User login
Heart on the right may sometimes be ‘right’
A 76-year-old man presented to the emergency department with right-sided exertional chest pain radiating to the right shoulder and arm associated with shortness of breath. His vital signs were normal. On clinical examination, the cardiac apex was palpated on the right side, 9 cm from the midsternal line in the fifth intercostal space.
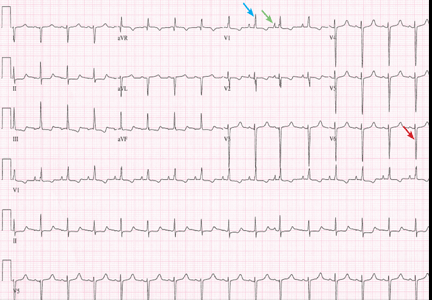
A standard left-sided 12-lead electrocardiogram (ECG) showed right-axis deviation and inverted P, QRS, and T waves in leads I and aVL (Figure 1). Although these changes are also seen when the right and left arm electrode wires are transposed, the precordial lead morphology in such a situation would usually be normal. In our patient, the precordial leads showed the absence or even slight reversal of R-wave progression, a feature indicative of dextrocardia.1,2
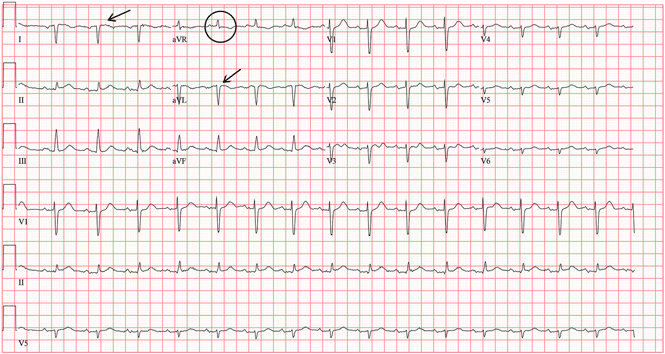
In patients with dextrocardia, right-sided hookup of the electrodes is usually necessary for proper interpretation of the ECG. When this was done in our patient, the ECG showed a normal cardiac axis, a negative QRS complex in lead aVR, a positive P wave and other complexes in lead I, and normal R-wave progression in the precordial leads—findings suggestive of dextrocardia (Figure 2).
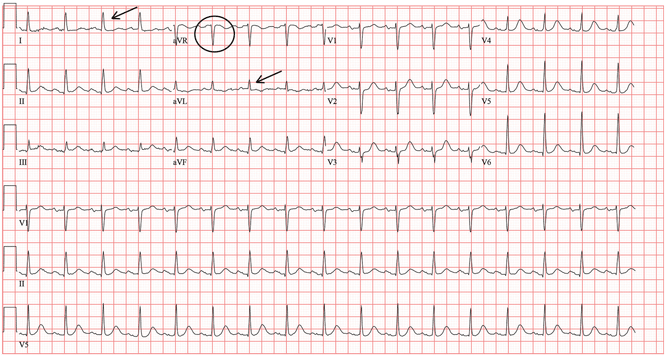
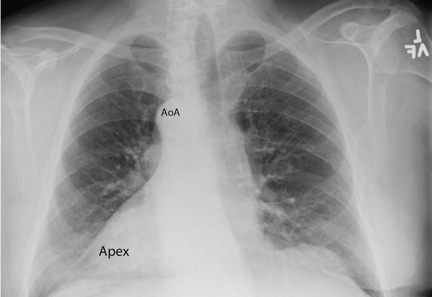
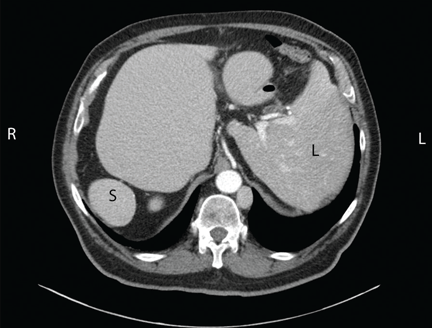
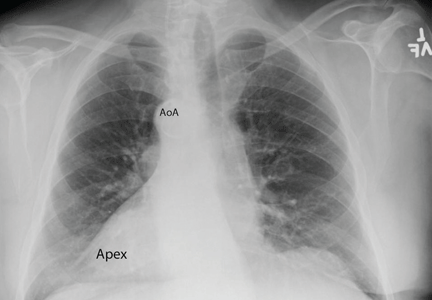
Chest radiography showed a right-sided cardiac silhouette (Figure 3), and computed tomography of the abdomen (Figure 4) revealed the liver positioned on the left side and the spleen on the right, confirming the diagnosis of situs inversus totalis. The ECG showed dextrocardia, but no other abnormalities. The patient eventually underwent coronary angiography, which showed nonobstructive coronary artery disease.
DEXTROCARDIA, OTHER CONGENITAL CARDIOVASCULAR MALFORMATIONS
Dextrocardia was first described in early 17th century.1 Situs solitus is the normal position of the heart and viscera, whereas situs inversus is a mirror-image anatomic arrangement of the organs. Situs inversus with dextrocardia, also called situs inversus totalis, is a rare condition (with a prevalence of 1 in 8,000) in which the heart and descending aorta are on the right and the thoracic and abdominal viscera are usually mirror images of the normal morphology.1,3,4 A mirror-image sinus node lies at the junction of the left superior vena cava and the left-sided (morphologic right) atrium.1 People with situs inversus with dextrocardia are usually asymptomatic and have a normal life expectancy.1,2 Situs inversus with levocardia is a rare condition in which the heart is in the normal position but the viscera are in the dextro-position. This anomaly has a prevalence of 1 in 22,000.5
Atrial situs almost always corresponds to visceral situs. However, when the alignment of the atria and viscera is inconsistent and situs cannot be determined clearly because of the malpositioning of organs, the condition is called “situs ambiguous.” This is very rare, with a prevalence of 1 in 40,000.6
Risk factors
The cause of congenital cardiovascular malformations such as these is not known, but risk factors include positive family history, maternal diabetes, and cocaine use in the first trimester.7
The prevalence of congenital heart disease in patients with situs inversus with dextrocardia is low and ranges from 2% to 5%. This is in contrast to situs solitus with dextrocardia (isolated dextrocardia), which is almost always associated with cardiovascular anomalies.2,4 Kartagener syndrome—the triad of situs inversus, sinusitis, and bronchiectasis—occurs in 25% of people with situs inversus with dextrocardia.4 Situs inversus with levocardia is also frequently associated with cardiac anomalies.5
The major features of dextrocardia on ECG are:
- Negative P wave, QRS complex, and T wave in lead I
- Positive QRS complex in aVR
- Right-axis deviation
- Reversal of R-wave progression in the precordial leads.
Ventricular activation and repolarization are reversed, resulting in a negative QRS complex and an inverted T wave in lead I. The absence of R-wave progression in the precordial leads helps differentiate mirror-image dextrocardia from erroneously reversed limb-electrode placement, which shows normal R-wave progression from V1 to V6 while showing similar features to those seen in dextrocardia in the limb leads.2 In right-sided hookup, the limb electrodes are reversed, and the chest electrodes are recorded from the right precordium.
CORONARY INTERVENTIONS REQUIRE SPECIAL CONSIDERATION
In patients with dextrocardia, coronary interventions can be challenging because of the mirror-image position of the coronary ostia and the aortic arch.8 These patients also need careful imaging, consideration of other associated congenital cardiac abnormalities, and detailed planning before cardiac surgery, including coronary artery bypass grafting.9
Patients with dextrocardia may present with cardiac symptoms localized to the right side of the body and have confusing clinical and diagnostic findings. Keeping dextrocardia and other such anomalies in mind can prevent delay in appropriately directed interventions. In a patient such as ours, the heart on the right side of the chest may indeed be “right.” Still, diagnostic tests to look for disorders encountered with dextrocardia may be necessary.
- Perloff JK. The cardiac malpositions. Am J Cardiol 2011; 108:1352–1361.
- Tanawuttiwat T, Vasaiwala S, Dia M. ECG image of the month. Mirror mirror. Am J Med 2010; 123:34–36.
- Douard R, Feldman A, Bargy F, Loric S, Delmas V. Anomalies of lateralization in man: a case of total situs in-versus. Surg Radiol Anat 2000; 22:293–297.
- Maldjian PD, Saric M. Approach to dextrocardia in adults: review. AJR Am J Roentgenol 2007; 188(suppl 6):S39–S49.
- Gindes L, Hegesh J, Barkai G, Jacobson JM, Achiron R. Isolated levocardia: prenatal diagnosis, clinical im-portance, and literature review. J Ultrasound Med 2007; 26:361–365.
- Abut E, Arman A, Güveli H, et al. Malposition of internal organs: a case of situs ambiguous anomaly in an adult. Turk J Gastroenterol 2003; 14:151–155.
- Kuehl KS, Loffredo C. Risk factors for heart disease associated with abnormal sidedness. Teratology 2002; 66:242–248.
- Aksoy S, Cam N, Gurkan U, Altay S, Bozbay M, Agirbasli M. Primary percutaneous intervention: for acute myo-cardial infarction in a patient with dextrocardia and situs inversus. Tex Heart Inst J 2012; 39:140–141.
- Murtuza B, Gupta P, Goli G, Lall KS. Coronary revascularization in adults with dextrocardia: surgical implications of the anatomic variants. Tex Heart Inst J 2010; 37:633–640.
A 76-year-old man presented to the emergency department with right-sided exertional chest pain radiating to the right shoulder and arm associated with shortness of breath. His vital signs were normal. On clinical examination, the cardiac apex was palpated on the right side, 9 cm from the midsternal line in the fifth intercostal space.
A standard left-sided 12-lead electrocardiogram (ECG) showed right-axis deviation and inverted P, QRS, and T waves in leads I and aVL (Figure 1). Although these changes are also seen when the right and left arm electrode wires are transposed, the precordial lead morphology in such a situation would usually be normal. In our patient, the precordial leads showed the absence or even slight reversal of R-wave progression, a feature indicative of dextrocardia.1,2
In patients with dextrocardia, right-sided hookup of the electrodes is usually necessary for proper interpretation of the ECG. When this was done in our patient, the ECG showed a normal cardiac axis, a negative QRS complex in lead aVR, a positive P wave and other complexes in lead I, and normal R-wave progression in the precordial leads—findings suggestive of dextrocardia (Figure 2).
Chest radiography showed a right-sided cardiac silhouette (Figure 3), and computed tomography of the abdomen (Figure 4) revealed the liver positioned on the left side and the spleen on the right, confirming the diagnosis of situs inversus totalis. The ECG showed dextrocardia, but no other abnormalities. The patient eventually underwent coronary angiography, which showed nonobstructive coronary artery disease.
DEXTROCARDIA, OTHER CONGENITAL CARDIOVASCULAR MALFORMATIONS
Dextrocardia was first described in early 17th century.1 Situs solitus is the normal position of the heart and viscera, whereas situs inversus is a mirror-image anatomic arrangement of the organs. Situs inversus with dextrocardia, also called situs inversus totalis, is a rare condition (with a prevalence of 1 in 8,000) in which the heart and descending aorta are on the right and the thoracic and abdominal viscera are usually mirror images of the normal morphology.1,3,4 A mirror-image sinus node lies at the junction of the left superior vena cava and the left-sided (morphologic right) atrium.1 People with situs inversus with dextrocardia are usually asymptomatic and have a normal life expectancy.1,2 Situs inversus with levocardia is a rare condition in which the heart is in the normal position but the viscera are in the dextro-position. This anomaly has a prevalence of 1 in 22,000.5
Atrial situs almost always corresponds to visceral situs. However, when the alignment of the atria and viscera is inconsistent and situs cannot be determined clearly because of the malpositioning of organs, the condition is called “situs ambiguous.” This is very rare, with a prevalence of 1 in 40,000.6
Risk factors
The cause of congenital cardiovascular malformations such as these is not known, but risk factors include positive family history, maternal diabetes, and cocaine use in the first trimester.7
The prevalence of congenital heart disease in patients with situs inversus with dextrocardia is low and ranges from 2% to 5%. This is in contrast to situs solitus with dextrocardia (isolated dextrocardia), which is almost always associated with cardiovascular anomalies.2,4 Kartagener syndrome—the triad of situs inversus, sinusitis, and bronchiectasis—occurs in 25% of people with situs inversus with dextrocardia.4 Situs inversus with levocardia is also frequently associated with cardiac anomalies.5
The major features of dextrocardia on ECG are:
- Negative P wave, QRS complex, and T wave in lead I
- Positive QRS complex in aVR
- Right-axis deviation
- Reversal of R-wave progression in the precordial leads.
Ventricular activation and repolarization are reversed, resulting in a negative QRS complex and an inverted T wave in lead I. The absence of R-wave progression in the precordial leads helps differentiate mirror-image dextrocardia from erroneously reversed limb-electrode placement, which shows normal R-wave progression from V1 to V6 while showing similar features to those seen in dextrocardia in the limb leads.2 In right-sided hookup, the limb electrodes are reversed, and the chest electrodes are recorded from the right precordium.
CORONARY INTERVENTIONS REQUIRE SPECIAL CONSIDERATION
In patients with dextrocardia, coronary interventions can be challenging because of the mirror-image position of the coronary ostia and the aortic arch.8 These patients also need careful imaging, consideration of other associated congenital cardiac abnormalities, and detailed planning before cardiac surgery, including coronary artery bypass grafting.9
Patients with dextrocardia may present with cardiac symptoms localized to the right side of the body and have confusing clinical and diagnostic findings. Keeping dextrocardia and other such anomalies in mind can prevent delay in appropriately directed interventions. In a patient such as ours, the heart on the right side of the chest may indeed be “right.” Still, diagnostic tests to look for disorders encountered with dextrocardia may be necessary.
A 76-year-old man presented to the emergency department with right-sided exertional chest pain radiating to the right shoulder and arm associated with shortness of breath. His vital signs were normal. On clinical examination, the cardiac apex was palpated on the right side, 9 cm from the midsternal line in the fifth intercostal space.
A standard left-sided 12-lead electrocardiogram (ECG) showed right-axis deviation and inverted P, QRS, and T waves in leads I and aVL (Figure 1). Although these changes are also seen when the right and left arm electrode wires are transposed, the precordial lead morphology in such a situation would usually be normal. In our patient, the precordial leads showed the absence or even slight reversal of R-wave progression, a feature indicative of dextrocardia.1,2
In patients with dextrocardia, right-sided hookup of the electrodes is usually necessary for proper interpretation of the ECG. When this was done in our patient, the ECG showed a normal cardiac axis, a negative QRS complex in lead aVR, a positive P wave and other complexes in lead I, and normal R-wave progression in the precordial leads—findings suggestive of dextrocardia (Figure 2).
Chest radiography showed a right-sided cardiac silhouette (Figure 3), and computed tomography of the abdomen (Figure 4) revealed the liver positioned on the left side and the spleen on the right, confirming the diagnosis of situs inversus totalis. The ECG showed dextrocardia, but no other abnormalities. The patient eventually underwent coronary angiography, which showed nonobstructive coronary artery disease.
DEXTROCARDIA, OTHER CONGENITAL CARDIOVASCULAR MALFORMATIONS
Dextrocardia was first described in early 17th century.1 Situs solitus is the normal position of the heart and viscera, whereas situs inversus is a mirror-image anatomic arrangement of the organs. Situs inversus with dextrocardia, also called situs inversus totalis, is a rare condition (with a prevalence of 1 in 8,000) in which the heart and descending aorta are on the right and the thoracic and abdominal viscera are usually mirror images of the normal morphology.1,3,4 A mirror-image sinus node lies at the junction of the left superior vena cava and the left-sided (morphologic right) atrium.1 People with situs inversus with dextrocardia are usually asymptomatic and have a normal life expectancy.1,2 Situs inversus with levocardia is a rare condition in which the heart is in the normal position but the viscera are in the dextro-position. This anomaly has a prevalence of 1 in 22,000.5
Atrial situs almost always corresponds to visceral situs. However, when the alignment of the atria and viscera is inconsistent and situs cannot be determined clearly because of the malpositioning of organs, the condition is called “situs ambiguous.” This is very rare, with a prevalence of 1 in 40,000.6
Risk factors
The cause of congenital cardiovascular malformations such as these is not known, but risk factors include positive family history, maternal diabetes, and cocaine use in the first trimester.7
The prevalence of congenital heart disease in patients with situs inversus with dextrocardia is low and ranges from 2% to 5%. This is in contrast to situs solitus with dextrocardia (isolated dextrocardia), which is almost always associated with cardiovascular anomalies.2,4 Kartagener syndrome—the triad of situs inversus, sinusitis, and bronchiectasis—occurs in 25% of people with situs inversus with dextrocardia.4 Situs inversus with levocardia is also frequently associated with cardiac anomalies.5
The major features of dextrocardia on ECG are:
- Negative P wave, QRS complex, and T wave in lead I
- Positive QRS complex in aVR
- Right-axis deviation
- Reversal of R-wave progression in the precordial leads.
Ventricular activation and repolarization are reversed, resulting in a negative QRS complex and an inverted T wave in lead I. The absence of R-wave progression in the precordial leads helps differentiate mirror-image dextrocardia from erroneously reversed limb-electrode placement, which shows normal R-wave progression from V1 to V6 while showing similar features to those seen in dextrocardia in the limb leads.2 In right-sided hookup, the limb electrodes are reversed, and the chest electrodes are recorded from the right precordium.
CORONARY INTERVENTIONS REQUIRE SPECIAL CONSIDERATION
In patients with dextrocardia, coronary interventions can be challenging because of the mirror-image position of the coronary ostia and the aortic arch.8 These patients also need careful imaging, consideration of other associated congenital cardiac abnormalities, and detailed planning before cardiac surgery, including coronary artery bypass grafting.9
Patients with dextrocardia may present with cardiac symptoms localized to the right side of the body and have confusing clinical and diagnostic findings. Keeping dextrocardia and other such anomalies in mind can prevent delay in appropriately directed interventions. In a patient such as ours, the heart on the right side of the chest may indeed be “right.” Still, diagnostic tests to look for disorders encountered with dextrocardia may be necessary.
- Perloff JK. The cardiac malpositions. Am J Cardiol 2011; 108:1352–1361.
- Tanawuttiwat T, Vasaiwala S, Dia M. ECG image of the month. Mirror mirror. Am J Med 2010; 123:34–36.
- Douard R, Feldman A, Bargy F, Loric S, Delmas V. Anomalies of lateralization in man: a case of total situs in-versus. Surg Radiol Anat 2000; 22:293–297.
- Maldjian PD, Saric M. Approach to dextrocardia in adults: review. AJR Am J Roentgenol 2007; 188(suppl 6):S39–S49.
- Gindes L, Hegesh J, Barkai G, Jacobson JM, Achiron R. Isolated levocardia: prenatal diagnosis, clinical im-portance, and literature review. J Ultrasound Med 2007; 26:361–365.
- Abut E, Arman A, Güveli H, et al. Malposition of internal organs: a case of situs ambiguous anomaly in an adult. Turk J Gastroenterol 2003; 14:151–155.
- Kuehl KS, Loffredo C. Risk factors for heart disease associated with abnormal sidedness. Teratology 2002; 66:242–248.
- Aksoy S, Cam N, Gurkan U, Altay S, Bozbay M, Agirbasli M. Primary percutaneous intervention: for acute myo-cardial infarction in a patient with dextrocardia and situs inversus. Tex Heart Inst J 2012; 39:140–141.
- Murtuza B, Gupta P, Goli G, Lall KS. Coronary revascularization in adults with dextrocardia: surgical implications of the anatomic variants. Tex Heart Inst J 2010; 37:633–640.
- Perloff JK. The cardiac malpositions. Am J Cardiol 2011; 108:1352–1361.
- Tanawuttiwat T, Vasaiwala S, Dia M. ECG image of the month. Mirror mirror. Am J Med 2010; 123:34–36.
- Douard R, Feldman A, Bargy F, Loric S, Delmas V. Anomalies of lateralization in man: a case of total situs in-versus. Surg Radiol Anat 2000; 22:293–297.
- Maldjian PD, Saric M. Approach to dextrocardia in adults: review. AJR Am J Roentgenol 2007; 188(suppl 6):S39–S49.
- Gindes L, Hegesh J, Barkai G, Jacobson JM, Achiron R. Isolated levocardia: prenatal diagnosis, clinical im-portance, and literature review. J Ultrasound Med 2007; 26:361–365.
- Abut E, Arman A, Güveli H, et al. Malposition of internal organs: a case of situs ambiguous anomaly in an adult. Turk J Gastroenterol 2003; 14:151–155.
- Kuehl KS, Loffredo C. Risk factors for heart disease associated with abnormal sidedness. Teratology 2002; 66:242–248.
- Aksoy S, Cam N, Gurkan U, Altay S, Bozbay M, Agirbasli M. Primary percutaneous intervention: for acute myo-cardial infarction in a patient with dextrocardia and situs inversus. Tex Heart Inst J 2012; 39:140–141.
- Murtuza B, Gupta P, Goli G, Lall KS. Coronary revascularization in adults with dextrocardia: surgical implications of the anatomic variants. Tex Heart Inst J 2010; 37:633–640.
Can an ARB be given to patients who have had angioedema on an ACE inhibitor?
Current evidence suggests no absolute contraindication to angiotensin receptor blockers (ARBs) in patients who have had angioedema attributable to an angiotensin-converting enzyme (ACE) inhibitor. However, since ARBs can also cause angioedema, they should be prescribed with extreme caution after a thorough risk-benefit analysis and after educating the patient to watch for signs of angioedema while taking the drug.
A GROWING PROBLEM
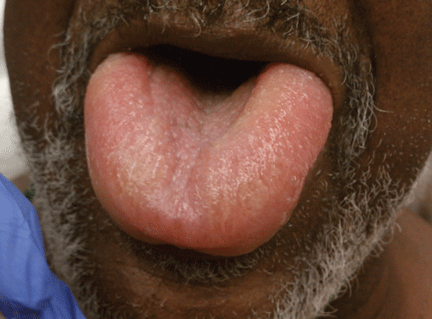
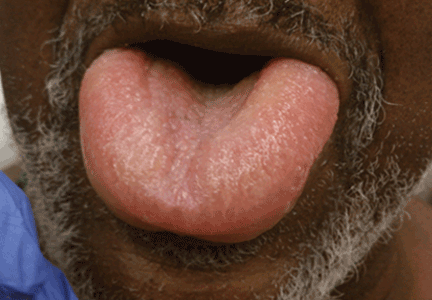
Angioedema is a potentially life-threatening swelling of the skin and subcutaneous tissues, often affecting the lips and tongue (Figure 1), and in some cases interfering with breathing and requiring tracheostomy.1 The incidence rate of angioedema in patients taking ACE inhibitors ranges from 0.1% to 0.7%.2–4 Although this rate may seem low, the widespread and growing use of ACE inhibitors and ARBs in patients with diabetes, diabetic nephropathy, and congestive heart failure5 makes angioedema fairly common in clinical practice.
ACE inhibitor-induced angioedema most commonly occurs within days of initiating therapy, but it also may occur weeks, months, or even years after the start of treatment.1 Patients who are over age 65, black, or female are at higher risk, as are renal transplant recipients taking mTOR inhibitors such as sirolimus. Diabetes appears to be associated with a lower risk.4,6,7 This adverse reaction to ACE inhibitors is thought to be a class side effect, and the future use of this class of drugs would be contraindicated.8,9
ACE inhibitors cause angioedema by direct interference with the degradation of bradykinin, thereby increasing bradykinin levels and potentiating its biologic effect, leading to increased vascular permeability, inflammation, and activation of nociceptors.8
EVIDENCE TO SUPPORT THE USE OF ARBs
ACE inhibitors and ARBs both block the renin-angiotensin-aldosterone pathway and confer similar advantages in patients with congestive heart failure, renal failure, and diabetes. But since ARBs directly inhibit the angiotensin receptor and do not interfere with bradykinin degradation, how they cause angioedema is unclear, and clinicians have questioned whether these agents might be used safely in patients who have had angioedema on an ACE inhibitor.
In a large meta-analysis of randomized clinical trials, Makani et al2 investigated the risk of angioedema with ARB use in 35,479 patients and compared this with other commonly used antihypertensive drugs. The weighted incidence of angioedema was 0.30% with an ACE inhibitor, 0.11% with an ARB, and 0.07% with placebo.2 In seven trials included in this study that compared ARBs with placebo, there was no significant difference in the risk of angioedema. Even in such a large study, the event rate was small, making definite conclusions difficult.
In a retrospective observational study of 4 million patients by Toh et al,3 patients on beta-blockers were used as a reference, and propensity scoring was used to estimate the hazard ratio of angioedema separately for drugs targeting the renin-angiotensin-aldosterone system, including ACE inhibitors and ARBs. The risk of angioedema, as measured by the cumulative incidence and incidence rate, was highest for ACE inhibitors and was similar between ARBs and beta-blockers. The risk of serious angioedema was three times higher with ACE inhibitors than with beta-blockers, and there was no higher risk of serious angioedema with ARBs than with beta-blockers.3
Looking specifically at the use of ARBs in patients who developed angioedema on an ACE inhibitor, Haymore et al10 performed a meta-analysis evaluating only three studies that showed the estimated risk of angioedema with an ARB was between 3.5% and 9.4% in patients with a history of ACE inhibitor-induced angioedema. Later, when the results of the Telmisartan Randomised Assessment Study in ACE Intolerant Subjects With Cardiovascular Disease trial11 were published, the previous meta-analysis was updated12: the risk of angioedema with an ARB was only 2.5% (95% confidence interval 0%–6.6%), and there was no statistically significant difference in the odds (odds ratio 1.1; 95% confidence interval 0.07–17) of angioedema between ARBs and placebo.10,12 Again, these results should be interpreted with caution, as only two patients in the ARB (telmisartan) group and three patients in the placebo group developed angioedema.
In another review, Beavers et al13 advised that the prescribing practitioner should carefully perform a risk-benefit analysis before substituting an ARB in patients with ACE inhibitor-induced angioedema. They concluded that an ARB could be considered in patients who are likely to have a large clinical benefit from an ARB, such as those with heart failure. They also suggested that angioedema related to ARBs was less severe and occurred earlier than with that linked to ACE inhibitors.
No large clinical trial has yet been specifically designed to address the use of ARBs in patients with a history of ACE inhibitor-induced angioedema. The package insert for the ARB losartan mentions that the risk of this adverse reaction might be higher in patients who have had angioedema on an ACE inhibitor. However, the issue of recurrent angioedema is not further addressed for this or other commonly used ARBs.
GENERAL RECOMMENDATIONS
The mechanisms of ARB-induced angioedema are yet unknown. However, studies have shown that the incidence of angioedema while on an ARB is low and is probably comparable to that of placebo.2,3,12–14 And since ARBs share many of the cardiac and renal protective effects of ACE inhibitors, ARBs may be beneficial for patients who discontinue an ACE inhibitor because of adverse effects including angioedema.9,15,16 Based on the discussion above, there is no clear evidence to suggest that ARBs are contraindicated in such patients, especially if there is a compelling indication for an ARB.
The National Kidney Foundation Kidney Disease Outcomes Quality Initiative (NKF KDOQI) guidelines on hypertension in chronic kidney disease recommend caution when substituting an ARB for an ACE inhibitor after angioedema.15 The joint guidelines of the American College of Cardiology and American Heart Association (ACC/AHA) for the diagnosis and management of heart failure in adults advise “extreme caution.”9,16
The risks and benefits of ARB therapy in this setting should be analyzed by the prescribing physician and discussed with the patient. The patient should be closely monitored for the recurrence of angioedema and should be given a clear plan of action should symptoms recur.
OUR ADVICE
In patients with ACE inhibitor-induced angioedema, we recommend the following:
- Determine that the patient truly has one of the evidence-based, compelling indications for an ARB. Carefully weigh the risks and benefits for the individual patient, and discuss the risk of angioedema based on age, race, sex, and medical history, and the availability of immediate medical care should angioedema occur.
- If there is an evidence-based indication for an ARB that outweighs the risk of angioedema, an ARB may be started with caution.
- Specifically discuss with the patient the possibility of recurrence of angioedema while on an ARB, and provide instructions on how to proceed if this should occur.
- Kaplan AP, Greaves MW. Angioedema. J Am Acad Dermatol 2005; 53:373–388.
- Makani H, Messerli FH, Romero J, et al. Meta-analysis of randomized trials of angioedema as an adverse event of renin-angiotensin system inhibitors. Am J Cardiol 2012; 110:383–391.
- Toh S, Reichman ME, Houstoun M, et al. Comparative risk for angioedema associated with the use of drugs that target the renin-angiotensin-aldosterone system. Arch Intern Med 2012; 172:1582–1589.
- Kostis JB, Kim HJ, Rusnak J, et al. Incidence and characteristics of angioedema associated with enalapril. Arch Intern Med 2005; 165:1637–1642.
- Taylor AA, Siragy H, Nesbitt S. Angiotensin receptor blockers: pharmacology, efficacy, and safety. J Clin Hypertens (Greenwich) 2011; 13:677–686.
- Duerr M, Glander P, Diekmann F, Dragun D, Neumayer HH, Budde K. Increased incidence of angioedema with ACE inhibitors in combination with mTOR inhibitors in kidney transplant recipients. Clin J Am Soc Nephrol 2010; 5:703–708.
- Byrd JB, Adam A, Brown NJ. Angiotensin-converting enzyme inhibitor-associated angioedema. Immunol Allergy Clin North Am 2006; 26:725–737.
- Inomata N. Recent advances in drug-induced angioedema. Allergol Int 2012; 61:545–557.
- Hunt SA, Abraham WT, Chin MH, et al; American College of Cardiology Foundation; American Heart Association. 2009 Focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines Developed in Collaboration With the International Society for Heart and Lung Transplantation. J Am Coll Cardiol 2009; 53:e1–e90.
- Haymore BR, Yoon J, Mikita CP, Klote MM, DeZee KJ. Risk of angioedema with angiotensin receptor blockers in patients with prior angioedema associated with angiotensin-converting enzyme inhibitors: a meta-analysis. Ann Allergy Asthma Immunol 2008; 101:495–499.
- Telmisartan Randomised Assessment Study in ACE Intolerant Subjects with Cardiovascular Disease (TRANSCEND) Investigators. Effects of the angiotensin-receptor blocker telmisartan on cardiovascular events in high-risk patients intolerant to angiotensin-converting enzyme inhibitors: a randomised controlled trial. Lancet 2008; 372:1174–1183.
- Haymore BR, DeZee KJ. Use of angiotensin receptor blockers after angioedema with an angiotensin-converting enzyme inhibitor. Ann Allergy Asthma Immunol 2009; 103:83–84.
- Beavers CJ, Dunn SP, Macaulay TE. The role of angiotensin receptor blockers in patients with angiotensin-converting enzyme inhibitor-induced angioedema. Ann Pharmacother 2011; 45:520–524.
- Caldeira D, David C, Sampaio C. Tolerability of angiotensin-receptor blockers in patients with intolerance to angiotensin-converting enzyme inhibitors: a systematic review and meta-analysis. Am J Cardiovasc Drugs 2012; 12:263–277.
- Kidney Disease Outcomes Quality Initiative (K/DOQI). K/DOQI clinical practice guidelines on hypertension and antihypertensive agents in chronic kidney disease. Am J Kidney Dis 2004; 43(suppl 1):S1–S290.
- Smith SC, Benjamin EJ, Bonow RO, et al. AHA/ACCF secondary prevention and risk reduction therapy for patients with coronary and other atherosclerotic vascular disease: 2011 update: a guideline from the American Heart Association and American College of Cardiology Foundation endorsed by the World Heart Federation and the Preventive Cardiovascular Nurses Association. J Am Coll Cardiol 2011; 58:2432–2446.
Current evidence suggests no absolute contraindication to angiotensin receptor blockers (ARBs) in patients who have had angioedema attributable to an angiotensin-converting enzyme (ACE) inhibitor. However, since ARBs can also cause angioedema, they should be prescribed with extreme caution after a thorough risk-benefit analysis and after educating the patient to watch for signs of angioedema while taking the drug.
A GROWING PROBLEM
Angioedema is a potentially life-threatening swelling of the skin and subcutaneous tissues, often affecting the lips and tongue (Figure 1), and in some cases interfering with breathing and requiring tracheostomy.1 The incidence rate of angioedema in patients taking ACE inhibitors ranges from 0.1% to 0.7%.2–4 Although this rate may seem low, the widespread and growing use of ACE inhibitors and ARBs in patients with diabetes, diabetic nephropathy, and congestive heart failure5 makes angioedema fairly common in clinical practice.
ACE inhibitor-induced angioedema most commonly occurs within days of initiating therapy, but it also may occur weeks, months, or even years after the start of treatment.1 Patients who are over age 65, black, or female are at higher risk, as are renal transplant recipients taking mTOR inhibitors such as sirolimus. Diabetes appears to be associated with a lower risk.4,6,7 This adverse reaction to ACE inhibitors is thought to be a class side effect, and the future use of this class of drugs would be contraindicated.8,9
ACE inhibitors cause angioedema by direct interference with the degradation of bradykinin, thereby increasing bradykinin levels and potentiating its biologic effect, leading to increased vascular permeability, inflammation, and activation of nociceptors.8
EVIDENCE TO SUPPORT THE USE OF ARBs
ACE inhibitors and ARBs both block the renin-angiotensin-aldosterone pathway and confer similar advantages in patients with congestive heart failure, renal failure, and diabetes. But since ARBs directly inhibit the angiotensin receptor and do not interfere with bradykinin degradation, how they cause angioedema is unclear, and clinicians have questioned whether these agents might be used safely in patients who have had angioedema on an ACE inhibitor.
In a large meta-analysis of randomized clinical trials, Makani et al2 investigated the risk of angioedema with ARB use in 35,479 patients and compared this with other commonly used antihypertensive drugs. The weighted incidence of angioedema was 0.30% with an ACE inhibitor, 0.11% with an ARB, and 0.07% with placebo.2 In seven trials included in this study that compared ARBs with placebo, there was no significant difference in the risk of angioedema. Even in such a large study, the event rate was small, making definite conclusions difficult.
In a retrospective observational study of 4 million patients by Toh et al,3 patients on beta-blockers were used as a reference, and propensity scoring was used to estimate the hazard ratio of angioedema separately for drugs targeting the renin-angiotensin-aldosterone system, including ACE inhibitors and ARBs. The risk of angioedema, as measured by the cumulative incidence and incidence rate, was highest for ACE inhibitors and was similar between ARBs and beta-blockers. The risk of serious angioedema was three times higher with ACE inhibitors than with beta-blockers, and there was no higher risk of serious angioedema with ARBs than with beta-blockers.3
Looking specifically at the use of ARBs in patients who developed angioedema on an ACE inhibitor, Haymore et al10 performed a meta-analysis evaluating only three studies that showed the estimated risk of angioedema with an ARB was between 3.5% and 9.4% in patients with a history of ACE inhibitor-induced angioedema. Later, when the results of the Telmisartan Randomised Assessment Study in ACE Intolerant Subjects With Cardiovascular Disease trial11 were published, the previous meta-analysis was updated12: the risk of angioedema with an ARB was only 2.5% (95% confidence interval 0%–6.6%), and there was no statistically significant difference in the odds (odds ratio 1.1; 95% confidence interval 0.07–17) of angioedema between ARBs and placebo.10,12 Again, these results should be interpreted with caution, as only two patients in the ARB (telmisartan) group and three patients in the placebo group developed angioedema.
In another review, Beavers et al13 advised that the prescribing practitioner should carefully perform a risk-benefit analysis before substituting an ARB in patients with ACE inhibitor-induced angioedema. They concluded that an ARB could be considered in patients who are likely to have a large clinical benefit from an ARB, such as those with heart failure. They also suggested that angioedema related to ARBs was less severe and occurred earlier than with that linked to ACE inhibitors.
No large clinical trial has yet been specifically designed to address the use of ARBs in patients with a history of ACE inhibitor-induced angioedema. The package insert for the ARB losartan mentions that the risk of this adverse reaction might be higher in patients who have had angioedema on an ACE inhibitor. However, the issue of recurrent angioedema is not further addressed for this or other commonly used ARBs.
GENERAL RECOMMENDATIONS
The mechanisms of ARB-induced angioedema are yet unknown. However, studies have shown that the incidence of angioedema while on an ARB is low and is probably comparable to that of placebo.2,3,12–14 And since ARBs share many of the cardiac and renal protective effects of ACE inhibitors, ARBs may be beneficial for patients who discontinue an ACE inhibitor because of adverse effects including angioedema.9,15,16 Based on the discussion above, there is no clear evidence to suggest that ARBs are contraindicated in such patients, especially if there is a compelling indication for an ARB.
The National Kidney Foundation Kidney Disease Outcomes Quality Initiative (NKF KDOQI) guidelines on hypertension in chronic kidney disease recommend caution when substituting an ARB for an ACE inhibitor after angioedema.15 The joint guidelines of the American College of Cardiology and American Heart Association (ACC/AHA) for the diagnosis and management of heart failure in adults advise “extreme caution.”9,16
The risks and benefits of ARB therapy in this setting should be analyzed by the prescribing physician and discussed with the patient. The patient should be closely monitored for the recurrence of angioedema and should be given a clear plan of action should symptoms recur.
OUR ADVICE
In patients with ACE inhibitor-induced angioedema, we recommend the following:
- Determine that the patient truly has one of the evidence-based, compelling indications for an ARB. Carefully weigh the risks and benefits for the individual patient, and discuss the risk of angioedema based on age, race, sex, and medical history, and the availability of immediate medical care should angioedema occur.
- If there is an evidence-based indication for an ARB that outweighs the risk of angioedema, an ARB may be started with caution.
- Specifically discuss with the patient the possibility of recurrence of angioedema while on an ARB, and provide instructions on how to proceed if this should occur.
Current evidence suggests no absolute contraindication to angiotensin receptor blockers (ARBs) in patients who have had angioedema attributable to an angiotensin-converting enzyme (ACE) inhibitor. However, since ARBs can also cause angioedema, they should be prescribed with extreme caution after a thorough risk-benefit analysis and after educating the patient to watch for signs of angioedema while taking the drug.
A GROWING PROBLEM
Angioedema is a potentially life-threatening swelling of the skin and subcutaneous tissues, often affecting the lips and tongue (Figure 1), and in some cases interfering with breathing and requiring tracheostomy.1 The incidence rate of angioedema in patients taking ACE inhibitors ranges from 0.1% to 0.7%.2–4 Although this rate may seem low, the widespread and growing use of ACE inhibitors and ARBs in patients with diabetes, diabetic nephropathy, and congestive heart failure5 makes angioedema fairly common in clinical practice.
ACE inhibitor-induced angioedema most commonly occurs within days of initiating therapy, but it also may occur weeks, months, or even years after the start of treatment.1 Patients who are over age 65, black, or female are at higher risk, as are renal transplant recipients taking mTOR inhibitors such as sirolimus. Diabetes appears to be associated with a lower risk.4,6,7 This adverse reaction to ACE inhibitors is thought to be a class side effect, and the future use of this class of drugs would be contraindicated.8,9
ACE inhibitors cause angioedema by direct interference with the degradation of bradykinin, thereby increasing bradykinin levels and potentiating its biologic effect, leading to increased vascular permeability, inflammation, and activation of nociceptors.8
EVIDENCE TO SUPPORT THE USE OF ARBs
ACE inhibitors and ARBs both block the renin-angiotensin-aldosterone pathway and confer similar advantages in patients with congestive heart failure, renal failure, and diabetes. But since ARBs directly inhibit the angiotensin receptor and do not interfere with bradykinin degradation, how they cause angioedema is unclear, and clinicians have questioned whether these agents might be used safely in patients who have had angioedema on an ACE inhibitor.
In a large meta-analysis of randomized clinical trials, Makani et al2 investigated the risk of angioedema with ARB use in 35,479 patients and compared this with other commonly used antihypertensive drugs. The weighted incidence of angioedema was 0.30% with an ACE inhibitor, 0.11% with an ARB, and 0.07% with placebo.2 In seven trials included in this study that compared ARBs with placebo, there was no significant difference in the risk of angioedema. Even in such a large study, the event rate was small, making definite conclusions difficult.
In a retrospective observational study of 4 million patients by Toh et al,3 patients on beta-blockers were used as a reference, and propensity scoring was used to estimate the hazard ratio of angioedema separately for drugs targeting the renin-angiotensin-aldosterone system, including ACE inhibitors and ARBs. The risk of angioedema, as measured by the cumulative incidence and incidence rate, was highest for ACE inhibitors and was similar between ARBs and beta-blockers. The risk of serious angioedema was three times higher with ACE inhibitors than with beta-blockers, and there was no higher risk of serious angioedema with ARBs than with beta-blockers.3
Looking specifically at the use of ARBs in patients who developed angioedema on an ACE inhibitor, Haymore et al10 performed a meta-analysis evaluating only three studies that showed the estimated risk of angioedema with an ARB was between 3.5% and 9.4% in patients with a history of ACE inhibitor-induced angioedema. Later, when the results of the Telmisartan Randomised Assessment Study in ACE Intolerant Subjects With Cardiovascular Disease trial11 were published, the previous meta-analysis was updated12: the risk of angioedema with an ARB was only 2.5% (95% confidence interval 0%–6.6%), and there was no statistically significant difference in the odds (odds ratio 1.1; 95% confidence interval 0.07–17) of angioedema between ARBs and placebo.10,12 Again, these results should be interpreted with caution, as only two patients in the ARB (telmisartan) group and three patients in the placebo group developed angioedema.
In another review, Beavers et al13 advised that the prescribing practitioner should carefully perform a risk-benefit analysis before substituting an ARB in patients with ACE inhibitor-induced angioedema. They concluded that an ARB could be considered in patients who are likely to have a large clinical benefit from an ARB, such as those with heart failure. They also suggested that angioedema related to ARBs was less severe and occurred earlier than with that linked to ACE inhibitors.
No large clinical trial has yet been specifically designed to address the use of ARBs in patients with a history of ACE inhibitor-induced angioedema. The package insert for the ARB losartan mentions that the risk of this adverse reaction might be higher in patients who have had angioedema on an ACE inhibitor. However, the issue of recurrent angioedema is not further addressed for this or other commonly used ARBs.
GENERAL RECOMMENDATIONS
The mechanisms of ARB-induced angioedema are yet unknown. However, studies have shown that the incidence of angioedema while on an ARB is low and is probably comparable to that of placebo.2,3,12–14 And since ARBs share many of the cardiac and renal protective effects of ACE inhibitors, ARBs may be beneficial for patients who discontinue an ACE inhibitor because of adverse effects including angioedema.9,15,16 Based on the discussion above, there is no clear evidence to suggest that ARBs are contraindicated in such patients, especially if there is a compelling indication for an ARB.
The National Kidney Foundation Kidney Disease Outcomes Quality Initiative (NKF KDOQI) guidelines on hypertension in chronic kidney disease recommend caution when substituting an ARB for an ACE inhibitor after angioedema.15 The joint guidelines of the American College of Cardiology and American Heart Association (ACC/AHA) for the diagnosis and management of heart failure in adults advise “extreme caution.”9,16
The risks and benefits of ARB therapy in this setting should be analyzed by the prescribing physician and discussed with the patient. The patient should be closely monitored for the recurrence of angioedema and should be given a clear plan of action should symptoms recur.
OUR ADVICE
In patients with ACE inhibitor-induced angioedema, we recommend the following:
- Determine that the patient truly has one of the evidence-based, compelling indications for an ARB. Carefully weigh the risks and benefits for the individual patient, and discuss the risk of angioedema based on age, race, sex, and medical history, and the availability of immediate medical care should angioedema occur.
- If there is an evidence-based indication for an ARB that outweighs the risk of angioedema, an ARB may be started with caution.
- Specifically discuss with the patient the possibility of recurrence of angioedema while on an ARB, and provide instructions on how to proceed if this should occur.
- Kaplan AP, Greaves MW. Angioedema. J Am Acad Dermatol 2005; 53:373–388.
- Makani H, Messerli FH, Romero J, et al. Meta-analysis of randomized trials of angioedema as an adverse event of renin-angiotensin system inhibitors. Am J Cardiol 2012; 110:383–391.
- Toh S, Reichman ME, Houstoun M, et al. Comparative risk for angioedema associated with the use of drugs that target the renin-angiotensin-aldosterone system. Arch Intern Med 2012; 172:1582–1589.
- Kostis JB, Kim HJ, Rusnak J, et al. Incidence and characteristics of angioedema associated with enalapril. Arch Intern Med 2005; 165:1637–1642.
- Taylor AA, Siragy H, Nesbitt S. Angiotensin receptor blockers: pharmacology, efficacy, and safety. J Clin Hypertens (Greenwich) 2011; 13:677–686.
- Duerr M, Glander P, Diekmann F, Dragun D, Neumayer HH, Budde K. Increased incidence of angioedema with ACE inhibitors in combination with mTOR inhibitors in kidney transplant recipients. Clin J Am Soc Nephrol 2010; 5:703–708.
- Byrd JB, Adam A, Brown NJ. Angiotensin-converting enzyme inhibitor-associated angioedema. Immunol Allergy Clin North Am 2006; 26:725–737.
- Inomata N. Recent advances in drug-induced angioedema. Allergol Int 2012; 61:545–557.
- Hunt SA, Abraham WT, Chin MH, et al; American College of Cardiology Foundation; American Heart Association. 2009 Focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines Developed in Collaboration With the International Society for Heart and Lung Transplantation. J Am Coll Cardiol 2009; 53:e1–e90.
- Haymore BR, Yoon J, Mikita CP, Klote MM, DeZee KJ. Risk of angioedema with angiotensin receptor blockers in patients with prior angioedema associated with angiotensin-converting enzyme inhibitors: a meta-analysis. Ann Allergy Asthma Immunol 2008; 101:495–499.
- Telmisartan Randomised Assessment Study in ACE Intolerant Subjects with Cardiovascular Disease (TRANSCEND) Investigators. Effects of the angiotensin-receptor blocker telmisartan on cardiovascular events in high-risk patients intolerant to angiotensin-converting enzyme inhibitors: a randomised controlled trial. Lancet 2008; 372:1174–1183.
- Haymore BR, DeZee KJ. Use of angiotensin receptor blockers after angioedema with an angiotensin-converting enzyme inhibitor. Ann Allergy Asthma Immunol 2009; 103:83–84.
- Beavers CJ, Dunn SP, Macaulay TE. The role of angiotensin receptor blockers in patients with angiotensin-converting enzyme inhibitor-induced angioedema. Ann Pharmacother 2011; 45:520–524.
- Caldeira D, David C, Sampaio C. Tolerability of angiotensin-receptor blockers in patients with intolerance to angiotensin-converting enzyme inhibitors: a systematic review and meta-analysis. Am J Cardiovasc Drugs 2012; 12:263–277.
- Kidney Disease Outcomes Quality Initiative (K/DOQI). K/DOQI clinical practice guidelines on hypertension and antihypertensive agents in chronic kidney disease. Am J Kidney Dis 2004; 43(suppl 1):S1–S290.
- Smith SC, Benjamin EJ, Bonow RO, et al. AHA/ACCF secondary prevention and risk reduction therapy for patients with coronary and other atherosclerotic vascular disease: 2011 update: a guideline from the American Heart Association and American College of Cardiology Foundation endorsed by the World Heart Federation and the Preventive Cardiovascular Nurses Association. J Am Coll Cardiol 2011; 58:2432–2446.
- Kaplan AP, Greaves MW. Angioedema. J Am Acad Dermatol 2005; 53:373–388.
- Makani H, Messerli FH, Romero J, et al. Meta-analysis of randomized trials of angioedema as an adverse event of renin-angiotensin system inhibitors. Am J Cardiol 2012; 110:383–391.
- Toh S, Reichman ME, Houstoun M, et al. Comparative risk for angioedema associated with the use of drugs that target the renin-angiotensin-aldosterone system. Arch Intern Med 2012; 172:1582–1589.
- Kostis JB, Kim HJ, Rusnak J, et al. Incidence and characteristics of angioedema associated with enalapril. Arch Intern Med 2005; 165:1637–1642.
- Taylor AA, Siragy H, Nesbitt S. Angiotensin receptor blockers: pharmacology, efficacy, and safety. J Clin Hypertens (Greenwich) 2011; 13:677–686.
- Duerr M, Glander P, Diekmann F, Dragun D, Neumayer HH, Budde K. Increased incidence of angioedema with ACE inhibitors in combination with mTOR inhibitors in kidney transplant recipients. Clin J Am Soc Nephrol 2010; 5:703–708.
- Byrd JB, Adam A, Brown NJ. Angiotensin-converting enzyme inhibitor-associated angioedema. Immunol Allergy Clin North Am 2006; 26:725–737.
- Inomata N. Recent advances in drug-induced angioedema. Allergol Int 2012; 61:545–557.
- Hunt SA, Abraham WT, Chin MH, et al; American College of Cardiology Foundation; American Heart Association. 2009 Focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines Developed in Collaboration With the International Society for Heart and Lung Transplantation. J Am Coll Cardiol 2009; 53:e1–e90.
- Haymore BR, Yoon J, Mikita CP, Klote MM, DeZee KJ. Risk of angioedema with angiotensin receptor blockers in patients with prior angioedema associated with angiotensin-converting enzyme inhibitors: a meta-analysis. Ann Allergy Asthma Immunol 2008; 101:495–499.
- Telmisartan Randomised Assessment Study in ACE Intolerant Subjects with Cardiovascular Disease (TRANSCEND) Investigators. Effects of the angiotensin-receptor blocker telmisartan on cardiovascular events in high-risk patients intolerant to angiotensin-converting enzyme inhibitors: a randomised controlled trial. Lancet 2008; 372:1174–1183.
- Haymore BR, DeZee KJ. Use of angiotensin receptor blockers after angioedema with an angiotensin-converting enzyme inhibitor. Ann Allergy Asthma Immunol 2009; 103:83–84.
- Beavers CJ, Dunn SP, Macaulay TE. The role of angiotensin receptor blockers in patients with angiotensin-converting enzyme inhibitor-induced angioedema. Ann Pharmacother 2011; 45:520–524.
- Caldeira D, David C, Sampaio C. Tolerability of angiotensin-receptor blockers in patients with intolerance to angiotensin-converting enzyme inhibitors: a systematic review and meta-analysis. Am J Cardiovasc Drugs 2012; 12:263–277.
- Kidney Disease Outcomes Quality Initiative (K/DOQI). K/DOQI clinical practice guidelines on hypertension and antihypertensive agents in chronic kidney disease. Am J Kidney Dis 2004; 43(suppl 1):S1–S290.
- Smith SC, Benjamin EJ, Bonow RO, et al. AHA/ACCF secondary prevention and risk reduction therapy for patients with coronary and other atherosclerotic vascular disease: 2011 update: a guideline from the American Heart Association and American College of Cardiology Foundation endorsed by the World Heart Federation and the Preventive Cardiovascular Nurses Association. J Am Coll Cardiol 2011; 58:2432–2446.
Implications of a prominent R wave in V1
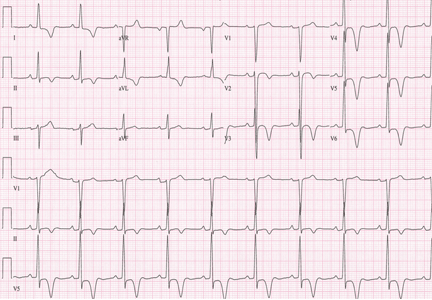
A 19-year-old woman with no significant cardiac or pulmonary history presented with exertional dyspnea, which had begun a few months earlier. Auscultation revealed a loud pulmonary component of the second heart sound and a diastolic murmur heard along the upper left sternal border. Her 12-lead electrocardiogram is shown in Figure 1.
Q: Which of the following can cause prominent R waves in lead V1?
- Normal variant in young adults
- Wolff-Parkinson-White syndrome
- Posterior wall myocardial infarction
- Right ventricular hypertrophy
- All of the above
A: The correct answer is all of the above.
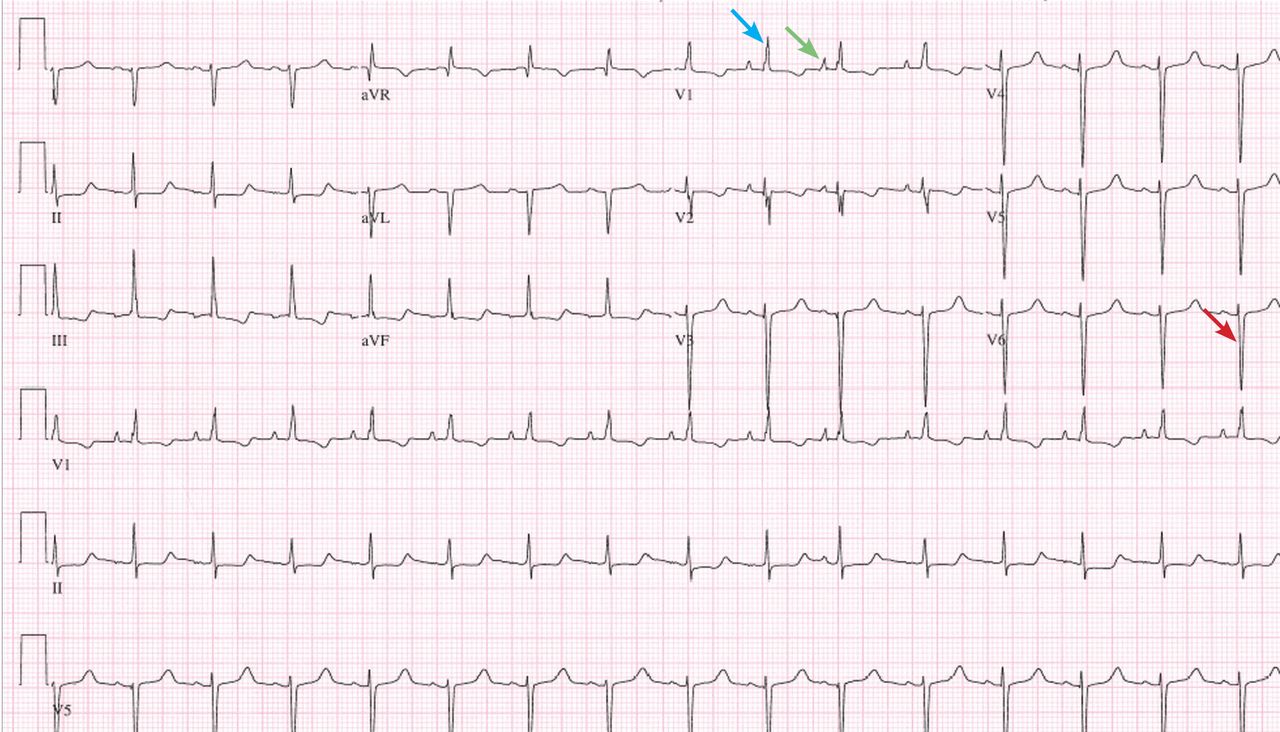
The patient’s electrocardiogram shows a right atrial abnormality and right ventricular hypertrophy. Right atrial enlargement is evidenced by a prominent initial P wave in V1 with an amplitude of at least 1.5 mm (0.15 mV). A P wave taller than 2.5 mm (0.25 mV) in lead II may also suggest a right atrial abnormality.1
Multiple criteria exist for the diagnosis of right ventricular hypertrophy. Tall R waves in V1 with an R/S ratio greater than 1 (ie, the R wave amplitude is more than the S wave depth) is commonly used.2 Deep S waves with an R/S ratio less than 1 in V6 is another criterion. Tall R waves of amplitude greater than 7 mm in V1 by themselves may represent right ventricular hypertrophy. Most of the electrocardiographic criteria are specific but not sensitive for this diagnosis.3

Other causes of tall R waves in V1 are given in Table 1.
Q: Which of the following diseases can present with an electrocardiographic pattern of right ventricular hypertrophy in young patients?
- Pulmonary hypertension
- Atrial septal defect
- Tetralogy of Fallot
- Pulmonary stenosis
- All of the above
A: The correct answer is all of the above.4
Our patient underwent multiple investigations. On echocardiography, her estimated right ventricular pressure was 80 mm Hg, and on cardiac catheterization her mean pulmonary arterial pressure was 55 mm Hg and her pulmonary capillary wedge pressure was 6 mm Hg. She was diagnosed with pulmonary arterial hypertension, which was the cause of her right ventricular hypertrophy. She eventually underwent bilateral lung transplantation.
- Hancock EW, Deal BJ, Mirvis DM, et al; American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; American College of Cardiology Foundation; Heart Rhythm Society. AHA/ ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part V: electrocardiogram changes associated with cardiac chamber hypertrophy: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society: endorsed by the International Society for Computerized Electrocardiology. Circulation 2009; 119:e251–e261.
- Milnor WR. Electrocardiogram and vectorcardiogram in right ventricular hypertrophy and right bundle-branch block. Circulation 1957; 16:348–367.
- Lehtonen J, Sutinen S, Ikäheimo M, Pääkkö P. Electrocardiographic criteria for the diagnosis of right ventricular hypertrophy verified at autopsy. Chest 1988; 93:839–842.
- Webb G, Gatzoulis MA. Atrial septal defects in the adult: recent progress and overview. Circulation 2006; 114:1645–1653.
A 19-year-old woman with no significant cardiac or pulmonary history presented with exertional dyspnea, which had begun a few months earlier. Auscultation revealed a loud pulmonary component of the second heart sound and a diastolic murmur heard along the upper left sternal border. Her 12-lead electrocardiogram is shown in Figure 1.
Q: Which of the following can cause prominent R waves in lead V1?
- Normal variant in young adults
- Wolff-Parkinson-White syndrome
- Posterior wall myocardial infarction
- Right ventricular hypertrophy
- All of the above
A: The correct answer is all of the above.
The patient’s electrocardiogram shows a right atrial abnormality and right ventricular hypertrophy. Right atrial enlargement is evidenced by a prominent initial P wave in V1 with an amplitude of at least 1.5 mm (0.15 mV). A P wave taller than 2.5 mm (0.25 mV) in lead II may also suggest a right atrial abnormality.1
Multiple criteria exist for the diagnosis of right ventricular hypertrophy. Tall R waves in V1 with an R/S ratio greater than 1 (ie, the R wave amplitude is more than the S wave depth) is commonly used.2 Deep S waves with an R/S ratio less than 1 in V6 is another criterion. Tall R waves of amplitude greater than 7 mm in V1 by themselves may represent right ventricular hypertrophy. Most of the electrocardiographic criteria are specific but not sensitive for this diagnosis.3
Other causes of tall R waves in V1 are given in Table 1.
Q: Which of the following diseases can present with an electrocardiographic pattern of right ventricular hypertrophy in young patients?
- Pulmonary hypertension
- Atrial septal defect
- Tetralogy of Fallot
- Pulmonary stenosis
- All of the above
A: The correct answer is all of the above.4
Our patient underwent multiple investigations. On echocardiography, her estimated right ventricular pressure was 80 mm Hg, and on cardiac catheterization her mean pulmonary arterial pressure was 55 mm Hg and her pulmonary capillary wedge pressure was 6 mm Hg. She was diagnosed with pulmonary arterial hypertension, which was the cause of her right ventricular hypertrophy. She eventually underwent bilateral lung transplantation.
A 19-year-old woman with no significant cardiac or pulmonary history presented with exertional dyspnea, which had begun a few months earlier. Auscultation revealed a loud pulmonary component of the second heart sound and a diastolic murmur heard along the upper left sternal border. Her 12-lead electrocardiogram is shown in Figure 1.
Q: Which of the following can cause prominent R waves in lead V1?
- Normal variant in young adults
- Wolff-Parkinson-White syndrome
- Posterior wall myocardial infarction
- Right ventricular hypertrophy
- All of the above
A: The correct answer is all of the above.
The patient’s electrocardiogram shows a right atrial abnormality and right ventricular hypertrophy. Right atrial enlargement is evidenced by a prominent initial P wave in V1 with an amplitude of at least 1.5 mm (0.15 mV). A P wave taller than 2.5 mm (0.25 mV) in lead II may also suggest a right atrial abnormality.1
Multiple criteria exist for the diagnosis of right ventricular hypertrophy. Tall R waves in V1 with an R/S ratio greater than 1 (ie, the R wave amplitude is more than the S wave depth) is commonly used.2 Deep S waves with an R/S ratio less than 1 in V6 is another criterion. Tall R waves of amplitude greater than 7 mm in V1 by themselves may represent right ventricular hypertrophy. Most of the electrocardiographic criteria are specific but not sensitive for this diagnosis.3
Other causes of tall R waves in V1 are given in Table 1.
Q: Which of the following diseases can present with an electrocardiographic pattern of right ventricular hypertrophy in young patients?
- Pulmonary hypertension
- Atrial septal defect
- Tetralogy of Fallot
- Pulmonary stenosis
- All of the above
A: The correct answer is all of the above.4
Our patient underwent multiple investigations. On echocardiography, her estimated right ventricular pressure was 80 mm Hg, and on cardiac catheterization her mean pulmonary arterial pressure was 55 mm Hg and her pulmonary capillary wedge pressure was 6 mm Hg. She was diagnosed with pulmonary arterial hypertension, which was the cause of her right ventricular hypertrophy. She eventually underwent bilateral lung transplantation.
- Hancock EW, Deal BJ, Mirvis DM, et al; American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; American College of Cardiology Foundation; Heart Rhythm Society. AHA/ ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part V: electrocardiogram changes associated with cardiac chamber hypertrophy: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society: endorsed by the International Society for Computerized Electrocardiology. Circulation 2009; 119:e251–e261.
- Milnor WR. Electrocardiogram and vectorcardiogram in right ventricular hypertrophy and right bundle-branch block. Circulation 1957; 16:348–367.
- Lehtonen J, Sutinen S, Ikäheimo M, Pääkkö P. Electrocardiographic criteria for the diagnosis of right ventricular hypertrophy verified at autopsy. Chest 1988; 93:839–842.
- Webb G, Gatzoulis MA. Atrial septal defects in the adult: recent progress and overview. Circulation 2006; 114:1645–1653.
- Hancock EW, Deal BJ, Mirvis DM, et al; American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; American College of Cardiology Foundation; Heart Rhythm Society. AHA/ ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part V: electrocardiogram changes associated with cardiac chamber hypertrophy: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society: endorsed by the International Society for Computerized Electrocardiology. Circulation 2009; 119:e251–e261.
- Milnor WR. Electrocardiogram and vectorcardiogram in right ventricular hypertrophy and right bundle-branch block. Circulation 1957; 16:348–367.
- Lehtonen J, Sutinen S, Ikäheimo M, Pääkkö P. Electrocardiographic criteria for the diagnosis of right ventricular hypertrophy verified at autopsy. Chest 1988; 93:839–842.
- Webb G, Gatzoulis MA. Atrial septal defects in the adult: recent progress and overview. Circulation 2006; 114:1645–1653.
Giant inverted T waves
A 48-year-old man with hypertension was being evaluated for a noncardiac issue (progressive multifocal leukoencephalopathy). He had been an active runner and did not have any cardiovascular symptoms at the time. The electrocardiogram (ECG) shown in Figure 1 was a routine study done as a part of that evaluation. His cardiovascular examination was unremarkable, without murmur, S3, or S4. His pulse was regular at 72 beats per minute, and his blood pressure was 112/76 mm Hg.
Q: Which of the following electrocardiographic findings suggest left ventricular hypertrophy?
- Sum of the S wave in V1 and the R wave in V6 ≥ 35 mm
- Sum of the S wave in V3 and the R wave in aVL > 28 mm (men)
- Sum of the S wave in V3 and the R wave in aVL > 20 mm (women)
- All of the above
A: The correct answer is all of the above.1,2
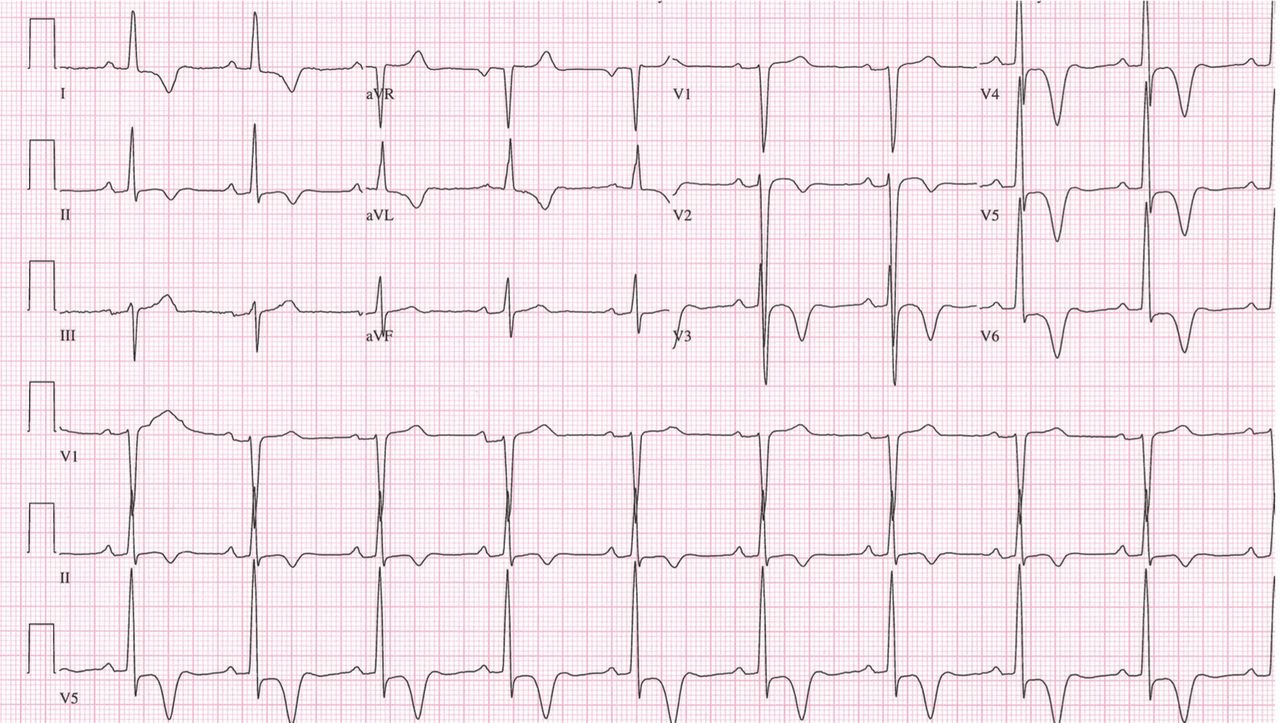
Our patient’s ECG shows sinus bradycardia and left ventricular hypertrophy, suggested by prominent voltage (sum of S in V1 and R in V6 ≥ 35 mm) and supported by ST-segment and T-wave changes in the lateral and midprecordial leads. Classic changes of left ventricular hypertrophy often include increased voltage and downsloping ST-segment depression with negative T waves in V5 and V6 (secondary repolarization changes or “strain” pattern).
Notable on this tracing are the large, asymmetric negative T waves in leads V3 through V6. Giant T waves are defined as negative T waves with voltage greater than 10 mm.3 Although there is no specific pattern of ventricular hypertrophy on an ECG that establishes the diagnosis of hypertrophic cardiomyopathy, left ventricular hypertrophy with T waves of this quality suggest the possibility of hypertrophic cardiomyopathy with apical hypertrophy.
Q: What are the other causes of giant negative T waves?
- Subarachnoid hemorrhage
- Complete heart block
- Non-Q-wave myocardial infarction
- All of the above

A: The correct answer is all of the above. Additional causes of dramatic T-wave inversion are listed in Table 1. Clinically, non-Q-wave myocardial infarction with T-wave changes and acute central nervous system injury are probably the most commonly seen.4
Echocardiography in this patient revealed severe apical hypertrophy of the ventricle with distal cavity obliteration. The left ventricular outflow-tract gradient was normal. The mitral valve appeared normal, and there was no resting systolic anterior motion.
Cardiac magnetic resonance imaging showed the apical variant of hypertrophic cardiomyopathy but no evidence of left ventricular noncompaction, which is a differential diagnosis of apical hypertrophic obstructive cardiomyopathy. This disease was first described in Japan by Yamaguchi et al5 and Sakamoto et al6 and is regarded as a subgroup of nonobstructive hypertrophic cardiomyopathy. The prognosis of apical hypertrophic cardiomyopathy with regard to sudden cardiac death is believed to be better than that of other forms of hypertrophic cardiomyopathy.3
- Sokolow M, Lyon TP. The ventricular complex in left ventricular hypertrophy as obtained by unipolar precordial and limb leads. 1949. Ann Noninvasive Electrocardiol 2001; 6:343–368.
- Casale PN, Devereux RB, Alonso DR, Campo E, Kligfield P. Improved sex-specific criteria of left ventricular hypertrophy for clinical and computer interpretation of electrocardiograms: validation with autopsy findings. Circulation 1987; 75:565–572.
- Eriksson MJ, Sonnenberg B, Woo A, et al. Long-term outcome in patients with apical hypertrophic cardiomyopathy. J Am Coll Cardiol 2002; 39:638–645.
- Jacobson D, Schrire V. Giant T wave inversion. Br Heart J 1966; 28:768–775.
- Yamaguchi H, Ishimura T, Nishiyama S, et al. Hypertrophic nonobstructive cardiomyopathy with giant negative T waves (apical hypertrophy): ventriculographic and echocardiographic features in 30 patients. Am J Cardiol 1979; 44:401–412.
- Sakamoto T, Tei C, Murayama M, Ichiyasu H, Hada Y. Giant T wave inversion as a manifestation of asymmetrical apical hypertrophy (AAH) of the left ventricle. Echocardiographic and ultrasonocardiotomographic study. Jpn Heart J 1976; 17:611–629.
A 48-year-old man with hypertension was being evaluated for a noncardiac issue (progressive multifocal leukoencephalopathy). He had been an active runner and did not have any cardiovascular symptoms at the time. The electrocardiogram (ECG) shown in Figure 1 was a routine study done as a part of that evaluation. His cardiovascular examination was unremarkable, without murmur, S3, or S4. His pulse was regular at 72 beats per minute, and his blood pressure was 112/76 mm Hg.
Q: Which of the following electrocardiographic findings suggest left ventricular hypertrophy?
- Sum of the S wave in V1 and the R wave in V6 ≥ 35 mm
- Sum of the S wave in V3 and the R wave in aVL > 28 mm (men)
- Sum of the S wave in V3 and the R wave in aVL > 20 mm (women)
- All of the above
A: The correct answer is all of the above.1,2
Our patient’s ECG shows sinus bradycardia and left ventricular hypertrophy, suggested by prominent voltage (sum of S in V1 and R in V6 ≥ 35 mm) and supported by ST-segment and T-wave changes in the lateral and midprecordial leads. Classic changes of left ventricular hypertrophy often include increased voltage and downsloping ST-segment depression with negative T waves in V5 and V6 (secondary repolarization changes or “strain” pattern).
Notable on this tracing are the large, asymmetric negative T waves in leads V3 through V6. Giant T waves are defined as negative T waves with voltage greater than 10 mm.3 Although there is no specific pattern of ventricular hypertrophy on an ECG that establishes the diagnosis of hypertrophic cardiomyopathy, left ventricular hypertrophy with T waves of this quality suggest the possibility of hypertrophic cardiomyopathy with apical hypertrophy.
Q: What are the other causes of giant negative T waves?
- Subarachnoid hemorrhage
- Complete heart block
- Non-Q-wave myocardial infarction
- All of the above
A: The correct answer is all of the above. Additional causes of dramatic T-wave inversion are listed in Table 1. Clinically, non-Q-wave myocardial infarction with T-wave changes and acute central nervous system injury are probably the most commonly seen.4
Echocardiography in this patient revealed severe apical hypertrophy of the ventricle with distal cavity obliteration. The left ventricular outflow-tract gradient was normal. The mitral valve appeared normal, and there was no resting systolic anterior motion.
Cardiac magnetic resonance imaging showed the apical variant of hypertrophic cardiomyopathy but no evidence of left ventricular noncompaction, which is a differential diagnosis of apical hypertrophic obstructive cardiomyopathy. This disease was first described in Japan by Yamaguchi et al5 and Sakamoto et al6 and is regarded as a subgroup of nonobstructive hypertrophic cardiomyopathy. The prognosis of apical hypertrophic cardiomyopathy with regard to sudden cardiac death is believed to be better than that of other forms of hypertrophic cardiomyopathy.3
A 48-year-old man with hypertension was being evaluated for a noncardiac issue (progressive multifocal leukoencephalopathy). He had been an active runner and did not have any cardiovascular symptoms at the time. The electrocardiogram (ECG) shown in Figure 1 was a routine study done as a part of that evaluation. His cardiovascular examination was unremarkable, without murmur, S3, or S4. His pulse was regular at 72 beats per minute, and his blood pressure was 112/76 mm Hg.
Q: Which of the following electrocardiographic findings suggest left ventricular hypertrophy?
- Sum of the S wave in V1 and the R wave in V6 ≥ 35 mm
- Sum of the S wave in V3 and the R wave in aVL > 28 mm (men)
- Sum of the S wave in V3 and the R wave in aVL > 20 mm (women)
- All of the above
A: The correct answer is all of the above.1,2
Our patient’s ECG shows sinus bradycardia and left ventricular hypertrophy, suggested by prominent voltage (sum of S in V1 and R in V6 ≥ 35 mm) and supported by ST-segment and T-wave changes in the lateral and midprecordial leads. Classic changes of left ventricular hypertrophy often include increased voltage and downsloping ST-segment depression with negative T waves in V5 and V6 (secondary repolarization changes or “strain” pattern).
Notable on this tracing are the large, asymmetric negative T waves in leads V3 through V6. Giant T waves are defined as negative T waves with voltage greater than 10 mm.3 Although there is no specific pattern of ventricular hypertrophy on an ECG that establishes the diagnosis of hypertrophic cardiomyopathy, left ventricular hypertrophy with T waves of this quality suggest the possibility of hypertrophic cardiomyopathy with apical hypertrophy.
Q: What are the other causes of giant negative T waves?
- Subarachnoid hemorrhage
- Complete heart block
- Non-Q-wave myocardial infarction
- All of the above
A: The correct answer is all of the above. Additional causes of dramatic T-wave inversion are listed in Table 1. Clinically, non-Q-wave myocardial infarction with T-wave changes and acute central nervous system injury are probably the most commonly seen.4
Echocardiography in this patient revealed severe apical hypertrophy of the ventricle with distal cavity obliteration. The left ventricular outflow-tract gradient was normal. The mitral valve appeared normal, and there was no resting systolic anterior motion.
Cardiac magnetic resonance imaging showed the apical variant of hypertrophic cardiomyopathy but no evidence of left ventricular noncompaction, which is a differential diagnosis of apical hypertrophic obstructive cardiomyopathy. This disease was first described in Japan by Yamaguchi et al5 and Sakamoto et al6 and is regarded as a subgroup of nonobstructive hypertrophic cardiomyopathy. The prognosis of apical hypertrophic cardiomyopathy with regard to sudden cardiac death is believed to be better than that of other forms of hypertrophic cardiomyopathy.3
- Sokolow M, Lyon TP. The ventricular complex in left ventricular hypertrophy as obtained by unipolar precordial and limb leads. 1949. Ann Noninvasive Electrocardiol 2001; 6:343–368.
- Casale PN, Devereux RB, Alonso DR, Campo E, Kligfield P. Improved sex-specific criteria of left ventricular hypertrophy for clinical and computer interpretation of electrocardiograms: validation with autopsy findings. Circulation 1987; 75:565–572.
- Eriksson MJ, Sonnenberg B, Woo A, et al. Long-term outcome in patients with apical hypertrophic cardiomyopathy. J Am Coll Cardiol 2002; 39:638–645.
- Jacobson D, Schrire V. Giant T wave inversion. Br Heart J 1966; 28:768–775.
- Yamaguchi H, Ishimura T, Nishiyama S, et al. Hypertrophic nonobstructive cardiomyopathy with giant negative T waves (apical hypertrophy): ventriculographic and echocardiographic features in 30 patients. Am J Cardiol 1979; 44:401–412.
- Sakamoto T, Tei C, Murayama M, Ichiyasu H, Hada Y. Giant T wave inversion as a manifestation of asymmetrical apical hypertrophy (AAH) of the left ventricle. Echocardiographic and ultrasonocardiotomographic study. Jpn Heart J 1976; 17:611–629.
- Sokolow M, Lyon TP. The ventricular complex in left ventricular hypertrophy as obtained by unipolar precordial and limb leads. 1949. Ann Noninvasive Electrocardiol 2001; 6:343–368.
- Casale PN, Devereux RB, Alonso DR, Campo E, Kligfield P. Improved sex-specific criteria of left ventricular hypertrophy for clinical and computer interpretation of electrocardiograms: validation with autopsy findings. Circulation 1987; 75:565–572.
- Eriksson MJ, Sonnenberg B, Woo A, et al. Long-term outcome in patients with apical hypertrophic cardiomyopathy. J Am Coll Cardiol 2002; 39:638–645.
- Jacobson D, Schrire V. Giant T wave inversion. Br Heart J 1966; 28:768–775.
- Yamaguchi H, Ishimura T, Nishiyama S, et al. Hypertrophic nonobstructive cardiomyopathy with giant negative T waves (apical hypertrophy): ventriculographic and echocardiographic features in 30 patients. Am J Cardiol 1979; 44:401–412.
- Sakamoto T, Tei C, Murayama M, Ichiyasu H, Hada Y. Giant T wave inversion as a manifestation of asymmetrical apical hypertrophy (AAH) of the left ventricle. Echocardiographic and ultrasonocardiotomographic study. Jpn Heart J 1976; 17:611–629.
V1: The most important lead in inferior STEMI
Q: Which would be the most appropriate diagnosis?
- Pericarditis
- Acute inferior and right ventricular myocardial infarction
- Anterior and inferior myocardial infarction
- None of the above
A: The correct answer is acute inferior and right ventricular myocardial infarction.
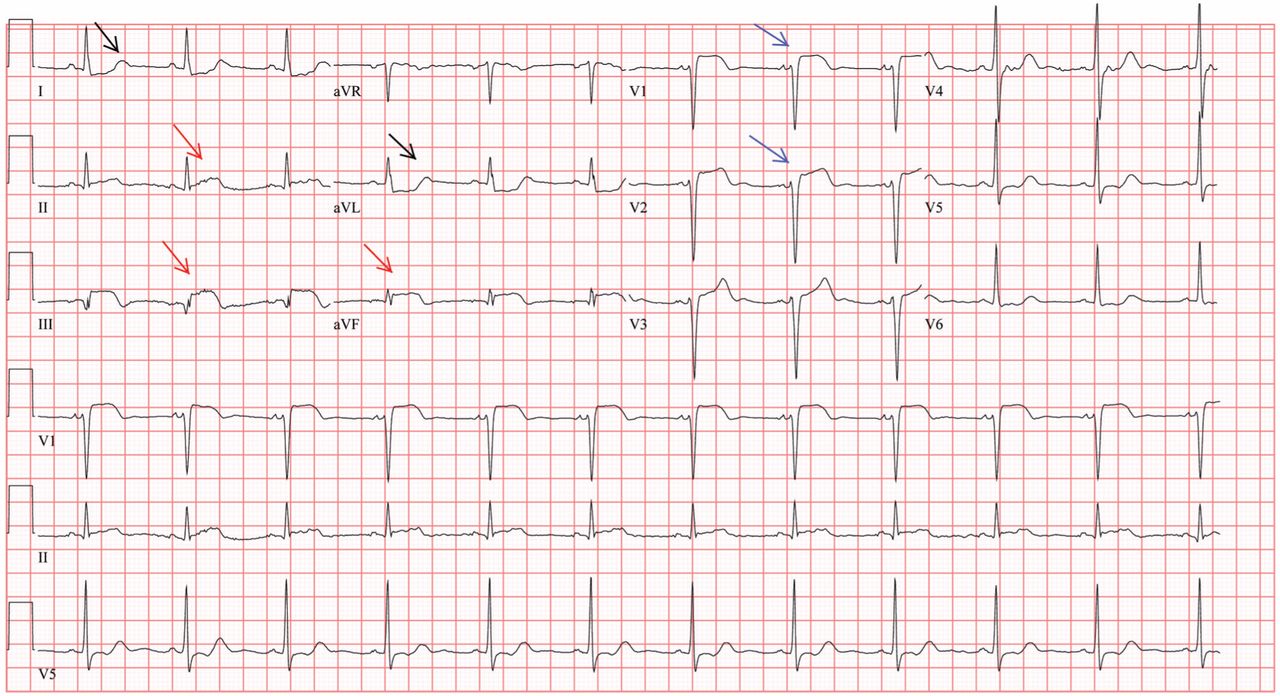
Her electrocardiogram showed sinus rhythm and inferior ST-segment elevation myocardial infarction (STEMI) evidenced by ST-segment elevation in leads II, III, and aVF. Hemodynamic instability or ST-segment elevation of more than 1 mm in lead V1 raises the suspicion of right ventricular myocardial infarction. In such patients, the American Heart Association guidelines recommend electrocardiography with right-sided precordial leads.1
A 1-mm ST-segment elevation in the right precordial lead V4R is one of the most predictive electrocardiographic findings in right ventricular infarction.2 The electrocardiographic changes in this type of myocardial infarction may be transient and resolve within 10 hours in up to 48% of cases.3
Echocardiography can also be used to confirm the possibility of right ventricular infarction.
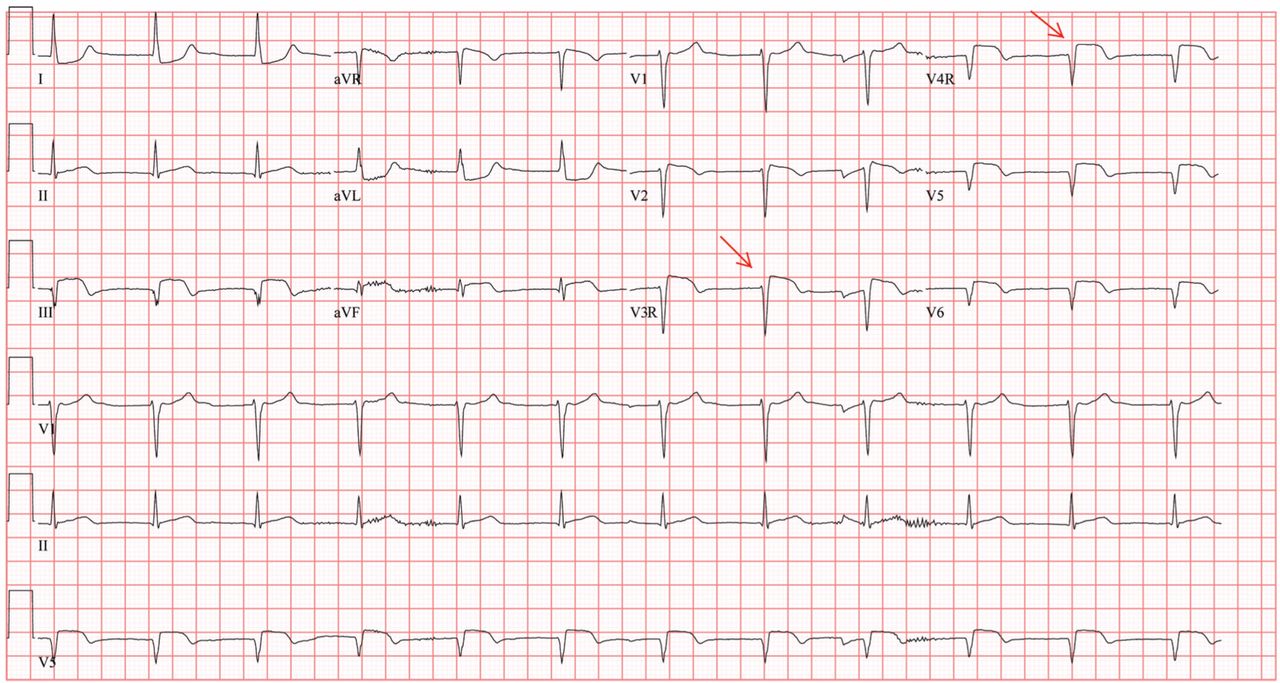
Q: Which clinical condition can occur as a complication of right ventricular myocardial infarction?
- Profound hypotension after nitrate administration
- High-degree heart block
- Atrial fibrillation
- All of the above
A: All of the conditions can occur.
Right ventricular involvement is very common, noted in up to 50% of patients with acute inferior STEMI in postmortem studies.4 However, hemodynamically significant right ventricular dysfunction is much less common.
Intravenous volume loading with normal saline is one of the first steps in the management of hypotension associated with right ventricular infarction. Patients with significant bradycardia or a high degree of atrioventricular block may require pacing. Early reperfusion should be achieved, if possible. Heightened suspicion is critical to the early diagnosis of this condition, since the prognosis is much worse than for isolated inferior STEMI.4
Our patient was found to have right coronary artery disease requiring percutaneous coronary intervention.
- Antman EM, Anbe DT, Armstrong PW, et al; American College of Cardiology. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Revise the 1999 Guidelines for the Management of Patients with Acute Myocardial Infarction). Circulation 2004; 110:e82–e292.
- Robalino BD, Whitlow PL, Underwood DA, Salcedo EE. Electrocardiographic manifestations of right ventricular infarction. Am Heart J 1989; 118:138–144.
- Braat SH, Brugada P, de Zwaan C, Coenegracht JM, Wellens HJ. Value of electrocardiogram in diagnosing right ventricular involvement in patients with an acute inferior wall myocardial infarction. Br Heart J 1983; 49:368–372.
- Zehender M, Kasper W, Kauder E, et al. Right ventricular infarction as an independent predictor of prognosis after acute inferior myocardial infarction. N Engl J Med 1993; 328:981–988.
Q: Which would be the most appropriate diagnosis?
- Pericarditis
- Acute inferior and right ventricular myocardial infarction
- Anterior and inferior myocardial infarction
- None of the above
A: The correct answer is acute inferior and right ventricular myocardial infarction.
Her electrocardiogram showed sinus rhythm and inferior ST-segment elevation myocardial infarction (STEMI) evidenced by ST-segment elevation in leads II, III, and aVF. Hemodynamic instability or ST-segment elevation of more than 1 mm in lead V1 raises the suspicion of right ventricular myocardial infarction. In such patients, the American Heart Association guidelines recommend electrocardiography with right-sided precordial leads.1
A 1-mm ST-segment elevation in the right precordial lead V4R is one of the most predictive electrocardiographic findings in right ventricular infarction.2 The electrocardiographic changes in this type of myocardial infarction may be transient and resolve within 10 hours in up to 48% of cases.3
Echocardiography can also be used to confirm the possibility of right ventricular infarction.
Q: Which clinical condition can occur as a complication of right ventricular myocardial infarction?
- Profound hypotension after nitrate administration
- High-degree heart block
- Atrial fibrillation
- All of the above
A: All of the conditions can occur.
Right ventricular involvement is very common, noted in up to 50% of patients with acute inferior STEMI in postmortem studies.4 However, hemodynamically significant right ventricular dysfunction is much less common.
Intravenous volume loading with normal saline is one of the first steps in the management of hypotension associated with right ventricular infarction. Patients with significant bradycardia or a high degree of atrioventricular block may require pacing. Early reperfusion should be achieved, if possible. Heightened suspicion is critical to the early diagnosis of this condition, since the prognosis is much worse than for isolated inferior STEMI.4
Our patient was found to have right coronary artery disease requiring percutaneous coronary intervention.
Q: Which would be the most appropriate diagnosis?
- Pericarditis
- Acute inferior and right ventricular myocardial infarction
- Anterior and inferior myocardial infarction
- None of the above
A: The correct answer is acute inferior and right ventricular myocardial infarction.
Her electrocardiogram showed sinus rhythm and inferior ST-segment elevation myocardial infarction (STEMI) evidenced by ST-segment elevation in leads II, III, and aVF. Hemodynamic instability or ST-segment elevation of more than 1 mm in lead V1 raises the suspicion of right ventricular myocardial infarction. In such patients, the American Heart Association guidelines recommend electrocardiography with right-sided precordial leads.1
A 1-mm ST-segment elevation in the right precordial lead V4R is one of the most predictive electrocardiographic findings in right ventricular infarction.2 The electrocardiographic changes in this type of myocardial infarction may be transient and resolve within 10 hours in up to 48% of cases.3
Echocardiography can also be used to confirm the possibility of right ventricular infarction.
Q: Which clinical condition can occur as a complication of right ventricular myocardial infarction?
- Profound hypotension after nitrate administration
- High-degree heart block
- Atrial fibrillation
- All of the above
A: All of the conditions can occur.
Right ventricular involvement is very common, noted in up to 50% of patients with acute inferior STEMI in postmortem studies.4 However, hemodynamically significant right ventricular dysfunction is much less common.
Intravenous volume loading with normal saline is one of the first steps in the management of hypotension associated with right ventricular infarction. Patients with significant bradycardia or a high degree of atrioventricular block may require pacing. Early reperfusion should be achieved, if possible. Heightened suspicion is critical to the early diagnosis of this condition, since the prognosis is much worse than for isolated inferior STEMI.4
Our patient was found to have right coronary artery disease requiring percutaneous coronary intervention.
- Antman EM, Anbe DT, Armstrong PW, et al; American College of Cardiology. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Revise the 1999 Guidelines for the Management of Patients with Acute Myocardial Infarction). Circulation 2004; 110:e82–e292.
- Robalino BD, Whitlow PL, Underwood DA, Salcedo EE. Electrocardiographic manifestations of right ventricular infarction. Am Heart J 1989; 118:138–144.
- Braat SH, Brugada P, de Zwaan C, Coenegracht JM, Wellens HJ. Value of electrocardiogram in diagnosing right ventricular involvement in patients with an acute inferior wall myocardial infarction. Br Heart J 1983; 49:368–372.
- Zehender M, Kasper W, Kauder E, et al. Right ventricular infarction as an independent predictor of prognosis after acute inferior myocardial infarction. N Engl J Med 1993; 328:981–988.
- Antman EM, Anbe DT, Armstrong PW, et al; American College of Cardiology. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Revise the 1999 Guidelines for the Management of Patients with Acute Myocardial Infarction). Circulation 2004; 110:e82–e292.
- Robalino BD, Whitlow PL, Underwood DA, Salcedo EE. Electrocardiographic manifestations of right ventricular infarction. Am Heart J 1989; 118:138–144.
- Braat SH, Brugada P, de Zwaan C, Coenegracht JM, Wellens HJ. Value of electrocardiogram in diagnosing right ventricular involvement in patients with an acute inferior wall myocardial infarction. Br Heart J 1983; 49:368–372.
- Zehender M, Kasper W, Kauder E, et al. Right ventricular infarction as an independent predictor of prognosis after acute inferior myocardial infarction. N Engl J Med 1993; 328:981–988.
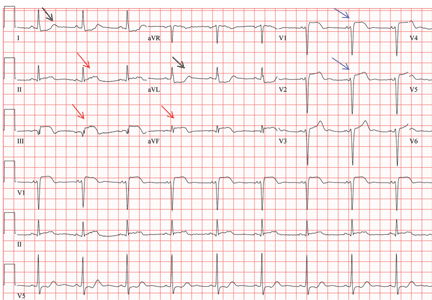
The role of aldosterone receptor antagonists in the management of heart failure: An update
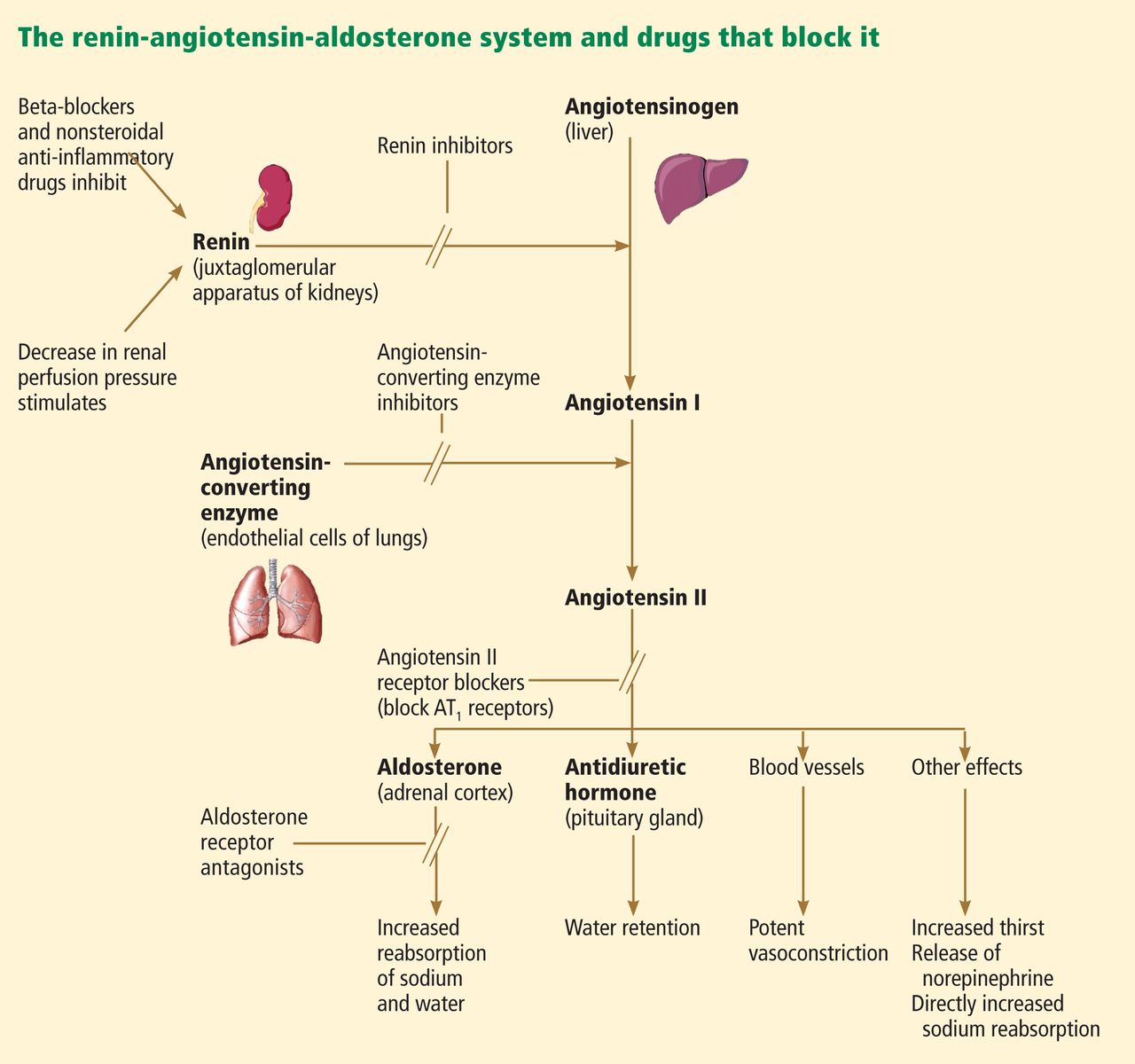
Over the past 30 years, the focus of treating heart failure has shifted from managing symptoms to prolonging lives. When the neurohormonal hypothesis (ie, the concept that neurohormonal dysregulation and not merely hemodynamic changes are responsible for the onset and progression of heart failure) was introduced, it brought a dramatic change that included new classes of drugs that interfere with the renin-angiotensin-aldosterone system, ie, angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), and, most recently, aldosterone receptor antagonists (ARAs) (Figure 1).
Evidence supporting the use of the ARAs spironolactone (Aldactone) and eplerenone (Inspra) in heart failure has been growing, as has evidence of their usefulness in treating diabetes and chronic renal disease. Still, these drugs must be used cautiously, as they can cause hyperkalemia.
This paper will review the clinical use of ARAs in symptomatic systolic heart failure, their side effects, the findings and implications of recent trials, and controversies in this area, notably whether there is any evidence favoring the use of one drug over another.
ALDOSTERONE IN HEART FAILURE
Aldosterone, a hormone secreted by the zona glomerulosa of the adrenal gland, was first isolated by Simpson and Tait more than half a century ago.1 Later, it was found to promote reabsorption of sodium and excretion of potassium in the kidneys and hence was categorized as a mineralocorticoid hormone.
Release of aldosterone is stimulated by decreased renal perfusion via angiotensin II, hyperkalemia, and possibly adrenocorticotropic hormone.2 Aldosterone exerts its effects by binding to mineralocorticoid receptors in renal epithelial cells.
Aldosterone has several deleterious effects on the failing heart, primarily sodium and fluid retention, but also endothelial dysfunction, left ventricular hypertrophy, and myocardial fibrosis.2,3 Plasma aldosterone levels can be markedly elevated in patients with heart failure, likely due to activation of the renin-angiotensin-aldosterone system. Elevated aldosterone and angiotensin II levels have been associated with higher mortality rates.4
ALDOSTERONE ‘ESCAPE’ BLUNTS THE EFFECT OF ACE INHIBITORS AND ARBs
ACE inhibitors and ARBs have become standards of care for patients with systolic heart failure, and for many years, it was believed that these drugs suppressed aldosterone levels sufficiently. But elevated aldosterone levels have been noted in up to 38% of patients on chronic ACE inhibitor therapy.5 In one study, patients on dual blockade, ie, on both an ACE inhibitor and an ARB, had significantly lower aldosterone levels at 17 weeks of therapy, but not at 43 weeks.6 This phenomenon is known as “aldosterone escape.”
Several mechanisms might explain this phenomenon. Angiotensin II, a potent inducer of aldosterone, is “reactivated” during long-term ACE inhibitor therapy. Interestingly, patients progress toward aldosterone escape regardless of whether the ACE inhibitor dose is low or high.7 There is evidence that some aldosterone is produced by endothelial cells and vascular smooth muscle in the heart and blood vessels,8 but ACE inhibitors and ARBs suppress only the aldosterone secreted by the adrenal glands.
Regardless of the mechanism, aldosterone escape can blunt the effects of ACE inhibitors and ARBs, reducing their favorable effects on the risk of death in heart failure patients. This is the rationale for also using ARAs.
ARAs IN HEART FAILURE
Aldosterone acts by regulating gene expression after binding to mineralocorticoid receptors. These receptors are found not only in epithelial tissue in the kidneys and glands, but also in nonepithelial tissues such as cardiomyocytes, vessel walls, and the hippocampus of the brain.9 The nonepithelial effects were first demonstrated 2 decades ago by Brilla et al,10 who noted that chronically elevated aldosterone levels in rats promoted cardiac fibroblast growth, collagen accumulation, and, hence, ventricular remodeling.
The hypertensive effect of aldosterone may also be mediated through mineralocorticoid receptors in the brain. Gomez-Sanchez et al11 found that infusing aldosterone into the cerebral ventricles caused significant hypertension. A selective mineralocorticoid antagonist inhibited this effect when infused into the cerebral ventricles but not when given systemically.
In 1959, Cella and Kagawa created spironolactone, a nonselective ARA, by combining elements of progesterone for its antimineralocorticoid effect and elements of digitoxin for its cardiotonic effect.12 Although spironolactone is very effective in treating hypertension and heart failure, its use is limited by progestational and antiandrogenic side effects. This led, in 1987, to the invention by de Gasparo et al of a newer molecule, a selective ARA now called eplerenone.13 Although eplerenone may be somewhat less potent than spironolactone in blocking mineralocorticoid receptors, no significant difference in efficacy has been noted in randomized clinical trials, and its antiandrogenic action is negligible.12
Although these drugs target aldosterone receptors, newer drugs may target different aspects of mineralocorticoid activities, and thus the term “mineralocorticoid receptor antagonist” has been proposed.
TRIALS OF ARAs IN HEART FAILURE
An online data supplement that accompanies this paper at provides a detailed comparison of the three major trials of ARAs in patients with heart failure.
The Randomized Aldactone Evaluation Study (RALES)
The first major clinical trial of an ARA was the Randomized Aldactone Evaluation Study (RALES),14 a randomized, double-blind, controlled comparison of spironolactone and placebo.
The 1,663 patients in the trial all had severe heart failure (New York Heart Association class [NYHA] III and ambulatory class IV symptoms) and a left ventricular ejection fraction of 35% or less. Most were on an ACE inhibitor, a loop diuretic, and digoxin, but only 10% of patients in both groups were on a beta-blocker. Patients with chronic renal failure (serum creatinine > 2.5 mg/dL) or hyperkalemia (potassium > 5.0 mmol/L) were excluded.
RALES was halted early when an interim analysis at a mean follow-up of 24 months showed that significantly fewer patients were dying in the spironolactone group; their all-cause mortality rate was 30% lower (relative risk [RR] 0.70, 95% confidence interval [CI] 0.60–0.82, P < .001), and their cardiac mortality rate was 31% lower (RR 0.69, 95% CI 0.58–0.82, P < .001). This was concordant with a lower risk of both sudden cardiac death and death from progressive heart failure. The risk of hospitalization for cardiac causes was also 30% lower for patients in the spironolactone group, who also experienced significant symptom improvement.
Gynecomastia and breast pain occurred in about 10% of patients in the spironolactone group, and adverse effects leading to study drug discontinuation occurred in 2%.14
The Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS)
The next landmark trial of an ARA was the Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS).15 A total of 6,632 patients were randomized to receive eplerenone or placebo in this multicenter, double-blind trial. To be enrolled, patients had to have acute myocardial infarction, a left ventricular ejection fraction of 40% or less, and either clinical signs of heart failure 3 to 14 days after the infarction or a history of diabetes mellitus. Patients were excluded if they had chronic kidney disease (defined as a serum creatinine > 2.5 mg/dL or an estimated glomerular filtration rate < 30 mL/min/1.73 m2) or hyperkalemia (a serum potassium > 5.0 mmol/L). All the patients received optimal medical therapy and reperfusion therapy, if warranted.
This event-driven trial was stopped when 1,012 deaths had occurred. During a mean follow-up of 16 months, there was a 15% lower rate of all-cause mortality in the eplerenone group (RR 0.85, 95% CI 0.75–0.96, P = .008) and a 13% lower rate of cardiovascular mortality (RR 0.83, 95% CI 0.72–0.94, P = .005). The reduction in the cardiovascular mortality rate was attributed to a 21% reduction in the rate of sudden cardiac deaths. The rate of heart failure hospitalization was also lower in the eplerenone group.
Serious hyperkalemia occurred significantly more frequently in the eplerenone group (5.5% vs 3.9%, P = .002), but similar rates of gynecomastia were observed. The incidence of hyperkalemia was higher in patients with a creatinine clearance less than 50 mL/min.
Further analyses revealed a 31% lower rate of all-cause mortality (95% CI 0.54–0.89, P = .004) and a 32% lower rate of cardiovascular mortality (95% CI 0.53–0.88, P = .003) at 30 days after randomization in the eplerenone group.16 Importantly, 25% of all deaths in the EPHESUS study during the 16-month follow-up period occurred in the first 30 days after randomization. The Kaplan-Meier survival curves showed separation as early as 5 days after randomization. Hence, the 30-day mortality results from EPHESUS further indicated that starting eplerenone early may be particularly beneficial.
The Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure (EMPHASIS-HF)
After RALES and EPHESUS, a gap remained in our knowledge, ie, how to use ARAs in patients with mild heart failure, who account for most cases. This led to the EMPHASIS-HF (Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure) trial, which expanded the indications for ARAs to patients with chronic systolic heart failure with mild symptoms.17
In this double-blind trial, 2,737 patients with NYHA class II heart failure with a left ventricular ejection fraction of 35% or less were randomized to receive oral eplerenone 25 mg or placebo once daily. All patients were already on a beta-blocker; they were also all on an ACE inhibitor, an ARB, or both at the recommended or maximal tolerated dose. Patients with a glomerular filtration rate between 30 and 49 mL/min were started on alternate-day dosing, and those with glomerular filtration rates below 30 mL/min were excluded.
To ensure that the event rate was high enough to give this trial sufficient power:
- Only patients age 55 years or older were included
- Patients with a left ventricular ejection fraction greater than 30% were enrolled only if the QRS duration was greater than 130 ms (only 3.5% of patients in both groups were enrolled based on this criterion)
- Patients either had to have been hospitalized for cardiovascular reasons in the 6 months before randomization or had to have elevated natriuretic peptides (B-type natriuretic peptide [BNP] level > 250 pg/mL or N-terminal pro-BNP > 500 pg/mL in men and > 750 pg/mL in women).
The study was stopped early at a median follow-up of 21 months after an interim analysis showed a significantly lower rate of the primary composite end point (death from a cardiovascular cause or hospitalization for heart failure) in the eplerenone group: 18.3% vs 25.9% (hazard ratio [HR] 0.63, 95% CI 0.54– 0.74, P < .001). The rates of all-cause mortality were 12.5% vs 15.5% (HR 0.76, 95% CI 0.62–0.93, P = .008), and the rates of cardiovascular mortality were 10.8% vs 13.5% (HR 0.76, 95% CI 0.61–0.94, P = .01). Kaplan-Meier curves for all-cause mortality showed significant separation only after 1 year, which was not the case in EPHESUS and RALES. But the curves for hospitalization separated within a few weeks after randomization.
The incidence of hyperkalemia (serum potassium level > 5.5 mmol/L) was significantly higher in the eplerenone group (11.8% vs 7.2%, P < .001), but there was no statistically significant difference between groups when potassium levels above 6 mmol/L were considered (2.5% vs 1.9%, P = .29). This is despite one-third of patients having an estimated glomerular filtration rate less than 60 mL/min/1.73 m2. Breast symptoms were very rare, occurring in 1% or fewer patients in both groups. The discontinuation rate of the study drug was similar in both groups.
HOW DO ARAs PREVENT DEATH?
Multiple studies show that spironolactone and eplerenone lower blood pressure in a dose-related manner.18 These drugs reduce fluid volume and pulmonary congestion, which could have been the primary mechanism for the reduction in heart failure hospitalizations in the EMPHASIS-HF trial. But other mechanisms might explain the reduction in cardiovascular mortality rates in the trials summarized above.
Transcardiac extraction of aldosterone was increased in a study of patients with heart failure. 19 The transcardiac gradient of plasma aldosterone correlated with levels of procollagen III N-terminal propeptide, a biochemical marker of myocardial fibrosis. This suggests that aldosterone could be a stimulant of myocardial fibrosis. Spironolactone inhibited the transcardiac extraction of aldosterone in the same study.19
In another study,20 spironolactone significantly suppressed elevation of procollagen III N-terminal propeptide after myocardial infarction. It was also demonstrated that spironolactone prevented left ventricular remodeling after infarction, even in patients receiving an ACE inhibitor. Similar results, ie, decreased left ventricular myocardial fibrosis and remodeling, were noted in another trial in which eplerenone was added to an ARB.21
Myocardial fibrosis is a known substrate for ventricular arrhythmias. In a randomized study in 35 patients, spironolactone decreased the incidence of ventricular arrhythmias.22 This finding correlates with the decreased incidence of sudden cardiac death in the RALES and EPHESUS trials.
ADVERSE EFFECTS OF ARAs
Hyperkalemia, hyperkalemia, hyperkalemia
Potassium excretion is physiologically regulated by the serum aldosterone concentration and by the delivery of sodium to the distal nephron. Aldosterone increases potassium excretion. As a result of decreased renal perfusion that occurs with heart failure, sodium is intensely reabsorbed in the proximal tubule, and very little sodium reaches the distal nephron. When aldosterone receptors are blocked by ARAs, the risk of hyperkalemia increases.23

Other electrolyte abnormalities associated with ARAs are hyponatremia and hyperchloremic metabolic acidosis (Table 1). There could be a reversible decline in the glomerular filtration rate as well.24 Of note, most patients with chronic systolic heart failure in the RALES and EMPHASIS-HF trials were already receiving a diuretic; thus, the adverse effect profile of ARAs in otherwise euvolemic (or even hypovolemic) patients is not well appreciated.
Failure to closely monitor electrolyte levels increases the risk of hyperkalemia and renal failure, so there is a need for regular follow-up visits for patients taking an ARA.25 This was made clear when a population-based analysis from Canada compared the rates of hyperkalemia-related hospitalization and death before and after the RALES trial was published. The prescription rate for spironolactone increased threefold, but the rate of hyperkalemia-related hospitalization increased fourfold and the rate of death increased sixfold.26
Although caution is recommended when starting a patient on an ARA, a recent trial conducted in 167 cardiology practices noted that ARAs were the most underused drugs for heart failure. In this study, an ARA was prescribed to only 35% of eligible patients. The prescription rate was not significantly higher even in dedicated heart failure clinics.27 Possible reasons suggested by the authors were drug side effects, the need for closer monitoring of laboratory values, and a lack of knowledge.
A population-based analysis from the United Kingdom found a significant increase over time in spironolactone prescriptions after the release of the RALES trial results, but there was no increase in the rate of serious hyperkalemia (serum potassium > 6 mmol/L) or hyperkalemia-related hospitalization.28 The authors suggested that careful monitoring could prevent hyperkalemia-related complications. They also observed that 75% of patients who had spironolactone-associated hyperkalemia were over 65 years old. Hence, we recommend closer monitoring when starting an elderly patient on an ARA.
Breast, gastrointestinal symptoms
The nonselective ARA spironolactone is associated with antiandrogenic side effects. In a smaller study in patients with resistant hypertension, Nishizaka et al noted that low-dose spironolactone (up to 50 mg/day) was associated with breast tenderness in about 10%.29 Breast symptoms with spironolactone are dose-related, and the incidence can be as high as 50% when the drug is used in dosages of 150 mg/day or higher.30
In one population-based case-control study, spironolactone was associated with a 2.7 times higher risk of gastrointestinal side effects (bleeding or ulcer).31
ARAs IN HEART FAILURE WITH PRESERVED EJECTION FRACTION
The concept of diastolic heart failure or “heart failure with preserved ejection fraction” has been growing. A significant proportion of patients with a diagnosis of heart failure have preserved left ventricular ejection fraction (≥ 50%) and diastolic dysfunction.
Despite multiple trials, no treatment has been shown to lower the mortality rate in heart failure with preserved ejection fraction.32,33 A recently published randomized controlled trial in 44 patients with this condition showed reduction in serum biochemical markers of collagen turnover and improvement in diastolic function with ARAs, but there was no difference in exercise capacity.34 A larger double-blind randomized control trial, Aldosterone Receptor Blockade in Diastolic Heart Failure (Aldo-DHF), is under way to evaluate the effects of ARAs on exercise capacity and diastolic function in patients with heart failure with preserved ejection fraction.35
In January 2012, the Trial of Aldosterone Antagonist Therapy in Adults With Preserved Ejection Fraction Congestive Heart Failure (TOPCAT) completed enrollment of 3,445 patients to study the effect of ARAs in reducing the composite end point of cardiovascular mortality, aborted cardiac arrest, and heart failure hospitalization. Long-term follow-up of this event-driven study is currently under way.
ARAs IN DIABETES MELLITUS AND CHRONIC KIDNEY DISEASE
Under physiologic conditions, the serum aldosterone level is regulated by volume status through the renin-angiotensin system. But in patients with chronic kidney disease, the serum aldosterone level could be elevated without renin-angiotensin system stimulation.36
High aldosterone levels were associated with proteinuria and glomerulosclerosis in rats.37 In a study in 83 patients, aldosterone receptor blockade was shown to decrease proteinuria and possibly to retard the progression of chronic kidney disease. In this trial, baseline serum aldosterone levels correlated with proteinuria.38 Animal studies suggest that adipocyte-derived factors may stimulate aldosterone, which may be relevant in patients who have both chronic kidney disease and metabolic syndrome.39
The impact of ARAs in patients with diabetes mellitus is often overlooked. In EPHESUS, diabetes mellitus was an inclusion criterion even in the absence of heart failure signs and symptoms in the postinfarction setting of impaired left ventricular ejection fraction.15
In patients with diabetic nephropathy, there is growing evidence that ARAs can decrease proteinuria, even if the serum aldosterone level is normal. For example, in a study in 20 patients with diabetic nephropathy, spironolactone reduced proteinuria by 32%. This reduction was independent of serum aldosterone levels.40
In diabetic rats, hyperglycemia was noted to cause podocyte injury through mineralocorticoid receptor-mediated production of reactive oxygen species, independently of serum aldosterone levels. Spironolactone decreased the production of reactive oxygen species, thereby potentially reducing proteinuria.41
RECOMMENDATIONS ARE BEING REVISED
The most recent joint guidelines of the American Heart Association and the American College of Cardiology for the management of heart failure42 were published in 2009, which was before the EMPHASIS-HF results. An update is expected soon. In the 2009 version, ARAs received a class I recommendation for patients with moderately severe to severe symptoms, decreased ejection fraction, normal renal function, and normal potassium levels. The guidelines also said that the risks of ARAs may outweigh their benefits if regular monitoring is not possible.
The recommended starting dosage is 12.5 mg/day of spironolactone or 25 mg/day of eplerenone; the dose can be doubled, if tolerated.
Close monitoring is recommended, ie, measuring serum potassium and renal function 3 and 7 days after starting therapy and then monthly for the first 3 months. Closer monitoring is needed if an ACE inhibitor or an ARB is added later. In elderly patients, the glomerular filtration rate is preferred over the serum creatinine level, and ARA therapy is not advisable if the glomerular filtration rate is less than 30 mL/min/1.73 m2.
Avoid concomitant use of the following:
- Potassium supplements (unless persistent hypokalemia is present)
- Nonsteroidal anti-inflammatory drugs
- An ACE inhibitor and an ARB in combination
- A high dose of an ACE inhibitor or ARB.
Conditions that can lead to dehydration (eg, diarrhea, excessive use of diuretics) or acute illness should warrant reduction (or even withholding) of ARAs. When to discontinue ARA therapy is not well described, nor is the safety of starting ARAs in the hospital. However, it is clear that many patients who are potentially eligible for ARAs are not prescribed them.43
The guidelines are currently being revised, and will likely incorporate the new data from EMPHASIS-HF to extend to a broader population. The benefits of ARAs can be met only if the risks are minimized.
WHICH ARA IS BETTER?
The pharmacologic differences between the two ARAs have been described earlier, and guidelines have advocated evidence-based use of ARAs for their respective indications. There have been no large-scale, head-to-head comparisons of spironolactone and eplerenone in the heart failure population, and in clinical practice the drugs are prescribed interchangeably in most patients.
A double-blind randomized controlled trial in 141 patients with hypertension and primary hyperaldosteronism found that spironolactone lowered diastolic blood pressure more, but it also caused antiandrogenic effects more often.44
There is some evidence to suggest that eplerenone has a better metabolic profile than spironolactone. The data came from a small randomized controlled trial in 107 stable outpatients with mild heart failure.45 Patients who were prescribed spironolactone had a higher cortisol level and hemoglobin A1c level 4 months after starting treatment. This effect was not seen in patients who were on eplerenone. However, these findings need to be confirmed in larger trials.
While the differences between the two drugs remain to be determined, the most important differences in clinical practice are selectivity for receptors (and hence their antiandrogenic side effects) and price. Even though it is available as a generic drug, eplerenone still costs at least three times more than spironolactone for the same dosage and indication.
- Simpson SA, Tait JF, Bush IE. Secretion of a salt-retaining hormone by the mammalian adrenal cortex. Lancet 1952; 2:226–228.
- Struthers AD, MacDonald TM. Review of aldosterone- and angiotensin II-induced target organ damage and prevention. Cardiovasc Res 2004; 61:663–670.
- Edelmann F, Schmidt AG, Gelbrich G, et al. Rationale and design of the “aldosterone receptor blockade in diastolic heart failure” trial: a double-blind, randomized, placebo-controlled, parallel group study to determine the effects of spironolactone on exercise capacity and diastolic function in patients with symptomatic diastolic heart failure (Aldo-DHF). Eur J Heart Fail 2010; 12:874–882.
- Swedberg K, Eneroth P, Kjekshus J, Wilhelmsen L. Hormones regulating cardiovascular function in patients with severe congestive heart failure and their relation to mortality. CONSENSUS Trial Study Group. Circulation 1990; 82:1730–1736.
- MacFadyen RJ, Lee AF, Morton JJ, Pringle SD, Struthers AD. How often are angiotensin II and aldosterone concentrations raised during chronic ACE inhibitor treatment in cardiac failure? Heart 1999; 82:57–61.
- McKelvie RS, Yusuf S, Pericak D, et al. Comparison of candesartan, enalapril, and their combination in congestive heart failure: randomized evaluation of strategies for left ventricular dysfunction (RESOLVD) pilot study. The RESOLVD Pilot Study Investigators. Circulation 1999; 100:1056–1064.
- Tang WH, Vagelos RH, Yee YG, et al. Neurohormonal and clinical responses to high- versus low-dose enalapril therapy in chronic heart failure. J Am Coll Cardiol 2002; 39:70–78.
- Weber KT. Aldosterone in congestive heart failure. N Engl J Med 2001; 345:1689–1697.
- Funder JW. The role of aldosterone and mineralocorticoid receptors in cardiovascular disease. Am J Cardiovasc Drugs 2007; 7:151–157.
- Brilla CG, Pick R, Tan LB, Janicki JS, Weber KT. Remodeling of the rat right and left ventricles in experimental hypertension. Circ Res 1990; 67:1355–1364.
- Gomez-Sanchez EP, Fort C, Thwaites D. Central mineralocorticoid receptor antagonism blocks hypertension in Dahl S/JR rats. Am J Physiol 1992; 262:E96–E99.
- Garthwaite SM, McMahon EG. The evolution of aldosterone antagonists. Mol Cell Endocrinol 2004; 217:27–31.
- de Gasparo M, Joss U, Ramjoué HP, et al. Three new epoxy-spirolactone derivatives: characterization in vivo and in vitro. J Pharmacol Exp Ther 1987; 240:650–656.
- Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999; 341:709–717.
- Pitt B, Remme W, Zannad F, et al; Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study Investigators. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 2003; 348:1309–1321.
- Pitt B, White H, Nicolau J, et al; EPHESUS Investigators. Eplerenone reduces mortality 30 days after randomization following acute myocardial infarction in patients with left ventricular systolic dysfunction and heart failure. J Am Coll Cardiol 2005; 46:425–431.
- Zannad F, McMurray JJ, Krum H, et al; EMPHASIS-HF Study Group. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011; 364:11–21.
- Weinberger MH, Roniker B, Krause SL, Weiss RJ. Eplerenone, a selective aldosterone blocker, in mild-to-moderate hypertension. Am J Hypertens 2002; 15:709–716.
- Tsutamoto T, Wada A, Maeda K, et al. Spironolactone inhibits the transcardiac extraction of aldosterone in patients with congestive heart failure. J Am Coll Cardiol 2000; 36:838–844.
- Hayashi M, Tsutamoto T, Wada A, et al. Immediate administration of mineralocorticoid receptor antagonist spironolactone prevents postinfarct left ventricular remodeling associated with suppression of a marker of myocardial collagen synthesis in patients with first anterior acute myocardial infarction. Circulation 2003; 107:2559–2565.
- Fraccarollo D, Galuppo P, Schmidt I, Ertl G, Bauersachs J. Additive amelioration of left ventricular remodeling and molecular alterations by combined aldosterone and angiotensin receptor blockade after myocardial infarction. Cardiovasc Res 2005; 67:97–105.
- Ramires FJ, Mansur A, Coelho O, et al. Effect of spironolactone on ventricular arrhythmias in congestive heart failure secondary to idiopathic dilated or to ischemic cardiomyopathy. Am J Cardiol 2000; 85:1207–1211.
- Palmer BF. Managing hyperkalemia caused by inhibitors of the reninangiotensin-aldosterone system. N Engl J Med 2004; 351:585–592.
- Sica DA. The risks and benefits of therapy with aldosterone receptor antagonist therapy. Curr Drug Saf 2007; 2:71–77.
- Shah KB, Rao K, Sawyer R, Gottlieb SS. The adequacy of laboratory monitoring in patients treated with spironolactone for congestive heart failure. J Am Coll Cardiol 2005; 46:845–849.
- Juurlink DN, Mamdani MM, Lee DS, et al. Rates of hyperkalemia after publication of the Randomized Aldactone Evaluation Study. N Engl J Med 2004; 351:543–551.
- Albert NM, Fonarow GC, Yancy CW, et al. Influence of dedicated heart failure clinics on delivery of recommended therapies in outpatient cardiology practices: findings from the Registry to Improve the Use of Evidence-Based Heart Failure Therapies in the Outpatient Setting (IMPROVE HF). Am Heart J 2010; 159:238–244.
- Wei L, Struthers AD, Fahey T, Watson AD, Macdonald TM. Spironolactone use and renal toxicity: population based longitudinal analysis. BMJ 2010; 340:c1768.
- Nishizaka MK, Zaman MA, Calhoun DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens 2003; 16:925–930.
- Jeunemaitre X, Chatellier G, Kreft-Jais C, et al. Efficacy and tolerance of spironolactone in essential hypertension. Am J Cardiol 1987; 60:820–825.
- Verhamme K, Mosis G, Dieleman J, Stricker B, Sturkenboom M. Spironolactone and risk of upper gastrointestinal events: population based case-control study. BMJ 2006; 333:330.
- Massie BM, Carson PE, McMurray JJ, et al; I-PRESERVE Investigators. Irbesartan in patients with heart failure and preserved ejection fraction. N Engl J Med 2008; 359:2456–2467.
- Yusuf S, Pfeffer MA, Swedberg K, et al; CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-Preserved Trial. Lancet 2003; 362:777–781.
- Deswal A, Richardson P, Bozkurt B, Mann DL. Results of the Randomized Aldosterone Antagonism in Heart Failure With Preserved Ejection Fraction Trial (RAAM-PEF). J Card Fail 2011; 17:634–642.
- Edelmann F, Schmidt AG, Gelbrich G, et al. Rationale and design of the ‘aldosterone receptor blockade in diastolic heart failure’ trial: a double-blind, randomized, placebo-controlled, parallel group study to determine the effects of spironolactone on exercise capacity and diastolic function in patients with symptomatic diastolic heart failure (Aldo-DHF). Eur J Heart Fail 2010; 12:874–882.
- Hené RJ, Boer P, Koomans HA, Mees EJ. Plasma aldosterone concentrations in chronic renal disease. Kidney Int 1982; 21:98–101.
- Greene EL, Kren S, Hostetter TH. Role of aldosterone in the remnant kidney model in the rat. J Clin Invest 1996; 98:1063–1068.
- Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int 2006; 70:2116–2123.
- Nagase M, Yoshida S, Shibata S, et al. Enhanced aldosterone signaling in the early nephropathy of rats with metabolic syndrome: possible contribution of fat-derived factors. J Am Soc Nephrol 2006; 17:3438–3446.
- Schjoedt KJ, Rossing K, Juhl TR, et al. Beneficial impact of spironolactone on nephrotic range albuminuria in diabetic nephropathy. Kidney Int 2006; 70:536–542.
- Toyonaga J, Tsuruya K, Ikeda H, et al. Spironolactone inhibits hyperglycemia-induced podocyte injury by attenuating ROS production. Nephrol Dial Transplant 2011; 26:2475–2484.
- Hunt SA, Abraham WT, Chin MH, et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 2009; 119:e391–e479.
- Albert NM, Yancy CW, Liang L, et al. Use of aldosterone antagonists in heart failure. JAMA 2009; 302:1658–1665.
- Parthasarathy HK, Ménard J, White WB, et al. A double-blind, randomized study comparing the antihypertensive effect of eplerenone and spironolactone in patients with hypertension and evidence of primary aldosteronism. J Hypertens 2011; 29:980–990.
- Yamaji M, Tsutamoto T, Kawahara C, et al. Effect of eplerenone versus spironolactone on cortisol and hemoglobin A1(c) levels in patients with chronic heart failure. Am Heart J 2010; 160:915–921.
Over the past 30 years, the focus of treating heart failure has shifted from managing symptoms to prolonging lives. When the neurohormonal hypothesis (ie, the concept that neurohormonal dysregulation and not merely hemodynamic changes are responsible for the onset and progression of heart failure) was introduced, it brought a dramatic change that included new classes of drugs that interfere with the renin-angiotensin-aldosterone system, ie, angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), and, most recently, aldosterone receptor antagonists (ARAs) (Figure 1).
Evidence supporting the use of the ARAs spironolactone (Aldactone) and eplerenone (Inspra) in heart failure has been growing, as has evidence of their usefulness in treating diabetes and chronic renal disease. Still, these drugs must be used cautiously, as they can cause hyperkalemia.
This paper will review the clinical use of ARAs in symptomatic systolic heart failure, their side effects, the findings and implications of recent trials, and controversies in this area, notably whether there is any evidence favoring the use of one drug over another.
ALDOSTERONE IN HEART FAILURE
Aldosterone, a hormone secreted by the zona glomerulosa of the adrenal gland, was first isolated by Simpson and Tait more than half a century ago.1 Later, it was found to promote reabsorption of sodium and excretion of potassium in the kidneys and hence was categorized as a mineralocorticoid hormone.
Release of aldosterone is stimulated by decreased renal perfusion via angiotensin II, hyperkalemia, and possibly adrenocorticotropic hormone.2 Aldosterone exerts its effects by binding to mineralocorticoid receptors in renal epithelial cells.
Aldosterone has several deleterious effects on the failing heart, primarily sodium and fluid retention, but also endothelial dysfunction, left ventricular hypertrophy, and myocardial fibrosis.2,3 Plasma aldosterone levels can be markedly elevated in patients with heart failure, likely due to activation of the renin-angiotensin-aldosterone system. Elevated aldosterone and angiotensin II levels have been associated with higher mortality rates.4
ALDOSTERONE ‘ESCAPE’ BLUNTS THE EFFECT OF ACE INHIBITORS AND ARBs
ACE inhibitors and ARBs have become standards of care for patients with systolic heart failure, and for many years, it was believed that these drugs suppressed aldosterone levels sufficiently. But elevated aldosterone levels have been noted in up to 38% of patients on chronic ACE inhibitor therapy.5 In one study, patients on dual blockade, ie, on both an ACE inhibitor and an ARB, had significantly lower aldosterone levels at 17 weeks of therapy, but not at 43 weeks.6 This phenomenon is known as “aldosterone escape.”
Several mechanisms might explain this phenomenon. Angiotensin II, a potent inducer of aldosterone, is “reactivated” during long-term ACE inhibitor therapy. Interestingly, patients progress toward aldosterone escape regardless of whether the ACE inhibitor dose is low or high.7 There is evidence that some aldosterone is produced by endothelial cells and vascular smooth muscle in the heart and blood vessels,8 but ACE inhibitors and ARBs suppress only the aldosterone secreted by the adrenal glands.
Regardless of the mechanism, aldosterone escape can blunt the effects of ACE inhibitors and ARBs, reducing their favorable effects on the risk of death in heart failure patients. This is the rationale for also using ARAs.
ARAs IN HEART FAILURE
Aldosterone acts by regulating gene expression after binding to mineralocorticoid receptors. These receptors are found not only in epithelial tissue in the kidneys and glands, but also in nonepithelial tissues such as cardiomyocytes, vessel walls, and the hippocampus of the brain.9 The nonepithelial effects were first demonstrated 2 decades ago by Brilla et al,10 who noted that chronically elevated aldosterone levels in rats promoted cardiac fibroblast growth, collagen accumulation, and, hence, ventricular remodeling.
The hypertensive effect of aldosterone may also be mediated through mineralocorticoid receptors in the brain. Gomez-Sanchez et al11 found that infusing aldosterone into the cerebral ventricles caused significant hypertension. A selective mineralocorticoid antagonist inhibited this effect when infused into the cerebral ventricles but not when given systemically.
In 1959, Cella and Kagawa created spironolactone, a nonselective ARA, by combining elements of progesterone for its antimineralocorticoid effect and elements of digitoxin for its cardiotonic effect.12 Although spironolactone is very effective in treating hypertension and heart failure, its use is limited by progestational and antiandrogenic side effects. This led, in 1987, to the invention by de Gasparo et al of a newer molecule, a selective ARA now called eplerenone.13 Although eplerenone may be somewhat less potent than spironolactone in blocking mineralocorticoid receptors, no significant difference in efficacy has been noted in randomized clinical trials, and its antiandrogenic action is negligible.12
Although these drugs target aldosterone receptors, newer drugs may target different aspects of mineralocorticoid activities, and thus the term “mineralocorticoid receptor antagonist” has been proposed.
TRIALS OF ARAs IN HEART FAILURE
An online data supplement that accompanies this paper at provides a detailed comparison of the three major trials of ARAs in patients with heart failure.
The Randomized Aldactone Evaluation Study (RALES)
The first major clinical trial of an ARA was the Randomized Aldactone Evaluation Study (RALES),14 a randomized, double-blind, controlled comparison of spironolactone and placebo.
The 1,663 patients in the trial all had severe heart failure (New York Heart Association class [NYHA] III and ambulatory class IV symptoms) and a left ventricular ejection fraction of 35% or less. Most were on an ACE inhibitor, a loop diuretic, and digoxin, but only 10% of patients in both groups were on a beta-blocker. Patients with chronic renal failure (serum creatinine > 2.5 mg/dL) or hyperkalemia (potassium > 5.0 mmol/L) were excluded.
RALES was halted early when an interim analysis at a mean follow-up of 24 months showed that significantly fewer patients were dying in the spironolactone group; their all-cause mortality rate was 30% lower (relative risk [RR] 0.70, 95% confidence interval [CI] 0.60–0.82, P < .001), and their cardiac mortality rate was 31% lower (RR 0.69, 95% CI 0.58–0.82, P < .001). This was concordant with a lower risk of both sudden cardiac death and death from progressive heart failure. The risk of hospitalization for cardiac causes was also 30% lower for patients in the spironolactone group, who also experienced significant symptom improvement.
Gynecomastia and breast pain occurred in about 10% of patients in the spironolactone group, and adverse effects leading to study drug discontinuation occurred in 2%.14
The Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS)
The next landmark trial of an ARA was the Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS).15 A total of 6,632 patients were randomized to receive eplerenone or placebo in this multicenter, double-blind trial. To be enrolled, patients had to have acute myocardial infarction, a left ventricular ejection fraction of 40% or less, and either clinical signs of heart failure 3 to 14 days after the infarction or a history of diabetes mellitus. Patients were excluded if they had chronic kidney disease (defined as a serum creatinine > 2.5 mg/dL or an estimated glomerular filtration rate < 30 mL/min/1.73 m2) or hyperkalemia (a serum potassium > 5.0 mmol/L). All the patients received optimal medical therapy and reperfusion therapy, if warranted.
This event-driven trial was stopped when 1,012 deaths had occurred. During a mean follow-up of 16 months, there was a 15% lower rate of all-cause mortality in the eplerenone group (RR 0.85, 95% CI 0.75–0.96, P = .008) and a 13% lower rate of cardiovascular mortality (RR 0.83, 95% CI 0.72–0.94, P = .005). The reduction in the cardiovascular mortality rate was attributed to a 21% reduction in the rate of sudden cardiac deaths. The rate of heart failure hospitalization was also lower in the eplerenone group.
Serious hyperkalemia occurred significantly more frequently in the eplerenone group (5.5% vs 3.9%, P = .002), but similar rates of gynecomastia were observed. The incidence of hyperkalemia was higher in patients with a creatinine clearance less than 50 mL/min.
Further analyses revealed a 31% lower rate of all-cause mortality (95% CI 0.54–0.89, P = .004) and a 32% lower rate of cardiovascular mortality (95% CI 0.53–0.88, P = .003) at 30 days after randomization in the eplerenone group.16 Importantly, 25% of all deaths in the EPHESUS study during the 16-month follow-up period occurred in the first 30 days after randomization. The Kaplan-Meier survival curves showed separation as early as 5 days after randomization. Hence, the 30-day mortality results from EPHESUS further indicated that starting eplerenone early may be particularly beneficial.
The Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure (EMPHASIS-HF)
After RALES and EPHESUS, a gap remained in our knowledge, ie, how to use ARAs in patients with mild heart failure, who account for most cases. This led to the EMPHASIS-HF (Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure) trial, which expanded the indications for ARAs to patients with chronic systolic heart failure with mild symptoms.17
In this double-blind trial, 2,737 patients with NYHA class II heart failure with a left ventricular ejection fraction of 35% or less were randomized to receive oral eplerenone 25 mg or placebo once daily. All patients were already on a beta-blocker; they were also all on an ACE inhibitor, an ARB, or both at the recommended or maximal tolerated dose. Patients with a glomerular filtration rate between 30 and 49 mL/min were started on alternate-day dosing, and those with glomerular filtration rates below 30 mL/min were excluded.
To ensure that the event rate was high enough to give this trial sufficient power:
- Only patients age 55 years or older were included
- Patients with a left ventricular ejection fraction greater than 30% were enrolled only if the QRS duration was greater than 130 ms (only 3.5% of patients in both groups were enrolled based on this criterion)
- Patients either had to have been hospitalized for cardiovascular reasons in the 6 months before randomization or had to have elevated natriuretic peptides (B-type natriuretic peptide [BNP] level > 250 pg/mL or N-terminal pro-BNP > 500 pg/mL in men and > 750 pg/mL in women).
The study was stopped early at a median follow-up of 21 months after an interim analysis showed a significantly lower rate of the primary composite end point (death from a cardiovascular cause or hospitalization for heart failure) in the eplerenone group: 18.3% vs 25.9% (hazard ratio [HR] 0.63, 95% CI 0.54– 0.74, P < .001). The rates of all-cause mortality were 12.5% vs 15.5% (HR 0.76, 95% CI 0.62–0.93, P = .008), and the rates of cardiovascular mortality were 10.8% vs 13.5% (HR 0.76, 95% CI 0.61–0.94, P = .01). Kaplan-Meier curves for all-cause mortality showed significant separation only after 1 year, which was not the case in EPHESUS and RALES. But the curves for hospitalization separated within a few weeks after randomization.
The incidence of hyperkalemia (serum potassium level > 5.5 mmol/L) was significantly higher in the eplerenone group (11.8% vs 7.2%, P < .001), but there was no statistically significant difference between groups when potassium levels above 6 mmol/L were considered (2.5% vs 1.9%, P = .29). This is despite one-third of patients having an estimated glomerular filtration rate less than 60 mL/min/1.73 m2. Breast symptoms were very rare, occurring in 1% or fewer patients in both groups. The discontinuation rate of the study drug was similar in both groups.
HOW DO ARAs PREVENT DEATH?
Multiple studies show that spironolactone and eplerenone lower blood pressure in a dose-related manner.18 These drugs reduce fluid volume and pulmonary congestion, which could have been the primary mechanism for the reduction in heart failure hospitalizations in the EMPHASIS-HF trial. But other mechanisms might explain the reduction in cardiovascular mortality rates in the trials summarized above.
Transcardiac extraction of aldosterone was increased in a study of patients with heart failure. 19 The transcardiac gradient of plasma aldosterone correlated with levels of procollagen III N-terminal propeptide, a biochemical marker of myocardial fibrosis. This suggests that aldosterone could be a stimulant of myocardial fibrosis. Spironolactone inhibited the transcardiac extraction of aldosterone in the same study.19
In another study,20 spironolactone significantly suppressed elevation of procollagen III N-terminal propeptide after myocardial infarction. It was also demonstrated that spironolactone prevented left ventricular remodeling after infarction, even in patients receiving an ACE inhibitor. Similar results, ie, decreased left ventricular myocardial fibrosis and remodeling, were noted in another trial in which eplerenone was added to an ARB.21
Myocardial fibrosis is a known substrate for ventricular arrhythmias. In a randomized study in 35 patients, spironolactone decreased the incidence of ventricular arrhythmias.22 This finding correlates with the decreased incidence of sudden cardiac death in the RALES and EPHESUS trials.
ADVERSE EFFECTS OF ARAs
Hyperkalemia, hyperkalemia, hyperkalemia
Potassium excretion is physiologically regulated by the serum aldosterone concentration and by the delivery of sodium to the distal nephron. Aldosterone increases potassium excretion. As a result of decreased renal perfusion that occurs with heart failure, sodium is intensely reabsorbed in the proximal tubule, and very little sodium reaches the distal nephron. When aldosterone receptors are blocked by ARAs, the risk of hyperkalemia increases.23
Other electrolyte abnormalities associated with ARAs are hyponatremia and hyperchloremic metabolic acidosis (Table 1). There could be a reversible decline in the glomerular filtration rate as well.24 Of note, most patients with chronic systolic heart failure in the RALES and EMPHASIS-HF trials were already receiving a diuretic; thus, the adverse effect profile of ARAs in otherwise euvolemic (or even hypovolemic) patients is not well appreciated.
Failure to closely monitor electrolyte levels increases the risk of hyperkalemia and renal failure, so there is a need for regular follow-up visits for patients taking an ARA.25 This was made clear when a population-based analysis from Canada compared the rates of hyperkalemia-related hospitalization and death before and after the RALES trial was published. The prescription rate for spironolactone increased threefold, but the rate of hyperkalemia-related hospitalization increased fourfold and the rate of death increased sixfold.26
Although caution is recommended when starting a patient on an ARA, a recent trial conducted in 167 cardiology practices noted that ARAs were the most underused drugs for heart failure. In this study, an ARA was prescribed to only 35% of eligible patients. The prescription rate was not significantly higher even in dedicated heart failure clinics.27 Possible reasons suggested by the authors were drug side effects, the need for closer monitoring of laboratory values, and a lack of knowledge.
A population-based analysis from the United Kingdom found a significant increase over time in spironolactone prescriptions after the release of the RALES trial results, but there was no increase in the rate of serious hyperkalemia (serum potassium > 6 mmol/L) or hyperkalemia-related hospitalization.28 The authors suggested that careful monitoring could prevent hyperkalemia-related complications. They also observed that 75% of patients who had spironolactone-associated hyperkalemia were over 65 years old. Hence, we recommend closer monitoring when starting an elderly patient on an ARA.
Breast, gastrointestinal symptoms
The nonselective ARA spironolactone is associated with antiandrogenic side effects. In a smaller study in patients with resistant hypertension, Nishizaka et al noted that low-dose spironolactone (up to 50 mg/day) was associated with breast tenderness in about 10%.29 Breast symptoms with spironolactone are dose-related, and the incidence can be as high as 50% when the drug is used in dosages of 150 mg/day or higher.30
In one population-based case-control study, spironolactone was associated with a 2.7 times higher risk of gastrointestinal side effects (bleeding or ulcer).31
ARAs IN HEART FAILURE WITH PRESERVED EJECTION FRACTION
The concept of diastolic heart failure or “heart failure with preserved ejection fraction” has been growing. A significant proportion of patients with a diagnosis of heart failure have preserved left ventricular ejection fraction (≥ 50%) and diastolic dysfunction.
Despite multiple trials, no treatment has been shown to lower the mortality rate in heart failure with preserved ejection fraction.32,33 A recently published randomized controlled trial in 44 patients with this condition showed reduction in serum biochemical markers of collagen turnover and improvement in diastolic function with ARAs, but there was no difference in exercise capacity.34 A larger double-blind randomized control trial, Aldosterone Receptor Blockade in Diastolic Heart Failure (Aldo-DHF), is under way to evaluate the effects of ARAs on exercise capacity and diastolic function in patients with heart failure with preserved ejection fraction.35
In January 2012, the Trial of Aldosterone Antagonist Therapy in Adults With Preserved Ejection Fraction Congestive Heart Failure (TOPCAT) completed enrollment of 3,445 patients to study the effect of ARAs in reducing the composite end point of cardiovascular mortality, aborted cardiac arrest, and heart failure hospitalization. Long-term follow-up of this event-driven study is currently under way.
ARAs IN DIABETES MELLITUS AND CHRONIC KIDNEY DISEASE
Under physiologic conditions, the serum aldosterone level is regulated by volume status through the renin-angiotensin system. But in patients with chronic kidney disease, the serum aldosterone level could be elevated without renin-angiotensin system stimulation.36
High aldosterone levels were associated with proteinuria and glomerulosclerosis in rats.37 In a study in 83 patients, aldosterone receptor blockade was shown to decrease proteinuria and possibly to retard the progression of chronic kidney disease. In this trial, baseline serum aldosterone levels correlated with proteinuria.38 Animal studies suggest that adipocyte-derived factors may stimulate aldosterone, which may be relevant in patients who have both chronic kidney disease and metabolic syndrome.39
The impact of ARAs in patients with diabetes mellitus is often overlooked. In EPHESUS, diabetes mellitus was an inclusion criterion even in the absence of heart failure signs and symptoms in the postinfarction setting of impaired left ventricular ejection fraction.15
In patients with diabetic nephropathy, there is growing evidence that ARAs can decrease proteinuria, even if the serum aldosterone level is normal. For example, in a study in 20 patients with diabetic nephropathy, spironolactone reduced proteinuria by 32%. This reduction was independent of serum aldosterone levels.40
In diabetic rats, hyperglycemia was noted to cause podocyte injury through mineralocorticoid receptor-mediated production of reactive oxygen species, independently of serum aldosterone levels. Spironolactone decreased the production of reactive oxygen species, thereby potentially reducing proteinuria.41
RECOMMENDATIONS ARE BEING REVISED
The most recent joint guidelines of the American Heart Association and the American College of Cardiology for the management of heart failure42 were published in 2009, which was before the EMPHASIS-HF results. An update is expected soon. In the 2009 version, ARAs received a class I recommendation for patients with moderately severe to severe symptoms, decreased ejection fraction, normal renal function, and normal potassium levels. The guidelines also said that the risks of ARAs may outweigh their benefits if regular monitoring is not possible.
The recommended starting dosage is 12.5 mg/day of spironolactone or 25 mg/day of eplerenone; the dose can be doubled, if tolerated.
Close monitoring is recommended, ie, measuring serum potassium and renal function 3 and 7 days after starting therapy and then monthly for the first 3 months. Closer monitoring is needed if an ACE inhibitor or an ARB is added later. In elderly patients, the glomerular filtration rate is preferred over the serum creatinine level, and ARA therapy is not advisable if the glomerular filtration rate is less than 30 mL/min/1.73 m2.
Avoid concomitant use of the following:
- Potassium supplements (unless persistent hypokalemia is present)
- Nonsteroidal anti-inflammatory drugs
- An ACE inhibitor and an ARB in combination
- A high dose of an ACE inhibitor or ARB.
Conditions that can lead to dehydration (eg, diarrhea, excessive use of diuretics) or acute illness should warrant reduction (or even withholding) of ARAs. When to discontinue ARA therapy is not well described, nor is the safety of starting ARAs in the hospital. However, it is clear that many patients who are potentially eligible for ARAs are not prescribed them.43
The guidelines are currently being revised, and will likely incorporate the new data from EMPHASIS-HF to extend to a broader population. The benefits of ARAs can be met only if the risks are minimized.
WHICH ARA IS BETTER?
The pharmacologic differences between the two ARAs have been described earlier, and guidelines have advocated evidence-based use of ARAs for their respective indications. There have been no large-scale, head-to-head comparisons of spironolactone and eplerenone in the heart failure population, and in clinical practice the drugs are prescribed interchangeably in most patients.
A double-blind randomized controlled trial in 141 patients with hypertension and primary hyperaldosteronism found that spironolactone lowered diastolic blood pressure more, but it also caused antiandrogenic effects more often.44
There is some evidence to suggest that eplerenone has a better metabolic profile than spironolactone. The data came from a small randomized controlled trial in 107 stable outpatients with mild heart failure.45 Patients who were prescribed spironolactone had a higher cortisol level and hemoglobin A1c level 4 months after starting treatment. This effect was not seen in patients who were on eplerenone. However, these findings need to be confirmed in larger trials.
While the differences between the two drugs remain to be determined, the most important differences in clinical practice are selectivity for receptors (and hence their antiandrogenic side effects) and price. Even though it is available as a generic drug, eplerenone still costs at least three times more than spironolactone for the same dosage and indication.
Over the past 30 years, the focus of treating heart failure has shifted from managing symptoms to prolonging lives. When the neurohormonal hypothesis (ie, the concept that neurohormonal dysregulation and not merely hemodynamic changes are responsible for the onset and progression of heart failure) was introduced, it brought a dramatic change that included new classes of drugs that interfere with the renin-angiotensin-aldosterone system, ie, angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), and, most recently, aldosterone receptor antagonists (ARAs) (Figure 1).
Evidence supporting the use of the ARAs spironolactone (Aldactone) and eplerenone (Inspra) in heart failure has been growing, as has evidence of their usefulness in treating diabetes and chronic renal disease. Still, these drugs must be used cautiously, as they can cause hyperkalemia.
This paper will review the clinical use of ARAs in symptomatic systolic heart failure, their side effects, the findings and implications of recent trials, and controversies in this area, notably whether there is any evidence favoring the use of one drug over another.
ALDOSTERONE IN HEART FAILURE
Aldosterone, a hormone secreted by the zona glomerulosa of the adrenal gland, was first isolated by Simpson and Tait more than half a century ago.1 Later, it was found to promote reabsorption of sodium and excretion of potassium in the kidneys and hence was categorized as a mineralocorticoid hormone.
Release of aldosterone is stimulated by decreased renal perfusion via angiotensin II, hyperkalemia, and possibly adrenocorticotropic hormone.2 Aldosterone exerts its effects by binding to mineralocorticoid receptors in renal epithelial cells.
Aldosterone has several deleterious effects on the failing heart, primarily sodium and fluid retention, but also endothelial dysfunction, left ventricular hypertrophy, and myocardial fibrosis.2,3 Plasma aldosterone levels can be markedly elevated in patients with heart failure, likely due to activation of the renin-angiotensin-aldosterone system. Elevated aldosterone and angiotensin II levels have been associated with higher mortality rates.4
ALDOSTERONE ‘ESCAPE’ BLUNTS THE EFFECT OF ACE INHIBITORS AND ARBs
ACE inhibitors and ARBs have become standards of care for patients with systolic heart failure, and for many years, it was believed that these drugs suppressed aldosterone levels sufficiently. But elevated aldosterone levels have been noted in up to 38% of patients on chronic ACE inhibitor therapy.5 In one study, patients on dual blockade, ie, on both an ACE inhibitor and an ARB, had significantly lower aldosterone levels at 17 weeks of therapy, but not at 43 weeks.6 This phenomenon is known as “aldosterone escape.”
Several mechanisms might explain this phenomenon. Angiotensin II, a potent inducer of aldosterone, is “reactivated” during long-term ACE inhibitor therapy. Interestingly, patients progress toward aldosterone escape regardless of whether the ACE inhibitor dose is low or high.7 There is evidence that some aldosterone is produced by endothelial cells and vascular smooth muscle in the heart and blood vessels,8 but ACE inhibitors and ARBs suppress only the aldosterone secreted by the adrenal glands.
Regardless of the mechanism, aldosterone escape can blunt the effects of ACE inhibitors and ARBs, reducing their favorable effects on the risk of death in heart failure patients. This is the rationale for also using ARAs.
ARAs IN HEART FAILURE
Aldosterone acts by regulating gene expression after binding to mineralocorticoid receptors. These receptors are found not only in epithelial tissue in the kidneys and glands, but also in nonepithelial tissues such as cardiomyocytes, vessel walls, and the hippocampus of the brain.9 The nonepithelial effects were first demonstrated 2 decades ago by Brilla et al,10 who noted that chronically elevated aldosterone levels in rats promoted cardiac fibroblast growth, collagen accumulation, and, hence, ventricular remodeling.
The hypertensive effect of aldosterone may also be mediated through mineralocorticoid receptors in the brain. Gomez-Sanchez et al11 found that infusing aldosterone into the cerebral ventricles caused significant hypertension. A selective mineralocorticoid antagonist inhibited this effect when infused into the cerebral ventricles but not when given systemically.
In 1959, Cella and Kagawa created spironolactone, a nonselective ARA, by combining elements of progesterone for its antimineralocorticoid effect and elements of digitoxin for its cardiotonic effect.12 Although spironolactone is very effective in treating hypertension and heart failure, its use is limited by progestational and antiandrogenic side effects. This led, in 1987, to the invention by de Gasparo et al of a newer molecule, a selective ARA now called eplerenone.13 Although eplerenone may be somewhat less potent than spironolactone in blocking mineralocorticoid receptors, no significant difference in efficacy has been noted in randomized clinical trials, and its antiandrogenic action is negligible.12
Although these drugs target aldosterone receptors, newer drugs may target different aspects of mineralocorticoid activities, and thus the term “mineralocorticoid receptor antagonist” has been proposed.
TRIALS OF ARAs IN HEART FAILURE
An online data supplement that accompanies this paper at provides a detailed comparison of the three major trials of ARAs in patients with heart failure.
The Randomized Aldactone Evaluation Study (RALES)
The first major clinical trial of an ARA was the Randomized Aldactone Evaluation Study (RALES),14 a randomized, double-blind, controlled comparison of spironolactone and placebo.
The 1,663 patients in the trial all had severe heart failure (New York Heart Association class [NYHA] III and ambulatory class IV symptoms) and a left ventricular ejection fraction of 35% or less. Most were on an ACE inhibitor, a loop diuretic, and digoxin, but only 10% of patients in both groups were on a beta-blocker. Patients with chronic renal failure (serum creatinine > 2.5 mg/dL) or hyperkalemia (potassium > 5.0 mmol/L) were excluded.
RALES was halted early when an interim analysis at a mean follow-up of 24 months showed that significantly fewer patients were dying in the spironolactone group; their all-cause mortality rate was 30% lower (relative risk [RR] 0.70, 95% confidence interval [CI] 0.60–0.82, P < .001), and their cardiac mortality rate was 31% lower (RR 0.69, 95% CI 0.58–0.82, P < .001). This was concordant with a lower risk of both sudden cardiac death and death from progressive heart failure. The risk of hospitalization for cardiac causes was also 30% lower for patients in the spironolactone group, who also experienced significant symptom improvement.
Gynecomastia and breast pain occurred in about 10% of patients in the spironolactone group, and adverse effects leading to study drug discontinuation occurred in 2%.14
The Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS)
The next landmark trial of an ARA was the Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS).15 A total of 6,632 patients were randomized to receive eplerenone or placebo in this multicenter, double-blind trial. To be enrolled, patients had to have acute myocardial infarction, a left ventricular ejection fraction of 40% or less, and either clinical signs of heart failure 3 to 14 days after the infarction or a history of diabetes mellitus. Patients were excluded if they had chronic kidney disease (defined as a serum creatinine > 2.5 mg/dL or an estimated glomerular filtration rate < 30 mL/min/1.73 m2) or hyperkalemia (a serum potassium > 5.0 mmol/L). All the patients received optimal medical therapy and reperfusion therapy, if warranted.
This event-driven trial was stopped when 1,012 deaths had occurred. During a mean follow-up of 16 months, there was a 15% lower rate of all-cause mortality in the eplerenone group (RR 0.85, 95% CI 0.75–0.96, P = .008) and a 13% lower rate of cardiovascular mortality (RR 0.83, 95% CI 0.72–0.94, P = .005). The reduction in the cardiovascular mortality rate was attributed to a 21% reduction in the rate of sudden cardiac deaths. The rate of heart failure hospitalization was also lower in the eplerenone group.
Serious hyperkalemia occurred significantly more frequently in the eplerenone group (5.5% vs 3.9%, P = .002), but similar rates of gynecomastia were observed. The incidence of hyperkalemia was higher in patients with a creatinine clearance less than 50 mL/min.
Further analyses revealed a 31% lower rate of all-cause mortality (95% CI 0.54–0.89, P = .004) and a 32% lower rate of cardiovascular mortality (95% CI 0.53–0.88, P = .003) at 30 days after randomization in the eplerenone group.16 Importantly, 25% of all deaths in the EPHESUS study during the 16-month follow-up period occurred in the first 30 days after randomization. The Kaplan-Meier survival curves showed separation as early as 5 days after randomization. Hence, the 30-day mortality results from EPHESUS further indicated that starting eplerenone early may be particularly beneficial.
The Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure (EMPHASIS-HF)
After RALES and EPHESUS, a gap remained in our knowledge, ie, how to use ARAs in patients with mild heart failure, who account for most cases. This led to the EMPHASIS-HF (Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure) trial, which expanded the indications for ARAs to patients with chronic systolic heart failure with mild symptoms.17
In this double-blind trial, 2,737 patients with NYHA class II heart failure with a left ventricular ejection fraction of 35% or less were randomized to receive oral eplerenone 25 mg or placebo once daily. All patients were already on a beta-blocker; they were also all on an ACE inhibitor, an ARB, or both at the recommended or maximal tolerated dose. Patients with a glomerular filtration rate between 30 and 49 mL/min were started on alternate-day dosing, and those with glomerular filtration rates below 30 mL/min were excluded.
To ensure that the event rate was high enough to give this trial sufficient power:
- Only patients age 55 years or older were included
- Patients with a left ventricular ejection fraction greater than 30% were enrolled only if the QRS duration was greater than 130 ms (only 3.5% of patients in both groups were enrolled based on this criterion)
- Patients either had to have been hospitalized for cardiovascular reasons in the 6 months before randomization or had to have elevated natriuretic peptides (B-type natriuretic peptide [BNP] level > 250 pg/mL or N-terminal pro-BNP > 500 pg/mL in men and > 750 pg/mL in women).
The study was stopped early at a median follow-up of 21 months after an interim analysis showed a significantly lower rate of the primary composite end point (death from a cardiovascular cause or hospitalization for heart failure) in the eplerenone group: 18.3% vs 25.9% (hazard ratio [HR] 0.63, 95% CI 0.54– 0.74, P < .001). The rates of all-cause mortality were 12.5% vs 15.5% (HR 0.76, 95% CI 0.62–0.93, P = .008), and the rates of cardiovascular mortality were 10.8% vs 13.5% (HR 0.76, 95% CI 0.61–0.94, P = .01). Kaplan-Meier curves for all-cause mortality showed significant separation only after 1 year, which was not the case in EPHESUS and RALES. But the curves for hospitalization separated within a few weeks after randomization.
The incidence of hyperkalemia (serum potassium level > 5.5 mmol/L) was significantly higher in the eplerenone group (11.8% vs 7.2%, P < .001), but there was no statistically significant difference between groups when potassium levels above 6 mmol/L were considered (2.5% vs 1.9%, P = .29). This is despite one-third of patients having an estimated glomerular filtration rate less than 60 mL/min/1.73 m2. Breast symptoms were very rare, occurring in 1% or fewer patients in both groups. The discontinuation rate of the study drug was similar in both groups.
HOW DO ARAs PREVENT DEATH?
Multiple studies show that spironolactone and eplerenone lower blood pressure in a dose-related manner.18 These drugs reduce fluid volume and pulmonary congestion, which could have been the primary mechanism for the reduction in heart failure hospitalizations in the EMPHASIS-HF trial. But other mechanisms might explain the reduction in cardiovascular mortality rates in the trials summarized above.
Transcardiac extraction of aldosterone was increased in a study of patients with heart failure. 19 The transcardiac gradient of plasma aldosterone correlated with levels of procollagen III N-terminal propeptide, a biochemical marker of myocardial fibrosis. This suggests that aldosterone could be a stimulant of myocardial fibrosis. Spironolactone inhibited the transcardiac extraction of aldosterone in the same study.19
In another study,20 spironolactone significantly suppressed elevation of procollagen III N-terminal propeptide after myocardial infarction. It was also demonstrated that spironolactone prevented left ventricular remodeling after infarction, even in patients receiving an ACE inhibitor. Similar results, ie, decreased left ventricular myocardial fibrosis and remodeling, were noted in another trial in which eplerenone was added to an ARB.21
Myocardial fibrosis is a known substrate for ventricular arrhythmias. In a randomized study in 35 patients, spironolactone decreased the incidence of ventricular arrhythmias.22 This finding correlates with the decreased incidence of sudden cardiac death in the RALES and EPHESUS trials.
ADVERSE EFFECTS OF ARAs
Hyperkalemia, hyperkalemia, hyperkalemia
Potassium excretion is physiologically regulated by the serum aldosterone concentration and by the delivery of sodium to the distal nephron. Aldosterone increases potassium excretion. As a result of decreased renal perfusion that occurs with heart failure, sodium is intensely reabsorbed in the proximal tubule, and very little sodium reaches the distal nephron. When aldosterone receptors are blocked by ARAs, the risk of hyperkalemia increases.23
Other electrolyte abnormalities associated with ARAs are hyponatremia and hyperchloremic metabolic acidosis (Table 1). There could be a reversible decline in the glomerular filtration rate as well.24 Of note, most patients with chronic systolic heart failure in the RALES and EMPHASIS-HF trials were already receiving a diuretic; thus, the adverse effect profile of ARAs in otherwise euvolemic (or even hypovolemic) patients is not well appreciated.
Failure to closely monitor electrolyte levels increases the risk of hyperkalemia and renal failure, so there is a need for regular follow-up visits for patients taking an ARA.25 This was made clear when a population-based analysis from Canada compared the rates of hyperkalemia-related hospitalization and death before and after the RALES trial was published. The prescription rate for spironolactone increased threefold, but the rate of hyperkalemia-related hospitalization increased fourfold and the rate of death increased sixfold.26
Although caution is recommended when starting a patient on an ARA, a recent trial conducted in 167 cardiology practices noted that ARAs were the most underused drugs for heart failure. In this study, an ARA was prescribed to only 35% of eligible patients. The prescription rate was not significantly higher even in dedicated heart failure clinics.27 Possible reasons suggested by the authors were drug side effects, the need for closer monitoring of laboratory values, and a lack of knowledge.
A population-based analysis from the United Kingdom found a significant increase over time in spironolactone prescriptions after the release of the RALES trial results, but there was no increase in the rate of serious hyperkalemia (serum potassium > 6 mmol/L) or hyperkalemia-related hospitalization.28 The authors suggested that careful monitoring could prevent hyperkalemia-related complications. They also observed that 75% of patients who had spironolactone-associated hyperkalemia were over 65 years old. Hence, we recommend closer monitoring when starting an elderly patient on an ARA.
Breast, gastrointestinal symptoms
The nonselective ARA spironolactone is associated with antiandrogenic side effects. In a smaller study in patients with resistant hypertension, Nishizaka et al noted that low-dose spironolactone (up to 50 mg/day) was associated with breast tenderness in about 10%.29 Breast symptoms with spironolactone are dose-related, and the incidence can be as high as 50% when the drug is used in dosages of 150 mg/day or higher.30
In one population-based case-control study, spironolactone was associated with a 2.7 times higher risk of gastrointestinal side effects (bleeding or ulcer).31
ARAs IN HEART FAILURE WITH PRESERVED EJECTION FRACTION
The concept of diastolic heart failure or “heart failure with preserved ejection fraction” has been growing. A significant proportion of patients with a diagnosis of heart failure have preserved left ventricular ejection fraction (≥ 50%) and diastolic dysfunction.
Despite multiple trials, no treatment has been shown to lower the mortality rate in heart failure with preserved ejection fraction.32,33 A recently published randomized controlled trial in 44 patients with this condition showed reduction in serum biochemical markers of collagen turnover and improvement in diastolic function with ARAs, but there was no difference in exercise capacity.34 A larger double-blind randomized control trial, Aldosterone Receptor Blockade in Diastolic Heart Failure (Aldo-DHF), is under way to evaluate the effects of ARAs on exercise capacity and diastolic function in patients with heart failure with preserved ejection fraction.35
In January 2012, the Trial of Aldosterone Antagonist Therapy in Adults With Preserved Ejection Fraction Congestive Heart Failure (TOPCAT) completed enrollment of 3,445 patients to study the effect of ARAs in reducing the composite end point of cardiovascular mortality, aborted cardiac arrest, and heart failure hospitalization. Long-term follow-up of this event-driven study is currently under way.
ARAs IN DIABETES MELLITUS AND CHRONIC KIDNEY DISEASE
Under physiologic conditions, the serum aldosterone level is regulated by volume status through the renin-angiotensin system. But in patients with chronic kidney disease, the serum aldosterone level could be elevated without renin-angiotensin system stimulation.36
High aldosterone levels were associated with proteinuria and glomerulosclerosis in rats.37 In a study in 83 patients, aldosterone receptor blockade was shown to decrease proteinuria and possibly to retard the progression of chronic kidney disease. In this trial, baseline serum aldosterone levels correlated with proteinuria.38 Animal studies suggest that adipocyte-derived factors may stimulate aldosterone, which may be relevant in patients who have both chronic kidney disease and metabolic syndrome.39
The impact of ARAs in patients with diabetes mellitus is often overlooked. In EPHESUS, diabetes mellitus was an inclusion criterion even in the absence of heart failure signs and symptoms in the postinfarction setting of impaired left ventricular ejection fraction.15
In patients with diabetic nephropathy, there is growing evidence that ARAs can decrease proteinuria, even if the serum aldosterone level is normal. For example, in a study in 20 patients with diabetic nephropathy, spironolactone reduced proteinuria by 32%. This reduction was independent of serum aldosterone levels.40
In diabetic rats, hyperglycemia was noted to cause podocyte injury through mineralocorticoid receptor-mediated production of reactive oxygen species, independently of serum aldosterone levels. Spironolactone decreased the production of reactive oxygen species, thereby potentially reducing proteinuria.41
RECOMMENDATIONS ARE BEING REVISED
The most recent joint guidelines of the American Heart Association and the American College of Cardiology for the management of heart failure42 were published in 2009, which was before the EMPHASIS-HF results. An update is expected soon. In the 2009 version, ARAs received a class I recommendation for patients with moderately severe to severe symptoms, decreased ejection fraction, normal renal function, and normal potassium levels. The guidelines also said that the risks of ARAs may outweigh their benefits if regular monitoring is not possible.
The recommended starting dosage is 12.5 mg/day of spironolactone or 25 mg/day of eplerenone; the dose can be doubled, if tolerated.
Close monitoring is recommended, ie, measuring serum potassium and renal function 3 and 7 days after starting therapy and then monthly for the first 3 months. Closer monitoring is needed if an ACE inhibitor or an ARB is added later. In elderly patients, the glomerular filtration rate is preferred over the serum creatinine level, and ARA therapy is not advisable if the glomerular filtration rate is less than 30 mL/min/1.73 m2.
Avoid concomitant use of the following:
- Potassium supplements (unless persistent hypokalemia is present)
- Nonsteroidal anti-inflammatory drugs
- An ACE inhibitor and an ARB in combination
- A high dose of an ACE inhibitor or ARB.
Conditions that can lead to dehydration (eg, diarrhea, excessive use of diuretics) or acute illness should warrant reduction (or even withholding) of ARAs. When to discontinue ARA therapy is not well described, nor is the safety of starting ARAs in the hospital. However, it is clear that many patients who are potentially eligible for ARAs are not prescribed them.43
The guidelines are currently being revised, and will likely incorporate the new data from EMPHASIS-HF to extend to a broader population. The benefits of ARAs can be met only if the risks are minimized.
WHICH ARA IS BETTER?
The pharmacologic differences between the two ARAs have been described earlier, and guidelines have advocated evidence-based use of ARAs for their respective indications. There have been no large-scale, head-to-head comparisons of spironolactone and eplerenone in the heart failure population, and in clinical practice the drugs are prescribed interchangeably in most patients.
A double-blind randomized controlled trial in 141 patients with hypertension and primary hyperaldosteronism found that spironolactone lowered diastolic blood pressure more, but it also caused antiandrogenic effects more often.44
There is some evidence to suggest that eplerenone has a better metabolic profile than spironolactone. The data came from a small randomized controlled trial in 107 stable outpatients with mild heart failure.45 Patients who were prescribed spironolactone had a higher cortisol level and hemoglobin A1c level 4 months after starting treatment. This effect was not seen in patients who were on eplerenone. However, these findings need to be confirmed in larger trials.
While the differences between the two drugs remain to be determined, the most important differences in clinical practice are selectivity for receptors (and hence their antiandrogenic side effects) and price. Even though it is available as a generic drug, eplerenone still costs at least three times more than spironolactone for the same dosage and indication.
- Simpson SA, Tait JF, Bush IE. Secretion of a salt-retaining hormone by the mammalian adrenal cortex. Lancet 1952; 2:226–228.
- Struthers AD, MacDonald TM. Review of aldosterone- and angiotensin II-induced target organ damage and prevention. Cardiovasc Res 2004; 61:663–670.
- Edelmann F, Schmidt AG, Gelbrich G, et al. Rationale and design of the “aldosterone receptor blockade in diastolic heart failure” trial: a double-blind, randomized, placebo-controlled, parallel group study to determine the effects of spironolactone on exercise capacity and diastolic function in patients with symptomatic diastolic heart failure (Aldo-DHF). Eur J Heart Fail 2010; 12:874–882.
- Swedberg K, Eneroth P, Kjekshus J, Wilhelmsen L. Hormones regulating cardiovascular function in patients with severe congestive heart failure and their relation to mortality. CONSENSUS Trial Study Group. Circulation 1990; 82:1730–1736.
- MacFadyen RJ, Lee AF, Morton JJ, Pringle SD, Struthers AD. How often are angiotensin II and aldosterone concentrations raised during chronic ACE inhibitor treatment in cardiac failure? Heart 1999; 82:57–61.
- McKelvie RS, Yusuf S, Pericak D, et al. Comparison of candesartan, enalapril, and their combination in congestive heart failure: randomized evaluation of strategies for left ventricular dysfunction (RESOLVD) pilot study. The RESOLVD Pilot Study Investigators. Circulation 1999; 100:1056–1064.
- Tang WH, Vagelos RH, Yee YG, et al. Neurohormonal and clinical responses to high- versus low-dose enalapril therapy in chronic heart failure. J Am Coll Cardiol 2002; 39:70–78.
- Weber KT. Aldosterone in congestive heart failure. N Engl J Med 2001; 345:1689–1697.
- Funder JW. The role of aldosterone and mineralocorticoid receptors in cardiovascular disease. Am J Cardiovasc Drugs 2007; 7:151–157.
- Brilla CG, Pick R, Tan LB, Janicki JS, Weber KT. Remodeling of the rat right and left ventricles in experimental hypertension. Circ Res 1990; 67:1355–1364.
- Gomez-Sanchez EP, Fort C, Thwaites D. Central mineralocorticoid receptor antagonism blocks hypertension in Dahl S/JR rats. Am J Physiol 1992; 262:E96–E99.
- Garthwaite SM, McMahon EG. The evolution of aldosterone antagonists. Mol Cell Endocrinol 2004; 217:27–31.
- de Gasparo M, Joss U, Ramjoué HP, et al. Three new epoxy-spirolactone derivatives: characterization in vivo and in vitro. J Pharmacol Exp Ther 1987; 240:650–656.
- Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999; 341:709–717.
- Pitt B, Remme W, Zannad F, et al; Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study Investigators. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 2003; 348:1309–1321.
- Pitt B, White H, Nicolau J, et al; EPHESUS Investigators. Eplerenone reduces mortality 30 days after randomization following acute myocardial infarction in patients with left ventricular systolic dysfunction and heart failure. J Am Coll Cardiol 2005; 46:425–431.
- Zannad F, McMurray JJ, Krum H, et al; EMPHASIS-HF Study Group. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011; 364:11–21.
- Weinberger MH, Roniker B, Krause SL, Weiss RJ. Eplerenone, a selective aldosterone blocker, in mild-to-moderate hypertension. Am J Hypertens 2002; 15:709–716.
- Tsutamoto T, Wada A, Maeda K, et al. Spironolactone inhibits the transcardiac extraction of aldosterone in patients with congestive heart failure. J Am Coll Cardiol 2000; 36:838–844.
- Hayashi M, Tsutamoto T, Wada A, et al. Immediate administration of mineralocorticoid receptor antagonist spironolactone prevents postinfarct left ventricular remodeling associated with suppression of a marker of myocardial collagen synthesis in patients with first anterior acute myocardial infarction. Circulation 2003; 107:2559–2565.
- Fraccarollo D, Galuppo P, Schmidt I, Ertl G, Bauersachs J. Additive amelioration of left ventricular remodeling and molecular alterations by combined aldosterone and angiotensin receptor blockade after myocardial infarction. Cardiovasc Res 2005; 67:97–105.
- Ramires FJ, Mansur A, Coelho O, et al. Effect of spironolactone on ventricular arrhythmias in congestive heart failure secondary to idiopathic dilated or to ischemic cardiomyopathy. Am J Cardiol 2000; 85:1207–1211.
- Palmer BF. Managing hyperkalemia caused by inhibitors of the reninangiotensin-aldosterone system. N Engl J Med 2004; 351:585–592.
- Sica DA. The risks and benefits of therapy with aldosterone receptor antagonist therapy. Curr Drug Saf 2007; 2:71–77.
- Shah KB, Rao K, Sawyer R, Gottlieb SS. The adequacy of laboratory monitoring in patients treated with spironolactone for congestive heart failure. J Am Coll Cardiol 2005; 46:845–849.
- Juurlink DN, Mamdani MM, Lee DS, et al. Rates of hyperkalemia after publication of the Randomized Aldactone Evaluation Study. N Engl J Med 2004; 351:543–551.
- Albert NM, Fonarow GC, Yancy CW, et al. Influence of dedicated heart failure clinics on delivery of recommended therapies in outpatient cardiology practices: findings from the Registry to Improve the Use of Evidence-Based Heart Failure Therapies in the Outpatient Setting (IMPROVE HF). Am Heart J 2010; 159:238–244.
- Wei L, Struthers AD, Fahey T, Watson AD, Macdonald TM. Spironolactone use and renal toxicity: population based longitudinal analysis. BMJ 2010; 340:c1768.
- Nishizaka MK, Zaman MA, Calhoun DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens 2003; 16:925–930.
- Jeunemaitre X, Chatellier G, Kreft-Jais C, et al. Efficacy and tolerance of spironolactone in essential hypertension. Am J Cardiol 1987; 60:820–825.
- Verhamme K, Mosis G, Dieleman J, Stricker B, Sturkenboom M. Spironolactone and risk of upper gastrointestinal events: population based case-control study. BMJ 2006; 333:330.
- Massie BM, Carson PE, McMurray JJ, et al; I-PRESERVE Investigators. Irbesartan in patients with heart failure and preserved ejection fraction. N Engl J Med 2008; 359:2456–2467.
- Yusuf S, Pfeffer MA, Swedberg K, et al; CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-Preserved Trial. Lancet 2003; 362:777–781.
- Deswal A, Richardson P, Bozkurt B, Mann DL. Results of the Randomized Aldosterone Antagonism in Heart Failure With Preserved Ejection Fraction Trial (RAAM-PEF). J Card Fail 2011; 17:634–642.
- Edelmann F, Schmidt AG, Gelbrich G, et al. Rationale and design of the ‘aldosterone receptor blockade in diastolic heart failure’ trial: a double-blind, randomized, placebo-controlled, parallel group study to determine the effects of spironolactone on exercise capacity and diastolic function in patients with symptomatic diastolic heart failure (Aldo-DHF). Eur J Heart Fail 2010; 12:874–882.
- Hené RJ, Boer P, Koomans HA, Mees EJ. Plasma aldosterone concentrations in chronic renal disease. Kidney Int 1982; 21:98–101.
- Greene EL, Kren S, Hostetter TH. Role of aldosterone in the remnant kidney model in the rat. J Clin Invest 1996; 98:1063–1068.
- Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int 2006; 70:2116–2123.
- Nagase M, Yoshida S, Shibata S, et al. Enhanced aldosterone signaling in the early nephropathy of rats with metabolic syndrome: possible contribution of fat-derived factors. J Am Soc Nephrol 2006; 17:3438–3446.
- Schjoedt KJ, Rossing K, Juhl TR, et al. Beneficial impact of spironolactone on nephrotic range albuminuria in diabetic nephropathy. Kidney Int 2006; 70:536–542.
- Toyonaga J, Tsuruya K, Ikeda H, et al. Spironolactone inhibits hyperglycemia-induced podocyte injury by attenuating ROS production. Nephrol Dial Transplant 2011; 26:2475–2484.
- Hunt SA, Abraham WT, Chin MH, et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 2009; 119:e391–e479.
- Albert NM, Yancy CW, Liang L, et al. Use of aldosterone antagonists in heart failure. JAMA 2009; 302:1658–1665.
- Parthasarathy HK, Ménard J, White WB, et al. A double-blind, randomized study comparing the antihypertensive effect of eplerenone and spironolactone in patients with hypertension and evidence of primary aldosteronism. J Hypertens 2011; 29:980–990.
- Yamaji M, Tsutamoto T, Kawahara C, et al. Effect of eplerenone versus spironolactone on cortisol and hemoglobin A1(c) levels in patients with chronic heart failure. Am Heart J 2010; 160:915–921.
- Simpson SA, Tait JF, Bush IE. Secretion of a salt-retaining hormone by the mammalian adrenal cortex. Lancet 1952; 2:226–228.
- Struthers AD, MacDonald TM. Review of aldosterone- and angiotensin II-induced target organ damage and prevention. Cardiovasc Res 2004; 61:663–670.
- Edelmann F, Schmidt AG, Gelbrich G, et al. Rationale and design of the “aldosterone receptor blockade in diastolic heart failure” trial: a double-blind, randomized, placebo-controlled, parallel group study to determine the effects of spironolactone on exercise capacity and diastolic function in patients with symptomatic diastolic heart failure (Aldo-DHF). Eur J Heart Fail 2010; 12:874–882.
- Swedberg K, Eneroth P, Kjekshus J, Wilhelmsen L. Hormones regulating cardiovascular function in patients with severe congestive heart failure and their relation to mortality. CONSENSUS Trial Study Group. Circulation 1990; 82:1730–1736.
- MacFadyen RJ, Lee AF, Morton JJ, Pringle SD, Struthers AD. How often are angiotensin II and aldosterone concentrations raised during chronic ACE inhibitor treatment in cardiac failure? Heart 1999; 82:57–61.
- McKelvie RS, Yusuf S, Pericak D, et al. Comparison of candesartan, enalapril, and their combination in congestive heart failure: randomized evaluation of strategies for left ventricular dysfunction (RESOLVD) pilot study. The RESOLVD Pilot Study Investigators. Circulation 1999; 100:1056–1064.
- Tang WH, Vagelos RH, Yee YG, et al. Neurohormonal and clinical responses to high- versus low-dose enalapril therapy in chronic heart failure. J Am Coll Cardiol 2002; 39:70–78.
- Weber KT. Aldosterone in congestive heart failure. N Engl J Med 2001; 345:1689–1697.
- Funder JW. The role of aldosterone and mineralocorticoid receptors in cardiovascular disease. Am J Cardiovasc Drugs 2007; 7:151–157.
- Brilla CG, Pick R, Tan LB, Janicki JS, Weber KT. Remodeling of the rat right and left ventricles in experimental hypertension. Circ Res 1990; 67:1355–1364.
- Gomez-Sanchez EP, Fort C, Thwaites D. Central mineralocorticoid receptor antagonism blocks hypertension in Dahl S/JR rats. Am J Physiol 1992; 262:E96–E99.
- Garthwaite SM, McMahon EG. The evolution of aldosterone antagonists. Mol Cell Endocrinol 2004; 217:27–31.
- de Gasparo M, Joss U, Ramjoué HP, et al. Three new epoxy-spirolactone derivatives: characterization in vivo and in vitro. J Pharmacol Exp Ther 1987; 240:650–656.
- Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999; 341:709–717.
- Pitt B, Remme W, Zannad F, et al; Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study Investigators. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 2003; 348:1309–1321.
- Pitt B, White H, Nicolau J, et al; EPHESUS Investigators. Eplerenone reduces mortality 30 days after randomization following acute myocardial infarction in patients with left ventricular systolic dysfunction and heart failure. J Am Coll Cardiol 2005; 46:425–431.
- Zannad F, McMurray JJ, Krum H, et al; EMPHASIS-HF Study Group. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011; 364:11–21.
- Weinberger MH, Roniker B, Krause SL, Weiss RJ. Eplerenone, a selective aldosterone blocker, in mild-to-moderate hypertension. Am J Hypertens 2002; 15:709–716.
- Tsutamoto T, Wada A, Maeda K, et al. Spironolactone inhibits the transcardiac extraction of aldosterone in patients with congestive heart failure. J Am Coll Cardiol 2000; 36:838–844.
- Hayashi M, Tsutamoto T, Wada A, et al. Immediate administration of mineralocorticoid receptor antagonist spironolactone prevents postinfarct left ventricular remodeling associated with suppression of a marker of myocardial collagen synthesis in patients with first anterior acute myocardial infarction. Circulation 2003; 107:2559–2565.
- Fraccarollo D, Galuppo P, Schmidt I, Ertl G, Bauersachs J. Additive amelioration of left ventricular remodeling and molecular alterations by combined aldosterone and angiotensin receptor blockade after myocardial infarction. Cardiovasc Res 2005; 67:97–105.
- Ramires FJ, Mansur A, Coelho O, et al. Effect of spironolactone on ventricular arrhythmias in congestive heart failure secondary to idiopathic dilated or to ischemic cardiomyopathy. Am J Cardiol 2000; 85:1207–1211.
- Palmer BF. Managing hyperkalemia caused by inhibitors of the reninangiotensin-aldosterone system. N Engl J Med 2004; 351:585–592.
- Sica DA. The risks and benefits of therapy with aldosterone receptor antagonist therapy. Curr Drug Saf 2007; 2:71–77.
- Shah KB, Rao K, Sawyer R, Gottlieb SS. The adequacy of laboratory monitoring in patients treated with spironolactone for congestive heart failure. J Am Coll Cardiol 2005; 46:845–849.
- Juurlink DN, Mamdani MM, Lee DS, et al. Rates of hyperkalemia after publication of the Randomized Aldactone Evaluation Study. N Engl J Med 2004; 351:543–551.
- Albert NM, Fonarow GC, Yancy CW, et al. Influence of dedicated heart failure clinics on delivery of recommended therapies in outpatient cardiology practices: findings from the Registry to Improve the Use of Evidence-Based Heart Failure Therapies in the Outpatient Setting (IMPROVE HF). Am Heart J 2010; 159:238–244.
- Wei L, Struthers AD, Fahey T, Watson AD, Macdonald TM. Spironolactone use and renal toxicity: population based longitudinal analysis. BMJ 2010; 340:c1768.
- Nishizaka MK, Zaman MA, Calhoun DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens 2003; 16:925–930.
- Jeunemaitre X, Chatellier G, Kreft-Jais C, et al. Efficacy and tolerance of spironolactone in essential hypertension. Am J Cardiol 1987; 60:820–825.
- Verhamme K, Mosis G, Dieleman J, Stricker B, Sturkenboom M. Spironolactone and risk of upper gastrointestinal events: population based case-control study. BMJ 2006; 333:330.
- Massie BM, Carson PE, McMurray JJ, et al; I-PRESERVE Investigators. Irbesartan in patients with heart failure and preserved ejection fraction. N Engl J Med 2008; 359:2456–2467.
- Yusuf S, Pfeffer MA, Swedberg K, et al; CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-Preserved Trial. Lancet 2003; 362:777–781.
- Deswal A, Richardson P, Bozkurt B, Mann DL. Results of the Randomized Aldosterone Antagonism in Heart Failure With Preserved Ejection Fraction Trial (RAAM-PEF). J Card Fail 2011; 17:634–642.
- Edelmann F, Schmidt AG, Gelbrich G, et al. Rationale and design of the ‘aldosterone receptor blockade in diastolic heart failure’ trial: a double-blind, randomized, placebo-controlled, parallel group study to determine the effects of spironolactone on exercise capacity and diastolic function in patients with symptomatic diastolic heart failure (Aldo-DHF). Eur J Heart Fail 2010; 12:874–882.
- Hené RJ, Boer P, Koomans HA, Mees EJ. Plasma aldosterone concentrations in chronic renal disease. Kidney Int 1982; 21:98–101.
- Greene EL, Kren S, Hostetter TH. Role of aldosterone in the remnant kidney model in the rat. J Clin Invest 1996; 98:1063–1068.
- Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int 2006; 70:2116–2123.
- Nagase M, Yoshida S, Shibata S, et al. Enhanced aldosterone signaling in the early nephropathy of rats with metabolic syndrome: possible contribution of fat-derived factors. J Am Soc Nephrol 2006; 17:3438–3446.
- Schjoedt KJ, Rossing K, Juhl TR, et al. Beneficial impact of spironolactone on nephrotic range albuminuria in diabetic nephropathy. Kidney Int 2006; 70:536–542.
- Toyonaga J, Tsuruya K, Ikeda H, et al. Spironolactone inhibits hyperglycemia-induced podocyte injury by attenuating ROS production. Nephrol Dial Transplant 2011; 26:2475–2484.
- Hunt SA, Abraham WT, Chin MH, et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 2009; 119:e391–e479.
- Albert NM, Yancy CW, Liang L, et al. Use of aldosterone antagonists in heart failure. JAMA 2009; 302:1658–1665.
- Parthasarathy HK, Ménard J, White WB, et al. A double-blind, randomized study comparing the antihypertensive effect of eplerenone and spironolactone in patients with hypertension and evidence of primary aldosteronism. J Hypertens 2011; 29:980–990.
- Yamaji M, Tsutamoto T, Kawahara C, et al. Effect of eplerenone versus spironolactone on cortisol and hemoglobin A1(c) levels in patients with chronic heart failure. Am Heart J 2010; 160:915–921.
KEY POINTS
- Although caution is advised in starting ARAs, these drugs are commonly underused in heart failure.
- Aldosterone “escape” can blunt the effects of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers. This is the rationale for also using ARAs.
- The major trials of ARAs in heart failure to date have been the Randomized Aldactone Evaluation Study (RALES), the Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS), and the Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure (EMPHASIS-HF).
- Close monitoring is essential when starting an ARA, as severe hyperkalemia and renal insufficiency can occur.