User login
Cardiac rehabilitation: A class 1 recommendation
Cardiac rehabilitation has a class 1 indication (ie, strong recommendation) after heart surgery, myocardial infarction, or coronary intervention, and for stable angina or peripheral artery disease. It has a class 2a indication (ie, moderate recommendation) for stable systolic heart failure. Yet it is still underutilized despite its demonstrated benefits, endorsement by most recognized cardiovascular societies, and coverage by the US Centers for Medicare and Medicaid Services (CMS).
Here, we review cardiac rehabilitation—its benefits, appropriate indications, barriers to referral and enrollment, and efforts to increase its use.
EXERCISE: SLOW TO BE ADOPTED
In 1772, William Heberden (also remembered today for describing swelling of the distal interphalangeal joints in osteoarthritis) described1 a patient with angina pectoris who “set himself a task of sawing wood for half an hour every day, and was nearly cured.”
Despite early clues, it would be some time before the medical community would recognize the benefits of exercise for cardiovascular health. Before the 1930s, immobilization and extended bedrest were encouraged for up to 6 weeks after a cardiovascular event, leading to significant deconditioning.2 Things slowly began to change in the 1940s with Levine’s introduction of up-to-chair therapy,3 and short daily walks were introduced in the 1950s. Over time, the link between a sedentary lifestyle and cardiovascular disease was studied and led to greater investigation into the benefits of exercise, propelling us into the modern era.4,5
CARDIAC REHABILITATION: COMPREHENSIVE RISK REDUCTION
The American Association of Cardiovascular and Pulmonary Rehabilitation (AACVPR) defines cardiac rehabilitation as the provision of comprehensive long-term services involving medical evaluation, prescriptive exercise, cardiac risk-factor modification, education, counseling, and behavioral interventions.6 CMS defines it as a physician-supervised program that furnishes physician-prescribed exercise, cardiac risk-factor modification (including education, counseling, and behavioral intervention), psychosocial assessment, outcomes assessment, and other items and services.7
In general, most cardiac rehabilitation programs provide medically supervised exercise and patient education designed to improve cardiac health and functional status. Risk factors are targeted to reduce disability and rates of morbidity and mortality, to improve functional capacity, and to alleviate activity-related symptoms.
FROM HOSPITAL TO SELF-MAINTENANCE

Phase 1: Inpatient rehabilitation
Phase 1 typically takes place in the inpatient setting, often after open heart surgery (eg, coronary artery bypass grafting, valve repair or replacement, heart transplant), myocardial infarction, or percutaneous coronary intervention. This phase may last only a few days, especially in the current era of short hospital stays.
During phase 1, patients discuss their health situation and goals with their primary provider or cardiologist and receive education about recovery and cardiovascular risk factors. Early mobilization to prepare for discharge and to resume simple activities of daily living is emphasized. Depending on the institution, phase 1 exercise may involve simple ambulation on the ward or using equipment such as a stationary bike or treadmill.6 Phase 2 enrollment ideally is set up before discharge.
Phase 2: Limited-time outpatient rehabilitation
Phase 2 traditionally takes place in a hospital-based outpatient facility and consists of a physician-supervised multidisciplinary program. Growing evidence shows that home-based cardiac rehabilitation may be as effective as a medical facility-based program and should be an option for patients who have difficulty getting access to a traditional program.8
A phase 2 program takes a threefold approach, consisting of exercise, aggressive risk-factor modification, and education classes. A Cochrane review9 included programs that also incorporated behavioral modification and psychosocial support as a means of secondary prevention, underscoring the evolving definition of cardiac rehabilitation.
During the initial phase 2 visit, an individualized treatment plan is developed, incorporating an exercise prescription and realistic goals for secondary prevention. Sessions typically take place 3 times a week for up to 36 sessions; usually, options are available for less frequent weekly attendance for a longer period to achieve a full course. In some cases, patients may qualify for up to 72 sessions, particularly if they have not progressed as expected.
Exercise. As part of the initial evaluation, AACVPR guidelines6 suggest an exercise test—eg, a symptom-limited exercise stress test, a 6-minute walk test, or use of a Rating of Perceived Exertion scale. Prescribed exercise generally targets moderate activity in the range of 50% to 70% of peak estimated functional capacity. In the appropriate clinical context, high-functioning patients can be offered high-intensity interval training instead of moderate exercise, as they confer similar benefits.10
Risk-factor reduction. Comprehensive risk-factor reduction can address smoking, hypertension, high cholesterol, diabetes, obesity, and diet, as well as psychosocial issues such as stress, anxiety, depression, and alcohol use. Sexual activity counseling may also be included.
Education classes are aimed at helping patients understand cardiovascular disease and empowering them to manage their medical treatment and lifestyle modifications.6
Phase 3: Lifetime maintenance
In phase 3, patients independently continue risk-factor modification and physical activity without cardiac monitoring. Most cardiac rehabilitation programs offer transition-to-maintenance classes after completion of phase 2; this may be a welcome option, particularly for those who have developed a good routine and rapport with the staff and other participants. Others may opt for an independent program, using their own home equipment or a local health club.
EXERCISE: MOSTLY SAFE, WITH PROVEN BENEFITS
The safety of cardiac rehabilitation is well established, with a low risk of major cardiovascular complications. A US study in the early 1980s of 167 cardiac rehabilitation programs found 1 cardiac arrest for every 111,996 exercise hours, 1 myocardial infarction per 293,990 exercise hours, and 1 fatality per 783,972 exercise hours.11 A 2006 study of more than 65 cardiac rehabilitation centers in France found 1 cardiac event per 8,484 exercise tests and 1.3 cardiac arrests per 1 million exercise hours.12
The benefits of cardiac rehabilitation are numerous and substantial.9,13–17 A 2016 Cochrane review and meta-analysis of 63 randomized controlled trials with 14,486 participants found a reduced rate of cardiovascular mortality (relative risk [RR] 0.74, 95% confidence interval [CI] 0.64–0.86), with a number needed to treat of 37, and fewer hospital readmissions (RR 0.82, 95% CI 0.70–0.96).9
Reductions in mortality rates are dose-dependent. A study of more than 30,000 Medicare beneficiaries who participated in cardiac rehabilitation found that those who attended more sessions had a lower rate of morbidity and death at 4 years, particularly if they participated in more than 11 sessions. Those who attended the full 36 sessions had a mortality rate 47% lower than those who attended a single session.17 There was a 15% reduction in mortality for those who attended 36 sessions compared with 24 sessions, a 28% lower risk with attending 36 sessions compared with 12. After adjustment, each additional 6 sessions was associated with a 6% reduction in mortality. The curves continued to separate up to 4 years.
The benefits of cardiac rehabilitation go beyond risk reduction and include improved functional capacity, greater ease with activities of daily living, and improved quality of life.9 Patients receive structure and support from the management team and other participants, which may provide an additional layer of friendship and psychosocial support for making lifestyle changes.
Is the overall mortality rate improved?
In the modern era, with access to optimal medical therapy and drug-eluting stents, one might expect only small additional benefit from cardiac rehabilitation. The 2016 Cochrane review and meta-analysis found that although cardiac rehabilitation contributed to improved cardiovascular mortality rates and health-related quality of life, no significant reduction was detected in the rate of death from all causes.8 But the analysis did not necessarily support removing the claim of reduced all-cause mortality for cardiac rehabilitation: only randomized controlled trials were examined, and the quality of evidence for each outcome was deemed to be low to moderate because of a general paucity of reports, including many small trials that followed patients for less than 12 months.
A large cohort analysis15 with more than 73,000 patients who had undergone cardiac rehabilitation found a relative reduction in mortality rate of 58% at 1 year and 21% to 34% at 5 years, with elderly women gaining the most benefit. In the Heart Failure: A Controlled Trial Investigating Outcomes of Exercise Training (HF-ACTION) trial, with more than 2,300 patients followed for a median of 2.5 years, exercise training for heart failure was associated with reduced rates of all-cause mortality or hospitalization (HR 0.89, 95% CI 0.81–0.99; P = .03) and of cardiovascular mortality or heart failure hospitalization (HR 0.85, 95% CI 0.74–0.99; P = .03).18
Regardless of the precise reduction in all-cause mortality, the cardiovascular and health-quality outcomes of cardiac rehabilitation clearly indicate benefit. More trials with follow-up longer than 1 year are needed to definitively determine the impact of cardiac rehabilitation on the all-cause mortality rate.
WHO SHOULD BE OFFERED CARDIAC REHABILITATION?
The 2006 CMS coverage criteria listed the indications for cardiac rehabilitation as myocardial infarction within the preceding 12 months, coronary artery bypass surgery, stable angina pectoris, heart valve repair or replacement, percutaneous coronary intervention, and heart or heart-lung transplant.

In 2014, stable chronic systolic heart failure was added to the list (Table 2). Qualifications include New York Heart Association class II (mild symptoms, slight limitation of activity) to class IV (severe limitations, symptoms at rest), an ejection fraction of 35% or less, and being stable on optimal medical therapy for at least 6 weeks.
In 2017, CMS approved supervised exercise therapy for peripheral arterial disease. Supervised exercise has a class 1 recommendation by the American Heart Association and American College of Cardiology for treating intermittent claudication. Supervised exercise therapy can increase walking distance by 180% and is superior to medical therapy alone. Unsupervised exercise has a class 2b recommendation.19,20
Other patients may not qualify for phase 2 cardiac rehabilitation according to CMS or private insurance but could benefit from an exercise prescription and enrollment in a local phase 3 or home exercise program. Indications might include diabetes, obesity, metabolic syndrome, atrial fibrillation, postural orthostatic tachycardia syndrome, and nonalcoholic steatohepatitis. The benefits of cardiac rehabilitation after newer, less-invasive procedures for transcatheter valve repair and replacement are not well established, and more research is needed in this area.
WHEN TO REFER
Ades et al have defined cardiac rehabilitation referral as a combination of electronic medical records order, patient-physician discussion, and receipt of an order by a cardiac rehabilitation program.21
Ideally, referral for outpatient cardiac rehabilitation should take place at the time of hospital discharge. The AACVPR endorses a “cardiovascular continuum of care” model that emphasizes a smooth transition from inpatient to outpatient programs.6 Inpatient referral is a strong predictor of cardiac rehabilitation enrollment, and lack of referral in phase 1 negatively affects enrollment rates.
Depending on the diagnosis, US and Canadian guidelines recommend cardiac rehabilitation starting within 1 to 4 weeks of the index event, with acceptable wait times up to 60 days.6,22 In the United Kingdom, referral is recommended within 24 hours of patient eligibility; assessment for a cardiovascular prevention and rehabilitation program, with a defined pathway and individual goals, is expected to be completed within 10 working days of referral.23 Such a standard is difficult to meet in the United States, where the time from hospital discharge to cardiac rehabilitation program enrollment averages 35 days.24,25
After an uncomplicated myocardial infarction or percutaneous coronary intervention, patients with a normal or mildly reduced left ventricular ejection fraction should start outpatient cardiac rehabilitation within 14 days of the index event. For such cases, cardiac rehabilitation has been shown to be safe within 1 to 2 weeks of hospital discharge and is associated with increased participation rates.

REHABILITATION IS STILL UNDERUSED
Despite its significant benefits, cardiac rehabilitation is underused for many reasons.
Referral rates vary
A study using the 1997 Medicare claims database showed national referral rates of only 14% after myocardial infarction and 31% after coronary artery bypass grafting.31

A later study using the National Cardiovascular Data Registry between 2009 and 2017 found that the situation had improved, with a referral rate of about 60% for patients undergoing percutaneous coronary intervention.32 Nevertheless, referral rates for cardiac rehabilitation remain highly variable and still lag behind other CMS quality measures for optimal medical therapy after acute myocardial infarction (Figure 1). Factors associated with higher referral rates included ST-segment elevation myocardial infarction, non-ST-segment elevation myocardial infarction, care in a high-volume center for percutaneous coronary intervention, and care in a private or community hospital in a Midwestern state. Small Midwestern hospitals generally had referral rates of over 80%, while major teaching hospitals and hospital systems on the East Coast and the West Coast had referral rates of less than 20%. Unlike some studies, this study found that insurance status had little bearing on referral rates.
Other studies found lower referral rates for women and patients with comorbidities such as previous coronary artery bypass grafting, diabetes, and heart failure.33,34
In the United Kingdom, patients with heart failure made up only 5% of patients in cardiac rehabilitation; only 7% to 20% of patients with a heart failure diagnosis were referred to cardiac rehabilitation from general and cardiology wards.35
Enrollment, completion rates even lower
Rates of referral for cardiac rehabilitation do not equate to rates of enrollment or participation. Enrollment was 50% in the United Kingdom in 2016.35 A 2015 US study evaluated 58,269 older patients eligible for cardiac rehabilitation after acute myocardial infarction; 62% were referred for cardiac rehabilitation at the time of discharge, but only 23% of the total attended at least 1 session, and just 5% of the total completed 36 or more sessions.36
BARRIERS, OPPORTUNITIES TO IMPROVE
The underuse of cardiac rehabilitation in the United States has led to an American Heart Association presidential advisory on the referral, enrollment, and delivery of cardiac rehabilitation.34 Dozens of barriers are mentioned, with several standing out as having the largest impact: lack of physician referral, weak endorsement by the prescribing provider, female sex of patients, lack of program availability, work-related hardship, low socioeconomic status, and lack of or limited healthcare insurance. Copayments have also become a major barrier, often ranging from $20 to $40 per session for patients with Medicare.
The Million Hearts Initiative has established a goal of 70% cardiac rehabilitation compliance for eligible patients by 2022, a goal they estimate could save 25,000 lives and prevent 180,000 hospitalizations annually.21
Lack of physician awareness and lack of referral may be the most modifiable factors with the capacity to have the largest impact. Increasing physician awareness is a top priority not only for primary care providers, but also for cardiologists. In 2014, CMS made referral for cardiac rehabilitation a quality measure that is trackable and reportable. CMS has also proposed models that would incentivize participation by increasing reimbursement for services provided, but these models have been halted.
Additional efforts to increase cardiac rehabilitation referral and participation include automated order sets, increased caregiver education, and early morning or late evening classes, single-sex classes, home or mobile-based exercise programs, and parking and transportation assistance.34 Grace et al37 reported that referral rates rose to 86% when a cardiac rehabilitation order was integrated into the electronic medical record and combined with a hospital liaison to educate patients about their need for cardiac rehabilitation. Lowering patient copayments would also be a good idea. We have recently seen some creative ways to reduce copayments, including philanthropy and grants.
- Herberden W. Classics in cardiology: description of angina pectoris by William Herberden. Heart Views 2006; 7(3):118–119. www.heartviews.org/text.asp?2006/7/3/118/63927. Accessed May 9, 2018.
- Mampuya WM. Cardiac rehabilitation past, present and future: an overview. Cardiovasc Diagn Ther 2012; 2(1):38–49. doi:10.3978/j.issn.2223-3652.2012.01.02
- Levine SA, Lown B. The “chair” treatment of acute thrombosis. Trans Assoc Am Physicians 1951; 64:316–327. pmid:14884265
- Morris JN, Everitt MG, Pollard R, Chave SP, Semmence AM. Vigorous exercise in leisure-time: protection against coronary heart disease. Lancet 1980; 2(8206):207–210. pmid:6108391
- Morris JN, Heady JA. Mortality in relation to the physical activity of work: a preliminary note on experience in middle age. Br J Ind Med 1953; 10(4):245–254. pmid:13106231
- American Association of Cardiovascular and Pulmonary Rehabilitation. Guidelines for cardiac rehabilitation and secondary prevention programs/American Association of Cardiovascular and Pulmonary Rehabilitation. 5th ed. Champaign, IL: Human Kinetics; 2013.
- Department of Health & Human Services (DHHS); Centers for Medicare & Medicaid Services (CMS). CMS manual system. Cardiac rehabilitation and intensive cardiac rehabilitation. www.cms.gov/Regulations-and-Guidance/Guidance/Transmittals/downloads/r126bp.pdf. Accessed May 9, 2018.
- Anderson L, Sharp GA, Norton RJ, et al. Home-based versus centre-based cardiac rehabilitation. Cochrane Database Syst Rev 2017; 6:CD007130. doi:10.1002/14651858.CD007130.pub4
- Anderson L, Oldridge N, Thompson DR, et al. Exercise-based cardiac rehabilitation for coronary heart disease: Cochrane systematic review and meta-analysis. J Am Coll Cardiol 2016; 67(1):1–12. doi:10.1016/j.jacc.2015.10.044
- Guiraud T, Nigam A, Gremeaux V, Meyer P, Juneau M, Bosquet L. High-intensity interval training in cardiac rehabilitation. Sports Med 2012; 42(7):587–605. doi:10.2165/11631910-000000000-00000
- Van Camp SP, Peterson RA. Cardiovascular complications of outpatient cardiac rehabilitation programs. JAMA 1986; 256(9):1160–1163. pmid:3735650
- Pavy B, Iliou MC, Meurin P, Tabet JY, Corone S; Functional Evaluation and Cardiac Rehabilitation Working Group of the French Society of Cardiology. Safety of exercise training for cardiac patients: results of the French registry of complications during cardiac rehabilitation. Arch Intern Med 2006; 166(21):2329–2334. doi:10.1001/archinte.166.21.2329
- Shaw LW. Effects of a prescribed supervised exercise program on mortality and cardiovascular morbidity in patients after a myocardial infarction: The National Exercise and Heart Disease Project. Am J Cardiol 1981; 48(1):39–46. pmid:6972693
- Sandesara PB, Lambert CT, Gordon NF, et al. Cardiac rehabilitation and risk reduction: time to “rebrand and reinvigorate.” J Am Coll Cardiol 2015; 65(4):389–395. doi:10.1016/j.jacc.2014.10.059
- Suaya JA, Stason WB, Ades PA, Normand SL, Shepard DS. Cardiac rehabilitation and survival in older coronary patients. J Am Coll Cardiol 2009; 54(1):25–33. doi:10.1016/j.jacc.2009.01.078
- Goel K, Lennon RJ, Tilbury RT, Squires RW, Thomas RJ. Impact of cardiac rehabilitation on mortality and cardiovascular events after percutaneous coronary intervention in the community. Circulation 2011: 123(21):2344–2352. doi:10.1161/CIRCULATIONAHA.110.983536
- Hammill BG, Curtis LH, Schulman KA, Whellan DJ. Relationship between cardiac rehabilitation and long-term risks of death and myocardial infarction among elderly Medicare beneficiaries. Circulation 2010; 121(1):63–70. doi:10.1161/CIRCULATIONAHA.109.876383
- O’Connor CM, Whellan DJ, Lee KL, et al; HF-ACTION Investigators. Efficacy and safety of exercise training in patients with chronic heart failure: HF-ACTION randomized controlled trial. JAMA 2009; 301(14):1439–1450. doi:10.1001/jama.2009.454
- Hirsch A, Haskal ZJ, Hertzer NR, et al. ACC/AHA 2005 practice guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic). Circulation 2006; 113(11):463–654. doi:10.1161/CIRCULATIONAHA.106.174526
- Ambrosetti M. Advances in exercise rehabilitation for patients with lower extremity peripheral artery disease. Monaldi Arch Chest Dis 2016; 86(1–2):752. doi:10.4081/monaldi.2016.752
- Ades PA, Keteyian SJ, Wright JS, et al. Increasing cardiac rehabilitation participation from 20% to 70%: a road map from the Million Hearts Cardiac Rehabilitation Collaborative. Mayo Clin Proc 2017; 92(2):234–242. doi:10.1016/j.mayocp.2016.10.014
- Dafoe W, Arthur H, Stokes H, Morrin L, Beaton L; Canadian Cardiovascular Society Access to Care Working Group on Cardiac Rehabilitation. Universal access: but when? Treating the right patient at the right time: access to cardiac rehabilitation. Can J Cardiol 2006; 22(11):905–911. pmid:16971975
- The British Association for Cardiovascular Prevention and Rehabilitation. The BACPR standards and core components for cardiovascular disease prevention and cardiac rehabilitation 2017. www.bacpr.com/resources/6A7_BACR_Standards_and_Core_Components_2017.pdf. Accessed May 9, 2018.
- Zullo MD, Jackson LW, Whalen CC, Dolansky MA. Evaluation of the recommended core components of cardiac rehabilitation practice: an opportunity for quality improvement. J Cardiopulm Rehabil Prev 2012; 32(1):32–40. doi:10.1097/HCR.0b013e31823be0e2
- Russell KL, Holloway TM, Brum M, Caruso V, Chessex C, Grace SL. Cardiac rehabilitation wait times: effect on enrollment. J Cardiopulm Rehabil Prev 2011; 31(6):373–377. doi:10.1097/HCR.0b013e318228a32f
- Soga Y, Yokoi H, Ando K, et al. Safety of early exercise training after elective coronary stenting in patients with stable coronary artery disease. Eur J Cardiovasc Prev Rehabil 2010; 17(2):230–234. doi:10.1097/HJR.0b013e3283359c4e
- Scheinowitz M, Harpaz D. Safety of cardiac rehabilitation in a medically supervised, community-based program. Cardiology 2005; 103(3):113–117. doi:10.1159/000083433
- Goto Y, Sumida H, Ueshima K, Adachi H, Nohara R, Itoh H. Safety and implementation of exercise testing and training after coronary stenting in patients with acute myocardial infarction. Circ J 2002; 66(10):930–936. pmid:12381088
- Parker K, Stone JA, Arena R, et al. An early cardiac access clinic significantly improves cardiac rehabilitation participation and completion rates in low-risk ST-elevation myocardial infarction patients. Can J Cardiol 2011; 27(5):619–627. doi:10.1016/j.cjca.2010.12.076
- Pack QR, Mansour M, Barboza JS, et al. An early appointment to outpatient cardiac rehabilitation at hospital discharge improves attendance at orientation: a randomized, single-blind, controlled trial. Circulation 2013; 127(3):349–355. doi:10.1161/CIRCULATIONAHA.112.121996
- Suaya JA, Shepard DS, Normand SL, Ades PA, Prottas J, Stason WB. Use of cardiac rehabilitation by Medicare beneficiaries after myocardial infarction or coronary bypass surgery. Circulation 2007; 116(15):1653–1662. doi:10.1161/CIRCULATIONAHA.107.701466
- Aragam KG, Dai D, Neely ML, et al. Gaps in referral to cardiac rehabilitation of patients undergoing percutaneous coronary intervention in the United States. J Am Coll Cardiol 2015; 65(19):2079–2088. doi:10.1016/j.jacc.2015.02.063
- Bittner V, Sanderson B, Breland J, Green D. Referral patterns to a university-based cardiac rehabilitation program. Am J Cardiol 1999; 83(2):252–255, A5. pmid:10073829
- Balady GJ, Ades PA, Bittner VA, et al. Referral, enrollment, and delivery of cardiac rehabilitation/secondary prevention programs at clinical centers and beyond. A presidential advisory from the American Heart Association. Circulation 2011; 124(25):2951–2960. doi:10.1161/CIR.0b013e31823b21e2
- British Heart Foundation. The national audit of cardiac rehabilitation annual statistical report 2016. www.cardiacrehabilitation.org.uk/docs/BHF_NACR_Report_2016.pdf. Accessed April 12, 2018.
- Doll JA, Hellkamp A, Ho PM, et al. Participation in cardiac rehabilitation programs among older patients after acute myocardial infarction. JAMA Intern Med 2015; 175(10):1700–1702. doi:10.1001/jamainternmed.2015.3819
- Grace SL, Russell KL, Reid RD, et al. Cardiac Rehabilitation Care Continuity Through Automatic Referral Evaluation (CRCARE) Investigators. Effect of cardiac rehabilitation referral strategies on utilization rates: a prospective, controlled study. Arch Intern Med 2011; 171(3):235–241. doi:10.1001/archinternmed.2010.501
Cardiac rehabilitation has a class 1 indication (ie, strong recommendation) after heart surgery, myocardial infarction, or coronary intervention, and for stable angina or peripheral artery disease. It has a class 2a indication (ie, moderate recommendation) for stable systolic heart failure. Yet it is still underutilized despite its demonstrated benefits, endorsement by most recognized cardiovascular societies, and coverage by the US Centers for Medicare and Medicaid Services (CMS).
Here, we review cardiac rehabilitation—its benefits, appropriate indications, barriers to referral and enrollment, and efforts to increase its use.
EXERCISE: SLOW TO BE ADOPTED
In 1772, William Heberden (also remembered today for describing swelling of the distal interphalangeal joints in osteoarthritis) described1 a patient with angina pectoris who “set himself a task of sawing wood for half an hour every day, and was nearly cured.”
Despite early clues, it would be some time before the medical community would recognize the benefits of exercise for cardiovascular health. Before the 1930s, immobilization and extended bedrest were encouraged for up to 6 weeks after a cardiovascular event, leading to significant deconditioning.2 Things slowly began to change in the 1940s with Levine’s introduction of up-to-chair therapy,3 and short daily walks were introduced in the 1950s. Over time, the link between a sedentary lifestyle and cardiovascular disease was studied and led to greater investigation into the benefits of exercise, propelling us into the modern era.4,5
CARDIAC REHABILITATION: COMPREHENSIVE RISK REDUCTION
The American Association of Cardiovascular and Pulmonary Rehabilitation (AACVPR) defines cardiac rehabilitation as the provision of comprehensive long-term services involving medical evaluation, prescriptive exercise, cardiac risk-factor modification, education, counseling, and behavioral interventions.6 CMS defines it as a physician-supervised program that furnishes physician-prescribed exercise, cardiac risk-factor modification (including education, counseling, and behavioral intervention), psychosocial assessment, outcomes assessment, and other items and services.7
In general, most cardiac rehabilitation programs provide medically supervised exercise and patient education designed to improve cardiac health and functional status. Risk factors are targeted to reduce disability and rates of morbidity and mortality, to improve functional capacity, and to alleviate activity-related symptoms.
FROM HOSPITAL TO SELF-MAINTENANCE
Phase 1: Inpatient rehabilitation
Phase 1 typically takes place in the inpatient setting, often after open heart surgery (eg, coronary artery bypass grafting, valve repair or replacement, heart transplant), myocardial infarction, or percutaneous coronary intervention. This phase may last only a few days, especially in the current era of short hospital stays.
During phase 1, patients discuss their health situation and goals with their primary provider or cardiologist and receive education about recovery and cardiovascular risk factors. Early mobilization to prepare for discharge and to resume simple activities of daily living is emphasized. Depending on the institution, phase 1 exercise may involve simple ambulation on the ward or using equipment such as a stationary bike or treadmill.6 Phase 2 enrollment ideally is set up before discharge.
Phase 2: Limited-time outpatient rehabilitation
Phase 2 traditionally takes place in a hospital-based outpatient facility and consists of a physician-supervised multidisciplinary program. Growing evidence shows that home-based cardiac rehabilitation may be as effective as a medical facility-based program and should be an option for patients who have difficulty getting access to a traditional program.8
A phase 2 program takes a threefold approach, consisting of exercise, aggressive risk-factor modification, and education classes. A Cochrane review9 included programs that also incorporated behavioral modification and psychosocial support as a means of secondary prevention, underscoring the evolving definition of cardiac rehabilitation.
During the initial phase 2 visit, an individualized treatment plan is developed, incorporating an exercise prescription and realistic goals for secondary prevention. Sessions typically take place 3 times a week for up to 36 sessions; usually, options are available for less frequent weekly attendance for a longer period to achieve a full course. In some cases, patients may qualify for up to 72 sessions, particularly if they have not progressed as expected.
Exercise. As part of the initial evaluation, AACVPR guidelines6 suggest an exercise test—eg, a symptom-limited exercise stress test, a 6-minute walk test, or use of a Rating of Perceived Exertion scale. Prescribed exercise generally targets moderate activity in the range of 50% to 70% of peak estimated functional capacity. In the appropriate clinical context, high-functioning patients can be offered high-intensity interval training instead of moderate exercise, as they confer similar benefits.10
Risk-factor reduction. Comprehensive risk-factor reduction can address smoking, hypertension, high cholesterol, diabetes, obesity, and diet, as well as psychosocial issues such as stress, anxiety, depression, and alcohol use. Sexual activity counseling may also be included.
Education classes are aimed at helping patients understand cardiovascular disease and empowering them to manage their medical treatment and lifestyle modifications.6
Phase 3: Lifetime maintenance
In phase 3, patients independently continue risk-factor modification and physical activity without cardiac monitoring. Most cardiac rehabilitation programs offer transition-to-maintenance classes after completion of phase 2; this may be a welcome option, particularly for those who have developed a good routine and rapport with the staff and other participants. Others may opt for an independent program, using their own home equipment or a local health club.
EXERCISE: MOSTLY SAFE, WITH PROVEN BENEFITS
The safety of cardiac rehabilitation is well established, with a low risk of major cardiovascular complications. A US study in the early 1980s of 167 cardiac rehabilitation programs found 1 cardiac arrest for every 111,996 exercise hours, 1 myocardial infarction per 293,990 exercise hours, and 1 fatality per 783,972 exercise hours.11 A 2006 study of more than 65 cardiac rehabilitation centers in France found 1 cardiac event per 8,484 exercise tests and 1.3 cardiac arrests per 1 million exercise hours.12
The benefits of cardiac rehabilitation are numerous and substantial.9,13–17 A 2016 Cochrane review and meta-analysis of 63 randomized controlled trials with 14,486 participants found a reduced rate of cardiovascular mortality (relative risk [RR] 0.74, 95% confidence interval [CI] 0.64–0.86), with a number needed to treat of 37, and fewer hospital readmissions (RR 0.82, 95% CI 0.70–0.96).9
Reductions in mortality rates are dose-dependent. A study of more than 30,000 Medicare beneficiaries who participated in cardiac rehabilitation found that those who attended more sessions had a lower rate of morbidity and death at 4 years, particularly if they participated in more than 11 sessions. Those who attended the full 36 sessions had a mortality rate 47% lower than those who attended a single session.17 There was a 15% reduction in mortality for those who attended 36 sessions compared with 24 sessions, a 28% lower risk with attending 36 sessions compared with 12. After adjustment, each additional 6 sessions was associated with a 6% reduction in mortality. The curves continued to separate up to 4 years.
The benefits of cardiac rehabilitation go beyond risk reduction and include improved functional capacity, greater ease with activities of daily living, and improved quality of life.9 Patients receive structure and support from the management team and other participants, which may provide an additional layer of friendship and psychosocial support for making lifestyle changes.
Is the overall mortality rate improved?
In the modern era, with access to optimal medical therapy and drug-eluting stents, one might expect only small additional benefit from cardiac rehabilitation. The 2016 Cochrane review and meta-analysis found that although cardiac rehabilitation contributed to improved cardiovascular mortality rates and health-related quality of life, no significant reduction was detected in the rate of death from all causes.8 But the analysis did not necessarily support removing the claim of reduced all-cause mortality for cardiac rehabilitation: only randomized controlled trials were examined, and the quality of evidence for each outcome was deemed to be low to moderate because of a general paucity of reports, including many small trials that followed patients for less than 12 months.
A large cohort analysis15 with more than 73,000 patients who had undergone cardiac rehabilitation found a relative reduction in mortality rate of 58% at 1 year and 21% to 34% at 5 years, with elderly women gaining the most benefit. In the Heart Failure: A Controlled Trial Investigating Outcomes of Exercise Training (HF-ACTION) trial, with more than 2,300 patients followed for a median of 2.5 years, exercise training for heart failure was associated with reduced rates of all-cause mortality or hospitalization (HR 0.89, 95% CI 0.81–0.99; P = .03) and of cardiovascular mortality or heart failure hospitalization (HR 0.85, 95% CI 0.74–0.99; P = .03).18
Regardless of the precise reduction in all-cause mortality, the cardiovascular and health-quality outcomes of cardiac rehabilitation clearly indicate benefit. More trials with follow-up longer than 1 year are needed to definitively determine the impact of cardiac rehabilitation on the all-cause mortality rate.
WHO SHOULD BE OFFERED CARDIAC REHABILITATION?
The 2006 CMS coverage criteria listed the indications for cardiac rehabilitation as myocardial infarction within the preceding 12 months, coronary artery bypass surgery, stable angina pectoris, heart valve repair or replacement, percutaneous coronary intervention, and heart or heart-lung transplant.
In 2014, stable chronic systolic heart failure was added to the list (Table 2). Qualifications include New York Heart Association class II (mild symptoms, slight limitation of activity) to class IV (severe limitations, symptoms at rest), an ejection fraction of 35% or less, and being stable on optimal medical therapy for at least 6 weeks.
In 2017, CMS approved supervised exercise therapy for peripheral arterial disease. Supervised exercise has a class 1 recommendation by the American Heart Association and American College of Cardiology for treating intermittent claudication. Supervised exercise therapy can increase walking distance by 180% and is superior to medical therapy alone. Unsupervised exercise has a class 2b recommendation.19,20
Other patients may not qualify for phase 2 cardiac rehabilitation according to CMS or private insurance but could benefit from an exercise prescription and enrollment in a local phase 3 or home exercise program. Indications might include diabetes, obesity, metabolic syndrome, atrial fibrillation, postural orthostatic tachycardia syndrome, and nonalcoholic steatohepatitis. The benefits of cardiac rehabilitation after newer, less-invasive procedures for transcatheter valve repair and replacement are not well established, and more research is needed in this area.
WHEN TO REFER
Ades et al have defined cardiac rehabilitation referral as a combination of electronic medical records order, patient-physician discussion, and receipt of an order by a cardiac rehabilitation program.21
Ideally, referral for outpatient cardiac rehabilitation should take place at the time of hospital discharge. The AACVPR endorses a “cardiovascular continuum of care” model that emphasizes a smooth transition from inpatient to outpatient programs.6 Inpatient referral is a strong predictor of cardiac rehabilitation enrollment, and lack of referral in phase 1 negatively affects enrollment rates.
Depending on the diagnosis, US and Canadian guidelines recommend cardiac rehabilitation starting within 1 to 4 weeks of the index event, with acceptable wait times up to 60 days.6,22 In the United Kingdom, referral is recommended within 24 hours of patient eligibility; assessment for a cardiovascular prevention and rehabilitation program, with a defined pathway and individual goals, is expected to be completed within 10 working days of referral.23 Such a standard is difficult to meet in the United States, where the time from hospital discharge to cardiac rehabilitation program enrollment averages 35 days.24,25
After an uncomplicated myocardial infarction or percutaneous coronary intervention, patients with a normal or mildly reduced left ventricular ejection fraction should start outpatient cardiac rehabilitation within 14 days of the index event. For such cases, cardiac rehabilitation has been shown to be safe within 1 to 2 weeks of hospital discharge and is associated with increased participation rates.
REHABILITATION IS STILL UNDERUSED
Despite its significant benefits, cardiac rehabilitation is underused for many reasons.
Referral rates vary
A study using the 1997 Medicare claims database showed national referral rates of only 14% after myocardial infarction and 31% after coronary artery bypass grafting.31
A later study using the National Cardiovascular Data Registry between 2009 and 2017 found that the situation had improved, with a referral rate of about 60% for patients undergoing percutaneous coronary intervention.32 Nevertheless, referral rates for cardiac rehabilitation remain highly variable and still lag behind other CMS quality measures for optimal medical therapy after acute myocardial infarction (Figure 1). Factors associated with higher referral rates included ST-segment elevation myocardial infarction, non-ST-segment elevation myocardial infarction, care in a high-volume center for percutaneous coronary intervention, and care in a private or community hospital in a Midwestern state. Small Midwestern hospitals generally had referral rates of over 80%, while major teaching hospitals and hospital systems on the East Coast and the West Coast had referral rates of less than 20%. Unlike some studies, this study found that insurance status had little bearing on referral rates.
Other studies found lower referral rates for women and patients with comorbidities such as previous coronary artery bypass grafting, diabetes, and heart failure.33,34
In the United Kingdom, patients with heart failure made up only 5% of patients in cardiac rehabilitation; only 7% to 20% of patients with a heart failure diagnosis were referred to cardiac rehabilitation from general and cardiology wards.35
Enrollment, completion rates even lower
Rates of referral for cardiac rehabilitation do not equate to rates of enrollment or participation. Enrollment was 50% in the United Kingdom in 2016.35 A 2015 US study evaluated 58,269 older patients eligible for cardiac rehabilitation after acute myocardial infarction; 62% were referred for cardiac rehabilitation at the time of discharge, but only 23% of the total attended at least 1 session, and just 5% of the total completed 36 or more sessions.36
BARRIERS, OPPORTUNITIES TO IMPROVE
The underuse of cardiac rehabilitation in the United States has led to an American Heart Association presidential advisory on the referral, enrollment, and delivery of cardiac rehabilitation.34 Dozens of barriers are mentioned, with several standing out as having the largest impact: lack of physician referral, weak endorsement by the prescribing provider, female sex of patients, lack of program availability, work-related hardship, low socioeconomic status, and lack of or limited healthcare insurance. Copayments have also become a major barrier, often ranging from $20 to $40 per session for patients with Medicare.
The Million Hearts Initiative has established a goal of 70% cardiac rehabilitation compliance for eligible patients by 2022, a goal they estimate could save 25,000 lives and prevent 180,000 hospitalizations annually.21
Lack of physician awareness and lack of referral may be the most modifiable factors with the capacity to have the largest impact. Increasing physician awareness is a top priority not only for primary care providers, but also for cardiologists. In 2014, CMS made referral for cardiac rehabilitation a quality measure that is trackable and reportable. CMS has also proposed models that would incentivize participation by increasing reimbursement for services provided, but these models have been halted.
Additional efforts to increase cardiac rehabilitation referral and participation include automated order sets, increased caregiver education, and early morning or late evening classes, single-sex classes, home or mobile-based exercise programs, and parking and transportation assistance.34 Grace et al37 reported that referral rates rose to 86% when a cardiac rehabilitation order was integrated into the electronic medical record and combined with a hospital liaison to educate patients about their need for cardiac rehabilitation. Lowering patient copayments would also be a good idea. We have recently seen some creative ways to reduce copayments, including philanthropy and grants.
Cardiac rehabilitation has a class 1 indication (ie, strong recommendation) after heart surgery, myocardial infarction, or coronary intervention, and for stable angina or peripheral artery disease. It has a class 2a indication (ie, moderate recommendation) for stable systolic heart failure. Yet it is still underutilized despite its demonstrated benefits, endorsement by most recognized cardiovascular societies, and coverage by the US Centers for Medicare and Medicaid Services (CMS).
Here, we review cardiac rehabilitation—its benefits, appropriate indications, barriers to referral and enrollment, and efforts to increase its use.
EXERCISE: SLOW TO BE ADOPTED
In 1772, William Heberden (also remembered today for describing swelling of the distal interphalangeal joints in osteoarthritis) described1 a patient with angina pectoris who “set himself a task of sawing wood for half an hour every day, and was nearly cured.”
Despite early clues, it would be some time before the medical community would recognize the benefits of exercise for cardiovascular health. Before the 1930s, immobilization and extended bedrest were encouraged for up to 6 weeks after a cardiovascular event, leading to significant deconditioning.2 Things slowly began to change in the 1940s with Levine’s introduction of up-to-chair therapy,3 and short daily walks were introduced in the 1950s. Over time, the link between a sedentary lifestyle and cardiovascular disease was studied and led to greater investigation into the benefits of exercise, propelling us into the modern era.4,5
CARDIAC REHABILITATION: COMPREHENSIVE RISK REDUCTION
The American Association of Cardiovascular and Pulmonary Rehabilitation (AACVPR) defines cardiac rehabilitation as the provision of comprehensive long-term services involving medical evaluation, prescriptive exercise, cardiac risk-factor modification, education, counseling, and behavioral interventions.6 CMS defines it as a physician-supervised program that furnishes physician-prescribed exercise, cardiac risk-factor modification (including education, counseling, and behavioral intervention), psychosocial assessment, outcomes assessment, and other items and services.7
In general, most cardiac rehabilitation programs provide medically supervised exercise and patient education designed to improve cardiac health and functional status. Risk factors are targeted to reduce disability and rates of morbidity and mortality, to improve functional capacity, and to alleviate activity-related symptoms.
FROM HOSPITAL TO SELF-MAINTENANCE
Phase 1: Inpatient rehabilitation
Phase 1 typically takes place in the inpatient setting, often after open heart surgery (eg, coronary artery bypass grafting, valve repair or replacement, heart transplant), myocardial infarction, or percutaneous coronary intervention. This phase may last only a few days, especially in the current era of short hospital stays.
During phase 1, patients discuss their health situation and goals with their primary provider or cardiologist and receive education about recovery and cardiovascular risk factors. Early mobilization to prepare for discharge and to resume simple activities of daily living is emphasized. Depending on the institution, phase 1 exercise may involve simple ambulation on the ward or using equipment such as a stationary bike or treadmill.6 Phase 2 enrollment ideally is set up before discharge.
Phase 2: Limited-time outpatient rehabilitation
Phase 2 traditionally takes place in a hospital-based outpatient facility and consists of a physician-supervised multidisciplinary program. Growing evidence shows that home-based cardiac rehabilitation may be as effective as a medical facility-based program and should be an option for patients who have difficulty getting access to a traditional program.8
A phase 2 program takes a threefold approach, consisting of exercise, aggressive risk-factor modification, and education classes. A Cochrane review9 included programs that also incorporated behavioral modification and psychosocial support as a means of secondary prevention, underscoring the evolving definition of cardiac rehabilitation.
During the initial phase 2 visit, an individualized treatment plan is developed, incorporating an exercise prescription and realistic goals for secondary prevention. Sessions typically take place 3 times a week for up to 36 sessions; usually, options are available for less frequent weekly attendance for a longer period to achieve a full course. In some cases, patients may qualify for up to 72 sessions, particularly if they have not progressed as expected.
Exercise. As part of the initial evaluation, AACVPR guidelines6 suggest an exercise test—eg, a symptom-limited exercise stress test, a 6-minute walk test, or use of a Rating of Perceived Exertion scale. Prescribed exercise generally targets moderate activity in the range of 50% to 70% of peak estimated functional capacity. In the appropriate clinical context, high-functioning patients can be offered high-intensity interval training instead of moderate exercise, as they confer similar benefits.10
Risk-factor reduction. Comprehensive risk-factor reduction can address smoking, hypertension, high cholesterol, diabetes, obesity, and diet, as well as psychosocial issues such as stress, anxiety, depression, and alcohol use. Sexual activity counseling may also be included.
Education classes are aimed at helping patients understand cardiovascular disease and empowering them to manage their medical treatment and lifestyle modifications.6
Phase 3: Lifetime maintenance
In phase 3, patients independently continue risk-factor modification and physical activity without cardiac monitoring. Most cardiac rehabilitation programs offer transition-to-maintenance classes after completion of phase 2; this may be a welcome option, particularly for those who have developed a good routine and rapport with the staff and other participants. Others may opt for an independent program, using their own home equipment or a local health club.
EXERCISE: MOSTLY SAFE, WITH PROVEN BENEFITS
The safety of cardiac rehabilitation is well established, with a low risk of major cardiovascular complications. A US study in the early 1980s of 167 cardiac rehabilitation programs found 1 cardiac arrest for every 111,996 exercise hours, 1 myocardial infarction per 293,990 exercise hours, and 1 fatality per 783,972 exercise hours.11 A 2006 study of more than 65 cardiac rehabilitation centers in France found 1 cardiac event per 8,484 exercise tests and 1.3 cardiac arrests per 1 million exercise hours.12
The benefits of cardiac rehabilitation are numerous and substantial.9,13–17 A 2016 Cochrane review and meta-analysis of 63 randomized controlled trials with 14,486 participants found a reduced rate of cardiovascular mortality (relative risk [RR] 0.74, 95% confidence interval [CI] 0.64–0.86), with a number needed to treat of 37, and fewer hospital readmissions (RR 0.82, 95% CI 0.70–0.96).9
Reductions in mortality rates are dose-dependent. A study of more than 30,000 Medicare beneficiaries who participated in cardiac rehabilitation found that those who attended more sessions had a lower rate of morbidity and death at 4 years, particularly if they participated in more than 11 sessions. Those who attended the full 36 sessions had a mortality rate 47% lower than those who attended a single session.17 There was a 15% reduction in mortality for those who attended 36 sessions compared with 24 sessions, a 28% lower risk with attending 36 sessions compared with 12. After adjustment, each additional 6 sessions was associated with a 6% reduction in mortality. The curves continued to separate up to 4 years.
The benefits of cardiac rehabilitation go beyond risk reduction and include improved functional capacity, greater ease with activities of daily living, and improved quality of life.9 Patients receive structure and support from the management team and other participants, which may provide an additional layer of friendship and psychosocial support for making lifestyle changes.
Is the overall mortality rate improved?
In the modern era, with access to optimal medical therapy and drug-eluting stents, one might expect only small additional benefit from cardiac rehabilitation. The 2016 Cochrane review and meta-analysis found that although cardiac rehabilitation contributed to improved cardiovascular mortality rates and health-related quality of life, no significant reduction was detected in the rate of death from all causes.8 But the analysis did not necessarily support removing the claim of reduced all-cause mortality for cardiac rehabilitation: only randomized controlled trials were examined, and the quality of evidence for each outcome was deemed to be low to moderate because of a general paucity of reports, including many small trials that followed patients for less than 12 months.
A large cohort analysis15 with more than 73,000 patients who had undergone cardiac rehabilitation found a relative reduction in mortality rate of 58% at 1 year and 21% to 34% at 5 years, with elderly women gaining the most benefit. In the Heart Failure: A Controlled Trial Investigating Outcomes of Exercise Training (HF-ACTION) trial, with more than 2,300 patients followed for a median of 2.5 years, exercise training for heart failure was associated with reduced rates of all-cause mortality or hospitalization (HR 0.89, 95% CI 0.81–0.99; P = .03) and of cardiovascular mortality or heart failure hospitalization (HR 0.85, 95% CI 0.74–0.99; P = .03).18
Regardless of the precise reduction in all-cause mortality, the cardiovascular and health-quality outcomes of cardiac rehabilitation clearly indicate benefit. More trials with follow-up longer than 1 year are needed to definitively determine the impact of cardiac rehabilitation on the all-cause mortality rate.
WHO SHOULD BE OFFERED CARDIAC REHABILITATION?
The 2006 CMS coverage criteria listed the indications for cardiac rehabilitation as myocardial infarction within the preceding 12 months, coronary artery bypass surgery, stable angina pectoris, heart valve repair or replacement, percutaneous coronary intervention, and heart or heart-lung transplant.
In 2014, stable chronic systolic heart failure was added to the list (Table 2). Qualifications include New York Heart Association class II (mild symptoms, slight limitation of activity) to class IV (severe limitations, symptoms at rest), an ejection fraction of 35% or less, and being stable on optimal medical therapy for at least 6 weeks.
In 2017, CMS approved supervised exercise therapy for peripheral arterial disease. Supervised exercise has a class 1 recommendation by the American Heart Association and American College of Cardiology for treating intermittent claudication. Supervised exercise therapy can increase walking distance by 180% and is superior to medical therapy alone. Unsupervised exercise has a class 2b recommendation.19,20
Other patients may not qualify for phase 2 cardiac rehabilitation according to CMS or private insurance but could benefit from an exercise prescription and enrollment in a local phase 3 or home exercise program. Indications might include diabetes, obesity, metabolic syndrome, atrial fibrillation, postural orthostatic tachycardia syndrome, and nonalcoholic steatohepatitis. The benefits of cardiac rehabilitation after newer, less-invasive procedures for transcatheter valve repair and replacement are not well established, and more research is needed in this area.
WHEN TO REFER
Ades et al have defined cardiac rehabilitation referral as a combination of electronic medical records order, patient-physician discussion, and receipt of an order by a cardiac rehabilitation program.21
Ideally, referral for outpatient cardiac rehabilitation should take place at the time of hospital discharge. The AACVPR endorses a “cardiovascular continuum of care” model that emphasizes a smooth transition from inpatient to outpatient programs.6 Inpatient referral is a strong predictor of cardiac rehabilitation enrollment, and lack of referral in phase 1 negatively affects enrollment rates.
Depending on the diagnosis, US and Canadian guidelines recommend cardiac rehabilitation starting within 1 to 4 weeks of the index event, with acceptable wait times up to 60 days.6,22 In the United Kingdom, referral is recommended within 24 hours of patient eligibility; assessment for a cardiovascular prevention and rehabilitation program, with a defined pathway and individual goals, is expected to be completed within 10 working days of referral.23 Such a standard is difficult to meet in the United States, where the time from hospital discharge to cardiac rehabilitation program enrollment averages 35 days.24,25
After an uncomplicated myocardial infarction or percutaneous coronary intervention, patients with a normal or mildly reduced left ventricular ejection fraction should start outpatient cardiac rehabilitation within 14 days of the index event. For such cases, cardiac rehabilitation has been shown to be safe within 1 to 2 weeks of hospital discharge and is associated with increased participation rates.
REHABILITATION IS STILL UNDERUSED
Despite its significant benefits, cardiac rehabilitation is underused for many reasons.
Referral rates vary
A study using the 1997 Medicare claims database showed national referral rates of only 14% after myocardial infarction and 31% after coronary artery bypass grafting.31
A later study using the National Cardiovascular Data Registry between 2009 and 2017 found that the situation had improved, with a referral rate of about 60% for patients undergoing percutaneous coronary intervention.32 Nevertheless, referral rates for cardiac rehabilitation remain highly variable and still lag behind other CMS quality measures for optimal medical therapy after acute myocardial infarction (Figure 1). Factors associated with higher referral rates included ST-segment elevation myocardial infarction, non-ST-segment elevation myocardial infarction, care in a high-volume center for percutaneous coronary intervention, and care in a private or community hospital in a Midwestern state. Small Midwestern hospitals generally had referral rates of over 80%, while major teaching hospitals and hospital systems on the East Coast and the West Coast had referral rates of less than 20%. Unlike some studies, this study found that insurance status had little bearing on referral rates.
Other studies found lower referral rates for women and patients with comorbidities such as previous coronary artery bypass grafting, diabetes, and heart failure.33,34
In the United Kingdom, patients with heart failure made up only 5% of patients in cardiac rehabilitation; only 7% to 20% of patients with a heart failure diagnosis were referred to cardiac rehabilitation from general and cardiology wards.35
Enrollment, completion rates even lower
Rates of referral for cardiac rehabilitation do not equate to rates of enrollment or participation. Enrollment was 50% in the United Kingdom in 2016.35 A 2015 US study evaluated 58,269 older patients eligible for cardiac rehabilitation after acute myocardial infarction; 62% were referred for cardiac rehabilitation at the time of discharge, but only 23% of the total attended at least 1 session, and just 5% of the total completed 36 or more sessions.36
BARRIERS, OPPORTUNITIES TO IMPROVE
The underuse of cardiac rehabilitation in the United States has led to an American Heart Association presidential advisory on the referral, enrollment, and delivery of cardiac rehabilitation.34 Dozens of barriers are mentioned, with several standing out as having the largest impact: lack of physician referral, weak endorsement by the prescribing provider, female sex of patients, lack of program availability, work-related hardship, low socioeconomic status, and lack of or limited healthcare insurance. Copayments have also become a major barrier, often ranging from $20 to $40 per session for patients with Medicare.
The Million Hearts Initiative has established a goal of 70% cardiac rehabilitation compliance for eligible patients by 2022, a goal they estimate could save 25,000 lives and prevent 180,000 hospitalizations annually.21
Lack of physician awareness and lack of referral may be the most modifiable factors with the capacity to have the largest impact. Increasing physician awareness is a top priority not only for primary care providers, but also for cardiologists. In 2014, CMS made referral for cardiac rehabilitation a quality measure that is trackable and reportable. CMS has also proposed models that would incentivize participation by increasing reimbursement for services provided, but these models have been halted.
Additional efforts to increase cardiac rehabilitation referral and participation include automated order sets, increased caregiver education, and early morning or late evening classes, single-sex classes, home or mobile-based exercise programs, and parking and transportation assistance.34 Grace et al37 reported that referral rates rose to 86% when a cardiac rehabilitation order was integrated into the electronic medical record and combined with a hospital liaison to educate patients about their need for cardiac rehabilitation. Lowering patient copayments would also be a good idea. We have recently seen some creative ways to reduce copayments, including philanthropy and grants.
- Herberden W. Classics in cardiology: description of angina pectoris by William Herberden. Heart Views 2006; 7(3):118–119. www.heartviews.org/text.asp?2006/7/3/118/63927. Accessed May 9, 2018.
- Mampuya WM. Cardiac rehabilitation past, present and future: an overview. Cardiovasc Diagn Ther 2012; 2(1):38–49. doi:10.3978/j.issn.2223-3652.2012.01.02
- Levine SA, Lown B. The “chair” treatment of acute thrombosis. Trans Assoc Am Physicians 1951; 64:316–327. pmid:14884265
- Morris JN, Everitt MG, Pollard R, Chave SP, Semmence AM. Vigorous exercise in leisure-time: protection against coronary heart disease. Lancet 1980; 2(8206):207–210. pmid:6108391
- Morris JN, Heady JA. Mortality in relation to the physical activity of work: a preliminary note on experience in middle age. Br J Ind Med 1953; 10(4):245–254. pmid:13106231
- American Association of Cardiovascular and Pulmonary Rehabilitation. Guidelines for cardiac rehabilitation and secondary prevention programs/American Association of Cardiovascular and Pulmonary Rehabilitation. 5th ed. Champaign, IL: Human Kinetics; 2013.
- Department of Health & Human Services (DHHS); Centers for Medicare & Medicaid Services (CMS). CMS manual system. Cardiac rehabilitation and intensive cardiac rehabilitation. www.cms.gov/Regulations-and-Guidance/Guidance/Transmittals/downloads/r126bp.pdf. Accessed May 9, 2018.
- Anderson L, Sharp GA, Norton RJ, et al. Home-based versus centre-based cardiac rehabilitation. Cochrane Database Syst Rev 2017; 6:CD007130. doi:10.1002/14651858.CD007130.pub4
- Anderson L, Oldridge N, Thompson DR, et al. Exercise-based cardiac rehabilitation for coronary heart disease: Cochrane systematic review and meta-analysis. J Am Coll Cardiol 2016; 67(1):1–12. doi:10.1016/j.jacc.2015.10.044
- Guiraud T, Nigam A, Gremeaux V, Meyer P, Juneau M, Bosquet L. High-intensity interval training in cardiac rehabilitation. Sports Med 2012; 42(7):587–605. doi:10.2165/11631910-000000000-00000
- Van Camp SP, Peterson RA. Cardiovascular complications of outpatient cardiac rehabilitation programs. JAMA 1986; 256(9):1160–1163. pmid:3735650
- Pavy B, Iliou MC, Meurin P, Tabet JY, Corone S; Functional Evaluation and Cardiac Rehabilitation Working Group of the French Society of Cardiology. Safety of exercise training for cardiac patients: results of the French registry of complications during cardiac rehabilitation. Arch Intern Med 2006; 166(21):2329–2334. doi:10.1001/archinte.166.21.2329
- Shaw LW. Effects of a prescribed supervised exercise program on mortality and cardiovascular morbidity in patients after a myocardial infarction: The National Exercise and Heart Disease Project. Am J Cardiol 1981; 48(1):39–46. pmid:6972693
- Sandesara PB, Lambert CT, Gordon NF, et al. Cardiac rehabilitation and risk reduction: time to “rebrand and reinvigorate.” J Am Coll Cardiol 2015; 65(4):389–395. doi:10.1016/j.jacc.2014.10.059
- Suaya JA, Stason WB, Ades PA, Normand SL, Shepard DS. Cardiac rehabilitation and survival in older coronary patients. J Am Coll Cardiol 2009; 54(1):25–33. doi:10.1016/j.jacc.2009.01.078
- Goel K, Lennon RJ, Tilbury RT, Squires RW, Thomas RJ. Impact of cardiac rehabilitation on mortality and cardiovascular events after percutaneous coronary intervention in the community. Circulation 2011: 123(21):2344–2352. doi:10.1161/CIRCULATIONAHA.110.983536
- Hammill BG, Curtis LH, Schulman KA, Whellan DJ. Relationship between cardiac rehabilitation and long-term risks of death and myocardial infarction among elderly Medicare beneficiaries. Circulation 2010; 121(1):63–70. doi:10.1161/CIRCULATIONAHA.109.876383
- O’Connor CM, Whellan DJ, Lee KL, et al; HF-ACTION Investigators. Efficacy and safety of exercise training in patients with chronic heart failure: HF-ACTION randomized controlled trial. JAMA 2009; 301(14):1439–1450. doi:10.1001/jama.2009.454
- Hirsch A, Haskal ZJ, Hertzer NR, et al. ACC/AHA 2005 practice guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic). Circulation 2006; 113(11):463–654. doi:10.1161/CIRCULATIONAHA.106.174526
- Ambrosetti M. Advances in exercise rehabilitation for patients with lower extremity peripheral artery disease. Monaldi Arch Chest Dis 2016; 86(1–2):752. doi:10.4081/monaldi.2016.752
- Ades PA, Keteyian SJ, Wright JS, et al. Increasing cardiac rehabilitation participation from 20% to 70%: a road map from the Million Hearts Cardiac Rehabilitation Collaborative. Mayo Clin Proc 2017; 92(2):234–242. doi:10.1016/j.mayocp.2016.10.014
- Dafoe W, Arthur H, Stokes H, Morrin L, Beaton L; Canadian Cardiovascular Society Access to Care Working Group on Cardiac Rehabilitation. Universal access: but when? Treating the right patient at the right time: access to cardiac rehabilitation. Can J Cardiol 2006; 22(11):905–911. pmid:16971975
- The British Association for Cardiovascular Prevention and Rehabilitation. The BACPR standards and core components for cardiovascular disease prevention and cardiac rehabilitation 2017. www.bacpr.com/resources/6A7_BACR_Standards_and_Core_Components_2017.pdf. Accessed May 9, 2018.
- Zullo MD, Jackson LW, Whalen CC, Dolansky MA. Evaluation of the recommended core components of cardiac rehabilitation practice: an opportunity for quality improvement. J Cardiopulm Rehabil Prev 2012; 32(1):32–40. doi:10.1097/HCR.0b013e31823be0e2
- Russell KL, Holloway TM, Brum M, Caruso V, Chessex C, Grace SL. Cardiac rehabilitation wait times: effect on enrollment. J Cardiopulm Rehabil Prev 2011; 31(6):373–377. doi:10.1097/HCR.0b013e318228a32f
- Soga Y, Yokoi H, Ando K, et al. Safety of early exercise training after elective coronary stenting in patients with stable coronary artery disease. Eur J Cardiovasc Prev Rehabil 2010; 17(2):230–234. doi:10.1097/HJR.0b013e3283359c4e
- Scheinowitz M, Harpaz D. Safety of cardiac rehabilitation in a medically supervised, community-based program. Cardiology 2005; 103(3):113–117. doi:10.1159/000083433
- Goto Y, Sumida H, Ueshima K, Adachi H, Nohara R, Itoh H. Safety and implementation of exercise testing and training after coronary stenting in patients with acute myocardial infarction. Circ J 2002; 66(10):930–936. pmid:12381088
- Parker K, Stone JA, Arena R, et al. An early cardiac access clinic significantly improves cardiac rehabilitation participation and completion rates in low-risk ST-elevation myocardial infarction patients. Can J Cardiol 2011; 27(5):619–627. doi:10.1016/j.cjca.2010.12.076
- Pack QR, Mansour M, Barboza JS, et al. An early appointment to outpatient cardiac rehabilitation at hospital discharge improves attendance at orientation: a randomized, single-blind, controlled trial. Circulation 2013; 127(3):349–355. doi:10.1161/CIRCULATIONAHA.112.121996
- Suaya JA, Shepard DS, Normand SL, Ades PA, Prottas J, Stason WB. Use of cardiac rehabilitation by Medicare beneficiaries after myocardial infarction or coronary bypass surgery. Circulation 2007; 116(15):1653–1662. doi:10.1161/CIRCULATIONAHA.107.701466
- Aragam KG, Dai D, Neely ML, et al. Gaps in referral to cardiac rehabilitation of patients undergoing percutaneous coronary intervention in the United States. J Am Coll Cardiol 2015; 65(19):2079–2088. doi:10.1016/j.jacc.2015.02.063
- Bittner V, Sanderson B, Breland J, Green D. Referral patterns to a university-based cardiac rehabilitation program. Am J Cardiol 1999; 83(2):252–255, A5. pmid:10073829
- Balady GJ, Ades PA, Bittner VA, et al. Referral, enrollment, and delivery of cardiac rehabilitation/secondary prevention programs at clinical centers and beyond. A presidential advisory from the American Heart Association. Circulation 2011; 124(25):2951–2960. doi:10.1161/CIR.0b013e31823b21e2
- British Heart Foundation. The national audit of cardiac rehabilitation annual statistical report 2016. www.cardiacrehabilitation.org.uk/docs/BHF_NACR_Report_2016.pdf. Accessed April 12, 2018.
- Doll JA, Hellkamp A, Ho PM, et al. Participation in cardiac rehabilitation programs among older patients after acute myocardial infarction. JAMA Intern Med 2015; 175(10):1700–1702. doi:10.1001/jamainternmed.2015.3819
- Grace SL, Russell KL, Reid RD, et al. Cardiac Rehabilitation Care Continuity Through Automatic Referral Evaluation (CRCARE) Investigators. Effect of cardiac rehabilitation referral strategies on utilization rates: a prospective, controlled study. Arch Intern Med 2011; 171(3):235–241. doi:10.1001/archinternmed.2010.501
- Herberden W. Classics in cardiology: description of angina pectoris by William Herberden. Heart Views 2006; 7(3):118–119. www.heartviews.org/text.asp?2006/7/3/118/63927. Accessed May 9, 2018.
- Mampuya WM. Cardiac rehabilitation past, present and future: an overview. Cardiovasc Diagn Ther 2012; 2(1):38–49. doi:10.3978/j.issn.2223-3652.2012.01.02
- Levine SA, Lown B. The “chair” treatment of acute thrombosis. Trans Assoc Am Physicians 1951; 64:316–327. pmid:14884265
- Morris JN, Everitt MG, Pollard R, Chave SP, Semmence AM. Vigorous exercise in leisure-time: protection against coronary heart disease. Lancet 1980; 2(8206):207–210. pmid:6108391
- Morris JN, Heady JA. Mortality in relation to the physical activity of work: a preliminary note on experience in middle age. Br J Ind Med 1953; 10(4):245–254. pmid:13106231
- American Association of Cardiovascular and Pulmonary Rehabilitation. Guidelines for cardiac rehabilitation and secondary prevention programs/American Association of Cardiovascular and Pulmonary Rehabilitation. 5th ed. Champaign, IL: Human Kinetics; 2013.
- Department of Health & Human Services (DHHS); Centers for Medicare & Medicaid Services (CMS). CMS manual system. Cardiac rehabilitation and intensive cardiac rehabilitation. www.cms.gov/Regulations-and-Guidance/Guidance/Transmittals/downloads/r126bp.pdf. Accessed May 9, 2018.
- Anderson L, Sharp GA, Norton RJ, et al. Home-based versus centre-based cardiac rehabilitation. Cochrane Database Syst Rev 2017; 6:CD007130. doi:10.1002/14651858.CD007130.pub4
- Anderson L, Oldridge N, Thompson DR, et al. Exercise-based cardiac rehabilitation for coronary heart disease: Cochrane systematic review and meta-analysis. J Am Coll Cardiol 2016; 67(1):1–12. doi:10.1016/j.jacc.2015.10.044
- Guiraud T, Nigam A, Gremeaux V, Meyer P, Juneau M, Bosquet L. High-intensity interval training in cardiac rehabilitation. Sports Med 2012; 42(7):587–605. doi:10.2165/11631910-000000000-00000
- Van Camp SP, Peterson RA. Cardiovascular complications of outpatient cardiac rehabilitation programs. JAMA 1986; 256(9):1160–1163. pmid:3735650
- Pavy B, Iliou MC, Meurin P, Tabet JY, Corone S; Functional Evaluation and Cardiac Rehabilitation Working Group of the French Society of Cardiology. Safety of exercise training for cardiac patients: results of the French registry of complications during cardiac rehabilitation. Arch Intern Med 2006; 166(21):2329–2334. doi:10.1001/archinte.166.21.2329
- Shaw LW. Effects of a prescribed supervised exercise program on mortality and cardiovascular morbidity in patients after a myocardial infarction: The National Exercise and Heart Disease Project. Am J Cardiol 1981; 48(1):39–46. pmid:6972693
- Sandesara PB, Lambert CT, Gordon NF, et al. Cardiac rehabilitation and risk reduction: time to “rebrand and reinvigorate.” J Am Coll Cardiol 2015; 65(4):389–395. doi:10.1016/j.jacc.2014.10.059
- Suaya JA, Stason WB, Ades PA, Normand SL, Shepard DS. Cardiac rehabilitation and survival in older coronary patients. J Am Coll Cardiol 2009; 54(1):25–33. doi:10.1016/j.jacc.2009.01.078
- Goel K, Lennon RJ, Tilbury RT, Squires RW, Thomas RJ. Impact of cardiac rehabilitation on mortality and cardiovascular events after percutaneous coronary intervention in the community. Circulation 2011: 123(21):2344–2352. doi:10.1161/CIRCULATIONAHA.110.983536
- Hammill BG, Curtis LH, Schulman KA, Whellan DJ. Relationship between cardiac rehabilitation and long-term risks of death and myocardial infarction among elderly Medicare beneficiaries. Circulation 2010; 121(1):63–70. doi:10.1161/CIRCULATIONAHA.109.876383
- O’Connor CM, Whellan DJ, Lee KL, et al; HF-ACTION Investigators. Efficacy and safety of exercise training in patients with chronic heart failure: HF-ACTION randomized controlled trial. JAMA 2009; 301(14):1439–1450. doi:10.1001/jama.2009.454
- Hirsch A, Haskal ZJ, Hertzer NR, et al. ACC/AHA 2005 practice guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic). Circulation 2006; 113(11):463–654. doi:10.1161/CIRCULATIONAHA.106.174526
- Ambrosetti M. Advances in exercise rehabilitation for patients with lower extremity peripheral artery disease. Monaldi Arch Chest Dis 2016; 86(1–2):752. doi:10.4081/monaldi.2016.752
- Ades PA, Keteyian SJ, Wright JS, et al. Increasing cardiac rehabilitation participation from 20% to 70%: a road map from the Million Hearts Cardiac Rehabilitation Collaborative. Mayo Clin Proc 2017; 92(2):234–242. doi:10.1016/j.mayocp.2016.10.014
- Dafoe W, Arthur H, Stokes H, Morrin L, Beaton L; Canadian Cardiovascular Society Access to Care Working Group on Cardiac Rehabilitation. Universal access: but when? Treating the right patient at the right time: access to cardiac rehabilitation. Can J Cardiol 2006; 22(11):905–911. pmid:16971975
- The British Association for Cardiovascular Prevention and Rehabilitation. The BACPR standards and core components for cardiovascular disease prevention and cardiac rehabilitation 2017. www.bacpr.com/resources/6A7_BACR_Standards_and_Core_Components_2017.pdf. Accessed May 9, 2018.
- Zullo MD, Jackson LW, Whalen CC, Dolansky MA. Evaluation of the recommended core components of cardiac rehabilitation practice: an opportunity for quality improvement. J Cardiopulm Rehabil Prev 2012; 32(1):32–40. doi:10.1097/HCR.0b013e31823be0e2
- Russell KL, Holloway TM, Brum M, Caruso V, Chessex C, Grace SL. Cardiac rehabilitation wait times: effect on enrollment. J Cardiopulm Rehabil Prev 2011; 31(6):373–377. doi:10.1097/HCR.0b013e318228a32f
- Soga Y, Yokoi H, Ando K, et al. Safety of early exercise training after elective coronary stenting in patients with stable coronary artery disease. Eur J Cardiovasc Prev Rehabil 2010; 17(2):230–234. doi:10.1097/HJR.0b013e3283359c4e
- Scheinowitz M, Harpaz D. Safety of cardiac rehabilitation in a medically supervised, community-based program. Cardiology 2005; 103(3):113–117. doi:10.1159/000083433
- Goto Y, Sumida H, Ueshima K, Adachi H, Nohara R, Itoh H. Safety and implementation of exercise testing and training after coronary stenting in patients with acute myocardial infarction. Circ J 2002; 66(10):930–936. pmid:12381088
- Parker K, Stone JA, Arena R, et al. An early cardiac access clinic significantly improves cardiac rehabilitation participation and completion rates in low-risk ST-elevation myocardial infarction patients. Can J Cardiol 2011; 27(5):619–627. doi:10.1016/j.cjca.2010.12.076
- Pack QR, Mansour M, Barboza JS, et al. An early appointment to outpatient cardiac rehabilitation at hospital discharge improves attendance at orientation: a randomized, single-blind, controlled trial. Circulation 2013; 127(3):349–355. doi:10.1161/CIRCULATIONAHA.112.121996
- Suaya JA, Shepard DS, Normand SL, Ades PA, Prottas J, Stason WB. Use of cardiac rehabilitation by Medicare beneficiaries after myocardial infarction or coronary bypass surgery. Circulation 2007; 116(15):1653–1662. doi:10.1161/CIRCULATIONAHA.107.701466
- Aragam KG, Dai D, Neely ML, et al. Gaps in referral to cardiac rehabilitation of patients undergoing percutaneous coronary intervention in the United States. J Am Coll Cardiol 2015; 65(19):2079–2088. doi:10.1016/j.jacc.2015.02.063
- Bittner V, Sanderson B, Breland J, Green D. Referral patterns to a university-based cardiac rehabilitation program. Am J Cardiol 1999; 83(2):252–255, A5. pmid:10073829
- Balady GJ, Ades PA, Bittner VA, et al. Referral, enrollment, and delivery of cardiac rehabilitation/secondary prevention programs at clinical centers and beyond. A presidential advisory from the American Heart Association. Circulation 2011; 124(25):2951–2960. doi:10.1161/CIR.0b013e31823b21e2
- British Heart Foundation. The national audit of cardiac rehabilitation annual statistical report 2016. www.cardiacrehabilitation.org.uk/docs/BHF_NACR_Report_2016.pdf. Accessed April 12, 2018.
- Doll JA, Hellkamp A, Ho PM, et al. Participation in cardiac rehabilitation programs among older patients after acute myocardial infarction. JAMA Intern Med 2015; 175(10):1700–1702. doi:10.1001/jamainternmed.2015.3819
- Grace SL, Russell KL, Reid RD, et al. Cardiac Rehabilitation Care Continuity Through Automatic Referral Evaluation (CRCARE) Investigators. Effect of cardiac rehabilitation referral strategies on utilization rates: a prospective, controlled study. Arch Intern Med 2011; 171(3):235–241. doi:10.1001/archinternmed.2010.501
KEY POINTS
- Cardiac rehabilitation should begin in the hospital after heart surgery or myocardial infarction, should continue with a hospital-centered 36-session program, and should be maintained independently by the patient for life.
- Exercise in a cardiac rehabilitation program entails little risk and many proven benefits.
- Cardiac rehabilitation is indicated and covered by the Centers for Medicare and Medicaid Services (CMS) for a number of cardiovascular conditions.
- Utilization of cardiac rehabilitation could be improved through CMS reimbursement incentives, electronic medical record prompts, lower copayments for participation, and home-based programs for patients who live far from medical centers.

New cholesterol guidelines: Worth the wait?
On November 12, 2013, a joint task force for the American College of Cardiology and American Heart Association released new guidelines for treating high blood cholesterol to reduce the risk of atherosclerotic cardiovascular disease (ASCVD) in adults.1
This document arrives after several years of intense deliberation, 12 years after the third Adult Treatment Panel (ATP III) guidelines,2 and 8 years after an ATP III update recommending that low-density lipoprotein cholesterol (LDL-C) levels be lowered aggressively (to less than 70 mg/dL) as an option in patients at high risk.3 It represents a major shift in the approach to and management of high blood cholesterol and has sparked considerable controversy.
In the following commentary, we summarize the new guidelines and the philosophy employed by the task force in generating them. We will also examine some advantages and what we believe to be several shortcomings of the new guidelines. These latter points are illustrated through case examples.
IN RANDOMIZED CONTROLLED TRIALS WE TRUST
In collaboration with the National Heart, Lung, and Blood Institute of the National Institutes of Health, the American College of Cardiology and American Heart Association formed an expert panel task force in 2008.

The task force elected to use only evidence from randomized controlled trials, systematic reviews, and meta-analyses of randomized controlled trials (and only predefined outcomes of the trials, not post hoc analyses) in formulating its recommendations, with the goal of providing the strongest possible evidence.
The authors state that “By using [randomized controlled trial] data to identify those most likely to benefit [emphasis in original] from cholesterol-lowering statin therapy, the recommendations will be of value to primary care clinicians as well as specialists concerned with ASCVD prevention. Importantly, the recommendations were designed to be easy to use in the clinical setting, facilitating the implementation of a strategy of risk assessment and treatment focused on the prevention of ASCVD.”3 They also state the guidelines are meant to “inform clinical judgment, not replace it” and that clinician judgment in addition to discussion with patients remains vital.
During the deliberations, the National Heart, Lung, and Blood Institute removed itself from participating, stating its mission no longer included drafting new guidelines. Additionally, other initial members of the task force removed themselves because of disagreement and concerns about the direction of the new guidelines.
These guidelines, and their accompanying new cardiovascular risk calculator,4 were released without a preliminary period to allow for open discussion, comment, and critique by physicians outside the panel. No attempt was made to harmonize the guidelines with previous versions (eg, ATP III) or with current international guidelines.
WHAT’S NEW IN THE GUIDELINES?
The following are the major changes in the new guidelines for treating high blood cholesterol:
- Treatment goals for LDL-C and non-high-density lipoprotein cholesterol (non-HDL-C) are no longer recommended.
- High-intensity and moderate-intensity statin treatment is emphasized, and low-intensity statin therapy is nearly eliminated.
- “ASCVD” now includes stroke in addition to coronary heart disease and peripheral arterial disease.
- Four groups are targeted for treatment (see below).
- Nonstatin therapies have been markedly de-emphasized.
- No guidelines are provided for treating high triglyceride levels.
The new guidelines emphasize lifestyle modification as the foundation for reducing risk, regardless of cholesterol therapy. No recommendations are given for patients with New York Heart Association class II, III, or IV heart failure or for hemodialysis patients, because there were insufficient data from randomized controlled trials to support recommendations. Similarly, the guidelines apply only to people between the ages of 40 and 75 (risk calculator ages 40–79), because the authors believed there was not enough evidence from randomized controlled trials to allow development of guidelines outside of this age range.
FOUR MAJOR STATIN TREATMENT GROUPS
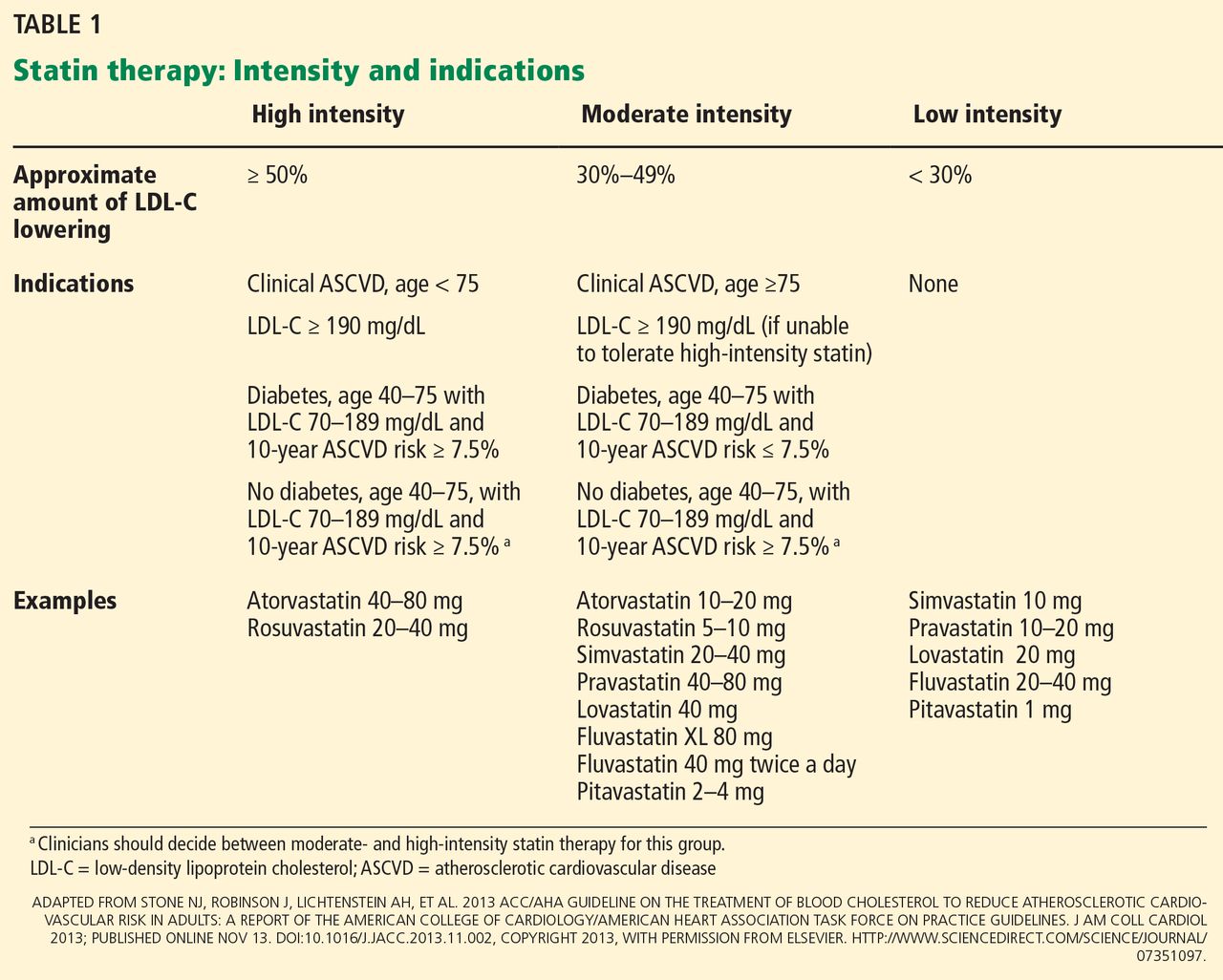
The new guidelines specify four groups that merit intensive or moderately intensive statin therapy (Table 1)1:
- People with clinical ASCVD
- People with LDL-C levels of 190 mg/dL or higher
- People with diabetes, age 40 to 75
- People without diabetes, age 40 to 75, with LDL-C levels 70–189 mg/dL, and a 10-year ASCVD risk of 7.5% or higher as determined by the new risk calculator4 (which also calculates the lifetime risk of ASCVD).
Below, we will address each of these four groups and provide case scenarios to consider. In general, our major disagreements with the new recommendations pertain to the first and fourth categories.
GROUP 1: PEOPLE WITH CLINICAL ASCVD
Advantages of the new guidelines
- They appropriately recommend statins in the highest tolerated doses as first-line treatment for this group at high risk.
- They designate all patients with ASCVD, including those with coronary, peripheral, and cerebrovascular disease, as a high-risk group.
- Without target LDL-C levels, treatment is simpler than before, requiring less monitoring of lipid levels. (This can also be seen as a limitation, as we discuss below.)
Limitations of the new guidelines
- They make follow-up LDL-C levels irrelevant, seeming to assume that there is no gradation in residual risk and, thus, no need to tailor therapy to the individual.
- Patients no longer have a goal to strive for or a way to monitor their progress.
- The guidelines ignore the pathophysiology of coronary artery disease and evidence of residual risk in patients on both moderate-intensity and high-intensity statin therapy.
- They also ignore the potential benefits of treating to lower LDL-C or non-HDL-C goals, thus eliminating consideration of multidrug therapy. They do not address patients with recurrent cardiovascular events already on maximal tolerated statin doses.
- They undermine the potential development and use of new therapies for dysplipidemia in patients with ASCVD.
Case 1: Is LDL-C 110 mg/dL low enough?
A 52-year-old African American man presents with newly discovered moderate coronary artery disease that is not severe enough to warrant stenting. He has no history of hypertension, diabetes mellitus, or smoking. His systolic blood pressure is 130 mm Hg, and his body mass index is 26 kg/m2. He exercises regularly and follows a low-cholesterol diet. He has the following fasting lipid values:
- Total cholesterol 290 mg/dL
- HDL-C 50 mg/dL
- Triglycerides 250 mg/dL
- Calculated LDL-C 190 mg/dL.
Two months later, after beginning atorvastatin 80 mg daily, meeting with a nutritionist, and redoubling his dietary efforts, his fasting lipid concentrations are:
- Total cholesterol 180 mg/dL
- HDL-C 55 mg/dL
- Triglycerides 75 mg/dL
- Calculated LDL-C 110 mg/dL.
Comment: Lack of LDL-C goals is a flaw
The new guidelines call for patients with known ASCVD, such as this patient, to receive intensive statin therapy—which he got.
However, once a patient is on therapy, the new guidelines do not encourage repeating the lipid panel other than to assess compliance. With intensive therapy, we expect a reduction in LDL-C of at least 50% (Table 1), but patient-to-patient differences in response to medications are common, and without repeat testing we would have no way of gauging this patient’s residual risk.
Further, the new guidelines emphasize the lack of hard outcome data supporting the addition of another lipid-lowering drug to a statin, although they do indicate that one can consider it. In a patient at high risk, such as this one, would you be comfortable with an LDL-C value of 110 mg/dL on maximum statin therapy? Would you consider adding a nonstatin drug?
A preponderance of data shows that LDL plays a causal role in ASCVD development and adverse events. Genetic data show that the LDL particle and the LDL receptor pathway are mechanistically linked to ASCVD pathogenesis, with lifetime exposure as a critical determinant of risk.5,6 Moreover, randomized controlled trials of statins and other studies of cholesterol-lowering show a reproducible relationship between the LDL-C level achieved and absolute risk (Figure 1).7–24 We believe the totality of data constitutes a strong rationale for targeting LDL-C and establishing goals for lowering its levels. For these reasons, we believe that removing LDL-C goals is a fundamental flaw of the new guidelines.
The reason for the lack of data from randomized controlled trials demonstrating benefits of adding therapies to statins (when LDL-C is still high) or benefits of treating to specific goals is that no such trials have been performed. Even trials of nonpharmacologic means of lowering LDL-C, such as ileal bypass, which was used in the Program on the Surgical Control of the Hyperlipidemias trial,20 provide independent evidence that lowering LDL-C reduces the risk of ASCVD (Figure 1).
In addition, trials of nonstatin drugs, such as the Coronary Drug Project,25 which tested niacin, also showed outcome benefits. On the other hand, studies such as the Atherothrombosis Intervention in Metabolic Syndrome With Low HDL/High Triglycerides: Impact on Global Health26 and Treatment of HDL to Reduce the Incidence of Vascular Events27 trials did not show additional risk reduction when niacin was added to statin therapy. However, the study designs arguably had flaws, including requirement of aggressive LDL-lowering with statins, with LDL-C levels below 70 to 80 mg/dL before randomization.
Therefore, these trials do not tell us what to do for a patient on maximal intensive therapy who has recurrent ASCVD events or who, like our patient, has an LDL-C level higher than previous targets.
For this patient, we would recommend adding a second medication to further lower his LDL-C, but discussing with him the absence of proven benefit in clinical trials and the risks of side effects. At present, lacking LDL-C goals in the new guidelines, we are keeping with the ATP III goals to help guide therapeutic choices and individualize patient management.
GROUP 2: PEOPLE WITH LDL-C ≥ 190
Advantages of the new guidelines
- They state that these patients should receive statins in the highest tolerated doses, which is universally accepted.
Limitations of the new guidelines
- The new guidelines mention only that one “may consider” adding a second agent if LDL-C remains above 190 mg/dL after maximum-dose therapy. Patients with familial hypercholesterolemia or other severe forms of hypercholesterolemia typically end up on multidrug therapy to further reduce LDL-C. The absence of randomized controlled trial data in this setting to show an additive value of second and third lipid-lowering agents does not mean these agents do not provide benefit.
GROUP 3: DIABETES, AGE 40–75, LDL-C 70–189, NO CLINICAL ASCVD
Advantages of the new guidelines
- They call for aggressive treatment of people with diabetes, a group at high risk that derives significant benefit from statin therapy, as shown in randomized controlled trials.
Limitations of the new guidelines
- Although high-intensity statin therapy is indicated for this group, we believe that, using the new risk calculator, some patients may receive overly aggressive treatment, thus increasing the possibility of statin side effects.
- The guidelines do not address patients younger than 40 or older than 75.
- Diabetic patients have a high residual risk of ASCVD events, even on statin therapy. Yet the guidelines ignore the potential benefits of more aggressive LDL-lowering or non-LDL secondary targets for therapy.
Case 2: How low is too low?
A 63-year-old white woman, a nonsmoker with recently diagnosed diabetes, is seen by her primary care physician. She has hypertension, for which she takes lisinopril 5 mg daily. Her fasting lipid values are:
- Total cholesterol 160 mg/dL
- HDL-C 64 mg/dL
- Triglycerides 100 mg/dL
- Calculated LDL-C 76 mg/dL.
Her systolic blood pressure is 129 mm Hg, and based on the new risk calculator, her 10-year risk of cardiovascular disease is 10.2%. According to the new guidelines, she should be started on high-intensity statin treatment (Table 1).
Although this is an acceptable initial course of action, it necessitates close vigilance, since it may actually drive her LDL-C level too low. Randomized controlled trials have typically used an LDL-C concentration of less than or equal to 25 mg/dL as the safety cutoff. With a typical LDL-C reduction of at least 50% on high-intensity statins, our patient’s expected LDL-C level will likely be in the low 30s. We believe this would be a good outcome, provided that she tolerates the medication without adverse effects. However, responses to statins vary from patient to patient.
High-intensity statin therapy may not be necessary to reduce risk adequately in all patients who have diabetes without preexisting vascular disease. The Collaborative Atorvastatin Diabetes Study12 compared atorvastatin 10 mg vs placebo in people with type 2 diabetes, age 40 to 75, who had one or more cardiovascular risk factors but no signs or symptoms of preexisting ASCVD and who had only average or below-average cholesterol levels—precisely like this patient. The trial was terminated early because of a clear benefit (a 37% reduction in the composite end point of major adverse cardiovascular events) in the intervention group. For our patient, we believe an alternative and acceptable approach would be to begin moderate-intensity statin therapy (eg, with atorvastatin 10 mg) (Table 1).
Alternatively, in a patient with diabetes and previous atherosclerotic vascular disease or with a high 10-year risk and high LDL-C, limiting treatment to high-intensity statin therapy by itself may deny them the potential benefits of combination therapies and targeting to lower LDL-C levels or non-HDL-C secondary targets. Guidelines from the American Diabetes Association28 and the American Association of Clinical Endocrinologists29 continue to recommend an LDL-C goal of less than 70 mg/dL in patients at high risk, a non-HDL-C less than 100 mg/dL, an apolipoprotein B less than 80 mg/dL, and an LDL particle number less than 1,000 nmol/L.
GROUP 4: AGE 40–75, LDL-C 70–189, NO ASCVD, BUT 10-YEAR RISK ≥ 7.5%
Advantages of the new guidelines
- They may reduce ASCVD events for patients at higher risk.
- The risk calculator is easy to use and focuses on global risk, ie, all forms of ASCVD.
- The guidelines promote discussion of risks and benefits between patients and providers.
Limitations of the new guidelines
- The new risk calculator is controversial (see below).
- There is potential for overtreatment, particularly in older patients.
- There is potential for undertreatment, particularly in patients with an elevated LDL-C but whose 10-year risk is less than 7.5% because they are young.
- The guidelines do not address patients younger than 40 or older than 75.
- They do not take into account some traditional risk factors, such as family history, and nontraditional risk factors such as C-reactive protein as measured by ultrasensitive assays, lipoprotein(a), and apolipoprotein B.
Risk calculator controversy
The new risk calculator has aroused strong opinions on both sides of the aisle.
Shortly after the new guidelines were released, cardiologists Dr. Paul Ridker and Dr. Nancy Cook from Brigham and Women’s Hospital in Boston published analyses30 showing that the new risk calculator, which was based on older data from several large cohorts such as the Atherosclerosis Risk in Communities study,31 the Cardiovascular Health Study,32 the Coronary Artery Risk Development in Young Adults study,33 and the Framingham Heart Study,34,35 was inaccurate in other cohorts. Specifically, in more-recent cohorts (the Women’s Health Study,36 Physicians’ Health Study,37 and Women’s Health Initiative38), the new calculator overestimates the 10-year risk of ASCVD by 75% to 150%.30 Using the new calculator would make approximately 30 million more Americans eligible for statin treatment. The concern is that patients at lower risk would be treated and exposed to potential side effects of statin therapy.
In addition, the risk calculator relies heavily on age and sex and does not include other factors such as triglyceride level, family history, C-reactive protein, or lipoprotein(a). Importantly, and somewhat ironically given the otherwise absolute adherence to randomized controlled trial data for guideline development, the risk calculator has never been verified in prospective studies to adequately show that using it reduces ASCVD events.
Case 3: Overtreating a primary prevention patient
Based on the risk calculator, essentially any African American man in his early 60s with no other risk factors has a 10-year risk of ASCVD of 7.5% or higher and, according to the new guidelines, should receive at least moderate-intensity statin therapy.
For example, consider a 64-year-old African American man whose systolic blood pressure is 129 mm Hg, who does not smoke, does not have diabetes, and does not have hypertension, and whose total cholesterol level is 180 mg/dL, HDL-C 70 mg/dL, triglycerides 130 mg/dL, and calculated LDL-C 84 mg/dL. His calculated 10-year risk is, surprisingly, 7.5%.
Alternatively, his twin brother is a two-pack-per-day smoker with untreated hypertension and systolic blood pressure 150 mm Hg, with fasting total cholesterol 153 mg/dL, HDL-C 70 mg/dL, triglycerides 60 mg/dL, and LDL-C 71 mg/dL. His calculated 10-year risk is 10.5%, so according to the new guidelines, he too should receive high-intensity statin therapy. Yet this patient clearly needs better blood pressure control and smoking cessation as his primary risk-reduction efforts, not a statin. While assessing global risk is important, a shortcoming of the new guidelines is that they can inappropriately lead to treating the risk score, not individualizing the treatment to the patient. Because of the errors inherent in the risk calculator, some experts have called for a temporary halt on implementing the new guidelines until the risk calculator can be further validated. In November 2013, the American Heart Association and the American College of Cardiology reaffirmed their support of the new guidelines and recommended that they be implemented as planned. As of the time this manuscript goes to print, there are no plans to halt implementation of the new guidelines.
Case 4: Undertreating a primary prevention patient
A 25-year-old white man with no medical history has a total cholesterol level of 310 mg/dL, HDL-C 50 mg/dL, triglycerides 400 mg/dL, and calculated LDL-C 180 mg/dL. He does not smoke or have hypertension or diabetes but has a strong family history of premature coronary disease (his father died of myocardial infarction at age 42). His body mass index is 25 kg/m2. Because he is less than 40 years old, the risk calculator does not apply to him.
If we assume he remains untreated and returns at age 40 with the same clinical factors and laboratory values, his calculated 10-year risk of an ASCVD event according to the new risk calculator will still be only 3.1%. Assuming his medical history remains unchanged as he continues to age, his 10-year risk would not reach 7.5% until he is 58. Would you feel comfortable waiting 33 years before starting statin therapy in this patient?
Waiting for dyslipidemic patients to reach middle age before starting LDL-C-lowering therapy is a failure of prevention. For practical reasons, there are no data from randomized controlled trials with hard outcomes in younger people. Nevertheless, a tenet of preventive cardiology is that cumulative exposure accelerates the “vascular age” ahead of the chronological age. This case illustrates why individualized recommendations guided by LDL-C goals as a target for therapy are needed. For this 25-year-old patient, we would recommend starting an intermediate- or high-potency statin.
Case 5: Rheumatoid arthritis
A 60-year-old postmenopausal white woman with severe rheumatoid arthritis presents for cholesterol evaluation. Her total cholesterol level is 235 mg/dL, HDL-C 50 mg/dL, and LDL-C 165 mg/dL. She does not smoke or have hypertension or diabetes. Her systolic blood pressure is 110 mm Hg. She has elevated C-reactive protein on an ultrasensitive assay and elevated lipoprotein(a).
Her calculated 10-year risk of ASCVD is 3.0%. Assuming her medical history remains the same, she would not reach a calculated 10-year risk of at least 7.5% until age 70. We suggest starting moderate- or high-dose statin therapy in this case, based on data (not from randomized controlled trials) showing an increased risk of ASCVD events in patients with rheumatologic disease, increased lipoprotein(a), and inflammatory markers like C-reactive protein. However, the current guidelines do not address this scenario, other than to suggest that clinician consideration can be given to other risk markers such as these, and that these findings should be discussed in detail with the patient. The Justification for the Use of Statins in Primary Prevention: An Intervention Trial Evaluating Rosuvastatin trial16 showed a dramatic ASCVD risk reduction in just such patients (Figure 1).
APPLAUSE—AND RESERVATIONS
The newest guidelines for treating high blood cholesterol represent a monumental shift away from using target levels of LDL-C and non-HDL-C and toward a focus on statin intensity for patients in the four highest-risk groups.
We applaud the expert panel for its idealistic approach of using only data from randomized controlled trials, for placing more emphasis on higher-intensity statin treatment, for including stroke in the new definition of ASCVD, and for focusing more attention on treating diabetic patients more aggressively. Simplifying the guidelines is a noble goal. Emphasizing moderate-to-high-intensity statin therapy in patients at moderate-to-high risk should have substantial long-term public health benefits.
However, as we have shown in the case examples, there are significant limitations, and some patients can end up being overtreated, while others may be undertreated.
Guidelines need to be crafted by looking at all the evidence, including the pathophysiology of the disease process, not just data from randomized controlled trials. It is difficult to implement a guideline that on one hand used randomized controlled trials exclusively for recommendations, but on the other hand used an untested risk calculator to guide therapy. Randomized controlled trials are not available for every scenario.
Further, absence of randomized controlled trial data in a given scenario should not be interpreted as evidence of lack of benefit. An example of this is a primary-prevention patient under age 40 with elevated LDL-C below the 190 mg/dL cutoff who otherwise is healthy and without risk factors (eg, Case 4). By disregarding all evidence that is not from randomized controlled trials, the expert panel fails to account for the extensive pathophysiology of ASCVD, which often begins at a young age and takes decades to develop.5,6,39 An entire generation of patients who have not reached the age of inclusion in most randomized controlled trials with hard outcomes is excluded (unless the LDL-C level is very high), potentially setting back decades of progress in the field of prevention. Prevention only works if started. With childhood and young adult obesity sharply rising, we should not fail to address the under-40-year-old patient population in our guidelines.
Guidelines are designed to be expert opinion, not to dictate practice. Focusing on the individual patient instead of the general population at risk, the expert panel appropriately emphasizes the “importance of clinician judgment, weighing potential benefits, adverse effects, drug-drug interactions and patient preferences.” However, by excluding all data that do not come from randomized controlled trials, the panel neglects a very large base of knowledge and leaves many clinicians without as much expert opinion as we had hoped for.
LDL-C goals are important: they provide a scorecard to help the patient with lifestyle and dietary changes. They provide the health care provider guidance in making treatment decisions and focusing on treatment of a single patient, not a population. Moreover, if a patient has difficulty taking standard doses of statins because of side effects, the absence of LDL-C goals makes decision-making nearly impossible. We hope physicians will rely on LDL-C goals in such situations, falling back on the ATP III recommendations, although many patients may simply go untreated until they present with ASCVD or until they “age in” to a higher risk category.
We suggest caution in strict adherence to the new guidelines and instead urge physicians to consider a hybrid of the old guidelines (using the ATP III LDL-C goals) and the new ones (emphasizing global risk assessment and high-intensity statin treatment).
- Stone NJ, Robinson J, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; published online Nov 13. DOI: 10.1016/j.jacc.2013.11.002.
- National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002; 106:3143–3421.
- Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004; 110:227–239.
- American Heart Association. 2013 Prevention guidelines tools. CV risk calculator. http://my.americanheart.org/professional/StatementsGuidelines/PreventionGuidelines/Prevention-Guidelines_UCM_457698_SubHomePage.jsp. Accessed December 10, 2013.
- Goldstein JL, Brown MS. The LDL receptor. Arterioscler Thromb Vasc Biol 2009; 29:431–438.
- Horton JD, Cohen JC, Hobbs HH. PCSK9: a convertase that coordinates LDL catabolism. J Lipid Res 2009; 50(suppl):S172–S177.
- Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 1994; 344:1383–1389.
- de Lemos JA, Blazing MA, Wiviott SD, et al; for the A to Z Investigators. Early intensive vs a delayed conservative simvastatin strategy in patients with acute coronary syndromes. Phase Z of the A to Z trial. JAMA 2004; 292:1307–1316.
- Downs JR, Clearfield M, Weis S, et al; for the AFCAPS/TexCAPS Research Group. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels. Results of AFCAPS/TexCAPS. JAMA 1998; 279:1615–1622.
- Koren MJ, Hunninghake DB, on behalf of the ALLIANCE investigators. Clinical outcomes in managed-care patients with coronary heart disease treated aggressively in lipid-lowering disease management clinics. J Am Coll Cardiol 2004; 44:1772–1779.
- Sever PS, Dahlof B, Poulter NR, et al; ASCOT investigators. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial - Lipid Lowering Arm (ASCOT-LLA): a multicentre randomised controlled trial. Lancet 2003; 361:1149–1158.
- Colhoun HM, Betteridge DJ, Durrington PN, et al; on behalf of the CARDS Investigators. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): multicentre randomised placebo-controlled trial. Lancet 2004; 364:685–696.
- Sacks FM, Pfeffer MA, Moye LA, et al; for the Cholesterol and Recurrent Events Trial Investigators. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. N Engl J Med 1996; 335:1001–1009.
- Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20 536 high-risk individuals: a randomised placebo-controlled trial. Lancet 2002; 360:7–22.
- Pedersen TR, Faegeman O, Kastelein JJ, et al. Incremental Decrease in End Points Through Aggressive Lipid Lowering Study Group. High-dose atorvastatin vs usual-dose simvastatin for secondary prevention after myocardial infarction: the IDEAL study: a randomized controlled trial. JAMA 2005; 294:2437–2445.
- Ridker PM, Danielson E, Fonseca FAH, et al; for the JUPITER Study Group. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- LIPID Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N Engl J Med 1998; 339:1349–1357.
- Nakamura H, Arakawa K, Itakura H, et al; for the MEGA Study Group. Primary prevention of cardiovascular disease with pravastatin Japan (MEGA Study): a prospective rabndomised controlled trial. Lancet 2006; 368:1155–1163.
- Schwartz GG, Olsson AG, Ezekowitz MD, et al. Myocardial Ischemia Reduction with Aggreessive Cholesterol Lowering (MIRACL) Study Investigators. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: the MIRACL study: a randomized controlled trial. JAMA 2001; 285:1711–1718.
- Buchwald H, Varco RL, Matts JP, et al. Effect of partial ileal bypass surgery on mortality and morbidity from coronary heart disease in patients with hypercholesterolemia: report of the Program on the Surgical Control of the Hyperlipidemias (POSCH). N Engl J Med 1990; 323:946–955.
- Cannon CP, Braunwald E, McCabe CH, et al; for the Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 Investigators. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- Baigent C, Landray MJ, Reith C, et al; SHARP Investigators. The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (Study of Heart and Renal Protection): a randomised placebo-controlled trial. Lancet 2011; 377:2181–2192.
- LaRosa JC, Grundy SM, Waters DD, et al. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med 2005; 352:1425–1435.
- Shepherd J, Cobbe SM, Ford I, et al; for the West of Scotland Coronary Prevention Study Group. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. N Engl J Med 1995; 333:1301–1308.
- Canner PL, Berge KG, Wenger NK, et al. Fifteen year mortality in Coronary Drug Project patients: long-term benefit with niacin. J Am Coll Cardiol 1989; 8:1245–1255.
- AIM-HIGH Investigators, Boden WE, Probstfield JL, Anderson T, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med 2011; 365:2255–2267.
- HPS2-Thrive Collaborative Group. HPS2-THRIVE randomized placebo-controlled trial in 25 673 high-risk patients of ER niacin/laropiprant: trial design, pre-specified muscle and liver outcomes, and reasons for stopping study treatment. Eur Heart J 2013; 34:1279–1291.
- American Diabetes Association. Standards of medical care in diabetes—2013. Diabetes Care 2013; 36(suppl 1):S11–S66.
- Garber AJ, Abrahamson MJ, Barzilay JI, et al. American Association of Clinical Endocrinologists’comprehensive diabetes management algorithm 2013 consensus statement—executive summary. Endocr Pract 2013; 19:536–557.
- Ridker PM, Cook NR. Statins: new American guidelines for prevention of cardiovascular disease. Lancet 2013doi: 10.1016/S0140-6736(13)62388-0. [Epub ahead of print]
- The ARIC investigators. The Atherosclerosis Risk in Communities (ARIC) study: design and objectives. Am J Epidemiol 1989; 129:687–702.
- Fried LP, Borhani NO, Enright P, et al. The Cardiovascular Health Study: design and rationale. Ann Epidemiol 1991; 1:263–276.
- Friedman GD, Cutter GR, Donahue RP, et al. CARDIA: study design, recruitment, and some characteristics of the examined subjects. J Clin Epidemiol 1988; 41:1105–1116.
- Dawber TR, Kannel WB, Lyell LP. An approach to longitudinal studies in a community: the Framingham study. Ann N Y Acad Sci 1963; 107:539–556.
- Kannel WB, Feinleib M, McNamara PM, Garrison RJ, Castelli WP. An investigation of coronary heart disease in families. The Framingham offspring study. Am J Epidemiol 1979; 110:281–290.
- Ridker PM, Cook NR, Lee IM, et al. A randomized trial of low-dose aspirin in the primary prevention of cardiovascular disease in women. N Engl J Med 2005; 352:1293–1304.
- Belancer C, Buring JE, Cook N, et al; The Steering Committee of the Physicians’ Health Study Research Group. Final report on the aspirin component of the ongoing Physicians’ Health Study. N Engl J Med 1989; 321:129–135.
- Langer R, White E, Lewis C, et al. The Women’s Health Initiative Observational Study: baseline characteristics of participants and reliability of baseline measures. Ann Epidemiol 2003; 13:S107–S121.
- Strong JP, Malcom GT, Oalmann MC, Wissler RW. The PDAY study: natural history, risk factors, and pathobiology. Ann N Y Acad Sci 1997; 811:226–235.
On November 12, 2013, a joint task force for the American College of Cardiology and American Heart Association released new guidelines for treating high blood cholesterol to reduce the risk of atherosclerotic cardiovascular disease (ASCVD) in adults.1
This document arrives after several years of intense deliberation, 12 years after the third Adult Treatment Panel (ATP III) guidelines,2 and 8 years after an ATP III update recommending that low-density lipoprotein cholesterol (LDL-C) levels be lowered aggressively (to less than 70 mg/dL) as an option in patients at high risk.3 It represents a major shift in the approach to and management of high blood cholesterol and has sparked considerable controversy.
In the following commentary, we summarize the new guidelines and the philosophy employed by the task force in generating them. We will also examine some advantages and what we believe to be several shortcomings of the new guidelines. These latter points are illustrated through case examples.
IN RANDOMIZED CONTROLLED TRIALS WE TRUST
In collaboration with the National Heart, Lung, and Blood Institute of the National Institutes of Health, the American College of Cardiology and American Heart Association formed an expert panel task force in 2008.
The task force elected to use only evidence from randomized controlled trials, systematic reviews, and meta-analyses of randomized controlled trials (and only predefined outcomes of the trials, not post hoc analyses) in formulating its recommendations, with the goal of providing the strongest possible evidence.
The authors state that “By using [randomized controlled trial] data to identify those most likely to benefit [emphasis in original] from cholesterol-lowering statin therapy, the recommendations will be of value to primary care clinicians as well as specialists concerned with ASCVD prevention. Importantly, the recommendations were designed to be easy to use in the clinical setting, facilitating the implementation of a strategy of risk assessment and treatment focused on the prevention of ASCVD.”3 They also state the guidelines are meant to “inform clinical judgment, not replace it” and that clinician judgment in addition to discussion with patients remains vital.
During the deliberations, the National Heart, Lung, and Blood Institute removed itself from participating, stating its mission no longer included drafting new guidelines. Additionally, other initial members of the task force removed themselves because of disagreement and concerns about the direction of the new guidelines.
These guidelines, and their accompanying new cardiovascular risk calculator,4 were released without a preliminary period to allow for open discussion, comment, and critique by physicians outside the panel. No attempt was made to harmonize the guidelines with previous versions (eg, ATP III) or with current international guidelines.
WHAT’S NEW IN THE GUIDELINES?
The following are the major changes in the new guidelines for treating high blood cholesterol:
- Treatment goals for LDL-C and non-high-density lipoprotein cholesterol (non-HDL-C) are no longer recommended.
- High-intensity and moderate-intensity statin treatment is emphasized, and low-intensity statin therapy is nearly eliminated.
- “ASCVD” now includes stroke in addition to coronary heart disease and peripheral arterial disease.
- Four groups are targeted for treatment (see below).
- Nonstatin therapies have been markedly de-emphasized.
- No guidelines are provided for treating high triglyceride levels.
The new guidelines emphasize lifestyle modification as the foundation for reducing risk, regardless of cholesterol therapy. No recommendations are given for patients with New York Heart Association class II, III, or IV heart failure or for hemodialysis patients, because there were insufficient data from randomized controlled trials to support recommendations. Similarly, the guidelines apply only to people between the ages of 40 and 75 (risk calculator ages 40–79), because the authors believed there was not enough evidence from randomized controlled trials to allow development of guidelines outside of this age range.
FOUR MAJOR STATIN TREATMENT GROUPS
The new guidelines specify four groups that merit intensive or moderately intensive statin therapy (Table 1)1:
- People with clinical ASCVD
- People with LDL-C levels of 190 mg/dL or higher
- People with diabetes, age 40 to 75
- People without diabetes, age 40 to 75, with LDL-C levels 70–189 mg/dL, and a 10-year ASCVD risk of 7.5% or higher as determined by the new risk calculator4 (which also calculates the lifetime risk of ASCVD).
Below, we will address each of these four groups and provide case scenarios to consider. In general, our major disagreements with the new recommendations pertain to the first and fourth categories.
GROUP 1: PEOPLE WITH CLINICAL ASCVD
Advantages of the new guidelines
- They appropriately recommend statins in the highest tolerated doses as first-line treatment for this group at high risk.
- They designate all patients with ASCVD, including those with coronary, peripheral, and cerebrovascular disease, as a high-risk group.
- Without target LDL-C levels, treatment is simpler than before, requiring less monitoring of lipid levels. (This can also be seen as a limitation, as we discuss below.)
Limitations of the new guidelines
- They make follow-up LDL-C levels irrelevant, seeming to assume that there is no gradation in residual risk and, thus, no need to tailor therapy to the individual.
- Patients no longer have a goal to strive for or a way to monitor their progress.
- The guidelines ignore the pathophysiology of coronary artery disease and evidence of residual risk in patients on both moderate-intensity and high-intensity statin therapy.
- They also ignore the potential benefits of treating to lower LDL-C or non-HDL-C goals, thus eliminating consideration of multidrug therapy. They do not address patients with recurrent cardiovascular events already on maximal tolerated statin doses.
- They undermine the potential development and use of new therapies for dysplipidemia in patients with ASCVD.
Case 1: Is LDL-C 110 mg/dL low enough?
A 52-year-old African American man presents with newly discovered moderate coronary artery disease that is not severe enough to warrant stenting. He has no history of hypertension, diabetes mellitus, or smoking. His systolic blood pressure is 130 mm Hg, and his body mass index is 26 kg/m2. He exercises regularly and follows a low-cholesterol diet. He has the following fasting lipid values:
- Total cholesterol 290 mg/dL
- HDL-C 50 mg/dL
- Triglycerides 250 mg/dL
- Calculated LDL-C 190 mg/dL.
Two months later, after beginning atorvastatin 80 mg daily, meeting with a nutritionist, and redoubling his dietary efforts, his fasting lipid concentrations are:
- Total cholesterol 180 mg/dL
- HDL-C 55 mg/dL
- Triglycerides 75 mg/dL
- Calculated LDL-C 110 mg/dL.
Comment: Lack of LDL-C goals is a flaw
The new guidelines call for patients with known ASCVD, such as this patient, to receive intensive statin therapy—which he got.
However, once a patient is on therapy, the new guidelines do not encourage repeating the lipid panel other than to assess compliance. With intensive therapy, we expect a reduction in LDL-C of at least 50% (Table 1), but patient-to-patient differences in response to medications are common, and without repeat testing we would have no way of gauging this patient’s residual risk.
Further, the new guidelines emphasize the lack of hard outcome data supporting the addition of another lipid-lowering drug to a statin, although they do indicate that one can consider it. In a patient at high risk, such as this one, would you be comfortable with an LDL-C value of 110 mg/dL on maximum statin therapy? Would you consider adding a nonstatin drug?
A preponderance of data shows that LDL plays a causal role in ASCVD development and adverse events. Genetic data show that the LDL particle and the LDL receptor pathway are mechanistically linked to ASCVD pathogenesis, with lifetime exposure as a critical determinant of risk.5,6 Moreover, randomized controlled trials of statins and other studies of cholesterol-lowering show a reproducible relationship between the LDL-C level achieved and absolute risk (Figure 1).7–24 We believe the totality of data constitutes a strong rationale for targeting LDL-C and establishing goals for lowering its levels. For these reasons, we believe that removing LDL-C goals is a fundamental flaw of the new guidelines.
The reason for the lack of data from randomized controlled trials demonstrating benefits of adding therapies to statins (when LDL-C is still high) or benefits of treating to specific goals is that no such trials have been performed. Even trials of nonpharmacologic means of lowering LDL-C, such as ileal bypass, which was used in the Program on the Surgical Control of the Hyperlipidemias trial,20 provide independent evidence that lowering LDL-C reduces the risk of ASCVD (Figure 1).
In addition, trials of nonstatin drugs, such as the Coronary Drug Project,25 which tested niacin, also showed outcome benefits. On the other hand, studies such as the Atherothrombosis Intervention in Metabolic Syndrome With Low HDL/High Triglycerides: Impact on Global Health26 and Treatment of HDL to Reduce the Incidence of Vascular Events27 trials did not show additional risk reduction when niacin was added to statin therapy. However, the study designs arguably had flaws, including requirement of aggressive LDL-lowering with statins, with LDL-C levels below 70 to 80 mg/dL before randomization.
Therefore, these trials do not tell us what to do for a patient on maximal intensive therapy who has recurrent ASCVD events or who, like our patient, has an LDL-C level higher than previous targets.
For this patient, we would recommend adding a second medication to further lower his LDL-C, but discussing with him the absence of proven benefit in clinical trials and the risks of side effects. At present, lacking LDL-C goals in the new guidelines, we are keeping with the ATP III goals to help guide therapeutic choices and individualize patient management.
GROUP 2: PEOPLE WITH LDL-C ≥ 190
Advantages of the new guidelines
- They state that these patients should receive statins in the highest tolerated doses, which is universally accepted.
Limitations of the new guidelines
- The new guidelines mention only that one “may consider” adding a second agent if LDL-C remains above 190 mg/dL after maximum-dose therapy. Patients with familial hypercholesterolemia or other severe forms of hypercholesterolemia typically end up on multidrug therapy to further reduce LDL-C. The absence of randomized controlled trial data in this setting to show an additive value of second and third lipid-lowering agents does not mean these agents do not provide benefit.
GROUP 3: DIABETES, AGE 40–75, LDL-C 70–189, NO CLINICAL ASCVD
Advantages of the new guidelines
- They call for aggressive treatment of people with diabetes, a group at high risk that derives significant benefit from statin therapy, as shown in randomized controlled trials.
Limitations of the new guidelines
- Although high-intensity statin therapy is indicated for this group, we believe that, using the new risk calculator, some patients may receive overly aggressive treatment, thus increasing the possibility of statin side effects.
- The guidelines do not address patients younger than 40 or older than 75.
- Diabetic patients have a high residual risk of ASCVD events, even on statin therapy. Yet the guidelines ignore the potential benefits of more aggressive LDL-lowering or non-LDL secondary targets for therapy.
Case 2: How low is too low?
A 63-year-old white woman, a nonsmoker with recently diagnosed diabetes, is seen by her primary care physician. She has hypertension, for which she takes lisinopril 5 mg daily. Her fasting lipid values are:
- Total cholesterol 160 mg/dL
- HDL-C 64 mg/dL
- Triglycerides 100 mg/dL
- Calculated LDL-C 76 mg/dL.
Her systolic blood pressure is 129 mm Hg, and based on the new risk calculator, her 10-year risk of cardiovascular disease is 10.2%. According to the new guidelines, she should be started on high-intensity statin treatment (Table 1).
Although this is an acceptable initial course of action, it necessitates close vigilance, since it may actually drive her LDL-C level too low. Randomized controlled trials have typically used an LDL-C concentration of less than or equal to 25 mg/dL as the safety cutoff. With a typical LDL-C reduction of at least 50% on high-intensity statins, our patient’s expected LDL-C level will likely be in the low 30s. We believe this would be a good outcome, provided that she tolerates the medication without adverse effects. However, responses to statins vary from patient to patient.
High-intensity statin therapy may not be necessary to reduce risk adequately in all patients who have diabetes without preexisting vascular disease. The Collaborative Atorvastatin Diabetes Study12 compared atorvastatin 10 mg vs placebo in people with type 2 diabetes, age 40 to 75, who had one or more cardiovascular risk factors but no signs or symptoms of preexisting ASCVD and who had only average or below-average cholesterol levels—precisely like this patient. The trial was terminated early because of a clear benefit (a 37% reduction in the composite end point of major adverse cardiovascular events) in the intervention group. For our patient, we believe an alternative and acceptable approach would be to begin moderate-intensity statin therapy (eg, with atorvastatin 10 mg) (Table 1).
Alternatively, in a patient with diabetes and previous atherosclerotic vascular disease or with a high 10-year risk and high LDL-C, limiting treatment to high-intensity statin therapy by itself may deny them the potential benefits of combination therapies and targeting to lower LDL-C levels or non-HDL-C secondary targets. Guidelines from the American Diabetes Association28 and the American Association of Clinical Endocrinologists29 continue to recommend an LDL-C goal of less than 70 mg/dL in patients at high risk, a non-HDL-C less than 100 mg/dL, an apolipoprotein B less than 80 mg/dL, and an LDL particle number less than 1,000 nmol/L.
GROUP 4: AGE 40–75, LDL-C 70–189, NO ASCVD, BUT 10-YEAR RISK ≥ 7.5%
Advantages of the new guidelines
- They may reduce ASCVD events for patients at higher risk.
- The risk calculator is easy to use and focuses on global risk, ie, all forms of ASCVD.
- The guidelines promote discussion of risks and benefits between patients and providers.
Limitations of the new guidelines
- The new risk calculator is controversial (see below).
- There is potential for overtreatment, particularly in older patients.
- There is potential for undertreatment, particularly in patients with an elevated LDL-C but whose 10-year risk is less than 7.5% because they are young.
- The guidelines do not address patients younger than 40 or older than 75.
- They do not take into account some traditional risk factors, such as family history, and nontraditional risk factors such as C-reactive protein as measured by ultrasensitive assays, lipoprotein(a), and apolipoprotein B.
Risk calculator controversy
The new risk calculator has aroused strong opinions on both sides of the aisle.
Shortly after the new guidelines were released, cardiologists Dr. Paul Ridker and Dr. Nancy Cook from Brigham and Women’s Hospital in Boston published analyses30 showing that the new risk calculator, which was based on older data from several large cohorts such as the Atherosclerosis Risk in Communities study,31 the Cardiovascular Health Study,32 the Coronary Artery Risk Development in Young Adults study,33 and the Framingham Heart Study,34,35 was inaccurate in other cohorts. Specifically, in more-recent cohorts (the Women’s Health Study,36 Physicians’ Health Study,37 and Women’s Health Initiative38), the new calculator overestimates the 10-year risk of ASCVD by 75% to 150%.30 Using the new calculator would make approximately 30 million more Americans eligible for statin treatment. The concern is that patients at lower risk would be treated and exposed to potential side effects of statin therapy.
In addition, the risk calculator relies heavily on age and sex and does not include other factors such as triglyceride level, family history, C-reactive protein, or lipoprotein(a). Importantly, and somewhat ironically given the otherwise absolute adherence to randomized controlled trial data for guideline development, the risk calculator has never been verified in prospective studies to adequately show that using it reduces ASCVD events.
Case 3: Overtreating a primary prevention patient
Based on the risk calculator, essentially any African American man in his early 60s with no other risk factors has a 10-year risk of ASCVD of 7.5% or higher and, according to the new guidelines, should receive at least moderate-intensity statin therapy.
For example, consider a 64-year-old African American man whose systolic blood pressure is 129 mm Hg, who does not smoke, does not have diabetes, and does not have hypertension, and whose total cholesterol level is 180 mg/dL, HDL-C 70 mg/dL, triglycerides 130 mg/dL, and calculated LDL-C 84 mg/dL. His calculated 10-year risk is, surprisingly, 7.5%.
Alternatively, his twin brother is a two-pack-per-day smoker with untreated hypertension and systolic blood pressure 150 mm Hg, with fasting total cholesterol 153 mg/dL, HDL-C 70 mg/dL, triglycerides 60 mg/dL, and LDL-C 71 mg/dL. His calculated 10-year risk is 10.5%, so according to the new guidelines, he too should receive high-intensity statin therapy. Yet this patient clearly needs better blood pressure control and smoking cessation as his primary risk-reduction efforts, not a statin. While assessing global risk is important, a shortcoming of the new guidelines is that they can inappropriately lead to treating the risk score, not individualizing the treatment to the patient. Because of the errors inherent in the risk calculator, some experts have called for a temporary halt on implementing the new guidelines until the risk calculator can be further validated. In November 2013, the American Heart Association and the American College of Cardiology reaffirmed their support of the new guidelines and recommended that they be implemented as planned. As of the time this manuscript goes to print, there are no plans to halt implementation of the new guidelines.
Case 4: Undertreating a primary prevention patient
A 25-year-old white man with no medical history has a total cholesterol level of 310 mg/dL, HDL-C 50 mg/dL, triglycerides 400 mg/dL, and calculated LDL-C 180 mg/dL. He does not smoke or have hypertension or diabetes but has a strong family history of premature coronary disease (his father died of myocardial infarction at age 42). His body mass index is 25 kg/m2. Because he is less than 40 years old, the risk calculator does not apply to him.
If we assume he remains untreated and returns at age 40 with the same clinical factors and laboratory values, his calculated 10-year risk of an ASCVD event according to the new risk calculator will still be only 3.1%. Assuming his medical history remains unchanged as he continues to age, his 10-year risk would not reach 7.5% until he is 58. Would you feel comfortable waiting 33 years before starting statin therapy in this patient?
Waiting for dyslipidemic patients to reach middle age before starting LDL-C-lowering therapy is a failure of prevention. For practical reasons, there are no data from randomized controlled trials with hard outcomes in younger people. Nevertheless, a tenet of preventive cardiology is that cumulative exposure accelerates the “vascular age” ahead of the chronological age. This case illustrates why individualized recommendations guided by LDL-C goals as a target for therapy are needed. For this 25-year-old patient, we would recommend starting an intermediate- or high-potency statin.
Case 5: Rheumatoid arthritis
A 60-year-old postmenopausal white woman with severe rheumatoid arthritis presents for cholesterol evaluation. Her total cholesterol level is 235 mg/dL, HDL-C 50 mg/dL, and LDL-C 165 mg/dL. She does not smoke or have hypertension or diabetes. Her systolic blood pressure is 110 mm Hg. She has elevated C-reactive protein on an ultrasensitive assay and elevated lipoprotein(a).
Her calculated 10-year risk of ASCVD is 3.0%. Assuming her medical history remains the same, she would not reach a calculated 10-year risk of at least 7.5% until age 70. We suggest starting moderate- or high-dose statin therapy in this case, based on data (not from randomized controlled trials) showing an increased risk of ASCVD events in patients with rheumatologic disease, increased lipoprotein(a), and inflammatory markers like C-reactive protein. However, the current guidelines do not address this scenario, other than to suggest that clinician consideration can be given to other risk markers such as these, and that these findings should be discussed in detail with the patient. The Justification for the Use of Statins in Primary Prevention: An Intervention Trial Evaluating Rosuvastatin trial16 showed a dramatic ASCVD risk reduction in just such patients (Figure 1).
APPLAUSE—AND RESERVATIONS
The newest guidelines for treating high blood cholesterol represent a monumental shift away from using target levels of LDL-C and non-HDL-C and toward a focus on statin intensity for patients in the four highest-risk groups.
We applaud the expert panel for its idealistic approach of using only data from randomized controlled trials, for placing more emphasis on higher-intensity statin treatment, for including stroke in the new definition of ASCVD, and for focusing more attention on treating diabetic patients more aggressively. Simplifying the guidelines is a noble goal. Emphasizing moderate-to-high-intensity statin therapy in patients at moderate-to-high risk should have substantial long-term public health benefits.
However, as we have shown in the case examples, there are significant limitations, and some patients can end up being overtreated, while others may be undertreated.
Guidelines need to be crafted by looking at all the evidence, including the pathophysiology of the disease process, not just data from randomized controlled trials. It is difficult to implement a guideline that on one hand used randomized controlled trials exclusively for recommendations, but on the other hand used an untested risk calculator to guide therapy. Randomized controlled trials are not available for every scenario.
Further, absence of randomized controlled trial data in a given scenario should not be interpreted as evidence of lack of benefit. An example of this is a primary-prevention patient under age 40 with elevated LDL-C below the 190 mg/dL cutoff who otherwise is healthy and without risk factors (eg, Case 4). By disregarding all evidence that is not from randomized controlled trials, the expert panel fails to account for the extensive pathophysiology of ASCVD, which often begins at a young age and takes decades to develop.5,6,39 An entire generation of patients who have not reached the age of inclusion in most randomized controlled trials with hard outcomes is excluded (unless the LDL-C level is very high), potentially setting back decades of progress in the field of prevention. Prevention only works if started. With childhood and young adult obesity sharply rising, we should not fail to address the under-40-year-old patient population in our guidelines.
Guidelines are designed to be expert opinion, not to dictate practice. Focusing on the individual patient instead of the general population at risk, the expert panel appropriately emphasizes the “importance of clinician judgment, weighing potential benefits, adverse effects, drug-drug interactions and patient preferences.” However, by excluding all data that do not come from randomized controlled trials, the panel neglects a very large base of knowledge and leaves many clinicians without as much expert opinion as we had hoped for.
LDL-C goals are important: they provide a scorecard to help the patient with lifestyle and dietary changes. They provide the health care provider guidance in making treatment decisions and focusing on treatment of a single patient, not a population. Moreover, if a patient has difficulty taking standard doses of statins because of side effects, the absence of LDL-C goals makes decision-making nearly impossible. We hope physicians will rely on LDL-C goals in such situations, falling back on the ATP III recommendations, although many patients may simply go untreated until they present with ASCVD or until they “age in” to a higher risk category.
We suggest caution in strict adherence to the new guidelines and instead urge physicians to consider a hybrid of the old guidelines (using the ATP III LDL-C goals) and the new ones (emphasizing global risk assessment and high-intensity statin treatment).
On November 12, 2013, a joint task force for the American College of Cardiology and American Heart Association released new guidelines for treating high blood cholesterol to reduce the risk of atherosclerotic cardiovascular disease (ASCVD) in adults.1
This document arrives after several years of intense deliberation, 12 years after the third Adult Treatment Panel (ATP III) guidelines,2 and 8 years after an ATP III update recommending that low-density lipoprotein cholesterol (LDL-C) levels be lowered aggressively (to less than 70 mg/dL) as an option in patients at high risk.3 It represents a major shift in the approach to and management of high blood cholesterol and has sparked considerable controversy.
In the following commentary, we summarize the new guidelines and the philosophy employed by the task force in generating them. We will also examine some advantages and what we believe to be several shortcomings of the new guidelines. These latter points are illustrated through case examples.
IN RANDOMIZED CONTROLLED TRIALS WE TRUST
In collaboration with the National Heart, Lung, and Blood Institute of the National Institutes of Health, the American College of Cardiology and American Heart Association formed an expert panel task force in 2008.
The task force elected to use only evidence from randomized controlled trials, systematic reviews, and meta-analyses of randomized controlled trials (and only predefined outcomes of the trials, not post hoc analyses) in formulating its recommendations, with the goal of providing the strongest possible evidence.
The authors state that “By using [randomized controlled trial] data to identify those most likely to benefit [emphasis in original] from cholesterol-lowering statin therapy, the recommendations will be of value to primary care clinicians as well as specialists concerned with ASCVD prevention. Importantly, the recommendations were designed to be easy to use in the clinical setting, facilitating the implementation of a strategy of risk assessment and treatment focused on the prevention of ASCVD.”3 They also state the guidelines are meant to “inform clinical judgment, not replace it” and that clinician judgment in addition to discussion with patients remains vital.
During the deliberations, the National Heart, Lung, and Blood Institute removed itself from participating, stating its mission no longer included drafting new guidelines. Additionally, other initial members of the task force removed themselves because of disagreement and concerns about the direction of the new guidelines.
These guidelines, and their accompanying new cardiovascular risk calculator,4 were released without a preliminary period to allow for open discussion, comment, and critique by physicians outside the panel. No attempt was made to harmonize the guidelines with previous versions (eg, ATP III) or with current international guidelines.
WHAT’S NEW IN THE GUIDELINES?
The following are the major changes in the new guidelines for treating high blood cholesterol:
- Treatment goals for LDL-C and non-high-density lipoprotein cholesterol (non-HDL-C) are no longer recommended.
- High-intensity and moderate-intensity statin treatment is emphasized, and low-intensity statin therapy is nearly eliminated.
- “ASCVD” now includes stroke in addition to coronary heart disease and peripheral arterial disease.
- Four groups are targeted for treatment (see below).
- Nonstatin therapies have been markedly de-emphasized.
- No guidelines are provided for treating high triglyceride levels.
The new guidelines emphasize lifestyle modification as the foundation for reducing risk, regardless of cholesterol therapy. No recommendations are given for patients with New York Heart Association class II, III, or IV heart failure or for hemodialysis patients, because there were insufficient data from randomized controlled trials to support recommendations. Similarly, the guidelines apply only to people between the ages of 40 and 75 (risk calculator ages 40–79), because the authors believed there was not enough evidence from randomized controlled trials to allow development of guidelines outside of this age range.
FOUR MAJOR STATIN TREATMENT GROUPS
The new guidelines specify four groups that merit intensive or moderately intensive statin therapy (Table 1)1:
- People with clinical ASCVD
- People with LDL-C levels of 190 mg/dL or higher
- People with diabetes, age 40 to 75
- People without diabetes, age 40 to 75, with LDL-C levels 70–189 mg/dL, and a 10-year ASCVD risk of 7.5% or higher as determined by the new risk calculator4 (which also calculates the lifetime risk of ASCVD).
Below, we will address each of these four groups and provide case scenarios to consider. In general, our major disagreements with the new recommendations pertain to the first and fourth categories.
GROUP 1: PEOPLE WITH CLINICAL ASCVD
Advantages of the new guidelines
- They appropriately recommend statins in the highest tolerated doses as first-line treatment for this group at high risk.
- They designate all patients with ASCVD, including those with coronary, peripheral, and cerebrovascular disease, as a high-risk group.
- Without target LDL-C levels, treatment is simpler than before, requiring less monitoring of lipid levels. (This can also be seen as a limitation, as we discuss below.)
Limitations of the new guidelines
- They make follow-up LDL-C levels irrelevant, seeming to assume that there is no gradation in residual risk and, thus, no need to tailor therapy to the individual.
- Patients no longer have a goal to strive for or a way to monitor their progress.
- The guidelines ignore the pathophysiology of coronary artery disease and evidence of residual risk in patients on both moderate-intensity and high-intensity statin therapy.
- They also ignore the potential benefits of treating to lower LDL-C or non-HDL-C goals, thus eliminating consideration of multidrug therapy. They do not address patients with recurrent cardiovascular events already on maximal tolerated statin doses.
- They undermine the potential development and use of new therapies for dysplipidemia in patients with ASCVD.
Case 1: Is LDL-C 110 mg/dL low enough?
A 52-year-old African American man presents with newly discovered moderate coronary artery disease that is not severe enough to warrant stenting. He has no history of hypertension, diabetes mellitus, or smoking. His systolic blood pressure is 130 mm Hg, and his body mass index is 26 kg/m2. He exercises regularly and follows a low-cholesterol diet. He has the following fasting lipid values:
- Total cholesterol 290 mg/dL
- HDL-C 50 mg/dL
- Triglycerides 250 mg/dL
- Calculated LDL-C 190 mg/dL.
Two months later, after beginning atorvastatin 80 mg daily, meeting with a nutritionist, and redoubling his dietary efforts, his fasting lipid concentrations are:
- Total cholesterol 180 mg/dL
- HDL-C 55 mg/dL
- Triglycerides 75 mg/dL
- Calculated LDL-C 110 mg/dL.
Comment: Lack of LDL-C goals is a flaw
The new guidelines call for patients with known ASCVD, such as this patient, to receive intensive statin therapy—which he got.
However, once a patient is on therapy, the new guidelines do not encourage repeating the lipid panel other than to assess compliance. With intensive therapy, we expect a reduction in LDL-C of at least 50% (Table 1), but patient-to-patient differences in response to medications are common, and without repeat testing we would have no way of gauging this patient’s residual risk.
Further, the new guidelines emphasize the lack of hard outcome data supporting the addition of another lipid-lowering drug to a statin, although they do indicate that one can consider it. In a patient at high risk, such as this one, would you be comfortable with an LDL-C value of 110 mg/dL on maximum statin therapy? Would you consider adding a nonstatin drug?
A preponderance of data shows that LDL plays a causal role in ASCVD development and adverse events. Genetic data show that the LDL particle and the LDL receptor pathway are mechanistically linked to ASCVD pathogenesis, with lifetime exposure as a critical determinant of risk.5,6 Moreover, randomized controlled trials of statins and other studies of cholesterol-lowering show a reproducible relationship between the LDL-C level achieved and absolute risk (Figure 1).7–24 We believe the totality of data constitutes a strong rationale for targeting LDL-C and establishing goals for lowering its levels. For these reasons, we believe that removing LDL-C goals is a fundamental flaw of the new guidelines.
The reason for the lack of data from randomized controlled trials demonstrating benefits of adding therapies to statins (when LDL-C is still high) or benefits of treating to specific goals is that no such trials have been performed. Even trials of nonpharmacologic means of lowering LDL-C, such as ileal bypass, which was used in the Program on the Surgical Control of the Hyperlipidemias trial,20 provide independent evidence that lowering LDL-C reduces the risk of ASCVD (Figure 1).
In addition, trials of nonstatin drugs, such as the Coronary Drug Project,25 which tested niacin, also showed outcome benefits. On the other hand, studies such as the Atherothrombosis Intervention in Metabolic Syndrome With Low HDL/High Triglycerides: Impact on Global Health26 and Treatment of HDL to Reduce the Incidence of Vascular Events27 trials did not show additional risk reduction when niacin was added to statin therapy. However, the study designs arguably had flaws, including requirement of aggressive LDL-lowering with statins, with LDL-C levels below 70 to 80 mg/dL before randomization.
Therefore, these trials do not tell us what to do for a patient on maximal intensive therapy who has recurrent ASCVD events or who, like our patient, has an LDL-C level higher than previous targets.
For this patient, we would recommend adding a second medication to further lower his LDL-C, but discussing with him the absence of proven benefit in clinical trials and the risks of side effects. At present, lacking LDL-C goals in the new guidelines, we are keeping with the ATP III goals to help guide therapeutic choices and individualize patient management.
GROUP 2: PEOPLE WITH LDL-C ≥ 190
Advantages of the new guidelines
- They state that these patients should receive statins in the highest tolerated doses, which is universally accepted.
Limitations of the new guidelines
- The new guidelines mention only that one “may consider” adding a second agent if LDL-C remains above 190 mg/dL after maximum-dose therapy. Patients with familial hypercholesterolemia or other severe forms of hypercholesterolemia typically end up on multidrug therapy to further reduce LDL-C. The absence of randomized controlled trial data in this setting to show an additive value of second and third lipid-lowering agents does not mean these agents do not provide benefit.
GROUP 3: DIABETES, AGE 40–75, LDL-C 70–189, NO CLINICAL ASCVD
Advantages of the new guidelines
- They call for aggressive treatment of people with diabetes, a group at high risk that derives significant benefit from statin therapy, as shown in randomized controlled trials.
Limitations of the new guidelines
- Although high-intensity statin therapy is indicated for this group, we believe that, using the new risk calculator, some patients may receive overly aggressive treatment, thus increasing the possibility of statin side effects.
- The guidelines do not address patients younger than 40 or older than 75.
- Diabetic patients have a high residual risk of ASCVD events, even on statin therapy. Yet the guidelines ignore the potential benefits of more aggressive LDL-lowering or non-LDL secondary targets for therapy.
Case 2: How low is too low?
A 63-year-old white woman, a nonsmoker with recently diagnosed diabetes, is seen by her primary care physician. She has hypertension, for which she takes lisinopril 5 mg daily. Her fasting lipid values are:
- Total cholesterol 160 mg/dL
- HDL-C 64 mg/dL
- Triglycerides 100 mg/dL
- Calculated LDL-C 76 mg/dL.
Her systolic blood pressure is 129 mm Hg, and based on the new risk calculator, her 10-year risk of cardiovascular disease is 10.2%. According to the new guidelines, she should be started on high-intensity statin treatment (Table 1).
Although this is an acceptable initial course of action, it necessitates close vigilance, since it may actually drive her LDL-C level too low. Randomized controlled trials have typically used an LDL-C concentration of less than or equal to 25 mg/dL as the safety cutoff. With a typical LDL-C reduction of at least 50% on high-intensity statins, our patient’s expected LDL-C level will likely be in the low 30s. We believe this would be a good outcome, provided that she tolerates the medication without adverse effects. However, responses to statins vary from patient to patient.
High-intensity statin therapy may not be necessary to reduce risk adequately in all patients who have diabetes without preexisting vascular disease. The Collaborative Atorvastatin Diabetes Study12 compared atorvastatin 10 mg vs placebo in people with type 2 diabetes, age 40 to 75, who had one or more cardiovascular risk factors but no signs or symptoms of preexisting ASCVD and who had only average or below-average cholesterol levels—precisely like this patient. The trial was terminated early because of a clear benefit (a 37% reduction in the composite end point of major adverse cardiovascular events) in the intervention group. For our patient, we believe an alternative and acceptable approach would be to begin moderate-intensity statin therapy (eg, with atorvastatin 10 mg) (Table 1).
Alternatively, in a patient with diabetes and previous atherosclerotic vascular disease or with a high 10-year risk and high LDL-C, limiting treatment to high-intensity statin therapy by itself may deny them the potential benefits of combination therapies and targeting to lower LDL-C levels or non-HDL-C secondary targets. Guidelines from the American Diabetes Association28 and the American Association of Clinical Endocrinologists29 continue to recommend an LDL-C goal of less than 70 mg/dL in patients at high risk, a non-HDL-C less than 100 mg/dL, an apolipoprotein B less than 80 mg/dL, and an LDL particle number less than 1,000 nmol/L.
GROUP 4: AGE 40–75, LDL-C 70–189, NO ASCVD, BUT 10-YEAR RISK ≥ 7.5%
Advantages of the new guidelines
- They may reduce ASCVD events for patients at higher risk.
- The risk calculator is easy to use and focuses on global risk, ie, all forms of ASCVD.
- The guidelines promote discussion of risks and benefits between patients and providers.
Limitations of the new guidelines
- The new risk calculator is controversial (see below).
- There is potential for overtreatment, particularly in older patients.
- There is potential for undertreatment, particularly in patients with an elevated LDL-C but whose 10-year risk is less than 7.5% because they are young.
- The guidelines do not address patients younger than 40 or older than 75.
- They do not take into account some traditional risk factors, such as family history, and nontraditional risk factors such as C-reactive protein as measured by ultrasensitive assays, lipoprotein(a), and apolipoprotein B.
Risk calculator controversy
The new risk calculator has aroused strong opinions on both sides of the aisle.
Shortly after the new guidelines were released, cardiologists Dr. Paul Ridker and Dr. Nancy Cook from Brigham and Women’s Hospital in Boston published analyses30 showing that the new risk calculator, which was based on older data from several large cohorts such as the Atherosclerosis Risk in Communities study,31 the Cardiovascular Health Study,32 the Coronary Artery Risk Development in Young Adults study,33 and the Framingham Heart Study,34,35 was inaccurate in other cohorts. Specifically, in more-recent cohorts (the Women’s Health Study,36 Physicians’ Health Study,37 and Women’s Health Initiative38), the new calculator overestimates the 10-year risk of ASCVD by 75% to 150%.30 Using the new calculator would make approximately 30 million more Americans eligible for statin treatment. The concern is that patients at lower risk would be treated and exposed to potential side effects of statin therapy.
In addition, the risk calculator relies heavily on age and sex and does not include other factors such as triglyceride level, family history, C-reactive protein, or lipoprotein(a). Importantly, and somewhat ironically given the otherwise absolute adherence to randomized controlled trial data for guideline development, the risk calculator has never been verified in prospective studies to adequately show that using it reduces ASCVD events.
Case 3: Overtreating a primary prevention patient
Based on the risk calculator, essentially any African American man in his early 60s with no other risk factors has a 10-year risk of ASCVD of 7.5% or higher and, according to the new guidelines, should receive at least moderate-intensity statin therapy.
For example, consider a 64-year-old African American man whose systolic blood pressure is 129 mm Hg, who does not smoke, does not have diabetes, and does not have hypertension, and whose total cholesterol level is 180 mg/dL, HDL-C 70 mg/dL, triglycerides 130 mg/dL, and calculated LDL-C 84 mg/dL. His calculated 10-year risk is, surprisingly, 7.5%.
Alternatively, his twin brother is a two-pack-per-day smoker with untreated hypertension and systolic blood pressure 150 mm Hg, with fasting total cholesterol 153 mg/dL, HDL-C 70 mg/dL, triglycerides 60 mg/dL, and LDL-C 71 mg/dL. His calculated 10-year risk is 10.5%, so according to the new guidelines, he too should receive high-intensity statin therapy. Yet this patient clearly needs better blood pressure control and smoking cessation as his primary risk-reduction efforts, not a statin. While assessing global risk is important, a shortcoming of the new guidelines is that they can inappropriately lead to treating the risk score, not individualizing the treatment to the patient. Because of the errors inherent in the risk calculator, some experts have called for a temporary halt on implementing the new guidelines until the risk calculator can be further validated. In November 2013, the American Heart Association and the American College of Cardiology reaffirmed their support of the new guidelines and recommended that they be implemented as planned. As of the time this manuscript goes to print, there are no plans to halt implementation of the new guidelines.
Case 4: Undertreating a primary prevention patient
A 25-year-old white man with no medical history has a total cholesterol level of 310 mg/dL, HDL-C 50 mg/dL, triglycerides 400 mg/dL, and calculated LDL-C 180 mg/dL. He does not smoke or have hypertension or diabetes but has a strong family history of premature coronary disease (his father died of myocardial infarction at age 42). His body mass index is 25 kg/m2. Because he is less than 40 years old, the risk calculator does not apply to him.
If we assume he remains untreated and returns at age 40 with the same clinical factors and laboratory values, his calculated 10-year risk of an ASCVD event according to the new risk calculator will still be only 3.1%. Assuming his medical history remains unchanged as he continues to age, his 10-year risk would not reach 7.5% until he is 58. Would you feel comfortable waiting 33 years before starting statin therapy in this patient?
Waiting for dyslipidemic patients to reach middle age before starting LDL-C-lowering therapy is a failure of prevention. For practical reasons, there are no data from randomized controlled trials with hard outcomes in younger people. Nevertheless, a tenet of preventive cardiology is that cumulative exposure accelerates the “vascular age” ahead of the chronological age. This case illustrates why individualized recommendations guided by LDL-C goals as a target for therapy are needed. For this 25-year-old patient, we would recommend starting an intermediate- or high-potency statin.
Case 5: Rheumatoid arthritis
A 60-year-old postmenopausal white woman with severe rheumatoid arthritis presents for cholesterol evaluation. Her total cholesterol level is 235 mg/dL, HDL-C 50 mg/dL, and LDL-C 165 mg/dL. She does not smoke or have hypertension or diabetes. Her systolic blood pressure is 110 mm Hg. She has elevated C-reactive protein on an ultrasensitive assay and elevated lipoprotein(a).
Her calculated 10-year risk of ASCVD is 3.0%. Assuming her medical history remains the same, she would not reach a calculated 10-year risk of at least 7.5% until age 70. We suggest starting moderate- or high-dose statin therapy in this case, based on data (not from randomized controlled trials) showing an increased risk of ASCVD events in patients with rheumatologic disease, increased lipoprotein(a), and inflammatory markers like C-reactive protein. However, the current guidelines do not address this scenario, other than to suggest that clinician consideration can be given to other risk markers such as these, and that these findings should be discussed in detail with the patient. The Justification for the Use of Statins in Primary Prevention: An Intervention Trial Evaluating Rosuvastatin trial16 showed a dramatic ASCVD risk reduction in just such patients (Figure 1).
APPLAUSE—AND RESERVATIONS
The newest guidelines for treating high blood cholesterol represent a monumental shift away from using target levels of LDL-C and non-HDL-C and toward a focus on statin intensity for patients in the four highest-risk groups.
We applaud the expert panel for its idealistic approach of using only data from randomized controlled trials, for placing more emphasis on higher-intensity statin treatment, for including stroke in the new definition of ASCVD, and for focusing more attention on treating diabetic patients more aggressively. Simplifying the guidelines is a noble goal. Emphasizing moderate-to-high-intensity statin therapy in patients at moderate-to-high risk should have substantial long-term public health benefits.
However, as we have shown in the case examples, there are significant limitations, and some patients can end up being overtreated, while others may be undertreated.
Guidelines need to be crafted by looking at all the evidence, including the pathophysiology of the disease process, not just data from randomized controlled trials. It is difficult to implement a guideline that on one hand used randomized controlled trials exclusively for recommendations, but on the other hand used an untested risk calculator to guide therapy. Randomized controlled trials are not available for every scenario.
Further, absence of randomized controlled trial data in a given scenario should not be interpreted as evidence of lack of benefit. An example of this is a primary-prevention patient under age 40 with elevated LDL-C below the 190 mg/dL cutoff who otherwise is healthy and without risk factors (eg, Case 4). By disregarding all evidence that is not from randomized controlled trials, the expert panel fails to account for the extensive pathophysiology of ASCVD, which often begins at a young age and takes decades to develop.5,6,39 An entire generation of patients who have not reached the age of inclusion in most randomized controlled trials with hard outcomes is excluded (unless the LDL-C level is very high), potentially setting back decades of progress in the field of prevention. Prevention only works if started. With childhood and young adult obesity sharply rising, we should not fail to address the under-40-year-old patient population in our guidelines.
Guidelines are designed to be expert opinion, not to dictate practice. Focusing on the individual patient instead of the general population at risk, the expert panel appropriately emphasizes the “importance of clinician judgment, weighing potential benefits, adverse effects, drug-drug interactions and patient preferences.” However, by excluding all data that do not come from randomized controlled trials, the panel neglects a very large base of knowledge and leaves many clinicians without as much expert opinion as we had hoped for.
LDL-C goals are important: they provide a scorecard to help the patient with lifestyle and dietary changes. They provide the health care provider guidance in making treatment decisions and focusing on treatment of a single patient, not a population. Moreover, if a patient has difficulty taking standard doses of statins because of side effects, the absence of LDL-C goals makes decision-making nearly impossible. We hope physicians will rely on LDL-C goals in such situations, falling back on the ATP III recommendations, although many patients may simply go untreated until they present with ASCVD or until they “age in” to a higher risk category.
We suggest caution in strict adherence to the new guidelines and instead urge physicians to consider a hybrid of the old guidelines (using the ATP III LDL-C goals) and the new ones (emphasizing global risk assessment and high-intensity statin treatment).
- Stone NJ, Robinson J, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; published online Nov 13. DOI: 10.1016/j.jacc.2013.11.002.
- National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002; 106:3143–3421.
- Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004; 110:227–239.
- American Heart Association. 2013 Prevention guidelines tools. CV risk calculator. http://my.americanheart.org/professional/StatementsGuidelines/PreventionGuidelines/Prevention-Guidelines_UCM_457698_SubHomePage.jsp. Accessed December 10, 2013.
- Goldstein JL, Brown MS. The LDL receptor. Arterioscler Thromb Vasc Biol 2009; 29:431–438.
- Horton JD, Cohen JC, Hobbs HH. PCSK9: a convertase that coordinates LDL catabolism. J Lipid Res 2009; 50(suppl):S172–S177.
- Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 1994; 344:1383–1389.
- de Lemos JA, Blazing MA, Wiviott SD, et al; for the A to Z Investigators. Early intensive vs a delayed conservative simvastatin strategy in patients with acute coronary syndromes. Phase Z of the A to Z trial. JAMA 2004; 292:1307–1316.
- Downs JR, Clearfield M, Weis S, et al; for the AFCAPS/TexCAPS Research Group. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels. Results of AFCAPS/TexCAPS. JAMA 1998; 279:1615–1622.
- Koren MJ, Hunninghake DB, on behalf of the ALLIANCE investigators. Clinical outcomes in managed-care patients with coronary heart disease treated aggressively in lipid-lowering disease management clinics. J Am Coll Cardiol 2004; 44:1772–1779.
- Sever PS, Dahlof B, Poulter NR, et al; ASCOT investigators. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial - Lipid Lowering Arm (ASCOT-LLA): a multicentre randomised controlled trial. Lancet 2003; 361:1149–1158.
- Colhoun HM, Betteridge DJ, Durrington PN, et al; on behalf of the CARDS Investigators. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): multicentre randomised placebo-controlled trial. Lancet 2004; 364:685–696.
- Sacks FM, Pfeffer MA, Moye LA, et al; for the Cholesterol and Recurrent Events Trial Investigators. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. N Engl J Med 1996; 335:1001–1009.
- Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20 536 high-risk individuals: a randomised placebo-controlled trial. Lancet 2002; 360:7–22.
- Pedersen TR, Faegeman O, Kastelein JJ, et al. Incremental Decrease in End Points Through Aggressive Lipid Lowering Study Group. High-dose atorvastatin vs usual-dose simvastatin for secondary prevention after myocardial infarction: the IDEAL study: a randomized controlled trial. JAMA 2005; 294:2437–2445.
- Ridker PM, Danielson E, Fonseca FAH, et al; for the JUPITER Study Group. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- LIPID Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N Engl J Med 1998; 339:1349–1357.
- Nakamura H, Arakawa K, Itakura H, et al; for the MEGA Study Group. Primary prevention of cardiovascular disease with pravastatin Japan (MEGA Study): a prospective rabndomised controlled trial. Lancet 2006; 368:1155–1163.
- Schwartz GG, Olsson AG, Ezekowitz MD, et al. Myocardial Ischemia Reduction with Aggreessive Cholesterol Lowering (MIRACL) Study Investigators. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: the MIRACL study: a randomized controlled trial. JAMA 2001; 285:1711–1718.
- Buchwald H, Varco RL, Matts JP, et al. Effect of partial ileal bypass surgery on mortality and morbidity from coronary heart disease in patients with hypercholesterolemia: report of the Program on the Surgical Control of the Hyperlipidemias (POSCH). N Engl J Med 1990; 323:946–955.
- Cannon CP, Braunwald E, McCabe CH, et al; for the Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 Investigators. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- Baigent C, Landray MJ, Reith C, et al; SHARP Investigators. The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (Study of Heart and Renal Protection): a randomised placebo-controlled trial. Lancet 2011; 377:2181–2192.
- LaRosa JC, Grundy SM, Waters DD, et al. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med 2005; 352:1425–1435.
- Shepherd J, Cobbe SM, Ford I, et al; for the West of Scotland Coronary Prevention Study Group. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. N Engl J Med 1995; 333:1301–1308.
- Canner PL, Berge KG, Wenger NK, et al. Fifteen year mortality in Coronary Drug Project patients: long-term benefit with niacin. J Am Coll Cardiol 1989; 8:1245–1255.
- AIM-HIGH Investigators, Boden WE, Probstfield JL, Anderson T, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med 2011; 365:2255–2267.
- HPS2-Thrive Collaborative Group. HPS2-THRIVE randomized placebo-controlled trial in 25 673 high-risk patients of ER niacin/laropiprant: trial design, pre-specified muscle and liver outcomes, and reasons for stopping study treatment. Eur Heart J 2013; 34:1279–1291.
- American Diabetes Association. Standards of medical care in diabetes—2013. Diabetes Care 2013; 36(suppl 1):S11–S66.
- Garber AJ, Abrahamson MJ, Barzilay JI, et al. American Association of Clinical Endocrinologists’comprehensive diabetes management algorithm 2013 consensus statement—executive summary. Endocr Pract 2013; 19:536–557.
- Ridker PM, Cook NR. Statins: new American guidelines for prevention of cardiovascular disease. Lancet 2013doi: 10.1016/S0140-6736(13)62388-0. [Epub ahead of print]
- The ARIC investigators. The Atherosclerosis Risk in Communities (ARIC) study: design and objectives. Am J Epidemiol 1989; 129:687–702.
- Fried LP, Borhani NO, Enright P, et al. The Cardiovascular Health Study: design and rationale. Ann Epidemiol 1991; 1:263–276.
- Friedman GD, Cutter GR, Donahue RP, et al. CARDIA: study design, recruitment, and some characteristics of the examined subjects. J Clin Epidemiol 1988; 41:1105–1116.
- Dawber TR, Kannel WB, Lyell LP. An approach to longitudinal studies in a community: the Framingham study. Ann N Y Acad Sci 1963; 107:539–556.
- Kannel WB, Feinleib M, McNamara PM, Garrison RJ, Castelli WP. An investigation of coronary heart disease in families. The Framingham offspring study. Am J Epidemiol 1979; 110:281–290.
- Ridker PM, Cook NR, Lee IM, et al. A randomized trial of low-dose aspirin in the primary prevention of cardiovascular disease in women. N Engl J Med 2005; 352:1293–1304.
- Belancer C, Buring JE, Cook N, et al; The Steering Committee of the Physicians’ Health Study Research Group. Final report on the aspirin component of the ongoing Physicians’ Health Study. N Engl J Med 1989; 321:129–135.
- Langer R, White E, Lewis C, et al. The Women’s Health Initiative Observational Study: baseline characteristics of participants and reliability of baseline measures. Ann Epidemiol 2003; 13:S107–S121.
- Strong JP, Malcom GT, Oalmann MC, Wissler RW. The PDAY study: natural history, risk factors, and pathobiology. Ann N Y Acad Sci 1997; 811:226–235.
- Stone NJ, Robinson J, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; published online Nov 13. DOI: 10.1016/j.jacc.2013.11.002.
- National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002; 106:3143–3421.
- Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004; 110:227–239.
- American Heart Association. 2013 Prevention guidelines tools. CV risk calculator. http://my.americanheart.org/professional/StatementsGuidelines/PreventionGuidelines/Prevention-Guidelines_UCM_457698_SubHomePage.jsp. Accessed December 10, 2013.
- Goldstein JL, Brown MS. The LDL receptor. Arterioscler Thromb Vasc Biol 2009; 29:431–438.
- Horton JD, Cohen JC, Hobbs HH. PCSK9: a convertase that coordinates LDL catabolism. J Lipid Res 2009; 50(suppl):S172–S177.
- Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 1994; 344:1383–1389.
- de Lemos JA, Blazing MA, Wiviott SD, et al; for the A to Z Investigators. Early intensive vs a delayed conservative simvastatin strategy in patients with acute coronary syndromes. Phase Z of the A to Z trial. JAMA 2004; 292:1307–1316.
- Downs JR, Clearfield M, Weis S, et al; for the AFCAPS/TexCAPS Research Group. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels. Results of AFCAPS/TexCAPS. JAMA 1998; 279:1615–1622.
- Koren MJ, Hunninghake DB, on behalf of the ALLIANCE investigators. Clinical outcomes in managed-care patients with coronary heart disease treated aggressively in lipid-lowering disease management clinics. J Am Coll Cardiol 2004; 44:1772–1779.
- Sever PS, Dahlof B, Poulter NR, et al; ASCOT investigators. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial - Lipid Lowering Arm (ASCOT-LLA): a multicentre randomised controlled trial. Lancet 2003; 361:1149–1158.
- Colhoun HM, Betteridge DJ, Durrington PN, et al; on behalf of the CARDS Investigators. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): multicentre randomised placebo-controlled trial. Lancet 2004; 364:685–696.
- Sacks FM, Pfeffer MA, Moye LA, et al; for the Cholesterol and Recurrent Events Trial Investigators. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. N Engl J Med 1996; 335:1001–1009.
- Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20 536 high-risk individuals: a randomised placebo-controlled trial. Lancet 2002; 360:7–22.
- Pedersen TR, Faegeman O, Kastelein JJ, et al. Incremental Decrease in End Points Through Aggressive Lipid Lowering Study Group. High-dose atorvastatin vs usual-dose simvastatin for secondary prevention after myocardial infarction: the IDEAL study: a randomized controlled trial. JAMA 2005; 294:2437–2445.
- Ridker PM, Danielson E, Fonseca FAH, et al; for the JUPITER Study Group. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- LIPID Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N Engl J Med 1998; 339:1349–1357.
- Nakamura H, Arakawa K, Itakura H, et al; for the MEGA Study Group. Primary prevention of cardiovascular disease with pravastatin Japan (MEGA Study): a prospective rabndomised controlled trial. Lancet 2006; 368:1155–1163.
- Schwartz GG, Olsson AG, Ezekowitz MD, et al. Myocardial Ischemia Reduction with Aggreessive Cholesterol Lowering (MIRACL) Study Investigators. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: the MIRACL study: a randomized controlled trial. JAMA 2001; 285:1711–1718.
- Buchwald H, Varco RL, Matts JP, et al. Effect of partial ileal bypass surgery on mortality and morbidity from coronary heart disease in patients with hypercholesterolemia: report of the Program on the Surgical Control of the Hyperlipidemias (POSCH). N Engl J Med 1990; 323:946–955.
- Cannon CP, Braunwald E, McCabe CH, et al; for the Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 Investigators. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- Baigent C, Landray MJ, Reith C, et al; SHARP Investigators. The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (Study of Heart and Renal Protection): a randomised placebo-controlled trial. Lancet 2011; 377:2181–2192.
- LaRosa JC, Grundy SM, Waters DD, et al. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med 2005; 352:1425–1435.
- Shepherd J, Cobbe SM, Ford I, et al; for the West of Scotland Coronary Prevention Study Group. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. N Engl J Med 1995; 333:1301–1308.
- Canner PL, Berge KG, Wenger NK, et al. Fifteen year mortality in Coronary Drug Project patients: long-term benefit with niacin. J Am Coll Cardiol 1989; 8:1245–1255.
- AIM-HIGH Investigators, Boden WE, Probstfield JL, Anderson T, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med 2011; 365:2255–2267.
- HPS2-Thrive Collaborative Group. HPS2-THRIVE randomized placebo-controlled trial in 25 673 high-risk patients of ER niacin/laropiprant: trial design, pre-specified muscle and liver outcomes, and reasons for stopping study treatment. Eur Heart J 2013; 34:1279–1291.
- American Diabetes Association. Standards of medical care in diabetes—2013. Diabetes Care 2013; 36(suppl 1):S11–S66.
- Garber AJ, Abrahamson MJ, Barzilay JI, et al. American Association of Clinical Endocrinologists’comprehensive diabetes management algorithm 2013 consensus statement—executive summary. Endocr Pract 2013; 19:536–557.
- Ridker PM, Cook NR. Statins: new American guidelines for prevention of cardiovascular disease. Lancet 2013doi: 10.1016/S0140-6736(13)62388-0. [Epub ahead of print]
- The ARIC investigators. The Atherosclerosis Risk in Communities (ARIC) study: design and objectives. Am J Epidemiol 1989; 129:687–702.
- Fried LP, Borhani NO, Enright P, et al. The Cardiovascular Health Study: design and rationale. Ann Epidemiol 1991; 1:263–276.
- Friedman GD, Cutter GR, Donahue RP, et al. CARDIA: study design, recruitment, and some characteristics of the examined subjects. J Clin Epidemiol 1988; 41:1105–1116.
- Dawber TR, Kannel WB, Lyell LP. An approach to longitudinal studies in a community: the Framingham study. Ann N Y Acad Sci 1963; 107:539–556.
- Kannel WB, Feinleib M, McNamara PM, Garrison RJ, Castelli WP. An investigation of coronary heart disease in families. The Framingham offspring study. Am J Epidemiol 1979; 110:281–290.
- Ridker PM, Cook NR, Lee IM, et al. A randomized trial of low-dose aspirin in the primary prevention of cardiovascular disease in women. N Engl J Med 2005; 352:1293–1304.
- Belancer C, Buring JE, Cook N, et al; The Steering Committee of the Physicians’ Health Study Research Group. Final report on the aspirin component of the ongoing Physicians’ Health Study. N Engl J Med 1989; 321:129–135.
- Langer R, White E, Lewis C, et al. The Women’s Health Initiative Observational Study: baseline characteristics of participants and reliability of baseline measures. Ann Epidemiol 2003; 13:S107–S121.
- Strong JP, Malcom GT, Oalmann MC, Wissler RW. The PDAY study: natural history, risk factors, and pathobiology. Ann N Y Acad Sci 1997; 811:226–235.
Biofeedback in coronary artery disease, type 2 diabetes, and multiple sclerosis
The homocysteine hypothesis: Still relevant to the prevention and treatment of cardiovascular disease?
Patients often ask primary care physicians and cardiologists about the measurement of biomarkers for cardiovascular disease and about the efficacy of preventive measures.
Although studies have shown that elevated homocysteine is a risk factor for cardiovascular and peripheral arterial disease1–3 and that supplementation with folic acid, vitamin B6, and vitamin B12 lowers homocysteine levels,4,5 it is unclear whether such supplementation prevents cardiovascular events. As a result, there is no consensus about whose homocysteine levels should be measured and who, if anyone, should receive homocysteine-lowering therapies.
The aim of this paper is to examine whether the evidence is sufficient to recommend homocysteine testing to guide the prevention and treatment of cardiovascular disease, or to recommend using folic acid, vitamin B6, and vitamin B12 for primary or secondary prevention of cardiovascular disease.
HISTORY OF HOMOCYSTEINE AS A RISK MARKER
Homocysteine is an amino acid formed from the metabolism of methionine, an essential amino acid derived from dietary protein. Although homocysteine was first isolated by Butz and du Vigneaud in 1932,6 it was not until 1964 that Gibson et al7 reported that patients with homocystinuria (more about this below) had vascular anomalies and arterial thrombosis. In 1969, McCully8 made the connection between elevated homocysteine levels and the risk of atherosclerosis.
Several possible mechanisms for the association between homocysteine and atherosclerosis have been demonstrated in experimental models. These include stimulation of smooth muscle growth, reduction in endothelial cell growth, impaired endothelial cell relaxation, decreased synthesis of high-density lipoprotein, promotion of autoimmune response, and accumulation of inflammatory monocytes in atherosclerotic plaques.3,9,10
In view of these findings, researchers have been evaluating whether homocysteine-lowering therapies decrease the risk of cardiovascular disease.
CAUSES OF ELEVATED PLASMA HOMOCYSTEINE

Vitamin deficiencies. Vitamin B6 (pyridoxine), vitamin B12 (cyanocobalamin), and folic acid are cofactors required for homocysteine metabolism, and deficiency in any or all of these leads to disruption of the relevant metabolic pathways.
Renal impairment. A low glomerular filtration rate has also been correlated with an elevated plasma homocysteine concentration. This makes sense, since the kidneys perform up to 70% of the clearance of homocysteine, although a cause-and-effect relationship is unclear.13
Inborn errors of homocysteine metabolism.Homocystinuria, ie, an abnormal elevation of homocysteine in the urine, is caused by several autosomal recessive disorders. People with these genetic variations have extremely high homocysteine levels.
A deficiency in the enzyme cystathionine beta-synthase is quite rare (the incidence in newborn babies has been found to be 1 in 344,000 worldwide and 1 in 65,000 in Ireland and Australia14), but leads to homocysteine levels greater than 100 μmol/L and often causes cardiovascular disease by the age of 30 years.15
A deficiency in the enzyme methylene tetrahydrofolate reductase (MTHFR) is a more common cause of mildly to moderately elevated plasma homocysteine levels.16 The MTHFR deficiency involves a variation at position 677 in the MTHFR gene in which cytosine is replaced by thymidine (thus called C677T or 677C>T).17 Ten percent of the population are homozygous for this variant (TT), 43% are heterozygous (CT), and 47% are unaffected (CC). Heterozygotes have slightly higher homocysteine levels than unaffected people, while people with the TT genotype have approximately 20% higher homocysteine levels.17
ELEVATED HOMOCYSTEINE IS COMMON
In a study of a population in Norway from 1992 to 1993, 8.5% had mild elevations in homocysteine (plasma levels 15–29.99 μmol/L), 0.8% had moderate elevations (30–99.99 μmol/L), and 0.02% had severe elevations (≥ 100 μmol/L).13,18 The prevalence of hyperhomocysteinemia in the United States is probably much lower, given that supplementation of white flour and cereal grains with folic acid has been mandatory since 1998, but this is not well described in the literature.
HOW GREAT IS THE RISK?
Studies over the past 10 to 20 years have shown that elevated homocysteine is a marker of risk of cardiovascular disease. The association was first noted in patients with cystathionine beta-synthase deficiency, who tend to have premature cardiovascular disease.
However, studies of patients with MTHFR 677C>T have yielded mixed results. Although several meta-analyses found up to a 42% higher rate of ischemic heart disease and stroke in patients homozygous for MTHFR 677C>T (the TT genotype) than in those with the CC genotype,17,19,20 two other large meta-analyses did not find an association between this variant and vascular risk.21,22
Nonetheless, in a meta-analysis of the association between homocysteine and cardiovascular disease, Wald et al17 found that for every 5-μmol/L increase in serum homocysteine concentration, the risk of ischemic heart disease increased 20% to 30%.
TRIALS OF HOMOCYSTEINE-LOWERING THERAPY HAVE HAD MIXED RESULTS
Primary prevention of cardiovascular disease
Given the finding that treatment with folic acid lowers homocysteine—initially noted in patients with homocystinuria—researchers hypothesized that treatment with folic acid, vitamin B6, and vitamin B12 would decrease the risk of cardiovascular disease.
Thus, in its recent evaluation of novel risk markers of cardiovascular disease, the United States Preventive Services Task Force 28,29 does not recommend measuring the plasma homocysteine level in the evaluation of either low-risk or intermediate-risk populations, finding no evidence that it adds any useful information in predicting major coronary events beyond what one could get from calculating the Framingham Risk Score. The task force also found no evidence that treating people who have elevated homocysteine levels decreases their risk of subsequent cardiovascular events.
In addition, a recent Cochrane Database review of eight randomized controlled trials in patients at low risk did not find a lower risk of myocardial infarction (fatal or nonfatal), stroke, or death from any cause in patients receiving B-complex vitamins.30
Secondary prevention of cardiovascular disease
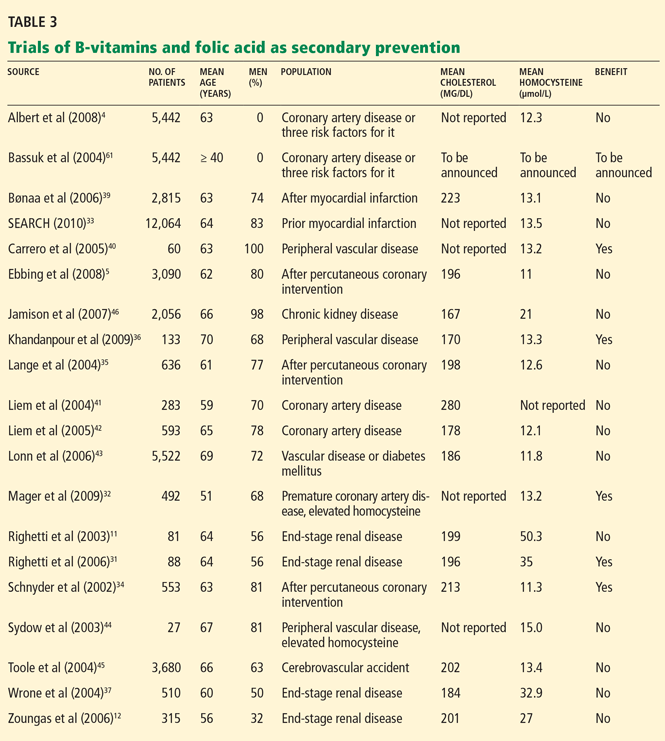
Bazzano et al,47 in a meta-analysis published in 2006, evaluated 12 randomized controlled trials of folic acid supplementation in patients with known cardiovascular disease and did not find that treated patients had better cardiovascular outcomes. The mean homocysteine level was elevated (> 15 μmol/L) at baseline in only 4 of the 12 trials. However, in 1 of these 4 trials, there was no difference in outcomes comparing those with and without elevated homocysteine.31
Albert et al4 more recently evaluated the effect of a combination pill containing folic acid, vitamin B6, and vitamin B12 on cardiovascular events in women at high risk, ie, those with a history of cardiovascular disease or having three or more coronary risk factors. Treatment did not decrease the rate of the composite outcome of cardiovascular disease mortality, stroke, myocardial infarction, or coronary revascularization, although the homocysteine level decreased by a mean of 30% in the treated group. However, only 27.7% of the participants had an elevated homocysteine level. One might not expect patients to benefit from such treatment if they had normal homocysteine levels to begin with.
Ebbing et al,5 in a trial published in 2008, investigated the effect of folic acid, vitamin B12, and vitamin B6 supplements on the risks of death from any cause and of cardiovascular events in patients undergoing coronary angiography. Outcomes were no better in the treatment group than in the control group, despite a mean decrease in homocysteine level of 19%. However, over 90% of the participants had a normal homocysteine level.
Mager et al,32 in a study published in 2009, looked specifically at whether patients with coronary artery disease and elevated homocysteine levels (> 15 μmol/L) would benefit from folate-based vitamin therapy. In this subset, the incidence of death from any cause was lower in the treated group than in the control group (4% vs 32%, P < .001), an association that was not present in patients with normal homocysteine levels.
The SEARCH trial (Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine),33 recently published, was a double-blind, randomized controlled trial of vitamin B12 and folic acid treatment in 12,064 patients who had survived a myocardial infarction. Although those who received the vitamin therapy had a 28% reduction in homocysteine level, no clinical benefit was demonstrated. Of note, 66% of the patients had a homocysteine level lower than 14 μmol/L at baseline.
Restenosis after angioplasty
Results are also mixed regarding whether folic acid supplements modify the risk of restenosis after coronary angioplasty.
Namazi et al48 evaluated the effect of folic acid supplementation on in-stent restenosis in 200 patients and found no difference between the treatment and placebo groups in the rates of either restenosis or target-vessel revascularization.
Schnyder et al49 evaluated the effect of folic acid, vitamin B6, and vitamin B12 treatment on the rate of coronary restenosis (in cases of balloon angioplasty) or in-stent restenosis (if a stent was used). Patients receiving treatment had lower rates of restenosis or instent restenosis (40% vs 48%, P = .01) and of need for target-vessel revascularization (11% vs 22%, P = .047). The mean homocysteine level was not elevated in this study either, and the researchers did not analyze the outcomes according to whether patients had high or normal homocysteine levels.
Lange et al35 also evaluated the effect of folic acid, vitamin B6, and vitamin B12 treatment on coronary in-stent restenosis. Paradoxically, the rate was higher with treatment in the overall group (mean homocysteine level 12.2 μmol/L), leading to a higher incidence of target-vessel revascularization. Patients who had a baseline elevation in homocysteine level had a nonsignificant trend toward a lower rate of in-stent restenosis.
Cerebrovascular and peripheral arterial disease
The evidence is also mixed for using folic acid and other B vitamins to prevent cerebrovascular disease and peripheral vascular disease. Although a 2007 meta-analysis found that folic acid supplementation decreased the risk of a first stroke by 18% (P = .045),50 a later meta-analysis contradicts this finding.51
A 2009 meta-analysis found that patients with peripheral arterial disease had higher homocysteine levels than controls, but it did not find any benefit from supplementation, owing to heterogeneity of the clinical end points used.52 Indeed, a 2009 Cochrane Database Systematic Review found that there were no adequate trials of the treatment of patients with peripheral vascular disease who have elevated plasma homocysteine.53
However, immediately after the Cochrane review was published, Khandanpour et al36 published the results of a trial of the effect of folic acid and 5-methyltetrahydrofolate (an active form of folic acid) supplementation on the ankle-brachial pressure index and the pulse-wave velocity in patients with peripheral arterial disease. These measures improved with 16 weeks of treatment. For the ankle-brachial pressure index, the P value was less than .01 for folic acid and .009 for 5-methyltetrahydrofolate; for the pulse-wave velocity, the P value was .051 for folic acid and .011 for 5-methyltetrahydrofolate.
Kidney disease
One could postulate that patients with end-stage renal disease or chronic kidney disease might benefit the most from folic acid supplementation, given the correlation of elevations in homocysteine levels with decline in glomerular filtration rate.
However, only one study found a lower rate of cardiovascular events with folic acid supplementation in dialysis patients, and the difference was not statistically significant (25% vs 36%, P < .08).31 Further, several studies found no benefit of folic acid supplementation in patients with chronic kidney disease.11,12,37
FUTURE DIRECTIONS AND RECOMMENDATIONS
Many experts have suggested that the existing evidence indicates that the homocysteine-lowering therapies folic acid, vitamin B6, and vitamin B12 do not lower the risk of cardiovascular disease.38,54–59 Indeed, the American Heart Association guidelines for cardiovascular disease prevention in women do not recommend folic acid supplementation to prevent cardiovascular disease.60 (Recommendations for men are the same as for women.) However, most of the clinical trials have not selected and treated patients with elevated homocysteine levels, but have instead included all patients regardless of homocysteine level.
At least two large ongoing trials are currently evaluating B-vitamin therapy for secondary prevention, but neither trial is looking specifically at patients with elevated homocysteine levels.61,62
Thus, instead of concluding that no patients could benefit from homocysteine-lowering treatment, future studies need to clarify:
- Whether patients with elevated homocysteine would benefit from such treatment
- At what level it would be appropriate to start treatment
- The appropriate target homocysteine level with treatment.
Particularly given the recent finding that folic acid supplementation may increase cancer risk,63 these questions need closer scrutiny.
- Humphrey LL, Fu R, Rogers K, Freeman M, Helfand M. Homocysteine level and coronary heart disease incidence: a systematic review and meta-analysis. Mayo Clin Proc 2008; 83:1203–1212.
- Boers GH. The case for mild hyperhomocysteinaemia as a risk factor. J Inherit Metab Dis 1997; 20:301–306.
- Austin RC, Lentz SR, Werstuck GH. Role of hyperhomocysteinemia in endothelial dysfunction and atherothrombotic disease. Cell Death Differ 2004; 11(suppl 1):S56–S64.
- Albert CM, Cook NR, Gaziano JM, et al. Effect of folic acid and B vitamins on risk of cardiovascular events and total mortality among women at high risk for cardiovascular disease: a randomized trial. JAMA 2008; 299:2027–2036.
- Ebbing M, Bleie Ø, Ueland PM, et al. Mortality and cardiovascular events in patients treated with homocysteine-lowering B vitamins after coronary angiography: a randomized controlled trial. JAMA 2008; 300:795–804.
- Butz LW, du Vigneaud V. The formation of a homologue of cystine by the decompensation of methionine with sulphuric acid. J Biol Chem 1932; 99:135–142.
- Gibson JB, Carson NA, Neill DW. Pathological findings in homocystinuria. J Clin Pathol 1964; 17:427–437.
- McCully KS. Vascular pathology of homocysteinemia: implications for the pathogenesis of arteriosclerosis. Am J Pathol 1969; 56:111–128.
- Zhang D, Jiang X, Fang P, et al. Hyperhomocysteinemia promotes inflammatory monocyte generation and accelerates atherosclerosis in transgenic cystathionine beta-synthase-deficient mice. Circulation 2009; 120:1893–1902.
- Woo KS, Chook P, Lolin YI, Sanderson JE, Metreweli C, Celermajer DS. Folic acid improves arterial endothelial function in adults with hyperhomocystinemia. J Am Coll Cardiol 1999; 34:2002–2006.
- Righetti M, Ferrario GM, Milani S, et al. Effects of folic acid treatment on homocysteine levels and vascular disease in hemodialysis patients. Med Sci Monit 2003; 9:PI19–PI24.
- Zoungas S, McGrath BP, Branley P, et al. Cardiovascular morbidity and mortality in the Atherosclerosis and Folic Acid Supplementation Trial (ASFAST) in chronic renal failure: a multicenter, randomized, controlled trial. J Am Coll Cardiol 2006; 47:1108–1116.
- Carmel R, Jacobsen DW, editors. Homocysteine in Health and Disease. Cambridge, UK: Cambridge University Press, 2001.
- Yap S, Boers GH, Wilcken B, et al. Vascular outcome in patients with homocystinuria due to cystathionine beta-synthase deficiency treated chronically: a multicenter observational study. Arterioscler Thromb Vasc Biol 2001; 21:2080–2085.
- McKusick V. 236200 Homocystinuria. In:McKusick V, editor. Mendelian Inheritance in Man. 10th ed. Baltimore, MD: The Johns Hopkins University Press, 1992:1444–1446.
- McKusick V. 236250 Homocystinuria due to deficiency of N(5,10)-methylenetetrahydrofolate reductase activity. In:McKusick V, editor. Mendelian Inheritance in Man. Baltimore, MD: The Johns Hopkins University Press, 1992:1447–1448.
- Wald DS, Law M, Morris JK. Homocysteine and cardiovascular disease: evidence on causality from a meta-analysis. BMJ 2002; 325:1202.
- Nygård O, Vollset SE, Refsum H, et al. Total plasma homocysteine and cardiovascular risk profile. The Hordaland Homocysteine Study. JAMA 1995; 274:1526–1533.
- Klerk M, Verhoef P, Clarke R, Blom HJ, Kok FJ, Schouten EG; MTHFR Studies Collaboration Group. MTHFR 677C-->T polymorphism and risk of coronary heart disease: a meta-analysis. JAMA 2002; 288:2023–2031.
- Kelly PJ, Rosand J, Kistler JP, et al. Homocysteine, MTHFR 677C-->T polymorphism, and risk of ischemic stroke: results of a meta-analysis. Neurology 2002; 59:529–536.
- Lewis SJ, Ebrahim S, Davey Smith G. Meta-analysis of MTHFR 677C->T polymorphism and coronary heart disease: does totality of evidence support causal role for homocysteine and preventive potential of folate? BMJ 2005; 331:1053.
- Brattström L, Wilcken DE, Ohrvik J, Brudin L. Common methylenetetrahydrofolate reductase gene mutation leads to hyperhomocysteinemia but not to vascular disease: the result of a meta-analysis. Circulation 1998; 98:2520–2526.
- Cui R, Iso H, Date C, Kikuchi S, Tamakoshi A; Japan Collaborative Cohort Study Group. Dietary folate and vitamin B6 and B12 intake in relation to mortality from cardiovascular diseases: Japan Collaborative Cohort Study. Stroke 2010; 41:1285–1289.
- Liu S, Stampfer MJ, Hu FB, et al. Whole-grain consumption and risk of coronary heart disease: results from the Nurses’ Health Study. Am J Clin Nutr 1999; 70:412–419.
- Liu S, Manson JE, Stampfer MJ, et al. Whole grain consumption and risk of ischemic stroke in women: a prospective study. JAMA 2000; 284:1534–1540.
- Merchant AT, Hu FB, Spiegelman D, Willett WC, Rimm EB, Ascherio A. The use of B vitamin supplements and peripheral arterial disease risk in men are inversely related. J Nutr 2003; 133:2863–2867.
- Rimm EB, Willett WC, Hu FB, et al. Folate and vitamin B6 from diet and supplements in relation to risk of coronary heart disease among women. JAMA 1998; 279:359–364.
- US Preventive Services Task Force. Using nontraditional risk factors in coronary heart disease risk assessment: US Preventive Services Task Force recommendation statement. Ann Intern Med 2009; 151:474–482.
- Helfand M, Buckley DI, Freeman M, et al. Emerging risk factors for coronary heart disease: a summary of systematic reviews conducted for the US Preventive Services Task Force. Ann Intern Med 2009; 151:496–507.
- Martí-Carvajal AJ, Solà I, Lathyris D, Salanti G. Homocysteine lowering interventions for preventing cardiovascular events. Cochrane Database Syst Rev 2009; ( 4):CD006612.
- Righetti M, Serbelloni P, Milani S, Ferrario G. Homocysteine-lowering vitamin B treatment decreases cardiovascular events in hemodialysis patients. Blood Purif 2006; 24:379–386.
- Mager A, Orvin K, Koren-Morag N, et al. Impact of homocysteine-lowering vitamin therapy on long-term outcome of patients with coronary artery disease. Am J Cardiol 2009; 104:745–749.
- Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) Collaborative Group; Armitage JM, Bowman L, Clarke RJ, et al. Effects of homocysteine-lowering with folic acid plus vitamin B12 vs placebo on mortality and major morbidity in myocardial infarction survivors: a randomized trial. JAMA 2010; 303:2486–2494.
- Schnyder G, Roffi M, Flammer Y, Pin R, Hess OM. Effect of homocysteine-lowering therapy with folic acid, vitamin B12, and percutaneous coronary intervention: the Swiss Heart Study: a randomized controlled trial. JAMA 2002; 288:973–979.
- Lange H, Suryapranata H, De Luca G, et al. Folate therapy and in-stent restenosis after coronary stenting. N Engl J Med 2004; 350:2673–2781.
- Khandanpour N, Armon MP, Jennings B, et al. Randomized clinical trial of folate supplementation in patients with peripheral arterial disease. Br J Surg 2009; 96:990–998.
- Wrone EM, Hornberger JM, Zehnder JL, McCann LM, Coplon NS, Fortmann SP. Randomized trial of folic acid for prevention of cardiovascular events in end-stage renal disease. J Am Soc Nephrol 2004; 15:420–426.
- Loscalzo J. Homocysteine trials—clear outcomes for complex reasons. N Engl J Med 2006; 354:1629–1632.
- Bønaa KH, Njølstad I, Ueland PM, et al; NORVIT Trial Investigators. Homocysteine lowering and cardiovascular events after acute myocardial infarction. N Engl J Med 2006; 354:1578–1588.
- Carrero JJ, López-Huertas E, Salmerón LM, Baró L, Ros E. Daily supplementation with (n-3) PUFAs, oleic acid, folic acid, and vitamins B-6 and E increases pain-free walking distance and improves risk factors in men with peripheral vascular disease. J Nutr 2005; 135:1393–1399.
- Liem AH, van Boven AJ, Veeger NJ, et al; Folic Acid on Risk Diminishment After Acute Myocarial Infarction Study Group. Efficacy of folic acid when added to statin therapy in patients with hypercholesterolemia following acute myocardial infarction: a randomised pilot trial. Int J Cardiol 2004; 93:175–179.
- Liem A, Reynierse-Buitenwerf GH, Zwinderman AH, Jukema JW, van Veldhuisen DJ. Secondary prevention with folic acid: results of the Goes extension study. Heart 2005; 91:1213–1214.
- Lonn E, Yusuf S, Arnold MJ, et al; Heart Outcomes Prevention Evaluation (HOPE) 2 Investigators. Homocysteine lowering with folic acid and B vitamins in vascular disease. N Engl J Med 2006; 354:1567–1577.
- Sydow K, Schwedhelm E, Arakawa N, et al. ADMA and oxidative stress are responsible for endothelial dysfunction in hyperhomocyst(e)inemia: effects of L-arginine and B vitamins. Cardiovasc Res 2003; 57:244–252.
- Toole JF, Malinow MR, Chambless LE, et al. Lowering homocysteine in patients with ischemic stroke to prevent recurrent stroke, myocardial infarction, and death: the Vitamin Intervention for Stroke Prevention (VISP) randomized controlled trial. JAMA 2004; 291:565–575.
- Jamison RL, Hartigan P, Kaufman JS, et al; Veterans Affairs Site Investigators. Effect of homocysteine lowering on mortality and vascular disease in advanced chronic kidney disease and end-stage renal disease: a randomized controlled trial. JAMA 2007; 2989:1163–1170. Erratum in JAMA 2008;300:170.
- Bazzano LA, Reynolds K, Holder KN, He J. Effect of folic acid supplementation on risk of cardiovascular diseases: a meta-analysis of randomized controlled trials. JAMA 2006; 296:2720–2726.
- Namazi MH, Motamedi MR, Safi M, Vakili H, Saadat H, Nazari N. Efficacy of folic acid therapy for prevention of in-stent restenosis: a randomized clinical trial. Arch Iran Med 2006; 9:108–110.
- Schnyder G, Roffi M, Pin R, et al. Decreased rate of coronary restenosis after lowering of plasma homocysteine levels. N Engl J Med 2001; 345:1593–1600.
- Wang X, Qin X, Demirtas H, et al. Efficacy of folic acid supplementation in stroke prevention: a meta-analysis. Lancet 2007; 369:1876–1882.
- Lee M, Hong KS, Chang SC, Saver JL. Efficacy of homocysteine-lowering therapy with folic Acid in stroke prevention: a meta-analysis. Stroke 2010; 41:1205–1212.
- Khandanpour N, Loke YK, Meyer FJ, Jennings B, Armon MP. Homocysteine and peripheral arterial disease: systematic review and meta-analysis. Eur J Vasc Endovasc Surg 2009; 38:316–322.
- Hansrani M, Stansby G. Homocysteine lowering interventions for peripheral arterial disease and bypass grafts. Cochrane Database Syst Rev 2002; ( 3):CD003285.
- Mosca L. Novel cardiovascular risk factors: do they add value to your practice? Am Fam Physician 2003; 67:264,266.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction. J Am Coll Cardiol 2007; 50:e1–e157.
- Lonn E. Homocysteine-lowering B vitamin therapy in cardiovascular prevention—wrong again? JAMA 2008; 299:2086–2087.
- Milani RV, Lavie CJ. Homocysteine: the Rubik’s cube of cardiovascular risk factors. Mayo Clin Proc 2008; 83:1200–1202.
- Bazzano LA. Folic acid supplementation and cardiovascular disease: the state of the art. Am J Med Sci 2009; 338:48–49.
- Ntaios G, Savopoulos C, Grekas D, Hatzitolios A. The controversial role of B-vitamins in cardiovascular risk: an update. Arch Cardiovasc Dis 2009; 102:847–854.
- Mosca L, Banka CL, Benjamin EJ; Expert Panel/Writing Group. Evidence-based guidelines for cardiovascular disease prevention in women: 2007 update. Circulation 2007; 115:1481–1501.
- Bassuk SS, Albert CM, Cook NR, et al. The Women’s Antioxidant Cardiovascular Study: design and baseline characteristics of participants. J Womens Health (Larchmt) 2004; 13:99–117.
- SEARCH Study Collaborative Group; Bowman L, Armitage J, Bulbulia R, Parish S, Collins R. Study of the effectiveness of additional reductions in cholesterol and homocysteine (SEARCH): characteristics of a randomized trial among 12064 myocardial infarction survivors. Am Heart J 2007; 154:815–823,823.e1–e6.
- Ebbing M, Bønaa KH, Nygård O, et al. Cancer incidence and mortality after treatment with folic acid and vitamin B12. JAMA 2009; 302:2119–2126.
Patients often ask primary care physicians and cardiologists about the measurement of biomarkers for cardiovascular disease and about the efficacy of preventive measures.
Although studies have shown that elevated homocysteine is a risk factor for cardiovascular and peripheral arterial disease1–3 and that supplementation with folic acid, vitamin B6, and vitamin B12 lowers homocysteine levels,4,5 it is unclear whether such supplementation prevents cardiovascular events. As a result, there is no consensus about whose homocysteine levels should be measured and who, if anyone, should receive homocysteine-lowering therapies.
The aim of this paper is to examine whether the evidence is sufficient to recommend homocysteine testing to guide the prevention and treatment of cardiovascular disease, or to recommend using folic acid, vitamin B6, and vitamin B12 for primary or secondary prevention of cardiovascular disease.
HISTORY OF HOMOCYSTEINE AS A RISK MARKER
Homocysteine is an amino acid formed from the metabolism of methionine, an essential amino acid derived from dietary protein. Although homocysteine was first isolated by Butz and du Vigneaud in 1932,6 it was not until 1964 that Gibson et al7 reported that patients with homocystinuria (more about this below) had vascular anomalies and arterial thrombosis. In 1969, McCully8 made the connection between elevated homocysteine levels and the risk of atherosclerosis.
Several possible mechanisms for the association between homocysteine and atherosclerosis have been demonstrated in experimental models. These include stimulation of smooth muscle growth, reduction in endothelial cell growth, impaired endothelial cell relaxation, decreased synthesis of high-density lipoprotein, promotion of autoimmune response, and accumulation of inflammatory monocytes in atherosclerotic plaques.3,9,10
In view of these findings, researchers have been evaluating whether homocysteine-lowering therapies decrease the risk of cardiovascular disease.
CAUSES OF ELEVATED PLASMA HOMOCYSTEINE
Vitamin deficiencies. Vitamin B6 (pyridoxine), vitamin B12 (cyanocobalamin), and folic acid are cofactors required for homocysteine metabolism, and deficiency in any or all of these leads to disruption of the relevant metabolic pathways.
Renal impairment. A low glomerular filtration rate has also been correlated with an elevated plasma homocysteine concentration. This makes sense, since the kidneys perform up to 70% of the clearance of homocysteine, although a cause-and-effect relationship is unclear.13
Inborn errors of homocysteine metabolism.Homocystinuria, ie, an abnormal elevation of homocysteine in the urine, is caused by several autosomal recessive disorders. People with these genetic variations have extremely high homocysteine levels.
A deficiency in the enzyme cystathionine beta-synthase is quite rare (the incidence in newborn babies has been found to be 1 in 344,000 worldwide and 1 in 65,000 in Ireland and Australia14), but leads to homocysteine levels greater than 100 μmol/L and often causes cardiovascular disease by the age of 30 years.15
A deficiency in the enzyme methylene tetrahydrofolate reductase (MTHFR) is a more common cause of mildly to moderately elevated plasma homocysteine levels.16 The MTHFR deficiency involves a variation at position 677 in the MTHFR gene in which cytosine is replaced by thymidine (thus called C677T or 677C>T).17 Ten percent of the population are homozygous for this variant (TT), 43% are heterozygous (CT), and 47% are unaffected (CC). Heterozygotes have slightly higher homocysteine levels than unaffected people, while people with the TT genotype have approximately 20% higher homocysteine levels.17
ELEVATED HOMOCYSTEINE IS COMMON
In a study of a population in Norway from 1992 to 1993, 8.5% had mild elevations in homocysteine (plasma levels 15–29.99 μmol/L), 0.8% had moderate elevations (30–99.99 μmol/L), and 0.02% had severe elevations (≥ 100 μmol/L).13,18 The prevalence of hyperhomocysteinemia in the United States is probably much lower, given that supplementation of white flour and cereal grains with folic acid has been mandatory since 1998, but this is not well described in the literature.
HOW GREAT IS THE RISK?
Studies over the past 10 to 20 years have shown that elevated homocysteine is a marker of risk of cardiovascular disease. The association was first noted in patients with cystathionine beta-synthase deficiency, who tend to have premature cardiovascular disease.
However, studies of patients with MTHFR 677C>T have yielded mixed results. Although several meta-analyses found up to a 42% higher rate of ischemic heart disease and stroke in patients homozygous for MTHFR 677C>T (the TT genotype) than in those with the CC genotype,17,19,20 two other large meta-analyses did not find an association between this variant and vascular risk.21,22
Nonetheless, in a meta-analysis of the association between homocysteine and cardiovascular disease, Wald et al17 found that for every 5-μmol/L increase in serum homocysteine concentration, the risk of ischemic heart disease increased 20% to 30%.
TRIALS OF HOMOCYSTEINE-LOWERING THERAPY HAVE HAD MIXED RESULTS
Primary prevention of cardiovascular disease
Given the finding that treatment with folic acid lowers homocysteine—initially noted in patients with homocystinuria—researchers hypothesized that treatment with folic acid, vitamin B6, and vitamin B12 would decrease the risk of cardiovascular disease.
Thus, in its recent evaluation of novel risk markers of cardiovascular disease, the United States Preventive Services Task Force 28,29 does not recommend measuring the plasma homocysteine level in the evaluation of either low-risk or intermediate-risk populations, finding no evidence that it adds any useful information in predicting major coronary events beyond what one could get from calculating the Framingham Risk Score. The task force also found no evidence that treating people who have elevated homocysteine levels decreases their risk of subsequent cardiovascular events.
In addition, a recent Cochrane Database review of eight randomized controlled trials in patients at low risk did not find a lower risk of myocardial infarction (fatal or nonfatal), stroke, or death from any cause in patients receiving B-complex vitamins.30
Secondary prevention of cardiovascular disease
Bazzano et al,47 in a meta-analysis published in 2006, evaluated 12 randomized controlled trials of folic acid supplementation in patients with known cardiovascular disease and did not find that treated patients had better cardiovascular outcomes. The mean homocysteine level was elevated (> 15 μmol/L) at baseline in only 4 of the 12 trials. However, in 1 of these 4 trials, there was no difference in outcomes comparing those with and without elevated homocysteine.31
Albert et al4 more recently evaluated the effect of a combination pill containing folic acid, vitamin B6, and vitamin B12 on cardiovascular events in women at high risk, ie, those with a history of cardiovascular disease or having three or more coronary risk factors. Treatment did not decrease the rate of the composite outcome of cardiovascular disease mortality, stroke, myocardial infarction, or coronary revascularization, although the homocysteine level decreased by a mean of 30% in the treated group. However, only 27.7% of the participants had an elevated homocysteine level. One might not expect patients to benefit from such treatment if they had normal homocysteine levels to begin with.
Ebbing et al,5 in a trial published in 2008, investigated the effect of folic acid, vitamin B12, and vitamin B6 supplements on the risks of death from any cause and of cardiovascular events in patients undergoing coronary angiography. Outcomes were no better in the treatment group than in the control group, despite a mean decrease in homocysteine level of 19%. However, over 90% of the participants had a normal homocysteine level.
Mager et al,32 in a study published in 2009, looked specifically at whether patients with coronary artery disease and elevated homocysteine levels (> 15 μmol/L) would benefit from folate-based vitamin therapy. In this subset, the incidence of death from any cause was lower in the treated group than in the control group (4% vs 32%, P < .001), an association that was not present in patients with normal homocysteine levels.
The SEARCH trial (Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine),33 recently published, was a double-blind, randomized controlled trial of vitamin B12 and folic acid treatment in 12,064 patients who had survived a myocardial infarction. Although those who received the vitamin therapy had a 28% reduction in homocysteine level, no clinical benefit was demonstrated. Of note, 66% of the patients had a homocysteine level lower than 14 μmol/L at baseline.
Restenosis after angioplasty
Results are also mixed regarding whether folic acid supplements modify the risk of restenosis after coronary angioplasty.
Namazi et al48 evaluated the effect of folic acid supplementation on in-stent restenosis in 200 patients and found no difference between the treatment and placebo groups in the rates of either restenosis or target-vessel revascularization.
Schnyder et al49 evaluated the effect of folic acid, vitamin B6, and vitamin B12 treatment on the rate of coronary restenosis (in cases of balloon angioplasty) or in-stent restenosis (if a stent was used). Patients receiving treatment had lower rates of restenosis or instent restenosis (40% vs 48%, P = .01) and of need for target-vessel revascularization (11% vs 22%, P = .047). The mean homocysteine level was not elevated in this study either, and the researchers did not analyze the outcomes according to whether patients had high or normal homocysteine levels.
Lange et al35 also evaluated the effect of folic acid, vitamin B6, and vitamin B12 treatment on coronary in-stent restenosis. Paradoxically, the rate was higher with treatment in the overall group (mean homocysteine level 12.2 μmol/L), leading to a higher incidence of target-vessel revascularization. Patients who had a baseline elevation in homocysteine level had a nonsignificant trend toward a lower rate of in-stent restenosis.
Cerebrovascular and peripheral arterial disease
The evidence is also mixed for using folic acid and other B vitamins to prevent cerebrovascular disease and peripheral vascular disease. Although a 2007 meta-analysis found that folic acid supplementation decreased the risk of a first stroke by 18% (P = .045),50 a later meta-analysis contradicts this finding.51
A 2009 meta-analysis found that patients with peripheral arterial disease had higher homocysteine levels than controls, but it did not find any benefit from supplementation, owing to heterogeneity of the clinical end points used.52 Indeed, a 2009 Cochrane Database Systematic Review found that there were no adequate trials of the treatment of patients with peripheral vascular disease who have elevated plasma homocysteine.53
However, immediately after the Cochrane review was published, Khandanpour et al36 published the results of a trial of the effect of folic acid and 5-methyltetrahydrofolate (an active form of folic acid) supplementation on the ankle-brachial pressure index and the pulse-wave velocity in patients with peripheral arterial disease. These measures improved with 16 weeks of treatment. For the ankle-brachial pressure index, the P value was less than .01 for folic acid and .009 for 5-methyltetrahydrofolate; for the pulse-wave velocity, the P value was .051 for folic acid and .011 for 5-methyltetrahydrofolate.
Kidney disease
One could postulate that patients with end-stage renal disease or chronic kidney disease might benefit the most from folic acid supplementation, given the correlation of elevations in homocysteine levels with decline in glomerular filtration rate.
However, only one study found a lower rate of cardiovascular events with folic acid supplementation in dialysis patients, and the difference was not statistically significant (25% vs 36%, P < .08).31 Further, several studies found no benefit of folic acid supplementation in patients with chronic kidney disease.11,12,37
FUTURE DIRECTIONS AND RECOMMENDATIONS
Many experts have suggested that the existing evidence indicates that the homocysteine-lowering therapies folic acid, vitamin B6, and vitamin B12 do not lower the risk of cardiovascular disease.38,54–59 Indeed, the American Heart Association guidelines for cardiovascular disease prevention in women do not recommend folic acid supplementation to prevent cardiovascular disease.60 (Recommendations for men are the same as for women.) However, most of the clinical trials have not selected and treated patients with elevated homocysteine levels, but have instead included all patients regardless of homocysteine level.
At least two large ongoing trials are currently evaluating B-vitamin therapy for secondary prevention, but neither trial is looking specifically at patients with elevated homocysteine levels.61,62
Thus, instead of concluding that no patients could benefit from homocysteine-lowering treatment, future studies need to clarify:
- Whether patients with elevated homocysteine would benefit from such treatment
- At what level it would be appropriate to start treatment
- The appropriate target homocysteine level with treatment.
Particularly given the recent finding that folic acid supplementation may increase cancer risk,63 these questions need closer scrutiny.
Patients often ask primary care physicians and cardiologists about the measurement of biomarkers for cardiovascular disease and about the efficacy of preventive measures.
Although studies have shown that elevated homocysteine is a risk factor for cardiovascular and peripheral arterial disease1–3 and that supplementation with folic acid, vitamin B6, and vitamin B12 lowers homocysteine levels,4,5 it is unclear whether such supplementation prevents cardiovascular events. As a result, there is no consensus about whose homocysteine levels should be measured and who, if anyone, should receive homocysteine-lowering therapies.
The aim of this paper is to examine whether the evidence is sufficient to recommend homocysteine testing to guide the prevention and treatment of cardiovascular disease, or to recommend using folic acid, vitamin B6, and vitamin B12 for primary or secondary prevention of cardiovascular disease.
HISTORY OF HOMOCYSTEINE AS A RISK MARKER
Homocysteine is an amino acid formed from the metabolism of methionine, an essential amino acid derived from dietary protein. Although homocysteine was first isolated by Butz and du Vigneaud in 1932,6 it was not until 1964 that Gibson et al7 reported that patients with homocystinuria (more about this below) had vascular anomalies and arterial thrombosis. In 1969, McCully8 made the connection between elevated homocysteine levels and the risk of atherosclerosis.
Several possible mechanisms for the association between homocysteine and atherosclerosis have been demonstrated in experimental models. These include stimulation of smooth muscle growth, reduction in endothelial cell growth, impaired endothelial cell relaxation, decreased synthesis of high-density lipoprotein, promotion of autoimmune response, and accumulation of inflammatory monocytes in atherosclerotic plaques.3,9,10
In view of these findings, researchers have been evaluating whether homocysteine-lowering therapies decrease the risk of cardiovascular disease.
CAUSES OF ELEVATED PLASMA HOMOCYSTEINE
Vitamin deficiencies. Vitamin B6 (pyridoxine), vitamin B12 (cyanocobalamin), and folic acid are cofactors required for homocysteine metabolism, and deficiency in any or all of these leads to disruption of the relevant metabolic pathways.
Renal impairment. A low glomerular filtration rate has also been correlated with an elevated plasma homocysteine concentration. This makes sense, since the kidneys perform up to 70% of the clearance of homocysteine, although a cause-and-effect relationship is unclear.13
Inborn errors of homocysteine metabolism.Homocystinuria, ie, an abnormal elevation of homocysteine in the urine, is caused by several autosomal recessive disorders. People with these genetic variations have extremely high homocysteine levels.
A deficiency in the enzyme cystathionine beta-synthase is quite rare (the incidence in newborn babies has been found to be 1 in 344,000 worldwide and 1 in 65,000 in Ireland and Australia14), but leads to homocysteine levels greater than 100 μmol/L and often causes cardiovascular disease by the age of 30 years.15
A deficiency in the enzyme methylene tetrahydrofolate reductase (MTHFR) is a more common cause of mildly to moderately elevated plasma homocysteine levels.16 The MTHFR deficiency involves a variation at position 677 in the MTHFR gene in which cytosine is replaced by thymidine (thus called C677T or 677C>T).17 Ten percent of the population are homozygous for this variant (TT), 43% are heterozygous (CT), and 47% are unaffected (CC). Heterozygotes have slightly higher homocysteine levels than unaffected people, while people with the TT genotype have approximately 20% higher homocysteine levels.17
ELEVATED HOMOCYSTEINE IS COMMON
In a study of a population in Norway from 1992 to 1993, 8.5% had mild elevations in homocysteine (plasma levels 15–29.99 μmol/L), 0.8% had moderate elevations (30–99.99 μmol/L), and 0.02% had severe elevations (≥ 100 μmol/L).13,18 The prevalence of hyperhomocysteinemia in the United States is probably much lower, given that supplementation of white flour and cereal grains with folic acid has been mandatory since 1998, but this is not well described in the literature.
HOW GREAT IS THE RISK?
Studies over the past 10 to 20 years have shown that elevated homocysteine is a marker of risk of cardiovascular disease. The association was first noted in patients with cystathionine beta-synthase deficiency, who tend to have premature cardiovascular disease.
However, studies of patients with MTHFR 677C>T have yielded mixed results. Although several meta-analyses found up to a 42% higher rate of ischemic heart disease and stroke in patients homozygous for MTHFR 677C>T (the TT genotype) than in those with the CC genotype,17,19,20 two other large meta-analyses did not find an association between this variant and vascular risk.21,22
Nonetheless, in a meta-analysis of the association between homocysteine and cardiovascular disease, Wald et al17 found that for every 5-μmol/L increase in serum homocysteine concentration, the risk of ischemic heart disease increased 20% to 30%.
TRIALS OF HOMOCYSTEINE-LOWERING THERAPY HAVE HAD MIXED RESULTS
Primary prevention of cardiovascular disease
Given the finding that treatment with folic acid lowers homocysteine—initially noted in patients with homocystinuria—researchers hypothesized that treatment with folic acid, vitamin B6, and vitamin B12 would decrease the risk of cardiovascular disease.
Thus, in its recent evaluation of novel risk markers of cardiovascular disease, the United States Preventive Services Task Force 28,29 does not recommend measuring the plasma homocysteine level in the evaluation of either low-risk or intermediate-risk populations, finding no evidence that it adds any useful information in predicting major coronary events beyond what one could get from calculating the Framingham Risk Score. The task force also found no evidence that treating people who have elevated homocysteine levels decreases their risk of subsequent cardiovascular events.
In addition, a recent Cochrane Database review of eight randomized controlled trials in patients at low risk did not find a lower risk of myocardial infarction (fatal or nonfatal), stroke, or death from any cause in patients receiving B-complex vitamins.30
Secondary prevention of cardiovascular disease
Bazzano et al,47 in a meta-analysis published in 2006, evaluated 12 randomized controlled trials of folic acid supplementation in patients with known cardiovascular disease and did not find that treated patients had better cardiovascular outcomes. The mean homocysteine level was elevated (> 15 μmol/L) at baseline in only 4 of the 12 trials. However, in 1 of these 4 trials, there was no difference in outcomes comparing those with and without elevated homocysteine.31
Albert et al4 more recently evaluated the effect of a combination pill containing folic acid, vitamin B6, and vitamin B12 on cardiovascular events in women at high risk, ie, those with a history of cardiovascular disease or having three or more coronary risk factors. Treatment did not decrease the rate of the composite outcome of cardiovascular disease mortality, stroke, myocardial infarction, or coronary revascularization, although the homocysteine level decreased by a mean of 30% in the treated group. However, only 27.7% of the participants had an elevated homocysteine level. One might not expect patients to benefit from such treatment if they had normal homocysteine levels to begin with.
Ebbing et al,5 in a trial published in 2008, investigated the effect of folic acid, vitamin B12, and vitamin B6 supplements on the risks of death from any cause and of cardiovascular events in patients undergoing coronary angiography. Outcomes were no better in the treatment group than in the control group, despite a mean decrease in homocysteine level of 19%. However, over 90% of the participants had a normal homocysteine level.
Mager et al,32 in a study published in 2009, looked specifically at whether patients with coronary artery disease and elevated homocysteine levels (> 15 μmol/L) would benefit from folate-based vitamin therapy. In this subset, the incidence of death from any cause was lower in the treated group than in the control group (4% vs 32%, P < .001), an association that was not present in patients with normal homocysteine levels.
The SEARCH trial (Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine),33 recently published, was a double-blind, randomized controlled trial of vitamin B12 and folic acid treatment in 12,064 patients who had survived a myocardial infarction. Although those who received the vitamin therapy had a 28% reduction in homocysteine level, no clinical benefit was demonstrated. Of note, 66% of the patients had a homocysteine level lower than 14 μmol/L at baseline.
Restenosis after angioplasty
Results are also mixed regarding whether folic acid supplements modify the risk of restenosis after coronary angioplasty.
Namazi et al48 evaluated the effect of folic acid supplementation on in-stent restenosis in 200 patients and found no difference between the treatment and placebo groups in the rates of either restenosis or target-vessel revascularization.
Schnyder et al49 evaluated the effect of folic acid, vitamin B6, and vitamin B12 treatment on the rate of coronary restenosis (in cases of balloon angioplasty) or in-stent restenosis (if a stent was used). Patients receiving treatment had lower rates of restenosis or instent restenosis (40% vs 48%, P = .01) and of need for target-vessel revascularization (11% vs 22%, P = .047). The mean homocysteine level was not elevated in this study either, and the researchers did not analyze the outcomes according to whether patients had high or normal homocysteine levels.
Lange et al35 also evaluated the effect of folic acid, vitamin B6, and vitamin B12 treatment on coronary in-stent restenosis. Paradoxically, the rate was higher with treatment in the overall group (mean homocysteine level 12.2 μmol/L), leading to a higher incidence of target-vessel revascularization. Patients who had a baseline elevation in homocysteine level had a nonsignificant trend toward a lower rate of in-stent restenosis.
Cerebrovascular and peripheral arterial disease
The evidence is also mixed for using folic acid and other B vitamins to prevent cerebrovascular disease and peripheral vascular disease. Although a 2007 meta-analysis found that folic acid supplementation decreased the risk of a first stroke by 18% (P = .045),50 a later meta-analysis contradicts this finding.51
A 2009 meta-analysis found that patients with peripheral arterial disease had higher homocysteine levels than controls, but it did not find any benefit from supplementation, owing to heterogeneity of the clinical end points used.52 Indeed, a 2009 Cochrane Database Systematic Review found that there were no adequate trials of the treatment of patients with peripheral vascular disease who have elevated plasma homocysteine.53
However, immediately after the Cochrane review was published, Khandanpour et al36 published the results of a trial of the effect of folic acid and 5-methyltetrahydrofolate (an active form of folic acid) supplementation on the ankle-brachial pressure index and the pulse-wave velocity in patients with peripheral arterial disease. These measures improved with 16 weeks of treatment. For the ankle-brachial pressure index, the P value was less than .01 for folic acid and .009 for 5-methyltetrahydrofolate; for the pulse-wave velocity, the P value was .051 for folic acid and .011 for 5-methyltetrahydrofolate.
Kidney disease
One could postulate that patients with end-stage renal disease or chronic kidney disease might benefit the most from folic acid supplementation, given the correlation of elevations in homocysteine levels with decline in glomerular filtration rate.
However, only one study found a lower rate of cardiovascular events with folic acid supplementation in dialysis patients, and the difference was not statistically significant (25% vs 36%, P < .08).31 Further, several studies found no benefit of folic acid supplementation in patients with chronic kidney disease.11,12,37
FUTURE DIRECTIONS AND RECOMMENDATIONS
Many experts have suggested that the existing evidence indicates that the homocysteine-lowering therapies folic acid, vitamin B6, and vitamin B12 do not lower the risk of cardiovascular disease.38,54–59 Indeed, the American Heart Association guidelines for cardiovascular disease prevention in women do not recommend folic acid supplementation to prevent cardiovascular disease.60 (Recommendations for men are the same as for women.) However, most of the clinical trials have not selected and treated patients with elevated homocysteine levels, but have instead included all patients regardless of homocysteine level.
At least two large ongoing trials are currently evaluating B-vitamin therapy for secondary prevention, but neither trial is looking specifically at patients with elevated homocysteine levels.61,62
Thus, instead of concluding that no patients could benefit from homocysteine-lowering treatment, future studies need to clarify:
- Whether patients with elevated homocysteine would benefit from such treatment
- At what level it would be appropriate to start treatment
- The appropriate target homocysteine level with treatment.
Particularly given the recent finding that folic acid supplementation may increase cancer risk,63 these questions need closer scrutiny.
- Humphrey LL, Fu R, Rogers K, Freeman M, Helfand M. Homocysteine level and coronary heart disease incidence: a systematic review and meta-analysis. Mayo Clin Proc 2008; 83:1203–1212.
- Boers GH. The case for mild hyperhomocysteinaemia as a risk factor. J Inherit Metab Dis 1997; 20:301–306.
- Austin RC, Lentz SR, Werstuck GH. Role of hyperhomocysteinemia in endothelial dysfunction and atherothrombotic disease. Cell Death Differ 2004; 11(suppl 1):S56–S64.
- Albert CM, Cook NR, Gaziano JM, et al. Effect of folic acid and B vitamins on risk of cardiovascular events and total mortality among women at high risk for cardiovascular disease: a randomized trial. JAMA 2008; 299:2027–2036.
- Ebbing M, Bleie Ø, Ueland PM, et al. Mortality and cardiovascular events in patients treated with homocysteine-lowering B vitamins after coronary angiography: a randomized controlled trial. JAMA 2008; 300:795–804.
- Butz LW, du Vigneaud V. The formation of a homologue of cystine by the decompensation of methionine with sulphuric acid. J Biol Chem 1932; 99:135–142.
- Gibson JB, Carson NA, Neill DW. Pathological findings in homocystinuria. J Clin Pathol 1964; 17:427–437.
- McCully KS. Vascular pathology of homocysteinemia: implications for the pathogenesis of arteriosclerosis. Am J Pathol 1969; 56:111–128.
- Zhang D, Jiang X, Fang P, et al. Hyperhomocysteinemia promotes inflammatory monocyte generation and accelerates atherosclerosis in transgenic cystathionine beta-synthase-deficient mice. Circulation 2009; 120:1893–1902.
- Woo KS, Chook P, Lolin YI, Sanderson JE, Metreweli C, Celermajer DS. Folic acid improves arterial endothelial function in adults with hyperhomocystinemia. J Am Coll Cardiol 1999; 34:2002–2006.
- Righetti M, Ferrario GM, Milani S, et al. Effects of folic acid treatment on homocysteine levels and vascular disease in hemodialysis patients. Med Sci Monit 2003; 9:PI19–PI24.
- Zoungas S, McGrath BP, Branley P, et al. Cardiovascular morbidity and mortality in the Atherosclerosis and Folic Acid Supplementation Trial (ASFAST) in chronic renal failure: a multicenter, randomized, controlled trial. J Am Coll Cardiol 2006; 47:1108–1116.
- Carmel R, Jacobsen DW, editors. Homocysteine in Health and Disease. Cambridge, UK: Cambridge University Press, 2001.
- Yap S, Boers GH, Wilcken B, et al. Vascular outcome in patients with homocystinuria due to cystathionine beta-synthase deficiency treated chronically: a multicenter observational study. Arterioscler Thromb Vasc Biol 2001; 21:2080–2085.
- McKusick V. 236200 Homocystinuria. In:McKusick V, editor. Mendelian Inheritance in Man. 10th ed. Baltimore, MD: The Johns Hopkins University Press, 1992:1444–1446.
- McKusick V. 236250 Homocystinuria due to deficiency of N(5,10)-methylenetetrahydrofolate reductase activity. In:McKusick V, editor. Mendelian Inheritance in Man. Baltimore, MD: The Johns Hopkins University Press, 1992:1447–1448.
- Wald DS, Law M, Morris JK. Homocysteine and cardiovascular disease: evidence on causality from a meta-analysis. BMJ 2002; 325:1202.
- Nygård O, Vollset SE, Refsum H, et al. Total plasma homocysteine and cardiovascular risk profile. The Hordaland Homocysteine Study. JAMA 1995; 274:1526–1533.
- Klerk M, Verhoef P, Clarke R, Blom HJ, Kok FJ, Schouten EG; MTHFR Studies Collaboration Group. MTHFR 677C-->T polymorphism and risk of coronary heart disease: a meta-analysis. JAMA 2002; 288:2023–2031.
- Kelly PJ, Rosand J, Kistler JP, et al. Homocysteine, MTHFR 677C-->T polymorphism, and risk of ischemic stroke: results of a meta-analysis. Neurology 2002; 59:529–536.
- Lewis SJ, Ebrahim S, Davey Smith G. Meta-analysis of MTHFR 677C->T polymorphism and coronary heart disease: does totality of evidence support causal role for homocysteine and preventive potential of folate? BMJ 2005; 331:1053.
- Brattström L, Wilcken DE, Ohrvik J, Brudin L. Common methylenetetrahydrofolate reductase gene mutation leads to hyperhomocysteinemia but not to vascular disease: the result of a meta-analysis. Circulation 1998; 98:2520–2526.
- Cui R, Iso H, Date C, Kikuchi S, Tamakoshi A; Japan Collaborative Cohort Study Group. Dietary folate and vitamin B6 and B12 intake in relation to mortality from cardiovascular diseases: Japan Collaborative Cohort Study. Stroke 2010; 41:1285–1289.
- Liu S, Stampfer MJ, Hu FB, et al. Whole-grain consumption and risk of coronary heart disease: results from the Nurses’ Health Study. Am J Clin Nutr 1999; 70:412–419.
- Liu S, Manson JE, Stampfer MJ, et al. Whole grain consumption and risk of ischemic stroke in women: a prospective study. JAMA 2000; 284:1534–1540.
- Merchant AT, Hu FB, Spiegelman D, Willett WC, Rimm EB, Ascherio A. The use of B vitamin supplements and peripheral arterial disease risk in men are inversely related. J Nutr 2003; 133:2863–2867.
- Rimm EB, Willett WC, Hu FB, et al. Folate and vitamin B6 from diet and supplements in relation to risk of coronary heart disease among women. JAMA 1998; 279:359–364.
- US Preventive Services Task Force. Using nontraditional risk factors in coronary heart disease risk assessment: US Preventive Services Task Force recommendation statement. Ann Intern Med 2009; 151:474–482.
- Helfand M, Buckley DI, Freeman M, et al. Emerging risk factors for coronary heart disease: a summary of systematic reviews conducted for the US Preventive Services Task Force. Ann Intern Med 2009; 151:496–507.
- Martí-Carvajal AJ, Solà I, Lathyris D, Salanti G. Homocysteine lowering interventions for preventing cardiovascular events. Cochrane Database Syst Rev 2009; ( 4):CD006612.
- Righetti M, Serbelloni P, Milani S, Ferrario G. Homocysteine-lowering vitamin B treatment decreases cardiovascular events in hemodialysis patients. Blood Purif 2006; 24:379–386.
- Mager A, Orvin K, Koren-Morag N, et al. Impact of homocysteine-lowering vitamin therapy on long-term outcome of patients with coronary artery disease. Am J Cardiol 2009; 104:745–749.
- Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) Collaborative Group; Armitage JM, Bowman L, Clarke RJ, et al. Effects of homocysteine-lowering with folic acid plus vitamin B12 vs placebo on mortality and major morbidity in myocardial infarction survivors: a randomized trial. JAMA 2010; 303:2486–2494.
- Schnyder G, Roffi M, Flammer Y, Pin R, Hess OM. Effect of homocysteine-lowering therapy with folic acid, vitamin B12, and percutaneous coronary intervention: the Swiss Heart Study: a randomized controlled trial. JAMA 2002; 288:973–979.
- Lange H, Suryapranata H, De Luca G, et al. Folate therapy and in-stent restenosis after coronary stenting. N Engl J Med 2004; 350:2673–2781.
- Khandanpour N, Armon MP, Jennings B, et al. Randomized clinical trial of folate supplementation in patients with peripheral arterial disease. Br J Surg 2009; 96:990–998.
- Wrone EM, Hornberger JM, Zehnder JL, McCann LM, Coplon NS, Fortmann SP. Randomized trial of folic acid for prevention of cardiovascular events in end-stage renal disease. J Am Soc Nephrol 2004; 15:420–426.
- Loscalzo J. Homocysteine trials—clear outcomes for complex reasons. N Engl J Med 2006; 354:1629–1632.
- Bønaa KH, Njølstad I, Ueland PM, et al; NORVIT Trial Investigators. Homocysteine lowering and cardiovascular events after acute myocardial infarction. N Engl J Med 2006; 354:1578–1588.
- Carrero JJ, López-Huertas E, Salmerón LM, Baró L, Ros E. Daily supplementation with (n-3) PUFAs, oleic acid, folic acid, and vitamins B-6 and E increases pain-free walking distance and improves risk factors in men with peripheral vascular disease. J Nutr 2005; 135:1393–1399.
- Liem AH, van Boven AJ, Veeger NJ, et al; Folic Acid on Risk Diminishment After Acute Myocarial Infarction Study Group. Efficacy of folic acid when added to statin therapy in patients with hypercholesterolemia following acute myocardial infarction: a randomised pilot trial. Int J Cardiol 2004; 93:175–179.
- Liem A, Reynierse-Buitenwerf GH, Zwinderman AH, Jukema JW, van Veldhuisen DJ. Secondary prevention with folic acid: results of the Goes extension study. Heart 2005; 91:1213–1214.
- Lonn E, Yusuf S, Arnold MJ, et al; Heart Outcomes Prevention Evaluation (HOPE) 2 Investigators. Homocysteine lowering with folic acid and B vitamins in vascular disease. N Engl J Med 2006; 354:1567–1577.
- Sydow K, Schwedhelm E, Arakawa N, et al. ADMA and oxidative stress are responsible for endothelial dysfunction in hyperhomocyst(e)inemia: effects of L-arginine and B vitamins. Cardiovasc Res 2003; 57:244–252.
- Toole JF, Malinow MR, Chambless LE, et al. Lowering homocysteine in patients with ischemic stroke to prevent recurrent stroke, myocardial infarction, and death: the Vitamin Intervention for Stroke Prevention (VISP) randomized controlled trial. JAMA 2004; 291:565–575.
- Jamison RL, Hartigan P, Kaufman JS, et al; Veterans Affairs Site Investigators. Effect of homocysteine lowering on mortality and vascular disease in advanced chronic kidney disease and end-stage renal disease: a randomized controlled trial. JAMA 2007; 2989:1163–1170. Erratum in JAMA 2008;300:170.
- Bazzano LA, Reynolds K, Holder KN, He J. Effect of folic acid supplementation on risk of cardiovascular diseases: a meta-analysis of randomized controlled trials. JAMA 2006; 296:2720–2726.
- Namazi MH, Motamedi MR, Safi M, Vakili H, Saadat H, Nazari N. Efficacy of folic acid therapy for prevention of in-stent restenosis: a randomized clinical trial. Arch Iran Med 2006; 9:108–110.
- Schnyder G, Roffi M, Pin R, et al. Decreased rate of coronary restenosis after lowering of plasma homocysteine levels. N Engl J Med 2001; 345:1593–1600.
- Wang X, Qin X, Demirtas H, et al. Efficacy of folic acid supplementation in stroke prevention: a meta-analysis. Lancet 2007; 369:1876–1882.
- Lee M, Hong KS, Chang SC, Saver JL. Efficacy of homocysteine-lowering therapy with folic Acid in stroke prevention: a meta-analysis. Stroke 2010; 41:1205–1212.
- Khandanpour N, Loke YK, Meyer FJ, Jennings B, Armon MP. Homocysteine and peripheral arterial disease: systematic review and meta-analysis. Eur J Vasc Endovasc Surg 2009; 38:316–322.
- Hansrani M, Stansby G. Homocysteine lowering interventions for peripheral arterial disease and bypass grafts. Cochrane Database Syst Rev 2002; ( 3):CD003285.
- Mosca L. Novel cardiovascular risk factors: do they add value to your practice? Am Fam Physician 2003; 67:264,266.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction. J Am Coll Cardiol 2007; 50:e1–e157.
- Lonn E. Homocysteine-lowering B vitamin therapy in cardiovascular prevention—wrong again? JAMA 2008; 299:2086–2087.
- Milani RV, Lavie CJ. Homocysteine: the Rubik’s cube of cardiovascular risk factors. Mayo Clin Proc 2008; 83:1200–1202.
- Bazzano LA. Folic acid supplementation and cardiovascular disease: the state of the art. Am J Med Sci 2009; 338:48–49.
- Ntaios G, Savopoulos C, Grekas D, Hatzitolios A. The controversial role of B-vitamins in cardiovascular risk: an update. Arch Cardiovasc Dis 2009; 102:847–854.
- Mosca L, Banka CL, Benjamin EJ; Expert Panel/Writing Group. Evidence-based guidelines for cardiovascular disease prevention in women: 2007 update. Circulation 2007; 115:1481–1501.
- Bassuk SS, Albert CM, Cook NR, et al. The Women’s Antioxidant Cardiovascular Study: design and baseline characteristics of participants. J Womens Health (Larchmt) 2004; 13:99–117.
- SEARCH Study Collaborative Group; Bowman L, Armitage J, Bulbulia R, Parish S, Collins R. Study of the effectiveness of additional reductions in cholesterol and homocysteine (SEARCH): characteristics of a randomized trial among 12064 myocardial infarction survivors. Am Heart J 2007; 154:815–823,823.e1–e6.
- Ebbing M, Bønaa KH, Nygård O, et al. Cancer incidence and mortality after treatment with folic acid and vitamin B12. JAMA 2009; 302:2119–2126.
- Humphrey LL, Fu R, Rogers K, Freeman M, Helfand M. Homocysteine level and coronary heart disease incidence: a systematic review and meta-analysis. Mayo Clin Proc 2008; 83:1203–1212.
- Boers GH. The case for mild hyperhomocysteinaemia as a risk factor. J Inherit Metab Dis 1997; 20:301–306.
- Austin RC, Lentz SR, Werstuck GH. Role of hyperhomocysteinemia in endothelial dysfunction and atherothrombotic disease. Cell Death Differ 2004; 11(suppl 1):S56–S64.
- Albert CM, Cook NR, Gaziano JM, et al. Effect of folic acid and B vitamins on risk of cardiovascular events and total mortality among women at high risk for cardiovascular disease: a randomized trial. JAMA 2008; 299:2027–2036.
- Ebbing M, Bleie Ø, Ueland PM, et al. Mortality and cardiovascular events in patients treated with homocysteine-lowering B vitamins after coronary angiography: a randomized controlled trial. JAMA 2008; 300:795–804.
- Butz LW, du Vigneaud V. The formation of a homologue of cystine by the decompensation of methionine with sulphuric acid. J Biol Chem 1932; 99:135–142.
- Gibson JB, Carson NA, Neill DW. Pathological findings in homocystinuria. J Clin Pathol 1964; 17:427–437.
- McCully KS. Vascular pathology of homocysteinemia: implications for the pathogenesis of arteriosclerosis. Am J Pathol 1969; 56:111–128.
- Zhang D, Jiang X, Fang P, et al. Hyperhomocysteinemia promotes inflammatory monocyte generation and accelerates atherosclerosis in transgenic cystathionine beta-synthase-deficient mice. Circulation 2009; 120:1893–1902.
- Woo KS, Chook P, Lolin YI, Sanderson JE, Metreweli C, Celermajer DS. Folic acid improves arterial endothelial function in adults with hyperhomocystinemia. J Am Coll Cardiol 1999; 34:2002–2006.
- Righetti M, Ferrario GM, Milani S, et al. Effects of folic acid treatment on homocysteine levels and vascular disease in hemodialysis patients. Med Sci Monit 2003; 9:PI19–PI24.
- Zoungas S, McGrath BP, Branley P, et al. Cardiovascular morbidity and mortality in the Atherosclerosis and Folic Acid Supplementation Trial (ASFAST) in chronic renal failure: a multicenter, randomized, controlled trial. J Am Coll Cardiol 2006; 47:1108–1116.
- Carmel R, Jacobsen DW, editors. Homocysteine in Health and Disease. Cambridge, UK: Cambridge University Press, 2001.
- Yap S, Boers GH, Wilcken B, et al. Vascular outcome in patients with homocystinuria due to cystathionine beta-synthase deficiency treated chronically: a multicenter observational study. Arterioscler Thromb Vasc Biol 2001; 21:2080–2085.
- McKusick V. 236200 Homocystinuria. In:McKusick V, editor. Mendelian Inheritance in Man. 10th ed. Baltimore, MD: The Johns Hopkins University Press, 1992:1444–1446.
- McKusick V. 236250 Homocystinuria due to deficiency of N(5,10)-methylenetetrahydrofolate reductase activity. In:McKusick V, editor. Mendelian Inheritance in Man. Baltimore, MD: The Johns Hopkins University Press, 1992:1447–1448.
- Wald DS, Law M, Morris JK. Homocysteine and cardiovascular disease: evidence on causality from a meta-analysis. BMJ 2002; 325:1202.
- Nygård O, Vollset SE, Refsum H, et al. Total plasma homocysteine and cardiovascular risk profile. The Hordaland Homocysteine Study. JAMA 1995; 274:1526–1533.
- Klerk M, Verhoef P, Clarke R, Blom HJ, Kok FJ, Schouten EG; MTHFR Studies Collaboration Group. MTHFR 677C-->T polymorphism and risk of coronary heart disease: a meta-analysis. JAMA 2002; 288:2023–2031.
- Kelly PJ, Rosand J, Kistler JP, et al. Homocysteine, MTHFR 677C-->T polymorphism, and risk of ischemic stroke: results of a meta-analysis. Neurology 2002; 59:529–536.
- Lewis SJ, Ebrahim S, Davey Smith G. Meta-analysis of MTHFR 677C->T polymorphism and coronary heart disease: does totality of evidence support causal role for homocysteine and preventive potential of folate? BMJ 2005; 331:1053.
- Brattström L, Wilcken DE, Ohrvik J, Brudin L. Common methylenetetrahydrofolate reductase gene mutation leads to hyperhomocysteinemia but not to vascular disease: the result of a meta-analysis. Circulation 1998; 98:2520–2526.
- Cui R, Iso H, Date C, Kikuchi S, Tamakoshi A; Japan Collaborative Cohort Study Group. Dietary folate and vitamin B6 and B12 intake in relation to mortality from cardiovascular diseases: Japan Collaborative Cohort Study. Stroke 2010; 41:1285–1289.
- Liu S, Stampfer MJ, Hu FB, et al. Whole-grain consumption and risk of coronary heart disease: results from the Nurses’ Health Study. Am J Clin Nutr 1999; 70:412–419.
- Liu S, Manson JE, Stampfer MJ, et al. Whole grain consumption and risk of ischemic stroke in women: a prospective study. JAMA 2000; 284:1534–1540.
- Merchant AT, Hu FB, Spiegelman D, Willett WC, Rimm EB, Ascherio A. The use of B vitamin supplements and peripheral arterial disease risk in men are inversely related. J Nutr 2003; 133:2863–2867.
- Rimm EB, Willett WC, Hu FB, et al. Folate and vitamin B6 from diet and supplements in relation to risk of coronary heart disease among women. JAMA 1998; 279:359–364.
- US Preventive Services Task Force. Using nontraditional risk factors in coronary heart disease risk assessment: US Preventive Services Task Force recommendation statement. Ann Intern Med 2009; 151:474–482.
- Helfand M, Buckley DI, Freeman M, et al. Emerging risk factors for coronary heart disease: a summary of systematic reviews conducted for the US Preventive Services Task Force. Ann Intern Med 2009; 151:496–507.
- Martí-Carvajal AJ, Solà I, Lathyris D, Salanti G. Homocysteine lowering interventions for preventing cardiovascular events. Cochrane Database Syst Rev 2009; ( 4):CD006612.
- Righetti M, Serbelloni P, Milani S, Ferrario G. Homocysteine-lowering vitamin B treatment decreases cardiovascular events in hemodialysis patients. Blood Purif 2006; 24:379–386.
- Mager A, Orvin K, Koren-Morag N, et al. Impact of homocysteine-lowering vitamin therapy on long-term outcome of patients with coronary artery disease. Am J Cardiol 2009; 104:745–749.
- Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) Collaborative Group; Armitage JM, Bowman L, Clarke RJ, et al. Effects of homocysteine-lowering with folic acid plus vitamin B12 vs placebo on mortality and major morbidity in myocardial infarction survivors: a randomized trial. JAMA 2010; 303:2486–2494.
- Schnyder G, Roffi M, Flammer Y, Pin R, Hess OM. Effect of homocysteine-lowering therapy with folic acid, vitamin B12, and percutaneous coronary intervention: the Swiss Heart Study: a randomized controlled trial. JAMA 2002; 288:973–979.
- Lange H, Suryapranata H, De Luca G, et al. Folate therapy and in-stent restenosis after coronary stenting. N Engl J Med 2004; 350:2673–2781.
- Khandanpour N, Armon MP, Jennings B, et al. Randomized clinical trial of folate supplementation in patients with peripheral arterial disease. Br J Surg 2009; 96:990–998.
- Wrone EM, Hornberger JM, Zehnder JL, McCann LM, Coplon NS, Fortmann SP. Randomized trial of folic acid for prevention of cardiovascular events in end-stage renal disease. J Am Soc Nephrol 2004; 15:420–426.
- Loscalzo J. Homocysteine trials—clear outcomes for complex reasons. N Engl J Med 2006; 354:1629–1632.
- Bønaa KH, Njølstad I, Ueland PM, et al; NORVIT Trial Investigators. Homocysteine lowering and cardiovascular events after acute myocardial infarction. N Engl J Med 2006; 354:1578–1588.
- Carrero JJ, López-Huertas E, Salmerón LM, Baró L, Ros E. Daily supplementation with (n-3) PUFAs, oleic acid, folic acid, and vitamins B-6 and E increases pain-free walking distance and improves risk factors in men with peripheral vascular disease. J Nutr 2005; 135:1393–1399.
- Liem AH, van Boven AJ, Veeger NJ, et al; Folic Acid on Risk Diminishment After Acute Myocarial Infarction Study Group. Efficacy of folic acid when added to statin therapy in patients with hypercholesterolemia following acute myocardial infarction: a randomised pilot trial. Int J Cardiol 2004; 93:175–179.
- Liem A, Reynierse-Buitenwerf GH, Zwinderman AH, Jukema JW, van Veldhuisen DJ. Secondary prevention with folic acid: results of the Goes extension study. Heart 2005; 91:1213–1214.
- Lonn E, Yusuf S, Arnold MJ, et al; Heart Outcomes Prevention Evaluation (HOPE) 2 Investigators. Homocysteine lowering with folic acid and B vitamins in vascular disease. N Engl J Med 2006; 354:1567–1577.
- Sydow K, Schwedhelm E, Arakawa N, et al. ADMA and oxidative stress are responsible for endothelial dysfunction in hyperhomocyst(e)inemia: effects of L-arginine and B vitamins. Cardiovasc Res 2003; 57:244–252.
- Toole JF, Malinow MR, Chambless LE, et al. Lowering homocysteine in patients with ischemic stroke to prevent recurrent stroke, myocardial infarction, and death: the Vitamin Intervention for Stroke Prevention (VISP) randomized controlled trial. JAMA 2004; 291:565–575.
- Jamison RL, Hartigan P, Kaufman JS, et al; Veterans Affairs Site Investigators. Effect of homocysteine lowering on mortality and vascular disease in advanced chronic kidney disease and end-stage renal disease: a randomized controlled trial. JAMA 2007; 2989:1163–1170. Erratum in JAMA 2008;300:170.
- Bazzano LA, Reynolds K, Holder KN, He J. Effect of folic acid supplementation on risk of cardiovascular diseases: a meta-analysis of randomized controlled trials. JAMA 2006; 296:2720–2726.
- Namazi MH, Motamedi MR, Safi M, Vakili H, Saadat H, Nazari N. Efficacy of folic acid therapy for prevention of in-stent restenosis: a randomized clinical trial. Arch Iran Med 2006; 9:108–110.
- Schnyder G, Roffi M, Pin R, et al. Decreased rate of coronary restenosis after lowering of plasma homocysteine levels. N Engl J Med 2001; 345:1593–1600.
- Wang X, Qin X, Demirtas H, et al. Efficacy of folic acid supplementation in stroke prevention: a meta-analysis. Lancet 2007; 369:1876–1882.
- Lee M, Hong KS, Chang SC, Saver JL. Efficacy of homocysteine-lowering therapy with folic Acid in stroke prevention: a meta-analysis. Stroke 2010; 41:1205–1212.
- Khandanpour N, Loke YK, Meyer FJ, Jennings B, Armon MP. Homocysteine and peripheral arterial disease: systematic review and meta-analysis. Eur J Vasc Endovasc Surg 2009; 38:316–322.
- Hansrani M, Stansby G. Homocysteine lowering interventions for peripheral arterial disease and bypass grafts. Cochrane Database Syst Rev 2002; ( 3):CD003285.
- Mosca L. Novel cardiovascular risk factors: do they add value to your practice? Am Fam Physician 2003; 67:264,266.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction. J Am Coll Cardiol 2007; 50:e1–e157.
- Lonn E. Homocysteine-lowering B vitamin therapy in cardiovascular prevention—wrong again? JAMA 2008; 299:2086–2087.
- Milani RV, Lavie CJ. Homocysteine: the Rubik’s cube of cardiovascular risk factors. Mayo Clin Proc 2008; 83:1200–1202.
- Bazzano LA. Folic acid supplementation and cardiovascular disease: the state of the art. Am J Med Sci 2009; 338:48–49.
- Ntaios G, Savopoulos C, Grekas D, Hatzitolios A. The controversial role of B-vitamins in cardiovascular risk: an update. Arch Cardiovasc Dis 2009; 102:847–854.
- Mosca L, Banka CL, Benjamin EJ; Expert Panel/Writing Group. Evidence-based guidelines for cardiovascular disease prevention in women: 2007 update. Circulation 2007; 115:1481–1501.
- Bassuk SS, Albert CM, Cook NR, et al. The Women’s Antioxidant Cardiovascular Study: design and baseline characteristics of participants. J Womens Health (Larchmt) 2004; 13:99–117.
- SEARCH Study Collaborative Group; Bowman L, Armitage J, Bulbulia R, Parish S, Collins R. Study of the effectiveness of additional reductions in cholesterol and homocysteine (SEARCH): characteristics of a randomized trial among 12064 myocardial infarction survivors. Am Heart J 2007; 154:815–823,823.e1–e6.
- Ebbing M, Bønaa KH, Nygård O, et al. Cancer incidence and mortality after treatment with folic acid and vitamin B12. JAMA 2009; 302:2119–2126.
KEY POINTS
- Factors that can cause the plasma homocysteine concentration to be high include deficiencies of vitamin B6, vitamin B12, and folic acid; renal insufficiency; and genetic variants in enzymes responsible for homocysteine metabolism.
- Higher plasma homocysteine levels are associated with a higher risk of cardiovascular, cerebrovascular, and peripheral arterial disease.
- Supplementation of B vitamins and folic acid can lower plasma homocysteine levels.
- Randomized controlled trials of supplementation to prevent cardiovascular events and other adverse outcomes have had mostly negative results. However, most patients in these trials had normal baseline plasma homocysteine levels.
- Needed are randomized trials to see if supplementation improves outcomes in patients with high homocysteine levels.
What can we expect from omega-3 fatty acids?
Many patients are taking fish oil supplements, which contain omega-3 fatty acids, either on their own initiative or on their physician’s advice. Driving this trend are accumulating data from observational and epidemiologic studies and clinical trials that these lipids actually reduce cardiovascular risk.
In the following article, we review available studies of omega-3 fatty acids in cardiovascular disease.
WHAT ARE OMEGA-3 FATTY ACIDS?
Omega-3 fatty acids are a class of polyunsaturated fatty acids. Their name means that they all have a double carbon-to-carbon bond in the third position from the omega (or methyl, or n) end of the fatty acid chain.
Most of the cardiovascular research on the omega-3 family has been on eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA), and alpha-linolenic acid (ALA). EPA and DHA are found primarily in fatty fish; ALA is abundant in flaxseed, walnuts, and soybeans.1 The human body can convert small amounts of ALA into EPA and DHA: only about 5% of ALA is converted to EPA and less than 0.5% is converted to DHA. Currently, it is not known whether ALA is active itself or only via these metabolites. In this review, the term omega-3 fatty acid refers to EPA and DHA only.
GETTING ENOUGH FISH OIL
Healthy people should consume fish (preferably oily fish) at least twice a week, according to the American Heart Association.1 However, not all fish contain the same amount of oil. Some, such as cod and catfish, contain only 0.2 g of EPA/DHA per 100-g serving; others, such as Atlantic salmon, contain about 10 times as much (Table 1).2
People with known coronary artery disease should take in 1 g of EPA/DHA per day, according to the American Heart Association.1 This recommendation is based on clinical trials that found omega-3 fatty acids to have beneficial effects.
For most people with coronary artery disease, this means taking supplements. However, buyers need to carefully examine the label of over-the-counter fish oil supplements to see if they contain the recommended amounts of both DHA and EPA. Generic 1-g fish oil supplements may contain variable amounts of DHA and EPA, and some may have less than 300 mg.
People with hypertriglyceridemia. The US Food and Drug Administration (FDA) has approved Lovaza (formerly Omacor), which contains EPA/DHA in higher concentrations than over-the-counter preparations, for the treatment of hypertriglyceridemia in people with triglyceride levels higher than 500 mg/ dL, along with a regimen of diet and regular exercise.3 It is currently the only FDA-approved prescription form of omega-3 fatty acid ethyl esters. Each 1-g capsule contains 375 mg of DHA and 465 mg of EPA; the recommended dose is 2 to 4 g/day. To take in an equivalent amount of these substances with over-the counter-preparations, patients might have to take many capsules a day.
Safety of omega-3 fatty acids
Generally, omega-3 fatty acids are very well tolerated, and their adverse effects are limited to gastrointestinal complaints (discomfort, upset stomach) and a fishy odor. Common ways to prevent these effects are to freeze the capsules or take them at bedtime or with meals.
Mercury advisory on fish. Nursing or pregnant women should limit their consumption of certain fish, as some fish (but not fish oil) contain high levels of mercury. The highest levels of mercury are usually found in the larger, older predatory fish such as swordfish, tilefish, and mackerel, and the FDA advises women who are nursing or pregnant to avoid these fish completely. Tuna, red snapper, and orange roughy are lower in mercury, but nursing or pregnant women should still limit consumption of these fish to 12 oz per week.4
Theoretical risk of bleeding. In theory, high doses of omega-3 fatty acids may increase the bleeding time by inhibiting the arachidonic acid pathway. Clinically, this effect is minimal. In a trial in 511 patients undergoing coronary artery bypass grafting who were receiving aspirin or warfarin (Coumadin), the bleeding time and the number of bleeding episodes were no higher in those who were randomized to receive 4 g/day of omega-3 fatty acids daily than in a control group.5
Harris6 reviewed 19 studies of omega-3 fatty acids in patients undergoing coronary artery bypass grafting, carotid endarterectomy, or femoral artery catheterization, and none of the studies found a significantly increased risk of bleeding.
HOW DO OMEGA-3 FATTY ACIDS REDUCE RISK?
After epidemiologic studies found that Greenland Eskimos (who consume diets rich in omega-3 fatty acids) have low rates of cardiovascular disease,7 omega-3 fatty acids were hypothesized to reduce cardiovascular risk. Over the past 3 decades, their potential benefit in lowering lipid levels, blood pressure, and the risk of death in patients with known heart disease has been widely researched.
Lower triglyceride levels
The growing problem of obesity in the United States has led to more patients presenting with hypertriglyceridemia, a risk factor for coronary heart disease.
In 2001, the National Cholesterol Education Program’s third Adult Treatment Panel (ATP III)8 redefined normal triglyceride levels as less than 150 mg/dL; previously, normal was defined as less than 200 mg/dL. For people with borderline-high triglyceride levels (150–200 mg/dL), the ATP III recommends focusing on lowering the level of low-density lipoprotein cholesterol (LDL-C). For those with high to very high triglyceride levels (> 500 mg/dL), the current treatment options are niacin, fibrates, and omega-3 fatty acids.
Hypertriglyceridemia is thought to increase the risk of coronary heart disease by two mechanisms. First, and more important, triglyceride-rich lipoproteins such as very-low-density lipoprotein (VLDL) and intermediate-density lipoprotein (IDL) are thought to be atherogenic. Secondly, triglyceride-lipoprotein metabolism involves competition with high-density lipoprotein (HDL), leading to a decrease in HDL production and to denser LDL particles.9
How omega-3 fatty acids lower triglyceride levels has been inferred from preclinical studies. One mechanism, seen in animal studies, is by decreasing hepatic synthesis and secretion of VLDL particles by inhibiting various enzyme transcription factors. Another proposed mechanism is that EPA and DHA increase the activity of lipoprotein lipase, leading to an increase in chylomicron clearance.10 This was validated by Khan et al,11 who showed that lipoprotein lipase activity increased in patients who received omega-3 fatty acids 3 g/day for 6 weeks.
How much do they lower triglycerides? Data from the makers of Lovaza3 indicate that in a patient population with a mean baseline triglyceride level of 816 mg/dL, 4 g/day of omega-3 fatty acids lowered triglyceride levels to 488 mg/dL, a 45% reduction (P < .0001). In addition, HDL cholesterol (HDL-C) levels increased by 9%.
The higher the dose and the higher the baseline triglyceride level, the greater the effect. Balk et al12 performed a meta-analysis of 25 randomized trials and calculated that each 1-g increase in fish oil dose per day lowered the triglyceride level by about 8 mg/dL. However, patients with high baseline triglyceride levels had more dramatic reduction of triglycerides with fish oil. The average reduction in triglyceride levels was 27 mg/dL, accompanied by an increase in HDL-C of 1.6 mg/dL, an increase in LDL-C of 6 mg/dL, and no change in total cholesterol levels.
Pownall et al13 report that, in 19 patients with hypertriglyceridemia (median baseline level 801 mg/dL), omega-3 fatty acids 4 g/day reduced triglyceride levels to 512 mg/dL, a 38.9% change (P = .001). In 21 patients receiving placebo, triglyceride levels decreased by 7.8% (P = .001 compared with active therapy). The effect on HDL-C was minimal, but the median LDL-C level increased by 16.7% (from 43 to 53 mg/dL, P = .007) with fish oil therapy.
Fish oil plus a statin may have advantages
Most patients seen in clinical practice present with mixed dyslipidemias. The current ATP III guidelines aim for stricter triglyceride and LDL-C targets than in the past, which monotherapy alone may not be able to achieve.
Statin therapy by itself effectively lowers LDL-C but has modest effects on triglycerides. Omega-3 fatty acids effectively reduce triglycerides but have been known to increase LDL-C levels. This net LDL-C increase averaged around 10 mg/dL as reported in a review by Harris et al,14 and 6 mg/dL as reported by Balk et al.12 However, despite the net effect of an increase in LDL-C, it is hypothesized that the larger LDL particles produced by omega-3 fatty acid treatment may be less atherogenic.15
The effectiveness of combined therapy in reducing triglycerides has been widely studied.
Chan et al,16 in a randomized, placebo-controlled trial, looked at the effectiveness of atorvastatin (Lipitor) and EPA/DHA. Fifty-two obese men were randomized to receive atorvastatin 40 mg/day, EPA/DHA 4 g/day, both in combination, or placebo. After 6 weeks, triglyceride levels had decreased by 26% from baseline in the atorvastatin group, 25% in the EPA/DHA group, and 40% in the combination therapy group (P = .002). LDL-C levels decreased to a similar degree with either atorvastatin monotherapy or combination therapy. Similar studies show similar results.
Combination therapy may also lower the rate of major coronary events (see below).
The Japan EPA Lipid Intervention Study (JELIS)17 randomized more than 18,000 patients to receive either a statin alone or a statin plus EPA 1,800 mg daily, in an open-label fashion. The statins used were pravastatin (Pravachol) 10 mg daily or simvastatin (Zocor) 5 mg daily; if hypercholesterolemia remained uncontrolled, these doses were doubled. The patients were 5,859 men and 12,786 postmenopausal women (mean age 61) with or without coronary artery disease who had total cholesterol levels of 251 mg/dL or greater. The mean baseline LDL-C level was 180 mg/ dL. People who had had an acute myocardial infarction in the past 6 months or unstable angina were excluded. The primary end point examined was any major coronary event, defined as sudden death, fatal or nonfatal myocardial infarction, unstable angina, angioplasty, or coronary artery bypass grafting.
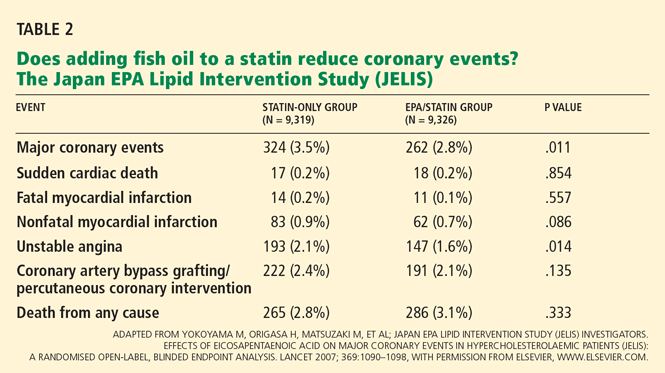
The JELIS trial showed that combination therapy may reduce the risk of coronary events, the aim of treating dyslipidemia. It was the largest randomized trial to date comparing statin use alone and in combination with omega-3 fatty acids. However, it was performed in Japan, where people already have a high intake of fatty fish, and the results may not be applicable to other countries.
May prevent arrhythmias
The Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico-Prevenzione (GISSI-Prevention) trial18 was the largest randomized trial to date of fish oil therapy as secondary prevention. In this trial, 11,323 patients who had had a myocardial infarction less than 3 months before enrollment were randomized to receive either EPA/DHA 850 mg daily, vitamin E, both, or no treatment. The primary end points were death from any cause, nonfatal myocardial infarction, and nonfatal stroke.
At 3 months, 63 (1.1%) of the patients in the EPA/DHA group had died, compared with 88 (1.6%) of those in the no-treatment group, for a relative risk of 0.59 (P = .037), and the benefit persisted for the duration of the study. However, the difference between the groups in the rates of nonfatal myocardial infarction did not reach statistical significance. Vitamin E seemed to have no effect.
EPA/DHA is thought to have prevented deaths in this study, not by reversing atherosclerosis, but rather by suppressing arrhythmias and inflammation. In support of this theory, Getz and Reardon19 noted that in GISSI the treatment showed its maximal benefit on the incidence of sudden death by 9 months, whereas statin treatment takes 1 to 2 years to reach its maximal effect. This point suggests that the role of omega-3 fatty acids in secondary prevention will be different from that of statins.
Extensive clinical studies have looked at the possibility of using omega-3 fatty acids as part of the treatment for reducing arrhythmic events. Several animal and human studies have shown that these drugs reduce the incidence of sudden death and ventricular fibrillation.20
Omega-3 fatty acids are thought to prevent arrhythmias by stabilizing the myocardial membrane through interaction with voltage-gated sodium and L-type calcium channels. During an ischemic event, the affected heart cells allow potassium ions to escape. Since potassium ions carry a positive charge, the resting membrane potential (ie, the difference in electrical charge between the inside and outside of the cell) is increased, lowering the threshold for initiating an action potential through sodium channels and increasing the risk of fatal arrhythmias. It is hypothesized that omega-3 fatty acids inhibit sodium channels by being incorporated into the membrane phospholipid bilayer, increasing its fluidity and thereby affecting the sodium channel. This reduces membrane excitability and arrhythmic potential.20
This premise was examined in three large randomized clinical trials specifically looking at ventricular arrhythmias in patients with an implanted cardioverter-defibrillator (ICD).21–23 The results were mixed.

In another study, Calo and colleagues25 randomized 160 patients to receive omega-3 fatty acids 2 g per day or placebo starting at least 5 days before elective coronary artery bypass surgery and continuing until discharge. The primary end point measured was the development of atrial fibrillation after surgery. The incidence of atrial fibrillation in the omega-3 fatty acid group was 15.2%, compared with 33% in the control group (P = .013).
Despite the differences in the results of these studies, experts generally believe that these agents reduce arrhythmic events. Nevertheless, we lack clear evidence of their clinical effectiveness, and their use for such purposes is off-label.
May reduce inflammation and platelet aggregation
Arachidonic acid is an omega-6 fatty acid that is metabolized into prostaglandins, leukotrienes, and thromboxanes, which are important for cell function. Many of these by-products (eg, leukotriene B4) have inflammatory effects, and others (eg, prostaglandin I2 E2) promote arrhythmias. EPA and DHA competitively inhibit the arachidonic acid cascade, leading to different by-products that promote vasodilation and inhibit platelet aggregation, among other effects.26 The impact of this effect in clinical practice is still unclear.
The evidence still conflicts as to whether omega-3 fatty acids reduce markers of inflammation such as C-reactive protein (CRP). Balk et al,12 in their meta-analysis, looked for studies that examined the effect of these agents on CRP and cardiovascular disease (either known risk factors or coronary artery disease). They excluded studies that were less than 4 weeks in duration, did not specify the dose of fish oil, or used doses higher than 6 g/day. Four trials were found that met their criteria, with dosages of omega-3 fatty acids ranging from 1.6 g/day to 5.9 g/day and from 12 to 20 patients in each study. Although baseline CRP levels in these studies varied, the net change in CRP was minimal, ranging from −0.15 to +1.7 mg/L.
May stabilize plaque
Thies et al27 randomized 188 patients to receive fish oil supplements before carotid endarterectomy. They found that the carotid plaque of patients who received the supplements had higher levels of EPA and DHA and had thicker fibrous caps and fewer signs of inflammation (eg, macrophages) compared with a control group and a group that received sunflower oil.
These findings show that omega-3 fatty acids are readily incorporated into atheromatous plaque and can help stabilize it. An inference from this study is that fish oil could also play a role in stabilizing coronary artery plaque.
No effect on restenosis
These agents, however, have no effect on restenosis rates after coronary angioplasty, as restenosis is mediated less by plaque formation than by intimal hyperplasia and negative remodeling within the endothelium. Even at high doses of 5 mg/day before angioplasty, omega-3 fatty acids failed to reduce the incidence of restenosis at 6 months.28
Modest effect on blood pressure
Omega-3 fatty acids are incorporated into the phospholipid bilayer of the endothelial membrane, increasing its fluidity and promoting vasodilation via an increase in nitric oxide production. These effects suggest they could be used to help control blood pressure, but studies have shown this effect to be minimal.
In a meta-analysis of 36 trials, Geleijnse et al29 estimated the reduction in blood pressure to be 2.1 mm Hg systolic and 1.6 mm Hg diastolic. The median intake of fish oil was 3.7 g/day. The largest reductions were in patients with known hypertension and those over age 45.
These findings seem consistent with the hypothesis that omega-3 fatty acids affect the endothelium, given that the arterial wall tends to become stiffer with age. Overall, however, the results of a number of studies show that fish oil supplementation is of limited clinical use in lowering blood pressure.
Confounding factors among studies
The variability in the results of different studies may be due to confounding factors such as the patients’ baseline diet, the doses of EPA and DHA given, the duration of treatment, and patient compliance. These factors must be considered when examining evidence supporting the use of omega-3 fatty acids.
- American Heart Association. Fish and Omega-3 Fatty Acids. www.americanheart.org/presenter.jhtml?identifier=4632. Accessed March 3, 2009.
- United States Department of Agriculture. Nutrient Data Laboratory. www.nal.usda.gov/fnic/foodcomp/search. Accessed March 3, 2009.
- GlaxoSmithKline. Patient Information: Lovaza. www.lovaza.com. Accessed March 3, 2009.
- US Food and Drug Administration. Mercury in fish: cause for concern? www.fda.gov/fdac/reprints/mercury.html. Accessed March 3, 2009.
- Eritsland J, Arnesen H, Seljeflot I, Kierulf P. Long-term effects of n-3 polyunsaturated fatty acids on haemostatic variables and bleeding episodes in patients with coronary artery disease. Blood Coagul Fibrinolysis 1995; 6:17–22.
- Harris WS. Expert opinion: omega-3 fatty acids and bleeding—cause for concern? Am J Cardiol 2007; 99:44C–46C.
- Bjerregaard P, Johansen LG. Mortality pattern in Greenland. Arctic Med Res 1987; 46:71–77.
- Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive Summary of the Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). JAMA 2001; 285:2486–2497.
- Jacobson TA. Secondary prevention of coronary artery disease with omega-3 fatty acids. Am J Cardiol 2006; 98:61i–70i.
- Harris WS, Miller M, Tighe AP, Davidson MH, Schaefer EJ. Omega-3 fatty acids and coronary heart disease risk: clinical and mechanistic perspectives. Atherosclerosis 2008; 197:12–24.
- Khan S, Minihane AM, Talmud PJ, et al. Dietary long-chain n-3 PUFAs increase LPL gene expression in adipose tissue of subjects with an atherogenic lipoprotein phenotype. J Lipid Res 2002; 43:979–985.
- Balk EM, Lichtenstein AH, Chung M, Kupelnick B, Chew P, Lau J. Effects of omega-3 fatty acids on serum markers of cardiovascular disease risk: a systematic review. Atherosclerosis 2006; 189:19–30.
- Pownall HJ, Brauchi D, Kilinc C, et al. Correlation of serum triglyceride and its reduction by omega-3 fatty acids with lipid transfer activity and the neutral lipid compositions of high-density and low-density lipoproteins. Atherosclerosis 1999; 143:285–297.
- Harris WS. N-3 fatty acids and serum lipoproteins: human studies. Am J Clin Nutr 1997; 65 suppl 5:1645S–1654S.
- Robinson JG, Stone NJ. Antiatherosclerotic and antithrombotic effects of omega-3 fatty acids. Am J Cardiol 2006; 98:39i–49i.
- Chan DC, Watts GF, Mori TA, Barrett PH, Beilin LJ, Redgrave TG. Factorial study of the effects of atorvastatin and fish oil on dyslipidaemia in visceral obesity. Eur J Clin Invest 2002; 32:429–436.
- Yokoyama M, Origasa H, Matsuzaki M, et al; Japan EPA lipid intervention study (JELIS) Investigators. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): a randomised open-label, blinded endpoint analysis. Lancet 2007; 369:1090–1098.
- Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: results of the GISSI-Prevenzione trial. Gruppo Italiano per lo Studio della Sopravvivenza Nell’Infarto Miocardico. Lancet 1999; 354:447–455.
- Getz GS, Reardon CA. Nutrition and cardiovascular disease. Arterioscler Thromb Vasc Biol 2007; 27:2499–2506.
- Reiffel JA, McDonald A. Antiarrhythmic effects of omega-3 fatty acids. Am J Cardiol 2006; 98:50i–60i.
- Raitt MH, Connor WE, Morris C, et al. Fish oil supplementation and risk of ventricular tachycardia and ventricular fibrillation in patients with implantable defibrillators: a randomized controlled trial. JAMA 2005; 293:2884–2891.
- Leaf A, Albert CM, Josephson M, et al; Fatty Acid Antiarrhythmia Trial Investigators. Prevention of fatal arrhythmias in high-risk subjects by fish oil n-3 fatty acid intake. Circulation 2005; 112:2762–2768.
- Brouwer IA, Zock PL, Wever EF, et al. Rationale and design of a randomised controlled clinical trial on supplemental intake of n-3 fatty acids and incidence of cardiac arrhythmia: SOFA. Eur J Clin Nutr 2003; 57:1323–1330.
- London B, Albert C, Anderson ME, et al. Omega-3 fatty acids and cardiac arrhythmias: prior studies and recommendations for future research: a report from the National Heart, Lung, and Blood Institute and Office of Dietary Supplements Omega-3 Fatty Acids and their Role in Cardiac Arrhythmogenesis Workshop. Circulation 2007; 116:e320–e335.
- Calo L, Bianconi L, Colivicchi F, et al. N-3 fatty acids for the prevention of atrial fibrillation after coronary artery bypass surgery: a randomized, controlled trial. J Am Coll Cardiol 2005; 45:1723–1728.
- Harris WS, Assaad B, Poston WC. Tissue omega-6/omega-3 fatty acid ratio and risk for coronary artery disease. Am J Cardiol 2006; 98:19i–26i.
- Thies F, Garry JM, Yaqoob P, et al. Association of n-3 polyunsaturated fatty acids with stability of atherosclerotic plaques: a randomised controlled trial. Lancet 2003; 361:477–485.
- Johansen O, Brekke M, Seljeflot I, Abdelnoor M, Arnesen H. N-3 fatty acids do not prevent restenosis after coronary angioplasty: results from the CART study. Coronary Angioplasty Restenosis Trial. J Am Coll Cardiol 1999; 33:1619–1626.
- Geleijnse JM, Giltay EJ, Grobbee DE, Donders AR, Kok FJ. Blood pressure response to fish oil supplementation: metaregression analysis of randomized trials. J Hypertens 2002; 20:1493–1499.
Many patients are taking fish oil supplements, which contain omega-3 fatty acids, either on their own initiative or on their physician’s advice. Driving this trend are accumulating data from observational and epidemiologic studies and clinical trials that these lipids actually reduce cardiovascular risk.
In the following article, we review available studies of omega-3 fatty acids in cardiovascular disease.
WHAT ARE OMEGA-3 FATTY ACIDS?
Omega-3 fatty acids are a class of polyunsaturated fatty acids. Their name means that they all have a double carbon-to-carbon bond in the third position from the omega (or methyl, or n) end of the fatty acid chain.
Most of the cardiovascular research on the omega-3 family has been on eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA), and alpha-linolenic acid (ALA). EPA and DHA are found primarily in fatty fish; ALA is abundant in flaxseed, walnuts, and soybeans.1 The human body can convert small amounts of ALA into EPA and DHA: only about 5% of ALA is converted to EPA and less than 0.5% is converted to DHA. Currently, it is not known whether ALA is active itself or only via these metabolites. In this review, the term omega-3 fatty acid refers to EPA and DHA only.
GETTING ENOUGH FISH OIL
Healthy people should consume fish (preferably oily fish) at least twice a week, according to the American Heart Association.1 However, not all fish contain the same amount of oil. Some, such as cod and catfish, contain only 0.2 g of EPA/DHA per 100-g serving; others, such as Atlantic salmon, contain about 10 times as much (Table 1).2
People with known coronary artery disease should take in 1 g of EPA/DHA per day, according to the American Heart Association.1 This recommendation is based on clinical trials that found omega-3 fatty acids to have beneficial effects.
For most people with coronary artery disease, this means taking supplements. However, buyers need to carefully examine the label of over-the-counter fish oil supplements to see if they contain the recommended amounts of both DHA and EPA. Generic 1-g fish oil supplements may contain variable amounts of DHA and EPA, and some may have less than 300 mg.
People with hypertriglyceridemia. The US Food and Drug Administration (FDA) has approved Lovaza (formerly Omacor), which contains EPA/DHA in higher concentrations than over-the-counter preparations, for the treatment of hypertriglyceridemia in people with triglyceride levels higher than 500 mg/ dL, along with a regimen of diet and regular exercise.3 It is currently the only FDA-approved prescription form of omega-3 fatty acid ethyl esters. Each 1-g capsule contains 375 mg of DHA and 465 mg of EPA; the recommended dose is 2 to 4 g/day. To take in an equivalent amount of these substances with over-the counter-preparations, patients might have to take many capsules a day.
Safety of omega-3 fatty acids
Generally, omega-3 fatty acids are very well tolerated, and their adverse effects are limited to gastrointestinal complaints (discomfort, upset stomach) and a fishy odor. Common ways to prevent these effects are to freeze the capsules or take them at bedtime or with meals.
Mercury advisory on fish. Nursing or pregnant women should limit their consumption of certain fish, as some fish (but not fish oil) contain high levels of mercury. The highest levels of mercury are usually found in the larger, older predatory fish such as swordfish, tilefish, and mackerel, and the FDA advises women who are nursing or pregnant to avoid these fish completely. Tuna, red snapper, and orange roughy are lower in mercury, but nursing or pregnant women should still limit consumption of these fish to 12 oz per week.4
Theoretical risk of bleeding. In theory, high doses of omega-3 fatty acids may increase the bleeding time by inhibiting the arachidonic acid pathway. Clinically, this effect is minimal. In a trial in 511 patients undergoing coronary artery bypass grafting who were receiving aspirin or warfarin (Coumadin), the bleeding time and the number of bleeding episodes were no higher in those who were randomized to receive 4 g/day of omega-3 fatty acids daily than in a control group.5
Harris6 reviewed 19 studies of omega-3 fatty acids in patients undergoing coronary artery bypass grafting, carotid endarterectomy, or femoral artery catheterization, and none of the studies found a significantly increased risk of bleeding.
HOW DO OMEGA-3 FATTY ACIDS REDUCE RISK?
After epidemiologic studies found that Greenland Eskimos (who consume diets rich in omega-3 fatty acids) have low rates of cardiovascular disease,7 omega-3 fatty acids were hypothesized to reduce cardiovascular risk. Over the past 3 decades, their potential benefit in lowering lipid levels, blood pressure, and the risk of death in patients with known heart disease has been widely researched.
Lower triglyceride levels
The growing problem of obesity in the United States has led to more patients presenting with hypertriglyceridemia, a risk factor for coronary heart disease.
In 2001, the National Cholesterol Education Program’s third Adult Treatment Panel (ATP III)8 redefined normal triglyceride levels as less than 150 mg/dL; previously, normal was defined as less than 200 mg/dL. For people with borderline-high triglyceride levels (150–200 mg/dL), the ATP III recommends focusing on lowering the level of low-density lipoprotein cholesterol (LDL-C). For those with high to very high triglyceride levels (> 500 mg/dL), the current treatment options are niacin, fibrates, and omega-3 fatty acids.
Hypertriglyceridemia is thought to increase the risk of coronary heart disease by two mechanisms. First, and more important, triglyceride-rich lipoproteins such as very-low-density lipoprotein (VLDL) and intermediate-density lipoprotein (IDL) are thought to be atherogenic. Secondly, triglyceride-lipoprotein metabolism involves competition with high-density lipoprotein (HDL), leading to a decrease in HDL production and to denser LDL particles.9
How omega-3 fatty acids lower triglyceride levels has been inferred from preclinical studies. One mechanism, seen in animal studies, is by decreasing hepatic synthesis and secretion of VLDL particles by inhibiting various enzyme transcription factors. Another proposed mechanism is that EPA and DHA increase the activity of lipoprotein lipase, leading to an increase in chylomicron clearance.10 This was validated by Khan et al,11 who showed that lipoprotein lipase activity increased in patients who received omega-3 fatty acids 3 g/day for 6 weeks.
How much do they lower triglycerides? Data from the makers of Lovaza3 indicate that in a patient population with a mean baseline triglyceride level of 816 mg/dL, 4 g/day of omega-3 fatty acids lowered triglyceride levels to 488 mg/dL, a 45% reduction (P < .0001). In addition, HDL cholesterol (HDL-C) levels increased by 9%.
The higher the dose and the higher the baseline triglyceride level, the greater the effect. Balk et al12 performed a meta-analysis of 25 randomized trials and calculated that each 1-g increase in fish oil dose per day lowered the triglyceride level by about 8 mg/dL. However, patients with high baseline triglyceride levels had more dramatic reduction of triglycerides with fish oil. The average reduction in triglyceride levels was 27 mg/dL, accompanied by an increase in HDL-C of 1.6 mg/dL, an increase in LDL-C of 6 mg/dL, and no change in total cholesterol levels.
Pownall et al13 report that, in 19 patients with hypertriglyceridemia (median baseline level 801 mg/dL), omega-3 fatty acids 4 g/day reduced triglyceride levels to 512 mg/dL, a 38.9% change (P = .001). In 21 patients receiving placebo, triglyceride levels decreased by 7.8% (P = .001 compared with active therapy). The effect on HDL-C was minimal, but the median LDL-C level increased by 16.7% (from 43 to 53 mg/dL, P = .007) with fish oil therapy.
Fish oil plus a statin may have advantages
Most patients seen in clinical practice present with mixed dyslipidemias. The current ATP III guidelines aim for stricter triglyceride and LDL-C targets than in the past, which monotherapy alone may not be able to achieve.
Statin therapy by itself effectively lowers LDL-C but has modest effects on triglycerides. Omega-3 fatty acids effectively reduce triglycerides but have been known to increase LDL-C levels. This net LDL-C increase averaged around 10 mg/dL as reported in a review by Harris et al,14 and 6 mg/dL as reported by Balk et al.12 However, despite the net effect of an increase in LDL-C, it is hypothesized that the larger LDL particles produced by omega-3 fatty acid treatment may be less atherogenic.15
The effectiveness of combined therapy in reducing triglycerides has been widely studied.
Chan et al,16 in a randomized, placebo-controlled trial, looked at the effectiveness of atorvastatin (Lipitor) and EPA/DHA. Fifty-two obese men were randomized to receive atorvastatin 40 mg/day, EPA/DHA 4 g/day, both in combination, or placebo. After 6 weeks, triglyceride levels had decreased by 26% from baseline in the atorvastatin group, 25% in the EPA/DHA group, and 40% in the combination therapy group (P = .002). LDL-C levels decreased to a similar degree with either atorvastatin monotherapy or combination therapy. Similar studies show similar results.
Combination therapy may also lower the rate of major coronary events (see below).
The Japan EPA Lipid Intervention Study (JELIS)17 randomized more than 18,000 patients to receive either a statin alone or a statin plus EPA 1,800 mg daily, in an open-label fashion. The statins used were pravastatin (Pravachol) 10 mg daily or simvastatin (Zocor) 5 mg daily; if hypercholesterolemia remained uncontrolled, these doses were doubled. The patients were 5,859 men and 12,786 postmenopausal women (mean age 61) with or without coronary artery disease who had total cholesterol levels of 251 mg/dL or greater. The mean baseline LDL-C level was 180 mg/ dL. People who had had an acute myocardial infarction in the past 6 months or unstable angina were excluded. The primary end point examined was any major coronary event, defined as sudden death, fatal or nonfatal myocardial infarction, unstable angina, angioplasty, or coronary artery bypass grafting.
The JELIS trial showed that combination therapy may reduce the risk of coronary events, the aim of treating dyslipidemia. It was the largest randomized trial to date comparing statin use alone and in combination with omega-3 fatty acids. However, it was performed in Japan, where people already have a high intake of fatty fish, and the results may not be applicable to other countries.
May prevent arrhythmias
The Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico-Prevenzione (GISSI-Prevention) trial18 was the largest randomized trial to date of fish oil therapy as secondary prevention. In this trial, 11,323 patients who had had a myocardial infarction less than 3 months before enrollment were randomized to receive either EPA/DHA 850 mg daily, vitamin E, both, or no treatment. The primary end points were death from any cause, nonfatal myocardial infarction, and nonfatal stroke.
At 3 months, 63 (1.1%) of the patients in the EPA/DHA group had died, compared with 88 (1.6%) of those in the no-treatment group, for a relative risk of 0.59 (P = .037), and the benefit persisted for the duration of the study. However, the difference between the groups in the rates of nonfatal myocardial infarction did not reach statistical significance. Vitamin E seemed to have no effect.
EPA/DHA is thought to have prevented deaths in this study, not by reversing atherosclerosis, but rather by suppressing arrhythmias and inflammation. In support of this theory, Getz and Reardon19 noted that in GISSI the treatment showed its maximal benefit on the incidence of sudden death by 9 months, whereas statin treatment takes 1 to 2 years to reach its maximal effect. This point suggests that the role of omega-3 fatty acids in secondary prevention will be different from that of statins.
Extensive clinical studies have looked at the possibility of using omega-3 fatty acids as part of the treatment for reducing arrhythmic events. Several animal and human studies have shown that these drugs reduce the incidence of sudden death and ventricular fibrillation.20
Omega-3 fatty acids are thought to prevent arrhythmias by stabilizing the myocardial membrane through interaction with voltage-gated sodium and L-type calcium channels. During an ischemic event, the affected heart cells allow potassium ions to escape. Since potassium ions carry a positive charge, the resting membrane potential (ie, the difference in electrical charge between the inside and outside of the cell) is increased, lowering the threshold for initiating an action potential through sodium channels and increasing the risk of fatal arrhythmias. It is hypothesized that omega-3 fatty acids inhibit sodium channels by being incorporated into the membrane phospholipid bilayer, increasing its fluidity and thereby affecting the sodium channel. This reduces membrane excitability and arrhythmic potential.20
This premise was examined in three large randomized clinical trials specifically looking at ventricular arrhythmias in patients with an implanted cardioverter-defibrillator (ICD).21–23 The results were mixed.
In another study, Calo and colleagues25 randomized 160 patients to receive omega-3 fatty acids 2 g per day or placebo starting at least 5 days before elective coronary artery bypass surgery and continuing until discharge. The primary end point measured was the development of atrial fibrillation after surgery. The incidence of atrial fibrillation in the omega-3 fatty acid group was 15.2%, compared with 33% in the control group (P = .013).
Despite the differences in the results of these studies, experts generally believe that these agents reduce arrhythmic events. Nevertheless, we lack clear evidence of their clinical effectiveness, and their use for such purposes is off-label.
May reduce inflammation and platelet aggregation
Arachidonic acid is an omega-6 fatty acid that is metabolized into prostaglandins, leukotrienes, and thromboxanes, which are important for cell function. Many of these by-products (eg, leukotriene B4) have inflammatory effects, and others (eg, prostaglandin I2 E2) promote arrhythmias. EPA and DHA competitively inhibit the arachidonic acid cascade, leading to different by-products that promote vasodilation and inhibit platelet aggregation, among other effects.26 The impact of this effect in clinical practice is still unclear.
The evidence still conflicts as to whether omega-3 fatty acids reduce markers of inflammation such as C-reactive protein (CRP). Balk et al,12 in their meta-analysis, looked for studies that examined the effect of these agents on CRP and cardiovascular disease (either known risk factors or coronary artery disease). They excluded studies that were less than 4 weeks in duration, did not specify the dose of fish oil, or used doses higher than 6 g/day. Four trials were found that met their criteria, with dosages of omega-3 fatty acids ranging from 1.6 g/day to 5.9 g/day and from 12 to 20 patients in each study. Although baseline CRP levels in these studies varied, the net change in CRP was minimal, ranging from −0.15 to +1.7 mg/L.
May stabilize plaque
Thies et al27 randomized 188 patients to receive fish oil supplements before carotid endarterectomy. They found that the carotid plaque of patients who received the supplements had higher levels of EPA and DHA and had thicker fibrous caps and fewer signs of inflammation (eg, macrophages) compared with a control group and a group that received sunflower oil.
These findings show that omega-3 fatty acids are readily incorporated into atheromatous plaque and can help stabilize it. An inference from this study is that fish oil could also play a role in stabilizing coronary artery plaque.
No effect on restenosis
These agents, however, have no effect on restenosis rates after coronary angioplasty, as restenosis is mediated less by plaque formation than by intimal hyperplasia and negative remodeling within the endothelium. Even at high doses of 5 mg/day before angioplasty, omega-3 fatty acids failed to reduce the incidence of restenosis at 6 months.28
Modest effect on blood pressure
Omega-3 fatty acids are incorporated into the phospholipid bilayer of the endothelial membrane, increasing its fluidity and promoting vasodilation via an increase in nitric oxide production. These effects suggest they could be used to help control blood pressure, but studies have shown this effect to be minimal.
In a meta-analysis of 36 trials, Geleijnse et al29 estimated the reduction in blood pressure to be 2.1 mm Hg systolic and 1.6 mm Hg diastolic. The median intake of fish oil was 3.7 g/day. The largest reductions were in patients with known hypertension and those over age 45.
These findings seem consistent with the hypothesis that omega-3 fatty acids affect the endothelium, given that the arterial wall tends to become stiffer with age. Overall, however, the results of a number of studies show that fish oil supplementation is of limited clinical use in lowering blood pressure.
Confounding factors among studies
The variability in the results of different studies may be due to confounding factors such as the patients’ baseline diet, the doses of EPA and DHA given, the duration of treatment, and patient compliance. These factors must be considered when examining evidence supporting the use of omega-3 fatty acids.
Many patients are taking fish oil supplements, which contain omega-3 fatty acids, either on their own initiative or on their physician’s advice. Driving this trend are accumulating data from observational and epidemiologic studies and clinical trials that these lipids actually reduce cardiovascular risk.
In the following article, we review available studies of omega-3 fatty acids in cardiovascular disease.
WHAT ARE OMEGA-3 FATTY ACIDS?
Omega-3 fatty acids are a class of polyunsaturated fatty acids. Their name means that they all have a double carbon-to-carbon bond in the third position from the omega (or methyl, or n) end of the fatty acid chain.
Most of the cardiovascular research on the omega-3 family has been on eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA), and alpha-linolenic acid (ALA). EPA and DHA are found primarily in fatty fish; ALA is abundant in flaxseed, walnuts, and soybeans.1 The human body can convert small amounts of ALA into EPA and DHA: only about 5% of ALA is converted to EPA and less than 0.5% is converted to DHA. Currently, it is not known whether ALA is active itself or only via these metabolites. In this review, the term omega-3 fatty acid refers to EPA and DHA only.
GETTING ENOUGH FISH OIL
Healthy people should consume fish (preferably oily fish) at least twice a week, according to the American Heart Association.1 However, not all fish contain the same amount of oil. Some, such as cod and catfish, contain only 0.2 g of EPA/DHA per 100-g serving; others, such as Atlantic salmon, contain about 10 times as much (Table 1).2
People with known coronary artery disease should take in 1 g of EPA/DHA per day, according to the American Heart Association.1 This recommendation is based on clinical trials that found omega-3 fatty acids to have beneficial effects.
For most people with coronary artery disease, this means taking supplements. However, buyers need to carefully examine the label of over-the-counter fish oil supplements to see if they contain the recommended amounts of both DHA and EPA. Generic 1-g fish oil supplements may contain variable amounts of DHA and EPA, and some may have less than 300 mg.
People with hypertriglyceridemia. The US Food and Drug Administration (FDA) has approved Lovaza (formerly Omacor), which contains EPA/DHA in higher concentrations than over-the-counter preparations, for the treatment of hypertriglyceridemia in people with triglyceride levels higher than 500 mg/ dL, along with a regimen of diet and regular exercise.3 It is currently the only FDA-approved prescription form of omega-3 fatty acid ethyl esters. Each 1-g capsule contains 375 mg of DHA and 465 mg of EPA; the recommended dose is 2 to 4 g/day. To take in an equivalent amount of these substances with over-the counter-preparations, patients might have to take many capsules a day.
Safety of omega-3 fatty acids
Generally, omega-3 fatty acids are very well tolerated, and their adverse effects are limited to gastrointestinal complaints (discomfort, upset stomach) and a fishy odor. Common ways to prevent these effects are to freeze the capsules or take them at bedtime or with meals.
Mercury advisory on fish. Nursing or pregnant women should limit their consumption of certain fish, as some fish (but not fish oil) contain high levels of mercury. The highest levels of mercury are usually found in the larger, older predatory fish such as swordfish, tilefish, and mackerel, and the FDA advises women who are nursing or pregnant to avoid these fish completely. Tuna, red snapper, and orange roughy are lower in mercury, but nursing or pregnant women should still limit consumption of these fish to 12 oz per week.4
Theoretical risk of bleeding. In theory, high doses of omega-3 fatty acids may increase the bleeding time by inhibiting the arachidonic acid pathway. Clinically, this effect is minimal. In a trial in 511 patients undergoing coronary artery bypass grafting who were receiving aspirin or warfarin (Coumadin), the bleeding time and the number of bleeding episodes were no higher in those who were randomized to receive 4 g/day of omega-3 fatty acids daily than in a control group.5
Harris6 reviewed 19 studies of omega-3 fatty acids in patients undergoing coronary artery bypass grafting, carotid endarterectomy, or femoral artery catheterization, and none of the studies found a significantly increased risk of bleeding.
HOW DO OMEGA-3 FATTY ACIDS REDUCE RISK?
After epidemiologic studies found that Greenland Eskimos (who consume diets rich in omega-3 fatty acids) have low rates of cardiovascular disease,7 omega-3 fatty acids were hypothesized to reduce cardiovascular risk. Over the past 3 decades, their potential benefit in lowering lipid levels, blood pressure, and the risk of death in patients with known heart disease has been widely researched.
Lower triglyceride levels
The growing problem of obesity in the United States has led to more patients presenting with hypertriglyceridemia, a risk factor for coronary heart disease.
In 2001, the National Cholesterol Education Program’s third Adult Treatment Panel (ATP III)8 redefined normal triglyceride levels as less than 150 mg/dL; previously, normal was defined as less than 200 mg/dL. For people with borderline-high triglyceride levels (150–200 mg/dL), the ATP III recommends focusing on lowering the level of low-density lipoprotein cholesterol (LDL-C). For those with high to very high triglyceride levels (> 500 mg/dL), the current treatment options are niacin, fibrates, and omega-3 fatty acids.
Hypertriglyceridemia is thought to increase the risk of coronary heart disease by two mechanisms. First, and more important, triglyceride-rich lipoproteins such as very-low-density lipoprotein (VLDL) and intermediate-density lipoprotein (IDL) are thought to be atherogenic. Secondly, triglyceride-lipoprotein metabolism involves competition with high-density lipoprotein (HDL), leading to a decrease in HDL production and to denser LDL particles.9
How omega-3 fatty acids lower triglyceride levels has been inferred from preclinical studies. One mechanism, seen in animal studies, is by decreasing hepatic synthesis and secretion of VLDL particles by inhibiting various enzyme transcription factors. Another proposed mechanism is that EPA and DHA increase the activity of lipoprotein lipase, leading to an increase in chylomicron clearance.10 This was validated by Khan et al,11 who showed that lipoprotein lipase activity increased in patients who received omega-3 fatty acids 3 g/day for 6 weeks.
How much do they lower triglycerides? Data from the makers of Lovaza3 indicate that in a patient population with a mean baseline triglyceride level of 816 mg/dL, 4 g/day of omega-3 fatty acids lowered triglyceride levels to 488 mg/dL, a 45% reduction (P < .0001). In addition, HDL cholesterol (HDL-C) levels increased by 9%.
The higher the dose and the higher the baseline triglyceride level, the greater the effect. Balk et al12 performed a meta-analysis of 25 randomized trials and calculated that each 1-g increase in fish oil dose per day lowered the triglyceride level by about 8 mg/dL. However, patients with high baseline triglyceride levels had more dramatic reduction of triglycerides with fish oil. The average reduction in triglyceride levels was 27 mg/dL, accompanied by an increase in HDL-C of 1.6 mg/dL, an increase in LDL-C of 6 mg/dL, and no change in total cholesterol levels.
Pownall et al13 report that, in 19 patients with hypertriglyceridemia (median baseline level 801 mg/dL), omega-3 fatty acids 4 g/day reduced triglyceride levels to 512 mg/dL, a 38.9% change (P = .001). In 21 patients receiving placebo, triglyceride levels decreased by 7.8% (P = .001 compared with active therapy). The effect on HDL-C was minimal, but the median LDL-C level increased by 16.7% (from 43 to 53 mg/dL, P = .007) with fish oil therapy.
Fish oil plus a statin may have advantages
Most patients seen in clinical practice present with mixed dyslipidemias. The current ATP III guidelines aim for stricter triglyceride and LDL-C targets than in the past, which monotherapy alone may not be able to achieve.
Statin therapy by itself effectively lowers LDL-C but has modest effects on triglycerides. Omega-3 fatty acids effectively reduce triglycerides but have been known to increase LDL-C levels. This net LDL-C increase averaged around 10 mg/dL as reported in a review by Harris et al,14 and 6 mg/dL as reported by Balk et al.12 However, despite the net effect of an increase in LDL-C, it is hypothesized that the larger LDL particles produced by omega-3 fatty acid treatment may be less atherogenic.15
The effectiveness of combined therapy in reducing triglycerides has been widely studied.
Chan et al,16 in a randomized, placebo-controlled trial, looked at the effectiveness of atorvastatin (Lipitor) and EPA/DHA. Fifty-two obese men were randomized to receive atorvastatin 40 mg/day, EPA/DHA 4 g/day, both in combination, or placebo. After 6 weeks, triglyceride levels had decreased by 26% from baseline in the atorvastatin group, 25% in the EPA/DHA group, and 40% in the combination therapy group (P = .002). LDL-C levels decreased to a similar degree with either atorvastatin monotherapy or combination therapy. Similar studies show similar results.
Combination therapy may also lower the rate of major coronary events (see below).
The Japan EPA Lipid Intervention Study (JELIS)17 randomized more than 18,000 patients to receive either a statin alone or a statin plus EPA 1,800 mg daily, in an open-label fashion. The statins used were pravastatin (Pravachol) 10 mg daily or simvastatin (Zocor) 5 mg daily; if hypercholesterolemia remained uncontrolled, these doses were doubled. The patients were 5,859 men and 12,786 postmenopausal women (mean age 61) with or without coronary artery disease who had total cholesterol levels of 251 mg/dL or greater. The mean baseline LDL-C level was 180 mg/ dL. People who had had an acute myocardial infarction in the past 6 months or unstable angina were excluded. The primary end point examined was any major coronary event, defined as sudden death, fatal or nonfatal myocardial infarction, unstable angina, angioplasty, or coronary artery bypass grafting.
The JELIS trial showed that combination therapy may reduce the risk of coronary events, the aim of treating dyslipidemia. It was the largest randomized trial to date comparing statin use alone and in combination with omega-3 fatty acids. However, it was performed in Japan, where people already have a high intake of fatty fish, and the results may not be applicable to other countries.
May prevent arrhythmias
The Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico-Prevenzione (GISSI-Prevention) trial18 was the largest randomized trial to date of fish oil therapy as secondary prevention. In this trial, 11,323 patients who had had a myocardial infarction less than 3 months before enrollment were randomized to receive either EPA/DHA 850 mg daily, vitamin E, both, or no treatment. The primary end points were death from any cause, nonfatal myocardial infarction, and nonfatal stroke.
At 3 months, 63 (1.1%) of the patients in the EPA/DHA group had died, compared with 88 (1.6%) of those in the no-treatment group, for a relative risk of 0.59 (P = .037), and the benefit persisted for the duration of the study. However, the difference between the groups in the rates of nonfatal myocardial infarction did not reach statistical significance. Vitamin E seemed to have no effect.
EPA/DHA is thought to have prevented deaths in this study, not by reversing atherosclerosis, but rather by suppressing arrhythmias and inflammation. In support of this theory, Getz and Reardon19 noted that in GISSI the treatment showed its maximal benefit on the incidence of sudden death by 9 months, whereas statin treatment takes 1 to 2 years to reach its maximal effect. This point suggests that the role of omega-3 fatty acids in secondary prevention will be different from that of statins.
Extensive clinical studies have looked at the possibility of using omega-3 fatty acids as part of the treatment for reducing arrhythmic events. Several animal and human studies have shown that these drugs reduce the incidence of sudden death and ventricular fibrillation.20
Omega-3 fatty acids are thought to prevent arrhythmias by stabilizing the myocardial membrane through interaction with voltage-gated sodium and L-type calcium channels. During an ischemic event, the affected heart cells allow potassium ions to escape. Since potassium ions carry a positive charge, the resting membrane potential (ie, the difference in electrical charge between the inside and outside of the cell) is increased, lowering the threshold for initiating an action potential through sodium channels and increasing the risk of fatal arrhythmias. It is hypothesized that omega-3 fatty acids inhibit sodium channels by being incorporated into the membrane phospholipid bilayer, increasing its fluidity and thereby affecting the sodium channel. This reduces membrane excitability and arrhythmic potential.20
This premise was examined in three large randomized clinical trials specifically looking at ventricular arrhythmias in patients with an implanted cardioverter-defibrillator (ICD).21–23 The results were mixed.
In another study, Calo and colleagues25 randomized 160 patients to receive omega-3 fatty acids 2 g per day or placebo starting at least 5 days before elective coronary artery bypass surgery and continuing until discharge. The primary end point measured was the development of atrial fibrillation after surgery. The incidence of atrial fibrillation in the omega-3 fatty acid group was 15.2%, compared with 33% in the control group (P = .013).
Despite the differences in the results of these studies, experts generally believe that these agents reduce arrhythmic events. Nevertheless, we lack clear evidence of their clinical effectiveness, and their use for such purposes is off-label.
May reduce inflammation and platelet aggregation
Arachidonic acid is an omega-6 fatty acid that is metabolized into prostaglandins, leukotrienes, and thromboxanes, which are important for cell function. Many of these by-products (eg, leukotriene B4) have inflammatory effects, and others (eg, prostaglandin I2 E2) promote arrhythmias. EPA and DHA competitively inhibit the arachidonic acid cascade, leading to different by-products that promote vasodilation and inhibit platelet aggregation, among other effects.26 The impact of this effect in clinical practice is still unclear.
The evidence still conflicts as to whether omega-3 fatty acids reduce markers of inflammation such as C-reactive protein (CRP). Balk et al,12 in their meta-analysis, looked for studies that examined the effect of these agents on CRP and cardiovascular disease (either known risk factors or coronary artery disease). They excluded studies that were less than 4 weeks in duration, did not specify the dose of fish oil, or used doses higher than 6 g/day. Four trials were found that met their criteria, with dosages of omega-3 fatty acids ranging from 1.6 g/day to 5.9 g/day and from 12 to 20 patients in each study. Although baseline CRP levels in these studies varied, the net change in CRP was minimal, ranging from −0.15 to +1.7 mg/L.
May stabilize plaque
Thies et al27 randomized 188 patients to receive fish oil supplements before carotid endarterectomy. They found that the carotid plaque of patients who received the supplements had higher levels of EPA and DHA and had thicker fibrous caps and fewer signs of inflammation (eg, macrophages) compared with a control group and a group that received sunflower oil.
These findings show that omega-3 fatty acids are readily incorporated into atheromatous plaque and can help stabilize it. An inference from this study is that fish oil could also play a role in stabilizing coronary artery plaque.
No effect on restenosis
These agents, however, have no effect on restenosis rates after coronary angioplasty, as restenosis is mediated less by plaque formation than by intimal hyperplasia and negative remodeling within the endothelium. Even at high doses of 5 mg/day before angioplasty, omega-3 fatty acids failed to reduce the incidence of restenosis at 6 months.28
Modest effect on blood pressure
Omega-3 fatty acids are incorporated into the phospholipid bilayer of the endothelial membrane, increasing its fluidity and promoting vasodilation via an increase in nitric oxide production. These effects suggest they could be used to help control blood pressure, but studies have shown this effect to be minimal.
In a meta-analysis of 36 trials, Geleijnse et al29 estimated the reduction in blood pressure to be 2.1 mm Hg systolic and 1.6 mm Hg diastolic. The median intake of fish oil was 3.7 g/day. The largest reductions were in patients with known hypertension and those over age 45.
These findings seem consistent with the hypothesis that omega-3 fatty acids affect the endothelium, given that the arterial wall tends to become stiffer with age. Overall, however, the results of a number of studies show that fish oil supplementation is of limited clinical use in lowering blood pressure.
Confounding factors among studies
The variability in the results of different studies may be due to confounding factors such as the patients’ baseline diet, the doses of EPA and DHA given, the duration of treatment, and patient compliance. These factors must be considered when examining evidence supporting the use of omega-3 fatty acids.
- American Heart Association. Fish and Omega-3 Fatty Acids. www.americanheart.org/presenter.jhtml?identifier=4632. Accessed March 3, 2009.
- United States Department of Agriculture. Nutrient Data Laboratory. www.nal.usda.gov/fnic/foodcomp/search. Accessed March 3, 2009.
- GlaxoSmithKline. Patient Information: Lovaza. www.lovaza.com. Accessed March 3, 2009.
- US Food and Drug Administration. Mercury in fish: cause for concern? www.fda.gov/fdac/reprints/mercury.html. Accessed March 3, 2009.
- Eritsland J, Arnesen H, Seljeflot I, Kierulf P. Long-term effects of n-3 polyunsaturated fatty acids on haemostatic variables and bleeding episodes in patients with coronary artery disease. Blood Coagul Fibrinolysis 1995; 6:17–22.
- Harris WS. Expert opinion: omega-3 fatty acids and bleeding—cause for concern? Am J Cardiol 2007; 99:44C–46C.
- Bjerregaard P, Johansen LG. Mortality pattern in Greenland. Arctic Med Res 1987; 46:71–77.
- Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive Summary of the Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). JAMA 2001; 285:2486–2497.
- Jacobson TA. Secondary prevention of coronary artery disease with omega-3 fatty acids. Am J Cardiol 2006; 98:61i–70i.
- Harris WS, Miller M, Tighe AP, Davidson MH, Schaefer EJ. Omega-3 fatty acids and coronary heart disease risk: clinical and mechanistic perspectives. Atherosclerosis 2008; 197:12–24.
- Khan S, Minihane AM, Talmud PJ, et al. Dietary long-chain n-3 PUFAs increase LPL gene expression in adipose tissue of subjects with an atherogenic lipoprotein phenotype. J Lipid Res 2002; 43:979–985.
- Balk EM, Lichtenstein AH, Chung M, Kupelnick B, Chew P, Lau J. Effects of omega-3 fatty acids on serum markers of cardiovascular disease risk: a systematic review. Atherosclerosis 2006; 189:19–30.
- Pownall HJ, Brauchi D, Kilinc C, et al. Correlation of serum triglyceride and its reduction by omega-3 fatty acids with lipid transfer activity and the neutral lipid compositions of high-density and low-density lipoproteins. Atherosclerosis 1999; 143:285–297.
- Harris WS. N-3 fatty acids and serum lipoproteins: human studies. Am J Clin Nutr 1997; 65 suppl 5:1645S–1654S.
- Robinson JG, Stone NJ. Antiatherosclerotic and antithrombotic effects of omega-3 fatty acids. Am J Cardiol 2006; 98:39i–49i.
- Chan DC, Watts GF, Mori TA, Barrett PH, Beilin LJ, Redgrave TG. Factorial study of the effects of atorvastatin and fish oil on dyslipidaemia in visceral obesity. Eur J Clin Invest 2002; 32:429–436.
- Yokoyama M, Origasa H, Matsuzaki M, et al; Japan EPA lipid intervention study (JELIS) Investigators. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): a randomised open-label, blinded endpoint analysis. Lancet 2007; 369:1090–1098.
- Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: results of the GISSI-Prevenzione trial. Gruppo Italiano per lo Studio della Sopravvivenza Nell’Infarto Miocardico. Lancet 1999; 354:447–455.
- Getz GS, Reardon CA. Nutrition and cardiovascular disease. Arterioscler Thromb Vasc Biol 2007; 27:2499–2506.
- Reiffel JA, McDonald A. Antiarrhythmic effects of omega-3 fatty acids. Am J Cardiol 2006; 98:50i–60i.
- Raitt MH, Connor WE, Morris C, et al. Fish oil supplementation and risk of ventricular tachycardia and ventricular fibrillation in patients with implantable defibrillators: a randomized controlled trial. JAMA 2005; 293:2884–2891.
- Leaf A, Albert CM, Josephson M, et al; Fatty Acid Antiarrhythmia Trial Investigators. Prevention of fatal arrhythmias in high-risk subjects by fish oil n-3 fatty acid intake. Circulation 2005; 112:2762–2768.
- Brouwer IA, Zock PL, Wever EF, et al. Rationale and design of a randomised controlled clinical trial on supplemental intake of n-3 fatty acids and incidence of cardiac arrhythmia: SOFA. Eur J Clin Nutr 2003; 57:1323–1330.
- London B, Albert C, Anderson ME, et al. Omega-3 fatty acids and cardiac arrhythmias: prior studies and recommendations for future research: a report from the National Heart, Lung, and Blood Institute and Office of Dietary Supplements Omega-3 Fatty Acids and their Role in Cardiac Arrhythmogenesis Workshop. Circulation 2007; 116:e320–e335.
- Calo L, Bianconi L, Colivicchi F, et al. N-3 fatty acids for the prevention of atrial fibrillation after coronary artery bypass surgery: a randomized, controlled trial. J Am Coll Cardiol 2005; 45:1723–1728.
- Harris WS, Assaad B, Poston WC. Tissue omega-6/omega-3 fatty acid ratio and risk for coronary artery disease. Am J Cardiol 2006; 98:19i–26i.
- Thies F, Garry JM, Yaqoob P, et al. Association of n-3 polyunsaturated fatty acids with stability of atherosclerotic plaques: a randomised controlled trial. Lancet 2003; 361:477–485.
- Johansen O, Brekke M, Seljeflot I, Abdelnoor M, Arnesen H. N-3 fatty acids do not prevent restenosis after coronary angioplasty: results from the CART study. Coronary Angioplasty Restenosis Trial. J Am Coll Cardiol 1999; 33:1619–1626.
- Geleijnse JM, Giltay EJ, Grobbee DE, Donders AR, Kok FJ. Blood pressure response to fish oil supplementation: metaregression analysis of randomized trials. J Hypertens 2002; 20:1493–1499.
- American Heart Association. Fish and Omega-3 Fatty Acids. www.americanheart.org/presenter.jhtml?identifier=4632. Accessed March 3, 2009.
- United States Department of Agriculture. Nutrient Data Laboratory. www.nal.usda.gov/fnic/foodcomp/search. Accessed March 3, 2009.
- GlaxoSmithKline. Patient Information: Lovaza. www.lovaza.com. Accessed March 3, 2009.
- US Food and Drug Administration. Mercury in fish: cause for concern? www.fda.gov/fdac/reprints/mercury.html. Accessed March 3, 2009.
- Eritsland J, Arnesen H, Seljeflot I, Kierulf P. Long-term effects of n-3 polyunsaturated fatty acids on haemostatic variables and bleeding episodes in patients with coronary artery disease. Blood Coagul Fibrinolysis 1995; 6:17–22.
- Harris WS. Expert opinion: omega-3 fatty acids and bleeding—cause for concern? Am J Cardiol 2007; 99:44C–46C.
- Bjerregaard P, Johansen LG. Mortality pattern in Greenland. Arctic Med Res 1987; 46:71–77.
- Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive Summary of the Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). JAMA 2001; 285:2486–2497.
- Jacobson TA. Secondary prevention of coronary artery disease with omega-3 fatty acids. Am J Cardiol 2006; 98:61i–70i.
- Harris WS, Miller M, Tighe AP, Davidson MH, Schaefer EJ. Omega-3 fatty acids and coronary heart disease risk: clinical and mechanistic perspectives. Atherosclerosis 2008; 197:12–24.
- Khan S, Minihane AM, Talmud PJ, et al. Dietary long-chain n-3 PUFAs increase LPL gene expression in adipose tissue of subjects with an atherogenic lipoprotein phenotype. J Lipid Res 2002; 43:979–985.
- Balk EM, Lichtenstein AH, Chung M, Kupelnick B, Chew P, Lau J. Effects of omega-3 fatty acids on serum markers of cardiovascular disease risk: a systematic review. Atherosclerosis 2006; 189:19–30.
- Pownall HJ, Brauchi D, Kilinc C, et al. Correlation of serum triglyceride and its reduction by omega-3 fatty acids with lipid transfer activity and the neutral lipid compositions of high-density and low-density lipoproteins. Atherosclerosis 1999; 143:285–297.
- Harris WS. N-3 fatty acids and serum lipoproteins: human studies. Am J Clin Nutr 1997; 65 suppl 5:1645S–1654S.
- Robinson JG, Stone NJ. Antiatherosclerotic and antithrombotic effects of omega-3 fatty acids. Am J Cardiol 2006; 98:39i–49i.
- Chan DC, Watts GF, Mori TA, Barrett PH, Beilin LJ, Redgrave TG. Factorial study of the effects of atorvastatin and fish oil on dyslipidaemia in visceral obesity. Eur J Clin Invest 2002; 32:429–436.
- Yokoyama M, Origasa H, Matsuzaki M, et al; Japan EPA lipid intervention study (JELIS) Investigators. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): a randomised open-label, blinded endpoint analysis. Lancet 2007; 369:1090–1098.
- Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: results of the GISSI-Prevenzione trial. Gruppo Italiano per lo Studio della Sopravvivenza Nell’Infarto Miocardico. Lancet 1999; 354:447–455.
- Getz GS, Reardon CA. Nutrition and cardiovascular disease. Arterioscler Thromb Vasc Biol 2007; 27:2499–2506.
- Reiffel JA, McDonald A. Antiarrhythmic effects of omega-3 fatty acids. Am J Cardiol 2006; 98:50i–60i.
- Raitt MH, Connor WE, Morris C, et al. Fish oil supplementation and risk of ventricular tachycardia and ventricular fibrillation in patients with implantable defibrillators: a randomized controlled trial. JAMA 2005; 293:2884–2891.
- Leaf A, Albert CM, Josephson M, et al; Fatty Acid Antiarrhythmia Trial Investigators. Prevention of fatal arrhythmias in high-risk subjects by fish oil n-3 fatty acid intake. Circulation 2005; 112:2762–2768.
- Brouwer IA, Zock PL, Wever EF, et al. Rationale and design of a randomised controlled clinical trial on supplemental intake of n-3 fatty acids and incidence of cardiac arrhythmia: SOFA. Eur J Clin Nutr 2003; 57:1323–1330.
- London B, Albert C, Anderson ME, et al. Omega-3 fatty acids and cardiac arrhythmias: prior studies and recommendations for future research: a report from the National Heart, Lung, and Blood Institute and Office of Dietary Supplements Omega-3 Fatty Acids and their Role in Cardiac Arrhythmogenesis Workshop. Circulation 2007; 116:e320–e335.
- Calo L, Bianconi L, Colivicchi F, et al. N-3 fatty acids for the prevention of atrial fibrillation after coronary artery bypass surgery: a randomized, controlled trial. J Am Coll Cardiol 2005; 45:1723–1728.
- Harris WS, Assaad B, Poston WC. Tissue omega-6/omega-3 fatty acid ratio and risk for coronary artery disease. Am J Cardiol 2006; 98:19i–26i.
- Thies F, Garry JM, Yaqoob P, et al. Association of n-3 polyunsaturated fatty acids with stability of atherosclerotic plaques: a randomised controlled trial. Lancet 2003; 361:477–485.
- Johansen O, Brekke M, Seljeflot I, Abdelnoor M, Arnesen H. N-3 fatty acids do not prevent restenosis after coronary angioplasty: results from the CART study. Coronary Angioplasty Restenosis Trial. J Am Coll Cardiol 1999; 33:1619–1626.
- Geleijnse JM, Giltay EJ, Grobbee DE, Donders AR, Kok FJ. Blood pressure response to fish oil supplementation: metaregression analysis of randomized trials. J Hypertens 2002; 20:1493–1499.
KEY POINTS
- The American Heart Association recommends that healthy people consume fatty fish at least twice a week. The recommendation for people with coronary artery disease is 1 g of eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) per day.
- A formulation of EPA 465 mg plus DHA 375 mg is available by prescription and is approved for treating triglyceridemia in excess of 500 mg/dL. The dose is 2 to 4 capsules per day.
- Experts generally believe that omega-3 fatty acids reduce arrhythmic events. Nevertheless, we lack clear evidence of their clinical effectiveness, and their use for such purposes is off-label.
- Overall, omega-3 fatty acids have minimal side effects.