User login
Hemorrhagic Lacrimation and Epistaxis: Rare Findings in Acute Hemorrhagic Edema of Infancy
To the Editor:
Hemorrhagic lacrimation and epistaxis are dramatic presentations with a narrow differential diagnosis. It rarely has been reported to present alongside the more typical features of acute hemorrhagic edema of infancy (AHEI), which is a benign self-limited leukocytoclastic vasculitis most often seen in children aged 4 months to 2 years. Extracutaneous involvement rarely is seen in AHEI, though joint, gastrointestinal tract, and renal involvement have been reported.1 Most patients present with edematous, annular, or cockade purpuric vasculitic lesions classically involving the face and distal extremities with relative sparing of the trunk. We present a case of a well-appearing, 10-month-old infant boy with hemorrhagic vasculitic lesions, acral edema, and an associated episode of hemorrhagic lacrimation and epistaxis.

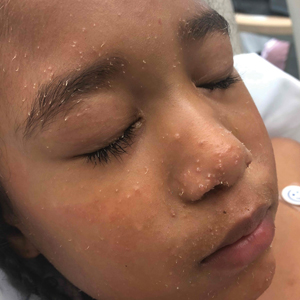
A 10-month-old infant boy who was otherwise healthy presented to the emergency department (ED) with an acute-onset, progressively worsening cutaneous eruption of 2 days’ duration. A thorough history revealed that the eruption initially had presented as several small, bright-red papules on the thighs. The eruption subsequently spread to involve the buttocks, legs, and arms (Figures 1 and 2). The parents also noted that the patient had experienced an episode of bloody tears and epistaxis that lasted a few minutes at the pediatrician’s office earlier that morning, a finding that prompted the urgent referral to the ED.

Dermatology was then consulted. A review of systems was notable for rhinorrhea and diarrhea during the week leading to the eruption. The patient’s parents denied fevers, decreased oral intake, or a recent course of antibiotics. The patient’s medical history was notable only for atopic dermatitis treated with emollients and occasional topical steroids. The parents denied recent travel or vaccinations. Physical examination showed an afebrile, well-appearing infant with multiple nontender, slightly edematous, circular, purpuric papules and plaques scattered on the buttocks and extremities with edema on the dorsal feet. The remainder of the patient’s workup in the ED was notable for mild elevations in C-reactive protein levels (1.4 mg/dL [reference range, 0–1.2 mg/dL]) and an elevated erythrocyte sedimentation rate (22 mm/h [reference range, 2–12 mm/h]). A complete blood cell count; liver function tests; urinalysis; and coagulation studies, including prothrombin, partial thromboplastin time, and international normalized ratio, were unremarkable. Acute hemorrhagic edema of infancy was diagnosed based on the clinical manifestations.
Acute hemorrhagic edema of infancy (also known as Finkelstein disease, medallionlike purpura, Seidemayer syndrome, infantile postinfectious irislike purpura and edema, and purpura en cocarde avec oedeme) is believed to result from an immune complex–related reaction, often in the setting of an upper respiratory tract infection; medications, especially antibiotics; or vaccinations. The condition previously was considered a benign form of Henoch-Schönlein purpura; however, it is now recognized as its own clinical entity. Acute hemorrhagic edema of infancy commonly affects children between the ages of 4 months and 2 years. The incidence peaks in the winter months, and males tend to be more affected than females.1
Acute hemorrhagic edema of infancy is clinically characterized by a triad of large purpuric lesions, low-grade fever, and peripheral acral edema. Edema can develop on the hands, feet, and genitalia. Importantly, facial edema has been noted to precede skin lesions.2 Coin-shaped or targetoid hemorrhagic and purpuric lesions in a cockade or rosette pattern with scalloped margins typically begin on the distal extremities and tend to spread proximally. The lesions are variable in size but have been reported to be as large as 5 cm in diameter. Although joint pain, bloody diarrhea, hematuria, and proteinuria can accompany AHEI, most cases are devoid of systemic symptoms.3 Hemorrhagic lacrimation and epistaxis—both present in our patient—are rare findings with AHEI. It is likely that most providers, including dermatologists, may be unfamiliar with these striking clinical findings. Although the pathophysiology of hemorrhagic lacrimation and epistaxis has not been formally investigated, we postulate that it likely is related to the formation of immune complexes that lead to small vessel vasculitis, underpinning the characteristic findings in AHEI.4,5 This reasoning is supported by the complete resolution of symptoms corresponding with clinical clearance of the cutaneous vasculitis in 2 prior cases4,5 as well as in our patient who did not have a relapse of symptoms following cessation of the cutaneous eruption at a pediatric follow-up appointment 2 weeks later.
Acute hemorrhagic edema of infancy is a clinical diagnosis; however, a skin biopsy can be performed to confirm the clinical suspicion and rule out more serious conditions. Histopathologic examination reveals a leukocytoclastic vasculitis involving the capillaries and postcapillary venules of the upper and mid dermis. Laboratory test results usually are nonspecific but can help distinguish AHEI from more serious diseases. The erythrocyte sedimentation rate and C-reactive protein level may be slightly elevated in infants with AHEI. Urinalysis and stool guaiac tests also can be performed to evaluate for any renal or gastrointestinal involvement.6
The differential diagnosis includes IgA vasculitis, erythema multiforme, acute meningococcemia, urticarial vasculitis, Kawasaki disease, and child abuse. IgA vasculitis often presents with more systemic involvement, with abdominal pain, vomiting, hematemesis, diarrhea, and hematochezia occurring in up to 50% of patients. The cutaneous findings of erythema multiforme classically are confined to the limbs and face, and edema of the extremities typically is not seen. Patients with acute meningococcemia appear toxic with high fevers, malaise, and possible septic shock.5
Acute hemorrhagic edema of infancy is a self-limited condition typically lasting 1 to 3 weeks and requires only supportive care.7 Antibiotics should be given to treat concurrent bacterial infections, and antihistamines and steroids may be useful for symptomatic relief. Importantly, however, systemic corticosteroids do not appear to conclusively alter the disease course.8
Acute hemorrhagic edema of infancy is a rare benign leukocytoclastic vasculitis with a striking presentation often seen following an upper respiratory tract infection or course of antibiotics. Our case demonstrates that on rare occasions, AHEI may be accompanied by hemorrhagic lacrimation and epistaxis—findings that can be quite alarming to both parents and medical providers. Nonetheless, patients and their caretakers should be assured that the condition is self-limited and resolves without permanent sequalae.
- Emerich PS, Prebianchi PA, Motta LL, et al. Acute hemorrhagic edema of infancy: report of three cases. An Bras Dermatol. 2011;86:1181-1184.
- Avhad G, Ghuge P, Jerajani H. Acute hemorrhagic edema of infancy. Indian Dermatol Online J. 2014;5:356-357.
- Krause I, Lazarov A, Rachmel A, et al. Acute haemorrhagic oedema of infancy, a benign variant of leucocytoclastic vasculitis. Acta Paediatr. 1996;85:114-117.
- Sneller H, Vega C, Zemel L, et al. Acute hemorrhagic edema of infancy with associated hemorrhagic lacrimation. Pediatr Emerg Care. 2021;37:E70-E72. doi:10.1097/PEC.0000000000001542
- Mreish S, Al-Tatari H. Hemorrhagic lacrimation and epistaxis in acute hemorrhagic edema of infancy. Case Rep Pediatr. 2016;2016:9762185. doi:10.1155/2016/9762185
- Savino F, Lupica MM, Tarasco V, et al. Acute hemorrhagic edema of infancy: a troubling cutaneous presentation with a self-limiting course. Pediatr Dermatol. 2013;30:E149-E152.
- Fiore E, Rizzi M, Ragazzi M, et al. Acute hemorrhagic edema of young children (cockade purpura and edema): a case series and systematic review. J Am Acad Dermatol. 2008;59:684-695.
- Acute hemorrhagic edema of young children: a concise narrative review. Eur J Pediatr. 2011;170:1507-1511.
To the Editor:
Hemorrhagic lacrimation and epistaxis are dramatic presentations with a narrow differential diagnosis. It rarely has been reported to present alongside the more typical features of acute hemorrhagic edema of infancy (AHEI), which is a benign self-limited leukocytoclastic vasculitis most often seen in children aged 4 months to 2 years. Extracutaneous involvement rarely is seen in AHEI, though joint, gastrointestinal tract, and renal involvement have been reported.1 Most patients present with edematous, annular, or cockade purpuric vasculitic lesions classically involving the face and distal extremities with relative sparing of the trunk. We present a case of a well-appearing, 10-month-old infant boy with hemorrhagic vasculitic lesions, acral edema, and an associated episode of hemorrhagic lacrimation and epistaxis.
A 10-month-old infant boy who was otherwise healthy presented to the emergency department (ED) with an acute-onset, progressively worsening cutaneous eruption of 2 days’ duration. A thorough history revealed that the eruption initially had presented as several small, bright-red papules on the thighs. The eruption subsequently spread to involve the buttocks, legs, and arms (Figures 1 and 2). The parents also noted that the patient had experienced an episode of bloody tears and epistaxis that lasted a few minutes at the pediatrician’s office earlier that morning, a finding that prompted the urgent referral to the ED.
Dermatology was then consulted. A review of systems was notable for rhinorrhea and diarrhea during the week leading to the eruption. The patient’s parents denied fevers, decreased oral intake, or a recent course of antibiotics. The patient’s medical history was notable only for atopic dermatitis treated with emollients and occasional topical steroids. The parents denied recent travel or vaccinations. Physical examination showed an afebrile, well-appearing infant with multiple nontender, slightly edematous, circular, purpuric papules and plaques scattered on the buttocks and extremities with edema on the dorsal feet. The remainder of the patient’s workup in the ED was notable for mild elevations in C-reactive protein levels (1.4 mg/dL [reference range, 0–1.2 mg/dL]) and an elevated erythrocyte sedimentation rate (22 mm/h [reference range, 2–12 mm/h]). A complete blood cell count; liver function tests; urinalysis; and coagulation studies, including prothrombin, partial thromboplastin time, and international normalized ratio, were unremarkable. Acute hemorrhagic edema of infancy was diagnosed based on the clinical manifestations.
Acute hemorrhagic edema of infancy (also known as Finkelstein disease, medallionlike purpura, Seidemayer syndrome, infantile postinfectious irislike purpura and edema, and purpura en cocarde avec oedeme) is believed to result from an immune complex–related reaction, often in the setting of an upper respiratory tract infection; medications, especially antibiotics; or vaccinations. The condition previously was considered a benign form of Henoch-Schönlein purpura; however, it is now recognized as its own clinical entity. Acute hemorrhagic edema of infancy commonly affects children between the ages of 4 months and 2 years. The incidence peaks in the winter months, and males tend to be more affected than females.1
Acute hemorrhagic edema of infancy is clinically characterized by a triad of large purpuric lesions, low-grade fever, and peripheral acral edema. Edema can develop on the hands, feet, and genitalia. Importantly, facial edema has been noted to precede skin lesions.2 Coin-shaped or targetoid hemorrhagic and purpuric lesions in a cockade or rosette pattern with scalloped margins typically begin on the distal extremities and tend to spread proximally. The lesions are variable in size but have been reported to be as large as 5 cm in diameter. Although joint pain, bloody diarrhea, hematuria, and proteinuria can accompany AHEI, most cases are devoid of systemic symptoms.3 Hemorrhagic lacrimation and epistaxis—both present in our patient—are rare findings with AHEI. It is likely that most providers, including dermatologists, may be unfamiliar with these striking clinical findings. Although the pathophysiology of hemorrhagic lacrimation and epistaxis has not been formally investigated, we postulate that it likely is related to the formation of immune complexes that lead to small vessel vasculitis, underpinning the characteristic findings in AHEI.4,5 This reasoning is supported by the complete resolution of symptoms corresponding with clinical clearance of the cutaneous vasculitis in 2 prior cases4,5 as well as in our patient who did not have a relapse of symptoms following cessation of the cutaneous eruption at a pediatric follow-up appointment 2 weeks later.
Acute hemorrhagic edema of infancy is a clinical diagnosis; however, a skin biopsy can be performed to confirm the clinical suspicion and rule out more serious conditions. Histopathologic examination reveals a leukocytoclastic vasculitis involving the capillaries and postcapillary venules of the upper and mid dermis. Laboratory test results usually are nonspecific but can help distinguish AHEI from more serious diseases. The erythrocyte sedimentation rate and C-reactive protein level may be slightly elevated in infants with AHEI. Urinalysis and stool guaiac tests also can be performed to evaluate for any renal or gastrointestinal involvement.6
The differential diagnosis includes IgA vasculitis, erythema multiforme, acute meningococcemia, urticarial vasculitis, Kawasaki disease, and child abuse. IgA vasculitis often presents with more systemic involvement, with abdominal pain, vomiting, hematemesis, diarrhea, and hematochezia occurring in up to 50% of patients. The cutaneous findings of erythema multiforme classically are confined to the limbs and face, and edema of the extremities typically is not seen. Patients with acute meningococcemia appear toxic with high fevers, malaise, and possible septic shock.5
Acute hemorrhagic edema of infancy is a self-limited condition typically lasting 1 to 3 weeks and requires only supportive care.7 Antibiotics should be given to treat concurrent bacterial infections, and antihistamines and steroids may be useful for symptomatic relief. Importantly, however, systemic corticosteroids do not appear to conclusively alter the disease course.8
Acute hemorrhagic edema of infancy is a rare benign leukocytoclastic vasculitis with a striking presentation often seen following an upper respiratory tract infection or course of antibiotics. Our case demonstrates that on rare occasions, AHEI may be accompanied by hemorrhagic lacrimation and epistaxis—findings that can be quite alarming to both parents and medical providers. Nonetheless, patients and their caretakers should be assured that the condition is self-limited and resolves without permanent sequalae.
To the Editor:
Hemorrhagic lacrimation and epistaxis are dramatic presentations with a narrow differential diagnosis. It rarely has been reported to present alongside the more typical features of acute hemorrhagic edema of infancy (AHEI), which is a benign self-limited leukocytoclastic vasculitis most often seen in children aged 4 months to 2 years. Extracutaneous involvement rarely is seen in AHEI, though joint, gastrointestinal tract, and renal involvement have been reported.1 Most patients present with edematous, annular, or cockade purpuric vasculitic lesions classically involving the face and distal extremities with relative sparing of the trunk. We present a case of a well-appearing, 10-month-old infant boy with hemorrhagic vasculitic lesions, acral edema, and an associated episode of hemorrhagic lacrimation and epistaxis.
A 10-month-old infant boy who was otherwise healthy presented to the emergency department (ED) with an acute-onset, progressively worsening cutaneous eruption of 2 days’ duration. A thorough history revealed that the eruption initially had presented as several small, bright-red papules on the thighs. The eruption subsequently spread to involve the buttocks, legs, and arms (Figures 1 and 2). The parents also noted that the patient had experienced an episode of bloody tears and epistaxis that lasted a few minutes at the pediatrician’s office earlier that morning, a finding that prompted the urgent referral to the ED.
Dermatology was then consulted. A review of systems was notable for rhinorrhea and diarrhea during the week leading to the eruption. The patient’s parents denied fevers, decreased oral intake, or a recent course of antibiotics. The patient’s medical history was notable only for atopic dermatitis treated with emollients and occasional topical steroids. The parents denied recent travel or vaccinations. Physical examination showed an afebrile, well-appearing infant with multiple nontender, slightly edematous, circular, purpuric papules and plaques scattered on the buttocks and extremities with edema on the dorsal feet. The remainder of the patient’s workup in the ED was notable for mild elevations in C-reactive protein levels (1.4 mg/dL [reference range, 0–1.2 mg/dL]) and an elevated erythrocyte sedimentation rate (22 mm/h [reference range, 2–12 mm/h]). A complete blood cell count; liver function tests; urinalysis; and coagulation studies, including prothrombin, partial thromboplastin time, and international normalized ratio, were unremarkable. Acute hemorrhagic edema of infancy was diagnosed based on the clinical manifestations.
Acute hemorrhagic edema of infancy (also known as Finkelstein disease, medallionlike purpura, Seidemayer syndrome, infantile postinfectious irislike purpura and edema, and purpura en cocarde avec oedeme) is believed to result from an immune complex–related reaction, often in the setting of an upper respiratory tract infection; medications, especially antibiotics; or vaccinations. The condition previously was considered a benign form of Henoch-Schönlein purpura; however, it is now recognized as its own clinical entity. Acute hemorrhagic edema of infancy commonly affects children between the ages of 4 months and 2 years. The incidence peaks in the winter months, and males tend to be more affected than females.1
Acute hemorrhagic edema of infancy is clinically characterized by a triad of large purpuric lesions, low-grade fever, and peripheral acral edema. Edema can develop on the hands, feet, and genitalia. Importantly, facial edema has been noted to precede skin lesions.2 Coin-shaped or targetoid hemorrhagic and purpuric lesions in a cockade or rosette pattern with scalloped margins typically begin on the distal extremities and tend to spread proximally. The lesions are variable in size but have been reported to be as large as 5 cm in diameter. Although joint pain, bloody diarrhea, hematuria, and proteinuria can accompany AHEI, most cases are devoid of systemic symptoms.3 Hemorrhagic lacrimation and epistaxis—both present in our patient—are rare findings with AHEI. It is likely that most providers, including dermatologists, may be unfamiliar with these striking clinical findings. Although the pathophysiology of hemorrhagic lacrimation and epistaxis has not been formally investigated, we postulate that it likely is related to the formation of immune complexes that lead to small vessel vasculitis, underpinning the characteristic findings in AHEI.4,5 This reasoning is supported by the complete resolution of symptoms corresponding with clinical clearance of the cutaneous vasculitis in 2 prior cases4,5 as well as in our patient who did not have a relapse of symptoms following cessation of the cutaneous eruption at a pediatric follow-up appointment 2 weeks later.
Acute hemorrhagic edema of infancy is a clinical diagnosis; however, a skin biopsy can be performed to confirm the clinical suspicion and rule out more serious conditions. Histopathologic examination reveals a leukocytoclastic vasculitis involving the capillaries and postcapillary venules of the upper and mid dermis. Laboratory test results usually are nonspecific but can help distinguish AHEI from more serious diseases. The erythrocyte sedimentation rate and C-reactive protein level may be slightly elevated in infants with AHEI. Urinalysis and stool guaiac tests also can be performed to evaluate for any renal or gastrointestinal involvement.6
The differential diagnosis includes IgA vasculitis, erythema multiforme, acute meningococcemia, urticarial vasculitis, Kawasaki disease, and child abuse. IgA vasculitis often presents with more systemic involvement, with abdominal pain, vomiting, hematemesis, diarrhea, and hematochezia occurring in up to 50% of patients. The cutaneous findings of erythema multiforme classically are confined to the limbs and face, and edema of the extremities typically is not seen. Patients with acute meningococcemia appear toxic with high fevers, malaise, and possible septic shock.5
Acute hemorrhagic edema of infancy is a self-limited condition typically lasting 1 to 3 weeks and requires only supportive care.7 Antibiotics should be given to treat concurrent bacterial infections, and antihistamines and steroids may be useful for symptomatic relief. Importantly, however, systemic corticosteroids do not appear to conclusively alter the disease course.8
Acute hemorrhagic edema of infancy is a rare benign leukocytoclastic vasculitis with a striking presentation often seen following an upper respiratory tract infection or course of antibiotics. Our case demonstrates that on rare occasions, AHEI may be accompanied by hemorrhagic lacrimation and epistaxis—findings that can be quite alarming to both parents and medical providers. Nonetheless, patients and their caretakers should be assured that the condition is self-limited and resolves without permanent sequalae.
- Emerich PS, Prebianchi PA, Motta LL, et al. Acute hemorrhagic edema of infancy: report of three cases. An Bras Dermatol. 2011;86:1181-1184.
- Avhad G, Ghuge P, Jerajani H. Acute hemorrhagic edema of infancy. Indian Dermatol Online J. 2014;5:356-357.
- Krause I, Lazarov A, Rachmel A, et al. Acute haemorrhagic oedema of infancy, a benign variant of leucocytoclastic vasculitis. Acta Paediatr. 1996;85:114-117.
- Sneller H, Vega C, Zemel L, et al. Acute hemorrhagic edema of infancy with associated hemorrhagic lacrimation. Pediatr Emerg Care. 2021;37:E70-E72. doi:10.1097/PEC.0000000000001542
- Mreish S, Al-Tatari H. Hemorrhagic lacrimation and epistaxis in acute hemorrhagic edema of infancy. Case Rep Pediatr. 2016;2016:9762185. doi:10.1155/2016/9762185
- Savino F, Lupica MM, Tarasco V, et al. Acute hemorrhagic edema of infancy: a troubling cutaneous presentation with a self-limiting course. Pediatr Dermatol. 2013;30:E149-E152.
- Fiore E, Rizzi M, Ragazzi M, et al. Acute hemorrhagic edema of young children (cockade purpura and edema): a case series and systematic review. J Am Acad Dermatol. 2008;59:684-695.
- Acute hemorrhagic edema of young children: a concise narrative review. Eur J Pediatr. 2011;170:1507-1511.
- Emerich PS, Prebianchi PA, Motta LL, et al. Acute hemorrhagic edema of infancy: report of three cases. An Bras Dermatol. 2011;86:1181-1184.
- Avhad G, Ghuge P, Jerajani H. Acute hemorrhagic edema of infancy. Indian Dermatol Online J. 2014;5:356-357.
- Krause I, Lazarov A, Rachmel A, et al. Acute haemorrhagic oedema of infancy, a benign variant of leucocytoclastic vasculitis. Acta Paediatr. 1996;85:114-117.
- Sneller H, Vega C, Zemel L, et al. Acute hemorrhagic edema of infancy with associated hemorrhagic lacrimation. Pediatr Emerg Care. 2021;37:E70-E72. doi:10.1097/PEC.0000000000001542
- Mreish S, Al-Tatari H. Hemorrhagic lacrimation and epistaxis in acute hemorrhagic edema of infancy. Case Rep Pediatr. 2016;2016:9762185. doi:10.1155/2016/9762185
- Savino F, Lupica MM, Tarasco V, et al. Acute hemorrhagic edema of infancy: a troubling cutaneous presentation with a self-limiting course. Pediatr Dermatol. 2013;30:E149-E152.
- Fiore E, Rizzi M, Ragazzi M, et al. Acute hemorrhagic edema of young children (cockade purpura and edema): a case series and systematic review. J Am Acad Dermatol. 2008;59:684-695.
- Acute hemorrhagic edema of young children: a concise narrative review. Eur J Pediatr. 2011;170:1507-1511.
PRACTICE POINTS
- Acute hemorrhagic edema of infancy (AHEI) is clinically characterized by a triad of large purpuric lesions, low-grade fever, and peripheral acral edema. Although joint pain, bloody diarrhea, hematuria, and proteinuria can accompany AHEI, most cases are devoid of systemic symptoms.
- It is a self-limited condition typically lasting 1 to 3 weeks and requires only supportive care.
- On rare occasions, AHEI may be accompanied by hemorrhagic lacrimation and epistaxis. Patients should be assured that the condition is self-limited and resolves without permanent sequalae.

A Fixed Drug Eruption to Medroxyprogesterone Acetate Injectable Suspension
To the Editor:
A fixed drug eruption (FDE) is a well-documented form of cutaneous hypersensitivity that typically manifests as a sharply demarcated, dusky, round to oval, edematous, red-violaceous macule or patch on the skin and mucous membranes. The lesion often resolves with residual postinflammatory hyperpigmentation, most commonly as a reaction to ingested drugs or drug components.1 Lesions generally occur at the same anatomic site with repeated exposure to the offending drug. Typically, a single site is affected, but additional sites with more generalized involvement have been reported to occur with subsequent exposure to the offending medication. The diagnosis usually is clinical, but histopathologic findings can help confirm the diagnosis in unusual presentations. We present a novel case of a patient with an FDE from medroxyprogesterone acetate, a contraceptive injection that contains the hormone progestin.
A 35-year-old woman presented to the dermatology clinic for evaluation of a lesion on the left lower buttock of 1 year’s duration. She reported periodic swelling and associated pruritus of the lesion. She denied any growth in size, and no other similar lesions were present. The patient reported a medication history of medroxyprogesterone acetate for birth control, but she denied any other prescription or over-the-counter medication, oral supplements, or recreational drug use. Upon further inquiry, she reported that the recurrence of symptoms appeared to coincide with each administration of medroxyprogesterone acetate, which occurred approximately every 3 months. The eruption cleared between injections and recurred in the same location following subsequent injections. The lesion appeared approximately 2 weeks after the first injection (approximately 1 year prior to presentation to dermatology) and within 2 to 3 days after each subsequent injection. Physical examination revealed a 2×2-cm, circular, slightly violaceous patch on the left buttock (Figure 1). A biopsy was recommended to aid in diagnosis, and the patient was offered a topical steroid for symptomatic relief. A punch biopsy revealed subtle interface dermatitis with superficial perivascular lymphoid infiltrate and marked pigmentary incontinence consistent with an FDE (Figure 2).

An FDE was first reported in 1889 by Bourns,2 and over time more implicated agents and varying clinical presentations have been linked to the disease. The FDE can be accompanied by symptoms of pruritus or paresthesia. Most cases are devoid of systemic symptoms. An FDE can be located anywhere on the body, but it most frequently manifests on the lips, face, hands, feet, and genitalia. Although the eruption often heals with residual postinflammatory hyperpigmentation, a nonpigmenting FDE due to pseudoephedrine has been reported.3

Common culprits include antibiotics (eg, sulfonamides, trimethoprim, fluoroquinolones, tetracyclines), nonsteroidal anti-inflammatory medications (eg, naproxen sodium, ibuprofen, celecoxib), barbiturates, antimalarials, and anticonvulsants. Rare cases of FDE induced by foods and food additives also have been reported.4 Oral fluconazole, levocetirizine dihydrochloride, loperamide, and multivitamin-mineral preparations are other rare inducers of FDE.5-8 In 2004, Ritter and Meffert9 described an FDE to the green dye used in inactive oral contraceptive pills. A similar case was reported by Rea et al10 that described an FDE from the inactive sugar pills in ethinyl estradiol and levonorgestrel, which is another combined oral contraceptive.
The time between ingestion of the offending agent and the manifestation of the disease usually is 1 to 2 weeks; however, upon subsequent exposure, the disease has been reported to manifest within hours.1 CD8+ memory T cells have been shown to be major players in the development of FDE and can be found along the dermoepidermal junction as part of a delayed type IV hypersensitivity reaction.11 Histopathology reveals superficial and deep interstitial and perivascular infiltrates consisting of lymphocytes with admixed eosinophils and possibly neutrophils in the dermis. In the epidermis, necrotic keratinocytes can be present. In rare cases, FDE may have atypical features, such as in generalized bullous FDE and nonpigmenting FDE, the latter of which more commonly is associated with pseudoephedrine.1
The differential diagnosis for FDE includes erythema multiforme, Stevens-Johnson syndrome/toxic epidermal necrolysis, autoimmune progesterone dermatitis, and large plaque parapsoriasis. The number and morphology of lesions in erythema multiforme help differentiate it from FDE, as erythema multiforme presents with multiple targetoid lesions. The lesions of generalized bullous FDE can be similar to those of Stevens-Johnson syndrome/toxic epidermal necrolysis, and the pigmented patches of FDE can resemble large plaque parapsoriasis.
It is important to consider any medication ingested in the 1- to 2-week period before FDE onset, including over-the-counter medications, health food supplements, and prescription medications. Discontinuation of the implicated medication or any medication potentially cross-reacting with another medication is the most important step in management. Wound care may be needed for any bullous or eroded lesions. Lesions typically resolve within a few days to weeks of stopping the offending agent. Importantly, patients should be counseled on the secondary pigment alterations that may be persistent for several months. Other treatment for FDEs is aimed at symptomatic relief and may include topical corticosteroids and oral antihistamines.1
Medroxyprogesterone acetate is a highly effective contraceptive drug with low rates of failure.12 It is a weak androgenic progestin that is administered as a single 150-mg intramuscular injection every 3 months and inhibits gonadotropins. Common side effects include local injection-site reactions, unscheduled bleeding, amenorrhea, weight gain, headache, and mood changes. However, FDE has not been reported as an adverse effect to medroxyprogesterone acetate, both in official US Food and Drug Administration information and in the current literature.12
Autoimmune progesterone dermatitis (also known as progestin hypersensitivity) is a well-characterized cyclic hypersensitivity reaction to the hormone progesterone that occurs during the luteal phase of the menstrual cycle. It is known to have a variable clinical presentation including urticaria, erythema multiforme, eczema, and angioedema.13 Autoimmune progesterone dermatitis also has been reported to present as an FDE.14-16 The onset of the cutaneous manifestation often starts a few days before the onset of menses, with spontaneous resolution occurring after the onset of menstruation. The mechanism by which endogenous progesterone or other secretory products become antigenic is unknown. It has been suggested that there is an alteration in the properties of the hormone that would predispose it to be antigenic as it would not be considered self. In 2001, Warin17 proposed the following diagnostic criteria for autoimmune progesterone dermatitis: (1) skin lesions associated with menstrual cycle (premenstrual flare); (2) a positive response to the progesterone intradermal or intramuscular test; and (3) symptomatic improvement after inhibiting progesterone secretion by suppressing ovulation.17 The treatment includes antiallergy medications, progesterone desensitization, omalizumab injection, and leuprolide acetate injection.
Our case represents FDE from medroxyprogesterone acetate. Although we did not formally investigate the antigenicity of the exogenous progesterone, we postulate that the pathophysiology likely is similar to an FDE associated with endogenous progesterone. This reasoning is supported by the time course of the patient’s lesion as well as the worsening of symptoms in the days following the administration of the medication. Additionally, the patient had no history of skin lesions prior to the initiation of medroxyprogesterone acetate or similar lesions associated with her menstrual cycles.
A careful and detailed review of medication history is necessary to evaluate FDEs. Our case emphasizes that not only endogenous but also exogenous forms of progesterone may cause hypersensitivity, leading to an FDE. With more than 2 million prescriptions of medroxyprogesterone acetate written every year, dermatologists should be aware of the rare but potential risk for an FDE in patients using this medication.18
- Bolognia J, Jorizzo JL, Rapini RP. Dermatology. 2nd ed. Mosby; 2008.
- Bourns DCG. Unusual effects of antipyrine. Br Med J. 1889;2:818-820.
- Shelley WB, Shelley ED. Nonpigmenting fixed drug eruption as a distinctive reaction pattern: examples caused by sensitivity to pseudoephedrine hydrochloride and tetrahydrozoline. J Am Acad Dermatol. 1987;17:403-407.
- Sohn KH, Kim BK, Kim JY, et al. Fixed food eruption caused by Actinidia arguta (hardy kiwi): a case report and literature review. Allergy Asthma Immunol Res. 2017;9:182-184.
- Nakai N, Katoh N. Fixed drug eruption caused by fluconazole: a case report and mini-review of the literature. Allergol Int. 2013;6:139-141.
- An I, Demir V, Ibiloglu I, et al. Fixed drug eruption induced by levocetirizine. Indian Dermatol Online J. 2017;8:276-278.
- Matarredona J, Borrás Blasco J, Navarro-Ruiz A, et al. Fixed drug eruption associated to loperamide [in Spanish]. Med Clin (Barc). 2005;124:198-199.
- Gohel D. Fixed drug eruption due to multi-vitamin multi-mineral preparation. J Assoc Physicians India. 2000;48:268.
- Ritter SE, Meffert J. A refractory fixed drug reaction to a dye used in an oral contraceptive. Cutis. 2004;74:243-244.
- Rea S, McMeniman E, Darch K, et al. A fixed drug eruption to the sugar pills of a combined oral contraceptive. Poster presented at: The Australasian College of Dermatologists 51st Annual Scientific Meeting; May 22, 2018; Queensland, Australia.
- Shiohara T, Mizukawa Y. Fixed drug eruption: a disease mediated by self-inflicted responses of intraepidermal T cells. Eur J Dermatol. 2007;17:201-208.
- Depo-Provera CI. Prescribing information. Pfizer; 2020. Accessed March 10, 2022. https://labeling.pfizer.com/ShowLabeling.aspx?format=PDF&id=522
- George R, Badawy SZ. Autoimmune progesterone dermatitis: a case report. Case Rep Obstet Gynecol. 2012;2012:757854.
- Mokhtari R, Sepaskhah M, Aslani FS, et al. Autoimmune progesterone dermatitis presenting as fixed drug eruption: a case report. Dermatol Online J. 2017;23:13030/qt685685p4.
- Asai J, Katoh N, Nakano M, et al. Case of autoimmune progesterone dermatitis presenting as fixed drug eruption. J Dermatol. 2009;36:643-645.
- Bhardwaj N, Jindal R, Chauhan P. Autoimmune progesterone dermatitis presenting as fixed drug eruption. BMJ Case Rep. 2019;12:E231873.
- Warin AP. Case 2. diagnosis: erythema multiforme as a presentation of autoimmune progesterone dermatitis. Clin Exp Dermatol. 2001;26:107-108.
- Medroxyprogesterone Drug Usage Statistics, United States, 2013-2019. ClinCalc website. Updated September 15, 2021. Accessed March 17, 2022. https://clincalc.com/DrugStats/Drugs/Medroxyprogesterone
To the Editor:
A fixed drug eruption (FDE) is a well-documented form of cutaneous hypersensitivity that typically manifests as a sharply demarcated, dusky, round to oval, edematous, red-violaceous macule or patch on the skin and mucous membranes. The lesion often resolves with residual postinflammatory hyperpigmentation, most commonly as a reaction to ingested drugs or drug components.1 Lesions generally occur at the same anatomic site with repeated exposure to the offending drug. Typically, a single site is affected, but additional sites with more generalized involvement have been reported to occur with subsequent exposure to the offending medication. The diagnosis usually is clinical, but histopathologic findings can help confirm the diagnosis in unusual presentations. We present a novel case of a patient with an FDE from medroxyprogesterone acetate, a contraceptive injection that contains the hormone progestin.
A 35-year-old woman presented to the dermatology clinic for evaluation of a lesion on the left lower buttock of 1 year’s duration. She reported periodic swelling and associated pruritus of the lesion. She denied any growth in size, and no other similar lesions were present. The patient reported a medication history of medroxyprogesterone acetate for birth control, but she denied any other prescription or over-the-counter medication, oral supplements, or recreational drug use. Upon further inquiry, she reported that the recurrence of symptoms appeared to coincide with each administration of medroxyprogesterone acetate, which occurred approximately every 3 months. The eruption cleared between injections and recurred in the same location following subsequent injections. The lesion appeared approximately 2 weeks after the first injection (approximately 1 year prior to presentation to dermatology) and within 2 to 3 days after each subsequent injection. Physical examination revealed a 2×2-cm, circular, slightly violaceous patch on the left buttock (Figure 1). A biopsy was recommended to aid in diagnosis, and the patient was offered a topical steroid for symptomatic relief. A punch biopsy revealed subtle interface dermatitis with superficial perivascular lymphoid infiltrate and marked pigmentary incontinence consistent with an FDE (Figure 2).
An FDE was first reported in 1889 by Bourns,2 and over time more implicated agents and varying clinical presentations have been linked to the disease. The FDE can be accompanied by symptoms of pruritus or paresthesia. Most cases are devoid of systemic symptoms. An FDE can be located anywhere on the body, but it most frequently manifests on the lips, face, hands, feet, and genitalia. Although the eruption often heals with residual postinflammatory hyperpigmentation, a nonpigmenting FDE due to pseudoephedrine has been reported.3
Common culprits include antibiotics (eg, sulfonamides, trimethoprim, fluoroquinolones, tetracyclines), nonsteroidal anti-inflammatory medications (eg, naproxen sodium, ibuprofen, celecoxib), barbiturates, antimalarials, and anticonvulsants. Rare cases of FDE induced by foods and food additives also have been reported.4 Oral fluconazole, levocetirizine dihydrochloride, loperamide, and multivitamin-mineral preparations are other rare inducers of FDE.5-8 In 2004, Ritter and Meffert9 described an FDE to the green dye used in inactive oral contraceptive pills. A similar case was reported by Rea et al10 that described an FDE from the inactive sugar pills in ethinyl estradiol and levonorgestrel, which is another combined oral contraceptive.
The time between ingestion of the offending agent and the manifestation of the disease usually is 1 to 2 weeks; however, upon subsequent exposure, the disease has been reported to manifest within hours.1 CD8+ memory T cells have been shown to be major players in the development of FDE and can be found along the dermoepidermal junction as part of a delayed type IV hypersensitivity reaction.11 Histopathology reveals superficial and deep interstitial and perivascular infiltrates consisting of lymphocytes with admixed eosinophils and possibly neutrophils in the dermis. In the epidermis, necrotic keratinocytes can be present. In rare cases, FDE may have atypical features, such as in generalized bullous FDE and nonpigmenting FDE, the latter of which more commonly is associated with pseudoephedrine.1
The differential diagnosis for FDE includes erythema multiforme, Stevens-Johnson syndrome/toxic epidermal necrolysis, autoimmune progesterone dermatitis, and large plaque parapsoriasis. The number and morphology of lesions in erythema multiforme help differentiate it from FDE, as erythema multiforme presents with multiple targetoid lesions. The lesions of generalized bullous FDE can be similar to those of Stevens-Johnson syndrome/toxic epidermal necrolysis, and the pigmented patches of FDE can resemble large plaque parapsoriasis.
It is important to consider any medication ingested in the 1- to 2-week period before FDE onset, including over-the-counter medications, health food supplements, and prescription medications. Discontinuation of the implicated medication or any medication potentially cross-reacting with another medication is the most important step in management. Wound care may be needed for any bullous or eroded lesions. Lesions typically resolve within a few days to weeks of stopping the offending agent. Importantly, patients should be counseled on the secondary pigment alterations that may be persistent for several months. Other treatment for FDEs is aimed at symptomatic relief and may include topical corticosteroids and oral antihistamines.1
Medroxyprogesterone acetate is a highly effective contraceptive drug with low rates of failure.12 It is a weak androgenic progestin that is administered as a single 150-mg intramuscular injection every 3 months and inhibits gonadotropins. Common side effects include local injection-site reactions, unscheduled bleeding, amenorrhea, weight gain, headache, and mood changes. However, FDE has not been reported as an adverse effect to medroxyprogesterone acetate, both in official US Food and Drug Administration information and in the current literature.12
Autoimmune progesterone dermatitis (also known as progestin hypersensitivity) is a well-characterized cyclic hypersensitivity reaction to the hormone progesterone that occurs during the luteal phase of the menstrual cycle. It is known to have a variable clinical presentation including urticaria, erythema multiforme, eczema, and angioedema.13 Autoimmune progesterone dermatitis also has been reported to present as an FDE.14-16 The onset of the cutaneous manifestation often starts a few days before the onset of menses, with spontaneous resolution occurring after the onset of menstruation. The mechanism by which endogenous progesterone or other secretory products become antigenic is unknown. It has been suggested that there is an alteration in the properties of the hormone that would predispose it to be antigenic as it would not be considered self. In 2001, Warin17 proposed the following diagnostic criteria for autoimmune progesterone dermatitis: (1) skin lesions associated with menstrual cycle (premenstrual flare); (2) a positive response to the progesterone intradermal or intramuscular test; and (3) symptomatic improvement after inhibiting progesterone secretion by suppressing ovulation.17 The treatment includes antiallergy medications, progesterone desensitization, omalizumab injection, and leuprolide acetate injection.
Our case represents FDE from medroxyprogesterone acetate. Although we did not formally investigate the antigenicity of the exogenous progesterone, we postulate that the pathophysiology likely is similar to an FDE associated with endogenous progesterone. This reasoning is supported by the time course of the patient’s lesion as well as the worsening of symptoms in the days following the administration of the medication. Additionally, the patient had no history of skin lesions prior to the initiation of medroxyprogesterone acetate or similar lesions associated with her menstrual cycles.
A careful and detailed review of medication history is necessary to evaluate FDEs. Our case emphasizes that not only endogenous but also exogenous forms of progesterone may cause hypersensitivity, leading to an FDE. With more than 2 million prescriptions of medroxyprogesterone acetate written every year, dermatologists should be aware of the rare but potential risk for an FDE in patients using this medication.18
To the Editor:
A fixed drug eruption (FDE) is a well-documented form of cutaneous hypersensitivity that typically manifests as a sharply demarcated, dusky, round to oval, edematous, red-violaceous macule or patch on the skin and mucous membranes. The lesion often resolves with residual postinflammatory hyperpigmentation, most commonly as a reaction to ingested drugs or drug components.1 Lesions generally occur at the same anatomic site with repeated exposure to the offending drug. Typically, a single site is affected, but additional sites with more generalized involvement have been reported to occur with subsequent exposure to the offending medication. The diagnosis usually is clinical, but histopathologic findings can help confirm the diagnosis in unusual presentations. We present a novel case of a patient with an FDE from medroxyprogesterone acetate, a contraceptive injection that contains the hormone progestin.
A 35-year-old woman presented to the dermatology clinic for evaluation of a lesion on the left lower buttock of 1 year’s duration. She reported periodic swelling and associated pruritus of the lesion. She denied any growth in size, and no other similar lesions were present. The patient reported a medication history of medroxyprogesterone acetate for birth control, but she denied any other prescription or over-the-counter medication, oral supplements, or recreational drug use. Upon further inquiry, she reported that the recurrence of symptoms appeared to coincide with each administration of medroxyprogesterone acetate, which occurred approximately every 3 months. The eruption cleared between injections and recurred in the same location following subsequent injections. The lesion appeared approximately 2 weeks after the first injection (approximately 1 year prior to presentation to dermatology) and within 2 to 3 days after each subsequent injection. Physical examination revealed a 2×2-cm, circular, slightly violaceous patch on the left buttock (Figure 1). A biopsy was recommended to aid in diagnosis, and the patient was offered a topical steroid for symptomatic relief. A punch biopsy revealed subtle interface dermatitis with superficial perivascular lymphoid infiltrate and marked pigmentary incontinence consistent with an FDE (Figure 2).
An FDE was first reported in 1889 by Bourns,2 and over time more implicated agents and varying clinical presentations have been linked to the disease. The FDE can be accompanied by symptoms of pruritus or paresthesia. Most cases are devoid of systemic symptoms. An FDE can be located anywhere on the body, but it most frequently manifests on the lips, face, hands, feet, and genitalia. Although the eruption often heals with residual postinflammatory hyperpigmentation, a nonpigmenting FDE due to pseudoephedrine has been reported.3
Common culprits include antibiotics (eg, sulfonamides, trimethoprim, fluoroquinolones, tetracyclines), nonsteroidal anti-inflammatory medications (eg, naproxen sodium, ibuprofen, celecoxib), barbiturates, antimalarials, and anticonvulsants. Rare cases of FDE induced by foods and food additives also have been reported.4 Oral fluconazole, levocetirizine dihydrochloride, loperamide, and multivitamin-mineral preparations are other rare inducers of FDE.5-8 In 2004, Ritter and Meffert9 described an FDE to the green dye used in inactive oral contraceptive pills. A similar case was reported by Rea et al10 that described an FDE from the inactive sugar pills in ethinyl estradiol and levonorgestrel, which is another combined oral contraceptive.
The time between ingestion of the offending agent and the manifestation of the disease usually is 1 to 2 weeks; however, upon subsequent exposure, the disease has been reported to manifest within hours.1 CD8+ memory T cells have been shown to be major players in the development of FDE and can be found along the dermoepidermal junction as part of a delayed type IV hypersensitivity reaction.11 Histopathology reveals superficial and deep interstitial and perivascular infiltrates consisting of lymphocytes with admixed eosinophils and possibly neutrophils in the dermis. In the epidermis, necrotic keratinocytes can be present. In rare cases, FDE may have atypical features, such as in generalized bullous FDE and nonpigmenting FDE, the latter of which more commonly is associated with pseudoephedrine.1
The differential diagnosis for FDE includes erythema multiforme, Stevens-Johnson syndrome/toxic epidermal necrolysis, autoimmune progesterone dermatitis, and large plaque parapsoriasis. The number and morphology of lesions in erythema multiforme help differentiate it from FDE, as erythema multiforme presents with multiple targetoid lesions. The lesions of generalized bullous FDE can be similar to those of Stevens-Johnson syndrome/toxic epidermal necrolysis, and the pigmented patches of FDE can resemble large plaque parapsoriasis.
It is important to consider any medication ingested in the 1- to 2-week period before FDE onset, including over-the-counter medications, health food supplements, and prescription medications. Discontinuation of the implicated medication or any medication potentially cross-reacting with another medication is the most important step in management. Wound care may be needed for any bullous or eroded lesions. Lesions typically resolve within a few days to weeks of stopping the offending agent. Importantly, patients should be counseled on the secondary pigment alterations that may be persistent for several months. Other treatment for FDEs is aimed at symptomatic relief and may include topical corticosteroids and oral antihistamines.1
Medroxyprogesterone acetate is a highly effective contraceptive drug with low rates of failure.12 It is a weak androgenic progestin that is administered as a single 150-mg intramuscular injection every 3 months and inhibits gonadotropins. Common side effects include local injection-site reactions, unscheduled bleeding, amenorrhea, weight gain, headache, and mood changes. However, FDE has not been reported as an adverse effect to medroxyprogesterone acetate, both in official US Food and Drug Administration information and in the current literature.12
Autoimmune progesterone dermatitis (also known as progestin hypersensitivity) is a well-characterized cyclic hypersensitivity reaction to the hormone progesterone that occurs during the luteal phase of the menstrual cycle. It is known to have a variable clinical presentation including urticaria, erythema multiforme, eczema, and angioedema.13 Autoimmune progesterone dermatitis also has been reported to present as an FDE.14-16 The onset of the cutaneous manifestation often starts a few days before the onset of menses, with spontaneous resolution occurring after the onset of menstruation. The mechanism by which endogenous progesterone or other secretory products become antigenic is unknown. It has been suggested that there is an alteration in the properties of the hormone that would predispose it to be antigenic as it would not be considered self. In 2001, Warin17 proposed the following diagnostic criteria for autoimmune progesterone dermatitis: (1) skin lesions associated with menstrual cycle (premenstrual flare); (2) a positive response to the progesterone intradermal or intramuscular test; and (3) symptomatic improvement after inhibiting progesterone secretion by suppressing ovulation.17 The treatment includes antiallergy medications, progesterone desensitization, omalizumab injection, and leuprolide acetate injection.
Our case represents FDE from medroxyprogesterone acetate. Although we did not formally investigate the antigenicity of the exogenous progesterone, we postulate that the pathophysiology likely is similar to an FDE associated with endogenous progesterone. This reasoning is supported by the time course of the patient’s lesion as well as the worsening of symptoms in the days following the administration of the medication. Additionally, the patient had no history of skin lesions prior to the initiation of medroxyprogesterone acetate or similar lesions associated with her menstrual cycles.
A careful and detailed review of medication history is necessary to evaluate FDEs. Our case emphasizes that not only endogenous but also exogenous forms of progesterone may cause hypersensitivity, leading to an FDE. With more than 2 million prescriptions of medroxyprogesterone acetate written every year, dermatologists should be aware of the rare but potential risk for an FDE in patients using this medication.18
- Bolognia J, Jorizzo JL, Rapini RP. Dermatology. 2nd ed. Mosby; 2008.
- Bourns DCG. Unusual effects of antipyrine. Br Med J. 1889;2:818-820.
- Shelley WB, Shelley ED. Nonpigmenting fixed drug eruption as a distinctive reaction pattern: examples caused by sensitivity to pseudoephedrine hydrochloride and tetrahydrozoline. J Am Acad Dermatol. 1987;17:403-407.
- Sohn KH, Kim BK, Kim JY, et al. Fixed food eruption caused by Actinidia arguta (hardy kiwi): a case report and literature review. Allergy Asthma Immunol Res. 2017;9:182-184.
- Nakai N, Katoh N. Fixed drug eruption caused by fluconazole: a case report and mini-review of the literature. Allergol Int. 2013;6:139-141.
- An I, Demir V, Ibiloglu I, et al. Fixed drug eruption induced by levocetirizine. Indian Dermatol Online J. 2017;8:276-278.
- Matarredona J, Borrás Blasco J, Navarro-Ruiz A, et al. Fixed drug eruption associated to loperamide [in Spanish]. Med Clin (Barc). 2005;124:198-199.
- Gohel D. Fixed drug eruption due to multi-vitamin multi-mineral preparation. J Assoc Physicians India. 2000;48:268.
- Ritter SE, Meffert J. A refractory fixed drug reaction to a dye used in an oral contraceptive. Cutis. 2004;74:243-244.
- Rea S, McMeniman E, Darch K, et al. A fixed drug eruption to the sugar pills of a combined oral contraceptive. Poster presented at: The Australasian College of Dermatologists 51st Annual Scientific Meeting; May 22, 2018; Queensland, Australia.
- Shiohara T, Mizukawa Y. Fixed drug eruption: a disease mediated by self-inflicted responses of intraepidermal T cells. Eur J Dermatol. 2007;17:201-208.
- Depo-Provera CI. Prescribing information. Pfizer; 2020. Accessed March 10, 2022. https://labeling.pfizer.com/ShowLabeling.aspx?format=PDF&id=522
- George R, Badawy SZ. Autoimmune progesterone dermatitis: a case report. Case Rep Obstet Gynecol. 2012;2012:757854.
- Mokhtari R, Sepaskhah M, Aslani FS, et al. Autoimmune progesterone dermatitis presenting as fixed drug eruption: a case report. Dermatol Online J. 2017;23:13030/qt685685p4.
- Asai J, Katoh N, Nakano M, et al. Case of autoimmune progesterone dermatitis presenting as fixed drug eruption. J Dermatol. 2009;36:643-645.
- Bhardwaj N, Jindal R, Chauhan P. Autoimmune progesterone dermatitis presenting as fixed drug eruption. BMJ Case Rep. 2019;12:E231873.
- Warin AP. Case 2. diagnosis: erythema multiforme as a presentation of autoimmune progesterone dermatitis. Clin Exp Dermatol. 2001;26:107-108.
- Medroxyprogesterone Drug Usage Statistics, United States, 2013-2019. ClinCalc website. Updated September 15, 2021. Accessed March 17, 2022. https://clincalc.com/DrugStats/Drugs/Medroxyprogesterone
- Bolognia J, Jorizzo JL, Rapini RP. Dermatology. 2nd ed. Mosby; 2008.
- Bourns DCG. Unusual effects of antipyrine. Br Med J. 1889;2:818-820.
- Shelley WB, Shelley ED. Nonpigmenting fixed drug eruption as a distinctive reaction pattern: examples caused by sensitivity to pseudoephedrine hydrochloride and tetrahydrozoline. J Am Acad Dermatol. 1987;17:403-407.
- Sohn KH, Kim BK, Kim JY, et al. Fixed food eruption caused by Actinidia arguta (hardy kiwi): a case report and literature review. Allergy Asthma Immunol Res. 2017;9:182-184.
- Nakai N, Katoh N. Fixed drug eruption caused by fluconazole: a case report and mini-review of the literature. Allergol Int. 2013;6:139-141.
- An I, Demir V, Ibiloglu I, et al. Fixed drug eruption induced by levocetirizine. Indian Dermatol Online J. 2017;8:276-278.
- Matarredona J, Borrás Blasco J, Navarro-Ruiz A, et al. Fixed drug eruption associated to loperamide [in Spanish]. Med Clin (Barc). 2005;124:198-199.
- Gohel D. Fixed drug eruption due to multi-vitamin multi-mineral preparation. J Assoc Physicians India. 2000;48:268.
- Ritter SE, Meffert J. A refractory fixed drug reaction to a dye used in an oral contraceptive. Cutis. 2004;74:243-244.
- Rea S, McMeniman E, Darch K, et al. A fixed drug eruption to the sugar pills of a combined oral contraceptive. Poster presented at: The Australasian College of Dermatologists 51st Annual Scientific Meeting; May 22, 2018; Queensland, Australia.
- Shiohara T, Mizukawa Y. Fixed drug eruption: a disease mediated by self-inflicted responses of intraepidermal T cells. Eur J Dermatol. 2007;17:201-208.
- Depo-Provera CI. Prescribing information. Pfizer; 2020. Accessed March 10, 2022. https://labeling.pfizer.com/ShowLabeling.aspx?format=PDF&id=522
- George R, Badawy SZ. Autoimmune progesterone dermatitis: a case report. Case Rep Obstet Gynecol. 2012;2012:757854.
- Mokhtari R, Sepaskhah M, Aslani FS, et al. Autoimmune progesterone dermatitis presenting as fixed drug eruption: a case report. Dermatol Online J. 2017;23:13030/qt685685p4.
- Asai J, Katoh N, Nakano M, et al. Case of autoimmune progesterone dermatitis presenting as fixed drug eruption. J Dermatol. 2009;36:643-645.
- Bhardwaj N, Jindal R, Chauhan P. Autoimmune progesterone dermatitis presenting as fixed drug eruption. BMJ Case Rep. 2019;12:E231873.
- Warin AP. Case 2. diagnosis: erythema multiforme as a presentation of autoimmune progesterone dermatitis. Clin Exp Dermatol. 2001;26:107-108.
- Medroxyprogesterone Drug Usage Statistics, United States, 2013-2019. ClinCalc website. Updated September 15, 2021. Accessed March 17, 2022. https://clincalc.com/DrugStats/Drugs/Medroxyprogesterone
Practice Points
- Exogenous progesterone from the administration of the contraceptive injectable medroxyprogesterone acetate has the potential to cause a cutaneous hypersensitivity reaction in the form of a fixed drug eruption (FDE).
- Dermatologists should perform a careful and detailed review of medication history to evaluate drug eruptions.

Pink, Scaly, Annular Plaques in Concentric Rings Localized to Vitiliginous Patches
The Diagnosis: Tinea Pseudoimbricata
Tinea pseudoimbricata and tinea indecisiva are synonyms describing cases of tinea corporis that manifest in scaly plaques in concentric rings evocative of those present in tinea imbricata. However, in contrast to tinea imbricata, cases of tinea pseudoimbricata are caused by dermatophytes other than Trichophyton concentricum. 1 Tinea pseudoimbricata usually presents in association with immunosuppression, either systemic or local, and can be produced by application of topical medications such as corticosteroids.2 Mask-Bull et al3 reported the case of a 21-year-old man in the United States with no history of immunosuppressive conditions who presented with scaly erythematous annular plaques on the lateral neck that resolved with 2 pulsed doses of terbinafine. Potassium hydroxide preparation and fungal culture were both consistent with Trichophyton tonsurans.3
Trichophyton concentricum is an anthropophilic species of dermatophyte endemic to areas within the South Pacific, Southeast Asia, and Central and South America. Infection with T concentricum produces tinea imbricata, which presents with concentric, scaly, annular rings. Cutaneous lesions of tinea imbricata caused by T concentricum have a more generalized distribution and more densely grouped, concentric circles than the cutaneous findings seen in patients with tinea pseudoimbricata.4 Affected patients typically demonstrate negative delayed-type hypersensitivity to T concentricum cytoplasmic antigen and T-lymphocyte hyporeactivity, which may contribute to the development of sequential waves of scaling observed in tinea imbricata.5
Trichophyton rubrum, the most common cause of tinea corporis, has been reported to cause some cases of tinea pseudoimbricata (indecisiva).1,2 It utilizes keratinases such as subtilisins (Sub3 and Sub4), leucine aminopeptidases (Lap1 and Lap2), and dipeptidyl peptidases (DppIV and DppV) to invade the skin. Once inside, mannans, glycoprotein constituents of the cell wall, are released and bind to the cell surface of mononuclear phagocytes, subsequently moving into the cell by phagocytosis, thereafter interfering with RNA synthesis that is necessary for presentation of antigens to appropriate T cells and allowing for initiation of chronic infection.6,7 The cytotoxic response to superficial dermatophyte infection is triggered by major histocompatibility complex class I molecule activation of CD8+ cells.6,8
Our case is of interest given the localization of the superficial dermatophyte infection to only vitiliginous skin. This distribution and appearance while undergoing narrowband UVB (NB-UVB) treatment is rare. We postulate that our patient likely represents a case of locus minoris resistentiae, a phenomenon in which an area of skin exhibits a compromised immune microenvironment that predisposes it to disease.9
In vitiligo, NB-UVB modulates the immune response by increasing IL-10, thereby promoting regulatory T-cell differentiation with suppression of autoreactive T cells and induction of direct T-lymphocyte apoptosis.10,11 Although the mechanism accounting for our patient’s presentation is unknown, we suspect NB-UVB–induced immunosuppression enabled persistence of the dermatophyte infection. The localization of the infection to the vitiliginous patches may result from the greater penetration of the UV light relative to the surrounding, normally pigmented skin. This relative difference in UV penetration would be expected to result in increased immunosuppression in the vitiliginous lesions and enhanced susceptibility to the fungal organisms.
Erythema annulare centrifugum is characterized by annular lesions with a trailing scale instead of the concentric rings seen in tinea pseudoimbricata. Erythema marginatum is seen in acute rheumatic fever and presents with a transient nonpruritic rash, usually on the trunk or extremities. Erythema migrans presents with fewer lesions that are less circinate in shape, and the patient often has a history of a tick bite. Tinea imbricata is caused by T concentricum, while tinea pseudoimbricata is caused by T tonsurans and other dermatophytes.
With the increasing use of immunosuppressant drugs, the prevalence of tinea pseudoimbricata is hypothesized to increase.1 The presence of tinea pseudoimbricata should alert dermatologists to the possible overuse of topical corticosteroids, and other forms of immunosuppression also should be considered.
- Lim SP, Smith AG. “Tinea pseudoimbricata”: tinea corporis in a renal transplant recipient mimicking the concentric rings of tinea imbricata. Clin Exp Dermatol. 2003;28:332-333.
- Batta K, Ramlogan D, Smith AG, et al. ‘Tinea indecisiva’ may mimic the concentric rings of tinea imbricata. Br J Dermatol. 2002;147:384.
- Mask-Bull L, Patel R, Tarbox MB. America’s first case of tinea pseudoimbricata. Am J Dermatol Venereol. 2015;4:15-17.
- Meena M, Mittal A. Tinea pseudo-imbricata. J Assoc Physicians India. 2018;66:79.
- Hay RJ, Reid S, Talwat E, et al. Immune responses of patients with tinea imbricata. Br J Dermatol. 1983;108:581-586.
- Dahl MV. Suppression of immunity and inflammation by products produced by dermatophytes. J Am Acad Dermatol. 1993;28(5 pt 1):S19-S23.
- Blutfield MS, Lohre JM, Pawich DA, et al. The immunologic response to Trichophyton rubrum in lower extremity fungal infections. J Fungi (Basel). 2015;1:130-137.
- De Hoog S, Monod M, Dawson T, et al. Skin fungi from colonization to infection [published online July 2017]. Microbiol Spectr. doi:10.1128/ microbiolspec.FUNK-0049-2016
- Lo Schiavo A, Ruocco E, Russo T, et al. Locus minoris resistentiae: an old but still valid way of thinking in medicine. Clin Dermatol. 2014;32:553-556.
- Ponsonby AL, Lucas RM, van der Mei IA. UVR, vitamin D and three autoimmune diseases—multiple sclerosis, type 1 diabetes, rheumatoid arthritis. Photochem Photobiol. 2005;81:1267-1275.
- Yazdani Abyaneh M, Griffith RD, Falto-Aizpurua L, et al. Narrowband ultraviolet B phototherapy in combination with other therapies for vitiligo: mechanisms and efficacies. J Eur Acad Dermatol Venereol. 2014;28:1610-1622.
The Diagnosis: Tinea Pseudoimbricata
Tinea pseudoimbricata and tinea indecisiva are synonyms describing cases of tinea corporis that manifest in scaly plaques in concentric rings evocative of those present in tinea imbricata. However, in contrast to tinea imbricata, cases of tinea pseudoimbricata are caused by dermatophytes other than Trichophyton concentricum. 1 Tinea pseudoimbricata usually presents in association with immunosuppression, either systemic or local, and can be produced by application of topical medications such as corticosteroids.2 Mask-Bull et al3 reported the case of a 21-year-old man in the United States with no history of immunosuppressive conditions who presented with scaly erythematous annular plaques on the lateral neck that resolved with 2 pulsed doses of terbinafine. Potassium hydroxide preparation and fungal culture were both consistent with Trichophyton tonsurans.3
Trichophyton concentricum is an anthropophilic species of dermatophyte endemic to areas within the South Pacific, Southeast Asia, and Central and South America. Infection with T concentricum produces tinea imbricata, which presents with concentric, scaly, annular rings. Cutaneous lesions of tinea imbricata caused by T concentricum have a more generalized distribution and more densely grouped, concentric circles than the cutaneous findings seen in patients with tinea pseudoimbricata.4 Affected patients typically demonstrate negative delayed-type hypersensitivity to T concentricum cytoplasmic antigen and T-lymphocyte hyporeactivity, which may contribute to the development of sequential waves of scaling observed in tinea imbricata.5
Trichophyton rubrum, the most common cause of tinea corporis, has been reported to cause some cases of tinea pseudoimbricata (indecisiva).1,2 It utilizes keratinases such as subtilisins (Sub3 and Sub4), leucine aminopeptidases (Lap1 and Lap2), and dipeptidyl peptidases (DppIV and DppV) to invade the skin. Once inside, mannans, glycoprotein constituents of the cell wall, are released and bind to the cell surface of mononuclear phagocytes, subsequently moving into the cell by phagocytosis, thereafter interfering with RNA synthesis that is necessary for presentation of antigens to appropriate T cells and allowing for initiation of chronic infection.6,7 The cytotoxic response to superficial dermatophyte infection is triggered by major histocompatibility complex class I molecule activation of CD8+ cells.6,8
Our case is of interest given the localization of the superficial dermatophyte infection to only vitiliginous skin. This distribution and appearance while undergoing narrowband UVB (NB-UVB) treatment is rare. We postulate that our patient likely represents a case of locus minoris resistentiae, a phenomenon in which an area of skin exhibits a compromised immune microenvironment that predisposes it to disease.9
In vitiligo, NB-UVB modulates the immune response by increasing IL-10, thereby promoting regulatory T-cell differentiation with suppression of autoreactive T cells and induction of direct T-lymphocyte apoptosis.10,11 Although the mechanism accounting for our patient’s presentation is unknown, we suspect NB-UVB–induced immunosuppression enabled persistence of the dermatophyte infection. The localization of the infection to the vitiliginous patches may result from the greater penetration of the UV light relative to the surrounding, normally pigmented skin. This relative difference in UV penetration would be expected to result in increased immunosuppression in the vitiliginous lesions and enhanced susceptibility to the fungal organisms.
Erythema annulare centrifugum is characterized by annular lesions with a trailing scale instead of the concentric rings seen in tinea pseudoimbricata. Erythema marginatum is seen in acute rheumatic fever and presents with a transient nonpruritic rash, usually on the trunk or extremities. Erythema migrans presents with fewer lesions that are less circinate in shape, and the patient often has a history of a tick bite. Tinea imbricata is caused by T concentricum, while tinea pseudoimbricata is caused by T tonsurans and other dermatophytes.
With the increasing use of immunosuppressant drugs, the prevalence of tinea pseudoimbricata is hypothesized to increase.1 The presence of tinea pseudoimbricata should alert dermatologists to the possible overuse of topical corticosteroids, and other forms of immunosuppression also should be considered.
The Diagnosis: Tinea Pseudoimbricata
Tinea pseudoimbricata and tinea indecisiva are synonyms describing cases of tinea corporis that manifest in scaly plaques in concentric rings evocative of those present in tinea imbricata. However, in contrast to tinea imbricata, cases of tinea pseudoimbricata are caused by dermatophytes other than Trichophyton concentricum. 1 Tinea pseudoimbricata usually presents in association with immunosuppression, either systemic or local, and can be produced by application of topical medications such as corticosteroids.2 Mask-Bull et al3 reported the case of a 21-year-old man in the United States with no history of immunosuppressive conditions who presented with scaly erythematous annular plaques on the lateral neck that resolved with 2 pulsed doses of terbinafine. Potassium hydroxide preparation and fungal culture were both consistent with Trichophyton tonsurans.3
Trichophyton concentricum is an anthropophilic species of dermatophyte endemic to areas within the South Pacific, Southeast Asia, and Central and South America. Infection with T concentricum produces tinea imbricata, which presents with concentric, scaly, annular rings. Cutaneous lesions of tinea imbricata caused by T concentricum have a more generalized distribution and more densely grouped, concentric circles than the cutaneous findings seen in patients with tinea pseudoimbricata.4 Affected patients typically demonstrate negative delayed-type hypersensitivity to T concentricum cytoplasmic antigen and T-lymphocyte hyporeactivity, which may contribute to the development of sequential waves of scaling observed in tinea imbricata.5
Trichophyton rubrum, the most common cause of tinea corporis, has been reported to cause some cases of tinea pseudoimbricata (indecisiva).1,2 It utilizes keratinases such as subtilisins (Sub3 and Sub4), leucine aminopeptidases (Lap1 and Lap2), and dipeptidyl peptidases (DppIV and DppV) to invade the skin. Once inside, mannans, glycoprotein constituents of the cell wall, are released and bind to the cell surface of mononuclear phagocytes, subsequently moving into the cell by phagocytosis, thereafter interfering with RNA synthesis that is necessary for presentation of antigens to appropriate T cells and allowing for initiation of chronic infection.6,7 The cytotoxic response to superficial dermatophyte infection is triggered by major histocompatibility complex class I molecule activation of CD8+ cells.6,8
Our case is of interest given the localization of the superficial dermatophyte infection to only vitiliginous skin. This distribution and appearance while undergoing narrowband UVB (NB-UVB) treatment is rare. We postulate that our patient likely represents a case of locus minoris resistentiae, a phenomenon in which an area of skin exhibits a compromised immune microenvironment that predisposes it to disease.9
In vitiligo, NB-UVB modulates the immune response by increasing IL-10, thereby promoting regulatory T-cell differentiation with suppression of autoreactive T cells and induction of direct T-lymphocyte apoptosis.10,11 Although the mechanism accounting for our patient’s presentation is unknown, we suspect NB-UVB–induced immunosuppression enabled persistence of the dermatophyte infection. The localization of the infection to the vitiliginous patches may result from the greater penetration of the UV light relative to the surrounding, normally pigmented skin. This relative difference in UV penetration would be expected to result in increased immunosuppression in the vitiliginous lesions and enhanced susceptibility to the fungal organisms.
Erythema annulare centrifugum is characterized by annular lesions with a trailing scale instead of the concentric rings seen in tinea pseudoimbricata. Erythema marginatum is seen in acute rheumatic fever and presents with a transient nonpruritic rash, usually on the trunk or extremities. Erythema migrans presents with fewer lesions that are less circinate in shape, and the patient often has a history of a tick bite. Tinea imbricata is caused by T concentricum, while tinea pseudoimbricata is caused by T tonsurans and other dermatophytes.
With the increasing use of immunosuppressant drugs, the prevalence of tinea pseudoimbricata is hypothesized to increase.1 The presence of tinea pseudoimbricata should alert dermatologists to the possible overuse of topical corticosteroids, and other forms of immunosuppression also should be considered.
- Lim SP, Smith AG. “Tinea pseudoimbricata”: tinea corporis in a renal transplant recipient mimicking the concentric rings of tinea imbricata. Clin Exp Dermatol. 2003;28:332-333.
- Batta K, Ramlogan D, Smith AG, et al. ‘Tinea indecisiva’ may mimic the concentric rings of tinea imbricata. Br J Dermatol. 2002;147:384.
- Mask-Bull L, Patel R, Tarbox MB. America’s first case of tinea pseudoimbricata. Am J Dermatol Venereol. 2015;4:15-17.
- Meena M, Mittal A. Tinea pseudo-imbricata. J Assoc Physicians India. 2018;66:79.
- Hay RJ, Reid S, Talwat E, et al. Immune responses of patients with tinea imbricata. Br J Dermatol. 1983;108:581-586.
- Dahl MV. Suppression of immunity and inflammation by products produced by dermatophytes. J Am Acad Dermatol. 1993;28(5 pt 1):S19-S23.
- Blutfield MS, Lohre JM, Pawich DA, et al. The immunologic response to Trichophyton rubrum in lower extremity fungal infections. J Fungi (Basel). 2015;1:130-137.
- De Hoog S, Monod M, Dawson T, et al. Skin fungi from colonization to infection [published online July 2017]. Microbiol Spectr. doi:10.1128/ microbiolspec.FUNK-0049-2016
- Lo Schiavo A, Ruocco E, Russo T, et al. Locus minoris resistentiae: an old but still valid way of thinking in medicine. Clin Dermatol. 2014;32:553-556.
- Ponsonby AL, Lucas RM, van der Mei IA. UVR, vitamin D and three autoimmune diseases—multiple sclerosis, type 1 diabetes, rheumatoid arthritis. Photochem Photobiol. 2005;81:1267-1275.
- Yazdani Abyaneh M, Griffith RD, Falto-Aizpurua L, et al. Narrowband ultraviolet B phototherapy in combination with other therapies for vitiligo: mechanisms and efficacies. J Eur Acad Dermatol Venereol. 2014;28:1610-1622.
- Lim SP, Smith AG. “Tinea pseudoimbricata”: tinea corporis in a renal transplant recipient mimicking the concentric rings of tinea imbricata. Clin Exp Dermatol. 2003;28:332-333.
- Batta K, Ramlogan D, Smith AG, et al. ‘Tinea indecisiva’ may mimic the concentric rings of tinea imbricata. Br J Dermatol. 2002;147:384.
- Mask-Bull L, Patel R, Tarbox MB. America’s first case of tinea pseudoimbricata. Am J Dermatol Venereol. 2015;4:15-17.
- Meena M, Mittal A. Tinea pseudo-imbricata. J Assoc Physicians India. 2018;66:79.
- Hay RJ, Reid S, Talwat E, et al. Immune responses of patients with tinea imbricata. Br J Dermatol. 1983;108:581-586.
- Dahl MV. Suppression of immunity and inflammation by products produced by dermatophytes. J Am Acad Dermatol. 1993;28(5 pt 1):S19-S23.
- Blutfield MS, Lohre JM, Pawich DA, et al. The immunologic response to Trichophyton rubrum in lower extremity fungal infections. J Fungi (Basel). 2015;1:130-137.
- De Hoog S, Monod M, Dawson T, et al. Skin fungi from colonization to infection [published online July 2017]. Microbiol Spectr. doi:10.1128/ microbiolspec.FUNK-0049-2016
- Lo Schiavo A, Ruocco E, Russo T, et al. Locus minoris resistentiae: an old but still valid way of thinking in medicine. Clin Dermatol. 2014;32:553-556.
- Ponsonby AL, Lucas RM, van der Mei IA. UVR, vitamin D and three autoimmune diseases—multiple sclerosis, type 1 diabetes, rheumatoid arthritis. Photochem Photobiol. 2005;81:1267-1275.
- Yazdani Abyaneh M, Griffith RD, Falto-Aizpurua L, et al. Narrowband ultraviolet B phototherapy in combination with other therapies for vitiligo: mechanisms and efficacies. J Eur Acad Dermatol Venereol. 2014;28:1610-1622.
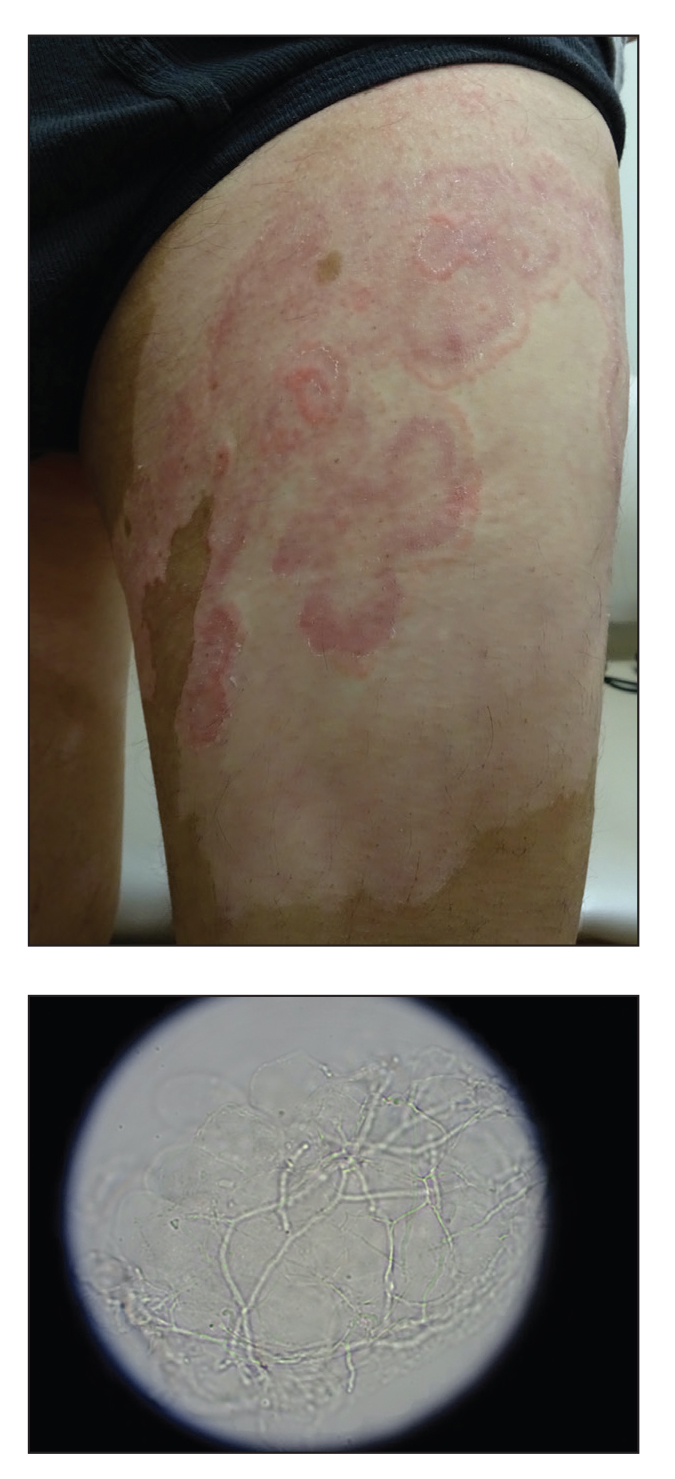
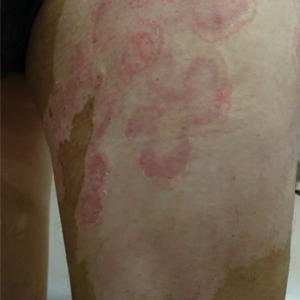
A 64-year-old man presented with generalized vitiligo. In addition to extensive depigmented macules, physical examination revealed the presence of onychomycosis and tinea corporis confirmed by microscopic examination of potassium hydroxide–treated superficial skin scrapings. Vitiligo treatment was postponed, and a 3-month course of oral terbinafine and naftifine cream was undertaken for the dermatophyte infections. Subsequent examination revealed that the patient’s tinea corporis had improved, though there were localized areas of persistence. Given the patient’s eagerness to treat his vitiligo, narrowband UVB phototherapy was started along with tolnaftate cream 1% for treatment of the residual tinea corporis. After 2 months of narrowband UVB, partial repigmentation of the vitiligo was observed; however, he had developed extensive pink, scaly, annular plaques in concentric rings within residual vitiliginous patches on the lower extremities (top). Repeat examination of potassium hydroxide–treated skin scrapings revealed numerous hyphae (bottom). A fungal culture identified Trichophyton rubrum.
Widespread Hyperkeratotic Papules in a Transplant Recipient
The Diagnosis: Trichodysplasia Spinulosa
Trichodysplasia spinulosa has been described in case reports over the last several decades, with its causative virus trichodysplasia spinulosa-associated polyomavirus (TSPyV) identified in 2010 by van der Meijden et al.1 Trichodysplasia spinulosa-associated polyomavirus is a small, nonenveloped, double-stranded DNA virus in the Polyomaviridae family, among several other known cutaneous polyomaviruses including Merkel cell polyomavirus, human polyomavirus (HPyV) 6, HPyV7, HPyV10, and possibly HPyV13.2 The primary target of TSPyV is follicular keratinocytes, and it is believed to cause trichodysplasia spinulosa by primary infection rather than by reactivation. Trichodysplasia spinulosa presents in immunosuppressed patients as a folliculocentric eruption of papules with keratinous spines on the face, often with concurrent alopecia, eventually spreading to the trunk and extremities.3 The diagnosis often is clinical, but a biopsy may be performed for histopathologic confirmation. Alternatively, lesional spicules can be painlessly collected manually and submitted for viral polymerase chain reaction (PCR).4 The diagnosis of trichodysplasia spinulosa can be difficult due to similarities with other more common conditions such as keratosis pilaris, milia, filiform warts, or lichen spinulosus.
Similar to trichodysplasia spinulosa, keratosis pilaris also presents with folliculocentric and often erythematous papules.5 Keratosis pilaris most frequently affects the posterior upper arms and thighs but also may affect the cheeks, as seen in trichodysplasia spinulosa. Differentiation between the 2 diagnoses can be made on a clinical basis, as keratosis pilaris lacks the characteristic keratinous spines and often spares the central face and nose, locations that commonly are affected in trichodysplasia spinulosa.3
Milia typically appear as white to yellow papules, often on the cheeks, eyelids, nose, and chin.6 Given their predilection for the face, milia can appear similarly to trichodysplasia spinulosa. Differentiation can be made clinically, as milia typically are not as numerous as the spiculed papules seen in trichodysplasia spinulosa. Morphologically, milia will present as smooth, dome-shaped papules as opposed to the keratinous spicules seen in trichodysplasia spinulosa. The diagnosis of milia can be confirmed by incision and removal of the white chalky keratin core, a feature absent in trichodysplasia spinulosa.
Filiform warts are benign epidermal proliferations caused by human papillomavirus infection that manifest as flesh-colored, verrucous, hyperkeratotic papules.7 They can appear on virtually any skin surface, including the face, and thus may be mistaken for trichodysplasia spinulosa. Close inspection usually will reveal tiny black dots that represent thrombosed capillaries, a feature lacking in trichodysplasia spinulosa. In long-standing lesions or immunocompromised patients, confluent verrucous plaques may develop.8 Diagnosis of filiform warts can be confirmed with biopsy, which will demonstrate a compact stratum corneum, coarse hypergranulosis, and papillomatosis curving inward, while biopsy of a trichodysplasia spinulosa lesion would show polyomavirus infection of the hair follicle and characteristic eosinophilic inclusion bodies.9
Lichen spinulosus may appear as multiple folliculocentric scaly papules with hairlike horny spines.10 Lichen spinulosus differs from trichodysplasia spinulosa in that it commonly appears on the neck, abdomen, trochanteric region, arms, elbows, or knees. Lichen spinulosus also classically appears as a concrete cluster of papules, often localized to a certain region, in contrast to trichodysplasia spinulosa, which will be widespread, often spreading over time. Finally, clinical history may help differentiate the 2 entities. Lichen spinulosus most often appears in children and adolescents and often has an indolent course, typically resolving during puberty, while trichodysplasia spinulosa is seen in immunocompromised patients.
In our patient, the dermatology team made a diagnosis of trichodysplasia spinulosa based on the characteristic clinical presentation, which was confirmed after approximately 10 lesional spicules were removed by tissue forceps and submitted for PCR analysis showing TSPyV (Figure). Two other cases utilized spicule PCR analysis for confirmation of TSPyV.11,12 This technique may represent a viable option for diagnostic confirmation in pediatric cases.
Although some articles have examined the molecular and biologic features of trichodysplasia spinulosa, literature on clinical presentation and management is limited to isolated case reports with no comprehensive studies to establish a standardized treatment. Of these reports, oral valganciclovir 900 mg daily, topical retinoids, cidofovir cream 1% to 3%, and decreasing or altering the immunosuppressive regimen all have been noted to provide clinical improvement.13,14 Other therapies including leflunomide and routine manual extraction of spicules also have shown effectiveness in the treatment of trichodysplasia spinulosa.15
In our patient, treatment included decreasing immunosuppression, as she was getting recurrent sinus and upper respiratory infections. Mycophenolate mofetil was discontinued, and the patient was continued solely on tacrolimus therapy. She demonstrated notable improvement after 3 months, with approximately 50% clearance of the eruption. A mutual decision was made at that visit to initiate therapy with compounded cidofovir cream 1% daily to the lesions until the next follow-up visit. Unfortunately, the patient did not return for her scheduled dermatology visits and was lost to long-term follow-up.
Acknowledgment
We thank Richard C. Wang, MD, PhD (Dallas, Texas), for his dermatologic expertise and assistance in analysis of lesional samples for TSPyV.
- van der Meijden E, Janssens RWA, Lauber C, et al. Discovery of a new human polyomavirus associated with trichodysplasia spinulosa in an immunocompromised patient. PLoS Pathog. 2010;6:E1001024.
- Sheu JC, Tran J, Rady PL, et al. Polyomaviruses of the skin: integrating molecular and clinical advances in an emerging class of viruses. Br J Dermatol. 2019;180:1302-1311.
- Sperling LC, Tomaszewski MM, Thomas DA. Viral-associated trichodysplasia in patients who are immunocompromised. J Am Acad Dermatol. 2004;50:318-322.
- Wu JH, Nguyen HP, Rady PL, et al. Molecular insight into the viral biology and clinical features of trichodysplasia spinulosa. Br J Dermatol. 2016;174:490-498.
- Hwang S, Schwartz RA. Keratosis pilaris: a common follicular hyperkeratosis. Cutis. 2008;82:177-180.
- Berk DR, Bayliss SJ. Milia: a review and classification. J Am Acad Dermatol. 2008;59:1050-1063.
- Micali G, Dall'Oglio F, Nasca MR, et al. Management of cutaneous warts: an evidence-based approach. Am J Clin Dermatol. 2004;5:311-317.
- Bolognia J, Schaffer JV, Cerroni L. Dermatology. 4th ed. Elsevier; 2018.
- Elston DM, Ferringer T, Ko CJ. Dermatopathology. 3rd ed. Elsevier; 2018.
- Tilly JJ, Drolet BA, Esterly NB. Lichenoid eruptions in children. J Am Acad Dermatol. 2004;51:606-624.
- Chamseddin BH, Tran BAPD, Lee EE, et al. Trichodysplasia spinulosa in a child: identification of trichodysplasia spinulosa-associated polyomavirus in skin, serum, and urine. Pediatr Dermatol. 2019;36:723-724.
- Sonstegard A, Grossman M, Garg A. Trichodysplasia spinulosa in a kidney transplant recipient. JAMA Dermatol. 2021;157:105.
- Leitenberger JJ, Abdelmalek M, Wang RC, et al. Two cases of trichodysplasia spinulosa responsive to compounded topical cidofovir 3% cream. JAAD Case Rep. 2015;1:S33-S35.
- DeCrescenzo AJ, Philips RC, Wilkerson MG. Trichodysplasia spinulosa: a rare complication of immunosuppression. JAAD Case Rep. 2016;2:307-309.
- Nguyen KD, Chamseddin BH, Cockerell CJ, et al. The biology and clinical features of cutaneous polyomaviruses. J Invest Dermatol. 2019;139:285-292.
The Diagnosis: Trichodysplasia Spinulosa
Trichodysplasia spinulosa has been described in case reports over the last several decades, with its causative virus trichodysplasia spinulosa-associated polyomavirus (TSPyV) identified in 2010 by van der Meijden et al.1 Trichodysplasia spinulosa-associated polyomavirus is a small, nonenveloped, double-stranded DNA virus in the Polyomaviridae family, among several other known cutaneous polyomaviruses including Merkel cell polyomavirus, human polyomavirus (HPyV) 6, HPyV7, HPyV10, and possibly HPyV13.2 The primary target of TSPyV is follicular keratinocytes, and it is believed to cause trichodysplasia spinulosa by primary infection rather than by reactivation. Trichodysplasia spinulosa presents in immunosuppressed patients as a folliculocentric eruption of papules with keratinous spines on the face, often with concurrent alopecia, eventually spreading to the trunk and extremities.3 The diagnosis often is clinical, but a biopsy may be performed for histopathologic confirmation. Alternatively, lesional spicules can be painlessly collected manually and submitted for viral polymerase chain reaction (PCR).4 The diagnosis of trichodysplasia spinulosa can be difficult due to similarities with other more common conditions such as keratosis pilaris, milia, filiform warts, or lichen spinulosus.
Similar to trichodysplasia spinulosa, keratosis pilaris also presents with folliculocentric and often erythematous papules.5 Keratosis pilaris most frequently affects the posterior upper arms and thighs but also may affect the cheeks, as seen in trichodysplasia spinulosa. Differentiation between the 2 diagnoses can be made on a clinical basis, as keratosis pilaris lacks the characteristic keratinous spines and often spares the central face and nose, locations that commonly are affected in trichodysplasia spinulosa.3
Milia typically appear as white to yellow papules, often on the cheeks, eyelids, nose, and chin.6 Given their predilection for the face, milia can appear similarly to trichodysplasia spinulosa. Differentiation can be made clinically, as milia typically are not as numerous as the spiculed papules seen in trichodysplasia spinulosa. Morphologically, milia will present as smooth, dome-shaped papules as opposed to the keratinous spicules seen in trichodysplasia spinulosa. The diagnosis of milia can be confirmed by incision and removal of the white chalky keratin core, a feature absent in trichodysplasia spinulosa.
Filiform warts are benign epidermal proliferations caused by human papillomavirus infection that manifest as flesh-colored, verrucous, hyperkeratotic papules.7 They can appear on virtually any skin surface, including the face, and thus may be mistaken for trichodysplasia spinulosa. Close inspection usually will reveal tiny black dots that represent thrombosed capillaries, a feature lacking in trichodysplasia spinulosa. In long-standing lesions or immunocompromised patients, confluent verrucous plaques may develop.8 Diagnosis of filiform warts can be confirmed with biopsy, which will demonstrate a compact stratum corneum, coarse hypergranulosis, and papillomatosis curving inward, while biopsy of a trichodysplasia spinulosa lesion would show polyomavirus infection of the hair follicle and characteristic eosinophilic inclusion bodies.9
Lichen spinulosus may appear as multiple folliculocentric scaly papules with hairlike horny spines.10 Lichen spinulosus differs from trichodysplasia spinulosa in that it commonly appears on the neck, abdomen, trochanteric region, arms, elbows, or knees. Lichen spinulosus also classically appears as a concrete cluster of papules, often localized to a certain region, in contrast to trichodysplasia spinulosa, which will be widespread, often spreading over time. Finally, clinical history may help differentiate the 2 entities. Lichen spinulosus most often appears in children and adolescents and often has an indolent course, typically resolving during puberty, while trichodysplasia spinulosa is seen in immunocompromised patients.
In our patient, the dermatology team made a diagnosis of trichodysplasia spinulosa based on the characteristic clinical presentation, which was confirmed after approximately 10 lesional spicules were removed by tissue forceps and submitted for PCR analysis showing TSPyV (Figure). Two other cases utilized spicule PCR analysis for confirmation of TSPyV.11,12 This technique may represent a viable option for diagnostic confirmation in pediatric cases.
Although some articles have examined the molecular and biologic features of trichodysplasia spinulosa, literature on clinical presentation and management is limited to isolated case reports with no comprehensive studies to establish a standardized treatment. Of these reports, oral valganciclovir 900 mg daily, topical retinoids, cidofovir cream 1% to 3%, and decreasing or altering the immunosuppressive regimen all have been noted to provide clinical improvement.13,14 Other therapies including leflunomide and routine manual extraction of spicules also have shown effectiveness in the treatment of trichodysplasia spinulosa.15
In our patient, treatment included decreasing immunosuppression, as she was getting recurrent sinus and upper respiratory infections. Mycophenolate mofetil was discontinued, and the patient was continued solely on tacrolimus therapy. She demonstrated notable improvement after 3 months, with approximately 50% clearance of the eruption. A mutual decision was made at that visit to initiate therapy with compounded cidofovir cream 1% daily to the lesions until the next follow-up visit. Unfortunately, the patient did not return for her scheduled dermatology visits and was lost to long-term follow-up.
Acknowledgment
We thank Richard C. Wang, MD, PhD (Dallas, Texas), for his dermatologic expertise and assistance in analysis of lesional samples for TSPyV.
The Diagnosis: Trichodysplasia Spinulosa
Trichodysplasia spinulosa has been described in case reports over the last several decades, with its causative virus trichodysplasia spinulosa-associated polyomavirus (TSPyV) identified in 2010 by van der Meijden et al.1 Trichodysplasia spinulosa-associated polyomavirus is a small, nonenveloped, double-stranded DNA virus in the Polyomaviridae family, among several other known cutaneous polyomaviruses including Merkel cell polyomavirus, human polyomavirus (HPyV) 6, HPyV7, HPyV10, and possibly HPyV13.2 The primary target of TSPyV is follicular keratinocytes, and it is believed to cause trichodysplasia spinulosa by primary infection rather than by reactivation. Trichodysplasia spinulosa presents in immunosuppressed patients as a folliculocentric eruption of papules with keratinous spines on the face, often with concurrent alopecia, eventually spreading to the trunk and extremities.3 The diagnosis often is clinical, but a biopsy may be performed for histopathologic confirmation. Alternatively, lesional spicules can be painlessly collected manually and submitted for viral polymerase chain reaction (PCR).4 The diagnosis of trichodysplasia spinulosa can be difficult due to similarities with other more common conditions such as keratosis pilaris, milia, filiform warts, or lichen spinulosus.
Similar to trichodysplasia spinulosa, keratosis pilaris also presents with folliculocentric and often erythematous papules.5 Keratosis pilaris most frequently affects the posterior upper arms and thighs but also may affect the cheeks, as seen in trichodysplasia spinulosa. Differentiation between the 2 diagnoses can be made on a clinical basis, as keratosis pilaris lacks the characteristic keratinous spines and often spares the central face and nose, locations that commonly are affected in trichodysplasia spinulosa.3
Milia typically appear as white to yellow papules, often on the cheeks, eyelids, nose, and chin.6 Given their predilection for the face, milia can appear similarly to trichodysplasia spinulosa. Differentiation can be made clinically, as milia typically are not as numerous as the spiculed papules seen in trichodysplasia spinulosa. Morphologically, milia will present as smooth, dome-shaped papules as opposed to the keratinous spicules seen in trichodysplasia spinulosa. The diagnosis of milia can be confirmed by incision and removal of the white chalky keratin core, a feature absent in trichodysplasia spinulosa.
Filiform warts are benign epidermal proliferations caused by human papillomavirus infection that manifest as flesh-colored, verrucous, hyperkeratotic papules.7 They can appear on virtually any skin surface, including the face, and thus may be mistaken for trichodysplasia spinulosa. Close inspection usually will reveal tiny black dots that represent thrombosed capillaries, a feature lacking in trichodysplasia spinulosa. In long-standing lesions or immunocompromised patients, confluent verrucous plaques may develop.8 Diagnosis of filiform warts can be confirmed with biopsy, which will demonstrate a compact stratum corneum, coarse hypergranulosis, and papillomatosis curving inward, while biopsy of a trichodysplasia spinulosa lesion would show polyomavirus infection of the hair follicle and characteristic eosinophilic inclusion bodies.9
Lichen spinulosus may appear as multiple folliculocentric scaly papules with hairlike horny spines.10 Lichen spinulosus differs from trichodysplasia spinulosa in that it commonly appears on the neck, abdomen, trochanteric region, arms, elbows, or knees. Lichen spinulosus also classically appears as a concrete cluster of papules, often localized to a certain region, in contrast to trichodysplasia spinulosa, which will be widespread, often spreading over time. Finally, clinical history may help differentiate the 2 entities. Lichen spinulosus most often appears in children and adolescents and often has an indolent course, typically resolving during puberty, while trichodysplasia spinulosa is seen in immunocompromised patients.
In our patient, the dermatology team made a diagnosis of trichodysplasia spinulosa based on the characteristic clinical presentation, which was confirmed after approximately 10 lesional spicules were removed by tissue forceps and submitted for PCR analysis showing TSPyV (Figure). Two other cases utilized spicule PCR analysis for confirmation of TSPyV.11,12 This technique may represent a viable option for diagnostic confirmation in pediatric cases.
Although some articles have examined the molecular and biologic features of trichodysplasia spinulosa, literature on clinical presentation and management is limited to isolated case reports with no comprehensive studies to establish a standardized treatment. Of these reports, oral valganciclovir 900 mg daily, topical retinoids, cidofovir cream 1% to 3%, and decreasing or altering the immunosuppressive regimen all have been noted to provide clinical improvement.13,14 Other therapies including leflunomide and routine manual extraction of spicules also have shown effectiveness in the treatment of trichodysplasia spinulosa.15
In our patient, treatment included decreasing immunosuppression, as she was getting recurrent sinus and upper respiratory infections. Mycophenolate mofetil was discontinued, and the patient was continued solely on tacrolimus therapy. She demonstrated notable improvement after 3 months, with approximately 50% clearance of the eruption. A mutual decision was made at that visit to initiate therapy with compounded cidofovir cream 1% daily to the lesions until the next follow-up visit. Unfortunately, the patient did not return for her scheduled dermatology visits and was lost to long-term follow-up.
Acknowledgment
We thank Richard C. Wang, MD, PhD (Dallas, Texas), for his dermatologic expertise and assistance in analysis of lesional samples for TSPyV.
- van der Meijden E, Janssens RWA, Lauber C, et al. Discovery of a new human polyomavirus associated with trichodysplasia spinulosa in an immunocompromised patient. PLoS Pathog. 2010;6:E1001024.
- Sheu JC, Tran J, Rady PL, et al. Polyomaviruses of the skin: integrating molecular and clinical advances in an emerging class of viruses. Br J Dermatol. 2019;180:1302-1311.
- Sperling LC, Tomaszewski MM, Thomas DA. Viral-associated trichodysplasia in patients who are immunocompromised. J Am Acad Dermatol. 2004;50:318-322.
- Wu JH, Nguyen HP, Rady PL, et al. Molecular insight into the viral biology and clinical features of trichodysplasia spinulosa. Br J Dermatol. 2016;174:490-498.
- Hwang S, Schwartz RA. Keratosis pilaris: a common follicular hyperkeratosis. Cutis. 2008;82:177-180.
- Berk DR, Bayliss SJ. Milia: a review and classification. J Am Acad Dermatol. 2008;59:1050-1063.
- Micali G, Dall'Oglio F, Nasca MR, et al. Management of cutaneous warts: an evidence-based approach. Am J Clin Dermatol. 2004;5:311-317.
- Bolognia J, Schaffer JV, Cerroni L. Dermatology. 4th ed. Elsevier; 2018.
- Elston DM, Ferringer T, Ko CJ. Dermatopathology. 3rd ed. Elsevier; 2018.
- Tilly JJ, Drolet BA, Esterly NB. Lichenoid eruptions in children. J Am Acad Dermatol. 2004;51:606-624.
- Chamseddin BH, Tran BAPD, Lee EE, et al. Trichodysplasia spinulosa in a child: identification of trichodysplasia spinulosa-associated polyomavirus in skin, serum, and urine. Pediatr Dermatol. 2019;36:723-724.
- Sonstegard A, Grossman M, Garg A. Trichodysplasia spinulosa in a kidney transplant recipient. JAMA Dermatol. 2021;157:105.
- Leitenberger JJ, Abdelmalek M, Wang RC, et al. Two cases of trichodysplasia spinulosa responsive to compounded topical cidofovir 3% cream. JAAD Case Rep. 2015;1:S33-S35.
- DeCrescenzo AJ, Philips RC, Wilkerson MG. Trichodysplasia spinulosa: a rare complication of immunosuppression. JAAD Case Rep. 2016;2:307-309.
- Nguyen KD, Chamseddin BH, Cockerell CJ, et al. The biology and clinical features of cutaneous polyomaviruses. J Invest Dermatol. 2019;139:285-292.
- van der Meijden E, Janssens RWA, Lauber C, et al. Discovery of a new human polyomavirus associated with trichodysplasia spinulosa in an immunocompromised patient. PLoS Pathog. 2010;6:E1001024.
- Sheu JC, Tran J, Rady PL, et al. Polyomaviruses of the skin: integrating molecular and clinical advances in an emerging class of viruses. Br J Dermatol. 2019;180:1302-1311.
- Sperling LC, Tomaszewski MM, Thomas DA. Viral-associated trichodysplasia in patients who are immunocompromised. J Am Acad Dermatol. 2004;50:318-322.
- Wu JH, Nguyen HP, Rady PL, et al. Molecular insight into the viral biology and clinical features of trichodysplasia spinulosa. Br J Dermatol. 2016;174:490-498.
- Hwang S, Schwartz RA. Keratosis pilaris: a common follicular hyperkeratosis. Cutis. 2008;82:177-180.
- Berk DR, Bayliss SJ. Milia: a review and classification. J Am Acad Dermatol. 2008;59:1050-1063.
- Micali G, Dall'Oglio F, Nasca MR, et al. Management of cutaneous warts: an evidence-based approach. Am J Clin Dermatol. 2004;5:311-317.
- Bolognia J, Schaffer JV, Cerroni L. Dermatology. 4th ed. Elsevier; 2018.
- Elston DM, Ferringer T, Ko CJ. Dermatopathology. 3rd ed. Elsevier; 2018.
- Tilly JJ, Drolet BA, Esterly NB. Lichenoid eruptions in children. J Am Acad Dermatol. 2004;51:606-624.
- Chamseddin BH, Tran BAPD, Lee EE, et al. Trichodysplasia spinulosa in a child: identification of trichodysplasia spinulosa-associated polyomavirus in skin, serum, and urine. Pediatr Dermatol. 2019;36:723-724.
- Sonstegard A, Grossman M, Garg A. Trichodysplasia spinulosa in a kidney transplant recipient. JAMA Dermatol. 2021;157:105.
- Leitenberger JJ, Abdelmalek M, Wang RC, et al. Two cases of trichodysplasia spinulosa responsive to compounded topical cidofovir 3% cream. JAAD Case Rep. 2015;1:S33-S35.
- DeCrescenzo AJ, Philips RC, Wilkerson MG. Trichodysplasia spinulosa: a rare complication of immunosuppression. JAAD Case Rep. 2016;2:307-309.
- Nguyen KD, Chamseddin BH, Cockerell CJ, et al. The biology and clinical features of cutaneous polyomaviruses. J Invest Dermatol. 2019;139:285-292.
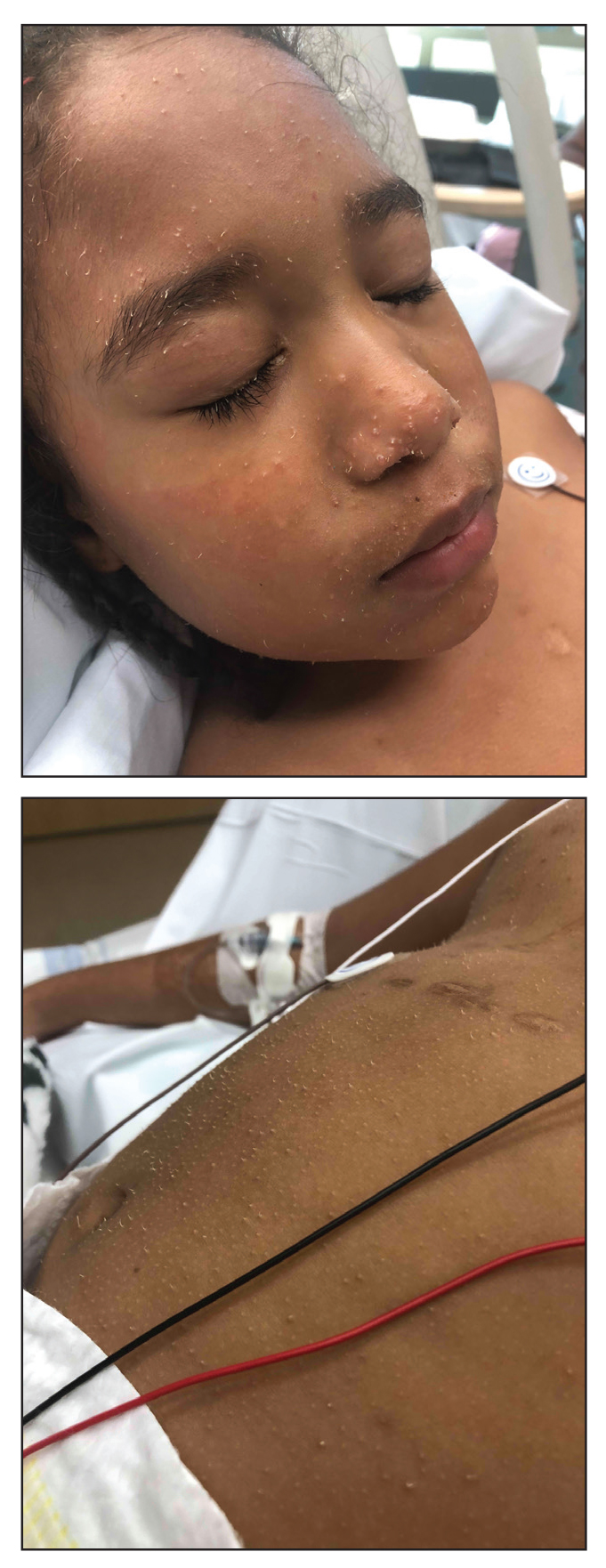
A 4-year-old girl with a history of cardiac transplantation 1 year prior for dilated cardiomyopathy presented to the dermatology consultation service with widespread hyperkeratotic papules of 2 months’ duration. The eruption initially had appeared on the face with subsequent involvement of the trunk and extremities. Her immunosuppressive medications included oral tacrolimus and mycophenolate mofetil. No over-the-counter or prescription treatments had been used for the eruption; the patient’s mother had been manually extracting the spicules from the nose, cheeks, and forehead with tweezers. The lesions were asymptomatic with only mild follicular erythema. Physical examination revealed multiple folliculocentric keratinous spicules on the nose, cheeks, forehead (top), trunk (bottom), arms, and legs.