User login
Upgrades to our CME test and to www.ccjm.org

As of this month, when you visit us at www.ccjm.org, you will notice some enhancements to our free CME test. Before taking the test you will log on to the Cleveland Clinic “myCME” system with a user name and a password. If you have taken a CCJM CME test since January 2004, we have already set up an account for most of you. Just click on a test link and follow the instructions. New users can quickly open an account. This means that to take the test you will first have to enter your user name and password. But it ultimately means less work: upon completing a test you will no longer have to enter your name and other information each time you take a different test. Plus, you can access your Cleveland Clinic myCME account at any time to see how many credits you have accrued, or to print out a duplicate certificate (as documentation if you are audited).
Later this year, we will revamp the CME test. Questions will be case-based and will be more challenging and more clinically relevant, and you will receive feedback about why incorrect answers are incorrect. We also plan to offer more credit hours via different online testing formats.
Finally, this fall we will launch a completely revamped Web site, in partnership with High Wire Press of Stanford University. High Wire currently hosts a number of highly visible journals, including the New England Journal of Medicine, JAMA, and Science. Our association with High Wire will provide our readers with a number of benefits, including free access to full-text versions of references from High Wire journals cited in the CCJM and enhanced search functionality. We will also finally have our articles available not only in PDF format, but also in HTML format, which is easier to read online. You will be able to forward articles of interest via e-mail, you can sign up for an e-mail alert when each new issue is published, and you can download citations to a Web-based citation management program.
These will be just the first of many improvements to www.ccjm.org. We expect that these online changes will enhance your educational interactions with the Journal. As always, we welcome your feedback.
As of this month, when you visit us at www.ccjm.org, you will notice some enhancements to our free CME test. Before taking the test you will log on to the Cleveland Clinic “myCME” system with a user name and a password. If you have taken a CCJM CME test since January 2004, we have already set up an account for most of you. Just click on a test link and follow the instructions. New users can quickly open an account. This means that to take the test you will first have to enter your user name and password. But it ultimately means less work: upon completing a test you will no longer have to enter your name and other information each time you take a different test. Plus, you can access your Cleveland Clinic myCME account at any time to see how many credits you have accrued, or to print out a duplicate certificate (as documentation if you are audited).
Later this year, we will revamp the CME test. Questions will be case-based and will be more challenging and more clinically relevant, and you will receive feedback about why incorrect answers are incorrect. We also plan to offer more credit hours via different online testing formats.
Finally, this fall we will launch a completely revamped Web site, in partnership with High Wire Press of Stanford University. High Wire currently hosts a number of highly visible journals, including the New England Journal of Medicine, JAMA, and Science. Our association with High Wire will provide our readers with a number of benefits, including free access to full-text versions of references from High Wire journals cited in the CCJM and enhanced search functionality. We will also finally have our articles available not only in PDF format, but also in HTML format, which is easier to read online. You will be able to forward articles of interest via e-mail, you can sign up for an e-mail alert when each new issue is published, and you can download citations to a Web-based citation management program.
These will be just the first of many improvements to www.ccjm.org. We expect that these online changes will enhance your educational interactions with the Journal. As always, we welcome your feedback.
As of this month, when you visit us at www.ccjm.org, you will notice some enhancements to our free CME test. Before taking the test you will log on to the Cleveland Clinic “myCME” system with a user name and a password. If you have taken a CCJM CME test since January 2004, we have already set up an account for most of you. Just click on a test link and follow the instructions. New users can quickly open an account. This means that to take the test you will first have to enter your user name and password. But it ultimately means less work: upon completing a test you will no longer have to enter your name and other information each time you take a different test. Plus, you can access your Cleveland Clinic myCME account at any time to see how many credits you have accrued, or to print out a duplicate certificate (as documentation if you are audited).
Later this year, we will revamp the CME test. Questions will be case-based and will be more challenging and more clinically relevant, and you will receive feedback about why incorrect answers are incorrect. We also plan to offer more credit hours via different online testing formats.
Finally, this fall we will launch a completely revamped Web site, in partnership with High Wire Press of Stanford University. High Wire currently hosts a number of highly visible journals, including the New England Journal of Medicine, JAMA, and Science. Our association with High Wire will provide our readers with a number of benefits, including free access to full-text versions of references from High Wire journals cited in the CCJM and enhanced search functionality. We will also finally have our articles available not only in PDF format, but also in HTML format, which is easier to read online. You will be able to forward articles of interest via e-mail, you can sign up for an e-mail alert when each new issue is published, and you can download citations to a Web-based citation management program.
These will be just the first of many improvements to www.ccjm.org. We expect that these online changes will enhance your educational interactions with the Journal. As always, we welcome your feedback.
Despite its treatability, gout remains a problem
In the spirit of full disclosure, I am a nonrecovering goutophile. Recent advances have even furthered my enthusiasm for the study of gout and boosted my optimism for improved management of patients with this disorder.
A curious clinical course that finally yields to insights
For years I have been fascinated by the clinical course of the gout—the explosive onset of attacks coupled with their spontaneous resolution. Attacks have been viewed as a relatively simple response to urate crystals, in contrast to the complex cascades that follow auto-antigen stimulation. Until recently, however, the nuances of the response to urate crystals were poorly defined. Several laboratories have contributed to our understanding of the mechanisms triggering the acute attack via activation by various cytokines of specific intracellular pathways involving inflammasomes. Models to explain the self-resolving nature of the attacks have also been developed.
For our patients, the concept that hyperuricemia plays a direct role in the development of hypertension and the progression of chronic kidney disease has been revitalized. Molecular studies have refined our insights into the renal handling of uric acid; we better understand how estrogen, diabetes, and certain medications affect renal uric acid reabsorption. Epidemiologic analyses, animal models of hyperuricemia, and human inter-ventional studies have reintroduced urate as an etiologic agent in cardiovascular disease.
The development of new agents for the treatment of hyperuricemia (a less immunogenic uricase preparation and a nonpurine inhibitor of xanthine oxidase) has stoked interest in—and funding for—research related to patients with hyperuricemia and gout.
Effective diagnosis and treatment: Achievable but not widespread
Gout can be definitively diagnosed by documenting the presence of urate crystals in the synovial fluid from affected joints. Most attacks of gouty arthritis can be readily and safely treated, assuming that attention is paid to the patient’s comorbid conditions. There is continuing discussion about which gouty patients need to have their urate levels reduced, but once the decision is made to treat, most patients can be effectively managed with urate-lowering therapies that will reduce the frequency of attacks and shrink tophi. So why does gout remain a problem?
Reviews of physician practice patterns and focus-group discussions show that despite the high prevalence of hyperuricemia and gouty arthritis, we do a suboptimal job at managing patients with these conditions. Conversations with clinical rheumatologists reveal the shared perception that gout often is misdiagnosed (or goes undiagnosed) because of failure to examine synovial fluids for crystals. There is an overreliance on serum urate levels to diagnose gout, despite the well-recognized lack of sensitivity and specificity of this measure. Plus, interpretation of the serum urate level is complicated by the fact that laboratory “normal” ranges typically include serum urate values above the biological solubility of urate (∼6.8 mg/dL).
It also seems that even when the decision is made to treat hyperuricemia, treatment is frequently suboptimal because of limited use of appropriate serum urate targets (ie, levels less than ∼6.0 mg/dL), insufficient monitoring of the urate level, and overly conservative drug escalation.
In other words, education in the management of gout and hyperuricemia is sorely needed.
A supplement conceived with nonspecialist feedback
In an effort to understand the difficulties faced by nonspecialists in managing patients with gout and hyperuricemia, I and several other rheumatologists with a special interest in clinical gout and continuing medical education took part in a symposium on this topic in Scottsdale, Ariz., on October 5, 2007. We made presentations to a group of invited internists and family practice physicians and gained feedback from them during breakout sessions that followed our talks.
This supplement is based on the series of formal talks presented at the symposium. The talks were transcribed and the authors developed their transcripts into the articles presented here, taking care to draw on questions and feedback from the breakout sessions to best address the educational needs of nonrheumatologists.
On behalf of my fellow authors, I hope we have succeeded in producing a readable and practical supplement that meets many of those needs and facilitates effective management of our patients with gout and hyperuricemia.
In the spirit of full disclosure, I am a nonrecovering goutophile. Recent advances have even furthered my enthusiasm for the study of gout and boosted my optimism for improved management of patients with this disorder.
A curious clinical course that finally yields to insights
For years I have been fascinated by the clinical course of the gout—the explosive onset of attacks coupled with their spontaneous resolution. Attacks have been viewed as a relatively simple response to urate crystals, in contrast to the complex cascades that follow auto-antigen stimulation. Until recently, however, the nuances of the response to urate crystals were poorly defined. Several laboratories have contributed to our understanding of the mechanisms triggering the acute attack via activation by various cytokines of specific intracellular pathways involving inflammasomes. Models to explain the self-resolving nature of the attacks have also been developed.
For our patients, the concept that hyperuricemia plays a direct role in the development of hypertension and the progression of chronic kidney disease has been revitalized. Molecular studies have refined our insights into the renal handling of uric acid; we better understand how estrogen, diabetes, and certain medications affect renal uric acid reabsorption. Epidemiologic analyses, animal models of hyperuricemia, and human inter-ventional studies have reintroduced urate as an etiologic agent in cardiovascular disease.
The development of new agents for the treatment of hyperuricemia (a less immunogenic uricase preparation and a nonpurine inhibitor of xanthine oxidase) has stoked interest in—and funding for—research related to patients with hyperuricemia and gout.
Effective diagnosis and treatment: Achievable but not widespread
Gout can be definitively diagnosed by documenting the presence of urate crystals in the synovial fluid from affected joints. Most attacks of gouty arthritis can be readily and safely treated, assuming that attention is paid to the patient’s comorbid conditions. There is continuing discussion about which gouty patients need to have their urate levels reduced, but once the decision is made to treat, most patients can be effectively managed with urate-lowering therapies that will reduce the frequency of attacks and shrink tophi. So why does gout remain a problem?
Reviews of physician practice patterns and focus-group discussions show that despite the high prevalence of hyperuricemia and gouty arthritis, we do a suboptimal job at managing patients with these conditions. Conversations with clinical rheumatologists reveal the shared perception that gout often is misdiagnosed (or goes undiagnosed) because of failure to examine synovial fluids for crystals. There is an overreliance on serum urate levels to diagnose gout, despite the well-recognized lack of sensitivity and specificity of this measure. Plus, interpretation of the serum urate level is complicated by the fact that laboratory “normal” ranges typically include serum urate values above the biological solubility of urate (∼6.8 mg/dL).
It also seems that even when the decision is made to treat hyperuricemia, treatment is frequently suboptimal because of limited use of appropriate serum urate targets (ie, levels less than ∼6.0 mg/dL), insufficient monitoring of the urate level, and overly conservative drug escalation.
In other words, education in the management of gout and hyperuricemia is sorely needed.
A supplement conceived with nonspecialist feedback
In an effort to understand the difficulties faced by nonspecialists in managing patients with gout and hyperuricemia, I and several other rheumatologists with a special interest in clinical gout and continuing medical education took part in a symposium on this topic in Scottsdale, Ariz., on October 5, 2007. We made presentations to a group of invited internists and family practice physicians and gained feedback from them during breakout sessions that followed our talks.
This supplement is based on the series of formal talks presented at the symposium. The talks were transcribed and the authors developed their transcripts into the articles presented here, taking care to draw on questions and feedback from the breakout sessions to best address the educational needs of nonrheumatologists.
On behalf of my fellow authors, I hope we have succeeded in producing a readable and practical supplement that meets many of those needs and facilitates effective management of our patients with gout and hyperuricemia.
In the spirit of full disclosure, I am a nonrecovering goutophile. Recent advances have even furthered my enthusiasm for the study of gout and boosted my optimism for improved management of patients with this disorder.
A curious clinical course that finally yields to insights
For years I have been fascinated by the clinical course of the gout—the explosive onset of attacks coupled with their spontaneous resolution. Attacks have been viewed as a relatively simple response to urate crystals, in contrast to the complex cascades that follow auto-antigen stimulation. Until recently, however, the nuances of the response to urate crystals were poorly defined. Several laboratories have contributed to our understanding of the mechanisms triggering the acute attack via activation by various cytokines of specific intracellular pathways involving inflammasomes. Models to explain the self-resolving nature of the attacks have also been developed.
For our patients, the concept that hyperuricemia plays a direct role in the development of hypertension and the progression of chronic kidney disease has been revitalized. Molecular studies have refined our insights into the renal handling of uric acid; we better understand how estrogen, diabetes, and certain medications affect renal uric acid reabsorption. Epidemiologic analyses, animal models of hyperuricemia, and human inter-ventional studies have reintroduced urate as an etiologic agent in cardiovascular disease.
The development of new agents for the treatment of hyperuricemia (a less immunogenic uricase preparation and a nonpurine inhibitor of xanthine oxidase) has stoked interest in—and funding for—research related to patients with hyperuricemia and gout.
Effective diagnosis and treatment: Achievable but not widespread
Gout can be definitively diagnosed by documenting the presence of urate crystals in the synovial fluid from affected joints. Most attacks of gouty arthritis can be readily and safely treated, assuming that attention is paid to the patient’s comorbid conditions. There is continuing discussion about which gouty patients need to have their urate levels reduced, but once the decision is made to treat, most patients can be effectively managed with urate-lowering therapies that will reduce the frequency of attacks and shrink tophi. So why does gout remain a problem?
Reviews of physician practice patterns and focus-group discussions show that despite the high prevalence of hyperuricemia and gouty arthritis, we do a suboptimal job at managing patients with these conditions. Conversations with clinical rheumatologists reveal the shared perception that gout often is misdiagnosed (or goes undiagnosed) because of failure to examine synovial fluids for crystals. There is an overreliance on serum urate levels to diagnose gout, despite the well-recognized lack of sensitivity and specificity of this measure. Plus, interpretation of the serum urate level is complicated by the fact that laboratory “normal” ranges typically include serum urate values above the biological solubility of urate (∼6.8 mg/dL).
It also seems that even when the decision is made to treat hyperuricemia, treatment is frequently suboptimal because of limited use of appropriate serum urate targets (ie, levels less than ∼6.0 mg/dL), insufficient monitoring of the urate level, and overly conservative drug escalation.
In other words, education in the management of gout and hyperuricemia is sorely needed.
A supplement conceived with nonspecialist feedback
In an effort to understand the difficulties faced by nonspecialists in managing patients with gout and hyperuricemia, I and several other rheumatologists with a special interest in clinical gout and continuing medical education took part in a symposium on this topic in Scottsdale, Ariz., on October 5, 2007. We made presentations to a group of invited internists and family practice physicians and gained feedback from them during breakout sessions that followed our talks.
This supplement is based on the series of formal talks presented at the symposium. The talks were transcribed and the authors developed their transcripts into the articles presented here, taking care to draw on questions and feedback from the breakout sessions to best address the educational needs of nonrheumatologists.
On behalf of my fellow authors, I hope we have succeeded in producing a readable and practical supplement that meets many of those needs and facilitates effective management of our patients with gout and hyperuricemia.
Clinical manifestations of hyperuricemia and gout
Uric acid—urate in most physiologic fluids—is an end product of purine degradation. The serum urate level in a given patient is determined by the amount of purines synthesized and ingested, the amount of urate produced from purines, and the amount of uric acid excreted by the kidney (and, to a lesser degree, from the gastrointestinal tract).1 A major source of circulating urate is the metabolized endogenous purine. Renal excretion is likely determined by genetic factors that dictate expression of uric acid transporters, as well as by the presence of organic acids, certain drugs, hormones, and the glomerular filtration rate. A small minority of patients will have increased production of urate as a result of enzymopathies, chronic hemolysis, or rapidly dividing tumors, psoriasis, or other disorders characterized by increased turnover of cells.
Humans do not have a functional enzyme (uricase) to break down urate into allantoin, which is more soluble and readily excreted. There may have been genetic pressures that explain why functional uricase was lost and why humans have relatively high urate levels compared with other species.2 If higher levels of serum urate are clinically detrimental, one would think that humans could have evolved an efficient way to excrete it. Instead, we excrete uric acid inefficiently as a result of active reabsorption in the proximal renal tubule. We have higher levels of serum urate than most other species, and we are predisposed to develop gouty arthritis and perhaps other sequelae of hyperuricemia, including hypertension, the metabolic syndrome, and coronary artery disease.
CLINICALLY SIGNIFICANT HYPERURICEMIA VS LAB-DEFINED HYPERURICEMIA
Clinically significant hyperuricemia is a serum urate level greater than 6.8 mg/dL, although the population-defined “normal” urate level indicated by the clinical laboratory is higher. At levels above 6.8 mg/dL, urate exceeds its solubility in most biological fluids.
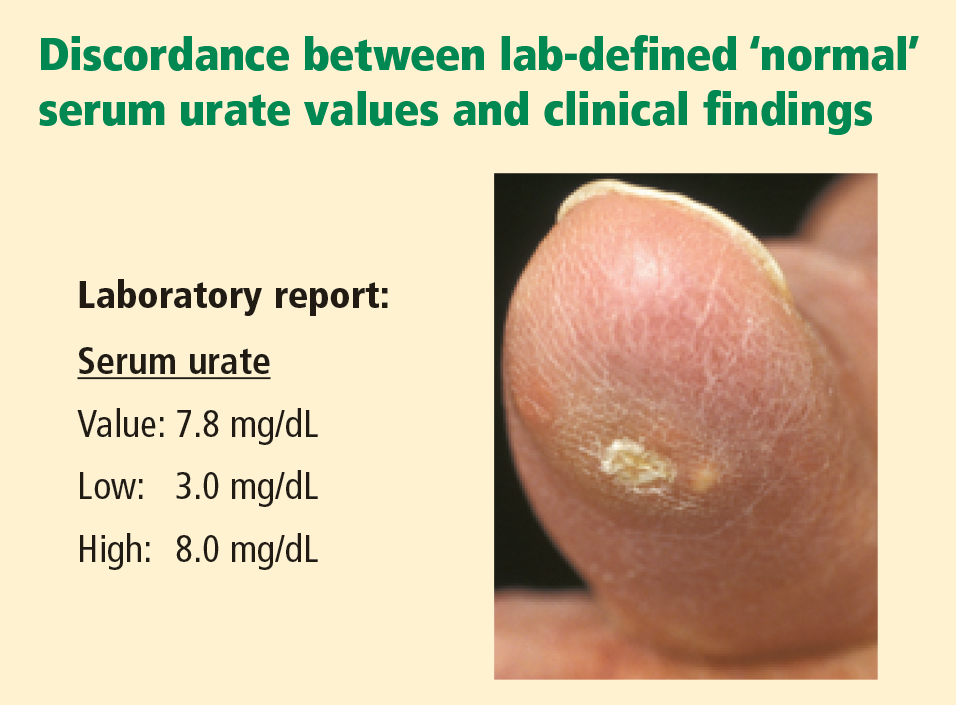
SERUM URATE CAN VARY BY SEX, AGE, DIET
Men generally have higher serum urate levels than premenopausal women; serum urate levels increase in women after menopause. For years these findings were attributed to an estrogen effect, but the mechanism was not well understood. Recently a specific transporter (urate transporter 1 [URAT1]) has been identified in the proximal tubule of the kidney3 that seems primarily responsible for the reabsorption of uric acid. Estrogen has a direct effect on the expression of this transporter. It also seems that the hypouricemic effects of probenecid and losartan, as well as the hyperuricemic effects of organic acids and high insulin levels, may be mediated via modulation of URAT1 activity.
Urate values tend to be lower in children, and urate levels are generally affected only modestly by diet.4 Epidemiologic studies, however, have linked increased ingestion of red meats and low ingestion of dairy foods with an increased incidence of gout.5 Acute alcohol ingestion can cause fluctuations in the serum urate levels and may precipitate acute gout attacks.
HYPERURICEMIA LEADING TO GOUT
Urate concentrations greater than 6.8 mg/dL may result in the deposition of urate crystals in the tissues around joints and in other soft tissue structures (tophi). Why this occurs in only some patients is not known. Crystals, when mobilized from these deposits, can provoke the acute gouty flare. The tophi are not usually hot or tender. Biopsy of a tophus reveals a chronic granulomatous inflammatory response around the sequestered crystals. However, the tophi are not inert; the uric acid can be mobilized by mass action effect if the urate in surrounding fluid is reduced. If tophi are adjacent to bone, erosion into bone may occur.
CLINICAL PROGRESSION OF HYPERURICEMIA AND GOUT: FOUR STAGES OF A CHRONIC DISEASE
Although there is significant heterogeneity in the expression of gout, we can conceptualize a prototypic progression from asymptomatic hyperuricemia to chronic gouty arthritis.
Stage 1: Asymptomatic hyperuricemia. At a serum urate concentration greater than 6.8 mg/dL, urate crystals may start to deposit. During this period of asymptomatic hyperuricemia, urate deposits may directly contribute to organ damage. This does not occur in everyone, however, and at present there is no evidence that treatment is warranted for asymptomatic hyperuricemia.
Stages 2 and 3: Acute gout and intercritical periods. If sufficient urate deposits develop around joints, and if the local milieu or some trauma triggers the release of crystals into the joint space, a patient will suffer acute attacks of gout. These flares are self-resolving but are likely to recur. The intervals between attacks are termed “intercritical periods.” During these periods, crystals may still be present at a low level in the fluid, and are certainly present in the periarticular and synovial tissue, providing a nidus for future attacks.
Stage 4: Advanced gout. If crystal deposits continue to accumulate, patients may develop chronically stiff and swollen joints. This advanced stage of gout is relatively uncommon but is avoidable with therapy.
Progression is variable
The progression from asymptomatic hyperuricemia to advanced gout is quite variable from person to person. In most people it takes many years to progress, if it does so at all. In patients treated with cyclosporine following an organ transplant, the progression can be accelerated, although the reasons are not fully understood.
ASYMPTOMATIC HYPERURICEMIA: TO TREAT OR NOT TO TREAT?
Clues for predicting the likelihood that an individual patient with asymptomatic hyperuricemia will develop articular gout are elusive. Campion and colleagues presented data on men without a history of gout who were grouped by serum urate level and followed over a 5-year period.6 The higher the patient’s urate level, the more likely that he would have a gouty attack during the 5 years. In this relatively young population of hyperuricemic men (average age of 42 years), less than 30% developed gout over this short period.
The dilemma is how to predict who is most likely to get gout and will benefit from early urate-lowering treatment, and who will not. Currently, clinicians have no reliable way of predicting the likelihood of gout development in a given hyperuricemic patient. A history of organ transplantation, the continued need for diuretics, an extremely high urate level, alcohol ingestion, low dietary consumption of dairy products, high consumption of meat and seafood, and a family history of gout at a young age suggest a higher risk of gouty arthritis. At present, treatment of asymptomatic hyperuricemia in order to prevent gouty arthritis is not generally recommended.
ACUTE GOUT FLARES: PAINFUL, UNPREDICTABLE, HIGHLY LIKELY TO RECUR
Acute flares of gouty arthritis are characterized by warmth, swelling, redness, and often severe pain. Pain frequently begins in the middle of the night or early morning. Many patients will describe awakening with pain in the foot that is so intense that they are unable to support their own weight. Patients may report fever and a flulike malaise. Fever and constitutional features are sequelae of the release of cytokines such as tumor necrosis factor, interleukin-1, and interleukin-6 following phagocytosis of crystals and activation of the intracellular inflammasome complex.7 Untreated, the initial attack will usually resolve in 3 to 14 days. Subsequent attacks tend to last longer and may involve more joints or tendons.
Where flares occur
It has been estimated that 90% of first attacks are monoarticular. However, the first recognized attack can be oligoarticular or even polyarticular. This seems particularly true in postmenopausal women and in transplant recipients. Gout attacks initially tend to occur in the lower extremities: midfoot, first metatarsophalangeal joint, ankle, or knee. Over time, gout tends to include additional joints, including those of the upper extremities. Axial joints are far less commonly involved. The initial (or subsequent) attack may be in the instep of the foot, not a well-defined joint. Patients may recall “ankle sprains,” often ascribed to an event such as “stepping off the curb wrong,” with delayed ankle swelling. These may have been attacks of gout that were not recognized by the patient and thus not reported to his or her physician. Bunion pain may be incorrectly attributed to gout (and vice versa). Therefore, we need to accept the limitations of historical recognition of gout attacks.
Acute flares also occur in periarticular structures, including bursae and tendons. The olecranon bursa, the tendons around the ankle, and the bursae around the knee are among the locations where acute attacks of gout can occur.
Risk of recurrence and implications for treatment
Based on historical data, the estimated flare recurrence rate is approximately 60% within 1 year after the initial attack, 78% within 2 years, and 84% within 3 years. Less than 10% of patients will not have a recurrence over a 10-year period. Untreated, some patients with gout will continue to have attacks and accrue chronic joint damage, stiffness, and tophi. However, that does not imply that published outcome data support treating every patient with urate-lowering therapy following an initial gout attack or even several attacks. There are no outcome data from appropriately controlled, long-term trials to validate such a treatment approach. Nonetheless, in some gouty patients, if hyperuricemia is not addressed, morbidity and joint damage will accrue. The decision as to when to intervene with urate-lowering therapy should be individualized, taking into consideration comorbidities, estimation of the likelihood of continued attacks, the impact of attacks on the patient’s lifestyle, and the potential complications of needing to use medications to treat acute attacks.
INTERCRITICAL PERIODS: CRYSTAL DEPOSITION CONTINUES SILENTLY
Immediately after an attack of gout, patients may be apt to have another if anti-inflammatory therapy is not provided for a long enough period, ie, until several days after an attack has completely resolved. Subsequently, there may be a prolonged period before another attack occurs. During this time, uric acid deposits may continue to increase silently. The factors that control the rate, location, and degree of ongoing deposition in a specific patient are not well defined. Crystals may still be found in the synovial fluid of previously involved joints until the serum urate level is reduced for a significant period to a level significantly less than 6.8 mg/dL.8
ADVANCED GOUT: DIFFERENTIATION FROM RHEUMATOID ARTHRITIS IS KEY
Tissue stores of urate may continue to increase if hyperuricemia persists at biologically significant levels (> 6.8 mg/dL). Crystal deposition can cause chronic polyarthritis. Some patients, especially as they age, develop rheumatoid factor positivity. Chronic gout, involving multiple joints, can mimic rheumatoid arthritis. Patients can develop subcutaneous tophi in areas of friction or trauma. These tophi, as well as periarticular ones, can be mistaken for rheumatoid nodules. It is unclear why only some people with hyperuricemia develop tophi. The presence of urate crystals in the aspirate of a nodule (tophus) or synovial fluid will distinguish gout from rheumatoid arthritis. Radiographs can also be of diagnostic use.
Unlike radiographic findings in rheumatoid arthritis, in gout there is a prominent, proliferative bony reaction, and tophi can cause bone destruction away from the joint. There may be a characteristic “overhanging edge” of proliferating bone surrounding a gout erosion (see Figure 3 in preceding article by Schumacher). These radiographic findings, although distinct from those of rheumatoid arthritis, can be confused with psoriatic arthritis, which also can be erosive with a proliferative bone response. Gout, however, is less likely to cause joint space narrowing than is either psoriatic arthritis or rheumatoid arthritis.
Intradermal tophi (Figure 1) are asymptomatic and frequently not recognized, yet are not that rare in severe untreated gout. Such tophi may be particularly common in transplant patients and appear as white or yellowish deposits with the overlying skin pulled taut.
POSTSCRIPT: GOUT IS NOT SO EASILY RECOGNIZED AFTER ALL
- Choi HK, Mount DB, Reginato AM. Pathogenesis of gout. Ann Intern Med 2005; 143:499–516.
- Oda M, Satta Y, Takenaka O, Takahata N. Loss of urate oxidase activity in hominoids and its evolutionary implications. Mol Biol Evol 2002; 19:640–653.
- Enomoto A, Kimura H, Chairoungdua A, et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature 2002; 417:447–452.
- Fam AG. Gout: excess calories, purines, and alcohol intake and beyond. Response to a urate-lowering diet. J Rheumatol 2005; 32:773–777.
- Choi HK, Atkinson K, Karlson EW, Willett W, Curhan G. Purine-rich foods, dairy and protein intake, and the risk of gout in men. N Engl J Med 2004; 350:1093–1103.
- Campion EW, Glynn RJ, DeLabry LO. Asymptomatic hyperuricemia. Risks and consequences in the Normative Aging Study. Am J Med 1987; 82:421–426.
- Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006; 440:237–241.
- Pascual E, Sivera F. Time required for disappearance of urate crystals from synovial fluid after successful hypouricaemic treatment relates to the duration of gout. Ann Rheum Dis 2007; 66:1056–1058.
Uric acid—urate in most physiologic fluids—is an end product of purine degradation. The serum urate level in a given patient is determined by the amount of purines synthesized and ingested, the amount of urate produced from purines, and the amount of uric acid excreted by the kidney (and, to a lesser degree, from the gastrointestinal tract).1 A major source of circulating urate is the metabolized endogenous purine. Renal excretion is likely determined by genetic factors that dictate expression of uric acid transporters, as well as by the presence of organic acids, certain drugs, hormones, and the glomerular filtration rate. A small minority of patients will have increased production of urate as a result of enzymopathies, chronic hemolysis, or rapidly dividing tumors, psoriasis, or other disorders characterized by increased turnover of cells.
Humans do not have a functional enzyme (uricase) to break down urate into allantoin, which is more soluble and readily excreted. There may have been genetic pressures that explain why functional uricase was lost and why humans have relatively high urate levels compared with other species.2 If higher levels of serum urate are clinically detrimental, one would think that humans could have evolved an efficient way to excrete it. Instead, we excrete uric acid inefficiently as a result of active reabsorption in the proximal renal tubule. We have higher levels of serum urate than most other species, and we are predisposed to develop gouty arthritis and perhaps other sequelae of hyperuricemia, including hypertension, the metabolic syndrome, and coronary artery disease.
CLINICALLY SIGNIFICANT HYPERURICEMIA VS LAB-DEFINED HYPERURICEMIA
Clinically significant hyperuricemia is a serum urate level greater than 6.8 mg/dL, although the population-defined “normal” urate level indicated by the clinical laboratory is higher. At levels above 6.8 mg/dL, urate exceeds its solubility in most biological fluids.
SERUM URATE CAN VARY BY SEX, AGE, DIET
Men generally have higher serum urate levels than premenopausal women; serum urate levels increase in women after menopause. For years these findings were attributed to an estrogen effect, but the mechanism was not well understood. Recently a specific transporter (urate transporter 1 [URAT1]) has been identified in the proximal tubule of the kidney3 that seems primarily responsible for the reabsorption of uric acid. Estrogen has a direct effect on the expression of this transporter. It also seems that the hypouricemic effects of probenecid and losartan, as well as the hyperuricemic effects of organic acids and high insulin levels, may be mediated via modulation of URAT1 activity.
Urate values tend to be lower in children, and urate levels are generally affected only modestly by diet.4 Epidemiologic studies, however, have linked increased ingestion of red meats and low ingestion of dairy foods with an increased incidence of gout.5 Acute alcohol ingestion can cause fluctuations in the serum urate levels and may precipitate acute gout attacks.
HYPERURICEMIA LEADING TO GOUT
Urate concentrations greater than 6.8 mg/dL may result in the deposition of urate crystals in the tissues around joints and in other soft tissue structures (tophi). Why this occurs in only some patients is not known. Crystals, when mobilized from these deposits, can provoke the acute gouty flare. The tophi are not usually hot or tender. Biopsy of a tophus reveals a chronic granulomatous inflammatory response around the sequestered crystals. However, the tophi are not inert; the uric acid can be mobilized by mass action effect if the urate in surrounding fluid is reduced. If tophi are adjacent to bone, erosion into bone may occur.
CLINICAL PROGRESSION OF HYPERURICEMIA AND GOUT: FOUR STAGES OF A CHRONIC DISEASE
Although there is significant heterogeneity in the expression of gout, we can conceptualize a prototypic progression from asymptomatic hyperuricemia to chronic gouty arthritis.
Stage 1: Asymptomatic hyperuricemia. At a serum urate concentration greater than 6.8 mg/dL, urate crystals may start to deposit. During this period of asymptomatic hyperuricemia, urate deposits may directly contribute to organ damage. This does not occur in everyone, however, and at present there is no evidence that treatment is warranted for asymptomatic hyperuricemia.
Stages 2 and 3: Acute gout and intercritical periods. If sufficient urate deposits develop around joints, and if the local milieu or some trauma triggers the release of crystals into the joint space, a patient will suffer acute attacks of gout. These flares are self-resolving but are likely to recur. The intervals between attacks are termed “intercritical periods.” During these periods, crystals may still be present at a low level in the fluid, and are certainly present in the periarticular and synovial tissue, providing a nidus for future attacks.
Stage 4: Advanced gout. If crystal deposits continue to accumulate, patients may develop chronically stiff and swollen joints. This advanced stage of gout is relatively uncommon but is avoidable with therapy.
Progression is variable
The progression from asymptomatic hyperuricemia to advanced gout is quite variable from person to person. In most people it takes many years to progress, if it does so at all. In patients treated with cyclosporine following an organ transplant, the progression can be accelerated, although the reasons are not fully understood.
ASYMPTOMATIC HYPERURICEMIA: TO TREAT OR NOT TO TREAT?
Clues for predicting the likelihood that an individual patient with asymptomatic hyperuricemia will develop articular gout are elusive. Campion and colleagues presented data on men without a history of gout who were grouped by serum urate level and followed over a 5-year period.6 The higher the patient’s urate level, the more likely that he would have a gouty attack during the 5 years. In this relatively young population of hyperuricemic men (average age of 42 years), less than 30% developed gout over this short period.
The dilemma is how to predict who is most likely to get gout and will benefit from early urate-lowering treatment, and who will not. Currently, clinicians have no reliable way of predicting the likelihood of gout development in a given hyperuricemic patient. A history of organ transplantation, the continued need for diuretics, an extremely high urate level, alcohol ingestion, low dietary consumption of dairy products, high consumption of meat and seafood, and a family history of gout at a young age suggest a higher risk of gouty arthritis. At present, treatment of asymptomatic hyperuricemia in order to prevent gouty arthritis is not generally recommended.
ACUTE GOUT FLARES: PAINFUL, UNPREDICTABLE, HIGHLY LIKELY TO RECUR
Acute flares of gouty arthritis are characterized by warmth, swelling, redness, and often severe pain. Pain frequently begins in the middle of the night or early morning. Many patients will describe awakening with pain in the foot that is so intense that they are unable to support their own weight. Patients may report fever and a flulike malaise. Fever and constitutional features are sequelae of the release of cytokines such as tumor necrosis factor, interleukin-1, and interleukin-6 following phagocytosis of crystals and activation of the intracellular inflammasome complex.7 Untreated, the initial attack will usually resolve in 3 to 14 days. Subsequent attacks tend to last longer and may involve more joints or tendons.
Where flares occur
It has been estimated that 90% of first attacks are monoarticular. However, the first recognized attack can be oligoarticular or even polyarticular. This seems particularly true in postmenopausal women and in transplant recipients. Gout attacks initially tend to occur in the lower extremities: midfoot, first metatarsophalangeal joint, ankle, or knee. Over time, gout tends to include additional joints, including those of the upper extremities. Axial joints are far less commonly involved. The initial (or subsequent) attack may be in the instep of the foot, not a well-defined joint. Patients may recall “ankle sprains,” often ascribed to an event such as “stepping off the curb wrong,” with delayed ankle swelling. These may have been attacks of gout that were not recognized by the patient and thus not reported to his or her physician. Bunion pain may be incorrectly attributed to gout (and vice versa). Therefore, we need to accept the limitations of historical recognition of gout attacks.
Acute flares also occur in periarticular structures, including bursae and tendons. The olecranon bursa, the tendons around the ankle, and the bursae around the knee are among the locations where acute attacks of gout can occur.
Risk of recurrence and implications for treatment
Based on historical data, the estimated flare recurrence rate is approximately 60% within 1 year after the initial attack, 78% within 2 years, and 84% within 3 years. Less than 10% of patients will not have a recurrence over a 10-year period. Untreated, some patients with gout will continue to have attacks and accrue chronic joint damage, stiffness, and tophi. However, that does not imply that published outcome data support treating every patient with urate-lowering therapy following an initial gout attack or even several attacks. There are no outcome data from appropriately controlled, long-term trials to validate such a treatment approach. Nonetheless, in some gouty patients, if hyperuricemia is not addressed, morbidity and joint damage will accrue. The decision as to when to intervene with urate-lowering therapy should be individualized, taking into consideration comorbidities, estimation of the likelihood of continued attacks, the impact of attacks on the patient’s lifestyle, and the potential complications of needing to use medications to treat acute attacks.
INTERCRITICAL PERIODS: CRYSTAL DEPOSITION CONTINUES SILENTLY
Immediately after an attack of gout, patients may be apt to have another if anti-inflammatory therapy is not provided for a long enough period, ie, until several days after an attack has completely resolved. Subsequently, there may be a prolonged period before another attack occurs. During this time, uric acid deposits may continue to increase silently. The factors that control the rate, location, and degree of ongoing deposition in a specific patient are not well defined. Crystals may still be found in the synovial fluid of previously involved joints until the serum urate level is reduced for a significant period to a level significantly less than 6.8 mg/dL.8
ADVANCED GOUT: DIFFERENTIATION FROM RHEUMATOID ARTHRITIS IS KEY
Tissue stores of urate may continue to increase if hyperuricemia persists at biologically significant levels (> 6.8 mg/dL). Crystal deposition can cause chronic polyarthritis. Some patients, especially as they age, develop rheumatoid factor positivity. Chronic gout, involving multiple joints, can mimic rheumatoid arthritis. Patients can develop subcutaneous tophi in areas of friction or trauma. These tophi, as well as periarticular ones, can be mistaken for rheumatoid nodules. It is unclear why only some people with hyperuricemia develop tophi. The presence of urate crystals in the aspirate of a nodule (tophus) or synovial fluid will distinguish gout from rheumatoid arthritis. Radiographs can also be of diagnostic use.
Unlike radiographic findings in rheumatoid arthritis, in gout there is a prominent, proliferative bony reaction, and tophi can cause bone destruction away from the joint. There may be a characteristic “overhanging edge” of proliferating bone surrounding a gout erosion (see Figure 3 in preceding article by Schumacher). These radiographic findings, although distinct from those of rheumatoid arthritis, can be confused with psoriatic arthritis, which also can be erosive with a proliferative bone response. Gout, however, is less likely to cause joint space narrowing than is either psoriatic arthritis or rheumatoid arthritis.
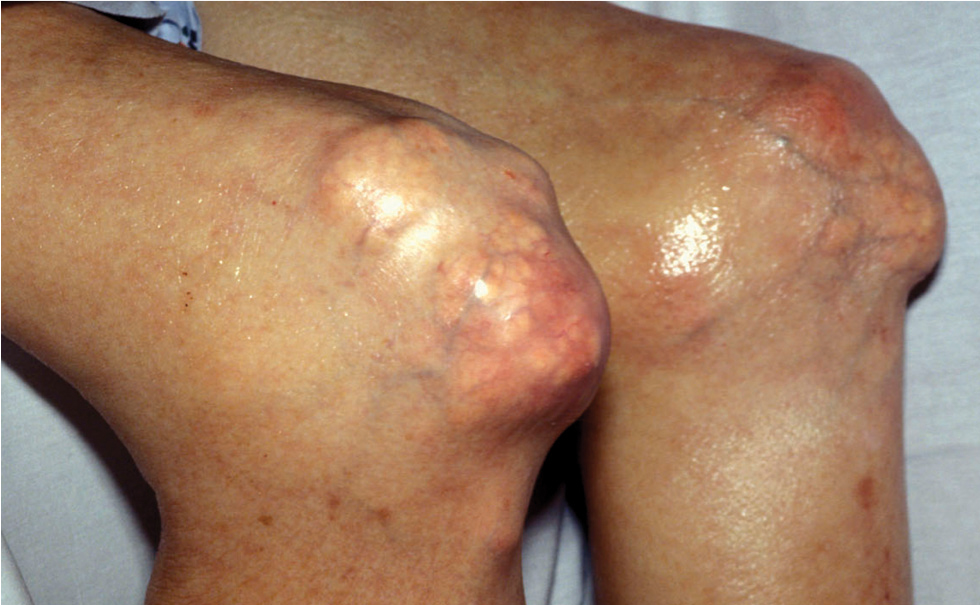
Intradermal tophi (Figure 1) are asymptomatic and frequently not recognized, yet are not that rare in severe untreated gout. Such tophi may be particularly common in transplant patients and appear as white or yellowish deposits with the overlying skin pulled taut.
POSTSCRIPT: GOUT IS NOT SO EASILY RECOGNIZED AFTER ALL
Uric acid—urate in most physiologic fluids—is an end product of purine degradation. The serum urate level in a given patient is determined by the amount of purines synthesized and ingested, the amount of urate produced from purines, and the amount of uric acid excreted by the kidney (and, to a lesser degree, from the gastrointestinal tract).1 A major source of circulating urate is the metabolized endogenous purine. Renal excretion is likely determined by genetic factors that dictate expression of uric acid transporters, as well as by the presence of organic acids, certain drugs, hormones, and the glomerular filtration rate. A small minority of patients will have increased production of urate as a result of enzymopathies, chronic hemolysis, or rapidly dividing tumors, psoriasis, or other disorders characterized by increased turnover of cells.
Humans do not have a functional enzyme (uricase) to break down urate into allantoin, which is more soluble and readily excreted. There may have been genetic pressures that explain why functional uricase was lost and why humans have relatively high urate levels compared with other species.2 If higher levels of serum urate are clinically detrimental, one would think that humans could have evolved an efficient way to excrete it. Instead, we excrete uric acid inefficiently as a result of active reabsorption in the proximal renal tubule. We have higher levels of serum urate than most other species, and we are predisposed to develop gouty arthritis and perhaps other sequelae of hyperuricemia, including hypertension, the metabolic syndrome, and coronary artery disease.
CLINICALLY SIGNIFICANT HYPERURICEMIA VS LAB-DEFINED HYPERURICEMIA
Clinically significant hyperuricemia is a serum urate level greater than 6.8 mg/dL, although the population-defined “normal” urate level indicated by the clinical laboratory is higher. At levels above 6.8 mg/dL, urate exceeds its solubility in most biological fluids.
SERUM URATE CAN VARY BY SEX, AGE, DIET
Men generally have higher serum urate levels than premenopausal women; serum urate levels increase in women after menopause. For years these findings were attributed to an estrogen effect, but the mechanism was not well understood. Recently a specific transporter (urate transporter 1 [URAT1]) has been identified in the proximal tubule of the kidney3 that seems primarily responsible for the reabsorption of uric acid. Estrogen has a direct effect on the expression of this transporter. It also seems that the hypouricemic effects of probenecid and losartan, as well as the hyperuricemic effects of organic acids and high insulin levels, may be mediated via modulation of URAT1 activity.
Urate values tend to be lower in children, and urate levels are generally affected only modestly by diet.4 Epidemiologic studies, however, have linked increased ingestion of red meats and low ingestion of dairy foods with an increased incidence of gout.5 Acute alcohol ingestion can cause fluctuations in the serum urate levels and may precipitate acute gout attacks.
HYPERURICEMIA LEADING TO GOUT
Urate concentrations greater than 6.8 mg/dL may result in the deposition of urate crystals in the tissues around joints and in other soft tissue structures (tophi). Why this occurs in only some patients is not known. Crystals, when mobilized from these deposits, can provoke the acute gouty flare. The tophi are not usually hot or tender. Biopsy of a tophus reveals a chronic granulomatous inflammatory response around the sequestered crystals. However, the tophi are not inert; the uric acid can be mobilized by mass action effect if the urate in surrounding fluid is reduced. If tophi are adjacent to bone, erosion into bone may occur.
CLINICAL PROGRESSION OF HYPERURICEMIA AND GOUT: FOUR STAGES OF A CHRONIC DISEASE
Although there is significant heterogeneity in the expression of gout, we can conceptualize a prototypic progression from asymptomatic hyperuricemia to chronic gouty arthritis.
Stage 1: Asymptomatic hyperuricemia. At a serum urate concentration greater than 6.8 mg/dL, urate crystals may start to deposit. During this period of asymptomatic hyperuricemia, urate deposits may directly contribute to organ damage. This does not occur in everyone, however, and at present there is no evidence that treatment is warranted for asymptomatic hyperuricemia.
Stages 2 and 3: Acute gout and intercritical periods. If sufficient urate deposits develop around joints, and if the local milieu or some trauma triggers the release of crystals into the joint space, a patient will suffer acute attacks of gout. These flares are self-resolving but are likely to recur. The intervals between attacks are termed “intercritical periods.” During these periods, crystals may still be present at a low level in the fluid, and are certainly present in the periarticular and synovial tissue, providing a nidus for future attacks.
Stage 4: Advanced gout. If crystal deposits continue to accumulate, patients may develop chronically stiff and swollen joints. This advanced stage of gout is relatively uncommon but is avoidable with therapy.
Progression is variable
The progression from asymptomatic hyperuricemia to advanced gout is quite variable from person to person. In most people it takes many years to progress, if it does so at all. In patients treated with cyclosporine following an organ transplant, the progression can be accelerated, although the reasons are not fully understood.
ASYMPTOMATIC HYPERURICEMIA: TO TREAT OR NOT TO TREAT?
Clues for predicting the likelihood that an individual patient with asymptomatic hyperuricemia will develop articular gout are elusive. Campion and colleagues presented data on men without a history of gout who were grouped by serum urate level and followed over a 5-year period.6 The higher the patient’s urate level, the more likely that he would have a gouty attack during the 5 years. In this relatively young population of hyperuricemic men (average age of 42 years), less than 30% developed gout over this short period.
The dilemma is how to predict who is most likely to get gout and will benefit from early urate-lowering treatment, and who will not. Currently, clinicians have no reliable way of predicting the likelihood of gout development in a given hyperuricemic patient. A history of organ transplantation, the continued need for diuretics, an extremely high urate level, alcohol ingestion, low dietary consumption of dairy products, high consumption of meat and seafood, and a family history of gout at a young age suggest a higher risk of gouty arthritis. At present, treatment of asymptomatic hyperuricemia in order to prevent gouty arthritis is not generally recommended.
ACUTE GOUT FLARES: PAINFUL, UNPREDICTABLE, HIGHLY LIKELY TO RECUR
Acute flares of gouty arthritis are characterized by warmth, swelling, redness, and often severe pain. Pain frequently begins in the middle of the night or early morning. Many patients will describe awakening with pain in the foot that is so intense that they are unable to support their own weight. Patients may report fever and a flulike malaise. Fever and constitutional features are sequelae of the release of cytokines such as tumor necrosis factor, interleukin-1, and interleukin-6 following phagocytosis of crystals and activation of the intracellular inflammasome complex.7 Untreated, the initial attack will usually resolve in 3 to 14 days. Subsequent attacks tend to last longer and may involve more joints or tendons.
Where flares occur
It has been estimated that 90% of first attacks are monoarticular. However, the first recognized attack can be oligoarticular or even polyarticular. This seems particularly true in postmenopausal women and in transplant recipients. Gout attacks initially tend to occur in the lower extremities: midfoot, first metatarsophalangeal joint, ankle, or knee. Over time, gout tends to include additional joints, including those of the upper extremities. Axial joints are far less commonly involved. The initial (or subsequent) attack may be in the instep of the foot, not a well-defined joint. Patients may recall “ankle sprains,” often ascribed to an event such as “stepping off the curb wrong,” with delayed ankle swelling. These may have been attacks of gout that were not recognized by the patient and thus not reported to his or her physician. Bunion pain may be incorrectly attributed to gout (and vice versa). Therefore, we need to accept the limitations of historical recognition of gout attacks.
Acute flares also occur in periarticular structures, including bursae and tendons. The olecranon bursa, the tendons around the ankle, and the bursae around the knee are among the locations where acute attacks of gout can occur.
Risk of recurrence and implications for treatment
Based on historical data, the estimated flare recurrence rate is approximately 60% within 1 year after the initial attack, 78% within 2 years, and 84% within 3 years. Less than 10% of patients will not have a recurrence over a 10-year period. Untreated, some patients with gout will continue to have attacks and accrue chronic joint damage, stiffness, and tophi. However, that does not imply that published outcome data support treating every patient with urate-lowering therapy following an initial gout attack or even several attacks. There are no outcome data from appropriately controlled, long-term trials to validate such a treatment approach. Nonetheless, in some gouty patients, if hyperuricemia is not addressed, morbidity and joint damage will accrue. The decision as to when to intervene with urate-lowering therapy should be individualized, taking into consideration comorbidities, estimation of the likelihood of continued attacks, the impact of attacks on the patient’s lifestyle, and the potential complications of needing to use medications to treat acute attacks.
INTERCRITICAL PERIODS: CRYSTAL DEPOSITION CONTINUES SILENTLY
Immediately after an attack of gout, patients may be apt to have another if anti-inflammatory therapy is not provided for a long enough period, ie, until several days after an attack has completely resolved. Subsequently, there may be a prolonged period before another attack occurs. During this time, uric acid deposits may continue to increase silently. The factors that control the rate, location, and degree of ongoing deposition in a specific patient are not well defined. Crystals may still be found in the synovial fluid of previously involved joints until the serum urate level is reduced for a significant period to a level significantly less than 6.8 mg/dL.8
ADVANCED GOUT: DIFFERENTIATION FROM RHEUMATOID ARTHRITIS IS KEY
Tissue stores of urate may continue to increase if hyperuricemia persists at biologically significant levels (> 6.8 mg/dL). Crystal deposition can cause chronic polyarthritis. Some patients, especially as they age, develop rheumatoid factor positivity. Chronic gout, involving multiple joints, can mimic rheumatoid arthritis. Patients can develop subcutaneous tophi in areas of friction or trauma. These tophi, as well as periarticular ones, can be mistaken for rheumatoid nodules. It is unclear why only some people with hyperuricemia develop tophi. The presence of urate crystals in the aspirate of a nodule (tophus) or synovial fluid will distinguish gout from rheumatoid arthritis. Radiographs can also be of diagnostic use.
Unlike radiographic findings in rheumatoid arthritis, in gout there is a prominent, proliferative bony reaction, and tophi can cause bone destruction away from the joint. There may be a characteristic “overhanging edge” of proliferating bone surrounding a gout erosion (see Figure 3 in preceding article by Schumacher). These radiographic findings, although distinct from those of rheumatoid arthritis, can be confused with psoriatic arthritis, which also can be erosive with a proliferative bone response. Gout, however, is less likely to cause joint space narrowing than is either psoriatic arthritis or rheumatoid arthritis.
Intradermal tophi (Figure 1) are asymptomatic and frequently not recognized, yet are not that rare in severe untreated gout. Such tophi may be particularly common in transplant patients and appear as white or yellowish deposits with the overlying skin pulled taut.
POSTSCRIPT: GOUT IS NOT SO EASILY RECOGNIZED AFTER ALL
- Choi HK, Mount DB, Reginato AM. Pathogenesis of gout. Ann Intern Med 2005; 143:499–516.
- Oda M, Satta Y, Takenaka O, Takahata N. Loss of urate oxidase activity in hominoids and its evolutionary implications. Mol Biol Evol 2002; 19:640–653.
- Enomoto A, Kimura H, Chairoungdua A, et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature 2002; 417:447–452.
- Fam AG. Gout: excess calories, purines, and alcohol intake and beyond. Response to a urate-lowering diet. J Rheumatol 2005; 32:773–777.
- Choi HK, Atkinson K, Karlson EW, Willett W, Curhan G. Purine-rich foods, dairy and protein intake, and the risk of gout in men. N Engl J Med 2004; 350:1093–1103.
- Campion EW, Glynn RJ, DeLabry LO. Asymptomatic hyperuricemia. Risks and consequences in the Normative Aging Study. Am J Med 1987; 82:421–426.
- Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006; 440:237–241.
- Pascual E, Sivera F. Time required for disappearance of urate crystals from synovial fluid after successful hypouricaemic treatment relates to the duration of gout. Ann Rheum Dis 2007; 66:1056–1058.
- Choi HK, Mount DB, Reginato AM. Pathogenesis of gout. Ann Intern Med 2005; 143:499–516.
- Oda M, Satta Y, Takenaka O, Takahata N. Loss of urate oxidase activity in hominoids and its evolutionary implications. Mol Biol Evol 2002; 19:640–653.
- Enomoto A, Kimura H, Chairoungdua A, et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature 2002; 417:447–452.
- Fam AG. Gout: excess calories, purines, and alcohol intake and beyond. Response to a urate-lowering diet. J Rheumatol 2005; 32:773–777.
- Choi HK, Atkinson K, Karlson EW, Willett W, Curhan G. Purine-rich foods, dairy and protein intake, and the risk of gout in men. N Engl J Med 2004; 350:1093–1103.
- Campion EW, Glynn RJ, DeLabry LO. Asymptomatic hyperuricemia. Risks and consequences in the Normative Aging Study. Am J Med 1987; 82:421–426.
- Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006; 440:237–241.
- Pascual E, Sivera F. Time required for disappearance of urate crystals from synovial fluid after successful hypouricaemic treatment relates to the duration of gout. Ann Rheum Dis 2007; 66:1056–1058.
KEY POINTS
- Clinically significant hyperuricemia includes serum urate levels that fall within the population-defined “normal” range of many clinical laboratories.
- There is no reliable way to predict the likelihood that gout will develop in a given hyperuricemic patient. Treatment of asymptomatic hyperuricemia is not generally recommended.
- Untreated, an initial acute gout attack resolves within 3 to 14 days. Subsequent attacks tend to last longer and may involve more joints.
- Chronic gout can mimic rheumatoid or psoriatic arthritis.
A drug, a concept, and a clinical trial on trial

Many trials are funded by industry and carried out by clinical investigators in academic and private practice. Drug companies must perform these trials to win approval from the US Food and Drug Administration (FDA) for their new drugs and package inserts, which dictates what they can and can’t say in their advertising. This latter requirement often leads to trials after a drug is approved in an effort to aid drug promotion and improve its position in the marketplace.
The FDA is increasingly demanding that new drug studies use “hard” measures of efficacy and less reliance on surrogate end points. This requires larger, longer, more expensive trials.
A recent trial that relied on surrogate end points was the ENHANCE trial, which evaluated the addition of a second approved cholesterol-lowering drug (ezetimibe) to a statin in a relatively small number of mostly pretreated patients. The surrogates were lipid-lowering and carotid intima-media thickness. At the time the study was designed, I’m sure it seemed obvious that lowering low-density lipoprotein cholesterol (LDL-C) or reducing the measured burden of atherosclerosis would reduce the consequences of hypercholesterolemia, including myocardial infarction and stroke. Therefore, the use of surrogate markers seemed an acceptable expediency.
However, the ENHANCE results hit the national news when the two surrogates didn’t coincide as anticipated. Although ezetimibe/simvastatin (Vytorin) lowered the LDL-C level more than simvastatin alone (Zocor), it did not reduce carotid intima-media thickness.
The response was intense. Trialists, drug safety pundits, industry representatives, politicians, and clinicians all weighed in. Some patients apparently stopped taking their lipid-lowering medications. Without any striking evidence of worse outcome, doubt has been cast on the safety and efficacy of the drug and—perhaps inappropriately—on the entire LDL-C hypothesis of atherosclerosis.
In this issue, Dr. Michael Davidson and Dr. Allen Taylor, two clinical experts in atherosclerosis, present widely divergent views on the conduct, results, and implications of the ENHANCE trial. Dr. Taylor was afforded the opportunity to read Dr. Davidson’s manuscript before writing his own editorial. I’m not sure their discussion will settle this intense debate, but they outline the issues clearly.
Many trials are funded by industry and carried out by clinical investigators in academic and private practice. Drug companies must perform these trials to win approval from the US Food and Drug Administration (FDA) for their new drugs and package inserts, which dictates what they can and can’t say in their advertising. This latter requirement often leads to trials after a drug is approved in an effort to aid drug promotion and improve its position in the marketplace.
The FDA is increasingly demanding that new drug studies use “hard” measures of efficacy and less reliance on surrogate end points. This requires larger, longer, more expensive trials.
A recent trial that relied on surrogate end points was the ENHANCE trial, which evaluated the addition of a second approved cholesterol-lowering drug (ezetimibe) to a statin in a relatively small number of mostly pretreated patients. The surrogates were lipid-lowering and carotid intima-media thickness. At the time the study was designed, I’m sure it seemed obvious that lowering low-density lipoprotein cholesterol (LDL-C) or reducing the measured burden of atherosclerosis would reduce the consequences of hypercholesterolemia, including myocardial infarction and stroke. Therefore, the use of surrogate markers seemed an acceptable expediency.
However, the ENHANCE results hit the national news when the two surrogates didn’t coincide as anticipated. Although ezetimibe/simvastatin (Vytorin) lowered the LDL-C level more than simvastatin alone (Zocor), it did not reduce carotid intima-media thickness.
The response was intense. Trialists, drug safety pundits, industry representatives, politicians, and clinicians all weighed in. Some patients apparently stopped taking their lipid-lowering medications. Without any striking evidence of worse outcome, doubt has been cast on the safety and efficacy of the drug and—perhaps inappropriately—on the entire LDL-C hypothesis of atherosclerosis.
In this issue, Dr. Michael Davidson and Dr. Allen Taylor, two clinical experts in atherosclerosis, present widely divergent views on the conduct, results, and implications of the ENHANCE trial. Dr. Taylor was afforded the opportunity to read Dr. Davidson’s manuscript before writing his own editorial. I’m not sure their discussion will settle this intense debate, but they outline the issues clearly.
Many trials are funded by industry and carried out by clinical investigators in academic and private practice. Drug companies must perform these trials to win approval from the US Food and Drug Administration (FDA) for their new drugs and package inserts, which dictates what they can and can’t say in their advertising. This latter requirement often leads to trials after a drug is approved in an effort to aid drug promotion and improve its position in the marketplace.
The FDA is increasingly demanding that new drug studies use “hard” measures of efficacy and less reliance on surrogate end points. This requires larger, longer, more expensive trials.
A recent trial that relied on surrogate end points was the ENHANCE trial, which evaluated the addition of a second approved cholesterol-lowering drug (ezetimibe) to a statin in a relatively small number of mostly pretreated patients. The surrogates were lipid-lowering and carotid intima-media thickness. At the time the study was designed, I’m sure it seemed obvious that lowering low-density lipoprotein cholesterol (LDL-C) or reducing the measured burden of atherosclerosis would reduce the consequences of hypercholesterolemia, including myocardial infarction and stroke. Therefore, the use of surrogate markers seemed an acceptable expediency.
However, the ENHANCE results hit the national news when the two surrogates didn’t coincide as anticipated. Although ezetimibe/simvastatin (Vytorin) lowered the LDL-C level more than simvastatin alone (Zocor), it did not reduce carotid intima-media thickness.
The response was intense. Trialists, drug safety pundits, industry representatives, politicians, and clinicians all weighed in. Some patients apparently stopped taking their lipid-lowering medications. Without any striking evidence of worse outcome, doubt has been cast on the safety and efficacy of the drug and—perhaps inappropriately—on the entire LDL-C hypothesis of atherosclerosis.
In this issue, Dr. Michael Davidson and Dr. Allen Taylor, two clinical experts in atherosclerosis, present widely divergent views on the conduct, results, and implications of the ENHANCE trial. Dr. Taylor was afforded the opportunity to read Dr. Davidson’s manuscript before writing his own editorial. I’m not sure their discussion will settle this intense debate, but they outline the issues clearly.
Meta-analyses, metaphysics, and reality

Dr. Jeffrey Aronson, in an editorial in the British Journal of Clinical Pharmacology,1 discussed the term meta-analysis and its link to Aristotle and metaphysics. It seems that Andronicus, an editor of Aristotle’s work, titled the first set of Aristotle’s papers on natural sciences The Physics, and a second set of papers The Metaphysics because they were written after The Physics. The Metaphysics, however, dealt more with philosophy, and thus the term metaphysics acquired over time the connotation of “not real” physics.
A meta-analysis is, in fact, a structured analysis of prior analyses (often randomized trials). In this issue of the Journal, Dr. Esteban Walker and colleagues2 clarify the specific rules that must be followed when doing a meta-analysis.
While I believe that meta-analyses often have significant issues that must be resolved before they can be translated into a change in clinical practice, a generic condemnation (or acceptance) of the tool is not appropriate. One study compared the results of meta-analyses with subsequently performed randomized clinical trials, and in only 12% were the conclusions significantly different.3
A goal of meta-analysis is to overcome limitations of small sample sizes by pooling results in an appropriate and orderly way. One problem has been the inability to access unpublished (usually “negative”) trials. With the Food and Drug Administration Amendments Act of 2007, all clinical trials beyond phase I studies will be posted on the Web, and because of this, much of the concern about publication bias may be overcome.
But the issue remains of how best to interpret the clinical significance of small effects or rare but serious events. As Walker et al note, clinicians need to be very careful about directly translating conclusions from meta-analyses into clinical practice. Patients (and the news media) would be wise to exercise the same caution.
- Aronson JK. Metameta-analysis [editorial]. Br J Clin Pharmacol 2005; 60:117–119.
- Walker E, Hernandez AV, Kattan MW. Meta-analysis: its strengths and limitations. Cleve Clin J Med 2008; 75:431–440.
- LeLorier J, Grégoire G, Benhaddad A, Lapierre J, Derderian F. Discrepancies between meta-analyses and subsequent large randomized, controlled trials. N Engl J Med 1997; 337:536–542.
Dr. Jeffrey Aronson, in an editorial in the British Journal of Clinical Pharmacology,1 discussed the term meta-analysis and its link to Aristotle and metaphysics. It seems that Andronicus, an editor of Aristotle’s work, titled the first set of Aristotle’s papers on natural sciences The Physics, and a second set of papers The Metaphysics because they were written after The Physics. The Metaphysics, however, dealt more with philosophy, and thus the term metaphysics acquired over time the connotation of “not real” physics.
A meta-analysis is, in fact, a structured analysis of prior analyses (often randomized trials). In this issue of the Journal, Dr. Esteban Walker and colleagues2 clarify the specific rules that must be followed when doing a meta-analysis.
While I believe that meta-analyses often have significant issues that must be resolved before they can be translated into a change in clinical practice, a generic condemnation (or acceptance) of the tool is not appropriate. One study compared the results of meta-analyses with subsequently performed randomized clinical trials, and in only 12% were the conclusions significantly different.3
A goal of meta-analysis is to overcome limitations of small sample sizes by pooling results in an appropriate and orderly way. One problem has been the inability to access unpublished (usually “negative”) trials. With the Food and Drug Administration Amendments Act of 2007, all clinical trials beyond phase I studies will be posted on the Web, and because of this, much of the concern about publication bias may be overcome.
But the issue remains of how best to interpret the clinical significance of small effects or rare but serious events. As Walker et al note, clinicians need to be very careful about directly translating conclusions from meta-analyses into clinical practice. Patients (and the news media) would be wise to exercise the same caution.
Dr. Jeffrey Aronson, in an editorial in the British Journal of Clinical Pharmacology,1 discussed the term meta-analysis and its link to Aristotle and metaphysics. It seems that Andronicus, an editor of Aristotle’s work, titled the first set of Aristotle’s papers on natural sciences The Physics, and a second set of papers The Metaphysics because they were written after The Physics. The Metaphysics, however, dealt more with philosophy, and thus the term metaphysics acquired over time the connotation of “not real” physics.
A meta-analysis is, in fact, a structured analysis of prior analyses (often randomized trials). In this issue of the Journal, Dr. Esteban Walker and colleagues2 clarify the specific rules that must be followed when doing a meta-analysis.
While I believe that meta-analyses often have significant issues that must be resolved before they can be translated into a change in clinical practice, a generic condemnation (or acceptance) of the tool is not appropriate. One study compared the results of meta-analyses with subsequently performed randomized clinical trials, and in only 12% were the conclusions significantly different.3
A goal of meta-analysis is to overcome limitations of small sample sizes by pooling results in an appropriate and orderly way. One problem has been the inability to access unpublished (usually “negative”) trials. With the Food and Drug Administration Amendments Act of 2007, all clinical trials beyond phase I studies will be posted on the Web, and because of this, much of the concern about publication bias may be overcome.
But the issue remains of how best to interpret the clinical significance of small effects or rare but serious events. As Walker et al note, clinicians need to be very careful about directly translating conclusions from meta-analyses into clinical practice. Patients (and the news media) would be wise to exercise the same caution.
- Aronson JK. Metameta-analysis [editorial]. Br J Clin Pharmacol 2005; 60:117–119.
- Walker E, Hernandez AV, Kattan MW. Meta-analysis: its strengths and limitations. Cleve Clin J Med 2008; 75:431–440.
- LeLorier J, Grégoire G, Benhaddad A, Lapierre J, Derderian F. Discrepancies between meta-analyses and subsequent large randomized, controlled trials. N Engl J Med 1997; 337:536–542.
- Aronson JK. Metameta-analysis [editorial]. Br J Clin Pharmacol 2005; 60:117–119.
- Walker E, Hernandez AV, Kattan MW. Meta-analysis: its strengths and limitations. Cleve Clin J Med 2008; 75:431–440.
- LeLorier J, Grégoire G, Benhaddad A, Lapierre J, Derderian F. Discrepancies between meta-analyses and subsequent large randomized, controlled trials. N Engl J Med 1997; 337:536–542.
‘Blood will have blood’

Erythropoiesis-stimulating agents (ESAs) changed the landscape, giving us the ability to normalize the hemoglobin level without giving blood products. Patients with renal failure who were making inadequate amounts of endogenous erythropoietin could be given exogenous ESAs. And patients with anemia characterized by resistance to erythropoietin could be given higher doses of an ESA, and the resistance could be overcome. Questions were occasionally raised about the outcomes when boosting the hemoglobin level above 10 g/dL, and seizures and hypertension were reported as complications of therapy, but cost was the major stumbling block to the expanded use of ESAs.
However, as Drs. Sevag Demirjian and Saul Nurko discuss in this issue of the Journal, striving to fully correct the anemia of chronic kidney disease with the use of ESAs may cause unexpected problems—such as more deaths and cardiovascular events.
Why should more patients with chronic kidney disease die if their hemoglobin levels are normalized? It could be another case of “messing with Mother Nature.” Perhaps the decreased erythropoietin production and anemia associated with renal failure are a protective reflex somehow beneficial to patients with decreased renal mass. On the other hand, it seems that the patients who had the most problems with ESAs in randomized trials were actually “resistant” to ESA therapy and were therefore probably given higher ESA doses. More blood may not be the problem, but, rather, too much ESA.
In an editorial in this issue, Dr. Alan Lichtin discusses additional concerns that have arisen with the use of ESAs when treating the anemia associated with malignancy. One issue relates to the expression of erythropoietin receptors by nonerythroid cells: some tumor cells express erythropoietin receptors, and giving high doses of ESAs might stimulate their growth. Dr. Lichtin concludes by saying the ESA story is far from over, and I believe him.
Erythropoiesis-stimulating agents (ESAs) changed the landscape, giving us the ability to normalize the hemoglobin level without giving blood products. Patients with renal failure who were making inadequate amounts of endogenous erythropoietin could be given exogenous ESAs. And patients with anemia characterized by resistance to erythropoietin could be given higher doses of an ESA, and the resistance could be overcome. Questions were occasionally raised about the outcomes when boosting the hemoglobin level above 10 g/dL, and seizures and hypertension were reported as complications of therapy, but cost was the major stumbling block to the expanded use of ESAs.
However, as Drs. Sevag Demirjian and Saul Nurko discuss in this issue of the Journal, striving to fully correct the anemia of chronic kidney disease with the use of ESAs may cause unexpected problems—such as more deaths and cardiovascular events.
Why should more patients with chronic kidney disease die if their hemoglobin levels are normalized? It could be another case of “messing with Mother Nature.” Perhaps the decreased erythropoietin production and anemia associated with renal failure are a protective reflex somehow beneficial to patients with decreased renal mass. On the other hand, it seems that the patients who had the most problems with ESAs in randomized trials were actually “resistant” to ESA therapy and were therefore probably given higher ESA doses. More blood may not be the problem, but, rather, too much ESA.
In an editorial in this issue, Dr. Alan Lichtin discusses additional concerns that have arisen with the use of ESAs when treating the anemia associated with malignancy. One issue relates to the expression of erythropoietin receptors by nonerythroid cells: some tumor cells express erythropoietin receptors, and giving high doses of ESAs might stimulate their growth. Dr. Lichtin concludes by saying the ESA story is far from over, and I believe him.
Erythropoiesis-stimulating agents (ESAs) changed the landscape, giving us the ability to normalize the hemoglobin level without giving blood products. Patients with renal failure who were making inadequate amounts of endogenous erythropoietin could be given exogenous ESAs. And patients with anemia characterized by resistance to erythropoietin could be given higher doses of an ESA, and the resistance could be overcome. Questions were occasionally raised about the outcomes when boosting the hemoglobin level above 10 g/dL, and seizures and hypertension were reported as complications of therapy, but cost was the major stumbling block to the expanded use of ESAs.
However, as Drs. Sevag Demirjian and Saul Nurko discuss in this issue of the Journal, striving to fully correct the anemia of chronic kidney disease with the use of ESAs may cause unexpected problems—such as more deaths and cardiovascular events.
Why should more patients with chronic kidney disease die if their hemoglobin levels are normalized? It could be another case of “messing with Mother Nature.” Perhaps the decreased erythropoietin production and anemia associated with renal failure are a protective reflex somehow beneficial to patients with decreased renal mass. On the other hand, it seems that the patients who had the most problems with ESAs in randomized trials were actually “resistant” to ESA therapy and were therefore probably given higher ESA doses. More blood may not be the problem, but, rather, too much ESA.
In an editorial in this issue, Dr. Alan Lichtin discusses additional concerns that have arisen with the use of ESAs when treating the anemia associated with malignancy. One issue relates to the expression of erythropoietin receptors by nonerythroid cells: some tumor cells express erythropoietin receptors, and giving high doses of ESAs might stimulate their growth. Dr. Lichtin concludes by saying the ESA story is far from over, and I believe him.
A new series, an old concept, continued value

The physical examination used to be a foundation of clinical practice, but it is under assault. A specialist in inpatient medicine here at Cleveland Clinic decried the inefficiency of time spent by residents performing and documenting the examination. How often, he asked, does the examination actually change the diagnostic workup? Academic provocateurs have done sensitivity and specificity analyses on components of the physical examination and found them to be imperfect. Imperfect, yes, but I would argue not worthless.
One reason for the imperfection is that skills of examination are not always appropriately emphasized during training, and then are not utilized in practice (especially in a 10-minute visit). Several published studies describe the attrition of examination skills. While doing teaching rounds as a visiting professor, I have found that some residents and medical students have difficulty distinguishing a murmur of aortic sclerosis from one of aortic insufficiency or detecting epitrochlear adenopathy.
The structured physical examination still fulfills a legitimate need. When approaching a patient with an ill-defined, potentially multisystem disorder, the examination should provide an initial “staging” of the disease process that contributes to the construction of the differential diagnosis, and thus refines the ordering of specific tests.
Adenopathy, enlarged lacrimal glands, and retinal exudates can all be asymptomatic and, if detected, may affect the differential diagnosis. Yet how many examinations focus attention on these areas? We often teach (and we were taught) the physical examination as a rote skill to be performed in toto. But the examination is not a static procedure. It needs to be tailored to the patient in front of us, in a reiterative, reflective manner. I am more likely to find adenopathy or an abdominal aneurysm if I am specifically looking for it, as opposed to performing a perfunctory examination. I am less likely to be struck by a pronounced second heart sound if I am not considering pulmonary hypertension as a possible explanation for the patient’s dyspnea.
I believe that a reflective physical examination is effective and valuable. Besides, our patients actually expect (and deserve) to be carefully examined.
The physical examination used to be a foundation of clinical practice, but it is under assault. A specialist in inpatient medicine here at Cleveland Clinic decried the inefficiency of time spent by residents performing and documenting the examination. How often, he asked, does the examination actually change the diagnostic workup? Academic provocateurs have done sensitivity and specificity analyses on components of the physical examination and found them to be imperfect. Imperfect, yes, but I would argue not worthless.
One reason for the imperfection is that skills of examination are not always appropriately emphasized during training, and then are not utilized in practice (especially in a 10-minute visit). Several published studies describe the attrition of examination skills. While doing teaching rounds as a visiting professor, I have found that some residents and medical students have difficulty distinguishing a murmur of aortic sclerosis from one of aortic insufficiency or detecting epitrochlear adenopathy.
The structured physical examination still fulfills a legitimate need. When approaching a patient with an ill-defined, potentially multisystem disorder, the examination should provide an initial “staging” of the disease process that contributes to the construction of the differential diagnosis, and thus refines the ordering of specific tests.
Adenopathy, enlarged lacrimal glands, and retinal exudates can all be asymptomatic and, if detected, may affect the differential diagnosis. Yet how many examinations focus attention on these areas? We often teach (and we were taught) the physical examination as a rote skill to be performed in toto. But the examination is not a static procedure. It needs to be tailored to the patient in front of us, in a reiterative, reflective manner. I am more likely to find adenopathy or an abdominal aneurysm if I am specifically looking for it, as opposed to performing a perfunctory examination. I am less likely to be struck by a pronounced second heart sound if I am not considering pulmonary hypertension as a possible explanation for the patient’s dyspnea.
I believe that a reflective physical examination is effective and valuable. Besides, our patients actually expect (and deserve) to be carefully examined.
The physical examination used to be a foundation of clinical practice, but it is under assault. A specialist in inpatient medicine here at Cleveland Clinic decried the inefficiency of time spent by residents performing and documenting the examination. How often, he asked, does the examination actually change the diagnostic workup? Academic provocateurs have done sensitivity and specificity analyses on components of the physical examination and found them to be imperfect. Imperfect, yes, but I would argue not worthless.
One reason for the imperfection is that skills of examination are not always appropriately emphasized during training, and then are not utilized in practice (especially in a 10-minute visit). Several published studies describe the attrition of examination skills. While doing teaching rounds as a visiting professor, I have found that some residents and medical students have difficulty distinguishing a murmur of aortic sclerosis from one of aortic insufficiency or detecting epitrochlear adenopathy.
The structured physical examination still fulfills a legitimate need. When approaching a patient with an ill-defined, potentially multisystem disorder, the examination should provide an initial “staging” of the disease process that contributes to the construction of the differential diagnosis, and thus refines the ordering of specific tests.
Adenopathy, enlarged lacrimal glands, and retinal exudates can all be asymptomatic and, if detected, may affect the differential diagnosis. Yet how many examinations focus attention on these areas? We often teach (and we were taught) the physical examination as a rote skill to be performed in toto. But the examination is not a static procedure. It needs to be tailored to the patient in front of us, in a reiterative, reflective manner. I am more likely to find adenopathy or an abdominal aneurysm if I am specifically looking for it, as opposed to performing a perfunctory examination. I am less likely to be struck by a pronounced second heart sound if I am not considering pulmonary hypertension as a possible explanation for the patient’s dyspnea.
I believe that a reflective physical examination is effective and valuable. Besides, our patients actually expect (and deserve) to be carefully examined.
A report from the department of staph affairs

Once, we worried about penicillin-resistant staph, but we had methicillin and several other effective antibiotics. Then, in the 1960s, MRSA started to appear. Intravenous drug abusers and then the chronically ill were favored hosts. Some hospitals became nests for MRSA. It began to acquire a reputation as a particularly nasty invader, associated with necrotizing pneumonia and resistant endocarditis. By 2004 more than 60% of staph isolates from critical care units were resistant to methicillin. But we had vancomycin.
Now, the hospital bugs are being reinforced by their community brethren—new strains from the suburbs that carry toxins that can damage tissue via stimulation of apoptosis of host cells. Most community-associated MRSA species are still sensitive to vancomycin (as well as to trimethoprim-sulfamethoxazole and clindamycin). But not all are. A growing community of bugs is relatively resistant to vancomycin and carries toxins—microbial suicide bombers with body armor.
The clinical presentation of necrotizing cellulitis, first appearing as a “spider bite,” is now seen in emergency wards around the country. In some cities, the overwhelming majority of deep skin infections evaluated in emergency rooms are due to MRSA. And patients with these infections, as well as asymptomatic nasal carriers of MRSA, are bringing these bugs into our hospitals.
The trends of emerging antibiotic resistance, discovery (and then perhaps overprescription) of new antibiotics, and changing patterns of staph infections are intimately intertwined. The evolutionary pressure that we humans are putting on S aureus with our antibiotics is taking this bug to a whole new place.
Once, we worried about penicillin-resistant staph, but we had methicillin and several other effective antibiotics. Then, in the 1960s, MRSA started to appear. Intravenous drug abusers and then the chronically ill were favored hosts. Some hospitals became nests for MRSA. It began to acquire a reputation as a particularly nasty invader, associated with necrotizing pneumonia and resistant endocarditis. By 2004 more than 60% of staph isolates from critical care units were resistant to methicillin. But we had vancomycin.
Now, the hospital bugs are being reinforced by their community brethren—new strains from the suburbs that carry toxins that can damage tissue via stimulation of apoptosis of host cells. Most community-associated MRSA species are still sensitive to vancomycin (as well as to trimethoprim-sulfamethoxazole and clindamycin). But not all are. A growing community of bugs is relatively resistant to vancomycin and carries toxins—microbial suicide bombers with body armor.
The clinical presentation of necrotizing cellulitis, first appearing as a “spider bite,” is now seen in emergency wards around the country. In some cities, the overwhelming majority of deep skin infections evaluated in emergency rooms are due to MRSA. And patients with these infections, as well as asymptomatic nasal carriers of MRSA, are bringing these bugs into our hospitals.
The trends of emerging antibiotic resistance, discovery (and then perhaps overprescription) of new antibiotics, and changing patterns of staph infections are intimately intertwined. The evolutionary pressure that we humans are putting on S aureus with our antibiotics is taking this bug to a whole new place.
Once, we worried about penicillin-resistant staph, but we had methicillin and several other effective antibiotics. Then, in the 1960s, MRSA started to appear. Intravenous drug abusers and then the chronically ill were favored hosts. Some hospitals became nests for MRSA. It began to acquire a reputation as a particularly nasty invader, associated with necrotizing pneumonia and resistant endocarditis. By 2004 more than 60% of staph isolates from critical care units were resistant to methicillin. But we had vancomycin.
Now, the hospital bugs are being reinforced by their community brethren—new strains from the suburbs that carry toxins that can damage tissue via stimulation of apoptosis of host cells. Most community-associated MRSA species are still sensitive to vancomycin (as well as to trimethoprim-sulfamethoxazole and clindamycin). But not all are. A growing community of bugs is relatively resistant to vancomycin and carries toxins—microbial suicide bombers with body armor.
The clinical presentation of necrotizing cellulitis, first appearing as a “spider bite,” is now seen in emergency wards around the country. In some cities, the overwhelming majority of deep skin infections evaluated in emergency rooms are due to MRSA. And patients with these infections, as well as asymptomatic nasal carriers of MRSA, are bringing these bugs into our hospitals.
The trends of emerging antibiotic resistance, discovery (and then perhaps overprescription) of new antibiotics, and changing patterns of staph infections are intimately intertwined. The evolutionary pressure that we humans are putting on S aureus with our antibiotics is taking this bug to a whole new place.
Surprisingly nonbenign imaging

As reviewed by Dr. Naim Issa and colleagues in this issue of the Journal, practices are changing with regard to choice of imaging techniques and contrast materials in patients with renal insufficiency. In 1 week, while on our inpatient rheumatology consultation service, I specifically commented in two patients’ charts that I would prefer to avoid the use of gadolinium because the patients had significant renal dysfunction. Since the patients did not yet need dialysis, standard contrast dye was also relatively contraindicated. It was a bit of a dilemma.
In an accompanying editorial, Dr. Jonathan Kay proposes that this pseudoscleroderma syndrome be called “gadolinium-associated systemic fibrosis (GASF).” Dr. Kay and colleagues have recently published an important study (Arthritis Rheum 2007; 56:3433–3441) in which they report that, with a focused physical examination, this often-unrecognized clinical syndrome can be diagnosed in 13% of patients undergoing chronic hemodialysis and that the diagnosis indicates a significant risk of death. They confirm the suggestion of earlier authors that exposure to gadolinium is a significant predisposing factor for the syndrome.
Gadolinium is not the first exogenous chemical trigger of a fibrosing syndrome to be identified: eg, bleomycin (Blenoxane) is a well-known trigger of pulmonary fibrosis. Pseudoscleroderma syndromes have been described after exposure to certain rapeseed oils and to impure preparations of tryptophan (the “eosinophilic-mayalgia syndrome”). The mechanisms of these reactions are not fully understood, and other than the accumulation of gadolinium in tissues in the setting of chronic renal disease, not much is known about the pathophysiology of GASF.
Guidelines will be proposed to try to limit the occurrence of this devastating syndrome. But we can only guess as to the glomerular filtration rate cutoff at which we should be most concerned, and we can only hope that acute dialysis after gadolinium exposure will be protective. In the meantime, we will need to revise our view that “MRI with contrast” is a benign test.
As reviewed by Dr. Naim Issa and colleagues in this issue of the Journal, practices are changing with regard to choice of imaging techniques and contrast materials in patients with renal insufficiency. In 1 week, while on our inpatient rheumatology consultation service, I specifically commented in two patients’ charts that I would prefer to avoid the use of gadolinium because the patients had significant renal dysfunction. Since the patients did not yet need dialysis, standard contrast dye was also relatively contraindicated. It was a bit of a dilemma.
In an accompanying editorial, Dr. Jonathan Kay proposes that this pseudoscleroderma syndrome be called “gadolinium-associated systemic fibrosis (GASF).” Dr. Kay and colleagues have recently published an important study (Arthritis Rheum 2007; 56:3433–3441) in which they report that, with a focused physical examination, this often-unrecognized clinical syndrome can be diagnosed in 13% of patients undergoing chronic hemodialysis and that the diagnosis indicates a significant risk of death. They confirm the suggestion of earlier authors that exposure to gadolinium is a significant predisposing factor for the syndrome.
Gadolinium is not the first exogenous chemical trigger of a fibrosing syndrome to be identified: eg, bleomycin (Blenoxane) is a well-known trigger of pulmonary fibrosis. Pseudoscleroderma syndromes have been described after exposure to certain rapeseed oils and to impure preparations of tryptophan (the “eosinophilic-mayalgia syndrome”). The mechanisms of these reactions are not fully understood, and other than the accumulation of gadolinium in tissues in the setting of chronic renal disease, not much is known about the pathophysiology of GASF.
Guidelines will be proposed to try to limit the occurrence of this devastating syndrome. But we can only guess as to the glomerular filtration rate cutoff at which we should be most concerned, and we can only hope that acute dialysis after gadolinium exposure will be protective. In the meantime, we will need to revise our view that “MRI with contrast” is a benign test.
As reviewed by Dr. Naim Issa and colleagues in this issue of the Journal, practices are changing with regard to choice of imaging techniques and contrast materials in patients with renal insufficiency. In 1 week, while on our inpatient rheumatology consultation service, I specifically commented in two patients’ charts that I would prefer to avoid the use of gadolinium because the patients had significant renal dysfunction. Since the patients did not yet need dialysis, standard contrast dye was also relatively contraindicated. It was a bit of a dilemma.
In an accompanying editorial, Dr. Jonathan Kay proposes that this pseudoscleroderma syndrome be called “gadolinium-associated systemic fibrosis (GASF).” Dr. Kay and colleagues have recently published an important study (Arthritis Rheum 2007; 56:3433–3441) in which they report that, with a focused physical examination, this often-unrecognized clinical syndrome can be diagnosed in 13% of patients undergoing chronic hemodialysis and that the diagnosis indicates a significant risk of death. They confirm the suggestion of earlier authors that exposure to gadolinium is a significant predisposing factor for the syndrome.
Gadolinium is not the first exogenous chemical trigger of a fibrosing syndrome to be identified: eg, bleomycin (Blenoxane) is a well-known trigger of pulmonary fibrosis. Pseudoscleroderma syndromes have been described after exposure to certain rapeseed oils and to impure preparations of tryptophan (the “eosinophilic-mayalgia syndrome”). The mechanisms of these reactions are not fully understood, and other than the accumulation of gadolinium in tissues in the setting of chronic renal disease, not much is known about the pathophysiology of GASF.
Guidelines will be proposed to try to limit the occurrence of this devastating syndrome. But we can only guess as to the glomerular filtration rate cutoff at which we should be most concerned, and we can only hope that acute dialysis after gadolinium exposure will be protective. In the meantime, we will need to revise our view that “MRI with contrast” is a benign test.
A New Year's toast

In this issue, Dr. Herbert Wiedemann, Chairman of the Department of Pulmonary, Allergy, and Critical Care Medicine at Cleveland Clinic, discusses the results of the Fluids and Catheters Treatment Trial (FACTT), of which he was cochair, and the impact that these results should have on the management of patients with acute lung injury and acute respiratory distress syndrome (ARDS).
The results are reasonably clear. Patients with ARDS should be treated with a strategy of low tidal volume ventilation (as previously shown) and also with fluid restriction and diuretics as needed in an attempt to reach the targeted low filling pressures (a central venous pressure < 4 mm Hg). But even with this aggressive fluid management approach, there was no consistent benefit from the early use of pulmonary artery catheters for pressure monitoring.
So my former attendings in the intensive care unit were right after all! I still remember them during my residency explaining how, with careful physical examination and vigilance, I could avoid the need for pulmonary catheters even as I attempted to avoid lung-stretching by “running the patient dry” and using lower ventilatory volumes. And they taught this on the basis of their knowledge of physiology and their clinical observations—the clinical trials had not yet been performed.
Multicenter networks of clinicians conducting trials such as FACTT represent major accomplishments of the medical system. These multicenter trials have the potential to generate useful information about complicated and sometimes uncommon diseases, and to change the way we practice medicine. But to me, the real accomplishments are made by the clinician in the trenches who, with eyes wide open,processes observational experiences and generates the questions that ultimately get tested in experimental trials.
The networks can accomplish patient recruitment, the “number crunchers” can generate the P values and perform their regression analyses, but it is the clinician with the bedside skills who often generates the meaningful questions.
So as we begin another year, certain to be filled with new and important research successes, I offer a toast to those clinicians who, despite the pressures of time, continue to hone their observational prowess, synthesize the published literature, develop the hypotheses that studies like FACTT address, and pass them on to their students—who may fondly remember them 25 years later.
In this issue, Dr. Herbert Wiedemann, Chairman of the Department of Pulmonary, Allergy, and Critical Care Medicine at Cleveland Clinic, discusses the results of the Fluids and Catheters Treatment Trial (FACTT), of which he was cochair, and the impact that these results should have on the management of patients with acute lung injury and acute respiratory distress syndrome (ARDS).
The results are reasonably clear. Patients with ARDS should be treated with a strategy of low tidal volume ventilation (as previously shown) and also with fluid restriction and diuretics as needed in an attempt to reach the targeted low filling pressures (a central venous pressure < 4 mm Hg). But even with this aggressive fluid management approach, there was no consistent benefit from the early use of pulmonary artery catheters for pressure monitoring.
So my former attendings in the intensive care unit were right after all! I still remember them during my residency explaining how, with careful physical examination and vigilance, I could avoid the need for pulmonary catheters even as I attempted to avoid lung-stretching by “running the patient dry” and using lower ventilatory volumes. And they taught this on the basis of their knowledge of physiology and their clinical observations—the clinical trials had not yet been performed.
Multicenter networks of clinicians conducting trials such as FACTT represent major accomplishments of the medical system. These multicenter trials have the potential to generate useful information about complicated and sometimes uncommon diseases, and to change the way we practice medicine. But to me, the real accomplishments are made by the clinician in the trenches who, with eyes wide open,processes observational experiences and generates the questions that ultimately get tested in experimental trials.
The networks can accomplish patient recruitment, the “number crunchers” can generate the P values and perform their regression analyses, but it is the clinician with the bedside skills who often generates the meaningful questions.
So as we begin another year, certain to be filled with new and important research successes, I offer a toast to those clinicians who, despite the pressures of time, continue to hone their observational prowess, synthesize the published literature, develop the hypotheses that studies like FACTT address, and pass them on to their students—who may fondly remember them 25 years later.
In this issue, Dr. Herbert Wiedemann, Chairman of the Department of Pulmonary, Allergy, and Critical Care Medicine at Cleveland Clinic, discusses the results of the Fluids and Catheters Treatment Trial (FACTT), of which he was cochair, and the impact that these results should have on the management of patients with acute lung injury and acute respiratory distress syndrome (ARDS).
The results are reasonably clear. Patients with ARDS should be treated with a strategy of low tidal volume ventilation (as previously shown) and also with fluid restriction and diuretics as needed in an attempt to reach the targeted low filling pressures (a central venous pressure < 4 mm Hg). But even with this aggressive fluid management approach, there was no consistent benefit from the early use of pulmonary artery catheters for pressure monitoring.
So my former attendings in the intensive care unit were right after all! I still remember them during my residency explaining how, with careful physical examination and vigilance, I could avoid the need for pulmonary catheters even as I attempted to avoid lung-stretching by “running the patient dry” and using lower ventilatory volumes. And they taught this on the basis of their knowledge of physiology and their clinical observations—the clinical trials had not yet been performed.
Multicenter networks of clinicians conducting trials such as FACTT represent major accomplishments of the medical system. These multicenter trials have the potential to generate useful information about complicated and sometimes uncommon diseases, and to change the way we practice medicine. But to me, the real accomplishments are made by the clinician in the trenches who, with eyes wide open,processes observational experiences and generates the questions that ultimately get tested in experimental trials.
The networks can accomplish patient recruitment, the “number crunchers” can generate the P values and perform their regression analyses, but it is the clinician with the bedside skills who often generates the meaningful questions.
So as we begin another year, certain to be filled with new and important research successes, I offer a toast to those clinicians who, despite the pressures of time, continue to hone their observational prowess, synthesize the published literature, develop the hypotheses that studies like FACTT address, and pass them on to their students—who may fondly remember them 25 years later.