User login
Welcome to Current Psychiatry, a leading source of information, online and in print, for practitioners of psychiatry and its related subspecialties, including addiction psychiatry, child and adolescent psychiatry, and geriatric psychiatry. This Web site contains evidence-based reviews of the prevention, diagnosis, and treatment of mental illness and psychological disorders; case reports; updates on psychopharmacology; news about the specialty of psychiatry; pearls for practice; and other topics of interest and use to this audience.
Dear Drupal User: You're seeing this because you're logged in to Drupal, and not redirected to MDedge.com/psychiatry.
Depression
adolescent depression
adolescent major depressive disorder
adolescent schizophrenia
adolescent with major depressive disorder
animals
autism
baby
brexpiprazole
child
child bipolar
child depression
child schizophrenia
children with bipolar disorder
children with depression
children with major depressive disorder
compulsive behaviors
cure
elderly bipolar
elderly depression
elderly major depressive disorder
elderly schizophrenia
elderly with dementia
first break
first episode
gambling
gaming
geriatric depression
geriatric major depressive disorder
geriatric schizophrenia
infant
kid
major depressive disorder
major depressive disorder in adolescents
major depressive disorder in children
parenting
pediatric
pediatric bipolar
pediatric depression
pediatric major depressive disorder
pediatric schizophrenia
pregnancy
pregnant
rexulti
skin care
teen
wine
section[contains(@class, 'nav-hidden')]
footer[@id='footer']
div[contains(@class, 'pane-pub-article-current-psychiatry')]
div[contains(@class, 'pane-pub-home-current-psychiatry')]
div[contains(@class, 'pane-pub-topic-current-psychiatry')]
div[contains(@class, 'panel-panel-inner')]
div[contains(@class, 'pane-node-field-article-topics')]
section[contains(@class, 'footer-nav-section-wrapper')]
Depressed and confused, and dizzy while walking the dog
CASE Light-headed
Mr. M, age 73, is a retired project manager who feels light-headed while walking his dog, causing him to go to the emergency department. His history is significant for hypertension, coronary artery disease (CAD), 3-vessel coronary artery bypass graft surgery (CABG), hyperlipidemia, erectile dysfunction, open-angle glaucoma, hemiretinal vein occlusion, symptoms suggesting rapid eye-movement behavior disorder (RBD), and major depressive disorder (MDD).
The psychiatry consultation-liaison service is asked to help manage Mr. M’s psychiatric medications in the context of orthostatic hypotension and cognitive deficits.
What could be causing Mr. M’s symptoms?
a) drug adverse effect
b) progressive cardiovascular disease
c) MDD
d) all of the above
HISTORY Depression, heart disease
15 years ago. Mr. M experienced his first major depressive episode. His primary care physician (PCP) commented on a history of falling asleep while driving and 1 episode of sleepwalking. His depression was treated to remission with fluoxetine and methylphenidate (dosages were not recorded), the latter also addressed his falling asleep while driving.
5 years ago. Mr. M had another depressive episode characterized by anxiety, difficulty sleeping, and irritability. He also described chest pain; a cardiac work-up revealed extensive CAD, which led to 3-vessel CABG later that year. He also reported dizziness upon standing, which was treated with compression stockings and an increase in sodium intake.
Mr. M continued to express feelings of depression. His cardiologist started him on paroxetine, 10 mg/d, which he took for 2 months and decided to stop because he felt better. He declined psychiatric referral.
4 years ago. Mr. M’s PCP referred him to a psychiatrist for depressed mood, anhedonia, decreased appetite, decreased energy, and difficulty concentrating. Immediate and delayed recall were found to be intact. The psychiatrist diagnosed MDD and Mr. M started escitalopram, 5 mg/d, titrated to 15 mg/d, and trazodone, 50 mg/d.
After starting treatment, Mr. M reported decreased libido. Sustained-release bupropion, 150 mg/d, was added to boost the effects of escitalopram and counteract sexual side effects.
At follow-up, Mr. M reported that his depressive symptoms and libido had improved, but that he had been experiencing unsteady gait when getting out of his car, which he had been noticing “for a while”—before he began trazodone. Mr. M was referred to his PCP, who attributed his symptoms to orthostasis. No treatment was indicated at the time because Mr. M’s lightheadedness had resolved.
3 years ago. Mr. M reported a syncopal attack and continued “dizziness.” His PCP prescribed fludrocortisone, 0.1 mg/d, later to be dosed 0.2 mg/d, and symptoms improved.
Although Mr. M had a history of orthostatic hypotension, he was later noted to have supine hypertension. Mr. M’s PCP was concerned that fludrocortisone could be causing the supine hypertension but that decreasing the dosage would cause his orthostatic hypotension to return.
The PCP also was concerned that the psychiatric medications (escitalopram, trazodone, and bupropion) could be causing orthostasis. There was discussion among Mr. M, his PCP, and his psychiatrist of stopping the psychotropics to see if the symptoms would remit; however, because of concerns about Mr. M’s depression, the medications were continued. Mr. M monitored his blood pressure at home and was referred to a neurologist for work-up of potential autonomic dysfunction.
Shortly afterward, Mr. M reported intermittent difficulty keeping track of his thoughts and finishing sentences. His psychiatrist ordered an MRI, which showed chronic small vessel ischemic changes, and started him on donepezil, 5 mg/d.
Neuropsychological testing revealed decreased processing speed and poor recognition memory; otherwise, results showed above-average intellectual ability and average or above-average performance in measures of language, attention, visuospatial/constructional functions, and executive functions—a pattern typically attributable to psychogenic factors, such as depression.
Mr. M reported to his neurologist that he forgets directions while driving but can focus better if he makes a conscious effort. Physical exam was significant hypotension; flat affect; deficits in concentration and short-term recall; mild impairment of Luria motor sequence (composed of a go/no-go and a reciprocal motor task); and vertical and horizontal saccades.1
Mr. M consulted with an ophthalmologist for anterior iridocyclitis and ocular hypertension, which was controlled with travoprost. He continued to experience trouble with his vision and was given a diagnosis of right inferior hemiretinal vein occlusion, macular edema, and suspected glaucoma. Subsequent notes recorded a history of Posner-Schlossman syndrome (a disease characterized by recurrent attacks of increased intraocular pressure in 1 eye with concomitant anterior chamber inflammation). His vision deteriorated until he was diagnosed with ocular hypertension, open-angle glaucoma, and dermatochalasis.
The authors’ observations
Involvement of multiple specialties in a patient’s care brings to question one’s philosophy on medical diagnosis. Interdisciplinary communication would seem to promote the principle of diagnostic parsimony, or Occam’s razor, which suggests a unifying diagnosis to explain all of the patient’s symptoms. Lack of communication might favor Hickam’s dictum, which states that “patients can have as many diseases as they damn well please.”
HISTORY Low energy, forgetfulness
2 years ago. Mr. M noticed low energy and motivation. He continued to work full-time but thought that it was taking him longer to get work done. He was tapered off escitalopram and started on desvenlafaxine, 50 mg/d; donepezil was increased to 10 mg/d.
The syncopal episodes resolved but blood pressure measured at home averaged 150/70 mm Hg. Mr. M was advised to decrease fludrocortisone from 0.2 mg/d to 0.1 mg/d. He tolerated the change and blood pressure measured at home dropped on average to 120 to 130/70 mm Hg.
1 year ago. Mr. M reported that his memory loss had become worse. He perceived having more stress because of forgetfulness and visual difficulties, which had led him to stop driving at night.
At a follow-up appointment with his psychiatrist, Mr. M reported that, first, he had not tapered escitalopram as discussed and, second, he forgot to increase the dosage of desvenlafaxine. A home blood pressure log revealed consistent hypotension; the psychiatrist was concerned that hypotension could be the cause of concentration difficulties and malaise. The psychiatrist advised Mr. M to follow-up with his PCP and neurologist.
Current admission. Shortly after the visit to the psychiatrist, Mr. M presented to the emergency department for increased syncopal events. Work-up was negative for a cardiac cause. A cosyntropin stimulation test was negative, showing that adrenal insufficiency did not cause his orthostatic hypotension. Chart review showed he had been having blood pressure problems for many years, independent of antidepressants. Physical exam revealed lower extremity ataxia and a bilateral extensor plantar reflex.
What diagnosis explains Mr. M’s symptoms?
a) Parkinson’s disease
b) multiple system atrophy (MSA)
c) depression due to a general medical condition
d) dementia
The authors’ observations
MSA, previously referred to as Shy-Drager syndrome, is a rare, rapidly progressive neurodegenerative disorder with an estimated prevalence of 3.7 cases for every 100,000 people worldwide.2 MSA primarily affects middle-aged patients; because it has no cure, most patients die in 7 to 10 years.3
MSA has 2 clinical variants4,5:
• parkinsonian type (MSA-P), characterized by striatonigral degeneration and increased spasticity
• cerebellar type (MSA-C), characterized by more autonomic dysfunction.
MSA has a range of symptoms, making it a challenging diagnosis (Table).6 Although psychiatric symptoms are not part of the diagnostic criteria, they can aid in its diagnosis. In Mr. M’s case, depression, anxiety, orthostatic hypotension, and ataxia support a diagnosis of MSA.
Gilman et al6 delineated 3 diagnostic categories for MSA: definite MSA, probable MSA, and possible MSA. Clinical criteria shared by the 3 diagnostic categories are sporadic and progressive onset after age 30.
Definite MSA requires “neuropathological findings of widespread and abundant CNS alpha-synuclein-positive glial cytoplasmic inclusions,” along with “neurodegenerative changes in striatonigral or olivopontocerebellar structures” at autopsy.6
Probable MSA. Without autopsy findings required for definite MSA, the next most specific diagnostic category is probable MSA. Probable MSA also specifies that the patient show either autonomic failure involving urinary incontinence—this includes erectile dysfunction in men—or, if autonomic failure is absent, orthostatic hypotension within 3 minutes of standing by at least 30 mm Hg systolic pressure or 15 mm Hg diastolic pressure.
Possible MSA has less stringent criteria for orthostatic hypotension. The category includes patients who have only 1 symptom that suggests autonomic failure. Criteria for possible MSA include parkinsonism or a cerebellar syndrome in addition to symptoms of MSA listed in the Table, whereas probable MSA has specific criteria of either a poorly levodopa-responsive parkinsonism (MSA-P) or a cerebellar syndrome (MSA-C). In addition to having parkinsonism or a cerebellar syndrome, and 1 sign of autonomic failure or orthostatic hypotension, patients also must have ≥1 additional feature to be assigned a diagnosis of possible MSA, including:
• rapidly progressive parkinsonism
• poor response to levodopa
• postural instability within 3 years of motor onset
• gait ataxia, cerebellar dysarthria, limb ataxia, or cerebellar oculomotor dysfunction
• dysphagia within 5 years of motor onset
• atrophy on MRI of putamen, middle cerebellar peduncle, pons, or cerebellum
• hypometabolism on fluorodeoxyglucose- PET in putamen, brainstem, or cerebellum.6
Diagnosing MSA can be challenging because its features are similar to those of many other disorders. Nonetheless, Gilman et al6 lists specific criteria for probable MSA, including autonomic dysfunction, orthostatic hypotension, and either parkinsonism or cerebellar syndrome symptoms. Although a definite MSA diagnosis only can be made by postmortem brain specimen analysis, Osaki et al7 found that a probable MSA diagnosis has a positive predictive value of 92% with a sensitivity of 22% for definite MSA.
Mr. M’s symptoms were consistent with a diagnosis of probable MSA, cerebellar type (Figure).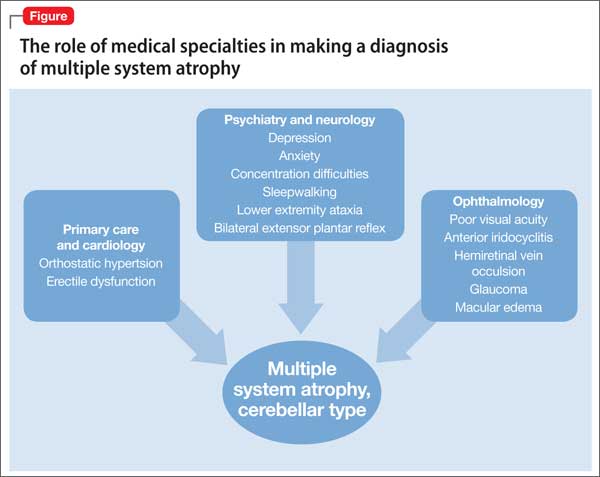
Psychiatric manifestations of MSA
There are a few case reports of depression identified early in patients who were later given a diagnosis of MSA.8
Depression. In a study by Benrud-Larson et al9 (N = 99), 49% of patients who had MSA reported moderate or severe depression, as indicated by a score of ≥17 on the Beck Depression Inventory (BDI); 80% reported at least mild depression (BDI ≥10, mean 17.0, standard deviation, 8.7).
In a similar study, by Balas et al,10 depression was reported as a common symptom and was statistically significant in MSA-P patients compared with controls (P = .013).
Anxiety, another symptom that was reported by Mr. M, is another psychiatric manifestation described by Balas et al10 and Chang et al.11 Balas et al10 noted that MSA-C and MSA-P patients had significantly more state anxiety (P = .009 and P = .022, respectively) compared with controls, although Chang et al11 noted higher anxiety scores in MSA-C patients compared with controls and MSA-P patients (P < .01).
Balas et al10 hypothesized that anxiety and depression contribute to cognitive decline; their study showed that MSA-C patients had difficulty learning new verbal information (P < .022) and controlling attention (P < .023). Mr. M exhibited some of these cognitive difficulties in his reports of losing track of conversations, forgetting the topic of a conversation when speaking, trouble focusing, and difficulty concentrating when driving.
Mr. M had depression and anxiety well before onset of autonomic dysfunction (orthostatic hypotension and erectile dysfunction), which eventually led to an MSA diagnosis. Psychiatrists should understand additional manifestations of MSA so that they can use psychiatric symptoms to identify these conditions in their patients. One of the most well-known and early manifestations of MSA is autonomic dysfunction; among men, another early sign is erectile dysfunction.6 Our patient also exhibited other less well-known symptoms linked to MSA and autonomic dysregulation, including RBD and ocular symptoms (iridocyclitis, glaucoma, decreased visual acuity).
Rapid eye-movement behavior disorder. Psychiatrists should consider screening for RBD during assessment of sleep problems. Identifying RBD is important because early studies have shown a strong association between RBD and development of a neurodegenerative disorder. Mr. M’s clinicians did not consider RBD, although his symptoms of sleepwalking and falling asleep while driving suggest a possible diagnosis. Also, considering this diagnosis would aid in diagnosing a synucleinopathy disorder because a higher incidence of RBD was noted in patients who developed synucleinopathy disorders (eg, Parkinson’s disease [PD] and dementia with Lewy bodies [DLB]) compared with patients who developed non-synucleinopathies (eg, frontotemporal dementia, corticobasal degeneration, progressive supranuclear palsy, mild cognitive impairment, primary progressive aphasia, and posterior cortical atrophy) or tauopathies (eg, Alzheimer’s disease).12
Zanigni et al13 reported similar findings in a later study that classified patients with RBD as having idiopathic RBD (IRBD) or RBD secondary to an underlying neurodegenerative disorder, particularly an α-synucleinopathy: PD, MSA, and DLB. Most IRBD patients developed 1 of the above mentioned neurodegenerative disorders as long as 10 years after a diagnosis of RBD.
In a study by Iranzo et al,14 patients with MSA were noted to have more severe RBD compared with PD patients. Severity is illustrated by greater periodic leg movements during sleep (P = .001), less total sleep time (P = .023), longer sleep onset latency (P = .023), and a higher percentage of REM sleep without atonia (RSWA, P = .001). McCarter et al15 also noted a higher incidence of RSWA in patients with MSA.
Patients with MSA might therefore be more likely to exhibit difficulty initiating and maintaining sleep and as having RSWA years before the MSA diagnosis.
Several psychotropics (eg, first-generation antipsychotics, tricyclic antidepressants, lithium, benzodiazepines, carbamazepine, topiramate, and selective serotonin reuptake inhibitors) can cause adverse ocular effects, such as closed-angle glaucoma in predisposed persons and retinopathy.16 Therefore, it is important for psychiatrists to ask about ocular symptoms because they might be an early sign of autonomic dysfunction.
Posner and Schlossman17 theorized a causal relationship between autonomic dysfunction and ocular diseases after studying a group of patients who had intermittent unilateral attacks of iridocyclitis and glaucoma (now known as Posner-Schlossman syndrome). They hypothesized that a central cause in the hypothalamus, combined with underlying autonomic dysregulation, could cause the intermittent attacks.
Gherghel et al18 noted a significant difference in ocular blood flow and blood pressure in patients with primary open-angle glaucoma (POAG) compared with controls. Patients with POAG did not show an increase in blood pressure or ocular blood flow when challenged by cold water, which should have increased their sympathetic activity. Gherghel et al18 concluded that this indicated possible systemic autonomic dysfunction in patients with POAG. In a study by Fischer et al,19 MSA patients also were noted to have significant loss of nasal retinal nerve fiber layer thickness vs controls (P < .05), leading to decreased peripheral vision sensitivity.
Bottom Line
Although psychiatric symptoms are not part of the diagnostic criteria for multiple system atrophy (MSA), they may serve as a clue to consider when they occur with other MSA symptoms. Evaluate the importance of psychiatric symptoms in terms of the whole picture of the patient. Although the diagnosis might not alter the patient’s course, it can allow family members to understand the patient’s condition and prepare for complications that will arise.
Related Resources
• The MSA Coalition. www.multiplesystematrophy.org.
• National Institute of Neurological Disorders and Stroke. Multiple system atrophy fact sheet. www.ninds.nih.gov/disorders/msa/detailmsa.htm.
• Wenning GK, Fanciulli A, eds. Multiple system atrophy. Vienna, Austria: Springer-Verlag Wien; 2014.
Drug Brand Names
Bupropion • Wellbutrin Lithium • Eskalith, Lithobid
Carbamazepine • Tegretol Methylphenidate • Ritalin
Desvenlafaxine • Pristiq Paroxetine • Paxil
Donepezil • Aricept Travoprost • Travatan
Escitalopram • Lexapro Trazodone • Desyrel, Oleptro
Fludrocortisone • Florinef Topiramate • Topamax
Fluoxetine • Prozac
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Weiner MF, Hynan LS, Rossetti H, et al. Luria’s three-step test: what is it and what does it tell us? Int Psychogeriatr. 2011;23(10):1602-1606.
2. Orphanet Report Series. Prevalence of rare diseases: bibliographic data. http://www.orpha.net/orphacom/ cahiers/docs/GB/Prevalence_of_rare_diseases_by_ alphabetical_list.pdf. Published May 2014. Accessed May 27, 2015.
3. National Institute of Neurological Disorders and Stroke. Multiple system atrophy with orthostatic hypotension information page. http://www.ninds.nih.gov/disorders/ msa_orthostatic_hypotension/msa_orthostatic_ hypotension.htm?css=print. Updated December 5, 2013. Accessed May 27, 2015.
4. Flaherty AW, Rost NS. The Massachusetts Hospital handbook of neurology. 2nd ed. Lippincott Williams & Wilkins: Boston, MA; 2007:79.
5. Hemingway J, Franco K, Chmelik E. Shy-Drager syndrome: multisystem atrophy with comorbid depression. Psychosomatics. 2005;46(1):73-76.
6. Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71(9):670-676.
7. Osaki Y, Wenning GK, Daniel SE, et al. Do published criteria improve clinical diagnostic accuracy in multiple system atrophy? Neurology. 2002;59(10):1486-1491.
8. Goto K, Ueki A, Shimode H, et al. Depression in multiple system atrophy: a case report. Psychiatry Clin Neurosci. 2000;54(4):507-511.
9. Benrud-Larson LM, Sandroni P, Schrag A, et al. Depressive symptoms and life satisfaction in patients with multiple system atrophy. Mov Disord. 2005;20(8):951-957.
10. Balas M, Balash Y, Giladi N, et al. Cognition in multiple system atrophy: neuropsychological profile and interaction with mood. J Neural Transm. 2010;117(3):369-375.
11. Chang CC, Chang YY, Chang WN, et al. Cognitive deficits in multiple system atrophy correlate with frontal atrophy and disease duration. Eur J Neurol. 2009;16(10):1144-1150.
12. Boeve BF, Silber MH, Parisi JE, et al. Synucleinopathy pathology and REM sleep behavior disorder plus dementia or parkinsonism. Neurology. 2003;61(1):40-45.
13. Zanigni S, Calandra-Buonaura G, Grimaldi D, et al. REM behaviour disorder and neurodegenerative diseases. Sleep Med. 2011;12(suppl 2):S54-S58.
14. Iranzo A, Santamaria J, Rye DB, et al. Characteristics of idiopathic REM sleep behavior disorder and that associated with MSA and PD. Neurology. 2005;65(2):247-252.
15. McCarter SJ, St. Louis EK, Boeve BF. REM sleep behavior disorder and REM sleep without atonia as early manifestation of degenerative neurological disease. Curr Neurol Neurosci Rep. 2012;12(2):182-192.
16. Richa S, Yazbek JC. Ocular adverse effects of common psychotropic agents: a review. CNS Drugs. 2010;24(6):501-526.
17. Posner A, Schlossman A. Syndrome of unilateral recurrent attacks of glaucoma with cyclitic symptoms. Arch Ophthal. 1948;39(4):517-535.
18. Gherghel D, Hosking SL, Cunliffe IA. Abnormal systemic and ocular vascular response to temperature provocation in primary open-angle glaucoma patients: a case for autonomic failure? Invest Ophthalmol Vis Sci. 2004;45(10):3546-3554.
19. Fischer MD, Synofzik M, Kernstock C, et al. Decreased retinal sensitivity and loss of retinal nerve fibers in multiple system atrophy. Graefes Arch Clin Exp Opthalmol. 2013;251(1):235-241.
CASE Light-headed
Mr. M, age 73, is a retired project manager who feels light-headed while walking his dog, causing him to go to the emergency department. His history is significant for hypertension, coronary artery disease (CAD), 3-vessel coronary artery bypass graft surgery (CABG), hyperlipidemia, erectile dysfunction, open-angle glaucoma, hemiretinal vein occlusion, symptoms suggesting rapid eye-movement behavior disorder (RBD), and major depressive disorder (MDD).
The psychiatry consultation-liaison service is asked to help manage Mr. M’s psychiatric medications in the context of orthostatic hypotension and cognitive deficits.
What could be causing Mr. M’s symptoms?
a) drug adverse effect
b) progressive cardiovascular disease
c) MDD
d) all of the above
HISTORY Depression, heart disease
15 years ago. Mr. M experienced his first major depressive episode. His primary care physician (PCP) commented on a history of falling asleep while driving and 1 episode of sleepwalking. His depression was treated to remission with fluoxetine and methylphenidate (dosages were not recorded), the latter also addressed his falling asleep while driving.
5 years ago. Mr. M had another depressive episode characterized by anxiety, difficulty sleeping, and irritability. He also described chest pain; a cardiac work-up revealed extensive CAD, which led to 3-vessel CABG later that year. He also reported dizziness upon standing, which was treated with compression stockings and an increase in sodium intake.
Mr. M continued to express feelings of depression. His cardiologist started him on paroxetine, 10 mg/d, which he took for 2 months and decided to stop because he felt better. He declined psychiatric referral.
4 years ago. Mr. M’s PCP referred him to a psychiatrist for depressed mood, anhedonia, decreased appetite, decreased energy, and difficulty concentrating. Immediate and delayed recall were found to be intact. The psychiatrist diagnosed MDD and Mr. M started escitalopram, 5 mg/d, titrated to 15 mg/d, and trazodone, 50 mg/d.
After starting treatment, Mr. M reported decreased libido. Sustained-release bupropion, 150 mg/d, was added to boost the effects of escitalopram and counteract sexual side effects.
At follow-up, Mr. M reported that his depressive symptoms and libido had improved, but that he had been experiencing unsteady gait when getting out of his car, which he had been noticing “for a while”—before he began trazodone. Mr. M was referred to his PCP, who attributed his symptoms to orthostasis. No treatment was indicated at the time because Mr. M’s lightheadedness had resolved.
3 years ago. Mr. M reported a syncopal attack and continued “dizziness.” His PCP prescribed fludrocortisone, 0.1 mg/d, later to be dosed 0.2 mg/d, and symptoms improved.
Although Mr. M had a history of orthostatic hypotension, he was later noted to have supine hypertension. Mr. M’s PCP was concerned that fludrocortisone could be causing the supine hypertension but that decreasing the dosage would cause his orthostatic hypotension to return.
The PCP also was concerned that the psychiatric medications (escitalopram, trazodone, and bupropion) could be causing orthostasis. There was discussion among Mr. M, his PCP, and his psychiatrist of stopping the psychotropics to see if the symptoms would remit; however, because of concerns about Mr. M’s depression, the medications were continued. Mr. M monitored his blood pressure at home and was referred to a neurologist for work-up of potential autonomic dysfunction.
Shortly afterward, Mr. M reported intermittent difficulty keeping track of his thoughts and finishing sentences. His psychiatrist ordered an MRI, which showed chronic small vessel ischemic changes, and started him on donepezil, 5 mg/d.
Neuropsychological testing revealed decreased processing speed and poor recognition memory; otherwise, results showed above-average intellectual ability and average or above-average performance in measures of language, attention, visuospatial/constructional functions, and executive functions—a pattern typically attributable to psychogenic factors, such as depression.
Mr. M reported to his neurologist that he forgets directions while driving but can focus better if he makes a conscious effort. Physical exam was significant hypotension; flat affect; deficits in concentration and short-term recall; mild impairment of Luria motor sequence (composed of a go/no-go and a reciprocal motor task); and vertical and horizontal saccades.1
Mr. M consulted with an ophthalmologist for anterior iridocyclitis and ocular hypertension, which was controlled with travoprost. He continued to experience trouble with his vision and was given a diagnosis of right inferior hemiretinal vein occlusion, macular edema, and suspected glaucoma. Subsequent notes recorded a history of Posner-Schlossman syndrome (a disease characterized by recurrent attacks of increased intraocular pressure in 1 eye with concomitant anterior chamber inflammation). His vision deteriorated until he was diagnosed with ocular hypertension, open-angle glaucoma, and dermatochalasis.
The authors’ observations
Involvement of multiple specialties in a patient’s care brings to question one’s philosophy on medical diagnosis. Interdisciplinary communication would seem to promote the principle of diagnostic parsimony, or Occam’s razor, which suggests a unifying diagnosis to explain all of the patient’s symptoms. Lack of communication might favor Hickam’s dictum, which states that “patients can have as many diseases as they damn well please.”
HISTORY Low energy, forgetfulness
2 years ago. Mr. M noticed low energy and motivation. He continued to work full-time but thought that it was taking him longer to get work done. He was tapered off escitalopram and started on desvenlafaxine, 50 mg/d; donepezil was increased to 10 mg/d.
The syncopal episodes resolved but blood pressure measured at home averaged 150/70 mm Hg. Mr. M was advised to decrease fludrocortisone from 0.2 mg/d to 0.1 mg/d. He tolerated the change and blood pressure measured at home dropped on average to 120 to 130/70 mm Hg.
1 year ago. Mr. M reported that his memory loss had become worse. He perceived having more stress because of forgetfulness and visual difficulties, which had led him to stop driving at night.
At a follow-up appointment with his psychiatrist, Mr. M reported that, first, he had not tapered escitalopram as discussed and, second, he forgot to increase the dosage of desvenlafaxine. A home blood pressure log revealed consistent hypotension; the psychiatrist was concerned that hypotension could be the cause of concentration difficulties and malaise. The psychiatrist advised Mr. M to follow-up with his PCP and neurologist.
Current admission. Shortly after the visit to the psychiatrist, Mr. M presented to the emergency department for increased syncopal events. Work-up was negative for a cardiac cause. A cosyntropin stimulation test was negative, showing that adrenal insufficiency did not cause his orthostatic hypotension. Chart review showed he had been having blood pressure problems for many years, independent of antidepressants. Physical exam revealed lower extremity ataxia and a bilateral extensor plantar reflex.
What diagnosis explains Mr. M’s symptoms?
a) Parkinson’s disease
b) multiple system atrophy (MSA)
c) depression due to a general medical condition
d) dementia
The authors’ observations
MSA, previously referred to as Shy-Drager syndrome, is a rare, rapidly progressive neurodegenerative disorder with an estimated prevalence of 3.7 cases for every 100,000 people worldwide.2 MSA primarily affects middle-aged patients; because it has no cure, most patients die in 7 to 10 years.3
MSA has 2 clinical variants4,5:
• parkinsonian type (MSA-P), characterized by striatonigral degeneration and increased spasticity
• cerebellar type (MSA-C), characterized by more autonomic dysfunction.
MSA has a range of symptoms, making it a challenging diagnosis (Table).6 Although psychiatric symptoms are not part of the diagnostic criteria, they can aid in its diagnosis. In Mr. M’s case, depression, anxiety, orthostatic hypotension, and ataxia support a diagnosis of MSA.
Gilman et al6 delineated 3 diagnostic categories for MSA: definite MSA, probable MSA, and possible MSA. Clinical criteria shared by the 3 diagnostic categories are sporadic and progressive onset after age 30.
Definite MSA requires “neuropathological findings of widespread and abundant CNS alpha-synuclein-positive glial cytoplasmic inclusions,” along with “neurodegenerative changes in striatonigral or olivopontocerebellar structures” at autopsy.6
Probable MSA. Without autopsy findings required for definite MSA, the next most specific diagnostic category is probable MSA. Probable MSA also specifies that the patient show either autonomic failure involving urinary incontinence—this includes erectile dysfunction in men—or, if autonomic failure is absent, orthostatic hypotension within 3 minutes of standing by at least 30 mm Hg systolic pressure or 15 mm Hg diastolic pressure.
Possible MSA has less stringent criteria for orthostatic hypotension. The category includes patients who have only 1 symptom that suggests autonomic failure. Criteria for possible MSA include parkinsonism or a cerebellar syndrome in addition to symptoms of MSA listed in the Table, whereas probable MSA has specific criteria of either a poorly levodopa-responsive parkinsonism (MSA-P) or a cerebellar syndrome (MSA-C). In addition to having parkinsonism or a cerebellar syndrome, and 1 sign of autonomic failure or orthostatic hypotension, patients also must have ≥1 additional feature to be assigned a diagnosis of possible MSA, including:
• rapidly progressive parkinsonism
• poor response to levodopa
• postural instability within 3 years of motor onset
• gait ataxia, cerebellar dysarthria, limb ataxia, or cerebellar oculomotor dysfunction
• dysphagia within 5 years of motor onset
• atrophy on MRI of putamen, middle cerebellar peduncle, pons, or cerebellum
• hypometabolism on fluorodeoxyglucose- PET in putamen, brainstem, or cerebellum.6
Diagnosing MSA can be challenging because its features are similar to those of many other disorders. Nonetheless, Gilman et al6 lists specific criteria for probable MSA, including autonomic dysfunction, orthostatic hypotension, and either parkinsonism or cerebellar syndrome symptoms. Although a definite MSA diagnosis only can be made by postmortem brain specimen analysis, Osaki et al7 found that a probable MSA diagnosis has a positive predictive value of 92% with a sensitivity of 22% for definite MSA.
Mr. M’s symptoms were consistent with a diagnosis of probable MSA, cerebellar type (Figure).
Psychiatric manifestations of MSA
There are a few case reports of depression identified early in patients who were later given a diagnosis of MSA.8
Depression. In a study by Benrud-Larson et al9 (N = 99), 49% of patients who had MSA reported moderate or severe depression, as indicated by a score of ≥17 on the Beck Depression Inventory (BDI); 80% reported at least mild depression (BDI ≥10, mean 17.0, standard deviation, 8.7).
In a similar study, by Balas et al,10 depression was reported as a common symptom and was statistically significant in MSA-P patients compared with controls (P = .013).
Anxiety, another symptom that was reported by Mr. M, is another psychiatric manifestation described by Balas et al10 and Chang et al.11 Balas et al10 noted that MSA-C and MSA-P patients had significantly more state anxiety (P = .009 and P = .022, respectively) compared with controls, although Chang et al11 noted higher anxiety scores in MSA-C patients compared with controls and MSA-P patients (P < .01).
Balas et al10 hypothesized that anxiety and depression contribute to cognitive decline; their study showed that MSA-C patients had difficulty learning new verbal information (P < .022) and controlling attention (P < .023). Mr. M exhibited some of these cognitive difficulties in his reports of losing track of conversations, forgetting the topic of a conversation when speaking, trouble focusing, and difficulty concentrating when driving.
Mr. M had depression and anxiety well before onset of autonomic dysfunction (orthostatic hypotension and erectile dysfunction), which eventually led to an MSA diagnosis. Psychiatrists should understand additional manifestations of MSA so that they can use psychiatric symptoms to identify these conditions in their patients. One of the most well-known and early manifestations of MSA is autonomic dysfunction; among men, another early sign is erectile dysfunction.6 Our patient also exhibited other less well-known symptoms linked to MSA and autonomic dysregulation, including RBD and ocular symptoms (iridocyclitis, glaucoma, decreased visual acuity).
Rapid eye-movement behavior disorder. Psychiatrists should consider screening for RBD during assessment of sleep problems. Identifying RBD is important because early studies have shown a strong association between RBD and development of a neurodegenerative disorder. Mr. M’s clinicians did not consider RBD, although his symptoms of sleepwalking and falling asleep while driving suggest a possible diagnosis. Also, considering this diagnosis would aid in diagnosing a synucleinopathy disorder because a higher incidence of RBD was noted in patients who developed synucleinopathy disorders (eg, Parkinson’s disease [PD] and dementia with Lewy bodies [DLB]) compared with patients who developed non-synucleinopathies (eg, frontotemporal dementia, corticobasal degeneration, progressive supranuclear palsy, mild cognitive impairment, primary progressive aphasia, and posterior cortical atrophy) or tauopathies (eg, Alzheimer’s disease).12
Zanigni et al13 reported similar findings in a later study that classified patients with RBD as having idiopathic RBD (IRBD) or RBD secondary to an underlying neurodegenerative disorder, particularly an α-synucleinopathy: PD, MSA, and DLB. Most IRBD patients developed 1 of the above mentioned neurodegenerative disorders as long as 10 years after a diagnosis of RBD.
In a study by Iranzo et al,14 patients with MSA were noted to have more severe RBD compared with PD patients. Severity is illustrated by greater periodic leg movements during sleep (P = .001), less total sleep time (P = .023), longer sleep onset latency (P = .023), and a higher percentage of REM sleep without atonia (RSWA, P = .001). McCarter et al15 also noted a higher incidence of RSWA in patients with MSA.
Patients with MSA might therefore be more likely to exhibit difficulty initiating and maintaining sleep and as having RSWA years before the MSA diagnosis.
Several psychotropics (eg, first-generation antipsychotics, tricyclic antidepressants, lithium, benzodiazepines, carbamazepine, topiramate, and selective serotonin reuptake inhibitors) can cause adverse ocular effects, such as closed-angle glaucoma in predisposed persons and retinopathy.16 Therefore, it is important for psychiatrists to ask about ocular symptoms because they might be an early sign of autonomic dysfunction.
Posner and Schlossman17 theorized a causal relationship between autonomic dysfunction and ocular diseases after studying a group of patients who had intermittent unilateral attacks of iridocyclitis and glaucoma (now known as Posner-Schlossman syndrome). They hypothesized that a central cause in the hypothalamus, combined with underlying autonomic dysregulation, could cause the intermittent attacks.
Gherghel et al18 noted a significant difference in ocular blood flow and blood pressure in patients with primary open-angle glaucoma (POAG) compared with controls. Patients with POAG did not show an increase in blood pressure or ocular blood flow when challenged by cold water, which should have increased their sympathetic activity. Gherghel et al18 concluded that this indicated possible systemic autonomic dysfunction in patients with POAG. In a study by Fischer et al,19 MSA patients also were noted to have significant loss of nasal retinal nerve fiber layer thickness vs controls (P < .05), leading to decreased peripheral vision sensitivity.
Bottom Line
Although psychiatric symptoms are not part of the diagnostic criteria for multiple system atrophy (MSA), they may serve as a clue to consider when they occur with other MSA symptoms. Evaluate the importance of psychiatric symptoms in terms of the whole picture of the patient. Although the diagnosis might not alter the patient’s course, it can allow family members to understand the patient’s condition and prepare for complications that will arise.
Related Resources
• The MSA Coalition. www.multiplesystematrophy.org.
• National Institute of Neurological Disorders and Stroke. Multiple system atrophy fact sheet. www.ninds.nih.gov/disorders/msa/detailmsa.htm.
• Wenning GK, Fanciulli A, eds. Multiple system atrophy. Vienna, Austria: Springer-Verlag Wien; 2014.
Drug Brand Names
Bupropion • Wellbutrin Lithium • Eskalith, Lithobid
Carbamazepine • Tegretol Methylphenidate • Ritalin
Desvenlafaxine • Pristiq Paroxetine • Paxil
Donepezil • Aricept Travoprost • Travatan
Escitalopram • Lexapro Trazodone • Desyrel, Oleptro
Fludrocortisone • Florinef Topiramate • Topamax
Fluoxetine • Prozac
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
CASE Light-headed
Mr. M, age 73, is a retired project manager who feels light-headed while walking his dog, causing him to go to the emergency department. His history is significant for hypertension, coronary artery disease (CAD), 3-vessel coronary artery bypass graft surgery (CABG), hyperlipidemia, erectile dysfunction, open-angle glaucoma, hemiretinal vein occlusion, symptoms suggesting rapid eye-movement behavior disorder (RBD), and major depressive disorder (MDD).
The psychiatry consultation-liaison service is asked to help manage Mr. M’s psychiatric medications in the context of orthostatic hypotension and cognitive deficits.
What could be causing Mr. M’s symptoms?
a) drug adverse effect
b) progressive cardiovascular disease
c) MDD
d) all of the above
HISTORY Depression, heart disease
15 years ago. Mr. M experienced his first major depressive episode. His primary care physician (PCP) commented on a history of falling asleep while driving and 1 episode of sleepwalking. His depression was treated to remission with fluoxetine and methylphenidate (dosages were not recorded), the latter also addressed his falling asleep while driving.
5 years ago. Mr. M had another depressive episode characterized by anxiety, difficulty sleeping, and irritability. He also described chest pain; a cardiac work-up revealed extensive CAD, which led to 3-vessel CABG later that year. He also reported dizziness upon standing, which was treated with compression stockings and an increase in sodium intake.
Mr. M continued to express feelings of depression. His cardiologist started him on paroxetine, 10 mg/d, which he took for 2 months and decided to stop because he felt better. He declined psychiatric referral.
4 years ago. Mr. M’s PCP referred him to a psychiatrist for depressed mood, anhedonia, decreased appetite, decreased energy, and difficulty concentrating. Immediate and delayed recall were found to be intact. The psychiatrist diagnosed MDD and Mr. M started escitalopram, 5 mg/d, titrated to 15 mg/d, and trazodone, 50 mg/d.
After starting treatment, Mr. M reported decreased libido. Sustained-release bupropion, 150 mg/d, was added to boost the effects of escitalopram and counteract sexual side effects.
At follow-up, Mr. M reported that his depressive symptoms and libido had improved, but that he had been experiencing unsteady gait when getting out of his car, which he had been noticing “for a while”—before he began trazodone. Mr. M was referred to his PCP, who attributed his symptoms to orthostasis. No treatment was indicated at the time because Mr. M’s lightheadedness had resolved.
3 years ago. Mr. M reported a syncopal attack and continued “dizziness.” His PCP prescribed fludrocortisone, 0.1 mg/d, later to be dosed 0.2 mg/d, and symptoms improved.
Although Mr. M had a history of orthostatic hypotension, he was later noted to have supine hypertension. Mr. M’s PCP was concerned that fludrocortisone could be causing the supine hypertension but that decreasing the dosage would cause his orthostatic hypotension to return.
The PCP also was concerned that the psychiatric medications (escitalopram, trazodone, and bupropion) could be causing orthostasis. There was discussion among Mr. M, his PCP, and his psychiatrist of stopping the psychotropics to see if the symptoms would remit; however, because of concerns about Mr. M’s depression, the medications were continued. Mr. M monitored his blood pressure at home and was referred to a neurologist for work-up of potential autonomic dysfunction.
Shortly afterward, Mr. M reported intermittent difficulty keeping track of his thoughts and finishing sentences. His psychiatrist ordered an MRI, which showed chronic small vessel ischemic changes, and started him on donepezil, 5 mg/d.
Neuropsychological testing revealed decreased processing speed and poor recognition memory; otherwise, results showed above-average intellectual ability and average or above-average performance in measures of language, attention, visuospatial/constructional functions, and executive functions—a pattern typically attributable to psychogenic factors, such as depression.
Mr. M reported to his neurologist that he forgets directions while driving but can focus better if he makes a conscious effort. Physical exam was significant hypotension; flat affect; deficits in concentration and short-term recall; mild impairment of Luria motor sequence (composed of a go/no-go and a reciprocal motor task); and vertical and horizontal saccades.1
Mr. M consulted with an ophthalmologist for anterior iridocyclitis and ocular hypertension, which was controlled with travoprost. He continued to experience trouble with his vision and was given a diagnosis of right inferior hemiretinal vein occlusion, macular edema, and suspected glaucoma. Subsequent notes recorded a history of Posner-Schlossman syndrome (a disease characterized by recurrent attacks of increased intraocular pressure in 1 eye with concomitant anterior chamber inflammation). His vision deteriorated until he was diagnosed with ocular hypertension, open-angle glaucoma, and dermatochalasis.
The authors’ observations
Involvement of multiple specialties in a patient’s care brings to question one’s philosophy on medical diagnosis. Interdisciplinary communication would seem to promote the principle of diagnostic parsimony, or Occam’s razor, which suggests a unifying diagnosis to explain all of the patient’s symptoms. Lack of communication might favor Hickam’s dictum, which states that “patients can have as many diseases as they damn well please.”
HISTORY Low energy, forgetfulness
2 years ago. Mr. M noticed low energy and motivation. He continued to work full-time but thought that it was taking him longer to get work done. He was tapered off escitalopram and started on desvenlafaxine, 50 mg/d; donepezil was increased to 10 mg/d.
The syncopal episodes resolved but blood pressure measured at home averaged 150/70 mm Hg. Mr. M was advised to decrease fludrocortisone from 0.2 mg/d to 0.1 mg/d. He tolerated the change and blood pressure measured at home dropped on average to 120 to 130/70 mm Hg.
1 year ago. Mr. M reported that his memory loss had become worse. He perceived having more stress because of forgetfulness and visual difficulties, which had led him to stop driving at night.
At a follow-up appointment with his psychiatrist, Mr. M reported that, first, he had not tapered escitalopram as discussed and, second, he forgot to increase the dosage of desvenlafaxine. A home blood pressure log revealed consistent hypotension; the psychiatrist was concerned that hypotension could be the cause of concentration difficulties and malaise. The psychiatrist advised Mr. M to follow-up with his PCP and neurologist.
Current admission. Shortly after the visit to the psychiatrist, Mr. M presented to the emergency department for increased syncopal events. Work-up was negative for a cardiac cause. A cosyntropin stimulation test was negative, showing that adrenal insufficiency did not cause his orthostatic hypotension. Chart review showed he had been having blood pressure problems for many years, independent of antidepressants. Physical exam revealed lower extremity ataxia and a bilateral extensor plantar reflex.
What diagnosis explains Mr. M’s symptoms?
a) Parkinson’s disease
b) multiple system atrophy (MSA)
c) depression due to a general medical condition
d) dementia
The authors’ observations
MSA, previously referred to as Shy-Drager syndrome, is a rare, rapidly progressive neurodegenerative disorder with an estimated prevalence of 3.7 cases for every 100,000 people worldwide.2 MSA primarily affects middle-aged patients; because it has no cure, most patients die in 7 to 10 years.3
MSA has 2 clinical variants4,5:
• parkinsonian type (MSA-P), characterized by striatonigral degeneration and increased spasticity
• cerebellar type (MSA-C), characterized by more autonomic dysfunction.
MSA has a range of symptoms, making it a challenging diagnosis (Table).6 Although psychiatric symptoms are not part of the diagnostic criteria, they can aid in its diagnosis. In Mr. M’s case, depression, anxiety, orthostatic hypotension, and ataxia support a diagnosis of MSA.
Gilman et al6 delineated 3 diagnostic categories for MSA: definite MSA, probable MSA, and possible MSA. Clinical criteria shared by the 3 diagnostic categories are sporadic and progressive onset after age 30.
Definite MSA requires “neuropathological findings of widespread and abundant CNS alpha-synuclein-positive glial cytoplasmic inclusions,” along with “neurodegenerative changes in striatonigral or olivopontocerebellar structures” at autopsy.6
Probable MSA. Without autopsy findings required for definite MSA, the next most specific diagnostic category is probable MSA. Probable MSA also specifies that the patient show either autonomic failure involving urinary incontinence—this includes erectile dysfunction in men—or, if autonomic failure is absent, orthostatic hypotension within 3 minutes of standing by at least 30 mm Hg systolic pressure or 15 mm Hg diastolic pressure.
Possible MSA has less stringent criteria for orthostatic hypotension. The category includes patients who have only 1 symptom that suggests autonomic failure. Criteria for possible MSA include parkinsonism or a cerebellar syndrome in addition to symptoms of MSA listed in the Table, whereas probable MSA has specific criteria of either a poorly levodopa-responsive parkinsonism (MSA-P) or a cerebellar syndrome (MSA-C). In addition to having parkinsonism or a cerebellar syndrome, and 1 sign of autonomic failure or orthostatic hypotension, patients also must have ≥1 additional feature to be assigned a diagnosis of possible MSA, including:
• rapidly progressive parkinsonism
• poor response to levodopa
• postural instability within 3 years of motor onset
• gait ataxia, cerebellar dysarthria, limb ataxia, or cerebellar oculomotor dysfunction
• dysphagia within 5 years of motor onset
• atrophy on MRI of putamen, middle cerebellar peduncle, pons, or cerebellum
• hypometabolism on fluorodeoxyglucose- PET in putamen, brainstem, or cerebellum.6
Diagnosing MSA can be challenging because its features are similar to those of many other disorders. Nonetheless, Gilman et al6 lists specific criteria for probable MSA, including autonomic dysfunction, orthostatic hypotension, and either parkinsonism or cerebellar syndrome symptoms. Although a definite MSA diagnosis only can be made by postmortem brain specimen analysis, Osaki et al7 found that a probable MSA diagnosis has a positive predictive value of 92% with a sensitivity of 22% for definite MSA.
Mr. M’s symptoms were consistent with a diagnosis of probable MSA, cerebellar type (Figure).
Psychiatric manifestations of MSA
There are a few case reports of depression identified early in patients who were later given a diagnosis of MSA.8
Depression. In a study by Benrud-Larson et al9 (N = 99), 49% of patients who had MSA reported moderate or severe depression, as indicated by a score of ≥17 on the Beck Depression Inventory (BDI); 80% reported at least mild depression (BDI ≥10, mean 17.0, standard deviation, 8.7).
In a similar study, by Balas et al,10 depression was reported as a common symptom and was statistically significant in MSA-P patients compared with controls (P = .013).
Anxiety, another symptom that was reported by Mr. M, is another psychiatric manifestation described by Balas et al10 and Chang et al.11 Balas et al10 noted that MSA-C and MSA-P patients had significantly more state anxiety (P = .009 and P = .022, respectively) compared with controls, although Chang et al11 noted higher anxiety scores in MSA-C patients compared with controls and MSA-P patients (P < .01).
Balas et al10 hypothesized that anxiety and depression contribute to cognitive decline; their study showed that MSA-C patients had difficulty learning new verbal information (P < .022) and controlling attention (P < .023). Mr. M exhibited some of these cognitive difficulties in his reports of losing track of conversations, forgetting the topic of a conversation when speaking, trouble focusing, and difficulty concentrating when driving.
Mr. M had depression and anxiety well before onset of autonomic dysfunction (orthostatic hypotension and erectile dysfunction), which eventually led to an MSA diagnosis. Psychiatrists should understand additional manifestations of MSA so that they can use psychiatric symptoms to identify these conditions in their patients. One of the most well-known and early manifestations of MSA is autonomic dysfunction; among men, another early sign is erectile dysfunction.6 Our patient also exhibited other less well-known symptoms linked to MSA and autonomic dysregulation, including RBD and ocular symptoms (iridocyclitis, glaucoma, decreased visual acuity).
Rapid eye-movement behavior disorder. Psychiatrists should consider screening for RBD during assessment of sleep problems. Identifying RBD is important because early studies have shown a strong association between RBD and development of a neurodegenerative disorder. Mr. M’s clinicians did not consider RBD, although his symptoms of sleepwalking and falling asleep while driving suggest a possible diagnosis. Also, considering this diagnosis would aid in diagnosing a synucleinopathy disorder because a higher incidence of RBD was noted in patients who developed synucleinopathy disorders (eg, Parkinson’s disease [PD] and dementia with Lewy bodies [DLB]) compared with patients who developed non-synucleinopathies (eg, frontotemporal dementia, corticobasal degeneration, progressive supranuclear palsy, mild cognitive impairment, primary progressive aphasia, and posterior cortical atrophy) or tauopathies (eg, Alzheimer’s disease).12
Zanigni et al13 reported similar findings in a later study that classified patients with RBD as having idiopathic RBD (IRBD) or RBD secondary to an underlying neurodegenerative disorder, particularly an α-synucleinopathy: PD, MSA, and DLB. Most IRBD patients developed 1 of the above mentioned neurodegenerative disorders as long as 10 years after a diagnosis of RBD.
In a study by Iranzo et al,14 patients with MSA were noted to have more severe RBD compared with PD patients. Severity is illustrated by greater periodic leg movements during sleep (P = .001), less total sleep time (P = .023), longer sleep onset latency (P = .023), and a higher percentage of REM sleep without atonia (RSWA, P = .001). McCarter et al15 also noted a higher incidence of RSWA in patients with MSA.
Patients with MSA might therefore be more likely to exhibit difficulty initiating and maintaining sleep and as having RSWA years before the MSA diagnosis.
Several psychotropics (eg, first-generation antipsychotics, tricyclic antidepressants, lithium, benzodiazepines, carbamazepine, topiramate, and selective serotonin reuptake inhibitors) can cause adverse ocular effects, such as closed-angle glaucoma in predisposed persons and retinopathy.16 Therefore, it is important for psychiatrists to ask about ocular symptoms because they might be an early sign of autonomic dysfunction.
Posner and Schlossman17 theorized a causal relationship between autonomic dysfunction and ocular diseases after studying a group of patients who had intermittent unilateral attacks of iridocyclitis and glaucoma (now known as Posner-Schlossman syndrome). They hypothesized that a central cause in the hypothalamus, combined with underlying autonomic dysregulation, could cause the intermittent attacks.
Gherghel et al18 noted a significant difference in ocular blood flow and blood pressure in patients with primary open-angle glaucoma (POAG) compared with controls. Patients with POAG did not show an increase in blood pressure or ocular blood flow when challenged by cold water, which should have increased their sympathetic activity. Gherghel et al18 concluded that this indicated possible systemic autonomic dysfunction in patients with POAG. In a study by Fischer et al,19 MSA patients also were noted to have significant loss of nasal retinal nerve fiber layer thickness vs controls (P < .05), leading to decreased peripheral vision sensitivity.
Bottom Line
Although psychiatric symptoms are not part of the diagnostic criteria for multiple system atrophy (MSA), they may serve as a clue to consider when they occur with other MSA symptoms. Evaluate the importance of psychiatric symptoms in terms of the whole picture of the patient. Although the diagnosis might not alter the patient’s course, it can allow family members to understand the patient’s condition and prepare for complications that will arise.
Related Resources
• The MSA Coalition. www.multiplesystematrophy.org.
• National Institute of Neurological Disorders and Stroke. Multiple system atrophy fact sheet. www.ninds.nih.gov/disorders/msa/detailmsa.htm.
• Wenning GK, Fanciulli A, eds. Multiple system atrophy. Vienna, Austria: Springer-Verlag Wien; 2014.
Drug Brand Names
Bupropion • Wellbutrin Lithium • Eskalith, Lithobid
Carbamazepine • Tegretol Methylphenidate • Ritalin
Desvenlafaxine • Pristiq Paroxetine • Paxil
Donepezil • Aricept Travoprost • Travatan
Escitalopram • Lexapro Trazodone • Desyrel, Oleptro
Fludrocortisone • Florinef Topiramate • Topamax
Fluoxetine • Prozac
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Weiner MF, Hynan LS, Rossetti H, et al. Luria’s three-step test: what is it and what does it tell us? Int Psychogeriatr. 2011;23(10):1602-1606.
2. Orphanet Report Series. Prevalence of rare diseases: bibliographic data. http://www.orpha.net/orphacom/ cahiers/docs/GB/Prevalence_of_rare_diseases_by_ alphabetical_list.pdf. Published May 2014. Accessed May 27, 2015.
3. National Institute of Neurological Disorders and Stroke. Multiple system atrophy with orthostatic hypotension information page. http://www.ninds.nih.gov/disorders/ msa_orthostatic_hypotension/msa_orthostatic_ hypotension.htm?css=print. Updated December 5, 2013. Accessed May 27, 2015.
4. Flaherty AW, Rost NS. The Massachusetts Hospital handbook of neurology. 2nd ed. Lippincott Williams & Wilkins: Boston, MA; 2007:79.
5. Hemingway J, Franco K, Chmelik E. Shy-Drager syndrome: multisystem atrophy with comorbid depression. Psychosomatics. 2005;46(1):73-76.
6. Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71(9):670-676.
7. Osaki Y, Wenning GK, Daniel SE, et al. Do published criteria improve clinical diagnostic accuracy in multiple system atrophy? Neurology. 2002;59(10):1486-1491.
8. Goto K, Ueki A, Shimode H, et al. Depression in multiple system atrophy: a case report. Psychiatry Clin Neurosci. 2000;54(4):507-511.
9. Benrud-Larson LM, Sandroni P, Schrag A, et al. Depressive symptoms and life satisfaction in patients with multiple system atrophy. Mov Disord. 2005;20(8):951-957.
10. Balas M, Balash Y, Giladi N, et al. Cognition in multiple system atrophy: neuropsychological profile and interaction with mood. J Neural Transm. 2010;117(3):369-375.
11. Chang CC, Chang YY, Chang WN, et al. Cognitive deficits in multiple system atrophy correlate with frontal atrophy and disease duration. Eur J Neurol. 2009;16(10):1144-1150.
12. Boeve BF, Silber MH, Parisi JE, et al. Synucleinopathy pathology and REM sleep behavior disorder plus dementia or parkinsonism. Neurology. 2003;61(1):40-45.
13. Zanigni S, Calandra-Buonaura G, Grimaldi D, et al. REM behaviour disorder and neurodegenerative diseases. Sleep Med. 2011;12(suppl 2):S54-S58.
14. Iranzo A, Santamaria J, Rye DB, et al. Characteristics of idiopathic REM sleep behavior disorder and that associated with MSA and PD. Neurology. 2005;65(2):247-252.
15. McCarter SJ, St. Louis EK, Boeve BF. REM sleep behavior disorder and REM sleep without atonia as early manifestation of degenerative neurological disease. Curr Neurol Neurosci Rep. 2012;12(2):182-192.
16. Richa S, Yazbek JC. Ocular adverse effects of common psychotropic agents: a review. CNS Drugs. 2010;24(6):501-526.
17. Posner A, Schlossman A. Syndrome of unilateral recurrent attacks of glaucoma with cyclitic symptoms. Arch Ophthal. 1948;39(4):517-535.
18. Gherghel D, Hosking SL, Cunliffe IA. Abnormal systemic and ocular vascular response to temperature provocation in primary open-angle glaucoma patients: a case for autonomic failure? Invest Ophthalmol Vis Sci. 2004;45(10):3546-3554.
19. Fischer MD, Synofzik M, Kernstock C, et al. Decreased retinal sensitivity and loss of retinal nerve fibers in multiple system atrophy. Graefes Arch Clin Exp Opthalmol. 2013;251(1):235-241.
1. Weiner MF, Hynan LS, Rossetti H, et al. Luria’s three-step test: what is it and what does it tell us? Int Psychogeriatr. 2011;23(10):1602-1606.
2. Orphanet Report Series. Prevalence of rare diseases: bibliographic data. http://www.orpha.net/orphacom/ cahiers/docs/GB/Prevalence_of_rare_diseases_by_ alphabetical_list.pdf. Published May 2014. Accessed May 27, 2015.
3. National Institute of Neurological Disorders and Stroke. Multiple system atrophy with orthostatic hypotension information page. http://www.ninds.nih.gov/disorders/ msa_orthostatic_hypotension/msa_orthostatic_ hypotension.htm?css=print. Updated December 5, 2013. Accessed May 27, 2015.
4. Flaherty AW, Rost NS. The Massachusetts Hospital handbook of neurology. 2nd ed. Lippincott Williams & Wilkins: Boston, MA; 2007:79.
5. Hemingway J, Franco K, Chmelik E. Shy-Drager syndrome: multisystem atrophy with comorbid depression. Psychosomatics. 2005;46(1):73-76.
6. Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71(9):670-676.
7. Osaki Y, Wenning GK, Daniel SE, et al. Do published criteria improve clinical diagnostic accuracy in multiple system atrophy? Neurology. 2002;59(10):1486-1491.
8. Goto K, Ueki A, Shimode H, et al. Depression in multiple system atrophy: a case report. Psychiatry Clin Neurosci. 2000;54(4):507-511.
9. Benrud-Larson LM, Sandroni P, Schrag A, et al. Depressive symptoms and life satisfaction in patients with multiple system atrophy. Mov Disord. 2005;20(8):951-957.
10. Balas M, Balash Y, Giladi N, et al. Cognition in multiple system atrophy: neuropsychological profile and interaction with mood. J Neural Transm. 2010;117(3):369-375.
11. Chang CC, Chang YY, Chang WN, et al. Cognitive deficits in multiple system atrophy correlate with frontal atrophy and disease duration. Eur J Neurol. 2009;16(10):1144-1150.
12. Boeve BF, Silber MH, Parisi JE, et al. Synucleinopathy pathology and REM sleep behavior disorder plus dementia or parkinsonism. Neurology. 2003;61(1):40-45.
13. Zanigni S, Calandra-Buonaura G, Grimaldi D, et al. REM behaviour disorder and neurodegenerative diseases. Sleep Med. 2011;12(suppl 2):S54-S58.
14. Iranzo A, Santamaria J, Rye DB, et al. Characteristics of idiopathic REM sleep behavior disorder and that associated with MSA and PD. Neurology. 2005;65(2):247-252.
15. McCarter SJ, St. Louis EK, Boeve BF. REM sleep behavior disorder and REM sleep without atonia as early manifestation of degenerative neurological disease. Curr Neurol Neurosci Rep. 2012;12(2):182-192.
16. Richa S, Yazbek JC. Ocular adverse effects of common psychotropic agents: a review. CNS Drugs. 2010;24(6):501-526.
17. Posner A, Schlossman A. Syndrome of unilateral recurrent attacks of glaucoma with cyclitic symptoms. Arch Ophthal. 1948;39(4):517-535.
18. Gherghel D, Hosking SL, Cunliffe IA. Abnormal systemic and ocular vascular response to temperature provocation in primary open-angle glaucoma patients: a case for autonomic failure? Invest Ophthalmol Vis Sci. 2004;45(10):3546-3554.
19. Fischer MD, Synofzik M, Kernstock C, et al. Decreased retinal sensitivity and loss of retinal nerve fibers in multiple system atrophy. Graefes Arch Clin Exp Opthalmol. 2013;251(1):235-241.
Managing first-episode psychosis: Rationale and evidence for nonstandard first-line treatments for schizophrenia
First-episode psychosis (FEP) in schizophrenia is characterized by high response rates to antipsychotic therapy, followed by frequent antipsychotic discontinuation and elevated relapse rates soon after maintenance treatment begins.1,2 With subsequent episodes, time to response progressively increases and likelihood of response decreases.3,4
To address these issues, this article—the second of 2 parts5—describes the rationale and evidence for using nonstandard first-line antipsychotic therapies to manage FEP. Specifically, we discuss when clinicians might consider monotherapy exceeding FDA-approved maximum dosages, combination therapy, long-acting injectable antipsychotics (LAIA), or clozapine.
Monotherapy beyond FDA-approved dosages
Treatment guidelines for FEP recommend oral antipsychotic dosages in the lower half of the treatment range and lower than those that are required for multi-episode schizophrenia.6-16 Ultimately, clinicians prescribe individualized dosages for their patients based on symptom improvement and tolerability. The optimal dosage at which to achieve a favorable D2 receptor occupancy likely will vary from patient to patient.17
To control symptoms, higher dosages may be needed than those used in FEP clinical trials, recommended by guidelines for FEP or multi-episode patients, or approved by the FDA. Patients seen in everyday practice may be more complicated (eg, have a comorbid condition or history of nonresponse) than study populations. Higher dosages also may be reasonable to overcome drug−drug interactions (eg, cigarette smoking-mediated cytochrome P450 1A2 induction, resulting in increased olanzapine metabolism),18 or to establish antipsychotic failure if adequate trials at lower dosages have resulted in a suboptimal response and the patient is not experiencing tolerability or safety concerns.
In a study of low-, full-, and high-dosage antipsychotic therapy in FEP, an additional 15% of patients responded to higher dosages of olanzapine and risperidone after failing to respond to a standard dosage.19 A study of data from the Recovery After an Initial Schizophrenia Episode Project’s Early Treatment Program (RAISE-ETP) found that, of participants identified who may benefit from therapy modification, 8.8% were prescribed an antipsychotic (often, olanzapine, risperidone, and haloperidol) at a higher-than-recommended dosage.20 Of note, only olanzapine was prescribed at higher than FDA-approved dosages.
Antipsychotic combination therapy
Prescribing combinations of antipsychotics—antipsychotic polypharmacy (APP)— has a negative connotation because of limited efficacy and safety data,21 and limited endorsement in schizophrenia treatment guidelines.9,13 Caution with APP is warranted; a complex medication regimen may increase the potential for adverse effects, poorer adherence, and adverse drug-drug interactions.9 APP has been shown to independently predict both shorter treatment duration and discontinuation before 1 year.22
Nonetheless, the clinician and patient may share the decision to implement APP and observe whether benefits outweigh risks in situations such as:
• to optimize neuroreceptor occupancy and targets (eg, attempting to achieve adequate D2 receptor blockade while minimizing side effects secondary to binding other receptors)
• to manage co-existing symptom domains (eg, mood changes, aggression, negative symptoms, disorganization, and cognitive deficits)
• to mitigate antipsychotic-induced side effects (eg, initiating aripiprazole to treat hyperprolactinemia induced by another antipsychotic to which the patient has achieved a favorable response).23
Clinicians report using APP to treat as many as 50% of patients with a history of multiple psychotic episodes.23 For FEP patients, 23% of participants in the RAISE-ETP trial who were identified as possibly benefiting from therapy modification were prescribed APP.20 Regrettably, researchers have not found evidence to support a reported rationale for using APP—that lower dosages of individual antipsychotics when used in combination may avoid high-dosage prescriptions.24
Before implementing APP, thoroughly explore and manage reasons for a patient’s suboptimal response to monotherapy.25 An adequate trial with any antipsychotic should be at the highest tolerated dosage for 12 to 16 weeks. Be mindful that response to an APP trial may be the result of additional time on the original antipsychotic.
Long-acting injectable antipsychotics in FEP
Guideline recommendations. Most older guidelines for schizophrenia treatment suggest LAIA after multiple relapses related to medication nonadherence or when a patient prefers injected medication (Table 1).6-13 Expert consensus guidelines also recommend considering LAIA in patients who lack insight into their illness. The Texas Medication Algorithm Project (TMAP) guidelines7 state LAIA can be considered for inadequate adherence at any stage, whereas the 2010 British Association for Psychopharmacology (BAP) guidelines9 express uncertainty about their use in FEP, because of limited evidence. Both the BAP and National Institute for Health and Care Excellence guidelines13 urge clinicians to consider LAIA when avoiding nonadherence is a treatment priority.
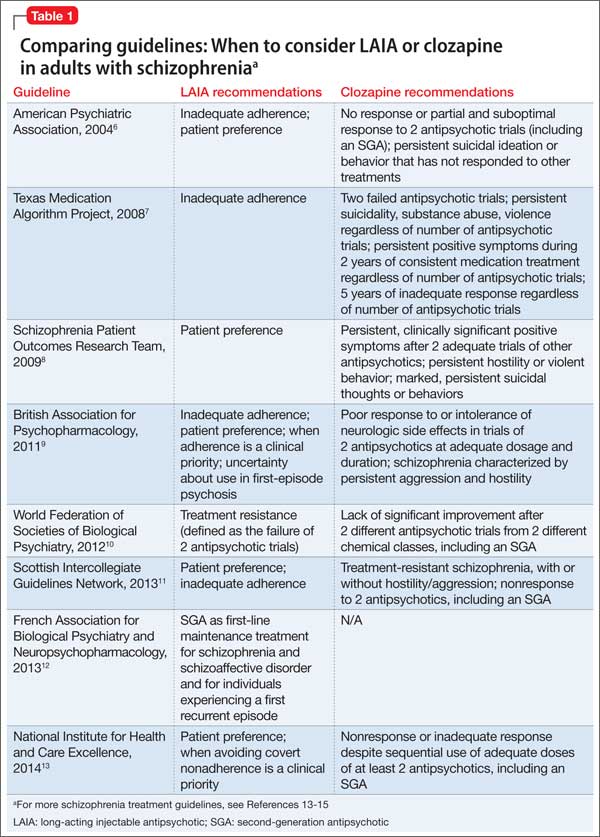
Recently, the French Association for Biological Psychiatry and Neuro-psychopharmacology (AFPBN) created expert consensus guidelines12 on using LAIA in practice. They recommend long-acting injectable second-generation antipsychotics (SGAs) as first-line maintenance treatment for schizophrenia and schizoaffective disorder and for individuals experiencing a first recurrent episode. The World Federation of Societies of Biological Psychiatry guidelines contain LAIA dosage recommendations for FEP (Table 2).10

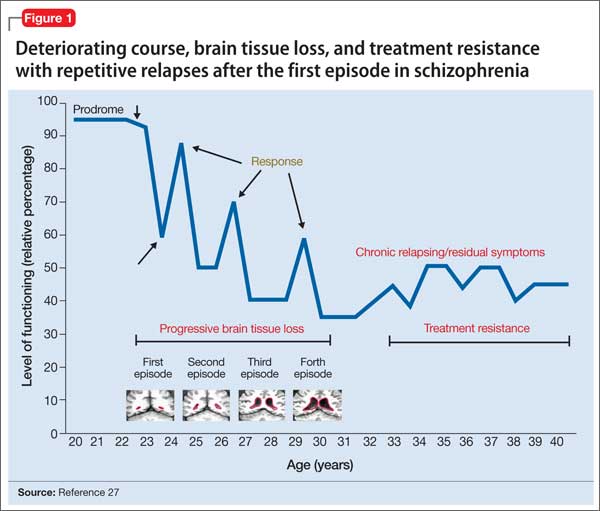
Advances have been made in understanding the serious neurobiological adverse effects of psychotic relapses, including neuroinflammation and oxidative stress, that may explain the atrophic changes observed with psychotic episodes starting with the FEP. Protecting the patient from a second episode has become a vital therapeutic management goal26 (Figure 127).
Concerns. Compared with oral antipsychotics, LAIA offers clinical advantages:
• improved pharmacokinetic profile (lower “peaks” and higher “valleys”)
• more consistent plasma concentrations (no variability related to administration timing or food effects)
• no first-pass metabolism, which can ease the process of finding the lowest effective and safe dosage
• reduced administration burden and objective tracking of adherence with typical dosing every 2 to 4 weeks
• less stigmatizing than oral medication for FEP patients, such as college students living in a dormitory.28,29
Barriers to LAIA use include:
• slow dosage titration and increased time to reach steady state drug level
• oral supplementation for some (eg, risperidone microspheres and aripiprazole long-acting injectable)
• logistical challenges for some (eg, 3-hour post-injection monitoring for delirium sedation syndrome with olanzapine pamoate)
• additional planning to coordinate care for scheduled injections
• higher expenses up front
• local injection site reactions
• dosage adjustment difficulties if adverse effects occur.28,29
Adoption rates of LAIA are low, especially for FEP.30 Most surveys indicate that (1) physicians believe LAIA treatment is ineffective for FEP31 and (2) patients do not prefer injectable to oral antipsychotics,32 despite evidence to the contrary.33,34 A survey of 198 psychiatrists identified 3 factors that influenced their decisions against using LAIA patients with FEP:
• limited availability of SGA depot formulations (4, to date, in the United States)
• frequent rejection by the patient when LAIA is offered without adequate explanation or encouragement
• skepticism of FEP patients (and their family) who lack experience with relapse.35
In reality, when SGA depots were introduced in the United Kingdom, prescribing rates of LAIA did not increase. As for patient rejection being a major reason for not prescribing LAIA, few patients (5% to 36%) are offered depot injections, particularly in FEP.29 Most patients using LAIA are chronic, multi-episode, violent people who are receiving medications involuntarily.29 Interestingly, this survey did not find 2 factors to be influential in psychiatrists’ decision not to use LAIA in FEP:
• guidelines do not explicitly recommend depot treatment in FEP
• treatment in FEP may be limited to 1 year, therefore depot administration is not worthwhile.35
Preliminary evidence. At least a dozen studies have explored LAIA treatment for FEP, with the use of fluphenazine decanoate,36 perphenazine enanthate37 (discontinued), and risperidone microspheres.37-48 The research demonstrates the efficacy and safety of LAIA in FEP as measured by these endpoints:
• improved symptom control38,40-43,46,48
• adherence43,44,48
• reduced relapse rates37,43 and rehospitalizations37,47
• lesser reductions in white matter brain volume45
• no differences in extrapyramidal side effects or prolactin-associated adverse effects.48
A few small studies demonstrate significant differences in outcomes between risperidone LAIA and oral comparator groups (Table 3).43-45 Ongoing studies of LAIA use in FEP are comparing paliperidone palmitate with risperidone microspheres and other oral antipsychotics.49-51 No studies are examining olanzapine pamoate in FEP, likely because several guidelines do not recommended its use. No studies have been published regarding aripiprazole long-acting injectable in FEP. This LAIA formulation was approved in February 2013, and robust studies of the oral formulation in FEP are limited.52

Discussion and recommendations. Psychiatrists relying on subjective measures of antipsychotic adherence may inaccurately assess whether patients meet this criterion for LAIA use.53 LAIA could combat the high relapse rate in FEP, yet depot antipsychotics are prescribed infrequently for FEP patients (eg, for only 9.5% of participants in the RAISE-ETP study).20 Most schizophrenia treatment guidelines do not discuss LAIA use specifically in FEP, although the AFPBN expert consensus guidelines published in 2013 do recommend SGA depot formulations in FEP.12 SGA LAIA may be preferable, given its neuroprotective effects, in contrast to the neurotoxicity concerns of FGA LAIA.54,55
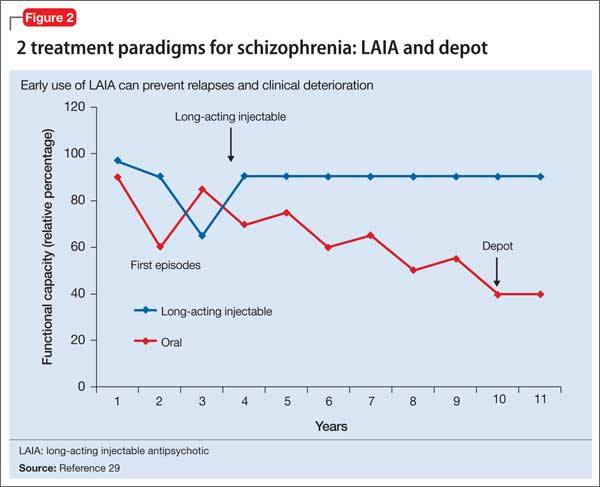
Relapses begin within a few months of illness stabilization after FEP, and >50% of patients relapse within 1 or 2 years2—the recommended minimum treatment duration for FEP.8,9,13 The use of LAIA is advisable in any patient with schizophrenia for whom long-term antipsychotic therapy is indicated.56 LAIA administration requirements objectively track medication adherence, which allows clinicians to be proactive in relapse prevention. Not using an intervention in FEP that improves adherence and decreases relapse rates contradicts our goal of instituting early, effective treatment to improve long-term functional outcomes (Figure 2).29
Considering clozapine in FEP
Guideline recommendations. Schizo-phrenia treatment guidelines and FDA labeling57 reserve clozapine for third-line treatment of refractory schizophrenia after 2 adequate antipsychotic trials have failed despite optimal dosing (Table 1).6-13 Some guidelines specify 1 of the 2 failed antipsychotic trials must include an SGA.6,7,10,11,13-16 Most say clozapine may be considered in patients with chronic aggression or hostility,7-9,14,16 or suicidal thoughts and behaviors.6-8,14,16 TMAP guidelines recommend a clozapine trial with concomitant substance abuse, persistent positive symptoms during 2 years of consistent medication treatment, and after 5 years of inadequate response (“treatment resistance”), regardless of the number of antipsychotic trials.7
Rationale and concerns. Clozapine is a superior choice for treatment-refractory delusions or hallucinations of schizophrenia, because it markedly enhances the response rate to antipsychotic therapy.58 Researchers therefore have investigated whether clozapine, compared with other antipsychotics, would yield more favorable initial and long-term outcomes when used first-line in FEP.
Preliminary evidence. Five studies have explored the use of clozapine as first-line therapy in FEP (Table 4).59-63 Interpreting the results is difficult because clozapine trials may be brief (mostly, 12 to 52 weeks); lack a comparator arm; suffer from a high attrition rate; enroll few patients; and lack potentially important outcome measures such as negative symptoms, suicidality, and functional assessment.
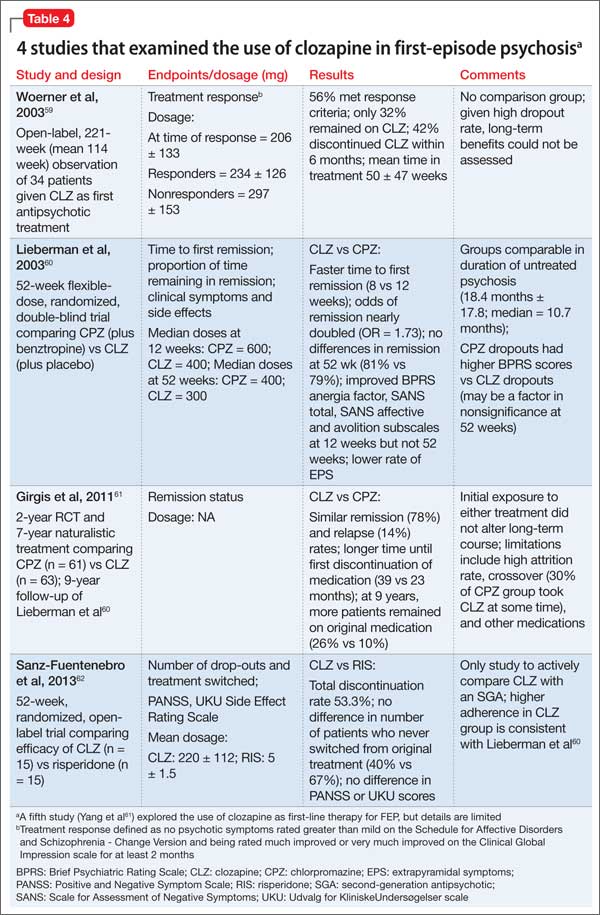
Overall, these studies demonstrate clozapine is as efficacious in this patient population as chlorpromazine (no difference in remission at 1-year, although clozapine-treated patients remitted faster and stayed in remission longer)60,61 or risperidone (no difference in Positive and Negative Syndrome Scale scores).62
At present, clozapine has not been shown superior to other antipsychotics as a first-line treatment for FEP. Research does underscore the importance of a clozapine trial as third-line treatment for FEP patients who have not responded well to 2 SGA trials.63 Many of these nonresponders (77%) have demonstrated a favorable response when promptly switched to clozapine.64
Discussion and recommendations. The limited evidence argues against using clozapine earlier than as third-line treatment in FEP. Perhaps the high treatment response that characterizes FEP creates a ceiling effect that obscures differences in antipsychotic efficacy at this stage.65 Clozapine use as first-line treatment should be re-evaluated with more robust methodology. One approach could be to assess its benefit in FEP by the duration of untreated psychosis.
The odds of achieving remission have been shown to decrease by 15% for each year that psychosis has not been treated.59 Studies exploring the use of clozapine as a second-line agent for FEP also are warranted, as antipsychotic response during subsequent trials is substantially reduced. In fact, the Scottish Intercollegiate Guidelines Network guidelines recommend this as an area for future research.11
For now, clozapine should continue to be reserved as second- or third-line treatment in a patient with FEP. The risks of clozapine’s potentially serious adverse effects (eg, agranulocytosis, seizures, obesity, diabetes, dyslipidemia, myocarditis, pancreatitis, hypotension, sialorrhea, severe sedation, ileus) can be justified only in the treatment of severe and persistent psychotic symptoms.57
Bottom Line
Nonstandard use of antipsychotic monotherapy dosages beyond the approved FDA limit and combination antipsychotic therapy may be reasonable for select first-episode psychosis (FEP) patients. Strongly consider long-acting injectable antipsychotics in FEP to proactively combat the high relapse rate and more easily identify antipsychotic failure. Continue to use clozapine as second- or third-line therapy in FEP: Studies have not found that it is more efficacious than other antipsychotics for first-line use.
Related Resource
• Recovery After an Initial Schizophrenia Episode (RAISE) Project Early Treatment Program. National Institute of Mental Health. http://raiseetp.org.
Drug Brand Names
Aripiprazole • Abilify, Abilify Maintena
Chlorpromazine • Thorazine
Clozapine • Clozaril
Fluphenazine decanoate • Prolixin-D
Haloperidol • Haldol
Haloperidol decanoate • Haldol-D
Olanzapine • Zyprexa
Olanzapine pamoate • Zyprexa Relprevv
Paliperidone palmitate • Invega Sustenna
Quetiapine • Seroquel
Risperidone • Risperdal
Risperidone microspheres • Risperdal Consta
Disclosures
Dr. Gardner reports no financial relationships with any companies whose products are mentioned in this article or with manufacturers of competing products. Dr. Nasrallah is a consultant to Acadia, Alkermes, Lundbeck, Janssen, Merck, Otsuka, and Sunovion, and is a speaker for Alkermes, Lundbeck, Janssen, Otsuka, and Sunovion.
1. Perkins DO, Gu H, Boteva K, et al. Relationship between duration of untreated psychosis and outcome in first-episode schizophrenia: a critical review and meta-analysis. Am J Psychiatry. 2005;162(10):1785-1804.
2. Bradford DW, Perkins DO, Lieberman JA. Pharmacological management of first-episode schizophrenia and related nonaffective psychoses. Drugs. 2003;63(21):2265-2283.
3. Lieberman JA, Koreen AR, Chakos M, et al. Factors influencing treatment response and outcome of first-episode schizophrenia: implications for understanding the pathophysiology of schizophrenia. J Clin Psychiatry. 1996;57(suppl 9):5-9.
4. Agid O, Arenovich T, Sajeev G, et al. An algorithm-based approach to first-episode schizophrenia: response rates over 3 prospective antipsychotic trials with a retrospective data analysis. J Clin Psychiatry. 2011;72(11):1439-1444.
5. Gardner KN, Nasrallah HA. Managing first-episode psychosis. An early stage of schizophrenia with distinct treatment needs. Current Psychiatry. 2015;14(5):32-34,36-40,42.
6. Lehman AF, Lieberman JA, Dixon LB, et al; American Psychiatric Association; Steering Committee on Practice Guidelines. Practice guideline for the treatment of patients with schizophrenia, second edition. Am J Psychiatry. 2004;161(suppl 2):1-56.
7. Texas Department of State Health Services. Texas Medication Algorithm Project (TMAP) Procedural Manual. Schizophrenia Treatment Algorithms. http://www.jpshealthnet.org/sites/default/files/ tmapalgorithmforschizophrenia.pdf. Updated April 2008. Accessed June 11, 2015.
8. Buchanan RW, Kreyenbuhl J, Kelly DL, et al; Schizophrenia Patient Outcomes Research Team (PORT). The 2009 schizophrenia PORT psychopharmacological treatment recommendations and summary statements. Schizophr Bull. 2010;36(1):71-93.
9. Barnes TR; Schizophrenia Consensus Group of British Association for Psychopharmacology. Evidence-based guidelines for the pharmacological treatment of schizophrenia: recommendations from the British Association for Psychopharmacology. J Psychopharmacol. 2011;25(5):567-620.
10. Hasan A, Falkai P, Wobrok T, et al; WFSBP Task force on Treatment Guidelines for Schizophrenia. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 2: update 2012 on the long-term treatment of schizophrenia and management of antipsychotic-induced side effects. World J Biol Psychiatry. 2013;14(1):2-44.
11. Scottish Intercollegiate Guidelines Network. SIGN 131: Management of schizophrenia. http://www.sign.ac.uk/ pdf/sign131.pdf. Published March 2013. Accessed June 11, 2015.
12. Llorca PM, Abbar M, Courtet P, et al. Guidelines for the use and management of long-acting injectable antipsychotics in serous mental illness. BMC Psychiatry. 2013;13:340.
13. National Institute for Health and Care Excellence. NICE clinical guideline 178: Psychosis and schizophrenia in adults: treatment and management. https://www.nice.org. uk/guidance/cg178/resources/guidance-psychosis-and-schizophrenia-in-adults-treatment-and-management-pdf. Updated March 2014. Accessed June 16, 2015.
14. Canadian Psychiatric Association. Clinical practice guidelines. Treatment of schizophrenia. Can J Psychiatry. 2005;50(13 suppl 1):7S-57S.
15. McEvoy JP, Scheifler PL, Frances A. The expert consensus guideline series: treatment of schizophrenia. J Clin Psychiatry. 1999;60(suppl 11):3-80.
16. Marder SR, Essock SM, Miller AL, et al. The Mount Sinai conference on the pharmacotherapy of schizophrenia. Schizophr Bull. 2002;28(1):5-16.
17. Kapur S, Zipursky R, Jones C, et al. Relationship between dopamine D(2) occupancy, clinical response, and side effects: a double-blind PET study of first-episode schizophrenia. Am J Psychiatry. 2000;157(4):514-520.
18. Fankhauser MP. Drug interactions with tobacco smoke: implications for patient care. Current Psychiatry. 2013;12(1):12-16.
19. Agid O, Schulze L, Arenovich T, et al. Antipsychotic response in first-episode schizophrenia: efficacy of high doses and switching. Eur Neuropsychopharmacol. 2013;23(9):1017-1022.
20. Robinson DG, Schooler NR, John M, et al. Prescription practices in the treatment of first-episode schizophrenia spectrum disorders: data from the national RAISE-ETP study. Am J Psychiatry. 2015;172(3):237-248.
21. Correll CU, Rummel-Kluge C, Corves C, et al. Antipsychotic combinations vs monotherapy in schizophrenia: a meta-analysis of randomized controlled trials. Schizophr Bull. 2009;35(2):443-457.
22. Fisher MD, Reilly K, Isenberg K, et al. Antipsychotic patterns of use in patients with schizophrenia: polypharmacy versus monotherapy. BMC Psychiatry. 2014;14(1):341.
23. Barnes TR, Paton C. Antipsychotic polypharmacy in schizophrenia: benefits and risks. CNS Drugs. 2011;25(5):383-399.
24. John AP, Dragovic M. Antipsychotic polypharmacy is not associated with reduced dose of individual antipsychotics in schizophrenia. J Clin Psychopharmacol. 2015;35(2):193-195.
25. Nasrallah HA. Treatment-resistant schizophrenia. Current Psychiatry. http://www.currentpsychiatry.com/specialty-focus/schizophrenia-other-psychotic-disorders/article/ treatment-resistant-schizophrenia/9be7bba3713d4a4cd68aa 8c92b79e5b1.html. Accessed June 16, 2015.
26. Alvarez-Jiménez M, Parker AG, Hetrick SE, et al. Preventing the second episode: a systematic review and meta-analysis of psychosocial and pharmacological trials in first-episode psychosis. Schizophr Bull. 2011;37(3):619-630.
27. Nasrallah HA, Smeltzer DJ. Contemporary diagnosis and management of the patient with schizophrenia. 2nd ed. Newton, PA: Handbooks in Health Care Co; 2011.
28. McEvoy JP. Risks versus benefits of different types of long-acting injectable antipsychotics. J Clin Psychiatry. 2006;67(suppl 5):15-18.
29. Agid O, Foussias G, Remington G. Long-acting injectable antipsychotics in the treatment of schizophrenia: their role in relapse prevention. Expert Opin Pharmacother. 2010;11(14):2301-2317.
30. Kirschner M, Theodoridou A, Fusar-Poli P, et al. Patients’ and clinicians’ attitude towards long-acting depot antipsychotics in subjects with a first episode psychosis. Ther Adv Psychophamacol. 2013;3(2):89-99.
31. Heres S, Hamann J, Mendel R, et al. Identifying the profile of optimal candidates for antipsychotic depot therapy: A cluster analysis. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(8):1987-1993.
32. Heres S, Lambert M, Vauth R. Treatment of early episode in patents with schizophrenia: the role of long acting antipsychotics. Eur Psychiatry. 2014;29(suppl 2):1409-1413.
33. Heres S, Schmitz FS, Leucht S, et al. The attitude of patients towards antipsychotic depot treatment. Int Clin Psychopharmacol. 2007;22(5):275-282.
34. Weiden PJ, Schooler NR, Weedon JC, et al. A randomized controlled trial of long-acting injectable risperidone vs continuation on oral atypical antipsychotics for first-episode schizophrenia patients: initial adherence outcome. J Clin Psychiatry. 2009;70(10):1397-1406.
35. Heres S, Reichhart T, Hamann J, et al. Psychiatrists’ attitude to antipsychotic depot treatment in patients with first-episode schizophrenia. Eur Psychiatry. 2011;26(5):297-301.
36. Kane JM, Rifkin A, Quitkin F, et al. Fluphenazine vs placebo in patients with remitted, acute first-episode schizophrenia. Arch Gen Psychiatry. 1982;39(1):70-73.
37. Tiihonen J, Wahlbeck K, Lönnqvist J, et al. Effectiveness of antipsychotic treatments in a nationwide cohort of patients in a community care after first hospitalization due to schizophrenia and schizoaffective disorder: observational follow-up study. BMJ. 2006;333(7561):224.
38. Parellada E, Andrezina R, Milanova V, et al. Patients in the early phases of schizophrenia and schizoaffective disorders effectively treated with risperidone long-acting injectable. J Psychopharmacol. 2005;19(suppl 5):5-14.
39. Malla A, Binder C, Chue P. Comparison of long-acting injectable risperidone and oral novel antipsychotic drugs for treatment in early phase of schizophrenia spectrum psychosis. Proceedings of the 61st Annual Convention Society of Biological Psychiatry; Toronto, Canada; 2006.
40. Lasser RA, Bossie CA, Zhu Y, et al. Long-acting risperidone in young adults with early schizophrenia or schizoaffective illness. Ann Clin Psychiatry. 2007;19(2):65-71.
41. Emsley R, Oosthuizen P, Koen L, et al. Remission in patients with first-episode schizophrenia receiving assured antipsychotic medication: a study with risperidone long-acting injection. Int Clin Psychopharmacol. 2008;23(6):325-331.
42. Emsley R, Oosthuizen P, Koen L, et al. Oral versus injectable antipsychotic treatment in early psychosis: post hoc comparison of two studies. Clin Ther. 2008;30(12):2378-2386.
43. Kim B, Lee SH, Choi TK, et al. Effectiveness of risperidone long-acting injection in first-episode schizophrenia: in naturalistic setting. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(5):1231-1235.
44. Weiden PJ, Schooler NJ, Weedon JC, et al. A randomized controlled trial of long-acting injectable risperidone vs continuation on oral atypical antipsychotics for first-episode schizophrenia patients: initial adherence outcome. J Clin Psychiatry. 2009;70(10):1397-1406.
45. Bartzokis G, Lu PH, Amar CP, et al. Long acting injection versus oral risperidone in first-episode schizophrenia: differential impact on white matter myelination trajectory. Schizophr Res. 2011;132(1):35-41.
46. Napryeyenko O, Burba B, Martinez G, et al. Risperidone long-acting injectable in recent-onset schizophrenia examined with clinician and patient self-report measures. J Clin Psychopharmacol. 2010;30(2):200-202.
47. Tiihonen J, Haukka J, Taylor M, et al. A nationwide cohort study of oral and depot antipsychotics after first hospitalization for schizophrenia. Am J Psychiatry. 2011;168(6):603-609.
48. Dubois V, Megens J, Mertens C, et al. Long-acting risperidone in early-episode schizophrenia. Acta Psychiatrica Belgica. 2011;111(1):9-21.
49. ClinicalTrials.gov. Oral risperidone versus injectable paliperidone palmitate for treating first-episode schizophrenia. https://clinicaltrials.gov/ct2/show/ NCT01451736. Accessed June 16, 2015.
50. ClinicalTrials.gov. Brain myelination effects of paliperidone palmitate versus oral risperidone in first episode schizophrenia. https://clinicaltrials.gov/ct2/ show/NCT01458379. Accessed June 16, 2015.
51. ClinicalTrials.gov. Effects of paliperidone palmitate versus oral antipsychotics on clinical outcomes and MRI measures. https://clinicaltrials.gov/ct2/show/NCT01359293. Accessed June 16, 2016.
52. U.S. Food and Drug Administration. Drugs@FDA. http:// www.accessdata.fda.gov/scripts/cder/drugsatfda. Accessed January 11, 2015.
53. Velligan DI, Weiden PJ, Sajatovic M, et al; Expert Consensus Panel on Adherence Problems in Serious and Persistent Mental Illness. The expert consensus guideline series: adherence problems in patients with serious and persistent mental illness. J Clin Psychiatry. 2009;70(suppl 4):1-46; quiz 47-48.
54. Nandra KS, Agius M. The difference between typical and atypical antipsychotics: the effects on neurogenesis. Psychiatr Danub. 2012;24(suppl 1):S95-S99.
55. Nasrallah HA. Haloperidol is clearly neurotoxic. Should it be banned? Current Psychiatry. 2013;12(7):7-8.
56. Kane JM, Garcia-Ribora C. Clinical guideline recommendations for antipsychotic long-acting injections. Br J Psychiatry. 2009;52:S63-S67.
57. Clozaril [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2014.
58. Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789-796.
59. Woerner MG, Robinson DG, Alvir JMJ, et al. Clozapine as a first treatment for schizophrenia. Am J Psychiatry. 2003;160(8):1514-1516.
60. Lieberman JA, Phillips M, Gu H, et al. Atypical and conventional antipsychotic drugs in treatment-naive first-episode schizophrenia: a 52-week randomized trial of clozapine vs chlorpromazine. Neuropsychopharmacology. 2003;28(5):995-1003.
61. Girgis RR, Phillips MR, Li X, et al. Clozapine v. chlorpromazine in treatment-naive, first-episode schizophrenia: 9-year outcomes of a randomised clinical trial. Br J Psychiatry. 2011;199(4):281-288.
62. Sanz-Fuentenebro J, Taboada D, Palomo T, et al. Randomized trial of clozapine vs. risperidone in treatment-naïve first-episode schizophrenia: results after one year. Schizophr Res. 2013;149(1-3):156-161.
63. Yang PD, Ji Z. The efficacy and related factors of clozapine on first-episode schizophrenia. Chin J Nerv Ment Dis. 1997;23:155-158.
64. Agid O, Schulze L, Arenovich T, et al. Antipsychotic response in first-episode schizophrenia: efficacy of high doses and switching. Eur Neuropsychopharmacol. 2013;23(9):1017-1022.
65. Remington G, Agid O, Foussias G, et al. Clozapine’s role in the treatment of first-episode schizophrenia. Am J Psychiatry. 2013;170(2):146-151.
First-episode psychosis (FEP) in schizophrenia is characterized by high response rates to antipsychotic therapy, followed by frequent antipsychotic discontinuation and elevated relapse rates soon after maintenance treatment begins.1,2 With subsequent episodes, time to response progressively increases and likelihood of response decreases.3,4
To address these issues, this article—the second of 2 parts5—describes the rationale and evidence for using nonstandard first-line antipsychotic therapies to manage FEP. Specifically, we discuss when clinicians might consider monotherapy exceeding FDA-approved maximum dosages, combination therapy, long-acting injectable antipsychotics (LAIA), or clozapine.
Monotherapy beyond FDA-approved dosages
Treatment guidelines for FEP recommend oral antipsychotic dosages in the lower half of the treatment range and lower than those that are required for multi-episode schizophrenia.6-16 Ultimately, clinicians prescribe individualized dosages for their patients based on symptom improvement and tolerability. The optimal dosage at which to achieve a favorable D2 receptor occupancy likely will vary from patient to patient.17
To control symptoms, higher dosages may be needed than those used in FEP clinical trials, recommended by guidelines for FEP or multi-episode patients, or approved by the FDA. Patients seen in everyday practice may be more complicated (eg, have a comorbid condition or history of nonresponse) than study populations. Higher dosages also may be reasonable to overcome drug−drug interactions (eg, cigarette smoking-mediated cytochrome P450 1A2 induction, resulting in increased olanzapine metabolism),18 or to establish antipsychotic failure if adequate trials at lower dosages have resulted in a suboptimal response and the patient is not experiencing tolerability or safety concerns.
In a study of low-, full-, and high-dosage antipsychotic therapy in FEP, an additional 15% of patients responded to higher dosages of olanzapine and risperidone after failing to respond to a standard dosage.19 A study of data from the Recovery After an Initial Schizophrenia Episode Project’s Early Treatment Program (RAISE-ETP) found that, of participants identified who may benefit from therapy modification, 8.8% were prescribed an antipsychotic (often, olanzapine, risperidone, and haloperidol) at a higher-than-recommended dosage.20 Of note, only olanzapine was prescribed at higher than FDA-approved dosages.
Antipsychotic combination therapy
Prescribing combinations of antipsychotics—antipsychotic polypharmacy (APP)— has a negative connotation because of limited efficacy and safety data,21 and limited endorsement in schizophrenia treatment guidelines.9,13 Caution with APP is warranted; a complex medication regimen may increase the potential for adverse effects, poorer adherence, and adverse drug-drug interactions.9 APP has been shown to independently predict both shorter treatment duration and discontinuation before 1 year.22
Nonetheless, the clinician and patient may share the decision to implement APP and observe whether benefits outweigh risks in situations such as:
• to optimize neuroreceptor occupancy and targets (eg, attempting to achieve adequate D2 receptor blockade while minimizing side effects secondary to binding other receptors)
• to manage co-existing symptom domains (eg, mood changes, aggression, negative symptoms, disorganization, and cognitive deficits)
• to mitigate antipsychotic-induced side effects (eg, initiating aripiprazole to treat hyperprolactinemia induced by another antipsychotic to which the patient has achieved a favorable response).23
Clinicians report using APP to treat as many as 50% of patients with a history of multiple psychotic episodes.23 For FEP patients, 23% of participants in the RAISE-ETP trial who were identified as possibly benefiting from therapy modification were prescribed APP.20 Regrettably, researchers have not found evidence to support a reported rationale for using APP—that lower dosages of individual antipsychotics when used in combination may avoid high-dosage prescriptions.24
Before implementing APP, thoroughly explore and manage reasons for a patient’s suboptimal response to monotherapy.25 An adequate trial with any antipsychotic should be at the highest tolerated dosage for 12 to 16 weeks. Be mindful that response to an APP trial may be the result of additional time on the original antipsychotic.
Long-acting injectable antipsychotics in FEP
Guideline recommendations. Most older guidelines for schizophrenia treatment suggest LAIA after multiple relapses related to medication nonadherence or when a patient prefers injected medication (Table 1).6-13 Expert consensus guidelines also recommend considering LAIA in patients who lack insight into their illness. The Texas Medication Algorithm Project (TMAP) guidelines7 state LAIA can be considered for inadequate adherence at any stage, whereas the 2010 British Association for Psychopharmacology (BAP) guidelines9 express uncertainty about their use in FEP, because of limited evidence. Both the BAP and National Institute for Health and Care Excellence guidelines13 urge clinicians to consider LAIA when avoiding nonadherence is a treatment priority.
Recently, the French Association for Biological Psychiatry and Neuro-psychopharmacology (AFPBN) created expert consensus guidelines12 on using LAIA in practice. They recommend long-acting injectable second-generation antipsychotics (SGAs) as first-line maintenance treatment for schizophrenia and schizoaffective disorder and for individuals experiencing a first recurrent episode. The World Federation of Societies of Biological Psychiatry guidelines contain LAIA dosage recommendations for FEP (Table 2).10
Advances have been made in understanding the serious neurobiological adverse effects of psychotic relapses, including neuroinflammation and oxidative stress, that may explain the atrophic changes observed with psychotic episodes starting with the FEP. Protecting the patient from a second episode has become a vital therapeutic management goal26 (Figure 127).
Concerns. Compared with oral antipsychotics, LAIA offers clinical advantages:
• improved pharmacokinetic profile (lower “peaks” and higher “valleys”)
• more consistent plasma concentrations (no variability related to administration timing or food effects)
• no first-pass metabolism, which can ease the process of finding the lowest effective and safe dosage
• reduced administration burden and objective tracking of adherence with typical dosing every 2 to 4 weeks
• less stigmatizing than oral medication for FEP patients, such as college students living in a dormitory.28,29
Barriers to LAIA use include:
• slow dosage titration and increased time to reach steady state drug level
• oral supplementation for some (eg, risperidone microspheres and aripiprazole long-acting injectable)
• logistical challenges for some (eg, 3-hour post-injection monitoring for delirium sedation syndrome with olanzapine pamoate)
• additional planning to coordinate care for scheduled injections
• higher expenses up front
• local injection site reactions
• dosage adjustment difficulties if adverse effects occur.28,29
Adoption rates of LAIA are low, especially for FEP.30 Most surveys indicate that (1) physicians believe LAIA treatment is ineffective for FEP31 and (2) patients do not prefer injectable to oral antipsychotics,32 despite evidence to the contrary.33,34 A survey of 198 psychiatrists identified 3 factors that influenced their decisions against using LAIA patients with FEP:
• limited availability of SGA depot formulations (4, to date, in the United States)
• frequent rejection by the patient when LAIA is offered without adequate explanation or encouragement
• skepticism of FEP patients (and their family) who lack experience with relapse.35
In reality, when SGA depots were introduced in the United Kingdom, prescribing rates of LAIA did not increase. As for patient rejection being a major reason for not prescribing LAIA, few patients (5% to 36%) are offered depot injections, particularly in FEP.29 Most patients using LAIA are chronic, multi-episode, violent people who are receiving medications involuntarily.29 Interestingly, this survey did not find 2 factors to be influential in psychiatrists’ decision not to use LAIA in FEP:
• guidelines do not explicitly recommend depot treatment in FEP
• treatment in FEP may be limited to 1 year, therefore depot administration is not worthwhile.35
Preliminary evidence. At least a dozen studies have explored LAIA treatment for FEP, with the use of fluphenazine decanoate,36 perphenazine enanthate37 (discontinued), and risperidone microspheres.37-48 The research demonstrates the efficacy and safety of LAIA in FEP as measured by these endpoints:
• improved symptom control38,40-43,46,48
• adherence43,44,48
• reduced relapse rates37,43 and rehospitalizations37,47
• lesser reductions in white matter brain volume45
• no differences in extrapyramidal side effects or prolactin-associated adverse effects.48
A few small studies demonstrate significant differences in outcomes between risperidone LAIA and oral comparator groups (Table 3).43-45 Ongoing studies of LAIA use in FEP are comparing paliperidone palmitate with risperidone microspheres and other oral antipsychotics.49-51 No studies are examining olanzapine pamoate in FEP, likely because several guidelines do not recommended its use. No studies have been published regarding aripiprazole long-acting injectable in FEP. This LAIA formulation was approved in February 2013, and robust studies of the oral formulation in FEP are limited.52
Discussion and recommendations. Psychiatrists relying on subjective measures of antipsychotic adherence may inaccurately assess whether patients meet this criterion for LAIA use.53 LAIA could combat the high relapse rate in FEP, yet depot antipsychotics are prescribed infrequently for FEP patients (eg, for only 9.5% of participants in the RAISE-ETP study).20 Most schizophrenia treatment guidelines do not discuss LAIA use specifically in FEP, although the AFPBN expert consensus guidelines published in 2013 do recommend SGA depot formulations in FEP.12 SGA LAIA may be preferable, given its neuroprotective effects, in contrast to the neurotoxicity concerns of FGA LAIA.54,55
Relapses begin within a few months of illness stabilization after FEP, and >50% of patients relapse within 1 or 2 years2—the recommended minimum treatment duration for FEP.8,9,13 The use of LAIA is advisable in any patient with schizophrenia for whom long-term antipsychotic therapy is indicated.56 LAIA administration requirements objectively track medication adherence, which allows clinicians to be proactive in relapse prevention. Not using an intervention in FEP that improves adherence and decreases relapse rates contradicts our goal of instituting early, effective treatment to improve long-term functional outcomes (Figure 2).29
Considering clozapine in FEP
Guideline recommendations. Schizo-phrenia treatment guidelines and FDA labeling57 reserve clozapine for third-line treatment of refractory schizophrenia after 2 adequate antipsychotic trials have failed despite optimal dosing (Table 1).6-13 Some guidelines specify 1 of the 2 failed antipsychotic trials must include an SGA.6,7,10,11,13-16 Most say clozapine may be considered in patients with chronic aggression or hostility,7-9,14,16 or suicidal thoughts and behaviors.6-8,14,16 TMAP guidelines recommend a clozapine trial with concomitant substance abuse, persistent positive symptoms during 2 years of consistent medication treatment, and after 5 years of inadequate response (“treatment resistance”), regardless of the number of antipsychotic trials.7
Rationale and concerns. Clozapine is a superior choice for treatment-refractory delusions or hallucinations of schizophrenia, because it markedly enhances the response rate to antipsychotic therapy.58 Researchers therefore have investigated whether clozapine, compared with other antipsychotics, would yield more favorable initial and long-term outcomes when used first-line in FEP.
Preliminary evidence. Five studies have explored the use of clozapine as first-line therapy in FEP (Table 4).59-63 Interpreting the results is difficult because clozapine trials may be brief (mostly, 12 to 52 weeks); lack a comparator arm; suffer from a high attrition rate; enroll few patients; and lack potentially important outcome measures such as negative symptoms, suicidality, and functional assessment.
Overall, these studies demonstrate clozapine is as efficacious in this patient population as chlorpromazine (no difference in remission at 1-year, although clozapine-treated patients remitted faster and stayed in remission longer)60,61 or risperidone (no difference in Positive and Negative Syndrome Scale scores).62
At present, clozapine has not been shown superior to other antipsychotics as a first-line treatment for FEP. Research does underscore the importance of a clozapine trial as third-line treatment for FEP patients who have not responded well to 2 SGA trials.63 Many of these nonresponders (77%) have demonstrated a favorable response when promptly switched to clozapine.64
Discussion and recommendations. The limited evidence argues against using clozapine earlier than as third-line treatment in FEP. Perhaps the high treatment response that characterizes FEP creates a ceiling effect that obscures differences in antipsychotic efficacy at this stage.65 Clozapine use as first-line treatment should be re-evaluated with more robust methodology. One approach could be to assess its benefit in FEP by the duration of untreated psychosis.
The odds of achieving remission have been shown to decrease by 15% for each year that psychosis has not been treated.59 Studies exploring the use of clozapine as a second-line agent for FEP also are warranted, as antipsychotic response during subsequent trials is substantially reduced. In fact, the Scottish Intercollegiate Guidelines Network guidelines recommend this as an area for future research.11
For now, clozapine should continue to be reserved as second- or third-line treatment in a patient with FEP. The risks of clozapine’s potentially serious adverse effects (eg, agranulocytosis, seizures, obesity, diabetes, dyslipidemia, myocarditis, pancreatitis, hypotension, sialorrhea, severe sedation, ileus) can be justified only in the treatment of severe and persistent psychotic symptoms.57
Bottom Line
Nonstandard use of antipsychotic monotherapy dosages beyond the approved FDA limit and combination antipsychotic therapy may be reasonable for select first-episode psychosis (FEP) patients. Strongly consider long-acting injectable antipsychotics in FEP to proactively combat the high relapse rate and more easily identify antipsychotic failure. Continue to use clozapine as second- or third-line therapy in FEP: Studies have not found that it is more efficacious than other antipsychotics for first-line use.
Related Resource
• Recovery After an Initial Schizophrenia Episode (RAISE) Project Early Treatment Program. National Institute of Mental Health. http://raiseetp.org.
Drug Brand Names
Aripiprazole • Abilify, Abilify Maintena
Chlorpromazine • Thorazine
Clozapine • Clozaril
Fluphenazine decanoate • Prolixin-D
Haloperidol • Haldol
Haloperidol decanoate • Haldol-D
Olanzapine • Zyprexa
Olanzapine pamoate • Zyprexa Relprevv
Paliperidone palmitate • Invega Sustenna
Quetiapine • Seroquel
Risperidone • Risperdal
Risperidone microspheres • Risperdal Consta
Disclosures
Dr. Gardner reports no financial relationships with any companies whose products are mentioned in this article or with manufacturers of competing products. Dr. Nasrallah is a consultant to Acadia, Alkermes, Lundbeck, Janssen, Merck, Otsuka, and Sunovion, and is a speaker for Alkermes, Lundbeck, Janssen, Otsuka, and Sunovion.
First-episode psychosis (FEP) in schizophrenia is characterized by high response rates to antipsychotic therapy, followed by frequent antipsychotic discontinuation and elevated relapse rates soon after maintenance treatment begins.1,2 With subsequent episodes, time to response progressively increases and likelihood of response decreases.3,4
To address these issues, this article—the second of 2 parts5—describes the rationale and evidence for using nonstandard first-line antipsychotic therapies to manage FEP. Specifically, we discuss when clinicians might consider monotherapy exceeding FDA-approved maximum dosages, combination therapy, long-acting injectable antipsychotics (LAIA), or clozapine.
Monotherapy beyond FDA-approved dosages
Treatment guidelines for FEP recommend oral antipsychotic dosages in the lower half of the treatment range and lower than those that are required for multi-episode schizophrenia.6-16 Ultimately, clinicians prescribe individualized dosages for their patients based on symptom improvement and tolerability. The optimal dosage at which to achieve a favorable D2 receptor occupancy likely will vary from patient to patient.17
To control symptoms, higher dosages may be needed than those used in FEP clinical trials, recommended by guidelines for FEP or multi-episode patients, or approved by the FDA. Patients seen in everyday practice may be more complicated (eg, have a comorbid condition or history of nonresponse) than study populations. Higher dosages also may be reasonable to overcome drug−drug interactions (eg, cigarette smoking-mediated cytochrome P450 1A2 induction, resulting in increased olanzapine metabolism),18 or to establish antipsychotic failure if adequate trials at lower dosages have resulted in a suboptimal response and the patient is not experiencing tolerability or safety concerns.
In a study of low-, full-, and high-dosage antipsychotic therapy in FEP, an additional 15% of patients responded to higher dosages of olanzapine and risperidone after failing to respond to a standard dosage.19 A study of data from the Recovery After an Initial Schizophrenia Episode Project’s Early Treatment Program (RAISE-ETP) found that, of participants identified who may benefit from therapy modification, 8.8% were prescribed an antipsychotic (often, olanzapine, risperidone, and haloperidol) at a higher-than-recommended dosage.20 Of note, only olanzapine was prescribed at higher than FDA-approved dosages.
Antipsychotic combination therapy
Prescribing combinations of antipsychotics—antipsychotic polypharmacy (APP)— has a negative connotation because of limited efficacy and safety data,21 and limited endorsement in schizophrenia treatment guidelines.9,13 Caution with APP is warranted; a complex medication regimen may increase the potential for adverse effects, poorer adherence, and adverse drug-drug interactions.9 APP has been shown to independently predict both shorter treatment duration and discontinuation before 1 year.22
Nonetheless, the clinician and patient may share the decision to implement APP and observe whether benefits outweigh risks in situations such as:
• to optimize neuroreceptor occupancy and targets (eg, attempting to achieve adequate D2 receptor blockade while minimizing side effects secondary to binding other receptors)
• to manage co-existing symptom domains (eg, mood changes, aggression, negative symptoms, disorganization, and cognitive deficits)
• to mitigate antipsychotic-induced side effects (eg, initiating aripiprazole to treat hyperprolactinemia induced by another antipsychotic to which the patient has achieved a favorable response).23
Clinicians report using APP to treat as many as 50% of patients with a history of multiple psychotic episodes.23 For FEP patients, 23% of participants in the RAISE-ETP trial who were identified as possibly benefiting from therapy modification were prescribed APP.20 Regrettably, researchers have not found evidence to support a reported rationale for using APP—that lower dosages of individual antipsychotics when used in combination may avoid high-dosage prescriptions.24
Before implementing APP, thoroughly explore and manage reasons for a patient’s suboptimal response to monotherapy.25 An adequate trial with any antipsychotic should be at the highest tolerated dosage for 12 to 16 weeks. Be mindful that response to an APP trial may be the result of additional time on the original antipsychotic.
Long-acting injectable antipsychotics in FEP
Guideline recommendations. Most older guidelines for schizophrenia treatment suggest LAIA after multiple relapses related to medication nonadherence or when a patient prefers injected medication (Table 1).6-13 Expert consensus guidelines also recommend considering LAIA in patients who lack insight into their illness. The Texas Medication Algorithm Project (TMAP) guidelines7 state LAIA can be considered for inadequate adherence at any stage, whereas the 2010 British Association for Psychopharmacology (BAP) guidelines9 express uncertainty about their use in FEP, because of limited evidence. Both the BAP and National Institute for Health and Care Excellence guidelines13 urge clinicians to consider LAIA when avoiding nonadherence is a treatment priority.
Recently, the French Association for Biological Psychiatry and Neuro-psychopharmacology (AFPBN) created expert consensus guidelines12 on using LAIA in practice. They recommend long-acting injectable second-generation antipsychotics (SGAs) as first-line maintenance treatment for schizophrenia and schizoaffective disorder and for individuals experiencing a first recurrent episode. The World Federation of Societies of Biological Psychiatry guidelines contain LAIA dosage recommendations for FEP (Table 2).10
Advances have been made in understanding the serious neurobiological adverse effects of psychotic relapses, including neuroinflammation and oxidative stress, that may explain the atrophic changes observed with psychotic episodes starting with the FEP. Protecting the patient from a second episode has become a vital therapeutic management goal26 (Figure 127).
Concerns. Compared with oral antipsychotics, LAIA offers clinical advantages:
• improved pharmacokinetic profile (lower “peaks” and higher “valleys”)
• more consistent plasma concentrations (no variability related to administration timing or food effects)
• no first-pass metabolism, which can ease the process of finding the lowest effective and safe dosage
• reduced administration burden and objective tracking of adherence with typical dosing every 2 to 4 weeks
• less stigmatizing than oral medication for FEP patients, such as college students living in a dormitory.28,29
Barriers to LAIA use include:
• slow dosage titration and increased time to reach steady state drug level
• oral supplementation for some (eg, risperidone microspheres and aripiprazole long-acting injectable)
• logistical challenges for some (eg, 3-hour post-injection monitoring for delirium sedation syndrome with olanzapine pamoate)
• additional planning to coordinate care for scheduled injections
• higher expenses up front
• local injection site reactions
• dosage adjustment difficulties if adverse effects occur.28,29
Adoption rates of LAIA are low, especially for FEP.30 Most surveys indicate that (1) physicians believe LAIA treatment is ineffective for FEP31 and (2) patients do not prefer injectable to oral antipsychotics,32 despite evidence to the contrary.33,34 A survey of 198 psychiatrists identified 3 factors that influenced their decisions against using LAIA patients with FEP:
• limited availability of SGA depot formulations (4, to date, in the United States)
• frequent rejection by the patient when LAIA is offered without adequate explanation or encouragement
• skepticism of FEP patients (and their family) who lack experience with relapse.35
In reality, when SGA depots were introduced in the United Kingdom, prescribing rates of LAIA did not increase. As for patient rejection being a major reason for not prescribing LAIA, few patients (5% to 36%) are offered depot injections, particularly in FEP.29 Most patients using LAIA are chronic, multi-episode, violent people who are receiving medications involuntarily.29 Interestingly, this survey did not find 2 factors to be influential in psychiatrists’ decision not to use LAIA in FEP:
• guidelines do not explicitly recommend depot treatment in FEP
• treatment in FEP may be limited to 1 year, therefore depot administration is not worthwhile.35
Preliminary evidence. At least a dozen studies have explored LAIA treatment for FEP, with the use of fluphenazine decanoate,36 perphenazine enanthate37 (discontinued), and risperidone microspheres.37-48 The research demonstrates the efficacy and safety of LAIA in FEP as measured by these endpoints:
• improved symptom control38,40-43,46,48
• adherence43,44,48
• reduced relapse rates37,43 and rehospitalizations37,47
• lesser reductions in white matter brain volume45
• no differences in extrapyramidal side effects or prolactin-associated adverse effects.48
A few small studies demonstrate significant differences in outcomes between risperidone LAIA and oral comparator groups (Table 3).43-45 Ongoing studies of LAIA use in FEP are comparing paliperidone palmitate with risperidone microspheres and other oral antipsychotics.49-51 No studies are examining olanzapine pamoate in FEP, likely because several guidelines do not recommended its use. No studies have been published regarding aripiprazole long-acting injectable in FEP. This LAIA formulation was approved in February 2013, and robust studies of the oral formulation in FEP are limited.52
Discussion and recommendations. Psychiatrists relying on subjective measures of antipsychotic adherence may inaccurately assess whether patients meet this criterion for LAIA use.53 LAIA could combat the high relapse rate in FEP, yet depot antipsychotics are prescribed infrequently for FEP patients (eg, for only 9.5% of participants in the RAISE-ETP study).20 Most schizophrenia treatment guidelines do not discuss LAIA use specifically in FEP, although the AFPBN expert consensus guidelines published in 2013 do recommend SGA depot formulations in FEP.12 SGA LAIA may be preferable, given its neuroprotective effects, in contrast to the neurotoxicity concerns of FGA LAIA.54,55
Relapses begin within a few months of illness stabilization after FEP, and >50% of patients relapse within 1 or 2 years2—the recommended minimum treatment duration for FEP.8,9,13 The use of LAIA is advisable in any patient with schizophrenia for whom long-term antipsychotic therapy is indicated.56 LAIA administration requirements objectively track medication adherence, which allows clinicians to be proactive in relapse prevention. Not using an intervention in FEP that improves adherence and decreases relapse rates contradicts our goal of instituting early, effective treatment to improve long-term functional outcomes (Figure 2).29
Considering clozapine in FEP
Guideline recommendations. Schizo-phrenia treatment guidelines and FDA labeling57 reserve clozapine for third-line treatment of refractory schizophrenia after 2 adequate antipsychotic trials have failed despite optimal dosing (Table 1).6-13 Some guidelines specify 1 of the 2 failed antipsychotic trials must include an SGA.6,7,10,11,13-16 Most say clozapine may be considered in patients with chronic aggression or hostility,7-9,14,16 or suicidal thoughts and behaviors.6-8,14,16 TMAP guidelines recommend a clozapine trial with concomitant substance abuse, persistent positive symptoms during 2 years of consistent medication treatment, and after 5 years of inadequate response (“treatment resistance”), regardless of the number of antipsychotic trials.7
Rationale and concerns. Clozapine is a superior choice for treatment-refractory delusions or hallucinations of schizophrenia, because it markedly enhances the response rate to antipsychotic therapy.58 Researchers therefore have investigated whether clozapine, compared with other antipsychotics, would yield more favorable initial and long-term outcomes when used first-line in FEP.
Preliminary evidence. Five studies have explored the use of clozapine as first-line therapy in FEP (Table 4).59-63 Interpreting the results is difficult because clozapine trials may be brief (mostly, 12 to 52 weeks); lack a comparator arm; suffer from a high attrition rate; enroll few patients; and lack potentially important outcome measures such as negative symptoms, suicidality, and functional assessment.
Overall, these studies demonstrate clozapine is as efficacious in this patient population as chlorpromazine (no difference in remission at 1-year, although clozapine-treated patients remitted faster and stayed in remission longer)60,61 or risperidone (no difference in Positive and Negative Syndrome Scale scores).62
At present, clozapine has not been shown superior to other antipsychotics as a first-line treatment for FEP. Research does underscore the importance of a clozapine trial as third-line treatment for FEP patients who have not responded well to 2 SGA trials.63 Many of these nonresponders (77%) have demonstrated a favorable response when promptly switched to clozapine.64
Discussion and recommendations. The limited evidence argues against using clozapine earlier than as third-line treatment in FEP. Perhaps the high treatment response that characterizes FEP creates a ceiling effect that obscures differences in antipsychotic efficacy at this stage.65 Clozapine use as first-line treatment should be re-evaluated with more robust methodology. One approach could be to assess its benefit in FEP by the duration of untreated psychosis.
The odds of achieving remission have been shown to decrease by 15% for each year that psychosis has not been treated.59 Studies exploring the use of clozapine as a second-line agent for FEP also are warranted, as antipsychotic response during subsequent trials is substantially reduced. In fact, the Scottish Intercollegiate Guidelines Network guidelines recommend this as an area for future research.11
For now, clozapine should continue to be reserved as second- or third-line treatment in a patient with FEP. The risks of clozapine’s potentially serious adverse effects (eg, agranulocytosis, seizures, obesity, diabetes, dyslipidemia, myocarditis, pancreatitis, hypotension, sialorrhea, severe sedation, ileus) can be justified only in the treatment of severe and persistent psychotic symptoms.57
Bottom Line
Nonstandard use of antipsychotic monotherapy dosages beyond the approved FDA limit and combination antipsychotic therapy may be reasonable for select first-episode psychosis (FEP) patients. Strongly consider long-acting injectable antipsychotics in FEP to proactively combat the high relapse rate and more easily identify antipsychotic failure. Continue to use clozapine as second- or third-line therapy in FEP: Studies have not found that it is more efficacious than other antipsychotics for first-line use.
Related Resource
• Recovery After an Initial Schizophrenia Episode (RAISE) Project Early Treatment Program. National Institute of Mental Health. http://raiseetp.org.
Drug Brand Names
Aripiprazole • Abilify, Abilify Maintena
Chlorpromazine • Thorazine
Clozapine • Clozaril
Fluphenazine decanoate • Prolixin-D
Haloperidol • Haldol
Haloperidol decanoate • Haldol-D
Olanzapine • Zyprexa
Olanzapine pamoate • Zyprexa Relprevv
Paliperidone palmitate • Invega Sustenna
Quetiapine • Seroquel
Risperidone • Risperdal
Risperidone microspheres • Risperdal Consta
Disclosures
Dr. Gardner reports no financial relationships with any companies whose products are mentioned in this article or with manufacturers of competing products. Dr. Nasrallah is a consultant to Acadia, Alkermes, Lundbeck, Janssen, Merck, Otsuka, and Sunovion, and is a speaker for Alkermes, Lundbeck, Janssen, Otsuka, and Sunovion.
1. Perkins DO, Gu H, Boteva K, et al. Relationship between duration of untreated psychosis and outcome in first-episode schizophrenia: a critical review and meta-analysis. Am J Psychiatry. 2005;162(10):1785-1804.
2. Bradford DW, Perkins DO, Lieberman JA. Pharmacological management of first-episode schizophrenia and related nonaffective psychoses. Drugs. 2003;63(21):2265-2283.
3. Lieberman JA, Koreen AR, Chakos M, et al. Factors influencing treatment response and outcome of first-episode schizophrenia: implications for understanding the pathophysiology of schizophrenia. J Clin Psychiatry. 1996;57(suppl 9):5-9.
4. Agid O, Arenovich T, Sajeev G, et al. An algorithm-based approach to first-episode schizophrenia: response rates over 3 prospective antipsychotic trials with a retrospective data analysis. J Clin Psychiatry. 2011;72(11):1439-1444.
5. Gardner KN, Nasrallah HA. Managing first-episode psychosis. An early stage of schizophrenia with distinct treatment needs. Current Psychiatry. 2015;14(5):32-34,36-40,42.
6. Lehman AF, Lieberman JA, Dixon LB, et al; American Psychiatric Association; Steering Committee on Practice Guidelines. Practice guideline for the treatment of patients with schizophrenia, second edition. Am J Psychiatry. 2004;161(suppl 2):1-56.
7. Texas Department of State Health Services. Texas Medication Algorithm Project (TMAP) Procedural Manual. Schizophrenia Treatment Algorithms. http://www.jpshealthnet.org/sites/default/files/ tmapalgorithmforschizophrenia.pdf. Updated April 2008. Accessed June 11, 2015.
8. Buchanan RW, Kreyenbuhl J, Kelly DL, et al; Schizophrenia Patient Outcomes Research Team (PORT). The 2009 schizophrenia PORT psychopharmacological treatment recommendations and summary statements. Schizophr Bull. 2010;36(1):71-93.
9. Barnes TR; Schizophrenia Consensus Group of British Association for Psychopharmacology. Evidence-based guidelines for the pharmacological treatment of schizophrenia: recommendations from the British Association for Psychopharmacology. J Psychopharmacol. 2011;25(5):567-620.
10. Hasan A, Falkai P, Wobrok T, et al; WFSBP Task force on Treatment Guidelines for Schizophrenia. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 2: update 2012 on the long-term treatment of schizophrenia and management of antipsychotic-induced side effects. World J Biol Psychiatry. 2013;14(1):2-44.
11. Scottish Intercollegiate Guidelines Network. SIGN 131: Management of schizophrenia. http://www.sign.ac.uk/ pdf/sign131.pdf. Published March 2013. Accessed June 11, 2015.
12. Llorca PM, Abbar M, Courtet P, et al. Guidelines for the use and management of long-acting injectable antipsychotics in serous mental illness. BMC Psychiatry. 2013;13:340.
13. National Institute for Health and Care Excellence. NICE clinical guideline 178: Psychosis and schizophrenia in adults: treatment and management. https://www.nice.org. uk/guidance/cg178/resources/guidance-psychosis-and-schizophrenia-in-adults-treatment-and-management-pdf. Updated March 2014. Accessed June 16, 2015.
14. Canadian Psychiatric Association. Clinical practice guidelines. Treatment of schizophrenia. Can J Psychiatry. 2005;50(13 suppl 1):7S-57S.
15. McEvoy JP, Scheifler PL, Frances A. The expert consensus guideline series: treatment of schizophrenia. J Clin Psychiatry. 1999;60(suppl 11):3-80.
16. Marder SR, Essock SM, Miller AL, et al. The Mount Sinai conference on the pharmacotherapy of schizophrenia. Schizophr Bull. 2002;28(1):5-16.
17. Kapur S, Zipursky R, Jones C, et al. Relationship between dopamine D(2) occupancy, clinical response, and side effects: a double-blind PET study of first-episode schizophrenia. Am J Psychiatry. 2000;157(4):514-520.
18. Fankhauser MP. Drug interactions with tobacco smoke: implications for patient care. Current Psychiatry. 2013;12(1):12-16.
19. Agid O, Schulze L, Arenovich T, et al. Antipsychotic response in first-episode schizophrenia: efficacy of high doses and switching. Eur Neuropsychopharmacol. 2013;23(9):1017-1022.
20. Robinson DG, Schooler NR, John M, et al. Prescription practices in the treatment of first-episode schizophrenia spectrum disorders: data from the national RAISE-ETP study. Am J Psychiatry. 2015;172(3):237-248.
21. Correll CU, Rummel-Kluge C, Corves C, et al. Antipsychotic combinations vs monotherapy in schizophrenia: a meta-analysis of randomized controlled trials. Schizophr Bull. 2009;35(2):443-457.
22. Fisher MD, Reilly K, Isenberg K, et al. Antipsychotic patterns of use in patients with schizophrenia: polypharmacy versus monotherapy. BMC Psychiatry. 2014;14(1):341.
23. Barnes TR, Paton C. Antipsychotic polypharmacy in schizophrenia: benefits and risks. CNS Drugs. 2011;25(5):383-399.
24. John AP, Dragovic M. Antipsychotic polypharmacy is not associated with reduced dose of individual antipsychotics in schizophrenia. J Clin Psychopharmacol. 2015;35(2):193-195.
25. Nasrallah HA. Treatment-resistant schizophrenia. Current Psychiatry. http://www.currentpsychiatry.com/specialty-focus/schizophrenia-other-psychotic-disorders/article/ treatment-resistant-schizophrenia/9be7bba3713d4a4cd68aa 8c92b79e5b1.html. Accessed June 16, 2015.
26. Alvarez-Jiménez M, Parker AG, Hetrick SE, et al. Preventing the second episode: a systematic review and meta-analysis of psychosocial and pharmacological trials in first-episode psychosis. Schizophr Bull. 2011;37(3):619-630.
27. Nasrallah HA, Smeltzer DJ. Contemporary diagnosis and management of the patient with schizophrenia. 2nd ed. Newton, PA: Handbooks in Health Care Co; 2011.
28. McEvoy JP. Risks versus benefits of different types of long-acting injectable antipsychotics. J Clin Psychiatry. 2006;67(suppl 5):15-18.
29. Agid O, Foussias G, Remington G. Long-acting injectable antipsychotics in the treatment of schizophrenia: their role in relapse prevention. Expert Opin Pharmacother. 2010;11(14):2301-2317.
30. Kirschner M, Theodoridou A, Fusar-Poli P, et al. Patients’ and clinicians’ attitude towards long-acting depot antipsychotics in subjects with a first episode psychosis. Ther Adv Psychophamacol. 2013;3(2):89-99.
31. Heres S, Hamann J, Mendel R, et al. Identifying the profile of optimal candidates for antipsychotic depot therapy: A cluster analysis. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(8):1987-1993.
32. Heres S, Lambert M, Vauth R. Treatment of early episode in patents with schizophrenia: the role of long acting antipsychotics. Eur Psychiatry. 2014;29(suppl 2):1409-1413.
33. Heres S, Schmitz FS, Leucht S, et al. The attitude of patients towards antipsychotic depot treatment. Int Clin Psychopharmacol. 2007;22(5):275-282.
34. Weiden PJ, Schooler NR, Weedon JC, et al. A randomized controlled trial of long-acting injectable risperidone vs continuation on oral atypical antipsychotics for first-episode schizophrenia patients: initial adherence outcome. J Clin Psychiatry. 2009;70(10):1397-1406.
35. Heres S, Reichhart T, Hamann J, et al. Psychiatrists’ attitude to antipsychotic depot treatment in patients with first-episode schizophrenia. Eur Psychiatry. 2011;26(5):297-301.
36. Kane JM, Rifkin A, Quitkin F, et al. Fluphenazine vs placebo in patients with remitted, acute first-episode schizophrenia. Arch Gen Psychiatry. 1982;39(1):70-73.
37. Tiihonen J, Wahlbeck K, Lönnqvist J, et al. Effectiveness of antipsychotic treatments in a nationwide cohort of patients in a community care after first hospitalization due to schizophrenia and schizoaffective disorder: observational follow-up study. BMJ. 2006;333(7561):224.
38. Parellada E, Andrezina R, Milanova V, et al. Patients in the early phases of schizophrenia and schizoaffective disorders effectively treated with risperidone long-acting injectable. J Psychopharmacol. 2005;19(suppl 5):5-14.
39. Malla A, Binder C, Chue P. Comparison of long-acting injectable risperidone and oral novel antipsychotic drugs for treatment in early phase of schizophrenia spectrum psychosis. Proceedings of the 61st Annual Convention Society of Biological Psychiatry; Toronto, Canada; 2006.
40. Lasser RA, Bossie CA, Zhu Y, et al. Long-acting risperidone in young adults with early schizophrenia or schizoaffective illness. Ann Clin Psychiatry. 2007;19(2):65-71.
41. Emsley R, Oosthuizen P, Koen L, et al. Remission in patients with first-episode schizophrenia receiving assured antipsychotic medication: a study with risperidone long-acting injection. Int Clin Psychopharmacol. 2008;23(6):325-331.
42. Emsley R, Oosthuizen P, Koen L, et al. Oral versus injectable antipsychotic treatment in early psychosis: post hoc comparison of two studies. Clin Ther. 2008;30(12):2378-2386.
43. Kim B, Lee SH, Choi TK, et al. Effectiveness of risperidone long-acting injection in first-episode schizophrenia: in naturalistic setting. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(5):1231-1235.
44. Weiden PJ, Schooler NJ, Weedon JC, et al. A randomized controlled trial of long-acting injectable risperidone vs continuation on oral atypical antipsychotics for first-episode schizophrenia patients: initial adherence outcome. J Clin Psychiatry. 2009;70(10):1397-1406.
45. Bartzokis G, Lu PH, Amar CP, et al. Long acting injection versus oral risperidone in first-episode schizophrenia: differential impact on white matter myelination trajectory. Schizophr Res. 2011;132(1):35-41.
46. Napryeyenko O, Burba B, Martinez G, et al. Risperidone long-acting injectable in recent-onset schizophrenia examined with clinician and patient self-report measures. J Clin Psychopharmacol. 2010;30(2):200-202.
47. Tiihonen J, Haukka J, Taylor M, et al. A nationwide cohort study of oral and depot antipsychotics after first hospitalization for schizophrenia. Am J Psychiatry. 2011;168(6):603-609.
48. Dubois V, Megens J, Mertens C, et al. Long-acting risperidone in early-episode schizophrenia. Acta Psychiatrica Belgica. 2011;111(1):9-21.
49. ClinicalTrials.gov. Oral risperidone versus injectable paliperidone palmitate for treating first-episode schizophrenia. https://clinicaltrials.gov/ct2/show/ NCT01451736. Accessed June 16, 2015.
50. ClinicalTrials.gov. Brain myelination effects of paliperidone palmitate versus oral risperidone in first episode schizophrenia. https://clinicaltrials.gov/ct2/ show/NCT01458379. Accessed June 16, 2015.
51. ClinicalTrials.gov. Effects of paliperidone palmitate versus oral antipsychotics on clinical outcomes and MRI measures. https://clinicaltrials.gov/ct2/show/NCT01359293. Accessed June 16, 2016.
52. U.S. Food and Drug Administration. Drugs@FDA. http:// www.accessdata.fda.gov/scripts/cder/drugsatfda. Accessed January 11, 2015.
53. Velligan DI, Weiden PJ, Sajatovic M, et al; Expert Consensus Panel on Adherence Problems in Serious and Persistent Mental Illness. The expert consensus guideline series: adherence problems in patients with serious and persistent mental illness. J Clin Psychiatry. 2009;70(suppl 4):1-46; quiz 47-48.
54. Nandra KS, Agius M. The difference between typical and atypical antipsychotics: the effects on neurogenesis. Psychiatr Danub. 2012;24(suppl 1):S95-S99.
55. Nasrallah HA. Haloperidol is clearly neurotoxic. Should it be banned? Current Psychiatry. 2013;12(7):7-8.
56. Kane JM, Garcia-Ribora C. Clinical guideline recommendations for antipsychotic long-acting injections. Br J Psychiatry. 2009;52:S63-S67.
57. Clozaril [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2014.
58. Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789-796.
59. Woerner MG, Robinson DG, Alvir JMJ, et al. Clozapine as a first treatment for schizophrenia. Am J Psychiatry. 2003;160(8):1514-1516.
60. Lieberman JA, Phillips M, Gu H, et al. Atypical and conventional antipsychotic drugs in treatment-naive first-episode schizophrenia: a 52-week randomized trial of clozapine vs chlorpromazine. Neuropsychopharmacology. 2003;28(5):995-1003.
61. Girgis RR, Phillips MR, Li X, et al. Clozapine v. chlorpromazine in treatment-naive, first-episode schizophrenia: 9-year outcomes of a randomised clinical trial. Br J Psychiatry. 2011;199(4):281-288.
62. Sanz-Fuentenebro J, Taboada D, Palomo T, et al. Randomized trial of clozapine vs. risperidone in treatment-naïve first-episode schizophrenia: results after one year. Schizophr Res. 2013;149(1-3):156-161.
63. Yang PD, Ji Z. The efficacy and related factors of clozapine on first-episode schizophrenia. Chin J Nerv Ment Dis. 1997;23:155-158.
64. Agid O, Schulze L, Arenovich T, et al. Antipsychotic response in first-episode schizophrenia: efficacy of high doses and switching. Eur Neuropsychopharmacol. 2013;23(9):1017-1022.
65. Remington G, Agid O, Foussias G, et al. Clozapine’s role in the treatment of first-episode schizophrenia. Am J Psychiatry. 2013;170(2):146-151.
1. Perkins DO, Gu H, Boteva K, et al. Relationship between duration of untreated psychosis and outcome in first-episode schizophrenia: a critical review and meta-analysis. Am J Psychiatry. 2005;162(10):1785-1804.
2. Bradford DW, Perkins DO, Lieberman JA. Pharmacological management of first-episode schizophrenia and related nonaffective psychoses. Drugs. 2003;63(21):2265-2283.
3. Lieberman JA, Koreen AR, Chakos M, et al. Factors influencing treatment response and outcome of first-episode schizophrenia: implications for understanding the pathophysiology of schizophrenia. J Clin Psychiatry. 1996;57(suppl 9):5-9.
4. Agid O, Arenovich T, Sajeev G, et al. An algorithm-based approach to first-episode schizophrenia: response rates over 3 prospective antipsychotic trials with a retrospective data analysis. J Clin Psychiatry. 2011;72(11):1439-1444.
5. Gardner KN, Nasrallah HA. Managing first-episode psychosis. An early stage of schizophrenia with distinct treatment needs. Current Psychiatry. 2015;14(5):32-34,36-40,42.
6. Lehman AF, Lieberman JA, Dixon LB, et al; American Psychiatric Association; Steering Committee on Practice Guidelines. Practice guideline for the treatment of patients with schizophrenia, second edition. Am J Psychiatry. 2004;161(suppl 2):1-56.
7. Texas Department of State Health Services. Texas Medication Algorithm Project (TMAP) Procedural Manual. Schizophrenia Treatment Algorithms. http://www.jpshealthnet.org/sites/default/files/ tmapalgorithmforschizophrenia.pdf. Updated April 2008. Accessed June 11, 2015.
8. Buchanan RW, Kreyenbuhl J, Kelly DL, et al; Schizophrenia Patient Outcomes Research Team (PORT). The 2009 schizophrenia PORT psychopharmacological treatment recommendations and summary statements. Schizophr Bull. 2010;36(1):71-93.
9. Barnes TR; Schizophrenia Consensus Group of British Association for Psychopharmacology. Evidence-based guidelines for the pharmacological treatment of schizophrenia: recommendations from the British Association for Psychopharmacology. J Psychopharmacol. 2011;25(5):567-620.
10. Hasan A, Falkai P, Wobrok T, et al; WFSBP Task force on Treatment Guidelines for Schizophrenia. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 2: update 2012 on the long-term treatment of schizophrenia and management of antipsychotic-induced side effects. World J Biol Psychiatry. 2013;14(1):2-44.
11. Scottish Intercollegiate Guidelines Network. SIGN 131: Management of schizophrenia. http://www.sign.ac.uk/ pdf/sign131.pdf. Published March 2013. Accessed June 11, 2015.
12. Llorca PM, Abbar M, Courtet P, et al. Guidelines for the use and management of long-acting injectable antipsychotics in serous mental illness. BMC Psychiatry. 2013;13:340.
13. National Institute for Health and Care Excellence. NICE clinical guideline 178: Psychosis and schizophrenia in adults: treatment and management. https://www.nice.org. uk/guidance/cg178/resources/guidance-psychosis-and-schizophrenia-in-adults-treatment-and-management-pdf. Updated March 2014. Accessed June 16, 2015.
14. Canadian Psychiatric Association. Clinical practice guidelines. Treatment of schizophrenia. Can J Psychiatry. 2005;50(13 suppl 1):7S-57S.
15. McEvoy JP, Scheifler PL, Frances A. The expert consensus guideline series: treatment of schizophrenia. J Clin Psychiatry. 1999;60(suppl 11):3-80.
16. Marder SR, Essock SM, Miller AL, et al. The Mount Sinai conference on the pharmacotherapy of schizophrenia. Schizophr Bull. 2002;28(1):5-16.
17. Kapur S, Zipursky R, Jones C, et al. Relationship between dopamine D(2) occupancy, clinical response, and side effects: a double-blind PET study of first-episode schizophrenia. Am J Psychiatry. 2000;157(4):514-520.
18. Fankhauser MP. Drug interactions with tobacco smoke: implications for patient care. Current Psychiatry. 2013;12(1):12-16.
19. Agid O, Schulze L, Arenovich T, et al. Antipsychotic response in first-episode schizophrenia: efficacy of high doses and switching. Eur Neuropsychopharmacol. 2013;23(9):1017-1022.
20. Robinson DG, Schooler NR, John M, et al. Prescription practices in the treatment of first-episode schizophrenia spectrum disorders: data from the national RAISE-ETP study. Am J Psychiatry. 2015;172(3):237-248.
21. Correll CU, Rummel-Kluge C, Corves C, et al. Antipsychotic combinations vs monotherapy in schizophrenia: a meta-analysis of randomized controlled trials. Schizophr Bull. 2009;35(2):443-457.
22. Fisher MD, Reilly K, Isenberg K, et al. Antipsychotic patterns of use in patients with schizophrenia: polypharmacy versus monotherapy. BMC Psychiatry. 2014;14(1):341.
23. Barnes TR, Paton C. Antipsychotic polypharmacy in schizophrenia: benefits and risks. CNS Drugs. 2011;25(5):383-399.
24. John AP, Dragovic M. Antipsychotic polypharmacy is not associated with reduced dose of individual antipsychotics in schizophrenia. J Clin Psychopharmacol. 2015;35(2):193-195.
25. Nasrallah HA. Treatment-resistant schizophrenia. Current Psychiatry. http://www.currentpsychiatry.com/specialty-focus/schizophrenia-other-psychotic-disorders/article/ treatment-resistant-schizophrenia/9be7bba3713d4a4cd68aa 8c92b79e5b1.html. Accessed June 16, 2015.
26. Alvarez-Jiménez M, Parker AG, Hetrick SE, et al. Preventing the second episode: a systematic review and meta-analysis of psychosocial and pharmacological trials in first-episode psychosis. Schizophr Bull. 2011;37(3):619-630.
27. Nasrallah HA, Smeltzer DJ. Contemporary diagnosis and management of the patient with schizophrenia. 2nd ed. Newton, PA: Handbooks in Health Care Co; 2011.
28. McEvoy JP. Risks versus benefits of different types of long-acting injectable antipsychotics. J Clin Psychiatry. 2006;67(suppl 5):15-18.
29. Agid O, Foussias G, Remington G. Long-acting injectable antipsychotics in the treatment of schizophrenia: their role in relapse prevention. Expert Opin Pharmacother. 2010;11(14):2301-2317.
30. Kirschner M, Theodoridou A, Fusar-Poli P, et al. Patients’ and clinicians’ attitude towards long-acting depot antipsychotics in subjects with a first episode psychosis. Ther Adv Psychophamacol. 2013;3(2):89-99.
31. Heres S, Hamann J, Mendel R, et al. Identifying the profile of optimal candidates for antipsychotic depot therapy: A cluster analysis. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(8):1987-1993.
32. Heres S, Lambert M, Vauth R. Treatment of early episode in patents with schizophrenia: the role of long acting antipsychotics. Eur Psychiatry. 2014;29(suppl 2):1409-1413.
33. Heres S, Schmitz FS, Leucht S, et al. The attitude of patients towards antipsychotic depot treatment. Int Clin Psychopharmacol. 2007;22(5):275-282.
34. Weiden PJ, Schooler NR, Weedon JC, et al. A randomized controlled trial of long-acting injectable risperidone vs continuation on oral atypical antipsychotics for first-episode schizophrenia patients: initial adherence outcome. J Clin Psychiatry. 2009;70(10):1397-1406.
35. Heres S, Reichhart T, Hamann J, et al. Psychiatrists’ attitude to antipsychotic depot treatment in patients with first-episode schizophrenia. Eur Psychiatry. 2011;26(5):297-301.
36. Kane JM, Rifkin A, Quitkin F, et al. Fluphenazine vs placebo in patients with remitted, acute first-episode schizophrenia. Arch Gen Psychiatry. 1982;39(1):70-73.
37. Tiihonen J, Wahlbeck K, Lönnqvist J, et al. Effectiveness of antipsychotic treatments in a nationwide cohort of patients in a community care after first hospitalization due to schizophrenia and schizoaffective disorder: observational follow-up study. BMJ. 2006;333(7561):224.
38. Parellada E, Andrezina R, Milanova V, et al. Patients in the early phases of schizophrenia and schizoaffective disorders effectively treated with risperidone long-acting injectable. J Psychopharmacol. 2005;19(suppl 5):5-14.
39. Malla A, Binder C, Chue P. Comparison of long-acting injectable risperidone and oral novel antipsychotic drugs for treatment in early phase of schizophrenia spectrum psychosis. Proceedings of the 61st Annual Convention Society of Biological Psychiatry; Toronto, Canada; 2006.
40. Lasser RA, Bossie CA, Zhu Y, et al. Long-acting risperidone in young adults with early schizophrenia or schizoaffective illness. Ann Clin Psychiatry. 2007;19(2):65-71.
41. Emsley R, Oosthuizen P, Koen L, et al. Remission in patients with first-episode schizophrenia receiving assured antipsychotic medication: a study with risperidone long-acting injection. Int Clin Psychopharmacol. 2008;23(6):325-331.
42. Emsley R, Oosthuizen P, Koen L, et al. Oral versus injectable antipsychotic treatment in early psychosis: post hoc comparison of two studies. Clin Ther. 2008;30(12):2378-2386.
43. Kim B, Lee SH, Choi TK, et al. Effectiveness of risperidone long-acting injection in first-episode schizophrenia: in naturalistic setting. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(5):1231-1235.
44. Weiden PJ, Schooler NJ, Weedon JC, et al. A randomized controlled trial of long-acting injectable risperidone vs continuation on oral atypical antipsychotics for first-episode schizophrenia patients: initial adherence outcome. J Clin Psychiatry. 2009;70(10):1397-1406.
45. Bartzokis G, Lu PH, Amar CP, et al. Long acting injection versus oral risperidone in first-episode schizophrenia: differential impact on white matter myelination trajectory. Schizophr Res. 2011;132(1):35-41.
46. Napryeyenko O, Burba B, Martinez G, et al. Risperidone long-acting injectable in recent-onset schizophrenia examined with clinician and patient self-report measures. J Clin Psychopharmacol. 2010;30(2):200-202.
47. Tiihonen J, Haukka J, Taylor M, et al. A nationwide cohort study of oral and depot antipsychotics after first hospitalization for schizophrenia. Am J Psychiatry. 2011;168(6):603-609.
48. Dubois V, Megens J, Mertens C, et al. Long-acting risperidone in early-episode schizophrenia. Acta Psychiatrica Belgica. 2011;111(1):9-21.
49. ClinicalTrials.gov. Oral risperidone versus injectable paliperidone palmitate for treating first-episode schizophrenia. https://clinicaltrials.gov/ct2/show/ NCT01451736. Accessed June 16, 2015.
50. ClinicalTrials.gov. Brain myelination effects of paliperidone palmitate versus oral risperidone in first episode schizophrenia. https://clinicaltrials.gov/ct2/ show/NCT01458379. Accessed June 16, 2015.
51. ClinicalTrials.gov. Effects of paliperidone palmitate versus oral antipsychotics on clinical outcomes and MRI measures. https://clinicaltrials.gov/ct2/show/NCT01359293. Accessed June 16, 2016.
52. U.S. Food and Drug Administration. Drugs@FDA. http:// www.accessdata.fda.gov/scripts/cder/drugsatfda. Accessed January 11, 2015.
53. Velligan DI, Weiden PJ, Sajatovic M, et al; Expert Consensus Panel on Adherence Problems in Serious and Persistent Mental Illness. The expert consensus guideline series: adherence problems in patients with serious and persistent mental illness. J Clin Psychiatry. 2009;70(suppl 4):1-46; quiz 47-48.
54. Nandra KS, Agius M. The difference between typical and atypical antipsychotics: the effects on neurogenesis. Psychiatr Danub. 2012;24(suppl 1):S95-S99.
55. Nasrallah HA. Haloperidol is clearly neurotoxic. Should it be banned? Current Psychiatry. 2013;12(7):7-8.
56. Kane JM, Garcia-Ribora C. Clinical guideline recommendations for antipsychotic long-acting injections. Br J Psychiatry. 2009;52:S63-S67.
57. Clozaril [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2014.
58. Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789-796.
59. Woerner MG, Robinson DG, Alvir JMJ, et al. Clozapine as a first treatment for schizophrenia. Am J Psychiatry. 2003;160(8):1514-1516.
60. Lieberman JA, Phillips M, Gu H, et al. Atypical and conventional antipsychotic drugs in treatment-naive first-episode schizophrenia: a 52-week randomized trial of clozapine vs chlorpromazine. Neuropsychopharmacology. 2003;28(5):995-1003.
61. Girgis RR, Phillips MR, Li X, et al. Clozapine v. chlorpromazine in treatment-naive, first-episode schizophrenia: 9-year outcomes of a randomised clinical trial. Br J Psychiatry. 2011;199(4):281-288.
62. Sanz-Fuentenebro J, Taboada D, Palomo T, et al. Randomized trial of clozapine vs. risperidone in treatment-naïve first-episode schizophrenia: results after one year. Schizophr Res. 2013;149(1-3):156-161.
63. Yang PD, Ji Z. The efficacy and related factors of clozapine on first-episode schizophrenia. Chin J Nerv Ment Dis. 1997;23:155-158.
64. Agid O, Schulze L, Arenovich T, et al. Antipsychotic response in first-episode schizophrenia: efficacy of high doses and switching. Eur Neuropsychopharmacol. 2013;23(9):1017-1022.
65. Remington G, Agid O, Foussias G, et al. Clozapine’s role in the treatment of first-episode schizophrenia. Am J Psychiatry. 2013;170(2):146-151.

Avoiding common drug−drug interactions
Mr. T, age 23, was given a diagnosis of bipolar disorder 1 year ago. After he experienced inadequate symptom relief with valproate, you switched him to extended-release lithium, 1,200 mg/d. Mr. T reported improved mood and stability with this medication adjustment. These positive changes led him to resume activities he enjoyed before onset of bipolar disorder, such as running, reading, and going out to dinner with friends.
Now, Mr. T’s mother calls your office to express concern about her son’s slight

hand tremor, which appeared after 2 days of gastrointestinal distress. She tells you that Mr. T sprained his ankle while running 1 week ago and has been taking over-the-counter ibuprofen for pain relief, which he did often in the past.
You suspect that Mr. T is experiencing lithium toxicity as a result of ibuprofen use.
Although mental health providers can easily recognize the drug−drug interaction between lithium and nonsteroidal anti-inflammatory drugs (NSAIDs) that Mr. T experienced, interpreting the safety of a medication regimen with respect to drug− drug interactions before prescribing often is more daunting. This article reviews the basics of drug−drug interactions, while briefly highlighting common examples in psychiatric medicine (Table 11-5). We also provide an outline of additional points to consider when reviewing your patients’ medication regimens and encountering unfamiliar drug−drug interactions.
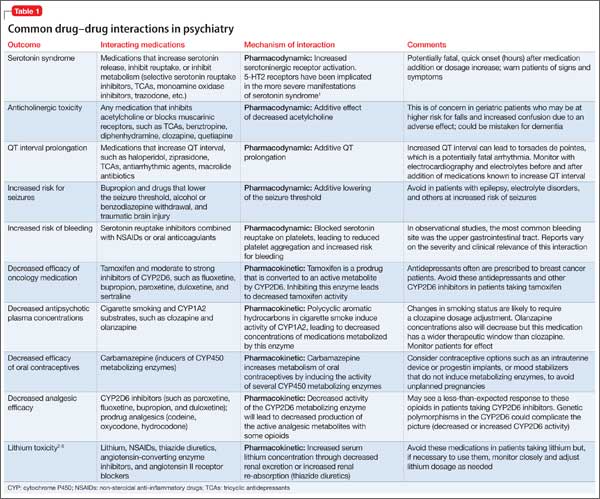
Types of drug−drug interactions
Drug−drug interactions fall into 2 categories: pharmacodynamic (PD) and pharmacokinetic (PK):
• PD interactions are a result of the combined impact of medications on the body when there is no direct effect on absorption, distribution, metabolism, or excretion characteristics, such as 2 medications that act at the same receptor or lead to similar or opposing pharmacologic effects.
• PK interactions occur when a drug affects the absorption, distribution, metabolism, or excretion characteristics of another drug.
Although it is possible that drug−drug interactions will have no clinical effect, when the impact of a PD or PK drug−drug interaction is evident, it likely is the result of additive, synergistic, or antagonistic consequences on the medications’ intended impact or side-effect profile.
Pharmacodynamic interactions
Serotonin syndrome. The potential for serotonin syndrome occurs when medications that increase synaptic serotonin concentration are used concomitantly.1 This can occur through several mechanisms, including increased serotonin release, decreased reuptake, or decreased serotonin metabolism. A high serotonin concentration in the CNS and in the periphery overstimulates serotonin receptors, leading to signs and symptoms that can include diarrhea, fever, delirium, coma, and potentially death.
QT prolongation and anticholinergic toxicity are further examples of additive PD drug−drug interactions. Anticholinergic toxicity is possible when multiple medications contribute to inhibition of the neuro-transmitter acetylcholine at muscarinic receptors. This leads to adverse effects such as dry mouth, constipation, confusion, and urinary retention.
QT prolongation, which can lead to arrhythmia, occurs when a patient is taking several medications that can increase the QT interval. Consider close monitoring and using alternative agents with less potential to increase the QT interval in patients at risk of arrhythmias (geriatric patients, those with an increased QT interval at baseline, etc.).
Decreased seizure threshold. The increased risk of seizures with bupropion and other medications that lower the seizure threshold is another example of an additive PD drug interaction. Bupropion can increase the risk of seizures in a dose-dependent manner, which increases when bupropion is taken with other drugs that lower the seizure threshold.6 Seizure risk associated with alcohol or benzodiazepine withdrawal also may increase the risk for this interaction.
Of note, the increased risk of seizures with the combination of bupropion and alcohol in the absence of withdrawal is not well studied in humans, but positive correlation has been seen in an animal study.6
Decreased platelet function. Another example of a PD drug−drug interaction is increased risk of bleeding when a selective serotonin reuptake inhibitor is used with a NSAID or oral anticoagulant. The proposed mechanism for this interaction is that blocking serotonin reuptake on platelets leads to decreased platelet function and an increased risk for prolonged bleeding.7 This is somewhat controversial because, first, it has been noted that drugs with the highest degree of serotonin reuptake inhibition do not always cause the highest risk of bleeding and, second, most of the evidence for this interaction is from observational studies.7
This potential interaction could be most important for patients who need an antidepressant, are on chronic NSAID or anticoagulant therapy, and are at high risk of bleeding.
Pharmacokinetic interactions
PK interactions in psychiatry often are caused by interference of drug metabolizing enzymes. The cytochrome P450 (CYP450) family of metabolizing enzymes in particular is important to the breakdown of medications in the body. Many drug−drug interactions involve medications that can inhibit or induce metabolism of other drugs through their effect on the CYP450 system.
Inhibition interactions. When a drug’s metabolism is inhibited, the result is usually increased serum concentration of that medication (because of less breakdown) and a more potent impact on the primary mechanism of action or adverse effects. Sometimes, inhibiting metabolism can lead to decreased clinical effect. Tamoxifen (an oral agent used to treat breast cancer) and certain analgesics when used in combination with moderate or strong inhibitors of the CYP2D6 subfamily of CYP450 metabolizing enzymes are 2 examples of metabolism inhibition leading to decreased efficacy.8 Both tamoxifen and the analgesics listed in Table 11-5 are prodrugs; that is, they must be metabolized to be active. When the enzymes that metabolize these drugs into their active form are inhibited, the concentration of active drug decreases.
Induction interactions. Alternatively, there is an increased rate of drug breakdown and resulting decrease in effect when drugs that induce the activity of metabolizing enzymes are used with medications that are substrates of the same enzyme. Carbamazepine is commonly involved in this type of drug interaction because it is a strong inducer of CYP 1A2, 2B6, 2C19, 2C9, and 3A4, and the p-glycoprotein drug efflux pump.9 As a result of this rampant induction, carbamazepine can decrease the serum concentration of oral contraceptives below a reliably effective level. Therefore, it is recommended that women of childbearing potential use other contraceptive methods, such as a progestin implant or an intrauterine device.10
In addition, the polycyclic aromatic hydrocarbons found in cigarettes induce activity of CYP1A2. Patients who smoke and use medications metabolized by this enzyme, such as clozapine and olanzapine, may need a higher dosage.
Drug elimination interactions
The last drug−drug interaction discussed here returns the discussion to Mr. T and involves drug elimination.2 The NSAIDs Mr. T was using for pain likely caused decreased renal excretion of lithium. Because lithium is primarily excreted through the kidneys, Mr. T’s NSAID use, possibly in combination with dehydration caused by gastrointestinal distress, resulted in lithium toxicity. This class of analgesics should be avoided or used cautiously in patients taking lithium.
Clinical applications
The relatively common drug−drug interactions discussed here are just a fraction of the potential interactions mental health practitioners see on a daily basis. Understanding the basics of PD and PK interactions in the setting of patient-specific factors can help to clarify the information found in drug−drug interaction databases, such as Micromedex, Lexicomp, Facts and Comparisons, and Epocrates. Table 2 lists additional insights into drug interactions.
Related Resources
• CredibleMeds. Online resource on QT prolonging drugs. http://crediblemeds.org.
• Madhusoodanan S, Velama U, Parmar J, et al. A current review of cytochrome P450 interactions of psychotropic drugs. Ann Clin Psychiatry. 2014;26(2):120-138.
Drug Brand Names
Benztropine • Cogentin Olanzapine • Zyprexa
Bupropion • Wellbutrin Oxycodone • Oxycontin
Carbamazepine • Tegretol Paroxetine • Paxil
Clozapine • Clozaril Quetiapine • Seroquel
Diphenhydramine • Benadryl Sertraline • Zoloft
Duloxetine • Cymbalta Tamoxifen • Soltamox
Fluoxetine • Prozac Trazodone • Desyrel
Lithium • Eskalith, Lithobid Valproate • Divalproex
Haloperidol • Haldol Ziprasidone • Geodon
Hydrocodone • Vicodin
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Buckley NA, Dawson AH, Isbister GK. Serotonin syndrome. BMJ. 2014;348:g1626. doi: 10.1136/bmj.g1626.
2. Eskalith [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2003.
3. Handler J. Lithium and antihypertensive medication: a potentially dangerous interaction. J Clin Hypertens (Greenwich). 2009;11(12):738-742.
4. Blanche P, Raynaud E, Kerob D, et al. Lithium intoxication in an elderly patient after combined treatment with losartan. Eur J Clin Pharmacol. 1997;52(6):501.
5. Atacand [package insert]. Wilmington, DE: AstraZeneca LP; 2013.
6. Silverstone PH, Williams R, McMahon L, et al. Alcohol significantly lowers the seizure threshold in mice when co-administered with bupropion hydrochloride. Ann Gen Psychiatry. 2008;7:11.
7. Spina E, Trifirò G, Caraci F. Clinically significant drug interactions with newer antidepressants. CNS Drugs. 2012;26(1):39-67.
8. Ereshefsky L, Sloan DM. Drug-drug interactions with the use of psychotropic medications. CNS Spectr. 2009;14(suppl Q and A forum 8):1-8.
9. Carbamazepine. Drug facts and comparisons database. St. Louis, MO: Wolters Kluwer Health Inc; November 2014.
10. Pennell PB. Pregnancy, epilepsy, and women’s issues. Continuum (Minneap Minn). 2013;19(3 Epilepsy):697-714.
Mr. T, age 23, was given a diagnosis of bipolar disorder 1 year ago. After he experienced inadequate symptom relief with valproate, you switched him to extended-release lithium, 1,200 mg/d. Mr. T reported improved mood and stability with this medication adjustment. These positive changes led him to resume activities he enjoyed before onset of bipolar disorder, such as running, reading, and going out to dinner with friends.
Now, Mr. T’s mother calls your office to express concern about her son’s slight
hand tremor, which appeared after 2 days of gastrointestinal distress. She tells you that Mr. T sprained his ankle while running 1 week ago and has been taking over-the-counter ibuprofen for pain relief, which he did often in the past.
You suspect that Mr. T is experiencing lithium toxicity as a result of ibuprofen use.
Although mental health providers can easily recognize the drug−drug interaction between lithium and nonsteroidal anti-inflammatory drugs (NSAIDs) that Mr. T experienced, interpreting the safety of a medication regimen with respect to drug− drug interactions before prescribing often is more daunting. This article reviews the basics of drug−drug interactions, while briefly highlighting common examples in psychiatric medicine (Table 11-5). We also provide an outline of additional points to consider when reviewing your patients’ medication regimens and encountering unfamiliar drug−drug interactions.
Types of drug−drug interactions
Drug−drug interactions fall into 2 categories: pharmacodynamic (PD) and pharmacokinetic (PK):
• PD interactions are a result of the combined impact of medications on the body when there is no direct effect on absorption, distribution, metabolism, or excretion characteristics, such as 2 medications that act at the same receptor or lead to similar or opposing pharmacologic effects.
• PK interactions occur when a drug affects the absorption, distribution, metabolism, or excretion characteristics of another drug.
Although it is possible that drug−drug interactions will have no clinical effect, when the impact of a PD or PK drug−drug interaction is evident, it likely is the result of additive, synergistic, or antagonistic consequences on the medications’ intended impact or side-effect profile.
Pharmacodynamic interactions
Serotonin syndrome. The potential for serotonin syndrome occurs when medications that increase synaptic serotonin concentration are used concomitantly.1 This can occur through several mechanisms, including increased serotonin release, decreased reuptake, or decreased serotonin metabolism. A high serotonin concentration in the CNS and in the periphery overstimulates serotonin receptors, leading to signs and symptoms that can include diarrhea, fever, delirium, coma, and potentially death.
QT prolongation and anticholinergic toxicity are further examples of additive PD drug−drug interactions. Anticholinergic toxicity is possible when multiple medications contribute to inhibition of the neuro-transmitter acetylcholine at muscarinic receptors. This leads to adverse effects such as dry mouth, constipation, confusion, and urinary retention.
QT prolongation, which can lead to arrhythmia, occurs when a patient is taking several medications that can increase the QT interval. Consider close monitoring and using alternative agents with less potential to increase the QT interval in patients at risk of arrhythmias (geriatric patients, those with an increased QT interval at baseline, etc.).
Decreased seizure threshold. The increased risk of seizures with bupropion and other medications that lower the seizure threshold is another example of an additive PD drug interaction. Bupropion can increase the risk of seizures in a dose-dependent manner, which increases when bupropion is taken with other drugs that lower the seizure threshold.6 Seizure risk associated with alcohol or benzodiazepine withdrawal also may increase the risk for this interaction.
Of note, the increased risk of seizures with the combination of bupropion and alcohol in the absence of withdrawal is not well studied in humans, but positive correlation has been seen in an animal study.6
Decreased platelet function. Another example of a PD drug−drug interaction is increased risk of bleeding when a selective serotonin reuptake inhibitor is used with a NSAID or oral anticoagulant. The proposed mechanism for this interaction is that blocking serotonin reuptake on platelets leads to decreased platelet function and an increased risk for prolonged bleeding.7 This is somewhat controversial because, first, it has been noted that drugs with the highest degree of serotonin reuptake inhibition do not always cause the highest risk of bleeding and, second, most of the evidence for this interaction is from observational studies.7
This potential interaction could be most important for patients who need an antidepressant, are on chronic NSAID or anticoagulant therapy, and are at high risk of bleeding.
Pharmacokinetic interactions
PK interactions in psychiatry often are caused by interference of drug metabolizing enzymes. The cytochrome P450 (CYP450) family of metabolizing enzymes in particular is important to the breakdown of medications in the body. Many drug−drug interactions involve medications that can inhibit or induce metabolism of other drugs through their effect on the CYP450 system.
Inhibition interactions. When a drug’s metabolism is inhibited, the result is usually increased serum concentration of that medication (because of less breakdown) and a more potent impact on the primary mechanism of action or adverse effects. Sometimes, inhibiting metabolism can lead to decreased clinical effect. Tamoxifen (an oral agent used to treat breast cancer) and certain analgesics when used in combination with moderate or strong inhibitors of the CYP2D6 subfamily of CYP450 metabolizing enzymes are 2 examples of metabolism inhibition leading to decreased efficacy.8 Both tamoxifen and the analgesics listed in Table 11-5 are prodrugs; that is, they must be metabolized to be active. When the enzymes that metabolize these drugs into their active form are inhibited, the concentration of active drug decreases.
Induction interactions. Alternatively, there is an increased rate of drug breakdown and resulting decrease in effect when drugs that induce the activity of metabolizing enzymes are used with medications that are substrates of the same enzyme. Carbamazepine is commonly involved in this type of drug interaction because it is a strong inducer of CYP 1A2, 2B6, 2C19, 2C9, and 3A4, and the p-glycoprotein drug efflux pump.9 As a result of this rampant induction, carbamazepine can decrease the serum concentration of oral contraceptives below a reliably effective level. Therefore, it is recommended that women of childbearing potential use other contraceptive methods, such as a progestin implant or an intrauterine device.10
In addition, the polycyclic aromatic hydrocarbons found in cigarettes induce activity of CYP1A2. Patients who smoke and use medications metabolized by this enzyme, such as clozapine and olanzapine, may need a higher dosage.
Drug elimination interactions
The last drug−drug interaction discussed here returns the discussion to Mr. T and involves drug elimination.2 The NSAIDs Mr. T was using for pain likely caused decreased renal excretion of lithium. Because lithium is primarily excreted through the kidneys, Mr. T’s NSAID use, possibly in combination with dehydration caused by gastrointestinal distress, resulted in lithium toxicity. This class of analgesics should be avoided or used cautiously in patients taking lithium.
Clinical applications
The relatively common drug−drug interactions discussed here are just a fraction of the potential interactions mental health practitioners see on a daily basis. Understanding the basics of PD and PK interactions in the setting of patient-specific factors can help to clarify the information found in drug−drug interaction databases, such as Micromedex, Lexicomp, Facts and Comparisons, and Epocrates. Table 2 lists additional insights into drug interactions.
Related Resources
• CredibleMeds. Online resource on QT prolonging drugs. http://crediblemeds.org.
• Madhusoodanan S, Velama U, Parmar J, et al. A current review of cytochrome P450 interactions of psychotropic drugs. Ann Clin Psychiatry. 2014;26(2):120-138.
Drug Brand Names
Benztropine • Cogentin Olanzapine • Zyprexa
Bupropion • Wellbutrin Oxycodone • Oxycontin
Carbamazepine • Tegretol Paroxetine • Paxil
Clozapine • Clozaril Quetiapine • Seroquel
Diphenhydramine • Benadryl Sertraline • Zoloft
Duloxetine • Cymbalta Tamoxifen • Soltamox
Fluoxetine • Prozac Trazodone • Desyrel
Lithium • Eskalith, Lithobid Valproate • Divalproex
Haloperidol • Haldol Ziprasidone • Geodon
Hydrocodone • Vicodin
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Mr. T, age 23, was given a diagnosis of bipolar disorder 1 year ago. After he experienced inadequate symptom relief with valproate, you switched him to extended-release lithium, 1,200 mg/d. Mr. T reported improved mood and stability with this medication adjustment. These positive changes led him to resume activities he enjoyed before onset of bipolar disorder, such as running, reading, and going out to dinner with friends.
Now, Mr. T’s mother calls your office to express concern about her son’s slight
hand tremor, which appeared after 2 days of gastrointestinal distress. She tells you that Mr. T sprained his ankle while running 1 week ago and has been taking over-the-counter ibuprofen for pain relief, which he did often in the past.
You suspect that Mr. T is experiencing lithium toxicity as a result of ibuprofen use.
Although mental health providers can easily recognize the drug−drug interaction between lithium and nonsteroidal anti-inflammatory drugs (NSAIDs) that Mr. T experienced, interpreting the safety of a medication regimen with respect to drug− drug interactions before prescribing often is more daunting. This article reviews the basics of drug−drug interactions, while briefly highlighting common examples in psychiatric medicine (Table 11-5). We also provide an outline of additional points to consider when reviewing your patients’ medication regimens and encountering unfamiliar drug−drug interactions.
Types of drug−drug interactions
Drug−drug interactions fall into 2 categories: pharmacodynamic (PD) and pharmacokinetic (PK):
• PD interactions are a result of the combined impact of medications on the body when there is no direct effect on absorption, distribution, metabolism, or excretion characteristics, such as 2 medications that act at the same receptor or lead to similar or opposing pharmacologic effects.
• PK interactions occur when a drug affects the absorption, distribution, metabolism, or excretion characteristics of another drug.
Although it is possible that drug−drug interactions will have no clinical effect, when the impact of a PD or PK drug−drug interaction is evident, it likely is the result of additive, synergistic, or antagonistic consequences on the medications’ intended impact or side-effect profile.
Pharmacodynamic interactions
Serotonin syndrome. The potential for serotonin syndrome occurs when medications that increase synaptic serotonin concentration are used concomitantly.1 This can occur through several mechanisms, including increased serotonin release, decreased reuptake, or decreased serotonin metabolism. A high serotonin concentration in the CNS and in the periphery overstimulates serotonin receptors, leading to signs and symptoms that can include diarrhea, fever, delirium, coma, and potentially death.
QT prolongation and anticholinergic toxicity are further examples of additive PD drug−drug interactions. Anticholinergic toxicity is possible when multiple medications contribute to inhibition of the neuro-transmitter acetylcholine at muscarinic receptors. This leads to adverse effects such as dry mouth, constipation, confusion, and urinary retention.
QT prolongation, which can lead to arrhythmia, occurs when a patient is taking several medications that can increase the QT interval. Consider close monitoring and using alternative agents with less potential to increase the QT interval in patients at risk of arrhythmias (geriatric patients, those with an increased QT interval at baseline, etc.).
Decreased seizure threshold. The increased risk of seizures with bupropion and other medications that lower the seizure threshold is another example of an additive PD drug interaction. Bupropion can increase the risk of seizures in a dose-dependent manner, which increases when bupropion is taken with other drugs that lower the seizure threshold.6 Seizure risk associated with alcohol or benzodiazepine withdrawal also may increase the risk for this interaction.
Of note, the increased risk of seizures with the combination of bupropion and alcohol in the absence of withdrawal is not well studied in humans, but positive correlation has been seen in an animal study.6
Decreased platelet function. Another example of a PD drug−drug interaction is increased risk of bleeding when a selective serotonin reuptake inhibitor is used with a NSAID or oral anticoagulant. The proposed mechanism for this interaction is that blocking serotonin reuptake on platelets leads to decreased platelet function and an increased risk for prolonged bleeding.7 This is somewhat controversial because, first, it has been noted that drugs with the highest degree of serotonin reuptake inhibition do not always cause the highest risk of bleeding and, second, most of the evidence for this interaction is from observational studies.7
This potential interaction could be most important for patients who need an antidepressant, are on chronic NSAID or anticoagulant therapy, and are at high risk of bleeding.
Pharmacokinetic interactions
PK interactions in psychiatry often are caused by interference of drug metabolizing enzymes. The cytochrome P450 (CYP450) family of metabolizing enzymes in particular is important to the breakdown of medications in the body. Many drug−drug interactions involve medications that can inhibit or induce metabolism of other drugs through their effect on the CYP450 system.
Inhibition interactions. When a drug’s metabolism is inhibited, the result is usually increased serum concentration of that medication (because of less breakdown) and a more potent impact on the primary mechanism of action or adverse effects. Sometimes, inhibiting metabolism can lead to decreased clinical effect. Tamoxifen (an oral agent used to treat breast cancer) and certain analgesics when used in combination with moderate or strong inhibitors of the CYP2D6 subfamily of CYP450 metabolizing enzymes are 2 examples of metabolism inhibition leading to decreased efficacy.8 Both tamoxifen and the analgesics listed in Table 11-5 are prodrugs; that is, they must be metabolized to be active. When the enzymes that metabolize these drugs into their active form are inhibited, the concentration of active drug decreases.
Induction interactions. Alternatively, there is an increased rate of drug breakdown and resulting decrease in effect when drugs that induce the activity of metabolizing enzymes are used with medications that are substrates of the same enzyme. Carbamazepine is commonly involved in this type of drug interaction because it is a strong inducer of CYP 1A2, 2B6, 2C19, 2C9, and 3A4, and the p-glycoprotein drug efflux pump.9 As a result of this rampant induction, carbamazepine can decrease the serum concentration of oral contraceptives below a reliably effective level. Therefore, it is recommended that women of childbearing potential use other contraceptive methods, such as a progestin implant or an intrauterine device.10
In addition, the polycyclic aromatic hydrocarbons found in cigarettes induce activity of CYP1A2. Patients who smoke and use medications metabolized by this enzyme, such as clozapine and olanzapine, may need a higher dosage.
Drug elimination interactions
The last drug−drug interaction discussed here returns the discussion to Mr. T and involves drug elimination.2 The NSAIDs Mr. T was using for pain likely caused decreased renal excretion of lithium. Because lithium is primarily excreted through the kidneys, Mr. T’s NSAID use, possibly in combination with dehydration caused by gastrointestinal distress, resulted in lithium toxicity. This class of analgesics should be avoided or used cautiously in patients taking lithium.
Clinical applications
The relatively common drug−drug interactions discussed here are just a fraction of the potential interactions mental health practitioners see on a daily basis. Understanding the basics of PD and PK interactions in the setting of patient-specific factors can help to clarify the information found in drug−drug interaction databases, such as Micromedex, Lexicomp, Facts and Comparisons, and Epocrates. Table 2 lists additional insights into drug interactions.
Related Resources
• CredibleMeds. Online resource on QT prolonging drugs. http://crediblemeds.org.
• Madhusoodanan S, Velama U, Parmar J, et al. A current review of cytochrome P450 interactions of psychotropic drugs. Ann Clin Psychiatry. 2014;26(2):120-138.
Drug Brand Names
Benztropine • Cogentin Olanzapine • Zyprexa
Bupropion • Wellbutrin Oxycodone • Oxycontin
Carbamazepine • Tegretol Paroxetine • Paxil
Clozapine • Clozaril Quetiapine • Seroquel
Diphenhydramine • Benadryl Sertraline • Zoloft
Duloxetine • Cymbalta Tamoxifen • Soltamox
Fluoxetine • Prozac Trazodone • Desyrel
Lithium • Eskalith, Lithobid Valproate • Divalproex
Haloperidol • Haldol Ziprasidone • Geodon
Hydrocodone • Vicodin
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Buckley NA, Dawson AH, Isbister GK. Serotonin syndrome. BMJ. 2014;348:g1626. doi: 10.1136/bmj.g1626.
2. Eskalith [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2003.
3. Handler J. Lithium and antihypertensive medication: a potentially dangerous interaction. J Clin Hypertens (Greenwich). 2009;11(12):738-742.
4. Blanche P, Raynaud E, Kerob D, et al. Lithium intoxication in an elderly patient after combined treatment with losartan. Eur J Clin Pharmacol. 1997;52(6):501.
5. Atacand [package insert]. Wilmington, DE: AstraZeneca LP; 2013.
6. Silverstone PH, Williams R, McMahon L, et al. Alcohol significantly lowers the seizure threshold in mice when co-administered with bupropion hydrochloride. Ann Gen Psychiatry. 2008;7:11.
7. Spina E, Trifirò G, Caraci F. Clinically significant drug interactions with newer antidepressants. CNS Drugs. 2012;26(1):39-67.
8. Ereshefsky L, Sloan DM. Drug-drug interactions with the use of psychotropic medications. CNS Spectr. 2009;14(suppl Q and A forum 8):1-8.
9. Carbamazepine. Drug facts and comparisons database. St. Louis, MO: Wolters Kluwer Health Inc; November 2014.
10. Pennell PB. Pregnancy, epilepsy, and women’s issues. Continuum (Minneap Minn). 2013;19(3 Epilepsy):697-714.
1. Buckley NA, Dawson AH, Isbister GK. Serotonin syndrome. BMJ. 2014;348:g1626. doi: 10.1136/bmj.g1626.
2. Eskalith [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2003.
3. Handler J. Lithium and antihypertensive medication: a potentially dangerous interaction. J Clin Hypertens (Greenwich). 2009;11(12):738-742.
4. Blanche P, Raynaud E, Kerob D, et al. Lithium intoxication in an elderly patient after combined treatment with losartan. Eur J Clin Pharmacol. 1997;52(6):501.
5. Atacand [package insert]. Wilmington, DE: AstraZeneca LP; 2013.
6. Silverstone PH, Williams R, McMahon L, et al. Alcohol significantly lowers the seizure threshold in mice when co-administered with bupropion hydrochloride. Ann Gen Psychiatry. 2008;7:11.
7. Spina E, Trifirò G, Caraci F. Clinically significant drug interactions with newer antidepressants. CNS Drugs. 2012;26(1):39-67.
8. Ereshefsky L, Sloan DM. Drug-drug interactions with the use of psychotropic medications. CNS Spectr. 2009;14(suppl Q and A forum 8):1-8.
9. Carbamazepine. Drug facts and comparisons database. St. Louis, MO: Wolters Kluwer Health Inc; November 2014.
10. Pennell PB. Pregnancy, epilepsy, and women’s issues. Continuum (Minneap Minn). 2013;19(3 Epilepsy):697-714.
Reducing medical comorbidity and mortality in severe mental illness
People with serious mental illness (SMI) have a life expectancy that is 25 years less than the general population, according to the Centers for Disease Control and Prevention.1 This disparity is partially a consequence of the lack of primary and preventive medical care for those with psychiatric illness. Decades of research have shown that people with SMI experience higher medical morbidity and mortality in addition to facing the stigma of mental illness.
This article aims to advance the idea that longitudinal “cross education” between primary care providers (PCPs) and behavioral health providers (BHPs) is essential in addressing this problem. BHPs include psychiatry clinics, which often are part of a university or large health systems; county-based community mental health programs; and independent mental health clinics that contract with public and private health plans to provide mental health services.
Although suicide and injury account for 40% of the excess mortality in schizophrenia, 60% can be attributed to cardiovascular disease, diabetes, respiratory diseases, and infection.2 Patients with SMI have 2 to 3 times the risk of diabetes, dyslipidemia, hypertension, and obesity.3,4 Furthermore, those with SMI consume more than one-third of tobacco products,5 and 50% to 80% of people with SMI smoke tobacco, an important reversible risk factor for cardiovascular disease.
Figure 1 shows that people with SMI are at higher risk of dying from a chronic medical condition, such as cardiovascular disease, diabetes, chronic obstructive pulmonary disease, and hepatitis C6-8—many of which can be managed by primary and preventive medical interventions. These and other conditions often are not diagnosed or effectively managed in patients with SMI.
The high prevalence of metabolic syndrome and tobacco dependence among people with SMI accelerates development of cardiovascular disease, as shown by several studies. Bobes et al9 found that the prevalence of metabolic syndrome and cardiovascular risk among patients with SMI is similar to what is found in the general population at 10 to 15 years of greater age. Osborn et al10 demonstrated that people with SMI age 18 to 49 had a higher relative risk of death from coronary heart disease, stroke, and lung cancer than age-matched controls (Figure 2).
It can be said, therefore, that patients with SMI seem to “age” and die prematurely. To reduce this disparity, primary and preventive medical care—especially for cardiovascular disease—must be delivered earlier in life for those with SMI.
Iatrogenic causes of morbidity
Many psychiatric medications, especially second-generation antipsychotics (SGAs), could exacerbate cardiovascular and metabolic conditions by increasing the risk of weight gain, insulin resistance, and dyslipidemia. Antipsychotics that generally are considered to be more effective for refractory psychotic illness (eg, clozapine and olanzapine) are associated with the highest risk of metabolic syndrome. Simon et al11 found a dose-response relationship between olanzapine and clozapine serum concentrations and worsening metabolic outcomes. Valproic acid also can cause significant weight gain and could require monitoring similar to what is done with to SGAs, although there has been less clinical and research attention to this mood stabilizer.
The American Diabetes Association et al12 have published guidelines on monitoring antipsychotic-induced obesity and diabetes, but adoption of these guidelines has been slow. Mackin et al13 found that providers are slow to recognize the elevated rate of obesity and dyslipidemia among psychiatric patients, possibly because of “an alarmingly poor rate of monitoring of metabolic parameters.”
Treating adverse metabolic outcomes also seems to lag behind. The same study13 found that physical health parameters among psychiatric patients continue to become worse even when appropriate health care professionals were notified. Rates of nontreatment for diabetes, dyslipidemia, and hypertension were 30%, 60%, and 88% respectively, according to Nasrallah et al.14
Randomized controlled studies have shown that obesity and metabolic syndrome can be effectively managed using lifestyle and pharmacotherapeutic approaches,15,16 but more research is needed to test long-term outcomes and how to best incorporate these interventions. Newcomer et al17 found that gradually switching an antipsychotic with high risk of metabolic adverse effects to one with lower risk could reduce adverse metabolic outcomes; however, some patients returned to their prior antipsychotic because other medications did not effectively treat their schizophrenia symptoms. Therefore, physicians must pay careful attention to the trade-off between benefits and risks of antipsychotics and make treatment decisions on an individual basis.
Barriers to medical care
Research has demonstrated that patients with SMI receive less screening and fewer preventive medical services, especially blood pressure monitoring, vaccinations, mammography, lipid monitoring, and osteoporosis screening, compared with the general population (Table).18 Some barriers to preventive services could exist because of demographic factors and medical insurance coverage19 or medical providers’ discomfort with symptoms of SMI,20 although Mitchell et al21 found that disparities in mammography screening could not be explained by the presence of emotional distress in women with SMI.

DiMatteo et al22 reported that patients with SMI are 3 times more likely to be noncompliant with medical treatment. These patients also are less likely to receive sec ondary preventive medical care and invasive medical procedures. Those with SMI who experience acute myocardial infarction are less likely to receive drug therapy, such as a thrombolytic, aspirin, beta blocker, or angiotensin-converting enzyme inhibitor.23 They also are less likely to receive invasive cardiovascular procedures, including cardiac catheterization, angioplasty, and coronary artery bypass grafting.24
Therefore, not only are patients with SMI less likely to receive preventive care, they are also less likely to receive potentially lifesaving treatments for SMI. Because those with SMI might not be able to advocate for themselves in these matters, psychiatric clinicians can improve their patients’ lives by advocating for appropriate medical care despite multiple barriers.
Bridging the gap: Managing mental health in primary care
Research from the 1970s and 1980s demonstrated that most persons who sought help for depression or anxiety received treatment from their PCP, many of whom felt limited by their lack of behavioral health training. Moreover, many patients failed to receive a psychiatric diagnosis or adequate treatment, despite efforts to educate primary care physicians on appropriate diagnosis and treatment of mental illness.
Katon et al25 at the University of Washington developed the collaborative care model in the early 1990s to help improve treatment of depression in primary care settings. This model involved:
• case load review by psychiatrists
• use of nurses and other support staff to help monitor patients’ adherence and treatment response
• use of standardized tools such as the Patient Health Questionnaire to monitor symptoms
• enhancement of patient education with pamphlets or classes.
Studies evaluating the success of collaborative care models found overall improved outcomes, making it the only evidence-based model for integration of behavioral health and primary care.26 As a result, the collaborative care model has been implemented across the United States in primary care clinics and specialty care settings, such as obstetrics and gynecology.27
Regrettably, access to primary care has been hampered by:
• population growth
• a shortage of PCPs
• enrollment of a flood of new patients into the health care marketplace as a result of mandates of the Affordable Care Act (ACA).
In many settings, a psychiatrist might be the patient’s only consistent care provider, and could be thought of as a “primary care psychiatrist.”
To resolve this predicament, mental health professionals need to recognize the unique medical conditions faced by people with SMI, and also might need to provide treatment of common medical conditions, either directly or through collaborative arrangements. Psychiatrists who are capable of managing core medical issues likely will witness improved psychiatric and overall health outcomes in their patients. Consequently, psychiatrists and mental health professionals are increasingly called on to be advocates to improve access to medical services in patients with SMI and to participate in health systems reform.
Managing medical conditions in mental health settings
Although traditional collaborative care involves mental health providers working at primary care sites, other models have emerged that manage chronic disease in behavioral health settings. Federally funded grants for primary behavioral health care integration have allowed community mental health centers to partner with federally qualified health centers to provide on-site primary care services.28
In these models, care managers in mental health clinics:
• link patients to primary care services
• encourage lifestyle changes to improve their overall health
• identify and overcome barriers to receiving care
• track clinical outcomes in a registry format.
Currently, 126 mental health sites in the United States have received these grants and are working toward greater integration of primary care.
In addition, the ACA provided funding for “health homes” in non-primary care settings, which includes SMI. These health homes cannot provide direct primary care, but can deliver comprehensive care management, care coordination, health promotion, comprehensive transitional care services between facilities, individual and family support, and referral to community social support services. In these health homes, a PCP can act as a consultant to help establish priorities for disease management and improving health status.29 The PCP consultant also can support psychiatric staff and collaborate with providers who want to provide some direct care of medical conditions.30
Last, some behavioral health sites are choosing to apply for Federally Qualified Health Clinic status or add primary care services to their clinics, with the hope that sustainable funding will become available. Without additional funding to cover the limited reimbursement provided by public payers, such as Medicaid and Medicare, these models might be unsustainable. Current innovations in health care funding reform hopefully will offer solutions for sites to provide medical care in the natural “medical home” of the SMI population.
Bottom Line
Psychiatric providers are in a favorable position to develop and oversee a partnership with primary care physicians with the goal of addressing significant and often lethal health disparities among those with mental illness. Psychiatric providers must use evidence-based practices that include assessment and prevention of cardiopulmonary, metabolic, infectious, and oncologic disorders. True primary care–behavioral health integration must include longitudinal “cross education” and changes in health care policy, with an emphasis on decreasing morbidity and mortality in psychiatric patients.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Colton CW, Manderscheid RW. Congruencies in increased mortality rates, years of potential life lost, and causes of death among public mental health clients in eight states. Prev Chronic Dis. 2006;3(2):A42.
2. Parks J, Svendsen D, Singer P, et al, eds. Morbidity and mortality in people with serious mental illness. Alexandria, VA: National Association of State Mental Health Program Directors (NASMHPD) Medical Directors Council; 2006.
3. Newcomer JW, Hennekens CH. Severe mental illness and risk of cardiovascular disease. JAMA. 2007;298(15):1794-1796.
4. McEvoy JP, Meyer JM, Goff DC, et al. Prevalence of the metabolic syndrome in patients with schizophrenia: baseline results from the Clinical Antipsychotic Trails of Intervention Effectiveness (CATIE) schizophrenia trial and comparison with national estimates from NHANES III. Schizophr Res. 2005;80(1):19-32.
5. Compton MT, Daumit GL, Druss BG. Cigarette smoking and overweight/obesity among individuals with serious mental illnesses: a preventive perspective. Harv Rev Psychiatry. 2006;14(2):212-222.
6. Saha S, Chant D, McGrath J. A systematic review of mortality in schizophrenia: is the differential mortality gap worsening over time? Arch Gen Psychiatry. 2007;64(10):1123-1131.
7. Roshanaei-Moghaddam B, Katon W. Premature mortality from general medical illnesses among persons with bipolar disorder: a review. Psychiatr Serv. 2009;60(2):147-156.
8. Carney CP, Jones L, Woolson RF. Medical comorbidity in women and men with schizophrenia: a population-based study. J Gen Intern Med. 2006;21(11):1133-1137.
9. Bobes J, Arango C, Aranda P, et al; CLAMORS Study Collaborative Group. Cardiovascular and metabolic risk in outpatients with schizoaffective disorder treated with antipsychotics; results from the CLAMORS study. Eur Psychiatry. 2012;27(4):267-274.
10. Osborn DP, Levy G, Nazareth I, et al. Relative risk of cardiovascular and cancer mortality in people with severe mental illness from the United Kingdom’s General Practice Research Database [Erratum in: Arch Gen Psychiatry. 2007;64(6):736]. Arch Gen Psychiatry. 2007;64(2):242-249.
11. Simon V, van Winkel R, De Hert M. Are weight gain and metabolic side effects of atypical antipsychotics dose dependent? A literature review. J Clin Psychiatry. 2009;70(7):1041-1050.
12. American Diabetes Association, American Psychiatric Association, American Association of Clinical Endocrinologists, et al. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601.
13. Mackin P, Bishop DR, Watkinson HM. A prospective study of monitoring practices for metabolic disease in antipsychotic-treated community psychiatric patients. BMC Psychiatry. 2007;7:28.
14. Nasrallah HA, Meyer JM, Goff DC, et al. Low rates of treatment for hypertension, dyslipidemia and diabetes in schizophrenia: data from the CATIE schizophrenia trial sample at baseline. Schizophr Res. 2006;86(1-3):15-22.
15. Alvarez-Jiménez M, Hetrick SE, González-Blanch C, et al. Non-pharmacological management of antipsychotic-induced weight gain: systematic review and meta-analysis of randomized controlled trials. Br J Psychiatry. 2008; 193(2):101-107.
16. Maayan L, Vakhrusheva J, Correll CU. Effectiveness of medication used to attenuate antipsychotic-related weight gain and metabolic abnormalities: a systematic review and meta-analysis. Neuropsychopharmacology. 2010;35(7):1520-1530.
17. Newcomer JW, Weiden PJ, Buchanan RW. Switching antipsychotic medications to reduce adverse event burden in schizophrenia: establishing evidence-based practice. J Clin Psychiatry. 2013;74(11):1108-1120.
18. Lord O, Malone D, Mitchell AJ. Receipt of preventive medical care and medical screening for patients with mental illness: a comparative analysis. Gen Hosp Psychiatry. 2010;32(5):519-543.
19. Xiong GL, Iosif AM, Bermudes RA, et al. Preventive medical services use among community mental health patients with severe mental illness: the influence of gender and insurance coverage. Prim Care Companion J Clin Psychiatry. 2010;12(5). doi: 10.4088/PCC.09m00927gre.
20. Daub S. Turning toward treating the seriously mentally ill in primary care. Fam Syst Health. 2014;32(1):12-13.
21. Mitchell A, Pereira IE, Yadegarfar M, et al. Breast cancer screening in women with mental illness: comparative meta-analysis of mammography uptake. Br J Psychiatry. 2014;205(6):428-435.
22. DiMatteo MR, Lepper HS, Croghan TW. Depression is a risk factor for noncompliance with medical treatment: meta-analysis of the effects of anxiety and depression on patient adherence. Arch Intern Med. 2000;160(14):2101-2107.
23. Druss BG, Bradford WD, Rosenheck RA, et al. Quality of medical care and excess mortality in older patients with mental disorders. Arch Gen Psychiatry. 2001;58(6):565-572.
24. Druss BG, Bradford DW, Rosenheck RA, et al. Mental disorders and use of cardiovascular procedures after myocardial infarction. JAMA. 2000;283(4):506-511.
25. Katon W, Unützer J, Wells K, et al. Collaborative depression care: history, evolution and ways to enhance dissemination and sustainability. Gen Hosp Psychiatry. 2010;32(5):456-464.
26. Archer J, Bower P, Gilbody S, et al. Collaborative care for depression and anxiety problems. Cochrane Database Syst Rev. 2012;10:CD006525.
27. Katon W, Russo J, Reed SD, et al. A randomized trial of collaborative depression care in obstetrics and gynecology clinics: socioeconomic disadvantage and treatment response. Am J Psychiatry. 2015;172(1):32-40.
28. Substance Abuse and Mental Health Services Administration. Request for Applications (RFA) No. SM- 09-011. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2009.
29. Parks J. Behavioral health homes. In: Integrated care: working at the interface of primary care and behavioral health. Raney LE, ed. Arlington, VA: American Psychiatric Publishing; 2015:195.
30. Raney L. Integrated care: the evolving role of psychiatry in the era of health care reform. Psychiatr Serv. 2013;64(11):1076-1078.
People with serious mental illness (SMI) have a life expectancy that is 25 years less than the general population, according to the Centers for Disease Control and Prevention.1 This disparity is partially a consequence of the lack of primary and preventive medical care for those with psychiatric illness. Decades of research have shown that people with SMI experience higher medical morbidity and mortality in addition to facing the stigma of mental illness.
This article aims to advance the idea that longitudinal “cross education” between primary care providers (PCPs) and behavioral health providers (BHPs) is essential in addressing this problem. BHPs include psychiatry clinics, which often are part of a university or large health systems; county-based community mental health programs; and independent mental health clinics that contract with public and private health plans to provide mental health services.
Although suicide and injury account for 40% of the excess mortality in schizophrenia, 60% can be attributed to cardiovascular disease, diabetes, respiratory diseases, and infection.2 Patients with SMI have 2 to 3 times the risk of diabetes, dyslipidemia, hypertension, and obesity.3,4 Furthermore, those with SMI consume more than one-third of tobacco products,5 and 50% to 80% of people with SMI smoke tobacco, an important reversible risk factor for cardiovascular disease.
Figure 1 shows that people with SMI are at higher risk of dying from a chronic medical condition, such as cardiovascular disease, diabetes, chronic obstructive pulmonary disease, and hepatitis C6-8—many of which can be managed by primary and preventive medical interventions. These and other conditions often are not diagnosed or effectively managed in patients with SMI.
The high prevalence of metabolic syndrome and tobacco dependence among people with SMI accelerates development of cardiovascular disease, as shown by several studies. Bobes et al9 found that the prevalence of metabolic syndrome and cardiovascular risk among patients with SMI is similar to what is found in the general population at 10 to 15 years of greater age. Osborn et al10 demonstrated that people with SMI age 18 to 49 had a higher relative risk of death from coronary heart disease, stroke, and lung cancer than age-matched controls (Figure 2).
It can be said, therefore, that patients with SMI seem to “age” and die prematurely. To reduce this disparity, primary and preventive medical care—especially for cardiovascular disease—must be delivered earlier in life for those with SMI.
Iatrogenic causes of morbidity
Many psychiatric medications, especially second-generation antipsychotics (SGAs), could exacerbate cardiovascular and metabolic conditions by increasing the risk of weight gain, insulin resistance, and dyslipidemia. Antipsychotics that generally are considered to be more effective for refractory psychotic illness (eg, clozapine and olanzapine) are associated with the highest risk of metabolic syndrome. Simon et al11 found a dose-response relationship between olanzapine and clozapine serum concentrations and worsening metabolic outcomes. Valproic acid also can cause significant weight gain and could require monitoring similar to what is done with to SGAs, although there has been less clinical and research attention to this mood stabilizer.
The American Diabetes Association et al12 have published guidelines on monitoring antipsychotic-induced obesity and diabetes, but adoption of these guidelines has been slow. Mackin et al13 found that providers are slow to recognize the elevated rate of obesity and dyslipidemia among psychiatric patients, possibly because of “an alarmingly poor rate of monitoring of metabolic parameters.”
Treating adverse metabolic outcomes also seems to lag behind. The same study13 found that physical health parameters among psychiatric patients continue to become worse even when appropriate health care professionals were notified. Rates of nontreatment for diabetes, dyslipidemia, and hypertension were 30%, 60%, and 88% respectively, according to Nasrallah et al.14
Randomized controlled studies have shown that obesity and metabolic syndrome can be effectively managed using lifestyle and pharmacotherapeutic approaches,15,16 but more research is needed to test long-term outcomes and how to best incorporate these interventions. Newcomer et al17 found that gradually switching an antipsychotic with high risk of metabolic adverse effects to one with lower risk could reduce adverse metabolic outcomes; however, some patients returned to their prior antipsychotic because other medications did not effectively treat their schizophrenia symptoms. Therefore, physicians must pay careful attention to the trade-off between benefits and risks of antipsychotics and make treatment decisions on an individual basis.
Barriers to medical care
Research has demonstrated that patients with SMI receive less screening and fewer preventive medical services, especially blood pressure monitoring, vaccinations, mammography, lipid monitoring, and osteoporosis screening, compared with the general population (Table).18 Some barriers to preventive services could exist because of demographic factors and medical insurance coverage19 or medical providers’ discomfort with symptoms of SMI,20 although Mitchell et al21 found that disparities in mammography screening could not be explained by the presence of emotional distress in women with SMI.
DiMatteo et al22 reported that patients with SMI are 3 times more likely to be noncompliant with medical treatment. These patients also are less likely to receive sec ondary preventive medical care and invasive medical procedures. Those with SMI who experience acute myocardial infarction are less likely to receive drug therapy, such as a thrombolytic, aspirin, beta blocker, or angiotensin-converting enzyme inhibitor.23 They also are less likely to receive invasive cardiovascular procedures, including cardiac catheterization, angioplasty, and coronary artery bypass grafting.24
Therefore, not only are patients with SMI less likely to receive preventive care, they are also less likely to receive potentially lifesaving treatments for SMI. Because those with SMI might not be able to advocate for themselves in these matters, psychiatric clinicians can improve their patients’ lives by advocating for appropriate medical care despite multiple barriers.
Bridging the gap: Managing mental health in primary care
Research from the 1970s and 1980s demonstrated that most persons who sought help for depression or anxiety received treatment from their PCP, many of whom felt limited by their lack of behavioral health training. Moreover, many patients failed to receive a psychiatric diagnosis or adequate treatment, despite efforts to educate primary care physicians on appropriate diagnosis and treatment of mental illness.
Katon et al25 at the University of Washington developed the collaborative care model in the early 1990s to help improve treatment of depression in primary care settings. This model involved:
• case load review by psychiatrists
• use of nurses and other support staff to help monitor patients’ adherence and treatment response
• use of standardized tools such as the Patient Health Questionnaire to monitor symptoms
• enhancement of patient education with pamphlets or classes.
Studies evaluating the success of collaborative care models found overall improved outcomes, making it the only evidence-based model for integration of behavioral health and primary care.26 As a result, the collaborative care model has been implemented across the United States in primary care clinics and specialty care settings, such as obstetrics and gynecology.27
Regrettably, access to primary care has been hampered by:
• population growth
• a shortage of PCPs
• enrollment of a flood of new patients into the health care marketplace as a result of mandates of the Affordable Care Act (ACA).
In many settings, a psychiatrist might be the patient’s only consistent care provider, and could be thought of as a “primary care psychiatrist.”
To resolve this predicament, mental health professionals need to recognize the unique medical conditions faced by people with SMI, and also might need to provide treatment of common medical conditions, either directly or through collaborative arrangements. Psychiatrists who are capable of managing core medical issues likely will witness improved psychiatric and overall health outcomes in their patients. Consequently, psychiatrists and mental health professionals are increasingly called on to be advocates to improve access to medical services in patients with SMI and to participate in health systems reform.
Managing medical conditions in mental health settings
Although traditional collaborative care involves mental health providers working at primary care sites, other models have emerged that manage chronic disease in behavioral health settings. Federally funded grants for primary behavioral health care integration have allowed community mental health centers to partner with federally qualified health centers to provide on-site primary care services.28
In these models, care managers in mental health clinics:
• link patients to primary care services
• encourage lifestyle changes to improve their overall health
• identify and overcome barriers to receiving care
• track clinical outcomes in a registry format.
Currently, 126 mental health sites in the United States have received these grants and are working toward greater integration of primary care.
In addition, the ACA provided funding for “health homes” in non-primary care settings, which includes SMI. These health homes cannot provide direct primary care, but can deliver comprehensive care management, care coordination, health promotion, comprehensive transitional care services between facilities, individual and family support, and referral to community social support services. In these health homes, a PCP can act as a consultant to help establish priorities for disease management and improving health status.29 The PCP consultant also can support psychiatric staff and collaborate with providers who want to provide some direct care of medical conditions.30
Last, some behavioral health sites are choosing to apply for Federally Qualified Health Clinic status or add primary care services to their clinics, with the hope that sustainable funding will become available. Without additional funding to cover the limited reimbursement provided by public payers, such as Medicaid and Medicare, these models might be unsustainable. Current innovations in health care funding reform hopefully will offer solutions for sites to provide medical care in the natural “medical home” of the SMI population.
Bottom Line
Psychiatric providers are in a favorable position to develop and oversee a partnership with primary care physicians with the goal of addressing significant and often lethal health disparities among those with mental illness. Psychiatric providers must use evidence-based practices that include assessment and prevention of cardiopulmonary, metabolic, infectious, and oncologic disorders. True primary care–behavioral health integration must include longitudinal “cross education” and changes in health care policy, with an emphasis on decreasing morbidity and mortality in psychiatric patients.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
People with serious mental illness (SMI) have a life expectancy that is 25 years less than the general population, according to the Centers for Disease Control and Prevention.1 This disparity is partially a consequence of the lack of primary and preventive medical care for those with psychiatric illness. Decades of research have shown that people with SMI experience higher medical morbidity and mortality in addition to facing the stigma of mental illness.
This article aims to advance the idea that longitudinal “cross education” between primary care providers (PCPs) and behavioral health providers (BHPs) is essential in addressing this problem. BHPs include psychiatry clinics, which often are part of a university or large health systems; county-based community mental health programs; and independent mental health clinics that contract with public and private health plans to provide mental health services.
Although suicide and injury account for 40% of the excess mortality in schizophrenia, 60% can be attributed to cardiovascular disease, diabetes, respiratory diseases, and infection.2 Patients with SMI have 2 to 3 times the risk of diabetes, dyslipidemia, hypertension, and obesity.3,4 Furthermore, those with SMI consume more than one-third of tobacco products,5 and 50% to 80% of people with SMI smoke tobacco, an important reversible risk factor for cardiovascular disease.
Figure 1 shows that people with SMI are at higher risk of dying from a chronic medical condition, such as cardiovascular disease, diabetes, chronic obstructive pulmonary disease, and hepatitis C6-8—many of which can be managed by primary and preventive medical interventions. These and other conditions often are not diagnosed or effectively managed in patients with SMI.
The high prevalence of metabolic syndrome and tobacco dependence among people with SMI accelerates development of cardiovascular disease, as shown by several studies. Bobes et al9 found that the prevalence of metabolic syndrome and cardiovascular risk among patients with SMI is similar to what is found in the general population at 10 to 15 years of greater age. Osborn et al10 demonstrated that people with SMI age 18 to 49 had a higher relative risk of death from coronary heart disease, stroke, and lung cancer than age-matched controls (Figure 2).
It can be said, therefore, that patients with SMI seem to “age” and die prematurely. To reduce this disparity, primary and preventive medical care—especially for cardiovascular disease—must be delivered earlier in life for those with SMI.
Iatrogenic causes of morbidity
Many psychiatric medications, especially second-generation antipsychotics (SGAs), could exacerbate cardiovascular and metabolic conditions by increasing the risk of weight gain, insulin resistance, and dyslipidemia. Antipsychotics that generally are considered to be more effective for refractory psychotic illness (eg, clozapine and olanzapine) are associated with the highest risk of metabolic syndrome. Simon et al11 found a dose-response relationship between olanzapine and clozapine serum concentrations and worsening metabolic outcomes. Valproic acid also can cause significant weight gain and could require monitoring similar to what is done with to SGAs, although there has been less clinical and research attention to this mood stabilizer.
The American Diabetes Association et al12 have published guidelines on monitoring antipsychotic-induced obesity and diabetes, but adoption of these guidelines has been slow. Mackin et al13 found that providers are slow to recognize the elevated rate of obesity and dyslipidemia among psychiatric patients, possibly because of “an alarmingly poor rate of monitoring of metabolic parameters.”
Treating adverse metabolic outcomes also seems to lag behind. The same study13 found that physical health parameters among psychiatric patients continue to become worse even when appropriate health care professionals were notified. Rates of nontreatment for diabetes, dyslipidemia, and hypertension were 30%, 60%, and 88% respectively, according to Nasrallah et al.14
Randomized controlled studies have shown that obesity and metabolic syndrome can be effectively managed using lifestyle and pharmacotherapeutic approaches,15,16 but more research is needed to test long-term outcomes and how to best incorporate these interventions. Newcomer et al17 found that gradually switching an antipsychotic with high risk of metabolic adverse effects to one with lower risk could reduce adverse metabolic outcomes; however, some patients returned to their prior antipsychotic because other medications did not effectively treat their schizophrenia symptoms. Therefore, physicians must pay careful attention to the trade-off between benefits and risks of antipsychotics and make treatment decisions on an individual basis.
Barriers to medical care
Research has demonstrated that patients with SMI receive less screening and fewer preventive medical services, especially blood pressure monitoring, vaccinations, mammography, lipid monitoring, and osteoporosis screening, compared with the general population (Table).18 Some barriers to preventive services could exist because of demographic factors and medical insurance coverage19 or medical providers’ discomfort with symptoms of SMI,20 although Mitchell et al21 found that disparities in mammography screening could not be explained by the presence of emotional distress in women with SMI.
DiMatteo et al22 reported that patients with SMI are 3 times more likely to be noncompliant with medical treatment. These patients also are less likely to receive sec ondary preventive medical care and invasive medical procedures. Those with SMI who experience acute myocardial infarction are less likely to receive drug therapy, such as a thrombolytic, aspirin, beta blocker, or angiotensin-converting enzyme inhibitor.23 They also are less likely to receive invasive cardiovascular procedures, including cardiac catheterization, angioplasty, and coronary artery bypass grafting.24
Therefore, not only are patients with SMI less likely to receive preventive care, they are also less likely to receive potentially lifesaving treatments for SMI. Because those with SMI might not be able to advocate for themselves in these matters, psychiatric clinicians can improve their patients’ lives by advocating for appropriate medical care despite multiple barriers.
Bridging the gap: Managing mental health in primary care
Research from the 1970s and 1980s demonstrated that most persons who sought help for depression or anxiety received treatment from their PCP, many of whom felt limited by their lack of behavioral health training. Moreover, many patients failed to receive a psychiatric diagnosis or adequate treatment, despite efforts to educate primary care physicians on appropriate diagnosis and treatment of mental illness.
Katon et al25 at the University of Washington developed the collaborative care model in the early 1990s to help improve treatment of depression in primary care settings. This model involved:
• case load review by psychiatrists
• use of nurses and other support staff to help monitor patients’ adherence and treatment response
• use of standardized tools such as the Patient Health Questionnaire to monitor symptoms
• enhancement of patient education with pamphlets or classes.
Studies evaluating the success of collaborative care models found overall improved outcomes, making it the only evidence-based model for integration of behavioral health and primary care.26 As a result, the collaborative care model has been implemented across the United States in primary care clinics and specialty care settings, such as obstetrics and gynecology.27
Regrettably, access to primary care has been hampered by:
• population growth
• a shortage of PCPs
• enrollment of a flood of new patients into the health care marketplace as a result of mandates of the Affordable Care Act (ACA).
In many settings, a psychiatrist might be the patient’s only consistent care provider, and could be thought of as a “primary care psychiatrist.”
To resolve this predicament, mental health professionals need to recognize the unique medical conditions faced by people with SMI, and also might need to provide treatment of common medical conditions, either directly or through collaborative arrangements. Psychiatrists who are capable of managing core medical issues likely will witness improved psychiatric and overall health outcomes in their patients. Consequently, psychiatrists and mental health professionals are increasingly called on to be advocates to improve access to medical services in patients with SMI and to participate in health systems reform.
Managing medical conditions in mental health settings
Although traditional collaborative care involves mental health providers working at primary care sites, other models have emerged that manage chronic disease in behavioral health settings. Federally funded grants for primary behavioral health care integration have allowed community mental health centers to partner with federally qualified health centers to provide on-site primary care services.28
In these models, care managers in mental health clinics:
• link patients to primary care services
• encourage lifestyle changes to improve their overall health
• identify and overcome barriers to receiving care
• track clinical outcomes in a registry format.
Currently, 126 mental health sites in the United States have received these grants and are working toward greater integration of primary care.
In addition, the ACA provided funding for “health homes” in non-primary care settings, which includes SMI. These health homes cannot provide direct primary care, but can deliver comprehensive care management, care coordination, health promotion, comprehensive transitional care services between facilities, individual and family support, and referral to community social support services. In these health homes, a PCP can act as a consultant to help establish priorities for disease management and improving health status.29 The PCP consultant also can support psychiatric staff and collaborate with providers who want to provide some direct care of medical conditions.30
Last, some behavioral health sites are choosing to apply for Federally Qualified Health Clinic status or add primary care services to their clinics, with the hope that sustainable funding will become available. Without additional funding to cover the limited reimbursement provided by public payers, such as Medicaid and Medicare, these models might be unsustainable. Current innovations in health care funding reform hopefully will offer solutions for sites to provide medical care in the natural “medical home” of the SMI population.
Bottom Line
Psychiatric providers are in a favorable position to develop and oversee a partnership with primary care physicians with the goal of addressing significant and often lethal health disparities among those with mental illness. Psychiatric providers must use evidence-based practices that include assessment and prevention of cardiopulmonary, metabolic, infectious, and oncologic disorders. True primary care–behavioral health integration must include longitudinal “cross education” and changes in health care policy, with an emphasis on decreasing morbidity and mortality in psychiatric patients.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Colton CW, Manderscheid RW. Congruencies in increased mortality rates, years of potential life lost, and causes of death among public mental health clients in eight states. Prev Chronic Dis. 2006;3(2):A42.
2. Parks J, Svendsen D, Singer P, et al, eds. Morbidity and mortality in people with serious mental illness. Alexandria, VA: National Association of State Mental Health Program Directors (NASMHPD) Medical Directors Council; 2006.
3. Newcomer JW, Hennekens CH. Severe mental illness and risk of cardiovascular disease. JAMA. 2007;298(15):1794-1796.
4. McEvoy JP, Meyer JM, Goff DC, et al. Prevalence of the metabolic syndrome in patients with schizophrenia: baseline results from the Clinical Antipsychotic Trails of Intervention Effectiveness (CATIE) schizophrenia trial and comparison with national estimates from NHANES III. Schizophr Res. 2005;80(1):19-32.
5. Compton MT, Daumit GL, Druss BG. Cigarette smoking and overweight/obesity among individuals with serious mental illnesses: a preventive perspective. Harv Rev Psychiatry. 2006;14(2):212-222.
6. Saha S, Chant D, McGrath J. A systematic review of mortality in schizophrenia: is the differential mortality gap worsening over time? Arch Gen Psychiatry. 2007;64(10):1123-1131.
7. Roshanaei-Moghaddam B, Katon W. Premature mortality from general medical illnesses among persons with bipolar disorder: a review. Psychiatr Serv. 2009;60(2):147-156.
8. Carney CP, Jones L, Woolson RF. Medical comorbidity in women and men with schizophrenia: a population-based study. J Gen Intern Med. 2006;21(11):1133-1137.
9. Bobes J, Arango C, Aranda P, et al; CLAMORS Study Collaborative Group. Cardiovascular and metabolic risk in outpatients with schizoaffective disorder treated with antipsychotics; results from the CLAMORS study. Eur Psychiatry. 2012;27(4):267-274.
10. Osborn DP, Levy G, Nazareth I, et al. Relative risk of cardiovascular and cancer mortality in people with severe mental illness from the United Kingdom’s General Practice Research Database [Erratum in: Arch Gen Psychiatry. 2007;64(6):736]. Arch Gen Psychiatry. 2007;64(2):242-249.
11. Simon V, van Winkel R, De Hert M. Are weight gain and metabolic side effects of atypical antipsychotics dose dependent? A literature review. J Clin Psychiatry. 2009;70(7):1041-1050.
12. American Diabetes Association, American Psychiatric Association, American Association of Clinical Endocrinologists, et al. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601.
13. Mackin P, Bishop DR, Watkinson HM. A prospective study of monitoring practices for metabolic disease in antipsychotic-treated community psychiatric patients. BMC Psychiatry. 2007;7:28.
14. Nasrallah HA, Meyer JM, Goff DC, et al. Low rates of treatment for hypertension, dyslipidemia and diabetes in schizophrenia: data from the CATIE schizophrenia trial sample at baseline. Schizophr Res. 2006;86(1-3):15-22.
15. Alvarez-Jiménez M, Hetrick SE, González-Blanch C, et al. Non-pharmacological management of antipsychotic-induced weight gain: systematic review and meta-analysis of randomized controlled trials. Br J Psychiatry. 2008; 193(2):101-107.
16. Maayan L, Vakhrusheva J, Correll CU. Effectiveness of medication used to attenuate antipsychotic-related weight gain and metabolic abnormalities: a systematic review and meta-analysis. Neuropsychopharmacology. 2010;35(7):1520-1530.
17. Newcomer JW, Weiden PJ, Buchanan RW. Switching antipsychotic medications to reduce adverse event burden in schizophrenia: establishing evidence-based practice. J Clin Psychiatry. 2013;74(11):1108-1120.
18. Lord O, Malone D, Mitchell AJ. Receipt of preventive medical care and medical screening for patients with mental illness: a comparative analysis. Gen Hosp Psychiatry. 2010;32(5):519-543.
19. Xiong GL, Iosif AM, Bermudes RA, et al. Preventive medical services use among community mental health patients with severe mental illness: the influence of gender and insurance coverage. Prim Care Companion J Clin Psychiatry. 2010;12(5). doi: 10.4088/PCC.09m00927gre.
20. Daub S. Turning toward treating the seriously mentally ill in primary care. Fam Syst Health. 2014;32(1):12-13.
21. Mitchell A, Pereira IE, Yadegarfar M, et al. Breast cancer screening in women with mental illness: comparative meta-analysis of mammography uptake. Br J Psychiatry. 2014;205(6):428-435.
22. DiMatteo MR, Lepper HS, Croghan TW. Depression is a risk factor for noncompliance with medical treatment: meta-analysis of the effects of anxiety and depression on patient adherence. Arch Intern Med. 2000;160(14):2101-2107.
23. Druss BG, Bradford WD, Rosenheck RA, et al. Quality of medical care and excess mortality in older patients with mental disorders. Arch Gen Psychiatry. 2001;58(6):565-572.
24. Druss BG, Bradford DW, Rosenheck RA, et al. Mental disorders and use of cardiovascular procedures after myocardial infarction. JAMA. 2000;283(4):506-511.
25. Katon W, Unützer J, Wells K, et al. Collaborative depression care: history, evolution and ways to enhance dissemination and sustainability. Gen Hosp Psychiatry. 2010;32(5):456-464.
26. Archer J, Bower P, Gilbody S, et al. Collaborative care for depression and anxiety problems. Cochrane Database Syst Rev. 2012;10:CD006525.
27. Katon W, Russo J, Reed SD, et al. A randomized trial of collaborative depression care in obstetrics and gynecology clinics: socioeconomic disadvantage and treatment response. Am J Psychiatry. 2015;172(1):32-40.
28. Substance Abuse and Mental Health Services Administration. Request for Applications (RFA) No. SM- 09-011. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2009.
29. Parks J. Behavioral health homes. In: Integrated care: working at the interface of primary care and behavioral health. Raney LE, ed. Arlington, VA: American Psychiatric Publishing; 2015:195.
30. Raney L. Integrated care: the evolving role of psychiatry in the era of health care reform. Psychiatr Serv. 2013;64(11):1076-1078.
1. Colton CW, Manderscheid RW. Congruencies in increased mortality rates, years of potential life lost, and causes of death among public mental health clients in eight states. Prev Chronic Dis. 2006;3(2):A42.
2. Parks J, Svendsen D, Singer P, et al, eds. Morbidity and mortality in people with serious mental illness. Alexandria, VA: National Association of State Mental Health Program Directors (NASMHPD) Medical Directors Council; 2006.
3. Newcomer JW, Hennekens CH. Severe mental illness and risk of cardiovascular disease. JAMA. 2007;298(15):1794-1796.
4. McEvoy JP, Meyer JM, Goff DC, et al. Prevalence of the metabolic syndrome in patients with schizophrenia: baseline results from the Clinical Antipsychotic Trails of Intervention Effectiveness (CATIE) schizophrenia trial and comparison with national estimates from NHANES III. Schizophr Res. 2005;80(1):19-32.
5. Compton MT, Daumit GL, Druss BG. Cigarette smoking and overweight/obesity among individuals with serious mental illnesses: a preventive perspective. Harv Rev Psychiatry. 2006;14(2):212-222.
6. Saha S, Chant D, McGrath J. A systematic review of mortality in schizophrenia: is the differential mortality gap worsening over time? Arch Gen Psychiatry. 2007;64(10):1123-1131.
7. Roshanaei-Moghaddam B, Katon W. Premature mortality from general medical illnesses among persons with bipolar disorder: a review. Psychiatr Serv. 2009;60(2):147-156.
8. Carney CP, Jones L, Woolson RF. Medical comorbidity in women and men with schizophrenia: a population-based study. J Gen Intern Med. 2006;21(11):1133-1137.
9. Bobes J, Arango C, Aranda P, et al; CLAMORS Study Collaborative Group. Cardiovascular and metabolic risk in outpatients with schizoaffective disorder treated with antipsychotics; results from the CLAMORS study. Eur Psychiatry. 2012;27(4):267-274.
10. Osborn DP, Levy G, Nazareth I, et al. Relative risk of cardiovascular and cancer mortality in people with severe mental illness from the United Kingdom’s General Practice Research Database [Erratum in: Arch Gen Psychiatry. 2007;64(6):736]. Arch Gen Psychiatry. 2007;64(2):242-249.
11. Simon V, van Winkel R, De Hert M. Are weight gain and metabolic side effects of atypical antipsychotics dose dependent? A literature review. J Clin Psychiatry. 2009;70(7):1041-1050.
12. American Diabetes Association, American Psychiatric Association, American Association of Clinical Endocrinologists, et al. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601.
13. Mackin P, Bishop DR, Watkinson HM. A prospective study of monitoring practices for metabolic disease in antipsychotic-treated community psychiatric patients. BMC Psychiatry. 2007;7:28.
14. Nasrallah HA, Meyer JM, Goff DC, et al. Low rates of treatment for hypertension, dyslipidemia and diabetes in schizophrenia: data from the CATIE schizophrenia trial sample at baseline. Schizophr Res. 2006;86(1-3):15-22.
15. Alvarez-Jiménez M, Hetrick SE, González-Blanch C, et al. Non-pharmacological management of antipsychotic-induced weight gain: systematic review and meta-analysis of randomized controlled trials. Br J Psychiatry. 2008; 193(2):101-107.
16. Maayan L, Vakhrusheva J, Correll CU. Effectiveness of medication used to attenuate antipsychotic-related weight gain and metabolic abnormalities: a systematic review and meta-analysis. Neuropsychopharmacology. 2010;35(7):1520-1530.
17. Newcomer JW, Weiden PJ, Buchanan RW. Switching antipsychotic medications to reduce adverse event burden in schizophrenia: establishing evidence-based practice. J Clin Psychiatry. 2013;74(11):1108-1120.
18. Lord O, Malone D, Mitchell AJ. Receipt of preventive medical care and medical screening for patients with mental illness: a comparative analysis. Gen Hosp Psychiatry. 2010;32(5):519-543.
19. Xiong GL, Iosif AM, Bermudes RA, et al. Preventive medical services use among community mental health patients with severe mental illness: the influence of gender and insurance coverage. Prim Care Companion J Clin Psychiatry. 2010;12(5). doi: 10.4088/PCC.09m00927gre.
20. Daub S. Turning toward treating the seriously mentally ill in primary care. Fam Syst Health. 2014;32(1):12-13.
21. Mitchell A, Pereira IE, Yadegarfar M, et al. Breast cancer screening in women with mental illness: comparative meta-analysis of mammography uptake. Br J Psychiatry. 2014;205(6):428-435.
22. DiMatteo MR, Lepper HS, Croghan TW. Depression is a risk factor for noncompliance with medical treatment: meta-analysis of the effects of anxiety and depression on patient adherence. Arch Intern Med. 2000;160(14):2101-2107.
23. Druss BG, Bradford WD, Rosenheck RA, et al. Quality of medical care and excess mortality in older patients with mental disorders. Arch Gen Psychiatry. 2001;58(6):565-572.
24. Druss BG, Bradford DW, Rosenheck RA, et al. Mental disorders and use of cardiovascular procedures after myocardial infarction. JAMA. 2000;283(4):506-511.
25. Katon W, Unützer J, Wells K, et al. Collaborative depression care: history, evolution and ways to enhance dissemination and sustainability. Gen Hosp Psychiatry. 2010;32(5):456-464.
26. Archer J, Bower P, Gilbody S, et al. Collaborative care for depression and anxiety problems. Cochrane Database Syst Rev. 2012;10:CD006525.
27. Katon W, Russo J, Reed SD, et al. A randomized trial of collaborative depression care in obstetrics and gynecology clinics: socioeconomic disadvantage and treatment response. Am J Psychiatry. 2015;172(1):32-40.
28. Substance Abuse and Mental Health Services Administration. Request for Applications (RFA) No. SM- 09-011. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2009.
29. Parks J. Behavioral health homes. In: Integrated care: working at the interface of primary care and behavioral health. Raney LE, ed. Arlington, VA: American Psychiatric Publishing; 2015:195.
30. Raney L. Integrated care: the evolving role of psychiatry in the era of health care reform. Psychiatr Serv. 2013;64(11):1076-1078.

Is there only 1 neurobiologic psychiatric disorder, with different clinical expressions?
In a report of a study that was published recently in a top-tier psychiatry journal,1 researchers describe a stunning finding that challenges the notion that there is a plethora of psychiatric brain disorders. They conducted a large meta-analysis of 193 published brain imaging studies of people with schizophrenia, bipolar disorder, major depression, obsessive-compulsive disorder (OCD), anxiety, and addiction. They found that those 6 supposedly discrete illnesses are all associated with a varying degree of shrinkage (atrophy or hypoplasia) of the same 3 brain regions:
• Dorsal anterior cingulate cortex. This region around the frontal part of the corpus callosum controls rational cognitive processes, reward anticipation, decision making, empathy, impulse control, and emotional response. Francis Crick, the Nobel laureate who first described the structure of DNA, hypothesized that the anterior cingulate sulcus might even be the center of what we call “free will.”
• Left insula and right insula. The insulae are the cortical regions deep inside the lateral sulcus, which is the fissure that separates the temporal lobe from the parietal and frontal lobes. The functions of the insulae include consciousness, emotions, perceptions, motor control, self-awareness, cognitive functioning, and interpersonal experience. (In addicts, the insular cortex is activated when they are exposed to environmental cues that trigger craving because the insulae are a target for the dopamine system. Notably, it has been reported that, when cigarette addicts suffer a stroke that damages the insulae, they stop smoking completely.)
The 3 regions of the brain, in which pathology extends across 6 DSM-5 diagnoses, work together to manage high-level executive functions, such as working memory, reasoning, and flexible thinking. The degree of dysfunction varies among the 6 clinical disorders, with schizophrenia having the highest severity.
Neurobiological commonality
The idea of shared neurobiological underpinnings among 6 distinct psychiatric disorders flies in the face of the entrenched DSM model, in which those 6 disorders are distinct disease entities. Other studies (including the Bipolar Schizophrenia Network on Intermediate Phenotypes) also found prominent biological similarities in varying degrees across schizophrenia, schizoaffective disorder, and bipolar disorder.2,3 The Research Domain Criteria of the National Institute of Mental Health also embraces the dimensional approach to neurobiological biomarkers across various psychiatric disorders.
A common genetic substrate also is emerging. A recently published genome-wide association study, conducted on 33,332 psychiatric patients and 27,888 controls,4 revealed that a number of genes are shared by 5 different psychiatric disorders: schizophrenia, autism, bipolar disorder, major depression, and attention-deficit/hyperactivity disorder. The genetic phenomenon of the same genes manifesting in different clinical phenotypes is called pleiotropy, and is consistent with shared neurobiological findings.
Genetic and brain structural commonalities among multiple DSM diagnostic categories might explain some well-known clinical observations:
• frequent comorbidity of certain psychiatric disorders, such as depression and addiction in schizophrenia; anxiety and OCD in bipolar disorder; depression with OCD and addictions; and so on
• the presence of intermediate phenotypes in unaffected family members, such as cognitive dysfunction in the parents of patients with schizophrenia, compared with parents of matched healthy controls5
• the much higher rate of psychopathology among family members of patients with a major psychiatric disorder, compared with the general population.6
Core inflexible thinking. So what about clinical features across those disorders that share genetic and neurobiologic similarities? Psychiatrists may agree that symptoms of schizophrenia, bipolar disorder, major depression, OCD, anxiety, and addiction appear very different. However, given that reasoning and flexible thinking are functions of the insulae, which are shrunken in all 6 disorders, one can postulate that inflexible thinking (fixed false beliefs also are called psychotic delusions) might be a common feature across all those disorders. Namely:
• schizophrenia is known for paranoid or implausible delusions
• bipolar disorder is characterized by grandiose delusions
• major depressive disorder is associated with a fixed false belief of worthlessness as well as hopelessness
• anxiety patients harbor the fixed false belief of impending doom or death (the plane will crash if they are a passenger on it)
• OCD manifests as ego-dystonic false beliefs (obsessions) that can progress into ego-syntonic delusions
• people with an alcohol or tobacco addiction are in delusional denial that they are not really addicted or that they will not be harmed by their drug of abuse. Pathologic gamblers harbor the false belief that they will soon reverse their fortunes and “win big.”
It seems that poor reality testing and impaired reasoning is a common feature of not only all 6 psychiatric disorders with shared neurobiology, but others, too, including anorexia nervosa, body dysmorphic disorder, delirium, and dementia.
From a thick volume to… a booklet?
Can you envision a day when psychiatric disorders are conceptualized as having a common genetic, neurobiological, and clinical core, with some variability in phenotype and behavior? If further brain research steers psychiatric nosology in that direction, we might end up with a DSM of 10 pages instead of almost 1,000, with an “Appendix” of genetic, neuroimaging, and other emerging biomarkers.
Bold scientific prophecies often sound delusional—until they come true….
1. Goodkind M, Eickhoff SB, Oathes DJ, et al. Identification of a common neurobiological substrate for mental illness. JAMA Psychiatry. 2015;72(4):305-315.
2. Hill SK, Reilly JL, Keefe RS, et al. Neuropsychological impairments in schizophrenia and psychotic bipolar disorder: findings from the Bipolar- Schizophrenia Network on Intermediate Phenotypes (B-SNIP) study. Am J Psychiatry. 2013;170(11):1275-1284.
3. Skudlarski P, Scretlen DJ, Thaker GK, et al. Diffusion tensor imaging white matter endophenotypes in patients with schizophrenia or psychotic bipolar disorder and their relatives. Am J Psychiatry. 2013;170(8):886-898.
4. Cross-Disorder Group of Psychiatric Genomics Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381(9875):1371-1379.
5. Appels MC, Sitskoorn MM, Westers P, et al. Cognitive dysfunction in parents of schizophrenia patients parallel the deficits found in patients. Schizophrenia Res. 2003;63(3):285-293.
6. Braff DL. The importance of endophenotypes in schizophrenia research. Schizophrenia Res. 2015;163(1-3):1-8.
In a report of a study that was published recently in a top-tier psychiatry journal,1 researchers describe a stunning finding that challenges the notion that there is a plethora of psychiatric brain disorders. They conducted a large meta-analysis of 193 published brain imaging studies of people with schizophrenia, bipolar disorder, major depression, obsessive-compulsive disorder (OCD), anxiety, and addiction. They found that those 6 supposedly discrete illnesses are all associated with a varying degree of shrinkage (atrophy or hypoplasia) of the same 3 brain regions:
• Dorsal anterior cingulate cortex. This region around the frontal part of the corpus callosum controls rational cognitive processes, reward anticipation, decision making, empathy, impulse control, and emotional response. Francis Crick, the Nobel laureate who first described the structure of DNA, hypothesized that the anterior cingulate sulcus might even be the center of what we call “free will.”
• Left insula and right insula. The insulae are the cortical regions deep inside the lateral sulcus, which is the fissure that separates the temporal lobe from the parietal and frontal lobes. The functions of the insulae include consciousness, emotions, perceptions, motor control, self-awareness, cognitive functioning, and interpersonal experience. (In addicts, the insular cortex is activated when they are exposed to environmental cues that trigger craving because the insulae are a target for the dopamine system. Notably, it has been reported that, when cigarette addicts suffer a stroke that damages the insulae, they stop smoking completely.)
The 3 regions of the brain, in which pathology extends across 6 DSM-5 diagnoses, work together to manage high-level executive functions, such as working memory, reasoning, and flexible thinking. The degree of dysfunction varies among the 6 clinical disorders, with schizophrenia having the highest severity.
Neurobiological commonality
The idea of shared neurobiological underpinnings among 6 distinct psychiatric disorders flies in the face of the entrenched DSM model, in which those 6 disorders are distinct disease entities. Other studies (including the Bipolar Schizophrenia Network on Intermediate Phenotypes) also found prominent biological similarities in varying degrees across schizophrenia, schizoaffective disorder, and bipolar disorder.2,3 The Research Domain Criteria of the National Institute of Mental Health also embraces the dimensional approach to neurobiological biomarkers across various psychiatric disorders.
A common genetic substrate also is emerging. A recently published genome-wide association study, conducted on 33,332 psychiatric patients and 27,888 controls,4 revealed that a number of genes are shared by 5 different psychiatric disorders: schizophrenia, autism, bipolar disorder, major depression, and attention-deficit/hyperactivity disorder. The genetic phenomenon of the same genes manifesting in different clinical phenotypes is called pleiotropy, and is consistent with shared neurobiological findings.
Genetic and brain structural commonalities among multiple DSM diagnostic categories might explain some well-known clinical observations:
• frequent comorbidity of certain psychiatric disorders, such as depression and addiction in schizophrenia; anxiety and OCD in bipolar disorder; depression with OCD and addictions; and so on
• the presence of intermediate phenotypes in unaffected family members, such as cognitive dysfunction in the parents of patients with schizophrenia, compared with parents of matched healthy controls5
• the much higher rate of psychopathology among family members of patients with a major psychiatric disorder, compared with the general population.6
Core inflexible thinking. So what about clinical features across those disorders that share genetic and neurobiologic similarities? Psychiatrists may agree that symptoms of schizophrenia, bipolar disorder, major depression, OCD, anxiety, and addiction appear very different. However, given that reasoning and flexible thinking are functions of the insulae, which are shrunken in all 6 disorders, one can postulate that inflexible thinking (fixed false beliefs also are called psychotic delusions) might be a common feature across all those disorders. Namely:
• schizophrenia is known for paranoid or implausible delusions
• bipolar disorder is characterized by grandiose delusions
• major depressive disorder is associated with a fixed false belief of worthlessness as well as hopelessness
• anxiety patients harbor the fixed false belief of impending doom or death (the plane will crash if they are a passenger on it)
• OCD manifests as ego-dystonic false beliefs (obsessions) that can progress into ego-syntonic delusions
• people with an alcohol or tobacco addiction are in delusional denial that they are not really addicted or that they will not be harmed by their drug of abuse. Pathologic gamblers harbor the false belief that they will soon reverse their fortunes and “win big.”
It seems that poor reality testing and impaired reasoning is a common feature of not only all 6 psychiatric disorders with shared neurobiology, but others, too, including anorexia nervosa, body dysmorphic disorder, delirium, and dementia.
From a thick volume to… a booklet?
Can you envision a day when psychiatric disorders are conceptualized as having a common genetic, neurobiological, and clinical core, with some variability in phenotype and behavior? If further brain research steers psychiatric nosology in that direction, we might end up with a DSM of 10 pages instead of almost 1,000, with an “Appendix” of genetic, neuroimaging, and other emerging biomarkers.
Bold scientific prophecies often sound delusional—until they come true….
In a report of a study that was published recently in a top-tier psychiatry journal,1 researchers describe a stunning finding that challenges the notion that there is a plethora of psychiatric brain disorders. They conducted a large meta-analysis of 193 published brain imaging studies of people with schizophrenia, bipolar disorder, major depression, obsessive-compulsive disorder (OCD), anxiety, and addiction. They found that those 6 supposedly discrete illnesses are all associated with a varying degree of shrinkage (atrophy or hypoplasia) of the same 3 brain regions:
• Dorsal anterior cingulate cortex. This region around the frontal part of the corpus callosum controls rational cognitive processes, reward anticipation, decision making, empathy, impulse control, and emotional response. Francis Crick, the Nobel laureate who first described the structure of DNA, hypothesized that the anterior cingulate sulcus might even be the center of what we call “free will.”
• Left insula and right insula. The insulae are the cortical regions deep inside the lateral sulcus, which is the fissure that separates the temporal lobe from the parietal and frontal lobes. The functions of the insulae include consciousness, emotions, perceptions, motor control, self-awareness, cognitive functioning, and interpersonal experience. (In addicts, the insular cortex is activated when they are exposed to environmental cues that trigger craving because the insulae are a target for the dopamine system. Notably, it has been reported that, when cigarette addicts suffer a stroke that damages the insulae, they stop smoking completely.)
The 3 regions of the brain, in which pathology extends across 6 DSM-5 diagnoses, work together to manage high-level executive functions, such as working memory, reasoning, and flexible thinking. The degree of dysfunction varies among the 6 clinical disorders, with schizophrenia having the highest severity.
Neurobiological commonality
The idea of shared neurobiological underpinnings among 6 distinct psychiatric disorders flies in the face of the entrenched DSM model, in which those 6 disorders are distinct disease entities. Other studies (including the Bipolar Schizophrenia Network on Intermediate Phenotypes) also found prominent biological similarities in varying degrees across schizophrenia, schizoaffective disorder, and bipolar disorder.2,3 The Research Domain Criteria of the National Institute of Mental Health also embraces the dimensional approach to neurobiological biomarkers across various psychiatric disorders.
A common genetic substrate also is emerging. A recently published genome-wide association study, conducted on 33,332 psychiatric patients and 27,888 controls,4 revealed that a number of genes are shared by 5 different psychiatric disorders: schizophrenia, autism, bipolar disorder, major depression, and attention-deficit/hyperactivity disorder. The genetic phenomenon of the same genes manifesting in different clinical phenotypes is called pleiotropy, and is consistent with shared neurobiological findings.
Genetic and brain structural commonalities among multiple DSM diagnostic categories might explain some well-known clinical observations:
• frequent comorbidity of certain psychiatric disorders, such as depression and addiction in schizophrenia; anxiety and OCD in bipolar disorder; depression with OCD and addictions; and so on
• the presence of intermediate phenotypes in unaffected family members, such as cognitive dysfunction in the parents of patients with schizophrenia, compared with parents of matched healthy controls5
• the much higher rate of psychopathology among family members of patients with a major psychiatric disorder, compared with the general population.6
Core inflexible thinking. So what about clinical features across those disorders that share genetic and neurobiologic similarities? Psychiatrists may agree that symptoms of schizophrenia, bipolar disorder, major depression, OCD, anxiety, and addiction appear very different. However, given that reasoning and flexible thinking are functions of the insulae, which are shrunken in all 6 disorders, one can postulate that inflexible thinking (fixed false beliefs also are called psychotic delusions) might be a common feature across all those disorders. Namely:
• schizophrenia is known for paranoid or implausible delusions
• bipolar disorder is characterized by grandiose delusions
• major depressive disorder is associated with a fixed false belief of worthlessness as well as hopelessness
• anxiety patients harbor the fixed false belief of impending doom or death (the plane will crash if they are a passenger on it)
• OCD manifests as ego-dystonic false beliefs (obsessions) that can progress into ego-syntonic delusions
• people with an alcohol or tobacco addiction are in delusional denial that they are not really addicted or that they will not be harmed by their drug of abuse. Pathologic gamblers harbor the false belief that they will soon reverse their fortunes and “win big.”
It seems that poor reality testing and impaired reasoning is a common feature of not only all 6 psychiatric disorders with shared neurobiology, but others, too, including anorexia nervosa, body dysmorphic disorder, delirium, and dementia.
From a thick volume to… a booklet?
Can you envision a day when psychiatric disorders are conceptualized as having a common genetic, neurobiological, and clinical core, with some variability in phenotype and behavior? If further brain research steers psychiatric nosology in that direction, we might end up with a DSM of 10 pages instead of almost 1,000, with an “Appendix” of genetic, neuroimaging, and other emerging biomarkers.
Bold scientific prophecies often sound delusional—until they come true….
1. Goodkind M, Eickhoff SB, Oathes DJ, et al. Identification of a common neurobiological substrate for mental illness. JAMA Psychiatry. 2015;72(4):305-315.
2. Hill SK, Reilly JL, Keefe RS, et al. Neuropsychological impairments in schizophrenia and psychotic bipolar disorder: findings from the Bipolar- Schizophrenia Network on Intermediate Phenotypes (B-SNIP) study. Am J Psychiatry. 2013;170(11):1275-1284.
3. Skudlarski P, Scretlen DJ, Thaker GK, et al. Diffusion tensor imaging white matter endophenotypes in patients with schizophrenia or psychotic bipolar disorder and their relatives. Am J Psychiatry. 2013;170(8):886-898.
4. Cross-Disorder Group of Psychiatric Genomics Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381(9875):1371-1379.
5. Appels MC, Sitskoorn MM, Westers P, et al. Cognitive dysfunction in parents of schizophrenia patients parallel the deficits found in patients. Schizophrenia Res. 2003;63(3):285-293.
6. Braff DL. The importance of endophenotypes in schizophrenia research. Schizophrenia Res. 2015;163(1-3):1-8.
1. Goodkind M, Eickhoff SB, Oathes DJ, et al. Identification of a common neurobiological substrate for mental illness. JAMA Psychiatry. 2015;72(4):305-315.
2. Hill SK, Reilly JL, Keefe RS, et al. Neuropsychological impairments in schizophrenia and psychotic bipolar disorder: findings from the Bipolar- Schizophrenia Network on Intermediate Phenotypes (B-SNIP) study. Am J Psychiatry. 2013;170(11):1275-1284.
3. Skudlarski P, Scretlen DJ, Thaker GK, et al. Diffusion tensor imaging white matter endophenotypes in patients with schizophrenia or psychotic bipolar disorder and their relatives. Am J Psychiatry. 2013;170(8):886-898.
4. Cross-Disorder Group of Psychiatric Genomics Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381(9875):1371-1379.
5. Appels MC, Sitskoorn MM, Westers P, et al. Cognitive dysfunction in parents of schizophrenia patients parallel the deficits found in patients. Schizophrenia Res. 2003;63(3):285-293.
6. Braff DL. The importance of endophenotypes in schizophrenia research. Schizophrenia Res. 2015;163(1-3):1-8.
The use of aripiprazole in the management of bipolar disorder during pregnancy
"This patient had presented 2-weeks postpartum in a manic state with psycotic features. She was screened by Ob-Gyn who collaborated with her care while she was admitted to the psychiatric inpatient unit. Patient had been non-compliant with prescribed medications prior to admission and she was started on aripiprazole from day one and the dose was tapered up to 15 mg BID by day 5. Patient's manic symptoms improved slowly as the days progressed by day 8 psychotic symptoms started to subside. As delivery was imminent, patient was transferred to Ob-Gyn service. She delivered a healthy but premature child via csection on day 12. Child did not exhibit any gross or anatomic malformations. She was continued on aripiprazole 15 mg BID after discharge and was seen weeks later in outpatient psychiatry."
Read more from the Poster Abstracts from the 2015 APA Annual Meeting
"This patient had presented 2-weeks postpartum in a manic state with psycotic features. She was screened by Ob-Gyn who collaborated with her care while she was admitted to the psychiatric inpatient unit. Patient had been non-compliant with prescribed medications prior to admission and she was started on aripiprazole from day one and the dose was tapered up to 15 mg BID by day 5. Patient's manic symptoms improved slowly as the days progressed by day 8 psychotic symptoms started to subside. As delivery was imminent, patient was transferred to Ob-Gyn service. She delivered a healthy but premature child via csection on day 12. Child did not exhibit any gross or anatomic malformations. She was continued on aripiprazole 15 mg BID after discharge and was seen weeks later in outpatient psychiatry."
Read more from the Poster Abstracts from the 2015 APA Annual Meeting
"This patient had presented 2-weeks postpartum in a manic state with psycotic features. She was screened by Ob-Gyn who collaborated with her care while she was admitted to the psychiatric inpatient unit. Patient had been non-compliant with prescribed medications prior to admission and she was started on aripiprazole from day one and the dose was tapered up to 15 mg BID by day 5. Patient's manic symptoms improved slowly as the days progressed by day 8 psychotic symptoms started to subside. As delivery was imminent, patient was transferred to Ob-Gyn service. She delivered a healthy but premature child via csection on day 12. Child did not exhibit any gross or anatomic malformations. She was continued on aripiprazole 15 mg BID after discharge and was seen weeks later in outpatient psychiatry."
Read more from the Poster Abstracts from the 2015 APA Annual Meeting
College students with depressive symptoms with and without fatigue: Differences in functioning, suicidality, anxiety, and depressive severity
Nyer et al examined whether fatigue was associated with greater symptomatic burden and functional impairment in 287 college students with depressive symptoms using data from the self-report Beck Depression Inventory (BDI). Students endorsing significant symptoms of depression (BDI score ≥13) were grouped into 3 levels: no fatigue, mild fatigue, or moderate/severe fatigue. Researchers compared the 3 levels of fatigue across a battery of psychiatric and functional outcome measures.
The study found that depressed college students with symptoms of fatigue demonstrated functional impairment and symptomatic burden that worsened with increasing levels of fatigue. The authors call for more attention to assessing and treating symptoms of fatigue within this population.
Nyer et al examined whether fatigue was associated with greater symptomatic burden and functional impairment in 287 college students with depressive symptoms using data from the self-report Beck Depression Inventory (BDI). Students endorsing significant symptoms of depression (BDI score ≥13) were grouped into 3 levels: no fatigue, mild fatigue, or moderate/severe fatigue. Researchers compared the 3 levels of fatigue across a battery of psychiatric and functional outcome measures.
The study found that depressed college students with symptoms of fatigue demonstrated functional impairment and symptomatic burden that worsened with increasing levels of fatigue. The authors call for more attention to assessing and treating symptoms of fatigue within this population.
Nyer et al examined whether fatigue was associated with greater symptomatic burden and functional impairment in 287 college students with depressive symptoms using data from the self-report Beck Depression Inventory (BDI). Students endorsing significant symptoms of depression (BDI score ≥13) were grouped into 3 levels: no fatigue, mild fatigue, or moderate/severe fatigue. Researchers compared the 3 levels of fatigue across a battery of psychiatric and functional outcome measures.
The study found that depressed college students with symptoms of fatigue demonstrated functional impairment and symptomatic burden that worsened with increasing levels of fatigue. The authors call for more attention to assessing and treating symptoms of fatigue within this population.
Rx: Preventive care
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel

Fatigue associated with depression

Recognizing autophonia in patients with anorexia nervosa
Anorexia nervosa can affect a number of systems of the body, including the otolaryngologic presentation of autophonia1,2—a rare hyperperception of an abnormally intense hearing of one’s own voice and respiratory sounds.2 The most common cause of autophonia in patients with anorexia is a patulous (patent) eustachian tube, which can be caused by extreme weight loss.2,3
Significant reduction in the quantity of fat tissue at the location of the eustachian tube can cause patency.3 This creates an abnormal connection between the nasopharynx and tympanic membrane, in which sounds are transmitted directly from the oral cavity to the middle ear, causing autophonia, tinnitus, or sound distortion.4
What are the symptoms?Patients often report hearing their own voice more loudly in the affected ear. This can be distressing, and they might become preoccupied with the sound of their voice—thus affecting quality of life.2,4
The intensity of symptoms varies: from a mild sensation of a clogged ear to extremely bothersome discomfort much like a middle-ear infection.2,4 Autophonia, however, cannot be relieved by conventional therapies for those conditions.2,3
A patulous eustachian tube is difficult to detect and can be misdiagnosed as another condition. Pregnancy, stress, fatigue, radiation therapy, hormonal therapy, and dramatic weight loss also can cause a patulous eustachian tube.2
How is the diagnosis made?The diagnosis of autophonia is clinical and begins with a detailed history. Symptoms often appear within the time frame of rapid weight loss and without evidence of infection or other illness.2,3 The clinical examination is otherwise unremarkable.2,4
Is there treatment?To improve the patient’s comfort and quality of life, intervention is required, best provided by an integrated team of medical specialists. Weight gain, of course, is the treatment goal in anorexia, but this is a complex process often marked by relapse; a detailed discussion of treatment strategies is beyond the scope of this “Pearl.” Symptoms usually diminish as fatty tissue is restored upon successful treatment of anorexia, which closes the abnormal eustachian tube opening.2,3
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Olthoff A, Laskawi R, Kruse E. Successful treatment of autophonia with botulinum toxin: case report. Ann Otol Rhinol Laryngol. 2007;116(8):594-598.
2. Godbole M, Key A. Autophonia in anorexia nervosa. Int J Eat Disord. 2010;43(5):480-482.
3. Karwautz A, Hafferl A, Ungar D, et al. Patulous eustachian tube in a case of adolescent anorexia nervosa. Int J Eat Disord. 1999;25(3):353-355.
4. Dornhoffer JL, Leuwer R, Schwager K, et al. A practical guide to the eustachian tube. New York, NY: Springer; 2014:23-41.
Anorexia nervosa can affect a number of systems of the body, including the otolaryngologic presentation of autophonia1,2—a rare hyperperception of an abnormally intense hearing of one’s own voice and respiratory sounds.2 The most common cause of autophonia in patients with anorexia is a patulous (patent) eustachian tube, which can be caused by extreme weight loss.2,3
Significant reduction in the quantity of fat tissue at the location of the eustachian tube can cause patency.3 This creates an abnormal connection between the nasopharynx and tympanic membrane, in which sounds are transmitted directly from the oral cavity to the middle ear, causing autophonia, tinnitus, or sound distortion.4
What are the symptoms?Patients often report hearing their own voice more loudly in the affected ear. This can be distressing, and they might become preoccupied with the sound of their voice—thus affecting quality of life.2,4
The intensity of symptoms varies: from a mild sensation of a clogged ear to extremely bothersome discomfort much like a middle-ear infection.2,4 Autophonia, however, cannot be relieved by conventional therapies for those conditions.2,3
A patulous eustachian tube is difficult to detect and can be misdiagnosed as another condition. Pregnancy, stress, fatigue, radiation therapy, hormonal therapy, and dramatic weight loss also can cause a patulous eustachian tube.2
How is the diagnosis made?The diagnosis of autophonia is clinical and begins with a detailed history. Symptoms often appear within the time frame of rapid weight loss and without evidence of infection or other illness.2,3 The clinical examination is otherwise unremarkable.2,4
Is there treatment?To improve the patient’s comfort and quality of life, intervention is required, best provided by an integrated team of medical specialists. Weight gain, of course, is the treatment goal in anorexia, but this is a complex process often marked by relapse; a detailed discussion of treatment strategies is beyond the scope of this “Pearl.” Symptoms usually diminish as fatty tissue is restored upon successful treatment of anorexia, which closes the abnormal eustachian tube opening.2,3
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Anorexia nervosa can affect a number of systems of the body, including the otolaryngologic presentation of autophonia1,2—a rare hyperperception of an abnormally intense hearing of one’s own voice and respiratory sounds.2 The most common cause of autophonia in patients with anorexia is a patulous (patent) eustachian tube, which can be caused by extreme weight loss.2,3
Significant reduction in the quantity of fat tissue at the location of the eustachian tube can cause patency.3 This creates an abnormal connection between the nasopharynx and tympanic membrane, in which sounds are transmitted directly from the oral cavity to the middle ear, causing autophonia, tinnitus, or sound distortion.4
What are the symptoms?Patients often report hearing their own voice more loudly in the affected ear. This can be distressing, and they might become preoccupied with the sound of their voice—thus affecting quality of life.2,4
The intensity of symptoms varies: from a mild sensation of a clogged ear to extremely bothersome discomfort much like a middle-ear infection.2,4 Autophonia, however, cannot be relieved by conventional therapies for those conditions.2,3
A patulous eustachian tube is difficult to detect and can be misdiagnosed as another condition. Pregnancy, stress, fatigue, radiation therapy, hormonal therapy, and dramatic weight loss also can cause a patulous eustachian tube.2
How is the diagnosis made?The diagnosis of autophonia is clinical and begins with a detailed history. Symptoms often appear within the time frame of rapid weight loss and without evidence of infection or other illness.2,3 The clinical examination is otherwise unremarkable.2,4
Is there treatment?To improve the patient’s comfort and quality of life, intervention is required, best provided by an integrated team of medical specialists. Weight gain, of course, is the treatment goal in anorexia, but this is a complex process often marked by relapse; a detailed discussion of treatment strategies is beyond the scope of this “Pearl.” Symptoms usually diminish as fatty tissue is restored upon successful treatment of anorexia, which closes the abnormal eustachian tube opening.2,3
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Olthoff A, Laskawi R, Kruse E. Successful treatment of autophonia with botulinum toxin: case report. Ann Otol Rhinol Laryngol. 2007;116(8):594-598.
2. Godbole M, Key A. Autophonia in anorexia nervosa. Int J Eat Disord. 2010;43(5):480-482.
3. Karwautz A, Hafferl A, Ungar D, et al. Patulous eustachian tube in a case of adolescent anorexia nervosa. Int J Eat Disord. 1999;25(3):353-355.
4. Dornhoffer JL, Leuwer R, Schwager K, et al. A practical guide to the eustachian tube. New York, NY: Springer; 2014:23-41.
1. Olthoff A, Laskawi R, Kruse E. Successful treatment of autophonia with botulinum toxin: case report. Ann Otol Rhinol Laryngol. 2007;116(8):594-598.
2. Godbole M, Key A. Autophonia in anorexia nervosa. Int J Eat Disord. 2010;43(5):480-482.
3. Karwautz A, Hafferl A, Ungar D, et al. Patulous eustachian tube in a case of adolescent anorexia nervosa. Int J Eat Disord. 1999;25(3):353-355.
4. Dornhoffer JL, Leuwer R, Schwager K, et al. A practical guide to the eustachian tube. New York, NY: Springer; 2014:23-41.