User login
CV outcomes data needed to expand approval of TG-lowering agent
SILVER SPRING, MD. – An approved formulation of an omega-3 fatty acid – icosapent ethyl – was effective in reducing triglycerides when added to a statin in adults with mixed dyslipidemia, at a high risk of coronary heart disease, but it should not be approved for treating such patients until the results of a cardiovascular outcomes trial become available, an expert panel has advised the Food and Drug Administration.
Icosapent ethyl is a purified formulation of ethyl eicosapentaenoic acid (EPA), derived from fish oil, which was approved by the FDA in 2012 as monotherapy for treatment of severe hypertriglyceridemia (500 mg/dL or higher), based on the premise that lowering triglyceride levels reduces the risk of pancreatitis, not that it affects cardiovascular outcomes, according to the FDA. It comes in a capsule formulation and is marketed as Vascepa by Amarin Pharmaceuticals Ireland Ltd.
On the basis of the results of the ANCHOR study, the manufacturer has proposed that Vascepa be approved as an adjunct to diet in combination with a statin to reduce triglycerides (TG), non-HDL cholesterol (non-HDL-C), apolipoprotein B (Apo B), LDL cholesterol (LDL-C), total cholesterol (TC) and very-LDL cholesterol (VLDL-C) "in adults with mixed dyslipidemia and coronary heart disease or a CHD risk equivalent."
However, at its meeting on Oct. 16, the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee voted 9-2 that the favorable effects of treatment with 4 g/day of Vascepa on triglycerides and other lipid and lipoprotein parameters over placebo after 12 weeks in the ANCHOR study were not sufficient to recommend approval. Those who voted against approval recommended waiting for the results of the REDUCE-IT study, a large CV outcomes trial of Vascepa currently underway, to determine if these effects translate into improved cardiovascular outcomes, before approving the expanded indication.
The ANCHOR study showed that Vascepa "effectively lowers triglycerides in the proposed population," but until the CV outcomes study is completed, "I just don’t know if it will reduce CV risk," said Dr. Ellen Seely, professor of medicine at Harvard University and an endocrinologist at Brigham and Women’s Hospital, Boston.
Another panel member, Dr. Brendan Everett, director of the general cardiology inpatient service at Brigham and Women’s, said that he was optimistic about the data and that treatment with Vascepa "could very well offer potential substantial benefits for patients with coronary heart disease or coronary heart disease risk equivalents." However, it’s not clear that reducing triglycerides will have an impact on that risk and "I am wary ... of approving a drug that has a potential market of tens of millions of people without any hard efficacy data," he added.
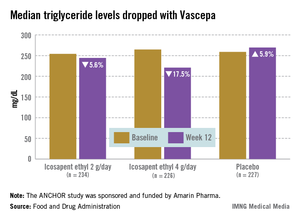
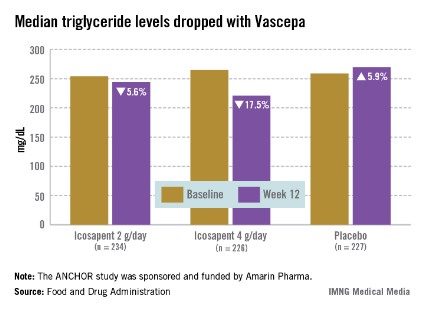
The ANCHOR study compared two doses of Vascepa to placebo in about 700 mostly white, male patients with mixed dyslipidemia and at high cardiovascular risk, who were taking a statin. Their mean age was 61 years and 73% had diabetes. After 12 weeks, the fasting TG value increased by a median of 5.9% from baseline among those on placebo, compared with a median 17.5% reduction among those on 4 g of Vascepa daily, the proposed dose – a 21.5% treatment difference that was highly statistically significant. Other changes among those on placebo went in the adverse direction, with the exception of HDL-C, which increased by a median of 4.8% vs. a median reduction of 1% among those on Vascepa.
Overall, Vascepa was well tolerated, with no new safety issues identified, according to the company. The most common adverse events associated with treatment were diarrhea, urinary tract infections, upper respiratory tract infections, and nausea, and the withdrawal rate due to adverse events was low (2% among those on 4 g/day). The FDA reviewers agreed with the company’s assessment of safety, although several panelists pointed out that the safety data are from a relatively small number of patients studied, and expressed some concerns about bleeding events (2.6% of those on 4 g/day vs. 1.7% of those on placebo).
The main issue discussed was the clinical significance of the effects of treatment with Vascepa, when administered with a statin, on triglycerides, and other changes in lipid and lipoproteins observed in the ANCHOR study, whether these changes would translate into a meaningful reduction ion CV risk, in the target population. The panelists agreed that Vascepa was effective in lowering TGs, but they were concerned about the possibility that the effects on TGs could be overstated because Vascepa was compared with placebo in the study, and they agreed that the impact on CV outcomes was uncertain, based on the data.
During the FDA review of the data, Dr. Mary Dunne Roberts, a clinical reviewer in the FDA’s Division of Metabolism and Endocrinology Products, referred to the ongoing debate over whether drug-induced modulation of TGs results in improved CV outcomes. Results of recent CV outcome trials with fenofibrate and niacin have raised questions about whether targeting lipids and lipoproteins other than LDL-cholesterol results in incremental cardiovascular benefits in patients on optimal statin therapy and LDL-C control, she pointed out.
In 2011, Amarin launched REDUCE-IT, which is comparing the impact of treatment with 4 g of Vascepa per day to placebo added to statin therapy in 8,000 patients with CV disease or CV risk, on a composite endpoint of CV death, nonfatal MI, nonfatal stroke, coronary revascularization, and unstable angina requiring hospitalization. At the beginning of October, about 6,000 patients with a median TG level of 202 mg/dL had been enrolled. The company expects the study to be completed in 2016 or 2017.
Panelists agreed that REDUCE-IT was a well-designed, important study, with an appropriate group of patients, which should provide answers about whether CV outcomes will be improved with Vascepa, and they were encouraged that the study was underway.
Panelist Dr. Peter Wilson, professor of medicine in the cardiology division of Emory University, Atlanta, said that there was no need to prescribe more agents "because we think they work," and he expressed concern about overprescribing. "There is already overuse of some omega-3s without proven benefit. We need the trial results in hand to guide decisions."
Vascepa is one of two omega-3 fatty acid formulations approved by the FDA; the other is Lovaza, a combination of EPA and docosahexaenoic acid (DHA). Many omega- 3 fatty acid products are available over the counter, with variable combinations and strengths.
The FDA usually follows the recommendations of its advisory panels. Members have been cleared of potential conflicts; occasionally, a panelist is given a waiver for a potential conflict, but not at this meeting.
SILVER SPRING, MD. – An approved formulation of an omega-3 fatty acid – icosapent ethyl – was effective in reducing triglycerides when added to a statin in adults with mixed dyslipidemia, at a high risk of coronary heart disease, but it should not be approved for treating such patients until the results of a cardiovascular outcomes trial become available, an expert panel has advised the Food and Drug Administration.
Icosapent ethyl is a purified formulation of ethyl eicosapentaenoic acid (EPA), derived from fish oil, which was approved by the FDA in 2012 as monotherapy for treatment of severe hypertriglyceridemia (500 mg/dL or higher), based on the premise that lowering triglyceride levels reduces the risk of pancreatitis, not that it affects cardiovascular outcomes, according to the FDA. It comes in a capsule formulation and is marketed as Vascepa by Amarin Pharmaceuticals Ireland Ltd.
On the basis of the results of the ANCHOR study, the manufacturer has proposed that Vascepa be approved as an adjunct to diet in combination with a statin to reduce triglycerides (TG), non-HDL cholesterol (non-HDL-C), apolipoprotein B (Apo B), LDL cholesterol (LDL-C), total cholesterol (TC) and very-LDL cholesterol (VLDL-C) "in adults with mixed dyslipidemia and coronary heart disease or a CHD risk equivalent."
However, at its meeting on Oct. 16, the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee voted 9-2 that the favorable effects of treatment with 4 g/day of Vascepa on triglycerides and other lipid and lipoprotein parameters over placebo after 12 weeks in the ANCHOR study were not sufficient to recommend approval. Those who voted against approval recommended waiting for the results of the REDUCE-IT study, a large CV outcomes trial of Vascepa currently underway, to determine if these effects translate into improved cardiovascular outcomes, before approving the expanded indication.
The ANCHOR study showed that Vascepa "effectively lowers triglycerides in the proposed population," but until the CV outcomes study is completed, "I just don’t know if it will reduce CV risk," said Dr. Ellen Seely, professor of medicine at Harvard University and an endocrinologist at Brigham and Women’s Hospital, Boston.
Another panel member, Dr. Brendan Everett, director of the general cardiology inpatient service at Brigham and Women’s, said that he was optimistic about the data and that treatment with Vascepa "could very well offer potential substantial benefits for patients with coronary heart disease or coronary heart disease risk equivalents." However, it’s not clear that reducing triglycerides will have an impact on that risk and "I am wary ... of approving a drug that has a potential market of tens of millions of people without any hard efficacy data," he added.
The ANCHOR study compared two doses of Vascepa to placebo in about 700 mostly white, male patients with mixed dyslipidemia and at high cardiovascular risk, who were taking a statin. Their mean age was 61 years and 73% had diabetes. After 12 weeks, the fasting TG value increased by a median of 5.9% from baseline among those on placebo, compared with a median 17.5% reduction among those on 4 g of Vascepa daily, the proposed dose – a 21.5% treatment difference that was highly statistically significant. Other changes among those on placebo went in the adverse direction, with the exception of HDL-C, which increased by a median of 4.8% vs. a median reduction of 1% among those on Vascepa.
Overall, Vascepa was well tolerated, with no new safety issues identified, according to the company. The most common adverse events associated with treatment were diarrhea, urinary tract infections, upper respiratory tract infections, and nausea, and the withdrawal rate due to adverse events was low (2% among those on 4 g/day). The FDA reviewers agreed with the company’s assessment of safety, although several panelists pointed out that the safety data are from a relatively small number of patients studied, and expressed some concerns about bleeding events (2.6% of those on 4 g/day vs. 1.7% of those on placebo).
The main issue discussed was the clinical significance of the effects of treatment with Vascepa, when administered with a statin, on triglycerides, and other changes in lipid and lipoproteins observed in the ANCHOR study, whether these changes would translate into a meaningful reduction ion CV risk, in the target population. The panelists agreed that Vascepa was effective in lowering TGs, but they were concerned about the possibility that the effects on TGs could be overstated because Vascepa was compared with placebo in the study, and they agreed that the impact on CV outcomes was uncertain, based on the data.
During the FDA review of the data, Dr. Mary Dunne Roberts, a clinical reviewer in the FDA’s Division of Metabolism and Endocrinology Products, referred to the ongoing debate over whether drug-induced modulation of TGs results in improved CV outcomes. Results of recent CV outcome trials with fenofibrate and niacin have raised questions about whether targeting lipids and lipoproteins other than LDL-cholesterol results in incremental cardiovascular benefits in patients on optimal statin therapy and LDL-C control, she pointed out.
In 2011, Amarin launched REDUCE-IT, which is comparing the impact of treatment with 4 g of Vascepa per day to placebo added to statin therapy in 8,000 patients with CV disease or CV risk, on a composite endpoint of CV death, nonfatal MI, nonfatal stroke, coronary revascularization, and unstable angina requiring hospitalization. At the beginning of October, about 6,000 patients with a median TG level of 202 mg/dL had been enrolled. The company expects the study to be completed in 2016 or 2017.
Panelists agreed that REDUCE-IT was a well-designed, important study, with an appropriate group of patients, which should provide answers about whether CV outcomes will be improved with Vascepa, and they were encouraged that the study was underway.
Panelist Dr. Peter Wilson, professor of medicine in the cardiology division of Emory University, Atlanta, said that there was no need to prescribe more agents "because we think they work," and he expressed concern about overprescribing. "There is already overuse of some omega-3s without proven benefit. We need the trial results in hand to guide decisions."
Vascepa is one of two omega-3 fatty acid formulations approved by the FDA; the other is Lovaza, a combination of EPA and docosahexaenoic acid (DHA). Many omega- 3 fatty acid products are available over the counter, with variable combinations and strengths.
The FDA usually follows the recommendations of its advisory panels. Members have been cleared of potential conflicts; occasionally, a panelist is given a waiver for a potential conflict, but not at this meeting.
SILVER SPRING, MD. – An approved formulation of an omega-3 fatty acid – icosapent ethyl – was effective in reducing triglycerides when added to a statin in adults with mixed dyslipidemia, at a high risk of coronary heart disease, but it should not be approved for treating such patients until the results of a cardiovascular outcomes trial become available, an expert panel has advised the Food and Drug Administration.
Icosapent ethyl is a purified formulation of ethyl eicosapentaenoic acid (EPA), derived from fish oil, which was approved by the FDA in 2012 as monotherapy for treatment of severe hypertriglyceridemia (500 mg/dL or higher), based on the premise that lowering triglyceride levels reduces the risk of pancreatitis, not that it affects cardiovascular outcomes, according to the FDA. It comes in a capsule formulation and is marketed as Vascepa by Amarin Pharmaceuticals Ireland Ltd.
On the basis of the results of the ANCHOR study, the manufacturer has proposed that Vascepa be approved as an adjunct to diet in combination with a statin to reduce triglycerides (TG), non-HDL cholesterol (non-HDL-C), apolipoprotein B (Apo B), LDL cholesterol (LDL-C), total cholesterol (TC) and very-LDL cholesterol (VLDL-C) "in adults with mixed dyslipidemia and coronary heart disease or a CHD risk equivalent."
However, at its meeting on Oct. 16, the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee voted 9-2 that the favorable effects of treatment with 4 g/day of Vascepa on triglycerides and other lipid and lipoprotein parameters over placebo after 12 weeks in the ANCHOR study were not sufficient to recommend approval. Those who voted against approval recommended waiting for the results of the REDUCE-IT study, a large CV outcomes trial of Vascepa currently underway, to determine if these effects translate into improved cardiovascular outcomes, before approving the expanded indication.
The ANCHOR study showed that Vascepa "effectively lowers triglycerides in the proposed population," but until the CV outcomes study is completed, "I just don’t know if it will reduce CV risk," said Dr. Ellen Seely, professor of medicine at Harvard University and an endocrinologist at Brigham and Women’s Hospital, Boston.
Another panel member, Dr. Brendan Everett, director of the general cardiology inpatient service at Brigham and Women’s, said that he was optimistic about the data and that treatment with Vascepa "could very well offer potential substantial benefits for patients with coronary heart disease or coronary heart disease risk equivalents." However, it’s not clear that reducing triglycerides will have an impact on that risk and "I am wary ... of approving a drug that has a potential market of tens of millions of people without any hard efficacy data," he added.
The ANCHOR study compared two doses of Vascepa to placebo in about 700 mostly white, male patients with mixed dyslipidemia and at high cardiovascular risk, who were taking a statin. Their mean age was 61 years and 73% had diabetes. After 12 weeks, the fasting TG value increased by a median of 5.9% from baseline among those on placebo, compared with a median 17.5% reduction among those on 4 g of Vascepa daily, the proposed dose – a 21.5% treatment difference that was highly statistically significant. Other changes among those on placebo went in the adverse direction, with the exception of HDL-C, which increased by a median of 4.8% vs. a median reduction of 1% among those on Vascepa.
Overall, Vascepa was well tolerated, with no new safety issues identified, according to the company. The most common adverse events associated with treatment were diarrhea, urinary tract infections, upper respiratory tract infections, and nausea, and the withdrawal rate due to adverse events was low (2% among those on 4 g/day). The FDA reviewers agreed with the company’s assessment of safety, although several panelists pointed out that the safety data are from a relatively small number of patients studied, and expressed some concerns about bleeding events (2.6% of those on 4 g/day vs. 1.7% of those on placebo).
The main issue discussed was the clinical significance of the effects of treatment with Vascepa, when administered with a statin, on triglycerides, and other changes in lipid and lipoproteins observed in the ANCHOR study, whether these changes would translate into a meaningful reduction ion CV risk, in the target population. The panelists agreed that Vascepa was effective in lowering TGs, but they were concerned about the possibility that the effects on TGs could be overstated because Vascepa was compared with placebo in the study, and they agreed that the impact on CV outcomes was uncertain, based on the data.
During the FDA review of the data, Dr. Mary Dunne Roberts, a clinical reviewer in the FDA’s Division of Metabolism and Endocrinology Products, referred to the ongoing debate over whether drug-induced modulation of TGs results in improved CV outcomes. Results of recent CV outcome trials with fenofibrate and niacin have raised questions about whether targeting lipids and lipoproteins other than LDL-cholesterol results in incremental cardiovascular benefits in patients on optimal statin therapy and LDL-C control, she pointed out.
In 2011, Amarin launched REDUCE-IT, which is comparing the impact of treatment with 4 g of Vascepa per day to placebo added to statin therapy in 8,000 patients with CV disease or CV risk, on a composite endpoint of CV death, nonfatal MI, nonfatal stroke, coronary revascularization, and unstable angina requiring hospitalization. At the beginning of October, about 6,000 patients with a median TG level of 202 mg/dL had been enrolled. The company expects the study to be completed in 2016 or 2017.
Panelists agreed that REDUCE-IT was a well-designed, important study, with an appropriate group of patients, which should provide answers about whether CV outcomes will be improved with Vascepa, and they were encouraged that the study was underway.
Panelist Dr. Peter Wilson, professor of medicine in the cardiology division of Emory University, Atlanta, said that there was no need to prescribe more agents "because we think they work," and he expressed concern about overprescribing. "There is already overuse of some omega-3s without proven benefit. We need the trial results in hand to guide decisions."
Vascepa is one of two omega-3 fatty acid formulations approved by the FDA; the other is Lovaza, a combination of EPA and docosahexaenoic acid (DHA). Many omega- 3 fatty acid products are available over the counter, with variable combinations and strengths.
The FDA usually follows the recommendations of its advisory panels. Members have been cleared of potential conflicts; occasionally, a panelist is given a waiver for a potential conflict, but not at this meeting.
FROM AN FDA ADVISORY PANEL MEETING
Isometric exercises among the recommendations for sports-related knee pain
WASHINGTON – For patients who come to the office complaining of knee pain, Dr. William Phillips advises physicians to always check for the motion of the hips, a common source of knee pain in athletes with overuse injuries.
At a practical pediatrics meeting sponsored by the American Academy of Pediatrics, Dr. Phillips, chief of the orthopedic service and orthopedic surgery clinic at Texas Children’s Hospital, Houston, recommended the following test to check for hip rotation: While the child is lying supine, flex their hips up to 90 degrees and flex their knees up "and try to internally rotate their legs, which means take their feet out and point the kneecaps in." X-rays are not necessary in these cases, he added.
Dr. Phillips also emphasized the value of encouraging patients with knee pain to start doing isometric exercises, which can be taught in the office and can be done at home. With a few exceptions, "the cure-all exercise for virtually every type of knee pain ... is isometric quadriceps strengthening exercises."
Repetitive overuse injuries occur when "an excessive, repetitive load overwhelms the body’s normal repair processes." Such injuries account for the majority of sports injuries seen by pediatricians – a result of the trend toward earlier specialization in sports, more-intensive involvement starting at younger ages, and year-round participation in one sport. These injuries can be soft-tissue or bony injuries, said Dr. Phillips, professor of pediatrics at Baylor College of Medicine, Houston.
In any evaluation, the history is the most important element, he said. The physical exam can be helpful, but imaging studies are less useful. He recommended asking patients when they experience pain, such as during an athletic activity; whether it limits their performance; what they are able to do; and how long they have been experiencing the pain.
Another question is whether there have been recent changes in their activity or sports practices, said Dr. Phillips. He described three cheerleaders from the same squad who came to his office complaining of the same type of knee pain around the same time. The cause turned out to be a new drill they were doing in practice.
An x-ray may be considered in situations that include a history of acute swelling, obvious limb asymmetry, limited range of motion, and certainly, when the patient is unable to bear weight. An x-ray "is usually not needed when the condition is bilateral ... and clearly the same on both sides," he said. But he added that he always offers the family the option of getting an x-ray if the child has not improved after a certain amount of time.
Injuries to the extensor mechanisms are probably the most common overuse injuries. "And again, knee pain can be referred from the hip," Dr. Phillips pointed out. Every year, he sees junior high school football players who have twisted their knees in a game and have been limping, but have a normal knee exam and turn out to have a slipped lateral femoral epiphysis. "So be sure to check the hips," he said, noting that the range of motion should be symmetrical, with at least 20 or 30 degrees of internal rotation, "and the feet go out, the kneecaps go in."
The most common causes of knee pain in young athletes are extensor patellofemoral problems, characterized by pain with such activities as running or climbing stairs. Another activity that elicits this type of pain is rising up from a sitting position, which "jams" the patellofemoral joint, he said. In such cases, there tends to be tenderness under the patella. "Usually, if you push the patella a little bit laterally, they will have pain."
Management of patellofemoral problems includes activity as tolerated; ice and analgesics; isometric quadriceps strengthening; and quadriceps and hamstring stretching. A brace – the type with a neoprene sleeve with a hole cut out, not a metal brace – is also acceptable to use if the patient likes it, but it is not essential, Dr. Phillips said.
Physical therapy is not usually necessary. Instead, he recommends teaching children who present with knee pain the following fundamental exercise: While lying down, bend the unaffected leg and keep that foot flat on the floor, then lift the affected leg 6-12 inches off the table with the knee fully extended, toe pointed to the ceiling – keeping it up for 20 seconds, down for 10 seconds, and repeating this for one or two sets of 20 repetitions, depending on the child and other factors. They should avoid lifting both legs up off the ground at the same time, which can hurt their backs.
For weights, they can use a knapsack, even their mother’s purse, starting with 1 or 2 pounds (or no weights if they are very sore). "This will go a long way toward helping them mend," he said.
Dr. Phillips advises avoiding weight machines where they kick their legs out, which is like getting up from a sitting position; squats; or stair climbing. He tells families that patellofemoral problems wax and wane, and "very rarely" require surgery, and he encourages a lot of rehabilitation before considering a recommendation for any type of surgery.
Dr. Phillips said he had no relevant disclosures.
WASHINGTON – For patients who come to the office complaining of knee pain, Dr. William Phillips advises physicians to always check for the motion of the hips, a common source of knee pain in athletes with overuse injuries.
At a practical pediatrics meeting sponsored by the American Academy of Pediatrics, Dr. Phillips, chief of the orthopedic service and orthopedic surgery clinic at Texas Children’s Hospital, Houston, recommended the following test to check for hip rotation: While the child is lying supine, flex their hips up to 90 degrees and flex their knees up "and try to internally rotate their legs, which means take their feet out and point the kneecaps in." X-rays are not necessary in these cases, he added.
Dr. Phillips also emphasized the value of encouraging patients with knee pain to start doing isometric exercises, which can be taught in the office and can be done at home. With a few exceptions, "the cure-all exercise for virtually every type of knee pain ... is isometric quadriceps strengthening exercises."
Repetitive overuse injuries occur when "an excessive, repetitive load overwhelms the body’s normal repair processes." Such injuries account for the majority of sports injuries seen by pediatricians – a result of the trend toward earlier specialization in sports, more-intensive involvement starting at younger ages, and year-round participation in one sport. These injuries can be soft-tissue or bony injuries, said Dr. Phillips, professor of pediatrics at Baylor College of Medicine, Houston.
In any evaluation, the history is the most important element, he said. The physical exam can be helpful, but imaging studies are less useful. He recommended asking patients when they experience pain, such as during an athletic activity; whether it limits their performance; what they are able to do; and how long they have been experiencing the pain.
Another question is whether there have been recent changes in their activity or sports practices, said Dr. Phillips. He described three cheerleaders from the same squad who came to his office complaining of the same type of knee pain around the same time. The cause turned out to be a new drill they were doing in practice.
An x-ray may be considered in situations that include a history of acute swelling, obvious limb asymmetry, limited range of motion, and certainly, when the patient is unable to bear weight. An x-ray "is usually not needed when the condition is bilateral ... and clearly the same on both sides," he said. But he added that he always offers the family the option of getting an x-ray if the child has not improved after a certain amount of time.
Injuries to the extensor mechanisms are probably the most common overuse injuries. "And again, knee pain can be referred from the hip," Dr. Phillips pointed out. Every year, he sees junior high school football players who have twisted their knees in a game and have been limping, but have a normal knee exam and turn out to have a slipped lateral femoral epiphysis. "So be sure to check the hips," he said, noting that the range of motion should be symmetrical, with at least 20 or 30 degrees of internal rotation, "and the feet go out, the kneecaps go in."
The most common causes of knee pain in young athletes are extensor patellofemoral problems, characterized by pain with such activities as running or climbing stairs. Another activity that elicits this type of pain is rising up from a sitting position, which "jams" the patellofemoral joint, he said. In such cases, there tends to be tenderness under the patella. "Usually, if you push the patella a little bit laterally, they will have pain."
Management of patellofemoral problems includes activity as tolerated; ice and analgesics; isometric quadriceps strengthening; and quadriceps and hamstring stretching. A brace – the type with a neoprene sleeve with a hole cut out, not a metal brace – is also acceptable to use if the patient likes it, but it is not essential, Dr. Phillips said.
Physical therapy is not usually necessary. Instead, he recommends teaching children who present with knee pain the following fundamental exercise: While lying down, bend the unaffected leg and keep that foot flat on the floor, then lift the affected leg 6-12 inches off the table with the knee fully extended, toe pointed to the ceiling – keeping it up for 20 seconds, down for 10 seconds, and repeating this for one or two sets of 20 repetitions, depending on the child and other factors. They should avoid lifting both legs up off the ground at the same time, which can hurt their backs.
For weights, they can use a knapsack, even their mother’s purse, starting with 1 or 2 pounds (or no weights if they are very sore). "This will go a long way toward helping them mend," he said.
Dr. Phillips advises avoiding weight machines where they kick their legs out, which is like getting up from a sitting position; squats; or stair climbing. He tells families that patellofemoral problems wax and wane, and "very rarely" require surgery, and he encourages a lot of rehabilitation before considering a recommendation for any type of surgery.
Dr. Phillips said he had no relevant disclosures.
WASHINGTON – For patients who come to the office complaining of knee pain, Dr. William Phillips advises physicians to always check for the motion of the hips, a common source of knee pain in athletes with overuse injuries.
At a practical pediatrics meeting sponsored by the American Academy of Pediatrics, Dr. Phillips, chief of the orthopedic service and orthopedic surgery clinic at Texas Children’s Hospital, Houston, recommended the following test to check for hip rotation: While the child is lying supine, flex their hips up to 90 degrees and flex their knees up "and try to internally rotate their legs, which means take their feet out and point the kneecaps in." X-rays are not necessary in these cases, he added.
Dr. Phillips also emphasized the value of encouraging patients with knee pain to start doing isometric exercises, which can be taught in the office and can be done at home. With a few exceptions, "the cure-all exercise for virtually every type of knee pain ... is isometric quadriceps strengthening exercises."
Repetitive overuse injuries occur when "an excessive, repetitive load overwhelms the body’s normal repair processes." Such injuries account for the majority of sports injuries seen by pediatricians – a result of the trend toward earlier specialization in sports, more-intensive involvement starting at younger ages, and year-round participation in one sport. These injuries can be soft-tissue or bony injuries, said Dr. Phillips, professor of pediatrics at Baylor College of Medicine, Houston.
In any evaluation, the history is the most important element, he said. The physical exam can be helpful, but imaging studies are less useful. He recommended asking patients when they experience pain, such as during an athletic activity; whether it limits their performance; what they are able to do; and how long they have been experiencing the pain.
Another question is whether there have been recent changes in their activity or sports practices, said Dr. Phillips. He described three cheerleaders from the same squad who came to his office complaining of the same type of knee pain around the same time. The cause turned out to be a new drill they were doing in practice.
An x-ray may be considered in situations that include a history of acute swelling, obvious limb asymmetry, limited range of motion, and certainly, when the patient is unable to bear weight. An x-ray "is usually not needed when the condition is bilateral ... and clearly the same on both sides," he said. But he added that he always offers the family the option of getting an x-ray if the child has not improved after a certain amount of time.
Injuries to the extensor mechanisms are probably the most common overuse injuries. "And again, knee pain can be referred from the hip," Dr. Phillips pointed out. Every year, he sees junior high school football players who have twisted their knees in a game and have been limping, but have a normal knee exam and turn out to have a slipped lateral femoral epiphysis. "So be sure to check the hips," he said, noting that the range of motion should be symmetrical, with at least 20 or 30 degrees of internal rotation, "and the feet go out, the kneecaps go in."
The most common causes of knee pain in young athletes are extensor patellofemoral problems, characterized by pain with such activities as running or climbing stairs. Another activity that elicits this type of pain is rising up from a sitting position, which "jams" the patellofemoral joint, he said. In such cases, there tends to be tenderness under the patella. "Usually, if you push the patella a little bit laterally, they will have pain."
Management of patellofemoral problems includes activity as tolerated; ice and analgesics; isometric quadriceps strengthening; and quadriceps and hamstring stretching. A brace – the type with a neoprene sleeve with a hole cut out, not a metal brace – is also acceptable to use if the patient likes it, but it is not essential, Dr. Phillips said.
Physical therapy is not usually necessary. Instead, he recommends teaching children who present with knee pain the following fundamental exercise: While lying down, bend the unaffected leg and keep that foot flat on the floor, then lift the affected leg 6-12 inches off the table with the knee fully extended, toe pointed to the ceiling – keeping it up for 20 seconds, down for 10 seconds, and repeating this for one or two sets of 20 repetitions, depending on the child and other factors. They should avoid lifting both legs up off the ground at the same time, which can hurt their backs.
For weights, they can use a knapsack, even their mother’s purse, starting with 1 or 2 pounds (or no weights if they are very sore). "This will go a long way toward helping them mend," he said.
Dr. Phillips advises avoiding weight machines where they kick their legs out, which is like getting up from a sitting position; squats; or stair climbing. He tells families that patellofemoral problems wax and wane, and "very rarely" require surgery, and he encourages a lot of rehabilitation before considering a recommendation for any type of surgery.
Dr. Phillips said he had no relevant disclosures.
EXPERT ANALYSIS FROM PRACTICAL PEDIATRICS
Reassurance, observation recommended for intoeing in children
WASHINGTON – Normal variations of the lower extremities in healthy children can be worrisome for parents, but they "virtually never" need to be treated in the first decade of life, Dr. William Phillips said.
These normal variations – which include intoeing and bowlegs – often lead to unnecessary referrals and treatments, said Dr. Phillips, chief of the orthopedic service and the orthopedic surgery clinic at Texas Children’s Hospital, Houston.
What is normal can vary with age, and usually resolves without any treatment, he said at a practical pediatrics meeting sponsored by the American Academy of Pediatrics.
Families can be reassured that it can take a few years, not months, for intoeing to improve. "The natural history of intoeing of any cause is that it gradually gets better," usually by age 8-10 years, he said, pointing out that 30% of 6-year-olds have intoeing, compared with less than 5% of adults. And only in rare cases is any treatment recommended, he said.
The Denis Browne bar (now used only for clubfoot) or special shoes are not effective; the only effective treatment is a derotation osteotomy, which he usually does not recommend, although he does perform this surgery for children with cerebral palsy.
The common causes of intoeing or out-toeing are metatarsus adductus, internal or external tibial torsion, or excessive femoral anteversion. Evaluating the torsional profile while the child is in a prone position makes it easier to identify these conditions; this includes looking at the lateral border of the foot, the thigh-foot angle, and hip rotation, said Dr. Phillips, who also is a professor of pediatrics at Baylor College of Medicine, Houston.
The lateral border of the foot is normally straight, but if it appears to be "bean-shaped," the child has metatarsus adductus, he noted. At the thigh-foot angle, most children have some external tibial torsion, but if the line from the second toe to the heel is pointing outward instead of inward, the child has internal tibial torsion.
A child with a normal exam with normal variations should be checked for short stature. If under the fifth percentile for age, he or she is more likely to have an underlying problem such as a skeletal dysplasia. It also helps to observe how the child stands, walks, and runs when barefoot and in shorts. Other factors that should be considered include family history and whether the child was full term or a breech delivery, as well as whether he or she is meeting developmental milestones and whether the intoeing is causing functional problems, Dr. Phillips added. Radiographs are not very helpful when evaluating children with intoeing.
Metatarsus adductus typically resolves spontaneously, and while it is occasionally treated with serial casting, this is not usually necessary, said Dr. Phillips, who has not recommended special shoes for these patients for many years.
Internal tibial torsion is seen in toddlers and also improves with time, although it often persists longer in children who have a tendency to sleep in a position similar to a fetal position, which delays remodeling, he said.
Children with external tibial torsion, which is much less common, tend to be awkward, clumsy runners. The only treatment for this is an osteotomy, which he usually does not recommend.
Excessive or increased femoral anteversion is most commonly seen in younger children, who present with an intoed gait that tends to get worse when they are tired; these children also tend to sit in a "W" position. Another sign of increased femoral anteversion is "kissing patellae" when a child lies down, Dr. Phillips said. There is no evidence that increased femoral anteversion increases the risk of osteoarthritis, although it can cause problems with the kneecap in a few cases – but not until adolescence, he added.
For children with intoeing, he recommends activities "where they can’t get very far if they turn their feet in all the way," such as roller and ice-skating, skiing, and ballet.
The wide range of normal variations of the lower extremities in children includes bowlegs and knock-knees, which are angular variations, Dr. Phillips said, pointing out that children at different ages have different alignments to their limbs. In the first year and a half, children tend to be bowlegged, followed by a "hyper–knock-kneed" phase, and, subsequently, their legs straighten out and they are mildly knock-kneed, he added.
But if a child is still bowlegged at age 3 years, however, he starts to investigate further.
He recommends radiographs for children with bowlegs or knock-knees when they are under the fifth percentile, because they are more likely to have a problem like skeletal dysplasia, and when there is a positive family history or they have an asymmetrical or worsening deformity. A special x-ray – a standing AP from the hips to the ankles – helps evaluate alignment as the child is standing with the kneecaps straight ahead.
While there are few pathologic torsional variations, there are some pathologic angular variations, Dr. Phillips pointed out. They include Blount’s disease, a growth disturbance of the medial proximal tibial epiphysis; metabolic bone disease, such as early renal failure or vitamin D–resistant rickets; and a posttraumatic deformity.
"What you ought to really be careful about is if the bowlegging is coming from below the knee," which can be a sign of congenital pseudoarthrosis of the tibia, he said.
Dr. Phillips had no disclosures.
WASHINGTON – Normal variations of the lower extremities in healthy children can be worrisome for parents, but they "virtually never" need to be treated in the first decade of life, Dr. William Phillips said.
These normal variations – which include intoeing and bowlegs – often lead to unnecessary referrals and treatments, said Dr. Phillips, chief of the orthopedic service and the orthopedic surgery clinic at Texas Children’s Hospital, Houston.
What is normal can vary with age, and usually resolves without any treatment, he said at a practical pediatrics meeting sponsored by the American Academy of Pediatrics.
Families can be reassured that it can take a few years, not months, for intoeing to improve. "The natural history of intoeing of any cause is that it gradually gets better," usually by age 8-10 years, he said, pointing out that 30% of 6-year-olds have intoeing, compared with less than 5% of adults. And only in rare cases is any treatment recommended, he said.
The Denis Browne bar (now used only for clubfoot) or special shoes are not effective; the only effective treatment is a derotation osteotomy, which he usually does not recommend, although he does perform this surgery for children with cerebral palsy.
The common causes of intoeing or out-toeing are metatarsus adductus, internal or external tibial torsion, or excessive femoral anteversion. Evaluating the torsional profile while the child is in a prone position makes it easier to identify these conditions; this includes looking at the lateral border of the foot, the thigh-foot angle, and hip rotation, said Dr. Phillips, who also is a professor of pediatrics at Baylor College of Medicine, Houston.
The lateral border of the foot is normally straight, but if it appears to be "bean-shaped," the child has metatarsus adductus, he noted. At the thigh-foot angle, most children have some external tibial torsion, but if the line from the second toe to the heel is pointing outward instead of inward, the child has internal tibial torsion.
A child with a normal exam with normal variations should be checked for short stature. If under the fifth percentile for age, he or she is more likely to have an underlying problem such as a skeletal dysplasia. It also helps to observe how the child stands, walks, and runs when barefoot and in shorts. Other factors that should be considered include family history and whether the child was full term or a breech delivery, as well as whether he or she is meeting developmental milestones and whether the intoeing is causing functional problems, Dr. Phillips added. Radiographs are not very helpful when evaluating children with intoeing.
Metatarsus adductus typically resolves spontaneously, and while it is occasionally treated with serial casting, this is not usually necessary, said Dr. Phillips, who has not recommended special shoes for these patients for many years.
Internal tibial torsion is seen in toddlers and also improves with time, although it often persists longer in children who have a tendency to sleep in a position similar to a fetal position, which delays remodeling, he said.
Children with external tibial torsion, which is much less common, tend to be awkward, clumsy runners. The only treatment for this is an osteotomy, which he usually does not recommend.
Excessive or increased femoral anteversion is most commonly seen in younger children, who present with an intoed gait that tends to get worse when they are tired; these children also tend to sit in a "W" position. Another sign of increased femoral anteversion is "kissing patellae" when a child lies down, Dr. Phillips said. There is no evidence that increased femoral anteversion increases the risk of osteoarthritis, although it can cause problems with the kneecap in a few cases – but not until adolescence, he added.
For children with intoeing, he recommends activities "where they can’t get very far if they turn their feet in all the way," such as roller and ice-skating, skiing, and ballet.
The wide range of normal variations of the lower extremities in children includes bowlegs and knock-knees, which are angular variations, Dr. Phillips said, pointing out that children at different ages have different alignments to their limbs. In the first year and a half, children tend to be bowlegged, followed by a "hyper–knock-kneed" phase, and, subsequently, their legs straighten out and they are mildly knock-kneed, he added.
But if a child is still bowlegged at age 3 years, however, he starts to investigate further.
He recommends radiographs for children with bowlegs or knock-knees when they are under the fifth percentile, because they are more likely to have a problem like skeletal dysplasia, and when there is a positive family history or they have an asymmetrical or worsening deformity. A special x-ray – a standing AP from the hips to the ankles – helps evaluate alignment as the child is standing with the kneecaps straight ahead.
While there are few pathologic torsional variations, there are some pathologic angular variations, Dr. Phillips pointed out. They include Blount’s disease, a growth disturbance of the medial proximal tibial epiphysis; metabolic bone disease, such as early renal failure or vitamin D–resistant rickets; and a posttraumatic deformity.
"What you ought to really be careful about is if the bowlegging is coming from below the knee," which can be a sign of congenital pseudoarthrosis of the tibia, he said.
Dr. Phillips had no disclosures.
WASHINGTON – Normal variations of the lower extremities in healthy children can be worrisome for parents, but they "virtually never" need to be treated in the first decade of life, Dr. William Phillips said.
These normal variations – which include intoeing and bowlegs – often lead to unnecessary referrals and treatments, said Dr. Phillips, chief of the orthopedic service and the orthopedic surgery clinic at Texas Children’s Hospital, Houston.
What is normal can vary with age, and usually resolves without any treatment, he said at a practical pediatrics meeting sponsored by the American Academy of Pediatrics.
Families can be reassured that it can take a few years, not months, for intoeing to improve. "The natural history of intoeing of any cause is that it gradually gets better," usually by age 8-10 years, he said, pointing out that 30% of 6-year-olds have intoeing, compared with less than 5% of adults. And only in rare cases is any treatment recommended, he said.
The Denis Browne bar (now used only for clubfoot) or special shoes are not effective; the only effective treatment is a derotation osteotomy, which he usually does not recommend, although he does perform this surgery for children with cerebral palsy.
The common causes of intoeing or out-toeing are metatarsus adductus, internal or external tibial torsion, or excessive femoral anteversion. Evaluating the torsional profile while the child is in a prone position makes it easier to identify these conditions; this includes looking at the lateral border of the foot, the thigh-foot angle, and hip rotation, said Dr. Phillips, who also is a professor of pediatrics at Baylor College of Medicine, Houston.
The lateral border of the foot is normally straight, but if it appears to be "bean-shaped," the child has metatarsus adductus, he noted. At the thigh-foot angle, most children have some external tibial torsion, but if the line from the second toe to the heel is pointing outward instead of inward, the child has internal tibial torsion.
A child with a normal exam with normal variations should be checked for short stature. If under the fifth percentile for age, he or she is more likely to have an underlying problem such as a skeletal dysplasia. It also helps to observe how the child stands, walks, and runs when barefoot and in shorts. Other factors that should be considered include family history and whether the child was full term or a breech delivery, as well as whether he or she is meeting developmental milestones and whether the intoeing is causing functional problems, Dr. Phillips added. Radiographs are not very helpful when evaluating children with intoeing.
Metatarsus adductus typically resolves spontaneously, and while it is occasionally treated with serial casting, this is not usually necessary, said Dr. Phillips, who has not recommended special shoes for these patients for many years.
Internal tibial torsion is seen in toddlers and also improves with time, although it often persists longer in children who have a tendency to sleep in a position similar to a fetal position, which delays remodeling, he said.
Children with external tibial torsion, which is much less common, tend to be awkward, clumsy runners. The only treatment for this is an osteotomy, which he usually does not recommend.
Excessive or increased femoral anteversion is most commonly seen in younger children, who present with an intoed gait that tends to get worse when they are tired; these children also tend to sit in a "W" position. Another sign of increased femoral anteversion is "kissing patellae" when a child lies down, Dr. Phillips said. There is no evidence that increased femoral anteversion increases the risk of osteoarthritis, although it can cause problems with the kneecap in a few cases – but not until adolescence, he added.
For children with intoeing, he recommends activities "where they can’t get very far if they turn their feet in all the way," such as roller and ice-skating, skiing, and ballet.
The wide range of normal variations of the lower extremities in children includes bowlegs and knock-knees, which are angular variations, Dr. Phillips said, pointing out that children at different ages have different alignments to their limbs. In the first year and a half, children tend to be bowlegged, followed by a "hyper–knock-kneed" phase, and, subsequently, their legs straighten out and they are mildly knock-kneed, he added.
But if a child is still bowlegged at age 3 years, however, he starts to investigate further.
He recommends radiographs for children with bowlegs or knock-knees when they are under the fifth percentile, because they are more likely to have a problem like skeletal dysplasia, and when there is a positive family history or they have an asymmetrical or worsening deformity. A special x-ray – a standing AP from the hips to the ankles – helps evaluate alignment as the child is standing with the kneecaps straight ahead.
While there are few pathologic torsional variations, there are some pathologic angular variations, Dr. Phillips pointed out. They include Blount’s disease, a growth disturbance of the medial proximal tibial epiphysis; metabolic bone disease, such as early renal failure or vitamin D–resistant rickets; and a posttraumatic deformity.
"What you ought to really be careful about is if the bowlegging is coming from below the knee," which can be a sign of congenital pseudoarthrosis of the tibia, he said.
Dr. Phillips had no disclosures.
EXPERT ANALYSIS FROM PRACTICAL PEDIATRICS
Consider MRSA in Skin and Soft Tissue Infections
WASHINGTON – Community-acquired methicillin-resistant Staphylococcus aureus should be considered in the differential diagnosis of skin and soft tissue infections, as well as neonatal eye infections, Dr. Morven Edwards said at a practical pediatrics meeting sponsored by the American Academy of Pediatrics.
Dr. Edwards, an infectious disease specialist at Texas Children’s Hospital, Houston, said that unlike a decade ago, she now considers community-acquired MRSA as a possible cause "in almost any manifestation of a problem with the skin or soft tissue, even including those entities that we used to think were caused by other microorganisms."
For example, in the past, perianal dermatitis in a young infant "was essentially a pathognomonic presentation for group A strep infection," which did not even require a culture for an accurate diagnosis and appropriate treatment, said Dr. Edwards, who is a professor of pediatrics at Baylor College of Medicine, Houston.
She referred to a small retrospective study of children between the ages of 5 months and 12 years with perianal erythema, which found that the dominant culture results were MRSA and methicillin-sensitive S. aureus (MSSA), some streptococci, and some mixed infections. In the past, staphylococcal skin and soft tissue infections were considered uncommon among healthy term newborns discharged home, "unless it was just a very mild diaper dermatitis," she noted.
In a 2006 study of healthy term babies who returned to the hospital within a month of discharge to Texas Children’s Hospital’s emergency department, cases of MRSA peaked at 8-12 days after discharge. Almost all were skin and soft tissue infections (SSTIs), and some of the babies had mild, pustular skin disease and were treated as outpatients with topical therapy and/or oral antistaphylococcal medications. But almost 40% required admission for incision and drainage and parenteral antibiotic therapy.
Dr. Edwards referred to a case of an SSTI that started with a stubbed toe and progressed to a disseminated staphylococcal infection and endocarditis – illustrating that "even with an apparently limited minor staph infection, it always should go through our minds that this could be one that’s more serious."
The healthy teenage boy had stubbed his toe during a wrestling match, then developed fever, fatigue, and buttock and back pain, along with swelling and pain in the toe 4 days later. A fracture was diagnosed at an urgent care clinic, and he was sent home, but 9 days after the initial injury, symptoms persisted and he was admitted to the hospital for treatment with broad-spectrum antimicrobials, including clindamycin.
On the fourth day in the hospital, he had a fever of 104° F, "massive" facial swelling, bibasilar crackles, a systolic murmur, left lower quadrant pain, and a large parietal abscess seen on MRI. He had vegetations on the anterior leaflet of the mitral valve and multiple microemboli to the brain. The abscess was drained, and he had a vegetectomy, where vegetation was "peeled" off the leaflet, leaving the valve intact. After 6 weeks of intravenous antimicrobial treatment, he did well and was discharged.
"The lesson is that a seemingly innocuous SSTI always has the potential to disseminate," Dr. Edwards said, adding that while this is very uncommon, especially in a healthy child, "it’s always something to keep in mind."
Transmitted by direct contact, risk factors for community-acquired MRSA include chronic skin conditions, participation in sports teams that involve close contact with other players, and a history of such infections in family members, she noted.
In infants, MRSA infection also should be included in the differential diagnosis of ophthalmia neonatorum, Dr. Edwards said. She described the case of a healthy term 4-day-old baby who presented with eye swelling and purulent discharge, but no other systemic findings. The mother’s pregnancy had been uncomplicated; maternal tests were negative for group B streptococcus, chlamydia, and Neisseria gonorrhoea; and the baby had been discharged home by the second day after a normal vaginal birth.
The cause turned out to be community-acquired MRSA infection. "We need to consider this diagnosis with a higher index of suspicion in young infants now," she said, noting that the peak onset is at age 4-6 days, and it is characterized by a purulent discharge not likely to be a gonococcal infection. The baby was admitted to the hospital and treated with parenteral antimicrobial therapy.
Dr. Edwards disclosed that she is a consultant for and has received research funds from Novartis related to the development of a group B streptococcus vaccine.
WASHINGTON – Community-acquired methicillin-resistant Staphylococcus aureus should be considered in the differential diagnosis of skin and soft tissue infections, as well as neonatal eye infections, Dr. Morven Edwards said at a practical pediatrics meeting sponsored by the American Academy of Pediatrics.
Dr. Edwards, an infectious disease specialist at Texas Children’s Hospital, Houston, said that unlike a decade ago, she now considers community-acquired MRSA as a possible cause "in almost any manifestation of a problem with the skin or soft tissue, even including those entities that we used to think were caused by other microorganisms."
For example, in the past, perianal dermatitis in a young infant "was essentially a pathognomonic presentation for group A strep infection," which did not even require a culture for an accurate diagnosis and appropriate treatment, said Dr. Edwards, who is a professor of pediatrics at Baylor College of Medicine, Houston.
She referred to a small retrospective study of children between the ages of 5 months and 12 years with perianal erythema, which found that the dominant culture results were MRSA and methicillin-sensitive S. aureus (MSSA), some streptococci, and some mixed infections. In the past, staphylococcal skin and soft tissue infections were considered uncommon among healthy term newborns discharged home, "unless it was just a very mild diaper dermatitis," she noted.
In a 2006 study of healthy term babies who returned to the hospital within a month of discharge to Texas Children’s Hospital’s emergency department, cases of MRSA peaked at 8-12 days after discharge. Almost all were skin and soft tissue infections (SSTIs), and some of the babies had mild, pustular skin disease and were treated as outpatients with topical therapy and/or oral antistaphylococcal medications. But almost 40% required admission for incision and drainage and parenteral antibiotic therapy.
Dr. Edwards referred to a case of an SSTI that started with a stubbed toe and progressed to a disseminated staphylococcal infection and endocarditis – illustrating that "even with an apparently limited minor staph infection, it always should go through our minds that this could be one that’s more serious."
The healthy teenage boy had stubbed his toe during a wrestling match, then developed fever, fatigue, and buttock and back pain, along with swelling and pain in the toe 4 days later. A fracture was diagnosed at an urgent care clinic, and he was sent home, but 9 days after the initial injury, symptoms persisted and he was admitted to the hospital for treatment with broad-spectrum antimicrobials, including clindamycin.
On the fourth day in the hospital, he had a fever of 104° F, "massive" facial swelling, bibasilar crackles, a systolic murmur, left lower quadrant pain, and a large parietal abscess seen on MRI. He had vegetations on the anterior leaflet of the mitral valve and multiple microemboli to the brain. The abscess was drained, and he had a vegetectomy, where vegetation was "peeled" off the leaflet, leaving the valve intact. After 6 weeks of intravenous antimicrobial treatment, he did well and was discharged.
"The lesson is that a seemingly innocuous SSTI always has the potential to disseminate," Dr. Edwards said, adding that while this is very uncommon, especially in a healthy child, "it’s always something to keep in mind."
Transmitted by direct contact, risk factors for community-acquired MRSA include chronic skin conditions, participation in sports teams that involve close contact with other players, and a history of such infections in family members, she noted.
In infants, MRSA infection also should be included in the differential diagnosis of ophthalmia neonatorum, Dr. Edwards said. She described the case of a healthy term 4-day-old baby who presented with eye swelling and purulent discharge, but no other systemic findings. The mother’s pregnancy had been uncomplicated; maternal tests were negative for group B streptococcus, chlamydia, and Neisseria gonorrhoea; and the baby had been discharged home by the second day after a normal vaginal birth.
The cause turned out to be community-acquired MRSA infection. "We need to consider this diagnosis with a higher index of suspicion in young infants now," she said, noting that the peak onset is at age 4-6 days, and it is characterized by a purulent discharge not likely to be a gonococcal infection. The baby was admitted to the hospital and treated with parenteral antimicrobial therapy.
Dr. Edwards disclosed that she is a consultant for and has received research funds from Novartis related to the development of a group B streptococcus vaccine.
WASHINGTON – Community-acquired methicillin-resistant Staphylococcus aureus should be considered in the differential diagnosis of skin and soft tissue infections, as well as neonatal eye infections, Dr. Morven Edwards said at a practical pediatrics meeting sponsored by the American Academy of Pediatrics.
Dr. Edwards, an infectious disease specialist at Texas Children’s Hospital, Houston, said that unlike a decade ago, she now considers community-acquired MRSA as a possible cause "in almost any manifestation of a problem with the skin or soft tissue, even including those entities that we used to think were caused by other microorganisms."
For example, in the past, perianal dermatitis in a young infant "was essentially a pathognomonic presentation for group A strep infection," which did not even require a culture for an accurate diagnosis and appropriate treatment, said Dr. Edwards, who is a professor of pediatrics at Baylor College of Medicine, Houston.
She referred to a small retrospective study of children between the ages of 5 months and 12 years with perianal erythema, which found that the dominant culture results were MRSA and methicillin-sensitive S. aureus (MSSA), some streptococci, and some mixed infections. In the past, staphylococcal skin and soft tissue infections were considered uncommon among healthy term newborns discharged home, "unless it was just a very mild diaper dermatitis," she noted.
In a 2006 study of healthy term babies who returned to the hospital within a month of discharge to Texas Children’s Hospital’s emergency department, cases of MRSA peaked at 8-12 days after discharge. Almost all were skin and soft tissue infections (SSTIs), and some of the babies had mild, pustular skin disease and were treated as outpatients with topical therapy and/or oral antistaphylococcal medications. But almost 40% required admission for incision and drainage and parenteral antibiotic therapy.
Dr. Edwards referred to a case of an SSTI that started with a stubbed toe and progressed to a disseminated staphylococcal infection and endocarditis – illustrating that "even with an apparently limited minor staph infection, it always should go through our minds that this could be one that’s more serious."
The healthy teenage boy had stubbed his toe during a wrestling match, then developed fever, fatigue, and buttock and back pain, along with swelling and pain in the toe 4 days later. A fracture was diagnosed at an urgent care clinic, and he was sent home, but 9 days after the initial injury, symptoms persisted and he was admitted to the hospital for treatment with broad-spectrum antimicrobials, including clindamycin.
On the fourth day in the hospital, he had a fever of 104° F, "massive" facial swelling, bibasilar crackles, a systolic murmur, left lower quadrant pain, and a large parietal abscess seen on MRI. He had vegetations on the anterior leaflet of the mitral valve and multiple microemboli to the brain. The abscess was drained, and he had a vegetectomy, where vegetation was "peeled" off the leaflet, leaving the valve intact. After 6 weeks of intravenous antimicrobial treatment, he did well and was discharged.
"The lesson is that a seemingly innocuous SSTI always has the potential to disseminate," Dr. Edwards said, adding that while this is very uncommon, especially in a healthy child, "it’s always something to keep in mind."
Transmitted by direct contact, risk factors for community-acquired MRSA include chronic skin conditions, participation in sports teams that involve close contact with other players, and a history of such infections in family members, she noted.
In infants, MRSA infection also should be included in the differential diagnosis of ophthalmia neonatorum, Dr. Edwards said. She described the case of a healthy term 4-day-old baby who presented with eye swelling and purulent discharge, but no other systemic findings. The mother’s pregnancy had been uncomplicated; maternal tests were negative for group B streptococcus, chlamydia, and Neisseria gonorrhoea; and the baby had been discharged home by the second day after a normal vaginal birth.
The cause turned out to be community-acquired MRSA infection. "We need to consider this diagnosis with a higher index of suspicion in young infants now," she said, noting that the peak onset is at age 4-6 days, and it is characterized by a purulent discharge not likely to be a gonococcal infection. The baby was admitted to the hospital and treated with parenteral antimicrobial therapy.
Dr. Edwards disclosed that she is a consultant for and has received research funds from Novartis related to the development of a group B streptococcus vaccine.
EXPERT ANALYSIS FROM PRACTICAL PEDIATRICS
Consider MRSA in skin and soft tissue, and newborn eye infections
WASHINGTON – Community-acquired methicillin-resistant Staphylococcus aureus should be considered in the differential diagnosis of skin and soft tissue infections, as well as neonatal eye infections, Dr. Morven Edwards said at a practical pediatrics meeting sponsored by the American Academy of Pediatrics.
Dr. Edwards, an infectious disease specialist at Texas Children’s Hospital, Houston, said that unlike a decade ago, she now considers community-acquired MRSA as a possible cause "in almost any manifestation of a problem with the skin or soft tissue, even including those entities that we used to think were caused by other microorganisms."
For example, in the past, perianal dermatitis in a young infant "was essentially a pathognomonic presentation for group A strep infection," which did not even require a culture for an accurate diagnosis and appropriate treatment, said Dr. Edwards, who is a professor of pediatrics at Baylor College of Medicine, Houston.
She referred to a small retrospective study of children between the ages of 5 months and 12 years with perianal erythema, which found that the dominant culture results were MRSA and methicillin-sensitive S. aureus (MSSA), some streptococci, and some mixed infections. In the past, staphylococcal skin and soft tissue infections were considered uncommon among healthy term newborns discharged home, "unless it was just a very mild diaper dermatitis," she noted.
In a 2006 study of healthy term babies who returned to the hospital within a month of discharge to Texas Children’s Hospital’s emergency department, cases of MRSA peaked at 8-12 days after discharge. Almost all were skin and soft tissue infections (SSTIs), and some of the babies had mild, pustular skin disease and were treated as outpatients with topical therapy and/or oral antistaphylococcal medications. But almost 40% required admission for incision and drainage and parenteral antibiotic therapy.
Dr. Edwards referred to a case of an SSTI that started with a stubbed toe and progressed to a disseminated staphylococcal infection and endocarditis – illustrating that "even with an apparently limited minor staph infection, it always should go through our minds that this could be one that’s more serious."
The healthy teenage boy had stubbed his toe during a wrestling match, then developed fever, fatigue, and buttock and back pain, along with swelling and pain in the toe 4 days later. A fracture was diagnosed at an urgent care clinic, and he was sent home, but 9 days after the initial injury, symptoms persisted and he was admitted to the hospital for treatment with broad-spectrum antimicrobials, including clindamycin.
On the fourth day in the hospital, he had a fever of 104° F, "massive" facial swelling, bibasilar crackles, a systolic murmur, left lower quadrant pain, and a large parietal abscess seen on MRI. He had vegetations on the anterior leaflet of the mitral valve and multiple microemboli to the brain. The abscess was drained, and he had a vegetectomy, where vegetation was "peeled" off the leaflet, leaving the valve intact. After 6 weeks of intravenous antimicrobial treatment, he did well and was discharged.
"The lesson is that a seemingly innocuous SSTI always has the potential to disseminate," Dr. Edwards said, adding that while this is very uncommon, especially in a healthy child, "it’s always something to keep in mind."
Transmitted by direct contact, risk factors for community-acquired MRSA include chronic skin conditions, participation in sports teams that involve close contact with other players, and a history of such infections in family members, she noted.
In infants, MRSA infection also should be included in the differential diagnosis of ophthalmia neonatorum, Dr. Edwards said. She described the case of a healthy term 4-day-old baby who presented with eye swelling and purulent discharge, but no other systemic findings. The mother’s pregnancy had been uncomplicated; maternal tests were negative for group B streptococcus, chlamydia, and Neisseria gonorrhoea; and the baby had been discharged home by the second day after a normal vaginal birth.
The cause turned out to be community-acquired MRSA infection. "We need to consider this diagnosis with a higher index of suspicion in young infants now," she said, noting that the peak onset is at age 4-6 days, and it is characterized by a purulent discharge not likely to be a gonococcal infection. The baby was admitted to the hospital and treated with parenteral antimicrobial therapy.
Dr. Edwards disclosed that she is a consultant for and has received research funds from Novartis related to the development of a group B streptococcus vaccine.
WASHINGTON – Community-acquired methicillin-resistant Staphylococcus aureus should be considered in the differential diagnosis of skin and soft tissue infections, as well as neonatal eye infections, Dr. Morven Edwards said at a practical pediatrics meeting sponsored by the American Academy of Pediatrics.
Dr. Edwards, an infectious disease specialist at Texas Children’s Hospital, Houston, said that unlike a decade ago, she now considers community-acquired MRSA as a possible cause "in almost any manifestation of a problem with the skin or soft tissue, even including those entities that we used to think were caused by other microorganisms."
For example, in the past, perianal dermatitis in a young infant "was essentially a pathognomonic presentation for group A strep infection," which did not even require a culture for an accurate diagnosis and appropriate treatment, said Dr. Edwards, who is a professor of pediatrics at Baylor College of Medicine, Houston.
She referred to a small retrospective study of children between the ages of 5 months and 12 years with perianal erythema, which found that the dominant culture results were MRSA and methicillin-sensitive S. aureus (MSSA), some streptococci, and some mixed infections. In the past, staphylococcal skin and soft tissue infections were considered uncommon among healthy term newborns discharged home, "unless it was just a very mild diaper dermatitis," she noted.
In a 2006 study of healthy term babies who returned to the hospital within a month of discharge to Texas Children’s Hospital’s emergency department, cases of MRSA peaked at 8-12 days after discharge. Almost all were skin and soft tissue infections (SSTIs), and some of the babies had mild, pustular skin disease and were treated as outpatients with topical therapy and/or oral antistaphylococcal medications. But almost 40% required admission for incision and drainage and parenteral antibiotic therapy.
Dr. Edwards referred to a case of an SSTI that started with a stubbed toe and progressed to a disseminated staphylococcal infection and endocarditis – illustrating that "even with an apparently limited minor staph infection, it always should go through our minds that this could be one that’s more serious."
The healthy teenage boy had stubbed his toe during a wrestling match, then developed fever, fatigue, and buttock and back pain, along with swelling and pain in the toe 4 days later. A fracture was diagnosed at an urgent care clinic, and he was sent home, but 9 days after the initial injury, symptoms persisted and he was admitted to the hospital for treatment with broad-spectrum antimicrobials, including clindamycin.
On the fourth day in the hospital, he had a fever of 104° F, "massive" facial swelling, bibasilar crackles, a systolic murmur, left lower quadrant pain, and a large parietal abscess seen on MRI. He had vegetations on the anterior leaflet of the mitral valve and multiple microemboli to the brain. The abscess was drained, and he had a vegetectomy, where vegetation was "peeled" off the leaflet, leaving the valve intact. After 6 weeks of intravenous antimicrobial treatment, he did well and was discharged.
"The lesson is that a seemingly innocuous SSTI always has the potential to disseminate," Dr. Edwards said, adding that while this is very uncommon, especially in a healthy child, "it’s always something to keep in mind."
Transmitted by direct contact, risk factors for community-acquired MRSA include chronic skin conditions, participation in sports teams that involve close contact with other players, and a history of such infections in family members, she noted.
In infants, MRSA infection also should be included in the differential diagnosis of ophthalmia neonatorum, Dr. Edwards said. She described the case of a healthy term 4-day-old baby who presented with eye swelling and purulent discharge, but no other systemic findings. The mother’s pregnancy had been uncomplicated; maternal tests were negative for group B streptococcus, chlamydia, and Neisseria gonorrhoea; and the baby had been discharged home by the second day after a normal vaginal birth.
The cause turned out to be community-acquired MRSA infection. "We need to consider this diagnosis with a higher index of suspicion in young infants now," she said, noting that the peak onset is at age 4-6 days, and it is characterized by a purulent discharge not likely to be a gonococcal infection. The baby was admitted to the hospital and treated with parenteral antimicrobial therapy.
Dr. Edwards disclosed that she is a consultant for and has received research funds from Novartis related to the development of a group B streptococcus vaccine.
WASHINGTON – Community-acquired methicillin-resistant Staphylococcus aureus should be considered in the differential diagnosis of skin and soft tissue infections, as well as neonatal eye infections, Dr. Morven Edwards said at a practical pediatrics meeting sponsored by the American Academy of Pediatrics.
Dr. Edwards, an infectious disease specialist at Texas Children’s Hospital, Houston, said that unlike a decade ago, she now considers community-acquired MRSA as a possible cause "in almost any manifestation of a problem with the skin or soft tissue, even including those entities that we used to think were caused by other microorganisms."
For example, in the past, perianal dermatitis in a young infant "was essentially a pathognomonic presentation for group A strep infection," which did not even require a culture for an accurate diagnosis and appropriate treatment, said Dr. Edwards, who is a professor of pediatrics at Baylor College of Medicine, Houston.
She referred to a small retrospective study of children between the ages of 5 months and 12 years with perianal erythema, which found that the dominant culture results were MRSA and methicillin-sensitive S. aureus (MSSA), some streptococci, and some mixed infections. In the past, staphylococcal skin and soft tissue infections were considered uncommon among healthy term newborns discharged home, "unless it was just a very mild diaper dermatitis," she noted.
In a 2006 study of healthy term babies who returned to the hospital within a month of discharge to Texas Children’s Hospital’s emergency department, cases of MRSA peaked at 8-12 days after discharge. Almost all were skin and soft tissue infections (SSTIs), and some of the babies had mild, pustular skin disease and were treated as outpatients with topical therapy and/or oral antistaphylococcal medications. But almost 40% required admission for incision and drainage and parenteral antibiotic therapy.
Dr. Edwards referred to a case of an SSTI that started with a stubbed toe and progressed to a disseminated staphylococcal infection and endocarditis – illustrating that "even with an apparently limited minor staph infection, it always should go through our minds that this could be one that’s more serious."
The healthy teenage boy had stubbed his toe during a wrestling match, then developed fever, fatigue, and buttock and back pain, along with swelling and pain in the toe 4 days later. A fracture was diagnosed at an urgent care clinic, and he was sent home, but 9 days after the initial injury, symptoms persisted and he was admitted to the hospital for treatment with broad-spectrum antimicrobials, including clindamycin.
On the fourth day in the hospital, he had a fever of 104° F, "massive" facial swelling, bibasilar crackles, a systolic murmur, left lower quadrant pain, and a large parietal abscess seen on MRI. He had vegetations on the anterior leaflet of the mitral valve and multiple microemboli to the brain. The abscess was drained, and he had a vegetectomy, where vegetation was "peeled" off the leaflet, leaving the valve intact. After 6 weeks of intravenous antimicrobial treatment, he did well and was discharged.
"The lesson is that a seemingly innocuous SSTI always has the potential to disseminate," Dr. Edwards said, adding that while this is very uncommon, especially in a healthy child, "it’s always something to keep in mind."
Transmitted by direct contact, risk factors for community-acquired MRSA include chronic skin conditions, participation in sports teams that involve close contact with other players, and a history of such infections in family members, she noted.
In infants, MRSA infection also should be included in the differential diagnosis of ophthalmia neonatorum, Dr. Edwards said. She described the case of a healthy term 4-day-old baby who presented with eye swelling and purulent discharge, but no other systemic findings. The mother’s pregnancy had been uncomplicated; maternal tests were negative for group B streptococcus, chlamydia, and Neisseria gonorrhoea; and the baby had been discharged home by the second day after a normal vaginal birth.
The cause turned out to be community-acquired MRSA infection. "We need to consider this diagnosis with a higher index of suspicion in young infants now," she said, noting that the peak onset is at age 4-6 days, and it is characterized by a purulent discharge not likely to be a gonococcal infection. The baby was admitted to the hospital and treated with parenteral antimicrobial therapy.
Dr. Edwards disclosed that she is a consultant for and has received research funds from Novartis related to the development of a group B streptococcus vaccine.
EXPERT ANALYSIS FROM PRACTICAL PEDIATRICS
Advisers support FDA approval of wireless HF monitoring device
GAITHERSBURG, MD. – An implantable wireless device that provides measurements of pulmonary arterial pressure and heart rate in patients with heart failure was supported for approval by a majority of a Food and Drug Administration advisory panel, almost 2 years after it was rejected for approval.
The Champion Heart Failure Monitoring System, a permanent implantable pressure-measurement system, includes a wireless sensor that is implanted in a small branch of the pulmonary artery during a right-heart cardiac catheterization, providing wireless PA measurements.
At a meeting of the FDA’s Circulatory System Devices Panel, the panel voted 6-4, with 1 abstention, that the benefits of the device outweighed its risks for monitoring patients who meet the criteria in the proposed indication, which is for wirelessly measuring and monitoring PA pressures and heart rate in patients with New York Heart Association (NYHA) class III heart failure who have been hospitalized for HF in the previous year.
The proposed indication includes the statement that the hemodynamic data "are used by physicians for heart failure management and to reduce heart failure hospitalizations" in hospital or office settings, and that it can also be used by patients at home or another location "to wirelessly obtain and send hemodynamic and PA pressure measurements" to a database to be reviewed and evaluated by their physicians. The manufacturer is CardioMEMS.
The panel was divided regarding the strength of the data supporting the effect the device had on reducing HF hospitalizations, the primary endpoint of studies, and this was reflected in the vote on the device’s effectiveness. The panel voted 7-4 that there was not reasonable assurance that the device was effective for use for the proposed indication. But they unanimously voted that there was reasonable assurance it was safe, based on the finding that most complications occurred in the 30 days after implantation.
However, no benefit was seen on HF hospitalizations in women, which panelists thought may have been due to the low number of women enrolled in the studies, and panelists recommended that the device needed to be studied further in women. Another recommendation was to drop the specific HF class in the indication because patients with HF tend to move from one class to the other.
The original study enrolled 550 patients with NYHA class III HF and a recent hospitalization for HF. All patients had the device implanted, but physicians had access to daily PA measurements for those randomized to the treatment group, and were able to adjust HF medications based on the values provided.
At 6 months, there were 84 HF hospitalizations in the treatment group (0.32 per patient), compared with 120 (0.44 per patient) in the control group, a highly significant difference that represented a 28% reduction in the rate of HF hospitalizations. In addition, 5.6% of the patients in the treatment group died, versus 7.1% in the control group, a nonsignificant 23% reduction in the relative risk of death.
In December 2011, the FDA’s Circulatory System Devices Panel met for the first time to review the device, and voted 6-4 that the risks outweighed the benefits. Like the FDA reviewers, the panel raised concerns that the nurse communications in this study may have influenced treatment decisions and biased the results, and the FDA did not approve the device.
In response, the company did not conduct another trial but conducted various analyses of HF hospitalizations and deaths during the open-access period of the study, which eliminated nurse input. Patients in the control and treatment groups in the randomized study were included in the study’s open-access period, where the PA pressures on all patients were provided to physicians.
Among the results was that HF hospitalization rates among those patients who were controls in the first part of the study dropped to a rate comparable to the rate among those who were in the original treatment group, when access to the PA measurements of the original controls were available. A third-party analysis showed that the possible effect the nurses’ input had on the outcomes in the randomized study was minimal, according to the company.
On the basis of these analyses, the company concluded that the effect on HF hospitalizations was a result of physicians knowing the PA measurements provided by the device, and was not related to the communications from nurses during the randomized segment of the study or during the open-access period.
Panelist Dr. Jeffrey Borer, chief of the division of cardiovascular medicine at the State University of New York Downstate Medical Center, Brooklyn, who voted yes on all three questions, said he found the ancillary studies very helpful in clarifying the utility of the device seen in the original study, and that this was "an approvable and probably very useful device." He, like other panelists, remained concerned about the use in women, noting that there was no benefit on HF hospitalizations in women who had the device implanted, but this may have been due to the low number of women enrolled in the study.
Dr. David Yuh, chief of cardiac surgery at Yale University, New Haven, Conn., said that while he was not surprised by the split vote on risk-benefit, he voted positively on this question. "I did feel this was an effective monitoring device that did facilitate closer monitoring of these very difficult patients for heart failure cardiologists."
Dr. Valluvan Jeevanandam, chief of cardiac and thoracic surgery at the University of Chicago, was one of two panelists who were not convinced by efficacy in terms of reducing HF hospitalizations; however, he voted in favor of the risk-benefit profile because of what he considered the device’s value as a diagnostic tool, which he said he would find useful in "innumerable" patients.
"I just got paged for two people who need right-heart catheterizations tomorrow, who wouldn’t need them" if they had this device, he said.
Voting no on the effectiveness and risk-benefit questions, Dr. David Milan, a cardiac electrophysiologist at Massachusetts General Hospital, Boston, said he believed the device was safe, but there were not enough data on effectiveness. The data analyses presented by the company were not valid or convincing, and the company should have conducted another randomized controlled study, he said.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. If approved, CardioMEMS has planned a postapproval study that would prospectively follow the safety and effectiveness of the device in a real-world setting in about 1,200 patients, including 420 women.
GAITHERSBURG, MD. – An implantable wireless device that provides measurements of pulmonary arterial pressure and heart rate in patients with heart failure was supported for approval by a majority of a Food and Drug Administration advisory panel, almost 2 years after it was rejected for approval.
The Champion Heart Failure Monitoring System, a permanent implantable pressure-measurement system, includes a wireless sensor that is implanted in a small branch of the pulmonary artery during a right-heart cardiac catheterization, providing wireless PA measurements.
At a meeting of the FDA’s Circulatory System Devices Panel, the panel voted 6-4, with 1 abstention, that the benefits of the device outweighed its risks for monitoring patients who meet the criteria in the proposed indication, which is for wirelessly measuring and monitoring PA pressures and heart rate in patients with New York Heart Association (NYHA) class III heart failure who have been hospitalized for HF in the previous year.
The proposed indication includes the statement that the hemodynamic data "are used by physicians for heart failure management and to reduce heart failure hospitalizations" in hospital or office settings, and that it can also be used by patients at home or another location "to wirelessly obtain and send hemodynamic and PA pressure measurements" to a database to be reviewed and evaluated by their physicians. The manufacturer is CardioMEMS.
The panel was divided regarding the strength of the data supporting the effect the device had on reducing HF hospitalizations, the primary endpoint of studies, and this was reflected in the vote on the device’s effectiveness. The panel voted 7-4 that there was not reasonable assurance that the device was effective for use for the proposed indication. But they unanimously voted that there was reasonable assurance it was safe, based on the finding that most complications occurred in the 30 days after implantation.
However, no benefit was seen on HF hospitalizations in women, which panelists thought may have been due to the low number of women enrolled in the studies, and panelists recommended that the device needed to be studied further in women. Another recommendation was to drop the specific HF class in the indication because patients with HF tend to move from one class to the other.
The original study enrolled 550 patients with NYHA class III HF and a recent hospitalization for HF. All patients had the device implanted, but physicians had access to daily PA measurements for those randomized to the treatment group, and were able to adjust HF medications based on the values provided.
At 6 months, there were 84 HF hospitalizations in the treatment group (0.32 per patient), compared with 120 (0.44 per patient) in the control group, a highly significant difference that represented a 28% reduction in the rate of HF hospitalizations. In addition, 5.6% of the patients in the treatment group died, versus 7.1% in the control group, a nonsignificant 23% reduction in the relative risk of death.
In December 2011, the FDA’s Circulatory System Devices Panel met for the first time to review the device, and voted 6-4 that the risks outweighed the benefits. Like the FDA reviewers, the panel raised concerns that the nurse communications in this study may have influenced treatment decisions and biased the results, and the FDA did not approve the device.
In response, the company did not conduct another trial but conducted various analyses of HF hospitalizations and deaths during the open-access period of the study, which eliminated nurse input. Patients in the control and treatment groups in the randomized study were included in the study’s open-access period, where the PA pressures on all patients were provided to physicians.
Among the results was that HF hospitalization rates among those patients who were controls in the first part of the study dropped to a rate comparable to the rate among those who were in the original treatment group, when access to the PA measurements of the original controls were available. A third-party analysis showed that the possible effect the nurses’ input had on the outcomes in the randomized study was minimal, according to the company.
On the basis of these analyses, the company concluded that the effect on HF hospitalizations was a result of physicians knowing the PA measurements provided by the device, and was not related to the communications from nurses during the randomized segment of the study or during the open-access period.
Panelist Dr. Jeffrey Borer, chief of the division of cardiovascular medicine at the State University of New York Downstate Medical Center, Brooklyn, who voted yes on all three questions, said he found the ancillary studies very helpful in clarifying the utility of the device seen in the original study, and that this was "an approvable and probably very useful device." He, like other panelists, remained concerned about the use in women, noting that there was no benefit on HF hospitalizations in women who had the device implanted, but this may have been due to the low number of women enrolled in the study.
Dr. David Yuh, chief of cardiac surgery at Yale University, New Haven, Conn., said that while he was not surprised by the split vote on risk-benefit, he voted positively on this question. "I did feel this was an effective monitoring device that did facilitate closer monitoring of these very difficult patients for heart failure cardiologists."
Dr. Valluvan Jeevanandam, chief of cardiac and thoracic surgery at the University of Chicago, was one of two panelists who were not convinced by efficacy in terms of reducing HF hospitalizations; however, he voted in favor of the risk-benefit profile because of what he considered the device’s value as a diagnostic tool, which he said he would find useful in "innumerable" patients.
"I just got paged for two people who need right-heart catheterizations tomorrow, who wouldn’t need them" if they had this device, he said.
Voting no on the effectiveness and risk-benefit questions, Dr. David Milan, a cardiac electrophysiologist at Massachusetts General Hospital, Boston, said he believed the device was safe, but there were not enough data on effectiveness. The data analyses presented by the company were not valid or convincing, and the company should have conducted another randomized controlled study, he said.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. If approved, CardioMEMS has planned a postapproval study that would prospectively follow the safety and effectiveness of the device in a real-world setting in about 1,200 patients, including 420 women.
GAITHERSBURG, MD. – An implantable wireless device that provides measurements of pulmonary arterial pressure and heart rate in patients with heart failure was supported for approval by a majority of a Food and Drug Administration advisory panel, almost 2 years after it was rejected for approval.
The Champion Heart Failure Monitoring System, a permanent implantable pressure-measurement system, includes a wireless sensor that is implanted in a small branch of the pulmonary artery during a right-heart cardiac catheterization, providing wireless PA measurements.
At a meeting of the FDA’s Circulatory System Devices Panel, the panel voted 6-4, with 1 abstention, that the benefits of the device outweighed its risks for monitoring patients who meet the criteria in the proposed indication, which is for wirelessly measuring and monitoring PA pressures and heart rate in patients with New York Heart Association (NYHA) class III heart failure who have been hospitalized for HF in the previous year.
The proposed indication includes the statement that the hemodynamic data "are used by physicians for heart failure management and to reduce heart failure hospitalizations" in hospital or office settings, and that it can also be used by patients at home or another location "to wirelessly obtain and send hemodynamic and PA pressure measurements" to a database to be reviewed and evaluated by their physicians. The manufacturer is CardioMEMS.
The panel was divided regarding the strength of the data supporting the effect the device had on reducing HF hospitalizations, the primary endpoint of studies, and this was reflected in the vote on the device’s effectiveness. The panel voted 7-4 that there was not reasonable assurance that the device was effective for use for the proposed indication. But they unanimously voted that there was reasonable assurance it was safe, based on the finding that most complications occurred in the 30 days after implantation.
However, no benefit was seen on HF hospitalizations in women, which panelists thought may have been due to the low number of women enrolled in the studies, and panelists recommended that the device needed to be studied further in women. Another recommendation was to drop the specific HF class in the indication because patients with HF tend to move from one class to the other.
The original study enrolled 550 patients with NYHA class III HF and a recent hospitalization for HF. All patients had the device implanted, but physicians had access to daily PA measurements for those randomized to the treatment group, and were able to adjust HF medications based on the values provided.
At 6 months, there were 84 HF hospitalizations in the treatment group (0.32 per patient), compared with 120 (0.44 per patient) in the control group, a highly significant difference that represented a 28% reduction in the rate of HF hospitalizations. In addition, 5.6% of the patients in the treatment group died, versus 7.1% in the control group, a nonsignificant 23% reduction in the relative risk of death.
In December 2011, the FDA’s Circulatory System Devices Panel met for the first time to review the device, and voted 6-4 that the risks outweighed the benefits. Like the FDA reviewers, the panel raised concerns that the nurse communications in this study may have influenced treatment decisions and biased the results, and the FDA did not approve the device.
In response, the company did not conduct another trial but conducted various analyses of HF hospitalizations and deaths during the open-access period of the study, which eliminated nurse input. Patients in the control and treatment groups in the randomized study were included in the study’s open-access period, where the PA pressures on all patients were provided to physicians.
Among the results was that HF hospitalization rates among those patients who were controls in the first part of the study dropped to a rate comparable to the rate among those who were in the original treatment group, when access to the PA measurements of the original controls were available. A third-party analysis showed that the possible effect the nurses’ input had on the outcomes in the randomized study was minimal, according to the company.
On the basis of these analyses, the company concluded that the effect on HF hospitalizations was a result of physicians knowing the PA measurements provided by the device, and was not related to the communications from nurses during the randomized segment of the study or during the open-access period.
Panelist Dr. Jeffrey Borer, chief of the division of cardiovascular medicine at the State University of New York Downstate Medical Center, Brooklyn, who voted yes on all three questions, said he found the ancillary studies very helpful in clarifying the utility of the device seen in the original study, and that this was "an approvable and probably very useful device." He, like other panelists, remained concerned about the use in women, noting that there was no benefit on HF hospitalizations in women who had the device implanted, but this may have been due to the low number of women enrolled in the study.
Dr. David Yuh, chief of cardiac surgery at Yale University, New Haven, Conn., said that while he was not surprised by the split vote on risk-benefit, he voted positively on this question. "I did feel this was an effective monitoring device that did facilitate closer monitoring of these very difficult patients for heart failure cardiologists."
Dr. Valluvan Jeevanandam, chief of cardiac and thoracic surgery at the University of Chicago, was one of two panelists who were not convinced by efficacy in terms of reducing HF hospitalizations; however, he voted in favor of the risk-benefit profile because of what he considered the device’s value as a diagnostic tool, which he said he would find useful in "innumerable" patients.
"I just got paged for two people who need right-heart catheterizations tomorrow, who wouldn’t need them" if they had this device, he said.
Voting no on the effectiveness and risk-benefit questions, Dr. David Milan, a cardiac electrophysiologist at Massachusetts General Hospital, Boston, said he believed the device was safe, but there were not enough data on effectiveness. The data analyses presented by the company were not valid or convincing, and the company should have conducted another randomized controlled study, he said.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. If approved, CardioMEMS has planned a postapproval study that would prospectively follow the safety and effectiveness of the device in a real-world setting in about 1,200 patients, including 420 women.
AT AN FDA ADVISORY PANEL MEETING
FDA panel narrowly backs expanding CRT devices’ indications
GAITHERSBURG, MD. – A Food and Drug Administration advisory panel narrowly voted to support expanding the approval of Medtronic’s Cardiac Resynchronization Therapy (CRT) devices to include patients with atrioventricular dysfunction and left ventricular systolic dysfunction.
At a meeting on Oct. 8, the Circulatory System Devices Panel voted 4-3, with one abstention, that the benefits of treatment with Medtronic’s CRT defibrillators and CRT pacemakers outweigh the risks for patients who meet the criteria in the company’s proposed indication for use – which the panel slightly modified before the vote.
Medtronic has proposed that approval of the company’s CRT-pacemaker (CRT-P) and CRT-defibrillator (CRT-D) devices be expanded to include treatment of patients who meet the following criteria: class I or class IIa indications for pacemaker implantation in accordance with American College of Cardiology/American Heart Association/Heart Rhythm Society guidelines, have New York Heart Association (NYHA) class I, II, or III heart failure, left ventricular ejection fraction (LVEF) less than or equal to 50%, with at least one of the following: third- or second-degree AV block; first-degree AV block with symptoms similar to pacemaker syndrome; or documented Wenckebach or PR interval greater than 300 ms when paced at 100 ppm. (Patients eligible for CRT-D also would need to be at risk of a life-threatening ventricular arrhythmia.)
The panel modified the proposed indication by dropping the segments on first-degree AV block and the documented Wenckebach or PR interval in response to issues that included uncertainty over whether this group of patients would benefit from treatment. This was changed to patients with first-degree AV block who are judged, with reliable confidence, to be in need of pacing most of the time.
The company’s proposed indication is based on the results of the BLOCK HF (Biventricular versus Right Ventricular Pacing in Heart Failure Patients with Atrioventricular Block) study, which randomized 691 patients with an indication for pacing with AV block; NYHA class I, II, or III heart failure; and a LVEF of 50% or less to right ventricular or biventricular pacing with a CRT-P or CRT-D.
The primary endpoint, a composite of death, an urgent care visit for HF that required intravenous therapy, or at least a 15% increase in the left ventricular end-systolic volume index (LVESVI), was met by 45.8% of those in the biventricular pacing arm vs. 55.8% of those in the right ventricular pacing arm, a statistically significant difference and a 27% reduction in risk of the primary outcome. The rate of left ventricular lead complications was 6.4% (N. Engl. J. Med. 2013;368:1585-93).
Currently, the CRT-P devices are approved for NYHA class III and IV patients who remain symptomatic despite stable, optimal medical therapy and have an LVEF of 35% or less and a prolonged QRS duration.
The CRT-D devices are approved for ventricular antitachycardia pacing and ventricular defibrillation for automated treatment of life-threatening ventricular arrhythmias, and for providing CRT in heart failure patients who remain symptomatic despite optimal medical therapy and meet any of the following classifications: NYHA class III or IV heart failure with an LVEF of 35% or less and a prolonged QRS duration; left bundle branch block with a QRS duration of 130 ms or more; an LVEF of 30% or less; and NYHA class II heart failure.
Panelists who said that the benefits did not outweigh the risks cited the potential long-term left ventricular lead complications as a concern. Those voting in favor also wrestled with this issue but agreed BLOCK HF was a well-conducted trial with results that some panelists found more compelling than others.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting.
GAITHERSBURG, MD. – A Food and Drug Administration advisory panel narrowly voted to support expanding the approval of Medtronic’s Cardiac Resynchronization Therapy (CRT) devices to include patients with atrioventricular dysfunction and left ventricular systolic dysfunction.
At a meeting on Oct. 8, the Circulatory System Devices Panel voted 4-3, with one abstention, that the benefits of treatment with Medtronic’s CRT defibrillators and CRT pacemakers outweigh the risks for patients who meet the criteria in the company’s proposed indication for use – which the panel slightly modified before the vote.
Medtronic has proposed that approval of the company’s CRT-pacemaker (CRT-P) and CRT-defibrillator (CRT-D) devices be expanded to include treatment of patients who meet the following criteria: class I or class IIa indications for pacemaker implantation in accordance with American College of Cardiology/American Heart Association/Heart Rhythm Society guidelines, have New York Heart Association (NYHA) class I, II, or III heart failure, left ventricular ejection fraction (LVEF) less than or equal to 50%, with at least one of the following: third- or second-degree AV block; first-degree AV block with symptoms similar to pacemaker syndrome; or documented Wenckebach or PR interval greater than 300 ms when paced at 100 ppm. (Patients eligible for CRT-D also would need to be at risk of a life-threatening ventricular arrhythmia.)
The panel modified the proposed indication by dropping the segments on first-degree AV block and the documented Wenckebach or PR interval in response to issues that included uncertainty over whether this group of patients would benefit from treatment. This was changed to patients with first-degree AV block who are judged, with reliable confidence, to be in need of pacing most of the time.
The company’s proposed indication is based on the results of the BLOCK HF (Biventricular versus Right Ventricular Pacing in Heart Failure Patients with Atrioventricular Block) study, which randomized 691 patients with an indication for pacing with AV block; NYHA class I, II, or III heart failure; and a LVEF of 50% or less to right ventricular or biventricular pacing with a CRT-P or CRT-D.
The primary endpoint, a composite of death, an urgent care visit for HF that required intravenous therapy, or at least a 15% increase in the left ventricular end-systolic volume index (LVESVI), was met by 45.8% of those in the biventricular pacing arm vs. 55.8% of those in the right ventricular pacing arm, a statistically significant difference and a 27% reduction in risk of the primary outcome. The rate of left ventricular lead complications was 6.4% (N. Engl. J. Med. 2013;368:1585-93).
Currently, the CRT-P devices are approved for NYHA class III and IV patients who remain symptomatic despite stable, optimal medical therapy and have an LVEF of 35% or less and a prolonged QRS duration.
The CRT-D devices are approved for ventricular antitachycardia pacing and ventricular defibrillation for automated treatment of life-threatening ventricular arrhythmias, and for providing CRT in heart failure patients who remain symptomatic despite optimal medical therapy and meet any of the following classifications: NYHA class III or IV heart failure with an LVEF of 35% or less and a prolonged QRS duration; left bundle branch block with a QRS duration of 130 ms or more; an LVEF of 30% or less; and NYHA class II heart failure.
Panelists who said that the benefits did not outweigh the risks cited the potential long-term left ventricular lead complications as a concern. Those voting in favor also wrestled with this issue but agreed BLOCK HF was a well-conducted trial with results that some panelists found more compelling than others.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting.
GAITHERSBURG, MD. – A Food and Drug Administration advisory panel narrowly voted to support expanding the approval of Medtronic’s Cardiac Resynchronization Therapy (CRT) devices to include patients with atrioventricular dysfunction and left ventricular systolic dysfunction.
At a meeting on Oct. 8, the Circulatory System Devices Panel voted 4-3, with one abstention, that the benefits of treatment with Medtronic’s CRT defibrillators and CRT pacemakers outweigh the risks for patients who meet the criteria in the company’s proposed indication for use – which the panel slightly modified before the vote.
Medtronic has proposed that approval of the company’s CRT-pacemaker (CRT-P) and CRT-defibrillator (CRT-D) devices be expanded to include treatment of patients who meet the following criteria: class I or class IIa indications for pacemaker implantation in accordance with American College of Cardiology/American Heart Association/Heart Rhythm Society guidelines, have New York Heart Association (NYHA) class I, II, or III heart failure, left ventricular ejection fraction (LVEF) less than or equal to 50%, with at least one of the following: third- or second-degree AV block; first-degree AV block with symptoms similar to pacemaker syndrome; or documented Wenckebach or PR interval greater than 300 ms when paced at 100 ppm. (Patients eligible for CRT-D also would need to be at risk of a life-threatening ventricular arrhythmia.)
The panel modified the proposed indication by dropping the segments on first-degree AV block and the documented Wenckebach or PR interval in response to issues that included uncertainty over whether this group of patients would benefit from treatment. This was changed to patients with first-degree AV block who are judged, with reliable confidence, to be in need of pacing most of the time.
The company’s proposed indication is based on the results of the BLOCK HF (Biventricular versus Right Ventricular Pacing in Heart Failure Patients with Atrioventricular Block) study, which randomized 691 patients with an indication for pacing with AV block; NYHA class I, II, or III heart failure; and a LVEF of 50% or less to right ventricular or biventricular pacing with a CRT-P or CRT-D.
The primary endpoint, a composite of death, an urgent care visit for HF that required intravenous therapy, or at least a 15% increase in the left ventricular end-systolic volume index (LVESVI), was met by 45.8% of those in the biventricular pacing arm vs. 55.8% of those in the right ventricular pacing arm, a statistically significant difference and a 27% reduction in risk of the primary outcome. The rate of left ventricular lead complications was 6.4% (N. Engl. J. Med. 2013;368:1585-93).
Currently, the CRT-P devices are approved for NYHA class III and IV patients who remain symptomatic despite stable, optimal medical therapy and have an LVEF of 35% or less and a prolonged QRS duration.
The CRT-D devices are approved for ventricular antitachycardia pacing and ventricular defibrillation for automated treatment of life-threatening ventricular arrhythmias, and for providing CRT in heart failure patients who remain symptomatic despite optimal medical therapy and meet any of the following classifications: NYHA class III or IV heart failure with an LVEF of 35% or less and a prolonged QRS duration; left bundle branch block with a QRS duration of 130 ms or more; an LVEF of 30% or less; and NYHA class II heart failure.
Panelists who said that the benefits did not outweigh the risks cited the potential long-term left ventricular lead complications as a concern. Those voting in favor also wrestled with this issue but agreed BLOCK HF was a well-conducted trial with results that some panelists found more compelling than others.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting.
AT AN FDA ADVISORY COMMITTEE MEETING
Novel oral vasodilator approved to treat pulmonary hypertension
Riociguat, a potent vasodilator that is the first in a new class of drugs, has been approved by the Food and Drug Administration for treating two types of pulmonary hypertension in adults.
The drug, which will be marketed as Adempas, was approved Oct. 8 to treat chronic thromboembolic pulmonary hypertension (CTEPH) and pulmonary arterial hypertension (PAH), the agency said in a statement.
Orally administered in tablet form, riociguat is a soluble guanylate cyclase (sGC) stimulator, the first drug in this class to be approved for pulmonary hypertension. It is also "the first drug of any class to be shown to be effective for patients with CTEPH," Dr. Norman Stockbridge, director of the Division of Cardiovascular and Renal Drug Products in the FDA’s Center for Drug Evaluation and Research, said in the statement.
Riociguat is a pregnancy category X drug and is available to women only through a Risk Evaluation and Mitigation Strategy (REMS) program.
The approved indications for the drug, which will be marketed by Bayer HealthCare Pharmaceuticals, are for persistent/recurrent CTEPH (WHO group 4) "after surgical treatment or inoperable CTEPH to improve exercise capacity and WHO functional class" or PAH (WHO group 1) "to improve exercise capacity, improve WHO functional class, and to delay clinical worsening."
Approval was based on the results of two international studies that found treatment resulted in significant improvements over placebo in the 6 minute walk test. Side effects of treatment include headache, dizziness, dyspepsia, peripheral edema, nausea, diarrhea, and vomiting, according to the FDA. Based on these results, an FDA advisory panel unanimously recommended approval of the two indications in August.
In the study of 380 patients with PAH, the change from baseline in the 6-minute walk test at 12 weeks improved by a mean of 30 m among those treated with riociguat, vs. a mean drop of 6 m in the placebo group. The WHO functional class improved in 21% of those on riociguat, compared with 14% in the placebo group, deteriorating in 4% and 14%, respectively.
In the study of 261 patients with CTEPH, the change from baseline in the 6-minute walk test at 16 weeks improved by a mean of 39 m among treated patients vs. a mean 6 m reduction in the placebo group at 16 weeks. WHO functional class improved in 33% of those on riociguat and 15% of those on placebo, deteriorating in 5% and 7%, respectively.
The two studies were published in the July 25 issue of the New England Journal of Medicine (2013;369:330-40; 2013;369:319-29).
The prescribing information includes a boxed warning about embryo-fetal toxicity. Women can receive the drug only through the REMS program.
Because of the risk of hypotension, the drug is contraindicated for use with nitrates or nitric oxide donors, such as amyl nitrate, and with phosphodiesterase inhibitors or nonspecific PDE inhibitors.
The wholesale cost of the drug is $7,500 for 30 days of treatment, with one tablet taken three times a day, according to a Bayer spokesperson. The company has set up a patient assistance program to help with coverage.
To date, riociguat has been approved in Canada for the treatment of inoperable or persistent/recurrent CTEPH after surgery in adults with WHO functional class II or III pulmonary hypertension, and it is under review in the European Union, according to the company spokesperson.
Riociguat, a potent vasodilator that is the first in a new class of drugs, has been approved by the Food and Drug Administration for treating two types of pulmonary hypertension in adults.
The drug, which will be marketed as Adempas, was approved Oct. 8 to treat chronic thromboembolic pulmonary hypertension (CTEPH) and pulmonary arterial hypertension (PAH), the agency said in a statement.
Orally administered in tablet form, riociguat is a soluble guanylate cyclase (sGC) stimulator, the first drug in this class to be approved for pulmonary hypertension. It is also "the first drug of any class to be shown to be effective for patients with CTEPH," Dr. Norman Stockbridge, director of the Division of Cardiovascular and Renal Drug Products in the FDA’s Center for Drug Evaluation and Research, said in the statement.
Riociguat is a pregnancy category X drug and is available to women only through a Risk Evaluation and Mitigation Strategy (REMS) program.
The approved indications for the drug, which will be marketed by Bayer HealthCare Pharmaceuticals, are for persistent/recurrent CTEPH (WHO group 4) "after surgical treatment or inoperable CTEPH to improve exercise capacity and WHO functional class" or PAH (WHO group 1) "to improve exercise capacity, improve WHO functional class, and to delay clinical worsening."
Approval was based on the results of two international studies that found treatment resulted in significant improvements over placebo in the 6 minute walk test. Side effects of treatment include headache, dizziness, dyspepsia, peripheral edema, nausea, diarrhea, and vomiting, according to the FDA. Based on these results, an FDA advisory panel unanimously recommended approval of the two indications in August.
In the study of 380 patients with PAH, the change from baseline in the 6-minute walk test at 12 weeks improved by a mean of 30 m among those treated with riociguat, vs. a mean drop of 6 m in the placebo group. The WHO functional class improved in 21% of those on riociguat, compared with 14% in the placebo group, deteriorating in 4% and 14%, respectively.
In the study of 261 patients with CTEPH, the change from baseline in the 6-minute walk test at 16 weeks improved by a mean of 39 m among treated patients vs. a mean 6 m reduction in the placebo group at 16 weeks. WHO functional class improved in 33% of those on riociguat and 15% of those on placebo, deteriorating in 5% and 7%, respectively.
The two studies were published in the July 25 issue of the New England Journal of Medicine (2013;369:330-40; 2013;369:319-29).
The prescribing information includes a boxed warning about embryo-fetal toxicity. Women can receive the drug only through the REMS program.
Because of the risk of hypotension, the drug is contraindicated for use with nitrates or nitric oxide donors, such as amyl nitrate, and with phosphodiesterase inhibitors or nonspecific PDE inhibitors.
The wholesale cost of the drug is $7,500 for 30 days of treatment, with one tablet taken three times a day, according to a Bayer spokesperson. The company has set up a patient assistance program to help with coverage.
To date, riociguat has been approved in Canada for the treatment of inoperable or persistent/recurrent CTEPH after surgery in adults with WHO functional class II or III pulmonary hypertension, and it is under review in the European Union, according to the company spokesperson.
Riociguat, a potent vasodilator that is the first in a new class of drugs, has been approved by the Food and Drug Administration for treating two types of pulmonary hypertension in adults.
The drug, which will be marketed as Adempas, was approved Oct. 8 to treat chronic thromboembolic pulmonary hypertension (CTEPH) and pulmonary arterial hypertension (PAH), the agency said in a statement.
Orally administered in tablet form, riociguat is a soluble guanylate cyclase (sGC) stimulator, the first drug in this class to be approved for pulmonary hypertension. It is also "the first drug of any class to be shown to be effective for patients with CTEPH," Dr. Norman Stockbridge, director of the Division of Cardiovascular and Renal Drug Products in the FDA’s Center for Drug Evaluation and Research, said in the statement.
Riociguat is a pregnancy category X drug and is available to women only through a Risk Evaluation and Mitigation Strategy (REMS) program.
The approved indications for the drug, which will be marketed by Bayer HealthCare Pharmaceuticals, are for persistent/recurrent CTEPH (WHO group 4) "after surgical treatment or inoperable CTEPH to improve exercise capacity and WHO functional class" or PAH (WHO group 1) "to improve exercise capacity, improve WHO functional class, and to delay clinical worsening."
Approval was based on the results of two international studies that found treatment resulted in significant improvements over placebo in the 6 minute walk test. Side effects of treatment include headache, dizziness, dyspepsia, peripheral edema, nausea, diarrhea, and vomiting, according to the FDA. Based on these results, an FDA advisory panel unanimously recommended approval of the two indications in August.
In the study of 380 patients with PAH, the change from baseline in the 6-minute walk test at 12 weeks improved by a mean of 30 m among those treated with riociguat, vs. a mean drop of 6 m in the placebo group. The WHO functional class improved in 21% of those on riociguat, compared with 14% in the placebo group, deteriorating in 4% and 14%, respectively.
In the study of 261 patients with CTEPH, the change from baseline in the 6-minute walk test at 16 weeks improved by a mean of 39 m among treated patients vs. a mean 6 m reduction in the placebo group at 16 weeks. WHO functional class improved in 33% of those on riociguat and 15% of those on placebo, deteriorating in 5% and 7%, respectively.
The two studies were published in the July 25 issue of the New England Journal of Medicine (2013;369:330-40; 2013;369:319-29).
The prescribing information includes a boxed warning about embryo-fetal toxicity. Women can receive the drug only through the REMS program.
Because of the risk of hypotension, the drug is contraindicated for use with nitrates or nitric oxide donors, such as amyl nitrate, and with phosphodiesterase inhibitors or nonspecific PDE inhibitors.
The wholesale cost of the drug is $7,500 for 30 days of treatment, with one tablet taken three times a day, according to a Bayer spokesperson. The company has set up a patient assistance program to help with coverage.
To date, riociguat has been approved in Canada for the treatment of inoperable or persistent/recurrent CTEPH after surgery in adults with WHO functional class II or III pulmonary hypertension, and it is under review in the European Union, according to the company spokesperson.
False-negative rate for sentinel nodes high after neoadjuvant chemotherapy
The false-negative rate of sentinel lymph node results was almost 13% after neoadjuvant chemotherapy, in a study of women initially presenting with biopsy-proven node-positive breast cancer, which is above the acceptable threshold, a study has shown.
Considering the threshold for a false-negative rate is 10%, "changes in approach and patient selection that result in greater sensitivity would be necessary" to support the use of sentinel lymph node (SLN) surgery "as an alternative" to axillary lymph node dissection in this population of patients with breast cancer, concluded Dr. Judy Boughey of the department of surgery, Mayo Clinic, Rochester, Minn., and her associates in the Alliance for Clinical Trials in Oncology.

The investigators also found that false-negative SLN biopsy results were significantly lower when two mapping agents were used; the false-negative rate was also significantly lower when at least three sentinel lymph nodes were sampled. The results of the study were presented at the annual clinical congress of the American Congress of Surgeons and simultaneously published online on Oct. 7, 2013, in JAMA (doi:10.1001/jama.2013.278932).
The phase II study addressed the false-negative rate of SLN biopsy after neoadjuvant chemotherapy, in women who initially presented with pathologically confirmed node-positive disease. The study enrolled 701 women from July 2009 to June 2011, at 136 institutions. The women had clinical stage T0 through T4, N1 through N2, M0 breast cancer; most (663) had cN1 disease (disease in movable axillary lymph nodes) and 38 had cN2 disease (disease in fixed or matted axillary lymph nodes). After chemotherapy was completed, the patients had surgery, involving SLN biopsy and axillary lymph node dissection.
Of the 701 women, 687 underwent both SLN biopsy and axillary lymph node dissection, 2 had only SLN biopsy, and 12 had axillary lymph node dissection only. Most of the 689 women who underwent SLN biopsy had the procedure performed with blue dye and radiolabeled colloid (79%), 116 (16.8%) had radiolabeled colloid only, and 28 (4.1%) had blue dye only.
Of the 689 women who had SLN biopsy, at least one SLN was detected in 639 (almost 93%). Among the 651 women with cN1 disease, at least one SLN was detected in 605 women (93%); and among the 38 women with cN2 disease, at least one SNL was detected in 34 (89.5%).
In 525 of the patients with cN1 disease, at least two SLNs were excised and axillary lymph node dissection was completed. There was no residual node disease found in 215 of these patients, for a nodal pathologic complete response rate of 41%.
Of the remaining 310 patients, nodal disease was found in the sentinel lymph nodes in 108 patients (20.6%), and in both axillary and sentinel nodes in 163 patients (31.1%).
Residual node disease was found in the axillary lymph nodes of only 39 (7.4%) of the patients. Therefore, the results of the SLN biopsy results were false negative in 39 of the 310 patients, for a false negative rate of 12.6%.
While the study results "suggest that surgeons cannot reliably detect all axillary lymph node metastases in patients with cN1 breast cancer following chemotherapy" with SLN procedures, "we did identify important factors influencing the likelihood of a false-negative SLN," the authors said. The false-negative rate for the SLN biopsy results was significantly decreased when both blue dye and radiolabeled colloid were used as mapping agents, and when at least three sentinel lymph nodes were biopsied.
When both blue dye and radiolabeled colloid were used, the false-negative rate was 10.8%, compared with 20.3% when only one agent was used. "Using two mapping agents with different molecule sizes and transit times is an important surgical standard that should be adhered to for SLN surgery after chemotherapy," they said, noting that after chemotherapy, "the axilla often has more fibrosis, making evaluation of lymphatic drainage and surgical dissection more challenging."
And when at least three SLNs were examined, the false negative rate was 9.1%, compared with 21.1% when two were sampled. "As the accuracy of any sampling test is dependent on the amount of material sampled, these results are not surprising," they said, noting that this has been found in other studies.
SLN is less invasive than axillary dissection and is considered a reliable way to check for axillary nodal disease in women who present with node-negative disease, but the use of SLN biopsy after neoadjuvant chemotherapy in women with cN1 disease "has been questioned," because the false-negative rate for this approach in this population has ranged from 7% to 25% in small studies, the only data available on this approach, according to the authors.
The study was supported by a grant from the National Cancer Institute to the American College of Surgeons Oncology Group (ACOSOG). The patients were in the ACOSOG Z1071 trial. Four of the authors reported having grants from the National Institutes of Health or Komen Foundation, having contracts with Galena BioPharma, or having been paid for lectures from LifeCell. The remaining 17 authors said they had no relevant financial disclosures.
"[T]he appropriateness of SLN biopsy in this setting remains uncertain," although the study provides important information, according to Dr. Monica Morrow and Dr. Chau Dang. The increasing number of targeted therapies for breast cancer has enabled physicians to "move away from the ‘one size fits all’ approach." As a result, "the prognostic information obtained from residual nodal disease after neoadjuvant therapy is likely to become increasingly important in determining the need for additional therapy." If true, "research in ways to improve the performance of the SLN biopsy after neoadjuvant therapy is needed for this approach to become a viable management strategy."
Dr. Morrow and Dr. Dang of Memorial Sloan-Kettering Cancer Center in New York made their remarks in an editorial accompanying the article (JAMA 2013 Oct. 7 [doi:10.l001/jama.2013.7844]). Dr. Dang disclosed having received grant funding from Genentech. Dr. Morrow said she had no relevant financial disclosures.
"[T]he appropriateness of SLN biopsy in this setting remains uncertain," although the study provides important information, according to Dr. Monica Morrow and Dr. Chau Dang. The increasing number of targeted therapies for breast cancer has enabled physicians to "move away from the ‘one size fits all’ approach." As a result, "the prognostic information obtained from residual nodal disease after neoadjuvant therapy is likely to become increasingly important in determining the need for additional therapy." If true, "research in ways to improve the performance of the SLN biopsy after neoadjuvant therapy is needed for this approach to become a viable management strategy."
Dr. Morrow and Dr. Dang of Memorial Sloan-Kettering Cancer Center in New York made their remarks in an editorial accompanying the article (JAMA 2013 Oct. 7 [doi:10.l001/jama.2013.7844]). Dr. Dang disclosed having received grant funding from Genentech. Dr. Morrow said she had no relevant financial disclosures.
"[T]he appropriateness of SLN biopsy in this setting remains uncertain," although the study provides important information, according to Dr. Monica Morrow and Dr. Chau Dang. The increasing number of targeted therapies for breast cancer has enabled physicians to "move away from the ‘one size fits all’ approach." As a result, "the prognostic information obtained from residual nodal disease after neoadjuvant therapy is likely to become increasingly important in determining the need for additional therapy." If true, "research in ways to improve the performance of the SLN biopsy after neoadjuvant therapy is needed for this approach to become a viable management strategy."
Dr. Morrow and Dr. Dang of Memorial Sloan-Kettering Cancer Center in New York made their remarks in an editorial accompanying the article (JAMA 2013 Oct. 7 [doi:10.l001/jama.2013.7844]). Dr. Dang disclosed having received grant funding from Genentech. Dr. Morrow said she had no relevant financial disclosures.
The false-negative rate of sentinel lymph node results was almost 13% after neoadjuvant chemotherapy, in a study of women initially presenting with biopsy-proven node-positive breast cancer, which is above the acceptable threshold, a study has shown.
Considering the threshold for a false-negative rate is 10%, "changes in approach and patient selection that result in greater sensitivity would be necessary" to support the use of sentinel lymph node (SLN) surgery "as an alternative" to axillary lymph node dissection in this population of patients with breast cancer, concluded Dr. Judy Boughey of the department of surgery, Mayo Clinic, Rochester, Minn., and her associates in the Alliance for Clinical Trials in Oncology.
The investigators also found that false-negative SLN biopsy results were significantly lower when two mapping agents were used; the false-negative rate was also significantly lower when at least three sentinel lymph nodes were sampled. The results of the study were presented at the annual clinical congress of the American Congress of Surgeons and simultaneously published online on Oct. 7, 2013, in JAMA (doi:10.1001/jama.2013.278932).
The phase II study addressed the false-negative rate of SLN biopsy after neoadjuvant chemotherapy, in women who initially presented with pathologically confirmed node-positive disease. The study enrolled 701 women from July 2009 to June 2011, at 136 institutions. The women had clinical stage T0 through T4, N1 through N2, M0 breast cancer; most (663) had cN1 disease (disease in movable axillary lymph nodes) and 38 had cN2 disease (disease in fixed or matted axillary lymph nodes). After chemotherapy was completed, the patients had surgery, involving SLN biopsy and axillary lymph node dissection.
Of the 701 women, 687 underwent both SLN biopsy and axillary lymph node dissection, 2 had only SLN biopsy, and 12 had axillary lymph node dissection only. Most of the 689 women who underwent SLN biopsy had the procedure performed with blue dye and radiolabeled colloid (79%), 116 (16.8%) had radiolabeled colloid only, and 28 (4.1%) had blue dye only.
Of the 689 women who had SLN biopsy, at least one SLN was detected in 639 (almost 93%). Among the 651 women with cN1 disease, at least one SLN was detected in 605 women (93%); and among the 38 women with cN2 disease, at least one SNL was detected in 34 (89.5%).
In 525 of the patients with cN1 disease, at least two SLNs were excised and axillary lymph node dissection was completed. There was no residual node disease found in 215 of these patients, for a nodal pathologic complete response rate of 41%.
Of the remaining 310 patients, nodal disease was found in the sentinel lymph nodes in 108 patients (20.6%), and in both axillary and sentinel nodes in 163 patients (31.1%).
Residual node disease was found in the axillary lymph nodes of only 39 (7.4%) of the patients. Therefore, the results of the SLN biopsy results were false negative in 39 of the 310 patients, for a false negative rate of 12.6%.
While the study results "suggest that surgeons cannot reliably detect all axillary lymph node metastases in patients with cN1 breast cancer following chemotherapy" with SLN procedures, "we did identify important factors influencing the likelihood of a false-negative SLN," the authors said. The false-negative rate for the SLN biopsy results was significantly decreased when both blue dye and radiolabeled colloid were used as mapping agents, and when at least three sentinel lymph nodes were biopsied.
When both blue dye and radiolabeled colloid were used, the false-negative rate was 10.8%, compared with 20.3% when only one agent was used. "Using two mapping agents with different molecule sizes and transit times is an important surgical standard that should be adhered to for SLN surgery after chemotherapy," they said, noting that after chemotherapy, "the axilla often has more fibrosis, making evaluation of lymphatic drainage and surgical dissection more challenging."
And when at least three SLNs were examined, the false negative rate was 9.1%, compared with 21.1% when two were sampled. "As the accuracy of any sampling test is dependent on the amount of material sampled, these results are not surprising," they said, noting that this has been found in other studies.
SLN is less invasive than axillary dissection and is considered a reliable way to check for axillary nodal disease in women who present with node-negative disease, but the use of SLN biopsy after neoadjuvant chemotherapy in women with cN1 disease "has been questioned," because the false-negative rate for this approach in this population has ranged from 7% to 25% in small studies, the only data available on this approach, according to the authors.
The study was supported by a grant from the National Cancer Institute to the American College of Surgeons Oncology Group (ACOSOG). The patients were in the ACOSOG Z1071 trial. Four of the authors reported having grants from the National Institutes of Health or Komen Foundation, having contracts with Galena BioPharma, or having been paid for lectures from LifeCell. The remaining 17 authors said they had no relevant financial disclosures.
The false-negative rate of sentinel lymph node results was almost 13% after neoadjuvant chemotherapy, in a study of women initially presenting with biopsy-proven node-positive breast cancer, which is above the acceptable threshold, a study has shown.
Considering the threshold for a false-negative rate is 10%, "changes in approach and patient selection that result in greater sensitivity would be necessary" to support the use of sentinel lymph node (SLN) surgery "as an alternative" to axillary lymph node dissection in this population of patients with breast cancer, concluded Dr. Judy Boughey of the department of surgery, Mayo Clinic, Rochester, Minn., and her associates in the Alliance for Clinical Trials in Oncology.
The investigators also found that false-negative SLN biopsy results were significantly lower when two mapping agents were used; the false-negative rate was also significantly lower when at least three sentinel lymph nodes were sampled. The results of the study were presented at the annual clinical congress of the American Congress of Surgeons and simultaneously published online on Oct. 7, 2013, in JAMA (doi:10.1001/jama.2013.278932).
The phase II study addressed the false-negative rate of SLN biopsy after neoadjuvant chemotherapy, in women who initially presented with pathologically confirmed node-positive disease. The study enrolled 701 women from July 2009 to June 2011, at 136 institutions. The women had clinical stage T0 through T4, N1 through N2, M0 breast cancer; most (663) had cN1 disease (disease in movable axillary lymph nodes) and 38 had cN2 disease (disease in fixed or matted axillary lymph nodes). After chemotherapy was completed, the patients had surgery, involving SLN biopsy and axillary lymph node dissection.
Of the 701 women, 687 underwent both SLN biopsy and axillary lymph node dissection, 2 had only SLN biopsy, and 12 had axillary lymph node dissection only. Most of the 689 women who underwent SLN biopsy had the procedure performed with blue dye and radiolabeled colloid (79%), 116 (16.8%) had radiolabeled colloid only, and 28 (4.1%) had blue dye only.
Of the 689 women who had SLN biopsy, at least one SLN was detected in 639 (almost 93%). Among the 651 women with cN1 disease, at least one SLN was detected in 605 women (93%); and among the 38 women with cN2 disease, at least one SNL was detected in 34 (89.5%).
In 525 of the patients with cN1 disease, at least two SLNs were excised and axillary lymph node dissection was completed. There was no residual node disease found in 215 of these patients, for a nodal pathologic complete response rate of 41%.
Of the remaining 310 patients, nodal disease was found in the sentinel lymph nodes in 108 patients (20.6%), and in both axillary and sentinel nodes in 163 patients (31.1%).
Residual node disease was found in the axillary lymph nodes of only 39 (7.4%) of the patients. Therefore, the results of the SLN biopsy results were false negative in 39 of the 310 patients, for a false negative rate of 12.6%.
While the study results "suggest that surgeons cannot reliably detect all axillary lymph node metastases in patients with cN1 breast cancer following chemotherapy" with SLN procedures, "we did identify important factors influencing the likelihood of a false-negative SLN," the authors said. The false-negative rate for the SLN biopsy results was significantly decreased when both blue dye and radiolabeled colloid were used as mapping agents, and when at least three sentinel lymph nodes were biopsied.
When both blue dye and radiolabeled colloid were used, the false-negative rate was 10.8%, compared with 20.3% when only one agent was used. "Using two mapping agents with different molecule sizes and transit times is an important surgical standard that should be adhered to for SLN surgery after chemotherapy," they said, noting that after chemotherapy, "the axilla often has more fibrosis, making evaluation of lymphatic drainage and surgical dissection more challenging."
And when at least three SLNs were examined, the false negative rate was 9.1%, compared with 21.1% when two were sampled. "As the accuracy of any sampling test is dependent on the amount of material sampled, these results are not surprising," they said, noting that this has been found in other studies.
SLN is less invasive than axillary dissection and is considered a reliable way to check for axillary nodal disease in women who present with node-negative disease, but the use of SLN biopsy after neoadjuvant chemotherapy in women with cN1 disease "has been questioned," because the false-negative rate for this approach in this population has ranged from 7% to 25% in small studies, the only data available on this approach, according to the authors.
The study was supported by a grant from the National Cancer Institute to the American College of Surgeons Oncology Group (ACOSOG). The patients were in the ACOSOG Z1071 trial. Four of the authors reported having grants from the National Institutes of Health or Komen Foundation, having contracts with Galena BioPharma, or having been paid for lectures from LifeCell. The remaining 17 authors said they had no relevant financial disclosures.
FROM THE ACS CLINICAL CONGRESS
Major finding: Sentinel lymph node findings had a false-negative rate of 12.6% among women with cN1 breast cancer, after neoadjuvant chemotherapy, above the acceptable threshold of 10%, but the false-negative rate dropped below this level when a dual mapping technique was used and when more than two sentinel nodes were biopsied.
Data source: A prospective, phase II multicenter study that evaluated the false-negative rate of sentinel lymph node surgery in more than 700 women with clinically node-positive breast cancer treated with chemotherapy before surgery.
Disclosures: The study was supported by a grant from the National Cancer Institute to the American College of Surgeons Oncology Group. Four authors reported having received grants from the National Institutes of Health or the Komen Foundation, having contracts with Galena BioPharma, or having been paid for lectures by LifeCell. The remaining authors said they had no relevant financial disclosures.

FDA approves vortioxetine for treating depression in adults
Vortioxetine, a novel antidepressant, has been approved for treating major depressive disorder in adults.
It will be marketed as Brintellix, by Takeda Pharmaceuticals and Lundbeck, and will be available in 5-mg, 10-mg, 15-mg, and 20-mg tablets. The Food and Drug Administration approved the drug Oct. 1.
The prescribing information and the Takeda/Lundbeck press release announcing the approval include the statement that the mechanism of vortioxetine’s antidepressant effect "is not fully understood."
"But it is thought to be related to its enhancement of serotonergic activity in the [central nervous system] through inhibition of the reuptake of serotonin (5-HT)," according to the prescribing information. The prescribing information adds: "It also has several other activities including 5-HT3 receptor antagonism and 5-HT1A receptor agonism. The contribution of these activities to vortioxetine’s antidepressant effect has not been established."
In the FDA’s statement, Dr. Mitchell Mathis said that since medications affect everyone differently, "it is important to have a variety of treatment options available for patients who suffer from depression." Dr. Mathis is acting director of the division of psychiatry products in the FDA’s Center for Drug Evaluation and Research.
Approval was based on six short-term, 6- to 8-week, randomized studies conducted in the United States and elsewhere, which found that treatment with at least one dose of the drug was more effective than placebo in improving depression symptoms. Five of the studies enrolled patients aged 18-75 years, and one enrolled patients aged 64-88 years.
In a long-term maintenance study, those treated with vortioxetine had a longer time to recurrence of depression than did those treated with placebo, a difference that was statistically significant. Nausea, vomiting, and constipation were the most common adverse effects associated with the drug in clinical trials, according to the FDA statement.
As with all antidepressant drugs, the prescribing information has a boxed warning and medication guide about the increased risk of suicidal thoughts and behavior in children, adolescents, and adults aged 18-24 years, during initial treatment. The warning includes the recommendation that people starting treatment with antidepressants be closely monitored for worsening depression and suicidal thoughts and behavior.
The drug is expected to be available in the United States by the end of 2013, according to Takeda. It has been submitted for approval in Europe.
Vortioxetine, a novel antidepressant, has been approved for treating major depressive disorder in adults.
It will be marketed as Brintellix, by Takeda Pharmaceuticals and Lundbeck, and will be available in 5-mg, 10-mg, 15-mg, and 20-mg tablets. The Food and Drug Administration approved the drug Oct. 1.
The prescribing information and the Takeda/Lundbeck press release announcing the approval include the statement that the mechanism of vortioxetine’s antidepressant effect "is not fully understood."
"But it is thought to be related to its enhancement of serotonergic activity in the [central nervous system] through inhibition of the reuptake of serotonin (5-HT)," according to the prescribing information. The prescribing information adds: "It also has several other activities including 5-HT3 receptor antagonism and 5-HT1A receptor agonism. The contribution of these activities to vortioxetine’s antidepressant effect has not been established."
In the FDA’s statement, Dr. Mitchell Mathis said that since medications affect everyone differently, "it is important to have a variety of treatment options available for patients who suffer from depression." Dr. Mathis is acting director of the division of psychiatry products in the FDA’s Center for Drug Evaluation and Research.
Approval was based on six short-term, 6- to 8-week, randomized studies conducted in the United States and elsewhere, which found that treatment with at least one dose of the drug was more effective than placebo in improving depression symptoms. Five of the studies enrolled patients aged 18-75 years, and one enrolled patients aged 64-88 years.
In a long-term maintenance study, those treated with vortioxetine had a longer time to recurrence of depression than did those treated with placebo, a difference that was statistically significant. Nausea, vomiting, and constipation were the most common adverse effects associated with the drug in clinical trials, according to the FDA statement.
As with all antidepressant drugs, the prescribing information has a boxed warning and medication guide about the increased risk of suicidal thoughts and behavior in children, adolescents, and adults aged 18-24 years, during initial treatment. The warning includes the recommendation that people starting treatment with antidepressants be closely monitored for worsening depression and suicidal thoughts and behavior.
The drug is expected to be available in the United States by the end of 2013, according to Takeda. It has been submitted for approval in Europe.
Vortioxetine, a novel antidepressant, has been approved for treating major depressive disorder in adults.
It will be marketed as Brintellix, by Takeda Pharmaceuticals and Lundbeck, and will be available in 5-mg, 10-mg, 15-mg, and 20-mg tablets. The Food and Drug Administration approved the drug Oct. 1.
The prescribing information and the Takeda/Lundbeck press release announcing the approval include the statement that the mechanism of vortioxetine’s antidepressant effect "is not fully understood."
"But it is thought to be related to its enhancement of serotonergic activity in the [central nervous system] through inhibition of the reuptake of serotonin (5-HT)," according to the prescribing information. The prescribing information adds: "It also has several other activities including 5-HT3 receptor antagonism and 5-HT1A receptor agonism. The contribution of these activities to vortioxetine’s antidepressant effect has not been established."
In the FDA’s statement, Dr. Mitchell Mathis said that since medications affect everyone differently, "it is important to have a variety of treatment options available for patients who suffer from depression." Dr. Mathis is acting director of the division of psychiatry products in the FDA’s Center for Drug Evaluation and Research.
Approval was based on six short-term, 6- to 8-week, randomized studies conducted in the United States and elsewhere, which found that treatment with at least one dose of the drug was more effective than placebo in improving depression symptoms. Five of the studies enrolled patients aged 18-75 years, and one enrolled patients aged 64-88 years.
In a long-term maintenance study, those treated with vortioxetine had a longer time to recurrence of depression than did those treated with placebo, a difference that was statistically significant. Nausea, vomiting, and constipation were the most common adverse effects associated with the drug in clinical trials, according to the FDA statement.
As with all antidepressant drugs, the prescribing information has a boxed warning and medication guide about the increased risk of suicidal thoughts and behavior in children, adolescents, and adults aged 18-24 years, during initial treatment. The warning includes the recommendation that people starting treatment with antidepressants be closely monitored for worsening depression and suicidal thoughts and behavior.
The drug is expected to be available in the United States by the end of 2013, according to Takeda. It has been submitted for approval in Europe.