User login
VIDEO: Stems cells may offer more equipoise in traumatic scar treatment
DANA POINT, CALIF. – It’s possible that burn victims and others with traumatic scarring will no longer need to have skin grafted from one part of their body, thus adding new scars, to mend the deeper scars, according to Dr. Jill Waibel, a speaker at Summit in Aesthetic Medicine 2014.
"As surgeons, we don’t have the right to take healthy tissue and create more scars," said Dr. Waibel, director of the Miami Dermatology and Laser Institute and a voluntary clinical professor of dermatology at the University of Miami.
Dr. Waibel and her colleagues are working with the Department of Defense to study whether stem cells can be applied directly to wounds to facilitate new skin tissue growth in service personnel with traumatic injuries from bomb blasts they suffered while fighting in Afghanistan.
She and her team also have been working with the DOD to refine the delivery system for these stem cells, using lasers and a hydrogel developed by the military that can used safely in vivo. Dr. Waibel discusses her hopes and concerns for stem cell use in traumatic scar treatment in this video from the meeting, which was held by Global Academy for Medical Education. GAME and this news organization are owned by Frontline Medical Communications.
Dr. Waibel disclosed she has financial relationships with Alma, Syneron/Candela, Sciton, Lutronics, and Lumenis.
On Twitter @whitneymcknight
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
DANA POINT, CALIF. – It’s possible that burn victims and others with traumatic scarring will no longer need to have skin grafted from one part of their body, thus adding new scars, to mend the deeper scars, according to Dr. Jill Waibel, a speaker at Summit in Aesthetic Medicine 2014.
"As surgeons, we don’t have the right to take healthy tissue and create more scars," said Dr. Waibel, director of the Miami Dermatology and Laser Institute and a voluntary clinical professor of dermatology at the University of Miami.
Dr. Waibel and her colleagues are working with the Department of Defense to study whether stem cells can be applied directly to wounds to facilitate new skin tissue growth in service personnel with traumatic injuries from bomb blasts they suffered while fighting in Afghanistan.
She and her team also have been working with the DOD to refine the delivery system for these stem cells, using lasers and a hydrogel developed by the military that can used safely in vivo. Dr. Waibel discusses her hopes and concerns for stem cell use in traumatic scar treatment in this video from the meeting, which was held by Global Academy for Medical Education. GAME and this news organization are owned by Frontline Medical Communications.
Dr. Waibel disclosed she has financial relationships with Alma, Syneron/Candela, Sciton, Lutronics, and Lumenis.
On Twitter @whitneymcknight
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
DANA POINT, CALIF. – It’s possible that burn victims and others with traumatic scarring will no longer need to have skin grafted from one part of their body, thus adding new scars, to mend the deeper scars, according to Dr. Jill Waibel, a speaker at Summit in Aesthetic Medicine 2014.
"As surgeons, we don’t have the right to take healthy tissue and create more scars," said Dr. Waibel, director of the Miami Dermatology and Laser Institute and a voluntary clinical professor of dermatology at the University of Miami.
Dr. Waibel and her colleagues are working with the Department of Defense to study whether stem cells can be applied directly to wounds to facilitate new skin tissue growth in service personnel with traumatic injuries from bomb blasts they suffered while fighting in Afghanistan.
She and her team also have been working with the DOD to refine the delivery system for these stem cells, using lasers and a hydrogel developed by the military that can used safely in vivo. Dr. Waibel discusses her hopes and concerns for stem cell use in traumatic scar treatment in this video from the meeting, which was held by Global Academy for Medical Education. GAME and this news organization are owned by Frontline Medical Communications.
Dr. Waibel disclosed she has financial relationships with Alma, Syneron/Candela, Sciton, Lutronics, and Lumenis.
On Twitter @whitneymcknight
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
EXPERT ANALYSIS FROM THE SUMMIT IN AESTHETIC MEDICINE

PODCAST: Chronic wound care offers insight into battling biofilms
DANA POINT, CALIF. – "Biofilms are ubiquitous," said Dr. Robert Galiano, a plastic and reconstructive surgeon at Northwestern Memorial Hospital in Chicago.
"They [bacteria] hijack the body’s healing mechanisms, so instead of healing, ... the body’s inflammatory cascade gets amplified to a degree where it actually becomes tissue damaging," he said at the Summit in Aesthetic Medicine 2014.
Much of what we know about how bacteria communicate with one another to form biofilms comes from research into the prevention and treatment of chronic wounds. More research is needed, but greater understanding of how bacteria communicate with one another to prevent antimicrobial action may lead to new therapeutic targets that could benefit many medical specialties, he added.
Potential therapeutic targets range from coatings for wound dressings to protective barriers on stents and implants of all kinds to extend their durability. In this interview, Dr. Galiano discusses current treatment strategies to manage bacteria, which patients are at greatest risk, and future directions for biofilm prevention.
The summit was held by Global Academy for Medical Education. GAME and this news organization are owned by Frontline Medical Communications. Dr. Galiano had no financial conflicts to disclose.
On Twitter @whitneymcknight
DANA POINT, CALIF. – "Biofilms are ubiquitous," said Dr. Robert Galiano, a plastic and reconstructive surgeon at Northwestern Memorial Hospital in Chicago.
"They [bacteria] hijack the body’s healing mechanisms, so instead of healing, ... the body’s inflammatory cascade gets amplified to a degree where it actually becomes tissue damaging," he said at the Summit in Aesthetic Medicine 2014.
Much of what we know about how bacteria communicate with one another to form biofilms comes from research into the prevention and treatment of chronic wounds. More research is needed, but greater understanding of how bacteria communicate with one another to prevent antimicrobial action may lead to new therapeutic targets that could benefit many medical specialties, he added.
Potential therapeutic targets range from coatings for wound dressings to protective barriers on stents and implants of all kinds to extend their durability. In this interview, Dr. Galiano discusses current treatment strategies to manage bacteria, which patients are at greatest risk, and future directions for biofilm prevention.
The summit was held by Global Academy for Medical Education. GAME and this news organization are owned by Frontline Medical Communications. Dr. Galiano had no financial conflicts to disclose.
On Twitter @whitneymcknight
DANA POINT, CALIF. – "Biofilms are ubiquitous," said Dr. Robert Galiano, a plastic and reconstructive surgeon at Northwestern Memorial Hospital in Chicago.
"They [bacteria] hijack the body’s healing mechanisms, so instead of healing, ... the body’s inflammatory cascade gets amplified to a degree where it actually becomes tissue damaging," he said at the Summit in Aesthetic Medicine 2014.
Much of what we know about how bacteria communicate with one another to form biofilms comes from research into the prevention and treatment of chronic wounds. More research is needed, but greater understanding of how bacteria communicate with one another to prevent antimicrobial action may lead to new therapeutic targets that could benefit many medical specialties, he added.
Potential therapeutic targets range from coatings for wound dressings to protective barriers on stents and implants of all kinds to extend their durability. In this interview, Dr. Galiano discusses current treatment strategies to manage bacteria, which patients are at greatest risk, and future directions for biofilm prevention.
The summit was held by Global Academy for Medical Education. GAME and this news organization are owned by Frontline Medical Communications. Dr. Galiano had no financial conflicts to disclose.
On Twitter @whitneymcknight
EXPERT ANALYSIS FROM THE SUMMIT IN AESTHETIC MEDICINE
Dissociation found to mediate ketamine’s antidepressive effects
HOLLYWOOD, FLA. – Dissociative side effects in patients given a ketamine infusion to treat either major depressive disorder or bipolar disorder predicted a more robust antidepressive response, according to a small secondary analysis.
"Patients who don’t have acute dissociation are more likely not to have antidepressant efficacy in the postinfusion period," Dr. Mark J. Niciu said during an interview discussing his poster presentation at a meeting of the American Society of Clinical Psychopharmacology, formerly known as the New Clinical Drug Evaluation Unit meeting.
"The patients with more disassociation might be the ones to have greater antidepressant efficacy, but they are also the patients we need to keep a closer clinical eye on because they’re having perceptual alterations during the postinfusion period," he said.
Dr. Niciu, a clinical research fellow at the National Institute of Mental Health in Bethesda, Md., reviewed data from 108 treatment-resistant inpatients who met criteria for major depressive disorder or bipolar I or II and were given a subanesthetic ketamine infusion. They examined whether dissociation and psychotic-like experiences, as measured by the Clinician Administered Dissociative States Scale (CADSS), the Brief Psychiatric Rating Scale (BPRS), and the Young Mania Rating Scale (YMRS), and vital sign changes correlated with improvements in the Hamilton Depression Rating Scale (HDRS) at 40 minutes and 230 minutes post infusion, and at 1 and 7 days post infusion.
Pearson correlations indicated that there was a significant association between increased CADSS scores at 40 minutes post infusion and improvement with ketamine in HDRS scores at 230 minutes (r = –0.35, P = .007). Changes in the YMRS or BPRS Positive Symptom score at 40 minutes did not significantly correlate with HDRS improvement at any time point with ketamine. Similarly, none of the vital signs analyzed (changes in systolic or diastolic blood pressure and pulse) significantly correlated to HDRS change.
The question of whether there was an "unblinding" effect was of concern to Dr. Niciu. "The subjects who received ketamine that had greater disassociation might also expect to have greater antidepressive efficacy post infusion," he said. To account for that possibility, he said some researchers are using more active placebos such as midazolam, but he did not think that it was a complete solution because the ideal active placebo would affect glutamate, dopamine, and noradrenaline without having an antidepressant effect.
The overall goal is to discover medications that have keen effects on glutamate receptors such as NMDA (N-methyl-D-aspartate) receptors, as ketamine does, but that do not also have dissociative side effects. "But maybe that’s not possible," Dr. Niciu said. "Maybe we need to have some degree of dissociation as a proxy for the strength of the NMDA receptor blockade because of its antidepressant effects downstream post infusion."
This study was funded by the Intramural Research Program at the National Institute of Mental Health, a NARSAD Independent Investigator Award, and a Brain and Behavior Mood Disorders Research Award. Both of the awards were given to Dr. Carlos A. Zarate.
On Twitter @whitneymcknight
HOLLYWOOD, FLA. – Dissociative side effects in patients given a ketamine infusion to treat either major depressive disorder or bipolar disorder predicted a more robust antidepressive response, according to a small secondary analysis.
"Patients who don’t have acute dissociation are more likely not to have antidepressant efficacy in the postinfusion period," Dr. Mark J. Niciu said during an interview discussing his poster presentation at a meeting of the American Society of Clinical Psychopharmacology, formerly known as the New Clinical Drug Evaluation Unit meeting.
"The patients with more disassociation might be the ones to have greater antidepressant efficacy, but they are also the patients we need to keep a closer clinical eye on because they’re having perceptual alterations during the postinfusion period," he said.
Dr. Niciu, a clinical research fellow at the National Institute of Mental Health in Bethesda, Md., reviewed data from 108 treatment-resistant inpatients who met criteria for major depressive disorder or bipolar I or II and were given a subanesthetic ketamine infusion. They examined whether dissociation and psychotic-like experiences, as measured by the Clinician Administered Dissociative States Scale (CADSS), the Brief Psychiatric Rating Scale (BPRS), and the Young Mania Rating Scale (YMRS), and vital sign changes correlated with improvements in the Hamilton Depression Rating Scale (HDRS) at 40 minutes and 230 minutes post infusion, and at 1 and 7 days post infusion.
Pearson correlations indicated that there was a significant association between increased CADSS scores at 40 minutes post infusion and improvement with ketamine in HDRS scores at 230 minutes (r = –0.35, P = .007). Changes in the YMRS or BPRS Positive Symptom score at 40 minutes did not significantly correlate with HDRS improvement at any time point with ketamine. Similarly, none of the vital signs analyzed (changes in systolic or diastolic blood pressure and pulse) significantly correlated to HDRS change.
The question of whether there was an "unblinding" effect was of concern to Dr. Niciu. "The subjects who received ketamine that had greater disassociation might also expect to have greater antidepressive efficacy post infusion," he said. To account for that possibility, he said some researchers are using more active placebos such as midazolam, but he did not think that it was a complete solution because the ideal active placebo would affect glutamate, dopamine, and noradrenaline without having an antidepressant effect.
The overall goal is to discover medications that have keen effects on glutamate receptors such as NMDA (N-methyl-D-aspartate) receptors, as ketamine does, but that do not also have dissociative side effects. "But maybe that’s not possible," Dr. Niciu said. "Maybe we need to have some degree of dissociation as a proxy for the strength of the NMDA receptor blockade because of its antidepressant effects downstream post infusion."
This study was funded by the Intramural Research Program at the National Institute of Mental Health, a NARSAD Independent Investigator Award, and a Brain and Behavior Mood Disorders Research Award. Both of the awards were given to Dr. Carlos A. Zarate.
On Twitter @whitneymcknight
HOLLYWOOD, FLA. – Dissociative side effects in patients given a ketamine infusion to treat either major depressive disorder or bipolar disorder predicted a more robust antidepressive response, according to a small secondary analysis.
"Patients who don’t have acute dissociation are more likely not to have antidepressant efficacy in the postinfusion period," Dr. Mark J. Niciu said during an interview discussing his poster presentation at a meeting of the American Society of Clinical Psychopharmacology, formerly known as the New Clinical Drug Evaluation Unit meeting.
"The patients with more disassociation might be the ones to have greater antidepressant efficacy, but they are also the patients we need to keep a closer clinical eye on because they’re having perceptual alterations during the postinfusion period," he said.
Dr. Niciu, a clinical research fellow at the National Institute of Mental Health in Bethesda, Md., reviewed data from 108 treatment-resistant inpatients who met criteria for major depressive disorder or bipolar I or II and were given a subanesthetic ketamine infusion. They examined whether dissociation and psychotic-like experiences, as measured by the Clinician Administered Dissociative States Scale (CADSS), the Brief Psychiatric Rating Scale (BPRS), and the Young Mania Rating Scale (YMRS), and vital sign changes correlated with improvements in the Hamilton Depression Rating Scale (HDRS) at 40 minutes and 230 minutes post infusion, and at 1 and 7 days post infusion.
Pearson correlations indicated that there was a significant association between increased CADSS scores at 40 minutes post infusion and improvement with ketamine in HDRS scores at 230 minutes (r = –0.35, P = .007). Changes in the YMRS or BPRS Positive Symptom score at 40 minutes did not significantly correlate with HDRS improvement at any time point with ketamine. Similarly, none of the vital signs analyzed (changes in systolic or diastolic blood pressure and pulse) significantly correlated to HDRS change.
The question of whether there was an "unblinding" effect was of concern to Dr. Niciu. "The subjects who received ketamine that had greater disassociation might also expect to have greater antidepressive efficacy post infusion," he said. To account for that possibility, he said some researchers are using more active placebos such as midazolam, but he did not think that it was a complete solution because the ideal active placebo would affect glutamate, dopamine, and noradrenaline without having an antidepressant effect.
The overall goal is to discover medications that have keen effects on glutamate receptors such as NMDA (N-methyl-D-aspartate) receptors, as ketamine does, but that do not also have dissociative side effects. "But maybe that’s not possible," Dr. Niciu said. "Maybe we need to have some degree of dissociation as a proxy for the strength of the NMDA receptor blockade because of its antidepressant effects downstream post infusion."
This study was funded by the Intramural Research Program at the National Institute of Mental Health, a NARSAD Independent Investigator Award, and a Brain and Behavior Mood Disorders Research Award. Both of the awards were given to Dr. Carlos A. Zarate.
On Twitter @whitneymcknight
AT THE ASCP ANNUAL MEETING
Key clinical point: Some degree of dissociation might be needed "as a proxy for the strength of the NMDA receptor blockade because of its antidepressant effects downstream post infusion."
Major finding: A significant association was found between increased CADSS scores at 40 minutes post infusion and improvement with ketamine in HDRS scores at 230 minutes (r = –0.35, P = .007).
Data source: Secondary analysis of 108 inpatients treated for MDD or BP I or II with ketamine infusion.
Disclosures: This study was funded by the Intramural Research Program at the National Institute of Mental Health, a NARSAD Independent Investigator Award, and a Brain and Behavior Mood Disorders Research Award. Both of the awards were given to Dr. Carlos A. Zarate.
No new safety concerns seen in combination treatment with dimethyl fumarate
DALLAS – The addition of oral delayed-release dimethyl fumarate to conventional treatments for relapsing-remitting multiple sclerosis had safety and tolerability profiles similar to monotherapy with the drug, according to results from a phase II, open-label study.
In the EXPLORE study, the side effects most often recorded in 104 MS patients with breakthrough disease who were given oral delayed-release dimethyl fumarate (Tecfidera) combined with either interferon-beta or glatiramer acetate were mild to moderate flushing (42% and 53%, respectively), diarrhea (32% and 15%), and abdominal pain (21% and 6%). Previous phase III, placebo-controlled studies (DEFINE and CONFIRM) indicated similar adverse event rates for dimethyl fumarate (DMF) as monotherapy.

"Adding Tecfidera to interferon-beta and glatiramer did not significantly increase the side effects of each of those drugs alone," Dr. Jonathan Calkwood said in an interview during a poster session at a meeting of the Consortium of Multiple Sclerosis Centers and the Americas Committee for Treatment and Research in Multiple Sclerosis.
"White blood cell count declined, generally starting at about 2 months," said Dr. Calkwood, executive director of the Schapiro Center for Multiple Sclerosis at the Minneapolis Clinic of Neurology in Golden Valley, Minn. "This mirrored what we saw in the phase III studies, where white blood cell count went down and kind of stayed down."
The lymphocyte count decreased from baseline to week 24 by 22% in the delayed-release DMF plus interferon-beta group, and by 7% in the delayed-release DMF plus glatiramer acetate group. "This was also consistent with the phase III trials, where it goes down and stays down, but not enough to discontinue the medication," he said.
No malignancies were observed, and there were no deaths. There was a transient increase in liver transaminases, primarily in the interferon group, but according to Dr. Calkwood, this did not meet Hy’s law criteria. With the exception of one Clostridium difficile infection, there "was no real signal on infectious disease," he said.
For the study, the investigators enrolled patients aged 18-55 years who met McDonald disease criteria and who had an Expanded Disease Status Scale score of less than 5. At the time of enrollment, patients had been taking the same dose of interferon-beta or glatiramer acetate for at least 12 months and had at least one relapse within the past 12 months or a gadolinium-enhancing lesion within 6 weeks. Patients continued on their prescribed MS therapy for the first 2 months of the study before starting oral delayed-release DMF 240 mg three times daily in addition to their prescribed MS therapy for 6 months.
"The study was not really long enough to show differences in efficacy, although MRIs were done monthly as a safety measure," Dr. Calkwood said. However, if the question had been that oral delayed-release DMF would somehow interfere with rather than enhance the platform therapies’ respective ability to protect against breakthrough disease, he said that the imaging performed during the add-on therapy phase of the study showed fewer active lesions than the wash-in monotherapy phase. "It further supported what we thought we already knew."
Dr. Calkwood disclosed that he has financial relationships with Biogen Idec, maker of Tecfidera, as well as Teva Neuroscience, Bayer HealthCare, and EMD Serono. Biogen Idec sponsored this study.
On Twitter @whitneymcknight
DALLAS – The addition of oral delayed-release dimethyl fumarate to conventional treatments for relapsing-remitting multiple sclerosis had safety and tolerability profiles similar to monotherapy with the drug, according to results from a phase II, open-label study.
In the EXPLORE study, the side effects most often recorded in 104 MS patients with breakthrough disease who were given oral delayed-release dimethyl fumarate (Tecfidera) combined with either interferon-beta or glatiramer acetate were mild to moderate flushing (42% and 53%, respectively), diarrhea (32% and 15%), and abdominal pain (21% and 6%). Previous phase III, placebo-controlled studies (DEFINE and CONFIRM) indicated similar adverse event rates for dimethyl fumarate (DMF) as monotherapy.
"Adding Tecfidera to interferon-beta and glatiramer did not significantly increase the side effects of each of those drugs alone," Dr. Jonathan Calkwood said in an interview during a poster session at a meeting of the Consortium of Multiple Sclerosis Centers and the Americas Committee for Treatment and Research in Multiple Sclerosis.
"White blood cell count declined, generally starting at about 2 months," said Dr. Calkwood, executive director of the Schapiro Center for Multiple Sclerosis at the Minneapolis Clinic of Neurology in Golden Valley, Minn. "This mirrored what we saw in the phase III studies, where white blood cell count went down and kind of stayed down."
The lymphocyte count decreased from baseline to week 24 by 22% in the delayed-release DMF plus interferon-beta group, and by 7% in the delayed-release DMF plus glatiramer acetate group. "This was also consistent with the phase III trials, where it goes down and stays down, but not enough to discontinue the medication," he said.
No malignancies were observed, and there were no deaths. There was a transient increase in liver transaminases, primarily in the interferon group, but according to Dr. Calkwood, this did not meet Hy’s law criteria. With the exception of one Clostridium difficile infection, there "was no real signal on infectious disease," he said.
For the study, the investigators enrolled patients aged 18-55 years who met McDonald disease criteria and who had an Expanded Disease Status Scale score of less than 5. At the time of enrollment, patients had been taking the same dose of interferon-beta or glatiramer acetate for at least 12 months and had at least one relapse within the past 12 months or a gadolinium-enhancing lesion within 6 weeks. Patients continued on their prescribed MS therapy for the first 2 months of the study before starting oral delayed-release DMF 240 mg three times daily in addition to their prescribed MS therapy for 6 months.
"The study was not really long enough to show differences in efficacy, although MRIs were done monthly as a safety measure," Dr. Calkwood said. However, if the question had been that oral delayed-release DMF would somehow interfere with rather than enhance the platform therapies’ respective ability to protect against breakthrough disease, he said that the imaging performed during the add-on therapy phase of the study showed fewer active lesions than the wash-in monotherapy phase. "It further supported what we thought we already knew."
Dr. Calkwood disclosed that he has financial relationships with Biogen Idec, maker of Tecfidera, as well as Teva Neuroscience, Bayer HealthCare, and EMD Serono. Biogen Idec sponsored this study.
On Twitter @whitneymcknight
DALLAS – The addition of oral delayed-release dimethyl fumarate to conventional treatments for relapsing-remitting multiple sclerosis had safety and tolerability profiles similar to monotherapy with the drug, according to results from a phase II, open-label study.
In the EXPLORE study, the side effects most often recorded in 104 MS patients with breakthrough disease who were given oral delayed-release dimethyl fumarate (Tecfidera) combined with either interferon-beta or glatiramer acetate were mild to moderate flushing (42% and 53%, respectively), diarrhea (32% and 15%), and abdominal pain (21% and 6%). Previous phase III, placebo-controlled studies (DEFINE and CONFIRM) indicated similar adverse event rates for dimethyl fumarate (DMF) as monotherapy.
"Adding Tecfidera to interferon-beta and glatiramer did not significantly increase the side effects of each of those drugs alone," Dr. Jonathan Calkwood said in an interview during a poster session at a meeting of the Consortium of Multiple Sclerosis Centers and the Americas Committee for Treatment and Research in Multiple Sclerosis.
"White blood cell count declined, generally starting at about 2 months," said Dr. Calkwood, executive director of the Schapiro Center for Multiple Sclerosis at the Minneapolis Clinic of Neurology in Golden Valley, Minn. "This mirrored what we saw in the phase III studies, where white blood cell count went down and kind of stayed down."
The lymphocyte count decreased from baseline to week 24 by 22% in the delayed-release DMF plus interferon-beta group, and by 7% in the delayed-release DMF plus glatiramer acetate group. "This was also consistent with the phase III trials, where it goes down and stays down, but not enough to discontinue the medication," he said.
No malignancies were observed, and there were no deaths. There was a transient increase in liver transaminases, primarily in the interferon group, but according to Dr. Calkwood, this did not meet Hy’s law criteria. With the exception of one Clostridium difficile infection, there "was no real signal on infectious disease," he said.
For the study, the investigators enrolled patients aged 18-55 years who met McDonald disease criteria and who had an Expanded Disease Status Scale score of less than 5. At the time of enrollment, patients had been taking the same dose of interferon-beta or glatiramer acetate for at least 12 months and had at least one relapse within the past 12 months or a gadolinium-enhancing lesion within 6 weeks. Patients continued on their prescribed MS therapy for the first 2 months of the study before starting oral delayed-release DMF 240 mg three times daily in addition to their prescribed MS therapy for 6 months.
"The study was not really long enough to show differences in efficacy, although MRIs were done monthly as a safety measure," Dr. Calkwood said. However, if the question had been that oral delayed-release DMF would somehow interfere with rather than enhance the platform therapies’ respective ability to protect against breakthrough disease, he said that the imaging performed during the add-on therapy phase of the study showed fewer active lesions than the wash-in monotherapy phase. "It further supported what we thought we already knew."
Dr. Calkwood disclosed that he has financial relationships with Biogen Idec, maker of Tecfidera, as well as Teva Neuroscience, Bayer HealthCare, and EMD Serono. Biogen Idec sponsored this study.
On Twitter @whitneymcknight
AT THE CMSC/ACTRIMS annual meeting
Key clinical point: Oral delayed-release DMF added to either interferon-beta or glatiramer acetate had adverse event rates similar to those of delayed-release DMF monotherapy.
Major finding: The most common adverse events observed with oral delayed-release DMF combined with either interferon-beta or glatiramer acetate were mild to moderate flushing (42% and 53%, respectively), diarrhea (32% and 15%), and abdominal pain (21% and 6%).
Data source: A phase II, open-label study (EXPLORE) of 104 adult relapsing-remitting multiple sclerosis patients who’d had 12 months of interferon-beta or glatiramer acetate monotherapy.
Disclosures: Dr. Calkwood disclosed that he has financial relationships with Biogen Idec, maker of Tecfidera, as well as Teva Neuroscience, Bayer HealthCare, and EMD Serono. Biogen Idec sponsored this study.

Low immunogenicity rates support peginterferon in MS
DALLAS – Subcutaneous administration of pegylated interferon every 2 or 4 weeks to patients with relapsing-remitting multiple sclerosis was associated with low rates of immunogenicity in a phase III, double-blind randomized trial.
At year 2 of the ADVANCE trial, persistent treatment-emergent antibodies occurred in 1% of patients who’d received pegylated interferon (PEG-INF) beta-1a (at 125 mcg delivered subcutaneously either every 2 weeks or 4 weeks. Patients given 1 year of placebo, followed by 1 year of dose-frequency–blinded PEG-INF beta-1a, had a similarly low rate.

Modification of a drug by adding polyethylene glycol molecules (pegylation) increases its half-life. It may also reduce the drug’s immunogenicity.
"Pegylated interferon may provide efficacy and safety to what we see with currently available therapies, with the added advantage of lower dosing frequencies per month, which may improve compliance for injectable [therapies], and possibly decrease immunogenicity," said Dr. Scott Newsome, director of the neurology outpatient infusion center at Johns Hopkins Hospital, Baltimore.
The overall incidence of antipeginterferon antibodies during the 2 years of observation was 7% in the total study population of 1,516 patients, with no difference between treatment arms (3% in the placebo arm vs. less than 1% in those given PEG-INF beta-1a every 2 weeks and 2% in those given it every 4 weeks).
During the 2-year observation of the blinded dosage, the incidence of binding antibodies was 4% in the group that got PEG-INF beta-1a every 2 weeks and 2% with PEG-INF beta-1a every 4 weeks. Antipeginterferon titers were 2% and 6%, respectively.
"I’d also like to point out that the incidence rate of interferon beta-1a neutralizing antibodies was exceedingly low, less than 1% across the entire study and across both treatment arms," Dr. Newsome said at a meeting of the Consortium of Multiple Sclerosis Centers and the Americas Committee for Treatment and Research in Multiple Sclerosis.
"No discernible impact on safety or tolerability was observed in this study," Dr. Newsome said, adding that the rate of adverse events such as injection-site reactions did not differ by antibody status or titer.
The findings strengthen PEG-INF’s profile as another potential interferon treatment for relapsing-remitting multiple sclerosis. In March of this year, the Food and Drug Administration extended its review of the Biogen IDEC product by 3 months; their decision is still forthcoming. If approved, the manufacturer plans to market the drug as Plegridy.
The results of the ADVANCE trial presented earlier this year at the annual meeting of the American Academy of Neurology, indicated that the drug met all primary and secondary efficacy endpoints by the end of year 1, and continued to show a similar efficacy and safety profile in year 2.
Dr. Newsome declared he has received funding from Biogen IDEC and Genzyme. Biogen IDEC sponsored this study.
On Twitter @whitneymcknight
DALLAS – Subcutaneous administration of pegylated interferon every 2 or 4 weeks to patients with relapsing-remitting multiple sclerosis was associated with low rates of immunogenicity in a phase III, double-blind randomized trial.
At year 2 of the ADVANCE trial, persistent treatment-emergent antibodies occurred in 1% of patients who’d received pegylated interferon (PEG-INF) beta-1a (at 125 mcg delivered subcutaneously either every 2 weeks or 4 weeks. Patients given 1 year of placebo, followed by 1 year of dose-frequency–blinded PEG-INF beta-1a, had a similarly low rate.
Modification of a drug by adding polyethylene glycol molecules (pegylation) increases its half-life. It may also reduce the drug’s immunogenicity.
"Pegylated interferon may provide efficacy and safety to what we see with currently available therapies, with the added advantage of lower dosing frequencies per month, which may improve compliance for injectable [therapies], and possibly decrease immunogenicity," said Dr. Scott Newsome, director of the neurology outpatient infusion center at Johns Hopkins Hospital, Baltimore.
The overall incidence of antipeginterferon antibodies during the 2 years of observation was 7% in the total study population of 1,516 patients, with no difference between treatment arms (3% in the placebo arm vs. less than 1% in those given PEG-INF beta-1a every 2 weeks and 2% in those given it every 4 weeks).
During the 2-year observation of the blinded dosage, the incidence of binding antibodies was 4% in the group that got PEG-INF beta-1a every 2 weeks and 2% with PEG-INF beta-1a every 4 weeks. Antipeginterferon titers were 2% and 6%, respectively.
"I’d also like to point out that the incidence rate of interferon beta-1a neutralizing antibodies was exceedingly low, less than 1% across the entire study and across both treatment arms," Dr. Newsome said at a meeting of the Consortium of Multiple Sclerosis Centers and the Americas Committee for Treatment and Research in Multiple Sclerosis.
"No discernible impact on safety or tolerability was observed in this study," Dr. Newsome said, adding that the rate of adverse events such as injection-site reactions did not differ by antibody status or titer.
The findings strengthen PEG-INF’s profile as another potential interferon treatment for relapsing-remitting multiple sclerosis. In March of this year, the Food and Drug Administration extended its review of the Biogen IDEC product by 3 months; their decision is still forthcoming. If approved, the manufacturer plans to market the drug as Plegridy.
The results of the ADVANCE trial presented earlier this year at the annual meeting of the American Academy of Neurology, indicated that the drug met all primary and secondary efficacy endpoints by the end of year 1, and continued to show a similar efficacy and safety profile in year 2.
Dr. Newsome declared he has received funding from Biogen IDEC and Genzyme. Biogen IDEC sponsored this study.
On Twitter @whitneymcknight
DALLAS – Subcutaneous administration of pegylated interferon every 2 or 4 weeks to patients with relapsing-remitting multiple sclerosis was associated with low rates of immunogenicity in a phase III, double-blind randomized trial.
At year 2 of the ADVANCE trial, persistent treatment-emergent antibodies occurred in 1% of patients who’d received pegylated interferon (PEG-INF) beta-1a (at 125 mcg delivered subcutaneously either every 2 weeks or 4 weeks. Patients given 1 year of placebo, followed by 1 year of dose-frequency–blinded PEG-INF beta-1a, had a similarly low rate.
Modification of a drug by adding polyethylene glycol molecules (pegylation) increases its half-life. It may also reduce the drug’s immunogenicity.
"Pegylated interferon may provide efficacy and safety to what we see with currently available therapies, with the added advantage of lower dosing frequencies per month, which may improve compliance for injectable [therapies], and possibly decrease immunogenicity," said Dr. Scott Newsome, director of the neurology outpatient infusion center at Johns Hopkins Hospital, Baltimore.
The overall incidence of antipeginterferon antibodies during the 2 years of observation was 7% in the total study population of 1,516 patients, with no difference between treatment arms (3% in the placebo arm vs. less than 1% in those given PEG-INF beta-1a every 2 weeks and 2% in those given it every 4 weeks).
During the 2-year observation of the blinded dosage, the incidence of binding antibodies was 4% in the group that got PEG-INF beta-1a every 2 weeks and 2% with PEG-INF beta-1a every 4 weeks. Antipeginterferon titers were 2% and 6%, respectively.
"I’d also like to point out that the incidence rate of interferon beta-1a neutralizing antibodies was exceedingly low, less than 1% across the entire study and across both treatment arms," Dr. Newsome said at a meeting of the Consortium of Multiple Sclerosis Centers and the Americas Committee for Treatment and Research in Multiple Sclerosis.
"No discernible impact on safety or tolerability was observed in this study," Dr. Newsome said, adding that the rate of adverse events such as injection-site reactions did not differ by antibody status or titer.
The findings strengthen PEG-INF’s profile as another potential interferon treatment for relapsing-remitting multiple sclerosis. In March of this year, the Food and Drug Administration extended its review of the Biogen IDEC product by 3 months; their decision is still forthcoming. If approved, the manufacturer plans to market the drug as Plegridy.
The results of the ADVANCE trial presented earlier this year at the annual meeting of the American Academy of Neurology, indicated that the drug met all primary and secondary efficacy endpoints by the end of year 1, and continued to show a similar efficacy and safety profile in year 2.
Dr. Newsome declared he has received funding from Biogen IDEC and Genzyme. Biogen IDEC sponsored this study.
On Twitter @whitneymcknight
AT THE CMSC/ACTRIMS ANNUAL MEETING
Key clinical point: The development of antibodies against PEG-INF beta-1a was low and had no discernible impact on safety or tolerability.
Major finding: There was no difference in the overall incidence of antipeginterferon antibodies during the 2 years of observation between treatment arms (3% in the placebo arm vs. less than 1% in those given PEG-INF beta-1a every 2 weeks and 2% in those given it every 4 weeks).
Data source: A phase III, double-blind, multicenter clinical trial (ADVANCE) involving 1,516 relapsing-remitting multiple sclerosis patients randomized to 1 year of placebo or subcutaneous PEG-INF beta-1a at 125 mcg either every 2 weeks or every 4 weeks followed by rerandomization of the placebo patients to PEG-INF beta-1a for the second year of treatment.
Disclosures: Dr. Newsome declared he has received funding from Biogen IDEC and Genzyme. Biogen IDEC sponsored this study.

Lurasidone monotherapy improves quality of life in bipolar I
HOLLYWOOD, FLA. – Lurasidone as monotherapy in the dosage range of either 20-60 mg/day* or 80-120 mg/day significantly improved functioning and quality-of-life in patients with bipolar depression, a post hoc analysis has shown.
In a previously published study of 318 patients randomized to either lurasidone dosage groups or placebo, the drug’s association at both dose ranges with reduced Montgomery-sberg Depression Scale total scores and Clinical Global Impressions scale for bipolar depression severity scores from baseline to week 6 was significant. The study also noted that both lurasidone groups had significant improvements in patient-reported measures of quality of life and functional impairment, compared with the placebo group (P less than .001 for each) (Am. J. Psychiatry 2014;171:160-8).
The findings led Dr. Terence A. Ketter, professor of psychiatry and chief of the bipolar disorders clinic at Stanford (Calif.) University, to wonder how the improvements in functionality and quality of life had happened in such relatively short order.
"It kind of makes sense that it’s related to the mood improvement," Dr. Ketter said in an interview during a poster session at a meeting of the American Society of Clinical Psychopharmacology, formerly known as the New Clinical Drug Evaluation Unit meeting.
To investigate further, Dr. Ketter and his colleagues performed a mediation regression analysis using data from phase III of the original trial. The results were that reduced depressive symptoms from baseline to week 6 mediated the effect of lurasidone on Sheehan Disability Scale functional recovery and Quality of Life Enjoyment and Satisfaction Questionnaire scores at week 6 (P less than .05 for each).
"This suggests that, at least in the first 6 weeks, most of the improvement [in functionality and quality of life] is related to the improvement in depressive symptoms," Dr. Ketter said. "Functional recovery is something that takes months, and it may be that beyond this by a month or two, you’re looking at a degree of mood improvement by duration interaction to get functional improvement." He added that there likely would be a "huge improvement" in cognition, simply because of the removal of depressive symptoms.
"Is it enough to be able to go out and get a job? Well, maybe not. Maybe you need things to integrate for months before you can go out and work." But, he added, over time, the less interference with one’s cognitive ability, the greater likelihood that person is employable.
In the original study in bipolar I patients, "the company wanted to know which was the right dose," Dr. Ketter said. "As it turned out, both worked. The higher one was a little bit harder to tolerate, but not horribly so."
As a result, Dr. Ketter said the drug has a "pretty flexible label." In his own practice, he said he starts patients on monotherapy with lurasidone 20 mg/day at dinner time, titrating upward in 20-mg increments each week until his patients reach the dosage that works best for them. "The average dose seems to be about 60 mg/day," he said.
Lurasidone, a second-generation antipsychotic, originally was approved by the Food and Drug Administration in 2010 to treat schizophrenia in adults; the indication was expanded in 2013 to include bipolar I, either as monotherapy or as an adjunct to lithium or valproate.
Because of its relatively low impact on metabolic function and its low sedative effect, compared with other second-generation antipsychotics, Dr. Ketter said he favors using lurasidone in his patients. "It’s like an easier to use quetiapine," he noted.
This study was supported by Sunovion Pharmaceuticals. Dr. Ketter disclosed that he has received funding from Sunovion, as well as from AstraZeneca Pharmaceuticals, Cephalon, Eli Lilly, and others.
On Twitter @whitneymcknight
*Correction, 7/2/2014: An earlier version of this story misstated the lurasidone dosage range.
HOLLYWOOD, FLA. – Lurasidone as monotherapy in the dosage range of either 20-60 mg/day* or 80-120 mg/day significantly improved functioning and quality-of-life in patients with bipolar depression, a post hoc analysis has shown.
In a previously published study of 318 patients randomized to either lurasidone dosage groups or placebo, the drug’s association at both dose ranges with reduced Montgomery-sberg Depression Scale total scores and Clinical Global Impressions scale for bipolar depression severity scores from baseline to week 6 was significant. The study also noted that both lurasidone groups had significant improvements in patient-reported measures of quality of life and functional impairment, compared with the placebo group (P less than .001 for each) (Am. J. Psychiatry 2014;171:160-8).
The findings led Dr. Terence A. Ketter, professor of psychiatry and chief of the bipolar disorders clinic at Stanford (Calif.) University, to wonder how the improvements in functionality and quality of life had happened in such relatively short order.
"It kind of makes sense that it’s related to the mood improvement," Dr. Ketter said in an interview during a poster session at a meeting of the American Society of Clinical Psychopharmacology, formerly known as the New Clinical Drug Evaluation Unit meeting.
To investigate further, Dr. Ketter and his colleagues performed a mediation regression analysis using data from phase III of the original trial. The results were that reduced depressive symptoms from baseline to week 6 mediated the effect of lurasidone on Sheehan Disability Scale functional recovery and Quality of Life Enjoyment and Satisfaction Questionnaire scores at week 6 (P less than .05 for each).
"This suggests that, at least in the first 6 weeks, most of the improvement [in functionality and quality of life] is related to the improvement in depressive symptoms," Dr. Ketter said. "Functional recovery is something that takes months, and it may be that beyond this by a month or two, you’re looking at a degree of mood improvement by duration interaction to get functional improvement." He added that there likely would be a "huge improvement" in cognition, simply because of the removal of depressive symptoms.
"Is it enough to be able to go out and get a job? Well, maybe not. Maybe you need things to integrate for months before you can go out and work." But, he added, over time, the less interference with one’s cognitive ability, the greater likelihood that person is employable.
In the original study in bipolar I patients, "the company wanted to know which was the right dose," Dr. Ketter said. "As it turned out, both worked. The higher one was a little bit harder to tolerate, but not horribly so."
As a result, Dr. Ketter said the drug has a "pretty flexible label." In his own practice, he said he starts patients on monotherapy with lurasidone 20 mg/day at dinner time, titrating upward in 20-mg increments each week until his patients reach the dosage that works best for them. "The average dose seems to be about 60 mg/day," he said.
Lurasidone, a second-generation antipsychotic, originally was approved by the Food and Drug Administration in 2010 to treat schizophrenia in adults; the indication was expanded in 2013 to include bipolar I, either as monotherapy or as an adjunct to lithium or valproate.
Because of its relatively low impact on metabolic function and its low sedative effect, compared with other second-generation antipsychotics, Dr. Ketter said he favors using lurasidone in his patients. "It’s like an easier to use quetiapine," he noted.
This study was supported by Sunovion Pharmaceuticals. Dr. Ketter disclosed that he has received funding from Sunovion, as well as from AstraZeneca Pharmaceuticals, Cephalon, Eli Lilly, and others.
On Twitter @whitneymcknight
*Correction, 7/2/2014: An earlier version of this story misstated the lurasidone dosage range.
HOLLYWOOD, FLA. – Lurasidone as monotherapy in the dosage range of either 20-60 mg/day* or 80-120 mg/day significantly improved functioning and quality-of-life in patients with bipolar depression, a post hoc analysis has shown.
In a previously published study of 318 patients randomized to either lurasidone dosage groups or placebo, the drug’s association at both dose ranges with reduced Montgomery-sberg Depression Scale total scores and Clinical Global Impressions scale for bipolar depression severity scores from baseline to week 6 was significant. The study also noted that both lurasidone groups had significant improvements in patient-reported measures of quality of life and functional impairment, compared with the placebo group (P less than .001 for each) (Am. J. Psychiatry 2014;171:160-8).
The findings led Dr. Terence A. Ketter, professor of psychiatry and chief of the bipolar disorders clinic at Stanford (Calif.) University, to wonder how the improvements in functionality and quality of life had happened in such relatively short order.
"It kind of makes sense that it’s related to the mood improvement," Dr. Ketter said in an interview during a poster session at a meeting of the American Society of Clinical Psychopharmacology, formerly known as the New Clinical Drug Evaluation Unit meeting.
To investigate further, Dr. Ketter and his colleagues performed a mediation regression analysis using data from phase III of the original trial. The results were that reduced depressive symptoms from baseline to week 6 mediated the effect of lurasidone on Sheehan Disability Scale functional recovery and Quality of Life Enjoyment and Satisfaction Questionnaire scores at week 6 (P less than .05 for each).
"This suggests that, at least in the first 6 weeks, most of the improvement [in functionality and quality of life] is related to the improvement in depressive symptoms," Dr. Ketter said. "Functional recovery is something that takes months, and it may be that beyond this by a month or two, you’re looking at a degree of mood improvement by duration interaction to get functional improvement." He added that there likely would be a "huge improvement" in cognition, simply because of the removal of depressive symptoms.
"Is it enough to be able to go out and get a job? Well, maybe not. Maybe you need things to integrate for months before you can go out and work." But, he added, over time, the less interference with one’s cognitive ability, the greater likelihood that person is employable.
In the original study in bipolar I patients, "the company wanted to know which was the right dose," Dr. Ketter said. "As it turned out, both worked. The higher one was a little bit harder to tolerate, but not horribly so."
As a result, Dr. Ketter said the drug has a "pretty flexible label." In his own practice, he said he starts patients on monotherapy with lurasidone 20 mg/day at dinner time, titrating upward in 20-mg increments each week until his patients reach the dosage that works best for them. "The average dose seems to be about 60 mg/day," he said.
Lurasidone, a second-generation antipsychotic, originally was approved by the Food and Drug Administration in 2010 to treat schizophrenia in adults; the indication was expanded in 2013 to include bipolar I, either as monotherapy or as an adjunct to lithium or valproate.
Because of its relatively low impact on metabolic function and its low sedative effect, compared with other second-generation antipsychotics, Dr. Ketter said he favors using lurasidone in his patients. "It’s like an easier to use quetiapine," he noted.
This study was supported by Sunovion Pharmaceuticals. Dr. Ketter disclosed that he has received funding from Sunovion, as well as from AstraZeneca Pharmaceuticals, Cephalon, Eli Lilly, and others.
On Twitter @whitneymcknight
*Correction, 7/2/2014: An earlier version of this story misstated the lurasidone dosage range.
AT THE ASCP ANNUAL MEETING
Key clinical point: Lurasidone appears to reduce depressive symptoms in patients with bipolar I depression without the metabolic complications characteristic of other second-generation antipsychotics.
Major finding: In 318 patients with bipolar I depression, reduced depressive symptoms from baseline to week 6 mediated the effect of lurasidone vs. placebo (P less than .05 in each) on functioning and quality of life.
Data source: A post hoc analysis of phase III data from a randomized, double-blind, placebo controlled trial in 318 intent-to-treat patients with bipolar I.
Disclosures: This study was supported by Sunovion Pharmaceuticals. Dr. Ketter disclosed he has received funding from Sunovion, as well as from AstraZeneca Pharmaceuticals, Cephalon, Eli Lilly, and others.
IOM: Military, veterans’ PTSD programs lack consistency, outcomes measures
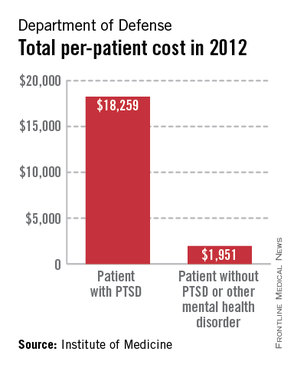
A lack of consistent outcome measures means there is no way to know whether the more than $3 billion spent on treating posttraumatic stress disorder by the Department of Defense and Veterans Affairs in 2012 yielded worthwhile results, according to a report released June 20.
"Given that the DOD and VA are responsible for serving millions of service members, families, and veterans, we found it surprising that no PTSD outcome measures are used consistently to know if these treatments are working," Dr. Sandro Galea, chair of the Institute of Medicine committee tasked by Congress to study PTSD treatment in military and veteran populations, said in a statement.

The report notes that currently, "neither the DOD nor the VA knows whether it is providing effective or adequate PTSD care, for which they spent $294 million and more than $3 billion, respectively, in 2012." Similar findings were reported by the IOM in 2012.
"What we found over and over again were really hardworking, well-intentioned people who wanted to do the best they could, but they either didn’t have an administrative structure to support them, or enough staff, or they had an overwhelming number of patients," committee member Dr. Elspeth Cameron Ritchie said during a press briefing.

In addition to better data collection and sharing, the report calls for the development of an adequate workforce to provide mental health care to this growing population.
Although tele-therapies and virtual reality therapies, for which the evidence base is growing, can provide some help, inadequate staffing still leads to a limitation in the number of evidence-based therapies available to patients, said Dr. Ritchie, a retired Army psychiatrist and current professor of psychiatry at Georgetown University in Washington. To wit, the report cited the VA’s failure in 2013 to provide the recommended eight sessions of psychotherapy within 14 weeks to nearly half of all Iraq and Afghanistan war veterans seeking care for a primary diagnosis of PTSD.
The report also calls for the development of evidenced-base treatments, including combination therapies of psychotherapies such as cognitive behavioral therapy, with medications such as SSRIs.
The report recommends that family members be involved in the treatment of PTSD; the recommendation was based on feedback from service members and veterans who said they wanted their loved ones to be actively included.
In addition, the report states that research into PTSD should be focused on current patient needs, and that both departments should actively collaborate with one another and with other government agencies, such as the National Institutes of Health, to fill knowledge gaps.
The number of veterans seeking care for PTSD from the VA has more than doubled from 190,000 (4.3% of all VA users) in 2003 to more than a half million (9.2%) in 2012. Although veterans of all eras are included in the increase, 23.6% (119,500) of those treated for PTSD by the VA in 2012 were veterans of the wars in Iraq and Afghanistan.

In 2013, 528,260 veterans made at least two visits to the VA for PTSD outpatient care; one-quarter were new patients. Although the overall incidence rate for PTSD across all service members is about 1%, the prevalence rose from 0.4% in 2004 to 5% in 2012, with an 8% increase in those who had been deployed previously, according to the report.
The committee said the DOD approach to PTSD treatment is "local, ad hoc, incremental, and crisis driven, with little planning." While VA programs benefits from better organization and consistency, the lack of data on either department’s delivery methods and outcomes means there is "no way of knowing whether the care they are providing is effective or whether DOD and VA’s expenditures are resulting in high-value health care," according to the report.
The report is based on 4 years of combing through data provided by the DOD and the VA, peer-reviewed literature, government documents, research databases, and testimonies from a variety of DOD and VA experts and providers at military bases and treatment facilities around the country, including six VA medical centers.
On Twitter @whitneymcknight
A lack of consistent outcome measures means there is no way to know whether the more than $3 billion spent on treating posttraumatic stress disorder by the Department of Defense and Veterans Affairs in 2012 yielded worthwhile results, according to a report released June 20.
"Given that the DOD and VA are responsible for serving millions of service members, families, and veterans, we found it surprising that no PTSD outcome measures are used consistently to know if these treatments are working," Dr. Sandro Galea, chair of the Institute of Medicine committee tasked by Congress to study PTSD treatment in military and veteran populations, said in a statement.
The report notes that currently, "neither the DOD nor the VA knows whether it is providing effective or adequate PTSD care, for which they spent $294 million and more than $3 billion, respectively, in 2012." Similar findings were reported by the IOM in 2012.
"What we found over and over again were really hardworking, well-intentioned people who wanted to do the best they could, but they either didn’t have an administrative structure to support them, or enough staff, or they had an overwhelming number of patients," committee member Dr. Elspeth Cameron Ritchie said during a press briefing.
In addition to better data collection and sharing, the report calls for the development of an adequate workforce to provide mental health care to this growing population.
Although tele-therapies and virtual reality therapies, for which the evidence base is growing, can provide some help, inadequate staffing still leads to a limitation in the number of evidence-based therapies available to patients, said Dr. Ritchie, a retired Army psychiatrist and current professor of psychiatry at Georgetown University in Washington. To wit, the report cited the VA’s failure in 2013 to provide the recommended eight sessions of psychotherapy within 14 weeks to nearly half of all Iraq and Afghanistan war veterans seeking care for a primary diagnosis of PTSD.
The report also calls for the development of evidenced-base treatments, including combination therapies of psychotherapies such as cognitive behavioral therapy, with medications such as SSRIs.
The report recommends that family members be involved in the treatment of PTSD; the recommendation was based on feedback from service members and veterans who said they wanted their loved ones to be actively included.
In addition, the report states that research into PTSD should be focused on current patient needs, and that both departments should actively collaborate with one another and with other government agencies, such as the National Institutes of Health, to fill knowledge gaps.
The number of veterans seeking care for PTSD from the VA has more than doubled from 190,000 (4.3% of all VA users) in 2003 to more than a half million (9.2%) in 2012. Although veterans of all eras are included in the increase, 23.6% (119,500) of those treated for PTSD by the VA in 2012 were veterans of the wars in Iraq and Afghanistan.
In 2013, 528,260 veterans made at least two visits to the VA for PTSD outpatient care; one-quarter were new patients. Although the overall incidence rate for PTSD across all service members is about 1%, the prevalence rose from 0.4% in 2004 to 5% in 2012, with an 8% increase in those who had been deployed previously, according to the report.
The committee said the DOD approach to PTSD treatment is "local, ad hoc, incremental, and crisis driven, with little planning." While VA programs benefits from better organization and consistency, the lack of data on either department’s delivery methods and outcomes means there is "no way of knowing whether the care they are providing is effective or whether DOD and VA’s expenditures are resulting in high-value health care," according to the report.
The report is based on 4 years of combing through data provided by the DOD and the VA, peer-reviewed literature, government documents, research databases, and testimonies from a variety of DOD and VA experts and providers at military bases and treatment facilities around the country, including six VA medical centers.
On Twitter @whitneymcknight
A lack of consistent outcome measures means there is no way to know whether the more than $3 billion spent on treating posttraumatic stress disorder by the Department of Defense and Veterans Affairs in 2012 yielded worthwhile results, according to a report released June 20.
"Given that the DOD and VA are responsible for serving millions of service members, families, and veterans, we found it surprising that no PTSD outcome measures are used consistently to know if these treatments are working," Dr. Sandro Galea, chair of the Institute of Medicine committee tasked by Congress to study PTSD treatment in military and veteran populations, said in a statement.
The report notes that currently, "neither the DOD nor the VA knows whether it is providing effective or adequate PTSD care, for which they spent $294 million and more than $3 billion, respectively, in 2012." Similar findings were reported by the IOM in 2012.
"What we found over and over again were really hardworking, well-intentioned people who wanted to do the best they could, but they either didn’t have an administrative structure to support them, or enough staff, or they had an overwhelming number of patients," committee member Dr. Elspeth Cameron Ritchie said during a press briefing.
In addition to better data collection and sharing, the report calls for the development of an adequate workforce to provide mental health care to this growing population.
Although tele-therapies and virtual reality therapies, for which the evidence base is growing, can provide some help, inadequate staffing still leads to a limitation in the number of evidence-based therapies available to patients, said Dr. Ritchie, a retired Army psychiatrist and current professor of psychiatry at Georgetown University in Washington. To wit, the report cited the VA’s failure in 2013 to provide the recommended eight sessions of psychotherapy within 14 weeks to nearly half of all Iraq and Afghanistan war veterans seeking care for a primary diagnosis of PTSD.
The report also calls for the development of evidenced-base treatments, including combination therapies of psychotherapies such as cognitive behavioral therapy, with medications such as SSRIs.
The report recommends that family members be involved in the treatment of PTSD; the recommendation was based on feedback from service members and veterans who said they wanted their loved ones to be actively included.
In addition, the report states that research into PTSD should be focused on current patient needs, and that both departments should actively collaborate with one another and with other government agencies, such as the National Institutes of Health, to fill knowledge gaps.
The number of veterans seeking care for PTSD from the VA has more than doubled from 190,000 (4.3% of all VA users) in 2003 to more than a half million (9.2%) in 2012. Although veterans of all eras are included in the increase, 23.6% (119,500) of those treated for PTSD by the VA in 2012 were veterans of the wars in Iraq and Afghanistan.
In 2013, 528,260 veterans made at least two visits to the VA for PTSD outpatient care; one-quarter were new patients. Although the overall incidence rate for PTSD across all service members is about 1%, the prevalence rose from 0.4% in 2004 to 5% in 2012, with an 8% increase in those who had been deployed previously, according to the report.
The committee said the DOD approach to PTSD treatment is "local, ad hoc, incremental, and crisis driven, with little planning." While VA programs benefits from better organization and consistency, the lack of data on either department’s delivery methods and outcomes means there is "no way of knowing whether the care they are providing is effective or whether DOD and VA’s expenditures are resulting in high-value health care," according to the report.
The report is based on 4 years of combing through data provided by the DOD and the VA, peer-reviewed literature, government documents, research databases, and testimonies from a variety of DOD and VA experts and providers at military bases and treatment facilities around the country, including six VA medical centers.
On Twitter @whitneymcknight

Comorbidities in migraine should guide treatment course
PHILADELPHIA – Anxiety and mood disorders, certain cardiovascular conditions, epilepsy, and poor sleep are all comorbidities in migraineurs that must be considered before choosing a patient’s treatment plan, according to Dr. Deborah I. Friedman.
Treating these comorbidities while minimizing risk factors is essential to chronic migraine prevention, Dr. Friedman told an audience at the Emerging Concepts in Headache Therapy session during this year’s annual meeting of the American Academy of Neurology.

Modifiable risk factors include the frequency of attack, caffeine intake, medication overuse, obesity, and sleep apnea.
Depression, anxiety, and suicide
Screening for anxiety and depression in migraine patients is important, given the close association between these psychiatric diagnoses and migraine, she said.
"Mood and anxiety disorders are anywhere from 2 to 10 times more prevalent in migraineurs," said Dr. Friedman, professor of neurology at the University of Texas Southwestern Medical Center at Dallas. "And 25% of patients with migraine meet [the] criteria for mood and anxiety disorders."
But teasing out which condition begets which comorbidity can be difficult; each might be a risk factor for the other.
"We know that depression, anxiety, and phobias are bidirectionally linked to migraine. If you have one, you’re more likely to have the other, and vice versa," Dr. Friedman said.
One possible mechanism for this is shared etiologic factors such as genetics. However, whether there is a confounder is still unknown. In the case of adverse childhood experiences and stressful life events, for example, "we know there is a large association of an unfortunate occurrence and migraine," Dr. Friedman said. "Is there a link between these things and posttraumatic stress disorder?"
However, patients with episodic migraine have less risk of developing mood and anxiety disorders, compared with chronic daily headache sufferers, Dr. Friedman said. "The comorbidity may relate to disabling and recurrent pain more than to the migraine itself."
In addition, a population-based cohort study of migraine patients, people with nonmigraine severe headache, and controls with no history of severe headache Dr. Friedman cited indicated statistically significant odds of attempted suicide in migraine when there is depression at baseline (odds ratio, 3.18); anxiety (OR, 4.78); depression and anxiety (OR, 12.1); and a previous suicide attempt (OR, 54.94) (Headache 2012;52:723-31).
Although the link between migraine and depression is high, Dr. Friedman said the link with anxiety is "probably much more common," occurring at a rate of five times more than would be expected in the general population.
For patients deemed to have a psychiatric comorbidity, Dr. Friedman said in an interview that cognitive-behavioral therapy is her preferred first-line therapy, particularly for anxiety and panic.
"It’s easy to reach for a pill to treat anxiety, but in the long haul, I am not sure it really benefits the patient that much because they don’t really gain insight into what’s causing their situation," Dr. Friedman said in the interview. It also might not be possible to "get two birds with one stone" by prescribing one pill to treat headache and depression and/or anxiety, Dr. Friedman said.
PFO and Raynaud’s
The odds that patients with migraine also have patent foramen ovale (PFO) or a right-left shunt are nearly eight times greater than in the general public, according to Dr. Friedman. The prevalence of PFO in migraine with aura is more than twice the rate of the general population.
Some nonrandomized studies have shown benefit to migraine from PFO closure, while the 2006 sham-controlled MIST (Migraine Intervention With STARFlex Technology) trial in the United Kingdom failed to achieve either its primary or secondary endpoints.
"Perhaps those endpoints were not realistic, because they were complete resolution of headache days at 3 months," Dr. Friedman said.
Some posit that the relationship between migraine and PFO might explain the increased risk of ischemic stroke and white matter intensities; however, since results from the PREMIUM and PRIMA studies on PFO and migraine are still pending, Dr. Friedman said there are no clinical recommendations for treating PFO in the context of just migraine at this time.
Raynaud’s phenomenon is another "chicken-egg" dilemma, Dr. Friedman said. Raynaud’s is five times more likely in those with migraine than in the general population, and migraine has similar occurrence rates in Raynaud’s.
Clinicians should take note that in addition to avoiding triptans and ergot derivatives in migraine patients with Raynaud’s, "beta-blockers can induce Raynaud’s in patients who have actually never experienced it," Dr. Friedman said, "and they can also make Raynaud’s worse."
However, particularly in patients with migraine who also have prominent autonomic symptoms, calcium channel blockers can treat both conditions, Dr. Friedman said.
Epilepsy and ‘migralepsy’
The incidence of migraine is nearly 2.5 times greater in persons with epilepsy than in those without it, according to Dr. Friedman.
Perhaps because they share a paroxysmal nature, Dr. Friedman said it is interesting to note that the comorbidities for epilepsy mirror those in migraine, including most major psychiatric diagnoses except psychosis, sleep and movement disorders, fibromyalgia, and asthma.
Seizures known as "migralepsy" that are triggered by migraine with aura can occur in patients either during or within 1 hour of a migraine attack, Dr. Friedman said. The mechanism is thought to be the cortical spread of depression.
Comorbid migraine in epilepsy decreases the likelihood of early treatment response, shortens remission periods, and is associated with intractable epilepsy, requiring polytherapy, Dr. Friedman said.
For those reasons, she recommended that patients not decrease their seizure threshold, either by their behaviors or their medications, and that clinicians select medications that can prevent both migraine and seizures. "Most of the medications we use for migraine do not affect the seizure threshold," she said in a follow-up interview. "Bupropion, venlafaxine, tramadol, [and] some of the antipsychotics and various stimulants (such as attention-deficit/hyperactivity disorder drugs) can do it. "The tricyclic antidepressants have long been associated with lowering the seizure threshold, but there is actually no good evidence that this is true."
Sleep disorders
"Sleep is like caffeine [in migraine]; it’s a double-edged sword," Dr. Friedman said. "Sleeping well can trigger a migraine, but it can also get rid of a migraine. Sleep interferes with pain, but pain interferes with sleep."
There are some data linking somnambulism, nightmares, and bruxism to migraine. "I have been surprised in my own practice how many of my patients tell me they used to sleep walk as a child," he said.
Referring to a 2010 study, Dr. Friedman said severe sleep disturbance was associated with a five times greater frequency of headache than in controls, and that insomnia, sleep initiation, and excessive daytime sleepiness were the most common complaints (J. Headache Pain 2010;11:197-206).
Although snoring and sleep apnea are not considered comorbidities of migraine, they do heighten the risk of episodic migraines becoming chronic, which is why Dr. Friedman recommended screening patients for sleep apnea. Asking patients about their sleep hygiene and sleep histories, including in childhood if they are adults, is important, as is reviewing what they ingest, from caffeine in food and drink to medications.
"Incorporating sleep questionnaires into your practice is very helpful," Dr. Friedman said.
She said she had no relevant financial disclosures.
On Twitter @whitneymcknight
PHILADELPHIA – Anxiety and mood disorders, certain cardiovascular conditions, epilepsy, and poor sleep are all comorbidities in migraineurs that must be considered before choosing a patient’s treatment plan, according to Dr. Deborah I. Friedman.
Treating these comorbidities while minimizing risk factors is essential to chronic migraine prevention, Dr. Friedman told an audience at the Emerging Concepts in Headache Therapy session during this year’s annual meeting of the American Academy of Neurology.
Modifiable risk factors include the frequency of attack, caffeine intake, medication overuse, obesity, and sleep apnea.
Depression, anxiety, and suicide
Screening for anxiety and depression in migraine patients is important, given the close association between these psychiatric diagnoses and migraine, she said.
"Mood and anxiety disorders are anywhere from 2 to 10 times more prevalent in migraineurs," said Dr. Friedman, professor of neurology at the University of Texas Southwestern Medical Center at Dallas. "And 25% of patients with migraine meet [the] criteria for mood and anxiety disorders."
But teasing out which condition begets which comorbidity can be difficult; each might be a risk factor for the other.
"We know that depression, anxiety, and phobias are bidirectionally linked to migraine. If you have one, you’re more likely to have the other, and vice versa," Dr. Friedman said.
One possible mechanism for this is shared etiologic factors such as genetics. However, whether there is a confounder is still unknown. In the case of adverse childhood experiences and stressful life events, for example, "we know there is a large association of an unfortunate occurrence and migraine," Dr. Friedman said. "Is there a link between these things and posttraumatic stress disorder?"
However, patients with episodic migraine have less risk of developing mood and anxiety disorders, compared with chronic daily headache sufferers, Dr. Friedman said. "The comorbidity may relate to disabling and recurrent pain more than to the migraine itself."
In addition, a population-based cohort study of migraine patients, people with nonmigraine severe headache, and controls with no history of severe headache Dr. Friedman cited indicated statistically significant odds of attempted suicide in migraine when there is depression at baseline (odds ratio, 3.18); anxiety (OR, 4.78); depression and anxiety (OR, 12.1); and a previous suicide attempt (OR, 54.94) (Headache 2012;52:723-31).
Although the link between migraine and depression is high, Dr. Friedman said the link with anxiety is "probably much more common," occurring at a rate of five times more than would be expected in the general population.
For patients deemed to have a psychiatric comorbidity, Dr. Friedman said in an interview that cognitive-behavioral therapy is her preferred first-line therapy, particularly for anxiety and panic.
"It’s easy to reach for a pill to treat anxiety, but in the long haul, I am not sure it really benefits the patient that much because they don’t really gain insight into what’s causing their situation," Dr. Friedman said in the interview. It also might not be possible to "get two birds with one stone" by prescribing one pill to treat headache and depression and/or anxiety, Dr. Friedman said.
PFO and Raynaud’s
The odds that patients with migraine also have patent foramen ovale (PFO) or a right-left shunt are nearly eight times greater than in the general public, according to Dr. Friedman. The prevalence of PFO in migraine with aura is more than twice the rate of the general population.
Some nonrandomized studies have shown benefit to migraine from PFO closure, while the 2006 sham-controlled MIST (Migraine Intervention With STARFlex Technology) trial in the United Kingdom failed to achieve either its primary or secondary endpoints.
"Perhaps those endpoints were not realistic, because they were complete resolution of headache days at 3 months," Dr. Friedman said.
Some posit that the relationship between migraine and PFO might explain the increased risk of ischemic stroke and white matter intensities; however, since results from the PREMIUM and PRIMA studies on PFO and migraine are still pending, Dr. Friedman said there are no clinical recommendations for treating PFO in the context of just migraine at this time.
Raynaud’s phenomenon is another "chicken-egg" dilemma, Dr. Friedman said. Raynaud’s is five times more likely in those with migraine than in the general population, and migraine has similar occurrence rates in Raynaud’s.
Clinicians should take note that in addition to avoiding triptans and ergot derivatives in migraine patients with Raynaud’s, "beta-blockers can induce Raynaud’s in patients who have actually never experienced it," Dr. Friedman said, "and they can also make Raynaud’s worse."
However, particularly in patients with migraine who also have prominent autonomic symptoms, calcium channel blockers can treat both conditions, Dr. Friedman said.
Epilepsy and ‘migralepsy’
The incidence of migraine is nearly 2.5 times greater in persons with epilepsy than in those without it, according to Dr. Friedman.
Perhaps because they share a paroxysmal nature, Dr. Friedman said it is interesting to note that the comorbidities for epilepsy mirror those in migraine, including most major psychiatric diagnoses except psychosis, sleep and movement disorders, fibromyalgia, and asthma.
Seizures known as "migralepsy" that are triggered by migraine with aura can occur in patients either during or within 1 hour of a migraine attack, Dr. Friedman said. The mechanism is thought to be the cortical spread of depression.
Comorbid migraine in epilepsy decreases the likelihood of early treatment response, shortens remission periods, and is associated with intractable epilepsy, requiring polytherapy, Dr. Friedman said.
For those reasons, she recommended that patients not decrease their seizure threshold, either by their behaviors or their medications, and that clinicians select medications that can prevent both migraine and seizures. "Most of the medications we use for migraine do not affect the seizure threshold," she said in a follow-up interview. "Bupropion, venlafaxine, tramadol, [and] some of the antipsychotics and various stimulants (such as attention-deficit/hyperactivity disorder drugs) can do it. "The tricyclic antidepressants have long been associated with lowering the seizure threshold, but there is actually no good evidence that this is true."
Sleep disorders
"Sleep is like caffeine [in migraine]; it’s a double-edged sword," Dr. Friedman said. "Sleeping well can trigger a migraine, but it can also get rid of a migraine. Sleep interferes with pain, but pain interferes with sleep."
There are some data linking somnambulism, nightmares, and bruxism to migraine. "I have been surprised in my own practice how many of my patients tell me they used to sleep walk as a child," he said.
Referring to a 2010 study, Dr. Friedman said severe sleep disturbance was associated with a five times greater frequency of headache than in controls, and that insomnia, sleep initiation, and excessive daytime sleepiness were the most common complaints (J. Headache Pain 2010;11:197-206).
Although snoring and sleep apnea are not considered comorbidities of migraine, they do heighten the risk of episodic migraines becoming chronic, which is why Dr. Friedman recommended screening patients for sleep apnea. Asking patients about their sleep hygiene and sleep histories, including in childhood if they are adults, is important, as is reviewing what they ingest, from caffeine in food and drink to medications.
"Incorporating sleep questionnaires into your practice is very helpful," Dr. Friedman said.
She said she had no relevant financial disclosures.
On Twitter @whitneymcknight
PHILADELPHIA – Anxiety and mood disorders, certain cardiovascular conditions, epilepsy, and poor sleep are all comorbidities in migraineurs that must be considered before choosing a patient’s treatment plan, according to Dr. Deborah I. Friedman.
Treating these comorbidities while minimizing risk factors is essential to chronic migraine prevention, Dr. Friedman told an audience at the Emerging Concepts in Headache Therapy session during this year’s annual meeting of the American Academy of Neurology.
Modifiable risk factors include the frequency of attack, caffeine intake, medication overuse, obesity, and sleep apnea.
Depression, anxiety, and suicide
Screening for anxiety and depression in migraine patients is important, given the close association between these psychiatric diagnoses and migraine, she said.
"Mood and anxiety disorders are anywhere from 2 to 10 times more prevalent in migraineurs," said Dr. Friedman, professor of neurology at the University of Texas Southwestern Medical Center at Dallas. "And 25% of patients with migraine meet [the] criteria for mood and anxiety disorders."
But teasing out which condition begets which comorbidity can be difficult; each might be a risk factor for the other.
"We know that depression, anxiety, and phobias are bidirectionally linked to migraine. If you have one, you’re more likely to have the other, and vice versa," Dr. Friedman said.
One possible mechanism for this is shared etiologic factors such as genetics. However, whether there is a confounder is still unknown. In the case of adverse childhood experiences and stressful life events, for example, "we know there is a large association of an unfortunate occurrence and migraine," Dr. Friedman said. "Is there a link between these things and posttraumatic stress disorder?"
However, patients with episodic migraine have less risk of developing mood and anxiety disorders, compared with chronic daily headache sufferers, Dr. Friedman said. "The comorbidity may relate to disabling and recurrent pain more than to the migraine itself."
In addition, a population-based cohort study of migraine patients, people with nonmigraine severe headache, and controls with no history of severe headache Dr. Friedman cited indicated statistically significant odds of attempted suicide in migraine when there is depression at baseline (odds ratio, 3.18); anxiety (OR, 4.78); depression and anxiety (OR, 12.1); and a previous suicide attempt (OR, 54.94) (Headache 2012;52:723-31).
Although the link between migraine and depression is high, Dr. Friedman said the link with anxiety is "probably much more common," occurring at a rate of five times more than would be expected in the general population.
For patients deemed to have a psychiatric comorbidity, Dr. Friedman said in an interview that cognitive-behavioral therapy is her preferred first-line therapy, particularly for anxiety and panic.
"It’s easy to reach for a pill to treat anxiety, but in the long haul, I am not sure it really benefits the patient that much because they don’t really gain insight into what’s causing their situation," Dr. Friedman said in the interview. It also might not be possible to "get two birds with one stone" by prescribing one pill to treat headache and depression and/or anxiety, Dr. Friedman said.
PFO and Raynaud’s
The odds that patients with migraine also have patent foramen ovale (PFO) or a right-left shunt are nearly eight times greater than in the general public, according to Dr. Friedman. The prevalence of PFO in migraine with aura is more than twice the rate of the general population.
Some nonrandomized studies have shown benefit to migraine from PFO closure, while the 2006 sham-controlled MIST (Migraine Intervention With STARFlex Technology) trial in the United Kingdom failed to achieve either its primary or secondary endpoints.
"Perhaps those endpoints were not realistic, because they were complete resolution of headache days at 3 months," Dr. Friedman said.
Some posit that the relationship between migraine and PFO might explain the increased risk of ischemic stroke and white matter intensities; however, since results from the PREMIUM and PRIMA studies on PFO and migraine are still pending, Dr. Friedman said there are no clinical recommendations for treating PFO in the context of just migraine at this time.
Raynaud’s phenomenon is another "chicken-egg" dilemma, Dr. Friedman said. Raynaud’s is five times more likely in those with migraine than in the general population, and migraine has similar occurrence rates in Raynaud’s.
Clinicians should take note that in addition to avoiding triptans and ergot derivatives in migraine patients with Raynaud’s, "beta-blockers can induce Raynaud’s in patients who have actually never experienced it," Dr. Friedman said, "and they can also make Raynaud’s worse."
However, particularly in patients with migraine who also have prominent autonomic symptoms, calcium channel blockers can treat both conditions, Dr. Friedman said.
Epilepsy and ‘migralepsy’
The incidence of migraine is nearly 2.5 times greater in persons with epilepsy than in those without it, according to Dr. Friedman.
Perhaps because they share a paroxysmal nature, Dr. Friedman said it is interesting to note that the comorbidities for epilepsy mirror those in migraine, including most major psychiatric diagnoses except psychosis, sleep and movement disorders, fibromyalgia, and asthma.
Seizures known as "migralepsy" that are triggered by migraine with aura can occur in patients either during or within 1 hour of a migraine attack, Dr. Friedman said. The mechanism is thought to be the cortical spread of depression.
Comorbid migraine in epilepsy decreases the likelihood of early treatment response, shortens remission periods, and is associated with intractable epilepsy, requiring polytherapy, Dr. Friedman said.
For those reasons, she recommended that patients not decrease their seizure threshold, either by their behaviors or their medications, and that clinicians select medications that can prevent both migraine and seizures. "Most of the medications we use for migraine do not affect the seizure threshold," she said in a follow-up interview. "Bupropion, venlafaxine, tramadol, [and] some of the antipsychotics and various stimulants (such as attention-deficit/hyperactivity disorder drugs) can do it. "The tricyclic antidepressants have long been associated with lowering the seizure threshold, but there is actually no good evidence that this is true."
Sleep disorders
"Sleep is like caffeine [in migraine]; it’s a double-edged sword," Dr. Friedman said. "Sleeping well can trigger a migraine, but it can also get rid of a migraine. Sleep interferes with pain, but pain interferes with sleep."
There are some data linking somnambulism, nightmares, and bruxism to migraine. "I have been surprised in my own practice how many of my patients tell me they used to sleep walk as a child," he said.
Referring to a 2010 study, Dr. Friedman said severe sleep disturbance was associated with a five times greater frequency of headache than in controls, and that insomnia, sleep initiation, and excessive daytime sleepiness were the most common complaints (J. Headache Pain 2010;11:197-206).
Although snoring and sleep apnea are not considered comorbidities of migraine, they do heighten the risk of episodic migraines becoming chronic, which is why Dr. Friedman recommended screening patients for sleep apnea. Asking patients about their sleep hygiene and sleep histories, including in childhood if they are adults, is important, as is reviewing what they ingest, from caffeine in food and drink to medications.
"Incorporating sleep questionnaires into your practice is very helpful," Dr. Friedman said.
She said she had no relevant financial disclosures.
On Twitter @whitneymcknight
EXPERT ANALYSIS AT THE AAN 2014 ANNUAL MEETING

EMS staff missed stroke 41% of the time, study shows
PHILADELPHIA – Emergency personnel miss stroke in 41% of patients in their prehospital care, a study shows. In light of this, physicians should have a high degree of suspicion for stroke when treating patients presenting with elevated systolic blood pressure, a dispatch call of stroke, prior stroke or diabetes history, and unconsciousness.
Prehospital identification and notification of stroke have been associated with both shorter door-to-needle times and increased thrombolysis rates, but "the sensitivity and specificity of prehospital care providers [are] widely variable," Dr. Ethan Brandler, of the emergency medicine department at SUNY Downstate Medical Center in Brooklyn, N.Y., told a Platform Blitz audience at the annual meeting of the American Academy of Neurology.

To assess the rates of proper stroke diagnosis by emergency medical services (EMS), Dr. Brandler and his associates reviewed 10,384 electronic prehospital care reports of all patients delivered by EMS to a single major metropolitan site between Jan. 1, 2010, and Dec. 31, 2011.
Using the American Heart Association’s Get With the Guidelines stroke database, the investigators determined that of the 75 confirmed cases of stroke, half of which were in men aged 19-49 years, EMS personnel accurately diagnosed 44 patients.
"Forty-one percent of all stroke patients were missed by EMS," Dr. Brandler said. "And 51 patients were misdiagnosed as stroke when they had something else: stroke-mimic, sepsis, infection, [and] hypoglycemia, among other neurologic and systemic problems."
The sensitivity level of 59% and specificity level of 99.5% (95% confidence interval, 47-69 and 99.1-99.6, respectively), combined with the positive likelihood ratio of 119 (95% CI, 85-165) and the negative likelihood ratio of 0.41 (95% CI, 0.32-0.54), meant that "if a paramedic called us to say a stroke was going on, the likelihood was extremely high it was a true stroke," Dr. Brandler said. "However, when they didn’t think it was a stroke, it didn’t mean much."
Stroke risk increased 83% with each increase in prehospital systolic blood pressure quartile (P = .04), Dr. Brandler and his colleagues found.
Independent predictors of stroke being the official diagnosis included an emergency services dispatcher who reported the call as a stroke, even if EMS personnel did not ultimately diagnose it that way (age-adjusted odds ratio, 9.8; 95% CI, 2.2-43.7; P = .003]. "Perhaps [the dispatcher’s] information is better than the crew’s because it is gathered in a more systematic fashion," Dr. Brandler said.
Other independent risk factors included a history of diabetes (AAOR, 2.2; 95% CI, 1.1-4.6; P = .026) and a history of stroke (AAOR, 4.4; 95% CI, 1.3-15; P = .017).
Patients who arrived unconscious with unknown cause also were considered at higher risk for having had a stroke (AAOR, 4.4; 95% CI, 1.3-15; P = .017). This was particularly the case when patients presented with basilar artery inclusions, Dr. Brandler said.
The study was funded by grants from the National Institutes of Health. Dr. Brandler did not report any other relevant disclosures.
On Twitter @whitneymcknight
PHILADELPHIA – Emergency personnel miss stroke in 41% of patients in their prehospital care, a study shows. In light of this, physicians should have a high degree of suspicion for stroke when treating patients presenting with elevated systolic blood pressure, a dispatch call of stroke, prior stroke or diabetes history, and unconsciousness.
Prehospital identification and notification of stroke have been associated with both shorter door-to-needle times and increased thrombolysis rates, but "the sensitivity and specificity of prehospital care providers [are] widely variable," Dr. Ethan Brandler, of the emergency medicine department at SUNY Downstate Medical Center in Brooklyn, N.Y., told a Platform Blitz audience at the annual meeting of the American Academy of Neurology.
To assess the rates of proper stroke diagnosis by emergency medical services (EMS), Dr. Brandler and his associates reviewed 10,384 electronic prehospital care reports of all patients delivered by EMS to a single major metropolitan site between Jan. 1, 2010, and Dec. 31, 2011.
Using the American Heart Association’s Get With the Guidelines stroke database, the investigators determined that of the 75 confirmed cases of stroke, half of which were in men aged 19-49 years, EMS personnel accurately diagnosed 44 patients.
"Forty-one percent of all stroke patients were missed by EMS," Dr. Brandler said. "And 51 patients were misdiagnosed as stroke when they had something else: stroke-mimic, sepsis, infection, [and] hypoglycemia, among other neurologic and systemic problems."
The sensitivity level of 59% and specificity level of 99.5% (95% confidence interval, 47-69 and 99.1-99.6, respectively), combined with the positive likelihood ratio of 119 (95% CI, 85-165) and the negative likelihood ratio of 0.41 (95% CI, 0.32-0.54), meant that "if a paramedic called us to say a stroke was going on, the likelihood was extremely high it was a true stroke," Dr. Brandler said. "However, when they didn’t think it was a stroke, it didn’t mean much."
Stroke risk increased 83% with each increase in prehospital systolic blood pressure quartile (P = .04), Dr. Brandler and his colleagues found.
Independent predictors of stroke being the official diagnosis included an emergency services dispatcher who reported the call as a stroke, even if EMS personnel did not ultimately diagnose it that way (age-adjusted odds ratio, 9.8; 95% CI, 2.2-43.7; P = .003]. "Perhaps [the dispatcher’s] information is better than the crew’s because it is gathered in a more systematic fashion," Dr. Brandler said.
Other independent risk factors included a history of diabetes (AAOR, 2.2; 95% CI, 1.1-4.6; P = .026) and a history of stroke (AAOR, 4.4; 95% CI, 1.3-15; P = .017).
Patients who arrived unconscious with unknown cause also were considered at higher risk for having had a stroke (AAOR, 4.4; 95% CI, 1.3-15; P = .017). This was particularly the case when patients presented with basilar artery inclusions, Dr. Brandler said.
The study was funded by grants from the National Institutes of Health. Dr. Brandler did not report any other relevant disclosures.
On Twitter @whitneymcknight
PHILADELPHIA – Emergency personnel miss stroke in 41% of patients in their prehospital care, a study shows. In light of this, physicians should have a high degree of suspicion for stroke when treating patients presenting with elevated systolic blood pressure, a dispatch call of stroke, prior stroke or diabetes history, and unconsciousness.
Prehospital identification and notification of stroke have been associated with both shorter door-to-needle times and increased thrombolysis rates, but "the sensitivity and specificity of prehospital care providers [are] widely variable," Dr. Ethan Brandler, of the emergency medicine department at SUNY Downstate Medical Center in Brooklyn, N.Y., told a Platform Blitz audience at the annual meeting of the American Academy of Neurology.
To assess the rates of proper stroke diagnosis by emergency medical services (EMS), Dr. Brandler and his associates reviewed 10,384 electronic prehospital care reports of all patients delivered by EMS to a single major metropolitan site between Jan. 1, 2010, and Dec. 31, 2011.
Using the American Heart Association’s Get With the Guidelines stroke database, the investigators determined that of the 75 confirmed cases of stroke, half of which were in men aged 19-49 years, EMS personnel accurately diagnosed 44 patients.
"Forty-one percent of all stroke patients were missed by EMS," Dr. Brandler said. "And 51 patients were misdiagnosed as stroke when they had something else: stroke-mimic, sepsis, infection, [and] hypoglycemia, among other neurologic and systemic problems."
The sensitivity level of 59% and specificity level of 99.5% (95% confidence interval, 47-69 and 99.1-99.6, respectively), combined with the positive likelihood ratio of 119 (95% CI, 85-165) and the negative likelihood ratio of 0.41 (95% CI, 0.32-0.54), meant that "if a paramedic called us to say a stroke was going on, the likelihood was extremely high it was a true stroke," Dr. Brandler said. "However, when they didn’t think it was a stroke, it didn’t mean much."
Stroke risk increased 83% with each increase in prehospital systolic blood pressure quartile (P = .04), Dr. Brandler and his colleagues found.
Independent predictors of stroke being the official diagnosis included an emergency services dispatcher who reported the call as a stroke, even if EMS personnel did not ultimately diagnose it that way (age-adjusted odds ratio, 9.8; 95% CI, 2.2-43.7; P = .003]. "Perhaps [the dispatcher’s] information is better than the crew’s because it is gathered in a more systematic fashion," Dr. Brandler said.
Other independent risk factors included a history of diabetes (AAOR, 2.2; 95% CI, 1.1-4.6; P = .026) and a history of stroke (AAOR, 4.4; 95% CI, 1.3-15; P = .017).
Patients who arrived unconscious with unknown cause also were considered at higher risk for having had a stroke (AAOR, 4.4; 95% CI, 1.3-15; P = .017). This was particularly the case when patients presented with basilar artery inclusions, Dr. Brandler said.
The study was funded by grants from the National Institutes of Health. Dr. Brandler did not report any other relevant disclosures.
On Twitter @whitneymcknight
AT THE AAN 2014 ANNUAL MEETING
Key clinical point: A higher degree of suspicion is warranted for stroke in patients with elevated systolic blood pressure, a dispatch call of stroke, prior stroke or diabetes history, and unconsciousness.
Major finding: EMS staff had 59% sensitivity and 99% specificity (95% confidence intervals, 47-69 and 99.4-99.6, respectively) when diagnosing stroke; and positive and negative likelihood ratios were 119 and 0.41 (95% CIs, 85-165 and 0.32-0.54). Stroke risk increased 83% with each increase in prehospital systolic blood pressure quartile (P = .04).
Data source: Two-year, retrospective analysis of data for 10,384 electronic prehospital care reports of patients brought by EMTs to a single metropolitan site.
Disclosures: The study was funded by grants from the National Institutes of Health. Dr. Brandler did not report any other relevant disclosures.

VIDEO: Team approach is best for successful oncofertility clinic
PHILADELPHIA – Whether you want to build an entire oncofertility clinic or just elevate the level of care your current facility offers, an integrated approach to care is key, according to Dr. Leslie A. Appiah, director of oncofertility at the University of Kentucky Medical Center, Lexington.
In a video interview at the annual meeting of the North American Society for Pediatric and Adolescent Gynecology, Dr. Appiah offers suggestions for how oncologists, gynecologists, reproductive endocrinologists, nurse managers, social workers, and others – even when they’re with other institutions – can work together to offer care that adheres to position statements on fertility rights for cancer patients from the American Society of Clinical Oncology and other associations.
Dr. Appiah also discusses how to achieve fluid communication so that fertility can be preserved without delaying cancer treatments, and best practices for oncofertility concerns in the pediatric setting.
On Twitter @whitneymcknight
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
PHILADELPHIA – Whether you want to build an entire oncofertility clinic or just elevate the level of care your current facility offers, an integrated approach to care is key, according to Dr. Leslie A. Appiah, director of oncofertility at the University of Kentucky Medical Center, Lexington.
In a video interview at the annual meeting of the North American Society for Pediatric and Adolescent Gynecology, Dr. Appiah offers suggestions for how oncologists, gynecologists, reproductive endocrinologists, nurse managers, social workers, and others – even when they’re with other institutions – can work together to offer care that adheres to position statements on fertility rights for cancer patients from the American Society of Clinical Oncology and other associations.
Dr. Appiah also discusses how to achieve fluid communication so that fertility can be preserved without delaying cancer treatments, and best practices for oncofertility concerns in the pediatric setting.
On Twitter @whitneymcknight
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
PHILADELPHIA – Whether you want to build an entire oncofertility clinic or just elevate the level of care your current facility offers, an integrated approach to care is key, according to Dr. Leslie A. Appiah, director of oncofertility at the University of Kentucky Medical Center, Lexington.
In a video interview at the annual meeting of the North American Society for Pediatric and Adolescent Gynecology, Dr. Appiah offers suggestions for how oncologists, gynecologists, reproductive endocrinologists, nurse managers, social workers, and others – even when they’re with other institutions – can work together to offer care that adheres to position statements on fertility rights for cancer patients from the American Society of Clinical Oncology and other associations.
Dr. Appiah also discusses how to achieve fluid communication so that fertility can be preserved without delaying cancer treatments, and best practices for oncofertility concerns in the pediatric setting.
On Twitter @whitneymcknight
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT THE NASPAG ANNUAL MEETING
