User login
Can Yoga Reduce Symptoms of Anxiety and Depression?
Yes, yoga can reduce symptoms of anxiety and depression (strength of recommendation [SOR]: B, systematic reviews of randomized controlled trials [RCTs] with significant heterogeneity). Across multiple RCTs using varied yoga interventions and diverse study populations, yoga typically improves overall symptom scores for anxiety and depression by about 40%, both by itself and as an adjunctive treatment. It produces no reported harmful side effects.
EVIDENCE SUMMARY
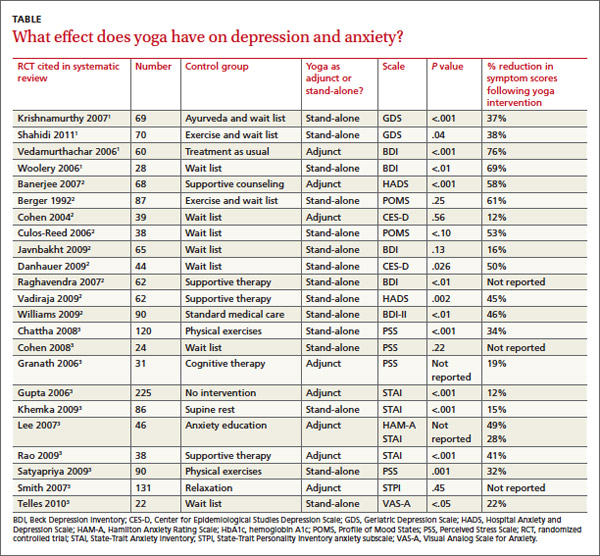
Across 3 systematic reviews of yoga for depression, anxiety, and stress, yoga produced overall reductions of symptoms between 12% and 76%, with an average of 39% net reduction in symptom scores across measures (TABLE).1-3 The RCTs included in the systematic reviews were too heterogeneous to allow quantitative analyses of effect sizes.
Yoga found to significantly reduce depression symptoms
Two 2012 systematic reviews of yoga for depression evaluated 13 RCTs with a total of 782 participants, ages 18 to 80 years with mild to moderate depression. In the 12 RCTs that reported gender, 82% of participants were female; in 6 RCTs a total of 313 patients had cancer.1,2
The RCTs compared yoga to wait-list controls, counseling, education, exercise, or usual care. They evaluated yoga both as a stand-alone intervention and an adjunct to usual care. Yoga sessions varied from 1 hour weekly to 90 minutes daily over 2 to 24 weeks and included physical postures, relaxation, and breathing techniques.
Eight moderate- to high-quality RCTs with a total of 483 participants reported statistically significant reductions in depression symptoms in the yoga groups compared with control groups. In 3 RCTs, yoga was equivalent to wait-list controls; 2 RCTs showed results equivalent to exercise and superior to wait-list controls.
Yoga alleviates anxiety and stress without adverse effects
A 2012 systematic review of yoga for stress and anxiety evaluated 10 RCTs with a total of 813 heterogeneous participants, ages 18 to 76 years, including pregnant women, breast cancer patients, flood survivors, healthy volunteers, patients with chronic illnesses, perimenopausal women, adults with metabolic syndrome, and people working in finance, all with a range of anxiety and stress symptoms.3 The RCTs compared yoga, as an adjunctive or stand-alone treatment, with wait-list controls, relaxation, therapy, anxiety education, rest, or exercise. Yoga regimens varied from a single 20-minute session to 16 weeks of daily 1-hour sessions, with most regimens lasting 6 to 10 weeks.
Of the 10 RCTs reviewed, 7 moderate- to high-quality studies with a total of 627 participants found statistically significant reductions in anxiety and stress in yoga groups compared with control groups. Of the remaining 3 studies, 1 found yoga equivalent to cognitive therapy; 1 found a nonsignificant benefit for yoga compared with wait-list controls; and 1 found no improvement with either yoga or relaxation.
Study limitations included a range of symptom severity, variable type and length of yoga, lack of participant blinding, wait-list rather than active-treatment controls, and a lack of consistent long-term follow-up data. The RCTs didn’t report any adverse effects of yoga, and yoga is considered safe when taught by a competent instructor.3,4
Continue for recommendations >>
recommendations
The Institute for Clinical Systems Improvement and the Canadian Network for Mood and Anxiety Treatments recommend yoga as an effective adjunctive treatment to decrease the severity of depression symptoms.5,6
The Veterans Health Administration and the US Department of Defense recommend yoga as a potential adjunctive treatment to manage the hyperarousal symptoms of post-traumatic stress disorder (PTSD).7
The Work Loss Data Institute recommends yoga as an intervention for workers compensation conditions including occupational stress, major depressive disorder, PTSD, and other mental disorders.8
1. Balasubramaniam M, Telles S, Doraiswamy PM. Yoga on our minds: a systematic review of yoga for neuropsychiatric disorders. Front Psychiatry. 2012;3:117.
2. D’Silva S, Poscablo C, Habousha R, et al. Mind-body medicine therapies for a range of depression severity: a systematic review. Psychosomatics. 2012;53:407-423.
3. Li AW, Goldsmith CA. The effects of yoga on anxiety and stress. Altern Med Rev. 2012;17:21-35.
4. Brown RP, Gerbarg PL. Sudarshan Kriya Yogic breathing in the treatment of stress, anxiety, and depression. Part II—clinical applications and guidelines. J Altern Complement Med. 2005;11:711-717.
5. Mitchell J, Trangle M, Degnan B, et al. Institute for Clinical Systems Improvement. Adult depression primary care. Available at: https://www.icsi.org/_asset/fnhdm3/Depr-Interactive0512b.pdf. Updated September 2013. Accessed March 6, 2014.
6. Ravindran AV, Lam RW, Filteau MJ, et al; Canadian Network for Mood and Anxiety Treatments (CANMAT). Canadian Network for Mood and Anxiety Treatments (CANMAT) clinical guidelines for the management of major depressive disorder in adults. V. Complementary and alternative medicine treatments. J Affect Disord. 2009;117(suppl 1):S54-S64.
7. US Department of Veterans Affairs. VA/DoD clinical practice guideline for management of post-traumatic stress disorder and acute stress reaction. US Department of Veterans Affairs Web site. Available at: http://www.healthquality.va.gov/ptsd/. Accessed March 6, 2014.
8. Agency for Healthcare Research and Quality. Mental illness & stress. Agency for Healthcare Research and Quality Web site. Available at: http://www.guideline.gov/content.aspx?id=47588. Updated May 2011. Accessed June 17, 2014
Yes, yoga can reduce symptoms of anxiety and depression (strength of recommendation [SOR]: B, systematic reviews of randomized controlled trials [RCTs] with significant heterogeneity). Across multiple RCTs using varied yoga interventions and diverse study populations, yoga typically improves overall symptom scores for anxiety and depression by about 40%, both by itself and as an adjunctive treatment. It produces no reported harmful side effects.
EVIDENCE SUMMARY
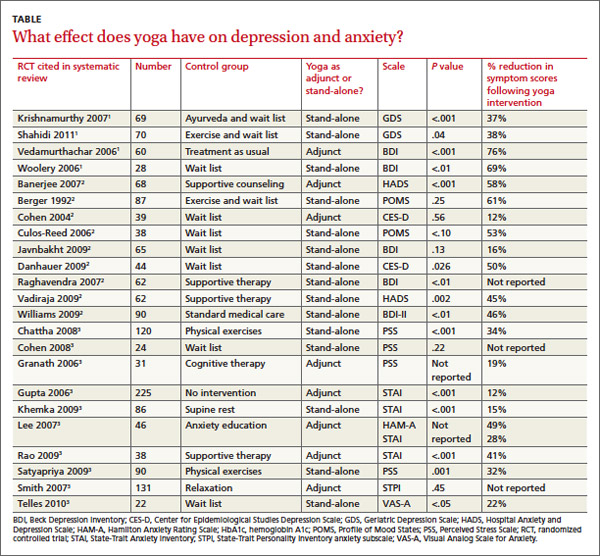
Across 3 systematic reviews of yoga for depression, anxiety, and stress, yoga produced overall reductions of symptoms between 12% and 76%, with an average of 39% net reduction in symptom scores across measures (TABLE).1-3 The RCTs included in the systematic reviews were too heterogeneous to allow quantitative analyses of effect sizes.
Yoga found to significantly reduce depression symptoms
Two 2012 systematic reviews of yoga for depression evaluated 13 RCTs with a total of 782 participants, ages 18 to 80 years with mild to moderate depression. In the 12 RCTs that reported gender, 82% of participants were female; in 6 RCTs a total of 313 patients had cancer.1,2
The RCTs compared yoga to wait-list controls, counseling, education, exercise, or usual care. They evaluated yoga both as a stand-alone intervention and an adjunct to usual care. Yoga sessions varied from 1 hour weekly to 90 minutes daily over 2 to 24 weeks and included physical postures, relaxation, and breathing techniques.
Eight moderate- to high-quality RCTs with a total of 483 participants reported statistically significant reductions in depression symptoms in the yoga groups compared with control groups. In 3 RCTs, yoga was equivalent to wait-list controls; 2 RCTs showed results equivalent to exercise and superior to wait-list controls.
Yoga alleviates anxiety and stress without adverse effects
A 2012 systematic review of yoga for stress and anxiety evaluated 10 RCTs with a total of 813 heterogeneous participants, ages 18 to 76 years, including pregnant women, breast cancer patients, flood survivors, healthy volunteers, patients with chronic illnesses, perimenopausal women, adults with metabolic syndrome, and people working in finance, all with a range of anxiety and stress symptoms.3 The RCTs compared yoga, as an adjunctive or stand-alone treatment, with wait-list controls, relaxation, therapy, anxiety education, rest, or exercise. Yoga regimens varied from a single 20-minute session to 16 weeks of daily 1-hour sessions, with most regimens lasting 6 to 10 weeks.
Of the 10 RCTs reviewed, 7 moderate- to high-quality studies with a total of 627 participants found statistically significant reductions in anxiety and stress in yoga groups compared with control groups. Of the remaining 3 studies, 1 found yoga equivalent to cognitive therapy; 1 found a nonsignificant benefit for yoga compared with wait-list controls; and 1 found no improvement with either yoga or relaxation.
Study limitations included a range of symptom severity, variable type and length of yoga, lack of participant blinding, wait-list rather than active-treatment controls, and a lack of consistent long-term follow-up data. The RCTs didn’t report any adverse effects of yoga, and yoga is considered safe when taught by a competent instructor.3,4
Continue for recommendations >>
recommendations
The Institute for Clinical Systems Improvement and the Canadian Network for Mood and Anxiety Treatments recommend yoga as an effective adjunctive treatment to decrease the severity of depression symptoms.5,6
The Veterans Health Administration and the US Department of Defense recommend yoga as a potential adjunctive treatment to manage the hyperarousal symptoms of post-traumatic stress disorder (PTSD).7
The Work Loss Data Institute recommends yoga as an intervention for workers compensation conditions including occupational stress, major depressive disorder, PTSD, and other mental disorders.8
Yes, yoga can reduce symptoms of anxiety and depression (strength of recommendation [SOR]: B, systematic reviews of randomized controlled trials [RCTs] with significant heterogeneity). Across multiple RCTs using varied yoga interventions and diverse study populations, yoga typically improves overall symptom scores for anxiety and depression by about 40%, both by itself and as an adjunctive treatment. It produces no reported harmful side effects.
EVIDENCE SUMMARY
Across 3 systematic reviews of yoga for depression, anxiety, and stress, yoga produced overall reductions of symptoms between 12% and 76%, with an average of 39% net reduction in symptom scores across measures (TABLE).1-3 The RCTs included in the systematic reviews were too heterogeneous to allow quantitative analyses of effect sizes.
Yoga found to significantly reduce depression symptoms
Two 2012 systematic reviews of yoga for depression evaluated 13 RCTs with a total of 782 participants, ages 18 to 80 years with mild to moderate depression. In the 12 RCTs that reported gender, 82% of participants were female; in 6 RCTs a total of 313 patients had cancer.1,2
The RCTs compared yoga to wait-list controls, counseling, education, exercise, or usual care. They evaluated yoga both as a stand-alone intervention and an adjunct to usual care. Yoga sessions varied from 1 hour weekly to 90 minutes daily over 2 to 24 weeks and included physical postures, relaxation, and breathing techniques.
Eight moderate- to high-quality RCTs with a total of 483 participants reported statistically significant reductions in depression symptoms in the yoga groups compared with control groups. In 3 RCTs, yoga was equivalent to wait-list controls; 2 RCTs showed results equivalent to exercise and superior to wait-list controls.
Yoga alleviates anxiety and stress without adverse effects
A 2012 systematic review of yoga for stress and anxiety evaluated 10 RCTs with a total of 813 heterogeneous participants, ages 18 to 76 years, including pregnant women, breast cancer patients, flood survivors, healthy volunteers, patients with chronic illnesses, perimenopausal women, adults with metabolic syndrome, and people working in finance, all with a range of anxiety and stress symptoms.3 The RCTs compared yoga, as an adjunctive or stand-alone treatment, with wait-list controls, relaxation, therapy, anxiety education, rest, or exercise. Yoga regimens varied from a single 20-minute session to 16 weeks of daily 1-hour sessions, with most regimens lasting 6 to 10 weeks.
Of the 10 RCTs reviewed, 7 moderate- to high-quality studies with a total of 627 participants found statistically significant reductions in anxiety and stress in yoga groups compared with control groups. Of the remaining 3 studies, 1 found yoga equivalent to cognitive therapy; 1 found a nonsignificant benefit for yoga compared with wait-list controls; and 1 found no improvement with either yoga or relaxation.
Study limitations included a range of symptom severity, variable type and length of yoga, lack of participant blinding, wait-list rather than active-treatment controls, and a lack of consistent long-term follow-up data. The RCTs didn’t report any adverse effects of yoga, and yoga is considered safe when taught by a competent instructor.3,4
Continue for recommendations >>
recommendations
The Institute for Clinical Systems Improvement and the Canadian Network for Mood and Anxiety Treatments recommend yoga as an effective adjunctive treatment to decrease the severity of depression symptoms.5,6
The Veterans Health Administration and the US Department of Defense recommend yoga as a potential adjunctive treatment to manage the hyperarousal symptoms of post-traumatic stress disorder (PTSD).7
The Work Loss Data Institute recommends yoga as an intervention for workers compensation conditions including occupational stress, major depressive disorder, PTSD, and other mental disorders.8
1. Balasubramaniam M, Telles S, Doraiswamy PM. Yoga on our minds: a systematic review of yoga for neuropsychiatric disorders. Front Psychiatry. 2012;3:117.
2. D’Silva S, Poscablo C, Habousha R, et al. Mind-body medicine therapies for a range of depression severity: a systematic review. Psychosomatics. 2012;53:407-423.
3. Li AW, Goldsmith CA. The effects of yoga on anxiety and stress. Altern Med Rev. 2012;17:21-35.
4. Brown RP, Gerbarg PL. Sudarshan Kriya Yogic breathing in the treatment of stress, anxiety, and depression. Part II—clinical applications and guidelines. J Altern Complement Med. 2005;11:711-717.
5. Mitchell J, Trangle M, Degnan B, et al. Institute for Clinical Systems Improvement. Adult depression primary care. Available at: https://www.icsi.org/_asset/fnhdm3/Depr-Interactive0512b.pdf. Updated September 2013. Accessed March 6, 2014.
6. Ravindran AV, Lam RW, Filteau MJ, et al; Canadian Network for Mood and Anxiety Treatments (CANMAT). Canadian Network for Mood and Anxiety Treatments (CANMAT) clinical guidelines for the management of major depressive disorder in adults. V. Complementary and alternative medicine treatments. J Affect Disord. 2009;117(suppl 1):S54-S64.
7. US Department of Veterans Affairs. VA/DoD clinical practice guideline for management of post-traumatic stress disorder and acute stress reaction. US Department of Veterans Affairs Web site. Available at: http://www.healthquality.va.gov/ptsd/. Accessed March 6, 2014.
8. Agency for Healthcare Research and Quality. Mental illness & stress. Agency for Healthcare Research and Quality Web site. Available at: http://www.guideline.gov/content.aspx?id=47588. Updated May 2011. Accessed June 17, 2014
1. Balasubramaniam M, Telles S, Doraiswamy PM. Yoga on our minds: a systematic review of yoga for neuropsychiatric disorders. Front Psychiatry. 2012;3:117.
2. D’Silva S, Poscablo C, Habousha R, et al. Mind-body medicine therapies for a range of depression severity: a systematic review. Psychosomatics. 2012;53:407-423.
3. Li AW, Goldsmith CA. The effects of yoga on anxiety and stress. Altern Med Rev. 2012;17:21-35.
4. Brown RP, Gerbarg PL. Sudarshan Kriya Yogic breathing in the treatment of stress, anxiety, and depression. Part II—clinical applications and guidelines. J Altern Complement Med. 2005;11:711-717.
5. Mitchell J, Trangle M, Degnan B, et al. Institute for Clinical Systems Improvement. Adult depression primary care. Available at: https://www.icsi.org/_asset/fnhdm3/Depr-Interactive0512b.pdf. Updated September 2013. Accessed March 6, 2014.
6. Ravindran AV, Lam RW, Filteau MJ, et al; Canadian Network for Mood and Anxiety Treatments (CANMAT). Canadian Network for Mood and Anxiety Treatments (CANMAT) clinical guidelines for the management of major depressive disorder in adults. V. Complementary and alternative medicine treatments. J Affect Disord. 2009;117(suppl 1):S54-S64.
7. US Department of Veterans Affairs. VA/DoD clinical practice guideline for management of post-traumatic stress disorder and acute stress reaction. US Department of Veterans Affairs Web site. Available at: http://www.healthquality.va.gov/ptsd/. Accessed March 6, 2014.
8. Agency for Healthcare Research and Quality. Mental illness & stress. Agency for Healthcare Research and Quality Web site. Available at: http://www.guideline.gov/content.aspx?id=47588. Updated May 2011. Accessed June 17, 2014
Can yoga reduce symptoms of anxiety and depression?
Yes, yoga can reduce symptoms of anxiety and depression (strength of recommendation [SOR]: B, systematic reviews of randomized controlled trials [RCTs] with significant heterogeneity). Across multiple RCTs using varied yoga interventions and diverse study populations, yoga typically improves overall symptom scores for anxiety and depression by about 40%, both by itself and as an adjunctive treatment. It produces no reported harmful side effects.
EVIDENCE SUMMARY
Across 3 systematic reviews of yoga for depression, anxiety, and stress, yoga produced overall reductions of symptoms between 12% and 76%, with an average of 39% net reduction in symptom scores across measures (TABLE).1-3 The RCTs included in the systematic reviews were too heterogeneous to allow quantitative analyses of effect sizes.
Yoga found to significantly reduce depression symptoms
Two 2012 systematic reviews of yoga for depression evaluated 13 RCTs with a total of 782 participants, ages 18 to 80 years with mild to moderate depression. In the 12 RCTs that reported gender, 82% of participants were female; in 6 RCTs a total of 313 patients had cancer.1,2
The RCTs compared yoga to wait-list controls, counseling, education, exercise, or usual care. They evaluated yoga both as a stand-alone intervention and an adjunct to usual care. Yoga sessions varied from 1 hour weekly to 90 minutes daily over 2 to 24 weeks and included physical postures, relaxation, and breathing techniques.
Eight moderate- to high-quality RCTs with a total of 483 participants reported statistically significant reductions in depression symptoms in the yoga groups compared with control groups. In 3 RCTs, yoga was equivalent to wait-list controls; 2 RCTs showed results equivalent to exercise and superior to wait-list controls.
Yoga alleviates anxiety and stress without adverse effects
A 2012 systematic review of yoga for stress and anxiety evaluated 10 RCTs with a total of 813 heterogeneous participants, ages 18 to 76 years, including pregnant women, breast cancer patients, flood survivors, healthy volunteers, patients with chronic illnesses, perimenopausal women, adults with metabolic syndrome, and people working in finance, all with a range of anxiety and stress symptoms.3 The RCTs compared yoga, as an adjunctive or stand-alone treatment, with wait-list controls, relaxation, therapy, anxiety education, rest, or exercise. Yoga regimens varied from a single 20-minute session to 16 weeks of daily 1-hour sessions, with most regimens lasting 6 to 10 weeks.
Of the 10 RCTs reviewed, 7 moderate- to high-quality studies with a total of 627 participants found statistically significant reductions in anxiety and stress in yoga groups compared with control groups. Of the remaining 3 studies, 1 found yoga equivalent to cognitive therapy; 1 found a nonsignificant benefit for yoga compared with wait-list controls; and 1 found no improvement with either yoga or relaxation.
Study limitations included a range of symptom severity, variable type and length of yoga, lack of participant blinding, wait-list rather than active-treatment controls, and a lack of consistent long-term follow-up data. The RCTs didn’t report any adverse effects of yoga, and yoga is considered safe when taught by a competent instructor.3,4
RECOMMENDATIONS
The Institute for Clinical Systems Improvement and the Canadian Network for Mood and Anxiety Treatments recommend yoga as an effective adjunctive treatment to decrease the severity of depression symptoms.5,6
The Veterans Health Administration and the US Department of Defense recommend yoga as a potential adjunctive treatment to manage the hyperarousal symptoms of post-traumatic stress disorder (PTSD).7
The Work Loss Data Institute recommends yoga as an intervention for workers compensation conditions including occupational stress, major depressive disorder, PTSD, and other mental disorders.8
1. Balasubramaniam M, Telles S, Doraiswamy PM. Yoga on our minds: a systematic review of yoga for neuropsychiatric disorders. Front Psychiatry. 2012;3:117.
2. D’Silva S, Poscablo C, Habousha R, et al. Mind-body medicine therapies for a range of depression severity: a systematic review. Psychosomatics. 2012;53:407-423.
3. Li AW, Goldsmith CA. The effects of yoga on anxiety and stress. Altern Med Rev. 2012;17:21-35.
4. Brown RP, Gerbarg PL. Sudarshan Kriya Yogic breathing in the treatment of stress, anxiety, and depression. Part II—clinical applications and guidelines. J Altern Complement Med. 2005;11:711-717.
5. Mitchell J, Trangle M, Degnan B, et al. Institute for Clinical Systems Improvement. Adult depression primary care. Available at: https://www.icsi.org/_asset/fnhdm3/Depr-Interactive0512b.pdf. Updated September 2013. Accessed March 6, 2014.
6. Ravindran AV, Lam RW, Filteau MJ, et al; Canadian Network for Mood and Anxiety Treatments (CANMAT). Canadian Network for Mood and Anxiety Treatments (CANMAT) clinical guidelines for the management of major depressive disorder in adults. V. Complementary and alternative medicine treatments. J Affect Disord. 2009;117(suppl 1):S54-S64.
7. US Department of Veterans Affairs. VA/DoD clinical practice guideline for management of post-traumatic stress disorder and acute stress reaction. US Department of Veterans Affairs Web site. Available at: http://www.healthquality.va.gov/ptsd/. Accessed March 6, 2014.
8. Agency for Healthcare Research and Quality. Mental illness & stress. Agency for Healthcare Research and Quality Web site. Available at: http://www.guideline.gov/content.aspx?id=47588. Updated May 2011. Accessed June 17, 2014
Yes, yoga can reduce symptoms of anxiety and depression (strength of recommendation [SOR]: B, systematic reviews of randomized controlled trials [RCTs] with significant heterogeneity). Across multiple RCTs using varied yoga interventions and diverse study populations, yoga typically improves overall symptom scores for anxiety and depression by about 40%, both by itself and as an adjunctive treatment. It produces no reported harmful side effects.
EVIDENCE SUMMARY
Across 3 systematic reviews of yoga for depression, anxiety, and stress, yoga produced overall reductions of symptoms between 12% and 76%, with an average of 39% net reduction in symptom scores across measures (TABLE).1-3 The RCTs included in the systematic reviews were too heterogeneous to allow quantitative analyses of effect sizes.
Yoga found to significantly reduce depression symptoms
Two 2012 systematic reviews of yoga for depression evaluated 13 RCTs with a total of 782 participants, ages 18 to 80 years with mild to moderate depression. In the 12 RCTs that reported gender, 82% of participants were female; in 6 RCTs a total of 313 patients had cancer.1,2
The RCTs compared yoga to wait-list controls, counseling, education, exercise, or usual care. They evaluated yoga both as a stand-alone intervention and an adjunct to usual care. Yoga sessions varied from 1 hour weekly to 90 minutes daily over 2 to 24 weeks and included physical postures, relaxation, and breathing techniques.
Eight moderate- to high-quality RCTs with a total of 483 participants reported statistically significant reductions in depression symptoms in the yoga groups compared with control groups. In 3 RCTs, yoga was equivalent to wait-list controls; 2 RCTs showed results equivalent to exercise and superior to wait-list controls.
Yoga alleviates anxiety and stress without adverse effects
A 2012 systematic review of yoga for stress and anxiety evaluated 10 RCTs with a total of 813 heterogeneous participants, ages 18 to 76 years, including pregnant women, breast cancer patients, flood survivors, healthy volunteers, patients with chronic illnesses, perimenopausal women, adults with metabolic syndrome, and people working in finance, all with a range of anxiety and stress symptoms.3 The RCTs compared yoga, as an adjunctive or stand-alone treatment, with wait-list controls, relaxation, therapy, anxiety education, rest, or exercise. Yoga regimens varied from a single 20-minute session to 16 weeks of daily 1-hour sessions, with most regimens lasting 6 to 10 weeks.
Of the 10 RCTs reviewed, 7 moderate- to high-quality studies with a total of 627 participants found statistically significant reductions in anxiety and stress in yoga groups compared with control groups. Of the remaining 3 studies, 1 found yoga equivalent to cognitive therapy; 1 found a nonsignificant benefit for yoga compared with wait-list controls; and 1 found no improvement with either yoga or relaxation.
Study limitations included a range of symptom severity, variable type and length of yoga, lack of participant blinding, wait-list rather than active-treatment controls, and a lack of consistent long-term follow-up data. The RCTs didn’t report any adverse effects of yoga, and yoga is considered safe when taught by a competent instructor.3,4
RECOMMENDATIONS
The Institute for Clinical Systems Improvement and the Canadian Network for Mood and Anxiety Treatments recommend yoga as an effective adjunctive treatment to decrease the severity of depression symptoms.5,6
The Veterans Health Administration and the US Department of Defense recommend yoga as a potential adjunctive treatment to manage the hyperarousal symptoms of post-traumatic stress disorder (PTSD).7
The Work Loss Data Institute recommends yoga as an intervention for workers compensation conditions including occupational stress, major depressive disorder, PTSD, and other mental disorders.8
Yes, yoga can reduce symptoms of anxiety and depression (strength of recommendation [SOR]: B, systematic reviews of randomized controlled trials [RCTs] with significant heterogeneity). Across multiple RCTs using varied yoga interventions and diverse study populations, yoga typically improves overall symptom scores for anxiety and depression by about 40%, both by itself and as an adjunctive treatment. It produces no reported harmful side effects.
EVIDENCE SUMMARY
Across 3 systematic reviews of yoga for depression, anxiety, and stress, yoga produced overall reductions of symptoms between 12% and 76%, with an average of 39% net reduction in symptom scores across measures (TABLE).1-3 The RCTs included in the systematic reviews were too heterogeneous to allow quantitative analyses of effect sizes.
Yoga found to significantly reduce depression symptoms
Two 2012 systematic reviews of yoga for depression evaluated 13 RCTs with a total of 782 participants, ages 18 to 80 years with mild to moderate depression. In the 12 RCTs that reported gender, 82% of participants were female; in 6 RCTs a total of 313 patients had cancer.1,2
The RCTs compared yoga to wait-list controls, counseling, education, exercise, or usual care. They evaluated yoga both as a stand-alone intervention and an adjunct to usual care. Yoga sessions varied from 1 hour weekly to 90 minutes daily over 2 to 24 weeks and included physical postures, relaxation, and breathing techniques.
Eight moderate- to high-quality RCTs with a total of 483 participants reported statistically significant reductions in depression symptoms in the yoga groups compared with control groups. In 3 RCTs, yoga was equivalent to wait-list controls; 2 RCTs showed results equivalent to exercise and superior to wait-list controls.
Yoga alleviates anxiety and stress without adverse effects
A 2012 systematic review of yoga for stress and anxiety evaluated 10 RCTs with a total of 813 heterogeneous participants, ages 18 to 76 years, including pregnant women, breast cancer patients, flood survivors, healthy volunteers, patients with chronic illnesses, perimenopausal women, adults with metabolic syndrome, and people working in finance, all with a range of anxiety and stress symptoms.3 The RCTs compared yoga, as an adjunctive or stand-alone treatment, with wait-list controls, relaxation, therapy, anxiety education, rest, or exercise. Yoga regimens varied from a single 20-minute session to 16 weeks of daily 1-hour sessions, with most regimens lasting 6 to 10 weeks.
Of the 10 RCTs reviewed, 7 moderate- to high-quality studies with a total of 627 participants found statistically significant reductions in anxiety and stress in yoga groups compared with control groups. Of the remaining 3 studies, 1 found yoga equivalent to cognitive therapy; 1 found a nonsignificant benefit for yoga compared with wait-list controls; and 1 found no improvement with either yoga or relaxation.
Study limitations included a range of symptom severity, variable type and length of yoga, lack of participant blinding, wait-list rather than active-treatment controls, and a lack of consistent long-term follow-up data. The RCTs didn’t report any adverse effects of yoga, and yoga is considered safe when taught by a competent instructor.3,4
RECOMMENDATIONS
The Institute for Clinical Systems Improvement and the Canadian Network for Mood and Anxiety Treatments recommend yoga as an effective adjunctive treatment to decrease the severity of depression symptoms.5,6
The Veterans Health Administration and the US Department of Defense recommend yoga as a potential adjunctive treatment to manage the hyperarousal symptoms of post-traumatic stress disorder (PTSD).7
The Work Loss Data Institute recommends yoga as an intervention for workers compensation conditions including occupational stress, major depressive disorder, PTSD, and other mental disorders.8
1. Balasubramaniam M, Telles S, Doraiswamy PM. Yoga on our minds: a systematic review of yoga for neuropsychiatric disorders. Front Psychiatry. 2012;3:117.
2. D’Silva S, Poscablo C, Habousha R, et al. Mind-body medicine therapies for a range of depression severity: a systematic review. Psychosomatics. 2012;53:407-423.
3. Li AW, Goldsmith CA. The effects of yoga on anxiety and stress. Altern Med Rev. 2012;17:21-35.
4. Brown RP, Gerbarg PL. Sudarshan Kriya Yogic breathing in the treatment of stress, anxiety, and depression. Part II—clinical applications and guidelines. J Altern Complement Med. 2005;11:711-717.
5. Mitchell J, Trangle M, Degnan B, et al. Institute for Clinical Systems Improvement. Adult depression primary care. Available at: https://www.icsi.org/_asset/fnhdm3/Depr-Interactive0512b.pdf. Updated September 2013. Accessed March 6, 2014.
6. Ravindran AV, Lam RW, Filteau MJ, et al; Canadian Network for Mood and Anxiety Treatments (CANMAT). Canadian Network for Mood and Anxiety Treatments (CANMAT) clinical guidelines for the management of major depressive disorder in adults. V. Complementary and alternative medicine treatments. J Affect Disord. 2009;117(suppl 1):S54-S64.
7. US Department of Veterans Affairs. VA/DoD clinical practice guideline for management of post-traumatic stress disorder and acute stress reaction. US Department of Veterans Affairs Web site. Available at: http://www.healthquality.va.gov/ptsd/. Accessed March 6, 2014.
8. Agency for Healthcare Research and Quality. Mental illness & stress. Agency for Healthcare Research and Quality Web site. Available at: http://www.guideline.gov/content.aspx?id=47588. Updated May 2011. Accessed June 17, 2014
1. Balasubramaniam M, Telles S, Doraiswamy PM. Yoga on our minds: a systematic review of yoga for neuropsychiatric disorders. Front Psychiatry. 2012;3:117.
2. D’Silva S, Poscablo C, Habousha R, et al. Mind-body medicine therapies for a range of depression severity: a systematic review. Psychosomatics. 2012;53:407-423.
3. Li AW, Goldsmith CA. The effects of yoga on anxiety and stress. Altern Med Rev. 2012;17:21-35.
4. Brown RP, Gerbarg PL. Sudarshan Kriya Yogic breathing in the treatment of stress, anxiety, and depression. Part II—clinical applications and guidelines. J Altern Complement Med. 2005;11:711-717.
5. Mitchell J, Trangle M, Degnan B, et al. Institute for Clinical Systems Improvement. Adult depression primary care. Available at: https://www.icsi.org/_asset/fnhdm3/Depr-Interactive0512b.pdf. Updated September 2013. Accessed March 6, 2014.
6. Ravindran AV, Lam RW, Filteau MJ, et al; Canadian Network for Mood and Anxiety Treatments (CANMAT). Canadian Network for Mood and Anxiety Treatments (CANMAT) clinical guidelines for the management of major depressive disorder in adults. V. Complementary and alternative medicine treatments. J Affect Disord. 2009;117(suppl 1):S54-S64.
7. US Department of Veterans Affairs. VA/DoD clinical practice guideline for management of post-traumatic stress disorder and acute stress reaction. US Department of Veterans Affairs Web site. Available at: http://www.healthquality.va.gov/ptsd/. Accessed March 6, 2014.
8. Agency for Healthcare Research and Quality. Mental illness & stress. Agency for Healthcare Research and Quality Web site. Available at: http://www.guideline.gov/content.aspx?id=47588. Updated May 2011. Accessed June 17, 2014
Evidence-based answers from the Family Physicians Inquiries Network
Why you shouldn’t start beta-blockers before surgery
Do not routinely initiate beta-blockers in patients undergoing intermediate- or high-risk noncardiac surgery. Beta-blockers appear to increase the 30-day risk of all-cause mortality.1
Strength of recommendation
A: Based on meta-analysis of 9 randomized controlled trials (RCTs).
Bouri S, Shun-Shin MJ, Cole GD, et al. Meta-analysis of secure randomised controlled trials of ß-blockade to prevent perioperative death in non-cardiac surgery. Heart. 2014;100:456-464.
Illustrative case
A 67-year-old woman with diabetes, hypertension, and hyperlipidemia comes to your office for an evaluation before undergoing a total hip arthroplasty. She is not taking a beta-blocker. Should you prescribe one?
Current guidelines from the American College of Cardiology Foundation (ACCF) and the American Heart Association (AHA) recommend starting beta-blockers to prevent cardiac events in patients about to undergo intermediate- or high-risk surgery or vascular surgery who have a history of inducible ischemia, coronary artery disease (CAD), or at least one risk factor for CAD.2 However, the majority of the evidence for these guidelines, which were published in 2009 and are in the process of being updated, came from the DECREASE (Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography) trials, which have been discredited due to serious methodological flaws, including falsified descriptions of how outcomes were determined and fictitious databases.3 A new meta-analysis by Bouri et al1 that excluded the DECREASE trials found that although preoperative beta-blockers reduce the rate of certain nonfatal outcomes, they increase the risk of death and stroke.
STUDY SUMMARY: Multiple RCTs find preop beta-blockers do more harm, than good
Bouri et al1 conducted a meta-analysis of published RCTs evaluating preoperative beta-blockers vs placebo for patients undergoing noncardiac surgery. Of the 11 studies that met eligibility criteria, 2 were the discredited DECREASE trials. Thus, Bouri et al1 analyzed 9 high-quality RCTs that included 10,529 patients.
Most studies included patients undergoing vascular surgery. Some studies also included intra-abdominal, intrathoracic, neurosurgical, orthopedic, urologic, and gynecologic surgeries. Beta-blockers were started no more than a day before surgery and were discontinued at hospital discharge or up to 30 days postop. Metoprolol was used in 5 trials, bisoprolol in one trial, atenolol in 2 trials, and propranolol in one trial. The primary endpoint was all-cause mortality within 30 days.
A total of 5264 patients were randomized to beta-blockers and 5265 to placebo. There were 162 deaths in the beta-blocker group and 129 deaths in the placebo group. Patients who received beta-blockers had a 27% increased risk of all-cause mortality (risk ratio [RR]=1.27; 95% confidence interval [CI], 1.01-1.60; P=.04). The number needed to harm was 160.
Six of the studies also evaluated rates of nonfatal myocardial infarction (MI), nonfatal stroke, and hypotension. Beta-blockers lowered the risk of nonfatal MI (RR=.73; 95% CI, .61-.88; P=.001), but increased the risk of nonfatal stroke (RR=1.73; 95% CI, 1.00-2.99; P=.05) and hypotension (RR=1.51; 95% CI, 1.37-1.67; P=.00001).
This meta-analysis was dominated by the 2008 Peri-Operative ISchemic Evaluation (POISE) trial, an RCT that compared placebo to extended-release metoprolol, 100 mg 2 to 4 hours before surgery followed by 200 mg/d for 30 days, in 8351 patients with, or at risk for, atherosclerotic disease.4 While beta-blockers reduced the risk of MI and atrial fibrillation, they increased the risk of mortality and stroke, likely due to drug-induced hypotension. The slightly larger-than-typical doses of beta-blockers used in the POISE study may have contributed to the excess mortality.
WHAT'S NEW: Avoiding beta-blockers in surgery patients will prevent deaths
Bouri et al1 found that while beta-blockers protect against nonfatal MIs, they increase the risk for nonfatal strokes and death. This new meta-analysis challenges the ACCF/AHA recommendations by suggesting that abandoning the use of beta-blockers for preoperative patients who aren’t already taking them will prevent a substantial number of perioperative deaths. Bouri et al1 estimate that in the United Kingdom, where 47,286 deaths occur annually within 30 days of intermediate or high-risk procedures, the number of iatrogenic deaths would drop by approximately 10,000 if beta-blockers were not used.1
CAVEATS: Don't stop beta-blockers in patients who already take them
This meta-analysis did not evaluate outcomes in patients who were already taking beta-blockers. Patients who are already on beta-blockers should continue to take them in the perioperative period, which is in line with current ACCF/AHA guidelines.
CHALLENGES TO IMPLEMENTATION: Some physician may be reluctant to disregard published guidelines
Some physicians may not be comfortable ignoring the current ACCF/AHA guidelines that make a Class IIa recommendation (it is reasonable to administer this treatment) for the use of preoperative beta-blockade for patients at risk of cardiovascular events who were not previously taking a beta-blocker. This updated meta-analysis excludes the discredited DECREASE trials and challenges us to act against these current guidelines while we wait for updated recommendations.
Acknowledgement
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Bouri S, Shun-Shin MJ, Cole GD, et al. Meta-analysis of secure randomised controlled trials of ß-blockade to prevent perioperative death in non-cardiac surgery. Heart. 2014;100:456-464.
2. American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines; American Society of Echocardiography; American Society of Nuclear Cardiology; Heart Rhythm Society; Society of Cardiovascular Anesthesiologists; Society for Cardiovascular Angiography and Interventions; Society for Vascular Medicine; Society for Vascular Surgery; Fleisher LA, Beckman JA, Brown KA, et al. 2009 ACCF/AHA focused update on perioperative beta blockade incorporated into the ACC/AHA 2007 guidelines on perioperative cardiovascular evaluation and care for noncardiac surgery. J Am Coll Cardiol. 2009;54:e13-e118.
3. Eramus Medical Center Follow-up Investigation Committee. Report on the 2012 follow-up investigation of possible breaches of academic integrity. CardioBrief Web site. Available at: http://cardiobrief.files.wordpress.com/2012/10/integrity-report-2012-10-english-translation.pdf. Published September 30, 2012. Accessed March 31, 2014.
4. POISE Study Group; Devereaux PJ; Yang H; Yusuf S; et al. Effects of extended-release metoprolol succinate in patients undergoing non-cardiac surgery (POISE trial): a randomised controlled trial. Lancet. 2008;371:1839-1847.
Do not routinely initiate beta-blockers in patients undergoing intermediate- or high-risk noncardiac surgery. Beta-blockers appear to increase the 30-day risk of all-cause mortality.1
Strength of recommendation
A: Based on meta-analysis of 9 randomized controlled trials (RCTs).
Bouri S, Shun-Shin MJ, Cole GD, et al. Meta-analysis of secure randomised controlled trials of ß-blockade to prevent perioperative death in non-cardiac surgery. Heart. 2014;100:456-464.
Illustrative case
A 67-year-old woman with diabetes, hypertension, and hyperlipidemia comes to your office for an evaluation before undergoing a total hip arthroplasty. She is not taking a beta-blocker. Should you prescribe one?
Current guidelines from the American College of Cardiology Foundation (ACCF) and the American Heart Association (AHA) recommend starting beta-blockers to prevent cardiac events in patients about to undergo intermediate- or high-risk surgery or vascular surgery who have a history of inducible ischemia, coronary artery disease (CAD), or at least one risk factor for CAD.2 However, the majority of the evidence for these guidelines, which were published in 2009 and are in the process of being updated, came from the DECREASE (Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography) trials, which have been discredited due to serious methodological flaws, including falsified descriptions of how outcomes were determined and fictitious databases.3 A new meta-analysis by Bouri et al1 that excluded the DECREASE trials found that although preoperative beta-blockers reduce the rate of certain nonfatal outcomes, they increase the risk of death and stroke.
STUDY SUMMARY: Multiple RCTs find preop beta-blockers do more harm, than good
Bouri et al1 conducted a meta-analysis of published RCTs evaluating preoperative beta-blockers vs placebo for patients undergoing noncardiac surgery. Of the 11 studies that met eligibility criteria, 2 were the discredited DECREASE trials. Thus, Bouri et al1 analyzed 9 high-quality RCTs that included 10,529 patients.
Most studies included patients undergoing vascular surgery. Some studies also included intra-abdominal, intrathoracic, neurosurgical, orthopedic, urologic, and gynecologic surgeries. Beta-blockers were started no more than a day before surgery and were discontinued at hospital discharge or up to 30 days postop. Metoprolol was used in 5 trials, bisoprolol in one trial, atenolol in 2 trials, and propranolol in one trial. The primary endpoint was all-cause mortality within 30 days.
A total of 5264 patients were randomized to beta-blockers and 5265 to placebo. There were 162 deaths in the beta-blocker group and 129 deaths in the placebo group. Patients who received beta-blockers had a 27% increased risk of all-cause mortality (risk ratio [RR]=1.27; 95% confidence interval [CI], 1.01-1.60; P=.04). The number needed to harm was 160.
Six of the studies also evaluated rates of nonfatal myocardial infarction (MI), nonfatal stroke, and hypotension. Beta-blockers lowered the risk of nonfatal MI (RR=.73; 95% CI, .61-.88; P=.001), but increased the risk of nonfatal stroke (RR=1.73; 95% CI, 1.00-2.99; P=.05) and hypotension (RR=1.51; 95% CI, 1.37-1.67; P=.00001).
This meta-analysis was dominated by the 2008 Peri-Operative ISchemic Evaluation (POISE) trial, an RCT that compared placebo to extended-release metoprolol, 100 mg 2 to 4 hours before surgery followed by 200 mg/d for 30 days, in 8351 patients with, or at risk for, atherosclerotic disease.4 While beta-blockers reduced the risk of MI and atrial fibrillation, they increased the risk of mortality and stroke, likely due to drug-induced hypotension. The slightly larger-than-typical doses of beta-blockers used in the POISE study may have contributed to the excess mortality.
WHAT'S NEW: Avoiding beta-blockers in surgery patients will prevent deaths
Bouri et al1 found that while beta-blockers protect against nonfatal MIs, they increase the risk for nonfatal strokes and death. This new meta-analysis challenges the ACCF/AHA recommendations by suggesting that abandoning the use of beta-blockers for preoperative patients who aren’t already taking them will prevent a substantial number of perioperative deaths. Bouri et al1 estimate that in the United Kingdom, where 47,286 deaths occur annually within 30 days of intermediate or high-risk procedures, the number of iatrogenic deaths would drop by approximately 10,000 if beta-blockers were not used.1
CAVEATS: Don't stop beta-blockers in patients who already take them
This meta-analysis did not evaluate outcomes in patients who were already taking beta-blockers. Patients who are already on beta-blockers should continue to take them in the perioperative period, which is in line with current ACCF/AHA guidelines.
CHALLENGES TO IMPLEMENTATION: Some physician may be reluctant to disregard published guidelines
Some physicians may not be comfortable ignoring the current ACCF/AHA guidelines that make a Class IIa recommendation (it is reasonable to administer this treatment) for the use of preoperative beta-blockade for patients at risk of cardiovascular events who were not previously taking a beta-blocker. This updated meta-analysis excludes the discredited DECREASE trials and challenges us to act against these current guidelines while we wait for updated recommendations.
Acknowledgement
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Do not routinely initiate beta-blockers in patients undergoing intermediate- or high-risk noncardiac surgery. Beta-blockers appear to increase the 30-day risk of all-cause mortality.1
Strength of recommendation
A: Based on meta-analysis of 9 randomized controlled trials (RCTs).
Bouri S, Shun-Shin MJ, Cole GD, et al. Meta-analysis of secure randomised controlled trials of ß-blockade to prevent perioperative death in non-cardiac surgery. Heart. 2014;100:456-464.
Illustrative case
A 67-year-old woman with diabetes, hypertension, and hyperlipidemia comes to your office for an evaluation before undergoing a total hip arthroplasty. She is not taking a beta-blocker. Should you prescribe one?
Current guidelines from the American College of Cardiology Foundation (ACCF) and the American Heart Association (AHA) recommend starting beta-blockers to prevent cardiac events in patients about to undergo intermediate- or high-risk surgery or vascular surgery who have a history of inducible ischemia, coronary artery disease (CAD), or at least one risk factor for CAD.2 However, the majority of the evidence for these guidelines, which were published in 2009 and are in the process of being updated, came from the DECREASE (Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography) trials, which have been discredited due to serious methodological flaws, including falsified descriptions of how outcomes were determined and fictitious databases.3 A new meta-analysis by Bouri et al1 that excluded the DECREASE trials found that although preoperative beta-blockers reduce the rate of certain nonfatal outcomes, they increase the risk of death and stroke.
STUDY SUMMARY: Multiple RCTs find preop beta-blockers do more harm, than good
Bouri et al1 conducted a meta-analysis of published RCTs evaluating preoperative beta-blockers vs placebo for patients undergoing noncardiac surgery. Of the 11 studies that met eligibility criteria, 2 were the discredited DECREASE trials. Thus, Bouri et al1 analyzed 9 high-quality RCTs that included 10,529 patients.
Most studies included patients undergoing vascular surgery. Some studies also included intra-abdominal, intrathoracic, neurosurgical, orthopedic, urologic, and gynecologic surgeries. Beta-blockers were started no more than a day before surgery and were discontinued at hospital discharge or up to 30 days postop. Metoprolol was used in 5 trials, bisoprolol in one trial, atenolol in 2 trials, and propranolol in one trial. The primary endpoint was all-cause mortality within 30 days.
A total of 5264 patients were randomized to beta-blockers and 5265 to placebo. There were 162 deaths in the beta-blocker group and 129 deaths in the placebo group. Patients who received beta-blockers had a 27% increased risk of all-cause mortality (risk ratio [RR]=1.27; 95% confidence interval [CI], 1.01-1.60; P=.04). The number needed to harm was 160.
Six of the studies also evaluated rates of nonfatal myocardial infarction (MI), nonfatal stroke, and hypotension. Beta-blockers lowered the risk of nonfatal MI (RR=.73; 95% CI, .61-.88; P=.001), but increased the risk of nonfatal stroke (RR=1.73; 95% CI, 1.00-2.99; P=.05) and hypotension (RR=1.51; 95% CI, 1.37-1.67; P=.00001).
This meta-analysis was dominated by the 2008 Peri-Operative ISchemic Evaluation (POISE) trial, an RCT that compared placebo to extended-release metoprolol, 100 mg 2 to 4 hours before surgery followed by 200 mg/d for 30 days, in 8351 patients with, or at risk for, atherosclerotic disease.4 While beta-blockers reduced the risk of MI and atrial fibrillation, they increased the risk of mortality and stroke, likely due to drug-induced hypotension. The slightly larger-than-typical doses of beta-blockers used in the POISE study may have contributed to the excess mortality.
WHAT'S NEW: Avoiding beta-blockers in surgery patients will prevent deaths
Bouri et al1 found that while beta-blockers protect against nonfatal MIs, they increase the risk for nonfatal strokes and death. This new meta-analysis challenges the ACCF/AHA recommendations by suggesting that abandoning the use of beta-blockers for preoperative patients who aren’t already taking them will prevent a substantial number of perioperative deaths. Bouri et al1 estimate that in the United Kingdom, where 47,286 deaths occur annually within 30 days of intermediate or high-risk procedures, the number of iatrogenic deaths would drop by approximately 10,000 if beta-blockers were not used.1
CAVEATS: Don't stop beta-blockers in patients who already take them
This meta-analysis did not evaluate outcomes in patients who were already taking beta-blockers. Patients who are already on beta-blockers should continue to take them in the perioperative period, which is in line with current ACCF/AHA guidelines.
CHALLENGES TO IMPLEMENTATION: Some physician may be reluctant to disregard published guidelines
Some physicians may not be comfortable ignoring the current ACCF/AHA guidelines that make a Class IIa recommendation (it is reasonable to administer this treatment) for the use of preoperative beta-blockade for patients at risk of cardiovascular events who were not previously taking a beta-blocker. This updated meta-analysis excludes the discredited DECREASE trials and challenges us to act against these current guidelines while we wait for updated recommendations.
Acknowledgement
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Bouri S, Shun-Shin MJ, Cole GD, et al. Meta-analysis of secure randomised controlled trials of ß-blockade to prevent perioperative death in non-cardiac surgery. Heart. 2014;100:456-464.
2. American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines; American Society of Echocardiography; American Society of Nuclear Cardiology; Heart Rhythm Society; Society of Cardiovascular Anesthesiologists; Society for Cardiovascular Angiography and Interventions; Society for Vascular Medicine; Society for Vascular Surgery; Fleisher LA, Beckman JA, Brown KA, et al. 2009 ACCF/AHA focused update on perioperative beta blockade incorporated into the ACC/AHA 2007 guidelines on perioperative cardiovascular evaluation and care for noncardiac surgery. J Am Coll Cardiol. 2009;54:e13-e118.
3. Eramus Medical Center Follow-up Investigation Committee. Report on the 2012 follow-up investigation of possible breaches of academic integrity. CardioBrief Web site. Available at: http://cardiobrief.files.wordpress.com/2012/10/integrity-report-2012-10-english-translation.pdf. Published September 30, 2012. Accessed March 31, 2014.
4. POISE Study Group; Devereaux PJ; Yang H; Yusuf S; et al. Effects of extended-release metoprolol succinate in patients undergoing non-cardiac surgery (POISE trial): a randomised controlled trial. Lancet. 2008;371:1839-1847.
1. Bouri S, Shun-Shin MJ, Cole GD, et al. Meta-analysis of secure randomised controlled trials of ß-blockade to prevent perioperative death in non-cardiac surgery. Heart. 2014;100:456-464.
2. American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines; American Society of Echocardiography; American Society of Nuclear Cardiology; Heart Rhythm Society; Society of Cardiovascular Anesthesiologists; Society for Cardiovascular Angiography and Interventions; Society for Vascular Medicine; Society for Vascular Surgery; Fleisher LA, Beckman JA, Brown KA, et al. 2009 ACCF/AHA focused update on perioperative beta blockade incorporated into the ACC/AHA 2007 guidelines on perioperative cardiovascular evaluation and care for noncardiac surgery. J Am Coll Cardiol. 2009;54:e13-e118.
3. Eramus Medical Center Follow-up Investigation Committee. Report on the 2012 follow-up investigation of possible breaches of academic integrity. CardioBrief Web site. Available at: http://cardiobrief.files.wordpress.com/2012/10/integrity-report-2012-10-english-translation.pdf. Published September 30, 2012. Accessed March 31, 2014.
4. POISE Study Group; Devereaux PJ; Yang H; Yusuf S; et al. Effects of extended-release metoprolol succinate in patients undergoing non-cardiac surgery (POISE trial): a randomised controlled trial. Lancet. 2008;371:1839-1847.
Copyright © 2014 Family Physicians Inquiries Network. All rights reserved.
Finally, a Way to Relieve Cancer-related Fatigue
PRACTICE CHANGER
Recommend American ginseng (1,000 mg bid) for four weeks to improve cancer-related fatigue in patients who are undergoing radiation or chemotherapy; no other treatment has been shown to be effective.1

STRENGTH OF RECOMMENDATION
B: Based on a single well-done randomized controlled trial (RCT).1
ILLUSTRATIVE CASE
A 54-year-old woman is receiving chemotherapy for adenocarcinoma of the right breast (T2N1M0) and has persistent, disabling fatigue. She has been unable to work or care for her family since starting chemotherapy. She says she gets enough sleep and denies being depressed or in pain. Lab testing for anemia and thyroid dysfunction is negative. Is there a safe and effective intervention?
On the next page: Study summary >>
Cancer-related fatigue is a common, distressing symptom that occurs in more than half of all patients undergoing chemotherapy and more than two-thirds of those receiving radiation therapy.2 For many cancer survivors, fatigue can persist for five to 10 years after treatment.3
Because no treatments have proven effective, many clinicians and patients accept fatigue as inevitable. In RCTs, psychostimulants (eg, methylphenidate) and antidepressants (eg, donepezil and paroxetine) have not been found effective.4-6 Dietary supplements, such as coenzyme Q10 and l-carnitine, also have not been found effective in placebo-controlled trials.7,8
The double-blind RCT reported on here looked at whether American ginseng might be effective in relieving cancer-related fatigue.
STUDY SUMMARY
Ginseng reduced fatigue after eight weeks
There are two major species of ginseng—Asian and American—and they have varying amounts, strengths, and varieties of ginsenosides, which are the active ingredients. In this eight-week, double-blind RCT, Barton et al1 randomly assigned more than 300 patients from 40 US cancer facilities to receive either 1,000 mg of American ginseng twice daily (in the morning and at noon) or matched placebo capsules.
Patients were either currently receiving treatment for cancer or were posttreatment but within two years of receiving a cancer diagnosis. All participants had experienced fatigue of at least a month’s duration that they rated as 4 or higher on a scale of 0 to 10. Patients with other causes of fatigue were excluded, as were those who had pain or insomnia rated 4 or higher, those with brain cancer or central nervous system (CNS) lymphoma, those taking systemic steroids or opioids, and those who were using, or had used, ginseng or other agents for fatigue.
Of the 364 randomized participants, 300 (147 ginseng patients, 153 placebo patients) remained in the study through the primary endpoint at four weeks, and 261 completed the entire eight-week study. There were no baseline differences between groups in demographic characteristics, time since cancer diagnosis, cancer type, current or prior treatment, and fatigue at baseline.
The primary outcome was a change in score on the Multidimensional Fatigue Symptom Inventory–Short Form (MFSI–SF) at four weeks. Secondary outcomes included a change in MFSI–SF score at eight weeks. The authors also conducted a subset analysis comparing ginseng and placebo in only those patients currently undergoing cancer treatment versus those who had completed treatment. To make it easier to compare results, all scores were converted to a 100-point scale; higher scores indicated less fatigue. Adverse events were documented by patient self-report questionnaires and also by researchers who called or visited patients every other week.
While ginseng did not appear to significantly impact fatigue scores versus placebo at four weeks (14.4 vs 8.2), fatigue scores at eight weeks were significantly improved (20 vs 10.3). Interestingly, though, there was a significant improvement in fatigue scores with ginseng at both four weeks and eight weeks when researchers looked at only those patients who were currently receiving cancer treatment. On the other hand, those patients who were not currently undergoing treatment did not show a significant improvement at either time cutoff.
There was no statistically significant difference in adverse events between the ginseng and placebo groups over the eight-week study.
On the next page: What's new >>
WHAT’S NEW
First evidence-based therapy
We now have good evidence that American ginseng (1,000 mg bid) is safe and effective for ameliorating cancer-related fatigue. Before this study, no other effective treatment had been identified.
CAVEATS
Ginseng may not help posttreatment
In this study, ginseng did not improve fatigue at four weeks, which was the primary outcome, although benefits were noted after eight weeks of treatment. Interestingly, though, participants who were receiving radiation and/or chemotherapy during the study experienced significant improvements at four and eight weeks, while those with previous (but not current) treatment did not significantly improve at either time point.
It may be that ginseng works best to ameliorate cancer-related fatigue in patients simultaneously receiving cancer treatment but not in those who have completed treatment. The findings also suggest that patients who have completed treatment may wish to try ginseng for longer than eight weeks to see if it offers any benefit.
Because this study excluded patients with brain cancer, CNS lymphoma, moderate to severe pain, or insomnia and those taking steroids, it is not known if ginseng would help them.
In one study, a low-dose methanolic extract of American ginseng caused a breast cancer cell line to proliferate; however, it was later discovered that this extract had been contaminated with Fusarium fungi containing zearalenone, which has strong estrogenic activity.9,10 However, higher doses of a similar methanolic extract, as well as other water-based extracts, have reduced proliferation of breast cancer cells.11
Proceed carefully if a patient is taking warfarin. Coadministration of ginseng and warfarin may reduce both warfarin concentrations and a patient’s international normalized ratio (INR).12 Therefore, carefully monitor INR in patients concurrently taking ginseng and warfarin. Furthermore, ginseng may lower blood glucose in patients with diabetes, so carefully monitor blood glucose in these patients when initiating or discontinuing ginseng.13
CHALLENGES TO IMPLEMENTATION
It’s hard to know exactly what you’re getting
Regulating dietary supplements, especially verifying ingredients and potency, has been a challenge for the FDA. Although ginseng commonly is adulterated, this is more common with the Asian species (Panax ginseng) than with the American species (Panax quinquefolius) used in this study.10 Clinicians who want to recommend ginseng for cancer-related fatigue should advise patients to use American ginseng root products produced in the US. Additionally, the products should contain at least 3% ginsenosides to match the dose used in this study.
REFERENCES
1. Barton DL, Liu H, Dakhil SR, et al. Wisconsin Ginseng (Panax quinquefolius) to improve cancer-related fatigue: a randomized, double-blind trial, N07C2. J Natl Cancer Inst. 2013;105(16):1230-1238.
2. Hofman M, Ryan JL, Figueroa-Moseley CD, et al. Cancer-related fatigue: the scale of the problem. Oncologist. 2007;12 (suppl 1):4-10.
3. Bower JE, Ganz PA, Desmond KA, et al. Fatigue in long-term breast carcinoma survivors: a longitudinal investigation. Cancer. 2006;106(4):751-758.
4. Moraska AR, Sood A, Dakhil SR, et al. Phase III, randomized, double-blind, placebo-controlled study of long-acting methylphenidate for cancer-related fatigue: North Central Cancer Treatment Group NCCTG-N05C7 trial. J Clin Oncol. 2010;28(23):3673-3679.
5. Bruera E, El Osta B, Valero V, et al. Donepezil for cancer fatigue: a double-blind, randomized, placebo-controlled trial. J Clin Oncol. 2007;25(23):3475-3481.
6. Morrow GR, Hickok JT, Roscoe JA, et al; University of Rochester Cancer Center Community Clinical Oncology Program. Differential effects of paroxetine on fatigue and depression: a randomized, double-blind trial from the University of Rochester Cancer Center Community Clinical Oncology Program. J Clin Oncol. 2003;21(24):4635-4641.
7. Lesser GJ, Case D, Stark N, et al; Wake Forest University Community Clinical Oncology Program Research Base. A randomized, double-blind, placebo-controlled study of oral coenzyme Q10 to relieve self-reported treatment-related fatigue in newly diagnosed patients with breast cancer. J Support Oncol. 2013;11(1):31-42.
8. Cruciani RA, Zhang JJ, Manola J, et al. L-carnitine supplementation for the management of fatigue in patients with cancer: an Eastern cooperative oncology group phase III, randomized, double-blind, placebo-controlled trial. J Clin Oncol. 2012;30(31):3864-3869.
9. Duda RB, Zhong Y, Navas V, et al. American ginseng and breast cancer therapeutic agents synergistically inhibit MCF-7
breast cancer cell growth. J Surg Oncol. 1999;72(4):230-239.
10. Upton R, ed. American ginseng root Panax quinquefolius, standards of analysis, quality control, and therapeutics. Scotts Valley, CA: American Herbal Pharmacopoeia; 2012.
11. King ML, Adler SR, Murphy LL. Extraction-dependent effects of American ginseng (Panax quinquefolium) on human breast cancer cell proliferation and estrogen receptor activation. Integr Cancer Ther. 2006;5(3): 236-243.
12. Yuan CS, Wei G, Dey L, et al. Brief communication: American ginseng reduces warfarin’s effect in healthy patients: a randomized, controlled trial. Ann Intern Med. 2004;141(1):23-27.
13. Vuksan V, Stavro MP, Sievenpiper JL, et al. Similar postprandial glycemic reductions with escalation of dose and administration time of American ginseng in type 2 diabetes. Diabetes Care. 2000;23(9):1221-1226.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center for Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
Copyright © 2014. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice. 2014;63(5):270-272.
PRACTICE CHANGER
Recommend American ginseng (1,000 mg bid) for four weeks to improve cancer-related fatigue in patients who are undergoing radiation or chemotherapy; no other treatment has been shown to be effective.1
STRENGTH OF RECOMMENDATION
B: Based on a single well-done randomized controlled trial (RCT).1
ILLUSTRATIVE CASE
A 54-year-old woman is receiving chemotherapy for adenocarcinoma of the right breast (T2N1M0) and has persistent, disabling fatigue. She has been unable to work or care for her family since starting chemotherapy. She says she gets enough sleep and denies being depressed or in pain. Lab testing for anemia and thyroid dysfunction is negative. Is there a safe and effective intervention?
On the next page: Study summary >>
Cancer-related fatigue is a common, distressing symptom that occurs in more than half of all patients undergoing chemotherapy and more than two-thirds of those receiving radiation therapy.2 For many cancer survivors, fatigue can persist for five to 10 years after treatment.3
Because no treatments have proven effective, many clinicians and patients accept fatigue as inevitable. In RCTs, psychostimulants (eg, methylphenidate) and antidepressants (eg, donepezil and paroxetine) have not been found effective.4-6 Dietary supplements, such as coenzyme Q10 and l-carnitine, also have not been found effective in placebo-controlled trials.7,8
The double-blind RCT reported on here looked at whether American ginseng might be effective in relieving cancer-related fatigue.
STUDY SUMMARY
Ginseng reduced fatigue after eight weeks
There are two major species of ginseng—Asian and American—and they have varying amounts, strengths, and varieties of ginsenosides, which are the active ingredients. In this eight-week, double-blind RCT, Barton et al1 randomly assigned more than 300 patients from 40 US cancer facilities to receive either 1,000 mg of American ginseng twice daily (in the morning and at noon) or matched placebo capsules.
Patients were either currently receiving treatment for cancer or were posttreatment but within two years of receiving a cancer diagnosis. All participants had experienced fatigue of at least a month’s duration that they rated as 4 or higher on a scale of 0 to 10. Patients with other causes of fatigue were excluded, as were those who had pain or insomnia rated 4 or higher, those with brain cancer or central nervous system (CNS) lymphoma, those taking systemic steroids or opioids, and those who were using, or had used, ginseng or other agents for fatigue.
Of the 364 randomized participants, 300 (147 ginseng patients, 153 placebo patients) remained in the study through the primary endpoint at four weeks, and 261 completed the entire eight-week study. There were no baseline differences between groups in demographic characteristics, time since cancer diagnosis, cancer type, current or prior treatment, and fatigue at baseline.
The primary outcome was a change in score on the Multidimensional Fatigue Symptom Inventory–Short Form (MFSI–SF) at four weeks. Secondary outcomes included a change in MFSI–SF score at eight weeks. The authors also conducted a subset analysis comparing ginseng and placebo in only those patients currently undergoing cancer treatment versus those who had completed treatment. To make it easier to compare results, all scores were converted to a 100-point scale; higher scores indicated less fatigue. Adverse events were documented by patient self-report questionnaires and also by researchers who called or visited patients every other week.
While ginseng did not appear to significantly impact fatigue scores versus placebo at four weeks (14.4 vs 8.2), fatigue scores at eight weeks were significantly improved (20 vs 10.3). Interestingly, though, there was a significant improvement in fatigue scores with ginseng at both four weeks and eight weeks when researchers looked at only those patients who were currently receiving cancer treatment. On the other hand, those patients who were not currently undergoing treatment did not show a significant improvement at either time cutoff.
There was no statistically significant difference in adverse events between the ginseng and placebo groups over the eight-week study.
On the next page: What's new >>
WHAT’S NEW
First evidence-based therapy
We now have good evidence that American ginseng (1,000 mg bid) is safe and effective for ameliorating cancer-related fatigue. Before this study, no other effective treatment had been identified.
CAVEATS
Ginseng may not help posttreatment
In this study, ginseng did not improve fatigue at four weeks, which was the primary outcome, although benefits were noted after eight weeks of treatment. Interestingly, though, participants who were receiving radiation and/or chemotherapy during the study experienced significant improvements at four and eight weeks, while those with previous (but not current) treatment did not significantly improve at either time point.
It may be that ginseng works best to ameliorate cancer-related fatigue in patients simultaneously receiving cancer treatment but not in those who have completed treatment. The findings also suggest that patients who have completed treatment may wish to try ginseng for longer than eight weeks to see if it offers any benefit.
Because this study excluded patients with brain cancer, CNS lymphoma, moderate to severe pain, or insomnia and those taking steroids, it is not known if ginseng would help them.
In one study, a low-dose methanolic extract of American ginseng caused a breast cancer cell line to proliferate; however, it was later discovered that this extract had been contaminated with Fusarium fungi containing zearalenone, which has strong estrogenic activity.9,10 However, higher doses of a similar methanolic extract, as well as other water-based extracts, have reduced proliferation of breast cancer cells.11
Proceed carefully if a patient is taking warfarin. Coadministration of ginseng and warfarin may reduce both warfarin concentrations and a patient’s international normalized ratio (INR).12 Therefore, carefully monitor INR in patients concurrently taking ginseng and warfarin. Furthermore, ginseng may lower blood glucose in patients with diabetes, so carefully monitor blood glucose in these patients when initiating or discontinuing ginseng.13
CHALLENGES TO IMPLEMENTATION
It’s hard to know exactly what you’re getting
Regulating dietary supplements, especially verifying ingredients and potency, has been a challenge for the FDA. Although ginseng commonly is adulterated, this is more common with the Asian species (Panax ginseng) than with the American species (Panax quinquefolius) used in this study.10 Clinicians who want to recommend ginseng for cancer-related fatigue should advise patients to use American ginseng root products produced in the US. Additionally, the products should contain at least 3% ginsenosides to match the dose used in this study.
REFERENCES
1. Barton DL, Liu H, Dakhil SR, et al. Wisconsin Ginseng (Panax quinquefolius) to improve cancer-related fatigue: a randomized, double-blind trial, N07C2. J Natl Cancer Inst. 2013;105(16):1230-1238.
2. Hofman M, Ryan JL, Figueroa-Moseley CD, et al. Cancer-related fatigue: the scale of the problem. Oncologist. 2007;12 (suppl 1):4-10.
3. Bower JE, Ganz PA, Desmond KA, et al. Fatigue in long-term breast carcinoma survivors: a longitudinal investigation. Cancer. 2006;106(4):751-758.
4. Moraska AR, Sood A, Dakhil SR, et al. Phase III, randomized, double-blind, placebo-controlled study of long-acting methylphenidate for cancer-related fatigue: North Central Cancer Treatment Group NCCTG-N05C7 trial. J Clin Oncol. 2010;28(23):3673-3679.
5. Bruera E, El Osta B, Valero V, et al. Donepezil for cancer fatigue: a double-blind, randomized, placebo-controlled trial. J Clin Oncol. 2007;25(23):3475-3481.
6. Morrow GR, Hickok JT, Roscoe JA, et al; University of Rochester Cancer Center Community Clinical Oncology Program. Differential effects of paroxetine on fatigue and depression: a randomized, double-blind trial from the University of Rochester Cancer Center Community Clinical Oncology Program. J Clin Oncol. 2003;21(24):4635-4641.
7. Lesser GJ, Case D, Stark N, et al; Wake Forest University Community Clinical Oncology Program Research Base. A randomized, double-blind, placebo-controlled study of oral coenzyme Q10 to relieve self-reported treatment-related fatigue in newly diagnosed patients with breast cancer. J Support Oncol. 2013;11(1):31-42.
8. Cruciani RA, Zhang JJ, Manola J, et al. L-carnitine supplementation for the management of fatigue in patients with cancer: an Eastern cooperative oncology group phase III, randomized, double-blind, placebo-controlled trial. J Clin Oncol. 2012;30(31):3864-3869.
9. Duda RB, Zhong Y, Navas V, et al. American ginseng and breast cancer therapeutic agents synergistically inhibit MCF-7
breast cancer cell growth. J Surg Oncol. 1999;72(4):230-239.
10. Upton R, ed. American ginseng root Panax quinquefolius, standards of analysis, quality control, and therapeutics. Scotts Valley, CA: American Herbal Pharmacopoeia; 2012.
11. King ML, Adler SR, Murphy LL. Extraction-dependent effects of American ginseng (Panax quinquefolium) on human breast cancer cell proliferation and estrogen receptor activation. Integr Cancer Ther. 2006;5(3): 236-243.
12. Yuan CS, Wei G, Dey L, et al. Brief communication: American ginseng reduces warfarin’s effect in healthy patients: a randomized, controlled trial. Ann Intern Med. 2004;141(1):23-27.
13. Vuksan V, Stavro MP, Sievenpiper JL, et al. Similar postprandial glycemic reductions with escalation of dose and administration time of American ginseng in type 2 diabetes. Diabetes Care. 2000;23(9):1221-1226.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center for Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
Copyright © 2014. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice. 2014;63(5):270-272.
PRACTICE CHANGER
Recommend American ginseng (1,000 mg bid) for four weeks to improve cancer-related fatigue in patients who are undergoing radiation or chemotherapy; no other treatment has been shown to be effective.1
STRENGTH OF RECOMMENDATION
B: Based on a single well-done randomized controlled trial (RCT).1
ILLUSTRATIVE CASE
A 54-year-old woman is receiving chemotherapy for adenocarcinoma of the right breast (T2N1M0) and has persistent, disabling fatigue. She has been unable to work or care for her family since starting chemotherapy. She says she gets enough sleep and denies being depressed or in pain. Lab testing for anemia and thyroid dysfunction is negative. Is there a safe and effective intervention?
On the next page: Study summary >>
Cancer-related fatigue is a common, distressing symptom that occurs in more than half of all patients undergoing chemotherapy and more than two-thirds of those receiving radiation therapy.2 For many cancer survivors, fatigue can persist for five to 10 years after treatment.3
Because no treatments have proven effective, many clinicians and patients accept fatigue as inevitable. In RCTs, psychostimulants (eg, methylphenidate) and antidepressants (eg, donepezil and paroxetine) have not been found effective.4-6 Dietary supplements, such as coenzyme Q10 and l-carnitine, also have not been found effective in placebo-controlled trials.7,8
The double-blind RCT reported on here looked at whether American ginseng might be effective in relieving cancer-related fatigue.
STUDY SUMMARY
Ginseng reduced fatigue after eight weeks
There are two major species of ginseng—Asian and American—and they have varying amounts, strengths, and varieties of ginsenosides, which are the active ingredients. In this eight-week, double-blind RCT, Barton et al1 randomly assigned more than 300 patients from 40 US cancer facilities to receive either 1,000 mg of American ginseng twice daily (in the morning and at noon) or matched placebo capsules.
Patients were either currently receiving treatment for cancer or were posttreatment but within two years of receiving a cancer diagnosis. All participants had experienced fatigue of at least a month’s duration that they rated as 4 or higher on a scale of 0 to 10. Patients with other causes of fatigue were excluded, as were those who had pain or insomnia rated 4 or higher, those with brain cancer or central nervous system (CNS) lymphoma, those taking systemic steroids or opioids, and those who were using, or had used, ginseng or other agents for fatigue.
Of the 364 randomized participants, 300 (147 ginseng patients, 153 placebo patients) remained in the study through the primary endpoint at four weeks, and 261 completed the entire eight-week study. There were no baseline differences between groups in demographic characteristics, time since cancer diagnosis, cancer type, current or prior treatment, and fatigue at baseline.
The primary outcome was a change in score on the Multidimensional Fatigue Symptom Inventory–Short Form (MFSI–SF) at four weeks. Secondary outcomes included a change in MFSI–SF score at eight weeks. The authors also conducted a subset analysis comparing ginseng and placebo in only those patients currently undergoing cancer treatment versus those who had completed treatment. To make it easier to compare results, all scores were converted to a 100-point scale; higher scores indicated less fatigue. Adverse events were documented by patient self-report questionnaires and also by researchers who called or visited patients every other week.
While ginseng did not appear to significantly impact fatigue scores versus placebo at four weeks (14.4 vs 8.2), fatigue scores at eight weeks were significantly improved (20 vs 10.3). Interestingly, though, there was a significant improvement in fatigue scores with ginseng at both four weeks and eight weeks when researchers looked at only those patients who were currently receiving cancer treatment. On the other hand, those patients who were not currently undergoing treatment did not show a significant improvement at either time cutoff.
There was no statistically significant difference in adverse events between the ginseng and placebo groups over the eight-week study.
On the next page: What's new >>
WHAT’S NEW
First evidence-based therapy
We now have good evidence that American ginseng (1,000 mg bid) is safe and effective for ameliorating cancer-related fatigue. Before this study, no other effective treatment had been identified.
CAVEATS
Ginseng may not help posttreatment
In this study, ginseng did not improve fatigue at four weeks, which was the primary outcome, although benefits were noted after eight weeks of treatment. Interestingly, though, participants who were receiving radiation and/or chemotherapy during the study experienced significant improvements at four and eight weeks, while those with previous (but not current) treatment did not significantly improve at either time point.
It may be that ginseng works best to ameliorate cancer-related fatigue in patients simultaneously receiving cancer treatment but not in those who have completed treatment. The findings also suggest that patients who have completed treatment may wish to try ginseng for longer than eight weeks to see if it offers any benefit.
Because this study excluded patients with brain cancer, CNS lymphoma, moderate to severe pain, or insomnia and those taking steroids, it is not known if ginseng would help them.
In one study, a low-dose methanolic extract of American ginseng caused a breast cancer cell line to proliferate; however, it was later discovered that this extract had been contaminated with Fusarium fungi containing zearalenone, which has strong estrogenic activity.9,10 However, higher doses of a similar methanolic extract, as well as other water-based extracts, have reduced proliferation of breast cancer cells.11
Proceed carefully if a patient is taking warfarin. Coadministration of ginseng and warfarin may reduce both warfarin concentrations and a patient’s international normalized ratio (INR).12 Therefore, carefully monitor INR in patients concurrently taking ginseng and warfarin. Furthermore, ginseng may lower blood glucose in patients with diabetes, so carefully monitor blood glucose in these patients when initiating or discontinuing ginseng.13
CHALLENGES TO IMPLEMENTATION
It’s hard to know exactly what you’re getting
Regulating dietary supplements, especially verifying ingredients and potency, has been a challenge for the FDA. Although ginseng commonly is adulterated, this is more common with the Asian species (Panax ginseng) than with the American species (Panax quinquefolius) used in this study.10 Clinicians who want to recommend ginseng for cancer-related fatigue should advise patients to use American ginseng root products produced in the US. Additionally, the products should contain at least 3% ginsenosides to match the dose used in this study.
REFERENCES
1. Barton DL, Liu H, Dakhil SR, et al. Wisconsin Ginseng (Panax quinquefolius) to improve cancer-related fatigue: a randomized, double-blind trial, N07C2. J Natl Cancer Inst. 2013;105(16):1230-1238.
2. Hofman M, Ryan JL, Figueroa-Moseley CD, et al. Cancer-related fatigue: the scale of the problem. Oncologist. 2007;12 (suppl 1):4-10.
3. Bower JE, Ganz PA, Desmond KA, et al. Fatigue in long-term breast carcinoma survivors: a longitudinal investigation. Cancer. 2006;106(4):751-758.
4. Moraska AR, Sood A, Dakhil SR, et al. Phase III, randomized, double-blind, placebo-controlled study of long-acting methylphenidate for cancer-related fatigue: North Central Cancer Treatment Group NCCTG-N05C7 trial. J Clin Oncol. 2010;28(23):3673-3679.
5. Bruera E, El Osta B, Valero V, et al. Donepezil for cancer fatigue: a double-blind, randomized, placebo-controlled trial. J Clin Oncol. 2007;25(23):3475-3481.
6. Morrow GR, Hickok JT, Roscoe JA, et al; University of Rochester Cancer Center Community Clinical Oncology Program. Differential effects of paroxetine on fatigue and depression: a randomized, double-blind trial from the University of Rochester Cancer Center Community Clinical Oncology Program. J Clin Oncol. 2003;21(24):4635-4641.
7. Lesser GJ, Case D, Stark N, et al; Wake Forest University Community Clinical Oncology Program Research Base. A randomized, double-blind, placebo-controlled study of oral coenzyme Q10 to relieve self-reported treatment-related fatigue in newly diagnosed patients with breast cancer. J Support Oncol. 2013;11(1):31-42.
8. Cruciani RA, Zhang JJ, Manola J, et al. L-carnitine supplementation for the management of fatigue in patients with cancer: an Eastern cooperative oncology group phase III, randomized, double-blind, placebo-controlled trial. J Clin Oncol. 2012;30(31):3864-3869.
9. Duda RB, Zhong Y, Navas V, et al. American ginseng and breast cancer therapeutic agents synergistically inhibit MCF-7
breast cancer cell growth. J Surg Oncol. 1999;72(4):230-239.
10. Upton R, ed. American ginseng root Panax quinquefolius, standards of analysis, quality control, and therapeutics. Scotts Valley, CA: American Herbal Pharmacopoeia; 2012.
11. King ML, Adler SR, Murphy LL. Extraction-dependent effects of American ginseng (Panax quinquefolium) on human breast cancer cell proliferation and estrogen receptor activation. Integr Cancer Ther. 2006;5(3): 236-243.
12. Yuan CS, Wei G, Dey L, et al. Brief communication: American ginseng reduces warfarin’s effect in healthy patients: a randomized, controlled trial. Ann Intern Med. 2004;141(1):23-27.
13. Vuksan V, Stavro MP, Sievenpiper JL, et al. Similar postprandial glycemic reductions with escalation of dose and administration time of American ginseng in type 2 diabetes. Diabetes Care. 2000;23(9):1221-1226.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center for Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
Copyright © 2014. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice. 2014;63(5):270-272.
Finally, a way to relieve cancer-related fatigue
Recommend American ginseng 1000 mg twice daily for 4 weeks to improve cancer-related fatigue for patients who are undergoing radiation or chemotherapy; no other treatment has been shown to be effective.1
Strength of recommendation
B: Based on a single well-done randomized controlled trial (RCT).
Barton DL, Liu H, Dakhil SR, et al. Wisconsin Ginseng (Panax quinquefolius) to improve cancer-related fatigue: a randomized, double-blind trial, N07C2. J Natl Cancer Inst. 2013;105:1230-1238.
Illustrative case
A 54-year-old woman is receiving chemotherapy for adenocarcinoma of the right breast (T2N1M0) and has persistent, disabling fatigue. She has been unable to work or care for her family since starting chemotherapy. She says she gets enough sleep and denies being depressed or in pain. Lab testing for anemia and thyroid dysfunction is negative.
Is there a safe and effective intervention that can reduce her fatigue?
Cancer-related fatigue is a common, distressing symptom that occurs in more than half of all patients undergoing chemotherapy and over two-thirds of those receiving radiation therapy.2 For many cancer survivors, fatigue can persist for 5 to 10 years after treatment.3 Because no treatments have been effective, many clinicians and patients accept it as inevitable. In RCTs, psychostimulants such as methylphenidate and antidepressants such as donepezil and paroxetine have not been found effective.4-6 Dietary supplements such as coenzyme Q10 and L-Carnitine also have not been found effective in placebo-controlled trials.7,8 The double-blind RCT reported on here looked at whether American ginseng might be effective in relieving cancer-related fatigue.
STUDY SUMMARY: Ginseng reduced fatigue after 8 weeks of treatment
There are 2 major species of ginseng—Asian and American—and they have varying amounts, strengths, and varieties of ginsenosides, which are the active ingredients. In this 8-week, double-blind RCT, Barton et al1 randomized more than 300 patients from 40 US cancer facilities to receive either 1000 mg of American ginseng twice daily (in the morning and at noon) or matched placebo capsules. Patients were either currently receiving treatment for cancer or were posttreatment, but within 2 years of receiving a cancer diagnosis. All participants had experienced fatigue for at least a month that they rated as ≥4 or higher on a scale of 0 to 10. Patients with other causes of fatigue were excluded, as were those who had pain or insomnia rated ≥4 on a scale of 0 to 10, those with brain cancer or central nervous system (CNS) lymphoma, those taking systemic steroids or opioids, and those who were using, or had used, ginseng or other agents for fatigue.
Of the 364 randomized participants, 300 (147 ginseng patients, 153 placebo patients) remained in the study through the primary endpoint at 4 weeks, and 261 completed the entire 8-week study. There were no baseline differences between groups in demographic characteristics, time since cancer diagnosis, cancer type, current or prior treatment, and fatigue at baseline.
The primary outcome was a change in score on the Multidimensional Fatigue Symptom Inventory–Short Form (MFSI-SF) at 4 weeks. Secondary outcomes included a change in MFSI-SF score at 8 weeks. The authors also conducted a subset analysis comparing ginseng vs placebo in just those patients currently undergoing cancer treatment vs those who had completed treatment. To make it easier to compare results, all scores were converted to a 100-point scale; higher scores indicated less fatigue. Adverse events were documented by patient self-report questionnaires and also by researchers who called or visited patients every other week.
While ginseng did not appear to significantly increase the change in fatigue scores over placebo at 4 weeks (14.4 vs 8.2; P=.07), fatigue scores at 8 weeks were significantly improved (20 vs 10.3; P=.003). Interestingly, though, there was a significant improvement in fatigue scores with ginseng at both 4 weeks (P=.02) and 8 weeks (P=.01) when researchers looked at only those patients who were currently receiving cancer treatment. On the other hand, those patients who were not currently undergoing treatment did not show a significant improvement at either time cutoff.
There was no statistically significant difference in adverse events between the ginseng and placebo groups over the 8-week study.
WHAT'S NEW: The first evidence-based therapy for cancer-related fatigue
We now have good evidence that American ginseng 1000 mg twice daily is safe and effective for ameliorating cancer-related fatigue. Before this study, no other effective treatments had been identified.
CAVEATS: Ginseng may not help patients who've finished cancer treatment
In this study, ginseng did not improve fatigue at 4 weeks, which was the primary outcome, although benefits were noted after 8 weeks of treatment. Interestingly, though, participants who were receiving radiation and/or chemotherapy during the study experienced significant improvements at 4 and 8 weeks, while those with previous (but not current) treatment did not significantly improve at either time point.
It may be that ginseng works best to ameliorate cancer-related fatigue in patients simultaneously receiving cancer treatment, but not in those who have completed treatment. The findings also suggest that patients who have completed treatment may wish to try ginseng for longer than 8 weeks to see if it offers any benefit.
Because this study excluded patients with brain cancer, CNS lymphoma, moderate to severe pain, or insomnia and those taking steroids, it is not known if ginseng would help them.
In one study, a low-dose methanolic extract of American ginseng caused a breast cancer cell line to proliferate; however, it was later discovered that this extract had been contaminated with Fusarium fungi containing zearalenone, which has strong estrogenic activity.9,10 However, higher doses of a similar methanolic extract, as well as other water-based extracts, have reduced proliferation of breast cancer cells.11
Proceed carefully if a patient is taking warfarin. Coadministration of ginseng and warfarin may reduce both warfarin concentrations and a patient’s international normalized ratio (INR).12 Therefore, carefully monitor INR in patients concurrently taking ginseng and warfarin. Furthermore, ginseng may lower blood glucose in patients with diabetes, so carefully monitor blood glucose in these patients when initiating or discontinuing ginseng.13
CHALLENGES TO IMPLEMENTATION: With ginseng, it's hard to know exactly what you're getting
Regulating dietary supplements has been a challenge for the US Food and Drug Administration, especially verifying ingredients and potency. Although ginseng commonly is adulterated, much of the adulteration occurs with the Asian species (Panax ginseng) rather than the American species (Panax quinquefolius) used in this study.10 Physicians who want to recommend ginseng for cancer-related fatigue should advise patients to use American ginseng root products produced in the United States. Additionally, ginseng products should contain at least 3% ginsenosides to match the dose used in this study.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Barton DL, Liu H, Dakhil SR, et al. Wisconsin Ginseng (Panax quinquefolius) to improve cancer-related fatigue: a randomized, double-blind trial, N07C2. J Natl Cancer Inst. 2013;105:1230-1238.
2. Hofman M, Ryan JL, Figueroa-Moseley CD, et al. Cancer-related fatigue: the scale of the problem. Oncologist. 2007;12 suppl 1:4-10.
3. Bower JE, Ganz PA, Desmond KA, et al. Fatigue in long-term breast carcinoma survivors: a longitudinal investigation. Cancer. 2006;106:751-758.
4. Moraska AR, Sood A, Dakhil SR, et al. Phase III, randomized, double-blind, placebo-controlled study of long-acting methylphenidate for cancer-related fatigue: North Central Cancer Treatment Group NCCTG-N05C7 trial. J Clin Oncol. 2010;28:3673-3679.
5. Bruera E, El Osta B, Valero V, et al. Donepezil for cancer fatigue: a double-blind, randomized, placebo-controlled trial. J Clin Oncol. 2007;25:3475-3481.
6. Morrow GR, Hickok JT, Roscoe JA, et al; University of Rochester Cancer Center Community Clinical Oncology Program. Differential effects of paroxetine on fatigue and depression: a randomized, double-blind trial from the University of Rochester Cancer Center Community Clinical Oncology Program. J Clin Oncol. 2003;21:4635-4641.
7. Lesser GJ, Case D, Stark N, et al; Wake Forest University Community Clinical Oncology Program Research Base. A randomized, double-blind, placebo-controlled study of oral coenzyme Q10 to relieve self-reported treatment-related fatigue in newly diagnosed patients with breast cancer. J Support Oncol. 2013;11:31-42.
8. Cruciani RA, Zhang JJ, Manola J, et al. L-carnitine supplementation for the management of fatigue in patients with cancer: an Eastern cooperative oncology group phase III, randomized, double-blind, placebo-controlled trial. J Clin Oncol. 2012;30:3864-3869.
9. Duda RB, Zhong Y, Navas V, et al. American ginseng and breast cancer therapeutic agents synergistically inhibit MCF-7 breast cancer cell growth. J Surg Oncol. 1999;72:230-239.
10. Upton, R (ed). American ginseng root panax quinquefolius, standards of analysis, quality control, and therapeutics. Scotts Valley, CA: American Herbal Pharmacopoeia; 2012.
11. King ML, Adler SR, Murphy LL. Extraction-dependent effects of American ginseng (Panax quinquefolium) on human breast cancer cell proliferation and estrogen receptor activation. Integr Cancer Ther. 2006;5:236-243.
12. Yuan CS, Wei G, Dey L, et al. Brief communication: American ginseng reduces warfarin’s effect in healthy patients: a randomized, controlled trial. Ann Intern Med. 2004;141:23-27.
13. Vuksan V, Stavro MP, Sievenpiper JL, et al. Similar postprandial glycemic reductions with escalation of dose and administration time of American ginseng in type 2 diabetes. Diabetes Care. 2000;23:1221-1226.
Recommend American ginseng 1000 mg twice daily for 4 weeks to improve cancer-related fatigue for patients who are undergoing radiation or chemotherapy; no other treatment has been shown to be effective.1
Strength of recommendation
B: Based on a single well-done randomized controlled trial (RCT).
Barton DL, Liu H, Dakhil SR, et al. Wisconsin Ginseng (Panax quinquefolius) to improve cancer-related fatigue: a randomized, double-blind trial, N07C2. J Natl Cancer Inst. 2013;105:1230-1238.
Illustrative case
A 54-year-old woman is receiving chemotherapy for adenocarcinoma of the right breast (T2N1M0) and has persistent, disabling fatigue. She has been unable to work or care for her family since starting chemotherapy. She says she gets enough sleep and denies being depressed or in pain. Lab testing for anemia and thyroid dysfunction is negative.
Is there a safe and effective intervention that can reduce her fatigue?
Cancer-related fatigue is a common, distressing symptom that occurs in more than half of all patients undergoing chemotherapy and over two-thirds of those receiving radiation therapy.2 For many cancer survivors, fatigue can persist for 5 to 10 years after treatment.3 Because no treatments have been effective, many clinicians and patients accept it as inevitable. In RCTs, psychostimulants such as methylphenidate and antidepressants such as donepezil and paroxetine have not been found effective.4-6 Dietary supplements such as coenzyme Q10 and L-Carnitine also have not been found effective in placebo-controlled trials.7,8 The double-blind RCT reported on here looked at whether American ginseng might be effective in relieving cancer-related fatigue.
STUDY SUMMARY: Ginseng reduced fatigue after 8 weeks of treatment
There are 2 major species of ginseng—Asian and American—and they have varying amounts, strengths, and varieties of ginsenosides, which are the active ingredients. In this 8-week, double-blind RCT, Barton et al1 randomized more than 300 patients from 40 US cancer facilities to receive either 1000 mg of American ginseng twice daily (in the morning and at noon) or matched placebo capsules. Patients were either currently receiving treatment for cancer or were posttreatment, but within 2 years of receiving a cancer diagnosis. All participants had experienced fatigue for at least a month that they rated as ≥4 or higher on a scale of 0 to 10. Patients with other causes of fatigue were excluded, as were those who had pain or insomnia rated ≥4 on a scale of 0 to 10, those with brain cancer or central nervous system (CNS) lymphoma, those taking systemic steroids or opioids, and those who were using, or had used, ginseng or other agents for fatigue.
Of the 364 randomized participants, 300 (147 ginseng patients, 153 placebo patients) remained in the study through the primary endpoint at 4 weeks, and 261 completed the entire 8-week study. There were no baseline differences between groups in demographic characteristics, time since cancer diagnosis, cancer type, current or prior treatment, and fatigue at baseline.
The primary outcome was a change in score on the Multidimensional Fatigue Symptom Inventory–Short Form (MFSI-SF) at 4 weeks. Secondary outcomes included a change in MFSI-SF score at 8 weeks. The authors also conducted a subset analysis comparing ginseng vs placebo in just those patients currently undergoing cancer treatment vs those who had completed treatment. To make it easier to compare results, all scores were converted to a 100-point scale; higher scores indicated less fatigue. Adverse events were documented by patient self-report questionnaires and also by researchers who called or visited patients every other week.
While ginseng did not appear to significantly increase the change in fatigue scores over placebo at 4 weeks (14.4 vs 8.2; P=.07), fatigue scores at 8 weeks were significantly improved (20 vs 10.3; P=.003). Interestingly, though, there was a significant improvement in fatigue scores with ginseng at both 4 weeks (P=.02) and 8 weeks (P=.01) when researchers looked at only those patients who were currently receiving cancer treatment. On the other hand, those patients who were not currently undergoing treatment did not show a significant improvement at either time cutoff.
There was no statistically significant difference in adverse events between the ginseng and placebo groups over the 8-week study.
WHAT'S NEW: The first evidence-based therapy for cancer-related fatigue
We now have good evidence that American ginseng 1000 mg twice daily is safe and effective for ameliorating cancer-related fatigue. Before this study, no other effective treatments had been identified.
CAVEATS: Ginseng may not help patients who've finished cancer treatment
In this study, ginseng did not improve fatigue at 4 weeks, which was the primary outcome, although benefits were noted after 8 weeks of treatment. Interestingly, though, participants who were receiving radiation and/or chemotherapy during the study experienced significant improvements at 4 and 8 weeks, while those with previous (but not current) treatment did not significantly improve at either time point.
It may be that ginseng works best to ameliorate cancer-related fatigue in patients simultaneously receiving cancer treatment, but not in those who have completed treatment. The findings also suggest that patients who have completed treatment may wish to try ginseng for longer than 8 weeks to see if it offers any benefit.
Because this study excluded patients with brain cancer, CNS lymphoma, moderate to severe pain, or insomnia and those taking steroids, it is not known if ginseng would help them.
In one study, a low-dose methanolic extract of American ginseng caused a breast cancer cell line to proliferate; however, it was later discovered that this extract had been contaminated with Fusarium fungi containing zearalenone, which has strong estrogenic activity.9,10 However, higher doses of a similar methanolic extract, as well as other water-based extracts, have reduced proliferation of breast cancer cells.11
Proceed carefully if a patient is taking warfarin. Coadministration of ginseng and warfarin may reduce both warfarin concentrations and a patient’s international normalized ratio (INR).12 Therefore, carefully monitor INR in patients concurrently taking ginseng and warfarin. Furthermore, ginseng may lower blood glucose in patients with diabetes, so carefully monitor blood glucose in these patients when initiating or discontinuing ginseng.13
CHALLENGES TO IMPLEMENTATION: With ginseng, it's hard to know exactly what you're getting
Regulating dietary supplements has been a challenge for the US Food and Drug Administration, especially verifying ingredients and potency. Although ginseng commonly is adulterated, much of the adulteration occurs with the Asian species (Panax ginseng) rather than the American species (Panax quinquefolius) used in this study.10 Physicians who want to recommend ginseng for cancer-related fatigue should advise patients to use American ginseng root products produced in the United States. Additionally, ginseng products should contain at least 3% ginsenosides to match the dose used in this study.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Recommend American ginseng 1000 mg twice daily for 4 weeks to improve cancer-related fatigue for patients who are undergoing radiation or chemotherapy; no other treatment has been shown to be effective.1
Strength of recommendation
B: Based on a single well-done randomized controlled trial (RCT).
Barton DL, Liu H, Dakhil SR, et al. Wisconsin Ginseng (Panax quinquefolius) to improve cancer-related fatigue: a randomized, double-blind trial, N07C2. J Natl Cancer Inst. 2013;105:1230-1238.
Illustrative case
A 54-year-old woman is receiving chemotherapy for adenocarcinoma of the right breast (T2N1M0) and has persistent, disabling fatigue. She has been unable to work or care for her family since starting chemotherapy. She says she gets enough sleep and denies being depressed or in pain. Lab testing for anemia and thyroid dysfunction is negative.
Is there a safe and effective intervention that can reduce her fatigue?
Cancer-related fatigue is a common, distressing symptom that occurs in more than half of all patients undergoing chemotherapy and over two-thirds of those receiving radiation therapy.2 For many cancer survivors, fatigue can persist for 5 to 10 years after treatment.3 Because no treatments have been effective, many clinicians and patients accept it as inevitable. In RCTs, psychostimulants such as methylphenidate and antidepressants such as donepezil and paroxetine have not been found effective.4-6 Dietary supplements such as coenzyme Q10 and L-Carnitine also have not been found effective in placebo-controlled trials.7,8 The double-blind RCT reported on here looked at whether American ginseng might be effective in relieving cancer-related fatigue.
STUDY SUMMARY: Ginseng reduced fatigue after 8 weeks of treatment
There are 2 major species of ginseng—Asian and American—and they have varying amounts, strengths, and varieties of ginsenosides, which are the active ingredients. In this 8-week, double-blind RCT, Barton et al1 randomized more than 300 patients from 40 US cancer facilities to receive either 1000 mg of American ginseng twice daily (in the morning and at noon) or matched placebo capsules. Patients were either currently receiving treatment for cancer or were posttreatment, but within 2 years of receiving a cancer diagnosis. All participants had experienced fatigue for at least a month that they rated as ≥4 or higher on a scale of 0 to 10. Patients with other causes of fatigue were excluded, as were those who had pain or insomnia rated ≥4 on a scale of 0 to 10, those with brain cancer or central nervous system (CNS) lymphoma, those taking systemic steroids or opioids, and those who were using, or had used, ginseng or other agents for fatigue.
Of the 364 randomized participants, 300 (147 ginseng patients, 153 placebo patients) remained in the study through the primary endpoint at 4 weeks, and 261 completed the entire 8-week study. There were no baseline differences between groups in demographic characteristics, time since cancer diagnosis, cancer type, current or prior treatment, and fatigue at baseline.
The primary outcome was a change in score on the Multidimensional Fatigue Symptom Inventory–Short Form (MFSI-SF) at 4 weeks. Secondary outcomes included a change in MFSI-SF score at 8 weeks. The authors also conducted a subset analysis comparing ginseng vs placebo in just those patients currently undergoing cancer treatment vs those who had completed treatment. To make it easier to compare results, all scores were converted to a 100-point scale; higher scores indicated less fatigue. Adverse events were documented by patient self-report questionnaires and also by researchers who called or visited patients every other week.
While ginseng did not appear to significantly increase the change in fatigue scores over placebo at 4 weeks (14.4 vs 8.2; P=.07), fatigue scores at 8 weeks were significantly improved (20 vs 10.3; P=.003). Interestingly, though, there was a significant improvement in fatigue scores with ginseng at both 4 weeks (P=.02) and 8 weeks (P=.01) when researchers looked at only those patients who were currently receiving cancer treatment. On the other hand, those patients who were not currently undergoing treatment did not show a significant improvement at either time cutoff.
There was no statistically significant difference in adverse events between the ginseng and placebo groups over the 8-week study.
WHAT'S NEW: The first evidence-based therapy for cancer-related fatigue
We now have good evidence that American ginseng 1000 mg twice daily is safe and effective for ameliorating cancer-related fatigue. Before this study, no other effective treatments had been identified.
CAVEATS: Ginseng may not help patients who've finished cancer treatment
In this study, ginseng did not improve fatigue at 4 weeks, which was the primary outcome, although benefits were noted after 8 weeks of treatment. Interestingly, though, participants who were receiving radiation and/or chemotherapy during the study experienced significant improvements at 4 and 8 weeks, while those with previous (but not current) treatment did not significantly improve at either time point.
It may be that ginseng works best to ameliorate cancer-related fatigue in patients simultaneously receiving cancer treatment, but not in those who have completed treatment. The findings also suggest that patients who have completed treatment may wish to try ginseng for longer than 8 weeks to see if it offers any benefit.
Because this study excluded patients with brain cancer, CNS lymphoma, moderate to severe pain, or insomnia and those taking steroids, it is not known if ginseng would help them.
In one study, a low-dose methanolic extract of American ginseng caused a breast cancer cell line to proliferate; however, it was later discovered that this extract had been contaminated with Fusarium fungi containing zearalenone, which has strong estrogenic activity.9,10 However, higher doses of a similar methanolic extract, as well as other water-based extracts, have reduced proliferation of breast cancer cells.11
Proceed carefully if a patient is taking warfarin. Coadministration of ginseng and warfarin may reduce both warfarin concentrations and a patient’s international normalized ratio (INR).12 Therefore, carefully monitor INR in patients concurrently taking ginseng and warfarin. Furthermore, ginseng may lower blood glucose in patients with diabetes, so carefully monitor blood glucose in these patients when initiating or discontinuing ginseng.13
CHALLENGES TO IMPLEMENTATION: With ginseng, it's hard to know exactly what you're getting
Regulating dietary supplements has been a challenge for the US Food and Drug Administration, especially verifying ingredients and potency. Although ginseng commonly is adulterated, much of the adulteration occurs with the Asian species (Panax ginseng) rather than the American species (Panax quinquefolius) used in this study.10 Physicians who want to recommend ginseng for cancer-related fatigue should advise patients to use American ginseng root products produced in the United States. Additionally, ginseng products should contain at least 3% ginsenosides to match the dose used in this study.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Barton DL, Liu H, Dakhil SR, et al. Wisconsin Ginseng (Panax quinquefolius) to improve cancer-related fatigue: a randomized, double-blind trial, N07C2. J Natl Cancer Inst. 2013;105:1230-1238.
2. Hofman M, Ryan JL, Figueroa-Moseley CD, et al. Cancer-related fatigue: the scale of the problem. Oncologist. 2007;12 suppl 1:4-10.
3. Bower JE, Ganz PA, Desmond KA, et al. Fatigue in long-term breast carcinoma survivors: a longitudinal investigation. Cancer. 2006;106:751-758.
4. Moraska AR, Sood A, Dakhil SR, et al. Phase III, randomized, double-blind, placebo-controlled study of long-acting methylphenidate for cancer-related fatigue: North Central Cancer Treatment Group NCCTG-N05C7 trial. J Clin Oncol. 2010;28:3673-3679.
5. Bruera E, El Osta B, Valero V, et al. Donepezil for cancer fatigue: a double-blind, randomized, placebo-controlled trial. J Clin Oncol. 2007;25:3475-3481.
6. Morrow GR, Hickok JT, Roscoe JA, et al; University of Rochester Cancer Center Community Clinical Oncology Program. Differential effects of paroxetine on fatigue and depression: a randomized, double-blind trial from the University of Rochester Cancer Center Community Clinical Oncology Program. J Clin Oncol. 2003;21:4635-4641.
7. Lesser GJ, Case D, Stark N, et al; Wake Forest University Community Clinical Oncology Program Research Base. A randomized, double-blind, placebo-controlled study of oral coenzyme Q10 to relieve self-reported treatment-related fatigue in newly diagnosed patients with breast cancer. J Support Oncol. 2013;11:31-42.
8. Cruciani RA, Zhang JJ, Manola J, et al. L-carnitine supplementation for the management of fatigue in patients with cancer: an Eastern cooperative oncology group phase III, randomized, double-blind, placebo-controlled trial. J Clin Oncol. 2012;30:3864-3869.
9. Duda RB, Zhong Y, Navas V, et al. American ginseng and breast cancer therapeutic agents synergistically inhibit MCF-7 breast cancer cell growth. J Surg Oncol. 1999;72:230-239.
10. Upton, R (ed). American ginseng root panax quinquefolius, standards of analysis, quality control, and therapeutics. Scotts Valley, CA: American Herbal Pharmacopoeia; 2012.
11. King ML, Adler SR, Murphy LL. Extraction-dependent effects of American ginseng (Panax quinquefolium) on human breast cancer cell proliferation and estrogen receptor activation. Integr Cancer Ther. 2006;5:236-243.
12. Yuan CS, Wei G, Dey L, et al. Brief communication: American ginseng reduces warfarin’s effect in healthy patients: a randomized, controlled trial. Ann Intern Med. 2004;141:23-27.
13. Vuksan V, Stavro MP, Sievenpiper JL, et al. Similar postprandial glycemic reductions with escalation of dose and administration time of American ginseng in type 2 diabetes. Diabetes Care. 2000;23:1221-1226.
1. Barton DL, Liu H, Dakhil SR, et al. Wisconsin Ginseng (Panax quinquefolius) to improve cancer-related fatigue: a randomized, double-blind trial, N07C2. J Natl Cancer Inst. 2013;105:1230-1238.
2. Hofman M, Ryan JL, Figueroa-Moseley CD, et al. Cancer-related fatigue: the scale of the problem. Oncologist. 2007;12 suppl 1:4-10.
3. Bower JE, Ganz PA, Desmond KA, et al. Fatigue in long-term breast carcinoma survivors: a longitudinal investigation. Cancer. 2006;106:751-758.
4. Moraska AR, Sood A, Dakhil SR, et al. Phase III, randomized, double-blind, placebo-controlled study of long-acting methylphenidate for cancer-related fatigue: North Central Cancer Treatment Group NCCTG-N05C7 trial. J Clin Oncol. 2010;28:3673-3679.
5. Bruera E, El Osta B, Valero V, et al. Donepezil for cancer fatigue: a double-blind, randomized, placebo-controlled trial. J Clin Oncol. 2007;25:3475-3481.
6. Morrow GR, Hickok JT, Roscoe JA, et al; University of Rochester Cancer Center Community Clinical Oncology Program. Differential effects of paroxetine on fatigue and depression: a randomized, double-blind trial from the University of Rochester Cancer Center Community Clinical Oncology Program. J Clin Oncol. 2003;21:4635-4641.
7. Lesser GJ, Case D, Stark N, et al; Wake Forest University Community Clinical Oncology Program Research Base. A randomized, double-blind, placebo-controlled study of oral coenzyme Q10 to relieve self-reported treatment-related fatigue in newly diagnosed patients with breast cancer. J Support Oncol. 2013;11:31-42.
8. Cruciani RA, Zhang JJ, Manola J, et al. L-carnitine supplementation for the management of fatigue in patients with cancer: an Eastern cooperative oncology group phase III, randomized, double-blind, placebo-controlled trial. J Clin Oncol. 2012;30:3864-3869.
9. Duda RB, Zhong Y, Navas V, et al. American ginseng and breast cancer therapeutic agents synergistically inhibit MCF-7 breast cancer cell growth. J Surg Oncol. 1999;72:230-239.
10. Upton, R (ed). American ginseng root panax quinquefolius, standards of analysis, quality control, and therapeutics. Scotts Valley, CA: American Herbal Pharmacopoeia; 2012.
11. King ML, Adler SR, Murphy LL. Extraction-dependent effects of American ginseng (Panax quinquefolium) on human breast cancer cell proliferation and estrogen receptor activation. Integr Cancer Ther. 2006;5:236-243.
12. Yuan CS, Wei G, Dey L, et al. Brief communication: American ginseng reduces warfarin’s effect in healthy patients: a randomized, controlled trial. Ann Intern Med. 2004;141:23-27.
13. Vuksan V, Stavro MP, Sievenpiper JL, et al. Similar postprandial glycemic reductions with escalation of dose and administration time of American ginseng in type 2 diabetes. Diabetes Care. 2000;23:1221-1226.
Copyright © 2014 Family Physicians Inquiries Network. All rights reserved.
Which prophylactic therapies best prevent gout attacks?
Allopurinol and febuxostat reduce the frequency of gout attacks equally after 8 weeks of treatment (strength of recommendation [SOR]: B, multiple randomized control trials [RCTs] with limitations).
Intravenous pegloticase decreases serum uric acid and gout attacks and improves quality of life (QOL) (SOR: A, 2 RCTs).
Colchicine reduces gout attacks when combined with probenecid or allopurinol at the start of urate-lowering therapy (SOR: B, 1 high-quality and 1 low-quality RCT).
EVIDENCE SUMMARY
A 28-week RCT compared the effects of placebo, allopurinol (300 mg/d), and febuxostat (80 mg, 120 mg, and 240 mg) on serum uric acid levels (sUA) and gout attacks in 1067 patients with gout and hyperuricemia (94% male, 78% white, 18 to 85 years of age with mean age ranging from 51 to 54 years ± 12 years in each group).1 Patients also received prophylaxis with either colchicine or naproxen during the first 8 weeks of the study.
During Weeks 1 through 8, investigators found no statistically significant differences in the percentage of patients requiring treatment for gout attacks between the febuxostat 80 mg, allopurinol, and placebo groups (28%, 23%, and 20%, respectively). During Weeks 8 through 28, no statistically significant differences in gout attack rates occurred between the allopurinol and febuxostat groups, although the study didn’t report specific attack rates for this period.
Both allopurinol and all doses of febuxostat reduced sUA to <6 mg/dL more effectively than placebo; more patients treated with febuxostat than allopurinol achieved a uric acid level of less than <6 mg/dL.
Another RCT of 762 mostly white, male patients (mean age 52 years) with gout and sUA >8 mg/dL—35% of whom had renal impairment, defined as creatinine clearance <80 mL/min/1.73m2—also concluded that febuxostat and allopurinol are equally effective in reducing gout attacks (incidence of gout flares during Weeks 9 to 52 was 64% with both febuxostat 80 mg and allopurinol 300 mg).2 The percentage of patients with sUA <6 mg/dL at the last 3 monthly visits was 53% in the febuxostat 80 mg group compared with 21% in the allopurinol 300 mg group (P<.001; number needed to treat [NNT]=4]).
One significant limitation of both RCTs was the fixed dose of allopurinol (300 mg/d). US Food and Drug Administration-approved dosing for allopurinol allows for titration to a maximum of 800 mg/d to achieve serum uric acid <6 mg/dL.
IV pegloticase decreases gout attacks after 3 months, improves quality of life
Pegloticase is an intravenously administered, recombinant form of uricase, the natural enzyme that converts uric acid to more soluble allantoin. Two RCTs compared pegloticase with placebo in a total of 212 patients with gout (mean age 54 to 59 years; 70% to 90% male) intolerant or refractory to allopurinol (defined as baseline sUA of ≥8 mg/dL and at least one of the following: ≥3 self-reported gout flares during the previous 18 months, ≥1 tophi, or gouty arthropathy.
These trials found that treatment with 8 mg of pegloticase every 2 weeks for 6 months initially increased gout flares during Months 1 to 3 (75% with pegloticase, 53% with placebo; P=.02; number needed to harm [NNH]=5) but then decreased the incidence of acute gout attacks during Months 4 to 6 (41% with pegloticase, 67% with placebo; P=.007; NNT=4).3 In addition, pegloticase resulted in statistically significant improvements in QOL measured at the final visit using the Health Assessment Questionnaire (HAQ) pain scale, the HAQ-Disability Index, and the 36-item Short Form Health Survey.
Colchicine plus probenecid or allopurinol reduces gout attacks
One small, low-quality RCT (N=38) found that colchicine 0.5 mg administered 3 times daily effectively prevented gout attacks when administered concomitantly with probenecid initiated to lower urate (gout attacks per month in colchicine and placebo-treated patients, respectively, were 0.19±0.05 and 0.48±0.12; P<.05).4
Another RCT that compared allopurinol with and without colchicine showed that coadministration of colchicine 0.6 mg twice daily reduced gout attacks: 33% of patients treated with colchicine experienced a gout flare compared with 77% of placebo-treated patients (P=.008; NNT=3 over 6 months).5
We identified no RCTs that evaluated the uricosuric agent probenecid and no studies that assessed the use of nonsteroidal anti-inflammatory drugs (NSAIDs) to prevent recurrent gout attacks.
RECOMMENDATIONS
The American College of Rheumatology (ACR) guidelines on managing gout recommend allopurinol or febuxostat as first-line pharmacologic urate-lowering therapy, with a goal of reducing sUA to <6 mg/dL. They recommend probenecid as an alternative if contraindications exist or the patient is intolerant to allopurinol and febuxostat.6 The guidelines note that allopurinol doses may exceed 300 mg/d, even in patients with chronic kidney disease.
The ACR recommends anti-inflammatory prophylaxis with colchicine or NSAIDs upon initiation of urate-lowering therapy. Anti-inflammatory prophylaxis should be continued as long as clinical evidence of continuing gout disease exists and until the sUA target has been acheived.7
1. Schumacher HR Jr, Becker MA, Wortmann RL, et al. Effects of febuxostat versus allopurinol and placebo in reducing serum urate in subjects with hyperuricemia and gout: a 28-week, phase III, randomized, double-blind, parallel-group trial. Arthritis Rheum. 2008;59:1540-1548.
2. Becker MA, Schumacher HR Jr, Wortmann RL, et al. Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N Engl J Med. 2005;353:2450-2461.
3. Sundy JS, Baraf HSB, Yood RA, et al. Efficacy and tolerability of pegloticase for the treatment of chronic gout in patients refractory to conventional treatment: two randomized controlled trials. JAMA. 2011;306:711-720.
4. Paulus HE, Schlosstein LH, Godfrey RG, et al. Prophylactic colchicine therapy of intercritical gout: a placebo-controlled study of probenecid-treated patients. Arthritis Rheum. 1974;17:609-614.
5. Borstad GC, Bryant LR, Abel MP, et al. Colchicine for prophylaxis of acute flares when initiating allopurinol for chronic gouty arthritis. J Rheumatol. 2004;31:2429-2432.
6. Khanna D, Fitzgerald JD, Khanna PP, et al; American College of Rheumatology. 2012 American College of Rheumatology guidelines for management of gout. Part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res (Hoboken). 2012;64:1431-1446.
7. Khanna D, Khanna PP, Fitzgerald JD, et al; American College of Rheumatology. 2012 American College of Rheumatology guidelines for management of gout. Part 2: therapy and anti-inflammatory prophylaxis of acute gouty arthritis. Arthritis Care Res (Hoboken). 2012;64:1447-1461.
Allopurinol and febuxostat reduce the frequency of gout attacks equally after 8 weeks of treatment (strength of recommendation [SOR]: B, multiple randomized control trials [RCTs] with limitations).
Intravenous pegloticase decreases serum uric acid and gout attacks and improves quality of life (QOL) (SOR: A, 2 RCTs).
Colchicine reduces gout attacks when combined with probenecid or allopurinol at the start of urate-lowering therapy (SOR: B, 1 high-quality and 1 low-quality RCT).
EVIDENCE SUMMARY
A 28-week RCT compared the effects of placebo, allopurinol (300 mg/d), and febuxostat (80 mg, 120 mg, and 240 mg) on serum uric acid levels (sUA) and gout attacks in 1067 patients with gout and hyperuricemia (94% male, 78% white, 18 to 85 years of age with mean age ranging from 51 to 54 years ± 12 years in each group).1 Patients also received prophylaxis with either colchicine or naproxen during the first 8 weeks of the study.
During Weeks 1 through 8, investigators found no statistically significant differences in the percentage of patients requiring treatment for gout attacks between the febuxostat 80 mg, allopurinol, and placebo groups (28%, 23%, and 20%, respectively). During Weeks 8 through 28, no statistically significant differences in gout attack rates occurred between the allopurinol and febuxostat groups, although the study didn’t report specific attack rates for this period.
Both allopurinol and all doses of febuxostat reduced sUA to <6 mg/dL more effectively than placebo; more patients treated with febuxostat than allopurinol achieved a uric acid level of less than <6 mg/dL.
Another RCT of 762 mostly white, male patients (mean age 52 years) with gout and sUA >8 mg/dL—35% of whom had renal impairment, defined as creatinine clearance <80 mL/min/1.73m2—also concluded that febuxostat and allopurinol are equally effective in reducing gout attacks (incidence of gout flares during Weeks 9 to 52 was 64% with both febuxostat 80 mg and allopurinol 300 mg).2 The percentage of patients with sUA <6 mg/dL at the last 3 monthly visits was 53% in the febuxostat 80 mg group compared with 21% in the allopurinol 300 mg group (P<.001; number needed to treat [NNT]=4]).
One significant limitation of both RCTs was the fixed dose of allopurinol (300 mg/d). US Food and Drug Administration-approved dosing for allopurinol allows for titration to a maximum of 800 mg/d to achieve serum uric acid <6 mg/dL.
IV pegloticase decreases gout attacks after 3 months, improves quality of life
Pegloticase is an intravenously administered, recombinant form of uricase, the natural enzyme that converts uric acid to more soluble allantoin. Two RCTs compared pegloticase with placebo in a total of 212 patients with gout (mean age 54 to 59 years; 70% to 90% male) intolerant or refractory to allopurinol (defined as baseline sUA of ≥8 mg/dL and at least one of the following: ≥3 self-reported gout flares during the previous 18 months, ≥1 tophi, or gouty arthropathy.
These trials found that treatment with 8 mg of pegloticase every 2 weeks for 6 months initially increased gout flares during Months 1 to 3 (75% with pegloticase, 53% with placebo; P=.02; number needed to harm [NNH]=5) but then decreased the incidence of acute gout attacks during Months 4 to 6 (41% with pegloticase, 67% with placebo; P=.007; NNT=4).3 In addition, pegloticase resulted in statistically significant improvements in QOL measured at the final visit using the Health Assessment Questionnaire (HAQ) pain scale, the HAQ-Disability Index, and the 36-item Short Form Health Survey.
Colchicine plus probenecid or allopurinol reduces gout attacks
One small, low-quality RCT (N=38) found that colchicine 0.5 mg administered 3 times daily effectively prevented gout attacks when administered concomitantly with probenecid initiated to lower urate (gout attacks per month in colchicine and placebo-treated patients, respectively, were 0.19±0.05 and 0.48±0.12; P<.05).4
Another RCT that compared allopurinol with and without colchicine showed that coadministration of colchicine 0.6 mg twice daily reduced gout attacks: 33% of patients treated with colchicine experienced a gout flare compared with 77% of placebo-treated patients (P=.008; NNT=3 over 6 months).5
We identified no RCTs that evaluated the uricosuric agent probenecid and no studies that assessed the use of nonsteroidal anti-inflammatory drugs (NSAIDs) to prevent recurrent gout attacks.
RECOMMENDATIONS
The American College of Rheumatology (ACR) guidelines on managing gout recommend allopurinol or febuxostat as first-line pharmacologic urate-lowering therapy, with a goal of reducing sUA to <6 mg/dL. They recommend probenecid as an alternative if contraindications exist or the patient is intolerant to allopurinol and febuxostat.6 The guidelines note that allopurinol doses may exceed 300 mg/d, even in patients with chronic kidney disease.
The ACR recommends anti-inflammatory prophylaxis with colchicine or NSAIDs upon initiation of urate-lowering therapy. Anti-inflammatory prophylaxis should be continued as long as clinical evidence of continuing gout disease exists and until the sUA target has been acheived.7
Allopurinol and febuxostat reduce the frequency of gout attacks equally after 8 weeks of treatment (strength of recommendation [SOR]: B, multiple randomized control trials [RCTs] with limitations).
Intravenous pegloticase decreases serum uric acid and gout attacks and improves quality of life (QOL) (SOR: A, 2 RCTs).
Colchicine reduces gout attacks when combined with probenecid or allopurinol at the start of urate-lowering therapy (SOR: B, 1 high-quality and 1 low-quality RCT).
EVIDENCE SUMMARY
A 28-week RCT compared the effects of placebo, allopurinol (300 mg/d), and febuxostat (80 mg, 120 mg, and 240 mg) on serum uric acid levels (sUA) and gout attacks in 1067 patients with gout and hyperuricemia (94% male, 78% white, 18 to 85 years of age with mean age ranging from 51 to 54 years ± 12 years in each group).1 Patients also received prophylaxis with either colchicine or naproxen during the first 8 weeks of the study.
During Weeks 1 through 8, investigators found no statistically significant differences in the percentage of patients requiring treatment for gout attacks between the febuxostat 80 mg, allopurinol, and placebo groups (28%, 23%, and 20%, respectively). During Weeks 8 through 28, no statistically significant differences in gout attack rates occurred between the allopurinol and febuxostat groups, although the study didn’t report specific attack rates for this period.
Both allopurinol and all doses of febuxostat reduced sUA to <6 mg/dL more effectively than placebo; more patients treated with febuxostat than allopurinol achieved a uric acid level of less than <6 mg/dL.
Another RCT of 762 mostly white, male patients (mean age 52 years) with gout and sUA >8 mg/dL—35% of whom had renal impairment, defined as creatinine clearance <80 mL/min/1.73m2—also concluded that febuxostat and allopurinol are equally effective in reducing gout attacks (incidence of gout flares during Weeks 9 to 52 was 64% with both febuxostat 80 mg and allopurinol 300 mg).2 The percentage of patients with sUA <6 mg/dL at the last 3 monthly visits was 53% in the febuxostat 80 mg group compared with 21% in the allopurinol 300 mg group (P<.001; number needed to treat [NNT]=4]).
One significant limitation of both RCTs was the fixed dose of allopurinol (300 mg/d). US Food and Drug Administration-approved dosing for allopurinol allows for titration to a maximum of 800 mg/d to achieve serum uric acid <6 mg/dL.
IV pegloticase decreases gout attacks after 3 months, improves quality of life
Pegloticase is an intravenously administered, recombinant form of uricase, the natural enzyme that converts uric acid to more soluble allantoin. Two RCTs compared pegloticase with placebo in a total of 212 patients with gout (mean age 54 to 59 years; 70% to 90% male) intolerant or refractory to allopurinol (defined as baseline sUA of ≥8 mg/dL and at least one of the following: ≥3 self-reported gout flares during the previous 18 months, ≥1 tophi, or gouty arthropathy.
These trials found that treatment with 8 mg of pegloticase every 2 weeks for 6 months initially increased gout flares during Months 1 to 3 (75% with pegloticase, 53% with placebo; P=.02; number needed to harm [NNH]=5) but then decreased the incidence of acute gout attacks during Months 4 to 6 (41% with pegloticase, 67% with placebo; P=.007; NNT=4).3 In addition, pegloticase resulted in statistically significant improvements in QOL measured at the final visit using the Health Assessment Questionnaire (HAQ) pain scale, the HAQ-Disability Index, and the 36-item Short Form Health Survey.
Colchicine plus probenecid or allopurinol reduces gout attacks
One small, low-quality RCT (N=38) found that colchicine 0.5 mg administered 3 times daily effectively prevented gout attacks when administered concomitantly with probenecid initiated to lower urate (gout attacks per month in colchicine and placebo-treated patients, respectively, were 0.19±0.05 and 0.48±0.12; P<.05).4
Another RCT that compared allopurinol with and without colchicine showed that coadministration of colchicine 0.6 mg twice daily reduced gout attacks: 33% of patients treated with colchicine experienced a gout flare compared with 77% of placebo-treated patients (P=.008; NNT=3 over 6 months).5
We identified no RCTs that evaluated the uricosuric agent probenecid and no studies that assessed the use of nonsteroidal anti-inflammatory drugs (NSAIDs) to prevent recurrent gout attacks.
RECOMMENDATIONS
The American College of Rheumatology (ACR) guidelines on managing gout recommend allopurinol or febuxostat as first-line pharmacologic urate-lowering therapy, with a goal of reducing sUA to <6 mg/dL. They recommend probenecid as an alternative if contraindications exist or the patient is intolerant to allopurinol and febuxostat.6 The guidelines note that allopurinol doses may exceed 300 mg/d, even in patients with chronic kidney disease.
The ACR recommends anti-inflammatory prophylaxis with colchicine or NSAIDs upon initiation of urate-lowering therapy. Anti-inflammatory prophylaxis should be continued as long as clinical evidence of continuing gout disease exists and until the sUA target has been acheived.7
1. Schumacher HR Jr, Becker MA, Wortmann RL, et al. Effects of febuxostat versus allopurinol and placebo in reducing serum urate in subjects with hyperuricemia and gout: a 28-week, phase III, randomized, double-blind, parallel-group trial. Arthritis Rheum. 2008;59:1540-1548.
2. Becker MA, Schumacher HR Jr, Wortmann RL, et al. Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N Engl J Med. 2005;353:2450-2461.
3. Sundy JS, Baraf HSB, Yood RA, et al. Efficacy and tolerability of pegloticase for the treatment of chronic gout in patients refractory to conventional treatment: two randomized controlled trials. JAMA. 2011;306:711-720.
4. Paulus HE, Schlosstein LH, Godfrey RG, et al. Prophylactic colchicine therapy of intercritical gout: a placebo-controlled study of probenecid-treated patients. Arthritis Rheum. 1974;17:609-614.
5. Borstad GC, Bryant LR, Abel MP, et al. Colchicine for prophylaxis of acute flares when initiating allopurinol for chronic gouty arthritis. J Rheumatol. 2004;31:2429-2432.
6. Khanna D, Fitzgerald JD, Khanna PP, et al; American College of Rheumatology. 2012 American College of Rheumatology guidelines for management of gout. Part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res (Hoboken). 2012;64:1431-1446.
7. Khanna D, Khanna PP, Fitzgerald JD, et al; American College of Rheumatology. 2012 American College of Rheumatology guidelines for management of gout. Part 2: therapy and anti-inflammatory prophylaxis of acute gouty arthritis. Arthritis Care Res (Hoboken). 2012;64:1447-1461.
1. Schumacher HR Jr, Becker MA, Wortmann RL, et al. Effects of febuxostat versus allopurinol and placebo in reducing serum urate in subjects with hyperuricemia and gout: a 28-week, phase III, randomized, double-blind, parallel-group trial. Arthritis Rheum. 2008;59:1540-1548.
2. Becker MA, Schumacher HR Jr, Wortmann RL, et al. Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N Engl J Med. 2005;353:2450-2461.
3. Sundy JS, Baraf HSB, Yood RA, et al. Efficacy and tolerability of pegloticase for the treatment of chronic gout in patients refractory to conventional treatment: two randomized controlled trials. JAMA. 2011;306:711-720.
4. Paulus HE, Schlosstein LH, Godfrey RG, et al. Prophylactic colchicine therapy of intercritical gout: a placebo-controlled study of probenecid-treated patients. Arthritis Rheum. 1974;17:609-614.
5. Borstad GC, Bryant LR, Abel MP, et al. Colchicine for prophylaxis of acute flares when initiating allopurinol for chronic gouty arthritis. J Rheumatol. 2004;31:2429-2432.
6. Khanna D, Fitzgerald JD, Khanna PP, et al; American College of Rheumatology. 2012 American College of Rheumatology guidelines for management of gout. Part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res (Hoboken). 2012;64:1431-1446.
7. Khanna D, Khanna PP, Fitzgerald JD, et al; American College of Rheumatology. 2012 American College of Rheumatology guidelines for management of gout. Part 2: therapy and anti-inflammatory prophylaxis of acute gouty arthritis. Arthritis Care Res (Hoboken). 2012;64:1447-1461.
Evidence-based answers from the Family Physicians Inquiries Network
Treating Migraine: The Case for Aspirin
PRACTICE CHANGER
Recommend aspirin 975 mg (three adult tablets) as a viable firstline treatment for acute migraine. Consider prescribing metoclopramide 10 mg to be taken with aspirin to markedly decrease associated nausea and help achieve maximum symptom relief.1
STRENGTH OF RECOMMENDATION
B: Based on a Cochrane meta-analysis of 13 good-quality, randomized controlled trials (RCTs).1
ILLUSTRATIVE CASE
During a routine physical, a 37-year-old patient asks you what she should take for occasional migraine. She describes a unilateral headache with associated nausea, vomiting, phonophobia, and photophobia. What medication should you recommend?
Migraine headache affects more than 37 million Americans.2 Women are three times more likely than men to experience migraine, with the highest prevalence among those ages 30 to 50.3,4 More than 50% of patients report that episodes cause severe impairment, resulting in an average loss of four to six workdays each year due to migraine.5,6
Do you recommend this low-cost option?
Although many patients try OTC headache remedies for migraine, when they do seek medical care for this condition, most (67%) turn to their primary care provider.7 But despite a 2010 Cochrane review showing aspirin’s efficacy for acute migraine,8 our experience—based on discussions with physicians at numerous residency programs—suggests that family practice providers are not likely to recommend it.
Further evidence of the underuse of aspirin for migraine comes from a 2013 review of national surveillance studies,5 which found that in 2009, triptans accounted for nearly 80% of antimigraine analgesics prescribed during office visits.5 Thus, when the Cochrane reviewers issued this update of the earlier meta-analysis, we welcomed the opportunity to feature a practice changer that might not be getting the “traction” it deserves.
Continue reading for the study summary...
STUDY SUMMARY
Multiple RCTs highlight aspirin’s efficacy
The 2013 Cochrane reviewers used the same 13 good-quality, double-blind RCTs involving 4,222 participants as the earlier review; no new studies that warranted inclusion were found. A total of 5,261 episodes of moderate-to-severe migraine were treated with either aspirin alone or aspirin plus the antiemetic metoclopramide.1
Five studies had placebo controls, four had active controls (eg, sumatriptan, zolmitriptan, ibuprofen, acetaminophen plus codeine, and ergotamine plus caffeine), and four had both active and placebo controls. Primary outcomes were painfree status at two hours and headache relief (defined as a reduction in pain from moderate/severe to none/mild without the use of rescue medication) at two hours. Sustained headache relief at 24 hours was a secondary outcome.
Patients self-assessed their headache pain, using either a four-point categorical scale (none, mild, moderate, or severe) or a 100-mm visual analog scale. On the analog scale, less than 30 mm was considered mild or no pain; 30 mm or more was considered moderate or severe.
Study participants were ages 18 to 65 (mean age range, 37 to 44), and their symptoms met International Headache Society criteria for migraine with or without aura.9 All participants had migraine symptoms for at least 12 months, with one to six attacks of moderate to severe intensity per month prior to the study period.
In six studies (n = 2,027), investigators compared either 900- or 1,000-mg aspirin alone with placebo. For both primary outcomes, aspirin alone was superior to placebo, with a number needed to treat (NNT) of 8.1 for two-hour painfree status and 4.9 for two-hour headache relief. In three studies (n = 1,142), aspirin was superior to placebo for 24-hour headache relief, with an NNT of 6.6.
Aspirin plus metoclopramide was also better than placebo for primary and secondary outcomes, with an NNT of 8.8 for two-hour painfree status, 3.3 for two-hour headache relief, and 6.2 for 24-hour headache relief. Based on subgroup analysis, aspirin plus metoclopramide was more effective than aspirin alone for two-hour headache relief but equivalent for two-hour painfree status and 24-hour headache relief. The addition of metoclopramide to aspirin significantly reduced nausea and vomiting.
In two studies (n = 726), aspirin alone was equivalent to sumatriptan 50 mg for reaching painfree and headache relief status at two hours. Two additional studies (n = 523) compared aspirin plus metoclopramide with sumatriptan 100 mg and found them to be equal for two-hour headache relief, but the aspirin combination was inferior to the triptan for painfree status at two hours (n = 528). Data were insufficient to compare the efficacy of aspirin with zolmitriptan, ibuprofen, or acetaminophen plus codeine.
There were no reports of gastrointestinal bleeding or other serious adverse events attributable to aspirin therapy. Most adverse effects were mild or moderate disturbances of the digestive and nervous systems, with a number needed to harm of 34 for aspirin (with or without metoclopramide) versus placebo.
WHAT’S NEW?
A reminder of aspirin’s efficacy in treating migraine
The update of this meta-analysis confirms that high-dose aspirin (900 to 1,000 mg) is an effective treatment for migraine headache in adults ages 18 to 65. The addition of metoclopramide reduces nausea and vomiting but offers little if any benefit for headache/pain relief.
Continue reading for the caveats and challenges to implementation...
CAVEATS
Lack of comparison with other treatments
Data were insufficient to compare the efficacy of aspirin with zolmitriptan, other NSAIDs alone, or acetaminophen plus codeine. Aspirin should be used with caution in patients who have chronic renal disease and/or a history of peptic ulcer disease.
CHALLENGES TO IMPLEMENTATION
Patients want a prescription
Patients often expect a prescription when they present with complaints of migraine headache and may feel shortchanged if they’re told to take an aspirin. Providing a prescription for the antiemetic metoclopramide, as well as a brief explanation of the evidence indicating that aspirin is effective for migraine, may adequately address such expectations.
Continue reading for the references...
REFERENCES
1. Kirthi V, Derry S, Moore RA. Aspirin with or without an antiemetic for acute migraine headaches in adults. Cochrane Database Syst Rev. 2013;(4):CD008041.
2. National Headache Foundation. Migraine. www.headaches.org/education/Headache_Topic_Sheets/Migraine. Accessed February 14, 2014.
3. Lipton RB, Stewart WF, Diamond S, et. al. Prevalence and burden of migraine in the United States: data from the American Migraine Study II. Headache. 2001;41:646-657.
4. Victor TW, Hu X, Campbell JC, et al. Migraine prevalence by age and sex in the United States: a life-span study. Cephalagia. 2010; 9:1065-1072.
5. Smitherman TA, Burch R, Sheikh H, et al. The prevalence, impact, and treatment of migraine and severe headaches in the United States: a review of statistics from national surveillance studies. Headache. 2013;53:427-436.
6. Hu XH, Markson LE, Lipton RB, et al. Burden of migraine in the United States: disability and economic costs. Arch Intern Med. 1999; 159:813-818.
7. Gibbs TS, Fleischer AB Jr, Feldman SR, et al. Health care utilization in patients with migraine: demographics and patterns of care in the ambulatory setting. Headache. 2003;43:330-335.
8. Kirthi V, Derry S, Moore RA, et al. Aspirin with or without an antiemetic for acute migraine headaches in adults. Cochrane Database Syst Rev. 2010;(4):CD008041.
9. The international classification of headache disorders. 2nd ed. Cephalalgia. 2004; 24 (suppl 1):S9-S160.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center for Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
Copyright © 2014. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice. 2014;63(2):94-96.
PRACTICE CHANGER
Recommend aspirin 975 mg (three adult tablets) as a viable firstline treatment for acute migraine. Consider prescribing metoclopramide 10 mg to be taken with aspirin to markedly decrease associated nausea and help achieve maximum symptom relief.1
STRENGTH OF RECOMMENDATION
B: Based on a Cochrane meta-analysis of 13 good-quality, randomized controlled trials (RCTs).1
ILLUSTRATIVE CASE
During a routine physical, a 37-year-old patient asks you what she should take for occasional migraine. She describes a unilateral headache with associated nausea, vomiting, phonophobia, and photophobia. What medication should you recommend?
Migraine headache affects more than 37 million Americans.2 Women are three times more likely than men to experience migraine, with the highest prevalence among those ages 30 to 50.3,4 More than 50% of patients report that episodes cause severe impairment, resulting in an average loss of four to six workdays each year due to migraine.5,6
Do you recommend this low-cost option?
Although many patients try OTC headache remedies for migraine, when they do seek medical care for this condition, most (67%) turn to their primary care provider.7 But despite a 2010 Cochrane review showing aspirin’s efficacy for acute migraine,8 our experience—based on discussions with physicians at numerous residency programs—suggests that family practice providers are not likely to recommend it.
Further evidence of the underuse of aspirin for migraine comes from a 2013 review of national surveillance studies,5 which found that in 2009, triptans accounted for nearly 80% of antimigraine analgesics prescribed during office visits.5 Thus, when the Cochrane reviewers issued this update of the earlier meta-analysis, we welcomed the opportunity to feature a practice changer that might not be getting the “traction” it deserves.
Continue reading for the study summary...
STUDY SUMMARY
Multiple RCTs highlight aspirin’s efficacy
The 2013 Cochrane reviewers used the same 13 good-quality, double-blind RCTs involving 4,222 participants as the earlier review; no new studies that warranted inclusion were found. A total of 5,261 episodes of moderate-to-severe migraine were treated with either aspirin alone or aspirin plus the antiemetic metoclopramide.1
Five studies had placebo controls, four had active controls (eg, sumatriptan, zolmitriptan, ibuprofen, acetaminophen plus codeine, and ergotamine plus caffeine), and four had both active and placebo controls. Primary outcomes were painfree status at two hours and headache relief (defined as a reduction in pain from moderate/severe to none/mild without the use of rescue medication) at two hours. Sustained headache relief at 24 hours was a secondary outcome.
Patients self-assessed their headache pain, using either a four-point categorical scale (none, mild, moderate, or severe) or a 100-mm visual analog scale. On the analog scale, less than 30 mm was considered mild or no pain; 30 mm or more was considered moderate or severe.
Study participants were ages 18 to 65 (mean age range, 37 to 44), and their symptoms met International Headache Society criteria for migraine with or without aura.9 All participants had migraine symptoms for at least 12 months, with one to six attacks of moderate to severe intensity per month prior to the study period.
In six studies (n = 2,027), investigators compared either 900- or 1,000-mg aspirin alone with placebo. For both primary outcomes, aspirin alone was superior to placebo, with a number needed to treat (NNT) of 8.1 for two-hour painfree status and 4.9 for two-hour headache relief. In three studies (n = 1,142), aspirin was superior to placebo for 24-hour headache relief, with an NNT of 6.6.
Aspirin plus metoclopramide was also better than placebo for primary and secondary outcomes, with an NNT of 8.8 for two-hour painfree status, 3.3 for two-hour headache relief, and 6.2 for 24-hour headache relief. Based on subgroup analysis, aspirin plus metoclopramide was more effective than aspirin alone for two-hour headache relief but equivalent for two-hour painfree status and 24-hour headache relief. The addition of metoclopramide to aspirin significantly reduced nausea and vomiting.
In two studies (n = 726), aspirin alone was equivalent to sumatriptan 50 mg for reaching painfree and headache relief status at two hours. Two additional studies (n = 523) compared aspirin plus metoclopramide with sumatriptan 100 mg and found them to be equal for two-hour headache relief, but the aspirin combination was inferior to the triptan for painfree status at two hours (n = 528). Data were insufficient to compare the efficacy of aspirin with zolmitriptan, ibuprofen, or acetaminophen plus codeine.
There were no reports of gastrointestinal bleeding or other serious adverse events attributable to aspirin therapy. Most adverse effects were mild or moderate disturbances of the digestive and nervous systems, with a number needed to harm of 34 for aspirin (with or without metoclopramide) versus placebo.
WHAT’S NEW?
A reminder of aspirin’s efficacy in treating migraine
The update of this meta-analysis confirms that high-dose aspirin (900 to 1,000 mg) is an effective treatment for migraine headache in adults ages 18 to 65. The addition of metoclopramide reduces nausea and vomiting but offers little if any benefit for headache/pain relief.
Continue reading for the caveats and challenges to implementation...
CAVEATS
Lack of comparison with other treatments
Data were insufficient to compare the efficacy of aspirin with zolmitriptan, other NSAIDs alone, or acetaminophen plus codeine. Aspirin should be used with caution in patients who have chronic renal disease and/or a history of peptic ulcer disease.
CHALLENGES TO IMPLEMENTATION
Patients want a prescription
Patients often expect a prescription when they present with complaints of migraine headache and may feel shortchanged if they’re told to take an aspirin. Providing a prescription for the antiemetic metoclopramide, as well as a brief explanation of the evidence indicating that aspirin is effective for migraine, may adequately address such expectations.
Continue reading for the references...
REFERENCES
1. Kirthi V, Derry S, Moore RA. Aspirin with or without an antiemetic for acute migraine headaches in adults. Cochrane Database Syst Rev. 2013;(4):CD008041.
2. National Headache Foundation. Migraine. www.headaches.org/education/Headache_Topic_Sheets/Migraine. Accessed February 14, 2014.
3. Lipton RB, Stewart WF, Diamond S, et. al. Prevalence and burden of migraine in the United States: data from the American Migraine Study II. Headache. 2001;41:646-657.
4. Victor TW, Hu X, Campbell JC, et al. Migraine prevalence by age and sex in the United States: a life-span study. Cephalagia. 2010; 9:1065-1072.
5. Smitherman TA, Burch R, Sheikh H, et al. The prevalence, impact, and treatment of migraine and severe headaches in the United States: a review of statistics from national surveillance studies. Headache. 2013;53:427-436.
6. Hu XH, Markson LE, Lipton RB, et al. Burden of migraine in the United States: disability and economic costs. Arch Intern Med. 1999; 159:813-818.
7. Gibbs TS, Fleischer AB Jr, Feldman SR, et al. Health care utilization in patients with migraine: demographics and patterns of care in the ambulatory setting. Headache. 2003;43:330-335.
8. Kirthi V, Derry S, Moore RA, et al. Aspirin with or without an antiemetic for acute migraine headaches in adults. Cochrane Database Syst Rev. 2010;(4):CD008041.
9. The international classification of headache disorders. 2nd ed. Cephalalgia. 2004; 24 (suppl 1):S9-S160.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center for Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
Copyright © 2014. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice. 2014;63(2):94-96.
PRACTICE CHANGER
Recommend aspirin 975 mg (three adult tablets) as a viable firstline treatment for acute migraine. Consider prescribing metoclopramide 10 mg to be taken with aspirin to markedly decrease associated nausea and help achieve maximum symptom relief.1
STRENGTH OF RECOMMENDATION
B: Based on a Cochrane meta-analysis of 13 good-quality, randomized controlled trials (RCTs).1
ILLUSTRATIVE CASE
During a routine physical, a 37-year-old patient asks you what she should take for occasional migraine. She describes a unilateral headache with associated nausea, vomiting, phonophobia, and photophobia. What medication should you recommend?
Migraine headache affects more than 37 million Americans.2 Women are three times more likely than men to experience migraine, with the highest prevalence among those ages 30 to 50.3,4 More than 50% of patients report that episodes cause severe impairment, resulting in an average loss of four to six workdays each year due to migraine.5,6
Do you recommend this low-cost option?
Although many patients try OTC headache remedies for migraine, when they do seek medical care for this condition, most (67%) turn to their primary care provider.7 But despite a 2010 Cochrane review showing aspirin’s efficacy for acute migraine,8 our experience—based on discussions with physicians at numerous residency programs—suggests that family practice providers are not likely to recommend it.
Further evidence of the underuse of aspirin for migraine comes from a 2013 review of national surveillance studies,5 which found that in 2009, triptans accounted for nearly 80% of antimigraine analgesics prescribed during office visits.5 Thus, when the Cochrane reviewers issued this update of the earlier meta-analysis, we welcomed the opportunity to feature a practice changer that might not be getting the “traction” it deserves.
Continue reading for the study summary...
STUDY SUMMARY
Multiple RCTs highlight aspirin’s efficacy
The 2013 Cochrane reviewers used the same 13 good-quality, double-blind RCTs involving 4,222 participants as the earlier review; no new studies that warranted inclusion were found. A total of 5,261 episodes of moderate-to-severe migraine were treated with either aspirin alone or aspirin plus the antiemetic metoclopramide.1
Five studies had placebo controls, four had active controls (eg, sumatriptan, zolmitriptan, ibuprofen, acetaminophen plus codeine, and ergotamine plus caffeine), and four had both active and placebo controls. Primary outcomes were painfree status at two hours and headache relief (defined as a reduction in pain from moderate/severe to none/mild without the use of rescue medication) at two hours. Sustained headache relief at 24 hours was a secondary outcome.
Patients self-assessed their headache pain, using either a four-point categorical scale (none, mild, moderate, or severe) or a 100-mm visual analog scale. On the analog scale, less than 30 mm was considered mild or no pain; 30 mm or more was considered moderate or severe.
Study participants were ages 18 to 65 (mean age range, 37 to 44), and their symptoms met International Headache Society criteria for migraine with or without aura.9 All participants had migraine symptoms for at least 12 months, with one to six attacks of moderate to severe intensity per month prior to the study period.
In six studies (n = 2,027), investigators compared either 900- or 1,000-mg aspirin alone with placebo. For both primary outcomes, aspirin alone was superior to placebo, with a number needed to treat (NNT) of 8.1 for two-hour painfree status and 4.9 for two-hour headache relief. In three studies (n = 1,142), aspirin was superior to placebo for 24-hour headache relief, with an NNT of 6.6.
Aspirin plus metoclopramide was also better than placebo for primary and secondary outcomes, with an NNT of 8.8 for two-hour painfree status, 3.3 for two-hour headache relief, and 6.2 for 24-hour headache relief. Based on subgroup analysis, aspirin plus metoclopramide was more effective than aspirin alone for two-hour headache relief but equivalent for two-hour painfree status and 24-hour headache relief. The addition of metoclopramide to aspirin significantly reduced nausea and vomiting.
In two studies (n = 726), aspirin alone was equivalent to sumatriptan 50 mg for reaching painfree and headache relief status at two hours. Two additional studies (n = 523) compared aspirin plus metoclopramide with sumatriptan 100 mg and found them to be equal for two-hour headache relief, but the aspirin combination was inferior to the triptan for painfree status at two hours (n = 528). Data were insufficient to compare the efficacy of aspirin with zolmitriptan, ibuprofen, or acetaminophen plus codeine.
There were no reports of gastrointestinal bleeding or other serious adverse events attributable to aspirin therapy. Most adverse effects were mild or moderate disturbances of the digestive and nervous systems, with a number needed to harm of 34 for aspirin (with or without metoclopramide) versus placebo.
WHAT’S NEW?
A reminder of aspirin’s efficacy in treating migraine
The update of this meta-analysis confirms that high-dose aspirin (900 to 1,000 mg) is an effective treatment for migraine headache in adults ages 18 to 65. The addition of metoclopramide reduces nausea and vomiting but offers little if any benefit for headache/pain relief.
Continue reading for the caveats and challenges to implementation...
CAVEATS
Lack of comparison with other treatments
Data were insufficient to compare the efficacy of aspirin with zolmitriptan, other NSAIDs alone, or acetaminophen plus codeine. Aspirin should be used with caution in patients who have chronic renal disease and/or a history of peptic ulcer disease.
CHALLENGES TO IMPLEMENTATION
Patients want a prescription
Patients often expect a prescription when they present with complaints of migraine headache and may feel shortchanged if they’re told to take an aspirin. Providing a prescription for the antiemetic metoclopramide, as well as a brief explanation of the evidence indicating that aspirin is effective for migraine, may adequately address such expectations.
Continue reading for the references...
REFERENCES
1. Kirthi V, Derry S, Moore RA. Aspirin with or without an antiemetic for acute migraine headaches in adults. Cochrane Database Syst Rev. 2013;(4):CD008041.
2. National Headache Foundation. Migraine. www.headaches.org/education/Headache_Topic_Sheets/Migraine. Accessed February 14, 2014.
3. Lipton RB, Stewart WF, Diamond S, et. al. Prevalence and burden of migraine in the United States: data from the American Migraine Study II. Headache. 2001;41:646-657.
4. Victor TW, Hu X, Campbell JC, et al. Migraine prevalence by age and sex in the United States: a life-span study. Cephalagia. 2010; 9:1065-1072.
5. Smitherman TA, Burch R, Sheikh H, et al. The prevalence, impact, and treatment of migraine and severe headaches in the United States: a review of statistics from national surveillance studies. Headache. 2013;53:427-436.
6. Hu XH, Markson LE, Lipton RB, et al. Burden of migraine in the United States: disability and economic costs. Arch Intern Med. 1999; 159:813-818.
7. Gibbs TS, Fleischer AB Jr, Feldman SR, et al. Health care utilization in patients with migraine: demographics and patterns of care in the ambulatory setting. Headache. 2003;43:330-335.
8. Kirthi V, Derry S, Moore RA, et al. Aspirin with or without an antiemetic for acute migraine headaches in adults. Cochrane Database Syst Rev. 2010;(4):CD008041.
9. The international classification of headache disorders. 2nd ed. Cephalalgia. 2004; 24 (suppl 1):S9-S160.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center for Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
Copyright © 2014. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice. 2014;63(2):94-96.
7 questions to ask when evaluating a noninferiority trial
The traditional clinical trial, designed to test whether a new treatment is better than a placebo or another active treatment, is known as a “superiority” trial—although rarely labeled as such. In contrast, the goal of a noninferiority trial is simply to demonstrate that a new treatment is not substantially less effective than the standard therapy.
Such trials are useful when a new therapy is thought to be safer, easier to administer, or less costly than the existing treatment, but not necessarily more effective. And, because it would be unethical to randomize patients with a serious condition for which there already is an effective treatment to placebo, a noninferiority trial is another means of determining if the new treatment is effective.
Noninferiority trials have unique design features and methodology and require a different analysis than traditional superiority trials. Yet many physicians know far less about them; many investigators appear to be less than proficient, as well. A review of 116 noninferiority trials and 46 equivalence trials found that only 20% fulfilled generally accepted quality criteria.1 To improve the quality of noninferiority trials, the CONSORT (Consolidated Standards of Reporting Trials) Group has published a checklist for trial design and reporting standards.2,3 Based on this checklist, we came up with 7 key questions to consider when evaluating a noninferiority trial. In the pages that follow, you’ll also find an at-a-glance guide (TABLE) and a methodology review using a hypothetical case (page E7).
1. Is a noninferiority trial appropriate?
The introduction to a noninferiority trial should provide the rationale for this design and the absence of a placebo control group. Look for a review of the evidence of the efficacy of the reference treatment that placebo-controlled trials have revealed, along with the effect size. The advantages of the new treatment over the standard treatment—eg, fewer adverse effects, easier administration, or lower cost—should be discussed, as well.

In the Randomized Evaluation of Long-term Anticoagulation Therapy (RE-LY)—a prominent noninferiority trial—investigators compared the standard anticoagulant (warfarin) for patients with atrial fibrillation (AF) at risk of stroke with a new agent, dabigatran.4 In the methods section of the abstract and the statistical analysis section of the main body, the authors clearly indicated that this was a noninferiority trial. They began by referring to the existing evidence of warfarin’s effectiveness, then detailed the qualities that make warfarin cumbersome to use, including the need for frequent laboratory monitoring. This was followed by evidence that many patients stop taking warfarin and that even for those who persist with treatment, adequate anticoagulation is difficult to maintain.
The authors went on to state that because dabigatran requires no long-term monitoring, it is easier to use. Therefore, if dabigatran could be shown to be no worse than warfarin in preventing strokes, it would be a reasonable alternative, leaving no doubt that this was an appropriate noninferiority trial.
2. Is the noninferiority margin based on clinical judgment and statistical reasoning?
The noninferiority margin should be based on clinical judgment as to how effective a new treatment must be in order to be declared not clinically inferior to the standard treatment. This can be based on several factors, including the severity of the outcome and the expected advantages of the new treatment. The margin should also take into account the size of the standard treatment’s effect vs placebo. In RELY, for example, the authors noted that the noninferiority margin was based on the desire to preserve at least 50% of the lower limit of the confidence interval (CI) of warfarin’s estimated effect; this was done using data from a previously published meta-analysis of 6 trials comparing warfarin with placebo for stroke prevention in patients with AF.4-6
3. Are the hypothesis and statistical analysis formulated correctly?
The clinical hypothesis in a noninferiority trial is that the new treatment is not worse than the standard treatment by a prespecified margin; therefore, the statistical null hypothesis to be tested is that the new treatment is worse than the reference treatment by more than that margin. Rejecting a true null hypothesis (for example, because the P value is <.05) is known as a type l error. In this setting, making a type I error would mean accepting a new treatment that is truly worse than the standard by at least the specified margin. Failure to reject a false null hypothesis is known as a type II error, which in this case would mean failing to identify a new treatment that is truly noninferior to the standard.7
In RE-LY, the authors stated that the upper limit of the one-sided 97.5% CI for the relative risk of a stroke with dabigatran vs warfarin had to fall below 1.46.4 (This is the same as testing the null hypothesis that the hazard ratio is ≥1.46.) Thus, the hypothesis was formulated correctly.
4. Is the sample size appropriate and justified?
The sample size in a noninferiority trial should provide high power to reject the null hypothesis that the difference (or relative risk) between groups is equal to or greater than the noninferiority margin under some clinically meaningful assumption about the true difference (or absolute risk reduction) between groups. A true difference of 0 (or a relative risk of 1) is typically assumed for sample size calculation. However, assuming that the new treatment is truly slightly better or slightly worse than the standard may be clinically appropriate in some cases. This would indicate a need for a smaller or larger sample size, respectively, than that required under the usual assumption of no difference.
When the justification for the sample size in a noninferiority trial is not provided or the number of participants is based on an inappropriate approach (eg, using superiority trial calculations for a noninferiority trial), questions about the quality of the trial arise. The primary concern is whether the noninferiority margin was actually selected before the trial began, as it should have been. And if the researchers used overly optimistic assumptions about the efficacy of the new treatment relative to the standard therapy, the failure to rule out the margin could be misleading. (As with superiority trials that fail to reject the null hypothesis, post hoc power calculations should be avoided.) After the study has ended, the resulting CIs should be used to evaluate whether the study was large enough to adequately assess the relative effectiveness of the treatments.
The RE-LY trial calculated the sample size that was expected to provide 84% power to rule out the prespecified hazard ratio of 1.46, assuming a true event rate of 1.6% per year (presumably for both groups), a recruitment period of 2 years, and at least one year of follow-up. The sample size was subsequently increased from 15,000 to 18,000 to maintain power in case of a low event rate.4,5
5. Is the noninferiority trial as similar as possible to the trial(s) comparing the standard treatment with placebo?
Characteristics of participants, setting, reference treatment, and outcomes used in a noninferiority trial should be as close as possible to those in the trial(s) comparing the treatment with placebo. This is known as the constancy assumption, and it is key to researchers’ ability to draw a conclusion about noninferiority.
The trials used to calculate the noninferiority margin and the RE-LY trial itself involved similar populations of patients with AF, and the outcome (stroke) was similar.
6. Is a per protocol analysis reported in the results?
In randomized controlled superiority trials, the participants should be analyzed in the groups to which they were originally allocated, regardless of whether they adhered to treatment during the entire follow-up period. Such intention-to-treat (ITT) analysis is important because it provides a more conservative estimate of treatment effect—taking into account that some people who are offered treatment will not accept it and others will discontinue treatment. An ITT analysis therefore tends to minimize treatment effects compared with a “per protocol” analysis, in which participants are analyzed according to the treatment they actually received and are often removed from the analysis if they discontinue or do not adhere to treatment.
In noninferiority trials, if patients in the intervention group cross over to the standard treatment group or those in the standard treatment group have poor adherence, an ITT analysis can increase the risk of wrongly claiming noninferiority.7 Therefore, a per protocol analysis should be included—and indeed may be preferable.
In RE-LY, ITT analyses were reported, and complete follow-up data were available for 99.9% of patients. However, the rates of treatment discontinuation at one year were about 15% for those on dabigatran and 10% for the warfarin group, and 21% and 17%, respectively, at 2 years.4,5 If the new treatment were truly less efficacious than the standard treatment, these moderate discontinuation rates could lead to more similar rates of stroke in the 2 groups than would be expected with higher continuation rates, biasing results towards the alternative of noninferiority. Although the original publication of trial results did not include a per protocol analysis, the RE-LY authors later reported that a per protocol analysis yielded similar results to the ITT analysis.

7. Are the overall design and execution of the trial high quality?
Because a poor quality noninferiority trial can appear to demonstrate noninferiority, looking at such studies critically is crucial. Appropriate randomization, concealed allocation, masking, and careful attention to participant flow must all be assessed.2,3
To continue with our example, the RE-LY trial was well conducted. Randomization was performed centrally via an automated telephone system and 2 doses of dabigatran were administered in a masked fashion, while warfarin was open-label. Remarkably, follow-up was achieved for 99.9% of participants over a median of 2 years, and independent adjudicators masked to treatment group assessed outcomes.4,5
CORRESPONDENCE
Anne Mounsey, MD, UNC Chapel Hill Department of Family Medicine, 590 Manning Drive, CB 7595, Chapel Hill, NC 27590; [email protected]
1. Le Henanff A, Giraudeau B, Baron G, et al. Quality of reporting of noninferiority and equivalence randomized trials. JAMA. 2006;295:1147-1151.
2. Piaggio G, Elbourne DR, Pocock SJ, et al; CONSORT Group. Reporting of noninferiority and equivalence randomized trials: extension of the CONSORT 2010 statement. JAMA. 2012;308:2594-2604.
3. Moher D, Schulz KF, Altman D; CONSORT Group (Consolidated Standards of Reporting Trials). The CONSORT statement: revised recommendations for improving the quality of reports of parallel-group randomized trials. JAMA. 2001;285:1987-1991.
4. Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med. 2009;361:1139-1151.
5. Ezekowitz MD, Connolly S, Parekh A, et al. Rationale and design of RE-LY: randomized evaluation of long-term anticoagulant therapy, warfarin, compared with dabigatran. Am Heart J. 2009;157:805-810, 810.e1-2.
6. Hart RG, Benavente O, McBride R, et al. Antithrombotic therapy to prevent stroke in patients with atrial fibrillation: a meta-analysis. Ann Intern Med. 1999;131:492-501.
7. US Department of Health and Human Services. Guidance for industry non-inferiority clinical trials. US Food and Drug Administration Web site. March 2010. Available at: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM202140.pdf. Accessed February 4, 2014.
The traditional clinical trial, designed to test whether a new treatment is better than a placebo or another active treatment, is known as a “superiority” trial—although rarely labeled as such. In contrast, the goal of a noninferiority trial is simply to demonstrate that a new treatment is not substantially less effective than the standard therapy.
Such trials are useful when a new therapy is thought to be safer, easier to administer, or less costly than the existing treatment, but not necessarily more effective. And, because it would be unethical to randomize patients with a serious condition for which there already is an effective treatment to placebo, a noninferiority trial is another means of determining if the new treatment is effective.
Noninferiority trials have unique design features and methodology and require a different analysis than traditional superiority trials. Yet many physicians know far less about them; many investigators appear to be less than proficient, as well. A review of 116 noninferiority trials and 46 equivalence trials found that only 20% fulfilled generally accepted quality criteria.1 To improve the quality of noninferiority trials, the CONSORT (Consolidated Standards of Reporting Trials) Group has published a checklist for trial design and reporting standards.2,3 Based on this checklist, we came up with 7 key questions to consider when evaluating a noninferiority trial. In the pages that follow, you’ll also find an at-a-glance guide (TABLE) and a methodology review using a hypothetical case (page E7).
1. Is a noninferiority trial appropriate?
The introduction to a noninferiority trial should provide the rationale for this design and the absence of a placebo control group. Look for a review of the evidence of the efficacy of the reference treatment that placebo-controlled trials have revealed, along with the effect size. The advantages of the new treatment over the standard treatment—eg, fewer adverse effects, easier administration, or lower cost—should be discussed, as well.
In the Randomized Evaluation of Long-term Anticoagulation Therapy (RE-LY)—a prominent noninferiority trial—investigators compared the standard anticoagulant (warfarin) for patients with atrial fibrillation (AF) at risk of stroke with a new agent, dabigatran.4 In the methods section of the abstract and the statistical analysis section of the main body, the authors clearly indicated that this was a noninferiority trial. They began by referring to the existing evidence of warfarin’s effectiveness, then detailed the qualities that make warfarin cumbersome to use, including the need for frequent laboratory monitoring. This was followed by evidence that many patients stop taking warfarin and that even for those who persist with treatment, adequate anticoagulation is difficult to maintain.
The authors went on to state that because dabigatran requires no long-term monitoring, it is easier to use. Therefore, if dabigatran could be shown to be no worse than warfarin in preventing strokes, it would be a reasonable alternative, leaving no doubt that this was an appropriate noninferiority trial.
2. Is the noninferiority margin based on clinical judgment and statistical reasoning?
The noninferiority margin should be based on clinical judgment as to how effective a new treatment must be in order to be declared not clinically inferior to the standard treatment. This can be based on several factors, including the severity of the outcome and the expected advantages of the new treatment. The margin should also take into account the size of the standard treatment’s effect vs placebo. In RELY, for example, the authors noted that the noninferiority margin was based on the desire to preserve at least 50% of the lower limit of the confidence interval (CI) of warfarin’s estimated effect; this was done using data from a previously published meta-analysis of 6 trials comparing warfarin with placebo for stroke prevention in patients with AF.4-6
3. Are the hypothesis and statistical analysis formulated correctly?
The clinical hypothesis in a noninferiority trial is that the new treatment is not worse than the standard treatment by a prespecified margin; therefore, the statistical null hypothesis to be tested is that the new treatment is worse than the reference treatment by more than that margin. Rejecting a true null hypothesis (for example, because the P value is <.05) is known as a type l error. In this setting, making a type I error would mean accepting a new treatment that is truly worse than the standard by at least the specified margin. Failure to reject a false null hypothesis is known as a type II error, which in this case would mean failing to identify a new treatment that is truly noninferior to the standard.7
In RE-LY, the authors stated that the upper limit of the one-sided 97.5% CI for the relative risk of a stroke with dabigatran vs warfarin had to fall below 1.46.4 (This is the same as testing the null hypothesis that the hazard ratio is ≥1.46.) Thus, the hypothesis was formulated correctly.
4. Is the sample size appropriate and justified?
The sample size in a noninferiority trial should provide high power to reject the null hypothesis that the difference (or relative risk) between groups is equal to or greater than the noninferiority margin under some clinically meaningful assumption about the true difference (or absolute risk reduction) between groups. A true difference of 0 (or a relative risk of 1) is typically assumed for sample size calculation. However, assuming that the new treatment is truly slightly better or slightly worse than the standard may be clinically appropriate in some cases. This would indicate a need for a smaller or larger sample size, respectively, than that required under the usual assumption of no difference.
When the justification for the sample size in a noninferiority trial is not provided or the number of participants is based on an inappropriate approach (eg, using superiority trial calculations for a noninferiority trial), questions about the quality of the trial arise. The primary concern is whether the noninferiority margin was actually selected before the trial began, as it should have been. And if the researchers used overly optimistic assumptions about the efficacy of the new treatment relative to the standard therapy, the failure to rule out the margin could be misleading. (As with superiority trials that fail to reject the null hypothesis, post hoc power calculations should be avoided.) After the study has ended, the resulting CIs should be used to evaluate whether the study was large enough to adequately assess the relative effectiveness of the treatments.
The RE-LY trial calculated the sample size that was expected to provide 84% power to rule out the prespecified hazard ratio of 1.46, assuming a true event rate of 1.6% per year (presumably for both groups), a recruitment period of 2 years, and at least one year of follow-up. The sample size was subsequently increased from 15,000 to 18,000 to maintain power in case of a low event rate.4,5
5. Is the noninferiority trial as similar as possible to the trial(s) comparing the standard treatment with placebo?
Characteristics of participants, setting, reference treatment, and outcomes used in a noninferiority trial should be as close as possible to those in the trial(s) comparing the treatment with placebo. This is known as the constancy assumption, and it is key to researchers’ ability to draw a conclusion about noninferiority.
The trials used to calculate the noninferiority margin and the RE-LY trial itself involved similar populations of patients with AF, and the outcome (stroke) was similar.
6. Is a per protocol analysis reported in the results?
In randomized controlled superiority trials, the participants should be analyzed in the groups to which they were originally allocated, regardless of whether they adhered to treatment during the entire follow-up period. Such intention-to-treat (ITT) analysis is important because it provides a more conservative estimate of treatment effect—taking into account that some people who are offered treatment will not accept it and others will discontinue treatment. An ITT analysis therefore tends to minimize treatment effects compared with a “per protocol” analysis, in which participants are analyzed according to the treatment they actually received and are often removed from the analysis if they discontinue or do not adhere to treatment.
In noninferiority trials, if patients in the intervention group cross over to the standard treatment group or those in the standard treatment group have poor adherence, an ITT analysis can increase the risk of wrongly claiming noninferiority.7 Therefore, a per protocol analysis should be included—and indeed may be preferable.
In RE-LY, ITT analyses were reported, and complete follow-up data were available for 99.9% of patients. However, the rates of treatment discontinuation at one year were about 15% for those on dabigatran and 10% for the warfarin group, and 21% and 17%, respectively, at 2 years.4,5 If the new treatment were truly less efficacious than the standard treatment, these moderate discontinuation rates could lead to more similar rates of stroke in the 2 groups than would be expected with higher continuation rates, biasing results towards the alternative of noninferiority. Although the original publication of trial results did not include a per protocol analysis, the RE-LY authors later reported that a per protocol analysis yielded similar results to the ITT analysis.
7. Are the overall design and execution of the trial high quality?
Because a poor quality noninferiority trial can appear to demonstrate noninferiority, looking at such studies critically is crucial. Appropriate randomization, concealed allocation, masking, and careful attention to participant flow must all be assessed.2,3
To continue with our example, the RE-LY trial was well conducted. Randomization was performed centrally via an automated telephone system and 2 doses of dabigatran were administered in a masked fashion, while warfarin was open-label. Remarkably, follow-up was achieved for 99.9% of participants over a median of 2 years, and independent adjudicators masked to treatment group assessed outcomes.4,5
CORRESPONDENCE
Anne Mounsey, MD, UNC Chapel Hill Department of Family Medicine, 590 Manning Drive, CB 7595, Chapel Hill, NC 27590; [email protected]
The traditional clinical trial, designed to test whether a new treatment is better than a placebo or another active treatment, is known as a “superiority” trial—although rarely labeled as such. In contrast, the goal of a noninferiority trial is simply to demonstrate that a new treatment is not substantially less effective than the standard therapy.
Such trials are useful when a new therapy is thought to be safer, easier to administer, or less costly than the existing treatment, but not necessarily more effective. And, because it would be unethical to randomize patients with a serious condition for which there already is an effective treatment to placebo, a noninferiority trial is another means of determining if the new treatment is effective.
Noninferiority trials have unique design features and methodology and require a different analysis than traditional superiority trials. Yet many physicians know far less about them; many investigators appear to be less than proficient, as well. A review of 116 noninferiority trials and 46 equivalence trials found that only 20% fulfilled generally accepted quality criteria.1 To improve the quality of noninferiority trials, the CONSORT (Consolidated Standards of Reporting Trials) Group has published a checklist for trial design and reporting standards.2,3 Based on this checklist, we came up with 7 key questions to consider when evaluating a noninferiority trial. In the pages that follow, you’ll also find an at-a-glance guide (TABLE) and a methodology review using a hypothetical case (page E7).
1. Is a noninferiority trial appropriate?
The introduction to a noninferiority trial should provide the rationale for this design and the absence of a placebo control group. Look for a review of the evidence of the efficacy of the reference treatment that placebo-controlled trials have revealed, along with the effect size. The advantages of the new treatment over the standard treatment—eg, fewer adverse effects, easier administration, or lower cost—should be discussed, as well.
In the Randomized Evaluation of Long-term Anticoagulation Therapy (RE-LY)—a prominent noninferiority trial—investigators compared the standard anticoagulant (warfarin) for patients with atrial fibrillation (AF) at risk of stroke with a new agent, dabigatran.4 In the methods section of the abstract and the statistical analysis section of the main body, the authors clearly indicated that this was a noninferiority trial. They began by referring to the existing evidence of warfarin’s effectiveness, then detailed the qualities that make warfarin cumbersome to use, including the need for frequent laboratory monitoring. This was followed by evidence that many patients stop taking warfarin and that even for those who persist with treatment, adequate anticoagulation is difficult to maintain.
The authors went on to state that because dabigatran requires no long-term monitoring, it is easier to use. Therefore, if dabigatran could be shown to be no worse than warfarin in preventing strokes, it would be a reasonable alternative, leaving no doubt that this was an appropriate noninferiority trial.
2. Is the noninferiority margin based on clinical judgment and statistical reasoning?
The noninferiority margin should be based on clinical judgment as to how effective a new treatment must be in order to be declared not clinically inferior to the standard treatment. This can be based on several factors, including the severity of the outcome and the expected advantages of the new treatment. The margin should also take into account the size of the standard treatment’s effect vs placebo. In RELY, for example, the authors noted that the noninferiority margin was based on the desire to preserve at least 50% of the lower limit of the confidence interval (CI) of warfarin’s estimated effect; this was done using data from a previously published meta-analysis of 6 trials comparing warfarin with placebo for stroke prevention in patients with AF.4-6
3. Are the hypothesis and statistical analysis formulated correctly?
The clinical hypothesis in a noninferiority trial is that the new treatment is not worse than the standard treatment by a prespecified margin; therefore, the statistical null hypothesis to be tested is that the new treatment is worse than the reference treatment by more than that margin. Rejecting a true null hypothesis (for example, because the P value is <.05) is known as a type l error. In this setting, making a type I error would mean accepting a new treatment that is truly worse than the standard by at least the specified margin. Failure to reject a false null hypothesis is known as a type II error, which in this case would mean failing to identify a new treatment that is truly noninferior to the standard.7
In RE-LY, the authors stated that the upper limit of the one-sided 97.5% CI for the relative risk of a stroke with dabigatran vs warfarin had to fall below 1.46.4 (This is the same as testing the null hypothesis that the hazard ratio is ≥1.46.) Thus, the hypothesis was formulated correctly.
4. Is the sample size appropriate and justified?
The sample size in a noninferiority trial should provide high power to reject the null hypothesis that the difference (or relative risk) between groups is equal to or greater than the noninferiority margin under some clinically meaningful assumption about the true difference (or absolute risk reduction) between groups. A true difference of 0 (or a relative risk of 1) is typically assumed for sample size calculation. However, assuming that the new treatment is truly slightly better or slightly worse than the standard may be clinically appropriate in some cases. This would indicate a need for a smaller or larger sample size, respectively, than that required under the usual assumption of no difference.
When the justification for the sample size in a noninferiority trial is not provided or the number of participants is based on an inappropriate approach (eg, using superiority trial calculations for a noninferiority trial), questions about the quality of the trial arise. The primary concern is whether the noninferiority margin was actually selected before the trial began, as it should have been. And if the researchers used overly optimistic assumptions about the efficacy of the new treatment relative to the standard therapy, the failure to rule out the margin could be misleading. (As with superiority trials that fail to reject the null hypothesis, post hoc power calculations should be avoided.) After the study has ended, the resulting CIs should be used to evaluate whether the study was large enough to adequately assess the relative effectiveness of the treatments.
The RE-LY trial calculated the sample size that was expected to provide 84% power to rule out the prespecified hazard ratio of 1.46, assuming a true event rate of 1.6% per year (presumably for both groups), a recruitment period of 2 years, and at least one year of follow-up. The sample size was subsequently increased from 15,000 to 18,000 to maintain power in case of a low event rate.4,5
5. Is the noninferiority trial as similar as possible to the trial(s) comparing the standard treatment with placebo?
Characteristics of participants, setting, reference treatment, and outcomes used in a noninferiority trial should be as close as possible to those in the trial(s) comparing the treatment with placebo. This is known as the constancy assumption, and it is key to researchers’ ability to draw a conclusion about noninferiority.
The trials used to calculate the noninferiority margin and the RE-LY trial itself involved similar populations of patients with AF, and the outcome (stroke) was similar.
6. Is a per protocol analysis reported in the results?
In randomized controlled superiority trials, the participants should be analyzed in the groups to which they were originally allocated, regardless of whether they adhered to treatment during the entire follow-up period. Such intention-to-treat (ITT) analysis is important because it provides a more conservative estimate of treatment effect—taking into account that some people who are offered treatment will not accept it and others will discontinue treatment. An ITT analysis therefore tends to minimize treatment effects compared with a “per protocol” analysis, in which participants are analyzed according to the treatment they actually received and are often removed from the analysis if they discontinue or do not adhere to treatment.
In noninferiority trials, if patients in the intervention group cross over to the standard treatment group or those in the standard treatment group have poor adherence, an ITT analysis can increase the risk of wrongly claiming noninferiority.7 Therefore, a per protocol analysis should be included—and indeed may be preferable.
In RE-LY, ITT analyses were reported, and complete follow-up data were available for 99.9% of patients. However, the rates of treatment discontinuation at one year were about 15% for those on dabigatran and 10% for the warfarin group, and 21% and 17%, respectively, at 2 years.4,5 If the new treatment were truly less efficacious than the standard treatment, these moderate discontinuation rates could lead to more similar rates of stroke in the 2 groups than would be expected with higher continuation rates, biasing results towards the alternative of noninferiority. Although the original publication of trial results did not include a per protocol analysis, the RE-LY authors later reported that a per protocol analysis yielded similar results to the ITT analysis.
7. Are the overall design and execution of the trial high quality?
Because a poor quality noninferiority trial can appear to demonstrate noninferiority, looking at such studies critically is crucial. Appropriate randomization, concealed allocation, masking, and careful attention to participant flow must all be assessed.2,3
To continue with our example, the RE-LY trial was well conducted. Randomization was performed centrally via an automated telephone system and 2 doses of dabigatran were administered in a masked fashion, while warfarin was open-label. Remarkably, follow-up was achieved for 99.9% of participants over a median of 2 years, and independent adjudicators masked to treatment group assessed outcomes.4,5
CORRESPONDENCE
Anne Mounsey, MD, UNC Chapel Hill Department of Family Medicine, 590 Manning Drive, CB 7595, Chapel Hill, NC 27590; [email protected]
1. Le Henanff A, Giraudeau B, Baron G, et al. Quality of reporting of noninferiority and equivalence randomized trials. JAMA. 2006;295:1147-1151.
2. Piaggio G, Elbourne DR, Pocock SJ, et al; CONSORT Group. Reporting of noninferiority and equivalence randomized trials: extension of the CONSORT 2010 statement. JAMA. 2012;308:2594-2604.
3. Moher D, Schulz KF, Altman D; CONSORT Group (Consolidated Standards of Reporting Trials). The CONSORT statement: revised recommendations for improving the quality of reports of parallel-group randomized trials. JAMA. 2001;285:1987-1991.
4. Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med. 2009;361:1139-1151.
5. Ezekowitz MD, Connolly S, Parekh A, et al. Rationale and design of RE-LY: randomized evaluation of long-term anticoagulant therapy, warfarin, compared with dabigatran. Am Heart J. 2009;157:805-810, 810.e1-2.
6. Hart RG, Benavente O, McBride R, et al. Antithrombotic therapy to prevent stroke in patients with atrial fibrillation: a meta-analysis. Ann Intern Med. 1999;131:492-501.
7. US Department of Health and Human Services. Guidance for industry non-inferiority clinical trials. US Food and Drug Administration Web site. March 2010. Available at: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM202140.pdf. Accessed February 4, 2014.
1. Le Henanff A, Giraudeau B, Baron G, et al. Quality of reporting of noninferiority and equivalence randomized trials. JAMA. 2006;295:1147-1151.
2. Piaggio G, Elbourne DR, Pocock SJ, et al; CONSORT Group. Reporting of noninferiority and equivalence randomized trials: extension of the CONSORT 2010 statement. JAMA. 2012;308:2594-2604.
3. Moher D, Schulz KF, Altman D; CONSORT Group (Consolidated Standards of Reporting Trials). The CONSORT statement: revised recommendations for improving the quality of reports of parallel-group randomized trials. JAMA. 2001;285:1987-1991.
4. Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med. 2009;361:1139-1151.
5. Ezekowitz MD, Connolly S, Parekh A, et al. Rationale and design of RE-LY: randomized evaluation of long-term anticoagulant therapy, warfarin, compared with dabigatran. Am Heart J. 2009;157:805-810, 810.e1-2.
6. Hart RG, Benavente O, McBride R, et al. Antithrombotic therapy to prevent stroke in patients with atrial fibrillation: a meta-analysis. Ann Intern Med. 1999;131:492-501.
7. US Department of Health and Human Services. Guidance for industry non-inferiority clinical trials. US Food and Drug Administration Web site. March 2010. Available at: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM202140.pdf. Accessed February 4, 2014.

When You Suspect ACS, Which Serologic Marker Is Best?
Measurement of troponin levels provides the most sensitive and accurate serologic information in evaluating a patient with acute coronary syndrome (ACS); troponin elevations are more sensitive than elevations of creatine kinase-MB (CK-MB). Isolated elevation of troponin levels increases the likelihood of myocardial infarction (MI) or death, whereas isolated elevation of CK-MB levels doesn’t. (Strength of recommendation [SOR] for all statements: A, multiple, large prospective cohort studies.)
Repeated measurement of troponin levels at presentation and then 3 and 6 hours afterward increases the diagnostic sensitivity for acute myocardial infarction (AMI) (SOR: A, multiple, small prospective studies).
EVIDENCE SUMMARY
Troponin I and T proteins are specific to cardiac myocytes and, unlike CK-MB, aren’t elevated by damage to skeletal muscle.
Measuring troponin levels increased the number of patients diagnosed with AMI
A multinational prospective cohort study of patients with suspected ACS (N=10,719) found that measuring troponin levels in addition to CK-MB levels improved the diagnosis of AMI.1 Investigators used elevation of any biomarker (CK, CK-MB, or troponin I or T) above the upper limit of normal as their diagnostic criterion. They found that measuring troponin increased the number of patients diagnosed with AMI by 10.4% over patients diagnosed using CK and CK-MB levels. Elevated troponin levels were associated with an inpatient mortality rate 1.5 to 3 times higher, regardless of the patient’s CK-MB status.
Troponin levels are more sensitive and specific than CK-MB
A prospective cohort study of 718 patients with suspected AMI calculated the area under curve (AUC) of the receiver operator curve—a measure of diagnostic accuracy in which an AUC value of 1 indicates 100% sensitivity and specificity—for troponin and CK-MB levels at initial presentation.2 Two independent cardiologists reviewed all available medical records and made the final diagnosis. The AUCs for troponin levels ranged from 0.94 to 0.96 compared with 0.88 for CK-MB.
Troponin levels and odds of MI or death
A prospective study of 1852 patients with suspected ACS from 3 trial populations evaluated the prognostic value of increased troponin levels vs CK-MB levels at initial presentation, compared with a reference group with normal troponin and CK-MB levels.3 Patients with isolated troponin elevation had an increased odds of MI or death at 24 hours (odds ratio [OR]=5.2; 95% confidence interval [CI], 2.2-11.9) and 30 days (OR=2.1; 95% CI, 1.4-3.0), whereas patients with isolated CK-MB elevations didn't. At 30 days, patients with isolated CK-MB elevations equaled the reference group odds for MI and death (OR=1.0; 95% CI, 0.6-1.6).
Serial troponin assessment boosts diagnostic sensitivity
A prospective cohort study found that serial measurements of troponin increased the diagnostic sensitivity for AMI.4 Investigators evaluated 1818 consecutive patients with new onset chest pain in 3 German chest-pain units with troponin levels on admission and at 3 and 6 hours later. The gold standard was diagnosis of AMI by 2 independent cardiologists. Troponin measurement produced an AUC of 0.96 at admission, increasing to 0.98 and 0.99 at 3 and 6 hours after admission, respectively.
Recommendations
The American College of Cardiology and American Heart Association recommend measuring biomarkers of cardiac injury in all patients who present with chest discomfort consistent with ACS.5 A cardiac-specific troponin is the preferred marker and should be measured in all patients. If troponin is not available, CK-MB is the best alternative. Cardiac biomarkers should be repeated 6 to 9 hours after presentation and, in patients with a high clinical suspicion of AMI, at 12 to 24 hours.6,7
1. Goodman SG, Steg PG, Eagle KA, et al; GRACE Investigators. The diagnostic and prognostic impact of the redefinition of acute myocardial infarction: lessons from the global registry of acute coronary events (GRACE). Am Heart J. 2006;151:654-660.
2. Reichlin T, Hochholzer W, Bassetti S, et al. Early diagnosis of myocardial infarction with sensitive cardiac troponin assays. N Engl J Med. 2009;361:858-867.
3. Rao SV, Ohman EM, Granger CB, et al. Prognostic value of isolated troponin elevation across the spectrum of chest pain syndromes. Am J Cardiol. 2003;91:936-940.
4. Keller T, Zeller T, Peetz D, et al. Sensitive troponin I assay in early diagnosis of acute myocardial infarction. N Engl J Med. 2009;361:868-877.
5. Anderson JL, Adams CD, Antman EM, et al. American College of Cardiology, American Heart Association Task Force on Practice Guidelines (Writing Committee, American College of Emergency Physicians, Society for Cardiovascular Angiography and Interventions, Society of Thoracic Surgeons, American Association of Cardiovascular and Pulmonary Rehabilitation, Society for Academic Emergency Medicine). ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction: a report of the American College of Cardiology. J Am Coll Cardiol. 2007;50:e1-e157.
6. Morrow DA, Cannon CP, Jesse RL, et al; National Academy of Clinical Biochemistry. National Academy of Clinical Biochemistry Laboratory Medicine practice guidelines: clinical characteristics and utilization of biochemical markers in acute coronary syndromes. Clin Chem. 2007;53:552-574.
7. Thygesen K, Alpert JS, White HD, et al. Universal definition of myocardial infarction. Circulation. 2007;116:2634-2653.
Measurement of troponin levels provides the most sensitive and accurate serologic information in evaluating a patient with acute coronary syndrome (ACS); troponin elevations are more sensitive than elevations of creatine kinase-MB (CK-MB). Isolated elevation of troponin levels increases the likelihood of myocardial infarction (MI) or death, whereas isolated elevation of CK-MB levels doesn’t. (Strength of recommendation [SOR] for all statements: A, multiple, large prospective cohort studies.)
Repeated measurement of troponin levels at presentation and then 3 and 6 hours afterward increases the diagnostic sensitivity for acute myocardial infarction (AMI) (SOR: A, multiple, small prospective studies).
EVIDENCE SUMMARY
Troponin I and T proteins are specific to cardiac myocytes and, unlike CK-MB, aren’t elevated by damage to skeletal muscle.
Measuring troponin levels increased the number of patients diagnosed with AMI
A multinational prospective cohort study of patients with suspected ACS (N=10,719) found that measuring troponin levels in addition to CK-MB levels improved the diagnosis of AMI.1 Investigators used elevation of any biomarker (CK, CK-MB, or troponin I or T) above the upper limit of normal as their diagnostic criterion. They found that measuring troponin increased the number of patients diagnosed with AMI by 10.4% over patients diagnosed using CK and CK-MB levels. Elevated troponin levels were associated with an inpatient mortality rate 1.5 to 3 times higher, regardless of the patient’s CK-MB status.
Troponin levels are more sensitive and specific than CK-MB
A prospective cohort study of 718 patients with suspected AMI calculated the area under curve (AUC) of the receiver operator curve—a measure of diagnostic accuracy in which an AUC value of 1 indicates 100% sensitivity and specificity—for troponin and CK-MB levels at initial presentation.2 Two independent cardiologists reviewed all available medical records and made the final diagnosis. The AUCs for troponin levels ranged from 0.94 to 0.96 compared with 0.88 for CK-MB.
Troponin levels and odds of MI or death
A prospective study of 1852 patients with suspected ACS from 3 trial populations evaluated the prognostic value of increased troponin levels vs CK-MB levels at initial presentation, compared with a reference group with normal troponin and CK-MB levels.3 Patients with isolated troponin elevation had an increased odds of MI or death at 24 hours (odds ratio [OR]=5.2; 95% confidence interval [CI], 2.2-11.9) and 30 days (OR=2.1; 95% CI, 1.4-3.0), whereas patients with isolated CK-MB elevations didn't. At 30 days, patients with isolated CK-MB elevations equaled the reference group odds for MI and death (OR=1.0; 95% CI, 0.6-1.6).
Serial troponin assessment boosts diagnostic sensitivity
A prospective cohort study found that serial measurements of troponin increased the diagnostic sensitivity for AMI.4 Investigators evaluated 1818 consecutive patients with new onset chest pain in 3 German chest-pain units with troponin levels on admission and at 3 and 6 hours later. The gold standard was diagnosis of AMI by 2 independent cardiologists. Troponin measurement produced an AUC of 0.96 at admission, increasing to 0.98 and 0.99 at 3 and 6 hours after admission, respectively.
Recommendations
The American College of Cardiology and American Heart Association recommend measuring biomarkers of cardiac injury in all patients who present with chest discomfort consistent with ACS.5 A cardiac-specific troponin is the preferred marker and should be measured in all patients. If troponin is not available, CK-MB is the best alternative. Cardiac biomarkers should be repeated 6 to 9 hours after presentation and, in patients with a high clinical suspicion of AMI, at 12 to 24 hours.6,7
Measurement of troponin levels provides the most sensitive and accurate serologic information in evaluating a patient with acute coronary syndrome (ACS); troponin elevations are more sensitive than elevations of creatine kinase-MB (CK-MB). Isolated elevation of troponin levels increases the likelihood of myocardial infarction (MI) or death, whereas isolated elevation of CK-MB levels doesn’t. (Strength of recommendation [SOR] for all statements: A, multiple, large prospective cohort studies.)
Repeated measurement of troponin levels at presentation and then 3 and 6 hours afterward increases the diagnostic sensitivity for acute myocardial infarction (AMI) (SOR: A, multiple, small prospective studies).
EVIDENCE SUMMARY
Troponin I and T proteins are specific to cardiac myocytes and, unlike CK-MB, aren’t elevated by damage to skeletal muscle.
Measuring troponin levels increased the number of patients diagnosed with AMI
A multinational prospective cohort study of patients with suspected ACS (N=10,719) found that measuring troponin levels in addition to CK-MB levels improved the diagnosis of AMI.1 Investigators used elevation of any biomarker (CK, CK-MB, or troponin I or T) above the upper limit of normal as their diagnostic criterion. They found that measuring troponin increased the number of patients diagnosed with AMI by 10.4% over patients diagnosed using CK and CK-MB levels. Elevated troponin levels were associated with an inpatient mortality rate 1.5 to 3 times higher, regardless of the patient’s CK-MB status.
Troponin levels are more sensitive and specific than CK-MB
A prospective cohort study of 718 patients with suspected AMI calculated the area under curve (AUC) of the receiver operator curve—a measure of diagnostic accuracy in which an AUC value of 1 indicates 100% sensitivity and specificity—for troponin and CK-MB levels at initial presentation.2 Two independent cardiologists reviewed all available medical records and made the final diagnosis. The AUCs for troponin levels ranged from 0.94 to 0.96 compared with 0.88 for CK-MB.
Troponin levels and odds of MI or death
A prospective study of 1852 patients with suspected ACS from 3 trial populations evaluated the prognostic value of increased troponin levels vs CK-MB levels at initial presentation, compared with a reference group with normal troponin and CK-MB levels.3 Patients with isolated troponin elevation had an increased odds of MI or death at 24 hours (odds ratio [OR]=5.2; 95% confidence interval [CI], 2.2-11.9) and 30 days (OR=2.1; 95% CI, 1.4-3.0), whereas patients with isolated CK-MB elevations didn't. At 30 days, patients with isolated CK-MB elevations equaled the reference group odds for MI and death (OR=1.0; 95% CI, 0.6-1.6).
Serial troponin assessment boosts diagnostic sensitivity
A prospective cohort study found that serial measurements of troponin increased the diagnostic sensitivity for AMI.4 Investigators evaluated 1818 consecutive patients with new onset chest pain in 3 German chest-pain units with troponin levels on admission and at 3 and 6 hours later. The gold standard was diagnosis of AMI by 2 independent cardiologists. Troponin measurement produced an AUC of 0.96 at admission, increasing to 0.98 and 0.99 at 3 and 6 hours after admission, respectively.
Recommendations
The American College of Cardiology and American Heart Association recommend measuring biomarkers of cardiac injury in all patients who present with chest discomfort consistent with ACS.5 A cardiac-specific troponin is the preferred marker and should be measured in all patients. If troponin is not available, CK-MB is the best alternative. Cardiac biomarkers should be repeated 6 to 9 hours after presentation and, in patients with a high clinical suspicion of AMI, at 12 to 24 hours.6,7
1. Goodman SG, Steg PG, Eagle KA, et al; GRACE Investigators. The diagnostic and prognostic impact of the redefinition of acute myocardial infarction: lessons from the global registry of acute coronary events (GRACE). Am Heart J. 2006;151:654-660.
2. Reichlin T, Hochholzer W, Bassetti S, et al. Early diagnosis of myocardial infarction with sensitive cardiac troponin assays. N Engl J Med. 2009;361:858-867.
3. Rao SV, Ohman EM, Granger CB, et al. Prognostic value of isolated troponin elevation across the spectrum of chest pain syndromes. Am J Cardiol. 2003;91:936-940.
4. Keller T, Zeller T, Peetz D, et al. Sensitive troponin I assay in early diagnosis of acute myocardial infarction. N Engl J Med. 2009;361:868-877.
5. Anderson JL, Adams CD, Antman EM, et al. American College of Cardiology, American Heart Association Task Force on Practice Guidelines (Writing Committee, American College of Emergency Physicians, Society for Cardiovascular Angiography and Interventions, Society of Thoracic Surgeons, American Association of Cardiovascular and Pulmonary Rehabilitation, Society for Academic Emergency Medicine). ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction: a report of the American College of Cardiology. J Am Coll Cardiol. 2007;50:e1-e157.
6. Morrow DA, Cannon CP, Jesse RL, et al; National Academy of Clinical Biochemistry. National Academy of Clinical Biochemistry Laboratory Medicine practice guidelines: clinical characteristics and utilization of biochemical markers in acute coronary syndromes. Clin Chem. 2007;53:552-574.
7. Thygesen K, Alpert JS, White HD, et al. Universal definition of myocardial infarction. Circulation. 2007;116:2634-2653.
1. Goodman SG, Steg PG, Eagle KA, et al; GRACE Investigators. The diagnostic and prognostic impact of the redefinition of acute myocardial infarction: lessons from the global registry of acute coronary events (GRACE). Am Heart J. 2006;151:654-660.
2. Reichlin T, Hochholzer W, Bassetti S, et al. Early diagnosis of myocardial infarction with sensitive cardiac troponin assays. N Engl J Med. 2009;361:858-867.
3. Rao SV, Ohman EM, Granger CB, et al. Prognostic value of isolated troponin elevation across the spectrum of chest pain syndromes. Am J Cardiol. 2003;91:936-940.
4. Keller T, Zeller T, Peetz D, et al. Sensitive troponin I assay in early diagnosis of acute myocardial infarction. N Engl J Med. 2009;361:868-877.
5. Anderson JL, Adams CD, Antman EM, et al. American College of Cardiology, American Heart Association Task Force on Practice Guidelines (Writing Committee, American College of Emergency Physicians, Society for Cardiovascular Angiography and Interventions, Society of Thoracic Surgeons, American Association of Cardiovascular and Pulmonary Rehabilitation, Society for Academic Emergency Medicine). ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction: a report of the American College of Cardiology. J Am Coll Cardiol. 2007;50:e1-e157.
6. Morrow DA, Cannon CP, Jesse RL, et al; National Academy of Clinical Biochemistry. National Academy of Clinical Biochemistry Laboratory Medicine practice guidelines: clinical characteristics and utilization of biochemical markers in acute coronary syndromes. Clin Chem. 2007;53:552-574.
7. Thygesen K, Alpert JS, White HD, et al. Universal definition of myocardial infarction. Circulation. 2007;116:2634-2653.
When you suspect ACS, which serologic marker is best?
Measurement of troponin levels provides the most sensitive and accurate serologic information in evaluating a patient with acute coronary syndrome (ACS); troponin elevations are more sensitive than elevations of creatine kinase-MB (CK-MB). Isolated elevation of troponin levels increases the likelihood of myocardial infarction (MI) or death, whereas isolated elevation of CK-MB levels doesn’t. (Strength of recommendation [SOR] for all statements: A, multiple, large prospective cohort studies.)
Repeated measurement of troponin levels at presentation and then 3 and 6 hours afterward increases the diagnostic sensitivity for acute myocardial infarction (AMI) (SOR: A, multiple, small prospective studies).
EVIDENCE SUMMARY
Troponin I and T proteins are specific to cardiac myocytes and, unlike CK-MB, aren’t elevated by damage to skeletal muscle.
Measuring troponin levels increased the number of patients diagnosed with AMI
A multinational prospective cohort study of patients with suspected ACS (N=10,719) found that measuring troponin levels in addition to CK-MB levels improved the diagnosis of AMI.1 Investigators used elevation of any biomarker (CK, CK-MB, or troponin I or T) above the upper limit of normal as their diagnostic criterion. They found that measuring troponin increased the number of patients diagnosed with AMI by 10.4% over patients diagnosed using CK and CK-MB levels. Elevated troponin levels were associated with an inpatient mortality rate 1.5 to 3 times higher, regardless of the patient’s CK-MB status.
Troponin levels are more sensitive and specific than CK-MB
A prospective cohort study of 718 patients with suspected AMI calculated the area under curve (AUC) of the receiver operator curve—a measure of diagnostic accuracy in which an AUC value of 1 indicates 100% sensitivity and specificity—for troponin and CK-MB levels at initial presentation.2 Two independent cardiologists reviewed all available medical records and made the final diagnosis. The AUCs for troponin levels ranged from 0.94 to 0.96 compared with 0.88 for CK-MB.
Troponin levels and odds of MI or death
A prospective study of 1852 patients with suspected ACS from 3 trial populations evaluated the prognostic value of increased troponin levels vs CK-MB levels at initial presentation, compared with a reference group with normal troponin and CK-MB levels.3 Patients with isolated troponin elevation had an increased odds of MI or death at 24 hours (odds ratio [OR]=5.2; 95% confidence interval [CI], 2.2-11.9) and 30 days (OR=2.1; 95% CI, 1.4-3.0), whereas patients with isolated CK-MB elevations didn't. At 30 days, patients with isolated CK-MB elevations equaled the reference group odds for MI and death (OR=1.0; 95% CI, 0.6-1.6).
Serial troponin assessment boosts diagnostic sensitivity
A prospective cohort study found that serial measurements of troponin increased the diagnostic sensitivity for AMI.4 Investigators evaluated 1818 consecutive patients with new onset chest pain in 3 German chest-pain units with troponin levels on admission and at 3 and 6 hours later. The gold standard was diagnosis of AMI by 2 independent cardiologists. Troponin measurement produced an AUC of 0.96 at admission, increasing to 0.98 and 0.99 at 3 and 6 hours after admission, respectively.
RECOMMENDATIONS
The American College of Cardiology and American Heart Association recommend measuring biomarkers of cardiac injury in all patients who present with chest discomfort consistent with ACS.5 A cardiac-specific troponin is the preferred marker and should be measured in all patients. If troponin is not available, CK-MB is the best alternative. Cardiac biomarkers should be repeated 6 to 9 hours after presentation and, in patients with a high clinical suspicion of AMI, at 12 to 24 hours.6,7
1. Goodman SG, Steg PG, Eagle KA, et al; GRACE Investigators. The diagnostic and prognostic impact of the redefinition of acute myocardial infarction: lessons from the global registry of acute coronary events (GRACE). Am Heart J. 2006;151:654-660.
2. Reichlin T, Hochholzer W, Bassetti S, et al. Early diagnosis of myocardial infarction with sensitive cardiac troponin assays. N Engl J Med. 2009;361:858-867.
3. Rao SV, Ohman EM, Granger CB, et al. Prognostic value of isolated troponin elevation across the spectrum of chest pain syndromes. Am J Cardiol. 2003;91:936-940.
4. Keller T, Zeller T, Peetz D, et al. Sensitive troponin I assay in early diagnosis of acute myocardial infarction. N Engl J Med. 2009;361:868-877.
5. Anderson JL, Adams CD, Antman EM, et al. American College of Cardiology, American Heart Association Task Force on Practice Guidelines (Writing Committee, American College of Emergency Physicians, Society for Cardiovascular Angiography and Interventions, Society of Thoracic Surgeons, American Association of Cardiovascular and Pulmonary Rehabilitation, Society for Academic Emergency Medicine). ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction: a report of the American College of Cardiology. J Am Coll Cardiol. 2007;50:e1-e157.
6. Morrow DA, Cannon CP, Jesse RL, et al; National Academy of Clinical Biochemistry. National Academy of Clinical Biochemistry Laboratory Medicine practice guidelines: clinical characteristics and utilization of biochemical markers in acute coronary syndromes. Clin Chem. 2007;53:552-574.
7. Thygesen K, Alpert JS, White HD, et al. Universal definition of myocardial infarction. Circulation. 2007;116:2634-2653.
Measurement of troponin levels provides the most sensitive and accurate serologic information in evaluating a patient with acute coronary syndrome (ACS); troponin elevations are more sensitive than elevations of creatine kinase-MB (CK-MB). Isolated elevation of troponin levels increases the likelihood of myocardial infarction (MI) or death, whereas isolated elevation of CK-MB levels doesn’t. (Strength of recommendation [SOR] for all statements: A, multiple, large prospective cohort studies.)
Repeated measurement of troponin levels at presentation and then 3 and 6 hours afterward increases the diagnostic sensitivity for acute myocardial infarction (AMI) (SOR: A, multiple, small prospective studies).
EVIDENCE SUMMARY
Troponin I and T proteins are specific to cardiac myocytes and, unlike CK-MB, aren’t elevated by damage to skeletal muscle.
Measuring troponin levels increased the number of patients diagnosed with AMI
A multinational prospective cohort study of patients with suspected ACS (N=10,719) found that measuring troponin levels in addition to CK-MB levels improved the diagnosis of AMI.1 Investigators used elevation of any biomarker (CK, CK-MB, or troponin I or T) above the upper limit of normal as their diagnostic criterion. They found that measuring troponin increased the number of patients diagnosed with AMI by 10.4% over patients diagnosed using CK and CK-MB levels. Elevated troponin levels were associated with an inpatient mortality rate 1.5 to 3 times higher, regardless of the patient’s CK-MB status.
Troponin levels are more sensitive and specific than CK-MB
A prospective cohort study of 718 patients with suspected AMI calculated the area under curve (AUC) of the receiver operator curve—a measure of diagnostic accuracy in which an AUC value of 1 indicates 100% sensitivity and specificity—for troponin and CK-MB levels at initial presentation.2 Two independent cardiologists reviewed all available medical records and made the final diagnosis. The AUCs for troponin levels ranged from 0.94 to 0.96 compared with 0.88 for CK-MB.
Troponin levels and odds of MI or death
A prospective study of 1852 patients with suspected ACS from 3 trial populations evaluated the prognostic value of increased troponin levels vs CK-MB levels at initial presentation, compared with a reference group with normal troponin and CK-MB levels.3 Patients with isolated troponin elevation had an increased odds of MI or death at 24 hours (odds ratio [OR]=5.2; 95% confidence interval [CI], 2.2-11.9) and 30 days (OR=2.1; 95% CI, 1.4-3.0), whereas patients with isolated CK-MB elevations didn't. At 30 days, patients with isolated CK-MB elevations equaled the reference group odds for MI and death (OR=1.0; 95% CI, 0.6-1.6).
Serial troponin assessment boosts diagnostic sensitivity
A prospective cohort study found that serial measurements of troponin increased the diagnostic sensitivity for AMI.4 Investigators evaluated 1818 consecutive patients with new onset chest pain in 3 German chest-pain units with troponin levels on admission and at 3 and 6 hours later. The gold standard was diagnosis of AMI by 2 independent cardiologists. Troponin measurement produced an AUC of 0.96 at admission, increasing to 0.98 and 0.99 at 3 and 6 hours after admission, respectively.
RECOMMENDATIONS
The American College of Cardiology and American Heart Association recommend measuring biomarkers of cardiac injury in all patients who present with chest discomfort consistent with ACS.5 A cardiac-specific troponin is the preferred marker and should be measured in all patients. If troponin is not available, CK-MB is the best alternative. Cardiac biomarkers should be repeated 6 to 9 hours after presentation and, in patients with a high clinical suspicion of AMI, at 12 to 24 hours.6,7
Measurement of troponin levels provides the most sensitive and accurate serologic information in evaluating a patient with acute coronary syndrome (ACS); troponin elevations are more sensitive than elevations of creatine kinase-MB (CK-MB). Isolated elevation of troponin levels increases the likelihood of myocardial infarction (MI) or death, whereas isolated elevation of CK-MB levels doesn’t. (Strength of recommendation [SOR] for all statements: A, multiple, large prospective cohort studies.)
Repeated measurement of troponin levels at presentation and then 3 and 6 hours afterward increases the diagnostic sensitivity for acute myocardial infarction (AMI) (SOR: A, multiple, small prospective studies).
EVIDENCE SUMMARY
Troponin I and T proteins are specific to cardiac myocytes and, unlike CK-MB, aren’t elevated by damage to skeletal muscle.
Measuring troponin levels increased the number of patients diagnosed with AMI
A multinational prospective cohort study of patients with suspected ACS (N=10,719) found that measuring troponin levels in addition to CK-MB levels improved the diagnosis of AMI.1 Investigators used elevation of any biomarker (CK, CK-MB, or troponin I or T) above the upper limit of normal as their diagnostic criterion. They found that measuring troponin increased the number of patients diagnosed with AMI by 10.4% over patients diagnosed using CK and CK-MB levels. Elevated troponin levels were associated with an inpatient mortality rate 1.5 to 3 times higher, regardless of the patient’s CK-MB status.
Troponin levels are more sensitive and specific than CK-MB
A prospective cohort study of 718 patients with suspected AMI calculated the area under curve (AUC) of the receiver operator curve—a measure of diagnostic accuracy in which an AUC value of 1 indicates 100% sensitivity and specificity—for troponin and CK-MB levels at initial presentation.2 Two independent cardiologists reviewed all available medical records and made the final diagnosis. The AUCs for troponin levels ranged from 0.94 to 0.96 compared with 0.88 for CK-MB.
Troponin levels and odds of MI or death
A prospective study of 1852 patients with suspected ACS from 3 trial populations evaluated the prognostic value of increased troponin levels vs CK-MB levels at initial presentation, compared with a reference group with normal troponin and CK-MB levels.3 Patients with isolated troponin elevation had an increased odds of MI or death at 24 hours (odds ratio [OR]=5.2; 95% confidence interval [CI], 2.2-11.9) and 30 days (OR=2.1; 95% CI, 1.4-3.0), whereas patients with isolated CK-MB elevations didn't. At 30 days, patients with isolated CK-MB elevations equaled the reference group odds for MI and death (OR=1.0; 95% CI, 0.6-1.6).
Serial troponin assessment boosts diagnostic sensitivity
A prospective cohort study found that serial measurements of troponin increased the diagnostic sensitivity for AMI.4 Investigators evaluated 1818 consecutive patients with new onset chest pain in 3 German chest-pain units with troponin levels on admission and at 3 and 6 hours later. The gold standard was diagnosis of AMI by 2 independent cardiologists. Troponin measurement produced an AUC of 0.96 at admission, increasing to 0.98 and 0.99 at 3 and 6 hours after admission, respectively.
RECOMMENDATIONS
The American College of Cardiology and American Heart Association recommend measuring biomarkers of cardiac injury in all patients who present with chest discomfort consistent with ACS.5 A cardiac-specific troponin is the preferred marker and should be measured in all patients. If troponin is not available, CK-MB is the best alternative. Cardiac biomarkers should be repeated 6 to 9 hours after presentation and, in patients with a high clinical suspicion of AMI, at 12 to 24 hours.6,7
1. Goodman SG, Steg PG, Eagle KA, et al; GRACE Investigators. The diagnostic and prognostic impact of the redefinition of acute myocardial infarction: lessons from the global registry of acute coronary events (GRACE). Am Heart J. 2006;151:654-660.
2. Reichlin T, Hochholzer W, Bassetti S, et al. Early diagnosis of myocardial infarction with sensitive cardiac troponin assays. N Engl J Med. 2009;361:858-867.
3. Rao SV, Ohman EM, Granger CB, et al. Prognostic value of isolated troponin elevation across the spectrum of chest pain syndromes. Am J Cardiol. 2003;91:936-940.
4. Keller T, Zeller T, Peetz D, et al. Sensitive troponin I assay in early diagnosis of acute myocardial infarction. N Engl J Med. 2009;361:868-877.
5. Anderson JL, Adams CD, Antman EM, et al. American College of Cardiology, American Heart Association Task Force on Practice Guidelines (Writing Committee, American College of Emergency Physicians, Society for Cardiovascular Angiography and Interventions, Society of Thoracic Surgeons, American Association of Cardiovascular and Pulmonary Rehabilitation, Society for Academic Emergency Medicine). ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction: a report of the American College of Cardiology. J Am Coll Cardiol. 2007;50:e1-e157.
6. Morrow DA, Cannon CP, Jesse RL, et al; National Academy of Clinical Biochemistry. National Academy of Clinical Biochemistry Laboratory Medicine practice guidelines: clinical characteristics and utilization of biochemical markers in acute coronary syndromes. Clin Chem. 2007;53:552-574.
7. Thygesen K, Alpert JS, White HD, et al. Universal definition of myocardial infarction. Circulation. 2007;116:2634-2653.
1. Goodman SG, Steg PG, Eagle KA, et al; GRACE Investigators. The diagnostic and prognostic impact of the redefinition of acute myocardial infarction: lessons from the global registry of acute coronary events (GRACE). Am Heart J. 2006;151:654-660.
2. Reichlin T, Hochholzer W, Bassetti S, et al. Early diagnosis of myocardial infarction with sensitive cardiac troponin assays. N Engl J Med. 2009;361:858-867.
3. Rao SV, Ohman EM, Granger CB, et al. Prognostic value of isolated troponin elevation across the spectrum of chest pain syndromes. Am J Cardiol. 2003;91:936-940.
4. Keller T, Zeller T, Peetz D, et al. Sensitive troponin I assay in early diagnosis of acute myocardial infarction. N Engl J Med. 2009;361:868-877.
5. Anderson JL, Adams CD, Antman EM, et al. American College of Cardiology, American Heart Association Task Force on Practice Guidelines (Writing Committee, American College of Emergency Physicians, Society for Cardiovascular Angiography and Interventions, Society of Thoracic Surgeons, American Association of Cardiovascular and Pulmonary Rehabilitation, Society for Academic Emergency Medicine). ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction: a report of the American College of Cardiology. J Am Coll Cardiol. 2007;50:e1-e157.
6. Morrow DA, Cannon CP, Jesse RL, et al; National Academy of Clinical Biochemistry. National Academy of Clinical Biochemistry Laboratory Medicine practice guidelines: clinical characteristics and utilization of biochemical markers in acute coronary syndromes. Clin Chem. 2007;53:552-574.
7. Thygesen K, Alpert JS, White HD, et al. Universal definition of myocardial infarction. Circulation. 2007;116:2634-2653.
Evidence-based answers from the Family Physicians Inquiries Network