User login
Sarcoidosis and Squamous Cell Carcinoma: A Connection Documented in a Case Series of 3 Patients
Sarcoidosis is a multisystem granulomatous disease of unknown etiology that most commonly affects the lungs, eyes, and skin. Cutaneous involvement is reported in 25% to 35% of patients with sarcoidosis and may occur in a variety of forms including macules, papules, plaques, and lupus pernio.1,2 Dermatologists commonly are confronted with the diagnosis and management of sarcoidosis because of its high incidence of cutaneous involvement. Due to the protean nature of the disease, skin biopsy plays a key role in confirming the diagnosis. Histological evidence of noncaseating granulomas in combination with an appropriate clinical and radiographic picture is necessary for the diagnosis of sarcoidosis.1,2 Brincker and Wilbek
We describe 3 patients with sarcoidosis who developed squamous cell carcinoma (SCC) of the skin, including 2 black patients, which highlights the potential for SCC development.
Case Reports
Patient 1
A black woman in her 60s with a history of sarcoidosis affecting the lungs and skin that was well controlled with biweekly adalimumab 40 mg subcutaneous injections presented with a new dark painful lesion on the right third finger. She reported the lesion had been present for 1 to 2 years prior to the current presentation and was increasing in size. She had no history of prior skin cancers.
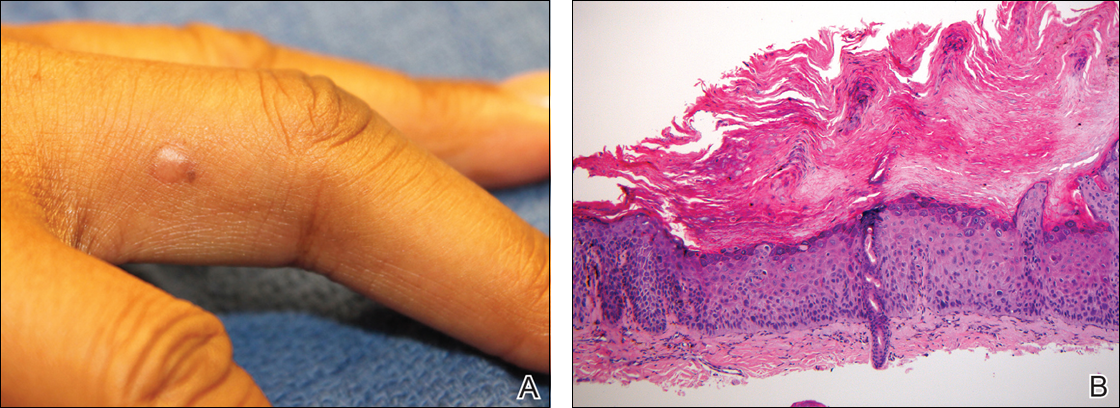
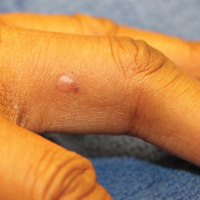
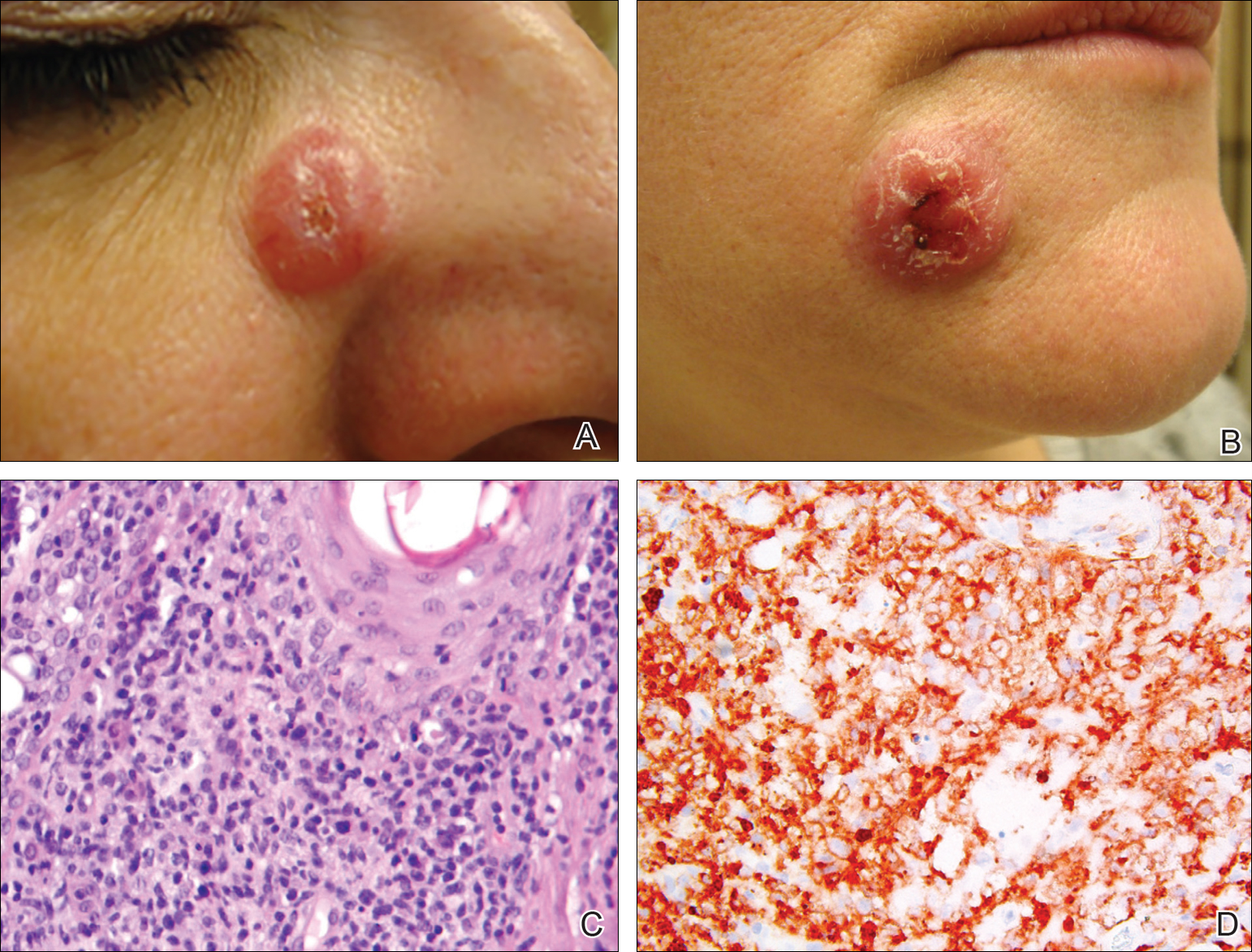
Physical examination revealed a waxy, brown-pigmented papule with overlying scale on the ulnar aspect of the right third digit near the web space (Figure 1A). A shave biopsy revealed atypical keratinocytes involving all layers of the epidermis along with associated parakeratotic scale consistent with a diagnosis of SCC in situ (Figure 1B). Human papillomavirus staining was negative. Due to the location of the lesion, the patient underwent Mohs micrographic surgery and the lesion was completely excised.
Patient 2
A black woman in her 60s with a history of cutaneous sarcoidosis that was maintained on minocycline 100 mg twice daily, chloroquine 250 mg daily, tacrolimus ointment 0.1%, tretinoin cream 0.025%, and intermittent intralesional triamcinolone acetonide injections to the nose, as well as quiescent pulmonary sarcoidosis, developed a new, growing, asymptomatic, hyperpigmented lesion on the left side of the submandibular neck over a period of a few months. A biopsy was performed and the lesion was found to be an SCC, which subsequently was completely excised.
Patient 3
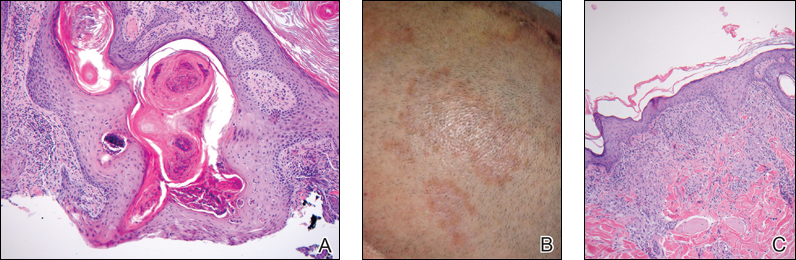
A white man in his 60s with a history of prior quiescent pulmonary sarcoidosis, remote melanoma, and multiple nonmelanoma skin cancers developed scaly papules on the scalp for months, one that was interpreted by an outside pathologist as an invasive SCC (Figure 2A). He was referred to our institution for Mohs micrographic surgery. On presentation when his scalp was shaved for surgery, he was noted to have several violaceous, annular, thin plaques on the scalp (Figure 2B). A biopsy of an annular plaque demonstrated several areas of granulomatous dermatitis consistent with a diagnosis of cutaneous sarcoidosis (Figure 2C). The patient had clinical lymphadenopathy of the neck and supraclavicular region. Given the patient’s history, the differential diagnosis for these lesions included metastatic SCC, lymphoma, and sarcoidosis. The patient underwent a positron emission tomography scan, which demonstrated fluorodeoxyglucose-positive regions in both lungs and the right side of the neck. After evaluation by the pulmonary and otorhinolaryngology departments, including a lymph node biopsy, the positron emission tomography–enhancing lesions were ultimately determined to be consistent with sarcoidosis.
The patient underwent Mohs micrographic surgery for treatment of the scalp SCC and was started on triamcinolone cream 0.1% for the body, clobetasol propionate foam 0.05% for the scalp, and hydroxychloroquine sulfate 400 mg daily for the cutaneous sarcoidosis. His annular scalp lesions resolved, but over the following 12 months the patient had numerous clinically suspicious skin lesions that were biopsied and were consistent with multiple basal cell carcinomas, actinic keratoses, and SCC in situ. They were treated with surgery, cryosurgical destruction with liquid nitrogen, and 5-fluorouracil cream.
Over the 3 years subsequent to initial presentation, the patient developed ocular inflammation attributed to his sarcoidosis and atrial fibrillation, which was determined to be unrelated. He also developed 5 scaly hyperkeratotic plaques on the vertex aspect of the scalp. Biopsy of 2 lesions revealed mild keratinocyte atypia and epidermal hyperplasia, favored to represent SCC over pseudoepitheliomatous hyperplasia overlying associated granulomatous inflammation. These lesions ultimately were believed to represent new SCCs, while biopsies of 2 other lesions revealed isolated granulomatous inflammation that was believed to represent hyperkeratotic cutaneous sarcoidosis clinically resembling his SCCs. The patient was again referred for Mohs micrographic surgery and the malignancies were completely removed, while the cutaneous sarcoidosis was again treated with topical corticosteroids with complete resolution.
Comment
The potential increased risk for malignancy in patients with sarcoidosis has been well documented.3-6 Brincker and Wilbek3 first reported this association after studying 2544 patients with pulmonary sarcoidosis from 1962 to 1971. In particular, they noted a difference between the expected and observed number of cases of malignancy, particularly lung cancer and lymphoma, in the sarcoidosis population.3 In a study of 10,037 hospitalized sarcoidosis patients from 1964 to 2004, Ji et al5 noted a 40% overall increase in the incidence of cancer and found that the risk for malignancy was highest in the year following hospitalization. Interestingly, they found that the risk for developing cutaneous SCC was elevated in sarcoidosis patients even after the first year following hospitalization.5 In a retrospective cohort study examining more than 9000 patients, Askling et al4 also confirmed the increased incidence of malignancy in sarcoidosis patients. Specifically, the authors found a higher than expected occurrence of skin cancer, both melanoma (standardized incidence ratio, 1.6; 95% confidence interval, 1.1-2.3) and nonmelanoma skin cancer (standardized incidence ratio, 2.8; 95% confidence interval, 2.0-3.8) in patients with sarcoidosis.4 Reich et al7 cross-matched 30,000 cases from the Kaiser Permanente Northwest Region Tumor Registry against a sarcoidosis registry of 243 cases to evaluate for evidence of linkage between sarcoidosis and malignancy. They concluded that there may be an etiologic relationship between sarcoidosis and malignancy in at least one-quarter of cases in which both are present and hypothesized that granulomas may be the result of a cell-mediated reaction to tumor antigens.7
Few published studies specifically address the incidence of malignancy in patients with primarily cutaneous sarcoidosis. Cutaneous sarcoidosis includes nonspecific lesions, such as erythema nodosum, as well as specific lesions, such as papules, plaques, nodules, and lupus pernio.8 Alexandrescu et al6 evaluated 110 patients with a diagnosis of both sarcoidosis (cutaneous and noncutaneous) and malignancy. Through their analysis, they found that cutaneous sarcoidosis is seen more commonly in patients presenting with sarcoidosis and malignancy (56.4%) than in the total sarcoidosis population (20%–25%). From these findings, the authors concluded that cutaneous sarcoidosis appears to be a subtype of sarcoidosis associated with cancer.6
We report 3 cases that specifically illustrate a link between cutaneous sarcoidosis and an increased risk for cutaneous SCC. Because sarcoidosis commonly affects the skin, patients often present to dermatologists for care. Once the initial diagnosis of cutaneous sarcoidosis is made via biopsy, it is natural to be tempted to attribute any new skin lesions to worsening or active disease; however, as cutaneous sarcoidosis may take on a variety of nonspecific forms, it is important to biopsy any unusual lesions. In our case series, patient 3 presented at several different points with scaly scalp lesions. Upon biopsy, several of these lesions were found to be SCCs, while others demonstrated regions of granulomatous inflammation consistent with a diagnosis of cutaneous sarcoidosis. On further review of pathology during the preparation of this manuscript after the initial diagnoses were made, it was further noted that it is challenging to distinguish granulomatous inflammation with reactive pseudoepitheliomatous hyperplasia from SCC. The fact that these lesions were clinically indistinguishable illustrates the critical importance of appropriate-depth biopsy in this situation, and the histopathologic challenges highlighted herein are important for pathologists to remember.
Patients 1 and 2 were both black women, and the fact that these patients both presented with cutaneous SCCs—one of whom was immunosuppressed due to treatment with adalimumab, the other without systemic immunosuppression—exemplifies the need for comprehensive skin examinations in sarcoidosis patients as well as for biopsies of new or unusual lesions.
The mechanism for the development of malignancy in patients with sarcoidosis is unknown and likely is multifactorial. Multiple theories have been proposed.1,2,5,6,8 Sarcoidosis is marked by the development of granulomas secondary to the interaction between CD4+ T cells and antigen-presenting cells, which is mediated by various cytokines and chemokines, including IL-2 and IFN-γ. Patients with sarcoidosis have been found to have oligoclonal T-cell lineages with a limited receptor repertoire, suggestive of selective immune system activation, as well as a deficiency of certain types of regulatory cells, namely natural killer cells.1,2 This immune dysregulation has been postulated to play an etiologic role in the development of malignancy in sarcoidosis patients.1,2,5 Furthermore, the chronic inflammation found in the organs commonly affected by both sarcoidosis and malignancy is another possible mechanism.6,8 Finally, immunosuppression and mutagenesis secondary to the treatment modalities used in sarcoidosis may be another contributing factor.6
Conclusion
An association between sarcoidosis and malignancy has been suggested for several decades. We specifically report 3 cases of patients with cutaneous sarcoidosis who presented with concurrent cutaneous SCCs. Given the varied and often nonspecific nature of cutaneous sarcoidosis, these cases highlight the importance of biopsy when sarcoidosis patients present with new and unusual skin lesions. Additionally, they illustrate the importance of thorough skin examinations in sarcoidosis patients as well as some of the challenges these patients pose for dermatologists.
- Iannuzzi MC, Rybicki BA, Teirsten AS. Sarcoidosis. N Engl J Med. 2007;357:2153-2165.
- Iannuzzi MC, Fontana JR. Sarcoidosis: clinical presentation, immunopathogenesis and therapeutics. JAMA. 2011;305:391-399.
- Brincker H, Wilbek E. The incidence of malignant tumours in patients with respiratory sarcoidosis. Br J Cancer. 1974;29:247-251.
- Askling J, Grunewald J, Eklund A, et al. Increased risk for cancer following sarcoidosis. Am J Respir Crit Care Med. 1999;160(5, pt 1):1668-1672.
- Ji J, Shu X, Li X, et al. Cancer risk in hospitalized sarcoidosis patients: a follow-up study in Sweden. Ann Oncol. 2009;20:1121-1126.
- Alexandrescu DT, Kauffman CL, Ichim TE, et al. Cutaneous sarcoidosis and malignancy: an association between sarcoidosis with skin manifestations and systemic neoplasia. Dermatol Online J. 2011;17:2.
- Reich JM, Mullooly JP, Johnson RE. Linkage analysis of malignancy-associated sarcoidosis. Chest. 1995;107:605-613.
- Cohen PR, Kurzrock R. Sarcoidosis and malignancy. Clin Dermatol. 2007;25:326-333.
Sarcoidosis is a multisystem granulomatous disease of unknown etiology that most commonly affects the lungs, eyes, and skin. Cutaneous involvement is reported in 25% to 35% of patients with sarcoidosis and may occur in a variety of forms including macules, papules, plaques, and lupus pernio.1,2 Dermatologists commonly are confronted with the diagnosis and management of sarcoidosis because of its high incidence of cutaneous involvement. Due to the protean nature of the disease, skin biopsy plays a key role in confirming the diagnosis. Histological evidence of noncaseating granulomas in combination with an appropriate clinical and radiographic picture is necessary for the diagnosis of sarcoidosis.1,2 Brincker and Wilbek
We describe 3 patients with sarcoidosis who developed squamous cell carcinoma (SCC) of the skin, including 2 black patients, which highlights the potential for SCC development.
Case Reports
Patient 1
A black woman in her 60s with a history of sarcoidosis affecting the lungs and skin that was well controlled with biweekly adalimumab 40 mg subcutaneous injections presented with a new dark painful lesion on the right third finger. She reported the lesion had been present for 1 to 2 years prior to the current presentation and was increasing in size. She had no history of prior skin cancers.
Physical examination revealed a waxy, brown-pigmented papule with overlying scale on the ulnar aspect of the right third digit near the web space (Figure 1A). A shave biopsy revealed atypical keratinocytes involving all layers of the epidermis along with associated parakeratotic scale consistent with a diagnosis of SCC in situ (Figure 1B). Human papillomavirus staining was negative. Due to the location of the lesion, the patient underwent Mohs micrographic surgery and the lesion was completely excised.
Patient 2
A black woman in her 60s with a history of cutaneous sarcoidosis that was maintained on minocycline 100 mg twice daily, chloroquine 250 mg daily, tacrolimus ointment 0.1%, tretinoin cream 0.025%, and intermittent intralesional triamcinolone acetonide injections to the nose, as well as quiescent pulmonary sarcoidosis, developed a new, growing, asymptomatic, hyperpigmented lesion on the left side of the submandibular neck over a period of a few months. A biopsy was performed and the lesion was found to be an SCC, which subsequently was completely excised.
Patient 3
A white man in his 60s with a history of prior quiescent pulmonary sarcoidosis, remote melanoma, and multiple nonmelanoma skin cancers developed scaly papules on the scalp for months, one that was interpreted by an outside pathologist as an invasive SCC (Figure 2A). He was referred to our institution for Mohs micrographic surgery. On presentation when his scalp was shaved for surgery, he was noted to have several violaceous, annular, thin plaques on the scalp (Figure 2B). A biopsy of an annular plaque demonstrated several areas of granulomatous dermatitis consistent with a diagnosis of cutaneous sarcoidosis (Figure 2C). The patient had clinical lymphadenopathy of the neck and supraclavicular region. Given the patient’s history, the differential diagnosis for these lesions included metastatic SCC, lymphoma, and sarcoidosis. The patient underwent a positron emission tomography scan, which demonstrated fluorodeoxyglucose-positive regions in both lungs and the right side of the neck. After evaluation by the pulmonary and otorhinolaryngology departments, including a lymph node biopsy, the positron emission tomography–enhancing lesions were ultimately determined to be consistent with sarcoidosis.
The patient underwent Mohs micrographic surgery for treatment of the scalp SCC and was started on triamcinolone cream 0.1% for the body, clobetasol propionate foam 0.05% for the scalp, and hydroxychloroquine sulfate 400 mg daily for the cutaneous sarcoidosis. His annular scalp lesions resolved, but over the following 12 months the patient had numerous clinically suspicious skin lesions that were biopsied and were consistent with multiple basal cell carcinomas, actinic keratoses, and SCC in situ. They were treated with surgery, cryosurgical destruction with liquid nitrogen, and 5-fluorouracil cream.
Over the 3 years subsequent to initial presentation, the patient developed ocular inflammation attributed to his sarcoidosis and atrial fibrillation, which was determined to be unrelated. He also developed 5 scaly hyperkeratotic plaques on the vertex aspect of the scalp. Biopsy of 2 lesions revealed mild keratinocyte atypia and epidermal hyperplasia, favored to represent SCC over pseudoepitheliomatous hyperplasia overlying associated granulomatous inflammation. These lesions ultimately were believed to represent new SCCs, while biopsies of 2 other lesions revealed isolated granulomatous inflammation that was believed to represent hyperkeratotic cutaneous sarcoidosis clinically resembling his SCCs. The patient was again referred for Mohs micrographic surgery and the malignancies were completely removed, while the cutaneous sarcoidosis was again treated with topical corticosteroids with complete resolution.
Comment
The potential increased risk for malignancy in patients with sarcoidosis has been well documented.3-6 Brincker and Wilbek3 first reported this association after studying 2544 patients with pulmonary sarcoidosis from 1962 to 1971. In particular, they noted a difference between the expected and observed number of cases of malignancy, particularly lung cancer and lymphoma, in the sarcoidosis population.3 In a study of 10,037 hospitalized sarcoidosis patients from 1964 to 2004, Ji et al5 noted a 40% overall increase in the incidence of cancer and found that the risk for malignancy was highest in the year following hospitalization. Interestingly, they found that the risk for developing cutaneous SCC was elevated in sarcoidosis patients even after the first year following hospitalization.5 In a retrospective cohort study examining more than 9000 patients, Askling et al4 also confirmed the increased incidence of malignancy in sarcoidosis patients. Specifically, the authors found a higher than expected occurrence of skin cancer, both melanoma (standardized incidence ratio, 1.6; 95% confidence interval, 1.1-2.3) and nonmelanoma skin cancer (standardized incidence ratio, 2.8; 95% confidence interval, 2.0-3.8) in patients with sarcoidosis.4 Reich et al7 cross-matched 30,000 cases from the Kaiser Permanente Northwest Region Tumor Registry against a sarcoidosis registry of 243 cases to evaluate for evidence of linkage between sarcoidosis and malignancy. They concluded that there may be an etiologic relationship between sarcoidosis and malignancy in at least one-quarter of cases in which both are present and hypothesized that granulomas may be the result of a cell-mediated reaction to tumor antigens.7
Few published studies specifically address the incidence of malignancy in patients with primarily cutaneous sarcoidosis. Cutaneous sarcoidosis includes nonspecific lesions, such as erythema nodosum, as well as specific lesions, such as papules, plaques, nodules, and lupus pernio.8 Alexandrescu et al6 evaluated 110 patients with a diagnosis of both sarcoidosis (cutaneous and noncutaneous) and malignancy. Through their analysis, they found that cutaneous sarcoidosis is seen more commonly in patients presenting with sarcoidosis and malignancy (56.4%) than in the total sarcoidosis population (20%–25%). From these findings, the authors concluded that cutaneous sarcoidosis appears to be a subtype of sarcoidosis associated with cancer.6
We report 3 cases that specifically illustrate a link between cutaneous sarcoidosis and an increased risk for cutaneous SCC. Because sarcoidosis commonly affects the skin, patients often present to dermatologists for care. Once the initial diagnosis of cutaneous sarcoidosis is made via biopsy, it is natural to be tempted to attribute any new skin lesions to worsening or active disease; however, as cutaneous sarcoidosis may take on a variety of nonspecific forms, it is important to biopsy any unusual lesions. In our case series, patient 3 presented at several different points with scaly scalp lesions. Upon biopsy, several of these lesions were found to be SCCs, while others demonstrated regions of granulomatous inflammation consistent with a diagnosis of cutaneous sarcoidosis. On further review of pathology during the preparation of this manuscript after the initial diagnoses were made, it was further noted that it is challenging to distinguish granulomatous inflammation with reactive pseudoepitheliomatous hyperplasia from SCC. The fact that these lesions were clinically indistinguishable illustrates the critical importance of appropriate-depth biopsy in this situation, and the histopathologic challenges highlighted herein are important for pathologists to remember.
Patients 1 and 2 were both black women, and the fact that these patients both presented with cutaneous SCCs—one of whom was immunosuppressed due to treatment with adalimumab, the other without systemic immunosuppression—exemplifies the need for comprehensive skin examinations in sarcoidosis patients as well as for biopsies of new or unusual lesions.
The mechanism for the development of malignancy in patients with sarcoidosis is unknown and likely is multifactorial. Multiple theories have been proposed.1,2,5,6,8 Sarcoidosis is marked by the development of granulomas secondary to the interaction between CD4+ T cells and antigen-presenting cells, which is mediated by various cytokines and chemokines, including IL-2 and IFN-γ. Patients with sarcoidosis have been found to have oligoclonal T-cell lineages with a limited receptor repertoire, suggestive of selective immune system activation, as well as a deficiency of certain types of regulatory cells, namely natural killer cells.1,2 This immune dysregulation has been postulated to play an etiologic role in the development of malignancy in sarcoidosis patients.1,2,5 Furthermore, the chronic inflammation found in the organs commonly affected by both sarcoidosis and malignancy is another possible mechanism.6,8 Finally, immunosuppression and mutagenesis secondary to the treatment modalities used in sarcoidosis may be another contributing factor.6
Conclusion
An association between sarcoidosis and malignancy has been suggested for several decades. We specifically report 3 cases of patients with cutaneous sarcoidosis who presented with concurrent cutaneous SCCs. Given the varied and often nonspecific nature of cutaneous sarcoidosis, these cases highlight the importance of biopsy when sarcoidosis patients present with new and unusual skin lesions. Additionally, they illustrate the importance of thorough skin examinations in sarcoidosis patients as well as some of the challenges these patients pose for dermatologists.
Sarcoidosis is a multisystem granulomatous disease of unknown etiology that most commonly affects the lungs, eyes, and skin. Cutaneous involvement is reported in 25% to 35% of patients with sarcoidosis and may occur in a variety of forms including macules, papules, plaques, and lupus pernio.1,2 Dermatologists commonly are confronted with the diagnosis and management of sarcoidosis because of its high incidence of cutaneous involvement. Due to the protean nature of the disease, skin biopsy plays a key role in confirming the diagnosis. Histological evidence of noncaseating granulomas in combination with an appropriate clinical and radiographic picture is necessary for the diagnosis of sarcoidosis.1,2 Brincker and Wilbek
We describe 3 patients with sarcoidosis who developed squamous cell carcinoma (SCC) of the skin, including 2 black patients, which highlights the potential for SCC development.
Case Reports
Patient 1
A black woman in her 60s with a history of sarcoidosis affecting the lungs and skin that was well controlled with biweekly adalimumab 40 mg subcutaneous injections presented with a new dark painful lesion on the right third finger. She reported the lesion had been present for 1 to 2 years prior to the current presentation and was increasing in size. She had no history of prior skin cancers.
Physical examination revealed a waxy, brown-pigmented papule with overlying scale on the ulnar aspect of the right third digit near the web space (Figure 1A). A shave biopsy revealed atypical keratinocytes involving all layers of the epidermis along with associated parakeratotic scale consistent with a diagnosis of SCC in situ (Figure 1B). Human papillomavirus staining was negative. Due to the location of the lesion, the patient underwent Mohs micrographic surgery and the lesion was completely excised.
Patient 2
A black woman in her 60s with a history of cutaneous sarcoidosis that was maintained on minocycline 100 mg twice daily, chloroquine 250 mg daily, tacrolimus ointment 0.1%, tretinoin cream 0.025%, and intermittent intralesional triamcinolone acetonide injections to the nose, as well as quiescent pulmonary sarcoidosis, developed a new, growing, asymptomatic, hyperpigmented lesion on the left side of the submandibular neck over a period of a few months. A biopsy was performed and the lesion was found to be an SCC, which subsequently was completely excised.
Patient 3
A white man in his 60s with a history of prior quiescent pulmonary sarcoidosis, remote melanoma, and multiple nonmelanoma skin cancers developed scaly papules on the scalp for months, one that was interpreted by an outside pathologist as an invasive SCC (Figure 2A). He was referred to our institution for Mohs micrographic surgery. On presentation when his scalp was shaved for surgery, he was noted to have several violaceous, annular, thin plaques on the scalp (Figure 2B). A biopsy of an annular plaque demonstrated several areas of granulomatous dermatitis consistent with a diagnosis of cutaneous sarcoidosis (Figure 2C). The patient had clinical lymphadenopathy of the neck and supraclavicular region. Given the patient’s history, the differential diagnosis for these lesions included metastatic SCC, lymphoma, and sarcoidosis. The patient underwent a positron emission tomography scan, which demonstrated fluorodeoxyglucose-positive regions in both lungs and the right side of the neck. After evaluation by the pulmonary and otorhinolaryngology departments, including a lymph node biopsy, the positron emission tomography–enhancing lesions were ultimately determined to be consistent with sarcoidosis.
The patient underwent Mohs micrographic surgery for treatment of the scalp SCC and was started on triamcinolone cream 0.1% for the body, clobetasol propionate foam 0.05% for the scalp, and hydroxychloroquine sulfate 400 mg daily for the cutaneous sarcoidosis. His annular scalp lesions resolved, but over the following 12 months the patient had numerous clinically suspicious skin lesions that were biopsied and were consistent with multiple basal cell carcinomas, actinic keratoses, and SCC in situ. They were treated with surgery, cryosurgical destruction with liquid nitrogen, and 5-fluorouracil cream.
Over the 3 years subsequent to initial presentation, the patient developed ocular inflammation attributed to his sarcoidosis and atrial fibrillation, which was determined to be unrelated. He also developed 5 scaly hyperkeratotic plaques on the vertex aspect of the scalp. Biopsy of 2 lesions revealed mild keratinocyte atypia and epidermal hyperplasia, favored to represent SCC over pseudoepitheliomatous hyperplasia overlying associated granulomatous inflammation. These lesions ultimately were believed to represent new SCCs, while biopsies of 2 other lesions revealed isolated granulomatous inflammation that was believed to represent hyperkeratotic cutaneous sarcoidosis clinically resembling his SCCs. The patient was again referred for Mohs micrographic surgery and the malignancies were completely removed, while the cutaneous sarcoidosis was again treated with topical corticosteroids with complete resolution.
Comment
The potential increased risk for malignancy in patients with sarcoidosis has been well documented.3-6 Brincker and Wilbek3 first reported this association after studying 2544 patients with pulmonary sarcoidosis from 1962 to 1971. In particular, they noted a difference between the expected and observed number of cases of malignancy, particularly lung cancer and lymphoma, in the sarcoidosis population.3 In a study of 10,037 hospitalized sarcoidosis patients from 1964 to 2004, Ji et al5 noted a 40% overall increase in the incidence of cancer and found that the risk for malignancy was highest in the year following hospitalization. Interestingly, they found that the risk for developing cutaneous SCC was elevated in sarcoidosis patients even after the first year following hospitalization.5 In a retrospective cohort study examining more than 9000 patients, Askling et al4 also confirmed the increased incidence of malignancy in sarcoidosis patients. Specifically, the authors found a higher than expected occurrence of skin cancer, both melanoma (standardized incidence ratio, 1.6; 95% confidence interval, 1.1-2.3) and nonmelanoma skin cancer (standardized incidence ratio, 2.8; 95% confidence interval, 2.0-3.8) in patients with sarcoidosis.4 Reich et al7 cross-matched 30,000 cases from the Kaiser Permanente Northwest Region Tumor Registry against a sarcoidosis registry of 243 cases to evaluate for evidence of linkage between sarcoidosis and malignancy. They concluded that there may be an etiologic relationship between sarcoidosis and malignancy in at least one-quarter of cases in which both are present and hypothesized that granulomas may be the result of a cell-mediated reaction to tumor antigens.7
Few published studies specifically address the incidence of malignancy in patients with primarily cutaneous sarcoidosis. Cutaneous sarcoidosis includes nonspecific lesions, such as erythema nodosum, as well as specific lesions, such as papules, plaques, nodules, and lupus pernio.8 Alexandrescu et al6 evaluated 110 patients with a diagnosis of both sarcoidosis (cutaneous and noncutaneous) and malignancy. Through their analysis, they found that cutaneous sarcoidosis is seen more commonly in patients presenting with sarcoidosis and malignancy (56.4%) than in the total sarcoidosis population (20%–25%). From these findings, the authors concluded that cutaneous sarcoidosis appears to be a subtype of sarcoidosis associated with cancer.6
We report 3 cases that specifically illustrate a link between cutaneous sarcoidosis and an increased risk for cutaneous SCC. Because sarcoidosis commonly affects the skin, patients often present to dermatologists for care. Once the initial diagnosis of cutaneous sarcoidosis is made via biopsy, it is natural to be tempted to attribute any new skin lesions to worsening or active disease; however, as cutaneous sarcoidosis may take on a variety of nonspecific forms, it is important to biopsy any unusual lesions. In our case series, patient 3 presented at several different points with scaly scalp lesions. Upon biopsy, several of these lesions were found to be SCCs, while others demonstrated regions of granulomatous inflammation consistent with a diagnosis of cutaneous sarcoidosis. On further review of pathology during the preparation of this manuscript after the initial diagnoses were made, it was further noted that it is challenging to distinguish granulomatous inflammation with reactive pseudoepitheliomatous hyperplasia from SCC. The fact that these lesions were clinically indistinguishable illustrates the critical importance of appropriate-depth biopsy in this situation, and the histopathologic challenges highlighted herein are important for pathologists to remember.
Patients 1 and 2 were both black women, and the fact that these patients both presented with cutaneous SCCs—one of whom was immunosuppressed due to treatment with adalimumab, the other without systemic immunosuppression—exemplifies the need for comprehensive skin examinations in sarcoidosis patients as well as for biopsies of new or unusual lesions.
The mechanism for the development of malignancy in patients with sarcoidosis is unknown and likely is multifactorial. Multiple theories have been proposed.1,2,5,6,8 Sarcoidosis is marked by the development of granulomas secondary to the interaction between CD4+ T cells and antigen-presenting cells, which is mediated by various cytokines and chemokines, including IL-2 and IFN-γ. Patients with sarcoidosis have been found to have oligoclonal T-cell lineages with a limited receptor repertoire, suggestive of selective immune system activation, as well as a deficiency of certain types of regulatory cells, namely natural killer cells.1,2 This immune dysregulation has been postulated to play an etiologic role in the development of malignancy in sarcoidosis patients.1,2,5 Furthermore, the chronic inflammation found in the organs commonly affected by both sarcoidosis and malignancy is another possible mechanism.6,8 Finally, immunosuppression and mutagenesis secondary to the treatment modalities used in sarcoidosis may be another contributing factor.6
Conclusion
An association between sarcoidosis and malignancy has been suggested for several decades. We specifically report 3 cases of patients with cutaneous sarcoidosis who presented with concurrent cutaneous SCCs. Given the varied and often nonspecific nature of cutaneous sarcoidosis, these cases highlight the importance of biopsy when sarcoidosis patients present with new and unusual skin lesions. Additionally, they illustrate the importance of thorough skin examinations in sarcoidosis patients as well as some of the challenges these patients pose for dermatologists.
- Iannuzzi MC, Rybicki BA, Teirsten AS. Sarcoidosis. N Engl J Med. 2007;357:2153-2165.
- Iannuzzi MC, Fontana JR. Sarcoidosis: clinical presentation, immunopathogenesis and therapeutics. JAMA. 2011;305:391-399.
- Brincker H, Wilbek E. The incidence of malignant tumours in patients with respiratory sarcoidosis. Br J Cancer. 1974;29:247-251.
- Askling J, Grunewald J, Eklund A, et al. Increased risk for cancer following sarcoidosis. Am J Respir Crit Care Med. 1999;160(5, pt 1):1668-1672.
- Ji J, Shu X, Li X, et al. Cancer risk in hospitalized sarcoidosis patients: a follow-up study in Sweden. Ann Oncol. 2009;20:1121-1126.
- Alexandrescu DT, Kauffman CL, Ichim TE, et al. Cutaneous sarcoidosis and malignancy: an association between sarcoidosis with skin manifestations and systemic neoplasia. Dermatol Online J. 2011;17:2.
- Reich JM, Mullooly JP, Johnson RE. Linkage analysis of malignancy-associated sarcoidosis. Chest. 1995;107:605-613.
- Cohen PR, Kurzrock R. Sarcoidosis and malignancy. Clin Dermatol. 2007;25:326-333.
- Iannuzzi MC, Rybicki BA, Teirsten AS. Sarcoidosis. N Engl J Med. 2007;357:2153-2165.
- Iannuzzi MC, Fontana JR. Sarcoidosis: clinical presentation, immunopathogenesis and therapeutics. JAMA. 2011;305:391-399.
- Brincker H, Wilbek E. The incidence of malignant tumours in patients with respiratory sarcoidosis. Br J Cancer. 1974;29:247-251.
- Askling J, Grunewald J, Eklund A, et al. Increased risk for cancer following sarcoidosis. Am J Respir Crit Care Med. 1999;160(5, pt 1):1668-1672.
- Ji J, Shu X, Li X, et al. Cancer risk in hospitalized sarcoidosis patients: a follow-up study in Sweden. Ann Oncol. 2009;20:1121-1126.
- Alexandrescu DT, Kauffman CL, Ichim TE, et al. Cutaneous sarcoidosis and malignancy: an association between sarcoidosis with skin manifestations and systemic neoplasia. Dermatol Online J. 2011;17:2.
- Reich JM, Mullooly JP, Johnson RE. Linkage analysis of malignancy-associated sarcoidosis. Chest. 1995;107:605-613.
- Cohen PR, Kurzrock R. Sarcoidosis and malignancy. Clin Dermatol. 2007;25:326-333.
Practice Points
- There may be an increased risk of skin cancer in patients with sarcoidosis.
- Sarcoidosis may present with multiple morphologies, including verrucous or hyperkeratotic lesions; superficial biopsy of this type of lesion may be mistaken for a squamous cell carcinoma.
- A biopsy diagnosis of squamous cell carcinoma in a black patient with sarcoidosis should be carefully reviewed for evidence of deeper granulomatous inflammation.
Low-Dose Radiotherapy for Primary Cutaneous Anaplastic Large-Cell Lymphoma While on Low-Dose Methotrexate
CD30+ primary cutaneous lymphoproliferative disorders (pcLPDs) are the second most common cause of cutaneous T-cell lymphoma, accounting for approximately 25% to 30% of cases.1 These disorders comprise a spectrum that includes primary cutaneous anaplastic large-cell lymphoma (pcALCL); lymphomatoid papulosis (LyP); and borderline lesions, which share clinicopathologic features of both pcALCL and LyP. Lymphomatoid papulosis is characterized as chronic, recurrent, papular or papulonodular skin lesions that typically are multifocal and regress spontaneously within weeks to months, only leaving small scars with atrophy and/or hyperpigmentation.2 Cutaneous anaplastic large-cell lymphoma typically presents as solitary or grouped nodules or tumors that may undergo spontaneous partial or complete regression in approximately 25% of cases3 but often persist if not treated. Patients may have an array of lesions comprising the spectrum of CD30 pcLPDs.4
There is no curative therapy for CD30+ pcLPDs. Although active treatment is not necessary for LyP, low-dose methotrexate (MTX)(10–50 mg weekly) or phototherapy are the preferred initial suppressive therapies for symptomatic patients with scarring, facial lesions, or multiple symptomatic lesions.5 Observation with expectant follow-up is an option in pcALCL, though spontaneous regression is less likely than in LyP. For single or grouped pcALCL lesions, local radiation is the first-line therapy.6 Multifocal pcALCL lesions also can be treated with low-dose MTX,2,5 as in LyP, or local radiation to selected areas. Although local radiotherapy is considered a first-line treatment in pcALCL, there is limited evidence on its clinical efficacy as well as the optimal dose and technique. We report the complete response of refractory pcALCL lesions to low-dose radiation while remaining on MTX weekly without any adverse effects.
Case Report
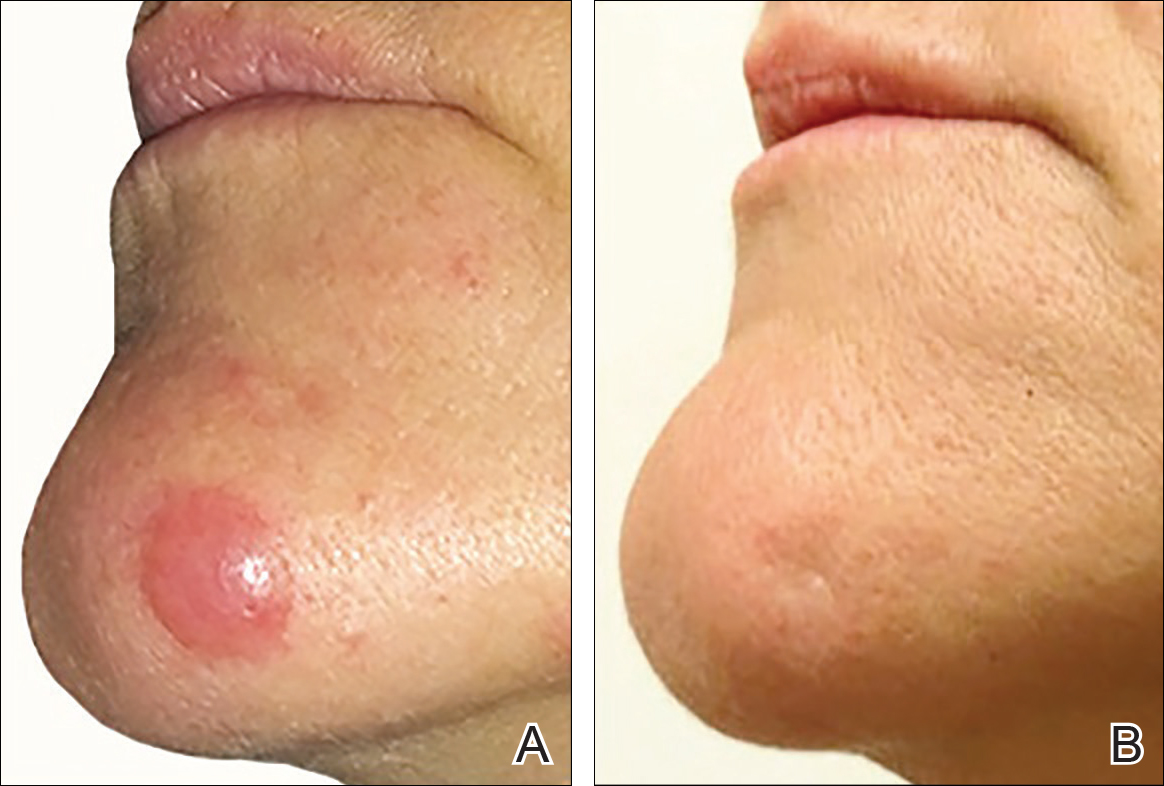
A 51-year-old woman presented with a 3-year history of CD30+ pcLPD manifesting primarily as pcALCL involving the head and neck, as well as LyP involving the head, arms, and trunk (T3N0M0). For 2 years her treatment regimen included clobetasol propionate cream 0.05% as needed for new lesions and 2 courses of standard-dose localized external beam radiation for larger pcALCL tumors on the right cheek and right side of the chin (Figure 1)(total dose for each course of treatment was 20 Gy and 36 Gy, respectively, each administered over 2–3 weeks). Because new unsightly papulonodules continued to develop on the patient’s face, she subsequently required low-dose oral MTX 30 mg once weekly for suppression of new lesions and was stable on this regimen for a year. However, she experienced an increase in LyP/pcALCL activity on the face during a 2-week break from MTX when she developed a herpes zoster infection on the right side of the forehead.
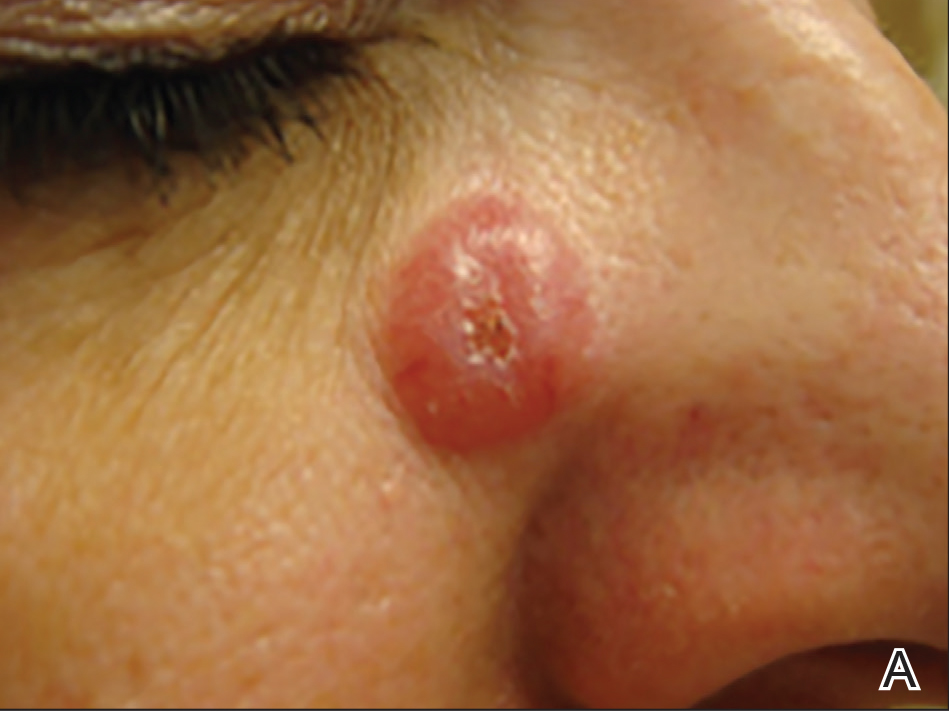
On physical examination 1 month later, 5 tiny pink papules scattered on the left eyebrow, left cheek, and left side of the chin were noted. She was advised to continue applying the clobetasol cream as needed and was restarted on MTX 10 mg once weekly. However, she developed 2 additional 1-cm nodules on the left side of the chin, neck, and shoulder. Methotrexate was increased to 30 mg once weekly over 2 weeks, which was the original dose prior to interruption, but the nodules grew to 1.5-cm in diameter. Due to their clinical appearance, the nodules were believed to be early pcALCL lesions (Figure 2A). Given the cosmetically sensitive location of the nodules, palliative radiotherapy was recommended rather than observe for possible regression. Based on a prior report by Neelis et al7 demonstrating efficacy of low-dose radiotherapy for cutaneous T-cell lymphoma and cutaneous B-cell lymphoma, we recommended starting with low radiation doses. Our patient was treated with 400 cGy twice to the left side of the chin and left side of the neck (800 cGy total at each site) while remaining on MTX 30 mg once weekly. This treatment was well tolerated without side effects and no evidence of radiation dermatitis. On follow-up examination 1 week later, the nodules had regressed and no new lesions were present (Figure 2B).
The patient has stayed on oral MTX and occasionally develops small lesions that quickly resolve with clobetasol cream. She has been followed for 3 years after radiotherapy and all 3 previously irradiated sites have remained recurrence free. Furthermore, she has not developed any new larger nodules or tumors and her MTX dose has been decreased to 15 mg once weekly.
Comment
Local radiotherapy is considered a first-line treatment of pcALCL; however, there is limited evidence on its clinical efficacy as well as the optimal dose and technique. Although no standard dose exists for pcALCL, the National Comprehensive Cancer Network guidelines8 recommend doses of 12 to 36 Gy in mycosis fungoides/Sézary syndrome subtypes of cutaneous T-cell lymphoma, which are consistent with guidelines published by the European Society for Medical Oncology.9 High complete response rates have been demonstrated in pcALCL at doses of 34 to 44 Gy6; however, lesions tend to recur elsewhere on the skin in 36% to 41% of patients despite treatment.2,10 Lower doses of radiation therapy would provide several advantages over higher-dose therapy if a complete response could be achieved without greatly increasing the local recurrence rate. In cases of local recurrence, low-dose radiation would more easily permit retreatment of lesions compared to higher doses of radiation. Similarly, in patients with multifocal pcALCL, lower doses of radiotherapy may allow for treatment of larger skin areas while limiting potential treatment risks. Furthermore, low-dose therapy would allow for treatments to be delivered more quickly and with less inconvenience to the patient who is likely to need multiple future treatments to other areas. Low-dose radiation has been described with a favorable efficacy profile for mycosis fungoides7,11 but has not been studied in patients with CD30+ pcLPDs.
Our case is notable because the patient remained on MTX during radiation therapy. B
Conclusion
We reported the use of low-dose radiation therapy for the treatment of localized pcALCL in a patient who remained on low-dose oral MTX. Additional studies will be necessary to more fully evaluate the efficacy of using low-dose radiation both as monotherapy and in combination with MTX for pcALCL.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Bekkenk MW, Geelen FA, van Voorst Vader PC, et al. Primary and secondary cutaneous CD30+ lymphoproliferative disorders: a report from the Dutch Cutaneous Lymphoma Group on the long-term follow-up data of 219 patients and guidelines for diagnosis and treatment. Blood. 2000;95:3653-3661.
- Willemze R, Beljaards RC. Spectrum of primary cutaneous CD30 (Ki-1)-positive lymphoproliferative disorders: a proposal for classification and guidelines for management and treatment. J Am Acad Dermatol. 1993;28:973-980.
- Kadin ME. The spectrum of Ki-1+ cutaneous lymphomas. Curr Probl Dermatol. 1990;19:132-143.
- Vonderheid EC, Sajjadian A, Kadin ME. Methotrexate is effective therapy for lymphomatoid papulosis and other primary cutaneous CD30-positive lymphoproliferative disorders. J Am Acad Dermatol. 1996;34:470-481.
- Yu JB, McNiff JM, Lund MW, et al. Treatment of primary cutaneous CD30+ anaplastic large-cell lymphoma with radiation therapy. Int J Radiat Oncol Biol Phys. 2008;70:1542-1545.
- Neelis KJ, Schimmel EC, Vermeer MH, et al. Low-dose palliative radiotherapy B-cell and T-cell lymphomas. Int J Radiat Oncol Biol Phys. 2009;74:154-158.
- National Comprehensive Cancer Network. CD30 lymphoproliferative disorders section in non-Hodgkin’s lymphoma (Version 3.2016). http://www.nccn.org/professionals/physician_gls/pdf/nhl.pdf. Accessed September 26, 2016.
- Willemze R, Hodak E, Zinzani PL, et al; ESMO Guidelines Working Group. Primary cutaneous lymphomas: EMSO clinical practice guidelines for diagnosis, treatment, and follow-up [published online July 17, 2013]. Ann Onc. 2013;24(suppl 6):vi149-vi154.
- Liu HL, Hoppe RT, Kohler S, et al. CD30+ cutaneous lymphoproliferative disorders: the Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. J Am Acad Dermatol. 2003;49:1049-1058.
- Harrison C, Young J, Navi D, et al. Revisiting low dose total skin electron beam radiotherapy in mycosis fungoides. Int J Radiat Oncol Biol Phys. 2011;81:651-657.
- Jaffe N, Farber S, Traggis D, et al. Favorable response of metastatic osteogenic sarcoma to pulse high-dose methotrexate with citrovorum rescue and radiation therapy. Cancer. 1973;31:1367-1373.
- Rosen G, Tefft M, Martinez A, et al. Combination chemotherapy and radiation therapy in the treatment of metastatic osteogenic sarcoma. Cancer. 1975;35:622-630.
- Kim YH, Aye MS, Fayos JV. Radiation necrosis of the scalp: a complication of cranial irradiation and methotrexate. Radiology. 1977;124:813-814.
CD30+ primary cutaneous lymphoproliferative disorders (pcLPDs) are the second most common cause of cutaneous T-cell lymphoma, accounting for approximately 25% to 30% of cases.1 These disorders comprise a spectrum that includes primary cutaneous anaplastic large-cell lymphoma (pcALCL); lymphomatoid papulosis (LyP); and borderline lesions, which share clinicopathologic features of both pcALCL and LyP. Lymphomatoid papulosis is characterized as chronic, recurrent, papular or papulonodular skin lesions that typically are multifocal and regress spontaneously within weeks to months, only leaving small scars with atrophy and/or hyperpigmentation.2 Cutaneous anaplastic large-cell lymphoma typically presents as solitary or grouped nodules or tumors that may undergo spontaneous partial or complete regression in approximately 25% of cases3 but often persist if not treated. Patients may have an array of lesions comprising the spectrum of CD30 pcLPDs.4
There is no curative therapy for CD30+ pcLPDs. Although active treatment is not necessary for LyP, low-dose methotrexate (MTX)(10–50 mg weekly) or phototherapy are the preferred initial suppressive therapies for symptomatic patients with scarring, facial lesions, or multiple symptomatic lesions.5 Observation with expectant follow-up is an option in pcALCL, though spontaneous regression is less likely than in LyP. For single or grouped pcALCL lesions, local radiation is the first-line therapy.6 Multifocal pcALCL lesions also can be treated with low-dose MTX,2,5 as in LyP, or local radiation to selected areas. Although local radiotherapy is considered a first-line treatment in pcALCL, there is limited evidence on its clinical efficacy as well as the optimal dose and technique. We report the complete response of refractory pcALCL lesions to low-dose radiation while remaining on MTX weekly without any adverse effects.
Case Report
A 51-year-old woman presented with a 3-year history of CD30+ pcLPD manifesting primarily as pcALCL involving the head and neck, as well as LyP involving the head, arms, and trunk (T3N0M0). For 2 years her treatment regimen included clobetasol propionate cream 0.05% as needed for new lesions and 2 courses of standard-dose localized external beam radiation for larger pcALCL tumors on the right cheek and right side of the chin (Figure 1)(total dose for each course of treatment was 20 Gy and 36 Gy, respectively, each administered over 2–3 weeks). Because new unsightly papulonodules continued to develop on the patient’s face, she subsequently required low-dose oral MTX 30 mg once weekly for suppression of new lesions and was stable on this regimen for a year. However, she experienced an increase in LyP/pcALCL activity on the face during a 2-week break from MTX when she developed a herpes zoster infection on the right side of the forehead.
On physical examination 1 month later, 5 tiny pink papules scattered on the left eyebrow, left cheek, and left side of the chin were noted. She was advised to continue applying the clobetasol cream as needed and was restarted on MTX 10 mg once weekly. However, she developed 2 additional 1-cm nodules on the left side of the chin, neck, and shoulder. Methotrexate was increased to 30 mg once weekly over 2 weeks, which was the original dose prior to interruption, but the nodules grew to 1.5-cm in diameter. Due to their clinical appearance, the nodules were believed to be early pcALCL lesions (Figure 2A). Given the cosmetically sensitive location of the nodules, palliative radiotherapy was recommended rather than observe for possible regression. Based on a prior report by Neelis et al7 demonstrating efficacy of low-dose radiotherapy for cutaneous T-cell lymphoma and cutaneous B-cell lymphoma, we recommended starting with low radiation doses. Our patient was treated with 400 cGy twice to the left side of the chin and left side of the neck (800 cGy total at each site) while remaining on MTX 30 mg once weekly. This treatment was well tolerated without side effects and no evidence of radiation dermatitis. On follow-up examination 1 week later, the nodules had regressed and no new lesions were present (Figure 2B).
The patient has stayed on oral MTX and occasionally develops small lesions that quickly resolve with clobetasol cream. She has been followed for 3 years after radiotherapy and all 3 previously irradiated sites have remained recurrence free. Furthermore, she has not developed any new larger nodules or tumors and her MTX dose has been decreased to 15 mg once weekly.
Comment
Local radiotherapy is considered a first-line treatment of pcALCL; however, there is limited evidence on its clinical efficacy as well as the optimal dose and technique. Although no standard dose exists for pcALCL, the National Comprehensive Cancer Network guidelines8 recommend doses of 12 to 36 Gy in mycosis fungoides/Sézary syndrome subtypes of cutaneous T-cell lymphoma, which are consistent with guidelines published by the European Society for Medical Oncology.9 High complete response rates have been demonstrated in pcALCL at doses of 34 to 44 Gy6; however, lesions tend to recur elsewhere on the skin in 36% to 41% of patients despite treatment.2,10 Lower doses of radiation therapy would provide several advantages over higher-dose therapy if a complete response could be achieved without greatly increasing the local recurrence rate. In cases of local recurrence, low-dose radiation would more easily permit retreatment of lesions compared to higher doses of radiation. Similarly, in patients with multifocal pcALCL, lower doses of radiotherapy may allow for treatment of larger skin areas while limiting potential treatment risks. Furthermore, low-dose therapy would allow for treatments to be delivered more quickly and with less inconvenience to the patient who is likely to need multiple future treatments to other areas. Low-dose radiation has been described with a favorable efficacy profile for mycosis fungoides7,11 but has not been studied in patients with CD30+ pcLPDs.
Our case is notable because the patient remained on MTX during radiation therapy. B
Conclusion
We reported the use of low-dose radiation therapy for the treatment of localized pcALCL in a patient who remained on low-dose oral MTX. Additional studies will be necessary to more fully evaluate the efficacy of using low-dose radiation both as monotherapy and in combination with MTX for pcALCL.
CD30+ primary cutaneous lymphoproliferative disorders (pcLPDs) are the second most common cause of cutaneous T-cell lymphoma, accounting for approximately 25% to 30% of cases.1 These disorders comprise a spectrum that includes primary cutaneous anaplastic large-cell lymphoma (pcALCL); lymphomatoid papulosis (LyP); and borderline lesions, which share clinicopathologic features of both pcALCL and LyP. Lymphomatoid papulosis is characterized as chronic, recurrent, papular or papulonodular skin lesions that typically are multifocal and regress spontaneously within weeks to months, only leaving small scars with atrophy and/or hyperpigmentation.2 Cutaneous anaplastic large-cell lymphoma typically presents as solitary or grouped nodules or tumors that may undergo spontaneous partial or complete regression in approximately 25% of cases3 but often persist if not treated. Patients may have an array of lesions comprising the spectrum of CD30 pcLPDs.4
There is no curative therapy for CD30+ pcLPDs. Although active treatment is not necessary for LyP, low-dose methotrexate (MTX)(10–50 mg weekly) or phototherapy are the preferred initial suppressive therapies for symptomatic patients with scarring, facial lesions, or multiple symptomatic lesions.5 Observation with expectant follow-up is an option in pcALCL, though spontaneous regression is less likely than in LyP. For single or grouped pcALCL lesions, local radiation is the first-line therapy.6 Multifocal pcALCL lesions also can be treated with low-dose MTX,2,5 as in LyP, or local radiation to selected areas. Although local radiotherapy is considered a first-line treatment in pcALCL, there is limited evidence on its clinical efficacy as well as the optimal dose and technique. We report the complete response of refractory pcALCL lesions to low-dose radiation while remaining on MTX weekly without any adverse effects.
Case Report
A 51-year-old woman presented with a 3-year history of CD30+ pcLPD manifesting primarily as pcALCL involving the head and neck, as well as LyP involving the head, arms, and trunk (T3N0M0). For 2 years her treatment regimen included clobetasol propionate cream 0.05% as needed for new lesions and 2 courses of standard-dose localized external beam radiation for larger pcALCL tumors on the right cheek and right side of the chin (Figure 1)(total dose for each course of treatment was 20 Gy and 36 Gy, respectively, each administered over 2–3 weeks). Because new unsightly papulonodules continued to develop on the patient’s face, she subsequently required low-dose oral MTX 30 mg once weekly for suppression of new lesions and was stable on this regimen for a year. However, she experienced an increase in LyP/pcALCL activity on the face during a 2-week break from MTX when she developed a herpes zoster infection on the right side of the forehead.
On physical examination 1 month later, 5 tiny pink papules scattered on the left eyebrow, left cheek, and left side of the chin were noted. She was advised to continue applying the clobetasol cream as needed and was restarted on MTX 10 mg once weekly. However, she developed 2 additional 1-cm nodules on the left side of the chin, neck, and shoulder. Methotrexate was increased to 30 mg once weekly over 2 weeks, which was the original dose prior to interruption, but the nodules grew to 1.5-cm in diameter. Due to their clinical appearance, the nodules were believed to be early pcALCL lesions (Figure 2A). Given the cosmetically sensitive location of the nodules, palliative radiotherapy was recommended rather than observe for possible regression. Based on a prior report by Neelis et al7 demonstrating efficacy of low-dose radiotherapy for cutaneous T-cell lymphoma and cutaneous B-cell lymphoma, we recommended starting with low radiation doses. Our patient was treated with 400 cGy twice to the left side of the chin and left side of the neck (800 cGy total at each site) while remaining on MTX 30 mg once weekly. This treatment was well tolerated without side effects and no evidence of radiation dermatitis. On follow-up examination 1 week later, the nodules had regressed and no new lesions were present (Figure 2B).
The patient has stayed on oral MTX and occasionally develops small lesions that quickly resolve with clobetasol cream. She has been followed for 3 years after radiotherapy and all 3 previously irradiated sites have remained recurrence free. Furthermore, she has not developed any new larger nodules or tumors and her MTX dose has been decreased to 15 mg once weekly.
Comment
Local radiotherapy is considered a first-line treatment of pcALCL; however, there is limited evidence on its clinical efficacy as well as the optimal dose and technique. Although no standard dose exists for pcALCL, the National Comprehensive Cancer Network guidelines8 recommend doses of 12 to 36 Gy in mycosis fungoides/Sézary syndrome subtypes of cutaneous T-cell lymphoma, which are consistent with guidelines published by the European Society for Medical Oncology.9 High complete response rates have been demonstrated in pcALCL at doses of 34 to 44 Gy6; however, lesions tend to recur elsewhere on the skin in 36% to 41% of patients despite treatment.2,10 Lower doses of radiation therapy would provide several advantages over higher-dose therapy if a complete response could be achieved without greatly increasing the local recurrence rate. In cases of local recurrence, low-dose radiation would more easily permit retreatment of lesions compared to higher doses of radiation. Similarly, in patients with multifocal pcALCL, lower doses of radiotherapy may allow for treatment of larger skin areas while limiting potential treatment risks. Furthermore, low-dose therapy would allow for treatments to be delivered more quickly and with less inconvenience to the patient who is likely to need multiple future treatments to other areas. Low-dose radiation has been described with a favorable efficacy profile for mycosis fungoides7,11 but has not been studied in patients with CD30+ pcLPDs.
Our case is notable because the patient remained on MTX during radiation therapy. B
Conclusion
We reported the use of low-dose radiation therapy for the treatment of localized pcALCL in a patient who remained on low-dose oral MTX. Additional studies will be necessary to more fully evaluate the efficacy of using low-dose radiation both as monotherapy and in combination with MTX for pcALCL.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Bekkenk MW, Geelen FA, van Voorst Vader PC, et al. Primary and secondary cutaneous CD30+ lymphoproliferative disorders: a report from the Dutch Cutaneous Lymphoma Group on the long-term follow-up data of 219 patients and guidelines for diagnosis and treatment. Blood. 2000;95:3653-3661.
- Willemze R, Beljaards RC. Spectrum of primary cutaneous CD30 (Ki-1)-positive lymphoproliferative disorders: a proposal for classification and guidelines for management and treatment. J Am Acad Dermatol. 1993;28:973-980.
- Kadin ME. The spectrum of Ki-1+ cutaneous lymphomas. Curr Probl Dermatol. 1990;19:132-143.
- Vonderheid EC, Sajjadian A, Kadin ME. Methotrexate is effective therapy for lymphomatoid papulosis and other primary cutaneous CD30-positive lymphoproliferative disorders. J Am Acad Dermatol. 1996;34:470-481.
- Yu JB, McNiff JM, Lund MW, et al. Treatment of primary cutaneous CD30+ anaplastic large-cell lymphoma with radiation therapy. Int J Radiat Oncol Biol Phys. 2008;70:1542-1545.
- Neelis KJ, Schimmel EC, Vermeer MH, et al. Low-dose palliative radiotherapy B-cell and T-cell lymphomas. Int J Radiat Oncol Biol Phys. 2009;74:154-158.
- National Comprehensive Cancer Network. CD30 lymphoproliferative disorders section in non-Hodgkin’s lymphoma (Version 3.2016). http://www.nccn.org/professionals/physician_gls/pdf/nhl.pdf. Accessed September 26, 2016.
- Willemze R, Hodak E, Zinzani PL, et al; ESMO Guidelines Working Group. Primary cutaneous lymphomas: EMSO clinical practice guidelines for diagnosis, treatment, and follow-up [published online July 17, 2013]. Ann Onc. 2013;24(suppl 6):vi149-vi154.
- Liu HL, Hoppe RT, Kohler S, et al. CD30+ cutaneous lymphoproliferative disorders: the Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. J Am Acad Dermatol. 2003;49:1049-1058.
- Harrison C, Young J, Navi D, et al. Revisiting low dose total skin electron beam radiotherapy in mycosis fungoides. Int J Radiat Oncol Biol Phys. 2011;81:651-657.
- Jaffe N, Farber S, Traggis D, et al. Favorable response of metastatic osteogenic sarcoma to pulse high-dose methotrexate with citrovorum rescue and radiation therapy. Cancer. 1973;31:1367-1373.
- Rosen G, Tefft M, Martinez A, et al. Combination chemotherapy and radiation therapy in the treatment of metastatic osteogenic sarcoma. Cancer. 1975;35:622-630.
- Kim YH, Aye MS, Fayos JV. Radiation necrosis of the scalp: a complication of cranial irradiation and methotrexate. Radiology. 1977;124:813-814.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Bekkenk MW, Geelen FA, van Voorst Vader PC, et al. Primary and secondary cutaneous CD30+ lymphoproliferative disorders: a report from the Dutch Cutaneous Lymphoma Group on the long-term follow-up data of 219 patients and guidelines for diagnosis and treatment. Blood. 2000;95:3653-3661.
- Willemze R, Beljaards RC. Spectrum of primary cutaneous CD30 (Ki-1)-positive lymphoproliferative disorders: a proposal for classification and guidelines for management and treatment. J Am Acad Dermatol. 1993;28:973-980.
- Kadin ME. The spectrum of Ki-1+ cutaneous lymphomas. Curr Probl Dermatol. 1990;19:132-143.
- Vonderheid EC, Sajjadian A, Kadin ME. Methotrexate is effective therapy for lymphomatoid papulosis and other primary cutaneous CD30-positive lymphoproliferative disorders. J Am Acad Dermatol. 1996;34:470-481.
- Yu JB, McNiff JM, Lund MW, et al. Treatment of primary cutaneous CD30+ anaplastic large-cell lymphoma with radiation therapy. Int J Radiat Oncol Biol Phys. 2008;70:1542-1545.
- Neelis KJ, Schimmel EC, Vermeer MH, et al. Low-dose palliative radiotherapy B-cell and T-cell lymphomas. Int J Radiat Oncol Biol Phys. 2009;74:154-158.
- National Comprehensive Cancer Network. CD30 lymphoproliferative disorders section in non-Hodgkin’s lymphoma (Version 3.2016). http://www.nccn.org/professionals/physician_gls/pdf/nhl.pdf. Accessed September 26, 2016.
- Willemze R, Hodak E, Zinzani PL, et al; ESMO Guidelines Working Group. Primary cutaneous lymphomas: EMSO clinical practice guidelines for diagnosis, treatment, and follow-up [published online July 17, 2013]. Ann Onc. 2013;24(suppl 6):vi149-vi154.
- Liu HL, Hoppe RT, Kohler S, et al. CD30+ cutaneous lymphoproliferative disorders: the Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. J Am Acad Dermatol. 2003;49:1049-1058.
- Harrison C, Young J, Navi D, et al. Revisiting low dose total skin electron beam radiotherapy in mycosis fungoides. Int J Radiat Oncol Biol Phys. 2011;81:651-657.
- Jaffe N, Farber S, Traggis D, et al. Favorable response of metastatic osteogenic sarcoma to pulse high-dose methotrexate with citrovorum rescue and radiation therapy. Cancer. 1973;31:1367-1373.
- Rosen G, Tefft M, Martinez A, et al. Combination chemotherapy and radiation therapy in the treatment of metastatic osteogenic sarcoma. Cancer. 1975;35:622-630.
- Kim YH, Aye MS, Fayos JV. Radiation necrosis of the scalp: a complication of cranial irradiation and methotrexate. Radiology. 1977;124:813-814.
Practice Points
- Cutaneous T-cell lymphoma tumors such as primary cutaneous anaplastic large-cell lymphoma can respond to low-dose radiation therapy, which enables future retreatment of sensitive sites.
- Low-dose radiation therapy requires a shorter course of therapy than traditional dosing, which is more convenient and less costly.
Acquired Port-wine Stain With Superimposed Eczema Following Penetrating Abdominal Trauma
Port-wine stains (PWSs) are common congenital capillary vascular malformations with an incidence of 3 per 1000 neonates.1 Rarely, acquired PWSs are seen, sometimes appearing following trauma.2-5 Port-wine stains are diagnosed clinically and present as painless, partially or entirely blanchable pink patches that respect the median (midline) plane.6 Although histopathologic examination is not necessary for diagnosis of PWS, typical findings include dilated, ectatic capillaries.7,8 Since it was first reported by Traub9 in 1939, more than 60 cases of acquired PWSs have been reported.10 A PubMed search of articles indexed for MEDLINE using the search terms acquired port-wine stain and port-wine stain and eczema yielded no cases of acquired PWS with associated eczematous changes and only 30 cases of congenital PWS with superimposed eczema.11-18 We report the case of an acquired PWS with superimposed eczema in an 18-year-old man following penetrating abdominal trauma.
Case Report
An otherwise healthy 18-year-old man presented to our dermatology office for evaluation of an eruption that had developed at the site of an abdominal stab wound he sustained 2 to 3 years prior. One year after he was stabbed, the patient developed a nonpruritic, painless red patch located 1 cm anterior to the healed wound on the left abdomen. The patch gradually grew larger to involve the entire left abdomen, extending to the left lower back. The site of the healed stab wound also became raised and pruritic, and the patient noted another pruritic plaque that formed within the larger patch. The patient reported no other skin conditions prior to the current eruption. His medical history was notable for seasonal allergies and asthma, but no childhood eczema.
Physical examination revealed a healthy, well-nourished man with Fitzpatrick skin type IV. A red, purpuric, coalescent patch with slightly arcuate borders extending from the mid abdomen to the left posterior flank was noted. The left lateral aspect of the patch blanched with pressure and respected the median plane. Within the larger patch, a 4-cm×2-cm lichenified, slightly macerated, hyperpigmented plaque was noted at the site of the stab wound (Figure 1). Based on these clinical findings, a presumptive diagnosis of an acquired PWS with superimposed eczema was made.
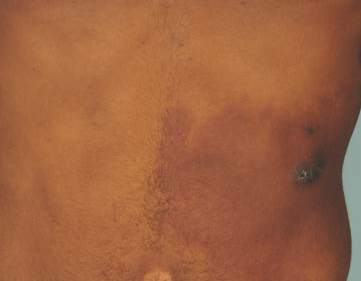
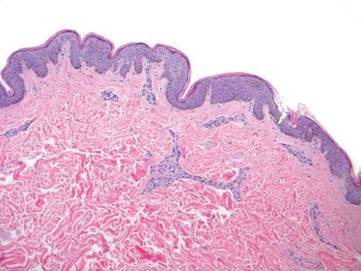
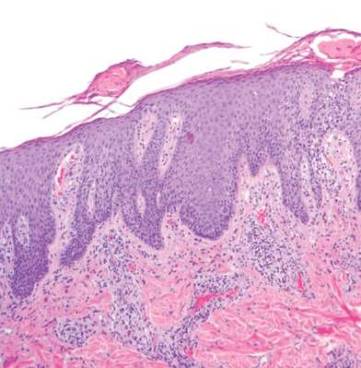
Punch biopsy specimens were taken from the large vascular patch and the smaller lichenified plaque. Histopathologic examination of the vascular patch showed an increased number of small vessels in the superficial dermis with thickened vessel walls, ectatic lumens, and no vasculopathy, consistent with a vascular malformation or a reactive vascular proliferation (Figure 2). On histopathology, the plaque showed epidermal spongiosis and hyperplasia with serum crust and a papillary dermis containing a mixed inflammatory infiltrate with occasional eosinophils, consistent with an eczematous dermatitis (Figure 3). The histologic findings confirmed the clinical diagnosis.
The pruritic, lichenified plaque improved with application of triamcinolone ointment 0.1% twice daily for 2 weeks. Magnetic resonance imaging to rule out an underlying arteriovenous malformation was recommended, but the patient declined.
Comment
The exact cause of PWS is unknown. There have been a multitude of genomic suspects for congenital lesions, including a somatic activating mutation (ie, a mutation acquired during fetal development) of the GNAQ (guanine nucleotide binding protein [G protein], q polypeptide) gene, which may contribute to abnormal cell proliferation including the regulation of blood vessels, and inactivating mutations in the RASA1 (RAS p21 protein activator [GTPase activating protein] 1) gene, which controls endothelial cell organization.19-22 Later mutations (ie, those occurring after the first trimester) may be more likely to result in isolated PWSs as opposed to syndromic PWSs.19 Whatever the source of genetic misinformation, it is thought that the diminished neuronal control of blood flow and the resulting alterations in dermal structure contribute to the pathogenesis of PWS and its associated histologic features.7,23
The clinical and histopathologic features of acquired PWSs are indistinguishable from those of congenital lesions, indicating that different processes may lead to the same presentation.4 Abnormal innervation and decreased supportive stroma have both been identified as contributing factors in the development of congenital and acquired PWSs.7,23-25 Rosen and Smoller23 found that diminished nerve density affects vascular tone and caliber in PWSs and had hypothesized in a prior report that decreased perivascular Schwann cells may indicate abnormal sympathetic innervation.7 Since then, PWS has been shown to lack both somatic and sensory innervation.24 Tsuji and Sawabe25 indicated that alterations to the perivascular stroma, whether congenital or as a result of trauma, decrease support for vessels, leading to ectasia.
In addition to an acquired PWS, our patient also had associated eczema within the PWS. Eczematous lesions were absent elsewhere, and he did not have a history of childhood eczema. Our review of the literature yielded 8 studies since 1996 that collectively described 30 cases of eczema within PWSs.11-18 Only 2 of these reports described adult patients with concomitant eczema and PWS and none described acquired PWS.13,18
Few studies have addressed the relationship between PWSs and eczema. It is unclear if concomitant PWS and localized eczema are collision dermatoses or if a PWS may predispose the affected skin to eczema.11-13 It has been hypothesized that the increased dermal vasculature in PWSs predisposes the skin to the development of eczema—more specifically, that ectasia may lead to increased inflammation.12,17 The concept of the “immunocompromised district” proposed by Ruocco et al26 is a unifying theory that may underlie the association noted between cases of trauma and later development of a PWS and superimposed eczematous dermatitis, such as in our case. Trauma is noted as one of a number of possible disruptive forces affecting both immunomodulation and neuromodulation within a local area of skin, leading to increased susceptibility of that district to various cutaneous diseases.26
Although our patient’s eczema responded to conservative treatment with a topical steroid, several case series have reported success with laser therapy in the treatment of PWS while preventing recurrence of associated eczematous dermatitis.12,17 Following the cessation of eczema treatment with topical steroid, which causes vasoconstriction, we suggest postponing laser therapy several weeks to allow resolution of vasoconstriction, thus providing enhanced therapeutic targeting with a vascular laser. Of particular relevance to our case, a recent study showed efficacy of the pulsed dye laser in treating PWSs in Fitzpatrick skin types IV and V.27
Conclusion
Although acquired PWS is rare, it can present later in life as an acquired lesion at a site of previous trauma.1-5 Congenital capillary malformations also can be associated with superimposed, localized eczema.11-18 We present a rarely reported case of an acquired PWS with superimposed, localized eczema. As in cases of congenital PWS with concomitant eczema, the associated eczema in our case was responsive to topical corticosteroid therapy. Additionally, pulsed dye laser has been shown to treat PWSs while preventing the recurrence of eczema, and it has been deemed effective for individuals with darker skin types.12,17, 27 Further studies are needed to explore the relationship between PWS and eczema.
- Jacobs AH, Walton RG. The incidence of birthmarks in the neonate. Pediatrics. 1976;58:218-222.
- Fegeler F. Naevus flammeus im trigeminusgebiet nach trauma im rahmen eines posttraumatisch-vegetativen syndroms. Arch Dermatol Syphilol. 1949;188:416-422.
- Kirkland CR, Mutasim DF. Acquired port-wine stain following repetitive trauma. J Am Acad Dermatol. 2011;65:462-463.
- Adams BB, Lucky AW. Acquired port-wine stains and antecedent trauma: case report and review of the literature. Arch Dermatol. 2000;136:897-899.
- Colver GB, Ryan TJ. Acquired port-wine stain. Arch Dermatol. 1986;122:1415-1416.
- Nigro J, Swerlick RA, Sepp NT, et al. Angiogenesis, vascular malformations and proliferations. In: Arndt KA, LeBoit PE, Robinson JK, Wintroub BU, eds. Cutaneous Medicine and Surgery: An Integrated Program in Dermatology. Philadelphia, PA: WB Saunders Co; 1996:1492-1521.
- Smoller BR, Rosen S. Port-wine stains. a disease of altered neural modulation of blood vessels? Arch Dermatol. 1986;122:177-179.
- Chang CJ, Yu JS, Nelson JS. Confocal microscopy study of neurovascular distribution in facial port wine stains(capillary malformation). J Formos Med Assoc. 2008;107:559-666.
- Traub EF. Naevus flammeus appearing at the age of twenty three. Arch Dermatol. 1939;39:752.
- Freysz M, Cribier B, Lipsker, D. Fegelers syndrome, acquired port-wine stain or acquired capillary malformation: three cases and a literature review [article in French]. Ann Dermatol Venereol. 2013;140:341-346.
- Tay YK, Morelli J, Weston WL. Inflammatory nuchal-occipital port-wine stains. J Am Acad Dermatol. 1996;35:811-813.
- Sidwell RU, Syed S, Harper JI. Port-wine stains and eczema. Br J Dermatol. 2001;144:1269-1270.
- Hofer T. Meyerson phenomenon within a nevus flammeus. Dermatology. 2002;205:180-183.
- Raff K, Landthaler M, Hoheleutner U. Port-wine stains with eczema. Phlebologie. 2003;32:15-17.
- Tsuboi H, Miyata T, Katsuoka K. Eczema in a port-wine stain. Clin Exp Dermatol. 2003;28:322-323.
- Rajan N, Natarahan S. Impetiginized eczema arising within a port-wine stain of the arm. J Eur Acad Dermatol Venereol. 2006;20:1009-1010.
- Fonder MA, Mamelak AJ, Kazin RA, et al. Port-wine-stain-associated dermatitis: implications for cutaneous vascular laser therapy. Pediatr Dermatol. 2007;24:376-379.
- Simon V, Wolfgan H, Katharina F. Meyerson-Phenomenon hides a nevus flammeus. J Dtsch Dermatol Ges. 2011;9:305-307.
- Shirley MD, Tang H, Gallione CJ, et al. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med. 2013;368:1971-1979.
- Hershkovitz D, Bercovich D, Sprecher E, et al. RASA1 mutations may cause hereditary capillary malformations without arteriovenous malformations. Br J Dermatol. 2008;158:1035-1040.
- Eerola I, Boon LM, Mulliken JB, et al. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet. 2003;73:1240-1249.
- Henkemeyer M, Rossi DJ, Holmyard DP, et al. Vascular system defects and neuronal apoptosis in mice lacking ras GTPase-activating protein. Nature. 1995;377:695-701.
- Rosen S, Smoller BR. Port-wine stains: a new hypothesis. J Am Acad Dermatol. 1987;17:164-166.
- Rydh M, Malm BM, Jernmeck J, et al. Ectatic blood vessels in port-wine stains lack innervation: possible role in pathogenesis. Plast Reconstr Surg. 1991;87:419-422.
- Tsuji T, Sawabe M. A new type of telangiectasia following trauma. J Cutan Pathol. 1988;15:22-26.
- Ruocco V, Ruocco E, Brunnetti G, et al. Opportunistic localization of skin lesions on vulnerable areas. Clin Dermatol. 2011;29:483-488.
- Thajudeheen CP, Jyothy K, Pryadarshi A. Treatment of port-wine stains with flash lamp pumped pulsed dye laser on Indian skin: a six year study. J Cutan Aesthet Surg. 2014;7:32-36.
Port-wine stains (PWSs) are common congenital capillary vascular malformations with an incidence of 3 per 1000 neonates.1 Rarely, acquired PWSs are seen, sometimes appearing following trauma.2-5 Port-wine stains are diagnosed clinically and present as painless, partially or entirely blanchable pink patches that respect the median (midline) plane.6 Although histopathologic examination is not necessary for diagnosis of PWS, typical findings include dilated, ectatic capillaries.7,8 Since it was first reported by Traub9 in 1939, more than 60 cases of acquired PWSs have been reported.10 A PubMed search of articles indexed for MEDLINE using the search terms acquired port-wine stain and port-wine stain and eczema yielded no cases of acquired PWS with associated eczematous changes and only 30 cases of congenital PWS with superimposed eczema.11-18 We report the case of an acquired PWS with superimposed eczema in an 18-year-old man following penetrating abdominal trauma.
Case Report
An otherwise healthy 18-year-old man presented to our dermatology office for evaluation of an eruption that had developed at the site of an abdominal stab wound he sustained 2 to 3 years prior. One year after he was stabbed, the patient developed a nonpruritic, painless red patch located 1 cm anterior to the healed wound on the left abdomen. The patch gradually grew larger to involve the entire left abdomen, extending to the left lower back. The site of the healed stab wound also became raised and pruritic, and the patient noted another pruritic plaque that formed within the larger patch. The patient reported no other skin conditions prior to the current eruption. His medical history was notable for seasonal allergies and asthma, but no childhood eczema.
Physical examination revealed a healthy, well-nourished man with Fitzpatrick skin type IV. A red, purpuric, coalescent patch with slightly arcuate borders extending from the mid abdomen to the left posterior flank was noted. The left lateral aspect of the patch blanched with pressure and respected the median plane. Within the larger patch, a 4-cm×2-cm lichenified, slightly macerated, hyperpigmented plaque was noted at the site of the stab wound (Figure 1). Based on these clinical findings, a presumptive diagnosis of an acquired PWS with superimposed eczema was made.
Punch biopsy specimens were taken from the large vascular patch and the smaller lichenified plaque. Histopathologic examination of the vascular patch showed an increased number of small vessels in the superficial dermis with thickened vessel walls, ectatic lumens, and no vasculopathy, consistent with a vascular malformation or a reactive vascular proliferation (Figure 2). On histopathology, the plaque showed epidermal spongiosis and hyperplasia with serum crust and a papillary dermis containing a mixed inflammatory infiltrate with occasional eosinophils, consistent with an eczematous dermatitis (Figure 3). The histologic findings confirmed the clinical diagnosis.
The pruritic, lichenified plaque improved with application of triamcinolone ointment 0.1% twice daily for 2 weeks. Magnetic resonance imaging to rule out an underlying arteriovenous malformation was recommended, but the patient declined.
Comment
The exact cause of PWS is unknown. There have been a multitude of genomic suspects for congenital lesions, including a somatic activating mutation (ie, a mutation acquired during fetal development) of the GNAQ (guanine nucleotide binding protein [G protein], q polypeptide) gene, which may contribute to abnormal cell proliferation including the regulation of blood vessels, and inactivating mutations in the RASA1 (RAS p21 protein activator [GTPase activating protein] 1) gene, which controls endothelial cell organization.19-22 Later mutations (ie, those occurring after the first trimester) may be more likely to result in isolated PWSs as opposed to syndromic PWSs.19 Whatever the source of genetic misinformation, it is thought that the diminished neuronal control of blood flow and the resulting alterations in dermal structure contribute to the pathogenesis of PWS and its associated histologic features.7,23
The clinical and histopathologic features of acquired PWSs are indistinguishable from those of congenital lesions, indicating that different processes may lead to the same presentation.4 Abnormal innervation and decreased supportive stroma have both been identified as contributing factors in the development of congenital and acquired PWSs.7,23-25 Rosen and Smoller23 found that diminished nerve density affects vascular tone and caliber in PWSs and had hypothesized in a prior report that decreased perivascular Schwann cells may indicate abnormal sympathetic innervation.7 Since then, PWS has been shown to lack both somatic and sensory innervation.24 Tsuji and Sawabe25 indicated that alterations to the perivascular stroma, whether congenital or as a result of trauma, decrease support for vessels, leading to ectasia.
In addition to an acquired PWS, our patient also had associated eczema within the PWS. Eczematous lesions were absent elsewhere, and he did not have a history of childhood eczema. Our review of the literature yielded 8 studies since 1996 that collectively described 30 cases of eczema within PWSs.11-18 Only 2 of these reports described adult patients with concomitant eczema and PWS and none described acquired PWS.13,18
Few studies have addressed the relationship between PWSs and eczema. It is unclear if concomitant PWS and localized eczema are collision dermatoses or if a PWS may predispose the affected skin to eczema.11-13 It has been hypothesized that the increased dermal vasculature in PWSs predisposes the skin to the development of eczema—more specifically, that ectasia may lead to increased inflammation.12,17 The concept of the “immunocompromised district” proposed by Ruocco et al26 is a unifying theory that may underlie the association noted between cases of trauma and later development of a PWS and superimposed eczematous dermatitis, such as in our case. Trauma is noted as one of a number of possible disruptive forces affecting both immunomodulation and neuromodulation within a local area of skin, leading to increased susceptibility of that district to various cutaneous diseases.26
Although our patient’s eczema responded to conservative treatment with a topical steroid, several case series have reported success with laser therapy in the treatment of PWS while preventing recurrence of associated eczematous dermatitis.12,17 Following the cessation of eczema treatment with topical steroid, which causes vasoconstriction, we suggest postponing laser therapy several weeks to allow resolution of vasoconstriction, thus providing enhanced therapeutic targeting with a vascular laser. Of particular relevance to our case, a recent study showed efficacy of the pulsed dye laser in treating PWSs in Fitzpatrick skin types IV and V.27
Conclusion
Although acquired PWS is rare, it can present later in life as an acquired lesion at a site of previous trauma.1-5 Congenital capillary malformations also can be associated with superimposed, localized eczema.11-18 We present a rarely reported case of an acquired PWS with superimposed, localized eczema. As in cases of congenital PWS with concomitant eczema, the associated eczema in our case was responsive to topical corticosteroid therapy. Additionally, pulsed dye laser has been shown to treat PWSs while preventing the recurrence of eczema, and it has been deemed effective for individuals with darker skin types.12,17, 27 Further studies are needed to explore the relationship between PWS and eczema.
Port-wine stains (PWSs) are common congenital capillary vascular malformations with an incidence of 3 per 1000 neonates.1 Rarely, acquired PWSs are seen, sometimes appearing following trauma.2-5 Port-wine stains are diagnosed clinically and present as painless, partially or entirely blanchable pink patches that respect the median (midline) plane.6 Although histopathologic examination is not necessary for diagnosis of PWS, typical findings include dilated, ectatic capillaries.7,8 Since it was first reported by Traub9 in 1939, more than 60 cases of acquired PWSs have been reported.10 A PubMed search of articles indexed for MEDLINE using the search terms acquired port-wine stain and port-wine stain and eczema yielded no cases of acquired PWS with associated eczematous changes and only 30 cases of congenital PWS with superimposed eczema.11-18 We report the case of an acquired PWS with superimposed eczema in an 18-year-old man following penetrating abdominal trauma.
Case Report
An otherwise healthy 18-year-old man presented to our dermatology office for evaluation of an eruption that had developed at the site of an abdominal stab wound he sustained 2 to 3 years prior. One year after he was stabbed, the patient developed a nonpruritic, painless red patch located 1 cm anterior to the healed wound on the left abdomen. The patch gradually grew larger to involve the entire left abdomen, extending to the left lower back. The site of the healed stab wound also became raised and pruritic, and the patient noted another pruritic plaque that formed within the larger patch. The patient reported no other skin conditions prior to the current eruption. His medical history was notable for seasonal allergies and asthma, but no childhood eczema.
Physical examination revealed a healthy, well-nourished man with Fitzpatrick skin type IV. A red, purpuric, coalescent patch with slightly arcuate borders extending from the mid abdomen to the left posterior flank was noted. The left lateral aspect of the patch blanched with pressure and respected the median plane. Within the larger patch, a 4-cm×2-cm lichenified, slightly macerated, hyperpigmented plaque was noted at the site of the stab wound (Figure 1). Based on these clinical findings, a presumptive diagnosis of an acquired PWS with superimposed eczema was made.
Punch biopsy specimens were taken from the large vascular patch and the smaller lichenified plaque. Histopathologic examination of the vascular patch showed an increased number of small vessels in the superficial dermis with thickened vessel walls, ectatic lumens, and no vasculopathy, consistent with a vascular malformation or a reactive vascular proliferation (Figure 2). On histopathology, the plaque showed epidermal spongiosis and hyperplasia with serum crust and a papillary dermis containing a mixed inflammatory infiltrate with occasional eosinophils, consistent with an eczematous dermatitis (Figure 3). The histologic findings confirmed the clinical diagnosis.
The pruritic, lichenified plaque improved with application of triamcinolone ointment 0.1% twice daily for 2 weeks. Magnetic resonance imaging to rule out an underlying arteriovenous malformation was recommended, but the patient declined.
Comment
The exact cause of PWS is unknown. There have been a multitude of genomic suspects for congenital lesions, including a somatic activating mutation (ie, a mutation acquired during fetal development) of the GNAQ (guanine nucleotide binding protein [G protein], q polypeptide) gene, which may contribute to abnormal cell proliferation including the regulation of blood vessels, and inactivating mutations in the RASA1 (RAS p21 protein activator [GTPase activating protein] 1) gene, which controls endothelial cell organization.19-22 Later mutations (ie, those occurring after the first trimester) may be more likely to result in isolated PWSs as opposed to syndromic PWSs.19 Whatever the source of genetic misinformation, it is thought that the diminished neuronal control of blood flow and the resulting alterations in dermal structure contribute to the pathogenesis of PWS and its associated histologic features.7,23
The clinical and histopathologic features of acquired PWSs are indistinguishable from those of congenital lesions, indicating that different processes may lead to the same presentation.4 Abnormal innervation and decreased supportive stroma have both been identified as contributing factors in the development of congenital and acquired PWSs.7,23-25 Rosen and Smoller23 found that diminished nerve density affects vascular tone and caliber in PWSs and had hypothesized in a prior report that decreased perivascular Schwann cells may indicate abnormal sympathetic innervation.7 Since then, PWS has been shown to lack both somatic and sensory innervation.24 Tsuji and Sawabe25 indicated that alterations to the perivascular stroma, whether congenital or as a result of trauma, decrease support for vessels, leading to ectasia.
In addition to an acquired PWS, our patient also had associated eczema within the PWS. Eczematous lesions were absent elsewhere, and he did not have a history of childhood eczema. Our review of the literature yielded 8 studies since 1996 that collectively described 30 cases of eczema within PWSs.11-18 Only 2 of these reports described adult patients with concomitant eczema and PWS and none described acquired PWS.13,18
Few studies have addressed the relationship between PWSs and eczema. It is unclear if concomitant PWS and localized eczema are collision dermatoses or if a PWS may predispose the affected skin to eczema.11-13 It has been hypothesized that the increased dermal vasculature in PWSs predisposes the skin to the development of eczema—more specifically, that ectasia may lead to increased inflammation.12,17 The concept of the “immunocompromised district” proposed by Ruocco et al26 is a unifying theory that may underlie the association noted between cases of trauma and later development of a PWS and superimposed eczematous dermatitis, such as in our case. Trauma is noted as one of a number of possible disruptive forces affecting both immunomodulation and neuromodulation within a local area of skin, leading to increased susceptibility of that district to various cutaneous diseases.26
Although our patient’s eczema responded to conservative treatment with a topical steroid, several case series have reported success with laser therapy in the treatment of PWS while preventing recurrence of associated eczematous dermatitis.12,17 Following the cessation of eczema treatment with topical steroid, which causes vasoconstriction, we suggest postponing laser therapy several weeks to allow resolution of vasoconstriction, thus providing enhanced therapeutic targeting with a vascular laser. Of particular relevance to our case, a recent study showed efficacy of the pulsed dye laser in treating PWSs in Fitzpatrick skin types IV and V.27
Conclusion
Although acquired PWS is rare, it can present later in life as an acquired lesion at a site of previous trauma.1-5 Congenital capillary malformations also can be associated with superimposed, localized eczema.11-18 We present a rarely reported case of an acquired PWS with superimposed, localized eczema. As in cases of congenital PWS with concomitant eczema, the associated eczema in our case was responsive to topical corticosteroid therapy. Additionally, pulsed dye laser has been shown to treat PWSs while preventing the recurrence of eczema, and it has been deemed effective for individuals with darker skin types.12,17, 27 Further studies are needed to explore the relationship between PWS and eczema.
- Jacobs AH, Walton RG. The incidence of birthmarks in the neonate. Pediatrics. 1976;58:218-222.
- Fegeler F. Naevus flammeus im trigeminusgebiet nach trauma im rahmen eines posttraumatisch-vegetativen syndroms. Arch Dermatol Syphilol. 1949;188:416-422.
- Kirkland CR, Mutasim DF. Acquired port-wine stain following repetitive trauma. J Am Acad Dermatol. 2011;65:462-463.
- Adams BB, Lucky AW. Acquired port-wine stains and antecedent trauma: case report and review of the literature. Arch Dermatol. 2000;136:897-899.
- Colver GB, Ryan TJ. Acquired port-wine stain. Arch Dermatol. 1986;122:1415-1416.
- Nigro J, Swerlick RA, Sepp NT, et al. Angiogenesis, vascular malformations and proliferations. In: Arndt KA, LeBoit PE, Robinson JK, Wintroub BU, eds. Cutaneous Medicine and Surgery: An Integrated Program in Dermatology. Philadelphia, PA: WB Saunders Co; 1996:1492-1521.
- Smoller BR, Rosen S. Port-wine stains. a disease of altered neural modulation of blood vessels? Arch Dermatol. 1986;122:177-179.
- Chang CJ, Yu JS, Nelson JS. Confocal microscopy study of neurovascular distribution in facial port wine stains(capillary malformation). J Formos Med Assoc. 2008;107:559-666.
- Traub EF. Naevus flammeus appearing at the age of twenty three. Arch Dermatol. 1939;39:752.
- Freysz M, Cribier B, Lipsker, D. Fegelers syndrome, acquired port-wine stain or acquired capillary malformation: three cases and a literature review [article in French]. Ann Dermatol Venereol. 2013;140:341-346.
- Tay YK, Morelli J, Weston WL. Inflammatory nuchal-occipital port-wine stains. J Am Acad Dermatol. 1996;35:811-813.
- Sidwell RU, Syed S, Harper JI. Port-wine stains and eczema. Br J Dermatol. 2001;144:1269-1270.
- Hofer T. Meyerson phenomenon within a nevus flammeus. Dermatology. 2002;205:180-183.
- Raff K, Landthaler M, Hoheleutner U. Port-wine stains with eczema. Phlebologie. 2003;32:15-17.
- Tsuboi H, Miyata T, Katsuoka K. Eczema in a port-wine stain. Clin Exp Dermatol. 2003;28:322-323.
- Rajan N, Natarahan S. Impetiginized eczema arising within a port-wine stain of the arm. J Eur Acad Dermatol Venereol. 2006;20:1009-1010.
- Fonder MA, Mamelak AJ, Kazin RA, et al. Port-wine-stain-associated dermatitis: implications for cutaneous vascular laser therapy. Pediatr Dermatol. 2007;24:376-379.
- Simon V, Wolfgan H, Katharina F. Meyerson-Phenomenon hides a nevus flammeus. J Dtsch Dermatol Ges. 2011;9:305-307.
- Shirley MD, Tang H, Gallione CJ, et al. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med. 2013;368:1971-1979.
- Hershkovitz D, Bercovich D, Sprecher E, et al. RASA1 mutations may cause hereditary capillary malformations without arteriovenous malformations. Br J Dermatol. 2008;158:1035-1040.
- Eerola I, Boon LM, Mulliken JB, et al. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet. 2003;73:1240-1249.
- Henkemeyer M, Rossi DJ, Holmyard DP, et al. Vascular system defects and neuronal apoptosis in mice lacking ras GTPase-activating protein. Nature. 1995;377:695-701.
- Rosen S, Smoller BR. Port-wine stains: a new hypothesis. J Am Acad Dermatol. 1987;17:164-166.
- Rydh M, Malm BM, Jernmeck J, et al. Ectatic blood vessels in port-wine stains lack innervation: possible role in pathogenesis. Plast Reconstr Surg. 1991;87:419-422.
- Tsuji T, Sawabe M. A new type of telangiectasia following trauma. J Cutan Pathol. 1988;15:22-26.
- Ruocco V, Ruocco E, Brunnetti G, et al. Opportunistic localization of skin lesions on vulnerable areas. Clin Dermatol. 2011;29:483-488.
- Thajudeheen CP, Jyothy K, Pryadarshi A. Treatment of port-wine stains with flash lamp pumped pulsed dye laser on Indian skin: a six year study. J Cutan Aesthet Surg. 2014;7:32-36.
- Jacobs AH, Walton RG. The incidence of birthmarks in the neonate. Pediatrics. 1976;58:218-222.
- Fegeler F. Naevus flammeus im trigeminusgebiet nach trauma im rahmen eines posttraumatisch-vegetativen syndroms. Arch Dermatol Syphilol. 1949;188:416-422.
- Kirkland CR, Mutasim DF. Acquired port-wine stain following repetitive trauma. J Am Acad Dermatol. 2011;65:462-463.
- Adams BB, Lucky AW. Acquired port-wine stains and antecedent trauma: case report and review of the literature. Arch Dermatol. 2000;136:897-899.
- Colver GB, Ryan TJ. Acquired port-wine stain. Arch Dermatol. 1986;122:1415-1416.
- Nigro J, Swerlick RA, Sepp NT, et al. Angiogenesis, vascular malformations and proliferations. In: Arndt KA, LeBoit PE, Robinson JK, Wintroub BU, eds. Cutaneous Medicine and Surgery: An Integrated Program in Dermatology. Philadelphia, PA: WB Saunders Co; 1996:1492-1521.
- Smoller BR, Rosen S. Port-wine stains. a disease of altered neural modulation of blood vessels? Arch Dermatol. 1986;122:177-179.
- Chang CJ, Yu JS, Nelson JS. Confocal microscopy study of neurovascular distribution in facial port wine stains(capillary malformation). J Formos Med Assoc. 2008;107:559-666.
- Traub EF. Naevus flammeus appearing at the age of twenty three. Arch Dermatol. 1939;39:752.
- Freysz M, Cribier B, Lipsker, D. Fegelers syndrome, acquired port-wine stain or acquired capillary malformation: three cases and a literature review [article in French]. Ann Dermatol Venereol. 2013;140:341-346.
- Tay YK, Morelli J, Weston WL. Inflammatory nuchal-occipital port-wine stains. J Am Acad Dermatol. 1996;35:811-813.
- Sidwell RU, Syed S, Harper JI. Port-wine stains and eczema. Br J Dermatol. 2001;144:1269-1270.
- Hofer T. Meyerson phenomenon within a nevus flammeus. Dermatology. 2002;205:180-183.
- Raff K, Landthaler M, Hoheleutner U. Port-wine stains with eczema. Phlebologie. 2003;32:15-17.
- Tsuboi H, Miyata T, Katsuoka K. Eczema in a port-wine stain. Clin Exp Dermatol. 2003;28:322-323.
- Rajan N, Natarahan S. Impetiginized eczema arising within a port-wine stain of the arm. J Eur Acad Dermatol Venereol. 2006;20:1009-1010.
- Fonder MA, Mamelak AJ, Kazin RA, et al. Port-wine-stain-associated dermatitis: implications for cutaneous vascular laser therapy. Pediatr Dermatol. 2007;24:376-379.
- Simon V, Wolfgan H, Katharina F. Meyerson-Phenomenon hides a nevus flammeus. J Dtsch Dermatol Ges. 2011;9:305-307.
- Shirley MD, Tang H, Gallione CJ, et al. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med. 2013;368:1971-1979.
- Hershkovitz D, Bercovich D, Sprecher E, et al. RASA1 mutations may cause hereditary capillary malformations without arteriovenous malformations. Br J Dermatol. 2008;158:1035-1040.
- Eerola I, Boon LM, Mulliken JB, et al. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet. 2003;73:1240-1249.
- Henkemeyer M, Rossi DJ, Holmyard DP, et al. Vascular system defects and neuronal apoptosis in mice lacking ras GTPase-activating protein. Nature. 1995;377:695-701.
- Rosen S, Smoller BR. Port-wine stains: a new hypothesis. J Am Acad Dermatol. 1987;17:164-166.
- Rydh M, Malm BM, Jernmeck J, et al. Ectatic blood vessels in port-wine stains lack innervation: possible role in pathogenesis. Plast Reconstr Surg. 1991;87:419-422.
- Tsuji T, Sawabe M. A new type of telangiectasia following trauma. J Cutan Pathol. 1988;15:22-26.
- Ruocco V, Ruocco E, Brunnetti G, et al. Opportunistic localization of skin lesions on vulnerable areas. Clin Dermatol. 2011;29:483-488.
- Thajudeheen CP, Jyothy K, Pryadarshi A. Treatment of port-wine stains with flash lamp pumped pulsed dye laser on Indian skin: a six year study. J Cutan Aesthet Surg. 2014;7:32-36.
Practice Points
- Port-wine stains (PWSs) most often are congenital lesions but can present later in life as acquired lesions with the same clinical and histologic findings.
- Magnetic resonance imaging of acquired PWSs should be considered to rule out underlying vascular anomalies (eg, deeper arteriovenous malformations).
- Pulsed dye laser therapy is safe for darker skin types and is the treatment of choice for acquired PWSs.