User login
Chronic constipation: Update on management
Chronic constipation has a variety of possible causes and mechanisms. Although traditional conservative treatments are still valid and first-line, if these fail, clinicians can choose from a growing list of new treatments, tailored to the cause in the individual patient.
This article discusses how defecation works (or doesn’t), the types of chronic constipation, the available diagnostic tools, and traditional and newer treatments, including some still in development.
THE EPIDEMIOLOGY OF CONSTIPATION
Chronic constipation is one of the most common gastrointestinal disorders, affecting about 15% of all adults and 30% of those over the age of 60.1 It can be a primary disorder or secondary to other factors.
Constipation is more prevalent in women and in institutionalized elderly people.2 It is associated with lower socioeconomic status, depression, less self-reported physical activity, certain medications, and stressful life events.3 Given its high prevalence and its impact on quality of life, it is also associated with significant utilization of healthcare resources.4
Constipation defined by Rome IV criteria
Physicians and patients may disagree about what constitutes constipation. Physicians primarily regard it as infrequent bowel movements, while patients tend to have a broader definition. According to the Rome IV criteria,5 chronic constipation is defined by the presence of the following for at least 3 months (with symptom onset at least 6 months prior to diagnosis):
(1) Two or more of the following for more than 25% of defecations:
- Straining
- Lumpy or hard stools
- Sensation of incomplete evacuation
- Sensation of anorectal obstruction or blockage
- Manual maneuvers to facilitate evacuation
- Fewer than 3 spontaneous bowel movements per week.
(2) Loose stools are rarely present without the use of laxatives.
(3) The patient does not meet the criteria for diagnosis of irritable bowel syndrome.
DEFECATION IS COMPLEX
Defecation begins when the rectum fills with stool, causing relaxation of the internal anal sphincter and the urge to defecate. The external anal sphincter, which is under voluntary control, can then either contract to delay defecation or relax to allow the stool to be expelled.6
Colonic muscles propel stool toward the rectum in repetitive localized contractions that help mix and promote absorption of the content, and larger coordinated (high-amplitude propagating) contractions that, in healthy individuals, move the stool forward from the proximal to the distal colon multiple times daily. These contractions usually occur in the morning and are accentuated by gastric distention from food and the resulting gastrocolic reflex.
Serotonin (5-HT) is released by enterochromaffin cells in response to distention of the gut wall. It mediates peristaltic movements of the gastrointestinal tract by binding to receptors (especially 5-HT4), stimulating release of neurotransmitters such as acetylcholine, causing smooth-muscle contraction behind the luminal contents and propelling them forward.
PRIMARY CONSTIPATION DISORDERS
The American Gastroenterological Association7 classifies constipation into 3 groups on the basis of colonic transit time and anorectal function:
Normal-transit constipation
Stool normally takes 20 to 72 hours to pass through the colon, with transit time affected by diet, drugs, level of physical activity, and emotional status.8
Normal-transit constipation is the most common type of constipation. The term is sometimes used interchangeably with constipation-predominant irritable bowel syndrome, but the latter is a distinct entity characterized by abdominal pain relieved by defecation as the primary symptom, as well as having occasional loose stools. These 2 conditions can be hard to tell apart, especially if the patient cannot describe the symptoms precisely.
Slow-transit constipation
Slow-transit constipation—also called delayed-transit constipation, colonoparesis, colonic inertia, and pseudo-obstruction—is defined as prolonged stool transit in the colon, ie, for more than 5 days.9 It can be the result of colonic smooth muscle dysfunction, compromised colonic neural pathways, or both, leading to slow colon peristalsis.
Factors that can affect colonic motility such as opioid use and hypothyroidism should be carefully considered in these patients. Opioids are notorious for causing constipation by decreasing bowel tone and contractility and thereby increasing colonic transit time. They also tighten up the anal sphincters, resulting in decreased rectal evacuation.10
Outlet dysfunction
Outlet dysfunction, also called pelvic floor dysfunction or defecatory disorder, is associated with incomplete rectal evacuation. It can be a consequence of weak rectal expulsion forces (slow colonic transit, rectal hyposensitivity), functional resistance to rectal evacuation (high anal resting pressure, anismus, incomplete relaxation of the anal sphincter, dyssynergic defecation), or structural outlet obstruction (excessive perineal descent, rectoceles, rectal intussusception). About 50% of patients with outlet dysfunction have concurrent slow-transit constipation.
Dyssynergic defecation is the most common outlet dysfunction disorder, accounting for about half of the cases referred to tertiary centers. It is defined as a paradoxical elevation in anal sphincter tone or less than 20% relaxation of the resting anal sphincter pressure with weak abdominal and pelvic propulsive forces.11 Anorectal biofeedback is a therapeutic option for dyssynergic defecation, as we discuss later in this article.
SECONDARY CONSTIPATION

Constipation can be secondary to several conditions and factors (Table 1), including:
- Neurologic disorders that affect gastrointestinal motility (eg, Hirschsprung disease, Parkinson disease, multiple sclerosis, spinal cord injury, stroke, spinal or ganglionic tumor, hypothyroidism, amyloidosis, diabetes mellitus, hypercalcemia)
- Drugs used to treat neurologic disorders
- Mechanical obstruction
- Diet (eg, low fiber, decreased fluid intake).
EVALUATION OF CONSTIPATION
It is crucial for physicians to efficiently use the available diagnostic tools for constipation to tailor the treatment to the patient.

Evaluation of chronic constipation begins with a thorough history and physical examination to rule out secondary constipation (Figure 1). Red flags such as unintentional weight loss, blood in the stool, rectal pain, fever, and iron-deficiency anemia should prompt referral for colonoscopy to evaluate for malignancy, colitis, or other potential colonic abnormalities.12
A detailed perineal and rectal examination can help diagnose defecatory disorders and should include evaluation of the resting anal tone and the sphincter during simulated evacuation.
Laboratory tests of thyroid function, electrolytes, and a complete blood cell count should be ordered if clinically indicated.13
Further tests
Further diagnostic tests can be considered if symptoms persist despite conservative treatment or if a defecatory disorder is suspected. These include anorectal manometry, colonic transit studies, defecography, and colonic manometry.
Anorectal manometry and the rectal balloon expulsion test are usually done first because of their high sensitivity (88%) and specificity (89%) for defecatory disorders.14 These tests measure the function of the internal and external anal sphincters at rest and with straining and assess rectal sensitivity and compliance. Anorectal manometry is also used in biofeedback therapy in patients with dyssynergic defecation.15
Colonic transit time can be measured if anorectal manometry and the balloon expulsion test are normal. The study uses radiopaque markers, radioisotopes, or wireless motility capsules to confirm slow-transit constipation and to identify areas of delayed transit in the colon.16
Defecography is usually the next step in diagnosis if anorectal manometry and balloon expulsion tests are inconclusive or if an anatomic abnormality of the pelvic floor is suspected. It can be done with a variety of techniques. Barium defecography can identify anatomic defects, scintigraphy can quantify evacuation of artificial stools, and magnetic resonance defecography visualizes anatomic landmarks to assess pelvic floor motion without exposing the patient to radiation.17,18
Colonic manometry is most useful in patients with refractory slow-transit constipation and can identify patients with isolated colonic motor dysfunction with no pelvic floor dysfunction who may benefit from subtotal colectomy and end-ileostomy.7
TRADITIONAL TREATMENTS STILL THE MAINSTAY

Nonpharmacologic treatments are the first-line options for patients with normal-transit and slow-transit constipation and should precede diagnostic testing. Lifestyle modifications and dietary changes (Table 2) aim to augment the known factors that stimulate the gastrocolic reflex and increase intestinal motility by high-amplitude propagated contractions.
Increasing physical activity increases intestinal gas clearance, decreases bloating, and lessens constipation.19,20
Toilet training is an integral part of lifestyle modifications.21
Diet. Drinking hot caffeinated beverages, eating breakfast within an hour of waking up, and consuming fiber in the morning (25–30 g of fiber daily) have traditionally been recommended as the first-line measures for chronic constipation. Dehydrated patients with constipation also benefit from increasing their fluid intake.22
LAXATIVES
Fiber (bulk-forming laxatives) for normal-transit constipation

Fiber remains a key part of the initial management of chronic constipation, as it is cheap, available, and safe. Increasing fiber intake is effective for normal-transit constipation, but patients with slow-transit constipation or refractory outlet dysfunction are less likely to benefit.23 Other laxatives are incorporated into the regimen if first-line nonpharmacologic interventions fail (Table 3).
Bulk-forming laxatives include insoluble fiber (wheat bran) and soluble fiber (psyllium, methylcellulose, inulin, calcium polycarbophil). Insoluble fiber, though often used, has little impact on symptoms of chronic constipation after 1 month of use, and up to 60% of patients report adverse effects from it.24 On the other hand, clinical trials have shown that soluble fiber such as psyllium facilitates defecation and improves functional bowel symptoms in patients with normal-transit constipation.25
Patients should be instructed to increase their dietary fiber intake gradually to avoid adverse effects and should be told to expect significant symptomatic improvement only after a few weeks. They should also be informed that increasing dietary fiber intake can cause bloating but that the bloating is temporary. If it continues, a different fiber can be tried.
Osmotic laxatives
Osmotic laxatives are often employed as a first- line laxative treatment option for patients with constipation. They draw water into the lumen by osmosis, helping to soften stool and speed intestinal transit. They include macrogols (inert polymers of ethylene glycol), nonabsorbable carbohydrates (lactulose, sorbitol), magnesium products, and sodium phosphate products.
Polyethylene glycol, the most studied osmotic laxative, has been shown to maintain therapeutic efficacy for up to 2 years, though it is not generally used this long.26 A meta-analysis of 10 randomized clinical trials found it to be superior to lactulose in improving stool consistency and frequency, and rates of adverse effects were similar to those with placebo.27
Lactulose and sorbitol are semisynthetic disaccharides that are not absorbed from the gastrointestinal tract. Apart from the osmotic effect of the disaccharide, these sugars are metabolized by colonic bacteria to acetic acid and other short-chain fatty acids, resulting in acidification of the stool, which exerts an osmotic effect in the colonic lumen.
Lactulose and sorbitol were shown to have similar efficacy in increasing the frequency of bowel movements in a small study, though patients taking lactulose had a higher rate of nausea.28
The usual recommended dose is 15 to 30 mL once or twice daily.
Adverse effects include gas, bloating, and abdominal distention (due to fermentation by colonic bacteria) and can limit long-term use.
Magnesium citrate and magnesium hydroxide are strong osmotic laxatives, but so far no clinical trial has been done to assess their efficacy in constipation. Although the risk of hypermagnesemia is low with magnesium-based products, this group of laxatives is generally avoided in patients with renal or cardiac disease.29
Sodium phosphate enemas (Fleet enemas) are used for bowel cleansing before certain procedures but have only limited use in constipation because of potential adverse effects such as hyperphosphatemia, hypocalcemia, and the rarer but more serious complication of acute phosphate nephropathy.30
Stimulant laxatives for short-term use only
Stimulant laxatives include glycerin, bisacodyl, senna, and sodium picosulfate. Sodium piosulfate and bisacodyl have been validated for treatment of chronic constipation for up to 4 weeks.31–33
Stimulant laxative suppositories should be used 30 minutes after meals to augment the physiologic gastrocolic reflex.
As more evidence is available for osmotic laxatives such as polyethylene glycol, they tend to be preferred over stimulant agents, especially for long-term use. Clinicians have traditionally hesitated to prescribe stimulant laxatives for long-term use, as they were thought to damage the enteric nervous system.34 Although more recent studies have not shown this potential effect,35 more research is warranted on the use of stimulant laxatives for longer than 4 weeks.
STOOL SOFTENERS: LITTLE EVIDENCE
Stool softeners enhance the interaction of stool and water, leading to softer stool and easier evacuation. Docusate sodium and docusate calcium are thought to facilitate the mixing of aqueous and fatty substances, thereby softening the stool.
However, there is little evidence to support the use of docusate for constipation in hospitalized adults or in ambulatory care. A recent review reported that docusate was no better than placebo in diminishing symptoms of constipation.36
INTESTINAL SECRETAGOGUES
The secretagogues include lubiprostone, linaclotide, and plecanatide. These medications are preferred therapy for patients with normal- or slow-transit constipation once conservative therapies have failed. Even though there is no current consensus, lifestyle measures and conservative treatment options should be tried for about 8 weeks.
Lubiprostone and linaclotide are approved by the US Food and Drug Administration (FDA) for both constipation and constipation-predominant irritable bowel syndrome. They activate chloride channels on the apical surface of enterocytes, increasing intestinal secretion of chloride, which in turn increases luminal sodium efflux to maintain electroneutrality, leading to secretion of water into the intestinal lumen. This eventually facilitates intestinal transit and increases the passage of stool.
Lubiprostone
Lubiprostone, a prostaglandin E1 derivative, is approved for treating chronic constipation, constipation-predominant irritable bowel syndrome in women, and opioid-induced constipation in patients with chronic noncancer pain.
Adverse effects in clinical trials were nausea (up to 30%) and headache.37,38
Linaclotide
Linaclotide, a minimally absorbed 14-amino acid peptide, increases intestinal secretion of chloride and bicarbonate, increasing intestinal fluid and promoting intestinal transit.39 It also decreases the firing rate of the visceral afferent pain fibers and helps reduce visceral pain, especially in patients with constipation-predominant irritable bowel syndrome.40 It is approved for chronic constipation and constipation-predominant irritable bowel syndrome.41–43
Dosage starts at 145 μg/day for chronic constipation, and can be titrated up to 290 μg if there is no response or if a diagnosis of constipation-predominant irritable bowel syndrome is under consideration. Linaclotide should be taken 30 to 60 minutes before breakfast to reduce the likelihood of diarrhea.44
Adverse effects. Diarrhea led to treatment discontinuation in 4.5% of patients in one study.42
Plecanatide
Plecanatide is a guanylate cyclase-c agonist with a mode of action similar to that of linaclotide. It was recently approved by the FDA for chronic idiopathic constipation in adults. The recommended dose is 3 mg once daily.
Data from phase 2 trials in chronic constipation showed improvement in straining, abdominal discomfort, and stool frequency after 14 days of treatment.45
A phase 3 trial showed that plecanatide was more effective than placebo when used for 12 weeks in 951 patients with chronic constipation (P = .009).46 The most common adverse effect reported was diarrhea.
SEROTONIN RECEPTOR AGONISTS
Activation of serotonin 5-HT4 receptors in the gut leads to release of acetylcholine, which in turn induces mucosal secretion by activating submucosal neurons and increasing gut motility.47
Two 5-HT4 receptor agonists were withdrawn from the market (cisapride in 2000 and tegaserod in 2007) due to serious cardiovascular adverse events (fatal arrhythmias, heart attacks, and strokes) resulting from their affinity for hERG-K+ cardiac channels.
The newer agents prucalopride,48 velusetrag, and naronapride are highly selective 5-HT4 agonists with low affinity for hERG-K+ receptors and do not have proarrhythmic properties, based on extensive assessment in clinical trials.
Prucalopride
Prucalopride has been shown to accelerate gastrointestinal and colonic transit in patients with chronic constipation, with improvement in bowel movements, symptoms of chronic constipation, and quality of life.49–52
Adverse effects reported with its use have been headache, nausea, abdominal pain, and cramps.
Prucalopride is approved in Europe and Canada for chronic constipation in women but is not yet approved in the United States.
Dosage is 2 mg orally once daily. Caution is advised in elderly patients, in whom the preferred maximum dose is 1 mg daily, as there are only limited data available on the safety of this medication in the elderly.
Velusetrag
Velusetrag has been shown to increase colonic motility and improve symptoms of chronic constipation. In a phase 2 trial,53 the most effective dose was 15 mg once daily. Higher doses were associated with a higher incidence of adverse effects such as diarrhea, headache, nausea, and vomiting.
Naronapride
Naronapride (ATI-7505) is in phase 2 trials for chronic constipation. Reported adverse effects were headache, diarrhea, nausea, and vomiting.54
BILE SALT ABSORPTION INHIBITORS
Bile acids exert prosecretory and prokinetic effects by increasing colonic secretion of water and electrolytes through the activation of adenylate cyclase. This happens as a result of their deconjugation after passage into the colon.
Elobixibat is an ileal bile acid transporter inhibitor that prevents absorption of nonconjugated bile salts in the distal ileum. It has few side effects because its systemic absorption is minimal. Phase 3 trials are under way. Dosage is 5 to 20 mg daily. Adverse effects are few because systemic absorption is minimal, but include abdominal pain and diarrhea.55,56
MANAGING OPIOID-INDUCED CONSTIPATION
Opioids cause constipation by binding to mu receptors in the enteric nervous system. Activation of these receptors decreases bowel tone and contractility, which increases transit time. Stimulation of these receptors also increases anal sphincter tone, resulting in decreased rectal evacuation.57
Though underrecognized, opioid-induced constipation affects 40% of patients who take these drugs for nonmalignant pain and 90% of those taking them for cancer pain. Patients with this condition were found to take more time off work and feel more impaired in their domestic and work-related obligations than patients who did not develop constipation with use of opioids.58
Initial management of opioid-induced constipation includes increasing intake of fluids and dietary fiber (fiber alone can worsen abdominal pain in this condition by increasing stool bulk without a concomitant improvement in peristalsis) and increasing physical activity. It is common clinical practice to use a stool softener along with a stimulant laxative if lifestyle modifications are inadequate.59 If these measures are ineffective, osmotic agents can be added.
If these conventional measures fail, a peripherally acting mu-opioid receptor antagonist such as methylnaltrexone or naloxegol should be considered.
Methylnaltrexone
Methylnaltrexone60,61 is a peripherally acting mu receptor antagonist with a rapid onset of action. It does not cross the blood-brain barrier, as it contains a methyl group. It was approved by the FDA in 2008 to treat opioid-induced constipation in adults with advanced illnesses when other approaches are ineffective.
Adverse effects. Although the mu receptor antagonist alvimopan had been shown to be associated with cardiovascular events hypothesized to be a consequence of opioid withdrawal, methylnaltrexone has been deemed to have a safe cardiovascular profile without any potential effects on platelets, corrected QT interval, metabolism, heart rate, or blood pressure.61 Side effects include abdominal pain, nausea, diarrhea, hot flashes, tremor, and chills.
Contraindications. Methylnaltrexone is contraindicated in patients with structural diseases of the gastrointestinal tract, ie, peptic ulcer disease, inflammatory bowel disease, diverticulitis, stomach or intestinal cancer) since it can increase the risk of perforation.
Dosing is 1 dose subcutaneously every other day, as needed, and no more than 1 dose in a 24-hour period. Dosage is based on weight: 0.15 mg/kg/dose for patients weighing less than 38 kg or more than 114 kg; 8 mg for those weighing 38 to 62 kg; and 12 mg for those weighing 62 to 114 kg.62
Naloxegol
Naloxegol, FDA-approved for treating opioid-induced constipation in 2014, consists of naloxone conjugated with polyethylene glycol, which prevents it from crossing the blood-brain barrier and diminishing the central effects of opioid-induced analgesia. Unlike methylnaltrexone, which is given by subcutaneous injection, naloxegol is taken orally.
Adverse effects reported in clinical trials63,64 were abdominal pain, diarrhea, nausea, headache, and flatulence. No clinically relevant association with QT and corrected QT interval prolongation or cardiac repolarization was noted.64
Dosing is 25 mg by mouth once daily, which can be decreased to 12.5 mg if the initial dose is difficult to tolerate. It should be taken on an empty stomach at least 1 hour before the first meal of the day or 2 hours after the meal. In patients with renal impairment (creatinine clearance < 60 mL/min), the dose is 12.5 mg once daily.65
CONSTIPATION-PREDOMINANT IRRITABLE BOWEL SYNDROME
Irritable bowel syndrome is the reason for 3.1 million office visits and 59 million prescriptions in the United States every year, with patients equally distributed between diarrhea-predominant, constipation-predominant, and mixed subtypes.66
To be diagnosed with constipation-predominant irritable bowel syndrome, patients must meet the Rome IV criteria, more than 25% of bowel movements should have Bristol stool form types 1 or 2, and less than 25% of bowel movements should have Bristol stool form types 6 or 7. In practice, patients reporting that their bowel movements are usually constipated often suffices to make the diagnosis.5
Osmotic laxatives are often tried first, but despite improving stool frequency and consistency, they have little efficacy in satisfying complaints of bloating or abdominal pain in patients with constipation-predominant irritable syndrome.67 Stimulant laxatives have not yet been tested in clinical trials. Lubiprostone and linaclotide are FDA-approved for this condition; in women, lubiprostone is approved only for those over age 18.
Antidepressant therapy
Patients often derive additional benefit from treatment with antidepressants. A meta-analysis demonstrated a number needed to treat of 4 for selective serotonin reuptake inhibitors and tricyclic antidepressants in managing abdominal pain associated with irritable bowel syndrome.68 The major limiting factor is usually adverse effects of these drugs.
For constipation-predominant irritable bowel syndrome, selective serotonin reuptake inhibitors are preferred over tricyclics because of their additional prokinetic properties. Starting at a low dose and titrating upward slowly avoids potential adverse effects.
Cognitive behavioral therapy has also been beneficial in treating irritable bowel syndrome.69
Adjunctive therapies
Adjunctive therapies including peppermint oil, probiotics (eg, Lactobacillus, Bifidobacterium), and acupuncture have also shown promise in managing irritable bowel syndrome, but more data are needed on the use of these therapies for constipation-predominant irritable bowel syndrome before any definite conclusions can be drawn.70 Other emerging pharmacologic therapies are plecanatide (discussed earlier) and tenapanor.
Peppermint oil is an antispasmodic that inhibits calcium channels, leading to relaxation of smooth muscles in the gastrointestinal tract. Different dosages and treatment durations have been studied—450 to 900 mg daily in 2 to 3 divided doses over 1 to 3 months.71,72 The most common adverse effect reported was gastroesophageal reflux, related in part to the oil’s relaxing effect on the lower esophageal sphincter. Observation of this led to the development of enteric-coated preparations that have the potential to bypass the upper gastrointestinal tract.73
Tenapanor inhibits the sodium-hydrogen exchanger 3 channel (a regulator of sodium and water uptake in intestinal lumen), which in turn leads to a higher sodium level in the entire gastrointestinal tract (whereas linaclotide’s action is limited to the duodenum and jejunem), resulting in more fluid volume and increased luminal transit.74 It was found effective in a phase 2 clinical trial,75 and the most effective dose was 50 mg twice daily.
Since tenapanor is minimally absorbed, it has few side effects, the major ones being diarrhea (11.2% vs 0% with placebo) and urinary tract infection (5.6% vs 4.4% with placebo).75 Further study is needed to confirm these findings.
Tenapanor also has the advantage of inhibiting luminal phosphorus absorption. This has led to exploration of its use as a phosphate binder in patients with end-stage renal disease.
DYSSYNERGIC DEFECATION AND ANORECTAL BIOFEEDBACK
According to the Rome IV criteria,5 dyssynergic defecation is present if the criteria for chronic constipation are met, if a dyssynergic pattern of defecation is confirmed by manometry, imaging, or electromyography, and if 1 or more of the following are present: inability to expel an artificial stool (a 50-mL water-filled balloon) within 1 minute, prolonged colonic transit time, inability to evacuate, or 50% or more retention of barium during defecography.5
Even though biofeedback has been controversial as a treatment for dyssynergic defecation because of conflicting results in older studies,76 3 trials have shown it to be better than placebo, laxatives, and muscle relaxants, with symptomatic improvement in 70% of patients.77–79
Biofeedback therapy involves an instrument-based auditory or visual tool (using electromyographic sensors or anorectal manometry) to help patients coordinate abdominal, rectal, puborectalis, and anal sphincter muscles and produce a propulsive force using their abdominal muscles to achieve complete evacuation. Important components of this therapy include:
Proper evacuation positioning (brace-pump technique, which involves sitting on the toilet leaning forward with forearms resting on thighs, shoulders relaxed, and feet placed on a small footstool
Breathing relaxation and training exercises during defecation (no straining, keeping a normal pattern of breathing, and avoiding holding the breath while defecating)
Use of the abdominal muscles by pushing the abdomen forward, along with relaxation of the anal sphincter.80
The anorectal feedback program usually consists of 6 weekly sessions of 45 to 60 minutes each. Limitations of this therapy include unavailability, lack of trained therapists, lack of insurance coverage, and inapplicability to certain patient groups, such as those with dementia or learning disabilities.
SURGERY FOR CHRONIC CONSTIPATION
Surgery for constipation is reserved for patients who continue to have symptoms despite optimal medical therapy.
Total abdominal colectomy and ileorectal anastomosis
Total abdominal colectomy with ileorectal anastomosis is a surgical option for medically intractable slow-transit constipation. Before considering surgery, complete diagnostic testing should be done, including colonic manometry and documentation of whether the patient also has outlet dysfunction.
Even though it has shown excellent outcomes and satisfaction rates as high as 100% in patients with pure slow-transit constipation,81–83 results in older studies in patients with mixed disorders (eg, slow-transit constipation with features of outlet dysfunction) were less predictable.84 More recent studies have reported comparable long-term morbidity and postoperative satisfaction rates in those with pure slow-transit constipation and those with a mixed disorder, indicating that careful patient selection is likely the key to a favorable outcome.85
Partial colectomies based on segmental colon transit time measurements can also be considered in some patients.86
Stapled transanal resection
Stapled transanal resection involves circumferential transanal stapling of the redundant rectal mucosa. It is an option for patients with defecatory disorders, specifically large rectoceles and rectal intussusception not amenable to therapy with pelvic floor retraining exercises.87
The efficacy of this procedure in controlling symptoms and improving quality of life is around 77% to 81% at 12 months, though complication rates as high as 46% and disappointing long-term outcomes have been a deterrent to its widespread acceptance in the United States.88–91
- Mugie SM, Benninga MA, Di Lorenzo C. Epidemiology of constipation in children and adults: a systematic review. Best Pract Res Clin Gastroenterol 2011; 25:3–18.
- Kinnunen O. Study of constipation in a geriatric hospital, day hospital, old people's home and at home. Aging (Milano) 1991; 3:161–170.
- Everhart JE, Go VL, Johannes RS, Fitzsimmons SC, Roth HP, White LR. A longitudinal survey of self-reported bowel habits in the United States. Dig Dis Sci 1989; 34:1153–1162.
- Shah ND, Chitkara DK, Locke GR, Meek PD, Talley NJ. Ambulatory care for constipation in the United States, 1993-2004. Am J Gastroenterol 2008; 103:1746–1753.
- Mearin F, Lacy BE, Chang L, et al. Bowel disorders. Gastroenterology 2016; 150:1393–1407.
- Bharucha AE. Pelvic floor: anatomy and function. Neurogastroenterol Motil 2006; 18:507–519.
- Bharucha AE, Pemberton JH, Locke GR 3rd. American Gastroenterological Association technical review on constipation. Gastroenterology 2013; 144:218–238.
- Grundy D, Al-Chaer ED, Aziz Q, et al. Fundamentals of neurogastroenterology: basic science. Gastroenterology 2006; 130:1391–1411.
- Gallegos-Orozco JF, Foxx-Orenstein AE, Sterler SM, Stoa JM. Chronic constipation in the elderly. Am J Gastroenterol 2012; 107:18–26.
- Mancini I, Bruera E. Constipation in advanced cancer patients. Support Care Cancer 1998; 6:356–364.
- Bassotti G, Chistolini F, Sietchiping-Nzepa F, de Roberto G, Morelli A, Chiarioni G. Biofeedback for pelvic floor dysfunction in constipation. BMJ 2004; 328:393–396.
- American Gastroenterological Association, Bharucha AE, Dorn SD, Lembo A, Pressman A. American Gastroenterological Association medical position statement on constipation. Gastroenterology 2013; 144:211–217.
- Costilla VC, Foxx-Orenstein AE. Constipation in adults: diagnosis and management. Curr Treat Options Gastroenterol 2014; 12:310–321.
- Rao SS, Singh S. Clinical utility of colonic and anorectal manometry in chronic constipation. J Clin Gastroenterol 2010; 44:597–609.
- Minguez M, Herreros B, Sanchiz V, et al. Predictive value of the balloon expulsion test for excluding the diagnosis of pelvic floor dyssynergia in constipation. Gastroenterology 2004; 126:57–62.
- Diamant NE, Kamm MA, Wald A, Whitehead WE. AGA technical review on anorectal testing techniques. Gastroenterology 1999; 116:735–760.
- Pezim ME, Pemberton JH, Levin KE, Litchy WJ, Phillips SF. Parameters of anorectal and colonic motility in health and in severe constipation. Dis Colon Rectum 1993; 36:484–491.
- Bharucha AE, Fletcher JG, Seide B, Riederer SJ, Zinsmeister AR. Phenotypic variation in functional disorders of defecation. Gastroenterology 2005; 128:1199–1210.
- De Schryver AM, Samsom M, Smout AI. Effects of a meal and bisacodyl on colonic motility in healthy volunteers and patients with slow-transit constipation. Dig Dis Sci 2003; 48:1206–1212.
- Villoria A, Serra J, Azpiroz F, Malagelada JR. Physical activity and intestinal gas clearance in patients with bloating. Am J Gastroenterol 2006; 101:2552–2557.
- Sikirov D. Comparison of straining during defecation in three positions: results and implications for human health. Dig Dis Sci 2003; 48:1201–1205.
- Muller-Lissner SA, Kamm MA, Scarpignato C, Wald A. Myths and misconceptions about chronic constipation. Am J Gastroenterol 2005; 100:232–242.
- Voderholzer WA, Schatke W, Muhldorfer BE, Klauser AG, Birkner B, Muller-Lissner SA. Clinical response to dietary fiber treatment of chronic constipation. Am J Gastroenterol 1997; 92:95–98.
- Bijkerk CJ, de Wit NJ, Muris JW, Whorwell PJ, Knottnerus JA, Hoes AW. Soluble or insoluble fibre in irritable bowel syndrome in primary care? Randomised placebo controlled trial. BMJ 2009; 339:b3154.
- Suares NC, Ford AC. Systematic review: the effects of fibre in the management of chronic idiopathic constipation. Aliment Pharmacol Ther 2011; 33:895–901.
- Dipalma JA, Cleveland MV, McGowan J, Herrera JL. A randomized, multicenter, placebo-controlled trial of polyethylene glycol laxative for chronic treatment of chronic constipation. Am J Gastroenterol 2007; 102:1436–1441.
- Lee-Robichaud H, Thomas K, Morgan J, Nelson RL. Lactulose versus polyethylene glycol for chronic constipation. Cochrane Database Syst Rev 2010; 7:CD007570.
- Lederle FA, Busch DL, Mattox KM, West MJ, Aske DM. Cost-effective treatment of constipation in the elderly: a randomized double-blind comparison of sorbitol and lactulose. Am J Med 1990; 89:597–601.
- Nyberg C, Hendel J, Nielsen OH. The safety of osmotically acting cathartics in colonic cleansing. Nat Rev Gastroenterol Hepatol 2010; 7:557–564.
- Ainley EJ, Winwood PJ, Begley JP. Measurement of serum electrolytes and phosphate after sodium phosphate colonoscopy bowel preparation: an evaluation. Dig Dis Sci 2005; 50:1319–1323.
- Kienzle-Horn S, Vix JM, Schuijt C, Peil H, Jordan CC, Kamm MA. Efficacy and safety of bisacodyl in the acute treatment of constipation: a double-blind, randomized, placebo-controlled study. Aliment Pharmacol Ther 2006; 23:1479–1488.
- Kienzle-Horn S, Vix JM, Schuijt C, Peil H, Jordan CC, Kamm MA. Comparison of bisacodyl and sodium picosulphate in the treatment of chronic constipation. Curr Med Res Opin 2007; 23:691–699.
- Mueller-Lissner S, Kamm MA, Wald A, et al. Multicenter, 4-week, double-blind, randomized, placebo-controlled trial of sodium picosulfate in patients with chronic constipation. Am J Gastroenterol 2010; 105:897–903.
- Smith B. Pathologic changes in the colon produced by anthraquinone purgatives. Dis Colon Rectum 1973; 16:455–458.
- Kiernan JA, Heinicke EA. Sennosides do not kill myenteric neurons in the colon of the rat or mouse. Neuroscience 1989; 30:837–842.
- Ottawa (ON): Canadian Agency for Drugs and Technologies in Health. Dioctyl sulfosuccinate or docusate (calcium or sodium) for the prevention or management of constipation: a review of the clinical effectiveness. www.ncbi.nlm.nih.gov/pubmedhealth/PMH0071207/. Accessed April 6, 2017.
- Saad R, Chey WD. Lubiprostone for chronic idiopathic constipation and irritable bowel syndrome with constipation. Expert Rev Gastroenterol Hepatol 2008; 2:497–508.
- Johanson JF, Morton D, Geenen J, Ueno R. Multicenter, 4-week, double-blind, randomized, placebo-controlled trial of lubiprostone, a locally-acting type-2 chloride channel activator, in patients with chronic constipation. Am J Gastroenterol 2008; 103:170–177.
- Harris LA, Crowell MD. Linaclotide, a new direction in the treatment of irritable bowel syndrome and chronic constipation. Curr Opin Mol Ther 2007; 9:403–410.
- Johnston JM, Kurtz CB, Macdougall JE, et al. Linaclotide improves abdominal pain and bowel habits in a phase IIb study of patients with irritable bowel syndrome with constipation. Gastroenterology 2010; 139:1877–1886.e2.
- Lembo AJ, Schneier HA, Shiff SJ, et al. Two randomized trials of linaclotide for chronic constipation. N Engl J Med 2011; 365:527–536.
- Chey WD, Lembo AJ, Lavins BJ, et al. Linaclotide for irritable bowel syndrome with constipation: a 26-week, randomized, double-blind, placebo-controlled trial to evaluate efficacy and safety. Am J Gastroenterol 2012; 107:1702–1712.
- Rao S, Lembo AJ, Shiff SJ, et al. A 12-week, randomized, controlled trial with a 4-week randomized withdrawal period to evaluate the efficacy and safety of linaclotide in irritable bowel syndrome with constipation. Am J Gastroenterol 2012; 107:1714–1725.
- Chey WD, Kurlander J, Eswaran S. Irritable bowel syndrome: a clinical review. JAMA 2015; 313:949–958.
- Shailubhai K, Talluto C, Comiskey S, Foss JA, Joslyn A, Jacob G. Phase II clinical evaluation of SP-304, a guanylate cyclase-C agonist, for treatment of chronic constipation. Am J Gastroenterol 2010; 105:S487–S488.
- Miner P, Surowitz R, Fogel R, et al. Plecanatide, a novel guanylate cyclase-C (GC-C) receptor agonist, is efficacious and safe in patients with chronic idiopathic constipation (CIC): results from a 951 patient, 12-week, multi-center trial (abstract). Gastroenterology 2013; 144:S163.
- Coss-Adame E, Rao SS. Brain and gut interactions in irritable bowel syndrome: new paradigms and new understandings. Curr Gastroenterol Rep 2014; 16:379.
- Mendzelevski B, Ausma J, Chanter DO, et al. Assessment of the cardiac safety of prucalopride in healthy volunteers: a randomized, double-blind, placebo- and positive-controlled thorough QT study. Br J Clin Pharmacol 2012; 73:203–209.
- Camilleri M, Kerstens R, Rykx A, Vandeplassche L. A placebo-controlled trial of prucalopride for severe chronic constipation. N Engl J Med 2008; 358:2344–2354.
- Tack J, van Outryve M, Beyens G, Kerstens R, Vandeplassche L. Prucalopride (Resolor) in the treatment of severe chronic constipation in patients dissatisfied with laxatives. Gut 2009; 58:357–365.
- Quigley EM, Vandeplassche L, Kerstens R, Ausma J. Clinical trial: the efficacy, impact on quality of life, and safety and tolerability of prucalopride in severe chronic constipation—a 12-week, randomized, double-blind, placebo-controlled study. Aliment Pharmacol Ther 2009; 29:315–328.
- Ford AC, Suares NC. Effect of laxatives and pharmacological therapies in chronic idiopathic constipation: systematic review and meta-analysis. Gut 2011; 60:209–218.
- Goldberg M, Li YP, Johanson JF, et al. Clinical trial: the efficacy and tolerability of velusetrag, a selective 5-HT4 agonist with high intrinsic activity, in chronic idiopathic constipation—a 4-week, randomized, double-blind, placebo-controlled, dose-response study. Aliment Pharmacol Ther 2010; 32:1102–1112.
- Palme M, Milner PG, Ellis DJ, Marmon T, Canafax DM. A novel gastrointestinal prokinetic, ATI-7505, increased spontaneous bowel movements (sbms) in a phase II, randomized, placebo-controlled study of patients with chronic idiopathic constipation (CIC). Gastroenterology 2010; 138:S-128–S-129.
- Chey WD, Camilleri M, Chang L, Rikner L, Graffner H. A randomized placebo-controlled phase IIb trial of a3309, a bile acid transporter inhibitor, for chronic idiopathic constipation. Am J Gastroenterol 2011; 106:1803–1812.
- Wong BS, Camilleri M, McKinzie S, Burton D, Graffner H, Zinsmeister AR. Effects of A3309, an ileal bile acid transporter inhibitor, on colonic transit and symptoms in females with functional constipation. Am J Gastroenterol 2011; 106:2154–2164.
- Pappagallo M. Incidence, prevalence, and management of opioid bowel dysfunction. Am J Surg 2001; 182(suppl):11S–18S.
- Bell T, Annunziata K, Leslie JB. Opioid-induced constipation negatively impacts pain management, productivity, and health-related quality of life: findings from the National Health and Wellness Survey. J Opioid Manag 2009; 5:137–144.
- Sykes NP. A volunteer model for the comparison of laxatives in opioid-related constipation. J Pain Symptom Manage 1996; 11:363–369.
- ClinicalTrials.gov. A multicenter, randomized, double-blind, placebo-controlled, parallel-group study of oral MOA-728 for the treatment of opioid- induced bowel dysfunction in subjects with chronic nonmalignant pain. ClinicalTrials.gov Identifier: NCT00547586. https://clinicaltrials.gov/ct2/show/NCT00547586. Accessed March 22, 2017.
- ClinicalTrials.gov. An open-label study to evaluate the long-term safety of subcutaneous MOA-728 for treatment of opioid-induced constipation in subjects with nonmalignant pain. ClinicalTrials.gov Identifier: NCT00804141. https://clinicaltrials.gov/ct2/show/NCT00804141. Accessed April 6, 2017.
- Wyeth Pharmaceuticals. Relistor package insert. http://labeling.pfizer.com/showlabeling.aspx?id=499. Accessed March 22, 2017.
- Webster L, Dhar S, Eldon M, Masuoka L, Lappalainen J, Sostek M. A phase 2, double-blind, randomized, placebo-controlled, dose-escalation study to evaluate the efficacy, safety, and tolerability of naloxegol in patients with opioid-induced constipation. Pain 2013; 154:1542–1550.
- Chey WD, Webster L, Sostek M, Lappalainen J, Barker PN, Tack J. Naloxegol for opioid-induced constipation in patients with noncancer pain. N Engl J Med 2014; 370:2387–2396.
- Jones R, Prommer E, Backstedt D. Naloxegol: a novel therapy in the management of opioid-induced constipation. Am J Hosp Palliat Care 2016; 33:875–880.
- Guilera M, Balboa A, Mearin F. Bowel habit subtypes and temporal patterns in irritable bowel syndrome: systematic review. Am J Gastroenterol 2005; 100:1174–1184.
- Chapman RW, Stanghellini V, Geraint M, Halphen M. Randomized clinical trial: macrogol/PEG 3350 plus electrolytes for treatment of patients with constipation associated with irritable bowel syndrome. Am J Gastroenterol 2013; 108:1508–1515.
- Ford AC, Quigley EM, Lacy BE, et al. Effect of antidepressants and psychological therapies, including hypnotherapy, in irritable bowel syndrome: systematic review and meta-analysis. Am J Gastroenterol 2014; 109:1350–1366.
- Ballou S, Keefer L. Psychological interventions for irritable bowel syndrome and inflammatory bowel diseases. Clin Transl Gastroenterol 2017; 8:e214.
- Ford AC, Moayyedi P, Lacy BE, et al; Task Force on the Management of Functional Bowel Disorders. American College of Gastroenterology monograph on the management of irritable bowel syndrome and chronic idiopathic constipation. Am J Gastroenterol 2014; 109(suppl 1):S2–S27.
- Ford AC, Talley NJ, Spiegel BM, et al. Effect of fibre, antispasmodics, and peppermint oil in the treatment of irritable bowel syndrome: systematic review and meta-analysis. BMJ 2008; 337:a2313.
- Wall GC, Bryant GA, Bottenberg MM, Maki ED, Miesner AR. Irritable bowel syndrome: a concise review of current treatment concepts. World J Gastroenterol 2014; 20:8796–8806.
- Kligler B, Chaudhary S. Peppermint oil. Am Fam Physician 2007; 75:1027–1030.
- Spencer AG, Labonte ED, Rosenbaum DP, et al. Intestinal inhibition of the Na+/H+ exchanger 3 prevents cardiorenal damage in rats and inhibits Na+ uptake in humans. Sci Transl Med 2014; 6:227ra36.
- Rosenbaum DP. A randomized, double-blind, placebo-controlled study to assess the safety and efficacy of AZD1722 for the treatment of constipation-predominant irritable bowel syndrome (IBS-C). 2014. https://clinicaltrials.gov/ct2/show/NCT01923428. Accessed April 6, 2017.
- Rao SS. Biofeedback therapy for dyssynergic (obstructive) defecation. J Clin Gastroenterol 2000; 30:115–116.
- Cadeddu F, Salis F, De Luca E, Ciangola I, Milito G. Efficacy of biofeedback plus transanal stimulation in the management of pelvic floor dyssynergia: a randomized trial. Tech Coloproctol 2015; 19:333–338.
- Chiarioni G, Whitehead WE, Pezza V, Morelli A, Bassotti G. Biofeedback is superior to laxatives for normal transit constipation due to pelvic floor dyssynergia. Gastroenterology 2006; 130:657–664.
- Chiarioni G, Heymen S, Whitehead WE. Biofeedback therapy for dyssynergic defecation. World J Gastroenterol 2006; 12:7069–7074.
- Rao SS. Biofeedback therapy for constipation in adults. Best Pract Res Clin Gastroenterol 2011; 25:159–166.
- Hassan I, Pemberton JH, Young-Fadok TM, et al. Ileorectal anastomosis for slow transit constipation: long-term functional and quality of life results. J Gastrointest Surg 2006; 10:1330–1337.
- You YT, Wang JY, Changchien CR, et al. Segmental colectomy in the management of colonic inertia. Am Surg 1998; 64:775–777.
- Nyam DC, Pemberton JH, Ilstrup DM, Rath DM. Long-term results of surgery for chronic constipation. Dis Colon Rectum 1997; 40:273–279.
- Pemberton JH, Rath DM, Ilstrup DM. Evaluation and surgical treatment of severe chronic constipation. Ann Surg 1991; 214:403–413.
- Reshef A, Alves-Ferreira P, Zutshi M, Hull T, Gurland B. Colectomy for slow transit constipation: effective for patients with coexistent obstructed defecation. Int J Colorectal Dis 2013; 28:841–847.
- Lundin E, Karlbom U, Pahlman L, Graf W. Outcome of segmental colonic resection for slow-transit constipation. Br J Surg 2002; 89:1270–1274.
- Schwandner O, Stuto A, Jayne D, et al. Decision-making algorithm for the STARR procedure in obstructed defecation syndrome: position statement of the group of STARR pioneers. Surg Innov 2008; 15:105–109.
- Titu LV, Riyad K, Carter H, Dixon AR. Stapled transanal rectal resection for obstructed defecation: a cautionary tale. Dis Colon Rectum 2009; 52:1716–1722.
- Goede AC, Glancy D, Carter H, Mills A, Mabey K, Dixon AR. Medium-term results of stapled transanal rectal resection (STARR) for obstructed defecation and symptomatic rectal-anal intussusception. Colorectal Dis 2011; 13:1052–1057.
- Jayne DG, Schwandner O, Stuto A. Stapled transanal rectal resection for obstructed defecation syndrome: one-year results of the european STARR registry. Dis Colon Rectum 2009; 52:1205–1214.
- Madbouly KM, Abbas KS, Hussein AM. Disappointing long-term outcomes after stapled transanal rectal resection for obstructed defecation. World J Surg 2010; 34:2191–2196.
Chronic constipation has a variety of possible causes and mechanisms. Although traditional conservative treatments are still valid and first-line, if these fail, clinicians can choose from a growing list of new treatments, tailored to the cause in the individual patient.
This article discusses how defecation works (or doesn’t), the types of chronic constipation, the available diagnostic tools, and traditional and newer treatments, including some still in development.
THE EPIDEMIOLOGY OF CONSTIPATION
Chronic constipation is one of the most common gastrointestinal disorders, affecting about 15% of all adults and 30% of those over the age of 60.1 It can be a primary disorder or secondary to other factors.
Constipation is more prevalent in women and in institutionalized elderly people.2 It is associated with lower socioeconomic status, depression, less self-reported physical activity, certain medications, and stressful life events.3 Given its high prevalence and its impact on quality of life, it is also associated with significant utilization of healthcare resources.4
Constipation defined by Rome IV criteria
Physicians and patients may disagree about what constitutes constipation. Physicians primarily regard it as infrequent bowel movements, while patients tend to have a broader definition. According to the Rome IV criteria,5 chronic constipation is defined by the presence of the following for at least 3 months (with symptom onset at least 6 months prior to diagnosis):
(1) Two or more of the following for more than 25% of defecations:
- Straining
- Lumpy or hard stools
- Sensation of incomplete evacuation
- Sensation of anorectal obstruction or blockage
- Manual maneuvers to facilitate evacuation
- Fewer than 3 spontaneous bowel movements per week.
(2) Loose stools are rarely present without the use of laxatives.
(3) The patient does not meet the criteria for diagnosis of irritable bowel syndrome.
DEFECATION IS COMPLEX
Defecation begins when the rectum fills with stool, causing relaxation of the internal anal sphincter and the urge to defecate. The external anal sphincter, which is under voluntary control, can then either contract to delay defecation or relax to allow the stool to be expelled.6
Colonic muscles propel stool toward the rectum in repetitive localized contractions that help mix and promote absorption of the content, and larger coordinated (high-amplitude propagating) contractions that, in healthy individuals, move the stool forward from the proximal to the distal colon multiple times daily. These contractions usually occur in the morning and are accentuated by gastric distention from food and the resulting gastrocolic reflex.
Serotonin (5-HT) is released by enterochromaffin cells in response to distention of the gut wall. It mediates peristaltic movements of the gastrointestinal tract by binding to receptors (especially 5-HT4), stimulating release of neurotransmitters such as acetylcholine, causing smooth-muscle contraction behind the luminal contents and propelling them forward.
PRIMARY CONSTIPATION DISORDERS
The American Gastroenterological Association7 classifies constipation into 3 groups on the basis of colonic transit time and anorectal function:
Normal-transit constipation
Stool normally takes 20 to 72 hours to pass through the colon, with transit time affected by diet, drugs, level of physical activity, and emotional status.8
Normal-transit constipation is the most common type of constipation. The term is sometimes used interchangeably with constipation-predominant irritable bowel syndrome, but the latter is a distinct entity characterized by abdominal pain relieved by defecation as the primary symptom, as well as having occasional loose stools. These 2 conditions can be hard to tell apart, especially if the patient cannot describe the symptoms precisely.
Slow-transit constipation
Slow-transit constipation—also called delayed-transit constipation, colonoparesis, colonic inertia, and pseudo-obstruction—is defined as prolonged stool transit in the colon, ie, for more than 5 days.9 It can be the result of colonic smooth muscle dysfunction, compromised colonic neural pathways, or both, leading to slow colon peristalsis.
Factors that can affect colonic motility such as opioid use and hypothyroidism should be carefully considered in these patients. Opioids are notorious for causing constipation by decreasing bowel tone and contractility and thereby increasing colonic transit time. They also tighten up the anal sphincters, resulting in decreased rectal evacuation.10
Outlet dysfunction
Outlet dysfunction, also called pelvic floor dysfunction or defecatory disorder, is associated with incomplete rectal evacuation. It can be a consequence of weak rectal expulsion forces (slow colonic transit, rectal hyposensitivity), functional resistance to rectal evacuation (high anal resting pressure, anismus, incomplete relaxation of the anal sphincter, dyssynergic defecation), or structural outlet obstruction (excessive perineal descent, rectoceles, rectal intussusception). About 50% of patients with outlet dysfunction have concurrent slow-transit constipation.
Dyssynergic defecation is the most common outlet dysfunction disorder, accounting for about half of the cases referred to tertiary centers. It is defined as a paradoxical elevation in anal sphincter tone or less than 20% relaxation of the resting anal sphincter pressure with weak abdominal and pelvic propulsive forces.11 Anorectal biofeedback is a therapeutic option for dyssynergic defecation, as we discuss later in this article.
SECONDARY CONSTIPATION
Constipation can be secondary to several conditions and factors (Table 1), including:
- Neurologic disorders that affect gastrointestinal motility (eg, Hirschsprung disease, Parkinson disease, multiple sclerosis, spinal cord injury, stroke, spinal or ganglionic tumor, hypothyroidism, amyloidosis, diabetes mellitus, hypercalcemia)
- Drugs used to treat neurologic disorders
- Mechanical obstruction
- Diet (eg, low fiber, decreased fluid intake).
EVALUATION OF CONSTIPATION
It is crucial for physicians to efficiently use the available diagnostic tools for constipation to tailor the treatment to the patient.
Evaluation of chronic constipation begins with a thorough history and physical examination to rule out secondary constipation (Figure 1). Red flags such as unintentional weight loss, blood in the stool, rectal pain, fever, and iron-deficiency anemia should prompt referral for colonoscopy to evaluate for malignancy, colitis, or other potential colonic abnormalities.12
A detailed perineal and rectal examination can help diagnose defecatory disorders and should include evaluation of the resting anal tone and the sphincter during simulated evacuation.
Laboratory tests of thyroid function, electrolytes, and a complete blood cell count should be ordered if clinically indicated.13
Further tests
Further diagnostic tests can be considered if symptoms persist despite conservative treatment or if a defecatory disorder is suspected. These include anorectal manometry, colonic transit studies, defecography, and colonic manometry.
Anorectal manometry and the rectal balloon expulsion test are usually done first because of their high sensitivity (88%) and specificity (89%) for defecatory disorders.14 These tests measure the function of the internal and external anal sphincters at rest and with straining and assess rectal sensitivity and compliance. Anorectal manometry is also used in biofeedback therapy in patients with dyssynergic defecation.15
Colonic transit time can be measured if anorectal manometry and the balloon expulsion test are normal. The study uses radiopaque markers, radioisotopes, or wireless motility capsules to confirm slow-transit constipation and to identify areas of delayed transit in the colon.16
Defecography is usually the next step in diagnosis if anorectal manometry and balloon expulsion tests are inconclusive or if an anatomic abnormality of the pelvic floor is suspected. It can be done with a variety of techniques. Barium defecography can identify anatomic defects, scintigraphy can quantify evacuation of artificial stools, and magnetic resonance defecography visualizes anatomic landmarks to assess pelvic floor motion without exposing the patient to radiation.17,18
Colonic manometry is most useful in patients with refractory slow-transit constipation and can identify patients with isolated colonic motor dysfunction with no pelvic floor dysfunction who may benefit from subtotal colectomy and end-ileostomy.7
TRADITIONAL TREATMENTS STILL THE MAINSTAY
Nonpharmacologic treatments are the first-line options for patients with normal-transit and slow-transit constipation and should precede diagnostic testing. Lifestyle modifications and dietary changes (Table 2) aim to augment the known factors that stimulate the gastrocolic reflex and increase intestinal motility by high-amplitude propagated contractions.
Increasing physical activity increases intestinal gas clearance, decreases bloating, and lessens constipation.19,20
Toilet training is an integral part of lifestyle modifications.21
Diet. Drinking hot caffeinated beverages, eating breakfast within an hour of waking up, and consuming fiber in the morning (25–30 g of fiber daily) have traditionally been recommended as the first-line measures for chronic constipation. Dehydrated patients with constipation also benefit from increasing their fluid intake.22
LAXATIVES
Fiber (bulk-forming laxatives) for normal-transit constipation
Fiber remains a key part of the initial management of chronic constipation, as it is cheap, available, and safe. Increasing fiber intake is effective for normal-transit constipation, but patients with slow-transit constipation or refractory outlet dysfunction are less likely to benefit.23 Other laxatives are incorporated into the regimen if first-line nonpharmacologic interventions fail (Table 3).
Bulk-forming laxatives include insoluble fiber (wheat bran) and soluble fiber (psyllium, methylcellulose, inulin, calcium polycarbophil). Insoluble fiber, though often used, has little impact on symptoms of chronic constipation after 1 month of use, and up to 60% of patients report adverse effects from it.24 On the other hand, clinical trials have shown that soluble fiber such as psyllium facilitates defecation and improves functional bowel symptoms in patients with normal-transit constipation.25
Patients should be instructed to increase their dietary fiber intake gradually to avoid adverse effects and should be told to expect significant symptomatic improvement only after a few weeks. They should also be informed that increasing dietary fiber intake can cause bloating but that the bloating is temporary. If it continues, a different fiber can be tried.
Osmotic laxatives
Osmotic laxatives are often employed as a first- line laxative treatment option for patients with constipation. They draw water into the lumen by osmosis, helping to soften stool and speed intestinal transit. They include macrogols (inert polymers of ethylene glycol), nonabsorbable carbohydrates (lactulose, sorbitol), magnesium products, and sodium phosphate products.
Polyethylene glycol, the most studied osmotic laxative, has been shown to maintain therapeutic efficacy for up to 2 years, though it is not generally used this long.26 A meta-analysis of 10 randomized clinical trials found it to be superior to lactulose in improving stool consistency and frequency, and rates of adverse effects were similar to those with placebo.27
Lactulose and sorbitol are semisynthetic disaccharides that are not absorbed from the gastrointestinal tract. Apart from the osmotic effect of the disaccharide, these sugars are metabolized by colonic bacteria to acetic acid and other short-chain fatty acids, resulting in acidification of the stool, which exerts an osmotic effect in the colonic lumen.
Lactulose and sorbitol were shown to have similar efficacy in increasing the frequency of bowel movements in a small study, though patients taking lactulose had a higher rate of nausea.28
The usual recommended dose is 15 to 30 mL once or twice daily.
Adverse effects include gas, bloating, and abdominal distention (due to fermentation by colonic bacteria) and can limit long-term use.
Magnesium citrate and magnesium hydroxide are strong osmotic laxatives, but so far no clinical trial has been done to assess their efficacy in constipation. Although the risk of hypermagnesemia is low with magnesium-based products, this group of laxatives is generally avoided in patients with renal or cardiac disease.29
Sodium phosphate enemas (Fleet enemas) are used for bowel cleansing before certain procedures but have only limited use in constipation because of potential adverse effects such as hyperphosphatemia, hypocalcemia, and the rarer but more serious complication of acute phosphate nephropathy.30
Stimulant laxatives for short-term use only
Stimulant laxatives include glycerin, bisacodyl, senna, and sodium picosulfate. Sodium piosulfate and bisacodyl have been validated for treatment of chronic constipation for up to 4 weeks.31–33
Stimulant laxative suppositories should be used 30 minutes after meals to augment the physiologic gastrocolic reflex.
As more evidence is available for osmotic laxatives such as polyethylene glycol, they tend to be preferred over stimulant agents, especially for long-term use. Clinicians have traditionally hesitated to prescribe stimulant laxatives for long-term use, as they were thought to damage the enteric nervous system.34 Although more recent studies have not shown this potential effect,35 more research is warranted on the use of stimulant laxatives for longer than 4 weeks.
STOOL SOFTENERS: LITTLE EVIDENCE
Stool softeners enhance the interaction of stool and water, leading to softer stool and easier evacuation. Docusate sodium and docusate calcium are thought to facilitate the mixing of aqueous and fatty substances, thereby softening the stool.
However, there is little evidence to support the use of docusate for constipation in hospitalized adults or in ambulatory care. A recent review reported that docusate was no better than placebo in diminishing symptoms of constipation.36
INTESTINAL SECRETAGOGUES
The secretagogues include lubiprostone, linaclotide, and plecanatide. These medications are preferred therapy for patients with normal- or slow-transit constipation once conservative therapies have failed. Even though there is no current consensus, lifestyle measures and conservative treatment options should be tried for about 8 weeks.
Lubiprostone and linaclotide are approved by the US Food and Drug Administration (FDA) for both constipation and constipation-predominant irritable bowel syndrome. They activate chloride channels on the apical surface of enterocytes, increasing intestinal secretion of chloride, which in turn increases luminal sodium efflux to maintain electroneutrality, leading to secretion of water into the intestinal lumen. This eventually facilitates intestinal transit and increases the passage of stool.
Lubiprostone
Lubiprostone, a prostaglandin E1 derivative, is approved for treating chronic constipation, constipation-predominant irritable bowel syndrome in women, and opioid-induced constipation in patients with chronic noncancer pain.
Adverse effects in clinical trials were nausea (up to 30%) and headache.37,38
Linaclotide
Linaclotide, a minimally absorbed 14-amino acid peptide, increases intestinal secretion of chloride and bicarbonate, increasing intestinal fluid and promoting intestinal transit.39 It also decreases the firing rate of the visceral afferent pain fibers and helps reduce visceral pain, especially in patients with constipation-predominant irritable bowel syndrome.40 It is approved for chronic constipation and constipation-predominant irritable bowel syndrome.41–43
Dosage starts at 145 μg/day for chronic constipation, and can be titrated up to 290 μg if there is no response or if a diagnosis of constipation-predominant irritable bowel syndrome is under consideration. Linaclotide should be taken 30 to 60 minutes before breakfast to reduce the likelihood of diarrhea.44
Adverse effects. Diarrhea led to treatment discontinuation in 4.5% of patients in one study.42
Plecanatide
Plecanatide is a guanylate cyclase-c agonist with a mode of action similar to that of linaclotide. It was recently approved by the FDA for chronic idiopathic constipation in adults. The recommended dose is 3 mg once daily.
Data from phase 2 trials in chronic constipation showed improvement in straining, abdominal discomfort, and stool frequency after 14 days of treatment.45
A phase 3 trial showed that plecanatide was more effective than placebo when used for 12 weeks in 951 patients with chronic constipation (P = .009).46 The most common adverse effect reported was diarrhea.
SEROTONIN RECEPTOR AGONISTS
Activation of serotonin 5-HT4 receptors in the gut leads to release of acetylcholine, which in turn induces mucosal secretion by activating submucosal neurons and increasing gut motility.47
Two 5-HT4 receptor agonists were withdrawn from the market (cisapride in 2000 and tegaserod in 2007) due to serious cardiovascular adverse events (fatal arrhythmias, heart attacks, and strokes) resulting from their affinity for hERG-K+ cardiac channels.
The newer agents prucalopride,48 velusetrag, and naronapride are highly selective 5-HT4 agonists with low affinity for hERG-K+ receptors and do not have proarrhythmic properties, based on extensive assessment in clinical trials.
Prucalopride
Prucalopride has been shown to accelerate gastrointestinal and colonic transit in patients with chronic constipation, with improvement in bowel movements, symptoms of chronic constipation, and quality of life.49–52
Adverse effects reported with its use have been headache, nausea, abdominal pain, and cramps.
Prucalopride is approved in Europe and Canada for chronic constipation in women but is not yet approved in the United States.
Dosage is 2 mg orally once daily. Caution is advised in elderly patients, in whom the preferred maximum dose is 1 mg daily, as there are only limited data available on the safety of this medication in the elderly.
Velusetrag
Velusetrag has been shown to increase colonic motility and improve symptoms of chronic constipation. In a phase 2 trial,53 the most effective dose was 15 mg once daily. Higher doses were associated with a higher incidence of adverse effects such as diarrhea, headache, nausea, and vomiting.
Naronapride
Naronapride (ATI-7505) is in phase 2 trials for chronic constipation. Reported adverse effects were headache, diarrhea, nausea, and vomiting.54
BILE SALT ABSORPTION INHIBITORS
Bile acids exert prosecretory and prokinetic effects by increasing colonic secretion of water and electrolytes through the activation of adenylate cyclase. This happens as a result of their deconjugation after passage into the colon.
Elobixibat is an ileal bile acid transporter inhibitor that prevents absorption of nonconjugated bile salts in the distal ileum. It has few side effects because its systemic absorption is minimal. Phase 3 trials are under way. Dosage is 5 to 20 mg daily. Adverse effects are few because systemic absorption is minimal, but include abdominal pain and diarrhea.55,56
MANAGING OPIOID-INDUCED CONSTIPATION
Opioids cause constipation by binding to mu receptors in the enteric nervous system. Activation of these receptors decreases bowel tone and contractility, which increases transit time. Stimulation of these receptors also increases anal sphincter tone, resulting in decreased rectal evacuation.57
Though underrecognized, opioid-induced constipation affects 40% of patients who take these drugs for nonmalignant pain and 90% of those taking them for cancer pain. Patients with this condition were found to take more time off work and feel more impaired in their domestic and work-related obligations than patients who did not develop constipation with use of opioids.58
Initial management of opioid-induced constipation includes increasing intake of fluids and dietary fiber (fiber alone can worsen abdominal pain in this condition by increasing stool bulk without a concomitant improvement in peristalsis) and increasing physical activity. It is common clinical practice to use a stool softener along with a stimulant laxative if lifestyle modifications are inadequate.59 If these measures are ineffective, osmotic agents can be added.
If these conventional measures fail, a peripherally acting mu-opioid receptor antagonist such as methylnaltrexone or naloxegol should be considered.
Methylnaltrexone
Methylnaltrexone60,61 is a peripherally acting mu receptor antagonist with a rapid onset of action. It does not cross the blood-brain barrier, as it contains a methyl group. It was approved by the FDA in 2008 to treat opioid-induced constipation in adults with advanced illnesses when other approaches are ineffective.
Adverse effects. Although the mu receptor antagonist alvimopan had been shown to be associated with cardiovascular events hypothesized to be a consequence of opioid withdrawal, methylnaltrexone has been deemed to have a safe cardiovascular profile without any potential effects on platelets, corrected QT interval, metabolism, heart rate, or blood pressure.61 Side effects include abdominal pain, nausea, diarrhea, hot flashes, tremor, and chills.
Contraindications. Methylnaltrexone is contraindicated in patients with structural diseases of the gastrointestinal tract, ie, peptic ulcer disease, inflammatory bowel disease, diverticulitis, stomach or intestinal cancer) since it can increase the risk of perforation.
Dosing is 1 dose subcutaneously every other day, as needed, and no more than 1 dose in a 24-hour period. Dosage is based on weight: 0.15 mg/kg/dose for patients weighing less than 38 kg or more than 114 kg; 8 mg for those weighing 38 to 62 kg; and 12 mg for those weighing 62 to 114 kg.62
Naloxegol
Naloxegol, FDA-approved for treating opioid-induced constipation in 2014, consists of naloxone conjugated with polyethylene glycol, which prevents it from crossing the blood-brain barrier and diminishing the central effects of opioid-induced analgesia. Unlike methylnaltrexone, which is given by subcutaneous injection, naloxegol is taken orally.
Adverse effects reported in clinical trials63,64 were abdominal pain, diarrhea, nausea, headache, and flatulence. No clinically relevant association with QT and corrected QT interval prolongation or cardiac repolarization was noted.64
Dosing is 25 mg by mouth once daily, which can be decreased to 12.5 mg if the initial dose is difficult to tolerate. It should be taken on an empty stomach at least 1 hour before the first meal of the day or 2 hours after the meal. In patients with renal impairment (creatinine clearance < 60 mL/min), the dose is 12.5 mg once daily.65
CONSTIPATION-PREDOMINANT IRRITABLE BOWEL SYNDROME
Irritable bowel syndrome is the reason for 3.1 million office visits and 59 million prescriptions in the United States every year, with patients equally distributed between diarrhea-predominant, constipation-predominant, and mixed subtypes.66
To be diagnosed with constipation-predominant irritable bowel syndrome, patients must meet the Rome IV criteria, more than 25% of bowel movements should have Bristol stool form types 1 or 2, and less than 25% of bowel movements should have Bristol stool form types 6 or 7. In practice, patients reporting that their bowel movements are usually constipated often suffices to make the diagnosis.5
Osmotic laxatives are often tried first, but despite improving stool frequency and consistency, they have little efficacy in satisfying complaints of bloating or abdominal pain in patients with constipation-predominant irritable syndrome.67 Stimulant laxatives have not yet been tested in clinical trials. Lubiprostone and linaclotide are FDA-approved for this condition; in women, lubiprostone is approved only for those over age 18.
Antidepressant therapy
Patients often derive additional benefit from treatment with antidepressants. A meta-analysis demonstrated a number needed to treat of 4 for selective serotonin reuptake inhibitors and tricyclic antidepressants in managing abdominal pain associated with irritable bowel syndrome.68 The major limiting factor is usually adverse effects of these drugs.
For constipation-predominant irritable bowel syndrome, selective serotonin reuptake inhibitors are preferred over tricyclics because of their additional prokinetic properties. Starting at a low dose and titrating upward slowly avoids potential adverse effects.
Cognitive behavioral therapy has also been beneficial in treating irritable bowel syndrome.69
Adjunctive therapies
Adjunctive therapies including peppermint oil, probiotics (eg, Lactobacillus, Bifidobacterium), and acupuncture have also shown promise in managing irritable bowel syndrome, but more data are needed on the use of these therapies for constipation-predominant irritable bowel syndrome before any definite conclusions can be drawn.70 Other emerging pharmacologic therapies are plecanatide (discussed earlier) and tenapanor.
Peppermint oil is an antispasmodic that inhibits calcium channels, leading to relaxation of smooth muscles in the gastrointestinal tract. Different dosages and treatment durations have been studied—450 to 900 mg daily in 2 to 3 divided doses over 1 to 3 months.71,72 The most common adverse effect reported was gastroesophageal reflux, related in part to the oil’s relaxing effect on the lower esophageal sphincter. Observation of this led to the development of enteric-coated preparations that have the potential to bypass the upper gastrointestinal tract.73
Tenapanor inhibits the sodium-hydrogen exchanger 3 channel (a regulator of sodium and water uptake in intestinal lumen), which in turn leads to a higher sodium level in the entire gastrointestinal tract (whereas linaclotide’s action is limited to the duodenum and jejunem), resulting in more fluid volume and increased luminal transit.74 It was found effective in a phase 2 clinical trial,75 and the most effective dose was 50 mg twice daily.
Since tenapanor is minimally absorbed, it has few side effects, the major ones being diarrhea (11.2% vs 0% with placebo) and urinary tract infection (5.6% vs 4.4% with placebo).75 Further study is needed to confirm these findings.
Tenapanor also has the advantage of inhibiting luminal phosphorus absorption. This has led to exploration of its use as a phosphate binder in patients with end-stage renal disease.
DYSSYNERGIC DEFECATION AND ANORECTAL BIOFEEDBACK
According to the Rome IV criteria,5 dyssynergic defecation is present if the criteria for chronic constipation are met, if a dyssynergic pattern of defecation is confirmed by manometry, imaging, or electromyography, and if 1 or more of the following are present: inability to expel an artificial stool (a 50-mL water-filled balloon) within 1 minute, prolonged colonic transit time, inability to evacuate, or 50% or more retention of barium during defecography.5
Even though biofeedback has been controversial as a treatment for dyssynergic defecation because of conflicting results in older studies,76 3 trials have shown it to be better than placebo, laxatives, and muscle relaxants, with symptomatic improvement in 70% of patients.77–79
Biofeedback therapy involves an instrument-based auditory or visual tool (using electromyographic sensors or anorectal manometry) to help patients coordinate abdominal, rectal, puborectalis, and anal sphincter muscles and produce a propulsive force using their abdominal muscles to achieve complete evacuation. Important components of this therapy include:
Proper evacuation positioning (brace-pump technique, which involves sitting on the toilet leaning forward with forearms resting on thighs, shoulders relaxed, and feet placed on a small footstool
Breathing relaxation and training exercises during defecation (no straining, keeping a normal pattern of breathing, and avoiding holding the breath while defecating)
Use of the abdominal muscles by pushing the abdomen forward, along with relaxation of the anal sphincter.80
The anorectal feedback program usually consists of 6 weekly sessions of 45 to 60 minutes each. Limitations of this therapy include unavailability, lack of trained therapists, lack of insurance coverage, and inapplicability to certain patient groups, such as those with dementia or learning disabilities.
SURGERY FOR CHRONIC CONSTIPATION
Surgery for constipation is reserved for patients who continue to have symptoms despite optimal medical therapy.
Total abdominal colectomy and ileorectal anastomosis
Total abdominal colectomy with ileorectal anastomosis is a surgical option for medically intractable slow-transit constipation. Before considering surgery, complete diagnostic testing should be done, including colonic manometry and documentation of whether the patient also has outlet dysfunction.
Even though it has shown excellent outcomes and satisfaction rates as high as 100% in patients with pure slow-transit constipation,81–83 results in older studies in patients with mixed disorders (eg, slow-transit constipation with features of outlet dysfunction) were less predictable.84 More recent studies have reported comparable long-term morbidity and postoperative satisfaction rates in those with pure slow-transit constipation and those with a mixed disorder, indicating that careful patient selection is likely the key to a favorable outcome.85
Partial colectomies based on segmental colon transit time measurements can also be considered in some patients.86
Stapled transanal resection
Stapled transanal resection involves circumferential transanal stapling of the redundant rectal mucosa. It is an option for patients with defecatory disorders, specifically large rectoceles and rectal intussusception not amenable to therapy with pelvic floor retraining exercises.87
The efficacy of this procedure in controlling symptoms and improving quality of life is around 77% to 81% at 12 months, though complication rates as high as 46% and disappointing long-term outcomes have been a deterrent to its widespread acceptance in the United States.88–91
Chronic constipation has a variety of possible causes and mechanisms. Although traditional conservative treatments are still valid and first-line, if these fail, clinicians can choose from a growing list of new treatments, tailored to the cause in the individual patient.
This article discusses how defecation works (or doesn’t), the types of chronic constipation, the available diagnostic tools, and traditional and newer treatments, including some still in development.
THE EPIDEMIOLOGY OF CONSTIPATION
Chronic constipation is one of the most common gastrointestinal disorders, affecting about 15% of all adults and 30% of those over the age of 60.1 It can be a primary disorder or secondary to other factors.
Constipation is more prevalent in women and in institutionalized elderly people.2 It is associated with lower socioeconomic status, depression, less self-reported physical activity, certain medications, and stressful life events.3 Given its high prevalence and its impact on quality of life, it is also associated with significant utilization of healthcare resources.4
Constipation defined by Rome IV criteria
Physicians and patients may disagree about what constitutes constipation. Physicians primarily regard it as infrequent bowel movements, while patients tend to have a broader definition. According to the Rome IV criteria,5 chronic constipation is defined by the presence of the following for at least 3 months (with symptom onset at least 6 months prior to diagnosis):
(1) Two or more of the following for more than 25% of defecations:
- Straining
- Lumpy or hard stools
- Sensation of incomplete evacuation
- Sensation of anorectal obstruction or blockage
- Manual maneuvers to facilitate evacuation
- Fewer than 3 spontaneous bowel movements per week.
(2) Loose stools are rarely present without the use of laxatives.
(3) The patient does not meet the criteria for diagnosis of irritable bowel syndrome.
DEFECATION IS COMPLEX
Defecation begins when the rectum fills with stool, causing relaxation of the internal anal sphincter and the urge to defecate. The external anal sphincter, which is under voluntary control, can then either contract to delay defecation or relax to allow the stool to be expelled.6
Colonic muscles propel stool toward the rectum in repetitive localized contractions that help mix and promote absorption of the content, and larger coordinated (high-amplitude propagating) contractions that, in healthy individuals, move the stool forward from the proximal to the distal colon multiple times daily. These contractions usually occur in the morning and are accentuated by gastric distention from food and the resulting gastrocolic reflex.
Serotonin (5-HT) is released by enterochromaffin cells in response to distention of the gut wall. It mediates peristaltic movements of the gastrointestinal tract by binding to receptors (especially 5-HT4), stimulating release of neurotransmitters such as acetylcholine, causing smooth-muscle contraction behind the luminal contents and propelling them forward.
PRIMARY CONSTIPATION DISORDERS
The American Gastroenterological Association7 classifies constipation into 3 groups on the basis of colonic transit time and anorectal function:
Normal-transit constipation
Stool normally takes 20 to 72 hours to pass through the colon, with transit time affected by diet, drugs, level of physical activity, and emotional status.8
Normal-transit constipation is the most common type of constipation. The term is sometimes used interchangeably with constipation-predominant irritable bowel syndrome, but the latter is a distinct entity characterized by abdominal pain relieved by defecation as the primary symptom, as well as having occasional loose stools. These 2 conditions can be hard to tell apart, especially if the patient cannot describe the symptoms precisely.
Slow-transit constipation
Slow-transit constipation—also called delayed-transit constipation, colonoparesis, colonic inertia, and pseudo-obstruction—is defined as prolonged stool transit in the colon, ie, for more than 5 days.9 It can be the result of colonic smooth muscle dysfunction, compromised colonic neural pathways, or both, leading to slow colon peristalsis.
Factors that can affect colonic motility such as opioid use and hypothyroidism should be carefully considered in these patients. Opioids are notorious for causing constipation by decreasing bowel tone and contractility and thereby increasing colonic transit time. They also tighten up the anal sphincters, resulting in decreased rectal evacuation.10
Outlet dysfunction
Outlet dysfunction, also called pelvic floor dysfunction or defecatory disorder, is associated with incomplete rectal evacuation. It can be a consequence of weak rectal expulsion forces (slow colonic transit, rectal hyposensitivity), functional resistance to rectal evacuation (high anal resting pressure, anismus, incomplete relaxation of the anal sphincter, dyssynergic defecation), or structural outlet obstruction (excessive perineal descent, rectoceles, rectal intussusception). About 50% of patients with outlet dysfunction have concurrent slow-transit constipation.
Dyssynergic defecation is the most common outlet dysfunction disorder, accounting for about half of the cases referred to tertiary centers. It is defined as a paradoxical elevation in anal sphincter tone or less than 20% relaxation of the resting anal sphincter pressure with weak abdominal and pelvic propulsive forces.11 Anorectal biofeedback is a therapeutic option for dyssynergic defecation, as we discuss later in this article.
SECONDARY CONSTIPATION
Constipation can be secondary to several conditions and factors (Table 1), including:
- Neurologic disorders that affect gastrointestinal motility (eg, Hirschsprung disease, Parkinson disease, multiple sclerosis, spinal cord injury, stroke, spinal or ganglionic tumor, hypothyroidism, amyloidosis, diabetes mellitus, hypercalcemia)
- Drugs used to treat neurologic disorders
- Mechanical obstruction
- Diet (eg, low fiber, decreased fluid intake).
EVALUATION OF CONSTIPATION
It is crucial for physicians to efficiently use the available diagnostic tools for constipation to tailor the treatment to the patient.
Evaluation of chronic constipation begins with a thorough history and physical examination to rule out secondary constipation (Figure 1). Red flags such as unintentional weight loss, blood in the stool, rectal pain, fever, and iron-deficiency anemia should prompt referral for colonoscopy to evaluate for malignancy, colitis, or other potential colonic abnormalities.12
A detailed perineal and rectal examination can help diagnose defecatory disorders and should include evaluation of the resting anal tone and the sphincter during simulated evacuation.
Laboratory tests of thyroid function, electrolytes, and a complete blood cell count should be ordered if clinically indicated.13
Further tests
Further diagnostic tests can be considered if symptoms persist despite conservative treatment or if a defecatory disorder is suspected. These include anorectal manometry, colonic transit studies, defecography, and colonic manometry.
Anorectal manometry and the rectal balloon expulsion test are usually done first because of their high sensitivity (88%) and specificity (89%) for defecatory disorders.14 These tests measure the function of the internal and external anal sphincters at rest and with straining and assess rectal sensitivity and compliance. Anorectal manometry is also used in biofeedback therapy in patients with dyssynergic defecation.15
Colonic transit time can be measured if anorectal manometry and the balloon expulsion test are normal. The study uses radiopaque markers, radioisotopes, or wireless motility capsules to confirm slow-transit constipation and to identify areas of delayed transit in the colon.16
Defecography is usually the next step in diagnosis if anorectal manometry and balloon expulsion tests are inconclusive or if an anatomic abnormality of the pelvic floor is suspected. It can be done with a variety of techniques. Barium defecography can identify anatomic defects, scintigraphy can quantify evacuation of artificial stools, and magnetic resonance defecography visualizes anatomic landmarks to assess pelvic floor motion without exposing the patient to radiation.17,18
Colonic manometry is most useful in patients with refractory slow-transit constipation and can identify patients with isolated colonic motor dysfunction with no pelvic floor dysfunction who may benefit from subtotal colectomy and end-ileostomy.7
TRADITIONAL TREATMENTS STILL THE MAINSTAY
Nonpharmacologic treatments are the first-line options for patients with normal-transit and slow-transit constipation and should precede diagnostic testing. Lifestyle modifications and dietary changes (Table 2) aim to augment the known factors that stimulate the gastrocolic reflex and increase intestinal motility by high-amplitude propagated contractions.
Increasing physical activity increases intestinal gas clearance, decreases bloating, and lessens constipation.19,20
Toilet training is an integral part of lifestyle modifications.21
Diet. Drinking hot caffeinated beverages, eating breakfast within an hour of waking up, and consuming fiber in the morning (25–30 g of fiber daily) have traditionally been recommended as the first-line measures for chronic constipation. Dehydrated patients with constipation also benefit from increasing their fluid intake.22
LAXATIVES
Fiber (bulk-forming laxatives) for normal-transit constipation
Fiber remains a key part of the initial management of chronic constipation, as it is cheap, available, and safe. Increasing fiber intake is effective for normal-transit constipation, but patients with slow-transit constipation or refractory outlet dysfunction are less likely to benefit.23 Other laxatives are incorporated into the regimen if first-line nonpharmacologic interventions fail (Table 3).
Bulk-forming laxatives include insoluble fiber (wheat bran) and soluble fiber (psyllium, methylcellulose, inulin, calcium polycarbophil). Insoluble fiber, though often used, has little impact on symptoms of chronic constipation after 1 month of use, and up to 60% of patients report adverse effects from it.24 On the other hand, clinical trials have shown that soluble fiber such as psyllium facilitates defecation and improves functional bowel symptoms in patients with normal-transit constipation.25
Patients should be instructed to increase their dietary fiber intake gradually to avoid adverse effects and should be told to expect significant symptomatic improvement only after a few weeks. They should also be informed that increasing dietary fiber intake can cause bloating but that the bloating is temporary. If it continues, a different fiber can be tried.
Osmotic laxatives
Osmotic laxatives are often employed as a first- line laxative treatment option for patients with constipation. They draw water into the lumen by osmosis, helping to soften stool and speed intestinal transit. They include macrogols (inert polymers of ethylene glycol), nonabsorbable carbohydrates (lactulose, sorbitol), magnesium products, and sodium phosphate products.
Polyethylene glycol, the most studied osmotic laxative, has been shown to maintain therapeutic efficacy for up to 2 years, though it is not generally used this long.26 A meta-analysis of 10 randomized clinical trials found it to be superior to lactulose in improving stool consistency and frequency, and rates of adverse effects were similar to those with placebo.27
Lactulose and sorbitol are semisynthetic disaccharides that are not absorbed from the gastrointestinal tract. Apart from the osmotic effect of the disaccharide, these sugars are metabolized by colonic bacteria to acetic acid and other short-chain fatty acids, resulting in acidification of the stool, which exerts an osmotic effect in the colonic lumen.
Lactulose and sorbitol were shown to have similar efficacy in increasing the frequency of bowel movements in a small study, though patients taking lactulose had a higher rate of nausea.28
The usual recommended dose is 15 to 30 mL once or twice daily.
Adverse effects include gas, bloating, and abdominal distention (due to fermentation by colonic bacteria) and can limit long-term use.
Magnesium citrate and magnesium hydroxide are strong osmotic laxatives, but so far no clinical trial has been done to assess their efficacy in constipation. Although the risk of hypermagnesemia is low with magnesium-based products, this group of laxatives is generally avoided in patients with renal or cardiac disease.29
Sodium phosphate enemas (Fleet enemas) are used for bowel cleansing before certain procedures but have only limited use in constipation because of potential adverse effects such as hyperphosphatemia, hypocalcemia, and the rarer but more serious complication of acute phosphate nephropathy.30
Stimulant laxatives for short-term use only
Stimulant laxatives include glycerin, bisacodyl, senna, and sodium picosulfate. Sodium piosulfate and bisacodyl have been validated for treatment of chronic constipation for up to 4 weeks.31–33
Stimulant laxative suppositories should be used 30 minutes after meals to augment the physiologic gastrocolic reflex.
As more evidence is available for osmotic laxatives such as polyethylene glycol, they tend to be preferred over stimulant agents, especially for long-term use. Clinicians have traditionally hesitated to prescribe stimulant laxatives for long-term use, as they were thought to damage the enteric nervous system.34 Although more recent studies have not shown this potential effect,35 more research is warranted on the use of stimulant laxatives for longer than 4 weeks.
STOOL SOFTENERS: LITTLE EVIDENCE
Stool softeners enhance the interaction of stool and water, leading to softer stool and easier evacuation. Docusate sodium and docusate calcium are thought to facilitate the mixing of aqueous and fatty substances, thereby softening the stool.
However, there is little evidence to support the use of docusate for constipation in hospitalized adults or in ambulatory care. A recent review reported that docusate was no better than placebo in diminishing symptoms of constipation.36
INTESTINAL SECRETAGOGUES
The secretagogues include lubiprostone, linaclotide, and plecanatide. These medications are preferred therapy for patients with normal- or slow-transit constipation once conservative therapies have failed. Even though there is no current consensus, lifestyle measures and conservative treatment options should be tried for about 8 weeks.
Lubiprostone and linaclotide are approved by the US Food and Drug Administration (FDA) for both constipation and constipation-predominant irritable bowel syndrome. They activate chloride channels on the apical surface of enterocytes, increasing intestinal secretion of chloride, which in turn increases luminal sodium efflux to maintain electroneutrality, leading to secretion of water into the intestinal lumen. This eventually facilitates intestinal transit and increases the passage of stool.
Lubiprostone
Lubiprostone, a prostaglandin E1 derivative, is approved for treating chronic constipation, constipation-predominant irritable bowel syndrome in women, and opioid-induced constipation in patients with chronic noncancer pain.
Adverse effects in clinical trials were nausea (up to 30%) and headache.37,38
Linaclotide
Linaclotide, a minimally absorbed 14-amino acid peptide, increases intestinal secretion of chloride and bicarbonate, increasing intestinal fluid and promoting intestinal transit.39 It also decreases the firing rate of the visceral afferent pain fibers and helps reduce visceral pain, especially in patients with constipation-predominant irritable bowel syndrome.40 It is approved for chronic constipation and constipation-predominant irritable bowel syndrome.41–43
Dosage starts at 145 μg/day for chronic constipation, and can be titrated up to 290 μg if there is no response or if a diagnosis of constipation-predominant irritable bowel syndrome is under consideration. Linaclotide should be taken 30 to 60 minutes before breakfast to reduce the likelihood of diarrhea.44
Adverse effects. Diarrhea led to treatment discontinuation in 4.5% of patients in one study.42
Plecanatide
Plecanatide is a guanylate cyclase-c agonist with a mode of action similar to that of linaclotide. It was recently approved by the FDA for chronic idiopathic constipation in adults. The recommended dose is 3 mg once daily.
Data from phase 2 trials in chronic constipation showed improvement in straining, abdominal discomfort, and stool frequency after 14 days of treatment.45
A phase 3 trial showed that plecanatide was more effective than placebo when used for 12 weeks in 951 patients with chronic constipation (P = .009).46 The most common adverse effect reported was diarrhea.
SEROTONIN RECEPTOR AGONISTS
Activation of serotonin 5-HT4 receptors in the gut leads to release of acetylcholine, which in turn induces mucosal secretion by activating submucosal neurons and increasing gut motility.47
Two 5-HT4 receptor agonists were withdrawn from the market (cisapride in 2000 and tegaserod in 2007) due to serious cardiovascular adverse events (fatal arrhythmias, heart attacks, and strokes) resulting from their affinity for hERG-K+ cardiac channels.
The newer agents prucalopride,48 velusetrag, and naronapride are highly selective 5-HT4 agonists with low affinity for hERG-K+ receptors and do not have proarrhythmic properties, based on extensive assessment in clinical trials.
Prucalopride
Prucalopride has been shown to accelerate gastrointestinal and colonic transit in patients with chronic constipation, with improvement in bowel movements, symptoms of chronic constipation, and quality of life.49–52
Adverse effects reported with its use have been headache, nausea, abdominal pain, and cramps.
Prucalopride is approved in Europe and Canada for chronic constipation in women but is not yet approved in the United States.
Dosage is 2 mg orally once daily. Caution is advised in elderly patients, in whom the preferred maximum dose is 1 mg daily, as there are only limited data available on the safety of this medication in the elderly.
Velusetrag
Velusetrag has been shown to increase colonic motility and improve symptoms of chronic constipation. In a phase 2 trial,53 the most effective dose was 15 mg once daily. Higher doses were associated with a higher incidence of adverse effects such as diarrhea, headache, nausea, and vomiting.
Naronapride
Naronapride (ATI-7505) is in phase 2 trials for chronic constipation. Reported adverse effects were headache, diarrhea, nausea, and vomiting.54
BILE SALT ABSORPTION INHIBITORS
Bile acids exert prosecretory and prokinetic effects by increasing colonic secretion of water and electrolytes through the activation of adenylate cyclase. This happens as a result of their deconjugation after passage into the colon.
Elobixibat is an ileal bile acid transporter inhibitor that prevents absorption of nonconjugated bile salts in the distal ileum. It has few side effects because its systemic absorption is minimal. Phase 3 trials are under way. Dosage is 5 to 20 mg daily. Adverse effects are few because systemic absorption is minimal, but include abdominal pain and diarrhea.55,56
MANAGING OPIOID-INDUCED CONSTIPATION
Opioids cause constipation by binding to mu receptors in the enteric nervous system. Activation of these receptors decreases bowel tone and contractility, which increases transit time. Stimulation of these receptors also increases anal sphincter tone, resulting in decreased rectal evacuation.57
Though underrecognized, opioid-induced constipation affects 40% of patients who take these drugs for nonmalignant pain and 90% of those taking them for cancer pain. Patients with this condition were found to take more time off work and feel more impaired in their domestic and work-related obligations than patients who did not develop constipation with use of opioids.58
Initial management of opioid-induced constipation includes increasing intake of fluids and dietary fiber (fiber alone can worsen abdominal pain in this condition by increasing stool bulk without a concomitant improvement in peristalsis) and increasing physical activity. It is common clinical practice to use a stool softener along with a stimulant laxative if lifestyle modifications are inadequate.59 If these measures are ineffective, osmotic agents can be added.
If these conventional measures fail, a peripherally acting mu-opioid receptor antagonist such as methylnaltrexone or naloxegol should be considered.
Methylnaltrexone
Methylnaltrexone60,61 is a peripherally acting mu receptor antagonist with a rapid onset of action. It does not cross the blood-brain barrier, as it contains a methyl group. It was approved by the FDA in 2008 to treat opioid-induced constipation in adults with advanced illnesses when other approaches are ineffective.
Adverse effects. Although the mu receptor antagonist alvimopan had been shown to be associated with cardiovascular events hypothesized to be a consequence of opioid withdrawal, methylnaltrexone has been deemed to have a safe cardiovascular profile without any potential effects on platelets, corrected QT interval, metabolism, heart rate, or blood pressure.61 Side effects include abdominal pain, nausea, diarrhea, hot flashes, tremor, and chills.
Contraindications. Methylnaltrexone is contraindicated in patients with structural diseases of the gastrointestinal tract, ie, peptic ulcer disease, inflammatory bowel disease, diverticulitis, stomach or intestinal cancer) since it can increase the risk of perforation.
Dosing is 1 dose subcutaneously every other day, as needed, and no more than 1 dose in a 24-hour period. Dosage is based on weight: 0.15 mg/kg/dose for patients weighing less than 38 kg or more than 114 kg; 8 mg for those weighing 38 to 62 kg; and 12 mg for those weighing 62 to 114 kg.62
Naloxegol
Naloxegol, FDA-approved for treating opioid-induced constipation in 2014, consists of naloxone conjugated with polyethylene glycol, which prevents it from crossing the blood-brain barrier and diminishing the central effects of opioid-induced analgesia. Unlike methylnaltrexone, which is given by subcutaneous injection, naloxegol is taken orally.
Adverse effects reported in clinical trials63,64 were abdominal pain, diarrhea, nausea, headache, and flatulence. No clinically relevant association with QT and corrected QT interval prolongation or cardiac repolarization was noted.64
Dosing is 25 mg by mouth once daily, which can be decreased to 12.5 mg if the initial dose is difficult to tolerate. It should be taken on an empty stomach at least 1 hour before the first meal of the day or 2 hours after the meal. In patients with renal impairment (creatinine clearance < 60 mL/min), the dose is 12.5 mg once daily.65
CONSTIPATION-PREDOMINANT IRRITABLE BOWEL SYNDROME
Irritable bowel syndrome is the reason for 3.1 million office visits and 59 million prescriptions in the United States every year, with patients equally distributed between diarrhea-predominant, constipation-predominant, and mixed subtypes.66
To be diagnosed with constipation-predominant irritable bowel syndrome, patients must meet the Rome IV criteria, more than 25% of bowel movements should have Bristol stool form types 1 or 2, and less than 25% of bowel movements should have Bristol stool form types 6 or 7. In practice, patients reporting that their bowel movements are usually constipated often suffices to make the diagnosis.5
Osmotic laxatives are often tried first, but despite improving stool frequency and consistency, they have little efficacy in satisfying complaints of bloating or abdominal pain in patients with constipation-predominant irritable syndrome.67 Stimulant laxatives have not yet been tested in clinical trials. Lubiprostone and linaclotide are FDA-approved for this condition; in women, lubiprostone is approved only for those over age 18.
Antidepressant therapy
Patients often derive additional benefit from treatment with antidepressants. A meta-analysis demonstrated a number needed to treat of 4 for selective serotonin reuptake inhibitors and tricyclic antidepressants in managing abdominal pain associated with irritable bowel syndrome.68 The major limiting factor is usually adverse effects of these drugs.
For constipation-predominant irritable bowel syndrome, selective serotonin reuptake inhibitors are preferred over tricyclics because of their additional prokinetic properties. Starting at a low dose and titrating upward slowly avoids potential adverse effects.
Cognitive behavioral therapy has also been beneficial in treating irritable bowel syndrome.69
Adjunctive therapies
Adjunctive therapies including peppermint oil, probiotics (eg, Lactobacillus, Bifidobacterium), and acupuncture have also shown promise in managing irritable bowel syndrome, but more data are needed on the use of these therapies for constipation-predominant irritable bowel syndrome before any definite conclusions can be drawn.70 Other emerging pharmacologic therapies are plecanatide (discussed earlier) and tenapanor.
Peppermint oil is an antispasmodic that inhibits calcium channels, leading to relaxation of smooth muscles in the gastrointestinal tract. Different dosages and treatment durations have been studied—450 to 900 mg daily in 2 to 3 divided doses over 1 to 3 months.71,72 The most common adverse effect reported was gastroesophageal reflux, related in part to the oil’s relaxing effect on the lower esophageal sphincter. Observation of this led to the development of enteric-coated preparations that have the potential to bypass the upper gastrointestinal tract.73
Tenapanor inhibits the sodium-hydrogen exchanger 3 channel (a regulator of sodium and water uptake in intestinal lumen), which in turn leads to a higher sodium level in the entire gastrointestinal tract (whereas linaclotide’s action is limited to the duodenum and jejunem), resulting in more fluid volume and increased luminal transit.74 It was found effective in a phase 2 clinical trial,75 and the most effective dose was 50 mg twice daily.
Since tenapanor is minimally absorbed, it has few side effects, the major ones being diarrhea (11.2% vs 0% with placebo) and urinary tract infection (5.6% vs 4.4% with placebo).75 Further study is needed to confirm these findings.
Tenapanor also has the advantage of inhibiting luminal phosphorus absorption. This has led to exploration of its use as a phosphate binder in patients with end-stage renal disease.
DYSSYNERGIC DEFECATION AND ANORECTAL BIOFEEDBACK
According to the Rome IV criteria,5 dyssynergic defecation is present if the criteria for chronic constipation are met, if a dyssynergic pattern of defecation is confirmed by manometry, imaging, or electromyography, and if 1 or more of the following are present: inability to expel an artificial stool (a 50-mL water-filled balloon) within 1 minute, prolonged colonic transit time, inability to evacuate, or 50% or more retention of barium during defecography.5
Even though biofeedback has been controversial as a treatment for dyssynergic defecation because of conflicting results in older studies,76 3 trials have shown it to be better than placebo, laxatives, and muscle relaxants, with symptomatic improvement in 70% of patients.77–79
Biofeedback therapy involves an instrument-based auditory or visual tool (using electromyographic sensors or anorectal manometry) to help patients coordinate abdominal, rectal, puborectalis, and anal sphincter muscles and produce a propulsive force using their abdominal muscles to achieve complete evacuation. Important components of this therapy include:
Proper evacuation positioning (brace-pump technique, which involves sitting on the toilet leaning forward with forearms resting on thighs, shoulders relaxed, and feet placed on a small footstool
Breathing relaxation and training exercises during defecation (no straining, keeping a normal pattern of breathing, and avoiding holding the breath while defecating)
Use of the abdominal muscles by pushing the abdomen forward, along with relaxation of the anal sphincter.80
The anorectal feedback program usually consists of 6 weekly sessions of 45 to 60 minutes each. Limitations of this therapy include unavailability, lack of trained therapists, lack of insurance coverage, and inapplicability to certain patient groups, such as those with dementia or learning disabilities.
SURGERY FOR CHRONIC CONSTIPATION
Surgery for constipation is reserved for patients who continue to have symptoms despite optimal medical therapy.
Total abdominal colectomy and ileorectal anastomosis
Total abdominal colectomy with ileorectal anastomosis is a surgical option for medically intractable slow-transit constipation. Before considering surgery, complete diagnostic testing should be done, including colonic manometry and documentation of whether the patient also has outlet dysfunction.
Even though it has shown excellent outcomes and satisfaction rates as high as 100% in patients with pure slow-transit constipation,81–83 results in older studies in patients with mixed disorders (eg, slow-transit constipation with features of outlet dysfunction) were less predictable.84 More recent studies have reported comparable long-term morbidity and postoperative satisfaction rates in those with pure slow-transit constipation and those with a mixed disorder, indicating that careful patient selection is likely the key to a favorable outcome.85
Partial colectomies based on segmental colon transit time measurements can also be considered in some patients.86
Stapled transanal resection
Stapled transanal resection involves circumferential transanal stapling of the redundant rectal mucosa. It is an option for patients with defecatory disorders, specifically large rectoceles and rectal intussusception not amenable to therapy with pelvic floor retraining exercises.87
The efficacy of this procedure in controlling symptoms and improving quality of life is around 77% to 81% at 12 months, though complication rates as high as 46% and disappointing long-term outcomes have been a deterrent to its widespread acceptance in the United States.88–91
- Mugie SM, Benninga MA, Di Lorenzo C. Epidemiology of constipation in children and adults: a systematic review. Best Pract Res Clin Gastroenterol 2011; 25:3–18.
- Kinnunen O. Study of constipation in a geriatric hospital, day hospital, old people's home and at home. Aging (Milano) 1991; 3:161–170.
- Everhart JE, Go VL, Johannes RS, Fitzsimmons SC, Roth HP, White LR. A longitudinal survey of self-reported bowel habits in the United States. Dig Dis Sci 1989; 34:1153–1162.
- Shah ND, Chitkara DK, Locke GR, Meek PD, Talley NJ. Ambulatory care for constipation in the United States, 1993-2004. Am J Gastroenterol 2008; 103:1746–1753.
- Mearin F, Lacy BE, Chang L, et al. Bowel disorders. Gastroenterology 2016; 150:1393–1407.
- Bharucha AE. Pelvic floor: anatomy and function. Neurogastroenterol Motil 2006; 18:507–519.
- Bharucha AE, Pemberton JH, Locke GR 3rd. American Gastroenterological Association technical review on constipation. Gastroenterology 2013; 144:218–238.
- Grundy D, Al-Chaer ED, Aziz Q, et al. Fundamentals of neurogastroenterology: basic science. Gastroenterology 2006; 130:1391–1411.
- Gallegos-Orozco JF, Foxx-Orenstein AE, Sterler SM, Stoa JM. Chronic constipation in the elderly. Am J Gastroenterol 2012; 107:18–26.
- Mancini I, Bruera E. Constipation in advanced cancer patients. Support Care Cancer 1998; 6:356–364.
- Bassotti G, Chistolini F, Sietchiping-Nzepa F, de Roberto G, Morelli A, Chiarioni G. Biofeedback for pelvic floor dysfunction in constipation. BMJ 2004; 328:393–396.
- American Gastroenterological Association, Bharucha AE, Dorn SD, Lembo A, Pressman A. American Gastroenterological Association medical position statement on constipation. Gastroenterology 2013; 144:211–217.
- Costilla VC, Foxx-Orenstein AE. Constipation in adults: diagnosis and management. Curr Treat Options Gastroenterol 2014; 12:310–321.
- Rao SS, Singh S. Clinical utility of colonic and anorectal manometry in chronic constipation. J Clin Gastroenterol 2010; 44:597–609.
- Minguez M, Herreros B, Sanchiz V, et al. Predictive value of the balloon expulsion test for excluding the diagnosis of pelvic floor dyssynergia in constipation. Gastroenterology 2004; 126:57–62.
- Diamant NE, Kamm MA, Wald A, Whitehead WE. AGA technical review on anorectal testing techniques. Gastroenterology 1999; 116:735–760.
- Pezim ME, Pemberton JH, Levin KE, Litchy WJ, Phillips SF. Parameters of anorectal and colonic motility in health and in severe constipation. Dis Colon Rectum 1993; 36:484–491.
- Bharucha AE, Fletcher JG, Seide B, Riederer SJ, Zinsmeister AR. Phenotypic variation in functional disorders of defecation. Gastroenterology 2005; 128:1199–1210.
- De Schryver AM, Samsom M, Smout AI. Effects of a meal and bisacodyl on colonic motility in healthy volunteers and patients with slow-transit constipation. Dig Dis Sci 2003; 48:1206–1212.
- Villoria A, Serra J, Azpiroz F, Malagelada JR. Physical activity and intestinal gas clearance in patients with bloating. Am J Gastroenterol 2006; 101:2552–2557.
- Sikirov D. Comparison of straining during defecation in three positions: results and implications for human health. Dig Dis Sci 2003; 48:1201–1205.
- Muller-Lissner SA, Kamm MA, Scarpignato C, Wald A. Myths and misconceptions about chronic constipation. Am J Gastroenterol 2005; 100:232–242.
- Voderholzer WA, Schatke W, Muhldorfer BE, Klauser AG, Birkner B, Muller-Lissner SA. Clinical response to dietary fiber treatment of chronic constipation. Am J Gastroenterol 1997; 92:95–98.
- Bijkerk CJ, de Wit NJ, Muris JW, Whorwell PJ, Knottnerus JA, Hoes AW. Soluble or insoluble fibre in irritable bowel syndrome in primary care? Randomised placebo controlled trial. BMJ 2009; 339:b3154.
- Suares NC, Ford AC. Systematic review: the effects of fibre in the management of chronic idiopathic constipation. Aliment Pharmacol Ther 2011; 33:895–901.
- Dipalma JA, Cleveland MV, McGowan J, Herrera JL. A randomized, multicenter, placebo-controlled trial of polyethylene glycol laxative for chronic treatment of chronic constipation. Am J Gastroenterol 2007; 102:1436–1441.
- Lee-Robichaud H, Thomas K, Morgan J, Nelson RL. Lactulose versus polyethylene glycol for chronic constipation. Cochrane Database Syst Rev 2010; 7:CD007570.
- Lederle FA, Busch DL, Mattox KM, West MJ, Aske DM. Cost-effective treatment of constipation in the elderly: a randomized double-blind comparison of sorbitol and lactulose. Am J Med 1990; 89:597–601.
- Nyberg C, Hendel J, Nielsen OH. The safety of osmotically acting cathartics in colonic cleansing. Nat Rev Gastroenterol Hepatol 2010; 7:557–564.
- Ainley EJ, Winwood PJ, Begley JP. Measurement of serum electrolytes and phosphate after sodium phosphate colonoscopy bowel preparation: an evaluation. Dig Dis Sci 2005; 50:1319–1323.
- Kienzle-Horn S, Vix JM, Schuijt C, Peil H, Jordan CC, Kamm MA. Efficacy and safety of bisacodyl in the acute treatment of constipation: a double-blind, randomized, placebo-controlled study. Aliment Pharmacol Ther 2006; 23:1479–1488.
- Kienzle-Horn S, Vix JM, Schuijt C, Peil H, Jordan CC, Kamm MA. Comparison of bisacodyl and sodium picosulphate in the treatment of chronic constipation. Curr Med Res Opin 2007; 23:691–699.
- Mueller-Lissner S, Kamm MA, Wald A, et al. Multicenter, 4-week, double-blind, randomized, placebo-controlled trial of sodium picosulfate in patients with chronic constipation. Am J Gastroenterol 2010; 105:897–903.
- Smith B. Pathologic changes in the colon produced by anthraquinone purgatives. Dis Colon Rectum 1973; 16:455–458.
- Kiernan JA, Heinicke EA. Sennosides do not kill myenteric neurons in the colon of the rat or mouse. Neuroscience 1989; 30:837–842.
- Ottawa (ON): Canadian Agency for Drugs and Technologies in Health. Dioctyl sulfosuccinate or docusate (calcium or sodium) for the prevention or management of constipation: a review of the clinical effectiveness. www.ncbi.nlm.nih.gov/pubmedhealth/PMH0071207/. Accessed April 6, 2017.
- Saad R, Chey WD. Lubiprostone for chronic idiopathic constipation and irritable bowel syndrome with constipation. Expert Rev Gastroenterol Hepatol 2008; 2:497–508.
- Johanson JF, Morton D, Geenen J, Ueno R. Multicenter, 4-week, double-blind, randomized, placebo-controlled trial of lubiprostone, a locally-acting type-2 chloride channel activator, in patients with chronic constipation. Am J Gastroenterol 2008; 103:170–177.
- Harris LA, Crowell MD. Linaclotide, a new direction in the treatment of irritable bowel syndrome and chronic constipation. Curr Opin Mol Ther 2007; 9:403–410.
- Johnston JM, Kurtz CB, Macdougall JE, et al. Linaclotide improves abdominal pain and bowel habits in a phase IIb study of patients with irritable bowel syndrome with constipation. Gastroenterology 2010; 139:1877–1886.e2.
- Lembo AJ, Schneier HA, Shiff SJ, et al. Two randomized trials of linaclotide for chronic constipation. N Engl J Med 2011; 365:527–536.
- Chey WD, Lembo AJ, Lavins BJ, et al. Linaclotide for irritable bowel syndrome with constipation: a 26-week, randomized, double-blind, placebo-controlled trial to evaluate efficacy and safety. Am J Gastroenterol 2012; 107:1702–1712.
- Rao S, Lembo AJ, Shiff SJ, et al. A 12-week, randomized, controlled trial with a 4-week randomized withdrawal period to evaluate the efficacy and safety of linaclotide in irritable bowel syndrome with constipation. Am J Gastroenterol 2012; 107:1714–1725.
- Chey WD, Kurlander J, Eswaran S. Irritable bowel syndrome: a clinical review. JAMA 2015; 313:949–958.
- Shailubhai K, Talluto C, Comiskey S, Foss JA, Joslyn A, Jacob G. Phase II clinical evaluation of SP-304, a guanylate cyclase-C agonist, for treatment of chronic constipation. Am J Gastroenterol 2010; 105:S487–S488.
- Miner P, Surowitz R, Fogel R, et al. Plecanatide, a novel guanylate cyclase-C (GC-C) receptor agonist, is efficacious and safe in patients with chronic idiopathic constipation (CIC): results from a 951 patient, 12-week, multi-center trial (abstract). Gastroenterology 2013; 144:S163.
- Coss-Adame E, Rao SS. Brain and gut interactions in irritable bowel syndrome: new paradigms and new understandings. Curr Gastroenterol Rep 2014; 16:379.
- Mendzelevski B, Ausma J, Chanter DO, et al. Assessment of the cardiac safety of prucalopride in healthy volunteers: a randomized, double-blind, placebo- and positive-controlled thorough QT study. Br J Clin Pharmacol 2012; 73:203–209.
- Camilleri M, Kerstens R, Rykx A, Vandeplassche L. A placebo-controlled trial of prucalopride for severe chronic constipation. N Engl J Med 2008; 358:2344–2354.
- Tack J, van Outryve M, Beyens G, Kerstens R, Vandeplassche L. Prucalopride (Resolor) in the treatment of severe chronic constipation in patients dissatisfied with laxatives. Gut 2009; 58:357–365.
- Quigley EM, Vandeplassche L, Kerstens R, Ausma J. Clinical trial: the efficacy, impact on quality of life, and safety and tolerability of prucalopride in severe chronic constipation—a 12-week, randomized, double-blind, placebo-controlled study. Aliment Pharmacol Ther 2009; 29:315–328.
- Ford AC, Suares NC. Effect of laxatives and pharmacological therapies in chronic idiopathic constipation: systematic review and meta-analysis. Gut 2011; 60:209–218.
- Goldberg M, Li YP, Johanson JF, et al. Clinical trial: the efficacy and tolerability of velusetrag, a selective 5-HT4 agonist with high intrinsic activity, in chronic idiopathic constipation—a 4-week, randomized, double-blind, placebo-controlled, dose-response study. Aliment Pharmacol Ther 2010; 32:1102–1112.
- Palme M, Milner PG, Ellis DJ, Marmon T, Canafax DM. A novel gastrointestinal prokinetic, ATI-7505, increased spontaneous bowel movements (sbms) in a phase II, randomized, placebo-controlled study of patients with chronic idiopathic constipation (CIC). Gastroenterology 2010; 138:S-128–S-129.
- Chey WD, Camilleri M, Chang L, Rikner L, Graffner H. A randomized placebo-controlled phase IIb trial of a3309, a bile acid transporter inhibitor, for chronic idiopathic constipation. Am J Gastroenterol 2011; 106:1803–1812.
- Wong BS, Camilleri M, McKinzie S, Burton D, Graffner H, Zinsmeister AR. Effects of A3309, an ileal bile acid transporter inhibitor, on colonic transit and symptoms in females with functional constipation. Am J Gastroenterol 2011; 106:2154–2164.
- Pappagallo M. Incidence, prevalence, and management of opioid bowel dysfunction. Am J Surg 2001; 182(suppl):11S–18S.
- Bell T, Annunziata K, Leslie JB. Opioid-induced constipation negatively impacts pain management, productivity, and health-related quality of life: findings from the National Health and Wellness Survey. J Opioid Manag 2009; 5:137–144.
- Sykes NP. A volunteer model for the comparison of laxatives in opioid-related constipation. J Pain Symptom Manage 1996; 11:363–369.
- ClinicalTrials.gov. A multicenter, randomized, double-blind, placebo-controlled, parallel-group study of oral MOA-728 for the treatment of opioid- induced bowel dysfunction in subjects with chronic nonmalignant pain. ClinicalTrials.gov Identifier: NCT00547586. https://clinicaltrials.gov/ct2/show/NCT00547586. Accessed March 22, 2017.
- ClinicalTrials.gov. An open-label study to evaluate the long-term safety of subcutaneous MOA-728 for treatment of opioid-induced constipation in subjects with nonmalignant pain. ClinicalTrials.gov Identifier: NCT00804141. https://clinicaltrials.gov/ct2/show/NCT00804141. Accessed April 6, 2017.
- Wyeth Pharmaceuticals. Relistor package insert. http://labeling.pfizer.com/showlabeling.aspx?id=499. Accessed March 22, 2017.
- Webster L, Dhar S, Eldon M, Masuoka L, Lappalainen J, Sostek M. A phase 2, double-blind, randomized, placebo-controlled, dose-escalation study to evaluate the efficacy, safety, and tolerability of naloxegol in patients with opioid-induced constipation. Pain 2013; 154:1542–1550.
- Chey WD, Webster L, Sostek M, Lappalainen J, Barker PN, Tack J. Naloxegol for opioid-induced constipation in patients with noncancer pain. N Engl J Med 2014; 370:2387–2396.
- Jones R, Prommer E, Backstedt D. Naloxegol: a novel therapy in the management of opioid-induced constipation. Am J Hosp Palliat Care 2016; 33:875–880.
- Guilera M, Balboa A, Mearin F. Bowel habit subtypes and temporal patterns in irritable bowel syndrome: systematic review. Am J Gastroenterol 2005; 100:1174–1184.
- Chapman RW, Stanghellini V, Geraint M, Halphen M. Randomized clinical trial: macrogol/PEG 3350 plus electrolytes for treatment of patients with constipation associated with irritable bowel syndrome. Am J Gastroenterol 2013; 108:1508–1515.
- Ford AC, Quigley EM, Lacy BE, et al. Effect of antidepressants and psychological therapies, including hypnotherapy, in irritable bowel syndrome: systematic review and meta-analysis. Am J Gastroenterol 2014; 109:1350–1366.
- Ballou S, Keefer L. Psychological interventions for irritable bowel syndrome and inflammatory bowel diseases. Clin Transl Gastroenterol 2017; 8:e214.
- Ford AC, Moayyedi P, Lacy BE, et al; Task Force on the Management of Functional Bowel Disorders. American College of Gastroenterology monograph on the management of irritable bowel syndrome and chronic idiopathic constipation. Am J Gastroenterol 2014; 109(suppl 1):S2–S27.
- Ford AC, Talley NJ, Spiegel BM, et al. Effect of fibre, antispasmodics, and peppermint oil in the treatment of irritable bowel syndrome: systematic review and meta-analysis. BMJ 2008; 337:a2313.
- Wall GC, Bryant GA, Bottenberg MM, Maki ED, Miesner AR. Irritable bowel syndrome: a concise review of current treatment concepts. World J Gastroenterol 2014; 20:8796–8806.
- Kligler B, Chaudhary S. Peppermint oil. Am Fam Physician 2007; 75:1027–1030.
- Spencer AG, Labonte ED, Rosenbaum DP, et al. Intestinal inhibition of the Na+/H+ exchanger 3 prevents cardiorenal damage in rats and inhibits Na+ uptake in humans. Sci Transl Med 2014; 6:227ra36.
- Rosenbaum DP. A randomized, double-blind, placebo-controlled study to assess the safety and efficacy of AZD1722 for the treatment of constipation-predominant irritable bowel syndrome (IBS-C). 2014. https://clinicaltrials.gov/ct2/show/NCT01923428. Accessed April 6, 2017.
- Rao SS. Biofeedback therapy for dyssynergic (obstructive) defecation. J Clin Gastroenterol 2000; 30:115–116.
- Cadeddu F, Salis F, De Luca E, Ciangola I, Milito G. Efficacy of biofeedback plus transanal stimulation in the management of pelvic floor dyssynergia: a randomized trial. Tech Coloproctol 2015; 19:333–338.
- Chiarioni G, Whitehead WE, Pezza V, Morelli A, Bassotti G. Biofeedback is superior to laxatives for normal transit constipation due to pelvic floor dyssynergia. Gastroenterology 2006; 130:657–664.
- Chiarioni G, Heymen S, Whitehead WE. Biofeedback therapy for dyssynergic defecation. World J Gastroenterol 2006; 12:7069–7074.
- Rao SS. Biofeedback therapy for constipation in adults. Best Pract Res Clin Gastroenterol 2011; 25:159–166.
- Hassan I, Pemberton JH, Young-Fadok TM, et al. Ileorectal anastomosis for slow transit constipation: long-term functional and quality of life results. J Gastrointest Surg 2006; 10:1330–1337.
- You YT, Wang JY, Changchien CR, et al. Segmental colectomy in the management of colonic inertia. Am Surg 1998; 64:775–777.
- Nyam DC, Pemberton JH, Ilstrup DM, Rath DM. Long-term results of surgery for chronic constipation. Dis Colon Rectum 1997; 40:273–279.
- Pemberton JH, Rath DM, Ilstrup DM. Evaluation and surgical treatment of severe chronic constipation. Ann Surg 1991; 214:403–413.
- Reshef A, Alves-Ferreira P, Zutshi M, Hull T, Gurland B. Colectomy for slow transit constipation: effective for patients with coexistent obstructed defecation. Int J Colorectal Dis 2013; 28:841–847.
- Lundin E, Karlbom U, Pahlman L, Graf W. Outcome of segmental colonic resection for slow-transit constipation. Br J Surg 2002; 89:1270–1274.
- Schwandner O, Stuto A, Jayne D, et al. Decision-making algorithm for the STARR procedure in obstructed defecation syndrome: position statement of the group of STARR pioneers. Surg Innov 2008; 15:105–109.
- Titu LV, Riyad K, Carter H, Dixon AR. Stapled transanal rectal resection for obstructed defecation: a cautionary tale. Dis Colon Rectum 2009; 52:1716–1722.
- Goede AC, Glancy D, Carter H, Mills A, Mabey K, Dixon AR. Medium-term results of stapled transanal rectal resection (STARR) for obstructed defecation and symptomatic rectal-anal intussusception. Colorectal Dis 2011; 13:1052–1057.
- Jayne DG, Schwandner O, Stuto A. Stapled transanal rectal resection for obstructed defecation syndrome: one-year results of the european STARR registry. Dis Colon Rectum 2009; 52:1205–1214.
- Madbouly KM, Abbas KS, Hussein AM. Disappointing long-term outcomes after stapled transanal rectal resection for obstructed defecation. World J Surg 2010; 34:2191–2196.
- Mugie SM, Benninga MA, Di Lorenzo C. Epidemiology of constipation in children and adults: a systematic review. Best Pract Res Clin Gastroenterol 2011; 25:3–18.
- Kinnunen O. Study of constipation in a geriatric hospital, day hospital, old people's home and at home. Aging (Milano) 1991; 3:161–170.
- Everhart JE, Go VL, Johannes RS, Fitzsimmons SC, Roth HP, White LR. A longitudinal survey of self-reported bowel habits in the United States. Dig Dis Sci 1989; 34:1153–1162.
- Shah ND, Chitkara DK, Locke GR, Meek PD, Talley NJ. Ambulatory care for constipation in the United States, 1993-2004. Am J Gastroenterol 2008; 103:1746–1753.
- Mearin F, Lacy BE, Chang L, et al. Bowel disorders. Gastroenterology 2016; 150:1393–1407.
- Bharucha AE. Pelvic floor: anatomy and function. Neurogastroenterol Motil 2006; 18:507–519.
- Bharucha AE, Pemberton JH, Locke GR 3rd. American Gastroenterological Association technical review on constipation. Gastroenterology 2013; 144:218–238.
- Grundy D, Al-Chaer ED, Aziz Q, et al. Fundamentals of neurogastroenterology: basic science. Gastroenterology 2006; 130:1391–1411.
- Gallegos-Orozco JF, Foxx-Orenstein AE, Sterler SM, Stoa JM. Chronic constipation in the elderly. Am J Gastroenterol 2012; 107:18–26.
- Mancini I, Bruera E. Constipation in advanced cancer patients. Support Care Cancer 1998; 6:356–364.
- Bassotti G, Chistolini F, Sietchiping-Nzepa F, de Roberto G, Morelli A, Chiarioni G. Biofeedback for pelvic floor dysfunction in constipation. BMJ 2004; 328:393–396.
- American Gastroenterological Association, Bharucha AE, Dorn SD, Lembo A, Pressman A. American Gastroenterological Association medical position statement on constipation. Gastroenterology 2013; 144:211–217.
- Costilla VC, Foxx-Orenstein AE. Constipation in adults: diagnosis and management. Curr Treat Options Gastroenterol 2014; 12:310–321.
- Rao SS, Singh S. Clinical utility of colonic and anorectal manometry in chronic constipation. J Clin Gastroenterol 2010; 44:597–609.
- Minguez M, Herreros B, Sanchiz V, et al. Predictive value of the balloon expulsion test for excluding the diagnosis of pelvic floor dyssynergia in constipation. Gastroenterology 2004; 126:57–62.
- Diamant NE, Kamm MA, Wald A, Whitehead WE. AGA technical review on anorectal testing techniques. Gastroenterology 1999; 116:735–760.
- Pezim ME, Pemberton JH, Levin KE, Litchy WJ, Phillips SF. Parameters of anorectal and colonic motility in health and in severe constipation. Dis Colon Rectum 1993; 36:484–491.
- Bharucha AE, Fletcher JG, Seide B, Riederer SJ, Zinsmeister AR. Phenotypic variation in functional disorders of defecation. Gastroenterology 2005; 128:1199–1210.
- De Schryver AM, Samsom M, Smout AI. Effects of a meal and bisacodyl on colonic motility in healthy volunteers and patients with slow-transit constipation. Dig Dis Sci 2003; 48:1206–1212.
- Villoria A, Serra J, Azpiroz F, Malagelada JR. Physical activity and intestinal gas clearance in patients with bloating. Am J Gastroenterol 2006; 101:2552–2557.
- Sikirov D. Comparison of straining during defecation in three positions: results and implications for human health. Dig Dis Sci 2003; 48:1201–1205.
- Muller-Lissner SA, Kamm MA, Scarpignato C, Wald A. Myths and misconceptions about chronic constipation. Am J Gastroenterol 2005; 100:232–242.
- Voderholzer WA, Schatke W, Muhldorfer BE, Klauser AG, Birkner B, Muller-Lissner SA. Clinical response to dietary fiber treatment of chronic constipation. Am J Gastroenterol 1997; 92:95–98.
- Bijkerk CJ, de Wit NJ, Muris JW, Whorwell PJ, Knottnerus JA, Hoes AW. Soluble or insoluble fibre in irritable bowel syndrome in primary care? Randomised placebo controlled trial. BMJ 2009; 339:b3154.
- Suares NC, Ford AC. Systematic review: the effects of fibre in the management of chronic idiopathic constipation. Aliment Pharmacol Ther 2011; 33:895–901.
- Dipalma JA, Cleveland MV, McGowan J, Herrera JL. A randomized, multicenter, placebo-controlled trial of polyethylene glycol laxative for chronic treatment of chronic constipation. Am J Gastroenterol 2007; 102:1436–1441.
- Lee-Robichaud H, Thomas K, Morgan J, Nelson RL. Lactulose versus polyethylene glycol for chronic constipation. Cochrane Database Syst Rev 2010; 7:CD007570.
- Lederle FA, Busch DL, Mattox KM, West MJ, Aske DM. Cost-effective treatment of constipation in the elderly: a randomized double-blind comparison of sorbitol and lactulose. Am J Med 1990; 89:597–601.
- Nyberg C, Hendel J, Nielsen OH. The safety of osmotically acting cathartics in colonic cleansing. Nat Rev Gastroenterol Hepatol 2010; 7:557–564.
- Ainley EJ, Winwood PJ, Begley JP. Measurement of serum electrolytes and phosphate after sodium phosphate colonoscopy bowel preparation: an evaluation. Dig Dis Sci 2005; 50:1319–1323.
- Kienzle-Horn S, Vix JM, Schuijt C, Peil H, Jordan CC, Kamm MA. Efficacy and safety of bisacodyl in the acute treatment of constipation: a double-blind, randomized, placebo-controlled study. Aliment Pharmacol Ther 2006; 23:1479–1488.
- Kienzle-Horn S, Vix JM, Schuijt C, Peil H, Jordan CC, Kamm MA. Comparison of bisacodyl and sodium picosulphate in the treatment of chronic constipation. Curr Med Res Opin 2007; 23:691–699.
- Mueller-Lissner S, Kamm MA, Wald A, et al. Multicenter, 4-week, double-blind, randomized, placebo-controlled trial of sodium picosulfate in patients with chronic constipation. Am J Gastroenterol 2010; 105:897–903.
- Smith B. Pathologic changes in the colon produced by anthraquinone purgatives. Dis Colon Rectum 1973; 16:455–458.
- Kiernan JA, Heinicke EA. Sennosides do not kill myenteric neurons in the colon of the rat or mouse. Neuroscience 1989; 30:837–842.
- Ottawa (ON): Canadian Agency for Drugs and Technologies in Health. Dioctyl sulfosuccinate or docusate (calcium or sodium) for the prevention or management of constipation: a review of the clinical effectiveness. www.ncbi.nlm.nih.gov/pubmedhealth/PMH0071207/. Accessed April 6, 2017.
- Saad R, Chey WD. Lubiprostone for chronic idiopathic constipation and irritable bowel syndrome with constipation. Expert Rev Gastroenterol Hepatol 2008; 2:497–508.
- Johanson JF, Morton D, Geenen J, Ueno R. Multicenter, 4-week, double-blind, randomized, placebo-controlled trial of lubiprostone, a locally-acting type-2 chloride channel activator, in patients with chronic constipation. Am J Gastroenterol 2008; 103:170–177.
- Harris LA, Crowell MD. Linaclotide, a new direction in the treatment of irritable bowel syndrome and chronic constipation. Curr Opin Mol Ther 2007; 9:403–410.
- Johnston JM, Kurtz CB, Macdougall JE, et al. Linaclotide improves abdominal pain and bowel habits in a phase IIb study of patients with irritable bowel syndrome with constipation. Gastroenterology 2010; 139:1877–1886.e2.
- Lembo AJ, Schneier HA, Shiff SJ, et al. Two randomized trials of linaclotide for chronic constipation. N Engl J Med 2011; 365:527–536.
- Chey WD, Lembo AJ, Lavins BJ, et al. Linaclotide for irritable bowel syndrome with constipation: a 26-week, randomized, double-blind, placebo-controlled trial to evaluate efficacy and safety. Am J Gastroenterol 2012; 107:1702–1712.
- Rao S, Lembo AJ, Shiff SJ, et al. A 12-week, randomized, controlled trial with a 4-week randomized withdrawal period to evaluate the efficacy and safety of linaclotide in irritable bowel syndrome with constipation. Am J Gastroenterol 2012; 107:1714–1725.
- Chey WD, Kurlander J, Eswaran S. Irritable bowel syndrome: a clinical review. JAMA 2015; 313:949–958.
- Shailubhai K, Talluto C, Comiskey S, Foss JA, Joslyn A, Jacob G. Phase II clinical evaluation of SP-304, a guanylate cyclase-C agonist, for treatment of chronic constipation. Am J Gastroenterol 2010; 105:S487–S488.
- Miner P, Surowitz R, Fogel R, et al. Plecanatide, a novel guanylate cyclase-C (GC-C) receptor agonist, is efficacious and safe in patients with chronic idiopathic constipation (CIC): results from a 951 patient, 12-week, multi-center trial (abstract). Gastroenterology 2013; 144:S163.
- Coss-Adame E, Rao SS. Brain and gut interactions in irritable bowel syndrome: new paradigms and new understandings. Curr Gastroenterol Rep 2014; 16:379.
- Mendzelevski B, Ausma J, Chanter DO, et al. Assessment of the cardiac safety of prucalopride in healthy volunteers: a randomized, double-blind, placebo- and positive-controlled thorough QT study. Br J Clin Pharmacol 2012; 73:203–209.
- Camilleri M, Kerstens R, Rykx A, Vandeplassche L. A placebo-controlled trial of prucalopride for severe chronic constipation. N Engl J Med 2008; 358:2344–2354.
- Tack J, van Outryve M, Beyens G, Kerstens R, Vandeplassche L. Prucalopride (Resolor) in the treatment of severe chronic constipation in patients dissatisfied with laxatives. Gut 2009; 58:357–365.
- Quigley EM, Vandeplassche L, Kerstens R, Ausma J. Clinical trial: the efficacy, impact on quality of life, and safety and tolerability of prucalopride in severe chronic constipation—a 12-week, randomized, double-blind, placebo-controlled study. Aliment Pharmacol Ther 2009; 29:315–328.
- Ford AC, Suares NC. Effect of laxatives and pharmacological therapies in chronic idiopathic constipation: systematic review and meta-analysis. Gut 2011; 60:209–218.
- Goldberg M, Li YP, Johanson JF, et al. Clinical trial: the efficacy and tolerability of velusetrag, a selective 5-HT4 agonist with high intrinsic activity, in chronic idiopathic constipation—a 4-week, randomized, double-blind, placebo-controlled, dose-response study. Aliment Pharmacol Ther 2010; 32:1102–1112.
- Palme M, Milner PG, Ellis DJ, Marmon T, Canafax DM. A novel gastrointestinal prokinetic, ATI-7505, increased spontaneous bowel movements (sbms) in a phase II, randomized, placebo-controlled study of patients with chronic idiopathic constipation (CIC). Gastroenterology 2010; 138:S-128–S-129.
- Chey WD, Camilleri M, Chang L, Rikner L, Graffner H. A randomized placebo-controlled phase IIb trial of a3309, a bile acid transporter inhibitor, for chronic idiopathic constipation. Am J Gastroenterol 2011; 106:1803–1812.
- Wong BS, Camilleri M, McKinzie S, Burton D, Graffner H, Zinsmeister AR. Effects of A3309, an ileal bile acid transporter inhibitor, on colonic transit and symptoms in females with functional constipation. Am J Gastroenterol 2011; 106:2154–2164.
- Pappagallo M. Incidence, prevalence, and management of opioid bowel dysfunction. Am J Surg 2001; 182(suppl):11S–18S.
- Bell T, Annunziata K, Leslie JB. Opioid-induced constipation negatively impacts pain management, productivity, and health-related quality of life: findings from the National Health and Wellness Survey. J Opioid Manag 2009; 5:137–144.
- Sykes NP. A volunteer model for the comparison of laxatives in opioid-related constipation. J Pain Symptom Manage 1996; 11:363–369.
- ClinicalTrials.gov. A multicenter, randomized, double-blind, placebo-controlled, parallel-group study of oral MOA-728 for the treatment of opioid- induced bowel dysfunction in subjects with chronic nonmalignant pain. ClinicalTrials.gov Identifier: NCT00547586. https://clinicaltrials.gov/ct2/show/NCT00547586. Accessed March 22, 2017.
- ClinicalTrials.gov. An open-label study to evaluate the long-term safety of subcutaneous MOA-728 for treatment of opioid-induced constipation in subjects with nonmalignant pain. ClinicalTrials.gov Identifier: NCT00804141. https://clinicaltrials.gov/ct2/show/NCT00804141. Accessed April 6, 2017.
- Wyeth Pharmaceuticals. Relistor package insert. http://labeling.pfizer.com/showlabeling.aspx?id=499. Accessed March 22, 2017.
- Webster L, Dhar S, Eldon M, Masuoka L, Lappalainen J, Sostek M. A phase 2, double-blind, randomized, placebo-controlled, dose-escalation study to evaluate the efficacy, safety, and tolerability of naloxegol in patients with opioid-induced constipation. Pain 2013; 154:1542–1550.
- Chey WD, Webster L, Sostek M, Lappalainen J, Barker PN, Tack J. Naloxegol for opioid-induced constipation in patients with noncancer pain. N Engl J Med 2014; 370:2387–2396.
- Jones R, Prommer E, Backstedt D. Naloxegol: a novel therapy in the management of opioid-induced constipation. Am J Hosp Palliat Care 2016; 33:875–880.
- Guilera M, Balboa A, Mearin F. Bowel habit subtypes and temporal patterns in irritable bowel syndrome: systematic review. Am J Gastroenterol 2005; 100:1174–1184.
- Chapman RW, Stanghellini V, Geraint M, Halphen M. Randomized clinical trial: macrogol/PEG 3350 plus electrolytes for treatment of patients with constipation associated with irritable bowel syndrome. Am J Gastroenterol 2013; 108:1508–1515.
- Ford AC, Quigley EM, Lacy BE, et al. Effect of antidepressants and psychological therapies, including hypnotherapy, in irritable bowel syndrome: systematic review and meta-analysis. Am J Gastroenterol 2014; 109:1350–1366.
- Ballou S, Keefer L. Psychological interventions for irritable bowel syndrome and inflammatory bowel diseases. Clin Transl Gastroenterol 2017; 8:e214.
- Ford AC, Moayyedi P, Lacy BE, et al; Task Force on the Management of Functional Bowel Disorders. American College of Gastroenterology monograph on the management of irritable bowel syndrome and chronic idiopathic constipation. Am J Gastroenterol 2014; 109(suppl 1):S2–S27.
- Ford AC, Talley NJ, Spiegel BM, et al. Effect of fibre, antispasmodics, and peppermint oil in the treatment of irritable bowel syndrome: systematic review and meta-analysis. BMJ 2008; 337:a2313.
- Wall GC, Bryant GA, Bottenberg MM, Maki ED, Miesner AR. Irritable bowel syndrome: a concise review of current treatment concepts. World J Gastroenterol 2014; 20:8796–8806.
- Kligler B, Chaudhary S. Peppermint oil. Am Fam Physician 2007; 75:1027–1030.
- Spencer AG, Labonte ED, Rosenbaum DP, et al. Intestinal inhibition of the Na+/H+ exchanger 3 prevents cardiorenal damage in rats and inhibits Na+ uptake in humans. Sci Transl Med 2014; 6:227ra36.
- Rosenbaum DP. A randomized, double-blind, placebo-controlled study to assess the safety and efficacy of AZD1722 for the treatment of constipation-predominant irritable bowel syndrome (IBS-C). 2014. https://clinicaltrials.gov/ct2/show/NCT01923428. Accessed April 6, 2017.
- Rao SS. Biofeedback therapy for dyssynergic (obstructive) defecation. J Clin Gastroenterol 2000; 30:115–116.
- Cadeddu F, Salis F, De Luca E, Ciangola I, Milito G. Efficacy of biofeedback plus transanal stimulation in the management of pelvic floor dyssynergia: a randomized trial. Tech Coloproctol 2015; 19:333–338.
- Chiarioni G, Whitehead WE, Pezza V, Morelli A, Bassotti G. Biofeedback is superior to laxatives for normal transit constipation due to pelvic floor dyssynergia. Gastroenterology 2006; 130:657–664.
- Chiarioni G, Heymen S, Whitehead WE. Biofeedback therapy for dyssynergic defecation. World J Gastroenterol 2006; 12:7069–7074.
- Rao SS. Biofeedback therapy for constipation in adults. Best Pract Res Clin Gastroenterol 2011; 25:159–166.
- Hassan I, Pemberton JH, Young-Fadok TM, et al. Ileorectal anastomosis for slow transit constipation: long-term functional and quality of life results. J Gastrointest Surg 2006; 10:1330–1337.
- You YT, Wang JY, Changchien CR, et al. Segmental colectomy in the management of colonic inertia. Am Surg 1998; 64:775–777.
- Nyam DC, Pemberton JH, Ilstrup DM, Rath DM. Long-term results of surgery for chronic constipation. Dis Colon Rectum 1997; 40:273–279.
- Pemberton JH, Rath DM, Ilstrup DM. Evaluation and surgical treatment of severe chronic constipation. Ann Surg 1991; 214:403–413.
- Reshef A, Alves-Ferreira P, Zutshi M, Hull T, Gurland B. Colectomy for slow transit constipation: effective for patients with coexistent obstructed defecation. Int J Colorectal Dis 2013; 28:841–847.
- Lundin E, Karlbom U, Pahlman L, Graf W. Outcome of segmental colonic resection for slow-transit constipation. Br J Surg 2002; 89:1270–1274.
- Schwandner O, Stuto A, Jayne D, et al. Decision-making algorithm for the STARR procedure in obstructed defecation syndrome: position statement of the group of STARR pioneers. Surg Innov 2008; 15:105–109.
- Titu LV, Riyad K, Carter H, Dixon AR. Stapled transanal rectal resection for obstructed defecation: a cautionary tale. Dis Colon Rectum 2009; 52:1716–1722.
- Goede AC, Glancy D, Carter H, Mills A, Mabey K, Dixon AR. Medium-term results of stapled transanal rectal resection (STARR) for obstructed defecation and symptomatic rectal-anal intussusception. Colorectal Dis 2011; 13:1052–1057.
- Jayne DG, Schwandner O, Stuto A. Stapled transanal rectal resection for obstructed defecation syndrome: one-year results of the european STARR registry. Dis Colon Rectum 2009; 52:1205–1214.
- Madbouly KM, Abbas KS, Hussein AM. Disappointing long-term outcomes after stapled transanal rectal resection for obstructed defecation. World J Surg 2010; 34:2191–2196.
KEY POINTS
- Although newer drugs are available, lifestyle modifications and laxatives continue to be the treatments of choice for chronic constipation, as they have high response rates and few adverse effects and are relatively affordable.
- Chronic constipation requires different management approaches depending on whether colonic transit time is normal or prolonged and whether outlet function is abnormal.
- Surgical treatments for constipation are reserved for patients whose symptoms persist despite maximal medical therapy.

Managing irritable bowel syndrome: The low-FODMAP diet
The role of diet in controlling symptoms of irritable bowel syndrome (IBS) has gained much traction over the years,1 but until recently, diet therapy for IBS has been hindered by a lack of quality evidence, in part because of the challenges of conducting dietary clinical trials.
Several clinical trials have now been done that support a diet low in fermentable oligosaccharides, disaccharides, monosaccharides, and polyols (FODMAPs) for managing IBS. Although restrictive and difficult to follow, the low-FODMAP diet is gaining popularity.
This article provides an overview of dietary interventions used to manage IBS, focusing on the low-FODMAP diet. We discuss mechanisms of malabsorption of FODMAPs and the role of FODMAPS in symptom induction; highlight clinical trials that provide evidence of benefits of the diet for IBS; and discuss the steps to implement it. We also address the nutritional adequacy of the diet and its potential effects on the gut microbiome.
IBS IS A COMMON FUNCTIONAL DISORDER
IBS is one of the most commonly diagnosed gastrointestinal disorders, and it has a significant impact on quality of life.2 It is a functional disorder characterized by chronic abdominal pain and altered bowel habits in the absence of a structural or organic cause.
The Rome IV diagnostic criteria define IBS by the following:
- Recurrent abdominal pain or discomfort at least 1 day a week in the last 3 months, associated with two or more of the following:
- Symptoms improved by defecation
- Onset associated with a change in frequency of stool
- Onset associated with a change in form or appearance of stool.
IBS mainly arises during young adulthood but can be diagnosed at any age.3
The pathophysiology of IBS involves mechanisms such as bowel distention, altered bowel motility, visceral hypersensitivity, and disruption of mucosal permeability.4 Several therapeutic modalities targeting these mechanisms have been implemented in IBS management, including antispasmodics, laxatives, antidepressants, antibiotics, and behavioral therapy. Diet is only one line of treatment and is most effective when part of a multipronged approach.
TRADITIONAL DIETARY MANAGEMENT
Diet is important in inducing the symptoms of IBS—and in controlling them. Patients identify eating as a common precipitator of symptoms, but the complex diet-symptom interaction is not fully understood and varies widely among patients. Traditional dietary advice for IBS includes adhering to a regular meal pattern, avoiding large meals, and reducing intake of fat, insoluble fibers, caffeine, spicy and gas-producing foods, and carbonated beverages.5,6
Increase soluble fiber
Fiber and fiber supplements, particularly soluble fibers such as psyllium, calcium polycarbophil, and ispaghula husk are often recommended. A meta-analysis7 found that soluble fiber but not insoluble fiber (eg, wheat bran) is associated with an improvement in IBS symptoms (relative risk [RR] 0.84, 95% confidence interval [CI] 0.73–0.94). By improving stool consistency and accelerating transit, soluble fiber is especially useful in constipation-predominant IBS while posing a low risk for adverse outcomes.7 Fiber should be started at a low dose and gradually increased over several weeks to as much as 20 to 30 g/day.
Avoid wheat
Only about 4% of patients with IBS also have celiac disease, but estimating the prevalence of nonceliac gluten sensitivity is confounded by overlapping symptoms. There is some evidence implicating gluten in IBS: celiac disease and IBS overlap in their symptoms, and symptoms are often precipitated by gluten-containing foods in patients with IBS.8 The pathogenesis of gluten-induced (or wheat-induced) symptoms in IBS is unclear, and studies have had conflicting results as to the benefits of gluten restriction in IBS.9
In a study of patients with IBS whose symptoms improved when they started a gluten-free and low-FODMAP diet, symptoms did not return when gluten was reintroduced, suggesting that it is the fructan (a FODMAP) component of wheat rather than gluten that contributes to symptoms in IBS.10
Probiotics
Probiotics are increasingly being recommended as dietary supplements for people with IBS, as awareness increases of the importance of the gut microbiota. In addition to their effects on the gut microbiota, probiotics in IBS have been shown to have anti-inflammatory effects, to alter gut motility, to modulate visceral hypersensitivity, and to restore epithelial integrity.
In a meta-analysis, Ford et al11 found that probiotics improved global IBS symptoms more than placebo (RR 0.79, 95% CI 0.70–0.89) and also reduced abdominal pain, bloating, and flatulence scores.
Which species and strains are most beneficial and the optimal dosing and duration of treatment are still unclear. Data from studies of prebiotics (nutrients that encourage the growth of probiotic bacteria) and synbiotics (combinations of prebiotics and probiotics) are limited and insufficient to draw conclusions.
FODMAPS ARE SHORT-CHAIN CARBOHYDRATES
The term FODMAPs was initially coined by researchers at Monash University in Australia to describe a collection of poorly absorbed short-chain fermentable carbohydrates that are natural components of many foods:
- Oligosaccharides, including fructans (which include inulins) and galacto-oligosaccharides
- Disaccharides, including lactose and sucrose
- Monosaccharides, including fructose
- Polyols, including sorbitol and mannitol.12
Intake of FODMAPs, especially fructose, has increased in Western diets over the past several decades from increased consumption of fruits and concentrated fruit juices, as well as from the widespread use of high-fructose corn syrup in processed foods and beverages.13
FODMAPs ARE POORLY ABSORBED

Different FODMAPs can be poorly absorbed for different reasons (Table 1). The poor absorption is related either to reduced or absent digestive enzymes (ie, hydrolases) or to slow transport across the intestinal mucosa. Excess FODMAPs in the distal small intestine and proximal colon exert osmotic pressure, drawing more water into the lumen. FODMAPs are also rapidly fermented by colonic bacteria, producing gas, bowel distention, and altered motility, all of which induce IBS symptoms.14
Fructans are fructose polymers that are not absorbed in human intestines. They have no intestinal hydrolases and no mechanisms for direct transport across the epithelium. However, a negligible amount may be absorbed after being degraded by microbes in the gut.15 Most dietary fructans are obtained from wheat and onion, which are actually low in fructans but tend to be consumed in large quantities.16
Galacto-oligosaccharides are available for colonic fermentation after ingestion due to lack of a human alpha-galactosidase. Common sources of galacto-oligosaccharides include legumes, nuts, seeds, some grains, dairy products, human milk, and some commercially produced forms added to infant formula.17,18
Lactose is poorly absorbed in people with lactase deficiency. It is mainly present in dairy products but is also added to commercial foods, including breads, cakes, and some diet products.19
Fructose is the most abundant FODMAP in the Western diet. It is either present as a free sugar or generated from the digestive breakdown of sucrose. In the intestine, it is absorbed via a direct low-capacity glucose transporter (GLUT)-5 and through GLUT-2, which is more efficient but requires the coexistence of glucose. Because of this requirement, fructose is more likely to be malabsorbed when present in excess of glucose, as in people with diminished sucrase activity. The main sources of fructose in the Western diet are fruits and fruit products, honey, and foods with added high-fructose sweeteners.13
Polyols such as sorbitol and mannitol are absorbed by slow passive diffusion because they have no active intestinal transport system. They are found in fruits and vegetables. Sugar-free chewing gum is a particularly rich source of sorbitol.20
QUANTIFYING FODMAP CONTENT
As interest in the low-FODMAP diet grew, studies were conducted to quantify FODMAPs in foods. One study used high-performance liquid chromatography to analyze FODMAP content in foods,21 and another evaluated fructan levels in a variety of fruits and vegetables using enzymatic hydrolysis.22 The Monash University low-FODMAP diet smartphone application provides patients and healthcare providers easy access to updated and detailed food analyses.23
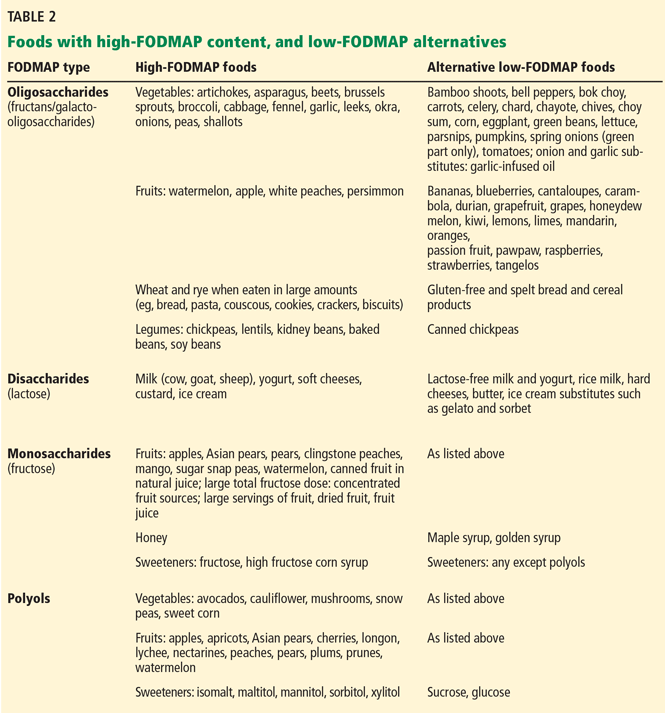
Table 2 lists foods high in FODMAPs along with low-FODMAP alternatives. Total FODMAP intake is important, as the effects are additive.24 Readers and patients can be directed to the following websites for more information on the low-FODMAP diet: www.med.monash.edu/cecs/gastro/fodmap or www.ibsfree.net/what-is-fodmap-diet.
LOW-FODMAP DIET REDUCES SYMPTOMS
The low-FODMAP diet was inspired by the results of several studies that evaluated the role of dietary carbohydrates in inducing IBS symptoms and found improvement with their restriction.25,26
One study found that 74% of patients with IBS had less bloating, nausea, abdominal pain, and diarrhea when they restricted their intake of fructose and fructans.27
A prospective trial randomized 41 patients with IBS to 4 weeks of either a low-FODMAP diet or their habitual diet.28 The low-FODMAP diet resulted in greater improvement in overall IBS symptoms (P < .05) and stool frequency (P = .008). This study was limited by different habitual diets between patients and by lack of standardization of the low-FODMAP diet.
Halmos et al,29 in a randomized crossover trial, compared gastrointestinal symptoms in IBS patients over 3 weeks on a low-FODMAP diet vs a moderate-FODMAP (ie, regular) diet, as well as in healthy controls. Food was provided by the study and was matched for all nutrients. Up to 70% of the IBS patients had significantly lower overall symptom scores while on a low-FODMAP diet vs IBS patients on a regular diet (P < .001); bloating, abdominal pain, and flatulence were reduced. Symptoms were minimal and unaffected by either diet in the healthy controls.
A double-blind trial30 randomly assigned 25 patients with IBS who initially responded to a low-FODMAP diet to be challenged by a graduated dose of fructose alone, fructans alone, a combination of both, or glucose. The severity of overall and individual symptoms was markedly more reduced with glucose consumption than with the other carbohydrates: 70% of patients receiving fructose, 77% of those receiving fructans, and 79% of those receiving a mixture of both reported that their symptoms were not adequately controlled, compared with 14% of patients receiving glucose (P ≤ .002).30
Murray et al31 evaluated the gastrointestinal tract after a carbohydrate challenge consisting of 0.5 L of water containing 40 g of glucose, fructose, or inulin or a combination of 40 g of glucose and 40 g of fructose in 16 healthy volunteers. Magnetic resonance imaging was performed hourly for 5 hours to assess the volume of gastric contents, small-bowel water content, and colonic gas. Breath hydrogen was also measured, and symptoms were recorded after each imaging session.
Fructose significantly increased small-bowel water content compared with glucose (mean difference 28 L/min, P < .001), but combined glucose and fructose lessened the effect. Inulin had no significant effect on small-bowel water content (mean difference with glucose 2 L/min, P > .7) but led to the greatest production of colonic gas compared with glucose alone (mean difference 15 L/min, P < .05) and combined glucose and fructose (mean difference 12 L/min, P < .05). Inulin also produced the most breath hydrogen: 81% of participants had a rise after drinking inulin compared with 50% after drinking fructose. Glucose did not affect breath hydrogen concentrations, and combined glucose and fructose significantly reduced the concentration measured vs fructose alone. In patients who reported “gas” symptoms, a correlation was observed between the volume of gas in the colon and gas symptoms (r = 0.59, P < .0001).31
The authors concluded31 that long-chain carbohydrates such as inulin have a greater effect on colonic gas production and little effect on small-bowel water content, whereas small-chain FODMAPs such as fructose are likely to cause luminal distention in both the small and large intestines. The study also showed that combining equal amounts of glucose and fructose reduces malabsorption of fructose in the small bowel and reduces the effect of fructose on small-bowel water content and breath hydrogen concentration.31
PROBIOTICS HELP
A Danish study32 randomized 123 patients with IBS to one of three treatments: a low-FODMAP diet, a normal diet with probiotics containing the strain Lactobacillus rhamnosus GG (two capsules daily), or no special intervention. Symptoms were recorded weekly. IBS severity scores at week 6 were lower in patients on either the low-FODMAP diet or probiotics compared with the control group (P < .01). Subgroup analysis determined that patients with primarily diarrheal symptoms were more likely to have improved quality of life with the low-FODMAP diet.
A LOW-FODMAP DIET MAY ALSO HELP IN INFLAMMATORY BOWEL DISEASE
The low-FODMAP diet has also been studied in patients with inflammatory bowel disease with functional gut symptoms. In a retrospective pilot study,33 overall symptoms improved in about half of such patients on a low-FODMAP diet. A controlled dietary intervention trial is needed to confirm these findings and define the role of the low-FODMAP approach for patients with inflammatory bowel disease.
Marsh et al34 performed a meta-analysis of six randomized clinical trials and 16 nonrandomized interventions of a low-FODMAP diet on improving functional gastrointestinal symptoms in patients with either IBS or inflammatory bowel disease. They found significant improvements in:
- IBS Symptoms Severity Scores in the randomized trials (odds ratio [OR] 0.44, 95% CI 0.25–0.76)
- IBS Symptoms Severity Scores in the nonrandomized interventions (OR 0.03, 95% CI 0.01–0.2)
- IBS Quality of Life scores in the randomized trials (OR 1.84, 95% CI 1.12–3.03)
- IBS Quality of Life scores in the nonrandomized interventions (OR 3.18, 95% CI 1.6–6.31)
- Overall symptom severity in the randomized trials (OR 1.81, 95% CI 1.11–2.95).
DIETARY COUNSELING IS RECOMMENDED
Adherence is a major factor in the success of the low-FODMAP diet in IBS management and is strongly correlated with improved symptoms.35 Patients should be counseled on the role of food in inducing their symptoms. Haphazard dietary advice can be detrimental to outcomes, as many diets restrict food groups, impairing the consumption of essential nutrients.36 The involvement of a knowledgeable dietitian is helpful, as physicians may lack sufficient training in dietary skills and knowledge of food composition.
Access to and cost of dietary counseling can be prohibitive for some patients. Group consultation, which can decrease costs to each patient, has been found to be as effective as one-on-one sessions when administering the low-FODMAP diet in functional bowel disorders.37
ELIMINATION, THEN REINTRODUCTION
Before embarking on the low-FODMAP diet, the patient’s interest in making dietary changes should be explored, a dietary history taken, and unusual food choices or dietary behaviors assessed. The patient’s ability to adopt a restricted diet should also be gauged.
The diet should be implemented in two phases. The initial phase involves strict elimination of foods high in FODMAPs, usually over 6 to 8 weeks.38 Symptom control should be assessed: failure to control symptoms requires assessment of adherence.
If symptoms are successfully controlled, then the second phase should begin with the aim of following a less-restricted version of the diet as tolerated. Foods should gradually be phased back in and symptoms monitored. This approach minimizes unnecessary dietary restriction and ensures that a maximum variety in the diet is achieved while maintaining adequate symptom control.39
LOW-FODMAP DIET ALTERS THE GUT MICROBIOTA
Multiple putative benefits of certain bacterial species for colonic health have been reported, including the production of short-chain fatty acids. Colonic luminal concentrations of short-chain fatty acids may be important to gut health, given their role in intestinal secretion, absorption, motility, and epithelial cell structure. Because short-chain fatty acids are products of bacterial fermentation, a change in the delivery of fermentable substrates to the colon would be expected to alter the concentrations and output of fecal short-chain fatty acids.18
Several studies evaluated the effect of the low-FODMAP diet on intestinal microbiota, finding a change in the bacterial profile in the stool of patients who adopt this diet. Staudacher et al28 found a marked reduction in luminal bifidobacteria concentration after 4 weeks of a low-FODMAP diet in patients with IBS.
A single-blind randomized crossover trial40 investigated the effects of a low-FODMAP diet vs a carefully matched typical Australian diet in 27 patients with IBS and 6 healthy controls. Marked differences in absolute and relative bacterial abundance and diversity were found between the diets, but not in short-chain fatty acids or gut transit time. Compared with fecal microbiota on the typical diet, low FODMAP intake was associated with reduced absolute abundance of bacteria, and the typical FODMAP diet had evidence of stimulation of the growth of bacterial groups with putative health benefits.
The authors concluded40 that the functional significance and health implications of such changes are reasons for caution when reducing FODMAP intake in the long term and recommended liberalizing FODMAP restriction to the level of adequate symptom control in IBS patients. The study also recommended that people without symptoms not go on the low-FODMAP diet.40
Molecular approaches to characterize the gut microbiota are also being explored in an effort to identify its association with diet.
The sustainability of changes in gut microbiota and the potential long-term impact on health of following a low-FODMAP diet require further evaluation. In the meantime, patients following this diet should have FODMAP foods reintroduced based on tolerance and should consider taking probiotic supplements.41
DIETARY ADEQUACY OF THE LOW-FODMAP DIET
Continual dietary counseling should minimize nutritional inadequacies and ensure that FODMAPS are restricted only enough to control symptoms. Because no single food group is completely eliminated in this diet, patients are unlikely to experience inadequate nutrition.
Ledochowski et al26 found that in the initial, strict phase of the diet, total intake of carbohydrates (eg, starches, sugars) was reduced but intake of total energy, protein, fat, and nonstarch polysaccharides was not affected. Calcium intake was reduced in those following a low-FODMAP diet for 4 weeks.
The diet can also reduce total fiber intake and subsequently worsen constipation-predominant IBS. For those patients, lightly fermented high-fiber alternatives like oat and rice bran can be used.
ACCUMULATING EVIDENCE
The low-FODMAP diet is accumulating quality evidence for its effectiveness in controlling the functional gastrointestinal symptoms in patients with IBS. It can be difficult to adhere to over the long term due to its restrictiveness, and it is important to gradually liberalize the diet while tailoring it to the individual patient and monitoring symptoms. Further clinical trials are needed to evaluate this diet in different IBS subtypes and other gastrointestinal disorders, while defining its nutritional adequacy and effects on the intestinal microbiota profile.
- Hayes P, Corish C, O’Mahony E, Quigley EM. A dietary survey of patients with irritable bowel syndrome. J Hum Nutr Diet 2014; 27(suppl 2):36–47.
- Pare P, Gray J, Lam S, et al. Health-related quality of life, work productivity, and health care resource utilization of subjects with irritable bowel syndrome: baseline results from LOGIC (Longitudinal Outcomes Study of Gastrointestinal Symptoms in Canada), a naturalistic study. Clin Ther 2006; 28:1726–1735; discussion 1710–1711.
- Lacey BE, Mearin F, Chang L, et al. Bowel disorders. Gastroenterology 2016; 150:1393–1407.
- Drossman DA, Camilleri M, Mayer EA, Whitehead WE. AGA technical review on irritable bowel syndrome. Gastroenterology 2002; 123:2108–2131.
- Floch MH, Narayan R. Diet in the irritable bowel syndrome. J Clin Gastroenterol 2002; 35(suppl 1):S45–S52.
- Reding KW, Cain KC, Jarrett ME, Eugenio MD, Heitkemper MM. Relationship between patterns of alcohol consumption and gastrointestinal symptoms among patients with irritable bowel syndrome. Am J Gastroenterol 2013; 108:270–276.
- Moayyedi P, Quigley EM, Lacy BE, et al. The effect of fiber supplementation on irritable bowel syndrome: a systematic review and meta-analysis. Am J Gastroenterol 2014; 109:1367–1374.
- Vazquez Roque MI, Camilleri M, Smyrk T, et al. A controlled trial of gluten-free diet in patients with irritable bowel syndrome-diarrhea: effects on bowel frequency and intestinal function. Gastroenterology 2013; 144:903–911.
- Biesiekierski JR, Newnham ED, Irving PM, et al. Gluten causes gastrointestinal symptoms in subjects without celiac disease: a double-blind randomized placebo-controlled trial. Am J Gastroenterol 2011; 106:508–515.
- Biesiekierski JR, Peters SL, Newnham ED, Rosella O, Muir JG, Gibson PR. No effects of gluten in patients with self-reported non-celiac gluten sensitivity after dietary reduction of fermentable, poorly absorbed, short-chain carbohydrates. Gastroenterology 2013; 145:320–328.
- Ford AC, Quigley EM, Lacy BE, et al. Efficacy of prebiotics, probiotics, and synbiotics in irritable bowel syndrome and chronic idiopathic constipation: systematic review and meta-analysis. Gastroenterology 2013; 145:320–328.e1–e3.
- Central Clinical School, Monash University and The Alfred Hospital. The Monash University Low FODMAP Diet. 4th ed. Melbourne, Australia: Monash University; 2012.
- Parker K, Salas M, Nwosu VC. High fructose corn syrup: production, uses and public health concerns. Biotechnol Mol Biol Rev 2010; 5:71–78.
- Clausen MR, Jorgensen J, Mortensen PB. Comparison of diarrhea induced by ingestion of fructooligosaccharide idolax and disaccharide lactulose: role of osmolarity versus fermentation of malabsorbed carbohydrate. Dig Dis Sci 1998; 43:2696–2707.
- Barrett JS, Gearry RB, Muir JG, et al. Dietary poorly absorbed, short-chain carbohydrates increase delivery of water and fermentable substrates to the proximal colon. Aliment Pharmacol Ther 2010; 31:874–882.
- Whelan K, Abrahmsohn O, David GJ, et al. Fructan content of commonly consumed wheat, rye and gluten-free breads. Int J Food Sci Nutr 2011; 62:498–503.
- Sangwan V, Tomar SK, Singh RR, Singh AK, Ali B. Galactooligosaccharides: novel components of designer foods. J Food Sci 2011; 76:R103–R111.
- Russell DA, Ross RP, Fitzgerald GF, Stanton C. Metabolic activities and probiotic potential of bifidobacteria. Int J Food Microbiol 2011; 149:88–105.
- Lomer MC, Parkes GC, Sanderson JD. Review article: lactose intolerance in clinical practice—myths and realities. Aliment Pharmacol Ther 2008; 27:93–103.
- Langkilde AM, Andersson H, Schweizer TF, Würsch P. Digestion and absorption of sorbitol, maltitol and isomalt from the small bowel. A study in ileostomy subjects. Eur J Clin Nutr 1994; 48:768–775.
- Muir JG, Rose R, Rosella O, et al. Measurement of short-chain carbohydrates in common Australian vegetables and fruits by high-performance liquid chromatography (HPLC). J Agric Food Chem 2009; 57:554–565.
- Muir JG, Shepherd SJ, Rosella O, Rose R, Barrett JS, Gibson PR. Fructan and free fructose content of common Australian vegetables and fruit. J Agric Food Chem 2007; 55:6619–6627.
- Monash University. Monash launches Low FODMAP Diet smartphone app. http://med.monash.edu.au/news/2012/fodmap-app.html. Accessed July 13, 2016.
- Fedewa A, Rao SS. Dietary fructose intolerance, fructan intolerance and FODMAPs. Curr Gastroenterol Rep 2014; 16:370.
- Born P, Vierling T, Barina W. Fructose malabsorption and the irritable bowel syndrome. Gastroenterology 1991; 101:1454.
- Ledochowski M, Widner B, Bair H, Probst T, Fuchs D. Fructose- and sorbitol-reduced diet improves mood and gastrointestinal disturbances in fructose malabsorbers. Scand J Gastroenterol 2000; 35:1048–1052.
- Shepherd SJ, Gibson PR. Fructose malabsorption and symptoms of irritable bowel syndrome: guidelines for effective dietary management. J Am Diet Assoc 2006; 106:1631–1639.
- Staudacher HM, Lomer MC, Anderson JL, et al. Fermentable carbohydrate restriction reduces luminal bifidobacteria and gastrointestinal symptoms in patients with irritable bowel syndrome. J Nutr 2012; 142:1510–1518.
- Halmos EP, Power VA, Shepherd SJ, Gibson PR, Muir JG. A diet low in FODMAPs reduces symptoms of irritable bowel syndrome. Gastroenterology 2014; 146:67–75.e5.
- Shepherd SJ, Parker FC, Muir JG, Gibson PR. Dietary triggers of abdominal symptoms in patients with irritable bowel syndrome: randomized placebo-controlled evidence. Clin Gastroenterol Hepatol 2008; 6:765–771.
- Murray K, Wilkinson-Smith V, Hoad C, et al. Differential effects of FODMAPs (fermentable oligo-, di-, mono-saccharides and polyols) on small and large intestinal contents in healthy subjects shown by MRI. Am J Gastroenterol 2014; 109:110–119.
- Pedersen N, Andersen NN, Vegh Z, et al. Ehealth: low FODMAP diet vs Lactobacillus rhamnosus GG in irritable bowel syndrome. World J Gastroenterol 2014; 20:16215–16226.
- Gearry RB, Irving PM, Barrett JS, Nathan DM, Shepherd SJ, Gibson PR. Reduction of dietary poorly absorbed short-chain carbohydrates (FODMAPs) improves abdominal symptoms in patients with inflammatory bowel disease-a pilot study. J Crohns Colitis 2009; 3:8–14.
- Marsh A, Eslick EM, Eslick GD. Does a diet low in FODMAPs reduce symptoms associated with functional gastrointestinal disorders? A comprehensive systematic review and meta-analysis. Eur J Nutr 2015 May 17. Epub ahead of print.
- de Roest RH, Dobbs BR, Chapman BA, et al. The low FODMAP diet improves gastrointestinal symptoms in patients with irritable bowel syndrome: a prospective study. Int J Clin Pract 2013; 67:895–903.
- Gibson PR, Barrett JS, Muir JG. Functional bowel symptoms and diet. Intern Med J 2013; 43:1067–1074.
- Whigham L, Joyce T, Harper G, et al. Clinical effectiveness and economic costs of group versus one-to-one education for short-chain fermentable carbohydrate restriction (low FODMAP diet) in the management of irritable bowel syndrome. J Hum Nutr Diet 2015; 28:687–696.
- Shepherd SJ, Lomer MC, Gibson PR. Short-chain carbohydrates and functional gastrointestinal disorders. Am J Gastroenterol 2013; 108:707–717.
- Shepherd SJ, Halmos E, Glance S. The role of FODMAPs in irritable bowel syndrome. Curr Opin Clin Nutr Metab Care 2014; 17:605–609.
- Halmos EP, Christophersen CT, Bird AR, Shepherd SJ, Gibson PR, Muir JG. Diets that differ in their FODMAP content alter the colonic luminal microenvironment. Gut 2015; 64:93–100.
- Staudacher HM, Irving PM, Lomer MC, Whelan K. Mechanisms and efficacy of dietary FODMAP restriction in IBS. Nat Rev Gastroenterol Hepatol 2014; 11:256–266.
The role of diet in controlling symptoms of irritable bowel syndrome (IBS) has gained much traction over the years,1 but until recently, diet therapy for IBS has been hindered by a lack of quality evidence, in part because of the challenges of conducting dietary clinical trials.
Several clinical trials have now been done that support a diet low in fermentable oligosaccharides, disaccharides, monosaccharides, and polyols (FODMAPs) for managing IBS. Although restrictive and difficult to follow, the low-FODMAP diet is gaining popularity.
This article provides an overview of dietary interventions used to manage IBS, focusing on the low-FODMAP diet. We discuss mechanisms of malabsorption of FODMAPs and the role of FODMAPS in symptom induction; highlight clinical trials that provide evidence of benefits of the diet for IBS; and discuss the steps to implement it. We also address the nutritional adequacy of the diet and its potential effects on the gut microbiome.
IBS IS A COMMON FUNCTIONAL DISORDER
IBS is one of the most commonly diagnosed gastrointestinal disorders, and it has a significant impact on quality of life.2 It is a functional disorder characterized by chronic abdominal pain and altered bowel habits in the absence of a structural or organic cause.
The Rome IV diagnostic criteria define IBS by the following:
- Recurrent abdominal pain or discomfort at least 1 day a week in the last 3 months, associated with two or more of the following:
- Symptoms improved by defecation
- Onset associated with a change in frequency of stool
- Onset associated with a change in form or appearance of stool.
IBS mainly arises during young adulthood but can be diagnosed at any age.3
The pathophysiology of IBS involves mechanisms such as bowel distention, altered bowel motility, visceral hypersensitivity, and disruption of mucosal permeability.4 Several therapeutic modalities targeting these mechanisms have been implemented in IBS management, including antispasmodics, laxatives, antidepressants, antibiotics, and behavioral therapy. Diet is only one line of treatment and is most effective when part of a multipronged approach.
TRADITIONAL DIETARY MANAGEMENT
Diet is important in inducing the symptoms of IBS—and in controlling them. Patients identify eating as a common precipitator of symptoms, but the complex diet-symptom interaction is not fully understood and varies widely among patients. Traditional dietary advice for IBS includes adhering to a regular meal pattern, avoiding large meals, and reducing intake of fat, insoluble fibers, caffeine, spicy and gas-producing foods, and carbonated beverages.5,6
Increase soluble fiber
Fiber and fiber supplements, particularly soluble fibers such as psyllium, calcium polycarbophil, and ispaghula husk are often recommended. A meta-analysis7 found that soluble fiber but not insoluble fiber (eg, wheat bran) is associated with an improvement in IBS symptoms (relative risk [RR] 0.84, 95% confidence interval [CI] 0.73–0.94). By improving stool consistency and accelerating transit, soluble fiber is especially useful in constipation-predominant IBS while posing a low risk for adverse outcomes.7 Fiber should be started at a low dose and gradually increased over several weeks to as much as 20 to 30 g/day.
Avoid wheat
Only about 4% of patients with IBS also have celiac disease, but estimating the prevalence of nonceliac gluten sensitivity is confounded by overlapping symptoms. There is some evidence implicating gluten in IBS: celiac disease and IBS overlap in their symptoms, and symptoms are often precipitated by gluten-containing foods in patients with IBS.8 The pathogenesis of gluten-induced (or wheat-induced) symptoms in IBS is unclear, and studies have had conflicting results as to the benefits of gluten restriction in IBS.9
In a study of patients with IBS whose symptoms improved when they started a gluten-free and low-FODMAP diet, symptoms did not return when gluten was reintroduced, suggesting that it is the fructan (a FODMAP) component of wheat rather than gluten that contributes to symptoms in IBS.10
Probiotics
Probiotics are increasingly being recommended as dietary supplements for people with IBS, as awareness increases of the importance of the gut microbiota. In addition to their effects on the gut microbiota, probiotics in IBS have been shown to have anti-inflammatory effects, to alter gut motility, to modulate visceral hypersensitivity, and to restore epithelial integrity.
In a meta-analysis, Ford et al11 found that probiotics improved global IBS symptoms more than placebo (RR 0.79, 95% CI 0.70–0.89) and also reduced abdominal pain, bloating, and flatulence scores.
Which species and strains are most beneficial and the optimal dosing and duration of treatment are still unclear. Data from studies of prebiotics (nutrients that encourage the growth of probiotic bacteria) and synbiotics (combinations of prebiotics and probiotics) are limited and insufficient to draw conclusions.
FODMAPS ARE SHORT-CHAIN CARBOHYDRATES
The term FODMAPs was initially coined by researchers at Monash University in Australia to describe a collection of poorly absorbed short-chain fermentable carbohydrates that are natural components of many foods:
- Oligosaccharides, including fructans (which include inulins) and galacto-oligosaccharides
- Disaccharides, including lactose and sucrose
- Monosaccharides, including fructose
- Polyols, including sorbitol and mannitol.12
Intake of FODMAPs, especially fructose, has increased in Western diets over the past several decades from increased consumption of fruits and concentrated fruit juices, as well as from the widespread use of high-fructose corn syrup in processed foods and beverages.13
FODMAPs ARE POORLY ABSORBED
Different FODMAPs can be poorly absorbed for different reasons (Table 1). The poor absorption is related either to reduced or absent digestive enzymes (ie, hydrolases) or to slow transport across the intestinal mucosa. Excess FODMAPs in the distal small intestine and proximal colon exert osmotic pressure, drawing more water into the lumen. FODMAPs are also rapidly fermented by colonic bacteria, producing gas, bowel distention, and altered motility, all of which induce IBS symptoms.14
Fructans are fructose polymers that are not absorbed in human intestines. They have no intestinal hydrolases and no mechanisms for direct transport across the epithelium. However, a negligible amount may be absorbed after being degraded by microbes in the gut.15 Most dietary fructans are obtained from wheat and onion, which are actually low in fructans but tend to be consumed in large quantities.16
Galacto-oligosaccharides are available for colonic fermentation after ingestion due to lack of a human alpha-galactosidase. Common sources of galacto-oligosaccharides include legumes, nuts, seeds, some grains, dairy products, human milk, and some commercially produced forms added to infant formula.17,18
Lactose is poorly absorbed in people with lactase deficiency. It is mainly present in dairy products but is also added to commercial foods, including breads, cakes, and some diet products.19
Fructose is the most abundant FODMAP in the Western diet. It is either present as a free sugar or generated from the digestive breakdown of sucrose. In the intestine, it is absorbed via a direct low-capacity glucose transporter (GLUT)-5 and through GLUT-2, which is more efficient but requires the coexistence of glucose. Because of this requirement, fructose is more likely to be malabsorbed when present in excess of glucose, as in people with diminished sucrase activity. The main sources of fructose in the Western diet are fruits and fruit products, honey, and foods with added high-fructose sweeteners.13
Polyols such as sorbitol and mannitol are absorbed by slow passive diffusion because they have no active intestinal transport system. They are found in fruits and vegetables. Sugar-free chewing gum is a particularly rich source of sorbitol.20
QUANTIFYING FODMAP CONTENT
As interest in the low-FODMAP diet grew, studies were conducted to quantify FODMAPs in foods. One study used high-performance liquid chromatography to analyze FODMAP content in foods,21 and another evaluated fructan levels in a variety of fruits and vegetables using enzymatic hydrolysis.22 The Monash University low-FODMAP diet smartphone application provides patients and healthcare providers easy access to updated and detailed food analyses.23
Table 2 lists foods high in FODMAPs along with low-FODMAP alternatives. Total FODMAP intake is important, as the effects are additive.24 Readers and patients can be directed to the following websites for more information on the low-FODMAP diet: www.med.monash.edu/cecs/gastro/fodmap or www.ibsfree.net/what-is-fodmap-diet.
LOW-FODMAP DIET REDUCES SYMPTOMS
The low-FODMAP diet was inspired by the results of several studies that evaluated the role of dietary carbohydrates in inducing IBS symptoms and found improvement with their restriction.25,26
One study found that 74% of patients with IBS had less bloating, nausea, abdominal pain, and diarrhea when they restricted their intake of fructose and fructans.27
A prospective trial randomized 41 patients with IBS to 4 weeks of either a low-FODMAP diet or their habitual diet.28 The low-FODMAP diet resulted in greater improvement in overall IBS symptoms (P < .05) and stool frequency (P = .008). This study was limited by different habitual diets between patients and by lack of standardization of the low-FODMAP diet.
Halmos et al,29 in a randomized crossover trial, compared gastrointestinal symptoms in IBS patients over 3 weeks on a low-FODMAP diet vs a moderate-FODMAP (ie, regular) diet, as well as in healthy controls. Food was provided by the study and was matched for all nutrients. Up to 70% of the IBS patients had significantly lower overall symptom scores while on a low-FODMAP diet vs IBS patients on a regular diet (P < .001); bloating, abdominal pain, and flatulence were reduced. Symptoms were minimal and unaffected by either diet in the healthy controls.
A double-blind trial30 randomly assigned 25 patients with IBS who initially responded to a low-FODMAP diet to be challenged by a graduated dose of fructose alone, fructans alone, a combination of both, or glucose. The severity of overall and individual symptoms was markedly more reduced with glucose consumption than with the other carbohydrates: 70% of patients receiving fructose, 77% of those receiving fructans, and 79% of those receiving a mixture of both reported that their symptoms were not adequately controlled, compared with 14% of patients receiving glucose (P ≤ .002).30
Murray et al31 evaluated the gastrointestinal tract after a carbohydrate challenge consisting of 0.5 L of water containing 40 g of glucose, fructose, or inulin or a combination of 40 g of glucose and 40 g of fructose in 16 healthy volunteers. Magnetic resonance imaging was performed hourly for 5 hours to assess the volume of gastric contents, small-bowel water content, and colonic gas. Breath hydrogen was also measured, and symptoms were recorded after each imaging session.
Fructose significantly increased small-bowel water content compared with glucose (mean difference 28 L/min, P < .001), but combined glucose and fructose lessened the effect. Inulin had no significant effect on small-bowel water content (mean difference with glucose 2 L/min, P > .7) but led to the greatest production of colonic gas compared with glucose alone (mean difference 15 L/min, P < .05) and combined glucose and fructose (mean difference 12 L/min, P < .05). Inulin also produced the most breath hydrogen: 81% of participants had a rise after drinking inulin compared with 50% after drinking fructose. Glucose did not affect breath hydrogen concentrations, and combined glucose and fructose significantly reduced the concentration measured vs fructose alone. In patients who reported “gas” symptoms, a correlation was observed between the volume of gas in the colon and gas symptoms (r = 0.59, P < .0001).31
The authors concluded31 that long-chain carbohydrates such as inulin have a greater effect on colonic gas production and little effect on small-bowel water content, whereas small-chain FODMAPs such as fructose are likely to cause luminal distention in both the small and large intestines. The study also showed that combining equal amounts of glucose and fructose reduces malabsorption of fructose in the small bowel and reduces the effect of fructose on small-bowel water content and breath hydrogen concentration.31
PROBIOTICS HELP
A Danish study32 randomized 123 patients with IBS to one of three treatments: a low-FODMAP diet, a normal diet with probiotics containing the strain Lactobacillus rhamnosus GG (two capsules daily), or no special intervention. Symptoms were recorded weekly. IBS severity scores at week 6 were lower in patients on either the low-FODMAP diet or probiotics compared with the control group (P < .01). Subgroup analysis determined that patients with primarily diarrheal symptoms were more likely to have improved quality of life with the low-FODMAP diet.
A LOW-FODMAP DIET MAY ALSO HELP IN INFLAMMATORY BOWEL DISEASE
The low-FODMAP diet has also been studied in patients with inflammatory bowel disease with functional gut symptoms. In a retrospective pilot study,33 overall symptoms improved in about half of such patients on a low-FODMAP diet. A controlled dietary intervention trial is needed to confirm these findings and define the role of the low-FODMAP approach for patients with inflammatory bowel disease.
Marsh et al34 performed a meta-analysis of six randomized clinical trials and 16 nonrandomized interventions of a low-FODMAP diet on improving functional gastrointestinal symptoms in patients with either IBS or inflammatory bowel disease. They found significant improvements in:
- IBS Symptoms Severity Scores in the randomized trials (odds ratio [OR] 0.44, 95% CI 0.25–0.76)
- IBS Symptoms Severity Scores in the nonrandomized interventions (OR 0.03, 95% CI 0.01–0.2)
- IBS Quality of Life scores in the randomized trials (OR 1.84, 95% CI 1.12–3.03)
- IBS Quality of Life scores in the nonrandomized interventions (OR 3.18, 95% CI 1.6–6.31)
- Overall symptom severity in the randomized trials (OR 1.81, 95% CI 1.11–2.95).
DIETARY COUNSELING IS RECOMMENDED
Adherence is a major factor in the success of the low-FODMAP diet in IBS management and is strongly correlated with improved symptoms.35 Patients should be counseled on the role of food in inducing their symptoms. Haphazard dietary advice can be detrimental to outcomes, as many diets restrict food groups, impairing the consumption of essential nutrients.36 The involvement of a knowledgeable dietitian is helpful, as physicians may lack sufficient training in dietary skills and knowledge of food composition.
Access to and cost of dietary counseling can be prohibitive for some patients. Group consultation, which can decrease costs to each patient, has been found to be as effective as one-on-one sessions when administering the low-FODMAP diet in functional bowel disorders.37
ELIMINATION, THEN REINTRODUCTION
Before embarking on the low-FODMAP diet, the patient’s interest in making dietary changes should be explored, a dietary history taken, and unusual food choices or dietary behaviors assessed. The patient’s ability to adopt a restricted diet should also be gauged.
The diet should be implemented in two phases. The initial phase involves strict elimination of foods high in FODMAPs, usually over 6 to 8 weeks.38 Symptom control should be assessed: failure to control symptoms requires assessment of adherence.
If symptoms are successfully controlled, then the second phase should begin with the aim of following a less-restricted version of the diet as tolerated. Foods should gradually be phased back in and symptoms monitored. This approach minimizes unnecessary dietary restriction and ensures that a maximum variety in the diet is achieved while maintaining adequate symptom control.39
LOW-FODMAP DIET ALTERS THE GUT MICROBIOTA
Multiple putative benefits of certain bacterial species for colonic health have been reported, including the production of short-chain fatty acids. Colonic luminal concentrations of short-chain fatty acids may be important to gut health, given their role in intestinal secretion, absorption, motility, and epithelial cell structure. Because short-chain fatty acids are products of bacterial fermentation, a change in the delivery of fermentable substrates to the colon would be expected to alter the concentrations and output of fecal short-chain fatty acids.18
Several studies evaluated the effect of the low-FODMAP diet on intestinal microbiota, finding a change in the bacterial profile in the stool of patients who adopt this diet. Staudacher et al28 found a marked reduction in luminal bifidobacteria concentration after 4 weeks of a low-FODMAP diet in patients with IBS.
A single-blind randomized crossover trial40 investigated the effects of a low-FODMAP diet vs a carefully matched typical Australian diet in 27 patients with IBS and 6 healthy controls. Marked differences in absolute and relative bacterial abundance and diversity were found between the diets, but not in short-chain fatty acids or gut transit time. Compared with fecal microbiota on the typical diet, low FODMAP intake was associated with reduced absolute abundance of bacteria, and the typical FODMAP diet had evidence of stimulation of the growth of bacterial groups with putative health benefits.
The authors concluded40 that the functional significance and health implications of such changes are reasons for caution when reducing FODMAP intake in the long term and recommended liberalizing FODMAP restriction to the level of adequate symptom control in IBS patients. The study also recommended that people without symptoms not go on the low-FODMAP diet.40
Molecular approaches to characterize the gut microbiota are also being explored in an effort to identify its association with diet.
The sustainability of changes in gut microbiota and the potential long-term impact on health of following a low-FODMAP diet require further evaluation. In the meantime, patients following this diet should have FODMAP foods reintroduced based on tolerance and should consider taking probiotic supplements.41
DIETARY ADEQUACY OF THE LOW-FODMAP DIET
Continual dietary counseling should minimize nutritional inadequacies and ensure that FODMAPS are restricted only enough to control symptoms. Because no single food group is completely eliminated in this diet, patients are unlikely to experience inadequate nutrition.
Ledochowski et al26 found that in the initial, strict phase of the diet, total intake of carbohydrates (eg, starches, sugars) was reduced but intake of total energy, protein, fat, and nonstarch polysaccharides was not affected. Calcium intake was reduced in those following a low-FODMAP diet for 4 weeks.
The diet can also reduce total fiber intake and subsequently worsen constipation-predominant IBS. For those patients, lightly fermented high-fiber alternatives like oat and rice bran can be used.
ACCUMULATING EVIDENCE
The low-FODMAP diet is accumulating quality evidence for its effectiveness in controlling the functional gastrointestinal symptoms in patients with IBS. It can be difficult to adhere to over the long term due to its restrictiveness, and it is important to gradually liberalize the diet while tailoring it to the individual patient and monitoring symptoms. Further clinical trials are needed to evaluate this diet in different IBS subtypes and other gastrointestinal disorders, while defining its nutritional adequacy and effects on the intestinal microbiota profile.
The role of diet in controlling symptoms of irritable bowel syndrome (IBS) has gained much traction over the years,1 but until recently, diet therapy for IBS has been hindered by a lack of quality evidence, in part because of the challenges of conducting dietary clinical trials.
Several clinical trials have now been done that support a diet low in fermentable oligosaccharides, disaccharides, monosaccharides, and polyols (FODMAPs) for managing IBS. Although restrictive and difficult to follow, the low-FODMAP diet is gaining popularity.
This article provides an overview of dietary interventions used to manage IBS, focusing on the low-FODMAP diet. We discuss mechanisms of malabsorption of FODMAPs and the role of FODMAPS in symptom induction; highlight clinical trials that provide evidence of benefits of the diet for IBS; and discuss the steps to implement it. We also address the nutritional adequacy of the diet and its potential effects on the gut microbiome.
IBS IS A COMMON FUNCTIONAL DISORDER
IBS is one of the most commonly diagnosed gastrointestinal disorders, and it has a significant impact on quality of life.2 It is a functional disorder characterized by chronic abdominal pain and altered bowel habits in the absence of a structural or organic cause.
The Rome IV diagnostic criteria define IBS by the following:
- Recurrent abdominal pain or discomfort at least 1 day a week in the last 3 months, associated with two or more of the following:
- Symptoms improved by defecation
- Onset associated with a change in frequency of stool
- Onset associated with a change in form or appearance of stool.
IBS mainly arises during young adulthood but can be diagnosed at any age.3
The pathophysiology of IBS involves mechanisms such as bowel distention, altered bowel motility, visceral hypersensitivity, and disruption of mucosal permeability.4 Several therapeutic modalities targeting these mechanisms have been implemented in IBS management, including antispasmodics, laxatives, antidepressants, antibiotics, and behavioral therapy. Diet is only one line of treatment and is most effective when part of a multipronged approach.
TRADITIONAL DIETARY MANAGEMENT
Diet is important in inducing the symptoms of IBS—and in controlling them. Patients identify eating as a common precipitator of symptoms, but the complex diet-symptom interaction is not fully understood and varies widely among patients. Traditional dietary advice for IBS includes adhering to a regular meal pattern, avoiding large meals, and reducing intake of fat, insoluble fibers, caffeine, spicy and gas-producing foods, and carbonated beverages.5,6
Increase soluble fiber
Fiber and fiber supplements, particularly soluble fibers such as psyllium, calcium polycarbophil, and ispaghula husk are often recommended. A meta-analysis7 found that soluble fiber but not insoluble fiber (eg, wheat bran) is associated with an improvement in IBS symptoms (relative risk [RR] 0.84, 95% confidence interval [CI] 0.73–0.94). By improving stool consistency and accelerating transit, soluble fiber is especially useful in constipation-predominant IBS while posing a low risk for adverse outcomes.7 Fiber should be started at a low dose and gradually increased over several weeks to as much as 20 to 30 g/day.
Avoid wheat
Only about 4% of patients with IBS also have celiac disease, but estimating the prevalence of nonceliac gluten sensitivity is confounded by overlapping symptoms. There is some evidence implicating gluten in IBS: celiac disease and IBS overlap in their symptoms, and symptoms are often precipitated by gluten-containing foods in patients with IBS.8 The pathogenesis of gluten-induced (or wheat-induced) symptoms in IBS is unclear, and studies have had conflicting results as to the benefits of gluten restriction in IBS.9
In a study of patients with IBS whose symptoms improved when they started a gluten-free and low-FODMAP diet, symptoms did not return when gluten was reintroduced, suggesting that it is the fructan (a FODMAP) component of wheat rather than gluten that contributes to symptoms in IBS.10
Probiotics
Probiotics are increasingly being recommended as dietary supplements for people with IBS, as awareness increases of the importance of the gut microbiota. In addition to their effects on the gut microbiota, probiotics in IBS have been shown to have anti-inflammatory effects, to alter gut motility, to modulate visceral hypersensitivity, and to restore epithelial integrity.
In a meta-analysis, Ford et al11 found that probiotics improved global IBS symptoms more than placebo (RR 0.79, 95% CI 0.70–0.89) and also reduced abdominal pain, bloating, and flatulence scores.
Which species and strains are most beneficial and the optimal dosing and duration of treatment are still unclear. Data from studies of prebiotics (nutrients that encourage the growth of probiotic bacteria) and synbiotics (combinations of prebiotics and probiotics) are limited and insufficient to draw conclusions.
FODMAPS ARE SHORT-CHAIN CARBOHYDRATES
The term FODMAPs was initially coined by researchers at Monash University in Australia to describe a collection of poorly absorbed short-chain fermentable carbohydrates that are natural components of many foods:
- Oligosaccharides, including fructans (which include inulins) and galacto-oligosaccharides
- Disaccharides, including lactose and sucrose
- Monosaccharides, including fructose
- Polyols, including sorbitol and mannitol.12
Intake of FODMAPs, especially fructose, has increased in Western diets over the past several decades from increased consumption of fruits and concentrated fruit juices, as well as from the widespread use of high-fructose corn syrup in processed foods and beverages.13
FODMAPs ARE POORLY ABSORBED
Different FODMAPs can be poorly absorbed for different reasons (Table 1). The poor absorption is related either to reduced or absent digestive enzymes (ie, hydrolases) or to slow transport across the intestinal mucosa. Excess FODMAPs in the distal small intestine and proximal colon exert osmotic pressure, drawing more water into the lumen. FODMAPs are also rapidly fermented by colonic bacteria, producing gas, bowel distention, and altered motility, all of which induce IBS symptoms.14
Fructans are fructose polymers that are not absorbed in human intestines. They have no intestinal hydrolases and no mechanisms for direct transport across the epithelium. However, a negligible amount may be absorbed after being degraded by microbes in the gut.15 Most dietary fructans are obtained from wheat and onion, which are actually low in fructans but tend to be consumed in large quantities.16
Galacto-oligosaccharides are available for colonic fermentation after ingestion due to lack of a human alpha-galactosidase. Common sources of galacto-oligosaccharides include legumes, nuts, seeds, some grains, dairy products, human milk, and some commercially produced forms added to infant formula.17,18
Lactose is poorly absorbed in people with lactase deficiency. It is mainly present in dairy products but is also added to commercial foods, including breads, cakes, and some diet products.19
Fructose is the most abundant FODMAP in the Western diet. It is either present as a free sugar or generated from the digestive breakdown of sucrose. In the intestine, it is absorbed via a direct low-capacity glucose transporter (GLUT)-5 and through GLUT-2, which is more efficient but requires the coexistence of glucose. Because of this requirement, fructose is more likely to be malabsorbed when present in excess of glucose, as in people with diminished sucrase activity. The main sources of fructose in the Western diet are fruits and fruit products, honey, and foods with added high-fructose sweeteners.13
Polyols such as sorbitol and mannitol are absorbed by slow passive diffusion because they have no active intestinal transport system. They are found in fruits and vegetables. Sugar-free chewing gum is a particularly rich source of sorbitol.20
QUANTIFYING FODMAP CONTENT
As interest in the low-FODMAP diet grew, studies were conducted to quantify FODMAPs in foods. One study used high-performance liquid chromatography to analyze FODMAP content in foods,21 and another evaluated fructan levels in a variety of fruits and vegetables using enzymatic hydrolysis.22 The Monash University low-FODMAP diet smartphone application provides patients and healthcare providers easy access to updated and detailed food analyses.23
Table 2 lists foods high in FODMAPs along with low-FODMAP alternatives. Total FODMAP intake is important, as the effects are additive.24 Readers and patients can be directed to the following websites for more information on the low-FODMAP diet: www.med.monash.edu/cecs/gastro/fodmap or www.ibsfree.net/what-is-fodmap-diet.
LOW-FODMAP DIET REDUCES SYMPTOMS
The low-FODMAP diet was inspired by the results of several studies that evaluated the role of dietary carbohydrates in inducing IBS symptoms and found improvement with their restriction.25,26
One study found that 74% of patients with IBS had less bloating, nausea, abdominal pain, and diarrhea when they restricted their intake of fructose and fructans.27
A prospective trial randomized 41 patients with IBS to 4 weeks of either a low-FODMAP diet or their habitual diet.28 The low-FODMAP diet resulted in greater improvement in overall IBS symptoms (P < .05) and stool frequency (P = .008). This study was limited by different habitual diets between patients and by lack of standardization of the low-FODMAP diet.
Halmos et al,29 in a randomized crossover trial, compared gastrointestinal symptoms in IBS patients over 3 weeks on a low-FODMAP diet vs a moderate-FODMAP (ie, regular) diet, as well as in healthy controls. Food was provided by the study and was matched for all nutrients. Up to 70% of the IBS patients had significantly lower overall symptom scores while on a low-FODMAP diet vs IBS patients on a regular diet (P < .001); bloating, abdominal pain, and flatulence were reduced. Symptoms were minimal and unaffected by either diet in the healthy controls.
A double-blind trial30 randomly assigned 25 patients with IBS who initially responded to a low-FODMAP diet to be challenged by a graduated dose of fructose alone, fructans alone, a combination of both, or glucose. The severity of overall and individual symptoms was markedly more reduced with glucose consumption than with the other carbohydrates: 70% of patients receiving fructose, 77% of those receiving fructans, and 79% of those receiving a mixture of both reported that their symptoms were not adequately controlled, compared with 14% of patients receiving glucose (P ≤ .002).30
Murray et al31 evaluated the gastrointestinal tract after a carbohydrate challenge consisting of 0.5 L of water containing 40 g of glucose, fructose, or inulin or a combination of 40 g of glucose and 40 g of fructose in 16 healthy volunteers. Magnetic resonance imaging was performed hourly for 5 hours to assess the volume of gastric contents, small-bowel water content, and colonic gas. Breath hydrogen was also measured, and symptoms were recorded after each imaging session.
Fructose significantly increased small-bowel water content compared with glucose (mean difference 28 L/min, P < .001), but combined glucose and fructose lessened the effect. Inulin had no significant effect on small-bowel water content (mean difference with glucose 2 L/min, P > .7) but led to the greatest production of colonic gas compared with glucose alone (mean difference 15 L/min, P < .05) and combined glucose and fructose (mean difference 12 L/min, P < .05). Inulin also produced the most breath hydrogen: 81% of participants had a rise after drinking inulin compared with 50% after drinking fructose. Glucose did not affect breath hydrogen concentrations, and combined glucose and fructose significantly reduced the concentration measured vs fructose alone. In patients who reported “gas” symptoms, a correlation was observed between the volume of gas in the colon and gas symptoms (r = 0.59, P < .0001).31
The authors concluded31 that long-chain carbohydrates such as inulin have a greater effect on colonic gas production and little effect on small-bowel water content, whereas small-chain FODMAPs such as fructose are likely to cause luminal distention in both the small and large intestines. The study also showed that combining equal amounts of glucose and fructose reduces malabsorption of fructose in the small bowel and reduces the effect of fructose on small-bowel water content and breath hydrogen concentration.31
PROBIOTICS HELP
A Danish study32 randomized 123 patients with IBS to one of three treatments: a low-FODMAP diet, a normal diet with probiotics containing the strain Lactobacillus rhamnosus GG (two capsules daily), or no special intervention. Symptoms were recorded weekly. IBS severity scores at week 6 were lower in patients on either the low-FODMAP diet or probiotics compared with the control group (P < .01). Subgroup analysis determined that patients with primarily diarrheal symptoms were more likely to have improved quality of life with the low-FODMAP diet.
A LOW-FODMAP DIET MAY ALSO HELP IN INFLAMMATORY BOWEL DISEASE
The low-FODMAP diet has also been studied in patients with inflammatory bowel disease with functional gut symptoms. In a retrospective pilot study,33 overall symptoms improved in about half of such patients on a low-FODMAP diet. A controlled dietary intervention trial is needed to confirm these findings and define the role of the low-FODMAP approach for patients with inflammatory bowel disease.
Marsh et al34 performed a meta-analysis of six randomized clinical trials and 16 nonrandomized interventions of a low-FODMAP diet on improving functional gastrointestinal symptoms in patients with either IBS or inflammatory bowel disease. They found significant improvements in:
- IBS Symptoms Severity Scores in the randomized trials (odds ratio [OR] 0.44, 95% CI 0.25–0.76)
- IBS Symptoms Severity Scores in the nonrandomized interventions (OR 0.03, 95% CI 0.01–0.2)
- IBS Quality of Life scores in the randomized trials (OR 1.84, 95% CI 1.12–3.03)
- IBS Quality of Life scores in the nonrandomized interventions (OR 3.18, 95% CI 1.6–6.31)
- Overall symptom severity in the randomized trials (OR 1.81, 95% CI 1.11–2.95).
DIETARY COUNSELING IS RECOMMENDED
Adherence is a major factor in the success of the low-FODMAP diet in IBS management and is strongly correlated with improved symptoms.35 Patients should be counseled on the role of food in inducing their symptoms. Haphazard dietary advice can be detrimental to outcomes, as many diets restrict food groups, impairing the consumption of essential nutrients.36 The involvement of a knowledgeable dietitian is helpful, as physicians may lack sufficient training in dietary skills and knowledge of food composition.
Access to and cost of dietary counseling can be prohibitive for some patients. Group consultation, which can decrease costs to each patient, has been found to be as effective as one-on-one sessions when administering the low-FODMAP diet in functional bowel disorders.37
ELIMINATION, THEN REINTRODUCTION
Before embarking on the low-FODMAP diet, the patient’s interest in making dietary changes should be explored, a dietary history taken, and unusual food choices or dietary behaviors assessed. The patient’s ability to adopt a restricted diet should also be gauged.
The diet should be implemented in two phases. The initial phase involves strict elimination of foods high in FODMAPs, usually over 6 to 8 weeks.38 Symptom control should be assessed: failure to control symptoms requires assessment of adherence.
If symptoms are successfully controlled, then the second phase should begin with the aim of following a less-restricted version of the diet as tolerated. Foods should gradually be phased back in and symptoms monitored. This approach minimizes unnecessary dietary restriction and ensures that a maximum variety in the diet is achieved while maintaining adequate symptom control.39
LOW-FODMAP DIET ALTERS THE GUT MICROBIOTA
Multiple putative benefits of certain bacterial species for colonic health have been reported, including the production of short-chain fatty acids. Colonic luminal concentrations of short-chain fatty acids may be important to gut health, given their role in intestinal secretion, absorption, motility, and epithelial cell structure. Because short-chain fatty acids are products of bacterial fermentation, a change in the delivery of fermentable substrates to the colon would be expected to alter the concentrations and output of fecal short-chain fatty acids.18
Several studies evaluated the effect of the low-FODMAP diet on intestinal microbiota, finding a change in the bacterial profile in the stool of patients who adopt this diet. Staudacher et al28 found a marked reduction in luminal bifidobacteria concentration after 4 weeks of a low-FODMAP diet in patients with IBS.
A single-blind randomized crossover trial40 investigated the effects of a low-FODMAP diet vs a carefully matched typical Australian diet in 27 patients with IBS and 6 healthy controls. Marked differences in absolute and relative bacterial abundance and diversity were found between the diets, but not in short-chain fatty acids or gut transit time. Compared with fecal microbiota on the typical diet, low FODMAP intake was associated with reduced absolute abundance of bacteria, and the typical FODMAP diet had evidence of stimulation of the growth of bacterial groups with putative health benefits.
The authors concluded40 that the functional significance and health implications of such changes are reasons for caution when reducing FODMAP intake in the long term and recommended liberalizing FODMAP restriction to the level of adequate symptom control in IBS patients. The study also recommended that people without symptoms not go on the low-FODMAP diet.40
Molecular approaches to characterize the gut microbiota are also being explored in an effort to identify its association with diet.
The sustainability of changes in gut microbiota and the potential long-term impact on health of following a low-FODMAP diet require further evaluation. In the meantime, patients following this diet should have FODMAP foods reintroduced based on tolerance and should consider taking probiotic supplements.41
DIETARY ADEQUACY OF THE LOW-FODMAP DIET
Continual dietary counseling should minimize nutritional inadequacies and ensure that FODMAPS are restricted only enough to control symptoms. Because no single food group is completely eliminated in this diet, patients are unlikely to experience inadequate nutrition.
Ledochowski et al26 found that in the initial, strict phase of the diet, total intake of carbohydrates (eg, starches, sugars) was reduced but intake of total energy, protein, fat, and nonstarch polysaccharides was not affected. Calcium intake was reduced in those following a low-FODMAP diet for 4 weeks.
The diet can also reduce total fiber intake and subsequently worsen constipation-predominant IBS. For those patients, lightly fermented high-fiber alternatives like oat and rice bran can be used.
ACCUMULATING EVIDENCE
The low-FODMAP diet is accumulating quality evidence for its effectiveness in controlling the functional gastrointestinal symptoms in patients with IBS. It can be difficult to adhere to over the long term due to its restrictiveness, and it is important to gradually liberalize the diet while tailoring it to the individual patient and monitoring symptoms. Further clinical trials are needed to evaluate this diet in different IBS subtypes and other gastrointestinal disorders, while defining its nutritional adequacy and effects on the intestinal microbiota profile.
- Hayes P, Corish C, O’Mahony E, Quigley EM. A dietary survey of patients with irritable bowel syndrome. J Hum Nutr Diet 2014; 27(suppl 2):36–47.
- Pare P, Gray J, Lam S, et al. Health-related quality of life, work productivity, and health care resource utilization of subjects with irritable bowel syndrome: baseline results from LOGIC (Longitudinal Outcomes Study of Gastrointestinal Symptoms in Canada), a naturalistic study. Clin Ther 2006; 28:1726–1735; discussion 1710–1711.
- Lacey BE, Mearin F, Chang L, et al. Bowel disorders. Gastroenterology 2016; 150:1393–1407.
- Drossman DA, Camilleri M, Mayer EA, Whitehead WE. AGA technical review on irritable bowel syndrome. Gastroenterology 2002; 123:2108–2131.
- Floch MH, Narayan R. Diet in the irritable bowel syndrome. J Clin Gastroenterol 2002; 35(suppl 1):S45–S52.
- Reding KW, Cain KC, Jarrett ME, Eugenio MD, Heitkemper MM. Relationship between patterns of alcohol consumption and gastrointestinal symptoms among patients with irritable bowel syndrome. Am J Gastroenterol 2013; 108:270–276.
- Moayyedi P, Quigley EM, Lacy BE, et al. The effect of fiber supplementation on irritable bowel syndrome: a systematic review and meta-analysis. Am J Gastroenterol 2014; 109:1367–1374.
- Vazquez Roque MI, Camilleri M, Smyrk T, et al. A controlled trial of gluten-free diet in patients with irritable bowel syndrome-diarrhea: effects on bowel frequency and intestinal function. Gastroenterology 2013; 144:903–911.
- Biesiekierski JR, Newnham ED, Irving PM, et al. Gluten causes gastrointestinal symptoms in subjects without celiac disease: a double-blind randomized placebo-controlled trial. Am J Gastroenterol 2011; 106:508–515.
- Biesiekierski JR, Peters SL, Newnham ED, Rosella O, Muir JG, Gibson PR. No effects of gluten in patients with self-reported non-celiac gluten sensitivity after dietary reduction of fermentable, poorly absorbed, short-chain carbohydrates. Gastroenterology 2013; 145:320–328.
- Ford AC, Quigley EM, Lacy BE, et al. Efficacy of prebiotics, probiotics, and synbiotics in irritable bowel syndrome and chronic idiopathic constipation: systematic review and meta-analysis. Gastroenterology 2013; 145:320–328.e1–e3.
- Central Clinical School, Monash University and The Alfred Hospital. The Monash University Low FODMAP Diet. 4th ed. Melbourne, Australia: Monash University; 2012.
- Parker K, Salas M, Nwosu VC. High fructose corn syrup: production, uses and public health concerns. Biotechnol Mol Biol Rev 2010; 5:71–78.
- Clausen MR, Jorgensen J, Mortensen PB. Comparison of diarrhea induced by ingestion of fructooligosaccharide idolax and disaccharide lactulose: role of osmolarity versus fermentation of malabsorbed carbohydrate. Dig Dis Sci 1998; 43:2696–2707.
- Barrett JS, Gearry RB, Muir JG, et al. Dietary poorly absorbed, short-chain carbohydrates increase delivery of water and fermentable substrates to the proximal colon. Aliment Pharmacol Ther 2010; 31:874–882.
- Whelan K, Abrahmsohn O, David GJ, et al. Fructan content of commonly consumed wheat, rye and gluten-free breads. Int J Food Sci Nutr 2011; 62:498–503.
- Sangwan V, Tomar SK, Singh RR, Singh AK, Ali B. Galactooligosaccharides: novel components of designer foods. J Food Sci 2011; 76:R103–R111.
- Russell DA, Ross RP, Fitzgerald GF, Stanton C. Metabolic activities and probiotic potential of bifidobacteria. Int J Food Microbiol 2011; 149:88–105.
- Lomer MC, Parkes GC, Sanderson JD. Review article: lactose intolerance in clinical practice—myths and realities. Aliment Pharmacol Ther 2008; 27:93–103.
- Langkilde AM, Andersson H, Schweizer TF, Würsch P. Digestion and absorption of sorbitol, maltitol and isomalt from the small bowel. A study in ileostomy subjects. Eur J Clin Nutr 1994; 48:768–775.
- Muir JG, Rose R, Rosella O, et al. Measurement of short-chain carbohydrates in common Australian vegetables and fruits by high-performance liquid chromatography (HPLC). J Agric Food Chem 2009; 57:554–565.
- Muir JG, Shepherd SJ, Rosella O, Rose R, Barrett JS, Gibson PR. Fructan and free fructose content of common Australian vegetables and fruit. J Agric Food Chem 2007; 55:6619–6627.
- Monash University. Monash launches Low FODMAP Diet smartphone app. http://med.monash.edu.au/news/2012/fodmap-app.html. Accessed July 13, 2016.
- Fedewa A, Rao SS. Dietary fructose intolerance, fructan intolerance and FODMAPs. Curr Gastroenterol Rep 2014; 16:370.
- Born P, Vierling T, Barina W. Fructose malabsorption and the irritable bowel syndrome. Gastroenterology 1991; 101:1454.
- Ledochowski M, Widner B, Bair H, Probst T, Fuchs D. Fructose- and sorbitol-reduced diet improves mood and gastrointestinal disturbances in fructose malabsorbers. Scand J Gastroenterol 2000; 35:1048–1052.
- Shepherd SJ, Gibson PR. Fructose malabsorption and symptoms of irritable bowel syndrome: guidelines for effective dietary management. J Am Diet Assoc 2006; 106:1631–1639.
- Staudacher HM, Lomer MC, Anderson JL, et al. Fermentable carbohydrate restriction reduces luminal bifidobacteria and gastrointestinal symptoms in patients with irritable bowel syndrome. J Nutr 2012; 142:1510–1518.
- Halmos EP, Power VA, Shepherd SJ, Gibson PR, Muir JG. A diet low in FODMAPs reduces symptoms of irritable bowel syndrome. Gastroenterology 2014; 146:67–75.e5.
- Shepherd SJ, Parker FC, Muir JG, Gibson PR. Dietary triggers of abdominal symptoms in patients with irritable bowel syndrome: randomized placebo-controlled evidence. Clin Gastroenterol Hepatol 2008; 6:765–771.
- Murray K, Wilkinson-Smith V, Hoad C, et al. Differential effects of FODMAPs (fermentable oligo-, di-, mono-saccharides and polyols) on small and large intestinal contents in healthy subjects shown by MRI. Am J Gastroenterol 2014; 109:110–119.
- Pedersen N, Andersen NN, Vegh Z, et al. Ehealth: low FODMAP diet vs Lactobacillus rhamnosus GG in irritable bowel syndrome. World J Gastroenterol 2014; 20:16215–16226.
- Gearry RB, Irving PM, Barrett JS, Nathan DM, Shepherd SJ, Gibson PR. Reduction of dietary poorly absorbed short-chain carbohydrates (FODMAPs) improves abdominal symptoms in patients with inflammatory bowel disease-a pilot study. J Crohns Colitis 2009; 3:8–14.
- Marsh A, Eslick EM, Eslick GD. Does a diet low in FODMAPs reduce symptoms associated with functional gastrointestinal disorders? A comprehensive systematic review and meta-analysis. Eur J Nutr 2015 May 17. Epub ahead of print.
- de Roest RH, Dobbs BR, Chapman BA, et al. The low FODMAP diet improves gastrointestinal symptoms in patients with irritable bowel syndrome: a prospective study. Int J Clin Pract 2013; 67:895–903.
- Gibson PR, Barrett JS, Muir JG. Functional bowel symptoms and diet. Intern Med J 2013; 43:1067–1074.
- Whigham L, Joyce T, Harper G, et al. Clinical effectiveness and economic costs of group versus one-to-one education for short-chain fermentable carbohydrate restriction (low FODMAP diet) in the management of irritable bowel syndrome. J Hum Nutr Diet 2015; 28:687–696.
- Shepherd SJ, Lomer MC, Gibson PR. Short-chain carbohydrates and functional gastrointestinal disorders. Am J Gastroenterol 2013; 108:707–717.
- Shepherd SJ, Halmos E, Glance S. The role of FODMAPs in irritable bowel syndrome. Curr Opin Clin Nutr Metab Care 2014; 17:605–609.
- Halmos EP, Christophersen CT, Bird AR, Shepherd SJ, Gibson PR, Muir JG. Diets that differ in their FODMAP content alter the colonic luminal microenvironment. Gut 2015; 64:93–100.
- Staudacher HM, Irving PM, Lomer MC, Whelan K. Mechanisms and efficacy of dietary FODMAP restriction in IBS. Nat Rev Gastroenterol Hepatol 2014; 11:256–266.
- Hayes P, Corish C, O’Mahony E, Quigley EM. A dietary survey of patients with irritable bowel syndrome. J Hum Nutr Diet 2014; 27(suppl 2):36–47.
- Pare P, Gray J, Lam S, et al. Health-related quality of life, work productivity, and health care resource utilization of subjects with irritable bowel syndrome: baseline results from LOGIC (Longitudinal Outcomes Study of Gastrointestinal Symptoms in Canada), a naturalistic study. Clin Ther 2006; 28:1726–1735; discussion 1710–1711.
- Lacey BE, Mearin F, Chang L, et al. Bowel disorders. Gastroenterology 2016; 150:1393–1407.
- Drossman DA, Camilleri M, Mayer EA, Whitehead WE. AGA technical review on irritable bowel syndrome. Gastroenterology 2002; 123:2108–2131.
- Floch MH, Narayan R. Diet in the irritable bowel syndrome. J Clin Gastroenterol 2002; 35(suppl 1):S45–S52.
- Reding KW, Cain KC, Jarrett ME, Eugenio MD, Heitkemper MM. Relationship between patterns of alcohol consumption and gastrointestinal symptoms among patients with irritable bowel syndrome. Am J Gastroenterol 2013; 108:270–276.
- Moayyedi P, Quigley EM, Lacy BE, et al. The effect of fiber supplementation on irritable bowel syndrome: a systematic review and meta-analysis. Am J Gastroenterol 2014; 109:1367–1374.
- Vazquez Roque MI, Camilleri M, Smyrk T, et al. A controlled trial of gluten-free diet in patients with irritable bowel syndrome-diarrhea: effects on bowel frequency and intestinal function. Gastroenterology 2013; 144:903–911.
- Biesiekierski JR, Newnham ED, Irving PM, et al. Gluten causes gastrointestinal symptoms in subjects without celiac disease: a double-blind randomized placebo-controlled trial. Am J Gastroenterol 2011; 106:508–515.
- Biesiekierski JR, Peters SL, Newnham ED, Rosella O, Muir JG, Gibson PR. No effects of gluten in patients with self-reported non-celiac gluten sensitivity after dietary reduction of fermentable, poorly absorbed, short-chain carbohydrates. Gastroenterology 2013; 145:320–328.
- Ford AC, Quigley EM, Lacy BE, et al. Efficacy of prebiotics, probiotics, and synbiotics in irritable bowel syndrome and chronic idiopathic constipation: systematic review and meta-analysis. Gastroenterology 2013; 145:320–328.e1–e3.
- Central Clinical School, Monash University and The Alfred Hospital. The Monash University Low FODMAP Diet. 4th ed. Melbourne, Australia: Monash University; 2012.
- Parker K, Salas M, Nwosu VC. High fructose corn syrup: production, uses and public health concerns. Biotechnol Mol Biol Rev 2010; 5:71–78.
- Clausen MR, Jorgensen J, Mortensen PB. Comparison of diarrhea induced by ingestion of fructooligosaccharide idolax and disaccharide lactulose: role of osmolarity versus fermentation of malabsorbed carbohydrate. Dig Dis Sci 1998; 43:2696–2707.
- Barrett JS, Gearry RB, Muir JG, et al. Dietary poorly absorbed, short-chain carbohydrates increase delivery of water and fermentable substrates to the proximal colon. Aliment Pharmacol Ther 2010; 31:874–882.
- Whelan K, Abrahmsohn O, David GJ, et al. Fructan content of commonly consumed wheat, rye and gluten-free breads. Int J Food Sci Nutr 2011; 62:498–503.
- Sangwan V, Tomar SK, Singh RR, Singh AK, Ali B. Galactooligosaccharides: novel components of designer foods. J Food Sci 2011; 76:R103–R111.
- Russell DA, Ross RP, Fitzgerald GF, Stanton C. Metabolic activities and probiotic potential of bifidobacteria. Int J Food Microbiol 2011; 149:88–105.
- Lomer MC, Parkes GC, Sanderson JD. Review article: lactose intolerance in clinical practice—myths and realities. Aliment Pharmacol Ther 2008; 27:93–103.
- Langkilde AM, Andersson H, Schweizer TF, Würsch P. Digestion and absorption of sorbitol, maltitol and isomalt from the small bowel. A study in ileostomy subjects. Eur J Clin Nutr 1994; 48:768–775.
- Muir JG, Rose R, Rosella O, et al. Measurement of short-chain carbohydrates in common Australian vegetables and fruits by high-performance liquid chromatography (HPLC). J Agric Food Chem 2009; 57:554–565.
- Muir JG, Shepherd SJ, Rosella O, Rose R, Barrett JS, Gibson PR. Fructan and free fructose content of common Australian vegetables and fruit. J Agric Food Chem 2007; 55:6619–6627.
- Monash University. Monash launches Low FODMAP Diet smartphone app. http://med.monash.edu.au/news/2012/fodmap-app.html. Accessed July 13, 2016.
- Fedewa A, Rao SS. Dietary fructose intolerance, fructan intolerance and FODMAPs. Curr Gastroenterol Rep 2014; 16:370.
- Born P, Vierling T, Barina W. Fructose malabsorption and the irritable bowel syndrome. Gastroenterology 1991; 101:1454.
- Ledochowski M, Widner B, Bair H, Probst T, Fuchs D. Fructose- and sorbitol-reduced diet improves mood and gastrointestinal disturbances in fructose malabsorbers. Scand J Gastroenterol 2000; 35:1048–1052.
- Shepherd SJ, Gibson PR. Fructose malabsorption and symptoms of irritable bowel syndrome: guidelines for effective dietary management. J Am Diet Assoc 2006; 106:1631–1639.
- Staudacher HM, Lomer MC, Anderson JL, et al. Fermentable carbohydrate restriction reduces luminal bifidobacteria and gastrointestinal symptoms in patients with irritable bowel syndrome. J Nutr 2012; 142:1510–1518.
- Halmos EP, Power VA, Shepherd SJ, Gibson PR, Muir JG. A diet low in FODMAPs reduces symptoms of irritable bowel syndrome. Gastroenterology 2014; 146:67–75.e5.
- Shepherd SJ, Parker FC, Muir JG, Gibson PR. Dietary triggers of abdominal symptoms in patients with irritable bowel syndrome: randomized placebo-controlled evidence. Clin Gastroenterol Hepatol 2008; 6:765–771.
- Murray K, Wilkinson-Smith V, Hoad C, et al. Differential effects of FODMAPs (fermentable oligo-, di-, mono-saccharides and polyols) on small and large intestinal contents in healthy subjects shown by MRI. Am J Gastroenterol 2014; 109:110–119.
- Pedersen N, Andersen NN, Vegh Z, et al. Ehealth: low FODMAP diet vs Lactobacillus rhamnosus GG in irritable bowel syndrome. World J Gastroenterol 2014; 20:16215–16226.
- Gearry RB, Irving PM, Barrett JS, Nathan DM, Shepherd SJ, Gibson PR. Reduction of dietary poorly absorbed short-chain carbohydrates (FODMAPs) improves abdominal symptoms in patients with inflammatory bowel disease-a pilot study. J Crohns Colitis 2009; 3:8–14.
- Marsh A, Eslick EM, Eslick GD. Does a diet low in FODMAPs reduce symptoms associated with functional gastrointestinal disorders? A comprehensive systematic review and meta-analysis. Eur J Nutr 2015 May 17. Epub ahead of print.
- de Roest RH, Dobbs BR, Chapman BA, et al. The low FODMAP diet improves gastrointestinal symptoms in patients with irritable bowel syndrome: a prospective study. Int J Clin Pract 2013; 67:895–903.
- Gibson PR, Barrett JS, Muir JG. Functional bowel symptoms and diet. Intern Med J 2013; 43:1067–1074.
- Whigham L, Joyce T, Harper G, et al. Clinical effectiveness and economic costs of group versus one-to-one education for short-chain fermentable carbohydrate restriction (low FODMAP diet) in the management of irritable bowel syndrome. J Hum Nutr Diet 2015; 28:687–696.
- Shepherd SJ, Lomer MC, Gibson PR. Short-chain carbohydrates and functional gastrointestinal disorders. Am J Gastroenterol 2013; 108:707–717.
- Shepherd SJ, Halmos E, Glance S. The role of FODMAPs in irritable bowel syndrome. Curr Opin Clin Nutr Metab Care 2014; 17:605–609.
- Halmos EP, Christophersen CT, Bird AR, Shepherd SJ, Gibson PR, Muir JG. Diets that differ in their FODMAP content alter the colonic luminal microenvironment. Gut 2015; 64:93–100.
- Staudacher HM, Irving PM, Lomer MC, Whelan K. Mechanisms and efficacy of dietary FODMAP restriction in IBS. Nat Rev Gastroenterol Hepatol 2014; 11:256–266.
KEY POINTS
- In clinical trials, the low-FODMAP diet has been found to improve symptoms in up to 70% of patients with IBS.
- FODMAPs are poorly absorbed for a variety of reasons.
- High-FODMAP foods include wheat, onions, legumes, dairy products, and many fruits and vegetables.
- The diet initially involves strict elimination of foods high in FODMAPs, after which they are gradually reintroduced as tolerated.
- A low-FODMAP diet may have negative effects on the gut microbiome. Therefore, we should be cautious about recommending this diet in the long term.
- Probiotics have a beneficial effect in IBS and can be taken concurrently with the diet.

In reply: Alcoholic hepatitis: An important consideration
In Reply: We thank Dr. Mirrakhimov for his interest in our article1 and for his comments on the importance of infection evaluation and treatment in patients with alcoholic hepatitis. We agree with the points he has raised and emphasized several of them in our article. We highlighted the need to evaluate for infections in these patients, as about a quarter of them are infected at the time of presentation.2
Importantly, patients with alcoholic hepatitis frequently have systemic inflammatory response syndrome criteria, which can be related to the overall inflammatory state of the disease itself or can reflect an active bacterial infection. Therefore, clinical monitoring for symptoms and signs of infection is crucial, and screening for infections is warranted on admission as well as repeatedly during the hospital stay for patients who experience clinical deterioration.3 Obtaining blood and urine cultures and performing paracentesis in patients with ascites to evaluate for bacterial peritonitis are required. Indeed, infections are a leading cause of death in patients with severe alcoholic hepatitis, both directly and indirectly by predisposing to multiorgan failure.4
Another factor to consider is the increased susceptibility to infection in these patients treated with corticosteroids. A study by Louvet et al2 showed that nonresponse to corticosteroids is the main factor contributing to the development of infection during treatment with corticosteroids, suggesting that infection is likely a consequence of the absence of improvement in liver function. More recently, results of the Steroids or Pentoxifylline for Alcoholic Hepatitis trial (which evaluated the treatment effect of prednisolone and pentoxifylline in the management of severe alcoholic hepatitis) showed that despite the higher rates of infections in patients treated with prednisolone, the mortality rates attributed to infections were similar across the treatment groups, regardless of whether prednisolone was administered.4
Finally, it is important to emphasize that criteria to initiate empiric antibiotics in patients with alcoholic hepatitis are currently lacking, and the decision to start antibiotics empirically in patients without a clear infection is largely based on the clinician’s assessment.
- Dugum M, Zein N, McCullough A, Hanouneh I. Alcoholic hepatitis: challenges in diagnosis and management. Cleve Clin J Med 2015; 82:226–236.
- Louvet A, Wartel F, Castel H, et al. Infection in patients with severe alcoholic hepatitis treated with steroids: early response to therapy is the key factor. Gastroenterology 2009; 137:541–548.
- European Association for the Study of Liver. EASL clinical practical guidelines: management of alcoholic liver disease. J Hepatol 2012; 57:399–420.
- Thursz MR, Richardson P, Allison M, et al. Prednisolone or pentoxifylline for alcoholic hepatitis. N Engl J Med 2015; 372:1619–1628.
In Reply: We thank Dr. Mirrakhimov for his interest in our article1 and for his comments on the importance of infection evaluation and treatment in patients with alcoholic hepatitis. We agree with the points he has raised and emphasized several of them in our article. We highlighted the need to evaluate for infections in these patients, as about a quarter of them are infected at the time of presentation.2
Importantly, patients with alcoholic hepatitis frequently have systemic inflammatory response syndrome criteria, which can be related to the overall inflammatory state of the disease itself or can reflect an active bacterial infection. Therefore, clinical monitoring for symptoms and signs of infection is crucial, and screening for infections is warranted on admission as well as repeatedly during the hospital stay for patients who experience clinical deterioration.3 Obtaining blood and urine cultures and performing paracentesis in patients with ascites to evaluate for bacterial peritonitis are required. Indeed, infections are a leading cause of death in patients with severe alcoholic hepatitis, both directly and indirectly by predisposing to multiorgan failure.4
Another factor to consider is the increased susceptibility to infection in these patients treated with corticosteroids. A study by Louvet et al2 showed that nonresponse to corticosteroids is the main factor contributing to the development of infection during treatment with corticosteroids, suggesting that infection is likely a consequence of the absence of improvement in liver function. More recently, results of the Steroids or Pentoxifylline for Alcoholic Hepatitis trial (which evaluated the treatment effect of prednisolone and pentoxifylline in the management of severe alcoholic hepatitis) showed that despite the higher rates of infections in patients treated with prednisolone, the mortality rates attributed to infections were similar across the treatment groups, regardless of whether prednisolone was administered.4
Finally, it is important to emphasize that criteria to initiate empiric antibiotics in patients with alcoholic hepatitis are currently lacking, and the decision to start antibiotics empirically in patients without a clear infection is largely based on the clinician’s assessment.
In Reply: We thank Dr. Mirrakhimov for his interest in our article1 and for his comments on the importance of infection evaluation and treatment in patients with alcoholic hepatitis. We agree with the points he has raised and emphasized several of them in our article. We highlighted the need to evaluate for infections in these patients, as about a quarter of them are infected at the time of presentation.2
Importantly, patients with alcoholic hepatitis frequently have systemic inflammatory response syndrome criteria, which can be related to the overall inflammatory state of the disease itself or can reflect an active bacterial infection. Therefore, clinical monitoring for symptoms and signs of infection is crucial, and screening for infections is warranted on admission as well as repeatedly during the hospital stay for patients who experience clinical deterioration.3 Obtaining blood and urine cultures and performing paracentesis in patients with ascites to evaluate for bacterial peritonitis are required. Indeed, infections are a leading cause of death in patients with severe alcoholic hepatitis, both directly and indirectly by predisposing to multiorgan failure.4
Another factor to consider is the increased susceptibility to infection in these patients treated with corticosteroids. A study by Louvet et al2 showed that nonresponse to corticosteroids is the main factor contributing to the development of infection during treatment with corticosteroids, suggesting that infection is likely a consequence of the absence of improvement in liver function. More recently, results of the Steroids or Pentoxifylline for Alcoholic Hepatitis trial (which evaluated the treatment effect of prednisolone and pentoxifylline in the management of severe alcoholic hepatitis) showed that despite the higher rates of infections in patients treated with prednisolone, the mortality rates attributed to infections were similar across the treatment groups, regardless of whether prednisolone was administered.4
Finally, it is important to emphasize that criteria to initiate empiric antibiotics in patients with alcoholic hepatitis are currently lacking, and the decision to start antibiotics empirically in patients without a clear infection is largely based on the clinician’s assessment.
- Dugum M, Zein N, McCullough A, Hanouneh I. Alcoholic hepatitis: challenges in diagnosis and management. Cleve Clin J Med 2015; 82:226–236.
- Louvet A, Wartel F, Castel H, et al. Infection in patients with severe alcoholic hepatitis treated with steroids: early response to therapy is the key factor. Gastroenterology 2009; 137:541–548.
- European Association for the Study of Liver. EASL clinical practical guidelines: management of alcoholic liver disease. J Hepatol 2012; 57:399–420.
- Thursz MR, Richardson P, Allison M, et al. Prednisolone or pentoxifylline for alcoholic hepatitis. N Engl J Med 2015; 372:1619–1628.
- Dugum M, Zein N, McCullough A, Hanouneh I. Alcoholic hepatitis: challenges in diagnosis and management. Cleve Clin J Med 2015; 82:226–236.
- Louvet A, Wartel F, Castel H, et al. Infection in patients with severe alcoholic hepatitis treated with steroids: early response to therapy is the key factor. Gastroenterology 2009; 137:541–548.
- European Association for the Study of Liver. EASL clinical practical guidelines: management of alcoholic liver disease. J Hepatol 2012; 57:399–420.
- Thursz MR, Richardson P, Allison M, et al. Prednisolone or pentoxifylline for alcoholic hepatitis. N Engl J Med 2015; 372:1619–1628.
Alcoholic hepatitis: Challenges in diagnosis and management
Alcoholic hepatitis, a severe manifestation of alcoholic liver disease, is rising in incidence. Complete abstinence from alcohol remains the cornerstone of treatment, while other specific interventions aim to decrease short-term mortality rates.
Despite current treatments, about 25% of patients with severe alcoholic hepatitis eventually die of it. For those who survive hospitalization, measures need to be taken to prevent recidivism. Although liver transplantation seems to hold promise, early transplantation is still largely experimental in alcoholic hepatitis and will likely be available to only a small subset of patients, especially in view of ethical issues and the possible wider implications for transplant centers.
New treatments will largely depend on a better understanding of the disease’s pathophysiology, and future clinical trials should evaluate therapies that improve short-term as well as long-term outcomes.
ACUTE HEPATIC DECOMPENSATION IN A HEAVY DRINKER
Excessive alcohol consumption is very common worldwide, is a major risk factor for liver disease, and is a leading cause of preventable death. Alcoholic cirrhosis is the eighth most common cause of death in the United States and in 2010 was responsible for nearly half of cirrhosis-related deaths worldwide.1

Alcoholic liver disease is a spectrum. Nearly all heavy drinkers (ie, those consuming 40 g or more of alcohol per day, Table 1) have fatty liver changes, 20% to 40% develop fibrosis, 10% to 20% progress to cirrhosis, and of those with cirrhosis, 1% to 2% are diagnosed with hepatocellular carcinoma every year.2
Within this spectrum, alcoholic hepatitis is a well-defined clinical syndrome characterized by acute hepatic decompensation that typically results from long-standing alcohol abuse. Binge drinkers may also be at risk for alcoholic hepatitis, but good data on the association between drinking patterns and the risk of alcoholic hepatitis are limited.
Alcoholic hepatitis varies in severity from mild to life-threatening.3 Although its exact incidence is unknown, its prevalence in alcoholics has been estimated at 20%.4 Nearly half of patients with alcoholic hepatitis have cirrhosis at the time of their acute presentation, and these patients generally have a poor prognosis, with a 28-day death rate as high as 50% in severe cases.5,6 Moreover, although alcoholic hepatitis develops in only a subset of patients with alcoholic liver disease, hospitalizations for it are increasing in the United States.7
Women are at higher risk of developing alcoholic hepatitis, an observation attributed to the effect of estrogens on oxidative stress and inflammation, lower gastric alcohol dehydrogenase levels resulting in slower first-pass metabolism of alcohol, and higher body fat content causing a lower volume of distribution for alcohol than in men.8 The incidence of alcoholic hepatitis is also influenced by a number of demographic and genetic factors as well as nutritional status and coexistence of other liver diseases.9 Most patients diagnosed with alcoholic hepatitis are active drinkers, but it can develop even after significantly reducing or stopping alcohol consumption.
FATTY ACIDS, ENZYMES, CYTOKINES, INFLAMMATION
Alcohol consumption induces fatty acid synthesis and inhibits fatty acid oxidation, thereby promoting fat deposition in the liver.
The major enzymes involved in alcohol metabolism are cytochrome P450 2E1 (CYP2E1) and alcohol dehydrogenase. CYP2E1 is inducible and is up-regulated when excess alcohol is ingested, while alcohol dehydrogen-
ase function is relatively stable. Oxidative degradation of alcohol by these enzymes generates reactive oxygen species and acetaldehyde, inducing liver injury.10 Interestingly, it has been proposed that variations in the genes for these enzymes influence alcohol consumption and dependency as well as alcohol-driven tissue damage.
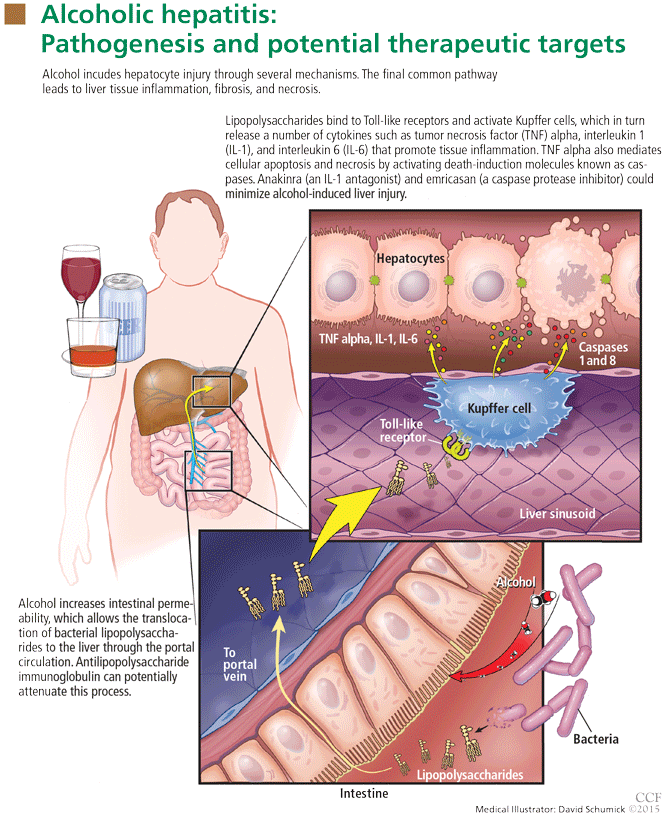
In addition, alcohol disrupts the intestinal mucosal barrier, allowing lipopolysaccharides from gram-negative bacteria to travel to the liver via the portal vein. These lipopolysaccharides then bind to and activate sinusoidal Kupffer cells, leading to production of several cytokines such as tumor necrosis factor alpha, interleukin 1, and transforming growth factor beta. These cytokines promote hepatocyte inflammation, apoptosis, and necrosis (Figure 1).11
Besides activating the innate immune system, the reactive oxygen species resulting from alcohol metabolism interact with cellular components, leading to production of protein adducts. These act as antigens that activate the adaptive immune response, followed by B- and T-lymphocyte infiltration, which in turn contribute to liver injury and inflammation.12
THE DIAGNOSIS IS MAINLY CLINICAL
The diagnosis of alcoholic hepatitis is mainly clinical. In its usual presentation, jaundice develops rapidly in a person with a known history of heavy alcohol use. Other symptoms and signs may include ascites, encephalopathy, and fever. On examination, the liver may be enlarged and tender, and a hepatic bruit has been reported.13
Other classic signs of liver disease such as parotid enlargement, Dupuytren contracture, dilated abdominal wall veins, and spider nevi can be present, but none is highly specific or sensitive for alcoholic hepatitis.
Elevated liver enzymes and other clues
Laboratory tests are important in evaluating potential alcoholic hepatitis, although no single laboratory marker can definitively establish alcohol as the cause of liver disease. To detect alcohol consumption, biochemical markers such as aspartate aminotransferase (AST), alanine aminotransferase (ALT), mean corpuscular volume, carbohydrate-deficient transferrin, and, more commonly, gamma-glutamyl transpeptidase are used.
In the acute setting, typical biochemical derangements in alcoholic hepatitis include elevated AST (up to 2 to 6 times the upper limit of normal; usually less than 300 IU/L) and elevated ALT to a lesser extent,14 with an AST-to-ALT ratio greater than 2. Neutrophilia, anemia, hyperbilirubinemia, and coagulopathy with an elevated international normalized ratio are common.
Patients with alcoholic hepatitis are also prone to develop bacterial infections, and about 7% develop hepatorenal syndrome, itself an ominous sign.15
Imaging studies are valuable in excluding other causes of abnormal liver test results in patients who abuse alcohol, such as biliary obstruction, infiltrative liver diseases, and hepatocellular carcinoma.
Screen for alcohol intake
During the initial evaluation of suspected alcoholic hepatitis, one should screen for excessive drinking. In a US Centers for Disease Control and Prevention study, only one of six US adults, including binge drinkers, said they had ever discussed alcohol consumption with a health professional.16 Many patients with alcoholic liver disease in general and alcoholic hepatitis in particular deny alcohol abuse or underreport their intake.17
Screening tests such as the CAGE questionnaire and the Alcohol Use Disorders Identification Test can be used to assess alcohol dependence or abuse.18,19 The CAGE questionnaire consists of four questions:
- Have you ever felt you should cut down on your drinking?
- Have people annoyed you by criticizing your drinking?
- Have you ever felt guilty about your drinking?
- Have you ever had a drink first thing in the morning (an eye-opener) to steady your nerves or to get rid of a hangover?
A yes answer to two or more questions is considered clinically significant.
Is liver biopsy always needed?
Although alcoholic hepatitis can be suspected on the basis of clinical and biochemical clues, liver biopsy remains the gold standard diagnostic tool. It confirms the clinical diagnosis of alcoholic hepatitis in about 85% of all patients and in up to 95% when significant hyperbilirubinemia is present.20
However, whether a particular patient needs a biopsy is not always clear. The American Association for the Study of Liver Diseases (AASLD) recommends biopsy in patients who have a clinical diagnosis of severe alcoholic hepatitis for whom medical treatment is being considered and in those with an uncertain underlying diagnosis.
Findings on liver biopsy in alcoholic hepatitis include steatosis, hepatocyte ballooning, neutrophilic infiltration, Mallory bodies (which represent aggregated cytokeratin intermediate filaments and other proteins), and scarring with a typical perivenular distribution as opposed to the periportal fibrosis seen in chronic viral hepatitis. Some histologic findings, such as centrilobular necrosis, may overlap alcoholic hepatitis and nonalcoholic steatohepatitis.
In addition to confirming the diagnosis and staging the disease, liver biopsy has prognostic value. The severity of inflammation and cholestatic changes correlates with poor prognosis and may also predict response to corticosteroid treatment in severe cases of alcoholic hepatitis.21
However, the utility of liver biopsy in confirming the diagnosis and assessing the prognosis of alcoholic hepatitis is controversial for several reasons. Coagulopathy, thrombocytopenia, and ascites are all common in patients with alcoholic hepatitis, often making percutaneous liver biopsy contraindicated. Trans-
jugular liver biopsy is not universally available outside tertiary care centers.
Needed is a minimally invasive test for assessing this disease. Breath analysis might be such a test, offering a noninvasive means to study the composition of volatile organic compounds and elemental gases and an attractive method to evaluate health and disease in a patient-friendly manner. Our group devised a model based on breath levels of trimethylamine and pentane. When we tested it, we found that it distinguishes patients with alcoholic hepatitis from those with acute liver decompensation from causes other than alcohol and controls without liver disease with up to 90% sensitivity and 80% specificity.22
ASSESSING THE SEVERITY OF ALCOHOLIC HEPATITIS
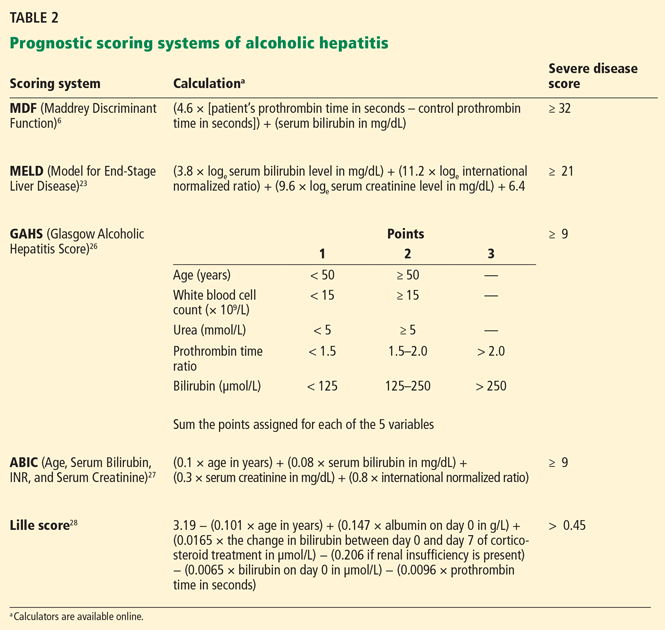
Several models have been developed to assess the severity of alcoholic hepatitis and guide treatment decisions (Table 2).
The MDF (Maddrey Discriminant Function)6 system was the first scoring system developed and is still the most widely used. A score of 32 or higher indicates severe alcoholic hepatitis and has been used as the threshold for starting treatment with corticosteroids.6
The MDF has limitations. Patients with a score lower than 32 are considered not to have severe alcoholic hepatitis, but up to 17% of them still die. Also, since it uses the prothrombin time, its results can vary considerably among laboratories, depending on the sensitivity of the thromboplastin reagent used.
The MELD (Model for End-stage Liver Disease) score. Sheth et al23 compared the MELD and the MDF scores in assessing the severity of alcoholic hepatitis. They found that the MELD performed as well as the MDF in predicting 30-day mortality. A MELD score of greater than 11 had a sensitivity in predicting 30-day mortality of 86% and a specificity of 81%, compared with 86% and 48%, respectively, for MDF scores greater than 32.
Another study found a MELD score of 21 to have the highest sensitivity and specificity in predicting mortality (an estimated 90-day death rate of 20%). Thus, a MELD score of 21 is an appropriate threshold for prompt consideration of specific therapies such as corticosteroids.24
The MELD score has become increasingly important in patients with alcoholic hepatitis, as some of them may become candidates for liver transplantation (see below). Also, serial MELD scores in hospitalized patients have prognostic implications, since an increase of 2 or more points in the first week has been shown to predict in-hospital mortality.25
The GAHS (Glasgow Alcoholic Hepatitis Score)26 was shown to identify patients with alcoholic hepatitis who have an especially poor prognosis and need corticosteroid therapy. In those with a GAHS of 9 or higher, the 28-day survival rate was 78% with corticosteroid treatment and 52% without corticosteroid treatment; survival rates at 84 days were 59% and 38%, respectively.26
The ABIC scoring system (Age, Serum Bilirubin, INR, and Serum Creatinine) stratifies patients by risk of death at 90 days27:
- Score less than 6.71: low risk (100% survival)
- A score 6.71–8.99: intermediate risk (70% survival)
- A score 9.0 or higher: high risk (25% survival).
Both the GAHS and ABIC score are limited by lack of external validation.
The Lille score.28 While the above scores are used to identify patients at risk of death from alcoholic hepatitis and to decide on starting corticosteroids, the Lille score is designed to assess response to corticosteroids after 1 week of treatment. It is calculated based on five pretreatment variables and the change in serum bilirubin level at day 7 of corticosteroid therapy. Lille scores range from 0 to 1; a score higher than 0.45 is associated with a 75% mortality rate at 6 months and indicates a lack of response to corticosteroids and that these drugs should be discontinued.28
MANAGEMENT
Supportive treatment
Abstinence from alcohol is the cornerstone of treatment of alcoholic hepatitis. Early management of alcohol abuse or dependence is, therefore, warranted in all patients with alcoholic hepatitis. Referral to addiction specialists, motivational therapies, and anticraving drugs such as baclofen can be utilized.
Treat alcohol withdrawal. Alcoholics who suddenly decrease or discontinue their alcohol use are at high risk of alcohol withdrawal syndrome. Within 24 hours after the last drink, patients can experience increases in their heart rate and blood pressure, along with irritability and hyperreflexia. Within the next few days, more dangerous complications including seizures and delirium tremens can arise.
Alcohol withdrawal symptoms should be treated with short-acting benzodiazepines or clomethiazole, keeping the risk of worsening encephalopathy in mind.29 If present, complications of cirrhosis such as encephalopathy, ascites, and variceal bleeding should be managed.
Nutritional support is important. Protein-calorie malnutrition is common in alcoholics, as are deficiencies of vitamin A, vitamin D, thiamine, folate, pyridoxine, and zinc.30 Although a randomized controlled trial comparing enteral nutrition (2,000 kcal/day) vs corticosteroids (prednisolone 40 mg/day) in patients with alcoholic hepatitis did not show any difference in the 28-day mortality rate, those who received nutritional support and survived the first month had a lower mortality rate than those treated with corticosteroids (8% vs 37%).31 A daily protein intake of 1.5 g per kilogram of body weight is therefore recommended, even in patients with hepatic encephalopathy.15
Combining enteral nutrition and corticosteroid treatment may have a synergistic effect but is yet to be investigated.
Screen for infection. Patients with alcoholic hepatitis should be screened for infection, as about 25% of those with severe alcoholic hepatitis have an infection at admission.32 Since many of these patients meet the criteria for systemic inflammatory response syndrome, infections can be particularly difficult to diagnose. Patients require close clinical monitoring as well as regular pancultures for early detection. Antibiotics are frequently started empirically even though we lack specific evidence-based guidelines on this practice.33
Corticosteroids
Various studies have evaluated the role of corticosteroids in treating alcoholic hepatitis, differing considerably in sample populations, methods, and end points. Although the results of individual trials differ, meta-analyses indicate that corticosteroids have a moderate beneficial effect in patients with severe alcoholic hepatitis.
For example, Rambaldi et al34 performed a meta-analysis that concluded the mortality rate was lower in alcoholic hepatitis patients with MDF scores of at least 32 or hepatic encephalopathy who were treated with corticosteroids than in controls (relative risk 0.37, 95% confidence interval 0.16–0.86).
Therefore, in the absence of contraindications, the AASLD recommends starting corticosteroids in patients with severe alcoholic hepatitis, defined as an MDF score of 32 or higher.21 The preferred agent is oral prednisolone 40 mg daily or parenteral methylprednisolone 32 mg daily for 4 weeks and then tapered over the next 2 to 4 weeks or abruptly discontinued. Because activation of prednisone is decreased in patients with liver disease, prednisolone (the active form) is preferred over prednisone (the inactive precursor).35 In alcoholic hepatitis, the number needed to treat with corticosteroids to prevent one death has been calculated36 at 5.
As mentioned, response to corticosteroids is commonly assessed at 1 week of treatment using the Lille score. A score higher than 0.45 predicts a poor response and should trigger discontinuation of corticosteroids, particularly in those classified as null responders (Lille score > 0.56).
Adverse effects of steroids include sepsis, gastrointestinal bleeding, and steroid psychosis. Of note, patients who have evidence of hepatorenal syndrome or gastrointestinal bleeding tend to have a less favorable response to corticosteroids. Also, while infections were once considered a contraindication to steroid therapy, recent evidence suggests that steroid use might not be precluded in infected patients after appropriate antibiotic therapy. Infections occur in about a quarter of all alcoholic hepatitis patients treated with steroids, more frequently in null responders (42.5%) than in responders (11.1%), which supports corticosteroid discontinuance at 1 week in null responders.32
Pentoxifylline
An oral phosphodiesterase inhibitor, pentoxifylline, also inhibits production of several cytokines, including tumor necrosis factor alpha. At a dose of 400 mg orally three times daily for 4 weeks, pentoxifylline has been used in treating severe alcoholic hepatitis (MDF score ≥ 32) and is recommended especially if corticosteroids are contraindicated, as with sepsis.21
An early double-blind clinical trial randomized patients with severe alcoholic hepatitis to receive either pentoxifylline 400 mg orally three times daily or placebo. Of the patients who received pentoxifylline, 24.5% died during the index hospitalization, compared with 46.1% of patients who received placebo. This survival benefit was mainly related to a markedly lower incidence of hepatorenal syndrome as the cause of death in the pentoxifylline group than in the placebo group (50% vs 91.7% of deaths).37
In a small clinical trial in patients with severe alcoholic hepatitis, pentoxifylline recipients had a higher 3-month survival rate than prednisolone recipients (35.29% vs 14.71%, P = .04).38 However, a larger trial showed no improvement in 6-month survival with the combination of prednisolone and pentoxifylline compared with prednisolone alone (69.9% vs 69.2%, P = .91).39 Also, a meta-analysis of five randomized clinical trials found no survival benefit with pentoxifylline therapy.40
Of note, in the unfortunate subgroup of patients who have a poor response to corticosteroids, no alternative treatment, including pentoxifylline, has been shown to be effective.41
Prednisone or pentoxifylline? Very recently, results of the Steroids or Pentoxifylline for Alcoholic Hepatitis (STOPAH) trial have been released.42 This is a large, multicenter, double-blinded clinical trial that aimed to provide a definitive answer to whether corticosteroids or pentoxifylline (or both) are beneficial in patients with alcoholic hepatitis. The study included 1,103 adult patients with severe alcoholic hepatitis (MDF score ≥ 32) who were randomized to monotherapy with prednisolone or pentoxifylline, combination therapy, or placebo. The primary end point was mortality at 28 days, and secondary end points included mortality at 90 days and at 1 year. Prednisolone reduced 28-day mortality by about 39%. In contrast, the 28-day mortality rate was similar in patients who received pentoxifylline and those who did not. Also, neither drug was significantly associated with a survival benefit beyond 28 days. The investigators concluded that pentoxifylline has no impact on disease progression and should not be used for the treatment of severe alcoholic hepatitis.42
Other tumor necrosis factor alpha inhibitors not recommended
Two other tumor necrosis factor alpha inhibitors, infliximab and etanercept, have been tested in clinical trials in alcoholic hepatitis. Unfortunately, the results were not encouraging, with no major reduction in mortality.43–45 In fact, these trials demonstrated a significantly increased risk of infections in the treatment groups. Therefore, these drugs are not recommended for treating alcoholic hepatitis.
A possible explanation is that tumor necrosis factor alpha plays an important role in liver regeneration, aiding in recovery from alcohol-induced liver injury, and inhibiting it can have deleterious consequences.
Other agents
A number of other agents have undergone clinical trials in alcoholic hepatitis.
N-acetylcysteine, an antioxidant that replenishes glutathione stores in hepatocytes, was evaluated in a randomized clinical trial in combination with prednisolone.46 Although the 1-month mortality rate was significantly lower in the combination group than in the prednisolone-only group (8% vs 24%, P = .006), 3-month and 6-month mortality rates were not. Nonetheless, the rates of infection and hepatorenal syndrome were lower in the combination group. Therefore, corticosteroids and N-acetylcysteine may have synergistic effects, but the optimum duration of N-acetylcysteine therapy needs to be determined in further studies.
Vitamin E, silymarin, propylthiouracil, colchicine, and oxandrolone (an anabolic steroid) have also been studied, but with no convincing benefit.21
Role of liver transplantation
Liver transplantation for alcoholic liver disease has been a topic of great medical and social controversy. The view that alcoholic patients are responsible for their own illness led to caution when contemplating liver transplantation. Many countries require 6 months of abstinence from alcohol before placing a patient on the liver transplant list, posing a major obstacle to patients with alcoholic hepatitis, as almost all are active drinkers at the time of presentation and many will die within 6 months. Reasons for this 6-month rule include donor shortage and risk of recidivism.47
With regard to survival following alcoholic hepatitis, a study utilizing the United Network for Organ Sharing database matched patients with alcoholic hepatitis and alcoholic cirrhosis who underwent liver transplantation. Rates of 5-year graft survival were 75% in those with alcoholic hepatitis and 73% in those with alcoholic cirrhosis (P = .97), and rates of patient survival were 80% and 78% (P = .90), respectively. Proportional regression analysis adjusting for other variables showed no impact of the etiology of liver disease on graft or patient survival. The investigators concluded that liver transplantation could be considered in a select group of patients with alcoholic hepatitis who do not improve with medical therapy.48
In a pivotal case-control prospective study,49 26 patients with Lille scores greater than 0.45 were listed for liver transplantation within a median of 13 days after nonresponse to medical therapy. The cumulative 6-month survival rate was higher in patients who received a liver transplant early than in those who did not (77% vs 23%, P < .001). This benefit was maintained through 2 years of follow-up (hazard ratio 6.08, P = .004). Of note, all these patients had supportive family members, no severe coexisting conditions, and a commitment to alcohol abstinence (although 3 patients resumed drinking after liver transplantation).49
Although these studies support early liver transplantation in carefully selected patients with severe alcoholic hepatitis, the criteria for transplantation in this group need to be refined. Views on alcoholism also need to be reconciled, as strong evidence is emerging that implicates genetic and environmental influences on alcohol dependence.
Management algorithm
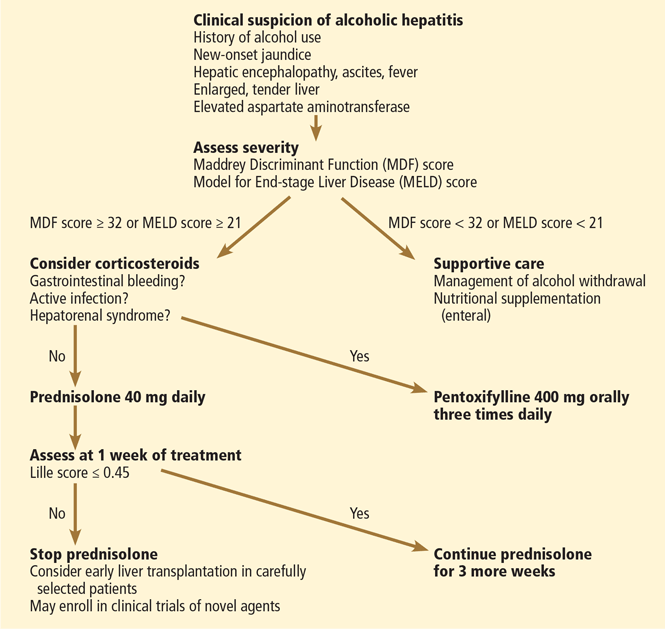
Figure 2 shows a suggested management algorithm for alcoholic hepatitis, adapted from the guidelines of the AASLD and European Association for the Study of the Liver.
NEW THERAPIES NEEDED
Novel therapies for severe alcoholic hepatitis are urgently needed to help combat this devastating condition. Advances in understanding its pathophysiology have uncovered several new therapeutic targets, and new agents are already being evaluated in clinical trials.
IMM 124-E, a hyperimmune bovine colostrum enriched with immunoglobulin G anti-lipopolysaccharide, is going to be evaluated in combination with prednisolone in patients with severe alcoholic hepatitis.
Anakinra, an interleukin 1 receptor antagonist, has significant anti-inflammatory activity and is used to treat rheumatoid arthritis. A clinical trial to evaluate its role in alcoholic hepatitis has been designed in which patients with severe alcoholic hepatitis (defined as a MELD score ≥ 21) will be randomized to receive either methylprednisolone or a combination of anakinra, pentoxifylline, and zinc (a mineral that improves gut integrity).
Emricasan, an orally active caspase protease inhibitor, is another agent currently being tested in a phase 2 clinical trial in patients with severe alcoholic hepatitis. Since caspases induce apoptosis, inhibiting them should theoretically dampen alcohol-induced hepatocyte injury.
Interleukin 22, a hepatoprotective cytokine, shows promise as a treatment and will soon be evaluated in alcoholic hepatitis.
- Rehm J, Samokhvalov AV, Shield KD. Global burden of alcoholic liver diseases. J Hepatol 2013; 59:160–168.
- Teli MR, Day CP, Burt AD, Bennett MK, James OF. Determinants of progression to cirrhosis or fibrosis in pure alcoholic fatty liver. Lancet 1995; 346:987–990.
- Alcoholic liver disease: morphological manifestations. Review by an international group. Lancet 1981; 1:707–711.
- Naveau S, Giraud V, Borotto E, Aubert A, Capron F, Chaput JC. Excess weight risk factor for alcoholic liver disease. Hepatology 1997; 25:108–111.
- Basra S, Anand BS. Definition, epidemiology and magnitude of alcoholic hepatitis. World J Hepatol 2011; 3:108–113.
- Maddrey WC, Boitnott JK, Bedine MS, Weber FL Jr, Mezey E, White RI Jr. Corticosteroid therapy of alcoholic hepatitis. Gastroenterology 1978; 75:193–199.
- Jinjuvadia R, Liangpunsakul S, for the Translational Research and Evolving Alcoholic Hepatitis Treatment Consortium. Trends in alcoholic hepatitis-related hospitalizations, financial burden, and mortality in the United States. J Clin Gastroenterol 2014 Jun 25 (Epub ahead of print).
- Sato N, Lindros KO, Baraona E, et al. Sex difference in alcohol-related organ injury. Alcohol Clin Exp Res 2001; 25(suppl s1):40S–45S.
- Singal AK, Kamath PS, Gores GJ, Shah VH. Alcoholic hepatitis: current challenges and future directions. Clin Gastroenterol Hepatol 2014; 12:555–564.
- Seitz HK, Stickel F. Risk factors and mechanisms of hepatocarcinogenesis with special emphasis on alcohol and oxidative stress. Biol Chem 2006; 387:349–360.
- Thurman RG. II. Alcoholic liver injury involves activation of Kupffer cells by endotoxin. Am J Physiol 1998; 275:G605–G611.
- Duddempudi AT. Immunology in alcoholic liver disease. Clin Liver Dis 2012; 16:687–698.
- Lischner MW, Alexander JF, Galambos JT. Natural history of alcoholic hepatitis. I. The acute disease. Am J Dig Dis 1971; 16:481–494.
- Cohen JA, Kaplan MM. The SGOT/SGPT ratio—an indicator of alcoholic liver disease. Dig Dis Sci 1979; 24:835–838.
- Lucey MR, Mathurin P, Morgan TR. Alcoholic hepatitis. N Engl J Med 2009; 360:2758–2769.
- McKnight-Eily LR, Liu Y, Brewer RD, et al; Centers for Disease Control and Prevention (CDC). Vital signs: communication between health professionals and their patients about alcohol use—44 states and the District of Columbia, 2011. MMWR Morb Mortal Wkly Rep 2014; 63:16–22.
- Grant BF. Barriers to alcoholism treatment: reasons for not seeking treatment in a general population sample. J Stud Alcohol 1997; 58:365–371.
- Aertgeerts B, Buntinx F, Kester A. The value of the CAGE in screening for alcohol abuse and alcohol dependence in general clinical populations: a diagnostic meta-analysis. J Clin Epidemiol 2004; 57:30–39.
- The Alcohol Use Disorders Identification Test Guidelines for Use in Primary Care. Second Edition. World Health Organization. Department of Mental Health and Substance Dependence. http://whqlibdoc.who.int/hq/2001/who_msd_msb_01.6a.pdf. Accessed February 3, 2015.
- Hamid R, Forrest EH. Is histology required for the diagnosis of alcoholic hepatitis? A review of published randomised controlled trials. Gut 2011; 60(suppl 1):A233.
- O’Shea RS, Dasarathy S, McCullough AJ; Practice Guideline Committee of the American Association for the Study of Liver Diseases; Practice Parameters Committee of the American College of Gastroenterology. Alcoholic liver disease. Hepatology 2010; 51:307–328.
- Hanouneh IA, Zein NN, Cikach F, et al. The breathprints in patients with liver disease identify novel breath biomarkers in alcoholic hepatitis. Clin Gastroenterol Hepatol 2014; 12:516–523.
- Sheth M, Riggs M, Patel T. Utility of the Mayo End-Stage Liver Disease (MELD) score in assessing prognosis of patients with alcoholic hepatitis. BMC Gastroenterol 2002; 2:2.
- Dunn W, Jamil LH, Brown LS, et al. MELD accurately predicts mortality in patients with alcoholic hepatitis. Hepatology 2005; 41:353–358.
- Srikureja W, Kyulo NL, Runyon BA, Hu KQ. MELD score is a better prognostic model than Child-Turcotte-Pugh score or Discriminant Function score in patients with alcoholic hepatitis. J Hepatol 2005; 42:700–706.
- Forrest EH, Morris AJ, Stewart S, et al. The Glasgow alcoholic hepatitis score identifies patients who may benefit from corticosteroids. Gut 2007; 56:1743–1746.
- Dominguez M, Rincón D, Abraldes JG, et al. A new scoring system for prognostic stratification of patients with alcoholic hepatitis. Am J Gastroenterol 2008; 103:2747–2756.
- Louvet A, Naveau S, Abdelnour M, et al. The Lille model: a new tool for therapeutic strategy in patients with severe alcoholic hepatitis treated with steroids. Hepatology 2007; 45:1348–1354.
- Mayo-Smith MF, Beecher LH, Fischer TL, et al; Working Group on the Management of Alcohol Withdrawal Delirium, Practice Guidelines Committee, American Society of Addiction Medicine. Management of alcohol withdrawal delirium. An evidence-based practice guideline. Arch Intern Med 2004; 164:1405–1412.
- Mezey E. Interaction between alcohol and nutrition in the pathogenesis of alcoholic liver disease. Semin Liver Dis 1991; 11:340–348.
- Cabré E, Rodríguez-Iglesias P, Caballería J, et al. Short- and long-term outcome of severe alcohol-induced hepatitis treated with steroids or enteral nutrition: a multicenter randomized trial. Hepatology 2000; 32:36–42.
- Louvet A, Wartel F, Castel H, et al. Infection in patients with severe alcoholic hepatitis treated with steroids: early response to therapy is the key factor. Gastroenterology 2009; 137:541–548.
- European Association for the Study of Liver. EASL clinical practical guidelines: management of alcoholic liver disease. J Hepatol 2012; 57:399–420.
- Rambaldi A, Saconato HH, Christensen E, Thorlund K, Wetterslev J, Gluud C. Systematic review: glucocorticosteroids for alcoholic hepatitis—a Cochrane Hepato-Biliary Group systematic review with meta-analyses and trial sequential analyses of randomized clinical trials. Aliment Pharmacol Ther 2008; 27:1167–1178.
- Powell LW, Axelsen E. Corticosteroids in liver disease: studies on the biological conversion of prednisone to prednisolone and plasma protein binding. Gut 1972; 13:690–696.
- Mathurin P, O’Grady J, Carithers RL, et al. Corticosteroids improve short-term survival in patients with severe alcoholic hepatitis: meta-analysis of individual patient data. Gut 2011; 60:255–260.
- Akriviadis E, Botla R, Briggs W, Han S, Reynolds T, Shakil O. Pentoxifylline improves short-term survival in severe acute alcoholic hepatitis: a double-blind, placebo-controlled trial. Gastroenterology 2000; 119:1637–1648.
- De BK, Gangopadhyay S, Dutta D, Baksi SD, Pani A, Ghosh P. Pentoxifylline versus prednisolone for severe alcoholic hepatitis: a randomized controlled trial. World J Gastroenterol 2009; 15:1613–1619.
- Mathurin P, Louvet A, Dao T, et al. Addition of pentoxifylline to prednisolone for severe alcoholic hepatitis does not improve 6-month survival: results of the CORPENTOX trial (abstract). Hepatology 2011; 54(suppl 1):81A.
- Whitfield K, Rambaldi A, Wetterslev J, Gluud C. Pentoxifylline for alcoholic hepatitis. Cochrane Database Syst Rev 2009; CD007339.
- Louvet A, Diaz E, Dharancy S, et al. Early switch to pentoxifylline in patients with severe alcoholic hepatitis is inefficient in non-responders to corticosteroids. J Hepatol 2008; 48:465–470.
- Thursz MR, Richardson P, Allison ME, et al. Steroids or pentoxifylline for alcoholic hepatitis: results of the STOPAH trial [abstract LB-1]. 65th Annual Meeting of the American Association for the Study of Liver Diseases; November 7–11, 2014; Boston, MA.
- Naveau S, Chollet-Martin S, Dharancy S, et al; Foie-Alcool group of the Association Française pour l’Etude du Foie. A double-blind randomized controlled trial of infliximab associated with prednisolone in acute alcoholic hepatitis. Hepatology 2004; 39:1390–1397.
- Menon KV, Stadheim L, Kamath PS, et al. A pilot study of the safety and tolerability of etanercept in patients with alcoholic hepatitis. Am J Gastroenterol 2004; 99:255–260.
- Boetticher NC, Peine CJ, Kwo P, et al. A randomized, double-blinded, placebo-controlled multicenter trial of etanercept in the treatment of alcoholic hepatitis. Gastroenterology 2008; 135:1953–1960.
- Nguyen-Khac E, Thevenot T, Piquet MA, et al; AAH-NAC Study Group. Glucocorticoids plus N-acetylcysteine in severe alcoholic hepatitis. N Engl J Med 2011; 365:1781–1789.
- Singal AK, Duchini A. Liver transplantation in acute alcoholic hepatitis: current status and future development. World J Hepatol 2011; 3:215–218.
- Singal AK, Bashar H, Anand BS, Jampana SC, Singal V, Kuo YF. Outcomes after liver transplantation for alcoholic hepatitis are similar to alcoholic cirrhosis: exploratory analysis from the UNOS database. Hepatology 2012; 55:1398–1405.
- Mathurin P, Moreno C, Samuel D, et al. Early liver transplantation for severe alcoholic hepatitis. N Engl J Med 2011; 365:1790–1800.
Alcoholic hepatitis, a severe manifestation of alcoholic liver disease, is rising in incidence. Complete abstinence from alcohol remains the cornerstone of treatment, while other specific interventions aim to decrease short-term mortality rates.
Despite current treatments, about 25% of patients with severe alcoholic hepatitis eventually die of it. For those who survive hospitalization, measures need to be taken to prevent recidivism. Although liver transplantation seems to hold promise, early transplantation is still largely experimental in alcoholic hepatitis and will likely be available to only a small subset of patients, especially in view of ethical issues and the possible wider implications for transplant centers.
New treatments will largely depend on a better understanding of the disease’s pathophysiology, and future clinical trials should evaluate therapies that improve short-term as well as long-term outcomes.
ACUTE HEPATIC DECOMPENSATION IN A HEAVY DRINKER
Excessive alcohol consumption is very common worldwide, is a major risk factor for liver disease, and is a leading cause of preventable death. Alcoholic cirrhosis is the eighth most common cause of death in the United States and in 2010 was responsible for nearly half of cirrhosis-related deaths worldwide.1
Alcoholic liver disease is a spectrum. Nearly all heavy drinkers (ie, those consuming 40 g or more of alcohol per day, Table 1) have fatty liver changes, 20% to 40% develop fibrosis, 10% to 20% progress to cirrhosis, and of those with cirrhosis, 1% to 2% are diagnosed with hepatocellular carcinoma every year.2
Within this spectrum, alcoholic hepatitis is a well-defined clinical syndrome characterized by acute hepatic decompensation that typically results from long-standing alcohol abuse. Binge drinkers may also be at risk for alcoholic hepatitis, but good data on the association between drinking patterns and the risk of alcoholic hepatitis are limited.
Alcoholic hepatitis varies in severity from mild to life-threatening.3 Although its exact incidence is unknown, its prevalence in alcoholics has been estimated at 20%.4 Nearly half of patients with alcoholic hepatitis have cirrhosis at the time of their acute presentation, and these patients generally have a poor prognosis, with a 28-day death rate as high as 50% in severe cases.5,6 Moreover, although alcoholic hepatitis develops in only a subset of patients with alcoholic liver disease, hospitalizations for it are increasing in the United States.7
Women are at higher risk of developing alcoholic hepatitis, an observation attributed to the effect of estrogens on oxidative stress and inflammation, lower gastric alcohol dehydrogenase levels resulting in slower first-pass metabolism of alcohol, and higher body fat content causing a lower volume of distribution for alcohol than in men.8 The incidence of alcoholic hepatitis is also influenced by a number of demographic and genetic factors as well as nutritional status and coexistence of other liver diseases.9 Most patients diagnosed with alcoholic hepatitis are active drinkers, but it can develop even after significantly reducing or stopping alcohol consumption.
FATTY ACIDS, ENZYMES, CYTOKINES, INFLAMMATION
Alcohol consumption induces fatty acid synthesis and inhibits fatty acid oxidation, thereby promoting fat deposition in the liver.
The major enzymes involved in alcohol metabolism are cytochrome P450 2E1 (CYP2E1) and alcohol dehydrogenase. CYP2E1 is inducible and is up-regulated when excess alcohol is ingested, while alcohol dehydrogen-
ase function is relatively stable. Oxidative degradation of alcohol by these enzymes generates reactive oxygen species and acetaldehyde, inducing liver injury.10 Interestingly, it has been proposed that variations in the genes for these enzymes influence alcohol consumption and dependency as well as alcohol-driven tissue damage.
In addition, alcohol disrupts the intestinal mucosal barrier, allowing lipopolysaccharides from gram-negative bacteria to travel to the liver via the portal vein. These lipopolysaccharides then bind to and activate sinusoidal Kupffer cells, leading to production of several cytokines such as tumor necrosis factor alpha, interleukin 1, and transforming growth factor beta. These cytokines promote hepatocyte inflammation, apoptosis, and necrosis (Figure 1).11
Besides activating the innate immune system, the reactive oxygen species resulting from alcohol metabolism interact with cellular components, leading to production of protein adducts. These act as antigens that activate the adaptive immune response, followed by B- and T-lymphocyte infiltration, which in turn contribute to liver injury and inflammation.12
THE DIAGNOSIS IS MAINLY CLINICAL
The diagnosis of alcoholic hepatitis is mainly clinical. In its usual presentation, jaundice develops rapidly in a person with a known history of heavy alcohol use. Other symptoms and signs may include ascites, encephalopathy, and fever. On examination, the liver may be enlarged and tender, and a hepatic bruit has been reported.13
Other classic signs of liver disease such as parotid enlargement, Dupuytren contracture, dilated abdominal wall veins, and spider nevi can be present, but none is highly specific or sensitive for alcoholic hepatitis.
Elevated liver enzymes and other clues
Laboratory tests are important in evaluating potential alcoholic hepatitis, although no single laboratory marker can definitively establish alcohol as the cause of liver disease. To detect alcohol consumption, biochemical markers such as aspartate aminotransferase (AST), alanine aminotransferase (ALT), mean corpuscular volume, carbohydrate-deficient transferrin, and, more commonly, gamma-glutamyl transpeptidase are used.
In the acute setting, typical biochemical derangements in alcoholic hepatitis include elevated AST (up to 2 to 6 times the upper limit of normal; usually less than 300 IU/L) and elevated ALT to a lesser extent,14 with an AST-to-ALT ratio greater than 2. Neutrophilia, anemia, hyperbilirubinemia, and coagulopathy with an elevated international normalized ratio are common.
Patients with alcoholic hepatitis are also prone to develop bacterial infections, and about 7% develop hepatorenal syndrome, itself an ominous sign.15
Imaging studies are valuable in excluding other causes of abnormal liver test results in patients who abuse alcohol, such as biliary obstruction, infiltrative liver diseases, and hepatocellular carcinoma.
Screen for alcohol intake
During the initial evaluation of suspected alcoholic hepatitis, one should screen for excessive drinking. In a US Centers for Disease Control and Prevention study, only one of six US adults, including binge drinkers, said they had ever discussed alcohol consumption with a health professional.16 Many patients with alcoholic liver disease in general and alcoholic hepatitis in particular deny alcohol abuse or underreport their intake.17
Screening tests such as the CAGE questionnaire and the Alcohol Use Disorders Identification Test can be used to assess alcohol dependence or abuse.18,19 The CAGE questionnaire consists of four questions:
- Have you ever felt you should cut down on your drinking?
- Have people annoyed you by criticizing your drinking?
- Have you ever felt guilty about your drinking?
- Have you ever had a drink first thing in the morning (an eye-opener) to steady your nerves or to get rid of a hangover?
A yes answer to two or more questions is considered clinically significant.
Is liver biopsy always needed?
Although alcoholic hepatitis can be suspected on the basis of clinical and biochemical clues, liver biopsy remains the gold standard diagnostic tool. It confirms the clinical diagnosis of alcoholic hepatitis in about 85% of all patients and in up to 95% when significant hyperbilirubinemia is present.20
However, whether a particular patient needs a biopsy is not always clear. The American Association for the Study of Liver Diseases (AASLD) recommends biopsy in patients who have a clinical diagnosis of severe alcoholic hepatitis for whom medical treatment is being considered and in those with an uncertain underlying diagnosis.
Findings on liver biopsy in alcoholic hepatitis include steatosis, hepatocyte ballooning, neutrophilic infiltration, Mallory bodies (which represent aggregated cytokeratin intermediate filaments and other proteins), and scarring with a typical perivenular distribution as opposed to the periportal fibrosis seen in chronic viral hepatitis. Some histologic findings, such as centrilobular necrosis, may overlap alcoholic hepatitis and nonalcoholic steatohepatitis.
In addition to confirming the diagnosis and staging the disease, liver biopsy has prognostic value. The severity of inflammation and cholestatic changes correlates with poor prognosis and may also predict response to corticosteroid treatment in severe cases of alcoholic hepatitis.21
However, the utility of liver biopsy in confirming the diagnosis and assessing the prognosis of alcoholic hepatitis is controversial for several reasons. Coagulopathy, thrombocytopenia, and ascites are all common in patients with alcoholic hepatitis, often making percutaneous liver biopsy contraindicated. Trans-
jugular liver biopsy is not universally available outside tertiary care centers.
Needed is a minimally invasive test for assessing this disease. Breath analysis might be such a test, offering a noninvasive means to study the composition of volatile organic compounds and elemental gases and an attractive method to evaluate health and disease in a patient-friendly manner. Our group devised a model based on breath levels of trimethylamine and pentane. When we tested it, we found that it distinguishes patients with alcoholic hepatitis from those with acute liver decompensation from causes other than alcohol and controls without liver disease with up to 90% sensitivity and 80% specificity.22
ASSESSING THE SEVERITY OF ALCOHOLIC HEPATITIS
Several models have been developed to assess the severity of alcoholic hepatitis and guide treatment decisions (Table 2).
The MDF (Maddrey Discriminant Function)6 system was the first scoring system developed and is still the most widely used. A score of 32 or higher indicates severe alcoholic hepatitis and has been used as the threshold for starting treatment with corticosteroids.6
The MDF has limitations. Patients with a score lower than 32 are considered not to have severe alcoholic hepatitis, but up to 17% of them still die. Also, since it uses the prothrombin time, its results can vary considerably among laboratories, depending on the sensitivity of the thromboplastin reagent used.
The MELD (Model for End-stage Liver Disease) score. Sheth et al23 compared the MELD and the MDF scores in assessing the severity of alcoholic hepatitis. They found that the MELD performed as well as the MDF in predicting 30-day mortality. A MELD score of greater than 11 had a sensitivity in predicting 30-day mortality of 86% and a specificity of 81%, compared with 86% and 48%, respectively, for MDF scores greater than 32.
Another study found a MELD score of 21 to have the highest sensitivity and specificity in predicting mortality (an estimated 90-day death rate of 20%). Thus, a MELD score of 21 is an appropriate threshold for prompt consideration of specific therapies such as corticosteroids.24
The MELD score has become increasingly important in patients with alcoholic hepatitis, as some of them may become candidates for liver transplantation (see below). Also, serial MELD scores in hospitalized patients have prognostic implications, since an increase of 2 or more points in the first week has been shown to predict in-hospital mortality.25
The GAHS (Glasgow Alcoholic Hepatitis Score)26 was shown to identify patients with alcoholic hepatitis who have an especially poor prognosis and need corticosteroid therapy. In those with a GAHS of 9 or higher, the 28-day survival rate was 78% with corticosteroid treatment and 52% without corticosteroid treatment; survival rates at 84 days were 59% and 38%, respectively.26
The ABIC scoring system (Age, Serum Bilirubin, INR, and Serum Creatinine) stratifies patients by risk of death at 90 days27:
- Score less than 6.71: low risk (100% survival)
- A score 6.71–8.99: intermediate risk (70% survival)
- A score 9.0 or higher: high risk (25% survival).
Both the GAHS and ABIC score are limited by lack of external validation.
The Lille score.28 While the above scores are used to identify patients at risk of death from alcoholic hepatitis and to decide on starting corticosteroids, the Lille score is designed to assess response to corticosteroids after 1 week of treatment. It is calculated based on five pretreatment variables and the change in serum bilirubin level at day 7 of corticosteroid therapy. Lille scores range from 0 to 1; a score higher than 0.45 is associated with a 75% mortality rate at 6 months and indicates a lack of response to corticosteroids and that these drugs should be discontinued.28
MANAGEMENT
Supportive treatment
Abstinence from alcohol is the cornerstone of treatment of alcoholic hepatitis. Early management of alcohol abuse or dependence is, therefore, warranted in all patients with alcoholic hepatitis. Referral to addiction specialists, motivational therapies, and anticraving drugs such as baclofen can be utilized.
Treat alcohol withdrawal. Alcoholics who suddenly decrease or discontinue their alcohol use are at high risk of alcohol withdrawal syndrome. Within 24 hours after the last drink, patients can experience increases in their heart rate and blood pressure, along with irritability and hyperreflexia. Within the next few days, more dangerous complications including seizures and delirium tremens can arise.
Alcohol withdrawal symptoms should be treated with short-acting benzodiazepines or clomethiazole, keeping the risk of worsening encephalopathy in mind.29 If present, complications of cirrhosis such as encephalopathy, ascites, and variceal bleeding should be managed.
Nutritional support is important. Protein-calorie malnutrition is common in alcoholics, as are deficiencies of vitamin A, vitamin D, thiamine, folate, pyridoxine, and zinc.30 Although a randomized controlled trial comparing enteral nutrition (2,000 kcal/day) vs corticosteroids (prednisolone 40 mg/day) in patients with alcoholic hepatitis did not show any difference in the 28-day mortality rate, those who received nutritional support and survived the first month had a lower mortality rate than those treated with corticosteroids (8% vs 37%).31 A daily protein intake of 1.5 g per kilogram of body weight is therefore recommended, even in patients with hepatic encephalopathy.15
Combining enteral nutrition and corticosteroid treatment may have a synergistic effect but is yet to be investigated.
Screen for infection. Patients with alcoholic hepatitis should be screened for infection, as about 25% of those with severe alcoholic hepatitis have an infection at admission.32 Since many of these patients meet the criteria for systemic inflammatory response syndrome, infections can be particularly difficult to diagnose. Patients require close clinical monitoring as well as regular pancultures for early detection. Antibiotics are frequently started empirically even though we lack specific evidence-based guidelines on this practice.33
Corticosteroids
Various studies have evaluated the role of corticosteroids in treating alcoholic hepatitis, differing considerably in sample populations, methods, and end points. Although the results of individual trials differ, meta-analyses indicate that corticosteroids have a moderate beneficial effect in patients with severe alcoholic hepatitis.
For example, Rambaldi et al34 performed a meta-analysis that concluded the mortality rate was lower in alcoholic hepatitis patients with MDF scores of at least 32 or hepatic encephalopathy who were treated with corticosteroids than in controls (relative risk 0.37, 95% confidence interval 0.16–0.86).
Therefore, in the absence of contraindications, the AASLD recommends starting corticosteroids in patients with severe alcoholic hepatitis, defined as an MDF score of 32 or higher.21 The preferred agent is oral prednisolone 40 mg daily or parenteral methylprednisolone 32 mg daily for 4 weeks and then tapered over the next 2 to 4 weeks or abruptly discontinued. Because activation of prednisone is decreased in patients with liver disease, prednisolone (the active form) is preferred over prednisone (the inactive precursor).35 In alcoholic hepatitis, the number needed to treat with corticosteroids to prevent one death has been calculated36 at 5.
As mentioned, response to corticosteroids is commonly assessed at 1 week of treatment using the Lille score. A score higher than 0.45 predicts a poor response and should trigger discontinuation of corticosteroids, particularly in those classified as null responders (Lille score > 0.56).
Adverse effects of steroids include sepsis, gastrointestinal bleeding, and steroid psychosis. Of note, patients who have evidence of hepatorenal syndrome or gastrointestinal bleeding tend to have a less favorable response to corticosteroids. Also, while infections were once considered a contraindication to steroid therapy, recent evidence suggests that steroid use might not be precluded in infected patients after appropriate antibiotic therapy. Infections occur in about a quarter of all alcoholic hepatitis patients treated with steroids, more frequently in null responders (42.5%) than in responders (11.1%), which supports corticosteroid discontinuance at 1 week in null responders.32
Pentoxifylline
An oral phosphodiesterase inhibitor, pentoxifylline, also inhibits production of several cytokines, including tumor necrosis factor alpha. At a dose of 400 mg orally three times daily for 4 weeks, pentoxifylline has been used in treating severe alcoholic hepatitis (MDF score ≥ 32) and is recommended especially if corticosteroids are contraindicated, as with sepsis.21
An early double-blind clinical trial randomized patients with severe alcoholic hepatitis to receive either pentoxifylline 400 mg orally three times daily or placebo. Of the patients who received pentoxifylline, 24.5% died during the index hospitalization, compared with 46.1% of patients who received placebo. This survival benefit was mainly related to a markedly lower incidence of hepatorenal syndrome as the cause of death in the pentoxifylline group than in the placebo group (50% vs 91.7% of deaths).37
In a small clinical trial in patients with severe alcoholic hepatitis, pentoxifylline recipients had a higher 3-month survival rate than prednisolone recipients (35.29% vs 14.71%, P = .04).38 However, a larger trial showed no improvement in 6-month survival with the combination of prednisolone and pentoxifylline compared with prednisolone alone (69.9% vs 69.2%, P = .91).39 Also, a meta-analysis of five randomized clinical trials found no survival benefit with pentoxifylline therapy.40
Of note, in the unfortunate subgroup of patients who have a poor response to corticosteroids, no alternative treatment, including pentoxifylline, has been shown to be effective.41
Prednisone or pentoxifylline? Very recently, results of the Steroids or Pentoxifylline for Alcoholic Hepatitis (STOPAH) trial have been released.42 This is a large, multicenter, double-blinded clinical trial that aimed to provide a definitive answer to whether corticosteroids or pentoxifylline (or both) are beneficial in patients with alcoholic hepatitis. The study included 1,103 adult patients with severe alcoholic hepatitis (MDF score ≥ 32) who were randomized to monotherapy with prednisolone or pentoxifylline, combination therapy, or placebo. The primary end point was mortality at 28 days, and secondary end points included mortality at 90 days and at 1 year. Prednisolone reduced 28-day mortality by about 39%. In contrast, the 28-day mortality rate was similar in patients who received pentoxifylline and those who did not. Also, neither drug was significantly associated with a survival benefit beyond 28 days. The investigators concluded that pentoxifylline has no impact on disease progression and should not be used for the treatment of severe alcoholic hepatitis.42
Other tumor necrosis factor alpha inhibitors not recommended
Two other tumor necrosis factor alpha inhibitors, infliximab and etanercept, have been tested in clinical trials in alcoholic hepatitis. Unfortunately, the results were not encouraging, with no major reduction in mortality.43–45 In fact, these trials demonstrated a significantly increased risk of infections in the treatment groups. Therefore, these drugs are not recommended for treating alcoholic hepatitis.
A possible explanation is that tumor necrosis factor alpha plays an important role in liver regeneration, aiding in recovery from alcohol-induced liver injury, and inhibiting it can have deleterious consequences.
Other agents
A number of other agents have undergone clinical trials in alcoholic hepatitis.
N-acetylcysteine, an antioxidant that replenishes glutathione stores in hepatocytes, was evaluated in a randomized clinical trial in combination with prednisolone.46 Although the 1-month mortality rate was significantly lower in the combination group than in the prednisolone-only group (8% vs 24%, P = .006), 3-month and 6-month mortality rates were not. Nonetheless, the rates of infection and hepatorenal syndrome were lower in the combination group. Therefore, corticosteroids and N-acetylcysteine may have synergistic effects, but the optimum duration of N-acetylcysteine therapy needs to be determined in further studies.
Vitamin E, silymarin, propylthiouracil, colchicine, and oxandrolone (an anabolic steroid) have also been studied, but with no convincing benefit.21
Role of liver transplantation
Liver transplantation for alcoholic liver disease has been a topic of great medical and social controversy. The view that alcoholic patients are responsible for their own illness led to caution when contemplating liver transplantation. Many countries require 6 months of abstinence from alcohol before placing a patient on the liver transplant list, posing a major obstacle to patients with alcoholic hepatitis, as almost all are active drinkers at the time of presentation and many will die within 6 months. Reasons for this 6-month rule include donor shortage and risk of recidivism.47
With regard to survival following alcoholic hepatitis, a study utilizing the United Network for Organ Sharing database matched patients with alcoholic hepatitis and alcoholic cirrhosis who underwent liver transplantation. Rates of 5-year graft survival were 75% in those with alcoholic hepatitis and 73% in those with alcoholic cirrhosis (P = .97), and rates of patient survival were 80% and 78% (P = .90), respectively. Proportional regression analysis adjusting for other variables showed no impact of the etiology of liver disease on graft or patient survival. The investigators concluded that liver transplantation could be considered in a select group of patients with alcoholic hepatitis who do not improve with medical therapy.48
In a pivotal case-control prospective study,49 26 patients with Lille scores greater than 0.45 were listed for liver transplantation within a median of 13 days after nonresponse to medical therapy. The cumulative 6-month survival rate was higher in patients who received a liver transplant early than in those who did not (77% vs 23%, P < .001). This benefit was maintained through 2 years of follow-up (hazard ratio 6.08, P = .004). Of note, all these patients had supportive family members, no severe coexisting conditions, and a commitment to alcohol abstinence (although 3 patients resumed drinking after liver transplantation).49
Although these studies support early liver transplantation in carefully selected patients with severe alcoholic hepatitis, the criteria for transplantation in this group need to be refined. Views on alcoholism also need to be reconciled, as strong evidence is emerging that implicates genetic and environmental influences on alcohol dependence.
Management algorithm
Figure 2 shows a suggested management algorithm for alcoholic hepatitis, adapted from the guidelines of the AASLD and European Association for the Study of the Liver.
NEW THERAPIES NEEDED
Novel therapies for severe alcoholic hepatitis are urgently needed to help combat this devastating condition. Advances in understanding its pathophysiology have uncovered several new therapeutic targets, and new agents are already being evaluated in clinical trials.
IMM 124-E, a hyperimmune bovine colostrum enriched with immunoglobulin G anti-lipopolysaccharide, is going to be evaluated in combination with prednisolone in patients with severe alcoholic hepatitis.
Anakinra, an interleukin 1 receptor antagonist, has significant anti-inflammatory activity and is used to treat rheumatoid arthritis. A clinical trial to evaluate its role in alcoholic hepatitis has been designed in which patients with severe alcoholic hepatitis (defined as a MELD score ≥ 21) will be randomized to receive either methylprednisolone or a combination of anakinra, pentoxifylline, and zinc (a mineral that improves gut integrity).
Emricasan, an orally active caspase protease inhibitor, is another agent currently being tested in a phase 2 clinical trial in patients with severe alcoholic hepatitis. Since caspases induce apoptosis, inhibiting them should theoretically dampen alcohol-induced hepatocyte injury.
Interleukin 22, a hepatoprotective cytokine, shows promise as a treatment and will soon be evaluated in alcoholic hepatitis.
Alcoholic hepatitis, a severe manifestation of alcoholic liver disease, is rising in incidence. Complete abstinence from alcohol remains the cornerstone of treatment, while other specific interventions aim to decrease short-term mortality rates.
Despite current treatments, about 25% of patients with severe alcoholic hepatitis eventually die of it. For those who survive hospitalization, measures need to be taken to prevent recidivism. Although liver transplantation seems to hold promise, early transplantation is still largely experimental in alcoholic hepatitis and will likely be available to only a small subset of patients, especially in view of ethical issues and the possible wider implications for transplant centers.
New treatments will largely depend on a better understanding of the disease’s pathophysiology, and future clinical trials should evaluate therapies that improve short-term as well as long-term outcomes.
ACUTE HEPATIC DECOMPENSATION IN A HEAVY DRINKER
Excessive alcohol consumption is very common worldwide, is a major risk factor for liver disease, and is a leading cause of preventable death. Alcoholic cirrhosis is the eighth most common cause of death in the United States and in 2010 was responsible for nearly half of cirrhosis-related deaths worldwide.1
Alcoholic liver disease is a spectrum. Nearly all heavy drinkers (ie, those consuming 40 g or more of alcohol per day, Table 1) have fatty liver changes, 20% to 40% develop fibrosis, 10% to 20% progress to cirrhosis, and of those with cirrhosis, 1% to 2% are diagnosed with hepatocellular carcinoma every year.2
Within this spectrum, alcoholic hepatitis is a well-defined clinical syndrome characterized by acute hepatic decompensation that typically results from long-standing alcohol abuse. Binge drinkers may also be at risk for alcoholic hepatitis, but good data on the association between drinking patterns and the risk of alcoholic hepatitis are limited.
Alcoholic hepatitis varies in severity from mild to life-threatening.3 Although its exact incidence is unknown, its prevalence in alcoholics has been estimated at 20%.4 Nearly half of patients with alcoholic hepatitis have cirrhosis at the time of their acute presentation, and these patients generally have a poor prognosis, with a 28-day death rate as high as 50% in severe cases.5,6 Moreover, although alcoholic hepatitis develops in only a subset of patients with alcoholic liver disease, hospitalizations for it are increasing in the United States.7
Women are at higher risk of developing alcoholic hepatitis, an observation attributed to the effect of estrogens on oxidative stress and inflammation, lower gastric alcohol dehydrogenase levels resulting in slower first-pass metabolism of alcohol, and higher body fat content causing a lower volume of distribution for alcohol than in men.8 The incidence of alcoholic hepatitis is also influenced by a number of demographic and genetic factors as well as nutritional status and coexistence of other liver diseases.9 Most patients diagnosed with alcoholic hepatitis are active drinkers, but it can develop even after significantly reducing or stopping alcohol consumption.
FATTY ACIDS, ENZYMES, CYTOKINES, INFLAMMATION
Alcohol consumption induces fatty acid synthesis and inhibits fatty acid oxidation, thereby promoting fat deposition in the liver.
The major enzymes involved in alcohol metabolism are cytochrome P450 2E1 (CYP2E1) and alcohol dehydrogenase. CYP2E1 is inducible and is up-regulated when excess alcohol is ingested, while alcohol dehydrogen-
ase function is relatively stable. Oxidative degradation of alcohol by these enzymes generates reactive oxygen species and acetaldehyde, inducing liver injury.10 Interestingly, it has been proposed that variations in the genes for these enzymes influence alcohol consumption and dependency as well as alcohol-driven tissue damage.
In addition, alcohol disrupts the intestinal mucosal barrier, allowing lipopolysaccharides from gram-negative bacteria to travel to the liver via the portal vein. These lipopolysaccharides then bind to and activate sinusoidal Kupffer cells, leading to production of several cytokines such as tumor necrosis factor alpha, interleukin 1, and transforming growth factor beta. These cytokines promote hepatocyte inflammation, apoptosis, and necrosis (Figure 1).11
Besides activating the innate immune system, the reactive oxygen species resulting from alcohol metabolism interact with cellular components, leading to production of protein adducts. These act as antigens that activate the adaptive immune response, followed by B- and T-lymphocyte infiltration, which in turn contribute to liver injury and inflammation.12
THE DIAGNOSIS IS MAINLY CLINICAL
The diagnosis of alcoholic hepatitis is mainly clinical. In its usual presentation, jaundice develops rapidly in a person with a known history of heavy alcohol use. Other symptoms and signs may include ascites, encephalopathy, and fever. On examination, the liver may be enlarged and tender, and a hepatic bruit has been reported.13
Other classic signs of liver disease such as parotid enlargement, Dupuytren contracture, dilated abdominal wall veins, and spider nevi can be present, but none is highly specific or sensitive for alcoholic hepatitis.
Elevated liver enzymes and other clues
Laboratory tests are important in evaluating potential alcoholic hepatitis, although no single laboratory marker can definitively establish alcohol as the cause of liver disease. To detect alcohol consumption, biochemical markers such as aspartate aminotransferase (AST), alanine aminotransferase (ALT), mean corpuscular volume, carbohydrate-deficient transferrin, and, more commonly, gamma-glutamyl transpeptidase are used.
In the acute setting, typical biochemical derangements in alcoholic hepatitis include elevated AST (up to 2 to 6 times the upper limit of normal; usually less than 300 IU/L) and elevated ALT to a lesser extent,14 with an AST-to-ALT ratio greater than 2. Neutrophilia, anemia, hyperbilirubinemia, and coagulopathy with an elevated international normalized ratio are common.
Patients with alcoholic hepatitis are also prone to develop bacterial infections, and about 7% develop hepatorenal syndrome, itself an ominous sign.15
Imaging studies are valuable in excluding other causes of abnormal liver test results in patients who abuse alcohol, such as biliary obstruction, infiltrative liver diseases, and hepatocellular carcinoma.
Screen for alcohol intake
During the initial evaluation of suspected alcoholic hepatitis, one should screen for excessive drinking. In a US Centers for Disease Control and Prevention study, only one of six US adults, including binge drinkers, said they had ever discussed alcohol consumption with a health professional.16 Many patients with alcoholic liver disease in general and alcoholic hepatitis in particular deny alcohol abuse or underreport their intake.17
Screening tests such as the CAGE questionnaire and the Alcohol Use Disorders Identification Test can be used to assess alcohol dependence or abuse.18,19 The CAGE questionnaire consists of four questions:
- Have you ever felt you should cut down on your drinking?
- Have people annoyed you by criticizing your drinking?
- Have you ever felt guilty about your drinking?
- Have you ever had a drink first thing in the morning (an eye-opener) to steady your nerves or to get rid of a hangover?
A yes answer to two or more questions is considered clinically significant.
Is liver biopsy always needed?
Although alcoholic hepatitis can be suspected on the basis of clinical and biochemical clues, liver biopsy remains the gold standard diagnostic tool. It confirms the clinical diagnosis of alcoholic hepatitis in about 85% of all patients and in up to 95% when significant hyperbilirubinemia is present.20
However, whether a particular patient needs a biopsy is not always clear. The American Association for the Study of Liver Diseases (AASLD) recommends biopsy in patients who have a clinical diagnosis of severe alcoholic hepatitis for whom medical treatment is being considered and in those with an uncertain underlying diagnosis.
Findings on liver biopsy in alcoholic hepatitis include steatosis, hepatocyte ballooning, neutrophilic infiltration, Mallory bodies (which represent aggregated cytokeratin intermediate filaments and other proteins), and scarring with a typical perivenular distribution as opposed to the periportal fibrosis seen in chronic viral hepatitis. Some histologic findings, such as centrilobular necrosis, may overlap alcoholic hepatitis and nonalcoholic steatohepatitis.
In addition to confirming the diagnosis and staging the disease, liver biopsy has prognostic value. The severity of inflammation and cholestatic changes correlates with poor prognosis and may also predict response to corticosteroid treatment in severe cases of alcoholic hepatitis.21
However, the utility of liver biopsy in confirming the diagnosis and assessing the prognosis of alcoholic hepatitis is controversial for several reasons. Coagulopathy, thrombocytopenia, and ascites are all common in patients with alcoholic hepatitis, often making percutaneous liver biopsy contraindicated. Trans-
jugular liver biopsy is not universally available outside tertiary care centers.
Needed is a minimally invasive test for assessing this disease. Breath analysis might be such a test, offering a noninvasive means to study the composition of volatile organic compounds and elemental gases and an attractive method to evaluate health and disease in a patient-friendly manner. Our group devised a model based on breath levels of trimethylamine and pentane. When we tested it, we found that it distinguishes patients with alcoholic hepatitis from those with acute liver decompensation from causes other than alcohol and controls without liver disease with up to 90% sensitivity and 80% specificity.22
ASSESSING THE SEVERITY OF ALCOHOLIC HEPATITIS
Several models have been developed to assess the severity of alcoholic hepatitis and guide treatment decisions (Table 2).
The MDF (Maddrey Discriminant Function)6 system was the first scoring system developed and is still the most widely used. A score of 32 or higher indicates severe alcoholic hepatitis and has been used as the threshold for starting treatment with corticosteroids.6
The MDF has limitations. Patients with a score lower than 32 are considered not to have severe alcoholic hepatitis, but up to 17% of them still die. Also, since it uses the prothrombin time, its results can vary considerably among laboratories, depending on the sensitivity of the thromboplastin reagent used.
The MELD (Model for End-stage Liver Disease) score. Sheth et al23 compared the MELD and the MDF scores in assessing the severity of alcoholic hepatitis. They found that the MELD performed as well as the MDF in predicting 30-day mortality. A MELD score of greater than 11 had a sensitivity in predicting 30-day mortality of 86% and a specificity of 81%, compared with 86% and 48%, respectively, for MDF scores greater than 32.
Another study found a MELD score of 21 to have the highest sensitivity and specificity in predicting mortality (an estimated 90-day death rate of 20%). Thus, a MELD score of 21 is an appropriate threshold for prompt consideration of specific therapies such as corticosteroids.24
The MELD score has become increasingly important in patients with alcoholic hepatitis, as some of them may become candidates for liver transplantation (see below). Also, serial MELD scores in hospitalized patients have prognostic implications, since an increase of 2 or more points in the first week has been shown to predict in-hospital mortality.25
The GAHS (Glasgow Alcoholic Hepatitis Score)26 was shown to identify patients with alcoholic hepatitis who have an especially poor prognosis and need corticosteroid therapy. In those with a GAHS of 9 or higher, the 28-day survival rate was 78% with corticosteroid treatment and 52% without corticosteroid treatment; survival rates at 84 days were 59% and 38%, respectively.26
The ABIC scoring system (Age, Serum Bilirubin, INR, and Serum Creatinine) stratifies patients by risk of death at 90 days27:
- Score less than 6.71: low risk (100% survival)
- A score 6.71–8.99: intermediate risk (70% survival)
- A score 9.0 or higher: high risk (25% survival).
Both the GAHS and ABIC score are limited by lack of external validation.
The Lille score.28 While the above scores are used to identify patients at risk of death from alcoholic hepatitis and to decide on starting corticosteroids, the Lille score is designed to assess response to corticosteroids after 1 week of treatment. It is calculated based on five pretreatment variables and the change in serum bilirubin level at day 7 of corticosteroid therapy. Lille scores range from 0 to 1; a score higher than 0.45 is associated with a 75% mortality rate at 6 months and indicates a lack of response to corticosteroids and that these drugs should be discontinued.28
MANAGEMENT
Supportive treatment
Abstinence from alcohol is the cornerstone of treatment of alcoholic hepatitis. Early management of alcohol abuse or dependence is, therefore, warranted in all patients with alcoholic hepatitis. Referral to addiction specialists, motivational therapies, and anticraving drugs such as baclofen can be utilized.
Treat alcohol withdrawal. Alcoholics who suddenly decrease or discontinue their alcohol use are at high risk of alcohol withdrawal syndrome. Within 24 hours after the last drink, patients can experience increases in their heart rate and blood pressure, along with irritability and hyperreflexia. Within the next few days, more dangerous complications including seizures and delirium tremens can arise.
Alcohol withdrawal symptoms should be treated with short-acting benzodiazepines or clomethiazole, keeping the risk of worsening encephalopathy in mind.29 If present, complications of cirrhosis such as encephalopathy, ascites, and variceal bleeding should be managed.
Nutritional support is important. Protein-calorie malnutrition is common in alcoholics, as are deficiencies of vitamin A, vitamin D, thiamine, folate, pyridoxine, and zinc.30 Although a randomized controlled trial comparing enteral nutrition (2,000 kcal/day) vs corticosteroids (prednisolone 40 mg/day) in patients with alcoholic hepatitis did not show any difference in the 28-day mortality rate, those who received nutritional support and survived the first month had a lower mortality rate than those treated with corticosteroids (8% vs 37%).31 A daily protein intake of 1.5 g per kilogram of body weight is therefore recommended, even in patients with hepatic encephalopathy.15
Combining enteral nutrition and corticosteroid treatment may have a synergistic effect but is yet to be investigated.
Screen for infection. Patients with alcoholic hepatitis should be screened for infection, as about 25% of those with severe alcoholic hepatitis have an infection at admission.32 Since many of these patients meet the criteria for systemic inflammatory response syndrome, infections can be particularly difficult to diagnose. Patients require close clinical monitoring as well as regular pancultures for early detection. Antibiotics are frequently started empirically even though we lack specific evidence-based guidelines on this practice.33
Corticosteroids
Various studies have evaluated the role of corticosteroids in treating alcoholic hepatitis, differing considerably in sample populations, methods, and end points. Although the results of individual trials differ, meta-analyses indicate that corticosteroids have a moderate beneficial effect in patients with severe alcoholic hepatitis.
For example, Rambaldi et al34 performed a meta-analysis that concluded the mortality rate was lower in alcoholic hepatitis patients with MDF scores of at least 32 or hepatic encephalopathy who were treated with corticosteroids than in controls (relative risk 0.37, 95% confidence interval 0.16–0.86).
Therefore, in the absence of contraindications, the AASLD recommends starting corticosteroids in patients with severe alcoholic hepatitis, defined as an MDF score of 32 or higher.21 The preferred agent is oral prednisolone 40 mg daily or parenteral methylprednisolone 32 mg daily for 4 weeks and then tapered over the next 2 to 4 weeks or abruptly discontinued. Because activation of prednisone is decreased in patients with liver disease, prednisolone (the active form) is preferred over prednisone (the inactive precursor).35 In alcoholic hepatitis, the number needed to treat with corticosteroids to prevent one death has been calculated36 at 5.
As mentioned, response to corticosteroids is commonly assessed at 1 week of treatment using the Lille score. A score higher than 0.45 predicts a poor response and should trigger discontinuation of corticosteroids, particularly in those classified as null responders (Lille score > 0.56).
Adverse effects of steroids include sepsis, gastrointestinal bleeding, and steroid psychosis. Of note, patients who have evidence of hepatorenal syndrome or gastrointestinal bleeding tend to have a less favorable response to corticosteroids. Also, while infections were once considered a contraindication to steroid therapy, recent evidence suggests that steroid use might not be precluded in infected patients after appropriate antibiotic therapy. Infections occur in about a quarter of all alcoholic hepatitis patients treated with steroids, more frequently in null responders (42.5%) than in responders (11.1%), which supports corticosteroid discontinuance at 1 week in null responders.32
Pentoxifylline
An oral phosphodiesterase inhibitor, pentoxifylline, also inhibits production of several cytokines, including tumor necrosis factor alpha. At a dose of 400 mg orally three times daily for 4 weeks, pentoxifylline has been used in treating severe alcoholic hepatitis (MDF score ≥ 32) and is recommended especially if corticosteroids are contraindicated, as with sepsis.21
An early double-blind clinical trial randomized patients with severe alcoholic hepatitis to receive either pentoxifylline 400 mg orally three times daily or placebo. Of the patients who received pentoxifylline, 24.5% died during the index hospitalization, compared with 46.1% of patients who received placebo. This survival benefit was mainly related to a markedly lower incidence of hepatorenal syndrome as the cause of death in the pentoxifylline group than in the placebo group (50% vs 91.7% of deaths).37
In a small clinical trial in patients with severe alcoholic hepatitis, pentoxifylline recipients had a higher 3-month survival rate than prednisolone recipients (35.29% vs 14.71%, P = .04).38 However, a larger trial showed no improvement in 6-month survival with the combination of prednisolone and pentoxifylline compared with prednisolone alone (69.9% vs 69.2%, P = .91).39 Also, a meta-analysis of five randomized clinical trials found no survival benefit with pentoxifylline therapy.40
Of note, in the unfortunate subgroup of patients who have a poor response to corticosteroids, no alternative treatment, including pentoxifylline, has been shown to be effective.41
Prednisone or pentoxifylline? Very recently, results of the Steroids or Pentoxifylline for Alcoholic Hepatitis (STOPAH) trial have been released.42 This is a large, multicenter, double-blinded clinical trial that aimed to provide a definitive answer to whether corticosteroids or pentoxifylline (or both) are beneficial in patients with alcoholic hepatitis. The study included 1,103 adult patients with severe alcoholic hepatitis (MDF score ≥ 32) who were randomized to monotherapy with prednisolone or pentoxifylline, combination therapy, or placebo. The primary end point was mortality at 28 days, and secondary end points included mortality at 90 days and at 1 year. Prednisolone reduced 28-day mortality by about 39%. In contrast, the 28-day mortality rate was similar in patients who received pentoxifylline and those who did not. Also, neither drug was significantly associated with a survival benefit beyond 28 days. The investigators concluded that pentoxifylline has no impact on disease progression and should not be used for the treatment of severe alcoholic hepatitis.42
Other tumor necrosis factor alpha inhibitors not recommended
Two other tumor necrosis factor alpha inhibitors, infliximab and etanercept, have been tested in clinical trials in alcoholic hepatitis. Unfortunately, the results were not encouraging, with no major reduction in mortality.43–45 In fact, these trials demonstrated a significantly increased risk of infections in the treatment groups. Therefore, these drugs are not recommended for treating alcoholic hepatitis.
A possible explanation is that tumor necrosis factor alpha plays an important role in liver regeneration, aiding in recovery from alcohol-induced liver injury, and inhibiting it can have deleterious consequences.
Other agents
A number of other agents have undergone clinical trials in alcoholic hepatitis.
N-acetylcysteine, an antioxidant that replenishes glutathione stores in hepatocytes, was evaluated in a randomized clinical trial in combination with prednisolone.46 Although the 1-month mortality rate was significantly lower in the combination group than in the prednisolone-only group (8% vs 24%, P = .006), 3-month and 6-month mortality rates were not. Nonetheless, the rates of infection and hepatorenal syndrome were lower in the combination group. Therefore, corticosteroids and N-acetylcysteine may have synergistic effects, but the optimum duration of N-acetylcysteine therapy needs to be determined in further studies.
Vitamin E, silymarin, propylthiouracil, colchicine, and oxandrolone (an anabolic steroid) have also been studied, but with no convincing benefit.21
Role of liver transplantation
Liver transplantation for alcoholic liver disease has been a topic of great medical and social controversy. The view that alcoholic patients are responsible for their own illness led to caution when contemplating liver transplantation. Many countries require 6 months of abstinence from alcohol before placing a patient on the liver transplant list, posing a major obstacle to patients with alcoholic hepatitis, as almost all are active drinkers at the time of presentation and many will die within 6 months. Reasons for this 6-month rule include donor shortage and risk of recidivism.47
With regard to survival following alcoholic hepatitis, a study utilizing the United Network for Organ Sharing database matched patients with alcoholic hepatitis and alcoholic cirrhosis who underwent liver transplantation. Rates of 5-year graft survival were 75% in those with alcoholic hepatitis and 73% in those with alcoholic cirrhosis (P = .97), and rates of patient survival were 80% and 78% (P = .90), respectively. Proportional regression analysis adjusting for other variables showed no impact of the etiology of liver disease on graft or patient survival. The investigators concluded that liver transplantation could be considered in a select group of patients with alcoholic hepatitis who do not improve with medical therapy.48
In a pivotal case-control prospective study,49 26 patients with Lille scores greater than 0.45 were listed for liver transplantation within a median of 13 days after nonresponse to medical therapy. The cumulative 6-month survival rate was higher in patients who received a liver transplant early than in those who did not (77% vs 23%, P < .001). This benefit was maintained through 2 years of follow-up (hazard ratio 6.08, P = .004). Of note, all these patients had supportive family members, no severe coexisting conditions, and a commitment to alcohol abstinence (although 3 patients resumed drinking after liver transplantation).49
Although these studies support early liver transplantation in carefully selected patients with severe alcoholic hepatitis, the criteria for transplantation in this group need to be refined. Views on alcoholism also need to be reconciled, as strong evidence is emerging that implicates genetic and environmental influences on alcohol dependence.
Management algorithm
Figure 2 shows a suggested management algorithm for alcoholic hepatitis, adapted from the guidelines of the AASLD and European Association for the Study of the Liver.
NEW THERAPIES NEEDED
Novel therapies for severe alcoholic hepatitis are urgently needed to help combat this devastating condition. Advances in understanding its pathophysiology have uncovered several new therapeutic targets, and new agents are already being evaluated in clinical trials.
IMM 124-E, a hyperimmune bovine colostrum enriched with immunoglobulin G anti-lipopolysaccharide, is going to be evaluated in combination with prednisolone in patients with severe alcoholic hepatitis.
Anakinra, an interleukin 1 receptor antagonist, has significant anti-inflammatory activity and is used to treat rheumatoid arthritis. A clinical trial to evaluate its role in alcoholic hepatitis has been designed in which patients with severe alcoholic hepatitis (defined as a MELD score ≥ 21) will be randomized to receive either methylprednisolone or a combination of anakinra, pentoxifylline, and zinc (a mineral that improves gut integrity).
Emricasan, an orally active caspase protease inhibitor, is another agent currently being tested in a phase 2 clinical trial in patients with severe alcoholic hepatitis. Since caspases induce apoptosis, inhibiting them should theoretically dampen alcohol-induced hepatocyte injury.
Interleukin 22, a hepatoprotective cytokine, shows promise as a treatment and will soon be evaluated in alcoholic hepatitis.
- Rehm J, Samokhvalov AV, Shield KD. Global burden of alcoholic liver diseases. J Hepatol 2013; 59:160–168.
- Teli MR, Day CP, Burt AD, Bennett MK, James OF. Determinants of progression to cirrhosis or fibrosis in pure alcoholic fatty liver. Lancet 1995; 346:987–990.
- Alcoholic liver disease: morphological manifestations. Review by an international group. Lancet 1981; 1:707–711.
- Naveau S, Giraud V, Borotto E, Aubert A, Capron F, Chaput JC. Excess weight risk factor for alcoholic liver disease. Hepatology 1997; 25:108–111.
- Basra S, Anand BS. Definition, epidemiology and magnitude of alcoholic hepatitis. World J Hepatol 2011; 3:108–113.
- Maddrey WC, Boitnott JK, Bedine MS, Weber FL Jr, Mezey E, White RI Jr. Corticosteroid therapy of alcoholic hepatitis. Gastroenterology 1978; 75:193–199.
- Jinjuvadia R, Liangpunsakul S, for the Translational Research and Evolving Alcoholic Hepatitis Treatment Consortium. Trends in alcoholic hepatitis-related hospitalizations, financial burden, and mortality in the United States. J Clin Gastroenterol 2014 Jun 25 (Epub ahead of print).
- Sato N, Lindros KO, Baraona E, et al. Sex difference in alcohol-related organ injury. Alcohol Clin Exp Res 2001; 25(suppl s1):40S–45S.
- Singal AK, Kamath PS, Gores GJ, Shah VH. Alcoholic hepatitis: current challenges and future directions. Clin Gastroenterol Hepatol 2014; 12:555–564.
- Seitz HK, Stickel F. Risk factors and mechanisms of hepatocarcinogenesis with special emphasis on alcohol and oxidative stress. Biol Chem 2006; 387:349–360.
- Thurman RG. II. Alcoholic liver injury involves activation of Kupffer cells by endotoxin. Am J Physiol 1998; 275:G605–G611.
- Duddempudi AT. Immunology in alcoholic liver disease. Clin Liver Dis 2012; 16:687–698.
- Lischner MW, Alexander JF, Galambos JT. Natural history of alcoholic hepatitis. I. The acute disease. Am J Dig Dis 1971; 16:481–494.
- Cohen JA, Kaplan MM. The SGOT/SGPT ratio—an indicator of alcoholic liver disease. Dig Dis Sci 1979; 24:835–838.
- Lucey MR, Mathurin P, Morgan TR. Alcoholic hepatitis. N Engl J Med 2009; 360:2758–2769.
- McKnight-Eily LR, Liu Y, Brewer RD, et al; Centers for Disease Control and Prevention (CDC). Vital signs: communication between health professionals and their patients about alcohol use—44 states and the District of Columbia, 2011. MMWR Morb Mortal Wkly Rep 2014; 63:16–22.
- Grant BF. Barriers to alcoholism treatment: reasons for not seeking treatment in a general population sample. J Stud Alcohol 1997; 58:365–371.
- Aertgeerts B, Buntinx F, Kester A. The value of the CAGE in screening for alcohol abuse and alcohol dependence in general clinical populations: a diagnostic meta-analysis. J Clin Epidemiol 2004; 57:30–39.
- The Alcohol Use Disorders Identification Test Guidelines for Use in Primary Care. Second Edition. World Health Organization. Department of Mental Health and Substance Dependence. http://whqlibdoc.who.int/hq/2001/who_msd_msb_01.6a.pdf. Accessed February 3, 2015.
- Hamid R, Forrest EH. Is histology required for the diagnosis of alcoholic hepatitis? A review of published randomised controlled trials. Gut 2011; 60(suppl 1):A233.
- O’Shea RS, Dasarathy S, McCullough AJ; Practice Guideline Committee of the American Association for the Study of Liver Diseases; Practice Parameters Committee of the American College of Gastroenterology. Alcoholic liver disease. Hepatology 2010; 51:307–328.
- Hanouneh IA, Zein NN, Cikach F, et al. The breathprints in patients with liver disease identify novel breath biomarkers in alcoholic hepatitis. Clin Gastroenterol Hepatol 2014; 12:516–523.
- Sheth M, Riggs M, Patel T. Utility of the Mayo End-Stage Liver Disease (MELD) score in assessing prognosis of patients with alcoholic hepatitis. BMC Gastroenterol 2002; 2:2.
- Dunn W, Jamil LH, Brown LS, et al. MELD accurately predicts mortality in patients with alcoholic hepatitis. Hepatology 2005; 41:353–358.
- Srikureja W, Kyulo NL, Runyon BA, Hu KQ. MELD score is a better prognostic model than Child-Turcotte-Pugh score or Discriminant Function score in patients with alcoholic hepatitis. J Hepatol 2005; 42:700–706.
- Forrest EH, Morris AJ, Stewart S, et al. The Glasgow alcoholic hepatitis score identifies patients who may benefit from corticosteroids. Gut 2007; 56:1743–1746.
- Dominguez M, Rincón D, Abraldes JG, et al. A new scoring system for prognostic stratification of patients with alcoholic hepatitis. Am J Gastroenterol 2008; 103:2747–2756.
- Louvet A, Naveau S, Abdelnour M, et al. The Lille model: a new tool for therapeutic strategy in patients with severe alcoholic hepatitis treated with steroids. Hepatology 2007; 45:1348–1354.
- Mayo-Smith MF, Beecher LH, Fischer TL, et al; Working Group on the Management of Alcohol Withdrawal Delirium, Practice Guidelines Committee, American Society of Addiction Medicine. Management of alcohol withdrawal delirium. An evidence-based practice guideline. Arch Intern Med 2004; 164:1405–1412.
- Mezey E. Interaction between alcohol and nutrition in the pathogenesis of alcoholic liver disease. Semin Liver Dis 1991; 11:340–348.
- Cabré E, Rodríguez-Iglesias P, Caballería J, et al. Short- and long-term outcome of severe alcohol-induced hepatitis treated with steroids or enteral nutrition: a multicenter randomized trial. Hepatology 2000; 32:36–42.
- Louvet A, Wartel F, Castel H, et al. Infection in patients with severe alcoholic hepatitis treated with steroids: early response to therapy is the key factor. Gastroenterology 2009; 137:541–548.
- European Association for the Study of Liver. EASL clinical practical guidelines: management of alcoholic liver disease. J Hepatol 2012; 57:399–420.
- Rambaldi A, Saconato HH, Christensen E, Thorlund K, Wetterslev J, Gluud C. Systematic review: glucocorticosteroids for alcoholic hepatitis—a Cochrane Hepato-Biliary Group systematic review with meta-analyses and trial sequential analyses of randomized clinical trials. Aliment Pharmacol Ther 2008; 27:1167–1178.
- Powell LW, Axelsen E. Corticosteroids in liver disease: studies on the biological conversion of prednisone to prednisolone and plasma protein binding. Gut 1972; 13:690–696.
- Mathurin P, O’Grady J, Carithers RL, et al. Corticosteroids improve short-term survival in patients with severe alcoholic hepatitis: meta-analysis of individual patient data. Gut 2011; 60:255–260.
- Akriviadis E, Botla R, Briggs W, Han S, Reynolds T, Shakil O. Pentoxifylline improves short-term survival in severe acute alcoholic hepatitis: a double-blind, placebo-controlled trial. Gastroenterology 2000; 119:1637–1648.
- De BK, Gangopadhyay S, Dutta D, Baksi SD, Pani A, Ghosh P. Pentoxifylline versus prednisolone for severe alcoholic hepatitis: a randomized controlled trial. World J Gastroenterol 2009; 15:1613–1619.
- Mathurin P, Louvet A, Dao T, et al. Addition of pentoxifylline to prednisolone for severe alcoholic hepatitis does not improve 6-month survival: results of the CORPENTOX trial (abstract). Hepatology 2011; 54(suppl 1):81A.
- Whitfield K, Rambaldi A, Wetterslev J, Gluud C. Pentoxifylline for alcoholic hepatitis. Cochrane Database Syst Rev 2009; CD007339.
- Louvet A, Diaz E, Dharancy S, et al. Early switch to pentoxifylline in patients with severe alcoholic hepatitis is inefficient in non-responders to corticosteroids. J Hepatol 2008; 48:465–470.
- Thursz MR, Richardson P, Allison ME, et al. Steroids or pentoxifylline for alcoholic hepatitis: results of the STOPAH trial [abstract LB-1]. 65th Annual Meeting of the American Association for the Study of Liver Diseases; November 7–11, 2014; Boston, MA.
- Naveau S, Chollet-Martin S, Dharancy S, et al; Foie-Alcool group of the Association Française pour l’Etude du Foie. A double-blind randomized controlled trial of infliximab associated with prednisolone in acute alcoholic hepatitis. Hepatology 2004; 39:1390–1397.
- Menon KV, Stadheim L, Kamath PS, et al. A pilot study of the safety and tolerability of etanercept in patients with alcoholic hepatitis. Am J Gastroenterol 2004; 99:255–260.
- Boetticher NC, Peine CJ, Kwo P, et al. A randomized, double-blinded, placebo-controlled multicenter trial of etanercept in the treatment of alcoholic hepatitis. Gastroenterology 2008; 135:1953–1960.
- Nguyen-Khac E, Thevenot T, Piquet MA, et al; AAH-NAC Study Group. Glucocorticoids plus N-acetylcysteine in severe alcoholic hepatitis. N Engl J Med 2011; 365:1781–1789.
- Singal AK, Duchini A. Liver transplantation in acute alcoholic hepatitis: current status and future development. World J Hepatol 2011; 3:215–218.
- Singal AK, Bashar H, Anand BS, Jampana SC, Singal V, Kuo YF. Outcomes after liver transplantation for alcoholic hepatitis are similar to alcoholic cirrhosis: exploratory analysis from the UNOS database. Hepatology 2012; 55:1398–1405.
- Mathurin P, Moreno C, Samuel D, et al. Early liver transplantation for severe alcoholic hepatitis. N Engl J Med 2011; 365:1790–1800.
- Rehm J, Samokhvalov AV, Shield KD. Global burden of alcoholic liver diseases. J Hepatol 2013; 59:160–168.
- Teli MR, Day CP, Burt AD, Bennett MK, James OF. Determinants of progression to cirrhosis or fibrosis in pure alcoholic fatty liver. Lancet 1995; 346:987–990.
- Alcoholic liver disease: morphological manifestations. Review by an international group. Lancet 1981; 1:707–711.
- Naveau S, Giraud V, Borotto E, Aubert A, Capron F, Chaput JC. Excess weight risk factor for alcoholic liver disease. Hepatology 1997; 25:108–111.
- Basra S, Anand BS. Definition, epidemiology and magnitude of alcoholic hepatitis. World J Hepatol 2011; 3:108–113.
- Maddrey WC, Boitnott JK, Bedine MS, Weber FL Jr, Mezey E, White RI Jr. Corticosteroid therapy of alcoholic hepatitis. Gastroenterology 1978; 75:193–199.
- Jinjuvadia R, Liangpunsakul S, for the Translational Research and Evolving Alcoholic Hepatitis Treatment Consortium. Trends in alcoholic hepatitis-related hospitalizations, financial burden, and mortality in the United States. J Clin Gastroenterol 2014 Jun 25 (Epub ahead of print).
- Sato N, Lindros KO, Baraona E, et al. Sex difference in alcohol-related organ injury. Alcohol Clin Exp Res 2001; 25(suppl s1):40S–45S.
- Singal AK, Kamath PS, Gores GJ, Shah VH. Alcoholic hepatitis: current challenges and future directions. Clin Gastroenterol Hepatol 2014; 12:555–564.
- Seitz HK, Stickel F. Risk factors and mechanisms of hepatocarcinogenesis with special emphasis on alcohol and oxidative stress. Biol Chem 2006; 387:349–360.
- Thurman RG. II. Alcoholic liver injury involves activation of Kupffer cells by endotoxin. Am J Physiol 1998; 275:G605–G611.
- Duddempudi AT. Immunology in alcoholic liver disease. Clin Liver Dis 2012; 16:687–698.
- Lischner MW, Alexander JF, Galambos JT. Natural history of alcoholic hepatitis. I. The acute disease. Am J Dig Dis 1971; 16:481–494.
- Cohen JA, Kaplan MM. The SGOT/SGPT ratio—an indicator of alcoholic liver disease. Dig Dis Sci 1979; 24:835–838.
- Lucey MR, Mathurin P, Morgan TR. Alcoholic hepatitis. N Engl J Med 2009; 360:2758–2769.
- McKnight-Eily LR, Liu Y, Brewer RD, et al; Centers for Disease Control and Prevention (CDC). Vital signs: communication between health professionals and their patients about alcohol use—44 states and the District of Columbia, 2011. MMWR Morb Mortal Wkly Rep 2014; 63:16–22.
- Grant BF. Barriers to alcoholism treatment: reasons for not seeking treatment in a general population sample. J Stud Alcohol 1997; 58:365–371.
- Aertgeerts B, Buntinx F, Kester A. The value of the CAGE in screening for alcohol abuse and alcohol dependence in general clinical populations: a diagnostic meta-analysis. J Clin Epidemiol 2004; 57:30–39.
- The Alcohol Use Disorders Identification Test Guidelines for Use in Primary Care. Second Edition. World Health Organization. Department of Mental Health and Substance Dependence. http://whqlibdoc.who.int/hq/2001/who_msd_msb_01.6a.pdf. Accessed February 3, 2015.
- Hamid R, Forrest EH. Is histology required for the diagnosis of alcoholic hepatitis? A review of published randomised controlled trials. Gut 2011; 60(suppl 1):A233.
- O’Shea RS, Dasarathy S, McCullough AJ; Practice Guideline Committee of the American Association for the Study of Liver Diseases; Practice Parameters Committee of the American College of Gastroenterology. Alcoholic liver disease. Hepatology 2010; 51:307–328.
- Hanouneh IA, Zein NN, Cikach F, et al. The breathprints in patients with liver disease identify novel breath biomarkers in alcoholic hepatitis. Clin Gastroenterol Hepatol 2014; 12:516–523.
- Sheth M, Riggs M, Patel T. Utility of the Mayo End-Stage Liver Disease (MELD) score in assessing prognosis of patients with alcoholic hepatitis. BMC Gastroenterol 2002; 2:2.
- Dunn W, Jamil LH, Brown LS, et al. MELD accurately predicts mortality in patients with alcoholic hepatitis. Hepatology 2005; 41:353–358.
- Srikureja W, Kyulo NL, Runyon BA, Hu KQ. MELD score is a better prognostic model than Child-Turcotte-Pugh score or Discriminant Function score in patients with alcoholic hepatitis. J Hepatol 2005; 42:700–706.
- Forrest EH, Morris AJ, Stewart S, et al. The Glasgow alcoholic hepatitis score identifies patients who may benefit from corticosteroids. Gut 2007; 56:1743–1746.
- Dominguez M, Rincón D, Abraldes JG, et al. A new scoring system for prognostic stratification of patients with alcoholic hepatitis. Am J Gastroenterol 2008; 103:2747–2756.
- Louvet A, Naveau S, Abdelnour M, et al. The Lille model: a new tool for therapeutic strategy in patients with severe alcoholic hepatitis treated with steroids. Hepatology 2007; 45:1348–1354.
- Mayo-Smith MF, Beecher LH, Fischer TL, et al; Working Group on the Management of Alcohol Withdrawal Delirium, Practice Guidelines Committee, American Society of Addiction Medicine. Management of alcohol withdrawal delirium. An evidence-based practice guideline. Arch Intern Med 2004; 164:1405–1412.
- Mezey E. Interaction between alcohol and nutrition in the pathogenesis of alcoholic liver disease. Semin Liver Dis 1991; 11:340–348.
- Cabré E, Rodríguez-Iglesias P, Caballería J, et al. Short- and long-term outcome of severe alcohol-induced hepatitis treated with steroids or enteral nutrition: a multicenter randomized trial. Hepatology 2000; 32:36–42.
- Louvet A, Wartel F, Castel H, et al. Infection in patients with severe alcoholic hepatitis treated with steroids: early response to therapy is the key factor. Gastroenterology 2009; 137:541–548.
- European Association for the Study of Liver. EASL clinical practical guidelines: management of alcoholic liver disease. J Hepatol 2012; 57:399–420.
- Rambaldi A, Saconato HH, Christensen E, Thorlund K, Wetterslev J, Gluud C. Systematic review: glucocorticosteroids for alcoholic hepatitis—a Cochrane Hepato-Biliary Group systematic review with meta-analyses and trial sequential analyses of randomized clinical trials. Aliment Pharmacol Ther 2008; 27:1167–1178.
- Powell LW, Axelsen E. Corticosteroids in liver disease: studies on the biological conversion of prednisone to prednisolone and plasma protein binding. Gut 1972; 13:690–696.
- Mathurin P, O’Grady J, Carithers RL, et al. Corticosteroids improve short-term survival in patients with severe alcoholic hepatitis: meta-analysis of individual patient data. Gut 2011; 60:255–260.
- Akriviadis E, Botla R, Briggs W, Han S, Reynolds T, Shakil O. Pentoxifylline improves short-term survival in severe acute alcoholic hepatitis: a double-blind, placebo-controlled trial. Gastroenterology 2000; 119:1637–1648.
- De BK, Gangopadhyay S, Dutta D, Baksi SD, Pani A, Ghosh P. Pentoxifylline versus prednisolone for severe alcoholic hepatitis: a randomized controlled trial. World J Gastroenterol 2009; 15:1613–1619.
- Mathurin P, Louvet A, Dao T, et al. Addition of pentoxifylline to prednisolone for severe alcoholic hepatitis does not improve 6-month survival: results of the CORPENTOX trial (abstract). Hepatology 2011; 54(suppl 1):81A.
- Whitfield K, Rambaldi A, Wetterslev J, Gluud C. Pentoxifylline for alcoholic hepatitis. Cochrane Database Syst Rev 2009; CD007339.
- Louvet A, Diaz E, Dharancy S, et al. Early switch to pentoxifylline in patients with severe alcoholic hepatitis is inefficient in non-responders to corticosteroids. J Hepatol 2008; 48:465–470.
- Thursz MR, Richardson P, Allison ME, et al. Steroids or pentoxifylline for alcoholic hepatitis: results of the STOPAH trial [abstract LB-1]. 65th Annual Meeting of the American Association for the Study of Liver Diseases; November 7–11, 2014; Boston, MA.
- Naveau S, Chollet-Martin S, Dharancy S, et al; Foie-Alcool group of the Association Française pour l’Etude du Foie. A double-blind randomized controlled trial of infliximab associated with prednisolone in acute alcoholic hepatitis. Hepatology 2004; 39:1390–1397.
- Menon KV, Stadheim L, Kamath PS, et al. A pilot study of the safety and tolerability of etanercept in patients with alcoholic hepatitis. Am J Gastroenterol 2004; 99:255–260.
- Boetticher NC, Peine CJ, Kwo P, et al. A randomized, double-blinded, placebo-controlled multicenter trial of etanercept in the treatment of alcoholic hepatitis. Gastroenterology 2008; 135:1953–1960.
- Nguyen-Khac E, Thevenot T, Piquet MA, et al; AAH-NAC Study Group. Glucocorticoids plus N-acetylcysteine in severe alcoholic hepatitis. N Engl J Med 2011; 365:1781–1789.
- Singal AK, Duchini A. Liver transplantation in acute alcoholic hepatitis: current status and future development. World J Hepatol 2011; 3:215–218.
- Singal AK, Bashar H, Anand BS, Jampana SC, Singal V, Kuo YF. Outcomes after liver transplantation for alcoholic hepatitis are similar to alcoholic cirrhosis: exploratory analysis from the UNOS database. Hepatology 2012; 55:1398–1405.
- Mathurin P, Moreno C, Samuel D, et al. Early liver transplantation for severe alcoholic hepatitis. N Engl J Med 2011; 365:1790–1800.
KEY POINTS
- One should assess the severity of alcoholic hepatitis, using defined scoring systems, to allocate resources and initiate appropriate therapy.
- Supportive care should focus on alcohol withdrawal and enteral nutrition while managing the complications of liver failure.
- Corticosteroids or pentoxifylline are commonly used, but increase the survival rate only by about 50%.
- Opinion is shifting toward allowing some patients with alcoholic hepatitis to receive liver transplants early in the course of their disease.
- Many new therapies are undergoing clinical trials.
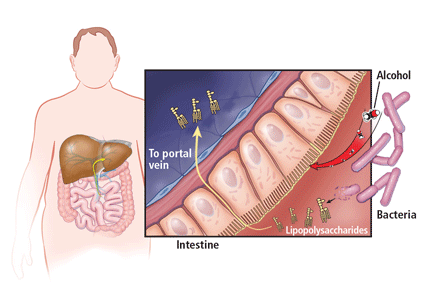
Hepatitis C virus: Here comes all-oral treatment
In late 2013, the US Food and Drug Administration (FDA) approved sofosbuvir and simeprevir, the newest direct-acting antiviral agents for treating chronic hepatitis C virus (HCV) infection. Multiple clinical trials have demonstrated dramatically improved treatment outcomes with these agents, opening the door to all-oral regimens or interferon-free regimens as the future standard of care for HCV.
In this article, we discuss the results of the trials that established the efficacy and safety of sofosbuvir and simeprevir and led to their FDA approval. We also summarize the importance of these agents and evaluate other direct-acting antivirals currently in the pipeline for HCV treatment.
HCV IS A RISING PROBLEM
Chronic HCV infection is a major clinical and public health problem, with the estimated number of people infected exceeding 170 million worldwide, including 3.2 million in the United States.1 It is a leading cause of cirrhosis, and its complications include hepatocellular carcinoma and liver failure. Cirrhosis due to HCV remains the leading indication for liver transplantation in the United States, accounting for nearly 40% of liver transplants in adults.2

The clinical impact of HCV will only continue to escalate, and in parallel, so will the cost to society. Models suggest that HCV-related deaths will double between 2010 and 2019, and considering only direct medical costs, the projected financial burden of treating HCV-related disease during this interval is estimated at between $6.5 and $13.6 billion.3
AN RNA VIRUS WITH SIX GENOTYPES

HCV, first identified in 1989, is an enveloped, single-stranded RNA flavivirus of the Hepacivirus genus measuring 50 to 60 nm in diameter.4 There are six viral genotypes, with genotype 1 being the most common in the United States and traditionally the most difficult to treat.
Once inside the host cell, the virus releases its RNA strand, which is translated into a single polyprotein of about 3,000 amino acids. This large molecule is then cleaved by proteases into several domains: three structural proteins (C, E1, and E2), a small protein called p7, and six nonstructural proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) (Figure 1).5 These nonstructural proteins enable the virus to replicate.
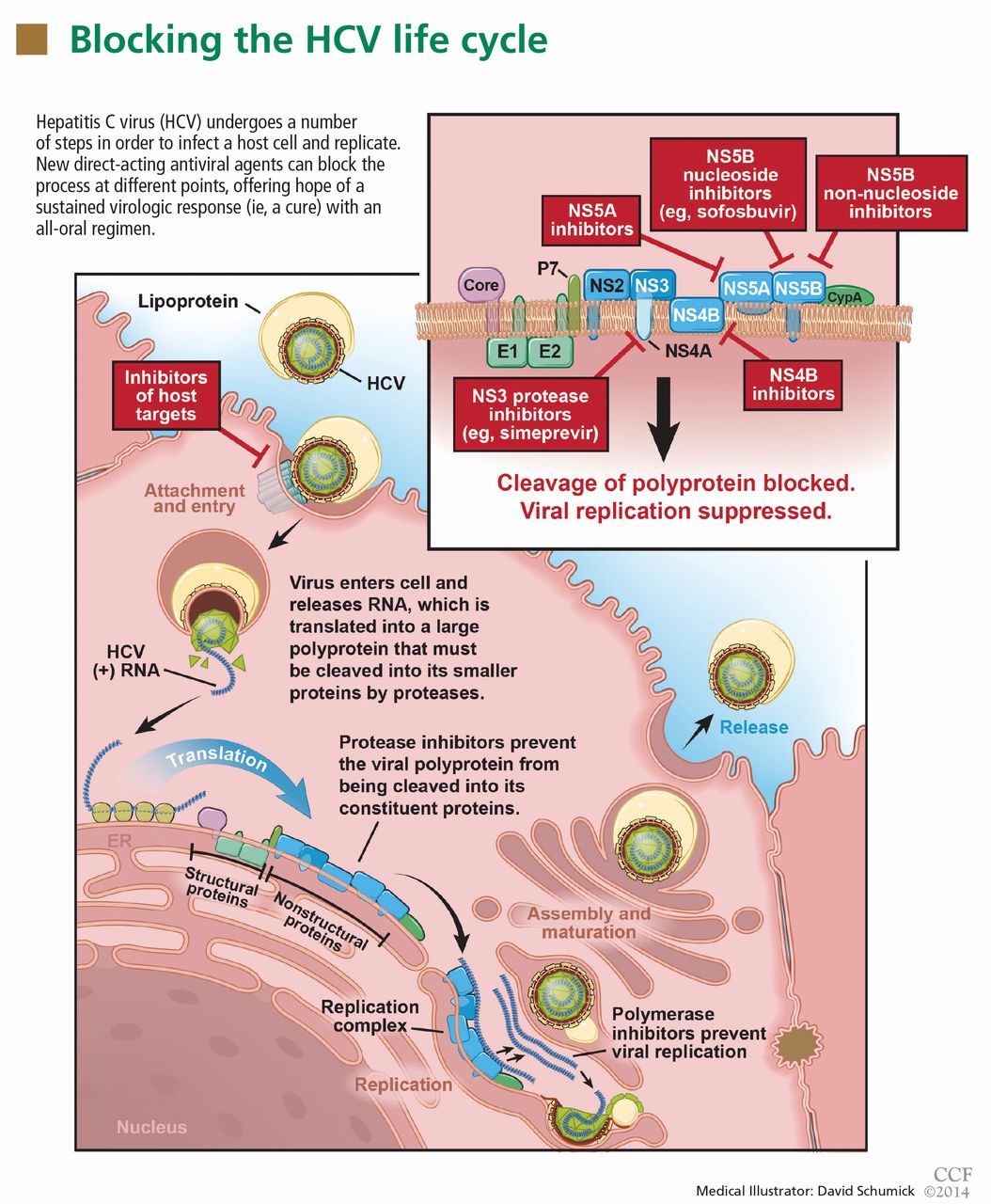
GOAL OF TREATING HCV: A SUSTAINED VIROLOGIC RESPONSE
The aim of HCV treatment is to achieve a sustained virologic response, defined as having no detectable viral RNA after completion of antiviral therapy. This is associated with substantially better clinical outcomes, lower rates of liver-related morbidity and all-cause mortality, and stabilization of or even improvement in liver histology.6,7 This end point has traditionally been assessed at 6 months after the end of therapy, but recent data suggest the rates at 12 weeks are essentially equivalent.

Table 1 summarizes the patterns of virologic response in treating HCV infection.
Interferon plus ribavirin: The standard of care for many years
HCV treatment has evolved over the past 20 years. Before 2011, the standard of care was a combination of interferon alfa-polyethylene glycol (peg-interferon), given as a weekly injection, and oral ribavirin. Neither drug has specific antiviral activity, and when they are used together they result in a sustained virologic response in fewer than 50% of patients with HCV genotype 1 and, at best, in 70% to 80% of patients with other genotypes.8
Nearly all patients receiving interferon experience side effects, which can be serious. Fatigue and flu-like symptoms are common, and the drug can also cause psychiatric symptoms (including depression or psychosis), weight loss, seizures, peripheral neuropathy, and bone marrow suppression. Ribavirin causes hemolysis and skin complications and is teratogenic.9
An important bit of information to know when using interferon is the patient’s IL28B genotype. This refers to a single-nucleotide polymorphism (C or T) on chromosome 19q13 (rs12979860) upstream of the IL28B gene encoding for interferon lambda-3. It is strongly associated with responsiveness to interferon: patients with the IL28B CC genotype have a much better chance of a sustained virologic response with interferon than do patients with CT or TT.
Boceprevir and telaprevir: First-generation protease inhibitors
In May 2011, the FDA approved the NS3/4A protease inhibitors boceprevir and telaprevir for treating HCV genotype 1, marking the beginning of the era of direct-acting antiviral agents.10 When these drugs are used in combination with peg-interferon alfa and ribavirin, up to 75% of patients with HCV genotype 1 who have had no previous treatment achieve a sustained virologic response.
But despite greatly improving the response rate, these first-generation protease inhibitors have substantial limitations. Twenty-five percent of patients with HCV genotype 1 who have received no previous treatment and 71% of patients who did not respond to previous treatment will not achieve a sustained virologic response with these agents.11 Further, they are effective only against HCV genotype 1, being highly specific for the amino acid target sequence of the NS3 region.
Also, they must be used in combination with interferon alfa and ribavirin because the virus needs to mutate only a little—a few amino-acid substitutions—to gain resistance to them.12 Therefore, patients are still exposed to interferon and ribavirin, with their toxicity. In addition, dysgeusia is seen with boceprevir, rash with telaprevir, and anemia with both.13,14
Finally, serious drug-drug interactions prompted the FDA to impose warnings for the use of these agents with other medications that interact with CYP3A4, the principal enzyme responsible for their metabolism. Thus, these significant adverse effects dampen the enthusiasm of patients contemplating a long course of treatment with these agents.
The need to improve the rate of sustained virologic response, shorten the duration of treatment, avoid serious side effects, improve efficacy in treating patients infected with genotypes other than 1, and, importantly, eliminate the need for interferon alfa and its serious adverse effects have driven the development of new direct-acting antiviral agents, including the two newly FDA-approved drugs, sofosbuvir and simeprevir.
SOFOSBUVIR: A POLYMERASE INHIBITOR
Sofosbuvir is a uridine nucleotide analogue that selectively inhibits the HCV NS5B RNA-dependent RNA polymerase (Figure 1). It targets the highly conserved nucleotide-binding pocket of this enzyme and functions as a chain terminator.15 While the protease inhibitors are genotype-dependent, inhibition of the highly conserved viral polymerase has an impact that spans genotypes.
Early clinical trials of sofosbuvir

Sofosbuvir has been tested in combination with interferon alfa and ribavirin, as well as in interferon-free regimens (Table 2).16–20
Rodriguez-Torres et al,15
- 56% with sofosbuvir 100 mg, peg-interferon, and ribavirin
- 83% with sofosbuvir 200 mg, peg-interferon, and ribavirin
- 80% with sofosbuvir 400 mg, peg-interferon, and ribavirin
- 43% with peg-interferon and ribavirin alone.
The ATOMIC trial16 tested the efficacy and safety of sofosbuvir in combination with peg-interferon and ribavirin in patients with HCV genotype 1, 4, or 6, without cirrhosis, who had not received any previous treatment. Patients with HCV genotype 1 were randomized to three treatments:
- Sofosbuvir 400 mg orally once daily plus peg-interferon and ribavirin for 12 weeks
- The same regimen, but for 24 weeks
- Sofosbuvir plus peg-interferon and ribavirin for 12 weeks, followed by 12 weeks of either sofosbuvir monotherapy or sofosbuvir plus ribavirin.
The rates of sustained virologic response were very high and were not significantly different among the three groups: 89%, 89%, and 87%, respectively. Patients who were able to complete a full course of therapy achieved even higher rates of sustained virologic response, ranging from 96% to 98%. The likelihood of response was not adversely affected by the usual markers of a poorer prognosis, such as a high viral load (≥ 800,000 IU/mL) or a non-CC IL28B genotype. Although patients with cirrhosis (another predictor of no response) were excluded from this study, the presence of bridging fibrosis did not seem to affect the rate of sustained virologic response. The results in patients with genotypes other than 1 were very encouraging, but the small number of patients enrolled precluded drawing firm conclusions in this group.
Important implications of the ATOMIC trial include the following:
There is no benefit in prolonging treatment with sofosbuvir beyond 12 weeks, since adverse events increased without any improvement in the rate of sustained virologic response.
There is a very low likelihood of developing viral resistance or mutation when using sofosbuvir.
There is no role for response-guided therapy, a concept used with protease inhibitor-based regimens in which patients who have complete clearance of the virus within the first 4 weeks of treatment (a rapid virologic response) and remain clear through 12 weeks of treatment (an extended rapid viral response) can be treated for a shorter duration without decreasing the likelihood of a sustained virologic response.
Lawitz et al17 conducted a randomized double-blind phase 2 trial to evaluate the effect of sofosbuvir dosing on response in noncirrhotic, previously untreated patients with HCV genotype 1, 2, or 3. Patients with HCV genotype 1 were randomized to one of three treatment groups in a 2:2:1 ratio: sofosbuvir 200 mg orally once daily, sofosbuvir 400 mg orally once daily, or placebo, all for 12 weeks in combination with peg-interferon (180 μg weekly) and ribavirin in a dosage based on weight. Depending on the viral response, patients continued peg-interferon and ribavirin for an additional 12 weeks if they achieved an extended rapid viral response, or 36 weeks if they did not achieve an extended rapid virologic response, and in all patients who received placebo. Patients with HCV genotype 2 or 3 were given sofosbuvir 400 mg once daily in combination with interferon and ribavirin for 12 weeks.
As in the ATOMIC trial, all patients treated with sofosbuvir had a very rapid reduction in viral load: 98% of patients with genotype 1 developed a rapid virologic response, and therefore almost all were eligible for the shorter treatment course of 24 weeks.17 The latter finding again suggested that response-guided treatment is not relevant with sofosbuvir-based regimens.
Very high rates of sustained virologic response were seen: 90% in patients with genotype 1 treated with sofosbuvir 200 mg, 91% in those with genotype 1 treated with 400 mg, and 92% in those with genotype 2 or 3. Although 6% of patients in the 200-mg group had virologic breakthrough after completing sofosbuvir treatment, no virologic breakthrough was observed in the 400-mg group, suggesting that the 400-mg dose might suppress the virus more effectively.17
The ELECTRON trial18 was a phase 2 study designed to evaluate the efficacy and safety of sofosbuvir and ribavirin in interferon-sparing and interferon-free regimens in patients with HCV genotype 1, 2, or 3 infection. Sofosbuvir was tested with peg-interferon and ribavirin, with ribavirin alone, and as monotherapy in previously untreated patients with genotype 2 or 3. A small number of patients with genotype 1 who were previously untreated and who were previously nonresponders were also treated with sofosbuvir and ribavirin.
All patients had a rapid virologic response, and viral suppression was sustained through the end of treatment. All patients with genotype 2 or 3 treated with double therapy (sofosbuvir and ribavirin) or triple therapy (sofosbuvir, peg-interferon, and ribavirin) achieved a sustained virologic response, compared with only 60% of patients treated with sofosbuvir monotherapy. The monotherapy group had an equal number of relapsers among those with genotype 2 or 3. Of the genotype 1 patients treated with sofosbuvir and ribavirin, 84% of those previously untreated developed a sustained virologic response, whereas only 10% of the previous nonresponders did.
Phase 3 clinical trials of sofosbuvir
The NEUTRINO trial19 studied the efficacy and safety of sofosbuvir in previously untreated patients with HCV genotype 1, 4, 5, or 6. In this phase 3 open-label study, all patients received sofosbuvir plus peg-interferon and weight-based ribavirin therapy for 12 weeks. Of the patients enrolled, 89% had genotype 1, while 9% had genotype 4 and 2% had genotype 5 or 6. Overall, 17% of the patients had cirrhosis.
The viral load rapidly decreased in all patients treated with sofosbuvir irrespective of the HCV genotype, IL28B status, race, or the presence or absence of cirrhosis. Ninety-nine percent of patients with genotype 1, 4, 5, or 6 achieved a rapid virologic response, and 90% achieved a sustained virologic response at 12 weeks after completion of treatment with sofosbuvir and ribavirin. Patients with cirrhosis had a slightly lower rate of sustained virologic response (80%, compared with 92% in patients without cirrhosis). Also, patients with non-CC IL28B genotypes had a lower rate of sustained virologic response (87% in non-CC allele vs 98% in patients with the favorable CC allele).
The FISSION trial19 recruited previously untreated patients with genotype 2 or 3 and randomized them to therapy with either sofosbuvir plus ribavirin in a weight-based dose for 12 weeks, or 24 weeks of interferon and ribavirin. In this study, 20% of patients in each treatment group had cirrhosis.
As in the NEUTRINO trial, the viral load rapidly decreased in all patients treated with sofosbuvir irrespective of HCV genotype, IL28B status, race, or the presence or absence of cirrhosis. Here, 100% of patients with genotype 2 or 3 who were treated with sofosbuvir and ribavirin achieved a rapid virologic response. Differences in outcome emerged based on genotype: 97% of those with genotype 2 and 56% of those with genotype 3 achieved a sustained virologic response. The overall rate was 67%, which was not different from patients treated with peg-interferon and ribavirin. In the subgroup of patients with cirrhosis, 47% of those treated with sofosbuvir and ribavirin achieved a sustained virologic response, vs 38% of those who received peg-interferon plus ribavirin.
In both the NEUTRINO and FISSION trials, few patients discontinued treatment, with higher rates of most adverse events occurring in patients treated with peg-interferon and ribavirin.
POSITRON,20 a phase 3 clinical trial, tested sofosbuvir in patients with HCV genotype 2 or 3 who were ineligible for peg-interferon, unwilling to take peg-interferon, or unable to tolerate peg-interferon (mainly because of clinically significant psychiatric disorders). Patients were randomized to two treatment groups for 12 weeks: sofosbuvir plus ribavirin, or placebo. About 50% of patients had HCV genotype 3, and 16% had cirrhosis.
The overall rate of sustained virologic response at 12 weeks after treatment was 78% in the sofosbuvir-and-ribavirin group (93% in genotype 2 patients and 61% in genotype 3 patients). Again, cirrhosis was associated with a lower rate of sustained virologic response (61% of patients with cirrhosis achieved a sustained virologic response vs 81% of patients without cirrhosis). None of the sofosbuvir-treated patients had virologic failure while on treatment.
FUSION,20 another phase 3 trial, evaluated sofosbuvir in patients infected with HCV genotype 2 or 3 for whom interferon-based treatment had failed. They were randomized to either 12 weeks or 16 weeks of sofosbuvir and weight-based ribavirin treatment. About 60% of patients had HCV genotype 3, and 34% had cirrhosis.
The overall sustained virologic response rate was 50% in the patients treated for 12 weeks and 73% in those treated for 16 weeks: specifically, 86% of patients with genotype 2 achieved a sustained virologic response at 12 weeks and 94% at 16 weeks, whereas in those with genotype 3 the rates were 30% at 12 weeks and 62% at 16 weeks.
Cirrhosis was again a predictor of lack of response to sofosbuvir. In the group treated for 12 weeks, 31% of those with cirrhosis achieved a sustained virologic response compared with 61% in those without cirrhosis. In the group treated for 16 weeks, 61% of those with cirrhosis achieved a sustained virologic response compared with 76% in those without cirrhosis.
In both the POSITRON and FUSION trials, relapse accounted for all treatment failures, and no virologic resistance was detected in patients who did not have a sustained virologic response. The investigators concluded that 12 weeks of treatment with sofosbuvir and ribavirin can be effective for HCV genotype 2 infection, but extending the treatment to 16 weeks may be beneficial for genotype 3. This may be especially important in patients with cirrhosis or those who did not have a response to peg-interferon-based treatment.
VALENCE,21 an ongoing phase 3 trial in Europe, is assessing the safety and efficacy of sofosbuvir 400 mg once daily and weight-based ribavirin in patients with HCV genotype 2 or 3. Eighty-five percent of the trial participants have received previous treatment, and 21% have cirrhosis. Patients were originally randomized in a 4:1 ratio to receive sofosbuvir plus ribavirin for 12 weeks or matching placebo, but as a result of emerging data suggesting that patients with genotype 3 would benefit from more than 12 weeks of treatment, the study was subsequently amended to extend treatment to 24 weeks for patients with genotype 3.
Overall rates of sustained virologic response were 93% in patients with genotype 2 and 85% in patients with genotype 3. In previously treated patients with genotype 2 who were treated for 12 weeks, the rates of sustained virologic response were 91% in those without cirrhosis vs 88% in those with cirrhosis. In previously treated patients with genotype 3, the rates in those treated for 24 weeks were 87% in patients without cirrhosis vs 60% with cirrhosis. The safety profile was consistent with that of ribavirin.
Side effects of sofosbuvir
In clinical trials, side effects occurred most often when sofosbuvir was combined with interferon and ribavirin and were consistent with the known side effects of the latter two agents. The most frequently reported side effects included fatigue, insomnia, nausea, rash, anemia, headache, and arthralgia, with most of these adverse events rated by treating clinicians as being mild in severity.15,20
In the ATOMIC trial, the most common events leading to drug discontinuation were anemia and neutropenia, both associated with interferon and ribavirin. Patients receiving sofosbuvir monotherapy after 12 weeks of triple therapy showed rapid improvement in hemoglobin levels and neutrophil counts, indicating that hematologic abnormalities attributed solely to sofosbuvir are minimal. In the FISSION trial, the incidence of adverse events was consistently lower in those receiving sofosbuvir-ribavirin than in patients receiving interferon-ribavirin without sofosbuvir.19
In the POSITRON trial, discontinuation of sofosbuvir because of adverse events was uncommon, and there were no differences in the incidence of adverse events and laboratory abnormalities between patients with and without cirrhosis when they received sofosbuvir and ribavirin.20
Sofosbuvir dosage and indications

Sofosbuvir is approved in an oral dose of 400 mg once daily in combination with ribavirin for patients infected with HCV genotype 2 or 3 and in combination with ribavirin and interferon alfa in patients infected with HCV genotype 1 or 4 (Table 3). It could be considered for HCV genotype 1 in combination with ribavirin alone for 24 weeks in patients who are ineligible for interferon.
Sofosbuvir is also recommended in combination with ribavirin in HCV-infected patients with hepatocellular carcinoma who are awaiting liver transplantation, for up to 48 weeks or until they receive a transplant, to prevent posttransplant reinfection with HCV.
Sofosbuvir is expensive
A course of therapy is expected to cost about $84,000, which is significantly more than the cost of previous triple therapy (peg-interferon, ribavirin, and either boceprevir or telaprevir).22 This high cost will undoubtedly lead to less widespread use in developing countries, and potentially even in the United States. As newer direct-acting antiviral agents become available, the price will likely come down, enhancing access to these drugs.
SIMEPREVIR: A SECOND-GENERATION PROTEASE INHIBITOR
Telaprevir and boceprevir are NS3/A4 protease inhibitors that belong to the alfa-ketoamid derivative class. Simeprevir belongs to the macrocyclic class and has a different way of binding to the target enzyme.23 Like sofosbuvir, simeprevir was recently approved by the FDA for the treatment of HCV genotype 1.
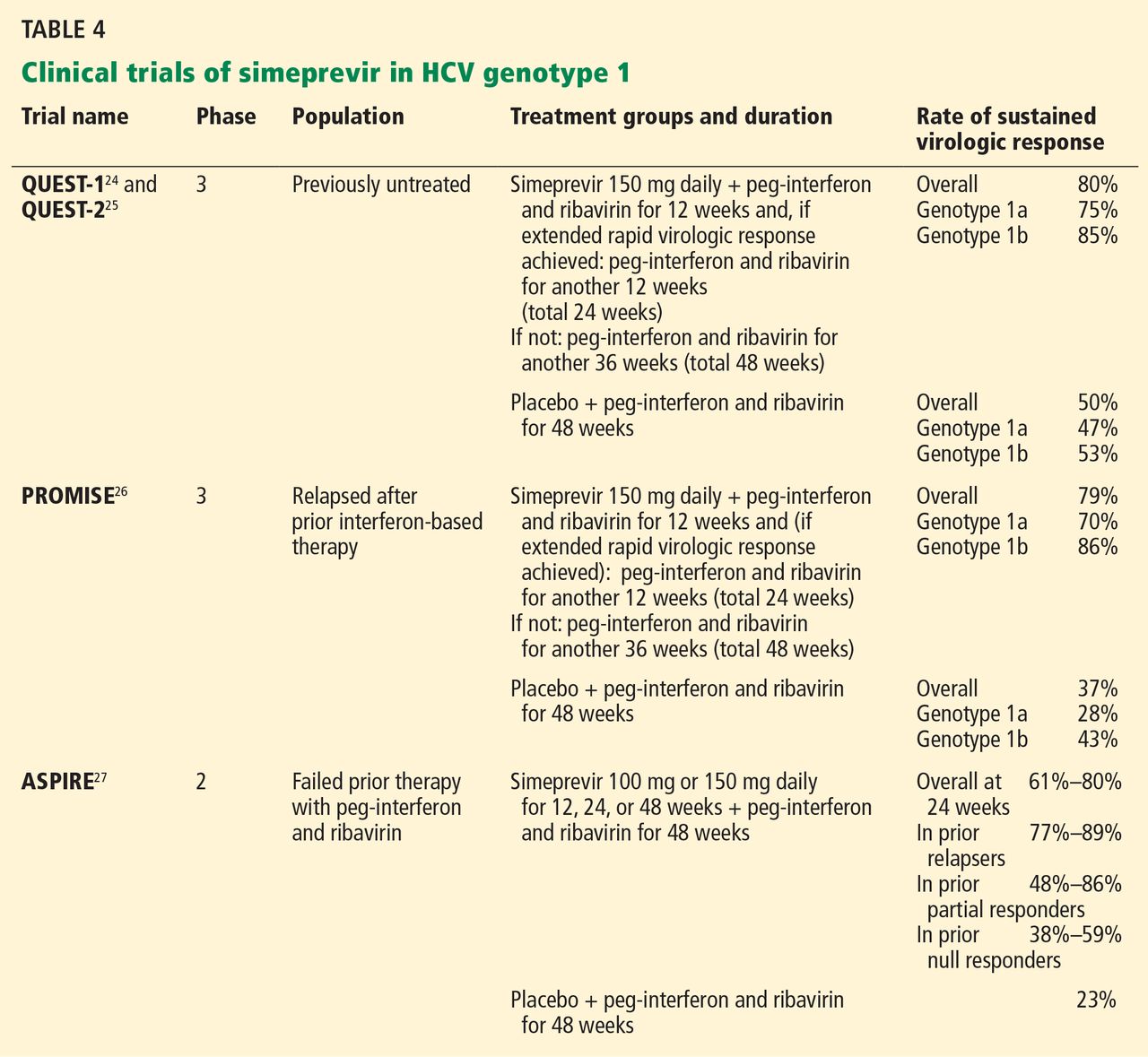
The therapeutic efficacy of simeprevir has been tested in several clinical trials (Table 4), including QUEST-124 and QUEST-225 (in previously untreated patients), PROMISE26 (in prior relapsers), and ASPIRE27 (in prior partial and null responders). Results from these trials showed high overall rates of sustained virologic response with triple therapy (ie, simeprevir combined with peg-interferon and ribavirin). It was generally well tolerated, and most adverse events reported during 12 weeks of treatment were of mild to moderate severity.
In QUEST-1 and QUEST-2, both double-blind phase 3 clinical trials, previously untreated patients infected with HCV genotype 1 were randomized in a 2:1 ratio to receive either simeprevir 150 mg daily or placebo for 12 weeks; both groups also received peg-interferon and ribavirin. Patients then received peg-interferon and ribavirin alone for 12 or 36 weeks in the simeprevir group (based on response) and for 36 weeks in the placebo group.
The overall rate of sustained virologic response at 12 weeks was 80% in the simeprevir group (75% in those with genotype 1a and 85% in those with genotype 1b) vs 50% in the placebo group (receiving peg-interferon and ribavirin alone).24,25
PROMISE,26 another double-blind randomized phase 3 clinical trial, evaluated simeprevir in patients with HCV genotype 1 who relapsed after previous interferon-based therapy. It had a similar design to QUEST-1 and QUEST-2, and 15% of all patients had cirrhosis.
The overall sustained virologic response rate at 12 weeks after treatment was 79% in the simeprevir group (70% in patients with genotype 1a and 86% in those with genotype 1b) vs 37% in the placebo group. Rates were similar in patients with absent to moderate fibrosis (82%), advanced fibrosis (73%), or cirrhosis (74%).
ASPIRE.27 Simeprevir efficacy in patients with HCV genotype 1 for whom previous therapy with peg-interferon and ribavirin had failed was tested in ASPIRE, a double-blind randomized phase 2 clinical trial. Patients were randomized to receive simeprevir (either 100 mg or 150 mg daily) for 12, 24, or 48 weeks in combination with 48 weeks of peg-interferon and ribavirin, or placebo plus peg-interferon and ribavirin for 48 weeks.
The primary end point was the rate of sustained virologic response at 24 weeks. Overall, rates were 61% to 80% for the simeprevir treatment groups compared with 23% with placebo, regardless of prior response to peg-interferon and ribavirin. By subgroup, rates were:
- 77% to 89% with simeprevir vs 37% with placebo in prior relapsers
- 48% to 86% with simeprevir vs 9% with placebo in prior partial responders
- 38% to 59% with placebo vs 19% for prior nonresponders.
The best rates of sustained viral response at 24 weeks were in the groups that received simeprevir 150 mg daily: 85% in prior relapsers, 75% in prior partial responders, and 51% in prior nonresponders.
Simeprevir vs other direct-acting antiviral drugs
Advantages of simeprevir over the earlier protease inhibitors include once-daily dosing, a lower rate of adverse events (the most common being fatigue, headache, rash, photosensitivity, and pruritus), a lower likelihood of discontinuation because of adverse events, and fewer drug-drug interactions (since it is a weak inhibitor of the CYP3A4 enzyme).
Unlike sofosbuvir, simeprevir was FDA-approved only for HCV genotype 1 and in combination with interferon alfa and ribavirin. Compared with sofosbuvir, the treatment duration with simeprevir regimens is longer overall (interferon alfa and ribavirin are given for 12 weeks in sofosbuvir-based regimens vs 24 to 48 weeks with simeprevir). As with sofosbuvir, the estimated cost of simeprevir is high, about $66,000 for a 12-week course.
Simeprevir dosage and indications
Simeprevir was approved at an oral dose of 150 mg once daily in combination with ribavirin and interferon alfa in patients with HCV genotype 1 (Table 5).
The approved regimens for simeprevir are fixed in total duration based on the patient’s treatment history. Specifically, all patients receive the drug in combination with peg-interferon and ribavirin for 12 weeks. Then, previously untreated patients and prior relapsers continue to receive peg-interferon and ribavirin alone for another 12 weeks, and those with a partial or null response continue with these drugs for another 36 weeks.
Patients infected with HCV genotype 1a should be screened for the NS3 Q80K polymorphism at baseline, as it has been associated with substantially reduced response to simeprevir.
Sofosbuvir and simeprevir in combination
The COSMOS trial.28 Given their differences in mechanism of action, sofosbuvir and simeprevir are being tested in combination. The COSMOS trial is an ongoing phase 2 randomized open-label study investigating the efficacy and safety of simeprevir and sofosbuvir in combination with and without ribavirin in patients with HCV genotype 1, including nonresponders and those with cirrhosis. Early results are promising, with very high rates of sustained virologic response with the sofosbuvir-simeprevir combination (93% to 100%) and indicate that the addition of ribavirin might not be needed to achieve sustained virologic response in this patient population.
THE FUTURE
The emergence of all-oral regimens for HCV treatment with increasingly sophisticated agents such as sofosbuvir and simeprevir will dramatically alter the management of HCV patients. In view of the improvement in sustained virologic response rates with these treatments, and since most HCV-infected persons have no symptoms, the US Centers for Disease Control and Prevention29 recently recommended one-time testing of the cohort in which the prevalence of HCV infection is highest: all persons born between 1945 and 1965. This undoubtedly will increase the detection of this infection—and the number of new patients expecting treatment.

Future drugs promise further improvements (Table 6).30–35 Advances in knowledge of the HCV molecular structure have led to the development of numerous direct-acting antiviral agents with very specific viral targets. A second wave of protease inhibitors and of nucleoside and nonnucleoside polymerase inhibitors will soon be available. Inhibitors of NS5A (a protein important in the assembly of the viral replication complex) such as daclatasvir and ledipasvir, are currently in phase 3 clinical trials. Other viral proteins involved in assembly of the virus, including the core protein and p7, are being explored as drug targets. In addition, inhibiting host targets such as cyclophilin A and miR122 has gained traction recently, with specific agents currently in phase 2 and 3 clinical trials.
Factors that previously were major determinants of response to treatment, such as IL28B genotype, viral load, race, age, extent of fibrosis, and genotype 1 subtypes, will become much less important with the introduction of highly potent direct-acting antiviral agents.
Many all-oral combinations are being evaluated in clinical trials. For example, the open-label, phase 2 LONESTAR trial tested the utility of combining sofosbuvir and ledipasvir (an NS5A inhibitor) with and without ribavirin for 8 or 12 weeks in previously untreated patients with HCV genotype 1, and for 12 weeks in patients with HCV genotype 1 who did not achieve a sustained virologic response after receiving a protease inhibitor-based regimen (half of whom had compensated cirrhosis).36 Sustained virologic response rates were very high (95% to 100%) in both previously treated and previously untreated patients, including those with cirrhosis. Similar rates were achieved by the 8-week and 12-week groups in noncirrhotic patients who had not been previously treated for HCV. The typical hematologic abnormalities associated with interferon were not observed except for mild anemia in patients who received ribavirin. These results suggest that the combination of sofosbuvir and ledipasvir could offer a very effective, short, all-oral treatment for patients with HCV genotype 1, including those with cirrhosis, who up to now have been difficult to treat.
Challenges remaining

The success of sofosbuvir and simeprevir paves the way for interferon-free regimens.37 For a long time, the treatment of HCV infection required close monitoring of patients while managing the side effects of interferon, but the current and emerging direct-acting antiviral agents will soon change this practice. Given the synergistic effects of combination therapy—targeting the virus at multiple locations, decreasing the likelihood of drug resistance, and improving efficacy—combination regimens seem to be the optimal solution to the HCV epidemic. Lower risk of side effects and shorter treatment duration will definitely improve the acceptance of any new regimen. New agents that act against conserved viral targets, thereby yielding activity across multiple genotypes, will be advantageous as well. Table 7 compares the rates of sustained virologic response of the different currently approved HCV treatment regimens.
Clinical challenges remain, including the management of special patient populations for whom data are still limited. These include patients with cirrhosis, chronic kidney disease, renal failure, and concurrent infection with human immunodeficiency virus, and patients who have undergone solid organ transplantation. Clinical trials are under way to evaluate the treatment options for these patients, who will likely need to wait for the emergence of additional agents before dramatic improvement in sustained virologic response rates may be expected.38
As the treatment of HCV becomes simpler, safer, and more effective, primary care physicians will increasingly be expected to manage it. Difficult-to-treat patients, including the special populations above, will require specialist management and individualized treatment regimens, at least until better therapies are available. The high projected cost of the new agents may limit access, at least initially. However, the dramatic improvement in sustained virologic response rates and all that that implies in terms of decreased risk of advanced liver disease and its complications will undoubtedly make these therapies cost-effective.39
- Averhoff FM, Glass N, Holtzman D. Global burden of hepatitis C: considerations for healthcare providers in the united states. Clin Infect Dis 2012; 55(suppl 1):S10–S15.
- Wiesner RH, Sorrell M, Villamil F; International Liver Transplantation Society Expert Panel. Report of the first international liver transplantation society expert panel consensus conference on liver transplantation and hepatitis C. Liver Transplant 2003; 9:S1–S9.
- Wong JB, McQuillan GM, McHutchison JG, Poynard T. Estimating future hepatitis C morbidity, mortality, and costs in the United States. Am J Public Health 2000; 90:1562–1569.
- Pawlotsky JM, Chevaliez S, McHutchison JG. The hepatitis C virus life cycle as a target for new antiviral therapies. Gastroenterology 2007; 132:1979–1998.
- Bartenschlager R, Lohmann V. Replication of hepatitis C virus. J Gen Virol 2000; 81:1631–1648.
- Singal AG, Volk ML, Jensen D, Di Bisceglie AM, Schoenfeld PS. A sustained viral response is associated with reduced liver-related morbidity and mortality in patients with hepatitis C virus. Clin Gastroenterol Hepatol 2010; 8:280–288,288.e1.
- Camma C, Di Bona D, Schepis F, et al. Effect of peginterferon alfa-2a on liver histology in chronic hepatitis C: a meta-analysis of individual patient data. Hepatology 2004; 39:333–342.
- Paeshuyse J, Dallmeier K, Neyts J. Ribavirin for the treatment of chronic hepatitis C virus infection: a review of the proposed mechanisms of action. Curr Opin Virol 2011; 1:590–598.
- Thomas E, Ghany MG, Liang TJ. The application and mechanism of action of ribavirin in therapy of hepatitis C. Antivir Chem Chemother 2012; 23:1–12.
- Ghany MG, Nelson DR, Strader DB, Thomas DL, Seeff LB; American Association for Study of Liver Diseases. An update on treatment of genotype 1 chronic hepatitis C virus infection: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 2011; 54:1433–1444.
- Soriano V, Vispo E, Poveda E, Labarga P, Barreiro P. Treatment failure with new hepatitis C drugs. Expert Opin Pharmacother 2012; 13:313–323.
- Asselah T, Marcellin P. Interferon free therapy with direct acting antivirals for HCV. Liver Int 2013; 33(suppl 1):93–104.
- Manns MP, McCone J, Davis MN, et al. Overall safety profile of boceprevir plus peginterferon alfa-2b and ribavirin in patients with chronic hepatitis C genotype 1: a combined analysis of 3 phase 2/3 clinical trials. Liver Int 2013; Aug 2. doi: 10.1111/liv.12300. [Epub ahead of print]
- Jacobson IM, McHutchison JG, Dusheiko G, et al. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364:2405–2416.
- Rodriguez-Torres M, Lawitz E, Kowdley KV, et al. Sofosbuvir (GS-7977) plus peginterferon/ribavirin in treatment-naive patients with HCV genotype 1: a randomized, 28-day, dose-ranging trial. J Hepatol 2013; 58:663–668.
- Kowdley KV, Lawitz E, Crespo I, et al. Sofosbuvir with pegylated interferon alfa-2a and ribavirin for treatment-naive patients with hepatitis C genotype-1 infection (ATOMIC): an open-label, randomised, multicentre phase 2 trial. Lancet 2013; 381:2100–2107.
- Lawitz E, Lalezari JP, Hassanein T, et al. Sofosbuvir in combination with peginterferon alfa-2a and ribavirin for non-cirrhotic, treatment-naive patients with genotypes 1, 2, and 3 hepatitis C infection: a randomised, double-blind, phase 2 trial. Lancet Infect Dis 2013; 13:401–408.
- Gane EJ, Stedman CA, Hyland RH, et al. Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N Engl J Med 2013; 368:34–44.
- Lawitz E, Mangia A, Wyles D, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 2013; 368:1878–1887.
- Jacobson IM, Gordon SC, Kowdley KV, et al. Sofosbuvir for hepatitis C genotype 2 or 3 in patients without treatment options. N Engl J Med 2013; 368:1867–1877.
- Zeuzem S, Dusheiko G, Salupere R, et al. Sofosbuvir + ribavirin for 12 or 24 weeks for patients with HCV genotype 2 or 3: the VALENCE trial [abstract no.1085]. 64th Annual Meeting of the American Association for the Study of Liver Diseases; November 1–5, 2013; Washington, DC.
- Soriano V, Vispo E, de Mendoza C, et al. Hepatitis C therapy with HCV NS5B polymerase inhibitors. Expert Opin Pharmacother 2013; 14:1161–1170.
- You DM, Pockros PJ. Simeprevir for the treatment of chronic hepatitis C. Expert Opin Pharmacother 2013; 14:2581–2589.
- Jacobson IM, Dore GJ, Foster G, et al. Simeprevir (TMC435) with peginterferon/ribavirin for chronic HCV genotype-1 infection in treatment-naive patients: results from Quest-1, a phase III trial [abstract no. 1425]. Annual Meeting of the European Association for the Study of the Liver; April 24–28, 2013; Amsterdam, Netherlands.
- Manns M, Marcellin P, Poordad FP, et al. Simeprevir (TMC435) with peginterferon/ribavirin for chronic HCV genotype-1 infection in treatment-naïve patients: results from QUEST-2, a phase III trial [abstract no. 1413]. Annual Meeting of the European Association for the Study of the Liver; April 24–28, 2013; Amsterdam, The Netherlands.
- Lawitz E, Forns X, Zeuzem S, et al. Simeprevir (TMC435) with peginterferon/ribavirin for treatment of chronic HCV genotype 1 infection in patients who relapsed after previous interferon-based therapy: results from promise, a phase III trial [abstract no. 869b]. Digestive Disease Week; May 18–21, 2013; Orlando, FL.
- Zeuzem S, Berg T, Gane E, et al. Simeprevir increases rate of sustained virologic response among treatment-experienced patients with HCV genotype-1 infection: a phase IIb trial. Gastroenterology epub Oct 31, 2013.
- Jacobson IM, Ghalib RM, Rodriguez-Torres M, et al. SVR results of a once-daily regimen of simeprevir (TMC435) plus sofosbuvir (GS-7977) with or without ribavirin in cirrhotic and non-cirrhotic HCV genotype 1 treatment-naive and prior null responder patients: the COSMOS study [abstract LB-3]. 64th Annual Meeting of the American Association for the Study of Liver Diseases; November 1–5, 2013; Washington, DC.
- Smith BD, Morgan RL, Beckett GA, et al. Recommendations for the identification of chronic hepatitis C virus infection among persons born during 1945–1965. MMWR Recomm Rep 2012; 61( RR-4):1–32.
- Sulkowski MS, Kang M, Matining R, et al. Safety and antiviral activity of the HCV entry inhibitor ITX5061 in treatment-naive HCV-infected adults: a randomized, double-blind, phase 1b study. J Infect Dis 2013 Oct 9. [Epub ahead of print]
- Pawlotsky JM. NS5A inhibitors in the treatment of hepatitis C. J Hepatol 2013; 59:375–382.
- Yu M, Corsa AC, Xu S, et al. In vitro efficacy of approved and experimental antivirals against novel genotype 3 hepatitis C virus subgenomic replicons. Antiviral Res 2013; 100:439–445.
- Aghemo A, De Francesco R. New horizons in hepatitis C antiviral therapy with direct-acting antivirals. Hepatology 2013; 58:428–438.
- Liang TJ, Ghany MG. Current and future therapies for hepatitis C virus infection. N Engl J Med 2013; 368:1907–1917.
- Flisiak R, Jaroszewicz J, Parfieniuk-Kowerda A. Emerging treatments for hepatitis C. Expert Opin Emerg Drugs 2013; 18:461–475.
- Lawitz E, Poordad FF, Pang PS, et al. Sofosbuvir and ledipasvir fixed-dose combination with and without ribavirin in treatment-naive and previously treated patients with genotype 1 hepatitis C virus infection (LONESTAR): an open-label, randomised, phase 2 trial. Lancet 2013 Nov 1. doi: 10.1016/S0140-6736(13)62121-2 [Epub ahead of print]
- Drenth JP. HCV treatment—no more room for interferonologists? N Engl J Med 2013; 368:1931–1932.
- Casey LC, Lee WM. Hepatitis C virus therapy update 2013. Curr Opin Gastroenterol 2013; 29:243–249.
- Afdhal NH, Zeuzem S, Schooley RT, et al. The new paradigm of hepatitis C therapy: integration of oral therapies into best practices. J Viral Hepatol 2013; 20:745–760.
In late 2013, the US Food and Drug Administration (FDA) approved sofosbuvir and simeprevir, the newest direct-acting antiviral agents for treating chronic hepatitis C virus (HCV) infection. Multiple clinical trials have demonstrated dramatically improved treatment outcomes with these agents, opening the door to all-oral regimens or interferon-free regimens as the future standard of care for HCV.
In this article, we discuss the results of the trials that established the efficacy and safety of sofosbuvir and simeprevir and led to their FDA approval. We also summarize the importance of these agents and evaluate other direct-acting antivirals currently in the pipeline for HCV treatment.
HCV IS A RISING PROBLEM
Chronic HCV infection is a major clinical and public health problem, with the estimated number of people infected exceeding 170 million worldwide, including 3.2 million in the United States.1 It is a leading cause of cirrhosis, and its complications include hepatocellular carcinoma and liver failure. Cirrhosis due to HCV remains the leading indication for liver transplantation in the United States, accounting for nearly 40% of liver transplants in adults.2
The clinical impact of HCV will only continue to escalate, and in parallel, so will the cost to society. Models suggest that HCV-related deaths will double between 2010 and 2019, and considering only direct medical costs, the projected financial burden of treating HCV-related disease during this interval is estimated at between $6.5 and $13.6 billion.3
AN RNA VIRUS WITH SIX GENOTYPES
HCV, first identified in 1989, is an enveloped, single-stranded RNA flavivirus of the Hepacivirus genus measuring 50 to 60 nm in diameter.4 There are six viral genotypes, with genotype 1 being the most common in the United States and traditionally the most difficult to treat.
Once inside the host cell, the virus releases its RNA strand, which is translated into a single polyprotein of about 3,000 amino acids. This large molecule is then cleaved by proteases into several domains: three structural proteins (C, E1, and E2), a small protein called p7, and six nonstructural proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) (Figure 1).5 These nonstructural proteins enable the virus to replicate.
GOAL OF TREATING HCV: A SUSTAINED VIROLOGIC RESPONSE
The aim of HCV treatment is to achieve a sustained virologic response, defined as having no detectable viral RNA after completion of antiviral therapy. This is associated with substantially better clinical outcomes, lower rates of liver-related morbidity and all-cause mortality, and stabilization of or even improvement in liver histology.6,7 This end point has traditionally been assessed at 6 months after the end of therapy, but recent data suggest the rates at 12 weeks are essentially equivalent.
Table 1 summarizes the patterns of virologic response in treating HCV infection.
Interferon plus ribavirin: The standard of care for many years
HCV treatment has evolved over the past 20 years. Before 2011, the standard of care was a combination of interferon alfa-polyethylene glycol (peg-interferon), given as a weekly injection, and oral ribavirin. Neither drug has specific antiviral activity, and when they are used together they result in a sustained virologic response in fewer than 50% of patients with HCV genotype 1 and, at best, in 70% to 80% of patients with other genotypes.8
Nearly all patients receiving interferon experience side effects, which can be serious. Fatigue and flu-like symptoms are common, and the drug can also cause psychiatric symptoms (including depression or psychosis), weight loss, seizures, peripheral neuropathy, and bone marrow suppression. Ribavirin causes hemolysis and skin complications and is teratogenic.9
An important bit of information to know when using interferon is the patient’s IL28B genotype. This refers to a single-nucleotide polymorphism (C or T) on chromosome 19q13 (rs12979860) upstream of the IL28B gene encoding for interferon lambda-3. It is strongly associated with responsiveness to interferon: patients with the IL28B CC genotype have a much better chance of a sustained virologic response with interferon than do patients with CT or TT.
Boceprevir and telaprevir: First-generation protease inhibitors
In May 2011, the FDA approved the NS3/4A protease inhibitors boceprevir and telaprevir for treating HCV genotype 1, marking the beginning of the era of direct-acting antiviral agents.10 When these drugs are used in combination with peg-interferon alfa and ribavirin, up to 75% of patients with HCV genotype 1 who have had no previous treatment achieve a sustained virologic response.
But despite greatly improving the response rate, these first-generation protease inhibitors have substantial limitations. Twenty-five percent of patients with HCV genotype 1 who have received no previous treatment and 71% of patients who did not respond to previous treatment will not achieve a sustained virologic response with these agents.11 Further, they are effective only against HCV genotype 1, being highly specific for the amino acid target sequence of the NS3 region.
Also, they must be used in combination with interferon alfa and ribavirin because the virus needs to mutate only a little—a few amino-acid substitutions—to gain resistance to them.12 Therefore, patients are still exposed to interferon and ribavirin, with their toxicity. In addition, dysgeusia is seen with boceprevir, rash with telaprevir, and anemia with both.13,14
Finally, serious drug-drug interactions prompted the FDA to impose warnings for the use of these agents with other medications that interact with CYP3A4, the principal enzyme responsible for their metabolism. Thus, these significant adverse effects dampen the enthusiasm of patients contemplating a long course of treatment with these agents.
The need to improve the rate of sustained virologic response, shorten the duration of treatment, avoid serious side effects, improve efficacy in treating patients infected with genotypes other than 1, and, importantly, eliminate the need for interferon alfa and its serious adverse effects have driven the development of new direct-acting antiviral agents, including the two newly FDA-approved drugs, sofosbuvir and simeprevir.
SOFOSBUVIR: A POLYMERASE INHIBITOR
Sofosbuvir is a uridine nucleotide analogue that selectively inhibits the HCV NS5B RNA-dependent RNA polymerase (Figure 1). It targets the highly conserved nucleotide-binding pocket of this enzyme and functions as a chain terminator.15 While the protease inhibitors are genotype-dependent, inhibition of the highly conserved viral polymerase has an impact that spans genotypes.
Early clinical trials of sofosbuvir
Sofosbuvir has been tested in combination with interferon alfa and ribavirin, as well as in interferon-free regimens (Table 2).16–20
Rodriguez-Torres et al,15
- 56% with sofosbuvir 100 mg, peg-interferon, and ribavirin
- 83% with sofosbuvir 200 mg, peg-interferon, and ribavirin
- 80% with sofosbuvir 400 mg, peg-interferon, and ribavirin
- 43% with peg-interferon and ribavirin alone.
The ATOMIC trial16 tested the efficacy and safety of sofosbuvir in combination with peg-interferon and ribavirin in patients with HCV genotype 1, 4, or 6, without cirrhosis, who had not received any previous treatment. Patients with HCV genotype 1 were randomized to three treatments:
- Sofosbuvir 400 mg orally once daily plus peg-interferon and ribavirin for 12 weeks
- The same regimen, but for 24 weeks
- Sofosbuvir plus peg-interferon and ribavirin for 12 weeks, followed by 12 weeks of either sofosbuvir monotherapy or sofosbuvir plus ribavirin.
The rates of sustained virologic response were very high and were not significantly different among the three groups: 89%, 89%, and 87%, respectively. Patients who were able to complete a full course of therapy achieved even higher rates of sustained virologic response, ranging from 96% to 98%. The likelihood of response was not adversely affected by the usual markers of a poorer prognosis, such as a high viral load (≥ 800,000 IU/mL) or a non-CC IL28B genotype. Although patients with cirrhosis (another predictor of no response) were excluded from this study, the presence of bridging fibrosis did not seem to affect the rate of sustained virologic response. The results in patients with genotypes other than 1 were very encouraging, but the small number of patients enrolled precluded drawing firm conclusions in this group.
Important implications of the ATOMIC trial include the following:
There is no benefit in prolonging treatment with sofosbuvir beyond 12 weeks, since adverse events increased without any improvement in the rate of sustained virologic response.
There is a very low likelihood of developing viral resistance or mutation when using sofosbuvir.
There is no role for response-guided therapy, a concept used with protease inhibitor-based regimens in which patients who have complete clearance of the virus within the first 4 weeks of treatment (a rapid virologic response) and remain clear through 12 weeks of treatment (an extended rapid viral response) can be treated for a shorter duration without decreasing the likelihood of a sustained virologic response.
Lawitz et al17 conducted a randomized double-blind phase 2 trial to evaluate the effect of sofosbuvir dosing on response in noncirrhotic, previously untreated patients with HCV genotype 1, 2, or 3. Patients with HCV genotype 1 were randomized to one of three treatment groups in a 2:2:1 ratio: sofosbuvir 200 mg orally once daily, sofosbuvir 400 mg orally once daily, or placebo, all for 12 weeks in combination with peg-interferon (180 μg weekly) and ribavirin in a dosage based on weight. Depending on the viral response, patients continued peg-interferon and ribavirin for an additional 12 weeks if they achieved an extended rapid viral response, or 36 weeks if they did not achieve an extended rapid virologic response, and in all patients who received placebo. Patients with HCV genotype 2 or 3 were given sofosbuvir 400 mg once daily in combination with interferon and ribavirin for 12 weeks.
As in the ATOMIC trial, all patients treated with sofosbuvir had a very rapid reduction in viral load: 98% of patients with genotype 1 developed a rapid virologic response, and therefore almost all were eligible for the shorter treatment course of 24 weeks.17 The latter finding again suggested that response-guided treatment is not relevant with sofosbuvir-based regimens.
Very high rates of sustained virologic response were seen: 90% in patients with genotype 1 treated with sofosbuvir 200 mg, 91% in those with genotype 1 treated with 400 mg, and 92% in those with genotype 2 or 3. Although 6% of patients in the 200-mg group had virologic breakthrough after completing sofosbuvir treatment, no virologic breakthrough was observed in the 400-mg group, suggesting that the 400-mg dose might suppress the virus more effectively.17
The ELECTRON trial18 was a phase 2 study designed to evaluate the efficacy and safety of sofosbuvir and ribavirin in interferon-sparing and interferon-free regimens in patients with HCV genotype 1, 2, or 3 infection. Sofosbuvir was tested with peg-interferon and ribavirin, with ribavirin alone, and as monotherapy in previously untreated patients with genotype 2 or 3. A small number of patients with genotype 1 who were previously untreated and who were previously nonresponders were also treated with sofosbuvir and ribavirin.
All patients had a rapid virologic response, and viral suppression was sustained through the end of treatment. All patients with genotype 2 or 3 treated with double therapy (sofosbuvir and ribavirin) or triple therapy (sofosbuvir, peg-interferon, and ribavirin) achieved a sustained virologic response, compared with only 60% of patients treated with sofosbuvir monotherapy. The monotherapy group had an equal number of relapsers among those with genotype 2 or 3. Of the genotype 1 patients treated with sofosbuvir and ribavirin, 84% of those previously untreated developed a sustained virologic response, whereas only 10% of the previous nonresponders did.
Phase 3 clinical trials of sofosbuvir
The NEUTRINO trial19 studied the efficacy and safety of sofosbuvir in previously untreated patients with HCV genotype 1, 4, 5, or 6. In this phase 3 open-label study, all patients received sofosbuvir plus peg-interferon and weight-based ribavirin therapy for 12 weeks. Of the patients enrolled, 89% had genotype 1, while 9% had genotype 4 and 2% had genotype 5 or 6. Overall, 17% of the patients had cirrhosis.
The viral load rapidly decreased in all patients treated with sofosbuvir irrespective of the HCV genotype, IL28B status, race, or the presence or absence of cirrhosis. Ninety-nine percent of patients with genotype 1, 4, 5, or 6 achieved a rapid virologic response, and 90% achieved a sustained virologic response at 12 weeks after completion of treatment with sofosbuvir and ribavirin. Patients with cirrhosis had a slightly lower rate of sustained virologic response (80%, compared with 92% in patients without cirrhosis). Also, patients with non-CC IL28B genotypes had a lower rate of sustained virologic response (87% in non-CC allele vs 98% in patients with the favorable CC allele).
The FISSION trial19 recruited previously untreated patients with genotype 2 or 3 and randomized them to therapy with either sofosbuvir plus ribavirin in a weight-based dose for 12 weeks, or 24 weeks of interferon and ribavirin. In this study, 20% of patients in each treatment group had cirrhosis.
As in the NEUTRINO trial, the viral load rapidly decreased in all patients treated with sofosbuvir irrespective of HCV genotype, IL28B status, race, or the presence or absence of cirrhosis. Here, 100% of patients with genotype 2 or 3 who were treated with sofosbuvir and ribavirin achieved a rapid virologic response. Differences in outcome emerged based on genotype: 97% of those with genotype 2 and 56% of those with genotype 3 achieved a sustained virologic response. The overall rate was 67%, which was not different from patients treated with peg-interferon and ribavirin. In the subgroup of patients with cirrhosis, 47% of those treated with sofosbuvir and ribavirin achieved a sustained virologic response, vs 38% of those who received peg-interferon plus ribavirin.
In both the NEUTRINO and FISSION trials, few patients discontinued treatment, with higher rates of most adverse events occurring in patients treated with peg-interferon and ribavirin.
POSITRON,20 a phase 3 clinical trial, tested sofosbuvir in patients with HCV genotype 2 or 3 who were ineligible for peg-interferon, unwilling to take peg-interferon, or unable to tolerate peg-interferon (mainly because of clinically significant psychiatric disorders). Patients were randomized to two treatment groups for 12 weeks: sofosbuvir plus ribavirin, or placebo. About 50% of patients had HCV genotype 3, and 16% had cirrhosis.
The overall rate of sustained virologic response at 12 weeks after treatment was 78% in the sofosbuvir-and-ribavirin group (93% in genotype 2 patients and 61% in genotype 3 patients). Again, cirrhosis was associated with a lower rate of sustained virologic response (61% of patients with cirrhosis achieved a sustained virologic response vs 81% of patients without cirrhosis). None of the sofosbuvir-treated patients had virologic failure while on treatment.
FUSION,20 another phase 3 trial, evaluated sofosbuvir in patients infected with HCV genotype 2 or 3 for whom interferon-based treatment had failed. They were randomized to either 12 weeks or 16 weeks of sofosbuvir and weight-based ribavirin treatment. About 60% of patients had HCV genotype 3, and 34% had cirrhosis.
The overall sustained virologic response rate was 50% in the patients treated for 12 weeks and 73% in those treated for 16 weeks: specifically, 86% of patients with genotype 2 achieved a sustained virologic response at 12 weeks and 94% at 16 weeks, whereas in those with genotype 3 the rates were 30% at 12 weeks and 62% at 16 weeks.
Cirrhosis was again a predictor of lack of response to sofosbuvir. In the group treated for 12 weeks, 31% of those with cirrhosis achieved a sustained virologic response compared with 61% in those without cirrhosis. In the group treated for 16 weeks, 61% of those with cirrhosis achieved a sustained virologic response compared with 76% in those without cirrhosis.
In both the POSITRON and FUSION trials, relapse accounted for all treatment failures, and no virologic resistance was detected in patients who did not have a sustained virologic response. The investigators concluded that 12 weeks of treatment with sofosbuvir and ribavirin can be effective for HCV genotype 2 infection, but extending the treatment to 16 weeks may be beneficial for genotype 3. This may be especially important in patients with cirrhosis or those who did not have a response to peg-interferon-based treatment.
VALENCE,21 an ongoing phase 3 trial in Europe, is assessing the safety and efficacy of sofosbuvir 400 mg once daily and weight-based ribavirin in patients with HCV genotype 2 or 3. Eighty-five percent of the trial participants have received previous treatment, and 21% have cirrhosis. Patients were originally randomized in a 4:1 ratio to receive sofosbuvir plus ribavirin for 12 weeks or matching placebo, but as a result of emerging data suggesting that patients with genotype 3 would benefit from more than 12 weeks of treatment, the study was subsequently amended to extend treatment to 24 weeks for patients with genotype 3.
Overall rates of sustained virologic response were 93% in patients with genotype 2 and 85% in patients with genotype 3. In previously treated patients with genotype 2 who were treated for 12 weeks, the rates of sustained virologic response were 91% in those without cirrhosis vs 88% in those with cirrhosis. In previously treated patients with genotype 3, the rates in those treated for 24 weeks were 87% in patients without cirrhosis vs 60% with cirrhosis. The safety profile was consistent with that of ribavirin.
Side effects of sofosbuvir
In clinical trials, side effects occurred most often when sofosbuvir was combined with interferon and ribavirin and were consistent with the known side effects of the latter two agents. The most frequently reported side effects included fatigue, insomnia, nausea, rash, anemia, headache, and arthralgia, with most of these adverse events rated by treating clinicians as being mild in severity.15,20
In the ATOMIC trial, the most common events leading to drug discontinuation were anemia and neutropenia, both associated with interferon and ribavirin. Patients receiving sofosbuvir monotherapy after 12 weeks of triple therapy showed rapid improvement in hemoglobin levels and neutrophil counts, indicating that hematologic abnormalities attributed solely to sofosbuvir are minimal. In the FISSION trial, the incidence of adverse events was consistently lower in those receiving sofosbuvir-ribavirin than in patients receiving interferon-ribavirin without sofosbuvir.19
In the POSITRON trial, discontinuation of sofosbuvir because of adverse events was uncommon, and there were no differences in the incidence of adverse events and laboratory abnormalities between patients with and without cirrhosis when they received sofosbuvir and ribavirin.20
Sofosbuvir dosage and indications
Sofosbuvir is approved in an oral dose of 400 mg once daily in combination with ribavirin for patients infected with HCV genotype 2 or 3 and in combination with ribavirin and interferon alfa in patients infected with HCV genotype 1 or 4 (Table 3). It could be considered for HCV genotype 1 in combination with ribavirin alone for 24 weeks in patients who are ineligible for interferon.
Sofosbuvir is also recommended in combination with ribavirin in HCV-infected patients with hepatocellular carcinoma who are awaiting liver transplantation, for up to 48 weeks or until they receive a transplant, to prevent posttransplant reinfection with HCV.
Sofosbuvir is expensive
A course of therapy is expected to cost about $84,000, which is significantly more than the cost of previous triple therapy (peg-interferon, ribavirin, and either boceprevir or telaprevir).22 This high cost will undoubtedly lead to less widespread use in developing countries, and potentially even in the United States. As newer direct-acting antiviral agents become available, the price will likely come down, enhancing access to these drugs.
SIMEPREVIR: A SECOND-GENERATION PROTEASE INHIBITOR
Telaprevir and boceprevir are NS3/A4 protease inhibitors that belong to the alfa-ketoamid derivative class. Simeprevir belongs to the macrocyclic class and has a different way of binding to the target enzyme.23 Like sofosbuvir, simeprevir was recently approved by the FDA for the treatment of HCV genotype 1.
The therapeutic efficacy of simeprevir has been tested in several clinical trials (Table 4), including QUEST-124 and QUEST-225 (in previously untreated patients), PROMISE26 (in prior relapsers), and ASPIRE27 (in prior partial and null responders). Results from these trials showed high overall rates of sustained virologic response with triple therapy (ie, simeprevir combined with peg-interferon and ribavirin). It was generally well tolerated, and most adverse events reported during 12 weeks of treatment were of mild to moderate severity.
In QUEST-1 and QUEST-2, both double-blind phase 3 clinical trials, previously untreated patients infected with HCV genotype 1 were randomized in a 2:1 ratio to receive either simeprevir 150 mg daily or placebo for 12 weeks; both groups also received peg-interferon and ribavirin. Patients then received peg-interferon and ribavirin alone for 12 or 36 weeks in the simeprevir group (based on response) and for 36 weeks in the placebo group.
The overall rate of sustained virologic response at 12 weeks was 80% in the simeprevir group (75% in those with genotype 1a and 85% in those with genotype 1b) vs 50% in the placebo group (receiving peg-interferon and ribavirin alone).24,25
PROMISE,26 another double-blind randomized phase 3 clinical trial, evaluated simeprevir in patients with HCV genotype 1 who relapsed after previous interferon-based therapy. It had a similar design to QUEST-1 and QUEST-2, and 15% of all patients had cirrhosis.
The overall sustained virologic response rate at 12 weeks after treatment was 79% in the simeprevir group (70% in patients with genotype 1a and 86% in those with genotype 1b) vs 37% in the placebo group. Rates were similar in patients with absent to moderate fibrosis (82%), advanced fibrosis (73%), or cirrhosis (74%).
ASPIRE.27 Simeprevir efficacy in patients with HCV genotype 1 for whom previous therapy with peg-interferon and ribavirin had failed was tested in ASPIRE, a double-blind randomized phase 2 clinical trial. Patients were randomized to receive simeprevir (either 100 mg or 150 mg daily) for 12, 24, or 48 weeks in combination with 48 weeks of peg-interferon and ribavirin, or placebo plus peg-interferon and ribavirin for 48 weeks.
The primary end point was the rate of sustained virologic response at 24 weeks. Overall, rates were 61% to 80% for the simeprevir treatment groups compared with 23% with placebo, regardless of prior response to peg-interferon and ribavirin. By subgroup, rates were:
- 77% to 89% with simeprevir vs 37% with placebo in prior relapsers
- 48% to 86% with simeprevir vs 9% with placebo in prior partial responders
- 38% to 59% with placebo vs 19% for prior nonresponders.
The best rates of sustained viral response at 24 weeks were in the groups that received simeprevir 150 mg daily: 85% in prior relapsers, 75% in prior partial responders, and 51% in prior nonresponders.
Simeprevir vs other direct-acting antiviral drugs
Advantages of simeprevir over the earlier protease inhibitors include once-daily dosing, a lower rate of adverse events (the most common being fatigue, headache, rash, photosensitivity, and pruritus), a lower likelihood of discontinuation because of adverse events, and fewer drug-drug interactions (since it is a weak inhibitor of the CYP3A4 enzyme).
Unlike sofosbuvir, simeprevir was FDA-approved only for HCV genotype 1 and in combination with interferon alfa and ribavirin. Compared with sofosbuvir, the treatment duration with simeprevir regimens is longer overall (interferon alfa and ribavirin are given for 12 weeks in sofosbuvir-based regimens vs 24 to 48 weeks with simeprevir). As with sofosbuvir, the estimated cost of simeprevir is high, about $66,000 for a 12-week course.
Simeprevir dosage and indications
Simeprevir was approved at an oral dose of 150 mg once daily in combination with ribavirin and interferon alfa in patients with HCV genotype 1 (Table 5).
The approved regimens for simeprevir are fixed in total duration based on the patient’s treatment history. Specifically, all patients receive the drug in combination with peg-interferon and ribavirin for 12 weeks. Then, previously untreated patients and prior relapsers continue to receive peg-interferon and ribavirin alone for another 12 weeks, and those with a partial or null response continue with these drugs for another 36 weeks.
Patients infected with HCV genotype 1a should be screened for the NS3 Q80K polymorphism at baseline, as it has been associated with substantially reduced response to simeprevir.
Sofosbuvir and simeprevir in combination
The COSMOS trial.28 Given their differences in mechanism of action, sofosbuvir and simeprevir are being tested in combination. The COSMOS trial is an ongoing phase 2 randomized open-label study investigating the efficacy and safety of simeprevir and sofosbuvir in combination with and without ribavirin in patients with HCV genotype 1, including nonresponders and those with cirrhosis. Early results are promising, with very high rates of sustained virologic response with the sofosbuvir-simeprevir combination (93% to 100%) and indicate that the addition of ribavirin might not be needed to achieve sustained virologic response in this patient population.
THE FUTURE
The emergence of all-oral regimens for HCV treatment with increasingly sophisticated agents such as sofosbuvir and simeprevir will dramatically alter the management of HCV patients. In view of the improvement in sustained virologic response rates with these treatments, and since most HCV-infected persons have no symptoms, the US Centers for Disease Control and Prevention29 recently recommended one-time testing of the cohort in which the prevalence of HCV infection is highest: all persons born between 1945 and 1965. This undoubtedly will increase the detection of this infection—and the number of new patients expecting treatment.
Future drugs promise further improvements (Table 6).30–35 Advances in knowledge of the HCV molecular structure have led to the development of numerous direct-acting antiviral agents with very specific viral targets. A second wave of protease inhibitors and of nucleoside and nonnucleoside polymerase inhibitors will soon be available. Inhibitors of NS5A (a protein important in the assembly of the viral replication complex) such as daclatasvir and ledipasvir, are currently in phase 3 clinical trials. Other viral proteins involved in assembly of the virus, including the core protein and p7, are being explored as drug targets. In addition, inhibiting host targets such as cyclophilin A and miR122 has gained traction recently, with specific agents currently in phase 2 and 3 clinical trials.
Factors that previously were major determinants of response to treatment, such as IL28B genotype, viral load, race, age, extent of fibrosis, and genotype 1 subtypes, will become much less important with the introduction of highly potent direct-acting antiviral agents.
Many all-oral combinations are being evaluated in clinical trials. For example, the open-label, phase 2 LONESTAR trial tested the utility of combining sofosbuvir and ledipasvir (an NS5A inhibitor) with and without ribavirin for 8 or 12 weeks in previously untreated patients with HCV genotype 1, and for 12 weeks in patients with HCV genotype 1 who did not achieve a sustained virologic response after receiving a protease inhibitor-based regimen (half of whom had compensated cirrhosis).36 Sustained virologic response rates were very high (95% to 100%) in both previously treated and previously untreated patients, including those with cirrhosis. Similar rates were achieved by the 8-week and 12-week groups in noncirrhotic patients who had not been previously treated for HCV. The typical hematologic abnormalities associated with interferon were not observed except for mild anemia in patients who received ribavirin. These results suggest that the combination of sofosbuvir and ledipasvir could offer a very effective, short, all-oral treatment for patients with HCV genotype 1, including those with cirrhosis, who up to now have been difficult to treat.
Challenges remaining
The success of sofosbuvir and simeprevir paves the way for interferon-free regimens.37 For a long time, the treatment of HCV infection required close monitoring of patients while managing the side effects of interferon, but the current and emerging direct-acting antiviral agents will soon change this practice. Given the synergistic effects of combination therapy—targeting the virus at multiple locations, decreasing the likelihood of drug resistance, and improving efficacy—combination regimens seem to be the optimal solution to the HCV epidemic. Lower risk of side effects and shorter treatment duration will definitely improve the acceptance of any new regimen. New agents that act against conserved viral targets, thereby yielding activity across multiple genotypes, will be advantageous as well. Table 7 compares the rates of sustained virologic response of the different currently approved HCV treatment regimens.
Clinical challenges remain, including the management of special patient populations for whom data are still limited. These include patients with cirrhosis, chronic kidney disease, renal failure, and concurrent infection with human immunodeficiency virus, and patients who have undergone solid organ transplantation. Clinical trials are under way to evaluate the treatment options for these patients, who will likely need to wait for the emergence of additional agents before dramatic improvement in sustained virologic response rates may be expected.38
As the treatment of HCV becomes simpler, safer, and more effective, primary care physicians will increasingly be expected to manage it. Difficult-to-treat patients, including the special populations above, will require specialist management and individualized treatment regimens, at least until better therapies are available. The high projected cost of the new agents may limit access, at least initially. However, the dramatic improvement in sustained virologic response rates and all that that implies in terms of decreased risk of advanced liver disease and its complications will undoubtedly make these therapies cost-effective.39
In late 2013, the US Food and Drug Administration (FDA) approved sofosbuvir and simeprevir, the newest direct-acting antiviral agents for treating chronic hepatitis C virus (HCV) infection. Multiple clinical trials have demonstrated dramatically improved treatment outcomes with these agents, opening the door to all-oral regimens or interferon-free regimens as the future standard of care for HCV.
In this article, we discuss the results of the trials that established the efficacy and safety of sofosbuvir and simeprevir and led to their FDA approval. We also summarize the importance of these agents and evaluate other direct-acting antivirals currently in the pipeline for HCV treatment.
HCV IS A RISING PROBLEM
Chronic HCV infection is a major clinical and public health problem, with the estimated number of people infected exceeding 170 million worldwide, including 3.2 million in the United States.1 It is a leading cause of cirrhosis, and its complications include hepatocellular carcinoma and liver failure. Cirrhosis due to HCV remains the leading indication for liver transplantation in the United States, accounting for nearly 40% of liver transplants in adults.2
The clinical impact of HCV will only continue to escalate, and in parallel, so will the cost to society. Models suggest that HCV-related deaths will double between 2010 and 2019, and considering only direct medical costs, the projected financial burden of treating HCV-related disease during this interval is estimated at between $6.5 and $13.6 billion.3
AN RNA VIRUS WITH SIX GENOTYPES
HCV, first identified in 1989, is an enveloped, single-stranded RNA flavivirus of the Hepacivirus genus measuring 50 to 60 nm in diameter.4 There are six viral genotypes, with genotype 1 being the most common in the United States and traditionally the most difficult to treat.
Once inside the host cell, the virus releases its RNA strand, which is translated into a single polyprotein of about 3,000 amino acids. This large molecule is then cleaved by proteases into several domains: three structural proteins (C, E1, and E2), a small protein called p7, and six nonstructural proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) (Figure 1).5 These nonstructural proteins enable the virus to replicate.
GOAL OF TREATING HCV: A SUSTAINED VIROLOGIC RESPONSE
The aim of HCV treatment is to achieve a sustained virologic response, defined as having no detectable viral RNA after completion of antiviral therapy. This is associated with substantially better clinical outcomes, lower rates of liver-related morbidity and all-cause mortality, and stabilization of or even improvement in liver histology.6,7 This end point has traditionally been assessed at 6 months after the end of therapy, but recent data suggest the rates at 12 weeks are essentially equivalent.
Table 1 summarizes the patterns of virologic response in treating HCV infection.
Interferon plus ribavirin: The standard of care for many years
HCV treatment has evolved over the past 20 years. Before 2011, the standard of care was a combination of interferon alfa-polyethylene glycol (peg-interferon), given as a weekly injection, and oral ribavirin. Neither drug has specific antiviral activity, and when they are used together they result in a sustained virologic response in fewer than 50% of patients with HCV genotype 1 and, at best, in 70% to 80% of patients with other genotypes.8
Nearly all patients receiving interferon experience side effects, which can be serious. Fatigue and flu-like symptoms are common, and the drug can also cause psychiatric symptoms (including depression or psychosis), weight loss, seizures, peripheral neuropathy, and bone marrow suppression. Ribavirin causes hemolysis and skin complications and is teratogenic.9
An important bit of information to know when using interferon is the patient’s IL28B genotype. This refers to a single-nucleotide polymorphism (C or T) on chromosome 19q13 (rs12979860) upstream of the IL28B gene encoding for interferon lambda-3. It is strongly associated with responsiveness to interferon: patients with the IL28B CC genotype have a much better chance of a sustained virologic response with interferon than do patients with CT or TT.
Boceprevir and telaprevir: First-generation protease inhibitors
In May 2011, the FDA approved the NS3/4A protease inhibitors boceprevir and telaprevir for treating HCV genotype 1, marking the beginning of the era of direct-acting antiviral agents.10 When these drugs are used in combination with peg-interferon alfa and ribavirin, up to 75% of patients with HCV genotype 1 who have had no previous treatment achieve a sustained virologic response.
But despite greatly improving the response rate, these first-generation protease inhibitors have substantial limitations. Twenty-five percent of patients with HCV genotype 1 who have received no previous treatment and 71% of patients who did not respond to previous treatment will not achieve a sustained virologic response with these agents.11 Further, they are effective only against HCV genotype 1, being highly specific for the amino acid target sequence of the NS3 region.
Also, they must be used in combination with interferon alfa and ribavirin because the virus needs to mutate only a little—a few amino-acid substitutions—to gain resistance to them.12 Therefore, patients are still exposed to interferon and ribavirin, with their toxicity. In addition, dysgeusia is seen with boceprevir, rash with telaprevir, and anemia with both.13,14
Finally, serious drug-drug interactions prompted the FDA to impose warnings for the use of these agents with other medications that interact with CYP3A4, the principal enzyme responsible for their metabolism. Thus, these significant adverse effects dampen the enthusiasm of patients contemplating a long course of treatment with these agents.
The need to improve the rate of sustained virologic response, shorten the duration of treatment, avoid serious side effects, improve efficacy in treating patients infected with genotypes other than 1, and, importantly, eliminate the need for interferon alfa and its serious adverse effects have driven the development of new direct-acting antiviral agents, including the two newly FDA-approved drugs, sofosbuvir and simeprevir.
SOFOSBUVIR: A POLYMERASE INHIBITOR
Sofosbuvir is a uridine nucleotide analogue that selectively inhibits the HCV NS5B RNA-dependent RNA polymerase (Figure 1). It targets the highly conserved nucleotide-binding pocket of this enzyme and functions as a chain terminator.15 While the protease inhibitors are genotype-dependent, inhibition of the highly conserved viral polymerase has an impact that spans genotypes.
Early clinical trials of sofosbuvir
Sofosbuvir has been tested in combination with interferon alfa and ribavirin, as well as in interferon-free regimens (Table 2).16–20
Rodriguez-Torres et al,15
- 56% with sofosbuvir 100 mg, peg-interferon, and ribavirin
- 83% with sofosbuvir 200 mg, peg-interferon, and ribavirin
- 80% with sofosbuvir 400 mg, peg-interferon, and ribavirin
- 43% with peg-interferon and ribavirin alone.
The ATOMIC trial16 tested the efficacy and safety of sofosbuvir in combination with peg-interferon and ribavirin in patients with HCV genotype 1, 4, or 6, without cirrhosis, who had not received any previous treatment. Patients with HCV genotype 1 were randomized to three treatments:
- Sofosbuvir 400 mg orally once daily plus peg-interferon and ribavirin for 12 weeks
- The same regimen, but for 24 weeks
- Sofosbuvir plus peg-interferon and ribavirin for 12 weeks, followed by 12 weeks of either sofosbuvir monotherapy or sofosbuvir plus ribavirin.
The rates of sustained virologic response were very high and were not significantly different among the three groups: 89%, 89%, and 87%, respectively. Patients who were able to complete a full course of therapy achieved even higher rates of sustained virologic response, ranging from 96% to 98%. The likelihood of response was not adversely affected by the usual markers of a poorer prognosis, such as a high viral load (≥ 800,000 IU/mL) or a non-CC IL28B genotype. Although patients with cirrhosis (another predictor of no response) were excluded from this study, the presence of bridging fibrosis did not seem to affect the rate of sustained virologic response. The results in patients with genotypes other than 1 were very encouraging, but the small number of patients enrolled precluded drawing firm conclusions in this group.
Important implications of the ATOMIC trial include the following:
There is no benefit in prolonging treatment with sofosbuvir beyond 12 weeks, since adverse events increased without any improvement in the rate of sustained virologic response.
There is a very low likelihood of developing viral resistance or mutation when using sofosbuvir.
There is no role for response-guided therapy, a concept used with protease inhibitor-based regimens in which patients who have complete clearance of the virus within the first 4 weeks of treatment (a rapid virologic response) and remain clear through 12 weeks of treatment (an extended rapid viral response) can be treated for a shorter duration without decreasing the likelihood of a sustained virologic response.
Lawitz et al17 conducted a randomized double-blind phase 2 trial to evaluate the effect of sofosbuvir dosing on response in noncirrhotic, previously untreated patients with HCV genotype 1, 2, or 3. Patients with HCV genotype 1 were randomized to one of three treatment groups in a 2:2:1 ratio: sofosbuvir 200 mg orally once daily, sofosbuvir 400 mg orally once daily, or placebo, all for 12 weeks in combination with peg-interferon (180 μg weekly) and ribavirin in a dosage based on weight. Depending on the viral response, patients continued peg-interferon and ribavirin for an additional 12 weeks if they achieved an extended rapid viral response, or 36 weeks if they did not achieve an extended rapid virologic response, and in all patients who received placebo. Patients with HCV genotype 2 or 3 were given sofosbuvir 400 mg once daily in combination with interferon and ribavirin for 12 weeks.
As in the ATOMIC trial, all patients treated with sofosbuvir had a very rapid reduction in viral load: 98% of patients with genotype 1 developed a rapid virologic response, and therefore almost all were eligible for the shorter treatment course of 24 weeks.17 The latter finding again suggested that response-guided treatment is not relevant with sofosbuvir-based regimens.
Very high rates of sustained virologic response were seen: 90% in patients with genotype 1 treated with sofosbuvir 200 mg, 91% in those with genotype 1 treated with 400 mg, and 92% in those with genotype 2 or 3. Although 6% of patients in the 200-mg group had virologic breakthrough after completing sofosbuvir treatment, no virologic breakthrough was observed in the 400-mg group, suggesting that the 400-mg dose might suppress the virus more effectively.17
The ELECTRON trial18 was a phase 2 study designed to evaluate the efficacy and safety of sofosbuvir and ribavirin in interferon-sparing and interferon-free regimens in patients with HCV genotype 1, 2, or 3 infection. Sofosbuvir was tested with peg-interferon and ribavirin, with ribavirin alone, and as monotherapy in previously untreated patients with genotype 2 or 3. A small number of patients with genotype 1 who were previously untreated and who were previously nonresponders were also treated with sofosbuvir and ribavirin.
All patients had a rapid virologic response, and viral suppression was sustained through the end of treatment. All patients with genotype 2 or 3 treated with double therapy (sofosbuvir and ribavirin) or triple therapy (sofosbuvir, peg-interferon, and ribavirin) achieved a sustained virologic response, compared with only 60% of patients treated with sofosbuvir monotherapy. The monotherapy group had an equal number of relapsers among those with genotype 2 or 3. Of the genotype 1 patients treated with sofosbuvir and ribavirin, 84% of those previously untreated developed a sustained virologic response, whereas only 10% of the previous nonresponders did.
Phase 3 clinical trials of sofosbuvir
The NEUTRINO trial19 studied the efficacy and safety of sofosbuvir in previously untreated patients with HCV genotype 1, 4, 5, or 6. In this phase 3 open-label study, all patients received sofosbuvir plus peg-interferon and weight-based ribavirin therapy for 12 weeks. Of the patients enrolled, 89% had genotype 1, while 9% had genotype 4 and 2% had genotype 5 or 6. Overall, 17% of the patients had cirrhosis.
The viral load rapidly decreased in all patients treated with sofosbuvir irrespective of the HCV genotype, IL28B status, race, or the presence or absence of cirrhosis. Ninety-nine percent of patients with genotype 1, 4, 5, or 6 achieved a rapid virologic response, and 90% achieved a sustained virologic response at 12 weeks after completion of treatment with sofosbuvir and ribavirin. Patients with cirrhosis had a slightly lower rate of sustained virologic response (80%, compared with 92% in patients without cirrhosis). Also, patients with non-CC IL28B genotypes had a lower rate of sustained virologic response (87% in non-CC allele vs 98% in patients with the favorable CC allele).
The FISSION trial19 recruited previously untreated patients with genotype 2 or 3 and randomized them to therapy with either sofosbuvir plus ribavirin in a weight-based dose for 12 weeks, or 24 weeks of interferon and ribavirin. In this study, 20% of patients in each treatment group had cirrhosis.
As in the NEUTRINO trial, the viral load rapidly decreased in all patients treated with sofosbuvir irrespective of HCV genotype, IL28B status, race, or the presence or absence of cirrhosis. Here, 100% of patients with genotype 2 or 3 who were treated with sofosbuvir and ribavirin achieved a rapid virologic response. Differences in outcome emerged based on genotype: 97% of those with genotype 2 and 56% of those with genotype 3 achieved a sustained virologic response. The overall rate was 67%, which was not different from patients treated with peg-interferon and ribavirin. In the subgroup of patients with cirrhosis, 47% of those treated with sofosbuvir and ribavirin achieved a sustained virologic response, vs 38% of those who received peg-interferon plus ribavirin.
In both the NEUTRINO and FISSION trials, few patients discontinued treatment, with higher rates of most adverse events occurring in patients treated with peg-interferon and ribavirin.
POSITRON,20 a phase 3 clinical trial, tested sofosbuvir in patients with HCV genotype 2 or 3 who were ineligible for peg-interferon, unwilling to take peg-interferon, or unable to tolerate peg-interferon (mainly because of clinically significant psychiatric disorders). Patients were randomized to two treatment groups for 12 weeks: sofosbuvir plus ribavirin, or placebo. About 50% of patients had HCV genotype 3, and 16% had cirrhosis.
The overall rate of sustained virologic response at 12 weeks after treatment was 78% in the sofosbuvir-and-ribavirin group (93% in genotype 2 patients and 61% in genotype 3 patients). Again, cirrhosis was associated with a lower rate of sustained virologic response (61% of patients with cirrhosis achieved a sustained virologic response vs 81% of patients without cirrhosis). None of the sofosbuvir-treated patients had virologic failure while on treatment.
FUSION,20 another phase 3 trial, evaluated sofosbuvir in patients infected with HCV genotype 2 or 3 for whom interferon-based treatment had failed. They were randomized to either 12 weeks or 16 weeks of sofosbuvir and weight-based ribavirin treatment. About 60% of patients had HCV genotype 3, and 34% had cirrhosis.
The overall sustained virologic response rate was 50% in the patients treated for 12 weeks and 73% in those treated for 16 weeks: specifically, 86% of patients with genotype 2 achieved a sustained virologic response at 12 weeks and 94% at 16 weeks, whereas in those with genotype 3 the rates were 30% at 12 weeks and 62% at 16 weeks.
Cirrhosis was again a predictor of lack of response to sofosbuvir. In the group treated for 12 weeks, 31% of those with cirrhosis achieved a sustained virologic response compared with 61% in those without cirrhosis. In the group treated for 16 weeks, 61% of those with cirrhosis achieved a sustained virologic response compared with 76% in those without cirrhosis.
In both the POSITRON and FUSION trials, relapse accounted for all treatment failures, and no virologic resistance was detected in patients who did not have a sustained virologic response. The investigators concluded that 12 weeks of treatment with sofosbuvir and ribavirin can be effective for HCV genotype 2 infection, but extending the treatment to 16 weeks may be beneficial for genotype 3. This may be especially important in patients with cirrhosis or those who did not have a response to peg-interferon-based treatment.
VALENCE,21 an ongoing phase 3 trial in Europe, is assessing the safety and efficacy of sofosbuvir 400 mg once daily and weight-based ribavirin in patients with HCV genotype 2 or 3. Eighty-five percent of the trial participants have received previous treatment, and 21% have cirrhosis. Patients were originally randomized in a 4:1 ratio to receive sofosbuvir plus ribavirin for 12 weeks or matching placebo, but as a result of emerging data suggesting that patients with genotype 3 would benefit from more than 12 weeks of treatment, the study was subsequently amended to extend treatment to 24 weeks for patients with genotype 3.
Overall rates of sustained virologic response were 93% in patients with genotype 2 and 85% in patients with genotype 3. In previously treated patients with genotype 2 who were treated for 12 weeks, the rates of sustained virologic response were 91% in those without cirrhosis vs 88% in those with cirrhosis. In previously treated patients with genotype 3, the rates in those treated for 24 weeks were 87% in patients without cirrhosis vs 60% with cirrhosis. The safety profile was consistent with that of ribavirin.
Side effects of sofosbuvir
In clinical trials, side effects occurred most often when sofosbuvir was combined with interferon and ribavirin and were consistent with the known side effects of the latter two agents. The most frequently reported side effects included fatigue, insomnia, nausea, rash, anemia, headache, and arthralgia, with most of these adverse events rated by treating clinicians as being mild in severity.15,20
In the ATOMIC trial, the most common events leading to drug discontinuation were anemia and neutropenia, both associated with interferon and ribavirin. Patients receiving sofosbuvir monotherapy after 12 weeks of triple therapy showed rapid improvement in hemoglobin levels and neutrophil counts, indicating that hematologic abnormalities attributed solely to sofosbuvir are minimal. In the FISSION trial, the incidence of adverse events was consistently lower in those receiving sofosbuvir-ribavirin than in patients receiving interferon-ribavirin without sofosbuvir.19
In the POSITRON trial, discontinuation of sofosbuvir because of adverse events was uncommon, and there were no differences in the incidence of adverse events and laboratory abnormalities between patients with and without cirrhosis when they received sofosbuvir and ribavirin.20
Sofosbuvir dosage and indications
Sofosbuvir is approved in an oral dose of 400 mg once daily in combination with ribavirin for patients infected with HCV genotype 2 or 3 and in combination with ribavirin and interferon alfa in patients infected with HCV genotype 1 or 4 (Table 3). It could be considered for HCV genotype 1 in combination with ribavirin alone for 24 weeks in patients who are ineligible for interferon.
Sofosbuvir is also recommended in combination with ribavirin in HCV-infected patients with hepatocellular carcinoma who are awaiting liver transplantation, for up to 48 weeks or until they receive a transplant, to prevent posttransplant reinfection with HCV.
Sofosbuvir is expensive
A course of therapy is expected to cost about $84,000, which is significantly more than the cost of previous triple therapy (peg-interferon, ribavirin, and either boceprevir or telaprevir).22 This high cost will undoubtedly lead to less widespread use in developing countries, and potentially even in the United States. As newer direct-acting antiviral agents become available, the price will likely come down, enhancing access to these drugs.
SIMEPREVIR: A SECOND-GENERATION PROTEASE INHIBITOR
Telaprevir and boceprevir are NS3/A4 protease inhibitors that belong to the alfa-ketoamid derivative class. Simeprevir belongs to the macrocyclic class and has a different way of binding to the target enzyme.23 Like sofosbuvir, simeprevir was recently approved by the FDA for the treatment of HCV genotype 1.
The therapeutic efficacy of simeprevir has been tested in several clinical trials (Table 4), including QUEST-124 and QUEST-225 (in previously untreated patients), PROMISE26 (in prior relapsers), and ASPIRE27 (in prior partial and null responders). Results from these trials showed high overall rates of sustained virologic response with triple therapy (ie, simeprevir combined with peg-interferon and ribavirin). It was generally well tolerated, and most adverse events reported during 12 weeks of treatment were of mild to moderate severity.
In QUEST-1 and QUEST-2, both double-blind phase 3 clinical trials, previously untreated patients infected with HCV genotype 1 were randomized in a 2:1 ratio to receive either simeprevir 150 mg daily or placebo for 12 weeks; both groups also received peg-interferon and ribavirin. Patients then received peg-interferon and ribavirin alone for 12 or 36 weeks in the simeprevir group (based on response) and for 36 weeks in the placebo group.
The overall rate of sustained virologic response at 12 weeks was 80% in the simeprevir group (75% in those with genotype 1a and 85% in those with genotype 1b) vs 50% in the placebo group (receiving peg-interferon and ribavirin alone).24,25
PROMISE,26 another double-blind randomized phase 3 clinical trial, evaluated simeprevir in patients with HCV genotype 1 who relapsed after previous interferon-based therapy. It had a similar design to QUEST-1 and QUEST-2, and 15% of all patients had cirrhosis.
The overall sustained virologic response rate at 12 weeks after treatment was 79% in the simeprevir group (70% in patients with genotype 1a and 86% in those with genotype 1b) vs 37% in the placebo group. Rates were similar in patients with absent to moderate fibrosis (82%), advanced fibrosis (73%), or cirrhosis (74%).
ASPIRE.27 Simeprevir efficacy in patients with HCV genotype 1 for whom previous therapy with peg-interferon and ribavirin had failed was tested in ASPIRE, a double-blind randomized phase 2 clinical trial. Patients were randomized to receive simeprevir (either 100 mg or 150 mg daily) for 12, 24, or 48 weeks in combination with 48 weeks of peg-interferon and ribavirin, or placebo plus peg-interferon and ribavirin for 48 weeks.
The primary end point was the rate of sustained virologic response at 24 weeks. Overall, rates were 61% to 80% for the simeprevir treatment groups compared with 23% with placebo, regardless of prior response to peg-interferon and ribavirin. By subgroup, rates were:
- 77% to 89% with simeprevir vs 37% with placebo in prior relapsers
- 48% to 86% with simeprevir vs 9% with placebo in prior partial responders
- 38% to 59% with placebo vs 19% for prior nonresponders.
The best rates of sustained viral response at 24 weeks were in the groups that received simeprevir 150 mg daily: 85% in prior relapsers, 75% in prior partial responders, and 51% in prior nonresponders.
Simeprevir vs other direct-acting antiviral drugs
Advantages of simeprevir over the earlier protease inhibitors include once-daily dosing, a lower rate of adverse events (the most common being fatigue, headache, rash, photosensitivity, and pruritus), a lower likelihood of discontinuation because of adverse events, and fewer drug-drug interactions (since it is a weak inhibitor of the CYP3A4 enzyme).
Unlike sofosbuvir, simeprevir was FDA-approved only for HCV genotype 1 and in combination with interferon alfa and ribavirin. Compared with sofosbuvir, the treatment duration with simeprevir regimens is longer overall (interferon alfa and ribavirin are given for 12 weeks in sofosbuvir-based regimens vs 24 to 48 weeks with simeprevir). As with sofosbuvir, the estimated cost of simeprevir is high, about $66,000 for a 12-week course.
Simeprevir dosage and indications
Simeprevir was approved at an oral dose of 150 mg once daily in combination with ribavirin and interferon alfa in patients with HCV genotype 1 (Table 5).
The approved regimens for simeprevir are fixed in total duration based on the patient’s treatment history. Specifically, all patients receive the drug in combination with peg-interferon and ribavirin for 12 weeks. Then, previously untreated patients and prior relapsers continue to receive peg-interferon and ribavirin alone for another 12 weeks, and those with a partial or null response continue with these drugs for another 36 weeks.
Patients infected with HCV genotype 1a should be screened for the NS3 Q80K polymorphism at baseline, as it has been associated with substantially reduced response to simeprevir.
Sofosbuvir and simeprevir in combination
The COSMOS trial.28 Given their differences in mechanism of action, sofosbuvir and simeprevir are being tested in combination. The COSMOS trial is an ongoing phase 2 randomized open-label study investigating the efficacy and safety of simeprevir and sofosbuvir in combination with and without ribavirin in patients with HCV genotype 1, including nonresponders and those with cirrhosis. Early results are promising, with very high rates of sustained virologic response with the sofosbuvir-simeprevir combination (93% to 100%) and indicate that the addition of ribavirin might not be needed to achieve sustained virologic response in this patient population.
THE FUTURE
The emergence of all-oral regimens for HCV treatment with increasingly sophisticated agents such as sofosbuvir and simeprevir will dramatically alter the management of HCV patients. In view of the improvement in sustained virologic response rates with these treatments, and since most HCV-infected persons have no symptoms, the US Centers for Disease Control and Prevention29 recently recommended one-time testing of the cohort in which the prevalence of HCV infection is highest: all persons born between 1945 and 1965. This undoubtedly will increase the detection of this infection—and the number of new patients expecting treatment.
Future drugs promise further improvements (Table 6).30–35 Advances in knowledge of the HCV molecular structure have led to the development of numerous direct-acting antiviral agents with very specific viral targets. A second wave of protease inhibitors and of nucleoside and nonnucleoside polymerase inhibitors will soon be available. Inhibitors of NS5A (a protein important in the assembly of the viral replication complex) such as daclatasvir and ledipasvir, are currently in phase 3 clinical trials. Other viral proteins involved in assembly of the virus, including the core protein and p7, are being explored as drug targets. In addition, inhibiting host targets such as cyclophilin A and miR122 has gained traction recently, with specific agents currently in phase 2 and 3 clinical trials.
Factors that previously were major determinants of response to treatment, such as IL28B genotype, viral load, race, age, extent of fibrosis, and genotype 1 subtypes, will become much less important with the introduction of highly potent direct-acting antiviral agents.
Many all-oral combinations are being evaluated in clinical trials. For example, the open-label, phase 2 LONESTAR trial tested the utility of combining sofosbuvir and ledipasvir (an NS5A inhibitor) with and without ribavirin for 8 or 12 weeks in previously untreated patients with HCV genotype 1, and for 12 weeks in patients with HCV genotype 1 who did not achieve a sustained virologic response after receiving a protease inhibitor-based regimen (half of whom had compensated cirrhosis).36 Sustained virologic response rates were very high (95% to 100%) in both previously treated and previously untreated patients, including those with cirrhosis. Similar rates were achieved by the 8-week and 12-week groups in noncirrhotic patients who had not been previously treated for HCV. The typical hematologic abnormalities associated with interferon were not observed except for mild anemia in patients who received ribavirin. These results suggest that the combination of sofosbuvir and ledipasvir could offer a very effective, short, all-oral treatment for patients with HCV genotype 1, including those with cirrhosis, who up to now have been difficult to treat.
Challenges remaining
The success of sofosbuvir and simeprevir paves the way for interferon-free regimens.37 For a long time, the treatment of HCV infection required close monitoring of patients while managing the side effects of interferon, but the current and emerging direct-acting antiviral agents will soon change this practice. Given the synergistic effects of combination therapy—targeting the virus at multiple locations, decreasing the likelihood of drug resistance, and improving efficacy—combination regimens seem to be the optimal solution to the HCV epidemic. Lower risk of side effects and shorter treatment duration will definitely improve the acceptance of any new regimen. New agents that act against conserved viral targets, thereby yielding activity across multiple genotypes, will be advantageous as well. Table 7 compares the rates of sustained virologic response of the different currently approved HCV treatment regimens.
Clinical challenges remain, including the management of special patient populations for whom data are still limited. These include patients with cirrhosis, chronic kidney disease, renal failure, and concurrent infection with human immunodeficiency virus, and patients who have undergone solid organ transplantation. Clinical trials are under way to evaluate the treatment options for these patients, who will likely need to wait for the emergence of additional agents before dramatic improvement in sustained virologic response rates may be expected.38
As the treatment of HCV becomes simpler, safer, and more effective, primary care physicians will increasingly be expected to manage it. Difficult-to-treat patients, including the special populations above, will require specialist management and individualized treatment regimens, at least until better therapies are available. The high projected cost of the new agents may limit access, at least initially. However, the dramatic improvement in sustained virologic response rates and all that that implies in terms of decreased risk of advanced liver disease and its complications will undoubtedly make these therapies cost-effective.39
- Averhoff FM, Glass N, Holtzman D. Global burden of hepatitis C: considerations for healthcare providers in the united states. Clin Infect Dis 2012; 55(suppl 1):S10–S15.
- Wiesner RH, Sorrell M, Villamil F; International Liver Transplantation Society Expert Panel. Report of the first international liver transplantation society expert panel consensus conference on liver transplantation and hepatitis C. Liver Transplant 2003; 9:S1–S9.
- Wong JB, McQuillan GM, McHutchison JG, Poynard T. Estimating future hepatitis C morbidity, mortality, and costs in the United States. Am J Public Health 2000; 90:1562–1569.
- Pawlotsky JM, Chevaliez S, McHutchison JG. The hepatitis C virus life cycle as a target for new antiviral therapies. Gastroenterology 2007; 132:1979–1998.
- Bartenschlager R, Lohmann V. Replication of hepatitis C virus. J Gen Virol 2000; 81:1631–1648.
- Singal AG, Volk ML, Jensen D, Di Bisceglie AM, Schoenfeld PS. A sustained viral response is associated with reduced liver-related morbidity and mortality in patients with hepatitis C virus. Clin Gastroenterol Hepatol 2010; 8:280–288,288.e1.
- Camma C, Di Bona D, Schepis F, et al. Effect of peginterferon alfa-2a on liver histology in chronic hepatitis C: a meta-analysis of individual patient data. Hepatology 2004; 39:333–342.
- Paeshuyse J, Dallmeier K, Neyts J. Ribavirin for the treatment of chronic hepatitis C virus infection: a review of the proposed mechanisms of action. Curr Opin Virol 2011; 1:590–598.
- Thomas E, Ghany MG, Liang TJ. The application and mechanism of action of ribavirin in therapy of hepatitis C. Antivir Chem Chemother 2012; 23:1–12.
- Ghany MG, Nelson DR, Strader DB, Thomas DL, Seeff LB; American Association for Study of Liver Diseases. An update on treatment of genotype 1 chronic hepatitis C virus infection: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 2011; 54:1433–1444.
- Soriano V, Vispo E, Poveda E, Labarga P, Barreiro P. Treatment failure with new hepatitis C drugs. Expert Opin Pharmacother 2012; 13:313–323.
- Asselah T, Marcellin P. Interferon free therapy with direct acting antivirals for HCV. Liver Int 2013; 33(suppl 1):93–104.
- Manns MP, McCone J, Davis MN, et al. Overall safety profile of boceprevir plus peginterferon alfa-2b and ribavirin in patients with chronic hepatitis C genotype 1: a combined analysis of 3 phase 2/3 clinical trials. Liver Int 2013; Aug 2. doi: 10.1111/liv.12300. [Epub ahead of print]
- Jacobson IM, McHutchison JG, Dusheiko G, et al. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364:2405–2416.
- Rodriguez-Torres M, Lawitz E, Kowdley KV, et al. Sofosbuvir (GS-7977) plus peginterferon/ribavirin in treatment-naive patients with HCV genotype 1: a randomized, 28-day, dose-ranging trial. J Hepatol 2013; 58:663–668.
- Kowdley KV, Lawitz E, Crespo I, et al. Sofosbuvir with pegylated interferon alfa-2a and ribavirin for treatment-naive patients with hepatitis C genotype-1 infection (ATOMIC): an open-label, randomised, multicentre phase 2 trial. Lancet 2013; 381:2100–2107.
- Lawitz E, Lalezari JP, Hassanein T, et al. Sofosbuvir in combination with peginterferon alfa-2a and ribavirin for non-cirrhotic, treatment-naive patients with genotypes 1, 2, and 3 hepatitis C infection: a randomised, double-blind, phase 2 trial. Lancet Infect Dis 2013; 13:401–408.
- Gane EJ, Stedman CA, Hyland RH, et al. Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N Engl J Med 2013; 368:34–44.
- Lawitz E, Mangia A, Wyles D, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 2013; 368:1878–1887.
- Jacobson IM, Gordon SC, Kowdley KV, et al. Sofosbuvir for hepatitis C genotype 2 or 3 in patients without treatment options. N Engl J Med 2013; 368:1867–1877.
- Zeuzem S, Dusheiko G, Salupere R, et al. Sofosbuvir + ribavirin for 12 or 24 weeks for patients with HCV genotype 2 or 3: the VALENCE trial [abstract no.1085]. 64th Annual Meeting of the American Association for the Study of Liver Diseases; November 1–5, 2013; Washington, DC.
- Soriano V, Vispo E, de Mendoza C, et al. Hepatitis C therapy with HCV NS5B polymerase inhibitors. Expert Opin Pharmacother 2013; 14:1161–1170.
- You DM, Pockros PJ. Simeprevir for the treatment of chronic hepatitis C. Expert Opin Pharmacother 2013; 14:2581–2589.
- Jacobson IM, Dore GJ, Foster G, et al. Simeprevir (TMC435) with peginterferon/ribavirin for chronic HCV genotype-1 infection in treatment-naive patients: results from Quest-1, a phase III trial [abstract no. 1425]. Annual Meeting of the European Association for the Study of the Liver; April 24–28, 2013; Amsterdam, Netherlands.
- Manns M, Marcellin P, Poordad FP, et al. Simeprevir (TMC435) with peginterferon/ribavirin for chronic HCV genotype-1 infection in treatment-naïve patients: results from QUEST-2, a phase III trial [abstract no. 1413]. Annual Meeting of the European Association for the Study of the Liver; April 24–28, 2013; Amsterdam, The Netherlands.
- Lawitz E, Forns X, Zeuzem S, et al. Simeprevir (TMC435) with peginterferon/ribavirin for treatment of chronic HCV genotype 1 infection in patients who relapsed after previous interferon-based therapy: results from promise, a phase III trial [abstract no. 869b]. Digestive Disease Week; May 18–21, 2013; Orlando, FL.
- Zeuzem S, Berg T, Gane E, et al. Simeprevir increases rate of sustained virologic response among treatment-experienced patients with HCV genotype-1 infection: a phase IIb trial. Gastroenterology epub Oct 31, 2013.
- Jacobson IM, Ghalib RM, Rodriguez-Torres M, et al. SVR results of a once-daily regimen of simeprevir (TMC435) plus sofosbuvir (GS-7977) with or without ribavirin in cirrhotic and non-cirrhotic HCV genotype 1 treatment-naive and prior null responder patients: the COSMOS study [abstract LB-3]. 64th Annual Meeting of the American Association for the Study of Liver Diseases; November 1–5, 2013; Washington, DC.
- Smith BD, Morgan RL, Beckett GA, et al. Recommendations for the identification of chronic hepatitis C virus infection among persons born during 1945–1965. MMWR Recomm Rep 2012; 61( RR-4):1–32.
- Sulkowski MS, Kang M, Matining R, et al. Safety and antiviral activity of the HCV entry inhibitor ITX5061 in treatment-naive HCV-infected adults: a randomized, double-blind, phase 1b study. J Infect Dis 2013 Oct 9. [Epub ahead of print]
- Pawlotsky JM. NS5A inhibitors in the treatment of hepatitis C. J Hepatol 2013; 59:375–382.
- Yu M, Corsa AC, Xu S, et al. In vitro efficacy of approved and experimental antivirals against novel genotype 3 hepatitis C virus subgenomic replicons. Antiviral Res 2013; 100:439–445.
- Aghemo A, De Francesco R. New horizons in hepatitis C antiviral therapy with direct-acting antivirals. Hepatology 2013; 58:428–438.
- Liang TJ, Ghany MG. Current and future therapies for hepatitis C virus infection. N Engl J Med 2013; 368:1907–1917.
- Flisiak R, Jaroszewicz J, Parfieniuk-Kowerda A. Emerging treatments for hepatitis C. Expert Opin Emerg Drugs 2013; 18:461–475.
- Lawitz E, Poordad FF, Pang PS, et al. Sofosbuvir and ledipasvir fixed-dose combination with and without ribavirin in treatment-naive and previously treated patients with genotype 1 hepatitis C virus infection (LONESTAR): an open-label, randomised, phase 2 trial. Lancet 2013 Nov 1. doi: 10.1016/S0140-6736(13)62121-2 [Epub ahead of print]
- Drenth JP. HCV treatment—no more room for interferonologists? N Engl J Med 2013; 368:1931–1932.
- Casey LC, Lee WM. Hepatitis C virus therapy update 2013. Curr Opin Gastroenterol 2013; 29:243–249.
- Afdhal NH, Zeuzem S, Schooley RT, et al. The new paradigm of hepatitis C therapy: integration of oral therapies into best practices. J Viral Hepatol 2013; 20:745–760.
- Averhoff FM, Glass N, Holtzman D. Global burden of hepatitis C: considerations for healthcare providers in the united states. Clin Infect Dis 2012; 55(suppl 1):S10–S15.
- Wiesner RH, Sorrell M, Villamil F; International Liver Transplantation Society Expert Panel. Report of the first international liver transplantation society expert panel consensus conference on liver transplantation and hepatitis C. Liver Transplant 2003; 9:S1–S9.
- Wong JB, McQuillan GM, McHutchison JG, Poynard T. Estimating future hepatitis C morbidity, mortality, and costs in the United States. Am J Public Health 2000; 90:1562–1569.
- Pawlotsky JM, Chevaliez S, McHutchison JG. The hepatitis C virus life cycle as a target for new antiviral therapies. Gastroenterology 2007; 132:1979–1998.
- Bartenschlager R, Lohmann V. Replication of hepatitis C virus. J Gen Virol 2000; 81:1631–1648.
- Singal AG, Volk ML, Jensen D, Di Bisceglie AM, Schoenfeld PS. A sustained viral response is associated with reduced liver-related morbidity and mortality in patients with hepatitis C virus. Clin Gastroenterol Hepatol 2010; 8:280–288,288.e1.
- Camma C, Di Bona D, Schepis F, et al. Effect of peginterferon alfa-2a on liver histology in chronic hepatitis C: a meta-analysis of individual patient data. Hepatology 2004; 39:333–342.
- Paeshuyse J, Dallmeier K, Neyts J. Ribavirin for the treatment of chronic hepatitis C virus infection: a review of the proposed mechanisms of action. Curr Opin Virol 2011; 1:590–598.
- Thomas E, Ghany MG, Liang TJ. The application and mechanism of action of ribavirin in therapy of hepatitis C. Antivir Chem Chemother 2012; 23:1–12.
- Ghany MG, Nelson DR, Strader DB, Thomas DL, Seeff LB; American Association for Study of Liver Diseases. An update on treatment of genotype 1 chronic hepatitis C virus infection: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 2011; 54:1433–1444.
- Soriano V, Vispo E, Poveda E, Labarga P, Barreiro P. Treatment failure with new hepatitis C drugs. Expert Opin Pharmacother 2012; 13:313–323.
- Asselah T, Marcellin P. Interferon free therapy with direct acting antivirals for HCV. Liver Int 2013; 33(suppl 1):93–104.
- Manns MP, McCone J, Davis MN, et al. Overall safety profile of boceprevir plus peginterferon alfa-2b and ribavirin in patients with chronic hepatitis C genotype 1: a combined analysis of 3 phase 2/3 clinical trials. Liver Int 2013; Aug 2. doi: 10.1111/liv.12300. [Epub ahead of print]
- Jacobson IM, McHutchison JG, Dusheiko G, et al. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364:2405–2416.
- Rodriguez-Torres M, Lawitz E, Kowdley KV, et al. Sofosbuvir (GS-7977) plus peginterferon/ribavirin in treatment-naive patients with HCV genotype 1: a randomized, 28-day, dose-ranging trial. J Hepatol 2013; 58:663–668.
- Kowdley KV, Lawitz E, Crespo I, et al. Sofosbuvir with pegylated interferon alfa-2a and ribavirin for treatment-naive patients with hepatitis C genotype-1 infection (ATOMIC): an open-label, randomised, multicentre phase 2 trial. Lancet 2013; 381:2100–2107.
- Lawitz E, Lalezari JP, Hassanein T, et al. Sofosbuvir in combination with peginterferon alfa-2a and ribavirin for non-cirrhotic, treatment-naive patients with genotypes 1, 2, and 3 hepatitis C infection: a randomised, double-blind, phase 2 trial. Lancet Infect Dis 2013; 13:401–408.
- Gane EJ, Stedman CA, Hyland RH, et al. Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N Engl J Med 2013; 368:34–44.
- Lawitz E, Mangia A, Wyles D, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 2013; 368:1878–1887.
- Jacobson IM, Gordon SC, Kowdley KV, et al. Sofosbuvir for hepatitis C genotype 2 or 3 in patients without treatment options. N Engl J Med 2013; 368:1867–1877.
- Zeuzem S, Dusheiko G, Salupere R, et al. Sofosbuvir + ribavirin for 12 or 24 weeks for patients with HCV genotype 2 or 3: the VALENCE trial [abstract no.1085]. 64th Annual Meeting of the American Association for the Study of Liver Diseases; November 1–5, 2013; Washington, DC.
- Soriano V, Vispo E, de Mendoza C, et al. Hepatitis C therapy with HCV NS5B polymerase inhibitors. Expert Opin Pharmacother 2013; 14:1161–1170.
- You DM, Pockros PJ. Simeprevir for the treatment of chronic hepatitis C. Expert Opin Pharmacother 2013; 14:2581–2589.
- Jacobson IM, Dore GJ, Foster G, et al. Simeprevir (TMC435) with peginterferon/ribavirin for chronic HCV genotype-1 infection in treatment-naive patients: results from Quest-1, a phase III trial [abstract no. 1425]. Annual Meeting of the European Association for the Study of the Liver; April 24–28, 2013; Amsterdam, Netherlands.
- Manns M, Marcellin P, Poordad FP, et al. Simeprevir (TMC435) with peginterferon/ribavirin for chronic HCV genotype-1 infection in treatment-naïve patients: results from QUEST-2, a phase III trial [abstract no. 1413]. Annual Meeting of the European Association for the Study of the Liver; April 24–28, 2013; Amsterdam, The Netherlands.
- Lawitz E, Forns X, Zeuzem S, et al. Simeprevir (TMC435) with peginterferon/ribavirin for treatment of chronic HCV genotype 1 infection in patients who relapsed after previous interferon-based therapy: results from promise, a phase III trial [abstract no. 869b]. Digestive Disease Week; May 18–21, 2013; Orlando, FL.
- Zeuzem S, Berg T, Gane E, et al. Simeprevir increases rate of sustained virologic response among treatment-experienced patients with HCV genotype-1 infection: a phase IIb trial. Gastroenterology epub Oct 31, 2013.
- Jacobson IM, Ghalib RM, Rodriguez-Torres M, et al. SVR results of a once-daily regimen of simeprevir (TMC435) plus sofosbuvir (GS-7977) with or without ribavirin in cirrhotic and non-cirrhotic HCV genotype 1 treatment-naive and prior null responder patients: the COSMOS study [abstract LB-3]. 64th Annual Meeting of the American Association for the Study of Liver Diseases; November 1–5, 2013; Washington, DC.
- Smith BD, Morgan RL, Beckett GA, et al. Recommendations for the identification of chronic hepatitis C virus infection among persons born during 1945–1965. MMWR Recomm Rep 2012; 61( RR-4):1–32.
- Sulkowski MS, Kang M, Matining R, et al. Safety and antiviral activity of the HCV entry inhibitor ITX5061 in treatment-naive HCV-infected adults: a randomized, double-blind, phase 1b study. J Infect Dis 2013 Oct 9. [Epub ahead of print]
- Pawlotsky JM. NS5A inhibitors in the treatment of hepatitis C. J Hepatol 2013; 59:375–382.
- Yu M, Corsa AC, Xu S, et al. In vitro efficacy of approved and experimental antivirals against novel genotype 3 hepatitis C virus subgenomic replicons. Antiviral Res 2013; 100:439–445.
- Aghemo A, De Francesco R. New horizons in hepatitis C antiviral therapy with direct-acting antivirals. Hepatology 2013; 58:428–438.
- Liang TJ, Ghany MG. Current and future therapies for hepatitis C virus infection. N Engl J Med 2013; 368:1907–1917.
- Flisiak R, Jaroszewicz J, Parfieniuk-Kowerda A. Emerging treatments for hepatitis C. Expert Opin Emerg Drugs 2013; 18:461–475.
- Lawitz E, Poordad FF, Pang PS, et al. Sofosbuvir and ledipasvir fixed-dose combination with and without ribavirin in treatment-naive and previously treated patients with genotype 1 hepatitis C virus infection (LONESTAR): an open-label, randomised, phase 2 trial. Lancet 2013 Nov 1. doi: 10.1016/S0140-6736(13)62121-2 [Epub ahead of print]
- Drenth JP. HCV treatment—no more room for interferonologists? N Engl J Med 2013; 368:1931–1932.
- Casey LC, Lee WM. Hepatitis C virus therapy update 2013. Curr Opin Gastroenterol 2013; 29:243–249.
- Afdhal NH, Zeuzem S, Schooley RT, et al. The new paradigm of hepatitis C therapy: integration of oral therapies into best practices. J Viral Hepatol 2013; 20:745–760.
KEY POINTS
- In clinical trials of treatment for chronic HCV infection, regimens that included a direct-acting antiviral agent were more effective than ones that did not.
- Sofosbuvir is approved in an oral dose of 400 mg once daily in combination with ribavirin for patients infected with HCV genotype 2 or 3, and in combination with ribavirin and interferon in patients infected with HCV genotype 1 or 4. It is also recommended in combination with ribavirin in HCV-infected patients with hepatocellular carcinoma who are awaiting liver transplantation.
- Simeprevir is approved in an oral dose of 150 mg once daily in combination with ribavirin and interferon for patients with HCV genotype 1.
- The new drugs are expensive, a potential barrier for many patients. As more direct-acting antiviral agents become available, their cost will likely decrease.
- Combinations of direct-acting antiviral agents of different classes may prove even more effective and could eliminate the need for interferon entirely.