User login
Controversial diabetes drug not associated with increased CV risk in patients with CAD
Treatment with rosiglitazone was not associated with an increased risk of major ischemic cardiovascular events in a large, international study of patients with type 2 diabetes and coronary artery disease.
"Our analyses did not detect any significant hazard of increased ischemic cardiovascular risk with rosiglitazone treatment despite its use in this particularly vulnerable, higher risk population," Dr. Richard Bach of Washington University, St. Louis, and the other authors concluded.
The study, an analysis of data from the Bypass Angioplasty Revascularization Investigation 2 Diabetes (BARI 2D) trial, was published online July 15 (doi: 10.1161/CIRCULATIONAHA.112.000678).

The primary outcomes of the study were published in 2009 (N. Engl. J. Med. 2009; 360:2503-15).
The cardiac safety of rosiglitazone, a thiazolidinedione (TZD) marketed as Avandia by GlaxoSmithKline, has been mired in controversy for several years because of analyses of data showing that rosiglitazone may increase the risk of MI or death. The Food and Drug Administration issued an alert about the safety of the drug and limited its use in 2010. The agency also halted a long-term prospective cardiovascular safety study of rosiglitazone. In 2010, the Europeans Medicines Agency took rosiglitazone off the market in Europe, citing the same data.
In June 2013, at an FDA advisory panel meeting held to review the readjudicated results of a cardiovascular safety trial of rosiglitazone – which did not identify a greater rate of adverse cardiovascular outcomes among those on the drug – the majority of the panel recommended loosening or lifting the restrictions that had been placed on prescribing the drug in 2010. (The FDA has not yet acted on the panel’s recommendations.)
In the post hoc analysis of the BARI 2D data, the investigators compared the rates – between those on rosiglitazone and those not on a TZD – of mortality, MI, stroke, and other outcomes, and composite outcomes among nearly 2,400 patients with type 2 diabetes and coronary artery disease in the study. Over an average of 4.5 years, about 990 patients were on rosiglitazone at some point during the study (for up to 3,025 patient-years of exposure) and 485 were treated with rosiglitazone for at least 80% of the time. About 1,200 were not on a TZD.
After adjustment for differences in multiple baseline characteristics and other antidiabetes medications, the results were as follows:
• The incidence of all-cause death was similar among those on rosiglitazone, compared with those not on a TZD (hazard ratio, 0.83), a statistically nonsignificant difference.
• The composite of death, MI, and stroke was significantly lower among those on rosiglitazone (HR, 0.72).
• The incidence of stroke alone was significantly lower among those on rosiglitazone (HR, 0.36).
• The incidence of MI was lower among those on rosiglitazone (HR, 0.77) and the incidence of heart failure was higher (HR, 1.22), but these differences were not statistically significant.
When compared with those not on a TZD, the incidence of fractures was higher in those on a TZD (HR, 1.62), a risk that appeared to be more pronounced among women.
The investigators did not find a significant interaction between rosiglitazone and coadministration with other antidiabetes and cardiovascular medications (insulin, metformin, gemfibrozil, other fibrates, sulfonylureas, nitrates, or ACE inhibitors) on the composite rate of death, MI and stroke, or on individual rates of death, MI, stroke, or fractures. There was a significant interaction, however, with rosiglitazone and metformin on the risk of heart failure associated with rosiglitazone, with a relative risk of heart failure significantly greater among those patients on rosiglitazone but not on metformin, than on those who were on both.
These results "provide no evidence that use of rosiglitazone is associated with increased rates of major adverse ischemic cardiovascular events among patients with type 2 diabetes and established CAD," the authors concluded.
While the study had limitations, the researchers noted that a possible difference between this study and studies that found adverse cardiovascular effects associated with rosiglitazone was that in BARI 2D, patients received intensive medical therapy for ischemic heart disease, angina, and/or lowering cholesterol and blood pressure. Therefore, they concluded, the results "may imply that, in the context of intensive treatment of cardiovascular risk factors, rosiglitazone may provide benefit with retained safety, alone or in combination with other hypoglycemic medications, for high-risk patients with type 2 diabetes and established CAD."
There are several lessons to be drawn from these results, "some generalizable and others very specific to this study," Dr. Daniel Einhorn, president of the American Association of Clinical Endocrinologists and medical director of the Scripps Whittier Diabetes Institute, said in an interview. First, "it takes time to digest data generated by large clinical trials," he said, noting that "nuances" of the population and concomitant treatments, subgroup analyses, and other factors "routinely refine our understanding of the findings of large studies."

BARI 2D reflects real-life situations for people with access to good care, since intensive treatment of CV risk factors is the goal of appropriate therapy in the United States, he noted. As with pioglitazone (the other TZD marketed in the United States), "there are plausible and extensively researched mechanisms whereby rosiglitazone would be expected to confer cardiovascular benefit" given its mechanism(s) of action, he said.
Finally, the previous FDA panel meetings on rosiglitazone were characterized by a "politicized and media grandstanding climate ... which is not conducive to the best science," while the recent panel meeting in June "appeared more dispassionate," he added.
But even with generic formulations available, neither TZD is likely to be used extensively "because of years of safety concerns, including those regarding bone and cancer, the effect of TZDs to cause weight gain and fluid retention, patient/family concerns raised by ads from lawyers, and the advent of newer agents with a seemingly less concerning safety profile."
Dr. Einhorn has been an adviser to Halozyme Therapeutics, MannKind, and Freedom Meditech, and a consultant to Eli Lilly, Novo Nordisk, Sanofi-Aventis, Bristol-Myers Squibb/AstraZeneca and Novartis.
The study was funded by the National Heart, Lung, and Blood Institute and the National Institute of Diabetes and Digestive and Kidney Diseases, with funding from companies that included rosiglitazone manufacturer GlaxoSmithKline. The lead author, Dr. Bach, disclosed ties to several pharmaceutical companies, but not GSK; one author disclosed having received honoraria from GSK.
The results of the post hoc analysis of the BARI 2D data were first reported at the American Diabetes Association meeting in 2009.
Rosiglitazone therapy in BARI 2D demonstrates rather reassuring efficacy and safety data. This supports the recommendation of a recent FDA advisory panel to modify rosiglitazone restrictions based on the RECORD results. It is critical to evaluate efficacy and safety removed from media hype and the vocal decrees of small numbers of individuals. Once a negative spin via the media is placed on a form of therapy, subsequent analysis, valid and reassuring as it may be, cannot undue the damage.
The issue of pancreatic disease and incretin based therapy is in front of us right now. Let’s get this one right the first time!
Dr. Paul Jellinger is an endocrinologist in Hollywood, Fla. He is on the speakers bureau for several manufacturers of diabetes drugs, but not GSK.
Rosiglitazone therapy in BARI 2D demonstrates rather reassuring efficacy and safety data. This supports the recommendation of a recent FDA advisory panel to modify rosiglitazone restrictions based on the RECORD results. It is critical to evaluate efficacy and safety removed from media hype and the vocal decrees of small numbers of individuals. Once a negative spin via the media is placed on a form of therapy, subsequent analysis, valid and reassuring as it may be, cannot undue the damage.
The issue of pancreatic disease and incretin based therapy is in front of us right now. Let’s get this one right the first time!
Dr. Paul Jellinger is an endocrinologist in Hollywood, Fla. He is on the speakers bureau for several manufacturers of diabetes drugs, but not GSK.
Rosiglitazone therapy in BARI 2D demonstrates rather reassuring efficacy and safety data. This supports the recommendation of a recent FDA advisory panel to modify rosiglitazone restrictions based on the RECORD results. It is critical to evaluate efficacy and safety removed from media hype and the vocal decrees of small numbers of individuals. Once a negative spin via the media is placed on a form of therapy, subsequent analysis, valid and reassuring as it may be, cannot undue the damage.
The issue of pancreatic disease and incretin based therapy is in front of us right now. Let’s get this one right the first time!
Dr. Paul Jellinger is an endocrinologist in Hollywood, Fla. He is on the speakers bureau for several manufacturers of diabetes drugs, but not GSK.
Treatment with rosiglitazone was not associated with an increased risk of major ischemic cardiovascular events in a large, international study of patients with type 2 diabetes and coronary artery disease.
"Our analyses did not detect any significant hazard of increased ischemic cardiovascular risk with rosiglitazone treatment despite its use in this particularly vulnerable, higher risk population," Dr. Richard Bach of Washington University, St. Louis, and the other authors concluded.
The study, an analysis of data from the Bypass Angioplasty Revascularization Investigation 2 Diabetes (BARI 2D) trial, was published online July 15 (doi: 10.1161/CIRCULATIONAHA.112.000678).
The primary outcomes of the study were published in 2009 (N. Engl. J. Med. 2009; 360:2503-15).
The cardiac safety of rosiglitazone, a thiazolidinedione (TZD) marketed as Avandia by GlaxoSmithKline, has been mired in controversy for several years because of analyses of data showing that rosiglitazone may increase the risk of MI or death. The Food and Drug Administration issued an alert about the safety of the drug and limited its use in 2010. The agency also halted a long-term prospective cardiovascular safety study of rosiglitazone. In 2010, the Europeans Medicines Agency took rosiglitazone off the market in Europe, citing the same data.
In June 2013, at an FDA advisory panel meeting held to review the readjudicated results of a cardiovascular safety trial of rosiglitazone – which did not identify a greater rate of adverse cardiovascular outcomes among those on the drug – the majority of the panel recommended loosening or lifting the restrictions that had been placed on prescribing the drug in 2010. (The FDA has not yet acted on the panel’s recommendations.)
In the post hoc analysis of the BARI 2D data, the investigators compared the rates – between those on rosiglitazone and those not on a TZD – of mortality, MI, stroke, and other outcomes, and composite outcomes among nearly 2,400 patients with type 2 diabetes and coronary artery disease in the study. Over an average of 4.5 years, about 990 patients were on rosiglitazone at some point during the study (for up to 3,025 patient-years of exposure) and 485 were treated with rosiglitazone for at least 80% of the time. About 1,200 were not on a TZD.
After adjustment for differences in multiple baseline characteristics and other antidiabetes medications, the results were as follows:
• The incidence of all-cause death was similar among those on rosiglitazone, compared with those not on a TZD (hazard ratio, 0.83), a statistically nonsignificant difference.
• The composite of death, MI, and stroke was significantly lower among those on rosiglitazone (HR, 0.72).
• The incidence of stroke alone was significantly lower among those on rosiglitazone (HR, 0.36).
• The incidence of MI was lower among those on rosiglitazone (HR, 0.77) and the incidence of heart failure was higher (HR, 1.22), but these differences were not statistically significant.
When compared with those not on a TZD, the incidence of fractures was higher in those on a TZD (HR, 1.62), a risk that appeared to be more pronounced among women.
The investigators did not find a significant interaction between rosiglitazone and coadministration with other antidiabetes and cardiovascular medications (insulin, metformin, gemfibrozil, other fibrates, sulfonylureas, nitrates, or ACE inhibitors) on the composite rate of death, MI and stroke, or on individual rates of death, MI, stroke, or fractures. There was a significant interaction, however, with rosiglitazone and metformin on the risk of heart failure associated with rosiglitazone, with a relative risk of heart failure significantly greater among those patients on rosiglitazone but not on metformin, than on those who were on both.
These results "provide no evidence that use of rosiglitazone is associated with increased rates of major adverse ischemic cardiovascular events among patients with type 2 diabetes and established CAD," the authors concluded.
While the study had limitations, the researchers noted that a possible difference between this study and studies that found adverse cardiovascular effects associated with rosiglitazone was that in BARI 2D, patients received intensive medical therapy for ischemic heart disease, angina, and/or lowering cholesterol and blood pressure. Therefore, they concluded, the results "may imply that, in the context of intensive treatment of cardiovascular risk factors, rosiglitazone may provide benefit with retained safety, alone or in combination with other hypoglycemic medications, for high-risk patients with type 2 diabetes and established CAD."
There are several lessons to be drawn from these results, "some generalizable and others very specific to this study," Dr. Daniel Einhorn, president of the American Association of Clinical Endocrinologists and medical director of the Scripps Whittier Diabetes Institute, said in an interview. First, "it takes time to digest data generated by large clinical trials," he said, noting that "nuances" of the population and concomitant treatments, subgroup analyses, and other factors "routinely refine our understanding of the findings of large studies."
BARI 2D reflects real-life situations for people with access to good care, since intensive treatment of CV risk factors is the goal of appropriate therapy in the United States, he noted. As with pioglitazone (the other TZD marketed in the United States), "there are plausible and extensively researched mechanisms whereby rosiglitazone would be expected to confer cardiovascular benefit" given its mechanism(s) of action, he said.
Finally, the previous FDA panel meetings on rosiglitazone were characterized by a "politicized and media grandstanding climate ... which is not conducive to the best science," while the recent panel meeting in June "appeared more dispassionate," he added.
But even with generic formulations available, neither TZD is likely to be used extensively "because of years of safety concerns, including those regarding bone and cancer, the effect of TZDs to cause weight gain and fluid retention, patient/family concerns raised by ads from lawyers, and the advent of newer agents with a seemingly less concerning safety profile."
Dr. Einhorn has been an adviser to Halozyme Therapeutics, MannKind, and Freedom Meditech, and a consultant to Eli Lilly, Novo Nordisk, Sanofi-Aventis, Bristol-Myers Squibb/AstraZeneca and Novartis.
The study was funded by the National Heart, Lung, and Blood Institute and the National Institute of Diabetes and Digestive and Kidney Diseases, with funding from companies that included rosiglitazone manufacturer GlaxoSmithKline. The lead author, Dr. Bach, disclosed ties to several pharmaceutical companies, but not GSK; one author disclosed having received honoraria from GSK.
The results of the post hoc analysis of the BARI 2D data were first reported at the American Diabetes Association meeting in 2009.
Treatment with rosiglitazone was not associated with an increased risk of major ischemic cardiovascular events in a large, international study of patients with type 2 diabetes and coronary artery disease.
"Our analyses did not detect any significant hazard of increased ischemic cardiovascular risk with rosiglitazone treatment despite its use in this particularly vulnerable, higher risk population," Dr. Richard Bach of Washington University, St. Louis, and the other authors concluded.
The study, an analysis of data from the Bypass Angioplasty Revascularization Investigation 2 Diabetes (BARI 2D) trial, was published online July 15 (doi: 10.1161/CIRCULATIONAHA.112.000678).
The primary outcomes of the study were published in 2009 (N. Engl. J. Med. 2009; 360:2503-15).
The cardiac safety of rosiglitazone, a thiazolidinedione (TZD) marketed as Avandia by GlaxoSmithKline, has been mired in controversy for several years because of analyses of data showing that rosiglitazone may increase the risk of MI or death. The Food and Drug Administration issued an alert about the safety of the drug and limited its use in 2010. The agency also halted a long-term prospective cardiovascular safety study of rosiglitazone. In 2010, the Europeans Medicines Agency took rosiglitazone off the market in Europe, citing the same data.
In June 2013, at an FDA advisory panel meeting held to review the readjudicated results of a cardiovascular safety trial of rosiglitazone – which did not identify a greater rate of adverse cardiovascular outcomes among those on the drug – the majority of the panel recommended loosening or lifting the restrictions that had been placed on prescribing the drug in 2010. (The FDA has not yet acted on the panel’s recommendations.)
In the post hoc analysis of the BARI 2D data, the investigators compared the rates – between those on rosiglitazone and those not on a TZD – of mortality, MI, stroke, and other outcomes, and composite outcomes among nearly 2,400 patients with type 2 diabetes and coronary artery disease in the study. Over an average of 4.5 years, about 990 patients were on rosiglitazone at some point during the study (for up to 3,025 patient-years of exposure) and 485 were treated with rosiglitazone for at least 80% of the time. About 1,200 were not on a TZD.
After adjustment for differences in multiple baseline characteristics and other antidiabetes medications, the results were as follows:
• The incidence of all-cause death was similar among those on rosiglitazone, compared with those not on a TZD (hazard ratio, 0.83), a statistically nonsignificant difference.
• The composite of death, MI, and stroke was significantly lower among those on rosiglitazone (HR, 0.72).
• The incidence of stroke alone was significantly lower among those on rosiglitazone (HR, 0.36).
• The incidence of MI was lower among those on rosiglitazone (HR, 0.77) and the incidence of heart failure was higher (HR, 1.22), but these differences were not statistically significant.
When compared with those not on a TZD, the incidence of fractures was higher in those on a TZD (HR, 1.62), a risk that appeared to be more pronounced among women.
The investigators did not find a significant interaction between rosiglitazone and coadministration with other antidiabetes and cardiovascular medications (insulin, metformin, gemfibrozil, other fibrates, sulfonylureas, nitrates, or ACE inhibitors) on the composite rate of death, MI and stroke, or on individual rates of death, MI, stroke, or fractures. There was a significant interaction, however, with rosiglitazone and metformin on the risk of heart failure associated with rosiglitazone, with a relative risk of heart failure significantly greater among those patients on rosiglitazone but not on metformin, than on those who were on both.
These results "provide no evidence that use of rosiglitazone is associated with increased rates of major adverse ischemic cardiovascular events among patients with type 2 diabetes and established CAD," the authors concluded.
While the study had limitations, the researchers noted that a possible difference between this study and studies that found adverse cardiovascular effects associated with rosiglitazone was that in BARI 2D, patients received intensive medical therapy for ischemic heart disease, angina, and/or lowering cholesterol and blood pressure. Therefore, they concluded, the results "may imply that, in the context of intensive treatment of cardiovascular risk factors, rosiglitazone may provide benefit with retained safety, alone or in combination with other hypoglycemic medications, for high-risk patients with type 2 diabetes and established CAD."
There are several lessons to be drawn from these results, "some generalizable and others very specific to this study," Dr. Daniel Einhorn, president of the American Association of Clinical Endocrinologists and medical director of the Scripps Whittier Diabetes Institute, said in an interview. First, "it takes time to digest data generated by large clinical trials," he said, noting that "nuances" of the population and concomitant treatments, subgroup analyses, and other factors "routinely refine our understanding of the findings of large studies."
BARI 2D reflects real-life situations for people with access to good care, since intensive treatment of CV risk factors is the goal of appropriate therapy in the United States, he noted. As with pioglitazone (the other TZD marketed in the United States), "there are plausible and extensively researched mechanisms whereby rosiglitazone would be expected to confer cardiovascular benefit" given its mechanism(s) of action, he said.
Finally, the previous FDA panel meetings on rosiglitazone were characterized by a "politicized and media grandstanding climate ... which is not conducive to the best science," while the recent panel meeting in June "appeared more dispassionate," he added.
But even with generic formulations available, neither TZD is likely to be used extensively "because of years of safety concerns, including those regarding bone and cancer, the effect of TZDs to cause weight gain and fluid retention, patient/family concerns raised by ads from lawyers, and the advent of newer agents with a seemingly less concerning safety profile."
Dr. Einhorn has been an adviser to Halozyme Therapeutics, MannKind, and Freedom Meditech, and a consultant to Eli Lilly, Novo Nordisk, Sanofi-Aventis, Bristol-Myers Squibb/AstraZeneca and Novartis.
The study was funded by the National Heart, Lung, and Blood Institute and the National Institute of Diabetes and Digestive and Kidney Diseases, with funding from companies that included rosiglitazone manufacturer GlaxoSmithKline. The lead author, Dr. Bach, disclosed ties to several pharmaceutical companies, but not GSK; one author disclosed having received honoraria from GSK.
The results of the post hoc analysis of the BARI 2D data were first reported at the American Diabetes Association meeting in 2009.
FROM CIRCULATION
Major finding: No increased risk of major cardiovascular events associated with rosiglitazone was identified in a study of patients with type 2 diabetes and coronary artery disease, including a nonsignificant lower risk (23%) of MI among those treated with the thiazolidinedione (TZD) compared with those who had not received a TZD.
Data source: A post hoc analysis of data from BARI 2D, a large, randomized international study of patients with type 2 diabetes and heart disease, which analyzed the CV outcomes among those treated with rosiglitazone and those who had not received a TZD.
Disclosures: The study was funded by the National Heart, Lung, and Blood Institute and the National Institute of Diabetes and Digestive and Kidney Diseases, with funding from companies that included rosiglitazone manufacturer GlaxoSmithKline. The lead author, Dr. Bach, disclosed ties to several pharmaceutical companies, but not GSK; one author disclosed having received honoraria from GSK.

FDA issues strong warning about oral ketoconazole
Ketoconazole tablets should not be used as a first-line treatment for fungal infections because treatment has been associated with an increased risk of adrenal insufficiency, potentially fatal hepatotoxicity, and drug interactions, the Food and Drug Administration has announced.
Marketed as Nizoral, oral ketoconazole is no longer indicated for the treatment of Candida and dermatophyte infections and "should be used only for the treatment of certain life-threatening mycoses when the potential benefits outweigh the risks and alternative therapeutic options are not available or tolerated," according to the MedWatch safety alert issued on July 26.
In addition, oral ketoconazole should not be used to treat fungal infections of the skin and nails, and is only indicated for the treatment of blastomycosis, coccidioidomycosis, histoplasmosis, chromomycosis, and paracoccidioidomycosis "in patients in whom other treatments have failed or who are intolerant to other therapies," according to the label, which has been modified to reflect these risks and recommendations.
There is now a Medication Guide that will be provided to patients with each filled prescription of oral ketoconazole, explaining the risks.
Because oral ketoconazole has been associated with hepatoxicity that can result in liver transplantation or death, it is now contraindicated in patients with acute or chronic liver disease. The label also now recommends that patients be assessed and monitored for liver toxicity. Monitoring of adrenal function also is now recommended in patients who take the oral formulation of the drug and have adrenal problems or "are under prolonged periods of stress such as those who have had a recent major surgery or who are under intensive care in the hospital."
In addition, coadministration of ketoconazole – a potent inhibitor of the cytochrome P450 3A4 isoenzyme (CYP3A4) – with certain drugs is either restricted or contraindicated because of the increase in drug concentrations and increased risk of QT prolongation and other serious reactions. Contraindicated drugs include dofetilide, quinidine, pimozide, and cisapride.
The FDA changes are based on risk-benefit analyses of data that include reports made to the FDA’s Adverse Events Reporting System.
On July 26, the European Medicines Agency’s Committee on Medicinal Products for Human Use (CHMP) announced that it has concluded that the risk of hepatoxicity with oral ketoconazole products was greater than the benefits in treating fungal infections and recommended that these products no longer be marketed in the European Union.
Creams, shampoos, and other topical ketoconazole formulations have not been associated with these problems, according to the FDA.
The updated label is available here. Serious adverse events associated with ketoconazole should be reported to the FDA at 800-332-1088 or MedWatch.
Ketoconazole tablets should not be used as a first-line treatment for fungal infections because treatment has been associated with an increased risk of adrenal insufficiency, potentially fatal hepatotoxicity, and drug interactions, the Food and Drug Administration has announced.
Marketed as Nizoral, oral ketoconazole is no longer indicated for the treatment of Candida and dermatophyte infections and "should be used only for the treatment of certain life-threatening mycoses when the potential benefits outweigh the risks and alternative therapeutic options are not available or tolerated," according to the MedWatch safety alert issued on July 26.
In addition, oral ketoconazole should not be used to treat fungal infections of the skin and nails, and is only indicated for the treatment of blastomycosis, coccidioidomycosis, histoplasmosis, chromomycosis, and paracoccidioidomycosis "in patients in whom other treatments have failed or who are intolerant to other therapies," according to the label, which has been modified to reflect these risks and recommendations.
There is now a Medication Guide that will be provided to patients with each filled prescription of oral ketoconazole, explaining the risks.
Because oral ketoconazole has been associated with hepatoxicity that can result in liver transplantation or death, it is now contraindicated in patients with acute or chronic liver disease. The label also now recommends that patients be assessed and monitored for liver toxicity. Monitoring of adrenal function also is now recommended in patients who take the oral formulation of the drug and have adrenal problems or "are under prolonged periods of stress such as those who have had a recent major surgery or who are under intensive care in the hospital."
In addition, coadministration of ketoconazole – a potent inhibitor of the cytochrome P450 3A4 isoenzyme (CYP3A4) – with certain drugs is either restricted or contraindicated because of the increase in drug concentrations and increased risk of QT prolongation and other serious reactions. Contraindicated drugs include dofetilide, quinidine, pimozide, and cisapride.
The FDA changes are based on risk-benefit analyses of data that include reports made to the FDA’s Adverse Events Reporting System.
On July 26, the European Medicines Agency’s Committee on Medicinal Products for Human Use (CHMP) announced that it has concluded that the risk of hepatoxicity with oral ketoconazole products was greater than the benefits in treating fungal infections and recommended that these products no longer be marketed in the European Union.
Creams, shampoos, and other topical ketoconazole formulations have not been associated with these problems, according to the FDA.
The updated label is available here. Serious adverse events associated with ketoconazole should be reported to the FDA at 800-332-1088 or MedWatch.
Ketoconazole tablets should not be used as a first-line treatment for fungal infections because treatment has been associated with an increased risk of adrenal insufficiency, potentially fatal hepatotoxicity, and drug interactions, the Food and Drug Administration has announced.
Marketed as Nizoral, oral ketoconazole is no longer indicated for the treatment of Candida and dermatophyte infections and "should be used only for the treatment of certain life-threatening mycoses when the potential benefits outweigh the risks and alternative therapeutic options are not available or tolerated," according to the MedWatch safety alert issued on July 26.
In addition, oral ketoconazole should not be used to treat fungal infections of the skin and nails, and is only indicated for the treatment of blastomycosis, coccidioidomycosis, histoplasmosis, chromomycosis, and paracoccidioidomycosis "in patients in whom other treatments have failed or who are intolerant to other therapies," according to the label, which has been modified to reflect these risks and recommendations.
There is now a Medication Guide that will be provided to patients with each filled prescription of oral ketoconazole, explaining the risks.
Because oral ketoconazole has been associated with hepatoxicity that can result in liver transplantation or death, it is now contraindicated in patients with acute or chronic liver disease. The label also now recommends that patients be assessed and monitored for liver toxicity. Monitoring of adrenal function also is now recommended in patients who take the oral formulation of the drug and have adrenal problems or "are under prolonged periods of stress such as those who have had a recent major surgery or who are under intensive care in the hospital."
In addition, coadministration of ketoconazole – a potent inhibitor of the cytochrome P450 3A4 isoenzyme (CYP3A4) – with certain drugs is either restricted or contraindicated because of the increase in drug concentrations and increased risk of QT prolongation and other serious reactions. Contraindicated drugs include dofetilide, quinidine, pimozide, and cisapride.
The FDA changes are based on risk-benefit analyses of data that include reports made to the FDA’s Adverse Events Reporting System.
On July 26, the European Medicines Agency’s Committee on Medicinal Products for Human Use (CHMP) announced that it has concluded that the risk of hepatoxicity with oral ketoconazole products was greater than the benefits in treating fungal infections and recommended that these products no longer be marketed in the European Union.
Creams, shampoos, and other topical ketoconazole formulations have not been associated with these problems, according to the FDA.
The updated label is available here. Serious adverse events associated with ketoconazole should be reported to the FDA at 800-332-1088 or MedWatch.
FDA issues strong warning about oral ketoconazole
Ketoconazole tablets should not be used as a first-line treatment for fungal infections because treatment has been associated with an increased risk of adrenal insufficiency, potentially fatal hepatotoxicity, and drug interactions, the Food and Drug Administration has announced.
Marketed as Nizoral, oral ketoconazole is no longer indicated for the treatment of Candida and dermatophyte infections and "should be used only for the treatment of certain life-threatening mycoses when the potential benefits outweigh the risks and alternative therapeutic options are not available or tolerated," according to the MedWatch safety alert issued on July 26.
In addition, oral ketoconazole should not be used to treat fungal infections of the skin and nails, and is only indicated for the treatment of blastomycosis, coccidioidomycosis, histoplasmosis, chromomycosis, and paracoccidioidomycosis "in patients in whom other treatments have failed or who are intolerant to other therapies," according to the label, which has been modified to reflect these risks and recommendations.
There is now a Medication Guide that will be provided to patients with each filled prescription of oral ketoconazole, explaining the risks.
Because oral ketoconazole has been associated with hepatoxicity that can result in liver transplantation or death, it is now contraindicated in patients with acute or chronic liver disease. The label also now recommends that patients be assessed and monitored for liver toxicity. Monitoring of adrenal function also is now recommended in patients who take the oral formulation of the drug and have adrenal problems or "are under prolonged periods of stress such as those who have had a recent major surgery or who are under intensive care in the hospital."
In addition, coadministration of ketoconazole – a potent inhibitor of the cytochrome P450 3A4 isoenzyme (CYP3A4) – with certain drugs is either restricted or contraindicated because of the increase in drug concentrations and increased risk of QT prolongation and other serious reactions. Contraindicated drugs include dofetilide, quinidine, pimozide, and cisapride.
The FDA changes are based on risk-benefit analyses of data that include reports made to the FDA’s Adverse Events Reporting System.
On July 26, the European Medicines Agency’s Committee on Medicinal Products for Human Use (CHMP) announced that it has concluded that the risk of hepatoxicity with oral ketoconazole products was greater than the benefits in treating fungal infections and recommended that these products no longer be marketed in the European Union.
Creams, shampoos, and other topical ketoconazole formulations have not been associated with these problems, according to the FDA.
The updated label is available here. Serious adverse events associated with ketoconazole should be reported to the FDA at 800-332-1088 or MedWatch.
Ketoconazole tablets should not be used as a first-line treatment for fungal infections because treatment has been associated with an increased risk of adrenal insufficiency, potentially fatal hepatotoxicity, and drug interactions, the Food and Drug Administration has announced.
Marketed as Nizoral, oral ketoconazole is no longer indicated for the treatment of Candida and dermatophyte infections and "should be used only for the treatment of certain life-threatening mycoses when the potential benefits outweigh the risks and alternative therapeutic options are not available or tolerated," according to the MedWatch safety alert issued on July 26.
In addition, oral ketoconazole should not be used to treat fungal infections of the skin and nails, and is only indicated for the treatment of blastomycosis, coccidioidomycosis, histoplasmosis, chromomycosis, and paracoccidioidomycosis "in patients in whom other treatments have failed or who are intolerant to other therapies," according to the label, which has been modified to reflect these risks and recommendations.
There is now a Medication Guide that will be provided to patients with each filled prescription of oral ketoconazole, explaining the risks.
Because oral ketoconazole has been associated with hepatoxicity that can result in liver transplantation or death, it is now contraindicated in patients with acute or chronic liver disease. The label also now recommends that patients be assessed and monitored for liver toxicity. Monitoring of adrenal function also is now recommended in patients who take the oral formulation of the drug and have adrenal problems or "are under prolonged periods of stress such as those who have had a recent major surgery or who are under intensive care in the hospital."
In addition, coadministration of ketoconazole – a potent inhibitor of the cytochrome P450 3A4 isoenzyme (CYP3A4) – with certain drugs is either restricted or contraindicated because of the increase in drug concentrations and increased risk of QT prolongation and other serious reactions. Contraindicated drugs include dofetilide, quinidine, pimozide, and cisapride.
The FDA changes are based on risk-benefit analyses of data that include reports made to the FDA’s Adverse Events Reporting System.
On July 26, the European Medicines Agency’s Committee on Medicinal Products for Human Use (CHMP) announced that it has concluded that the risk of hepatoxicity with oral ketoconazole products was greater than the benefits in treating fungal infections and recommended that these products no longer be marketed in the European Union.
Creams, shampoos, and other topical ketoconazole formulations have not been associated with these problems, according to the FDA.
The updated label is available here. Serious adverse events associated with ketoconazole should be reported to the FDA at 800-332-1088 or MedWatch.
Ketoconazole tablets should not be used as a first-line treatment for fungal infections because treatment has been associated with an increased risk of adrenal insufficiency, potentially fatal hepatotoxicity, and drug interactions, the Food and Drug Administration has announced.
Marketed as Nizoral, oral ketoconazole is no longer indicated for the treatment of Candida and dermatophyte infections and "should be used only for the treatment of certain life-threatening mycoses when the potential benefits outweigh the risks and alternative therapeutic options are not available or tolerated," according to the MedWatch safety alert issued on July 26.
In addition, oral ketoconazole should not be used to treat fungal infections of the skin and nails, and is only indicated for the treatment of blastomycosis, coccidioidomycosis, histoplasmosis, chromomycosis, and paracoccidioidomycosis "in patients in whom other treatments have failed or who are intolerant to other therapies," according to the label, which has been modified to reflect these risks and recommendations.
There is now a Medication Guide that will be provided to patients with each filled prescription of oral ketoconazole, explaining the risks.
Because oral ketoconazole has been associated with hepatoxicity that can result in liver transplantation or death, it is now contraindicated in patients with acute or chronic liver disease. The label also now recommends that patients be assessed and monitored for liver toxicity. Monitoring of adrenal function also is now recommended in patients who take the oral formulation of the drug and have adrenal problems or "are under prolonged periods of stress such as those who have had a recent major surgery or who are under intensive care in the hospital."
In addition, coadministration of ketoconazole – a potent inhibitor of the cytochrome P450 3A4 isoenzyme (CYP3A4) – with certain drugs is either restricted or contraindicated because of the increase in drug concentrations and increased risk of QT prolongation and other serious reactions. Contraindicated drugs include dofetilide, quinidine, pimozide, and cisapride.
The FDA changes are based on risk-benefit analyses of data that include reports made to the FDA’s Adverse Events Reporting System.
On July 26, the European Medicines Agency’s Committee on Medicinal Products for Human Use (CHMP) announced that it has concluded that the risk of hepatoxicity with oral ketoconazole products was greater than the benefits in treating fungal infections and recommended that these products no longer be marketed in the European Union.
Creams, shampoos, and other topical ketoconazole formulations have not been associated with these problems, according to the FDA.
The updated label is available here. Serious adverse events associated with ketoconazole should be reported to the FDA at 800-332-1088 or MedWatch.
CDC investigates Cyclospora outbreak
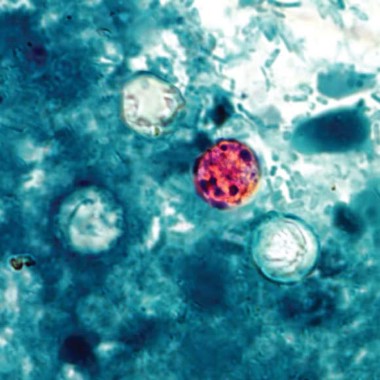
Patients with watery diarrhea that does not resolve over several days should be tested specifically for Cyclospora infection, according to Dr. Tom Frieden, director of the Centers for Disease Control and Prevention.
The CDC is investigating an outbreak of Cyclospora infections in conjunction with federal and local partners, including the Food and Drug Administration. The first lab-confirmed cases reported to the CDC were in Iowa in two people who had not traveled internationally for 14 days before the onset of the illness.
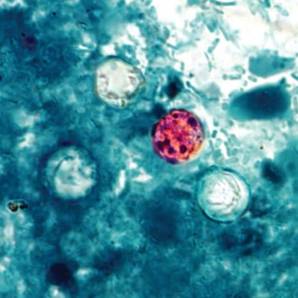
As of July 24, the CDC had been notified of 285 cases of Cyclospora infection in multiple states, including Iowa, Nebraska, Texas, Wisconsin, Georgia, Minnesota, Connecticut, New Jersey, and Ohio, Dr. Frieden said during a CDC telebriefing on July 25. Additionally, single cases were reported in both Illinois and Kansas that may have been acquired in another state.
Most patients became ill in June or early July, and it is too soon to say whether the outbreak is ongoing or subsiding, he said. "We have not yet identified a source, although I am confident we will," he added.
Infection with the parasite Cyclospora cayetanensis typically causes watery diarrhea. "People who have illness that hasn’t gone away on its own in several days should see their health care provider, and discuss the possibility of Cyclospora," he said. Testing of stool samples for Cyclospora "should be specifically requested in people who’ve had diarrheal illness that hasn’t gone away in several days."
While the cause of the current outbreak has not yet been identified, Dr. Frieden said that the best way to prevent infection is to avoid food or water that may have been contaminated with the parasite and to carefully wash fresh produce before eating.
Cyclospora can stick to some foods, though, so washing produce can be helpful but does not eliminate the risk, he noted.
Patients with watery diarrhea that does not resolve over several days should be tested specifically for Cyclospora infection, according to Dr. Tom Frieden, director of the Centers for Disease Control and Prevention.
The CDC is investigating an outbreak of Cyclospora infections in conjunction with federal and local partners, including the Food and Drug Administration. The first lab-confirmed cases reported to the CDC were in Iowa in two people who had not traveled internationally for 14 days before the onset of the illness.
As of July 24, the CDC had been notified of 285 cases of Cyclospora infection in multiple states, including Iowa, Nebraska, Texas, Wisconsin, Georgia, Minnesota, Connecticut, New Jersey, and Ohio, Dr. Frieden said during a CDC telebriefing on July 25. Additionally, single cases were reported in both Illinois and Kansas that may have been acquired in another state.
Most patients became ill in June or early July, and it is too soon to say whether the outbreak is ongoing or subsiding, he said. "We have not yet identified a source, although I am confident we will," he added.
Infection with the parasite Cyclospora cayetanensis typically causes watery diarrhea. "People who have illness that hasn’t gone away on its own in several days should see their health care provider, and discuss the possibility of Cyclospora," he said. Testing of stool samples for Cyclospora "should be specifically requested in people who’ve had diarrheal illness that hasn’t gone away in several days."
While the cause of the current outbreak has not yet been identified, Dr. Frieden said that the best way to prevent infection is to avoid food or water that may have been contaminated with the parasite and to carefully wash fresh produce before eating.
Cyclospora can stick to some foods, though, so washing produce can be helpful but does not eliminate the risk, he noted.
Patients with watery diarrhea that does not resolve over several days should be tested specifically for Cyclospora infection, according to Dr. Tom Frieden, director of the Centers for Disease Control and Prevention.
The CDC is investigating an outbreak of Cyclospora infections in conjunction with federal and local partners, including the Food and Drug Administration. The first lab-confirmed cases reported to the CDC were in Iowa in two people who had not traveled internationally for 14 days before the onset of the illness.
As of July 24, the CDC had been notified of 285 cases of Cyclospora infection in multiple states, including Iowa, Nebraska, Texas, Wisconsin, Georgia, Minnesota, Connecticut, New Jersey, and Ohio, Dr. Frieden said during a CDC telebriefing on July 25. Additionally, single cases were reported in both Illinois and Kansas that may have been acquired in another state.
Most patients became ill in June or early July, and it is too soon to say whether the outbreak is ongoing or subsiding, he said. "We have not yet identified a source, although I am confident we will," he added.
Infection with the parasite Cyclospora cayetanensis typically causes watery diarrhea. "People who have illness that hasn’t gone away on its own in several days should see their health care provider, and discuss the possibility of Cyclospora," he said. Testing of stool samples for Cyclospora "should be specifically requested in people who’ve had diarrheal illness that hasn’t gone away in several days."
While the cause of the current outbreak has not yet been identified, Dr. Frieden said that the best way to prevent infection is to avoid food or water that may have been contaminated with the parasite and to carefully wash fresh produce before eating.
Cyclospora can stick to some foods, though, so washing produce can be helpful but does not eliminate the risk, he noted.
FROM A PRESS BRIEFING HELD BY THE CDC
HPV vaccination coverage among girls is low, CDC reports
Clinicians can do more to improve human papillomavirus vaccination coverage rates among adolescent females, including emphasizing to parents that the vaccine prevents cancer, according to Dr. Thomas Frieden, the director of the Centers for Disease Control and Prevention.
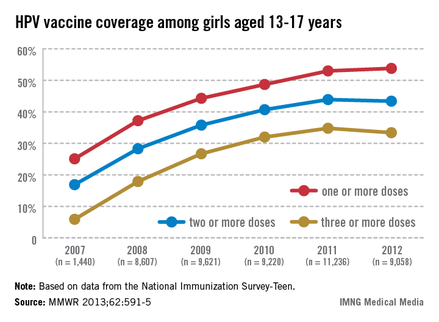
In 2011, only 35% of girls aged 13-17 years had received the full three doses of the HPV vaccine, which dropped slightly in 2012, which is "a huge disappointment," Dr. Frieden said during a July 25 telebriefing. The proportion of adolescent girls who received at least one, two, and the full three-dose HPV vaccine series increased every year from 2007 to 2011, but stalled between 2011 and 2012 and remains at unacceptably low levels – while missed opportunities for vaccination in this population have increased markedly, according to the data (MMWR 2013;62:591-5).
But rates can be increased if clinicians take every possible opportunity to vaccinate adolescents with the HPV vaccine, including when they receive other vaccines, he said during the briefing. While administering a three-dose series was thought to be difficult and was behind the low rates, the data indicated that if the HPV vaccine was given every time the adolescents received another vaccine, the completion of the three-dose series could increase to 93%, he said. According to the report, "Every health care visit, whether for back-to-school evaluations or acute problems, should be used to assess teenagers’ immunization status and provide recommended vaccines if indicated."
"If we get the three-dose series to 80%, an estimated 53,000 cases of cervical cancer could be prevented over the lifetimes of girls aged 12 [years] and younger," Dr. Frieden said.
A three-dose series of Gardasil – the quadrivalent HPV vaccine directed against HPV-16 and -18 (which cause most cervical cancers) and HPV-6 and -11 (which cause most genital warts) – was licensed and recommended by the CDC as a routine vaccine in 2006 for females aged 11-12 years and for females aged 13-26 years who had not been vaccinated. In 2009, the bivalent vaccine Cervarix, directed against HPV-16 and -18, was licensed for use in females aged 10-25 years and added to the recommendation in 2009.
The American Academy of Pediatrics "shares CDC’s disappointment and concern that we are not providing this lifesaving vaccine at the same rates as we do with other immunizations," Dr. Thomas K. McInerny, president of the AAP, said during the briefing.
"Use every opportunity to vaccinate your adolescent patients at both illness and well-child visits," as well as visits for sports physicals, he advised. He also recommended using alerts in electronic medical record systems so vaccination status is reviewed at every patient visit, as well as reminding parents with automated postcards or phone calls, having nurses check vaccine status when they bring patients into the exam room, and implementing standing order policies so that patients can receive vaccines without a physician’s exam or a physician’s order.
The vaccination coverage data were from the National Immunization Survey–Teen (NIS-Teen), which collects data on vaccination among adolescents aged 13-17 years in the 50 states, the District of Columbia, and certain areas of the country, including New York City, and Chicago. Data on HPV vaccination rates among adolescent males will be reported in the future.
In 2007, 25.1% of adolescent girls aged 13-17 years had received at least one dose of the HPV vaccine, which increased to 53% in 2011 and to only 53.8% in 2012. The proportion of adolescent girls who had received the full three-dose series was even lower, increasing from 5.9% in 2007 to 34.8% in 2011 and dropping slightly in 2012 to 33.4%.
The report also provides estimates of missed opportunities for vaccinating adolescent girls, defined as "a health-care encounter occurring on or after a girl’s 11th birthday," and on or after March 23, 2007. Missed opportunities increased from 20.8% in 2007 to 84% in 2012. (March 23, 2007, was when the CDC’s Advisory Committee on Immunization Practices (ACIP) recommendation on the quadrivalent vaccine was published).
The survey found that 23% of parents said they did not plan to get their daughters vaccinated in the next year. The most common reason cited by these parents is that they did not believe the vaccine was necessary (19.1%), followed by concerns about safety (13.1%), vaccine not recommended (14.2%), lack of knowledge of the vaccine or the disease (12.6%), and their daughter was not sexually active (10.1%). Cost did not appear to be an issue.
The report also included safety data, based on the estimated 56 million doses of the quadrivalent vaccine distributed in the United States from June 2006 through March 2013, which did not identify any new safety issues associated with the HPV vaccine, Dr. Frieden said. (Safety focused on the quadrivalent vaccine because most of the vaccine used has been this vaccine).
Most – 92.1% – of the 21,194 adverse event reports received by the Vaccine Adverse Event Reporting System (VAERS) were classified as "nonserious," and included syncope, dizziness, nausea, headache, fever, and urticaria. Local effects included injection-site pain, redness and swelling. Only 8% of the reports were classified as serious and included headache, vomiting, fatigue, dizziness, syncope, and generalized weakness.
The report also points out that no serious safety concerns have been identified in three population-based U.S. studies of the quadrivalent vaccine safety, although one observational study since the vaccine was licensed found an increased syncope risk.
Dr. Frieden noted that an increased risk of syncope may not be specific to the HPV vaccine because syncope is associated with other vaccines among adolescents, and that a 15-minute observation period after the vaccine is administered is recommended.
He referred to a report released in June in the Journal of Infectious Diseases, which found that the infections with the HPV types in the quadrivalent HPV vaccine dropped by almost 60% among females aged 14-19 years during the 4-year period after the vaccine became available and was recommended as a routine vaccine. (J. Inf. Dis. 2013; 208:385-93).
In the United States every year, an estimated 14 million people will become infected with HPV and about 26,200 will develop new cancers from HPV: 17,400 of those cancers are among females, 10,300 of which are cervical cancers, and 8,800 of the cancers are among males, of which 6,700 are oropharyngeal cancers.
In 2011, the recommendation for the quadrivalent vaccine was expanded to include boys aged 11-12 years, and for unvaccinated males up to age 26 years.
For those senior pediatricians such as me, the importance of vaccines to
child and adolescent health such as Hemophilus influenzae, pneumococcal
vaccine and others is a "no brainer." We recall the devastating
diseases that are no longer commonplace, because of prevention by
immunization. In the United States, 14 million people will become
infected with HPV every year and 26,200 will develop new cancers from
HPV, almost half of these cervical cancer. As Dr. Frieden pointed out in
the telebriefing, even the small percentage of girls who have received
with the HPV vaccine has made a difference, reducing HPV infections by
almost 60% among females aged 14-19 years in just 4 years.
The HPV vaccine series has met with an initial difficult reception from parents, patients, and providers. Although, at first, the cost of approximately $380 for the three-shot series was a barrier, over time this has become less of an obstacle, and interestingly, this issue was not reported to be a barrier to receiving the vaccine series in the Centers for Disease Control and Prevention's Advisory Committee on Immunization Practices survey on the quadrivalent series. Current lassitude has resulted in only 33.4% of 13- to 17-year-olds being fully immunized in 2012, as reported in the MMWR.
In the same survey, missed opportunities to provide this vaccine after a girl's 11th birthday increased from 20.8% to 84%. Clearly, recommendations to refine systems of delivery in physicians' offices and the use of electronic health records (EHR) to prime providers to immunize at every opportunity in the clinical setting are good ideas. But an undercurrent that is often not openly discussed is that physicians often don't ask or wish to "take on" negative parental reaction to this important vaccine that can prevent cancer but is often linked to reproductive behavior. The link with acquisition of this virus through sexual activity is one that parents often don't want to consider, and frequently the provider acquiesces. In the ACIP survey, the most common reason cited by parents is that they do not believe the vaccine was necessary, followed by concerns of safety, that the vaccine was not recommended, lack of knowledge about the disease, and, finally, that their daughter was not sexually active.
Pediatricians must use every visit to review immunization status. When in an exam room, HPV should be included in the vaccines that are given at a health visit to prevent meningitis and pertussis. For example, say, "Maggie is due to receive these shots today; they are HPV, Menactra, and Tdap."* Too often, other physicians and I timidly approach the HPV vaccine series and allow parents' lack of knowledge - or worse, media hearsay - to dictate a youngster's care and, they opt to not immunize. Moreover, we often don't even have the conversation!
In the clinical trenches, if a parent questions why her daughter or son needs the vaccine, a directed but not overly lengthy response of the importance of this vaccine in preventing cervical and other cancers will inform the parent and teen, and is generally well received. Often, asking if an older family member or friend has suffered with cervical cancer will allow the parent to acknowledge that this is a reality that they do not want their teen to experience. When we fail to recommend the HPV series or say its fine "to wait" because of an inability and unwillingness to raise the link of acquisition of HPV and sexual activity, an opportunity is missed.
The media has done much to promote positive health behaviors, but sadly, parents often recall only the negatives associated with the HPV vaccine. Both clinicians and parents must repurpose themselves to protecting children and adolescents. Parents and youth rely on their pediatrician's recommendations and knowledge. In brief, pediatricians must pin a Post It note to their EHR to cue them to educate, but then vaccinate! In short, "Do Ask, Do Tell." No opportunity to protect our youth should be lost!
Dr. Susan Jay is program director of adolescent health and medicine at Children's Hospital of Wisconsin in Wauwatosa, and a professor of pediatrics at the Medical College of Wisconsin. Dr. Jay has no relevant financial disclosures.
*Correction 8/5/2013: The vaccine was misstated as DTap; teens receive Tdap.
This story was updated. 8/5/2013
For those senior pediatricians such as me, the importance of vaccines to
child and adolescent health such as Hemophilus influenzae, pneumococcal
vaccine and others is a "no brainer." We recall the devastating
diseases that are no longer commonplace, because of prevention by
immunization. In the United States, 14 million people will become
infected with HPV every year and 26,200 will develop new cancers from
HPV, almost half of these cervical cancer. As Dr. Frieden pointed out in
the telebriefing, even the small percentage of girls who have received
with the HPV vaccine has made a difference, reducing HPV infections by
almost 60% among females aged 14-19 years in just 4 years.
The HPV vaccine series has met with an initial difficult reception from parents, patients, and providers. Although, at first, the cost of approximately $380 for the three-shot series was a barrier, over time this has become less of an obstacle, and interestingly, this issue was not reported to be a barrier to receiving the vaccine series in the Centers for Disease Control and Prevention's Advisory Committee on Immunization Practices survey on the quadrivalent series. Current lassitude has resulted in only 33.4% of 13- to 17-year-olds being fully immunized in 2012, as reported in the MMWR.
In the same survey, missed opportunities to provide this vaccine after a girl's 11th birthday increased from 20.8% to 84%. Clearly, recommendations to refine systems of delivery in physicians' offices and the use of electronic health records (EHR) to prime providers to immunize at every opportunity in the clinical setting are good ideas. But an undercurrent that is often not openly discussed is that physicians often don't ask or wish to "take on" negative parental reaction to this important vaccine that can prevent cancer but is often linked to reproductive behavior. The link with acquisition of this virus through sexual activity is one that parents often don't want to consider, and frequently the provider acquiesces. In the ACIP survey, the most common reason cited by parents is that they do not believe the vaccine was necessary, followed by concerns of safety, that the vaccine was not recommended, lack of knowledge about the disease, and, finally, that their daughter was not sexually active.
Pediatricians must use every visit to review immunization status. When in an exam room, HPV should be included in the vaccines that are given at a health visit to prevent meningitis and pertussis. For example, say, "Maggie is due to receive these shots today; they are HPV, Menactra, and Tdap."* Too often, other physicians and I timidly approach the HPV vaccine series and allow parents' lack of knowledge - or worse, media hearsay - to dictate a youngster's care and, they opt to not immunize. Moreover, we often don't even have the conversation!
In the clinical trenches, if a parent questions why her daughter or son needs the vaccine, a directed but not overly lengthy response of the importance of this vaccine in preventing cervical and other cancers will inform the parent and teen, and is generally well received. Often, asking if an older family member or friend has suffered with cervical cancer will allow the parent to acknowledge that this is a reality that they do not want their teen to experience. When we fail to recommend the HPV series or say its fine "to wait" because of an inability and unwillingness to raise the link of acquisition of HPV and sexual activity, an opportunity is missed.
The media has done much to promote positive health behaviors, but sadly, parents often recall only the negatives associated with the HPV vaccine. Both clinicians and parents must repurpose themselves to protecting children and adolescents. Parents and youth rely on their pediatrician's recommendations and knowledge. In brief, pediatricians must pin a Post It note to their EHR to cue them to educate, but then vaccinate! In short, "Do Ask, Do Tell." No opportunity to protect our youth should be lost!
Dr. Susan Jay is program director of adolescent health and medicine at Children's Hospital of Wisconsin in Wauwatosa, and a professor of pediatrics at the Medical College of Wisconsin. Dr. Jay has no relevant financial disclosures.
*Correction 8/5/2013: The vaccine was misstated as DTap; teens receive Tdap.
This story was updated. 8/5/2013
For those senior pediatricians such as me, the importance of vaccines to
child and adolescent health such as Hemophilus influenzae, pneumococcal
vaccine and others is a "no brainer." We recall the devastating
diseases that are no longer commonplace, because of prevention by
immunization. In the United States, 14 million people will become
infected with HPV every year and 26,200 will develop new cancers from
HPV, almost half of these cervical cancer. As Dr. Frieden pointed out in
the telebriefing, even the small percentage of girls who have received
with the HPV vaccine has made a difference, reducing HPV infections by
almost 60% among females aged 14-19 years in just 4 years.
The HPV vaccine series has met with an initial difficult reception from parents, patients, and providers. Although, at first, the cost of approximately $380 for the three-shot series was a barrier, over time this has become less of an obstacle, and interestingly, this issue was not reported to be a barrier to receiving the vaccine series in the Centers for Disease Control and Prevention's Advisory Committee on Immunization Practices survey on the quadrivalent series. Current lassitude has resulted in only 33.4% of 13- to 17-year-olds being fully immunized in 2012, as reported in the MMWR.
In the same survey, missed opportunities to provide this vaccine after a girl's 11th birthday increased from 20.8% to 84%. Clearly, recommendations to refine systems of delivery in physicians' offices and the use of electronic health records (EHR) to prime providers to immunize at every opportunity in the clinical setting are good ideas. But an undercurrent that is often not openly discussed is that physicians often don't ask or wish to "take on" negative parental reaction to this important vaccine that can prevent cancer but is often linked to reproductive behavior. The link with acquisition of this virus through sexual activity is one that parents often don't want to consider, and frequently the provider acquiesces. In the ACIP survey, the most common reason cited by parents is that they do not believe the vaccine was necessary, followed by concerns of safety, that the vaccine was not recommended, lack of knowledge about the disease, and, finally, that their daughter was not sexually active.
Pediatricians must use every visit to review immunization status. When in an exam room, HPV should be included in the vaccines that are given at a health visit to prevent meningitis and pertussis. For example, say, "Maggie is due to receive these shots today; they are HPV, Menactra, and Tdap."* Too often, other physicians and I timidly approach the HPV vaccine series and allow parents' lack of knowledge - or worse, media hearsay - to dictate a youngster's care and, they opt to not immunize. Moreover, we often don't even have the conversation!
In the clinical trenches, if a parent questions why her daughter or son needs the vaccine, a directed but not overly lengthy response of the importance of this vaccine in preventing cervical and other cancers will inform the parent and teen, and is generally well received. Often, asking if an older family member or friend has suffered with cervical cancer will allow the parent to acknowledge that this is a reality that they do not want their teen to experience. When we fail to recommend the HPV series or say its fine "to wait" because of an inability and unwillingness to raise the link of acquisition of HPV and sexual activity, an opportunity is missed.
The media has done much to promote positive health behaviors, but sadly, parents often recall only the negatives associated with the HPV vaccine. Both clinicians and parents must repurpose themselves to protecting children and adolescents. Parents and youth rely on their pediatrician's recommendations and knowledge. In brief, pediatricians must pin a Post It note to their EHR to cue them to educate, but then vaccinate! In short, "Do Ask, Do Tell." No opportunity to protect our youth should be lost!
Dr. Susan Jay is program director of adolescent health and medicine at Children's Hospital of Wisconsin in Wauwatosa, and a professor of pediatrics at the Medical College of Wisconsin. Dr. Jay has no relevant financial disclosures.
*Correction 8/5/2013: The vaccine was misstated as DTap; teens receive Tdap.
This story was updated. 8/5/2013
Clinicians can do more to improve human papillomavirus vaccination coverage rates among adolescent females, including emphasizing to parents that the vaccine prevents cancer, according to Dr. Thomas Frieden, the director of the Centers for Disease Control and Prevention.
In 2011, only 35% of girls aged 13-17 years had received the full three doses of the HPV vaccine, which dropped slightly in 2012, which is "a huge disappointment," Dr. Frieden said during a July 25 telebriefing. The proportion of adolescent girls who received at least one, two, and the full three-dose HPV vaccine series increased every year from 2007 to 2011, but stalled between 2011 and 2012 and remains at unacceptably low levels – while missed opportunities for vaccination in this population have increased markedly, according to the data (MMWR 2013;62:591-5).
But rates can be increased if clinicians take every possible opportunity to vaccinate adolescents with the HPV vaccine, including when they receive other vaccines, he said during the briefing. While administering a three-dose series was thought to be difficult and was behind the low rates, the data indicated that if the HPV vaccine was given every time the adolescents received another vaccine, the completion of the three-dose series could increase to 93%, he said. According to the report, "Every health care visit, whether for back-to-school evaluations or acute problems, should be used to assess teenagers’ immunization status and provide recommended vaccines if indicated."
"If we get the three-dose series to 80%, an estimated 53,000 cases of cervical cancer could be prevented over the lifetimes of girls aged 12 [years] and younger," Dr. Frieden said.
A three-dose series of Gardasil – the quadrivalent HPV vaccine directed against HPV-16 and -18 (which cause most cervical cancers) and HPV-6 and -11 (which cause most genital warts) – was licensed and recommended by the CDC as a routine vaccine in 2006 for females aged 11-12 years and for females aged 13-26 years who had not been vaccinated. In 2009, the bivalent vaccine Cervarix, directed against HPV-16 and -18, was licensed for use in females aged 10-25 years and added to the recommendation in 2009.
The American Academy of Pediatrics "shares CDC’s disappointment and concern that we are not providing this lifesaving vaccine at the same rates as we do with other immunizations," Dr. Thomas K. McInerny, president of the AAP, said during the briefing.
"Use every opportunity to vaccinate your adolescent patients at both illness and well-child visits," as well as visits for sports physicals, he advised. He also recommended using alerts in electronic medical record systems so vaccination status is reviewed at every patient visit, as well as reminding parents with automated postcards or phone calls, having nurses check vaccine status when they bring patients into the exam room, and implementing standing order policies so that patients can receive vaccines without a physician’s exam or a physician’s order.
The vaccination coverage data were from the National Immunization Survey–Teen (NIS-Teen), which collects data on vaccination among adolescents aged 13-17 years in the 50 states, the District of Columbia, and certain areas of the country, including New York City, and Chicago. Data on HPV vaccination rates among adolescent males will be reported in the future.
In 2007, 25.1% of adolescent girls aged 13-17 years had received at least one dose of the HPV vaccine, which increased to 53% in 2011 and to only 53.8% in 2012. The proportion of adolescent girls who had received the full three-dose series was even lower, increasing from 5.9% in 2007 to 34.8% in 2011 and dropping slightly in 2012 to 33.4%.
The report also provides estimates of missed opportunities for vaccinating adolescent girls, defined as "a health-care encounter occurring on or after a girl’s 11th birthday," and on or after March 23, 2007. Missed opportunities increased from 20.8% in 2007 to 84% in 2012. (March 23, 2007, was when the CDC’s Advisory Committee on Immunization Practices (ACIP) recommendation on the quadrivalent vaccine was published).
The survey found that 23% of parents said they did not plan to get their daughters vaccinated in the next year. The most common reason cited by these parents is that they did not believe the vaccine was necessary (19.1%), followed by concerns about safety (13.1%), vaccine not recommended (14.2%), lack of knowledge of the vaccine or the disease (12.6%), and their daughter was not sexually active (10.1%). Cost did not appear to be an issue.
The report also included safety data, based on the estimated 56 million doses of the quadrivalent vaccine distributed in the United States from June 2006 through March 2013, which did not identify any new safety issues associated with the HPV vaccine, Dr. Frieden said. (Safety focused on the quadrivalent vaccine because most of the vaccine used has been this vaccine).
Most – 92.1% – of the 21,194 adverse event reports received by the Vaccine Adverse Event Reporting System (VAERS) were classified as "nonserious," and included syncope, dizziness, nausea, headache, fever, and urticaria. Local effects included injection-site pain, redness and swelling. Only 8% of the reports were classified as serious and included headache, vomiting, fatigue, dizziness, syncope, and generalized weakness.
The report also points out that no serious safety concerns have been identified in three population-based U.S. studies of the quadrivalent vaccine safety, although one observational study since the vaccine was licensed found an increased syncope risk.
Dr. Frieden noted that an increased risk of syncope may not be specific to the HPV vaccine because syncope is associated with other vaccines among adolescents, and that a 15-minute observation period after the vaccine is administered is recommended.
He referred to a report released in June in the Journal of Infectious Diseases, which found that the infections with the HPV types in the quadrivalent HPV vaccine dropped by almost 60% among females aged 14-19 years during the 4-year period after the vaccine became available and was recommended as a routine vaccine. (J. Inf. Dis. 2013; 208:385-93).
In the United States every year, an estimated 14 million people will become infected with HPV and about 26,200 will develop new cancers from HPV: 17,400 of those cancers are among females, 10,300 of which are cervical cancers, and 8,800 of the cancers are among males, of which 6,700 are oropharyngeal cancers.
In 2011, the recommendation for the quadrivalent vaccine was expanded to include boys aged 11-12 years, and for unvaccinated males up to age 26 years.
Clinicians can do more to improve human papillomavirus vaccination coverage rates among adolescent females, including emphasizing to parents that the vaccine prevents cancer, according to Dr. Thomas Frieden, the director of the Centers for Disease Control and Prevention.
In 2011, only 35% of girls aged 13-17 years had received the full three doses of the HPV vaccine, which dropped slightly in 2012, which is "a huge disappointment," Dr. Frieden said during a July 25 telebriefing. The proportion of adolescent girls who received at least one, two, and the full three-dose HPV vaccine series increased every year from 2007 to 2011, but stalled between 2011 and 2012 and remains at unacceptably low levels – while missed opportunities for vaccination in this population have increased markedly, according to the data (MMWR 2013;62:591-5).
But rates can be increased if clinicians take every possible opportunity to vaccinate adolescents with the HPV vaccine, including when they receive other vaccines, he said during the briefing. While administering a three-dose series was thought to be difficult and was behind the low rates, the data indicated that if the HPV vaccine was given every time the adolescents received another vaccine, the completion of the three-dose series could increase to 93%, he said. According to the report, "Every health care visit, whether for back-to-school evaluations or acute problems, should be used to assess teenagers’ immunization status and provide recommended vaccines if indicated."
"If we get the three-dose series to 80%, an estimated 53,000 cases of cervical cancer could be prevented over the lifetimes of girls aged 12 [years] and younger," Dr. Frieden said.
A three-dose series of Gardasil – the quadrivalent HPV vaccine directed against HPV-16 and -18 (which cause most cervical cancers) and HPV-6 and -11 (which cause most genital warts) – was licensed and recommended by the CDC as a routine vaccine in 2006 for females aged 11-12 years and for females aged 13-26 years who had not been vaccinated. In 2009, the bivalent vaccine Cervarix, directed against HPV-16 and -18, was licensed for use in females aged 10-25 years and added to the recommendation in 2009.
The American Academy of Pediatrics "shares CDC’s disappointment and concern that we are not providing this lifesaving vaccine at the same rates as we do with other immunizations," Dr. Thomas K. McInerny, president of the AAP, said during the briefing.
"Use every opportunity to vaccinate your adolescent patients at both illness and well-child visits," as well as visits for sports physicals, he advised. He also recommended using alerts in electronic medical record systems so vaccination status is reviewed at every patient visit, as well as reminding parents with automated postcards or phone calls, having nurses check vaccine status when they bring patients into the exam room, and implementing standing order policies so that patients can receive vaccines without a physician’s exam or a physician’s order.
The vaccination coverage data were from the National Immunization Survey–Teen (NIS-Teen), which collects data on vaccination among adolescents aged 13-17 years in the 50 states, the District of Columbia, and certain areas of the country, including New York City, and Chicago. Data on HPV vaccination rates among adolescent males will be reported in the future.
In 2007, 25.1% of adolescent girls aged 13-17 years had received at least one dose of the HPV vaccine, which increased to 53% in 2011 and to only 53.8% in 2012. The proportion of adolescent girls who had received the full three-dose series was even lower, increasing from 5.9% in 2007 to 34.8% in 2011 and dropping slightly in 2012 to 33.4%.
The report also provides estimates of missed opportunities for vaccinating adolescent girls, defined as "a health-care encounter occurring on or after a girl’s 11th birthday," and on or after March 23, 2007. Missed opportunities increased from 20.8% in 2007 to 84% in 2012. (March 23, 2007, was when the CDC’s Advisory Committee on Immunization Practices (ACIP) recommendation on the quadrivalent vaccine was published).
The survey found that 23% of parents said they did not plan to get their daughters vaccinated in the next year. The most common reason cited by these parents is that they did not believe the vaccine was necessary (19.1%), followed by concerns about safety (13.1%), vaccine not recommended (14.2%), lack of knowledge of the vaccine or the disease (12.6%), and their daughter was not sexually active (10.1%). Cost did not appear to be an issue.
The report also included safety data, based on the estimated 56 million doses of the quadrivalent vaccine distributed in the United States from June 2006 through March 2013, which did not identify any new safety issues associated with the HPV vaccine, Dr. Frieden said. (Safety focused on the quadrivalent vaccine because most of the vaccine used has been this vaccine).
Most – 92.1% – of the 21,194 adverse event reports received by the Vaccine Adverse Event Reporting System (VAERS) were classified as "nonserious," and included syncope, dizziness, nausea, headache, fever, and urticaria. Local effects included injection-site pain, redness and swelling. Only 8% of the reports were classified as serious and included headache, vomiting, fatigue, dizziness, syncope, and generalized weakness.
The report also points out that no serious safety concerns have been identified in three population-based U.S. studies of the quadrivalent vaccine safety, although one observational study since the vaccine was licensed found an increased syncope risk.
Dr. Frieden noted that an increased risk of syncope may not be specific to the HPV vaccine because syncope is associated with other vaccines among adolescents, and that a 15-minute observation period after the vaccine is administered is recommended.
He referred to a report released in June in the Journal of Infectious Diseases, which found that the infections with the HPV types in the quadrivalent HPV vaccine dropped by almost 60% among females aged 14-19 years during the 4-year period after the vaccine became available and was recommended as a routine vaccine. (J. Inf. Dis. 2013; 208:385-93).
In the United States every year, an estimated 14 million people will become infected with HPV and about 26,200 will develop new cancers from HPV: 17,400 of those cancers are among females, 10,300 of which are cervical cancers, and 8,800 of the cancers are among males, of which 6,700 are oropharyngeal cancers.
In 2011, the recommendation for the quadrivalent vaccine was expanded to include boys aged 11-12 years, and for unvaccinated males up to age 26 years.
FROM MORBIDITY AND MORTALITY WEEKLY REPORT
Major finding: Missed opportunities increased from 20.8% in 2007 to 84% in 2012.
Data source: National Immunization Survey–Teen (NIS-Teen) survey results from 2007 to 2012
Disclosures: CDC did not provide any disclosures.
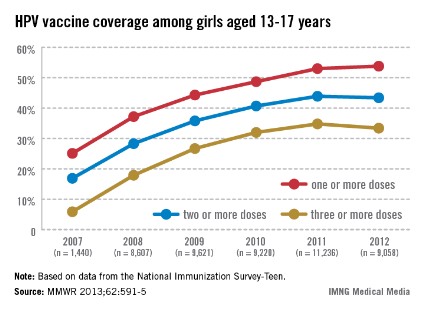
Proposed axial spondyloarthritis indication for certolizumab divides FDA panel
SILVER SPRING, MD. – A Food and Drug Administration advisory panel split its vote on whether to recommend the approval of certolizumab as a treatment for axial spondyloarthritis, based on a study of 325 patients with ankylosing spondylitis or nonradiographic axial spondyloarthritis.
At a July 23 meeting, the FDA’s Arthritis Advisory Committee voted 7-6, with one abstention, to recommend approval of the tumor necrosis factor (TNF) blocker for the manufacturer’s proposed indication: treatment of "active axial spondyloarthritis, including patients with ankylosing spondylitis."
Certolizumab, a TNF blocker, was approved in 2008 for treating Crohn’s disease and in 2009 for treating rheumatoid arthritis (RA). It is marketed as Cimzia by UCB and is administered by subcutaneous injection every 4 weeks for Crohn’s disease and every other week or every 4 weeks for RA, as maintenance therapy. Currently, four other TNF inhibitors are approved for ankylosing spondylitis (AS) but none are approved for treating nonradiographic axial spondyloarthritis (nr-axSpA).
The pivotal study compared two dosing regimens of certolizumab against placebo in patients with active axial spondyloarthritis who had an inadequate response to NSAIDs or a contraindication to NSAIDs (178 patients with AS, and 147 patients with nr-axSpA). The dosing regimens were the same as those currently approved for rheumatoid arthritis: a loading dose of 400 mg in weeks 0, 2, and 4, followed by either 200 mg every 2 weeks (Q2W) or 400 mg every 4 weeks (Q4W). Requirements for enrollment included chronic back pain for at least 3 months; onset before age 45 years; and imaging criteria that included sacroiliitis (on MRI or X-ray) with at least one spondyloarthritis feature, such as inflammation, back pain, arthritis, uveitis, or dactylitis.
At 12 weeks, 58% of patients on the Q2W regimen and 64% on the Q4W regimen had achieved an ASAS (Assessment of SpondyloArthritis international Society) 20 response, the primary endpoint, compared with 38% of those on placebo, both of which were statistically significant differences. The differences remained significant at 24 weeks. ASAS20 responses in the two subpopulations, those with AS and those with nr-axSpA, also favored those treated with certolizumab.
Treatment with certolizumab also resulted in significant improvements in function and spinal mobility, with similar responses at both dosage regimens, compared with placebo, as well as improvements in spinal and sacroiliac joint inflammation, productivity, and health-related quality of life, according to UCB.
The rates of serious adverse events were similar in the certolizumab-treated and placebo groups, the safety profile of certolizumab was consistent with the known safety profile of TNF inhibitors, and no new safety signals were identified. The rate of serious infections, a known risk, was higher among those on certolizumab.
The main issue raised by the FDA was whether the data from this study were adequate to support the indication, especially for patients with nr-axSpA, which would be a new indication for an approved treatment, according to Dr. Janet Maynard, acting clinical team leader in the FDA’s Division of Pulmonary, Allergy, and Rheumatology Products. But the FDA’s analysis of the data did not suggest that the efficacy results were driven by any subgroup, and the efficacy data for AS "appear reasonable," she said at the meeting.
The panel had no substantial safety concerns – voting 13-1 that the safety profile of certolizumab was adequate to support approval of the new indication – and agreed there was evidence of efficacy. Those voting against approval, however, thought that the indication was too broad and had concerns that included the small number of patients for a new indication (nr-axSpA), the lack of a clear definition of active disease in the indication, and the possibility that primary care physicians would prescribe the biologic drug for patients with low back pain.
Panelists supporting approval said that adequate prescribing information would result in appropriate use for patients with active disease in clinical practice.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting; occasionally, a panelist might be given a waiver, but not at this meeting.
Certolizumab, a TNF blocker, Crohn’s disease, rheumatoid arthritis (RA), Cimzia, UCB, subcutaneous injection, Dr. Janet Maynard, FDA’s Division of Pulmonary, Allergy, and Rheumatology Products,
SILVER SPRING, MD. – A Food and Drug Administration advisory panel split its vote on whether to recommend the approval of certolizumab as a treatment for axial spondyloarthritis, based on a study of 325 patients with ankylosing spondylitis or nonradiographic axial spondyloarthritis.
At a July 23 meeting, the FDA’s Arthritis Advisory Committee voted 7-6, with one abstention, to recommend approval of the tumor necrosis factor (TNF) blocker for the manufacturer’s proposed indication: treatment of "active axial spondyloarthritis, including patients with ankylosing spondylitis."
Certolizumab, a TNF blocker, was approved in 2008 for treating Crohn’s disease and in 2009 for treating rheumatoid arthritis (RA). It is marketed as Cimzia by UCB and is administered by subcutaneous injection every 4 weeks for Crohn’s disease and every other week or every 4 weeks for RA, as maintenance therapy. Currently, four other TNF inhibitors are approved for ankylosing spondylitis (AS) but none are approved for treating nonradiographic axial spondyloarthritis (nr-axSpA).
The pivotal study compared two dosing regimens of certolizumab against placebo in patients with active axial spondyloarthritis who had an inadequate response to NSAIDs or a contraindication to NSAIDs (178 patients with AS, and 147 patients with nr-axSpA). The dosing regimens were the same as those currently approved for rheumatoid arthritis: a loading dose of 400 mg in weeks 0, 2, and 4, followed by either 200 mg every 2 weeks (Q2W) or 400 mg every 4 weeks (Q4W). Requirements for enrollment included chronic back pain for at least 3 months; onset before age 45 years; and imaging criteria that included sacroiliitis (on MRI or X-ray) with at least one spondyloarthritis feature, such as inflammation, back pain, arthritis, uveitis, or dactylitis.
At 12 weeks, 58% of patients on the Q2W regimen and 64% on the Q4W regimen had achieved an ASAS (Assessment of SpondyloArthritis international Society) 20 response, the primary endpoint, compared with 38% of those on placebo, both of which were statistically significant differences. The differences remained significant at 24 weeks. ASAS20 responses in the two subpopulations, those with AS and those with nr-axSpA, also favored those treated with certolizumab.
Treatment with certolizumab also resulted in significant improvements in function and spinal mobility, with similar responses at both dosage regimens, compared with placebo, as well as improvements in spinal and sacroiliac joint inflammation, productivity, and health-related quality of life, according to UCB.
The rates of serious adverse events were similar in the certolizumab-treated and placebo groups, the safety profile of certolizumab was consistent with the known safety profile of TNF inhibitors, and no new safety signals were identified. The rate of serious infections, a known risk, was higher among those on certolizumab.
The main issue raised by the FDA was whether the data from this study were adequate to support the indication, especially for patients with nr-axSpA, which would be a new indication for an approved treatment, according to Dr. Janet Maynard, acting clinical team leader in the FDA’s Division of Pulmonary, Allergy, and Rheumatology Products. But the FDA’s analysis of the data did not suggest that the efficacy results were driven by any subgroup, and the efficacy data for AS "appear reasonable," she said at the meeting.
The panel had no substantial safety concerns – voting 13-1 that the safety profile of certolizumab was adequate to support approval of the new indication – and agreed there was evidence of efficacy. Those voting against approval, however, thought that the indication was too broad and had concerns that included the small number of patients for a new indication (nr-axSpA), the lack of a clear definition of active disease in the indication, and the possibility that primary care physicians would prescribe the biologic drug for patients with low back pain.
Panelists supporting approval said that adequate prescribing information would result in appropriate use for patients with active disease in clinical practice.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting; occasionally, a panelist might be given a waiver, but not at this meeting.
SILVER SPRING, MD. – A Food and Drug Administration advisory panel split its vote on whether to recommend the approval of certolizumab as a treatment for axial spondyloarthritis, based on a study of 325 patients with ankylosing spondylitis or nonradiographic axial spondyloarthritis.
At a July 23 meeting, the FDA’s Arthritis Advisory Committee voted 7-6, with one abstention, to recommend approval of the tumor necrosis factor (TNF) blocker for the manufacturer’s proposed indication: treatment of "active axial spondyloarthritis, including patients with ankylosing spondylitis."
Certolizumab, a TNF blocker, was approved in 2008 for treating Crohn’s disease and in 2009 for treating rheumatoid arthritis (RA). It is marketed as Cimzia by UCB and is administered by subcutaneous injection every 4 weeks for Crohn’s disease and every other week or every 4 weeks for RA, as maintenance therapy. Currently, four other TNF inhibitors are approved for ankylosing spondylitis (AS) but none are approved for treating nonradiographic axial spondyloarthritis (nr-axSpA).
The pivotal study compared two dosing regimens of certolizumab against placebo in patients with active axial spondyloarthritis who had an inadequate response to NSAIDs or a contraindication to NSAIDs (178 patients with AS, and 147 patients with nr-axSpA). The dosing regimens were the same as those currently approved for rheumatoid arthritis: a loading dose of 400 mg in weeks 0, 2, and 4, followed by either 200 mg every 2 weeks (Q2W) or 400 mg every 4 weeks (Q4W). Requirements for enrollment included chronic back pain for at least 3 months; onset before age 45 years; and imaging criteria that included sacroiliitis (on MRI or X-ray) with at least one spondyloarthritis feature, such as inflammation, back pain, arthritis, uveitis, or dactylitis.
At 12 weeks, 58% of patients on the Q2W regimen and 64% on the Q4W regimen had achieved an ASAS (Assessment of SpondyloArthritis international Society) 20 response, the primary endpoint, compared with 38% of those on placebo, both of which were statistically significant differences. The differences remained significant at 24 weeks. ASAS20 responses in the two subpopulations, those with AS and those with nr-axSpA, also favored those treated with certolizumab.
Treatment with certolizumab also resulted in significant improvements in function and spinal mobility, with similar responses at both dosage regimens, compared with placebo, as well as improvements in spinal and sacroiliac joint inflammation, productivity, and health-related quality of life, according to UCB.
The rates of serious adverse events were similar in the certolizumab-treated and placebo groups, the safety profile of certolizumab was consistent with the known safety profile of TNF inhibitors, and no new safety signals were identified. The rate of serious infections, a known risk, was higher among those on certolizumab.
The main issue raised by the FDA was whether the data from this study were adequate to support the indication, especially for patients with nr-axSpA, which would be a new indication for an approved treatment, according to Dr. Janet Maynard, acting clinical team leader in the FDA’s Division of Pulmonary, Allergy, and Rheumatology Products. But the FDA’s analysis of the data did not suggest that the efficacy results were driven by any subgroup, and the efficacy data for AS "appear reasonable," she said at the meeting.
The panel had no substantial safety concerns – voting 13-1 that the safety profile of certolizumab was adequate to support approval of the new indication – and agreed there was evidence of efficacy. Those voting against approval, however, thought that the indication was too broad and had concerns that included the small number of patients for a new indication (nr-axSpA), the lack of a clear definition of active disease in the indication, and the possibility that primary care physicians would prescribe the biologic drug for patients with low back pain.
Panelists supporting approval said that adequate prescribing information would result in appropriate use for patients with active disease in clinical practice.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting; occasionally, a panelist might be given a waiver, but not at this meeting.
Certolizumab, a TNF blocker, Crohn’s disease, rheumatoid arthritis (RA), Cimzia, UCB, subcutaneous injection, Dr. Janet Maynard, FDA’s Division of Pulmonary, Allergy, and Rheumatology Products,
Certolizumab, a TNF blocker, Crohn’s disease, rheumatoid arthritis (RA), Cimzia, UCB, subcutaneous injection, Dr. Janet Maynard, FDA’s Division of Pulmonary, Allergy, and Rheumatology Products,
AT THE FDA ADVISORY COMMITTEE MEETING
FDA panel votes against new AS indication for TNF-blocker
SILVER SPRING, MD. – Citing uncertainties about the efficacy data in a pivotal trial, the majority of a Food and Drug Administration advisory panel recommended against expanding the approval of adalimumab to include patients with axial spondyloarthritis who do not have radiographic evidence of ankylosing spondylitis.
At a July 23 meeting, the FDA’s Arthritis Advisory Committee voted 12-1, with 1 abstention, against recommending approval of adalimumab for the indication proposed by the manufacturer, AbbVie Inc.: reducing signs and symptoms in adults with active nonradiographic axial spondyloarthritis (nr-axSpA) with objective signs of inflammation (elevated C-reactive protein or MRI findings) and who have had an inadequate response to, or are intolerant of nonsteroidal anti-inflammatory drugs.
Adalimumab is a monoclonal antibody against tumor necrosis factor (TNF) that was initially approved by the FDA in December 2002 as a treatment for rheumatoid arthritis in adults and was subsequently approved in 2006 for reducing the signs and symptoms of active ankylosing spondylitis (AS) in adults. It is also approved for psoriatic arthritis, juvenile idiopathic arthritis, Crohn’s disease, ulcerative colitis, and plaque psoriasis. It is administered by subcutaneous injection every other week.
Axial spondyloarthritis (axial SpA) includes AS and nr-axSpA; there are no FDA-approved treatments for people with nr-axSpA.
The majority of the panel agreed that no new safety signals were seen in the pivotal study and that the safety profile of adalimumab would not be expected to be different in patients with nr-axSpA and those with RA and other approved indications. But the panel voted that the manufacturer’s data did not provide substantial evidence that the drug resulted in a "clinically meaningful beneficial effect" for these patients, the efficacy criteria needed for approval.
The study compared adalimumab to placebo in 192 patients with active nr-axSpA, who had an inadequate response to NSAIDs, could not tolerate NSAIDs, or had a contraindication to NSAIDs. Patients were able to continue treatment with DMARDs, including sulfasalazine and methotrexate. The primary endpoint used to evaluate clinical responses was the ASAS (Assessment of SpondyloArthritis International Society) 40 response, which is defined as an improvement of at least 40% and absolute improvement of at least two units from baseline in at least three of four domains with no deterioration in the remaining domain (pain, function, morning stiffness, and Patient’s Global Assessment of Disease Activity).
At 12 weeks, 36% of those on adalimumab at a dose of 40 mg every other week had achieved an ASAS40 response, compared with 15% of those on placebo, a statistically significant difference. About 20% of those on adalimumab were in clinical remission at week 12, and efficacy was maintained or improved further among those who continued adalimumab in the open-label period through 68 weeks, according to the company. Adverse events were similar to those associated with the use of adalimumab in patients with AS, psoriatic arthritis, and rheumatoid arthritis.
But based on patient x-rays read at a central facility, a substantial proportion of patients in the trial had radiographic evidence of AS, an approved indication for adalimumab, which the FDA reviewer said may have affected the efficacy results. The FDA’s analysis of ASAS40 responses identified a markedly greater beneficial effect among those who had AS and had positive findings on x-rays, compared with those who had negative x-ray results and had nr-axSpA. In addition, the estimated effect of treatment in the nr-axSpA patients was described as "modest" by the FDA.
Panelists said that there is an unmet need for treatments in patients with this group, and encouraged the company to conduct another, better-designed study.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting; occasionally, a panelist might be given a waiver, but not at this meeting.
In July 2012, adalimumab was approved in the European Union for nr-axSpA.
SILVER SPRING, MD. – Citing uncertainties about the efficacy data in a pivotal trial, the majority of a Food and Drug Administration advisory panel recommended against expanding the approval of adalimumab to include patients with axial spondyloarthritis who do not have radiographic evidence of ankylosing spondylitis.
At a July 23 meeting, the FDA’s Arthritis Advisory Committee voted 12-1, with 1 abstention, against recommending approval of adalimumab for the indication proposed by the manufacturer, AbbVie Inc.: reducing signs and symptoms in adults with active nonradiographic axial spondyloarthritis (nr-axSpA) with objective signs of inflammation (elevated C-reactive protein or MRI findings) and who have had an inadequate response to, or are intolerant of nonsteroidal anti-inflammatory drugs.
Adalimumab is a monoclonal antibody against tumor necrosis factor (TNF) that was initially approved by the FDA in December 2002 as a treatment for rheumatoid arthritis in adults and was subsequently approved in 2006 for reducing the signs and symptoms of active ankylosing spondylitis (AS) in adults. It is also approved for psoriatic arthritis, juvenile idiopathic arthritis, Crohn’s disease, ulcerative colitis, and plaque psoriasis. It is administered by subcutaneous injection every other week.
Axial spondyloarthritis (axial SpA) includes AS and nr-axSpA; there are no FDA-approved treatments for people with nr-axSpA.
The majority of the panel agreed that no new safety signals were seen in the pivotal study and that the safety profile of adalimumab would not be expected to be different in patients with nr-axSpA and those with RA and other approved indications. But the panel voted that the manufacturer’s data did not provide substantial evidence that the drug resulted in a "clinically meaningful beneficial effect" for these patients, the efficacy criteria needed for approval.
The study compared adalimumab to placebo in 192 patients with active nr-axSpA, who had an inadequate response to NSAIDs, could not tolerate NSAIDs, or had a contraindication to NSAIDs. Patients were able to continue treatment with DMARDs, including sulfasalazine and methotrexate. The primary endpoint used to evaluate clinical responses was the ASAS (Assessment of SpondyloArthritis International Society) 40 response, which is defined as an improvement of at least 40% and absolute improvement of at least two units from baseline in at least three of four domains with no deterioration in the remaining domain (pain, function, morning stiffness, and Patient’s Global Assessment of Disease Activity).
At 12 weeks, 36% of those on adalimumab at a dose of 40 mg every other week had achieved an ASAS40 response, compared with 15% of those on placebo, a statistically significant difference. About 20% of those on adalimumab were in clinical remission at week 12, and efficacy was maintained or improved further among those who continued adalimumab in the open-label period through 68 weeks, according to the company. Adverse events were similar to those associated with the use of adalimumab in patients with AS, psoriatic arthritis, and rheumatoid arthritis.
But based on patient x-rays read at a central facility, a substantial proportion of patients in the trial had radiographic evidence of AS, an approved indication for adalimumab, which the FDA reviewer said may have affected the efficacy results. The FDA’s analysis of ASAS40 responses identified a markedly greater beneficial effect among those who had AS and had positive findings on x-rays, compared with those who had negative x-ray results and had nr-axSpA. In addition, the estimated effect of treatment in the nr-axSpA patients was described as "modest" by the FDA.
Panelists said that there is an unmet need for treatments in patients with this group, and encouraged the company to conduct another, better-designed study.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting; occasionally, a panelist might be given a waiver, but not at this meeting.
In July 2012, adalimumab was approved in the European Union for nr-axSpA.
SILVER SPRING, MD. – Citing uncertainties about the efficacy data in a pivotal trial, the majority of a Food and Drug Administration advisory panel recommended against expanding the approval of adalimumab to include patients with axial spondyloarthritis who do not have radiographic evidence of ankylosing spondylitis.
At a July 23 meeting, the FDA’s Arthritis Advisory Committee voted 12-1, with 1 abstention, against recommending approval of adalimumab for the indication proposed by the manufacturer, AbbVie Inc.: reducing signs and symptoms in adults with active nonradiographic axial spondyloarthritis (nr-axSpA) with objective signs of inflammation (elevated C-reactive protein or MRI findings) and who have had an inadequate response to, or are intolerant of nonsteroidal anti-inflammatory drugs.
Adalimumab is a monoclonal antibody against tumor necrosis factor (TNF) that was initially approved by the FDA in December 2002 as a treatment for rheumatoid arthritis in adults and was subsequently approved in 2006 for reducing the signs and symptoms of active ankylosing spondylitis (AS) in adults. It is also approved for psoriatic arthritis, juvenile idiopathic arthritis, Crohn’s disease, ulcerative colitis, and plaque psoriasis. It is administered by subcutaneous injection every other week.
Axial spondyloarthritis (axial SpA) includes AS and nr-axSpA; there are no FDA-approved treatments for people with nr-axSpA.
The majority of the panel agreed that no new safety signals were seen in the pivotal study and that the safety profile of adalimumab would not be expected to be different in patients with nr-axSpA and those with RA and other approved indications. But the panel voted that the manufacturer’s data did not provide substantial evidence that the drug resulted in a "clinically meaningful beneficial effect" for these patients, the efficacy criteria needed for approval.
The study compared adalimumab to placebo in 192 patients with active nr-axSpA, who had an inadequate response to NSAIDs, could not tolerate NSAIDs, or had a contraindication to NSAIDs. Patients were able to continue treatment with DMARDs, including sulfasalazine and methotrexate. The primary endpoint used to evaluate clinical responses was the ASAS (Assessment of SpondyloArthritis International Society) 40 response, which is defined as an improvement of at least 40% and absolute improvement of at least two units from baseline in at least three of four domains with no deterioration in the remaining domain (pain, function, morning stiffness, and Patient’s Global Assessment of Disease Activity).
At 12 weeks, 36% of those on adalimumab at a dose of 40 mg every other week had achieved an ASAS40 response, compared with 15% of those on placebo, a statistically significant difference. About 20% of those on adalimumab were in clinical remission at week 12, and efficacy was maintained or improved further among those who continued adalimumab in the open-label period through 68 weeks, according to the company. Adverse events were similar to those associated with the use of adalimumab in patients with AS, psoriatic arthritis, and rheumatoid arthritis.
But based on patient x-rays read at a central facility, a substantial proportion of patients in the trial had radiographic evidence of AS, an approved indication for adalimumab, which the FDA reviewer said may have affected the efficacy results. The FDA’s analysis of ASAS40 responses identified a markedly greater beneficial effect among those who had AS and had positive findings on x-rays, compared with those who had negative x-ray results and had nr-axSpA. In addition, the estimated effect of treatment in the nr-axSpA patients was described as "modest" by the FDA.
Panelists said that there is an unmet need for treatments in patients with this group, and encouraged the company to conduct another, better-designed study.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting; occasionally, a panelist might be given a waiver, but not at this meeting.
In July 2012, adalimumab was approved in the European Union for nr-axSpA.
AT AN FDA ADVISORY COMMITTEE MEETING
Roche halts studies of experimental type 2 diabetes drug
Citing "safety signals and lack of efficacy," Roche has announced that it is terminating clinical trials of aleglitazar, a dual PPAR agonist that the company had been pursuing as a treatment for type 2 diabetes that lowered cardiovascular risk in addition to controlling glucose.
In a statement on July 10, the company said that the decision was based on a recommendation of the independent Data and Safety Monitoring Board (DSMB) to halt the AleCardio study, a phase III study of aleglitazar in patients with type 2 diabetes and a recent acute coronary syndrome event. The company also has decided to terminate all other studies of the drug.
"While phase II data supported the investigation of the potential benefit of aleglitazar in reducing cardiovascular events in type II diabetes, a lack of efficacy and an increased rate of adverse events including fractures, renal impairment and heart failure were seen in the AleCardio trial, resulting in an unfavorable benefit/risk profile," a company spokesperson said.
"We are disappointed by this outcome as we hoped that aleglitazar would provide significant benefit for patients with type 2 diabetes who are at risk of cardiovascular disease," Dr. Hal Barron, Roche’s chief medical officer and head of global product development, said in the statement. The company "is working with investigators to support the management of patients and their transition from aleglitazar treatment to other blood sugar control therapies," he added.
When the company announced the launch of phase III trials of aleglitazar in June 2009, the drug was described in a Roche statement as "uniquely designed to reduce cardiovascular morbidity and mortality" in high-risk patients with type 2 diabetes. The statement said that the results of SYNCHRONY, a phase II, placebo-controlled dose-ranging study, showed that aleglitazar "had a balanced synergistic effect on both lipid and glucose control with a good safety and tolerability profile" in type 2 diabetes patients.
Roche plans to analyze the data from the AleCardio study and will present the results at a medical meeting, according to the statement announcing the end of the study.
Aleglitazar is a peroxisome proliferator–activated receptor (PPAR) co-agonist, activating both PPAR-alpha and PPAR-gamma. As of July 10, nine studies of aleglitazar were listed as either recruiting or as active and not recruiting on clinicaltrials.gov.
The results of the SYNCHRONY study were published in 2009 (Lancet 2009;374:126-35).
Citing "safety signals and lack of efficacy," Roche has announced that it is terminating clinical trials of aleglitazar, a dual PPAR agonist that the company had been pursuing as a treatment for type 2 diabetes that lowered cardiovascular risk in addition to controlling glucose.
In a statement on July 10, the company said that the decision was based on a recommendation of the independent Data and Safety Monitoring Board (DSMB) to halt the AleCardio study, a phase III study of aleglitazar in patients with type 2 diabetes and a recent acute coronary syndrome event. The company also has decided to terminate all other studies of the drug.
"While phase II data supported the investigation of the potential benefit of aleglitazar in reducing cardiovascular events in type II diabetes, a lack of efficacy and an increased rate of adverse events including fractures, renal impairment and heart failure were seen in the AleCardio trial, resulting in an unfavorable benefit/risk profile," a company spokesperson said.
"We are disappointed by this outcome as we hoped that aleglitazar would provide significant benefit for patients with type 2 diabetes who are at risk of cardiovascular disease," Dr. Hal Barron, Roche’s chief medical officer and head of global product development, said in the statement. The company "is working with investigators to support the management of patients and their transition from aleglitazar treatment to other blood sugar control therapies," he added.
When the company announced the launch of phase III trials of aleglitazar in June 2009, the drug was described in a Roche statement as "uniquely designed to reduce cardiovascular morbidity and mortality" in high-risk patients with type 2 diabetes. The statement said that the results of SYNCHRONY, a phase II, placebo-controlled dose-ranging study, showed that aleglitazar "had a balanced synergistic effect on both lipid and glucose control with a good safety and tolerability profile" in type 2 diabetes patients.
Roche plans to analyze the data from the AleCardio study and will present the results at a medical meeting, according to the statement announcing the end of the study.
Aleglitazar is a peroxisome proliferator–activated receptor (PPAR) co-agonist, activating both PPAR-alpha and PPAR-gamma. As of July 10, nine studies of aleglitazar were listed as either recruiting or as active and not recruiting on clinicaltrials.gov.
The results of the SYNCHRONY study were published in 2009 (Lancet 2009;374:126-35).
Citing "safety signals and lack of efficacy," Roche has announced that it is terminating clinical trials of aleglitazar, a dual PPAR agonist that the company had been pursuing as a treatment for type 2 diabetes that lowered cardiovascular risk in addition to controlling glucose.
In a statement on July 10, the company said that the decision was based on a recommendation of the independent Data and Safety Monitoring Board (DSMB) to halt the AleCardio study, a phase III study of aleglitazar in patients with type 2 diabetes and a recent acute coronary syndrome event. The company also has decided to terminate all other studies of the drug.
"While phase II data supported the investigation of the potential benefit of aleglitazar in reducing cardiovascular events in type II diabetes, a lack of efficacy and an increased rate of adverse events including fractures, renal impairment and heart failure were seen in the AleCardio trial, resulting in an unfavorable benefit/risk profile," a company spokesperson said.
"We are disappointed by this outcome as we hoped that aleglitazar would provide significant benefit for patients with type 2 diabetes who are at risk of cardiovascular disease," Dr. Hal Barron, Roche’s chief medical officer and head of global product development, said in the statement. The company "is working with investigators to support the management of patients and their transition from aleglitazar treatment to other blood sugar control therapies," he added.
When the company announced the launch of phase III trials of aleglitazar in June 2009, the drug was described in a Roche statement as "uniquely designed to reduce cardiovascular morbidity and mortality" in high-risk patients with type 2 diabetes. The statement said that the results of SYNCHRONY, a phase II, placebo-controlled dose-ranging study, showed that aleglitazar "had a balanced synergistic effect on both lipid and glucose control with a good safety and tolerability profile" in type 2 diabetes patients.
Roche plans to analyze the data from the AleCardio study and will present the results at a medical meeting, according to the statement announcing the end of the study.
Aleglitazar is a peroxisome proliferator–activated receptor (PPAR) co-agonist, activating both PPAR-alpha and PPAR-gamma. As of July 10, nine studies of aleglitazar were listed as either recruiting or as active and not recruiting on clinicaltrials.gov.
The results of the SYNCHRONY study were published in 2009 (Lancet 2009;374:126-35).
No safety issues detected in HPV vaccine pregnancy registry
The analysis of the Gardasil pregnancy registry data has provided "reassuring" results regarding the potential risks of exposure to the vaccine during pregnancy, Dr. Fabio Lievano reported at a meetingof the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices.
Based on more than 2,500 pregnancies reported to Merck and enrolled in the registry over a 6-year period, the overall rates of spontaneous abortion, fetal deaths, and congenital anomalies were "at or below background rates," and no pattern in the type of birth defects has been identified, said Dr. Lievano, executive director of clinical risk management at Merck Research Laboratories. Merck is the manufacturer of the quadrivalent HPV vaccine, which provides coverage against HPV types 6, 11, 16, and 18. The vaccine was approved in the United States in 2006, is marketed as Gardasil, and is administered in a three-dose series.
Gardasil is not recommended for use in pregnancy, and as part of the company’s postmarketing safety commitments for the Food and Drug Administration and European and Canadian regulatory authorities, Merck established a pregnancy registry to monitor outcomes associated with inadvertent pregnancy exposures. Women were enrolled in the pregnancy registry if they were living in the United States, Canada, or France and had received a dose of Gardasil within 1 month of their last menstrual period or at any time during pregnancy.
Between June 1, 2006, and May 31, 2012, 2,802 pregnancies exposed to the vaccine were enrolled in the registry, reported from health care providers and vaccinees. (Enrollment closed on Dec. 31, 2012.) The main outcomes the company monitored were infant outcomes (congenital anomalies) and pregnancy outcomes, which included elective abortions, spontaneous abortions (before 20 weeks’ gestation), fetal deaths (at 20 weeks or later), and live births.
Of the 2,440 prospective pregnancy reports (received before the pregnancy outcome was known), about 91% indicated exposure to the vaccine occurred before the end of the first trimester. Of the total exposed pregnancies, there were 102 (3.6%) elective abortions (including 1 associated with anencephaly and hypoplastic heart) and 105 (3.7%) spontaneous abortions (including 1 triplet pregnancy and 1 with a chromosomal anomaly).
Among the 1,460 newborns, 1,381 (95%) had normal outcomes. Of the remainder, 34 had major congenital anomalies and 45 had minor congenital anomalies.
The rate of spontaneous abortions was 6.7/100 outcomes, Dr. Lievano said, noting that the background rate among clinically recognized pregnancies is 15%. The rate of fetal deaths was 0.8/100 outcomes (live births plus fetal deaths) compared with the background rate of 0.62-1/100.
The overall rate of major congenital anomalies was 2.5/100 live born infants, compared with the background rate of 2.67/100 live born infants. The rate of congenital anomalies appears to be similar to the background rates, and the anomalies reported in the registry have varied in type and etiology, Dr. Lievano said, concluding that the analysis "does not support a causal relationship" between exposure to the vaccine in pregnancy and the birth defects reported in the registry,
Of the 362 retrospective reports of pregnancy exposures in the registry (those with outcomes that were known at the time of enrollment), there were 319 known pregnancy outcomes, which included 61 spontaneous abortions and 8 fetal deaths. Among the retrospective reports, 25 infants had major congenital anomalies, including 13 with an isolated anomaly, 4 with two anomalies each, 3 with multiple anomalies, and 2 with multiple anomalies as part of a chromosomal abnormality, he said.
As of April, the FDA, as well as the European Medicines Agency and Health Canada, consider that the company has met the postmarketing pregnancy registry commitment. The results will be published and will be added to the drug’s label. Merck will continue to update the registry with reports of exposures and will provide periodic safety updates to the regulatory agencies, Dr. Lievano said.
The updated ACIP statement on HPV vaccine, which is being planned, will advise that pregnancy exposures continue to be reported to Merck and to the FDA and CDC’s Vaccine Adverse Event Reporting System (VAERS).
The updated Gardasil pregnancy registry website says that cases of exposure to Gardasil during pregnancy should be reported to Merck at 877-888-4231.
The analysis of the Gardasil pregnancy registry data has provided "reassuring" results regarding the potential risks of exposure to the vaccine during pregnancy, Dr. Fabio Lievano reported at a meetingof the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices.
Based on more than 2,500 pregnancies reported to Merck and enrolled in the registry over a 6-year period, the overall rates of spontaneous abortion, fetal deaths, and congenital anomalies were "at or below background rates," and no pattern in the type of birth defects has been identified, said Dr. Lievano, executive director of clinical risk management at Merck Research Laboratories. Merck is the manufacturer of the quadrivalent HPV vaccine, which provides coverage against HPV types 6, 11, 16, and 18. The vaccine was approved in the United States in 2006, is marketed as Gardasil, and is administered in a three-dose series.
Gardasil is not recommended for use in pregnancy, and as part of the company’s postmarketing safety commitments for the Food and Drug Administration and European and Canadian regulatory authorities, Merck established a pregnancy registry to monitor outcomes associated with inadvertent pregnancy exposures. Women were enrolled in the pregnancy registry if they were living in the United States, Canada, or France and had received a dose of Gardasil within 1 month of their last menstrual period or at any time during pregnancy.
Between June 1, 2006, and May 31, 2012, 2,802 pregnancies exposed to the vaccine were enrolled in the registry, reported from health care providers and vaccinees. (Enrollment closed on Dec. 31, 2012.) The main outcomes the company monitored were infant outcomes (congenital anomalies) and pregnancy outcomes, which included elective abortions, spontaneous abortions (before 20 weeks’ gestation), fetal deaths (at 20 weeks or later), and live births.
Of the 2,440 prospective pregnancy reports (received before the pregnancy outcome was known), about 91% indicated exposure to the vaccine occurred before the end of the first trimester. Of the total exposed pregnancies, there were 102 (3.6%) elective abortions (including 1 associated with anencephaly and hypoplastic heart) and 105 (3.7%) spontaneous abortions (including 1 triplet pregnancy and 1 with a chromosomal anomaly).
Among the 1,460 newborns, 1,381 (95%) had normal outcomes. Of the remainder, 34 had major congenital anomalies and 45 had minor congenital anomalies.
The rate of spontaneous abortions was 6.7/100 outcomes, Dr. Lievano said, noting that the background rate among clinically recognized pregnancies is 15%. The rate of fetal deaths was 0.8/100 outcomes (live births plus fetal deaths) compared with the background rate of 0.62-1/100.
The overall rate of major congenital anomalies was 2.5/100 live born infants, compared with the background rate of 2.67/100 live born infants. The rate of congenital anomalies appears to be similar to the background rates, and the anomalies reported in the registry have varied in type and etiology, Dr. Lievano said, concluding that the analysis "does not support a causal relationship" between exposure to the vaccine in pregnancy and the birth defects reported in the registry,
Of the 362 retrospective reports of pregnancy exposures in the registry (those with outcomes that were known at the time of enrollment), there were 319 known pregnancy outcomes, which included 61 spontaneous abortions and 8 fetal deaths. Among the retrospective reports, 25 infants had major congenital anomalies, including 13 with an isolated anomaly, 4 with two anomalies each, 3 with multiple anomalies, and 2 with multiple anomalies as part of a chromosomal abnormality, he said.
As of April, the FDA, as well as the European Medicines Agency and Health Canada, consider that the company has met the postmarketing pregnancy registry commitment. The results will be published and will be added to the drug’s label. Merck will continue to update the registry with reports of exposures and will provide periodic safety updates to the regulatory agencies, Dr. Lievano said.
The updated ACIP statement on HPV vaccine, which is being planned, will advise that pregnancy exposures continue to be reported to Merck and to the FDA and CDC’s Vaccine Adverse Event Reporting System (VAERS).
The updated Gardasil pregnancy registry website says that cases of exposure to Gardasil during pregnancy should be reported to Merck at 877-888-4231.
The analysis of the Gardasil pregnancy registry data has provided "reassuring" results regarding the potential risks of exposure to the vaccine during pregnancy, Dr. Fabio Lievano reported at a meetingof the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices.
Based on more than 2,500 pregnancies reported to Merck and enrolled in the registry over a 6-year period, the overall rates of spontaneous abortion, fetal deaths, and congenital anomalies were "at or below background rates," and no pattern in the type of birth defects has been identified, said Dr. Lievano, executive director of clinical risk management at Merck Research Laboratories. Merck is the manufacturer of the quadrivalent HPV vaccine, which provides coverage against HPV types 6, 11, 16, and 18. The vaccine was approved in the United States in 2006, is marketed as Gardasil, and is administered in a three-dose series.
Gardasil is not recommended for use in pregnancy, and as part of the company’s postmarketing safety commitments for the Food and Drug Administration and European and Canadian regulatory authorities, Merck established a pregnancy registry to monitor outcomes associated with inadvertent pregnancy exposures. Women were enrolled in the pregnancy registry if they were living in the United States, Canada, or France and had received a dose of Gardasil within 1 month of their last menstrual period or at any time during pregnancy.
Between June 1, 2006, and May 31, 2012, 2,802 pregnancies exposed to the vaccine were enrolled in the registry, reported from health care providers and vaccinees. (Enrollment closed on Dec. 31, 2012.) The main outcomes the company monitored were infant outcomes (congenital anomalies) and pregnancy outcomes, which included elective abortions, spontaneous abortions (before 20 weeks’ gestation), fetal deaths (at 20 weeks or later), and live births.
Of the 2,440 prospective pregnancy reports (received before the pregnancy outcome was known), about 91% indicated exposure to the vaccine occurred before the end of the first trimester. Of the total exposed pregnancies, there were 102 (3.6%) elective abortions (including 1 associated with anencephaly and hypoplastic heart) and 105 (3.7%) spontaneous abortions (including 1 triplet pregnancy and 1 with a chromosomal anomaly).
Among the 1,460 newborns, 1,381 (95%) had normal outcomes. Of the remainder, 34 had major congenital anomalies and 45 had minor congenital anomalies.
The rate of spontaneous abortions was 6.7/100 outcomes, Dr. Lievano said, noting that the background rate among clinically recognized pregnancies is 15%. The rate of fetal deaths was 0.8/100 outcomes (live births plus fetal deaths) compared with the background rate of 0.62-1/100.
The overall rate of major congenital anomalies was 2.5/100 live born infants, compared with the background rate of 2.67/100 live born infants. The rate of congenital anomalies appears to be similar to the background rates, and the anomalies reported in the registry have varied in type and etiology, Dr. Lievano said, concluding that the analysis "does not support a causal relationship" between exposure to the vaccine in pregnancy and the birth defects reported in the registry,
Of the 362 retrospective reports of pregnancy exposures in the registry (those with outcomes that were known at the time of enrollment), there were 319 known pregnancy outcomes, which included 61 spontaneous abortions and 8 fetal deaths. Among the retrospective reports, 25 infants had major congenital anomalies, including 13 with an isolated anomaly, 4 with two anomalies each, 3 with multiple anomalies, and 2 with multiple anomalies as part of a chromosomal abnormality, he said.
As of April, the FDA, as well as the European Medicines Agency and Health Canada, consider that the company has met the postmarketing pregnancy registry commitment. The results will be published and will be added to the drug’s label. Merck will continue to update the registry with reports of exposures and will provide periodic safety updates to the regulatory agencies, Dr. Lievano said.
The updated ACIP statement on HPV vaccine, which is being planned, will advise that pregnancy exposures continue to be reported to Merck and to the FDA and CDC’s Vaccine Adverse Event Reporting System (VAERS).
The updated Gardasil pregnancy registry website says that cases of exposure to Gardasil during pregnancy should be reported to Merck at 877-888-4231.
FROM A CDC ADVISORY COMMITTEE MEETING
FDA approves sublingual buprenorphine-naloxone combo for opioid dependence
A sublingual formulation of the partial opioid agonist buprenorphine, combined with naloxone to reduce the potential for misuse and diversion, has been approved by the Food and Drug Administration, the manufacturer announced.
The combination is approved for the maintenance treatment of opioid dependence, as part of a complete treatment plan that should include counseling and psychosocial support.
The manufacturer, Orexo US, will market the product as Zubsolv; it will be available in two dosage strengths: a tablet containing 1.4 mg of buprenorphine and 0.36 mg of naloxone, and a tablet with 5.7 mg of buprenorphine and 1.4 mg of naloxone.
It should be administered once a day and should be used as maintenance for patients who "have been initially inducted using buprenorphine sublingual tablets," according to the prescribing information. The prescribing information includes a chart that provides the corresponding dosage strengths for the two formulations, as guidance when going from induction to maintenance treatment.
A sublingual formulation of the partial opioid agonist buprenorphine, combined with naloxone to reduce the potential for misuse and diversion, has been approved by the Food and Drug Administration, the manufacturer announced.
The combination is approved for the maintenance treatment of opioid dependence, as part of a complete treatment plan that should include counseling and psychosocial support.
The manufacturer, Orexo US, will market the product as Zubsolv; it will be available in two dosage strengths: a tablet containing 1.4 mg of buprenorphine and 0.36 mg of naloxone, and a tablet with 5.7 mg of buprenorphine and 1.4 mg of naloxone.
It should be administered once a day and should be used as maintenance for patients who "have been initially inducted using buprenorphine sublingual tablets," according to the prescribing information. The prescribing information includes a chart that provides the corresponding dosage strengths for the two formulations, as guidance when going from induction to maintenance treatment.
A sublingual formulation of the partial opioid agonist buprenorphine, combined with naloxone to reduce the potential for misuse and diversion, has been approved by the Food and Drug Administration, the manufacturer announced.
The combination is approved for the maintenance treatment of opioid dependence, as part of a complete treatment plan that should include counseling and psychosocial support.
The manufacturer, Orexo US, will market the product as Zubsolv; it will be available in two dosage strengths: a tablet containing 1.4 mg of buprenorphine and 0.36 mg of naloxone, and a tablet with 5.7 mg of buprenorphine and 1.4 mg of naloxone.
It should be administered once a day and should be used as maintenance for patients who "have been initially inducted using buprenorphine sublingual tablets," according to the prescribing information. The prescribing information includes a chart that provides the corresponding dosage strengths for the two formulations, as guidance when going from induction to maintenance treatment.