User login
Treatment-related AML rises in non-Hodgkin's lymphoma
The risks of chemotherapy-related acute myeloid leukemia have changed significantly over the last 30 years, increasing in people treated for non-Hodgkin’s lymphoma and decreasing in people treated for ovarian cancer and myeloma.
Further, treatment-associated acute myeloid leukemia (t-AML) has increased since 2000 in people treated for esophageal, prostate, and cervical cancers, and in those treated since the 1990s for bone and joint cancers and endometrial cancers, reported Lindsay Morton, Ph.D., and her associates online Feb. 14 in Blood (2013 Feb. 14 [doi: 10.1182/blood-2012-08-448068]).
The incidence of t-AML is likely to increase because more patients have been treated with cytotoxic agents over the last 10 years, wrote Dr. Morton, of the division of Cancer Epidemiology and Genetics at the National Cancer Institute, and her colleagues. More research is needed "to evaluate the leukemogenicity of new targeted and immunomodulatory agents introduced into the clinic, and increasing use of (neo) adjuvant and maintenance therapies," they said.
"It will be interesting to evaluate the role of a patient’s genetic susceptibility to treatment-related leukemia," Dr. Morton said in a press release. "If certain inherited traits predispose patients to a second malignancy as a result of treatment, clinicians could more accurately weigh the risk of leukemia against the established benefits of chemotherapy."
The researchers based their conclusions on cancer registry data from nine Surveillance, Epidemiology, and End Results (SEER) registries for more than 400,000 adults treated from 1975 to 2008 with chemotherapy for their first primary malignancy. Despite major changes in cancer treatments, no large published study has attempted to quantify the risks of AML after chemotherapy for different types of primary malignancies in adults since 1984, they pointed out.
No data on specific drugs and doses were available in the SEER registries, but "our results are consistent with changing treatment practices and differential leukemogenicity of specific treatments, with increasing t-AML risks after chemotherapy likely associated with increasing use of (neo) adjuvant therapy, multiple courses of therapy, duration/intensity of treatment, and newly introduced agents," the authors said.
Among the 426,068 patients treated between 1975 and 2008, 801 subsequently developed AML. That rate is 4.7 times higher than the 170.5 cases expected for the general population. The risks of t-AML during this time were "especially pronounced" among those treated for cancers of the bone/joints (19.25 times greater risk than the general population), soft tissue (14.41), ovary (8.68), lung (6.82), non-Hodgkin’s lymphoma (16.65), and myeloma (10.43).
The "striking changes" in the risk in t-AML after initial treatment for non-Hodgkin’s lymphoma "may be explained by more widespread use of subsequent therapy for persistent or relapsed/refractory non-Hodgkin’s lymphoma," they wrote.
The risk of t-AML after chemotherapy for ovarian cancer and myeloma dropped significantly over time. The risk associated with lung cancer treatment also declined, but the drop was not statistically significant. The drop in t-AML associated with treatment for ovarian cancer is "consistent" with the shift from the alkylating agent melphalan to platinum-based chemotherapy in the early 1980s, the authors said. The drop in t-AML in myeloma patients may be due to decreases in the dose and/or length of treatment with melphalan. A possible drop in t-AML associated with lung cancer may reflect "modest advances in treatment," they added.
The risk of t-AML was significantly increased among patients treated during 1999-2008 for cancers of the esophagus, anus, cervix, and prostate, although this conclusion was based on small numbers.
For breast cancer, the risks was greatest during 1975-1978 and significantly lower in the 1980s, with a nonsignificant increase in the 1990s, reflecting the higher t-AML risk that is "concentrated in the initial years" after a diagnosis of breast cancer.
The risks were not significantly higher for cancers of the stomach, colon, and rectum.
The study was supported by the Intramural Program of the National Cancer Institute. Dr. Morton’s coauthors were from the NCI, the Veterans Affairs Medical Center in Oklahoma City, and the department of pediatrics at the University of Chicago. The authors had no disclosures.
Commentary: Could transplants account for the increase?
Dr. Steven Coutré comments: While interesting, this report provides little insight into possible reasons for an observed increase in treatment-associated acute myeloid leukemia. As the authors point out, t-AML has declined in patients treated for ovarian cancer, likely due to a change in the standard chemotherapy used. For non-Hodgkin’s lymphoma, one would like to know if there has been a similar change in treatment, in the opposite direction, that accounts for an increase in t-AML. Is there a greater use of transplant, which is known to be associated with t-AML? Have these patients been treated more intensively with alkylator-based regimens?
Dr. Coutré is associate professor, division of hematology, and director of the hematology clinic at Stanford (Calif.) University. He had no disclosures.
While interesting, this report provides little insight into possible reasons for an observed increase in treatment-associated acute myeloid leukemia. As the authors point out, t-AML has declined in patients treated for ovarian cancer, likely due to a change in the standard chemotherapy used. For non-Hodgkin’s lymphoma, one would like to know if there has been a similar change in treatment, in the opposite direction, that accounts for an increase in t-AML. Is there a greater use of transplant, which is known to be associated with t-AML? Have these patients been treated more intensively with alkylator-based regimens?
Dr. Steven Coutré is associate professor, division of hematology, and director of the hematology clinic at Stanford (Calif.) University. He had no disclosures.
While interesting, this report provides little insight into possible reasons for an observed increase in treatment-associated acute myeloid leukemia. As the authors point out, t-AML has declined in patients treated for ovarian cancer, likely due to a change in the standard chemotherapy used. For non-Hodgkin’s lymphoma, one would like to know if there has been a similar change in treatment, in the opposite direction, that accounts for an increase in t-AML. Is there a greater use of transplant, which is known to be associated with t-AML? Have these patients been treated more intensively with alkylator-based regimens?
Dr. Steven Coutré is associate professor, division of hematology, and director of the hematology clinic at Stanford (Calif.) University. He had no disclosures.
While interesting, this report provides little insight into possible reasons for an observed increase in treatment-associated acute myeloid leukemia. As the authors point out, t-AML has declined in patients treated for ovarian cancer, likely due to a change in the standard chemotherapy used. For non-Hodgkin’s lymphoma, one would like to know if there has been a similar change in treatment, in the opposite direction, that accounts for an increase in t-AML. Is there a greater use of transplant, which is known to be associated with t-AML? Have these patients been treated more intensively with alkylator-based regimens?
Dr. Steven Coutré is associate professor, division of hematology, and director of the hematology clinic at Stanford (Calif.) University. He had no disclosures.
The risks of chemotherapy-related acute myeloid leukemia have changed significantly over the last 30 years, increasing in people treated for non-Hodgkin’s lymphoma and decreasing in people treated for ovarian cancer and myeloma.
Further, treatment-associated acute myeloid leukemia (t-AML) has increased since 2000 in people treated for esophageal, prostate, and cervical cancers, and in those treated since the 1990s for bone and joint cancers and endometrial cancers, reported Lindsay Morton, Ph.D., and her associates online Feb. 14 in Blood (2013 Feb. 14 [doi: 10.1182/blood-2012-08-448068]).
The incidence of t-AML is likely to increase because more patients have been treated with cytotoxic agents over the last 10 years, wrote Dr. Morton, of the division of Cancer Epidemiology and Genetics at the National Cancer Institute, and her colleagues. More research is needed "to evaluate the leukemogenicity of new targeted and immunomodulatory agents introduced into the clinic, and increasing use of (neo) adjuvant and maintenance therapies," they said.
"It will be interesting to evaluate the role of a patient’s genetic susceptibility to treatment-related leukemia," Dr. Morton said in a press release. "If certain inherited traits predispose patients to a second malignancy as a result of treatment, clinicians could more accurately weigh the risk of leukemia against the established benefits of chemotherapy."
The researchers based their conclusions on cancer registry data from nine Surveillance, Epidemiology, and End Results (SEER) registries for more than 400,000 adults treated from 1975 to 2008 with chemotherapy for their first primary malignancy. Despite major changes in cancer treatments, no large published study has attempted to quantify the risks of AML after chemotherapy for different types of primary malignancies in adults since 1984, they pointed out.
No data on specific drugs and doses were available in the SEER registries, but "our results are consistent with changing treatment practices and differential leukemogenicity of specific treatments, with increasing t-AML risks after chemotherapy likely associated with increasing use of (neo) adjuvant therapy, multiple courses of therapy, duration/intensity of treatment, and newly introduced agents," the authors said.
Among the 426,068 patients treated between 1975 and 2008, 801 subsequently developed AML. That rate is 4.7 times higher than the 170.5 cases expected for the general population. The risks of t-AML during this time were "especially pronounced" among those treated for cancers of the bone/joints (19.25 times greater risk than the general population), soft tissue (14.41), ovary (8.68), lung (6.82), non-Hodgkin’s lymphoma (16.65), and myeloma (10.43).
The "striking changes" in the risk in t-AML after initial treatment for non-Hodgkin’s lymphoma "may be explained by more widespread use of subsequent therapy for persistent or relapsed/refractory non-Hodgkin’s lymphoma," they wrote.
The risk of t-AML after chemotherapy for ovarian cancer and myeloma dropped significantly over time. The risk associated with lung cancer treatment also declined, but the drop was not statistically significant. The drop in t-AML associated with treatment for ovarian cancer is "consistent" with the shift from the alkylating agent melphalan to platinum-based chemotherapy in the early 1980s, the authors said. The drop in t-AML in myeloma patients may be due to decreases in the dose and/or length of treatment with melphalan. A possible drop in t-AML associated with lung cancer may reflect "modest advances in treatment," they added.
The risk of t-AML was significantly increased among patients treated during 1999-2008 for cancers of the esophagus, anus, cervix, and prostate, although this conclusion was based on small numbers.
For breast cancer, the risks was greatest during 1975-1978 and significantly lower in the 1980s, with a nonsignificant increase in the 1990s, reflecting the higher t-AML risk that is "concentrated in the initial years" after a diagnosis of breast cancer.
The risks were not significantly higher for cancers of the stomach, colon, and rectum.
The study was supported by the Intramural Program of the National Cancer Institute. Dr. Morton’s coauthors were from the NCI, the Veterans Affairs Medical Center in Oklahoma City, and the department of pediatrics at the University of Chicago. The authors had no disclosures.
Commentary: Could transplants account for the increase?
Dr. Steven Coutré comments: While interesting, this report provides little insight into possible reasons for an observed increase in treatment-associated acute myeloid leukemia. As the authors point out, t-AML has declined in patients treated for ovarian cancer, likely due to a change in the standard chemotherapy used. For non-Hodgkin’s lymphoma, one would like to know if there has been a similar change in treatment, in the opposite direction, that accounts for an increase in t-AML. Is there a greater use of transplant, which is known to be associated with t-AML? Have these patients been treated more intensively with alkylator-based regimens?
Dr. Coutré is associate professor, division of hematology, and director of the hematology clinic at Stanford (Calif.) University. He had no disclosures.
The risks of chemotherapy-related acute myeloid leukemia have changed significantly over the last 30 years, increasing in people treated for non-Hodgkin’s lymphoma and decreasing in people treated for ovarian cancer and myeloma.
Further, treatment-associated acute myeloid leukemia (t-AML) has increased since 2000 in people treated for esophageal, prostate, and cervical cancers, and in those treated since the 1990s for bone and joint cancers and endometrial cancers, reported Lindsay Morton, Ph.D., and her associates online Feb. 14 in Blood (2013 Feb. 14 [doi: 10.1182/blood-2012-08-448068]).
The incidence of t-AML is likely to increase because more patients have been treated with cytotoxic agents over the last 10 years, wrote Dr. Morton, of the division of Cancer Epidemiology and Genetics at the National Cancer Institute, and her colleagues. More research is needed "to evaluate the leukemogenicity of new targeted and immunomodulatory agents introduced into the clinic, and increasing use of (neo) adjuvant and maintenance therapies," they said.
"It will be interesting to evaluate the role of a patient’s genetic susceptibility to treatment-related leukemia," Dr. Morton said in a press release. "If certain inherited traits predispose patients to a second malignancy as a result of treatment, clinicians could more accurately weigh the risk of leukemia against the established benefits of chemotherapy."
The researchers based their conclusions on cancer registry data from nine Surveillance, Epidemiology, and End Results (SEER) registries for more than 400,000 adults treated from 1975 to 2008 with chemotherapy for their first primary malignancy. Despite major changes in cancer treatments, no large published study has attempted to quantify the risks of AML after chemotherapy for different types of primary malignancies in adults since 1984, they pointed out.
No data on specific drugs and doses were available in the SEER registries, but "our results are consistent with changing treatment practices and differential leukemogenicity of specific treatments, with increasing t-AML risks after chemotherapy likely associated with increasing use of (neo) adjuvant therapy, multiple courses of therapy, duration/intensity of treatment, and newly introduced agents," the authors said.
Among the 426,068 patients treated between 1975 and 2008, 801 subsequently developed AML. That rate is 4.7 times higher than the 170.5 cases expected for the general population. The risks of t-AML during this time were "especially pronounced" among those treated for cancers of the bone/joints (19.25 times greater risk than the general population), soft tissue (14.41), ovary (8.68), lung (6.82), non-Hodgkin’s lymphoma (16.65), and myeloma (10.43).
The "striking changes" in the risk in t-AML after initial treatment for non-Hodgkin’s lymphoma "may be explained by more widespread use of subsequent therapy for persistent or relapsed/refractory non-Hodgkin’s lymphoma," they wrote.
The risk of t-AML after chemotherapy for ovarian cancer and myeloma dropped significantly over time. The risk associated with lung cancer treatment also declined, but the drop was not statistically significant. The drop in t-AML associated with treatment for ovarian cancer is "consistent" with the shift from the alkylating agent melphalan to platinum-based chemotherapy in the early 1980s, the authors said. The drop in t-AML in myeloma patients may be due to decreases in the dose and/or length of treatment with melphalan. A possible drop in t-AML associated with lung cancer may reflect "modest advances in treatment," they added.
The risk of t-AML was significantly increased among patients treated during 1999-2008 for cancers of the esophagus, anus, cervix, and prostate, although this conclusion was based on small numbers.
For breast cancer, the risks was greatest during 1975-1978 and significantly lower in the 1980s, with a nonsignificant increase in the 1990s, reflecting the higher t-AML risk that is "concentrated in the initial years" after a diagnosis of breast cancer.
The risks were not significantly higher for cancers of the stomach, colon, and rectum.
The study was supported by the Intramural Program of the National Cancer Institute. Dr. Morton’s coauthors were from the NCI, the Veterans Affairs Medical Center in Oklahoma City, and the department of pediatrics at the University of Chicago. The authors had no disclosures.
Commentary: Could transplants account for the increase?
Dr. Steven Coutré comments: While interesting, this report provides little insight into possible reasons for an observed increase in treatment-associated acute myeloid leukemia. As the authors point out, t-AML has declined in patients treated for ovarian cancer, likely due to a change in the standard chemotherapy used. For non-Hodgkin’s lymphoma, one would like to know if there has been a similar change in treatment, in the opposite direction, that accounts for an increase in t-AML. Is there a greater use of transplant, which is known to be associated with t-AML? Have these patients been treated more intensively with alkylator-based regimens?
Dr. Coutré is associate professor, division of hematology, and director of the hematology clinic at Stanford (Calif.) University. He had no disclosures.
FROM BLOOD
Major finding: Among the 426,068 patients treated between 1975 and 2008 with chemotherapy for their first primary malignancy, 801 subsequently developed AML. That rate is 4.7 times higher than the 170.5 cases expected for the general population.
Data source: Cancer registry data from nine Surveillance, Epidemiology, and End Results (SEER) registries.
Disclosures: The study was supported by the Intramural Program of the National Cancer Institute. Dr. Morton’s coauthors were from the NCI, the Veterans Affairs Medical Center in Oklahoma City, and the department of pediatrics at the University of Chicago. The authors had no disclosures.
Kadcyla wins FDA approval for late-stage breast cancer
Ado-trastuzumab emtansine, a HER-2 targeted antibody-drug conjugate, has been approved by the Food and Drug Administration for treatment of women with HER2-positive, late-stage metastatic breast cancer, the agency announced Feb. 22.
Marketed as Kadcyla by Genentech, the treatment is a combination of an HER2-targeted antibody, trastuzumab, and DM1, a microtubule inhibitor, and is indicated as a single agent "for the treatment of patients with HER2-positive, metastatic breast cancer who previously received trastuzumab and a taxane, separately or in combination," according to the prescribing information. Patients should have either received previous therapy for metastatic disease, or had disease recurrence during or within 6 months of completing adjuvant therapy, according to the indication. The treatment is administered in an intravenous infusion every 3 weeks.
"Kadcyla is trastuzumab connected to a drug called DM1 that interferes with cancer cell growth," Dr. Richard Pazdur, director of the office of hematology and oncology products in the FDA’s Center for Drug Evaluation and Research, said in a statement. "Kadcyla delivers the drug to the cancer site to shrink the tumor, slow disease progression, and prolong survival," he added.
Kadcyla is the fourth FDA-approved breast cancer treatment that targets the HER2 protein. The other three drugs approved for treating HER2-positive breast cancer are trastuzumab (Herceptin), approved in 1998; lapatinib (Tykerb), approved in 2007; and pertuzumab (Perjeta), approved in 2012.
Approval of Kadcyla was based on a randomized, open label, phase III study of 991 patients with HER2-positive, locally-advanced breast cancer or metastatic breast cancer who had previously been treated with trastuzumab and a taxane chemotherapy. The median progression-free survival was 9.6 months among those treated with Kadcyla, compared with 6.4 months in patients treated with lapatinib plus capecitabine. The median overall survival was 30.9 months among those treated with Kadcyla, compared with 25.1 months among those treated with lapatinib and capecitabine, according to statements from the FDA and Genentech. Overall survival and progression-free survival were the study’s primary endpoints.
The most common side effects associated with treatment (affecting more than 25% of patients) were fatigue, nausea, musculoskeletal pain, thrombocytopenia, headache, increased transaminases, and constipation.
This accelerated approval was completed in 6 months instead of the usual 12-month review period, which is used for drugs that may provide safer and effective treatment when there is no satisfactory alternative treatment or when the treatment provides significant improvements over available treatments. Ado-trastuzumab emtansine has been referred to as T-DM1 during clinical trials.
Kadcyla will be available within 2 weeks of approval, according to the statement from Genentech, a member of the Roche group. The agent is currently under review by the European Medicines Agency for the same indication.
Serious adverse events associated with Kadcyla should be reported to the FDA’s MedWatch program at 800-332-1088 or www.fda.gov/medwatch. The prescribing information is available at www.accessdata.fda.gov/drugsatfda_docs/label/2013/125427lbl.pdf.
Ado-trastuzumab emtansine, a HER-2 targeted antibody-drug conjugate, has been approved by the Food and Drug Administration for treatment of women with HER2-positive, late-stage metastatic breast cancer, the agency announced Feb. 22.
Marketed as Kadcyla by Genentech, the treatment is a combination of an HER2-targeted antibody, trastuzumab, and DM1, a microtubule inhibitor, and is indicated as a single agent "for the treatment of patients with HER2-positive, metastatic breast cancer who previously received trastuzumab and a taxane, separately or in combination," according to the prescribing information. Patients should have either received previous therapy for metastatic disease, or had disease recurrence during or within 6 months of completing adjuvant therapy, according to the indication. The treatment is administered in an intravenous infusion every 3 weeks.
"Kadcyla is trastuzumab connected to a drug called DM1 that interferes with cancer cell growth," Dr. Richard Pazdur, director of the office of hematology and oncology products in the FDA’s Center for Drug Evaluation and Research, said in a statement. "Kadcyla delivers the drug to the cancer site to shrink the tumor, slow disease progression, and prolong survival," he added.
Kadcyla is the fourth FDA-approved breast cancer treatment that targets the HER2 protein. The other three drugs approved for treating HER2-positive breast cancer are trastuzumab (Herceptin), approved in 1998; lapatinib (Tykerb), approved in 2007; and pertuzumab (Perjeta), approved in 2012.
Approval of Kadcyla was based on a randomized, open label, phase III study of 991 patients with HER2-positive, locally-advanced breast cancer or metastatic breast cancer who had previously been treated with trastuzumab and a taxane chemotherapy. The median progression-free survival was 9.6 months among those treated with Kadcyla, compared with 6.4 months in patients treated with lapatinib plus capecitabine. The median overall survival was 30.9 months among those treated with Kadcyla, compared with 25.1 months among those treated with lapatinib and capecitabine, according to statements from the FDA and Genentech. Overall survival and progression-free survival were the study’s primary endpoints.
The most common side effects associated with treatment (affecting more than 25% of patients) were fatigue, nausea, musculoskeletal pain, thrombocytopenia, headache, increased transaminases, and constipation.
This accelerated approval was completed in 6 months instead of the usual 12-month review period, which is used for drugs that may provide safer and effective treatment when there is no satisfactory alternative treatment or when the treatment provides significant improvements over available treatments. Ado-trastuzumab emtansine has been referred to as T-DM1 during clinical trials.
Kadcyla will be available within 2 weeks of approval, according to the statement from Genentech, a member of the Roche group. The agent is currently under review by the European Medicines Agency for the same indication.
Serious adverse events associated with Kadcyla should be reported to the FDA’s MedWatch program at 800-332-1088 or www.fda.gov/medwatch. The prescribing information is available at www.accessdata.fda.gov/drugsatfda_docs/label/2013/125427lbl.pdf.
Ado-trastuzumab emtansine, a HER-2 targeted antibody-drug conjugate, has been approved by the Food and Drug Administration for treatment of women with HER2-positive, late-stage metastatic breast cancer, the agency announced Feb. 22.
Marketed as Kadcyla by Genentech, the treatment is a combination of an HER2-targeted antibody, trastuzumab, and DM1, a microtubule inhibitor, and is indicated as a single agent "for the treatment of patients with HER2-positive, metastatic breast cancer who previously received trastuzumab and a taxane, separately or in combination," according to the prescribing information. Patients should have either received previous therapy for metastatic disease, or had disease recurrence during or within 6 months of completing adjuvant therapy, according to the indication. The treatment is administered in an intravenous infusion every 3 weeks.
"Kadcyla is trastuzumab connected to a drug called DM1 that interferes with cancer cell growth," Dr. Richard Pazdur, director of the office of hematology and oncology products in the FDA’s Center for Drug Evaluation and Research, said in a statement. "Kadcyla delivers the drug to the cancer site to shrink the tumor, slow disease progression, and prolong survival," he added.
Kadcyla is the fourth FDA-approved breast cancer treatment that targets the HER2 protein. The other three drugs approved for treating HER2-positive breast cancer are trastuzumab (Herceptin), approved in 1998; lapatinib (Tykerb), approved in 2007; and pertuzumab (Perjeta), approved in 2012.
Approval of Kadcyla was based on a randomized, open label, phase III study of 991 patients with HER2-positive, locally-advanced breast cancer or metastatic breast cancer who had previously been treated with trastuzumab and a taxane chemotherapy. The median progression-free survival was 9.6 months among those treated with Kadcyla, compared with 6.4 months in patients treated with lapatinib plus capecitabine. The median overall survival was 30.9 months among those treated with Kadcyla, compared with 25.1 months among those treated with lapatinib and capecitabine, according to statements from the FDA and Genentech. Overall survival and progression-free survival were the study’s primary endpoints.
The most common side effects associated with treatment (affecting more than 25% of patients) were fatigue, nausea, musculoskeletal pain, thrombocytopenia, headache, increased transaminases, and constipation.
This accelerated approval was completed in 6 months instead of the usual 12-month review period, which is used for drugs that may provide safer and effective treatment when there is no satisfactory alternative treatment or when the treatment provides significant improvements over available treatments. Ado-trastuzumab emtansine has been referred to as T-DM1 during clinical trials.
Kadcyla will be available within 2 weeks of approval, according to the statement from Genentech, a member of the Roche group. The agent is currently under review by the European Medicines Agency for the same indication.
Serious adverse events associated with Kadcyla should be reported to the FDA’s MedWatch program at 800-332-1088 or www.fda.gov/medwatch. The prescribing information is available at www.accessdata.fda.gov/drugsatfda_docs/label/2013/125427lbl.pdf.
ACIP backs routine PCV13 vaccine in high risk-pediatric group
Vaccination with the 13-valent pneumococcal conjugate vaccine should be routine for certain high-risk children aged 6-18 years, panel members unanimously agreed at a meeting of the Centers for Disease Control and Prevention Advisory Committee on Immunization Practices.
Specifically, the panel recommended expanding the indication for this vaccine to include a group of older children – those between 6 and 18 years of age – who are at high risk for invasive pneumococcal disease (IPD) because of immunocompromising conditions, functional or anatomic asplenia, cerebrospinal fluid leaks, or cochlear implants, regardless or whether they have previously received the PCV7 or pneumococcal polysaccaride vaccine (PPSV23) vaccines. The 13-valent pneumococcal conjugate vaccine (PCV13) is already recommended by ACIP for high risk children 24-71 months of age who have these underlying conditions.*
The ACIP panel voted 15-0 in favor of this recommendation proposed by ACIP’s pneumococcal vaccines work group at its Feb. 20 meeting.
The inclusion of the older pediatric age group will harmonize the PCV13 pediatric recommendation and may improve vaccine uptake in these groups, Tamara Pilishvili, M.P.H., a member of the CDC’s pneumococcal vaccines work group, said at the meeting. Previously, the recommendation for this group was that a dose of PCV13 "may be administered" to children and adolescents 6 through 18 years of age who have certain medical conditions.
Initially approved in 2010, PCV13 (marketed by Pfizer as Prevnar 13) was approved in January 2013 for children aged 6 through 17 years for preventing invasive disease caused by the Streptococcus pneumoniae serotypes contained in the vaccine (1, 3, 4, 5, 6A, 6B, 7F, 9V, 14, 18C, 19A, 19F, and 23F). PCV13 contains the seven strains contained in PCV7 (4, 6B, 9V, 14, 18C, 19F, and 23F), plus six more (1, 3, 5, 6A, 7F, and 19A).
PCV13 was added to the most recent updated adult schedule, released in January, for PCV13-naive adults 19 years and older who have immunocompromising conditions such as chronic renal failure, functional or anatomic asplenia, cerebrospinal fluid leaks, or cochlear implants and are immunocompromised (such as HIV infection).
After a review of the available evidence, the working group concluded that the burden of disease is "extremely high" among immunocompromised children aged 6-18 years and that the vaccine would probably be effective and the benefits would likely outweigh the risks in this group, Ms. Pilishvili of the respiratory diseases branch in the CDC’s National Center for Immunization and Respiratory Diseases, said at the meeting.
When compared with children without underlying conditions, the relative risk of IPD is over 1,000 times higher among children with a hematologic malignancy, almost 44-fold higher among children with sickle cell disease, and 158-fold higher among those with HIV/AIDS, she said, citing unpublished CDC data.
In addition, the use of both PCV13 and PPSV23 would provide broader serotype coverage for high-risk children in this age group, she said. In 2011, 38% of the IPD cases among high-risk children aged 6-18 years were caused by serotypes included in the PCV13 vaccine, 33% were the result of non-PCV13 serotypes in the PPSV23 vaccine, and 29% were caused by serotypes not included in either vaccine, she said.
Studies reviewed by the working group indicated that vaccination with other PCV vaccines reduced IPD, which included vaccine efficacy studies of PCV7 and PCV9, which, when combined, were estimated to be 69% effective in reducing IPD from serotypes included in the vaccines, Other evidence reviewed by the working group included immunogenicity data of PCV13 in people with sickle cell disease who had previously been vaccinated with PPSV23, in an open-label, single-arm study of children aged 6-7 years, as well as HIV-infected children.
Experts in immunization-related fields are members of the ACIP committee, which develops written recommendations for the routine administration of vaccines to children and adults in the civilian population.
*Updated 2/22/13
Vaccination with the 13-valent pneumococcal conjugate vaccine should be routine for certain high-risk children aged 6-18 years, panel members unanimously agreed at a meeting of the Centers for Disease Control and Prevention Advisory Committee on Immunization Practices.
Specifically, the panel recommended expanding the indication for this vaccine to include a group of older children – those between 6 and 18 years of age – who are at high risk for invasive pneumococcal disease (IPD) because of immunocompromising conditions, functional or anatomic asplenia, cerebrospinal fluid leaks, or cochlear implants, regardless or whether they have previously received the PCV7 or pneumococcal polysaccaride vaccine (PPSV23) vaccines. The 13-valent pneumococcal conjugate vaccine (PCV13) is already recommended by ACIP for high risk children 24-71 months of age who have these underlying conditions.*
The ACIP panel voted 15-0 in favor of this recommendation proposed by ACIP’s pneumococcal vaccines work group at its Feb. 20 meeting.
The inclusion of the older pediatric age group will harmonize the PCV13 pediatric recommendation and may improve vaccine uptake in these groups, Tamara Pilishvili, M.P.H., a member of the CDC’s pneumococcal vaccines work group, said at the meeting. Previously, the recommendation for this group was that a dose of PCV13 "may be administered" to children and adolescents 6 through 18 years of age who have certain medical conditions.
Initially approved in 2010, PCV13 (marketed by Pfizer as Prevnar 13) was approved in January 2013 for children aged 6 through 17 years for preventing invasive disease caused by the Streptococcus pneumoniae serotypes contained in the vaccine (1, 3, 4, 5, 6A, 6B, 7F, 9V, 14, 18C, 19A, 19F, and 23F). PCV13 contains the seven strains contained in PCV7 (4, 6B, 9V, 14, 18C, 19F, and 23F), plus six more (1, 3, 5, 6A, 7F, and 19A).
PCV13 was added to the most recent updated adult schedule, released in January, for PCV13-naive adults 19 years and older who have immunocompromising conditions such as chronic renal failure, functional or anatomic asplenia, cerebrospinal fluid leaks, or cochlear implants and are immunocompromised (such as HIV infection).
After a review of the available evidence, the working group concluded that the burden of disease is "extremely high" among immunocompromised children aged 6-18 years and that the vaccine would probably be effective and the benefits would likely outweigh the risks in this group, Ms. Pilishvili of the respiratory diseases branch in the CDC’s National Center for Immunization and Respiratory Diseases, said at the meeting.
When compared with children without underlying conditions, the relative risk of IPD is over 1,000 times higher among children with a hematologic malignancy, almost 44-fold higher among children with sickle cell disease, and 158-fold higher among those with HIV/AIDS, she said, citing unpublished CDC data.
In addition, the use of both PCV13 and PPSV23 would provide broader serotype coverage for high-risk children in this age group, she said. In 2011, 38% of the IPD cases among high-risk children aged 6-18 years were caused by serotypes included in the PCV13 vaccine, 33% were the result of non-PCV13 serotypes in the PPSV23 vaccine, and 29% were caused by serotypes not included in either vaccine, she said.
Studies reviewed by the working group indicated that vaccination with other PCV vaccines reduced IPD, which included vaccine efficacy studies of PCV7 and PCV9, which, when combined, were estimated to be 69% effective in reducing IPD from serotypes included in the vaccines, Other evidence reviewed by the working group included immunogenicity data of PCV13 in people with sickle cell disease who had previously been vaccinated with PPSV23, in an open-label, single-arm study of children aged 6-7 years, as well as HIV-infected children.
Experts in immunization-related fields are members of the ACIP committee, which develops written recommendations for the routine administration of vaccines to children and adults in the civilian population.
*Updated 2/22/13
Vaccination with the 13-valent pneumococcal conjugate vaccine should be routine for certain high-risk children aged 6-18 years, panel members unanimously agreed at a meeting of the Centers for Disease Control and Prevention Advisory Committee on Immunization Practices.
Specifically, the panel recommended expanding the indication for this vaccine to include a group of older children – those between 6 and 18 years of age – who are at high risk for invasive pneumococcal disease (IPD) because of immunocompromising conditions, functional or anatomic asplenia, cerebrospinal fluid leaks, or cochlear implants, regardless or whether they have previously received the PCV7 or pneumococcal polysaccaride vaccine (PPSV23) vaccines. The 13-valent pneumococcal conjugate vaccine (PCV13) is already recommended by ACIP for high risk children 24-71 months of age who have these underlying conditions.*
The ACIP panel voted 15-0 in favor of this recommendation proposed by ACIP’s pneumococcal vaccines work group at its Feb. 20 meeting.
The inclusion of the older pediatric age group will harmonize the PCV13 pediatric recommendation and may improve vaccine uptake in these groups, Tamara Pilishvili, M.P.H., a member of the CDC’s pneumococcal vaccines work group, said at the meeting. Previously, the recommendation for this group was that a dose of PCV13 "may be administered" to children and adolescents 6 through 18 years of age who have certain medical conditions.
Initially approved in 2010, PCV13 (marketed by Pfizer as Prevnar 13) was approved in January 2013 for children aged 6 through 17 years for preventing invasive disease caused by the Streptococcus pneumoniae serotypes contained in the vaccine (1, 3, 4, 5, 6A, 6B, 7F, 9V, 14, 18C, 19A, 19F, and 23F). PCV13 contains the seven strains contained in PCV7 (4, 6B, 9V, 14, 18C, 19F, and 23F), plus six more (1, 3, 5, 6A, 7F, and 19A).
PCV13 was added to the most recent updated adult schedule, released in January, for PCV13-naive adults 19 years and older who have immunocompromising conditions such as chronic renal failure, functional or anatomic asplenia, cerebrospinal fluid leaks, or cochlear implants and are immunocompromised (such as HIV infection).
After a review of the available evidence, the working group concluded that the burden of disease is "extremely high" among immunocompromised children aged 6-18 years and that the vaccine would probably be effective and the benefits would likely outweigh the risks in this group, Ms. Pilishvili of the respiratory diseases branch in the CDC’s National Center for Immunization and Respiratory Diseases, said at the meeting.
When compared with children without underlying conditions, the relative risk of IPD is over 1,000 times higher among children with a hematologic malignancy, almost 44-fold higher among children with sickle cell disease, and 158-fold higher among those with HIV/AIDS, she said, citing unpublished CDC data.
In addition, the use of both PCV13 and PPSV23 would provide broader serotype coverage for high-risk children in this age group, she said. In 2011, 38% of the IPD cases among high-risk children aged 6-18 years were caused by serotypes included in the PCV13 vaccine, 33% were the result of non-PCV13 serotypes in the PPSV23 vaccine, and 29% were caused by serotypes not included in either vaccine, she said.
Studies reviewed by the working group indicated that vaccination with other PCV vaccines reduced IPD, which included vaccine efficacy studies of PCV7 and PCV9, which, when combined, were estimated to be 69% effective in reducing IPD from serotypes included in the vaccines, Other evidence reviewed by the working group included immunogenicity data of PCV13 in people with sickle cell disease who had previously been vaccinated with PPSV23, in an open-label, single-arm study of children aged 6-7 years, as well as HIV-infected children.
Experts in immunization-related fields are members of the ACIP committee, which develops written recommendations for the routine administration of vaccines to children and adults in the civilian population.
*Updated 2/22/13
FROM AN ACIP MEETING
IOM report addresses global problem of poor-quality drugs
Falsified and substandard drugs are a global public health threat that needs to be addressed cooperatively by governments, manufacturers, and other health organizations, according to an Institute of Medicine report issued Feb. 13.
This is "a grave public health problem because [falsified and substandard drugs] are ineffective, promote drug resistance, and even cause severe illness and death, particularly in developing countries where they regularly flood the market," chair of the report committee, Lawrence Gostin, O’Neill Professor of Global Health Law, Georgetown University Law Center in Washington, said in a statement.
"Given the international nature of modern manufacturing and trade, every nation has a stake and a role to play in ensuring the production and sale of high-quality medications," he said.
The IOM committee is calling on the World Health Organization, other international organizations, governments, and stakeholders to collaborate on "a global code of practice, build national regulatory capabilities, and promote international cooperation."
The committee was convened by the IOM at the request of the Food and Drug Administration. In a statement, Commissioner Margaret A. Hamburg noted that the FDA is transforming from a "predominantly domestically focused agency to one that is fully prepared to help ensure product safety and quality within a globalized world," and that many of the recommendations in the IOM report are already underway, including having a presence in 12 countries in seven regions of the world.
Quite simply, the first thing that needs to be done, according to the report, is for the World Health Assembly to adopt consistent definitions, using "substandard" to describe drugs that do not meet pharmacopeia or manufacturer specifications, and "falsified" for those products with a "false representation" of identity and/or source.
To avoid confusion, the report recommends against using the term counterfeit, which, although widely used to refer to drugs that do not contain what they claim to, has a narrow legal definition related to infringement of registered trademarks.
Available in street markets, unregulated websites, and other unreliable sources, falsified and substandard drugs cause problems ranging from a lack of a therapeutic effect and resistance to antimalarial and tuberculosis treatments to frank poisoning.
While it is difficult to quantify the magnitude of the problem, the report cites some data, including the finding by 25 major pharmaceutical companies that in 2011, falsified or substandard drugs were sold in at least 124 countries. And although poorer countries are disproportionately affected, richer countries are impacted too. The report specifically cites the recent case of the Massachusetts compounding company that produced contaminated injectable steroid products, which caused many cases of meningitis, including 44 cases that were fatal in a 5-month period.
"Lack of clarity about the relative authority of the FDA and state pharmacy councils to regulate compounding pharmacies contributed to the outbreak," the report noted.
Another key recommendation: Establish in the United States a mandatory drug tracking system, and strengthen the requirements for licensing medication wholesalers. The report describes secondary wholesalers as the "weakest point" in the U.S. drug distribution chain. The latter should include the requirement that state licensing boards in the United States only license wholesalers that meet accreditation standards of the National Association of Boards of Pharmacy (NABP), and that the FDA and state licensing boards establish a public database with information on wholesalers with revoked or suspended licenses.
To facilitate the changes, Congress should authorize and fund the FDA to create a mandatory "track and trace system" using unique product serial numbers to track packages of medications from the factory to the consumer, according to the report.
The IOM committee also recommended better training for pharmacy staff in developing countries and improved communication and training programs to educate health care workers and consumers about poor-quality medications and how to report suspected cases.
Funding from international investment agencies should be used to help pharmaceutical manufacturers upgrade to international standards, and for governments in low- and middle-income countries to help their regulatory agencies comply with international manufacturing and quality control standards.
The study was supported by a contract between the National Academy of Sciences and the FDA.
Falsified and substandard drugs are a global public health threat that needs to be addressed cooperatively by governments, manufacturers, and other health organizations, according to an Institute of Medicine report issued Feb. 13.
This is "a grave public health problem because [falsified and substandard drugs] are ineffective, promote drug resistance, and even cause severe illness and death, particularly in developing countries where they regularly flood the market," chair of the report committee, Lawrence Gostin, O’Neill Professor of Global Health Law, Georgetown University Law Center in Washington, said in a statement.
"Given the international nature of modern manufacturing and trade, every nation has a stake and a role to play in ensuring the production and sale of high-quality medications," he said.
The IOM committee is calling on the World Health Organization, other international organizations, governments, and stakeholders to collaborate on "a global code of practice, build national regulatory capabilities, and promote international cooperation."
The committee was convened by the IOM at the request of the Food and Drug Administration. In a statement, Commissioner Margaret A. Hamburg noted that the FDA is transforming from a "predominantly domestically focused agency to one that is fully prepared to help ensure product safety and quality within a globalized world," and that many of the recommendations in the IOM report are already underway, including having a presence in 12 countries in seven regions of the world.
Quite simply, the first thing that needs to be done, according to the report, is for the World Health Assembly to adopt consistent definitions, using "substandard" to describe drugs that do not meet pharmacopeia or manufacturer specifications, and "falsified" for those products with a "false representation" of identity and/or source.
To avoid confusion, the report recommends against using the term counterfeit, which, although widely used to refer to drugs that do not contain what they claim to, has a narrow legal definition related to infringement of registered trademarks.
Available in street markets, unregulated websites, and other unreliable sources, falsified and substandard drugs cause problems ranging from a lack of a therapeutic effect and resistance to antimalarial and tuberculosis treatments to frank poisoning.
While it is difficult to quantify the magnitude of the problem, the report cites some data, including the finding by 25 major pharmaceutical companies that in 2011, falsified or substandard drugs were sold in at least 124 countries. And although poorer countries are disproportionately affected, richer countries are impacted too. The report specifically cites the recent case of the Massachusetts compounding company that produced contaminated injectable steroid products, which caused many cases of meningitis, including 44 cases that were fatal in a 5-month period.
"Lack of clarity about the relative authority of the FDA and state pharmacy councils to regulate compounding pharmacies contributed to the outbreak," the report noted.
Another key recommendation: Establish in the United States a mandatory drug tracking system, and strengthen the requirements for licensing medication wholesalers. The report describes secondary wholesalers as the "weakest point" in the U.S. drug distribution chain. The latter should include the requirement that state licensing boards in the United States only license wholesalers that meet accreditation standards of the National Association of Boards of Pharmacy (NABP), and that the FDA and state licensing boards establish a public database with information on wholesalers with revoked or suspended licenses.
To facilitate the changes, Congress should authorize and fund the FDA to create a mandatory "track and trace system" using unique product serial numbers to track packages of medications from the factory to the consumer, according to the report.
The IOM committee also recommended better training for pharmacy staff in developing countries and improved communication and training programs to educate health care workers and consumers about poor-quality medications and how to report suspected cases.
Funding from international investment agencies should be used to help pharmaceutical manufacturers upgrade to international standards, and for governments in low- and middle-income countries to help their regulatory agencies comply with international manufacturing and quality control standards.
The study was supported by a contract between the National Academy of Sciences and the FDA.
Falsified and substandard drugs are a global public health threat that needs to be addressed cooperatively by governments, manufacturers, and other health organizations, according to an Institute of Medicine report issued Feb. 13.
This is "a grave public health problem because [falsified and substandard drugs] are ineffective, promote drug resistance, and even cause severe illness and death, particularly in developing countries where they regularly flood the market," chair of the report committee, Lawrence Gostin, O’Neill Professor of Global Health Law, Georgetown University Law Center in Washington, said in a statement.
"Given the international nature of modern manufacturing and trade, every nation has a stake and a role to play in ensuring the production and sale of high-quality medications," he said.
The IOM committee is calling on the World Health Organization, other international organizations, governments, and stakeholders to collaborate on "a global code of practice, build national regulatory capabilities, and promote international cooperation."
The committee was convened by the IOM at the request of the Food and Drug Administration. In a statement, Commissioner Margaret A. Hamburg noted that the FDA is transforming from a "predominantly domestically focused agency to one that is fully prepared to help ensure product safety and quality within a globalized world," and that many of the recommendations in the IOM report are already underway, including having a presence in 12 countries in seven regions of the world.
Quite simply, the first thing that needs to be done, according to the report, is for the World Health Assembly to adopt consistent definitions, using "substandard" to describe drugs that do not meet pharmacopeia or manufacturer specifications, and "falsified" for those products with a "false representation" of identity and/or source.
To avoid confusion, the report recommends against using the term counterfeit, which, although widely used to refer to drugs that do not contain what they claim to, has a narrow legal definition related to infringement of registered trademarks.
Available in street markets, unregulated websites, and other unreliable sources, falsified and substandard drugs cause problems ranging from a lack of a therapeutic effect and resistance to antimalarial and tuberculosis treatments to frank poisoning.
While it is difficult to quantify the magnitude of the problem, the report cites some data, including the finding by 25 major pharmaceutical companies that in 2011, falsified or substandard drugs were sold in at least 124 countries. And although poorer countries are disproportionately affected, richer countries are impacted too. The report specifically cites the recent case of the Massachusetts compounding company that produced contaminated injectable steroid products, which caused many cases of meningitis, including 44 cases that were fatal in a 5-month period.
"Lack of clarity about the relative authority of the FDA and state pharmacy councils to regulate compounding pharmacies contributed to the outbreak," the report noted.
Another key recommendation: Establish in the United States a mandatory drug tracking system, and strengthen the requirements for licensing medication wholesalers. The report describes secondary wholesalers as the "weakest point" in the U.S. drug distribution chain. The latter should include the requirement that state licensing boards in the United States only license wholesalers that meet accreditation standards of the National Association of Boards of Pharmacy (NABP), and that the FDA and state licensing boards establish a public database with information on wholesalers with revoked or suspended licenses.
To facilitate the changes, Congress should authorize and fund the FDA to create a mandatory "track and trace system" using unique product serial numbers to track packages of medications from the factory to the consumer, according to the report.
The IOM committee also recommended better training for pharmacy staff in developing countries and improved communication and training programs to educate health care workers and consumers about poor-quality medications and how to report suspected cases.
Funding from international investment agencies should be used to help pharmaceutical manufacturers upgrade to international standards, and for governments in low- and middle-income countries to help their regulatory agencies comply with international manufacturing and quality control standards.
The study was supported by a contract between the National Academy of Sciences and the FDA.
FROM THE INSTITUTE OF MEDICINE
FDA withholds insulin degludec approval; wants safety data
The Food and Drug Administration has declined to approve insulin degludec, calling for more cardiovascular safety data on the long-acting basal insulin analogue, setting back the timeline for its availability by at least a year.
In a statement released on Feb. 11, the manufacturer, Novo Nordisk, said that in a complete response letter, the FDA has requested "additional cardiovascular data from a dedicated cardiovascular outcomes trial" before the review of insulin degludec (IDeg) and insulin degludec combined with insulin aspart can be completed. The decision applies to both IDeg alone and IDeg combined with the fast-acting insulin analogue insulin aspart. In September 2011, the company filed for approval of these products to improve glycemic control in adults with type 1 and type 2 diabetes.
The FDA issues complete response letters when a product is not ready to be approved, outlining deficiencies in the application, and, when possible, including recommendations on what is needed to complete the approval process. The FDA does not announce these letters publicly, but the companies are free to announce them.
"Novo Nordisk is evaluating the content of the Complete Response Letter and will work closely with the FDA to provide the requested data," according to the statement, which adds that the company does not expect to have the data available in 2013. Approval is also dependent on the resolution of violations in good manufacturing practice (CGMP) at manufacturing facility, outlined in a Dec. 12 warning letter from the FDA, the company said.
In November 2012, the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee unanimously recommended that Novo Nordisk conduct a cardiovascular outcomes study to investigate a cardiovascular safety signal detected in a meta-analysis of clinical trials, an increase in a composite major cardiovascular event (MACE) endpoint of unstable angina pectoris, cardiovascular death, stroke, and acute coronary syndrome. But the panel was less united on whether the data should be required before or after approval, voting 8-4 to recommend approval.
Based on a 2008 guidance issued by the FDA on cardiovascular risk assessments of new drugs for type 2 diabetes, preapproval safety analyses should provide reassuring estimates of risk.
IDeg forms multihexamers after injection, resulting in a soluble depot from which there is a slow, continuous, and extended release of IDeg, and a flat stable profile, according to Novo Nordisk, which markets insulin aspart as Novolog. If approved, the company plans to market insulin degludec as Tresiba and the combination product as Ryzodeg. Both have been approved in the European Union, Mexico, and Japan.
The Food and Drug Administration has declined to approve insulin degludec, calling for more cardiovascular safety data on the long-acting basal insulin analogue, setting back the timeline for its availability by at least a year.
In a statement released on Feb. 11, the manufacturer, Novo Nordisk, said that in a complete response letter, the FDA has requested "additional cardiovascular data from a dedicated cardiovascular outcomes trial" before the review of insulin degludec (IDeg) and insulin degludec combined with insulin aspart can be completed. The decision applies to both IDeg alone and IDeg combined with the fast-acting insulin analogue insulin aspart. In September 2011, the company filed for approval of these products to improve glycemic control in adults with type 1 and type 2 diabetes.
The FDA issues complete response letters when a product is not ready to be approved, outlining deficiencies in the application, and, when possible, including recommendations on what is needed to complete the approval process. The FDA does not announce these letters publicly, but the companies are free to announce them.
"Novo Nordisk is evaluating the content of the Complete Response Letter and will work closely with the FDA to provide the requested data," according to the statement, which adds that the company does not expect to have the data available in 2013. Approval is also dependent on the resolution of violations in good manufacturing practice (CGMP) at manufacturing facility, outlined in a Dec. 12 warning letter from the FDA, the company said.
In November 2012, the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee unanimously recommended that Novo Nordisk conduct a cardiovascular outcomes study to investigate a cardiovascular safety signal detected in a meta-analysis of clinical trials, an increase in a composite major cardiovascular event (MACE) endpoint of unstable angina pectoris, cardiovascular death, stroke, and acute coronary syndrome. But the panel was less united on whether the data should be required before or after approval, voting 8-4 to recommend approval.
Based on a 2008 guidance issued by the FDA on cardiovascular risk assessments of new drugs for type 2 diabetes, preapproval safety analyses should provide reassuring estimates of risk.
IDeg forms multihexamers after injection, resulting in a soluble depot from which there is a slow, continuous, and extended release of IDeg, and a flat stable profile, according to Novo Nordisk, which markets insulin aspart as Novolog. If approved, the company plans to market insulin degludec as Tresiba and the combination product as Ryzodeg. Both have been approved in the European Union, Mexico, and Japan.
The Food and Drug Administration has declined to approve insulin degludec, calling for more cardiovascular safety data on the long-acting basal insulin analogue, setting back the timeline for its availability by at least a year.
In a statement released on Feb. 11, the manufacturer, Novo Nordisk, said that in a complete response letter, the FDA has requested "additional cardiovascular data from a dedicated cardiovascular outcomes trial" before the review of insulin degludec (IDeg) and insulin degludec combined with insulin aspart can be completed. The decision applies to both IDeg alone and IDeg combined with the fast-acting insulin analogue insulin aspart. In September 2011, the company filed for approval of these products to improve glycemic control in adults with type 1 and type 2 diabetes.
The FDA issues complete response letters when a product is not ready to be approved, outlining deficiencies in the application, and, when possible, including recommendations on what is needed to complete the approval process. The FDA does not announce these letters publicly, but the companies are free to announce them.
"Novo Nordisk is evaluating the content of the Complete Response Letter and will work closely with the FDA to provide the requested data," according to the statement, which adds that the company does not expect to have the data available in 2013. Approval is also dependent on the resolution of violations in good manufacturing practice (CGMP) at manufacturing facility, outlined in a Dec. 12 warning letter from the FDA, the company said.
In November 2012, the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee unanimously recommended that Novo Nordisk conduct a cardiovascular outcomes study to investigate a cardiovascular safety signal detected in a meta-analysis of clinical trials, an increase in a composite major cardiovascular event (MACE) endpoint of unstable angina pectoris, cardiovascular death, stroke, and acute coronary syndrome. But the panel was less united on whether the data should be required before or after approval, voting 8-4 to recommend approval.
Based on a 2008 guidance issued by the FDA on cardiovascular risk assessments of new drugs for type 2 diabetes, preapproval safety analyses should provide reassuring estimates of risk.
IDeg forms multihexamers after injection, resulting in a soluble depot from which there is a slow, continuous, and extended release of IDeg, and a flat stable profile, according to Novo Nordisk, which markets insulin aspart as Novolog. If approved, the company plans to market insulin degludec as Tresiba and the combination product as Ryzodeg. Both have been approved in the European Union, Mexico, and Japan.
No benefit of endovascular therapy added to TPA for stroke
Functional outcomes in patients treated with intravenous tissue plasminogen activator with or without endovascular therapy after a moderate to severe acute ischemic stroke were not significantly different, and safety outcomes were similar, in a study that was stopped early because of these results.
In the IMS (Interventional Management of Stroke) III study, 40.8% of patients randomized to receive endovascular therapy plus intravenous TPA met the primary endpoint, a measure of functional independence – a modified Rankin score of 2 or less at 90 days – compared with 38.7% among those who had intravenous TPA alone, a difference that was not statistically significant, reported Dr. Joseph Broderick of the University of Cincinnati Neuroscience Institute, and the other IMS III investigators.

Mortality and other safety outcomes were also not significantly different between the two groups of patients in the study, which was stopped early because of futility after 656 of the planned 900 patients had been randomized. The study is being published online on Feb. 7 to coincide with the presentation of the results at the International Stroke Conference (N. Engl. J. Med. 2013 [doi:10.1056/NEJMoa1214300]).
Referring to the lack of randomized clinical trial data, the authors pointed out that it is uncertain whether endovascular therapy (which includes endovascular pharmacologic thrombolysis and, more recently, the use of stent retrievers) alone or combined with intravenous TPA is a more effective treatment of acute stroke than intravenous TPA alone, "the only proven reperfusion therapy for acute ischemic stroke."
In the study, conducted at 58 centers in the United States, Canada, Australia, and Europe, 434 patients were randomized to endovascular therapy plus intravenous TPA and 222 were randomized to standard treatment with intravenous TPA alone (started within 3 hours of stroke onset). The median age of those enrolled was 68-69 years (range, 23-89 years), a little over half were men, about 14% were black or Hispanic, and the median the National Institute of Health Stroke Scale (NIHSS) score was 16-17 (8-19 is a moderately severe stroke and 20 or greater is a severe stroke At the beginning of the study, only one thrombectomy device had been cleared by the Food and Drug Administration and, as the trial continued, other devices were used as they became cleared for use in the different countries.
In addition to the main finding, there were no differences in the primary outcome among those patients with an NIHSS score of 20 or more, and those with a score of 19 or lower, said the authors, who had hypothesized that endovascular therapy would have greater efficacy in patients with more-severe strokes since they "have the highest likelihood of occlusion in a major intracranial artery and the greatest volume of ischemic brain at risk."
They had also hypothesized that receiving endovascular therapy earlier would be associated with a greater benefit, but this was also not a significant factor in outcomes.
Mortality at 90 days was 19.1% in the endovascular therapy group and 21.6% in the intravenous TPA–alone group. Within 30 hours of TPA initiation, 6.2% of those on endovascular therapy and 5.9% of those on TPA alone had a symptomatic intracerebral hemorrhage. The differences in mortality at 7 days and in parenchymal hematoma rates were also not significantly different between the two groups. The rate of asymptomatic intracerebral hemorrhage, however, was significantly higher in the endovascular group.
Outcomes consistently trended better with combined therapy in patients with strokes involving larger artery occlusions and those with the shortest times from stroke onset to initiation of treatment, although because of small patient numbers the differences didn’t achieve statistical significance. These will be the subgroups that ought to be the focus of future clinical trials, Dr. Broderick said in a press briefing at the conference.
The underlying rationale for combined therapy is that intravenous TPA can quickly be started in the emergency department while the endovascular device therapy team is assembling, often at another hospital, which entails time-consuming patient transfer.
Intravenous TPA is the only proven therapy for acute ischemic stroke, but endovascular therapy is more effective at achieving recanalization. The study results bore this out: for example, the rate of partial or complete recanalization at 24 hours for an occlusion in the internal carotid artery was 81% with combined therapy compared to 35% with intravenous TPA alone. Yet this higher recanalization rate bore no clinical benefit, possibly because recanalization occurred too late, after ischemia had turned into infarction, Dr. Broderick explained.
"ISM III is going to be disappointing for a lot of people who are proponents of endovascular therapy. However, there is a light at the end of the tunnel in that there are these subgroups who may benefit," Dr. Brian Silver, who was not involved in the trial, said in an interview.
"The most critical feature is to treat the patients as soon as possible when they arrive in the emergency department, perhaps within 90 minutes. I think that’s the best chance for recovery. We are nowhere near what’s being done in cardiology, where there are door-to-balloon times of an hour. We need to do that in stroke. Since we’re dealing with an organ that’s more sensitive than the heart to ischemia, we probably need to be even faster than what’s being done in cardiology. There is definitely room for improvement in our systems, perhaps by having the endovascular team stay in the hospital. Expense will be the limitation," according to Dr. Silver, director of the stroke center at Brown University, Providence, R.I.
ISM III investigator and interventional neuroradiologist Dr. Thomas A. Tomsick said in an interview that the study results won’t change his own clinical practice.
"ISM III is by no means the final word on combined therapy. In Cincinnati tomorrow, if a patient with a large NIH Stroke Severity score shows up and we’re treating him with IV TPA at 2 hours from stroke onset, we’re not going to do a CT angiogram to evaluate that patient. He’s going to the cath lab for angiography to see if there’s a clot suitable for endovascular therapy," said Dr. Tomsick, professor of radiology at the University of Cincinnati.
Five different endovascular device therapies were utilized in IMS III. As new devices reached clinical practice, their use was allowed by investigators in order to keep the randomized trial clinically relevant. But recruitment for the study was slow because so many clinicians were already convinced by anecdotal experience that combined therapy is better. So the endovascular therapies used most frequently in IMS III aren’t the ones widely used in clinical practice today. Major new randomized trials are now getting underway comparing combined therapy using state-of-the-art, more effective stent clot retriever devices to intravenous TPA alone, he added.
In the New England Journal of Medicine report, the authors noted that "the use of randomization in ongoing and future stroke trials, rather than the treatment of eligible patients with endovascular therapy outside any trial, and minimization of the time to treatment will be essential for assessing the potential benefit of endovascular therapy for acute ischemic stroke."
No matter how future trials of combined therapy turn out, endovascular therapy is not going away, Dr. Broderick observed.
"It’s a very good tool. The reason why is there are patients who can’t get TPA. For example, roughly 5% of patients who undergo coronary artery bypass surgery have a stroke. If you have somebody with a big stroke 2 days after having their chest cracked, you can’t use TPA. In that case, those endovascular devices are the way we can get up in there and get rid of the clot," he explained.
In an editorial accompanying the report in the New England Journal of Medicine (doi:10.1056/NEJMe1215730), Dr. Marc I. Chimowitz declared that the clinical implication of IMS III is that endovascular therapy remains unproven and IV-tPA should continue to be the first-line treatment for patients with acute ischemic stroke within 4.5 hours after stroke onset.
While new clinical trials featuring more effective IV clot busters, such as tenecteplase, and next-generation endovascular devices are urgently needed in an effort to improve stroke outcomes, patient recruitment is likely to continue to be a challenge in the current environment. This could be overcome if Medicare were to place a moratorium on reimbursement for endovascular therapy of acute ischemic stroke except as part of a randomized trial, according to Dr. Chimowitz, professor of neurology at the Medical University of South Carolina, Charleston.
The study was supported with grants from NIH and the National Institute of Neurological Disorders and Stroke; and by Genentech (which supplied the TPA); and EKOS, Concentric Medical, and Cordis Neurovascular (which supplied catheters); and Actilyse (alteplase) manufacturer Boehringer Ingelheim (which, along with Genentech and EKOS, provided support for investigator meetings). Dr. Broderick disclosed consulting fees from PhotoThera. Of the 28 other authors, disclosures for 14 were listed and included having received consulting fees, grant support, and/or lecture fees from a variety of device and pharmaceutical companies that include Genentech. Dr. Chimowitz, Dr. Silver, and Dr. Tomsick reported having no financial conflicts.
* This story was updated February 8, 2013.
Functional outcomes in patients treated with intravenous tissue plasminogen activator with or without endovascular therapy after a moderate to severe acute ischemic stroke were not significantly different, and safety outcomes were similar, in a study that was stopped early because of these results.
In the IMS (Interventional Management of Stroke) III study, 40.8% of patients randomized to receive endovascular therapy plus intravenous TPA met the primary endpoint, a measure of functional independence – a modified Rankin score of 2 or less at 90 days – compared with 38.7% among those who had intravenous TPA alone, a difference that was not statistically significant, reported Dr. Joseph Broderick of the University of Cincinnati Neuroscience Institute, and the other IMS III investigators.
Mortality and other safety outcomes were also not significantly different between the two groups of patients in the study, which was stopped early because of futility after 656 of the planned 900 patients had been randomized. The study is being published online on Feb. 7 to coincide with the presentation of the results at the International Stroke Conference (N. Engl. J. Med. 2013 [doi:10.1056/NEJMoa1214300]).
Referring to the lack of randomized clinical trial data, the authors pointed out that it is uncertain whether endovascular therapy (which includes endovascular pharmacologic thrombolysis and, more recently, the use of stent retrievers) alone or combined with intravenous TPA is a more effective treatment of acute stroke than intravenous TPA alone, "the only proven reperfusion therapy for acute ischemic stroke."
In the study, conducted at 58 centers in the United States, Canada, Australia, and Europe, 434 patients were randomized to endovascular therapy plus intravenous TPA and 222 were randomized to standard treatment with intravenous TPA alone (started within 3 hours of stroke onset). The median age of those enrolled was 68-69 years (range, 23-89 years), a little over half were men, about 14% were black or Hispanic, and the median the National Institute of Health Stroke Scale (NIHSS) score was 16-17 (8-19 is a moderately severe stroke and 20 or greater is a severe stroke At the beginning of the study, only one thrombectomy device had been cleared by the Food and Drug Administration and, as the trial continued, other devices were used as they became cleared for use in the different countries.
In addition to the main finding, there were no differences in the primary outcome among those patients with an NIHSS score of 20 or more, and those with a score of 19 or lower, said the authors, who had hypothesized that endovascular therapy would have greater efficacy in patients with more-severe strokes since they "have the highest likelihood of occlusion in a major intracranial artery and the greatest volume of ischemic brain at risk."
They had also hypothesized that receiving endovascular therapy earlier would be associated with a greater benefit, but this was also not a significant factor in outcomes.
Mortality at 90 days was 19.1% in the endovascular therapy group and 21.6% in the intravenous TPA–alone group. Within 30 hours of TPA initiation, 6.2% of those on endovascular therapy and 5.9% of those on TPA alone had a symptomatic intracerebral hemorrhage. The differences in mortality at 7 days and in parenchymal hematoma rates were also not significantly different between the two groups. The rate of asymptomatic intracerebral hemorrhage, however, was significantly higher in the endovascular group.
Outcomes consistently trended better with combined therapy in patients with strokes involving larger artery occlusions and those with the shortest times from stroke onset to initiation of treatment, although because of small patient numbers the differences didn’t achieve statistical significance. These will be the subgroups that ought to be the focus of future clinical trials, Dr. Broderick said in a press briefing at the conference.
The underlying rationale for combined therapy is that intravenous TPA can quickly be started in the emergency department while the endovascular device therapy team is assembling, often at another hospital, which entails time-consuming patient transfer.
Intravenous TPA is the only proven therapy for acute ischemic stroke, but endovascular therapy is more effective at achieving recanalization. The study results bore this out: for example, the rate of partial or complete recanalization at 24 hours for an occlusion in the internal carotid artery was 81% with combined therapy compared to 35% with intravenous TPA alone. Yet this higher recanalization rate bore no clinical benefit, possibly because recanalization occurred too late, after ischemia had turned into infarction, Dr. Broderick explained.
"ISM III is going to be disappointing for a lot of people who are proponents of endovascular therapy. However, there is a light at the end of the tunnel in that there are these subgroups who may benefit," Dr. Brian Silver, who was not involved in the trial, said in an interview.
"The most critical feature is to treat the patients as soon as possible when they arrive in the emergency department, perhaps within 90 minutes. I think that’s the best chance for recovery. We are nowhere near what’s being done in cardiology, where there are door-to-balloon times of an hour. We need to do that in stroke. Since we’re dealing with an organ that’s more sensitive than the heart to ischemia, we probably need to be even faster than what’s being done in cardiology. There is definitely room for improvement in our systems, perhaps by having the endovascular team stay in the hospital. Expense will be the limitation," according to Dr. Silver, director of the stroke center at Brown University, Providence, R.I.
ISM III investigator and interventional neuroradiologist Dr. Thomas A. Tomsick said in an interview that the study results won’t change his own clinical practice.
"ISM III is by no means the final word on combined therapy. In Cincinnati tomorrow, if a patient with a large NIH Stroke Severity score shows up and we’re treating him with IV TPA at 2 hours from stroke onset, we’re not going to do a CT angiogram to evaluate that patient. He’s going to the cath lab for angiography to see if there’s a clot suitable for endovascular therapy," said Dr. Tomsick, professor of radiology at the University of Cincinnati.
Five different endovascular device therapies were utilized in IMS III. As new devices reached clinical practice, their use was allowed by investigators in order to keep the randomized trial clinically relevant. But recruitment for the study was slow because so many clinicians were already convinced by anecdotal experience that combined therapy is better. So the endovascular therapies used most frequently in IMS III aren’t the ones widely used in clinical practice today. Major new randomized trials are now getting underway comparing combined therapy using state-of-the-art, more effective stent clot retriever devices to intravenous TPA alone, he added.
In the New England Journal of Medicine report, the authors noted that "the use of randomization in ongoing and future stroke trials, rather than the treatment of eligible patients with endovascular therapy outside any trial, and minimization of the time to treatment will be essential for assessing the potential benefit of endovascular therapy for acute ischemic stroke."
No matter how future trials of combined therapy turn out, endovascular therapy is not going away, Dr. Broderick observed.
"It’s a very good tool. The reason why is there are patients who can’t get TPA. For example, roughly 5% of patients who undergo coronary artery bypass surgery have a stroke. If you have somebody with a big stroke 2 days after having their chest cracked, you can’t use TPA. In that case, those endovascular devices are the way we can get up in there and get rid of the clot," he explained.
In an editorial accompanying the report in the New England Journal of Medicine (doi:10.1056/NEJMe1215730), Dr. Marc I. Chimowitz declared that the clinical implication of IMS III is that endovascular therapy remains unproven and IV-tPA should continue to be the first-line treatment for patients with acute ischemic stroke within 4.5 hours after stroke onset.
While new clinical trials featuring more effective IV clot busters, such as tenecteplase, and next-generation endovascular devices are urgently needed in an effort to improve stroke outcomes, patient recruitment is likely to continue to be a challenge in the current environment. This could be overcome if Medicare were to place a moratorium on reimbursement for endovascular therapy of acute ischemic stroke except as part of a randomized trial, according to Dr. Chimowitz, professor of neurology at the Medical University of South Carolina, Charleston.
The study was supported with grants from NIH and the National Institute of Neurological Disorders and Stroke; and by Genentech (which supplied the TPA); and EKOS, Concentric Medical, and Cordis Neurovascular (which supplied catheters); and Actilyse (alteplase) manufacturer Boehringer Ingelheim (which, along with Genentech and EKOS, provided support for investigator meetings). Dr. Broderick disclosed consulting fees from PhotoThera. Of the 28 other authors, disclosures for 14 were listed and included having received consulting fees, grant support, and/or lecture fees from a variety of device and pharmaceutical companies that include Genentech. Dr. Chimowitz, Dr. Silver, and Dr. Tomsick reported having no financial conflicts.
* This story was updated February 8, 2013.
Functional outcomes in patients treated with intravenous tissue plasminogen activator with or without endovascular therapy after a moderate to severe acute ischemic stroke were not significantly different, and safety outcomes were similar, in a study that was stopped early because of these results.
In the IMS (Interventional Management of Stroke) III study, 40.8% of patients randomized to receive endovascular therapy plus intravenous TPA met the primary endpoint, a measure of functional independence – a modified Rankin score of 2 or less at 90 days – compared with 38.7% among those who had intravenous TPA alone, a difference that was not statistically significant, reported Dr. Joseph Broderick of the University of Cincinnati Neuroscience Institute, and the other IMS III investigators.
Mortality and other safety outcomes were also not significantly different between the two groups of patients in the study, which was stopped early because of futility after 656 of the planned 900 patients had been randomized. The study is being published online on Feb. 7 to coincide with the presentation of the results at the International Stroke Conference (N. Engl. J. Med. 2013 [doi:10.1056/NEJMoa1214300]).
Referring to the lack of randomized clinical trial data, the authors pointed out that it is uncertain whether endovascular therapy (which includes endovascular pharmacologic thrombolysis and, more recently, the use of stent retrievers) alone or combined with intravenous TPA is a more effective treatment of acute stroke than intravenous TPA alone, "the only proven reperfusion therapy for acute ischemic stroke."
In the study, conducted at 58 centers in the United States, Canada, Australia, and Europe, 434 patients were randomized to endovascular therapy plus intravenous TPA and 222 were randomized to standard treatment with intravenous TPA alone (started within 3 hours of stroke onset). The median age of those enrolled was 68-69 years (range, 23-89 years), a little over half were men, about 14% were black or Hispanic, and the median the National Institute of Health Stroke Scale (NIHSS) score was 16-17 (8-19 is a moderately severe stroke and 20 or greater is a severe stroke At the beginning of the study, only one thrombectomy device had been cleared by the Food and Drug Administration and, as the trial continued, other devices were used as they became cleared for use in the different countries.
In addition to the main finding, there were no differences in the primary outcome among those patients with an NIHSS score of 20 or more, and those with a score of 19 or lower, said the authors, who had hypothesized that endovascular therapy would have greater efficacy in patients with more-severe strokes since they "have the highest likelihood of occlusion in a major intracranial artery and the greatest volume of ischemic brain at risk."
They had also hypothesized that receiving endovascular therapy earlier would be associated with a greater benefit, but this was also not a significant factor in outcomes.
Mortality at 90 days was 19.1% in the endovascular therapy group and 21.6% in the intravenous TPA–alone group. Within 30 hours of TPA initiation, 6.2% of those on endovascular therapy and 5.9% of those on TPA alone had a symptomatic intracerebral hemorrhage. The differences in mortality at 7 days and in parenchymal hematoma rates were also not significantly different between the two groups. The rate of asymptomatic intracerebral hemorrhage, however, was significantly higher in the endovascular group.
Outcomes consistently trended better with combined therapy in patients with strokes involving larger artery occlusions and those with the shortest times from stroke onset to initiation of treatment, although because of small patient numbers the differences didn’t achieve statistical significance. These will be the subgroups that ought to be the focus of future clinical trials, Dr. Broderick said in a press briefing at the conference.
The underlying rationale for combined therapy is that intravenous TPA can quickly be started in the emergency department while the endovascular device therapy team is assembling, often at another hospital, which entails time-consuming patient transfer.
Intravenous TPA is the only proven therapy for acute ischemic stroke, but endovascular therapy is more effective at achieving recanalization. The study results bore this out: for example, the rate of partial or complete recanalization at 24 hours for an occlusion in the internal carotid artery was 81% with combined therapy compared to 35% with intravenous TPA alone. Yet this higher recanalization rate bore no clinical benefit, possibly because recanalization occurred too late, after ischemia had turned into infarction, Dr. Broderick explained.
"ISM III is going to be disappointing for a lot of people who are proponents of endovascular therapy. However, there is a light at the end of the tunnel in that there are these subgroups who may benefit," Dr. Brian Silver, who was not involved in the trial, said in an interview.
"The most critical feature is to treat the patients as soon as possible when they arrive in the emergency department, perhaps within 90 minutes. I think that’s the best chance for recovery. We are nowhere near what’s being done in cardiology, where there are door-to-balloon times of an hour. We need to do that in stroke. Since we’re dealing with an organ that’s more sensitive than the heart to ischemia, we probably need to be even faster than what’s being done in cardiology. There is definitely room for improvement in our systems, perhaps by having the endovascular team stay in the hospital. Expense will be the limitation," according to Dr. Silver, director of the stroke center at Brown University, Providence, R.I.
ISM III investigator and interventional neuroradiologist Dr. Thomas A. Tomsick said in an interview that the study results won’t change his own clinical practice.
"ISM III is by no means the final word on combined therapy. In Cincinnati tomorrow, if a patient with a large NIH Stroke Severity score shows up and we’re treating him with IV TPA at 2 hours from stroke onset, we’re not going to do a CT angiogram to evaluate that patient. He’s going to the cath lab for angiography to see if there’s a clot suitable for endovascular therapy," said Dr. Tomsick, professor of radiology at the University of Cincinnati.
Five different endovascular device therapies were utilized in IMS III. As new devices reached clinical practice, their use was allowed by investigators in order to keep the randomized trial clinically relevant. But recruitment for the study was slow because so many clinicians were already convinced by anecdotal experience that combined therapy is better. So the endovascular therapies used most frequently in IMS III aren’t the ones widely used in clinical practice today. Major new randomized trials are now getting underway comparing combined therapy using state-of-the-art, more effective stent clot retriever devices to intravenous TPA alone, he added.
In the New England Journal of Medicine report, the authors noted that "the use of randomization in ongoing and future stroke trials, rather than the treatment of eligible patients with endovascular therapy outside any trial, and minimization of the time to treatment will be essential for assessing the potential benefit of endovascular therapy for acute ischemic stroke."
No matter how future trials of combined therapy turn out, endovascular therapy is not going away, Dr. Broderick observed.
"It’s a very good tool. The reason why is there are patients who can’t get TPA. For example, roughly 5% of patients who undergo coronary artery bypass surgery have a stroke. If you have somebody with a big stroke 2 days after having their chest cracked, you can’t use TPA. In that case, those endovascular devices are the way we can get up in there and get rid of the clot," he explained.
In an editorial accompanying the report in the New England Journal of Medicine (doi:10.1056/NEJMe1215730), Dr. Marc I. Chimowitz declared that the clinical implication of IMS III is that endovascular therapy remains unproven and IV-tPA should continue to be the first-line treatment for patients with acute ischemic stroke within 4.5 hours after stroke onset.
While new clinical trials featuring more effective IV clot busters, such as tenecteplase, and next-generation endovascular devices are urgently needed in an effort to improve stroke outcomes, patient recruitment is likely to continue to be a challenge in the current environment. This could be overcome if Medicare were to place a moratorium on reimbursement for endovascular therapy of acute ischemic stroke except as part of a randomized trial, according to Dr. Chimowitz, professor of neurology at the Medical University of South Carolina, Charleston.
The study was supported with grants from NIH and the National Institute of Neurological Disorders and Stroke; and by Genentech (which supplied the TPA); and EKOS, Concentric Medical, and Cordis Neurovascular (which supplied catheters); and Actilyse (alteplase) manufacturer Boehringer Ingelheim (which, along with Genentech and EKOS, provided support for investigator meetings). Dr. Broderick disclosed consulting fees from PhotoThera. Of the 28 other authors, disclosures for 14 were listed and included having received consulting fees, grant support, and/or lecture fees from a variety of device and pharmaceutical companies that include Genentech. Dr. Chimowitz, Dr. Silver, and Dr. Tomsick reported having no financial conflicts.
* This story was updated February 8, 2013.
FROM THE INTERNATIONAL STROKE CONFERENCE
Major Finding: Endovascular therapy plus intravenous TPA showed no added outcome benefits, compared with TPA alone, with 40.8% and 38.7% of patients, respectively, reaching functional independence at 90 days.
Data Source: An international, phase III study of 656 patients with an acute moderate to severe ischemic stroke randomized 2:1 to IV TPA with endovascular therapy or IV TPA alone.
Disclosures: The study was supported with grants from NIH and the National Institute of Neurological Disorders and Stroke, and by Genentech (supplier of TPA), and EKOS, Concentric Medical, and Cordis Neurovascular (which supplied catheters). Dr. Broderick's disclosed consulting fees from PhotoThera. Of the 28 other authors, disclosures for 14 were listed and included having received consulting fees, grant support, and/or lecture fees from a variety of device and pharmaceutical companies that included Genentech.

Nitrogen-binding agent approved for treating urea cycle disorders
Glycerol phenylbutyrate, a nitrogen-binding agent, has been approved for the chronic treatment of adults and children aged 2 years and older with urea cycle disorders, who cannot be managed with a protein-restricted diet or amino acid supplements alone, the Food and Drug Administration announced on Feb. 1.
The product, in a liquid formulation, helps eliminate excess ammonia that results from the accumulation of nitrogen in people with urea cycle disorders, which are inherited metabolic disorders. The drug is taken three times a day with a protein-restricted diet, according to the FDA statement announcing the approval. In some cases, it is used with dietary supplements, such as amino acids or arginine.
It will be marketed under the trade name Ravicti by Hyperion Therapeutics.
Approval was based on a study of 44 adults with urea cycle disorders, which found that glycerol phenylbutyrate was as effective as sodium phenylbutyrate (Buphenyl), in controlling ammonia levels, according to the FDA. In the study, patients were randomized to treatment with sodium phenylbutyrate, an approved treatment for urea cycle disorders, or glycerol phenylbutyrate for 2 weeks, then were switched to the other treatment for 2 weeks. Another three studies in children and adults "provided evidence supporting the long-term safety and effectiveness of Ravicti in patients 2 years and older," the FDA said.
Diarrhea, flatulence, and headache were the most common side effects associated with treatment, according to the FDA.
The manufacturer expects to launch the product by the end of April 2013 and will distribute it through two specialty pharmacies, according to a Hyperion statement.
The product, in a liquid formulation, helps eliminate excess ammonia that results from the accumulation of nitrogen in people with urea cycle disorders, which are inherited metabolic disorders. The drug is taken three times a day with a protein-restricted diet, according to the FDA statement announcing the approval. In some cases, it is used with dietary supplements, such as amino acids or arginine.
Glycerol phenylbutyrate, a nitrogen-binding agent, has been approved for the chronic treatment of adults and children aged 2 years and older with urea cycle disorders, who cannot be managed with a protein-restricted diet or amino acid supplements alone, the Food and Drug Administration announced on Feb. 1.
The product, in a liquid formulation, helps eliminate excess ammonia that results from the accumulation of nitrogen in people with urea cycle disorders, which are inherited metabolic disorders. The drug is taken three times a day with a protein-restricted diet, according to the FDA statement announcing the approval. In some cases, it is used with dietary supplements, such as amino acids or arginine.
It will be marketed under the trade name Ravicti by Hyperion Therapeutics.
Approval was based on a study of 44 adults with urea cycle disorders, which found that glycerol phenylbutyrate was as effective as sodium phenylbutyrate (Buphenyl), in controlling ammonia levels, according to the FDA. In the study, patients were randomized to treatment with sodium phenylbutyrate, an approved treatment for urea cycle disorders, or glycerol phenylbutyrate for 2 weeks, then were switched to the other treatment for 2 weeks. Another three studies in children and adults "provided evidence supporting the long-term safety and effectiveness of Ravicti in patients 2 years and older," the FDA said.
Diarrhea, flatulence, and headache were the most common side effects associated with treatment, according to the FDA.
The manufacturer expects to launch the product by the end of April 2013 and will distribute it through two specialty pharmacies, according to a Hyperion statement.
Glycerol phenylbutyrate, a nitrogen-binding agent, has been approved for the chronic treatment of adults and children aged 2 years and older with urea cycle disorders, who cannot be managed with a protein-restricted diet or amino acid supplements alone, the Food and Drug Administration announced on Feb. 1.
The product, in a liquid formulation, helps eliminate excess ammonia that results from the accumulation of nitrogen in people with urea cycle disorders, which are inherited metabolic disorders. The drug is taken three times a day with a protein-restricted diet, according to the FDA statement announcing the approval. In some cases, it is used with dietary supplements, such as amino acids or arginine.
It will be marketed under the trade name Ravicti by Hyperion Therapeutics.
Approval was based on a study of 44 adults with urea cycle disorders, which found that glycerol phenylbutyrate was as effective as sodium phenylbutyrate (Buphenyl), in controlling ammonia levels, according to the FDA. In the study, patients were randomized to treatment with sodium phenylbutyrate, an approved treatment for urea cycle disorders, or glycerol phenylbutyrate for 2 weeks, then were switched to the other treatment for 2 weeks. Another three studies in children and adults "provided evidence supporting the long-term safety and effectiveness of Ravicti in patients 2 years and older," the FDA said.
Diarrhea, flatulence, and headache were the most common side effects associated with treatment, according to the FDA.
The manufacturer expects to launch the product by the end of April 2013 and will distribute it through two specialty pharmacies, according to a Hyperion statement.
The product, in a liquid formulation, helps eliminate excess ammonia that results from the accumulation of nitrogen in people with urea cycle disorders, which are inherited metabolic disorders. The drug is taken three times a day with a protein-restricted diet, according to the FDA statement announcing the approval. In some cases, it is used with dietary supplements, such as amino acids or arginine.
The product, in a liquid formulation, helps eliminate excess ammonia that results from the accumulation of nitrogen in people with urea cycle disorders, which are inherited metabolic disorders. The drug is taken three times a day with a protein-restricted diet, according to the FDA statement announcing the approval. In some cases, it is used with dietary supplements, such as amino acids or arginine.
CDC: Address high smoking rate among mentally ill
The rate of cigarette smoking among mentally ill adults is 70% higher than among adults without a mental illness and is particularly high in certain groups, including young adults, according to a report released Feb. 5 by the Centers for Disease Control and Prevention and the Substance Abuse and Mental Health Services Administration.
The estimates are based on data from SAMHSA’s 2009-2011 National Survey on Drug Use and Health, which calculated the rates of cigarette smoking among people aged 18 years and older in the United States who reported having "any mental illness," defined as "a diagnosable mental, behavioral, or emotional disorder, excluding developmental and substance use disorders" in the past year.
During this period, 36.1% of adults with a mental illness were current smokers, compared with 21.4% of adults with no mental illness. Those with a mental illness who smoked were heavier smokers, smoking an average of 331 cigarettes a month, compared with 310 a month among adult smokers who do not have a mental illness.
Wide variations were found in the proportion of people with a mental illness who smoked across states, ranging from a low of 18.2% in Utah to almost 50% in West Virginia. In addition to younger adults, the gap in smoking rates was particularly stark among certain populations, including American Indians and Alaska natives (54.7% of those with a mental illness smoked vs. 30.5% of those who did not have a mental illness) and people living below the poverty line (48% vs. 33%). Rates were also higher among people with a mental illness who had lower levels of education (47% among those with less than a high school education and 40.2% of those with a high school education, vs. 19% of college grads).
During a telebriefing held to announce the results, CDC director Dr. Thomas Frieden emphasized that although adults with mental illness smoke more and are less likely to quit, smoking cessation programs work but are underused in this population, and more efforts should be directed toward helping adults with mental illness quit successfully.
"People with mental illness who smoke want to and can quit, and more needs to be done to provide them with the resources and services to help them quit successfully," including making mental health facilities tobacco- and smoke-free, he said at the telebriefing, which was sponsored by the CDC.
The CDC report refers to the activities developed by SAMHSA and the Smoking Cessation Leadership Center to promote smoking cessation efforts in behavioral health care settings. These activities include expansion of the "100 Pioneers for Smoking Cessation Campaign," which provides support for mental health facilities and organizations, according to the CDC press release.
The CDC report is available here. Information on quitting smoking, including a link for health care professionals, is available at www.smokefree.gov.
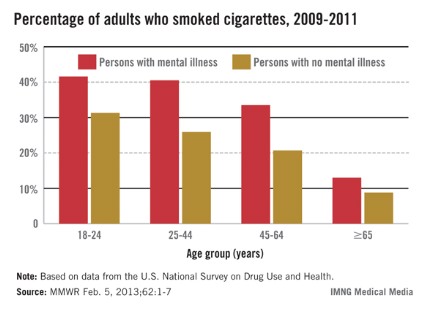
The rate of cigarette smoking among mentally ill adults is 70% higher than among adults without a mental illness and is particularly high in certain groups, including young adults, according to a report released Feb. 5 by the Centers for Disease Control and Prevention and the Substance Abuse and Mental Health Services Administration.
The estimates are based on data from SAMHSA’s 2009-2011 National Survey on Drug Use and Health, which calculated the rates of cigarette smoking among people aged 18 years and older in the United States who reported having "any mental illness," defined as "a diagnosable mental, behavioral, or emotional disorder, excluding developmental and substance use disorders" in the past year.
During this period, 36.1% of adults with a mental illness were current smokers, compared with 21.4% of adults with no mental illness. Those with a mental illness who smoked were heavier smokers, smoking an average of 331 cigarettes a month, compared with 310 a month among adult smokers who do not have a mental illness.
Wide variations were found in the proportion of people with a mental illness who smoked across states, ranging from a low of 18.2% in Utah to almost 50% in West Virginia. In addition to younger adults, the gap in smoking rates was particularly stark among certain populations, including American Indians and Alaska natives (54.7% of those with a mental illness smoked vs. 30.5% of those who did not have a mental illness) and people living below the poverty line (48% vs. 33%). Rates were also higher among people with a mental illness who had lower levels of education (47% among those with less than a high school education and 40.2% of those with a high school education, vs. 19% of college grads).
During a telebriefing held to announce the results, CDC director Dr. Thomas Frieden emphasized that although adults with mental illness smoke more and are less likely to quit, smoking cessation programs work but are underused in this population, and more efforts should be directed toward helping adults with mental illness quit successfully.
"People with mental illness who smoke want to and can quit, and more needs to be done to provide them with the resources and services to help them quit successfully," including making mental health facilities tobacco- and smoke-free, he said at the telebriefing, which was sponsored by the CDC.
The CDC report refers to the activities developed by SAMHSA and the Smoking Cessation Leadership Center to promote smoking cessation efforts in behavioral health care settings. These activities include expansion of the "100 Pioneers for Smoking Cessation Campaign," which provides support for mental health facilities and organizations, according to the CDC press release.
The CDC report is available here. Information on quitting smoking, including a link for health care professionals, is available at www.smokefree.gov.
The rate of cigarette smoking among mentally ill adults is 70% higher than among adults without a mental illness and is particularly high in certain groups, including young adults, according to a report released Feb. 5 by the Centers for Disease Control and Prevention and the Substance Abuse and Mental Health Services Administration.
The estimates are based on data from SAMHSA’s 2009-2011 National Survey on Drug Use and Health, which calculated the rates of cigarette smoking among people aged 18 years and older in the United States who reported having "any mental illness," defined as "a diagnosable mental, behavioral, or emotional disorder, excluding developmental and substance use disorders" in the past year.
During this period, 36.1% of adults with a mental illness were current smokers, compared with 21.4% of adults with no mental illness. Those with a mental illness who smoked were heavier smokers, smoking an average of 331 cigarettes a month, compared with 310 a month among adult smokers who do not have a mental illness.
Wide variations were found in the proportion of people with a mental illness who smoked across states, ranging from a low of 18.2% in Utah to almost 50% in West Virginia. In addition to younger adults, the gap in smoking rates was particularly stark among certain populations, including American Indians and Alaska natives (54.7% of those with a mental illness smoked vs. 30.5% of those who did not have a mental illness) and people living below the poverty line (48% vs. 33%). Rates were also higher among people with a mental illness who had lower levels of education (47% among those with less than a high school education and 40.2% of those with a high school education, vs. 19% of college grads).
During a telebriefing held to announce the results, CDC director Dr. Thomas Frieden emphasized that although adults with mental illness smoke more and are less likely to quit, smoking cessation programs work but are underused in this population, and more efforts should be directed toward helping adults with mental illness quit successfully.
"People with mental illness who smoke want to and can quit, and more needs to be done to provide them with the resources and services to help them quit successfully," including making mental health facilities tobacco- and smoke-free, he said at the telebriefing, which was sponsored by the CDC.
The CDC report refers to the activities developed by SAMHSA and the Smoking Cessation Leadership Center to promote smoking cessation efforts in behavioral health care settings. These activities include expansion of the "100 Pioneers for Smoking Cessation Campaign," which provides support for mental health facilities and organizations, according to the CDC press release.
The CDC report is available here. Information on quitting smoking, including a link for health care professionals, is available at www.smokefree.gov.
FROM A TELEBRIEFING SPONSORED BY THE CENTERS FOR DISEASE CONTROL AND PREVENTION
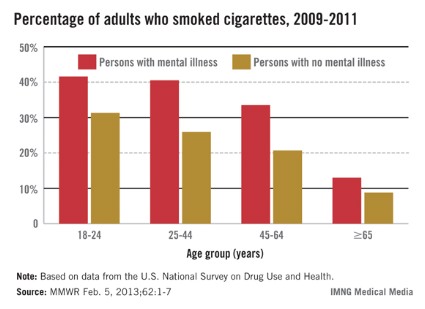
Injectable 'antisense' drug approved for familial hypercholesterolemia
Mipomersen, an apolipoprotein B synthesis inhibitor administered as a subcutaneous injection once a week, has been approved for treatment of homozygous familial hypercholesterolemia, with a plan that addresses the risk of hepatoxicity associated with treatment.
On Jan. 29, the Food and Drug Administration announced the approval of mipomersen as an adjunct to lipid-lowering medications and diet to reduce LDL cholesterol, apolipoprotein B (apo B), total cholesterol, and non-HDL cholesterol in patients with homozygous familial hypercholesterolemia (HoFH). Because the disease affects fewer than 200,000 people in the United States, it was approved as an orphan drug and will be marketed as Kynamro by Genzyme Corp., which developed the drug with Isis Pharmaceuticals.
This follows the approval of lomitapide (Juxtapid), a microsomal triglyceride transfer protein (MTP) inhibitor approved in December, to reduce LDL cholesterol, total cholesterol, apolipoprotein B, and non-HDL cholesterol in patients with HoFH.
Approval of mipomersen was based on a study of 51 patients with HoFH, according to the FDA announcement. Patients in the study, aged 12-53 years (mean 32 years), had a mean LDL cholesterol level at baseline of 400-439 mg/dL; over 26 weeks, those levels dropped by a mean of almost 25% among those on mipomersen, compared with about 3% in those on placebo, a statistically significant difference. There were also significant reductions in apo B, total cholesterol, and non-HDL cholesterol. The most common adverse reactions in the clinical trial included injection site reactions, flu-like symptoms, nausea, headache, and elevations in serum transaminases,
The label includes a boxed warning regarding the risk of hepatoxicity with treatment, because of elevations in serum transaminases associated with treatment. The warning also states that treatment has been associated with hepatic steatosis, with or without concomitant increases in transaminases, which "may be a risk factor for progressive liver disease, including steatohepatitis and cirrhosis."
The approval comes with a Risk Evaluation and Mitigation Strategy (REMS) with "elements to assure safe use," aimed at educating prescribers about the hepatoxicity risks, the need to monitor patients, and restrict access to the drug to patients with a clinical or laboratory diagnosis of HoFH, according to the FDA. Requirements include certification of prescribers and pharmacies to prescribe and dispense the drug, and a prescription authorization form for each new prescription.
The company is also required to conduct postmarketing studies, including a patient registry to evaluate long-term safety, and an enhanced pharmacovigilance program to monitor malignancies, immune-mediated reactions, and hepatic abnormalities in treated patients.
Mipomersen is the first systemic "antisense" drug to be marketed, according to the Genzyme statement announcing the approval. In the prescribing information, it is described as an "antisense oligonucleotide targeted to human messenger ribonucleic 399 acid (mRNA) for apo B-100, the principal apolipoprotein of LDL and its metabolic 400 precursor, [very-low-density lipoprotein]."
Genzyme has priced mipomersen at about $3,300 per week, according to a company spokesperson. The company has a program to help with reimbursement, including out-of-pocket costs for those who qualify,
In October, an FDA advisory panel voted to support approval of mipomersen for the approved indication, but panelists voting on both sides expressed concerns about the drug’s safety profile, mainly the potential for drug-induced liver injury.
Mipomersen, an apolipoprotein B synthesis inhibitor administered as a subcutaneous injection once a week, has been approved for treatment of homozygous familial hypercholesterolemia, with a plan that addresses the risk of hepatoxicity associated with treatment.
On Jan. 29, the Food and Drug Administration announced the approval of mipomersen as an adjunct to lipid-lowering medications and diet to reduce LDL cholesterol, apolipoprotein B (apo B), total cholesterol, and non-HDL cholesterol in patients with homozygous familial hypercholesterolemia (HoFH). Because the disease affects fewer than 200,000 people in the United States, it was approved as an orphan drug and will be marketed as Kynamro by Genzyme Corp., which developed the drug with Isis Pharmaceuticals.
This follows the approval of lomitapide (Juxtapid), a microsomal triglyceride transfer protein (MTP) inhibitor approved in December, to reduce LDL cholesterol, total cholesterol, apolipoprotein B, and non-HDL cholesterol in patients with HoFH.
Approval of mipomersen was based on a study of 51 patients with HoFH, according to the FDA announcement. Patients in the study, aged 12-53 years (mean 32 years), had a mean LDL cholesterol level at baseline of 400-439 mg/dL; over 26 weeks, those levels dropped by a mean of almost 25% among those on mipomersen, compared with about 3% in those on placebo, a statistically significant difference. There were also significant reductions in apo B, total cholesterol, and non-HDL cholesterol. The most common adverse reactions in the clinical trial included injection site reactions, flu-like symptoms, nausea, headache, and elevations in serum transaminases,
The label includes a boxed warning regarding the risk of hepatoxicity with treatment, because of elevations in serum transaminases associated with treatment. The warning also states that treatment has been associated with hepatic steatosis, with or without concomitant increases in transaminases, which "may be a risk factor for progressive liver disease, including steatohepatitis and cirrhosis."
The approval comes with a Risk Evaluation and Mitigation Strategy (REMS) with "elements to assure safe use," aimed at educating prescribers about the hepatoxicity risks, the need to monitor patients, and restrict access to the drug to patients with a clinical or laboratory diagnosis of HoFH, according to the FDA. Requirements include certification of prescribers and pharmacies to prescribe and dispense the drug, and a prescription authorization form for each new prescription.
The company is also required to conduct postmarketing studies, including a patient registry to evaluate long-term safety, and an enhanced pharmacovigilance program to monitor malignancies, immune-mediated reactions, and hepatic abnormalities in treated patients.
Mipomersen is the first systemic "antisense" drug to be marketed, according to the Genzyme statement announcing the approval. In the prescribing information, it is described as an "antisense oligonucleotide targeted to human messenger ribonucleic 399 acid (mRNA) for apo B-100, the principal apolipoprotein of LDL and its metabolic 400 precursor, [very-low-density lipoprotein]."
Genzyme has priced mipomersen at about $3,300 per week, according to a company spokesperson. The company has a program to help with reimbursement, including out-of-pocket costs for those who qualify,
In October, an FDA advisory panel voted to support approval of mipomersen for the approved indication, but panelists voting on both sides expressed concerns about the drug’s safety profile, mainly the potential for drug-induced liver injury.
Mipomersen, an apolipoprotein B synthesis inhibitor administered as a subcutaneous injection once a week, has been approved for treatment of homozygous familial hypercholesterolemia, with a plan that addresses the risk of hepatoxicity associated with treatment.
On Jan. 29, the Food and Drug Administration announced the approval of mipomersen as an adjunct to lipid-lowering medications and diet to reduce LDL cholesterol, apolipoprotein B (apo B), total cholesterol, and non-HDL cholesterol in patients with homozygous familial hypercholesterolemia (HoFH). Because the disease affects fewer than 200,000 people in the United States, it was approved as an orphan drug and will be marketed as Kynamro by Genzyme Corp., which developed the drug with Isis Pharmaceuticals.
This follows the approval of lomitapide (Juxtapid), a microsomal triglyceride transfer protein (MTP) inhibitor approved in December, to reduce LDL cholesterol, total cholesterol, apolipoprotein B, and non-HDL cholesterol in patients with HoFH.
Approval of mipomersen was based on a study of 51 patients with HoFH, according to the FDA announcement. Patients in the study, aged 12-53 years (mean 32 years), had a mean LDL cholesterol level at baseline of 400-439 mg/dL; over 26 weeks, those levels dropped by a mean of almost 25% among those on mipomersen, compared with about 3% in those on placebo, a statistically significant difference. There were also significant reductions in apo B, total cholesterol, and non-HDL cholesterol. The most common adverse reactions in the clinical trial included injection site reactions, flu-like symptoms, nausea, headache, and elevations in serum transaminases,
The label includes a boxed warning regarding the risk of hepatoxicity with treatment, because of elevations in serum transaminases associated with treatment. The warning also states that treatment has been associated with hepatic steatosis, with or without concomitant increases in transaminases, which "may be a risk factor for progressive liver disease, including steatohepatitis and cirrhosis."
The approval comes with a Risk Evaluation and Mitigation Strategy (REMS) with "elements to assure safe use," aimed at educating prescribers about the hepatoxicity risks, the need to monitor patients, and restrict access to the drug to patients with a clinical or laboratory diagnosis of HoFH, according to the FDA. Requirements include certification of prescribers and pharmacies to prescribe and dispense the drug, and a prescription authorization form for each new prescription.
The company is also required to conduct postmarketing studies, including a patient registry to evaluate long-term safety, and an enhanced pharmacovigilance program to monitor malignancies, immune-mediated reactions, and hepatic abnormalities in treated patients.
Mipomersen is the first systemic "antisense" drug to be marketed, according to the Genzyme statement announcing the approval. In the prescribing information, it is described as an "antisense oligonucleotide targeted to human messenger ribonucleic 399 acid (mRNA) for apo B-100, the principal apolipoprotein of LDL and its metabolic 400 precursor, [very-low-density lipoprotein]."
Genzyme has priced mipomersen at about $3,300 per week, according to a company spokesperson. The company has a program to help with reimbursement, including out-of-pocket costs for those who qualify,
In October, an FDA advisory panel voted to support approval of mipomersen for the approved indication, but panelists voting on both sides expressed concerns about the drug’s safety profile, mainly the potential for drug-induced liver injury.
FDA approves generic doxorubicin to address shortage
The Food and Drug Administration has approved a generic formulation of doxorubicin hydrochloride liposome injection to address the current shortage of the cancer drug, the agency announced Feb. 4.
The generic formulation is manufactured by Sun Pharma Global FZE and will be available in 20-mg and 50-mg vials.
Doxil, the brand version of doxorubicin that was first approved in 1995 and is marketed by Janssen Products, is on the FDA’s Current Drug Shortages list, with the note that limited supplies are available.
To address the shortage, the FDA announced in February 2012 that it would allow Lipodox, a version of doxorubicin manufactured by Sun that was not approved in the United States, to be imported on a temporary basis to avoid interruption of the supply. The agency also allowed the one lot of the Janssen product manufactured through an unapproved process to be released for use.
"Once supplies of Sun’s generic doxorubicin hydrochloride liposome injection are sufficient to meet projected demand, the FDA expects to stop exercising enforcement discretion for any unapproved doxorubicin HCl liposomal product," an FDA statement said.
"The agency is committed to doing everything we can to address drug shortages so that patients can get the medicines they need when they need them," Capt. Valerie Jensen, R.Ph., director of the Drug Shortage staff at the FDA’s Center for Drug Evaluation and Research, noted in the statement. The agency is giving priority review to generic products on the drug shortage list.
Doxil is approved for treating ovarian cancer, multiple myeloma, and AIDS-related Kaposi’s sarcoma.
The Food and Drug Administration has approved a generic formulation of doxorubicin hydrochloride liposome injection to address the current shortage of the cancer drug, the agency announced Feb. 4.
The generic formulation is manufactured by Sun Pharma Global FZE and will be available in 20-mg and 50-mg vials.
Doxil, the brand version of doxorubicin that was first approved in 1995 and is marketed by Janssen Products, is on the FDA’s Current Drug Shortages list, with the note that limited supplies are available.
To address the shortage, the FDA announced in February 2012 that it would allow Lipodox, a version of doxorubicin manufactured by Sun that was not approved in the United States, to be imported on a temporary basis to avoid interruption of the supply. The agency also allowed the one lot of the Janssen product manufactured through an unapproved process to be released for use.
"Once supplies of Sun’s generic doxorubicin hydrochloride liposome injection are sufficient to meet projected demand, the FDA expects to stop exercising enforcement discretion for any unapproved doxorubicin HCl liposomal product," an FDA statement said.
"The agency is committed to doing everything we can to address drug shortages so that patients can get the medicines they need when they need them," Capt. Valerie Jensen, R.Ph., director of the Drug Shortage staff at the FDA’s Center for Drug Evaluation and Research, noted in the statement. The agency is giving priority review to generic products on the drug shortage list.
Doxil is approved for treating ovarian cancer, multiple myeloma, and AIDS-related Kaposi’s sarcoma.
The Food and Drug Administration has approved a generic formulation of doxorubicin hydrochloride liposome injection to address the current shortage of the cancer drug, the agency announced Feb. 4.
The generic formulation is manufactured by Sun Pharma Global FZE and will be available in 20-mg and 50-mg vials.
Doxil, the brand version of doxorubicin that was first approved in 1995 and is marketed by Janssen Products, is on the FDA’s Current Drug Shortages list, with the note that limited supplies are available.
To address the shortage, the FDA announced in February 2012 that it would allow Lipodox, a version of doxorubicin manufactured by Sun that was not approved in the United States, to be imported on a temporary basis to avoid interruption of the supply. The agency also allowed the one lot of the Janssen product manufactured through an unapproved process to be released for use.
"Once supplies of Sun’s generic doxorubicin hydrochloride liposome injection are sufficient to meet projected demand, the FDA expects to stop exercising enforcement discretion for any unapproved doxorubicin HCl liposomal product," an FDA statement said.
"The agency is committed to doing everything we can to address drug shortages so that patients can get the medicines they need when they need them," Capt. Valerie Jensen, R.Ph., director of the Drug Shortage staff at the FDA’s Center for Drug Evaluation and Research, noted in the statement. The agency is giving priority review to generic products on the drug shortage list.
Doxil is approved for treating ovarian cancer, multiple myeloma, and AIDS-related Kaposi’s sarcoma.