User login
FDA panel backs ICS/LABA inhaler for COPD
SILVER SPRING, MD. – The majority of a Food and Drug Administration advisory panel supported the approval of an inhaled powder formulation of fluticasone furoate and vilanterol as a treatment for chronic obstructive pulmonary disease, based on studies of almost 4,000 patients.
At a meeting on April 17, the FDA’s Pulmonary-Allergy Drugs Advisory Committee voted that the results of the studies provided "substantial evidence" to support approval of the fixed-dose combination of 100 mcg of fluticasone furoate (FF) with 25 mcg of vilanterol (VI), administered once a day, for the long-term maintenance treatment of airflow obstruction (in a 9 to 4 vote), and for reducing COPD exacerbations (in an 8 to 5 vote).
The inhaled corticosteroid/long-acting beta-2 agonist (ICS/LABA) combination is formulated in a dry powder, and is administered once a day in a new inhaler that provides 30 doses, according to GlaxoSmithKline, which is developing the product with Theravance.
If the formulation is approved, it would be the first ICS/LABA product that is dosed once a day for COPD, and would be the first such product that would only be available in the combined formulation, since the companies have no plans to pursue approval of the VI component alone. GSK also markets Advair Diskus, the combination of fluticasone propionate and salmeterol, one of the two inhaled ICS/LABA treatments that is FDA approved for the treatment of COPD. The other is the combination of budesonide and formoterol (Symbicort). Both are administered twice a day, and salmeterol and formoterol are both available as separate products for COPD.
The safety and efficacy of three different dosage strengths of FF – 200, 100, and 50 mcg – combined with 25 mcg of VI were evaluated in four pivotal phase III studies of 3,852 mostly Caucasian people (mean age, 62-63 years) with COPD. All patients had a forced expiratory volume in 1 second (FEV1) less than 70% predicted. About half were current smokers.
In two 24-week studies of 2,254 patients evaluating the effect on lung function, the three FF/VI dosage strengths were compared with placebo, FF 100 mcg alone, and VI 25 mcg alone for two primary endpoints: weighted mean FEV1 at 0-4 hours and change from baseline in trough FEV1.
At 24 weeks, there were significant improvements in the weighted mean FEV1 and in trough FEV1 for all three strengths of FF/VI and for VI 25 mcg, compared with placebo. There were also significant improvements in the mean FEV1 (0-4 hours) for all three strengths of FF/VI, compared with FF alone. Improvement in trough FEV1, however, was not significantly better with any of the three doses, when compared with VI 25 mcg alone.
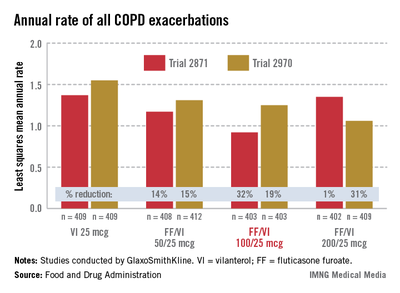
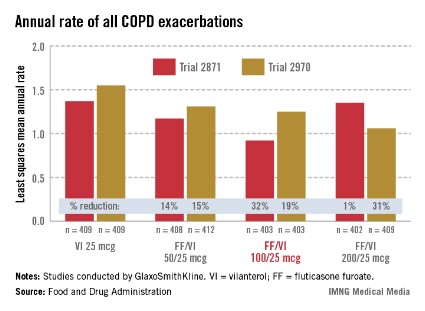
In the two 52-week exacerbation trials of 1,622 patients who had had at least one COPD exacerbation in the previous year, the rate of moderate and severe COPD exacerbations in 1 year was reduced by 21% and 34%, respectively, among those on the FF/VI 100/25 dose over those on VI 25 mcg alone. In one study, the reduction in exacerbations was significant for all three doses, but in the other study, the effect of the proposed 100/25 dose on exacerbations was only numerically, not significantly, improved over the effect of VI 25, the FDA reviewers pointed out. And overall, for the 100/25 dose, the rate of moderate and severe exacerbations was reduced only "by a fraction of an event in 1 year," said Kiya Hamilton, Ph.D., a biostatistician reviewer at the FDA.
Treatment was associated with an increase in local steroid effects, such as oral candidiasis, as expected. In the exacerbation studies, there were more cases of pneumonia and an increase in pneumonia-related deaths with increasing FF doses, and more fractures among those on the FF/VI combination. Bone loss and pneumonia are recognized as adverse effects of ICS in COPD patients, and with one exception, all the deaths were among those on 200/25 mcg dose, which is not going to be marketed, the FDA said.
In a 12 to 1 vote, the panel agreed that the efficacy data provided substantial evidence that the proposed dose provided "a clinically meaningful benefit" for the long-term maintenance treatment of airflow obstruction in COPD. But they were less united on the impact on exacerbations, voting 8 to 5 that the data provided substantial evidence that the reduction in exacerbations seen in the study was clinically meaningful. The panel also voted 10 to 3 that the safety of the 100/25 mcg dose had been adequately demonstrated for the COPD indication.
Panelists supporting approval cited the totality of the database and the utility of having a once a day dose as among the reasons for their votes. Dr. James Stoller, the Jean Wall Bennett Professor of Medicine at the Cleveland Clinic, supported approval and said that the safety profile was acceptable as long as the formulation was not used to treat patients with mild COPD.
Like other panelists who did not support approval, Dr. Paula Carvalho, professor of medicine in the division of pulmonary and critical care medicine, University of Washington, Seattle, said the benefit-risk profile was not adequate. She added that she was more impressed by the effect that treatment with VI 25 mcg alone had on COPD exacerbations than with the combination. Also voting against approval was Peter Peduzzi, Ph.D., professor of public health at the Yale School of Public Health in New Haven, Conn., who said that the impact on trough FEV1 appeared to be driven by the VI component, and like others voting against approval, he cited safety issues.
If approved, GSK plans to market the product as Breo Ellipta. The FDA’s deadline for making a decision on the approval is May 12. The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist might be given a waiver, but not at this meeting.
The product is under review for both COPD and asthma in the European Union and in Japan. It is not under review for asthma in the United States.
SILVER SPRING, MD. – The majority of a Food and Drug Administration advisory panel supported the approval of an inhaled powder formulation of fluticasone furoate and vilanterol as a treatment for chronic obstructive pulmonary disease, based on studies of almost 4,000 patients.
At a meeting on April 17, the FDA’s Pulmonary-Allergy Drugs Advisory Committee voted that the results of the studies provided "substantial evidence" to support approval of the fixed-dose combination of 100 mcg of fluticasone furoate (FF) with 25 mcg of vilanterol (VI), administered once a day, for the long-term maintenance treatment of airflow obstruction (in a 9 to 4 vote), and for reducing COPD exacerbations (in an 8 to 5 vote).
The inhaled corticosteroid/long-acting beta-2 agonist (ICS/LABA) combination is formulated in a dry powder, and is administered once a day in a new inhaler that provides 30 doses, according to GlaxoSmithKline, which is developing the product with Theravance.
If the formulation is approved, it would be the first ICS/LABA product that is dosed once a day for COPD, and would be the first such product that would only be available in the combined formulation, since the companies have no plans to pursue approval of the VI component alone. GSK also markets Advair Diskus, the combination of fluticasone propionate and salmeterol, one of the two inhaled ICS/LABA treatments that is FDA approved for the treatment of COPD. The other is the combination of budesonide and formoterol (Symbicort). Both are administered twice a day, and salmeterol and formoterol are both available as separate products for COPD.
The safety and efficacy of three different dosage strengths of FF – 200, 100, and 50 mcg – combined with 25 mcg of VI were evaluated in four pivotal phase III studies of 3,852 mostly Caucasian people (mean age, 62-63 years) with COPD. All patients had a forced expiratory volume in 1 second (FEV1) less than 70% predicted. About half were current smokers.
In two 24-week studies of 2,254 patients evaluating the effect on lung function, the three FF/VI dosage strengths were compared with placebo, FF 100 mcg alone, and VI 25 mcg alone for two primary endpoints: weighted mean FEV1 at 0-4 hours and change from baseline in trough FEV1.
At 24 weeks, there were significant improvements in the weighted mean FEV1 and in trough FEV1 for all three strengths of FF/VI and for VI 25 mcg, compared with placebo. There were also significant improvements in the mean FEV1 (0-4 hours) for all three strengths of FF/VI, compared with FF alone. Improvement in trough FEV1, however, was not significantly better with any of the three doses, when compared with VI 25 mcg alone.
In the two 52-week exacerbation trials of 1,622 patients who had had at least one COPD exacerbation in the previous year, the rate of moderate and severe COPD exacerbations in 1 year was reduced by 21% and 34%, respectively, among those on the FF/VI 100/25 dose over those on VI 25 mcg alone. In one study, the reduction in exacerbations was significant for all three doses, but in the other study, the effect of the proposed 100/25 dose on exacerbations was only numerically, not significantly, improved over the effect of VI 25, the FDA reviewers pointed out. And overall, for the 100/25 dose, the rate of moderate and severe exacerbations was reduced only "by a fraction of an event in 1 year," said Kiya Hamilton, Ph.D., a biostatistician reviewer at the FDA.
Treatment was associated with an increase in local steroid effects, such as oral candidiasis, as expected. In the exacerbation studies, there were more cases of pneumonia and an increase in pneumonia-related deaths with increasing FF doses, and more fractures among those on the FF/VI combination. Bone loss and pneumonia are recognized as adverse effects of ICS in COPD patients, and with one exception, all the deaths were among those on 200/25 mcg dose, which is not going to be marketed, the FDA said.
In a 12 to 1 vote, the panel agreed that the efficacy data provided substantial evidence that the proposed dose provided "a clinically meaningful benefit" for the long-term maintenance treatment of airflow obstruction in COPD. But they were less united on the impact on exacerbations, voting 8 to 5 that the data provided substantial evidence that the reduction in exacerbations seen in the study was clinically meaningful. The panel also voted 10 to 3 that the safety of the 100/25 mcg dose had been adequately demonstrated for the COPD indication.
Panelists supporting approval cited the totality of the database and the utility of having a once a day dose as among the reasons for their votes. Dr. James Stoller, the Jean Wall Bennett Professor of Medicine at the Cleveland Clinic, supported approval and said that the safety profile was acceptable as long as the formulation was not used to treat patients with mild COPD.
Like other panelists who did not support approval, Dr. Paula Carvalho, professor of medicine in the division of pulmonary and critical care medicine, University of Washington, Seattle, said the benefit-risk profile was not adequate. She added that she was more impressed by the effect that treatment with VI 25 mcg alone had on COPD exacerbations than with the combination. Also voting against approval was Peter Peduzzi, Ph.D., professor of public health at the Yale School of Public Health in New Haven, Conn., who said that the impact on trough FEV1 appeared to be driven by the VI component, and like others voting against approval, he cited safety issues.
If approved, GSK plans to market the product as Breo Ellipta. The FDA’s deadline for making a decision on the approval is May 12. The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist might be given a waiver, but not at this meeting.
The product is under review for both COPD and asthma in the European Union and in Japan. It is not under review for asthma in the United States.
SILVER SPRING, MD. – The majority of a Food and Drug Administration advisory panel supported the approval of an inhaled powder formulation of fluticasone furoate and vilanterol as a treatment for chronic obstructive pulmonary disease, based on studies of almost 4,000 patients.
At a meeting on April 17, the FDA’s Pulmonary-Allergy Drugs Advisory Committee voted that the results of the studies provided "substantial evidence" to support approval of the fixed-dose combination of 100 mcg of fluticasone furoate (FF) with 25 mcg of vilanterol (VI), administered once a day, for the long-term maintenance treatment of airflow obstruction (in a 9 to 4 vote), and for reducing COPD exacerbations (in an 8 to 5 vote).
The inhaled corticosteroid/long-acting beta-2 agonist (ICS/LABA) combination is formulated in a dry powder, and is administered once a day in a new inhaler that provides 30 doses, according to GlaxoSmithKline, which is developing the product with Theravance.
If the formulation is approved, it would be the first ICS/LABA product that is dosed once a day for COPD, and would be the first such product that would only be available in the combined formulation, since the companies have no plans to pursue approval of the VI component alone. GSK also markets Advair Diskus, the combination of fluticasone propionate and salmeterol, one of the two inhaled ICS/LABA treatments that is FDA approved for the treatment of COPD. The other is the combination of budesonide and formoterol (Symbicort). Both are administered twice a day, and salmeterol and formoterol are both available as separate products for COPD.
The safety and efficacy of three different dosage strengths of FF – 200, 100, and 50 mcg – combined with 25 mcg of VI were evaluated in four pivotal phase III studies of 3,852 mostly Caucasian people (mean age, 62-63 years) with COPD. All patients had a forced expiratory volume in 1 second (FEV1) less than 70% predicted. About half were current smokers.
In two 24-week studies of 2,254 patients evaluating the effect on lung function, the three FF/VI dosage strengths were compared with placebo, FF 100 mcg alone, and VI 25 mcg alone for two primary endpoints: weighted mean FEV1 at 0-4 hours and change from baseline in trough FEV1.
At 24 weeks, there were significant improvements in the weighted mean FEV1 and in trough FEV1 for all three strengths of FF/VI and for VI 25 mcg, compared with placebo. There were also significant improvements in the mean FEV1 (0-4 hours) for all three strengths of FF/VI, compared with FF alone. Improvement in trough FEV1, however, was not significantly better with any of the three doses, when compared with VI 25 mcg alone.
In the two 52-week exacerbation trials of 1,622 patients who had had at least one COPD exacerbation in the previous year, the rate of moderate and severe COPD exacerbations in 1 year was reduced by 21% and 34%, respectively, among those on the FF/VI 100/25 dose over those on VI 25 mcg alone. In one study, the reduction in exacerbations was significant for all three doses, but in the other study, the effect of the proposed 100/25 dose on exacerbations was only numerically, not significantly, improved over the effect of VI 25, the FDA reviewers pointed out. And overall, for the 100/25 dose, the rate of moderate and severe exacerbations was reduced only "by a fraction of an event in 1 year," said Kiya Hamilton, Ph.D., a biostatistician reviewer at the FDA.
Treatment was associated with an increase in local steroid effects, such as oral candidiasis, as expected. In the exacerbation studies, there were more cases of pneumonia and an increase in pneumonia-related deaths with increasing FF doses, and more fractures among those on the FF/VI combination. Bone loss and pneumonia are recognized as adverse effects of ICS in COPD patients, and with one exception, all the deaths were among those on 200/25 mcg dose, which is not going to be marketed, the FDA said.
In a 12 to 1 vote, the panel agreed that the efficacy data provided substantial evidence that the proposed dose provided "a clinically meaningful benefit" for the long-term maintenance treatment of airflow obstruction in COPD. But they were less united on the impact on exacerbations, voting 8 to 5 that the data provided substantial evidence that the reduction in exacerbations seen in the study was clinically meaningful. The panel also voted 10 to 3 that the safety of the 100/25 mcg dose had been adequately demonstrated for the COPD indication.
Panelists supporting approval cited the totality of the database and the utility of having a once a day dose as among the reasons for their votes. Dr. James Stoller, the Jean Wall Bennett Professor of Medicine at the Cleveland Clinic, supported approval and said that the safety profile was acceptable as long as the formulation was not used to treat patients with mild COPD.
Like other panelists who did not support approval, Dr. Paula Carvalho, professor of medicine in the division of pulmonary and critical care medicine, University of Washington, Seattle, said the benefit-risk profile was not adequate. She added that she was more impressed by the effect that treatment with VI 25 mcg alone had on COPD exacerbations than with the combination. Also voting against approval was Peter Peduzzi, Ph.D., professor of public health at the Yale School of Public Health in New Haven, Conn., who said that the impact on trough FEV1 appeared to be driven by the VI component, and like others voting against approval, he cited safety issues.
If approved, GSK plans to market the product as Breo Ellipta. The FDA’s deadline for making a decision on the approval is May 12. The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist might be given a waiver, but not at this meeting.
The product is under review for both COPD and asthma in the European Union and in Japan. It is not under review for asthma in the United States.
AT AN FDA ADVISORY PANEL MEETING
Study finds anti-NGF drug effective for hip OA
Treatment with the anti–nerve growth factor tanezumab was associated with significant improvements over placebo in pain, physical function, and Patient’s Global Assessment of osteoarthritis results, after 16 weeks in a phase III study of patients with osteoarthritis of the hip.
In the randomized, double-blind, placebo-controlled study of 621 patients with painful hip OA, treatment with the anti–nerve growth factor (anti-NGF) was also well tolerated, according to Dr. Mark Brown and his associates. The study was funded by Pfizer, the manufacturer of tanezumab, and Dr. Brown and three of the five other authors are full-time employees of the company.

Tanezumab "may eventually be useful in OA patients who are intolerant of or nonresponsive to nonopioid treatment," the authors concluded. The study was published online (Arthritis Rheum. 2013 April 1 [doi:10.1002/art.37950]).
Anti-NGFs are monoclonal antibodies directed against nerve growth factor, which "modulates pain processing and sensitivity via nociceptor sensitization," and increased NGF levels are associated with injury, inflammation, and chronic pain, according to the authors.
Tanezumab is one of several anti-NGFs being studied for OA and other chronic pain conditions and is the furthest along in development. In 2010, the Food and Drug Administration placed a partial hold on anti-NGF clinical trials after cases of osteonecrosis and avascular necrosis were reported in patients treated with tanezumab and in those treated with another anti-NGF, including cases in nonindex joints and people with no history of OA. Dr. Brown and his coauthors wrote that some of these cases were reported in phase III hip and knee OA studies, after their study was completed.
In March 2012, an FDA advisory panel of outside experts agreed that the anti-NGFs showed promise as analgesics in studies and unanimously recommended that clinical development should continue, with close monitoring of joint-related adverse effects and adequate informed consent. The authors noted that the clinical hold on tanezumab trials was lifted in August 2012. (The FDA could not provide a comment on this issue because it relates to a drug application under review, an agency spokesperson said.)
The development of the anti-NGFs "is an exciting potential addition to our present inadequate control of pain," and research is resuming with a careful review of the risk of bone and joint changes, Dr. Roy Altman said in an interview. In his view, "the known risks would be acceptable to many people with poorly controlled pain." Dr. Altman, professor of medicine, rheumatology, at the University of California, Los Angeles, was not an investigator in the study.
The Pfizer study compared three tanezumab doses (2.5 mg, 5 mg, and 10 mg) administered intravenously at baseline, 8 weeks, and 16 weeks, with placebo, in 621 people with OA of the hip (mean age, 62-63 years) who could not or did not want to take nonopiate pain medications, did not get enough relief from these medications, and/or were candidates for hip surgery or other invasive treatments. Their baseline Western Ontario and McMaster Universities OA Index (WOMAC) pain and physical function subscale scores were at least 5 at baseline and Patient’s Global Assessment of OA (PGA OA) was "fair," "poor," or "very poor." Almost half were candidates for invasive treatment.
At 16 weeks, compared with placebo, treatment with tanezumab, at all three doses, "produced greater, clinically meaningful, and statistically significant improvements" in each of the three primary efficacy endpoints: changes in the WOMAC pain subscale, the WOMAC physical function subscale, and the PGA OA. Differences between placebo and treatment were greatest for the two higher doses.
On the WOMAC pain subscale, mean baseline scores of all groups ranged from 7.2 to 7.3. Placebo patients’ scores improved by a mean of -1.62 points, but in comparison, improvements were significantly better on progressively higher doses of tanezumab: –2.90 for 2.5 mg, –3.31 for 5 mg, and –3.37 for 10 mg. Similar results occurred on the WOMAC physical function subscale scores, with mean improvements of –1.39 for placebo, –2.57 for 2.5 mg, –2.88 mg for 5 mg, and –3.00 mg for 10 mg.
Other findings included a significantly greater proportion of patients on the treatment who achieved at least 30%, 50%, 70%, and 90% reductions in the WOMAC pain subscale, compared with placebo at 16 weeks. About two-thirds of the patients on treatment remained in the study through week 24 (considered the end of the study because of tanezumab’s long half-life), compared with 38% of those on placebo.
Overall, more patients on tanezumab reported adverse effects (55%-58%, vs. 44% on placebo). Serious adverse events occurred in 3%-4.5% of patients in the treatment groups.
The rate of total hip replacements was similar in the treatment and placebo groups, with a total of eight patients who had a total hip replacement (three in the placebo group, two each in the 2.5-mg and 5-mg groups, and one in the 10-mg group). These included two patients on tanezumab who had osteonecrosis thought to be related to tanezumab. The authors pointed out that an outside adjudication committee, however, did not confirm osteonecrosis in either patient, and concluded that the reasons for joint replacements in the eight patients were inflammatory arthropathy, end-stage OA, rapidly progressive OA, or worsening OA.
Dr. Altman said that he has consulted with Abbott and its now discontinued program on anti-NGF drugs.
Treatment with the anti–nerve growth factor tanezumab was associated with significant improvements over placebo in pain, physical function, and Patient’s Global Assessment of osteoarthritis results, after 16 weeks in a phase III study of patients with osteoarthritis of the hip.
In the randomized, double-blind, placebo-controlled study of 621 patients with painful hip OA, treatment with the anti–nerve growth factor (anti-NGF) was also well tolerated, according to Dr. Mark Brown and his associates. The study was funded by Pfizer, the manufacturer of tanezumab, and Dr. Brown and three of the five other authors are full-time employees of the company.
Tanezumab "may eventually be useful in OA patients who are intolerant of or nonresponsive to nonopioid treatment," the authors concluded. The study was published online (Arthritis Rheum. 2013 April 1 [doi:10.1002/art.37950]).
Anti-NGFs are monoclonal antibodies directed against nerve growth factor, which "modulates pain processing and sensitivity via nociceptor sensitization," and increased NGF levels are associated with injury, inflammation, and chronic pain, according to the authors.
Tanezumab is one of several anti-NGFs being studied for OA and other chronic pain conditions and is the furthest along in development. In 2010, the Food and Drug Administration placed a partial hold on anti-NGF clinical trials after cases of osteonecrosis and avascular necrosis were reported in patients treated with tanezumab and in those treated with another anti-NGF, including cases in nonindex joints and people with no history of OA. Dr. Brown and his coauthors wrote that some of these cases were reported in phase III hip and knee OA studies, after their study was completed.
In March 2012, an FDA advisory panel of outside experts agreed that the anti-NGFs showed promise as analgesics in studies and unanimously recommended that clinical development should continue, with close monitoring of joint-related adverse effects and adequate informed consent. The authors noted that the clinical hold on tanezumab trials was lifted in August 2012. (The FDA could not provide a comment on this issue because it relates to a drug application under review, an agency spokesperson said.)
The development of the anti-NGFs "is an exciting potential addition to our present inadequate control of pain," and research is resuming with a careful review of the risk of bone and joint changes, Dr. Roy Altman said in an interview. In his view, "the known risks would be acceptable to many people with poorly controlled pain." Dr. Altman, professor of medicine, rheumatology, at the University of California, Los Angeles, was not an investigator in the study.
The Pfizer study compared three tanezumab doses (2.5 mg, 5 mg, and 10 mg) administered intravenously at baseline, 8 weeks, and 16 weeks, with placebo, in 621 people with OA of the hip (mean age, 62-63 years) who could not or did not want to take nonopiate pain medications, did not get enough relief from these medications, and/or were candidates for hip surgery or other invasive treatments. Their baseline Western Ontario and McMaster Universities OA Index (WOMAC) pain and physical function subscale scores were at least 5 at baseline and Patient’s Global Assessment of OA (PGA OA) was "fair," "poor," or "very poor." Almost half were candidates for invasive treatment.
At 16 weeks, compared with placebo, treatment with tanezumab, at all three doses, "produced greater, clinically meaningful, and statistically significant improvements" in each of the three primary efficacy endpoints: changes in the WOMAC pain subscale, the WOMAC physical function subscale, and the PGA OA. Differences between placebo and treatment were greatest for the two higher doses.
On the WOMAC pain subscale, mean baseline scores of all groups ranged from 7.2 to 7.3. Placebo patients’ scores improved by a mean of -1.62 points, but in comparison, improvements were significantly better on progressively higher doses of tanezumab: –2.90 for 2.5 mg, –3.31 for 5 mg, and –3.37 for 10 mg. Similar results occurred on the WOMAC physical function subscale scores, with mean improvements of –1.39 for placebo, –2.57 for 2.5 mg, –2.88 mg for 5 mg, and –3.00 mg for 10 mg.
Other findings included a significantly greater proportion of patients on the treatment who achieved at least 30%, 50%, 70%, and 90% reductions in the WOMAC pain subscale, compared with placebo at 16 weeks. About two-thirds of the patients on treatment remained in the study through week 24 (considered the end of the study because of tanezumab’s long half-life), compared with 38% of those on placebo.
Overall, more patients on tanezumab reported adverse effects (55%-58%, vs. 44% on placebo). Serious adverse events occurred in 3%-4.5% of patients in the treatment groups.
The rate of total hip replacements was similar in the treatment and placebo groups, with a total of eight patients who had a total hip replacement (three in the placebo group, two each in the 2.5-mg and 5-mg groups, and one in the 10-mg group). These included two patients on tanezumab who had osteonecrosis thought to be related to tanezumab. The authors pointed out that an outside adjudication committee, however, did not confirm osteonecrosis in either patient, and concluded that the reasons for joint replacements in the eight patients were inflammatory arthropathy, end-stage OA, rapidly progressive OA, or worsening OA.
Dr. Altman said that he has consulted with Abbott and its now discontinued program on anti-NGF drugs.
Treatment with the anti–nerve growth factor tanezumab was associated with significant improvements over placebo in pain, physical function, and Patient’s Global Assessment of osteoarthritis results, after 16 weeks in a phase III study of patients with osteoarthritis of the hip.
In the randomized, double-blind, placebo-controlled study of 621 patients with painful hip OA, treatment with the anti–nerve growth factor (anti-NGF) was also well tolerated, according to Dr. Mark Brown and his associates. The study was funded by Pfizer, the manufacturer of tanezumab, and Dr. Brown and three of the five other authors are full-time employees of the company.
Tanezumab "may eventually be useful in OA patients who are intolerant of or nonresponsive to nonopioid treatment," the authors concluded. The study was published online (Arthritis Rheum. 2013 April 1 [doi:10.1002/art.37950]).
Anti-NGFs are monoclonal antibodies directed against nerve growth factor, which "modulates pain processing and sensitivity via nociceptor sensitization," and increased NGF levels are associated with injury, inflammation, and chronic pain, according to the authors.
Tanezumab is one of several anti-NGFs being studied for OA and other chronic pain conditions and is the furthest along in development. In 2010, the Food and Drug Administration placed a partial hold on anti-NGF clinical trials after cases of osteonecrosis and avascular necrosis were reported in patients treated with tanezumab and in those treated with another anti-NGF, including cases in nonindex joints and people with no history of OA. Dr. Brown and his coauthors wrote that some of these cases were reported in phase III hip and knee OA studies, after their study was completed.
In March 2012, an FDA advisory panel of outside experts agreed that the anti-NGFs showed promise as analgesics in studies and unanimously recommended that clinical development should continue, with close monitoring of joint-related adverse effects and adequate informed consent. The authors noted that the clinical hold on tanezumab trials was lifted in August 2012. (The FDA could not provide a comment on this issue because it relates to a drug application under review, an agency spokesperson said.)
The development of the anti-NGFs "is an exciting potential addition to our present inadequate control of pain," and research is resuming with a careful review of the risk of bone and joint changes, Dr. Roy Altman said in an interview. In his view, "the known risks would be acceptable to many people with poorly controlled pain." Dr. Altman, professor of medicine, rheumatology, at the University of California, Los Angeles, was not an investigator in the study.
The Pfizer study compared three tanezumab doses (2.5 mg, 5 mg, and 10 mg) administered intravenously at baseline, 8 weeks, and 16 weeks, with placebo, in 621 people with OA of the hip (mean age, 62-63 years) who could not or did not want to take nonopiate pain medications, did not get enough relief from these medications, and/or were candidates for hip surgery or other invasive treatments. Their baseline Western Ontario and McMaster Universities OA Index (WOMAC) pain and physical function subscale scores were at least 5 at baseline and Patient’s Global Assessment of OA (PGA OA) was "fair," "poor," or "very poor." Almost half were candidates for invasive treatment.
At 16 weeks, compared with placebo, treatment with tanezumab, at all three doses, "produced greater, clinically meaningful, and statistically significant improvements" in each of the three primary efficacy endpoints: changes in the WOMAC pain subscale, the WOMAC physical function subscale, and the PGA OA. Differences between placebo and treatment were greatest for the two higher doses.
On the WOMAC pain subscale, mean baseline scores of all groups ranged from 7.2 to 7.3. Placebo patients’ scores improved by a mean of -1.62 points, but in comparison, improvements were significantly better on progressively higher doses of tanezumab: –2.90 for 2.5 mg, –3.31 for 5 mg, and –3.37 for 10 mg. Similar results occurred on the WOMAC physical function subscale scores, with mean improvements of –1.39 for placebo, –2.57 for 2.5 mg, –2.88 mg for 5 mg, and –3.00 mg for 10 mg.
Other findings included a significantly greater proportion of patients on the treatment who achieved at least 30%, 50%, 70%, and 90% reductions in the WOMAC pain subscale, compared with placebo at 16 weeks. About two-thirds of the patients on treatment remained in the study through week 24 (considered the end of the study because of tanezumab’s long half-life), compared with 38% of those on placebo.
Overall, more patients on tanezumab reported adverse effects (55%-58%, vs. 44% on placebo). Serious adverse events occurred in 3%-4.5% of patients in the treatment groups.
The rate of total hip replacements was similar in the treatment and placebo groups, with a total of eight patients who had a total hip replacement (three in the placebo group, two each in the 2.5-mg and 5-mg groups, and one in the 10-mg group). These included two patients on tanezumab who had osteonecrosis thought to be related to tanezumab. The authors pointed out that an outside adjudication committee, however, did not confirm osteonecrosis in either patient, and concluded that the reasons for joint replacements in the eight patients were inflammatory arthropathy, end-stage OA, rapidly progressive OA, or worsening OA.
Dr. Altman said that he has consulted with Abbott and its now discontinued program on anti-NGF drugs.
FROM ARTHRITIS & RHEUMATISM
Major finding: Mean scores on the WOMAC pain subscale at 16 weeks improved by a mean of –1.62 points on placebo, but in comparison, improvements were significantly better on progressively higher doses of tanezumab: –2.90 for 2.5 mg, –3.31 for 5 mg, and –3.37 for 10 mg.
Data source: A 16-week, randomized, double-blind placebo-controlled study of 621 patients with painful hip OA.
Disclosures: The study was funded by Pfizer and four of the six authors are full-time Pfizer employees. Dr. Altman said that he has consulted with Abbott and its now discontinued program on anti-NGF drugs.

'Mobility shoes' potentially benefit knee OA
A pilot study involving 16 people with osteoarthritis of the knee found that wearing specially designed shoes for 6 months significantly reduced knee loading, providing evidence that specialized footwear might be useful in the management of patients with OA of the knee, according to the investigators.
The improvements seen with the use of the shoes suggest "that footwear can be used as a mechanical device to achieve beneficial adaptations and alteration of gait with chronic use," said Dr. Najia Shakoor of the rheumatology section at Rush University, Chicago, and her associates.

In addition, after wearing the shoes for 6 months, the reduced load was evident even when the participants wore conventional shoes, suggesting "the presence of a gait adaptation with reductions in loading present even once the mobility shoes are removed," they added (Arthritis Rheum. 2013 April 10 [doi:10.1002/art.37896]).
Invented by Dr. Shakoor and one of her coauthors, Dr. Roy Lidtke, who is also at Rush, the mobility shoe has grooves that are "strategically placed at major flexion points of the foot to allow for natural, ‘barefoot-like’ movement of the foot," they said. In a previous study, they showed that the shoe mimicked the unloading effect of walking barefoot. They referred to evidence that elevated load on the knee joint in people with knee OA is associated with "radiographic severity, disease progression, and pain," and therefore, "reduction of loading at the knee may yield significant symptomatic benefits."
The patent for the shoe is owned by Rush University Medical Center, and if successful, the university will be paid for the licensing agreement and Dr. Shakoor and Dr. Lidtke also will receive some of the payments.
The study evaluated the effects of the shoes on the kinematics of gait in patients with medial compartment knee OA who were a mean age of 57 years. In the more symptomatic knee of each patient, the investigators measured the knee adduction moment (KAM) at 6, 12, and 24 weeks. KAM is a validated measure of medial compartment knee loading that has been associated with pain and progression of knee OA. Patients wore the shoes for at least 6 hours a day, 6 days a week (mean was 7 hours a day). Overall, 10 patients completed the study; the other 6 either dropped out because of a lack of effect or did not return for later visits; their results were included in the analysis.
At 24 weeks, the KAM was 18% lower when compared with measurements obtained at baseline, when the participants were wearing their regular shoes – a statistically significant difference. In addition, at 24 weeks, compared with baseline, the KAM was 11% lower while they were wearing their own shoes and 10% lower when walking barefoot, indicating that wearing the shoes may have "retrained" their gait, according to Dr. Shakoor and her associates.
Other findings included significant improvements in the pain of the affected knee, as measured by the Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC).
This proof-of-principle study "provides support for the importance of footwear choice in the management of knee OA," the authors concluded. In previous studies, the authors showed that walking barefoot and wearing footwear designed to mimic barefoot mechanics was associated with reductions in knee loading, compared with wearing conventional footwear, but they had not determined the effects of the shoes over a longer period of time.
The study had limitations, including its sample size and follow-up duration of only 6 months, as well as the fact that the complete benefit of the shoes may not be clear until they are worn in a larger group of people for a longer period of time, the authors pointed out.
The study was supported by the Arthritis Foundation.
A pilot study involving 16 people with osteoarthritis of the knee found that wearing specially designed shoes for 6 months significantly reduced knee loading, providing evidence that specialized footwear might be useful in the management of patients with OA of the knee, according to the investigators.
The improvements seen with the use of the shoes suggest "that footwear can be used as a mechanical device to achieve beneficial adaptations and alteration of gait with chronic use," said Dr. Najia Shakoor of the rheumatology section at Rush University, Chicago, and her associates.
In addition, after wearing the shoes for 6 months, the reduced load was evident even when the participants wore conventional shoes, suggesting "the presence of a gait adaptation with reductions in loading present even once the mobility shoes are removed," they added (Arthritis Rheum. 2013 April 10 [doi:10.1002/art.37896]).
Invented by Dr. Shakoor and one of her coauthors, Dr. Roy Lidtke, who is also at Rush, the mobility shoe has grooves that are "strategically placed at major flexion points of the foot to allow for natural, ‘barefoot-like’ movement of the foot," they said. In a previous study, they showed that the shoe mimicked the unloading effect of walking barefoot. They referred to evidence that elevated load on the knee joint in people with knee OA is associated with "radiographic severity, disease progression, and pain," and therefore, "reduction of loading at the knee may yield significant symptomatic benefits."
The patent for the shoe is owned by Rush University Medical Center, and if successful, the university will be paid for the licensing agreement and Dr. Shakoor and Dr. Lidtke also will receive some of the payments.
The study evaluated the effects of the shoes on the kinematics of gait in patients with medial compartment knee OA who were a mean age of 57 years. In the more symptomatic knee of each patient, the investigators measured the knee adduction moment (KAM) at 6, 12, and 24 weeks. KAM is a validated measure of medial compartment knee loading that has been associated with pain and progression of knee OA. Patients wore the shoes for at least 6 hours a day, 6 days a week (mean was 7 hours a day). Overall, 10 patients completed the study; the other 6 either dropped out because of a lack of effect or did not return for later visits; their results were included in the analysis.
At 24 weeks, the KAM was 18% lower when compared with measurements obtained at baseline, when the participants were wearing their regular shoes – a statistically significant difference. In addition, at 24 weeks, compared with baseline, the KAM was 11% lower while they were wearing their own shoes and 10% lower when walking barefoot, indicating that wearing the shoes may have "retrained" their gait, according to Dr. Shakoor and her associates.
Other findings included significant improvements in the pain of the affected knee, as measured by the Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC).
This proof-of-principle study "provides support for the importance of footwear choice in the management of knee OA," the authors concluded. In previous studies, the authors showed that walking barefoot and wearing footwear designed to mimic barefoot mechanics was associated with reductions in knee loading, compared with wearing conventional footwear, but they had not determined the effects of the shoes over a longer period of time.
The study had limitations, including its sample size and follow-up duration of only 6 months, as well as the fact that the complete benefit of the shoes may not be clear until they are worn in a larger group of people for a longer period of time, the authors pointed out.
The study was supported by the Arthritis Foundation.
A pilot study involving 16 people with osteoarthritis of the knee found that wearing specially designed shoes for 6 months significantly reduced knee loading, providing evidence that specialized footwear might be useful in the management of patients with OA of the knee, according to the investigators.
The improvements seen with the use of the shoes suggest "that footwear can be used as a mechanical device to achieve beneficial adaptations and alteration of gait with chronic use," said Dr. Najia Shakoor of the rheumatology section at Rush University, Chicago, and her associates.
In addition, after wearing the shoes for 6 months, the reduced load was evident even when the participants wore conventional shoes, suggesting "the presence of a gait adaptation with reductions in loading present even once the mobility shoes are removed," they added (Arthritis Rheum. 2013 April 10 [doi:10.1002/art.37896]).
Invented by Dr. Shakoor and one of her coauthors, Dr. Roy Lidtke, who is also at Rush, the mobility shoe has grooves that are "strategically placed at major flexion points of the foot to allow for natural, ‘barefoot-like’ movement of the foot," they said. In a previous study, they showed that the shoe mimicked the unloading effect of walking barefoot. They referred to evidence that elevated load on the knee joint in people with knee OA is associated with "radiographic severity, disease progression, and pain," and therefore, "reduction of loading at the knee may yield significant symptomatic benefits."
The patent for the shoe is owned by Rush University Medical Center, and if successful, the university will be paid for the licensing agreement and Dr. Shakoor and Dr. Lidtke also will receive some of the payments.
The study evaluated the effects of the shoes on the kinematics of gait in patients with medial compartment knee OA who were a mean age of 57 years. In the more symptomatic knee of each patient, the investigators measured the knee adduction moment (KAM) at 6, 12, and 24 weeks. KAM is a validated measure of medial compartment knee loading that has been associated with pain and progression of knee OA. Patients wore the shoes for at least 6 hours a day, 6 days a week (mean was 7 hours a day). Overall, 10 patients completed the study; the other 6 either dropped out because of a lack of effect or did not return for later visits; their results were included in the analysis.
At 24 weeks, the KAM was 18% lower when compared with measurements obtained at baseline, when the participants were wearing their regular shoes – a statistically significant difference. In addition, at 24 weeks, compared with baseline, the KAM was 11% lower while they were wearing their own shoes and 10% lower when walking barefoot, indicating that wearing the shoes may have "retrained" their gait, according to Dr. Shakoor and her associates.
Other findings included significant improvements in the pain of the affected knee, as measured by the Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC).
This proof-of-principle study "provides support for the importance of footwear choice in the management of knee OA," the authors concluded. In previous studies, the authors showed that walking barefoot and wearing footwear designed to mimic barefoot mechanics was associated with reductions in knee loading, compared with wearing conventional footwear, but they had not determined the effects of the shoes over a longer period of time.
The study had limitations, including its sample size and follow-up duration of only 6 months, as well as the fact that the complete benefit of the shoes may not be clear until they are worn in a larger group of people for a longer period of time, the authors pointed out.
The study was supported by the Arthritis Foundation.
FROM ARTHRITIS & RHEUMATISM
Major finding: Wearing specially designed shoes for 6 months resulted in an 18% reduction in the load on the knee (the knee adduction moment).
Data source: A proof-of-concept study involving 16 patients with medial compartment knee OA.
Disclosures: The patent for the shoe is owned by Rush University Medical Center, and if successful, the university will receive payment for the licensing agreement and Dr. Shakoor and Dr. Lidtke also will receive some of the payment. The study was supported by the Arthritis Foundation.

FDA approves once-popular morning sickness drug
For the first time in 30 years, clinicians will be able to prescribe a Food and Drug Administration-approved treatment for their pregnant patients who are experiencing nausea and vomiting and have not responded to changes in their diet or other conservative measures.
On April 9, the FDA announced the approval of a delayed-release, fixed-dose formulation of 10 mg of doxylamine, an antihistamine; and 10 mg of pyridoxine, a vitamin B6 analog, for "the treatment of nausea and vomiting of pregnancy in women who do not respond to conservative management."
This combination was widely used as an effective treatment for nausea and vomiting of pregnancy (NVP) in the United States and Canada in the 1960s and 1970s, and was marketed as Bendectin. But it was voluntarily taken off the U.S. market by the manufacturer in 1983, amid allegations and lawsuits that the drug caused birth defects – which were never substantiated with scientific data. The combination, which has been available as separate components in the United States, was recommended as a first-line treatment for NVP in a 2004 American College of Obstetricians and Gynecologists practice statement that was reaffirmed in 2011 (Obstet. Gynecol. 2004;103:803-15).
In a clear sign that the issue has been resolved, the FDA has assigned Diclegis a category A pregnancy risk rating, with a statement in the label summarizing the epidemiologic studies indicating that the combination of the two active ingredients has not been associated with increased risks to the fetus.
"Diclegis is now the only FDA-approved treatment for nausea and vomiting due to pregnancy, providing a therapeutic option for pregnant women seeking relief from these symptoms," Dr. Hylton V. Joffe, director of the division of reproductive and urologic products in the FDA’s Center for Drug Evaluation and Research, said in the FDA statement. It is expected to be widely available in the United States by the end of May, according to the Canadian manufacturer, Duchesnay Inc., which markets the same product in Canada.
The approval "is a big victory for American women and health care professionals, especially obstetricians and family physicians, because over the last 3 decades, American women have not had an FDA-approved medication for the most common condition of pregnancy, morning sickness," said Dr. Gideon Koren, the head of the Research Leadership for Better Pharmacotherapy During Pregnancy and Lactation at the Hospital for Sick Children, Toronto. After Bendectin was removed from the market, the rate of hospitalizations for hyperemesis gravidarum markedly increased in the United States, "clearly showing that when you remove a drug that a very large number of women need, a void is created and women suffer," he added.
The FDA approval was based on a study of 261 women with NVP in the United States who were at least 18 years-of-age and who were 7-14 weeks pregnant (mean gestational age was 9.3 weeks). Those randomized to receive treatment with Diclegis for 2 weeks had greater improvements in nausea and vomiting, compared with those who received a placebo, as reflected in changes in a score that incorporated the number of daily episodes of vomiting and heaves, and hours of nausea. The most common side effect associated with Diclegis was drowsiness, which can be severe, according to the FDA.
Diclegis is taken daily and is not used as an as-needed treatment for symptoms. The recommended dosage regimen is an initial dose of two tablets at bedtime on the first day. If symptoms persist through the afternoon of the second day, the woman should take two tablets at bedtime and one tablet the following morning (day 3). If symptoms are still present on the fourth day, she should take one tablet in the morning, one tablet in the middle of the afternoon and two tablets at bedtime. Four tablets are the maximum recommended daily dose. In the study, 19% of the women treated with Diclegis took two tablets a day, 21% took three tablets a day, and 60% took four tablets a day.
The prescribing information includes a statement that the combination of doxylamine and pyridoxine "has been the subject of many epidemiologic studies (cohort, case control, and meta-analyses) designed to detect possible teratogenicity," including studies published between 1963 and 1991, which did not report evidence of fetal abnormalities associated with first trimester exposure.
In an interview, Dr. Koren, professor of pediatrics, pharmacology, pharmacy, medicine, and medical genetics at the University of Toronto, said that the safety studies include data on more than 300,000 mother-child pairs. When Bendectin was taken off the U.S. market, it was already available as a generic in Canada as Diclectin and was never taken off the market. The continuous experience with the product in Canada should also make U.S. practitioners confident with its safety, he added. Dr. Koren was the primary investigator in the U.S. study (Am. J. Obstet. Gynecol. 2010;203:571.e1-7).
Since NVP usually improves after the first trimester, "health care professionals should reassess their patients for continued need for Diclegis as pregnancy progresses," the FDA statement says. In addition, women should not use Diclegis "when engaging in activities requiring mental alertness, such as driving or operating heavy machinery, until cleared to do so by their health care provider."
The label states that Diclegis has not been studied in women with hyperemesis gravidarum and that women should not breastfeed while on the drug.
Dr. Koren has served as a consultant for Duschesnay.
The prescribing information for Diclegis is available here. Adverse events associated with this product should be reported to the FDA’s MedWatch program or by calling 800-332-1088.
For the first time in 30 years, clinicians will be able to prescribe a Food and Drug Administration-approved treatment for their pregnant patients who are experiencing nausea and vomiting and have not responded to changes in their diet or other conservative measures.
On April 9, the FDA announced the approval of a delayed-release, fixed-dose formulation of 10 mg of doxylamine, an antihistamine; and 10 mg of pyridoxine, a vitamin B6 analog, for "the treatment of nausea and vomiting of pregnancy in women who do not respond to conservative management."
This combination was widely used as an effective treatment for nausea and vomiting of pregnancy (NVP) in the United States and Canada in the 1960s and 1970s, and was marketed as Bendectin. But it was voluntarily taken off the U.S. market by the manufacturer in 1983, amid allegations and lawsuits that the drug caused birth defects – which were never substantiated with scientific data. The combination, which has been available as separate components in the United States, was recommended as a first-line treatment for NVP in a 2004 American College of Obstetricians and Gynecologists practice statement that was reaffirmed in 2011 (Obstet. Gynecol. 2004;103:803-15).
In a clear sign that the issue has been resolved, the FDA has assigned Diclegis a category A pregnancy risk rating, with a statement in the label summarizing the epidemiologic studies indicating that the combination of the two active ingredients has not been associated with increased risks to the fetus.
"Diclegis is now the only FDA-approved treatment for nausea and vomiting due to pregnancy, providing a therapeutic option for pregnant women seeking relief from these symptoms," Dr. Hylton V. Joffe, director of the division of reproductive and urologic products in the FDA’s Center for Drug Evaluation and Research, said in the FDA statement. It is expected to be widely available in the United States by the end of May, according to the Canadian manufacturer, Duchesnay Inc., which markets the same product in Canada.
The approval "is a big victory for American women and health care professionals, especially obstetricians and family physicians, because over the last 3 decades, American women have not had an FDA-approved medication for the most common condition of pregnancy, morning sickness," said Dr. Gideon Koren, the head of the Research Leadership for Better Pharmacotherapy During Pregnancy and Lactation at the Hospital for Sick Children, Toronto. After Bendectin was removed from the market, the rate of hospitalizations for hyperemesis gravidarum markedly increased in the United States, "clearly showing that when you remove a drug that a very large number of women need, a void is created and women suffer," he added.
The FDA approval was based on a study of 261 women with NVP in the United States who were at least 18 years-of-age and who were 7-14 weeks pregnant (mean gestational age was 9.3 weeks). Those randomized to receive treatment with Diclegis for 2 weeks had greater improvements in nausea and vomiting, compared with those who received a placebo, as reflected in changes in a score that incorporated the number of daily episodes of vomiting and heaves, and hours of nausea. The most common side effect associated with Diclegis was drowsiness, which can be severe, according to the FDA.
Diclegis is taken daily and is not used as an as-needed treatment for symptoms. The recommended dosage regimen is an initial dose of two tablets at bedtime on the first day. If symptoms persist through the afternoon of the second day, the woman should take two tablets at bedtime and one tablet the following morning (day 3). If symptoms are still present on the fourth day, she should take one tablet in the morning, one tablet in the middle of the afternoon and two tablets at bedtime. Four tablets are the maximum recommended daily dose. In the study, 19% of the women treated with Diclegis took two tablets a day, 21% took three tablets a day, and 60% took four tablets a day.
The prescribing information includes a statement that the combination of doxylamine and pyridoxine "has been the subject of many epidemiologic studies (cohort, case control, and meta-analyses) designed to detect possible teratogenicity," including studies published between 1963 and 1991, which did not report evidence of fetal abnormalities associated with first trimester exposure.
In an interview, Dr. Koren, professor of pediatrics, pharmacology, pharmacy, medicine, and medical genetics at the University of Toronto, said that the safety studies include data on more than 300,000 mother-child pairs. When Bendectin was taken off the U.S. market, it was already available as a generic in Canada as Diclectin and was never taken off the market. The continuous experience with the product in Canada should also make U.S. practitioners confident with its safety, he added. Dr. Koren was the primary investigator in the U.S. study (Am. J. Obstet. Gynecol. 2010;203:571.e1-7).
Since NVP usually improves after the first trimester, "health care professionals should reassess their patients for continued need for Diclegis as pregnancy progresses," the FDA statement says. In addition, women should not use Diclegis "when engaging in activities requiring mental alertness, such as driving or operating heavy machinery, until cleared to do so by their health care provider."
The label states that Diclegis has not been studied in women with hyperemesis gravidarum and that women should not breastfeed while on the drug.
Dr. Koren has served as a consultant for Duschesnay.
The prescribing information for Diclegis is available here. Adverse events associated with this product should be reported to the FDA’s MedWatch program or by calling 800-332-1088.
For the first time in 30 years, clinicians will be able to prescribe a Food and Drug Administration-approved treatment for their pregnant patients who are experiencing nausea and vomiting and have not responded to changes in their diet or other conservative measures.
On April 9, the FDA announced the approval of a delayed-release, fixed-dose formulation of 10 mg of doxylamine, an antihistamine; and 10 mg of pyridoxine, a vitamin B6 analog, for "the treatment of nausea and vomiting of pregnancy in women who do not respond to conservative management."
This combination was widely used as an effective treatment for nausea and vomiting of pregnancy (NVP) in the United States and Canada in the 1960s and 1970s, and was marketed as Bendectin. But it was voluntarily taken off the U.S. market by the manufacturer in 1983, amid allegations and lawsuits that the drug caused birth defects – which were never substantiated with scientific data. The combination, which has been available as separate components in the United States, was recommended as a first-line treatment for NVP in a 2004 American College of Obstetricians and Gynecologists practice statement that was reaffirmed in 2011 (Obstet. Gynecol. 2004;103:803-15).
In a clear sign that the issue has been resolved, the FDA has assigned Diclegis a category A pregnancy risk rating, with a statement in the label summarizing the epidemiologic studies indicating that the combination of the two active ingredients has not been associated with increased risks to the fetus.
"Diclegis is now the only FDA-approved treatment for nausea and vomiting due to pregnancy, providing a therapeutic option for pregnant women seeking relief from these symptoms," Dr. Hylton V. Joffe, director of the division of reproductive and urologic products in the FDA’s Center for Drug Evaluation and Research, said in the FDA statement. It is expected to be widely available in the United States by the end of May, according to the Canadian manufacturer, Duchesnay Inc., which markets the same product in Canada.
The approval "is a big victory for American women and health care professionals, especially obstetricians and family physicians, because over the last 3 decades, American women have not had an FDA-approved medication for the most common condition of pregnancy, morning sickness," said Dr. Gideon Koren, the head of the Research Leadership for Better Pharmacotherapy During Pregnancy and Lactation at the Hospital for Sick Children, Toronto. After Bendectin was removed from the market, the rate of hospitalizations for hyperemesis gravidarum markedly increased in the United States, "clearly showing that when you remove a drug that a very large number of women need, a void is created and women suffer," he added.
The FDA approval was based on a study of 261 women with NVP in the United States who were at least 18 years-of-age and who were 7-14 weeks pregnant (mean gestational age was 9.3 weeks). Those randomized to receive treatment with Diclegis for 2 weeks had greater improvements in nausea and vomiting, compared with those who received a placebo, as reflected in changes in a score that incorporated the number of daily episodes of vomiting and heaves, and hours of nausea. The most common side effect associated with Diclegis was drowsiness, which can be severe, according to the FDA.
Diclegis is taken daily and is not used as an as-needed treatment for symptoms. The recommended dosage regimen is an initial dose of two tablets at bedtime on the first day. If symptoms persist through the afternoon of the second day, the woman should take two tablets at bedtime and one tablet the following morning (day 3). If symptoms are still present on the fourth day, she should take one tablet in the morning, one tablet in the middle of the afternoon and two tablets at bedtime. Four tablets are the maximum recommended daily dose. In the study, 19% of the women treated with Diclegis took two tablets a day, 21% took three tablets a day, and 60% took four tablets a day.
The prescribing information includes a statement that the combination of doxylamine and pyridoxine "has been the subject of many epidemiologic studies (cohort, case control, and meta-analyses) designed to detect possible teratogenicity," including studies published between 1963 and 1991, which did not report evidence of fetal abnormalities associated with first trimester exposure.
In an interview, Dr. Koren, professor of pediatrics, pharmacology, pharmacy, medicine, and medical genetics at the University of Toronto, said that the safety studies include data on more than 300,000 mother-child pairs. When Bendectin was taken off the U.S. market, it was already available as a generic in Canada as Diclectin and was never taken off the market. The continuous experience with the product in Canada should also make U.S. practitioners confident with its safety, he added. Dr. Koren was the primary investigator in the U.S. study (Am. J. Obstet. Gynecol. 2010;203:571.e1-7).
Since NVP usually improves after the first trimester, "health care professionals should reassess their patients for continued need for Diclegis as pregnancy progresses," the FDA statement says. In addition, women should not use Diclegis "when engaging in activities requiring mental alertness, such as driving or operating heavy machinery, until cleared to do so by their health care provider."
The label states that Diclegis has not been studied in women with hyperemesis gravidarum and that women should not breastfeed while on the drug.
Dr. Koren has served as a consultant for Duschesnay.
The prescribing information for Diclegis is available here. Adverse events associated with this product should be reported to the FDA’s MedWatch program or by calling 800-332-1088.
High selenium exposure may lower incidence of advanced prostate cancer
Increased exposure to selenium was associated with a significantly reduced risk of advanced prostate cancer, according to the results of a large, prospective cohort study of men in the Netherlands.
The results of the study, conducted in men with low to moderate selenium levels, "suggest that selenium may prevent advanced, clinically relevant, prostate cancer," noted Milan Geybels, M.Sc., at the annual meeting of the American Association for Cancer Research. And since there is little evidence on risk factors that can modify prostate cancer risk, "any compound that would prevent the incidence of advanced, clinically relevant prostate cancer would have a substantial impact on public health," Mr. Geybels said in an interview.
The men were enrolled in the Netherlands Cohort Study, a study of diet and cancer in 58,279 men between the ages of 55 and 69 years at entry. Over 17 years, 898 men were diagnosed with advanced prostate cancer and were compared with 1,203 men also in the study. Using the concentration of selenium in the men’s toenails, an indication of long-term exposure, the investigators determined that the risk of advanced prostate cancer was significantly reduced as selenium levels increased. The risk for advanced prostate cancer was more than 60% lower in men with the highest levels of toenail selenium, when compared with those with the lowest levels, and the association was "more pronounced" among the men diagnosed later in the follow-up period, noted Mr. Geybels, a doctoral candidate in cancer epidemiology at Maastricht University, the Netherlands.
Previous prospective studies on the possible link between selenium and prostate cancer risk have produced conflicting results. Many of the studies included men with moderate to high selenium levels. Also, they did not exclusively study advanced prostate cancer, and "almost exclusively" used blood levels of selenium, which reflect recent exposure, he noted.
SELECT (Selenium and Vitamin E Cancer Prevention Trial) found no relationship between selenium supplementation and prostate cancer risk. However, "a possible explanation for the null finding is that selenium intake among SELECT participants was adequate ... [so], further selenium supplementation had no effect on prostate cancer incidence," he said.
The intake of selenium varies worldwide as a result of variations in soil content and the related variability in the selenium content in foods; in the Netherlands, low selenium is common.
The results need to be replicated in more prospective studies, and if confirmed, "A prevention trial of selenium and advanced prostate cancer in a low selenium population may be justified," said Mr. Geybels. Whether levels higher than those detected in the men in the study are associated with a further reduction in risk is unknown.
He and his coauthors had no disclosures to report.
Increased exposure to selenium was associated with a significantly reduced risk of advanced prostate cancer, according to the results of a large, prospective cohort study of men in the Netherlands.
The results of the study, conducted in men with low to moderate selenium levels, "suggest that selenium may prevent advanced, clinically relevant, prostate cancer," noted Milan Geybels, M.Sc., at the annual meeting of the American Association for Cancer Research. And since there is little evidence on risk factors that can modify prostate cancer risk, "any compound that would prevent the incidence of advanced, clinically relevant prostate cancer would have a substantial impact on public health," Mr. Geybels said in an interview.
The men were enrolled in the Netherlands Cohort Study, a study of diet and cancer in 58,279 men between the ages of 55 and 69 years at entry. Over 17 years, 898 men were diagnosed with advanced prostate cancer and were compared with 1,203 men also in the study. Using the concentration of selenium in the men’s toenails, an indication of long-term exposure, the investigators determined that the risk of advanced prostate cancer was significantly reduced as selenium levels increased. The risk for advanced prostate cancer was more than 60% lower in men with the highest levels of toenail selenium, when compared with those with the lowest levels, and the association was "more pronounced" among the men diagnosed later in the follow-up period, noted Mr. Geybels, a doctoral candidate in cancer epidemiology at Maastricht University, the Netherlands.
Previous prospective studies on the possible link between selenium and prostate cancer risk have produced conflicting results. Many of the studies included men with moderate to high selenium levels. Also, they did not exclusively study advanced prostate cancer, and "almost exclusively" used blood levels of selenium, which reflect recent exposure, he noted.
SELECT (Selenium and Vitamin E Cancer Prevention Trial) found no relationship between selenium supplementation and prostate cancer risk. However, "a possible explanation for the null finding is that selenium intake among SELECT participants was adequate ... [so], further selenium supplementation had no effect on prostate cancer incidence," he said.
The intake of selenium varies worldwide as a result of variations in soil content and the related variability in the selenium content in foods; in the Netherlands, low selenium is common.
The results need to be replicated in more prospective studies, and if confirmed, "A prevention trial of selenium and advanced prostate cancer in a low selenium population may be justified," said Mr. Geybels. Whether levels higher than those detected in the men in the study are associated with a further reduction in risk is unknown.
He and his coauthors had no disclosures to report.
Increased exposure to selenium was associated with a significantly reduced risk of advanced prostate cancer, according to the results of a large, prospective cohort study of men in the Netherlands.
The results of the study, conducted in men with low to moderate selenium levels, "suggest that selenium may prevent advanced, clinically relevant, prostate cancer," noted Milan Geybels, M.Sc., at the annual meeting of the American Association for Cancer Research. And since there is little evidence on risk factors that can modify prostate cancer risk, "any compound that would prevent the incidence of advanced, clinically relevant prostate cancer would have a substantial impact on public health," Mr. Geybels said in an interview.
The men were enrolled in the Netherlands Cohort Study, a study of diet and cancer in 58,279 men between the ages of 55 and 69 years at entry. Over 17 years, 898 men were diagnosed with advanced prostate cancer and were compared with 1,203 men also in the study. Using the concentration of selenium in the men’s toenails, an indication of long-term exposure, the investigators determined that the risk of advanced prostate cancer was significantly reduced as selenium levels increased. The risk for advanced prostate cancer was more than 60% lower in men with the highest levels of toenail selenium, when compared with those with the lowest levels, and the association was "more pronounced" among the men diagnosed later in the follow-up period, noted Mr. Geybels, a doctoral candidate in cancer epidemiology at Maastricht University, the Netherlands.
Previous prospective studies on the possible link between selenium and prostate cancer risk have produced conflicting results. Many of the studies included men with moderate to high selenium levels. Also, they did not exclusively study advanced prostate cancer, and "almost exclusively" used blood levels of selenium, which reflect recent exposure, he noted.
SELECT (Selenium and Vitamin E Cancer Prevention Trial) found no relationship between selenium supplementation and prostate cancer risk. However, "a possible explanation for the null finding is that selenium intake among SELECT participants was adequate ... [so], further selenium supplementation had no effect on prostate cancer incidence," he said.
The intake of selenium varies worldwide as a result of variations in soil content and the related variability in the selenium content in foods; in the Netherlands, low selenium is common.
The results need to be replicated in more prospective studies, and if confirmed, "A prevention trial of selenium and advanced prostate cancer in a low selenium population may be justified," said Mr. Geybels. Whether levels higher than those detected in the men in the study are associated with a further reduction in risk is unknown.
He and his coauthors had no disclosures to report.
FROM THE ANNUAL AACR MEETING
Major finding: The risk for advanced prostate cancer was more than 60% lower in men with the highest levels of toenail selenium, when compared with those with the lowest levels.
Data source: Men in the Netherlands Cohort Study diagnosed with advanced prostate cancer (898) were compared with 1,203 men also in the study.
Disclosures: Mr. Geybels and his colleagues had no disclosures to report.
Ruling allows all ages access to OTC emergency contraceptives
Levonorgestrel-based emergency contraceptives now will be able to be placed on pharmacy shelves, with no age restrictions on who can purchase the products over-the-counter, as a result of a U.S. District Court judge’s ruling on April 5.
Restrictions must be lifted within 30 days, according to the ruling.
The ruling addresses a controversial decision made in 2011 by Health and Human Services Secretary Kathleen Sebelius. Ms. Sebelius overruled a Food and Drug Administration decision to extend approval of the levonorgestrel-based Plan B One-Step emergency contraceptive as an over-the-counter product for women of all ages; she cited a lack of data on women of all age groups in studies submitted to the FDA for approval.
As a result, products have been kept behind pharmacy counters, and a prescription has been required for teenagers under age 17 – limiting access to ECs, particularly when pharmacies are closed.
The court’s "decision was a long time coming and is based on sound scientific evidence" and represents "a major victory for women’s health, removing barriers to teens’ and other women’s access to a needed medication by making it truly over-the-counter," Dr. Eve Espey, professor of obstetrics and gynecology, University of New Mexico, Albuquerque, said in an interview. Emergency contraception "will be on the shelf instead of behind-the-counter, where it’s easier for all women to get, and emergency contraception may be available in stores other than pharmacies since it’s over-the-counter."
For more than a decade, women’s health advocacy groups have been working to make emergency contraception available without restrictions. The effort dates back to 2001, when several groups including the Center for Reproductive Rights filed a citizen’s petition with the FDA to make emergency contraceptives available over-the-counter. In 2006, the FDA approved Plan B as an OTC product for women aged 17 years and older, who could provide proof of their age with a government-issued ID, and as a prescription product for younger women.
That approval came several years after an FDA advisory panel recommended OTC approval for women of all age groups.
In a December 2011 statement, FDA Commissioner Margaret Hamburg said that the agency’s review had concluded that there was "adequate and reasonable, well-supported, and science-based evidence that Plan B One-Step is safe and effective and should be approved for nonprescription use for all females of childbearing potential." But she announced that she had been directed by Ms. Sebelius, who disagreed, to notify the manufacturer, Teva Pharmaceuticals, that a prescription would still be required for women under age 17.
In the April 5 ruling, Federal District Judge Edward Korman, of the Eastern District of New York, lifted the age restrictions, and in his opinion he referred to Ms. Sebelius’ decision and the FDA decision to deny the citizen’s petition, as "arbitrary, capricious, and unreasonable."
"The obstructions in the path of those adolescents in obtaining levonorgestrel-based emergency contraceptives under the current behind-the-counter regime have the practical effect of making the contraceptives unavailable without a doctor’s prescription," the ruling said. The ruling reverses the FDA’s decision to deny the citizen’s petition and remanded the case to the FDA "with the instruction to grant the citizen’s petition and make levonorgestrel-based emergency contraceptives available without a prescription and without point-of-sale or age restrictions within 30 days."
During a press briefing April 5, lawyers and representatives of women’s health advocacy groups lauded the ruling as an example of science prevailing over politics.
Nancy Northrup, president and CEO of the Center for Reproductive Rights, said that "despite mountains of evidence that emergency contraception is safe and effective for all ages, the FDA has kept it locked behind the pharmacy counter, subject to ID checks, doctors’ prescriptions for younger women, and the pharmacy hours of operation."
Because of the court order, emergency contraception will no longer be denied to women who do not have a government-issued ID, young women will no longer have to get a prescription, "and no longer will any woman rush into an all-night drugstore only to find the pharmacy gates closed and emergency contraception, which is most effective when taken immediately, just out of her reach," she said.
Speaking on behalf of the American Academy of Pediatrics during the briefing, Dr. Cora Collette Breuner, professor of adolescent medicine and pediatrics at Seattle Children’s Hospital and the University of Washington, Seattle, said that the ruling was long overdue and that it was welcomed by the AAP, which issued a policy statement in December 2012 supporting teen use of emergency contraception as an important backup contraceptive method. Dr. Breuner, a coauthor of that statement, said that even with the lifted restrictions, it was important to ensure that the OTC ECs would remain available to all women of all income levels, and that the cost not be prohibitive for younger teens. The current retail cost is about $40-$50.
The American College of Obstetricians and Gynecologists and the Society for Adolescent Health and Medicine also commented on the ruling in a statement.
But in a statement issued by the Family Research Council, Anna Higgins, a lawyer and director of the council’s Center for Human Dignity, said that the ruling "places the health of young girls at risk. Making Plan B available for girls under the age of 17 without a prescription flies in the face of medical information and sound judgment. I am very troubled that the court has not fully taken into account the concerns expressed by HHS Secretary Kathleen Sebelius and many public health advocates that there is not enough data on the health effects of Plan B on young girls."
Concerns expressed by opponents of teens’ access to OTC emergency contraceptives include the risk that OTC access would encourage repeated use of ECs without medical supervision and that teens might be coerced into taking ECs.
During the briefing, Dr. Breuner said there are no scientific data showing that repeated EC use is harmful to young women, but she emphasized that it was important that young women discuss more effective and consistent forms of contraception with their health care providers. Coercion is another concern and should also be discussed with health care providers, she said.
During the briefing, Susannah Baruch, interim president and CEO of the Reproductive Health Technologies Project, added that there are studies indicating that teenaged and adult women understand that emergency contraception is intended as a backup method, and that while it may be difficult for some to accept the fact that a teenage couple may be having sex, "if they are, we think it’s important that they have access to safe options."
The FDA or Health and Human Services department had no comment on the ruling.
Plan B One-Step, which contains one 1.5-mg levonorgestrel tablet, was approved in 2009. Plan B, which contains two 0.75-mg levonorgestrel tablets, approved in 1999, is no longer marketed but generic versions are available.
The plaintiffs in the case, Tummino vs. Hamburg, include the Association of Reproductive Health Professionals.
Levonorgestrel-based emergency contraceptives now will be able to be placed on pharmacy shelves, with no age restrictions on who can purchase the products over-the-counter, as a result of a U.S. District Court judge’s ruling on April 5.
Restrictions must be lifted within 30 days, according to the ruling.
The ruling addresses a controversial decision made in 2011 by Health and Human Services Secretary Kathleen Sebelius. Ms. Sebelius overruled a Food and Drug Administration decision to extend approval of the levonorgestrel-based Plan B One-Step emergency contraceptive as an over-the-counter product for women of all ages; she cited a lack of data on women of all age groups in studies submitted to the FDA for approval.
As a result, products have been kept behind pharmacy counters, and a prescription has been required for teenagers under age 17 – limiting access to ECs, particularly when pharmacies are closed.
The court’s "decision was a long time coming and is based on sound scientific evidence" and represents "a major victory for women’s health, removing barriers to teens’ and other women’s access to a needed medication by making it truly over-the-counter," Dr. Eve Espey, professor of obstetrics and gynecology, University of New Mexico, Albuquerque, said in an interview. Emergency contraception "will be on the shelf instead of behind-the-counter, where it’s easier for all women to get, and emergency contraception may be available in stores other than pharmacies since it’s over-the-counter."
For more than a decade, women’s health advocacy groups have been working to make emergency contraception available without restrictions. The effort dates back to 2001, when several groups including the Center for Reproductive Rights filed a citizen’s petition with the FDA to make emergency contraceptives available over-the-counter. In 2006, the FDA approved Plan B as an OTC product for women aged 17 years and older, who could provide proof of their age with a government-issued ID, and as a prescription product for younger women.
That approval came several years after an FDA advisory panel recommended OTC approval for women of all age groups.
In a December 2011 statement, FDA Commissioner Margaret Hamburg said that the agency’s review had concluded that there was "adequate and reasonable, well-supported, and science-based evidence that Plan B One-Step is safe and effective and should be approved for nonprescription use for all females of childbearing potential." But she announced that she had been directed by Ms. Sebelius, who disagreed, to notify the manufacturer, Teva Pharmaceuticals, that a prescription would still be required for women under age 17.
In the April 5 ruling, Federal District Judge Edward Korman, of the Eastern District of New York, lifted the age restrictions, and in his opinion he referred to Ms. Sebelius’ decision and the FDA decision to deny the citizen’s petition, as "arbitrary, capricious, and unreasonable."
"The obstructions in the path of those adolescents in obtaining levonorgestrel-based emergency contraceptives under the current behind-the-counter regime have the practical effect of making the contraceptives unavailable without a doctor’s prescription," the ruling said. The ruling reverses the FDA’s decision to deny the citizen’s petition and remanded the case to the FDA "with the instruction to grant the citizen’s petition and make levonorgestrel-based emergency contraceptives available without a prescription and without point-of-sale or age restrictions within 30 days."
During a press briefing April 5, lawyers and representatives of women’s health advocacy groups lauded the ruling as an example of science prevailing over politics.
Nancy Northrup, president and CEO of the Center for Reproductive Rights, said that "despite mountains of evidence that emergency contraception is safe and effective for all ages, the FDA has kept it locked behind the pharmacy counter, subject to ID checks, doctors’ prescriptions for younger women, and the pharmacy hours of operation."
Because of the court order, emergency contraception will no longer be denied to women who do not have a government-issued ID, young women will no longer have to get a prescription, "and no longer will any woman rush into an all-night drugstore only to find the pharmacy gates closed and emergency contraception, which is most effective when taken immediately, just out of her reach," she said.
Speaking on behalf of the American Academy of Pediatrics during the briefing, Dr. Cora Collette Breuner, professor of adolescent medicine and pediatrics at Seattle Children’s Hospital and the University of Washington, Seattle, said that the ruling was long overdue and that it was welcomed by the AAP, which issued a policy statement in December 2012 supporting teen use of emergency contraception as an important backup contraceptive method. Dr. Breuner, a coauthor of that statement, said that even with the lifted restrictions, it was important to ensure that the OTC ECs would remain available to all women of all income levels, and that the cost not be prohibitive for younger teens. The current retail cost is about $40-$50.
The American College of Obstetricians and Gynecologists and the Society for Adolescent Health and Medicine also commented on the ruling in a statement.
But in a statement issued by the Family Research Council, Anna Higgins, a lawyer and director of the council’s Center for Human Dignity, said that the ruling "places the health of young girls at risk. Making Plan B available for girls under the age of 17 without a prescription flies in the face of medical information and sound judgment. I am very troubled that the court has not fully taken into account the concerns expressed by HHS Secretary Kathleen Sebelius and many public health advocates that there is not enough data on the health effects of Plan B on young girls."
Concerns expressed by opponents of teens’ access to OTC emergency contraceptives include the risk that OTC access would encourage repeated use of ECs without medical supervision and that teens might be coerced into taking ECs.
During the briefing, Dr. Breuner said there are no scientific data showing that repeated EC use is harmful to young women, but she emphasized that it was important that young women discuss more effective and consistent forms of contraception with their health care providers. Coercion is another concern and should also be discussed with health care providers, she said.
During the briefing, Susannah Baruch, interim president and CEO of the Reproductive Health Technologies Project, added that there are studies indicating that teenaged and adult women understand that emergency contraception is intended as a backup method, and that while it may be difficult for some to accept the fact that a teenage couple may be having sex, "if they are, we think it’s important that they have access to safe options."
The FDA or Health and Human Services department had no comment on the ruling.
Plan B One-Step, which contains one 1.5-mg levonorgestrel tablet, was approved in 2009. Plan B, which contains two 0.75-mg levonorgestrel tablets, approved in 1999, is no longer marketed but generic versions are available.
The plaintiffs in the case, Tummino vs. Hamburg, include the Association of Reproductive Health Professionals.
Levonorgestrel-based emergency contraceptives now will be able to be placed on pharmacy shelves, with no age restrictions on who can purchase the products over-the-counter, as a result of a U.S. District Court judge’s ruling on April 5.
Restrictions must be lifted within 30 days, according to the ruling.
The ruling addresses a controversial decision made in 2011 by Health and Human Services Secretary Kathleen Sebelius. Ms. Sebelius overruled a Food and Drug Administration decision to extend approval of the levonorgestrel-based Plan B One-Step emergency contraceptive as an over-the-counter product for women of all ages; she cited a lack of data on women of all age groups in studies submitted to the FDA for approval.
As a result, products have been kept behind pharmacy counters, and a prescription has been required for teenagers under age 17 – limiting access to ECs, particularly when pharmacies are closed.
The court’s "decision was a long time coming and is based on sound scientific evidence" and represents "a major victory for women’s health, removing barriers to teens’ and other women’s access to a needed medication by making it truly over-the-counter," Dr. Eve Espey, professor of obstetrics and gynecology, University of New Mexico, Albuquerque, said in an interview. Emergency contraception "will be on the shelf instead of behind-the-counter, where it’s easier for all women to get, and emergency contraception may be available in stores other than pharmacies since it’s over-the-counter."
For more than a decade, women’s health advocacy groups have been working to make emergency contraception available without restrictions. The effort dates back to 2001, when several groups including the Center for Reproductive Rights filed a citizen’s petition with the FDA to make emergency contraceptives available over-the-counter. In 2006, the FDA approved Plan B as an OTC product for women aged 17 years and older, who could provide proof of their age with a government-issued ID, and as a prescription product for younger women.
That approval came several years after an FDA advisory panel recommended OTC approval for women of all age groups.
In a December 2011 statement, FDA Commissioner Margaret Hamburg said that the agency’s review had concluded that there was "adequate and reasonable, well-supported, and science-based evidence that Plan B One-Step is safe and effective and should be approved for nonprescription use for all females of childbearing potential." But she announced that she had been directed by Ms. Sebelius, who disagreed, to notify the manufacturer, Teva Pharmaceuticals, that a prescription would still be required for women under age 17.
In the April 5 ruling, Federal District Judge Edward Korman, of the Eastern District of New York, lifted the age restrictions, and in his opinion he referred to Ms. Sebelius’ decision and the FDA decision to deny the citizen’s petition, as "arbitrary, capricious, and unreasonable."
"The obstructions in the path of those adolescents in obtaining levonorgestrel-based emergency contraceptives under the current behind-the-counter regime have the practical effect of making the contraceptives unavailable without a doctor’s prescription," the ruling said. The ruling reverses the FDA’s decision to deny the citizen’s petition and remanded the case to the FDA "with the instruction to grant the citizen’s petition and make levonorgestrel-based emergency contraceptives available without a prescription and without point-of-sale or age restrictions within 30 days."
During a press briefing April 5, lawyers and representatives of women’s health advocacy groups lauded the ruling as an example of science prevailing over politics.
Nancy Northrup, president and CEO of the Center for Reproductive Rights, said that "despite mountains of evidence that emergency contraception is safe and effective for all ages, the FDA has kept it locked behind the pharmacy counter, subject to ID checks, doctors’ prescriptions for younger women, and the pharmacy hours of operation."
Because of the court order, emergency contraception will no longer be denied to women who do not have a government-issued ID, young women will no longer have to get a prescription, "and no longer will any woman rush into an all-night drugstore only to find the pharmacy gates closed and emergency contraception, which is most effective when taken immediately, just out of her reach," she said.
Speaking on behalf of the American Academy of Pediatrics during the briefing, Dr. Cora Collette Breuner, professor of adolescent medicine and pediatrics at Seattle Children’s Hospital and the University of Washington, Seattle, said that the ruling was long overdue and that it was welcomed by the AAP, which issued a policy statement in December 2012 supporting teen use of emergency contraception as an important backup contraceptive method. Dr. Breuner, a coauthor of that statement, said that even with the lifted restrictions, it was important to ensure that the OTC ECs would remain available to all women of all income levels, and that the cost not be prohibitive for younger teens. The current retail cost is about $40-$50.
The American College of Obstetricians and Gynecologists and the Society for Adolescent Health and Medicine also commented on the ruling in a statement.
But in a statement issued by the Family Research Council, Anna Higgins, a lawyer and director of the council’s Center for Human Dignity, said that the ruling "places the health of young girls at risk. Making Plan B available for girls under the age of 17 without a prescription flies in the face of medical information and sound judgment. I am very troubled that the court has not fully taken into account the concerns expressed by HHS Secretary Kathleen Sebelius and many public health advocates that there is not enough data on the health effects of Plan B on young girls."
Concerns expressed by opponents of teens’ access to OTC emergency contraceptives include the risk that OTC access would encourage repeated use of ECs without medical supervision and that teens might be coerced into taking ECs.
During the briefing, Dr. Breuner said there are no scientific data showing that repeated EC use is harmful to young women, but she emphasized that it was important that young women discuss more effective and consistent forms of contraception with their health care providers. Coercion is another concern and should also be discussed with health care providers, she said.
During the briefing, Susannah Baruch, interim president and CEO of the Reproductive Health Technologies Project, added that there are studies indicating that teenaged and adult women understand that emergency contraception is intended as a backup method, and that while it may be difficult for some to accept the fact that a teenage couple may be having sex, "if they are, we think it’s important that they have access to safe options."
The FDA or Health and Human Services department had no comment on the ruling.
Plan B One-Step, which contains one 1.5-mg levonorgestrel tablet, was approved in 2009. Plan B, which contains two 0.75-mg levonorgestrel tablets, approved in 1999, is no longer marketed but generic versions are available.
The plaintiffs in the case, Tummino vs. Hamburg, include the Association of Reproductive Health Professionals.
Panel backs approval of implants for addiction treatment
SILVER SPRING, MD. – A four-rod subdermal implant that slowly releases buprenorphine over 6 months should be approved for maintenance treatment of opioid dependence, although more work is needed to determine optimal dosing strategies and how to address the potential risks of the treatment, the majority of a Food and Drug Administration advisory panel recommended.
At a meeting on March 21, the FDA’s Psychopharmacologic Drugs Advisory Committee voted 10-4, with 1 abstention, to recommend approval, based on the efficacy, safety, and risk-benefit profile in opioid-addicted adults treated with the implants, in two phase III placebo-controlled studies. In those studies, the mean proportion of negative urine tests over 24 weeks, the primary endpoint, was significantly higher among those who received the buprenorphine implant, compared with those who had a placebo implant.
The panel agreed the treatment had a beneficial effect on reducing illicit opioid use, but those voting on both sides pointed out that more information on dosing was needed. The product is indicated for patients who are maintained on 12 mg-16 mg of oral buprenorphine a day. But as addiction treatment specialists on the panel pointed out, some patients are maintained on lower oral doses, and there were no data on how to use the product in such patients. The manufacturer, Titan Pharmaceuticals, plans to study that issue further.
The panel also voted 12-2 with 1 abstention, that the company had adequately characterized the safety profile of treatment with the buprenorphine implants, which was associated with the known side effects of the oral formulation in the trials. But there are also the potential complications associated with surgical implantation and removal of the rods every 6 months, as well as the potential for abuse, misuse and diversion, and accidental exposure, which is addressed in a complex risk management program proposed by the manufacturer.
Using similar technology as contraceptive implants, the product consists of four rods, each measuring 26 mm by 2.5 mm and containing 80 mg of the buprenorphine, a partial mu-opioid receptor agonist that has been available in oral formulations since 2002 for the treatment of opioid dependence. The rods are subdermally implanted in the upper arm with a single-use application by a trained health care provider in an office-based surgical procedure with local anesthesia after the patient has been titrated with an oral dose. The rods stay in place for up to 6 months, at which time they are removed and replaced at another site.
"Clearly, the data need to be further assessed and the safety and dosing improved upon," said Dr. Christopher J. Kratochvil, professor of psychiatry and pediatrics, University of Nebraska, Omaha. But he voted in favor of approval, commenting, "This is will at least be an incremental step forward for a disorder that has very tragic consequences."
"Overall, I thought that benefit was shown and there might be a particular subset of patients who respond best to this intervention," said one of the two panelists with expertise on contraceptive implants, Dr. Geri D. Hewitt, an obstetrician-gynecologist at Ohio State University, Columbus, who voted for approval. "I did not see any evidence of significant harm; I do have questions about dosing and which physicians will be willing to place this and ... it’s going to be important to keep track of adverse event reporting," she added.
Voting against approval, Dr.. Laura F. McNicholas of the Center for Studies of Addiction at the University of Pennsylvania, Philadelphia, said she thought the product was approvable, but that approval before a dose-ranging study was conducted would be premature. A major concern she had was running out of sites to implant the rods for patients on long-term treatment, since many of her patients have been taking buprenorphine continuously for years. Many of her patients also are on doses under 12 mg a day, and there was no information on how to dose such patients with the implant, for which there is currently only one dose, she added.
So far, the company has identified four sites on the upper arms as appropriate for implantation and is studying this further.

The two studies enrolled 450 adults (mean age, mid-30s) addicted to an opioid, randomized to treatment with the buprenorphine implant or a placebo implant. The most common drug of abuse was heroin in 52% to 67%, followed by prescription analgesics in 33% to 48%.Over 24 weeks, the mean percentage of opioid-negative urine samples was statistically significantly greater among those who were treated with the buprenorphine implant (about 40% and about 35% among those who received the active drug vs. about 30% and 15%, respectively, among those on the placebo implant.)
About 80% of the patients were adequately treated with four implants, but in some patients, a fifth rod or supplemental treatment with sublingual buprenorphine was needed. Completion rates were lower among those on placebo (31% and 26%, vs. 64% and 66%, respectively). In the two studies, the most common implant site–related adverse events included pruritus, pain, erythema, edema, and hematoma.
The company’s Risk Evaluation and Mitigation Strategy (REMS) includes plans for restricted distribution, a specialty pharmacy only, and a training-certification program for health care providers who implant the rods. (The FDA requires a REMS for a drug when it determines that a risk management strategies beyond the label are needed to ensure the benefits of a drug outweigh its risks.) Both the panel and the FDA reviewers said the REMS needs to be improved.
When asked about the panel’s recommendation, Dr. Robert L DuPont said in an interview that it is a major development in medication-assisted treatment of opiate dependence in light of problems with diversion and nonmedical use of buprenorphine. "This novel dosing strategy removes the risk of diversion in a large population of drug abusing patients that poses a high risk of diversion and misuse," said Dr. DuPont, the first director of the National Institute on Drug Abuse.
"The development of abuse-resistant formulations of controlled substances is one of the best new ideas to turn back the major epidemic of prescription drug abuse."
If approved, the panel recommended that other potential problems with the device should be monitored closely, including removal of implants by nonmedical personnel for diversion (which was not seen in the studies) and the risk of long-term exposure to the components if they are not removed. The panel urged study of whether implants can be inserted in previous implant sites.
Buprenorphine can be administered in an office setting as opposed to a methadone clinic. The currently marketed buprenorphine products are Subutex and Suboxone (buprenorphine with naloxone), both available in generic formulations; and a sublingual formulation approved in 2010. In 2012, 10.7 million prescriptions were dispensed for buprenorphine products for an estimated 1 million patients, mostly the buprenorphine/naloxone combination, according to the FDA. Between January 2003 and December 2012, an estimated 40 million buprenorphine prescriptions were written. The top three categories of prescribers were general practitioners/family physicians/doctors of osteopathy (one category), psychiatrists, and internists.
If approved, the manufacturer plans to market it as Probuphine. The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist might be given a waiver, but not at this meeting.
SILVER SPRING, MD. – A four-rod subdermal implant that slowly releases buprenorphine over 6 months should be approved for maintenance treatment of opioid dependence, although more work is needed to determine optimal dosing strategies and how to address the potential risks of the treatment, the majority of a Food and Drug Administration advisory panel recommended.
At a meeting on March 21, the FDA’s Psychopharmacologic Drugs Advisory Committee voted 10-4, with 1 abstention, to recommend approval, based on the efficacy, safety, and risk-benefit profile in opioid-addicted adults treated with the implants, in two phase III placebo-controlled studies. In those studies, the mean proportion of negative urine tests over 24 weeks, the primary endpoint, was significantly higher among those who received the buprenorphine implant, compared with those who had a placebo implant.
The panel agreed the treatment had a beneficial effect on reducing illicit opioid use, but those voting on both sides pointed out that more information on dosing was needed. The product is indicated for patients who are maintained on 12 mg-16 mg of oral buprenorphine a day. But as addiction treatment specialists on the panel pointed out, some patients are maintained on lower oral doses, and there were no data on how to use the product in such patients. The manufacturer, Titan Pharmaceuticals, plans to study that issue further.
The panel also voted 12-2 with 1 abstention, that the company had adequately characterized the safety profile of treatment with the buprenorphine implants, which was associated with the known side effects of the oral formulation in the trials. But there are also the potential complications associated with surgical implantation and removal of the rods every 6 months, as well as the potential for abuse, misuse and diversion, and accidental exposure, which is addressed in a complex risk management program proposed by the manufacturer.
Using similar technology as contraceptive implants, the product consists of four rods, each measuring 26 mm by 2.5 mm and containing 80 mg of the buprenorphine, a partial mu-opioid receptor agonist that has been available in oral formulations since 2002 for the treatment of opioid dependence. The rods are subdermally implanted in the upper arm with a single-use application by a trained health care provider in an office-based surgical procedure with local anesthesia after the patient has been titrated with an oral dose. The rods stay in place for up to 6 months, at which time they are removed and replaced at another site.
"Clearly, the data need to be further assessed and the safety and dosing improved upon," said Dr. Christopher J. Kratochvil, professor of psychiatry and pediatrics, University of Nebraska, Omaha. But he voted in favor of approval, commenting, "This is will at least be an incremental step forward for a disorder that has very tragic consequences."
"Overall, I thought that benefit was shown and there might be a particular subset of patients who respond best to this intervention," said one of the two panelists with expertise on contraceptive implants, Dr. Geri D. Hewitt, an obstetrician-gynecologist at Ohio State University, Columbus, who voted for approval. "I did not see any evidence of significant harm; I do have questions about dosing and which physicians will be willing to place this and ... it’s going to be important to keep track of adverse event reporting," she added.
Voting against approval, Dr.. Laura F. McNicholas of the Center for Studies of Addiction at the University of Pennsylvania, Philadelphia, said she thought the product was approvable, but that approval before a dose-ranging study was conducted would be premature. A major concern she had was running out of sites to implant the rods for patients on long-term treatment, since many of her patients have been taking buprenorphine continuously for years. Many of her patients also are on doses under 12 mg a day, and there was no information on how to dose such patients with the implant, for which there is currently only one dose, she added.
So far, the company has identified four sites on the upper arms as appropriate for implantation and is studying this further.
The two studies enrolled 450 adults (mean age, mid-30s) addicted to an opioid, randomized to treatment with the buprenorphine implant or a placebo implant. The most common drug of abuse was heroin in 52% to 67%, followed by prescription analgesics in 33% to 48%.Over 24 weeks, the mean percentage of opioid-negative urine samples was statistically significantly greater among those who were treated with the buprenorphine implant (about 40% and about 35% among those who received the active drug vs. about 30% and 15%, respectively, among those on the placebo implant.)
About 80% of the patients were adequately treated with four implants, but in some patients, a fifth rod or supplemental treatment with sublingual buprenorphine was needed. Completion rates were lower among those on placebo (31% and 26%, vs. 64% and 66%, respectively). In the two studies, the most common implant site–related adverse events included pruritus, pain, erythema, edema, and hematoma.
The company’s Risk Evaluation and Mitigation Strategy (REMS) includes plans for restricted distribution, a specialty pharmacy only, and a training-certification program for health care providers who implant the rods. (The FDA requires a REMS for a drug when it determines that a risk management strategies beyond the label are needed to ensure the benefits of a drug outweigh its risks.) Both the panel and the FDA reviewers said the REMS needs to be improved.
When asked about the panel’s recommendation, Dr. Robert L DuPont said in an interview that it is a major development in medication-assisted treatment of opiate dependence in light of problems with diversion and nonmedical use of buprenorphine. "This novel dosing strategy removes the risk of diversion in a large population of drug abusing patients that poses a high risk of diversion and misuse," said Dr. DuPont, the first director of the National Institute on Drug Abuse.
"The development of abuse-resistant formulations of controlled substances is one of the best new ideas to turn back the major epidemic of prescription drug abuse."
If approved, the panel recommended that other potential problems with the device should be monitored closely, including removal of implants by nonmedical personnel for diversion (which was not seen in the studies) and the risk of long-term exposure to the components if they are not removed. The panel urged study of whether implants can be inserted in previous implant sites.
Buprenorphine can be administered in an office setting as opposed to a methadone clinic. The currently marketed buprenorphine products are Subutex and Suboxone (buprenorphine with naloxone), both available in generic formulations; and a sublingual formulation approved in 2010. In 2012, 10.7 million prescriptions were dispensed for buprenorphine products for an estimated 1 million patients, mostly the buprenorphine/naloxone combination, according to the FDA. Between January 2003 and December 2012, an estimated 40 million buprenorphine prescriptions were written. The top three categories of prescribers were general practitioners/family physicians/doctors of osteopathy (one category), psychiatrists, and internists.
If approved, the manufacturer plans to market it as Probuphine. The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist might be given a waiver, but not at this meeting.
SILVER SPRING, MD. – A four-rod subdermal implant that slowly releases buprenorphine over 6 months should be approved for maintenance treatment of opioid dependence, although more work is needed to determine optimal dosing strategies and how to address the potential risks of the treatment, the majority of a Food and Drug Administration advisory panel recommended.
At a meeting on March 21, the FDA’s Psychopharmacologic Drugs Advisory Committee voted 10-4, with 1 abstention, to recommend approval, based on the efficacy, safety, and risk-benefit profile in opioid-addicted adults treated with the implants, in two phase III placebo-controlled studies. In those studies, the mean proportion of negative urine tests over 24 weeks, the primary endpoint, was significantly higher among those who received the buprenorphine implant, compared with those who had a placebo implant.
The panel agreed the treatment had a beneficial effect on reducing illicit opioid use, but those voting on both sides pointed out that more information on dosing was needed. The product is indicated for patients who are maintained on 12 mg-16 mg of oral buprenorphine a day. But as addiction treatment specialists on the panel pointed out, some patients are maintained on lower oral doses, and there were no data on how to use the product in such patients. The manufacturer, Titan Pharmaceuticals, plans to study that issue further.
The panel also voted 12-2 with 1 abstention, that the company had adequately characterized the safety profile of treatment with the buprenorphine implants, which was associated with the known side effects of the oral formulation in the trials. But there are also the potential complications associated with surgical implantation and removal of the rods every 6 months, as well as the potential for abuse, misuse and diversion, and accidental exposure, which is addressed in a complex risk management program proposed by the manufacturer.
Using similar technology as contraceptive implants, the product consists of four rods, each measuring 26 mm by 2.5 mm and containing 80 mg of the buprenorphine, a partial mu-opioid receptor agonist that has been available in oral formulations since 2002 for the treatment of opioid dependence. The rods are subdermally implanted in the upper arm with a single-use application by a trained health care provider in an office-based surgical procedure with local anesthesia after the patient has been titrated with an oral dose. The rods stay in place for up to 6 months, at which time they are removed and replaced at another site.
"Clearly, the data need to be further assessed and the safety and dosing improved upon," said Dr. Christopher J. Kratochvil, professor of psychiatry and pediatrics, University of Nebraska, Omaha. But he voted in favor of approval, commenting, "This is will at least be an incremental step forward for a disorder that has very tragic consequences."
"Overall, I thought that benefit was shown and there might be a particular subset of patients who respond best to this intervention," said one of the two panelists with expertise on contraceptive implants, Dr. Geri D. Hewitt, an obstetrician-gynecologist at Ohio State University, Columbus, who voted for approval. "I did not see any evidence of significant harm; I do have questions about dosing and which physicians will be willing to place this and ... it’s going to be important to keep track of adverse event reporting," she added.
Voting against approval, Dr.. Laura F. McNicholas of the Center for Studies of Addiction at the University of Pennsylvania, Philadelphia, said she thought the product was approvable, but that approval before a dose-ranging study was conducted would be premature. A major concern she had was running out of sites to implant the rods for patients on long-term treatment, since many of her patients have been taking buprenorphine continuously for years. Many of her patients also are on doses under 12 mg a day, and there was no information on how to dose such patients with the implant, for which there is currently only one dose, she added.
So far, the company has identified four sites on the upper arms as appropriate for implantation and is studying this further.
The two studies enrolled 450 adults (mean age, mid-30s) addicted to an opioid, randomized to treatment with the buprenorphine implant or a placebo implant. The most common drug of abuse was heroin in 52% to 67%, followed by prescription analgesics in 33% to 48%.Over 24 weeks, the mean percentage of opioid-negative urine samples was statistically significantly greater among those who were treated with the buprenorphine implant (about 40% and about 35% among those who received the active drug vs. about 30% and 15%, respectively, among those on the placebo implant.)
About 80% of the patients were adequately treated with four implants, but in some patients, a fifth rod or supplemental treatment with sublingual buprenorphine was needed. Completion rates were lower among those on placebo (31% and 26%, vs. 64% and 66%, respectively). In the two studies, the most common implant site–related adverse events included pruritus, pain, erythema, edema, and hematoma.
The company’s Risk Evaluation and Mitigation Strategy (REMS) includes plans for restricted distribution, a specialty pharmacy only, and a training-certification program for health care providers who implant the rods. (The FDA requires a REMS for a drug when it determines that a risk management strategies beyond the label are needed to ensure the benefits of a drug outweigh its risks.) Both the panel and the FDA reviewers said the REMS needs to be improved.
When asked about the panel’s recommendation, Dr. Robert L DuPont said in an interview that it is a major development in medication-assisted treatment of opiate dependence in light of problems with diversion and nonmedical use of buprenorphine. "This novel dosing strategy removes the risk of diversion in a large population of drug abusing patients that poses a high risk of diversion and misuse," said Dr. DuPont, the first director of the National Institute on Drug Abuse.
"The development of abuse-resistant formulations of controlled substances is one of the best new ideas to turn back the major epidemic of prescription drug abuse."
If approved, the panel recommended that other potential problems with the device should be monitored closely, including removal of implants by nonmedical personnel for diversion (which was not seen in the studies) and the risk of long-term exposure to the components if they are not removed. The panel urged study of whether implants can be inserted in previous implant sites.
Buprenorphine can be administered in an office setting as opposed to a methadone clinic. The currently marketed buprenorphine products are Subutex and Suboxone (buprenorphine with naloxone), both available in generic formulations; and a sublingual formulation approved in 2010. In 2012, 10.7 million prescriptions were dispensed for buprenorphine products for an estimated 1 million patients, mostly the buprenorphine/naloxone combination, according to the FDA. Between January 2003 and December 2012, an estimated 40 million buprenorphine prescriptions were written. The top three categories of prescribers were general practitioners/family physicians/doctors of osteopathy (one category), psychiatrists, and internists.
If approved, the manufacturer plans to market it as Probuphine. The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist might be given a waiver, but not at this meeting.
FROM AN FDA ADVISORY PANEL MEETING
Panel backs approval of percutaneous mitral valve repair device
GAITHERSBURG, MD – An expert panel narrowly voted in favor of recommending Food and Drug Administration approval of a novel, percutaneously implanted device that repairs the mitral valve for use in selected patients with significant symptomatic mitral regurgitation who are poor surgical candidates.
At a meeting on March 20, the FDA’s Circulatory Systems Devices panel, it voted 5-3 that the benefits of the MitraClip Delivery System outweighed the risks for patients who met the criteria specified in the indication under review: "percutaneous reduction of significant symptomatic mitral regurgitation [moderate to severe (3+)MR] in patients who have been determined by a cardiac surgeon to be too high risk for open mitral valve surgery and in whom existing co-morbidities would not preclude the expected benefit from correction of the mitral regurgitation."
The panel was more comfortable with the safety data, unanimously voting that there was "reasonable assurance" that the device was safe for patients who met the criteria in the indication.
Largely because of limitations of the studies submitted by manufacturer Abbott Vascular – which included combining nonpivotal data from two high-risk patient registries – the panel was less confident with the efficacy data, voting 5-4 that there was not reasonable assurance that the device was effective. (The extra vote was the panel chair, who votes in case of a tie, and he voted no.)
The panelists generally agreed with the FDA’s conclusions that there were numerous problems with the data – the basis of the FDA’s conclusion that the device should not be approved at this time – but they said that they had a sense there were patients who could benefit from treatment.
Dr. Craig Selzman, a cardiothoracic surgeon at the University of Utah, Salt Lake City, voted in favor of approval "with trepidation" because he believed the benefit-risk ratio was likely favorable, based on the safety profile and the apparent benefit. He stressed, however, that the device should be limited to "truly inoperable patients" and warned about "indication creep," when a device is rapidly used for treating patients outside the approved indication once it is marketed. He also recommended that the patient should be on optimal medical treatment and that an experienced mitral valve surgeon should identify which patients are candidates for treatment.
Also voting in favor of approval, Dr. George Vetrovec, professor of medicine and director of the adult cardiac catheterization laboratory at the Medical College of Virginia, Richmond, like several others, cited the improvements in New York Heart Association class in those treated with the device, "which got so much better it couldn’t just be happenstance. ... It seemed to me that had to be real and overall risks seemed to be reasonable for a very selected, limited population."
Those voting against approval said that treatment with the device appeared to be potentially beneficial but that it was difficult to identify which patients would benefit from treatment. Therefore, a randomized controlled trial was needed before approval.
The MitraClip system includes a delivery catheter and the MitraClip device, a mechanical clip designed to reduce regurgitation by clipping together the leaflets of the mitral valve.
In its application, Abbott combined two studies of nonpivotal data – a single-arm study of 78 high-risk patients that was an adjunct to EVEREST II (a randomized controlled study of patients with mitral regurgitation who were surgical candidates that did not find a benefit of MitraClip over surgery) and another registry of 273 high-risk patients (REALISM). The 351 patients had functional or degenerative MR, their mean age was 76 years, mean ejection fraction was 48%, 85% were NYHA functional class III/IV, and their mean predicted surgical mortality risk was 18%.
Implantation was successful in about 95% of patients, and the major effectiveness end point, change in left ventricular size from baseline to 1 year, significantly improved, with about a 10% reduction in LV volumes and LV dimensions. Other changes included improvements in NYHA class (including a two-class improvement in 36% of patients at 1 year) and significant improvement in a quality-of-life questionnaire. There were also reductions in heart failure hospitalization rates in the year after implantation, compared with the year before; the mean hospital stay was about 3 days. The primary safety end point, 30-day mortality, was 4.8%, which was lower than the mean predicted mortality risk of 18%, according to the company. Three of the 17 deaths within the first 30 days were device related.
The company also compared mortality in a Duke University cardiovascular database of high-surgical-risk patients who were treated medically (31% at 1 year), with mortality in a matched cohort of 211 MitraClip patients in the registry studies (24%), which the company concluded indicated that there was no increased mortality of the procedure compared with the natural history of the disease.
The FDA reviewers concluded that the device could not be approved, citing multiple problems with the registry data, which were not intended to be used as pivotal data, were difficult to interpret, and could only be considered "hypothesis-generating" at this point.
Abbott recently started two studies – one in the United States involving patients with symptomatic functional mitral regurgitation who are not good surgical candidates, the other a postmarketing study in Europe of patients with severe heart failure – and has plans for postmarketing studies in the United States, including 5-year follow-up of the patients in the combined high-risk registry trials, and a national MitraClip patient registry.
The device received the equivalent of approval in the European Union in 2008 and is marketed in about 30 countries, according to Abbott.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, and at this meeting, two such waivers were granted to the panel chair, Dr. Jeffrey Borer, professor of medicine at the State University of New York, Downstate Medical Center in Brooklyn, and Dr. Vetrovec. Dr. Borer also is an adjunct professor at Cornell University, a clinical site of a MitraClip study, but he is not one of the investigators. Dr. Vetrovec reported stockholdings in Abbott and in two firms that make competing products.
GAITHERSBURG, MD – An expert panel narrowly voted in favor of recommending Food and Drug Administration approval of a novel, percutaneously implanted device that repairs the mitral valve for use in selected patients with significant symptomatic mitral regurgitation who are poor surgical candidates.
At a meeting on March 20, the FDA’s Circulatory Systems Devices panel, it voted 5-3 that the benefits of the MitraClip Delivery System outweighed the risks for patients who met the criteria specified in the indication under review: "percutaneous reduction of significant symptomatic mitral regurgitation [moderate to severe (3+)MR] in patients who have been determined by a cardiac surgeon to be too high risk for open mitral valve surgery and in whom existing co-morbidities would not preclude the expected benefit from correction of the mitral regurgitation."
The panel was more comfortable with the safety data, unanimously voting that there was "reasonable assurance" that the device was safe for patients who met the criteria in the indication.
Largely because of limitations of the studies submitted by manufacturer Abbott Vascular – which included combining nonpivotal data from two high-risk patient registries – the panel was less confident with the efficacy data, voting 5-4 that there was not reasonable assurance that the device was effective. (The extra vote was the panel chair, who votes in case of a tie, and he voted no.)
The panelists generally agreed with the FDA’s conclusions that there were numerous problems with the data – the basis of the FDA’s conclusion that the device should not be approved at this time – but they said that they had a sense there were patients who could benefit from treatment.
Dr. Craig Selzman, a cardiothoracic surgeon at the University of Utah, Salt Lake City, voted in favor of approval "with trepidation" because he believed the benefit-risk ratio was likely favorable, based on the safety profile and the apparent benefit. He stressed, however, that the device should be limited to "truly inoperable patients" and warned about "indication creep," when a device is rapidly used for treating patients outside the approved indication once it is marketed. He also recommended that the patient should be on optimal medical treatment and that an experienced mitral valve surgeon should identify which patients are candidates for treatment.
Also voting in favor of approval, Dr. George Vetrovec, professor of medicine and director of the adult cardiac catheterization laboratory at the Medical College of Virginia, Richmond, like several others, cited the improvements in New York Heart Association class in those treated with the device, "which got so much better it couldn’t just be happenstance. ... It seemed to me that had to be real and overall risks seemed to be reasonable for a very selected, limited population."
Those voting against approval said that treatment with the device appeared to be potentially beneficial but that it was difficult to identify which patients would benefit from treatment. Therefore, a randomized controlled trial was needed before approval.
The MitraClip system includes a delivery catheter and the MitraClip device, a mechanical clip designed to reduce regurgitation by clipping together the leaflets of the mitral valve.
In its application, Abbott combined two studies of nonpivotal data – a single-arm study of 78 high-risk patients that was an adjunct to EVEREST II (a randomized controlled study of patients with mitral regurgitation who were surgical candidates that did not find a benefit of MitraClip over surgery) and another registry of 273 high-risk patients (REALISM). The 351 patients had functional or degenerative MR, their mean age was 76 years, mean ejection fraction was 48%, 85% were NYHA functional class III/IV, and their mean predicted surgical mortality risk was 18%.
Implantation was successful in about 95% of patients, and the major effectiveness end point, change in left ventricular size from baseline to 1 year, significantly improved, with about a 10% reduction in LV volumes and LV dimensions. Other changes included improvements in NYHA class (including a two-class improvement in 36% of patients at 1 year) and significant improvement in a quality-of-life questionnaire. There were also reductions in heart failure hospitalization rates in the year after implantation, compared with the year before; the mean hospital stay was about 3 days. The primary safety end point, 30-day mortality, was 4.8%, which was lower than the mean predicted mortality risk of 18%, according to the company. Three of the 17 deaths within the first 30 days were device related.
The company also compared mortality in a Duke University cardiovascular database of high-surgical-risk patients who were treated medically (31% at 1 year), with mortality in a matched cohort of 211 MitraClip patients in the registry studies (24%), which the company concluded indicated that there was no increased mortality of the procedure compared with the natural history of the disease.
The FDA reviewers concluded that the device could not be approved, citing multiple problems with the registry data, which were not intended to be used as pivotal data, were difficult to interpret, and could only be considered "hypothesis-generating" at this point.
Abbott recently started two studies – one in the United States involving patients with symptomatic functional mitral regurgitation who are not good surgical candidates, the other a postmarketing study in Europe of patients with severe heart failure – and has plans for postmarketing studies in the United States, including 5-year follow-up of the patients in the combined high-risk registry trials, and a national MitraClip patient registry.
The device received the equivalent of approval in the European Union in 2008 and is marketed in about 30 countries, according to Abbott.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, and at this meeting, two such waivers were granted to the panel chair, Dr. Jeffrey Borer, professor of medicine at the State University of New York, Downstate Medical Center in Brooklyn, and Dr. Vetrovec. Dr. Borer also is an adjunct professor at Cornell University, a clinical site of a MitraClip study, but he is not one of the investigators. Dr. Vetrovec reported stockholdings in Abbott and in two firms that make competing products.
GAITHERSBURG, MD – An expert panel narrowly voted in favor of recommending Food and Drug Administration approval of a novel, percutaneously implanted device that repairs the mitral valve for use in selected patients with significant symptomatic mitral regurgitation who are poor surgical candidates.
At a meeting on March 20, the FDA’s Circulatory Systems Devices panel, it voted 5-3 that the benefits of the MitraClip Delivery System outweighed the risks for patients who met the criteria specified in the indication under review: "percutaneous reduction of significant symptomatic mitral regurgitation [moderate to severe (3+)MR] in patients who have been determined by a cardiac surgeon to be too high risk for open mitral valve surgery and in whom existing co-morbidities would not preclude the expected benefit from correction of the mitral regurgitation."
The panel was more comfortable with the safety data, unanimously voting that there was "reasonable assurance" that the device was safe for patients who met the criteria in the indication.
Largely because of limitations of the studies submitted by manufacturer Abbott Vascular – which included combining nonpivotal data from two high-risk patient registries – the panel was less confident with the efficacy data, voting 5-4 that there was not reasonable assurance that the device was effective. (The extra vote was the panel chair, who votes in case of a tie, and he voted no.)
The panelists generally agreed with the FDA’s conclusions that there were numerous problems with the data – the basis of the FDA’s conclusion that the device should not be approved at this time – but they said that they had a sense there were patients who could benefit from treatment.
Dr. Craig Selzman, a cardiothoracic surgeon at the University of Utah, Salt Lake City, voted in favor of approval "with trepidation" because he believed the benefit-risk ratio was likely favorable, based on the safety profile and the apparent benefit. He stressed, however, that the device should be limited to "truly inoperable patients" and warned about "indication creep," when a device is rapidly used for treating patients outside the approved indication once it is marketed. He also recommended that the patient should be on optimal medical treatment and that an experienced mitral valve surgeon should identify which patients are candidates for treatment.
Also voting in favor of approval, Dr. George Vetrovec, professor of medicine and director of the adult cardiac catheterization laboratory at the Medical College of Virginia, Richmond, like several others, cited the improvements in New York Heart Association class in those treated with the device, "which got so much better it couldn’t just be happenstance. ... It seemed to me that had to be real and overall risks seemed to be reasonable for a very selected, limited population."
Those voting against approval said that treatment with the device appeared to be potentially beneficial but that it was difficult to identify which patients would benefit from treatment. Therefore, a randomized controlled trial was needed before approval.
The MitraClip system includes a delivery catheter and the MitraClip device, a mechanical clip designed to reduce regurgitation by clipping together the leaflets of the mitral valve.
In its application, Abbott combined two studies of nonpivotal data – a single-arm study of 78 high-risk patients that was an adjunct to EVEREST II (a randomized controlled study of patients with mitral regurgitation who were surgical candidates that did not find a benefit of MitraClip over surgery) and another registry of 273 high-risk patients (REALISM). The 351 patients had functional or degenerative MR, their mean age was 76 years, mean ejection fraction was 48%, 85% were NYHA functional class III/IV, and their mean predicted surgical mortality risk was 18%.
Implantation was successful in about 95% of patients, and the major effectiveness end point, change in left ventricular size from baseline to 1 year, significantly improved, with about a 10% reduction in LV volumes and LV dimensions. Other changes included improvements in NYHA class (including a two-class improvement in 36% of patients at 1 year) and significant improvement in a quality-of-life questionnaire. There were also reductions in heart failure hospitalization rates in the year after implantation, compared with the year before; the mean hospital stay was about 3 days. The primary safety end point, 30-day mortality, was 4.8%, which was lower than the mean predicted mortality risk of 18%, according to the company. Three of the 17 deaths within the first 30 days were device related.
The company also compared mortality in a Duke University cardiovascular database of high-surgical-risk patients who were treated medically (31% at 1 year), with mortality in a matched cohort of 211 MitraClip patients in the registry studies (24%), which the company concluded indicated that there was no increased mortality of the procedure compared with the natural history of the disease.
The FDA reviewers concluded that the device could not be approved, citing multiple problems with the registry data, which were not intended to be used as pivotal data, were difficult to interpret, and could only be considered "hypothesis-generating" at this point.
Abbott recently started two studies – one in the United States involving patients with symptomatic functional mitral regurgitation who are not good surgical candidates, the other a postmarketing study in Europe of patients with severe heart failure – and has plans for postmarketing studies in the United States, including 5-year follow-up of the patients in the combined high-risk registry trials, and a national MitraClip patient registry.
The device received the equivalent of approval in the European Union in 2008 and is marketed in about 30 countries, according to Abbott.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, and at this meeting, two such waivers were granted to the panel chair, Dr. Jeffrey Borer, professor of medicine at the State University of New York, Downstate Medical Center in Brooklyn, and Dr. Vetrovec. Dr. Borer also is an adjunct professor at Cornell University, a clinical site of a MitraClip study, but he is not one of the investigators. Dr. Vetrovec reported stockholdings in Abbott and in two firms that make competing products.
FROM AN FDA ADVISORY COMMITTEE MEETING

Breast cancer: Cardiac risk increases with radiation dose to heart
The risk of major ischemic coronary events was significantly and proportionately associated with the estimated mean radiation dose to the heart in a study of women in Sweden and Denmark who received radiotherapy for breast cancer over a 43-year period.
"The risk of a major coronary event increased linearly with the mean dose to the heart," reported Sarah Darby, Ph.D., of the University of Oxford (England), and her associates. The risk began to increase within the first 5 years of treatment and continued to increase for at least 20 years.

The findings make it possible for a woman to estimate her absolute risk of radiation-related ischemic heart disease, the authors wrote. "This absolute risk can be weighed against the probable absolute reduction in her risk of recurrence or death from breast cancer that would be achieved without radiotherapy" (N. Engl. J. Med. 2013;368:987-98 [doi: 10.1056/NEJMoa1209825]).
The population-based study included 2,168 women who had been treated with external-beam radiation for invasive breast cancer between 1958 and 2001, and were enrolled in the Swedish National Cancer Register or the Danish Breast Cancer Cooperative Group. The 963 women who were subsequently diagnosed with a major coronary event (myocardial infarction, coronary revascularization, or death from ischemic heart disease, but not angina) were compared with 1,205 controls.
The major coronary events were diagnosed in the first decade after breast cancer diagnosis in 44% of patients; 33% of events were diagnosed 10-19 years after breast cancer diagnosis; and 23% occurred 20 or more years later. Of the cases, 54% died of ischemic heart disease.
The estimated mean radiation dose to the heart overall was 4.9 Gy (range, 0.03-27.72 Gy). For those with cancer in their left breast, the mean dose exposure to the heart was 6.6 Gy; for those with right-breast tumors, it was 2.9 Gy. Major coronary events were significantly higher among the women with radiation to the left breast.
The estimated dose to the heart of women who are currently treated with radiotherapy ranges from 1 to 5 Gy, the authors said.
For each 1-Gy increase in the mean dose of radiation to the heart, the rate of major coronary events increased by 7.4%, which was a highly statistically significant finding. Compared with controls who had no cardiac dose, the rate of major coronary events increased by 10% among those exposed to a mean radiation dose of less than 2 Gy, by 30% among those exposed to 2-4 Gy, by 40% among those exposed to 5-9 Gy, and by 116% in those exposed to 10 Gy or more.
Among women with a history of ischemic heart disease, the risk of major coronary events was almost sevenfold higher than it was in women with no history of ischemic heart disease. This risk was increased by about 13-fold during the first 10 years after treatment and was about twofold higher in later years.
"Absolute increases in risk for a given dose to the heart were larger for women with preexisting risk factors," they wrote, so "clinicians may wish to consider cardiac dose and cardiac risk factors as well as tumor control when making decisions about the use of radiotherapy for breast cancer."
Among the strengths of the study was that the analysis included all women who were documented as having received radiotherapy for breast cancer in the two countries during the time period studied. The authors cautioned against applying the results to breast cancer patients who are treated before age 30 because few women in this age group were included in the study.
The study was supported by the Oxford University Clinical Trial Service Unit from Cancer Research UK, the British Heart Foundation, and the UK Medical Research Council.
While the results of the study are interesting, they likely overestimate the risk of coronary events associated with contemporary radiation therapy. Of greater concern, the findings could be misinterpreted and could deter women from having potentially lifesaving treatment.
Increases in the rate of major coronary events – about 20% higher per 1 Gy – were far greater among the women diagnosed in the 1980s, who drive much of the increase in risk. There was barely an increase in the rate of events – 0.85% per 1 Gy – among those diagnosed in the 1990s, for example.
Most of the data are taken from a time when radiation was administered by techniques that differ from those used today, which are associated with a lot less scatter to the heart. Three-dimensional, CT-based planning was not used for the women in the study, which the authors acknowledged was a limitation. Also, the dose was estimated using radiotherapy charts, which are notoriously inaccurate.
Further, there is now a better understanding of which patients are likely to have a survival benefit from radiation. Correctly targeted radiation therapy plays an incredibly important role in the excellent results we see today, with the majority of breast cancer patients surviving their disease.
Dr. Hope Rugo is professor of medicine at the University of California, San Francisco, and director of breast oncology and clinical trials education at the UCSF Helen Diller Family Comprehensive Cancer Center. She had no relevant financial disclosures.
While the results of the study are interesting, they likely overestimate the risk of coronary events associated with contemporary radiation therapy. Of greater concern, the findings could be misinterpreted and could deter women from having potentially lifesaving treatment.
Increases in the rate of major coronary events – about 20% higher per 1 Gy – were far greater among the women diagnosed in the 1980s, who drive much of the increase in risk. There was barely an increase in the rate of events – 0.85% per 1 Gy – among those diagnosed in the 1990s, for example.
Most of the data are taken from a time when radiation was administered by techniques that differ from those used today, which are associated with a lot less scatter to the heart. Three-dimensional, CT-based planning was not used for the women in the study, which the authors acknowledged was a limitation. Also, the dose was estimated using radiotherapy charts, which are notoriously inaccurate.
Further, there is now a better understanding of which patients are likely to have a survival benefit from radiation. Correctly targeted radiation therapy plays an incredibly important role in the excellent results we see today, with the majority of breast cancer patients surviving their disease.
Dr. Hope Rugo is professor of medicine at the University of California, San Francisco, and director of breast oncology and clinical trials education at the UCSF Helen Diller Family Comprehensive Cancer Center. She had no relevant financial disclosures.
While the results of the study are interesting, they likely overestimate the risk of coronary events associated with contemporary radiation therapy. Of greater concern, the findings could be misinterpreted and could deter women from having potentially lifesaving treatment.
Increases in the rate of major coronary events – about 20% higher per 1 Gy – were far greater among the women diagnosed in the 1980s, who drive much of the increase in risk. There was barely an increase in the rate of events – 0.85% per 1 Gy – among those diagnosed in the 1990s, for example.
Most of the data are taken from a time when radiation was administered by techniques that differ from those used today, which are associated with a lot less scatter to the heart. Three-dimensional, CT-based planning was not used for the women in the study, which the authors acknowledged was a limitation. Also, the dose was estimated using radiotherapy charts, which are notoriously inaccurate.
Further, there is now a better understanding of which patients are likely to have a survival benefit from radiation. Correctly targeted radiation therapy plays an incredibly important role in the excellent results we see today, with the majority of breast cancer patients surviving their disease.
Dr. Hope Rugo is professor of medicine at the University of California, San Francisco, and director of breast oncology and clinical trials education at the UCSF Helen Diller Family Comprehensive Cancer Center. She had no relevant financial disclosures.
The risk of major ischemic coronary events was significantly and proportionately associated with the estimated mean radiation dose to the heart in a study of women in Sweden and Denmark who received radiotherapy for breast cancer over a 43-year period.
"The risk of a major coronary event increased linearly with the mean dose to the heart," reported Sarah Darby, Ph.D., of the University of Oxford (England), and her associates. The risk began to increase within the first 5 years of treatment and continued to increase for at least 20 years.
The findings make it possible for a woman to estimate her absolute risk of radiation-related ischemic heart disease, the authors wrote. "This absolute risk can be weighed against the probable absolute reduction in her risk of recurrence or death from breast cancer that would be achieved without radiotherapy" (N. Engl. J. Med. 2013;368:987-98 [doi: 10.1056/NEJMoa1209825]).
The population-based study included 2,168 women who had been treated with external-beam radiation for invasive breast cancer between 1958 and 2001, and were enrolled in the Swedish National Cancer Register or the Danish Breast Cancer Cooperative Group. The 963 women who were subsequently diagnosed with a major coronary event (myocardial infarction, coronary revascularization, or death from ischemic heart disease, but not angina) were compared with 1,205 controls.
The major coronary events were diagnosed in the first decade after breast cancer diagnosis in 44% of patients; 33% of events were diagnosed 10-19 years after breast cancer diagnosis; and 23% occurred 20 or more years later. Of the cases, 54% died of ischemic heart disease.
The estimated mean radiation dose to the heart overall was 4.9 Gy (range, 0.03-27.72 Gy). For those with cancer in their left breast, the mean dose exposure to the heart was 6.6 Gy; for those with right-breast tumors, it was 2.9 Gy. Major coronary events were significantly higher among the women with radiation to the left breast.
The estimated dose to the heart of women who are currently treated with radiotherapy ranges from 1 to 5 Gy, the authors said.
For each 1-Gy increase in the mean dose of radiation to the heart, the rate of major coronary events increased by 7.4%, which was a highly statistically significant finding. Compared with controls who had no cardiac dose, the rate of major coronary events increased by 10% among those exposed to a mean radiation dose of less than 2 Gy, by 30% among those exposed to 2-4 Gy, by 40% among those exposed to 5-9 Gy, and by 116% in those exposed to 10 Gy or more.
Among women with a history of ischemic heart disease, the risk of major coronary events was almost sevenfold higher than it was in women with no history of ischemic heart disease. This risk was increased by about 13-fold during the first 10 years after treatment and was about twofold higher in later years.
"Absolute increases in risk for a given dose to the heart were larger for women with preexisting risk factors," they wrote, so "clinicians may wish to consider cardiac dose and cardiac risk factors as well as tumor control when making decisions about the use of radiotherapy for breast cancer."
Among the strengths of the study was that the analysis included all women who were documented as having received radiotherapy for breast cancer in the two countries during the time period studied. The authors cautioned against applying the results to breast cancer patients who are treated before age 30 because few women in this age group were included in the study.
The study was supported by the Oxford University Clinical Trial Service Unit from Cancer Research UK, the British Heart Foundation, and the UK Medical Research Council.
The risk of major ischemic coronary events was significantly and proportionately associated with the estimated mean radiation dose to the heart in a study of women in Sweden and Denmark who received radiotherapy for breast cancer over a 43-year period.
"The risk of a major coronary event increased linearly with the mean dose to the heart," reported Sarah Darby, Ph.D., of the University of Oxford (England), and her associates. The risk began to increase within the first 5 years of treatment and continued to increase for at least 20 years.
The findings make it possible for a woman to estimate her absolute risk of radiation-related ischemic heart disease, the authors wrote. "This absolute risk can be weighed against the probable absolute reduction in her risk of recurrence or death from breast cancer that would be achieved without radiotherapy" (N. Engl. J. Med. 2013;368:987-98 [doi: 10.1056/NEJMoa1209825]).
The population-based study included 2,168 women who had been treated with external-beam radiation for invasive breast cancer between 1958 and 2001, and were enrolled in the Swedish National Cancer Register or the Danish Breast Cancer Cooperative Group. The 963 women who were subsequently diagnosed with a major coronary event (myocardial infarction, coronary revascularization, or death from ischemic heart disease, but not angina) were compared with 1,205 controls.
The major coronary events were diagnosed in the first decade after breast cancer diagnosis in 44% of patients; 33% of events were diagnosed 10-19 years after breast cancer diagnosis; and 23% occurred 20 or more years later. Of the cases, 54% died of ischemic heart disease.
The estimated mean radiation dose to the heart overall was 4.9 Gy (range, 0.03-27.72 Gy). For those with cancer in their left breast, the mean dose exposure to the heart was 6.6 Gy; for those with right-breast tumors, it was 2.9 Gy. Major coronary events were significantly higher among the women with radiation to the left breast.
The estimated dose to the heart of women who are currently treated with radiotherapy ranges from 1 to 5 Gy, the authors said.
For each 1-Gy increase in the mean dose of radiation to the heart, the rate of major coronary events increased by 7.4%, which was a highly statistically significant finding. Compared with controls who had no cardiac dose, the rate of major coronary events increased by 10% among those exposed to a mean radiation dose of less than 2 Gy, by 30% among those exposed to 2-4 Gy, by 40% among those exposed to 5-9 Gy, and by 116% in those exposed to 10 Gy or more.
Among women with a history of ischemic heart disease, the risk of major coronary events was almost sevenfold higher than it was in women with no history of ischemic heart disease. This risk was increased by about 13-fold during the first 10 years after treatment and was about twofold higher in later years.
"Absolute increases in risk for a given dose to the heart were larger for women with preexisting risk factors," they wrote, so "clinicians may wish to consider cardiac dose and cardiac risk factors as well as tumor control when making decisions about the use of radiotherapy for breast cancer."
Among the strengths of the study was that the analysis included all women who were documented as having received radiotherapy for breast cancer in the two countries during the time period studied. The authors cautioned against applying the results to breast cancer patients who are treated before age 30 because few women in this age group were included in the study.
The study was supported by the Oxford University Clinical Trial Service Unit from Cancer Research UK, the British Heart Foundation, and the UK Medical Research Council.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Major finding: The risk of major coronary events increased by 7.4% per 1 Gy of radiation received as treatment for breast cancer.
Data source: A population-based, case-control study in 2,168 women treated with external-beam radiation therapy for invasive breast cancer in Sweden and Denmark between 1958 and 2001.
Disclosures: The study was supported by the Oxford University Clinical Trial Service Unit from Cancer Research UK, the British Heart Foundation, and the UK Medical Research Council.

Parkinson's symptoms improved in new drug trials
Parkinson’s disease symptoms that affect the quality of life of patients with the condition improved in studies of drugs that are under investigational status or have already been approved, according to new research coming from the annual meeting of the American Academy of Neurology.*
The reports include phase II studies evaluating a novel, nondopaminergic adjunctive treatment with levodopa and an oral precursor to norepinephrine to treat neurogenic orthostatic hypotension, as well as a phase IV study testing an already approved therapy as an add-on treatment to patients not well controlled on a dopamine agonist.
"All of these treatments are promising news for people with Parkinson’s disease," said Dr. Robert Hauser, in a statement issued by the AAN. He is an author in all three studies and is a neurologist at the University of South Florida, Tampa.
In the first of the phase II studies, patients with PD treated with tozadenant as an adjunct to levodopa experienced significant reductions in off-time without worsening dyskinesia, compared with those on placebo. Tozadenant is an oral, selective adenosine 2-alpha receptor antagonist.
The international, 12-week, double-blind study enrolled patients who had had PD for about 9 years, were on stable doses of levodopa, and had at least 2.5 hours of off-time per day. A total of 337 patients (mean age, 63 years) completed treatment. Comparisons against placebo for four different doses of the drug taken twice daily showed that those on the 120-mg and 180-mg twice-daily doses had a mean 1.1-1.2 hours’ reduction in off-time over placebo.
Scores on the Unified Parkinson’s Disease Rating Scale (UPDRS) motor subscale also improved significantly among those on these two doses, with a mean reduction of 2.2 and 2.5 among people on the 120-mg and 180-mg twice-daily doses, respectively, compared with placebo. The Patient Global Impression of Improvement (PGI-I) scores also significantly improved among those on 120 mg twice daily.
Dyskinesia, nausea, dizziness, constipation, and worsening of PD were the most common adverse events among all the patients on tozadenant.
"With our most effective doses, we were able to show significant reductions in off-time in comparison with placebo, but importantly, we were able to do that without seeing a worsening of dyskinesia," said the lead author, Dr. C. Warren Olanow. There were also some improvements in motor skills, he added in an interview.
While it is too early to make a statement on how tozadenant compares with other drugs, "the fact that it is nondopaminergic and well tolerated is very promising," said Dr. Olanow, the Henry P. and Georgette Goldschmidt professor of neurology and professor of neuroscience at Mount Sinai School of Medicine, New York.
"Now that we have shown benefit and identified what we think are the good doses," the next step is to conduct phase III confirmatory studies, which will evaluate the two doses that were effective in the phase II study, he said.
In another phase II randomized, double-blind study, 225 people with PD received droxidopa, an oral precursor of norepinephrine, or placebo to treat symptomatic neurogenic orthostatic hypotension. The 10-week study evaluated changes in the OHQ (Orthostatic Hypotension Questionnaire), dizziness/light-headedness (based on the Orthostatic Hypotension Symptom Assessment Item 1), standing systolic blood pressure, and frequency of falls.
The dosing regimen for droxidopa was titrated to 100-600 mg three times a day over a 2-week period. After 1 week on the drug, those on droxidopa had significant improvements in dizziness and light-headedness, compared with those on placebo. At 8 weeks, there was a trend toward improvement over placebo, but the difference was no longer significant, according to the investigators. Significant improvement in standing systolic blood pressure at 1 week (a 6.8 mm Hg difference over placebo) also did not hold up at 8 weeks (a 2.2 mm Hg improvement over placebo).
In the treated patients, there was also a drop in the number of falls per patient per week (0.38 fewer among those on placebo vs. 1.73 fewer among those on droxidopa), which was not statistically significant. Headache, dizziness, hypertension, nausea, and fatigue were each reported in more than 5% of patients taking droxidopa and in more than 5% of untreated patients.
A third study, called ANDANTE, evaluated rasagiline as add-on therapy to a dopamine agonist in 321 patients with early PD whose symptoms were not well controlled on the dopamine agonist. The patients were on stable doses of a dopamine agonist and were randomized to receive 1 mg of rasagiline per day or placebo, while remaining on stable doses of the dopamine agonist (ropinirole or pramipexole).
Approved in 2006, rasagiline (Azilect), a selective irreversible MAO-B inhibitor, is indicated for the treatment of the signs and symptoms of idiopathic Parkinson’s disease as initial monotherapy and as adjunct therapy to levodopa.
Over the first 18 weeks of the postmarketing, randomized, double-blind study, those treated with rasagiline had a significantly greater change from the baseline in the total UPDRS score (a mean reduction of 2.4 vs. placebo), the primary endpoint. There were also significant improvements in UPDRS motor scores among those on rasagiline, but no significant differences between the two groups in UPDRS activities of daily living scores. The rates of adverse events and serious adverse events were similar between the two groups: Among those on rasagiline, the overall rate of adverse events was 64% (5% were serious), compared with 61% (3% serious) for the placebo group.
Dr. Olanow disclosed that he is a consultant for almost all manufacturers of drugs for PD, including Biotie Therapies, the manufacturer of tozadenant, which also supported the study. The droxidopa study was supported by Chelsea Therapeutics, the drug’s manufacturer. The ANDANTE study was supported by Teva Pharmaceuticals, the manufacturer of rasagiline.
* This article was revised on 3/15/13.
tozadenant, levodopa, Tozadenant, selective adenosine 2-alpha receptor antagonist, Unified Parkinson’s Disease Rating Scale,
Parkinson’s disease symptoms that affect the quality of life of patients with the condition improved in studies of drugs that are under investigational status or have already been approved, according to new research coming from the annual meeting of the American Academy of Neurology.*
The reports include phase II studies evaluating a novel, nondopaminergic adjunctive treatment with levodopa and an oral precursor to norepinephrine to treat neurogenic orthostatic hypotension, as well as a phase IV study testing an already approved therapy as an add-on treatment to patients not well controlled on a dopamine agonist.
"All of these treatments are promising news for people with Parkinson’s disease," said Dr. Robert Hauser, in a statement issued by the AAN. He is an author in all three studies and is a neurologist at the University of South Florida, Tampa.
In the first of the phase II studies, patients with PD treated with tozadenant as an adjunct to levodopa experienced significant reductions in off-time without worsening dyskinesia, compared with those on placebo. Tozadenant is an oral, selective adenosine 2-alpha receptor antagonist.
The international, 12-week, double-blind study enrolled patients who had had PD for about 9 years, were on stable doses of levodopa, and had at least 2.5 hours of off-time per day. A total of 337 patients (mean age, 63 years) completed treatment. Comparisons against placebo for four different doses of the drug taken twice daily showed that those on the 120-mg and 180-mg twice-daily doses had a mean 1.1-1.2 hours’ reduction in off-time over placebo.
Scores on the Unified Parkinson’s Disease Rating Scale (UPDRS) motor subscale also improved significantly among those on these two doses, with a mean reduction of 2.2 and 2.5 among people on the 120-mg and 180-mg twice-daily doses, respectively, compared with placebo. The Patient Global Impression of Improvement (PGI-I) scores also significantly improved among those on 120 mg twice daily.
Dyskinesia, nausea, dizziness, constipation, and worsening of PD were the most common adverse events among all the patients on tozadenant.
"With our most effective doses, we were able to show significant reductions in off-time in comparison with placebo, but importantly, we were able to do that without seeing a worsening of dyskinesia," said the lead author, Dr. C. Warren Olanow. There were also some improvements in motor skills, he added in an interview.
While it is too early to make a statement on how tozadenant compares with other drugs, "the fact that it is nondopaminergic and well tolerated is very promising," said Dr. Olanow, the Henry P. and Georgette Goldschmidt professor of neurology and professor of neuroscience at Mount Sinai School of Medicine, New York.
"Now that we have shown benefit and identified what we think are the good doses," the next step is to conduct phase III confirmatory studies, which will evaluate the two doses that were effective in the phase II study, he said.
In another phase II randomized, double-blind study, 225 people with PD received droxidopa, an oral precursor of norepinephrine, or placebo to treat symptomatic neurogenic orthostatic hypotension. The 10-week study evaluated changes in the OHQ (Orthostatic Hypotension Questionnaire), dizziness/light-headedness (based on the Orthostatic Hypotension Symptom Assessment Item 1), standing systolic blood pressure, and frequency of falls.
The dosing regimen for droxidopa was titrated to 100-600 mg three times a day over a 2-week period. After 1 week on the drug, those on droxidopa had significant improvements in dizziness and light-headedness, compared with those on placebo. At 8 weeks, there was a trend toward improvement over placebo, but the difference was no longer significant, according to the investigators. Significant improvement in standing systolic blood pressure at 1 week (a 6.8 mm Hg difference over placebo) also did not hold up at 8 weeks (a 2.2 mm Hg improvement over placebo).
In the treated patients, there was also a drop in the number of falls per patient per week (0.38 fewer among those on placebo vs. 1.73 fewer among those on droxidopa), which was not statistically significant. Headache, dizziness, hypertension, nausea, and fatigue were each reported in more than 5% of patients taking droxidopa and in more than 5% of untreated patients.
A third study, called ANDANTE, evaluated rasagiline as add-on therapy to a dopamine agonist in 321 patients with early PD whose symptoms were not well controlled on the dopamine agonist. The patients were on stable doses of a dopamine agonist and were randomized to receive 1 mg of rasagiline per day or placebo, while remaining on stable doses of the dopamine agonist (ropinirole or pramipexole).
Approved in 2006, rasagiline (Azilect), a selective irreversible MAO-B inhibitor, is indicated for the treatment of the signs and symptoms of idiopathic Parkinson’s disease as initial monotherapy and as adjunct therapy to levodopa.
Over the first 18 weeks of the postmarketing, randomized, double-blind study, those treated with rasagiline had a significantly greater change from the baseline in the total UPDRS score (a mean reduction of 2.4 vs. placebo), the primary endpoint. There were also significant improvements in UPDRS motor scores among those on rasagiline, but no significant differences between the two groups in UPDRS activities of daily living scores. The rates of adverse events and serious adverse events were similar between the two groups: Among those on rasagiline, the overall rate of adverse events was 64% (5% were serious), compared with 61% (3% serious) for the placebo group.
Dr. Olanow disclosed that he is a consultant for almost all manufacturers of drugs for PD, including Biotie Therapies, the manufacturer of tozadenant, which also supported the study. The droxidopa study was supported by Chelsea Therapeutics, the drug’s manufacturer. The ANDANTE study was supported by Teva Pharmaceuticals, the manufacturer of rasagiline.
* This article was revised on 3/15/13.
Parkinson’s disease symptoms that affect the quality of life of patients with the condition improved in studies of drugs that are under investigational status or have already been approved, according to new research coming from the annual meeting of the American Academy of Neurology.*
The reports include phase II studies evaluating a novel, nondopaminergic adjunctive treatment with levodopa and an oral precursor to norepinephrine to treat neurogenic orthostatic hypotension, as well as a phase IV study testing an already approved therapy as an add-on treatment to patients not well controlled on a dopamine agonist.
"All of these treatments are promising news for people with Parkinson’s disease," said Dr. Robert Hauser, in a statement issued by the AAN. He is an author in all three studies and is a neurologist at the University of South Florida, Tampa.
In the first of the phase II studies, patients with PD treated with tozadenant as an adjunct to levodopa experienced significant reductions in off-time without worsening dyskinesia, compared with those on placebo. Tozadenant is an oral, selective adenosine 2-alpha receptor antagonist.
The international, 12-week, double-blind study enrolled patients who had had PD for about 9 years, were on stable doses of levodopa, and had at least 2.5 hours of off-time per day. A total of 337 patients (mean age, 63 years) completed treatment. Comparisons against placebo for four different doses of the drug taken twice daily showed that those on the 120-mg and 180-mg twice-daily doses had a mean 1.1-1.2 hours’ reduction in off-time over placebo.
Scores on the Unified Parkinson’s Disease Rating Scale (UPDRS) motor subscale also improved significantly among those on these two doses, with a mean reduction of 2.2 and 2.5 among people on the 120-mg and 180-mg twice-daily doses, respectively, compared with placebo. The Patient Global Impression of Improvement (PGI-I) scores also significantly improved among those on 120 mg twice daily.
Dyskinesia, nausea, dizziness, constipation, and worsening of PD were the most common adverse events among all the patients on tozadenant.
"With our most effective doses, we were able to show significant reductions in off-time in comparison with placebo, but importantly, we were able to do that without seeing a worsening of dyskinesia," said the lead author, Dr. C. Warren Olanow. There were also some improvements in motor skills, he added in an interview.
While it is too early to make a statement on how tozadenant compares with other drugs, "the fact that it is nondopaminergic and well tolerated is very promising," said Dr. Olanow, the Henry P. and Georgette Goldschmidt professor of neurology and professor of neuroscience at Mount Sinai School of Medicine, New York.
"Now that we have shown benefit and identified what we think are the good doses," the next step is to conduct phase III confirmatory studies, which will evaluate the two doses that were effective in the phase II study, he said.
In another phase II randomized, double-blind study, 225 people with PD received droxidopa, an oral precursor of norepinephrine, or placebo to treat symptomatic neurogenic orthostatic hypotension. The 10-week study evaluated changes in the OHQ (Orthostatic Hypotension Questionnaire), dizziness/light-headedness (based on the Orthostatic Hypotension Symptom Assessment Item 1), standing systolic blood pressure, and frequency of falls.
The dosing regimen for droxidopa was titrated to 100-600 mg three times a day over a 2-week period. After 1 week on the drug, those on droxidopa had significant improvements in dizziness and light-headedness, compared with those on placebo. At 8 weeks, there was a trend toward improvement over placebo, but the difference was no longer significant, according to the investigators. Significant improvement in standing systolic blood pressure at 1 week (a 6.8 mm Hg difference over placebo) also did not hold up at 8 weeks (a 2.2 mm Hg improvement over placebo).
In the treated patients, there was also a drop in the number of falls per patient per week (0.38 fewer among those on placebo vs. 1.73 fewer among those on droxidopa), which was not statistically significant. Headache, dizziness, hypertension, nausea, and fatigue were each reported in more than 5% of patients taking droxidopa and in more than 5% of untreated patients.
A third study, called ANDANTE, evaluated rasagiline as add-on therapy to a dopamine agonist in 321 patients with early PD whose symptoms were not well controlled on the dopamine agonist. The patients were on stable doses of a dopamine agonist and were randomized to receive 1 mg of rasagiline per day or placebo, while remaining on stable doses of the dopamine agonist (ropinirole or pramipexole).
Approved in 2006, rasagiline (Azilect), a selective irreversible MAO-B inhibitor, is indicated for the treatment of the signs and symptoms of idiopathic Parkinson’s disease as initial monotherapy and as adjunct therapy to levodopa.
Over the first 18 weeks of the postmarketing, randomized, double-blind study, those treated with rasagiline had a significantly greater change from the baseline in the total UPDRS score (a mean reduction of 2.4 vs. placebo), the primary endpoint. There were also significant improvements in UPDRS motor scores among those on rasagiline, but no significant differences between the two groups in UPDRS activities of daily living scores. The rates of adverse events and serious adverse events were similar between the two groups: Among those on rasagiline, the overall rate of adverse events was 64% (5% were serious), compared with 61% (3% serious) for the placebo group.
Dr. Olanow disclosed that he is a consultant for almost all manufacturers of drugs for PD, including Biotie Therapies, the manufacturer of tozadenant, which also supported the study. The droxidopa study was supported by Chelsea Therapeutics, the drug’s manufacturer. The ANDANTE study was supported by Teva Pharmaceuticals, the manufacturer of rasagiline.
* This article was revised on 3/15/13.
tozadenant, levodopa, Tozadenant, selective adenosine 2-alpha receptor antagonist, Unified Parkinson’s Disease Rating Scale,
tozadenant, levodopa, Tozadenant, selective adenosine 2-alpha receptor antagonist, Unified Parkinson’s Disease Rating Scale,
FROM THE 2013 AAN ANNUAL MEETING