20 points with saw palmetto and by 2.99 points with placebo.
Data Source: A randomized, double-blind, placebo-controlled clinical trial involving 369 men with urinary symptoms due to BPH.
Disclosures: This study was supported by the National Institutes of Health, the National Institute of Diabetes and Digestive and Kidney Diseases, the National Center for Complementary and Alternative Medicine, and the NIH Office of Dietary Supplements. Saw palmetto extract and matching placebo gelcaps were donated by Rottapharm/Madaus, Cologne, Germany. This study was conducted under an Investigational New Drug Application from the Food and Drug Administration. Dr. Barry’s associates reported ties to numerous industry sources.
ESRD Linked to Risk for Pneumonia Hospitalization
CHICAGO – Patients with end-stage renal disease have sharply elevated rates of hospitalization for pneumonia throughout the renal transplantation trajectory, researchers reported at the annual Interscience Conference on Antimicrobial Agents and Chemotherapy.
The findings underscore the importance of vaccinating this group against pneumococcal and other diseases, lead investigator Lise Haubjerg Nielsen recommended in an interview. Pneumonia "is a big economic burden for society and it is a huge [source of] mortality for these patients."
In a Danish nationwide population-based cohort study among more than 90,000 individuals, those with end-stage renal disease (ESRD) had an 8- to 14-fold higher incidence of such hospitalization, depending on whether they were wait-listed, posttransplant, or post–graft failure, when compared with their counterparts in the general population.
About one-third of the posttransplant group was hospitalized for pneumonia. Male sex and older age were among the significant risk factors for pneumonia hospitalization at this stage. On the other hand, risk fell after the first year posttransplant.
The marked increase in posttransplant risk was expected, given patients’ use of immunosuppressants, according to Ms. Nielsen, who is a medical student undertaking a research year in the department of infectious diseases at Aarhus University Hospital, Skejby. However, the fact that the elevations seen before and after transplantation were even greater was surprising, she said at the meeting, which was sponsored by the American Society for Microbiology.
The increase in pretransplant risk was probably caused by patients’ uremic state, while that post–graft failure "could also be just [a reflection of] these patients being more sick than the general population," she speculated.
The investigators analyzed data from the Danish National Hospital Registry, identifying all hospitalizations since 1977 having a discharge diagnosis of pneumonia, regardless of whether the infection was community or hospital acquired.
They assessed first hospitalizations for pneumonia (excluding those caused by Pneumocystis jiroveci) occurring during 1990-2009. Patients with ESRD who were wait-listed for and/or underwent transplantation were matched by age and sex with up to 19 unaffected individuals from the general population. Analyses were based on 4,973 individuals with and 85,899 individuals without ESRD.
The incidence of first pneumonia hospitalization was 46, 32, and 63 per 1,000 person-years among wait-listed patients, renal transplant recipients, and patients who experienced graft loss, respectively.
These groups had corresponding 10-, 9, and 14-fold increases in the incidence of such hospitalization compared with the general population, according to Ms. Nielsen.
In an analysis of risk factors among the renal transplant recipients, the adjusted incidence rate of pneumonia hospitalization was significantly higher for men; patients aged 50 years or older; those who underwent 1-3 years of dialysis pretransplantation versus none; and those whose renal disease was associated with diabetes, chronic interstitial nephritis, or polycystic kidney disease as compared with glomerulonephritis. The risk of pneumonia hospitalization was significantly lower for those who were at least 1 year out from transplantation.
Ms. Nielsen reported having no conflicts of interest.
CHICAGO – Patients with end-stage renal disease have sharply elevated rates of hospitalization for pneumonia throughout the renal transplantation trajectory, researchers reported at the annual Interscience Conference on Antimicrobial Agents and Chemotherapy.
The findings underscore the importance of vaccinating this group against pneumococcal and other diseases, lead investigator Lise Haubjerg Nielsen recommended in an interview. Pneumonia "is a big economic burden for society and it is a huge [source of] mortality for these patients."
In a Danish nationwide population-based cohort study among more than 90,000 individuals, those with end-stage renal disease (ESRD) had an 8- to 14-fold higher incidence of such hospitalization, depending on whether they were wait-listed, posttransplant, or post–graft failure, when compared with their counterparts in the general population.
About one-third of the posttransplant group was hospitalized for pneumonia. Male sex and older age were among the significant risk factors for pneumonia hospitalization at this stage. On the other hand, risk fell after the first year posttransplant.
The marked increase in posttransplant risk was expected, given patients’ use of immunosuppressants, according to Ms. Nielsen, who is a medical student undertaking a research year in the department of infectious diseases at Aarhus University Hospital, Skejby. However, the fact that the elevations seen before and after transplantation were even greater was surprising, she said at the meeting, which was sponsored by the American Society for Microbiology.
The increase in pretransplant risk was probably caused by patients’ uremic state, while that post–graft failure "could also be just [a reflection of] these patients being more sick than the general population," she speculated.
The investigators analyzed data from the Danish National Hospital Registry, identifying all hospitalizations since 1977 having a discharge diagnosis of pneumonia, regardless of whether the infection was community or hospital acquired.
They assessed first hospitalizations for pneumonia (excluding those caused by Pneumocystis jiroveci) occurring during 1990-2009. Patients with ESRD who were wait-listed for and/or underwent transplantation were matched by age and sex with up to 19 unaffected individuals from the general population. Analyses were based on 4,973 individuals with and 85,899 individuals without ESRD.
The incidence of first pneumonia hospitalization was 46, 32, and 63 per 1,000 person-years among wait-listed patients, renal transplant recipients, and patients who experienced graft loss, respectively.
These groups had corresponding 10-, 9, and 14-fold increases in the incidence of such hospitalization compared with the general population, according to Ms. Nielsen.
In an analysis of risk factors among the renal transplant recipients, the adjusted incidence rate of pneumonia hospitalization was significantly higher for men; patients aged 50 years or older; those who underwent 1-3 years of dialysis pretransplantation versus none; and those whose renal disease was associated with diabetes, chronic interstitial nephritis, or polycystic kidney disease as compared with glomerulonephritis. The risk of pneumonia hospitalization was significantly lower for those who were at least 1 year out from transplantation.
Ms. Nielsen reported having no conflicts of interest.
CHICAGO – Patients with end-stage renal disease have sharply elevated rates of hospitalization for pneumonia throughout the renal transplantation trajectory, researchers reported at the annual Interscience Conference on Antimicrobial Agents and Chemotherapy.
The findings underscore the importance of vaccinating this group against pneumococcal and other diseases, lead investigator Lise Haubjerg Nielsen recommended in an interview. Pneumonia "is a big economic burden for society and it is a huge [source of] mortality for these patients."
In a Danish nationwide population-based cohort study among more than 90,000 individuals, those with end-stage renal disease (ESRD) had an 8- to 14-fold higher incidence of such hospitalization, depending on whether they were wait-listed, posttransplant, or post–graft failure, when compared with their counterparts in the general population.
About one-third of the posttransplant group was hospitalized for pneumonia. Male sex and older age were among the significant risk factors for pneumonia hospitalization at this stage. On the other hand, risk fell after the first year posttransplant.
The marked increase in posttransplant risk was expected, given patients’ use of immunosuppressants, according to Ms. Nielsen, who is a medical student undertaking a research year in the department of infectious diseases at Aarhus University Hospital, Skejby. However, the fact that the elevations seen before and after transplantation were even greater was surprising, she said at the meeting, which was sponsored by the American Society for Microbiology.
The increase in pretransplant risk was probably caused by patients’ uremic state, while that post–graft failure "could also be just [a reflection of] these patients being more sick than the general population," she speculated.
The investigators analyzed data from the Danish National Hospital Registry, identifying all hospitalizations since 1977 having a discharge diagnosis of pneumonia, regardless of whether the infection was community or hospital acquired.
They assessed first hospitalizations for pneumonia (excluding those caused by Pneumocystis jiroveci) occurring during 1990-2009. Patients with ESRD who were wait-listed for and/or underwent transplantation were matched by age and sex with up to 19 unaffected individuals from the general population. Analyses were based on 4,973 individuals with and 85,899 individuals without ESRD.
The incidence of first pneumonia hospitalization was 46, 32, and 63 per 1,000 person-years among wait-listed patients, renal transplant recipients, and patients who experienced graft loss, respectively.
These groups had corresponding 10-, 9, and 14-fold increases in the incidence of such hospitalization compared with the general population, according to Ms. Nielsen.
In an analysis of risk factors among the renal transplant recipients, the adjusted incidence rate of pneumonia hospitalization was significantly higher for men; patients aged 50 years or older; those who underwent 1-3 years of dialysis pretransplantation versus none; and those whose renal disease was associated with diabetes, chronic interstitial nephritis, or polycystic kidney disease as compared with glomerulonephritis. The risk of pneumonia hospitalization was significantly lower for those who were at least 1 year out from transplantation.
Ms. Nielsen reported having no conflicts of interest.
FROM THE ANNUAL INTERSCIENCE CONFERENCE ON ANTIMICROBIAL AGENTS AND CHEMOTHERAPY
Major Finding: Patients who were wait-listed for renal transplant, underwent transplantation, and experienced graft loss had 10-, 9-, and 14-fold increases, respectively, in the incidence of pneumonia hospitalization compared with the general population.
Data Source: A nationwide, population-based cohort study of 4,973 individuals with and 85,899 individuals without end-stage renal disease
Disclosures: Ms. Nielsen reported that she had no relevant conflicts of interest.
Reducing Cardiovascular Risks Lessens Erectile Dysfunction
Lifestyle modifications and pharmacotherapy to reduce the risk of cardiovascular disease can also improve sexual function in men who have erectile dysfunction, according to findings from a meta-analysis first posted online Sept. 12 in Archives of Internal Medicine.
Erectile dysfunction (ED), with a prevalence ranging from 12% of men younger than 59 years of age to 42% of men aged 40-70, shares modifiable risks factors with atherosclerosis and coronary artery disease. These factors include hypertension, diabetes, dyslipidemia, cigarette smoking, obesity, metabolic syndrome, and sedentary behavior. ED has a high prevalence in individuals with multiple risk factors for cardiovascular (CV) disease, and its presence may be an early predictor or marker for cardiovascular events.
While clinical trials have shown that modifying lifestyle risks led to improvement in ED, many are limited by a small sample size and single geographic location and have not studied both lifestyle modifications and pharmacotherapy on ED.
So, Dr. Bhanu P. Gupta and colleagues with the Mayo Clinic, Rochester, Minn., conducted a meta-analysis of six previous randomized controlled trials from four countries to evaluate the relationship between lifestyle intervention and pharmaceutical treatment of cardiovascular risk factors and the severity of ED (Arch. Intern. Med. 2011 Sept 12 [doi:10.1001/archinternmed.2011.440]). The six trials, published between 2004 and 2010 examined in the meta-analysis, included a total of 740 participants (374 who received intervention and 366 control subjects), with the number of participants per trial ranging from 12 to 372. Average age of the participants was 55.4 years, and the study duration ranged from 12 to 104 weeks. All studies included in the analysis showed lessening of ED with adoption of a more healthful lifestyle and improvement in blood lipid parameters.
The meta-analysis showed that improvement in CV risk factors was associated with statistically significant improvement in sexual function, as measured by the Internal Index of Erectile Function, or IIEF-5 score, in which a score of 22- 25 points indicates normal erectile function, 17-21 indicates mild ED, 12-16 indicates mild to moderate ED, 8-11 indicates moderate ED, and 7 and below indicates severe ED.
Meta-analysis of all six trials showed a 2.7-point improvement in mean IIEF-5 score. When excluding studies that included use of statin medications, there was a 2.4-point improvement on the IIEF-5 score. Pharmacotherapy targeting CV risk factors demonstrated improvement of 3.1 points.
Typically, a 4-point improvement in the IIEF-5 score is considered the minimal clinically important difference (MCID). However, the MCID varies significantly according to baseline ED severity, ranging from 2.0 for mild ED to 7.0 for severe ED. "Therefore, the results of this analysis regarding the pooled IIEF-5 score improvement of 2.7 points might not translate into clinically important differences for moderate and severe ED," the researchers say. "Nevertheless, the overall weighted mean difference of 2.7 in IIEF-5 score improvement is consistent with significant improvement in mild ED and lesser improvement in more advanced ED."
"The results of the present meta-analysis add to and strengthen existing knowledge that healthy dietary habits and increased physical activity are important components of health to improve quality of life in men by improving sexual health," the researchers say.
The authors had no financial disclosures to report.
Studies have shown a link between unhealthful lifestyles and a poor quality of life. Despite the benefits of lifestyle modification, however, cardiac risk factors are rampant and increasing in Western societies.
Many Americans seek treatment for erectile dysfunction (ED), which may result from vascular, neurological, psychological, and other factors. ED, known to be related to cardiovascular risk factors, may be a marker of cardiac disease. Ischemic stroke, hemorrhagic stroke, congestive heart failure, and ED are among the various lifestyle-related diseases.
The meta-analysis conducted by Dr. Bhanu P. Gupta and colleagues shows how a healthful lifestyle and pharmacotherapy could improve the severity of ED in men as well as the incidence of cerebral vascular disease (Arch. Intern. Med. 2011 Sept 12 [doi:10.1001/archinternmed.2011.440]).
The increasing epidemic of obesity in the United States should serve as a call to physicians to increase their efforts to motivate their patients and the public at large to make even small changes toward healthier lifestyles. These new associations between healthy lifestyles and reducing incidence of stroke, congestive heart failure, and ED can be powerful tools of persuasion.
Dr. Militza Moreno and Dr. Thomas A. Pearson are in the department of community and preventive medicine at the University of Rochester (N.Y.). The authors had no financial disclosures to report, but they are supported, in part by an Institutional Research Career Development Award from the National Heart, Lung, and Blood Institute, a division of the U.S. National Institutes of Health.
Studies have shown a link between unhealthful lifestyles and a poor quality of life. Despite the benefits of lifestyle modification, however, cardiac risk factors are rampant and increasing in Western societies.
Many Americans seek treatment for erectile dysfunction (ED), which may result from vascular, neurological, psychological, and other factors. ED, known to be related to cardiovascular risk factors, may be a marker of cardiac disease. Ischemic stroke, hemorrhagic stroke, congestive heart failure, and ED are among the various lifestyle-related diseases.
The meta-analysis conducted by Dr. Bhanu P. Gupta and colleagues shows how a healthful lifestyle and pharmacotherapy could improve the severity of ED in men as well as the incidence of cerebral vascular disease (Arch. Intern. Med. 2011 Sept 12 [doi:10.1001/archinternmed.2011.440]).
The increasing epidemic of obesity in the United States should serve as a call to physicians to increase their efforts to motivate their patients and the public at large to make even small changes toward healthier lifestyles. These new associations between healthy lifestyles and reducing incidence of stroke, congestive heart failure, and ED can be powerful tools of persuasion.
Dr. Militza Moreno and Dr. Thomas A. Pearson are in the department of community and preventive medicine at the University of Rochester (N.Y.). The authors had no financial disclosures to report, but they are supported, in part by an Institutional Research Career Development Award from the National Heart, Lung, and Blood Institute, a division of the U.S. National Institutes of Health.
Studies have shown a link between unhealthful lifestyles and a poor quality of life. Despite the benefits of lifestyle modification, however, cardiac risk factors are rampant and increasing in Western societies.
Many Americans seek treatment for erectile dysfunction (ED), which may result from vascular, neurological, psychological, and other factors. ED, known to be related to cardiovascular risk factors, may be a marker of cardiac disease. Ischemic stroke, hemorrhagic stroke, congestive heart failure, and ED are among the various lifestyle-related diseases.
The meta-analysis conducted by Dr. Bhanu P. Gupta and colleagues shows how a healthful lifestyle and pharmacotherapy could improve the severity of ED in men as well as the incidence of cerebral vascular disease (Arch. Intern. Med. 2011 Sept 12 [doi:10.1001/archinternmed.2011.440]).
The increasing epidemic of obesity in the United States should serve as a call to physicians to increase their efforts to motivate their patients and the public at large to make even small changes toward healthier lifestyles. These new associations between healthy lifestyles and reducing incidence of stroke, congestive heart failure, and ED can be powerful tools of persuasion.
Dr. Militza Moreno and Dr. Thomas A. Pearson are in the department of community and preventive medicine at the University of Rochester (N.Y.). The authors had no financial disclosures to report, but they are supported, in part by an Institutional Research Career Development Award from the National Heart, Lung, and Blood Institute, a division of the U.S. National Institutes of Health.
Lifestyle modifications and pharmacotherapy to reduce the risk of cardiovascular disease can also improve sexual function in men who have erectile dysfunction, according to findings from a meta-analysis first posted online Sept. 12 in Archives of Internal Medicine.
Erectile dysfunction (ED), with a prevalence ranging from 12% of men younger than 59 years of age to 42% of men aged 40-70, shares modifiable risks factors with atherosclerosis and coronary artery disease. These factors include hypertension, diabetes, dyslipidemia, cigarette smoking, obesity, metabolic syndrome, and sedentary behavior. ED has a high prevalence in individuals with multiple risk factors for cardiovascular (CV) disease, and its presence may be an early predictor or marker for cardiovascular events.
While clinical trials have shown that modifying lifestyle risks led to improvement in ED, many are limited by a small sample size and single geographic location and have not studied both lifestyle modifications and pharmacotherapy on ED.
So, Dr. Bhanu P. Gupta and colleagues with the Mayo Clinic, Rochester, Minn., conducted a meta-analysis of six previous randomized controlled trials from four countries to evaluate the relationship between lifestyle intervention and pharmaceutical treatment of cardiovascular risk factors and the severity of ED (Arch. Intern. Med. 2011 Sept 12 [doi:10.1001/archinternmed.2011.440]). The six trials, published between 2004 and 2010 examined in the meta-analysis, included a total of 740 participants (374 who received intervention and 366 control subjects), with the number of participants per trial ranging from 12 to 372. Average age of the participants was 55.4 years, and the study duration ranged from 12 to 104 weeks. All studies included in the analysis showed lessening of ED with adoption of a more healthful lifestyle and improvement in blood lipid parameters.
The meta-analysis showed that improvement in CV risk factors was associated with statistically significant improvement in sexual function, as measured by the Internal Index of Erectile Function, or IIEF-5 score, in which a score of 22- 25 points indicates normal erectile function, 17-21 indicates mild ED, 12-16 indicates mild to moderate ED, 8-11 indicates moderate ED, and 7 and below indicates severe ED.
Meta-analysis of all six trials showed a 2.7-point improvement in mean IIEF-5 score. When excluding studies that included use of statin medications, there was a 2.4-point improvement on the IIEF-5 score. Pharmacotherapy targeting CV risk factors demonstrated improvement of 3.1 points.
Typically, a 4-point improvement in the IIEF-5 score is considered the minimal clinically important difference (MCID). However, the MCID varies significantly according to baseline ED severity, ranging from 2.0 for mild ED to 7.0 for severe ED. "Therefore, the results of this analysis regarding the pooled IIEF-5 score improvement of 2.7 points might not translate into clinically important differences for moderate and severe ED," the researchers say. "Nevertheless, the overall weighted mean difference of 2.7 in IIEF-5 score improvement is consistent with significant improvement in mild ED and lesser improvement in more advanced ED."
"The results of the present meta-analysis add to and strengthen existing knowledge that healthy dietary habits and increased physical activity are important components of health to improve quality of life in men by improving sexual health," the researchers say.
The authors had no financial disclosures to report.
Lifestyle modifications and pharmacotherapy to reduce the risk of cardiovascular disease can also improve sexual function in men who have erectile dysfunction, according to findings from a meta-analysis first posted online Sept. 12 in Archives of Internal Medicine.
Erectile dysfunction (ED), with a prevalence ranging from 12% of men younger than 59 years of age to 42% of men aged 40-70, shares modifiable risks factors with atherosclerosis and coronary artery disease. These factors include hypertension, diabetes, dyslipidemia, cigarette smoking, obesity, metabolic syndrome, and sedentary behavior. ED has a high prevalence in individuals with multiple risk factors for cardiovascular (CV) disease, and its presence may be an early predictor or marker for cardiovascular events.
While clinical trials have shown that modifying lifestyle risks led to improvement in ED, many are limited by a small sample size and single geographic location and have not studied both lifestyle modifications and pharmacotherapy on ED.
So, Dr. Bhanu P. Gupta and colleagues with the Mayo Clinic, Rochester, Minn., conducted a meta-analysis of six previous randomized controlled trials from four countries to evaluate the relationship between lifestyle intervention and pharmaceutical treatment of cardiovascular risk factors and the severity of ED (Arch. Intern. Med. 2011 Sept 12 [doi:10.1001/archinternmed.2011.440]). The six trials, published between 2004 and 2010 examined in the meta-analysis, included a total of 740 participants (374 who received intervention and 366 control subjects), with the number of participants per trial ranging from 12 to 372. Average age of the participants was 55.4 years, and the study duration ranged from 12 to 104 weeks. All studies included in the analysis showed lessening of ED with adoption of a more healthful lifestyle and improvement in blood lipid parameters.
The meta-analysis showed that improvement in CV risk factors was associated with statistically significant improvement in sexual function, as measured by the Internal Index of Erectile Function, or IIEF-5 score, in which a score of 22- 25 points indicates normal erectile function, 17-21 indicates mild ED, 12-16 indicates mild to moderate ED, 8-11 indicates moderate ED, and 7 and below indicates severe ED.
Meta-analysis of all six trials showed a 2.7-point improvement in mean IIEF-5 score. When excluding studies that included use of statin medications, there was a 2.4-point improvement on the IIEF-5 score. Pharmacotherapy targeting CV risk factors demonstrated improvement of 3.1 points.
Typically, a 4-point improvement in the IIEF-5 score is considered the minimal clinically important difference (MCID). However, the MCID varies significantly according to baseline ED severity, ranging from 2.0 for mild ED to 7.0 for severe ED. "Therefore, the results of this analysis regarding the pooled IIEF-5 score improvement of 2.7 points might not translate into clinically important differences for moderate and severe ED," the researchers say. "Nevertheless, the overall weighted mean difference of 2.7 in IIEF-5 score improvement is consistent with significant improvement in mild ED and lesser improvement in more advanced ED."
"The results of the present meta-analysis add to and strengthen existing knowledge that healthy dietary habits and increased physical activity are important components of health to improve quality of life in men by improving sexual health," the researchers say.
The authors had no financial disclosures to report.
FROM ARCHIVES OF INTERNAL MEDICINE
Major Finding: Lifestyle modifications and pharmacotherapy not only reduce the risk of cardiovascular disease; they can lead to improved sexual functioning in men with erectile dysfunction.
Data Source: Meta-analysis of six randomized controlled clinical trials in four countries.
Disclosures: The authors had no financial disclosures to report.
Genetic Discovery Shows Pathway of Kidney Disease in Blacks
BOSTON – The recent identification of two gene mutations in a cohort of African Americans with nondiabetic kidney disease helps explain the disproportionately higher rates of kidney disease in this population and represents a disease-mechanism pathway that could lead to new treatments and possibly a cure, Dr. David J. Friedman said at the annual meeting of the International Society on Hypertension in Blacks.
Dr. Friedman of Beth-Israel Deaconess Medical Center, Boston, and his colleagues recently reported the association between two independent variants in the apolipoprotein L1 (APOL1) gene on chromosome 22 and focal segmental glomerulosclerosis (FSGS) and hypertension-attributed end-stage kidney disease in blacks (Science 2010;329:841-5). Not only do the investigators believe that APOL1 is very important to the understanding of nondiabetic renal disease in blacks, "we think the variants in the gene are among the most powerful that have been discovered to date," Dr. Friedman stressed.
The disparity between the rates of end-stage renal disease (ESRD) in blacks and whites in the United States is "incredible," Dr. Friedman stated, noting that the incidence rate is four to five times higher in blacks, according to the 2010 United States Renal Data System annual report. "People have been debating for decades whether the major cause of this disparity is genes or environment. No doubt both are important, but given how strongly this phenotype travels in families, I think we can say with certainty that genes play an important role."
The APOL1 discovery came on the heels of an earlier association linking FSGS, nondiabetic ESRD, and HIV nephropathy in blacks with the MYH9 gene located on the same chromosome, Dr. Friedman explained. "This was quite striking, because we used to think of the three conditions as entirely different diseases, yet each one had exactly the same locus."
Despite the strong association and several years spent looking for causal mutations using fine mapping sequences, the causal variants remained elusive until Dr. Friedman and his colleagues approached the problem from a different perspective.
"We asked, ‘How could any disease gene that’s this deleterious become so common in a population?’ We assumed there was something in this [genetic region] that was beneficial once upon a time to human evolution in Africa," he said. Using mathematical techniques, "we realized that because of the effects of natural selection, the disease gene interval was much larger than anyone thought and probably contained at least five genes." Consequently, the investigators tested new variants in other genes for association with renal disease in African Americans, looking specifically for variants that had not yet been documented, he said.
In a cohort of 205 African Americans with biopsy-proven FSGS and no family history of the disease and 180 African American control subjects, "we saw that variants in the neighboring APOL1 gene were much more strongly associated with renal disease, and unlike the MYH9 variants, which were located in regions of the gene that did not encode for protein, the APOL1 variants were protein-coding sequences."
The investigators determined that the top two variants almost always co-occurred on the same chromosome and each changed an amino acid somewhere on the protein. "We called this the g1 risk allele, and when we controlled for it, a new variant popped up, which we called the g2 allele," he said. Controlling for both the g1 and g2 alleles, "the entire association of this region disappeared and there was no signal left for MYH9."
The investigators also tested the genetic variants in hypertension-associated ESRD in a larger cohort of 1,030 African Americans with the disease and 1,025 geographically matched control subjects and found that the same two variants had a tremendous impact on the development of the disease.
"When combined together, the P value was on the order of 10 to the minus 60, or 35 orders of magnitude greater than the very best MYH9 [result]," Dr. Friedman said. Surprisingly, he noted, we found that these disease variants follow a recessive pattern and together the odds ratio was on the order of 7-10, while the very largest effect sizes of the common variants that affect hypertension or diabetes will confer odds ratios of about 1.4-1.5."
The APOL1 gene and these variants "tend to fall into a different category that we’ve all been familiar with in the past," Dr. Friedman explained. "Most disease variants are either very rare with powerful effects or common with relatively modest effects. The APOL1 variants have a surprising combination of effect size and frequency such that 50%-60% of African Americans carry g1 and/or g2 risk alleles, and 50% are risk homozygous, meaning they are in the highest risk for kidney disease: That translates into about 3.5 million individuals."
Further, while the odds ratios for the more common forms of nondiabetic kidney disease in this population range from 7 to 10, "we’re starting to see odds ratios in the range of 30 for diseases like HIV nephropathy."
Comparing Kidney Disease Rates Between Races
To determine how much of nondiabetic kidney disease can be explained by the genetic variants, the investigators reviewed data from the prospective population-based Dallas Heart Study and compared the outcomes of European American and Caucasian patients, in whom the renal risk alleles are essentially nonexistent, with those of African Americans with zero or one risk allele and those with both risk alleles. Looking at urine protein levels, an indicator of renal microvascular disease, "we found that black individuals with zero or one copy of the risk allele had rates more similar to whites than to blacks with two alleles," Dr. Friedman reported.
The results were even more striking for actual hard measures of renal function, he said. "Rates of chronic kidney disease or impaired renal function, indicated by glomerular filtration rates less than 60 mL/min per 1.73 m2, were essentially the same among blacks with zero or one allele and whites, whereas individuals with a risk genotype had a fourfold increase in impaired renal function." Although the study does not include many individuals with ESRD, the investigators hope to look more closely into such patients in future studies, he said.
In their preliminary review of the data, "we can’t tell any difference between African Americans with zero or one copy of the allele and Caucasians, but African Americans with two renal risk alleles have at least 10-fold increase in kidney failure," Dr. Friedman stated. "To our surprise, this really only applies to nondiabetic kidney disease. The alleles have essentially no effect that we can detect on diabetic renal disease."
This realization led the investigators to revisit the issue of natural selection. It turns out, according to Dr. Friedman, "APOL1 is the genetic source for the immunity factor that protected people from African sleeping sickness, a parasitic infection caused by Trypanosoma brucei gambiense." Similar to selection for the gene variants associated with sickle cell anemia, he explained, "inheriting one copy of the APOL1 gene risk variant provides protection from the parasite, while two copies seems to render individuals increases the risk of kidney disease up to 10-fold." Through natural selection, as more people survived African sleeping sickness, the percentage of the population with kidney disease risk variants increased, he said.
The investigators are currently studying the risk variants intensively to figure out how they work. "We think they may differentially regulate processes such as apoptosis and cell repair may function as a chloride channel in mammalian systems in the same way it doses in lysosomes and may affect biological function," Dr. Friedman hypothesized.
In addition to exploring the underlying mechanisms, the potential clinical value of the genetic discovery is also being considered. "This may help us improve risk stratification," Dr. Friedman said. "It’s one thing to say that African Americans have a fourfold increased risk of kidney disease. It’s better to find the tag SNP [single nucleotide polymorphism] that will tell if an individual might have an increased risk. If you can actually find the causal variant, then you can potentially predict with much higher success who is and is not at risk for kidney failure," he stated. "The problem is that it works pretty well in Western African populations, such as Nigerians, but not as well in East Africans, such as Ethiopians."
One of the main questions that Dr. Friedman and his colleagues currently are pursuing is whether hypertension causes kidney disease in these at-risk individuals or whether hypertension is the result of primary renal vascular disease.
"To us, the fact that the very same genetic variants cause hypertension-associated ESRD and FSGS, a primary renal microvascular disease, suggests that these may be the same disease process that we are either catching at different stages or that have different modifiers, and that hypertension in these patients may just be a symptom and not a cause of kidney failure," Dr. Friedman said. Additionally, the investigators are trying to determine why only some people with two risk alleles develop kidney disease and why APOL1 risk variants have little to no effect on diabetic nephropathy, which may offer some clues to how the molecules work, he said.
A cure for nondiabetic kidney disease, which accounts for more than $8.2 billion annually in dialysis coasts, may directly result from the APOL1 finding, Dr. Friedman said. "It’s that important."
Dr. Friedman reported no financial conflicts of interested related to his presentation.
BOSTON – The recent identification of two gene mutations in a cohort of African Americans with nondiabetic kidney disease helps explain the disproportionately higher rates of kidney disease in this population and represents a disease-mechanism pathway that could lead to new treatments and possibly a cure, Dr. David J. Friedman said at the annual meeting of the International Society on Hypertension in Blacks.
Dr. Friedman of Beth-Israel Deaconess Medical Center, Boston, and his colleagues recently reported the association between two independent variants in the apolipoprotein L1 (APOL1) gene on chromosome 22 and focal segmental glomerulosclerosis (FSGS) and hypertension-attributed end-stage kidney disease in blacks (Science 2010;329:841-5). Not only do the investigators believe that APOL1 is very important to the understanding of nondiabetic renal disease in blacks, "we think the variants in the gene are among the most powerful that have been discovered to date," Dr. Friedman stressed.
The disparity between the rates of end-stage renal disease (ESRD) in blacks and whites in the United States is "incredible," Dr. Friedman stated, noting that the incidence rate is four to five times higher in blacks, according to the 2010 United States Renal Data System annual report. "People have been debating for decades whether the major cause of this disparity is genes or environment. No doubt both are important, but given how strongly this phenotype travels in families, I think we can say with certainty that genes play an important role."
The APOL1 discovery came on the heels of an earlier association linking FSGS, nondiabetic ESRD, and HIV nephropathy in blacks with the MYH9 gene located on the same chromosome, Dr. Friedman explained. "This was quite striking, because we used to think of the three conditions as entirely different diseases, yet each one had exactly the same locus."
Despite the strong association and several years spent looking for causal mutations using fine mapping sequences, the causal variants remained elusive until Dr. Friedman and his colleagues approached the problem from a different perspective.
"We asked, ‘How could any disease gene that’s this deleterious become so common in a population?’ We assumed there was something in this [genetic region] that was beneficial once upon a time to human evolution in Africa," he said. Using mathematical techniques, "we realized that because of the effects of natural selection, the disease gene interval was much larger than anyone thought and probably contained at least five genes." Consequently, the investigators tested new variants in other genes for association with renal disease in African Americans, looking specifically for variants that had not yet been documented, he said.
In a cohort of 205 African Americans with biopsy-proven FSGS and no family history of the disease and 180 African American control subjects, "we saw that variants in the neighboring APOL1 gene were much more strongly associated with renal disease, and unlike the MYH9 variants, which were located in regions of the gene that did not encode for protein, the APOL1 variants were protein-coding sequences."
The investigators determined that the top two variants almost always co-occurred on the same chromosome and each changed an amino acid somewhere on the protein. "We called this the g1 risk allele, and when we controlled for it, a new variant popped up, which we called the g2 allele," he said. Controlling for both the g1 and g2 alleles, "the entire association of this region disappeared and there was no signal left for MYH9."
The investigators also tested the genetic variants in hypertension-associated ESRD in a larger cohort of 1,030 African Americans with the disease and 1,025 geographically matched control subjects and found that the same two variants had a tremendous impact on the development of the disease.
"When combined together, the P value was on the order of 10 to the minus 60, or 35 orders of magnitude greater than the very best MYH9 [result]," Dr. Friedman said. Surprisingly, he noted, we found that these disease variants follow a recessive pattern and together the odds ratio was on the order of 7-10, while the very largest effect sizes of the common variants that affect hypertension or diabetes will confer odds ratios of about 1.4-1.5."
The APOL1 gene and these variants "tend to fall into a different category that we’ve all been familiar with in the past," Dr. Friedman explained. "Most disease variants are either very rare with powerful effects or common with relatively modest effects. The APOL1 variants have a surprising combination of effect size and frequency such that 50%-60% of African Americans carry g1 and/or g2 risk alleles, and 50% are risk homozygous, meaning they are in the highest risk for kidney disease: That translates into about 3.5 million individuals."
Further, while the odds ratios for the more common forms of nondiabetic kidney disease in this population range from 7 to 10, "we’re starting to see odds ratios in the range of 30 for diseases like HIV nephropathy."
Comparing Kidney Disease Rates Between Races
To determine how much of nondiabetic kidney disease can be explained by the genetic variants, the investigators reviewed data from the prospective population-based Dallas Heart Study and compared the outcomes of European American and Caucasian patients, in whom the renal risk alleles are essentially nonexistent, with those of African Americans with zero or one risk allele and those with both risk alleles. Looking at urine protein levels, an indicator of renal microvascular disease, "we found that black individuals with zero or one copy of the risk allele had rates more similar to whites than to blacks with two alleles," Dr. Friedman reported.
The results were even more striking for actual hard measures of renal function, he said. "Rates of chronic kidney disease or impaired renal function, indicated by glomerular filtration rates less than 60 mL/min per 1.73 m2, were essentially the same among blacks with zero or one allele and whites, whereas individuals with a risk genotype had a fourfold increase in impaired renal function." Although the study does not include many individuals with ESRD, the investigators hope to look more closely into such patients in future studies, he said.
In their preliminary review of the data, "we can’t tell any difference between African Americans with zero or one copy of the allele and Caucasians, but African Americans with two renal risk alleles have at least 10-fold increase in kidney failure," Dr. Friedman stated. "To our surprise, this really only applies to nondiabetic kidney disease. The alleles have essentially no effect that we can detect on diabetic renal disease."
This realization led the investigators to revisit the issue of natural selection. It turns out, according to Dr. Friedman, "APOL1 is the genetic source for the immunity factor that protected people from African sleeping sickness, a parasitic infection caused by Trypanosoma brucei gambiense." Similar to selection for the gene variants associated with sickle cell anemia, he explained, "inheriting one copy of the APOL1 gene risk variant provides protection from the parasite, while two copies seems to render individuals increases the risk of kidney disease up to 10-fold." Through natural selection, as more people survived African sleeping sickness, the percentage of the population with kidney disease risk variants increased, he said.
The investigators are currently studying the risk variants intensively to figure out how they work. "We think they may differentially regulate processes such as apoptosis and cell repair may function as a chloride channel in mammalian systems in the same way it doses in lysosomes and may affect biological function," Dr. Friedman hypothesized.
In addition to exploring the underlying mechanisms, the potential clinical value of the genetic discovery is also being considered. "This may help us improve risk stratification," Dr. Friedman said. "It’s one thing to say that African Americans have a fourfold increased risk of kidney disease. It’s better to find the tag SNP [single nucleotide polymorphism] that will tell if an individual might have an increased risk. If you can actually find the causal variant, then you can potentially predict with much higher success who is and is not at risk for kidney failure," he stated. "The problem is that it works pretty well in Western African populations, such as Nigerians, but not as well in East Africans, such as Ethiopians."
One of the main questions that Dr. Friedman and his colleagues currently are pursuing is whether hypertension causes kidney disease in these at-risk individuals or whether hypertension is the result of primary renal vascular disease.
"To us, the fact that the very same genetic variants cause hypertension-associated ESRD and FSGS, a primary renal microvascular disease, suggests that these may be the same disease process that we are either catching at different stages or that have different modifiers, and that hypertension in these patients may just be a symptom and not a cause of kidney failure," Dr. Friedman said. Additionally, the investigators are trying to determine why only some people with two risk alleles develop kidney disease and why APOL1 risk variants have little to no effect on diabetic nephropathy, which may offer some clues to how the molecules work, he said.
A cure for nondiabetic kidney disease, which accounts for more than $8.2 billion annually in dialysis coasts, may directly result from the APOL1 finding, Dr. Friedman said. "It’s that important."
Dr. Friedman reported no financial conflicts of interested related to his presentation.
BOSTON – The recent identification of two gene mutations in a cohort of African Americans with nondiabetic kidney disease helps explain the disproportionately higher rates of kidney disease in this population and represents a disease-mechanism pathway that could lead to new treatments and possibly a cure, Dr. David J. Friedman said at the annual meeting of the International Society on Hypertension in Blacks.
Dr. Friedman of Beth-Israel Deaconess Medical Center, Boston, and his colleagues recently reported the association between two independent variants in the apolipoprotein L1 (APOL1) gene on chromosome 22 and focal segmental glomerulosclerosis (FSGS) and hypertension-attributed end-stage kidney disease in blacks (Science 2010;329:841-5). Not only do the investigators believe that APOL1 is very important to the understanding of nondiabetic renal disease in blacks, "we think the variants in the gene are among the most powerful that have been discovered to date," Dr. Friedman stressed.
The disparity between the rates of end-stage renal disease (ESRD) in blacks and whites in the United States is "incredible," Dr. Friedman stated, noting that the incidence rate is four to five times higher in blacks, according to the 2010 United States Renal Data System annual report. "People have been debating for decades whether the major cause of this disparity is genes or environment. No doubt both are important, but given how strongly this phenotype travels in families, I think we can say with certainty that genes play an important role."
The APOL1 discovery came on the heels of an earlier association linking FSGS, nondiabetic ESRD, and HIV nephropathy in blacks with the MYH9 gene located on the same chromosome, Dr. Friedman explained. "This was quite striking, because we used to think of the three conditions as entirely different diseases, yet each one had exactly the same locus."
Despite the strong association and several years spent looking for causal mutations using fine mapping sequences, the causal variants remained elusive until Dr. Friedman and his colleagues approached the problem from a different perspective.
"We asked, ‘How could any disease gene that’s this deleterious become so common in a population?’ We assumed there was something in this [genetic region] that was beneficial once upon a time to human evolution in Africa," he said. Using mathematical techniques, "we realized that because of the effects of natural selection, the disease gene interval was much larger than anyone thought and probably contained at least five genes." Consequently, the investigators tested new variants in other genes for association with renal disease in African Americans, looking specifically for variants that had not yet been documented, he said.
In a cohort of 205 African Americans with biopsy-proven FSGS and no family history of the disease and 180 African American control subjects, "we saw that variants in the neighboring APOL1 gene were much more strongly associated with renal disease, and unlike the MYH9 variants, which were located in regions of the gene that did not encode for protein, the APOL1 variants were protein-coding sequences."
The investigators determined that the top two variants almost always co-occurred on the same chromosome and each changed an amino acid somewhere on the protein. "We called this the g1 risk allele, and when we controlled for it, a new variant popped up, which we called the g2 allele," he said. Controlling for both the g1 and g2 alleles, "the entire association of this region disappeared and there was no signal left for MYH9."
The investigators also tested the genetic variants in hypertension-associated ESRD in a larger cohort of 1,030 African Americans with the disease and 1,025 geographically matched control subjects and found that the same two variants had a tremendous impact on the development of the disease.
"When combined together, the P value was on the order of 10 to the minus 60, or 35 orders of magnitude greater than the very best MYH9 [result]," Dr. Friedman said. Surprisingly, he noted, we found that these disease variants follow a recessive pattern and together the odds ratio was on the order of 7-10, while the very largest effect sizes of the common variants that affect hypertension or diabetes will confer odds ratios of about 1.4-1.5."
The APOL1 gene and these variants "tend to fall into a different category that we’ve all been familiar with in the past," Dr. Friedman explained. "Most disease variants are either very rare with powerful effects or common with relatively modest effects. The APOL1 variants have a surprising combination of effect size and frequency such that 50%-60% of African Americans carry g1 and/or g2 risk alleles, and 50% are risk homozygous, meaning they are in the highest risk for kidney disease: That translates into about 3.5 million individuals."
Further, while the odds ratios for the more common forms of nondiabetic kidney disease in this population range from 7 to 10, "we’re starting to see odds ratios in the range of 30 for diseases like HIV nephropathy."
Comparing Kidney Disease Rates Between Races
To determine how much of nondiabetic kidney disease can be explained by the genetic variants, the investigators reviewed data from the prospective population-based Dallas Heart Study and compared the outcomes of European American and Caucasian patients, in whom the renal risk alleles are essentially nonexistent, with those of African Americans with zero or one risk allele and those with both risk alleles. Looking at urine protein levels, an indicator of renal microvascular disease, "we found that black individuals with zero or one copy of the risk allele had rates more similar to whites than to blacks with two alleles," Dr. Friedman reported.
The results were even more striking for actual hard measures of renal function, he said. "Rates of chronic kidney disease or impaired renal function, indicated by glomerular filtration rates less than 60 mL/min per 1.73 m2, were essentially the same among blacks with zero or one allele and whites, whereas individuals with a risk genotype had a fourfold increase in impaired renal function." Although the study does not include many individuals with ESRD, the investigators hope to look more closely into such patients in future studies, he said.
In their preliminary review of the data, "we can’t tell any difference between African Americans with zero or one copy of the allele and Caucasians, but African Americans with two renal risk alleles have at least 10-fold increase in kidney failure," Dr. Friedman stated. "To our surprise, this really only applies to nondiabetic kidney disease. The alleles have essentially no effect that we can detect on diabetic renal disease."
This realization led the investigators to revisit the issue of natural selection. It turns out, according to Dr. Friedman, "APOL1 is the genetic source for the immunity factor that protected people from African sleeping sickness, a parasitic infection caused by Trypanosoma brucei gambiense." Similar to selection for the gene variants associated with sickle cell anemia, he explained, "inheriting one copy of the APOL1 gene risk variant provides protection from the parasite, while two copies seems to render individuals increases the risk of kidney disease up to 10-fold." Through natural selection, as more people survived African sleeping sickness, the percentage of the population with kidney disease risk variants increased, he said.
The investigators are currently studying the risk variants intensively to figure out how they work. "We think they may differentially regulate processes such as apoptosis and cell repair may function as a chloride channel in mammalian systems in the same way it doses in lysosomes and may affect biological function," Dr. Friedman hypothesized.
In addition to exploring the underlying mechanisms, the potential clinical value of the genetic discovery is also being considered. "This may help us improve risk stratification," Dr. Friedman said. "It’s one thing to say that African Americans have a fourfold increased risk of kidney disease. It’s better to find the tag SNP [single nucleotide polymorphism] that will tell if an individual might have an increased risk. If you can actually find the causal variant, then you can potentially predict with much higher success who is and is not at risk for kidney failure," he stated. "The problem is that it works pretty well in Western African populations, such as Nigerians, but not as well in East Africans, such as Ethiopians."
One of the main questions that Dr. Friedman and his colleagues currently are pursuing is whether hypertension causes kidney disease in these at-risk individuals or whether hypertension is the result of primary renal vascular disease.
"To us, the fact that the very same genetic variants cause hypertension-associated ESRD and FSGS, a primary renal microvascular disease, suggests that these may be the same disease process that we are either catching at different stages or that have different modifiers, and that hypertension in these patients may just be a symptom and not a cause of kidney failure," Dr. Friedman said. Additionally, the investigators are trying to determine why only some people with two risk alleles develop kidney disease and why APOL1 risk variants have little to no effect on diabetic nephropathy, which may offer some clues to how the molecules work, he said.
A cure for nondiabetic kidney disease, which accounts for more than $8.2 billion annually in dialysis coasts, may directly result from the APOL1 finding, Dr. Friedman said. "It’s that important."
Dr. Friedman reported no financial conflicts of interested related to his presentation.
EXPERT ANALYSIS FROM THE ANNUAL MEETING OF THE INTERNATIONAL SOCIETY ON HYPERTENSION IN BLACKS
Microscopic Hematuria in Youth Signals High ESRD Risk
Persistent asymptomatic isolated microscopic hematuria in adolescence and young adulthood appears to be a strong predictor of end-stage renal disease in later adulthood, independent of other risk factors, according to a report in the Aug. 17 JAMA.
Such hematuria is frequently an incidental finding on routine examination in this age group, but its significance has been unclear. Short-term prognosis is favorable, and the condition is generally considered benign. But long-term data are lacking, and uncertainty about the implications has prompted "considerable controversy over appropriate evaluation, management, and prognosis," said Dr. Asaf Vivante of the Israeli Defense Forces Medical Corps and the Edmond and Lily Safra Children’s Hospital, Tel Hashomer, Israel, and his associates.
"The most recent American Academy of Pediatrics guidelines rescinded the recommendation for urine screening during the second decade of life," they noted.
They conducted a nationwide retrospective cohort study to assess the long-term outcomes of persistent asymptomatic isolated microscopic hematuria, which by definition is unaccompanied by proteinuria or kidney abnormalities and unrelated to any systemic condition. By using mandatory military service records, the researchers assessed medical data on over 1.2 million Israelis who were aged 16-25 years at induction in 1975-1997 and who were followed for about 22 years.
Study subjects were initially screened by urinary dipstick test. In those with positive results for hematuria, urinary sediment was examined by microscopy. A total of 3,690 of these young men and women (0.3% of the cohort) were found to have persistent asymptomatic isolated hematuria.
The investigators then used a national end-stage renal disease (ESRD) database to identify all patients receiving any form of renal replacement therapy from 1980 through 2010. During follow-up, 565 members of the study cohort were treated for ESRD.
The incidence of ESRD was 34 per 100,000 person-years for subjects who had had hematuria in adolescence and young adulthood, a strikingly higher rate than the 2.05 cases per 100,000 person-years for subjects who had not had hematuria, Dr. Vivante and his associates said (JAMA 2011;306:729-36).
A total of 0.7% of subjects with hematuria in adolescence developed ESRD, compared with 0.04% of those without hematuria, yielding an unadjusted hazard ratio of 19.5. When the data were adjusted to account for factors that might influence kidney function, such as subject age, sex, BMI, and blood pressure, there was no significant change in the estimated HR (18.5).
In addition, study subjects who had hematuria in adolescence were considerably younger at the onset of ESRD (34 years) than were subjects without hematuria during their youth (38 years).
When the cases of ESRD were categorized by nine possible causes – diabetes, hypertension, glomerulonephritis, hereditary nephritis, interstitial nephritis, cystic kidney disease, secondary glomerulonephritis, drug-induced, and other causes – the clear majority of cases among subjects who had hematuria in adolescence were found to be due to glomerular disease.
"Our findings suggest that persistent asymptomatic isolated microscopic hematuria detected during adolescence and young adulthood is an early marker for primary glomerular injury and may be the first sign of an occult renal disease," the researchers said.
They added that follow-up in this study ended well before subjects reached the age at which ESRD incidence peaks, so their calculations likely underestimate the true significance of hematuria as a predictor for the disease.
Since this study involved only Jewish subjects, the results may not be generalizable to other racial/ethnic groups, and confirmation of these results should be sought in other populations.
"Future studies [also] are warranted to evaluate the utility of population screening in improving clinical outcomes," they noted.
This study was supported by the Israel Defense Forces Medical Corps and the Israeli Ministry of Health. No financial conflicts of interest were reported.
"The time may have arrived for routine urine dipstick screening in adolescents and adults, at least at all initial examinations and perhaps every 5 to 10 years thereafter," said Dr. Robert S. Brown.
"Prior to the study by Vivante et al, patients with isolated microscopic hematuria and a negative evaluation were usually considered to have benign hematuria and required no follow-up. Now it seems reasonable to reevaluate such patients every 1 to 2 years for a possible increased incidence of proteinuria, hypertension, or renal insufficiency," he said.
dr. brown is in the department of medicine at Beth Israel Deaconess Medical Center and Harvard Medical School, Boston. He reported having no financial conflicts of interest. These remarks were taken from his editorial accompanying Dr. Vivante’s report (JAMA 2011;306:764-5).
"The time may have arrived for routine urine dipstick screening in adolescents and adults, at least at all initial examinations and perhaps every 5 to 10 years thereafter," said Dr. Robert S. Brown.
"Prior to the study by Vivante et al, patients with isolated microscopic hematuria and a negative evaluation were usually considered to have benign hematuria and required no follow-up. Now it seems reasonable to reevaluate such patients every 1 to 2 years for a possible increased incidence of proteinuria, hypertension, or renal insufficiency," he said.
dr. brown is in the department of medicine at Beth Israel Deaconess Medical Center and Harvard Medical School, Boston. He reported having no financial conflicts of interest. These remarks were taken from his editorial accompanying Dr. Vivante’s report (JAMA 2011;306:764-5).
"The time may have arrived for routine urine dipstick screening in adolescents and adults, at least at all initial examinations and perhaps every 5 to 10 years thereafter," said Dr. Robert S. Brown.
"Prior to the study by Vivante et al, patients with isolated microscopic hematuria and a negative evaluation were usually considered to have benign hematuria and required no follow-up. Now it seems reasonable to reevaluate such patients every 1 to 2 years for a possible increased incidence of proteinuria, hypertension, or renal insufficiency," he said.
dr. brown is in the department of medicine at Beth Israel Deaconess Medical Center and Harvard Medical School, Boston. He reported having no financial conflicts of interest. These remarks were taken from his editorial accompanying Dr. Vivante’s report (JAMA 2011;306:764-5).
Persistent asymptomatic isolated microscopic hematuria in adolescence and young adulthood appears to be a strong predictor of end-stage renal disease in later adulthood, independent of other risk factors, according to a report in the Aug. 17 JAMA.
Such hematuria is frequently an incidental finding on routine examination in this age group, but its significance has been unclear. Short-term prognosis is favorable, and the condition is generally considered benign. But long-term data are lacking, and uncertainty about the implications has prompted "considerable controversy over appropriate evaluation, management, and prognosis," said Dr. Asaf Vivante of the Israeli Defense Forces Medical Corps and the Edmond and Lily Safra Children’s Hospital, Tel Hashomer, Israel, and his associates.
"The most recent American Academy of Pediatrics guidelines rescinded the recommendation for urine screening during the second decade of life," they noted.
They conducted a nationwide retrospective cohort study to assess the long-term outcomes of persistent asymptomatic isolated microscopic hematuria, which by definition is unaccompanied by proteinuria or kidney abnormalities and unrelated to any systemic condition. By using mandatory military service records, the researchers assessed medical data on over 1.2 million Israelis who were aged 16-25 years at induction in 1975-1997 and who were followed for about 22 years.
Study subjects were initially screened by urinary dipstick test. In those with positive results for hematuria, urinary sediment was examined by microscopy. A total of 3,690 of these young men and women (0.3% of the cohort) were found to have persistent asymptomatic isolated hematuria.
The investigators then used a national end-stage renal disease (ESRD) database to identify all patients receiving any form of renal replacement therapy from 1980 through 2010. During follow-up, 565 members of the study cohort were treated for ESRD.
The incidence of ESRD was 34 per 100,000 person-years for subjects who had had hematuria in adolescence and young adulthood, a strikingly higher rate than the 2.05 cases per 100,000 person-years for subjects who had not had hematuria, Dr. Vivante and his associates said (JAMA 2011;306:729-36).
A total of 0.7% of subjects with hematuria in adolescence developed ESRD, compared with 0.04% of those without hematuria, yielding an unadjusted hazard ratio of 19.5. When the data were adjusted to account for factors that might influence kidney function, such as subject age, sex, BMI, and blood pressure, there was no significant change in the estimated HR (18.5).
In addition, study subjects who had hematuria in adolescence were considerably younger at the onset of ESRD (34 years) than were subjects without hematuria during their youth (38 years).
When the cases of ESRD were categorized by nine possible causes – diabetes, hypertension, glomerulonephritis, hereditary nephritis, interstitial nephritis, cystic kidney disease, secondary glomerulonephritis, drug-induced, and other causes – the clear majority of cases among subjects who had hematuria in adolescence were found to be due to glomerular disease.
"Our findings suggest that persistent asymptomatic isolated microscopic hematuria detected during adolescence and young adulthood is an early marker for primary glomerular injury and may be the first sign of an occult renal disease," the researchers said.
They added that follow-up in this study ended well before subjects reached the age at which ESRD incidence peaks, so their calculations likely underestimate the true significance of hematuria as a predictor for the disease.
Since this study involved only Jewish subjects, the results may not be generalizable to other racial/ethnic groups, and confirmation of these results should be sought in other populations.
"Future studies [also] are warranted to evaluate the utility of population screening in improving clinical outcomes," they noted.
This study was supported by the Israel Defense Forces Medical Corps and the Israeli Ministry of Health. No financial conflicts of interest were reported.
Persistent asymptomatic isolated microscopic hematuria in adolescence and young adulthood appears to be a strong predictor of end-stage renal disease in later adulthood, independent of other risk factors, according to a report in the Aug. 17 JAMA.
Such hematuria is frequently an incidental finding on routine examination in this age group, but its significance has been unclear. Short-term prognosis is favorable, and the condition is generally considered benign. But long-term data are lacking, and uncertainty about the implications has prompted "considerable controversy over appropriate evaluation, management, and prognosis," said Dr. Asaf Vivante of the Israeli Defense Forces Medical Corps and the Edmond and Lily Safra Children’s Hospital, Tel Hashomer, Israel, and his associates.
"The most recent American Academy of Pediatrics guidelines rescinded the recommendation for urine screening during the second decade of life," they noted.
They conducted a nationwide retrospective cohort study to assess the long-term outcomes of persistent asymptomatic isolated microscopic hematuria, which by definition is unaccompanied by proteinuria or kidney abnormalities and unrelated to any systemic condition. By using mandatory military service records, the researchers assessed medical data on over 1.2 million Israelis who were aged 16-25 years at induction in 1975-1997 and who were followed for about 22 years.
Study subjects were initially screened by urinary dipstick test. In those with positive results for hematuria, urinary sediment was examined by microscopy. A total of 3,690 of these young men and women (0.3% of the cohort) were found to have persistent asymptomatic isolated hematuria.
The investigators then used a national end-stage renal disease (ESRD) database to identify all patients receiving any form of renal replacement therapy from 1980 through 2010. During follow-up, 565 members of the study cohort were treated for ESRD.
The incidence of ESRD was 34 per 100,000 person-years for subjects who had had hematuria in adolescence and young adulthood, a strikingly higher rate than the 2.05 cases per 100,000 person-years for subjects who had not had hematuria, Dr. Vivante and his associates said (JAMA 2011;306:729-36).
A total of 0.7% of subjects with hematuria in adolescence developed ESRD, compared with 0.04% of those without hematuria, yielding an unadjusted hazard ratio of 19.5. When the data were adjusted to account for factors that might influence kidney function, such as subject age, sex, BMI, and blood pressure, there was no significant change in the estimated HR (18.5).
In addition, study subjects who had hematuria in adolescence were considerably younger at the onset of ESRD (34 years) than were subjects without hematuria during their youth (38 years).
When the cases of ESRD were categorized by nine possible causes – diabetes, hypertension, glomerulonephritis, hereditary nephritis, interstitial nephritis, cystic kidney disease, secondary glomerulonephritis, drug-induced, and other causes – the clear majority of cases among subjects who had hematuria in adolescence were found to be due to glomerular disease.
"Our findings suggest that persistent asymptomatic isolated microscopic hematuria detected during adolescence and young adulthood is an early marker for primary glomerular injury and may be the first sign of an occult renal disease," the researchers said.
They added that follow-up in this study ended well before subjects reached the age at which ESRD incidence peaks, so their calculations likely underestimate the true significance of hematuria as a predictor for the disease.
Since this study involved only Jewish subjects, the results may not be generalizable to other racial/ethnic groups, and confirmation of these results should be sought in other populations.
"Future studies [also] are warranted to evaluate the utility of population screening in improving clinical outcomes," they noted.
This study was supported by the Israel Defense Forces Medical Corps and the Israeli Ministry of Health. No financial conflicts of interest were reported.
FROM JAMA
Major Finding: Among males and females who had persistent asymptomatic isolated microscopic hematuria at ages 16-25, the incidence of ESRD during 22 years of follow-up was 34 per 100,000 person-years, a strikingly higher rate than the 2.05 cases per 100,000 person-years for subjects who had not had hematuria in adolescence and young adulthood.
Data Source: A retrospective nationwide cohort study involving 1.2 million Israeli adolescents and young adults who underwent urinary dipstick testing for hematuria as part of compulsory medical exams and whose ESRD status was followed for 22 years.
Disclosures: This study was supported by the Israel Defense Forces Medical Corps and the Israeli Ministry of Health. No financial conflicts of interest were reported.