User login
Welcome to Current Psychiatry, a leading source of information, online and in print, for practitioners of psychiatry and its related subspecialties, including addiction psychiatry, child and adolescent psychiatry, and geriatric psychiatry. This Web site contains evidence-based reviews of the prevention, diagnosis, and treatment of mental illness and psychological disorders; case reports; updates on psychopharmacology; news about the specialty of psychiatry; pearls for practice; and other topics of interest and use to this audience.
Dear Drupal User: You're seeing this because you're logged in to Drupal, and not redirected to MDedge.com/psychiatry.
Depression
adolescent depression
adolescent major depressive disorder
adolescent schizophrenia
adolescent with major depressive disorder
animals
autism
baby
brexpiprazole
child
child bipolar
child depression
child schizophrenia
children with bipolar disorder
children with depression
children with major depressive disorder
compulsive behaviors
cure
elderly bipolar
elderly depression
elderly major depressive disorder
elderly schizophrenia
elderly with dementia
first break
first episode
gambling
gaming
geriatric depression
geriatric major depressive disorder
geriatric schizophrenia
infant
kid
major depressive disorder
major depressive disorder in adolescents
major depressive disorder in children
parenting
pediatric
pediatric bipolar
pediatric depression
pediatric major depressive disorder
pediatric schizophrenia
pregnancy
pregnant
rexulti
skin care
teen
wine
section[contains(@class, 'nav-hidden')]
footer[@id='footer']
div[contains(@class, 'pane-pub-article-current-psychiatry')]
div[contains(@class, 'pane-pub-home-current-psychiatry')]
div[contains(@class, 'pane-pub-topic-current-psychiatry')]
div[contains(@class, 'panel-panel-inner')]
div[contains(@class, 'pane-node-field-article-topics')]
section[contains(@class, 'footer-nav-section-wrapper')]
Lithium for bipolar disorder: A re-emerging treatment for mood instability
Lithium is among the most effective therapies for bipolar disorder (BD), and enthusiasm for this simple molecule is waxing. The history of lithium is fascinating,1 and recent considerations include that this element, the third on the periodic table, has few, if any, industry champions. The recent renaissance is caused by a groundswell of appreciation for the clinical efficacy of lithium and an increasing number of providers who are willing to manage patients with lithium.
Target: Bipolar disorder
The target illness for lithium is BD, a spectrum of mood disorders with characteristic features of unstable mood and affect. Shifts in mood include recurrent episodes of mania, which are pathologically energized states with misguided volition and behavior with intoxicating euphoria (or irritability).2 Psychomotor activity is elevated and out of character; speech and body movements are revved up, with a diminished need for sleep. The social, personal, and vocational consequences often are disastrous.
The most common mood state of BD is depression. Depressive episodes consist of pathologically compromised energy and volition with a slowing of bodily functions, most prominently cognition and concentration; a pervasive depressed or sad mood is common but not always present. Presence of mixed states, when features of depression and mania are present simultaneously, is one of the many challenges of treating BD; an elevated volitional or energized state may occur with a depressed, dysphoric mood.
Evidence for lithium
Efficacy studies of lithium have focused on managing mood disorders, treating mania and depression, and prevention or maintenance care.3 Most were performed during the 1970s and 1980s,3 but recent studies have been comparing lithium with other mood stabilizers4-7 and searching for a genetic basis for lithium response.8-10 Other researchers have examined the use of lithium to prevent suicide.11 Some have suggested a neuroprotective effect of lithium, which may have profound implications for neuropsychiatry if valid.12-14 Results of additional studies, which are at different stages of completion, will clarify lithium use,15,16 and characterize the genetic makeup of individuals who respond to lithium.17 The primary evidence for lithium, however, is for maintenance treatment of BD and for preventing manic and depressive episodes.
Biochemistry and physiology of lithium. The biochemical and physiological effects of lithium are complex, wide-ranging, and likely to affect hundreds, if not thousands, of genes and gene products. The mechanisms of action remain a focus of academic pursuit (for a review of hypotheses related to these mechanisms see Goodwin and Jamison2 and Can et al18) Lithium is involved in cell signaling pathways that involve complex molecular mechanisms of inter- and intracellular communication19; some neural receptors are down-regulated20 and others show inhibition,21 which is thought to be a mechanism of lithium. The hypothesized neuroprotective effect of lithium22 may be mediated through an increased level of brain-derived neurotrophic factor in brain tissue.14 Recently, investigators using induced pluripotent stem cell derived neurons have shown that patterns of calcium-related cell signaling in bipolar neurons are affected specifically by lithium in the culture media.23 There likely are many mechanisms through which lithium’s effects are mediated, including a series of dynamic pathways that vary over time and in reaction to the internal and external environments of the cell and person.
The lithium renaissance
In the past decade, there has been an increase in interest and use of lithium because clinicians recognize its efficacy and advantages and can monitor serum levels and gauge the patient’s response and side effects24 against the lithium level. This is important because balancing effi cacy and side effects depends on the serum level. Efficacy often is not immediate, although side effects may emerge early. All systems of the body may show effects that could be related to lithium use. It is helpful to be aware of the side effects in chronological order, because some immediate effects may be associated with starting at higher dosages (Table 1). Common side effects in the short term include:
• GI distress, such as nausea, vomiting, diarrhea, and abdominal discomfort
• a fine neurologic tremor, which may be seen with accentuation upon deliberate movement
• prominent thirst with polyuria
• drowsiness and clouded thinking, which can be upsetting to the patient and family.
In the longer term, adverse effects on kidney and thyroid function are common. Management must include monitoring of the serum level.
Lithium is FDA-approved for acute and maintenance treatment of mania in BD. There are reports that discuss most variants of mood disorders, including BD I, BD II, unipolar depression, rapid cycling, and even alcohol abuse.25-29 Lithium could help manage mood dysregulation in the context of temperament and personality.30 There is evidence that lithium has an antidepressant effect31-33 and has shown efficacy as an adjunctive treatment for depression.31-33 There are data that suggest that lithium, with its neuroprotective mechanisms, may prevent progression of mild cognitive impairment.34
Is there an ideal lithium candidate?
Mood instability is the characteristic feature of a lithium responder. The instability may be over the course of the day, such as a dysregulated temperament that often is associated with DSM-IV personality categories, shorter-term fluctuations (within days with BD II), or in the context of episodic shifts of mood states over weeks and months, which are characteristic of BD I. The hallmark of mood instability is fluctuation from depression to elevated mood states and charged emotions with increased energy.
The patient considered ideal for lithium treatment has BD I with recurrent severe euphoric manic episodes, absence of significant comorbid disorders such as substance abuse, and a family history of lithium response. However, any patient with a clinically significant and unstable mood disorder, regardless of the DSM diagnosis, should be considered for lithium treatment.
When considering a lithium trial for a patient with significant mood instability, it is critical to establish the target symptoms and behavior that will help you gauge the efficacy of the intervention. Measurement-based care utilizes clinician and self-report instruments to provide data on the illness course and response to intervention. Commonly used clinician driven assessments include the Young Mania Rating Scale35 and the Quick Inventory of Depressive Symptoms,36 while the self-report assessments are the Patient Health Questionnaire37 and the Altman Self- Rating Mania Scale.38
During acute mania or depression, lithium often is used in combination with another medications such as an antipsychotic or antidepressant. Used in the outpatient and non-acute setting, lithium may be an “add-on” or monotherapy for preventing recurrence of episodes. Response in early acute manic symptoms are predictive of later response and remission.39
Dosing strategies
An initial problem with lithium is side effects that emerge when beginning treatment, which may discourage the patient and family from using this agent. Starting with 150 mg/d for the first 2 or 3 doses is unlikely to produce any adverse effects and can show the patient that there is a high likelihood that he will be able to tolerate the medication. Gradual titration over several days—or even weeks—to the target dosage and serum levels will enhance patient compliance. Rate of dosage increase is best guided by tolerance to the medication. The general consensus is that lithium is most effective at levels of 0.6 to 0.8 mEq/L,40 although a lower level (0.5 mEq/L) over a 2-year period also can be effective.41 Lithium may be used in to treat acute mania at higher serum levels (0.8 to 1.2 mEq/L), however, the acute phase often requires urgent management, usually with an antipsychotic.
Emerging consensus
Although there is a need to gather and analyze longer observational periods to clarify the clinical and biological characteristics of persons who respond to lithium, there are several points of consensus. Management will be guided by patient characteristics such as age, comorbidities, and other therapies. Most studies that address the effect of lithium level focus on high vs low serum levels. There are 3 categories of lithium serum levels, low (<0.6 mEq/L), mid-range (0.6 to 0.8 mEq/L), and high (>0.8 mEq/L), each has risk-benefit considerations.
The LiTMUS study42 compared low-level lithium augmentation with optimized personal treatment without lithium. Both groups had similar outcomes but the lithium-treated group had significantly lower use of atypical antipsychotics. This may be important when considering the long-term risk of the metabolic syndrome because the tolerability and side-effect profile of lithium at lower levels is more favorable than that of atypical antipsychotics. As lithium levels increase, there seems to be concomitant increase in efficacy and side effects. Many patients will benefit with low-level lithium use; yet clearly some individuals require higher dosages for effective maintenance therapy.
Dosing and monitoring. In patients age >50 or those with comorbid medical conditions, use a lower level of lithium (<0.6 mEq/L). Most individuals with BD likely will benefit from the mid-range level strategy (0.6 to 0.8 mEq/L); however, there will be those who require a higher level. When beginning lithium, start at a low dosage (150 mg/d) and increase as tolerated to the desired serum level. With acute mania, temporary use of an antipsychotic will be required.
There are no tests available to determine whether a patient will do well at any of these lithium serum levels. Breakthrough mania in an adherent patient with a serum lithium level of 0.7 mEq/L indicates the need to obtain a higher lithium level. A major deficit in lithium research is the lack of long-term data (>5 years) on outcomes, clinical and biological features with lithium levels because of a lack of pharmaceutical company support.3,17 Monitoring mood symptoms using detailed mood charts, whether clinician-administered or self-reported, is an effective way to monitor outcomes, provided the clinician uses the same scales or methods to record a patient’s moods. If a patient wants to discontinue lithium, taper the drug over an extended period (months) to minimize the likelihood of emerging manic or depressive episodes related to drug discontinuation.
Managing side effects
Consider lithium’s side effects in the context of their short-, intermediate-, and long-term presence (Table 2). Gradually increasing the lithium dosage often will prevent side effects that manifest in the short term. If side effects emerge at low dosages, proceed slowly with lithium and manage symptoms with other medications. When a patient shows a change in side effects, obtain lithium and electrolytes levels; a change in mental status with confusion will require an acute lithium level. 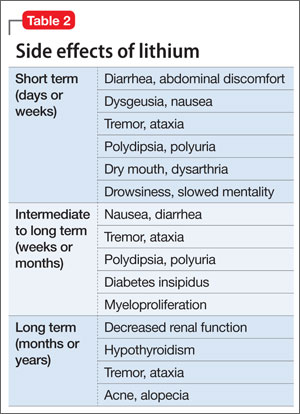
A diary of symptoms or clinically relevant matters such as fluid intake or frequency of GI- or neurological-related events will help the clinician monitor the frequency and severity of side effects. The patient and clinician should not be discouraged by emerging side effects in the short term, because they may dissipate or become minimally intrusive.
Several strategies can alleviate immediate GI effects, such as dosing with meals, enteric-coated formulations, multiple dose strategies, and short-term use of antidiarrheal medicine as needed. Side effects that disrupt a patient’s fluid and electrolyte balance (diabetes insipidus) to the point of clouding mental status will require discontinuing the medication until mental status improves, then reconsideration of the treatment regime, which will include managing diabetes insipidus with amiloride. Managing side effects may require consultation with specialty services. Likewise, some patients might experience neurologic side effects, such as profound tremor, that interferes with their ability to function. However, many side effects can be managed symptomatically with practical strategies (eg, a sugar-free lozenge for dry mouth or dysgeusia). Consider lower lithium dosages and serum levels because patients may experience benefits with lower therapeutic levels.
Long-term side effects include decreased renal function, hypothyroidism, persistent tremor, and dermatologic effects of acne and alopecia. Monitor renal and thyroid function annually in stable patients and more frequently when making changes in the treatment plan.
Before discontinuing lithium, consider discussing the medical issues with a specialist who has experience with complications of lithium.
Bottom Line
Lithium is an effective and under used medication for managing bipolar disorder. Initial prejudices and side effects often deter patients and prescribers from proceeding with a therapeutic trial of lithium. Although the mid-range lithium level of 0.6 to 0.8 mEq/L is desirable, many patients will experience significant benefits with lower levels. Initial strategies include the use of low-dose preparations that are unlikely to have uncomfortable side effects.
Related Resources
• Andreasen A, Ellingrod VL. Lithium-induced diabetes insipidus: prevention and management. Current Psychiatry. 2013;12(7):42-45.
• Cipriani A, Hawton K, Stockton S, et al. Lithium in the prevention of suicide in mood disorders: updated systematic review and meta-analysis. BMJ. 2013;346:f3646. doi: 10.1136/bmj.f3646.
Drug Brand Names
Amiloride • Midamor Lithium • Eskalith, Lithobid
Disclosure
Dr. McInnis reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Shorter E. The history of lithium therapy. Bipolar Disord. 2009;11(11 suppl 2):4-9.
2. Goodwin FK, Jamison KR. Manic-depressive illness: bipolar disorders and recurrent depression. 2nd ed. New York, NY: Oxford University Press; 2007.
3. Burgess S, Geddes J, Hawton K, et al. Lithium for maintenance treatment of mood disorders. Cochrane Database Syst Rev. 2001:CD003013.
4. Bowden CL, Calabrese JR, McElroy SL, et al. A randomized, placebo-controlled 12-month trial of divalproex and lithium in treatment of outpatients with bipolar I disorder. Divalproex maintenance study group. Arch Gen Psychiatry. 2000;57(5):481-489.
5. Bowden CL, Calabrese JR, Sachs G, et al; Lamictal 606 Study Group. A placebo-controlled 18-month trial of lamotrigine and lithium maintenance treatment in recently manic or hypomanic patients with bipolar I disorder. Arch Gen Psychiatry. 2003;60(4):392-400.
6. Swann AC, Bowden CL, Calabrese JR, et al. Pattern of response to divalproex, lithium, or placebo in four naturalistic subtypes of mania. Neuropsychopharmacology. 2002;26(4):530-536.
7. Tohen M, Chengappa KN, Suppes T, et al. Efficacy of olanzapine in combination with valproate or lithium in the treatment of mania in patients partially nonresponsive to valproate or lithium monotherapy. Arch Gen Psychiatry. 2002;59(1):62-69.
8. Perlis RH, Smoller JW, Ferreira MA, et al. A genomewide association study of response to lithium for prevention of recurrence in bipolar disorder. Am J Psychiatry. 2009; 166(6):718-725.
9. Grof P, Duffy A, Cavazzoni P, et al. Is response to prophylactic lithium a familial trait? J Clin Psychiatry. 2002;63(10): 942-947.
10. Duffy A, Alda M, Kutcher S, et al. A prospective study of the offspring of bipolar parents responsive and nonresponsive to lithium treatment. J Clin Psychiatry. 2002;63(12): 1171-1178.
11. Goodwin FK, Fireman B, Simon GE, et al. Suicide risk in bipolar disorder during treatment with lithium and divalproex. JAMA. 2003;290(11):1467-1473.
12. Quiroz JA, Machado-Vieira R, Zarate CA Jr, et al. Novel insights into lithium’s mechanism of action: neurotrophic and neuroprotective effects. Neuropsychobiology. 2010; 62(1):50-60.
13. Forlenza OV, Diniz BS, Radanovic M, et al. Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: randomised controlled trial. Br J Psychiatry. 2011;198(5):351-356.
14. de Sousa RT, van de Bilt MT, Diniz BS, et al. Lithium increases plasma brain-derived neurotrophic factor in acute bipolar mania: a preliminary 4-week study. Neurosci Lett. 2011;494(1):54-56.
15. Nierenberg AA, Sylvia LG, Leon AC, et al; LiTMUS Study Group. Lithium treatment–moderate dose use study (LiTMUS) for bipolar disorder: rationale and design. Clin Trials. 2009;6(6):637-648.
16. Sylvia LG, Reilly-Harrington NA, Leon AC, et al. Methods to limit attrition in longitudinal comparative effectiveness trials: lessons from the Lithium Treatment - Moderate dose Use Study (LiTMUS) for bipolar disorder. Clin Trials. 2012;9(1):94-101.
17. McCarthy MJ, Leckband SG, Kelsoe JR. Pharmacogenetics of lithium response in bipolar disorder. Pharmacogenomics. 2010;11(10):1439-1465.
18. Can A, Schulze TG, Gould TD. Molecular actions and clinical pharmacogenetics of lithium therapy [published online February 15, 2014]. Pharmacol Biochem Behav. doi: 10.1016/j.pbb.2014.02.004.
19. Berridge MJ. Unlocking the secrets of cell signaling. Annu Rev Physiol. 2005;67:1-21.
20. Devaki R, Shankar Rao S, Nadgir SM. The effect of lithium on the adrenoceptor-mediated second messenger system in the rat brain. J Psychiatry Neurosci. 2006;31(4):246-252.
21. Pan JQ, Lewis MC, Ketterman JK, et al. AKT kinase activity is required for lithium to modulate mood-related behaviors in mice. Neuropsychopharmacology. 2011;36(7):1397-1411.
22. Hu LW, Kawamoto EM, Brietzke E, et al. The role of Wnt signaling and its interaction with diverse mechanisms of cellular apoptosis in the pathophysiology of bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35(1):11-17.
23. Chen HM, DeLong CJ, Bame M, et al. Transcripts involved in calcium signaling and telencephalic neuronal fate are altered in induced pluripotent stem cells from bipolar disorder patients [published online March 25, 2014]. Transl Psychiatry. doi:10.1038/tp.2014.12.
24. Jefferson JW. Lithium. In: Aronson JK, ed. Side effects of drugs annual, volume 26. Amsterdam, The Netherlands: Elsevier Science; 2003:19-29.
25. Baldessarini RJ, Tondo L, Floris G, et al. Effects of rapid cycling on response to lithium maintenance treatment in 360 bipolar I and II disorder patients. J Affect Disord. 2000;61(2):13-22.
26. Baldessarini RJ, Tondo L, Hennen J, et al. Latency and episodes before treatment: response to lithium maintenance in bipolar I and II disorders. Bipolar Disord. 1999;1(2): 91-97.
27. Fieve RR, Kumbaraci T, Dunner DL. Lithium prophylaxis of depression in bipolar I, bipolar II, and unipolar patients. Am J Psychiatry. 1976;133(8):925-929.
28. Peck CC, Pond SM, Becker CE, et al. An evaluation of the effects of lithium in the treatment of chronic alcoholism. II. Assessment of the two-period crossover design. Alcohol Clin Exp Res. 1981;5(2):252-255.
29. Peselow ED, Dunner DL, Fieve RR, et al. Lithium prophylaxis of depression in unipolar, bipolar II, and cyclothymic patients. Am J Psychiatry. 1982;139(6):747-752.
30. Bellino S, Paradiso E, Bogetto F. Efficacy and tolerability of pharmacotherapies for borderline personality disorder. CNS Drugs. 2008;22(8):671-692.
31. Alevizos B, Alevizos E, Leonardou A, et al. Low dosage lithium augmentation in venlafaxine resistant depression: an open-label study. Psychiatrike. 2012;23(2):143-148.
32. Goldberg JF, Sacks MH, Kocsis JH. Low-dose lithium augmentation of divalproex in geriatric mania. J Clin Psychiatry. 2000;61(4):304.
33. Saunders KE, Goodwin GM. New approaches in the treatment of bipolar depression. Curr Top Behav Neurosci. 2013;14:291-307.
34. Forlenza OV, Diniz BS, Radanovic M, et al. Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: randomised controlled trial. Br J Psychiatry. 2011;198(5):351-356.
35. Young RC, Biggs JT, Ziegler VE, et al. A rating scale for mania: reliability, validity and sensitivity. Br J Psychiatry. 1978;133:429-435.
36. Trivedi MH, Rush AJ, Ibrahim HM, et al. The Inventory of Depressive Symptomatology, Clinician Rating (IDS-C) and Self-Report (IDS-SR), and the Quick Inventory of Depressive Symptomatology, Clinician Rating (QIDS-C) and Self-Report (QIDS-SR) in public sector patients with mood disorders: a psychometric evaluation. Psychol Med. 2004;34(1):73-82.
37. Kroenke K, Spitzer RL, Williams JB. The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med. 2001;16(9):606-613.
38. Altman EG, Hedeker D, Peterson JL, et al. The Altman Self- Rating Mania Scale. Biol Psychiatry. 1997;42(10):948-955.
39. Machado-Vieira R, Luckenbaugh DA, Soeiro-de-Souza MG, et al. Early improvement with lithium in classic mania and its association with later response. J Affect Disord. 2013;144(1-2):160-164.
40. Severus WE, Lipkovich IA, Licht RW, et al. In search of optimal lithium levels and olanzapine doses in the long-term treatment of bipolar I disorder. A post-hoc analysis of the maintenance study by Tohen et al. 2005. Eur Psychiatry. 2010;25(8):443-449.
41. Vestergaard P, Licht RW, Brodersen A, et al. Outcome of lithium prophylaxis: a prospective follow-up of affective disorder patients assigned to high and low serum lithium levels. Acta Psychiatr Scand. 1998;98(4):310-315.
42. Nierenberg AA, Friedman ES, Bowden CL, et al. Lithium treatment moderate-dose use study (LiTMUS) for bipolar disorder: a randomized comparative effectiveness trial of optimized personalized treatment with and without lithium. Am J Psychiatry. 2013;170(1):102-111.
Lithium is among the most effective therapies for bipolar disorder (BD), and enthusiasm for this simple molecule is waxing. The history of lithium is fascinating,1 and recent considerations include that this element, the third on the periodic table, has few, if any, industry champions. The recent renaissance is caused by a groundswell of appreciation for the clinical efficacy of lithium and an increasing number of providers who are willing to manage patients with lithium.
Target: Bipolar disorder
The target illness for lithium is BD, a spectrum of mood disorders with characteristic features of unstable mood and affect. Shifts in mood include recurrent episodes of mania, which are pathologically energized states with misguided volition and behavior with intoxicating euphoria (or irritability).2 Psychomotor activity is elevated and out of character; speech and body movements are revved up, with a diminished need for sleep. The social, personal, and vocational consequences often are disastrous.
The most common mood state of BD is depression. Depressive episodes consist of pathologically compromised energy and volition with a slowing of bodily functions, most prominently cognition and concentration; a pervasive depressed or sad mood is common but not always present. Presence of mixed states, when features of depression and mania are present simultaneously, is one of the many challenges of treating BD; an elevated volitional or energized state may occur with a depressed, dysphoric mood.
Evidence for lithium
Efficacy studies of lithium have focused on managing mood disorders, treating mania and depression, and prevention or maintenance care.3 Most were performed during the 1970s and 1980s,3 but recent studies have been comparing lithium with other mood stabilizers4-7 and searching for a genetic basis for lithium response.8-10 Other researchers have examined the use of lithium to prevent suicide.11 Some have suggested a neuroprotective effect of lithium, which may have profound implications for neuropsychiatry if valid.12-14 Results of additional studies, which are at different stages of completion, will clarify lithium use,15,16 and characterize the genetic makeup of individuals who respond to lithium.17 The primary evidence for lithium, however, is for maintenance treatment of BD and for preventing manic and depressive episodes.
Biochemistry and physiology of lithium. The biochemical and physiological effects of lithium are complex, wide-ranging, and likely to affect hundreds, if not thousands, of genes and gene products. The mechanisms of action remain a focus of academic pursuit (for a review of hypotheses related to these mechanisms see Goodwin and Jamison2 and Can et al18) Lithium is involved in cell signaling pathways that involve complex molecular mechanisms of inter- and intracellular communication19; some neural receptors are down-regulated20 and others show inhibition,21 which is thought to be a mechanism of lithium. The hypothesized neuroprotective effect of lithium22 may be mediated through an increased level of brain-derived neurotrophic factor in brain tissue.14 Recently, investigators using induced pluripotent stem cell derived neurons have shown that patterns of calcium-related cell signaling in bipolar neurons are affected specifically by lithium in the culture media.23 There likely are many mechanisms through which lithium’s effects are mediated, including a series of dynamic pathways that vary over time and in reaction to the internal and external environments of the cell and person.
The lithium renaissance
In the past decade, there has been an increase in interest and use of lithium because clinicians recognize its efficacy and advantages and can monitor serum levels and gauge the patient’s response and side effects24 against the lithium level. This is important because balancing effi cacy and side effects depends on the serum level. Efficacy often is not immediate, although side effects may emerge early. All systems of the body may show effects that could be related to lithium use. It is helpful to be aware of the side effects in chronological order, because some immediate effects may be associated with starting at higher dosages (Table 1). Common side effects in the short term include:
• GI distress, such as nausea, vomiting, diarrhea, and abdominal discomfort
• a fine neurologic tremor, which may be seen with accentuation upon deliberate movement
• prominent thirst with polyuria
• drowsiness and clouded thinking, which can be upsetting to the patient and family.
In the longer term, adverse effects on kidney and thyroid function are common. Management must include monitoring of the serum level.
Lithium is FDA-approved for acute and maintenance treatment of mania in BD. There are reports that discuss most variants of mood disorders, including BD I, BD II, unipolar depression, rapid cycling, and even alcohol abuse.25-29 Lithium could help manage mood dysregulation in the context of temperament and personality.30 There is evidence that lithium has an antidepressant effect31-33 and has shown efficacy as an adjunctive treatment for depression.31-33 There are data that suggest that lithium, with its neuroprotective mechanisms, may prevent progression of mild cognitive impairment.34
Is there an ideal lithium candidate?
Mood instability is the characteristic feature of a lithium responder. The instability may be over the course of the day, such as a dysregulated temperament that often is associated with DSM-IV personality categories, shorter-term fluctuations (within days with BD II), or in the context of episodic shifts of mood states over weeks and months, which are characteristic of BD I. The hallmark of mood instability is fluctuation from depression to elevated mood states and charged emotions with increased energy.
The patient considered ideal for lithium treatment has BD I with recurrent severe euphoric manic episodes, absence of significant comorbid disorders such as substance abuse, and a family history of lithium response. However, any patient with a clinically significant and unstable mood disorder, regardless of the DSM diagnosis, should be considered for lithium treatment.
When considering a lithium trial for a patient with significant mood instability, it is critical to establish the target symptoms and behavior that will help you gauge the efficacy of the intervention. Measurement-based care utilizes clinician and self-report instruments to provide data on the illness course and response to intervention. Commonly used clinician driven assessments include the Young Mania Rating Scale35 and the Quick Inventory of Depressive Symptoms,36 while the self-report assessments are the Patient Health Questionnaire37 and the Altman Self- Rating Mania Scale.38
During acute mania or depression, lithium often is used in combination with another medications such as an antipsychotic or antidepressant. Used in the outpatient and non-acute setting, lithium may be an “add-on” or monotherapy for preventing recurrence of episodes. Response in early acute manic symptoms are predictive of later response and remission.39
Dosing strategies
An initial problem with lithium is side effects that emerge when beginning treatment, which may discourage the patient and family from using this agent. Starting with 150 mg/d for the first 2 or 3 doses is unlikely to produce any adverse effects and can show the patient that there is a high likelihood that he will be able to tolerate the medication. Gradual titration over several days—or even weeks—to the target dosage and serum levels will enhance patient compliance. Rate of dosage increase is best guided by tolerance to the medication. The general consensus is that lithium is most effective at levels of 0.6 to 0.8 mEq/L,40 although a lower level (0.5 mEq/L) over a 2-year period also can be effective.41 Lithium may be used in to treat acute mania at higher serum levels (0.8 to 1.2 mEq/L), however, the acute phase often requires urgent management, usually with an antipsychotic.
Emerging consensus
Although there is a need to gather and analyze longer observational periods to clarify the clinical and biological characteristics of persons who respond to lithium, there are several points of consensus. Management will be guided by patient characteristics such as age, comorbidities, and other therapies. Most studies that address the effect of lithium level focus on high vs low serum levels. There are 3 categories of lithium serum levels, low (<0.6 mEq/L), mid-range (0.6 to 0.8 mEq/L), and high (>0.8 mEq/L), each has risk-benefit considerations.
The LiTMUS study42 compared low-level lithium augmentation with optimized personal treatment without lithium. Both groups had similar outcomes but the lithium-treated group had significantly lower use of atypical antipsychotics. This may be important when considering the long-term risk of the metabolic syndrome because the tolerability and side-effect profile of lithium at lower levels is more favorable than that of atypical antipsychotics. As lithium levels increase, there seems to be concomitant increase in efficacy and side effects. Many patients will benefit with low-level lithium use; yet clearly some individuals require higher dosages for effective maintenance therapy.
Dosing and monitoring. In patients age >50 or those with comorbid medical conditions, use a lower level of lithium (<0.6 mEq/L). Most individuals with BD likely will benefit from the mid-range level strategy (0.6 to 0.8 mEq/L); however, there will be those who require a higher level. When beginning lithium, start at a low dosage (150 mg/d) and increase as tolerated to the desired serum level. With acute mania, temporary use of an antipsychotic will be required.
There are no tests available to determine whether a patient will do well at any of these lithium serum levels. Breakthrough mania in an adherent patient with a serum lithium level of 0.7 mEq/L indicates the need to obtain a higher lithium level. A major deficit in lithium research is the lack of long-term data (>5 years) on outcomes, clinical and biological features with lithium levels because of a lack of pharmaceutical company support.3,17 Monitoring mood symptoms using detailed mood charts, whether clinician-administered or self-reported, is an effective way to monitor outcomes, provided the clinician uses the same scales or methods to record a patient’s moods. If a patient wants to discontinue lithium, taper the drug over an extended period (months) to minimize the likelihood of emerging manic or depressive episodes related to drug discontinuation.
Managing side effects
Consider lithium’s side effects in the context of their short-, intermediate-, and long-term presence (Table 2). Gradually increasing the lithium dosage often will prevent side effects that manifest in the short term. If side effects emerge at low dosages, proceed slowly with lithium and manage symptoms with other medications. When a patient shows a change in side effects, obtain lithium and electrolytes levels; a change in mental status with confusion will require an acute lithium level.
A diary of symptoms or clinically relevant matters such as fluid intake or frequency of GI- or neurological-related events will help the clinician monitor the frequency and severity of side effects. The patient and clinician should not be discouraged by emerging side effects in the short term, because they may dissipate or become minimally intrusive.
Several strategies can alleviate immediate GI effects, such as dosing with meals, enteric-coated formulations, multiple dose strategies, and short-term use of antidiarrheal medicine as needed. Side effects that disrupt a patient’s fluid and electrolyte balance (diabetes insipidus) to the point of clouding mental status will require discontinuing the medication until mental status improves, then reconsideration of the treatment regime, which will include managing diabetes insipidus with amiloride. Managing side effects may require consultation with specialty services. Likewise, some patients might experience neurologic side effects, such as profound tremor, that interferes with their ability to function. However, many side effects can be managed symptomatically with practical strategies (eg, a sugar-free lozenge for dry mouth or dysgeusia). Consider lower lithium dosages and serum levels because patients may experience benefits with lower therapeutic levels.
Long-term side effects include decreased renal function, hypothyroidism, persistent tremor, and dermatologic effects of acne and alopecia. Monitor renal and thyroid function annually in stable patients and more frequently when making changes in the treatment plan.
Before discontinuing lithium, consider discussing the medical issues with a specialist who has experience with complications of lithium.
Bottom Line
Lithium is an effective and under used medication for managing bipolar disorder. Initial prejudices and side effects often deter patients and prescribers from proceeding with a therapeutic trial of lithium. Although the mid-range lithium level of 0.6 to 0.8 mEq/L is desirable, many patients will experience significant benefits with lower levels. Initial strategies include the use of low-dose preparations that are unlikely to have uncomfortable side effects.
Related Resources
• Andreasen A, Ellingrod VL. Lithium-induced diabetes insipidus: prevention and management. Current Psychiatry. 2013;12(7):42-45.
• Cipriani A, Hawton K, Stockton S, et al. Lithium in the prevention of suicide in mood disorders: updated systematic review and meta-analysis. BMJ. 2013;346:f3646. doi: 10.1136/bmj.f3646.
Drug Brand Names
Amiloride • Midamor Lithium • Eskalith, Lithobid
Disclosure
Dr. McInnis reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Lithium is among the most effective therapies for bipolar disorder (BD), and enthusiasm for this simple molecule is waxing. The history of lithium is fascinating,1 and recent considerations include that this element, the third on the periodic table, has few, if any, industry champions. The recent renaissance is caused by a groundswell of appreciation for the clinical efficacy of lithium and an increasing number of providers who are willing to manage patients with lithium.
Target: Bipolar disorder
The target illness for lithium is BD, a spectrum of mood disorders with characteristic features of unstable mood and affect. Shifts in mood include recurrent episodes of mania, which are pathologically energized states with misguided volition and behavior with intoxicating euphoria (or irritability).2 Psychomotor activity is elevated and out of character; speech and body movements are revved up, with a diminished need for sleep. The social, personal, and vocational consequences often are disastrous.
The most common mood state of BD is depression. Depressive episodes consist of pathologically compromised energy and volition with a slowing of bodily functions, most prominently cognition and concentration; a pervasive depressed or sad mood is common but not always present. Presence of mixed states, when features of depression and mania are present simultaneously, is one of the many challenges of treating BD; an elevated volitional or energized state may occur with a depressed, dysphoric mood.
Evidence for lithium
Efficacy studies of lithium have focused on managing mood disorders, treating mania and depression, and prevention or maintenance care.3 Most were performed during the 1970s and 1980s,3 but recent studies have been comparing lithium with other mood stabilizers4-7 and searching for a genetic basis for lithium response.8-10 Other researchers have examined the use of lithium to prevent suicide.11 Some have suggested a neuroprotective effect of lithium, which may have profound implications for neuropsychiatry if valid.12-14 Results of additional studies, which are at different stages of completion, will clarify lithium use,15,16 and characterize the genetic makeup of individuals who respond to lithium.17 The primary evidence for lithium, however, is for maintenance treatment of BD and for preventing manic and depressive episodes.
Biochemistry and physiology of lithium. The biochemical and physiological effects of lithium are complex, wide-ranging, and likely to affect hundreds, if not thousands, of genes and gene products. The mechanisms of action remain a focus of academic pursuit (for a review of hypotheses related to these mechanisms see Goodwin and Jamison2 and Can et al18) Lithium is involved in cell signaling pathways that involve complex molecular mechanisms of inter- and intracellular communication19; some neural receptors are down-regulated20 and others show inhibition,21 which is thought to be a mechanism of lithium. The hypothesized neuroprotective effect of lithium22 may be mediated through an increased level of brain-derived neurotrophic factor in brain tissue.14 Recently, investigators using induced pluripotent stem cell derived neurons have shown that patterns of calcium-related cell signaling in bipolar neurons are affected specifically by lithium in the culture media.23 There likely are many mechanisms through which lithium’s effects are mediated, including a series of dynamic pathways that vary over time and in reaction to the internal and external environments of the cell and person.
The lithium renaissance
In the past decade, there has been an increase in interest and use of lithium because clinicians recognize its efficacy and advantages and can monitor serum levels and gauge the patient’s response and side effects24 against the lithium level. This is important because balancing effi cacy and side effects depends on the serum level. Efficacy often is not immediate, although side effects may emerge early. All systems of the body may show effects that could be related to lithium use. It is helpful to be aware of the side effects in chronological order, because some immediate effects may be associated with starting at higher dosages (Table 1). Common side effects in the short term include:
• GI distress, such as nausea, vomiting, diarrhea, and abdominal discomfort
• a fine neurologic tremor, which may be seen with accentuation upon deliberate movement
• prominent thirst with polyuria
• drowsiness and clouded thinking, which can be upsetting to the patient and family.
In the longer term, adverse effects on kidney and thyroid function are common. Management must include monitoring of the serum level.
Lithium is FDA-approved for acute and maintenance treatment of mania in BD. There are reports that discuss most variants of mood disorders, including BD I, BD II, unipolar depression, rapid cycling, and even alcohol abuse.25-29 Lithium could help manage mood dysregulation in the context of temperament and personality.30 There is evidence that lithium has an antidepressant effect31-33 and has shown efficacy as an adjunctive treatment for depression.31-33 There are data that suggest that lithium, with its neuroprotective mechanisms, may prevent progression of mild cognitive impairment.34
Is there an ideal lithium candidate?
Mood instability is the characteristic feature of a lithium responder. The instability may be over the course of the day, such as a dysregulated temperament that often is associated with DSM-IV personality categories, shorter-term fluctuations (within days with BD II), or in the context of episodic shifts of mood states over weeks and months, which are characteristic of BD I. The hallmark of mood instability is fluctuation from depression to elevated mood states and charged emotions with increased energy.
The patient considered ideal for lithium treatment has BD I with recurrent severe euphoric manic episodes, absence of significant comorbid disorders such as substance abuse, and a family history of lithium response. However, any patient with a clinically significant and unstable mood disorder, regardless of the DSM diagnosis, should be considered for lithium treatment.
When considering a lithium trial for a patient with significant mood instability, it is critical to establish the target symptoms and behavior that will help you gauge the efficacy of the intervention. Measurement-based care utilizes clinician and self-report instruments to provide data on the illness course and response to intervention. Commonly used clinician driven assessments include the Young Mania Rating Scale35 and the Quick Inventory of Depressive Symptoms,36 while the self-report assessments are the Patient Health Questionnaire37 and the Altman Self- Rating Mania Scale.38
During acute mania or depression, lithium often is used in combination with another medications such as an antipsychotic or antidepressant. Used in the outpatient and non-acute setting, lithium may be an “add-on” or monotherapy for preventing recurrence of episodes. Response in early acute manic symptoms are predictive of later response and remission.39
Dosing strategies
An initial problem with lithium is side effects that emerge when beginning treatment, which may discourage the patient and family from using this agent. Starting with 150 mg/d for the first 2 or 3 doses is unlikely to produce any adverse effects and can show the patient that there is a high likelihood that he will be able to tolerate the medication. Gradual titration over several days—or even weeks—to the target dosage and serum levels will enhance patient compliance. Rate of dosage increase is best guided by tolerance to the medication. The general consensus is that lithium is most effective at levels of 0.6 to 0.8 mEq/L,40 although a lower level (0.5 mEq/L) over a 2-year period also can be effective.41 Lithium may be used in to treat acute mania at higher serum levels (0.8 to 1.2 mEq/L), however, the acute phase often requires urgent management, usually with an antipsychotic.
Emerging consensus
Although there is a need to gather and analyze longer observational periods to clarify the clinical and biological characteristics of persons who respond to lithium, there are several points of consensus. Management will be guided by patient characteristics such as age, comorbidities, and other therapies. Most studies that address the effect of lithium level focus on high vs low serum levels. There are 3 categories of lithium serum levels, low (<0.6 mEq/L), mid-range (0.6 to 0.8 mEq/L), and high (>0.8 mEq/L), each has risk-benefit considerations.
The LiTMUS study42 compared low-level lithium augmentation with optimized personal treatment without lithium. Both groups had similar outcomes but the lithium-treated group had significantly lower use of atypical antipsychotics. This may be important when considering the long-term risk of the metabolic syndrome because the tolerability and side-effect profile of lithium at lower levels is more favorable than that of atypical antipsychotics. As lithium levels increase, there seems to be concomitant increase in efficacy and side effects. Many patients will benefit with low-level lithium use; yet clearly some individuals require higher dosages for effective maintenance therapy.
Dosing and monitoring. In patients age >50 or those with comorbid medical conditions, use a lower level of lithium (<0.6 mEq/L). Most individuals with BD likely will benefit from the mid-range level strategy (0.6 to 0.8 mEq/L); however, there will be those who require a higher level. When beginning lithium, start at a low dosage (150 mg/d) and increase as tolerated to the desired serum level. With acute mania, temporary use of an antipsychotic will be required.
There are no tests available to determine whether a patient will do well at any of these lithium serum levels. Breakthrough mania in an adherent patient with a serum lithium level of 0.7 mEq/L indicates the need to obtain a higher lithium level. A major deficit in lithium research is the lack of long-term data (>5 years) on outcomes, clinical and biological features with lithium levels because of a lack of pharmaceutical company support.3,17 Monitoring mood symptoms using detailed mood charts, whether clinician-administered or self-reported, is an effective way to monitor outcomes, provided the clinician uses the same scales or methods to record a patient’s moods. If a patient wants to discontinue lithium, taper the drug over an extended period (months) to minimize the likelihood of emerging manic or depressive episodes related to drug discontinuation.
Managing side effects
Consider lithium’s side effects in the context of their short-, intermediate-, and long-term presence (Table 2). Gradually increasing the lithium dosage often will prevent side effects that manifest in the short term. If side effects emerge at low dosages, proceed slowly with lithium and manage symptoms with other medications. When a patient shows a change in side effects, obtain lithium and electrolytes levels; a change in mental status with confusion will require an acute lithium level.
A diary of symptoms or clinically relevant matters such as fluid intake or frequency of GI- or neurological-related events will help the clinician monitor the frequency and severity of side effects. The patient and clinician should not be discouraged by emerging side effects in the short term, because they may dissipate or become minimally intrusive.
Several strategies can alleviate immediate GI effects, such as dosing with meals, enteric-coated formulations, multiple dose strategies, and short-term use of antidiarrheal medicine as needed. Side effects that disrupt a patient’s fluid and electrolyte balance (diabetes insipidus) to the point of clouding mental status will require discontinuing the medication until mental status improves, then reconsideration of the treatment regime, which will include managing diabetes insipidus with amiloride. Managing side effects may require consultation with specialty services. Likewise, some patients might experience neurologic side effects, such as profound tremor, that interferes with their ability to function. However, many side effects can be managed symptomatically with practical strategies (eg, a sugar-free lozenge for dry mouth or dysgeusia). Consider lower lithium dosages and serum levels because patients may experience benefits with lower therapeutic levels.
Long-term side effects include decreased renal function, hypothyroidism, persistent tremor, and dermatologic effects of acne and alopecia. Monitor renal and thyroid function annually in stable patients and more frequently when making changes in the treatment plan.
Before discontinuing lithium, consider discussing the medical issues with a specialist who has experience with complications of lithium.
Bottom Line
Lithium is an effective and under used medication for managing bipolar disorder. Initial prejudices and side effects often deter patients and prescribers from proceeding with a therapeutic trial of lithium. Although the mid-range lithium level of 0.6 to 0.8 mEq/L is desirable, many patients will experience significant benefits with lower levels. Initial strategies include the use of low-dose preparations that are unlikely to have uncomfortable side effects.
Related Resources
• Andreasen A, Ellingrod VL. Lithium-induced diabetes insipidus: prevention and management. Current Psychiatry. 2013;12(7):42-45.
• Cipriani A, Hawton K, Stockton S, et al. Lithium in the prevention of suicide in mood disorders: updated systematic review and meta-analysis. BMJ. 2013;346:f3646. doi: 10.1136/bmj.f3646.
Drug Brand Names
Amiloride • Midamor Lithium • Eskalith, Lithobid
Disclosure
Dr. McInnis reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Shorter E. The history of lithium therapy. Bipolar Disord. 2009;11(11 suppl 2):4-9.
2. Goodwin FK, Jamison KR. Manic-depressive illness: bipolar disorders and recurrent depression. 2nd ed. New York, NY: Oxford University Press; 2007.
3. Burgess S, Geddes J, Hawton K, et al. Lithium for maintenance treatment of mood disorders. Cochrane Database Syst Rev. 2001:CD003013.
4. Bowden CL, Calabrese JR, McElroy SL, et al. A randomized, placebo-controlled 12-month trial of divalproex and lithium in treatment of outpatients with bipolar I disorder. Divalproex maintenance study group. Arch Gen Psychiatry. 2000;57(5):481-489.
5. Bowden CL, Calabrese JR, Sachs G, et al; Lamictal 606 Study Group. A placebo-controlled 18-month trial of lamotrigine and lithium maintenance treatment in recently manic or hypomanic patients with bipolar I disorder. Arch Gen Psychiatry. 2003;60(4):392-400.
6. Swann AC, Bowden CL, Calabrese JR, et al. Pattern of response to divalproex, lithium, or placebo in four naturalistic subtypes of mania. Neuropsychopharmacology. 2002;26(4):530-536.
7. Tohen M, Chengappa KN, Suppes T, et al. Efficacy of olanzapine in combination with valproate or lithium in the treatment of mania in patients partially nonresponsive to valproate or lithium monotherapy. Arch Gen Psychiatry. 2002;59(1):62-69.
8. Perlis RH, Smoller JW, Ferreira MA, et al. A genomewide association study of response to lithium for prevention of recurrence in bipolar disorder. Am J Psychiatry. 2009; 166(6):718-725.
9. Grof P, Duffy A, Cavazzoni P, et al. Is response to prophylactic lithium a familial trait? J Clin Psychiatry. 2002;63(10): 942-947.
10. Duffy A, Alda M, Kutcher S, et al. A prospective study of the offspring of bipolar parents responsive and nonresponsive to lithium treatment. J Clin Psychiatry. 2002;63(12): 1171-1178.
11. Goodwin FK, Fireman B, Simon GE, et al. Suicide risk in bipolar disorder during treatment with lithium and divalproex. JAMA. 2003;290(11):1467-1473.
12. Quiroz JA, Machado-Vieira R, Zarate CA Jr, et al. Novel insights into lithium’s mechanism of action: neurotrophic and neuroprotective effects. Neuropsychobiology. 2010; 62(1):50-60.
13. Forlenza OV, Diniz BS, Radanovic M, et al. Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: randomised controlled trial. Br J Psychiatry. 2011;198(5):351-356.
14. de Sousa RT, van de Bilt MT, Diniz BS, et al. Lithium increases plasma brain-derived neurotrophic factor in acute bipolar mania: a preliminary 4-week study. Neurosci Lett. 2011;494(1):54-56.
15. Nierenberg AA, Sylvia LG, Leon AC, et al; LiTMUS Study Group. Lithium treatment–moderate dose use study (LiTMUS) for bipolar disorder: rationale and design. Clin Trials. 2009;6(6):637-648.
16. Sylvia LG, Reilly-Harrington NA, Leon AC, et al. Methods to limit attrition in longitudinal comparative effectiveness trials: lessons from the Lithium Treatment - Moderate dose Use Study (LiTMUS) for bipolar disorder. Clin Trials. 2012;9(1):94-101.
17. McCarthy MJ, Leckband SG, Kelsoe JR. Pharmacogenetics of lithium response in bipolar disorder. Pharmacogenomics. 2010;11(10):1439-1465.
18. Can A, Schulze TG, Gould TD. Molecular actions and clinical pharmacogenetics of lithium therapy [published online February 15, 2014]. Pharmacol Biochem Behav. doi: 10.1016/j.pbb.2014.02.004.
19. Berridge MJ. Unlocking the secrets of cell signaling. Annu Rev Physiol. 2005;67:1-21.
20. Devaki R, Shankar Rao S, Nadgir SM. The effect of lithium on the adrenoceptor-mediated second messenger system in the rat brain. J Psychiatry Neurosci. 2006;31(4):246-252.
21. Pan JQ, Lewis MC, Ketterman JK, et al. AKT kinase activity is required for lithium to modulate mood-related behaviors in mice. Neuropsychopharmacology. 2011;36(7):1397-1411.
22. Hu LW, Kawamoto EM, Brietzke E, et al. The role of Wnt signaling and its interaction with diverse mechanisms of cellular apoptosis in the pathophysiology of bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35(1):11-17.
23. Chen HM, DeLong CJ, Bame M, et al. Transcripts involved in calcium signaling and telencephalic neuronal fate are altered in induced pluripotent stem cells from bipolar disorder patients [published online March 25, 2014]. Transl Psychiatry. doi:10.1038/tp.2014.12.
24. Jefferson JW. Lithium. In: Aronson JK, ed. Side effects of drugs annual, volume 26. Amsterdam, The Netherlands: Elsevier Science; 2003:19-29.
25. Baldessarini RJ, Tondo L, Floris G, et al. Effects of rapid cycling on response to lithium maintenance treatment in 360 bipolar I and II disorder patients. J Affect Disord. 2000;61(2):13-22.
26. Baldessarini RJ, Tondo L, Hennen J, et al. Latency and episodes before treatment: response to lithium maintenance in bipolar I and II disorders. Bipolar Disord. 1999;1(2): 91-97.
27. Fieve RR, Kumbaraci T, Dunner DL. Lithium prophylaxis of depression in bipolar I, bipolar II, and unipolar patients. Am J Psychiatry. 1976;133(8):925-929.
28. Peck CC, Pond SM, Becker CE, et al. An evaluation of the effects of lithium in the treatment of chronic alcoholism. II. Assessment of the two-period crossover design. Alcohol Clin Exp Res. 1981;5(2):252-255.
29. Peselow ED, Dunner DL, Fieve RR, et al. Lithium prophylaxis of depression in unipolar, bipolar II, and cyclothymic patients. Am J Psychiatry. 1982;139(6):747-752.
30. Bellino S, Paradiso E, Bogetto F. Efficacy and tolerability of pharmacotherapies for borderline personality disorder. CNS Drugs. 2008;22(8):671-692.
31. Alevizos B, Alevizos E, Leonardou A, et al. Low dosage lithium augmentation in venlafaxine resistant depression: an open-label study. Psychiatrike. 2012;23(2):143-148.
32. Goldberg JF, Sacks MH, Kocsis JH. Low-dose lithium augmentation of divalproex in geriatric mania. J Clin Psychiatry. 2000;61(4):304.
33. Saunders KE, Goodwin GM. New approaches in the treatment of bipolar depression. Curr Top Behav Neurosci. 2013;14:291-307.
34. Forlenza OV, Diniz BS, Radanovic M, et al. Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: randomised controlled trial. Br J Psychiatry. 2011;198(5):351-356.
35. Young RC, Biggs JT, Ziegler VE, et al. A rating scale for mania: reliability, validity and sensitivity. Br J Psychiatry. 1978;133:429-435.
36. Trivedi MH, Rush AJ, Ibrahim HM, et al. The Inventory of Depressive Symptomatology, Clinician Rating (IDS-C) and Self-Report (IDS-SR), and the Quick Inventory of Depressive Symptomatology, Clinician Rating (QIDS-C) and Self-Report (QIDS-SR) in public sector patients with mood disorders: a psychometric evaluation. Psychol Med. 2004;34(1):73-82.
37. Kroenke K, Spitzer RL, Williams JB. The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med. 2001;16(9):606-613.
38. Altman EG, Hedeker D, Peterson JL, et al. The Altman Self- Rating Mania Scale. Biol Psychiatry. 1997;42(10):948-955.
39. Machado-Vieira R, Luckenbaugh DA, Soeiro-de-Souza MG, et al. Early improvement with lithium in classic mania and its association with later response. J Affect Disord. 2013;144(1-2):160-164.
40. Severus WE, Lipkovich IA, Licht RW, et al. In search of optimal lithium levels and olanzapine doses in the long-term treatment of bipolar I disorder. A post-hoc analysis of the maintenance study by Tohen et al. 2005. Eur Psychiatry. 2010;25(8):443-449.
41. Vestergaard P, Licht RW, Brodersen A, et al. Outcome of lithium prophylaxis: a prospective follow-up of affective disorder patients assigned to high and low serum lithium levels. Acta Psychiatr Scand. 1998;98(4):310-315.
42. Nierenberg AA, Friedman ES, Bowden CL, et al. Lithium treatment moderate-dose use study (LiTMUS) for bipolar disorder: a randomized comparative effectiveness trial of optimized personalized treatment with and without lithium. Am J Psychiatry. 2013;170(1):102-111.
1. Shorter E. The history of lithium therapy. Bipolar Disord. 2009;11(11 suppl 2):4-9.
2. Goodwin FK, Jamison KR. Manic-depressive illness: bipolar disorders and recurrent depression. 2nd ed. New York, NY: Oxford University Press; 2007.
3. Burgess S, Geddes J, Hawton K, et al. Lithium for maintenance treatment of mood disorders. Cochrane Database Syst Rev. 2001:CD003013.
4. Bowden CL, Calabrese JR, McElroy SL, et al. A randomized, placebo-controlled 12-month trial of divalproex and lithium in treatment of outpatients with bipolar I disorder. Divalproex maintenance study group. Arch Gen Psychiatry. 2000;57(5):481-489.
5. Bowden CL, Calabrese JR, Sachs G, et al; Lamictal 606 Study Group. A placebo-controlled 18-month trial of lamotrigine and lithium maintenance treatment in recently manic or hypomanic patients with bipolar I disorder. Arch Gen Psychiatry. 2003;60(4):392-400.
6. Swann AC, Bowden CL, Calabrese JR, et al. Pattern of response to divalproex, lithium, or placebo in four naturalistic subtypes of mania. Neuropsychopharmacology. 2002;26(4):530-536.
7. Tohen M, Chengappa KN, Suppes T, et al. Efficacy of olanzapine in combination with valproate or lithium in the treatment of mania in patients partially nonresponsive to valproate or lithium monotherapy. Arch Gen Psychiatry. 2002;59(1):62-69.
8. Perlis RH, Smoller JW, Ferreira MA, et al. A genomewide association study of response to lithium for prevention of recurrence in bipolar disorder. Am J Psychiatry. 2009; 166(6):718-725.
9. Grof P, Duffy A, Cavazzoni P, et al. Is response to prophylactic lithium a familial trait? J Clin Psychiatry. 2002;63(10): 942-947.
10. Duffy A, Alda M, Kutcher S, et al. A prospective study of the offspring of bipolar parents responsive and nonresponsive to lithium treatment. J Clin Psychiatry. 2002;63(12): 1171-1178.
11. Goodwin FK, Fireman B, Simon GE, et al. Suicide risk in bipolar disorder during treatment with lithium and divalproex. JAMA. 2003;290(11):1467-1473.
12. Quiroz JA, Machado-Vieira R, Zarate CA Jr, et al. Novel insights into lithium’s mechanism of action: neurotrophic and neuroprotective effects. Neuropsychobiology. 2010; 62(1):50-60.
13. Forlenza OV, Diniz BS, Radanovic M, et al. Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: randomised controlled trial. Br J Psychiatry. 2011;198(5):351-356.
14. de Sousa RT, van de Bilt MT, Diniz BS, et al. Lithium increases plasma brain-derived neurotrophic factor in acute bipolar mania: a preliminary 4-week study. Neurosci Lett. 2011;494(1):54-56.
15. Nierenberg AA, Sylvia LG, Leon AC, et al; LiTMUS Study Group. Lithium treatment–moderate dose use study (LiTMUS) for bipolar disorder: rationale and design. Clin Trials. 2009;6(6):637-648.
16. Sylvia LG, Reilly-Harrington NA, Leon AC, et al. Methods to limit attrition in longitudinal comparative effectiveness trials: lessons from the Lithium Treatment - Moderate dose Use Study (LiTMUS) for bipolar disorder. Clin Trials. 2012;9(1):94-101.
17. McCarthy MJ, Leckband SG, Kelsoe JR. Pharmacogenetics of lithium response in bipolar disorder. Pharmacogenomics. 2010;11(10):1439-1465.
18. Can A, Schulze TG, Gould TD. Molecular actions and clinical pharmacogenetics of lithium therapy [published online February 15, 2014]. Pharmacol Biochem Behav. doi: 10.1016/j.pbb.2014.02.004.
19. Berridge MJ. Unlocking the secrets of cell signaling. Annu Rev Physiol. 2005;67:1-21.
20. Devaki R, Shankar Rao S, Nadgir SM. The effect of lithium on the adrenoceptor-mediated second messenger system in the rat brain. J Psychiatry Neurosci. 2006;31(4):246-252.
21. Pan JQ, Lewis MC, Ketterman JK, et al. AKT kinase activity is required for lithium to modulate mood-related behaviors in mice. Neuropsychopharmacology. 2011;36(7):1397-1411.
22. Hu LW, Kawamoto EM, Brietzke E, et al. The role of Wnt signaling and its interaction with diverse mechanisms of cellular apoptosis in the pathophysiology of bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35(1):11-17.
23. Chen HM, DeLong CJ, Bame M, et al. Transcripts involved in calcium signaling and telencephalic neuronal fate are altered in induced pluripotent stem cells from bipolar disorder patients [published online March 25, 2014]. Transl Psychiatry. doi:10.1038/tp.2014.12.
24. Jefferson JW. Lithium. In: Aronson JK, ed. Side effects of drugs annual, volume 26. Amsterdam, The Netherlands: Elsevier Science; 2003:19-29.
25. Baldessarini RJ, Tondo L, Floris G, et al. Effects of rapid cycling on response to lithium maintenance treatment in 360 bipolar I and II disorder patients. J Affect Disord. 2000;61(2):13-22.
26. Baldessarini RJ, Tondo L, Hennen J, et al. Latency and episodes before treatment: response to lithium maintenance in bipolar I and II disorders. Bipolar Disord. 1999;1(2): 91-97.
27. Fieve RR, Kumbaraci T, Dunner DL. Lithium prophylaxis of depression in bipolar I, bipolar II, and unipolar patients. Am J Psychiatry. 1976;133(8):925-929.
28. Peck CC, Pond SM, Becker CE, et al. An evaluation of the effects of lithium in the treatment of chronic alcoholism. II. Assessment of the two-period crossover design. Alcohol Clin Exp Res. 1981;5(2):252-255.
29. Peselow ED, Dunner DL, Fieve RR, et al. Lithium prophylaxis of depression in unipolar, bipolar II, and cyclothymic patients. Am J Psychiatry. 1982;139(6):747-752.
30. Bellino S, Paradiso E, Bogetto F. Efficacy and tolerability of pharmacotherapies for borderline personality disorder. CNS Drugs. 2008;22(8):671-692.
31. Alevizos B, Alevizos E, Leonardou A, et al. Low dosage lithium augmentation in venlafaxine resistant depression: an open-label study. Psychiatrike. 2012;23(2):143-148.
32. Goldberg JF, Sacks MH, Kocsis JH. Low-dose lithium augmentation of divalproex in geriatric mania. J Clin Psychiatry. 2000;61(4):304.
33. Saunders KE, Goodwin GM. New approaches in the treatment of bipolar depression. Curr Top Behav Neurosci. 2013;14:291-307.
34. Forlenza OV, Diniz BS, Radanovic M, et al. Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: randomised controlled trial. Br J Psychiatry. 2011;198(5):351-356.
35. Young RC, Biggs JT, Ziegler VE, et al. A rating scale for mania: reliability, validity and sensitivity. Br J Psychiatry. 1978;133:429-435.
36. Trivedi MH, Rush AJ, Ibrahim HM, et al. The Inventory of Depressive Symptomatology, Clinician Rating (IDS-C) and Self-Report (IDS-SR), and the Quick Inventory of Depressive Symptomatology, Clinician Rating (QIDS-C) and Self-Report (QIDS-SR) in public sector patients with mood disorders: a psychometric evaluation. Psychol Med. 2004;34(1):73-82.
37. Kroenke K, Spitzer RL, Williams JB. The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med. 2001;16(9):606-613.
38. Altman EG, Hedeker D, Peterson JL, et al. The Altman Self- Rating Mania Scale. Biol Psychiatry. 1997;42(10):948-955.
39. Machado-Vieira R, Luckenbaugh DA, Soeiro-de-Souza MG, et al. Early improvement with lithium in classic mania and its association with later response. J Affect Disord. 2013;144(1-2):160-164.
40. Severus WE, Lipkovich IA, Licht RW, et al. In search of optimal lithium levels and olanzapine doses in the long-term treatment of bipolar I disorder. A post-hoc analysis of the maintenance study by Tohen et al. 2005. Eur Psychiatry. 2010;25(8):443-449.
41. Vestergaard P, Licht RW, Brodersen A, et al. Outcome of lithium prophylaxis: a prospective follow-up of affective disorder patients assigned to high and low serum lithium levels. Acta Psychiatr Scand. 1998;98(4):310-315.
42. Nierenberg AA, Friedman ES, Bowden CL, et al. Lithium treatment moderate-dose use study (LiTMUS) for bipolar disorder: a randomized comparative effectiveness trial of optimized personalized treatment with and without lithium. Am J Psychiatry. 2013;170(1):102-111.

BPD and the broader landscape of neuropsychiatric illness
Dr. Henry A. Nasrallah’s recent Editorial on borderline personality disorder (BPD) (Current Psychiatry, From the Editor, April 2014, p. 19-20, 32 [http://bit.ly/1e8yAwE]) describes BPD as a heritable brain disease. I have been arguing this point for many years, often finding support from my colleague, Hagop Akiskal, MD, and opposition from my psychoanalytic colleagues.
In recent papers1,2 on brain changes in BPD and the connection between BPD and bipolar disorders, I wrote that there often is a heritable aspect to the condition. There are exceptions to such heritability, as in the setting of a horrific environment (eg, father-daughter incest, parental brutality), where the same symptoms seen in BPD develop primarily from post-natal influences. Dr. Akiskal and I were discussing this a long time back, before MRI. Now I feel vindicated, with generous help from someone of Dr. Nasrallah’s prestige and influence.
There also is electrophysiological (including evoked potential) evidence for neural pathology in BPD, as well as data derived from single photon emission CT scanning. The burgeoning literature on MRI and functional MRI studies of BPD is in good agreement about the brain changes most relevant to BPD and that are found with regularity in this condition.
Particularly when BPD is diagnosed in people (usually women) who do not have a history of neglect, sexual molestation, parental humiliation or cruelty, or head injury, what else is there, if not genetically predisposed alterations in the frontolimbic structures (and maybe the periaqueductal gray) that underlie the so-called “personality disorder,” and, not surprisingly, bipolar disorders, especially bipolar II disorder, which often is the other side of the coin as BPD, and amenable to the same combination of medication and psychotherapy?
Michael H. Stone, MD
Professor of Clinical Psychiatry Columbia College of Physicians and Surgeons
New York, New York
----------------------------------------------------------------------------------------------------------
As a psychiatrist/psychoanalyst who works with BPD patients, I read Dr. Nasrallah’s April 2014 Editorial with great interest and enthusiasm. Over the past 10 years, I have been impressed with the number of patients with BPD whose nonverbal learning disorders and auditory and visual processing disorders have gone undiagnosed. Recently, I lectured on this topic to the staff of a school for children with a range of neuropsychiatric disorders; the staff found my observations about such comorbidity consistent with their observations. These dysfunctions, or neurological variations—unknown to the parent and the child—interfere with early object-relation formation, attachment capacity, and learning. Neuropsychiatry and psychological development are, in fact, part of the same system.
An example: For 12 years, I have been treating a patient who has auditory processing and working memory problems, meaning that she could not process the connections among different ideas. This difficulty frustrated her parents, who, in their frustration, criticized her for not paying attention. She was labeled “bad” and assumed the role of the “black sheep” in her family. Although she was intelligent, she was often wrong in her judgments and choices, and easily frustrated. In therapy, as I realized what part of her problem was, I changed my technique.
When my patient asked me to tell her the sequence of understandings that we had just put together, I invited her to take my pad and write down her sense of it. As she described each part of that sequence to me, we would discuss it and I would remind her of lost fragments. Gradually, she learned to put ideas together; however, I also watched her struggle to hold these ideas in working memory and to use them.
Over time, she has improved and is more functional. After several years of disability, she returned to work, although she still struggles interpersonally.
With many of such patients, I have had to modify traditional techniques of psychotherapy. I am fascinated by, and enjoy, such intensive psychotherapy. I am also amazed to see the impact of previously unknown neuropathologic variations on development. The more I learn about the impact of neuropsychiatry on psychological development, the more I can help my patients.
Howard Wishnie, MD
Cambridge, Massachusetts
Dr. Nasrallah responds
I appreciate Dr. Stone’s kind words and concurrence with my thinking about BPD. It would have been appropriate to include discussion of neurophysiological findings in my Editorial, but I opted to use my limited space to focus on structural and functional neuroimaging and genetics.
Henry A. Nasrallah, MD
Professor and Chairman Department of Neurology & Psychiatry
Saint Louis University School of Medicine
St. Louis, Missouri
1. Stone MH. The spectrum of borderline personality disorder: a neurophysiological view. Neuropsychiatric Electrophysiology. In press.
2. Stone MH. A new look at borderline personality disorder and related disorders: hyper-activity in the limbic system and lower centers. Psychodyn Psychiatry. 2013;41(3):437-466.
Dr. Henry A. Nasrallah’s recent Editorial on borderline personality disorder (BPD) (Current Psychiatry, From the Editor, April 2014, p. 19-20, 32 [http://bit.ly/1e8yAwE]) describes BPD as a heritable brain disease. I have been arguing this point for many years, often finding support from my colleague, Hagop Akiskal, MD, and opposition from my psychoanalytic colleagues.
In recent papers1,2 on brain changes in BPD and the connection between BPD and bipolar disorders, I wrote that there often is a heritable aspect to the condition. There are exceptions to such heritability, as in the setting of a horrific environment (eg, father-daughter incest, parental brutality), where the same symptoms seen in BPD develop primarily from post-natal influences. Dr. Akiskal and I were discussing this a long time back, before MRI. Now I feel vindicated, with generous help from someone of Dr. Nasrallah’s prestige and influence.
There also is electrophysiological (including evoked potential) evidence for neural pathology in BPD, as well as data derived from single photon emission CT scanning. The burgeoning literature on MRI and functional MRI studies of BPD is in good agreement about the brain changes most relevant to BPD and that are found with regularity in this condition.
Particularly when BPD is diagnosed in people (usually women) who do not have a history of neglect, sexual molestation, parental humiliation or cruelty, or head injury, what else is there, if not genetically predisposed alterations in the frontolimbic structures (and maybe the periaqueductal gray) that underlie the so-called “personality disorder,” and, not surprisingly, bipolar disorders, especially bipolar II disorder, which often is the other side of the coin as BPD, and amenable to the same combination of medication and psychotherapy?
Michael H. Stone, MD
Professor of Clinical Psychiatry Columbia College of Physicians and Surgeons
New York, New York
----------------------------------------------------------------------------------------------------------
As a psychiatrist/psychoanalyst who works with BPD patients, I read Dr. Nasrallah’s April 2014 Editorial with great interest and enthusiasm. Over the past 10 years, I have been impressed with the number of patients with BPD whose nonverbal learning disorders and auditory and visual processing disorders have gone undiagnosed. Recently, I lectured on this topic to the staff of a school for children with a range of neuropsychiatric disorders; the staff found my observations about such comorbidity consistent with their observations. These dysfunctions, or neurological variations—unknown to the parent and the child—interfere with early object-relation formation, attachment capacity, and learning. Neuropsychiatry and psychological development are, in fact, part of the same system.
An example: For 12 years, I have been treating a patient who has auditory processing and working memory problems, meaning that she could not process the connections among different ideas. This difficulty frustrated her parents, who, in their frustration, criticized her for not paying attention. She was labeled “bad” and assumed the role of the “black sheep” in her family. Although she was intelligent, she was often wrong in her judgments and choices, and easily frustrated. In therapy, as I realized what part of her problem was, I changed my technique.
When my patient asked me to tell her the sequence of understandings that we had just put together, I invited her to take my pad and write down her sense of it. As she described each part of that sequence to me, we would discuss it and I would remind her of lost fragments. Gradually, she learned to put ideas together; however, I also watched her struggle to hold these ideas in working memory and to use them.
Over time, she has improved and is more functional. After several years of disability, she returned to work, although she still struggles interpersonally.
With many of such patients, I have had to modify traditional techniques of psychotherapy. I am fascinated by, and enjoy, such intensive psychotherapy. I am also amazed to see the impact of previously unknown neuropathologic variations on development. The more I learn about the impact of neuropsychiatry on psychological development, the more I can help my patients.
Howard Wishnie, MD
Cambridge, Massachusetts
Dr. Nasrallah responds
I appreciate Dr. Stone’s kind words and concurrence with my thinking about BPD. It would have been appropriate to include discussion of neurophysiological findings in my Editorial, but I opted to use my limited space to focus on structural and functional neuroimaging and genetics.
Henry A. Nasrallah, MD
Professor and Chairman Department of Neurology & Psychiatry
Saint Louis University School of Medicine
St. Louis, Missouri
Dr. Henry A. Nasrallah’s recent Editorial on borderline personality disorder (BPD) (Current Psychiatry, From the Editor, April 2014, p. 19-20, 32 [http://bit.ly/1e8yAwE]) describes BPD as a heritable brain disease. I have been arguing this point for many years, often finding support from my colleague, Hagop Akiskal, MD, and opposition from my psychoanalytic colleagues.
In recent papers1,2 on brain changes in BPD and the connection between BPD and bipolar disorders, I wrote that there often is a heritable aspect to the condition. There are exceptions to such heritability, as in the setting of a horrific environment (eg, father-daughter incest, parental brutality), where the same symptoms seen in BPD develop primarily from post-natal influences. Dr. Akiskal and I were discussing this a long time back, before MRI. Now I feel vindicated, with generous help from someone of Dr. Nasrallah’s prestige and influence.
There also is electrophysiological (including evoked potential) evidence for neural pathology in BPD, as well as data derived from single photon emission CT scanning. The burgeoning literature on MRI and functional MRI studies of BPD is in good agreement about the brain changes most relevant to BPD and that are found with regularity in this condition.
Particularly when BPD is diagnosed in people (usually women) who do not have a history of neglect, sexual molestation, parental humiliation or cruelty, or head injury, what else is there, if not genetically predisposed alterations in the frontolimbic structures (and maybe the periaqueductal gray) that underlie the so-called “personality disorder,” and, not surprisingly, bipolar disorders, especially bipolar II disorder, which often is the other side of the coin as BPD, and amenable to the same combination of medication and psychotherapy?
Michael H. Stone, MD
Professor of Clinical Psychiatry Columbia College of Physicians and Surgeons
New York, New York
----------------------------------------------------------------------------------------------------------
As a psychiatrist/psychoanalyst who works with BPD patients, I read Dr. Nasrallah’s April 2014 Editorial with great interest and enthusiasm. Over the past 10 years, I have been impressed with the number of patients with BPD whose nonverbal learning disorders and auditory and visual processing disorders have gone undiagnosed. Recently, I lectured on this topic to the staff of a school for children with a range of neuropsychiatric disorders; the staff found my observations about such comorbidity consistent with their observations. These dysfunctions, or neurological variations—unknown to the parent and the child—interfere with early object-relation formation, attachment capacity, and learning. Neuropsychiatry and psychological development are, in fact, part of the same system.
An example: For 12 years, I have been treating a patient who has auditory processing and working memory problems, meaning that she could not process the connections among different ideas. This difficulty frustrated her parents, who, in their frustration, criticized her for not paying attention. She was labeled “bad” and assumed the role of the “black sheep” in her family. Although she was intelligent, she was often wrong in her judgments and choices, and easily frustrated. In therapy, as I realized what part of her problem was, I changed my technique.
When my patient asked me to tell her the sequence of understandings that we had just put together, I invited her to take my pad and write down her sense of it. As she described each part of that sequence to me, we would discuss it and I would remind her of lost fragments. Gradually, she learned to put ideas together; however, I also watched her struggle to hold these ideas in working memory and to use them.
Over time, she has improved and is more functional. After several years of disability, she returned to work, although she still struggles interpersonally.
With many of such patients, I have had to modify traditional techniques of psychotherapy. I am fascinated by, and enjoy, such intensive psychotherapy. I am also amazed to see the impact of previously unknown neuropathologic variations on development. The more I learn about the impact of neuropsychiatry on psychological development, the more I can help my patients.
Howard Wishnie, MD
Cambridge, Massachusetts
Dr. Nasrallah responds
I appreciate Dr. Stone’s kind words and concurrence with my thinking about BPD. It would have been appropriate to include discussion of neurophysiological findings in my Editorial, but I opted to use my limited space to focus on structural and functional neuroimaging and genetics.
Henry A. Nasrallah, MD
Professor and Chairman Department of Neurology & Psychiatry
Saint Louis University School of Medicine
St. Louis, Missouri
1. Stone MH. The spectrum of borderline personality disorder: a neurophysiological view. Neuropsychiatric Electrophysiology. In press.
2. Stone MH. A new look at borderline personality disorder and related disorders: hyper-activity in the limbic system and lower centers. Psychodyn Psychiatry. 2013;41(3):437-466.
1. Stone MH. The spectrum of borderline personality disorder: a neurophysiological view. Neuropsychiatric Electrophysiology. In press.
2. Stone MH. A new look at borderline personality disorder and related disorders: hyper-activity in the limbic system and lower centers. Psychodyn Psychiatry. 2013;41(3):437-466.
Could ‘Rx: Pet therapy’ come back to bite you?
Dear Dr. Mossman,
My patient, Ms. A, asked me to write a letter to her landlord (who has a “no pets” policy) stating that she needed to keep her dog in her apartment for “therapeutic” purposes—to provide comfort and reduce her posttraumatic stress (PTSD) and anxiety. I hesitated. Could my written statement make me liable if her dog bit someone?
Submitted by “Dr. B”
Studies showing that animals can help outpatients manage psychiatric conditions have received a lot of publicity lately. As a result, more patients are asking physicians to provide documentation to support having pets in their apartments or letting their pets accompany them on planes and buses and at restaurants and shopping malls.
But sometimes, animals hurt people. The Centers for Disease Control and Prevention reports that dogs bite 4.5 million Americans each year and that one-fifth of dog bites cause injury that requires medical attention; in 2012, more than 27,000 dog-bite victims needed reconstructive surgery.1 If Dr. B writes a letter to support letting Ms. A keep a dog in her apartment, how likely is Dr. B to incur professional liability?
To answer this question, let’s examine:
• the history and background of “pet therapy”
• types of assistance animals
• potential liability for owners, landlords, and clinicians.
History and background
Using animals to improve hospitalized patients’ mental well-being dates back to the 18th century.2 In the late 1980s, medical publications began to document systematically how service dogs whose primary role was to help physically disabled individuals to navigate independently also provided social and emotional benefits.3-7 Since the 1990s, accessibility mandates in Title III of the Americans with Disabilities Act (ADA) (Table 18) have led to the gradual acceptance of service animals in public places where their presence was previously frowned upon or prohibited.9,10

If service dogs help people with physical problems feel better, it only makes sense that dogs and other animals might lessen emotional ailments, too.11-13 Florence Nightingale and Sigmund Freud both recognized that involving pets in treatment reduced patients’ depression and anxiety,14 but credit for formally introducing animals into therapy usually goes to psychologist Boris Levinson, whose 1969 book described how his dog Jingles helped troubled children communicate.15 Over the past decade, using animals— trained and untrained—for psychological assistance has become an increasingly popular therapeutic maneuver for diverse mental disorders, including autism, anxiety, schizophrenia, and PTSD.16-19
Terminology
Because animals can provide many types of assistance and support, a variety of terms are used to refer to them: service animals, companion animals, therapy pets, and so on. In certain situations (including the one described by Dr. B), carefully delineating animals’ roles and functions can reduce confusion and misinterpretation by patients, health care professionals, policy makers, and regulators.
Parenti et al20 have proposed a “taxonomy” for assistance dogs based on variables that include:
• performing task related to a disability
• the skill level required of the dog
• who uses the dog
• applicable training standards
• legal protections for the dog and its handler.
Table 220 summarizes this classification system and key variables that differentiate types of assistance dogs.
Certification
Health care facilities often require that visiting dogs have some form of “certification” that they are well behaved, and the ADA and many state statutes require that service dogs and some other animals be “certified” to perform their roles. Yet no federal or state statutes lay out explicit training standards or requirements for certification. Therapy Dogs International21 and Pet Partners22 are 2 organizations that provide certifications accepted by many agencies and organizations.
Assistance Dogs International, an assistance animal advocacy group, has proposed “minimum standards” for training and deployment of service dogs. These include responding to basic obedience commands from the client in public and at home, being able to perform at least 3 tasks to mitigate the client’s disability, teaching the client about dog training and health care, and scheduled follow-ups for skill maintenance. Dogs also should be spayed or neutered, properly vaccinated, nonaggressive, clean, and continent in public places.23
Liability laws
Most U.S. jurisdictions make owners liable for animal-caused injuries, including injuries caused by service dogs.24 In many states (eg, Minnesota25), an owner can be liable for dog-bite injury even if the owner did nothing wrong and had no reason to suspect from prior behavior that the dog might bite someone. Other jurisdictions require evidence of owner negligence, or they allow liability only when bites occur off the owner’s premises26 or if the owner let the dog run loose.27 Many homeowners’ insurance policies include liability coverage for dog bites, and a few companies offer a special canine liability policy.
Landlords often try to bar tenants from having a dog, partly to avoid liability for dog bites. Most states have case law stating that, if a tenant’s apparently friendly dog bites someone, the landlord is not liable for the injury28,29; landlords can be liable only if they know about a dangerous dog and do nothing about it.30 In a recent decision, however, the Kentucky Supreme Court made landlords statutory owners with potential liability for dog bites if they give tenants permission to have dogs “on or about” the rental premises.31
Clinicians and liability
Asking tenants to provide documentation about their need for therapeutic pets has become standard operating procedure for landlords in many states, so Ms. A’s request to Dr. B sounds reasonable. But could Dr. B’s written statement lead to liability if Ms. A’s dog bit and injured someone else?
The best answer is, “It’s conceivable, but really unlikely.” Donna Vanderpool, MBA, JD, an author and attorney who develops and implements risk management services for psychiatrists, has not seen any claims or case reports on litigation blaming mental health clinicians for injury caused by emotional support pets after the clinicians had written a letter for housing purposes (oral and written communications, April 7-13, 2014).
Dr. B might wonder whether writing a letter for Ms. A would imply that he had evaluated the dog and Ms. A’s ability to control it. Psychiatrists don’t usually discuss—let alone evaluate—the temperament or behavior of their patients’ pets; even if they did they aren’t experts on pet training. Recognizing this, Dr. B’s letter could include a statement to the effect that he was not vouching for the dog’s behavior, but only for how the dog would help Ms. A.
Dr. B also might talk with Ms. A about her need for the dog and whether she had obtained appropriate certification, as discussed above. The ADA provisions pertaining to use and presence of service animals do not apply to dogs that are merely patients’ pets, notwithstanding the genuine emotional benefits that a dog’s companionship might provide. Stating that a patient needs an animal to treat an illness might be fraud if the doctor knew the pet was just a buddy.
Bottom Line
Psychiatrists can expect that more and more patients will ask them for letters to support having pets accompany them at home or in public. Although liability seems unlikely, cautious psychiatrists can state in such letters that they have not evaluated the animal in question, only the potential benefits that the patient might derive from it.
Disclosure
Dr. Mossman reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing articles.
1. Dog Bites. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/homeandrecreationalsafety/ dog-bites/index.html. Updated October 25, 2013. Accessed April 22, 2014.
2. Serpell JA. Animal-assisted interventions in historical perspective. In Fine AH, ed. Handbook on animal-assisted therapy: theoretical foundations and guidelines for practice. 3rd ed. Burlington, MA: Academic Press; 2010:17-32.
3. Eddy J, Hart LA, Boltz RP. The effects of service dogs on social acknowledgments of people in wheelchairs. J Psychol. 1988;122(1):39-45.
4. Mader B, Hart LA, Bergin B. Social acknowledgments for children with disabilities: effects of service dogs. Child Dev. 1989;60(6):1529-1534.
5. Allen K, Blascovich J. The value of service dogs for people with severe ambulatory disabilities. A randomized controlled trial. JAMA. 1996;275(13):1001-1006.
6. Camp MM. The use of service dogs as an adaptive strategy: a qualitative study. Am J Occup Ther. 2001;55(5):509-517.
7. Allen K, Shykoff BE, Izzo JL Jr. Pet ownership, but not ace inhibitor therapy, blunts home blood pressure responses to mental stress. Hypertension. 2001;38(4):815-820.
8. ADA requirements: service animals. United States Department of Justice Civil Rights Division, Disability Rights Section Web site. http://www.ada.gov/service_ animals_2010.htm. Published September 15, 2010. Accessed April 22, 2014.
9. Eames E, Eames T. Interpreting legal mandates. Assistance dogs in medical facilities. Nurs Manage. 1997;28(6):49-51.
10. Houghtalen RP, Doody J. After the ADA: service dogs on inpatient psychiatric units. Bull Am Acad Psychiatry Law. 1995;23(2):211-217.
11. Wenthold N, Savage TA. Ethical issues with service animals. Top Stroke Rehabil. 2007;14(2):68-74.
12. DiSalvo H, Haiduven D, Johnson N, et al. Who let the dogs out? Infection control did: utility of dogs in health care settings and infection control aspects. Am J Infect Control. 2006;34:301-307.
13. Collins DM, Fitzgerald SG, Sachs-Ericsson N, et al. Psychosocial well-being and community participation of service dog partners. Disabil Rehabil Assist Technol. 2006;1(1-2):41-48.
14. Coren S. Foreward. In: Fine AH, ed. Handbook on animal-assisted therapy: theoretical foundations and guidelines for practice. 3rd ed. Burlington, MA: Academic Press; 2010: xv-xviii.
15. Levinson BM, Mallon GP. Pet-oriented child psychotherapy. 2nd ed. Springfield IL: Charles C Thomas Publisher, Ltd; 1997.
16. Esnayra J. Help from man’s best friend. Psychiatric service dogs are helping consumers deal with the symptoms of mental illness. Behav Healthc. 2007;27(7):30-32.
17. Barak Y, Savorai O, Mavashev S, et al. Animal-assisted therapy for elderly schizophrenic patients: a one year controlled trial. Am J Geriatr Psychiatry. 2001;9(4):439-442.
18. Burrows KE, Adams CL, Millman ST. Factors affecting behavior and welfare of service dogs for children with autism spectrum disorder. J Appl Anim Welf Sci. 2008;11(1):42-62.
19. Yount RA, Olmert MD, Lee MR. Service dog training program for treatment of posttraumatic stress in service members. US Army Med Dep J. 2012:63-69.
20. Parenti L, Foreman A, Meade BJ, et al. A revised taxonomy of assistance animals. J Rehabil Res Dev. 2013;50(6):745-756.
21. Testing Requirements. Therapy Dogs International. http:// www.tdi-dog.org/images/TestingBrochure.pdf. Accessed April 22, 2014.
22. How to become a registered therapy animal team. Pet Partners. http://www.petpartners.org/TAPinfo. Accessed April 22, 2014.
23. ADI Guide to Assistance Dog Laws. Assistance Dogs International. http://www.assistancedogsinternational. org/access-and-laws/adi-guide-to-assistance-dog-laws. Accessed April 22, 2014.
24. Id Stat §56-704.
25. Seim v Garavalia, 306 NW2d 806 (Minn 1981).
26. ME Rev Stat title 7, §3961.
27. Chadbourne v Kappaz, 2001 779 A2d 293 (DC App).
28. Stokes v Lyddy, 2002 75 (Conn App 252).
29. Georgianna v Gizzy, 483 NYS2d 892 (NY 1984).
30. Linebaugh v Hyndman, 516 A2d 638 (NJ 1986).
31. Benningfield v Zinsmeister, 367 SW3d 561 (Ky 2012).
Dear Dr. Mossman,
My patient, Ms. A, asked me to write a letter to her landlord (who has a “no pets” policy) stating that she needed to keep her dog in her apartment for “therapeutic” purposes—to provide comfort and reduce her posttraumatic stress (PTSD) and anxiety. I hesitated. Could my written statement make me liable if her dog bit someone?
Submitted by “Dr. B”
Studies showing that animals can help outpatients manage psychiatric conditions have received a lot of publicity lately. As a result, more patients are asking physicians to provide documentation to support having pets in their apartments or letting their pets accompany them on planes and buses and at restaurants and shopping malls.
But sometimes, animals hurt people. The Centers for Disease Control and Prevention reports that dogs bite 4.5 million Americans each year and that one-fifth of dog bites cause injury that requires medical attention; in 2012, more than 27,000 dog-bite victims needed reconstructive surgery.1 If Dr. B writes a letter to support letting Ms. A keep a dog in her apartment, how likely is Dr. B to incur professional liability?
To answer this question, let’s examine:
• the history and background of “pet therapy”
• types of assistance animals
• potential liability for owners, landlords, and clinicians.
History and background
Using animals to improve hospitalized patients’ mental well-being dates back to the 18th century.2 In the late 1980s, medical publications began to document systematically how service dogs whose primary role was to help physically disabled individuals to navigate independently also provided social and emotional benefits.3-7 Since the 1990s, accessibility mandates in Title III of the Americans with Disabilities Act (ADA) (Table 18) have led to the gradual acceptance of service animals in public places where their presence was previously frowned upon or prohibited.9,10
If service dogs help people with physical problems feel better, it only makes sense that dogs and other animals might lessen emotional ailments, too.11-13 Florence Nightingale and Sigmund Freud both recognized that involving pets in treatment reduced patients’ depression and anxiety,14 but credit for formally introducing animals into therapy usually goes to psychologist Boris Levinson, whose 1969 book described how his dog Jingles helped troubled children communicate.15 Over the past decade, using animals— trained and untrained—for psychological assistance has become an increasingly popular therapeutic maneuver for diverse mental disorders, including autism, anxiety, schizophrenia, and PTSD.16-19
Terminology
Because animals can provide many types of assistance and support, a variety of terms are used to refer to them: service animals, companion animals, therapy pets, and so on. In certain situations (including the one described by Dr. B), carefully delineating animals’ roles and functions can reduce confusion and misinterpretation by patients, health care professionals, policy makers, and regulators.
Parenti et al20 have proposed a “taxonomy” for assistance dogs based on variables that include:
• performing task related to a disability
• the skill level required of the dog
• who uses the dog
• applicable training standards
• legal protections for the dog and its handler.
Table 220 summarizes this classification system and key variables that differentiate types of assistance dogs.
Certification
Health care facilities often require that visiting dogs have some form of “certification” that they are well behaved, and the ADA and many state statutes require that service dogs and some other animals be “certified” to perform their roles. Yet no federal or state statutes lay out explicit training standards or requirements for certification. Therapy Dogs International21 and Pet Partners22 are 2 organizations that provide certifications accepted by many agencies and organizations.
Assistance Dogs International, an assistance animal advocacy group, has proposed “minimum standards” for training and deployment of service dogs. These include responding to basic obedience commands from the client in public and at home, being able to perform at least 3 tasks to mitigate the client’s disability, teaching the client about dog training and health care, and scheduled follow-ups for skill maintenance. Dogs also should be spayed or neutered, properly vaccinated, nonaggressive, clean, and continent in public places.23
Liability laws
Most U.S. jurisdictions make owners liable for animal-caused injuries, including injuries caused by service dogs.24 In many states (eg, Minnesota25), an owner can be liable for dog-bite injury even if the owner did nothing wrong and had no reason to suspect from prior behavior that the dog might bite someone. Other jurisdictions require evidence of owner negligence, or they allow liability only when bites occur off the owner’s premises26 or if the owner let the dog run loose.27 Many homeowners’ insurance policies include liability coverage for dog bites, and a few companies offer a special canine liability policy.
Landlords often try to bar tenants from having a dog, partly to avoid liability for dog bites. Most states have case law stating that, if a tenant’s apparently friendly dog bites someone, the landlord is not liable for the injury28,29; landlords can be liable only if they know about a dangerous dog and do nothing about it.30 In a recent decision, however, the Kentucky Supreme Court made landlords statutory owners with potential liability for dog bites if they give tenants permission to have dogs “on or about” the rental premises.31
Clinicians and liability
Asking tenants to provide documentation about their need for therapeutic pets has become standard operating procedure for landlords in many states, so Ms. A’s request to Dr. B sounds reasonable. But could Dr. B’s written statement lead to liability if Ms. A’s dog bit and injured someone else?
The best answer is, “It’s conceivable, but really unlikely.” Donna Vanderpool, MBA, JD, an author and attorney who develops and implements risk management services for psychiatrists, has not seen any claims or case reports on litigation blaming mental health clinicians for injury caused by emotional support pets after the clinicians had written a letter for housing purposes (oral and written communications, April 7-13, 2014).
Dr. B might wonder whether writing a letter for Ms. A would imply that he had evaluated the dog and Ms. A’s ability to control it. Psychiatrists don’t usually discuss—let alone evaluate—the temperament or behavior of their patients’ pets; even if they did they aren’t experts on pet training. Recognizing this, Dr. B’s letter could include a statement to the effect that he was not vouching for the dog’s behavior, but only for how the dog would help Ms. A.
Dr. B also might talk with Ms. A about her need for the dog and whether she had obtained appropriate certification, as discussed above. The ADA provisions pertaining to use and presence of service animals do not apply to dogs that are merely patients’ pets, notwithstanding the genuine emotional benefits that a dog’s companionship might provide. Stating that a patient needs an animal to treat an illness might be fraud if the doctor knew the pet was just a buddy.
Bottom Line
Psychiatrists can expect that more and more patients will ask them for letters to support having pets accompany them at home or in public. Although liability seems unlikely, cautious psychiatrists can state in such letters that they have not evaluated the animal in question, only the potential benefits that the patient might derive from it.
Disclosure
Dr. Mossman reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing articles.
Dear Dr. Mossman,
My patient, Ms. A, asked me to write a letter to her landlord (who has a “no pets” policy) stating that she needed to keep her dog in her apartment for “therapeutic” purposes—to provide comfort and reduce her posttraumatic stress (PTSD) and anxiety. I hesitated. Could my written statement make me liable if her dog bit someone?
Submitted by “Dr. B”
Studies showing that animals can help outpatients manage psychiatric conditions have received a lot of publicity lately. As a result, more patients are asking physicians to provide documentation to support having pets in their apartments or letting their pets accompany them on planes and buses and at restaurants and shopping malls.
But sometimes, animals hurt people. The Centers for Disease Control and Prevention reports that dogs bite 4.5 million Americans each year and that one-fifth of dog bites cause injury that requires medical attention; in 2012, more than 27,000 dog-bite victims needed reconstructive surgery.1 If Dr. B writes a letter to support letting Ms. A keep a dog in her apartment, how likely is Dr. B to incur professional liability?
To answer this question, let’s examine:
• the history and background of “pet therapy”
• types of assistance animals
• potential liability for owners, landlords, and clinicians.
History and background
Using animals to improve hospitalized patients’ mental well-being dates back to the 18th century.2 In the late 1980s, medical publications began to document systematically how service dogs whose primary role was to help physically disabled individuals to navigate independently also provided social and emotional benefits.3-7 Since the 1990s, accessibility mandates in Title III of the Americans with Disabilities Act (ADA) (Table 18) have led to the gradual acceptance of service animals in public places where their presence was previously frowned upon or prohibited.9,10
If service dogs help people with physical problems feel better, it only makes sense that dogs and other animals might lessen emotional ailments, too.11-13 Florence Nightingale and Sigmund Freud both recognized that involving pets in treatment reduced patients’ depression and anxiety,14 but credit for formally introducing animals into therapy usually goes to psychologist Boris Levinson, whose 1969 book described how his dog Jingles helped troubled children communicate.15 Over the past decade, using animals— trained and untrained—for psychological assistance has become an increasingly popular therapeutic maneuver for diverse mental disorders, including autism, anxiety, schizophrenia, and PTSD.16-19
Terminology
Because animals can provide many types of assistance and support, a variety of terms are used to refer to them: service animals, companion animals, therapy pets, and so on. In certain situations (including the one described by Dr. B), carefully delineating animals’ roles and functions can reduce confusion and misinterpretation by patients, health care professionals, policy makers, and regulators.
Parenti et al20 have proposed a “taxonomy” for assistance dogs based on variables that include:
• performing task related to a disability
• the skill level required of the dog
• who uses the dog
• applicable training standards
• legal protections for the dog and its handler.
Table 220 summarizes this classification system and key variables that differentiate types of assistance dogs.
Certification
Health care facilities often require that visiting dogs have some form of “certification” that they are well behaved, and the ADA and many state statutes require that service dogs and some other animals be “certified” to perform their roles. Yet no federal or state statutes lay out explicit training standards or requirements for certification. Therapy Dogs International21 and Pet Partners22 are 2 organizations that provide certifications accepted by many agencies and organizations.
Assistance Dogs International, an assistance animal advocacy group, has proposed “minimum standards” for training and deployment of service dogs. These include responding to basic obedience commands from the client in public and at home, being able to perform at least 3 tasks to mitigate the client’s disability, teaching the client about dog training and health care, and scheduled follow-ups for skill maintenance. Dogs also should be spayed or neutered, properly vaccinated, nonaggressive, clean, and continent in public places.23
Liability laws
Most U.S. jurisdictions make owners liable for animal-caused injuries, including injuries caused by service dogs.24 In many states (eg, Minnesota25), an owner can be liable for dog-bite injury even if the owner did nothing wrong and had no reason to suspect from prior behavior that the dog might bite someone. Other jurisdictions require evidence of owner negligence, or they allow liability only when bites occur off the owner’s premises26 or if the owner let the dog run loose.27 Many homeowners’ insurance policies include liability coverage for dog bites, and a few companies offer a special canine liability policy.
Landlords often try to bar tenants from having a dog, partly to avoid liability for dog bites. Most states have case law stating that, if a tenant’s apparently friendly dog bites someone, the landlord is not liable for the injury28,29; landlords can be liable only if they know about a dangerous dog and do nothing about it.30 In a recent decision, however, the Kentucky Supreme Court made landlords statutory owners with potential liability for dog bites if they give tenants permission to have dogs “on or about” the rental premises.31
Clinicians and liability
Asking tenants to provide documentation about their need for therapeutic pets has become standard operating procedure for landlords in many states, so Ms. A’s request to Dr. B sounds reasonable. But could Dr. B’s written statement lead to liability if Ms. A’s dog bit and injured someone else?
The best answer is, “It’s conceivable, but really unlikely.” Donna Vanderpool, MBA, JD, an author and attorney who develops and implements risk management services for psychiatrists, has not seen any claims or case reports on litigation blaming mental health clinicians for injury caused by emotional support pets after the clinicians had written a letter for housing purposes (oral and written communications, April 7-13, 2014).
Dr. B might wonder whether writing a letter for Ms. A would imply that he had evaluated the dog and Ms. A’s ability to control it. Psychiatrists don’t usually discuss—let alone evaluate—the temperament or behavior of their patients’ pets; even if they did they aren’t experts on pet training. Recognizing this, Dr. B’s letter could include a statement to the effect that he was not vouching for the dog’s behavior, but only for how the dog would help Ms. A.
Dr. B also might talk with Ms. A about her need for the dog and whether she had obtained appropriate certification, as discussed above. The ADA provisions pertaining to use and presence of service animals do not apply to dogs that are merely patients’ pets, notwithstanding the genuine emotional benefits that a dog’s companionship might provide. Stating that a patient needs an animal to treat an illness might be fraud if the doctor knew the pet was just a buddy.
Bottom Line
Psychiatrists can expect that more and more patients will ask them for letters to support having pets accompany them at home or in public. Although liability seems unlikely, cautious psychiatrists can state in such letters that they have not evaluated the animal in question, only the potential benefits that the patient might derive from it.
Disclosure
Dr. Mossman reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing articles.
1. Dog Bites. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/homeandrecreationalsafety/ dog-bites/index.html. Updated October 25, 2013. Accessed April 22, 2014.
2. Serpell JA. Animal-assisted interventions in historical perspective. In Fine AH, ed. Handbook on animal-assisted therapy: theoretical foundations and guidelines for practice. 3rd ed. Burlington, MA: Academic Press; 2010:17-32.
3. Eddy J, Hart LA, Boltz RP. The effects of service dogs on social acknowledgments of people in wheelchairs. J Psychol. 1988;122(1):39-45.
4. Mader B, Hart LA, Bergin B. Social acknowledgments for children with disabilities: effects of service dogs. Child Dev. 1989;60(6):1529-1534.
5. Allen K, Blascovich J. The value of service dogs for people with severe ambulatory disabilities. A randomized controlled trial. JAMA. 1996;275(13):1001-1006.
6. Camp MM. The use of service dogs as an adaptive strategy: a qualitative study. Am J Occup Ther. 2001;55(5):509-517.
7. Allen K, Shykoff BE, Izzo JL Jr. Pet ownership, but not ace inhibitor therapy, blunts home blood pressure responses to mental stress. Hypertension. 2001;38(4):815-820.
8. ADA requirements: service animals. United States Department of Justice Civil Rights Division, Disability Rights Section Web site. http://www.ada.gov/service_ animals_2010.htm. Published September 15, 2010. Accessed April 22, 2014.
9. Eames E, Eames T. Interpreting legal mandates. Assistance dogs in medical facilities. Nurs Manage. 1997;28(6):49-51.
10. Houghtalen RP, Doody J. After the ADA: service dogs on inpatient psychiatric units. Bull Am Acad Psychiatry Law. 1995;23(2):211-217.
11. Wenthold N, Savage TA. Ethical issues with service animals. Top Stroke Rehabil. 2007;14(2):68-74.
12. DiSalvo H, Haiduven D, Johnson N, et al. Who let the dogs out? Infection control did: utility of dogs in health care settings and infection control aspects. Am J Infect Control. 2006;34:301-307.
13. Collins DM, Fitzgerald SG, Sachs-Ericsson N, et al. Psychosocial well-being and community participation of service dog partners. Disabil Rehabil Assist Technol. 2006;1(1-2):41-48.
14. Coren S. Foreward. In: Fine AH, ed. Handbook on animal-assisted therapy: theoretical foundations and guidelines for practice. 3rd ed. Burlington, MA: Academic Press; 2010: xv-xviii.
15. Levinson BM, Mallon GP. Pet-oriented child psychotherapy. 2nd ed. Springfield IL: Charles C Thomas Publisher, Ltd; 1997.
16. Esnayra J. Help from man’s best friend. Psychiatric service dogs are helping consumers deal with the symptoms of mental illness. Behav Healthc. 2007;27(7):30-32.
17. Barak Y, Savorai O, Mavashev S, et al. Animal-assisted therapy for elderly schizophrenic patients: a one year controlled trial. Am J Geriatr Psychiatry. 2001;9(4):439-442.
18. Burrows KE, Adams CL, Millman ST. Factors affecting behavior and welfare of service dogs for children with autism spectrum disorder. J Appl Anim Welf Sci. 2008;11(1):42-62.
19. Yount RA, Olmert MD, Lee MR. Service dog training program for treatment of posttraumatic stress in service members. US Army Med Dep J. 2012:63-69.
20. Parenti L, Foreman A, Meade BJ, et al. A revised taxonomy of assistance animals. J Rehabil Res Dev. 2013;50(6):745-756.
21. Testing Requirements. Therapy Dogs International. http:// www.tdi-dog.org/images/TestingBrochure.pdf. Accessed April 22, 2014.
22. How to become a registered therapy animal team. Pet Partners. http://www.petpartners.org/TAPinfo. Accessed April 22, 2014.
23. ADI Guide to Assistance Dog Laws. Assistance Dogs International. http://www.assistancedogsinternational. org/access-and-laws/adi-guide-to-assistance-dog-laws. Accessed April 22, 2014.
24. Id Stat §56-704.
25. Seim v Garavalia, 306 NW2d 806 (Minn 1981).
26. ME Rev Stat title 7, §3961.
27. Chadbourne v Kappaz, 2001 779 A2d 293 (DC App).
28. Stokes v Lyddy, 2002 75 (Conn App 252).
29. Georgianna v Gizzy, 483 NYS2d 892 (NY 1984).
30. Linebaugh v Hyndman, 516 A2d 638 (NJ 1986).
31. Benningfield v Zinsmeister, 367 SW3d 561 (Ky 2012).
1. Dog Bites. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/homeandrecreationalsafety/ dog-bites/index.html. Updated October 25, 2013. Accessed April 22, 2014.
2. Serpell JA. Animal-assisted interventions in historical perspective. In Fine AH, ed. Handbook on animal-assisted therapy: theoretical foundations and guidelines for practice. 3rd ed. Burlington, MA: Academic Press; 2010:17-32.
3. Eddy J, Hart LA, Boltz RP. The effects of service dogs on social acknowledgments of people in wheelchairs. J Psychol. 1988;122(1):39-45.
4. Mader B, Hart LA, Bergin B. Social acknowledgments for children with disabilities: effects of service dogs. Child Dev. 1989;60(6):1529-1534.
5. Allen K, Blascovich J. The value of service dogs for people with severe ambulatory disabilities. A randomized controlled trial. JAMA. 1996;275(13):1001-1006.
6. Camp MM. The use of service dogs as an adaptive strategy: a qualitative study. Am J Occup Ther. 2001;55(5):509-517.
7. Allen K, Shykoff BE, Izzo JL Jr. Pet ownership, but not ace inhibitor therapy, blunts home blood pressure responses to mental stress. Hypertension. 2001;38(4):815-820.
8. ADA requirements: service animals. United States Department of Justice Civil Rights Division, Disability Rights Section Web site. http://www.ada.gov/service_ animals_2010.htm. Published September 15, 2010. Accessed April 22, 2014.
9. Eames E, Eames T. Interpreting legal mandates. Assistance dogs in medical facilities. Nurs Manage. 1997;28(6):49-51.
10. Houghtalen RP, Doody J. After the ADA: service dogs on inpatient psychiatric units. Bull Am Acad Psychiatry Law. 1995;23(2):211-217.
11. Wenthold N, Savage TA. Ethical issues with service animals. Top Stroke Rehabil. 2007;14(2):68-74.
12. DiSalvo H, Haiduven D, Johnson N, et al. Who let the dogs out? Infection control did: utility of dogs in health care settings and infection control aspects. Am J Infect Control. 2006;34:301-307.
13. Collins DM, Fitzgerald SG, Sachs-Ericsson N, et al. Psychosocial well-being and community participation of service dog partners. Disabil Rehabil Assist Technol. 2006;1(1-2):41-48.
14. Coren S. Foreward. In: Fine AH, ed. Handbook on animal-assisted therapy: theoretical foundations and guidelines for practice. 3rd ed. Burlington, MA: Academic Press; 2010: xv-xviii.
15. Levinson BM, Mallon GP. Pet-oriented child psychotherapy. 2nd ed. Springfield IL: Charles C Thomas Publisher, Ltd; 1997.
16. Esnayra J. Help from man’s best friend. Psychiatric service dogs are helping consumers deal with the symptoms of mental illness. Behav Healthc. 2007;27(7):30-32.
17. Barak Y, Savorai O, Mavashev S, et al. Animal-assisted therapy for elderly schizophrenic patients: a one year controlled trial. Am J Geriatr Psychiatry. 2001;9(4):439-442.
18. Burrows KE, Adams CL, Millman ST. Factors affecting behavior and welfare of service dogs for children with autism spectrum disorder. J Appl Anim Welf Sci. 2008;11(1):42-62.
19. Yount RA, Olmert MD, Lee MR. Service dog training program for treatment of posttraumatic stress in service members. US Army Med Dep J. 2012:63-69.
20. Parenti L, Foreman A, Meade BJ, et al. A revised taxonomy of assistance animals. J Rehabil Res Dev. 2013;50(6):745-756.
21. Testing Requirements. Therapy Dogs International. http:// www.tdi-dog.org/images/TestingBrochure.pdf. Accessed April 22, 2014.
22. How to become a registered therapy animal team. Pet Partners. http://www.petpartners.org/TAPinfo. Accessed April 22, 2014.
23. ADI Guide to Assistance Dog Laws. Assistance Dogs International. http://www.assistancedogsinternational. org/access-and-laws/adi-guide-to-assistance-dog-laws. Accessed April 22, 2014.
24. Id Stat §56-704.
25. Seim v Garavalia, 306 NW2d 806 (Minn 1981).
26. ME Rev Stat title 7, §3961.
27. Chadbourne v Kappaz, 2001 779 A2d 293 (DC App).
28. Stokes v Lyddy, 2002 75 (Conn App 252).
29. Georgianna v Gizzy, 483 NYS2d 892 (NY 1984).
30. Linebaugh v Hyndman, 516 A2d 638 (NJ 1986).
31. Benningfield v Zinsmeister, 367 SW3d 561 (Ky 2012).
The transient truths of medical ‘progress’
I might have a jaundiced view of progress but, across most medical specialties, diseases are still managed, not cured. Chronicity is almost ubiquitous among medical ailments, and no specialty can boast that it restores function completely and fully restores patients’ quality of life.
Psychiatry has had its share of missteps
Prefrontal lobotomy is perhaps the most infamous of many discredited treatments that were introduced as a great solution to severe brain disorders such as schizophrenia.1 Prefrontal lobotomy (leucotomy) was initially heralded as a major medical advance in 1935; its originator, neurosurgeon António Egas Moniz, shared the Nobel Prize in Medicine or Physiology in 1949 for what is now regarded as mayhem. Prefrontal lobotomy was widely used for many conditions—not just for psychosis—but it fell from favor rapidly after the discovery of anti-psychotic drugs.
A similar fate befell other treatments that were introduced to psychiatry:
• malaria therapy (1917) for general paresis of the insane (the condition was later recognized as tertiary syphilis)
• deep sleep therapy (1920) for schizophrenia
• insulin shock therapy (1933), also for schizophrenia.
Those discredited therapies were lauded as significant advances, only to be shunned later as harmful, even barbaric.
Treating addiction is another saga of false steps. Fifty-nine different treatments for addiction have been introduced over the past few decades, many later discredited as “psychoquackery.”2 In the breathless rush to cure desperate conditions, there often is the risk that pseudoscience will masquerade as science. Many patients suffer needlessly before the medical community examines the accumulated evidence and discredits useless or harmful treatments.
Psychiatry isn’t alone in lacking cures
A fitting slogan of many non-psychiatric medical specialties is “to treat, perchance to cure.” Consider some examples:
• In cardiology, congestive heart failure, a chronic illness, is managed but rarely cured, and leads to early mortality.
• Nephrologists struggle to maintain a semblance of kidney function in renal failure patients, before placing them on the long waiting list for a kidney transplant.
• Gastroenterologists can only hope to maintain liver function in severe hepatitis, or to alleviate the misery of ulcerative colitis.
• Rheumatologists do what they can to relieve the debilitating symptoms of rheumatoid arthritis, systemic lupus erythematosus, and Sjögren’s syndrome.
• Pulmonologists know they can never restore normal lung function for their patients with chronic obstructive pulmonary disease; they can only help them hang on with partial function.
• Oncologists valiantly fight aggressive cancers with the hope of prolonging life for a few months or years.
• Neurologists valiantly try to manage multiple sclerosis, Parkinson’s disease, Alzheimer’s disease, epilepsy, stroke, myasthenia gravis, and amyotrophic lateral sclerosis—often with limited, if any, success at achieving remission.
Internal medicine has had its share of discredited therapies, too—ones that were withdrawn because they caused harm or were of dubious or inconclusive efficacy.3 Thanks to careful analysis of the efficacy and safety of medical procedures introduced during the past decade, we know that 40% of 146 procedures examined were eventually discredited and withdrawn. (That kind of analysis should be undertaken in psychotherapy, where evidence-based therapies can be counted on one hand but dozens more are promoted as legitimate.4 Psychotherapy can be harmful.5)
As with patients in psychiatry, patients of all these specialties are at risk of suffering disruptive iatrogenic side effects that, at times, approach torture—just to have progression of disease halted but not necessarily to deliver full remission. The quality of life for patients who have a chronic disease ranges from barely tolerable to poor, but is rarely good or optimal—and that is the case across all of medicine, including psychiatry.
Desperation often drives dubious innovation
There are numerous “desperate” diseases across all medical specialties, including psychiatry. Radical and harmful measures are sometimes proposed and marketed to treat many of those conditions; more often, useless, ineffective, futile “treatments” are introduced, and it might take years before they are discredited and withdrawn.6,7 Useless treatments can be harmful, too, because they delay the use of potentially effective procedures.
Move forward with caution!
What does this brief look at the missteps of medicine tell us? First, medical progress is like the mambo: We take steps forward but then step backward again; and, as Karl Popper noted, science learns more from its failures than from its successes.8 Second, all physicians must be judicious and guided by evidence when they select treatments.
For your patients’ sake, choose wisely!9
1. Valenstein ES. Great and desperate cures: The rise and decline of psychosurgery and other radical treatments for mental illness. New York, NY: Basic Books; 1986.
2. Norcross JC, Koocher GP, Fala NC, et al. What does not work? Expert consensus on discredited treatments in the addictions. J Addic Med. 2010;4(3):174-180.
3. Prasad V, Vandross A, Toomey C, et al. A decade of reversal: an analysis of 146 contradicted medical practices. Mayo Clin Proc. 2013;88(8):790-798.
4. Corsini RJ. Handbook of innovative therapy. Philadelphia, PA: John Wiley & Sons, Inc; 2001.
5. Berk M, Parker G. The elephant on the couch: side-effects of psychotherapy. Austr N Z J Psychiatry. 2009;43(9):787-794.
6. Iaonnidis JP. Contradicted and initially stronger effects in highly cited clinical research. JAMA. 2005;294(2): 218-228.
7. Elshang AG, Watt AM, Mundy L, et al. Over 150 potentially low-value health care practices: an Australian study. Med J Aust. 2012;197(10):556-560.
8. Popper K. The logic of scientific discovery. London, United Kingdom: Hutchinson & Co.; 1959.
9. Cassel CK, Guest JA. Choosing wisely: helping physicians and patients make smart decision about their care. JAMA. 2012;307(17):1801-1802.
I might have a jaundiced view of progress but, across most medical specialties, diseases are still managed, not cured. Chronicity is almost ubiquitous among medical ailments, and no specialty can boast that it restores function completely and fully restores patients’ quality of life.
Psychiatry has had its share of missteps
Prefrontal lobotomy is perhaps the most infamous of many discredited treatments that were introduced as a great solution to severe brain disorders such as schizophrenia.1 Prefrontal lobotomy (leucotomy) was initially heralded as a major medical advance in 1935; its originator, neurosurgeon António Egas Moniz, shared the Nobel Prize in Medicine or Physiology in 1949 for what is now regarded as mayhem. Prefrontal lobotomy was widely used for many conditions—not just for psychosis—but it fell from favor rapidly after the discovery of anti-psychotic drugs.
A similar fate befell other treatments that were introduced to psychiatry:
• malaria therapy (1917) for general paresis of the insane (the condition was later recognized as tertiary syphilis)
• deep sleep therapy (1920) for schizophrenia
• insulin shock therapy (1933), also for schizophrenia.
Those discredited therapies were lauded as significant advances, only to be shunned later as harmful, even barbaric.
Treating addiction is another saga of false steps. Fifty-nine different treatments for addiction have been introduced over the past few decades, many later discredited as “psychoquackery.”2 In the breathless rush to cure desperate conditions, there often is the risk that pseudoscience will masquerade as science. Many patients suffer needlessly before the medical community examines the accumulated evidence and discredits useless or harmful treatments.
Psychiatry isn’t alone in lacking cures
A fitting slogan of many non-psychiatric medical specialties is “to treat, perchance to cure.” Consider some examples:
• In cardiology, congestive heart failure, a chronic illness, is managed but rarely cured, and leads to early mortality.
• Nephrologists struggle to maintain a semblance of kidney function in renal failure patients, before placing them on the long waiting list for a kidney transplant.
• Gastroenterologists can only hope to maintain liver function in severe hepatitis, or to alleviate the misery of ulcerative colitis.
• Rheumatologists do what they can to relieve the debilitating symptoms of rheumatoid arthritis, systemic lupus erythematosus, and Sjögren’s syndrome.
• Pulmonologists know they can never restore normal lung function for their patients with chronic obstructive pulmonary disease; they can only help them hang on with partial function.
• Oncologists valiantly fight aggressive cancers with the hope of prolonging life for a few months or years.
• Neurologists valiantly try to manage multiple sclerosis, Parkinson’s disease, Alzheimer’s disease, epilepsy, stroke, myasthenia gravis, and amyotrophic lateral sclerosis—often with limited, if any, success at achieving remission.
Internal medicine has had its share of discredited therapies, too—ones that were withdrawn because they caused harm or were of dubious or inconclusive efficacy.3 Thanks to careful analysis of the efficacy and safety of medical procedures introduced during the past decade, we know that 40% of 146 procedures examined were eventually discredited and withdrawn. (That kind of analysis should be undertaken in psychotherapy, where evidence-based therapies can be counted on one hand but dozens more are promoted as legitimate.4 Psychotherapy can be harmful.5)
As with patients in psychiatry, patients of all these specialties are at risk of suffering disruptive iatrogenic side effects that, at times, approach torture—just to have progression of disease halted but not necessarily to deliver full remission. The quality of life for patients who have a chronic disease ranges from barely tolerable to poor, but is rarely good or optimal—and that is the case across all of medicine, including psychiatry.
Desperation often drives dubious innovation
There are numerous “desperate” diseases across all medical specialties, including psychiatry. Radical and harmful measures are sometimes proposed and marketed to treat many of those conditions; more often, useless, ineffective, futile “treatments” are introduced, and it might take years before they are discredited and withdrawn.6,7 Useless treatments can be harmful, too, because they delay the use of potentially effective procedures.
Move forward with caution!
What does this brief look at the missteps of medicine tell us? First, medical progress is like the mambo: We take steps forward but then step backward again; and, as Karl Popper noted, science learns more from its failures than from its successes.8 Second, all physicians must be judicious and guided by evidence when they select treatments.
For your patients’ sake, choose wisely!9
I might have a jaundiced view of progress but, across most medical specialties, diseases are still managed, not cured. Chronicity is almost ubiquitous among medical ailments, and no specialty can boast that it restores function completely and fully restores patients’ quality of life.
Psychiatry has had its share of missteps
Prefrontal lobotomy is perhaps the most infamous of many discredited treatments that were introduced as a great solution to severe brain disorders such as schizophrenia.1 Prefrontal lobotomy (leucotomy) was initially heralded as a major medical advance in 1935; its originator, neurosurgeon António Egas Moniz, shared the Nobel Prize in Medicine or Physiology in 1949 for what is now regarded as mayhem. Prefrontal lobotomy was widely used for many conditions—not just for psychosis—but it fell from favor rapidly after the discovery of anti-psychotic drugs.
A similar fate befell other treatments that were introduced to psychiatry:
• malaria therapy (1917) for general paresis of the insane (the condition was later recognized as tertiary syphilis)
• deep sleep therapy (1920) for schizophrenia
• insulin shock therapy (1933), also for schizophrenia.
Those discredited therapies were lauded as significant advances, only to be shunned later as harmful, even barbaric.
Treating addiction is another saga of false steps. Fifty-nine different treatments for addiction have been introduced over the past few decades, many later discredited as “psychoquackery.”2 In the breathless rush to cure desperate conditions, there often is the risk that pseudoscience will masquerade as science. Many patients suffer needlessly before the medical community examines the accumulated evidence and discredits useless or harmful treatments.
Psychiatry isn’t alone in lacking cures
A fitting slogan of many non-psychiatric medical specialties is “to treat, perchance to cure.” Consider some examples:
• In cardiology, congestive heart failure, a chronic illness, is managed but rarely cured, and leads to early mortality.
• Nephrologists struggle to maintain a semblance of kidney function in renal failure patients, before placing them on the long waiting list for a kidney transplant.
• Gastroenterologists can only hope to maintain liver function in severe hepatitis, or to alleviate the misery of ulcerative colitis.
• Rheumatologists do what they can to relieve the debilitating symptoms of rheumatoid arthritis, systemic lupus erythematosus, and Sjögren’s syndrome.
• Pulmonologists know they can never restore normal lung function for their patients with chronic obstructive pulmonary disease; they can only help them hang on with partial function.
• Oncologists valiantly fight aggressive cancers with the hope of prolonging life for a few months or years.
• Neurologists valiantly try to manage multiple sclerosis, Parkinson’s disease, Alzheimer’s disease, epilepsy, stroke, myasthenia gravis, and amyotrophic lateral sclerosis—often with limited, if any, success at achieving remission.
Internal medicine has had its share of discredited therapies, too—ones that were withdrawn because they caused harm or were of dubious or inconclusive efficacy.3 Thanks to careful analysis of the efficacy and safety of medical procedures introduced during the past decade, we know that 40% of 146 procedures examined were eventually discredited and withdrawn. (That kind of analysis should be undertaken in psychotherapy, where evidence-based therapies can be counted on one hand but dozens more are promoted as legitimate.4 Psychotherapy can be harmful.5)
As with patients in psychiatry, patients of all these specialties are at risk of suffering disruptive iatrogenic side effects that, at times, approach torture—just to have progression of disease halted but not necessarily to deliver full remission. The quality of life for patients who have a chronic disease ranges from barely tolerable to poor, but is rarely good or optimal—and that is the case across all of medicine, including psychiatry.
Desperation often drives dubious innovation
There are numerous “desperate” diseases across all medical specialties, including psychiatry. Radical and harmful measures are sometimes proposed and marketed to treat many of those conditions; more often, useless, ineffective, futile “treatments” are introduced, and it might take years before they are discredited and withdrawn.6,7 Useless treatments can be harmful, too, because they delay the use of potentially effective procedures.
Move forward with caution!
What does this brief look at the missteps of medicine tell us? First, medical progress is like the mambo: We take steps forward but then step backward again; and, as Karl Popper noted, science learns more from its failures than from its successes.8 Second, all physicians must be judicious and guided by evidence when they select treatments.
For your patients’ sake, choose wisely!9
1. Valenstein ES. Great and desperate cures: The rise and decline of psychosurgery and other radical treatments for mental illness. New York, NY: Basic Books; 1986.
2. Norcross JC, Koocher GP, Fala NC, et al. What does not work? Expert consensus on discredited treatments in the addictions. J Addic Med. 2010;4(3):174-180.
3. Prasad V, Vandross A, Toomey C, et al. A decade of reversal: an analysis of 146 contradicted medical practices. Mayo Clin Proc. 2013;88(8):790-798.
4. Corsini RJ. Handbook of innovative therapy. Philadelphia, PA: John Wiley & Sons, Inc; 2001.
5. Berk M, Parker G. The elephant on the couch: side-effects of psychotherapy. Austr N Z J Psychiatry. 2009;43(9):787-794.
6. Iaonnidis JP. Contradicted and initially stronger effects in highly cited clinical research. JAMA. 2005;294(2): 218-228.
7. Elshang AG, Watt AM, Mundy L, et al. Over 150 potentially low-value health care practices: an Australian study. Med J Aust. 2012;197(10):556-560.
8. Popper K. The logic of scientific discovery. London, United Kingdom: Hutchinson & Co.; 1959.
9. Cassel CK, Guest JA. Choosing wisely: helping physicians and patients make smart decision about their care. JAMA. 2012;307(17):1801-1802.
1. Valenstein ES. Great and desperate cures: The rise and decline of psychosurgery and other radical treatments for mental illness. New York, NY: Basic Books; 1986.
2. Norcross JC, Koocher GP, Fala NC, et al. What does not work? Expert consensus on discredited treatments in the addictions. J Addic Med. 2010;4(3):174-180.
3. Prasad V, Vandross A, Toomey C, et al. A decade of reversal: an analysis of 146 contradicted medical practices. Mayo Clin Proc. 2013;88(8):790-798.
4. Corsini RJ. Handbook of innovative therapy. Philadelphia, PA: John Wiley & Sons, Inc; 2001.
5. Berk M, Parker G. The elephant on the couch: side-effects of psychotherapy. Austr N Z J Psychiatry. 2009;43(9):787-794.
6. Iaonnidis JP. Contradicted and initially stronger effects in highly cited clinical research. JAMA. 2005;294(2): 218-228.
7. Elshang AG, Watt AM, Mundy L, et al. Over 150 potentially low-value health care practices: an Australian study. Med J Aust. 2012;197(10):556-560.
8. Popper K. The logic of scientific discovery. London, United Kingdom: Hutchinson & Co.; 1959.
9. Cassel CK, Guest JA. Choosing wisely: helping physicians and patients make smart decision about their care. JAMA. 2012;307(17):1801-1802.
Sleep restriction therapy
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel

Epilepsy and psychiatric illness

Pills to powder: A clinician’s reference for crushable psychotropic medications
Many patients experience difficulty swallowing pills, for various reasons:
• discomfort (particularly pediatric and geriatric patients)
• postsurgical need for an alternate route of enteral intake (nasogastric tube, gastrostomy, jejunostomy)
• dysphagia due to a neurologic disorder (multiple sclerosis, impaired gag reflex, dementing processes)
• odynophagia (pain upon swallowing) due to gastroesophageal reflux or a structural abnormality
• a structural abnormality of the head or neck that impairs swallowing.1
If these difficulties are not addressed, they can interfere with medication adherence. In those instances, using an alternative dosage form or manipulating an available formulation might be required.
Crushing guidelines
There are limited data on crushed-form products and their impact on efficacy. Therefore, when patients have difficulty taking pills, switching to liquid solution or orally disintegrating forms is recommended. However, most psychotropics are available only as tablets or capsules. Patients can crush their pills immediately before administration for easier intake. The following are some general guidelines for doing so:2
• Scored tablets typically can be crushed.
• Crushing sublingual and buccal tablets can alter their effectiveness.
• Crushing sustained-release medications can eliminate the sustained-release action.3
• Enteric-coated medications should not be crushed, because this can alter drug absorption.
• Capsules can generally be opened to administer powdered contents, unless the capsule has time-release properties or an enteric coating.
The accompanying Table, organized by drug class, indicates whether a drug can be crushed to a powdered form, which usually is mixed with food or liquid for easier intake. The Table also lists liquid and orally disintegrating forms available, and other routes, including injectable immediate and long-acting formulations. Helping patients find a medication formulation that suits their needs strengthens adherence and the therapeutic relationship.
1. Schiele JT, Quinzler R, Klimm HD, et al. Difficulties swallowing solid oral dosage forms in a general practice population: prevalence, causes, and relationship to dosage forms. Eur J Clin Pharmacol. 2013;69(4): 937-948.
2. PL Detail-Document, Meds That Should Not Be Crushed. Pharmacist’s Letter/Prescriber’s Letter. July 2012.
3. Mitchell JF. Oral dosage forms that should not be crushed. http://www.ismp. org/tools/donotcrush.pdf. Updated January 2014. Accessed March 13, 2014.
Many patients experience difficulty swallowing pills, for various reasons:
• discomfort (particularly pediatric and geriatric patients)
• postsurgical need for an alternate route of enteral intake (nasogastric tube, gastrostomy, jejunostomy)
• dysphagia due to a neurologic disorder (multiple sclerosis, impaired gag reflex, dementing processes)
• odynophagia (pain upon swallowing) due to gastroesophageal reflux or a structural abnormality
• a structural abnormality of the head or neck that impairs swallowing.1
If these difficulties are not addressed, they can interfere with medication adherence. In those instances, using an alternative dosage form or manipulating an available formulation might be required.
Crushing guidelines
There are limited data on crushed-form products and their impact on efficacy. Therefore, when patients have difficulty taking pills, switching to liquid solution or orally disintegrating forms is recommended. However, most psychotropics are available only as tablets or capsules. Patients can crush their pills immediately before administration for easier intake. The following are some general guidelines for doing so:2
• Scored tablets typically can be crushed.
• Crushing sublingual and buccal tablets can alter their effectiveness.
• Crushing sustained-release medications can eliminate the sustained-release action.3
• Enteric-coated medications should not be crushed, because this can alter drug absorption.
• Capsules can generally be opened to administer powdered contents, unless the capsule has time-release properties or an enteric coating.
The accompanying Table, organized by drug class, indicates whether a drug can be crushed to a powdered form, which usually is mixed with food or liquid for easier intake. The Table also lists liquid and orally disintegrating forms available, and other routes, including injectable immediate and long-acting formulations. Helping patients find a medication formulation that suits their needs strengthens adherence and the therapeutic relationship.
Many patients experience difficulty swallowing pills, for various reasons:
• discomfort (particularly pediatric and geriatric patients)
• postsurgical need for an alternate route of enteral intake (nasogastric tube, gastrostomy, jejunostomy)
• dysphagia due to a neurologic disorder (multiple sclerosis, impaired gag reflex, dementing processes)
• odynophagia (pain upon swallowing) due to gastroesophageal reflux or a structural abnormality
• a structural abnormality of the head or neck that impairs swallowing.1
If these difficulties are not addressed, they can interfere with medication adherence. In those instances, using an alternative dosage form or manipulating an available formulation might be required.
Crushing guidelines
There are limited data on crushed-form products and their impact on efficacy. Therefore, when patients have difficulty taking pills, switching to liquid solution or orally disintegrating forms is recommended. However, most psychotropics are available only as tablets or capsules. Patients can crush their pills immediately before administration for easier intake. The following are some general guidelines for doing so:2
• Scored tablets typically can be crushed.
• Crushing sublingual and buccal tablets can alter their effectiveness.
• Crushing sustained-release medications can eliminate the sustained-release action.3
• Enteric-coated medications should not be crushed, because this can alter drug absorption.
• Capsules can generally be opened to administer powdered contents, unless the capsule has time-release properties or an enteric coating.
The accompanying Table, organized by drug class, indicates whether a drug can be crushed to a powdered form, which usually is mixed with food or liquid for easier intake. The Table also lists liquid and orally disintegrating forms available, and other routes, including injectable immediate and long-acting formulations. Helping patients find a medication formulation that suits their needs strengthens adherence and the therapeutic relationship.
1. Schiele JT, Quinzler R, Klimm HD, et al. Difficulties swallowing solid oral dosage forms in a general practice population: prevalence, causes, and relationship to dosage forms. Eur J Clin Pharmacol. 2013;69(4): 937-948.
2. PL Detail-Document, Meds That Should Not Be Crushed. Pharmacist’s Letter/Prescriber’s Letter. July 2012.
3. Mitchell JF. Oral dosage forms that should not be crushed. http://www.ismp. org/tools/donotcrush.pdf. Updated January 2014. Accessed March 13, 2014.
1. Schiele JT, Quinzler R, Klimm HD, et al. Difficulties swallowing solid oral dosage forms in a general practice population: prevalence, causes, and relationship to dosage forms. Eur J Clin Pharmacol. 2013;69(4): 937-948.
2. PL Detail-Document, Meds That Should Not Be Crushed. Pharmacist’s Letter/Prescriber’s Letter. July 2012.
3. Mitchell JF. Oral dosage forms that should not be crushed. http://www.ismp. org/tools/donotcrush.pdf. Updated January 2014. Accessed March 13, 2014.
6 ‘M’s to keep in mind when you next see a patient with anorexia nervosa
Anorexia nervosa is associated with comorbid psychiatric disorders, severe physical complications, and high mortality. To help you remember important clinical information when working with patients with anorexia, we propose this “6 M” model for screening, treatment, and prognosis.
Monitor closely. Anorexia can go undiagnosed and untreated for years. During your patients’ office visits, ask about body image, exercise habits, and menstrual irregularities, especially when seeing at-risk youth. During physical examination, reluctance to be weighed, vital sign abnormalities (eg, orthostatic hypotension, variability in pulse), skin abnormalities (lanugo hair, dryness), and marks indicating self-harm can serve as diagnostic indicators. Consider hospitalization for patients at <75% of their ideal body weight, who refuse to eat, or who show vital signs and laboratory abnormalities.
Media. By providing information on healthy eating and nutrition, the Internet can be an excellent resource for people with an eating disorder; however, you should also be aware of the impact of so-called pro-ana Web sites. People with anorexia use these Web sites to discuss their illness, but the sites sometimes glorify eating disorders as a lifestyle choice, and can be a place to share tips and tricks on extreme dieting, and might promote what is known as “thinspiration” in popular culture.
Meals. The American Dietetic Association recommends that anorexic patients begin oral intake at no more than 30 to 40 kcal/kg/day, and then gradually increase it, with a weight gain goal of 0.5 to 1 lb per week.
This graduated weight gain is done to prevent refeeding syndrome. After chronic starvation, intracellular phosphate stores are depleted and once carbohydrate intake resumes, insulin release causes phosphate to enter cells, thereby leading to hypophosphatemia. This electrolyte abnormality can result in cardiac failure. As a result, consider regular monitoring of phosphate levels, especially during the first week of reintroducing food.
Multimodal therapy. Despite being notoriously difficult to treat, patients with anorexia might respond to psychotherapy—especially family therapy—with an increased remission rate and faster return to health, compared with other forms of treatment. With a multimodal regimen involving proper refeeding techniques, family therapy, and medications as appropriate, recovery is possible.
Medications might be a helpful adjunct in patients who do not gain weight despite psychotherapy and proper nutritional measures. For example:
• There is some research on medications such as olanzapine and anxiolytics for treating anorexia.
• A low-dose anxiolytic might benefit patients with preprandial anxiety.
• Comorbid psychiatric disorders might improve during treatment of the eating disorder.
• Selective serotonin reuptake inhibitors and second-generation antipsychotics might help manage severe comorbid psychiatric disorders.
Because of low body weight and altered plasma protein binding, start medications at a low dosage. The risk of adverse effects can increase because more “free” medication is available. Consider avoiding medications such as bupropion and tricyclic antidepressants, because they carry an increased risk of seizures and cardiac effects, respectively.
Morbidity and mortality. Untreated anorexia has the highest mortality among psychiatric disorders: approximately 5.1 deaths for every 1,000 people.1 Recent meta-analyses show that patients with anorexia may have a 5.86 times greater risk of death than the general population.1 Serious sequelae include cardiac complications; osteoporosis; infertility; and comorbid psychiatric conditions such as substance abuse, depression, and obsessive-compulsive disorder.2
1. Arcelus J, Mitchell AJ, Wales J, et al. Mortality rates in patients with anorexia nervosa and other eating disorders. A meta-analysis of 36 studies. Arch Gen Psychiatry. 2011; 68(7):724-731.
2. Yager J, Andersen AE. Clinical practice. Anorexia nervosa. N Engl J Med. 2005;353(14):1481-1488.
Anorexia nervosa is associated with comorbid psychiatric disorders, severe physical complications, and high mortality. To help you remember important clinical information when working with patients with anorexia, we propose this “6 M” model for screening, treatment, and prognosis.
Monitor closely. Anorexia can go undiagnosed and untreated for years. During your patients’ office visits, ask about body image, exercise habits, and menstrual irregularities, especially when seeing at-risk youth. During physical examination, reluctance to be weighed, vital sign abnormalities (eg, orthostatic hypotension, variability in pulse), skin abnormalities (lanugo hair, dryness), and marks indicating self-harm can serve as diagnostic indicators. Consider hospitalization for patients at <75% of their ideal body weight, who refuse to eat, or who show vital signs and laboratory abnormalities.
Media. By providing information on healthy eating and nutrition, the Internet can be an excellent resource for people with an eating disorder; however, you should also be aware of the impact of so-called pro-ana Web sites. People with anorexia use these Web sites to discuss their illness, but the sites sometimes glorify eating disorders as a lifestyle choice, and can be a place to share tips and tricks on extreme dieting, and might promote what is known as “thinspiration” in popular culture.
Meals. The American Dietetic Association recommends that anorexic patients begin oral intake at no more than 30 to 40 kcal/kg/day, and then gradually increase it, with a weight gain goal of 0.5 to 1 lb per week.
This graduated weight gain is done to prevent refeeding syndrome. After chronic starvation, intracellular phosphate stores are depleted and once carbohydrate intake resumes, insulin release causes phosphate to enter cells, thereby leading to hypophosphatemia. This electrolyte abnormality can result in cardiac failure. As a result, consider regular monitoring of phosphate levels, especially during the first week of reintroducing food.
Multimodal therapy. Despite being notoriously difficult to treat, patients with anorexia might respond to psychotherapy—especially family therapy—with an increased remission rate and faster return to health, compared with other forms of treatment. With a multimodal regimen involving proper refeeding techniques, family therapy, and medications as appropriate, recovery is possible.
Medications might be a helpful adjunct in patients who do not gain weight despite psychotherapy and proper nutritional measures. For example:
• There is some research on medications such as olanzapine and anxiolytics for treating anorexia.
• A low-dose anxiolytic might benefit patients with preprandial anxiety.
• Comorbid psychiatric disorders might improve during treatment of the eating disorder.
• Selective serotonin reuptake inhibitors and second-generation antipsychotics might help manage severe comorbid psychiatric disorders.
Because of low body weight and altered plasma protein binding, start medications at a low dosage. The risk of adverse effects can increase because more “free” medication is available. Consider avoiding medications such as bupropion and tricyclic antidepressants, because they carry an increased risk of seizures and cardiac effects, respectively.
Morbidity and mortality. Untreated anorexia has the highest mortality among psychiatric disorders: approximately 5.1 deaths for every 1,000 people.1 Recent meta-analyses show that patients with anorexia may have a 5.86 times greater risk of death than the general population.1 Serious sequelae include cardiac complications; osteoporosis; infertility; and comorbid psychiatric conditions such as substance abuse, depression, and obsessive-compulsive disorder.2
Anorexia nervosa is associated with comorbid psychiatric disorders, severe physical complications, and high mortality. To help you remember important clinical information when working with patients with anorexia, we propose this “6 M” model for screening, treatment, and prognosis.
Monitor closely. Anorexia can go undiagnosed and untreated for years. During your patients’ office visits, ask about body image, exercise habits, and menstrual irregularities, especially when seeing at-risk youth. During physical examination, reluctance to be weighed, vital sign abnormalities (eg, orthostatic hypotension, variability in pulse), skin abnormalities (lanugo hair, dryness), and marks indicating self-harm can serve as diagnostic indicators. Consider hospitalization for patients at <75% of their ideal body weight, who refuse to eat, or who show vital signs and laboratory abnormalities.
Media. By providing information on healthy eating and nutrition, the Internet can be an excellent resource for people with an eating disorder; however, you should also be aware of the impact of so-called pro-ana Web sites. People with anorexia use these Web sites to discuss their illness, but the sites sometimes glorify eating disorders as a lifestyle choice, and can be a place to share tips and tricks on extreme dieting, and might promote what is known as “thinspiration” in popular culture.
Meals. The American Dietetic Association recommends that anorexic patients begin oral intake at no more than 30 to 40 kcal/kg/day, and then gradually increase it, with a weight gain goal of 0.5 to 1 lb per week.
This graduated weight gain is done to prevent refeeding syndrome. After chronic starvation, intracellular phosphate stores are depleted and once carbohydrate intake resumes, insulin release causes phosphate to enter cells, thereby leading to hypophosphatemia. This electrolyte abnormality can result in cardiac failure. As a result, consider regular monitoring of phosphate levels, especially during the first week of reintroducing food.
Multimodal therapy. Despite being notoriously difficult to treat, patients with anorexia might respond to psychotherapy—especially family therapy—with an increased remission rate and faster return to health, compared with other forms of treatment. With a multimodal regimen involving proper refeeding techniques, family therapy, and medications as appropriate, recovery is possible.
Medications might be a helpful adjunct in patients who do not gain weight despite psychotherapy and proper nutritional measures. For example:
• There is some research on medications such as olanzapine and anxiolytics for treating anorexia.
• A low-dose anxiolytic might benefit patients with preprandial anxiety.
• Comorbid psychiatric disorders might improve during treatment of the eating disorder.
• Selective serotonin reuptake inhibitors and second-generation antipsychotics might help manage severe comorbid psychiatric disorders.
Because of low body weight and altered plasma protein binding, start medications at a low dosage. The risk of adverse effects can increase because more “free” medication is available. Consider avoiding medications such as bupropion and tricyclic antidepressants, because they carry an increased risk of seizures and cardiac effects, respectively.
Morbidity and mortality. Untreated anorexia has the highest mortality among psychiatric disorders: approximately 5.1 deaths for every 1,000 people.1 Recent meta-analyses show that patients with anorexia may have a 5.86 times greater risk of death than the general population.1 Serious sequelae include cardiac complications; osteoporosis; infertility; and comorbid psychiatric conditions such as substance abuse, depression, and obsessive-compulsive disorder.2
1. Arcelus J, Mitchell AJ, Wales J, et al. Mortality rates in patients with anorexia nervosa and other eating disorders. A meta-analysis of 36 studies. Arch Gen Psychiatry. 2011; 68(7):724-731.
2. Yager J, Andersen AE. Clinical practice. Anorexia nervosa. N Engl J Med. 2005;353(14):1481-1488.
1. Arcelus J, Mitchell AJ, Wales J, et al. Mortality rates in patients with anorexia nervosa and other eating disorders. A meta-analysis of 36 studies. Arch Gen Psychiatry. 2011; 68(7):724-731.
2. Yager J, Andersen AE. Clinical practice. Anorexia nervosa. N Engl J Med. 2005;353(14):1481-1488.
What to do when your patient who takes clozapine enters a smoke-free facility
Mr. D, age 30, has a 12-year history of schizophrenia and is experiencing worsening auditory hallucinations despite reported medication adherence. He has been taking clozapine, maintenance dosages 500 to 700 mg/d, for 4 years and smokes 2 packs of cigarettes a day. When Mr. D is admitted to a nonsmoking inpatient psychiatric facility, he receives nicotine transdermal patches, 21 mg/d, for nicotine withdrawal. Mr. D’s most recent outpatient clozapine dosage, 700 mg/d, is resumed. All laboratory tests, including complete blood count with differential, are within normal limits at admission.
Five days later Mr. D is tachycardic with a heart rate of 109 beats per minute. When assessing Mr. D, we notice he has alogia and that, when he does speak, his speech is slowed with a 4 to 5 second delay in response. He also appears sedated. We observe occasional mild jerking of his shoulder and lower legs.
Mr. D reports that his auditory hallucinations have lessened since his admission, but complains of difficulty remembering information and feeling tired during the day. The treatment team suspects clozapine toxicity; his trough clozapine level is 1,350 ng/mL (therapeutic range, 350 to 1,000 ng/mL).
It is well documented that cigarette smoke can induce cytochrome P450 (CYP) isoenzymes, specifically CYP1A1, CYP1A2, and CYP2E1. Because clozapine is primarily metabolized by CYP1A2 (approximately 70%), smoking can induce clozapine metabolism and abruptly stopping smoking can increase clozapine levels.1 The polycyclic aromatic hydrocarbons, not the nicotine, found in cigarettes are thought to be responsible for CYP1A2 induction; therefore, use of a nicotine replacement product did not prevent the increase in Mr. D’s clozapine levels.
Examining the evidence

Meyer1 evaluated clozapine levels before and after implementation of a hospital-wide smoking ban (N = 11). Clozapine dosages were not adjusted at the time of the smoking ban, which resulted in a mean 72% increase in clozapine levels after a minimum of 2 weeks as nonsmokers. Even after eliminating 2 outliers, the mean increase in clozapine levels was 36.1%. Murayama-Sung et al2 reported a statistically significant increase in the level of clozapine (46%, P = .004) and the level of norclozapine (23%, P = .02) after a hospital-wide smoking ban was instituted (N = 14). However, the pre-change and post-change in the ratio of clozapine to norclozapine level was not found to be statistically significant. Haslemo et al3 found that smoking as few as 7 to 12 cigarettes a day was sufficient for maximum induction of CYP1A2. Because Mr. D was smoking 2 packs of cigarettes a day (40 cigarettes) with an clozapine dosage 700 mg/d as an outpatient, he likely experienced significant induction of clozapine metabolism through CYP1A2, which was no longer present when he stopped smoking.
Therapeutic clozapine concentrations are typically above 350 and 420 ng/mL.4 Concentrations >700 ng/mL are associated with increased adverse effects, but generally are not associated with a higher response; levels >900 ng/mL have been associated with toxicity.4 Clozapine-treated patients on a stable dosage who smoke can experience clozapine-related adverse effects after admission to a smoke-free facility secondary to an increase in the clozapine concentration (Table 1).4

Five days after admission to the facility, Mr. D was noted to have myoclonus, somnolence, and tachycardia, with a clozapine level of 1,350 ng/mL. Additional adverse effects that can be seen include orthostatic hypotension, sialorrhea, worsening psychiatric symptoms (eg, hallucinations), and seizures.5 Although there is variability in the timing of the decrease in CYP1A2 activity after smoking cessation, practitioners should begin to monitor for clozapine-related adverse effects 1 or 2 days after smoking cessation.6
Treatment recommendations
Monitoring of the clozapine concentration and adjustment of the dosage might be needed to account for the fluctuation seen with smoking cessation to maintain efficacy and minimize adverse effects. However, a test of the clozapine level may not be available at all facilities, often requiring that the specimen be sent to an outside laboratory, taking 3 to 7 days to receive results.
Faber and Fuhr6 recommended reducing the dosage of a CYP1A2 substrate medication, such as clozapine, olanzapine, or theophylline, by 10% each day until the dosage has been reduced by 40% in patients who stop smoking. Lowe and Ackman5 proposed reducing the clozapine dosage by 30% to 40% to achieve a pre-cessation serum concentration at 1 week. For Mr. D, this would mean decreasing the clozapine dosage to 425 to 500 mg/d.
Assuming that Mr. D’s clozapine dosage is decreased during his hospitalization and that he resumes smoking after discharge, it is likely the dosage will need to be increased. It may take several weeks to see maximal induction, because new CYP enzymes need to be synthesized when the patient resumes smoking.7 One recommendation is to increase the clozapine dosage by a factor of 1.5 over 2 to 4 weeks, with close monitoring of the clozapine concentration and adverse effects because this increase is approximate.7 Depending on when Mr. D’s follow-up appointment is scheduled, the practitioner may need to plan a dosage adjustment to prevent a decrease in his clozapine level caused by smoking to prevent a worsening of symptoms and rehospitalization.
This case emphasizes the importance of asking clozapine-treated patients about their smoking history when they are admitted to a smoke-free facility. For several reasons, >60% of patients with schizophrenia smoke cigarettes8 (Table 2).9-14 Patients who smoke and are on a stable dosage of clozapine might require a dosage reduction when they are admitted to a smoke-free facility to avoid adverse effects. If the dosage is not adjusted, a patient may experience clozapine-induced adverse effects, such as tachycardia, sedation, and seizures. It is likely that patients such as Mr. D will experience fluctuation in the clozapine level and possibly changes in efficacy and tolerability transitioning between inpatient and outpatient settings if the dosage is not adjusted.
Related Resources
• Kroon LA. Drug interactions with smoking. Am J Health Syst Pharm. 2007;64(18):1917-1921.
• Fankhauser MP. Drug interactions with tobacco smoke: Implications for patient care. Current Psychiatry. 2013; 12(1):12-16.
• Greenwood-Smith C, Lubman DI, Castle DJ. Serum clozapine levels: a review of their clinical utility. J Psychopharmacol. 2003;17(2):234-248.
• Olesen OV, Thomsen K, Jensen PN, et al. Clozapine serum levels and side effects during steady state treatment of schizophrenic patients: a cross sectional study. Psychopharmacology (Berl). 1995;117(3):371-378.
Drug Brand Names
Clozapine • Clozaril Theophylline • Theo-Dur
Olanzapine • Zyprexa
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Meyer JM. Individual changes in clozapine levels after smoking cessation: results and a predictive model. J Clin Psychopharmacol. 2001;21(6):569-574.
2. Murayama-Sung L, Ahmed I, Goebert D, et al. The impact of hospital smoking ban on clozapine and norclozapine levels. J Clin Psychopharmacol. 2011;31(1):124-126.
3. Haslemo T, Eikeseth PH, Tanum L, et al. The effect of variable cigarette consumption on the interaction with clozapine and olanzapine. Eur J Clin Psychopharmacol. 2006;62(12): 1049-1053.
4. Nielsen J, Damkier P, Lublin H, et al. Optimizing clozapine treatment. Acta Psychiatr Scand. 2011;123(6):411-422.
5. Lowe EJ, Ackman ML. Impact of tobacco smoking cessation on stable clozapine and olanzapine treatment. Ann Pharmacother. 2010;44(4):727-732.
6. Faber MS, Fuhr U. Time response of cytochrome P450 1A2 activity on cessation of heavy smoking. Clin Pharmacol Ther. 2004;76(2):178-184.
7. de Leon J. Atypical antipsychotic dosing: the effect of smoking and caffeine. Psychiatr Serv. 2004;55(5):491-493.
8. Dickerson F, Stallings CR, Origoni AE, et al. Cigarette smoking among persons with schizophrenia or bipolar disorder in routine clinical settings, 1999-2011. Psychiatr Serv. 2013;64(1):44-50.
9. Esterberg ML, Compton MT. Smoking behavior in persons with a schizophrenia-spectrum disorder: a qualitative investigation of the transtheoretical model. Soc Sci Med. 2005;61(2):293-303.
10. Barr RS, Culhane MA, Jubelt LE, et al. The effects of transdermal nicotine on cognition in nonsmokers with schizophrenia and nonpsychiatric controls. Neuropsychopharmacology. 2008; 33(3):480-490.
11. Adler LE, Hoffer LD, Wiser A, et al. Normalization of auditory physiology by cigarette smoking in schizophrenic patients. Am J Psychiatry. 1993;150(12):1856-1861.
12. Sallette J, Pons S, Devillers-Thiery A, et al. Nicotine upregulates its own receptors through enhanced intracellular maturation. Neuron. 2005;46(4):595-607.
13. Breese CR, Lee MJ, Adams CE, et al. Abnormal regulation of high affinity nicotinic receptors in subjects with schizophrenia. Neuropsychopharmacology. 2000;23(4):351-364.
14. Miller DD, Kelly MW, Perry PJ, et al. The influence of cigarette smoking on haloperidol pharmacokinetics. J Clin Psychiatry. 1990;28(6):529-231.
Mr. D, age 30, has a 12-year history of schizophrenia and is experiencing worsening auditory hallucinations despite reported medication adherence. He has been taking clozapine, maintenance dosages 500 to 700 mg/d, for 4 years and smokes 2 packs of cigarettes a day. When Mr. D is admitted to a nonsmoking inpatient psychiatric facility, he receives nicotine transdermal patches, 21 mg/d, for nicotine withdrawal. Mr. D’s most recent outpatient clozapine dosage, 700 mg/d, is resumed. All laboratory tests, including complete blood count with differential, are within normal limits at admission.
Five days later Mr. D is tachycardic with a heart rate of 109 beats per minute. When assessing Mr. D, we notice he has alogia and that, when he does speak, his speech is slowed with a 4 to 5 second delay in response. He also appears sedated. We observe occasional mild jerking of his shoulder and lower legs.
Mr. D reports that his auditory hallucinations have lessened since his admission, but complains of difficulty remembering information and feeling tired during the day. The treatment team suspects clozapine toxicity; his trough clozapine level is 1,350 ng/mL (therapeutic range, 350 to 1,000 ng/mL).
It is well documented that cigarette smoke can induce cytochrome P450 (CYP) isoenzymes, specifically CYP1A1, CYP1A2, and CYP2E1. Because clozapine is primarily metabolized by CYP1A2 (approximately 70%), smoking can induce clozapine metabolism and abruptly stopping smoking can increase clozapine levels.1 The polycyclic aromatic hydrocarbons, not the nicotine, found in cigarettes are thought to be responsible for CYP1A2 induction; therefore, use of a nicotine replacement product did not prevent the increase in Mr. D’s clozapine levels.
Examining the evidence
Meyer1 evaluated clozapine levels before and after implementation of a hospital-wide smoking ban (N = 11). Clozapine dosages were not adjusted at the time of the smoking ban, which resulted in a mean 72% increase in clozapine levels after a minimum of 2 weeks as nonsmokers. Even after eliminating 2 outliers, the mean increase in clozapine levels was 36.1%. Murayama-Sung et al2 reported a statistically significant increase in the level of clozapine (46%, P = .004) and the level of norclozapine (23%, P = .02) after a hospital-wide smoking ban was instituted (N = 14). However, the pre-change and post-change in the ratio of clozapine to norclozapine level was not found to be statistically significant. Haslemo et al3 found that smoking as few as 7 to 12 cigarettes a day was sufficient for maximum induction of CYP1A2. Because Mr. D was smoking 2 packs of cigarettes a day (40 cigarettes) with an clozapine dosage 700 mg/d as an outpatient, he likely experienced significant induction of clozapine metabolism through CYP1A2, which was no longer present when he stopped smoking.
Therapeutic clozapine concentrations are typically above 350 and 420 ng/mL.4 Concentrations >700 ng/mL are associated with increased adverse effects, but generally are not associated with a higher response; levels >900 ng/mL have been associated with toxicity.4 Clozapine-treated patients on a stable dosage who smoke can experience clozapine-related adverse effects after admission to a smoke-free facility secondary to an increase in the clozapine concentration (Table 1).4
Five days after admission to the facility, Mr. D was noted to have myoclonus, somnolence, and tachycardia, with a clozapine level of 1,350 ng/mL. Additional adverse effects that can be seen include orthostatic hypotension, sialorrhea, worsening psychiatric symptoms (eg, hallucinations), and seizures.5 Although there is variability in the timing of the decrease in CYP1A2 activity after smoking cessation, practitioners should begin to monitor for clozapine-related adverse effects 1 or 2 days after smoking cessation.6
Treatment recommendations
Monitoring of the clozapine concentration and adjustment of the dosage might be needed to account for the fluctuation seen with smoking cessation to maintain efficacy and minimize adverse effects. However, a test of the clozapine level may not be available at all facilities, often requiring that the specimen be sent to an outside laboratory, taking 3 to 7 days to receive results.
Faber and Fuhr6 recommended reducing the dosage of a CYP1A2 substrate medication, such as clozapine, olanzapine, or theophylline, by 10% each day until the dosage has been reduced by 40% in patients who stop smoking. Lowe and Ackman5 proposed reducing the clozapine dosage by 30% to 40% to achieve a pre-cessation serum concentration at 1 week. For Mr. D, this would mean decreasing the clozapine dosage to 425 to 500 mg/d.
Assuming that Mr. D’s clozapine dosage is decreased during his hospitalization and that he resumes smoking after discharge, it is likely the dosage will need to be increased. It may take several weeks to see maximal induction, because new CYP enzymes need to be synthesized when the patient resumes smoking.7 One recommendation is to increase the clozapine dosage by a factor of 1.5 over 2 to 4 weeks, with close monitoring of the clozapine concentration and adverse effects because this increase is approximate.7 Depending on when Mr. D’s follow-up appointment is scheduled, the practitioner may need to plan a dosage adjustment to prevent a decrease in his clozapine level caused by smoking to prevent a worsening of symptoms and rehospitalization.
This case emphasizes the importance of asking clozapine-treated patients about their smoking history when they are admitted to a smoke-free facility. For several reasons, >60% of patients with schizophrenia smoke cigarettes8 (Table 2).9-14 Patients who smoke and are on a stable dosage of clozapine might require a dosage reduction when they are admitted to a smoke-free facility to avoid adverse effects. If the dosage is not adjusted, a patient may experience clozapine-induced adverse effects, such as tachycardia, sedation, and seizures. It is likely that patients such as Mr. D will experience fluctuation in the clozapine level and possibly changes in efficacy and tolerability transitioning between inpatient and outpatient settings if the dosage is not adjusted.
Related Resources
• Kroon LA. Drug interactions with smoking. Am J Health Syst Pharm. 2007;64(18):1917-1921.
• Fankhauser MP. Drug interactions with tobacco smoke: Implications for patient care. Current Psychiatry. 2013; 12(1):12-16.
• Greenwood-Smith C, Lubman DI, Castle DJ. Serum clozapine levels: a review of their clinical utility. J Psychopharmacol. 2003;17(2):234-248.
• Olesen OV, Thomsen K, Jensen PN, et al. Clozapine serum levels and side effects during steady state treatment of schizophrenic patients: a cross sectional study. Psychopharmacology (Berl). 1995;117(3):371-378.
Drug Brand Names
Clozapine • Clozaril Theophylline • Theo-Dur
Olanzapine • Zyprexa
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Mr. D, age 30, has a 12-year history of schizophrenia and is experiencing worsening auditory hallucinations despite reported medication adherence. He has been taking clozapine, maintenance dosages 500 to 700 mg/d, for 4 years and smokes 2 packs of cigarettes a day. When Mr. D is admitted to a nonsmoking inpatient psychiatric facility, he receives nicotine transdermal patches, 21 mg/d, for nicotine withdrawal. Mr. D’s most recent outpatient clozapine dosage, 700 mg/d, is resumed. All laboratory tests, including complete blood count with differential, are within normal limits at admission.
Five days later Mr. D is tachycardic with a heart rate of 109 beats per minute. When assessing Mr. D, we notice he has alogia and that, when he does speak, his speech is slowed with a 4 to 5 second delay in response. He also appears sedated. We observe occasional mild jerking of his shoulder and lower legs.
Mr. D reports that his auditory hallucinations have lessened since his admission, but complains of difficulty remembering information and feeling tired during the day. The treatment team suspects clozapine toxicity; his trough clozapine level is 1,350 ng/mL (therapeutic range, 350 to 1,000 ng/mL).
It is well documented that cigarette smoke can induce cytochrome P450 (CYP) isoenzymes, specifically CYP1A1, CYP1A2, and CYP2E1. Because clozapine is primarily metabolized by CYP1A2 (approximately 70%), smoking can induce clozapine metabolism and abruptly stopping smoking can increase clozapine levels.1 The polycyclic aromatic hydrocarbons, not the nicotine, found in cigarettes are thought to be responsible for CYP1A2 induction; therefore, use of a nicotine replacement product did not prevent the increase in Mr. D’s clozapine levels.
Examining the evidence
Meyer1 evaluated clozapine levels before and after implementation of a hospital-wide smoking ban (N = 11). Clozapine dosages were not adjusted at the time of the smoking ban, which resulted in a mean 72% increase in clozapine levels after a minimum of 2 weeks as nonsmokers. Even after eliminating 2 outliers, the mean increase in clozapine levels was 36.1%. Murayama-Sung et al2 reported a statistically significant increase in the level of clozapine (46%, P = .004) and the level of norclozapine (23%, P = .02) after a hospital-wide smoking ban was instituted (N = 14). However, the pre-change and post-change in the ratio of clozapine to norclozapine level was not found to be statistically significant. Haslemo et al3 found that smoking as few as 7 to 12 cigarettes a day was sufficient for maximum induction of CYP1A2. Because Mr. D was smoking 2 packs of cigarettes a day (40 cigarettes) with an clozapine dosage 700 mg/d as an outpatient, he likely experienced significant induction of clozapine metabolism through CYP1A2, which was no longer present when he stopped smoking.
Therapeutic clozapine concentrations are typically above 350 and 420 ng/mL.4 Concentrations >700 ng/mL are associated with increased adverse effects, but generally are not associated with a higher response; levels >900 ng/mL have been associated with toxicity.4 Clozapine-treated patients on a stable dosage who smoke can experience clozapine-related adverse effects after admission to a smoke-free facility secondary to an increase in the clozapine concentration (Table 1).4
Five days after admission to the facility, Mr. D was noted to have myoclonus, somnolence, and tachycardia, with a clozapine level of 1,350 ng/mL. Additional adverse effects that can be seen include orthostatic hypotension, sialorrhea, worsening psychiatric symptoms (eg, hallucinations), and seizures.5 Although there is variability in the timing of the decrease in CYP1A2 activity after smoking cessation, practitioners should begin to monitor for clozapine-related adverse effects 1 or 2 days after smoking cessation.6
Treatment recommendations
Monitoring of the clozapine concentration and adjustment of the dosage might be needed to account for the fluctuation seen with smoking cessation to maintain efficacy and minimize adverse effects. However, a test of the clozapine level may not be available at all facilities, often requiring that the specimen be sent to an outside laboratory, taking 3 to 7 days to receive results.
Faber and Fuhr6 recommended reducing the dosage of a CYP1A2 substrate medication, such as clozapine, olanzapine, or theophylline, by 10% each day until the dosage has been reduced by 40% in patients who stop smoking. Lowe and Ackman5 proposed reducing the clozapine dosage by 30% to 40% to achieve a pre-cessation serum concentration at 1 week. For Mr. D, this would mean decreasing the clozapine dosage to 425 to 500 mg/d.
Assuming that Mr. D’s clozapine dosage is decreased during his hospitalization and that he resumes smoking after discharge, it is likely the dosage will need to be increased. It may take several weeks to see maximal induction, because new CYP enzymes need to be synthesized when the patient resumes smoking.7 One recommendation is to increase the clozapine dosage by a factor of 1.5 over 2 to 4 weeks, with close monitoring of the clozapine concentration and adverse effects because this increase is approximate.7 Depending on when Mr. D’s follow-up appointment is scheduled, the practitioner may need to plan a dosage adjustment to prevent a decrease in his clozapine level caused by smoking to prevent a worsening of symptoms and rehospitalization.
This case emphasizes the importance of asking clozapine-treated patients about their smoking history when they are admitted to a smoke-free facility. For several reasons, >60% of patients with schizophrenia smoke cigarettes8 (Table 2).9-14 Patients who smoke and are on a stable dosage of clozapine might require a dosage reduction when they are admitted to a smoke-free facility to avoid adverse effects. If the dosage is not adjusted, a patient may experience clozapine-induced adverse effects, such as tachycardia, sedation, and seizures. It is likely that patients such as Mr. D will experience fluctuation in the clozapine level and possibly changes in efficacy and tolerability transitioning between inpatient and outpatient settings if the dosage is not adjusted.
Related Resources
• Kroon LA. Drug interactions with smoking. Am J Health Syst Pharm. 2007;64(18):1917-1921.
• Fankhauser MP. Drug interactions with tobacco smoke: Implications for patient care. Current Psychiatry. 2013; 12(1):12-16.
• Greenwood-Smith C, Lubman DI, Castle DJ. Serum clozapine levels: a review of their clinical utility. J Psychopharmacol. 2003;17(2):234-248.
• Olesen OV, Thomsen K, Jensen PN, et al. Clozapine serum levels and side effects during steady state treatment of schizophrenic patients: a cross sectional study. Psychopharmacology (Berl). 1995;117(3):371-378.
Drug Brand Names
Clozapine • Clozaril Theophylline • Theo-Dur
Olanzapine • Zyprexa
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Meyer JM. Individual changes in clozapine levels after smoking cessation: results and a predictive model. J Clin Psychopharmacol. 2001;21(6):569-574.
2. Murayama-Sung L, Ahmed I, Goebert D, et al. The impact of hospital smoking ban on clozapine and norclozapine levels. J Clin Psychopharmacol. 2011;31(1):124-126.
3. Haslemo T, Eikeseth PH, Tanum L, et al. The effect of variable cigarette consumption on the interaction with clozapine and olanzapine. Eur J Clin Psychopharmacol. 2006;62(12): 1049-1053.
4. Nielsen J, Damkier P, Lublin H, et al. Optimizing clozapine treatment. Acta Psychiatr Scand. 2011;123(6):411-422.
5. Lowe EJ, Ackman ML. Impact of tobacco smoking cessation on stable clozapine and olanzapine treatment. Ann Pharmacother. 2010;44(4):727-732.
6. Faber MS, Fuhr U. Time response of cytochrome P450 1A2 activity on cessation of heavy smoking. Clin Pharmacol Ther. 2004;76(2):178-184.
7. de Leon J. Atypical antipsychotic dosing: the effect of smoking and caffeine. Psychiatr Serv. 2004;55(5):491-493.
8. Dickerson F, Stallings CR, Origoni AE, et al. Cigarette smoking among persons with schizophrenia or bipolar disorder in routine clinical settings, 1999-2011. Psychiatr Serv. 2013;64(1):44-50.
9. Esterberg ML, Compton MT. Smoking behavior in persons with a schizophrenia-spectrum disorder: a qualitative investigation of the transtheoretical model. Soc Sci Med. 2005;61(2):293-303.
10. Barr RS, Culhane MA, Jubelt LE, et al. The effects of transdermal nicotine on cognition in nonsmokers with schizophrenia and nonpsychiatric controls. Neuropsychopharmacology. 2008; 33(3):480-490.
11. Adler LE, Hoffer LD, Wiser A, et al. Normalization of auditory physiology by cigarette smoking in schizophrenic patients. Am J Psychiatry. 1993;150(12):1856-1861.
12. Sallette J, Pons S, Devillers-Thiery A, et al. Nicotine upregulates its own receptors through enhanced intracellular maturation. Neuron. 2005;46(4):595-607.
13. Breese CR, Lee MJ, Adams CE, et al. Abnormal regulation of high affinity nicotinic receptors in subjects with schizophrenia. Neuropsychopharmacology. 2000;23(4):351-364.
14. Miller DD, Kelly MW, Perry PJ, et al. The influence of cigarette smoking on haloperidol pharmacokinetics. J Clin Psychiatry. 1990;28(6):529-231.
1. Meyer JM. Individual changes in clozapine levels after smoking cessation: results and a predictive model. J Clin Psychopharmacol. 2001;21(6):569-574.
2. Murayama-Sung L, Ahmed I, Goebert D, et al. The impact of hospital smoking ban on clozapine and norclozapine levels. J Clin Psychopharmacol. 2011;31(1):124-126.
3. Haslemo T, Eikeseth PH, Tanum L, et al. The effect of variable cigarette consumption on the interaction with clozapine and olanzapine. Eur J Clin Psychopharmacol. 2006;62(12): 1049-1053.
4. Nielsen J, Damkier P, Lublin H, et al. Optimizing clozapine treatment. Acta Psychiatr Scand. 2011;123(6):411-422.
5. Lowe EJ, Ackman ML. Impact of tobacco smoking cessation on stable clozapine and olanzapine treatment. Ann Pharmacother. 2010;44(4):727-732.
6. Faber MS, Fuhr U. Time response of cytochrome P450 1A2 activity on cessation of heavy smoking. Clin Pharmacol Ther. 2004;76(2):178-184.
7. de Leon J. Atypical antipsychotic dosing: the effect of smoking and caffeine. Psychiatr Serv. 2004;55(5):491-493.
8. Dickerson F, Stallings CR, Origoni AE, et al. Cigarette smoking among persons with schizophrenia or bipolar disorder in routine clinical settings, 1999-2011. Psychiatr Serv. 2013;64(1):44-50.
9. Esterberg ML, Compton MT. Smoking behavior in persons with a schizophrenia-spectrum disorder: a qualitative investigation of the transtheoretical model. Soc Sci Med. 2005;61(2):293-303.
10. Barr RS, Culhane MA, Jubelt LE, et al. The effects of transdermal nicotine on cognition in nonsmokers with schizophrenia and nonpsychiatric controls. Neuropsychopharmacology. 2008; 33(3):480-490.
11. Adler LE, Hoffer LD, Wiser A, et al. Normalization of auditory physiology by cigarette smoking in schizophrenic patients. Am J Psychiatry. 1993;150(12):1856-1861.
12. Sallette J, Pons S, Devillers-Thiery A, et al. Nicotine upregulates its own receptors through enhanced intracellular maturation. Neuron. 2005;46(4):595-607.
13. Breese CR, Lee MJ, Adams CE, et al. Abnormal regulation of high affinity nicotinic receptors in subjects with schizophrenia. Neuropsychopharmacology. 2000;23(4):351-364.
14. Miller DD, Kelly MW, Perry PJ, et al. The influence of cigarette smoking on haloperidol pharmacokinetics. J Clin Psychiatry. 1990;28(6):529-231.
Second of 2 parts: A practical approach to subtyping depression among your patients
Depression—sad, empty, or irritable mood accompanied by somatic or cognitive changes—is not a homogeneous condition. Recognizing subtypes of depressive illness can guide treatment and relieve your patient’s suffering. In this 2-part article [April and May 2014 issues], I summarize information about clinically distinct subtypes of depression, as they are recognized within diagnostic systems or as descriptors of treatment outcomes for particular subgroups of patients. My focus is on practical considerations for assessing and managing depression. Because many forms of the disorder respond inadequately to initial antidepressant treatment, optimal “next-step” pharmacotherapy, after nonresponse or partial response, often hinges on clinical subtyping.
The second part of this article examines “situational,” treatment-resistant, melancholic, agitated, anxious, and atypical depression; depression occurring with a substance use disorder; premenstrual dysphoric disorder; and seasonal affective disorder. Treatments for these subtypes for which there is evidence, or a clinical rationale, are given in the Table.
‘Situational depression’
In recent decades, the phenomenon of nonsyndromal depression after a life stress has undergone many name changes but little conceptional revision: “situational,” “reactive,” and “neurotic” labels for depression that were used before DSM-III became “adjustment disorders” in DSM-IV-TR and then “stress response syndromes” in DSM-5. These names all connote presentations of depressed mood after an environmental stressor, without either the full constellation of symptoms that define major depression or the chronicity of dysthymic disorder.
Paucity of guidance. There has been little research to identify vulnerability variables for adjustment disorders in the aftermath of particular stressors. Similarly, extensive data are lacking on 1) the likely progression of such disorders to a syndromal form of depression or 2) protective factors against developing clinically significant depression after a life stress. The extent to which adjustment disorders lie on a continuum with major mood disorders is not well-established, although subthreshold levels of depression can predispose to major depression or suicidal behaviors.1
Models of behavioral sensitization posit that stressful life events more often precede first or early episodes of depression than subsequent recurrences.2 At the same time, non-melancholic depressions that are preceded by “situational stresses” tend to recur in similar fashion.3
Medical therapy of value? Psychotherapy without medication—apart from occasional sedative−hypnotic drugs as needed for insomnia, anxiety, or distress—is considered the standard of care for treating an adjustment disorder. No drug has demonstrated superiority to placebo for alleviating symptoms of an adjustment disorder, but some clinicians nonetheless sometimes feel compelled to “up-code” the diagnosis of an adjustment disorder to the status of a major affective disorder, even when syndromal criteria for major depressive disorder (MDD) or dysthymia are absent.
Treatment-resistant depression
Disease staging models for depression and other psychiatric disordersa make note that, elsewhere in medicine, distinct clinical entities often are identified based on their responsivity to treatment (eg, classifying infections as antibiotic-sensitive or -resistant). Within the study and management of mood disorders, “treatment resistance” sometimes is a catch-all description of situations in which past treatment 1) yielded no improvement or partial improvement or 2) was marked by intolerance. Poor outcomes due to past medication intolerance or an aborted trial often are commingled with cases of true lack of improvement after an adequate treatment trial.
aSee “Staging psychiatric disorders: A clinico-biologic model,” Current Psychiatry, May 2013, at CurrentPsychiatry.com.
It is useful, therefore, to define terminology precisely when describing “treatment-resistant depression” and “treatment-refractory depression.” True past nonresponse to appropriate treatment often carries prognostic importance and bears on future treatment decisions.
Few interventions are FDA approved for treatment-resistant depression (Table). 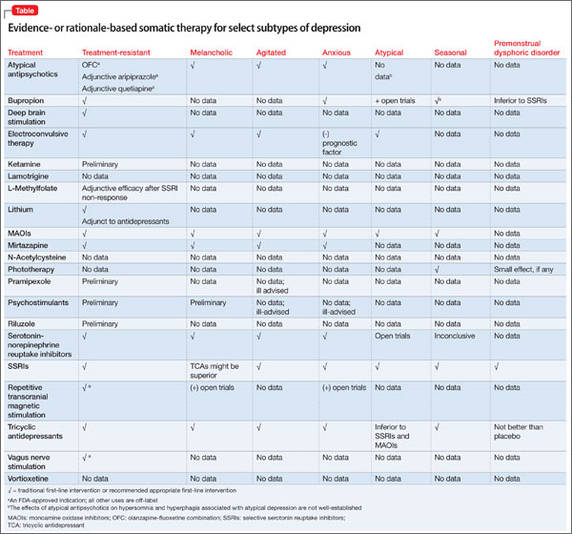
Neuromodulation techniques are attracting interest in this area, although repetitive transcranial magnetic stimulation appears inferior to electroconvulsive therapy (ECT) for this indication.4
Melancholic depression
Melancholia involves the cardinal symptoms of anhedonia and lack of mood reactivity, alongside such features as distinct quality of mood, diurnal variation, excessive guilt, and severe weight loss. It most closely approximates pre-DSM-III “endogenous depression” and can involve 1) greater genetic loading5 and 2) structural and functional abnormalities in frontostriatal pathways.6,7
Melancholic features do not necessarily recur across successive episodes8 but carry an increased risk of psychosis9 and high-lethality suicidal behavior.10 Melancholia implies necessity for pharmacotherapy or ECT rather than psychosocial treatment alone; some researchers have suggested that tricyclic antidepressants (TCAs) might yield better results than selective serotonin reuptake inhibitors (SSRIs).11
Agitated depression
The Research Diagnostic Criteria (a forerunner in the 1970s to DSM-III) described agitated depression, but the disorder was not included in any DSM editions—although it is a “clinical modification” for MDD in the 10th revision of the International Statistical Classification of Diseases and Related Health Problems.
Agitated depression refers to a major depressive episode involving motor or psychic agitation, intense inner tension, and racing or “crowded” thoughts. Some experts believe that it represents a variant of psychotic depression or a bipolar mixed state, but the construct does not specify that criteria for a full manic or hypomanic episode exist.
Recovery from agitated depression tends to be slower than in non-agitated depression. Treatment usually entails an antidepressant plus an antipsychotic, although some believe that antidepressants can exacerbate, not alleviate, symptoms and, instead, favor antipsychotics, mood stabilizers, or ECT.12
Anxious depression
Anxiety symptoms or syndromes occur in at least one-half of outpatients who have major depression, and might account for a substantial percentage of nonresponse to first-line antidepressant therapies.13 The construct of a mixed anxiety−depressive disorder is, in fact, well-represented in the literature, particularly in primary care medicine, but its poor inter-rater reliability in DSM-5 field trials led to its exclusion there as a formal diagnosis.14
Serotonergic antidepressants remain the mainstay of treatment for depression with anxiety, although (contrary to popular perception) bupropion exerts an anxiolytic effect that is comparable to the effect of SSRIs.15 Notably, high somatic anxiety during depression might predict a poor outcome from ECT.16
Atypical depression
Often closely linked with early onset and chronicity, the construct of atypical depression has been defined in the literature by the symptom constellation of:
• mood reactiveness to environmental circumstances (unlike melancholia)
• heightened interpersonal sensitivity
• hypersomnia
• hyperphagia
• profound fatigue or a sense of physical heaviness.
Some authorities regard atypical features as being especially common in bipolar depression, or in depression among people who have borderline personality disorder.
Particular interest in this construct grew from studies that suggested that atypical depression is more responsive to a monoamine oxidase inhibitor (MAOI) than to a TCA, but also that SSRIs are not clearly superior to MAOIs.17 Response to ECT might also be better in atypical than in typical depression.18
Depression with a substance use disorder
Although not a distinct diagnostic entity, depression with a coexisting substance use disorder poses special challenges with regard to the source of symptom emergence (that is, when does depression lead to drug or alcohol use to “self medicate,” and when does drug use cause depression?) and treatment. Debate continues about whether 1) medicines that treat depression are effective and worthwhile in the setting of active substance use or 2) aggressive treatment of substance misuse is a prerequisite for subsequent pharmacotherapy for depression that is “uncontaminated” by the psychotoxic effects of concurrent substances of abuse.
Meta-analysis of controlled trials of antidepressants for patients who have MDD or a dysthymic disorder plus a comorbid alcohol use disorder found that antidepressants were, overall, superior to placebo unless a patient is actively drinking.19 Of the various classes of antidepressants, TCAs and nefazodone were found to be superior to placebo but, surprisingly, SSRIs were not. Another meta-analysis of adjunctive antidepressant outcomes for opiate-dependent, depressed patients who are receiving methadone maintenance therapy found no difference between antidepressants and placebo in their effect on depression symptom outcomes.20
Premenstrual dysphoric disorder
A new category in DSM-5, premenstrual dysphoric disorder (PMDD) represents a variant of premenstrual syndrome that arises during the luteal phase and ends with menstruation. Symptoms include several of those identified with MDD (without duration criteria), as well as mood swings, panic attacks, and physical complaints.
SSRIs—but not bupropion21 or TCAs22— and, sometimes, low-estrogen oral contraceptives are mainstays of treatment; so is cognitive-behavioral therapy, as well as lifestyle modifications (eg, exercise and changes to diet). Phototherapy has not shown robust efficacy for PMDD.23
Secondary depression
In DSM-5, depressive episodes that arise secondary to a general medical condition (eg, hypothyroidism and other endocrinopathies, cerebrovascular accidents, malignancies) or iatrogenically from medications (eg, corticosteroids, some anticonvulsants, interinterferon) are viewed as distinct from MDD in regard to risk of recurrence, genetic underpinnings, and possible neurodegenerative pathophysiology.b Unlike MDD, patient-specific risk factors are poorly defined for anticipating that a secondary depression is more or less likely to develop in the context of an exogenous substance or medical illness.
bFor further discussion, see “Is a medical illness causing your patient’s depression? Current Psychiatry, August 2009, at CurrentPsychiatry.com.
Treating secondary depression involves addressing the underlying condition and might include antidepressant medication.
Seasonal affective disorder
DSM-5 identifies “with seasonal pattern” as a specifier for recurrent major depression. Phototherapy remains a standard treatment, although a Cochrane Review identified comparable outcomes with fluoxetine, but inconclusive data for other, newer antidepressants.24 Small open trials have suggested that MAOIs and TCAs can be efficacious.
Note: Phototherapy lacks demonstrated efficacy in non-seasonal forms of depression.25
What does the future hold for classifying depressive disorders?
Recent initiatives have attempted to classify depression less by traditional clinical signs and more by focusing on possible underlying neurobiological substrates.c In the future, subtyping of mood disorders might focus on such constructs as:
• positive and negative valence systems and attentional domains
• treatment-responsivity relative to genotypic variants (for example, the serotonin transporter gene locus [SLC6A4] or prediction of L-methylfolate-responsive depression based on the genotype of the methylenetetrahydrofolate reductase [MTHFR] polymorphism)
• disrupted neural plasticity in brain circuits believed to regulate emotion.
cAn example is the Research Domain Criteria [RDoC],www. nimh.nih.gov/research-priorities/rdoc/index.shtml.
Until robust biomarkers for depression are identified and validated, however, such advances in nosology remain experimental and speculative.
BOTTOM LINE
Depressive disorders comprise a range of conditions that can be viewed along many dimensions, including “situational,” treatment-resistant, melancholic, agitated, anxious, and atypical depression; depression occurring with a substance use disorder; premenstrual dysphoric disorder; and seasonal affective disorder, among other classifications. Clinical characteristics vary across subtypes—as do corresponding preferred treatments, which should be tailored to the needs of your patients.
Editor’s note: The first part of Dr. Goldberg’s review of depression subtypes—focusing on major and minor depression, chronicity, polarity, severity, and psychosis—appeared in the April 2014 issue.
Related Resources
• Kosinski EC, Rothschild AJ. Monoamine oxidase inhibitors: Forgotten treatment for depression. Current Psychiatry. 2012;11(12):20-26.
• Rodgers S, Grosse Holtforth M, Müller M, et al. Symptom-based subtypes of depression and their psychosocial correlates: a person-centered approach focusing on the influence of sex. J Affect Disord. 2014;156:92-103.
Drug Brand Names
Aripiprazole • Abilify Mirtazapine • Remeron
Bupropion • Wellbutrin Nefazodone • Serzone
Fluoxetine • Prozac Olanzapine/fluoxetine • Symbyax
Ketamine • Ketalar Pramipexole • Mirapex
L-Methylfolate • Deplin Quetiapine • Seroquel
Lamotrigine • Lamictal Riluzole • Rilutek
Lithium • Eskalith, Lithobid Vortioxetine • Brintellix
Methadone • Dolophine
Disclosure
Dr. Goldberg has been a consultant to Avanir Pharmaceuticals and Merck; has served on the speakers’ bureau for AstraZeneca, Merck, Novartis, Sunovion Pharmaceuticals, Takeda-Lundbeck; and has received royalties from American Psychiatric Publishing and honoraria from Medscape and WebMD.
1. Fergusson DM, Horwood LJ, Ridder EM, et al. Subthreshold depression in adolescence and mental health outcomes in adulthood. Arch Gen Psychiatry. 2005;62(1):66-72.
2. Mitchell PB, Parker GB, Gladstone GL, et al. Severity of stressful life events in first and subsequent episodes of depression: the relevance of depressive subtypes. J Affect Disord. 2003;73(3):245-252.
3. Coryell W, Winokur G, Maser JD, et al. Recurrently situational (reactive) depression: a study of course, phenomenology and familial psychopathology. J Affect Disord. 1994;31(3):203-210.
4. Slotema CW, Blom JD, Hoek HW, et al. Should we expand the toolbox of psychiatric treatment methods to include Repetitive Transcranial Magnetic Stimulation (rTMS)? A meta-analysis of the efficacy of rTMS in psychiatric disorders. J Clin Psychiatry. 2010;71(7):873-884.
5. Kendler KS. The diagnostic validity of melancholic major depression in a population-based sample of female twins. Arch Gen Psychiatry. 1997;54(4):299-304.
6. Bracht T, Horn H, Strik W, et al. White matter microstructure alterations of the medial forebrain bundle in melancholic depression. J Affect Disord. 2014;155:186-193.
7. Pizzagalli DA, Oakes TR, Fox AS, et al. Functional but not structural subgenual prefrontal cortex abnormalities in melancholia. Mol Psychiatry. 2004;9(4):325, 393-405.
8. Melartin T, Leskelä U, Rytsälä H, et al. Co-morbidity and stability of melancholic features in DSM-IV major depressive disorder. Psychol Med. 2004;34(8):1443-1452.
9. Caldieraro MA, Baeza FL, Pinheiro DO, et al. Prevalence of psychotic symptoms in those with melancholic and nonmelancholic depression. J Nerv Ment Dis. 2013;201(10):855-859.
10. Grunebaum MF, Galfalvy HC, Oquendo MA, et al. Melancholia and the probability and lethality of suicide attempts. Br J Psychiatry. 2004;184:534-535.
11. Roose SP, Glassman AH, Attia E, et al. Comparative efficacy of selective serotonin reuptake inhibitors and tricyclics in the treatment of melancholia. Am J Psychiatry. 1994;151(12):1735-1739.
12. Koukopoulos A, Sani G, Koukopoulos AE, et al. Melancholia agitata and mixed depression. Acta Psychiatr Scand Suppl. 2007;(433):50-57.
13. Fava M, Rush AJ, Alpert JE, et al. Difference in treatment outcome in outpatients with anxious versus nonanxious depression: a STAR*D report. Am J Psychiatry. 2008; 165(3):342-351.
14. Regier DA, Narrow WE, Clarke DE, et al. DSM-5 field trials in the United States and Canada, Part II: test-retest reliability of selected categorical diagnoses. Am J Psychiatry. 2013;170(1):59-70.
15. Rush AJ, Trivedi MH, Carmody TJ, et al. Response in relation to baseline anxiety levels in major depressive disorder treated with bupropion sustained release or sertraline. Neuropsychopharmacology. 2001;25(1):131-138.
16. Rasmussen KG, Snyder KA, Knapp RG, et al. Relationship between somatization and remission with ECT. Psychiatry Res. 2004;129(3):293-295.
17. Henkel V, Mergl R, Allgaier AK, et al. Treatment of depression with atypical features: a meta-analytic approach. Psychiatry Res. 2006;141(1):89-101.
18. Husain MM, McClintock SM, Rush AJ, et al. The efficacy of acute electroconvulsive therapy in atypical depression. J Clin Psychiatry. 2008;69(3):406-411.
19. Iovieno N, Tedeschini E, Bentley KH, et al. Antidepressants for major depressive disorder and dysthymic disorder in patients with comorbid alcohol use disorders: a meta-analysis of placebo-controlled randomized trials. J Clin Psychiatry. 2011;72(8):1144-1151.
20. Pedrelli P, Iovieno N, Vitali M, et al. Treatment of major depressive disorder and dysthymic disorder with antidepressants in patients with comorbid opiate use disorders enrolled in methadone maintenance therapy: a meta-analysis. J Clin Psychopharmacol. 2011;31(5):582-586.
21. Pearlstein TB, Stone AB, Lund SA, et al. Comparison of fluoxetine, bupropion, and placebo in the treatment of premenstrual dysphoric disorder. J Clin Psychopharmacol. 1997;17(4):261-266.
22. Freeman EW, Rickels K, Sondheimer SJ, et al. Differential response to antidepressants in women with premenstrual syndrome/premenstrual dysphoric disorder: a randomized controlled trial. Arch Gen Psychiatry. 1999;56(10):932-939.
23. Krasnik C, Montori VM, Guyatt GH, et al. The effect of bright light therapy on depression associated with premenstrual dysphoric disorder. Am J Obstet Gynecol. 2005;193(3, pt 1):658-661.
24. Thaler K, Delivuk M, Chapman A, et al. Second-generation antidepressants for seasonal affective disorder. Cochrane Database Syst Rev. 2011;7(12):CD008591.
25. Thalén BE, Kjellman BF, Mørkid L, et al. Light treatment in seasonal and nonseasonal depression. Acta Psychiatr Scand. 1995;91(5):352-360.
Depression—sad, empty, or irritable mood accompanied by somatic or cognitive changes—is not a homogeneous condition. Recognizing subtypes of depressive illness can guide treatment and relieve your patient’s suffering. In this 2-part article [April and May 2014 issues], I summarize information about clinically distinct subtypes of depression, as they are recognized within diagnostic systems or as descriptors of treatment outcomes for particular subgroups of patients. My focus is on practical considerations for assessing and managing depression. Because many forms of the disorder respond inadequately to initial antidepressant treatment, optimal “next-step” pharmacotherapy, after nonresponse or partial response, often hinges on clinical subtyping.
The second part of this article examines “situational,” treatment-resistant, melancholic, agitated, anxious, and atypical depression; depression occurring with a substance use disorder; premenstrual dysphoric disorder; and seasonal affective disorder. Treatments for these subtypes for which there is evidence, or a clinical rationale, are given in the Table.
‘Situational depression’
In recent decades, the phenomenon of nonsyndromal depression after a life stress has undergone many name changes but little conceptional revision: “situational,” “reactive,” and “neurotic” labels for depression that were used before DSM-III became “adjustment disorders” in DSM-IV-TR and then “stress response syndromes” in DSM-5. These names all connote presentations of depressed mood after an environmental stressor, without either the full constellation of symptoms that define major depression or the chronicity of dysthymic disorder.
Paucity of guidance. There has been little research to identify vulnerability variables for adjustment disorders in the aftermath of particular stressors. Similarly, extensive data are lacking on 1) the likely progression of such disorders to a syndromal form of depression or 2) protective factors against developing clinically significant depression after a life stress. The extent to which adjustment disorders lie on a continuum with major mood disorders is not well-established, although subthreshold levels of depression can predispose to major depression or suicidal behaviors.1
Models of behavioral sensitization posit that stressful life events more often precede first or early episodes of depression than subsequent recurrences.2 At the same time, non-melancholic depressions that are preceded by “situational stresses” tend to recur in similar fashion.3
Medical therapy of value? Psychotherapy without medication—apart from occasional sedative−hypnotic drugs as needed for insomnia, anxiety, or distress—is considered the standard of care for treating an adjustment disorder. No drug has demonstrated superiority to placebo for alleviating symptoms of an adjustment disorder, but some clinicians nonetheless sometimes feel compelled to “up-code” the diagnosis of an adjustment disorder to the status of a major affective disorder, even when syndromal criteria for major depressive disorder (MDD) or dysthymia are absent.
Treatment-resistant depression
Disease staging models for depression and other psychiatric disordersa make note that, elsewhere in medicine, distinct clinical entities often are identified based on their responsivity to treatment (eg, classifying infections as antibiotic-sensitive or -resistant). Within the study and management of mood disorders, “treatment resistance” sometimes is a catch-all description of situations in which past treatment 1) yielded no improvement or partial improvement or 2) was marked by intolerance. Poor outcomes due to past medication intolerance or an aborted trial often are commingled with cases of true lack of improvement after an adequate treatment trial.
aSee “Staging psychiatric disorders: A clinico-biologic model,” Current Psychiatry, May 2013, at CurrentPsychiatry.com.
It is useful, therefore, to define terminology precisely when describing “treatment-resistant depression” and “treatment-refractory depression.” True past nonresponse to appropriate treatment often carries prognostic importance and bears on future treatment decisions.
Few interventions are FDA approved for treatment-resistant depression (Table).
Neuromodulation techniques are attracting interest in this area, although repetitive transcranial magnetic stimulation appears inferior to electroconvulsive therapy (ECT) for this indication.4
Melancholic depression
Melancholia involves the cardinal symptoms of anhedonia and lack of mood reactivity, alongside such features as distinct quality of mood, diurnal variation, excessive guilt, and severe weight loss. It most closely approximates pre-DSM-III “endogenous depression” and can involve 1) greater genetic loading5 and 2) structural and functional abnormalities in frontostriatal pathways.6,7
Melancholic features do not necessarily recur across successive episodes8 but carry an increased risk of psychosis9 and high-lethality suicidal behavior.10 Melancholia implies necessity for pharmacotherapy or ECT rather than psychosocial treatment alone; some researchers have suggested that tricyclic antidepressants (TCAs) might yield better results than selective serotonin reuptake inhibitors (SSRIs).11
Agitated depression
The Research Diagnostic Criteria (a forerunner in the 1970s to DSM-III) described agitated depression, but the disorder was not included in any DSM editions—although it is a “clinical modification” for MDD in the 10th revision of the International Statistical Classification of Diseases and Related Health Problems.
Agitated depression refers to a major depressive episode involving motor or psychic agitation, intense inner tension, and racing or “crowded” thoughts. Some experts believe that it represents a variant of psychotic depression or a bipolar mixed state, but the construct does not specify that criteria for a full manic or hypomanic episode exist.
Recovery from agitated depression tends to be slower than in non-agitated depression. Treatment usually entails an antidepressant plus an antipsychotic, although some believe that antidepressants can exacerbate, not alleviate, symptoms and, instead, favor antipsychotics, mood stabilizers, or ECT.12
Anxious depression
Anxiety symptoms or syndromes occur in at least one-half of outpatients who have major depression, and might account for a substantial percentage of nonresponse to first-line antidepressant therapies.13 The construct of a mixed anxiety−depressive disorder is, in fact, well-represented in the literature, particularly in primary care medicine, but its poor inter-rater reliability in DSM-5 field trials led to its exclusion there as a formal diagnosis.14
Serotonergic antidepressants remain the mainstay of treatment for depression with anxiety, although (contrary to popular perception) bupropion exerts an anxiolytic effect that is comparable to the effect of SSRIs.15 Notably, high somatic anxiety during depression might predict a poor outcome from ECT.16
Atypical depression
Often closely linked with early onset and chronicity, the construct of atypical depression has been defined in the literature by the symptom constellation of:
• mood reactiveness to environmental circumstances (unlike melancholia)
• heightened interpersonal sensitivity
• hypersomnia
• hyperphagia
• profound fatigue or a sense of physical heaviness.
Some authorities regard atypical features as being especially common in bipolar depression, or in depression among people who have borderline personality disorder.
Particular interest in this construct grew from studies that suggested that atypical depression is more responsive to a monoamine oxidase inhibitor (MAOI) than to a TCA, but also that SSRIs are not clearly superior to MAOIs.17 Response to ECT might also be better in atypical than in typical depression.18
Depression with a substance use disorder
Although not a distinct diagnostic entity, depression with a coexisting substance use disorder poses special challenges with regard to the source of symptom emergence (that is, when does depression lead to drug or alcohol use to “self medicate,” and when does drug use cause depression?) and treatment. Debate continues about whether 1) medicines that treat depression are effective and worthwhile in the setting of active substance use or 2) aggressive treatment of substance misuse is a prerequisite for subsequent pharmacotherapy for depression that is “uncontaminated” by the psychotoxic effects of concurrent substances of abuse.
Meta-analysis of controlled trials of antidepressants for patients who have MDD or a dysthymic disorder plus a comorbid alcohol use disorder found that antidepressants were, overall, superior to placebo unless a patient is actively drinking.19 Of the various classes of antidepressants, TCAs and nefazodone were found to be superior to placebo but, surprisingly, SSRIs were not. Another meta-analysis of adjunctive antidepressant outcomes for opiate-dependent, depressed patients who are receiving methadone maintenance therapy found no difference between antidepressants and placebo in their effect on depression symptom outcomes.20
Premenstrual dysphoric disorder
A new category in DSM-5, premenstrual dysphoric disorder (PMDD) represents a variant of premenstrual syndrome that arises during the luteal phase and ends with menstruation. Symptoms include several of those identified with MDD (without duration criteria), as well as mood swings, panic attacks, and physical complaints.
SSRIs—but not bupropion21 or TCAs22— and, sometimes, low-estrogen oral contraceptives are mainstays of treatment; so is cognitive-behavioral therapy, as well as lifestyle modifications (eg, exercise and changes to diet). Phototherapy has not shown robust efficacy for PMDD.23
Secondary depression
In DSM-5, depressive episodes that arise secondary to a general medical condition (eg, hypothyroidism and other endocrinopathies, cerebrovascular accidents, malignancies) or iatrogenically from medications (eg, corticosteroids, some anticonvulsants, interinterferon) are viewed as distinct from MDD in regard to risk of recurrence, genetic underpinnings, and possible neurodegenerative pathophysiology.b Unlike MDD, patient-specific risk factors are poorly defined for anticipating that a secondary depression is more or less likely to develop in the context of an exogenous substance or medical illness.
bFor further discussion, see “Is a medical illness causing your patient’s depression? Current Psychiatry, August 2009, at CurrentPsychiatry.com.
Treating secondary depression involves addressing the underlying condition and might include antidepressant medication.
Seasonal affective disorder
DSM-5 identifies “with seasonal pattern” as a specifier for recurrent major depression. Phototherapy remains a standard treatment, although a Cochrane Review identified comparable outcomes with fluoxetine, but inconclusive data for other, newer antidepressants.24 Small open trials have suggested that MAOIs and TCAs can be efficacious.
Note: Phototherapy lacks demonstrated efficacy in non-seasonal forms of depression.25
What does the future hold for classifying depressive disorders?
Recent initiatives have attempted to classify depression less by traditional clinical signs and more by focusing on possible underlying neurobiological substrates.c In the future, subtyping of mood disorders might focus on such constructs as:
• positive and negative valence systems and attentional domains
• treatment-responsivity relative to genotypic variants (for example, the serotonin transporter gene locus [SLC6A4] or prediction of L-methylfolate-responsive depression based on the genotype of the methylenetetrahydrofolate reductase [MTHFR] polymorphism)
• disrupted neural plasticity in brain circuits believed to regulate emotion.
cAn example is the Research Domain Criteria [RDoC],www. nimh.nih.gov/research-priorities/rdoc/index.shtml.
Until robust biomarkers for depression are identified and validated, however, such advances in nosology remain experimental and speculative.
BOTTOM LINE
Depressive disorders comprise a range of conditions that can be viewed along many dimensions, including “situational,” treatment-resistant, melancholic, agitated, anxious, and atypical depression; depression occurring with a substance use disorder; premenstrual dysphoric disorder; and seasonal affective disorder, among other classifications. Clinical characteristics vary across subtypes—as do corresponding preferred treatments, which should be tailored to the needs of your patients.
Editor’s note: The first part of Dr. Goldberg’s review of depression subtypes—focusing on major and minor depression, chronicity, polarity, severity, and psychosis—appeared in the April 2014 issue.
Related Resources
• Kosinski EC, Rothschild AJ. Monoamine oxidase inhibitors: Forgotten treatment for depression. Current Psychiatry. 2012;11(12):20-26.
• Rodgers S, Grosse Holtforth M, Müller M, et al. Symptom-based subtypes of depression and their psychosocial correlates: a person-centered approach focusing on the influence of sex. J Affect Disord. 2014;156:92-103.
Drug Brand Names
Aripiprazole • Abilify Mirtazapine • Remeron
Bupropion • Wellbutrin Nefazodone • Serzone
Fluoxetine • Prozac Olanzapine/fluoxetine • Symbyax
Ketamine • Ketalar Pramipexole • Mirapex
L-Methylfolate • Deplin Quetiapine • Seroquel
Lamotrigine • Lamictal Riluzole • Rilutek
Lithium • Eskalith, Lithobid Vortioxetine • Brintellix
Methadone • Dolophine
Disclosure
Dr. Goldberg has been a consultant to Avanir Pharmaceuticals and Merck; has served on the speakers’ bureau for AstraZeneca, Merck, Novartis, Sunovion Pharmaceuticals, Takeda-Lundbeck; and has received royalties from American Psychiatric Publishing and honoraria from Medscape and WebMD.
Depression—sad, empty, or irritable mood accompanied by somatic or cognitive changes—is not a homogeneous condition. Recognizing subtypes of depressive illness can guide treatment and relieve your patient’s suffering. In this 2-part article [April and May 2014 issues], I summarize information about clinically distinct subtypes of depression, as they are recognized within diagnostic systems or as descriptors of treatment outcomes for particular subgroups of patients. My focus is on practical considerations for assessing and managing depression. Because many forms of the disorder respond inadequately to initial antidepressant treatment, optimal “next-step” pharmacotherapy, after nonresponse or partial response, often hinges on clinical subtyping.
The second part of this article examines “situational,” treatment-resistant, melancholic, agitated, anxious, and atypical depression; depression occurring with a substance use disorder; premenstrual dysphoric disorder; and seasonal affective disorder. Treatments for these subtypes for which there is evidence, or a clinical rationale, are given in the Table.
‘Situational depression’
In recent decades, the phenomenon of nonsyndromal depression after a life stress has undergone many name changes but little conceptional revision: “situational,” “reactive,” and “neurotic” labels for depression that were used before DSM-III became “adjustment disorders” in DSM-IV-TR and then “stress response syndromes” in DSM-5. These names all connote presentations of depressed mood after an environmental stressor, without either the full constellation of symptoms that define major depression or the chronicity of dysthymic disorder.
Paucity of guidance. There has been little research to identify vulnerability variables for adjustment disorders in the aftermath of particular stressors. Similarly, extensive data are lacking on 1) the likely progression of such disorders to a syndromal form of depression or 2) protective factors against developing clinically significant depression after a life stress. The extent to which adjustment disorders lie on a continuum with major mood disorders is not well-established, although subthreshold levels of depression can predispose to major depression or suicidal behaviors.1
Models of behavioral sensitization posit that stressful life events more often precede first or early episodes of depression than subsequent recurrences.2 At the same time, non-melancholic depressions that are preceded by “situational stresses” tend to recur in similar fashion.3
Medical therapy of value? Psychotherapy without medication—apart from occasional sedative−hypnotic drugs as needed for insomnia, anxiety, or distress—is considered the standard of care for treating an adjustment disorder. No drug has demonstrated superiority to placebo for alleviating symptoms of an adjustment disorder, but some clinicians nonetheless sometimes feel compelled to “up-code” the diagnosis of an adjustment disorder to the status of a major affective disorder, even when syndromal criteria for major depressive disorder (MDD) or dysthymia are absent.
Treatment-resistant depression
Disease staging models for depression and other psychiatric disordersa make note that, elsewhere in medicine, distinct clinical entities often are identified based on their responsivity to treatment (eg, classifying infections as antibiotic-sensitive or -resistant). Within the study and management of mood disorders, “treatment resistance” sometimes is a catch-all description of situations in which past treatment 1) yielded no improvement or partial improvement or 2) was marked by intolerance. Poor outcomes due to past medication intolerance or an aborted trial often are commingled with cases of true lack of improvement after an adequate treatment trial.
aSee “Staging psychiatric disorders: A clinico-biologic model,” Current Psychiatry, May 2013, at CurrentPsychiatry.com.
It is useful, therefore, to define terminology precisely when describing “treatment-resistant depression” and “treatment-refractory depression.” True past nonresponse to appropriate treatment often carries prognostic importance and bears on future treatment decisions.
Few interventions are FDA approved for treatment-resistant depression (Table).
Neuromodulation techniques are attracting interest in this area, although repetitive transcranial magnetic stimulation appears inferior to electroconvulsive therapy (ECT) for this indication.4
Melancholic depression
Melancholia involves the cardinal symptoms of anhedonia and lack of mood reactivity, alongside such features as distinct quality of mood, diurnal variation, excessive guilt, and severe weight loss. It most closely approximates pre-DSM-III “endogenous depression” and can involve 1) greater genetic loading5 and 2) structural and functional abnormalities in frontostriatal pathways.6,7
Melancholic features do not necessarily recur across successive episodes8 but carry an increased risk of psychosis9 and high-lethality suicidal behavior.10 Melancholia implies necessity for pharmacotherapy or ECT rather than psychosocial treatment alone; some researchers have suggested that tricyclic antidepressants (TCAs) might yield better results than selective serotonin reuptake inhibitors (SSRIs).11
Agitated depression
The Research Diagnostic Criteria (a forerunner in the 1970s to DSM-III) described agitated depression, but the disorder was not included in any DSM editions—although it is a “clinical modification” for MDD in the 10th revision of the International Statistical Classification of Diseases and Related Health Problems.
Agitated depression refers to a major depressive episode involving motor or psychic agitation, intense inner tension, and racing or “crowded” thoughts. Some experts believe that it represents a variant of psychotic depression or a bipolar mixed state, but the construct does not specify that criteria for a full manic or hypomanic episode exist.
Recovery from agitated depression tends to be slower than in non-agitated depression. Treatment usually entails an antidepressant plus an antipsychotic, although some believe that antidepressants can exacerbate, not alleviate, symptoms and, instead, favor antipsychotics, mood stabilizers, or ECT.12
Anxious depression
Anxiety symptoms or syndromes occur in at least one-half of outpatients who have major depression, and might account for a substantial percentage of nonresponse to first-line antidepressant therapies.13 The construct of a mixed anxiety−depressive disorder is, in fact, well-represented in the literature, particularly in primary care medicine, but its poor inter-rater reliability in DSM-5 field trials led to its exclusion there as a formal diagnosis.14
Serotonergic antidepressants remain the mainstay of treatment for depression with anxiety, although (contrary to popular perception) bupropion exerts an anxiolytic effect that is comparable to the effect of SSRIs.15 Notably, high somatic anxiety during depression might predict a poor outcome from ECT.16
Atypical depression
Often closely linked with early onset and chronicity, the construct of atypical depression has been defined in the literature by the symptom constellation of:
• mood reactiveness to environmental circumstances (unlike melancholia)
• heightened interpersonal sensitivity
• hypersomnia
• hyperphagia
• profound fatigue or a sense of physical heaviness.
Some authorities regard atypical features as being especially common in bipolar depression, or in depression among people who have borderline personality disorder.
Particular interest in this construct grew from studies that suggested that atypical depression is more responsive to a monoamine oxidase inhibitor (MAOI) than to a TCA, but also that SSRIs are not clearly superior to MAOIs.17 Response to ECT might also be better in atypical than in typical depression.18
Depression with a substance use disorder
Although not a distinct diagnostic entity, depression with a coexisting substance use disorder poses special challenges with regard to the source of symptom emergence (that is, when does depression lead to drug or alcohol use to “self medicate,” and when does drug use cause depression?) and treatment. Debate continues about whether 1) medicines that treat depression are effective and worthwhile in the setting of active substance use or 2) aggressive treatment of substance misuse is a prerequisite for subsequent pharmacotherapy for depression that is “uncontaminated” by the psychotoxic effects of concurrent substances of abuse.
Meta-analysis of controlled trials of antidepressants for patients who have MDD or a dysthymic disorder plus a comorbid alcohol use disorder found that antidepressants were, overall, superior to placebo unless a patient is actively drinking.19 Of the various classes of antidepressants, TCAs and nefazodone were found to be superior to placebo but, surprisingly, SSRIs were not. Another meta-analysis of adjunctive antidepressant outcomes for opiate-dependent, depressed patients who are receiving methadone maintenance therapy found no difference between antidepressants and placebo in their effect on depression symptom outcomes.20
Premenstrual dysphoric disorder
A new category in DSM-5, premenstrual dysphoric disorder (PMDD) represents a variant of premenstrual syndrome that arises during the luteal phase and ends with menstruation. Symptoms include several of those identified with MDD (without duration criteria), as well as mood swings, panic attacks, and physical complaints.
SSRIs—but not bupropion21 or TCAs22— and, sometimes, low-estrogen oral contraceptives are mainstays of treatment; so is cognitive-behavioral therapy, as well as lifestyle modifications (eg, exercise and changes to diet). Phototherapy has not shown robust efficacy for PMDD.23
Secondary depression
In DSM-5, depressive episodes that arise secondary to a general medical condition (eg, hypothyroidism and other endocrinopathies, cerebrovascular accidents, malignancies) or iatrogenically from medications (eg, corticosteroids, some anticonvulsants, interinterferon) are viewed as distinct from MDD in regard to risk of recurrence, genetic underpinnings, and possible neurodegenerative pathophysiology.b Unlike MDD, patient-specific risk factors are poorly defined for anticipating that a secondary depression is more or less likely to develop in the context of an exogenous substance or medical illness.
bFor further discussion, see “Is a medical illness causing your patient’s depression? Current Psychiatry, August 2009, at CurrentPsychiatry.com.
Treating secondary depression involves addressing the underlying condition and might include antidepressant medication.
Seasonal affective disorder
DSM-5 identifies “with seasonal pattern” as a specifier for recurrent major depression. Phototherapy remains a standard treatment, although a Cochrane Review identified comparable outcomes with fluoxetine, but inconclusive data for other, newer antidepressants.24 Small open trials have suggested that MAOIs and TCAs can be efficacious.
Note: Phototherapy lacks demonstrated efficacy in non-seasonal forms of depression.25
What does the future hold for classifying depressive disorders?
Recent initiatives have attempted to classify depression less by traditional clinical signs and more by focusing on possible underlying neurobiological substrates.c In the future, subtyping of mood disorders might focus on such constructs as:
• positive and negative valence systems and attentional domains
• treatment-responsivity relative to genotypic variants (for example, the serotonin transporter gene locus [SLC6A4] or prediction of L-methylfolate-responsive depression based on the genotype of the methylenetetrahydrofolate reductase [MTHFR] polymorphism)
• disrupted neural plasticity in brain circuits believed to regulate emotion.
cAn example is the Research Domain Criteria [RDoC],www. nimh.nih.gov/research-priorities/rdoc/index.shtml.
Until robust biomarkers for depression are identified and validated, however, such advances in nosology remain experimental and speculative.
BOTTOM LINE
Depressive disorders comprise a range of conditions that can be viewed along many dimensions, including “situational,” treatment-resistant, melancholic, agitated, anxious, and atypical depression; depression occurring with a substance use disorder; premenstrual dysphoric disorder; and seasonal affective disorder, among other classifications. Clinical characteristics vary across subtypes—as do corresponding preferred treatments, which should be tailored to the needs of your patients.
Editor’s note: The first part of Dr. Goldberg’s review of depression subtypes—focusing on major and minor depression, chronicity, polarity, severity, and psychosis—appeared in the April 2014 issue.
Related Resources
• Kosinski EC, Rothschild AJ. Monoamine oxidase inhibitors: Forgotten treatment for depression. Current Psychiatry. 2012;11(12):20-26.
• Rodgers S, Grosse Holtforth M, Müller M, et al. Symptom-based subtypes of depression and their psychosocial correlates: a person-centered approach focusing on the influence of sex. J Affect Disord. 2014;156:92-103.
Drug Brand Names
Aripiprazole • Abilify Mirtazapine • Remeron
Bupropion • Wellbutrin Nefazodone • Serzone
Fluoxetine • Prozac Olanzapine/fluoxetine • Symbyax
Ketamine • Ketalar Pramipexole • Mirapex
L-Methylfolate • Deplin Quetiapine • Seroquel
Lamotrigine • Lamictal Riluzole • Rilutek
Lithium • Eskalith, Lithobid Vortioxetine • Brintellix
Methadone • Dolophine
Disclosure
Dr. Goldberg has been a consultant to Avanir Pharmaceuticals and Merck; has served on the speakers’ bureau for AstraZeneca, Merck, Novartis, Sunovion Pharmaceuticals, Takeda-Lundbeck; and has received royalties from American Psychiatric Publishing and honoraria from Medscape and WebMD.
1. Fergusson DM, Horwood LJ, Ridder EM, et al. Subthreshold depression in adolescence and mental health outcomes in adulthood. Arch Gen Psychiatry. 2005;62(1):66-72.
2. Mitchell PB, Parker GB, Gladstone GL, et al. Severity of stressful life events in first and subsequent episodes of depression: the relevance of depressive subtypes. J Affect Disord. 2003;73(3):245-252.
3. Coryell W, Winokur G, Maser JD, et al. Recurrently situational (reactive) depression: a study of course, phenomenology and familial psychopathology. J Affect Disord. 1994;31(3):203-210.
4. Slotema CW, Blom JD, Hoek HW, et al. Should we expand the toolbox of psychiatric treatment methods to include Repetitive Transcranial Magnetic Stimulation (rTMS)? A meta-analysis of the efficacy of rTMS in psychiatric disorders. J Clin Psychiatry. 2010;71(7):873-884.
5. Kendler KS. The diagnostic validity of melancholic major depression in a population-based sample of female twins. Arch Gen Psychiatry. 1997;54(4):299-304.
6. Bracht T, Horn H, Strik W, et al. White matter microstructure alterations of the medial forebrain bundle in melancholic depression. J Affect Disord. 2014;155:186-193.
7. Pizzagalli DA, Oakes TR, Fox AS, et al. Functional but not structural subgenual prefrontal cortex abnormalities in melancholia. Mol Psychiatry. 2004;9(4):325, 393-405.
8. Melartin T, Leskelä U, Rytsälä H, et al. Co-morbidity and stability of melancholic features in DSM-IV major depressive disorder. Psychol Med. 2004;34(8):1443-1452.
9. Caldieraro MA, Baeza FL, Pinheiro DO, et al. Prevalence of psychotic symptoms in those with melancholic and nonmelancholic depression. J Nerv Ment Dis. 2013;201(10):855-859.
10. Grunebaum MF, Galfalvy HC, Oquendo MA, et al. Melancholia and the probability and lethality of suicide attempts. Br J Psychiatry. 2004;184:534-535.
11. Roose SP, Glassman AH, Attia E, et al. Comparative efficacy of selective serotonin reuptake inhibitors and tricyclics in the treatment of melancholia. Am J Psychiatry. 1994;151(12):1735-1739.
12. Koukopoulos A, Sani G, Koukopoulos AE, et al. Melancholia agitata and mixed depression. Acta Psychiatr Scand Suppl. 2007;(433):50-57.
13. Fava M, Rush AJ, Alpert JE, et al. Difference in treatment outcome in outpatients with anxious versus nonanxious depression: a STAR*D report. Am J Psychiatry. 2008; 165(3):342-351.
14. Regier DA, Narrow WE, Clarke DE, et al. DSM-5 field trials in the United States and Canada, Part II: test-retest reliability of selected categorical diagnoses. Am J Psychiatry. 2013;170(1):59-70.
15. Rush AJ, Trivedi MH, Carmody TJ, et al. Response in relation to baseline anxiety levels in major depressive disorder treated with bupropion sustained release or sertraline. Neuropsychopharmacology. 2001;25(1):131-138.
16. Rasmussen KG, Snyder KA, Knapp RG, et al. Relationship between somatization and remission with ECT. Psychiatry Res. 2004;129(3):293-295.
17. Henkel V, Mergl R, Allgaier AK, et al. Treatment of depression with atypical features: a meta-analytic approach. Psychiatry Res. 2006;141(1):89-101.
18. Husain MM, McClintock SM, Rush AJ, et al. The efficacy of acute electroconvulsive therapy in atypical depression. J Clin Psychiatry. 2008;69(3):406-411.
19. Iovieno N, Tedeschini E, Bentley KH, et al. Antidepressants for major depressive disorder and dysthymic disorder in patients with comorbid alcohol use disorders: a meta-analysis of placebo-controlled randomized trials. J Clin Psychiatry. 2011;72(8):1144-1151.
20. Pedrelli P, Iovieno N, Vitali M, et al. Treatment of major depressive disorder and dysthymic disorder with antidepressants in patients with comorbid opiate use disorders enrolled in methadone maintenance therapy: a meta-analysis. J Clin Psychopharmacol. 2011;31(5):582-586.
21. Pearlstein TB, Stone AB, Lund SA, et al. Comparison of fluoxetine, bupropion, and placebo in the treatment of premenstrual dysphoric disorder. J Clin Psychopharmacol. 1997;17(4):261-266.
22. Freeman EW, Rickels K, Sondheimer SJ, et al. Differential response to antidepressants in women with premenstrual syndrome/premenstrual dysphoric disorder: a randomized controlled trial. Arch Gen Psychiatry. 1999;56(10):932-939.
23. Krasnik C, Montori VM, Guyatt GH, et al. The effect of bright light therapy on depression associated with premenstrual dysphoric disorder. Am J Obstet Gynecol. 2005;193(3, pt 1):658-661.
24. Thaler K, Delivuk M, Chapman A, et al. Second-generation antidepressants for seasonal affective disorder. Cochrane Database Syst Rev. 2011;7(12):CD008591.
25. Thalén BE, Kjellman BF, Mørkid L, et al. Light treatment in seasonal and nonseasonal depression. Acta Psychiatr Scand. 1995;91(5):352-360.
1. Fergusson DM, Horwood LJ, Ridder EM, et al. Subthreshold depression in adolescence and mental health outcomes in adulthood. Arch Gen Psychiatry. 2005;62(1):66-72.
2. Mitchell PB, Parker GB, Gladstone GL, et al. Severity of stressful life events in first and subsequent episodes of depression: the relevance of depressive subtypes. J Affect Disord. 2003;73(3):245-252.
3. Coryell W, Winokur G, Maser JD, et al. Recurrently situational (reactive) depression: a study of course, phenomenology and familial psychopathology. J Affect Disord. 1994;31(3):203-210.
4. Slotema CW, Blom JD, Hoek HW, et al. Should we expand the toolbox of psychiatric treatment methods to include Repetitive Transcranial Magnetic Stimulation (rTMS)? A meta-analysis of the efficacy of rTMS in psychiatric disorders. J Clin Psychiatry. 2010;71(7):873-884.
5. Kendler KS. The diagnostic validity of melancholic major depression in a population-based sample of female twins. Arch Gen Psychiatry. 1997;54(4):299-304.
6. Bracht T, Horn H, Strik W, et al. White matter microstructure alterations of the medial forebrain bundle in melancholic depression. J Affect Disord. 2014;155:186-193.
7. Pizzagalli DA, Oakes TR, Fox AS, et al. Functional but not structural subgenual prefrontal cortex abnormalities in melancholia. Mol Psychiatry. 2004;9(4):325, 393-405.
8. Melartin T, Leskelä U, Rytsälä H, et al. Co-morbidity and stability of melancholic features in DSM-IV major depressive disorder. Psychol Med. 2004;34(8):1443-1452.
9. Caldieraro MA, Baeza FL, Pinheiro DO, et al. Prevalence of psychotic symptoms in those with melancholic and nonmelancholic depression. J Nerv Ment Dis. 2013;201(10):855-859.
10. Grunebaum MF, Galfalvy HC, Oquendo MA, et al. Melancholia and the probability and lethality of suicide attempts. Br J Psychiatry. 2004;184:534-535.
11. Roose SP, Glassman AH, Attia E, et al. Comparative efficacy of selective serotonin reuptake inhibitors and tricyclics in the treatment of melancholia. Am J Psychiatry. 1994;151(12):1735-1739.
12. Koukopoulos A, Sani G, Koukopoulos AE, et al. Melancholia agitata and mixed depression. Acta Psychiatr Scand Suppl. 2007;(433):50-57.
13. Fava M, Rush AJ, Alpert JE, et al. Difference in treatment outcome in outpatients with anxious versus nonanxious depression: a STAR*D report. Am J Psychiatry. 2008; 165(3):342-351.
14. Regier DA, Narrow WE, Clarke DE, et al. DSM-5 field trials in the United States and Canada, Part II: test-retest reliability of selected categorical diagnoses. Am J Psychiatry. 2013;170(1):59-70.
15. Rush AJ, Trivedi MH, Carmody TJ, et al. Response in relation to baseline anxiety levels in major depressive disorder treated with bupropion sustained release or sertraline. Neuropsychopharmacology. 2001;25(1):131-138.
16. Rasmussen KG, Snyder KA, Knapp RG, et al. Relationship between somatization and remission with ECT. Psychiatry Res. 2004;129(3):293-295.
17. Henkel V, Mergl R, Allgaier AK, et al. Treatment of depression with atypical features: a meta-analytic approach. Psychiatry Res. 2006;141(1):89-101.
18. Husain MM, McClintock SM, Rush AJ, et al. The efficacy of acute electroconvulsive therapy in atypical depression. J Clin Psychiatry. 2008;69(3):406-411.
19. Iovieno N, Tedeschini E, Bentley KH, et al. Antidepressants for major depressive disorder and dysthymic disorder in patients with comorbid alcohol use disorders: a meta-analysis of placebo-controlled randomized trials. J Clin Psychiatry. 2011;72(8):1144-1151.
20. Pedrelli P, Iovieno N, Vitali M, et al. Treatment of major depressive disorder and dysthymic disorder with antidepressants in patients with comorbid opiate use disorders enrolled in methadone maintenance therapy: a meta-analysis. J Clin Psychopharmacol. 2011;31(5):582-586.
21. Pearlstein TB, Stone AB, Lund SA, et al. Comparison of fluoxetine, bupropion, and placebo in the treatment of premenstrual dysphoric disorder. J Clin Psychopharmacol. 1997;17(4):261-266.
22. Freeman EW, Rickels K, Sondheimer SJ, et al. Differential response to antidepressants in women with premenstrual syndrome/premenstrual dysphoric disorder: a randomized controlled trial. Arch Gen Psychiatry. 1999;56(10):932-939.
23. Krasnik C, Montori VM, Guyatt GH, et al. The effect of bright light therapy on depression associated with premenstrual dysphoric disorder. Am J Obstet Gynecol. 2005;193(3, pt 1):658-661.
24. Thaler K, Delivuk M, Chapman A, et al. Second-generation antidepressants for seasonal affective disorder. Cochrane Database Syst Rev. 2011;7(12):CD008591.
25. Thalén BE, Kjellman BF, Mørkid L, et al. Light treatment in seasonal and nonseasonal depression. Acta Psychiatr Scand. 1995;91(5):352-360.
