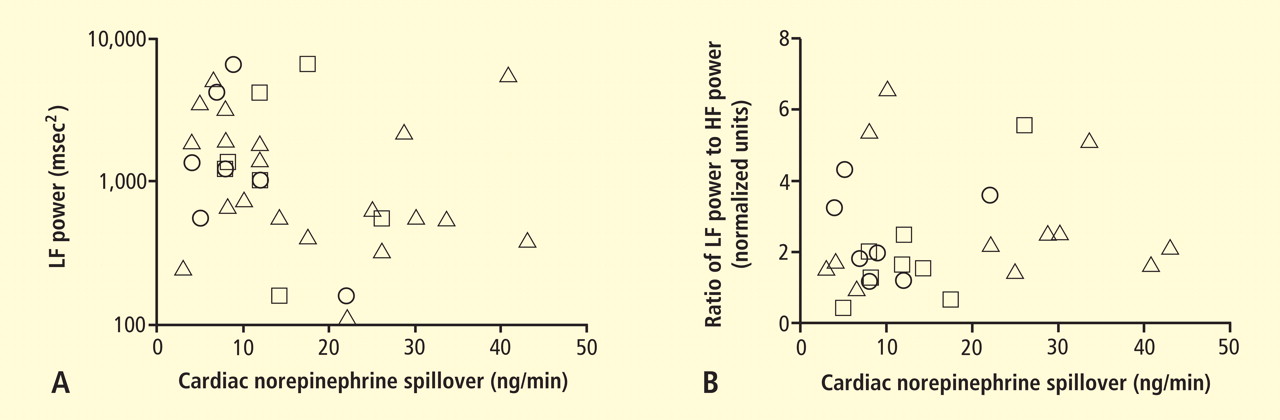
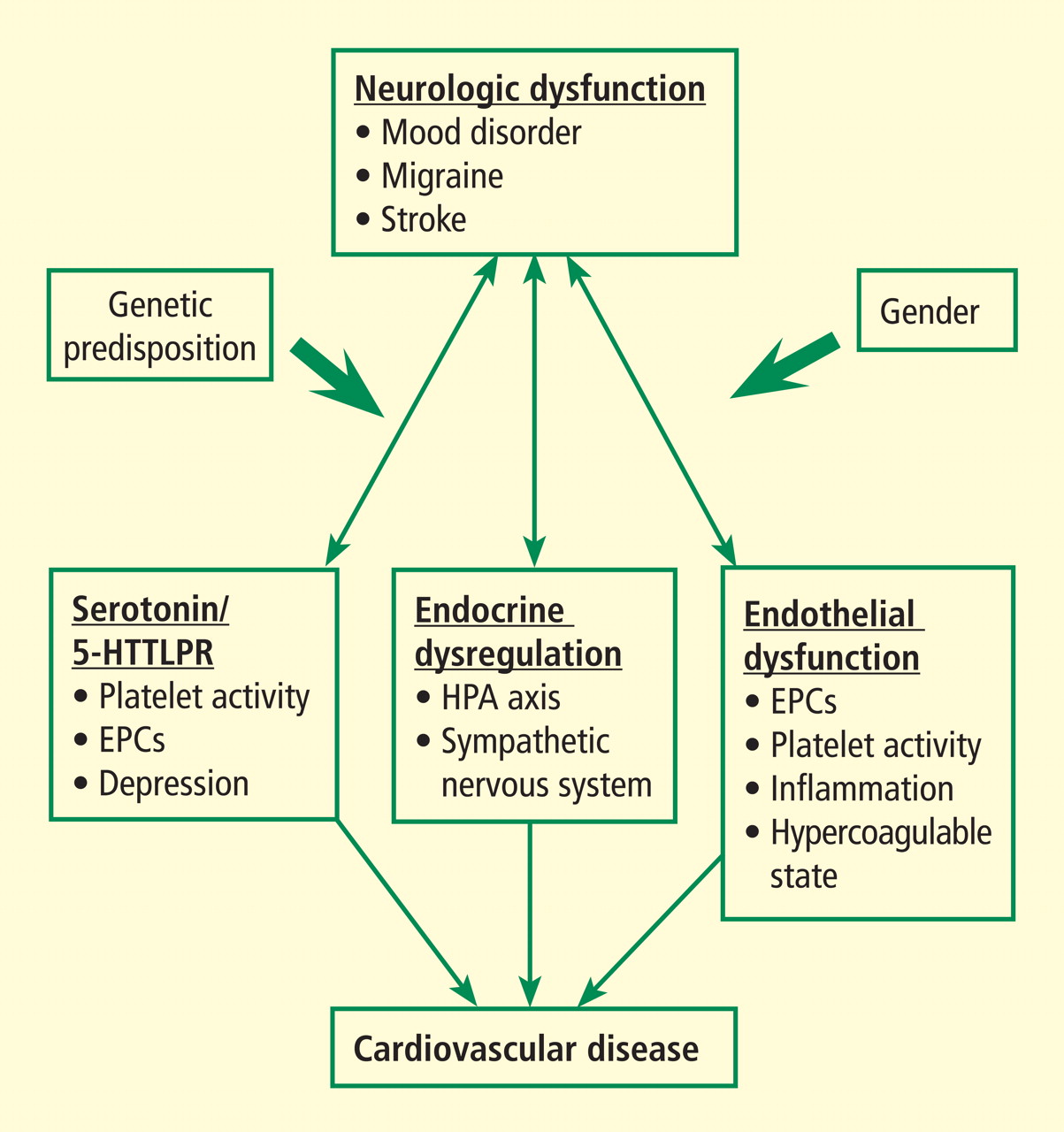
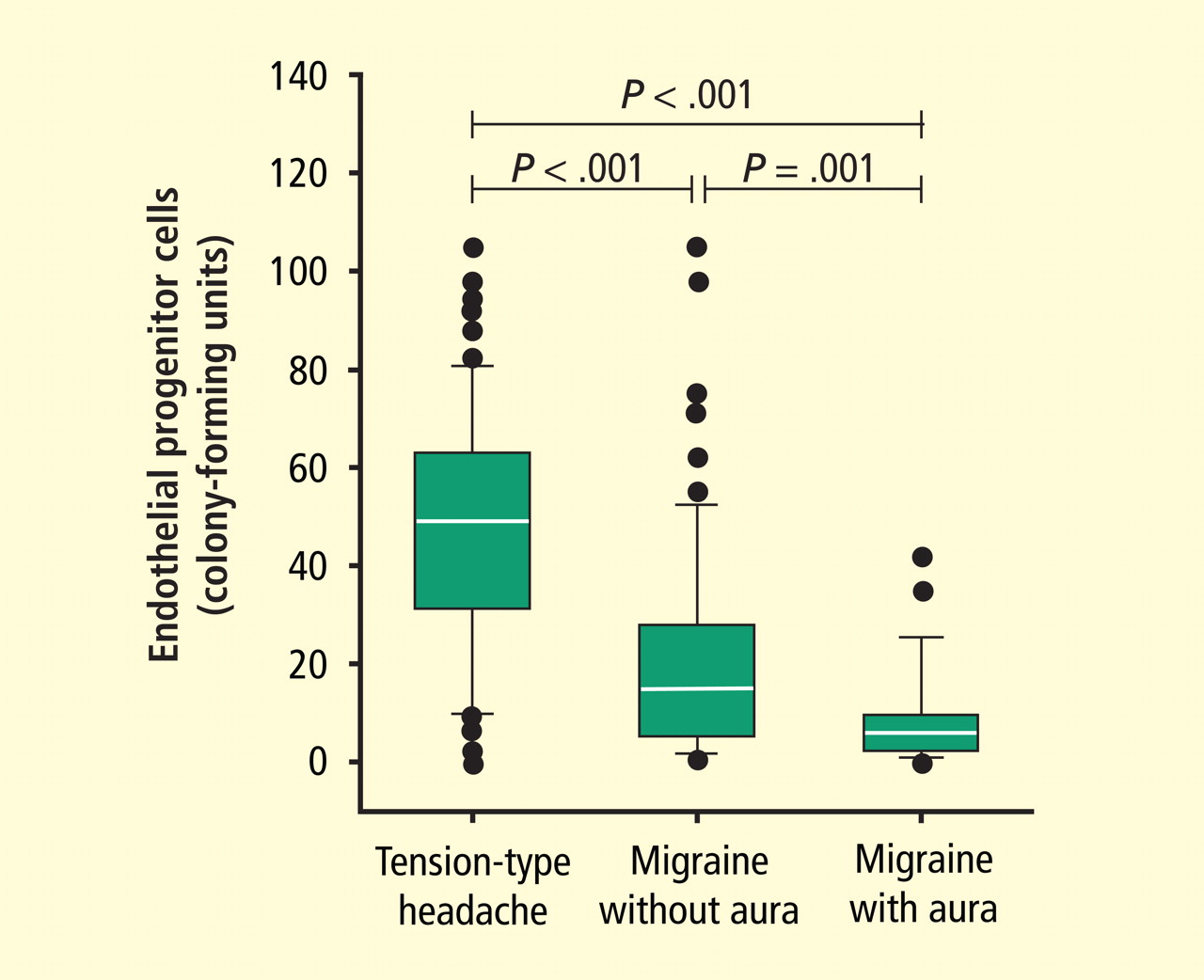
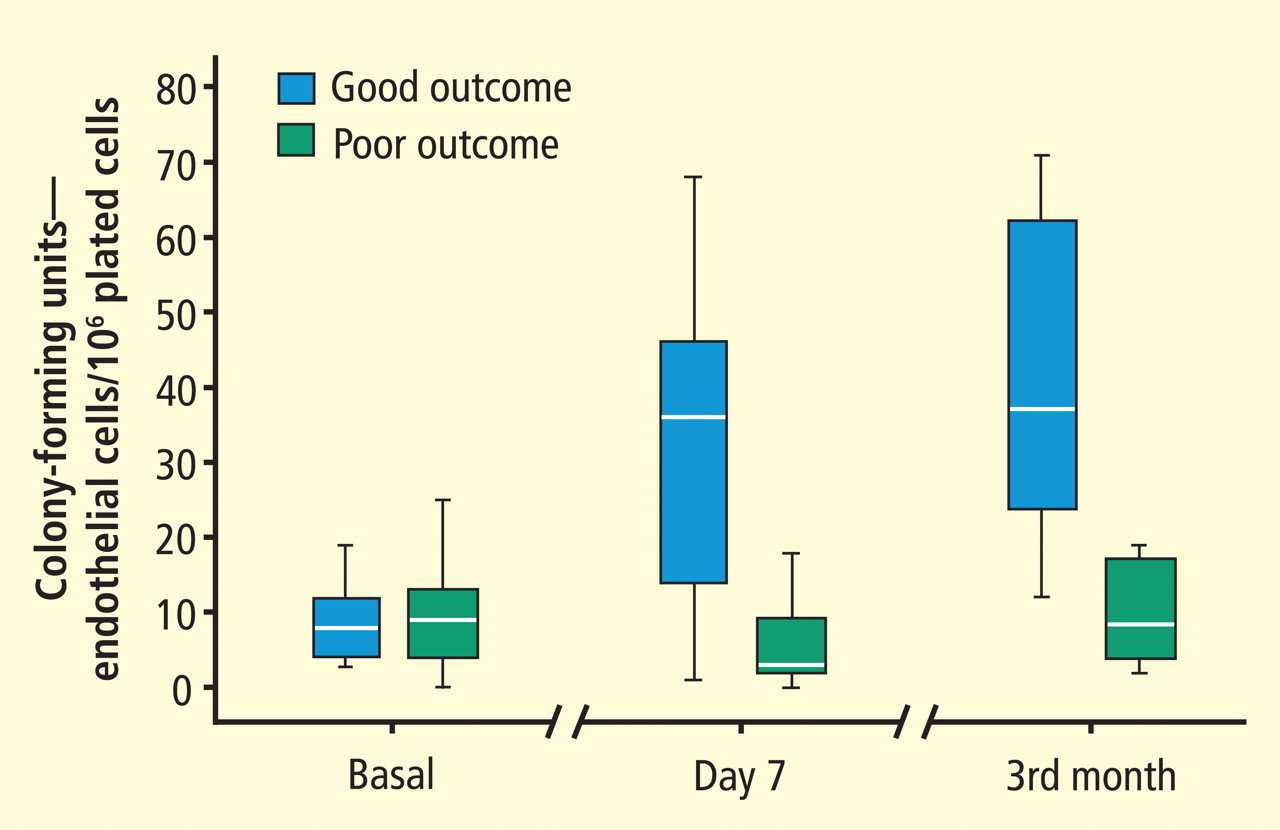
CCJM delivers practical clinical articles relevant to internists, cardiologists, endocrinologists, and other specialists, all written by known experts.
In 2008, the American Heart Association (AHA) published a science advisory on depression and coronary heart disease (CHD).1 Since its publication, the advisory has evoked substantial commentary. The purpose of this article is threefold: (1) to explain the aims of the AHA science advisory, (2) to briefly discuss its content, and (3) to examine some of the comments it has provoked.
WHAT THE ADVISORY SET OUT TO DO
The purpose of an AHA science advisory is to provide rapid, clear, and consistent AHA positioning on a scientific issue. Advisories are statements on an evolving, prominent scientific issue of great interest to the public and health professionals. All AHA science advisories undergo peer review and are also reviewed and approved by the AHA Science Advisory and Coordinating Committee, AHA’s highest science body. Because this particular advisory addressed the interaction of cardiovascular and mental health, the AHA asked the American Psychiatric Association (APA) to review the document; the APA endorsed the AHA advisory.
Two points are worth emphasizing:
An AHA science advisory is not a treatment guideline.
Advisories usually are brief and therefore do not exhaustively discuss their topic.
After discussing epidemiologic studies that elucidated the relationships between depression and CHD, the AHA advisory on depression and CHD focuses on screening, referral, and treatment of depression from a cardiology perspective.
The 1-year prevalence of major depressive disorder in the US general population is 7%, and the lifetime prevalence is about 16%.2 Depression in otherwise healthy persons almost doubles the risk of developing CHD.3 About 20% of patients hospitalized for acute coronary syndromes (ACS) have major depressive disorder on admission or within a few weeks thereafter, and these patients have about 2.5 times the mortality rate as patients who are not depressed, after adjusting for infarct severity and cardiovascular risk factors.4–6
ASSESSMENT OF DEPRESSION AND DEPRESSIVE SYMPTOMS
The AHA advisory discusses use of the 2-question Patient Health Questionnaire (PHQ-2) as the first step in screening for depression.7,8 The PHQ-2 inquires about the frequency of depressed mood and anhedonia by asking the following:
Over the past 2 weeks, how often have you been bothered by either of the following problems?
(1) Little interest or pleasure in doing things
(2) Feeling down, depressed, or hopeless.
For each item the response options are “not at all” (scored as 0), “several days” (scored as 1), “more than half the days” (scored as 2), and “nearly every day” (scored as 3). Thus, the total PHQ-2 score can range from 0 to 6.
Using a structured psychiatric interview as the standard, a total PHQ-2 score of 3 or greater has been shown to have a sensitivity of 83% and a specificity of 92% for major depression.7 A PHQ-2 score of 3 is the optimal cut point for screening purposes. A PHQ-2 score of 0 virtually excludes depression.
If a patient’s PHQ-2 score is 3 or greater, it is recommended that answers be obtained for a full 9-item PHQ. The PHQ-9 provides the sensitivity and specificity suitable for assigning a provisional diagnosis of major depressive disorder and a symptom severity score that can be used to identify patients for further evaluation and to make decisions about therapy.9–11
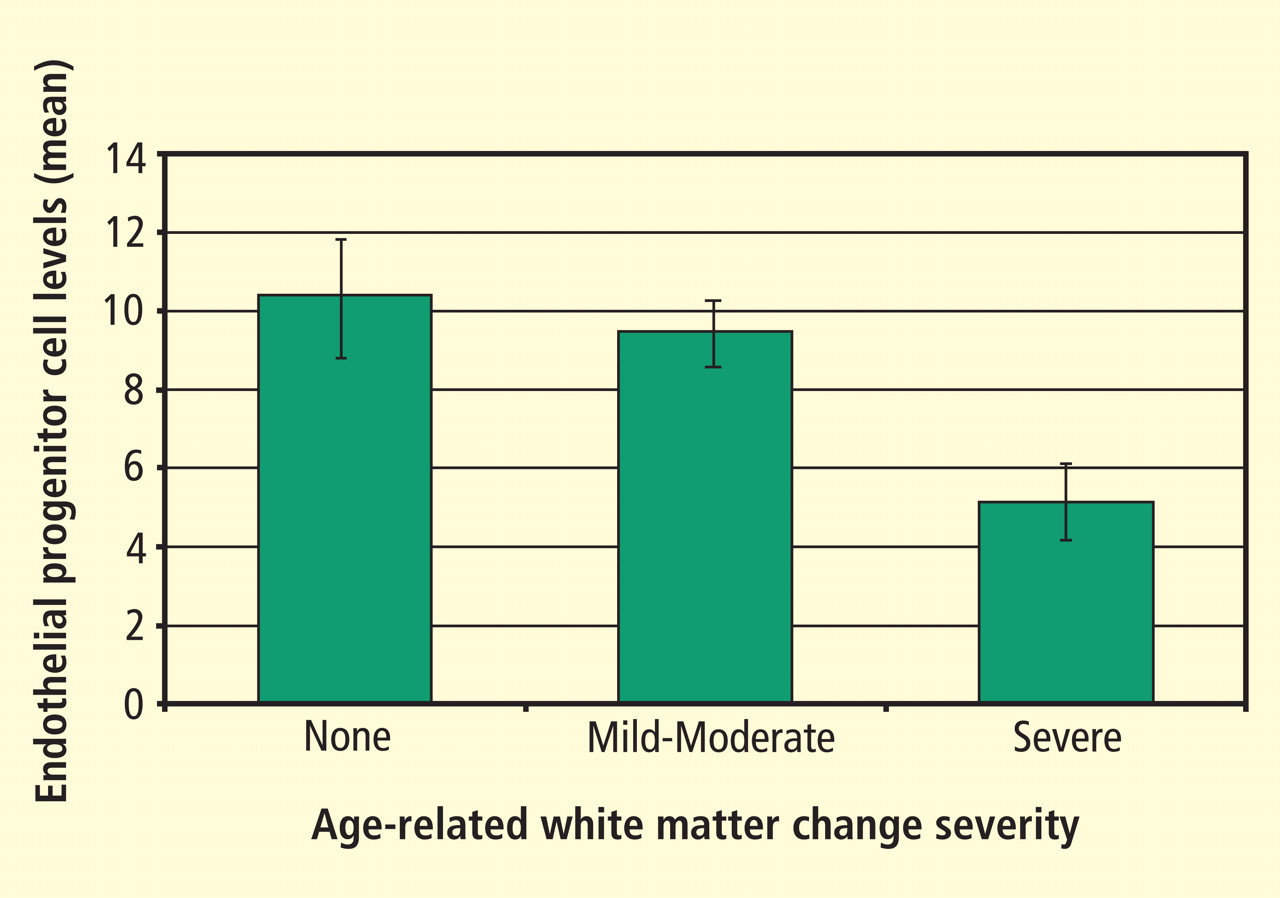
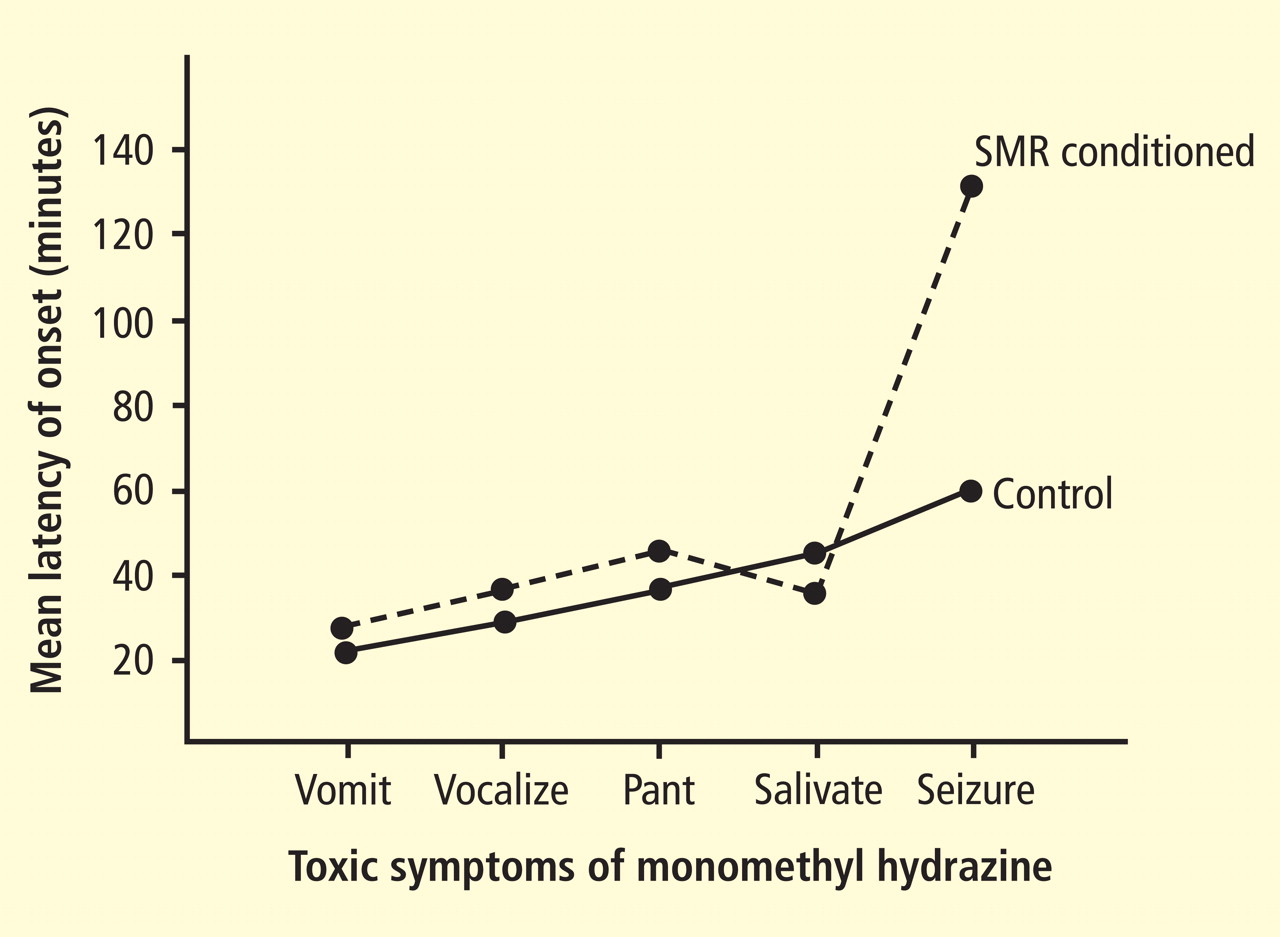
Adapted from the 2008 American Heart Association science advisory on depression and coronary heart disease.1 Source: American Heart Association, Inc.
Figure 1. Screening for depression in patients with coronary heart disease.The AHA advisory’s section on assessment of depression and depressive symptoms discusses briefly the principles enunciated by the MacArthur Initiative on Depression and Primary Care.12–14 The advisory carefully provides practical guidance specifically for cardiologists (Figure 1).1 The section on assessment points out that screening for depression coupled with therapy has not been proven to improve cardiovascular outcomes but that some antidepressant drugs and/or psychotherapy have proved safe and can improve quality of life, reduce depressive symptoms, improve compliance with lifestyle advice, and improve adherence to prescribed cardiovascular medications.1,15,16
The US Preventive Services Task Force (USPSTF) currently recommends that clinicians screen adults for depression in clinical practices that have systems in place to assure accurate diagnosis, effective treatment, and follow-up.12 The USPSTF concluded that there is good evidence that screening improves the accurate identification of depressed patients in primary care settings and that treating depressed adults identified in such settings reduces clinical morbidity. Results of trials that have directly evaluated the effect of screening on clinical outcomes depend on follow-up. Limited benefits have been demonstrated in studies that simply feed screening results back to clinicians. Larger benefits have been seen in studies in which the communication of screening results is coordinated with effective follow-up and treatment. The USPSTF concluded that the benefits of screening and treating are likely to outweigh any potential harms.12
DEPRESSION TREATMENT
Drug therapy
The safety of fluoxetine, sertraline (SADHART, ENRICHD), citalopram (CREATE), and mirtazapine (MIND-IT) has been evaluated in clinical trials.4,17–21 By far the strongest evidence of safety is for selective serotonin reuptake inhibitors (SSRIs), especially sertraline.
The Sertraline Antidepressant Heart Attack Randomized Trial (SADHART; N = 369), a randomized study of depression after ACS, found no difference in cardiovascular adverse events between the sertraline and placebo groups after 16 weeks of therapy, and there were no significant differences in left ventricular ejection fraction, heart rate, blood pressure, ventricular premature complexes, or electrocardiogram changes.17 In the group assigned to sertraline, life-threatening cardiovascular events occurred less frequently (15% vs 22%, P = NS).
The Enhancing Recovery in Coronary Heart Disease study (ENRICHD) was a large trial (N = 2,481) to evaluate the effect of cognitive behavioral therapy (CBT) on depression or low perceived social support in patients enrolled within 28 days of myocardial infarction (MI).4 The ENRICHD protocol required patients randomized to CBT with Hamilton Depression Rating Scale (HAM-D) scores greater than 24, or who showed less than 50% reduction in Beck Depression Inventory (BDI) scores after 5 weeks of CBT, to be referred to a study psychiatrist for consideration of pharmacotherapy, usually sertraline. Of the overall ENRICHD population, 1,834 participants (74%) had a diagnosis of depression, and 446 of these participants (24%) were treated with antidepressant drugs, 301 with an SSRI and 145 with other antidepressants.18 During mean follow-up of 29 months, the SSRI-treated group had a statistically significant 43% reduction in death or MI.4
The Canadian Cardiac Randomized Evaluation of Antidepressant and Psychotherapy Efficacy (CREATE) recruited 284 patients with chronic CHD, major depressive disorder, and a 24-item HAM-D score of 20 or greater and randomly assigned half to citalopram (another SSRI) and half to placebo.19 Citalopram showed antidepressant efficacy and no evidence of harm.
The Myocardial Infarction and Depression Intervention Trial (MIND-IT) recruited 91 patients within 30 days of hospital admission for MI with depression and randomized them in a 1:1 ratio to mirtazapine or placebo. Mirtazapine showed some evidence of antidepressant efficacy and no evidence of harm.20
Strik et al in the Netherlands recruited 54 patients who had major depression after a first MI and randomized them 1:1 to the SSRI fluoxetine or placebo 3 months after the MI.21 The fluoxetine group showed a trend toward antidepressant efficacy, and no cardiovascular safety problems were detected clinically or by either electrocardiogram or echocardiogram.
Psychotherapy
APA practice guidelines for major depressive disorder indicate that among psycho therapeutic approaches, CBT and interpersonal psychotherapy have the best-documented efficacy for treatment of major depressive disorder.22 CBT aims to solve problems related to dysfunctional emotions, behaviors, and cognition and is an umbrella term for various techniques that share a theoretical basis in behavioristic learning theory and cognitive psychology. Aaron T. Beck proposed that depressed people are quick to make negative evaluations of themselves and the world, and he designed treatment to reduce these negative cognitions.23 Interpersonal psychotherapy stems from the work of Harry Stack Sullivan, who believed that emotional reactions were triggered by interpersonal behaviors.24 Gerald Klerman and Myrna Weissman used this method to treat adults diagnosed with moderate or severe nondelusional clinical depression.25
CBT was used in ENRICHD,4 interpersonal psychotherapy in CREATE,19 and problem-solving therapy in the Coronary Psychosocial Evaluation Studies (COPES).26,27 Unintended therapy can also be a confounder in antidepressant drug trials; education and supportive care (eg, frequent visits or telephone calls with monitoring of depressive symptoms and counseling) are often provided for both the intervention and control (placebo) groups. If a study is blinded, education, supportive care, and attention will be identical for both groups and thus may reduce the likelihood of finding a difference between the drug and placebo, if one exists.
Psychotherapy can be helpful for depression, is preferred over antidepressant drugs by some patients, and can be combined with drugs to increase antidepressant efficacy. We still have much to learn about timing and choice of therapy, as well as about sequencing and combination of antidepressant drugs and psychotherapy.
Physical activity and exercise
Aerobic exercise28 and cardiac rehabilitation29 can reduce depressive symptoms in addition to improving cardiovascular fitness. Depression can serve as a barrier to participation in cardiac rehabilitation and exercise programs, but cardiologists can help depressed patients overcome this barrier by offering encouragement and follow-up contacts. Cardiologists also should enlist the help of spouses or other family members and friends to promote adherence. The prescription of exercise needs to be based on the cardiac status and exercise capacity of each individual.30
SUMMARY OF ADVISORY RECOMMENDATIONS
The AHA advisory1 summarized its recommendations as follows:
1. Routine screening for depression in patients with CHD should be considered in a variety of settings, including the hospital, the physician’s office, clinics, and cardiac rehabilitation centers. The opportunity to screen for and treat depression in cardiac patients should not be missed, as effective depression treatment may improve health outcomes.
2. Patients with positive screening results should be evaluated by a professional qualified in diagnosis and management of depression. Such a clinician can determine whether depression is present and needs treatment, as well as how to connect a patient to an effective care program in the local area.
3. Patients with heart disease who are being treated for depression should undergo careful monitoring for adherence to their medical care and for the efficacy and safety of drug therapy for their medical and mental health conditions.
4. Coordination of care among health care providers is essential for patients with coexisting medical and mental health issues.
COMMENTS ON THE ADVISORY
Since AHA advisories usually address evolving scientific issues, the knowledge base on these issues is constantly growing and a range of opinions and hypotheses are tenable, pending new information. Soon after the AHA science advisory on depression and CHD was issued, a systematic review on depression screening and patient outcomes in cardiac care was published.31 This review posed three key questions that are explored below.
Three key questions
Key question 1: What’s the accuracy of screening instruments for depression in cardiovascular care populations? To answer this question, a case-finding method such as a questionnaire (eg, BDI or PHQ) must be compared with a structured interview by mental health personnel as the “gold standard” for diagnosis (ie, the truth).
To estimate the accuracy of clinical diagnosis by general practitioners, Mitchell et al pooled data from 50,371 primary care patients across 41 studies in Europe or the United States to evaluate general practitioners’ ability to make an unassisted diagnosis of depression (ie, without specific help from severity scales, diagnostic instruments, education programs, etc).32 They reported a sensitivity of about 50% and a specificity of 81% when the prevalence of depression was about 20%. In other words, general practitioners missed about half the cases of depression when no case-finding tool was used. These researchers pointed out that a low prevalence of depression favors identification of nondepressed cases (false-positive diagnoses), whereas a high prevalence favors diagnosis of depression (true-positive diagnoses).32
Cardiologists focused on treatment of ACS are probably less likely to make a clinical diagnosis of depression and may attribute emotional symptoms to rapidly evolving cardiovascular events. The simple 2-question PHQ-2 case-finding instrument takes only a few minutes to administer and is recommended for use by primary care physicians or cardiologists to evaluate patients at high risk for depression or who manifest symptoms suggestive of depression.7
In the United States, most patients with depression are cared for in primary care venues. The AHA advisory recommends referring ACS patients who screen positive on a PHQ to a professional qualified in the diagnosis and management of depression.1 Enhanced care—using outreach, monitoring, adjustment of therapy, and psychiatric backup—produces significant improvement in depression.33
Key question 2: Is treatment of depression in cardiovascular care patients effective in improving depression? Cardiac outcomes? The evidence for benefit of SSRI therapy for depression detected at the time of ACS is consistent and was discussed above.4,17–21 However, the antidepressant effect of SSRIs is modest in placebo-controlled trials. Most such trials excluded patients who were taking antidepressant drugs when screened, and many patients recruited for antidepressant clinical trials had no previous episodes of depression, had relatively mild symptoms of short duration, and were recruited by screening (ie, they were not seeking treatment for depression).34 Patients with brief and short episodes are likely to remit spontaneously and to respond to psychotherapy or supportive care.34 Moreover, in most antidepressant trials, both the intervention and “control” groups received elements of enhanced depression care, which will reduce the apparent benefit of antidepressant drugs. For instance, because the primary goal of SADHART was to evaluate the safety of sertraline in patients with ACS, monitoring for adverse effects included six or seven visits during 16 weeks of follow-up plus six or seven phone calls, providing several elements of enhanced depression care, including face-to-face education, frequent follow-up, support by a case manager (research coordinator) with a mental health background, and support from a psychiatrist or psychologist.17 Randomization to sertraline or placebo in SADHART was blinded, so the frequent contact and support was the same in both groups.
Whether SSRI treatment for depression will improve survival and cardiovascular outcomes is not established by adequately powered randomized trials. Most randomized (SADHART, CREATE) or nonrandomized (ENRICHD) studies suggest that SSRIs reduce cardiovascular events, but only ENRICHD produced a statistically significant result. SSRIs—certainly sertraline and citalopram—are not associated with significant cardiovascular adverse effects, even during ACS, when the cardiovascular system is unstable and multiple drugs are being started and titrated. Patients who do not improve significantly during antidepressant drug therapy or psychotherapy have a two-to threefold increase in cardiovascular events compared with patients who do improve substantially.35
Key question 3: Is systematic screening for depression more effective than usual care for identifying patients with depression? Facilitating treatment of depression? Reducing depressive symptoms? Improving cardiac outcomes? Pignone et al conducted a literature review and meta-analysis on behalf of the USPSTF to clarify whether screening adults for depression in primary care venues improves recognition, treatment, and clinical outcomes.36 They reviewed randomized trials conducted in primary care settings that evaluated the effect of screening for depression on identification, treatment, or health outcomes, including trials that examined integrated, systematic support for treatment after identification of depression. The meta-analysis suggested that screening and feedback of screening results reduced the risk of persistent depression. Stronger effects were observed with programs that integrated interventions to improve recognition and treatment of depressed patients and that incorporated quality improvements into clinic systems as compared with programs that provided only screening and feedback.
Screening patients with CHD, especially those with ACS, is more effective than usual care (no screening). Indeed, many depressed patients go undetected unless identified by screening. Not surprisingly, those identified by screening tend to have milder depression. For example, half the participants in SADHART had a HAM-D score less than 18 (≥ 25 is considered severe). However, a considerable number identified by screening had above-average HAM-D scores but either viewed their symptoms as appropriate for their medical condition or were simply denying their psychiatric illness. If patients had not been screened, their depression would not have been detected or treated. Even among SADHART participants with baseline HAM-D scores less than 18, those whose depression failed to respond to sertraline/placebo had twice the 7-year mortality rate as did those whose depression remitted.16
The Prevention of Suicide in Primary Care Elderly: Collaborative Trial (PROSPECT) examined the impact of a care management intervention on suicidal ideation and depression in older primary care patients and reported outcomes over a 2-year follow-up.37 PROSPECT screened 9,072 patients in 20 primary care practices to find 599 study participants aged 60 years or older with major or minor depression (not all had cardiovascular disease). Participants were randomly assigned to either usual care or the PROSPECT intervention, which consisted of services rendered by trained care managers who offered algorithm-based recommendations to physicians and helped patients adhere to treatment during the 24-month trial. Compared with patients receiving usual care, those receiving the intervention had a greater likelihood of receiving antidepressant drugs and/or psychotherapy (85%–89% vs 49%–62%) and had a 2.2-fold greater decline in suicidal ideation over 24 months. Treatment response started sooner in the intervention group and continued to improve at 18 and 24 months; no appreciable increase in treatment response was observed in the usual-care group during the same period. Among patients with major depression, a significantly greater percentage of the intervention group achieved remission compared with the usual-care group at 4, 8, and 24 months. Patients with minor depression had favorable mental health outcomes regardless of treatment assignment. Sustained collaborative care maintained high utilization of depression treatment, reduced suicidal ideation, and improved the outcomes of major depression during 2 years of follow-up.37
No randomized trial of SSRIs has been designed to show an effect on mortality or cardiovascular events. ENRICHD has produced the strongest evidence that an SSRI can reduce mortality or cardiovascular outcomes. The study was designed with approximately 85% power to detect a 25% to 30% reduction in the primary end point (death or MI) as a result of CBT; as noted above, 2,481 patients (1,834 with depression) were randomized to usual care or CBT within 28 days of MI.4 Depression significantly improved with CBT, but the rate of death or MI did not: during mean follow-up of 29 months, death or MI occurred in 299 patients in the CBT group versus 300 in the usual-care group.4 A post hoc analysis specific to the 1,834 ENRICHD participants who had depression aimed to determine the effects of antidepressant drugs on morbidity and mortality.18 The protocol required patients in the intervention group with scores of 25 or higher on the 17-item HAM-D, or those who had less than a 50% reduction in BDI scores after 5 weeks of treatment, to be referred to study psychiatrists for consideration of pharmacotherapy. Study psychiatrists met with patients who were being treated with antidepressant drugs to monitor medication use. Unless contraindicated or previously ineffective or poorly tolerated, sertraline was the first antidepressant used. Using a time-dependent multivariable Cox proportional hazards regression model to adjust for baseline depression score and cardiac risk factors, the researchers found SSRI use to be associated with a statistically significant 43% lower risk of death or nonfatal MI. Like other SSRI studies, ENRICHD found SSRIs safe for post-MI patients and to possibly, but not certainly, reduce death and MI.18
Medication adherence
One obvious but important way antidepressant drug therapy could prevent death or MI is by improving adherence to post-MI cardiovascular drugs. Four or five classes of cardiac drugs have each been proven to improve survival following ACS (aspirin, beta-blockers, statins, and angiotensin-converting enzyme inhibitors/angiotensin receptor blockers), and when all are taken regularly, mortality is reduced by about half.38,39 In addition, antihypertensive and antidiabetic drugs are often needed to control blood pressure or blood glucose. However, patients have to actually take these drugs to receive their benefit. Depression in the setting of CHD, especially ACS, is a risk indicator for lack of adherence to medical therapy, mental health therapy, or both.
DiMatteo et al conducted a meta-analysis to test the hypothesis that anxiety and depression might explain poor adherence to treatment recommendations and result in poor medical outcomes.15 Of the 25 trials that met the inclusion criteria, 13 studied anxiety and 12 studied depression. The associations between anxiety and nonadherence were small and not statistically significant, but depression was strongly associated with nonadherence to medications (odds ratio = 3.03; 95% confidence interval, 1.96–4.89). In other words, depressed patients were three times as likely as nondepressed patients to be nonadherent to treatment recommendations. The authors speculated that depression might increase nonadherence in the following ways: (1) the hopelessness of depression might reduce patients’ hope in the therapy, (2) depression may cause withdrawal from family and social networks that otherwise would provide support and assistance, or (3) the impaired cognitive dysfunction associated with depression may impair memory and follow through on treatment recommendations.15
The findings from this meta-analysis could reflect depression causing medication nonadherence or vice versa. To establish the sequence, Rieckmann et al measured adherence to aspirin therapy during a 3-month period in a consecutive cohort of 172 patients (25 to 85 years old) recruited within 1 week of hospitalization for ACS.40 Severity of depressive symptoms was quantified using the BDI during hospitalization and at 1 and 3 months after discharge. Adherence was defined as taking aspirin as prescribed on at least 80% of days. Using an electronic monitoring system that recorded the date and time when the aspirin bottle cap was opened, the study found that more than 30% of patients with post-ACS depression were nonadherent to their aspirin therapy compared with only 15% of nondepressed patients.40 The more severe patients’ depressive symptoms were, the greater the nonadherence to aspirin therapy. Moreover, a lagged correlation statistical model determined that improvement in depression preceded improvement in medication adherence.
SADHART was conducted under the new drug application for sertraline,16 which required that the use of trial drugs be under strict compliance to protocol, sponsor monitoring, and auditing by the US Food and Drug Administration. Drug use data were complete in 98.1% of participants. Adherence was measured using tablet counts. Depressed patients who had a large improvement in depression during blinded drug therapy (sertraline or placebo) showed improved adherence to the blinded therapy. To determine whether depression improved before medication adherence improved, researchers compared responders’ medication adherence before and after their improvement in depression. Medication adherence increased following remission of depression in 128 of 187 participants (68.4%) who remitted on trial medication (remission was defined as a Clinical Global Impression–Improvement score of 1). This sequence of change (improved depression before improved medication adherence) occurred significantly more often than would be expected by chance (P < .001). This finding suggests that improvement in depression is driving improved medication adherence.
Because persistent depression is associated with increased mortality rates and reduced medication adherence, physicians need to not only aggressively treat depression but also diligently promote adherence to guideline-defined cardiovascular drug therapy. If depression doesn’t improve, additional measures should be initiated not only to improve depression but also to achieve adherence to cardiovascular drug therapy (eg, assistance from spouse, child, or visiting nurse; calls by case manager; electronic health record monitoring of drug prescription refills). When depression is found during clinical encounters or by screening, nonadherence to drug treatment is much more likely and should be sought vigilantly.
Adherence to lifestyle recommendations
Ziegelstein et al have shown that depressed patients have poorer adherence to lifestyle recommendations (diet, exercise, smoking cessation).41 The Heart and Soul Study, a prospective cohort study of 1,017 outpatients with stable CHD, attempted to determine why depressive symptoms (as determined by PHQ-9 self-report) are associated with an increased risk of cardiovascular events.42 Participants were predominantly older men, about half of whom were recruited from Veterans Administration hospitals. A total of 341 cardiovascular events occurred during a mean follow-up of 4.8 years. Participants with baseline depressive symptoms had a 50% greater rate of cardiovascular events during the study period compared with participants without depressive symptoms. However, no significant association between depressive symptoms and cardiovascular events remained after adjustment for health behaviors—most strikingly, physical activity.42 This finding was consistent with an earlier study that found that exercise therapy plus antidepressant medication could reduce the risk of cardiovascular events in patients with depression.43 The ongoing Understanding Prognostic Benefits of Exercise and Antidepressant Therapy (UPBEAT) study is comparing the effects of exercise and antidepressant medication on depression and biomarkers of cardiovascular risk in patients with depressive symptoms and CHD.44 The study’s longer-term goal is to identify an intervention that improves both depression and cardiovascular disease outcomes.
CONCLUSIONS
The USPSTF recommends screening for depression in adults. The PHQ-2 is an efficient first-step screening tool that can identify many depressed patients who would otherwise go undetected. It is clear that SSRIs are safe in cardiac patients, can reduce depression, and can improve medication adherence, but it is not enough to screen and report depression. Optimal benefit depends on having (1) a primary care provider who is familiar with managing depression, (2) a case manager with a mental health background to follow and support patients, and (3) regular supervision of the case manager by a psychiatrist or psychologist. Cardiologists see large numbers of patients with chronic CHD, ACS, or recent coronary artery bypass graft surgery who are at high risk for depression. The AHA advisory recommends a care model that is practical for CHD patients with depression. Such a care model must be based on detection of depression and referral to a practice that has resources and knowledge to manage it well.
References
Lichtman JH, Bigger JT, Blumenthal JA, et al Depression and coronary heart disease: recommendations for screening, referral, and treatment. A science advisory from the American Heart Association Prevention Committee of the Council on Cardiovascular Nursing, Council on Clinical Cardiology, Council on Epidemiology and Prevention, and Interdisciplinary Council on Quality of Care and Outcomes Research; endorsed by the American Psychiatric Association. Circulation2008; 118:1768–1775.
Kessler RC, Merikangas KR, Wang PS. Prevalence, comorbidity, and service utilization for mood disorders in the United States at the beginning of the twenty-first century. Annu Rev Clin Psychol2007; 3:137–158.
Wulsin LR, Singal BM. Do depressive symptoms increase the risk for the onset of coronary disease? A systematic quantitative review. Psychosom Med2003; 65:201–210.
Berkman LF, Blumenthal J, Burg M, et al Effects of treating depression and low perceived social support on clinical events after myocardial infarction: the Enhancing Recovery in Coronary Heart Disease Patients (ENRICHD) randomized trial. JAMA2003; 289:3106–3116.
Thombs BD, Bass EB, Ford DE, et al Prevalence of depression in survivors of acute myocardial infarction. J Gen Intern Med2006; 21:30–38.
van Melle JP, de Jonge P, Spijkerman TA, et al Prognostic association of depression following myocardial infarction with mortality and cardiovascular events: a meta-analysis. Psychosom Med2004; 66:814–822.
Kroenke K, Spitzer RL, Williams JB. The Patient Health Questionnaire-2: validity of a two-item depression screener. Med Care2003; 41:1284–1292.
Whooley M, Avins AL, Miranda J, Brown WS. Case-finding instruments for depression: two questions are as good as many. J Gen Intern Med1997; 12:289–290.
Gilbody S, Richards D, Brealey S, Hewitt C. Screening for depression in medical settings with the Patient Health Questionnaire (PHQ): a diagnostic meta-analysis. J Gen Intern Med2007; 22:1596–1602.
Stafford L, Berk M, Jackson HJ. Validity of the Hospital Anxiety and Depression Scale and Patient Health Questionnaire-9 to screen for depression in patients with coronary artery disease. Gen Hosp Psychiatry2007; 29:417–424.
McManus D, Pipkin SS, Whooley MA. Screening for depression in patients with coronary heart disease (data from the Heart and Soul Study). Am J Cardiol2005; 96:1076–1081.
U.S. Preventive Services Task Force. Screening for depression: recommendations and rationale. Rockville, MD: Agency for Healthcare Research and Quality; May2002. http://www.ahrq.gov/clinic/3rduspstf/depression/depressrr.htm. Accessed January 8, 2010.
Depression Guideline Panel. Clinical Practice Guideline Number 5: Depression in Primary Care, 1: Detection and Diagnosis. Rockville, MD: US Department of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research; April1993. AHCPR publication 93-0550.
Depression Guideline Panel. Clinical Practice Guideline Number 5: Depression in Primary Care, 2: Treatment of Major Depression. Rockville, MD: US Department of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research; April1993. AHCPR publication 93-0551.
DiMatteo MR, Lepper HS, Croghan TW. Depression is a risk factor for noncompliance with medical treatment: meta-analysis of the effects of anxiety and depression on patient adherence. Arch Intern Med2000; 160:2101–2107.
Glassman AH, Bigger JT, Gaffney M. Psychiatric characteristics associated with long-term mortality among 361 patients having an acute coronary syndrome and major depression: seven-year follow-up of SADHART participants. Arch Gen Psychiatry2009; 66:1022–1029.
Glassman AH, O’Connor CM, Califf RM, et al Sertraline treatment of major depression in patients with acute MI or unstable angina. JAMA2002; 288:701–709.
Taylor CB, Youngblood ME, Catellier D, et al Effects of antidepressant medication on morbidity and mortality in depressed patients after myocardial infarction. Arch Gen Psychiatry2005; 62:792–798.
Lesperance F, Frasure-Smith N, Koszycki D, et al Effects of citalopram and interpersonal psychotherapy on depression in patients with coronary artery disease: the Canadian Cardiac Randomized Evaluation of Antidepressant and Psychotherapy Efficacy (CREATE) trial. JAMA2007; 297:367–379.
Honig A, Kuyper A, Schene AH, et al Treatment of post-myocardial infarction depressive disorder: a randomized, placebo-controlled trial with mirtazapine. Psychosom Med2007; 69:606–613.
Strik J, Honig A, Lousberg R, et al Efficacy and safety of fluoxetine in the treatment of patients with major depression after first myocardial infarction: findings from a double-blind, placebo-controlled trial. Psychosom Med2000; 62:783–789.
American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder (revision). Am J Psychiatry2000; 157( suppl 4):1–45.
Beck AT. Cognitive Therapy and the Emotional Disorders. Madison, CT: International Universities Press Inc; 1975.
Weissman MM. A brief history of interpersonal psychotherapy. Psychiatr Ann2006; 36:553–557.
Klerman GL, Weissmann MM. Interpersonal psychotherapy (IPT) and drugs in the treatment of depression. Pharmacopsychiatry1987; 20:3–7.
Burg MM, Lespérance F, Rieckmann N, Clemow L, Skotzko C, Davidson KW. Treating persistent depressive symptoms in post-ACS patients: the project COPES phase-I randomized controlled trial. Contemp Clin Trials2008; 29:231–240.
Davidson KW, Rieckmann N, Clemow L, et al Collaborative depression care for acute coronary syndrome patients with persistent depression: Coronary Psychosocial Evaluation Studies (COPES) randomized controlled trial. Arch Intern Med. In press.
Brosse AL, Sheets ES, Lett HS, Blumenthal JA. Exercise and the treatment of clinical depression in adults: recent findings and future directions. Sports Med2002; 32:741–760.
Milani RV, Lavie CJ. Impact of cardiac rehabilitation on depression and its associated mortality. Am J Med2007; 120:799–806.
Smith SC, Allen J, Blair SN, et al AHA/ACC guidelines for secondary prevention for patients with coronary and other atherosclerotic vascular disease: 2006 update; endorsed by the National Heart, Lung, and Blood Institute. Circulation2006; 113:2363–2372.
Thombs BD, de Jonge P, Coyne JC, et al Ziegelstein RC. Depression screening and patient outcomes in cardiovascular care: a systematic review. JAMA2008; 300:2161–2171.
Mitchell AJ, Vaze A, Rao S. Clinical diagnosis of depression in primary care: a meta-analysis. Lancet2009; 374:609–619.
Gilbody S, Bower P, Fletcher J, Richards D, Sutton AJ. Collaborative care for depression: a cumulative meta-analysis and review of longer-term outcomes. Arch Intern Med2006; 166:2314–2321.
Glassman AH, Bigger JT, Gaffney M, Shapiro PA, Swenson JR. Onset of major depression associated with acute coronary syndromes: relationship of onset, major depressive disorder history, and episode severity to sertraline benefit. Arch Gen Psychiatry2006; 63:283–288.
Carney RM, Freedland KE. Treatment-resistant depression and mortality after acute coronary syndrome. Am J Psychiatry2009; 166:410–417.
Pignone MP, Gaynes BN, Rushton JL, et al Screening for depression in adults: a summary of the evidence for the U.S. Preventive Services Task Force. Ann Intern Med2002; 136:765–776.
Alexopoulos GS, Reynolds CF, Bruce ML, et al Reducing suicidal ideation and depression in older primary care patients: 24-month outcomes of the PROSPECT study. Am J Psychiatry2009; 166:882–890.
Mukherjee D, Fang J, Chetcuti S, Moscucci M, Kline-Rogers E, Eagle KA. Impact of combination evidence-based medical therapy on mortality in patients with acute coronary syndromes. Circulation2004; 109:745–749.
Eagle KA, Kline-Rogers E, Goodman SG, et al Adherence to evidence-based therapies after discharge for acute coronary syndromes: an ongoing prospective, observational study. Am J Med2004; 117:73–81.
Rieckmann N, Gerin W, Kronish IA, et al Course of depressive symptoms and medication adherence after acute coronary syndromes: an electronic medication monitoring study. J Am Coll Cardiol2006; 48:2218–2222.
Ziegelstein RC, Fauerbach JA, Stevens SS, Romanelli J, Richter DP, Bush DE. Patients with depression are less likely to follow recommendations to reduce cardiac risk during recovery from a myocardial infarction. Arch Intern Med2000; 160:1818–1823.
Whooley MA, de Jonge P, Vittinghoff E, et al Depressive symptoms, health behaviors, and risk of cardiovascular events in patients with coronary heart disease. JAMA2008; 300:2379–2388.
Blumenthal JA, Babyak MA, Doraiswamy PM, et al Exercise and pharmacotherapy in the treatment of major depressive disorder. Psychosom Med2007; 69:587–596.
Blumenthal JA, Sherwood A, Rogers SD, et al Understanding prognostic benefits of exercise and antidepressant therapy for persons with depression and heart disease: the UPBEAT study—rationale, design, and methodological issues. Clin Trials2007; 4:548–559.
J. Thomas Bigger, MD Department of Medicine, Columbia University College of Physicians and Surgeons, New York, NY
Alexander H. Glassman, MD Department of Psychiatry, Columbia University College of Physicians and Surgeons, New York, NY
Correspondence: J. Thomas Bigger, MD, Department of Medicine, Columbia University Medical Center, 630 West 168th Street, PH-9-103, New York, NY 10032; [email protected]
Dr. Bigger reported that he has no financial relationships that could be perceived as a potential conflict of interest with this article. Dr. Glassman reported that he has received consulting fees and speaking honoraria from Pfizer Inc.
This work was supported by grant 5-UL1-RR024156 from the National Center for Research Resources, National Institutes of Health, and by the Suzanne C. Murphy Foundation.
J. Thomas Bigger, MD Department of Medicine, Columbia University College of Physicians and Surgeons, New York, NY
Alexander H. Glassman, MD Department of Psychiatry, Columbia University College of Physicians and Surgeons, New York, NY
Correspondence: J. Thomas Bigger, MD, Department of Medicine, Columbia University Medical Center, 630 West 168th Street, PH-9-103, New York, NY 10032; [email protected]
Dr. Bigger reported that he has no financial relationships that could be perceived as a potential conflict of interest with this article. Dr. Glassman reported that he has received consulting fees and speaking honoraria from Pfizer Inc.
This work was supported by grant 5-UL1-RR024156 from the National Center for Research Resources, National Institutes of Health, and by the Suzanne C. Murphy Foundation.
Author and Disclosure Information
J. Thomas Bigger, MD Department of Medicine, Columbia University College of Physicians and Surgeons, New York, NY
Alexander H. Glassman, MD Department of Psychiatry, Columbia University College of Physicians and Surgeons, New York, NY
Correspondence: J. Thomas Bigger, MD, Department of Medicine, Columbia University Medical Center, 630 West 168th Street, PH-9-103, New York, NY 10032; [email protected]
Dr. Bigger reported that he has no financial relationships that could be perceived as a potential conflict of interest with this article. Dr. Glassman reported that he has received consulting fees and speaking honoraria from Pfizer Inc.
This work was supported by grant 5-UL1-RR024156 from the National Center for Research Resources, National Institutes of Health, and by the Suzanne C. Murphy Foundation.
In 2008, the American Heart Association (AHA) published a science advisory on depression and coronary heart disease (CHD).1 Since its publication, the advisory has evoked substantial commentary. The purpose of this article is threefold: (1) to explain the aims of the AHA science advisory, (2) to briefly discuss its content, and (3) to examine some of the comments it has provoked.
WHAT THE ADVISORY SET OUT TO DO
The purpose of an AHA science advisory is to provide rapid, clear, and consistent AHA positioning on a scientific issue. Advisories are statements on an evolving, prominent scientific issue of great interest to the public and health professionals. All AHA science advisories undergo peer review and are also reviewed and approved by the AHA Science Advisory and Coordinating Committee, AHA’s highest science body. Because this particular advisory addressed the interaction of cardiovascular and mental health, the AHA asked the American Psychiatric Association (APA) to review the document; the APA endorsed the AHA advisory.
Two points are worth emphasizing:
An AHA science advisory is not a treatment guideline.
Advisories usually are brief and therefore do not exhaustively discuss their topic.
After discussing epidemiologic studies that elucidated the relationships between depression and CHD, the AHA advisory on depression and CHD focuses on screening, referral, and treatment of depression from a cardiology perspective.
The 1-year prevalence of major depressive disorder in the US general population is 7%, and the lifetime prevalence is about 16%.2 Depression in otherwise healthy persons almost doubles the risk of developing CHD.3 About 20% of patients hospitalized for acute coronary syndromes (ACS) have major depressive disorder on admission or within a few weeks thereafter, and these patients have about 2.5 times the mortality rate as patients who are not depressed, after adjusting for infarct severity and cardiovascular risk factors.4–6
ASSESSMENT OF DEPRESSION AND DEPRESSIVE SYMPTOMS
The AHA advisory discusses use of the 2-question Patient Health Questionnaire (PHQ-2) as the first step in screening for depression.7,8 The PHQ-2 inquires about the frequency of depressed mood and anhedonia by asking the following:
Over the past 2 weeks, how often have you been bothered by either of the following problems?
(1) Little interest or pleasure in doing things
(2) Feeling down, depressed, or hopeless.
For each item the response options are “not at all” (scored as 0), “several days” (scored as 1), “more than half the days” (scored as 2), and “nearly every day” (scored as 3). Thus, the total PHQ-2 score can range from 0 to 6.
Using a structured psychiatric interview as the standard, a total PHQ-2 score of 3 or greater has been shown to have a sensitivity of 83% and a specificity of 92% for major depression.7 A PHQ-2 score of 3 is the optimal cut point for screening purposes. A PHQ-2 score of 0 virtually excludes depression.
If a patient’s PHQ-2 score is 3 or greater, it is recommended that answers be obtained for a full 9-item PHQ. The PHQ-9 provides the sensitivity and specificity suitable for assigning a provisional diagnosis of major depressive disorder and a symptom severity score that can be used to identify patients for further evaluation and to make decisions about therapy.9–11
Adapted from the 2008 American Heart Association science advisory on depression and coronary heart disease.1 Source: American Heart Association, Inc.
Figure 1. Screening for depression in patients with coronary heart disease.The AHA advisory’s section on assessment of depression and depressive symptoms discusses briefly the principles enunciated by the MacArthur Initiative on Depression and Primary Care.12–14 The advisory carefully provides practical guidance specifically for cardiologists (Figure 1).1 The section on assessment points out that screening for depression coupled with therapy has not been proven to improve cardiovascular outcomes but that some antidepressant drugs and/or psychotherapy have proved safe and can improve quality of life, reduce depressive symptoms, improve compliance with lifestyle advice, and improve adherence to prescribed cardiovascular medications.1,15,16
The US Preventive Services Task Force (USPSTF) currently recommends that clinicians screen adults for depression in clinical practices that have systems in place to assure accurate diagnosis, effective treatment, and follow-up.12 The USPSTF concluded that there is good evidence that screening improves the accurate identification of depressed patients in primary care settings and that treating depressed adults identified in such settings reduces clinical morbidity. Results of trials that have directly evaluated the effect of screening on clinical outcomes depend on follow-up. Limited benefits have been demonstrated in studies that simply feed screening results back to clinicians. Larger benefits have been seen in studies in which the communication of screening results is coordinated with effective follow-up and treatment. The USPSTF concluded that the benefits of screening and treating are likely to outweigh any potential harms.12
DEPRESSION TREATMENT
Drug therapy
The safety of fluoxetine, sertraline (SADHART, ENRICHD), citalopram (CREATE), and mirtazapine (MIND-IT) has been evaluated in clinical trials.4,17–21 By far the strongest evidence of safety is for selective serotonin reuptake inhibitors (SSRIs), especially sertraline.
The Sertraline Antidepressant Heart Attack Randomized Trial (SADHART; N = 369), a randomized study of depression after ACS, found no difference in cardiovascular adverse events between the sertraline and placebo groups after 16 weeks of therapy, and there were no significant differences in left ventricular ejection fraction, heart rate, blood pressure, ventricular premature complexes, or electrocardiogram changes.17 In the group assigned to sertraline, life-threatening cardiovascular events occurred less frequently (15% vs 22%, P = NS).
The Enhancing Recovery in Coronary Heart Disease study (ENRICHD) was a large trial (N = 2,481) to evaluate the effect of cognitive behavioral therapy (CBT) on depression or low perceived social support in patients enrolled within 28 days of myocardial infarction (MI).4 The ENRICHD protocol required patients randomized to CBT with Hamilton Depression Rating Scale (HAM-D) scores greater than 24, or who showed less than 50% reduction in Beck Depression Inventory (BDI) scores after 5 weeks of CBT, to be referred to a study psychiatrist for consideration of pharmacotherapy, usually sertraline. Of the overall ENRICHD population, 1,834 participants (74%) had a diagnosis of depression, and 446 of these participants (24%) were treated with antidepressant drugs, 301 with an SSRI and 145 with other antidepressants.18 During mean follow-up of 29 months, the SSRI-treated group had a statistically significant 43% reduction in death or MI.4
The Canadian Cardiac Randomized Evaluation of Antidepressant and Psychotherapy Efficacy (CREATE) recruited 284 patients with chronic CHD, major depressive disorder, and a 24-item HAM-D score of 20 or greater and randomly assigned half to citalopram (another SSRI) and half to placebo.19 Citalopram showed antidepressant efficacy and no evidence of harm.
The Myocardial Infarction and Depression Intervention Trial (MIND-IT) recruited 91 patients within 30 days of hospital admission for MI with depression and randomized them in a 1:1 ratio to mirtazapine or placebo. Mirtazapine showed some evidence of antidepressant efficacy and no evidence of harm.20
Strik et al in the Netherlands recruited 54 patients who had major depression after a first MI and randomized them 1:1 to the SSRI fluoxetine or placebo 3 months after the MI.21 The fluoxetine group showed a trend toward antidepressant efficacy, and no cardiovascular safety problems were detected clinically or by either electrocardiogram or echocardiogram.
Psychotherapy
APA practice guidelines for major depressive disorder indicate that among psycho therapeutic approaches, CBT and interpersonal psychotherapy have the best-documented efficacy for treatment of major depressive disorder.22 CBT aims to solve problems related to dysfunctional emotions, behaviors, and cognition and is an umbrella term for various techniques that share a theoretical basis in behavioristic learning theory and cognitive psychology. Aaron T. Beck proposed that depressed people are quick to make negative evaluations of themselves and the world, and he designed treatment to reduce these negative cognitions.23 Interpersonal psychotherapy stems from the work of Harry Stack Sullivan, who believed that emotional reactions were triggered by interpersonal behaviors.24 Gerald Klerman and Myrna Weissman used this method to treat adults diagnosed with moderate or severe nondelusional clinical depression.25
CBT was used in ENRICHD,4 interpersonal psychotherapy in CREATE,19 and problem-solving therapy in the Coronary Psychosocial Evaluation Studies (COPES).26,27 Unintended therapy can also be a confounder in antidepressant drug trials; education and supportive care (eg, frequent visits or telephone calls with monitoring of depressive symptoms and counseling) are often provided for both the intervention and control (placebo) groups. If a study is blinded, education, supportive care, and attention will be identical for both groups and thus may reduce the likelihood of finding a difference between the drug and placebo, if one exists.
Psychotherapy can be helpful for depression, is preferred over antidepressant drugs by some patients, and can be combined with drugs to increase antidepressant efficacy. We still have much to learn about timing and choice of therapy, as well as about sequencing and combination of antidepressant drugs and psychotherapy.
Physical activity and exercise
Aerobic exercise28 and cardiac rehabilitation29 can reduce depressive symptoms in addition to improving cardiovascular fitness. Depression can serve as a barrier to participation in cardiac rehabilitation and exercise programs, but cardiologists can help depressed patients overcome this barrier by offering encouragement and follow-up contacts. Cardiologists also should enlist the help of spouses or other family members and friends to promote adherence. The prescription of exercise needs to be based on the cardiac status and exercise capacity of each individual.30
SUMMARY OF ADVISORY RECOMMENDATIONS
The AHA advisory1 summarized its recommendations as follows:
1. Routine screening for depression in patients with CHD should be considered in a variety of settings, including the hospital, the physician’s office, clinics, and cardiac rehabilitation centers. The opportunity to screen for and treat depression in cardiac patients should not be missed, as effective depression treatment may improve health outcomes.
2. Patients with positive screening results should be evaluated by a professional qualified in diagnosis and management of depression. Such a clinician can determine whether depression is present and needs treatment, as well as how to connect a patient to an effective care program in the local area.
3. Patients with heart disease who are being treated for depression should undergo careful monitoring for adherence to their medical care and for the efficacy and safety of drug therapy for their medical and mental health conditions.
4. Coordination of care among health care providers is essential for patients with coexisting medical and mental health issues.
COMMENTS ON THE ADVISORY
Since AHA advisories usually address evolving scientific issues, the knowledge base on these issues is constantly growing and a range of opinions and hypotheses are tenable, pending new information. Soon after the AHA science advisory on depression and CHD was issued, a systematic review on depression screening and patient outcomes in cardiac care was published.31 This review posed three key questions that are explored below.
Three key questions
Key question 1: What’s the accuracy of screening instruments for depression in cardiovascular care populations? To answer this question, a case-finding method such as a questionnaire (eg, BDI or PHQ) must be compared with a structured interview by mental health personnel as the “gold standard” for diagnosis (ie, the truth).
To estimate the accuracy of clinical diagnosis by general practitioners, Mitchell et al pooled data from 50,371 primary care patients across 41 studies in Europe or the United States to evaluate general practitioners’ ability to make an unassisted diagnosis of depression (ie, without specific help from severity scales, diagnostic instruments, education programs, etc).32 They reported a sensitivity of about 50% and a specificity of 81% when the prevalence of depression was about 20%. In other words, general practitioners missed about half the cases of depression when no case-finding tool was used. These researchers pointed out that a low prevalence of depression favors identification of nondepressed cases (false-positive diagnoses), whereas a high prevalence favors diagnosis of depression (true-positive diagnoses).32
Cardiologists focused on treatment of ACS are probably less likely to make a clinical diagnosis of depression and may attribute emotional symptoms to rapidly evolving cardiovascular events. The simple 2-question PHQ-2 case-finding instrument takes only a few minutes to administer and is recommended for use by primary care physicians or cardiologists to evaluate patients at high risk for depression or who manifest symptoms suggestive of depression.7
In the United States, most patients with depression are cared for in primary care venues. The AHA advisory recommends referring ACS patients who screen positive on a PHQ to a professional qualified in the diagnosis and management of depression.1 Enhanced care—using outreach, monitoring, adjustment of therapy, and psychiatric backup—produces significant improvement in depression.33
Key question 2: Is treatment of depression in cardiovascular care patients effective in improving depression? Cardiac outcomes? The evidence for benefit of SSRI therapy for depression detected at the time of ACS is consistent and was discussed above.4,17–21 However, the antidepressant effect of SSRIs is modest in placebo-controlled trials. Most such trials excluded patients who were taking antidepressant drugs when screened, and many patients recruited for antidepressant clinical trials had no previous episodes of depression, had relatively mild symptoms of short duration, and were recruited by screening (ie, they were not seeking treatment for depression).34 Patients with brief and short episodes are likely to remit spontaneously and to respond to psychotherapy or supportive care.34 Moreover, in most antidepressant trials, both the intervention and “control” groups received elements of enhanced depression care, which will reduce the apparent benefit of antidepressant drugs. For instance, because the primary goal of SADHART was to evaluate the safety of sertraline in patients with ACS, monitoring for adverse effects included six or seven visits during 16 weeks of follow-up plus six or seven phone calls, providing several elements of enhanced depression care, including face-to-face education, frequent follow-up, support by a case manager (research coordinator) with a mental health background, and support from a psychiatrist or psychologist.17 Randomization to sertraline or placebo in SADHART was blinded, so the frequent contact and support was the same in both groups.
Whether SSRI treatment for depression will improve survival and cardiovascular outcomes is not established by adequately powered randomized trials. Most randomized (SADHART, CREATE) or nonrandomized (ENRICHD) studies suggest that SSRIs reduce cardiovascular events, but only ENRICHD produced a statistically significant result. SSRIs—certainly sertraline and citalopram—are not associated with significant cardiovascular adverse effects, even during ACS, when the cardiovascular system is unstable and multiple drugs are being started and titrated. Patients who do not improve significantly during antidepressant drug therapy or psychotherapy have a two-to threefold increase in cardiovascular events compared with patients who do improve substantially.35
Key question 3: Is systematic screening for depression more effective than usual care for identifying patients with depression? Facilitating treatment of depression? Reducing depressive symptoms? Improving cardiac outcomes? Pignone et al conducted a literature review and meta-analysis on behalf of the USPSTF to clarify whether screening adults for depression in primary care venues improves recognition, treatment, and clinical outcomes.36 They reviewed randomized trials conducted in primary care settings that evaluated the effect of screening for depression on identification, treatment, or health outcomes, including trials that examined integrated, systematic support for treatment after identification of depression. The meta-analysis suggested that screening and feedback of screening results reduced the risk of persistent depression. Stronger effects were observed with programs that integrated interventions to improve recognition and treatment of depressed patients and that incorporated quality improvements into clinic systems as compared with programs that provided only screening and feedback.
Screening patients with CHD, especially those with ACS, is more effective than usual care (no screening). Indeed, many depressed patients go undetected unless identified by screening. Not surprisingly, those identified by screening tend to have milder depression. For example, half the participants in SADHART had a HAM-D score less than 18 (≥ 25 is considered severe). However, a considerable number identified by screening had above-average HAM-D scores but either viewed their symptoms as appropriate for their medical condition or were simply denying their psychiatric illness. If patients had not been screened, their depression would not have been detected or treated. Even among SADHART participants with baseline HAM-D scores less than 18, those whose depression failed to respond to sertraline/placebo had twice the 7-year mortality rate as did those whose depression remitted.16
The Prevention of Suicide in Primary Care Elderly: Collaborative Trial (PROSPECT) examined the impact of a care management intervention on suicidal ideation and depression in older primary care patients and reported outcomes over a 2-year follow-up.37 PROSPECT screened 9,072 patients in 20 primary care practices to find 599 study participants aged 60 years or older with major or minor depression (not all had cardiovascular disease). Participants were randomly assigned to either usual care or the PROSPECT intervention, which consisted of services rendered by trained care managers who offered algorithm-based recommendations to physicians and helped patients adhere to treatment during the 24-month trial. Compared with patients receiving usual care, those receiving the intervention had a greater likelihood of receiving antidepressant drugs and/or psychotherapy (85%–89% vs 49%–62%) and had a 2.2-fold greater decline in suicidal ideation over 24 months. Treatment response started sooner in the intervention group and continued to improve at 18 and 24 months; no appreciable increase in treatment response was observed in the usual-care group during the same period. Among patients with major depression, a significantly greater percentage of the intervention group achieved remission compared with the usual-care group at 4, 8, and 24 months. Patients with minor depression had favorable mental health outcomes regardless of treatment assignment. Sustained collaborative care maintained high utilization of depression treatment, reduced suicidal ideation, and improved the outcomes of major depression during 2 years of follow-up.37
No randomized trial of SSRIs has been designed to show an effect on mortality or cardiovascular events. ENRICHD has produced the strongest evidence that an SSRI can reduce mortality or cardiovascular outcomes. The study was designed with approximately 85% power to detect a 25% to 30% reduction in the primary end point (death or MI) as a result of CBT; as noted above, 2,481 patients (1,834 with depression) were randomized to usual care or CBT within 28 days of MI.4 Depression significantly improved with CBT, but the rate of death or MI did not: during mean follow-up of 29 months, death or MI occurred in 299 patients in the CBT group versus 300 in the usual-care group.4 A post hoc analysis specific to the 1,834 ENRICHD participants who had depression aimed to determine the effects of antidepressant drugs on morbidity and mortality.18 The protocol required patients in the intervention group with scores of 25 or higher on the 17-item HAM-D, or those who had less than a 50% reduction in BDI scores after 5 weeks of treatment, to be referred to study psychiatrists for consideration of pharmacotherapy. Study psychiatrists met with patients who were being treated with antidepressant drugs to monitor medication use. Unless contraindicated or previously ineffective or poorly tolerated, sertraline was the first antidepressant used. Using a time-dependent multivariable Cox proportional hazards regression model to adjust for baseline depression score and cardiac risk factors, the researchers found SSRI use to be associated with a statistically significant 43% lower risk of death or nonfatal MI. Like other SSRI studies, ENRICHD found SSRIs safe for post-MI patients and to possibly, but not certainly, reduce death and MI.18
Medication adherence
One obvious but important way antidepressant drug therapy could prevent death or MI is by improving adherence to post-MI cardiovascular drugs. Four or five classes of cardiac drugs have each been proven to improve survival following ACS (aspirin, beta-blockers, statins, and angiotensin-converting enzyme inhibitors/angiotensin receptor blockers), and when all are taken regularly, mortality is reduced by about half.38,39 In addition, antihypertensive and antidiabetic drugs are often needed to control blood pressure or blood glucose. However, patients have to actually take these drugs to receive their benefit. Depression in the setting of CHD, especially ACS, is a risk indicator for lack of adherence to medical therapy, mental health therapy, or both.
DiMatteo et al conducted a meta-analysis to test the hypothesis that anxiety and depression might explain poor adherence to treatment recommendations and result in poor medical outcomes.15 Of the 25 trials that met the inclusion criteria, 13 studied anxiety and 12 studied depression. The associations between anxiety and nonadherence were small and not statistically significant, but depression was strongly associated with nonadherence to medications (odds ratio = 3.03; 95% confidence interval, 1.96–4.89). In other words, depressed patients were three times as likely as nondepressed patients to be nonadherent to treatment recommendations. The authors speculated that depression might increase nonadherence in the following ways: (1) the hopelessness of depression might reduce patients’ hope in the therapy, (2) depression may cause withdrawal from family and social networks that otherwise would provide support and assistance, or (3) the impaired cognitive dysfunction associated with depression may impair memory and follow through on treatment recommendations.15
The findings from this meta-analysis could reflect depression causing medication nonadherence or vice versa. To establish the sequence, Rieckmann et al measured adherence to aspirin therapy during a 3-month period in a consecutive cohort of 172 patients (25 to 85 years old) recruited within 1 week of hospitalization for ACS.40 Severity of depressive symptoms was quantified using the BDI during hospitalization and at 1 and 3 months after discharge. Adherence was defined as taking aspirin as prescribed on at least 80% of days. Using an electronic monitoring system that recorded the date and time when the aspirin bottle cap was opened, the study found that more than 30% of patients with post-ACS depression were nonadherent to their aspirin therapy compared with only 15% of nondepressed patients.40 The more severe patients’ depressive symptoms were, the greater the nonadherence to aspirin therapy. Moreover, a lagged correlation statistical model determined that improvement in depression preceded improvement in medication adherence.
SADHART was conducted under the new drug application for sertraline,16 which required that the use of trial drugs be under strict compliance to protocol, sponsor monitoring, and auditing by the US Food and Drug Administration. Drug use data were complete in 98.1% of participants. Adherence was measured using tablet counts. Depressed patients who had a large improvement in depression during blinded drug therapy (sertraline or placebo) showed improved adherence to the blinded therapy. To determine whether depression improved before medication adherence improved, researchers compared responders’ medication adherence before and after their improvement in depression. Medication adherence increased following remission of depression in 128 of 187 participants (68.4%) who remitted on trial medication (remission was defined as a Clinical Global Impression–Improvement score of 1). This sequence of change (improved depression before improved medication adherence) occurred significantly more often than would be expected by chance (P < .001). This finding suggests that improvement in depression is driving improved medication adherence.
Because persistent depression is associated with increased mortality rates and reduced medication adherence, physicians need to not only aggressively treat depression but also diligently promote adherence to guideline-defined cardiovascular drug therapy. If depression doesn’t improve, additional measures should be initiated not only to improve depression but also to achieve adherence to cardiovascular drug therapy (eg, assistance from spouse, child, or visiting nurse; calls by case manager; electronic health record monitoring of drug prescription refills). When depression is found during clinical encounters or by screening, nonadherence to drug treatment is much more likely and should be sought vigilantly.
Adherence to lifestyle recommendations
Ziegelstein et al have shown that depressed patients have poorer adherence to lifestyle recommendations (diet, exercise, smoking cessation).41 The Heart and Soul Study, a prospective cohort study of 1,017 outpatients with stable CHD, attempted to determine why depressive symptoms (as determined by PHQ-9 self-report) are associated with an increased risk of cardiovascular events.42 Participants were predominantly older men, about half of whom were recruited from Veterans Administration hospitals. A total of 341 cardiovascular events occurred during a mean follow-up of 4.8 years. Participants with baseline depressive symptoms had a 50% greater rate of cardiovascular events during the study period compared with participants without depressive symptoms. However, no significant association between depressive symptoms and cardiovascular events remained after adjustment for health behaviors—most strikingly, physical activity.42 This finding was consistent with an earlier study that found that exercise therapy plus antidepressant medication could reduce the risk of cardiovascular events in patients with depression.43 The ongoing Understanding Prognostic Benefits of Exercise and Antidepressant Therapy (UPBEAT) study is comparing the effects of exercise and antidepressant medication on depression and biomarkers of cardiovascular risk in patients with depressive symptoms and CHD.44 The study’s longer-term goal is to identify an intervention that improves both depression and cardiovascular disease outcomes.
CONCLUSIONS
The USPSTF recommends screening for depression in adults. The PHQ-2 is an efficient first-step screening tool that can identify many depressed patients who would otherwise go undetected. It is clear that SSRIs are safe in cardiac patients, can reduce depression, and can improve medication adherence, but it is not enough to screen and report depression. Optimal benefit depends on having (1) a primary care provider who is familiar with managing depression, (2) a case manager with a mental health background to follow and support patients, and (3) regular supervision of the case manager by a psychiatrist or psychologist. Cardiologists see large numbers of patients with chronic CHD, ACS, or recent coronary artery bypass graft surgery who are at high risk for depression. The AHA advisory recommends a care model that is practical for CHD patients with depression. Such a care model must be based on detection of depression and referral to a practice that has resources and knowledge to manage it well.
In 2008, the American Heart Association (AHA) published a science advisory on depression and coronary heart disease (CHD).1 Since its publication, the advisory has evoked substantial commentary. The purpose of this article is threefold: (1) to explain the aims of the AHA science advisory, (2) to briefly discuss its content, and (3) to examine some of the comments it has provoked.
WHAT THE ADVISORY SET OUT TO DO
The purpose of an AHA science advisory is to provide rapid, clear, and consistent AHA positioning on a scientific issue. Advisories are statements on an evolving, prominent scientific issue of great interest to the public and health professionals. All AHA science advisories undergo peer review and are also reviewed and approved by the AHA Science Advisory and Coordinating Committee, AHA’s highest science body. Because this particular advisory addressed the interaction of cardiovascular and mental health, the AHA asked the American Psychiatric Association (APA) to review the document; the APA endorsed the AHA advisory.
Two points are worth emphasizing:
An AHA science advisory is not a treatment guideline.
Advisories usually are brief and therefore do not exhaustively discuss their topic.
After discussing epidemiologic studies that elucidated the relationships between depression and CHD, the AHA advisory on depression and CHD focuses on screening, referral, and treatment of depression from a cardiology perspective.
The 1-year prevalence of major depressive disorder in the US general population is 7%, and the lifetime prevalence is about 16%.2 Depression in otherwise healthy persons almost doubles the risk of developing CHD.3 About 20% of patients hospitalized for acute coronary syndromes (ACS) have major depressive disorder on admission or within a few weeks thereafter, and these patients have about 2.5 times the mortality rate as patients who are not depressed, after adjusting for infarct severity and cardiovascular risk factors.4–6
ASSESSMENT OF DEPRESSION AND DEPRESSIVE SYMPTOMS
The AHA advisory discusses use of the 2-question Patient Health Questionnaire (PHQ-2) as the first step in screening for depression.7,8 The PHQ-2 inquires about the frequency of depressed mood and anhedonia by asking the following:
Over the past 2 weeks, how often have you been bothered by either of the following problems?
(1) Little interest or pleasure in doing things
(2) Feeling down, depressed, or hopeless.
For each item the response options are “not at all” (scored as 0), “several days” (scored as 1), “more than half the days” (scored as 2), and “nearly every day” (scored as 3). Thus, the total PHQ-2 score can range from 0 to 6.
Using a structured psychiatric interview as the standard, a total PHQ-2 score of 3 or greater has been shown to have a sensitivity of 83% and a specificity of 92% for major depression.7 A PHQ-2 score of 3 is the optimal cut point for screening purposes. A PHQ-2 score of 0 virtually excludes depression.
If a patient’s PHQ-2 score is 3 or greater, it is recommended that answers be obtained for a full 9-item PHQ. The PHQ-9 provides the sensitivity and specificity suitable for assigning a provisional diagnosis of major depressive disorder and a symptom severity score that can be used to identify patients for further evaluation and to make decisions about therapy.9–11
Adapted from the 2008 American Heart Association science advisory on depression and coronary heart disease.1 Source: American Heart Association, Inc.
Figure 1. Screening for depression in patients with coronary heart disease.The AHA advisory’s section on assessment of depression and depressive symptoms discusses briefly the principles enunciated by the MacArthur Initiative on Depression and Primary Care.12–14 The advisory carefully provides practical guidance specifically for cardiologists (Figure 1).1 The section on assessment points out that screening for depression coupled with therapy has not been proven to improve cardiovascular outcomes but that some antidepressant drugs and/or psychotherapy have proved safe and can improve quality of life, reduce depressive symptoms, improve compliance with lifestyle advice, and improve adherence to prescribed cardiovascular medications.1,15,16
The US Preventive Services Task Force (USPSTF) currently recommends that clinicians screen adults for depression in clinical practices that have systems in place to assure accurate diagnosis, effective treatment, and follow-up.12 The USPSTF concluded that there is good evidence that screening improves the accurate identification of depressed patients in primary care settings and that treating depressed adults identified in such settings reduces clinical morbidity. Results of trials that have directly evaluated the effect of screening on clinical outcomes depend on follow-up. Limited benefits have been demonstrated in studies that simply feed screening results back to clinicians. Larger benefits have been seen in studies in which the communication of screening results is coordinated with effective follow-up and treatment. The USPSTF concluded that the benefits of screening and treating are likely to outweigh any potential harms.12
DEPRESSION TREATMENT
Drug therapy
The safety of fluoxetine, sertraline (SADHART, ENRICHD), citalopram (CREATE), and mirtazapine (MIND-IT) has been evaluated in clinical trials.4,17–21 By far the strongest evidence of safety is for selective serotonin reuptake inhibitors (SSRIs), especially sertraline.
The Sertraline Antidepressant Heart Attack Randomized Trial (SADHART; N = 369), a randomized study of depression after ACS, found no difference in cardiovascular adverse events between the sertraline and placebo groups after 16 weeks of therapy, and there were no significant differences in left ventricular ejection fraction, heart rate, blood pressure, ventricular premature complexes, or electrocardiogram changes.17 In the group assigned to sertraline, life-threatening cardiovascular events occurred less frequently (15% vs 22%, P = NS).
The Enhancing Recovery in Coronary Heart Disease study (ENRICHD) was a large trial (N = 2,481) to evaluate the effect of cognitive behavioral therapy (CBT) on depression or low perceived social support in patients enrolled within 28 days of myocardial infarction (MI).4 The ENRICHD protocol required patients randomized to CBT with Hamilton Depression Rating Scale (HAM-D) scores greater than 24, or who showed less than 50% reduction in Beck Depression Inventory (BDI) scores after 5 weeks of CBT, to be referred to a study psychiatrist for consideration of pharmacotherapy, usually sertraline. Of the overall ENRICHD population, 1,834 participants (74%) had a diagnosis of depression, and 446 of these participants (24%) were treated with antidepressant drugs, 301 with an SSRI and 145 with other antidepressants.18 During mean follow-up of 29 months, the SSRI-treated group had a statistically significant 43% reduction in death or MI.4
The Canadian Cardiac Randomized Evaluation of Antidepressant and Psychotherapy Efficacy (CREATE) recruited 284 patients with chronic CHD, major depressive disorder, and a 24-item HAM-D score of 20 or greater and randomly assigned half to citalopram (another SSRI) and half to placebo.19 Citalopram showed antidepressant efficacy and no evidence of harm.
The Myocardial Infarction and Depression Intervention Trial (MIND-IT) recruited 91 patients within 30 days of hospital admission for MI with depression and randomized them in a 1:1 ratio to mirtazapine or placebo. Mirtazapine showed some evidence of antidepressant efficacy and no evidence of harm.20
Strik et al in the Netherlands recruited 54 patients who had major depression after a first MI and randomized them 1:1 to the SSRI fluoxetine or placebo 3 months after the MI.21 The fluoxetine group showed a trend toward antidepressant efficacy, and no cardiovascular safety problems were detected clinically or by either electrocardiogram or echocardiogram.
Psychotherapy
APA practice guidelines for major depressive disorder indicate that among psycho therapeutic approaches, CBT and interpersonal psychotherapy have the best-documented efficacy for treatment of major depressive disorder.22 CBT aims to solve problems related to dysfunctional emotions, behaviors, and cognition and is an umbrella term for various techniques that share a theoretical basis in behavioristic learning theory and cognitive psychology. Aaron T. Beck proposed that depressed people are quick to make negative evaluations of themselves and the world, and he designed treatment to reduce these negative cognitions.23 Interpersonal psychotherapy stems from the work of Harry Stack Sullivan, who believed that emotional reactions were triggered by interpersonal behaviors.24 Gerald Klerman and Myrna Weissman used this method to treat adults diagnosed with moderate or severe nondelusional clinical depression.25
CBT was used in ENRICHD,4 interpersonal psychotherapy in CREATE,19 and problem-solving therapy in the Coronary Psychosocial Evaluation Studies (COPES).26,27 Unintended therapy can also be a confounder in antidepressant drug trials; education and supportive care (eg, frequent visits or telephone calls with monitoring of depressive symptoms and counseling) are often provided for both the intervention and control (placebo) groups. If a study is blinded, education, supportive care, and attention will be identical for both groups and thus may reduce the likelihood of finding a difference between the drug and placebo, if one exists.
Psychotherapy can be helpful for depression, is preferred over antidepressant drugs by some patients, and can be combined with drugs to increase antidepressant efficacy. We still have much to learn about timing and choice of therapy, as well as about sequencing and combination of antidepressant drugs and psychotherapy.
Physical activity and exercise
Aerobic exercise28 and cardiac rehabilitation29 can reduce depressive symptoms in addition to improving cardiovascular fitness. Depression can serve as a barrier to participation in cardiac rehabilitation and exercise programs, but cardiologists can help depressed patients overcome this barrier by offering encouragement and follow-up contacts. Cardiologists also should enlist the help of spouses or other family members and friends to promote adherence. The prescription of exercise needs to be based on the cardiac status and exercise capacity of each individual.30
SUMMARY OF ADVISORY RECOMMENDATIONS
The AHA advisory1 summarized its recommendations as follows:
1. Routine screening for depression in patients with CHD should be considered in a variety of settings, including the hospital, the physician’s office, clinics, and cardiac rehabilitation centers. The opportunity to screen for and treat depression in cardiac patients should not be missed, as effective depression treatment may improve health outcomes.
2. Patients with positive screening results should be evaluated by a professional qualified in diagnosis and management of depression. Such a clinician can determine whether depression is present and needs treatment, as well as how to connect a patient to an effective care program in the local area.
3. Patients with heart disease who are being treated for depression should undergo careful monitoring for adherence to their medical care and for the efficacy and safety of drug therapy for their medical and mental health conditions.
4. Coordination of care among health care providers is essential for patients with coexisting medical and mental health issues.
COMMENTS ON THE ADVISORY
Since AHA advisories usually address evolving scientific issues, the knowledge base on these issues is constantly growing and a range of opinions and hypotheses are tenable, pending new information. Soon after the AHA science advisory on depression and CHD was issued, a systematic review on depression screening and patient outcomes in cardiac care was published.31 This review posed three key questions that are explored below.
Three key questions
Key question 1: What’s the accuracy of screening instruments for depression in cardiovascular care populations? To answer this question, a case-finding method such as a questionnaire (eg, BDI or PHQ) must be compared with a structured interview by mental health personnel as the “gold standard” for diagnosis (ie, the truth).
To estimate the accuracy of clinical diagnosis by general practitioners, Mitchell et al pooled data from 50,371 primary care patients across 41 studies in Europe or the United States to evaluate general practitioners’ ability to make an unassisted diagnosis of depression (ie, without specific help from severity scales, diagnostic instruments, education programs, etc).32 They reported a sensitivity of about 50% and a specificity of 81% when the prevalence of depression was about 20%. In other words, general practitioners missed about half the cases of depression when no case-finding tool was used. These researchers pointed out that a low prevalence of depression favors identification of nondepressed cases (false-positive diagnoses), whereas a high prevalence favors diagnosis of depression (true-positive diagnoses).32
Cardiologists focused on treatment of ACS are probably less likely to make a clinical diagnosis of depression and may attribute emotional symptoms to rapidly evolving cardiovascular events. The simple 2-question PHQ-2 case-finding instrument takes only a few minutes to administer and is recommended for use by primary care physicians or cardiologists to evaluate patients at high risk for depression or who manifest symptoms suggestive of depression.7
In the United States, most patients with depression are cared for in primary care venues. The AHA advisory recommends referring ACS patients who screen positive on a PHQ to a professional qualified in the diagnosis and management of depression.1 Enhanced care—using outreach, monitoring, adjustment of therapy, and psychiatric backup—produces significant improvement in depression.33
Key question 2: Is treatment of depression in cardiovascular care patients effective in improving depression? Cardiac outcomes? The evidence for benefit of SSRI therapy for depression detected at the time of ACS is consistent and was discussed above.4,17–21 However, the antidepressant effect of SSRIs is modest in placebo-controlled trials. Most such trials excluded patients who were taking antidepressant drugs when screened, and many patients recruited for antidepressant clinical trials had no previous episodes of depression, had relatively mild symptoms of short duration, and were recruited by screening (ie, they were not seeking treatment for depression).34 Patients with brief and short episodes are likely to remit spontaneously and to respond to psychotherapy or supportive care.34 Moreover, in most antidepressant trials, both the intervention and “control” groups received elements of enhanced depression care, which will reduce the apparent benefit of antidepressant drugs. For instance, because the primary goal of SADHART was to evaluate the safety of sertraline in patients with ACS, monitoring for adverse effects included six or seven visits during 16 weeks of follow-up plus six or seven phone calls, providing several elements of enhanced depression care, including face-to-face education, frequent follow-up, support by a case manager (research coordinator) with a mental health background, and support from a psychiatrist or psychologist.17 Randomization to sertraline or placebo in SADHART was blinded, so the frequent contact and support was the same in both groups.
Whether SSRI treatment for depression will improve survival and cardiovascular outcomes is not established by adequately powered randomized trials. Most randomized (SADHART, CREATE) or nonrandomized (ENRICHD) studies suggest that SSRIs reduce cardiovascular events, but only ENRICHD produced a statistically significant result. SSRIs—certainly sertraline and citalopram—are not associated with significant cardiovascular adverse effects, even during ACS, when the cardiovascular system is unstable and multiple drugs are being started and titrated. Patients who do not improve significantly during antidepressant drug therapy or psychotherapy have a two-to threefold increase in cardiovascular events compared with patients who do improve substantially.35
Key question 3: Is systematic screening for depression more effective than usual care for identifying patients with depression? Facilitating treatment of depression? Reducing depressive symptoms? Improving cardiac outcomes? Pignone et al conducted a literature review and meta-analysis on behalf of the USPSTF to clarify whether screening adults for depression in primary care venues improves recognition, treatment, and clinical outcomes.36 They reviewed randomized trials conducted in primary care settings that evaluated the effect of screening for depression on identification, treatment, or health outcomes, including trials that examined integrated, systematic support for treatment after identification of depression. The meta-analysis suggested that screening and feedback of screening results reduced the risk of persistent depression. Stronger effects were observed with programs that integrated interventions to improve recognition and treatment of depressed patients and that incorporated quality improvements into clinic systems as compared with programs that provided only screening and feedback.
Screening patients with CHD, especially those with ACS, is more effective than usual care (no screening). Indeed, many depressed patients go undetected unless identified by screening. Not surprisingly, those identified by screening tend to have milder depression. For example, half the participants in SADHART had a HAM-D score less than 18 (≥ 25 is considered severe). However, a considerable number identified by screening had above-average HAM-D scores but either viewed their symptoms as appropriate for their medical condition or were simply denying their psychiatric illness. If patients had not been screened, their depression would not have been detected or treated. Even among SADHART participants with baseline HAM-D scores less than 18, those whose depression failed to respond to sertraline/placebo had twice the 7-year mortality rate as did those whose depression remitted.16
The Prevention of Suicide in Primary Care Elderly: Collaborative Trial (PROSPECT) examined the impact of a care management intervention on suicidal ideation and depression in older primary care patients and reported outcomes over a 2-year follow-up.37 PROSPECT screened 9,072 patients in 20 primary care practices to find 599 study participants aged 60 years or older with major or minor depression (not all had cardiovascular disease). Participants were randomly assigned to either usual care or the PROSPECT intervention, which consisted of services rendered by trained care managers who offered algorithm-based recommendations to physicians and helped patients adhere to treatment during the 24-month trial. Compared with patients receiving usual care, those receiving the intervention had a greater likelihood of receiving antidepressant drugs and/or psychotherapy (85%–89% vs 49%–62%) and had a 2.2-fold greater decline in suicidal ideation over 24 months. Treatment response started sooner in the intervention group and continued to improve at 18 and 24 months; no appreciable increase in treatment response was observed in the usual-care group during the same period. Among patients with major depression, a significantly greater percentage of the intervention group achieved remission compared with the usual-care group at 4, 8, and 24 months. Patients with minor depression had favorable mental health outcomes regardless of treatment assignment. Sustained collaborative care maintained high utilization of depression treatment, reduced suicidal ideation, and improved the outcomes of major depression during 2 years of follow-up.37
No randomized trial of SSRIs has been designed to show an effect on mortality or cardiovascular events. ENRICHD has produced the strongest evidence that an SSRI can reduce mortality or cardiovascular outcomes. The study was designed with approximately 85% power to detect a 25% to 30% reduction in the primary end point (death or MI) as a result of CBT; as noted above, 2,481 patients (1,834 with depression) were randomized to usual care or CBT within 28 days of MI.4 Depression significantly improved with CBT, but the rate of death or MI did not: during mean follow-up of 29 months, death or MI occurred in 299 patients in the CBT group versus 300 in the usual-care group.4 A post hoc analysis specific to the 1,834 ENRICHD participants who had depression aimed to determine the effects of antidepressant drugs on morbidity and mortality.18 The protocol required patients in the intervention group with scores of 25 or higher on the 17-item HAM-D, or those who had less than a 50% reduction in BDI scores after 5 weeks of treatment, to be referred to study psychiatrists for consideration of pharmacotherapy. Study psychiatrists met with patients who were being treated with antidepressant drugs to monitor medication use. Unless contraindicated or previously ineffective or poorly tolerated, sertraline was the first antidepressant used. Using a time-dependent multivariable Cox proportional hazards regression model to adjust for baseline depression score and cardiac risk factors, the researchers found SSRI use to be associated with a statistically significant 43% lower risk of death or nonfatal MI. Like other SSRI studies, ENRICHD found SSRIs safe for post-MI patients and to possibly, but not certainly, reduce death and MI.18
Medication adherence
One obvious but important way antidepressant drug therapy could prevent death or MI is by improving adherence to post-MI cardiovascular drugs. Four or five classes of cardiac drugs have each been proven to improve survival following ACS (aspirin, beta-blockers, statins, and angiotensin-converting enzyme inhibitors/angiotensin receptor blockers), and when all are taken regularly, mortality is reduced by about half.38,39 In addition, antihypertensive and antidiabetic drugs are often needed to control blood pressure or blood glucose. However, patients have to actually take these drugs to receive their benefit. Depression in the setting of CHD, especially ACS, is a risk indicator for lack of adherence to medical therapy, mental health therapy, or both.
DiMatteo et al conducted a meta-analysis to test the hypothesis that anxiety and depression might explain poor adherence to treatment recommendations and result in poor medical outcomes.15 Of the 25 trials that met the inclusion criteria, 13 studied anxiety and 12 studied depression. The associations between anxiety and nonadherence were small and not statistically significant, but depression was strongly associated with nonadherence to medications (odds ratio = 3.03; 95% confidence interval, 1.96–4.89). In other words, depressed patients were three times as likely as nondepressed patients to be nonadherent to treatment recommendations. The authors speculated that depression might increase nonadherence in the following ways: (1) the hopelessness of depression might reduce patients’ hope in the therapy, (2) depression may cause withdrawal from family and social networks that otherwise would provide support and assistance, or (3) the impaired cognitive dysfunction associated with depression may impair memory and follow through on treatment recommendations.15
The findings from this meta-analysis could reflect depression causing medication nonadherence or vice versa. To establish the sequence, Rieckmann et al measured adherence to aspirin therapy during a 3-month period in a consecutive cohort of 172 patients (25 to 85 years old) recruited within 1 week of hospitalization for ACS.40 Severity of depressive symptoms was quantified using the BDI during hospitalization and at 1 and 3 months after discharge. Adherence was defined as taking aspirin as prescribed on at least 80% of days. Using an electronic monitoring system that recorded the date and time when the aspirin bottle cap was opened, the study found that more than 30% of patients with post-ACS depression were nonadherent to their aspirin therapy compared with only 15% of nondepressed patients.40 The more severe patients’ depressive symptoms were, the greater the nonadherence to aspirin therapy. Moreover, a lagged correlation statistical model determined that improvement in depression preceded improvement in medication adherence.
SADHART was conducted under the new drug application for sertraline,16 which required that the use of trial drugs be under strict compliance to protocol, sponsor monitoring, and auditing by the US Food and Drug Administration. Drug use data were complete in 98.1% of participants. Adherence was measured using tablet counts. Depressed patients who had a large improvement in depression during blinded drug therapy (sertraline or placebo) showed improved adherence to the blinded therapy. To determine whether depression improved before medication adherence improved, researchers compared responders’ medication adherence before and after their improvement in depression. Medication adherence increased following remission of depression in 128 of 187 participants (68.4%) who remitted on trial medication (remission was defined as a Clinical Global Impression–Improvement score of 1). This sequence of change (improved depression before improved medication adherence) occurred significantly more often than would be expected by chance (P < .001). This finding suggests that improvement in depression is driving improved medication adherence.
Because persistent depression is associated with increased mortality rates and reduced medication adherence, physicians need to not only aggressively treat depression but also diligently promote adherence to guideline-defined cardiovascular drug therapy. If depression doesn’t improve, additional measures should be initiated not only to improve depression but also to achieve adherence to cardiovascular drug therapy (eg, assistance from spouse, child, or visiting nurse; calls by case manager; electronic health record monitoring of drug prescription refills). When depression is found during clinical encounters or by screening, nonadherence to drug treatment is much more likely and should be sought vigilantly.
Adherence to lifestyle recommendations
Ziegelstein et al have shown that depressed patients have poorer adherence to lifestyle recommendations (diet, exercise, smoking cessation).41 The Heart and Soul Study, a prospective cohort study of 1,017 outpatients with stable CHD, attempted to determine why depressive symptoms (as determined by PHQ-9 self-report) are associated with an increased risk of cardiovascular events.42 Participants were predominantly older men, about half of whom were recruited from Veterans Administration hospitals. A total of 341 cardiovascular events occurred during a mean follow-up of 4.8 years. Participants with baseline depressive symptoms had a 50% greater rate of cardiovascular events during the study period compared with participants without depressive symptoms. However, no significant association between depressive symptoms and cardiovascular events remained after adjustment for health behaviors—most strikingly, physical activity.42 This finding was consistent with an earlier study that found that exercise therapy plus antidepressant medication could reduce the risk of cardiovascular events in patients with depression.43 The ongoing Understanding Prognostic Benefits of Exercise and Antidepressant Therapy (UPBEAT) study is comparing the effects of exercise and antidepressant medication on depression and biomarkers of cardiovascular risk in patients with depressive symptoms and CHD.44 The study’s longer-term goal is to identify an intervention that improves both depression and cardiovascular disease outcomes.
CONCLUSIONS
The USPSTF recommends screening for depression in adults. The PHQ-2 is an efficient first-step screening tool that can identify many depressed patients who would otherwise go undetected. It is clear that SSRIs are safe in cardiac patients, can reduce depression, and can improve medication adherence, but it is not enough to screen and report depression. Optimal benefit depends on having (1) a primary care provider who is familiar with managing depression, (2) a case manager with a mental health background to follow and support patients, and (3) regular supervision of the case manager by a psychiatrist or psychologist. Cardiologists see large numbers of patients with chronic CHD, ACS, or recent coronary artery bypass graft surgery who are at high risk for depression. The AHA advisory recommends a care model that is practical for CHD patients with depression. Such a care model must be based on detection of depression and referral to a practice that has resources and knowledge to manage it well.
References
Lichtman JH, Bigger JT, Blumenthal JA, et al Depression and coronary heart disease: recommendations for screening, referral, and treatment. A science advisory from the American Heart Association Prevention Committee of the Council on Cardiovascular Nursing, Council on Clinical Cardiology, Council on Epidemiology and Prevention, and Interdisciplinary Council on Quality of Care and Outcomes Research; endorsed by the American Psychiatric Association. Circulation2008; 118:1768–1775.
Kessler RC, Merikangas KR, Wang PS. Prevalence, comorbidity, and service utilization for mood disorders in the United States at the beginning of the twenty-first century. Annu Rev Clin Psychol2007; 3:137–158.
Wulsin LR, Singal BM. Do depressive symptoms increase the risk for the onset of coronary disease? A systematic quantitative review. Psychosom Med2003; 65:201–210.
Berkman LF, Blumenthal J, Burg M, et al Effects of treating depression and low perceived social support on clinical events after myocardial infarction: the Enhancing Recovery in Coronary Heart Disease Patients (ENRICHD) randomized trial. JAMA2003; 289:3106–3116.
Thombs BD, Bass EB, Ford DE, et al Prevalence of depression in survivors of acute myocardial infarction. J Gen Intern Med2006; 21:30–38.
van Melle JP, de Jonge P, Spijkerman TA, et al Prognostic association of depression following myocardial infarction with mortality and cardiovascular events: a meta-analysis. Psychosom Med2004; 66:814–822.
Kroenke K, Spitzer RL, Williams JB. The Patient Health Questionnaire-2: validity of a two-item depression screener. Med Care2003; 41:1284–1292.
Whooley M, Avins AL, Miranda J, Brown WS. Case-finding instruments for depression: two questions are as good as many. J Gen Intern Med1997; 12:289–290.
Gilbody S, Richards D, Brealey S, Hewitt C. Screening for depression in medical settings with the Patient Health Questionnaire (PHQ): a diagnostic meta-analysis. J Gen Intern Med2007; 22:1596–1602.
Stafford L, Berk M, Jackson HJ. Validity of the Hospital Anxiety and Depression Scale and Patient Health Questionnaire-9 to screen for depression in patients with coronary artery disease. Gen Hosp Psychiatry2007; 29:417–424.
McManus D, Pipkin SS, Whooley MA. Screening for depression in patients with coronary heart disease (data from the Heart and Soul Study). Am J Cardiol2005; 96:1076–1081.
U.S. Preventive Services Task Force. Screening for depression: recommendations and rationale. Rockville, MD: Agency for Healthcare Research and Quality; May2002. http://www.ahrq.gov/clinic/3rduspstf/depression/depressrr.htm. Accessed January 8, 2010.
Depression Guideline Panel. Clinical Practice Guideline Number 5: Depression in Primary Care, 1: Detection and Diagnosis. Rockville, MD: US Department of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research; April1993. AHCPR publication 93-0550.
Depression Guideline Panel. Clinical Practice Guideline Number 5: Depression in Primary Care, 2: Treatment of Major Depression. Rockville, MD: US Department of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research; April1993. AHCPR publication 93-0551.
DiMatteo MR, Lepper HS, Croghan TW. Depression is a risk factor for noncompliance with medical treatment: meta-analysis of the effects of anxiety and depression on patient adherence. Arch Intern Med2000; 160:2101–2107.
Glassman AH, Bigger JT, Gaffney M. Psychiatric characteristics associated with long-term mortality among 361 patients having an acute coronary syndrome and major depression: seven-year follow-up of SADHART participants. Arch Gen Psychiatry2009; 66:1022–1029.
Glassman AH, O’Connor CM, Califf RM, et al Sertraline treatment of major depression in patients with acute MI or unstable angina. JAMA2002; 288:701–709.
Taylor CB, Youngblood ME, Catellier D, et al Effects of antidepressant medication on morbidity and mortality in depressed patients after myocardial infarction. Arch Gen Psychiatry2005; 62:792–798.
Lesperance F, Frasure-Smith N, Koszycki D, et al Effects of citalopram and interpersonal psychotherapy on depression in patients with coronary artery disease: the Canadian Cardiac Randomized Evaluation of Antidepressant and Psychotherapy Efficacy (CREATE) trial. JAMA2007; 297:367–379.
Honig A, Kuyper A, Schene AH, et al Treatment of post-myocardial infarction depressive disorder: a randomized, placebo-controlled trial with mirtazapine. Psychosom Med2007; 69:606–613.
Strik J, Honig A, Lousberg R, et al Efficacy and safety of fluoxetine in the treatment of patients with major depression after first myocardial infarction: findings from a double-blind, placebo-controlled trial. Psychosom Med2000; 62:783–789.
American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder (revision). Am J Psychiatry2000; 157( suppl 4):1–45.
Beck AT. Cognitive Therapy and the Emotional Disorders. Madison, CT: International Universities Press Inc; 1975.
Weissman MM. A brief history of interpersonal psychotherapy. Psychiatr Ann2006; 36:553–557.
Klerman GL, Weissmann MM. Interpersonal psychotherapy (IPT) and drugs in the treatment of depression. Pharmacopsychiatry1987; 20:3–7.
Burg MM, Lespérance F, Rieckmann N, Clemow L, Skotzko C, Davidson KW. Treating persistent depressive symptoms in post-ACS patients: the project COPES phase-I randomized controlled trial. Contemp Clin Trials2008; 29:231–240.
Davidson KW, Rieckmann N, Clemow L, et al Collaborative depression care for acute coronary syndrome patients with persistent depression: Coronary Psychosocial Evaluation Studies (COPES) randomized controlled trial. Arch Intern Med. In press.
Brosse AL, Sheets ES, Lett HS, Blumenthal JA. Exercise and the treatment of clinical depression in adults: recent findings and future directions. Sports Med2002; 32:741–760.
Milani RV, Lavie CJ. Impact of cardiac rehabilitation on depression and its associated mortality. Am J Med2007; 120:799–806.
Smith SC, Allen J, Blair SN, et al AHA/ACC guidelines for secondary prevention for patients with coronary and other atherosclerotic vascular disease: 2006 update; endorsed by the National Heart, Lung, and Blood Institute. Circulation2006; 113:2363–2372.
Thombs BD, de Jonge P, Coyne JC, et al Ziegelstein RC. Depression screening and patient outcomes in cardiovascular care: a systematic review. JAMA2008; 300:2161–2171.
Mitchell AJ, Vaze A, Rao S. Clinical diagnosis of depression in primary care: a meta-analysis. Lancet2009; 374:609–619.
Gilbody S, Bower P, Fletcher J, Richards D, Sutton AJ. Collaborative care for depression: a cumulative meta-analysis and review of longer-term outcomes. Arch Intern Med2006; 166:2314–2321.
Glassman AH, Bigger JT, Gaffney M, Shapiro PA, Swenson JR. Onset of major depression associated with acute coronary syndromes: relationship of onset, major depressive disorder history, and episode severity to sertraline benefit. Arch Gen Psychiatry2006; 63:283–288.
Carney RM, Freedland KE. Treatment-resistant depression and mortality after acute coronary syndrome. Am J Psychiatry2009; 166:410–417.
Pignone MP, Gaynes BN, Rushton JL, et al Screening for depression in adults: a summary of the evidence for the U.S. Preventive Services Task Force. Ann Intern Med2002; 136:765–776.
Alexopoulos GS, Reynolds CF, Bruce ML, et al Reducing suicidal ideation and depression in older primary care patients: 24-month outcomes of the PROSPECT study. Am J Psychiatry2009; 166:882–890.
Mukherjee D, Fang J, Chetcuti S, Moscucci M, Kline-Rogers E, Eagle KA. Impact of combination evidence-based medical therapy on mortality in patients with acute coronary syndromes. Circulation2004; 109:745–749.
Eagle KA, Kline-Rogers E, Goodman SG, et al Adherence to evidence-based therapies after discharge for acute coronary syndromes: an ongoing prospective, observational study. Am J Med2004; 117:73–81.
Rieckmann N, Gerin W, Kronish IA, et al Course of depressive symptoms and medication adherence after acute coronary syndromes: an electronic medication monitoring study. J Am Coll Cardiol2006; 48:2218–2222.
Ziegelstein RC, Fauerbach JA, Stevens SS, Romanelli J, Richter DP, Bush DE. Patients with depression are less likely to follow recommendations to reduce cardiac risk during recovery from a myocardial infarction. Arch Intern Med2000; 160:1818–1823.
Whooley MA, de Jonge P, Vittinghoff E, et al Depressive symptoms, health behaviors, and risk of cardiovascular events in patients with coronary heart disease. JAMA2008; 300:2379–2388.
Blumenthal JA, Babyak MA, Doraiswamy PM, et al Exercise and pharmacotherapy in the treatment of major depressive disorder. Psychosom Med2007; 69:587–596.
Blumenthal JA, Sherwood A, Rogers SD, et al Understanding prognostic benefits of exercise and antidepressant therapy for persons with depression and heart disease: the UPBEAT study—rationale, design, and methodological issues. Clin Trials2007; 4:548–559.
References
Lichtman JH, Bigger JT, Blumenthal JA, et al Depression and coronary heart disease: recommendations for screening, referral, and treatment. A science advisory from the American Heart Association Prevention Committee of the Council on Cardiovascular Nursing, Council on Clinical Cardiology, Council on Epidemiology and Prevention, and Interdisciplinary Council on Quality of Care and Outcomes Research; endorsed by the American Psychiatric Association. Circulation2008; 118:1768–1775.
Kessler RC, Merikangas KR, Wang PS. Prevalence, comorbidity, and service utilization for mood disorders in the United States at the beginning of the twenty-first century. Annu Rev Clin Psychol2007; 3:137–158.
Wulsin LR, Singal BM. Do depressive symptoms increase the risk for the onset of coronary disease? A systematic quantitative review. Psychosom Med2003; 65:201–210.
Berkman LF, Blumenthal J, Burg M, et al Effects of treating depression and low perceived social support on clinical events after myocardial infarction: the Enhancing Recovery in Coronary Heart Disease Patients (ENRICHD) randomized trial. JAMA2003; 289:3106–3116.
Thombs BD, Bass EB, Ford DE, et al Prevalence of depression in survivors of acute myocardial infarction. J Gen Intern Med2006; 21:30–38.
van Melle JP, de Jonge P, Spijkerman TA, et al Prognostic association of depression following myocardial infarction with mortality and cardiovascular events: a meta-analysis. Psychosom Med2004; 66:814–822.
Kroenke K, Spitzer RL, Williams JB. The Patient Health Questionnaire-2: validity of a two-item depression screener. Med Care2003; 41:1284–1292.
Whooley M, Avins AL, Miranda J, Brown WS. Case-finding instruments for depression: two questions are as good as many. J Gen Intern Med1997; 12:289–290.
Gilbody S, Richards D, Brealey S, Hewitt C. Screening for depression in medical settings with the Patient Health Questionnaire (PHQ): a diagnostic meta-analysis. J Gen Intern Med2007; 22:1596–1602.
Stafford L, Berk M, Jackson HJ. Validity of the Hospital Anxiety and Depression Scale and Patient Health Questionnaire-9 to screen for depression in patients with coronary artery disease. Gen Hosp Psychiatry2007; 29:417–424.
McManus D, Pipkin SS, Whooley MA. Screening for depression in patients with coronary heart disease (data from the Heart and Soul Study). Am J Cardiol2005; 96:1076–1081.
U.S. Preventive Services Task Force. Screening for depression: recommendations and rationale. Rockville, MD: Agency for Healthcare Research and Quality; May2002. http://www.ahrq.gov/clinic/3rduspstf/depression/depressrr.htm. Accessed January 8, 2010.
Depression Guideline Panel. Clinical Practice Guideline Number 5: Depression in Primary Care, 1: Detection and Diagnosis. Rockville, MD: US Department of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research; April1993. AHCPR publication 93-0550.
Depression Guideline Panel. Clinical Practice Guideline Number 5: Depression in Primary Care, 2: Treatment of Major Depression. Rockville, MD: US Department of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research; April1993. AHCPR publication 93-0551.
DiMatteo MR, Lepper HS, Croghan TW. Depression is a risk factor for noncompliance with medical treatment: meta-analysis of the effects of anxiety and depression on patient adherence. Arch Intern Med2000; 160:2101–2107.
Glassman AH, Bigger JT, Gaffney M. Psychiatric characteristics associated with long-term mortality among 361 patients having an acute coronary syndrome and major depression: seven-year follow-up of SADHART participants. Arch Gen Psychiatry2009; 66:1022–1029.
Glassman AH, O’Connor CM, Califf RM, et al Sertraline treatment of major depression in patients with acute MI or unstable angina. JAMA2002; 288:701–709.
Taylor CB, Youngblood ME, Catellier D, et al Effects of antidepressant medication on morbidity and mortality in depressed patients after myocardial infarction. Arch Gen Psychiatry2005; 62:792–798.
Lesperance F, Frasure-Smith N, Koszycki D, et al Effects of citalopram and interpersonal psychotherapy on depression in patients with coronary artery disease: the Canadian Cardiac Randomized Evaluation of Antidepressant and Psychotherapy Efficacy (CREATE) trial. JAMA2007; 297:367–379.
Honig A, Kuyper A, Schene AH, et al Treatment of post-myocardial infarction depressive disorder: a randomized, placebo-controlled trial with mirtazapine. Psychosom Med2007; 69:606–613.
Strik J, Honig A, Lousberg R, et al Efficacy and safety of fluoxetine in the treatment of patients with major depression after first myocardial infarction: findings from a double-blind, placebo-controlled trial. Psychosom Med2000; 62:783–789.
American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder (revision). Am J Psychiatry2000; 157( suppl 4):1–45.
Beck AT. Cognitive Therapy and the Emotional Disorders. Madison, CT: International Universities Press Inc; 1975.
Weissman MM. A brief history of interpersonal psychotherapy. Psychiatr Ann2006; 36:553–557.
Klerman GL, Weissmann MM. Interpersonal psychotherapy (IPT) and drugs in the treatment of depression. Pharmacopsychiatry1987; 20:3–7.
Burg MM, Lespérance F, Rieckmann N, Clemow L, Skotzko C, Davidson KW. Treating persistent depressive symptoms in post-ACS patients: the project COPES phase-I randomized controlled trial. Contemp Clin Trials2008; 29:231–240.
Davidson KW, Rieckmann N, Clemow L, et al Collaborative depression care for acute coronary syndrome patients with persistent depression: Coronary Psychosocial Evaluation Studies (COPES) randomized controlled trial. Arch Intern Med. In press.
Brosse AL, Sheets ES, Lett HS, Blumenthal JA. Exercise and the treatment of clinical depression in adults: recent findings and future directions. Sports Med2002; 32:741–760.
Milani RV, Lavie CJ. Impact of cardiac rehabilitation on depression and its associated mortality. Am J Med2007; 120:799–806.
Smith SC, Allen J, Blair SN, et al AHA/ACC guidelines for secondary prevention for patients with coronary and other atherosclerotic vascular disease: 2006 update; endorsed by the National Heart, Lung, and Blood Institute. Circulation2006; 113:2363–2372.
Thombs BD, de Jonge P, Coyne JC, et al Ziegelstein RC. Depression screening and patient outcomes in cardiovascular care: a systematic review. JAMA2008; 300:2161–2171.
Mitchell AJ, Vaze A, Rao S. Clinical diagnosis of depression in primary care: a meta-analysis. Lancet2009; 374:609–619.
Gilbody S, Bower P, Fletcher J, Richards D, Sutton AJ. Collaborative care for depression: a cumulative meta-analysis and review of longer-term outcomes. Arch Intern Med2006; 166:2314–2321.
Glassman AH, Bigger JT, Gaffney M, Shapiro PA, Swenson JR. Onset of major depression associated with acute coronary syndromes: relationship of onset, major depressive disorder history, and episode severity to sertraline benefit. Arch Gen Psychiatry2006; 63:283–288.
Carney RM, Freedland KE. Treatment-resistant depression and mortality after acute coronary syndrome. Am J Psychiatry2009; 166:410–417.
Pignone MP, Gaynes BN, Rushton JL, et al Screening for depression in adults: a summary of the evidence for the U.S. Preventive Services Task Force. Ann Intern Med2002; 136:765–776.
Alexopoulos GS, Reynolds CF, Bruce ML, et al Reducing suicidal ideation and depression in older primary care patients: 24-month outcomes of the PROSPECT study. Am J Psychiatry2009; 166:882–890.
Mukherjee D, Fang J, Chetcuti S, Moscucci M, Kline-Rogers E, Eagle KA. Impact of combination evidence-based medical therapy on mortality in patients with acute coronary syndromes. Circulation2004; 109:745–749.
Eagle KA, Kline-Rogers E, Goodman SG, et al Adherence to evidence-based therapies after discharge for acute coronary syndromes: an ongoing prospective, observational study. Am J Med2004; 117:73–81.
Rieckmann N, Gerin W, Kronish IA, et al Course of depressive symptoms and medication adherence after acute coronary syndromes: an electronic medication monitoring study. J Am Coll Cardiol2006; 48:2218–2222.
Ziegelstein RC, Fauerbach JA, Stevens SS, Romanelli J, Richter DP, Bush DE. Patients with depression are less likely to follow recommendations to reduce cardiac risk during recovery from a myocardial infarction. Arch Intern Med2000; 160:1818–1823.
Whooley MA, de Jonge P, Vittinghoff E, et al Depressive symptoms, health behaviors, and risk of cardiovascular events in patients with coronary heart disease. JAMA2008; 300:2379–2388.
Blumenthal JA, Babyak MA, Doraiswamy PM, et al Exercise and pharmacotherapy in the treatment of major depressive disorder. Psychosom Med2007; 69:587–596.
Blumenthal JA, Sherwood A, Rogers SD, et al Understanding prognostic benefits of exercise and antidepressant therapy for persons with depression and heart disease: the UPBEAT study—rationale, design, and methodological issues. Clin Trials2007; 4:548–559.
Many advances in the understanding of the relationship between depression and cardiovascular disease (CVD) were reported in 2009. As the study of this relationship encompasses cardiology, psychiatry, behavioral medicine, and many other fields, it is difficult to keep abreast of new developments. Relevant papers are found in a variety of journals. Therefore, we systematically searched the empirical research on depression and CVD published in English in 2009. Our search yielded nearly 500 articles. We review here a few of the most provocative and potentially influential findings. We begin with an overview of the methodology of our systematic review and then summarize the key findings and controversies we identified from the 2009 literature before exploring each key finding in detail.
METHODOLOGY OF OUR SYSTEMATIC SEARCH AND RATIONALE FOR FINDINGS REVIEWED
Previous evidence demonstrated that the presence of depressive symptoms or a diagnosis of a depressive disorder predicts poor prognosis and reduced survival rates after any coronary artery disease diagnosis, including myocardial infarction (MI) and unstable angina, as well as after coronary artery bypass graft surgery (CABG).1
We aimed to see how this evidence base was expanded in 2009 by using a systematic search strategy to retrieve the most relevant articles about depression and coronary heart disease (CHD) from the MEDLINE and Psyc-INFO (Ovid interface) databases. The most relevant subject headings and free text terms were identified and combined with “or.” The two sets were then combined with “and.” Terms included “depression,” “depressive disorder,” “depress$,” “coronary artery disease” (CAD) “coronary disease,” “acute coronary syndrome” (ACS), “cardiovascular disease” (CVD), “coronary heart disease,” and “heart diseas$.” The final set was limited to the English-language literature and identified 494 unique articles published during 2009.
Closer inspection of titles and abstracts revealed well more than 100 articles directly relevant to the science and management of patients with CVD and depression or pronounced depressive symptoms. In light of this quantity, a thorough review of all new findings, editorials, and reviews is not feasible. Thus, we review here a few exciting articles in several distinct topical areas that could influence views of the relationship between depression and CVD as well as how we screen for and treat depression in patients with CVD. As with any review that is not strictly evidence-based, our choice of articles is subjective and incomplete, but we hope it will stimulate discussion and further exploration.
SUMMARY OF KEY FINDINGS AND CONTROVERSIES
The following numbered topics emerged as common themes or controversies from our survey of the 2009 literature. The remainder of this article will review findings in each of these areas in detail and discuss important clinical implications as appropriate.
1. Antidepressant use and adverse cardiac events. In surprising findings, antidepressant use was associated with increased risk of incident stroke, CVD, and sudden cardiac death in multiple large observational cohort studies. It is not known if the association is caused by unmeasured confounders or depressive symptom severity, although controlling for symptom severity did not reduce this elevated risk in some studies. Another interesting possibility is that the treatment-resistant depressed patient is at particularly high risk of CVD and death.
2. Effects of depression intervention in CVD patients. Four exciting randomized controlled trials on depression intervention in patients with CVD reported important efficacy results and suggested future directions for larger, definitive trials of depression treatment for these patients.
3. Depression screening and treatment in CVD patients. In the absence of large randomized controlled trials, the debate continues on whether depression screening or any type of depression treatment is beneficial, harmless, or harmful to patients with CVD. Continuation of this debate does not serve the health and well-being of patients or the public. Less controversial—and thus less discussed—is the important insight that psychiatric patients with depression should be routinely screened for cardiac disease and risk factors, as they are clearly at risk of CVD. We await clinical trials in this area to ensure that screening leads to improved CVD outcomes.
OBSERVATIONAL EVIDENCE ON ANTIDEPRESSANT USE AND CVD OUTCOMES
In an analysis of the Nurses’ Health Study, Whang et al examined the relationship of depressive symptoms and antidepressant use with sudden cardiac death and adverse cardiac events in 63,469 women without CVD.2 Depressive symptoms were assessed using the Mental Health Index, a five-item subscale of the Short Form-36 Health Survey. Among women who reported antidepressant use, most (61%) were taking a selective serotonin reuptake inhibitor (SSRI; sertraline, fluoxetine, paroxetine, or citalopram), while 39% reported use of other antidepressants. Women taking antidepressants were more likely to suffer sudden cardiac death, with a fully adjusted hazard ratio (HR) of 3.34 (95% confidence interval [CI], 2.03–5.50).
Krantz et al examined psychotropic medication use and risk of adverse cardiovascular events in 519 women from the Women’s Ischemia Syndrome Evaluation.3 Enrolled women underwent coronary angiography, were separated into four groups according to their psychotropic medication use (none, anxiolytics only, antidepressants only, or both anxiolytics and antidepressants), and were observed for a median of 5.9 years. Results revealed that women who received both medications had a higher risk for adverse cardiovascular events and higher all-cause mortality compared with those using neither medication, even after controlling for anxious and depressive symptoms. In addition, whereas the use of antidepressant medication was associated with a doubling of risk for subsequent CVD events (HR = 2.16; 95% CI, 1.21–3.93) and all-cause mortality (HR = 2.15; 95% CI, 1.16–3.98), use of anxiolytic medication alone was not. While this study did not examine cause of death, cardiac death is likely to have constituted a large proportion of total mortality in this cohort selected for likelihood of CAD.
Although CVD and death have long been outcomes of interest for those studying the effects of depression, additional end points have recently been investigated as well. In a prospective cohort study of 136,293 community-dwelling postmenopausal women in the Women’s Health Initiative, Smoller et al found that new antidepressant use was significantly associated with increased incidence of stroke and all-cause mortality but not with incidence of CHD.4 The rate of stroke per 1,000 person-years was 2.99 for subjects with no antidepressant use versus 4.16 for patients with new SSRI use; the rate of all-cause death was 7.79 versus 12.77, respectively. The rate of all-cause death for subjects with new tricyclic antidepressant use was 14.14 per 1,000 person-years. To address potential confounding by indication, the researchers obtained a propensity score from a logistic regression model to predict any new antidepressant use from demographic, lifestyle, risk factor, and comorbidity variables measured at baseline. New SSRI use was associated with a doubling of the risk of incident hemorrhagic stroke as well as fatal stroke. There were no significant interactions between use of SSRIs and use of statins or aspirin in terms of risk of hemorrhagic stroke.
In an interesting observational study of 7,709 patients with confirmed CAD but without a diagnosis of heart failure or depression (and without current antidepressant use), May et al found that a subsequent diagnosis of depression was associated with a significant 50% increase in the risk of heart failure.5 There was no difference, however, between depressed patients who were using antidepressants and those who were not.
Increased risk of bleeding with SSRI use, particularly in patients with CAD, has also been a concern. Kim et al evaluated 1,380 adults who received any anti depressant before CABG for in-hospital mortality or any bleeding events.6 After controlling for the percentage of patients taking SSRIs (78%), there were no significant differences between those taking SSRIs and those taking non-SSRIs in the rate of any bleeding events (6.5% vs 7.2%; odds ratio [OR] = 0.93; 95% CI, 0.50–1.76) or in-hospital mortality (3.1% vs 2.3%; OR = 0.88; 95% CI, 0.47–1.65). There was no increased risk of bleeding associated with SSRI use when the analysis was restricted to patients who received antiplatelet and anticoagulant therapy. Thus, compared with patients who received non-SSRI antidepressants, patients who received SSRIs preoperatively had no increased risk of bleeding or in-hospital mortality after CABG; however, this study did not evaluate the effect of no antidepressant use.
Another study hypothesized that the use of any drug with the potential to prolong cardiac repolarization would be associated with an increased risk of sudden death.7 Use of individual drugs was analyzed among 1,010 cases of sudden unexplained death and 3,030 living primary care controls, all from the community. SSRI use was associated with a doubling of risk of sudden death (OR = 2.21; 95% CI, 1.61–3.05), and tricyclic antidepressant use was associated with a nonsignificant trend toward increased risk (OR = 1.44; 95% CI, 0.96–2.13). Further analysis that stratified patients according to prior CVD showed that most of the association of SSRIs with sudden death was in those with existing CVD and not in those without CVD. Other drugs found to raise sudden death risk included the typical and atypical antipsychotics.
Summary and clinical implications
The Nurses’ Health Study analysis by Whang et al suggested that antidepressant use triples the risk of sudden cardiac death in healthy women, and the authors suggested that the association between fatal ventricular arrhythmias and antidepressant use be examined further. 2 The analysis of the Women’s Health Initiative by Smoller et al found that use of SSRIs and tricyclic antidepressants doubled the risk of fatal stroke in healthy women.4 The analysis of the Women’s Ischemia Syndrome Evaluation by Krantz et al revealed a doubling of the risk of CVD and death in women taking antidepressants who had been referred for coronary angiography.3 At the same time, May et al found no increase in the risk of heart failure conversion with antidepressant use in patients with CAD,5 and Kim et al found no increase in the risk of bleeding with use of SSRIs compared with non-SSRI antidepressants in patients with CAD undergoing CABG.6 Finally, in a population- and community-based case-control study, SSRI use was associated with an increased risk of sudden death, particularly in patients with CVD.7 So what are we to make of these findings?8
In all observational studies (including those reviewed above), unmeasured confounders pose a threat to the validity of any causal conclusions. A study recently tested some of the proposed confounders that might have existed in the above studies. Waldman et al examined racial differences in depressive symptoms and antidepressant treatment among a cohort of 864 consecutive patients with CHD undergoing diagnostic coronary angiography (727 white and 137 African American).9 While levels of depression were similar between the white and African American patients, the African Americans were less likely than their white counter-parts to receive antidepressant medications. Patients with only some high school, men, and patients with more severe depressive symptoms were significantly more likely to receive a prescription for antidepressants. Clearly, low education, male sex, and elevated depressive symptoms are related to poor prognosis for CHD, and the simple interpretation that antidepressant use is causing poorer outcomes is problematic.8
Two additional interpretations of the observational findings should be considered. First, confounding by indication (depressive symptom severity) might exist in these studies.10 In other words, patients who are prescribed antidepressant medication may be those with the most severe depressive illness, and it could be this severity, rather than the antidepressant use, that is causally implicated in the CVD incidence.11 However, all of the studies reviewed above either directly controlled for depressive symptom severity (at least as obtained at baseline) or used propensity scores or stratified subjects based on depression severity. The results showed an increased risk among those who were taking antidepressants. However, none of the studies examined depressive symptom severity during or at the end of the study or depression diagnosis and severity before antidepressant use; these data are needed for a clearer understanding of whether the results were confounded by indication.
Second, these findings are also consistent with a treatment-resistant depression phenotype.12 Krantz et al caution that it is not clear from their observational study3 whether medication use itself or depression refractory to treatment is implicated in the increased risk of CVD events and mortality. Depression that is refractory to treatment may be the type of depression that places patients at risk for sudden death, stroke, or CHD recurrence, so it may not be the antidepressant use per se that is associated with this risk. This phenomenon was documented in a secondary analysis13 of the largest-to-date randomized controlled trial of patients with MI undergoing treatment for depression (ENRICHD).14 It showed that those whose depressive symptoms did not respond to treatment had a higher risk of late mortality (ie, death ≥ 6 months after acute MI). This finding was replicated in 2009 in an important follow-up15 of the SADHART trial16; among patients with MI and major depression, treatment-resistant depression (ie, depression that failed to improve substantially during treatment with either sertraline or placebo) was strongly and independently associated with long-term mortality (HR = 2.39; 95% CI, 1.39–2.44; P < .001).15
What is needed next? Testing the alternative hypothesis— ie, that an unmeasured confounder may exist—is difficult, requiring new observational studies and measurement of the putative third common causes or previously unmeasured confounder. Other putative confounders would then be hypothesized and would need to be included in additional observational studies. To properly test the putative confounding by depressive symptom severity, future observational studies should examine initial depressive symptom severity prior to antidepressant use and then collect data on depressive symptom severity and antidepressant use as time-varying covariates to CVD outcomes. To test whether treatment-resistant depression is the phenotype driving the spurious observational association between antidepressant use and increased risk of CVD, the phenotype and its underlying causal mechanisms need to be better understood. Of course, rigorous and adequately powered randomized controlled trials of antidepressant use in patients with CVD would be a more straightforward way to test the observational association between antidepressant use and increased risk of CVD. We turn now to the recently published randomized controlled trials in this field.
NEW EVIDENCE FROM RANDOMIZED TRIALS IN PATIENTS WITH CVD AND DEPRESSION
Concerns have been voiced for some time about the ability to effectively treat depression and whether an effective depression treatment will affect the risk of CVD recurrence and mortality.8 Adding to these concerns is our limited knowledge of the causal pathways and behavioral and biologic mechanisms implicated in this risk association.1 For these reasons, results from new randomized controlled trials, such as the four summarized below, are important.
Rollman et al compared the effectiveness of telephone-delivered collaborative care (treatment group) and usual physician care (control group) for improving mental health quality of life and reducing depressive symptoms in 302 patients with depression after CABG.17 Patients were observed for 8 months following randomization. Mental health quality of life and depressive symptoms were both significantly improved in the treatment group relative to the control group. Significantly more patients in the treatment group had a 50% or greater reduction in depressive symptoms (50.0% vs 29.6% in control group, P < .001; number needed to treat = 4.9 [95% CI, 3.2–10.4]). Men particularly benefited from the treatment. This trial suggests that collaborative care can be delivered effectively (and potentially cost-efficiently) over the phone.
Freedland et al also evaluated depression treatment in 123 patients with major or minor depression who underwent CABG.18 Their primary objective was to determine the efficacy of two behavioral treatments (cognitive behavioral therapy [CBT] or supportive stress management) compared with usual care. Significantly more patients in both the CBT group (71%) and the stress management group (57%) had a low score (indicating less severe depressive symptoms) on the clinician-based Hamilton Rating Scale for Depression compared with the usual care group (33%). These results were maintained 6 months after the end of the trial. Secondary measures of depressive symptoms, anxiety, and quality of life were also significantly improved in the depression treatment groups compared with the usual care group. This trial is important for the following reasons:
The use of a second control group, the stress management group, represents a strict, high-quality design that controls for professional attention, generic or placebo therapy effect, and time or effort on the part of the patient.
The second control group also provides treatment options for the patient, as both CBT and stress management were beneficial.
Outcome assessors were blinded to treatment assignment, an important design feature in behavioral trials.
In a rigorously conducted randomized, double-blind, placebo-controlled trial, Carney et al tested whether 2 g/day of omega-3 acid ethyl esters (eicosapentaenoic acid [EPA] and docosahexaenoic acid [DHA]) improved depressive symptoms in 122 patients with major depression and CHD.19 Patients in both the omega-3 and placebo groups received sertraline (50 mg/day) during the 10-week trial. A deficiency of omega-3 fatty acids has been implicated in both depression and CHD and is a possible causal link between the two diseases. Also, there is some evidence that the efficacy of antidepressants is increased by the addition of omega-3 supplementation. Unfortunately, there were no differences in self-reported or clinician-assessed depressive symptoms or in predefined depression remission at the study’s end. The trial included a 2-week adherence run-in period, ensuring that medication adherence in the trial was excellent (97%), and concluded that omega-3 supplementation, at least at these dosage levels, does not improve depression outcomes.
In the Coronary Psychosocial Evaluation Studies (COPES) randomized controlled trial, Davidson et al compared 6 months of an enhanced care intervention with usual depression care among 157 patients with ACS and persistently elevated depressive symptoms.20 Under the enhanced care intervention, patients received either problem-solving therapy or antidepressant medication, depending on their preference, and then had the option of later augmentation with the other treatment, intensification of the initial treatment, or switching of treatments, if indicated by depressive symptom severity (stepped care). The purpose of the trial was to determine the acceptability and efficacy of depression treatment among patients with ACS, who often neither agree with the diagnosis of depression nor had been seeking treatment for depression. Significantly more patients were satisfied with their depression care in the intervention arm, and depressive symptoms and major adverse cardiac events (nonfatal MI, hospitalization for unstable angina, or all-cause mortality) were significantly reduced in the intervention arm compared with the usual care arm. The absolute numbers were very small; at the end of the trial, 3 patients in the intervention group and 10 patients in the usual care group had major adverse cardiac events (4% and 13%, respectively; log-rank test, χ21 = 3.93; P = .047). The results suggested that involving patients in the type of depression care they receive (medication and/or psychotherapy) and stepping treatments aggressively may be methods to improve the treatment of depression in patients with CHD. In addition, persistently depressed patients may be an interesting patient group to select for future trials, as usual care has resulted in large reductions in depressive symptoms in some previous trials but not in this study of patients with persistent depression.
Summary and clinical implications
Each of these four efficacy trials adds critical information to the evidence base. Depressed patients who have undergone CABG can be effectively treated in primary care settings with integrative care,17 and CBT is also extremely effective for these patients.18 Additional studies of omega-3 supplementation should not be pursued at this time, but using a run-in period to better identify patients who are prepared to engage in treatment is a prudent idea and should be used in future trials in this area.19 Patients with CHD and persistent depressive symptoms are a promising group to target for depression therapy, and asking patients to choose their type of depression treatment may improve response to therapy for both depression and CHD.20
DEPRESSION SCREENING, REFERRAL, AND TREATMENT IN PATIENTS WITH CVD
We finish with the least evidence-based and most controversial issue in the area of depression and CVD. This controversy started in 2008 when the American Heart Association recommended in an advisory (endorsed by the American Psychiatric Association) that “screening tests for depressive symptoms should be applied to identify patients who may require further assessment and treatment” if appropriate referral for further depression assessment and treatment is available.21 Partly in response to this advisory, Thombs et al conducted a systematic review of the evidence on whether screening or treatment improves outcomes of depression or CVD in patients with CVD.22 They found no trial that tested whether depression screening was beneficial in patients with CVD, and the randomized controlled trials of depression treatment provided evidence of only mild improvement of depressive symptoms and no improvement in CVD outcome. Therefore, they questioned whether routine depression screening was appropriate.22
In at least eight editorials, letters, and reviews published on this subject in 2009, investigators continued to debate this issue.23–30 Below we provide a simplified list of reasons presented for and against screening and subsequent treatment raised in these articles.
Arguments for depression screening and treatment
The proponents of screening contend that depression is highly prevalent in patients with CVD and is clearly a risk marker for increased adverse events, reduced quality of life, and poorer adherence to treatment.24 They argue that since there are plausible biologic and behavioral mechanisms for this association, and since SSRI use improves depressive symptoms in other patient populations and is safe in patients with CVD, health care providers should not hesitate to screen and refer patients for appropriate depression treatment. At the same time, they have cautioned that SSRIs interact with anti coagulants and that bleeding should be monitored closely in patients with CVD who are taking SSRIs.24
Whooley28 noted that although there are controversial findings in this area, depression screening provided in conjunction with collaborative care depression management is cost-effective and has a documented positive impact on depression, if not on CVD outcomes.17,31 She observed that there are some costs to screening, such as false-positive findings (resulting in stigma for patients incorrectly diagnosed) and diversion of resources from other health care needs. However, Whooley suggested that primary care providers, rather than cardiologists, should conduct depression screening and that patients should undergo screening only when an established collaborative care treatment protocol exists.28
Carney et al argued that depression, like age, clearly marks CVD risk, and that health care providers should aggressively treat readily modifiable CVD risk factors.23 They added that because of the strong association between depression and medication nonadherence,32 providers should carefully monitor patient adherence to life-saving therapies.
Taking another tack, Shemesh and colleagues advocated the importance of documenting the prevalence of suicidal ideation and intent if recommendations to screen for depression in CVD patient populations were implemented.25 Using a sample of more than 1,000 patients with CVD, they determined the prevalence of suicidal ideation (12.0%) and the number of patients who required hospitalization for risk of suicide (0.5%) when routine depression screening occurred in a large cardiology clinic. They concluded that identification and stabilization of imminently suicidal patients would be a benefit of universal screening and that there is a high societal cost to neglecting suicidal ideation, intent, and risk in patients with CVD. However, more patients would need immediate thorough psychiatric evaluations for safety, which would affect resource allocation and cost in cardiology clinics.
Arguments against depression screening and treatment
The main argument against screening for and treating depression in patients with CVD is that there are neither randomized controlled trials nor systematic evidence-based reviews showing that screening for depression and/or referring for additional treatment sufficiently improves outcomes for depression or CVD, and that existing evidence does not support the recommendation to screen all patients with CVD.22,30 Furthermore, antidepressant use is associated with only mild improvement in depressive symptoms, even in other patient populations,33 and publication bias (“the file-drawer problem”) has prevented the publication of antidepressant trials with null results, thereby skewing the evidence base.34 In addition, considerable health care resources would be needed to mount such a large screening effort, and these resources would come at the expense of other efforts. Finally, the adverse effects of medications and the inevitability of some false-positive screening results must be weighed against any benefit that might occur with universal screening.35
In addition to the arguments above, Ziegelstein et al,29 in commenting on the American Heart Association advisory,21 wryly observed that there is far greater observational evidence that depressed patients seen in mental health settings are at risk for incident and recurrent CVD and that there should be universal screening and referral for CVD in patients with depression. They contended as well that the evidence is insufficient to recommend that patients with CVD undergo universal depression screening and referral.
Summary and clinical implications
Although we were hesitant to raise this tense and often emotional issue, we are in favor of routine, algorithm-based depression screening by all cardiologists, with the critical proviso that a nationwide and/or Centers for Medicare and Medicaid Services–coordinated randomized controlled trial be conducted to evaluate this practice. All patients with pronounced depressive symptoms should be referred to the trial, and two depression treatments should be evaluated, such as usual referral versus telephone-based collaborative care17 or enhanced depression care.20 Such a trial would allow us to ensure that data are collected on the cost,36 the benefit, and even the possible harms associated with routine depression screening for patients with CVD, and we could ascertain if there is an acceptable, beneficial treatment for depression that can be delivered and definitively tested.
References
Carney RM, Freedland KE. Depression in patients with coronary heart disease. Am J Med2008; 121( 11 suppl 2):S20–S27.
Whang W, Kubzansky LD, Kawachi I, et al Depression and risk of sudden cardiac death and coronary heart disease in women: results from the Nurses’ Health Study. J Am Coll Cardiol2009; 53:950–958.
Krantz DS, Whittaker KS, Francis JL, et al Psychotropic medication use and risk of adverse cardiovascular events in women with suspected coronary artery disease: outcomes from the Women’s Ischemia Syndrome Evaluation (WISE) study. Heart2009; 95:1901–1906.
Smoller JW, Allison M, Cochrane BB, et al Antidepressant use and risk of incident cardiovascular morbidity and mortality among postmenopausal women in the Women’s Health Initiative study. Arch Intern Med2009; 169:2128–2139.
May HT, Horne BD, Carlquist JF, Sheng X, Joy E, Catinella AP. Depression after coronary artery disease is associated with heart failure. J Am Coll Cardiol2009; 53:1440–1447.
Kim DH, Daskalakis C, Whellan DJ, et al Safety of selective serotonin reuptake inhibitor in adults undergoing coronary artery bypass grafting. Am J Cardiol2009; 103:1391–1395.
Jolly K, Gammage MD, Cheng KK, Bradburn P, Banting MV, Langman MJ. Sudden death in patients receiving drugs tending to prolong the QT interval. Br J Clin Pharmacol2009; 68:743–751.
Jolly K, Langman MJ. Psychotropic medication: curing illness or creating problems?Heart2009; 95:1893–1894.
Waldman SV, Blumenthal JA, Babyak MA, et al Ethnic differences in the treatment of depression in patients with ischemic heart disease. Am Heart J2009; 157:77–83.
Salas M, Hofman A, Stricker BH. Confounding by indication: an example of variation in the use of epidemiologic terminology. Am J Epidemiol1999; 149:981–983.
Carney RM, Freedland KE. Treatment-resistant depression and sudden cardiac death [letter]. J Am Coll Cardiol2009; 54:958–959.
Carney RM, Freedland KE. Treatment-resistant depression and mortality after acute coronary syndrome. Am J Psychiatry2009; 166:410–417.
Carney RM, Blumenthal JA, Freedland KE, et al Depression and late mortality after myocardial infarction in the Enhancing Recovery in Coronary Heart Disease (ENRICHD) study. Psychosom Med2004; 66:466–474.
Berkman LF, Blumenthal J, Burg M, et al Effects of treating depression and low perceived social support on clinical events after myocardial infarction: the Enhancing Recovery in Coronary Heart Disease Patients (ENRICHD) randomized trial. JAMA2003; 289:3106–3116.
Glassman AH, Bigger JT, Gaffney M. Psychiatric characteristics associated with long-term mortality among 361 patients having an acute coronary syndrome and major depression: seven-year follow-up of SADHART participants. Arch Gen Psychiatry2009; 66:1022–1029.
Glassman AH, O’Connor CM, Califf RM, et al Sertraline treatment of major depression in patients with acute MI or unstable angina. JAMA2002; 288:701–709.
Rollman BL, Belnap BH, LeMenager MS, et al Telephone-delivered collaborative care for treating post-CABG depression: a randomized controlled trial. JAMA2009; 302:2095–2103.
Freedland KE, Skala JA, Carney RM, et al Treatment of depression after coronary artery bypass surgery: a randomized controlled trial. Arch Gen Psychiatry2009; 66:387–396.
Carney RM, Freedland KE, Rubin EH, Rich MW, Steinmeyer BC, Harris WS. Omega-3 augmentation of sertraline in treatment of depression in patients with coronary heart disease: a randomized controlled trial. JAMA2009; 302:1651–1657.
Davidson KW, Rieckmann N, Clemow L, et al Enhanced depression care for patients with acute coronary syndrome and persistent depressive symptoms: Coronary Psychosocial Evaluation Studies randomized controlled trial. Arch Intern Med2010; 170:600–608.
Lichtman JH, Bigger JT, Blumenthal JA, et al Depression and coronary heart disease: recommendations for screening, referral, and treatment: a science advisory from the American Heart Association Prevention Committee of the Council on Cardiovascular Nursing, Council on Clinical Cardiology, Council on Epidemiology and Prevention, and Interdisciplinary Council on Quality of Care and Outcomes Research: endorsed by the American Psychiatric Association. Circulation2008; 118:1768–1775.
Thombs BD, de Jonge P, Coyne JC, et al Depression screening and patient outcomes in cardiovascular care: a systematic review. JAMA2008; 300:2161–2171.
Carney RM, Freedland KE, Jaffe AS. Depression screening in patients with heart disease [letter]. JAMA2009; 301:1337–1338.
Pozuelo L, Zhang J, Franco K, Tesar G, Penn M, Jiang W. Depression and heart disease: what do we know, and where are we headed?Cleve Clin J Med2009; 76:59–70.
Shemesh E, Annunziato RA, Rubinstein D, et al Screening for depression and suicidality in patients with cardiovascular illnesses. Am J Cardiol2009; 104:1194–1197.
Thombs BD, Adeponle AB, Kirmayer LJ, Rousseau C, Ziegelstein RC. More antidepressants for African Americans with coronary heart disease? Maybe—maybe not [letter]. Am Heart J2009; 157:e33; author reply,e35–e37.
Whang W, Davidson KW. Is it time to treat depression in patients with cardiovascular disease?Circulation2009; 120:99–100.
Whooley MA. To screen or not to screen? Depression in patients with cardiovascular disease. J Am Coll Cardiol2009; 54:891–893.
Ziegelstein RC, Thombs BD, Coyne JC, de Jonge P. Routine screening for depression in patients with coronary heart disease: never mind. J Am Coll Cardiol2009; 54:886–890.
Thombs BD, Jewett LR, Knafo R, Coyne JC, Ziegelstein RC. Learning from history: a commentary on the American Heart Association Science Advisory on depression screening. Am Heart J2009; 158:503–505.
Unutzer J, Katon W, Callahan CM, et al Collaborative care management of late-life depression in the primary care setting: a randomized controlled trial. JAMA2002; 288:2836–2845.
Rieckmann N, Gerin W, Kronish IM, et al Course of depressive symptoms and medication adherence after acute coronary syndromes: an electronic medication monitoring study. J Am Coll Cardiol2006; 48:2218–2222.
Kirsch I, Deacon BJ, Huedo-Medina TB, et al Initial severity and antidepressant benefits: a meta-analysis of data submitted to the Food and Drug Administration. PLoS Med2008; 5:e45.
Turner EH, Matthews AM, Linardatos E, Tell RA, Rosenthal R. Selective publication of antidepressant trials and its influence on apparent efficacy. N Engl J Med2008; 358:252–260.
Thombs BD, de Jonge P, Ziegelstein RC. Depression screening in patients with heart disease [reply to letter]. JAMA2009; 301:1338.
Frasure-Smith N, Lespérance F. Depression and cardiac risk: present status and future directions. Heart2010; 96:173–176.
Karina W. Davidson, PhD Department of Medicine, Columbia University College of Physicians and Surgeons, New York, NY
Maya Rom Korin, PhD Department of Medicine, Columbia University College of Physicians and Surgeons, New York, NY
Correspondence: Karina W. Davidson, PhD, Department of Medicine, Columbia University College of Physicians and Surgeons, Room 948, PH9 Center, 622 W. 168th St, New York, NY 10032; [email protected]
Drs. Davidson and Korin reported that they have no financial relationships that pose a potential conflict of interest with this article.
This work was supported by grants HL-088117, HC-25197, HL-076857, HL-080665, and HL-084034 from the National Institutes of Health, Bethesda, MD, and by grant UL1 RR024156 from the National Center for Research Resources, Bethesda, MD, a component of the National Institutes of Health and National Institutes of Health Roadmap for Medical Research.
Karina W. Davidson, PhD Department of Medicine, Columbia University College of Physicians and Surgeons, New York, NY
Maya Rom Korin, PhD Department of Medicine, Columbia University College of Physicians and Surgeons, New York, NY
Correspondence: Karina W. Davidson, PhD, Department of Medicine, Columbia University College of Physicians and Surgeons, Room 948, PH9 Center, 622 W. 168th St, New York, NY 10032; [email protected]
Drs. Davidson and Korin reported that they have no financial relationships that pose a potential conflict of interest with this article.
This work was supported by grants HL-088117, HC-25197, HL-076857, HL-080665, and HL-084034 from the National Institutes of Health, Bethesda, MD, and by grant UL1 RR024156 from the National Center for Research Resources, Bethesda, MD, a component of the National Institutes of Health and National Institutes of Health Roadmap for Medical Research.
Author and Disclosure Information
Karina W. Davidson, PhD Department of Medicine, Columbia University College of Physicians and Surgeons, New York, NY
Maya Rom Korin, PhD Department of Medicine, Columbia University College of Physicians and Surgeons, New York, NY
Correspondence: Karina W. Davidson, PhD, Department of Medicine, Columbia University College of Physicians and Surgeons, Room 948, PH9 Center, 622 W. 168th St, New York, NY 10032; [email protected]
Drs. Davidson and Korin reported that they have no financial relationships that pose a potential conflict of interest with this article.
This work was supported by grants HL-088117, HC-25197, HL-076857, HL-080665, and HL-084034 from the National Institutes of Health, Bethesda, MD, and by grant UL1 RR024156 from the National Center for Research Resources, Bethesda, MD, a component of the National Institutes of Health and National Institutes of Health Roadmap for Medical Research.
Many advances in the understanding of the relationship between depression and cardiovascular disease (CVD) were reported in 2009. As the study of this relationship encompasses cardiology, psychiatry, behavioral medicine, and many other fields, it is difficult to keep abreast of new developments. Relevant papers are found in a variety of journals. Therefore, we systematically searched the empirical research on depression and CVD published in English in 2009. Our search yielded nearly 500 articles. We review here a few of the most provocative and potentially influential findings. We begin with an overview of the methodology of our systematic review and then summarize the key findings and controversies we identified from the 2009 literature before exploring each key finding in detail.
METHODOLOGY OF OUR SYSTEMATIC SEARCH AND RATIONALE FOR FINDINGS REVIEWED
Previous evidence demonstrated that the presence of depressive symptoms or a diagnosis of a depressive disorder predicts poor prognosis and reduced survival rates after any coronary artery disease diagnosis, including myocardial infarction (MI) and unstable angina, as well as after coronary artery bypass graft surgery (CABG).1
We aimed to see how this evidence base was expanded in 2009 by using a systematic search strategy to retrieve the most relevant articles about depression and coronary heart disease (CHD) from the MEDLINE and Psyc-INFO (Ovid interface) databases. The most relevant subject headings and free text terms were identified and combined with “or.” The two sets were then combined with “and.” Terms included “depression,” “depressive disorder,” “depress$,” “coronary artery disease” (CAD) “coronary disease,” “acute coronary syndrome” (ACS), “cardiovascular disease” (CVD), “coronary heart disease,” and “heart diseas$.” The final set was limited to the English-language literature and identified 494 unique articles published during 2009.
Closer inspection of titles and abstracts revealed well more than 100 articles directly relevant to the science and management of patients with CVD and depression or pronounced depressive symptoms. In light of this quantity, a thorough review of all new findings, editorials, and reviews is not feasible. Thus, we review here a few exciting articles in several distinct topical areas that could influence views of the relationship between depression and CVD as well as how we screen for and treat depression in patients with CVD. As with any review that is not strictly evidence-based, our choice of articles is subjective and incomplete, but we hope it will stimulate discussion and further exploration.
SUMMARY OF KEY FINDINGS AND CONTROVERSIES
The following numbered topics emerged as common themes or controversies from our survey of the 2009 literature. The remainder of this article will review findings in each of these areas in detail and discuss important clinical implications as appropriate.
1. Antidepressant use and adverse cardiac events. In surprising findings, antidepressant use was associated with increased risk of incident stroke, CVD, and sudden cardiac death in multiple large observational cohort studies. It is not known if the association is caused by unmeasured confounders or depressive symptom severity, although controlling for symptom severity did not reduce this elevated risk in some studies. Another interesting possibility is that the treatment-resistant depressed patient is at particularly high risk of CVD and death.
2. Effects of depression intervention in CVD patients. Four exciting randomized controlled trials on depression intervention in patients with CVD reported important efficacy results and suggested future directions for larger, definitive trials of depression treatment for these patients.
3. Depression screening and treatment in CVD patients. In the absence of large randomized controlled trials, the debate continues on whether depression screening or any type of depression treatment is beneficial, harmless, or harmful to patients with CVD. Continuation of this debate does not serve the health and well-being of patients or the public. Less controversial—and thus less discussed—is the important insight that psychiatric patients with depression should be routinely screened for cardiac disease and risk factors, as they are clearly at risk of CVD. We await clinical trials in this area to ensure that screening leads to improved CVD outcomes.
OBSERVATIONAL EVIDENCE ON ANTIDEPRESSANT USE AND CVD OUTCOMES
In an analysis of the Nurses’ Health Study, Whang et al examined the relationship of depressive symptoms and antidepressant use with sudden cardiac death and adverse cardiac events in 63,469 women without CVD.2 Depressive symptoms were assessed using the Mental Health Index, a five-item subscale of the Short Form-36 Health Survey. Among women who reported antidepressant use, most (61%) were taking a selective serotonin reuptake inhibitor (SSRI; sertraline, fluoxetine, paroxetine, or citalopram), while 39% reported use of other antidepressants. Women taking antidepressants were more likely to suffer sudden cardiac death, with a fully adjusted hazard ratio (HR) of 3.34 (95% confidence interval [CI], 2.03–5.50).
Krantz et al examined psychotropic medication use and risk of adverse cardiovascular events in 519 women from the Women’s Ischemia Syndrome Evaluation.3 Enrolled women underwent coronary angiography, were separated into four groups according to their psychotropic medication use (none, anxiolytics only, antidepressants only, or both anxiolytics and antidepressants), and were observed for a median of 5.9 years. Results revealed that women who received both medications had a higher risk for adverse cardiovascular events and higher all-cause mortality compared with those using neither medication, even after controlling for anxious and depressive symptoms. In addition, whereas the use of antidepressant medication was associated with a doubling of risk for subsequent CVD events (HR = 2.16; 95% CI, 1.21–3.93) and all-cause mortality (HR = 2.15; 95% CI, 1.16–3.98), use of anxiolytic medication alone was not. While this study did not examine cause of death, cardiac death is likely to have constituted a large proportion of total mortality in this cohort selected for likelihood of CAD.
Although CVD and death have long been outcomes of interest for those studying the effects of depression, additional end points have recently been investigated as well. In a prospective cohort study of 136,293 community-dwelling postmenopausal women in the Women’s Health Initiative, Smoller et al found that new antidepressant use was significantly associated with increased incidence of stroke and all-cause mortality but not with incidence of CHD.4 The rate of stroke per 1,000 person-years was 2.99 for subjects with no antidepressant use versus 4.16 for patients with new SSRI use; the rate of all-cause death was 7.79 versus 12.77, respectively. The rate of all-cause death for subjects with new tricyclic antidepressant use was 14.14 per 1,000 person-years. To address potential confounding by indication, the researchers obtained a propensity score from a logistic regression model to predict any new antidepressant use from demographic, lifestyle, risk factor, and comorbidity variables measured at baseline. New SSRI use was associated with a doubling of the risk of incident hemorrhagic stroke as well as fatal stroke. There were no significant interactions between use of SSRIs and use of statins or aspirin in terms of risk of hemorrhagic stroke.
In an interesting observational study of 7,709 patients with confirmed CAD but without a diagnosis of heart failure or depression (and without current antidepressant use), May et al found that a subsequent diagnosis of depression was associated with a significant 50% increase in the risk of heart failure.5 There was no difference, however, between depressed patients who were using antidepressants and those who were not.
Increased risk of bleeding with SSRI use, particularly in patients with CAD, has also been a concern. Kim et al evaluated 1,380 adults who received any anti depressant before CABG for in-hospital mortality or any bleeding events.6 After controlling for the percentage of patients taking SSRIs (78%), there were no significant differences between those taking SSRIs and those taking non-SSRIs in the rate of any bleeding events (6.5% vs 7.2%; odds ratio [OR] = 0.93; 95% CI, 0.50–1.76) or in-hospital mortality (3.1% vs 2.3%; OR = 0.88; 95% CI, 0.47–1.65). There was no increased risk of bleeding associated with SSRI use when the analysis was restricted to patients who received antiplatelet and anticoagulant therapy. Thus, compared with patients who received non-SSRI antidepressants, patients who received SSRIs preoperatively had no increased risk of bleeding or in-hospital mortality after CABG; however, this study did not evaluate the effect of no antidepressant use.
Another study hypothesized that the use of any drug with the potential to prolong cardiac repolarization would be associated with an increased risk of sudden death.7 Use of individual drugs was analyzed among 1,010 cases of sudden unexplained death and 3,030 living primary care controls, all from the community. SSRI use was associated with a doubling of risk of sudden death (OR = 2.21; 95% CI, 1.61–3.05), and tricyclic antidepressant use was associated with a nonsignificant trend toward increased risk (OR = 1.44; 95% CI, 0.96–2.13). Further analysis that stratified patients according to prior CVD showed that most of the association of SSRIs with sudden death was in those with existing CVD and not in those without CVD. Other drugs found to raise sudden death risk included the typical and atypical antipsychotics.
Summary and clinical implications
The Nurses’ Health Study analysis by Whang et al suggested that antidepressant use triples the risk of sudden cardiac death in healthy women, and the authors suggested that the association between fatal ventricular arrhythmias and antidepressant use be examined further. 2 The analysis of the Women’s Health Initiative by Smoller et al found that use of SSRIs and tricyclic antidepressants doubled the risk of fatal stroke in healthy women.4 The analysis of the Women’s Ischemia Syndrome Evaluation by Krantz et al revealed a doubling of the risk of CVD and death in women taking antidepressants who had been referred for coronary angiography.3 At the same time, May et al found no increase in the risk of heart failure conversion with antidepressant use in patients with CAD,5 and Kim et al found no increase in the risk of bleeding with use of SSRIs compared with non-SSRI antidepressants in patients with CAD undergoing CABG.6 Finally, in a population- and community-based case-control study, SSRI use was associated with an increased risk of sudden death, particularly in patients with CVD.7 So what are we to make of these findings?8
In all observational studies (including those reviewed above), unmeasured confounders pose a threat to the validity of any causal conclusions. A study recently tested some of the proposed confounders that might have existed in the above studies. Waldman et al examined racial differences in depressive symptoms and antidepressant treatment among a cohort of 864 consecutive patients with CHD undergoing diagnostic coronary angiography (727 white and 137 African American).9 While levels of depression were similar between the white and African American patients, the African Americans were less likely than their white counter-parts to receive antidepressant medications. Patients with only some high school, men, and patients with more severe depressive symptoms were significantly more likely to receive a prescription for antidepressants. Clearly, low education, male sex, and elevated depressive symptoms are related to poor prognosis for CHD, and the simple interpretation that antidepressant use is causing poorer outcomes is problematic.8
Two additional interpretations of the observational findings should be considered. First, confounding by indication (depressive symptom severity) might exist in these studies.10 In other words, patients who are prescribed antidepressant medication may be those with the most severe depressive illness, and it could be this severity, rather than the antidepressant use, that is causally implicated in the CVD incidence.11 However, all of the studies reviewed above either directly controlled for depressive symptom severity (at least as obtained at baseline) or used propensity scores or stratified subjects based on depression severity. The results showed an increased risk among those who were taking antidepressants. However, none of the studies examined depressive symptom severity during or at the end of the study or depression diagnosis and severity before antidepressant use; these data are needed for a clearer understanding of whether the results were confounded by indication.
Second, these findings are also consistent with a treatment-resistant depression phenotype.12 Krantz et al caution that it is not clear from their observational study3 whether medication use itself or depression refractory to treatment is implicated in the increased risk of CVD events and mortality. Depression that is refractory to treatment may be the type of depression that places patients at risk for sudden death, stroke, or CHD recurrence, so it may not be the antidepressant use per se that is associated with this risk. This phenomenon was documented in a secondary analysis13 of the largest-to-date randomized controlled trial of patients with MI undergoing treatment for depression (ENRICHD).14 It showed that those whose depressive symptoms did not respond to treatment had a higher risk of late mortality (ie, death ≥ 6 months after acute MI). This finding was replicated in 2009 in an important follow-up15 of the SADHART trial16; among patients with MI and major depression, treatment-resistant depression (ie, depression that failed to improve substantially during treatment with either sertraline or placebo) was strongly and independently associated with long-term mortality (HR = 2.39; 95% CI, 1.39–2.44; P < .001).15
What is needed next? Testing the alternative hypothesis— ie, that an unmeasured confounder may exist—is difficult, requiring new observational studies and measurement of the putative third common causes or previously unmeasured confounder. Other putative confounders would then be hypothesized and would need to be included in additional observational studies. To properly test the putative confounding by depressive symptom severity, future observational studies should examine initial depressive symptom severity prior to antidepressant use and then collect data on depressive symptom severity and antidepressant use as time-varying covariates to CVD outcomes. To test whether treatment-resistant depression is the phenotype driving the spurious observational association between antidepressant use and increased risk of CVD, the phenotype and its underlying causal mechanisms need to be better understood. Of course, rigorous and adequately powered randomized controlled trials of antidepressant use in patients with CVD would be a more straightforward way to test the observational association between antidepressant use and increased risk of CVD. We turn now to the recently published randomized controlled trials in this field.
NEW EVIDENCE FROM RANDOMIZED TRIALS IN PATIENTS WITH CVD AND DEPRESSION
Concerns have been voiced for some time about the ability to effectively treat depression and whether an effective depression treatment will affect the risk of CVD recurrence and mortality.8 Adding to these concerns is our limited knowledge of the causal pathways and behavioral and biologic mechanisms implicated in this risk association.1 For these reasons, results from new randomized controlled trials, such as the four summarized below, are important.
Rollman et al compared the effectiveness of telephone-delivered collaborative care (treatment group) and usual physician care (control group) for improving mental health quality of life and reducing depressive symptoms in 302 patients with depression after CABG.17 Patients were observed for 8 months following randomization. Mental health quality of life and depressive symptoms were both significantly improved in the treatment group relative to the control group. Significantly more patients in the treatment group had a 50% or greater reduction in depressive symptoms (50.0% vs 29.6% in control group, P < .001; number needed to treat = 4.9 [95% CI, 3.2–10.4]). Men particularly benefited from the treatment. This trial suggests that collaborative care can be delivered effectively (and potentially cost-efficiently) over the phone.
Freedland et al also evaluated depression treatment in 123 patients with major or minor depression who underwent CABG.18 Their primary objective was to determine the efficacy of two behavioral treatments (cognitive behavioral therapy [CBT] or supportive stress management) compared with usual care. Significantly more patients in both the CBT group (71%) and the stress management group (57%) had a low score (indicating less severe depressive symptoms) on the clinician-based Hamilton Rating Scale for Depression compared with the usual care group (33%). These results were maintained 6 months after the end of the trial. Secondary measures of depressive symptoms, anxiety, and quality of life were also significantly improved in the depression treatment groups compared with the usual care group. This trial is important for the following reasons:
The use of a second control group, the stress management group, represents a strict, high-quality design that controls for professional attention, generic or placebo therapy effect, and time or effort on the part of the patient.
The second control group also provides treatment options for the patient, as both CBT and stress management were beneficial.
Outcome assessors were blinded to treatment assignment, an important design feature in behavioral trials.
In a rigorously conducted randomized, double-blind, placebo-controlled trial, Carney et al tested whether 2 g/day of omega-3 acid ethyl esters (eicosapentaenoic acid [EPA] and docosahexaenoic acid [DHA]) improved depressive symptoms in 122 patients with major depression and CHD.19 Patients in both the omega-3 and placebo groups received sertraline (50 mg/day) during the 10-week trial. A deficiency of omega-3 fatty acids has been implicated in both depression and CHD and is a possible causal link between the two diseases. Also, there is some evidence that the efficacy of antidepressants is increased by the addition of omega-3 supplementation. Unfortunately, there were no differences in self-reported or clinician-assessed depressive symptoms or in predefined depression remission at the study’s end. The trial included a 2-week adherence run-in period, ensuring that medication adherence in the trial was excellent (97%), and concluded that omega-3 supplementation, at least at these dosage levels, does not improve depression outcomes.
In the Coronary Psychosocial Evaluation Studies (COPES) randomized controlled trial, Davidson et al compared 6 months of an enhanced care intervention with usual depression care among 157 patients with ACS and persistently elevated depressive symptoms.20 Under the enhanced care intervention, patients received either problem-solving therapy or antidepressant medication, depending on their preference, and then had the option of later augmentation with the other treatment, intensification of the initial treatment, or switching of treatments, if indicated by depressive symptom severity (stepped care). The purpose of the trial was to determine the acceptability and efficacy of depression treatment among patients with ACS, who often neither agree with the diagnosis of depression nor had been seeking treatment for depression. Significantly more patients were satisfied with their depression care in the intervention arm, and depressive symptoms and major adverse cardiac events (nonfatal MI, hospitalization for unstable angina, or all-cause mortality) were significantly reduced in the intervention arm compared with the usual care arm. The absolute numbers were very small; at the end of the trial, 3 patients in the intervention group and 10 patients in the usual care group had major adverse cardiac events (4% and 13%, respectively; log-rank test, χ21 = 3.93; P = .047). The results suggested that involving patients in the type of depression care they receive (medication and/or psychotherapy) and stepping treatments aggressively may be methods to improve the treatment of depression in patients with CHD. In addition, persistently depressed patients may be an interesting patient group to select for future trials, as usual care has resulted in large reductions in depressive symptoms in some previous trials but not in this study of patients with persistent depression.
Summary and clinical implications
Each of these four efficacy trials adds critical information to the evidence base. Depressed patients who have undergone CABG can be effectively treated in primary care settings with integrative care,17 and CBT is also extremely effective for these patients.18 Additional studies of omega-3 supplementation should not be pursued at this time, but using a run-in period to better identify patients who are prepared to engage in treatment is a prudent idea and should be used in future trials in this area.19 Patients with CHD and persistent depressive symptoms are a promising group to target for depression therapy, and asking patients to choose their type of depression treatment may improve response to therapy for both depression and CHD.20
DEPRESSION SCREENING, REFERRAL, AND TREATMENT IN PATIENTS WITH CVD
We finish with the least evidence-based and most controversial issue in the area of depression and CVD. This controversy started in 2008 when the American Heart Association recommended in an advisory (endorsed by the American Psychiatric Association) that “screening tests for depressive symptoms should be applied to identify patients who may require further assessment and treatment” if appropriate referral for further depression assessment and treatment is available.21 Partly in response to this advisory, Thombs et al conducted a systematic review of the evidence on whether screening or treatment improves outcomes of depression or CVD in patients with CVD.22 They found no trial that tested whether depression screening was beneficial in patients with CVD, and the randomized controlled trials of depression treatment provided evidence of only mild improvement of depressive symptoms and no improvement in CVD outcome. Therefore, they questioned whether routine depression screening was appropriate.22
In at least eight editorials, letters, and reviews published on this subject in 2009, investigators continued to debate this issue.23–30 Below we provide a simplified list of reasons presented for and against screening and subsequent treatment raised in these articles.
Arguments for depression screening and treatment
The proponents of screening contend that depression is highly prevalent in patients with CVD and is clearly a risk marker for increased adverse events, reduced quality of life, and poorer adherence to treatment.24 They argue that since there are plausible biologic and behavioral mechanisms for this association, and since SSRI use improves depressive symptoms in other patient populations and is safe in patients with CVD, health care providers should not hesitate to screen and refer patients for appropriate depression treatment. At the same time, they have cautioned that SSRIs interact with anti coagulants and that bleeding should be monitored closely in patients with CVD who are taking SSRIs.24
Whooley28 noted that although there are controversial findings in this area, depression screening provided in conjunction with collaborative care depression management is cost-effective and has a documented positive impact on depression, if not on CVD outcomes.17,31 She observed that there are some costs to screening, such as false-positive findings (resulting in stigma for patients incorrectly diagnosed) and diversion of resources from other health care needs. However, Whooley suggested that primary care providers, rather than cardiologists, should conduct depression screening and that patients should undergo screening only when an established collaborative care treatment protocol exists.28
Carney et al argued that depression, like age, clearly marks CVD risk, and that health care providers should aggressively treat readily modifiable CVD risk factors.23 They added that because of the strong association between depression and medication nonadherence,32 providers should carefully monitor patient adherence to life-saving therapies.
Taking another tack, Shemesh and colleagues advocated the importance of documenting the prevalence of suicidal ideation and intent if recommendations to screen for depression in CVD patient populations were implemented.25 Using a sample of more than 1,000 patients with CVD, they determined the prevalence of suicidal ideation (12.0%) and the number of patients who required hospitalization for risk of suicide (0.5%) when routine depression screening occurred in a large cardiology clinic. They concluded that identification and stabilization of imminently suicidal patients would be a benefit of universal screening and that there is a high societal cost to neglecting suicidal ideation, intent, and risk in patients with CVD. However, more patients would need immediate thorough psychiatric evaluations for safety, which would affect resource allocation and cost in cardiology clinics.
Arguments against depression screening and treatment
The main argument against screening for and treating depression in patients with CVD is that there are neither randomized controlled trials nor systematic evidence-based reviews showing that screening for depression and/or referring for additional treatment sufficiently improves outcomes for depression or CVD, and that existing evidence does not support the recommendation to screen all patients with CVD.22,30 Furthermore, antidepressant use is associated with only mild improvement in depressive symptoms, even in other patient populations,33 and publication bias (“the file-drawer problem”) has prevented the publication of antidepressant trials with null results, thereby skewing the evidence base.34 In addition, considerable health care resources would be needed to mount such a large screening effort, and these resources would come at the expense of other efforts. Finally, the adverse effects of medications and the inevitability of some false-positive screening results must be weighed against any benefit that might occur with universal screening.35
In addition to the arguments above, Ziegelstein et al,29 in commenting on the American Heart Association advisory,21 wryly observed that there is far greater observational evidence that depressed patients seen in mental health settings are at risk for incident and recurrent CVD and that there should be universal screening and referral for CVD in patients with depression. They contended as well that the evidence is insufficient to recommend that patients with CVD undergo universal depression screening and referral.
Summary and clinical implications
Although we were hesitant to raise this tense and often emotional issue, we are in favor of routine, algorithm-based depression screening by all cardiologists, with the critical proviso that a nationwide and/or Centers for Medicare and Medicaid Services–coordinated randomized controlled trial be conducted to evaluate this practice. All patients with pronounced depressive symptoms should be referred to the trial, and two depression treatments should be evaluated, such as usual referral versus telephone-based collaborative care17 or enhanced depression care.20 Such a trial would allow us to ensure that data are collected on the cost,36 the benefit, and even the possible harms associated with routine depression screening for patients with CVD, and we could ascertain if there is an acceptable, beneficial treatment for depression that can be delivered and definitively tested.
Many advances in the understanding of the relationship between depression and cardiovascular disease (CVD) were reported in 2009. As the study of this relationship encompasses cardiology, psychiatry, behavioral medicine, and many other fields, it is difficult to keep abreast of new developments. Relevant papers are found in a variety of journals. Therefore, we systematically searched the empirical research on depression and CVD published in English in 2009. Our search yielded nearly 500 articles. We review here a few of the most provocative and potentially influential findings. We begin with an overview of the methodology of our systematic review and then summarize the key findings and controversies we identified from the 2009 literature before exploring each key finding in detail.
METHODOLOGY OF OUR SYSTEMATIC SEARCH AND RATIONALE FOR FINDINGS REVIEWED
Previous evidence demonstrated that the presence of depressive symptoms or a diagnosis of a depressive disorder predicts poor prognosis and reduced survival rates after any coronary artery disease diagnosis, including myocardial infarction (MI) and unstable angina, as well as after coronary artery bypass graft surgery (CABG).1
We aimed to see how this evidence base was expanded in 2009 by using a systematic search strategy to retrieve the most relevant articles about depression and coronary heart disease (CHD) from the MEDLINE and Psyc-INFO (Ovid interface) databases. The most relevant subject headings and free text terms were identified and combined with “or.” The two sets were then combined with “and.” Terms included “depression,” “depressive disorder,” “depress$,” “coronary artery disease” (CAD) “coronary disease,” “acute coronary syndrome” (ACS), “cardiovascular disease” (CVD), “coronary heart disease,” and “heart diseas$.” The final set was limited to the English-language literature and identified 494 unique articles published during 2009.
Closer inspection of titles and abstracts revealed well more than 100 articles directly relevant to the science and management of patients with CVD and depression or pronounced depressive symptoms. In light of this quantity, a thorough review of all new findings, editorials, and reviews is not feasible. Thus, we review here a few exciting articles in several distinct topical areas that could influence views of the relationship between depression and CVD as well as how we screen for and treat depression in patients with CVD. As with any review that is not strictly evidence-based, our choice of articles is subjective and incomplete, but we hope it will stimulate discussion and further exploration.
SUMMARY OF KEY FINDINGS AND CONTROVERSIES
The following numbered topics emerged as common themes or controversies from our survey of the 2009 literature. The remainder of this article will review findings in each of these areas in detail and discuss important clinical implications as appropriate.
1. Antidepressant use and adverse cardiac events. In surprising findings, antidepressant use was associated with increased risk of incident stroke, CVD, and sudden cardiac death in multiple large observational cohort studies. It is not known if the association is caused by unmeasured confounders or depressive symptom severity, although controlling for symptom severity did not reduce this elevated risk in some studies. Another interesting possibility is that the treatment-resistant depressed patient is at particularly high risk of CVD and death.
2. Effects of depression intervention in CVD patients. Four exciting randomized controlled trials on depression intervention in patients with CVD reported important efficacy results and suggested future directions for larger, definitive trials of depression treatment for these patients.
3. Depression screening and treatment in CVD patients. In the absence of large randomized controlled trials, the debate continues on whether depression screening or any type of depression treatment is beneficial, harmless, or harmful to patients with CVD. Continuation of this debate does not serve the health and well-being of patients or the public. Less controversial—and thus less discussed—is the important insight that psychiatric patients with depression should be routinely screened for cardiac disease and risk factors, as they are clearly at risk of CVD. We await clinical trials in this area to ensure that screening leads to improved CVD outcomes.
OBSERVATIONAL EVIDENCE ON ANTIDEPRESSANT USE AND CVD OUTCOMES
In an analysis of the Nurses’ Health Study, Whang et al examined the relationship of depressive symptoms and antidepressant use with sudden cardiac death and adverse cardiac events in 63,469 women without CVD.2 Depressive symptoms were assessed using the Mental Health Index, a five-item subscale of the Short Form-36 Health Survey. Among women who reported antidepressant use, most (61%) were taking a selective serotonin reuptake inhibitor (SSRI; sertraline, fluoxetine, paroxetine, or citalopram), while 39% reported use of other antidepressants. Women taking antidepressants were more likely to suffer sudden cardiac death, with a fully adjusted hazard ratio (HR) of 3.34 (95% confidence interval [CI], 2.03–5.50).
Krantz et al examined psychotropic medication use and risk of adverse cardiovascular events in 519 women from the Women’s Ischemia Syndrome Evaluation.3 Enrolled women underwent coronary angiography, were separated into four groups according to their psychotropic medication use (none, anxiolytics only, antidepressants only, or both anxiolytics and antidepressants), and were observed for a median of 5.9 years. Results revealed that women who received both medications had a higher risk for adverse cardiovascular events and higher all-cause mortality compared with those using neither medication, even after controlling for anxious and depressive symptoms. In addition, whereas the use of antidepressant medication was associated with a doubling of risk for subsequent CVD events (HR = 2.16; 95% CI, 1.21–3.93) and all-cause mortality (HR = 2.15; 95% CI, 1.16–3.98), use of anxiolytic medication alone was not. While this study did not examine cause of death, cardiac death is likely to have constituted a large proportion of total mortality in this cohort selected for likelihood of CAD.
Although CVD and death have long been outcomes of interest for those studying the effects of depression, additional end points have recently been investigated as well. In a prospective cohort study of 136,293 community-dwelling postmenopausal women in the Women’s Health Initiative, Smoller et al found that new antidepressant use was significantly associated with increased incidence of stroke and all-cause mortality but not with incidence of CHD.4 The rate of stroke per 1,000 person-years was 2.99 for subjects with no antidepressant use versus 4.16 for patients with new SSRI use; the rate of all-cause death was 7.79 versus 12.77, respectively. The rate of all-cause death for subjects with new tricyclic antidepressant use was 14.14 per 1,000 person-years. To address potential confounding by indication, the researchers obtained a propensity score from a logistic regression model to predict any new antidepressant use from demographic, lifestyle, risk factor, and comorbidity variables measured at baseline. New SSRI use was associated with a doubling of the risk of incident hemorrhagic stroke as well as fatal stroke. There were no significant interactions between use of SSRIs and use of statins or aspirin in terms of risk of hemorrhagic stroke.
In an interesting observational study of 7,709 patients with confirmed CAD but without a diagnosis of heart failure or depression (and without current antidepressant use), May et al found that a subsequent diagnosis of depression was associated with a significant 50% increase in the risk of heart failure.5 There was no difference, however, between depressed patients who were using antidepressants and those who were not.
Increased risk of bleeding with SSRI use, particularly in patients with CAD, has also been a concern. Kim et al evaluated 1,380 adults who received any anti depressant before CABG for in-hospital mortality or any bleeding events.6 After controlling for the percentage of patients taking SSRIs (78%), there were no significant differences between those taking SSRIs and those taking non-SSRIs in the rate of any bleeding events (6.5% vs 7.2%; odds ratio [OR] = 0.93; 95% CI, 0.50–1.76) or in-hospital mortality (3.1% vs 2.3%; OR = 0.88; 95% CI, 0.47–1.65). There was no increased risk of bleeding associated with SSRI use when the analysis was restricted to patients who received antiplatelet and anticoagulant therapy. Thus, compared with patients who received non-SSRI antidepressants, patients who received SSRIs preoperatively had no increased risk of bleeding or in-hospital mortality after CABG; however, this study did not evaluate the effect of no antidepressant use.
Another study hypothesized that the use of any drug with the potential to prolong cardiac repolarization would be associated with an increased risk of sudden death.7 Use of individual drugs was analyzed among 1,010 cases of sudden unexplained death and 3,030 living primary care controls, all from the community. SSRI use was associated with a doubling of risk of sudden death (OR = 2.21; 95% CI, 1.61–3.05), and tricyclic antidepressant use was associated with a nonsignificant trend toward increased risk (OR = 1.44; 95% CI, 0.96–2.13). Further analysis that stratified patients according to prior CVD showed that most of the association of SSRIs with sudden death was in those with existing CVD and not in those without CVD. Other drugs found to raise sudden death risk included the typical and atypical antipsychotics.
Summary and clinical implications
The Nurses’ Health Study analysis by Whang et al suggested that antidepressant use triples the risk of sudden cardiac death in healthy women, and the authors suggested that the association between fatal ventricular arrhythmias and antidepressant use be examined further. 2 The analysis of the Women’s Health Initiative by Smoller et al found that use of SSRIs and tricyclic antidepressants doubled the risk of fatal stroke in healthy women.4 The analysis of the Women’s Ischemia Syndrome Evaluation by Krantz et al revealed a doubling of the risk of CVD and death in women taking antidepressants who had been referred for coronary angiography.3 At the same time, May et al found no increase in the risk of heart failure conversion with antidepressant use in patients with CAD,5 and Kim et al found no increase in the risk of bleeding with use of SSRIs compared with non-SSRI antidepressants in patients with CAD undergoing CABG.6 Finally, in a population- and community-based case-control study, SSRI use was associated with an increased risk of sudden death, particularly in patients with CVD.7 So what are we to make of these findings?8
In all observational studies (including those reviewed above), unmeasured confounders pose a threat to the validity of any causal conclusions. A study recently tested some of the proposed confounders that might have existed in the above studies. Waldman et al examined racial differences in depressive symptoms and antidepressant treatment among a cohort of 864 consecutive patients with CHD undergoing diagnostic coronary angiography (727 white and 137 African American).9 While levels of depression were similar between the white and African American patients, the African Americans were less likely than their white counter-parts to receive antidepressant medications. Patients with only some high school, men, and patients with more severe depressive symptoms were significantly more likely to receive a prescription for antidepressants. Clearly, low education, male sex, and elevated depressive symptoms are related to poor prognosis for CHD, and the simple interpretation that antidepressant use is causing poorer outcomes is problematic.8
Two additional interpretations of the observational findings should be considered. First, confounding by indication (depressive symptom severity) might exist in these studies.10 In other words, patients who are prescribed antidepressant medication may be those with the most severe depressive illness, and it could be this severity, rather than the antidepressant use, that is causally implicated in the CVD incidence.11 However, all of the studies reviewed above either directly controlled for depressive symptom severity (at least as obtained at baseline) or used propensity scores or stratified subjects based on depression severity. The results showed an increased risk among those who were taking antidepressants. However, none of the studies examined depressive symptom severity during or at the end of the study or depression diagnosis and severity before antidepressant use; these data are needed for a clearer understanding of whether the results were confounded by indication.
Second, these findings are also consistent with a treatment-resistant depression phenotype.12 Krantz et al caution that it is not clear from their observational study3 whether medication use itself or depression refractory to treatment is implicated in the increased risk of CVD events and mortality. Depression that is refractory to treatment may be the type of depression that places patients at risk for sudden death, stroke, or CHD recurrence, so it may not be the antidepressant use per se that is associated with this risk. This phenomenon was documented in a secondary analysis13 of the largest-to-date randomized controlled trial of patients with MI undergoing treatment for depression (ENRICHD).14 It showed that those whose depressive symptoms did not respond to treatment had a higher risk of late mortality (ie, death ≥ 6 months after acute MI). This finding was replicated in 2009 in an important follow-up15 of the SADHART trial16; among patients with MI and major depression, treatment-resistant depression (ie, depression that failed to improve substantially during treatment with either sertraline or placebo) was strongly and independently associated with long-term mortality (HR = 2.39; 95% CI, 1.39–2.44; P < .001).15
What is needed next? Testing the alternative hypothesis— ie, that an unmeasured confounder may exist—is difficult, requiring new observational studies and measurement of the putative third common causes or previously unmeasured confounder. Other putative confounders would then be hypothesized and would need to be included in additional observational studies. To properly test the putative confounding by depressive symptom severity, future observational studies should examine initial depressive symptom severity prior to antidepressant use and then collect data on depressive symptom severity and antidepressant use as time-varying covariates to CVD outcomes. To test whether treatment-resistant depression is the phenotype driving the spurious observational association between antidepressant use and increased risk of CVD, the phenotype and its underlying causal mechanisms need to be better understood. Of course, rigorous and adequately powered randomized controlled trials of antidepressant use in patients with CVD would be a more straightforward way to test the observational association between antidepressant use and increased risk of CVD. We turn now to the recently published randomized controlled trials in this field.
NEW EVIDENCE FROM RANDOMIZED TRIALS IN PATIENTS WITH CVD AND DEPRESSION
Concerns have been voiced for some time about the ability to effectively treat depression and whether an effective depression treatment will affect the risk of CVD recurrence and mortality.8 Adding to these concerns is our limited knowledge of the causal pathways and behavioral and biologic mechanisms implicated in this risk association.1 For these reasons, results from new randomized controlled trials, such as the four summarized below, are important.
Rollman et al compared the effectiveness of telephone-delivered collaborative care (treatment group) and usual physician care (control group) for improving mental health quality of life and reducing depressive symptoms in 302 patients with depression after CABG.17 Patients were observed for 8 months following randomization. Mental health quality of life and depressive symptoms were both significantly improved in the treatment group relative to the control group. Significantly more patients in the treatment group had a 50% or greater reduction in depressive symptoms (50.0% vs 29.6% in control group, P < .001; number needed to treat = 4.9 [95% CI, 3.2–10.4]). Men particularly benefited from the treatment. This trial suggests that collaborative care can be delivered effectively (and potentially cost-efficiently) over the phone.
Freedland et al also evaluated depression treatment in 123 patients with major or minor depression who underwent CABG.18 Their primary objective was to determine the efficacy of two behavioral treatments (cognitive behavioral therapy [CBT] or supportive stress management) compared with usual care. Significantly more patients in both the CBT group (71%) and the stress management group (57%) had a low score (indicating less severe depressive symptoms) on the clinician-based Hamilton Rating Scale for Depression compared with the usual care group (33%). These results were maintained 6 months after the end of the trial. Secondary measures of depressive symptoms, anxiety, and quality of life were also significantly improved in the depression treatment groups compared with the usual care group. This trial is important for the following reasons:
The use of a second control group, the stress management group, represents a strict, high-quality design that controls for professional attention, generic or placebo therapy effect, and time or effort on the part of the patient.
The second control group also provides treatment options for the patient, as both CBT and stress management were beneficial.
Outcome assessors were blinded to treatment assignment, an important design feature in behavioral trials.
In a rigorously conducted randomized, double-blind, placebo-controlled trial, Carney et al tested whether 2 g/day of omega-3 acid ethyl esters (eicosapentaenoic acid [EPA] and docosahexaenoic acid [DHA]) improved depressive symptoms in 122 patients with major depression and CHD.19 Patients in both the omega-3 and placebo groups received sertraline (50 mg/day) during the 10-week trial. A deficiency of omega-3 fatty acids has been implicated in both depression and CHD and is a possible causal link between the two diseases. Also, there is some evidence that the efficacy of antidepressants is increased by the addition of omega-3 supplementation. Unfortunately, there were no differences in self-reported or clinician-assessed depressive symptoms or in predefined depression remission at the study’s end. The trial included a 2-week adherence run-in period, ensuring that medication adherence in the trial was excellent (97%), and concluded that omega-3 supplementation, at least at these dosage levels, does not improve depression outcomes.
In the Coronary Psychosocial Evaluation Studies (COPES) randomized controlled trial, Davidson et al compared 6 months of an enhanced care intervention with usual depression care among 157 patients with ACS and persistently elevated depressive symptoms.20 Under the enhanced care intervention, patients received either problem-solving therapy or antidepressant medication, depending on their preference, and then had the option of later augmentation with the other treatment, intensification of the initial treatment, or switching of treatments, if indicated by depressive symptom severity (stepped care). The purpose of the trial was to determine the acceptability and efficacy of depression treatment among patients with ACS, who often neither agree with the diagnosis of depression nor had been seeking treatment for depression. Significantly more patients were satisfied with their depression care in the intervention arm, and depressive symptoms and major adverse cardiac events (nonfatal MI, hospitalization for unstable angina, or all-cause mortality) were significantly reduced in the intervention arm compared with the usual care arm. The absolute numbers were very small; at the end of the trial, 3 patients in the intervention group and 10 patients in the usual care group had major adverse cardiac events (4% and 13%, respectively; log-rank test, χ21 = 3.93; P = .047). The results suggested that involving patients in the type of depression care they receive (medication and/or psychotherapy) and stepping treatments aggressively may be methods to improve the treatment of depression in patients with CHD. In addition, persistently depressed patients may be an interesting patient group to select for future trials, as usual care has resulted in large reductions in depressive symptoms in some previous trials but not in this study of patients with persistent depression.
Summary and clinical implications
Each of these four efficacy trials adds critical information to the evidence base. Depressed patients who have undergone CABG can be effectively treated in primary care settings with integrative care,17 and CBT is also extremely effective for these patients.18 Additional studies of omega-3 supplementation should not be pursued at this time, but using a run-in period to better identify patients who are prepared to engage in treatment is a prudent idea and should be used in future trials in this area.19 Patients with CHD and persistent depressive symptoms are a promising group to target for depression therapy, and asking patients to choose their type of depression treatment may improve response to therapy for both depression and CHD.20
DEPRESSION SCREENING, REFERRAL, AND TREATMENT IN PATIENTS WITH CVD
We finish with the least evidence-based and most controversial issue in the area of depression and CVD. This controversy started in 2008 when the American Heart Association recommended in an advisory (endorsed by the American Psychiatric Association) that “screening tests for depressive symptoms should be applied to identify patients who may require further assessment and treatment” if appropriate referral for further depression assessment and treatment is available.21 Partly in response to this advisory, Thombs et al conducted a systematic review of the evidence on whether screening or treatment improves outcomes of depression or CVD in patients with CVD.22 They found no trial that tested whether depression screening was beneficial in patients with CVD, and the randomized controlled trials of depression treatment provided evidence of only mild improvement of depressive symptoms and no improvement in CVD outcome. Therefore, they questioned whether routine depression screening was appropriate.22
In at least eight editorials, letters, and reviews published on this subject in 2009, investigators continued to debate this issue.23–30 Below we provide a simplified list of reasons presented for and against screening and subsequent treatment raised in these articles.
Arguments for depression screening and treatment
The proponents of screening contend that depression is highly prevalent in patients with CVD and is clearly a risk marker for increased adverse events, reduced quality of life, and poorer adherence to treatment.24 They argue that since there are plausible biologic and behavioral mechanisms for this association, and since SSRI use improves depressive symptoms in other patient populations and is safe in patients with CVD, health care providers should not hesitate to screen and refer patients for appropriate depression treatment. At the same time, they have cautioned that SSRIs interact with anti coagulants and that bleeding should be monitored closely in patients with CVD who are taking SSRIs.24
Whooley28 noted that although there are controversial findings in this area, depression screening provided in conjunction with collaborative care depression management is cost-effective and has a documented positive impact on depression, if not on CVD outcomes.17,31 She observed that there are some costs to screening, such as false-positive findings (resulting in stigma for patients incorrectly diagnosed) and diversion of resources from other health care needs. However, Whooley suggested that primary care providers, rather than cardiologists, should conduct depression screening and that patients should undergo screening only when an established collaborative care treatment protocol exists.28
Carney et al argued that depression, like age, clearly marks CVD risk, and that health care providers should aggressively treat readily modifiable CVD risk factors.23 They added that because of the strong association between depression and medication nonadherence,32 providers should carefully monitor patient adherence to life-saving therapies.
Taking another tack, Shemesh and colleagues advocated the importance of documenting the prevalence of suicidal ideation and intent if recommendations to screen for depression in CVD patient populations were implemented.25 Using a sample of more than 1,000 patients with CVD, they determined the prevalence of suicidal ideation (12.0%) and the number of patients who required hospitalization for risk of suicide (0.5%) when routine depression screening occurred in a large cardiology clinic. They concluded that identification and stabilization of imminently suicidal patients would be a benefit of universal screening and that there is a high societal cost to neglecting suicidal ideation, intent, and risk in patients with CVD. However, more patients would need immediate thorough psychiatric evaluations for safety, which would affect resource allocation and cost in cardiology clinics.
Arguments against depression screening and treatment
The main argument against screening for and treating depression in patients with CVD is that there are neither randomized controlled trials nor systematic evidence-based reviews showing that screening for depression and/or referring for additional treatment sufficiently improves outcomes for depression or CVD, and that existing evidence does not support the recommendation to screen all patients with CVD.22,30 Furthermore, antidepressant use is associated with only mild improvement in depressive symptoms, even in other patient populations,33 and publication bias (“the file-drawer problem”) has prevented the publication of antidepressant trials with null results, thereby skewing the evidence base.34 In addition, considerable health care resources would be needed to mount such a large screening effort, and these resources would come at the expense of other efforts. Finally, the adverse effects of medications and the inevitability of some false-positive screening results must be weighed against any benefit that might occur with universal screening.35
In addition to the arguments above, Ziegelstein et al,29 in commenting on the American Heart Association advisory,21 wryly observed that there is far greater observational evidence that depressed patients seen in mental health settings are at risk for incident and recurrent CVD and that there should be universal screening and referral for CVD in patients with depression. They contended as well that the evidence is insufficient to recommend that patients with CVD undergo universal depression screening and referral.
Summary and clinical implications
Although we were hesitant to raise this tense and often emotional issue, we are in favor of routine, algorithm-based depression screening by all cardiologists, with the critical proviso that a nationwide and/or Centers for Medicare and Medicaid Services–coordinated randomized controlled trial be conducted to evaluate this practice. All patients with pronounced depressive symptoms should be referred to the trial, and two depression treatments should be evaluated, such as usual referral versus telephone-based collaborative care17 or enhanced depression care.20 Such a trial would allow us to ensure that data are collected on the cost,36 the benefit, and even the possible harms associated with routine depression screening for patients with CVD, and we could ascertain if there is an acceptable, beneficial treatment for depression that can be delivered and definitively tested.
References
Carney RM, Freedland KE. Depression in patients with coronary heart disease. Am J Med2008; 121( 11 suppl 2):S20–S27.
Whang W, Kubzansky LD, Kawachi I, et al Depression and risk of sudden cardiac death and coronary heart disease in women: results from the Nurses’ Health Study. J Am Coll Cardiol2009; 53:950–958.
Krantz DS, Whittaker KS, Francis JL, et al Psychotropic medication use and risk of adverse cardiovascular events in women with suspected coronary artery disease: outcomes from the Women’s Ischemia Syndrome Evaluation (WISE) study. Heart2009; 95:1901–1906.
Smoller JW, Allison M, Cochrane BB, et al Antidepressant use and risk of incident cardiovascular morbidity and mortality among postmenopausal women in the Women’s Health Initiative study. Arch Intern Med2009; 169:2128–2139.
May HT, Horne BD, Carlquist JF, Sheng X, Joy E, Catinella AP. Depression after coronary artery disease is associated with heart failure. J Am Coll Cardiol2009; 53:1440–1447.
Kim DH, Daskalakis C, Whellan DJ, et al Safety of selective serotonin reuptake inhibitor in adults undergoing coronary artery bypass grafting. Am J Cardiol2009; 103:1391–1395.
Jolly K, Gammage MD, Cheng KK, Bradburn P, Banting MV, Langman MJ. Sudden death in patients receiving drugs tending to prolong the QT interval. Br J Clin Pharmacol2009; 68:743–751.
Jolly K, Langman MJ. Psychotropic medication: curing illness or creating problems?Heart2009; 95:1893–1894.
Waldman SV, Blumenthal JA, Babyak MA, et al Ethnic differences in the treatment of depression in patients with ischemic heart disease. Am Heart J2009; 157:77–83.
Salas M, Hofman A, Stricker BH. Confounding by indication: an example of variation in the use of epidemiologic terminology. Am J Epidemiol1999; 149:981–983.
Carney RM, Freedland KE. Treatment-resistant depression and sudden cardiac death [letter]. J Am Coll Cardiol2009; 54:958–959.
Carney RM, Freedland KE. Treatment-resistant depression and mortality after acute coronary syndrome. Am J Psychiatry2009; 166:410–417.
Carney RM, Blumenthal JA, Freedland KE, et al Depression and late mortality after myocardial infarction in the Enhancing Recovery in Coronary Heart Disease (ENRICHD) study. Psychosom Med2004; 66:466–474.
Berkman LF, Blumenthal J, Burg M, et al Effects of treating depression and low perceived social support on clinical events after myocardial infarction: the Enhancing Recovery in Coronary Heart Disease Patients (ENRICHD) randomized trial. JAMA2003; 289:3106–3116.
Glassman AH, Bigger JT, Gaffney M. Psychiatric characteristics associated with long-term mortality among 361 patients having an acute coronary syndrome and major depression: seven-year follow-up of SADHART participants. Arch Gen Psychiatry2009; 66:1022–1029.
Glassman AH, O’Connor CM, Califf RM, et al Sertraline treatment of major depression in patients with acute MI or unstable angina. JAMA2002; 288:701–709.
Rollman BL, Belnap BH, LeMenager MS, et al Telephone-delivered collaborative care for treating post-CABG depression: a randomized controlled trial. JAMA2009; 302:2095–2103.
Freedland KE, Skala JA, Carney RM, et al Treatment of depression after coronary artery bypass surgery: a randomized controlled trial. Arch Gen Psychiatry2009; 66:387–396.
Carney RM, Freedland KE, Rubin EH, Rich MW, Steinmeyer BC, Harris WS. Omega-3 augmentation of sertraline in treatment of depression in patients with coronary heart disease: a randomized controlled trial. JAMA2009; 302:1651–1657.
Davidson KW, Rieckmann N, Clemow L, et al Enhanced depression care for patients with acute coronary syndrome and persistent depressive symptoms: Coronary Psychosocial Evaluation Studies randomized controlled trial. Arch Intern Med2010; 170:600–608.
Lichtman JH, Bigger JT, Blumenthal JA, et al Depression and coronary heart disease: recommendations for screening, referral, and treatment: a science advisory from the American Heart Association Prevention Committee of the Council on Cardiovascular Nursing, Council on Clinical Cardiology, Council on Epidemiology and Prevention, and Interdisciplinary Council on Quality of Care and Outcomes Research: endorsed by the American Psychiatric Association. Circulation2008; 118:1768–1775.
Thombs BD, de Jonge P, Coyne JC, et al Depression screening and patient outcomes in cardiovascular care: a systematic review. JAMA2008; 300:2161–2171.
Carney RM, Freedland KE, Jaffe AS. Depression screening in patients with heart disease [letter]. JAMA2009; 301:1337–1338.
Pozuelo L, Zhang J, Franco K, Tesar G, Penn M, Jiang W. Depression and heart disease: what do we know, and where are we headed?Cleve Clin J Med2009; 76:59–70.
Shemesh E, Annunziato RA, Rubinstein D, et al Screening for depression and suicidality in patients with cardiovascular illnesses. Am J Cardiol2009; 104:1194–1197.
Thombs BD, Adeponle AB, Kirmayer LJ, Rousseau C, Ziegelstein RC. More antidepressants for African Americans with coronary heart disease? Maybe—maybe not [letter]. Am Heart J2009; 157:e33; author reply,e35–e37.
Whang W, Davidson KW. Is it time to treat depression in patients with cardiovascular disease?Circulation2009; 120:99–100.
Whooley MA. To screen or not to screen? Depression in patients with cardiovascular disease. J Am Coll Cardiol2009; 54:891–893.
Ziegelstein RC, Thombs BD, Coyne JC, de Jonge P. Routine screening for depression in patients with coronary heart disease: never mind. J Am Coll Cardiol2009; 54:886–890.
Thombs BD, Jewett LR, Knafo R, Coyne JC, Ziegelstein RC. Learning from history: a commentary on the American Heart Association Science Advisory on depression screening. Am Heart J2009; 158:503–505.
Unutzer J, Katon W, Callahan CM, et al Collaborative care management of late-life depression in the primary care setting: a randomized controlled trial. JAMA2002; 288:2836–2845.
Rieckmann N, Gerin W, Kronish IM, et al Course of depressive symptoms and medication adherence after acute coronary syndromes: an electronic medication monitoring study. J Am Coll Cardiol2006; 48:2218–2222.
Kirsch I, Deacon BJ, Huedo-Medina TB, et al Initial severity and antidepressant benefits: a meta-analysis of data submitted to the Food and Drug Administration. PLoS Med2008; 5:e45.
Turner EH, Matthews AM, Linardatos E, Tell RA, Rosenthal R. Selective publication of antidepressant trials and its influence on apparent efficacy. N Engl J Med2008; 358:252–260.
Thombs BD, de Jonge P, Ziegelstein RC. Depression screening in patients with heart disease [reply to letter]. JAMA2009; 301:1338.
Frasure-Smith N, Lespérance F. Depression and cardiac risk: present status and future directions. Heart2010; 96:173–176.
References
Carney RM, Freedland KE. Depression in patients with coronary heart disease. Am J Med2008; 121( 11 suppl 2):S20–S27.
Whang W, Kubzansky LD, Kawachi I, et al Depression and risk of sudden cardiac death and coronary heart disease in women: results from the Nurses’ Health Study. J Am Coll Cardiol2009; 53:950–958.
Krantz DS, Whittaker KS, Francis JL, et al Psychotropic medication use and risk of adverse cardiovascular events in women with suspected coronary artery disease: outcomes from the Women’s Ischemia Syndrome Evaluation (WISE) study. Heart2009; 95:1901–1906.
Smoller JW, Allison M, Cochrane BB, et al Antidepressant use and risk of incident cardiovascular morbidity and mortality among postmenopausal women in the Women’s Health Initiative study. Arch Intern Med2009; 169:2128–2139.
May HT, Horne BD, Carlquist JF, Sheng X, Joy E, Catinella AP. Depression after coronary artery disease is associated with heart failure. J Am Coll Cardiol2009; 53:1440–1447.
Kim DH, Daskalakis C, Whellan DJ, et al Safety of selective serotonin reuptake inhibitor in adults undergoing coronary artery bypass grafting. Am J Cardiol2009; 103:1391–1395.
Jolly K, Gammage MD, Cheng KK, Bradburn P, Banting MV, Langman MJ. Sudden death in patients receiving drugs tending to prolong the QT interval. Br J Clin Pharmacol2009; 68:743–751.
Jolly K, Langman MJ. Psychotropic medication: curing illness or creating problems?Heart2009; 95:1893–1894.
Waldman SV, Blumenthal JA, Babyak MA, et al Ethnic differences in the treatment of depression in patients with ischemic heart disease. Am Heart J2009; 157:77–83.
Salas M, Hofman A, Stricker BH. Confounding by indication: an example of variation in the use of epidemiologic terminology. Am J Epidemiol1999; 149:981–983.
Carney RM, Freedland KE. Treatment-resistant depression and sudden cardiac death [letter]. J Am Coll Cardiol2009; 54:958–959.
Carney RM, Freedland KE. Treatment-resistant depression and mortality after acute coronary syndrome. Am J Psychiatry2009; 166:410–417.
Carney RM, Blumenthal JA, Freedland KE, et al Depression and late mortality after myocardial infarction in the Enhancing Recovery in Coronary Heart Disease (ENRICHD) study. Psychosom Med2004; 66:466–474.
Berkman LF, Blumenthal J, Burg M, et al Effects of treating depression and low perceived social support on clinical events after myocardial infarction: the Enhancing Recovery in Coronary Heart Disease Patients (ENRICHD) randomized trial. JAMA2003; 289:3106–3116.
Glassman AH, Bigger JT, Gaffney M. Psychiatric characteristics associated with long-term mortality among 361 patients having an acute coronary syndrome and major depression: seven-year follow-up of SADHART participants. Arch Gen Psychiatry2009; 66:1022–1029.
Glassman AH, O’Connor CM, Califf RM, et al Sertraline treatment of major depression in patients with acute MI or unstable angina. JAMA2002; 288:701–709.
Rollman BL, Belnap BH, LeMenager MS, et al Telephone-delivered collaborative care for treating post-CABG depression: a randomized controlled trial. JAMA2009; 302:2095–2103.
Freedland KE, Skala JA, Carney RM, et al Treatment of depression after coronary artery bypass surgery: a randomized controlled trial. Arch Gen Psychiatry2009; 66:387–396.
Carney RM, Freedland KE, Rubin EH, Rich MW, Steinmeyer BC, Harris WS. Omega-3 augmentation of sertraline in treatment of depression in patients with coronary heart disease: a randomized controlled trial. JAMA2009; 302:1651–1657.
Davidson KW, Rieckmann N, Clemow L, et al Enhanced depression care for patients with acute coronary syndrome and persistent depressive symptoms: Coronary Psychosocial Evaluation Studies randomized controlled trial. Arch Intern Med2010; 170:600–608.
Lichtman JH, Bigger JT, Blumenthal JA, et al Depression and coronary heart disease: recommendations for screening, referral, and treatment: a science advisory from the American Heart Association Prevention Committee of the Council on Cardiovascular Nursing, Council on Clinical Cardiology, Council on Epidemiology and Prevention, and Interdisciplinary Council on Quality of Care and Outcomes Research: endorsed by the American Psychiatric Association. Circulation2008; 118:1768–1775.
Thombs BD, de Jonge P, Coyne JC, et al Depression screening and patient outcomes in cardiovascular care: a systematic review. JAMA2008; 300:2161–2171.
Carney RM, Freedland KE, Jaffe AS. Depression screening in patients with heart disease [letter]. JAMA2009; 301:1337–1338.
Pozuelo L, Zhang J, Franco K, Tesar G, Penn M, Jiang W. Depression and heart disease: what do we know, and where are we headed?Cleve Clin J Med2009; 76:59–70.
Shemesh E, Annunziato RA, Rubinstein D, et al Screening for depression and suicidality in patients with cardiovascular illnesses. Am J Cardiol2009; 104:1194–1197.
Thombs BD, Adeponle AB, Kirmayer LJ, Rousseau C, Ziegelstein RC. More antidepressants for African Americans with coronary heart disease? Maybe—maybe not [letter]. Am Heart J2009; 157:e33; author reply,e35–e37.
Whang W, Davidson KW. Is it time to treat depression in patients with cardiovascular disease?Circulation2009; 120:99–100.
Whooley MA. To screen or not to screen? Depression in patients with cardiovascular disease. J Am Coll Cardiol2009; 54:891–893.
Ziegelstein RC, Thombs BD, Coyne JC, de Jonge P. Routine screening for depression in patients with coronary heart disease: never mind. J Am Coll Cardiol2009; 54:886–890.
Thombs BD, Jewett LR, Knafo R, Coyne JC, Ziegelstein RC. Learning from history: a commentary on the American Heart Association Science Advisory on depression screening. Am Heart J2009; 158:503–505.
Unutzer J, Katon W, Callahan CM, et al Collaborative care management of late-life depression in the primary care setting: a randomized controlled trial. JAMA2002; 288:2836–2845.
Rieckmann N, Gerin W, Kronish IM, et al Course of depressive symptoms and medication adherence after acute coronary syndromes: an electronic medication monitoring study. J Am Coll Cardiol2006; 48:2218–2222.
Kirsch I, Deacon BJ, Huedo-Medina TB, et al Initial severity and antidepressant benefits: a meta-analysis of data submitted to the Food and Drug Administration. PLoS Med2008; 5:e45.
Turner EH, Matthews AM, Linardatos E, Tell RA, Rosenthal R. Selective publication of antidepressant trials and its influence on apparent efficacy. N Engl J Med2008; 358:252–260.
Thombs BD, de Jonge P, Ziegelstein RC. Depression screening in patients with heart disease [reply to letter]. JAMA2009; 301:1338.
Frasure-Smith N, Lespérance F. Depression and cardiac risk: present status and future directions. Heart2010; 96:173–176.
WHY WE NEED TO UNDERSTAND MECHANISMS OF RECOVERY AFTER SEVERE BRAIN INJURY
The problem of recovery of consciousness after severe brain injury is one that easily captures the imagination of both the lay public and the professional. Puzzling reports continue to arise of late recovery of speech, language, memory, and other higher cognitive functions in rare patients, yet a scientific framework for the systematic assessment of these phenomena has been lacking.1–3 Some of these cases provide intriguing hints to the possible role of various medications (such as dopaminergic, serotoninergic, and noradrenergic agents) as well as spontaneous changes in brain function arising over time. As discussed below, the varying levels of recovery following coma seen after multifocal traumatic or nontraumatic brain injuries may share some common underlying mechanisms at the “circuit” level. Severe brain injuries producing coma have many causes (see Posner et al4 for a comprehensive review), but careful review reveals an overlap of structural pathologies and functional disturbances isolated to specific cerebral structures across several clinical syndromes grouped under the framework of “disorders of consciousness,” 5 with an emphasis on the role of particular substructures. Perhaps most important is a consideration of the pathologic, anatomic, and pathophysiologic role of the anterior forebrain, particularly the relationships of the brainstem and basal forebrain arousal systems, the central thalamus, and frontostriatal pathways, as reviewed below.
Figure 1. Correspondence of cognitive and motor impairment across outcomes following severe brain injuries. Impairment ranges from coma and vegetative state to minimally conscious state to locked-in state, which is not a disorder of consciousness. The gray box shows the large region of diagnostic uncertainty in establishing the true cognitive level of patients who behaviorally cannot reliably signal through controlled goal-directed movements (horizontal dashed line).Figure 1 indexes neurologic disorders of consciousness on a two-dimensional grid that highlights the independence of the degree of impairment of cognitive function and motor function that may be encountered in a patient. In the bottom left corner of the figure, coma and vegetative state are both considered unconscious brain states as judged by the bedside behavioral examination in the context of appropriate neurologic history. In both coma and vegetative state, patients do not demonstrate responses to environmental stimuli or initiate goal-directed behaviors. Comatose patients also show no state variation and usually remain close-eyed. In vegetative state, an observable cycling of irregular periods of eye opening and eye closure is evident, but this cyclical variation in behavioral state does not correlate with identifiable electroencephalographic features of either sleep or normal wakefulness.6 To the right of vegetative state in the figure is the minimally conscious state.7 Patients in minimally conscious state show unequivocal but inconsistent evidence of awareness of self or the environment through a wide range of behavioral response patterns that can be elicited by bedside examination.8 Patients may track objects with their eyes, exhibit stereotyped automatic motor behaviors, follow simple commands with small motor movements, or intermittently communicate through verbal or gestural means. The functional boundary indicating emergence from minimally conscious state is the demonstration of reliable verbal or gestural communication. Operationalizing this level of function is a topic of current research, as even simple “yes” versus “no” communication can be unreliable in brain-injured patients who recover past the level of minimally conscious state.9
The large gray box in Figure 1 indicates the disquietingly high degree of uncertainty in assessing cognitive level in some patients who lack controllable motor output channels. The locked-in state (bottom right corner of figure) defines patients who retain total preservation of cognitive function but otherwise may appear no different from those in deep coma. Although locked-in state often arises in the context of neurologic injuries that selectively damage motor output pathways distal from their cortical origins or that slowly reduce primary motor neuron function, this syndrome and closely similar conditions arise in patients with complex brain injuries. Such patients likely retain full or nearly normal consciousness but unfortunately are unable to produce consistent goal-directed movements that allow for communication. In principle, such patients could retain significant cognitive capacity near the normal range of cognitive function yet be indistinguishable from patients in minimally conscious state.
ROLE OF THE CENTRAL THALAMUS IN SEVERE BRAIN INJURIES
Recent studies have yielded evidence for common anatomic pathologies following severe injuries associated with vegetative state10 and minimally conscious state11 as well as pathologies underlying severe to moderate cognitive disability.12 Autopsy studies of patients remaining in vegetative state at the time of death have identified widespread neuronal death throughout the thalamus as the common finding following either anoxia or diffuse axonal injury that produces widespread disruption of white matter connections.10 The severe bilateral thalamic damage after either trauma or anoxia seen in permanent vegetative state is not, however, invariably associated with diffuse neocortical neuronal cell death. This is particularly true of traumatic brain injury, in which only approximately 10% of brains at autopsy show widespread neocortical cell death.10 Specific subnuclei of the thalamus show the greatest neuronal cell loss following global and multifocal cerebral injuries produced by traumatic brain injuries.13 In particular, the central thalamic nuclei (intralaminar nuclei and related paralaminar nuclei) demonstrate progressive neuronal loss following severe traumatic brain injuries,13 and there is some evidence that a similar pattern might be identified in hypoxic-ischemic injuries.14
Progressively severe disability grades with neuronal loss along a rostrocaudal axis: the anterior intralaminar and surrounding regions initially show volume loss associated with moderate disability, while neuronal loss in the ventral and lateral nuclei of the central thalamus (posterior intralaminar group) appears with worsening disability associated with minimally conscious state and vegetative state.13 This progressive and relatively specific involvement of the nuclei of the central thalamus likely results from the unique geometry of these neurons, which have wide point-to-point connectivity across the cerebral hemisphere.15,16 The marked neuronal volume loss in these cells is likely due to their integration of the effects of neuronal cell death across large cerebral territories after diffuse trauma, hypoxia, and other nonselective severe brain injuries.
Importantly, however, focal bilateral injuries to these regions of the central thalamus are also associated with global disorders of consciousness (coma, vegetative state, and minimally conscious state).5,17 This observation indicates that these neurons also play a causal role in the production of disorders of consciousness. Abrupt injuries of the central thalamus on both sides of the brain are associated with acute coma, reflecting these cells’ key contribution to normal mechanisms of arousal regulation (reviewed by Schiff18). The central thalamus receives ascending projections from the brainstem/basal forebrain “arousal systems” that control the activity of many cortical and thalamic neurons during the sleep-wake cycle. Importantly, the central thalamus is strongly innervated by the cholinergic, serotoninergic, and noradrenergic afferents of the brainstem arousal systems (see Schiff18 for review). These same neurons also are innervated by descending projections from frontal cortical regions supporting “executive” functions that underlie goal-directed behaviors. Collectively, these ascending and descending influences on the central thalamus appear to modulate the level of arousal associated with generalized alertness and variations in cognitive effort, stress, sleep deprivation, and other variables affecting the wakeful state.15,18–22
Neuroimaging and electrophysiologic studies offer further evidence that the anatomic specializations of the central thalamus play an important role in regulating brain activation during attentive wakefulness. The central thalamus shows selective activation in normal subjects performing tasks requiring a short-term shift of attention,19,23 sustained cognitive demands of high vigilance,22 or memory holds over extended time periods.23,24 Central thalamic activation associated with varying levels of vigilance correlates with global cerebral blood flow19 and specifically covaries within the anterior cingulate cortex and pontomesencephalon. 22 Brain activity in the anterior cingulate cortex grades with increasing cognitive load and is recruited by a wide range of cognitive tasks, apparently reciprocally increasing activity along with the central thalamus in response to increasing demands of cognitive effort.20,22
CIRCUIT MECHANISMS UNDERLYING RECOVERY AFTER SEVERE BRAIN INJURY
Figure 2. Proposed “mesocircuit”25 linking behavioral fluctuations following severe brain injuries and improvements in response to interventions. The model focuses on the modulatory role of the anterior forebrain in overall corticothalamic dynamics. The anterior forebrain (frontal/prefrontal cortical-striatopallidal thalamocortical loop system) is particularly vulnerable to large-scale dysfunction following multifocal brain injuries that produce widespread deafferentation or neuronal cell loss. At least two mechanisms related to this mesocircuit appear to play a key role after severe injuries: (1) the high demands of striatum output neurons for background activity and dopaminergic innervations and (2) the anatomic connections and physiologic specializations of the thalamocortical projections from the central thalamus. These central thalamic neurons have a potent activating role strongly driving both cortical and striatal neurons and wide point-to-point connections that make them more sensitive reporters of global neuronal loss than other thalamic nuclei. Withdrawal of thalamocortical transmission from the central thalamus is known to associate with coma and other disorders of consciousness (see Schiff and Plum5). The thalamostriatal projection from the central thalamus contacts the medium spiny neurons (MSNs) of the striatum, forming axodendritic (centromedian32) or axospinous (central lateral, parafascicular31) synapses. The MSNs, in turn, send inhibitory projections to the globus pallidus interna (GPi); without MSN output, the GPi tonically inhibits the central thalamus.33 Thus, a suppression of MSN output resulting from a loss of dopaminergic modulation or marked reduction in background synaptic activity can potentially catalyze a shutdown of the anterior forebrain. The mesocircuit model economically accounts for several clinical observations and aspects of normal physiology (see text for further discussion). (DBS = deep brain stimulation)In addition to the studies reviewed above providing evidence for the role of specific disconnection of neurons within the central thalamus in disorders of consciousness, Figure 2 illustrates a key vulnerability of the anterior forebrain in the setting of the widespread deafferentation and neuronal cell loss seen with severe brain injuries. This vulnerability places the role of the central thalamus in a wider context. A “mesocircuit”-level25 model has been proposed that suggests that functional alterations across very large connected neuronal populations of the anterior forebrain may arise primarily as a result of global reductions of excitatory neurotransmission.26,27 The majority of etiologies associated with coma and related disorders of consciousness diagrammed in Figure 1 effectively produce a broad decrease in background synaptic activity and excitatory neurotransmission (eg, diffuse axonal injury, anoxia, hypoxia-ischemia, cerebral vasospasm with strokes; see Posner et al4 for review).
In addition to the wide point-to-point connections of the central thalamus with the cerebral cortex (predominantly connections to frontal and prefrontal cortices; see Van der Werf et al,15 Groenewegen and Berendse,28 Morel et al29), these neurons have important projections to the striatum that return via projections from the globus pallidus. 30 These projections from the central thalamus (both central lateral nucleus and parafascicularis nucleus) diffusely innervate the striatum and project onto the medium spiny neurons (MSNs), the output neuron of structure.31 Because the specific thalamostriatal projections from these central thalamic neurons use glutamate transmitters with a high probability of synaptic release,32 they likely also have a strong role in modulating background activity in the striatum.
The MSNs represent an important point of vulnerability in this anterior forebrain mesocircuit, as they have a key role in maintaining activity in the anterior forebrain through their inhibitory projections to the globus pallidus interna, which in turn inhibits the central thalamus. 33 MSNs have instrinsic cell membrane properties that keep them below their firing threshold unless a high level of spontaneous background synaptic activity arising from excitatory corticostriatal and thalamostriatal inputs is present in concert with sufficient concentrations of the neurotransmitter dopamine.33 In the setting of diffuse deafferentation or neuronal loss following severe brain injury of any type, it is expected that background excitatory synaptic activity is considerably reduced. Under these circumstances, a broad withdrawal of direct excitatory striatal projections from the central thalamus and corticostriatal inputs is likely to cause MSN output to shut down. Observations of regional changes in brain metabolism following severe brain injuries, specific responses to pharmacologic and electrophysiologic interventions in brain-injured subjects, and normal variations in brain state are all consistent with this mesocircuit model (see Schiff26 for comprehensive review). Similarly, a consistent pattern of selective metabolic downregulation within the anterior forebrain has been shown to specifically grade with severity of behavioral impairment following diffuse axonal injury,34 and application of dopaminergic agents in such patients often will produce behavioral facilitation.35,36 These medications may facilitate the output of the MSNs and directly modulate mesial frontal cortical neurons, possibly restoring anterior forebrain activity within the loop connections of the frontal cortex, striatum, pallidum, and central thalamus.
This model provides context for understanding another paradoxical observation—ie, the association of the sedative zolpidem (Ambien), a nonbenzodiazepine hypnotic that potentiates GABAA receptors, with behavioral improvement of alertness and interactive behavior in severely brain-injured patients.37–41 Zolpidem’s primary direct action in patient responders, as originally proposed by Schiff and Posner,27 may be upon on the globus pallidus interna, producing a release of tonic inhibition of the central thalamus in the setting of a broad reduction in background excitatory neurotransmission (as seen, for example, following diffuse hypoxic-ischemic injury) and leading to a shutdown of the inhibitory projection of the MSNs. The GABAA alpha-1 subunit is expressed in large quantity in the globus pallidus interna, and experimental studies support this mechanism of action.42
SINGLE-SUBJECT STUDY OF CENTRAL THALAMIC STIMULATION IN MINIMALLY CONSCIOUS STATE
A further implication of the mesocircuit model is that direct activation of the central thalamus is expected to be the causal step in reactivating a downregulated anterior forebrain system, suggesting that direct modulation of the central thalamus might facilitate behavioral responsiveness in some patients with severe brain injuries. A recent study offers evidence that direct electrical stimulation of the central thalamus can produce behavioral facilitation.
In this single-subject study of central thalamic deep brain stimulation (DBS), a 38-year-old man remained in minimally conscious state for 6 years following a severe closed head injury following blunt trauma to the right frontal lobe.43 After 3 months in a vegetative state, the patient exhibited the first evidence of clear behaviors in response to sensory stimulation consistent with minimally conscious state and advanced to eventually demonstrating a best behavioral response of inconsistent command-following and communication using eye movements. This behavioral level remained unchanged at the start of the DBS study 4 years later, as confirmed by evaluation with the Coma Recovery Scale–Revised (CRS-R), a formal behavioral assessment tool.
Reprinted from Schiff et al.43
Figure 3. Overview of a 6-month alternating crossover study of central thalamic deep brain stimulation (DBS) in a patient in minimally conscious state following severe traumatic brain injury.43 (A) Study timeline. (B) Electrode lead placements within central thalamus of patient’s right (R) and left (L) hemispheres displayed in T1-weighted coronal magnetic resonance image. (C) The patient’s responsiveness on six cognitive and functional measures at presurgical baseline (“Pre”) and during periods when DBS was on (“On”) and off (“Off”) during the crossover phase. Responsiveness measures are shown with 95% confidence intervals (whiskers). Asterisks denote measures for which there were statistically significant differences between DBS “on” versus “off” periods. Dagger is present after “Oral feeding” to note that oral feeding data were not available before titration (hence no “Pre” value for this measure); also, oral feeding scores 1 and 2 are combined for dichotomy. See text for further explanation (including explanations of outcome measures).The patient entered into a study of central thalamic DBS according to the timeline in Figure 3A. An initial 4-month quantitative behavioral assessment was completed prior to placement of the DBS electrodes, which were implanted bilaterally in the anterior intralaminar thalamic nuclei (central lateral nucleus and adjacent paralaminar regions of the thalamus; Figure 3B). Following electrode placement and brief contact-by-contact testing of the electrodes, 2 months of behavioral testing was conducted with the electrodes remaining off to reassess the patient’s postsurgical behavioral baseline, which had not changed as a result of electrode placement. Two subsequent phases of the study focused on evaluation of DBS effects. A 5-month titration phase focused on establishing tolerance of DBS and evaluating several combinations of different electrical stimulation parameters (contact geometry, frequency, intensity) as well as the duration of the stimulation period. Following this titration phase, the patient entered a 6-month double-blind alternating crossover study. Through all phases of the study, the patient received standard rehabilitation efforts amounting to 3 hours a day, 4 days per week.43
Figure 3C summarizes results of the alternating crossover study and compares the prestimulation baseline assessments of various behaviors with the “on” versus “off” testing of the DBS electrodes during the crossover phase. The results demonstrate the overall impact of DBS compared with approximately 6 months of ongoing rehabilitation efforts in the absence of DBS exposure. Overall the findings show marked improvement in behavioral responsiveness compared with prestimulation frequencies of the highest-level behavioral response across six categories. The primary outcome assessments were prospectively chosen from subscales of the CRS-R, which is a well-validated psychometric tool used in patients with disorders of consciousness. CRS-R subscale items that had shown variation during the presurgical baseline assessment were chosen prospectively as the primary outcome measures. Notably, the CRS-R oral motor subscale was not chosen because no variation in this measure had been identified during the baseline assessment period. In addition, an object-naming scale and two other tailored secondary measures were developed later, during the titration phase, as the patient’s behavior changed, and were calibrated to be tested using these secondary measurement scales. All six measures showed marked change from prestimulation baseline levels, with five of the six measures showing higher-level behaviors than those seen prior to stimulation, regardless of whether the electrodes were on or off.43
As shown in Figure 3C, the behaviors captured by secondary measures had never occurred before the titration phase of the study; ie, the patient initially lacked a capacity for object naming, oral feeding, and the complex controlled goal-directed movements captured in the secondary limb movement measure, thus setting a prestimulation baseline frequency of 0 for these measures (see supplementary material in Schiff et al43). Three outcome measures—one primary (CRS-R arousal subscale) and two secondary (oral feeding and limb control)—showed statistically significant dependence on DBS during the 6-month period, as indicated by a significantly higher frequency of maximal score rating during “on” versus “off” periods (Figure 3C). The continuation of improvements during the “off” periods of the crossover trial (relative to the prestimulation baseline assessments) showed that the DBS effects produced carryover changes that remained after the extensive exposure to DBS during the titration period (for further analysis of the dynamic of these data, see supplementary material in Schiff et al43).
Importantly, these observations are limited to a single human subject and do not provide a guide to their generalizability, 44,45 although they are consistent with the proposed mesocircuit model reviewed above. While the precise mechanism underlying this patient’s improved behavioral responsiveness with central thalamic DBS is unknown, it is likely that DBS served to partially reverse the markedly depressed cerebral global metabolism earlier measured in this patient using fluorodeoxyglucose position emission tomography (FDG-PET)46 and also seen in other patients in minimally conscious state.47 The depressed cerebral metabolism seen in minimally conscious state likely reflects volume loss of neurons, deafferentation of remaining neurons, and neuronal functional impairments. All of these mechanisms may result in low firing rates of neurons in the neocortex, thalamus, and striatum. The mesocircuit model in Figure 2 suggests that direct activation of the central thalamus in patients with such chronically downregulated background synaptic activity may produce excitatory output from central thalamic neurons that acts to partially normalize firing rates and possibly firing patterns within the corticostriatopallidal-thalamocortical system.
SPECULATIONS ON THE IMPORTANCE OF HEART-BRAIN RESEARCH IN FUTURE STUDIES OF RECOVERY OF CONSCIOUSNESS
As this conference is focused on the heart-brain interface, it is appropriate to consider the relevance of heart-brain research to the general set of problems reviewed above. In fact, the linkage is quite natural, and classical physiologic psychology research has shown that cerebral arousal regulation is associated with patterned modulation of cardiac rhythm and autonomic function linked to the behavioral state.48,49 Among the most relevant observations are demonstrations that sustained focused attention is associated with several stereotyped cardiac and autonomic changes, including anticipatory bradycardia,50,51 pupillary dilatation, 52 and others (eg, galvanic skin response). Neurologic cases have shown that such couplings of effort to reflex bradycardia, pupillary dilatation, and other autonomic markers are altered by focal cerebral lesions in the right frontal lobe53 and left anterior cingulate cortex.54
In the single-subject central thalamic DBS study reviewed above,43 there were several unpublished observations that are potentially relevant to these mechanisms. During initial bedside testing of the individual DBS electrode contacts in first 2 postoperative days, electrical stimulation above threshold voltages associated with visible arousal response (for details, see supplementary material in Schiff et al43) consistently produced marked changes in heart rate and audible modulations of heart rhythm during interactions with the patient. Notably, the patient’s basal heart rate rose from a stable level of approximately 50 to 55 beats per minute to approximately 70 to 75 beats per minute—a nearly 50% increase. While increases in blood pressure and heart rate typically accompany arousal, the heart rate change observed here may reflect a marked change in cerebral metabolic rates. Earlier quantitative FDG-PET imaging in this patient revealed a global metabolic rate across the brain of approximately half the normal level.46 Considering that the brain consumes approximately 23% of the cardiac output,55 the increased heart rate observed in this setting may reflect an increase in demand in cardiac output, possibly as much as 100%. At the same time that these changes occurred, there was an audible cardiac deceleration noted when the patient was attentionally engaged by the examiner (this occurred without scoreable variation in most of the quantitative neurobehavioral metrics; see supplementary material in Schiff et al43). Of note, although the patient had suffered a complex severe brain injury, the right ventral frontal lobe showed the largest structural lesion46; injuries to the right hemisphere are associated with loss of such anticipatory changes in heart rate during attentional task performance.53
These anecdotal observations suggest that future studies that include measures to track patterns of heart rate variation during recovery of consciousness might provide an indirect index of increasing brain demand for allocation of cardiac output or emergent neural control of mechanisms linking cardiovascular response to attentive behavior. Ongoing coupling of electroencephalographic measures to autonomic and basal cardiac rhythms may be particularly interesting to examine during social interactions,56 as it is likely that behavioral responsiveness is linked to social stimuli. Emotional reactivity has been proposed as an essential component of arousal per se,49 and although not formally studied in the DBS trial reviewed above, emotional reengagement seems to be a clear concomitant of the collection of gestural and verbal behavioral improvements operationally tracked using quantitative behavioral scales. Beyond tracking heart-brain interactions as an index of brain recovery, it is possible that the integrity of heart-brain interactions may also be a target for optimization in support of recovery of consciousness after nonprogressive brain injury. Moreover, studies of optimization of cardiac function in severely brain-injured patients may provide insight into the recovery process as well.
References
Blake R. The Day Donny Herbert Woke Up: A True Story. New York, NY: Harmony Press; 2007.
Schiff ND, Fins JJ. Hope for “comatose” patients. Cerebrum2003; 5:7–24.
Posner J, Saper C, Schiff N, Plum F. Plum and Posner’s Diagnosis of Stupor and Coma. 4th ed.New York: Oxford University Press; 2007.
Schiff ND, Plum F. The role of arousal and ‘gating’ systems in the neurology of impaired consciousness. J Clin Neurophysiol2000; 17:438–452.
Kobylarz EJ, Schiff ND. Neurophysiological correlates of persistent vegetative and minimally conscious states. Neuropsychol Rehabil2005; 15:323–332.
Giacino JT, Ashwal S, Childs N, et al The minimally conscious state: definition and diagnostic criteria. Neurology2002; 58:349–353.
Giacino JT, Whyte J. The vegetative and minimally conscious states: current knowledge and remaining questions. J Head Trauma Rehabil2005; 20:30–50.
Nakase-Richardson R, Yablon SA, Sherer M, Nick TG, Evans CC. Emergence from minimally conscious state: insights from evaluation of posttraumatic confusion. Neurology2009; 73:1120–1126.
Adams JH, Graham DI, Jennett B. The neuropathology of the vegetative state after acute insult. Brain2000; 123:1327–1338.
Jennett B, Adams JH, Murray LS, Graham DI. Neuropathology in vegetative and severely disabled patients after head injury. Neurology2001; 56:486–490.
Adams JH, Graham DI, Jennett B. The structural basis of moderate disability after traumatic brain damage. J Neurol Neurosurg Psychiatry2001; 71:521–524.
Maxwell WL, MacKinnon MA, Smith DH, McIntosh TK, Graham DI. Thalamic nuclei after human blunt head injury. J Neuropathol Exp Neurol2006; 65:478–488.
Kinney HC, Korein J, Panigrahy A, Dikkes P, Goode R. Neuropathological findings in the brain of Karen Ann Quinlan. The role of the thalamus in the persistent vegetative state. N Engl J Med1994; 330:1469–1475.
Van der Werf YD, Witter MP, Groenewegen HJ. The intralaminar and midline nuclei of the thalamus. Anatomical and functional evidence for participation in processes of arousal and awareness. Brain Res Brain Res Rev2002; 39:107–140.
Scannell JW, Burns GA, Hilgetag CC, O’Neil MA, Young MP. The connectional organization of the cortico-thalamic system of the cat. Cereb Cortex1999; 9:277–299.
Castaigne P, Lhermitte F, Buge A, Escourolle R, Hauw JJ, Lyon-Caen O. Paramedian thalamic and midbrain infarct: clinical and neuropathological study. Ann Neurol1981; 10:127–148.
Schiff ND. Central thalamic contributions to arousal regulation and neurological disorders of consciousness. Ann N Y Acad Sci2008; 1129:105–118.
Kinomura S, Larsson J, Gulyás B, Roland PE. Activation by attention of the human reticular formation and thalamic intralaminar nuclei. Science1996; 271:512–515.
Paus T, Koski L, Caramanos Z, Westbury C. Regional differences in the effects of task difficulty and motor output on blood flow response in the human anterior cingulate cortex: a review of 107 PET activation studies. Neuroreport1998; 9:R37–47.
Nagai Y, Critchley HD, Featherstone E, Fenwick PB, Trimble MR, Dolan RJ. Brain activity relating to the contingent negative variation: an fMRI investigation. Neuroimage2004; 21:1232–1241.
Paus T, Zatorre RJ, Hofle N, et al Time-related changes in neural systems underlying attention and arousal during the performance of an auditory vigilance task. J Cogn Neurosci1997; 9:392–408.
Shah SA, Baker JL, Ryou JW, Purpura KP, Schiff ND. Modulation of arousal regulation with central thalamic deep brain stimulation. Conf Proc IEEE Eng Med Biol Soc2009; 2009:3314–3317.
Wyder MT, Massoglia DP, Stanford TR. Contextual modulation of central thalamic delay-period activity: representation of visual and saccadic goals. J Neurophysiol2004; 91:2628–2648.
Bohland JW, Wu C, Barbas H, et al A proposal for a coordinated effort for the determination of brainwide neuroanatomical connectivity in model organisms at a mesoscopic scale. PLoS Comput Biol2009; 5:e1000334.
Schiff ND. Recovery of consciousness after brain injury: a mesocircuit hypothesis. Trends Neurosci2010; 33:1–9.
Schiff ND, Posner JP. Another “Awakenings.”Ann Neurol2007; 62:5–7.
Groenewegen HJ, Berendse HW. The specificity of the ‘nonspecific’ midline and intralaminar thalamic nuclei. Trends Neurosci1994; 17:52–57.
Morel A, Liu J, Wannier T, Jeanmonod D, Rouiller EM. Divergence and convergence of thalamocortical projections to premotor and supplementary motor cortex: a multiple tracing study in the macaque monkey. Eur J Neurosci2005; 21:1007–1029.
Deschenes M, Bourassa J, Parent A. Striatal and cortical projections of single neurons from the central lateral thalamic nucleus in the rat. Neuroscience1996; 72:679–687.
Lacey CJ, Bolam JP, Magill PJ. Novel and distinct operational principles of intralaminar thalamic neurons and their striatal projections. J Neurosci2007; 27:4374–4384.
Smith Y, Raju D, Nanda B, Pare JF, Galvan A, Wichmann T. The thalamostriatal systems: anatomical and functional organization in normal and parkinsonian states. Brain Res Bull2009; 78:60–68.
Grillner S, Hellgren J, Ménard A, Saitoh K, Wikström MA. Mechanisms for selection of basic motor programs—roles for the striatum and pallidum. Trends Neurosci2005; 28:364–370.
Kato T, Nakayama N, Yasokawa Y, Okumura A, Shinoda J, Iwama T. Statistical image analysis of cerebral glucose metabolism in patients with cognitive impairment following diffuse traumatic brain injury. J Neurotrauma2007; 24:919–926.
Matsuda W, Matsumura A, Komatsu Y, Yanaka K, Nose T. Awakenings from persistent vegetative state: report of three cases with parkinsonism and brain stem lesions on MRI. J Neurol Neurosurg Psychiatry2003; 74:1571–1573.
Meythaler JM, Brunner RC, Johnson A, Novack TA. Amantadine to improve neurorecovery in traumatic brain injury–associated diffuse axonal injury: a pilot double-blind randomized trial. J Head Trauma Rehabil2002; 17:300–313.
Brefel-Courbon C, Payoux P, Ory F, et al Clinical and imaging evidence of zolpidem effect in hypoxic encephalopathy. Ann Neurol2007; 62:102–105.
Whyte J, Myers R. Incidence of clinically significant responses to zolpidem among patients with disorders of consciousness: a preliminary placebo controlled trial. Am J Phys Med Rehabil2009; 88:410–418.
Shames JL, Ring H. Transient reversal of anoxic brain injury–related minimally conscious state after zolpidem administration: a case report. Arch Phys Med Rehabil2008; 89:386–388.
Cohen SI, Duong TT. Increased arousal in a patient with anoxic brain injury after administration of zolpidem. Am J Phys Med Rehabil2008; 87:229–231.
Williams ST, Conte MM, Kobylarz EJ, Hersh JE, Victor JD, Schiff ND. Quantitative neurophysiologic characterization of a paradoxical response to zolpidem in a severely brain-injured human subject. Program No. 541.6/R9. 2009Neuroscience Meeting Planner. Chicago, IL: Society for Neuroscience, 2009. Online.
Chen L, Savio Chan C, Yung WH. Electrophysiological and behavioral effects of zolpidem in rat globus pallidus. Exp Neurol2004; 186:212–220.
Schiff ND, Giacino JT, Kalmar K, et al Behavioral improvements with thalamic stimulation after severe traumatic brain injury. Nature2007; 448:600–603.
Victor JD, Schiff ND. Meeting rigorous statistical standards in case reports. Ann Neurol2008; 64:592.
Schiff ND, Giacino JT, Fins JJ. Deep brain stimulation, neuroethics, and the minimally conscious state: moving beyond proof of principle. Arch Neurol2009; 66:697–702.
Schiff ND, Rodriguez-Moreno D, Kamal A, et al fMRI reveals large-scale network activation in minimally conscious patients. Neurology2005; 64:514–523.
Laureys S, Owen AM, Schiff ND. Brain function in coma, vegetative state, and related disorders. Lancet Neurol2004; 3:537–546.
Obrist PA. Presidential address, 1975. The cardiovascular-behavioral interaction—as it appears today. Psychophysiology1976; 13:95–107.
Pfaff D. Brain Arousal and Information Theory: Neural and Genetic Mechanisms. Cambridge, MA: Harvard University Press; 2006.
Obrist PA, Light KC, Langer AW, Gringnolo A, McCubbin JA. Behavioural-cardiac interactions: the psychosomatic hypothesis. J Psychosom Res1978; 22:301–325.
Kahneman D. Attention and Effort. Englewood Cliffs, NJ: Prentice-Hall; 1973.
Yokoyama K, Jennings R, Ackles P, Hood P, Boller F. Lack of heart rate changes during an attention-demanding task after right hemisphere lesions. Neurology1987; 37:624–630.
Naccache L, Dehaene S, Cohen L, et al Effortless control: executive attention and conscious feeling of mental effort are dissociable. Neuropsychologia2005; 43:1318–1328.
Roland P. Brain Activation. New York, NY: Wiley Press; 1997.
Porges SW. The polyvagal theory: new insights into adaptive reactions of the autonomic nervous system. Cleve Clin J Med2009; 76( suppl 2):S86–S90.
Nicholas D. Schiff, MD Department of Neurology and Neuroscience, Weill Cornell Medical College, New York, NY
Correspondence: Nicholas D. Schiff, MD, Associate Professor of Neurology and Neuroscience, and Director, Laboratory of Cognitive Neuromodulation, Department of Neurology and Neuroscience, Weill Medical College of Cornell University, New York, NY 10065; [email protected]
The author gratefully acknowledges the support of the Charles A. Dana Foundation, the National Institute of Neurological Disorders and Stroke, the National Institute of Child Health and Human Development, and IntElect Medical, Inc.
Dr. Schiff reported that he is a listed inventor on patents for deep brain stimulation in the central thalamus for cognitive neuromodulation issued to Cornell University and licensed to IntElect Medical, Inc., a start-up company formed by Cleveland Clinic and Cornell University. He is also a paid consultant and advisor to IntElect Medical, Inc. Published research described in this article received partial support from IntElect Medical, Inc.
Nicholas D. Schiff, MD Department of Neurology and Neuroscience, Weill Cornell Medical College, New York, NY
Correspondence: Nicholas D. Schiff, MD, Associate Professor of Neurology and Neuroscience, and Director, Laboratory of Cognitive Neuromodulation, Department of Neurology and Neuroscience, Weill Medical College of Cornell University, New York, NY 10065; [email protected]
The author gratefully acknowledges the support of the Charles A. Dana Foundation, the National Institute of Neurological Disorders and Stroke, the National Institute of Child Health and Human Development, and IntElect Medical, Inc.
Dr. Schiff reported that he is a listed inventor on patents for deep brain stimulation in the central thalamus for cognitive neuromodulation issued to Cornell University and licensed to IntElect Medical, Inc., a start-up company formed by Cleveland Clinic and Cornell University. He is also a paid consultant and advisor to IntElect Medical, Inc. Published research described in this article received partial support from IntElect Medical, Inc.
Author and Disclosure Information
Nicholas D. Schiff, MD Department of Neurology and Neuroscience, Weill Cornell Medical College, New York, NY
Correspondence: Nicholas D. Schiff, MD, Associate Professor of Neurology and Neuroscience, and Director, Laboratory of Cognitive Neuromodulation, Department of Neurology and Neuroscience, Weill Medical College of Cornell University, New York, NY 10065; [email protected]
The author gratefully acknowledges the support of the Charles A. Dana Foundation, the National Institute of Neurological Disorders and Stroke, the National Institute of Child Health and Human Development, and IntElect Medical, Inc.
Dr. Schiff reported that he is a listed inventor on patents for deep brain stimulation in the central thalamus for cognitive neuromodulation issued to Cornell University and licensed to IntElect Medical, Inc., a start-up company formed by Cleveland Clinic and Cornell University. He is also a paid consultant and advisor to IntElect Medical, Inc. Published research described in this article received partial support from IntElect Medical, Inc.
WHY WE NEED TO UNDERSTAND MECHANISMS OF RECOVERY AFTER SEVERE BRAIN INJURY
The problem of recovery of consciousness after severe brain injury is one that easily captures the imagination of both the lay public and the professional. Puzzling reports continue to arise of late recovery of speech, language, memory, and other higher cognitive functions in rare patients, yet a scientific framework for the systematic assessment of these phenomena has been lacking.1–3 Some of these cases provide intriguing hints to the possible role of various medications (such as dopaminergic, serotoninergic, and noradrenergic agents) as well as spontaneous changes in brain function arising over time. As discussed below, the varying levels of recovery following coma seen after multifocal traumatic or nontraumatic brain injuries may share some common underlying mechanisms at the “circuit” level. Severe brain injuries producing coma have many causes (see Posner et al4 for a comprehensive review), but careful review reveals an overlap of structural pathologies and functional disturbances isolated to specific cerebral structures across several clinical syndromes grouped under the framework of “disorders of consciousness,” 5 with an emphasis on the role of particular substructures. Perhaps most important is a consideration of the pathologic, anatomic, and pathophysiologic role of the anterior forebrain, particularly the relationships of the brainstem and basal forebrain arousal systems, the central thalamus, and frontostriatal pathways, as reviewed below.
Figure 1. Correspondence of cognitive and motor impairment across outcomes following severe brain injuries. Impairment ranges from coma and vegetative state to minimally conscious state to locked-in state, which is not a disorder of consciousness. The gray box shows the large region of diagnostic uncertainty in establishing the true cognitive level of patients who behaviorally cannot reliably signal through controlled goal-directed movements (horizontal dashed line).Figure 1 indexes neurologic disorders of consciousness on a two-dimensional grid that highlights the independence of the degree of impairment of cognitive function and motor function that may be encountered in a patient. In the bottom left corner of the figure, coma and vegetative state are both considered unconscious brain states as judged by the bedside behavioral examination in the context of appropriate neurologic history. In both coma and vegetative state, patients do not demonstrate responses to environmental stimuli or initiate goal-directed behaviors. Comatose patients also show no state variation and usually remain close-eyed. In vegetative state, an observable cycling of irregular periods of eye opening and eye closure is evident, but this cyclical variation in behavioral state does not correlate with identifiable electroencephalographic features of either sleep or normal wakefulness.6 To the right of vegetative state in the figure is the minimally conscious state.7 Patients in minimally conscious state show unequivocal but inconsistent evidence of awareness of self or the environment through a wide range of behavioral response patterns that can be elicited by bedside examination.8 Patients may track objects with their eyes, exhibit stereotyped automatic motor behaviors, follow simple commands with small motor movements, or intermittently communicate through verbal or gestural means. The functional boundary indicating emergence from minimally conscious state is the demonstration of reliable verbal or gestural communication. Operationalizing this level of function is a topic of current research, as even simple “yes” versus “no” communication can be unreliable in brain-injured patients who recover past the level of minimally conscious state.9
The large gray box in Figure 1 indicates the disquietingly high degree of uncertainty in assessing cognitive level in some patients who lack controllable motor output channels. The locked-in state (bottom right corner of figure) defines patients who retain total preservation of cognitive function but otherwise may appear no different from those in deep coma. Although locked-in state often arises in the context of neurologic injuries that selectively damage motor output pathways distal from their cortical origins or that slowly reduce primary motor neuron function, this syndrome and closely similar conditions arise in patients with complex brain injuries. Such patients likely retain full or nearly normal consciousness but unfortunately are unable to produce consistent goal-directed movements that allow for communication. In principle, such patients could retain significant cognitive capacity near the normal range of cognitive function yet be indistinguishable from patients in minimally conscious state.
ROLE OF THE CENTRAL THALAMUS IN SEVERE BRAIN INJURIES
Recent studies have yielded evidence for common anatomic pathologies following severe injuries associated with vegetative state10 and minimally conscious state11 as well as pathologies underlying severe to moderate cognitive disability.12 Autopsy studies of patients remaining in vegetative state at the time of death have identified widespread neuronal death throughout the thalamus as the common finding following either anoxia or diffuse axonal injury that produces widespread disruption of white matter connections.10 The severe bilateral thalamic damage after either trauma or anoxia seen in permanent vegetative state is not, however, invariably associated with diffuse neocortical neuronal cell death. This is particularly true of traumatic brain injury, in which only approximately 10% of brains at autopsy show widespread neocortical cell death.10 Specific subnuclei of the thalamus show the greatest neuronal cell loss following global and multifocal cerebral injuries produced by traumatic brain injuries.13 In particular, the central thalamic nuclei (intralaminar nuclei and related paralaminar nuclei) demonstrate progressive neuronal loss following severe traumatic brain injuries,13 and there is some evidence that a similar pattern might be identified in hypoxic-ischemic injuries.14
Progressively severe disability grades with neuronal loss along a rostrocaudal axis: the anterior intralaminar and surrounding regions initially show volume loss associated with moderate disability, while neuronal loss in the ventral and lateral nuclei of the central thalamus (posterior intralaminar group) appears with worsening disability associated with minimally conscious state and vegetative state.13 This progressive and relatively specific involvement of the nuclei of the central thalamus likely results from the unique geometry of these neurons, which have wide point-to-point connectivity across the cerebral hemisphere.15,16 The marked neuronal volume loss in these cells is likely due to their integration of the effects of neuronal cell death across large cerebral territories after diffuse trauma, hypoxia, and other nonselective severe brain injuries.
Importantly, however, focal bilateral injuries to these regions of the central thalamus are also associated with global disorders of consciousness (coma, vegetative state, and minimally conscious state).5,17 This observation indicates that these neurons also play a causal role in the production of disorders of consciousness. Abrupt injuries of the central thalamus on both sides of the brain are associated with acute coma, reflecting these cells’ key contribution to normal mechanisms of arousal regulation (reviewed by Schiff18). The central thalamus receives ascending projections from the brainstem/basal forebrain “arousal systems” that control the activity of many cortical and thalamic neurons during the sleep-wake cycle. Importantly, the central thalamus is strongly innervated by the cholinergic, serotoninergic, and noradrenergic afferents of the brainstem arousal systems (see Schiff18 for review). These same neurons also are innervated by descending projections from frontal cortical regions supporting “executive” functions that underlie goal-directed behaviors. Collectively, these ascending and descending influences on the central thalamus appear to modulate the level of arousal associated with generalized alertness and variations in cognitive effort, stress, sleep deprivation, and other variables affecting the wakeful state.15,18–22
Neuroimaging and electrophysiologic studies offer further evidence that the anatomic specializations of the central thalamus play an important role in regulating brain activation during attentive wakefulness. The central thalamus shows selective activation in normal subjects performing tasks requiring a short-term shift of attention,19,23 sustained cognitive demands of high vigilance,22 or memory holds over extended time periods.23,24 Central thalamic activation associated with varying levels of vigilance correlates with global cerebral blood flow19 and specifically covaries within the anterior cingulate cortex and pontomesencephalon. 22 Brain activity in the anterior cingulate cortex grades with increasing cognitive load and is recruited by a wide range of cognitive tasks, apparently reciprocally increasing activity along with the central thalamus in response to increasing demands of cognitive effort.20,22
CIRCUIT MECHANISMS UNDERLYING RECOVERY AFTER SEVERE BRAIN INJURY
Figure 2. Proposed “mesocircuit”25 linking behavioral fluctuations following severe brain injuries and improvements in response to interventions. The model focuses on the modulatory role of the anterior forebrain in overall corticothalamic dynamics. The anterior forebrain (frontal/prefrontal cortical-striatopallidal thalamocortical loop system) is particularly vulnerable to large-scale dysfunction following multifocal brain injuries that produce widespread deafferentation or neuronal cell loss. At least two mechanisms related to this mesocircuit appear to play a key role after severe injuries: (1) the high demands of striatum output neurons for background activity and dopaminergic innervations and (2) the anatomic connections and physiologic specializations of the thalamocortical projections from the central thalamus. These central thalamic neurons have a potent activating role strongly driving both cortical and striatal neurons and wide point-to-point connections that make them more sensitive reporters of global neuronal loss than other thalamic nuclei. Withdrawal of thalamocortical transmission from the central thalamus is known to associate with coma and other disorders of consciousness (see Schiff and Plum5). The thalamostriatal projection from the central thalamus contacts the medium spiny neurons (MSNs) of the striatum, forming axodendritic (centromedian32) or axospinous (central lateral, parafascicular31) synapses. The MSNs, in turn, send inhibitory projections to the globus pallidus interna (GPi); without MSN output, the GPi tonically inhibits the central thalamus.33 Thus, a suppression of MSN output resulting from a loss of dopaminergic modulation or marked reduction in background synaptic activity can potentially catalyze a shutdown of the anterior forebrain. The mesocircuit model economically accounts for several clinical observations and aspects of normal physiology (see text for further discussion). (DBS = deep brain stimulation)In addition to the studies reviewed above providing evidence for the role of specific disconnection of neurons within the central thalamus in disorders of consciousness, Figure 2 illustrates a key vulnerability of the anterior forebrain in the setting of the widespread deafferentation and neuronal cell loss seen with severe brain injuries. This vulnerability places the role of the central thalamus in a wider context. A “mesocircuit”-level25 model has been proposed that suggests that functional alterations across very large connected neuronal populations of the anterior forebrain may arise primarily as a result of global reductions of excitatory neurotransmission.26,27 The majority of etiologies associated with coma and related disorders of consciousness diagrammed in Figure 1 effectively produce a broad decrease in background synaptic activity and excitatory neurotransmission (eg, diffuse axonal injury, anoxia, hypoxia-ischemia, cerebral vasospasm with strokes; see Posner et al4 for review).
In addition to the wide point-to-point connections of the central thalamus with the cerebral cortex (predominantly connections to frontal and prefrontal cortices; see Van der Werf et al,15 Groenewegen and Berendse,28 Morel et al29), these neurons have important projections to the striatum that return via projections from the globus pallidus. 30 These projections from the central thalamus (both central lateral nucleus and parafascicularis nucleus) diffusely innervate the striatum and project onto the medium spiny neurons (MSNs), the output neuron of structure.31 Because the specific thalamostriatal projections from these central thalamic neurons use glutamate transmitters with a high probability of synaptic release,32 they likely also have a strong role in modulating background activity in the striatum.
The MSNs represent an important point of vulnerability in this anterior forebrain mesocircuit, as they have a key role in maintaining activity in the anterior forebrain through their inhibitory projections to the globus pallidus interna, which in turn inhibits the central thalamus. 33 MSNs have instrinsic cell membrane properties that keep them below their firing threshold unless a high level of spontaneous background synaptic activity arising from excitatory corticostriatal and thalamostriatal inputs is present in concert with sufficient concentrations of the neurotransmitter dopamine.33 In the setting of diffuse deafferentation or neuronal loss following severe brain injury of any type, it is expected that background excitatory synaptic activity is considerably reduced. Under these circumstances, a broad withdrawal of direct excitatory striatal projections from the central thalamus and corticostriatal inputs is likely to cause MSN output to shut down. Observations of regional changes in brain metabolism following severe brain injuries, specific responses to pharmacologic and electrophysiologic interventions in brain-injured subjects, and normal variations in brain state are all consistent with this mesocircuit model (see Schiff26 for comprehensive review). Similarly, a consistent pattern of selective metabolic downregulation within the anterior forebrain has been shown to specifically grade with severity of behavioral impairment following diffuse axonal injury,34 and application of dopaminergic agents in such patients often will produce behavioral facilitation.35,36 These medications may facilitate the output of the MSNs and directly modulate mesial frontal cortical neurons, possibly restoring anterior forebrain activity within the loop connections of the frontal cortex, striatum, pallidum, and central thalamus.
This model provides context for understanding another paradoxical observation—ie, the association of the sedative zolpidem (Ambien), a nonbenzodiazepine hypnotic that potentiates GABAA receptors, with behavioral improvement of alertness and interactive behavior in severely brain-injured patients.37–41 Zolpidem’s primary direct action in patient responders, as originally proposed by Schiff and Posner,27 may be upon on the globus pallidus interna, producing a release of tonic inhibition of the central thalamus in the setting of a broad reduction in background excitatory neurotransmission (as seen, for example, following diffuse hypoxic-ischemic injury) and leading to a shutdown of the inhibitory projection of the MSNs. The GABAA alpha-1 subunit is expressed in large quantity in the globus pallidus interna, and experimental studies support this mechanism of action.42
SINGLE-SUBJECT STUDY OF CENTRAL THALAMIC STIMULATION IN MINIMALLY CONSCIOUS STATE
A further implication of the mesocircuit model is that direct activation of the central thalamus is expected to be the causal step in reactivating a downregulated anterior forebrain system, suggesting that direct modulation of the central thalamus might facilitate behavioral responsiveness in some patients with severe brain injuries. A recent study offers evidence that direct electrical stimulation of the central thalamus can produce behavioral facilitation.
In this single-subject study of central thalamic deep brain stimulation (DBS), a 38-year-old man remained in minimally conscious state for 6 years following a severe closed head injury following blunt trauma to the right frontal lobe.43 After 3 months in a vegetative state, the patient exhibited the first evidence of clear behaviors in response to sensory stimulation consistent with minimally conscious state and advanced to eventually demonstrating a best behavioral response of inconsistent command-following and communication using eye movements. This behavioral level remained unchanged at the start of the DBS study 4 years later, as confirmed by evaluation with the Coma Recovery Scale–Revised (CRS-R), a formal behavioral assessment tool.
Reprinted from Schiff et al.43
Figure 3. Overview of a 6-month alternating crossover study of central thalamic deep brain stimulation (DBS) in a patient in minimally conscious state following severe traumatic brain injury.43 (A) Study timeline. (B) Electrode lead placements within central thalamus of patient’s right (R) and left (L) hemispheres displayed in T1-weighted coronal magnetic resonance image. (C) The patient’s responsiveness on six cognitive and functional measures at presurgical baseline (“Pre”) and during periods when DBS was on (“On”) and off (“Off”) during the crossover phase. Responsiveness measures are shown with 95% confidence intervals (whiskers). Asterisks denote measures for which there were statistically significant differences between DBS “on” versus “off” periods. Dagger is present after “Oral feeding” to note that oral feeding data were not available before titration (hence no “Pre” value for this measure); also, oral feeding scores 1 and 2 are combined for dichotomy. See text for further explanation (including explanations of outcome measures).The patient entered into a study of central thalamic DBS according to the timeline in Figure 3A. An initial 4-month quantitative behavioral assessment was completed prior to placement of the DBS electrodes, which were implanted bilaterally in the anterior intralaminar thalamic nuclei (central lateral nucleus and adjacent paralaminar regions of the thalamus; Figure 3B). Following electrode placement and brief contact-by-contact testing of the electrodes, 2 months of behavioral testing was conducted with the electrodes remaining off to reassess the patient’s postsurgical behavioral baseline, which had not changed as a result of electrode placement. Two subsequent phases of the study focused on evaluation of DBS effects. A 5-month titration phase focused on establishing tolerance of DBS and evaluating several combinations of different electrical stimulation parameters (contact geometry, frequency, intensity) as well as the duration of the stimulation period. Following this titration phase, the patient entered a 6-month double-blind alternating crossover study. Through all phases of the study, the patient received standard rehabilitation efforts amounting to 3 hours a day, 4 days per week.43
Figure 3C summarizes results of the alternating crossover study and compares the prestimulation baseline assessments of various behaviors with the “on” versus “off” testing of the DBS electrodes during the crossover phase. The results demonstrate the overall impact of DBS compared with approximately 6 months of ongoing rehabilitation efforts in the absence of DBS exposure. Overall the findings show marked improvement in behavioral responsiveness compared with prestimulation frequencies of the highest-level behavioral response across six categories. The primary outcome assessments were prospectively chosen from subscales of the CRS-R, which is a well-validated psychometric tool used in patients with disorders of consciousness. CRS-R subscale items that had shown variation during the presurgical baseline assessment were chosen prospectively as the primary outcome measures. Notably, the CRS-R oral motor subscale was not chosen because no variation in this measure had been identified during the baseline assessment period. In addition, an object-naming scale and two other tailored secondary measures were developed later, during the titration phase, as the patient’s behavior changed, and were calibrated to be tested using these secondary measurement scales. All six measures showed marked change from prestimulation baseline levels, with five of the six measures showing higher-level behaviors than those seen prior to stimulation, regardless of whether the electrodes were on or off.43
As shown in Figure 3C, the behaviors captured by secondary measures had never occurred before the titration phase of the study; ie, the patient initially lacked a capacity for object naming, oral feeding, and the complex controlled goal-directed movements captured in the secondary limb movement measure, thus setting a prestimulation baseline frequency of 0 for these measures (see supplementary material in Schiff et al43). Three outcome measures—one primary (CRS-R arousal subscale) and two secondary (oral feeding and limb control)—showed statistically significant dependence on DBS during the 6-month period, as indicated by a significantly higher frequency of maximal score rating during “on” versus “off” periods (Figure 3C). The continuation of improvements during the “off” periods of the crossover trial (relative to the prestimulation baseline assessments) showed that the DBS effects produced carryover changes that remained after the extensive exposure to DBS during the titration period (for further analysis of the dynamic of these data, see supplementary material in Schiff et al43).
Importantly, these observations are limited to a single human subject and do not provide a guide to their generalizability, 44,45 although they are consistent with the proposed mesocircuit model reviewed above. While the precise mechanism underlying this patient’s improved behavioral responsiveness with central thalamic DBS is unknown, it is likely that DBS served to partially reverse the markedly depressed cerebral global metabolism earlier measured in this patient using fluorodeoxyglucose position emission tomography (FDG-PET)46 and also seen in other patients in minimally conscious state.47 The depressed cerebral metabolism seen in minimally conscious state likely reflects volume loss of neurons, deafferentation of remaining neurons, and neuronal functional impairments. All of these mechanisms may result in low firing rates of neurons in the neocortex, thalamus, and striatum. The mesocircuit model in Figure 2 suggests that direct activation of the central thalamus in patients with such chronically downregulated background synaptic activity may produce excitatory output from central thalamic neurons that acts to partially normalize firing rates and possibly firing patterns within the corticostriatopallidal-thalamocortical system.
SPECULATIONS ON THE IMPORTANCE OF HEART-BRAIN RESEARCH IN FUTURE STUDIES OF RECOVERY OF CONSCIOUSNESS
As this conference is focused on the heart-brain interface, it is appropriate to consider the relevance of heart-brain research to the general set of problems reviewed above. In fact, the linkage is quite natural, and classical physiologic psychology research has shown that cerebral arousal regulation is associated with patterned modulation of cardiac rhythm and autonomic function linked to the behavioral state.48,49 Among the most relevant observations are demonstrations that sustained focused attention is associated with several stereotyped cardiac and autonomic changes, including anticipatory bradycardia,50,51 pupillary dilatation, 52 and others (eg, galvanic skin response). Neurologic cases have shown that such couplings of effort to reflex bradycardia, pupillary dilatation, and other autonomic markers are altered by focal cerebral lesions in the right frontal lobe53 and left anterior cingulate cortex.54
In the single-subject central thalamic DBS study reviewed above,43 there were several unpublished observations that are potentially relevant to these mechanisms. During initial bedside testing of the individual DBS electrode contacts in first 2 postoperative days, electrical stimulation above threshold voltages associated with visible arousal response (for details, see supplementary material in Schiff et al43) consistently produced marked changes in heart rate and audible modulations of heart rhythm during interactions with the patient. Notably, the patient’s basal heart rate rose from a stable level of approximately 50 to 55 beats per minute to approximately 70 to 75 beats per minute—a nearly 50% increase. While increases in blood pressure and heart rate typically accompany arousal, the heart rate change observed here may reflect a marked change in cerebral metabolic rates. Earlier quantitative FDG-PET imaging in this patient revealed a global metabolic rate across the brain of approximately half the normal level.46 Considering that the brain consumes approximately 23% of the cardiac output,55 the increased heart rate observed in this setting may reflect an increase in demand in cardiac output, possibly as much as 100%. At the same time that these changes occurred, there was an audible cardiac deceleration noted when the patient was attentionally engaged by the examiner (this occurred without scoreable variation in most of the quantitative neurobehavioral metrics; see supplementary material in Schiff et al43). Of note, although the patient had suffered a complex severe brain injury, the right ventral frontal lobe showed the largest structural lesion46; injuries to the right hemisphere are associated with loss of such anticipatory changes in heart rate during attentional task performance.53
These anecdotal observations suggest that future studies that include measures to track patterns of heart rate variation during recovery of consciousness might provide an indirect index of increasing brain demand for allocation of cardiac output or emergent neural control of mechanisms linking cardiovascular response to attentive behavior. Ongoing coupling of electroencephalographic measures to autonomic and basal cardiac rhythms may be particularly interesting to examine during social interactions,56 as it is likely that behavioral responsiveness is linked to social stimuli. Emotional reactivity has been proposed as an essential component of arousal per se,49 and although not formally studied in the DBS trial reviewed above, emotional reengagement seems to be a clear concomitant of the collection of gestural and verbal behavioral improvements operationally tracked using quantitative behavioral scales. Beyond tracking heart-brain interactions as an index of brain recovery, it is possible that the integrity of heart-brain interactions may also be a target for optimization in support of recovery of consciousness after nonprogressive brain injury. Moreover, studies of optimization of cardiac function in severely brain-injured patients may provide insight into the recovery process as well.
WHY WE NEED TO UNDERSTAND MECHANISMS OF RECOVERY AFTER SEVERE BRAIN INJURY
The problem of recovery of consciousness after severe brain injury is one that easily captures the imagination of both the lay public and the professional. Puzzling reports continue to arise of late recovery of speech, language, memory, and other higher cognitive functions in rare patients, yet a scientific framework for the systematic assessment of these phenomena has been lacking.1–3 Some of these cases provide intriguing hints to the possible role of various medications (such as dopaminergic, serotoninergic, and noradrenergic agents) as well as spontaneous changes in brain function arising over time. As discussed below, the varying levels of recovery following coma seen after multifocal traumatic or nontraumatic brain injuries may share some common underlying mechanisms at the “circuit” level. Severe brain injuries producing coma have many causes (see Posner et al4 for a comprehensive review), but careful review reveals an overlap of structural pathologies and functional disturbances isolated to specific cerebral structures across several clinical syndromes grouped under the framework of “disorders of consciousness,” 5 with an emphasis on the role of particular substructures. Perhaps most important is a consideration of the pathologic, anatomic, and pathophysiologic role of the anterior forebrain, particularly the relationships of the brainstem and basal forebrain arousal systems, the central thalamus, and frontostriatal pathways, as reviewed below.
Figure 1. Correspondence of cognitive and motor impairment across outcomes following severe brain injuries. Impairment ranges from coma and vegetative state to minimally conscious state to locked-in state, which is not a disorder of consciousness. The gray box shows the large region of diagnostic uncertainty in establishing the true cognitive level of patients who behaviorally cannot reliably signal through controlled goal-directed movements (horizontal dashed line).Figure 1 indexes neurologic disorders of consciousness on a two-dimensional grid that highlights the independence of the degree of impairment of cognitive function and motor function that may be encountered in a patient. In the bottom left corner of the figure, coma and vegetative state are both considered unconscious brain states as judged by the bedside behavioral examination in the context of appropriate neurologic history. In both coma and vegetative state, patients do not demonstrate responses to environmental stimuli or initiate goal-directed behaviors. Comatose patients also show no state variation and usually remain close-eyed. In vegetative state, an observable cycling of irregular periods of eye opening and eye closure is evident, but this cyclical variation in behavioral state does not correlate with identifiable electroencephalographic features of either sleep or normal wakefulness.6 To the right of vegetative state in the figure is the minimally conscious state.7 Patients in minimally conscious state show unequivocal but inconsistent evidence of awareness of self or the environment through a wide range of behavioral response patterns that can be elicited by bedside examination.8 Patients may track objects with their eyes, exhibit stereotyped automatic motor behaviors, follow simple commands with small motor movements, or intermittently communicate through verbal or gestural means. The functional boundary indicating emergence from minimally conscious state is the demonstration of reliable verbal or gestural communication. Operationalizing this level of function is a topic of current research, as even simple “yes” versus “no” communication can be unreliable in brain-injured patients who recover past the level of minimally conscious state.9
The large gray box in Figure 1 indicates the disquietingly high degree of uncertainty in assessing cognitive level in some patients who lack controllable motor output channels. The locked-in state (bottom right corner of figure) defines patients who retain total preservation of cognitive function but otherwise may appear no different from those in deep coma. Although locked-in state often arises in the context of neurologic injuries that selectively damage motor output pathways distal from their cortical origins or that slowly reduce primary motor neuron function, this syndrome and closely similar conditions arise in patients with complex brain injuries. Such patients likely retain full or nearly normal consciousness but unfortunately are unable to produce consistent goal-directed movements that allow for communication. In principle, such patients could retain significant cognitive capacity near the normal range of cognitive function yet be indistinguishable from patients in minimally conscious state.
ROLE OF THE CENTRAL THALAMUS IN SEVERE BRAIN INJURIES
Recent studies have yielded evidence for common anatomic pathologies following severe injuries associated with vegetative state10 and minimally conscious state11 as well as pathologies underlying severe to moderate cognitive disability.12 Autopsy studies of patients remaining in vegetative state at the time of death have identified widespread neuronal death throughout the thalamus as the common finding following either anoxia or diffuse axonal injury that produces widespread disruption of white matter connections.10 The severe bilateral thalamic damage after either trauma or anoxia seen in permanent vegetative state is not, however, invariably associated with diffuse neocortical neuronal cell death. This is particularly true of traumatic brain injury, in which only approximately 10% of brains at autopsy show widespread neocortical cell death.10 Specific subnuclei of the thalamus show the greatest neuronal cell loss following global and multifocal cerebral injuries produced by traumatic brain injuries.13 In particular, the central thalamic nuclei (intralaminar nuclei and related paralaminar nuclei) demonstrate progressive neuronal loss following severe traumatic brain injuries,13 and there is some evidence that a similar pattern might be identified in hypoxic-ischemic injuries.14
Progressively severe disability grades with neuronal loss along a rostrocaudal axis: the anterior intralaminar and surrounding regions initially show volume loss associated with moderate disability, while neuronal loss in the ventral and lateral nuclei of the central thalamus (posterior intralaminar group) appears with worsening disability associated with minimally conscious state and vegetative state.13 This progressive and relatively specific involvement of the nuclei of the central thalamus likely results from the unique geometry of these neurons, which have wide point-to-point connectivity across the cerebral hemisphere.15,16 The marked neuronal volume loss in these cells is likely due to their integration of the effects of neuronal cell death across large cerebral territories after diffuse trauma, hypoxia, and other nonselective severe brain injuries.
Importantly, however, focal bilateral injuries to these regions of the central thalamus are also associated with global disorders of consciousness (coma, vegetative state, and minimally conscious state).5,17 This observation indicates that these neurons also play a causal role in the production of disorders of consciousness. Abrupt injuries of the central thalamus on both sides of the brain are associated with acute coma, reflecting these cells’ key contribution to normal mechanisms of arousal regulation (reviewed by Schiff18). The central thalamus receives ascending projections from the brainstem/basal forebrain “arousal systems” that control the activity of many cortical and thalamic neurons during the sleep-wake cycle. Importantly, the central thalamus is strongly innervated by the cholinergic, serotoninergic, and noradrenergic afferents of the brainstem arousal systems (see Schiff18 for review). These same neurons also are innervated by descending projections from frontal cortical regions supporting “executive” functions that underlie goal-directed behaviors. Collectively, these ascending and descending influences on the central thalamus appear to modulate the level of arousal associated with generalized alertness and variations in cognitive effort, stress, sleep deprivation, and other variables affecting the wakeful state.15,18–22
Neuroimaging and electrophysiologic studies offer further evidence that the anatomic specializations of the central thalamus play an important role in regulating brain activation during attentive wakefulness. The central thalamus shows selective activation in normal subjects performing tasks requiring a short-term shift of attention,19,23 sustained cognitive demands of high vigilance,22 or memory holds over extended time periods.23,24 Central thalamic activation associated with varying levels of vigilance correlates with global cerebral blood flow19 and specifically covaries within the anterior cingulate cortex and pontomesencephalon. 22 Brain activity in the anterior cingulate cortex grades with increasing cognitive load and is recruited by a wide range of cognitive tasks, apparently reciprocally increasing activity along with the central thalamus in response to increasing demands of cognitive effort.20,22
CIRCUIT MECHANISMS UNDERLYING RECOVERY AFTER SEVERE BRAIN INJURY
Figure 2. Proposed “mesocircuit”25 linking behavioral fluctuations following severe brain injuries and improvements in response to interventions. The model focuses on the modulatory role of the anterior forebrain in overall corticothalamic dynamics. The anterior forebrain (frontal/prefrontal cortical-striatopallidal thalamocortical loop system) is particularly vulnerable to large-scale dysfunction following multifocal brain injuries that produce widespread deafferentation or neuronal cell loss. At least two mechanisms related to this mesocircuit appear to play a key role after severe injuries: (1) the high demands of striatum output neurons for background activity and dopaminergic innervations and (2) the anatomic connections and physiologic specializations of the thalamocortical projections from the central thalamus. These central thalamic neurons have a potent activating role strongly driving both cortical and striatal neurons and wide point-to-point connections that make them more sensitive reporters of global neuronal loss than other thalamic nuclei. Withdrawal of thalamocortical transmission from the central thalamus is known to associate with coma and other disorders of consciousness (see Schiff and Plum5). The thalamostriatal projection from the central thalamus contacts the medium spiny neurons (MSNs) of the striatum, forming axodendritic (centromedian32) or axospinous (central lateral, parafascicular31) synapses. The MSNs, in turn, send inhibitory projections to the globus pallidus interna (GPi); without MSN output, the GPi tonically inhibits the central thalamus.33 Thus, a suppression of MSN output resulting from a loss of dopaminergic modulation or marked reduction in background synaptic activity can potentially catalyze a shutdown of the anterior forebrain. The mesocircuit model economically accounts for several clinical observations and aspects of normal physiology (see text for further discussion). (DBS = deep brain stimulation)In addition to the studies reviewed above providing evidence for the role of specific disconnection of neurons within the central thalamus in disorders of consciousness, Figure 2 illustrates a key vulnerability of the anterior forebrain in the setting of the widespread deafferentation and neuronal cell loss seen with severe brain injuries. This vulnerability places the role of the central thalamus in a wider context. A “mesocircuit”-level25 model has been proposed that suggests that functional alterations across very large connected neuronal populations of the anterior forebrain may arise primarily as a result of global reductions of excitatory neurotransmission.26,27 The majority of etiologies associated with coma and related disorders of consciousness diagrammed in Figure 1 effectively produce a broad decrease in background synaptic activity and excitatory neurotransmission (eg, diffuse axonal injury, anoxia, hypoxia-ischemia, cerebral vasospasm with strokes; see Posner et al4 for review).
In addition to the wide point-to-point connections of the central thalamus with the cerebral cortex (predominantly connections to frontal and prefrontal cortices; see Van der Werf et al,15 Groenewegen and Berendse,28 Morel et al29), these neurons have important projections to the striatum that return via projections from the globus pallidus. 30 These projections from the central thalamus (both central lateral nucleus and parafascicularis nucleus) diffusely innervate the striatum and project onto the medium spiny neurons (MSNs), the output neuron of structure.31 Because the specific thalamostriatal projections from these central thalamic neurons use glutamate transmitters with a high probability of synaptic release,32 they likely also have a strong role in modulating background activity in the striatum.
The MSNs represent an important point of vulnerability in this anterior forebrain mesocircuit, as they have a key role in maintaining activity in the anterior forebrain through their inhibitory projections to the globus pallidus interna, which in turn inhibits the central thalamus. 33 MSNs have instrinsic cell membrane properties that keep them below their firing threshold unless a high level of spontaneous background synaptic activity arising from excitatory corticostriatal and thalamostriatal inputs is present in concert with sufficient concentrations of the neurotransmitter dopamine.33 In the setting of diffuse deafferentation or neuronal loss following severe brain injury of any type, it is expected that background excitatory synaptic activity is considerably reduced. Under these circumstances, a broad withdrawal of direct excitatory striatal projections from the central thalamus and corticostriatal inputs is likely to cause MSN output to shut down. Observations of regional changes in brain metabolism following severe brain injuries, specific responses to pharmacologic and electrophysiologic interventions in brain-injured subjects, and normal variations in brain state are all consistent with this mesocircuit model (see Schiff26 for comprehensive review). Similarly, a consistent pattern of selective metabolic downregulation within the anterior forebrain has been shown to specifically grade with severity of behavioral impairment following diffuse axonal injury,34 and application of dopaminergic agents in such patients often will produce behavioral facilitation.35,36 These medications may facilitate the output of the MSNs and directly modulate mesial frontal cortical neurons, possibly restoring anterior forebrain activity within the loop connections of the frontal cortex, striatum, pallidum, and central thalamus.
This model provides context for understanding another paradoxical observation—ie, the association of the sedative zolpidem (Ambien), a nonbenzodiazepine hypnotic that potentiates GABAA receptors, with behavioral improvement of alertness and interactive behavior in severely brain-injured patients.37–41 Zolpidem’s primary direct action in patient responders, as originally proposed by Schiff and Posner,27 may be upon on the globus pallidus interna, producing a release of tonic inhibition of the central thalamus in the setting of a broad reduction in background excitatory neurotransmission (as seen, for example, following diffuse hypoxic-ischemic injury) and leading to a shutdown of the inhibitory projection of the MSNs. The GABAA alpha-1 subunit is expressed in large quantity in the globus pallidus interna, and experimental studies support this mechanism of action.42
SINGLE-SUBJECT STUDY OF CENTRAL THALAMIC STIMULATION IN MINIMALLY CONSCIOUS STATE
A further implication of the mesocircuit model is that direct activation of the central thalamus is expected to be the causal step in reactivating a downregulated anterior forebrain system, suggesting that direct modulation of the central thalamus might facilitate behavioral responsiveness in some patients with severe brain injuries. A recent study offers evidence that direct electrical stimulation of the central thalamus can produce behavioral facilitation.
In this single-subject study of central thalamic deep brain stimulation (DBS), a 38-year-old man remained in minimally conscious state for 6 years following a severe closed head injury following blunt trauma to the right frontal lobe.43 After 3 months in a vegetative state, the patient exhibited the first evidence of clear behaviors in response to sensory stimulation consistent with minimally conscious state and advanced to eventually demonstrating a best behavioral response of inconsistent command-following and communication using eye movements. This behavioral level remained unchanged at the start of the DBS study 4 years later, as confirmed by evaluation with the Coma Recovery Scale–Revised (CRS-R), a formal behavioral assessment tool.
Reprinted from Schiff et al.43
Figure 3. Overview of a 6-month alternating crossover study of central thalamic deep brain stimulation (DBS) in a patient in minimally conscious state following severe traumatic brain injury.43 (A) Study timeline. (B) Electrode lead placements within central thalamus of patient’s right (R) and left (L) hemispheres displayed in T1-weighted coronal magnetic resonance image. (C) The patient’s responsiveness on six cognitive and functional measures at presurgical baseline (“Pre”) and during periods when DBS was on (“On”) and off (“Off”) during the crossover phase. Responsiveness measures are shown with 95% confidence intervals (whiskers). Asterisks denote measures for which there were statistically significant differences between DBS “on” versus “off” periods. Dagger is present after “Oral feeding” to note that oral feeding data were not available before titration (hence no “Pre” value for this measure); also, oral feeding scores 1 and 2 are combined for dichotomy. See text for further explanation (including explanations of outcome measures).The patient entered into a study of central thalamic DBS according to the timeline in Figure 3A. An initial 4-month quantitative behavioral assessment was completed prior to placement of the DBS electrodes, which were implanted bilaterally in the anterior intralaminar thalamic nuclei (central lateral nucleus and adjacent paralaminar regions of the thalamus; Figure 3B). Following electrode placement and brief contact-by-contact testing of the electrodes, 2 months of behavioral testing was conducted with the electrodes remaining off to reassess the patient’s postsurgical behavioral baseline, which had not changed as a result of electrode placement. Two subsequent phases of the study focused on evaluation of DBS effects. A 5-month titration phase focused on establishing tolerance of DBS and evaluating several combinations of different electrical stimulation parameters (contact geometry, frequency, intensity) as well as the duration of the stimulation period. Following this titration phase, the patient entered a 6-month double-blind alternating crossover study. Through all phases of the study, the patient received standard rehabilitation efforts amounting to 3 hours a day, 4 days per week.43
Figure 3C summarizes results of the alternating crossover study and compares the prestimulation baseline assessments of various behaviors with the “on” versus “off” testing of the DBS electrodes during the crossover phase. The results demonstrate the overall impact of DBS compared with approximately 6 months of ongoing rehabilitation efforts in the absence of DBS exposure. Overall the findings show marked improvement in behavioral responsiveness compared with prestimulation frequencies of the highest-level behavioral response across six categories. The primary outcome assessments were prospectively chosen from subscales of the CRS-R, which is a well-validated psychometric tool used in patients with disorders of consciousness. CRS-R subscale items that had shown variation during the presurgical baseline assessment were chosen prospectively as the primary outcome measures. Notably, the CRS-R oral motor subscale was not chosen because no variation in this measure had been identified during the baseline assessment period. In addition, an object-naming scale and two other tailored secondary measures were developed later, during the titration phase, as the patient’s behavior changed, and were calibrated to be tested using these secondary measurement scales. All six measures showed marked change from prestimulation baseline levels, with five of the six measures showing higher-level behaviors than those seen prior to stimulation, regardless of whether the electrodes were on or off.43
As shown in Figure 3C, the behaviors captured by secondary measures had never occurred before the titration phase of the study; ie, the patient initially lacked a capacity for object naming, oral feeding, and the complex controlled goal-directed movements captured in the secondary limb movement measure, thus setting a prestimulation baseline frequency of 0 for these measures (see supplementary material in Schiff et al43). Three outcome measures—one primary (CRS-R arousal subscale) and two secondary (oral feeding and limb control)—showed statistically significant dependence on DBS during the 6-month period, as indicated by a significantly higher frequency of maximal score rating during “on” versus “off” periods (Figure 3C). The continuation of improvements during the “off” periods of the crossover trial (relative to the prestimulation baseline assessments) showed that the DBS effects produced carryover changes that remained after the extensive exposure to DBS during the titration period (for further analysis of the dynamic of these data, see supplementary material in Schiff et al43).
Importantly, these observations are limited to a single human subject and do not provide a guide to their generalizability, 44,45 although they are consistent with the proposed mesocircuit model reviewed above. While the precise mechanism underlying this patient’s improved behavioral responsiveness with central thalamic DBS is unknown, it is likely that DBS served to partially reverse the markedly depressed cerebral global metabolism earlier measured in this patient using fluorodeoxyglucose position emission tomography (FDG-PET)46 and also seen in other patients in minimally conscious state.47 The depressed cerebral metabolism seen in minimally conscious state likely reflects volume loss of neurons, deafferentation of remaining neurons, and neuronal functional impairments. All of these mechanisms may result in low firing rates of neurons in the neocortex, thalamus, and striatum. The mesocircuit model in Figure 2 suggests that direct activation of the central thalamus in patients with such chronically downregulated background synaptic activity may produce excitatory output from central thalamic neurons that acts to partially normalize firing rates and possibly firing patterns within the corticostriatopallidal-thalamocortical system.
SPECULATIONS ON THE IMPORTANCE OF HEART-BRAIN RESEARCH IN FUTURE STUDIES OF RECOVERY OF CONSCIOUSNESS
As this conference is focused on the heart-brain interface, it is appropriate to consider the relevance of heart-brain research to the general set of problems reviewed above. In fact, the linkage is quite natural, and classical physiologic psychology research has shown that cerebral arousal regulation is associated with patterned modulation of cardiac rhythm and autonomic function linked to the behavioral state.48,49 Among the most relevant observations are demonstrations that sustained focused attention is associated with several stereotyped cardiac and autonomic changes, including anticipatory bradycardia,50,51 pupillary dilatation, 52 and others (eg, galvanic skin response). Neurologic cases have shown that such couplings of effort to reflex bradycardia, pupillary dilatation, and other autonomic markers are altered by focal cerebral lesions in the right frontal lobe53 and left anterior cingulate cortex.54
In the single-subject central thalamic DBS study reviewed above,43 there were several unpublished observations that are potentially relevant to these mechanisms. During initial bedside testing of the individual DBS electrode contacts in first 2 postoperative days, electrical stimulation above threshold voltages associated with visible arousal response (for details, see supplementary material in Schiff et al43) consistently produced marked changes in heart rate and audible modulations of heart rhythm during interactions with the patient. Notably, the patient’s basal heart rate rose from a stable level of approximately 50 to 55 beats per minute to approximately 70 to 75 beats per minute—a nearly 50% increase. While increases in blood pressure and heart rate typically accompany arousal, the heart rate change observed here may reflect a marked change in cerebral metabolic rates. Earlier quantitative FDG-PET imaging in this patient revealed a global metabolic rate across the brain of approximately half the normal level.46 Considering that the brain consumes approximately 23% of the cardiac output,55 the increased heart rate observed in this setting may reflect an increase in demand in cardiac output, possibly as much as 100%. At the same time that these changes occurred, there was an audible cardiac deceleration noted when the patient was attentionally engaged by the examiner (this occurred without scoreable variation in most of the quantitative neurobehavioral metrics; see supplementary material in Schiff et al43). Of note, although the patient had suffered a complex severe brain injury, the right ventral frontal lobe showed the largest structural lesion46; injuries to the right hemisphere are associated with loss of such anticipatory changes in heart rate during attentional task performance.53
These anecdotal observations suggest that future studies that include measures to track patterns of heart rate variation during recovery of consciousness might provide an indirect index of increasing brain demand for allocation of cardiac output or emergent neural control of mechanisms linking cardiovascular response to attentive behavior. Ongoing coupling of electroencephalographic measures to autonomic and basal cardiac rhythms may be particularly interesting to examine during social interactions,56 as it is likely that behavioral responsiveness is linked to social stimuli. Emotional reactivity has been proposed as an essential component of arousal per se,49 and although not formally studied in the DBS trial reviewed above, emotional reengagement seems to be a clear concomitant of the collection of gestural and verbal behavioral improvements operationally tracked using quantitative behavioral scales. Beyond tracking heart-brain interactions as an index of brain recovery, it is possible that the integrity of heart-brain interactions may also be a target for optimization in support of recovery of consciousness after nonprogressive brain injury. Moreover, studies of optimization of cardiac function in severely brain-injured patients may provide insight into the recovery process as well.
References
Blake R. The Day Donny Herbert Woke Up: A True Story. New York, NY: Harmony Press; 2007.
Schiff ND, Fins JJ. Hope for “comatose” patients. Cerebrum2003; 5:7–24.
Posner J, Saper C, Schiff N, Plum F. Plum and Posner’s Diagnosis of Stupor and Coma. 4th ed.New York: Oxford University Press; 2007.
Schiff ND, Plum F. The role of arousal and ‘gating’ systems in the neurology of impaired consciousness. J Clin Neurophysiol2000; 17:438–452.
Kobylarz EJ, Schiff ND. Neurophysiological correlates of persistent vegetative and minimally conscious states. Neuropsychol Rehabil2005; 15:323–332.
Giacino JT, Ashwal S, Childs N, et al The minimally conscious state: definition and diagnostic criteria. Neurology2002; 58:349–353.
Giacino JT, Whyte J. The vegetative and minimally conscious states: current knowledge and remaining questions. J Head Trauma Rehabil2005; 20:30–50.
Nakase-Richardson R, Yablon SA, Sherer M, Nick TG, Evans CC. Emergence from minimally conscious state: insights from evaluation of posttraumatic confusion. Neurology2009; 73:1120–1126.
Adams JH, Graham DI, Jennett B. The neuropathology of the vegetative state after acute insult. Brain2000; 123:1327–1338.
Jennett B, Adams JH, Murray LS, Graham DI. Neuropathology in vegetative and severely disabled patients after head injury. Neurology2001; 56:486–490.
Adams JH, Graham DI, Jennett B. The structural basis of moderate disability after traumatic brain damage. J Neurol Neurosurg Psychiatry2001; 71:521–524.
Maxwell WL, MacKinnon MA, Smith DH, McIntosh TK, Graham DI. Thalamic nuclei after human blunt head injury. J Neuropathol Exp Neurol2006; 65:478–488.
Kinney HC, Korein J, Panigrahy A, Dikkes P, Goode R. Neuropathological findings in the brain of Karen Ann Quinlan. The role of the thalamus in the persistent vegetative state. N Engl J Med1994; 330:1469–1475.
Van der Werf YD, Witter MP, Groenewegen HJ. The intralaminar and midline nuclei of the thalamus. Anatomical and functional evidence for participation in processes of arousal and awareness. Brain Res Brain Res Rev2002; 39:107–140.
Scannell JW, Burns GA, Hilgetag CC, O’Neil MA, Young MP. The connectional organization of the cortico-thalamic system of the cat. Cereb Cortex1999; 9:277–299.
Castaigne P, Lhermitte F, Buge A, Escourolle R, Hauw JJ, Lyon-Caen O. Paramedian thalamic and midbrain infarct: clinical and neuropathological study. Ann Neurol1981; 10:127–148.
Schiff ND. Central thalamic contributions to arousal regulation and neurological disorders of consciousness. Ann N Y Acad Sci2008; 1129:105–118.
Kinomura S, Larsson J, Gulyás B, Roland PE. Activation by attention of the human reticular formation and thalamic intralaminar nuclei. Science1996; 271:512–515.
Paus T, Koski L, Caramanos Z, Westbury C. Regional differences in the effects of task difficulty and motor output on blood flow response in the human anterior cingulate cortex: a review of 107 PET activation studies. Neuroreport1998; 9:R37–47.
Nagai Y, Critchley HD, Featherstone E, Fenwick PB, Trimble MR, Dolan RJ. Brain activity relating to the contingent negative variation: an fMRI investigation. Neuroimage2004; 21:1232–1241.
Paus T, Zatorre RJ, Hofle N, et al Time-related changes in neural systems underlying attention and arousal during the performance of an auditory vigilance task. J Cogn Neurosci1997; 9:392–408.
Shah SA, Baker JL, Ryou JW, Purpura KP, Schiff ND. Modulation of arousal regulation with central thalamic deep brain stimulation. Conf Proc IEEE Eng Med Biol Soc2009; 2009:3314–3317.
Wyder MT, Massoglia DP, Stanford TR. Contextual modulation of central thalamic delay-period activity: representation of visual and saccadic goals. J Neurophysiol2004; 91:2628–2648.
Bohland JW, Wu C, Barbas H, et al A proposal for a coordinated effort for the determination of brainwide neuroanatomical connectivity in model organisms at a mesoscopic scale. PLoS Comput Biol2009; 5:e1000334.
Schiff ND. Recovery of consciousness after brain injury: a mesocircuit hypothesis. Trends Neurosci2010; 33:1–9.
Schiff ND, Posner JP. Another “Awakenings.”Ann Neurol2007; 62:5–7.
Groenewegen HJ, Berendse HW. The specificity of the ‘nonspecific’ midline and intralaminar thalamic nuclei. Trends Neurosci1994; 17:52–57.
Morel A, Liu J, Wannier T, Jeanmonod D, Rouiller EM. Divergence and convergence of thalamocortical projections to premotor and supplementary motor cortex: a multiple tracing study in the macaque monkey. Eur J Neurosci2005; 21:1007–1029.
Deschenes M, Bourassa J, Parent A. Striatal and cortical projections of single neurons from the central lateral thalamic nucleus in the rat. Neuroscience1996; 72:679–687.
Lacey CJ, Bolam JP, Magill PJ. Novel and distinct operational principles of intralaminar thalamic neurons and their striatal projections. J Neurosci2007; 27:4374–4384.
Smith Y, Raju D, Nanda B, Pare JF, Galvan A, Wichmann T. The thalamostriatal systems: anatomical and functional organization in normal and parkinsonian states. Brain Res Bull2009; 78:60–68.
Grillner S, Hellgren J, Ménard A, Saitoh K, Wikström MA. Mechanisms for selection of basic motor programs—roles for the striatum and pallidum. Trends Neurosci2005; 28:364–370.
Kato T, Nakayama N, Yasokawa Y, Okumura A, Shinoda J, Iwama T. Statistical image analysis of cerebral glucose metabolism in patients with cognitive impairment following diffuse traumatic brain injury. J Neurotrauma2007; 24:919–926.
Matsuda W, Matsumura A, Komatsu Y, Yanaka K, Nose T. Awakenings from persistent vegetative state: report of three cases with parkinsonism and brain stem lesions on MRI. J Neurol Neurosurg Psychiatry2003; 74:1571–1573.
Meythaler JM, Brunner RC, Johnson A, Novack TA. Amantadine to improve neurorecovery in traumatic brain injury–associated diffuse axonal injury: a pilot double-blind randomized trial. J Head Trauma Rehabil2002; 17:300–313.
Brefel-Courbon C, Payoux P, Ory F, et al Clinical and imaging evidence of zolpidem effect in hypoxic encephalopathy. Ann Neurol2007; 62:102–105.
Whyte J, Myers R. Incidence of clinically significant responses to zolpidem among patients with disorders of consciousness: a preliminary placebo controlled trial. Am J Phys Med Rehabil2009; 88:410–418.
Shames JL, Ring H. Transient reversal of anoxic brain injury–related minimally conscious state after zolpidem administration: a case report. Arch Phys Med Rehabil2008; 89:386–388.
Cohen SI, Duong TT. Increased arousal in a patient with anoxic brain injury after administration of zolpidem. Am J Phys Med Rehabil2008; 87:229–231.
Williams ST, Conte MM, Kobylarz EJ, Hersh JE, Victor JD, Schiff ND. Quantitative neurophysiologic characterization of a paradoxical response to zolpidem in a severely brain-injured human subject. Program No. 541.6/R9. 2009Neuroscience Meeting Planner. Chicago, IL: Society for Neuroscience, 2009. Online.
Chen L, Savio Chan C, Yung WH. Electrophysiological and behavioral effects of zolpidem in rat globus pallidus. Exp Neurol2004; 186:212–220.
Schiff ND, Giacino JT, Kalmar K, et al Behavioral improvements with thalamic stimulation after severe traumatic brain injury. Nature2007; 448:600–603.
Victor JD, Schiff ND. Meeting rigorous statistical standards in case reports. Ann Neurol2008; 64:592.
Schiff ND, Giacino JT, Fins JJ. Deep brain stimulation, neuroethics, and the minimally conscious state: moving beyond proof of principle. Arch Neurol2009; 66:697–702.
Schiff ND, Rodriguez-Moreno D, Kamal A, et al fMRI reveals large-scale network activation in minimally conscious patients. Neurology2005; 64:514–523.
Laureys S, Owen AM, Schiff ND. Brain function in coma, vegetative state, and related disorders. Lancet Neurol2004; 3:537–546.
Obrist PA. Presidential address, 1975. The cardiovascular-behavioral interaction—as it appears today. Psychophysiology1976; 13:95–107.
Pfaff D. Brain Arousal and Information Theory: Neural and Genetic Mechanisms. Cambridge, MA: Harvard University Press; 2006.
Obrist PA, Light KC, Langer AW, Gringnolo A, McCubbin JA. Behavioural-cardiac interactions: the psychosomatic hypothesis. J Psychosom Res1978; 22:301–325.
Kahneman D. Attention and Effort. Englewood Cliffs, NJ: Prentice-Hall; 1973.
Yokoyama K, Jennings R, Ackles P, Hood P, Boller F. Lack of heart rate changes during an attention-demanding task after right hemisphere lesions. Neurology1987; 37:624–630.
Naccache L, Dehaene S, Cohen L, et al Effortless control: executive attention and conscious feeling of mental effort are dissociable. Neuropsychologia2005; 43:1318–1328.
Roland P. Brain Activation. New York, NY: Wiley Press; 1997.
Porges SW. The polyvagal theory: new insights into adaptive reactions of the autonomic nervous system. Cleve Clin J Med2009; 76( suppl 2):S86–S90.
References
Blake R. The Day Donny Herbert Woke Up: A True Story. New York, NY: Harmony Press; 2007.
Schiff ND, Fins JJ. Hope for “comatose” patients. Cerebrum2003; 5:7–24.
Posner J, Saper C, Schiff N, Plum F. Plum and Posner’s Diagnosis of Stupor and Coma. 4th ed.New York: Oxford University Press; 2007.
Schiff ND, Plum F. The role of arousal and ‘gating’ systems in the neurology of impaired consciousness. J Clin Neurophysiol2000; 17:438–452.
Kobylarz EJ, Schiff ND. Neurophysiological correlates of persistent vegetative and minimally conscious states. Neuropsychol Rehabil2005; 15:323–332.
Giacino JT, Ashwal S, Childs N, et al The minimally conscious state: definition and diagnostic criteria. Neurology2002; 58:349–353.
Giacino JT, Whyte J. The vegetative and minimally conscious states: current knowledge and remaining questions. J Head Trauma Rehabil2005; 20:30–50.
Nakase-Richardson R, Yablon SA, Sherer M, Nick TG, Evans CC. Emergence from minimally conscious state: insights from evaluation of posttraumatic confusion. Neurology2009; 73:1120–1126.
Adams JH, Graham DI, Jennett B. The neuropathology of the vegetative state after acute insult. Brain2000; 123:1327–1338.
Jennett B, Adams JH, Murray LS, Graham DI. Neuropathology in vegetative and severely disabled patients after head injury. Neurology2001; 56:486–490.
Adams JH, Graham DI, Jennett B. The structural basis of moderate disability after traumatic brain damage. J Neurol Neurosurg Psychiatry2001; 71:521–524.
Maxwell WL, MacKinnon MA, Smith DH, McIntosh TK, Graham DI. Thalamic nuclei after human blunt head injury. J Neuropathol Exp Neurol2006; 65:478–488.
Kinney HC, Korein J, Panigrahy A, Dikkes P, Goode R. Neuropathological findings in the brain of Karen Ann Quinlan. The role of the thalamus in the persistent vegetative state. N Engl J Med1994; 330:1469–1475.
Van der Werf YD, Witter MP, Groenewegen HJ. The intralaminar and midline nuclei of the thalamus. Anatomical and functional evidence for participation in processes of arousal and awareness. Brain Res Brain Res Rev2002; 39:107–140.
Scannell JW, Burns GA, Hilgetag CC, O’Neil MA, Young MP. The connectional organization of the cortico-thalamic system of the cat. Cereb Cortex1999; 9:277–299.
Castaigne P, Lhermitte F, Buge A, Escourolle R, Hauw JJ, Lyon-Caen O. Paramedian thalamic and midbrain infarct: clinical and neuropathological study. Ann Neurol1981; 10:127–148.
Schiff ND. Central thalamic contributions to arousal regulation and neurological disorders of consciousness. Ann N Y Acad Sci2008; 1129:105–118.
Kinomura S, Larsson J, Gulyás B, Roland PE. Activation by attention of the human reticular formation and thalamic intralaminar nuclei. Science1996; 271:512–515.
Paus T, Koski L, Caramanos Z, Westbury C. Regional differences in the effects of task difficulty and motor output on blood flow response in the human anterior cingulate cortex: a review of 107 PET activation studies. Neuroreport1998; 9:R37–47.
Nagai Y, Critchley HD, Featherstone E, Fenwick PB, Trimble MR, Dolan RJ. Brain activity relating to the contingent negative variation: an fMRI investigation. Neuroimage2004; 21:1232–1241.
Paus T, Zatorre RJ, Hofle N, et al Time-related changes in neural systems underlying attention and arousal during the performance of an auditory vigilance task. J Cogn Neurosci1997; 9:392–408.
Shah SA, Baker JL, Ryou JW, Purpura KP, Schiff ND. Modulation of arousal regulation with central thalamic deep brain stimulation. Conf Proc IEEE Eng Med Biol Soc2009; 2009:3314–3317.
Wyder MT, Massoglia DP, Stanford TR. Contextual modulation of central thalamic delay-period activity: representation of visual and saccadic goals. J Neurophysiol2004; 91:2628–2648.
Bohland JW, Wu C, Barbas H, et al A proposal for a coordinated effort for the determination of brainwide neuroanatomical connectivity in model organisms at a mesoscopic scale. PLoS Comput Biol2009; 5:e1000334.
Schiff ND. Recovery of consciousness after brain injury: a mesocircuit hypothesis. Trends Neurosci2010; 33:1–9.
Schiff ND, Posner JP. Another “Awakenings.”Ann Neurol2007; 62:5–7.
Groenewegen HJ, Berendse HW. The specificity of the ‘nonspecific’ midline and intralaminar thalamic nuclei. Trends Neurosci1994; 17:52–57.
Morel A, Liu J, Wannier T, Jeanmonod D, Rouiller EM. Divergence and convergence of thalamocortical projections to premotor and supplementary motor cortex: a multiple tracing study in the macaque monkey. Eur J Neurosci2005; 21:1007–1029.
Deschenes M, Bourassa J, Parent A. Striatal and cortical projections of single neurons from the central lateral thalamic nucleus in the rat. Neuroscience1996; 72:679–687.
Lacey CJ, Bolam JP, Magill PJ. Novel and distinct operational principles of intralaminar thalamic neurons and their striatal projections. J Neurosci2007; 27:4374–4384.
Smith Y, Raju D, Nanda B, Pare JF, Galvan A, Wichmann T. The thalamostriatal systems: anatomical and functional organization in normal and parkinsonian states. Brain Res Bull2009; 78:60–68.
Grillner S, Hellgren J, Ménard A, Saitoh K, Wikström MA. Mechanisms for selection of basic motor programs—roles for the striatum and pallidum. Trends Neurosci2005; 28:364–370.
Kato T, Nakayama N, Yasokawa Y, Okumura A, Shinoda J, Iwama T. Statistical image analysis of cerebral glucose metabolism in patients with cognitive impairment following diffuse traumatic brain injury. J Neurotrauma2007; 24:919–926.
Matsuda W, Matsumura A, Komatsu Y, Yanaka K, Nose T. Awakenings from persistent vegetative state: report of three cases with parkinsonism and brain stem lesions on MRI. J Neurol Neurosurg Psychiatry2003; 74:1571–1573.
Meythaler JM, Brunner RC, Johnson A, Novack TA. Amantadine to improve neurorecovery in traumatic brain injury–associated diffuse axonal injury: a pilot double-blind randomized trial. J Head Trauma Rehabil2002; 17:300–313.
Brefel-Courbon C, Payoux P, Ory F, et al Clinical and imaging evidence of zolpidem effect in hypoxic encephalopathy. Ann Neurol2007; 62:102–105.
Whyte J, Myers R. Incidence of clinically significant responses to zolpidem among patients with disorders of consciousness: a preliminary placebo controlled trial. Am J Phys Med Rehabil2009; 88:410–418.
Shames JL, Ring H. Transient reversal of anoxic brain injury–related minimally conscious state after zolpidem administration: a case report. Arch Phys Med Rehabil2008; 89:386–388.
Cohen SI, Duong TT. Increased arousal in a patient with anoxic brain injury after administration of zolpidem. Am J Phys Med Rehabil2008; 87:229–231.
Williams ST, Conte MM, Kobylarz EJ, Hersh JE, Victor JD, Schiff ND. Quantitative neurophysiologic characterization of a paradoxical response to zolpidem in a severely brain-injured human subject. Program No. 541.6/R9. 2009Neuroscience Meeting Planner. Chicago, IL: Society for Neuroscience, 2009. Online.
Chen L, Savio Chan C, Yung WH. Electrophysiological and behavioral effects of zolpidem in rat globus pallidus. Exp Neurol2004; 186:212–220.
Schiff ND, Giacino JT, Kalmar K, et al Behavioral improvements with thalamic stimulation after severe traumatic brain injury. Nature2007; 448:600–603.
Victor JD, Schiff ND. Meeting rigorous statistical standards in case reports. Ann Neurol2008; 64:592.
Schiff ND, Giacino JT, Fins JJ. Deep brain stimulation, neuroethics, and the minimally conscious state: moving beyond proof of principle. Arch Neurol2009; 66:697–702.
Schiff ND, Rodriguez-Moreno D, Kamal A, et al fMRI reveals large-scale network activation in minimally conscious patients. Neurology2005; 64:514–523.
Laureys S, Owen AM, Schiff ND. Brain function in coma, vegetative state, and related disorders. Lancet Neurol2004; 3:537–546.
Obrist PA. Presidential address, 1975. The cardiovascular-behavioral interaction—as it appears today. Psychophysiology1976; 13:95–107.
Pfaff D. Brain Arousal and Information Theory: Neural and Genetic Mechanisms. Cambridge, MA: Harvard University Press; 2006.
Obrist PA, Light KC, Langer AW, Gringnolo A, McCubbin JA. Behavioural-cardiac interactions: the psychosomatic hypothesis. J Psychosom Res1978; 22:301–325.
Kahneman D. Attention and Effort. Englewood Cliffs, NJ: Prentice-Hall; 1973.
Yokoyama K, Jennings R, Ackles P, Hood P, Boller F. Lack of heart rate changes during an attention-demanding task after right hemisphere lesions. Neurology1987; 37:624–630.
Naccache L, Dehaene S, Cohen L, et al Effortless control: executive attention and conscious feeling of mental effort are dissociable. Neuropsychologia2005; 43:1318–1328.
Roland P. Brain Activation. New York, NY: Wiley Press; 1997.
Porges SW. The polyvagal theory: new insights into adaptive reactions of the autonomic nervous system. Cleve Clin J Med2009; 76( suppl 2):S86–S90.
This review highlights important recent publications in the area of neuroscience and heart-brain medicine. Abnormalities of regulation of the circulation by catecholamine systems figure as a general theme of the topics highlighted. These topics, which are reviewed in turn below, are (1) mechanisms of cardiac sympathetic denervation in Parkinson disease (PD), (2) cytoplasmic monoamine metabolites as autotoxins, and (3) the validity of power spectral analysis of heart rate variability to indicate cardiac sympathetic tone.
MECHANISMS OF CARDIAC SYMPATHETIC DENERVATION IN PARKINSON DISEASE
The movement disorder component of PD is well recognized as resulting from loss of dopaminergic neurons in the nigrostriatal system of the brain. The finding of low myocardial 6-[18F]fluorodopamine–derived radioactivity by positron emission tomography provided the first neuroimaging evidence for loss of catecholaminergic neurons outside the brain in PD.1 Many reports using 123I-metaiodobenzylguanidine scanning have concurred with this finding. Beginning in the early 2000s, post-mortem neuropathologic studies demonstrated virtually absent immunoreactivity for tyrosine hydroxylase, the rate-limiting enzyme in norepinephrine biosynthesis, in epicardial nerves in PD.2,3 These results provided clues to the mechanism of autonomic dysfunction in PD, a prominent nonmotor manifestation of the disease.
With kind permission from Springer Science+Business Media: Acta Neuropathologica,
Figure 1. Tyrosine hydroxylase immunoreactivity (THir) in epicardial nerve from (A) a control subject and (B) a patient with familial Parkinson disease due to duplication of the gene encoding alpha-synuclein (PARK4).6Alpha-synuclein is a key protein in the pathogenesis of PD. It is abundant in Lewy bodies and Lewy neurites, and mutations or multiplications of the gene that encodes it cause rare inherited forms of PD. In 2001 we reported evidence for cardiac sympathetic denervation, neurogenic orthostatic hypotension, and baroreflex failure in familial PD from mutation of the gene encoding alpha-synuclein.4 Subsequently we reported analogous denervation in familial PD from triplication of the normal gene.5 This past year Orimo’s group in Tokyo provided the first pathological confirmation of cardiac sympathetic denervation in familial PD from inherited alpha-synucleinopathy, based on severely decreased epicardial neuronal tyrosine hydroxylase immunoreactivity (Figure 1).6 In contrast, patients with familial PD from parkin gene mutation, which is not thought to be a Lewy body disease, have been found to have normal cardiac 123I-metaiodobenzyl-guanidine–derived radioactivity and normal epicardial neuronal tyrosine hydroxylase immunoreactivity.7 These findings establish a link between alpha-synucleinopathy and cardiac sympathetic denervation.
Some individuals who die without clinical parkinsonism have Lewy bodies detected pathologically. Growing evidence shows that incidental Lewy body disease represents early, presymptomatic PD.8 Orimo’s group therefore studied cardiac tissues and paravertebral sympathetic ganglia from patients with incidental Lewy body disease.9 Postmortem tissues were likewise obtained from comparison subjects with multiple system atrophy and from control subjects. Immunohistochemical analyses were performed using antibodies against tyrosine hydroxylase, phosphorylated neurofilament as a marker of axons, and phosphorylated alpha-synuclein as a marker of abnormal alpha-synuclein deposits. Key findings from this study9 were as follows:
Reprinted from Brain (Orimo S, et al. Axonal α-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain 2008; 131:642–650) by permission of Oxford University Press.
Figure 2. Concept diagram of the pathogenetic sequence of cardiac sympathetic denervation. In incidental Lewy body disease with preserved tyrosine hydroxylase–immunoreactive (THir) axons (a), alpha-synuclein aggregates (black shading) accumulate abundantly in the distal axons but sparsely in the paravertebral sympathetic ganglia. In contrast, in incidental Lewy body disease with decreased THir axons (b), alpha-synuclein aggregates diminish in the distal axons but increase in the paravertebral sympathetic ganglia. In Parkinson disease, alpha-synuclein aggregates disappear in the distal axons and accumulate much more abundantly in the paravertebral sympathetic ganglia. In multiple system atrophy, alpha-synuclein aggregates are generally not observed (as in controls), with a few exceptions. Dotted lines indicate degeneration of THir axons.9Alpha-synuclein aggregates in distal epicardial nerve fascicles were more abundant in incidental Lewy body disease with preserved tyrosine hydroxylase–immunoreactive (THir) axons than in incidental Lewy body disease with decreased THir axons (Figure 2).
Alpha-synuclein aggregates in the epicardial nerve fibers were closely related to the disappearance of THir axons.
In incidental Lewy body disease with preserved THir axons, alpha-synuclein aggregates were consistently more abundant in the epicardial nerves than in the paravertebral sympathetic ganglia (Figure 2).
Distally dominant accumulation of alpha-synuclein aggregates was reversed in incidental Lewy body disease with decreased THir axons and in PD, because both conditions involve fewer alpha-synuclein aggregates in axons and more abundant aggregates in the paravertebral sympathetic ganglia (Figure 2).
Thus, accumulation of alpha-synuclein aggregates in distal cardiac sympathetic axons precedes aggregation in neuronal somata or ganglionic neurites, heralding centripetal degeneration of cardiac sympathetic nerves in PD. This chronological and dynamic relationship between alpha-synuclein aggregation and distally dominant degeneration of cardiac noradrenergic nerves may represent the pathological mechanism behind a common degenerative process in PD.
In conclusion, cardiac noradrenergic denervation in Lewy body diseases, even in early stages, accounts for reduced cardiac uptake of 123I-metaiodobenzylguanidine and 6-[18F]fluorodopamine in PD. Alpha-synuclein aggregation appears to be intimately involved in the cardiac noradrenergic denervation that attends Lewy body diseases. The pathogenetic process seems to proceed in a centripetal, retrograde direction.
CYTOPLASMIC MONOAMINE METABOLITES AS AUTOTOXINS
Current concepts about mechanisms of PD emphasize pathologic alpha-synuclein accumulation, oxidative injury, impaired proteasomal or mitochondrial functions, neuroinflammation, or abnormal kinase signaling. These concepts do not explain relatively selective nigrostriatal dopaminergic and cardiac noradrenergic denervation in PD.
Figure 3. According to the monoamine aldehyde hypothesis, interference with the vesicular recycling of cytoplasmic monoamines (dopamine [DA], norepinephrine [NE], and serotonin [5-HT]) augments formation of toxic aldehydes. For instance, DA that leaks from vesicles (V) into the cytoplasm (C) or that is taken up via the cell membrane DA transporter (DAT) and escapes vesicular reuptake via the vesicular monoamine transporter (VMAT) is subject to oxidative deamination catalyzed by monoamine oxidase (MAO) to form the catecholaldehyde DOPAL, which is toxic. DOPAL is detoxified by ALDH to form DOPAC, the major metabolic route, or by AR to form DOPET, the minor metabolic route. Analogously, NE is converted to DOPEGAL, and 5-HT is converted to 5-HT-aldehyde (5-HTAld).
A potential explanation is that cytoplasmic catecholamine metabolites are autotoxins (Figure 3). The mechanisms of autotoxicity include spontaneous auto-oxidation, to form quinones and chromes leading to increased production of reactive oxygen species, and enzymatic oxidation.
Catecholamines in the neuronal cytoplasm undergo enzymatic oxidative deamination to form catecholaldehydes (dihydroxyphenylacetaldehyde [DOPAL] from dopamine), which are cytotoxic, as predicted by Blaschko more than a half century ago.10 DOPAL is detoxified mainly by aldehyde dehydrogenase (ALDH). In the substantia nigra, aldehyde dehydrogenase 1A1 (ALDH1A1) is the main isoform of ALDH, and postmortem studies have noted decreased nigral ALDH1A1 gene expression11,12 and protein content13 in PD patients.
All neurons express alpha-synuclein. Current concepts about mechanisms also do not explain the relatively selective aggregation of alpha-synuclein in catecholaminergic neurons. Alpha-synuclein appears to play a role in the cycling of catecholamines across vesicular and cell membranes.14
Figure 4. Cell survival and cytoplasmic dopamine are inversely related, according to a murine model by Mosharov et al.16 Graph shows the dependence of cell survival under l-dopa–induced stress on the cytoplasmic dopamine (DAcyt) dose in mouse neurons. The DAcyt dose was estimated as: [DAcyt]×TExposure = [DAcyt]×Ln([L-dopa]/K0.5)/k,where [DAcyt] is the concentration of cytosolic DA in cells treated with a saturating level (> 50 μM) of l-dopa for 1 hour, where [l-dopa] is the initial drug concentration, and where K0.5 = 9.7 μM and k = 0.15 hr−1 are the kinetic constants. TExposure approximates the time during which extracellular l-dopa remained higher than K0.5. The data points are (from left to right): filled circles—ventral midbrain cultures treated with 25, 100, 250, 500, and 1,000 μM l-dopa alone; open circles—ventral midbrain neurons treated with 250 μM l-dopa in the presence of benserazide, methamphetamine, reserpine, pargyline, and pargyline reserpine; diamonds—ventral tegmental area and substantia nigra neurons; triangles—striatal and cortical neurons treated with 250 μM l-dopa. Dotted lines and shaded boxes represent mean ± SEM in untreated cells. The solid line is the linear fit of all data points, excluding striatal and cortical neurons and the two data points indicated by the asterisk. Treatments to the right of this line are neuroprotective, as the same level of cell death is achieved with higher DAcyt doses; treatments to the left of this line are more susceptible to DAcyt stress.In the past year, a few important studies have been published related to autotoxicity of cytoplasmic catecholamine metabolites and to pathogenic interactions with alpha-synuclein. In 2006, Mosharov et al reported that alpha-synuclein overexpression increases cytoplasmic dopamine concentrations in rat pheochromocytoma PC-12 cells.15 Recently, the same group, using intracellular patch electrochemistry, directly measured cytoplasmic dopamine in cultured midbrain neurons and found that increases in dopamine and its metabolites are neurotoxic, whereas manipulations that reduce cytoplasmic dopamine are neuroprotective (Figure 4).16 Levodopa (l-dopa) increased cytoplasmic dopamine more in substantia nigra neurons than in ventral tegmental neurons, suggesting that this difference might help explain the greater susceptibility of nigral neurons to the pathogenetic process. The greater buildup of cytoplasmic dopamine seemed to depend on dihydropyridine-sensitive calcium (Ca2+) channels. Finally, dopaminergic neurons lacking alpha-synuclein were resistant to l-dopa–induced cell death. These findings led the authors to propose a “multiple-hit” model (Figure 5) in which interactions between intracellular ionized calcium, cytoplasmic dopamine, and alpha-synuclein underlie susceptibility of nigral neurons in PD.16
Figure 5. The “multiple-hit” model of Parkinson disease pathogenesis,16 which holds that neurotoxicity is a result of multiple factors, including the presence of alpha-synuclein (α-syn), elevation of cytoplasmic calcium (Ca2+), and buildup of cytoplasmic dopamine (DAcyt) and its metabolites. Nonexclusive toxic steps may result from (1) mechanisms that require direct interaction between DA or its metabolites with α-syn, such as DA-modified stabilization of α-syn protofibrils or inhibition of chaperone-mediated autophagy, or (2) cumulative damage from multiple independent sources. Reducing the levels of any of the three players provides neuroprotection. (AADC = aromatic l-amino acid decarboxylase; DOPAL = dihydroxyphenylacetaldehyde; TH = tyrosine hydroxylase)Burke et al added a potentially important clue, demonstrating that DOPAL potently oligomerizes and aggregates alpha-synuclein.17 This finding introduces the possibility of multiple pathogenetic positive feedback loops.
Under resting conditions, most catecholamine turnover results from leakage from vesicular stores into the cytoplasm and subsequent oxidative deamination by monoamine oxidase. Ordinarily, however, catecholamines in the cytoplasm are efficiently recycled back into the vesicles via the type 2 vesicular monoamine transporter (VMAT-2). Accordingly, interference with VMAT functions would be expected to tend to build up cytoplasmic catecholamines, with potentially cytotoxic consequences. In 2007, Caudle et al reported that mice with severely decreased VMAT-2 have aging-associated decreases in striatal dopamine that begin in the terminal fields, alpha-synuclein deposition in substantia nigra neurons, and l-dopa–responsive behavioral deficits.18 More recently the same group noted nonmotor signs associated with PD in VMAT-2–deficient mice, such as anosmia, gastrointestinal hypomotility, sleep disturbances, anxiety, and depression.19 Since VMAT-2 serves to recycle not only dopamine but also norepinephrine and serotonin, this single abnormality could help explain loss of all three types of monoaminergic neurons in PD.
Finally, Pena-Silva et al recently tested whether serotonin induces oxidative stress in human heart valves.20 They showed that in heart valves from explanted human hearts not used for transplantation, incubation of homogenates of cardiac valves and blood vessels with serotonin increased generation of the superoxide free radical. Inhibitors of monoamine oxidase prevented this effect. Dopamine also increased superoxide levels in heart valves, and this effect was also attenuated by monoamine oxidase inhibition. These findings fit with the concept that the aldehydes produced by the action of monoamine oxidase on cytoplasmic monoamines generate toxic free radicals.
VALIDITY OF POWER SPECTRAL ANALYSIS OF HEART RATE VARIABILITY TO INDICATE CARDIAC SYMPATHETIC TONE
Power spectral analysis of heart rate variability is simple, relatively inexpensive, noninvasive, and widely used to indicate cardiac sympathetic “tone” or sympathovagal “balance.” Almost 2,000 studies to date have used this modality. Relatively increased cardiac sympathetic tone, reflected by low-frequency (LF) power or the ratio of LF power to high-frequency (HF) power, is an adverse prognostic sign in a variety of conditions. Nevertheless, the validity of LF power, or the LF:HF ratio, as an index of cardiac sympathetic tone remains unsettled.
In 2007 we assessed the validity of power spectral analysis rather directly, by taking advantage of our ability to delineate cardiac sympathetic innervation. We compared LF power in patients with cardiac sympathetic denervation, indicated by low myocardial levels of 6-[18F]fluorodopamine–derived radioactivity or low rates of norepinephrine entry into coronary sinus plasma (cardiac norepinephrine spillover), with values in patients with intact innervation. LF power was unrelated to myocardial 6-[18F]fluorodopamine–derived radioactivity or cardiac norepinephrine spillover, but it was related to baroreflex-cardiovagal gain. Patients with a low baroreflex-cardiovagal gain had low LF power, regardless of cardiac innervation. From these findings we concluded that LF power reflects baroreflex function, not cardiac sympathetic innervation.21
Figure 6. Relationships of heart rate variability indices with cardiac norepinephrine spillover. Graphs show time and frequency domain heart rate variability measures (LF = low frequency; HF = high frequency) versus cardiac norepinephrine in healthy subjects (squares) and patients with major depression (triangles) and panic disorder (circles).22 Recently Baumert et al also examined the relationship between indices from power spectral analysis of heart rate variability and cardiac norepinephrine spillover.22 They found, as we did, that none of the standard heart rate variability parameters was correlated with cardiac norepinephrine spillover (Figure 6). The same group reported a positive correlation between the heart rate–corrected QT interval and cardiac norepinephrine spillover.23 Among patients with major depression, the distribution of cardiac norepinephrine spillover seemed bimodal. Overall, cardiac norepinephrine spillover was not increased, although a subgroup had clearly increased spillover.
In congestive heart failure, baroreflex-cardiovagal gain tends to be low and cardiac sympathetic outflow markedly increased, yet the LF:HF ratio is not increased during supine rest.24,25 It therefore appears that power spectral analysis of heart rate variability may provide a measure of baroreflexive modulation of autonomic outflows to the heart but not a measure of those outflows themselves. The search continues for a valid, noninvasive means to assess cardiac sympathetic function.
References
Goldstein DS, Holmes C, Cannon RO, Eisenhofer G, Kopin IJ. Sympathetic cardioneuropathy in dysautonomias. N Engl J Med1997; 336:696–702.
Orimo S, Ozawa E, Oka T, et al Different histopathology accounting for a decrease in myocardial MIBG uptake in PD and MSA. Neurology2001; 57:1140–1141.
Amino T, Orimo S, Takahashi A, Uchihara T, Mizusawa H. Profound cardiac sympathetic denervation occurs in Parkinson disease. Brain Path2005; 15:29–34.
Goldstein DS, Li S-T, Kopin IJ. Sympathetic neurocirculatory failure in Parkinson disease: evidence for an etiologic role of α-synuclein. Ann Intern Med2001; 135:1010–1011.
Singleton A, Gwinn-Hardy K, Sharabi Y, et al Association between cardiac denervation and parkinsonism caused by alpha-synuclein gene triplication. Brain2004; 127:768–772.
Orimo S, Uchihara T, Nakamura A, et al Cardiac sympathetic denervation in Parkinson’s disease linked to SNCA duplication. Acta Neuropathol2008; 116:575–577.
Orimo S, Amino T, Yokochi M, et al Preserved cardiac sympathetic nerve accounts for normal cardiac uptake of MIBG in PARK2. Mov Disord2005; 20:1350–1353.
Dickson DW, Fujishiro H, DelleDonne A, et al Evidence that incidental Lewy body disease is pre-symptomatic Parkinson’s disease. Acta Neuropathol2008; 115:437–444.
Orimo S, Uchihara T, Nakamura A, et al Axonal alpha-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain2008; 131:642–650.
Blaschko H. Amine oxidase and amine metabolism. Pharmacol Rev1952; 4:415–458.
Galter D, Buervenich S, Carmine A, Anvret M, Olson L. ALDH1 mRNA: presence in human dopamine neurons and decreases in substantia nigra in Parkinson’s disease and in the ventral tegmental area in schizophrenia. Neurobiol Dis2003; 14:637–647.
Mandel S, Grunblatt E, Riederer P, et al Gene expression profiling of sporadic Parkinson’s disease substantia nigra pars compacta reveals impairment of ubiquitin-proteasome subunits, SKP1A, aldehyde dehydrogenase, and chaperone HSC-70. Ann N Y Acad Sci2005; 1053:356–375.
Werner CJ, Heyny-von Haussen R, Mall G, Wolf S. Proteome analysis of human substantia nigra in Parkinson’s disease. Proteome Sci2008; 6:8.
Larsen KE, Schmitz Y, Troyer MD, et al Alpha-synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J Neurosci2006; 26:11915–11922.
Mosharov EV, Staal RG, Bove J, et al Alpha-synuclein overexpression increases cytosolic catecholamine concentration. J Neurosci2006; 26:9304–9311.
Mosharov EV, Larsen KE, Kanter E, et al Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron2009; 62:218–229.
Burke WJ, Kumar VB, Pandey N, et al Aggregation of alpha-synuclein by DOPAL, the monoamine oxidase metabolite of dopamine. Acta Neuropathol2008; 115:193–203.
Caudle WM, Richardson JR, Wang MZ, et al Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. J Neurosci2007; 27:8138–8148.
Taylor TN, Caudle WM, Shepherd KR, et al Nonmotor symptoms of Parkinson’s disease revealed in an animal model with reduced monoamine storage capacity. J Neurosci2009; 29:8103–8113.
Pena-Silva RA, Miller JD, Chu Y, Heistad DD. Serotonin produces monoamine oxidase-dependent oxidative stress in human heart valves. Am J Physiol Heart Circ Physiol2009; 297:H1354–H1360.
Moak JP, Goldstein DS, Eldadah BA, et al Supine low-frequency power of heart rate variability reflects baroreflex function, not cardiac sympathetic innervation. Heart Rhythm2007; 4:1523–1529.
Baumert M, Lambert GW, Dawood T, et al Short-term heart rate variability and cardiac norepinephrine spillover in patients with depression and panic disorder. Am J Physiol Heart Circ Physiol2009; 297:H674–H679.
Baumert M, Lambert GW, Dawood T, et al QT interval variability and cardiac norepinephrine spillover in patients with depression and panic disorder. Am J Physiol Heart Circ Physiol2008; 295:H962–H968.
Tygesen H, Rundqvist B, Waagstein F, Wennerblom B. Heart rate variability measurement correlates with cardiac norepinephrine spillover in congestive heart failure. Am J Cardiol2001; 87:1308–1311.
Kingwell BA, Thompson JM, Kaye DM, McPherson GA, Jennings GL, Esler MD. Heart rate spectral analysis, cardiac norepinephrine spillover, and muscle sympathetic nerve activity during human sympathetic nervous activation and failure. Circulation1994; 90:234–240.
David S. Goldstein, MD, PhD Clinical Neurocardiology Section, Clinical Neurosciences Program, Division of Intramural Research, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD
Correspondence: David S. Goldstein, MD, PhD, Building 10, Room 5N220, 10 Center Drive, MSC-1620, Bethesda, MD 20892–1620; [email protected]
Dr. Goldstein reported that he has no financial relationships that pose a potential conflict of interest with this article.
Some of the research summarized here was supported by the intramural research program of the National Institutes of Health.
David S. Goldstein, MD, PhD Clinical Neurocardiology Section, Clinical Neurosciences Program, Division of Intramural Research, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD
Correspondence: David S. Goldstein, MD, PhD, Building 10, Room 5N220, 10 Center Drive, MSC-1620, Bethesda, MD 20892–1620; [email protected]
Dr. Goldstein reported that he has no financial relationships that pose a potential conflict of interest with this article.
Some of the research summarized here was supported by the intramural research program of the National Institutes of Health.
Author and Disclosure Information
David S. Goldstein, MD, PhD Clinical Neurocardiology Section, Clinical Neurosciences Program, Division of Intramural Research, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD
Correspondence: David S. Goldstein, MD, PhD, Building 10, Room 5N220, 10 Center Drive, MSC-1620, Bethesda, MD 20892–1620; [email protected]
Dr. Goldstein reported that he has no financial relationships that pose a potential conflict of interest with this article.
Some of the research summarized here was supported by the intramural research program of the National Institutes of Health.
This review highlights important recent publications in the area of neuroscience and heart-brain medicine. Abnormalities of regulation of the circulation by catecholamine systems figure as a general theme of the topics highlighted. These topics, which are reviewed in turn below, are (1) mechanisms of cardiac sympathetic denervation in Parkinson disease (PD), (2) cytoplasmic monoamine metabolites as autotoxins, and (3) the validity of power spectral analysis of heart rate variability to indicate cardiac sympathetic tone.
MECHANISMS OF CARDIAC SYMPATHETIC DENERVATION IN PARKINSON DISEASE
The movement disorder component of PD is well recognized as resulting from loss of dopaminergic neurons in the nigrostriatal system of the brain. The finding of low myocardial 6-[18F]fluorodopamine–derived radioactivity by positron emission tomography provided the first neuroimaging evidence for loss of catecholaminergic neurons outside the brain in PD.1 Many reports using 123I-metaiodobenzylguanidine scanning have concurred with this finding. Beginning in the early 2000s, post-mortem neuropathologic studies demonstrated virtually absent immunoreactivity for tyrosine hydroxylase, the rate-limiting enzyme in norepinephrine biosynthesis, in epicardial nerves in PD.2,3 These results provided clues to the mechanism of autonomic dysfunction in PD, a prominent nonmotor manifestation of the disease.
With kind permission from Springer Science+Business Media: Acta Neuropathologica,
Figure 1. Tyrosine hydroxylase immunoreactivity (THir) in epicardial nerve from (A) a control subject and (B) a patient with familial Parkinson disease due to duplication of the gene encoding alpha-synuclein (PARK4).6Alpha-synuclein is a key protein in the pathogenesis of PD. It is abundant in Lewy bodies and Lewy neurites, and mutations or multiplications of the gene that encodes it cause rare inherited forms of PD. In 2001 we reported evidence for cardiac sympathetic denervation, neurogenic orthostatic hypotension, and baroreflex failure in familial PD from mutation of the gene encoding alpha-synuclein.4 Subsequently we reported analogous denervation in familial PD from triplication of the normal gene.5 This past year Orimo’s group in Tokyo provided the first pathological confirmation of cardiac sympathetic denervation in familial PD from inherited alpha-synucleinopathy, based on severely decreased epicardial neuronal tyrosine hydroxylase immunoreactivity (Figure 1).6 In contrast, patients with familial PD from parkin gene mutation, which is not thought to be a Lewy body disease, have been found to have normal cardiac 123I-metaiodobenzyl-guanidine–derived radioactivity and normal epicardial neuronal tyrosine hydroxylase immunoreactivity.7 These findings establish a link between alpha-synucleinopathy and cardiac sympathetic denervation.
Some individuals who die without clinical parkinsonism have Lewy bodies detected pathologically. Growing evidence shows that incidental Lewy body disease represents early, presymptomatic PD.8 Orimo’s group therefore studied cardiac tissues and paravertebral sympathetic ganglia from patients with incidental Lewy body disease.9 Postmortem tissues were likewise obtained from comparison subjects with multiple system atrophy and from control subjects. Immunohistochemical analyses were performed using antibodies against tyrosine hydroxylase, phosphorylated neurofilament as a marker of axons, and phosphorylated alpha-synuclein as a marker of abnormal alpha-synuclein deposits. Key findings from this study9 were as follows:
Reprinted from Brain (Orimo S, et al. Axonal α-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain 2008; 131:642–650) by permission of Oxford University Press.
Figure 2. Concept diagram of the pathogenetic sequence of cardiac sympathetic denervation. In incidental Lewy body disease with preserved tyrosine hydroxylase–immunoreactive (THir) axons (a), alpha-synuclein aggregates (black shading) accumulate abundantly in the distal axons but sparsely in the paravertebral sympathetic ganglia. In contrast, in incidental Lewy body disease with decreased THir axons (b), alpha-synuclein aggregates diminish in the distal axons but increase in the paravertebral sympathetic ganglia. In Parkinson disease, alpha-synuclein aggregates disappear in the distal axons and accumulate much more abundantly in the paravertebral sympathetic ganglia. In multiple system atrophy, alpha-synuclein aggregates are generally not observed (as in controls), with a few exceptions. Dotted lines indicate degeneration of THir axons.9Alpha-synuclein aggregates in distal epicardial nerve fascicles were more abundant in incidental Lewy body disease with preserved tyrosine hydroxylase–immunoreactive (THir) axons than in incidental Lewy body disease with decreased THir axons (Figure 2).
Alpha-synuclein aggregates in the epicardial nerve fibers were closely related to the disappearance of THir axons.
In incidental Lewy body disease with preserved THir axons, alpha-synuclein aggregates were consistently more abundant in the epicardial nerves than in the paravertebral sympathetic ganglia (Figure 2).
Distally dominant accumulation of alpha-synuclein aggregates was reversed in incidental Lewy body disease with decreased THir axons and in PD, because both conditions involve fewer alpha-synuclein aggregates in axons and more abundant aggregates in the paravertebral sympathetic ganglia (Figure 2).
Thus, accumulation of alpha-synuclein aggregates in distal cardiac sympathetic axons precedes aggregation in neuronal somata or ganglionic neurites, heralding centripetal degeneration of cardiac sympathetic nerves in PD. This chronological and dynamic relationship between alpha-synuclein aggregation and distally dominant degeneration of cardiac noradrenergic nerves may represent the pathological mechanism behind a common degenerative process in PD.
In conclusion, cardiac noradrenergic denervation in Lewy body diseases, even in early stages, accounts for reduced cardiac uptake of 123I-metaiodobenzylguanidine and 6-[18F]fluorodopamine in PD. Alpha-synuclein aggregation appears to be intimately involved in the cardiac noradrenergic denervation that attends Lewy body diseases. The pathogenetic process seems to proceed in a centripetal, retrograde direction.
CYTOPLASMIC MONOAMINE METABOLITES AS AUTOTOXINS
Current concepts about mechanisms of PD emphasize pathologic alpha-synuclein accumulation, oxidative injury, impaired proteasomal or mitochondrial functions, neuroinflammation, or abnormal kinase signaling. These concepts do not explain relatively selective nigrostriatal dopaminergic and cardiac noradrenergic denervation in PD.
Figure 3. According to the monoamine aldehyde hypothesis, interference with the vesicular recycling of cytoplasmic monoamines (dopamine [DA], norepinephrine [NE], and serotonin [5-HT]) augments formation of toxic aldehydes. For instance, DA that leaks from vesicles (V) into the cytoplasm (C) or that is taken up via the cell membrane DA transporter (DAT) and escapes vesicular reuptake via the vesicular monoamine transporter (VMAT) is subject to oxidative deamination catalyzed by monoamine oxidase (MAO) to form the catecholaldehyde DOPAL, which is toxic. DOPAL is detoxified by ALDH to form DOPAC, the major metabolic route, or by AR to form DOPET, the minor metabolic route. Analogously, NE is converted to DOPEGAL, and 5-HT is converted to 5-HT-aldehyde (5-HTAld).
A potential explanation is that cytoplasmic catecholamine metabolites are autotoxins (Figure 3). The mechanisms of autotoxicity include spontaneous auto-oxidation, to form quinones and chromes leading to increased production of reactive oxygen species, and enzymatic oxidation.
Catecholamines in the neuronal cytoplasm undergo enzymatic oxidative deamination to form catecholaldehydes (dihydroxyphenylacetaldehyde [DOPAL] from dopamine), which are cytotoxic, as predicted by Blaschko more than a half century ago.10 DOPAL is detoxified mainly by aldehyde dehydrogenase (ALDH). In the substantia nigra, aldehyde dehydrogenase 1A1 (ALDH1A1) is the main isoform of ALDH, and postmortem studies have noted decreased nigral ALDH1A1 gene expression11,12 and protein content13 in PD patients.
All neurons express alpha-synuclein. Current concepts about mechanisms also do not explain the relatively selective aggregation of alpha-synuclein in catecholaminergic neurons. Alpha-synuclein appears to play a role in the cycling of catecholamines across vesicular and cell membranes.14
Figure 4. Cell survival and cytoplasmic dopamine are inversely related, according to a murine model by Mosharov et al.16 Graph shows the dependence of cell survival under l-dopa–induced stress on the cytoplasmic dopamine (DAcyt) dose in mouse neurons. The DAcyt dose was estimated as: [DAcyt]×TExposure = [DAcyt]×Ln([L-dopa]/K0.5)/k,where [DAcyt] is the concentration of cytosolic DA in cells treated with a saturating level (> 50 μM) of l-dopa for 1 hour, where [l-dopa] is the initial drug concentration, and where K0.5 = 9.7 μM and k = 0.15 hr−1 are the kinetic constants. TExposure approximates the time during which extracellular l-dopa remained higher than K0.5. The data points are (from left to right): filled circles—ventral midbrain cultures treated with 25, 100, 250, 500, and 1,000 μM l-dopa alone; open circles—ventral midbrain neurons treated with 250 μM l-dopa in the presence of benserazide, methamphetamine, reserpine, pargyline, and pargyline reserpine; diamonds—ventral tegmental area and substantia nigra neurons; triangles—striatal and cortical neurons treated with 250 μM l-dopa. Dotted lines and shaded boxes represent mean ± SEM in untreated cells. The solid line is the linear fit of all data points, excluding striatal and cortical neurons and the two data points indicated by the asterisk. Treatments to the right of this line are neuroprotective, as the same level of cell death is achieved with higher DAcyt doses; treatments to the left of this line are more susceptible to DAcyt stress.In the past year, a few important studies have been published related to autotoxicity of cytoplasmic catecholamine metabolites and to pathogenic interactions with alpha-synuclein. In 2006, Mosharov et al reported that alpha-synuclein overexpression increases cytoplasmic dopamine concentrations in rat pheochromocytoma PC-12 cells.15 Recently, the same group, using intracellular patch electrochemistry, directly measured cytoplasmic dopamine in cultured midbrain neurons and found that increases in dopamine and its metabolites are neurotoxic, whereas manipulations that reduce cytoplasmic dopamine are neuroprotective (Figure 4).16 Levodopa (l-dopa) increased cytoplasmic dopamine more in substantia nigra neurons than in ventral tegmental neurons, suggesting that this difference might help explain the greater susceptibility of nigral neurons to the pathogenetic process. The greater buildup of cytoplasmic dopamine seemed to depend on dihydropyridine-sensitive calcium (Ca2+) channels. Finally, dopaminergic neurons lacking alpha-synuclein were resistant to l-dopa–induced cell death. These findings led the authors to propose a “multiple-hit” model (Figure 5) in which interactions between intracellular ionized calcium, cytoplasmic dopamine, and alpha-synuclein underlie susceptibility of nigral neurons in PD.16
Figure 5. The “multiple-hit” model of Parkinson disease pathogenesis,16 which holds that neurotoxicity is a result of multiple factors, including the presence of alpha-synuclein (α-syn), elevation of cytoplasmic calcium (Ca2+), and buildup of cytoplasmic dopamine (DAcyt) and its metabolites. Nonexclusive toxic steps may result from (1) mechanisms that require direct interaction between DA or its metabolites with α-syn, such as DA-modified stabilization of α-syn protofibrils or inhibition of chaperone-mediated autophagy, or (2) cumulative damage from multiple independent sources. Reducing the levels of any of the three players provides neuroprotection. (AADC = aromatic l-amino acid decarboxylase; DOPAL = dihydroxyphenylacetaldehyde; TH = tyrosine hydroxylase)Burke et al added a potentially important clue, demonstrating that DOPAL potently oligomerizes and aggregates alpha-synuclein.17 This finding introduces the possibility of multiple pathogenetic positive feedback loops.
Under resting conditions, most catecholamine turnover results from leakage from vesicular stores into the cytoplasm and subsequent oxidative deamination by monoamine oxidase. Ordinarily, however, catecholamines in the cytoplasm are efficiently recycled back into the vesicles via the type 2 vesicular monoamine transporter (VMAT-2). Accordingly, interference with VMAT functions would be expected to tend to build up cytoplasmic catecholamines, with potentially cytotoxic consequences. In 2007, Caudle et al reported that mice with severely decreased VMAT-2 have aging-associated decreases in striatal dopamine that begin in the terminal fields, alpha-synuclein deposition in substantia nigra neurons, and l-dopa–responsive behavioral deficits.18 More recently the same group noted nonmotor signs associated with PD in VMAT-2–deficient mice, such as anosmia, gastrointestinal hypomotility, sleep disturbances, anxiety, and depression.19 Since VMAT-2 serves to recycle not only dopamine but also norepinephrine and serotonin, this single abnormality could help explain loss of all three types of monoaminergic neurons in PD.
Finally, Pena-Silva et al recently tested whether serotonin induces oxidative stress in human heart valves.20 They showed that in heart valves from explanted human hearts not used for transplantation, incubation of homogenates of cardiac valves and blood vessels with serotonin increased generation of the superoxide free radical. Inhibitors of monoamine oxidase prevented this effect. Dopamine also increased superoxide levels in heart valves, and this effect was also attenuated by monoamine oxidase inhibition. These findings fit with the concept that the aldehydes produced by the action of monoamine oxidase on cytoplasmic monoamines generate toxic free radicals.
VALIDITY OF POWER SPECTRAL ANALYSIS OF HEART RATE VARIABILITY TO INDICATE CARDIAC SYMPATHETIC TONE
Power spectral analysis of heart rate variability is simple, relatively inexpensive, noninvasive, and widely used to indicate cardiac sympathetic “tone” or sympathovagal “balance.” Almost 2,000 studies to date have used this modality. Relatively increased cardiac sympathetic tone, reflected by low-frequency (LF) power or the ratio of LF power to high-frequency (HF) power, is an adverse prognostic sign in a variety of conditions. Nevertheless, the validity of LF power, or the LF:HF ratio, as an index of cardiac sympathetic tone remains unsettled.
In 2007 we assessed the validity of power spectral analysis rather directly, by taking advantage of our ability to delineate cardiac sympathetic innervation. We compared LF power in patients with cardiac sympathetic denervation, indicated by low myocardial levels of 6-[18F]fluorodopamine–derived radioactivity or low rates of norepinephrine entry into coronary sinus plasma (cardiac norepinephrine spillover), with values in patients with intact innervation. LF power was unrelated to myocardial 6-[18F]fluorodopamine–derived radioactivity or cardiac norepinephrine spillover, but it was related to baroreflex-cardiovagal gain. Patients with a low baroreflex-cardiovagal gain had low LF power, regardless of cardiac innervation. From these findings we concluded that LF power reflects baroreflex function, not cardiac sympathetic innervation.21
Figure 6. Relationships of heart rate variability indices with cardiac norepinephrine spillover. Graphs show time and frequency domain heart rate variability measures (LF = low frequency; HF = high frequency) versus cardiac norepinephrine in healthy subjects (squares) and patients with major depression (triangles) and panic disorder (circles).22 Recently Baumert et al also examined the relationship between indices from power spectral analysis of heart rate variability and cardiac norepinephrine spillover.22 They found, as we did, that none of the standard heart rate variability parameters was correlated with cardiac norepinephrine spillover (Figure 6). The same group reported a positive correlation between the heart rate–corrected QT interval and cardiac norepinephrine spillover.23 Among patients with major depression, the distribution of cardiac norepinephrine spillover seemed bimodal. Overall, cardiac norepinephrine spillover was not increased, although a subgroup had clearly increased spillover.
In congestive heart failure, baroreflex-cardiovagal gain tends to be low and cardiac sympathetic outflow markedly increased, yet the LF:HF ratio is not increased during supine rest.24,25 It therefore appears that power spectral analysis of heart rate variability may provide a measure of baroreflexive modulation of autonomic outflows to the heart but not a measure of those outflows themselves. The search continues for a valid, noninvasive means to assess cardiac sympathetic function.
This review highlights important recent publications in the area of neuroscience and heart-brain medicine. Abnormalities of regulation of the circulation by catecholamine systems figure as a general theme of the topics highlighted. These topics, which are reviewed in turn below, are (1) mechanisms of cardiac sympathetic denervation in Parkinson disease (PD), (2) cytoplasmic monoamine metabolites as autotoxins, and (3) the validity of power spectral analysis of heart rate variability to indicate cardiac sympathetic tone.
MECHANISMS OF CARDIAC SYMPATHETIC DENERVATION IN PARKINSON DISEASE
The movement disorder component of PD is well recognized as resulting from loss of dopaminergic neurons in the nigrostriatal system of the brain. The finding of low myocardial 6-[18F]fluorodopamine–derived radioactivity by positron emission tomography provided the first neuroimaging evidence for loss of catecholaminergic neurons outside the brain in PD.1 Many reports using 123I-metaiodobenzylguanidine scanning have concurred with this finding. Beginning in the early 2000s, post-mortem neuropathologic studies demonstrated virtually absent immunoreactivity for tyrosine hydroxylase, the rate-limiting enzyme in norepinephrine biosynthesis, in epicardial nerves in PD.2,3 These results provided clues to the mechanism of autonomic dysfunction in PD, a prominent nonmotor manifestation of the disease.
With kind permission from Springer Science+Business Media: Acta Neuropathologica,
Figure 1. Tyrosine hydroxylase immunoreactivity (THir) in epicardial nerve from (A) a control subject and (B) a patient with familial Parkinson disease due to duplication of the gene encoding alpha-synuclein (PARK4).6Alpha-synuclein is a key protein in the pathogenesis of PD. It is abundant in Lewy bodies and Lewy neurites, and mutations or multiplications of the gene that encodes it cause rare inherited forms of PD. In 2001 we reported evidence for cardiac sympathetic denervation, neurogenic orthostatic hypotension, and baroreflex failure in familial PD from mutation of the gene encoding alpha-synuclein.4 Subsequently we reported analogous denervation in familial PD from triplication of the normal gene.5 This past year Orimo’s group in Tokyo provided the first pathological confirmation of cardiac sympathetic denervation in familial PD from inherited alpha-synucleinopathy, based on severely decreased epicardial neuronal tyrosine hydroxylase immunoreactivity (Figure 1).6 In contrast, patients with familial PD from parkin gene mutation, which is not thought to be a Lewy body disease, have been found to have normal cardiac 123I-metaiodobenzyl-guanidine–derived radioactivity and normal epicardial neuronal tyrosine hydroxylase immunoreactivity.7 These findings establish a link between alpha-synucleinopathy and cardiac sympathetic denervation.
Some individuals who die without clinical parkinsonism have Lewy bodies detected pathologically. Growing evidence shows that incidental Lewy body disease represents early, presymptomatic PD.8 Orimo’s group therefore studied cardiac tissues and paravertebral sympathetic ganglia from patients with incidental Lewy body disease.9 Postmortem tissues were likewise obtained from comparison subjects with multiple system atrophy and from control subjects. Immunohistochemical analyses were performed using antibodies against tyrosine hydroxylase, phosphorylated neurofilament as a marker of axons, and phosphorylated alpha-synuclein as a marker of abnormal alpha-synuclein deposits. Key findings from this study9 were as follows:
Reprinted from Brain (Orimo S, et al. Axonal α-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain 2008; 131:642–650) by permission of Oxford University Press.
Figure 2. Concept diagram of the pathogenetic sequence of cardiac sympathetic denervation. In incidental Lewy body disease with preserved tyrosine hydroxylase–immunoreactive (THir) axons (a), alpha-synuclein aggregates (black shading) accumulate abundantly in the distal axons but sparsely in the paravertebral sympathetic ganglia. In contrast, in incidental Lewy body disease with decreased THir axons (b), alpha-synuclein aggregates diminish in the distal axons but increase in the paravertebral sympathetic ganglia. In Parkinson disease, alpha-synuclein aggregates disappear in the distal axons and accumulate much more abundantly in the paravertebral sympathetic ganglia. In multiple system atrophy, alpha-synuclein aggregates are generally not observed (as in controls), with a few exceptions. Dotted lines indicate degeneration of THir axons.9Alpha-synuclein aggregates in distal epicardial nerve fascicles were more abundant in incidental Lewy body disease with preserved tyrosine hydroxylase–immunoreactive (THir) axons than in incidental Lewy body disease with decreased THir axons (Figure 2).
Alpha-synuclein aggregates in the epicardial nerve fibers were closely related to the disappearance of THir axons.
In incidental Lewy body disease with preserved THir axons, alpha-synuclein aggregates were consistently more abundant in the epicardial nerves than in the paravertebral sympathetic ganglia (Figure 2).
Distally dominant accumulation of alpha-synuclein aggregates was reversed in incidental Lewy body disease with decreased THir axons and in PD, because both conditions involve fewer alpha-synuclein aggregates in axons and more abundant aggregates in the paravertebral sympathetic ganglia (Figure 2).
Thus, accumulation of alpha-synuclein aggregates in distal cardiac sympathetic axons precedes aggregation in neuronal somata or ganglionic neurites, heralding centripetal degeneration of cardiac sympathetic nerves in PD. This chronological and dynamic relationship between alpha-synuclein aggregation and distally dominant degeneration of cardiac noradrenergic nerves may represent the pathological mechanism behind a common degenerative process in PD.
In conclusion, cardiac noradrenergic denervation in Lewy body diseases, even in early stages, accounts for reduced cardiac uptake of 123I-metaiodobenzylguanidine and 6-[18F]fluorodopamine in PD. Alpha-synuclein aggregation appears to be intimately involved in the cardiac noradrenergic denervation that attends Lewy body diseases. The pathogenetic process seems to proceed in a centripetal, retrograde direction.
CYTOPLASMIC MONOAMINE METABOLITES AS AUTOTOXINS
Current concepts about mechanisms of PD emphasize pathologic alpha-synuclein accumulation, oxidative injury, impaired proteasomal or mitochondrial functions, neuroinflammation, or abnormal kinase signaling. These concepts do not explain relatively selective nigrostriatal dopaminergic and cardiac noradrenergic denervation in PD.
Figure 3. According to the monoamine aldehyde hypothesis, interference with the vesicular recycling of cytoplasmic monoamines (dopamine [DA], norepinephrine [NE], and serotonin [5-HT]) augments formation of toxic aldehydes. For instance, DA that leaks from vesicles (V) into the cytoplasm (C) or that is taken up via the cell membrane DA transporter (DAT) and escapes vesicular reuptake via the vesicular monoamine transporter (VMAT) is subject to oxidative deamination catalyzed by monoamine oxidase (MAO) to form the catecholaldehyde DOPAL, which is toxic. DOPAL is detoxified by ALDH to form DOPAC, the major metabolic route, or by AR to form DOPET, the minor metabolic route. Analogously, NE is converted to DOPEGAL, and 5-HT is converted to 5-HT-aldehyde (5-HTAld).
A potential explanation is that cytoplasmic catecholamine metabolites are autotoxins (Figure 3). The mechanisms of autotoxicity include spontaneous auto-oxidation, to form quinones and chromes leading to increased production of reactive oxygen species, and enzymatic oxidation.
Catecholamines in the neuronal cytoplasm undergo enzymatic oxidative deamination to form catecholaldehydes (dihydroxyphenylacetaldehyde [DOPAL] from dopamine), which are cytotoxic, as predicted by Blaschko more than a half century ago.10 DOPAL is detoxified mainly by aldehyde dehydrogenase (ALDH). In the substantia nigra, aldehyde dehydrogenase 1A1 (ALDH1A1) is the main isoform of ALDH, and postmortem studies have noted decreased nigral ALDH1A1 gene expression11,12 and protein content13 in PD patients.
All neurons express alpha-synuclein. Current concepts about mechanisms also do not explain the relatively selective aggregation of alpha-synuclein in catecholaminergic neurons. Alpha-synuclein appears to play a role in the cycling of catecholamines across vesicular and cell membranes.14
Figure 4. Cell survival and cytoplasmic dopamine are inversely related, according to a murine model by Mosharov et al.16 Graph shows the dependence of cell survival under l-dopa–induced stress on the cytoplasmic dopamine (DAcyt) dose in mouse neurons. The DAcyt dose was estimated as: [DAcyt]×TExposure = [DAcyt]×Ln([L-dopa]/K0.5)/k,where [DAcyt] is the concentration of cytosolic DA in cells treated with a saturating level (> 50 μM) of l-dopa for 1 hour, where [l-dopa] is the initial drug concentration, and where K0.5 = 9.7 μM and k = 0.15 hr−1 are the kinetic constants. TExposure approximates the time during which extracellular l-dopa remained higher than K0.5. The data points are (from left to right): filled circles—ventral midbrain cultures treated with 25, 100, 250, 500, and 1,000 μM l-dopa alone; open circles—ventral midbrain neurons treated with 250 μM l-dopa in the presence of benserazide, methamphetamine, reserpine, pargyline, and pargyline reserpine; diamonds—ventral tegmental area and substantia nigra neurons; triangles—striatal and cortical neurons treated with 250 μM l-dopa. Dotted lines and shaded boxes represent mean ± SEM in untreated cells. The solid line is the linear fit of all data points, excluding striatal and cortical neurons and the two data points indicated by the asterisk. Treatments to the right of this line are neuroprotective, as the same level of cell death is achieved with higher DAcyt doses; treatments to the left of this line are more susceptible to DAcyt stress.In the past year, a few important studies have been published related to autotoxicity of cytoplasmic catecholamine metabolites and to pathogenic interactions with alpha-synuclein. In 2006, Mosharov et al reported that alpha-synuclein overexpression increases cytoplasmic dopamine concentrations in rat pheochromocytoma PC-12 cells.15 Recently, the same group, using intracellular patch electrochemistry, directly measured cytoplasmic dopamine in cultured midbrain neurons and found that increases in dopamine and its metabolites are neurotoxic, whereas manipulations that reduce cytoplasmic dopamine are neuroprotective (Figure 4).16 Levodopa (l-dopa) increased cytoplasmic dopamine more in substantia nigra neurons than in ventral tegmental neurons, suggesting that this difference might help explain the greater susceptibility of nigral neurons to the pathogenetic process. The greater buildup of cytoplasmic dopamine seemed to depend on dihydropyridine-sensitive calcium (Ca2+) channels. Finally, dopaminergic neurons lacking alpha-synuclein were resistant to l-dopa–induced cell death. These findings led the authors to propose a “multiple-hit” model (Figure 5) in which interactions between intracellular ionized calcium, cytoplasmic dopamine, and alpha-synuclein underlie susceptibility of nigral neurons in PD.16
Figure 5. The “multiple-hit” model of Parkinson disease pathogenesis,16 which holds that neurotoxicity is a result of multiple factors, including the presence of alpha-synuclein (α-syn), elevation of cytoplasmic calcium (Ca2+), and buildup of cytoplasmic dopamine (DAcyt) and its metabolites. Nonexclusive toxic steps may result from (1) mechanisms that require direct interaction between DA or its metabolites with α-syn, such as DA-modified stabilization of α-syn protofibrils or inhibition of chaperone-mediated autophagy, or (2) cumulative damage from multiple independent sources. Reducing the levels of any of the three players provides neuroprotection. (AADC = aromatic l-amino acid decarboxylase; DOPAL = dihydroxyphenylacetaldehyde; TH = tyrosine hydroxylase)Burke et al added a potentially important clue, demonstrating that DOPAL potently oligomerizes and aggregates alpha-synuclein.17 This finding introduces the possibility of multiple pathogenetic positive feedback loops.
Under resting conditions, most catecholamine turnover results from leakage from vesicular stores into the cytoplasm and subsequent oxidative deamination by monoamine oxidase. Ordinarily, however, catecholamines in the cytoplasm are efficiently recycled back into the vesicles via the type 2 vesicular monoamine transporter (VMAT-2). Accordingly, interference with VMAT functions would be expected to tend to build up cytoplasmic catecholamines, with potentially cytotoxic consequences. In 2007, Caudle et al reported that mice with severely decreased VMAT-2 have aging-associated decreases in striatal dopamine that begin in the terminal fields, alpha-synuclein deposition in substantia nigra neurons, and l-dopa–responsive behavioral deficits.18 More recently the same group noted nonmotor signs associated with PD in VMAT-2–deficient mice, such as anosmia, gastrointestinal hypomotility, sleep disturbances, anxiety, and depression.19 Since VMAT-2 serves to recycle not only dopamine but also norepinephrine and serotonin, this single abnormality could help explain loss of all three types of monoaminergic neurons in PD.
Finally, Pena-Silva et al recently tested whether serotonin induces oxidative stress in human heart valves.20 They showed that in heart valves from explanted human hearts not used for transplantation, incubation of homogenates of cardiac valves and blood vessels with serotonin increased generation of the superoxide free radical. Inhibitors of monoamine oxidase prevented this effect. Dopamine also increased superoxide levels in heart valves, and this effect was also attenuated by monoamine oxidase inhibition. These findings fit with the concept that the aldehydes produced by the action of monoamine oxidase on cytoplasmic monoamines generate toxic free radicals.
VALIDITY OF POWER SPECTRAL ANALYSIS OF HEART RATE VARIABILITY TO INDICATE CARDIAC SYMPATHETIC TONE
Power spectral analysis of heart rate variability is simple, relatively inexpensive, noninvasive, and widely used to indicate cardiac sympathetic “tone” or sympathovagal “balance.” Almost 2,000 studies to date have used this modality. Relatively increased cardiac sympathetic tone, reflected by low-frequency (LF) power or the ratio of LF power to high-frequency (HF) power, is an adverse prognostic sign in a variety of conditions. Nevertheless, the validity of LF power, or the LF:HF ratio, as an index of cardiac sympathetic tone remains unsettled.
In 2007 we assessed the validity of power spectral analysis rather directly, by taking advantage of our ability to delineate cardiac sympathetic innervation. We compared LF power in patients with cardiac sympathetic denervation, indicated by low myocardial levels of 6-[18F]fluorodopamine–derived radioactivity or low rates of norepinephrine entry into coronary sinus plasma (cardiac norepinephrine spillover), with values in patients with intact innervation. LF power was unrelated to myocardial 6-[18F]fluorodopamine–derived radioactivity or cardiac norepinephrine spillover, but it was related to baroreflex-cardiovagal gain. Patients with a low baroreflex-cardiovagal gain had low LF power, regardless of cardiac innervation. From these findings we concluded that LF power reflects baroreflex function, not cardiac sympathetic innervation.21
Figure 6. Relationships of heart rate variability indices with cardiac norepinephrine spillover. Graphs show time and frequency domain heart rate variability measures (LF = low frequency; HF = high frequency) versus cardiac norepinephrine in healthy subjects (squares) and patients with major depression (triangles) and panic disorder (circles).22 Recently Baumert et al also examined the relationship between indices from power spectral analysis of heart rate variability and cardiac norepinephrine spillover.22 They found, as we did, that none of the standard heart rate variability parameters was correlated with cardiac norepinephrine spillover (Figure 6). The same group reported a positive correlation between the heart rate–corrected QT interval and cardiac norepinephrine spillover.23 Among patients with major depression, the distribution of cardiac norepinephrine spillover seemed bimodal. Overall, cardiac norepinephrine spillover was not increased, although a subgroup had clearly increased spillover.
In congestive heart failure, baroreflex-cardiovagal gain tends to be low and cardiac sympathetic outflow markedly increased, yet the LF:HF ratio is not increased during supine rest.24,25 It therefore appears that power spectral analysis of heart rate variability may provide a measure of baroreflexive modulation of autonomic outflows to the heart but not a measure of those outflows themselves. The search continues for a valid, noninvasive means to assess cardiac sympathetic function.
References
Goldstein DS, Holmes C, Cannon RO, Eisenhofer G, Kopin IJ. Sympathetic cardioneuropathy in dysautonomias. N Engl J Med1997; 336:696–702.
Orimo S, Ozawa E, Oka T, et al Different histopathology accounting for a decrease in myocardial MIBG uptake in PD and MSA. Neurology2001; 57:1140–1141.
Amino T, Orimo S, Takahashi A, Uchihara T, Mizusawa H. Profound cardiac sympathetic denervation occurs in Parkinson disease. Brain Path2005; 15:29–34.
Goldstein DS, Li S-T, Kopin IJ. Sympathetic neurocirculatory failure in Parkinson disease: evidence for an etiologic role of α-synuclein. Ann Intern Med2001; 135:1010–1011.
Singleton A, Gwinn-Hardy K, Sharabi Y, et al Association between cardiac denervation and parkinsonism caused by alpha-synuclein gene triplication. Brain2004; 127:768–772.
Orimo S, Uchihara T, Nakamura A, et al Cardiac sympathetic denervation in Parkinson’s disease linked to SNCA duplication. Acta Neuropathol2008; 116:575–577.
Orimo S, Amino T, Yokochi M, et al Preserved cardiac sympathetic nerve accounts for normal cardiac uptake of MIBG in PARK2. Mov Disord2005; 20:1350–1353.
Dickson DW, Fujishiro H, DelleDonne A, et al Evidence that incidental Lewy body disease is pre-symptomatic Parkinson’s disease. Acta Neuropathol2008; 115:437–444.
Orimo S, Uchihara T, Nakamura A, et al Axonal alpha-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain2008; 131:642–650.
Blaschko H. Amine oxidase and amine metabolism. Pharmacol Rev1952; 4:415–458.
Galter D, Buervenich S, Carmine A, Anvret M, Olson L. ALDH1 mRNA: presence in human dopamine neurons and decreases in substantia nigra in Parkinson’s disease and in the ventral tegmental area in schizophrenia. Neurobiol Dis2003; 14:637–647.
Mandel S, Grunblatt E, Riederer P, et al Gene expression profiling of sporadic Parkinson’s disease substantia nigra pars compacta reveals impairment of ubiquitin-proteasome subunits, SKP1A, aldehyde dehydrogenase, and chaperone HSC-70. Ann N Y Acad Sci2005; 1053:356–375.
Werner CJ, Heyny-von Haussen R, Mall G, Wolf S. Proteome analysis of human substantia nigra in Parkinson’s disease. Proteome Sci2008; 6:8.
Larsen KE, Schmitz Y, Troyer MD, et al Alpha-synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J Neurosci2006; 26:11915–11922.
Mosharov EV, Staal RG, Bove J, et al Alpha-synuclein overexpression increases cytosolic catecholamine concentration. J Neurosci2006; 26:9304–9311.
Mosharov EV, Larsen KE, Kanter E, et al Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron2009; 62:218–229.
Burke WJ, Kumar VB, Pandey N, et al Aggregation of alpha-synuclein by DOPAL, the monoamine oxidase metabolite of dopamine. Acta Neuropathol2008; 115:193–203.
Caudle WM, Richardson JR, Wang MZ, et al Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. J Neurosci2007; 27:8138–8148.
Taylor TN, Caudle WM, Shepherd KR, et al Nonmotor symptoms of Parkinson’s disease revealed in an animal model with reduced monoamine storage capacity. J Neurosci2009; 29:8103–8113.
Pena-Silva RA, Miller JD, Chu Y, Heistad DD. Serotonin produces monoamine oxidase-dependent oxidative stress in human heart valves. Am J Physiol Heart Circ Physiol2009; 297:H1354–H1360.
Moak JP, Goldstein DS, Eldadah BA, et al Supine low-frequency power of heart rate variability reflects baroreflex function, not cardiac sympathetic innervation. Heart Rhythm2007; 4:1523–1529.
Baumert M, Lambert GW, Dawood T, et al Short-term heart rate variability and cardiac norepinephrine spillover in patients with depression and panic disorder. Am J Physiol Heart Circ Physiol2009; 297:H674–H679.
Baumert M, Lambert GW, Dawood T, et al QT interval variability and cardiac norepinephrine spillover in patients with depression and panic disorder. Am J Physiol Heart Circ Physiol2008; 295:H962–H968.
Tygesen H, Rundqvist B, Waagstein F, Wennerblom B. Heart rate variability measurement correlates with cardiac norepinephrine spillover in congestive heart failure. Am J Cardiol2001; 87:1308–1311.
Kingwell BA, Thompson JM, Kaye DM, McPherson GA, Jennings GL, Esler MD. Heart rate spectral analysis, cardiac norepinephrine spillover, and muscle sympathetic nerve activity during human sympathetic nervous activation and failure. Circulation1994; 90:234–240.
References
Goldstein DS, Holmes C, Cannon RO, Eisenhofer G, Kopin IJ. Sympathetic cardioneuropathy in dysautonomias. N Engl J Med1997; 336:696–702.
Orimo S, Ozawa E, Oka T, et al Different histopathology accounting for a decrease in myocardial MIBG uptake in PD and MSA. Neurology2001; 57:1140–1141.
Amino T, Orimo S, Takahashi A, Uchihara T, Mizusawa H. Profound cardiac sympathetic denervation occurs in Parkinson disease. Brain Path2005; 15:29–34.
Goldstein DS, Li S-T, Kopin IJ. Sympathetic neurocirculatory failure in Parkinson disease: evidence for an etiologic role of α-synuclein. Ann Intern Med2001; 135:1010–1011.
Singleton A, Gwinn-Hardy K, Sharabi Y, et al Association between cardiac denervation and parkinsonism caused by alpha-synuclein gene triplication. Brain2004; 127:768–772.
Orimo S, Uchihara T, Nakamura A, et al Cardiac sympathetic denervation in Parkinson’s disease linked to SNCA duplication. Acta Neuropathol2008; 116:575–577.
Orimo S, Amino T, Yokochi M, et al Preserved cardiac sympathetic nerve accounts for normal cardiac uptake of MIBG in PARK2. Mov Disord2005; 20:1350–1353.
Dickson DW, Fujishiro H, DelleDonne A, et al Evidence that incidental Lewy body disease is pre-symptomatic Parkinson’s disease. Acta Neuropathol2008; 115:437–444.
Orimo S, Uchihara T, Nakamura A, et al Axonal alpha-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain2008; 131:642–650.
Blaschko H. Amine oxidase and amine metabolism. Pharmacol Rev1952; 4:415–458.
Galter D, Buervenich S, Carmine A, Anvret M, Olson L. ALDH1 mRNA: presence in human dopamine neurons and decreases in substantia nigra in Parkinson’s disease and in the ventral tegmental area in schizophrenia. Neurobiol Dis2003; 14:637–647.
Mandel S, Grunblatt E, Riederer P, et al Gene expression profiling of sporadic Parkinson’s disease substantia nigra pars compacta reveals impairment of ubiquitin-proteasome subunits, SKP1A, aldehyde dehydrogenase, and chaperone HSC-70. Ann N Y Acad Sci2005; 1053:356–375.
Werner CJ, Heyny-von Haussen R, Mall G, Wolf S. Proteome analysis of human substantia nigra in Parkinson’s disease. Proteome Sci2008; 6:8.
Larsen KE, Schmitz Y, Troyer MD, et al Alpha-synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J Neurosci2006; 26:11915–11922.
Mosharov EV, Staal RG, Bove J, et al Alpha-synuclein overexpression increases cytosolic catecholamine concentration. J Neurosci2006; 26:9304–9311.
Mosharov EV, Larsen KE, Kanter E, et al Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron2009; 62:218–229.
Burke WJ, Kumar VB, Pandey N, et al Aggregation of alpha-synuclein by DOPAL, the monoamine oxidase metabolite of dopamine. Acta Neuropathol2008; 115:193–203.
Caudle WM, Richardson JR, Wang MZ, et al Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. J Neurosci2007; 27:8138–8148.
Taylor TN, Caudle WM, Shepherd KR, et al Nonmotor symptoms of Parkinson’s disease revealed in an animal model with reduced monoamine storage capacity. J Neurosci2009; 29:8103–8113.
Pena-Silva RA, Miller JD, Chu Y, Heistad DD. Serotonin produces monoamine oxidase-dependent oxidative stress in human heart valves. Am J Physiol Heart Circ Physiol2009; 297:H1354–H1360.
Moak JP, Goldstein DS, Eldadah BA, et al Supine low-frequency power of heart rate variability reflects baroreflex function, not cardiac sympathetic innervation. Heart Rhythm2007; 4:1523–1529.
Baumert M, Lambert GW, Dawood T, et al Short-term heart rate variability and cardiac norepinephrine spillover in patients with depression and panic disorder. Am J Physiol Heart Circ Physiol2009; 297:H674–H679.
Baumert M, Lambert GW, Dawood T, et al QT interval variability and cardiac norepinephrine spillover in patients with depression and panic disorder. Am J Physiol Heart Circ Physiol2008; 295:H962–H968.
Tygesen H, Rundqvist B, Waagstein F, Wennerblom B. Heart rate variability measurement correlates with cardiac norepinephrine spillover in congestive heart failure. Am J Cardiol2001; 87:1308–1311.
Kingwell BA, Thompson JM, Kaye DM, McPherson GA, Jennings GL, Esler MD. Heart rate spectral analysis, cardiac norepinephrine spillover, and muscle sympathetic nerve activity during human sympathetic nervous activation and failure. Circulation1994; 90:234–240.
The complex interaction between the nervous system and cardiovascular (CV) health is well recognized. In addition to stroke, migraine, and other disorders, the focus has shifted to depression and related negative-affect states including anxiety, chronic stress, posttraumatic stress disorder, and social isolation. Although all of these conditions have been linked to poor CV health, the mechanisms responsible for this brain-cardiovascular interaction have been poorly understood until recently.
Figure 1. Neurologic and psychological disorders share some overlapping pathophysiologic mechanisms with vascular disorders. (5-HTTLPR = serotonin transporter-linked promoter region; EPCs = endothelial progenitor cells; HPA = hypothalamic-pituitary-adrenal)At first, it appeared that the link between CV disease and such states as depression and anxiety was mediated by a clustering of shared risk conditions such as smoking, high low-density lipoprotein cholesterol, and obesity. As more research uncovered the pathophysiologic mechanisms underlying these negative-affect states, however, it became apparent that some of these mechanisms overlapped with those leading to vascular dysfunction (Figure 1). For instance, regulation of neurotransmitters such as serotonin is known to play a key role in mood dysfunction as well as in sleep and appetite, but only recently has this regulation been identified as an important component in moderating platelet function as well. Several other common pathways have been identified, including the hypothalamic-pituitary-adrenal (HPA) axis, endothelial progenitor cell (EPC) regulation, and inflammatory cell dysfunction. The role of genetic predisposition as a common factor leading to psychological and vascular dysfunction has been studied as well. In addition, research has recently expanded to examine other forms of central neurologic dysfunction besides mood, including stroke and migraine, and their connection with CV health.
This paper reviews a sample of the more recent advances in our understanding the pathophysiologic mechanisms underlying this complex brain-vascular interaction—including the roles of genetic predisposition, endothelial cell dysfunction, and endocrine dysregulation—and discusses future directions for research in this area.
GENETIC PREDISPOSITION
Increasing evidence highlights the importance of genetic predisposition in both psychological dysfunction and CV disease. The importance of common genetic variability in these two conditions originated from studies of twins that established that both depression and coronary artery disease (CAD) tended to run in families. However, recent studies focused on identifying specific genes that may link depression/negative affect and CAD through various pathways related to platelet reactivity, inflammation, and autonomic nervous system regulation, among many others. A candidate gene study by McCaffery et al1 sought to identify specific genes influencing depressive symptoms in CAD patients. Genes were selected based on their role in biologic pathways involved in inflammation, platelet activation, and the HPA axis and sympathetic nervous system. Among the 59 genes analyzed, the strongest associations were between markers involved in endothelial dysfunction and platelet aggregation—specifically, the involvement of von Willebrand factor in platelet recruitment and of vascular cell adhesion molecule–1 in recruitment and adhesion of inflammatory cells to injured endothelium. Other recent studies have analyzed the role of various genes in mediating neurologic and CV dysfunction.
Serotonin: A multitude of cardiovascular effects
Serotonin (5-HT) is best known as a neurotransmitter involved in mood regulation, but it also directly affects endothelial cells and vascular smooth muscle. Human brain microvascular endothelial cells have 5-HT2A receptors, which are thought to be involved in blood-brain trafficking.2 Polymorphisms in this receptor have been associated with impaired glucose tolerance and type 2 diabetes.3
However, the role of serotonin within the CV system has only recently been realized. Along with its associated serotonin transporter (SERT), serotonin has been best studied within the CV system for its role in development of pulmonary hypertension via vasoconstricton. However, serotonin and SERT exhibit other effects on the CV system, including regulation of factors involved in vascular integrity, such as platelet activity, endothelial dysfunction, and smooth muscle cell and endothelial cell mitogenesis. Serotonin is stored peripherally in platelets and, when released at sites of endothelial damage, promotes platelet aggregation. Cardiomyocytes also are a source of serotonin within the heart, resulting in positive chronotropy, positive inotropy, and activation of mitogen activity.4
Serotonin also affects endothelium through effects on bone marrow production of EPCs. It has been shown in mice to increase the proliferative activity of hematopoietic stem cells in the bone marrow via 5-HT2 receptors.5 Serotonin is a mitogen for canine and bovine endothelial cells6 and has also been shown to enhance ex vivo expansion of CD34+ hematopoietic stem cells in mice.7 It also seems to have an effect on regulation of inflammatory cytokines by regulating different secretory pathways. Activity of several different serotonin receptors, including 5-HT2A, 5-HT4, and 5-HT7, inhibits tumor necrosis factor production in peripheral cells.8
Insights from genetic analysis of SERT
These areas of recent research continue to highlight the importance of serotonin within the CV system, and its overlapping role in depression and other forms of psychological dysfunction underscores the importance of understanding its regulation.
The best-studied component of serotonin regulation is SERT, a sodium-dependent transporter that removes serotonin from outside the cell, bringing it back into the cell for repackaging. It is recognized as the site of action of the selective serotonin reuptake inhibitors (SSRIs).
Analysis of the serotonin transporter gene has traditionally focused on polymorphisms within the promoter region (5-HTTLPR). Two common alleles, the long (L) and short (S) variants, are the best characterized. The S variant is associated with a lower expression of SERT, leading to reduced uptake and release of serotonin. The SS genotype of SERT has been linked to major depressive disorder9 and to increased risk of subsequent cardiac events after myocardial infarction (MI).10
A study of the effects of environmental stress and gender on associations between depression and 5-HTTLPR found that this link varied according to gender and stressful life events.11 Specifically, women with the SS genotype, which leads to less transcriptional activity of 5-HTTLPR, tended to be more susceptible to depression under stressful life conditions. However, in men the LL genotype was associated with increased transcriptional activity and the same increased susceptibility to depression. 11 Expression of the LL genotype also resulted in upregulation of SERT, leading to higher MI risk.12
Another study investigated the relationship between genetic variability in two serotonin-related gene polymorphisms and found that the 5-HTTLPR gene polymorphism was associated with adverse cardiac events after coronary artery bypass graft surgery in combination with depression, specifically in patients with the L allele compared with SS.13
Humans who possess the L variant of the SERT protein have more rapid platelet serotonin uptake.14 Inhibitors of SERT have been shown to reduce platelet serotonin content, leading to disruption of thrombosis and increased bleeding due to inhibition of serotonin reuptake. A study in middle-aged men also found that genetic variability within SERT was associated with increased depressive symptoms and elevated levels of interleukin-6, a marker of inflammation.15 This indicates a common source of genetic vulnerability accounting for both depression and inflammation, and could help to explain the increased risk of CV disease in patients with depression.15
Mental stress–induced myocardial ischemia
The association between mental stress and activation of the sympathetic nervous system is well established. Stress leads to increases in blood pressure and heart rate. In patients with CAD, mental stress–induced myocardial ischemia (MSIMI) is a well-characterized phenomenon whereby ischemia is provoked by psychological stress. Sympathetic nervous system activation from stressful life events can increase vulnerability to CV outcomes such as MI, arrhythmias, and sudden cardiac death. MSIMI has been identified as a risk factor for poor outcomes in patients with known CAD. Proposed mechanisms include mismatch of myocardial blood supply and demand, as well as coronary spasm.
A recent study by Hassan et al evaluated associations of beta1-adrenergic receptor gene (ADBR1) polymorphisms with MSIMI in CAD patients.16 The researchers found a threefold increase in MSIMI in patients with a particular allele of the ADBR1 gene. This is the first report to highlight a specific genetic predisposition to susceptibility to MSIMI; it could help explain why some patients are at increased risk and also suggest targeting specific behavioral or pharmacologic therapies to reduce MSIMI.
ENDOTHELIAL DYSFUNCTION
Dysfunctional endothelium is important in the link between neurologic dysfunction and CV disease. Circulating EPCs have been recognized as playing an important role in maintaining vascular health. These cells originate in the bone marrow and may be identified by their surface markers and various functional characteristics. They have been shown to be a key part of the process involved in repair of biologic risk factor–mediated damage to endothelium and are reduced and/or dysfunctional in patients with CV risk factors such as tobacco use, diabetes, and hypertension. Low EPC levels have also been found in patients with cerebrovascular disease and are key in vascular neogenesis after ischemic insult.
Genetic effects on endothelium in migraine and stroke
A more recent area of interest in brain and cardiovascular health has been the role of EPCs in migraine, which is often debilitating. The association between migraine and increased risk of depression,17 as well as stroke and MI, has been well recognized. A recent study by MacClellan et al evaluated whether polymorphisms in genes regulating endothelial function and vascular tone influenced susceptibility to migraine and stroke in a large subset of young women.18 Analyzed genes included endothelin-1 (EDN), endothelin receptor type B (EDNRB), and nitric oxide synthase-3 (NOS3). Several polymorphisms within these genes were associated with stroke in white women but not in black women. However, the study did not show whether the association between migraine and stroke was mediated by the polymorphisms studied from the candidate genes. Others have found no association with EDNRB but found that the homozygous minor genotype (present in 5% of cases) of the EDNRA SNP rs2048894 showed association with migraine with aura (odds ratio [OR] = 1.61, 95% confidence interval [CI], 1.12–2.32; P = .010] when adjusted for gender and sample origin.19 Early age at onset (P = .011) in the pooled sample.19 Studies on larger samples will be needed to confirm these findings.
Depression may alter endothelial homeostasis
One mechanism by which depression may lead to increased CV disease risk is through effects on endothelial homeostasis. Several studies have found an association between depression and attenuated flow-mediated vasodilation. However, it has been postulated that this effect was mediated by atherosclerosis, as these studies were performed in older cohorts. To evaluate this association without the possibility of confounding severe atherosclerosis, a recent study evaluated this association in adolescent women with no known health problems.20 Regression analysis demonstrated a significant inverse relationship between depression and endothelial function as measured by pulse-wave amplitude. Most patients in this study had evidence of subclinical depression, suggesting that even those with mild symptoms can have endothelial dysfunction.20 These findings provide evidence to suggest that there may be some factors that underlie the vascular dysfunction associated with both depression and early subclinical atherosclerosis.
Focus on endothelial progenitor cells
EPCs from bone marrow and likely other sites (eg, spleen, perivascular omentum, liver, mesentery) play an important role in maintaining vascular integrity. The interaction between the bone marrow, nervous system, HPA axis, and progenitor cells, together with the effect of this interaction on vascular integrity, has become an area of increasing research. Specifically, the sympathetic nervous system has been identified as playing an important role in progenitor cell egress from the bone marrow. Mice with abnormal nerve conduction produce virtually no progenitor cells when treated with granulocyte colony-stimulating factor.21
Psychosocial factors influence activation of the sympathetic nervous system. An innovative recent study examined the effect of psychosocial determinants on bone marrow–derived progenitor cells; the psychosocial variables and progenitor cell counts were associated independently from traditional biological and behavioral risk factors.22 This is one of the first studies to examine the effect of psychosocial stressors on progenitor cells and should open the way for further research exploring this association and its effects on CV health.
Reprinted, with permission, from Neurology (Lee S-T, et al. Decreased number and function of endothelial progenitor cells in patients with migraine. Neurology 2008; 70:1510–1517).
Figure 2. Endothelial progenitor cell counts in headache patients. The counts were lowest in patients with migraine with aura, followed by migraine without aura, followed by tension headaches. Box plots show the median count (white lines), interquartile ranges (green boxes), 5% to 95% percentiles (whiskers), and outliers (dots). P values were calculated using a two-tailed Student t test.24Another area of interest has been the ability of progenitor cells to aid in repair after neurologic damage following vascular insult. Two areas related to progenitor cells have been stroke and migraine, which is a risk factor for stroke as well as depression.23 Compared with patients with tension headaches, patients with migraines (with and without aura) had lower levels of EPCs, with the lowest levels observed in migraine patients with aura (Figure2).24 The EPCs in patients with migraine with and without aura also showed reduced migratory capacity and increased senescence.24
Figure 3. Temporal profile of number of circulating endothelial progenitor cells in stroke patients by stroke outcome at 3 months. Box plots show the median number (white lines) and interquartile ranges (boxes).25Another study examined the relationship between EPCs and ischemic stroke, finding that an increase in circulating EPCs after acute ischemic stroke was associated with better outcomes and reduced infarct growth (Figure 3).25 This suggests that EPCs may play an important role in neurovascular repair after ischemic insult.
Figure 4. Endothelial progenitor cell levels and age-related white matter change severity as measured by computed tomography.26A different study evaluated EPC levels in individuals with age-related white matter changes.26 These white matter changes, measured on computed tomography or magnetic resonance imaging, correlate with microvascular pathology mainly within the elderly and are associated with increased risk of stroke and cognitive impairment such as dementia. In the study, circulating levels of EPCs were found to be significantly lower in patients with severe age-related white matter changes (Figure 4).26 This suggests that defects in endothelial repair are linked to small-vessel cerebrovascular disease.
Platelet activity
Platelets play a critical role in endothelial hemostasis. Alterations in platelet function have been hypothesized as a mechanism by which depression may lead to CV disease.27 A recent study analyzed the effects of persistent depressive symptoms on platelet activation in a cohort of spousal dementia caregivers.28 P-selectin, measured as an index of platelet activation, and depression, mainly in the subclinical range, were associated with elevated platelet reactivity and recovery. This may be one mechanism by which elderly caregivers are at risk of CV disease even without evidence of clinical depression.
Serotonin is also known for its effect on platelet function and vascular tone and is one of the main targets of antidepressant therapy. Several studies have analyzed the effect of depression treatment with SSRIs on platelet function. An initial analysis from the SADHART trial in 2003 found that in depressed patients treated with sertraline after acute coronary syndrome, reductions in platelet/endothelial activation occurred despite concurrent treatment with antiplatelet drugs, including aspirin and clopidogrel.29 This suggests that the antiplatelet properties of sertraline differ from those of aspirin and other antiplatelet therapies. A study by van Zyl et al30 yielded similar results with a different SSRI, citalopram, along with enhanced production of nitric oxide.
HORMONES, NEGATIVE AFFECT, AND CV DISEASE
Patients with CAD have different patterns of cortisol excretion compared with controls. A dysfunctional HPA axis has been implicated, leading to a failure in containing inflammatory activity.31 In healthy older patients without known CAD, heightened cortisol reactivity is associated with a greater extent of coronary artery calcification.32 Cynical hostility is also associated with CAD, but the mechanism is unclear. Higher levels of cynical hostility were associated with attenuation of the decreasing phase of the cortisol awakening response.33 Another recent study also found a higher cortisol awakening response and a larger ratio of total cholesterol to high-density lipoprotein cholesterol in response to stress in socially isolated men.34
Although cortisol is the most studied end product of the HPA axis, aldosterone is also released. Recent research has focused on the role of aldosterone in CV injury through its increase in superoxide generation and its upregulation of genes involved in inflammation, fibrosis, and atherosclerosis.35,36 Several studies have demonstrated the increased incidence of CV disease, including atrial fibrillation, stroke, and MI, among patients with primary hyperaldosteronism compared with patients with similar blood pressure elevations.37,38 Continued evidence supports the importance of the endocrine pathways in modulation of CV disease, and future research should continue to explore the role of various hormones in this interaction.
DIRECTIONS FOR FUTURE RESEARCH
Although advances have been made in understanding the pathophysiologic mechanisms involved in interactions between the nervous system and CV disease, many factors have yet to be completely understood. For example, the role of gender in the interaction between psychological distress and heart disease is an area of increasing discussion. CV disease is a significant cause of morbidity and mortality among women, who are also at increased risk of depression and anxiety as well as stroke and migraine. Recent literature supports the concept that significant biologic differences exist in the effect of stress and depression on men and women. A recent study in mice found that a lifelong increase in SERT function decreased constitutive cerebral metabolism in a number of brain regions, and that this effect was significant only in females.39 Gender differences also appear to exist in the efficacy of antidepressant therapy.11 Although these differences have been observed, it remains unclear whether they primarily are due to obvious hormonal differences or to differences in genetic predisposition.
Stem cell therapy—for stroke and perhaps even for migraine and other disorders—is another area of potential future research. In CV disease, bone marrow–derived EPCs have been used in the post-MI setting and for heart failure. Injured endothelium presents a similar target for endothelial repair with cell therapy in stroke and migraine as well. Continued research in these areas will provide new insights into this brain-vascular interaction and could help provide new directions for treatment in the future.
References
McCaffery JM, Duan QL, Frasure-Smith N, et al Genetic predictors of depressive symptoms in cardiac patients. Am J Med Genet B Neuropsychiatr Genet2009; 150B:381–388.
Cohen Z, Bouchelet I, Olivier A, et al Multiple microvascular and astroglial 5-hydroxytryptamine receptor subtypes in human brain: molecular and pharmacologic characterization. J Cereb Blood Flow Metab1999; 19:908–917.
Kring SI, Werge T, Holst C, et al Polymorphisms of serotonin receptor 2A and 2C genes and COMT in relation to obesity and type 2 diabetes. PLoS One2009; 4:e6696.
Ni W, Watts SW. 5-Hydroxytryptamine in the cardiovascular system: focus on the serotonin transporter (SERT). Clin Exp Pharmacol Physiol2006; 33:575–583.
Nefedov VP, Nefedova VV, Kononykhina VS, et al The effect of serotonin on the hematopoietic stem cells of the bone marrow [in Russian]. Biull Eksp Biol Med1991; 112:488–489.
Pakala R, Willerson JT, Benedict CR. Mitogenic effect of serotonin on vascular endothelial cells. Circulation1994; 90:1919–1926.
Yang M, Li K, Ng PC, et al Promoting effects of serotonin on hematopoiesis: ex vivo expansion of cord blood CD34+ stem/progenitor cells, proliferation of bone marrow stromal cells, and antiapoptosis. Stem Cells2007; 25:1800–1806.
Cloez-Tayarani I, Petit-Bertron AF, Venters HD, et al Differential effect of serotonin on cytokine production in lipopolysaccharide-stimulated human peripheral blood mononuclear cells: involvement of 5-hydroxytryptamine2A receptors. Int Immunol2003; 15:233–240.
Hoefgen B, Schulze TG, Ohlraun S, et al The power of sample size and homogenous sampling: association between the 5-HTTLPR serotonin transporter polymorphism and major depressive disorder. Biol Psychiatry2005; 57:247–251.
Nakatani D, Sato H, Sakata Y, et al Influence of serotonin transporter gene polymorphism on depressive symptoms and new cardiac events after acute myocardial infarction. Am Heart J2005; 150:652–658.
Brummett BH, Boyle SH, Siegler IC, et al Effects of environmental stress and gender on associations among symptoms of depression and the serotonin transporter gene linked polymorphic region (5-HTTLPR). Behav Genet2008; 38:34–43.
Fumeron F, Betoulle D, Nicaud V, et al Serotonin transporter gene polymorphism and myocardial infarction: Etude Cas-Témoins de l’Infarctus du Myocarde (ECTIM). Circulation2002; 105:2943–2945.
Phillips-Bute B, Mathew JP, Blumenthal JA, et al Relationship of genetic variability and depressive symptoms to adverse events after coronary artery bypass graft surgery. Psychosom Med2008; 70:953–959.
Greenberg BD, Tolliver TJ, Huang SJ, et al Genetic variation in the serotonin transporter promoter region affects serotonin uptake in human blood platelets. Am J Med Genet1999; 88:83–87.
Su S, Zhao J, Bremner JD, et al Serotonin transporter gene, depressive symptoms, and interleukin-6. Circ Cardiovasc Genet2009; 2:614–620.
Hassan M, York KM, Li H, et al Association of beta1-adrenergic receptor genetic polymorphism with mental stress-induced myocardial ischemia in patients with coronary artery disease. Arch Intern Med2008; 168:763–770.
Victor TW, Hu X, Campbell J, et al Association between migraine, anxiety and depression. Cephalalgia2009; Jul9[Epub ahead of print]
MacClellan LR, Howard TD, Cole JW, et al Relation of candidate genes that encode for endothelial function to migraine and stroke: the Stroke Prevention in Young Women study. Stroke2009; 40:e550–e557.
Tikka-Kleemola P, Kaunisto MA, Hamalainen E, et al Genetic association study of endothelin-1 and its receptors EDNRA and EDNRB in migraine with aura. Cephalalgia2009; 29:1224–1231.
Tomfohr LM, Martin TM, Miller GE. Symptoms of depression and impaired endothelial function in healthy adolescent women. J Behav Med2008; 31:137–143.
Katayama Y, Battista M, Kao WM, et al Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell2006; 124:407–421.
Fischer JC, Kudielka BM, von Kanel R, et al Bone-marrow derived progenitor cells are associated with psychosocial determinants of health after controlling for classical biological and behavioral cardiovascular risk factors. Brain Behav Immun2009; 23:419–426.
Baskin SM, Smitherman TA. Migraine and psychiatric disorders: comorbidities, mechanisms, and clinical applications. Neurol Sci2009; 30( suppl 1):S61–S65.
Lee S-T, Chu K, Jung KH, et al Decreased number and function of endothelial progenitor cells in patients with migraine. Neurology2008; 70:1510–1517.
Sobrino T, Hurtado O, Moro MA, et al The increase of circulating endothelial progenitor cells after acute ischemic stroke is associated with good outcome. Stroke2007; 38:2759–2764.
Jickling G, Salam A, Mohammad A, et al Circulating endothelial progenitor cells and age-related white matter changes. Stroke2009; 40:3191–3196.
Parakh K, Sakhuja A, Bhat U, et al Platelet function in patients with depression. South Med J2008; 101:612–617.
Aschbacher K, Mills PJ, von Känel R, et al Effects of depressive and anxious symptoms on norepinephrine and platelet P-selectin responses to acute psychological stress among elderly caregivers. Brain Behav Immun2008; 22:493–502.
Serebruany VL, Glassman AH, Malinin AI, et al Platelet/endothelial biomarkers in depressed patients treated with the selective serotonin reuptake inhibitor sertraline after acute coronary events: the Sertraline AntiDepressant Heart Attack Randomized Trial (SADHART) platelet substudy. Circulation2003; 108:939–944.
van Zyl LT, Lesperance F, Frasure-Smith N, et al Platelet and endothelial activity in comorbid major depression and coronary artery disease patients treated with citalopram: the Canadian Cardiac Randomized Evaluation of Antidepressant and Psychotherapy Efficacy Trial (CREATE) biomarker sub-study. J Thromb Thrombolysis2009; 27:48–56.
Nijm J, Kristenson M, Olsson AG, et al Impaired cortisol response to acute stressors in patients with coronary disease: implications for inflammatory activity. J Intern Med2007; 262:375–384.
Hamer M, O’Donnell K, Lahiri A, Steptoe A. Salivary cortisol responses to mental stress are associated with coronary artery calcification in healthy men and women. Eur Heart J2010; 31:424–429.
Ranjit N, Diez-Roux AV, Sanchez B, et al Association of salivary cortisol circadian pattern with cynical hostility: multi-ethnic study of atherosclerosis. Psychosom Med2009; 71:748–755.
Grant N, Hamer M, Steptoe A. Social isolation and stress-related cardiovascular, lipid, and cortisol responses. Ann Behav Med2009; 37:29–37.
Jaffe IZ, Mendelsohn ME. Angiotensin II and aldosterone regulate gene transcription via functional mineralocortocoid receptors in human coronary artery smooth muscle cells. Circ Res2005; 96:643–650.
Kubzansky LD, Adler GK. Aldosterone: a forgotten mediator of the relationship between psychological stress and heart disease. Neurosci Biobehav Rev2010; 34:80–86.
Milliez P, Girerd X, Plouin PF, et al Evidence for an increased rate of cardiovascular events in patients with primary aldosteronism. J Am Coll Cardiol2005; 45:1243–1248.
Duprez DA, Bauwens FR, De Buyzere ML, et al Influence of arterial blood pressure and aldosterone on left ventricular hypertrophy in moderate essential hypertension. Am J Cardiol1993; 71:17A–20A.
Dawson N, Ferrington L, Olverman HJ, et al Sex influences the effect of a lifelong increase in serotonin transporter function on cerebral metabolism. J Neurosci Res2009; 87:2375–2385.
Ki E. Park, MD Division of Cardiovascular Medicine, Department of Medicine, University of Florida, Gainesville, FL
Carl J. Pepine, MD Division of Cardiovascular Medicine, Department of Medicine, University of Florida, Gainesville, FL
Correspondence: Carl J. Pepine, MD, Division of Cardiovascular Medicine, Department of Medicine, University of Florida College of Medicine, 1600 SW Archer Road, P.O. Box 100277, Gainesville, FL 32610–0277; [email protected]
Both authors reported that they have no financial relationships that pose a potential conflict of interest with this article.
Ki E. Park, MD Division of Cardiovascular Medicine, Department of Medicine, University of Florida, Gainesville, FL
Carl J. Pepine, MD Division of Cardiovascular Medicine, Department of Medicine, University of Florida, Gainesville, FL
Correspondence: Carl J. Pepine, MD, Division of Cardiovascular Medicine, Department of Medicine, University of Florida College of Medicine, 1600 SW Archer Road, P.O. Box 100277, Gainesville, FL 32610–0277; [email protected]
Both authors reported that they have no financial relationships that pose a potential conflict of interest with this article.
Author and Disclosure Information
Ki E. Park, MD Division of Cardiovascular Medicine, Department of Medicine, University of Florida, Gainesville, FL
Carl J. Pepine, MD Division of Cardiovascular Medicine, Department of Medicine, University of Florida, Gainesville, FL
Correspondence: Carl J. Pepine, MD, Division of Cardiovascular Medicine, Department of Medicine, University of Florida College of Medicine, 1600 SW Archer Road, P.O. Box 100277, Gainesville, FL 32610–0277; [email protected]
Both authors reported that they have no financial relationships that pose a potential conflict of interest with this article.
The complex interaction between the nervous system and cardiovascular (CV) health is well recognized. In addition to stroke, migraine, and other disorders, the focus has shifted to depression and related negative-affect states including anxiety, chronic stress, posttraumatic stress disorder, and social isolation. Although all of these conditions have been linked to poor CV health, the mechanisms responsible for this brain-cardiovascular interaction have been poorly understood until recently.
Figure 1. Neurologic and psychological disorders share some overlapping pathophysiologic mechanisms with vascular disorders. (5-HTTLPR = serotonin transporter-linked promoter region; EPCs = endothelial progenitor cells; HPA = hypothalamic-pituitary-adrenal)At first, it appeared that the link between CV disease and such states as depression and anxiety was mediated by a clustering of shared risk conditions such as smoking, high low-density lipoprotein cholesterol, and obesity. As more research uncovered the pathophysiologic mechanisms underlying these negative-affect states, however, it became apparent that some of these mechanisms overlapped with those leading to vascular dysfunction (Figure 1). For instance, regulation of neurotransmitters such as serotonin is known to play a key role in mood dysfunction as well as in sleep and appetite, but only recently has this regulation been identified as an important component in moderating platelet function as well. Several other common pathways have been identified, including the hypothalamic-pituitary-adrenal (HPA) axis, endothelial progenitor cell (EPC) regulation, and inflammatory cell dysfunction. The role of genetic predisposition as a common factor leading to psychological and vascular dysfunction has been studied as well. In addition, research has recently expanded to examine other forms of central neurologic dysfunction besides mood, including stroke and migraine, and their connection with CV health.
This paper reviews a sample of the more recent advances in our understanding the pathophysiologic mechanisms underlying this complex brain-vascular interaction—including the roles of genetic predisposition, endothelial cell dysfunction, and endocrine dysregulation—and discusses future directions for research in this area.
GENETIC PREDISPOSITION
Increasing evidence highlights the importance of genetic predisposition in both psychological dysfunction and CV disease. The importance of common genetic variability in these two conditions originated from studies of twins that established that both depression and coronary artery disease (CAD) tended to run in families. However, recent studies focused on identifying specific genes that may link depression/negative affect and CAD through various pathways related to platelet reactivity, inflammation, and autonomic nervous system regulation, among many others. A candidate gene study by McCaffery et al1 sought to identify specific genes influencing depressive symptoms in CAD patients. Genes were selected based on their role in biologic pathways involved in inflammation, platelet activation, and the HPA axis and sympathetic nervous system. Among the 59 genes analyzed, the strongest associations were between markers involved in endothelial dysfunction and platelet aggregation—specifically, the involvement of von Willebrand factor in platelet recruitment and of vascular cell adhesion molecule–1 in recruitment and adhesion of inflammatory cells to injured endothelium. Other recent studies have analyzed the role of various genes in mediating neurologic and CV dysfunction.
Serotonin: A multitude of cardiovascular effects
Serotonin (5-HT) is best known as a neurotransmitter involved in mood regulation, but it also directly affects endothelial cells and vascular smooth muscle. Human brain microvascular endothelial cells have 5-HT2A receptors, which are thought to be involved in blood-brain trafficking.2 Polymorphisms in this receptor have been associated with impaired glucose tolerance and type 2 diabetes.3
However, the role of serotonin within the CV system has only recently been realized. Along with its associated serotonin transporter (SERT), serotonin has been best studied within the CV system for its role in development of pulmonary hypertension via vasoconstricton. However, serotonin and SERT exhibit other effects on the CV system, including regulation of factors involved in vascular integrity, such as platelet activity, endothelial dysfunction, and smooth muscle cell and endothelial cell mitogenesis. Serotonin is stored peripherally in platelets and, when released at sites of endothelial damage, promotes platelet aggregation. Cardiomyocytes also are a source of serotonin within the heart, resulting in positive chronotropy, positive inotropy, and activation of mitogen activity.4
Serotonin also affects endothelium through effects on bone marrow production of EPCs. It has been shown in mice to increase the proliferative activity of hematopoietic stem cells in the bone marrow via 5-HT2 receptors.5 Serotonin is a mitogen for canine and bovine endothelial cells6 and has also been shown to enhance ex vivo expansion of CD34+ hematopoietic stem cells in mice.7 It also seems to have an effect on regulation of inflammatory cytokines by regulating different secretory pathways. Activity of several different serotonin receptors, including 5-HT2A, 5-HT4, and 5-HT7, inhibits tumor necrosis factor production in peripheral cells.8
Insights from genetic analysis of SERT
These areas of recent research continue to highlight the importance of serotonin within the CV system, and its overlapping role in depression and other forms of psychological dysfunction underscores the importance of understanding its regulation.
The best-studied component of serotonin regulation is SERT, a sodium-dependent transporter that removes serotonin from outside the cell, bringing it back into the cell for repackaging. It is recognized as the site of action of the selective serotonin reuptake inhibitors (SSRIs).
Analysis of the serotonin transporter gene has traditionally focused on polymorphisms within the promoter region (5-HTTLPR). Two common alleles, the long (L) and short (S) variants, are the best characterized. The S variant is associated with a lower expression of SERT, leading to reduced uptake and release of serotonin. The SS genotype of SERT has been linked to major depressive disorder9 and to increased risk of subsequent cardiac events after myocardial infarction (MI).10
A study of the effects of environmental stress and gender on associations between depression and 5-HTTLPR found that this link varied according to gender and stressful life events.11 Specifically, women with the SS genotype, which leads to less transcriptional activity of 5-HTTLPR, tended to be more susceptible to depression under stressful life conditions. However, in men the LL genotype was associated with increased transcriptional activity and the same increased susceptibility to depression. 11 Expression of the LL genotype also resulted in upregulation of SERT, leading to higher MI risk.12
Another study investigated the relationship between genetic variability in two serotonin-related gene polymorphisms and found that the 5-HTTLPR gene polymorphism was associated with adverse cardiac events after coronary artery bypass graft surgery in combination with depression, specifically in patients with the L allele compared with SS.13
Humans who possess the L variant of the SERT protein have more rapid platelet serotonin uptake.14 Inhibitors of SERT have been shown to reduce platelet serotonin content, leading to disruption of thrombosis and increased bleeding due to inhibition of serotonin reuptake. A study in middle-aged men also found that genetic variability within SERT was associated with increased depressive symptoms and elevated levels of interleukin-6, a marker of inflammation.15 This indicates a common source of genetic vulnerability accounting for both depression and inflammation, and could help to explain the increased risk of CV disease in patients with depression.15
Mental stress–induced myocardial ischemia
The association between mental stress and activation of the sympathetic nervous system is well established. Stress leads to increases in blood pressure and heart rate. In patients with CAD, mental stress–induced myocardial ischemia (MSIMI) is a well-characterized phenomenon whereby ischemia is provoked by psychological stress. Sympathetic nervous system activation from stressful life events can increase vulnerability to CV outcomes such as MI, arrhythmias, and sudden cardiac death. MSIMI has been identified as a risk factor for poor outcomes in patients with known CAD. Proposed mechanisms include mismatch of myocardial blood supply and demand, as well as coronary spasm.
A recent study by Hassan et al evaluated associations of beta1-adrenergic receptor gene (ADBR1) polymorphisms with MSIMI in CAD patients.16 The researchers found a threefold increase in MSIMI in patients with a particular allele of the ADBR1 gene. This is the first report to highlight a specific genetic predisposition to susceptibility to MSIMI; it could help explain why some patients are at increased risk and also suggest targeting specific behavioral or pharmacologic therapies to reduce MSIMI.
ENDOTHELIAL DYSFUNCTION
Dysfunctional endothelium is important in the link between neurologic dysfunction and CV disease. Circulating EPCs have been recognized as playing an important role in maintaining vascular health. These cells originate in the bone marrow and may be identified by their surface markers and various functional characteristics. They have been shown to be a key part of the process involved in repair of biologic risk factor–mediated damage to endothelium and are reduced and/or dysfunctional in patients with CV risk factors such as tobacco use, diabetes, and hypertension. Low EPC levels have also been found in patients with cerebrovascular disease and are key in vascular neogenesis after ischemic insult.
Genetic effects on endothelium in migraine and stroke
A more recent area of interest in brain and cardiovascular health has been the role of EPCs in migraine, which is often debilitating. The association between migraine and increased risk of depression,17 as well as stroke and MI, has been well recognized. A recent study by MacClellan et al evaluated whether polymorphisms in genes regulating endothelial function and vascular tone influenced susceptibility to migraine and stroke in a large subset of young women.18 Analyzed genes included endothelin-1 (EDN), endothelin receptor type B (EDNRB), and nitric oxide synthase-3 (NOS3). Several polymorphisms within these genes were associated with stroke in white women but not in black women. However, the study did not show whether the association between migraine and stroke was mediated by the polymorphisms studied from the candidate genes. Others have found no association with EDNRB but found that the homozygous minor genotype (present in 5% of cases) of the EDNRA SNP rs2048894 showed association with migraine with aura (odds ratio [OR] = 1.61, 95% confidence interval [CI], 1.12–2.32; P = .010] when adjusted for gender and sample origin.19 Early age at onset (P = .011) in the pooled sample.19 Studies on larger samples will be needed to confirm these findings.
Depression may alter endothelial homeostasis
One mechanism by which depression may lead to increased CV disease risk is through effects on endothelial homeostasis. Several studies have found an association between depression and attenuated flow-mediated vasodilation. However, it has been postulated that this effect was mediated by atherosclerosis, as these studies were performed in older cohorts. To evaluate this association without the possibility of confounding severe atherosclerosis, a recent study evaluated this association in adolescent women with no known health problems.20 Regression analysis demonstrated a significant inverse relationship between depression and endothelial function as measured by pulse-wave amplitude. Most patients in this study had evidence of subclinical depression, suggesting that even those with mild symptoms can have endothelial dysfunction.20 These findings provide evidence to suggest that there may be some factors that underlie the vascular dysfunction associated with both depression and early subclinical atherosclerosis.
Focus on endothelial progenitor cells
EPCs from bone marrow and likely other sites (eg, spleen, perivascular omentum, liver, mesentery) play an important role in maintaining vascular integrity. The interaction between the bone marrow, nervous system, HPA axis, and progenitor cells, together with the effect of this interaction on vascular integrity, has become an area of increasing research. Specifically, the sympathetic nervous system has been identified as playing an important role in progenitor cell egress from the bone marrow. Mice with abnormal nerve conduction produce virtually no progenitor cells when treated with granulocyte colony-stimulating factor.21
Psychosocial factors influence activation of the sympathetic nervous system. An innovative recent study examined the effect of psychosocial determinants on bone marrow–derived progenitor cells; the psychosocial variables and progenitor cell counts were associated independently from traditional biological and behavioral risk factors.22 This is one of the first studies to examine the effect of psychosocial stressors on progenitor cells and should open the way for further research exploring this association and its effects on CV health.
Reprinted, with permission, from Neurology (Lee S-T, et al. Decreased number and function of endothelial progenitor cells in patients with migraine. Neurology 2008; 70:1510–1517).
Figure 2. Endothelial progenitor cell counts in headache patients. The counts were lowest in patients with migraine with aura, followed by migraine without aura, followed by tension headaches. Box plots show the median count (white lines), interquartile ranges (green boxes), 5% to 95% percentiles (whiskers), and outliers (dots). P values were calculated using a two-tailed Student t test.24Another area of interest has been the ability of progenitor cells to aid in repair after neurologic damage following vascular insult. Two areas related to progenitor cells have been stroke and migraine, which is a risk factor for stroke as well as depression.23 Compared with patients with tension headaches, patients with migraines (with and without aura) had lower levels of EPCs, with the lowest levels observed in migraine patients with aura (Figure2).24 The EPCs in patients with migraine with and without aura also showed reduced migratory capacity and increased senescence.24
Figure 3. Temporal profile of number of circulating endothelial progenitor cells in stroke patients by stroke outcome at 3 months. Box plots show the median number (white lines) and interquartile ranges (boxes).25Another study examined the relationship between EPCs and ischemic stroke, finding that an increase in circulating EPCs after acute ischemic stroke was associated with better outcomes and reduced infarct growth (Figure 3).25 This suggests that EPCs may play an important role in neurovascular repair after ischemic insult.
Figure 4. Endothelial progenitor cell levels and age-related white matter change severity as measured by computed tomography.26A different study evaluated EPC levels in individuals with age-related white matter changes.26 These white matter changes, measured on computed tomography or magnetic resonance imaging, correlate with microvascular pathology mainly within the elderly and are associated with increased risk of stroke and cognitive impairment such as dementia. In the study, circulating levels of EPCs were found to be significantly lower in patients with severe age-related white matter changes (Figure 4).26 This suggests that defects in endothelial repair are linked to small-vessel cerebrovascular disease.
Platelet activity
Platelets play a critical role in endothelial hemostasis. Alterations in platelet function have been hypothesized as a mechanism by which depression may lead to CV disease.27 A recent study analyzed the effects of persistent depressive symptoms on platelet activation in a cohort of spousal dementia caregivers.28 P-selectin, measured as an index of platelet activation, and depression, mainly in the subclinical range, were associated with elevated platelet reactivity and recovery. This may be one mechanism by which elderly caregivers are at risk of CV disease even without evidence of clinical depression.
Serotonin is also known for its effect on platelet function and vascular tone and is one of the main targets of antidepressant therapy. Several studies have analyzed the effect of depression treatment with SSRIs on platelet function. An initial analysis from the SADHART trial in 2003 found that in depressed patients treated with sertraline after acute coronary syndrome, reductions in platelet/endothelial activation occurred despite concurrent treatment with antiplatelet drugs, including aspirin and clopidogrel.29 This suggests that the antiplatelet properties of sertraline differ from those of aspirin and other antiplatelet therapies. A study by van Zyl et al30 yielded similar results with a different SSRI, citalopram, along with enhanced production of nitric oxide.
HORMONES, NEGATIVE AFFECT, AND CV DISEASE
Patients with CAD have different patterns of cortisol excretion compared with controls. A dysfunctional HPA axis has been implicated, leading to a failure in containing inflammatory activity.31 In healthy older patients without known CAD, heightened cortisol reactivity is associated with a greater extent of coronary artery calcification.32 Cynical hostility is also associated with CAD, but the mechanism is unclear. Higher levels of cynical hostility were associated with attenuation of the decreasing phase of the cortisol awakening response.33 Another recent study also found a higher cortisol awakening response and a larger ratio of total cholesterol to high-density lipoprotein cholesterol in response to stress in socially isolated men.34
Although cortisol is the most studied end product of the HPA axis, aldosterone is also released. Recent research has focused on the role of aldosterone in CV injury through its increase in superoxide generation and its upregulation of genes involved in inflammation, fibrosis, and atherosclerosis.35,36 Several studies have demonstrated the increased incidence of CV disease, including atrial fibrillation, stroke, and MI, among patients with primary hyperaldosteronism compared with patients with similar blood pressure elevations.37,38 Continued evidence supports the importance of the endocrine pathways in modulation of CV disease, and future research should continue to explore the role of various hormones in this interaction.
DIRECTIONS FOR FUTURE RESEARCH
Although advances have been made in understanding the pathophysiologic mechanisms involved in interactions between the nervous system and CV disease, many factors have yet to be completely understood. For example, the role of gender in the interaction between psychological distress and heart disease is an area of increasing discussion. CV disease is a significant cause of morbidity and mortality among women, who are also at increased risk of depression and anxiety as well as stroke and migraine. Recent literature supports the concept that significant biologic differences exist in the effect of stress and depression on men and women. A recent study in mice found that a lifelong increase in SERT function decreased constitutive cerebral metabolism in a number of brain regions, and that this effect was significant only in females.39 Gender differences also appear to exist in the efficacy of antidepressant therapy.11 Although these differences have been observed, it remains unclear whether they primarily are due to obvious hormonal differences or to differences in genetic predisposition.
Stem cell therapy—for stroke and perhaps even for migraine and other disorders—is another area of potential future research. In CV disease, bone marrow–derived EPCs have been used in the post-MI setting and for heart failure. Injured endothelium presents a similar target for endothelial repair with cell therapy in stroke and migraine as well. Continued research in these areas will provide new insights into this brain-vascular interaction and could help provide new directions for treatment in the future.
The complex interaction between the nervous system and cardiovascular (CV) health is well recognized. In addition to stroke, migraine, and other disorders, the focus has shifted to depression and related negative-affect states including anxiety, chronic stress, posttraumatic stress disorder, and social isolation. Although all of these conditions have been linked to poor CV health, the mechanisms responsible for this brain-cardiovascular interaction have been poorly understood until recently.
Figure 1. Neurologic and psychological disorders share some overlapping pathophysiologic mechanisms with vascular disorders. (5-HTTLPR = serotonin transporter-linked promoter region; EPCs = endothelial progenitor cells; HPA = hypothalamic-pituitary-adrenal)At first, it appeared that the link between CV disease and such states as depression and anxiety was mediated by a clustering of shared risk conditions such as smoking, high low-density lipoprotein cholesterol, and obesity. As more research uncovered the pathophysiologic mechanisms underlying these negative-affect states, however, it became apparent that some of these mechanisms overlapped with those leading to vascular dysfunction (Figure 1). For instance, regulation of neurotransmitters such as serotonin is known to play a key role in mood dysfunction as well as in sleep and appetite, but only recently has this regulation been identified as an important component in moderating platelet function as well. Several other common pathways have been identified, including the hypothalamic-pituitary-adrenal (HPA) axis, endothelial progenitor cell (EPC) regulation, and inflammatory cell dysfunction. The role of genetic predisposition as a common factor leading to psychological and vascular dysfunction has been studied as well. In addition, research has recently expanded to examine other forms of central neurologic dysfunction besides mood, including stroke and migraine, and their connection with CV health.
This paper reviews a sample of the more recent advances in our understanding the pathophysiologic mechanisms underlying this complex brain-vascular interaction—including the roles of genetic predisposition, endothelial cell dysfunction, and endocrine dysregulation—and discusses future directions for research in this area.
GENETIC PREDISPOSITION
Increasing evidence highlights the importance of genetic predisposition in both psychological dysfunction and CV disease. The importance of common genetic variability in these two conditions originated from studies of twins that established that both depression and coronary artery disease (CAD) tended to run in families. However, recent studies focused on identifying specific genes that may link depression/negative affect and CAD through various pathways related to platelet reactivity, inflammation, and autonomic nervous system regulation, among many others. A candidate gene study by McCaffery et al1 sought to identify specific genes influencing depressive symptoms in CAD patients. Genes were selected based on their role in biologic pathways involved in inflammation, platelet activation, and the HPA axis and sympathetic nervous system. Among the 59 genes analyzed, the strongest associations were between markers involved in endothelial dysfunction and platelet aggregation—specifically, the involvement of von Willebrand factor in platelet recruitment and of vascular cell adhesion molecule–1 in recruitment and adhesion of inflammatory cells to injured endothelium. Other recent studies have analyzed the role of various genes in mediating neurologic and CV dysfunction.
Serotonin: A multitude of cardiovascular effects
Serotonin (5-HT) is best known as a neurotransmitter involved in mood regulation, but it also directly affects endothelial cells and vascular smooth muscle. Human brain microvascular endothelial cells have 5-HT2A receptors, which are thought to be involved in blood-brain trafficking.2 Polymorphisms in this receptor have been associated with impaired glucose tolerance and type 2 diabetes.3
However, the role of serotonin within the CV system has only recently been realized. Along with its associated serotonin transporter (SERT), serotonin has been best studied within the CV system for its role in development of pulmonary hypertension via vasoconstricton. However, serotonin and SERT exhibit other effects on the CV system, including regulation of factors involved in vascular integrity, such as platelet activity, endothelial dysfunction, and smooth muscle cell and endothelial cell mitogenesis. Serotonin is stored peripherally in platelets and, when released at sites of endothelial damage, promotes platelet aggregation. Cardiomyocytes also are a source of serotonin within the heart, resulting in positive chronotropy, positive inotropy, and activation of mitogen activity.4
Serotonin also affects endothelium through effects on bone marrow production of EPCs. It has been shown in mice to increase the proliferative activity of hematopoietic stem cells in the bone marrow via 5-HT2 receptors.5 Serotonin is a mitogen for canine and bovine endothelial cells6 and has also been shown to enhance ex vivo expansion of CD34+ hematopoietic stem cells in mice.7 It also seems to have an effect on regulation of inflammatory cytokines by regulating different secretory pathways. Activity of several different serotonin receptors, including 5-HT2A, 5-HT4, and 5-HT7, inhibits tumor necrosis factor production in peripheral cells.8
Insights from genetic analysis of SERT
These areas of recent research continue to highlight the importance of serotonin within the CV system, and its overlapping role in depression and other forms of psychological dysfunction underscores the importance of understanding its regulation.
The best-studied component of serotonin regulation is SERT, a sodium-dependent transporter that removes serotonin from outside the cell, bringing it back into the cell for repackaging. It is recognized as the site of action of the selective serotonin reuptake inhibitors (SSRIs).
Analysis of the serotonin transporter gene has traditionally focused on polymorphisms within the promoter region (5-HTTLPR). Two common alleles, the long (L) and short (S) variants, are the best characterized. The S variant is associated with a lower expression of SERT, leading to reduced uptake and release of serotonin. The SS genotype of SERT has been linked to major depressive disorder9 and to increased risk of subsequent cardiac events after myocardial infarction (MI).10
A study of the effects of environmental stress and gender on associations between depression and 5-HTTLPR found that this link varied according to gender and stressful life events.11 Specifically, women with the SS genotype, which leads to less transcriptional activity of 5-HTTLPR, tended to be more susceptible to depression under stressful life conditions. However, in men the LL genotype was associated with increased transcriptional activity and the same increased susceptibility to depression. 11 Expression of the LL genotype also resulted in upregulation of SERT, leading to higher MI risk.12
Another study investigated the relationship between genetic variability in two serotonin-related gene polymorphisms and found that the 5-HTTLPR gene polymorphism was associated with adverse cardiac events after coronary artery bypass graft surgery in combination with depression, specifically in patients with the L allele compared with SS.13
Humans who possess the L variant of the SERT protein have more rapid platelet serotonin uptake.14 Inhibitors of SERT have been shown to reduce platelet serotonin content, leading to disruption of thrombosis and increased bleeding due to inhibition of serotonin reuptake. A study in middle-aged men also found that genetic variability within SERT was associated with increased depressive symptoms and elevated levels of interleukin-6, a marker of inflammation.15 This indicates a common source of genetic vulnerability accounting for both depression and inflammation, and could help to explain the increased risk of CV disease in patients with depression.15
Mental stress–induced myocardial ischemia
The association between mental stress and activation of the sympathetic nervous system is well established. Stress leads to increases in blood pressure and heart rate. In patients with CAD, mental stress–induced myocardial ischemia (MSIMI) is a well-characterized phenomenon whereby ischemia is provoked by psychological stress. Sympathetic nervous system activation from stressful life events can increase vulnerability to CV outcomes such as MI, arrhythmias, and sudden cardiac death. MSIMI has been identified as a risk factor for poor outcomes in patients with known CAD. Proposed mechanisms include mismatch of myocardial blood supply and demand, as well as coronary spasm.
A recent study by Hassan et al evaluated associations of beta1-adrenergic receptor gene (ADBR1) polymorphisms with MSIMI in CAD patients.16 The researchers found a threefold increase in MSIMI in patients with a particular allele of the ADBR1 gene. This is the first report to highlight a specific genetic predisposition to susceptibility to MSIMI; it could help explain why some patients are at increased risk and also suggest targeting specific behavioral or pharmacologic therapies to reduce MSIMI.
ENDOTHELIAL DYSFUNCTION
Dysfunctional endothelium is important in the link between neurologic dysfunction and CV disease. Circulating EPCs have been recognized as playing an important role in maintaining vascular health. These cells originate in the bone marrow and may be identified by their surface markers and various functional characteristics. They have been shown to be a key part of the process involved in repair of biologic risk factor–mediated damage to endothelium and are reduced and/or dysfunctional in patients with CV risk factors such as tobacco use, diabetes, and hypertension. Low EPC levels have also been found in patients with cerebrovascular disease and are key in vascular neogenesis after ischemic insult.
Genetic effects on endothelium in migraine and stroke
A more recent area of interest in brain and cardiovascular health has been the role of EPCs in migraine, which is often debilitating. The association between migraine and increased risk of depression,17 as well as stroke and MI, has been well recognized. A recent study by MacClellan et al evaluated whether polymorphisms in genes regulating endothelial function and vascular tone influenced susceptibility to migraine and stroke in a large subset of young women.18 Analyzed genes included endothelin-1 (EDN), endothelin receptor type B (EDNRB), and nitric oxide synthase-3 (NOS3). Several polymorphisms within these genes were associated with stroke in white women but not in black women. However, the study did not show whether the association between migraine and stroke was mediated by the polymorphisms studied from the candidate genes. Others have found no association with EDNRB but found that the homozygous minor genotype (present in 5% of cases) of the EDNRA SNP rs2048894 showed association with migraine with aura (odds ratio [OR] = 1.61, 95% confidence interval [CI], 1.12–2.32; P = .010] when adjusted for gender and sample origin.19 Early age at onset (P = .011) in the pooled sample.19 Studies on larger samples will be needed to confirm these findings.
Depression may alter endothelial homeostasis
One mechanism by which depression may lead to increased CV disease risk is through effects on endothelial homeostasis. Several studies have found an association between depression and attenuated flow-mediated vasodilation. However, it has been postulated that this effect was mediated by atherosclerosis, as these studies were performed in older cohorts. To evaluate this association without the possibility of confounding severe atherosclerosis, a recent study evaluated this association in adolescent women with no known health problems.20 Regression analysis demonstrated a significant inverse relationship between depression and endothelial function as measured by pulse-wave amplitude. Most patients in this study had evidence of subclinical depression, suggesting that even those with mild symptoms can have endothelial dysfunction.20 These findings provide evidence to suggest that there may be some factors that underlie the vascular dysfunction associated with both depression and early subclinical atherosclerosis.
Focus on endothelial progenitor cells
EPCs from bone marrow and likely other sites (eg, spleen, perivascular omentum, liver, mesentery) play an important role in maintaining vascular integrity. The interaction between the bone marrow, nervous system, HPA axis, and progenitor cells, together with the effect of this interaction on vascular integrity, has become an area of increasing research. Specifically, the sympathetic nervous system has been identified as playing an important role in progenitor cell egress from the bone marrow. Mice with abnormal nerve conduction produce virtually no progenitor cells when treated with granulocyte colony-stimulating factor.21
Psychosocial factors influence activation of the sympathetic nervous system. An innovative recent study examined the effect of psychosocial determinants on bone marrow–derived progenitor cells; the psychosocial variables and progenitor cell counts were associated independently from traditional biological and behavioral risk factors.22 This is one of the first studies to examine the effect of psychosocial stressors on progenitor cells and should open the way for further research exploring this association and its effects on CV health.
Reprinted, with permission, from Neurology (Lee S-T, et al. Decreased number and function of endothelial progenitor cells in patients with migraine. Neurology 2008; 70:1510–1517).
Figure 2. Endothelial progenitor cell counts in headache patients. The counts were lowest in patients with migraine with aura, followed by migraine without aura, followed by tension headaches. Box plots show the median count (white lines), interquartile ranges (green boxes), 5% to 95% percentiles (whiskers), and outliers (dots). P values were calculated using a two-tailed Student t test.24Another area of interest has been the ability of progenitor cells to aid in repair after neurologic damage following vascular insult. Two areas related to progenitor cells have been stroke and migraine, which is a risk factor for stroke as well as depression.23 Compared with patients with tension headaches, patients with migraines (with and without aura) had lower levels of EPCs, with the lowest levels observed in migraine patients with aura (Figure2).24 The EPCs in patients with migraine with and without aura also showed reduced migratory capacity and increased senescence.24
Figure 3. Temporal profile of number of circulating endothelial progenitor cells in stroke patients by stroke outcome at 3 months. Box plots show the median number (white lines) and interquartile ranges (boxes).25Another study examined the relationship between EPCs and ischemic stroke, finding that an increase in circulating EPCs after acute ischemic stroke was associated with better outcomes and reduced infarct growth (Figure 3).25 This suggests that EPCs may play an important role in neurovascular repair after ischemic insult.
Figure 4. Endothelial progenitor cell levels and age-related white matter change severity as measured by computed tomography.26A different study evaluated EPC levels in individuals with age-related white matter changes.26 These white matter changes, measured on computed tomography or magnetic resonance imaging, correlate with microvascular pathology mainly within the elderly and are associated with increased risk of stroke and cognitive impairment such as dementia. In the study, circulating levels of EPCs were found to be significantly lower in patients with severe age-related white matter changes (Figure 4).26 This suggests that defects in endothelial repair are linked to small-vessel cerebrovascular disease.
Platelet activity
Platelets play a critical role in endothelial hemostasis. Alterations in platelet function have been hypothesized as a mechanism by which depression may lead to CV disease.27 A recent study analyzed the effects of persistent depressive symptoms on platelet activation in a cohort of spousal dementia caregivers.28 P-selectin, measured as an index of platelet activation, and depression, mainly in the subclinical range, were associated with elevated platelet reactivity and recovery. This may be one mechanism by which elderly caregivers are at risk of CV disease even without evidence of clinical depression.
Serotonin is also known for its effect on platelet function and vascular tone and is one of the main targets of antidepressant therapy. Several studies have analyzed the effect of depression treatment with SSRIs on platelet function. An initial analysis from the SADHART trial in 2003 found that in depressed patients treated with sertraline after acute coronary syndrome, reductions in platelet/endothelial activation occurred despite concurrent treatment with antiplatelet drugs, including aspirin and clopidogrel.29 This suggests that the antiplatelet properties of sertraline differ from those of aspirin and other antiplatelet therapies. A study by van Zyl et al30 yielded similar results with a different SSRI, citalopram, along with enhanced production of nitric oxide.
HORMONES, NEGATIVE AFFECT, AND CV DISEASE
Patients with CAD have different patterns of cortisol excretion compared with controls. A dysfunctional HPA axis has been implicated, leading to a failure in containing inflammatory activity.31 In healthy older patients without known CAD, heightened cortisol reactivity is associated with a greater extent of coronary artery calcification.32 Cynical hostility is also associated with CAD, but the mechanism is unclear. Higher levels of cynical hostility were associated with attenuation of the decreasing phase of the cortisol awakening response.33 Another recent study also found a higher cortisol awakening response and a larger ratio of total cholesterol to high-density lipoprotein cholesterol in response to stress in socially isolated men.34
Although cortisol is the most studied end product of the HPA axis, aldosterone is also released. Recent research has focused on the role of aldosterone in CV injury through its increase in superoxide generation and its upregulation of genes involved in inflammation, fibrosis, and atherosclerosis.35,36 Several studies have demonstrated the increased incidence of CV disease, including atrial fibrillation, stroke, and MI, among patients with primary hyperaldosteronism compared with patients with similar blood pressure elevations.37,38 Continued evidence supports the importance of the endocrine pathways in modulation of CV disease, and future research should continue to explore the role of various hormones in this interaction.
DIRECTIONS FOR FUTURE RESEARCH
Although advances have been made in understanding the pathophysiologic mechanisms involved in interactions between the nervous system and CV disease, many factors have yet to be completely understood. For example, the role of gender in the interaction between psychological distress and heart disease is an area of increasing discussion. CV disease is a significant cause of morbidity and mortality among women, who are also at increased risk of depression and anxiety as well as stroke and migraine. Recent literature supports the concept that significant biologic differences exist in the effect of stress and depression on men and women. A recent study in mice found that a lifelong increase in SERT function decreased constitutive cerebral metabolism in a number of brain regions, and that this effect was significant only in females.39 Gender differences also appear to exist in the efficacy of antidepressant therapy.11 Although these differences have been observed, it remains unclear whether they primarily are due to obvious hormonal differences or to differences in genetic predisposition.
Stem cell therapy—for stroke and perhaps even for migraine and other disorders—is another area of potential future research. In CV disease, bone marrow–derived EPCs have been used in the post-MI setting and for heart failure. Injured endothelium presents a similar target for endothelial repair with cell therapy in stroke and migraine as well. Continued research in these areas will provide new insights into this brain-vascular interaction and could help provide new directions for treatment in the future.
References
McCaffery JM, Duan QL, Frasure-Smith N, et al Genetic predictors of depressive symptoms in cardiac patients. Am J Med Genet B Neuropsychiatr Genet2009; 150B:381–388.
Cohen Z, Bouchelet I, Olivier A, et al Multiple microvascular and astroglial 5-hydroxytryptamine receptor subtypes in human brain: molecular and pharmacologic characterization. J Cereb Blood Flow Metab1999; 19:908–917.
Kring SI, Werge T, Holst C, et al Polymorphisms of serotonin receptor 2A and 2C genes and COMT in relation to obesity and type 2 diabetes. PLoS One2009; 4:e6696.
Ni W, Watts SW. 5-Hydroxytryptamine in the cardiovascular system: focus on the serotonin transporter (SERT). Clin Exp Pharmacol Physiol2006; 33:575–583.
Nefedov VP, Nefedova VV, Kononykhina VS, et al The effect of serotonin on the hematopoietic stem cells of the bone marrow [in Russian]. Biull Eksp Biol Med1991; 112:488–489.
Pakala R, Willerson JT, Benedict CR. Mitogenic effect of serotonin on vascular endothelial cells. Circulation1994; 90:1919–1926.
Yang M, Li K, Ng PC, et al Promoting effects of serotonin on hematopoiesis: ex vivo expansion of cord blood CD34+ stem/progenitor cells, proliferation of bone marrow stromal cells, and antiapoptosis. Stem Cells2007; 25:1800–1806.
Cloez-Tayarani I, Petit-Bertron AF, Venters HD, et al Differential effect of serotonin on cytokine production in lipopolysaccharide-stimulated human peripheral blood mononuclear cells: involvement of 5-hydroxytryptamine2A receptors. Int Immunol2003; 15:233–240.
Hoefgen B, Schulze TG, Ohlraun S, et al The power of sample size and homogenous sampling: association between the 5-HTTLPR serotonin transporter polymorphism and major depressive disorder. Biol Psychiatry2005; 57:247–251.
Nakatani D, Sato H, Sakata Y, et al Influence of serotonin transporter gene polymorphism on depressive symptoms and new cardiac events after acute myocardial infarction. Am Heart J2005; 150:652–658.
Brummett BH, Boyle SH, Siegler IC, et al Effects of environmental stress and gender on associations among symptoms of depression and the serotonin transporter gene linked polymorphic region (5-HTTLPR). Behav Genet2008; 38:34–43.
Fumeron F, Betoulle D, Nicaud V, et al Serotonin transporter gene polymorphism and myocardial infarction: Etude Cas-Témoins de l’Infarctus du Myocarde (ECTIM). Circulation2002; 105:2943–2945.
Phillips-Bute B, Mathew JP, Blumenthal JA, et al Relationship of genetic variability and depressive symptoms to adverse events after coronary artery bypass graft surgery. Psychosom Med2008; 70:953–959.
Greenberg BD, Tolliver TJ, Huang SJ, et al Genetic variation in the serotonin transporter promoter region affects serotonin uptake in human blood platelets. Am J Med Genet1999; 88:83–87.
Su S, Zhao J, Bremner JD, et al Serotonin transporter gene, depressive symptoms, and interleukin-6. Circ Cardiovasc Genet2009; 2:614–620.
Hassan M, York KM, Li H, et al Association of beta1-adrenergic receptor genetic polymorphism with mental stress-induced myocardial ischemia in patients with coronary artery disease. Arch Intern Med2008; 168:763–770.
Victor TW, Hu X, Campbell J, et al Association between migraine, anxiety and depression. Cephalalgia2009; Jul9[Epub ahead of print]
MacClellan LR, Howard TD, Cole JW, et al Relation of candidate genes that encode for endothelial function to migraine and stroke: the Stroke Prevention in Young Women study. Stroke2009; 40:e550–e557.
Tikka-Kleemola P, Kaunisto MA, Hamalainen E, et al Genetic association study of endothelin-1 and its receptors EDNRA and EDNRB in migraine with aura. Cephalalgia2009; 29:1224–1231.
Tomfohr LM, Martin TM, Miller GE. Symptoms of depression and impaired endothelial function in healthy adolescent women. J Behav Med2008; 31:137–143.
Katayama Y, Battista M, Kao WM, et al Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell2006; 124:407–421.
Fischer JC, Kudielka BM, von Kanel R, et al Bone-marrow derived progenitor cells are associated with psychosocial determinants of health after controlling for classical biological and behavioral cardiovascular risk factors. Brain Behav Immun2009; 23:419–426.
Baskin SM, Smitherman TA. Migraine and psychiatric disorders: comorbidities, mechanisms, and clinical applications. Neurol Sci2009; 30( suppl 1):S61–S65.
Lee S-T, Chu K, Jung KH, et al Decreased number and function of endothelial progenitor cells in patients with migraine. Neurology2008; 70:1510–1517.
Sobrino T, Hurtado O, Moro MA, et al The increase of circulating endothelial progenitor cells after acute ischemic stroke is associated with good outcome. Stroke2007; 38:2759–2764.
Jickling G, Salam A, Mohammad A, et al Circulating endothelial progenitor cells and age-related white matter changes. Stroke2009; 40:3191–3196.
Parakh K, Sakhuja A, Bhat U, et al Platelet function in patients with depression. South Med J2008; 101:612–617.
Aschbacher K, Mills PJ, von Känel R, et al Effects of depressive and anxious symptoms on norepinephrine and platelet P-selectin responses to acute psychological stress among elderly caregivers. Brain Behav Immun2008; 22:493–502.
Serebruany VL, Glassman AH, Malinin AI, et al Platelet/endothelial biomarkers in depressed patients treated with the selective serotonin reuptake inhibitor sertraline after acute coronary events: the Sertraline AntiDepressant Heart Attack Randomized Trial (SADHART) platelet substudy. Circulation2003; 108:939–944.
van Zyl LT, Lesperance F, Frasure-Smith N, et al Platelet and endothelial activity in comorbid major depression and coronary artery disease patients treated with citalopram: the Canadian Cardiac Randomized Evaluation of Antidepressant and Psychotherapy Efficacy Trial (CREATE) biomarker sub-study. J Thromb Thrombolysis2009; 27:48–56.
Nijm J, Kristenson M, Olsson AG, et al Impaired cortisol response to acute stressors in patients with coronary disease: implications for inflammatory activity. J Intern Med2007; 262:375–384.
Hamer M, O’Donnell K, Lahiri A, Steptoe A. Salivary cortisol responses to mental stress are associated with coronary artery calcification in healthy men and women. Eur Heart J2010; 31:424–429.
Ranjit N, Diez-Roux AV, Sanchez B, et al Association of salivary cortisol circadian pattern with cynical hostility: multi-ethnic study of atherosclerosis. Psychosom Med2009; 71:748–755.
Grant N, Hamer M, Steptoe A. Social isolation and stress-related cardiovascular, lipid, and cortisol responses. Ann Behav Med2009; 37:29–37.
Jaffe IZ, Mendelsohn ME. Angiotensin II and aldosterone regulate gene transcription via functional mineralocortocoid receptors in human coronary artery smooth muscle cells. Circ Res2005; 96:643–650.
Kubzansky LD, Adler GK. Aldosterone: a forgotten mediator of the relationship between psychological stress and heart disease. Neurosci Biobehav Rev2010; 34:80–86.
Milliez P, Girerd X, Plouin PF, et al Evidence for an increased rate of cardiovascular events in patients with primary aldosteronism. J Am Coll Cardiol2005; 45:1243–1248.
Duprez DA, Bauwens FR, De Buyzere ML, et al Influence of arterial blood pressure and aldosterone on left ventricular hypertrophy in moderate essential hypertension. Am J Cardiol1993; 71:17A–20A.
Dawson N, Ferrington L, Olverman HJ, et al Sex influences the effect of a lifelong increase in serotonin transporter function on cerebral metabolism. J Neurosci Res2009; 87:2375–2385.
References
McCaffery JM, Duan QL, Frasure-Smith N, et al Genetic predictors of depressive symptoms in cardiac patients. Am J Med Genet B Neuropsychiatr Genet2009; 150B:381–388.
Cohen Z, Bouchelet I, Olivier A, et al Multiple microvascular and astroglial 5-hydroxytryptamine receptor subtypes in human brain: molecular and pharmacologic characterization. J Cereb Blood Flow Metab1999; 19:908–917.
Kring SI, Werge T, Holst C, et al Polymorphisms of serotonin receptor 2A and 2C genes and COMT in relation to obesity and type 2 diabetes. PLoS One2009; 4:e6696.
Ni W, Watts SW. 5-Hydroxytryptamine in the cardiovascular system: focus on the serotonin transporter (SERT). Clin Exp Pharmacol Physiol2006; 33:575–583.
Nefedov VP, Nefedova VV, Kononykhina VS, et al The effect of serotonin on the hematopoietic stem cells of the bone marrow [in Russian]. Biull Eksp Biol Med1991; 112:488–489.
Pakala R, Willerson JT, Benedict CR. Mitogenic effect of serotonin on vascular endothelial cells. Circulation1994; 90:1919–1926.
Yang M, Li K, Ng PC, et al Promoting effects of serotonin on hematopoiesis: ex vivo expansion of cord blood CD34+ stem/progenitor cells, proliferation of bone marrow stromal cells, and antiapoptosis. Stem Cells2007; 25:1800–1806.
Cloez-Tayarani I, Petit-Bertron AF, Venters HD, et al Differential effect of serotonin on cytokine production in lipopolysaccharide-stimulated human peripheral blood mononuclear cells: involvement of 5-hydroxytryptamine2A receptors. Int Immunol2003; 15:233–240.
Hoefgen B, Schulze TG, Ohlraun S, et al The power of sample size and homogenous sampling: association between the 5-HTTLPR serotonin transporter polymorphism and major depressive disorder. Biol Psychiatry2005; 57:247–251.
Nakatani D, Sato H, Sakata Y, et al Influence of serotonin transporter gene polymorphism on depressive symptoms and new cardiac events after acute myocardial infarction. Am Heart J2005; 150:652–658.
Brummett BH, Boyle SH, Siegler IC, et al Effects of environmental stress and gender on associations among symptoms of depression and the serotonin transporter gene linked polymorphic region (5-HTTLPR). Behav Genet2008; 38:34–43.
Fumeron F, Betoulle D, Nicaud V, et al Serotonin transporter gene polymorphism and myocardial infarction: Etude Cas-Témoins de l’Infarctus du Myocarde (ECTIM). Circulation2002; 105:2943–2945.
Phillips-Bute B, Mathew JP, Blumenthal JA, et al Relationship of genetic variability and depressive symptoms to adverse events after coronary artery bypass graft surgery. Psychosom Med2008; 70:953–959.
Greenberg BD, Tolliver TJ, Huang SJ, et al Genetic variation in the serotonin transporter promoter region affects serotonin uptake in human blood platelets. Am J Med Genet1999; 88:83–87.
Su S, Zhao J, Bremner JD, et al Serotonin transporter gene, depressive symptoms, and interleukin-6. Circ Cardiovasc Genet2009; 2:614–620.
Hassan M, York KM, Li H, et al Association of beta1-adrenergic receptor genetic polymorphism with mental stress-induced myocardial ischemia in patients with coronary artery disease. Arch Intern Med2008; 168:763–770.
Victor TW, Hu X, Campbell J, et al Association between migraine, anxiety and depression. Cephalalgia2009; Jul9[Epub ahead of print]
MacClellan LR, Howard TD, Cole JW, et al Relation of candidate genes that encode for endothelial function to migraine and stroke: the Stroke Prevention in Young Women study. Stroke2009; 40:e550–e557.
Tikka-Kleemola P, Kaunisto MA, Hamalainen E, et al Genetic association study of endothelin-1 and its receptors EDNRA and EDNRB in migraine with aura. Cephalalgia2009; 29:1224–1231.
Tomfohr LM, Martin TM, Miller GE. Symptoms of depression and impaired endothelial function in healthy adolescent women. J Behav Med2008; 31:137–143.
Katayama Y, Battista M, Kao WM, et al Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell2006; 124:407–421.
Fischer JC, Kudielka BM, von Kanel R, et al Bone-marrow derived progenitor cells are associated with psychosocial determinants of health after controlling for classical biological and behavioral cardiovascular risk factors. Brain Behav Immun2009; 23:419–426.
Baskin SM, Smitherman TA. Migraine and psychiatric disorders: comorbidities, mechanisms, and clinical applications. Neurol Sci2009; 30( suppl 1):S61–S65.
Lee S-T, Chu K, Jung KH, et al Decreased number and function of endothelial progenitor cells in patients with migraine. Neurology2008; 70:1510–1517.
Sobrino T, Hurtado O, Moro MA, et al The increase of circulating endothelial progenitor cells after acute ischemic stroke is associated with good outcome. Stroke2007; 38:2759–2764.
Jickling G, Salam A, Mohammad A, et al Circulating endothelial progenitor cells and age-related white matter changes. Stroke2009; 40:3191–3196.
Parakh K, Sakhuja A, Bhat U, et al Platelet function in patients with depression. South Med J2008; 101:612–617.
Aschbacher K, Mills PJ, von Känel R, et al Effects of depressive and anxious symptoms on norepinephrine and platelet P-selectin responses to acute psychological stress among elderly caregivers. Brain Behav Immun2008; 22:493–502.
Serebruany VL, Glassman AH, Malinin AI, et al Platelet/endothelial biomarkers in depressed patients treated with the selective serotonin reuptake inhibitor sertraline after acute coronary events: the Sertraline AntiDepressant Heart Attack Randomized Trial (SADHART) platelet substudy. Circulation2003; 108:939–944.
van Zyl LT, Lesperance F, Frasure-Smith N, et al Platelet and endothelial activity in comorbid major depression and coronary artery disease patients treated with citalopram: the Canadian Cardiac Randomized Evaluation of Antidepressant and Psychotherapy Efficacy Trial (CREATE) biomarker sub-study. J Thromb Thrombolysis2009; 27:48–56.
Nijm J, Kristenson M, Olsson AG, et al Impaired cortisol response to acute stressors in patients with coronary disease: implications for inflammatory activity. J Intern Med2007; 262:375–384.
Hamer M, O’Donnell K, Lahiri A, Steptoe A. Salivary cortisol responses to mental stress are associated with coronary artery calcification in healthy men and women. Eur Heart J2010; 31:424–429.
Ranjit N, Diez-Roux AV, Sanchez B, et al Association of salivary cortisol circadian pattern with cynical hostility: multi-ethnic study of atherosclerosis. Psychosom Med2009; 71:748–755.
Grant N, Hamer M, Steptoe A. Social isolation and stress-related cardiovascular, lipid, and cortisol responses. Ann Behav Med2009; 37:29–37.
Jaffe IZ, Mendelsohn ME. Angiotensin II and aldosterone regulate gene transcription via functional mineralocortocoid receptors in human coronary artery smooth muscle cells. Circ Res2005; 96:643–650.
Kubzansky LD, Adler GK. Aldosterone: a forgotten mediator of the relationship between psychological stress and heart disease. Neurosci Biobehav Rev2010; 34:80–86.
Milliez P, Girerd X, Plouin PF, et al Evidence for an increased rate of cardiovascular events in patients with primary aldosteronism. J Am Coll Cardiol2005; 45:1243–1248.
Duprez DA, Bauwens FR, De Buyzere ML, et al Influence of arterial blood pressure and aldosterone on left ventricular hypertrophy in moderate essential hypertension. Am J Cardiol1993; 71:17A–20A.
Dawson N, Ferrington L, Olverman HJ, et al Sex influences the effect of a lifelong increase in serotonin transporter function on cerebral metabolism. J Neurosci Res2009; 87:2375–2385.
Biomedical engineering is a rapidly growing field, as indicated by a recent threefold increase in the number of students enrolled in the more than 80 programs granting biomedical engineering degrees.1 The field is known primarily for contributing to the development of devices that aid diagnosis (such as chemical sensors and medical imagers) or help to restore lost function (such as pacemakers, cochlear implants, and artificial limbs). At the same time, biomedical engineering has also contributed to the understanding of physiology and is now a participant in the more recent molecular- and cellular-based discoveries and their potential clinical applications.
This article reviews the contributions of biomedical engineering to heart-brain medicine and looks ahead to where its future contributions may be expected. The focus is on the autonomic control of the heart, with special emphasis on stimulating or blocking the activity of peripheral nerves for therapeutic purposes.
AUTONOMIC MECHANISMS
One way biomedical engineering has contributed to heart-brain medicine is through systems physiology, attempting to characterize complex cardiac control mechanisms by mathematical models ranging from the relatively simple to the very complex.2 In particular, investigation of the effect of the baroreceptor reflex (baroreflex) on heart rate led to recording of efferent vagal activity, demonstrating that the respiratory variations in heart rate are attributable to complete stoppage of vagal efferent activity, at least in anesthetized dogs.3 The results suggested that respiratory sinus arrhythmia was a measure of parasympathetic cardiac control.4 Extensive further work investigated heart rate variability in humans, resulting in the generally accepted concept that rapid variations in heart rate are primarily due to the parasympathetic nervous system, while slower variations are primarily due to sympathetic contributions. 5 It has been amply demonstrated that in a variety disease states, a high degree of parasympathetic control is correlated with improved outcome.6,7
Heart rate variability: Much is still unknown
Because heart rate variability is derived through analysis of easily recorded single-lead electrocardiograms (ECGs), techniques are still being described for determining the degree of variability.8–10 Measurements may be based on heart rate or heart period (interbeat interval); they may be made during spontaneous, deep, or timed periodic breathing; and the recordings may last for a few minutes or for 24 hours. The results are rarely reported for different states, yet variability depends on whether the subjects are awake, in quiet sleep, or in REM sleep.11
The present incomplete understanding of the physiological basis and clinical significance of heart rate variations makes it difficult to judge what the optimum measurement is. For example, recent publications have still debated whether the respiratory variations are primarily of central origin or a result of the baroreflex.12,13 The reviewed evidence leaves little doubt that most respiratory variations are caused by vagal modulation induced by breathing, independently of the baroreflex. On the other hand, breathing affects blood pressure and thus must have some influence on heart rate through the baroreflex as well. These effects are likely to be important when considering low-frequency variations.14 It also has been suggested that the branch of the vagus that contributes to the slow variations may be different from the one responsible for the rapid changes.15
A role for mathematical modeling
Untangling the above relationships requires at least an approximate physiologically based quantitative model of the entire system. What is the “entire system”? It includes all components that significantly affect the clinical condition or physiological/biological phenomenon studied. Developing such models is challenging since they can be misleading if they are too simple. However, if they are too complex, they may obscure rather than illuminate. Even though mathematical modeling has been a major component of biomedical engineering for decades, there is still great need for physiological and clinical studies that are aided by biomedical engineers with mathematical skills and a discerning eye toward the life sciences.
For example, following numerous previous efforts to develop models of cardiorespiratory control systems, a model has been developed to describe changes in heart rate, stroke volume, and blood pressure caused by disordered breathing during sleep.16 The model was initially tested in animals but recently has been evaluated in children with obstructive sleep apnea.17 It was found to be more effective in identifying and characterizing cardiac control abnormalities than was spectrum-derived variability of heart rate or blood pressure alone.
To enhance the usefulness of heart rate variability as a clinical tool, it is necessary to go beyond observing that decreased high-frequency heart rate variations are ominous in a particular disease state because there is an “imbalance” of autonomic control. This gives the physician little guidance as to what to do. Should he or she treat the brain to get more parasympathetic outflow? Should attention be concentrated on making the heart more tolerant to the imbalance? Or should both approaches be tried?
In addition to the building of models, biomedical engineers can also contribute to heart-brain medicine by developing sensors that can measure appropriate physiological variables in animal experiments—and eventually in humans. Such variables include neural activity and chemical signals controlling the heart, as well as neural and chemical signals that arise from the heart and are sensed by the brain. These variables must be included in any model for a comprehensive characterization of heart-brain interactions.
NEURAL INTERVENTIONS
A second and complementary way in which bioengineering can contribute to heart-brain medicine is through the development and evaluation of technology that applies selective electrical, chemical, or mechanical stimulation to the physiological system. External interventions may have therapeutic effects even though the underlying physiological mechanisms are not fully understood. For example, deep brain electrical stimulation is increasingly used or explored to treat epilepsy, Parkinson disease, and depression.18,19 The development of technology to deliver drugs locally is advancing rapidly and is almost certain to play a major role in exploring heart-brain interactions. The remainder of this paper concentrates on interventions applied through peripheral nerves.
Figure 1. The vagus nerve innervates numerous organs in addition to the heart.A major recent development is the demonstration that vagus nerve stimulation may have beneficial effects. The vagus nerve is large, easily accessible, and well studied. Its afferents carry sensory information from the periphery to the brain, while its efferents provide central control of numerous organs, including the heart (Figure 1). Because of its accessibility, it can be easily stimulated.
Vagal stimulation is being used to treat epilepsy, but now it is also being explored for the treatment of drug-resistant depression and chronic migraine.20,21 The intensity, frequency, pulse width, and train duration of the apparently single-channel stimulation are set telemetrically. Preliminary data indicate a long-term success rate of about 20% for depression20 and improvement in 2 of 4 patients with chronic migraine.21 Side effects include discomfort caused by the electrical stimulation and vocalization impairments.
Figure 2. Schematic structure of a peripheral nerve.The modesty of the success in these trials was likely due in large part to the method of stimulation. Current preferentially stimulates large nerve fibers as well as those closest to the electrode. Yet the vagus contains a variety of afferent and efferent nerves that have varying fiber sizes at different distances from the stimulating electrode. The complex structure of peripheral nerves such as the vagus is schematically illustrated in Figure 2. Simple electrodes cannot control the location and function of the nerves that are actually stimulated.
The selective stimulation of nerves has received much attention in rehabilitation engineering since electrical activation of peripheral motor fibers can restore at least some function in patents with spinal cord injury. The stimulation must be selective: different muscles to restore movement need to contract in the appropriate sequence and with appropriate intensity. Thus, fascicles of motor nerves innervating these muscles must be stimulated accordingly.
Electrodes have been developed in a variety of configurations for selectively activating the desired fibers in a nerve bundle.22 Multiple electrodes around the nerve allow targeting of the desired fascicles and preferential stimulation of small nerve fibers.23,24 Gently compressing the nerve using a flat electrode sleeve enhances selectivity by increasing the surface area of the stimulated nerve.25 A tripolar sleeve electrode along the nerve (one cathode at the middle and an anode on each side) may be used to preferentially stimulate small fibers by “anodal blockade” of the propagation of activity in large fibers.26 Mathematical models of nerve excitation, combined with models of the tissue surrounding the nerve fibers, show that positioning the anodes at different distances from the cathode can generate unidirectional propagation.27 Mathematical analysis also shows that the stimulating pulse should have a slowly decaying trailing edge to assure effectiveness of the blockade.27,28
Similar technologies are likely necessary for stimulating the vagus for specific purposes. If the goal is to induce central effects, appropriate afferents should be stimulated. If the goal is to influence the heart directly, cardiac vagal efferents need to be stimulated without confounding the effect by also stimulating afferents. Since cardiac efferent fibers are small and constitute only a small portion of vagal trunk,3 their stimulation requires special care to reverse the normal “largest first” recruitment of stimulated fibers.
The recently reported first pilot study of vagal stimulation in heart failure patients used an electrode that seems to partially satisfy such requirements.29,30 Although the details appear to be proprietary, the implantable electrical stimulator has multiple electrodes and induces anodal blockade to preferentially stimulate efferent rather than afferent nerve fibers.
In the pilot study, 8 patients received intermittent vagal stimulation (2 to 10 sec “on” and 6 to 30 sec “off”) of the right cervical vagus using a pulse delivered 70 msec after each R wave of the ECG. The stimulating current, limited by a threshold or the onset of side effects, was adjusted to achieve a heart rate reduction of 5 to 10 beats/min. Patients were evaluated up to 6 months; no permanent side effects were reported. There was a modest improvement of cardiac function as judged by a reduction in left ventricular volumes, as well as a clear improvement in a measure of quality of life. The study shows feasibility and suggests further investigations.
The primary task seems to be optimization of stimulation parameters. Since natural vagal impulses are distributed through the cardiac cycle,3 artificial stimulation might be more effective if it emulated the natural firing pattern. Since long-term heart rate reduction was minimal, parameters might be tuned further to stimulate small cardiac efferent fibers that may be far from the electrodes. Measurements of heart rate variability during controlled conditions may reveal the pacemaker’s responsiveness to vagal stimulation. Comparison of duty cycles (duration of “on” and “off”) may show whether the study’s choice, to some extent already mimicking the breathing-induced modulation of natural vagal activity, is most effective. Such studies to optimize effectiveness may be best performed in chronically instrumented animals.
Baroreceptor stimulation
An extensively studied mechanism that modulates the autonomic nervous system is the baroreceptor reflex that is known to be depressed in heart failure. Former efforts to use this reflex therapeutically were recently revived in both animal experiments and human studies.31 For example, bilateral carotid sinus stimulation nearly doubled the survival time of dogs with pacing-induced heart failure.32 Although measures of left ventricular function, obtained while the stimulator was turned off, were similar in dogs with stimulated and unstimulated carotid sinuses, plasma norepinephrine was lower in the animals receiving stimulation. This suggests that carotid sinus stimulation led to a general decrease in sympathetic activation. It is noteworthy that the stimulation was applied intermittently (9 min “on”, 1 min “off”) to avoid the resetting (or adaptation) of baroreceptors, a phenomenon that had led to the now-doubted traditional belief that the baroreflex regulates only acute rather than long-term changes of blood pressure.2
Chronic bilateral baroreceptor stimulation was also applied in 21 patients with essential hypertension that could not be controlled by medication.33 Measurements were taken 1 month after implantation with the stimulator turned off and 3 months after chronic stimulation with the stimulator on. Stimulation moderately reduced both blood pressure and heart rate; heart rate variability suggested an increase in parasympathetic activity and a decrease in sympathetic activity.
Renal sympathetic ablation
Technology-based approaches encompass not only the stimulation of nerves but also the abolition of nerve activity. In an exploratory study, bilateral sympathetic denervation was performed in 45 patients with drugresistant hypertension.34 A catheter, introduced through the femoral artery, was positioned at the entrance of each renal artery. Sympathetic denervation was produced by radiofrequeny energy applied for a maximum of 2 minutes. The details of the technology appear proprietary, complete ablation could not be ascertained, and both efferents and afferents are likely to have been stimulated. Nevertheless, the procedure appeared safe and resulted in a significant reduction in both diastolic and systolic pressures over 12 months. In addition to lowering blood pressure, catheter-based sympathetic denervation might also prove therapeutic in heart failure and chronic kidney disease.
AUTOMATED CONTROL OF PHYSIOLOGIC VARIABLES
These initial experiences with baroreceptor stimulation, together with the results obtained by stimulating the vagus and ablating renal sympathetic nerves, indicate that device-based approaches may be useful additions to the treatment of drug-resistant hypertension and heart failure. The incorporation of control features that automatically respond to the changes in physiological states are natural extensions.35 As a recent example, in anesthetized dogs with heart failure and paced heart, systemic arterial pressure, cardiac output, and left atrial pressure were automatically regulated at set levels by a model-based infusion of nitroprusside, dobutamine, and volume expanders.36 When the heart rate was reduced, cardiac energetics (based on a reduction in left ventricular oxygen consumption) improved while the hemodynamic variables remained constant.
CONCLUSIONS
Biomedical engineering played a major role in linking measures of heart rate variation to sympathetic and parasympathetic contributions to cardiac control, as well as in demonstrating that the balance of control was correlated with a variety of disease states of heart and brain. However, much work remains to be done to fully realize the field’s potential for aiding clinicians in preventing or treating diseases. Further studies, some to be performed in chronically prepared animals, are needed to quantitatively characterize the many interacting mechanisms that determine cardiac function. Such studies would benefit from recording naturally occurring neural activity to and from the brain, and are already starting to benefit from the artificial electrical stimulation of nerves in both experimental animals and preliminary trials in patients.
The appropriate use of mathematical modeling is an essential tool for gaining in-depth understanding of physiological function. Mathematical modeling is also essential for developing stimulators that selectively stimulate those nerves that control the function to be influenced. The determination of stimulation parameters, based on understanding rather than on trial and error, is highly desirable. The use of “intelligent” stimulators, automatically controlled by appropriate physiological measurements, is an ambitious but achievable goal for improving human health.
References
Katona PG. Biomedical engineering and The Whitaker Foundation: a thirty-year partnership. Ann Biomed Eng2006; 34:904–916.
Guyton AC, Coleman TG, Cowley AW, et al Systems analysis of arterial pressure regulation and hypertension. Ann Biomed Eng1972; 1:254–281.
Katona PG, Poitras JW, Barnett GO, Terry BS. Cardiac vagal efferent activity and heart period in the carotid sinus reflex. Am J Physiol1979; 218:1030–1037.
Katona PG, Jih F. Respiratory sinus arrhythmia: noninvasive measure of parasympathetic cardiac control. J Appl Physiol1975; 39:801–805.
Akselrod S, Gordon D, Ubel FA, et al Power spectrum analysis of heart rate fluctuation: a quantitative probe of beat-to-beat cardiovascular control. Science1981; 213:220–222.
Carney RM, Freedland KE. Depression and heart rate variability in patients with coronary heart disease. Cleve Clin J Med2009; 76( suppl 2):S13–S17.
Lauer MS. Autonomic function and prognosis. Cleve Clin J Med2009; 76( suppl 2):S18–S22.
Cnockaert L, Migeotte P-F, Daubigny L, et al A method for the analysis of respiratory sinus arrhythmia using continuous wavelet transforms. IEEE Trans Biomed Eng2008; 55:1640–1642.
Castiglioni P, Parati G, Civijian A, et al Local scale exponents of blood pressure and heart rate variability by detrended fluctuation analysis: effects of posture, exercise, and aging. IEEE Trans Biomed Eng2009; 56:675–684.
Chen Z, Brown EN, Barbieri R. Assessment of autonomic control and respiratory sinus arrhythmia using point process models of human heart beat dynamics. IEEE Trans Biomed Eng2009; 56:1791–1802.
Schmitt DT, Stein PK, Ivanov PC. Stratification pattern of static and scale-invariant dynamic measures of heartbeat fluctuations across sleep stages in young and elderly. IEEE Trans Biomed Eng2009; 56:1564–1573.
Eckberg DL. Point:counterpoint: respiratory sinus arrhythmia is due to a central mechanism vs. respiratory sinus arrhythmia is due to the baroreflex mechanism. J Appl Physiol2009; 16:1740–1742.
Karemaker JM. Counterpoint: respiratory sinus arrhythmia is due to the baroreflex mechanism. J Appl Physiol2009; 106:1742–1743.
Moak JP, Goldstein DS, Eldadah BA, et al Supine low-frequency power of heart rate variability reflects baroreflex function, not cardiac sympathetic innervation. Heart Rhythm2007; 4:1523–1529.
Porges SW. The polyvagal theory: new insights into adaptive reactions of the autonomic nervous system. Cleve Clin J Med2009; 76( suppl 2):S86–S90.
Khoo MC. Modeling of autonomic control in sleep-disordered breathing. Cardiovasc Eng2008; 8:30–41.
Chaicharn J, Kin Z, Chen ML, et al Model-based assessment of cardiovascular autonomic control in children with obstructive sleep apnea. Sleep2009; 32:927–938.
Vitek JL. Deep brain stimulation: how does it work?Cleve Clin J Med2008; 75( suppl 2):S59–S65.
Lee KH, Blaha CD, Garrris PA. Evolution of deep brain stimulation: human electrometer and smart devices supporting the next generation of therapy. Neuromodulation: Technol Neural Interface2009; 12:85–103.
Fitzgerald PB, Daskalaris ZJ. The use of repetitive transcranial magnetic stimulation and vagal nerve stimulation in the treatment of depression. Curr Opin Psych2008; 21:25–29.
Cecchini AP, Mea E, Tullo V, et al Vagus nerve stimulation in drug-resistant daily chronic migraine with depression: preliminary data. Neurol Sci2009; 30( suppl 1):S101–S104.
Grill WM, Norman SE, Bellamkonda RV. Implanted neural interfaces: biochallenges and engineered solutions. Annu Rev Biomed Eng2009; 11:1–24.
Tarler MD, Mortimer JT. Selective and independent activation of four motor fascicles using a four-contact nerve-cuff electrode. IEEE Trans Neural Syst Rehab Eng2004; 12:251–257.
Lertmanorat Z, Gustafson KJ, Durand DM. Electrode array for reversing the recruitment order of peripheral nerve stimulation: experimental studies. Ann Biomed Eng2006; 34:152–160.
Tyler DJ, Durand DM. Functionally selective peripheral nerve stimulation with a flat interface nerve electrode. IEEE Trans Neural Syst Rehab Eng2002; 10:294–303.
Fang ZP, Mortimer JT. Selective activation of small motor axons by quasitrapezoidal current pulses. IEEE Trans Biomed Eng1991; 38:168–174.
Rijkoff NJM, Holsheimer J, Koledwijn EL, et al Selective stimulation of sacral nerve roots for bladder control: a study in computer modeling. IEEE Trans Biomed Eng1994; 41:413–424.
Tosato M, Yoshida K, Toft E, et al Quazi-trapezoidal pulses to selectively block the activation of intrinsic laryngeal muscles during vagal nerve stimulation. J Neural Eng2007; 41:205–212.
Schwartz PK, De Ferrari GM, Sanzo A, et al Long term vagal stimulation in patients with advanced heart failure: first experience in man. Eur J Heart Failure2008; 10:884–891.
De Ferrari GM, Sanzo A, Borggrefe M, et al Chronic vagus nerve stimulation in patients with chronic heart failure is feasible and appears beneficial. Circulation2008; 118( suppl):S721. Abstract 2412.
Durand MT, Fazan R, Salgado MCO, et al Acute and chronic electrical activation of baroreceptor afferents in awake and anesthetized subjects. Braz J Med Biol Res2009; 42:53–60.
Zucker IH, Hackley JF, Cornish KG, et al Chronic baroreceptor activation enhances survival in dogs with pacing-induced heart failure. Hypertension2007; 50:904–910.
Wustman K, Kucera JP, Scheffers I, et al Effects of chronic baroreceptor stimulation on the autonomic cardiovascular regulation in patients with drug-resistant arterial hypertension. Hypertension2009; 54:530–536.
Krum H, Schlaih M, Whitbourn R, et al Catheter-based renal denervation for resistant hypertension: a multicentre safety and proof-of-principle study. Lancet2009; 373:1228–1239.
Kawada T, Sugamichi M. Artifical neural interfaces for bionic cardio vascular treatments. J Artific Organs2009; 12:17–22.
Uemura K, Sunagawa K, Sugimachi M. Computationally managed bradycardia improved cardiac energetics while restoring normal hemodynamics in heart failure. Ann Biomed Eng2009; 37:82–93.
Peter G. Katona, ScD Department of Electrical and Computer Engineering, George Mason University, Fairfax, VA
Correspondence: Peter G. Katona, ScD, Department of Electrical and Computer Engineering, Nguyen Engineering Building, George Mason University, 4400 University Drive MS 1G5, Fairfax, VA 22030; [email protected]
Dr. Katona reported that he has no financial relationships that pose a potential conflict of interest with this article.
Peter G. Katona, ScD Department of Electrical and Computer Engineering, George Mason University, Fairfax, VA
Correspondence: Peter G. Katona, ScD, Department of Electrical and Computer Engineering, Nguyen Engineering Building, George Mason University, 4400 University Drive MS 1G5, Fairfax, VA 22030; [email protected]
Dr. Katona reported that he has no financial relationships that pose a potential conflict of interest with this article.
Author and Disclosure Information
Peter G. Katona, ScD Department of Electrical and Computer Engineering, George Mason University, Fairfax, VA
Correspondence: Peter G. Katona, ScD, Department of Electrical and Computer Engineering, Nguyen Engineering Building, George Mason University, 4400 University Drive MS 1G5, Fairfax, VA 22030; [email protected]
Dr. Katona reported that he has no financial relationships that pose a potential conflict of interest with this article.
Biomedical engineering is a rapidly growing field, as indicated by a recent threefold increase in the number of students enrolled in the more than 80 programs granting biomedical engineering degrees.1 The field is known primarily for contributing to the development of devices that aid diagnosis (such as chemical sensors and medical imagers) or help to restore lost function (such as pacemakers, cochlear implants, and artificial limbs). At the same time, biomedical engineering has also contributed to the understanding of physiology and is now a participant in the more recent molecular- and cellular-based discoveries and their potential clinical applications.
This article reviews the contributions of biomedical engineering to heart-brain medicine and looks ahead to where its future contributions may be expected. The focus is on the autonomic control of the heart, with special emphasis on stimulating or blocking the activity of peripheral nerves for therapeutic purposes.
AUTONOMIC MECHANISMS
One way biomedical engineering has contributed to heart-brain medicine is through systems physiology, attempting to characterize complex cardiac control mechanisms by mathematical models ranging from the relatively simple to the very complex.2 In particular, investigation of the effect of the baroreceptor reflex (baroreflex) on heart rate led to recording of efferent vagal activity, demonstrating that the respiratory variations in heart rate are attributable to complete stoppage of vagal efferent activity, at least in anesthetized dogs.3 The results suggested that respiratory sinus arrhythmia was a measure of parasympathetic cardiac control.4 Extensive further work investigated heart rate variability in humans, resulting in the generally accepted concept that rapid variations in heart rate are primarily due to the parasympathetic nervous system, while slower variations are primarily due to sympathetic contributions. 5 It has been amply demonstrated that in a variety disease states, a high degree of parasympathetic control is correlated with improved outcome.6,7
Heart rate variability: Much is still unknown
Because heart rate variability is derived through analysis of easily recorded single-lead electrocardiograms (ECGs), techniques are still being described for determining the degree of variability.8–10 Measurements may be based on heart rate or heart period (interbeat interval); they may be made during spontaneous, deep, or timed periodic breathing; and the recordings may last for a few minutes or for 24 hours. The results are rarely reported for different states, yet variability depends on whether the subjects are awake, in quiet sleep, or in REM sleep.11
The present incomplete understanding of the physiological basis and clinical significance of heart rate variations makes it difficult to judge what the optimum measurement is. For example, recent publications have still debated whether the respiratory variations are primarily of central origin or a result of the baroreflex.12,13 The reviewed evidence leaves little doubt that most respiratory variations are caused by vagal modulation induced by breathing, independently of the baroreflex. On the other hand, breathing affects blood pressure and thus must have some influence on heart rate through the baroreflex as well. These effects are likely to be important when considering low-frequency variations.14 It also has been suggested that the branch of the vagus that contributes to the slow variations may be different from the one responsible for the rapid changes.15
A role for mathematical modeling
Untangling the above relationships requires at least an approximate physiologically based quantitative model of the entire system. What is the “entire system”? It includes all components that significantly affect the clinical condition or physiological/biological phenomenon studied. Developing such models is challenging since they can be misleading if they are too simple. However, if they are too complex, they may obscure rather than illuminate. Even though mathematical modeling has been a major component of biomedical engineering for decades, there is still great need for physiological and clinical studies that are aided by biomedical engineers with mathematical skills and a discerning eye toward the life sciences.
For example, following numerous previous efforts to develop models of cardiorespiratory control systems, a model has been developed to describe changes in heart rate, stroke volume, and blood pressure caused by disordered breathing during sleep.16 The model was initially tested in animals but recently has been evaluated in children with obstructive sleep apnea.17 It was found to be more effective in identifying and characterizing cardiac control abnormalities than was spectrum-derived variability of heart rate or blood pressure alone.
To enhance the usefulness of heart rate variability as a clinical tool, it is necessary to go beyond observing that decreased high-frequency heart rate variations are ominous in a particular disease state because there is an “imbalance” of autonomic control. This gives the physician little guidance as to what to do. Should he or she treat the brain to get more parasympathetic outflow? Should attention be concentrated on making the heart more tolerant to the imbalance? Or should both approaches be tried?
In addition to the building of models, biomedical engineers can also contribute to heart-brain medicine by developing sensors that can measure appropriate physiological variables in animal experiments—and eventually in humans. Such variables include neural activity and chemical signals controlling the heart, as well as neural and chemical signals that arise from the heart and are sensed by the brain. These variables must be included in any model for a comprehensive characterization of heart-brain interactions.
NEURAL INTERVENTIONS
A second and complementary way in which bioengineering can contribute to heart-brain medicine is through the development and evaluation of technology that applies selective electrical, chemical, or mechanical stimulation to the physiological system. External interventions may have therapeutic effects even though the underlying physiological mechanisms are not fully understood. For example, deep brain electrical stimulation is increasingly used or explored to treat epilepsy, Parkinson disease, and depression.18,19 The development of technology to deliver drugs locally is advancing rapidly and is almost certain to play a major role in exploring heart-brain interactions. The remainder of this paper concentrates on interventions applied through peripheral nerves.
Figure 1. The vagus nerve innervates numerous organs in addition to the heart.A major recent development is the demonstration that vagus nerve stimulation may have beneficial effects. The vagus nerve is large, easily accessible, and well studied. Its afferents carry sensory information from the periphery to the brain, while its efferents provide central control of numerous organs, including the heart (Figure 1). Because of its accessibility, it can be easily stimulated.
Vagal stimulation is being used to treat epilepsy, but now it is also being explored for the treatment of drug-resistant depression and chronic migraine.20,21 The intensity, frequency, pulse width, and train duration of the apparently single-channel stimulation are set telemetrically. Preliminary data indicate a long-term success rate of about 20% for depression20 and improvement in 2 of 4 patients with chronic migraine.21 Side effects include discomfort caused by the electrical stimulation and vocalization impairments.
Figure 2. Schematic structure of a peripheral nerve.The modesty of the success in these trials was likely due in large part to the method of stimulation. Current preferentially stimulates large nerve fibers as well as those closest to the electrode. Yet the vagus contains a variety of afferent and efferent nerves that have varying fiber sizes at different distances from the stimulating electrode. The complex structure of peripheral nerves such as the vagus is schematically illustrated in Figure 2. Simple electrodes cannot control the location and function of the nerves that are actually stimulated.
The selective stimulation of nerves has received much attention in rehabilitation engineering since electrical activation of peripheral motor fibers can restore at least some function in patents with spinal cord injury. The stimulation must be selective: different muscles to restore movement need to contract in the appropriate sequence and with appropriate intensity. Thus, fascicles of motor nerves innervating these muscles must be stimulated accordingly.
Electrodes have been developed in a variety of configurations for selectively activating the desired fibers in a nerve bundle.22 Multiple electrodes around the nerve allow targeting of the desired fascicles and preferential stimulation of small nerve fibers.23,24 Gently compressing the nerve using a flat electrode sleeve enhances selectivity by increasing the surface area of the stimulated nerve.25 A tripolar sleeve electrode along the nerve (one cathode at the middle and an anode on each side) may be used to preferentially stimulate small fibers by “anodal blockade” of the propagation of activity in large fibers.26 Mathematical models of nerve excitation, combined with models of the tissue surrounding the nerve fibers, show that positioning the anodes at different distances from the cathode can generate unidirectional propagation.27 Mathematical analysis also shows that the stimulating pulse should have a slowly decaying trailing edge to assure effectiveness of the blockade.27,28
Similar technologies are likely necessary for stimulating the vagus for specific purposes. If the goal is to induce central effects, appropriate afferents should be stimulated. If the goal is to influence the heart directly, cardiac vagal efferents need to be stimulated without confounding the effect by also stimulating afferents. Since cardiac efferent fibers are small and constitute only a small portion of vagal trunk,3 their stimulation requires special care to reverse the normal “largest first” recruitment of stimulated fibers.
The recently reported first pilot study of vagal stimulation in heart failure patients used an electrode that seems to partially satisfy such requirements.29,30 Although the details appear to be proprietary, the implantable electrical stimulator has multiple electrodes and induces anodal blockade to preferentially stimulate efferent rather than afferent nerve fibers.
In the pilot study, 8 patients received intermittent vagal stimulation (2 to 10 sec “on” and 6 to 30 sec “off”) of the right cervical vagus using a pulse delivered 70 msec after each R wave of the ECG. The stimulating current, limited by a threshold or the onset of side effects, was adjusted to achieve a heart rate reduction of 5 to 10 beats/min. Patients were evaluated up to 6 months; no permanent side effects were reported. There was a modest improvement of cardiac function as judged by a reduction in left ventricular volumes, as well as a clear improvement in a measure of quality of life. The study shows feasibility and suggests further investigations.
The primary task seems to be optimization of stimulation parameters. Since natural vagal impulses are distributed through the cardiac cycle,3 artificial stimulation might be more effective if it emulated the natural firing pattern. Since long-term heart rate reduction was minimal, parameters might be tuned further to stimulate small cardiac efferent fibers that may be far from the electrodes. Measurements of heart rate variability during controlled conditions may reveal the pacemaker’s responsiveness to vagal stimulation. Comparison of duty cycles (duration of “on” and “off”) may show whether the study’s choice, to some extent already mimicking the breathing-induced modulation of natural vagal activity, is most effective. Such studies to optimize effectiveness may be best performed in chronically instrumented animals.
Baroreceptor stimulation
An extensively studied mechanism that modulates the autonomic nervous system is the baroreceptor reflex that is known to be depressed in heart failure. Former efforts to use this reflex therapeutically were recently revived in both animal experiments and human studies.31 For example, bilateral carotid sinus stimulation nearly doubled the survival time of dogs with pacing-induced heart failure.32 Although measures of left ventricular function, obtained while the stimulator was turned off, were similar in dogs with stimulated and unstimulated carotid sinuses, plasma norepinephrine was lower in the animals receiving stimulation. This suggests that carotid sinus stimulation led to a general decrease in sympathetic activation. It is noteworthy that the stimulation was applied intermittently (9 min “on”, 1 min “off”) to avoid the resetting (or adaptation) of baroreceptors, a phenomenon that had led to the now-doubted traditional belief that the baroreflex regulates only acute rather than long-term changes of blood pressure.2
Chronic bilateral baroreceptor stimulation was also applied in 21 patients with essential hypertension that could not be controlled by medication.33 Measurements were taken 1 month after implantation with the stimulator turned off and 3 months after chronic stimulation with the stimulator on. Stimulation moderately reduced both blood pressure and heart rate; heart rate variability suggested an increase in parasympathetic activity and a decrease in sympathetic activity.
Renal sympathetic ablation
Technology-based approaches encompass not only the stimulation of nerves but also the abolition of nerve activity. In an exploratory study, bilateral sympathetic denervation was performed in 45 patients with drugresistant hypertension.34 A catheter, introduced through the femoral artery, was positioned at the entrance of each renal artery. Sympathetic denervation was produced by radiofrequeny energy applied for a maximum of 2 minutes. The details of the technology appear proprietary, complete ablation could not be ascertained, and both efferents and afferents are likely to have been stimulated. Nevertheless, the procedure appeared safe and resulted in a significant reduction in both diastolic and systolic pressures over 12 months. In addition to lowering blood pressure, catheter-based sympathetic denervation might also prove therapeutic in heart failure and chronic kidney disease.
AUTOMATED CONTROL OF PHYSIOLOGIC VARIABLES
These initial experiences with baroreceptor stimulation, together with the results obtained by stimulating the vagus and ablating renal sympathetic nerves, indicate that device-based approaches may be useful additions to the treatment of drug-resistant hypertension and heart failure. The incorporation of control features that automatically respond to the changes in physiological states are natural extensions.35 As a recent example, in anesthetized dogs with heart failure and paced heart, systemic arterial pressure, cardiac output, and left atrial pressure were automatically regulated at set levels by a model-based infusion of nitroprusside, dobutamine, and volume expanders.36 When the heart rate was reduced, cardiac energetics (based on a reduction in left ventricular oxygen consumption) improved while the hemodynamic variables remained constant.
CONCLUSIONS
Biomedical engineering played a major role in linking measures of heart rate variation to sympathetic and parasympathetic contributions to cardiac control, as well as in demonstrating that the balance of control was correlated with a variety of disease states of heart and brain. However, much work remains to be done to fully realize the field’s potential for aiding clinicians in preventing or treating diseases. Further studies, some to be performed in chronically prepared animals, are needed to quantitatively characterize the many interacting mechanisms that determine cardiac function. Such studies would benefit from recording naturally occurring neural activity to and from the brain, and are already starting to benefit from the artificial electrical stimulation of nerves in both experimental animals and preliminary trials in patients.
The appropriate use of mathematical modeling is an essential tool for gaining in-depth understanding of physiological function. Mathematical modeling is also essential for developing stimulators that selectively stimulate those nerves that control the function to be influenced. The determination of stimulation parameters, based on understanding rather than on trial and error, is highly desirable. The use of “intelligent” stimulators, automatically controlled by appropriate physiological measurements, is an ambitious but achievable goal for improving human health.
Biomedical engineering is a rapidly growing field, as indicated by a recent threefold increase in the number of students enrolled in the more than 80 programs granting biomedical engineering degrees.1 The field is known primarily for contributing to the development of devices that aid diagnosis (such as chemical sensors and medical imagers) or help to restore lost function (such as pacemakers, cochlear implants, and artificial limbs). At the same time, biomedical engineering has also contributed to the understanding of physiology and is now a participant in the more recent molecular- and cellular-based discoveries and their potential clinical applications.
This article reviews the contributions of biomedical engineering to heart-brain medicine and looks ahead to where its future contributions may be expected. The focus is on the autonomic control of the heart, with special emphasis on stimulating or blocking the activity of peripheral nerves for therapeutic purposes.
AUTONOMIC MECHANISMS
One way biomedical engineering has contributed to heart-brain medicine is through systems physiology, attempting to characterize complex cardiac control mechanisms by mathematical models ranging from the relatively simple to the very complex.2 In particular, investigation of the effect of the baroreceptor reflex (baroreflex) on heart rate led to recording of efferent vagal activity, demonstrating that the respiratory variations in heart rate are attributable to complete stoppage of vagal efferent activity, at least in anesthetized dogs.3 The results suggested that respiratory sinus arrhythmia was a measure of parasympathetic cardiac control.4 Extensive further work investigated heart rate variability in humans, resulting in the generally accepted concept that rapid variations in heart rate are primarily due to the parasympathetic nervous system, while slower variations are primarily due to sympathetic contributions. 5 It has been amply demonstrated that in a variety disease states, a high degree of parasympathetic control is correlated with improved outcome.6,7
Heart rate variability: Much is still unknown
Because heart rate variability is derived through analysis of easily recorded single-lead electrocardiograms (ECGs), techniques are still being described for determining the degree of variability.8–10 Measurements may be based on heart rate or heart period (interbeat interval); they may be made during spontaneous, deep, or timed periodic breathing; and the recordings may last for a few minutes or for 24 hours. The results are rarely reported for different states, yet variability depends on whether the subjects are awake, in quiet sleep, or in REM sleep.11
The present incomplete understanding of the physiological basis and clinical significance of heart rate variations makes it difficult to judge what the optimum measurement is. For example, recent publications have still debated whether the respiratory variations are primarily of central origin or a result of the baroreflex.12,13 The reviewed evidence leaves little doubt that most respiratory variations are caused by vagal modulation induced by breathing, independently of the baroreflex. On the other hand, breathing affects blood pressure and thus must have some influence on heart rate through the baroreflex as well. These effects are likely to be important when considering low-frequency variations.14 It also has been suggested that the branch of the vagus that contributes to the slow variations may be different from the one responsible for the rapid changes.15
A role for mathematical modeling
Untangling the above relationships requires at least an approximate physiologically based quantitative model of the entire system. What is the “entire system”? It includes all components that significantly affect the clinical condition or physiological/biological phenomenon studied. Developing such models is challenging since they can be misleading if they are too simple. However, if they are too complex, they may obscure rather than illuminate. Even though mathematical modeling has been a major component of biomedical engineering for decades, there is still great need for physiological and clinical studies that are aided by biomedical engineers with mathematical skills and a discerning eye toward the life sciences.
For example, following numerous previous efforts to develop models of cardiorespiratory control systems, a model has been developed to describe changes in heart rate, stroke volume, and blood pressure caused by disordered breathing during sleep.16 The model was initially tested in animals but recently has been evaluated in children with obstructive sleep apnea.17 It was found to be more effective in identifying and characterizing cardiac control abnormalities than was spectrum-derived variability of heart rate or blood pressure alone.
To enhance the usefulness of heart rate variability as a clinical tool, it is necessary to go beyond observing that decreased high-frequency heart rate variations are ominous in a particular disease state because there is an “imbalance” of autonomic control. This gives the physician little guidance as to what to do. Should he or she treat the brain to get more parasympathetic outflow? Should attention be concentrated on making the heart more tolerant to the imbalance? Or should both approaches be tried?
In addition to the building of models, biomedical engineers can also contribute to heart-brain medicine by developing sensors that can measure appropriate physiological variables in animal experiments—and eventually in humans. Such variables include neural activity and chemical signals controlling the heart, as well as neural and chemical signals that arise from the heart and are sensed by the brain. These variables must be included in any model for a comprehensive characterization of heart-brain interactions.
NEURAL INTERVENTIONS
A second and complementary way in which bioengineering can contribute to heart-brain medicine is through the development and evaluation of technology that applies selective electrical, chemical, or mechanical stimulation to the physiological system. External interventions may have therapeutic effects even though the underlying physiological mechanisms are not fully understood. For example, deep brain electrical stimulation is increasingly used or explored to treat epilepsy, Parkinson disease, and depression.18,19 The development of technology to deliver drugs locally is advancing rapidly and is almost certain to play a major role in exploring heart-brain interactions. The remainder of this paper concentrates on interventions applied through peripheral nerves.
Figure 1. The vagus nerve innervates numerous organs in addition to the heart.A major recent development is the demonstration that vagus nerve stimulation may have beneficial effects. The vagus nerve is large, easily accessible, and well studied. Its afferents carry sensory information from the periphery to the brain, while its efferents provide central control of numerous organs, including the heart (Figure 1). Because of its accessibility, it can be easily stimulated.
Vagal stimulation is being used to treat epilepsy, but now it is also being explored for the treatment of drug-resistant depression and chronic migraine.20,21 The intensity, frequency, pulse width, and train duration of the apparently single-channel stimulation are set telemetrically. Preliminary data indicate a long-term success rate of about 20% for depression20 and improvement in 2 of 4 patients with chronic migraine.21 Side effects include discomfort caused by the electrical stimulation and vocalization impairments.
Figure 2. Schematic structure of a peripheral nerve.The modesty of the success in these trials was likely due in large part to the method of stimulation. Current preferentially stimulates large nerve fibers as well as those closest to the electrode. Yet the vagus contains a variety of afferent and efferent nerves that have varying fiber sizes at different distances from the stimulating electrode. The complex structure of peripheral nerves such as the vagus is schematically illustrated in Figure 2. Simple electrodes cannot control the location and function of the nerves that are actually stimulated.
The selective stimulation of nerves has received much attention in rehabilitation engineering since electrical activation of peripheral motor fibers can restore at least some function in patents with spinal cord injury. The stimulation must be selective: different muscles to restore movement need to contract in the appropriate sequence and with appropriate intensity. Thus, fascicles of motor nerves innervating these muscles must be stimulated accordingly.
Electrodes have been developed in a variety of configurations for selectively activating the desired fibers in a nerve bundle.22 Multiple electrodes around the nerve allow targeting of the desired fascicles and preferential stimulation of small nerve fibers.23,24 Gently compressing the nerve using a flat electrode sleeve enhances selectivity by increasing the surface area of the stimulated nerve.25 A tripolar sleeve electrode along the nerve (one cathode at the middle and an anode on each side) may be used to preferentially stimulate small fibers by “anodal blockade” of the propagation of activity in large fibers.26 Mathematical models of nerve excitation, combined with models of the tissue surrounding the nerve fibers, show that positioning the anodes at different distances from the cathode can generate unidirectional propagation.27 Mathematical analysis also shows that the stimulating pulse should have a slowly decaying trailing edge to assure effectiveness of the blockade.27,28
Similar technologies are likely necessary for stimulating the vagus for specific purposes. If the goal is to induce central effects, appropriate afferents should be stimulated. If the goal is to influence the heart directly, cardiac vagal efferents need to be stimulated without confounding the effect by also stimulating afferents. Since cardiac efferent fibers are small and constitute only a small portion of vagal trunk,3 their stimulation requires special care to reverse the normal “largest first” recruitment of stimulated fibers.
The recently reported first pilot study of vagal stimulation in heart failure patients used an electrode that seems to partially satisfy such requirements.29,30 Although the details appear to be proprietary, the implantable electrical stimulator has multiple electrodes and induces anodal blockade to preferentially stimulate efferent rather than afferent nerve fibers.
In the pilot study, 8 patients received intermittent vagal stimulation (2 to 10 sec “on” and 6 to 30 sec “off”) of the right cervical vagus using a pulse delivered 70 msec after each R wave of the ECG. The stimulating current, limited by a threshold or the onset of side effects, was adjusted to achieve a heart rate reduction of 5 to 10 beats/min. Patients were evaluated up to 6 months; no permanent side effects were reported. There was a modest improvement of cardiac function as judged by a reduction in left ventricular volumes, as well as a clear improvement in a measure of quality of life. The study shows feasibility and suggests further investigations.
The primary task seems to be optimization of stimulation parameters. Since natural vagal impulses are distributed through the cardiac cycle,3 artificial stimulation might be more effective if it emulated the natural firing pattern. Since long-term heart rate reduction was minimal, parameters might be tuned further to stimulate small cardiac efferent fibers that may be far from the electrodes. Measurements of heart rate variability during controlled conditions may reveal the pacemaker’s responsiveness to vagal stimulation. Comparison of duty cycles (duration of “on” and “off”) may show whether the study’s choice, to some extent already mimicking the breathing-induced modulation of natural vagal activity, is most effective. Such studies to optimize effectiveness may be best performed in chronically instrumented animals.
Baroreceptor stimulation
An extensively studied mechanism that modulates the autonomic nervous system is the baroreceptor reflex that is known to be depressed in heart failure. Former efforts to use this reflex therapeutically were recently revived in both animal experiments and human studies.31 For example, bilateral carotid sinus stimulation nearly doubled the survival time of dogs with pacing-induced heart failure.32 Although measures of left ventricular function, obtained while the stimulator was turned off, were similar in dogs with stimulated and unstimulated carotid sinuses, plasma norepinephrine was lower in the animals receiving stimulation. This suggests that carotid sinus stimulation led to a general decrease in sympathetic activation. It is noteworthy that the stimulation was applied intermittently (9 min “on”, 1 min “off”) to avoid the resetting (or adaptation) of baroreceptors, a phenomenon that had led to the now-doubted traditional belief that the baroreflex regulates only acute rather than long-term changes of blood pressure.2
Chronic bilateral baroreceptor stimulation was also applied in 21 patients with essential hypertension that could not be controlled by medication.33 Measurements were taken 1 month after implantation with the stimulator turned off and 3 months after chronic stimulation with the stimulator on. Stimulation moderately reduced both blood pressure and heart rate; heart rate variability suggested an increase in parasympathetic activity and a decrease in sympathetic activity.
Renal sympathetic ablation
Technology-based approaches encompass not only the stimulation of nerves but also the abolition of nerve activity. In an exploratory study, bilateral sympathetic denervation was performed in 45 patients with drugresistant hypertension.34 A catheter, introduced through the femoral artery, was positioned at the entrance of each renal artery. Sympathetic denervation was produced by radiofrequeny energy applied for a maximum of 2 minutes. The details of the technology appear proprietary, complete ablation could not be ascertained, and both efferents and afferents are likely to have been stimulated. Nevertheless, the procedure appeared safe and resulted in a significant reduction in both diastolic and systolic pressures over 12 months. In addition to lowering blood pressure, catheter-based sympathetic denervation might also prove therapeutic in heart failure and chronic kidney disease.
AUTOMATED CONTROL OF PHYSIOLOGIC VARIABLES
These initial experiences with baroreceptor stimulation, together with the results obtained by stimulating the vagus and ablating renal sympathetic nerves, indicate that device-based approaches may be useful additions to the treatment of drug-resistant hypertension and heart failure. The incorporation of control features that automatically respond to the changes in physiological states are natural extensions.35 As a recent example, in anesthetized dogs with heart failure and paced heart, systemic arterial pressure, cardiac output, and left atrial pressure were automatically regulated at set levels by a model-based infusion of nitroprusside, dobutamine, and volume expanders.36 When the heart rate was reduced, cardiac energetics (based on a reduction in left ventricular oxygen consumption) improved while the hemodynamic variables remained constant.
CONCLUSIONS
Biomedical engineering played a major role in linking measures of heart rate variation to sympathetic and parasympathetic contributions to cardiac control, as well as in demonstrating that the balance of control was correlated with a variety of disease states of heart and brain. However, much work remains to be done to fully realize the field’s potential for aiding clinicians in preventing or treating diseases. Further studies, some to be performed in chronically prepared animals, are needed to quantitatively characterize the many interacting mechanisms that determine cardiac function. Such studies would benefit from recording naturally occurring neural activity to and from the brain, and are already starting to benefit from the artificial electrical stimulation of nerves in both experimental animals and preliminary trials in patients.
The appropriate use of mathematical modeling is an essential tool for gaining in-depth understanding of physiological function. Mathematical modeling is also essential for developing stimulators that selectively stimulate those nerves that control the function to be influenced. The determination of stimulation parameters, based on understanding rather than on trial and error, is highly desirable. The use of “intelligent” stimulators, automatically controlled by appropriate physiological measurements, is an ambitious but achievable goal for improving human health.
References
Katona PG. Biomedical engineering and The Whitaker Foundation: a thirty-year partnership. Ann Biomed Eng2006; 34:904–916.
Guyton AC, Coleman TG, Cowley AW, et al Systems analysis of arterial pressure regulation and hypertension. Ann Biomed Eng1972; 1:254–281.
Katona PG, Poitras JW, Barnett GO, Terry BS. Cardiac vagal efferent activity and heart period in the carotid sinus reflex. Am J Physiol1979; 218:1030–1037.
Katona PG, Jih F. Respiratory sinus arrhythmia: noninvasive measure of parasympathetic cardiac control. J Appl Physiol1975; 39:801–805.
Akselrod S, Gordon D, Ubel FA, et al Power spectrum analysis of heart rate fluctuation: a quantitative probe of beat-to-beat cardiovascular control. Science1981; 213:220–222.
Carney RM, Freedland KE. Depression and heart rate variability in patients with coronary heart disease. Cleve Clin J Med2009; 76( suppl 2):S13–S17.
Lauer MS. Autonomic function and prognosis. Cleve Clin J Med2009; 76( suppl 2):S18–S22.
Cnockaert L, Migeotte P-F, Daubigny L, et al A method for the analysis of respiratory sinus arrhythmia using continuous wavelet transforms. IEEE Trans Biomed Eng2008; 55:1640–1642.
Castiglioni P, Parati G, Civijian A, et al Local scale exponents of blood pressure and heart rate variability by detrended fluctuation analysis: effects of posture, exercise, and aging. IEEE Trans Biomed Eng2009; 56:675–684.
Chen Z, Brown EN, Barbieri R. Assessment of autonomic control and respiratory sinus arrhythmia using point process models of human heart beat dynamics. IEEE Trans Biomed Eng2009; 56:1791–1802.
Schmitt DT, Stein PK, Ivanov PC. Stratification pattern of static and scale-invariant dynamic measures of heartbeat fluctuations across sleep stages in young and elderly. IEEE Trans Biomed Eng2009; 56:1564–1573.
Eckberg DL. Point:counterpoint: respiratory sinus arrhythmia is due to a central mechanism vs. respiratory sinus arrhythmia is due to the baroreflex mechanism. J Appl Physiol2009; 16:1740–1742.
Karemaker JM. Counterpoint: respiratory sinus arrhythmia is due to the baroreflex mechanism. J Appl Physiol2009; 106:1742–1743.
Moak JP, Goldstein DS, Eldadah BA, et al Supine low-frequency power of heart rate variability reflects baroreflex function, not cardiac sympathetic innervation. Heart Rhythm2007; 4:1523–1529.
Porges SW. The polyvagal theory: new insights into adaptive reactions of the autonomic nervous system. Cleve Clin J Med2009; 76( suppl 2):S86–S90.
Khoo MC. Modeling of autonomic control in sleep-disordered breathing. Cardiovasc Eng2008; 8:30–41.
Chaicharn J, Kin Z, Chen ML, et al Model-based assessment of cardiovascular autonomic control in children with obstructive sleep apnea. Sleep2009; 32:927–938.
Vitek JL. Deep brain stimulation: how does it work?Cleve Clin J Med2008; 75( suppl 2):S59–S65.
Lee KH, Blaha CD, Garrris PA. Evolution of deep brain stimulation: human electrometer and smart devices supporting the next generation of therapy. Neuromodulation: Technol Neural Interface2009; 12:85–103.
Fitzgerald PB, Daskalaris ZJ. The use of repetitive transcranial magnetic stimulation and vagal nerve stimulation in the treatment of depression. Curr Opin Psych2008; 21:25–29.
Cecchini AP, Mea E, Tullo V, et al Vagus nerve stimulation in drug-resistant daily chronic migraine with depression: preliminary data. Neurol Sci2009; 30( suppl 1):S101–S104.
Grill WM, Norman SE, Bellamkonda RV. Implanted neural interfaces: biochallenges and engineered solutions. Annu Rev Biomed Eng2009; 11:1–24.
Tarler MD, Mortimer JT. Selective and independent activation of four motor fascicles using a four-contact nerve-cuff electrode. IEEE Trans Neural Syst Rehab Eng2004; 12:251–257.
Lertmanorat Z, Gustafson KJ, Durand DM. Electrode array for reversing the recruitment order of peripheral nerve stimulation: experimental studies. Ann Biomed Eng2006; 34:152–160.
Tyler DJ, Durand DM. Functionally selective peripheral nerve stimulation with a flat interface nerve electrode. IEEE Trans Neural Syst Rehab Eng2002; 10:294–303.
Fang ZP, Mortimer JT. Selective activation of small motor axons by quasitrapezoidal current pulses. IEEE Trans Biomed Eng1991; 38:168–174.
Rijkoff NJM, Holsheimer J, Koledwijn EL, et al Selective stimulation of sacral nerve roots for bladder control: a study in computer modeling. IEEE Trans Biomed Eng1994; 41:413–424.
Tosato M, Yoshida K, Toft E, et al Quazi-trapezoidal pulses to selectively block the activation of intrinsic laryngeal muscles during vagal nerve stimulation. J Neural Eng2007; 41:205–212.
Schwartz PK, De Ferrari GM, Sanzo A, et al Long term vagal stimulation in patients with advanced heart failure: first experience in man. Eur J Heart Failure2008; 10:884–891.
De Ferrari GM, Sanzo A, Borggrefe M, et al Chronic vagus nerve stimulation in patients with chronic heart failure is feasible and appears beneficial. Circulation2008; 118( suppl):S721. Abstract 2412.
Durand MT, Fazan R, Salgado MCO, et al Acute and chronic electrical activation of baroreceptor afferents in awake and anesthetized subjects. Braz J Med Biol Res2009; 42:53–60.
Zucker IH, Hackley JF, Cornish KG, et al Chronic baroreceptor activation enhances survival in dogs with pacing-induced heart failure. Hypertension2007; 50:904–910.
Wustman K, Kucera JP, Scheffers I, et al Effects of chronic baroreceptor stimulation on the autonomic cardiovascular regulation in patients with drug-resistant arterial hypertension. Hypertension2009; 54:530–536.
Krum H, Schlaih M, Whitbourn R, et al Catheter-based renal denervation for resistant hypertension: a multicentre safety and proof-of-principle study. Lancet2009; 373:1228–1239.
Kawada T, Sugamichi M. Artifical neural interfaces for bionic cardio vascular treatments. J Artific Organs2009; 12:17–22.
Uemura K, Sunagawa K, Sugimachi M. Computationally managed bradycardia improved cardiac energetics while restoring normal hemodynamics in heart failure. Ann Biomed Eng2009; 37:82–93.
References
Katona PG. Biomedical engineering and The Whitaker Foundation: a thirty-year partnership. Ann Biomed Eng2006; 34:904–916.
Guyton AC, Coleman TG, Cowley AW, et al Systems analysis of arterial pressure regulation and hypertension. Ann Biomed Eng1972; 1:254–281.
Katona PG, Poitras JW, Barnett GO, Terry BS. Cardiac vagal efferent activity and heart period in the carotid sinus reflex. Am J Physiol1979; 218:1030–1037.
Katona PG, Jih F. Respiratory sinus arrhythmia: noninvasive measure of parasympathetic cardiac control. J Appl Physiol1975; 39:801–805.
Akselrod S, Gordon D, Ubel FA, et al Power spectrum analysis of heart rate fluctuation: a quantitative probe of beat-to-beat cardiovascular control. Science1981; 213:220–222.
Carney RM, Freedland KE. Depression and heart rate variability in patients with coronary heart disease. Cleve Clin J Med2009; 76( suppl 2):S13–S17.
Lauer MS. Autonomic function and prognosis. Cleve Clin J Med2009; 76( suppl 2):S18–S22.
Cnockaert L, Migeotte P-F, Daubigny L, et al A method for the analysis of respiratory sinus arrhythmia using continuous wavelet transforms. IEEE Trans Biomed Eng2008; 55:1640–1642.
Castiglioni P, Parati G, Civijian A, et al Local scale exponents of blood pressure and heart rate variability by detrended fluctuation analysis: effects of posture, exercise, and aging. IEEE Trans Biomed Eng2009; 56:675–684.
Chen Z, Brown EN, Barbieri R. Assessment of autonomic control and respiratory sinus arrhythmia using point process models of human heart beat dynamics. IEEE Trans Biomed Eng2009; 56:1791–1802.
Schmitt DT, Stein PK, Ivanov PC. Stratification pattern of static and scale-invariant dynamic measures of heartbeat fluctuations across sleep stages in young and elderly. IEEE Trans Biomed Eng2009; 56:1564–1573.
Eckberg DL. Point:counterpoint: respiratory sinus arrhythmia is due to a central mechanism vs. respiratory sinus arrhythmia is due to the baroreflex mechanism. J Appl Physiol2009; 16:1740–1742.
Karemaker JM. Counterpoint: respiratory sinus arrhythmia is due to the baroreflex mechanism. J Appl Physiol2009; 106:1742–1743.
Moak JP, Goldstein DS, Eldadah BA, et al Supine low-frequency power of heart rate variability reflects baroreflex function, not cardiac sympathetic innervation. Heart Rhythm2007; 4:1523–1529.
Porges SW. The polyvagal theory: new insights into adaptive reactions of the autonomic nervous system. Cleve Clin J Med2009; 76( suppl 2):S86–S90.
Khoo MC. Modeling of autonomic control in sleep-disordered breathing. Cardiovasc Eng2008; 8:30–41.
Chaicharn J, Kin Z, Chen ML, et al Model-based assessment of cardiovascular autonomic control in children with obstructive sleep apnea. Sleep2009; 32:927–938.
Vitek JL. Deep brain stimulation: how does it work?Cleve Clin J Med2008; 75( suppl 2):S59–S65.
Lee KH, Blaha CD, Garrris PA. Evolution of deep brain stimulation: human electrometer and smart devices supporting the next generation of therapy. Neuromodulation: Technol Neural Interface2009; 12:85–103.
Fitzgerald PB, Daskalaris ZJ. The use of repetitive transcranial magnetic stimulation and vagal nerve stimulation in the treatment of depression. Curr Opin Psych2008; 21:25–29.
Cecchini AP, Mea E, Tullo V, et al Vagus nerve stimulation in drug-resistant daily chronic migraine with depression: preliminary data. Neurol Sci2009; 30( suppl 1):S101–S104.
Grill WM, Norman SE, Bellamkonda RV. Implanted neural interfaces: biochallenges and engineered solutions. Annu Rev Biomed Eng2009; 11:1–24.
Tarler MD, Mortimer JT. Selective and independent activation of four motor fascicles using a four-contact nerve-cuff electrode. IEEE Trans Neural Syst Rehab Eng2004; 12:251–257.
Lertmanorat Z, Gustafson KJ, Durand DM. Electrode array for reversing the recruitment order of peripheral nerve stimulation: experimental studies. Ann Biomed Eng2006; 34:152–160.
Tyler DJ, Durand DM. Functionally selective peripheral nerve stimulation with a flat interface nerve electrode. IEEE Trans Neural Syst Rehab Eng2002; 10:294–303.
Fang ZP, Mortimer JT. Selective activation of small motor axons by quasitrapezoidal current pulses. IEEE Trans Biomed Eng1991; 38:168–174.
Rijkoff NJM, Holsheimer J, Koledwijn EL, et al Selective stimulation of sacral nerve roots for bladder control: a study in computer modeling. IEEE Trans Biomed Eng1994; 41:413–424.
Tosato M, Yoshida K, Toft E, et al Quazi-trapezoidal pulses to selectively block the activation of intrinsic laryngeal muscles during vagal nerve stimulation. J Neural Eng2007; 41:205–212.
Schwartz PK, De Ferrari GM, Sanzo A, et al Long term vagal stimulation in patients with advanced heart failure: first experience in man. Eur J Heart Failure2008; 10:884–891.
De Ferrari GM, Sanzo A, Borggrefe M, et al Chronic vagus nerve stimulation in patients with chronic heart failure is feasible and appears beneficial. Circulation2008; 118( suppl):S721. Abstract 2412.
Durand MT, Fazan R, Salgado MCO, et al Acute and chronic electrical activation of baroreceptor afferents in awake and anesthetized subjects. Braz J Med Biol Res2009; 42:53–60.
Zucker IH, Hackley JF, Cornish KG, et al Chronic baroreceptor activation enhances survival in dogs with pacing-induced heart failure. Hypertension2007; 50:904–910.
Wustman K, Kucera JP, Scheffers I, et al Effects of chronic baroreceptor stimulation on the autonomic cardiovascular regulation in patients with drug-resistant arterial hypertension. Hypertension2009; 54:530–536.
Krum H, Schlaih M, Whitbourn R, et al Catheter-based renal denervation for resistant hypertension: a multicentre safety and proof-of-principle study. Lancet2009; 373:1228–1239.
Kawada T, Sugamichi M. Artifical neural interfaces for bionic cardio vascular treatments. J Artific Organs2009; 12:17–22.
Uemura K, Sunagawa K, Sugimachi M. Computationally managed bradycardia improved cardiac energetics while restoring normal hemodynamics in heart failure. Ann Biomed Eng2009; 37:82–93.
Sudden unexpected death in epilepsy (SUDEP) is most often defined as the sudden, unexpected, nontraumatic, and nondrowning death of patients with epilepsy. The death may be either witnessed or unwitnessed, and may occur with or without evidence of a seizure, excluding documented status epilepticus. In cases of definite SUDEP, postmortem examination does not reveal a structural or toxicologic cause of death.1
The reported incidence of SUDEP ranges from 0.7 to 1.3 cases per 1,000 patient-years in large cohorts of epilepsy patients2,3 and from 3.5 to 9.3 cases per 1,000 patient-years in antiepileptic drug registries, medical device registries, and epilepsy surgery programs.4–6 SUDEP is currently accepted as the most important epilepsy-related mode of death5,7 and is associated with standardized mortality ratios in patients with ongoing seizures as high as 24 to 40 times those of the general population.8 The exact mechanism or mechanisms leading to SUDEP remain unknown,1,5,9,10 and despite the identification of several potential risk factors, elimination of seizures still represents the main intervention correlating with risk reduction.5,11
This review will first discuss evidence for a cardiac mechanism of SUDEP, then review data traditionally used to support freedom from seizures following epilepsy surgery as a method for reducing SUDEP risk, and finally examine recent evidence suggesting that both SUDEP and seizure outcomes following epilepsy surgery are governed by a common pathway with certain cardiac manifestations.
CENTRAL AUTONOMIC NETWORK
Figure 1. The cardiovascular autonomic system. Afferent pathways (violet tracings): The afferent loop of the cardiac autonomic system receives input from chemo- and baroreceptors in the carotid sinus (cranial nerve [CN] IX) and the aortic arch (CN X). Incoming visceral sensations are projected via lamina I of the spinal cord to the nucleus tractus solitarius and, from there, via the parabrachial region to the primary viscerosensitive cortex located in the insula. Efferent pathways (red tracings): The anterior cingulate and orbitofrontal cortex send projections to the hypothalamus and amygdala, but also to the autonomic centers within the brainstem: the periaqueductal gray matter, parabrachial nucleus, nucleus tractus solitarius, nucleus ambiguus, and rostral ventrolateral medulla. Parasympathetic influence on the heart arises primarily from the nucleus ambiguus. Sympathetic output receives tonic excitation from the ventrolateral medulla and projects from the intermediolateral cell columns to the cardiac conduction system and ventricle. Integrated response to emotion and stress (green arrows): The amygdala has reciprocal connections with the cerebral cortex and mediates autonomic response to emotions via projections to the hypothalamus and brainstem. The paraventricular nucleus of the hypothalamus controls internal homeostasis and innervates the autonomic relay centers in the rostral ventrolateral medulla, nucleus tractus solitarius, parabrachial nucleus, and preganglionic vagal and sympathetic neurons. Reprinted from Schuele et al,12 which was modified from Britton and Benarroch.13A tightly interconnected neuronal network controls various elements of the cardiovascular autonomic system. Consideration of a cardiac pathophysiology of SUDEP requires a fundamental understanding of this network (Figure 1).7,12,13 The insula, the anterior cingulate gyrus, and the ventromedial prefrontal cortex are key to central cortical control of autonomic function. The insula represents the primary viscerosensory cortex, whereas the cingulate gyrus and prefrontal cortices constitute the premotor autonomic region. At the subcortical level, the hypothalamus provides an interface with endocrine stimuli and triggers autonomic responses to maintain homeostasis. The amygdala, which is an integral component of the limbic system that links the previously mentioned cortical and subcortical centers, mediates the autonomic response to emotions. Beyond their centrality to autonomic control, the insula, amygdala, cingulate gyrus, and prefrontal cortex also represent the most common foci of partial epilepsy, a finding that might explain the frequent observation of autonomic changes in relation to epileptic seizures.14
EVIDENCE FOR A CARDIAC MECHANISM OF SUDEP
The most significant and broadly discussed cardiac mechanism of SUDEP is cardiac arrhythmia brought about by seizure discharges acting through the autonomic nervous system.5,7,15,16
Clinical evidence
A wide spectrum of cardiac arrhythmias—from ictal asystole to atrial fibrillation to repolarization abnormalities to bundle branch blocks—has been reported during seizures. 14–19 In one study, ictal cardiac arrhythmias occurred in 42% of hospitalized epilepsy patients, the most common being an irregular series of abrupt rate changes near the end of the electroencephalographic (EEG) seizure discharge.17 In another study, R-R interval analysis during the first 10-second period of EEG discharge showed a significant early heart rate increase in 49% of seizures and an early heart rate reduction in 25.5%.18
Certain clinical seizure characteristics have been correlated with the occurrence of ictal electrocardiographic (ECG) abnormalities. One study found that mean seizure duration was longer in patients with ECG abnormalities than in those without such changes.16 Others observed that ictal ECG abnormalities occurred more often and were more severe in generalized tonic-clonic seizures than in complex partial seizures.15,16,19 These same clinical seizure characteristics were correlated with a higher risk of SUDEP,20 which suggests an interrelation among seizure semiology, ECG abnormalities, and SUDEP.
Additionally, direct evidence of seizure-related cardiac changes occurring specifically in SUDEP victims has come from a study that compared EEG and ECG data between 21 SUDEP patients and 43 clinically similar historical controls with refractory partial epilepsy.15 The patients who eventually succumbed to SUDEP had a higher ictal maximal heart rate, a greater increase in heart rate with seizures arising from sleep, and a higher incidence of ictal cardiac repolarization and rhythm abnormalities.15
Experimental evidence
Electrical brain stimulation of the limbic system and insular cortex has repeatedly been shown to provoke heart rate changes, including bradycardia, tachycardia, and asystole.14 Some studies have even suggested a lateralized influence of the insulae on cardiovascular autonomic control, with intraoperative stimulation of the left posterior insula eliciting a cardioinhibitory response and hypotension, and with stimulation of the right anterior insula eliciting tachycardia and hypertension.7 Other studies have suggested that the limbic system has a localization-related influence on cardiovascular responses, with stimulation of the amygdala alone being insufficient to produce the ictal tachycardia so commonly seen in epileptic seizures, which suggests that cortical involvement is essential for the increase in heart rate.21 Such cortical stimulation–induced heart rate changes may explain how massive seizure-related discharges can affect cardiac rhythm during the seizure itself.
There is also, however, evidence of a baseline epilepsy-related autonomic dysfunction. Abnormalities in cardiac uptake of meta-iodobenzylguanidine (MIBG) have been demonstrated interictally in patients with chronic temporal lobe epilepsy relative to controls.22 A recent study specifically showed a pronounced reduction in cardiac MIBG uptake in patients who had ictal asystole compared with other epileptic patients and nonepileptic controls, a finding that suggests a postganglionic cardiac catecholamine disturbance in patients with epilepsy.23 The authors proposed that epilepsy-related impairment of sympathetic cardiac innervation limits adjustment and heart rate modulation, and may thus increase the risk of asystole and, ultimately, SUDEP.23
EPILEPSY SURGERY AND SUDEP RISK
The above findings make it reasonable to postulate that recurrent uncontrolled seizures may lead to significant cardiac changes—namely, arrhythmias—which may then lead to a higher risk of SUDEP. Successful epilepsy surgery, the treatment of choice for uncontrolled partial epilepsy, would eliminate seizures and would thus be expected to reduce SUDEP risk.
Several studies support this hypothesis. Reductions in all-cause mortality and in SUDEP following successful epilepsy surgery have been observed when patients who became seizure-free after surgery were compared either with patients who had ongoing postoperative seizures or with patients with intractable epilepsy who did not undergo epilepsy surgery.11,24 In one study with a mean follow-up of 3.82 years, no deaths occurred among 199 patients who were seizure-free following epilepsy surgery, whereas there were 11 deaths—including 6 cases of SUDEP—among 194 patients who had persistent seizures following epilepsy surgery.11 A separate study compared 202 patients who underwent epilepsy surgery with 46 patients with medically treated intractable epilepsy over a mean follow-up period of greater than 5.7 years.24 In this study, death occurred in only 7% of the surgical patients compared with 20% of the medically treated patients, which suggests that epilepsy surgery (or lack thereof) may be an independent predictor of mortality. Favorable outcome was linked closely to seizure control, as 81% of the patients who died had 2 or more seizures per year at last follow-up, compared with only 47% of survivors in the overall cohort.24
Complete seizure resolution—not just reduction—seems necessary for SUDEP risk reduction
In our experience at the Cleveland Clinic Epilepsy Center, we have found that a reduction in seizure frequency following epilepsy surgery is not enough to eliminate SUDEP risk—complete freedom from seizures is required. Of 37 SUDEP cases identified so far in a cohort of 3,481 patients evaluated in our epilepsy monitoring unit between 1990 and 2005 (Jehi et al, in preparation), 7 patients had undergone epilepsy surgery. None of these 7 patients was seizure-free at the time of death; four had a greater than 50% reduction in seizure frequency. No cases of SUDEP occurred in patients who were seizure-free after surgery.
Our findings mirror earlier results reported by Sperling et al,11 who also found that epilepsy surgery improves mortality only when seizures resolve completely, not when they merely “improve.” It seems plausible, therefore, that elimination of seizures postoperatively eliminates the risk for the several seizure-related cardiac arrhythmogenic changes discussed above, thereby eliminating the risk for the series of events that eventually leads to SUDEP.
A role for stabilized baseline autonomic control?
Alternatively, some data suggest that the “autonomic” impacts of epilepsy surgery may extend beyond the immediate seizure-related manifestations to the baseline interictal period in epilepsy patients. One study found that surgery for temporal lobe epilepsy is followed by a reduction of sympathetic cardiovascular modulation and baroreflex sensitivity.25 The authors proposed that this finding may be attributable to decreased influences of interictal epileptogenic discharges on brain areas involved in cardiovascular autonomic control. These researchers continue to postulate that surgery for temporal lobe epilepsy seems to stabilize cardiovascular control in epilepsy patients by reducing the risk of sympathetically mediated tachyarrhythmias and excessive bradycardiac counterregulation, potentially lowering SUDEP risk.25
CARDIAC FINDINGS, SUDEP, AND SEIZURE OUTCOMES
Whether it is the elimination of seizure-related cardiac arrhythmias achieved by rendering patients seizure-free after surgery, or whether it is the stabilization of baseline autonomic cardiac control by reducing interictal epileptiform discharges, this line of thought assumes that the autonomic dysfunction contributing to SUDEP is caused by epilepsy and that freedom from seizures following epilepsy surgery should therefore be responsible for reducing the risk of death. An alternative hypothesis for the infrequent occurrence of SUDEP in seizure-free patients may be that seizure outcomes following epilepsy surgery and SUDEP risk are actually both governed by the same underlying biologic process. This would suggest that the same patient cohort is at a higher baseline mortality risk and in a prognostically poorer seizure outcome group following epilepsy surgery.
This idea is supported by a recent prospective study among 21 consecutive candidates for temporal lobe epilepsy surgery in which spectral analysis of heart rate variability performed preoperatively was correlated with seizure control 1 year after surgery.26 The study found that patients with poor seizure outcomes (Engel class II to IV, signifying ongoing postoperative seizures) had significantly lower power in all domains of heart rate variability than did patients with favorable seizure outcomes.26
In another study, heart rate was recorded in 16 patients before and after temporal lobe epilepsy surgery, and sympathetic and parasympathetic cardiac modulation was determined as powers of low-frequency (LF, 0.04–0.15 Hz) and high-frequency (HF, 0.15–0.5 Hz) heart rate oscillations.27 The LF/HF ratio was calculated as an index of sympathovagal balance. Cardiac MIBG uptake was measured with MIBG single-photon emission computed tomography and compared with control data. Baseline sympathetic LF power and LF/HF ratio were higher in patients who eventually had persistent seizures than in those who became seizure-free. Following surgery, both measures decreased in seizure-free patients but increased in patients with persistent seizures. MIBG uptake was lower in patients than in controls and even lower yet in patients who had persistent seizures. In this subgroup, MIBG uptake declined further after surgery.
Essentially, both of the above studies26,27 demonstrate findings of autonomic cardiac abnormalities that predated epilepsy surgery and reliably predicted the eventual surgical outcome in terms of seizure continuance. This suggests that “poor candidates” for epilepsy surgery—ie, those with lower chances of achieving seizure freedom with surgery—may a priori have a higher SUDEP risk. A possible explanation for these findings may be epileptogenic zones, including the insula and other components of the central autonomic network, or molecular/genetic diffuse abnormalities that extend beyond a limited surgically removable seizure focus and involve the heart, increasing the risk for cardiac conduction abnormalities.
CONCLUSIONS
SUDEP is the most common cause of death in epilepsy patients. A significant body of literature suggests that a cardiac mechanism contributes to its occurrence. Although the exact relationship between seizure outcomes following epilepsy surgery and SUDEP risk is still being investigated, it is accepted that seizure control correlates with reduced mortality. Cardiac changes and autonomic dysregulation seem to be at the “heart” of the problem.
References
Nashef L, Hindocha N, Makoff A. Risk factors in sudden death in epilepsy (SUDEP): the quest for mechanisms. Epilepsia2007; 48:859–871.
Nilsson L, Tomson T, Farahmand BY, et al Cause-specific mortality in epilepsy: a cohort study of more than 9,000 patients once hospitalized for epilepsy. Epilepsia1997; 38:1062–1068.
Tennis P, Cole TB, Annegers JF, et al Cohort study of incidence of sudden unexplained death in persons with seizure disorder treated with antiepileptic drugs in Saskatchewan, Canada. Epilepsia1995; 36:29–36.
Leestma JE, Annegers JF, Brodie MJ, et al Sudden unexplained death in epilepsy: observations from a large clinical development program. Epilepsia1997; 38:47–55.
Tomson T, Nashef L, Ryvlin P. Sudden unexpected death in epilepsy: current knowledge and future directions. Lancet Neurol2008; 7:1021–1031.
Nashef L, Fish DR, Sander JW, Shorvon SD. Incidence of sudden unexpected death in an adult outpatient cohort with epilepsy at a tertiary referral centre. J Neurol Neurosurg Psychiatry1995; 58:462–464.
Jehi L, Najm IM. Sudden unexpected death in epilepsy: impact, mechanisms, and prevention. Cleve Clin J Med2008; 75( suppl 2):S66–S70.
Lhatoo SD, Johnson AL, Goodridge DM, et al Mortality in epilepsy in the first 11 to 14 years after diagnosis: multivariate analysis of a long-term, prospective, population-based cohort. Ann Neurol2001; 49:336–344.
Pedley TA, Hauser WA. Sudden death in epilepsy: a wake-up call for management. Lancet2002; 359:1790–1791.
Schraeder PL, Delin K, McClelland RL, So EL. Coroner and medical examiner documentation of sudden unexplained deaths in epilepsy. Epilepsy Res2006; 68:137–143.
Sperling MR, Feldman H, Kinman J, et al Seizure control and mortality in epilepsy. Ann Neurol1999; 46:45–50.
Schuele SU, Widdess-Walsh P, Bermeo A, Lüders HO. Sudden unexplained death in epilepsy: the role of the heart. Cleve Clin J Med2007; 74( suppl 1):S121–S127.
Britton JW, Benarroch E. Seizures and syncope: anatomic basis and diagnostic considerations. Clin Auton Res2006; 16:18–28.
Leung H, Kwan P, Elger CE. Finding the missing link between ictal bradyarrhythmia, ictal asystole, and sudden unexpected death in epilepsy. Epilepsy Behav2006; 9:19–30.
Nei M, Ho RT, Abou-Khalil BW, et al EEG and ECG in sudden unexplained death in epilepsy. Epilepsia2004; 45:338–345.
Nei M, Ho RT, Sperling MR. EKG abnormalities during partial seizures in refractory epilepsy. Epilepsia2000; 41:542–548.
Blumhardt LD, Smith PE, Owen L. Electrocardiographic accompaniments of temporal lobe epileptic seizures. Lancet1986; 1:1051–1056.
Galimberti CA, Marchioni E, Barzizza F, et al Partial epileptic seizures of different origin variably affect cardiac rhythm. Epilepsia1996; 37:742–747.
Opherk C, Coromilas J, Hirsch LJ. Heart rate and EKG changes in 102 seizures: analysis of influencing factors. Epilepsy Res2002; 52:117–127.
Langan Y, Nashef L, Sander JW. Case-control study of SUDEP. Neurology2005; 64:1131–1133.
Keilson MJ, Hauser WA, Magrill JP, Goldman M. ECG abnormalities in patients with epilepsy. Neurology1987; 37:1624–1626.
Druschky A, Hilz MJ, Hopp P, et al Interictal cardiac autonomic dysfunction in temporal lobe epilepsy demonstrated by [123I] metaiodobenzylguanidine-SPECT. Brain2001; 124:2372–2382.
Kerling F, Dütsch M, Linke R, Kuwert T, Stefan H, Hilz MJ. Relation between ictal asystole and cardiac sympathetic dysfunction shown by MIBG-SPECT. Acta Neurol Scand2009; 120:123–129.
Vickrey BG, Hays RD, Rausch R, et al Outcomes in 248 patients who had diagnostic evaluations for epilepsy surgery. Lancet1995; 346:1445–1449.
Hilz MJ, Devinsky O, Doyle W, et al Decrease of sympathetic cardiovascular modulation after temporal lobe epilepsy surgery. Brain2002; 125:985–995.
Persson H, Kumlien E, Ericson M, Tomson T. Preoperative heart rate variability in relation to surgery outcome in refractory epilepsy. Neurology2005; 65:1021–1025.
Hilz MJ, Platsch G, Druschky K, et al Outcome of epilepsy surgery correlates with sympathetic modulation and neuroimaging of the heart. J Neurol Sci2003; 216:153–162.
Sudden unexpected death in epilepsy (SUDEP) is most often defined as the sudden, unexpected, nontraumatic, and nondrowning death of patients with epilepsy. The death may be either witnessed or unwitnessed, and may occur with or without evidence of a seizure, excluding documented status epilepticus. In cases of definite SUDEP, postmortem examination does not reveal a structural or toxicologic cause of death.1
The reported incidence of SUDEP ranges from 0.7 to 1.3 cases per 1,000 patient-years in large cohorts of epilepsy patients2,3 and from 3.5 to 9.3 cases per 1,000 patient-years in antiepileptic drug registries, medical device registries, and epilepsy surgery programs.4–6 SUDEP is currently accepted as the most important epilepsy-related mode of death5,7 and is associated with standardized mortality ratios in patients with ongoing seizures as high as 24 to 40 times those of the general population.8 The exact mechanism or mechanisms leading to SUDEP remain unknown,1,5,9,10 and despite the identification of several potential risk factors, elimination of seizures still represents the main intervention correlating with risk reduction.5,11
This review will first discuss evidence for a cardiac mechanism of SUDEP, then review data traditionally used to support freedom from seizures following epilepsy surgery as a method for reducing SUDEP risk, and finally examine recent evidence suggesting that both SUDEP and seizure outcomes following epilepsy surgery are governed by a common pathway with certain cardiac manifestations.
CENTRAL AUTONOMIC NETWORK
Figure 1. The cardiovascular autonomic system. Afferent pathways (violet tracings): The afferent loop of the cardiac autonomic system receives input from chemo- and baroreceptors in the carotid sinus (cranial nerve [CN] IX) and the aortic arch (CN X). Incoming visceral sensations are projected via lamina I of the spinal cord to the nucleus tractus solitarius and, from there, via the parabrachial region to the primary viscerosensitive cortex located in the insula. Efferent pathways (red tracings): The anterior cingulate and orbitofrontal cortex send projections to the hypothalamus and amygdala, but also to the autonomic centers within the brainstem: the periaqueductal gray matter, parabrachial nucleus, nucleus tractus solitarius, nucleus ambiguus, and rostral ventrolateral medulla. Parasympathetic influence on the heart arises primarily from the nucleus ambiguus. Sympathetic output receives tonic excitation from the ventrolateral medulla and projects from the intermediolateral cell columns to the cardiac conduction system and ventricle. Integrated response to emotion and stress (green arrows): The amygdala has reciprocal connections with the cerebral cortex and mediates autonomic response to emotions via projections to the hypothalamus and brainstem. The paraventricular nucleus of the hypothalamus controls internal homeostasis and innervates the autonomic relay centers in the rostral ventrolateral medulla, nucleus tractus solitarius, parabrachial nucleus, and preganglionic vagal and sympathetic neurons. Reprinted from Schuele et al,12 which was modified from Britton and Benarroch.13A tightly interconnected neuronal network controls various elements of the cardiovascular autonomic system. Consideration of a cardiac pathophysiology of SUDEP requires a fundamental understanding of this network (Figure 1).7,12,13 The insula, the anterior cingulate gyrus, and the ventromedial prefrontal cortex are key to central cortical control of autonomic function. The insula represents the primary viscerosensory cortex, whereas the cingulate gyrus and prefrontal cortices constitute the premotor autonomic region. At the subcortical level, the hypothalamus provides an interface with endocrine stimuli and triggers autonomic responses to maintain homeostasis. The amygdala, which is an integral component of the limbic system that links the previously mentioned cortical and subcortical centers, mediates the autonomic response to emotions. Beyond their centrality to autonomic control, the insula, amygdala, cingulate gyrus, and prefrontal cortex also represent the most common foci of partial epilepsy, a finding that might explain the frequent observation of autonomic changes in relation to epileptic seizures.14
EVIDENCE FOR A CARDIAC MECHANISM OF SUDEP
The most significant and broadly discussed cardiac mechanism of SUDEP is cardiac arrhythmia brought about by seizure discharges acting through the autonomic nervous system.5,7,15,16
Clinical evidence
A wide spectrum of cardiac arrhythmias—from ictal asystole to atrial fibrillation to repolarization abnormalities to bundle branch blocks—has been reported during seizures. 14–19 In one study, ictal cardiac arrhythmias occurred in 42% of hospitalized epilepsy patients, the most common being an irregular series of abrupt rate changes near the end of the electroencephalographic (EEG) seizure discharge.17 In another study, R-R interval analysis during the first 10-second period of EEG discharge showed a significant early heart rate increase in 49% of seizures and an early heart rate reduction in 25.5%.18
Certain clinical seizure characteristics have been correlated with the occurrence of ictal electrocardiographic (ECG) abnormalities. One study found that mean seizure duration was longer in patients with ECG abnormalities than in those without such changes.16 Others observed that ictal ECG abnormalities occurred more often and were more severe in generalized tonic-clonic seizures than in complex partial seizures.15,16,19 These same clinical seizure characteristics were correlated with a higher risk of SUDEP,20 which suggests an interrelation among seizure semiology, ECG abnormalities, and SUDEP.
Additionally, direct evidence of seizure-related cardiac changes occurring specifically in SUDEP victims has come from a study that compared EEG and ECG data between 21 SUDEP patients and 43 clinically similar historical controls with refractory partial epilepsy.15 The patients who eventually succumbed to SUDEP had a higher ictal maximal heart rate, a greater increase in heart rate with seizures arising from sleep, and a higher incidence of ictal cardiac repolarization and rhythm abnormalities.15
Experimental evidence
Electrical brain stimulation of the limbic system and insular cortex has repeatedly been shown to provoke heart rate changes, including bradycardia, tachycardia, and asystole.14 Some studies have even suggested a lateralized influence of the insulae on cardiovascular autonomic control, with intraoperative stimulation of the left posterior insula eliciting a cardioinhibitory response and hypotension, and with stimulation of the right anterior insula eliciting tachycardia and hypertension.7 Other studies have suggested that the limbic system has a localization-related influence on cardiovascular responses, with stimulation of the amygdala alone being insufficient to produce the ictal tachycardia so commonly seen in epileptic seizures, which suggests that cortical involvement is essential for the increase in heart rate.21 Such cortical stimulation–induced heart rate changes may explain how massive seizure-related discharges can affect cardiac rhythm during the seizure itself.
There is also, however, evidence of a baseline epilepsy-related autonomic dysfunction. Abnormalities in cardiac uptake of meta-iodobenzylguanidine (MIBG) have been demonstrated interictally in patients with chronic temporal lobe epilepsy relative to controls.22 A recent study specifically showed a pronounced reduction in cardiac MIBG uptake in patients who had ictal asystole compared with other epileptic patients and nonepileptic controls, a finding that suggests a postganglionic cardiac catecholamine disturbance in patients with epilepsy.23 The authors proposed that epilepsy-related impairment of sympathetic cardiac innervation limits adjustment and heart rate modulation, and may thus increase the risk of asystole and, ultimately, SUDEP.23
EPILEPSY SURGERY AND SUDEP RISK
The above findings make it reasonable to postulate that recurrent uncontrolled seizures may lead to significant cardiac changes—namely, arrhythmias—which may then lead to a higher risk of SUDEP. Successful epilepsy surgery, the treatment of choice for uncontrolled partial epilepsy, would eliminate seizures and would thus be expected to reduce SUDEP risk.
Several studies support this hypothesis. Reductions in all-cause mortality and in SUDEP following successful epilepsy surgery have been observed when patients who became seizure-free after surgery were compared either with patients who had ongoing postoperative seizures or with patients with intractable epilepsy who did not undergo epilepsy surgery.11,24 In one study with a mean follow-up of 3.82 years, no deaths occurred among 199 patients who were seizure-free following epilepsy surgery, whereas there were 11 deaths—including 6 cases of SUDEP—among 194 patients who had persistent seizures following epilepsy surgery.11 A separate study compared 202 patients who underwent epilepsy surgery with 46 patients with medically treated intractable epilepsy over a mean follow-up period of greater than 5.7 years.24 In this study, death occurred in only 7% of the surgical patients compared with 20% of the medically treated patients, which suggests that epilepsy surgery (or lack thereof) may be an independent predictor of mortality. Favorable outcome was linked closely to seizure control, as 81% of the patients who died had 2 or more seizures per year at last follow-up, compared with only 47% of survivors in the overall cohort.24
Complete seizure resolution—not just reduction—seems necessary for SUDEP risk reduction
In our experience at the Cleveland Clinic Epilepsy Center, we have found that a reduction in seizure frequency following epilepsy surgery is not enough to eliminate SUDEP risk—complete freedom from seizures is required. Of 37 SUDEP cases identified so far in a cohort of 3,481 patients evaluated in our epilepsy monitoring unit between 1990 and 2005 (Jehi et al, in preparation), 7 patients had undergone epilepsy surgery. None of these 7 patients was seizure-free at the time of death; four had a greater than 50% reduction in seizure frequency. No cases of SUDEP occurred in patients who were seizure-free after surgery.
Our findings mirror earlier results reported by Sperling et al,11 who also found that epilepsy surgery improves mortality only when seizures resolve completely, not when they merely “improve.” It seems plausible, therefore, that elimination of seizures postoperatively eliminates the risk for the several seizure-related cardiac arrhythmogenic changes discussed above, thereby eliminating the risk for the series of events that eventually leads to SUDEP.
A role for stabilized baseline autonomic control?
Alternatively, some data suggest that the “autonomic” impacts of epilepsy surgery may extend beyond the immediate seizure-related manifestations to the baseline interictal period in epilepsy patients. One study found that surgery for temporal lobe epilepsy is followed by a reduction of sympathetic cardiovascular modulation and baroreflex sensitivity.25 The authors proposed that this finding may be attributable to decreased influences of interictal epileptogenic discharges on brain areas involved in cardiovascular autonomic control. These researchers continue to postulate that surgery for temporal lobe epilepsy seems to stabilize cardiovascular control in epilepsy patients by reducing the risk of sympathetically mediated tachyarrhythmias and excessive bradycardiac counterregulation, potentially lowering SUDEP risk.25
CARDIAC FINDINGS, SUDEP, AND SEIZURE OUTCOMES
Whether it is the elimination of seizure-related cardiac arrhythmias achieved by rendering patients seizure-free after surgery, or whether it is the stabilization of baseline autonomic cardiac control by reducing interictal epileptiform discharges, this line of thought assumes that the autonomic dysfunction contributing to SUDEP is caused by epilepsy and that freedom from seizures following epilepsy surgery should therefore be responsible for reducing the risk of death. An alternative hypothesis for the infrequent occurrence of SUDEP in seizure-free patients may be that seizure outcomes following epilepsy surgery and SUDEP risk are actually both governed by the same underlying biologic process. This would suggest that the same patient cohort is at a higher baseline mortality risk and in a prognostically poorer seizure outcome group following epilepsy surgery.
This idea is supported by a recent prospective study among 21 consecutive candidates for temporal lobe epilepsy surgery in which spectral analysis of heart rate variability performed preoperatively was correlated with seizure control 1 year after surgery.26 The study found that patients with poor seizure outcomes (Engel class II to IV, signifying ongoing postoperative seizures) had significantly lower power in all domains of heart rate variability than did patients with favorable seizure outcomes.26
In another study, heart rate was recorded in 16 patients before and after temporal lobe epilepsy surgery, and sympathetic and parasympathetic cardiac modulation was determined as powers of low-frequency (LF, 0.04–0.15 Hz) and high-frequency (HF, 0.15–0.5 Hz) heart rate oscillations.27 The LF/HF ratio was calculated as an index of sympathovagal balance. Cardiac MIBG uptake was measured with MIBG single-photon emission computed tomography and compared with control data. Baseline sympathetic LF power and LF/HF ratio were higher in patients who eventually had persistent seizures than in those who became seizure-free. Following surgery, both measures decreased in seizure-free patients but increased in patients with persistent seizures. MIBG uptake was lower in patients than in controls and even lower yet in patients who had persistent seizures. In this subgroup, MIBG uptake declined further after surgery.
Essentially, both of the above studies26,27 demonstrate findings of autonomic cardiac abnormalities that predated epilepsy surgery and reliably predicted the eventual surgical outcome in terms of seizure continuance. This suggests that “poor candidates” for epilepsy surgery—ie, those with lower chances of achieving seizure freedom with surgery—may a priori have a higher SUDEP risk. A possible explanation for these findings may be epileptogenic zones, including the insula and other components of the central autonomic network, or molecular/genetic diffuse abnormalities that extend beyond a limited surgically removable seizure focus and involve the heart, increasing the risk for cardiac conduction abnormalities.
CONCLUSIONS
SUDEP is the most common cause of death in epilepsy patients. A significant body of literature suggests that a cardiac mechanism contributes to its occurrence. Although the exact relationship between seizure outcomes following epilepsy surgery and SUDEP risk is still being investigated, it is accepted that seizure control correlates with reduced mortality. Cardiac changes and autonomic dysregulation seem to be at the “heart” of the problem.
Sudden unexpected death in epilepsy (SUDEP) is most often defined as the sudden, unexpected, nontraumatic, and nondrowning death of patients with epilepsy. The death may be either witnessed or unwitnessed, and may occur with or without evidence of a seizure, excluding documented status epilepticus. In cases of definite SUDEP, postmortem examination does not reveal a structural or toxicologic cause of death.1
The reported incidence of SUDEP ranges from 0.7 to 1.3 cases per 1,000 patient-years in large cohorts of epilepsy patients2,3 and from 3.5 to 9.3 cases per 1,000 patient-years in antiepileptic drug registries, medical device registries, and epilepsy surgery programs.4–6 SUDEP is currently accepted as the most important epilepsy-related mode of death5,7 and is associated with standardized mortality ratios in patients with ongoing seizures as high as 24 to 40 times those of the general population.8 The exact mechanism or mechanisms leading to SUDEP remain unknown,1,5,9,10 and despite the identification of several potential risk factors, elimination of seizures still represents the main intervention correlating with risk reduction.5,11
This review will first discuss evidence for a cardiac mechanism of SUDEP, then review data traditionally used to support freedom from seizures following epilepsy surgery as a method for reducing SUDEP risk, and finally examine recent evidence suggesting that both SUDEP and seizure outcomes following epilepsy surgery are governed by a common pathway with certain cardiac manifestations.
CENTRAL AUTONOMIC NETWORK
Figure 1. The cardiovascular autonomic system. Afferent pathways (violet tracings): The afferent loop of the cardiac autonomic system receives input from chemo- and baroreceptors in the carotid sinus (cranial nerve [CN] IX) and the aortic arch (CN X). Incoming visceral sensations are projected via lamina I of the spinal cord to the nucleus tractus solitarius and, from there, via the parabrachial region to the primary viscerosensitive cortex located in the insula. Efferent pathways (red tracings): The anterior cingulate and orbitofrontal cortex send projections to the hypothalamus and amygdala, but also to the autonomic centers within the brainstem: the periaqueductal gray matter, parabrachial nucleus, nucleus tractus solitarius, nucleus ambiguus, and rostral ventrolateral medulla. Parasympathetic influence on the heart arises primarily from the nucleus ambiguus. Sympathetic output receives tonic excitation from the ventrolateral medulla and projects from the intermediolateral cell columns to the cardiac conduction system and ventricle. Integrated response to emotion and stress (green arrows): The amygdala has reciprocal connections with the cerebral cortex and mediates autonomic response to emotions via projections to the hypothalamus and brainstem. The paraventricular nucleus of the hypothalamus controls internal homeostasis and innervates the autonomic relay centers in the rostral ventrolateral medulla, nucleus tractus solitarius, parabrachial nucleus, and preganglionic vagal and sympathetic neurons. Reprinted from Schuele et al,12 which was modified from Britton and Benarroch.13A tightly interconnected neuronal network controls various elements of the cardiovascular autonomic system. Consideration of a cardiac pathophysiology of SUDEP requires a fundamental understanding of this network (Figure 1).7,12,13 The insula, the anterior cingulate gyrus, and the ventromedial prefrontal cortex are key to central cortical control of autonomic function. The insula represents the primary viscerosensory cortex, whereas the cingulate gyrus and prefrontal cortices constitute the premotor autonomic region. At the subcortical level, the hypothalamus provides an interface with endocrine stimuli and triggers autonomic responses to maintain homeostasis. The amygdala, which is an integral component of the limbic system that links the previously mentioned cortical and subcortical centers, mediates the autonomic response to emotions. Beyond their centrality to autonomic control, the insula, amygdala, cingulate gyrus, and prefrontal cortex also represent the most common foci of partial epilepsy, a finding that might explain the frequent observation of autonomic changes in relation to epileptic seizures.14
EVIDENCE FOR A CARDIAC MECHANISM OF SUDEP
The most significant and broadly discussed cardiac mechanism of SUDEP is cardiac arrhythmia brought about by seizure discharges acting through the autonomic nervous system.5,7,15,16
Clinical evidence
A wide spectrum of cardiac arrhythmias—from ictal asystole to atrial fibrillation to repolarization abnormalities to bundle branch blocks—has been reported during seizures. 14–19 In one study, ictal cardiac arrhythmias occurred in 42% of hospitalized epilepsy patients, the most common being an irregular series of abrupt rate changes near the end of the electroencephalographic (EEG) seizure discharge.17 In another study, R-R interval analysis during the first 10-second period of EEG discharge showed a significant early heart rate increase in 49% of seizures and an early heart rate reduction in 25.5%.18
Certain clinical seizure characteristics have been correlated with the occurrence of ictal electrocardiographic (ECG) abnormalities. One study found that mean seizure duration was longer in patients with ECG abnormalities than in those without such changes.16 Others observed that ictal ECG abnormalities occurred more often and were more severe in generalized tonic-clonic seizures than in complex partial seizures.15,16,19 These same clinical seizure characteristics were correlated with a higher risk of SUDEP,20 which suggests an interrelation among seizure semiology, ECG abnormalities, and SUDEP.
Additionally, direct evidence of seizure-related cardiac changes occurring specifically in SUDEP victims has come from a study that compared EEG and ECG data between 21 SUDEP patients and 43 clinically similar historical controls with refractory partial epilepsy.15 The patients who eventually succumbed to SUDEP had a higher ictal maximal heart rate, a greater increase in heart rate with seizures arising from sleep, and a higher incidence of ictal cardiac repolarization and rhythm abnormalities.15
Experimental evidence
Electrical brain stimulation of the limbic system and insular cortex has repeatedly been shown to provoke heart rate changes, including bradycardia, tachycardia, and asystole.14 Some studies have even suggested a lateralized influence of the insulae on cardiovascular autonomic control, with intraoperative stimulation of the left posterior insula eliciting a cardioinhibitory response and hypotension, and with stimulation of the right anterior insula eliciting tachycardia and hypertension.7 Other studies have suggested that the limbic system has a localization-related influence on cardiovascular responses, with stimulation of the amygdala alone being insufficient to produce the ictal tachycardia so commonly seen in epileptic seizures, which suggests that cortical involvement is essential for the increase in heart rate.21 Such cortical stimulation–induced heart rate changes may explain how massive seizure-related discharges can affect cardiac rhythm during the seizure itself.
There is also, however, evidence of a baseline epilepsy-related autonomic dysfunction. Abnormalities in cardiac uptake of meta-iodobenzylguanidine (MIBG) have been demonstrated interictally in patients with chronic temporal lobe epilepsy relative to controls.22 A recent study specifically showed a pronounced reduction in cardiac MIBG uptake in patients who had ictal asystole compared with other epileptic patients and nonepileptic controls, a finding that suggests a postganglionic cardiac catecholamine disturbance in patients with epilepsy.23 The authors proposed that epilepsy-related impairment of sympathetic cardiac innervation limits adjustment and heart rate modulation, and may thus increase the risk of asystole and, ultimately, SUDEP.23
EPILEPSY SURGERY AND SUDEP RISK
The above findings make it reasonable to postulate that recurrent uncontrolled seizures may lead to significant cardiac changes—namely, arrhythmias—which may then lead to a higher risk of SUDEP. Successful epilepsy surgery, the treatment of choice for uncontrolled partial epilepsy, would eliminate seizures and would thus be expected to reduce SUDEP risk.
Several studies support this hypothesis. Reductions in all-cause mortality and in SUDEP following successful epilepsy surgery have been observed when patients who became seizure-free after surgery were compared either with patients who had ongoing postoperative seizures or with patients with intractable epilepsy who did not undergo epilepsy surgery.11,24 In one study with a mean follow-up of 3.82 years, no deaths occurred among 199 patients who were seizure-free following epilepsy surgery, whereas there were 11 deaths—including 6 cases of SUDEP—among 194 patients who had persistent seizures following epilepsy surgery.11 A separate study compared 202 patients who underwent epilepsy surgery with 46 patients with medically treated intractable epilepsy over a mean follow-up period of greater than 5.7 years.24 In this study, death occurred in only 7% of the surgical patients compared with 20% of the medically treated patients, which suggests that epilepsy surgery (or lack thereof) may be an independent predictor of mortality. Favorable outcome was linked closely to seizure control, as 81% of the patients who died had 2 or more seizures per year at last follow-up, compared with only 47% of survivors in the overall cohort.24
Complete seizure resolution—not just reduction—seems necessary for SUDEP risk reduction
In our experience at the Cleveland Clinic Epilepsy Center, we have found that a reduction in seizure frequency following epilepsy surgery is not enough to eliminate SUDEP risk—complete freedom from seizures is required. Of 37 SUDEP cases identified so far in a cohort of 3,481 patients evaluated in our epilepsy monitoring unit between 1990 and 2005 (Jehi et al, in preparation), 7 patients had undergone epilepsy surgery. None of these 7 patients was seizure-free at the time of death; four had a greater than 50% reduction in seizure frequency. No cases of SUDEP occurred in patients who were seizure-free after surgery.
Our findings mirror earlier results reported by Sperling et al,11 who also found that epilepsy surgery improves mortality only when seizures resolve completely, not when they merely “improve.” It seems plausible, therefore, that elimination of seizures postoperatively eliminates the risk for the several seizure-related cardiac arrhythmogenic changes discussed above, thereby eliminating the risk for the series of events that eventually leads to SUDEP.
A role for stabilized baseline autonomic control?
Alternatively, some data suggest that the “autonomic” impacts of epilepsy surgery may extend beyond the immediate seizure-related manifestations to the baseline interictal period in epilepsy patients. One study found that surgery for temporal lobe epilepsy is followed by a reduction of sympathetic cardiovascular modulation and baroreflex sensitivity.25 The authors proposed that this finding may be attributable to decreased influences of interictal epileptogenic discharges on brain areas involved in cardiovascular autonomic control. These researchers continue to postulate that surgery for temporal lobe epilepsy seems to stabilize cardiovascular control in epilepsy patients by reducing the risk of sympathetically mediated tachyarrhythmias and excessive bradycardiac counterregulation, potentially lowering SUDEP risk.25
CARDIAC FINDINGS, SUDEP, AND SEIZURE OUTCOMES
Whether it is the elimination of seizure-related cardiac arrhythmias achieved by rendering patients seizure-free after surgery, or whether it is the stabilization of baseline autonomic cardiac control by reducing interictal epileptiform discharges, this line of thought assumes that the autonomic dysfunction contributing to SUDEP is caused by epilepsy and that freedom from seizures following epilepsy surgery should therefore be responsible for reducing the risk of death. An alternative hypothesis for the infrequent occurrence of SUDEP in seizure-free patients may be that seizure outcomes following epilepsy surgery and SUDEP risk are actually both governed by the same underlying biologic process. This would suggest that the same patient cohort is at a higher baseline mortality risk and in a prognostically poorer seizure outcome group following epilepsy surgery.
This idea is supported by a recent prospective study among 21 consecutive candidates for temporal lobe epilepsy surgery in which spectral analysis of heart rate variability performed preoperatively was correlated with seizure control 1 year after surgery.26 The study found that patients with poor seizure outcomes (Engel class II to IV, signifying ongoing postoperative seizures) had significantly lower power in all domains of heart rate variability than did patients with favorable seizure outcomes.26
In another study, heart rate was recorded in 16 patients before and after temporal lobe epilepsy surgery, and sympathetic and parasympathetic cardiac modulation was determined as powers of low-frequency (LF, 0.04–0.15 Hz) and high-frequency (HF, 0.15–0.5 Hz) heart rate oscillations.27 The LF/HF ratio was calculated as an index of sympathovagal balance. Cardiac MIBG uptake was measured with MIBG single-photon emission computed tomography and compared with control data. Baseline sympathetic LF power and LF/HF ratio were higher in patients who eventually had persistent seizures than in those who became seizure-free. Following surgery, both measures decreased in seizure-free patients but increased in patients with persistent seizures. MIBG uptake was lower in patients than in controls and even lower yet in patients who had persistent seizures. In this subgroup, MIBG uptake declined further after surgery.
Essentially, both of the above studies26,27 demonstrate findings of autonomic cardiac abnormalities that predated epilepsy surgery and reliably predicted the eventual surgical outcome in terms of seizure continuance. This suggests that “poor candidates” for epilepsy surgery—ie, those with lower chances of achieving seizure freedom with surgery—may a priori have a higher SUDEP risk. A possible explanation for these findings may be epileptogenic zones, including the insula and other components of the central autonomic network, or molecular/genetic diffuse abnormalities that extend beyond a limited surgically removable seizure focus and involve the heart, increasing the risk for cardiac conduction abnormalities.
CONCLUSIONS
SUDEP is the most common cause of death in epilepsy patients. A significant body of literature suggests that a cardiac mechanism contributes to its occurrence. Although the exact relationship between seizure outcomes following epilepsy surgery and SUDEP risk is still being investigated, it is accepted that seizure control correlates with reduced mortality. Cardiac changes and autonomic dysregulation seem to be at the “heart” of the problem.
References
Nashef L, Hindocha N, Makoff A. Risk factors in sudden death in epilepsy (SUDEP): the quest for mechanisms. Epilepsia2007; 48:859–871.
Nilsson L, Tomson T, Farahmand BY, et al Cause-specific mortality in epilepsy: a cohort study of more than 9,000 patients once hospitalized for epilepsy. Epilepsia1997; 38:1062–1068.
Tennis P, Cole TB, Annegers JF, et al Cohort study of incidence of sudden unexplained death in persons with seizure disorder treated with antiepileptic drugs in Saskatchewan, Canada. Epilepsia1995; 36:29–36.
Leestma JE, Annegers JF, Brodie MJ, et al Sudden unexplained death in epilepsy: observations from a large clinical development program. Epilepsia1997; 38:47–55.
Tomson T, Nashef L, Ryvlin P. Sudden unexpected death in epilepsy: current knowledge and future directions. Lancet Neurol2008; 7:1021–1031.
Nashef L, Fish DR, Sander JW, Shorvon SD. Incidence of sudden unexpected death in an adult outpatient cohort with epilepsy at a tertiary referral centre. J Neurol Neurosurg Psychiatry1995; 58:462–464.
Jehi L, Najm IM. Sudden unexpected death in epilepsy: impact, mechanisms, and prevention. Cleve Clin J Med2008; 75( suppl 2):S66–S70.
Lhatoo SD, Johnson AL, Goodridge DM, et al Mortality in epilepsy in the first 11 to 14 years after diagnosis: multivariate analysis of a long-term, prospective, population-based cohort. Ann Neurol2001; 49:336–344.
Pedley TA, Hauser WA. Sudden death in epilepsy: a wake-up call for management. Lancet2002; 359:1790–1791.
Schraeder PL, Delin K, McClelland RL, So EL. Coroner and medical examiner documentation of sudden unexplained deaths in epilepsy. Epilepsy Res2006; 68:137–143.
Sperling MR, Feldman H, Kinman J, et al Seizure control and mortality in epilepsy. Ann Neurol1999; 46:45–50.
Schuele SU, Widdess-Walsh P, Bermeo A, Lüders HO. Sudden unexplained death in epilepsy: the role of the heart. Cleve Clin J Med2007; 74( suppl 1):S121–S127.
Britton JW, Benarroch E. Seizures and syncope: anatomic basis and diagnostic considerations. Clin Auton Res2006; 16:18–28.
Leung H, Kwan P, Elger CE. Finding the missing link between ictal bradyarrhythmia, ictal asystole, and sudden unexpected death in epilepsy. Epilepsy Behav2006; 9:19–30.
Nei M, Ho RT, Abou-Khalil BW, et al EEG and ECG in sudden unexplained death in epilepsy. Epilepsia2004; 45:338–345.
Nei M, Ho RT, Sperling MR. EKG abnormalities during partial seizures in refractory epilepsy. Epilepsia2000; 41:542–548.
Blumhardt LD, Smith PE, Owen L. Electrocardiographic accompaniments of temporal lobe epileptic seizures. Lancet1986; 1:1051–1056.
Galimberti CA, Marchioni E, Barzizza F, et al Partial epileptic seizures of different origin variably affect cardiac rhythm. Epilepsia1996; 37:742–747.
Opherk C, Coromilas J, Hirsch LJ. Heart rate and EKG changes in 102 seizures: analysis of influencing factors. Epilepsy Res2002; 52:117–127.
Langan Y, Nashef L, Sander JW. Case-control study of SUDEP. Neurology2005; 64:1131–1133.
Keilson MJ, Hauser WA, Magrill JP, Goldman M. ECG abnormalities in patients with epilepsy. Neurology1987; 37:1624–1626.
Druschky A, Hilz MJ, Hopp P, et al Interictal cardiac autonomic dysfunction in temporal lobe epilepsy demonstrated by [123I] metaiodobenzylguanidine-SPECT. Brain2001; 124:2372–2382.
Kerling F, Dütsch M, Linke R, Kuwert T, Stefan H, Hilz MJ. Relation between ictal asystole and cardiac sympathetic dysfunction shown by MIBG-SPECT. Acta Neurol Scand2009; 120:123–129.
Vickrey BG, Hays RD, Rausch R, et al Outcomes in 248 patients who had diagnostic evaluations for epilepsy surgery. Lancet1995; 346:1445–1449.
Hilz MJ, Devinsky O, Doyle W, et al Decrease of sympathetic cardiovascular modulation after temporal lobe epilepsy surgery. Brain2002; 125:985–995.
Persson H, Kumlien E, Ericson M, Tomson T. Preoperative heart rate variability in relation to surgery outcome in refractory epilepsy. Neurology2005; 65:1021–1025.
Hilz MJ, Platsch G, Druschky K, et al Outcome of epilepsy surgery correlates with sympathetic modulation and neuroimaging of the heart. J Neurol Sci2003; 216:153–162.
References
Nashef L, Hindocha N, Makoff A. Risk factors in sudden death in epilepsy (SUDEP): the quest for mechanisms. Epilepsia2007; 48:859–871.
Nilsson L, Tomson T, Farahmand BY, et al Cause-specific mortality in epilepsy: a cohort study of more than 9,000 patients once hospitalized for epilepsy. Epilepsia1997; 38:1062–1068.
Tennis P, Cole TB, Annegers JF, et al Cohort study of incidence of sudden unexplained death in persons with seizure disorder treated with antiepileptic drugs in Saskatchewan, Canada. Epilepsia1995; 36:29–36.
Leestma JE, Annegers JF, Brodie MJ, et al Sudden unexplained death in epilepsy: observations from a large clinical development program. Epilepsia1997; 38:47–55.
Tomson T, Nashef L, Ryvlin P. Sudden unexpected death in epilepsy: current knowledge and future directions. Lancet Neurol2008; 7:1021–1031.
Nashef L, Fish DR, Sander JW, Shorvon SD. Incidence of sudden unexpected death in an adult outpatient cohort with epilepsy at a tertiary referral centre. J Neurol Neurosurg Psychiatry1995; 58:462–464.
Jehi L, Najm IM. Sudden unexpected death in epilepsy: impact, mechanisms, and prevention. Cleve Clin J Med2008; 75( suppl 2):S66–S70.
Lhatoo SD, Johnson AL, Goodridge DM, et al Mortality in epilepsy in the first 11 to 14 years after diagnosis: multivariate analysis of a long-term, prospective, population-based cohort. Ann Neurol2001; 49:336–344.
Pedley TA, Hauser WA. Sudden death in epilepsy: a wake-up call for management. Lancet2002; 359:1790–1791.
Schraeder PL, Delin K, McClelland RL, So EL. Coroner and medical examiner documentation of sudden unexplained deaths in epilepsy. Epilepsy Res2006; 68:137–143.
Sperling MR, Feldman H, Kinman J, et al Seizure control and mortality in epilepsy. Ann Neurol1999; 46:45–50.
Schuele SU, Widdess-Walsh P, Bermeo A, Lüders HO. Sudden unexplained death in epilepsy: the role of the heart. Cleve Clin J Med2007; 74( suppl 1):S121–S127.
Britton JW, Benarroch E. Seizures and syncope: anatomic basis and diagnostic considerations. Clin Auton Res2006; 16:18–28.
Leung H, Kwan P, Elger CE. Finding the missing link between ictal bradyarrhythmia, ictal asystole, and sudden unexpected death in epilepsy. Epilepsy Behav2006; 9:19–30.
Nei M, Ho RT, Abou-Khalil BW, et al EEG and ECG in sudden unexplained death in epilepsy. Epilepsia2004; 45:338–345.
Nei M, Ho RT, Sperling MR. EKG abnormalities during partial seizures in refractory epilepsy. Epilepsia2000; 41:542–548.
Blumhardt LD, Smith PE, Owen L. Electrocardiographic accompaniments of temporal lobe epileptic seizures. Lancet1986; 1:1051–1056.
Galimberti CA, Marchioni E, Barzizza F, et al Partial epileptic seizures of different origin variably affect cardiac rhythm. Epilepsia1996; 37:742–747.
Opherk C, Coromilas J, Hirsch LJ. Heart rate and EKG changes in 102 seizures: analysis of influencing factors. Epilepsy Res2002; 52:117–127.
Langan Y, Nashef L, Sander JW. Case-control study of SUDEP. Neurology2005; 64:1131–1133.
Keilson MJ, Hauser WA, Magrill JP, Goldman M. ECG abnormalities in patients with epilepsy. Neurology1987; 37:1624–1626.
Druschky A, Hilz MJ, Hopp P, et al Interictal cardiac autonomic dysfunction in temporal lobe epilepsy demonstrated by [123I] metaiodobenzylguanidine-SPECT. Brain2001; 124:2372–2382.
Kerling F, Dütsch M, Linke R, Kuwert T, Stefan H, Hilz MJ. Relation between ictal asystole and cardiac sympathetic dysfunction shown by MIBG-SPECT. Acta Neurol Scand2009; 120:123–129.
Vickrey BG, Hays RD, Rausch R, et al Outcomes in 248 patients who had diagnostic evaluations for epilepsy surgery. Lancet1995; 346:1445–1449.
Hilz MJ, Devinsky O, Doyle W, et al Decrease of sympathetic cardiovascular modulation after temporal lobe epilepsy surgery. Brain2002; 125:985–995.
Persson H, Kumlien E, Ericson M, Tomson T. Preoperative heart rate variability in relation to surgery outcome in refractory epilepsy. Neurology2005; 65:1021–1025.
Hilz MJ, Platsch G, Druschky K, et al Outcome of epilepsy surgery correlates with sympathetic modulation and neuroimaging of the heart. J Neurol Sci2003; 216:153–162.
Biofeedback is a self-regulation therapy that aims to teach individuals the skills that will allow them to change their physiology in healthy directions.1–3 Biofeedback involves a client, a trained biofeedback coach, and appropriate instrumentation. Sensors are connected to the client, and various physiologic parameters (such as heart rate, blood pressure, and digital peripheral temperature) are displayed on a computer screen. The client is guided through a brief mental stress test and a relaxation exercise to learn to recognize differences between hyperarousal and a more relaxed physiology. Biofeedback training involves a series of sessions in which the goal is to help the client gain control of his or her own physiology by learning relaxation techniques such as deep breathing, progressive muscle relaxation, and guided imagery.1,3,4 Although biofeedback can be used solely as operant conditioning, it is more commonly and more effectively combined with techniques of stress management.
Biofeedback training is commonly (although not exclusively) used to decrease activation of the sympathetic branch of the autonomic nervous system (the “fight or flight” response). The reduction in sympathetic nervous system (SNS) activity is manifest as an increase in digital peripheral temperature and decreases in skin conductance, heart rate, and blood pressure, as well as changes in the frequency distribution of heart rate variability. While the SNS is becoming less activated, the parasympathetic portion of the autonomic nervous system (“rest and digest”) is becoming more involved in regulating body functions. More parasympathetic nervous system (PNS) activation and less SNS activation produces a healthier physiologic state, and thus biofeedback can be used to move the body in the direction of health and wellness.1–4
HEART FAILURE: BIOLOGIC MECHANISMS OF INJURY
Heart failure is the end result of most untreated cardiovascular diseases. Heart failure involves inadequate cardiac pump function, such that appropriate perfusion of end organs does not occur. The process of developing heart failure is a gradual one that begins with compensatory processes. In response to an injury or insult, such as chronic high blood pressure or long-standing coronary artery blockage, the heart compensates by activating various neurohormonal pathways in an attempt to preserve cardiac function and end-organ perfusion.5 When these pathways are activated, they initially help the heart to compensate for the ongoing challenge of increased pressure or decreased tissue oxygenation and allow the cardiovascular system to pump sufficient blood. Over time, however, these compensatory processes become maladaptive. Cellular signaling pathways, which were activated in order to help the heart compensate, actually become as much of a problem as the decreased cardiac function.5
Hyperactivation of the SNS
Chief among these pathways is the SNS, which is the most powerful means by which cardiac function can be augmented.5 In response to decreased cardiac function, cardiac sympathetic nerves are activated, releasing norepinephrine locally, and both norepinephrine and epinephrine increase in the circulating blood. Beta-adrenergic receptors on cardiac myocytes and on vascular smooth muscle cells are stimulated, and the resulting augmentation of cardiac contraction helps the heart to overcome an immediate challenge. If the insult or injury to the heart is acute and time-limited, this system compensates and the situation is resolved. However, chronic activation of the SNS creates more problems than it solves for the failing heart,5,6 including the following:
Myocardial cells are challenged by the need for increased energy production to support the chronic stimulation
Oxidative stress ensues
Receptors are downregulated
Pathways that result in necrosis and apoptosis are activated
Myofilament proteins respond to chronically elevated intracellular calcium.
As a result, the heart begins to spiral more quickly into a decompensated state. The toxicity of SNS overactivation is the reason for the success of beta-adrenergic blocking drugs in treating heart failure, but this situation is complicated further by adrenergic receptor polymorphisms and nonhomogeneous responses to beta-blocking agents.6 It is safe to say that the goal of much heart failure therapy is inactivation of the once-compensatory SNS and its resulting biologic effects.
Hypoactivation of the PNS
In addition to hyperactivation of the SNS, heart failure is also accompanied by a decrease in the role of the PNS. Under normal resting conditions, the human heart is governed more by the PNS than the SNS, with the SNS becoming a major source of cardiac control only during periods of decreased cardiac function. In heart failure, however, this relationship is reversed, with the SNS taking over the governing role and PNS input becoming less significant. Studies have suggested that the lack of contribution of the PNS to cardiac regulation in heart failure may be as deleterious as overactivation of the SNS.7 Most recently, stimulation of the vagal nerve has been shown to be beneficial in both animal models8 and humans with heart failure,9 confirming that augmenting PNS activity may be as important as inhibiting SNS activity. Although vagal nerve stimulation may be the first heart failure therapy aimed specifically at the PNS, it is likely that the future will hold more therapies with this goal.
PNS as regulator of inflammation of the failing heart?
It has recently been suggested that beyond its role in regulating cardiac function under baseline conditions, the PNS may participate in regulating the inflammatory state of the failing heart. It has been established since the observations of Packer and colleagues in the early 1990s10 that proinflammatory cytokines such as tumor necrosis factor–alpha, interleukin-6, and interleukin-1 are elevated in the circulation of heart failure patients, that these cytokines are correlated with clinical prognosis, and that they play a role in the activation of deleterious cardiac signaling pathways.11,12 Trials of antiinflammatory therapies in heart failure have been less than successful, but this may be because the complexity of the activation has been underestimated.11 In elegant work reported several years ago, Kevin Tracey’s group showed that stimulation of the vagus nerve could inhibit inflammatory processes associated with sepsis.13,14 Since that time, the reflex activation and inactivation of inflammatory processes by the PNS have become more widely accepted. Although it has not yet been directly demonstrated, it is possible that part of the benefit of vagal nerve stimulation in heart failure will prove to be due to its ability to reduce the chronic inflammatory state of the failing heart.
BIOFEEDBACK IN HEART FAILURE: RATIONALE
The failing heart is characterized by autonomic imbalance (hyperactivation of the SNS and hypoactivation of the PNS) and by a chronic inflammatory state. It has been hypothesized both directly and indirectly that these two major pathophysiologic processes may be intertwined.13–15 Biofeedback-assisted stress management is a therapy that has the potential to interfere with both processes. If the patient with heart failure can be trained to reduce activation of the SNS and to increase control by the PNS, it is likely that the negative consequences of autonomic imbalance will be decreased or possibly even reversed. Whether these effects are limited to quality of life and clinical status, or whether they extend to an effect on myocardial remodeling processes, remains to be established. Since the chronic inflammatory state can also be affected by increasing PNS control of the cardiovascular system, we further hypothesize that biofeedback training may have a direct effect on the inflammatory processes involved in the downward spiral of heart failure.
BIOFEEDBACK IN HEART FAILURE: STUDIES
We are certainly not the first group to hypothesize that self-regulation may have a role in the treatment of cardiovascular diseases in general or heart failure in particular. It has been shown that patients with heart failure manage their disease better and experience less emotional distress when they have a greater sense of control over their condition.16 In addition to giving patients a greater sense of control, some mind-body therapies have been shown to be beneficial in those with heart failure. Pischke et al showed as part of the Multicenter Lifestyle Demonstration Project that patients with left ventricular ejection fractions in the range associated with heart failure (≤ 40%) were able to learn and benefit from stress management techniques equally as well as those with more normal cardiac function.17 Both relaxation training18,19 and meditation20 have been shown to improve quality of life in heart failure patients, but meditation also reduced circulating norepinephrine, a marker of SNS activation.20 Mindfulness training improved clinical symptoms of heart failure and also reduced both anxiety and depression in patients with heart failure.21 Training heart failure patients to breathe more slowly is an intervention that is normally part of biofeedback training, but even when used alone it has resulted in decreased dyspnea,22 increased oxygen saturation,23 and improved exercise tolerance.22,23
To our knowledge, three studies to date have specifically used biofeedback training in patients with documented heart failure. As early as 1997, Moser and colleagues showed that heart failure patients were able to raise their finger temperature in spite of disease-related vascular changes, and that a single session of finger temperature biofeedback resulted in meaningful clinical improvement.24 Luskin et al randomized 33 heart failure patients to either biofeedback-assisted stress management or a control group, and showed improvement with the intervention in perceived stress, emotional distress, exercise tolerance, and depression.25 Most recently, Swanson and colleagues demonstrated improved exercise tolerance after cardiorespiratory biofeedback in patients with higher left ventricular ejection fractions (≥ 31%), although improvement could not be accomplished in those with ejection fractions below 30%.26
ONGOING STUDY IN END-STAGE HEART FAILURE AND FUTURE DIRECTIONS
We are currently involved at Cleveland Clinic in a study of end-stage heart failure patients who are awaiting cardiac transplantation. Each patient is provided with eight sessions of biofeedback training, including respiratory rate, digital peripheral temperature, muscle tension, and heart rate variability. Clinical status, quality of life, and heart failure–specific symptoms are being monitored throughout the training period. Success with biofeedback training is being analyzed, and we are testing the hypothesis that the degree of success in learning self-regulation will predict change in clinical status, quality of life, and the biology of the heart. What is unique to our study is that we will obtain the heart tissue at explant, when the patient receives a cardiac transplant, and we will conduct experiments to determine whether the cellular and molecular phenotype of the heart have been changed by the intervention, particularly components of the SNS, PNS, and inflammatory pathways.
We have been studying human heart failure for many years, and we have previously shown the changes in receptors and signaling pathways that occur in the failing human heart.27–30 We were also among the first to demonstrate that the cellular and molecular changes that occur in the failing human heart are not actually irreversible but can be changed by interventions such as a left ventricular assist device.31–33 Thus we hypothesize that biofeedback training, by interfering with overactivation of the SNS and by allowing the PNS to more adequately contribute to cardiac regulation, will have a meaningful effect on the biology of the failing human heart in addition to improving clinical status and quality of life. We hope to be among the first to demonstrate that effect.
References
McKee MG. Biofeedback: an overview in the context of heart-brain medicine. Cleve Clin J Med2008; 75( suppl 2):S31–S34.
Moravec CS. Biofeedback therapy in cardiovascular disease: rationale and research overview. Cleve Clin J Med2008; 75( suppl 2):S35–S38.
Moss D, McGrady A, Davies TC, Wickramasekera I. Handbook of Mind-Body Medicine for Primary Care. London: Sage Publications; 2003.
Schwartz MS, Andrasik F. Biofeedback: A Practitioner’s Guide. 3rd ed. New York, NY: Guilford Press; 2003.
Triposkiadis F, Karayannis G, Giamouzis G, Skoularigis J, Louridas G, Butler J. The sympathetic nervous system in heart failure. J Am Coll Cardiol2009; 54:1747–1762.
Floras JS. Sympathetic nervous system activation in human heart failure. J Am Coll Cardiol2009; 54:375–385.
Binkley PF, Nunziata E, Haas GJ, Nelson SD, Cody RJ. Parasympathetic withdrawal is an integral component of autonomic imbalance in congestive heart failure: demonstration in human subjects and verification in a paced canine model of ventricular failure. J Am Coll Cardiol1991; 18:464–472.
Li M, Zheng C, Sato T, Kawada T, Sugimachi M, Sunugawa K. Vagal nerve stimulation markedly improves long-term survival after chronic heart failure in rats. Circulation2004; 109:120–124.
Schwartz PJ, DeFerrari GM, Sanzo A, et al Long term vagal stimulation in patients with advanced heart failure: first experience in man. Eur J Heart Failure2008; 10:884–891.
Levine B, Kalman J, Mayer L, Fillit M, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med1990; 323:236–241.
Mann DL. Inflammatory mediators and the failing heart: past, present and the foreseeable future. Circ Res2002; 91:988–998.
Parish RC, Evans JD. Inflammation in chronic heart failure. Ann Pharmacother2008; 42:1002–1016.
Tracey KJ. The inflammatory reflex. Nature2002; 420:853–859.
Tracey KJ. Reflex control of immunity. Nat Rev Immunol2009; 9:418–428.
Dracup K, Westlake C, Erickson VS, Moser DK, Caldwell ML, Hamilton MA. Perceived control reduces emotional stress in patients with heart failure. J Heart Lung Transplant2003; 22:90–93.
Pischke CR, Weidner G, Elliott-Eller M, Ornish D. Lifestyle changes and clinical profile in coronary heart disease patients with an ejection fraction of ≤ 40% or > 40% in the Multicenter Lifestyle Demonstration Project. Eur J Heart Failure2007; 9:928–934.
Chang BH, Henricks A, Zhao Y, Rothendler JA, LoCastro JS, Slawsky MT. A relaxation response randomized trial on patients with chronic heart failure. J Cardiopulm Rehab2005; 25:149–157.
Yu DSF, Lee DTF, Woo J. Effects of relaxation therapy on psychologic distress and symptom status in older Chinese patients with heart failure. Psychosom Res2007;427–437.
Curiati JA, Bocchi E, Freire JO, et al Meditation reduces sympathetic activation and improves the quality of life in elderly patients with optimally treated heart failure: a prospective randomized study. J Altern Complement Med2005; 11:465–472.
Sullivan MJ, Wood L, Terry J, et al The support, education and research in chronic heart failure study (SEARCH): a mindfulness-based psychoeducational intervention improves depression and clinical symptoms in patients with chronic heart failure. Am Heart J2009; 157:84–90.
Weiner P, Waizman J, Magadle R, Berar-Yanay N, Pelled B. The effect of specific inspiratory muscle training on the sensation of dyspnea and exercise tolerance in patients with congestive heart failure. Clin Cardiol1999; 22:727–732.
Bernardi L, Porta C, Spicuzza L, et al Slow breathing increases arterial baroreflex sensitivity in patients with chronic heart failure. Circulation2002; 105:143–145.
Moser DK, Dracup K, Woo MA, Stevenson LW. Voluntary control of vascular tone by using skin-temperature biofeedback-relaxation in patients with advanced heart failure. Altern Ther Health Med1997; 3:51–60.
Luskin F, Reitz M, Newell K, Quinn TG, Haskell W. A controlled pilot study of stress management training of elderly patients with congestive heart failure. Prev Cardiol2002; 5:168–172.
Swanson KS, Gevirtz RN, Brown M, Spira J, Guarneri E, Stoletniy L. The effect of biofeedback on function in patients with heart failure. Appl Psychophysiol Biofeedback2009; 34:71–91.
Razeghi P, Mukhopadhyay M, Myers TJ, et al Myocardial tumor necrosis factor alpha expression does not correlate with clinical indices of heart failure in patients on left ventricular assist device support. Ann Thorac Surg2001; 72:2044–2050.
Matteo R, Moravec CS. Expression and immunolocalization of annexins IV, V and VI in the failing and non-failing human heart. Cardiovasc Res2000; 45:961–970.
Dash R, Frank K, Carr AN, Moravec CS, Kranias EG. Gender influences on sarcoplasmic reticulum calcium handling in the failing human myocardium. J Mol Cell Cardiol2001; 33:1345–1353.
DiPaola NR, Sweet WE, Stull LB, Francis GS, Moravec CS. Beta-adrenergic receptors and calcium cycling proteins in normal, hypertrophied and failing human hearts: transition from hypertrophy to failure. J Mol Cell Cardiol2001; 33:1283–1295.
Aquila LA, McCarthy PM, Smedira NG, Young JB, Moravec CS. Cytoskeletal structure and recovery in single human cardiac myocytes. J Heart Lung Transplant2004; 23:954–963.
Fedak PWM, Moravec CS, McCarthy PM, et al Altered expression of disintegrin metalloproteinases and their inhibitor in human dilated cardiomyopathy. Circulation2006; 113:238–245.
Ogletree-Hughes ML, Stull LB, Sweet WE, Smedira NG, McCarthy PM, Moravec CS. Mechanical unloading restores beta adrenergic responsiveness and reverses receptor downregulation in the failing human heart. Circulation2001; 104:881–886.
Michael G. McKee, PhD Department of Psychiatry and Psychology, Cleveland Clinic, Cleveland, OH
Christine S. Moravec, PhD Department of Cardiovascular Medicine, Cleveland Clinic, Cleveland, OH
Correspondence: Michael G. McKee, PhD, Department of Psychiatry and Psychology, Cleveland Clinic, 9500 Euclid Avenue, P57, Cleveland, OH 44195; [email protected]
Both authors reported that they have no financial relationships that pose a potential conflict of interest with this article.
Michael G. McKee, PhD Department of Psychiatry and Psychology, Cleveland Clinic, Cleveland, OH
Christine S. Moravec, PhD Department of Cardiovascular Medicine, Cleveland Clinic, Cleveland, OH
Correspondence: Michael G. McKee, PhD, Department of Psychiatry and Psychology, Cleveland Clinic, 9500 Euclid Avenue, P57, Cleveland, OH 44195; [email protected]
Both authors reported that they have no financial relationships that pose a potential conflict of interest with this article.
Author and Disclosure Information
Michael G. McKee, PhD Department of Psychiatry and Psychology, Cleveland Clinic, Cleveland, OH
Christine S. Moravec, PhD Department of Cardiovascular Medicine, Cleveland Clinic, Cleveland, OH
Correspondence: Michael G. McKee, PhD, Department of Psychiatry and Psychology, Cleveland Clinic, 9500 Euclid Avenue, P57, Cleveland, OH 44195; [email protected]
Both authors reported that they have no financial relationships that pose a potential conflict of interest with this article.
Biofeedback is a self-regulation therapy that aims to teach individuals the skills that will allow them to change their physiology in healthy directions.1–3 Biofeedback involves a client, a trained biofeedback coach, and appropriate instrumentation. Sensors are connected to the client, and various physiologic parameters (such as heart rate, blood pressure, and digital peripheral temperature) are displayed on a computer screen. The client is guided through a brief mental stress test and a relaxation exercise to learn to recognize differences between hyperarousal and a more relaxed physiology. Biofeedback training involves a series of sessions in which the goal is to help the client gain control of his or her own physiology by learning relaxation techniques such as deep breathing, progressive muscle relaxation, and guided imagery.1,3,4 Although biofeedback can be used solely as operant conditioning, it is more commonly and more effectively combined with techniques of stress management.
Biofeedback training is commonly (although not exclusively) used to decrease activation of the sympathetic branch of the autonomic nervous system (the “fight or flight” response). The reduction in sympathetic nervous system (SNS) activity is manifest as an increase in digital peripheral temperature and decreases in skin conductance, heart rate, and blood pressure, as well as changes in the frequency distribution of heart rate variability. While the SNS is becoming less activated, the parasympathetic portion of the autonomic nervous system (“rest and digest”) is becoming more involved in regulating body functions. More parasympathetic nervous system (PNS) activation and less SNS activation produces a healthier physiologic state, and thus biofeedback can be used to move the body in the direction of health and wellness.1–4
HEART FAILURE: BIOLOGIC MECHANISMS OF INJURY
Heart failure is the end result of most untreated cardiovascular diseases. Heart failure involves inadequate cardiac pump function, such that appropriate perfusion of end organs does not occur. The process of developing heart failure is a gradual one that begins with compensatory processes. In response to an injury or insult, such as chronic high blood pressure or long-standing coronary artery blockage, the heart compensates by activating various neurohormonal pathways in an attempt to preserve cardiac function and end-organ perfusion.5 When these pathways are activated, they initially help the heart to compensate for the ongoing challenge of increased pressure or decreased tissue oxygenation and allow the cardiovascular system to pump sufficient blood. Over time, however, these compensatory processes become maladaptive. Cellular signaling pathways, which were activated in order to help the heart compensate, actually become as much of a problem as the decreased cardiac function.5
Hyperactivation of the SNS
Chief among these pathways is the SNS, which is the most powerful means by which cardiac function can be augmented.5 In response to decreased cardiac function, cardiac sympathetic nerves are activated, releasing norepinephrine locally, and both norepinephrine and epinephrine increase in the circulating blood. Beta-adrenergic receptors on cardiac myocytes and on vascular smooth muscle cells are stimulated, and the resulting augmentation of cardiac contraction helps the heart to overcome an immediate challenge. If the insult or injury to the heart is acute and time-limited, this system compensates and the situation is resolved. However, chronic activation of the SNS creates more problems than it solves for the failing heart,5,6 including the following:
Myocardial cells are challenged by the need for increased energy production to support the chronic stimulation
Oxidative stress ensues
Receptors are downregulated
Pathways that result in necrosis and apoptosis are activated
Myofilament proteins respond to chronically elevated intracellular calcium.
As a result, the heart begins to spiral more quickly into a decompensated state. The toxicity of SNS overactivation is the reason for the success of beta-adrenergic blocking drugs in treating heart failure, but this situation is complicated further by adrenergic receptor polymorphisms and nonhomogeneous responses to beta-blocking agents.6 It is safe to say that the goal of much heart failure therapy is inactivation of the once-compensatory SNS and its resulting biologic effects.
Hypoactivation of the PNS
In addition to hyperactivation of the SNS, heart failure is also accompanied by a decrease in the role of the PNS. Under normal resting conditions, the human heart is governed more by the PNS than the SNS, with the SNS becoming a major source of cardiac control only during periods of decreased cardiac function. In heart failure, however, this relationship is reversed, with the SNS taking over the governing role and PNS input becoming less significant. Studies have suggested that the lack of contribution of the PNS to cardiac regulation in heart failure may be as deleterious as overactivation of the SNS.7 Most recently, stimulation of the vagal nerve has been shown to be beneficial in both animal models8 and humans with heart failure,9 confirming that augmenting PNS activity may be as important as inhibiting SNS activity. Although vagal nerve stimulation may be the first heart failure therapy aimed specifically at the PNS, it is likely that the future will hold more therapies with this goal.
PNS as regulator of inflammation of the failing heart?
It has recently been suggested that beyond its role in regulating cardiac function under baseline conditions, the PNS may participate in regulating the inflammatory state of the failing heart. It has been established since the observations of Packer and colleagues in the early 1990s10 that proinflammatory cytokines such as tumor necrosis factor–alpha, interleukin-6, and interleukin-1 are elevated in the circulation of heart failure patients, that these cytokines are correlated with clinical prognosis, and that they play a role in the activation of deleterious cardiac signaling pathways.11,12 Trials of antiinflammatory therapies in heart failure have been less than successful, but this may be because the complexity of the activation has been underestimated.11 In elegant work reported several years ago, Kevin Tracey’s group showed that stimulation of the vagus nerve could inhibit inflammatory processes associated with sepsis.13,14 Since that time, the reflex activation and inactivation of inflammatory processes by the PNS have become more widely accepted. Although it has not yet been directly demonstrated, it is possible that part of the benefit of vagal nerve stimulation in heart failure will prove to be due to its ability to reduce the chronic inflammatory state of the failing heart.
BIOFEEDBACK IN HEART FAILURE: RATIONALE
The failing heart is characterized by autonomic imbalance (hyperactivation of the SNS and hypoactivation of the PNS) and by a chronic inflammatory state. It has been hypothesized both directly and indirectly that these two major pathophysiologic processes may be intertwined.13–15 Biofeedback-assisted stress management is a therapy that has the potential to interfere with both processes. If the patient with heart failure can be trained to reduce activation of the SNS and to increase control by the PNS, it is likely that the negative consequences of autonomic imbalance will be decreased or possibly even reversed. Whether these effects are limited to quality of life and clinical status, or whether they extend to an effect on myocardial remodeling processes, remains to be established. Since the chronic inflammatory state can also be affected by increasing PNS control of the cardiovascular system, we further hypothesize that biofeedback training may have a direct effect on the inflammatory processes involved in the downward spiral of heart failure.
BIOFEEDBACK IN HEART FAILURE: STUDIES
We are certainly not the first group to hypothesize that self-regulation may have a role in the treatment of cardiovascular diseases in general or heart failure in particular. It has been shown that patients with heart failure manage their disease better and experience less emotional distress when they have a greater sense of control over their condition.16 In addition to giving patients a greater sense of control, some mind-body therapies have been shown to be beneficial in those with heart failure. Pischke et al showed as part of the Multicenter Lifestyle Demonstration Project that patients with left ventricular ejection fractions in the range associated with heart failure (≤ 40%) were able to learn and benefit from stress management techniques equally as well as those with more normal cardiac function.17 Both relaxation training18,19 and meditation20 have been shown to improve quality of life in heart failure patients, but meditation also reduced circulating norepinephrine, a marker of SNS activation.20 Mindfulness training improved clinical symptoms of heart failure and also reduced both anxiety and depression in patients with heart failure.21 Training heart failure patients to breathe more slowly is an intervention that is normally part of biofeedback training, but even when used alone it has resulted in decreased dyspnea,22 increased oxygen saturation,23 and improved exercise tolerance.22,23
To our knowledge, three studies to date have specifically used biofeedback training in patients with documented heart failure. As early as 1997, Moser and colleagues showed that heart failure patients were able to raise their finger temperature in spite of disease-related vascular changes, and that a single session of finger temperature biofeedback resulted in meaningful clinical improvement.24 Luskin et al randomized 33 heart failure patients to either biofeedback-assisted stress management or a control group, and showed improvement with the intervention in perceived stress, emotional distress, exercise tolerance, and depression.25 Most recently, Swanson and colleagues demonstrated improved exercise tolerance after cardiorespiratory biofeedback in patients with higher left ventricular ejection fractions (≥ 31%), although improvement could not be accomplished in those with ejection fractions below 30%.26
ONGOING STUDY IN END-STAGE HEART FAILURE AND FUTURE DIRECTIONS
We are currently involved at Cleveland Clinic in a study of end-stage heart failure patients who are awaiting cardiac transplantation. Each patient is provided with eight sessions of biofeedback training, including respiratory rate, digital peripheral temperature, muscle tension, and heart rate variability. Clinical status, quality of life, and heart failure–specific symptoms are being monitored throughout the training period. Success with biofeedback training is being analyzed, and we are testing the hypothesis that the degree of success in learning self-regulation will predict change in clinical status, quality of life, and the biology of the heart. What is unique to our study is that we will obtain the heart tissue at explant, when the patient receives a cardiac transplant, and we will conduct experiments to determine whether the cellular and molecular phenotype of the heart have been changed by the intervention, particularly components of the SNS, PNS, and inflammatory pathways.
We have been studying human heart failure for many years, and we have previously shown the changes in receptors and signaling pathways that occur in the failing human heart.27–30 We were also among the first to demonstrate that the cellular and molecular changes that occur in the failing human heart are not actually irreversible but can be changed by interventions such as a left ventricular assist device.31–33 Thus we hypothesize that biofeedback training, by interfering with overactivation of the SNS and by allowing the PNS to more adequately contribute to cardiac regulation, will have a meaningful effect on the biology of the failing human heart in addition to improving clinical status and quality of life. We hope to be among the first to demonstrate that effect.
BIOFEEDBACK: AN OVERVIEW
Biofeedback is a self-regulation therapy that aims to teach individuals the skills that will allow them to change their physiology in healthy directions.1–3 Biofeedback involves a client, a trained biofeedback coach, and appropriate instrumentation. Sensors are connected to the client, and various physiologic parameters (such as heart rate, blood pressure, and digital peripheral temperature) are displayed on a computer screen. The client is guided through a brief mental stress test and a relaxation exercise to learn to recognize differences between hyperarousal and a more relaxed physiology. Biofeedback training involves a series of sessions in which the goal is to help the client gain control of his or her own physiology by learning relaxation techniques such as deep breathing, progressive muscle relaxation, and guided imagery.1,3,4 Although biofeedback can be used solely as operant conditioning, it is more commonly and more effectively combined with techniques of stress management.
Biofeedback training is commonly (although not exclusively) used to decrease activation of the sympathetic branch of the autonomic nervous system (the “fight or flight” response). The reduction in sympathetic nervous system (SNS) activity is manifest as an increase in digital peripheral temperature and decreases in skin conductance, heart rate, and blood pressure, as well as changes in the frequency distribution of heart rate variability. While the SNS is becoming less activated, the parasympathetic portion of the autonomic nervous system (“rest and digest”) is becoming more involved in regulating body functions. More parasympathetic nervous system (PNS) activation and less SNS activation produces a healthier physiologic state, and thus biofeedback can be used to move the body in the direction of health and wellness.1–4
HEART FAILURE: BIOLOGIC MECHANISMS OF INJURY
Heart failure is the end result of most untreated cardiovascular diseases. Heart failure involves inadequate cardiac pump function, such that appropriate perfusion of end organs does not occur. The process of developing heart failure is a gradual one that begins with compensatory processes. In response to an injury or insult, such as chronic high blood pressure or long-standing coronary artery blockage, the heart compensates by activating various neurohormonal pathways in an attempt to preserve cardiac function and end-organ perfusion.5 When these pathways are activated, they initially help the heart to compensate for the ongoing challenge of increased pressure or decreased tissue oxygenation and allow the cardiovascular system to pump sufficient blood. Over time, however, these compensatory processes become maladaptive. Cellular signaling pathways, which were activated in order to help the heart compensate, actually become as much of a problem as the decreased cardiac function.5
Hyperactivation of the SNS
Chief among these pathways is the SNS, which is the most powerful means by which cardiac function can be augmented.5 In response to decreased cardiac function, cardiac sympathetic nerves are activated, releasing norepinephrine locally, and both norepinephrine and epinephrine increase in the circulating blood. Beta-adrenergic receptors on cardiac myocytes and on vascular smooth muscle cells are stimulated, and the resulting augmentation of cardiac contraction helps the heart to overcome an immediate challenge. If the insult or injury to the heart is acute and time-limited, this system compensates and the situation is resolved. However, chronic activation of the SNS creates more problems than it solves for the failing heart,5,6 including the following:
Myocardial cells are challenged by the need for increased energy production to support the chronic stimulation
Oxidative stress ensues
Receptors are downregulated
Pathways that result in necrosis and apoptosis are activated
Myofilament proteins respond to chronically elevated intracellular calcium.
As a result, the heart begins to spiral more quickly into a decompensated state. The toxicity of SNS overactivation is the reason for the success of beta-adrenergic blocking drugs in treating heart failure, but this situation is complicated further by adrenergic receptor polymorphisms and nonhomogeneous responses to beta-blocking agents.6 It is safe to say that the goal of much heart failure therapy is inactivation of the once-compensatory SNS and its resulting biologic effects.
Hypoactivation of the PNS
In addition to hyperactivation of the SNS, heart failure is also accompanied by a decrease in the role of the PNS. Under normal resting conditions, the human heart is governed more by the PNS than the SNS, with the SNS becoming a major source of cardiac control only during periods of decreased cardiac function. In heart failure, however, this relationship is reversed, with the SNS taking over the governing role and PNS input becoming less significant. Studies have suggested that the lack of contribution of the PNS to cardiac regulation in heart failure may be as deleterious as overactivation of the SNS.7 Most recently, stimulation of the vagal nerve has been shown to be beneficial in both animal models8 and humans with heart failure,9 confirming that augmenting PNS activity may be as important as inhibiting SNS activity. Although vagal nerve stimulation may be the first heart failure therapy aimed specifically at the PNS, it is likely that the future will hold more therapies with this goal.
PNS as regulator of inflammation of the failing heart?
It has recently been suggested that beyond its role in regulating cardiac function under baseline conditions, the PNS may participate in regulating the inflammatory state of the failing heart. It has been established since the observations of Packer and colleagues in the early 1990s10 that proinflammatory cytokines such as tumor necrosis factor–alpha, interleukin-6, and interleukin-1 are elevated in the circulation of heart failure patients, that these cytokines are correlated with clinical prognosis, and that they play a role in the activation of deleterious cardiac signaling pathways.11,12 Trials of antiinflammatory therapies in heart failure have been less than successful, but this may be because the complexity of the activation has been underestimated.11 In elegant work reported several years ago, Kevin Tracey’s group showed that stimulation of the vagus nerve could inhibit inflammatory processes associated with sepsis.13,14 Since that time, the reflex activation and inactivation of inflammatory processes by the PNS have become more widely accepted. Although it has not yet been directly demonstrated, it is possible that part of the benefit of vagal nerve stimulation in heart failure will prove to be due to its ability to reduce the chronic inflammatory state of the failing heart.
BIOFEEDBACK IN HEART FAILURE: RATIONALE
The failing heart is characterized by autonomic imbalance (hyperactivation of the SNS and hypoactivation of the PNS) and by a chronic inflammatory state. It has been hypothesized both directly and indirectly that these two major pathophysiologic processes may be intertwined.13–15 Biofeedback-assisted stress management is a therapy that has the potential to interfere with both processes. If the patient with heart failure can be trained to reduce activation of the SNS and to increase control by the PNS, it is likely that the negative consequences of autonomic imbalance will be decreased or possibly even reversed. Whether these effects are limited to quality of life and clinical status, or whether they extend to an effect on myocardial remodeling processes, remains to be established. Since the chronic inflammatory state can also be affected by increasing PNS control of the cardiovascular system, we further hypothesize that biofeedback training may have a direct effect on the inflammatory processes involved in the downward spiral of heart failure.
BIOFEEDBACK IN HEART FAILURE: STUDIES
We are certainly not the first group to hypothesize that self-regulation may have a role in the treatment of cardiovascular diseases in general or heart failure in particular. It has been shown that patients with heart failure manage their disease better and experience less emotional distress when they have a greater sense of control over their condition.16 In addition to giving patients a greater sense of control, some mind-body therapies have been shown to be beneficial in those with heart failure. Pischke et al showed as part of the Multicenter Lifestyle Demonstration Project that patients with left ventricular ejection fractions in the range associated with heart failure (≤ 40%) were able to learn and benefit from stress management techniques equally as well as those with more normal cardiac function.17 Both relaxation training18,19 and meditation20 have been shown to improve quality of life in heart failure patients, but meditation also reduced circulating norepinephrine, a marker of SNS activation.20 Mindfulness training improved clinical symptoms of heart failure and also reduced both anxiety and depression in patients with heart failure.21 Training heart failure patients to breathe more slowly is an intervention that is normally part of biofeedback training, but even when used alone it has resulted in decreased dyspnea,22 increased oxygen saturation,23 and improved exercise tolerance.22,23
To our knowledge, three studies to date have specifically used biofeedback training in patients with documented heart failure. As early as 1997, Moser and colleagues showed that heart failure patients were able to raise their finger temperature in spite of disease-related vascular changes, and that a single session of finger temperature biofeedback resulted in meaningful clinical improvement.24 Luskin et al randomized 33 heart failure patients to either biofeedback-assisted stress management or a control group, and showed improvement with the intervention in perceived stress, emotional distress, exercise tolerance, and depression.25 Most recently, Swanson and colleagues demonstrated improved exercise tolerance after cardiorespiratory biofeedback in patients with higher left ventricular ejection fractions (≥ 31%), although improvement could not be accomplished in those with ejection fractions below 30%.26
ONGOING STUDY IN END-STAGE HEART FAILURE AND FUTURE DIRECTIONS
We are currently involved at Cleveland Clinic in a study of end-stage heart failure patients who are awaiting cardiac transplantation. Each patient is provided with eight sessions of biofeedback training, including respiratory rate, digital peripheral temperature, muscle tension, and heart rate variability. Clinical status, quality of life, and heart failure–specific symptoms are being monitored throughout the training period. Success with biofeedback training is being analyzed, and we are testing the hypothesis that the degree of success in learning self-regulation will predict change in clinical status, quality of life, and the biology of the heart. What is unique to our study is that we will obtain the heart tissue at explant, when the patient receives a cardiac transplant, and we will conduct experiments to determine whether the cellular and molecular phenotype of the heart have been changed by the intervention, particularly components of the SNS, PNS, and inflammatory pathways.
We have been studying human heart failure for many years, and we have previously shown the changes in receptors and signaling pathways that occur in the failing human heart.27–30 We were also among the first to demonstrate that the cellular and molecular changes that occur in the failing human heart are not actually irreversible but can be changed by interventions such as a left ventricular assist device.31–33 Thus we hypothesize that biofeedback training, by interfering with overactivation of the SNS and by allowing the PNS to more adequately contribute to cardiac regulation, will have a meaningful effect on the biology of the failing human heart in addition to improving clinical status and quality of life. We hope to be among the first to demonstrate that effect.
References
McKee MG. Biofeedback: an overview in the context of heart-brain medicine. Cleve Clin J Med2008; 75( suppl 2):S31–S34.
Moravec CS. Biofeedback therapy in cardiovascular disease: rationale and research overview. Cleve Clin J Med2008; 75( suppl 2):S35–S38.
Moss D, McGrady A, Davies TC, Wickramasekera I. Handbook of Mind-Body Medicine for Primary Care. London: Sage Publications; 2003.
Schwartz MS, Andrasik F. Biofeedback: A Practitioner’s Guide. 3rd ed. New York, NY: Guilford Press; 2003.
Triposkiadis F, Karayannis G, Giamouzis G, Skoularigis J, Louridas G, Butler J. The sympathetic nervous system in heart failure. J Am Coll Cardiol2009; 54:1747–1762.
Floras JS. Sympathetic nervous system activation in human heart failure. J Am Coll Cardiol2009; 54:375–385.
Binkley PF, Nunziata E, Haas GJ, Nelson SD, Cody RJ. Parasympathetic withdrawal is an integral component of autonomic imbalance in congestive heart failure: demonstration in human subjects and verification in a paced canine model of ventricular failure. J Am Coll Cardiol1991; 18:464–472.
Li M, Zheng C, Sato T, Kawada T, Sugimachi M, Sunugawa K. Vagal nerve stimulation markedly improves long-term survival after chronic heart failure in rats. Circulation2004; 109:120–124.
Schwartz PJ, DeFerrari GM, Sanzo A, et al Long term vagal stimulation in patients with advanced heart failure: first experience in man. Eur J Heart Failure2008; 10:884–891.
Levine B, Kalman J, Mayer L, Fillit M, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med1990; 323:236–241.
Mann DL. Inflammatory mediators and the failing heart: past, present and the foreseeable future. Circ Res2002; 91:988–998.
Parish RC, Evans JD. Inflammation in chronic heart failure. Ann Pharmacother2008; 42:1002–1016.
Tracey KJ. The inflammatory reflex. Nature2002; 420:853–859.
Tracey KJ. Reflex control of immunity. Nat Rev Immunol2009; 9:418–428.
Dracup K, Westlake C, Erickson VS, Moser DK, Caldwell ML, Hamilton MA. Perceived control reduces emotional stress in patients with heart failure. J Heart Lung Transplant2003; 22:90–93.
Pischke CR, Weidner G, Elliott-Eller M, Ornish D. Lifestyle changes and clinical profile in coronary heart disease patients with an ejection fraction of ≤ 40% or > 40% in the Multicenter Lifestyle Demonstration Project. Eur J Heart Failure2007; 9:928–934.
Chang BH, Henricks A, Zhao Y, Rothendler JA, LoCastro JS, Slawsky MT. A relaxation response randomized trial on patients with chronic heart failure. J Cardiopulm Rehab2005; 25:149–157.
Yu DSF, Lee DTF, Woo J. Effects of relaxation therapy on psychologic distress and symptom status in older Chinese patients with heart failure. Psychosom Res2007;427–437.
Curiati JA, Bocchi E, Freire JO, et al Meditation reduces sympathetic activation and improves the quality of life in elderly patients with optimally treated heart failure: a prospective randomized study. J Altern Complement Med2005; 11:465–472.
Sullivan MJ, Wood L, Terry J, et al The support, education and research in chronic heart failure study (SEARCH): a mindfulness-based psychoeducational intervention improves depression and clinical symptoms in patients with chronic heart failure. Am Heart J2009; 157:84–90.
Weiner P, Waizman J, Magadle R, Berar-Yanay N, Pelled B. The effect of specific inspiratory muscle training on the sensation of dyspnea and exercise tolerance in patients with congestive heart failure. Clin Cardiol1999; 22:727–732.
Bernardi L, Porta C, Spicuzza L, et al Slow breathing increases arterial baroreflex sensitivity in patients with chronic heart failure. Circulation2002; 105:143–145.
Moser DK, Dracup K, Woo MA, Stevenson LW. Voluntary control of vascular tone by using skin-temperature biofeedback-relaxation in patients with advanced heart failure. Altern Ther Health Med1997; 3:51–60.
Luskin F, Reitz M, Newell K, Quinn TG, Haskell W. A controlled pilot study of stress management training of elderly patients with congestive heart failure. Prev Cardiol2002; 5:168–172.
Swanson KS, Gevirtz RN, Brown M, Spira J, Guarneri E, Stoletniy L. The effect of biofeedback on function in patients with heart failure. Appl Psychophysiol Biofeedback2009; 34:71–91.
Razeghi P, Mukhopadhyay M, Myers TJ, et al Myocardial tumor necrosis factor alpha expression does not correlate with clinical indices of heart failure in patients on left ventricular assist device support. Ann Thorac Surg2001; 72:2044–2050.
Matteo R, Moravec CS. Expression and immunolocalization of annexins IV, V and VI in the failing and non-failing human heart. Cardiovasc Res2000; 45:961–970.
Dash R, Frank K, Carr AN, Moravec CS, Kranias EG. Gender influences on sarcoplasmic reticulum calcium handling in the failing human myocardium. J Mol Cell Cardiol2001; 33:1345–1353.
DiPaola NR, Sweet WE, Stull LB, Francis GS, Moravec CS. Beta-adrenergic receptors and calcium cycling proteins in normal, hypertrophied and failing human hearts: transition from hypertrophy to failure. J Mol Cell Cardiol2001; 33:1283–1295.
Aquila LA, McCarthy PM, Smedira NG, Young JB, Moravec CS. Cytoskeletal structure and recovery in single human cardiac myocytes. J Heart Lung Transplant2004; 23:954–963.
Fedak PWM, Moravec CS, McCarthy PM, et al Altered expression of disintegrin metalloproteinases and their inhibitor in human dilated cardiomyopathy. Circulation2006; 113:238–245.
Ogletree-Hughes ML, Stull LB, Sweet WE, Smedira NG, McCarthy PM, Moravec CS. Mechanical unloading restores beta adrenergic responsiveness and reverses receptor downregulation in the failing human heart. Circulation2001; 104:881–886.
References
McKee MG. Biofeedback: an overview in the context of heart-brain medicine. Cleve Clin J Med2008; 75( suppl 2):S31–S34.
Moravec CS. Biofeedback therapy in cardiovascular disease: rationale and research overview. Cleve Clin J Med2008; 75( suppl 2):S35–S38.
Moss D, McGrady A, Davies TC, Wickramasekera I. Handbook of Mind-Body Medicine for Primary Care. London: Sage Publications; 2003.
Schwartz MS, Andrasik F. Biofeedback: A Practitioner’s Guide. 3rd ed. New York, NY: Guilford Press; 2003.
Triposkiadis F, Karayannis G, Giamouzis G, Skoularigis J, Louridas G, Butler J. The sympathetic nervous system in heart failure. J Am Coll Cardiol2009; 54:1747–1762.
Floras JS. Sympathetic nervous system activation in human heart failure. J Am Coll Cardiol2009; 54:375–385.
Binkley PF, Nunziata E, Haas GJ, Nelson SD, Cody RJ. Parasympathetic withdrawal is an integral component of autonomic imbalance in congestive heart failure: demonstration in human subjects and verification in a paced canine model of ventricular failure. J Am Coll Cardiol1991; 18:464–472.
Li M, Zheng C, Sato T, Kawada T, Sugimachi M, Sunugawa K. Vagal nerve stimulation markedly improves long-term survival after chronic heart failure in rats. Circulation2004; 109:120–124.
Schwartz PJ, DeFerrari GM, Sanzo A, et al Long term vagal stimulation in patients with advanced heart failure: first experience in man. Eur J Heart Failure2008; 10:884–891.
Levine B, Kalman J, Mayer L, Fillit M, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med1990; 323:236–241.
Mann DL. Inflammatory mediators and the failing heart: past, present and the foreseeable future. Circ Res2002; 91:988–998.
Parish RC, Evans JD. Inflammation in chronic heart failure. Ann Pharmacother2008; 42:1002–1016.
Tracey KJ. The inflammatory reflex. Nature2002; 420:853–859.
Tracey KJ. Reflex control of immunity. Nat Rev Immunol2009; 9:418–428.
Dracup K, Westlake C, Erickson VS, Moser DK, Caldwell ML, Hamilton MA. Perceived control reduces emotional stress in patients with heart failure. J Heart Lung Transplant2003; 22:90–93.
Pischke CR, Weidner G, Elliott-Eller M, Ornish D. Lifestyle changes and clinical profile in coronary heart disease patients with an ejection fraction of ≤ 40% or > 40% in the Multicenter Lifestyle Demonstration Project. Eur J Heart Failure2007; 9:928–934.
Chang BH, Henricks A, Zhao Y, Rothendler JA, LoCastro JS, Slawsky MT. A relaxation response randomized trial on patients with chronic heart failure. J Cardiopulm Rehab2005; 25:149–157.
Yu DSF, Lee DTF, Woo J. Effects of relaxation therapy on psychologic distress and symptom status in older Chinese patients with heart failure. Psychosom Res2007;427–437.
Curiati JA, Bocchi E, Freire JO, et al Meditation reduces sympathetic activation and improves the quality of life in elderly patients with optimally treated heart failure: a prospective randomized study. J Altern Complement Med2005; 11:465–472.
Sullivan MJ, Wood L, Terry J, et al The support, education and research in chronic heart failure study (SEARCH): a mindfulness-based psychoeducational intervention improves depression and clinical symptoms in patients with chronic heart failure. Am Heart J2009; 157:84–90.
Weiner P, Waizman J, Magadle R, Berar-Yanay N, Pelled B. The effect of specific inspiratory muscle training on the sensation of dyspnea and exercise tolerance in patients with congestive heart failure. Clin Cardiol1999; 22:727–732.
Bernardi L, Porta C, Spicuzza L, et al Slow breathing increases arterial baroreflex sensitivity in patients with chronic heart failure. Circulation2002; 105:143–145.
Moser DK, Dracup K, Woo MA, Stevenson LW. Voluntary control of vascular tone by using skin-temperature biofeedback-relaxation in patients with advanced heart failure. Altern Ther Health Med1997; 3:51–60.
Luskin F, Reitz M, Newell K, Quinn TG, Haskell W. A controlled pilot study of stress management training of elderly patients with congestive heart failure. Prev Cardiol2002; 5:168–172.
Swanson KS, Gevirtz RN, Brown M, Spira J, Guarneri E, Stoletniy L. The effect of biofeedback on function in patients with heart failure. Appl Psychophysiol Biofeedback2009; 34:71–91.
Razeghi P, Mukhopadhyay M, Myers TJ, et al Myocardial tumor necrosis factor alpha expression does not correlate with clinical indices of heart failure in patients on left ventricular assist device support. Ann Thorac Surg2001; 72:2044–2050.
Matteo R, Moravec CS. Expression and immunolocalization of annexins IV, V and VI in the failing and non-failing human heart. Cardiovasc Res2000; 45:961–970.
Dash R, Frank K, Carr AN, Moravec CS, Kranias EG. Gender influences on sarcoplasmic reticulum calcium handling in the failing human myocardium. J Mol Cell Cardiol2001; 33:1345–1353.
DiPaola NR, Sweet WE, Stull LB, Francis GS, Moravec CS. Beta-adrenergic receptors and calcium cycling proteins in normal, hypertrophied and failing human hearts: transition from hypertrophy to failure. J Mol Cell Cardiol2001; 33:1283–1295.
Aquila LA, McCarthy PM, Smedira NG, Young JB, Moravec CS. Cytoskeletal structure and recovery in single human cardiac myocytes. J Heart Lung Transplant2004; 23:954–963.
Fedak PWM, Moravec CS, McCarthy PM, et al Altered expression of disintegrin metalloproteinases and their inhibitor in human dilated cardiomyopathy. Circulation2006; 113:238–245.
Ogletree-Hughes ML, Stull LB, Sweet WE, Smedira NG, McCarthy PM, Moravec CS. Mechanical unloading restores beta adrenergic responsiveness and reverses receptor downregulation in the failing human heart. Circulation2001; 104:881–886.
The attempt to alter electroencephalographic (EEG) frequency/amplitude patterns and their underlying brain mechanisms using contingent operant conditioning methods is today referred to variously as EEG biofeedback, neurofeedback, or neurotherapy. This article traces the history of the clinical application of EEG operant conditioning from empirical animal investigations to its emergence as a treatment option for major seizure types. In light of the diversity of the clinical applications of this method in general, and the complexity of seizure disorders in particular, I also present an overview of specific methods used in our EEG biofeedback program.
Figure 1. A carefully documented 6-year seizure data log from an adult female subject (aged 23 years at the start of the log) with nocturnal tonic-clonic seizures, often with incontinence.2 The log starts 1 year before initiation of electroencephalographic (EEG) feedback training (“Pre-SMR”), continues through 2.5 years of twice-weekly EEG training sessions (“Post-SMR”), and continues through 2.5 years after wi thdrawal from this training (“Withdrawal”). Training consisted of auditory and visual reward for increased 12- to 15-Hz EEG activity over the left sensorimotor cortex, which has been labeled the sensorimotor rhythm (SMR). Medications were held constant during training and adjusted downward after withdrawal from training. In 1977 this patient was issued a California driver’s license.This application was officially added to the broader field of biofeedback with the publication of a 1972 paper by Sterman and Friar titled, “Suppression of seizures in an epileptic following sensorimotor EEG feedback training.” 1 In this paper we documented a sustained and progressive reduction of generalized nocturnal tonic-clonic seizures in a 23-year-old female epileptic with a 7-year history of frequent and medically refractory seizures of unknown origin. The patient’s clinical EEG showed left sensorimotor cortex spikes and slow 5- to 7-Hz activity. Seizure reduction occurred in response to an experimental course of EEG operant conditioning aimed at increasing 12- to 15-Hz EEG activity in the left sensorimotor cortex while suppressing slower activity at this same site. The 12- to 15-Hz EEG rhythm was discovered in animal research and labeled as the sensorimotor rhythm (SMR). Although the patient had previously been worked up and treated unsuccessfully with anticonvulsant medications at several prestigious medical institutions, over the course of 2.5 years of twice-weekly EEG feedback training sessions she became essentially seizure free (Figure 1)2 and was ultimately issued a California driver’s license.
BACKDROP TO THE CLINICAL APPLICATION: KEY ANIMAL STUDIES
Figure reprinted from Sterman et al (Science 1970; 167:1146–1148).3
Figure 2. Bipolar electroencephalographic (EEG) samples from sensorimotor and parietal cortex in the cat during quiet (motionless) wakefulness (left) and quiet (non-REM) sleep (right). Both states are associated with bursts of 12- to 15-Hz EEG rhythmic activity in sensorimotor cortex. During sleep these bursts are higher in amplitude and associated with slower rhythmic patterns in parietal cortex.The above landmark study was predicated on the observation of a discrete 11- to 19-Hz EEG rhythmic pattern in cats, which occurred intermittently over the sensorimotor cortex during behavioral quiescence. When animals were trained to suppress a learned bar-press response for food if a tone was sounded in the chamber, a 12- to 15-Hz version of this EEG pattern always accompanied inhibition of the bar-press response. If animals later fell asleep, a similar rhythmic EEG pattern, known as the sleep spindle, was localized to the same cortical area at the same frequency (Figure 2).3 Our interest at the time was in the neurophysiological control of sleep. Because both of these patterns occurred uniquely in the absence of movement, we sought to determine if the underlying neural mechanisms were related.
To accomplish this, we attempted to facilitate the SMR during wakefulness using an operant conditioning paradigm with a liquid food reward, and then study any resulting changes in sleep spindle activity and sleep structure. Necessary quality controls included alternate training to suppress this rhythm and a counterbalanced design employing two separate groups of cats. Six weeks of three training sessions per week to satiation led to profound and differential changes in sleep EEG and sleep architecture. SMR training, whether it preceded or followed suppression training, led to a significant increase in EEG sleep spindle density, as well as a significant reduction in sleep period fragmentation due to arousals. No changes occurred in the control condition.3
A more profound finding in the cat
Figure modified from Sterman.5
Figure 3. The sequence of prodromal events preceding generalized convulsions in two groups of 10 cats, all of which were injected intra-abdominally with 9 mg/kg of GABA-depleting monomethyl hydrazine. One group (dashed tracing) had received 6 weeks of electroencephalographic feedback training for sensorimotor rhythm (SMR) enhancement with food reward (see text). The two groups did not differ statistically in the latency to prodromal symptoms. All control animals seized reliably at approximately 60 minutes, as had been previously documented. In contrast, the SMR-trained group had a significantly prolonged mean latency to seizures (130 minutes), and several did not seize within the 4-hour test period.As interesting as this finding was, the most profound outcome of the study emerged later. A different cat study under way in our laboratory, funded by the US Air Force, was seeking to determine the effects on behavior of low-dose exposure to monomethyl hydrazine (MMH).4 This compound is a highly toxic component of the liquid rocket fuel used for launching virtually all space vehicles. Significant MMH exposure via any route causes pro-found nausea and gradual onset of convulsions, which are lethal at adequate doses. The mechanism for this effect was ultimately determined to be a disruption of the synthesis of gamma-aminobutyric acid, the primary inhibitory neurotransmitter in the central nervous system. We were investigating the effects of low-dose exposure to determine the possible disruption of cognitive functions such exposure might cause in flight crews. Our first objective for studies in cats was to establish the doseresponse curve for convulsive effects in that species. We had succeeded in determining a curve showing that 9 mg/kg of MMH was the threshold dose for reliably producing nonlethal convulsions after a prodrome of approximately 40 to 67 minutes. This prodrome consisted of a sequence of reliable autonomic and behavioral events. When data from animals provided with SMR operant conditioning as the final training procedure were added to this curve, the same prodrome was observed but there were no seizures at 60 minutes. Instead, the latency to seizures was delayed to a range of 80 to 220 minutes, and several animals failed to seize at all.4 A subsequent systematic study of this effect with animals as their own controls in a counterbalanced design confirmed this effect (Figure 3).5 This finding then led to the test in the human epileptic subject described above.1
Platform for a dual research approach
These two studies provided several interesting conclusions that directed our subsequent scientific efforts. First, in the cat study we observed a common prodrome in both SMR-trained and control animals even though the SMR-trained animals had acquired protection against seizures. This suggested a direct effect on the seizure process and not on MMH toxicity in general. Second, in our human epileptic patient, the seizures that were suppressed arose out of the unconscious state of sleep, a fact that eliminated the possibility of any voluntary countermeasure and again indicated a direct effect on the seizure mechanism. Accordingly, we undertook a dual approach to understanding the basis of this effect, involving both additional animal electrophysiologic and human clinical studies.
Animal studies evaluated motor behavior, motor reflexes, motor and thalamic unit firing, and somatosensory pathway correlates of the SMR response. Clinical studies, as reviewed in the following section, sought to further document the anticonvulsant effects of SMR operant conditioning and examine this effect on various seizure types. Possible alternative explanations, such as altered medication compliance and placebo effects, were also addressed in several comprehensive studies. Additionally, by this time other laboratories were beginning to add to the research literature in this new field.
Reproduced, with permission of the American Academy of Sleep Medicine, from Sleep (Sterman MB, et al. Sensorimotor electroencephalogram rhythmic activity: a functional gate mechanism. Sleep 1981; 4(4):408–422).
Figure 4. Trained sensorimotor rhythm (SMR) responses are associated with changes in both afferent and efferent pathways of the sensorimotor system. These include decreased red nucleus (RN) activity, stretch reflex excitability, and muscle tone. These changes produce reduced somatic afferent (SA) discharge and lead to thalamic hyperpolarization and reciprocal oscillatory burst activity between the ventrobasal (vPL) and reticular (nRt) nuclei of the thalamus. This burst activity is propagated to sensorimotor cortex (S1) and initiates corresponding bursts of SMR activity.6Neurophysiologic studies in cats revealed a convergent pattern of changes that were directly correlated with the SMR pattern in the EEG and clearly indicated reduced motor excitability. These included a specific attenuation of cellular activity and reflex excitability in the motor pathway, a reduction in muscle tone and associated motor unit firing, and cessation of behavioral movements. Further, unit studies in afferent nuclei of the somatosensory pathway revealed evidence of reduced somatic afferent firing and the onset of reciprocal burst oscillation between the thalamic reticular nucleus and the adjacent ventrobasal relay nucleus. This oscillation provides the thalamic source of the cortical SMR pattern. These findings are summarized in Figure 4.6 Details of the studies and resulting publications are provided in recent review articles.7–9 They represent empirical evidence for significant reorganization of neuronal function when SMR activity appears in the sensorimotor EEG.
CLINICAL STUDIES
A series of human studies followed our initial clinical report, including group studies involving crossover and placebo-controlled designs. These studies consistently reported significant seizure reductions in epileptic patients in response to reward for increasing sensorimotor EEG rhythmic activity.
Two independent meta-analyses of the peer-reviewed papers in this literature have appeared in the last decade.8,10 In a review of 24 studies involving 243 patients with predominantly partial complex seizures provided with central cortical SMR feedback training, Sterman determined that 82% of these subjects registered seizure reductions greater than 50%.8 More recently, Tan and colleagues evaluated data from 63 studies and selected for comprehensive analysis 10 studies that met stringent criteria for controls and population and seizure details.10 They reported that 79% of the patients treated with SMR feedback training experienced a statistically significant reduction in seizure frequency despite a collective history of failed medication therapy.
Data are from Lantz and Sterman.11
Figure 5. Reported seizure rates in three experimental groups of randomly assigned patients with medication-refractory complex partial seizures.11 Each group of 8 subjects received 6 weeks of treatment consisting of either (1) detailed tabulation of seizures, (2) non-contingent sensorimotor rhythm (SMR) training (“yoked control”), or (3) contingent SMR training. Following this initial 6-week period, all 24 subjects were combined into one contingent SMR training group for 6 additional weeks and then gradually withdrawn from training. A final 6-week follow-up seizure tabulation period completed the analysis. Data are plotted against group baselines. A significant reduction in seizures was registered after contingent training only, and this effect increased progressively across subsequent conditions.Data from one of the studies11 evaluated in both of these systematic reviews are summarized in Figure 5. In this study, 24 subjects with complex partial seizures, many with seizure foci confirmed through depth recordings, were randomly assigned to three experimental treatment groups:
One group simply tabulated their seizure experiences for 6 weeks using a comprehensive logging method.
The second group received EEG feedback training for 1 hour three times a week for 6 weeks; however, the EEG signal responsible for reward was previously recorded from a different individual. This noncontingent feedback constituted a “yoked control” group.
The third group received 6 weeks of contingent training for increasing SMR activity in somatosensory cortex while simultaneously suppressing slower 4- to 8-Hz activity.
After the initial 6 weeks, all 24 subjects were combined into one group that received 6 more weeks of contingent training only. This was followed by a 4-week period of gradual withdrawal from training and then by a final tabulation of seizure incidence during a 6-week period after training was terminated. As can be seen in Figure 5, the seizure tabulation control was associated with an increased seizure count and the “yoked control” noncontingent SMR training was associated with no significant change in seizure incidence, whereas contingent SMR training was associated with a statistically significant reduction in seizures. The statistical significance of this reduction increased progressively as subjects from the other two groups were added to a second 6-week period of contingent training, and after an additional 6 weeks following withdrawal from training. In addition to this exclusive seizure reduction after SMR contingent training, pre-/post-training neuropsychological testing showed that responding SMR-trained subjects also improved significantly in performance of tasks specific to the hemisphere contralateral to their frontotemporal lesion, indicating a reduced corrosive disturbance from the seizure focus.11
EEG BIOFEEDBACK IN PRACTICE: PROFILE OF THE AUTHOR’S PROGRAM
EEG operant conditioning methods for biofeedback training have diversified as various hardware and software products have emerged and as individuals with differing backgrounds and credentials have entered the field. A lack of methodologic standards and professional regulations has contributed to an undesirable inconsistency in the competence and effectiveness of therapeutic applications. Nevertheless, abundant peer-reviewed research by qualified investigators has proven the worth of this method as a viable alternative treatment for seizure disorders, so I will attempt to provide some idea of a systematic and evidence-guided approach to treatment as used in our program.
Figure 6. This 12-year-old girl has suffered since early childhood from frequent multiple seizure types and myoclonic jerks that are unresponsive to pharmacologic treatments. She currently functions at about third-grade level but is aware and behaviorally compliant. Here she is responding to visual feedback in the context of sensorimotor rhythm training. Her mother assists by providing raisin and candy rewards when certain response criteria are achieved. Her seizures have declined in frequency and severity.Patients are subjected to a quantitative multi-channel EEG evaluation (QEEG) using hardware and software complying with both technical and learning-theory principles critical to valid data collection and operant conditioning applications. Data obtained from this study are combined with medical reports from other studies and information gained in a comprehensive intake interview. QEEG and background information guide the design of an empirical protocol, often with several training components, that is used consistently throughout the treatment period, which consists of one or two 60- to 90-minute treatment sessions per week for at least 20 weeks. Subjects are seated in front of a large-monitor screen and instructed on the requirements for reward. Reinforcement consists of visual images and tones, as well as a numeric display of scores achieved and the time remaining in a trial. On rare occasion a committed parent may be seated next to a more challenged patient and provide additional reinforcement in the form of earned treats, such as raisins and pieces of candy (Figure 6).
Figure 7. Primary display used in our sensorimotor rhythm biofeedback program. The display conforms strictly to operant conditioning principles while still promoting cognitive engagement in the human subject. Reward here is for two different EEG frequencies at two different cortical sites. The far left “green” site shows reinforced 12- to 15-Hz bandpass activity at the C3 electrode site. Low-frequency suppression of abnormal 3- to 5-Hz slow activity at Fz is addressed here through “successive approximation” and consumes the final three display units from left to right. See text for more details. Display results are for the subject depicted in Figure 6.The display that subjects see can varies within limits but must always be as simple as possible and must provide information exclusively relevant to achieving the desired EEG changes. One such display is shown in Figure 7. It consists of a series of four rectangular boxes, each with a segment of band-passed EEG data for selected frequency bands and enclosed by reward threshold guidelines. If the objective is to increase the amplitude and/or incidence of a particular frequency band, the band-pass display must exceed the upper threshold guideline. If the objective is to suppress that frequency band, the display must drop below the threshold line. The duration of the required response can be adjusted and is typically 0.25 to 0.5 seconds. When the desired response is achieved a small horizontal bar at the upper right of each band-pass display turns from red to green, and a large blue ball appears above, together with a chime or other tone. The display is frozen for 2 seconds and then becomes active again, thus providing for discrete trials. A yellow score bar at the bottom of the screen advances by one unit. The timing of each performance set (typically 3 minutes) is indicated by a moving blue bar at the bottom of the screen.
With each box monitoring the same electrode site and each frequency tuned to the same band, thresholds can be set to promote facilitation or suppression through “successive approximation,” or sequencing from left to right with sequentially more difficult thresholds. Numerous other configurations are possible. In the case shown in Figure 7, the band-pass at the far left is set at 12 to 15 Hz (SMR) for the C3 electrode site, and the remaining three bands to the right are set to 3 to 5 Hz at the left medial frontal location Fz, with successively lower thresholds to promote suppression of this band at this site.
Figure 8. Performance plot from the patient represented in Figures 6 and 7. The plot registers the scoring rate per 3-minute trial at top, along with corresponding smoothed amplitude output in the band-passed frequencies set for each electrode placement. In this case the patient was rewarded for increasing 12- to 15-Hz sensorimotor rhythm (SMR) activity (top EEG trace) and decreasing 3- to 5-Hz slow activity at Fz (bottom EEG trace) The subject showed response acquisition both within and across 3-minute feedback trials, together with a stabilization of the SMR frequency and a reduction in frontal slow activity.Performance outcome is measured systematically by tracking the scoring rate per trial, together with associated EEG patterns. Data from the 12-year-old female subject described above provide an example. The top of Figure 8 shows a plot of reward rate across four successive 3-minute EEG feedback trials. The patient was rewarded for simultaneously increasing 12- to 15-Hz SMR activity at C3 and reducing 3- to 5-Hz activity at Fz, as described above. Smoothed EEG plots for the targeted frequency bands are shown below these reward curves, starting with the C3 12- to 15-Hz channel. Activity in this band became increasingly stable across trials. Data from three frontal recording sites are also shown, with the targeted Fz 3- to 5-Hz band output at the bottom. Amplitudes decreased progressively at all frontal sites but most markedly at the bottom Fz location. Thus, SMR stabilization and simultaneously suppressed frontal slow activity resulted in a progressive pattern of incremental reward both within trials and across the session. The resulting profiles are indicative of learning.
A RATIONAL MODEL FROM RECENT NEUROIMAGING STUDIES
While it is difficult to evaluate neurophysiologic changes in human subjects to a degree similar to that in animals, certain parallels can be drawn. Further, new imaging methods allow for assessment of localized metabolic changes in the human brain during and after EEG feedback training. Behaviorally, during successful SMR training, human subjects become behaviorally quiet and direct their attention to the task. It is safe to presume that the SMR response develops as a result of reduced motor excitation and resulting intrathalamic ventrobasal oscillations, since this mechanism is well established as a basis for mammalian sensorimotor EEG rhythm generation.12 These changes, as well as others documented in our animal studies, set the stage for the development of SMR activity and are likely collectively initiated by altered input from some other executive system.
Reprinted from Lévesque J, et al. Effect of neurofeedback training on the neural substrates of selective attention in children with attention-deficit/hyperactivity disorder. Neurosci Lett 2006;394:216-221. Copyright 2006 with permission from Elsevier.
Figure 9. Functional magnetic resonance images before (left) and after (right) sensorimotor rhythm (SMR) feedback training in a study of the effect of SMR training in learning-disabled children.13 The images are sagittal sections for the data averaged across subjects, who either received SMR feedback training (experimental group) or did not (control group). In the pretraining condition, significant loci of activation were noted in the left superior parietal lobe for both groups. In the posttraining condition, activations were again seen in this cortical region for both groups. In addition, the experimental group also showed stronger and statistically significant loci of activation in the left striatum and substantia nigra.Several recent studies have suggested a specific pattern of motor inhibition output from the striatum of the basal ganglia as the source of these changes. Birbaumer observed increased striatal metabolic activity with functional magnetic resonance imaging (fMRI) analysis in subjects producing SMR activity (personal communication, 2005). Further, Lévesque and colleagues studied pre-/post-fMRI blood oxygenation level–dependent response patterns in learning-disabled children trained to increase SMR activity and found a specific increase in the metabolic activity of the striatum and substantia nigra (Figure 9).13 The SMR-trained subjects showed significant academic improvement as well.13
These facts provide a rational model for a threshold-altering process that could affect seizure discharge propagation to motor networks. Although there are many different neurotransmitters used within the basal ganglia (principally acetylcholine, gamma-aminobutyric acid, and dopamine), the overall effect on thalamus and premotor networks in the mesencephalic tegmentum and superior colliculus is inhibitory.14–16 If activation of these inhibitory basal ganglia networks can become labeled by the SMR through contingent feedback training, and if responsible circuits can be potentiated by this association, motor inhibitory regulation would be generally facilitated.
CONCLUSIONS
Despite the encouraging findings and concepts reviewed here, there are significant issues at virtually every step of the thinking and practice behind this new therapy. This method depends on a comprehensive understanding of the EEG signal and the technical requirements of valid quantitative analysis and feedback applications. This includes a basic knowledge of the principles essential for effective operant conditioning. Further, in light of the complexity of seizure disorders, accurate history and seizure classification must be evaluated and understood.
Alternative explanations for therapeutic results include such considerations as short-lasting expectation effects and changes in patient behavior. However, it must again be noted that the prolonged anticonvulsant effect documented in our animal studies, as well as in relation to nocturnal seizures arising out of sleep in a human subject, would seem to rule out placebo or nonspecific effects. This conclusion is supported further by the finding of improved neuropsychological performance after SMR training in tasks mediated by the hemisphere contralateral to disrupting localized epileptogenic lesions. Additionally, an alternative explanation for improved seizure control based on increased medication compliance has been rejected through studies that carefully monitored blood levels of prescribed anticonvulsant drugs before, during, and after training.
Finally, the epileptic patients who have demonstrated clinical improvement in EEG biofeedback research studies, along with many who seek this treatment today, represent unquestionable failures of anticonvulsant drug therapy. Notably, positive outcomes have frequently been achieved in patients with complex-partial seizures, an extremely difficult-to-treat seizure type. It is therefore unfortunate that some professionals still criticize neurofeedback therapy for a lack of more consistent or successful outcomes. On the contrary, as noted here, evidence has shown that most of these difficult-to-treat patients benefit beyond any chance or placebo outcome, in some cases dramatically so. In light of the frequent adverse effects and costs associated with lifelong pharmacotherapy, we view EEG biofeedback therapy not as a “last resort” option to be restricted solely to pharmacotherapy-resistant cases but rather as a generally viable consideration for any patient suffering from seizures. Moreover, in contrast to drug-dependent management approaches, the altered modulation of striatal and thalamocortical inhibition that is possible through neurofeedback training may sufficiently raise seizure thresholds to greatly increase the prospects for the long-term nondependent management of epilepsy.
References
Sterman MB, Friar L. Suppression of seizures in an epileptic following sensorimotor EEG feedback training. Electroencephalogr Clin Neurophysiol1972; 33:89–95.
Sterman MB. Effects of sensorimotor EEG feedback training on sleep and clinical manifestations of epilepsy. In:Beatty J, Legewie H, eds. Biofeedback and Behavior. New York, NY: Plenum Press; 1977:167–200.
Sterman MB, Howe RD, Macdonald LR. Facilitation of spindle-burst sleep by conditioning of electroencephalographic activity while awake. Science1970; 167:1146–1148.
Sterman MB, LoPresti RW, Fairchild MD. Electroencephalographic and behavioral studies of monomethyl hydrazine toxicity in the cat. Aerospace Medical Research Laboratory1969; AMRL-TR-69-3:1–8.
Sterman MB. Studies of EEG biofeedback training in man and cats. In: Veterans Administration Studies in Mental Health and Behavioral Sciences: Highlights of the 17th Annual Conference. Perry Point, MD: US Veterans Administration; 1972:50–60.
Sterman MB. Physiological origins and functional correlates of EEG rhythmic activities: implications for self-regulation. Biofeedack Self Reg1996; 21:3–33.
Sterman MB. Basic concepts and clinical findings in the treatment of seizure disorders with EEG operant conditioning. Clin Electroencephalogr2000; 31:45–55.
Egner T, Sterman MB. Neurofeedback treatment of epilepsy: from basic rationale to practical application. Expert Rev Neurother2006; 6:247–257.
Tan G, Thornby J, Hammond D, et al Meta-analysis of EEG biofeedback in treating epilepsy. Clin EEG Neurosci2009; 40:173–179.
Lantz D, Sterman MB. Neuropsychological assessment of subjects with uncontrolled epilepsy: effects of EEG biofeedback training. Epilepsia1988; 29:163–171.
Steriade M, McCormick DA, Seinowski TJ. Thalamocortical oscillations in the sleeping and aroused brain. Science1993; 262:679–685.
Lévesque J, Beauregard M, Mensour B. Effect of neurofeedback training on the neural substrates of selective attention in children with attention-deficit/hyperactivity disorder: a functional magnetic resonance imaging study. Neurosci Lett2006; 394:216–221.
Brodal P. The basal ganglia. In: The Central Nervous System: Structure and Function. New York, NY: Oxford University Press; 1992:246–261.
Chevalier G, Deniau JM. Disinhibition as a basic process in the expression of striatal functions. Trends Neurosci1990; 13:277–280.
DeLong MR. Primate models of movement disorders of basal ganglia origin. Trends Neurosci1990; 13:281–285.
M. Barry Sterman, PhD Professor Emeritus, Departments of Neurobiology and Biobehavioral Psychiatry, Geffen School of Medicine, University of California, Los Angeles, CA
Correspondence: M.B. Sterman, PhD, 9773 Blantyre Drive, Beverly Hills, CA 90210; [email protected]
Dr. Sterman reported that he has intellectual property rights and ownership in Sterman-Kaiser Imaging Laboratory, for which he also is a member of the board of directors.
M. Barry Sterman, PhD Professor Emeritus, Departments of Neurobiology and Biobehavioral Psychiatry, Geffen School of Medicine, University of California, Los Angeles, CA
Correspondence: M.B. Sterman, PhD, 9773 Blantyre Drive, Beverly Hills, CA 90210; [email protected]
Dr. Sterman reported that he has intellectual property rights and ownership in Sterman-Kaiser Imaging Laboratory, for which he also is a member of the board of directors.
Author and Disclosure Information
M. Barry Sterman, PhD Professor Emeritus, Departments of Neurobiology and Biobehavioral Psychiatry, Geffen School of Medicine, University of California, Los Angeles, CA
Correspondence: M.B. Sterman, PhD, 9773 Blantyre Drive, Beverly Hills, CA 90210; [email protected]
Dr. Sterman reported that he has intellectual property rights and ownership in Sterman-Kaiser Imaging Laboratory, for which he also is a member of the board of directors.
The attempt to alter electroencephalographic (EEG) frequency/amplitude patterns and their underlying brain mechanisms using contingent operant conditioning methods is today referred to variously as EEG biofeedback, neurofeedback, or neurotherapy. This article traces the history of the clinical application of EEG operant conditioning from empirical animal investigations to its emergence as a treatment option for major seizure types. In light of the diversity of the clinical applications of this method in general, and the complexity of seizure disorders in particular, I also present an overview of specific methods used in our EEG biofeedback program.
Figure 1. A carefully documented 6-year seizure data log from an adult female subject (aged 23 years at the start of the log) with nocturnal tonic-clonic seizures, often with incontinence.2 The log starts 1 year before initiation of electroencephalographic (EEG) feedback training (“Pre-SMR”), continues through 2.5 years of twice-weekly EEG training sessions (“Post-SMR”), and continues through 2.5 years after wi thdrawal from this training (“Withdrawal”). Training consisted of auditory and visual reward for increased 12- to 15-Hz EEG activity over the left sensorimotor cortex, which has been labeled the sensorimotor rhythm (SMR). Medications were held constant during training and adjusted downward after withdrawal from training. In 1977 this patient was issued a California driver’s license.This application was officially added to the broader field of biofeedback with the publication of a 1972 paper by Sterman and Friar titled, “Suppression of seizures in an epileptic following sensorimotor EEG feedback training.” 1 In this paper we documented a sustained and progressive reduction of generalized nocturnal tonic-clonic seizures in a 23-year-old female epileptic with a 7-year history of frequent and medically refractory seizures of unknown origin. The patient’s clinical EEG showed left sensorimotor cortex spikes and slow 5- to 7-Hz activity. Seizure reduction occurred in response to an experimental course of EEG operant conditioning aimed at increasing 12- to 15-Hz EEG activity in the left sensorimotor cortex while suppressing slower activity at this same site. The 12- to 15-Hz EEG rhythm was discovered in animal research and labeled as the sensorimotor rhythm (SMR). Although the patient had previously been worked up and treated unsuccessfully with anticonvulsant medications at several prestigious medical institutions, over the course of 2.5 years of twice-weekly EEG feedback training sessions she became essentially seizure free (Figure 1)2 and was ultimately issued a California driver’s license.
BACKDROP TO THE CLINICAL APPLICATION: KEY ANIMAL STUDIES
Figure reprinted from Sterman et al (Science 1970; 167:1146–1148).3
Figure 2. Bipolar electroencephalographic (EEG) samples from sensorimotor and parietal cortex in the cat during quiet (motionless) wakefulness (left) and quiet (non-REM) sleep (right). Both states are associated with bursts of 12- to 15-Hz EEG rhythmic activity in sensorimotor cortex. During sleep these bursts are higher in amplitude and associated with slower rhythmic patterns in parietal cortex.The above landmark study was predicated on the observation of a discrete 11- to 19-Hz EEG rhythmic pattern in cats, which occurred intermittently over the sensorimotor cortex during behavioral quiescence. When animals were trained to suppress a learned bar-press response for food if a tone was sounded in the chamber, a 12- to 15-Hz version of this EEG pattern always accompanied inhibition of the bar-press response. If animals later fell asleep, a similar rhythmic EEG pattern, known as the sleep spindle, was localized to the same cortical area at the same frequency (Figure 2).3 Our interest at the time was in the neurophysiological control of sleep. Because both of these patterns occurred uniquely in the absence of movement, we sought to determine if the underlying neural mechanisms were related.
To accomplish this, we attempted to facilitate the SMR during wakefulness using an operant conditioning paradigm with a liquid food reward, and then study any resulting changes in sleep spindle activity and sleep structure. Necessary quality controls included alternate training to suppress this rhythm and a counterbalanced design employing two separate groups of cats. Six weeks of three training sessions per week to satiation led to profound and differential changes in sleep EEG and sleep architecture. SMR training, whether it preceded or followed suppression training, led to a significant increase in EEG sleep spindle density, as well as a significant reduction in sleep period fragmentation due to arousals. No changes occurred in the control condition.3
A more profound finding in the cat
Figure modified from Sterman.5
Figure 3. The sequence of prodromal events preceding generalized convulsions in two groups of 10 cats, all of which were injected intra-abdominally with 9 mg/kg of GABA-depleting monomethyl hydrazine. One group (dashed tracing) had received 6 weeks of electroencephalographic feedback training for sensorimotor rhythm (SMR) enhancement with food reward (see text). The two groups did not differ statistically in the latency to prodromal symptoms. All control animals seized reliably at approximately 60 minutes, as had been previously documented. In contrast, the SMR-trained group had a significantly prolonged mean latency to seizures (130 minutes), and several did not seize within the 4-hour test period.As interesting as this finding was, the most profound outcome of the study emerged later. A different cat study under way in our laboratory, funded by the US Air Force, was seeking to determine the effects on behavior of low-dose exposure to monomethyl hydrazine (MMH).4 This compound is a highly toxic component of the liquid rocket fuel used for launching virtually all space vehicles. Significant MMH exposure via any route causes pro-found nausea and gradual onset of convulsions, which are lethal at adequate doses. The mechanism for this effect was ultimately determined to be a disruption of the synthesis of gamma-aminobutyric acid, the primary inhibitory neurotransmitter in the central nervous system. We were investigating the effects of low-dose exposure to determine the possible disruption of cognitive functions such exposure might cause in flight crews. Our first objective for studies in cats was to establish the doseresponse curve for convulsive effects in that species. We had succeeded in determining a curve showing that 9 mg/kg of MMH was the threshold dose for reliably producing nonlethal convulsions after a prodrome of approximately 40 to 67 minutes. This prodrome consisted of a sequence of reliable autonomic and behavioral events. When data from animals provided with SMR operant conditioning as the final training procedure were added to this curve, the same prodrome was observed but there were no seizures at 60 minutes. Instead, the latency to seizures was delayed to a range of 80 to 220 minutes, and several animals failed to seize at all.4 A subsequent systematic study of this effect with animals as their own controls in a counterbalanced design confirmed this effect (Figure 3).5 This finding then led to the test in the human epileptic subject described above.1
Platform for a dual research approach
These two studies provided several interesting conclusions that directed our subsequent scientific efforts. First, in the cat study we observed a common prodrome in both SMR-trained and control animals even though the SMR-trained animals had acquired protection against seizures. This suggested a direct effect on the seizure process and not on MMH toxicity in general. Second, in our human epileptic patient, the seizures that were suppressed arose out of the unconscious state of sleep, a fact that eliminated the possibility of any voluntary countermeasure and again indicated a direct effect on the seizure mechanism. Accordingly, we undertook a dual approach to understanding the basis of this effect, involving both additional animal electrophysiologic and human clinical studies.
Animal studies evaluated motor behavior, motor reflexes, motor and thalamic unit firing, and somatosensory pathway correlates of the SMR response. Clinical studies, as reviewed in the following section, sought to further document the anticonvulsant effects of SMR operant conditioning and examine this effect on various seizure types. Possible alternative explanations, such as altered medication compliance and placebo effects, were also addressed in several comprehensive studies. Additionally, by this time other laboratories were beginning to add to the research literature in this new field.
Reproduced, with permission of the American Academy of Sleep Medicine, from Sleep (Sterman MB, et al. Sensorimotor electroencephalogram rhythmic activity: a functional gate mechanism. Sleep 1981; 4(4):408–422).
Figure 4. Trained sensorimotor rhythm (SMR) responses are associated with changes in both afferent and efferent pathways of the sensorimotor system. These include decreased red nucleus (RN) activity, stretch reflex excitability, and muscle tone. These changes produce reduced somatic afferent (SA) discharge and lead to thalamic hyperpolarization and reciprocal oscillatory burst activity between the ventrobasal (vPL) and reticular (nRt) nuclei of the thalamus. This burst activity is propagated to sensorimotor cortex (S1) and initiates corresponding bursts of SMR activity.6Neurophysiologic studies in cats revealed a convergent pattern of changes that were directly correlated with the SMR pattern in the EEG and clearly indicated reduced motor excitability. These included a specific attenuation of cellular activity and reflex excitability in the motor pathway, a reduction in muscle tone and associated motor unit firing, and cessation of behavioral movements. Further, unit studies in afferent nuclei of the somatosensory pathway revealed evidence of reduced somatic afferent firing and the onset of reciprocal burst oscillation between the thalamic reticular nucleus and the adjacent ventrobasal relay nucleus. This oscillation provides the thalamic source of the cortical SMR pattern. These findings are summarized in Figure 4.6 Details of the studies and resulting publications are provided in recent review articles.7–9 They represent empirical evidence for significant reorganization of neuronal function when SMR activity appears in the sensorimotor EEG.
CLINICAL STUDIES
A series of human studies followed our initial clinical report, including group studies involving crossover and placebo-controlled designs. These studies consistently reported significant seizure reductions in epileptic patients in response to reward for increasing sensorimotor EEG rhythmic activity.
Two independent meta-analyses of the peer-reviewed papers in this literature have appeared in the last decade.8,10 In a review of 24 studies involving 243 patients with predominantly partial complex seizures provided with central cortical SMR feedback training, Sterman determined that 82% of these subjects registered seizure reductions greater than 50%.8 More recently, Tan and colleagues evaluated data from 63 studies and selected for comprehensive analysis 10 studies that met stringent criteria for controls and population and seizure details.10 They reported that 79% of the patients treated with SMR feedback training experienced a statistically significant reduction in seizure frequency despite a collective history of failed medication therapy.
Data are from Lantz and Sterman.11
Figure 5. Reported seizure rates in three experimental groups of randomly assigned patients with medication-refractory complex partial seizures.11 Each group of 8 subjects received 6 weeks of treatment consisting of either (1) detailed tabulation of seizures, (2) non-contingent sensorimotor rhythm (SMR) training (“yoked control”), or (3) contingent SMR training. Following this initial 6-week period, all 24 subjects were combined into one contingent SMR training group for 6 additional weeks and then gradually withdrawn from training. A final 6-week follow-up seizure tabulation period completed the analysis. Data are plotted against group baselines. A significant reduction in seizures was registered after contingent training only, and this effect increased progressively across subsequent conditions.Data from one of the studies11 evaluated in both of these systematic reviews are summarized in Figure 5. In this study, 24 subjects with complex partial seizures, many with seizure foci confirmed through depth recordings, were randomly assigned to three experimental treatment groups:
One group simply tabulated their seizure experiences for 6 weeks using a comprehensive logging method.
The second group received EEG feedback training for 1 hour three times a week for 6 weeks; however, the EEG signal responsible for reward was previously recorded from a different individual. This noncontingent feedback constituted a “yoked control” group.
The third group received 6 weeks of contingent training for increasing SMR activity in somatosensory cortex while simultaneously suppressing slower 4- to 8-Hz activity.
After the initial 6 weeks, all 24 subjects were combined into one group that received 6 more weeks of contingent training only. This was followed by a 4-week period of gradual withdrawal from training and then by a final tabulation of seizure incidence during a 6-week period after training was terminated. As can be seen in Figure 5, the seizure tabulation control was associated with an increased seizure count and the “yoked control” noncontingent SMR training was associated with no significant change in seizure incidence, whereas contingent SMR training was associated with a statistically significant reduction in seizures. The statistical significance of this reduction increased progressively as subjects from the other two groups were added to a second 6-week period of contingent training, and after an additional 6 weeks following withdrawal from training. In addition to this exclusive seizure reduction after SMR contingent training, pre-/post-training neuropsychological testing showed that responding SMR-trained subjects also improved significantly in performance of tasks specific to the hemisphere contralateral to their frontotemporal lesion, indicating a reduced corrosive disturbance from the seizure focus.11
EEG BIOFEEDBACK IN PRACTICE: PROFILE OF THE AUTHOR’S PROGRAM
EEG operant conditioning methods for biofeedback training have diversified as various hardware and software products have emerged and as individuals with differing backgrounds and credentials have entered the field. A lack of methodologic standards and professional regulations has contributed to an undesirable inconsistency in the competence and effectiveness of therapeutic applications. Nevertheless, abundant peer-reviewed research by qualified investigators has proven the worth of this method as a viable alternative treatment for seizure disorders, so I will attempt to provide some idea of a systematic and evidence-guided approach to treatment as used in our program.
Figure 6. This 12-year-old girl has suffered since early childhood from frequent multiple seizure types and myoclonic jerks that are unresponsive to pharmacologic treatments. She currently functions at about third-grade level but is aware and behaviorally compliant. Here she is responding to visual feedback in the context of sensorimotor rhythm training. Her mother assists by providing raisin and candy rewards when certain response criteria are achieved. Her seizures have declined in frequency and severity.Patients are subjected to a quantitative multi-channel EEG evaluation (QEEG) using hardware and software complying with both technical and learning-theory principles critical to valid data collection and operant conditioning applications. Data obtained from this study are combined with medical reports from other studies and information gained in a comprehensive intake interview. QEEG and background information guide the design of an empirical protocol, often with several training components, that is used consistently throughout the treatment period, which consists of one or two 60- to 90-minute treatment sessions per week for at least 20 weeks. Subjects are seated in front of a large-monitor screen and instructed on the requirements for reward. Reinforcement consists of visual images and tones, as well as a numeric display of scores achieved and the time remaining in a trial. On rare occasion a committed parent may be seated next to a more challenged patient and provide additional reinforcement in the form of earned treats, such as raisins and pieces of candy (Figure 6).
Figure 7. Primary display used in our sensorimotor rhythm biofeedback program. The display conforms strictly to operant conditioning principles while still promoting cognitive engagement in the human subject. Reward here is for two different EEG frequencies at two different cortical sites. The far left “green” site shows reinforced 12- to 15-Hz bandpass activity at the C3 electrode site. Low-frequency suppression of abnormal 3- to 5-Hz slow activity at Fz is addressed here through “successive approximation” and consumes the final three display units from left to right. See text for more details. Display results are for the subject depicted in Figure 6.The display that subjects see can varies within limits but must always be as simple as possible and must provide information exclusively relevant to achieving the desired EEG changes. One such display is shown in Figure 7. It consists of a series of four rectangular boxes, each with a segment of band-passed EEG data for selected frequency bands and enclosed by reward threshold guidelines. If the objective is to increase the amplitude and/or incidence of a particular frequency band, the band-pass display must exceed the upper threshold guideline. If the objective is to suppress that frequency band, the display must drop below the threshold line. The duration of the required response can be adjusted and is typically 0.25 to 0.5 seconds. When the desired response is achieved a small horizontal bar at the upper right of each band-pass display turns from red to green, and a large blue ball appears above, together with a chime or other tone. The display is frozen for 2 seconds and then becomes active again, thus providing for discrete trials. A yellow score bar at the bottom of the screen advances by one unit. The timing of each performance set (typically 3 minutes) is indicated by a moving blue bar at the bottom of the screen.
With each box monitoring the same electrode site and each frequency tuned to the same band, thresholds can be set to promote facilitation or suppression through “successive approximation,” or sequencing from left to right with sequentially more difficult thresholds. Numerous other configurations are possible. In the case shown in Figure 7, the band-pass at the far left is set at 12 to 15 Hz (SMR) for the C3 electrode site, and the remaining three bands to the right are set to 3 to 5 Hz at the left medial frontal location Fz, with successively lower thresholds to promote suppression of this band at this site.
Figure 8. Performance plot from the patient represented in Figures 6 and 7. The plot registers the scoring rate per 3-minute trial at top, along with corresponding smoothed amplitude output in the band-passed frequencies set for each electrode placement. In this case the patient was rewarded for increasing 12- to 15-Hz sensorimotor rhythm (SMR) activity (top EEG trace) and decreasing 3- to 5-Hz slow activity at Fz (bottom EEG trace) The subject showed response acquisition both within and across 3-minute feedback trials, together with a stabilization of the SMR frequency and a reduction in frontal slow activity.Performance outcome is measured systematically by tracking the scoring rate per trial, together with associated EEG patterns. Data from the 12-year-old female subject described above provide an example. The top of Figure 8 shows a plot of reward rate across four successive 3-minute EEG feedback trials. The patient was rewarded for simultaneously increasing 12- to 15-Hz SMR activity at C3 and reducing 3- to 5-Hz activity at Fz, as described above. Smoothed EEG plots for the targeted frequency bands are shown below these reward curves, starting with the C3 12- to 15-Hz channel. Activity in this band became increasingly stable across trials. Data from three frontal recording sites are also shown, with the targeted Fz 3- to 5-Hz band output at the bottom. Amplitudes decreased progressively at all frontal sites but most markedly at the bottom Fz location. Thus, SMR stabilization and simultaneously suppressed frontal slow activity resulted in a progressive pattern of incremental reward both within trials and across the session. The resulting profiles are indicative of learning.
A RATIONAL MODEL FROM RECENT NEUROIMAGING STUDIES
While it is difficult to evaluate neurophysiologic changes in human subjects to a degree similar to that in animals, certain parallels can be drawn. Further, new imaging methods allow for assessment of localized metabolic changes in the human brain during and after EEG feedback training. Behaviorally, during successful SMR training, human subjects become behaviorally quiet and direct their attention to the task. It is safe to presume that the SMR response develops as a result of reduced motor excitation and resulting intrathalamic ventrobasal oscillations, since this mechanism is well established as a basis for mammalian sensorimotor EEG rhythm generation.12 These changes, as well as others documented in our animal studies, set the stage for the development of SMR activity and are likely collectively initiated by altered input from some other executive system.
Reprinted from Lévesque J, et al. Effect of neurofeedback training on the neural substrates of selective attention in children with attention-deficit/hyperactivity disorder. Neurosci Lett 2006;394:216-221. Copyright 2006 with permission from Elsevier.
Figure 9. Functional magnetic resonance images before (left) and after (right) sensorimotor rhythm (SMR) feedback training in a study of the effect of SMR training in learning-disabled children.13 The images are sagittal sections for the data averaged across subjects, who either received SMR feedback training (experimental group) or did not (control group). In the pretraining condition, significant loci of activation were noted in the left superior parietal lobe for both groups. In the posttraining condition, activations were again seen in this cortical region for both groups. In addition, the experimental group also showed stronger and statistically significant loci of activation in the left striatum and substantia nigra.Several recent studies have suggested a specific pattern of motor inhibition output from the striatum of the basal ganglia as the source of these changes. Birbaumer observed increased striatal metabolic activity with functional magnetic resonance imaging (fMRI) analysis in subjects producing SMR activity (personal communication, 2005). Further, Lévesque and colleagues studied pre-/post-fMRI blood oxygenation level–dependent response patterns in learning-disabled children trained to increase SMR activity and found a specific increase in the metabolic activity of the striatum and substantia nigra (Figure 9).13 The SMR-trained subjects showed significant academic improvement as well.13
These facts provide a rational model for a threshold-altering process that could affect seizure discharge propagation to motor networks. Although there are many different neurotransmitters used within the basal ganglia (principally acetylcholine, gamma-aminobutyric acid, and dopamine), the overall effect on thalamus and premotor networks in the mesencephalic tegmentum and superior colliculus is inhibitory.14–16 If activation of these inhibitory basal ganglia networks can become labeled by the SMR through contingent feedback training, and if responsible circuits can be potentiated by this association, motor inhibitory regulation would be generally facilitated.
CONCLUSIONS
Despite the encouraging findings and concepts reviewed here, there are significant issues at virtually every step of the thinking and practice behind this new therapy. This method depends on a comprehensive understanding of the EEG signal and the technical requirements of valid quantitative analysis and feedback applications. This includes a basic knowledge of the principles essential for effective operant conditioning. Further, in light of the complexity of seizure disorders, accurate history and seizure classification must be evaluated and understood.
Alternative explanations for therapeutic results include such considerations as short-lasting expectation effects and changes in patient behavior. However, it must again be noted that the prolonged anticonvulsant effect documented in our animal studies, as well as in relation to nocturnal seizures arising out of sleep in a human subject, would seem to rule out placebo or nonspecific effects. This conclusion is supported further by the finding of improved neuropsychological performance after SMR training in tasks mediated by the hemisphere contralateral to disrupting localized epileptogenic lesions. Additionally, an alternative explanation for improved seizure control based on increased medication compliance has been rejected through studies that carefully monitored blood levels of prescribed anticonvulsant drugs before, during, and after training.
Finally, the epileptic patients who have demonstrated clinical improvement in EEG biofeedback research studies, along with many who seek this treatment today, represent unquestionable failures of anticonvulsant drug therapy. Notably, positive outcomes have frequently been achieved in patients with complex-partial seizures, an extremely difficult-to-treat seizure type. It is therefore unfortunate that some professionals still criticize neurofeedback therapy for a lack of more consistent or successful outcomes. On the contrary, as noted here, evidence has shown that most of these difficult-to-treat patients benefit beyond any chance or placebo outcome, in some cases dramatically so. In light of the frequent adverse effects and costs associated with lifelong pharmacotherapy, we view EEG biofeedback therapy not as a “last resort” option to be restricted solely to pharmacotherapy-resistant cases but rather as a generally viable consideration for any patient suffering from seizures. Moreover, in contrast to drug-dependent management approaches, the altered modulation of striatal and thalamocortical inhibition that is possible through neurofeedback training may sufficiently raise seizure thresholds to greatly increase the prospects for the long-term nondependent management of epilepsy.
The attempt to alter electroencephalographic (EEG) frequency/amplitude patterns and their underlying brain mechanisms using contingent operant conditioning methods is today referred to variously as EEG biofeedback, neurofeedback, or neurotherapy. This article traces the history of the clinical application of EEG operant conditioning from empirical animal investigations to its emergence as a treatment option for major seizure types. In light of the diversity of the clinical applications of this method in general, and the complexity of seizure disorders in particular, I also present an overview of specific methods used in our EEG biofeedback program.
Figure 1. A carefully documented 6-year seizure data log from an adult female subject (aged 23 years at the start of the log) with nocturnal tonic-clonic seizures, often with incontinence.2 The log starts 1 year before initiation of electroencephalographic (EEG) feedback training (“Pre-SMR”), continues through 2.5 years of twice-weekly EEG training sessions (“Post-SMR”), and continues through 2.5 years after wi thdrawal from this training (“Withdrawal”). Training consisted of auditory and visual reward for increased 12- to 15-Hz EEG activity over the left sensorimotor cortex, which has been labeled the sensorimotor rhythm (SMR). Medications were held constant during training and adjusted downward after withdrawal from training. In 1977 this patient was issued a California driver’s license.This application was officially added to the broader field of biofeedback with the publication of a 1972 paper by Sterman and Friar titled, “Suppression of seizures in an epileptic following sensorimotor EEG feedback training.” 1 In this paper we documented a sustained and progressive reduction of generalized nocturnal tonic-clonic seizures in a 23-year-old female epileptic with a 7-year history of frequent and medically refractory seizures of unknown origin. The patient’s clinical EEG showed left sensorimotor cortex spikes and slow 5- to 7-Hz activity. Seizure reduction occurred in response to an experimental course of EEG operant conditioning aimed at increasing 12- to 15-Hz EEG activity in the left sensorimotor cortex while suppressing slower activity at this same site. The 12- to 15-Hz EEG rhythm was discovered in animal research and labeled as the sensorimotor rhythm (SMR). Although the patient had previously been worked up and treated unsuccessfully with anticonvulsant medications at several prestigious medical institutions, over the course of 2.5 years of twice-weekly EEG feedback training sessions she became essentially seizure free (Figure 1)2 and was ultimately issued a California driver’s license.
BACKDROP TO THE CLINICAL APPLICATION: KEY ANIMAL STUDIES
Figure reprinted from Sterman et al (Science 1970; 167:1146–1148).3
Figure 2. Bipolar electroencephalographic (EEG) samples from sensorimotor and parietal cortex in the cat during quiet (motionless) wakefulness (left) and quiet (non-REM) sleep (right). Both states are associated with bursts of 12- to 15-Hz EEG rhythmic activity in sensorimotor cortex. During sleep these bursts are higher in amplitude and associated with slower rhythmic patterns in parietal cortex.The above landmark study was predicated on the observation of a discrete 11- to 19-Hz EEG rhythmic pattern in cats, which occurred intermittently over the sensorimotor cortex during behavioral quiescence. When animals were trained to suppress a learned bar-press response for food if a tone was sounded in the chamber, a 12- to 15-Hz version of this EEG pattern always accompanied inhibition of the bar-press response. If animals later fell asleep, a similar rhythmic EEG pattern, known as the sleep spindle, was localized to the same cortical area at the same frequency (Figure 2).3 Our interest at the time was in the neurophysiological control of sleep. Because both of these patterns occurred uniquely in the absence of movement, we sought to determine if the underlying neural mechanisms were related.
To accomplish this, we attempted to facilitate the SMR during wakefulness using an operant conditioning paradigm with a liquid food reward, and then study any resulting changes in sleep spindle activity and sleep structure. Necessary quality controls included alternate training to suppress this rhythm and a counterbalanced design employing two separate groups of cats. Six weeks of three training sessions per week to satiation led to profound and differential changes in sleep EEG and sleep architecture. SMR training, whether it preceded or followed suppression training, led to a significant increase in EEG sleep spindle density, as well as a significant reduction in sleep period fragmentation due to arousals. No changes occurred in the control condition.3
A more profound finding in the cat
Figure modified from Sterman.5
Figure 3. The sequence of prodromal events preceding generalized convulsions in two groups of 10 cats, all of which were injected intra-abdominally with 9 mg/kg of GABA-depleting monomethyl hydrazine. One group (dashed tracing) had received 6 weeks of electroencephalographic feedback training for sensorimotor rhythm (SMR) enhancement with food reward (see text). The two groups did not differ statistically in the latency to prodromal symptoms. All control animals seized reliably at approximately 60 minutes, as had been previously documented. In contrast, the SMR-trained group had a significantly prolonged mean latency to seizures (130 minutes), and several did not seize within the 4-hour test period.As interesting as this finding was, the most profound outcome of the study emerged later. A different cat study under way in our laboratory, funded by the US Air Force, was seeking to determine the effects on behavior of low-dose exposure to monomethyl hydrazine (MMH).4 This compound is a highly toxic component of the liquid rocket fuel used for launching virtually all space vehicles. Significant MMH exposure via any route causes pro-found nausea and gradual onset of convulsions, which are lethal at adequate doses. The mechanism for this effect was ultimately determined to be a disruption of the synthesis of gamma-aminobutyric acid, the primary inhibitory neurotransmitter in the central nervous system. We were investigating the effects of low-dose exposure to determine the possible disruption of cognitive functions such exposure might cause in flight crews. Our first objective for studies in cats was to establish the doseresponse curve for convulsive effects in that species. We had succeeded in determining a curve showing that 9 mg/kg of MMH was the threshold dose for reliably producing nonlethal convulsions after a prodrome of approximately 40 to 67 minutes. This prodrome consisted of a sequence of reliable autonomic and behavioral events. When data from animals provided with SMR operant conditioning as the final training procedure were added to this curve, the same prodrome was observed but there were no seizures at 60 minutes. Instead, the latency to seizures was delayed to a range of 80 to 220 minutes, and several animals failed to seize at all.4 A subsequent systematic study of this effect with animals as their own controls in a counterbalanced design confirmed this effect (Figure 3).5 This finding then led to the test in the human epileptic subject described above.1
Platform for a dual research approach
These two studies provided several interesting conclusions that directed our subsequent scientific efforts. First, in the cat study we observed a common prodrome in both SMR-trained and control animals even though the SMR-trained animals had acquired protection against seizures. This suggested a direct effect on the seizure process and not on MMH toxicity in general. Second, in our human epileptic patient, the seizures that were suppressed arose out of the unconscious state of sleep, a fact that eliminated the possibility of any voluntary countermeasure and again indicated a direct effect on the seizure mechanism. Accordingly, we undertook a dual approach to understanding the basis of this effect, involving both additional animal electrophysiologic and human clinical studies.
Animal studies evaluated motor behavior, motor reflexes, motor and thalamic unit firing, and somatosensory pathway correlates of the SMR response. Clinical studies, as reviewed in the following section, sought to further document the anticonvulsant effects of SMR operant conditioning and examine this effect on various seizure types. Possible alternative explanations, such as altered medication compliance and placebo effects, were also addressed in several comprehensive studies. Additionally, by this time other laboratories were beginning to add to the research literature in this new field.
Reproduced, with permission of the American Academy of Sleep Medicine, from Sleep (Sterman MB, et al. Sensorimotor electroencephalogram rhythmic activity: a functional gate mechanism. Sleep 1981; 4(4):408–422).
Figure 4. Trained sensorimotor rhythm (SMR) responses are associated with changes in both afferent and efferent pathways of the sensorimotor system. These include decreased red nucleus (RN) activity, stretch reflex excitability, and muscle tone. These changes produce reduced somatic afferent (SA) discharge and lead to thalamic hyperpolarization and reciprocal oscillatory burst activity between the ventrobasal (vPL) and reticular (nRt) nuclei of the thalamus. This burst activity is propagated to sensorimotor cortex (S1) and initiates corresponding bursts of SMR activity.6Neurophysiologic studies in cats revealed a convergent pattern of changes that were directly correlated with the SMR pattern in the EEG and clearly indicated reduced motor excitability. These included a specific attenuation of cellular activity and reflex excitability in the motor pathway, a reduction in muscle tone and associated motor unit firing, and cessation of behavioral movements. Further, unit studies in afferent nuclei of the somatosensory pathway revealed evidence of reduced somatic afferent firing and the onset of reciprocal burst oscillation between the thalamic reticular nucleus and the adjacent ventrobasal relay nucleus. This oscillation provides the thalamic source of the cortical SMR pattern. These findings are summarized in Figure 4.6 Details of the studies and resulting publications are provided in recent review articles.7–9 They represent empirical evidence for significant reorganization of neuronal function when SMR activity appears in the sensorimotor EEG.
CLINICAL STUDIES
A series of human studies followed our initial clinical report, including group studies involving crossover and placebo-controlled designs. These studies consistently reported significant seizure reductions in epileptic patients in response to reward for increasing sensorimotor EEG rhythmic activity.
Two independent meta-analyses of the peer-reviewed papers in this literature have appeared in the last decade.8,10 In a review of 24 studies involving 243 patients with predominantly partial complex seizures provided with central cortical SMR feedback training, Sterman determined that 82% of these subjects registered seizure reductions greater than 50%.8 More recently, Tan and colleagues evaluated data from 63 studies and selected for comprehensive analysis 10 studies that met stringent criteria for controls and population and seizure details.10 They reported that 79% of the patients treated with SMR feedback training experienced a statistically significant reduction in seizure frequency despite a collective history of failed medication therapy.
Data are from Lantz and Sterman.11
Figure 5. Reported seizure rates in three experimental groups of randomly assigned patients with medication-refractory complex partial seizures.11 Each group of 8 subjects received 6 weeks of treatment consisting of either (1) detailed tabulation of seizures, (2) non-contingent sensorimotor rhythm (SMR) training (“yoked control”), or (3) contingent SMR training. Following this initial 6-week period, all 24 subjects were combined into one contingent SMR training group for 6 additional weeks and then gradually withdrawn from training. A final 6-week follow-up seizure tabulation period completed the analysis. Data are plotted against group baselines. A significant reduction in seizures was registered after contingent training only, and this effect increased progressively across subsequent conditions.Data from one of the studies11 evaluated in both of these systematic reviews are summarized in Figure 5. In this study, 24 subjects with complex partial seizures, many with seizure foci confirmed through depth recordings, were randomly assigned to three experimental treatment groups:
One group simply tabulated their seizure experiences for 6 weeks using a comprehensive logging method.
The second group received EEG feedback training for 1 hour three times a week for 6 weeks; however, the EEG signal responsible for reward was previously recorded from a different individual. This noncontingent feedback constituted a “yoked control” group.
The third group received 6 weeks of contingent training for increasing SMR activity in somatosensory cortex while simultaneously suppressing slower 4- to 8-Hz activity.
After the initial 6 weeks, all 24 subjects were combined into one group that received 6 more weeks of contingent training only. This was followed by a 4-week period of gradual withdrawal from training and then by a final tabulation of seizure incidence during a 6-week period after training was terminated. As can be seen in Figure 5, the seizure tabulation control was associated with an increased seizure count and the “yoked control” noncontingent SMR training was associated with no significant change in seizure incidence, whereas contingent SMR training was associated with a statistically significant reduction in seizures. The statistical significance of this reduction increased progressively as subjects from the other two groups were added to a second 6-week period of contingent training, and after an additional 6 weeks following withdrawal from training. In addition to this exclusive seizure reduction after SMR contingent training, pre-/post-training neuropsychological testing showed that responding SMR-trained subjects also improved significantly in performance of tasks specific to the hemisphere contralateral to their frontotemporal lesion, indicating a reduced corrosive disturbance from the seizure focus.11
EEG BIOFEEDBACK IN PRACTICE: PROFILE OF THE AUTHOR’S PROGRAM
EEG operant conditioning methods for biofeedback training have diversified as various hardware and software products have emerged and as individuals with differing backgrounds and credentials have entered the field. A lack of methodologic standards and professional regulations has contributed to an undesirable inconsistency in the competence and effectiveness of therapeutic applications. Nevertheless, abundant peer-reviewed research by qualified investigators has proven the worth of this method as a viable alternative treatment for seizure disorders, so I will attempt to provide some idea of a systematic and evidence-guided approach to treatment as used in our program.
Figure 6. This 12-year-old girl has suffered since early childhood from frequent multiple seizure types and myoclonic jerks that are unresponsive to pharmacologic treatments. She currently functions at about third-grade level but is aware and behaviorally compliant. Here she is responding to visual feedback in the context of sensorimotor rhythm training. Her mother assists by providing raisin and candy rewards when certain response criteria are achieved. Her seizures have declined in frequency and severity.Patients are subjected to a quantitative multi-channel EEG evaluation (QEEG) using hardware and software complying with both technical and learning-theory principles critical to valid data collection and operant conditioning applications. Data obtained from this study are combined with medical reports from other studies and information gained in a comprehensive intake interview. QEEG and background information guide the design of an empirical protocol, often with several training components, that is used consistently throughout the treatment period, which consists of one or two 60- to 90-minute treatment sessions per week for at least 20 weeks. Subjects are seated in front of a large-monitor screen and instructed on the requirements for reward. Reinforcement consists of visual images and tones, as well as a numeric display of scores achieved and the time remaining in a trial. On rare occasion a committed parent may be seated next to a more challenged patient and provide additional reinforcement in the form of earned treats, such as raisins and pieces of candy (Figure 6).
Figure 7. Primary display used in our sensorimotor rhythm biofeedback program. The display conforms strictly to operant conditioning principles while still promoting cognitive engagement in the human subject. Reward here is for two different EEG frequencies at two different cortical sites. The far left “green” site shows reinforced 12- to 15-Hz bandpass activity at the C3 electrode site. Low-frequency suppression of abnormal 3- to 5-Hz slow activity at Fz is addressed here through “successive approximation” and consumes the final three display units from left to right. See text for more details. Display results are for the subject depicted in Figure 6.The display that subjects see can varies within limits but must always be as simple as possible and must provide information exclusively relevant to achieving the desired EEG changes. One such display is shown in Figure 7. It consists of a series of four rectangular boxes, each with a segment of band-passed EEG data for selected frequency bands and enclosed by reward threshold guidelines. If the objective is to increase the amplitude and/or incidence of a particular frequency band, the band-pass display must exceed the upper threshold guideline. If the objective is to suppress that frequency band, the display must drop below the threshold line. The duration of the required response can be adjusted and is typically 0.25 to 0.5 seconds. When the desired response is achieved a small horizontal bar at the upper right of each band-pass display turns from red to green, and a large blue ball appears above, together with a chime or other tone. The display is frozen for 2 seconds and then becomes active again, thus providing for discrete trials. A yellow score bar at the bottom of the screen advances by one unit. The timing of each performance set (typically 3 minutes) is indicated by a moving blue bar at the bottom of the screen.
With each box monitoring the same electrode site and each frequency tuned to the same band, thresholds can be set to promote facilitation or suppression through “successive approximation,” or sequencing from left to right with sequentially more difficult thresholds. Numerous other configurations are possible. In the case shown in Figure 7, the band-pass at the far left is set at 12 to 15 Hz (SMR) for the C3 electrode site, and the remaining three bands to the right are set to 3 to 5 Hz at the left medial frontal location Fz, with successively lower thresholds to promote suppression of this band at this site.
Figure 8. Performance plot from the patient represented in Figures 6 and 7. The plot registers the scoring rate per 3-minute trial at top, along with corresponding smoothed amplitude output in the band-passed frequencies set for each electrode placement. In this case the patient was rewarded for increasing 12- to 15-Hz sensorimotor rhythm (SMR) activity (top EEG trace) and decreasing 3- to 5-Hz slow activity at Fz (bottom EEG trace) The subject showed response acquisition both within and across 3-minute feedback trials, together with a stabilization of the SMR frequency and a reduction in frontal slow activity.Performance outcome is measured systematically by tracking the scoring rate per trial, together with associated EEG patterns. Data from the 12-year-old female subject described above provide an example. The top of Figure 8 shows a plot of reward rate across four successive 3-minute EEG feedback trials. The patient was rewarded for simultaneously increasing 12- to 15-Hz SMR activity at C3 and reducing 3- to 5-Hz activity at Fz, as described above. Smoothed EEG plots for the targeted frequency bands are shown below these reward curves, starting with the C3 12- to 15-Hz channel. Activity in this band became increasingly stable across trials. Data from three frontal recording sites are also shown, with the targeted Fz 3- to 5-Hz band output at the bottom. Amplitudes decreased progressively at all frontal sites but most markedly at the bottom Fz location. Thus, SMR stabilization and simultaneously suppressed frontal slow activity resulted in a progressive pattern of incremental reward both within trials and across the session. The resulting profiles are indicative of learning.
A RATIONAL MODEL FROM RECENT NEUROIMAGING STUDIES
While it is difficult to evaluate neurophysiologic changes in human subjects to a degree similar to that in animals, certain parallels can be drawn. Further, new imaging methods allow for assessment of localized metabolic changes in the human brain during and after EEG feedback training. Behaviorally, during successful SMR training, human subjects become behaviorally quiet and direct their attention to the task. It is safe to presume that the SMR response develops as a result of reduced motor excitation and resulting intrathalamic ventrobasal oscillations, since this mechanism is well established as a basis for mammalian sensorimotor EEG rhythm generation.12 These changes, as well as others documented in our animal studies, set the stage for the development of SMR activity and are likely collectively initiated by altered input from some other executive system.
Reprinted from Lévesque J, et al. Effect of neurofeedback training on the neural substrates of selective attention in children with attention-deficit/hyperactivity disorder. Neurosci Lett 2006;394:216-221. Copyright 2006 with permission from Elsevier.
Figure 9. Functional magnetic resonance images before (left) and after (right) sensorimotor rhythm (SMR) feedback training in a study of the effect of SMR training in learning-disabled children.13 The images are sagittal sections for the data averaged across subjects, who either received SMR feedback training (experimental group) or did not (control group). In the pretraining condition, significant loci of activation were noted in the left superior parietal lobe for both groups. In the posttraining condition, activations were again seen in this cortical region for both groups. In addition, the experimental group also showed stronger and statistically significant loci of activation in the left striatum and substantia nigra.Several recent studies have suggested a specific pattern of motor inhibition output from the striatum of the basal ganglia as the source of these changes. Birbaumer observed increased striatal metabolic activity with functional magnetic resonance imaging (fMRI) analysis in subjects producing SMR activity (personal communication, 2005). Further, Lévesque and colleagues studied pre-/post-fMRI blood oxygenation level–dependent response patterns in learning-disabled children trained to increase SMR activity and found a specific increase in the metabolic activity of the striatum and substantia nigra (Figure 9).13 The SMR-trained subjects showed significant academic improvement as well.13
These facts provide a rational model for a threshold-altering process that could affect seizure discharge propagation to motor networks. Although there are many different neurotransmitters used within the basal ganglia (principally acetylcholine, gamma-aminobutyric acid, and dopamine), the overall effect on thalamus and premotor networks in the mesencephalic tegmentum and superior colliculus is inhibitory.14–16 If activation of these inhibitory basal ganglia networks can become labeled by the SMR through contingent feedback training, and if responsible circuits can be potentiated by this association, motor inhibitory regulation would be generally facilitated.
CONCLUSIONS
Despite the encouraging findings and concepts reviewed here, there are significant issues at virtually every step of the thinking and practice behind this new therapy. This method depends on a comprehensive understanding of the EEG signal and the technical requirements of valid quantitative analysis and feedback applications. This includes a basic knowledge of the principles essential for effective operant conditioning. Further, in light of the complexity of seizure disorders, accurate history and seizure classification must be evaluated and understood.
Alternative explanations for therapeutic results include such considerations as short-lasting expectation effects and changes in patient behavior. However, it must again be noted that the prolonged anticonvulsant effect documented in our animal studies, as well as in relation to nocturnal seizures arising out of sleep in a human subject, would seem to rule out placebo or nonspecific effects. This conclusion is supported further by the finding of improved neuropsychological performance after SMR training in tasks mediated by the hemisphere contralateral to disrupting localized epileptogenic lesions. Additionally, an alternative explanation for improved seizure control based on increased medication compliance has been rejected through studies that carefully monitored blood levels of prescribed anticonvulsant drugs before, during, and after training.
Finally, the epileptic patients who have demonstrated clinical improvement in EEG biofeedback research studies, along with many who seek this treatment today, represent unquestionable failures of anticonvulsant drug therapy. Notably, positive outcomes have frequently been achieved in patients with complex-partial seizures, an extremely difficult-to-treat seizure type. It is therefore unfortunate that some professionals still criticize neurofeedback therapy for a lack of more consistent or successful outcomes. On the contrary, as noted here, evidence has shown that most of these difficult-to-treat patients benefit beyond any chance or placebo outcome, in some cases dramatically so. In light of the frequent adverse effects and costs associated with lifelong pharmacotherapy, we view EEG biofeedback therapy not as a “last resort” option to be restricted solely to pharmacotherapy-resistant cases but rather as a generally viable consideration for any patient suffering from seizures. Moreover, in contrast to drug-dependent management approaches, the altered modulation of striatal and thalamocortical inhibition that is possible through neurofeedback training may sufficiently raise seizure thresholds to greatly increase the prospects for the long-term nondependent management of epilepsy.
References
Sterman MB, Friar L. Suppression of seizures in an epileptic following sensorimotor EEG feedback training. Electroencephalogr Clin Neurophysiol1972; 33:89–95.
Sterman MB. Effects of sensorimotor EEG feedback training on sleep and clinical manifestations of epilepsy. In:Beatty J, Legewie H, eds. Biofeedback and Behavior. New York, NY: Plenum Press; 1977:167–200.
Sterman MB, Howe RD, Macdonald LR. Facilitation of spindle-burst sleep by conditioning of electroencephalographic activity while awake. Science1970; 167:1146–1148.
Sterman MB, LoPresti RW, Fairchild MD. Electroencephalographic and behavioral studies of monomethyl hydrazine toxicity in the cat. Aerospace Medical Research Laboratory1969; AMRL-TR-69-3:1–8.
Sterman MB. Studies of EEG biofeedback training in man and cats. In: Veterans Administration Studies in Mental Health and Behavioral Sciences: Highlights of the 17th Annual Conference. Perry Point, MD: US Veterans Administration; 1972:50–60.
Sterman MB. Physiological origins and functional correlates of EEG rhythmic activities: implications for self-regulation. Biofeedack Self Reg1996; 21:3–33.
Sterman MB. Basic concepts and clinical findings in the treatment of seizure disorders with EEG operant conditioning. Clin Electroencephalogr2000; 31:45–55.
Egner T, Sterman MB. Neurofeedback treatment of epilepsy: from basic rationale to practical application. Expert Rev Neurother2006; 6:247–257.
Tan G, Thornby J, Hammond D, et al Meta-analysis of EEG biofeedback in treating epilepsy. Clin EEG Neurosci2009; 40:173–179.
Lantz D, Sterman MB. Neuropsychological assessment of subjects with uncontrolled epilepsy: effects of EEG biofeedback training. Epilepsia1988; 29:163–171.
Steriade M, McCormick DA, Seinowski TJ. Thalamocortical oscillations in the sleeping and aroused brain. Science1993; 262:679–685.
Lévesque J, Beauregard M, Mensour B. Effect of neurofeedback training on the neural substrates of selective attention in children with attention-deficit/hyperactivity disorder: a functional magnetic resonance imaging study. Neurosci Lett2006; 394:216–221.
Brodal P. The basal ganglia. In: The Central Nervous System: Structure and Function. New York, NY: Oxford University Press; 1992:246–261.
Chevalier G, Deniau JM. Disinhibition as a basic process in the expression of striatal functions. Trends Neurosci1990; 13:277–280.
DeLong MR. Primate models of movement disorders of basal ganglia origin. Trends Neurosci1990; 13:281–285.
References
Sterman MB, Friar L. Suppression of seizures in an epileptic following sensorimotor EEG feedback training. Electroencephalogr Clin Neurophysiol1972; 33:89–95.
Sterman MB. Effects of sensorimotor EEG feedback training on sleep and clinical manifestations of epilepsy. In:Beatty J, Legewie H, eds. Biofeedback and Behavior. New York, NY: Plenum Press; 1977:167–200.
Sterman MB, Howe RD, Macdonald LR. Facilitation of spindle-burst sleep by conditioning of electroencephalographic activity while awake. Science1970; 167:1146–1148.
Sterman MB, LoPresti RW, Fairchild MD. Electroencephalographic and behavioral studies of monomethyl hydrazine toxicity in the cat. Aerospace Medical Research Laboratory1969; AMRL-TR-69-3:1–8.
Sterman MB. Studies of EEG biofeedback training in man and cats. In: Veterans Administration Studies in Mental Health and Behavioral Sciences: Highlights of the 17th Annual Conference. Perry Point, MD: US Veterans Administration; 1972:50–60.
Sterman MB. Physiological origins and functional correlates of EEG rhythmic activities: implications for self-regulation. Biofeedack Self Reg1996; 21:3–33.
Sterman MB. Basic concepts and clinical findings in the treatment of seizure disorders with EEG operant conditioning. Clin Electroencephalogr2000; 31:45–55.
Egner T, Sterman MB. Neurofeedback treatment of epilepsy: from basic rationale to practical application. Expert Rev Neurother2006; 6:247–257.
Tan G, Thornby J, Hammond D, et al Meta-analysis of EEG biofeedback in treating epilepsy. Clin EEG Neurosci2009; 40:173–179.
Lantz D, Sterman MB. Neuropsychological assessment of subjects with uncontrolled epilepsy: effects of EEG biofeedback training. Epilepsia1988; 29:163–171.
Steriade M, McCormick DA, Seinowski TJ. Thalamocortical oscillations in the sleeping and aroused brain. Science1993; 262:679–685.
Lévesque J, Beauregard M, Mensour B. Effect of neurofeedback training on the neural substrates of selective attention in children with attention-deficit/hyperactivity disorder: a functional magnetic resonance imaging study. Neurosci Lett2006; 394:216–221.
Brodal P. The basal ganglia. In: The Central Nervous System: Structure and Function. New York, NY: Oxford University Press; 1992:246–261.
Chevalier G, Deniau JM. Disinhibition as a basic process in the expression of striatal functions. Trends Neurosci1990; 13:277–280.
DeLong MR. Primate models of movement disorders of basal ganglia origin. Trends Neurosci1990; 13:281–285.
Type 2 diabetes, essential hypertension, obesity, and hyperlipidemia are the major components of the metabolic syndrome. Understanding the psychophysiologic basis of the metabolic syndrome is important since its prevalence has been increasing dramatically over the last decade. In the past, type 2 diabetes was diagnosed almost exclusively in persons in their 40s or older. Health care providers are now reporting emergence of type 2 diabetes and the metabolic syndrome in individuals in their 30s and even their late 20s.1,2
This article outlines the psychophysiologic bases for components of the metabolic syndrome and reviews the application of biofeedback and other psychophysiologic interventions on the two components for which such interventions have been most studied—diabetes and essential hypertension.
ETIOLOGY OF METABOLIC SYNDROME: THE INTERSECTION OF BIOLOGY, LIFESTYLE, STRESS
The disorders that constitute the metabolic syndrome share several etiologic factors. First, genetic predisposition increases the risk for diabetes, hypertension, hyperlipidemia, and obesity.3,4 Second, patients’ own behaviors—their choice of activity or inactivity, their food preferences, and their appetite—lead to gradual loss of control over body weight, blood glucose, blood pressure, and lipid levels. Third, chronic stress and its coincident psychological burden contribute to the etiology of various components of the metabolic syndrome.5 As life events accumulate and individuals lose their ability to cope, the stress response system maintains a higher than optimal level of activation.5–8
Chronic stress affects multiple organ systems, including the two master systems—nervous and endocrine. The biologic effects of stress include disordered breathing, increased activation of the renin-angiotension system, vascular constriction, tachycardia, decreased heart rate variability, inflammation, and sleep disruption.9 The mechanisms involved in acute stress responses are purpose-driven and adaptive. In contrast, chronically activated stress response systems involving increased sympathetic activity, decreased parasympathetic activity, and release of stress hormones have serious deleterious effects.10 Psychobiologic systems fail to adapt, delay recovery, or become exhausted.11
Role of psychological factors
As summarized in a review by Goldbacher and Matthews, 12 psychological factors have been related to increased risk for the metabolic syndrome. Depression has probably been most studied in the settings of cardiovascular disease and diabetes, whereas the psychological states of anger, hostility, and anxiety have been identified as salient etiologic factors in hypertension. In particular, depressed mood has been linked to decreased heart rate variability during the stress response.13 Anxiety affects blood pressure and blood glucose in normal individuals as part of the adaptive stress response, and the effects of anxiety are exacerbated in persons with the metabolic syndrome.14
Importance of sleep
Sleep disruption is often ignored in discussions of the mind-body interface in hypertension and diabetes. However, Knutson and Van Cauter15 suggested that sleep quality and sleep length have important effects on leptin levels and risk for diabetes. Very short sleepers have stronger appetites, as a result of lower leptin concentrations, and are much more likely to be obese compared with long sleepers (≥ 10 hours). This indicates that sleep length and quality affect metabolism. With regard to hypertension, a very important reduction of blood pressure occurs during the night, and a lack of nighttime blood pressure “dipping” is one of the markers for sustained blood pressure elevation.16
Factors overlap and begin to affect self-care
In addition to the effects of stress on mood and anxiety, repeated necessary demands for adaptation have marked effects on self-care behavior. Patients who suffer from anxiety are less efficient in managing their time and may be distracted from monitoring blood glucose and blood pressure. Anxious people often turn to the use of high-calorie comfort foods to soothe themselves during stressful times. Alcohol may be chosen as a means of reducing worry and tension. Depressed people lack the energy needed to maintain medical regimens and tend to be poor adherents to treatment recommendations. They also may choose comfort foods and addictive substances instead of nutritious, high-quality food and drink.17
Both anxiety and depression affect sleep routines and efficiency. Anxious people have trouble getting to sleep and may wake up often during the night, while depressed individuals frequently wake up early and cannot get back to sleep. Additionally, psychological distress influences social behaviors. Overt depressive and anxious symptoms tend not to foster social interactions with family and friends. Lack of social support and a scarcity of personal resources eventually contribute to the risk for diabetes and hypertension.18,19
In short, the metabolic syndrome is most likely to emerge when there is a combination of genetic factors, chronic stress, negative emotion, and unhealthy habits. The application of psychophysiologic interventions to diabetes and hypertension is based on our understanding of the etiology of these disorders, particularly the roles of psychological distress and behavior on blood glucose and blood pressure.
BIOFEEDBACK IN TYPE 2 DIABETES
Diabetes is characterized by elevated blood glucose and resistance of cell membranes to insulin, such that glucose is impeded from crossing from the blood into the cells. Standard treatment consists of oral antihyperglycemic agents, exogenous insulin, diet, and exercise.20 Type 2 diabetes may be the most behaviorally demanding of all chronic illnesses because patients must take an active role in daily management. Typical requirements are to measure blood glucose and take oral medicine, perhaps along with insulin, as well as to exercise, monitor diet, and adjust calories depending on activity level.
Therapy goals and a sampling of evidence
The goal of psychophysiologic therapy is not to replace standard treatment with relaxation training or biofeedback but rather to use biofeedback-assisted relaxation therapy to improve control of blood glucose. For example, McGinnis and colleagues compared the effects of 10 sessions of biofeedback (both surface electromyography and thermal feedback) and relaxation therapy versus three sessions of education in a sample of 30 patients with type 2 diabetes.21 No medicines were changed unless medically necessary. Patients kept daily logs of blood glucose, and had their hemoglobin A1c measured before and after treatment. Significant between-group differences in hemoglobin A1c and average blood glucose emerged in favor of the biofeedback group.21 However, patients with high scores on the Beck Depression Inventory22 (indicating more severe depressive symptoms) tended to drop out of the study or did not do as well as patients who were not symptomatic.
Another application of biofeedback in type 2 diabetes has been demonstrated by Rice and Schindler23 and Fiero et al.24 These investigators showed that patients with peripheral neuropathy, a common long-term complication of diabetes, were able to warm their hands and feet with the use of thermal biofeedback. Increased peripheral blood flow mediated the decrease in neuropathic pain.
Possible mechanisms of biofeedback in diabetes
Several explanations can be suggested to account for the results of biofeedback on blood glucose levels. Forehead muscle tension feedback (surface electromyography) helps patients to reduce facial tension and relax skeletal muscles, while increased finger temperature is an indicator of general relaxation. In the patients who completed the above study by McGinnis et al,21 both depression and anxiety scores decreased, which suggests a psychological mechanism for blood glucose reduction. Patients also reported improved sleep duration and quality with the use of relaxation therapy at bedtime.
BIOFEEDBACK IN ESSENTIAL HYPERTENSION
Biofeedback-assisted relaxation therapy has also been applied to control essential hypertension. The definition of hypertension, according to the Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC7),25 is systolic blood pressure greater than 139 mm Hg and diastolic blood pressure greater than 90 mm Hg. Prehypertension refers to systolic blood pressures between 130 and 139 mm Hg and diastolic pressures between 80 and 89 mm Hg. Standard treatment for established hypertension is antihypertensive medications, diet, and exercise. For patients in the prehypertensive blood pressure range, lifestyle changes are the primary intervention, unless the patient has multiple risk factors.25
Representative clinical evidence
Linden et al reported on the effects of 10 weeks of individualized psychophysiologic treatment on ambulatory blood pressure in patients with essential hypertension.26 Patients were initially screened for anxiety, depression, and anger, after which a program was designed for each patient based on his or her psychological risk factors. All patients received some form of relaxation therapy, and some received biofeedback. Over time significant reductions in ambulatory systolic and diastolic blood pressure were observed.26 In a separate study, Yucha et al provided a multimodal training program to hypertensive individuals and also reported significant decreases in blood pressure.27
Elliott et al trained hypertensive patients to use the RESPeRATE™ device to achieve the slow, deep breathing associated with the “relaxation response” sought in relaxation training.28 After initial training, patients were instructed to practice this device-guided breathing technique at home. Significant reductions in systolic blood pressure were observed over 8 weeks in the patients who used the device compared with controls who simply monitored their blood pressure at home. A maximum mean systolic blood pressure reduction of 15 mm Hg was achieved in the group of patients who practiced device-guided breathing for the greatest number of minutes during the 8-week study. Similar results with device-guided breathing using this device have been reported in two separate studies.29,30
More general stress reduction programs have also achieved success when offered to patients with essential hypertension in the clinic or the workplace. Studies of programs focusing on meditation and repeated practicing of centered breathing and relaxation responses, without use of biofeedback, have reported reductions of approximately 10.7 mm Hg in systolic pressure and 6.4 mm Hg in diastolic pressure.31,32 McCraty and colleagues provided a stress management program to hypertensive individuals at their place of work,33 based on the premise that individuals’ work demands are a source of chronic stress and thus create an ideal setting for the application of new coping skills. In this study, stress reduction training was associated with significant reductions both in blood pressure and in global measures of distress.33
Prehypertensive patients: An ideal target population
Although meta-analyses demonstrate that there is support for the efficacy of biofeedback in patients with essential hypertension,34,35 the field has been handicapped by the reality that most patients with hypertension are already being treated pharmacologically, which means that their blood pressure levels when starting biofeedback treatment are often low,36 limiting the potential effects of the intervention. The new category of patients with prehypertension may thus be the ideal population for stress management therapies, since their blood pressure is elevated, but not elevated enough to have prompted medication prescriptions in most cases. Lifestyle modifications, which could certainly include stress management, are the recommended first-line therapies for these prehypertensive patients.25
Possible mechanisms of biofeedback in hypertension
One can hypothesize on the mechanisms of action of relaxation-based therapies in hypertension. Relaxing the muscles of the face via electromyography biofeedback and increasing finger temperature facilitates whole-body relaxation and decreased sympathetic adrenergic activity. Parasympathetic dominance is facilitated by the use of breathing techniques to increase heart rate variability. 37,38 The improved deep sleep that results from relaxation may also reduce blood pressure by restoration of nighttime blood pressure dipping.16
IDENTIFYING THE BEST CANDIDATES IS NOT EASY
Some individuals are excellent candidates for biofeedback, while others do not benefit despite their best efforts.39,40 The likelihood of response is generally associated with adherence to medical recommendations and willingness and ability to follow instructions for home practice of relaxation. Nevertheless, some patients who attend sessions and practice still do not succeed, perhaps because they have few signs of overarousal in the system, such as a high degree of sympathetic activation, muscle tension, or low heart rate variability. Further, patients must be able to demonstrate that they learned the skill that was trained, such as consistent warming of the hands. If the training was for heart rate variability, the patient should be in the optimal range of heart rate variability and be able to demonstrate high-frequency waves.34 Patients with specific characteristics, such as stress sensitivity, may benefit more than those whose blood pressure and blood glucose are chronically elevated with few fluctuations.
CONCLUSIONS
The etiology of the metabolic syndrome is complex and multifactorial. Psychophysiologic interventions such as biofeedback and relaxation training are sometimes warranted for multiple aspects of metabolic syndrome, and they target several specific associated disruptions, particularly chronic stress, negative mood, and behavior. Initial patient evaluation should aim to assess the patient’s readiness for change, which must be present to a sufficient degree before continuing with biofeedback or relaxation techniques. Use of motivational interviewing techniques is recommended to increase patients’ preparedness for change.41 Understanding patients’ characteristic responses to stress will guide decisions on the type of biofeedback and relaxation therapies to use and whether or not psychotherapy will be necessary. Specific modalities of biofeedback or particular types of relaxation do not appear to be as critical as the total package of individualized psychophysiologic therapy.
Mayer-Davis EJ. Type 2 diabetes in youth: epidemiology and current research toward prevention and treatment. J Am Diet Assoc2008; 108( 4 suppl 1):S45–S51.
Groop L. Genetics of the metabolic syndrome. Br J Nutr2000; 83( suppl 1):S39–S48.
Pasquali R, Vicennati V. Activity of the hypothalamic-pituitary-adrenal axis in different obesity phenotypes. Int J Obes Relat Metab2000; 24( suppl 2):S47–S49.
Chandola T, Brunner E, Marmot M. Chronic stress at work and the metabolic syndrome: prospective study. BMJ2006; 332:521–525.
Jonas BS, Lando JF. Negative affect as a prospective risk factor for hypertension. Psychosom Med2000; 62:188–196.
McGrady A, Bourey R, Bailey B. The metabolic syndrome: obesity, type 2 diabetes, hypertension, and hyperlipidemia. In:Moss D, McGrady A, Davies TC, Wickramasekera I, eds. Handbook of Mind-Body Medicine for Primary Care. Thousand Oaks, CA: Sage Publications; 2003:275–297.
Branth S, Ronquist G, Stridsberg M, et al Development of abdominal fat and incipient metabolic syndrome in young healthy men exposed to long-term stress. Nutr Metab Cardiovasc Dis2007; 17:427–435.
Ganong WF. Review of Medical Physiology. 22nd ed. New York, NY: Lange Medical; 2005.
McGrady A. Psychophysiological foundations of the mind-body therapies. In:Moss D, McGrady A, Davies TC, Wickramasekera I, eds. Handbook of Mind-Body Medicine for Primary Care. Thousand Oaks, CA: Sage Publications; 2003:43–56.
McEwen B. The End of Stress As We Know It. Washington, DC: Joseph Henry Press; 2002.
Goldbacher EM, Matthews KA. Are psychological characteristics related to risk of the metabolic syndrome? A review of the literature. Ann Behav Med2007; 34:240–252.
Hughes JW, Stoney CM. Depressed mood is related to high-frequency heart rate variability during stressors. Psychosom Med2000; 62:796–803.
Brook RD, Julius S. Autonomic imbalance, hypertension, and cardiovascular risk. Am J Hypertens2000; 13( 6 Pt 2):112S–122S.
Knutson KL, Van Cauter E. Associations between sleep loss and increased risk of obesity and diabetes. Ann N Y Acad Sci2008; 1129:287–304.
Linden W, Klassen K, Phillips M. Can psychological factors account for a lack of nocturnal blood pressure dipping?Ann Behav Med2008; 36:253–258.
Fisher EB, Thorpe CT, DeVellis BM, DeVellis RF. Healthy coping, negative emotions, and diabetes management: a systematic review and appraisal. Diabetes Educ2007; 33:1080–1103.
Vitaliano PP, Scanlan JM, Zhang J, Savage MV, Hirsch IB, Siegler IC. A path model of chronic stress, the metabolic syndrome, and coronary heart disease. Psychosom Med2002; 64:418–435.
Flaa A, Aksnes TA, Kjeldsen SE, Eide I, Rostrup M. Increased sympathetic reactivity may predict insulin resistance: an 18-year follow-up study. Metab Clin Exp2008; 57:1422–1427.
American Diabetes Association. Standards of medical care in diabetes—2009. Diabetes Care2009; 32( suppl 1):S13–S61.
McGinnis R, McGrady A, Cox S, Grower-Dowling K. Biofeedback-assisted relaxation in type 2 diabetes mellitus. Diabetes Care2005; 28:2145–2149.
Rice BI, Schindler JV. Effect of thermal biofeedback–assisted relaxation training on blood circulation in the lower extremities of a population with diabetes. Diabetes Care1992; 15:853–859.
Fiero PL, Galper DI, Cox DJ, Phillips LH, Fryburg DA. Thermal biofeedback and lower extremity blood flow in adults with diabetes: is neuropathy a limiting factor?Appl Psychophysiol Biofeedback2003; 28:193–203.
Chobanian AV, Bakris GL, Black HR, et al The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA2003; 289:2560–2572.
Linden W, Lenz JW, Con AH. Individualized stress management for primary hypertension: a randomized trial. Arch Intern Med2001; 161:1071–1080.
Yucha CB, Clark L, Smith M, Uris P, LaFleur B, Duval S. The effect of biofeedback in hypertension. Appl Nurs Res2001; 14:29–35.
Elliott W, Izzo J, White WB, et al Graded blood pressure reduction in hypertensive outpatients associated with use of a device to assist with slow breathing. J Clin Hypertens2004; 6:553–559.
Schein M, Gavish B, Herz M, et al Treating hypertension with a device that slows and regularizes breathing: a randomized double-blind controlled study. J Hum Hypertens2001; 15:271–278.
Giannattasio C, Failla M, Meles E, Gentile G, Grappiolo A, Mancia G. Efficacy of self-treatment of hypertension at home with device-guided breathing. Am J Hypertens2002; 15( 4 Pt 2):186A. Abstract P-425.
Schneider RH, Alexander CN, Staggers F, et al Long-term effects of stress reduction on mortality in persons ≥ 55 years of age with systemic hypertension. Am J Cardiol2005; 95:1060–1064.
Paul-Labrador M, Polk D, Dwyer JH, et al Effects of randomized controlled trial of transcendental meditation on components of the metabolic syndrome in subjects with coronary heart disease. Arch Intern Med2006; 166:1218–1224.
McCraty R, Atkinson M, Tomasino D. Impact of a workplace stress reduction program on blood pressure and emotional health in hypertensive employees. J Altern Complement Med2003; 9:355–369.
Rainforth MV, Schneider RH, Nidich SI, Gaylord-King C, Salerno JW, Anderson JW. Stress reduction programs in patients with elevated blood pressure: a systematic review and meta-analysis. Curr Hypertens Rep2007; 9:520–528.
Nakao M, Yano E, Nomura S, Kuboki T. Blood pressure lowering effects of biofeedback treatment in hypertension: a meta-analysis of randomized controlled trials. Hypertens Res2003; 26:37–46.
Linden W, Chambers LA. Clinical effectiveness of non-drug therapies for hypertension: a meta-analysis. Ann Behav Med1994; 16:35–45.
McGrady AV, Linden W. Biobehavioral treatments of essential hypertension. In:Schwartz M, Andrasik F, eds. Biofeedback: A Practitioner’s Guide. New York, NY: Guilford Press; 2003:382–408.
Lehrer P. Biofeedback training to increase heart rate variability. In:Lehrer P, Woolfolk R, Sime W, eds. Principles and Practice of Stress Management. New York, NY: Guilford Press; 2007:227–248.
McGrady AV, Higgins JT. Prediction of response to biofeedback-assisted relaxation in hypertensives: development of a hypertensive predictor file (HYPP). Psychosom Med1989; 51:277–284.
Yucha CB, Tsai PS, Calderson KS, Tian L. Biofeedback-assisted relaxation training for essential hypertension: who is most likely to benefit?J Cardiovasc Nurs2005; 20:198–205.
Miller WR, Rollnick S. Motivational Interviewing. 2nd ed. New York, NY: Guilford Press; 2002.
Angele McGrady, PhD, MEd, LPCC Professor, Department of Psychiatry, University of Toledo, Toledo, OH
Correspondence: Angele McGrady, PhD, MEd, LPCC, Professor, Department of Psychiatry, University of Toledo, 3000 Arlington Avenue, MS #1193, Toledo, OH 43614–2598; [email protected]
Dr. McGrady reported that she has no financial relationships that pose a potential conflict of interest with this article.
Angele McGrady, PhD, MEd, LPCC Professor, Department of Psychiatry, University of Toledo, Toledo, OH
Correspondence: Angele McGrady, PhD, MEd, LPCC, Professor, Department of Psychiatry, University of Toledo, 3000 Arlington Avenue, MS #1193, Toledo, OH 43614–2598; [email protected]
Dr. McGrady reported that she has no financial relationships that pose a potential conflict of interest with this article.
Author and Disclosure Information
Angele McGrady, PhD, MEd, LPCC Professor, Department of Psychiatry, University of Toledo, Toledo, OH
Correspondence: Angele McGrady, PhD, MEd, LPCC, Professor, Department of Psychiatry, University of Toledo, 3000 Arlington Avenue, MS #1193, Toledo, OH 43614–2598; [email protected]
Dr. McGrady reported that she has no financial relationships that pose a potential conflict of interest with this article.
Type 2 diabetes, essential hypertension, obesity, and hyperlipidemia are the major components of the metabolic syndrome. Understanding the psychophysiologic basis of the metabolic syndrome is important since its prevalence has been increasing dramatically over the last decade. In the past, type 2 diabetes was diagnosed almost exclusively in persons in their 40s or older. Health care providers are now reporting emergence of type 2 diabetes and the metabolic syndrome in individuals in their 30s and even their late 20s.1,2
This article outlines the psychophysiologic bases for components of the metabolic syndrome and reviews the application of biofeedback and other psychophysiologic interventions on the two components for which such interventions have been most studied—diabetes and essential hypertension.
ETIOLOGY OF METABOLIC SYNDROME: THE INTERSECTION OF BIOLOGY, LIFESTYLE, STRESS
The disorders that constitute the metabolic syndrome share several etiologic factors. First, genetic predisposition increases the risk for diabetes, hypertension, hyperlipidemia, and obesity.3,4 Second, patients’ own behaviors—their choice of activity or inactivity, their food preferences, and their appetite—lead to gradual loss of control over body weight, blood glucose, blood pressure, and lipid levels. Third, chronic stress and its coincident psychological burden contribute to the etiology of various components of the metabolic syndrome.5 As life events accumulate and individuals lose their ability to cope, the stress response system maintains a higher than optimal level of activation.5–8
Chronic stress affects multiple organ systems, including the two master systems—nervous and endocrine. The biologic effects of stress include disordered breathing, increased activation of the renin-angiotension system, vascular constriction, tachycardia, decreased heart rate variability, inflammation, and sleep disruption.9 The mechanisms involved in acute stress responses are purpose-driven and adaptive. In contrast, chronically activated stress response systems involving increased sympathetic activity, decreased parasympathetic activity, and release of stress hormones have serious deleterious effects.10 Psychobiologic systems fail to adapt, delay recovery, or become exhausted.11
Role of psychological factors
As summarized in a review by Goldbacher and Matthews, 12 psychological factors have been related to increased risk for the metabolic syndrome. Depression has probably been most studied in the settings of cardiovascular disease and diabetes, whereas the psychological states of anger, hostility, and anxiety have been identified as salient etiologic factors in hypertension. In particular, depressed mood has been linked to decreased heart rate variability during the stress response.13 Anxiety affects blood pressure and blood glucose in normal individuals as part of the adaptive stress response, and the effects of anxiety are exacerbated in persons with the metabolic syndrome.14
Importance of sleep
Sleep disruption is often ignored in discussions of the mind-body interface in hypertension and diabetes. However, Knutson and Van Cauter15 suggested that sleep quality and sleep length have important effects on leptin levels and risk for diabetes. Very short sleepers have stronger appetites, as a result of lower leptin concentrations, and are much more likely to be obese compared with long sleepers (≥ 10 hours). This indicates that sleep length and quality affect metabolism. With regard to hypertension, a very important reduction of blood pressure occurs during the night, and a lack of nighttime blood pressure “dipping” is one of the markers for sustained blood pressure elevation.16
Factors overlap and begin to affect self-care
In addition to the effects of stress on mood and anxiety, repeated necessary demands for adaptation have marked effects on self-care behavior. Patients who suffer from anxiety are less efficient in managing their time and may be distracted from monitoring blood glucose and blood pressure. Anxious people often turn to the use of high-calorie comfort foods to soothe themselves during stressful times. Alcohol may be chosen as a means of reducing worry and tension. Depressed people lack the energy needed to maintain medical regimens and tend to be poor adherents to treatment recommendations. They also may choose comfort foods and addictive substances instead of nutritious, high-quality food and drink.17
Both anxiety and depression affect sleep routines and efficiency. Anxious people have trouble getting to sleep and may wake up often during the night, while depressed individuals frequently wake up early and cannot get back to sleep. Additionally, psychological distress influences social behaviors. Overt depressive and anxious symptoms tend not to foster social interactions with family and friends. Lack of social support and a scarcity of personal resources eventually contribute to the risk for diabetes and hypertension.18,19
In short, the metabolic syndrome is most likely to emerge when there is a combination of genetic factors, chronic stress, negative emotion, and unhealthy habits. The application of psychophysiologic interventions to diabetes and hypertension is based on our understanding of the etiology of these disorders, particularly the roles of psychological distress and behavior on blood glucose and blood pressure.
BIOFEEDBACK IN TYPE 2 DIABETES
Diabetes is characterized by elevated blood glucose and resistance of cell membranes to insulin, such that glucose is impeded from crossing from the blood into the cells. Standard treatment consists of oral antihyperglycemic agents, exogenous insulin, diet, and exercise.20 Type 2 diabetes may be the most behaviorally demanding of all chronic illnesses because patients must take an active role in daily management. Typical requirements are to measure blood glucose and take oral medicine, perhaps along with insulin, as well as to exercise, monitor diet, and adjust calories depending on activity level.
Therapy goals and a sampling of evidence
The goal of psychophysiologic therapy is not to replace standard treatment with relaxation training or biofeedback but rather to use biofeedback-assisted relaxation therapy to improve control of blood glucose. For example, McGinnis and colleagues compared the effects of 10 sessions of biofeedback (both surface electromyography and thermal feedback) and relaxation therapy versus three sessions of education in a sample of 30 patients with type 2 diabetes.21 No medicines were changed unless medically necessary. Patients kept daily logs of blood glucose, and had their hemoglobin A1c measured before and after treatment. Significant between-group differences in hemoglobin A1c and average blood glucose emerged in favor of the biofeedback group.21 However, patients with high scores on the Beck Depression Inventory22 (indicating more severe depressive symptoms) tended to drop out of the study or did not do as well as patients who were not symptomatic.
Another application of biofeedback in type 2 diabetes has been demonstrated by Rice and Schindler23 and Fiero et al.24 These investigators showed that patients with peripheral neuropathy, a common long-term complication of diabetes, were able to warm their hands and feet with the use of thermal biofeedback. Increased peripheral blood flow mediated the decrease in neuropathic pain.
Possible mechanisms of biofeedback in diabetes
Several explanations can be suggested to account for the results of biofeedback on blood glucose levels. Forehead muscle tension feedback (surface electromyography) helps patients to reduce facial tension and relax skeletal muscles, while increased finger temperature is an indicator of general relaxation. In the patients who completed the above study by McGinnis et al,21 both depression and anxiety scores decreased, which suggests a psychological mechanism for blood glucose reduction. Patients also reported improved sleep duration and quality with the use of relaxation therapy at bedtime.
BIOFEEDBACK IN ESSENTIAL HYPERTENSION
Biofeedback-assisted relaxation therapy has also been applied to control essential hypertension. The definition of hypertension, according to the Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC7),25 is systolic blood pressure greater than 139 mm Hg and diastolic blood pressure greater than 90 mm Hg. Prehypertension refers to systolic blood pressures between 130 and 139 mm Hg and diastolic pressures between 80 and 89 mm Hg. Standard treatment for established hypertension is antihypertensive medications, diet, and exercise. For patients in the prehypertensive blood pressure range, lifestyle changes are the primary intervention, unless the patient has multiple risk factors.25
Representative clinical evidence
Linden et al reported on the effects of 10 weeks of individualized psychophysiologic treatment on ambulatory blood pressure in patients with essential hypertension.26 Patients were initially screened for anxiety, depression, and anger, after which a program was designed for each patient based on his or her psychological risk factors. All patients received some form of relaxation therapy, and some received biofeedback. Over time significant reductions in ambulatory systolic and diastolic blood pressure were observed.26 In a separate study, Yucha et al provided a multimodal training program to hypertensive individuals and also reported significant decreases in blood pressure.27
Elliott et al trained hypertensive patients to use the RESPeRATE™ device to achieve the slow, deep breathing associated with the “relaxation response” sought in relaxation training.28 After initial training, patients were instructed to practice this device-guided breathing technique at home. Significant reductions in systolic blood pressure were observed over 8 weeks in the patients who used the device compared with controls who simply monitored their blood pressure at home. A maximum mean systolic blood pressure reduction of 15 mm Hg was achieved in the group of patients who practiced device-guided breathing for the greatest number of minutes during the 8-week study. Similar results with device-guided breathing using this device have been reported in two separate studies.29,30
More general stress reduction programs have also achieved success when offered to patients with essential hypertension in the clinic or the workplace. Studies of programs focusing on meditation and repeated practicing of centered breathing and relaxation responses, without use of biofeedback, have reported reductions of approximately 10.7 mm Hg in systolic pressure and 6.4 mm Hg in diastolic pressure.31,32 McCraty and colleagues provided a stress management program to hypertensive individuals at their place of work,33 based on the premise that individuals’ work demands are a source of chronic stress and thus create an ideal setting for the application of new coping skills. In this study, stress reduction training was associated with significant reductions both in blood pressure and in global measures of distress.33
Prehypertensive patients: An ideal target population
Although meta-analyses demonstrate that there is support for the efficacy of biofeedback in patients with essential hypertension,34,35 the field has been handicapped by the reality that most patients with hypertension are already being treated pharmacologically, which means that their blood pressure levels when starting biofeedback treatment are often low,36 limiting the potential effects of the intervention. The new category of patients with prehypertension may thus be the ideal population for stress management therapies, since their blood pressure is elevated, but not elevated enough to have prompted medication prescriptions in most cases. Lifestyle modifications, which could certainly include stress management, are the recommended first-line therapies for these prehypertensive patients.25
Possible mechanisms of biofeedback in hypertension
One can hypothesize on the mechanisms of action of relaxation-based therapies in hypertension. Relaxing the muscles of the face via electromyography biofeedback and increasing finger temperature facilitates whole-body relaxation and decreased sympathetic adrenergic activity. Parasympathetic dominance is facilitated by the use of breathing techniques to increase heart rate variability. 37,38 The improved deep sleep that results from relaxation may also reduce blood pressure by restoration of nighttime blood pressure dipping.16
IDENTIFYING THE BEST CANDIDATES IS NOT EASY
Some individuals are excellent candidates for biofeedback, while others do not benefit despite their best efforts.39,40 The likelihood of response is generally associated with adherence to medical recommendations and willingness and ability to follow instructions for home practice of relaxation. Nevertheless, some patients who attend sessions and practice still do not succeed, perhaps because they have few signs of overarousal in the system, such as a high degree of sympathetic activation, muscle tension, or low heart rate variability. Further, patients must be able to demonstrate that they learned the skill that was trained, such as consistent warming of the hands. If the training was for heart rate variability, the patient should be in the optimal range of heart rate variability and be able to demonstrate high-frequency waves.34 Patients with specific characteristics, such as stress sensitivity, may benefit more than those whose blood pressure and blood glucose are chronically elevated with few fluctuations.
CONCLUSIONS
The etiology of the metabolic syndrome is complex and multifactorial. Psychophysiologic interventions such as biofeedback and relaxation training are sometimes warranted for multiple aspects of metabolic syndrome, and they target several specific associated disruptions, particularly chronic stress, negative mood, and behavior. Initial patient evaluation should aim to assess the patient’s readiness for change, which must be present to a sufficient degree before continuing with biofeedback or relaxation techniques. Use of motivational interviewing techniques is recommended to increase patients’ preparedness for change.41 Understanding patients’ characteristic responses to stress will guide decisions on the type of biofeedback and relaxation therapies to use and whether or not psychotherapy will be necessary. Specific modalities of biofeedback or particular types of relaxation do not appear to be as critical as the total package of individualized psychophysiologic therapy.
Type 2 diabetes, essential hypertension, obesity, and hyperlipidemia are the major components of the metabolic syndrome. Understanding the psychophysiologic basis of the metabolic syndrome is important since its prevalence has been increasing dramatically over the last decade. In the past, type 2 diabetes was diagnosed almost exclusively in persons in their 40s or older. Health care providers are now reporting emergence of type 2 diabetes and the metabolic syndrome in individuals in their 30s and even their late 20s.1,2
This article outlines the psychophysiologic bases for components of the metabolic syndrome and reviews the application of biofeedback and other psychophysiologic interventions on the two components for which such interventions have been most studied—diabetes and essential hypertension.
ETIOLOGY OF METABOLIC SYNDROME: THE INTERSECTION OF BIOLOGY, LIFESTYLE, STRESS
The disorders that constitute the metabolic syndrome share several etiologic factors. First, genetic predisposition increases the risk for diabetes, hypertension, hyperlipidemia, and obesity.3,4 Second, patients’ own behaviors—their choice of activity or inactivity, their food preferences, and their appetite—lead to gradual loss of control over body weight, blood glucose, blood pressure, and lipid levels. Third, chronic stress and its coincident psychological burden contribute to the etiology of various components of the metabolic syndrome.5 As life events accumulate and individuals lose their ability to cope, the stress response system maintains a higher than optimal level of activation.5–8
Chronic stress affects multiple organ systems, including the two master systems—nervous and endocrine. The biologic effects of stress include disordered breathing, increased activation of the renin-angiotension system, vascular constriction, tachycardia, decreased heart rate variability, inflammation, and sleep disruption.9 The mechanisms involved in acute stress responses are purpose-driven and adaptive. In contrast, chronically activated stress response systems involving increased sympathetic activity, decreased parasympathetic activity, and release of stress hormones have serious deleterious effects.10 Psychobiologic systems fail to adapt, delay recovery, or become exhausted.11
Role of psychological factors
As summarized in a review by Goldbacher and Matthews, 12 psychological factors have been related to increased risk for the metabolic syndrome. Depression has probably been most studied in the settings of cardiovascular disease and diabetes, whereas the psychological states of anger, hostility, and anxiety have been identified as salient etiologic factors in hypertension. In particular, depressed mood has been linked to decreased heart rate variability during the stress response.13 Anxiety affects blood pressure and blood glucose in normal individuals as part of the adaptive stress response, and the effects of anxiety are exacerbated in persons with the metabolic syndrome.14
Importance of sleep
Sleep disruption is often ignored in discussions of the mind-body interface in hypertension and diabetes. However, Knutson and Van Cauter15 suggested that sleep quality and sleep length have important effects on leptin levels and risk for diabetes. Very short sleepers have stronger appetites, as a result of lower leptin concentrations, and are much more likely to be obese compared with long sleepers (≥ 10 hours). This indicates that sleep length and quality affect metabolism. With regard to hypertension, a very important reduction of blood pressure occurs during the night, and a lack of nighttime blood pressure “dipping” is one of the markers for sustained blood pressure elevation.16
Factors overlap and begin to affect self-care
In addition to the effects of stress on mood and anxiety, repeated necessary demands for adaptation have marked effects on self-care behavior. Patients who suffer from anxiety are less efficient in managing their time and may be distracted from monitoring blood glucose and blood pressure. Anxious people often turn to the use of high-calorie comfort foods to soothe themselves during stressful times. Alcohol may be chosen as a means of reducing worry and tension. Depressed people lack the energy needed to maintain medical regimens and tend to be poor adherents to treatment recommendations. They also may choose comfort foods and addictive substances instead of nutritious, high-quality food and drink.17
Both anxiety and depression affect sleep routines and efficiency. Anxious people have trouble getting to sleep and may wake up often during the night, while depressed individuals frequently wake up early and cannot get back to sleep. Additionally, psychological distress influences social behaviors. Overt depressive and anxious symptoms tend not to foster social interactions with family and friends. Lack of social support and a scarcity of personal resources eventually contribute to the risk for diabetes and hypertension.18,19
In short, the metabolic syndrome is most likely to emerge when there is a combination of genetic factors, chronic stress, negative emotion, and unhealthy habits. The application of psychophysiologic interventions to diabetes and hypertension is based on our understanding of the etiology of these disorders, particularly the roles of psychological distress and behavior on blood glucose and blood pressure.
BIOFEEDBACK IN TYPE 2 DIABETES
Diabetes is characterized by elevated blood glucose and resistance of cell membranes to insulin, such that glucose is impeded from crossing from the blood into the cells. Standard treatment consists of oral antihyperglycemic agents, exogenous insulin, diet, and exercise.20 Type 2 diabetes may be the most behaviorally demanding of all chronic illnesses because patients must take an active role in daily management. Typical requirements are to measure blood glucose and take oral medicine, perhaps along with insulin, as well as to exercise, monitor diet, and adjust calories depending on activity level.
Therapy goals and a sampling of evidence
The goal of psychophysiologic therapy is not to replace standard treatment with relaxation training or biofeedback but rather to use biofeedback-assisted relaxation therapy to improve control of blood glucose. For example, McGinnis and colleagues compared the effects of 10 sessions of biofeedback (both surface electromyography and thermal feedback) and relaxation therapy versus three sessions of education in a sample of 30 patients with type 2 diabetes.21 No medicines were changed unless medically necessary. Patients kept daily logs of blood glucose, and had their hemoglobin A1c measured before and after treatment. Significant between-group differences in hemoglobin A1c and average blood glucose emerged in favor of the biofeedback group.21 However, patients with high scores on the Beck Depression Inventory22 (indicating more severe depressive symptoms) tended to drop out of the study or did not do as well as patients who were not symptomatic.
Another application of biofeedback in type 2 diabetes has been demonstrated by Rice and Schindler23 and Fiero et al.24 These investigators showed that patients with peripheral neuropathy, a common long-term complication of diabetes, were able to warm their hands and feet with the use of thermal biofeedback. Increased peripheral blood flow mediated the decrease in neuropathic pain.
Possible mechanisms of biofeedback in diabetes
Several explanations can be suggested to account for the results of biofeedback on blood glucose levels. Forehead muscle tension feedback (surface electromyography) helps patients to reduce facial tension and relax skeletal muscles, while increased finger temperature is an indicator of general relaxation. In the patients who completed the above study by McGinnis et al,21 both depression and anxiety scores decreased, which suggests a psychological mechanism for blood glucose reduction. Patients also reported improved sleep duration and quality with the use of relaxation therapy at bedtime.
BIOFEEDBACK IN ESSENTIAL HYPERTENSION
Biofeedback-assisted relaxation therapy has also been applied to control essential hypertension. The definition of hypertension, according to the Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC7),25 is systolic blood pressure greater than 139 mm Hg and diastolic blood pressure greater than 90 mm Hg. Prehypertension refers to systolic blood pressures between 130 and 139 mm Hg and diastolic pressures between 80 and 89 mm Hg. Standard treatment for established hypertension is antihypertensive medications, diet, and exercise. For patients in the prehypertensive blood pressure range, lifestyle changes are the primary intervention, unless the patient has multiple risk factors.25
Representative clinical evidence
Linden et al reported on the effects of 10 weeks of individualized psychophysiologic treatment on ambulatory blood pressure in patients with essential hypertension.26 Patients were initially screened for anxiety, depression, and anger, after which a program was designed for each patient based on his or her psychological risk factors. All patients received some form of relaxation therapy, and some received biofeedback. Over time significant reductions in ambulatory systolic and diastolic blood pressure were observed.26 In a separate study, Yucha et al provided a multimodal training program to hypertensive individuals and also reported significant decreases in blood pressure.27
Elliott et al trained hypertensive patients to use the RESPeRATE™ device to achieve the slow, deep breathing associated with the “relaxation response” sought in relaxation training.28 After initial training, patients were instructed to practice this device-guided breathing technique at home. Significant reductions in systolic blood pressure were observed over 8 weeks in the patients who used the device compared with controls who simply monitored their blood pressure at home. A maximum mean systolic blood pressure reduction of 15 mm Hg was achieved in the group of patients who practiced device-guided breathing for the greatest number of minutes during the 8-week study. Similar results with device-guided breathing using this device have been reported in two separate studies.29,30
More general stress reduction programs have also achieved success when offered to patients with essential hypertension in the clinic or the workplace. Studies of programs focusing on meditation and repeated practicing of centered breathing and relaxation responses, without use of biofeedback, have reported reductions of approximately 10.7 mm Hg in systolic pressure and 6.4 mm Hg in diastolic pressure.31,32 McCraty and colleagues provided a stress management program to hypertensive individuals at their place of work,33 based on the premise that individuals’ work demands are a source of chronic stress and thus create an ideal setting for the application of new coping skills. In this study, stress reduction training was associated with significant reductions both in blood pressure and in global measures of distress.33
Prehypertensive patients: An ideal target population
Although meta-analyses demonstrate that there is support for the efficacy of biofeedback in patients with essential hypertension,34,35 the field has been handicapped by the reality that most patients with hypertension are already being treated pharmacologically, which means that their blood pressure levels when starting biofeedback treatment are often low,36 limiting the potential effects of the intervention. The new category of patients with prehypertension may thus be the ideal population for stress management therapies, since their blood pressure is elevated, but not elevated enough to have prompted medication prescriptions in most cases. Lifestyle modifications, which could certainly include stress management, are the recommended first-line therapies for these prehypertensive patients.25
Possible mechanisms of biofeedback in hypertension
One can hypothesize on the mechanisms of action of relaxation-based therapies in hypertension. Relaxing the muscles of the face via electromyography biofeedback and increasing finger temperature facilitates whole-body relaxation and decreased sympathetic adrenergic activity. Parasympathetic dominance is facilitated by the use of breathing techniques to increase heart rate variability. 37,38 The improved deep sleep that results from relaxation may also reduce blood pressure by restoration of nighttime blood pressure dipping.16
IDENTIFYING THE BEST CANDIDATES IS NOT EASY
Some individuals are excellent candidates for biofeedback, while others do not benefit despite their best efforts.39,40 The likelihood of response is generally associated with adherence to medical recommendations and willingness and ability to follow instructions for home practice of relaxation. Nevertheless, some patients who attend sessions and practice still do not succeed, perhaps because they have few signs of overarousal in the system, such as a high degree of sympathetic activation, muscle tension, or low heart rate variability. Further, patients must be able to demonstrate that they learned the skill that was trained, such as consistent warming of the hands. If the training was for heart rate variability, the patient should be in the optimal range of heart rate variability and be able to demonstrate high-frequency waves.34 Patients with specific characteristics, such as stress sensitivity, may benefit more than those whose blood pressure and blood glucose are chronically elevated with few fluctuations.
CONCLUSIONS
The etiology of the metabolic syndrome is complex and multifactorial. Psychophysiologic interventions such as biofeedback and relaxation training are sometimes warranted for multiple aspects of metabolic syndrome, and they target several specific associated disruptions, particularly chronic stress, negative mood, and behavior. Initial patient evaluation should aim to assess the patient’s readiness for change, which must be present to a sufficient degree before continuing with biofeedback or relaxation techniques. Use of motivational interviewing techniques is recommended to increase patients’ preparedness for change.41 Understanding patients’ characteristic responses to stress will guide decisions on the type of biofeedback and relaxation therapies to use and whether or not psychotherapy will be necessary. Specific modalities of biofeedback or particular types of relaxation do not appear to be as critical as the total package of individualized psychophysiologic therapy.
Mayer-Davis EJ. Type 2 diabetes in youth: epidemiology and current research toward prevention and treatment. J Am Diet Assoc2008; 108( 4 suppl 1):S45–S51.
Groop L. Genetics of the metabolic syndrome. Br J Nutr2000; 83( suppl 1):S39–S48.
Pasquali R, Vicennati V. Activity of the hypothalamic-pituitary-adrenal axis in different obesity phenotypes. Int J Obes Relat Metab2000; 24( suppl 2):S47–S49.
Chandola T, Brunner E, Marmot M. Chronic stress at work and the metabolic syndrome: prospective study. BMJ2006; 332:521–525.
Jonas BS, Lando JF. Negative affect as a prospective risk factor for hypertension. Psychosom Med2000; 62:188–196.
McGrady A, Bourey R, Bailey B. The metabolic syndrome: obesity, type 2 diabetes, hypertension, and hyperlipidemia. In:Moss D, McGrady A, Davies TC, Wickramasekera I, eds. Handbook of Mind-Body Medicine for Primary Care. Thousand Oaks, CA: Sage Publications; 2003:275–297.
Branth S, Ronquist G, Stridsberg M, et al Development of abdominal fat and incipient metabolic syndrome in young healthy men exposed to long-term stress. Nutr Metab Cardiovasc Dis2007; 17:427–435.
Ganong WF. Review of Medical Physiology. 22nd ed. New York, NY: Lange Medical; 2005.
McGrady A. Psychophysiological foundations of the mind-body therapies. In:Moss D, McGrady A, Davies TC, Wickramasekera I, eds. Handbook of Mind-Body Medicine for Primary Care. Thousand Oaks, CA: Sage Publications; 2003:43–56.
McEwen B. The End of Stress As We Know It. Washington, DC: Joseph Henry Press; 2002.
Goldbacher EM, Matthews KA. Are psychological characteristics related to risk of the metabolic syndrome? A review of the literature. Ann Behav Med2007; 34:240–252.
Hughes JW, Stoney CM. Depressed mood is related to high-frequency heart rate variability during stressors. Psychosom Med2000; 62:796–803.
Brook RD, Julius S. Autonomic imbalance, hypertension, and cardiovascular risk. Am J Hypertens2000; 13( 6 Pt 2):112S–122S.
Knutson KL, Van Cauter E. Associations between sleep loss and increased risk of obesity and diabetes. Ann N Y Acad Sci2008; 1129:287–304.
Linden W, Klassen K, Phillips M. Can psychological factors account for a lack of nocturnal blood pressure dipping?Ann Behav Med2008; 36:253–258.
Fisher EB, Thorpe CT, DeVellis BM, DeVellis RF. Healthy coping, negative emotions, and diabetes management: a systematic review and appraisal. Diabetes Educ2007; 33:1080–1103.
Vitaliano PP, Scanlan JM, Zhang J, Savage MV, Hirsch IB, Siegler IC. A path model of chronic stress, the metabolic syndrome, and coronary heart disease. Psychosom Med2002; 64:418–435.
Flaa A, Aksnes TA, Kjeldsen SE, Eide I, Rostrup M. Increased sympathetic reactivity may predict insulin resistance: an 18-year follow-up study. Metab Clin Exp2008; 57:1422–1427.
American Diabetes Association. Standards of medical care in diabetes—2009. Diabetes Care2009; 32( suppl 1):S13–S61.
McGinnis R, McGrady A, Cox S, Grower-Dowling K. Biofeedback-assisted relaxation in type 2 diabetes mellitus. Diabetes Care2005; 28:2145–2149.
Rice BI, Schindler JV. Effect of thermal biofeedback–assisted relaxation training on blood circulation in the lower extremities of a population with diabetes. Diabetes Care1992; 15:853–859.
Fiero PL, Galper DI, Cox DJ, Phillips LH, Fryburg DA. Thermal biofeedback and lower extremity blood flow in adults with diabetes: is neuropathy a limiting factor?Appl Psychophysiol Biofeedback2003; 28:193–203.
Chobanian AV, Bakris GL, Black HR, et al The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA2003; 289:2560–2572.
Linden W, Lenz JW, Con AH. Individualized stress management for primary hypertension: a randomized trial. Arch Intern Med2001; 161:1071–1080.
Yucha CB, Clark L, Smith M, Uris P, LaFleur B, Duval S. The effect of biofeedback in hypertension. Appl Nurs Res2001; 14:29–35.
Elliott W, Izzo J, White WB, et al Graded blood pressure reduction in hypertensive outpatients associated with use of a device to assist with slow breathing. J Clin Hypertens2004; 6:553–559.
Schein M, Gavish B, Herz M, et al Treating hypertension with a device that slows and regularizes breathing: a randomized double-blind controlled study. J Hum Hypertens2001; 15:271–278.
Giannattasio C, Failla M, Meles E, Gentile G, Grappiolo A, Mancia G. Efficacy of self-treatment of hypertension at home with device-guided breathing. Am J Hypertens2002; 15( 4 Pt 2):186A. Abstract P-425.
Schneider RH, Alexander CN, Staggers F, et al Long-term effects of stress reduction on mortality in persons ≥ 55 years of age with systemic hypertension. Am J Cardiol2005; 95:1060–1064.
Paul-Labrador M, Polk D, Dwyer JH, et al Effects of randomized controlled trial of transcendental meditation on components of the metabolic syndrome in subjects with coronary heart disease. Arch Intern Med2006; 166:1218–1224.
McCraty R, Atkinson M, Tomasino D. Impact of a workplace stress reduction program on blood pressure and emotional health in hypertensive employees. J Altern Complement Med2003; 9:355–369.
Rainforth MV, Schneider RH, Nidich SI, Gaylord-King C, Salerno JW, Anderson JW. Stress reduction programs in patients with elevated blood pressure: a systematic review and meta-analysis. Curr Hypertens Rep2007; 9:520–528.
Nakao M, Yano E, Nomura S, Kuboki T. Blood pressure lowering effects of biofeedback treatment in hypertension: a meta-analysis of randomized controlled trials. Hypertens Res2003; 26:37–46.
Linden W, Chambers LA. Clinical effectiveness of non-drug therapies for hypertension: a meta-analysis. Ann Behav Med1994; 16:35–45.
McGrady AV, Linden W. Biobehavioral treatments of essential hypertension. In:Schwartz M, Andrasik F, eds. Biofeedback: A Practitioner’s Guide. New York, NY: Guilford Press; 2003:382–408.
Lehrer P. Biofeedback training to increase heart rate variability. In:Lehrer P, Woolfolk R, Sime W, eds. Principles and Practice of Stress Management. New York, NY: Guilford Press; 2007:227–248.
McGrady AV, Higgins JT. Prediction of response to biofeedback-assisted relaxation in hypertensives: development of a hypertensive predictor file (HYPP). Psychosom Med1989; 51:277–284.
Yucha CB, Tsai PS, Calderson KS, Tian L. Biofeedback-assisted relaxation training for essential hypertension: who is most likely to benefit?J Cardiovasc Nurs2005; 20:198–205.
Miller WR, Rollnick S. Motivational Interviewing. 2nd ed. New York, NY: Guilford Press; 2002.
Mayer-Davis EJ. Type 2 diabetes in youth: epidemiology and current research toward prevention and treatment. J Am Diet Assoc2008; 108( 4 suppl 1):S45–S51.
Groop L. Genetics of the metabolic syndrome. Br J Nutr2000; 83( suppl 1):S39–S48.
Pasquali R, Vicennati V. Activity of the hypothalamic-pituitary-adrenal axis in different obesity phenotypes. Int J Obes Relat Metab2000; 24( suppl 2):S47–S49.
Chandola T, Brunner E, Marmot M. Chronic stress at work and the metabolic syndrome: prospective study. BMJ2006; 332:521–525.
Jonas BS, Lando JF. Negative affect as a prospective risk factor for hypertension. Psychosom Med2000; 62:188–196.
McGrady A, Bourey R, Bailey B. The metabolic syndrome: obesity, type 2 diabetes, hypertension, and hyperlipidemia. In:Moss D, McGrady A, Davies TC, Wickramasekera I, eds. Handbook of Mind-Body Medicine for Primary Care. Thousand Oaks, CA: Sage Publications; 2003:275–297.
Branth S, Ronquist G, Stridsberg M, et al Development of abdominal fat and incipient metabolic syndrome in young healthy men exposed to long-term stress. Nutr Metab Cardiovasc Dis2007; 17:427–435.
Ganong WF. Review of Medical Physiology. 22nd ed. New York, NY: Lange Medical; 2005.
McGrady A. Psychophysiological foundations of the mind-body therapies. In:Moss D, McGrady A, Davies TC, Wickramasekera I, eds. Handbook of Mind-Body Medicine for Primary Care. Thousand Oaks, CA: Sage Publications; 2003:43–56.
McEwen B. The End of Stress As We Know It. Washington, DC: Joseph Henry Press; 2002.
Goldbacher EM, Matthews KA. Are psychological characteristics related to risk of the metabolic syndrome? A review of the literature. Ann Behav Med2007; 34:240–252.
Hughes JW, Stoney CM. Depressed mood is related to high-frequency heart rate variability during stressors. Psychosom Med2000; 62:796–803.
Brook RD, Julius S. Autonomic imbalance, hypertension, and cardiovascular risk. Am J Hypertens2000; 13( 6 Pt 2):112S–122S.
Knutson KL, Van Cauter E. Associations between sleep loss and increased risk of obesity and diabetes. Ann N Y Acad Sci2008; 1129:287–304.
Linden W, Klassen K, Phillips M. Can psychological factors account for a lack of nocturnal blood pressure dipping?Ann Behav Med2008; 36:253–258.
Fisher EB, Thorpe CT, DeVellis BM, DeVellis RF. Healthy coping, negative emotions, and diabetes management: a systematic review and appraisal. Diabetes Educ2007; 33:1080–1103.
Vitaliano PP, Scanlan JM, Zhang J, Savage MV, Hirsch IB, Siegler IC. A path model of chronic stress, the metabolic syndrome, and coronary heart disease. Psychosom Med2002; 64:418–435.
Flaa A, Aksnes TA, Kjeldsen SE, Eide I, Rostrup M. Increased sympathetic reactivity may predict insulin resistance: an 18-year follow-up study. Metab Clin Exp2008; 57:1422–1427.
American Diabetes Association. Standards of medical care in diabetes—2009. Diabetes Care2009; 32( suppl 1):S13–S61.
McGinnis R, McGrady A, Cox S, Grower-Dowling K. Biofeedback-assisted relaxation in type 2 diabetes mellitus. Diabetes Care2005; 28:2145–2149.
Rice BI, Schindler JV. Effect of thermal biofeedback–assisted relaxation training on blood circulation in the lower extremities of a population with diabetes. Diabetes Care1992; 15:853–859.
Fiero PL, Galper DI, Cox DJ, Phillips LH, Fryburg DA. Thermal biofeedback and lower extremity blood flow in adults with diabetes: is neuropathy a limiting factor?Appl Psychophysiol Biofeedback2003; 28:193–203.
Chobanian AV, Bakris GL, Black HR, et al The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA2003; 289:2560–2572.
Linden W, Lenz JW, Con AH. Individualized stress management for primary hypertension: a randomized trial. Arch Intern Med2001; 161:1071–1080.
Yucha CB, Clark L, Smith M, Uris P, LaFleur B, Duval S. The effect of biofeedback in hypertension. Appl Nurs Res2001; 14:29–35.
Elliott W, Izzo J, White WB, et al Graded blood pressure reduction in hypertensive outpatients associated with use of a device to assist with slow breathing. J Clin Hypertens2004; 6:553–559.
Schein M, Gavish B, Herz M, et al Treating hypertension with a device that slows and regularizes breathing: a randomized double-blind controlled study. J Hum Hypertens2001; 15:271–278.
Giannattasio C, Failla M, Meles E, Gentile G, Grappiolo A, Mancia G. Efficacy of self-treatment of hypertension at home with device-guided breathing. Am J Hypertens2002; 15( 4 Pt 2):186A. Abstract P-425.
Schneider RH, Alexander CN, Staggers F, et al Long-term effects of stress reduction on mortality in persons ≥ 55 years of age with systemic hypertension. Am J Cardiol2005; 95:1060–1064.
Paul-Labrador M, Polk D, Dwyer JH, et al Effects of randomized controlled trial of transcendental meditation on components of the metabolic syndrome in subjects with coronary heart disease. Arch Intern Med2006; 166:1218–1224.
McCraty R, Atkinson M, Tomasino D. Impact of a workplace stress reduction program on blood pressure and emotional health in hypertensive employees. J Altern Complement Med2003; 9:355–369.
Rainforth MV, Schneider RH, Nidich SI, Gaylord-King C, Salerno JW, Anderson JW. Stress reduction programs in patients with elevated blood pressure: a systematic review and meta-analysis. Curr Hypertens Rep2007; 9:520–528.
Nakao M, Yano E, Nomura S, Kuboki T. Blood pressure lowering effects of biofeedback treatment in hypertension: a meta-analysis of randomized controlled trials. Hypertens Res2003; 26:37–46.
Linden W, Chambers LA. Clinical effectiveness of non-drug therapies for hypertension: a meta-analysis. Ann Behav Med1994; 16:35–45.
McGrady AV, Linden W. Biobehavioral treatments of essential hypertension. In:Schwartz M, Andrasik F, eds. Biofeedback: A Practitioner’s Guide. New York, NY: Guilford Press; 2003:382–408.
Lehrer P. Biofeedback training to increase heart rate variability. In:Lehrer P, Woolfolk R, Sime W, eds. Principles and Practice of Stress Management. New York, NY: Guilford Press; 2007:227–248.
McGrady AV, Higgins JT. Prediction of response to biofeedback-assisted relaxation in hypertensives: development of a hypertensive predictor file (HYPP). Psychosom Med1989; 51:277–284.
Yucha CB, Tsai PS, Calderson KS, Tian L. Biofeedback-assisted relaxation training for essential hypertension: who is most likely to benefit?J Cardiovasc Nurs2005; 20:198–205.
Miller WR, Rollnick S. Motivational Interviewing. 2nd ed. New York, NY: Guilford Press; 2002.
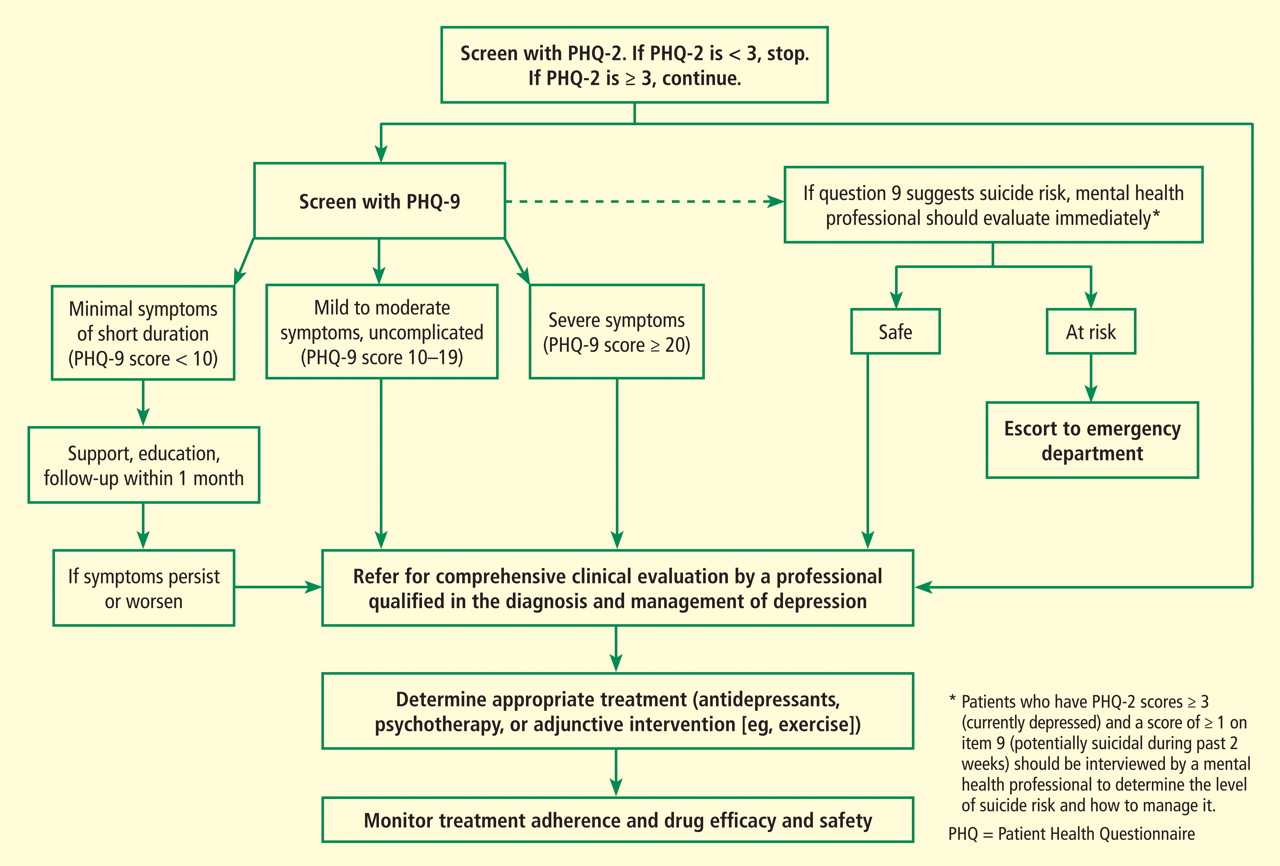
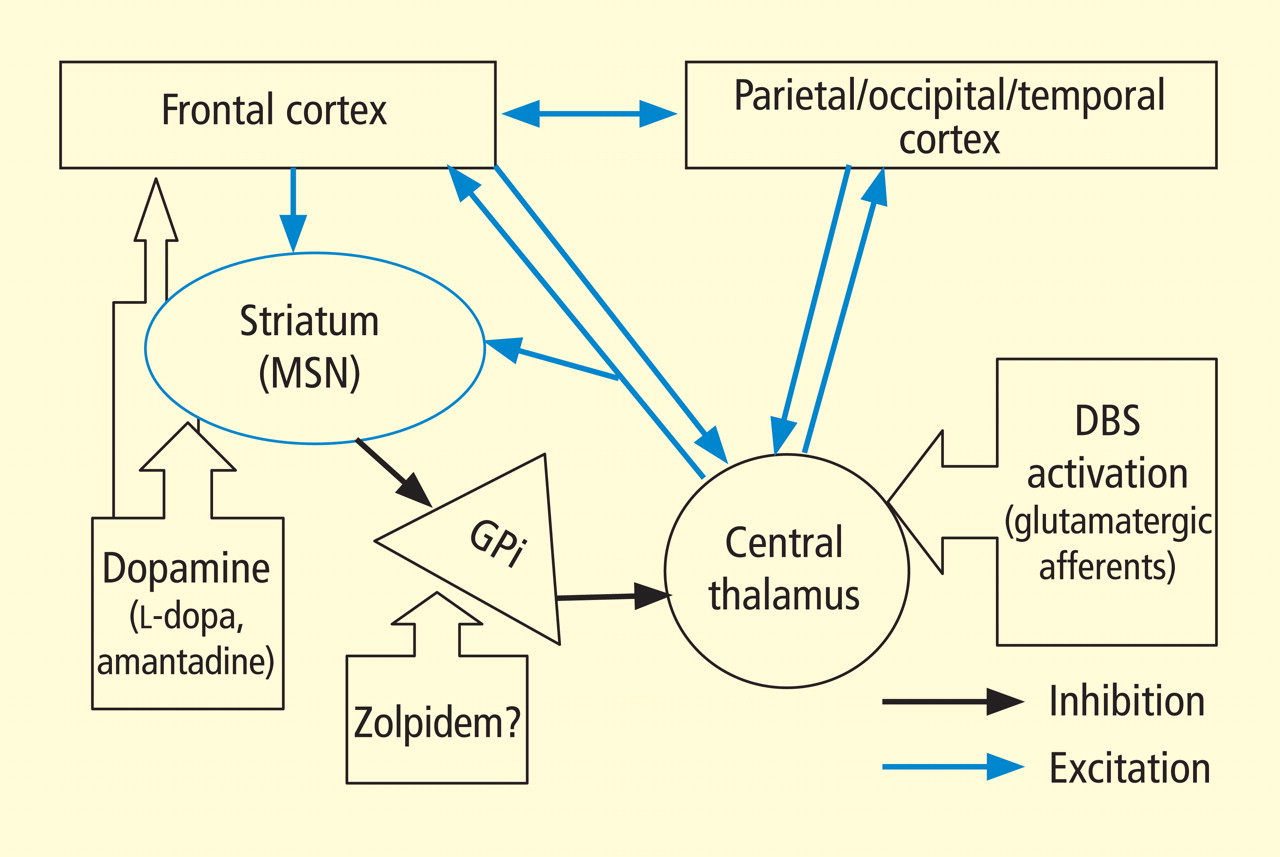
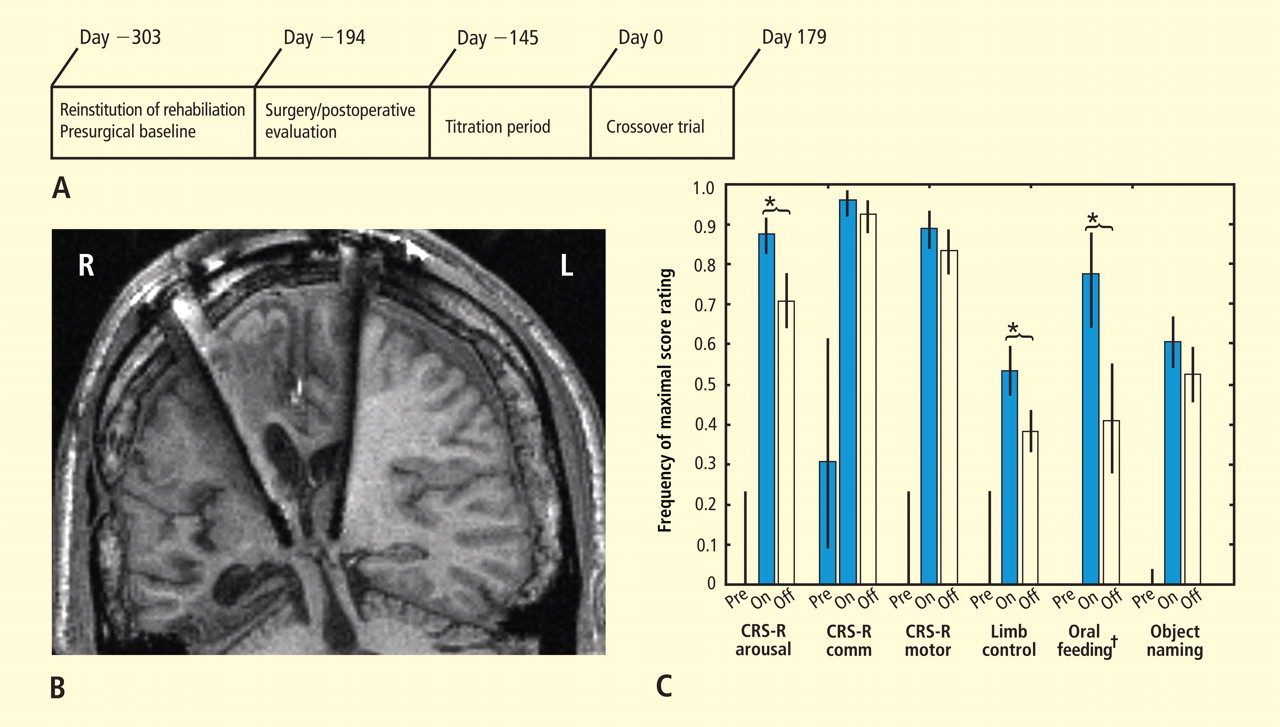

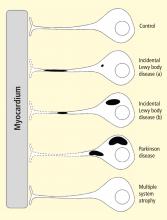 Reprinted from Brain (Orimo S, et al. Axonal α-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain 2008; 131:642–650) by permission of Oxford University Press.
Reprinted from Brain (Orimo S, et al. Axonal α-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain 2008; 131:642–650) by permission of Oxford University Press.