User login
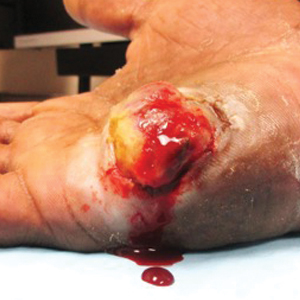
Well-Circumscribed Tumor on the Hand
The Diagnosis: Nodular Kaposi Sarcoma
Epidemic Kaposi sarcoma (KS) primarily affects patients with human immunodeficiency virus (HIV) infection. Kaposi sarcoma can appear as brown, red, or blue-black macules, plaques, patches, nodules, or tumors, and it often is observed as multifocal cutaneous lesions located on the head, neck, and upper aspects of the trunk in a fulminant manner. Kaposi sarcoma portends a poor prognosis and is an AIDS-defining malignancy.1-3 Importantly, antiretroviral therapy does not preclude its consideration in those without AIDS-defining CD4 cell counts and undetectable HIV viremia presenting with cutaneous manifestations.2,3 A retrospective review by Daly et al4 reported KS lesions in patients with CD4 lymphocyte counts greater than 300 cells/µL, most of whom were antiretroviral therapy-naïve patients. Also, those with higher CD4 counts tended to have a solitary KS lesion at presentation, while those with CD4 counts less than 300 cells/µL tended to present with multiple foci.4 Epidemic KS lesions are clinically indistinguishable from other common cutaneous conditions in the differential diagnosis of KS, necessitating biopsy for histopathologic examination. Light microscopy findings help to delineate the diagnosis of KS. Immunohistochemical staining to the latent nuclear antigen 1 of human herpesvirus 8 (HHV-8) confirms the KS diagnosis.5,6 Our patient's presentation as a solitary acral lesion was atypical for KS.
Light microscopy of our patient's biopsy demonstrated a large tumor on the acral surface of the right hand. Dermal collections of basophilic spindled cells clustered with small slitlike vascular spaces with abundant erythrocyte extravasation and numerous large ectatic vessels at the periphery were seen (Figure, A). At higher magnification, interlaced bundles of spindle cells with slitlike vessels with scattered lymphocytes and plasma cells were seen (Figure, B). An immunohistochemical stain for HHV-8 was positive and largely confined to spindle cells (Figure, C). These findings confirmed KS and met AIDS-defining criteria. Awareness of these histopathologic features is key in differentiating KS from other conditions in the differential diagnosis.

The patient's history of late latent syphilis coinfected with HIV and persistently elevated rapid plasma reagin that was recalcitrant to therapy placed an atypical nodular presentation within reason for the differential diagnosis. Deviations from the typical papulosquamous presentation with acral involvement in an immunocompromised patient mandates a consideration for syphilis with an atypical presentation. Atypical presentations include nodular, annular, pustular, lues maligna, frambesiform, corymbose, and photosensitive distributions.7,8 Notably, coinfection with HIV modifies the clinical presentation, serology, and efficacy of treatment.7-10 Atypical presentations are more common in coinfected HIV-positive patients, mandating a high degree of suspicion. Nodular secondary syphilis and the noduloulcerative form (lues maligna) often spare the palmar and plantar surfaces, and patients often have constitutional symptoms accompanying the cutaneous eruptions. In questionable cases, a biopsy lends clarification. Light microscopy on hematoxylin and eosin (H&E) staining may display acanthosis, superficial and deep perivascular swelling, plasma, histiocyte infiltrates, dermoepidermal junction changes, mixed patterns, epidermal hyperplasia, and dermal vascular thickening.7-9,11 Spirochetes may be observed on Warthin-Starry stain; however, artifact obscuration from melanin granules and reticular fibers or paucity of organisms can make identification difficult. Immunohistochemical staining may prove useful when H&E stains are atypical or have a paucity of organisms or plasma cells or when silver stains have artifactual obscuration.9 Our patient's solitary palmar lesion without constitutional symptoms made an atypical nodular secondary syphilis presentation less likely. Ultimately, the histopathologic findings were consistent with KS.
Bacillary angiomatosis (BA) is caused by Bartonella species and results in vascular proliferation with cutaneous manifestation. It frequently is observed in patients with HIV or other immunosuppressive conditions as well as patients with exposure to mammals or their vectors. Protean cutaneous manifestations and distributions of BA exist. The number of lesions can be singular to thousands. Solitary superficial pyogenic granuloma-like lesions can be clinically indistinguishable from both KS and pyogenic granuloma (PG). Superficial lesions often begin as red, violaceous, or flesh-colored papules that hemorrhage easily with trauma. The morphology of the papule can progress to be exophytic with dome-shaped or ulcerative surface features and is rubbery on palpation.12 Biopsy is required to differentiate BA from KS. Bacillary angiomatosis on light microscopy with H&E shows protuberant, lobulated, round vessels with plump endothelial cells with or without necrosis. A neutrophil infiltrate in close proximity to bacilli may be noted. Warthin-Starry stain demonstrates numerous bacilli juxtaposed to these endothelial cells. The lack of immunohistochemical staining for HHV-8 also differentiates BA from KS.12,13
Pyogenic granuloma is resultant from proliferation of endothelial cells with a lobular architecture. Pyogenic granulomas are benign, rapidly progressive, acquired lesions presenting in the skin and mucous membranes. Pyogenic granuloma often presents as a single painless papule or nodule with a glistening red-violaceous color that occasionally appears with a perilesional collarette. The lesions are friable and easily hemorrhage. Pyogenic granuloma has been associated with local skin trauma and estrogen hormones. Histopathologic examination of PG assists with differentiation from other nodular lesions. Light microscopy with standard H&E staining demonstrates a network of capillaries arranged into a lobule surrounded by a fibrous matrix. Endothelial cells appear round and protrude into the vascular lamina. Mitotic activity is increased. Lack of findings on Warthin-Starry stain assists with differentiating PG from BA, while the microscopy architecture and immunohistochemical staining differentiates PG from KS.6,13,14
Squamous cell carcinoma (SCC) is the primary malignant cancer of the hand. The dorsal aspect of the hand is the most common location; SCC less commonly is located on the palmar surface, fingers, nail bed, or intertriginous areas.15-17 Chakrabarti et al16 found that these lesions were invasive SCC when located on the palmar surface. Morphologically, SCC takes an exophytic papular, nodular, or scaly appearance with a red to flesh-colored appearance and poor demarcation of the borders. Progression to large ulcerated or secondarily infected lesions also can occur. The inflammatory reaction may cause tenderness to palpation and hemorrhage with trauma. Histopathologic examination of invasive SCC reveals atypical keratinocytes violating the basement membrane and abundant cytoplasm. Our patient's clinical presentation placed invasive SCC low on the differential diagnosis, and the histopathologic and immunohistochemical results eliminated SCC as the diagnosis.
- Antman K, Chang Y. Kaposi's sarcoma. N Engl J Med. 2000;342:1027-1038.
- Pipette WW. The incidence of second malignancies in subsets of Kaposi's sarcoma. J Am Acad Dermatol. 1987;16:855-861.
- Shiels MS, Engels EA. Evolving epidemiology of HIV-associated malignancies. Curr Opin HIV AIDS. 2017;12:6-11.
- Daly ML, Fogo A, McDonald C, et al. Kaposi sarcoma: no longer an AIDS-defining illness? a retrospective study of Kaposi sarcoma cases with CD4 counts above 300/mm³ at presentation. Clin Exp Dermatol. 2014;39:7-12.
- Broccolo F, Tassan Din C, Viganò MG, et al. HHV-8 DNA replication correlates with the clinical status in AIDS-related Kaposi's sarcoma. J Clin Virol. 2016;78:47-52.
- Pereira PF, Cuzzi T, Galhardo MC. Immunohistochemical detection of the latent nuclear antigen-1 of the human herpesvirus type 8 to differentiate cutaneous epidemic Kaposi sarcoma and its histological simulators. An Bras Dermatol. 2013;88:243-246.
- Gevorgyan O, Owen BD, Balavenkataraman A, et al. A nodular-ulcerative form of secondary syphilis in AIDS. Proc (Bayl Univ Med Cent). 2017;30:80-82.
- Balagula Y, Mattei PL, Wisco OJ, et al. The great imitator revisited: the spectrum of atypical cutaneous manifestations of secondary syphilis. Int J Dermatol. 2014;53:1434-1441.
- Hoang MP, High WA, Molberg KH. Secondary syphilis: a histologic and immunohistochemical evaluation. J Cutan Pathol. 2004;31:595-599.
- Yayli S, della Torre R, Hegyi I, et al. Late secondary syphilis with nodular lesions mimicking Kaposi sarcoma in a patient with human immunodeficiency virus. Int J Dermatol. 2014;53:E71-E73.
- Jeerapaet P, Ackerman AB. Histologic patterns of secondary syphilis. Arch Dermatol. 1973;107:373-377.
- Cockerell CJ, LeBoit PE. Bacillary angiomatosis: a newly characterized, pseudoneoplastic, infectious, cutaneous vascular disorder. J Am Acad Dermatol. 1990;22:501-512.
- Forrestel AK, Naujokas A, Martin JN, et al. Bacillary angiomatosis masquerading as Kaposi's sarcoma in East Africa. J Int Assoc Provid AIDS Care. 2015;14:21-25.
- Fortna RR, Junkins-Hopkins JM. A case of lobular capillary hemangioma (pyogenic granuloma), localized to the subcutaneous tissue, and a review of the literature. Am J Dermatopathol. 2007;29:408-411.
- Marks R. Squamous cell carcinoma. Lancet. 1996;347:735-738.
- Chakrabarti I, Watson JD, Dorrance H. Skin tumours of the hand. a 10-year review. J Hand Surg Br. 1993;18:484-486.
- Sobanko JF, Dagum AB, Davis IC, et al. Soft tissue tumors of the hand. 2. malignant. Dermatol Surg. 2007;33:771-785.
The Diagnosis: Nodular Kaposi Sarcoma
Epidemic Kaposi sarcoma (KS) primarily affects patients with human immunodeficiency virus (HIV) infection. Kaposi sarcoma can appear as brown, red, or blue-black macules, plaques, patches, nodules, or tumors, and it often is observed as multifocal cutaneous lesions located on the head, neck, and upper aspects of the trunk in a fulminant manner. Kaposi sarcoma portends a poor prognosis and is an AIDS-defining malignancy.1-3 Importantly, antiretroviral therapy does not preclude its consideration in those without AIDS-defining CD4 cell counts and undetectable HIV viremia presenting with cutaneous manifestations.2,3 A retrospective review by Daly et al4 reported KS lesions in patients with CD4 lymphocyte counts greater than 300 cells/µL, most of whom were antiretroviral therapy-naïve patients. Also, those with higher CD4 counts tended to have a solitary KS lesion at presentation, while those with CD4 counts less than 300 cells/µL tended to present with multiple foci.4 Epidemic KS lesions are clinically indistinguishable from other common cutaneous conditions in the differential diagnosis of KS, necessitating biopsy for histopathologic examination. Light microscopy findings help to delineate the diagnosis of KS. Immunohistochemical staining to the latent nuclear antigen 1 of human herpesvirus 8 (HHV-8) confirms the KS diagnosis.5,6 Our patient's presentation as a solitary acral lesion was atypical for KS.
Light microscopy of our patient's biopsy demonstrated a large tumor on the acral surface of the right hand. Dermal collections of basophilic spindled cells clustered with small slitlike vascular spaces with abundant erythrocyte extravasation and numerous large ectatic vessels at the periphery were seen (Figure, A). At higher magnification, interlaced bundles of spindle cells with slitlike vessels with scattered lymphocytes and plasma cells were seen (Figure, B). An immunohistochemical stain for HHV-8 was positive and largely confined to spindle cells (Figure, C). These findings confirmed KS and met AIDS-defining criteria. Awareness of these histopathologic features is key in differentiating KS from other conditions in the differential diagnosis.
The patient's history of late latent syphilis coinfected with HIV and persistently elevated rapid plasma reagin that was recalcitrant to therapy placed an atypical nodular presentation within reason for the differential diagnosis. Deviations from the typical papulosquamous presentation with acral involvement in an immunocompromised patient mandates a consideration for syphilis with an atypical presentation. Atypical presentations include nodular, annular, pustular, lues maligna, frambesiform, corymbose, and photosensitive distributions.7,8 Notably, coinfection with HIV modifies the clinical presentation, serology, and efficacy of treatment.7-10 Atypical presentations are more common in coinfected HIV-positive patients, mandating a high degree of suspicion. Nodular secondary syphilis and the noduloulcerative form (lues maligna) often spare the palmar and plantar surfaces, and patients often have constitutional symptoms accompanying the cutaneous eruptions. In questionable cases, a biopsy lends clarification. Light microscopy on hematoxylin and eosin (H&E) staining may display acanthosis, superficial and deep perivascular swelling, plasma, histiocyte infiltrates, dermoepidermal junction changes, mixed patterns, epidermal hyperplasia, and dermal vascular thickening.7-9,11 Spirochetes may be observed on Warthin-Starry stain; however, artifact obscuration from melanin granules and reticular fibers or paucity of organisms can make identification difficult. Immunohistochemical staining may prove useful when H&E stains are atypical or have a paucity of organisms or plasma cells or when silver stains have artifactual obscuration.9 Our patient's solitary palmar lesion without constitutional symptoms made an atypical nodular secondary syphilis presentation less likely. Ultimately, the histopathologic findings were consistent with KS.
Bacillary angiomatosis (BA) is caused by Bartonella species and results in vascular proliferation with cutaneous manifestation. It frequently is observed in patients with HIV or other immunosuppressive conditions as well as patients with exposure to mammals or their vectors. Protean cutaneous manifestations and distributions of BA exist. The number of lesions can be singular to thousands. Solitary superficial pyogenic granuloma-like lesions can be clinically indistinguishable from both KS and pyogenic granuloma (PG). Superficial lesions often begin as red, violaceous, or flesh-colored papules that hemorrhage easily with trauma. The morphology of the papule can progress to be exophytic with dome-shaped or ulcerative surface features and is rubbery on palpation.12 Biopsy is required to differentiate BA from KS. Bacillary angiomatosis on light microscopy with H&E shows protuberant, lobulated, round vessels with plump endothelial cells with or without necrosis. A neutrophil infiltrate in close proximity to bacilli may be noted. Warthin-Starry stain demonstrates numerous bacilli juxtaposed to these endothelial cells. The lack of immunohistochemical staining for HHV-8 also differentiates BA from KS.12,13
Pyogenic granuloma is resultant from proliferation of endothelial cells with a lobular architecture. Pyogenic granulomas are benign, rapidly progressive, acquired lesions presenting in the skin and mucous membranes. Pyogenic granuloma often presents as a single painless papule or nodule with a glistening red-violaceous color that occasionally appears with a perilesional collarette. The lesions are friable and easily hemorrhage. Pyogenic granuloma has been associated with local skin trauma and estrogen hormones. Histopathologic examination of PG assists with differentiation from other nodular lesions. Light microscopy with standard H&E staining demonstrates a network of capillaries arranged into a lobule surrounded by a fibrous matrix. Endothelial cells appear round and protrude into the vascular lamina. Mitotic activity is increased. Lack of findings on Warthin-Starry stain assists with differentiating PG from BA, while the microscopy architecture and immunohistochemical staining differentiates PG from KS.6,13,14
Squamous cell carcinoma (SCC) is the primary malignant cancer of the hand. The dorsal aspect of the hand is the most common location; SCC less commonly is located on the palmar surface, fingers, nail bed, or intertriginous areas.15-17 Chakrabarti et al16 found that these lesions were invasive SCC when located on the palmar surface. Morphologically, SCC takes an exophytic papular, nodular, or scaly appearance with a red to flesh-colored appearance and poor demarcation of the borders. Progression to large ulcerated or secondarily infected lesions also can occur. The inflammatory reaction may cause tenderness to palpation and hemorrhage with trauma. Histopathologic examination of invasive SCC reveals atypical keratinocytes violating the basement membrane and abundant cytoplasm. Our patient's clinical presentation placed invasive SCC low on the differential diagnosis, and the histopathologic and immunohistochemical results eliminated SCC as the diagnosis.
The Diagnosis: Nodular Kaposi Sarcoma
Epidemic Kaposi sarcoma (KS) primarily affects patients with human immunodeficiency virus (HIV) infection. Kaposi sarcoma can appear as brown, red, or blue-black macules, plaques, patches, nodules, or tumors, and it often is observed as multifocal cutaneous lesions located on the head, neck, and upper aspects of the trunk in a fulminant manner. Kaposi sarcoma portends a poor prognosis and is an AIDS-defining malignancy.1-3 Importantly, antiretroviral therapy does not preclude its consideration in those without AIDS-defining CD4 cell counts and undetectable HIV viremia presenting with cutaneous manifestations.2,3 A retrospective review by Daly et al4 reported KS lesions in patients with CD4 lymphocyte counts greater than 300 cells/µL, most of whom were antiretroviral therapy-naïve patients. Also, those with higher CD4 counts tended to have a solitary KS lesion at presentation, while those with CD4 counts less than 300 cells/µL tended to present with multiple foci.4 Epidemic KS lesions are clinically indistinguishable from other common cutaneous conditions in the differential diagnosis of KS, necessitating biopsy for histopathologic examination. Light microscopy findings help to delineate the diagnosis of KS. Immunohistochemical staining to the latent nuclear antigen 1 of human herpesvirus 8 (HHV-8) confirms the KS diagnosis.5,6 Our patient's presentation as a solitary acral lesion was atypical for KS.
Light microscopy of our patient's biopsy demonstrated a large tumor on the acral surface of the right hand. Dermal collections of basophilic spindled cells clustered with small slitlike vascular spaces with abundant erythrocyte extravasation and numerous large ectatic vessels at the periphery were seen (Figure, A). At higher magnification, interlaced bundles of spindle cells with slitlike vessels with scattered lymphocytes and plasma cells were seen (Figure, B). An immunohistochemical stain for HHV-8 was positive and largely confined to spindle cells (Figure, C). These findings confirmed KS and met AIDS-defining criteria. Awareness of these histopathologic features is key in differentiating KS from other conditions in the differential diagnosis.
The patient's history of late latent syphilis coinfected with HIV and persistently elevated rapid plasma reagin that was recalcitrant to therapy placed an atypical nodular presentation within reason for the differential diagnosis. Deviations from the typical papulosquamous presentation with acral involvement in an immunocompromised patient mandates a consideration for syphilis with an atypical presentation. Atypical presentations include nodular, annular, pustular, lues maligna, frambesiform, corymbose, and photosensitive distributions.7,8 Notably, coinfection with HIV modifies the clinical presentation, serology, and efficacy of treatment.7-10 Atypical presentations are more common in coinfected HIV-positive patients, mandating a high degree of suspicion. Nodular secondary syphilis and the noduloulcerative form (lues maligna) often spare the palmar and plantar surfaces, and patients often have constitutional symptoms accompanying the cutaneous eruptions. In questionable cases, a biopsy lends clarification. Light microscopy on hematoxylin and eosin (H&E) staining may display acanthosis, superficial and deep perivascular swelling, plasma, histiocyte infiltrates, dermoepidermal junction changes, mixed patterns, epidermal hyperplasia, and dermal vascular thickening.7-9,11 Spirochetes may be observed on Warthin-Starry stain; however, artifact obscuration from melanin granules and reticular fibers or paucity of organisms can make identification difficult. Immunohistochemical staining may prove useful when H&E stains are atypical or have a paucity of organisms or plasma cells or when silver stains have artifactual obscuration.9 Our patient's solitary palmar lesion without constitutional symptoms made an atypical nodular secondary syphilis presentation less likely. Ultimately, the histopathologic findings were consistent with KS.
Bacillary angiomatosis (BA) is caused by Bartonella species and results in vascular proliferation with cutaneous manifestation. It frequently is observed in patients with HIV or other immunosuppressive conditions as well as patients with exposure to mammals or their vectors. Protean cutaneous manifestations and distributions of BA exist. The number of lesions can be singular to thousands. Solitary superficial pyogenic granuloma-like lesions can be clinically indistinguishable from both KS and pyogenic granuloma (PG). Superficial lesions often begin as red, violaceous, or flesh-colored papules that hemorrhage easily with trauma. The morphology of the papule can progress to be exophytic with dome-shaped or ulcerative surface features and is rubbery on palpation.12 Biopsy is required to differentiate BA from KS. Bacillary angiomatosis on light microscopy with H&E shows protuberant, lobulated, round vessels with plump endothelial cells with or without necrosis. A neutrophil infiltrate in close proximity to bacilli may be noted. Warthin-Starry stain demonstrates numerous bacilli juxtaposed to these endothelial cells. The lack of immunohistochemical staining for HHV-8 also differentiates BA from KS.12,13
Pyogenic granuloma is resultant from proliferation of endothelial cells with a lobular architecture. Pyogenic granulomas are benign, rapidly progressive, acquired lesions presenting in the skin and mucous membranes. Pyogenic granuloma often presents as a single painless papule or nodule with a glistening red-violaceous color that occasionally appears with a perilesional collarette. The lesions are friable and easily hemorrhage. Pyogenic granuloma has been associated with local skin trauma and estrogen hormones. Histopathologic examination of PG assists with differentiation from other nodular lesions. Light microscopy with standard H&E staining demonstrates a network of capillaries arranged into a lobule surrounded by a fibrous matrix. Endothelial cells appear round and protrude into the vascular lamina. Mitotic activity is increased. Lack of findings on Warthin-Starry stain assists with differentiating PG from BA, while the microscopy architecture and immunohistochemical staining differentiates PG from KS.6,13,14
Squamous cell carcinoma (SCC) is the primary malignant cancer of the hand. The dorsal aspect of the hand is the most common location; SCC less commonly is located on the palmar surface, fingers, nail bed, or intertriginous areas.15-17 Chakrabarti et al16 found that these lesions were invasive SCC when located on the palmar surface. Morphologically, SCC takes an exophytic papular, nodular, or scaly appearance with a red to flesh-colored appearance and poor demarcation of the borders. Progression to large ulcerated or secondarily infected lesions also can occur. The inflammatory reaction may cause tenderness to palpation and hemorrhage with trauma. Histopathologic examination of invasive SCC reveals atypical keratinocytes violating the basement membrane and abundant cytoplasm. Our patient's clinical presentation placed invasive SCC low on the differential diagnosis, and the histopathologic and immunohistochemical results eliminated SCC as the diagnosis.
- Antman K, Chang Y. Kaposi's sarcoma. N Engl J Med. 2000;342:1027-1038.
- Pipette WW. The incidence of second malignancies in subsets of Kaposi's sarcoma. J Am Acad Dermatol. 1987;16:855-861.
- Shiels MS, Engels EA. Evolving epidemiology of HIV-associated malignancies. Curr Opin HIV AIDS. 2017;12:6-11.
- Daly ML, Fogo A, McDonald C, et al. Kaposi sarcoma: no longer an AIDS-defining illness? a retrospective study of Kaposi sarcoma cases with CD4 counts above 300/mm³ at presentation. Clin Exp Dermatol. 2014;39:7-12.
- Broccolo F, Tassan Din C, Viganò MG, et al. HHV-8 DNA replication correlates with the clinical status in AIDS-related Kaposi's sarcoma. J Clin Virol. 2016;78:47-52.
- Pereira PF, Cuzzi T, Galhardo MC. Immunohistochemical detection of the latent nuclear antigen-1 of the human herpesvirus type 8 to differentiate cutaneous epidemic Kaposi sarcoma and its histological simulators. An Bras Dermatol. 2013;88:243-246.
- Gevorgyan O, Owen BD, Balavenkataraman A, et al. A nodular-ulcerative form of secondary syphilis in AIDS. Proc (Bayl Univ Med Cent). 2017;30:80-82.
- Balagula Y, Mattei PL, Wisco OJ, et al. The great imitator revisited: the spectrum of atypical cutaneous manifestations of secondary syphilis. Int J Dermatol. 2014;53:1434-1441.
- Hoang MP, High WA, Molberg KH. Secondary syphilis: a histologic and immunohistochemical evaluation. J Cutan Pathol. 2004;31:595-599.
- Yayli S, della Torre R, Hegyi I, et al. Late secondary syphilis with nodular lesions mimicking Kaposi sarcoma in a patient with human immunodeficiency virus. Int J Dermatol. 2014;53:E71-E73.
- Jeerapaet P, Ackerman AB. Histologic patterns of secondary syphilis. Arch Dermatol. 1973;107:373-377.
- Cockerell CJ, LeBoit PE. Bacillary angiomatosis: a newly characterized, pseudoneoplastic, infectious, cutaneous vascular disorder. J Am Acad Dermatol. 1990;22:501-512.
- Forrestel AK, Naujokas A, Martin JN, et al. Bacillary angiomatosis masquerading as Kaposi's sarcoma in East Africa. J Int Assoc Provid AIDS Care. 2015;14:21-25.
- Fortna RR, Junkins-Hopkins JM. A case of lobular capillary hemangioma (pyogenic granuloma), localized to the subcutaneous tissue, and a review of the literature. Am J Dermatopathol. 2007;29:408-411.
- Marks R. Squamous cell carcinoma. Lancet. 1996;347:735-738.
- Chakrabarti I, Watson JD, Dorrance H. Skin tumours of the hand. a 10-year review. J Hand Surg Br. 1993;18:484-486.
- Sobanko JF, Dagum AB, Davis IC, et al. Soft tissue tumors of the hand. 2. malignant. Dermatol Surg. 2007;33:771-785.
- Antman K, Chang Y. Kaposi's sarcoma. N Engl J Med. 2000;342:1027-1038.
- Pipette WW. The incidence of second malignancies in subsets of Kaposi's sarcoma. J Am Acad Dermatol. 1987;16:855-861.
- Shiels MS, Engels EA. Evolving epidemiology of HIV-associated malignancies. Curr Opin HIV AIDS. 2017;12:6-11.
- Daly ML, Fogo A, McDonald C, et al. Kaposi sarcoma: no longer an AIDS-defining illness? a retrospective study of Kaposi sarcoma cases with CD4 counts above 300/mm³ at presentation. Clin Exp Dermatol. 2014;39:7-12.
- Broccolo F, Tassan Din C, Viganò MG, et al. HHV-8 DNA replication correlates with the clinical status in AIDS-related Kaposi's sarcoma. J Clin Virol. 2016;78:47-52.
- Pereira PF, Cuzzi T, Galhardo MC. Immunohistochemical detection of the latent nuclear antigen-1 of the human herpesvirus type 8 to differentiate cutaneous epidemic Kaposi sarcoma and its histological simulators. An Bras Dermatol. 2013;88:243-246.
- Gevorgyan O, Owen BD, Balavenkataraman A, et al. A nodular-ulcerative form of secondary syphilis in AIDS. Proc (Bayl Univ Med Cent). 2017;30:80-82.
- Balagula Y, Mattei PL, Wisco OJ, et al. The great imitator revisited: the spectrum of atypical cutaneous manifestations of secondary syphilis. Int J Dermatol. 2014;53:1434-1441.
- Hoang MP, High WA, Molberg KH. Secondary syphilis: a histologic and immunohistochemical evaluation. J Cutan Pathol. 2004;31:595-599.
- Yayli S, della Torre R, Hegyi I, et al. Late secondary syphilis with nodular lesions mimicking Kaposi sarcoma in a patient with human immunodeficiency virus. Int J Dermatol. 2014;53:E71-E73.
- Jeerapaet P, Ackerman AB. Histologic patterns of secondary syphilis. Arch Dermatol. 1973;107:373-377.
- Cockerell CJ, LeBoit PE. Bacillary angiomatosis: a newly characterized, pseudoneoplastic, infectious, cutaneous vascular disorder. J Am Acad Dermatol. 1990;22:501-512.
- Forrestel AK, Naujokas A, Martin JN, et al. Bacillary angiomatosis masquerading as Kaposi's sarcoma in East Africa. J Int Assoc Provid AIDS Care. 2015;14:21-25.
- Fortna RR, Junkins-Hopkins JM. A case of lobular capillary hemangioma (pyogenic granuloma), localized to the subcutaneous tissue, and a review of the literature. Am J Dermatopathol. 2007;29:408-411.
- Marks R. Squamous cell carcinoma. Lancet. 1996;347:735-738.
- Chakrabarti I, Watson JD, Dorrance H. Skin tumours of the hand. a 10-year review. J Hand Surg Br. 1993;18:484-486.
- Sobanko JF, Dagum AB, Davis IC, et al. Soft tissue tumors of the hand. 2. malignant. Dermatol Surg. 2007;33:771-785.

A 52-year-old man presented to the dermatology clinic with a 2×3-cm, fungating, dome-shaped, ulcerative, moist, well-circumscribed tumor with peripheral maceration on the volar aspect of the right hand of 3 months’ duration. The tumor was malodorous, painful, and hemorrhaged easily with minimal trauma. The patient’s medical history was notable for human immunodeficiency virus and latent syphilis, with elevated rapid plasma reagin titers and a positive Treponema palladium antibody on chemiluminescent immunoassay, that was refractory to 3 treatments with penicillin. The patient was not on antiretroviral therapy. He had a CD4+ lymphocyte count of 980 cells/µL (reference range, 359–1519 cells/µL) and a viral load of 8560 copies/mL (reference range, <200 copies/mL). No other skin or systemic concerns were noted, and the patient denied any recent travel, exposure to animals, or constitutional symptoms. A deep shave biopsy of the lesion was performed.
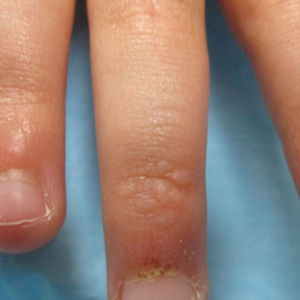
Papules and Telangiectases on the Distal Fingers of a Child
The Diagnosis: Juvenile Dermatomyositis
Juvenile dermatomyositis (JDM) is a rare idiopathic inflammatory myopathy of childhood that is autoimmune in nature with an annual incidence ranging from 2.5 to 4.1 cases per million children. Its peak incidence is between 5 and 10 years of age, and it affects girls more than boys at a 2-fold to 5-fold greater rate.1 Juvenile dermatomyositis is characterized by skeletal muscle weakness in the presence of distinctive rashes, including Gottron papules and heliotrope erythema. Muscle weakness typically is proximal and symmetrical, and eventually patients may have trouble rising from a seated position or lifting objects overhead. Other skin manifestations include nail fold capillary changes, calcinosis cutis, and less commonly ulcerations signifying vasculopathy of the skin.2 A subset of patients will present with juvenile amyopathic dermatomyositis. These children have the characteristic skin changes without the muscle weakness or elevated muscle enzymes for more than 6 months; however, one-quarter may go on to develop mysositis.3
Diagnosis of JDM traditionally was based on the following 5 diagnostic criteria: characteristic skin rash, proximal muscle weakness, elevated muscle enzymes, myopathic changes on electromyogram, and typical muscle biopsy.1 Current practice shows a broadening of diagnostic criteria using new techniques in the diagnosis of JDM. To make the diagnosis, the patient must have the characteristic skin manifestations with a minimum of 3 other criteria.4 A 2006 international consensus survey expanded the list of criteria to include typical findings on magnetic resonance imaging (MRI), nail fold capillaroscopy abnormalities, calcinosis, and
dysphonia.5
To assess muscle disease, MRI is utilized because it is a reliable noninvasive tool to assess muscle inflammation. Muscle biopsy is only recommended if the diagnosis is unclear.5 The results of the MRI in our patient displayed symmetric mild fatty atrophy of the gluteus maximus muscle, as well as edema in the right rectus femoris and left vastus lateralis muscles, suggesting early findings of myositis. Muscle enzymes may not be diagnostic because they are not always elevated at diagnosis. Our patient had a normal creatinine kinase level (92 U/L [reference range, <190 U/L]), and both aldolase and lactate dehydrogenase also were within reference range. Conversely, antinuclear antibodies frequently are positive in patients with JDM, such as in our patient at a 1:320 dilution, but are nonspecific and nondiagnostic. It is recommended to include nail fold capillaroscopy to evaluate periungual capillary changes because nailfold capillary density is a sensitive measure of both skin and muscle disease.5 Using dermoscopy, nail fold capillary dilation was observed in our patient.
Other differential diagnoses can have somewhat similar clinical features to JDM. Infantile papular acrodermatitis, commonly referred to as Gianotti-Crosti syndrome, is a viral exanthem that affects children (median age, 2 years).6 The rash appears as monomorphous, flat-topped, pink to brown papules affecting the face, buttocks, and arms; it typically spontaneously resolves in 10 days.6
Juvenile-onset lupus is a chronic autoimmune disorder that can involve any organ system and typically affects children aged 11 to 12 years with a female preponderance. Skin manifestations are similar to adult-onset lupus and include malar rash, discoid rash, oral ulcerations, petechiae, palpable purpura, and digital telangiectasia and ulcers. 7
Juvenile scleroderma is rare connective-tissue disorder that also has multiple organ involvement. Cutaneous involvement can range from isolated morphealike plaques to diffuse sclerotic lesions with growth disturbances, contractures, and facial atrophy.8
Verrucae planae, commonly referred to as flat warts, are papules caused primarily by human papillomavirus types 3, 10, 28, and 41. Children and young adults commonly are affected, and warts can appear on the hands, as in our patient.6
Treatment of JDM depends on disease severity at initial presentation and requires a multidisciplinary approach. The mainstay of treatment is high-dose oral prednisone in combination with disease-modifying drugs such as methotrexate and cyclosporin A. Patients with more severe presentations (eg, ulcerative skin disease) or life-threatening organ involvement are treated with cyclophosphamide, usually in combination with high-dose glucocorticoids.9
Early detection with aggressive treatment is vital to reduce morbidity and mortality from organ damage and disease complications. Mortality rates have dropped to 3%10 in recent decades with the use of systemic glucocorticoids. Delayed treatment is associated with a prolonged disease course and poorer outcomes. Disease complications in children with JDM include osteoporosis, calcinosis, and intestinal perforation; however, with early treatment, children with JDM can expect full recovery and to live a normal life as compared to adults with dermatomyositis.10
Prior to our patient's diagnosis, the family was assigned to move to an overseas location through the US Military with no direct access to advanced medical care. Early detection and diagnosis of JDM through an astute clinical examination allowed the patient and her family to remain in the continental United States to continue receiving specialty care.
- Mendez EP, Lipton R, Ramsey-Goldman R, et al. US incidence of juvenile dermatomyositis,1995-1998: results from the National Institute of Arthritis and Musculoskeletal and Skin Diseases Registry. Arthritis Rheum. 2003;49:300-305.
- Shah M, Mamyrova G, Targoff IN, et al. The clinical phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine. 2013;92:25-41.
- Gerami P, Walling HW, Lewis J, et al. A systematic review of juvenile-onset clinically amyopathic dermatomyositis. Br J Dermatol. 2007;57:637-644.
- Enders FB, Bader-Meunier B, Baildam E, et al. Consensus-based recommendations for the management of juvenile dermatomyositis. Ann Rheum Dis. 2017;76:329-340.
- Brown VE, Pilkington CA, Feldman BM, et al. An international consensus survey of the diagnostic criteria for juvenile dermatomyositis (JDM). Rheumatology (Oxford). 2006;45:990-993.
- William JD, Berger TG, Elston DM. Viral diseases. In: William JD, Berger TG, Elston DM. Andrews' Diseases of the Skin: Clinical Dermatology. 11th ed. China: Saunders Elsevier; 2011:360-413.
- Levy DM, Kamphuis S. Systemic lupus erythematosus in children and adolescents. Pediatr Clin North Am. 2012;59:345-364.
- Li SC, Torok KS, Pope E, et al; Childhood Arthritis and Rheumatology Research Alliance (CARRA) Localized Scleroderma Workgroup. Development of consensus treatment plans for juvenile localized scleroderma: a roadmap toward comparative effectiveness studies in juvenile localized scleroderma. Arthritis Care Res (Hoboken). 2012;64:1175-1185.
- Stringer E, Ota S, Bohnsack J, et al. Treatment approaches to juvenile dermatomyositis (JDM) across North America: the Childhood Arthritis and Rheumatology Research Alliance (CARRA) JDM treatment study. J Rhematol. 2010;37:S1953-S1961.
- Huber AM, Feldman BM. Long-term outcomes in juvenile dermatomyositis: how did we get here and where are we going? Curr Rheumatol Rep. 2005;7:441-446.
The Diagnosis: Juvenile Dermatomyositis
Juvenile dermatomyositis (JDM) is a rare idiopathic inflammatory myopathy of childhood that is autoimmune in nature with an annual incidence ranging from 2.5 to 4.1 cases per million children. Its peak incidence is between 5 and 10 years of age, and it affects girls more than boys at a 2-fold to 5-fold greater rate.1 Juvenile dermatomyositis is characterized by skeletal muscle weakness in the presence of distinctive rashes, including Gottron papules and heliotrope erythema. Muscle weakness typically is proximal and symmetrical, and eventually patients may have trouble rising from a seated position or lifting objects overhead. Other skin manifestations include nail fold capillary changes, calcinosis cutis, and less commonly ulcerations signifying vasculopathy of the skin.2 A subset of patients will present with juvenile amyopathic dermatomyositis. These children have the characteristic skin changes without the muscle weakness or elevated muscle enzymes for more than 6 months; however, one-quarter may go on to develop mysositis.3
Diagnosis of JDM traditionally was based on the following 5 diagnostic criteria: characteristic skin rash, proximal muscle weakness, elevated muscle enzymes, myopathic changes on electromyogram, and typical muscle biopsy.1 Current practice shows a broadening of diagnostic criteria using new techniques in the diagnosis of JDM. To make the diagnosis, the patient must have the characteristic skin manifestations with a minimum of 3 other criteria.4 A 2006 international consensus survey expanded the list of criteria to include typical findings on magnetic resonance imaging (MRI), nail fold capillaroscopy abnormalities, calcinosis, and
dysphonia.5
To assess muscle disease, MRI is utilized because it is a reliable noninvasive tool to assess muscle inflammation. Muscle biopsy is only recommended if the diagnosis is unclear.5 The results of the MRI in our patient displayed symmetric mild fatty atrophy of the gluteus maximus muscle, as well as edema in the right rectus femoris and left vastus lateralis muscles, suggesting early findings of myositis. Muscle enzymes may not be diagnostic because they are not always elevated at diagnosis. Our patient had a normal creatinine kinase level (92 U/L [reference range, <190 U/L]), and both aldolase and lactate dehydrogenase also were within reference range. Conversely, antinuclear antibodies frequently are positive in patients with JDM, such as in our patient at a 1:320 dilution, but are nonspecific and nondiagnostic. It is recommended to include nail fold capillaroscopy to evaluate periungual capillary changes because nailfold capillary density is a sensitive measure of both skin and muscle disease.5 Using dermoscopy, nail fold capillary dilation was observed in our patient.
Other differential diagnoses can have somewhat similar clinical features to JDM. Infantile papular acrodermatitis, commonly referred to as Gianotti-Crosti syndrome, is a viral exanthem that affects children (median age, 2 years).6 The rash appears as monomorphous, flat-topped, pink to brown papules affecting the face, buttocks, and arms; it typically spontaneously resolves in 10 days.6
Juvenile-onset lupus is a chronic autoimmune disorder that can involve any organ system and typically affects children aged 11 to 12 years with a female preponderance. Skin manifestations are similar to adult-onset lupus and include malar rash, discoid rash, oral ulcerations, petechiae, palpable purpura, and digital telangiectasia and ulcers. 7
Juvenile scleroderma is rare connective-tissue disorder that also has multiple organ involvement. Cutaneous involvement can range from isolated morphealike plaques to diffuse sclerotic lesions with growth disturbances, contractures, and facial atrophy.8
Verrucae planae, commonly referred to as flat warts, are papules caused primarily by human papillomavirus types 3, 10, 28, and 41. Children and young adults commonly are affected, and warts can appear on the hands, as in our patient.6
Treatment of JDM depends on disease severity at initial presentation and requires a multidisciplinary approach. The mainstay of treatment is high-dose oral prednisone in combination with disease-modifying drugs such as methotrexate and cyclosporin A. Patients with more severe presentations (eg, ulcerative skin disease) or life-threatening organ involvement are treated with cyclophosphamide, usually in combination with high-dose glucocorticoids.9
Early detection with aggressive treatment is vital to reduce morbidity and mortality from organ damage and disease complications. Mortality rates have dropped to 3%10 in recent decades with the use of systemic glucocorticoids. Delayed treatment is associated with a prolonged disease course and poorer outcomes. Disease complications in children with JDM include osteoporosis, calcinosis, and intestinal perforation; however, with early treatment, children with JDM can expect full recovery and to live a normal life as compared to adults with dermatomyositis.10
Prior to our patient's diagnosis, the family was assigned to move to an overseas location through the US Military with no direct access to advanced medical care. Early detection and diagnosis of JDM through an astute clinical examination allowed the patient and her family to remain in the continental United States to continue receiving specialty care.
The Diagnosis: Juvenile Dermatomyositis
Juvenile dermatomyositis (JDM) is a rare idiopathic inflammatory myopathy of childhood that is autoimmune in nature with an annual incidence ranging from 2.5 to 4.1 cases per million children. Its peak incidence is between 5 and 10 years of age, and it affects girls more than boys at a 2-fold to 5-fold greater rate.1 Juvenile dermatomyositis is characterized by skeletal muscle weakness in the presence of distinctive rashes, including Gottron papules and heliotrope erythema. Muscle weakness typically is proximal and symmetrical, and eventually patients may have trouble rising from a seated position or lifting objects overhead. Other skin manifestations include nail fold capillary changes, calcinosis cutis, and less commonly ulcerations signifying vasculopathy of the skin.2 A subset of patients will present with juvenile amyopathic dermatomyositis. These children have the characteristic skin changes without the muscle weakness or elevated muscle enzymes for more than 6 months; however, one-quarter may go on to develop mysositis.3
Diagnosis of JDM traditionally was based on the following 5 diagnostic criteria: characteristic skin rash, proximal muscle weakness, elevated muscle enzymes, myopathic changes on electromyogram, and typical muscle biopsy.1 Current practice shows a broadening of diagnostic criteria using new techniques in the diagnosis of JDM. To make the diagnosis, the patient must have the characteristic skin manifestations with a minimum of 3 other criteria.4 A 2006 international consensus survey expanded the list of criteria to include typical findings on magnetic resonance imaging (MRI), nail fold capillaroscopy abnormalities, calcinosis, and
dysphonia.5
To assess muscle disease, MRI is utilized because it is a reliable noninvasive tool to assess muscle inflammation. Muscle biopsy is only recommended if the diagnosis is unclear.5 The results of the MRI in our patient displayed symmetric mild fatty atrophy of the gluteus maximus muscle, as well as edema in the right rectus femoris and left vastus lateralis muscles, suggesting early findings of myositis. Muscle enzymes may not be diagnostic because they are not always elevated at diagnosis. Our patient had a normal creatinine kinase level (92 U/L [reference range, <190 U/L]), and both aldolase and lactate dehydrogenase also were within reference range. Conversely, antinuclear antibodies frequently are positive in patients with JDM, such as in our patient at a 1:320 dilution, but are nonspecific and nondiagnostic. It is recommended to include nail fold capillaroscopy to evaluate periungual capillary changes because nailfold capillary density is a sensitive measure of both skin and muscle disease.5 Using dermoscopy, nail fold capillary dilation was observed in our patient.
Other differential diagnoses can have somewhat similar clinical features to JDM. Infantile papular acrodermatitis, commonly referred to as Gianotti-Crosti syndrome, is a viral exanthem that affects children (median age, 2 years).6 The rash appears as monomorphous, flat-topped, pink to brown papules affecting the face, buttocks, and arms; it typically spontaneously resolves in 10 days.6
Juvenile-onset lupus is a chronic autoimmune disorder that can involve any organ system and typically affects children aged 11 to 12 years with a female preponderance. Skin manifestations are similar to adult-onset lupus and include malar rash, discoid rash, oral ulcerations, petechiae, palpable purpura, and digital telangiectasia and ulcers. 7
Juvenile scleroderma is rare connective-tissue disorder that also has multiple organ involvement. Cutaneous involvement can range from isolated morphealike plaques to diffuse sclerotic lesions with growth disturbances, contractures, and facial atrophy.8
Verrucae planae, commonly referred to as flat warts, are papules caused primarily by human papillomavirus types 3, 10, 28, and 41. Children and young adults commonly are affected, and warts can appear on the hands, as in our patient.6
Treatment of JDM depends on disease severity at initial presentation and requires a multidisciplinary approach. The mainstay of treatment is high-dose oral prednisone in combination with disease-modifying drugs such as methotrexate and cyclosporin A. Patients with more severe presentations (eg, ulcerative skin disease) or life-threatening organ involvement are treated with cyclophosphamide, usually in combination with high-dose glucocorticoids.9
Early detection with aggressive treatment is vital to reduce morbidity and mortality from organ damage and disease complications. Mortality rates have dropped to 3%10 in recent decades with the use of systemic glucocorticoids. Delayed treatment is associated with a prolonged disease course and poorer outcomes. Disease complications in children with JDM include osteoporosis, calcinosis, and intestinal perforation; however, with early treatment, children with JDM can expect full recovery and to live a normal life as compared to adults with dermatomyositis.10
Prior to our patient's diagnosis, the family was assigned to move to an overseas location through the US Military with no direct access to advanced medical care. Early detection and diagnosis of JDM through an astute clinical examination allowed the patient and her family to remain in the continental United States to continue receiving specialty care.
- Mendez EP, Lipton R, Ramsey-Goldman R, et al. US incidence of juvenile dermatomyositis,1995-1998: results from the National Institute of Arthritis and Musculoskeletal and Skin Diseases Registry. Arthritis Rheum. 2003;49:300-305.
- Shah M, Mamyrova G, Targoff IN, et al. The clinical phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine. 2013;92:25-41.
- Gerami P, Walling HW, Lewis J, et al. A systematic review of juvenile-onset clinically amyopathic dermatomyositis. Br J Dermatol. 2007;57:637-644.
- Enders FB, Bader-Meunier B, Baildam E, et al. Consensus-based recommendations for the management of juvenile dermatomyositis. Ann Rheum Dis. 2017;76:329-340.
- Brown VE, Pilkington CA, Feldman BM, et al. An international consensus survey of the diagnostic criteria for juvenile dermatomyositis (JDM). Rheumatology (Oxford). 2006;45:990-993.
- William JD, Berger TG, Elston DM. Viral diseases. In: William JD, Berger TG, Elston DM. Andrews' Diseases of the Skin: Clinical Dermatology. 11th ed. China: Saunders Elsevier; 2011:360-413.
- Levy DM, Kamphuis S. Systemic lupus erythematosus in children and adolescents. Pediatr Clin North Am. 2012;59:345-364.
- Li SC, Torok KS, Pope E, et al; Childhood Arthritis and Rheumatology Research Alliance (CARRA) Localized Scleroderma Workgroup. Development of consensus treatment plans for juvenile localized scleroderma: a roadmap toward comparative effectiveness studies in juvenile localized scleroderma. Arthritis Care Res (Hoboken). 2012;64:1175-1185.
- Stringer E, Ota S, Bohnsack J, et al. Treatment approaches to juvenile dermatomyositis (JDM) across North America: the Childhood Arthritis and Rheumatology Research Alliance (CARRA) JDM treatment study. J Rhematol. 2010;37:S1953-S1961.
- Huber AM, Feldman BM. Long-term outcomes in juvenile dermatomyositis: how did we get here and where are we going? Curr Rheumatol Rep. 2005;7:441-446.
- Mendez EP, Lipton R, Ramsey-Goldman R, et al. US incidence of juvenile dermatomyositis,1995-1998: results from the National Institute of Arthritis and Musculoskeletal and Skin Diseases Registry. Arthritis Rheum. 2003;49:300-305.
- Shah M, Mamyrova G, Targoff IN, et al. The clinical phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine. 2013;92:25-41.
- Gerami P, Walling HW, Lewis J, et al. A systematic review of juvenile-onset clinically amyopathic dermatomyositis. Br J Dermatol. 2007;57:637-644.
- Enders FB, Bader-Meunier B, Baildam E, et al. Consensus-based recommendations for the management of juvenile dermatomyositis. Ann Rheum Dis. 2017;76:329-340.
- Brown VE, Pilkington CA, Feldman BM, et al. An international consensus survey of the diagnostic criteria for juvenile dermatomyositis (JDM). Rheumatology (Oxford). 2006;45:990-993.
- William JD, Berger TG, Elston DM. Viral diseases. In: William JD, Berger TG, Elston DM. Andrews' Diseases of the Skin: Clinical Dermatology. 11th ed. China: Saunders Elsevier; 2011:360-413.
- Levy DM, Kamphuis S. Systemic lupus erythematosus in children and adolescents. Pediatr Clin North Am. 2012;59:345-364.
- Li SC, Torok KS, Pope E, et al; Childhood Arthritis and Rheumatology Research Alliance (CARRA) Localized Scleroderma Workgroup. Development of consensus treatment plans for juvenile localized scleroderma: a roadmap toward comparative effectiveness studies in juvenile localized scleroderma. Arthritis Care Res (Hoboken). 2012;64:1175-1185.
- Stringer E, Ota S, Bohnsack J, et al. Treatment approaches to juvenile dermatomyositis (JDM) across North America: the Childhood Arthritis and Rheumatology Research Alliance (CARRA) JDM treatment study. J Rhematol. 2010;37:S1953-S1961.
- Huber AM, Feldman BM. Long-term outcomes in juvenile dermatomyositis: how did we get here and where are we going? Curr Rheumatol Rep. 2005;7:441-446.
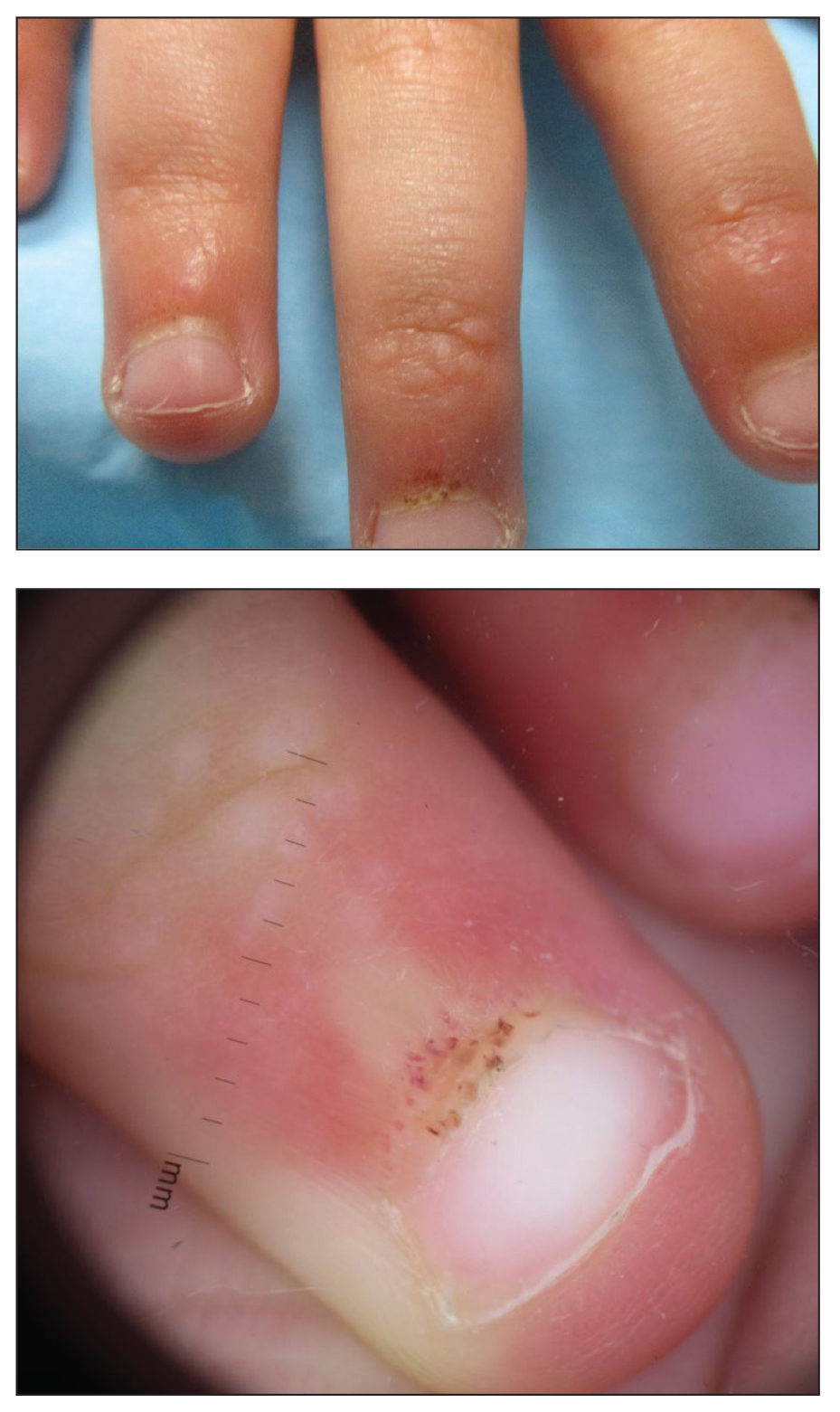
A 4-year-old girl presented to our dermatology clinic with asymptomatic flesh-colored bumps on the fingers of 2 to 3 months’ duration. Prior to presentation the patient was otherwise healthy with normal growth and development. She was referred to dermatology for recommended treatment options for suspected flat warts. On physical examination, grouped 1- to 3-mm, smooth, flat-topped papules were found on the dorsal aspects of the distal interphalangeal joints of all fingers (top). The papules were nonpruritic. Additionally, there were nail findings of ragged cuticles and dilated capillary loops in the proximal nail folds (bottom). The patient did not bite her nails, per the mother’s report, and no other rashes were noted. There were no systemic symptoms or reports of muscle fatigue. She was positive for antinuclear antibodies at 1:320 dilution. Magnetic resonance imaging of the thighs and pelvis was ordered.
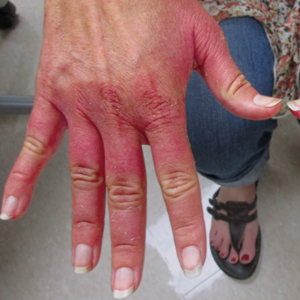
Erythematous Edematous Plaques on the Dorsal Aspects of the Hands
The Diagnosis: Phytophotodermatitis
Initially, there was concern for autoimmune or connective tissue disease because of the edematous plaques localized over sun-exposed regions of the hands with marked sparing of the knuckles. Lupus erythematosus (LE), mixed connective tissue disease, CREST (calcinosis, Raynaud phenomenon, esophageal motility disorders, sclerodactyly, telangiectasia) syndrome, dermatomyositis (DM), and erythromelalgia all were considered. Common disorders such as contact dermatitis and phytophotodermatitis remained in the differential diagnosis, though the patient adamantly denied any recent exposures. As part of the initial workup, laboratory studies including a complete blood cell count, comprehensive metabolic panel, serum lactate dehydrogenase, serum creatinine kinase, erythrocyte sedimentation rate, and an antinuclear antibody panel were performed. Additionally, a punch biopsy at the border of the lesion was performed.
Lupus erythematosus was considered given the patient’s age and sex and the photoexposed location of the plaques. The photosensitive rash of LE classically affects the dorsal aspects of the hands while sparing the interphalangeal joints.1,2 However, the patient had no nail fold findings consistent with systemic LE with no evidence of erythema or dilated tortuous vessels.3 Furthermore, there were no other cutaneous symptoms, and there was a negative review of systems, including malar/discoid rash, oral ulcers, photosensitivity, history of hematologic abnormalities, and end organ damage.4,5 A negative antinuclear antibody serologic panel combined with a negative review of systems made the diagnosis of LE less likely.
Given the presenting clinical appearance, DM also was considered. Dermatomyositis traditionally displays ragged cuticular dystrophy with nail fold telangiectasia, mechanic hands, and involvement of the dorsal aspects of the hands with violaceous accentuation of the knuckles.6 The patient reported pruritus, which is common among DM patients; however, the nail folds were unaffected.7 Finally, she demonstrated sparing rather than involvement of the knuckles, which would be an unlikely presentation for DM.6
CREST syndrome, systemic sclerosis, and syndromes with overlapping features such as mixed connective tissue disease also were considered. The cutaneous features of CREST syndrome are characterized by initial edema of the digits with a subsequent taut and shiny indurated phase. Flexion contractures, ulceration, tapering of the digits, and loss of cutaneous fat pads can progressively occur.8,9 Raynaud phenomenon is a common early finding in CREST syndrome or systemic sclerosis, and patients may develop ice pick digital infarcts and calcinosis in progressed disease.8 Common nail fold findings include periungual telangiectasia with dropout areas.10,11 The marked edema and white discoloration of the knuckles in this patient could be mistaken for Raynaud phenomenon; however, she lacked pain or cold sensitivity and her discoloration was static.12 Without sclerodermoid changes, nail fold findings, matted telangiectasia, taut skin, or systemic findings, a diagnosis of CREST syndrome, scleroderma, or other mixed connective tissue disease would be unlikely.8
Erythromelalgia is a clinical syndrome characterized by burning pain, erythema, and increased skin temperature that intermittently affects both the arms and legs. This rare disorder can be further classified into type 1 (associated with thrombocytopenia), type 2 (primary or idiopathic), and type 3 (associated with other medical cause excluding thrombocytopenia).1,13 The patient endorsed some discomfort from the lesions but denied any subjective feeling of burning pain or increased skin temperature. Additionally, she had no family history of inheritable skin disorders and no personal history of polycythemia. Consequently, erythromelalgia remained less likely on the differential diagnosis.
The histology of the acral skin revealed mild focal spongiosis with no increase in dermal mucin on colloidal iron or mucopolysaccharide stains (Figure). After receiving the biopsy results and additional questioning of the patient, it was discovered that 2 days prior to her initial presentation she had juiced numerous limes by hand and subsequently spent a long period of time outside with sunlight exposure. Upon discovery of this additional historical information, the diagnosis of phytophotodermatitis was made.
Phytophotodermatitis is an erythematous inflammatory reaction that occurs on the skin after exposure to a plant-derived photosensitizer followed by UVA light radiation.14 This phenomenon was first described by the ancient Egyptians as a treatment for vitiligo.1 The most common plant families that can cause this nonimmune cutaneous reaction include Apiaceae eg, hogweed, celery, dill, fennel) and Rutaceae (eg, citrus plants, rue).14 The psoralens or furocoumarins found in these plants bind loosely to DNA at their ground state but covalently bond to pyrimidine bases during photoexcitation with UVA, resulting in DNA damage and subsequent local inflammation.14 Given the patient’s clinical examination, pathology findings, and history, phytophotodermatitis secondary to lime juice exposure was confirmed. Two weeks after applying clobetasol ointment twice daily, the patient’s hands had returned to baseline with complete resolution of the erythematous lesions.
Although lime phytophotodermatitis is a routine diagnosis, this clinical case stands as an important reminder to demonstrate how common diseases can masquerade as more exotic cutaneous disorders. There often is a clinical desire to seek out more complicated diagnoses, particularly during residency training; however, this case reinforces the invaluable importance of collecting a thorough patient history, as it can ultimately minimize excessive testing and in some cases prevent unnecessary therapy.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China:Elsevier Saunders; 2012.
- Uva L, Miguel D, Pinheiro C, et al. Cutaneous manifestations of systemiclupus erythematosus. Autoimmune Dis. 2012;2012:834291.
- Furtado R, Pucinelli M, Cristo V, et al. Scleroderma-like nailfold capillaroscopicabnormalities are associated with anti-U1-RNP antibodies and Raynaud’s phenomenon in SLE patients. Lupus. 2002;11:35-41.
- Wenzel J, Zahn S, Tuting T. Pathogenesis of cutaneous lupus erythematosus:common and different features in distinct subsets. Lupus. 2010;19:1020-1028.
- Avilés Izquierdo JA, Cano Martínez N, Lázaro Ochaita P. Epidemiologicalcharacteristics of patients with cutaneous lupus erythematosus.Actas Dermosifiliogr. 2014;105:69-73.
- Marvi U, Chung L, Fiorentino DF. Clinical presentation and evaluation of dermatomyositis. Indian J Dermatol. 2012;57:375-381.
- Shirani Z, Kucenic MJ, Carroll CL, et al. Pruritus in adult dermatomyositis. Clin Exp Dermatol. 2004;29:273-276.
- Krieg T, Takehara K. Skin disease: a cardinal feature of systemic sclerosis. Rheumatology (Oxford). 2009;48(suppl 3):14-18.
- Mizutani H, Mizutani T, Okada H, et al. Round fingerpad sign: an early sign of scleroderma. J Am Acad Dermatol. 1991;24:67-69.
- Baran R, Dawber RP, Haneke E, et al, eds. A Text Atlas of Nail Disorders Techniques in Investigation and Diagnosis. 3rd ed. Boca Raton, FL: CRC Press; 2005.
- Ghali FE, Stein LD, Fine J, et al. Gingival telangiectases: an underappreciated physical sign of juvenile dermatomyositis. Arch Dermatol. 1999;135:1370-1374.
- Grader-Beck T, Wigley FM. Raynaud’s phenomenon in mixed connective tissue disease. Rheum Dis Clin North Am. 2005;31:465-481.
- Davis MD, Weenig RH, Genebriera J, et al. Histopathologic findings in primary erythromelalgia are nonspecific: special studies show a decrease in small nerve fiber density. J Am Acad Dermatol. 2006;55:519-522.
- Sasseville D. Clinical patterns of phytophotodermatitis. Dermatol Clin. 2009;27:299-308.
The Diagnosis: Phytophotodermatitis
Initially, there was concern for autoimmune or connective tissue disease because of the edematous plaques localized over sun-exposed regions of the hands with marked sparing of the knuckles. Lupus erythematosus (LE), mixed connective tissue disease, CREST (calcinosis, Raynaud phenomenon, esophageal motility disorders, sclerodactyly, telangiectasia) syndrome, dermatomyositis (DM), and erythromelalgia all were considered. Common disorders such as contact dermatitis and phytophotodermatitis remained in the differential diagnosis, though the patient adamantly denied any recent exposures. As part of the initial workup, laboratory studies including a complete blood cell count, comprehensive metabolic panel, serum lactate dehydrogenase, serum creatinine kinase, erythrocyte sedimentation rate, and an antinuclear antibody panel were performed. Additionally, a punch biopsy at the border of the lesion was performed.
Lupus erythematosus was considered given the patient’s age and sex and the photoexposed location of the plaques. The photosensitive rash of LE classically affects the dorsal aspects of the hands while sparing the interphalangeal joints.1,2 However, the patient had no nail fold findings consistent with systemic LE with no evidence of erythema or dilated tortuous vessels.3 Furthermore, there were no other cutaneous symptoms, and there was a negative review of systems, including malar/discoid rash, oral ulcers, photosensitivity, history of hematologic abnormalities, and end organ damage.4,5 A negative antinuclear antibody serologic panel combined with a negative review of systems made the diagnosis of LE less likely.
Given the presenting clinical appearance, DM also was considered. Dermatomyositis traditionally displays ragged cuticular dystrophy with nail fold telangiectasia, mechanic hands, and involvement of the dorsal aspects of the hands with violaceous accentuation of the knuckles.6 The patient reported pruritus, which is common among DM patients; however, the nail folds were unaffected.7 Finally, she demonstrated sparing rather than involvement of the knuckles, which would be an unlikely presentation for DM.6
CREST syndrome, systemic sclerosis, and syndromes with overlapping features such as mixed connective tissue disease also were considered. The cutaneous features of CREST syndrome are characterized by initial edema of the digits with a subsequent taut and shiny indurated phase. Flexion contractures, ulceration, tapering of the digits, and loss of cutaneous fat pads can progressively occur.8,9 Raynaud phenomenon is a common early finding in CREST syndrome or systemic sclerosis, and patients may develop ice pick digital infarcts and calcinosis in progressed disease.8 Common nail fold findings include periungual telangiectasia with dropout areas.10,11 The marked edema and white discoloration of the knuckles in this patient could be mistaken for Raynaud phenomenon; however, she lacked pain or cold sensitivity and her discoloration was static.12 Without sclerodermoid changes, nail fold findings, matted telangiectasia, taut skin, or systemic findings, a diagnosis of CREST syndrome, scleroderma, or other mixed connective tissue disease would be unlikely.8
Erythromelalgia is a clinical syndrome characterized by burning pain, erythema, and increased skin temperature that intermittently affects both the arms and legs. This rare disorder can be further classified into type 1 (associated with thrombocytopenia), type 2 (primary or idiopathic), and type 3 (associated with other medical cause excluding thrombocytopenia).1,13 The patient endorsed some discomfort from the lesions but denied any subjective feeling of burning pain or increased skin temperature. Additionally, she had no family history of inheritable skin disorders and no personal history of polycythemia. Consequently, erythromelalgia remained less likely on the differential diagnosis.
The histology of the acral skin revealed mild focal spongiosis with no increase in dermal mucin on colloidal iron or mucopolysaccharide stains (Figure). After receiving the biopsy results and additional questioning of the patient, it was discovered that 2 days prior to her initial presentation she had juiced numerous limes by hand and subsequently spent a long period of time outside with sunlight exposure. Upon discovery of this additional historical information, the diagnosis of phytophotodermatitis was made.
Phytophotodermatitis is an erythematous inflammatory reaction that occurs on the skin after exposure to a plant-derived photosensitizer followed by UVA light radiation.14 This phenomenon was first described by the ancient Egyptians as a treatment for vitiligo.1 The most common plant families that can cause this nonimmune cutaneous reaction include Apiaceae eg, hogweed, celery, dill, fennel) and Rutaceae (eg, citrus plants, rue).14 The psoralens or furocoumarins found in these plants bind loosely to DNA at their ground state but covalently bond to pyrimidine bases during photoexcitation with UVA, resulting in DNA damage and subsequent local inflammation.14 Given the patient’s clinical examination, pathology findings, and history, phytophotodermatitis secondary to lime juice exposure was confirmed. Two weeks after applying clobetasol ointment twice daily, the patient’s hands had returned to baseline with complete resolution of the erythematous lesions.
Although lime phytophotodermatitis is a routine diagnosis, this clinical case stands as an important reminder to demonstrate how common diseases can masquerade as more exotic cutaneous disorders. There often is a clinical desire to seek out more complicated diagnoses, particularly during residency training; however, this case reinforces the invaluable importance of collecting a thorough patient history, as it can ultimately minimize excessive testing and in some cases prevent unnecessary therapy.
The Diagnosis: Phytophotodermatitis
Initially, there was concern for autoimmune or connective tissue disease because of the edematous plaques localized over sun-exposed regions of the hands with marked sparing of the knuckles. Lupus erythematosus (LE), mixed connective tissue disease, CREST (calcinosis, Raynaud phenomenon, esophageal motility disorders, sclerodactyly, telangiectasia) syndrome, dermatomyositis (DM), and erythromelalgia all were considered. Common disorders such as contact dermatitis and phytophotodermatitis remained in the differential diagnosis, though the patient adamantly denied any recent exposures. As part of the initial workup, laboratory studies including a complete blood cell count, comprehensive metabolic panel, serum lactate dehydrogenase, serum creatinine kinase, erythrocyte sedimentation rate, and an antinuclear antibody panel were performed. Additionally, a punch biopsy at the border of the lesion was performed.
Lupus erythematosus was considered given the patient’s age and sex and the photoexposed location of the plaques. The photosensitive rash of LE classically affects the dorsal aspects of the hands while sparing the interphalangeal joints.1,2 However, the patient had no nail fold findings consistent with systemic LE with no evidence of erythema or dilated tortuous vessels.3 Furthermore, there were no other cutaneous symptoms, and there was a negative review of systems, including malar/discoid rash, oral ulcers, photosensitivity, history of hematologic abnormalities, and end organ damage.4,5 A negative antinuclear antibody serologic panel combined with a negative review of systems made the diagnosis of LE less likely.
Given the presenting clinical appearance, DM also was considered. Dermatomyositis traditionally displays ragged cuticular dystrophy with nail fold telangiectasia, mechanic hands, and involvement of the dorsal aspects of the hands with violaceous accentuation of the knuckles.6 The patient reported pruritus, which is common among DM patients; however, the nail folds were unaffected.7 Finally, she demonstrated sparing rather than involvement of the knuckles, which would be an unlikely presentation for DM.6
CREST syndrome, systemic sclerosis, and syndromes with overlapping features such as mixed connective tissue disease also were considered. The cutaneous features of CREST syndrome are characterized by initial edema of the digits with a subsequent taut and shiny indurated phase. Flexion contractures, ulceration, tapering of the digits, and loss of cutaneous fat pads can progressively occur.8,9 Raynaud phenomenon is a common early finding in CREST syndrome or systemic sclerosis, and patients may develop ice pick digital infarcts and calcinosis in progressed disease.8 Common nail fold findings include periungual telangiectasia with dropout areas.10,11 The marked edema and white discoloration of the knuckles in this patient could be mistaken for Raynaud phenomenon; however, she lacked pain or cold sensitivity and her discoloration was static.12 Without sclerodermoid changes, nail fold findings, matted telangiectasia, taut skin, or systemic findings, a diagnosis of CREST syndrome, scleroderma, or other mixed connective tissue disease would be unlikely.8
Erythromelalgia is a clinical syndrome characterized by burning pain, erythema, and increased skin temperature that intermittently affects both the arms and legs. This rare disorder can be further classified into type 1 (associated with thrombocytopenia), type 2 (primary or idiopathic), and type 3 (associated with other medical cause excluding thrombocytopenia).1,13 The patient endorsed some discomfort from the lesions but denied any subjective feeling of burning pain or increased skin temperature. Additionally, she had no family history of inheritable skin disorders and no personal history of polycythemia. Consequently, erythromelalgia remained less likely on the differential diagnosis.
The histology of the acral skin revealed mild focal spongiosis with no increase in dermal mucin on colloidal iron or mucopolysaccharide stains (Figure). After receiving the biopsy results and additional questioning of the patient, it was discovered that 2 days prior to her initial presentation she had juiced numerous limes by hand and subsequently spent a long period of time outside with sunlight exposure. Upon discovery of this additional historical information, the diagnosis of phytophotodermatitis was made.
Phytophotodermatitis is an erythematous inflammatory reaction that occurs on the skin after exposure to a plant-derived photosensitizer followed by UVA light radiation.14 This phenomenon was first described by the ancient Egyptians as a treatment for vitiligo.1 The most common plant families that can cause this nonimmune cutaneous reaction include Apiaceae eg, hogweed, celery, dill, fennel) and Rutaceae (eg, citrus plants, rue).14 The psoralens or furocoumarins found in these plants bind loosely to DNA at their ground state but covalently bond to pyrimidine bases during photoexcitation with UVA, resulting in DNA damage and subsequent local inflammation.14 Given the patient’s clinical examination, pathology findings, and history, phytophotodermatitis secondary to lime juice exposure was confirmed. Two weeks after applying clobetasol ointment twice daily, the patient’s hands had returned to baseline with complete resolution of the erythematous lesions.
Although lime phytophotodermatitis is a routine diagnosis, this clinical case stands as an important reminder to demonstrate how common diseases can masquerade as more exotic cutaneous disorders. There often is a clinical desire to seek out more complicated diagnoses, particularly during residency training; however, this case reinforces the invaluable importance of collecting a thorough patient history, as it can ultimately minimize excessive testing and in some cases prevent unnecessary therapy.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China:Elsevier Saunders; 2012.
- Uva L, Miguel D, Pinheiro C, et al. Cutaneous manifestations of systemiclupus erythematosus. Autoimmune Dis. 2012;2012:834291.
- Furtado R, Pucinelli M, Cristo V, et al. Scleroderma-like nailfold capillaroscopicabnormalities are associated with anti-U1-RNP antibodies and Raynaud’s phenomenon in SLE patients. Lupus. 2002;11:35-41.
- Wenzel J, Zahn S, Tuting T. Pathogenesis of cutaneous lupus erythematosus:common and different features in distinct subsets. Lupus. 2010;19:1020-1028.
- Avilés Izquierdo JA, Cano Martínez N, Lázaro Ochaita P. Epidemiologicalcharacteristics of patients with cutaneous lupus erythematosus.Actas Dermosifiliogr. 2014;105:69-73.
- Marvi U, Chung L, Fiorentino DF. Clinical presentation and evaluation of dermatomyositis. Indian J Dermatol. 2012;57:375-381.
- Shirani Z, Kucenic MJ, Carroll CL, et al. Pruritus in adult dermatomyositis. Clin Exp Dermatol. 2004;29:273-276.
- Krieg T, Takehara K. Skin disease: a cardinal feature of systemic sclerosis. Rheumatology (Oxford). 2009;48(suppl 3):14-18.
- Mizutani H, Mizutani T, Okada H, et al. Round fingerpad sign: an early sign of scleroderma. J Am Acad Dermatol. 1991;24:67-69.
- Baran R, Dawber RP, Haneke E, et al, eds. A Text Atlas of Nail Disorders Techniques in Investigation and Diagnosis. 3rd ed. Boca Raton, FL: CRC Press; 2005.
- Ghali FE, Stein LD, Fine J, et al. Gingival telangiectases: an underappreciated physical sign of juvenile dermatomyositis. Arch Dermatol. 1999;135:1370-1374.
- Grader-Beck T, Wigley FM. Raynaud’s phenomenon in mixed connective tissue disease. Rheum Dis Clin North Am. 2005;31:465-481.
- Davis MD, Weenig RH, Genebriera J, et al. Histopathologic findings in primary erythromelalgia are nonspecific: special studies show a decrease in small nerve fiber density. J Am Acad Dermatol. 2006;55:519-522.
- Sasseville D. Clinical patterns of phytophotodermatitis. Dermatol Clin. 2009;27:299-308.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China:Elsevier Saunders; 2012.
- Uva L, Miguel D, Pinheiro C, et al. Cutaneous manifestations of systemiclupus erythematosus. Autoimmune Dis. 2012;2012:834291.
- Furtado R, Pucinelli M, Cristo V, et al. Scleroderma-like nailfold capillaroscopicabnormalities are associated with anti-U1-RNP antibodies and Raynaud’s phenomenon in SLE patients. Lupus. 2002;11:35-41.
- Wenzel J, Zahn S, Tuting T. Pathogenesis of cutaneous lupus erythematosus:common and different features in distinct subsets. Lupus. 2010;19:1020-1028.
- Avilés Izquierdo JA, Cano Martínez N, Lázaro Ochaita P. Epidemiologicalcharacteristics of patients with cutaneous lupus erythematosus.Actas Dermosifiliogr. 2014;105:69-73.
- Marvi U, Chung L, Fiorentino DF. Clinical presentation and evaluation of dermatomyositis. Indian J Dermatol. 2012;57:375-381.
- Shirani Z, Kucenic MJ, Carroll CL, et al. Pruritus in adult dermatomyositis. Clin Exp Dermatol. 2004;29:273-276.
- Krieg T, Takehara K. Skin disease: a cardinal feature of systemic sclerosis. Rheumatology (Oxford). 2009;48(suppl 3):14-18.
- Mizutani H, Mizutani T, Okada H, et al. Round fingerpad sign: an early sign of scleroderma. J Am Acad Dermatol. 1991;24:67-69.
- Baran R, Dawber RP, Haneke E, et al, eds. A Text Atlas of Nail Disorders Techniques in Investigation and Diagnosis. 3rd ed. Boca Raton, FL: CRC Press; 2005.
- Ghali FE, Stein LD, Fine J, et al. Gingival telangiectases: an underappreciated physical sign of juvenile dermatomyositis. Arch Dermatol. 1999;135:1370-1374.
- Grader-Beck T, Wigley FM. Raynaud’s phenomenon in mixed connective tissue disease. Rheum Dis Clin North Am. 2005;31:465-481.
- Davis MD, Weenig RH, Genebriera J, et al. Histopathologic findings in primary erythromelalgia are nonspecific: special studies show a decrease in small nerve fiber density. J Am Acad Dermatol. 2006;55:519-522.
- Sasseville D. Clinical patterns of phytophotodermatitis. Dermatol Clin. 2009;27:299-308.
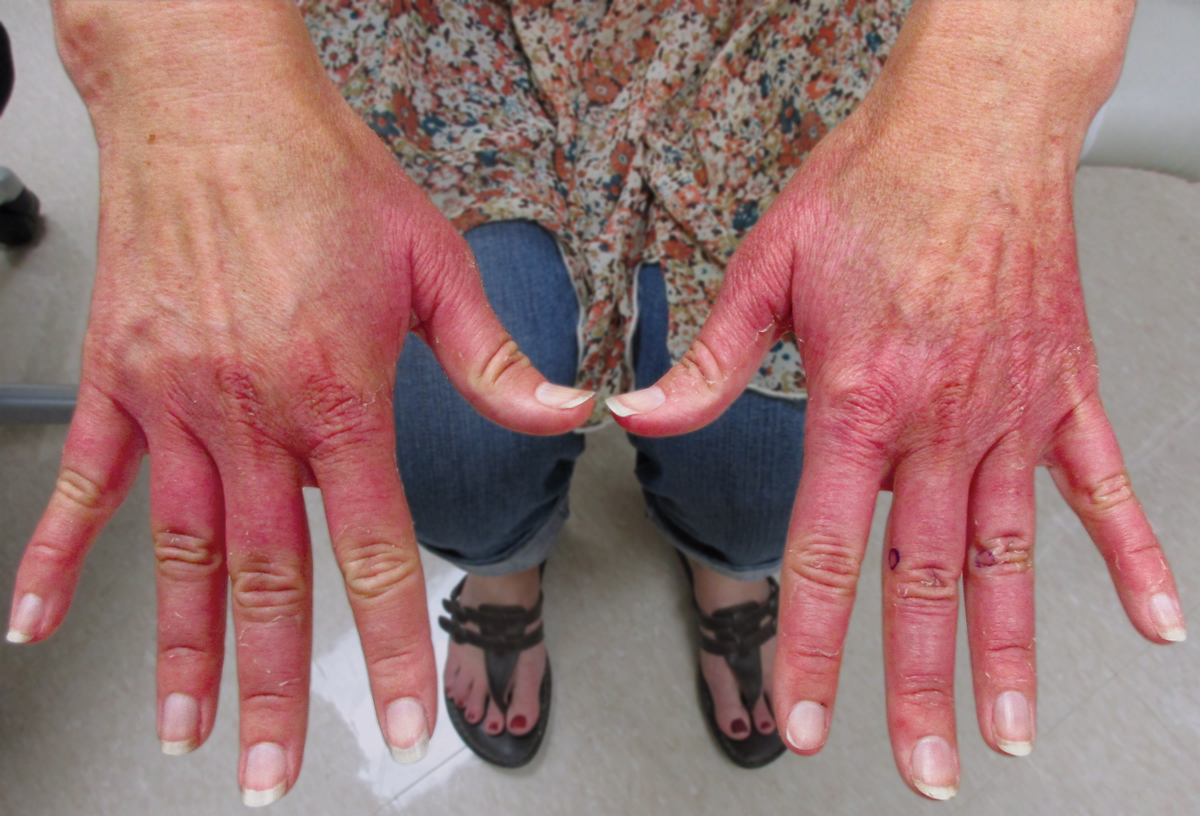
A 48-year-old woman presented with erythematous swelling of the dorsal aspects of the bilateral hands followed by desquamation and pruritus of 2 weeks’ duration. She denied any recent contact with plants, chemicals, or topical products or use of over-the-counter medications. A 6-day course of prednisone provided by her primary care physician relieved the swelling and pruritus; however, the erythema persisted. Physical examination revealed clearly demarcated, erythematous to violaceous, edematous plaques with peripheral scaling that involved all digits. There was notable sparing of the proximal interphalangeal joints and volar aspects of the hands extending proximally to the metacarpophalangeal joints.