User login
FDA issues warning on azithromycin arrhythmia risk
Use of the antibiotic azithromycin is associated with an increased risk for fatal arrhythmia, according to a warning issued by the Food and Drug Administration on Mar. 12.
The FDA has taken the step to strengthen the existing warning on the drug’s label about the risk of QT interval prolongation and torsades de pointes. In general, the people at greatest risk are those with known risk factors such as existing QT interval prolongation, low blood levels of potassium or magnesium, a slower than normal heart rate, or use of certain drugs used to treat abnormal heart rhythms or arrhythmias, according to the FDA.

Macrolides or nonmacrolides such as fluoroquinolones are among the antibiotics that physicians might consider using as alternatives to azithromycin, but there is no easy answer to which antibiotic to use in at-risk patients since these agents carry their own increased risk for QT prolongation, according to the FDA.
The FDA’s statement is a result of the agency’s review of a study showing that the risk of cardiovascular deaths, and the risk of death from any cause, was increased among those treated with a 5-day course of azithromycin, compared with people treated with amoxicillin, ciprofloxacin, levofloxacin, or no drug (N. Engl. J. Med. 2012;366:1881-90).
When compared with levofloxacin, the risk of cardiovascular death associated with azithromycin was similar. When compared with those who took no antibiotic, the risk of cardiovascular death was increased by 2.88 and the risk of death from any cause was increased by 1.85 among those treated with azithromycin, both statistically significant effects.
Although this study had limitations, it was "methodologically sound and supports the validity of the overall findings," and the excess risk of cardiovascular death, "especially of sudden death, is consistent with arrhythmias from drug-related QT prolongation," the FDA said.
In formulating its warning, the FDA also considered findings from a clinical QT study conducted by the manufacturer. The results of the manufacturer’s study, which have been added to the drug label, indicated that azithromycin prolonged the QTc interval, according to the FDA statement.
The FDA statement includes a list of specific groups at increased risk for torsades de pointes, including those with known prolongation of the QT interval, history of torsades de pointes, congenital long QT syndrome, bradyarrhythmias, or uncompensated heart failure, as well as those who are on drugs known to prolong the QT interval.
Also at risk are people with ongoing proarrhythmic conditions, including uncorrected hypokalemia or hypomagnesemia; those with clinically significant bradycardia; and patients treated with class IA (quinidine, procainamide) or class III (dofetilide, amiodarone, sotalol) antiarrhythmic drugs. Elderly patients and those with cardiac disease "may be more susceptible to the effects of arrhythmogenic drugs on the QT interval," the statement adds.
This information has been added to the warnings and precautions section of the labels for azithromycin products – marketed as Zithromax and Zmax – which are approved for indications that include acute bacterial exacerbations of COPD, acute bacterial sinusitis, community-acquired pneumonia, pharyngitis/tonsillitis, uncomplicated skin and skin structure infections, and urethritis and cervicitis.
Serious adverse events associated with azithromycin should be reported to the FDA’s MedWatch program at 800-332-1088 or www.fda.gov/medwatch/.
Use of the antibiotic azithromycin is associated with an increased risk for fatal arrhythmia, according to a warning issued by the Food and Drug Administration on Mar. 12.
The FDA has taken the step to strengthen the existing warning on the drug’s label about the risk of QT interval prolongation and torsades de pointes. In general, the people at greatest risk are those with known risk factors such as existing QT interval prolongation, low blood levels of potassium or magnesium, a slower than normal heart rate, or use of certain drugs used to treat abnormal heart rhythms or arrhythmias, according to the FDA.
Macrolides or nonmacrolides such as fluoroquinolones are among the antibiotics that physicians might consider using as alternatives to azithromycin, but there is no easy answer to which antibiotic to use in at-risk patients since these agents carry their own increased risk for QT prolongation, according to the FDA.
The FDA’s statement is a result of the agency’s review of a study showing that the risk of cardiovascular deaths, and the risk of death from any cause, was increased among those treated with a 5-day course of azithromycin, compared with people treated with amoxicillin, ciprofloxacin, levofloxacin, or no drug (N. Engl. J. Med. 2012;366:1881-90).
When compared with levofloxacin, the risk of cardiovascular death associated with azithromycin was similar. When compared with those who took no antibiotic, the risk of cardiovascular death was increased by 2.88 and the risk of death from any cause was increased by 1.85 among those treated with azithromycin, both statistically significant effects.
Although this study had limitations, it was "methodologically sound and supports the validity of the overall findings," and the excess risk of cardiovascular death, "especially of sudden death, is consistent with arrhythmias from drug-related QT prolongation," the FDA said.
In formulating its warning, the FDA also considered findings from a clinical QT study conducted by the manufacturer. The results of the manufacturer’s study, which have been added to the drug label, indicated that azithromycin prolonged the QTc interval, according to the FDA statement.
The FDA statement includes a list of specific groups at increased risk for torsades de pointes, including those with known prolongation of the QT interval, history of torsades de pointes, congenital long QT syndrome, bradyarrhythmias, or uncompensated heart failure, as well as those who are on drugs known to prolong the QT interval.
Also at risk are people with ongoing proarrhythmic conditions, including uncorrected hypokalemia or hypomagnesemia; those with clinically significant bradycardia; and patients treated with class IA (quinidine, procainamide) or class III (dofetilide, amiodarone, sotalol) antiarrhythmic drugs. Elderly patients and those with cardiac disease "may be more susceptible to the effects of arrhythmogenic drugs on the QT interval," the statement adds.
This information has been added to the warnings and precautions section of the labels for azithromycin products – marketed as Zithromax and Zmax – which are approved for indications that include acute bacterial exacerbations of COPD, acute bacterial sinusitis, community-acquired pneumonia, pharyngitis/tonsillitis, uncomplicated skin and skin structure infections, and urethritis and cervicitis.
Serious adverse events associated with azithromycin should be reported to the FDA’s MedWatch program at 800-332-1088 or www.fda.gov/medwatch/.
Use of the antibiotic azithromycin is associated with an increased risk for fatal arrhythmia, according to a warning issued by the Food and Drug Administration on Mar. 12.
The FDA has taken the step to strengthen the existing warning on the drug’s label about the risk of QT interval prolongation and torsades de pointes. In general, the people at greatest risk are those with known risk factors such as existing QT interval prolongation, low blood levels of potassium or magnesium, a slower than normal heart rate, or use of certain drugs used to treat abnormal heart rhythms or arrhythmias, according to the FDA.
Macrolides or nonmacrolides such as fluoroquinolones are among the antibiotics that physicians might consider using as alternatives to azithromycin, but there is no easy answer to which antibiotic to use in at-risk patients since these agents carry their own increased risk for QT prolongation, according to the FDA.
The FDA’s statement is a result of the agency’s review of a study showing that the risk of cardiovascular deaths, and the risk of death from any cause, was increased among those treated with a 5-day course of azithromycin, compared with people treated with amoxicillin, ciprofloxacin, levofloxacin, or no drug (N. Engl. J. Med. 2012;366:1881-90).
When compared with levofloxacin, the risk of cardiovascular death associated with azithromycin was similar. When compared with those who took no antibiotic, the risk of cardiovascular death was increased by 2.88 and the risk of death from any cause was increased by 1.85 among those treated with azithromycin, both statistically significant effects.
Although this study had limitations, it was "methodologically sound and supports the validity of the overall findings," and the excess risk of cardiovascular death, "especially of sudden death, is consistent with arrhythmias from drug-related QT prolongation," the FDA said.
In formulating its warning, the FDA also considered findings from a clinical QT study conducted by the manufacturer. The results of the manufacturer’s study, which have been added to the drug label, indicated that azithromycin prolonged the QTc interval, according to the FDA statement.
The FDA statement includes a list of specific groups at increased risk for torsades de pointes, including those with known prolongation of the QT interval, history of torsades de pointes, congenital long QT syndrome, bradyarrhythmias, or uncompensated heart failure, as well as those who are on drugs known to prolong the QT interval.
Also at risk are people with ongoing proarrhythmic conditions, including uncorrected hypokalemia or hypomagnesemia; those with clinically significant bradycardia; and patients treated with class IA (quinidine, procainamide) or class III (dofetilide, amiodarone, sotalol) antiarrhythmic drugs. Elderly patients and those with cardiac disease "may be more susceptible to the effects of arrhythmogenic drugs on the QT interval," the statement adds.
This information has been added to the warnings and precautions section of the labels for azithromycin products – marketed as Zithromax and Zmax – which are approved for indications that include acute bacterial exacerbations of COPD, acute bacterial sinusitis, community-acquired pneumonia, pharyngitis/tonsillitis, uncomplicated skin and skin structure infections, and urethritis and cervicitis.
Serious adverse events associated with azithromycin should be reported to the FDA’s MedWatch program at 800-332-1088 or www.fda.gov/medwatch/.

Rivaroxaban ACS indication remains unapproved, manufacturer says
The approval of the oral anticoagulant rivaroxaban for patients with acute coronary syndrome has been delayed for a second time by the Food and Drug Administration, according to the manufacturer, Janssen Pharmaceuticals.
In a March 4 statement, the company announced that the FDA has issued a second Complete Response Letter regarding the application for approval of rivaroxaban, a factor Xa inhibitor marketed as Xarelto, for reducing the risk of cardiovascular events in people with acute coronary syndrome (ACS). The statement says that in response to the FDA’s first Complete Response Letter, issued in June 2012, the company had submitted some of the missing data from the ATLAS ACS 2 TIMI 51 (Anti-Xa Therapy to Lower Cardiovascular Events in Addition to Aspirin With/Without Thienopyridine Therapy in Subjects With Acute Coronary Syndrome) trial. The data were on 843 (63%) of the people enrolled in the study whose vital status was unknown, which, when included, did not affect the mortality benefit observed in the study, according to the statement.
The phase III ATLAS ACS 2 TIMI 51 results were submitted as part of the application filed in December 2011 for approval of rivaroxaban for reducing the risk of thrombotic cardiovascular events "in patients with ACS [ST-elevation myocardial infarction (STEMI), non–ST-elevation myocardial infarction (NSTEMI), or unstable angina] in combination with aspirin alone or with aspirin plus clopidogrel or ticlopidine," at a 2.5-mg twice-daily dose.
But at a meeting in May 2012, an FDA advisory panel voted 6 to 4, with 1 abstention, against approval of this indication, with those voting no citing a large amount of missing data and safety concerns among the reasons for their votes.
The company statement does not specify the FDA’s reasons for not approving the ACS indication. The FDA issues Complete Response Letters for a drug when there are outstanding issues that need to be resolved before approval, but does not announce when Complete Response Letters are issued. Like the last time, Janssen said that the company is evaluating the FDA’s letter and will continue to work with the FDA to respond to the agency’s questions.
In an interview, Dr. Sanjay Kaul, one of the FDA panelists who voted against approval at the May 2012 panel meeting, said that the company did "a commendable job" of obtaining vital status information in over 60% of those missing this information. However, data are missing in 3.2%, which "is still much higher than that reported in contemporary ACS trials such as TRITON, PLATO, APPRAISE II, or TRACER," he added.

Moreover, although concerns about the missing data dominated the panel deliberations, "it was not the only issue with potential relevance to the approvability of the expanded indication for rivaroxaban," said Dr. Kaul, a professor at the University of California, Los Angeles. Those issues included the lack of a dose response with the 2.5-mg, but not the 5-mg dose, yielding benefit; and "divergent effects" of the two doses on ischemic endpoints (reduction of CV mortality, but not MI, with 2.5 mg and reduction of MI, but not CV mortality, with the 5-mg dose).
Others issues raised at the meeting were the lack of external evidence supporting the role of dual antiplatelet regimen plus novel anticoagulant therapy and the lack of a statistically persuasive treatment benefit, as defined by a robust P value below .001, to allow approval on the basis of one trial, he said.
He said that while he could not speculate as to why the Complete Response Letter was issued, he would not be surprised "if any one or all of these issues played a major role in the FDA’s decision."
Dr. Kaul, who is also attending cardiologist at Cedars-Sinai Medical Center, Los Angeles, disclosed that he owns stock in Johnson & Johnson, of which Janssen is a subsidiary.
First approved in July 2011 for prophylaxis of deep vein thrombosis or pulmonary embolism in patients undergoing hip or knee replacement surgery, rivaroxaban is now approved for six indications, including the treatment of DVT or PE and for reducing the recurrence of DVT and PE after initial treatment.
The approval of the oral anticoagulant rivaroxaban for patients with acute coronary syndrome has been delayed for a second time by the Food and Drug Administration, according to the manufacturer, Janssen Pharmaceuticals.
In a March 4 statement, the company announced that the FDA has issued a second Complete Response Letter regarding the application for approval of rivaroxaban, a factor Xa inhibitor marketed as Xarelto, for reducing the risk of cardiovascular events in people with acute coronary syndrome (ACS). The statement says that in response to the FDA’s first Complete Response Letter, issued in June 2012, the company had submitted some of the missing data from the ATLAS ACS 2 TIMI 51 (Anti-Xa Therapy to Lower Cardiovascular Events in Addition to Aspirin With/Without Thienopyridine Therapy in Subjects With Acute Coronary Syndrome) trial. The data were on 843 (63%) of the people enrolled in the study whose vital status was unknown, which, when included, did not affect the mortality benefit observed in the study, according to the statement.
The phase III ATLAS ACS 2 TIMI 51 results were submitted as part of the application filed in December 2011 for approval of rivaroxaban for reducing the risk of thrombotic cardiovascular events "in patients with ACS [ST-elevation myocardial infarction (STEMI), non–ST-elevation myocardial infarction (NSTEMI), or unstable angina] in combination with aspirin alone or with aspirin plus clopidogrel or ticlopidine," at a 2.5-mg twice-daily dose.
But at a meeting in May 2012, an FDA advisory panel voted 6 to 4, with 1 abstention, against approval of this indication, with those voting no citing a large amount of missing data and safety concerns among the reasons for their votes.
The company statement does not specify the FDA’s reasons for not approving the ACS indication. The FDA issues Complete Response Letters for a drug when there are outstanding issues that need to be resolved before approval, but does not announce when Complete Response Letters are issued. Like the last time, Janssen said that the company is evaluating the FDA’s letter and will continue to work with the FDA to respond to the agency’s questions.
In an interview, Dr. Sanjay Kaul, one of the FDA panelists who voted against approval at the May 2012 panel meeting, said that the company did "a commendable job" of obtaining vital status information in over 60% of those missing this information. However, data are missing in 3.2%, which "is still much higher than that reported in contemporary ACS trials such as TRITON, PLATO, APPRAISE II, or TRACER," he added.
Moreover, although concerns about the missing data dominated the panel deliberations, "it was not the only issue with potential relevance to the approvability of the expanded indication for rivaroxaban," said Dr. Kaul, a professor at the University of California, Los Angeles. Those issues included the lack of a dose response with the 2.5-mg, but not the 5-mg dose, yielding benefit; and "divergent effects" of the two doses on ischemic endpoints (reduction of CV mortality, but not MI, with 2.5 mg and reduction of MI, but not CV mortality, with the 5-mg dose).
Others issues raised at the meeting were the lack of external evidence supporting the role of dual antiplatelet regimen plus novel anticoagulant therapy and the lack of a statistically persuasive treatment benefit, as defined by a robust P value below .001, to allow approval on the basis of one trial, he said.
He said that while he could not speculate as to why the Complete Response Letter was issued, he would not be surprised "if any one or all of these issues played a major role in the FDA’s decision."
Dr. Kaul, who is also attending cardiologist at Cedars-Sinai Medical Center, Los Angeles, disclosed that he owns stock in Johnson & Johnson, of which Janssen is a subsidiary.
First approved in July 2011 for prophylaxis of deep vein thrombosis or pulmonary embolism in patients undergoing hip or knee replacement surgery, rivaroxaban is now approved for six indications, including the treatment of DVT or PE and for reducing the recurrence of DVT and PE after initial treatment.
The approval of the oral anticoagulant rivaroxaban for patients with acute coronary syndrome has been delayed for a second time by the Food and Drug Administration, according to the manufacturer, Janssen Pharmaceuticals.
In a March 4 statement, the company announced that the FDA has issued a second Complete Response Letter regarding the application for approval of rivaroxaban, a factor Xa inhibitor marketed as Xarelto, for reducing the risk of cardiovascular events in people with acute coronary syndrome (ACS). The statement says that in response to the FDA’s first Complete Response Letter, issued in June 2012, the company had submitted some of the missing data from the ATLAS ACS 2 TIMI 51 (Anti-Xa Therapy to Lower Cardiovascular Events in Addition to Aspirin With/Without Thienopyridine Therapy in Subjects With Acute Coronary Syndrome) trial. The data were on 843 (63%) of the people enrolled in the study whose vital status was unknown, which, when included, did not affect the mortality benefit observed in the study, according to the statement.
The phase III ATLAS ACS 2 TIMI 51 results were submitted as part of the application filed in December 2011 for approval of rivaroxaban for reducing the risk of thrombotic cardiovascular events "in patients with ACS [ST-elevation myocardial infarction (STEMI), non–ST-elevation myocardial infarction (NSTEMI), or unstable angina] in combination with aspirin alone or with aspirin plus clopidogrel or ticlopidine," at a 2.5-mg twice-daily dose.
But at a meeting in May 2012, an FDA advisory panel voted 6 to 4, with 1 abstention, against approval of this indication, with those voting no citing a large amount of missing data and safety concerns among the reasons for their votes.
The company statement does not specify the FDA’s reasons for not approving the ACS indication. The FDA issues Complete Response Letters for a drug when there are outstanding issues that need to be resolved before approval, but does not announce when Complete Response Letters are issued. Like the last time, Janssen said that the company is evaluating the FDA’s letter and will continue to work with the FDA to respond to the agency’s questions.
In an interview, Dr. Sanjay Kaul, one of the FDA panelists who voted against approval at the May 2012 panel meeting, said that the company did "a commendable job" of obtaining vital status information in over 60% of those missing this information. However, data are missing in 3.2%, which "is still much higher than that reported in contemporary ACS trials such as TRITON, PLATO, APPRAISE II, or TRACER," he added.
Moreover, although concerns about the missing data dominated the panel deliberations, "it was not the only issue with potential relevance to the approvability of the expanded indication for rivaroxaban," said Dr. Kaul, a professor at the University of California, Los Angeles. Those issues included the lack of a dose response with the 2.5-mg, but not the 5-mg dose, yielding benefit; and "divergent effects" of the two doses on ischemic endpoints (reduction of CV mortality, but not MI, with 2.5 mg and reduction of MI, but not CV mortality, with the 5-mg dose).
Others issues raised at the meeting were the lack of external evidence supporting the role of dual antiplatelet regimen plus novel anticoagulant therapy and the lack of a statistically persuasive treatment benefit, as defined by a robust P value below .001, to allow approval on the basis of one trial, he said.
He said that while he could not speculate as to why the Complete Response Letter was issued, he would not be surprised "if any one or all of these issues played a major role in the FDA’s decision."
Dr. Kaul, who is also attending cardiologist at Cedars-Sinai Medical Center, Los Angeles, disclosed that he owns stock in Johnson & Johnson, of which Janssen is a subsidiary.
First approved in July 2011 for prophylaxis of deep vein thrombosis or pulmonary embolism in patients undergoing hip or knee replacement surgery, rivaroxaban is now approved for six indications, including the treatment of DVT or PE and for reducing the recurrence of DVT and PE after initial treatment.

FDA panel rejects SSRI's approval as a hot flash treatment
SILVER SPRING, MD. – The majority of a Food and Drug Administration advisory panel recommended against approval of a low-dose formulation of paroxetine mesylate as a treatment for menopausal vasomotor symptoms, citing minimal benefits over placebo in clinical trials and the potential risks associated with treatment.
At the March 4 meeting, the FDA’s Reproductive Health Drugs Advisory Committee voted 10 to 4 that the overall risk-benefit profile of paroxetine did not support approval of the indication proposed by the manufacturer, Noven Therapeutics, as a treatment for moderate to severe vasomotor symptoms associated with menopause, at a dose of 7.5 mg once a day at bedtime.
The 7.5-mg dose is lower than the doses approved for psychiatric indications of paroxetine, which range from an initial dose of 10 mg to a maximum of 60 mg per day, depending on the indication.
Currently, paroxetine, a selective serotonin reuptake inhibitor (SSRI), is used off-label to treat vasomotor symptoms (VMS), and if approved, this could be the first nonhormonal treatment for VMS associated with menopause. Earlier in the day, the same panel voted against recommending approval for the antiepileptic drug gabapentin, another drug used off-label for VMS, for the same indication, citing the drug’s modest effects on VMS and its potential risks. In studies of both drugs, there was a marked placebo effect, which complicated the evaluations of the effectiveness.
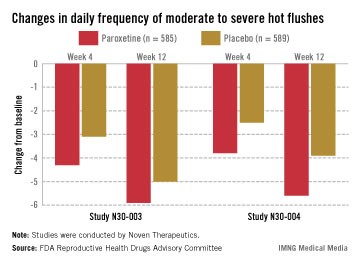
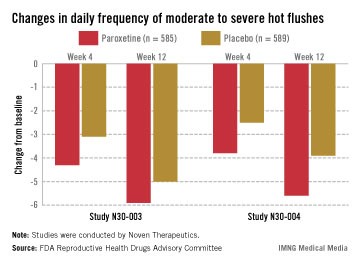
In two pivotal studies conducted in the United States, 1,180 women (mean age, 54-55 years) with natural or surgical menopause who had at least 7 hot flashes per day or at least 50 per week were randomized to receive paroxetine or placebo, after a 12-week run-in period. Electronic diaries were used to document hot flashes. The primary endpoints were the mean change from baseline in hot flash frequency, and the mean change from baseline in hot flash severity, at weeks 4 and 12.
Those on paroxetine had statistically significant reductions in the daily number of hot flashes from baseline, compared with those on placebo at weeks 4 and 12 in both studies, and at week 24 in one of the two studies, which was a 24-week study. The reductions in the daily severity of hot flashes also were statistically significant at week 4 in both studies, and at week 12 in one study but not in the other study, according to the FDA.
However, the women on placebo had notable responses, not unexpected for a nonhormonal treatment, and the difference in the median number of hot flashes per day between the two groups was less than 2: In one study, the median number of hot flashes a day dropped by 4.29 by week 4 and 5.93 at week 12 among those on paroxetine, versus 3.13 and 5, respectively, among those on placebo. In the other study, the median number of hot flashes a day dropped by 3.79 at week 4 and 5.57 by week 12 among those on paroxetine, versus 2.5 and 3.86, respectively, among those on placebo.
The safety profile in the studies was consistent with the known safety profile of paroxetine, with no new or unexpected findings, according to Noven. Fatigue, nausea, and dizziness were the most common side effects in people treated with paroxetine, but they occurred mostly in the first 4 weeks of treatment and were mild to moderate Serious adverse events were higher among those on paroxetine (2.2% vs. 1.4%). In the 24-week study, there were three cases of suicidal ideation and one suicide attempt among those on paroxetine, all after 12 weeks of treatment. The one death in the studies was in a woman on paroxetine, but the death not considered related to the drug, according to the company.
Paroxetine was first approved in 1992 as paroxetine hydrochloride. Paroxetine mesylate was first approved in 2003 and is marketed as Pexeva; it is approved for major depressive disorder, obsessive compulsive disorder, panic disorder, and generalized anxiety disorder. As with all antidepressants, it has a boxed warning about the risk of suicidality and serotonin syndrome associated with treatment.
Noven refers to low-dose paroxetine as LDMP, for low-dose mesylate salt of paroxetine.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
psychiatric indications of paroxetine, selective serotonin reuptake inhibitor, SSRI,
SILVER SPRING, MD. – The majority of a Food and Drug Administration advisory panel recommended against approval of a low-dose formulation of paroxetine mesylate as a treatment for menopausal vasomotor symptoms, citing minimal benefits over placebo in clinical trials and the potential risks associated with treatment.
At the March 4 meeting, the FDA’s Reproductive Health Drugs Advisory Committee voted 10 to 4 that the overall risk-benefit profile of paroxetine did not support approval of the indication proposed by the manufacturer, Noven Therapeutics, as a treatment for moderate to severe vasomotor symptoms associated with menopause, at a dose of 7.5 mg once a day at bedtime.
The 7.5-mg dose is lower than the doses approved for psychiatric indications of paroxetine, which range from an initial dose of 10 mg to a maximum of 60 mg per day, depending on the indication.
Currently, paroxetine, a selective serotonin reuptake inhibitor (SSRI), is used off-label to treat vasomotor symptoms (VMS), and if approved, this could be the first nonhormonal treatment for VMS associated with menopause. Earlier in the day, the same panel voted against recommending approval for the antiepileptic drug gabapentin, another drug used off-label for VMS, for the same indication, citing the drug’s modest effects on VMS and its potential risks. In studies of both drugs, there was a marked placebo effect, which complicated the evaluations of the effectiveness.
In two pivotal studies conducted in the United States, 1,180 women (mean age, 54-55 years) with natural or surgical menopause who had at least 7 hot flashes per day or at least 50 per week were randomized to receive paroxetine or placebo, after a 12-week run-in period. Electronic diaries were used to document hot flashes. The primary endpoints were the mean change from baseline in hot flash frequency, and the mean change from baseline in hot flash severity, at weeks 4 and 12.
Those on paroxetine had statistically significant reductions in the daily number of hot flashes from baseline, compared with those on placebo at weeks 4 and 12 in both studies, and at week 24 in one of the two studies, which was a 24-week study. The reductions in the daily severity of hot flashes also were statistically significant at week 4 in both studies, and at week 12 in one study but not in the other study, according to the FDA.
However, the women on placebo had notable responses, not unexpected for a nonhormonal treatment, and the difference in the median number of hot flashes per day between the two groups was less than 2: In one study, the median number of hot flashes a day dropped by 4.29 by week 4 and 5.93 at week 12 among those on paroxetine, versus 3.13 and 5, respectively, among those on placebo. In the other study, the median number of hot flashes a day dropped by 3.79 at week 4 and 5.57 by week 12 among those on paroxetine, versus 2.5 and 3.86, respectively, among those on placebo.
The safety profile in the studies was consistent with the known safety profile of paroxetine, with no new or unexpected findings, according to Noven. Fatigue, nausea, and dizziness were the most common side effects in people treated with paroxetine, but they occurred mostly in the first 4 weeks of treatment and were mild to moderate Serious adverse events were higher among those on paroxetine (2.2% vs. 1.4%). In the 24-week study, there were three cases of suicidal ideation and one suicide attempt among those on paroxetine, all after 12 weeks of treatment. The one death in the studies was in a woman on paroxetine, but the death not considered related to the drug, according to the company.
Paroxetine was first approved in 1992 as paroxetine hydrochloride. Paroxetine mesylate was first approved in 2003 and is marketed as Pexeva; it is approved for major depressive disorder, obsessive compulsive disorder, panic disorder, and generalized anxiety disorder. As with all antidepressants, it has a boxed warning about the risk of suicidality and serotonin syndrome associated with treatment.
Noven refers to low-dose paroxetine as LDMP, for low-dose mesylate salt of paroxetine.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
SILVER SPRING, MD. – The majority of a Food and Drug Administration advisory panel recommended against approval of a low-dose formulation of paroxetine mesylate as a treatment for menopausal vasomotor symptoms, citing minimal benefits over placebo in clinical trials and the potential risks associated with treatment.
At the March 4 meeting, the FDA’s Reproductive Health Drugs Advisory Committee voted 10 to 4 that the overall risk-benefit profile of paroxetine did not support approval of the indication proposed by the manufacturer, Noven Therapeutics, as a treatment for moderate to severe vasomotor symptoms associated with menopause, at a dose of 7.5 mg once a day at bedtime.
The 7.5-mg dose is lower than the doses approved for psychiatric indications of paroxetine, which range from an initial dose of 10 mg to a maximum of 60 mg per day, depending on the indication.
Currently, paroxetine, a selective serotonin reuptake inhibitor (SSRI), is used off-label to treat vasomotor symptoms (VMS), and if approved, this could be the first nonhormonal treatment for VMS associated with menopause. Earlier in the day, the same panel voted against recommending approval for the antiepileptic drug gabapentin, another drug used off-label for VMS, for the same indication, citing the drug’s modest effects on VMS and its potential risks. In studies of both drugs, there was a marked placebo effect, which complicated the evaluations of the effectiveness.
In two pivotal studies conducted in the United States, 1,180 women (mean age, 54-55 years) with natural or surgical menopause who had at least 7 hot flashes per day or at least 50 per week were randomized to receive paroxetine or placebo, after a 12-week run-in period. Electronic diaries were used to document hot flashes. The primary endpoints were the mean change from baseline in hot flash frequency, and the mean change from baseline in hot flash severity, at weeks 4 and 12.
Those on paroxetine had statistically significant reductions in the daily number of hot flashes from baseline, compared with those on placebo at weeks 4 and 12 in both studies, and at week 24 in one of the two studies, which was a 24-week study. The reductions in the daily severity of hot flashes also were statistically significant at week 4 in both studies, and at week 12 in one study but not in the other study, according to the FDA.
However, the women on placebo had notable responses, not unexpected for a nonhormonal treatment, and the difference in the median number of hot flashes per day between the two groups was less than 2: In one study, the median number of hot flashes a day dropped by 4.29 by week 4 and 5.93 at week 12 among those on paroxetine, versus 3.13 and 5, respectively, among those on placebo. In the other study, the median number of hot flashes a day dropped by 3.79 at week 4 and 5.57 by week 12 among those on paroxetine, versus 2.5 and 3.86, respectively, among those on placebo.
The safety profile in the studies was consistent with the known safety profile of paroxetine, with no new or unexpected findings, according to Noven. Fatigue, nausea, and dizziness were the most common side effects in people treated with paroxetine, but they occurred mostly in the first 4 weeks of treatment and were mild to moderate Serious adverse events were higher among those on paroxetine (2.2% vs. 1.4%). In the 24-week study, there were three cases of suicidal ideation and one suicide attempt among those on paroxetine, all after 12 weeks of treatment. The one death in the studies was in a woman on paroxetine, but the death not considered related to the drug, according to the company.
Paroxetine was first approved in 1992 as paroxetine hydrochloride. Paroxetine mesylate was first approved in 2003 and is marketed as Pexeva; it is approved for major depressive disorder, obsessive compulsive disorder, panic disorder, and generalized anxiety disorder. As with all antidepressants, it has a boxed warning about the risk of suicidality and serotonin syndrome associated with treatment.
Noven refers to low-dose paroxetine as LDMP, for low-dose mesylate salt of paroxetine.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
psychiatric indications of paroxetine, selective serotonin reuptake inhibitor, SSRI,
psychiatric indications of paroxetine, selective serotonin reuptake inhibitor, SSRI,
FROM AN FDA ADVISORY COMMITTEE MEETING
FDA panel recommends against gabapentin's approval for hot flashes
SILVER SPRING, MD. – Women need a nonhormonal treatment for moderate to severe vasomotor symptoms in menopause. But gabapentin is not that drug, according to the majority of a Food and Drug Administration advisory panel, who recommended against approval of a sustained release formulation of the antiepileptic drug on March 3.
At the meeting, the FDA’s Reproductive Health Drugs Advisory Committee voted 12-2 that the risk-benefit profile of gabapentin did not support approval of the proposed indication, citing the limited, modest effect on vasomotor symptoms in three clinical trials. Also working against recommendation of approval were the known risks of the medication, particularly effects on cognition and other central nervous system effects. The sustained release formulation, manufactured by Depomed Inc., stays in the stomach for 8-10 hours, prolonging release of the drug. Were it to be approved despite the panel’s recommendation, it would be the first nonhormonal drug approved for treating vasomotor symptoms. The manufacturer recommends that the drug be dosed by titration to a total daily dose of 1,800 mg, with a 600-mg dose taken with the morning meal and 1,200 mg taken with the evening meal.
"We all recognize the critical need for nonhormonal medications [for vasomotor symptoms], and our patients have clearly been asking for such a product," said the panel chair Julia Johnson, professor and chair of the department of obstetrics and gynecology, University of Massachusetts, Worcester. "However, the risk of the medication cannot be ignored for a treatment with marginal effectiveness on hot flashes and gabapentin’s adverse event profile, she noted.
Another panelist, Kathryn Curtis, Ph.D., in the Centers for Disease Control and Prevention’s division of reproductive health, said that despite the "huge" need for a nonhormonal treatment for vasomotor symptoms, the drug had a very limited, modest effect and its approval would be "misleading" to women who need the treatment.
Depomed conducted three phase III studies of postmenopausal women (mean ages 53-54 years), who had at least 7 moderate to severe hot flashes per day or at least 50 per week during the previous 30 days. The studies compared gabapentin with placebo. There was a statistically significant difference in the frequency of hot flashes among those treated with 1,800 mg/day, compared with those on placebo at week 4, but not at week 12, in all three studies. Changes from baseline in the severity of hot flashes was statistically significant at week 4 in all three studies and remained statistically significant in two of the three studies at week 12. But while statically significant, the benefits over placebo were modest.
The most common adverse events associated with treatment were dizziness and vertigo, and somnolence and sedation, as expected.
Gabapentin is widely prescribed off-label for vasomotor symptoms. The Depomed formulation was approved in 2011 for postherpetic neuralgia and is marketed as Gralise for this indication. The labeling includes a warning about suicidality risk, a class labeling for antiepileptic drugs.
The immediate-release formulation of gabapentin (Neurontin) was first approved in 1993 as an adjunctive treatment of partial seizures, and has been used off label for vasomotor symptoms. Gabapentin encarbil, a prodrug of gabapentin, marketed as Horizant, was approved in 2011 for restless legs syndrome and postherpetic neuralgia.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
SILVER SPRING, MD. – Women need a nonhormonal treatment for moderate to severe vasomotor symptoms in menopause. But gabapentin is not that drug, according to the majority of a Food and Drug Administration advisory panel, who recommended against approval of a sustained release formulation of the antiepileptic drug on March 3.
At the meeting, the FDA’s Reproductive Health Drugs Advisory Committee voted 12-2 that the risk-benefit profile of gabapentin did not support approval of the proposed indication, citing the limited, modest effect on vasomotor symptoms in three clinical trials. Also working against recommendation of approval were the known risks of the medication, particularly effects on cognition and other central nervous system effects. The sustained release formulation, manufactured by Depomed Inc., stays in the stomach for 8-10 hours, prolonging release of the drug. Were it to be approved despite the panel’s recommendation, it would be the first nonhormonal drug approved for treating vasomotor symptoms. The manufacturer recommends that the drug be dosed by titration to a total daily dose of 1,800 mg, with a 600-mg dose taken with the morning meal and 1,200 mg taken with the evening meal.
"We all recognize the critical need for nonhormonal medications [for vasomotor symptoms], and our patients have clearly been asking for such a product," said the panel chair Julia Johnson, professor and chair of the department of obstetrics and gynecology, University of Massachusetts, Worcester. "However, the risk of the medication cannot be ignored for a treatment with marginal effectiveness on hot flashes and gabapentin’s adverse event profile, she noted.
Another panelist, Kathryn Curtis, Ph.D., in the Centers for Disease Control and Prevention’s division of reproductive health, said that despite the "huge" need for a nonhormonal treatment for vasomotor symptoms, the drug had a very limited, modest effect and its approval would be "misleading" to women who need the treatment.
Depomed conducted three phase III studies of postmenopausal women (mean ages 53-54 years), who had at least 7 moderate to severe hot flashes per day or at least 50 per week during the previous 30 days. The studies compared gabapentin with placebo. There was a statistically significant difference in the frequency of hot flashes among those treated with 1,800 mg/day, compared with those on placebo at week 4, but not at week 12, in all three studies. Changes from baseline in the severity of hot flashes was statistically significant at week 4 in all three studies and remained statistically significant in two of the three studies at week 12. But while statically significant, the benefits over placebo were modest.
The most common adverse events associated with treatment were dizziness and vertigo, and somnolence and sedation, as expected.
Gabapentin is widely prescribed off-label for vasomotor symptoms. The Depomed formulation was approved in 2011 for postherpetic neuralgia and is marketed as Gralise for this indication. The labeling includes a warning about suicidality risk, a class labeling for antiepileptic drugs.
The immediate-release formulation of gabapentin (Neurontin) was first approved in 1993 as an adjunctive treatment of partial seizures, and has been used off label for vasomotor symptoms. Gabapentin encarbil, a prodrug of gabapentin, marketed as Horizant, was approved in 2011 for restless legs syndrome and postherpetic neuralgia.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
SILVER SPRING, MD. – Women need a nonhormonal treatment for moderate to severe vasomotor symptoms in menopause. But gabapentin is not that drug, according to the majority of a Food and Drug Administration advisory panel, who recommended against approval of a sustained release formulation of the antiepileptic drug on March 3.
At the meeting, the FDA’s Reproductive Health Drugs Advisory Committee voted 12-2 that the risk-benefit profile of gabapentin did not support approval of the proposed indication, citing the limited, modest effect on vasomotor symptoms in three clinical trials. Also working against recommendation of approval were the known risks of the medication, particularly effects on cognition and other central nervous system effects. The sustained release formulation, manufactured by Depomed Inc., stays in the stomach for 8-10 hours, prolonging release of the drug. Were it to be approved despite the panel’s recommendation, it would be the first nonhormonal drug approved for treating vasomotor symptoms. The manufacturer recommends that the drug be dosed by titration to a total daily dose of 1,800 mg, with a 600-mg dose taken with the morning meal and 1,200 mg taken with the evening meal.
"We all recognize the critical need for nonhormonal medications [for vasomotor symptoms], and our patients have clearly been asking for such a product," said the panel chair Julia Johnson, professor and chair of the department of obstetrics and gynecology, University of Massachusetts, Worcester. "However, the risk of the medication cannot be ignored for a treatment with marginal effectiveness on hot flashes and gabapentin’s adverse event profile, she noted.
Another panelist, Kathryn Curtis, Ph.D., in the Centers for Disease Control and Prevention’s division of reproductive health, said that despite the "huge" need for a nonhormonal treatment for vasomotor symptoms, the drug had a very limited, modest effect and its approval would be "misleading" to women who need the treatment.
Depomed conducted three phase III studies of postmenopausal women (mean ages 53-54 years), who had at least 7 moderate to severe hot flashes per day or at least 50 per week during the previous 30 days. The studies compared gabapentin with placebo. There was a statistically significant difference in the frequency of hot flashes among those treated with 1,800 mg/day, compared with those on placebo at week 4, but not at week 12, in all three studies. Changes from baseline in the severity of hot flashes was statistically significant at week 4 in all three studies and remained statistically significant in two of the three studies at week 12. But while statically significant, the benefits over placebo were modest.
The most common adverse events associated with treatment were dizziness and vertigo, and somnolence and sedation, as expected.
Gabapentin is widely prescribed off-label for vasomotor symptoms. The Depomed formulation was approved in 2011 for postherpetic neuralgia and is marketed as Gralise for this indication. The labeling includes a warning about suicidality risk, a class labeling for antiepileptic drugs.
The immediate-release formulation of gabapentin (Neurontin) was first approved in 1993 as an adjunctive treatment of partial seizures, and has been used off label for vasomotor symptoms. Gabapentin encarbil, a prodrug of gabapentin, marketed as Horizant, was approved in 2011 for restless legs syndrome and postherpetic neuralgia.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
FROM AN FDA ADVISORY COMMITTEE MEETING
Everolimus approval now includes prevention of liver transplant rejection
Food and Drug Administration approval of the immunosuppressant drug everolimus has been expanded to include prophylaxis of organ rejection in adults undergoing a liver transplant.
The approval, on Feb. 15, was announced by the manufacturer, Novartis. Everolimus, a mammalian target of rapamycin (mTOR) inhibitor, was first approved for prophylaxis of organ rejection in adults undergoing a kidney transplant in 2010, and is taken twice a day by mouth.
The approved indication states that in liver transplant recipients, everolimus is used in combination with the calcineurin inhibitor tacrolimus (at a reduced dose) and corticosteroids, and that it should be administered no earlier than 30 days post transplant. Safety and efficacy have not been established in pediatric patients, according to the prescribing information.
FDA approval was based on the 12-month results of a phase III, open-label international study of 719 liver transplant recipients (mean age, 54 years). At 12 months, the rate of the "efficacy failure" endpoint (defined as biopsy-proven acute rejection, graft loss, death, or loss to follow-up) was "comparable" between those patients treated with the reduced dose of everolimus, started 30 days after transplantation (9%), and the patients treated with the standard dose of tacrolimus, started 30 days after transplantation (13.6%), according to the Novartis statement and prescribing information. All patients were treated with corticosteroids.
Everolimus is the first mTOR inhibitor approved for liver transplant recipients, and is the first immunosuppressant approved by the FDA for use in liver transplant recipients in more than 10 years, according to Novartis.
Novartis markets everolimus in the United States as Zortress. It was approved in the European Union in the fourth quarter of 2012 for adult liver transplant recipients; it is marketed as Certican in Europe.
The new label is available here. Serious adverse events associated with everolimus should be reported to the FDA’s MedWatch program at 800-332-1088 or www.fda.gov/medwatch/.
Food and Drug Administration approval of the immunosuppressant drug everolimus has been expanded to include prophylaxis of organ rejection in adults undergoing a liver transplant.
The approval, on Feb. 15, was announced by the manufacturer, Novartis. Everolimus, a mammalian target of rapamycin (mTOR) inhibitor, was first approved for prophylaxis of organ rejection in adults undergoing a kidney transplant in 2010, and is taken twice a day by mouth.
The approved indication states that in liver transplant recipients, everolimus is used in combination with the calcineurin inhibitor tacrolimus (at a reduced dose) and corticosteroids, and that it should be administered no earlier than 30 days post transplant. Safety and efficacy have not been established in pediatric patients, according to the prescribing information.
FDA approval was based on the 12-month results of a phase III, open-label international study of 719 liver transplant recipients (mean age, 54 years). At 12 months, the rate of the "efficacy failure" endpoint (defined as biopsy-proven acute rejection, graft loss, death, or loss to follow-up) was "comparable" between those patients treated with the reduced dose of everolimus, started 30 days after transplantation (9%), and the patients treated with the standard dose of tacrolimus, started 30 days after transplantation (13.6%), according to the Novartis statement and prescribing information. All patients were treated with corticosteroids.
Everolimus is the first mTOR inhibitor approved for liver transplant recipients, and is the first immunosuppressant approved by the FDA for use in liver transplant recipients in more than 10 years, according to Novartis.
Novartis markets everolimus in the United States as Zortress. It was approved in the European Union in the fourth quarter of 2012 for adult liver transplant recipients; it is marketed as Certican in Europe.
The new label is available here. Serious adverse events associated with everolimus should be reported to the FDA’s MedWatch program at 800-332-1088 or www.fda.gov/medwatch/.
Food and Drug Administration approval of the immunosuppressant drug everolimus has been expanded to include prophylaxis of organ rejection in adults undergoing a liver transplant.
The approval, on Feb. 15, was announced by the manufacturer, Novartis. Everolimus, a mammalian target of rapamycin (mTOR) inhibitor, was first approved for prophylaxis of organ rejection in adults undergoing a kidney transplant in 2010, and is taken twice a day by mouth.
The approved indication states that in liver transplant recipients, everolimus is used in combination with the calcineurin inhibitor tacrolimus (at a reduced dose) and corticosteroids, and that it should be administered no earlier than 30 days post transplant. Safety and efficacy have not been established in pediatric patients, according to the prescribing information.
FDA approval was based on the 12-month results of a phase III, open-label international study of 719 liver transplant recipients (mean age, 54 years). At 12 months, the rate of the "efficacy failure" endpoint (defined as biopsy-proven acute rejection, graft loss, death, or loss to follow-up) was "comparable" between those patients treated with the reduced dose of everolimus, started 30 days after transplantation (9%), and the patients treated with the standard dose of tacrolimus, started 30 days after transplantation (13.6%), according to the Novartis statement and prescribing information. All patients were treated with corticosteroids.
Everolimus is the first mTOR inhibitor approved for liver transplant recipients, and is the first immunosuppressant approved by the FDA for use in liver transplant recipients in more than 10 years, according to Novartis.
Novartis markets everolimus in the United States as Zortress. It was approved in the European Union in the fourth quarter of 2012 for adult liver transplant recipients; it is marketed as Certican in Europe.
The new label is available here. Serious adverse events associated with everolimus should be reported to the FDA’s MedWatch program at 800-332-1088 or www.fda.gov/medwatch/.
FDA panel picks strains for next flu season's vaccine
The trivalent influenza vaccine for the 2013-2014 season in the United States should retain one of the two influenza A strains included in the current vaccine, but the second influenza A strain and the influenza B strain should be replaced, a Food and Drug Administration expert panel agreed on Feb. 27.
At a meeting of the FDA’s Vaccines and Related Biological Products Advisory Committee, members voted 17-0 that next season’s influenza vaccine should retain the current influenza A (H1N1) strain, an A/California/7/2009 (H1N1)-like virus. They also voted 16-0 with one abstention to replace the influenza A (H3N2) component with an A (H3N2) virus described as "antigenically like the cell-propagated prototype virus A/Victoria/361/2011."
For the influenza B component, the panel voted 17-0 to replace the current strain with another B/Yamagata lineage strain, a B/Massachusetts/2/2012-like virus.
And since quadrivalent influenza vaccines will be available for the first time for the coming season, the panel selected the B/Victoria lineage strain (a B/Brisbane/60/2008-like virus) included in recent vaccines as the second B strain. That vote was 16-0, with one abstention.
The FDA panel meets at the same time each year to review influenza surveillance and epidemiology data in North America and globally, antigenic characteristics of influenza virus strains currently circulating in human populations, and serologic responses to circulating influenza viruses of people immunized with the current approved vaccine.
The panel’s recommendations echo those of the World Health Organization, which were made earlier in February.
The coming flu season will be the first time a quadrivalent vaccine is available. MedImmune will supply only the quadrivalent form of FluMist for the 2013-2014 season, discontinuing the trivalent form, according to a representative.
GlaxoSmithKline will manufacture both the trivalent and quadrivalent forms of Fluarix for next season, a company representative said.
Dr. David Greenberg of Sanofi Pasteur said that the company expects its quadrivalent influenza vaccine, currently being reviewed by the FDA, to be licensed in the summer. The company plans to launch a limited supply for the next season, but most of the GSK 2013-2014 vaccine will be the trivalent version, he added.
The current influenza season started about 4 weeks earlier than usual in the United States with "moderately high" activity. Activity increased in late November and peaked in late December, according to Dr. Lynn Finelli of the influenza surveillance and outbreak response team at the Centers for Disease Control and Prevention.
At the meeting, Dr. Finelli said that influenza activity has dropped in most parts of the United States, with moderate activity continuing only in parts of the West. As of Feb. 16, a total of 78 influenza-associated pediatric deaths had been reported. In comparison, 34 pediatric deaths were reported in the 2011-2012 season, 122 during 2010-2011, and 282 in 2009-2010.
Dr. Finelli also presented data that indicate suboptimal vaccine effectiveness against A (H3N2) among adults aged 65 years and older – only about 9%, compared with 46%-58% for younger age groups.
Her presentation led to a discussion of why the influenza vaccine is less effective in the older population, including whether the high-dose version of Fluzone, licensed for people 65 years and older, was more effective. High-dose Fluzone was licensed as an accelerated approval, and a trial to confirm its clinical benefit in the indicated age group is underway.
Sanofi Pasteur’s Dr. Greenberg, who was in the audience, said that the results of the study, which compares high-dose Fluzone to Fluzone, are expected in 2014 or 2015.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
The trivalent influenza vaccine for the 2013-2014 season in the United States should retain one of the two influenza A strains included in the current vaccine, but the second influenza A strain and the influenza B strain should be replaced, a Food and Drug Administration expert panel agreed on Feb. 27.
At a meeting of the FDA’s Vaccines and Related Biological Products Advisory Committee, members voted 17-0 that next season’s influenza vaccine should retain the current influenza A (H1N1) strain, an A/California/7/2009 (H1N1)-like virus. They also voted 16-0 with one abstention to replace the influenza A (H3N2) component with an A (H3N2) virus described as "antigenically like the cell-propagated prototype virus A/Victoria/361/2011."
For the influenza B component, the panel voted 17-0 to replace the current strain with another B/Yamagata lineage strain, a B/Massachusetts/2/2012-like virus.
And since quadrivalent influenza vaccines will be available for the first time for the coming season, the panel selected the B/Victoria lineage strain (a B/Brisbane/60/2008-like virus) included in recent vaccines as the second B strain. That vote was 16-0, with one abstention.
The FDA panel meets at the same time each year to review influenza surveillance and epidemiology data in North America and globally, antigenic characteristics of influenza virus strains currently circulating in human populations, and serologic responses to circulating influenza viruses of people immunized with the current approved vaccine.
The panel’s recommendations echo those of the World Health Organization, which were made earlier in February.
The coming flu season will be the first time a quadrivalent vaccine is available. MedImmune will supply only the quadrivalent form of FluMist for the 2013-2014 season, discontinuing the trivalent form, according to a representative.
GlaxoSmithKline will manufacture both the trivalent and quadrivalent forms of Fluarix for next season, a company representative said.
Dr. David Greenberg of Sanofi Pasteur said that the company expects its quadrivalent influenza vaccine, currently being reviewed by the FDA, to be licensed in the summer. The company plans to launch a limited supply for the next season, but most of the GSK 2013-2014 vaccine will be the trivalent version, he added.
The current influenza season started about 4 weeks earlier than usual in the United States with "moderately high" activity. Activity increased in late November and peaked in late December, according to Dr. Lynn Finelli of the influenza surveillance and outbreak response team at the Centers for Disease Control and Prevention.
At the meeting, Dr. Finelli said that influenza activity has dropped in most parts of the United States, with moderate activity continuing only in parts of the West. As of Feb. 16, a total of 78 influenza-associated pediatric deaths had been reported. In comparison, 34 pediatric deaths were reported in the 2011-2012 season, 122 during 2010-2011, and 282 in 2009-2010.
Dr. Finelli also presented data that indicate suboptimal vaccine effectiveness against A (H3N2) among adults aged 65 years and older – only about 9%, compared with 46%-58% for younger age groups.
Her presentation led to a discussion of why the influenza vaccine is less effective in the older population, including whether the high-dose version of Fluzone, licensed for people 65 years and older, was more effective. High-dose Fluzone was licensed as an accelerated approval, and a trial to confirm its clinical benefit in the indicated age group is underway.
Sanofi Pasteur’s Dr. Greenberg, who was in the audience, said that the results of the study, which compares high-dose Fluzone to Fluzone, are expected in 2014 or 2015.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
The trivalent influenza vaccine for the 2013-2014 season in the United States should retain one of the two influenza A strains included in the current vaccine, but the second influenza A strain and the influenza B strain should be replaced, a Food and Drug Administration expert panel agreed on Feb. 27.
At a meeting of the FDA’s Vaccines and Related Biological Products Advisory Committee, members voted 17-0 that next season’s influenza vaccine should retain the current influenza A (H1N1) strain, an A/California/7/2009 (H1N1)-like virus. They also voted 16-0 with one abstention to replace the influenza A (H3N2) component with an A (H3N2) virus described as "antigenically like the cell-propagated prototype virus A/Victoria/361/2011."
For the influenza B component, the panel voted 17-0 to replace the current strain with another B/Yamagata lineage strain, a B/Massachusetts/2/2012-like virus.
And since quadrivalent influenza vaccines will be available for the first time for the coming season, the panel selected the B/Victoria lineage strain (a B/Brisbane/60/2008-like virus) included in recent vaccines as the second B strain. That vote was 16-0, with one abstention.
The FDA panel meets at the same time each year to review influenza surveillance and epidemiology data in North America and globally, antigenic characteristics of influenza virus strains currently circulating in human populations, and serologic responses to circulating influenza viruses of people immunized with the current approved vaccine.
The panel’s recommendations echo those of the World Health Organization, which were made earlier in February.
The coming flu season will be the first time a quadrivalent vaccine is available. MedImmune will supply only the quadrivalent form of FluMist for the 2013-2014 season, discontinuing the trivalent form, according to a representative.
GlaxoSmithKline will manufacture both the trivalent and quadrivalent forms of Fluarix for next season, a company representative said.
Dr. David Greenberg of Sanofi Pasteur said that the company expects its quadrivalent influenza vaccine, currently being reviewed by the FDA, to be licensed in the summer. The company plans to launch a limited supply for the next season, but most of the GSK 2013-2014 vaccine will be the trivalent version, he added.
The current influenza season started about 4 weeks earlier than usual in the United States with "moderately high" activity. Activity increased in late November and peaked in late December, according to Dr. Lynn Finelli of the influenza surveillance and outbreak response team at the Centers for Disease Control and Prevention.
At the meeting, Dr. Finelli said that influenza activity has dropped in most parts of the United States, with moderate activity continuing only in parts of the West. As of Feb. 16, a total of 78 influenza-associated pediatric deaths had been reported. In comparison, 34 pediatric deaths were reported in the 2011-2012 season, 122 during 2010-2011, and 282 in 2009-2010.
Dr. Finelli also presented data that indicate suboptimal vaccine effectiveness against A (H3N2) among adults aged 65 years and older – only about 9%, compared with 46%-58% for younger age groups.
Her presentation led to a discussion of why the influenza vaccine is less effective in the older population, including whether the high-dose version of Fluzone, licensed for people 65 years and older, was more effective. High-dose Fluzone was licensed as an accelerated approval, and a trial to confirm its clinical benefit in the indicated age group is underway.
Sanofi Pasteur’s Dr. Greenberg, who was in the audience, said that the results of the study, which compares high-dose Fluzone to Fluzone, are expected in 2014 or 2015.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
AT AN FDA ADVISORY COMMITTEE MEETING
Meta-analysis bolsters strength of tuberculosis assay
The sensitivity and specificity of a diagnostic test that rapidly detects tuberculosis support its use as an initial diagnostic test in adults suspected of having TB, including multidrug- resistant or HIV-associated disease, based on a meta-analysis of studies evaluating the test, researchers say.
In addition, the meta-analysis results suggest that the test, the Xpert assay, "may also be valuable as an add-on test following a negative smear microscopy result in patients suspected of having TB," said Karen Steingart, of the University of Washington School of Public Health, Seattle, and her associates. The researchers were from the Cochrane Infectious Diseases Group; McGill University, Montreal; and the Foundation for Innovative New Diagnostics (FIND), Geneva. The study was published in the Cochrane Database of Systematic Reviews (doi:10.1002/14651858.CD009593.pub2).
Xpert, manufactured by Cepheid Inc., is an automated polymerase chain reaction test that detects Mycobacterium tuberculosis complex and rifampin resistance within two hours using a sputum sample, used outside of a reference lab "with minimal hands-on technical time," as opposed to conventional sputum smear microscopy, which is cheaper, but requires a microscope in a lab and does not detect drug resistance.
In 2010, Xpert was endorsed by the World Health Organization for use as the initial diagnostic test on people suspected of having MDR-TB or HIV-associated TB. That year, WHO reported that there were 8.8 million new cases of TB worldwide; and that 1.1 million people who did not have HIV and 350,000 people with HIV died of TB.
The study reviewed 18 studies of more than 7,500 adults, mostly in low and middle-income countries, with a high burden of TB, which evaluated the test as an initial test used as a replacement for microscopy, and as an add-on test when a microscopy smear was negative, in adults suspected of having in pulmonary TB, or multidrug-resistant TB, with or without HIV infection.
The main results of the test are as follows:
• A "modest" sensitivity of 88% and a "high" specificity of 98% for detecting TB when used it as an initial test that replaced smear microscopy.
• A sensitivity of 67% and a specificity of 98% when used as an add-on follow-up test to detect TB, after a negative smear microscopy.
• A sensitivity of 80% for detecting TB in people with HIV and 89% in people without HIV.
•A sensitivity of 94% and a specificity of 98% for detecting rifampin resistance (an indicator of multidrug resistances) when used as a replacement for culture based-drug susceptibility testing
The rate of indeterminate results was a "very low" 1%, they said.
The results "lend support to the WHO recommendation on the use of Xpert as an initial diagnostic test for TB detection and rifampicin resistance detection in patients suspected of having MDR-TB or HIV-associated TB," the authors concluded. Future studies should include those that evaluate the results of the test in children, as those data are starting to accumulate, and result of the test in different settings, such as TB screening centers and antiretroviral clinics, they added.
The review "provides high quality evidence that reinforces WHO’s endorsement of this test," Dr. Karin Weyer, the coordinator of Diagnostics and Drug Resistance Laboratories at WHO, said in a written statement. "Recent price reductions have greatly facilitated roll-out of this technology," with 1.4 million test cartridges and the instruments needed to perform the test to be distributed in 21 countries with a high TB burden, she added.
In the United States, TB has been declining, since a peak in 1992, with 10,528 TB cases reported in 2011, almost a 6% drop from the number of reports in 2010, according to the Centers for Disease Control and Prevention. In 2009, the last year mortality data are available, there were 529 TB deaths, a 10% drop from 2008, when there were 590 deaths.
The editorial base of the Cochrane Infectious Diseases Group (CIDG), which provided part of the funding for the review, is funded by the U.K. Department for International Development "for the benefit of developing countries." One study author disclosed having conducted studies and published on the Xpert test in a project with FIND, Cepheid, and academic partners. Others reported having no relevant financial conflicts.
The sensitivity and specificity of a diagnostic test that rapidly detects tuberculosis support its use as an initial diagnostic test in adults suspected of having TB, including multidrug- resistant or HIV-associated disease, based on a meta-analysis of studies evaluating the test, researchers say.
In addition, the meta-analysis results suggest that the test, the Xpert assay, "may also be valuable as an add-on test following a negative smear microscopy result in patients suspected of having TB," said Karen Steingart, of the University of Washington School of Public Health, Seattle, and her associates. The researchers were from the Cochrane Infectious Diseases Group; McGill University, Montreal; and the Foundation for Innovative New Diagnostics (FIND), Geneva. The study was published in the Cochrane Database of Systematic Reviews (doi:10.1002/14651858.CD009593.pub2).
Xpert, manufactured by Cepheid Inc., is an automated polymerase chain reaction test that detects Mycobacterium tuberculosis complex and rifampin resistance within two hours using a sputum sample, used outside of a reference lab "with minimal hands-on technical time," as opposed to conventional sputum smear microscopy, which is cheaper, but requires a microscope in a lab and does not detect drug resistance.
In 2010, Xpert was endorsed by the World Health Organization for use as the initial diagnostic test on people suspected of having MDR-TB or HIV-associated TB. That year, WHO reported that there were 8.8 million new cases of TB worldwide; and that 1.1 million people who did not have HIV and 350,000 people with HIV died of TB.
The study reviewed 18 studies of more than 7,500 adults, mostly in low and middle-income countries, with a high burden of TB, which evaluated the test as an initial test used as a replacement for microscopy, and as an add-on test when a microscopy smear was negative, in adults suspected of having in pulmonary TB, or multidrug-resistant TB, with or without HIV infection.
The main results of the test are as follows:
• A "modest" sensitivity of 88% and a "high" specificity of 98% for detecting TB when used it as an initial test that replaced smear microscopy.
• A sensitivity of 67% and a specificity of 98% when used as an add-on follow-up test to detect TB, after a negative smear microscopy.
• A sensitivity of 80% for detecting TB in people with HIV and 89% in people without HIV.
•A sensitivity of 94% and a specificity of 98% for detecting rifampin resistance (an indicator of multidrug resistances) when used as a replacement for culture based-drug susceptibility testing
The rate of indeterminate results was a "very low" 1%, they said.
The results "lend support to the WHO recommendation on the use of Xpert as an initial diagnostic test for TB detection and rifampicin resistance detection in patients suspected of having MDR-TB or HIV-associated TB," the authors concluded. Future studies should include those that evaluate the results of the test in children, as those data are starting to accumulate, and result of the test in different settings, such as TB screening centers and antiretroviral clinics, they added.
The review "provides high quality evidence that reinforces WHO’s endorsement of this test," Dr. Karin Weyer, the coordinator of Diagnostics and Drug Resistance Laboratories at WHO, said in a written statement. "Recent price reductions have greatly facilitated roll-out of this technology," with 1.4 million test cartridges and the instruments needed to perform the test to be distributed in 21 countries with a high TB burden, she added.
In the United States, TB has been declining, since a peak in 1992, with 10,528 TB cases reported in 2011, almost a 6% drop from the number of reports in 2010, according to the Centers for Disease Control and Prevention. In 2009, the last year mortality data are available, there were 529 TB deaths, a 10% drop from 2008, when there were 590 deaths.
The editorial base of the Cochrane Infectious Diseases Group (CIDG), which provided part of the funding for the review, is funded by the U.K. Department for International Development "for the benefit of developing countries." One study author disclosed having conducted studies and published on the Xpert test in a project with FIND, Cepheid, and academic partners. Others reported having no relevant financial conflicts.
The sensitivity and specificity of a diagnostic test that rapidly detects tuberculosis support its use as an initial diagnostic test in adults suspected of having TB, including multidrug- resistant or HIV-associated disease, based on a meta-analysis of studies evaluating the test, researchers say.
In addition, the meta-analysis results suggest that the test, the Xpert assay, "may also be valuable as an add-on test following a negative smear microscopy result in patients suspected of having TB," said Karen Steingart, of the University of Washington School of Public Health, Seattle, and her associates. The researchers were from the Cochrane Infectious Diseases Group; McGill University, Montreal; and the Foundation for Innovative New Diagnostics (FIND), Geneva. The study was published in the Cochrane Database of Systematic Reviews (doi:10.1002/14651858.CD009593.pub2).
Xpert, manufactured by Cepheid Inc., is an automated polymerase chain reaction test that detects Mycobacterium tuberculosis complex and rifampin resistance within two hours using a sputum sample, used outside of a reference lab "with minimal hands-on technical time," as opposed to conventional sputum smear microscopy, which is cheaper, but requires a microscope in a lab and does not detect drug resistance.
In 2010, Xpert was endorsed by the World Health Organization for use as the initial diagnostic test on people suspected of having MDR-TB or HIV-associated TB. That year, WHO reported that there were 8.8 million new cases of TB worldwide; and that 1.1 million people who did not have HIV and 350,000 people with HIV died of TB.
The study reviewed 18 studies of more than 7,500 adults, mostly in low and middle-income countries, with a high burden of TB, which evaluated the test as an initial test used as a replacement for microscopy, and as an add-on test when a microscopy smear was negative, in adults suspected of having in pulmonary TB, or multidrug-resistant TB, with or without HIV infection.
The main results of the test are as follows:
• A "modest" sensitivity of 88% and a "high" specificity of 98% for detecting TB when used it as an initial test that replaced smear microscopy.
• A sensitivity of 67% and a specificity of 98% when used as an add-on follow-up test to detect TB, after a negative smear microscopy.
• A sensitivity of 80% for detecting TB in people with HIV and 89% in people without HIV.
•A sensitivity of 94% and a specificity of 98% for detecting rifampin resistance (an indicator of multidrug resistances) when used as a replacement for culture based-drug susceptibility testing
The rate of indeterminate results was a "very low" 1%, they said.
The results "lend support to the WHO recommendation on the use of Xpert as an initial diagnostic test for TB detection and rifampicin resistance detection in patients suspected of having MDR-TB or HIV-associated TB," the authors concluded. Future studies should include those that evaluate the results of the test in children, as those data are starting to accumulate, and result of the test in different settings, such as TB screening centers and antiretroviral clinics, they added.
The review "provides high quality evidence that reinforces WHO’s endorsement of this test," Dr. Karin Weyer, the coordinator of Diagnostics and Drug Resistance Laboratories at WHO, said in a written statement. "Recent price reductions have greatly facilitated roll-out of this technology," with 1.4 million test cartridges and the instruments needed to perform the test to be distributed in 21 countries with a high TB burden, she added.
In the United States, TB has been declining, since a peak in 1992, with 10,528 TB cases reported in 2011, almost a 6% drop from the number of reports in 2010, according to the Centers for Disease Control and Prevention. In 2009, the last year mortality data are available, there were 529 TB deaths, a 10% drop from 2008, when there were 590 deaths.
The editorial base of the Cochrane Infectious Diseases Group (CIDG), which provided part of the funding for the review, is funded by the U.K. Department for International Development "for the benefit of developing countries." One study author disclosed having conducted studies and published on the Xpert test in a project with FIND, Cepheid, and academic partners. Others reported having no relevant financial conflicts.
FROM THE COCHRANE LIBRARY
Major Finding: A rapid automated polymerase chain reaction test that detects Mycobacterium tuberculosis complex and rifampin resistance using a sputum sample had a "modest" sensitivity of 88% and a "high" specificity of 98% for detecting tuberculosis when used it as an initial test that replaced smear microscopy.
Data Source: A meta-analysis of 18 studies of more than 7,500 adults.
Disclosures: The editorial base of the Cochrane Infectious Diseases Group, funded by the U.K. Department for International Development, provided part of the funding. The authors had no financial disclosures.
Third drug approved for metastatic, treatment-resistant GIST
Regorafenib, a multikinase inhibitor, has been approved as a treatment for locally advanced, unresectable, or metastatic gastrointestinal stromal tumor in people who have been treated with imatinib and sunitinib, the other two treatments approved for GIST, the Food and Drug Administration announced on Feb. 26.
Regorafenib was first approved in September as a treatment for metastatic colorectal cancer, and "provides an important new treatment option for patients with GIST in which other approved drugs are no longer effective," Dr. Richard Pazdur, director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, said in the statement. The recommended dose is 160 mg orally once a day for the first 21 days of each 28-day cycle, according to the prescribing information for regorafenib, which is marketed as Stivarga by Bayer HealthCare Pharmaceuticals.
Approval was based on the interim results of the phase III GRID (GIST – Regorafenib In Progressive Disease) study, comparing placebo plus best supportive care (BSC) to regorafenib plus BSC in 199 patients with locally advanced, unresectable, or metastatic GIST, previously treated with imatinib and sunitinib, according to the FDA statement, as well as the statement issued by the manufacturer. The median progression-free survival (the primary endpoint) was 4.8 months among those on regorafenib, compared with 0.8 months among those on placebo, a statistically significant difference (Lancet 381;9863:295-302). At the time of the planned interim analysis, there was no statistically significant difference in overall survival.
The most common adverse events associated with treatment, reported by at least 30% of those treated, included hand-foot syndrome, diarrhea, mucositis, dysphonia, asthenia/fatigue, hypertension, reduced appetite and food intake, and rash. Serious adverse events, affecting less than 1% of patients, included hepatotoxicity, severe bleeding, blistering and peeling of skin, very high blood pressures requiring emergency treatment, heart attacks, and intestinal perforations. The regorafenib label includes a boxed warning about the risk of hepatotoxicity associated with treatment, noting that severe and sometimes fatal hepatotoxicity has been reported in clinical trials, and that hepatic function should be monitored before and during treatment.
Regorafenib inhibits multiple kinases that are involved in normal cellular functions, as well as oncogenesis, tumor angiogenesis, and maintenance of the tumor microenvironment, according to the manufacturer.
Regorafenib was the focus of the FDA’s priority review program, which evaluates the drug in 6 months instead of the usual 12 months, and is designated for products "that may provide safe and effective therapy when no satisfactory alternative therapy exists, or offer significant improvement compared to marketed products," according to the FDA statement.
The FDA cites a National Cancer Institute estimate that 3,300-6,000 new cases of GIST are diagnosed every year in the United States, affecting mostly older adults. The previously approved colorectal cancer indication is for people who have metastatic colorectal cancer, who have been previously treated with fluoropyrimidine-, oxaliplatin- and irinotecan-based chemotherapy, an anti-VEGF therapy, and, if KRAS wild type, an anti-EGFR therapy.
Imatinib (Gleevec) and sunitinib (Sutent) are both orally administered kinase inhibitors.
Regorafenib, a multikinase inhibitor, has been approved as a treatment for locally advanced, unresectable, or metastatic gastrointestinal stromal tumor in people who have been treated with imatinib and sunitinib, the other two treatments approved for GIST, the Food and Drug Administration announced on Feb. 26.
Regorafenib was first approved in September as a treatment for metastatic colorectal cancer, and "provides an important new treatment option for patients with GIST in which other approved drugs are no longer effective," Dr. Richard Pazdur, director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, said in the statement. The recommended dose is 160 mg orally once a day for the first 21 days of each 28-day cycle, according to the prescribing information for regorafenib, which is marketed as Stivarga by Bayer HealthCare Pharmaceuticals.
Approval was based on the interim results of the phase III GRID (GIST – Regorafenib In Progressive Disease) study, comparing placebo plus best supportive care (BSC) to regorafenib plus BSC in 199 patients with locally advanced, unresectable, or metastatic GIST, previously treated with imatinib and sunitinib, according to the FDA statement, as well as the statement issued by the manufacturer. The median progression-free survival (the primary endpoint) was 4.8 months among those on regorafenib, compared with 0.8 months among those on placebo, a statistically significant difference (Lancet 381;9863:295-302). At the time of the planned interim analysis, there was no statistically significant difference in overall survival.
The most common adverse events associated with treatment, reported by at least 30% of those treated, included hand-foot syndrome, diarrhea, mucositis, dysphonia, asthenia/fatigue, hypertension, reduced appetite and food intake, and rash. Serious adverse events, affecting less than 1% of patients, included hepatotoxicity, severe bleeding, blistering and peeling of skin, very high blood pressures requiring emergency treatment, heart attacks, and intestinal perforations. The regorafenib label includes a boxed warning about the risk of hepatotoxicity associated with treatment, noting that severe and sometimes fatal hepatotoxicity has been reported in clinical trials, and that hepatic function should be monitored before and during treatment.
Regorafenib inhibits multiple kinases that are involved in normal cellular functions, as well as oncogenesis, tumor angiogenesis, and maintenance of the tumor microenvironment, according to the manufacturer.
Regorafenib was the focus of the FDA’s priority review program, which evaluates the drug in 6 months instead of the usual 12 months, and is designated for products "that may provide safe and effective therapy when no satisfactory alternative therapy exists, or offer significant improvement compared to marketed products," according to the FDA statement.
The FDA cites a National Cancer Institute estimate that 3,300-6,000 new cases of GIST are diagnosed every year in the United States, affecting mostly older adults. The previously approved colorectal cancer indication is for people who have metastatic colorectal cancer, who have been previously treated with fluoropyrimidine-, oxaliplatin- and irinotecan-based chemotherapy, an anti-VEGF therapy, and, if KRAS wild type, an anti-EGFR therapy.
Imatinib (Gleevec) and sunitinib (Sutent) are both orally administered kinase inhibitors.
Regorafenib, a multikinase inhibitor, has been approved as a treatment for locally advanced, unresectable, or metastatic gastrointestinal stromal tumor in people who have been treated with imatinib and sunitinib, the other two treatments approved for GIST, the Food and Drug Administration announced on Feb. 26.
Regorafenib was first approved in September as a treatment for metastatic colorectal cancer, and "provides an important new treatment option for patients with GIST in which other approved drugs are no longer effective," Dr. Richard Pazdur, director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, said in the statement. The recommended dose is 160 mg orally once a day for the first 21 days of each 28-day cycle, according to the prescribing information for regorafenib, which is marketed as Stivarga by Bayer HealthCare Pharmaceuticals.
Approval was based on the interim results of the phase III GRID (GIST – Regorafenib In Progressive Disease) study, comparing placebo plus best supportive care (BSC) to regorafenib plus BSC in 199 patients with locally advanced, unresectable, or metastatic GIST, previously treated with imatinib and sunitinib, according to the FDA statement, as well as the statement issued by the manufacturer. The median progression-free survival (the primary endpoint) was 4.8 months among those on regorafenib, compared with 0.8 months among those on placebo, a statistically significant difference (Lancet 381;9863:295-302). At the time of the planned interim analysis, there was no statistically significant difference in overall survival.
The most common adverse events associated with treatment, reported by at least 30% of those treated, included hand-foot syndrome, diarrhea, mucositis, dysphonia, asthenia/fatigue, hypertension, reduced appetite and food intake, and rash. Serious adverse events, affecting less than 1% of patients, included hepatotoxicity, severe bleeding, blistering and peeling of skin, very high blood pressures requiring emergency treatment, heart attacks, and intestinal perforations. The regorafenib label includes a boxed warning about the risk of hepatotoxicity associated with treatment, noting that severe and sometimes fatal hepatotoxicity has been reported in clinical trials, and that hepatic function should be monitored before and during treatment.
Regorafenib inhibits multiple kinases that are involved in normal cellular functions, as well as oncogenesis, tumor angiogenesis, and maintenance of the tumor microenvironment, according to the manufacturer.
Regorafenib was the focus of the FDA’s priority review program, which evaluates the drug in 6 months instead of the usual 12 months, and is designated for products "that may provide safe and effective therapy when no satisfactory alternative therapy exists, or offer significant improvement compared to marketed products," according to the FDA statement.
The FDA cites a National Cancer Institute estimate that 3,300-6,000 new cases of GIST are diagnosed every year in the United States, affecting mostly older adults. The previously approved colorectal cancer indication is for people who have metastatic colorectal cancer, who have been previously treated with fluoropyrimidine-, oxaliplatin- and irinotecan-based chemotherapy, an anti-VEGF therapy, and, if KRAS wild type, an anti-EGFR therapy.
Imatinib (Gleevec) and sunitinib (Sutent) are both orally administered kinase inhibitors.
New influenza vaccines provide more options for next season
The Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices unanimously voted in favor of adding various recently licensed influenza vaccines to the list of acceptable vaccines for the prevention of influenza at the most recent meeting of the committee on Feb. 21.
The four influenza vaccines licensed by the Food and Drug Administration over the past year are two quadrivalent vaccines that include two influenza B strains and two influenza A strains; a cell-culture-based vaccine; and an egg-free, recombinant hemagglutinin vaccine manufactured without the use of influenza viruses.
These vaccines are expected to be available during the 2013-2014 influenza season, along with previously available vaccines, Dr. Lisa Grohskopf of the influenza division in the CDC’s National Center for Immunization and Respiratory Diseases said at the meeting.
She also provided an overview of new influenza vaccine abbreviations, reflecting the availability of the new vaccines. With the introduction of the quadrivalent vaccines, "we’ve had to replace our time-honored TIV abbreviation," replacing the abbreviation for trivalent inactivated vaccine (TIV) with IIV (inactivated influenza vaccine), she said.
The IIV class includes trivalent (IIV3) vaccines (those that are egg based and those that are cell-culture based) and quadrivalent (IIV4) inactivated vaccines. Cell-culture-based IIV is referred to as ccIIV or ccIIV3m. RIV3 refers to the trivalent recombinant hemagglutinin influenza vaccine, and LAIV and LAIV4 refer to live attenuated influenza vaccine.
The new vaccines "are acceptable alternatives to the other licensed vaccine products within specified indications," she said. But there are no formal recommendations proposed for using one vaccine over another when a provider has more than one vaccine that is available and appropriate for a given recipient, she added. Moreover, during her presentation Dr. Grohskopf emphasized repeatedly that providers should not delay vaccinating a patient in order to wait for a specific product.
The four new influenza vaccines are:
Flumist Quadrivalent (MedImmune). This quadrivalent live-attenuated influenza vaccine (LAIV4) was licensed in February 2012 for the same indication as the trivalent LAIV for healthy, nonpregnant people aged 2-49 years. Based on evidence from studies comparing the trivalent LAIV with IIV3 that indicate LAIV is more effective in children, providers "may consider use of LAIV4 rather than IIV for children in situations in which both vaccines are available and are otherwise appropriate," Dr. Grohskopf said. But vaccination should not be delayed if the LAIV4 is not available, she added.
• Fluarix Quadrivalent (GSK). This quadrivalent inactivated influenza vaccine (IIV4) was licensed in December 2012 for people aged 3 years and older. This vaccine is "an acceptable alternative" to other licensed products, used within indications. Both the trivalent version (IIV3) and IIV4 will be available in 2103-2014, but IIV3 will make up most of the supply. Either is acceptable, and since IIV4 "may provide broader coverage of circulating influenza viruses, providers who have access to both IIV4 and IIV3 may wish to consider IIV4 for individuals with conditions that confer high risk for influenza complications, when either vaccine is otherwise appropriate," Dr. Grohskopf said.
• Flucelvax (Novartis). This cell-culture-based inactivated influenza vaccine (ccIIV3) was licensed in November 2012 for people aged 18 years and older. It is also an acceptable alternative to other licensed products "used within indications," Dr. Grohskopf said. Although the vaccine viruses are not propagated in eggs, this vaccine cannot be considered completely egg free, but it is expected to contain "considerably less egg protein than other IIVs." Providers "may consider use" of ccIIV3 for egg-allergic patients aged 18 years and older, but "vaccination of mildly egg-allergic individuals should not be delayed if ccIIV3 is not available," when any licensed IIV should be administered, she noted.
• Flublok (Protein Sciences). This recombinant hemagglutinin vaccine (RIV3) – the first purified, recombinant hemagglutinin (HA) protein influenza vaccine, manufactured without the use of influenza virus – was licensed for adults aged 18-49 years in January 2013. The vaccine contains 45 mcg of each HA component, more than other vaccines, and contains no egg protein, preservative, or latex, Dr. Lisa M. Dunkle, chief medical officer at Protein Sciences, said at the meeting. She said that 3-5 million doses of Flublok, which is contained in single-use vials, are expected to be available for the 2013-2014 season. The company is working on a quadrivalent version of Flublok.
RIV3 is truly egg free, and in a situation where both IIV and RIV3 are available and "otherwise appropriate," a provider could consider using RIV3 for people aged 18-49 years who have a mild egg allergy, Dr. Grohskopf said. But vaccination should not be delayed if RIV3 is not available. RIV3 is not contraindicated in people with anaphylaxis to egg.
In 2011, the Advisory Committee on Immunization Practices (ACIP) recommended that people with a mild egg allergy receive IIV. Noting that egg allergy is most prevalent in younger children, she pointed out that Flucelvax or Flublok are not approved in children, and that there is no recommendation for off-label use of these vaccines in egg-allergic children.
Dr. Dunkle said that Protein Sciences is conducting additional studies in younger and older people, and the company expects that licensure of the vaccine will be expanded to include adults aged 50 years and older for the 2013-2014 season. At the FDA’s request, the company is planning a Flublok pregnancy registry. Reproductive toxicology tests of the vaccine in rats were negative, and during clinical trials, 20 women who received Flublok became pregnant, and with 75% follow-up, there have been no birth defects among the live births or vaccine-related adverse events, she said.
The panel also reaffirmed the recommendation for annual influenza vaccination for people aged 6 months and older, with the caveat that younger children through age 8 years receive two doses at least 20 days apart.
There are 15 experts in immunization-related fields on ACIP, which develops written recommendations for the routine administration of vaccines to children and adults in the civilian population.
Information on approved influenza vaccines is available here.
The Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices unanimously voted in favor of adding various recently licensed influenza vaccines to the list of acceptable vaccines for the prevention of influenza at the most recent meeting of the committee on Feb. 21.
The four influenza vaccines licensed by the Food and Drug Administration over the past year are two quadrivalent vaccines that include two influenza B strains and two influenza A strains; a cell-culture-based vaccine; and an egg-free, recombinant hemagglutinin vaccine manufactured without the use of influenza viruses.
These vaccines are expected to be available during the 2013-2014 influenza season, along with previously available vaccines, Dr. Lisa Grohskopf of the influenza division in the CDC’s National Center for Immunization and Respiratory Diseases said at the meeting.
She also provided an overview of new influenza vaccine abbreviations, reflecting the availability of the new vaccines. With the introduction of the quadrivalent vaccines, "we’ve had to replace our time-honored TIV abbreviation," replacing the abbreviation for trivalent inactivated vaccine (TIV) with IIV (inactivated influenza vaccine), she said.
The IIV class includes trivalent (IIV3) vaccines (those that are egg based and those that are cell-culture based) and quadrivalent (IIV4) inactivated vaccines. Cell-culture-based IIV is referred to as ccIIV or ccIIV3m. RIV3 refers to the trivalent recombinant hemagglutinin influenza vaccine, and LAIV and LAIV4 refer to live attenuated influenza vaccine.
The new vaccines "are acceptable alternatives to the other licensed vaccine products within specified indications," she said. But there are no formal recommendations proposed for using one vaccine over another when a provider has more than one vaccine that is available and appropriate for a given recipient, she added. Moreover, during her presentation Dr. Grohskopf emphasized repeatedly that providers should not delay vaccinating a patient in order to wait for a specific product.
The four new influenza vaccines are:
Flumist Quadrivalent (MedImmune). This quadrivalent live-attenuated influenza vaccine (LAIV4) was licensed in February 2012 for the same indication as the trivalent LAIV for healthy, nonpregnant people aged 2-49 years. Based on evidence from studies comparing the trivalent LAIV with IIV3 that indicate LAIV is more effective in children, providers "may consider use of LAIV4 rather than IIV for children in situations in which both vaccines are available and are otherwise appropriate," Dr. Grohskopf said. But vaccination should not be delayed if the LAIV4 is not available, she added.
• Fluarix Quadrivalent (GSK). This quadrivalent inactivated influenza vaccine (IIV4) was licensed in December 2012 for people aged 3 years and older. This vaccine is "an acceptable alternative" to other licensed products, used within indications. Both the trivalent version (IIV3) and IIV4 will be available in 2103-2014, but IIV3 will make up most of the supply. Either is acceptable, and since IIV4 "may provide broader coverage of circulating influenza viruses, providers who have access to both IIV4 and IIV3 may wish to consider IIV4 for individuals with conditions that confer high risk for influenza complications, when either vaccine is otherwise appropriate," Dr. Grohskopf said.
• Flucelvax (Novartis). This cell-culture-based inactivated influenza vaccine (ccIIV3) was licensed in November 2012 for people aged 18 years and older. It is also an acceptable alternative to other licensed products "used within indications," Dr. Grohskopf said. Although the vaccine viruses are not propagated in eggs, this vaccine cannot be considered completely egg free, but it is expected to contain "considerably less egg protein than other IIVs." Providers "may consider use" of ccIIV3 for egg-allergic patients aged 18 years and older, but "vaccination of mildly egg-allergic individuals should not be delayed if ccIIV3 is not available," when any licensed IIV should be administered, she noted.
• Flublok (Protein Sciences). This recombinant hemagglutinin vaccine (RIV3) – the first purified, recombinant hemagglutinin (HA) protein influenza vaccine, manufactured without the use of influenza virus – was licensed for adults aged 18-49 years in January 2013. The vaccine contains 45 mcg of each HA component, more than other vaccines, and contains no egg protein, preservative, or latex, Dr. Lisa M. Dunkle, chief medical officer at Protein Sciences, said at the meeting. She said that 3-5 million doses of Flublok, which is contained in single-use vials, are expected to be available for the 2013-2014 season. The company is working on a quadrivalent version of Flublok.
RIV3 is truly egg free, and in a situation where both IIV and RIV3 are available and "otherwise appropriate," a provider could consider using RIV3 for people aged 18-49 years who have a mild egg allergy, Dr. Grohskopf said. But vaccination should not be delayed if RIV3 is not available. RIV3 is not contraindicated in people with anaphylaxis to egg.
In 2011, the Advisory Committee on Immunization Practices (ACIP) recommended that people with a mild egg allergy receive IIV. Noting that egg allergy is most prevalent in younger children, she pointed out that Flucelvax or Flublok are not approved in children, and that there is no recommendation for off-label use of these vaccines in egg-allergic children.
Dr. Dunkle said that Protein Sciences is conducting additional studies in younger and older people, and the company expects that licensure of the vaccine will be expanded to include adults aged 50 years and older for the 2013-2014 season. At the FDA’s request, the company is planning a Flublok pregnancy registry. Reproductive toxicology tests of the vaccine in rats were negative, and during clinical trials, 20 women who received Flublok became pregnant, and with 75% follow-up, there have been no birth defects among the live births or vaccine-related adverse events, she said.
The panel also reaffirmed the recommendation for annual influenza vaccination for people aged 6 months and older, with the caveat that younger children through age 8 years receive two doses at least 20 days apart.
There are 15 experts in immunization-related fields on ACIP, which develops written recommendations for the routine administration of vaccines to children and adults in the civilian population.
Information on approved influenza vaccines is available here.
The Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices unanimously voted in favor of adding various recently licensed influenza vaccines to the list of acceptable vaccines for the prevention of influenza at the most recent meeting of the committee on Feb. 21.
The four influenza vaccines licensed by the Food and Drug Administration over the past year are two quadrivalent vaccines that include two influenza B strains and two influenza A strains; a cell-culture-based vaccine; and an egg-free, recombinant hemagglutinin vaccine manufactured without the use of influenza viruses.
These vaccines are expected to be available during the 2013-2014 influenza season, along with previously available vaccines, Dr. Lisa Grohskopf of the influenza division in the CDC’s National Center for Immunization and Respiratory Diseases said at the meeting.
She also provided an overview of new influenza vaccine abbreviations, reflecting the availability of the new vaccines. With the introduction of the quadrivalent vaccines, "we’ve had to replace our time-honored TIV abbreviation," replacing the abbreviation for trivalent inactivated vaccine (TIV) with IIV (inactivated influenza vaccine), she said.
The IIV class includes trivalent (IIV3) vaccines (those that are egg based and those that are cell-culture based) and quadrivalent (IIV4) inactivated vaccines. Cell-culture-based IIV is referred to as ccIIV or ccIIV3m. RIV3 refers to the trivalent recombinant hemagglutinin influenza vaccine, and LAIV and LAIV4 refer to live attenuated influenza vaccine.
The new vaccines "are acceptable alternatives to the other licensed vaccine products within specified indications," she said. But there are no formal recommendations proposed for using one vaccine over another when a provider has more than one vaccine that is available and appropriate for a given recipient, she added. Moreover, during her presentation Dr. Grohskopf emphasized repeatedly that providers should not delay vaccinating a patient in order to wait for a specific product.
The four new influenza vaccines are:
Flumist Quadrivalent (MedImmune). This quadrivalent live-attenuated influenza vaccine (LAIV4) was licensed in February 2012 for the same indication as the trivalent LAIV for healthy, nonpregnant people aged 2-49 years. Based on evidence from studies comparing the trivalent LAIV with IIV3 that indicate LAIV is more effective in children, providers "may consider use of LAIV4 rather than IIV for children in situations in which both vaccines are available and are otherwise appropriate," Dr. Grohskopf said. But vaccination should not be delayed if the LAIV4 is not available, she added.
• Fluarix Quadrivalent (GSK). This quadrivalent inactivated influenza vaccine (IIV4) was licensed in December 2012 for people aged 3 years and older. This vaccine is "an acceptable alternative" to other licensed products, used within indications. Both the trivalent version (IIV3) and IIV4 will be available in 2103-2014, but IIV3 will make up most of the supply. Either is acceptable, and since IIV4 "may provide broader coverage of circulating influenza viruses, providers who have access to both IIV4 and IIV3 may wish to consider IIV4 for individuals with conditions that confer high risk for influenza complications, when either vaccine is otherwise appropriate," Dr. Grohskopf said.
• Flucelvax (Novartis). This cell-culture-based inactivated influenza vaccine (ccIIV3) was licensed in November 2012 for people aged 18 years and older. It is also an acceptable alternative to other licensed products "used within indications," Dr. Grohskopf said. Although the vaccine viruses are not propagated in eggs, this vaccine cannot be considered completely egg free, but it is expected to contain "considerably less egg protein than other IIVs." Providers "may consider use" of ccIIV3 for egg-allergic patients aged 18 years and older, but "vaccination of mildly egg-allergic individuals should not be delayed if ccIIV3 is not available," when any licensed IIV should be administered, she noted.
• Flublok (Protein Sciences). This recombinant hemagglutinin vaccine (RIV3) – the first purified, recombinant hemagglutinin (HA) protein influenza vaccine, manufactured without the use of influenza virus – was licensed for adults aged 18-49 years in January 2013. The vaccine contains 45 mcg of each HA component, more than other vaccines, and contains no egg protein, preservative, or latex, Dr. Lisa M. Dunkle, chief medical officer at Protein Sciences, said at the meeting. She said that 3-5 million doses of Flublok, which is contained in single-use vials, are expected to be available for the 2013-2014 season. The company is working on a quadrivalent version of Flublok.
RIV3 is truly egg free, and in a situation where both IIV and RIV3 are available and "otherwise appropriate," a provider could consider using RIV3 for people aged 18-49 years who have a mild egg allergy, Dr. Grohskopf said. But vaccination should not be delayed if RIV3 is not available. RIV3 is not contraindicated in people with anaphylaxis to egg.
In 2011, the Advisory Committee on Immunization Practices (ACIP) recommended that people with a mild egg allergy receive IIV. Noting that egg allergy is most prevalent in younger children, she pointed out that Flucelvax or Flublok are not approved in children, and that there is no recommendation for off-label use of these vaccines in egg-allergic children.
Dr. Dunkle said that Protein Sciences is conducting additional studies in younger and older people, and the company expects that licensure of the vaccine will be expanded to include adults aged 50 years and older for the 2013-2014 season. At the FDA’s request, the company is planning a Flublok pregnancy registry. Reproductive toxicology tests of the vaccine in rats were negative, and during clinical trials, 20 women who received Flublok became pregnant, and with 75% follow-up, there have been no birth defects among the live births or vaccine-related adverse events, she said.
The panel also reaffirmed the recommendation for annual influenza vaccination for people aged 6 months and older, with the caveat that younger children through age 8 years receive two doses at least 20 days apart.
There are 15 experts in immunization-related fields on ACIP, which develops written recommendations for the routine administration of vaccines to children and adults in the civilian population.
Information on approved influenza vaccines is available here.
FROM AN ACIP MEETING
CDC committee affirms update of 1993 Hib statement
The first update in almost 20 years to the Centers for Disease Control and Prevention’s statement on Haemophilus influenzae type b will reflect changes in the epidemiology of the disease and the vaccines available to prevent it.
The CDC’s Advisory Committee on Immunization Practices unanimously approved a draft of the updated statement on Haemophilus influenzae type b (Hib) at the committee’s meeting Feb. 20.
Dr. Elizabeth C. Briere* summarized the draft statement, which lists the currently available and licensed vaccines, with no changes to previously published recommendations on routine use. But the statement includes guidance for special populations not included in the 1993 statement, consistent with the 2013 IDSA Clinical Practice Guideline for Vaccination of the Immunocompromised, which has not yet been published; as well as 2012 Red Book recommendations, 2011 ACIP General Recommendations on Immunizations, and 2009 ACIP Guidelines for the Prevention and Treatment of Opportunistic Infections for HIV-Infected Adults and Adolescents.In the years since the first Hib vaccine –a polysaccharide vaccine – was licensed in 1985, and the first conjugate Hib vaccine was licensed in 1989, the incidence of invasive Hib infection among children aged under 5 years has dropped dramatically, and has remained low through the 2000s, Dr. Briere of the CDC’s National Center for Immunization and Respiratory Diseases, pointed out at the meeting.
The special populations listed are Alaskan natives and American Indians, children aged under 24 months with invasive Hib, preterm infants, and high-risk groups (those with functional or anatomic asplenia, HIV, IgG deficiency, early component complement deficiency, and recipients of hematopoietic stem cell transplant and chemotherapy).
The guidance for high risk groups includes the recommendation to provide one dose of Hib vaccine to asplenic children aged 60 months and older and asplenic adults who are unimmunized**; and for HIV-infected children aged 60 months and older, who are unimmunized, she said. The Hib vaccination is not recommended for HIV-infected adults.
For patients undergoing an elective splenectomy, who are aged 15 months and older and are unimmunized, one dose of vaccine prior to the procedure is recommended. For children undergoing chemotherapy or radiation therapy who are aged younger than 59 months, revaccination is not required if routine Hib doses were administered 14 or more days before starting treatment. But if the dose is given within 14 days of starting treatment – or during therapy – the recommendation in the guidance is to repeat doses starting at least 3 months following the completion of treatment, Dr. Briere said.
In the updated statement, the guidance for chemoprophylaxis – which was limited in the old statement – is consistent with the guidance in the 2012 Red Book, and includes recommendations for the use of rifampin as chemoprophylaxis, she added.
The draft of the statement was updated after a review of Hib vaccine recommendations, including a draft of IDSA clinical practice guidelines for vaccination of the immunocompromised host, peer-reviewed literature, and surveillance data; and was reviewed by ACIP’s meningococcal and Haemophilus influenzae type b vaccine work group and ACIP voting members, providing comments, prior to the ACIP meeting.
There are 15 experts in immunization-related fields on the ACIP committee, which develops written recommendations for the routine administration of vaccines to children and adults in the civilian population.
*Updated: 2/26/13
**CORRECTION: A previous version of this story incorrectly described the asplenic patients who should receive the Hib vaccine.
The first update in almost 20 years to the Centers for Disease Control and Prevention’s statement on Haemophilus influenzae type b will reflect changes in the epidemiology of the disease and the vaccines available to prevent it.
The CDC’s Advisory Committee on Immunization Practices unanimously approved a draft of the updated statement on Haemophilus influenzae type b (Hib) at the committee’s meeting Feb. 20.
Dr. Elizabeth C. Briere* summarized the draft statement, which lists the currently available and licensed vaccines, with no changes to previously published recommendations on routine use. But the statement includes guidance for special populations not included in the 1993 statement, consistent with the 2013 IDSA Clinical Practice Guideline for Vaccination of the Immunocompromised, which has not yet been published; as well as 2012 Red Book recommendations, 2011 ACIP General Recommendations on Immunizations, and 2009 ACIP Guidelines for the Prevention and Treatment of Opportunistic Infections for HIV-Infected Adults and Adolescents.In the years since the first Hib vaccine –a polysaccharide vaccine – was licensed in 1985, and the first conjugate Hib vaccine was licensed in 1989, the incidence of invasive Hib infection among children aged under 5 years has dropped dramatically, and has remained low through the 2000s, Dr. Briere of the CDC’s National Center for Immunization and Respiratory Diseases, pointed out at the meeting.
The special populations listed are Alaskan natives and American Indians, children aged under 24 months with invasive Hib, preterm infants, and high-risk groups (those with functional or anatomic asplenia, HIV, IgG deficiency, early component complement deficiency, and recipients of hematopoietic stem cell transplant and chemotherapy).
The guidance for high risk groups includes the recommendation to provide one dose of Hib vaccine to asplenic children aged 60 months and older and asplenic adults who are unimmunized**; and for HIV-infected children aged 60 months and older, who are unimmunized, she said. The Hib vaccination is not recommended for HIV-infected adults.
For patients undergoing an elective splenectomy, who are aged 15 months and older and are unimmunized, one dose of vaccine prior to the procedure is recommended. For children undergoing chemotherapy or radiation therapy who are aged younger than 59 months, revaccination is not required if routine Hib doses were administered 14 or more days before starting treatment. But if the dose is given within 14 days of starting treatment – or during therapy – the recommendation in the guidance is to repeat doses starting at least 3 months following the completion of treatment, Dr. Briere said.
In the updated statement, the guidance for chemoprophylaxis – which was limited in the old statement – is consistent with the guidance in the 2012 Red Book, and includes recommendations for the use of rifampin as chemoprophylaxis, she added.
The draft of the statement was updated after a review of Hib vaccine recommendations, including a draft of IDSA clinical practice guidelines for vaccination of the immunocompromised host, peer-reviewed literature, and surveillance data; and was reviewed by ACIP’s meningococcal and Haemophilus influenzae type b vaccine work group and ACIP voting members, providing comments, prior to the ACIP meeting.
There are 15 experts in immunization-related fields on the ACIP committee, which develops written recommendations for the routine administration of vaccines to children and adults in the civilian population.
*Updated: 2/26/13
**CORRECTION: A previous version of this story incorrectly described the asplenic patients who should receive the Hib vaccine.
The first update in almost 20 years to the Centers for Disease Control and Prevention’s statement on Haemophilus influenzae type b will reflect changes in the epidemiology of the disease and the vaccines available to prevent it.
The CDC’s Advisory Committee on Immunization Practices unanimously approved a draft of the updated statement on Haemophilus influenzae type b (Hib) at the committee’s meeting Feb. 20.
Dr. Elizabeth C. Briere* summarized the draft statement, which lists the currently available and licensed vaccines, with no changes to previously published recommendations on routine use. But the statement includes guidance for special populations not included in the 1993 statement, consistent with the 2013 IDSA Clinical Practice Guideline for Vaccination of the Immunocompromised, which has not yet been published; as well as 2012 Red Book recommendations, 2011 ACIP General Recommendations on Immunizations, and 2009 ACIP Guidelines for the Prevention and Treatment of Opportunistic Infections for HIV-Infected Adults and Adolescents.In the years since the first Hib vaccine –a polysaccharide vaccine – was licensed in 1985, and the first conjugate Hib vaccine was licensed in 1989, the incidence of invasive Hib infection among children aged under 5 years has dropped dramatically, and has remained low through the 2000s, Dr. Briere of the CDC’s National Center for Immunization and Respiratory Diseases, pointed out at the meeting.
The special populations listed are Alaskan natives and American Indians, children aged under 24 months with invasive Hib, preterm infants, and high-risk groups (those with functional or anatomic asplenia, HIV, IgG deficiency, early component complement deficiency, and recipients of hematopoietic stem cell transplant and chemotherapy).
The guidance for high risk groups includes the recommendation to provide one dose of Hib vaccine to asplenic children aged 60 months and older and asplenic adults who are unimmunized**; and for HIV-infected children aged 60 months and older, who are unimmunized, she said. The Hib vaccination is not recommended for HIV-infected adults.
For patients undergoing an elective splenectomy, who are aged 15 months and older and are unimmunized, one dose of vaccine prior to the procedure is recommended. For children undergoing chemotherapy or radiation therapy who are aged younger than 59 months, revaccination is not required if routine Hib doses were administered 14 or more days before starting treatment. But if the dose is given within 14 days of starting treatment – or during therapy – the recommendation in the guidance is to repeat doses starting at least 3 months following the completion of treatment, Dr. Briere said.
In the updated statement, the guidance for chemoprophylaxis – which was limited in the old statement – is consistent with the guidance in the 2012 Red Book, and includes recommendations for the use of rifampin as chemoprophylaxis, she added.
The draft of the statement was updated after a review of Hib vaccine recommendations, including a draft of IDSA clinical practice guidelines for vaccination of the immunocompromised host, peer-reviewed literature, and surveillance data; and was reviewed by ACIP’s meningococcal and Haemophilus influenzae type b vaccine work group and ACIP voting members, providing comments, prior to the ACIP meeting.
There are 15 experts in immunization-related fields on the ACIP committee, which develops written recommendations for the routine administration of vaccines to children and adults in the civilian population.
*Updated: 2/26/13
**CORRECTION: A previous version of this story incorrectly described the asplenic patients who should receive the Hib vaccine.
FROM AN ACIP MEETING